KR20140131677A - 설포닐 화합물을 이용한 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물의 제조방법 - Google Patents
설포닐 화합물을 이용한 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물의 제조방법 Download PDFInfo
- Publication number
- KR20140131677A KR20140131677A KR20130050545A KR20130050545A KR20140131677A KR 20140131677 A KR20140131677 A KR 20140131677A KR 20130050545 A KR20130050545 A KR 20130050545A KR 20130050545 A KR20130050545 A KR 20130050545A KR 20140131677 A KR20140131677 A KR 20140131677A
- Authority
- KR
- South Korea
- Prior art keywords
- phenylsulfonyl
- oxo
- triphenyl
- compound
- phosphanylidene
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/535—Organo-phosphoranes
- C07F9/5352—Phosphoranes containing the structure P=C-
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
Abstract
본 발명은 신규한 설포닐 화합물과 할로젠화 알킬 화합물을 사용하여 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물을 제조하는 방법에 관한 것이다. 본 발명에 따르면, α-케토 아미드 및 α-케토 에스테르 작용기를 도입하기 위한 핵심 중간체인 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물을 상업적 및 합성적으로 용이하게 입수 가능한 물질들을 사용하여 용이하게 고수율로 제조할 수 있다.
Description
본 발명은 설포닐 화합물을 이용한 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물의 제조방법에 관한 것이다. 보다 구체적으로, 본 발명은 신규한 설포닐 화합물과 할로젠화 알킬 화합물을 출발물질로 사용하여 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물을 제조하는 방법, 이를 위한 신규한 설포닐 화합물인 3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴 및 이의 제조방법에 관한 것이다.
α-케토 아미드(α-keto amide) 및 α-케토 에스테르(α-keto ester) 작용기는 생리활성이 있는 많은 천연물질에서 종종 발견되고 있는 약리 작용과 직결되는 중요한 작용기로서, 유기합성 분야에서도 자주 반응의 중간체로 활용되기도 하며, 더욱이 최근에는 합성 펩티드 화합물에 약리 작용과 관련하여 인위적으로 도입되고 있는 등 유기합성 및 약학 분야에서 많은 관심을 끌고 있는 중요한 전자 결핍성 친전자성 작용기이다 [참고문헌: Otto, H. H.; Schirmeister, T. Chem. Rev. 1997, 97, 133; Babine, R. E.; Bender, S. L. Chem. Rev. 1997, 97, 1359; Cooper, A. J. E.; Ginos, J. Z.; Meister, A. Chem. Rev. 1983, 83, 321]. 따라서, α-케토 아미드 및 α-케토 에스테르 작용기를 도입하는 많은 합성법들이 문헌에 보고되어 있는데, 특히 미국 특허 제5,834,588호에는 하기 반응식 1에서와 같이, α-케토 (시아노메틸렌)트리페닐포스포레인 (α-Keto (cyanomethylene)triphenylphosphoranes, 7)을 핵심 중간체로 활용하여 α-케토 아미드 및 α-케토 에스테르 작용기를 도입하는 합성법이 보고되어 있다 [참고문헌: Wasserman, H. H.; Ho, W.-B. J. Org. Chem. 1994, 59, 4364-4366].
[반응식 1]

상기 합성법은 온화한 반응조건, 우수한 수렴성 및 넓은 적용 범위를 가지는 등, α-케토 아미드 및 α-케토 에스테르 작용기를 도입할 수 있는 매우 우수한 방법이라 할 수 있다. 그러나, 상기 합성법에서 필수적인 핵심 중간체인 α-케토 (시아노메틸렌)트리페닐포스포레인 (7)의 합성에는 카르복실산 또는 카르복실산에서 유도된 염화 아실 화합물 (acyl chlorides)이 반드시 필요한데, 카르복실산은 종종 상업적으로 구입하기 불가능한 경우가 있으며, 또한 염화 아실 화합물은 일반적으로 반응성이 매우 큰 시약으로 쉽게 가수분해되는 등 제조 및 사용에 큰 어려움이 있다.
이러한 문제점을 극복하기 위하여 본 발명자는 하기 반응식 2에서와 같이 새로운 호르너-워스워스-에몬스 위티히 시약 (Horner-Wadsworth-Emmons Wittig Reagent)인 디에틸 [3-시아노-2-옥소-3-(트리페닐포스포라닐리덴)프로필]포스포네이트 (9)를 사용하여 카르보닐 화합물 (알데히드 또는 케톤, 10)으로부터 상기한 와서맨(Wasserman) 합성법의 핵심 중간체인 α-케토 (시아노메틸렌)트리페닐포스포레인 (11, 12)을 합성할 수 있는 새로운 제조방법을 개발한 바 있다 [참고문헌: Kieseung Lee, Bull. Korean Chem. Soc., 2007, 28, 1641-1642; 대한민국 특허 제10-0911357호].
[반응식 2]
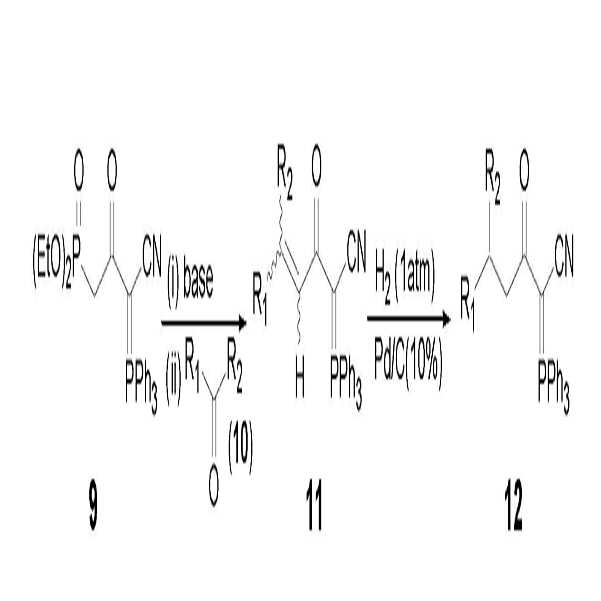
또한, 본 발명자는 하기 반응식 3에서와 같이 신규한 설피닐 화합물 (13)과 상업적 구입 및 제조가 용이한 다양한 형태의 브롬화 알킬 화합물 (alkyl bromides, 4)을 출발 물질로 사용하여 다양한 α-케토 (시아노메틸렌)트리페닐포스포레인 (14, 15)을 효과적으로 합성할 수 있는 새로운 제조방법을 개발한 바 있다 [참고문헌: Kieseung Lee, Bull. Korean Chem. Soc., 2009, 30, 2521-2522; 대한민국 특허 제10-1085202호].
[반응식 3]

상기한 카르보닐 화합물 또는 브롬화 알킬 화합물에서 α-케토 (시아노메틸렌)트리페닐포스포레인을 제조할 수 있는 방법이 상기 와서맨(Wasserman) 합성법의 한계점을 극복하고 적용 범위를 넓히고는 있지만, β,γ-포화 α-케토 (시아노메틸렌)트리페닐포스포레인 (β,γ-saturated α-keto (cyanomethylene)triphenylphosphoranes, 12, 15)을 제조하고자 하는 경우 β,γ-불포화 α-케토 (시아노메틸렌)트리페닐포스포레인 (β,γ-unsaturated α-keto (cyanomethylene)triphenylphosphoranes, 11, 14)을 합성한 다음, 수소화 반응을 진행하여야 하므로 공정이 길어지고 수율 및 순도가 감소되는 문제점이 있다.
따라서, 상업적으로 염가에 구입할 수 있고 동시에 용이하게 제조할 수 있는 화합물을 출발물질로 사용하면서 수소화 공정을 거치지 않고 β,γ-포화 α-케토 (시아노메틸렌)트리페닐포스포레인을 제조할 수 있는 새로운 합성법의 개발이 절실히 요구되어 왔다.
본 발명자는 β,γ-포화 α-케토 (시아노메틸렌)트리페닐포스포레인을 수소화 공정 없이 보다 효과적으로 제조할 수 있는 방법을 개발하기 위해 예의 연구 검토한 결과, 신규한 설포닐 화합물과 상업적 구입 및 제조가 용이한 다양한 형태의 할로젠화 알킬 화합물 (alkyl halides)을 출발 물질로 사용하여 다양한 β,γ-포화 α-케토 (시아노메틸렌)트리페닐포스포레인을 수소화 공정 없이 효과적으로 제조할 수 있음을 알아내고 본 발명을 완성하게 되었다.
따라서, 본 발명의 목적은 신규한 설포닐 화합물과 할로젠화 알킬 화합물을 사용하여 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물을 제조하는 방법을 제공하는 것이다.
본 발명의 다른 목적은 α-케토 (시아노메틸렌)트리페닐포스포레인의 제조를 위한 신규한 설포닐 화합물을 제공하는 것이다.
본 발명의 또 다른 목적은 상기 설포닐 화합물의 제조방법을 제공하는 것이다.
본 발명의 일 실시형태는 신규한 설포닐 화합물과 할로젠화 알킬 화합물을 이용한 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물의 제조방법에 관한 것으로서, 본 발명의 제조방법은
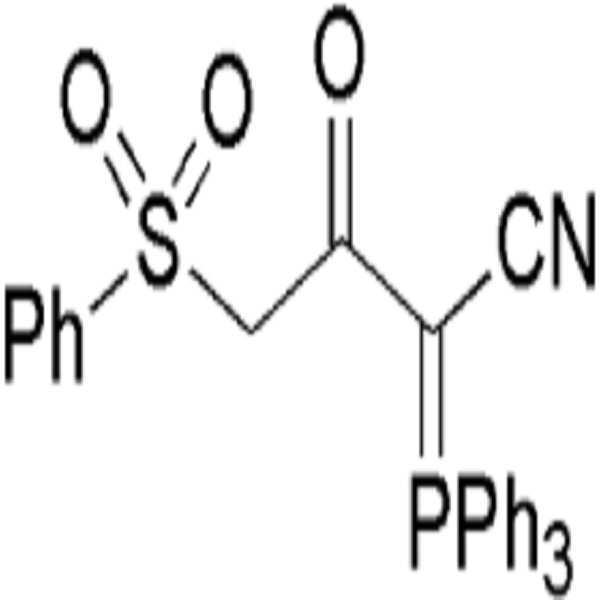
(a) 하기 화학식 I의 3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴을 하기 화학식 IV의 할로젠화 알킬 화합물로 알킬화 반응시켜 하기 화학식 V의 알킬화된 설포닐 화합물을 수득하는 단계; 및
(b) 하기 화학식 V의 알킬화된 설포닐 화합물을 환원적 탈설포닐화 반응시켜 하기 화학식 VI의 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물을 수득하는 단계를 포함한다.
[화학식 I]
[화학식 IV]
[화학식 V]

[화학식 VI]
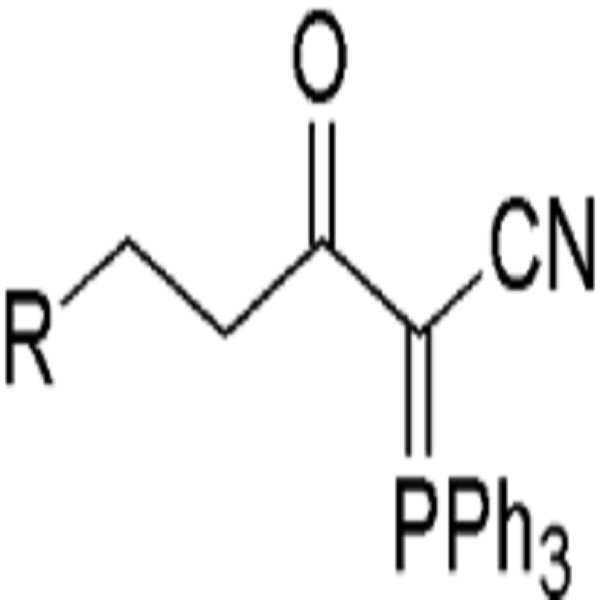
상기 식에서,
R은 C1-C10의 알킬기, C2-C10의 알케닐기 또는 아릴기이고,
X는 할로젠이다.
본 명세서에서 사용되는 C1-C10의 알킬기는 탄소수 1 내지 10개로 구성된 직쇄형 또는 분지형 탄화수소를 의미하며, 예를 들어 메틸, 에틸, n-프로필, i-프로필, n-부틸, n-펜틸, n-헥실, n-헵틸 등이 포함되나 이에 한정되는 것은 아니다.
본 명세서에서 사용되는 C2-C10의 알케닐기는 하나 이상의 탄소-탄소 이중결합을 갖는 탄소수 2 내지 10개로 구성된 직쇄형 또는 분지형 불포화 탄화수소를 의미하며, 예를 들어 에틸렌일, 프로펜일, 부텐일 등이 포함되나 이에 한정되는 것은 아니다.
본 명세서에서 사용되는 아릴기는 아로메틱기와 헤테로아로메틱기 및 그들의 부분적으로 환원된 유도체를 모두 포함한다. 상기 아로메틱기는 5 내지 15각형으로 이루어진 단순 또는 융합 고리형이며, 헤테로아로메틱기는 산소, 황 또는 질소를 하나 이상 포함하는 아로메틱기를 의미한다. 대표적인 아릴기의 예로는 페닐, 나프틸, 피리디닐(pyridinyl), 푸라닐(furanyl), 티오페닐(thiophenyl), 인돌릴(indolyl), 퀴놀리닐(quinolinyl), 이미다졸리닐(imidazolinyl), 옥사졸릴(oxazolyl), 티아졸릴(thiazolyl), 테트라히드로나프틸 등이 있으나 이에 한정되는 것은 아니다.
상기 C1-C10의 알킬기, C2-C10의 알케닐기 및 아릴기는 한 개 또는 그 이상의 수소가 C1-C5의 알킬기, C2-C6의 알케닐기, C2-C6의 알키닐기, C3-C10의 시클로알킬기, C3-C10의 헤테로시클로알킬기, C3-C10의 헤테로시클로알킬옥시, C1-C5의 할로알킬기, C1-C5의 알콕시기, C1-C5의 티오알콕시기, 아릴기, 아실기, 히드록시, 티오(thio), 할로젠, 아미노, 알콕시카보닐, 카복시, 카바모일, 시아노, 니트로 등으로 치환될 수 있다.
본 발명의 일 실시형태에서, R은 페닐, C3-C10의 헤테로시클로알킬기 및 C3-C10의 헤테로시클로알킬옥시로 구성된 군으로부터 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않은 C1-C10의 알킬기 또는 C2-C10의 알케닐기; C1-C5의 알킬기로 치환되거나 치환되지 않은 페닐; 또는 티오페닐이다. R의 예로는 페닐, 2-메틸페닐, n-헵틸, 2-페닐에틸, 2-페닐에텐일, 2-티오페닐, (테트라히드로-2H-피란-2-일옥시)메틸, (1,3-디옥산-2-일)메틸 등이 있으나 이에 한정되는 것은 아니다.
본 발명의 일 실시형태에서, X는 염소, 브롬 또는 요오드이다.
이하, 본 발명의 제조방법을 하기 반응식 4를 참조로 보다 상세히 설명하고자 한다. 하기에 기재된 방법은 대표적으로 사용된 방법을 예시한 것일 뿐 반응시약, 반응조건 등은 경우에 따라 얼마든지 변경될 수 있다.
[반응식 4]

알킬화된 설포닐 화합물 (5)은 3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴 (1)을 할로젠화 알킬 화합물 (4)로 알킬화 반응시켜 수득한다. 구체적으로 3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴 (1)을 온화한 반응 조건에서 염기로 처리하여 이놀레이트형 설포닐 음이온을 형성하고, 이를 할로젠화 알킬 화합물 (4)과 반응시켜 안정한 형태의 알킬화된 설포닐 화합물 (5)를 수득한다. 이때 사용되는 염기로는 수소화나트륨(NaH), 탄산칼륨(K2CO3) 등이 있으나 이에 한정되는 것은 아니다. 상기 염기의 사용량은 1.0 내지 3.0 당량이 바람직하다. 반응 용매로는 디메틸포름아미드 (DMF) 또는 테트라히드로퓨란 (THF)을 사용하는 것이 바람직하다.
수득한 알킬화된 설포닐 화합물 (5)을 온화한 반응 조건에서 환원적 탈설포닐화 반응 (reductive desulfonylation)시키면 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물 (6)을 고수율로 수득할 수 있다. 이때 환원적 탈설포닐화 반응은 나트륨 아말감 (Na(Hg)) 및 인산수소이나트륨(Na2HPO4)의 존재 하에 수행하는 것이 바람직하다. 상기 나트륨 아말감 (Na(Hg)) 및 인산수소이나트륨(Na2HPO4)의 사용량은 3.0 내지 5.0 당량이 바람직하다. 반응 용매로는 메탄올 (MeOH) 또는 메탄올과 디메틸포름아미드 (DMF)의 혼합용매를 사용하는 것이 바람직하다.
본 발명의 또 다른 실시형태는 신규한 설포닐 화합물인 하기 화학식 I의 3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴에 관한 것이다.
[화학식 I]

본 발명의 설포닐 화합물은 상기 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물을 제조하기 위한 출발물질로 유용하게 사용될 수 있다.
본 발명의 또 다른 실시형태는 상기 화학식 I의 3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴의 제조방법에 관한 것으로, 본 발명의 제조방법은 하기 화학식 II의 페닐설포닐 아세트산과 하기 화학식 III의 (트리페닐포스포라닐리덴)아세토니트릴을 축합 반응시키는 단계를 포함한다.
[화학식 II]
[화학식 III]
이하, 상기 설포닐 화합물의 제조방법을 하기 반응식 5를 참조로 보다 상세히 설명하고자 한다. 하기에 기재된 방법은 대표적으로 사용된 방법을 예시한 것일 뿐 반응시약, 반응조건 등은 경우에 따라 얼마든지 변경될 수 있다.
[반응식 5]

3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴 (1)은 페닐설포닐 아세트산 (2)과 (트리페닐포스포라닐리덴)아세토니트릴 (3)을 1-[3-(디메틸아미노)프로필]-3-에틸카보디이마이드·HCl(EDC) 및 4-디메틸아미노피리딘(DMAP)을 사용하여 축합 반응시켜 제조할 수 있다.
상기 페닐설포닐 아세트산 (2)과 (트리페닐포스포라닐리덴)아세토니트릴 (3)은 상업적 구입이 가능하다.
본 발명의 제조방법에 따르면, α-케토 아미드 및 α-케토 에스테르 작용기를 도입하기 위한 핵심 중간체인 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물을 신규한 설포닐 화합물인 3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴과 상업적 구입 및 제조가 용이한 할로젠화 알킬 화합물로부터 수소화 공정 없이 용이하게 고수율로 제조할 수 있다.
아울러, 신규한 설포닐 화합물인 3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴도 상업적 구입 및 제조가 용이한 시약들을 사용하여 용이하게 고수율로 제조할 수 있다.
이하, 실시예에 의해 본 발명을 보다 구체적으로 설명하고자 한다. 이들 실시예는 오직 본 발명을 설명하기 위한 것으로 본 발명의 범위가 이들 실시예에 국한되지 않는다는 것은 당업자에게 있어서 자명하다.
실시예 1: 3-옥소-4-(페닐설포닐)-2-(트리페닐-λ
5
-포스파닐리덴)부탄니트릴 (3-Oxo-4-(phenylsulfonyl)-2-(triphenyl-λ
5
-phosphanylidene)butanenitrile, 1)의 제조
무수 CH2Cl2 (30 mL)에 페닐설포닐 아세트산 (1.42 g, 7.10 mmol)과 (트리페닐포스포라닐리덴)아세토니트릴 (2.14 g, 1.0 당량)을 녹인 후, 생성된 용액을 0 oC로 냉각한 다음, 1-[3-(디메틸아미노)프로필]-3-에틸카보디이마이드·HCl (1.36 g, 1.0 당량)와 4-디메틸아미노피리딘 (86.7 mg, 0.1 당량)을 첨가하고 0 oC에서 1시간, 상온에서 12시간 동안 아르곤 기체 하에서 교반하였다. 반응종결 후 반응 용액에 물 (20 mL)을 가하여 흔들어 준 후, 분액 깔대기로 하부 유기층은 분리하고 상부 수용액층을 CH2Cl2 (10 mL)로 2회 더 추출하였다. 분리된 유기층을 전부 합한 다음 무수 황산마그네슘 (5.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 용매를 제거하여 얻어진 고체 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 5/1)로 분리하여 표제 화합물 (1, 2.95 g, 86%)을 흰색 고체 상태로 수득하였다.
녹는점 (mp) 232-235 oC;
IR (KBr) 2174, 1597, 1307, 1147 cm-1;
1H NMR (CDCl3, 400 MHz) δ 4.47 (s, 2H), 7.42-7.92 (m, 20H).
실시예 2: 3-옥소-5-페닐-4-(페닐설포닐)-2-(트리페닐-λ
5
-포스파닐리덴)펜탄니트릴 (3-Oxo-5-phenyl-4-(phenylsulfonyl)-2-(triphenyl-λ
5
-phosphanylidene)pentanenitrile, 5a)의 제조
3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴 (1, 120.9 mg, 0.25 mmol)을 함유한 무수 DMF (3 mL) 용액에 NaH (13.0 mg, 60% in mineral oil, 1.3 당량)을 가하고 아르곤 기체 속에서 상온에서 20분, 0 oC 에서 20분 동안 교반하였다. 이 반응 용액에 벤질 클로라이드 (37.4 mL, 1.3 당량)를 가하고 아르곤 기체 하에서 0 oC 에서 1시간, 상온에서 3시간 동안 교반한 다음, CH2Cl2 (20 mL)와 물 (10 mL)을 순차적으로 가하고 다시 5분 동안 교반하였다. 분액 깔대기를 이용하여 유기층은 분리하고 수용액층을 CH2Cl2 (10 mL)로 2회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 용매를 제거하였다. 증류되지 않은 잔류 DMF를 다시 감압 하에서 진공 증류하여 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 20/1)로 정제하여 흰색 고체 상태의 순수한 표제 화합물 (5a, 107.7 mg, 75%)을 수득하였다.
녹는점 263-265 oC;
IR (KBr) 1128, 1300, 1596, 2177 cm-1;
1H NMR (CDCl3, 400 MHz) δ 3.15 (bt, 1H, J = 12.7 Hz), 3.29 (dd, 1H, J 1 = 13.4 Hz, J 2 = 3.1 Hz), 5.22 (dd, 1H, J 1 = 12.7 Hz, J 2 = 3.1 Hz), 7.15-7.82 (m, 25H).
실시예 3: 3-옥소-5-페닐-4-(페닐설포닐)-2-(트리페닐-λ
5
-포스파닐리덴)펜탄니트릴 (3-Oxo-5-phenyl-4-(phenylsulfonyl)-2-(triphenyl-λ
5
-phosphanylidene)pentanenitrile, 5a)의 제조
3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴 (1, 120.9 mg, 0.25 mmol)을 함유한 무수 DMF (3 mL) 용액에 NaH (13.0 mg, 60% in mineral oil, 1.3 당량)을 가하고 아르곤 기체 속에서 상온에서 20분, 0 oC 에서 20분 동안 교반하였다. 이 반응 용액에 벤질 브로마이드 (38.6 mL, 1.3 당량)를 가하고 아르곤 기체 하에서 0 oC 에서 1시간, 상온에서 3시간 동안 교반한 다음, CH2Cl2 (20 mL)와 물 (10 mL)을 순차적으로 가하고 다시 5분 동안 교반하였다. 분액 깔대기를 이용하여 유기층은 분리하고 수용액층을 CH2Cl2 (10 mL)로 2회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 용매를 제거하였다. 증류되지 않은 잔류 DMF를 다시 감압 하에서 진공 증류하여 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 20/1)로 정제하여 흰색 고체 상태의 순수한 표제 화합물 (5a, 126.5 mg, 88%)을 수득하였다.
실시예 4: 3-옥소-5-페닐-4-(페닐설포닐)-2-(트리페닐-λ
5
-포스파닐리덴)펜탄니트릴 (3-Oxo-5-phenyl-4-(phenylsulfonyl)-2-(triphenyl-λ
5
-phosphanylidene)pentanenitrile, 5a)의 제조
3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴 (1, 120.9 mg, 0.25 mmol)을 함유한 무수 DMF (3 mL) 용액에 K2CO3 (103.7 mg, 3.0 당량)와 벤질 브로마이드 (59.4 mL, 2.0 당량)를 가하고 아르곤 기체 하에서 상온에서 24시간 동안 교반한 다음, CH2Cl2 (20 mL)와 물 (10 mL)을 순차적으로 가하고 다시 5분 동안 교반하였다. 분액 깔대기를 이용하여 유기층은 분리하고 수용액층을 CH2Cl2 (10 mL)로 2회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 용매를 제거하였다. 증류되지 않은 잔류 DMF를 다시 감압 하에서 진공 증류하여 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 20/1)로 정제하여 흰색 고체 상태의 순수한 표제 화합물 (5a, 123.6 mg, 86%)을 수득하였다.
실시예 5: 5-(2-메틸페닐)-3-옥소-4-(페닐설포닐)-2-(트리페닐-λ
5
-포스파닐리덴)펜탄니트릴 (5-(2-Methylphenyl)-3-oxo-4-(phenylsulfonyl)-2-(triphenyl-λ
5
-phosphanylidene)pentanenitrile, 5b)의 제조
3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴 (1, 120.9 mg, 0.25 mmol)을 함유한 무수 DMF (3 mL) 용액에 NaH (13.0 mg, 60% in mineral oil, 1.3 당량)을 가하고 아르곤 기체 하에서 상온에서 20분, 0 oC 에서 20분 동안 교반하였다. 이 반응 용액에 2-메틸벤질 브로마이드 (43.4 mL, 1.3 당량)를 가하고 아르곤 기체 하에서 0 oC 에서 1시간, 상온에서 3시간 동안 교반한 다음, CH2Cl2 (20 mL)와 물 (10 mL)을 순차적으로 가하고 다시 5분 동안 교반하였다. 분액 깔대기를 이용하여 유기층은 분리하고 수용액층을 CH2Cl2 (10 mL)로 2회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 용매를 제거하였다. 증류되지 않은 잔류 DMF를 다시 감압 하에서 진공 증류하여 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 20/1)로 정제하여 흰색 고체 상태의 순수한 표제 화합물 (5b, 120.9 mg, 82%)을 수득하였다.
녹는점 224-225 oC;
IR (KBr) 1126, 1306, 1591, 2177 cm-1;
1H NMR (CDCl3, 400 MHz) δ 2.18 (s, 3H), 3.19 (dd, 1H, J 1 = 13.4 Hz, J 2 = 2.9 Hz), 3.27 (bt, 1H, J = 12.4 Hz), 5.18 (dd, 1H, J 1 = 12.0 Hz, J 2 = 2.9 Hz), 7.03-7.91 (m, 24H).
실시예 6: 3-옥소-4-(페닐설포닐)-2-(트리페닐-λ
5
-포스파닐리덴)도데칸니트릴 (3-Oxo-4-(phenylsulfonyl)-2-(triphenyl-λ
5
-phosphanylidene)dodecanenitrile, 5c)의 제조
3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴 (1, 120.9 mg, 0.25 mmol)을 함유한 무수 DMF (3 mL) 용액에 NaH (13.0 mg, 60% in mineral oil, 1.3 당량)을 가하고 아르곤 기체 속에서 상온에서 20분, 0 oC 에서 20분 동안 교반하였다. 이 반응 용액에 1-브로모옥탄 (56.5 mL, 1.3 당량)를 가하고 아르곤 기체 속에서 0 oC에서 1시간, 상온에서 3시간 동안 교반한 후, CH2Cl2 (20 mL)와 물 (10 mL)을 순차적으로 가하고 다시 5분 동안 교반하였다. 분액 깔대기를 이용하여 유기층은 분리하고 수용액층을 CH2Cl2 (10 mL)로 2회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 용매를 제거하였다. 증류되지 않은 잔류 DMF를 다시 감압 하에서 진공 증류하여 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 15/1)로 정제하여 고체 상태의 순수한 표제 화합물 (5c, 126.4 mg, 85%)을 수득하였다.
녹는점 138-140 oC;
IR (KBr) 1132, 1306, 1592, 2178 cm-1;
1H NMR (CDCl3, 400 MHz) δ 0.87 (t, 3H, J = 6.3 Hz), 1.15-1.39 (m, 12H), 1.77-1.99 (m, 2H), 4.81 (dd, 1H, J 1 = 11.0 Hz, J 2 = 3.2 Hz), 7.36-7.78 (m, 20H).
실시예 7: 3-옥소-7-페닐-4-(페닐설포닐)-2-(트리페닐-λ
5
-포스파닐리덴)헵탄니트릴 (3-Oxo-7-phenyl-4-(phenylsulfonyl)-2-(triphenyl-λ
5
-phosphanylidene)heptanenitrile, 5d)의 제조
3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴 (1, 120.9 mg, 0.25 mmol)을 함유한 무수 DMF (3 mL) 용액에 NaH (13.0 mg, 60% in mineral oil, 1.3 당량)을 가하고 아르곤 기체 속에서 상온에서 20분, 0 oC 에서 20분 동안 교반하였다. 이 반응 용액에 1-브로모-3-페닐프로판 (49.4 mL, 1.3 당량)를 가하고 아르곤 기체 속에서 0 oC에서 1시간, 상온에서 3시간 동안 교반한 후, CH2Cl2 (20 mL)와 물 (10 mL)을 순차적으로 가하고 다시 5분 동안 교반하였다. 분액 깔대기를 이용하여 유기층은 분리하고 수용액층을 CH2Cl2 (10 mL)로 2회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 용매를 제거하였다. 증류되지 않은 잔류 DMF를 다시 감압 하에서 진공 증류하여 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 15/1)로 정제하여 흰색 고체 상태의 순수한 표제 화합물 (5d, 129.1 mg, 86%)을 수득하였다.
녹는점 183-185 oC;
IR (KBr) 1146, 1306, 1593, 2177 cm-1;
1H NMR (CDCl3, 400 MHz) δ 1.50-1.73 (m, 2H), 1.82-2.09 (m, 2H), 2.47-2.68 (m, 2H), 4.86 (dd, 1H, J 1 = 11.2 Hz, J 2 = 2.9 Hz), 7.02-7.82 (m, 25H).
실시예 8: (
6E
)-3-옥소-7-페닐-4-(페닐설포닐)-2-(트리페닐-λ
5
-포스파닐리덴)헵트-6-엔니트릴 ((
6E
)-3-Oxo-7-phenyl-4-(phenylsulfonyl)-2-(triphenyl-λ
5
-phosphanylidene)hept-6-enenitrile, 5e)의 제조
3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴 (1, 120.9 mg, 0.25 mmol)을 함유한 무수 DMF (3 mL) 용액에 NaH (13.0 mg, 60% in mineral oil, 1.3 당량)을 가하고 아르곤 기체 속에서 상온에서 20분, 0 oC 에서 20분 동안 교반하였다. 이 반응 용액에 3-브로모-1-페닐-1-프로펜 (48.1 mL, 1.3 당량)를 가하고 아르곤 기체 속에서 0 oC에서 1시간, 상온에서 3시간 동안 교반한 후, CH2Cl2 (20 mL)와 물 (10 mL)을 순차적으로 가하고 다시 5분 동안 교반하였다. 분액 깔대기를 이용하여 유기층은 분리하고 수용액층을 CH2Cl2 (10 mL)로 2회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 용매를 제거하였다. 증류되지 않은 잔류 DMF를 다시 감압 하에서 진공 증류하여 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 15/1)로 정제하여 흰색 고체 상태의 순수한 표제 화합물 (5e, 126.4 mg, 84%)을 수득하였다.
녹는점 230-232 oC;
IR (KBr) 2180, 1596, 1305, 1108 cm-1;
1H NMR (CDCl3, 400 MHz) δ 2.72-2.90 (m, 2H), 5.01 (dd, 1H, J 1 = 10.5 Hz, J 2 = 4.2 Hz), 6.07-6.18 (m, 1H), 6.42 (d, 1H, J 1 = 15.6 Hz), 7.22-7.82 (m, 25H).
실시예 9: 3-옥소-4-(페닐설포닐)-5-(티오펜-2-일)-2-(트리페닐-λ
5
-포스파닐리덴)펜탄니트릴 (3-Oxo-4-(phenylsulfonyl)-5-(thiophen-2-yl)-2-(triphenyl-λ
5
-phosphanylidene)pentanenitrile, 5f)의 제조
3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴 (1, 120.9 mg, 0.25 mmol)을 함유한 무수 DMF (3 mL) 용액에 NaH (13.0 mg, 60% in mineral oil, 1.3 당량)을 가하고 아르곤 기체 속에서 상온에서 20분, 0 oC 에서 20분 동안 교반하였다. 이 반응 용액에 2-브로모메틸티오펜 (57.5 mg, 1.3 당량)를 가하고 아르곤 기체 속에서 0 oC에서 1시간, 상온에서 3시간 동안 교반한 후, CH2Cl2 (20 mL)와 물 (10 mL)을 순차적으로 가하고 다시 5분 동안 교반하였다. 분액 깔대기를 이용하여 유기층은 분리하고 수용액층을 CH2Cl2 (10 mL)로 2회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 용매를 제거하였다. 증류되지 않은 잔류 DMF를 다시 감압 하에서 진공 증류하여 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 20/1)로 정제하여 흰색 고체 상태의 순수한 표제 화합물 (5f, 119.7 mg, 83%)을 수득하였다.
녹는점 265-267 oC;
IR (KBr) 2177, 1595, 1308, 1109 cm-1;
1H NMR (CDCl3, 400 MHz) δ 3.38 (dd, 1H, J 1 = 14.3 Hz, J 2 = 3.7 Hz), 3.47 (dd, 1H, J 1 = 14.3 Hz, J 2 = 11.7 Hz), 5.20 (dd, 1H, J 1 = 11.7 Hz, J 2 = 3.7 Hz), 6.84 (d, 1H, J = 3.4 Hz), 6.95 (dd, 1H, J 1 = 5.0 Hz, J 2 = 3.4 Hz), 7.19 (dd, 1H, J 1 = 5.0 Hz, J 2 = 1.2 Hz), 7.37-7.82 (m, 20H).
실시예 10: 3-옥소-4-(페닐설포닐)-6-(테트라히드로-2
H
-피란-2-일옥시)-2-(트리페닐-λ
5
-포스파닐리덴)헥산니트릴 (3-Oxo-4-(phenylsulfonyl)-6-(tetrahydro-2
H
-pyran-2-yloxy)-2-(triphenyl-λ
5
-phosphanylidene)hexanenitrile, 5g)의 제조
3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴 (1, 120.9 mg, 0.25 mmol)을 함유한 무수 DMF (3 mL) 용액에 NaH (13.0 mg, 60% in mineral oil, 1.3 당량)을 가하고 아르곤 기체 속에서 상온에서 20분, 0 oC 에서 20분 동안 교반하였다. 이 반응 용액에 2-(2-요오도에톡시)테트라히드로-2H-피란 (83.2 mg, 1.3 당량)을 가하고 아르곤 기체 속에서 0 oC에서 1시간, 상온에서 3시간 동안 교반한 후, CH2Cl2 (20 mL)와 물 (10 mL)을 순차적으로 가하고 다시 5분 동안 교반하였다. 분액 깔대기를 이용하여 유기층은 분리하고 수용액층을 CH2Cl2 (10 mL)로 2회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 용매를 제거하였다. 증류되지 않은 잔류 DMF를 다시 감압 하에서 진공 증류하여 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 5/1)로 정제하여 흰색 고체 상태의 순수한 표제 화합물 (5g, 121.0 mg, 79%)을 수득하였다.
녹는점 157-159 oC;
IR (KBr) 2179, 1592, 1307, 1109 cm-1;
1H NMR (CDCl3, 400 MHz) δ 1.40-1.93 (m, 6H), 2.11-2.25 (m, 2H), 3.18-3.21 (m, 0.5H), 3.36-3.51 (m, 1.5H), 3.61-3.94 (m, 2H), 4.54 (t, 0.5H, J = 3.4 Hz), 4.60 (t, 0.5H, J = 3.4 Hz), 5.02-5.13 (m, 1H), 7.37-7.72 (m, 20H).
실시예 11: 6-(1,3-디옥산-2-일)-3-옥소-4-(페닐설포닐)-2-(트리페닐-λ
5
-포스파닐리덴)헥산니트릴 (6-(1,3-Dioxan-2-yl)-3-oxo-4-(phenylsulfonyl)-2-(triphenyl-λ
5
-phosphanylidene)hexanenitrile, 5h)의 제조
3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴 (1, 120.9 mg, 0.25 mmol)을 함유한 무수 DMF (3 mL) 용액에 NaH (13.0 mg, 60% in mineral oil, 1.3 당량)을 가하고 아르곤 기체 속에서 상온에서 20분, 0 oC 에서 20분 동안 교반하였다. 이 반응 용액에 2-(2-요오도에틸)-1,3-디옥산 (78.7 mg, 1.3 당량)을 가하고 아르곤 기체 속에서 0 oC에서 1시간, 상온에서 3시간 동안 교반한 후, CH2Cl2 (20 mL)와 물 (10 mL)을 순차적으로 가하고 다시 5분 동안 교반하였다. 분액 깔대기를 이용하여 유기층은 분리하고 수용액층을 CH2Cl2 (10 mL)로 2회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 용매를 제거하였다. 증류되지 않은 잔류 DMF를 다시 감압 하에서 진공 증류하여 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 5/1)로 정제하여 흰색 거품형 고체 상태의 순수한 표제 화합물 (5h, 121.5 mg, 81%)을 수득하였다.
녹는점 82-103 oC;
IR (KBr) 2178, 1592, 1307, 1108 cm-1;
1H NMR (CDCl3, 400 MHz) δ 1.20-1.34 (m, 1H), 1.50-1.72 (m, 2H), 1.91-2.12 (m, 3H), 3.61-3.79 (m, 2H), 3.95-4.18 (m, 2H), 4.52 (t, 1H, J = 4.9 Hz), 4.90-5.03 (m, 1H), 7.36-7.82 (m, 20H).
실시예 12: 3-옥소-5-페닐-2-(트리페닐-λ
5
-포스파닐리덴)펜탄니트릴 (3-Oxo-5-phenyl-2-(triphenyl-λ
5
-phosphanylidene)pentanenitrile, 6a)의 제조
실시예 2에서 수득한 3-옥소-5-페닐-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)펜탄니트릴 (5a, 114.7 mg, 0.20 mmol)을 무수 혼합용매 (DMF: 14 mL, MeOH: 2 mL)에 녹이고 0 oC로 냉각한 후, Na2HPO4 (114.0 mg, 4.0 당량) 및 Na(Hg) (368.0 mg, 5%, 4.0 당량)을 순차적으로 가하고 아르곤 기체 속에서 0 oC에서 5시간 동안 교반하였다. 이 반응 용액에 에틸 아세테이트 (20 mL)와 물 (10 mL)를 순차적으로 가하고 5분 동안 교반한 후, 분액 깔대기로 상부 유기층을 분리하고 수용액층은 에틸 아세테이트 (10 mL)로 3회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 농축하였다. 잔류 DMF를 다시 감압 하에서 진공 증류시켜 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 20/1)로 정제하여 순수한 표제 화합물 (6a, 71.3 mg, 82%)을 흰색 고체로 수득하였다.
녹는점 170-172 oC;
IR (KBr) 2172, 1584 cm-1;
1H NMR (CDCl3, 400 MHz) δ 2.99 (t, 2H, J = 6.6 Hz), 3.05 (t, 2H, J = 6.6 Hz), 7.15-7.32 (m, 5H), 7.42-7.68 (m, 15H).
실시예 13: 5-(2-메틸페닐)-3-옥소-2-(트리페닐-λ
5
-포스파닐리덴)펜탄니트릴 (5-(2-Methylphenyl)-3-oxo-2-(triphenyl-λ
5
-phosphanylidene)pentanenitrile, 6b)의 제조
실시예 5에서 수득한 5-(2-메틸페닐)-3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)펜탄니트릴 (5b, 117.5 mg, 0.20 mmol)을 무수 혼합용매 (DMF: 14 mL, MeOH: 2 mL)에 녹이고 0 oC로 냉각한 후, Na2HPO4 (114.0 mg, 4.0 당량) 및 Na(Hg) (368.0 mg, 5%, 4.0 당량)을 순차적으로 가하고 아르곤 기체 속에서 0 oC에서 5 시간 동안 교반하였다. 이 반응 용액에 에틸 아세테이트 (20 mL)와 물 (10 mL)를 순차적으로 가하고 5분 동안 교반한 후, 분액 깔대기로 상부 유기층을 분리하고 수용액층은 에틸 아세테이트 (10 mL)로 3회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 농축하였다. 잔류 DMF를 다시 감압 하에서 진공 증류시켜 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 20/1)로 정제하여 순수한 표제 화합물 (6b, 73.3 mg, 82%)을 흰색 고체로 수득하였다.
녹는점 163-165 oC;
IR (KBr) 2170, 1579 cm-1;
1H NMR (CDCl3, 400 MHz) δ 2.34 (s, 3H), 2.98 (bs, 4H), 7.08-7.26 (m, 4H), 7.46-7.66 (m, 15H).
실시예 14: 3-옥소-2-(트리페닐-λ
5
-포스파닐리덴)도데칸니트릴 (3-Oxo-2-(triphenyl-λ
5
-phosphanylidene)dodecanenitrile, 6c)의 제조
실시예 6에서 수득한 3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)도데칸니트릴 (5c, 119.1 mg, 0.20 mmol)을 무수 메탄올 (10 mL)에 녹이고 0 oC로 냉각한 후, Na2HPO4 (114.0 mg, 4.0 당량) 및 Na(Hg) (368.0 mg, 5%, 4.0 당량)을 순차적으로 가하고 아르곤 기체 속에서 0 oC에서 5시간 동안 교반하였다. 이 반응 용액에 에틸 아세테이트 (20 mL)와 물 (10 mL)를 순차적으로 가하고 5분 동안 교반한 후, 분액 깔대기로 상부 유기층을 분리하고 수용액층은 에틸 아세테이트 (10 mL)로 3회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 농축하였다. 잔류 DMF를 다시 감압 하에서 진공 증류시켜 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 20/1)로 정제하여 순수한 표제 화합물 (6c, 80.0 mg, 88%)을 흰색 고체로 수득하였다.
녹는점 115-116 oC;
IR (KBr) 2173, 1582 cm-1;
1H NMR (CDCl3, 400 MHz) δ 0.88 (t, 3H, J = 6.8 Hz), 1.18-1.40 (m, 12H), 1.57-1.72 (m, 2H), 2.68 (t, 2H, J = 7.6 Hz), 7.47-7.67 (m, 15H).
실시예 15: 3-옥소-7-페닐-2-(트리페닐-λ
5
-포스파닐리덴)헵탄니트릴 (3-Oxo-7-phenyl-2-(triphenyl-λ
5
-phosphanylidene)heptanenitrile, 6d)의 제조
실시예 7에서 수득한 3-옥소-7-페닐-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)헵탄니트릴 (5d, 120.3 mg, 0.20 mmol)을 무수 혼합용매 (DMF: 14 mL, MeOH: 2 mL)에 녹이고 0 oC로 냉각한 후, Na2HPO4 (114.0 mg, 4.0 당량) 및 Na(Hg) (368.0 mg, 5%, 4.0 당량)을 순차적으로 가하고 아르곤 기체 속에서 0 oC에서 5시간 동안 교반하였다. 이 반응 용액에 에틸 아세테이트 (20 mL)와 물 (10 mL)를 순차적으로 가하고 5분 동안 교반한 후, 분액 깔대기로 상부 유기층을 분리하고 수용액층은 에틸 아세테이트 (10 mL)로 3회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 농축하였다. 잔류 DMF를 다시 감압 하에서 진공 증류시켜 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 20/1)로 정제하여 순수한 표제 화합물 (6d, 79.0 mg, 86%)을 흰색 고체로 수득하였다.
녹는점 124-126 oC;
IR (KBr) 2169, 1592 cm-1;
1H NMR (CDCl3, 400 MHz) δ 1.60-1.80 (m, 4H), 2.63 (t, 2H, J = 7.3 Hz), 2.73 (t, 2H, J = 7.3 Hz), 7.13-7.30 (m, 5H), 7.46-7.66 (m, 15H).
실시예 16: (
6E
)-3-옥소-7-페닐-2-(트리페닐-λ
5
-포스파닐리덴)헵트-6-엔니트릴 ((
6E
)-3-Oxo-7-phenyl-2-(triphenyl-λ
5
-phosphanylidene)hept-6-enenitrile, 6e)의 제조
실시예 8에서 수득한 (6E)-3-옥소-7-페닐-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)헵트-6-엔니트릴 (5e, 119.9 mg, 0.20 mmol)을 무수 혼합용매 (DMF: 14 mL, MeOH: 2 mL)에 녹이고 0 oC로 냉각한 후, Na2HPO4 (114.0 mg, 4.0 당량) 및 Na(Hg) (368.0 mg, 5%, 4.0 당량)을 순차적으로 가하고 아르곤 기체 속에서 0 oC에서 5시간 동안 교반하였다. 이 반응 용액에 에틸 아세테이트 (20 mL)와 물 (10 mL)를 순차적으로 가하고 5분 동안 교반한 후, 분액 깔대기로 상부 유기층을 분리하고 수용액층은 에틸 아세테이트 (10 mL)로 3회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 농축하였다. 잔류 DMF를 다시 감압 하에서 진공 증류시켜 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 20/1)로 정제하여 순수한 표제 화합물 (6e, 77.9 mg, 85%)을 흰색 고체로 수득하였다.
녹는점 154-156 oC;
IR (KBr) 2175, 1582 cm-1;
1H NMR (CDCl3, 400 MHz) δ 2.59 (bq, 2H, J = 6.8 Hz), 2.89 (t, 2H, J = 7.1 Hz), 6.30 (dt, 1H, J 1 = 16.1 Hz, J 2 = 6.8 Hz), 6.46 (d, 1H, J = 16.1 Hz), 7.17-7.66 (m, 20H).
실시예 17: 3-옥소-5-(티오펜-2-일)-2-(트리페닐-λ
5
-포스파닐리덴)펜탄니트릴 (3-Oxo-5-(thiophen-2-yl)-2-(triphenyl-λ
5
-phosphanylidene)pentanenitrile, 6f)의 제조
실시예 9에서 수득한 3-옥소-4-(페닐설포닐)-5-(티오펜-2-일)-2-(트리페닐-λ5-포스파닐리덴)펜탄니트릴 (5f, 115.9 mg, 0.20 mmol)을 무수 혼합용매 (DMF: 20 mL, MeOH: 2 mL)에 녹이고 0 oC로 냉각한 후, Na2HPO4 (114.0 mg, 4.0 당량) 및 Na(Hg) (368.0 mg, 5%, 4.0 당량)을 순차적으로 가하고 아르곤 기체 속에서 0 oC에서 5시간 동안 교반하였다. 이 반응 용액에 에틸 아세테이트 (30 mL)와 물 (20 mL)를 순차적으로 가하고 5분 동안 교반한 후, 분액 깔대기로 상부 유기층을 분리하고 수용액 층은 에틸 아세테이트 (10 mL)로 3회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 농축하였다. 잔류 DMF를 다시 감압 하에서 진공 증류시켜 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 20/1)로 정제하여 순수한 표제 화합물 (6f, 75.5 mg, 86%)을 은회색 고체로 수득하였다.
녹는점 151-153 oC;
IR (KBr) 2174, 1583 cm-1;
1H NMR (CDCl3, 400 MHz) δ 3.10 (t, 2H, J = 6.8 Hz), 3.19 (t, 2H, J = 6.8 Hz), 6.87 (bd, 1H, J = 2.9 Hz), 6.94 (dd, 1H, J 1 = 4.9 Hz, J 2 = 3.4 Hz), 7.14 (dd, 1H, J 1 = 4.9 Hz, J 2 = 1.0 Hz), 7.45-7.68 (m, 15H).
실시예 18: 3-옥소-6-(테트라히드로-2
H
-피란-2-일옥시)-2-(트리페닐-λ
5
-포스파닐리덴)헥산니트릴 (3-Oxo-6-(tetrahydro-2
H
-pyran-2-yloxy)-2-(triphenyl-λ
5
-phosphanylidene)hexanenitrile, 6g)의 제조
실시예 10에서 수득한 3-옥소-4-(페닐설포닐)-6-(테트라히드로-2H-피란-2-일옥시)-2-(트리페닐-λ5-포스파닐리덴)헥산니트릴 (5g, 122.3 mg, 0.20 mmol)을 무수 혼합용매 (DMF: 14 mL, MeOH: 2 mL)에 녹이고 0 oC로 냉각한 후, Na2HPO4 (114.0 mg, 4.0 당량) 및 Na(Hg) (368.0 mg, 5%, 4.0 당량)을 순차적으로 가하고 아르곤 기체 속에서 0 oC에서 5시간 동안 교반하였다. 이 반응 용액에 에틸 아세테이트 (20 mL)와 물 (10 mL)를 순차적으로 가하고 5분 동안 교반한 후, 분액 깔대기로 상부 유기층을 분리하고 수용액층은 에틸 아세테이트 (10 mL)로 3회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 농축하였다. 잔류 DMF를 다시 감압 하에서 진공 증류시켜 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 3/1)로 정제하여 순수한 표제 화합물 (6g, 80.5 mg, 85%)을 은회색 고체로 수득하였다.
녹는점 127-129 oC;
IR (KBr) 2171, 1595 cm-1;
1H NMR (CDCl3, 400 MHz) δ 1.45-1.75 (m, 5H), 1.77-1.89 (m, 1H), 1.91-2.02 (m, 2H), 2.80 (t, 2H, J = 7.6 Hz), 3.38-3.54 (m, 2H), 3.71-3.81 (m, 1H), 3.82-3.93 (m, 1H), 4.61 (t, 1H, J = 3.4 Hz), 7.47-7.67 (m, 15H).
실시예 19: 6-(1,3-디옥산-2-일)-3-옥소-2-(트리페닐-λ
5
-포스파닐리덴)헥산니트릴 (6-(1,3-Dioxan-2-yl)-3-oxo-2-(triphenyl-λ
5
-phosphanylidene)hexanenitrile, 6h)의 제조
실시예 11에서 수득한 6-(1,3-디옥산-2-일)-3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)헥산니트릴 (5h, 119.5 mg, 0.20 mmol)을 무수 혼합용매 (DMF: 14 mL, MeOH: 2 mL)에 녹이고 0 oC로 냉각한 후, Na2HPO4 (114.0 mg, 4.0 당량) 및 Na(Hg) (368.0 mg, 5%, 4.0 당량)을 순차적으로 가하고 아르곤 기체 속에서 0 oC에서 5시간 동안 교반하였다. 이 반응 용액에 에틸 아세테이트 (20 mL)와 물 (10 mL)를 순차적으로 가하고 5분 동안 교반한 후, 분액 깔대기로 상부 유기층을 분리하고 수용액층은 에틸 아세테이트 (10 mL)로 3회 더 추출하였다. 분리한 유기층을 전부 합하여 무수 황산마그네슘 (3.0 g)으로 처리하고 여과한 후, 회전 감압 증발기로 농축하였다. 잔류 DMF를 다시 감압 하에서 진공 증류시켜 제거하면 은회색의 고체 잔류물이 얻어지며, 이 잔류물을 속성 크로마토그래피 (실리카젤: Merck 70-230, 이동상: CH2Cl2/EtOAc = 2/1)로 정제하여 순수한 표제 화합물 (6h, 73.0 mg, 80%)을 흰색 고체로 수득하였다.
녹는점 142-144 oC;
IR (KBr) 2177, 1578 cm-1;
1H NMR (CDCl3, 400 MHz) δ 1.21-1.37 (m, 1H), 1.55-1.85 (m, 4H), 1.97-2.15 (m, 1H), 2.73 (t, 2H, J = 7.3 Hz), 3.75 (td, 2H, J 1 = 12.2 Hz, J 2 = 2.4 Hz), 4.09 (dd, 2H, J 1 = 10.7 Hz, J 2 = 4.9 Hz), 4.55 (t, 1H, J = 4.9 Hz), 7.46-7.69 (m, 15H).
Claims (10)
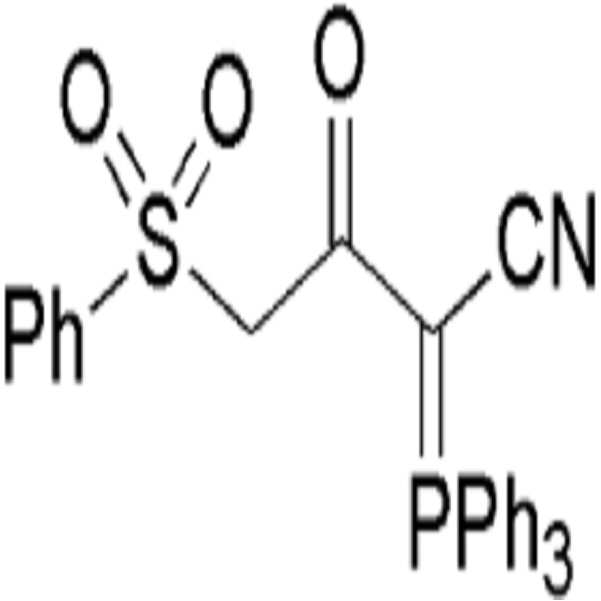
- (a) 하기 화학식 I의 3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴을 하기 화학식 IV의 할로젠화 알킬 화합물로 알킬화 반응시켜 하기 화학식 V의 알킬화된 설포닐 화합물을 수득하는 단계; 및
(b) 하기 화학식 V의 알킬화된 설포닐 화합물을 환원적 탈설포닐화 반응시키는 단계를 포함하는 하기 화학식 VI의 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물의 제조방법:
[화학식 I]
[화학식 IV]

[화학식 V]

[화학식 VI]

상기 식에서,
R은 C1-C10의 알킬기, C2-C10의 알케닐기 또는 아릴기이고,
X는 할로젠이다. - 제1항에 있어서, R이 페닐, C3-C10의 헤테로시클로알킬기 및 C3-C10의 헤테로시클로알킬옥시로 구성된 군으로부터 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않은 C1-C10의 알킬기 또는 C2-C10의 알케닐기; C1-C5의 알킬기로 치환되거나 치환되지 않은 페닐; 또는 티오페닐인 것을 특징으로 하는 제조방법.
- 제1항에 있어서, R이 페닐, 2-메틸페닐, n-헵틸, 2-페닐에틸, 2-페닐에텐일, 2-티오페닐, (테트라히드로-2H-피란-2-일옥시)메틸 또는 (1,3-디옥산-2-일)메틸인 것을 특징으로 하는 제조방법.
- 제1항에 있어서, X가 염소, 브롬 또는 요오드인 것을 특징으로 하는 제조방법.
- 제1항에 있어서, 알킬화 반응이 염기의 존재 하에 수행되는 것을 특징으로 하는 제조방법.
- 제5항에 있어서, 염기가 수소화나트륨(NaH) 또는 탄산칼륨(K2CO3)인 것을 특징으로 하는 제조방법.
- 제1항에 있어서, 환원적 탈설포닐화 반응이 나트륨 아말감 (Na(Hg)) 및 인산수소이나트륨(Na2HPO4)의 존재 하에 수행되는 것을 특징으로 하는 제조방법.
- 하기 화학식 I의 3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴:
[화학식 I]

- 하기 화학식 II의 페닐설포닐 아세트산과 하기 화학식 III의 (트리페닐포스포라닐리덴)아세토니트릴을 축합 반응시키는 단계를 포함하는 하기 화학식 I의 3-옥소-4-(페닐설포닐)-2-(트리페닐-λ5-포스파닐리덴)부탄니트릴의 제조방법:
[화학식 II]

[화학식 III]

[화학식 I]

- 제9항에 있어서, 축합 반응이 1-[3-(디메틸아미노)프로필]-3-에틸카보디이마이드·HCl (EDC) 및 4-디메틸아미노피리딘(DMAP)을 사용하여 수행되는 것을 특징으로 하는 제조방법.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR20130050545A KR101483977B1 (ko) | 2013-05-06 | 2013-05-06 | 설포닐 화합물을 이용한 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물의 제조방법 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR20130050545A KR101483977B1 (ko) | 2013-05-06 | 2013-05-06 | 설포닐 화합물을 이용한 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물의 제조방법 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR20140131677A true KR20140131677A (ko) | 2014-11-14 |
| KR101483977B1 KR101483977B1 (ko) | 2015-01-19 |
Family
ID=52452965
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR20130050545A KR101483977B1 (ko) | 2013-05-06 | 2013-05-06 | 설포닐 화합물을 이용한 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물의 제조방법 |
Country Status (1)
| Country | Link |
|---|---|
| KR (1) | KR101483977B1 (ko) |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5834588A (en) * | 1995-07-14 | 1998-11-10 | Yale University | (Cyanomethylene) phosphoranes as carbonyl 1,1-dipole synthons for use in constructing combinatorial libraries |
-
2013
- 2013-05-06 KR KR20130050545A patent/KR101483977B1/ko not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| KR101483977B1 (ko) | 2015-01-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI454471B (zh) | 製備細胞凋亡促進劑之方法 | |
| CZ124697A3 (en) | (4r-cis)-1,1-dimethylethyl-6-arylsulfonyloxymethyl-2,2-dimethyl-1,3-di- oxan-4-acetates and synthesis process thereof | |
| JP2007326784A (ja) | 1−置換−5−フルオロアルキルピラゾール−4−カルボン酸エステルの製造方法 | |
| Liu et al. | Concise asymmetric synthesis of (−)-deoxoprosophylline | |
| KR101728443B1 (ko) | 2-아미노니코틴산벤질에스테르 유도체의 제조 방법 | |
| KR100788529B1 (ko) | 3-(1-히드록시-펜틸리덴)-5-니트로-3h-벤조푸란-2-온,그의 제조 방법 및 용도 | |
| KR101483977B1 (ko) | 설포닐 화합물을 이용한 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물의 제조방법 | |
| CN104163777B (zh) | 一种合成甲腈化合物的方法及其在伊伐布雷定合成中的应用 | |
| JP4879907B2 (ja) | フェニル2−ピリミジニルケトン類の製造方法及びその新規中間体 | |
| CN101410386B (zh) | 2-链烯基-3-氨基噻吩衍生物及其制备方法 | |
| JP2005504019A (ja) | イソクマリンを調製するための方法 | |
| JP4258658B2 (ja) | アセチレン化合物の製造方法 | |
| JP4899385B2 (ja) | 3−アミノメチルオキセタン化合物の製法 | |
| KR101085202B1 (ko) | 브롬화 알킬 화합물로부터 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물의 제조방법 | |
| KR100968576B1 (ko) | 2-아실-3-아미노-2-알케노에이트의 제조방법 | |
| JP5205971B2 (ja) | テトラヒドロピラン化合物の製造方法 | |
| KR101621754B1 (ko) | 잔테이트 화합물을 이용한 α-케토 (시아노메틸렌)트리페닐포스포레인 화합물의 제조방법 | |
| JP2005015402A (ja) | 光学活性3,5−ジヒドロ−4H−ジナフト[2,1−c:1’,2’−e]アゼピンおよびそのシュウ酸塩の製造方法 | |
| JP2011057575A (ja) | 4−ヒドロキシベンゾチオフェン誘導体の製造方法 | |
| KR101590592B1 (ko) | 디피롤 케톤의 제조 방법 및 이에 의하여 제조된 디피롤 케톤 | |
| JP4973210B2 (ja) | 新規合成方法 | |
| JP2013129642A (ja) | 光学活性3,4−ビス(アルキルオキシカルボニル)−1,6−ヘキサン二酸誘導体の製造法 | |
| JP2015071572A (ja) | クロメントリフロン類,クマリントリフロン類及びその製造法 | |
| CN115210220A (zh) | 用于制备5-氯-3-烷基硫烷基-吡啶-2-甲酸酰胺和甲酸酯的方法 | |
| JP3918468B2 (ja) | 3,3−ビス(アルコキシカルボニル−メチルチオ)プロピオニトリル及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A201 | Request for examination | ||
| E902 | Notification of reason for refusal | ||
| E701 | Decision to grant or registration of patent right | ||
| GRNT | Written decision to grant | ||
| FPAY | Annual fee payment |
Payment date: 20180112 Year of fee payment: 4 |
|
| LAPS | Lapse due to unpaid annual fee |