KR20120084298A - 펩티드 제거제 - Google Patents
펩티드 제거제 Download PDFInfo
- Publication number
- KR20120084298A KR20120084298A KR1020127010173A KR20127010173A KR20120084298A KR 20120084298 A KR20120084298 A KR 20120084298A KR 1020127010173 A KR1020127010173 A KR 1020127010173A KR 20127010173 A KR20127010173 A KR 20127010173A KR 20120084298 A KR20120084298 A KR 20120084298A
- Authority
- KR
- South Korea
- Prior art keywords
- peptide
- binding
- enzyme
- peptides
- cpg2
- Prior art date
Links
- 108090000765 processed proteins & peptides Proteins 0.000 title claims abstract description 277
- 230000027455 binding Effects 0.000 claims abstract description 177
- 102000004190 Enzymes Human genes 0.000 claims abstract description 155
- 108090000790 Enzymes Proteins 0.000 claims abstract description 155
- 102000004196 processed proteins & peptides Human genes 0.000 claims abstract description 75
- 150000001413 amino acids Chemical group 0.000 claims abstract description 55
- 102000005427 Asialoglycoprotein Receptor Human genes 0.000 claims abstract description 30
- 108010006523 asialoglycoprotein receptor Proteins 0.000 claims abstract description 30
- 210000005229 liver cell Anatomy 0.000 claims abstract description 29
- 230000002255 enzymatic effect Effects 0.000 claims abstract description 19
- 210000004185 liver Anatomy 0.000 claims abstract description 19
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 claims abstract description 18
- 108010016626 Dipeptides Proteins 0.000 claims abstract description 8
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 claims abstract description 5
- CXOWHCCVISNMIX-UHFFFAOYSA-N 2-aminonaphthalene-1-carboxylic acid Chemical compound C1=CC=CC2=C(C(O)=O)C(N)=CC=C21 CXOWHCCVISNMIX-UHFFFAOYSA-N 0.000 claims abstract description 4
- 108010062699 gamma-Glutamyl Hydrolase Proteins 0.000 claims description 57
- 238000000034 method Methods 0.000 claims description 43
- 239000000427 antigen Substances 0.000 claims description 34
- 102000036639 antigens Human genes 0.000 claims description 34
- 108091007433 antigens Proteins 0.000 claims description 34
- 229940002612 prodrug Drugs 0.000 claims description 31
- 239000000651 prodrug Substances 0.000 claims description 31
- 239000000758 substrate Substances 0.000 claims description 28
- 230000000694 effects Effects 0.000 claims description 26
- 229940079593 drug Drugs 0.000 claims description 19
- 239000003814 drug Substances 0.000 claims description 19
- 239000002516 radical scavenger Substances 0.000 claims description 14
- 238000012360 testing method Methods 0.000 claims description 13
- 238000002560 therapeutic procedure Methods 0.000 claims description 13
- -1 beta-glucoronidase Proteins 0.000 claims description 12
- 238000010494 dissociation reaction Methods 0.000 claims description 12
- 230000005593 dissociations Effects 0.000 claims description 12
- 108090000623 proteins and genes Proteins 0.000 claims description 10
- 102000004169 proteins and genes Human genes 0.000 claims description 8
- 238000012216 screening Methods 0.000 claims description 8
- 238000006467 substitution reaction Methods 0.000 claims description 8
- 230000008878 coupling Effects 0.000 claims description 7
- 238000010168 coupling process Methods 0.000 claims description 7
- 238000005859 coupling reaction Methods 0.000 claims description 7
- 238000004519 manufacturing process Methods 0.000 claims description 7
- 230000013595 glycosylation Effects 0.000 claims description 6
- 238000006206 glycosylation reaction Methods 0.000 claims description 6
- GNFTZDOKVXKIBK-UHFFFAOYSA-N 3-(2-methoxyethoxy)benzohydrazide Chemical compound COCCOC1=CC=CC(C(=O)NN)=C1 GNFTZDOKVXKIBK-UHFFFAOYSA-N 0.000 claims description 5
- 102000002260 Alkaline Phosphatase Human genes 0.000 claims description 4
- 108020004774 Alkaline Phosphatase Proteins 0.000 claims description 4
- 108010067902 Peptide Library Proteins 0.000 claims description 4
- 230000004071 biological effect Effects 0.000 claims description 4
- 108091005601 modified peptides Proteins 0.000 claims description 4
- 230000009870 specific binding Effects 0.000 claims description 4
- 210000004881 tumor cell Anatomy 0.000 claims description 4
- 238000003491 array Methods 0.000 claims description 3
- 230000002163 immunogen Effects 0.000 claims description 3
- 230000006872 improvement Effects 0.000 claims description 3
- 108090000204 Dipeptidase 1 Proteins 0.000 claims description 2
- 101001133631 Lysinibacillus sphaericus Penicillin acylase Proteins 0.000 claims description 2
- 102000004459 Nitroreductase Human genes 0.000 claims description 2
- 102000006995 beta-Glucosidase Human genes 0.000 claims description 2
- 108010047754 beta-Glucosidase Proteins 0.000 claims description 2
- 102000006635 beta-lactamase Human genes 0.000 claims description 2
- 230000001279 glycosylating effect Effects 0.000 claims description 2
- 108020001162 nitroreductase Proteins 0.000 claims description 2
- 238000002360 preparation method Methods 0.000 claims description 2
- 230000001737 promoting effect Effects 0.000 claims description 2
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 claims description 2
- 238000002651 drug therapy Methods 0.000 claims 1
- 102000005962 receptors Human genes 0.000 abstract description 4
- 108020003175 receptors Proteins 0.000 abstract description 4
- 108090000288 Glycoproteins Proteins 0.000 abstract description 2
- 102000003886 Glycoproteins Human genes 0.000 abstract description 2
- 101000624947 Homo sapiens Nesprin-1 Proteins 0.000 abstract 1
- 102100023306 Nesprin-1 Human genes 0.000 abstract 1
- 210000004027 cell Anatomy 0.000 description 47
- 229940024606 amino acid Drugs 0.000 description 42
- 235000001014 amino acid Nutrition 0.000 description 42
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 26
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 25
- 238000003556 assay Methods 0.000 description 23
- 239000000243 solution Substances 0.000 description 21
- 206010028980 Neoplasm Diseases 0.000 description 16
- 235000019152 folic acid Nutrition 0.000 description 15
- 239000011724 folic acid Substances 0.000 description 15
- 229940049906 glutamate Drugs 0.000 description 15
- 102000037865 fusion proteins Human genes 0.000 description 14
- 108020001507 fusion proteins Proteins 0.000 description 14
- PKYCWFICOKSIHZ-UHFFFAOYSA-N 1-(3,7-dihydroxyphenoxazin-10-yl)ethanone Chemical compound OC1=CC=C2N(C(=O)C)C3=CC=C(O)C=C3OC2=C1 PKYCWFICOKSIHZ-UHFFFAOYSA-N 0.000 description 12
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 12
- 229960000304 folic acid Drugs 0.000 description 12
- 239000012131 assay buffer Substances 0.000 description 11
- 238000000684 flow cytometry Methods 0.000 description 11
- 229930195712 glutamate Natural products 0.000 description 10
- 230000004048 modification Effects 0.000 description 10
- 238000012986 modification Methods 0.000 description 10
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 9
- 239000011230 binding agent Substances 0.000 description 9
- 238000006243 chemical reaction Methods 0.000 description 9
- 230000005764 inhibitory process Effects 0.000 description 9
- 239000007787 solid Substances 0.000 description 9
- VDIQZIVYOLXOTG-VCGPICOLSA-N (2s)-3-hydroxy-2-[[(3r,4s,5r,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]amino]propanoic acid Chemical group OC[C@@H](C(O)=O)NC1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O VDIQZIVYOLXOTG-VCGPICOLSA-N 0.000 description 8
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 8
- 201000011510 cancer Diseases 0.000 description 8
- 229960000485 methotrexate Drugs 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- 230000002494 anti-cea effect Effects 0.000 description 7
- 239000000047 product Substances 0.000 description 7
- 235000018102 proteins Nutrition 0.000 description 7
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- 239000000872 buffer Substances 0.000 description 6
- WHUUTDBJXJRKMK-VKHMYHEASA-L glutamate group Chemical group N[C@@H](CCC(=O)[O-])C(=O)[O-] WHUUTDBJXJRKMK-VKHMYHEASA-L 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- 238000002823 phage display Methods 0.000 description 6
- 230000003389 potentiating effect Effects 0.000 description 6
- 239000011347 resin Substances 0.000 description 6
- 229920005989 resin Polymers 0.000 description 6
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- UCMIRNVEIXFBKS-UHFFFAOYSA-N beta-alanine Chemical compound NCCC(O)=O UCMIRNVEIXFBKS-UHFFFAOYSA-N 0.000 description 5
- 238000010828 elution Methods 0.000 description 5
- 239000003112 inhibitor Substances 0.000 description 5
- 239000002953 phosphate buffered saline Substances 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 230000002829 reductive effect Effects 0.000 description 5
- FSYKKLYZXJSNPZ-UHFFFAOYSA-N sarcosine Chemical compound C[NH2+]CC([O-])=O FSYKKLYZXJSNPZ-UHFFFAOYSA-N 0.000 description 5
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 4
- OYIFNHCXNCRBQI-UHFFFAOYSA-N 2-aminoadipic acid Chemical compound OC(=O)C(N)CCCC(O)=O OYIFNHCXNCRBQI-UHFFFAOYSA-N 0.000 description 4
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 4
- KSPIYJQBLVDRRI-UHFFFAOYSA-N N-methylisoleucine Chemical compound CCC(C)C(NC)C(O)=O KSPIYJQBLVDRRI-UHFFFAOYSA-N 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 229930182830 galactose Natural products 0.000 description 4
- 125000003147 glycosyl group Chemical group 0.000 description 4
- 210000003494 hepatocyte Anatomy 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- 210000001161 mammalian embryo Anatomy 0.000 description 4
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 241000283707 Capra Species 0.000 description 3
- 150000008574 D-amino acids Chemical class 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 3
- 150000008575 L-amino acids Chemical class 0.000 description 3
- 238000002835 absorbance Methods 0.000 description 3
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 3
- QWCKQJZIFLGMSD-UHFFFAOYSA-N alpha-aminobutyric acid Chemical compound CCC(N)C(O)=O QWCKQJZIFLGMSD-UHFFFAOYSA-N 0.000 description 3
- 125000003118 aryl group Chemical group 0.000 description 3
- 229960002685 biotin Drugs 0.000 description 3
- 239000011616 biotin Substances 0.000 description 3
- 210000004899 c-terminal region Anatomy 0.000 description 3
- 150000001875 compounds Chemical class 0.000 description 3
- PMMYEEVYMWASQN-UHFFFAOYSA-N dl-hydroxyproline Natural products OC1C[NH2+]C(C([O-])=O)C1 PMMYEEVYMWASQN-UHFFFAOYSA-N 0.000 description 3
- 229940014144 folate Drugs 0.000 description 3
- 229960002989 glutamic acid Drugs 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 230000003993 interaction Effects 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 239000000203 mixture Substances 0.000 description 3
- 239000006225 natural substrate Substances 0.000 description 3
- 238000010647 peptide synthesis reaction Methods 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- XJMOSONTPMZWPB-UHFFFAOYSA-M propidium iodide Chemical compound [I-].[I-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CCC[N+](C)(CC)CC)=C1C1=CC=CC=C1 XJMOSONTPMZWPB-UHFFFAOYSA-M 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- FUOOLUPWFVMBKG-UHFFFAOYSA-N 2-Aminoisobutyric acid Chemical compound CC(C)(N)C(O)=O FUOOLUPWFVMBKG-UHFFFAOYSA-N 0.000 description 2
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 2
- RDFMDVXONNIGBC-UHFFFAOYSA-N 2-aminoheptanoic acid Chemical compound CCCCCC(N)C(O)=O RDFMDVXONNIGBC-UHFFFAOYSA-N 0.000 description 2
- PECYZEOJVXMISF-UHFFFAOYSA-N 3-aminoalanine Chemical compound [NH3+]CC(N)C([O-])=O PECYZEOJVXMISF-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 201000009030 Carcinoma Diseases 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 2
- SNDPXSYFESPGGJ-UHFFFAOYSA-N L-norVal-OH Natural products CCCC(N)C(O)=O SNDPXSYFESPGGJ-UHFFFAOYSA-N 0.000 description 2
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 2
- OLNLSTNFRUFTLM-UHFFFAOYSA-N N-ethylasparagine Chemical compound CCNC(C(O)=O)CC(N)=O OLNLSTNFRUFTLM-UHFFFAOYSA-N 0.000 description 2
- YPIGGYHFMKJNKV-UHFFFAOYSA-N N-ethylglycine Chemical compound CC[NH2+]CC([O-])=O YPIGGYHFMKJNKV-UHFFFAOYSA-N 0.000 description 2
- 108010065338 N-ethylglycine Proteins 0.000 description 2
- AKCRVYNORCOYQT-YFKPBYRVSA-N N-methyl-L-valine Chemical compound CN[C@@H](C(C)C)C(O)=O AKCRVYNORCOYQT-YFKPBYRVSA-N 0.000 description 2
- 108090000854 Oxidoreductases Proteins 0.000 description 2
- 102000004316 Oxidoreductases Human genes 0.000 description 2
- 241001494479 Pecora Species 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- 102000035195 Peptidases Human genes 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 108010077895 Sarcosine Proteins 0.000 description 2
- 108010003723 Single-Domain Antibodies Proteins 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- 108010090804 Streptavidin Proteins 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 235000020958 biotin Nutrition 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 238000001311 chemical methods and process Methods 0.000 description 2
- 201000010989 colorectal carcinoma Diseases 0.000 description 2
- 231100000599 cytotoxic agent Toxicity 0.000 description 2
- YSMODUONRAFBET-UHFFFAOYSA-N delta-DL-hydroxylysine Natural products NCC(O)CCC(N)C(O)=O YSMODUONRAFBET-UHFFFAOYSA-N 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 239000000539 dimer Substances 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 230000004927 fusion Effects 0.000 description 2
- 125000002519 galactosyl group Chemical group C1([C@H](O)[C@@H](O)[C@@H](O)[C@H](O1)CO)* 0.000 description 2
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 235000013922 glutamic acid Nutrition 0.000 description 2
- 239000004220 glutamic acid Substances 0.000 description 2
- 125000003630 glycyl group Chemical group [H]N([H])C([H])([H])C(*)=O 0.000 description 2
- 229940127121 immunoconjugate Drugs 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 230000003278 mimic effect Effects 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 230000009897 systematic effect Effects 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 239000011592 zinc chloride Substances 0.000 description 2
- 235000005074 zinc chloride Nutrition 0.000 description 2
- BJBUEDPLEOHJGE-UHFFFAOYSA-N (2R,3S)-3-Hydroxy-2-pyrolidinecarboxylic acid Natural products OC1CCNC1C(O)=O BJBUEDPLEOHJGE-UHFFFAOYSA-N 0.000 description 1
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- JZTKZVJMSCONAK-INIZCTEOSA-N (2s)-2-(9h-fluoren-9-ylmethoxycarbonylamino)-3-hydroxypropanoic acid Chemical compound C1=CC=C2C(COC(=O)N[C@@H](CO)C(O)=O)C3=CC=CC=C3C2=C1 JZTKZVJMSCONAK-INIZCTEOSA-N 0.000 description 1
- XABCFXXGZPWJQP-BYPYZUCNSA-N (S)-3-aminoadipic acid Chemical compound OC(=O)C[C@@H](N)CCC(O)=O XABCFXXGZPWJQP-BYPYZUCNSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- JHTPBGFVWWSHDL-UHFFFAOYSA-N 1,4-dichloro-2-isothiocyanatobenzene Chemical compound ClC1=CC=C(Cl)C(N=C=S)=C1 JHTPBGFVWWSHDL-UHFFFAOYSA-N 0.000 description 1
- OGNSCSPNOLGXSM-UHFFFAOYSA-N 2,4-diaminobutyric acid Chemical compound NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- KPGXRSRHYNQIFN-UHFFFAOYSA-N 2-oxoglutaric acid Chemical compound OC(=O)CCC(=O)C(O)=O KPGXRSRHYNQIFN-UHFFFAOYSA-N 0.000 description 1
- UMCMPZBLKLEWAF-BCTGSCMUSA-N 3-[(3-cholamidopropyl)dimethylammonio]propane-1-sulfonate Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCC[N+](C)(C)CCCS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 UMCMPZBLKLEWAF-BCTGSCMUSA-N 0.000 description 1
- QCHPKSFMDHPSNR-UHFFFAOYSA-N 3-aminoisobutyric acid Chemical compound NCC(C)C(O)=O QCHPKSFMDHPSNR-UHFFFAOYSA-N 0.000 description 1
- BTJIUGUIPKRLHP-UHFFFAOYSA-N 4-nitrophenol Chemical compound OC1=CC=C([N+]([O-])=O)C=C1 BTJIUGUIPKRLHP-UHFFFAOYSA-N 0.000 description 1
- SLXKOJJOQWFEFD-UHFFFAOYSA-N 6-aminohexanoic acid Chemical compound NCCCCCC(O)=O SLXKOJJOQWFEFD-UHFFFAOYSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 108010002913 Asialoglycoproteins Proteins 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 1
- 108090001008 Avidin Proteins 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N Benzoic acid Natural products OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- MOKPKEKFPXEHDS-UHFFFAOYSA-N C(=O)(OC(C)(C)C)C=1C(=C(C2=CC=CC=C2C1)C(=O)O)N Chemical compound C(=O)(OC(C)(C)C)C=1C(=C(C2=CC=CC=C2C1)C(=O)O)N MOKPKEKFPXEHDS-UHFFFAOYSA-N 0.000 description 1
- 101710112752 Cytotoxin Proteins 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 102000004157 Hydrolases Human genes 0.000 description 1
- 108090000604 Hydrolases Proteins 0.000 description 1
- LCWXJXMHJVIJFK-UHFFFAOYSA-N Hydroxylysine Natural products NCC(O)CC(N)CC(O)=O LCWXJXMHJVIJFK-UHFFFAOYSA-N 0.000 description 1
- PMMYEEVYMWASQN-DMTCNVIQSA-N Hydroxyproline Chemical compound O[C@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-DMTCNVIQSA-N 0.000 description 1
- 108010093096 Immobilized Enzymes Proteins 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 1
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 1
- SNDPXSYFESPGGJ-BYPYZUCNSA-N L-2-aminopentanoic acid Chemical compound CCC[C@H](N)C(O)=O SNDPXSYFESPGGJ-BYPYZUCNSA-N 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- AGPKZVBTJJNPAG-UHNVWZDZSA-N L-allo-Isoleucine Chemical compound CC[C@@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-UHNVWZDZSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- 229930195714 L-glutamate Natural products 0.000 description 1
- 108010069325 L-glutamate oxidase Proteins 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 1
- 102000004856 Lectins Human genes 0.000 description 1
- 108090001090 Lectins Proteins 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- PQNASZJZHFPQLE-LURJTMIESA-N N(6)-methyl-L-lysine Chemical compound CNCCCC[C@H](N)C(O)=O PQNASZJZHFPQLE-LURJTMIESA-N 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 1
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- SSJMZMUVNKEENT-IMJSIDKUSA-N Ser-Ala Chemical compound OC(=O)[C@H](C)NC(=O)[C@@H](N)CO SSJMZMUVNKEENT-IMJSIDKUSA-N 0.000 description 1
- 244000191761 Sida cordifolia Species 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 229960002684 aminocaproic acid Drugs 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 239000012491 analyte Substances 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 230000005875 antibody response Effects 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 235000009582 asparagine Nutrition 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 150000001540 azides Chemical class 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229940000635 beta-alanine Drugs 0.000 description 1
- 230000008033 biological extinction Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 238000010382 chemical cross-linking Methods 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 101150071577 chi2 gene Proteins 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 239000002254 cytotoxic agent Substances 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 239000002619 cytotoxin Substances 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- NTUGPDFKMVHCCJ-VIFPVBQESA-N ditert-butyl (2s)-2-aminopentanedioate Chemical compound CC(C)(C)OC(=O)CC[C@H](N)C(=O)OC(C)(C)C NTUGPDFKMVHCCJ-VIFPVBQESA-N 0.000 description 1
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- YSMODUONRAFBET-UHNVWZDZSA-N erythro-5-hydroxy-L-lysine Chemical compound NC[C@H](O)CC[C@H](N)C(O)=O YSMODUONRAFBET-UHNVWZDZSA-N 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 1
- 238000002523 gelfiltration Methods 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 125000003563 glycoside group Chemical group 0.000 description 1
- PJJJBBJSCAKJQF-UHFFFAOYSA-N guanidinium chloride Chemical compound [Cl-].NC(N)=[NH2+] PJJJBBJSCAKJQF-UHFFFAOYSA-N 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- QJHBJHUKURJDLG-UHFFFAOYSA-N hydroxy-L-lysine Natural products NCCCCC(NO)C(O)=O QJHBJHUKURJDLG-UHFFFAOYSA-N 0.000 description 1
- 229960002591 hydroxyproline Drugs 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 230000000415 inactivating effect Effects 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 229960000310 isoleucine Drugs 0.000 description 1
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 1
- 239000002523 lectin Substances 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 230000009871 nonspecific binding Effects 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 230000000771 oncological effect Effects 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- 230000007030 peptide scission Effects 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 235000019833 protease Nutrition 0.000 description 1
- 235000019419 proteases Nutrition 0.000 description 1
- 239000008213 purified water Substances 0.000 description 1
- 239000012557 regeneration buffer Substances 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- HSSLDCABUXLXKM-UHFFFAOYSA-N resorufin Chemical compound C1=CC(=O)C=C2OC3=CC(O)=CC=C3N=C21 HSSLDCABUXLXKM-UHFFFAOYSA-N 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 229940043230 sarcosine Drugs 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- YSMODUONRAFBET-WHFBIAKZSA-N threo-5-hydroxy-L-lysine Chemical compound NC[C@@H](O)CC[C@H](N)C(O)=O YSMODUONRAFBET-WHFBIAKZSA-N 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- BJBUEDPLEOHJGE-IMJSIDKUSA-N trans-3-hydroxy-L-proline Chemical compound O[C@H]1CC[NH2+][C@@H]1C([O-])=O BJBUEDPLEOHJGE-IMJSIDKUSA-N 0.000 description 1
- 238000010396 two-hybrid screening Methods 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 239000004474 valine Substances 0.000 description 1
- 239000003039 volatile agent Substances 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Images
















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/549—Sugars, nucleosides, nucleotides or nucleic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/66—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid the modifying agent being a pre-targeting system involving a peptide or protein for targeting specific cells
- A61K47/665—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid the modifying agent being a pre-targeting system involving a peptide or protein for targeting specific cells the pre-targeting system, clearing therapy or rescue therapy involving biotin-(strept) avidin systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82Y—SPECIFIC USES OR APPLICATIONS OF NANOSTRUCTURES; MEASUREMENT OR ANALYSIS OF NANOSTRUCTURES; MANUFACTURE OR TREATMENT OF NANOSTRUCTURES
- B82Y5/00—Nanobiotechnology or nanomedicine, e.g. protein engineering or drug delivery
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/02—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/02—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link
- C07K5/0202—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link containing the structure -NH-X-X-C(=0)-, X being an optionally substituted carbon atom or a heteroatom, e.g. beta-amino acids
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Pharmacology & Pharmacy (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Epidemiology (AREA)
- Nanotechnology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Genetics & Genomics (AREA)
- General Engineering & Computer Science (AREA)
- Medical Informatics (AREA)
- Biotechnology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Cell Biology (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Medicinal Preparation (AREA)
Abstract
펩티드 제거제는 대상체에서 효소 및 표적 위치에 특이적으로 결합하는 결합 분자의 접합체를 비-표적 위치로부터 제거하기 위해 제공되는 것이다. 펩티드 제거제는 효소의 활성 부위에 결합한다. 또한 펩티드는 간 세포에 의해 발현되는 아시알로당단백질 수용체에 결합함으로써 간을 통한 제거가 촉진된다. 펩티드는 아시알로당단백질 수용체를 발현하는 간 세포에의 결합에 의해 이루어지는 간을 통한 제거가 촉진되도록 글리코실화될 수 있다. 전형적으로 펩티드는 효소에 결합시 효소 활성을 방지하거나 또는 억제하고, 효소 활성에 의해 실질적으로 변형되지 않는다. 펩티드는 디펩티드 아미노-나프토산 (ANA)-글루타메이트 (Glu)를 기초로 할 수 있고, 아미노산 서열 세린 (Ser)-알라닌 (Ala)-아미노-나프토산 (ANA)-글루타메이트 (Glu)를 포함할 수 있다. 이러한 경우, 관심 대상 효소는 전형적으로 CPG2이다.
Description
본 발명은 대상체에서 비-표적 위치로부터 효소를 제거하는 작용을 하는 펩티드 제거제에 관한 것이다. 본 발명은 또한 이러한 펩티드 제거제를 제조하는 방법에 관한 것이다.
항체 지향 효소 전구약물 치료법 (antibody directed enzyme prodrug therapy; ADEPT)의 목표는 세포독성 약물의 선택성을 개선시키는 것이다. 전구약물을 활성 약물로 전환시키는 역할을 하는 효소가 사용된다. 종양 관련 항원에 결합하는 항체의 사용을 통해 약물의 작용은 관심 대상 부위로 국소화된다. 이러한 항체는 전구약물에 대해 작용하는 효소에 접합되거나, 또는 그와 융합 단백질을 형성함으로써 전구약물의 전환이 주로 관심 대상 부위에서 확실하게 일어날 수 있도록 한다.
그러나, 잔류하는 효소-항체 접합체, 및 종양으로부터의 접합체의 누출 (이는 종양 세포로부터 순환계로의 항원 소실에 기인하는 것일 수 있다)로 인해 ADEPT의 치료학적 효능 및 특이성은 정상 조직에서의 전구약물의 전환에 의해 제한되었다. 이러한 문제를 처리하기 위한 시도로, 잔류하는 효소 활성을 제거하여 상기 요법의 부작용을 최소화시키는 효소 제거 단계가 개발되었다. 이는 상기 효소에 결합하는 추가 항체에 의존하였다. 이러한 항체는 간을 통해 이루어지는 제거가 촉진되도록 글리코실화된다 (1).
US 2003-0068322 (한센(Hansen))에는 순환성 표적 단백질-효소 접합체를 제거하는 항체 기반 제거제가 기재되어 있다. 한 실시양태에서, 제거제는 효소에 결합한다. 그러나, 제거제는 효소 활성을 방해하지 않는 부위에 결합한다. WO 96/40245 및 US 5,958,408은 유사한 개시내용을 제시한다.
문헌 [Napier et al., - Clinical Cancer Research Vol. 6, 765 to 772, March 2000]에는 암배아 항원을 발현하는 결장직장 암종을 앓는 10명의 환자가 카르복시펩티다제 G2 (CPG2)에 접합된, 암배아 항원에 대한 A5B7 F(ab')2 항체를 사용한 항체 지향 효소 전구약물 치료법을 받은 임상 시험이 기재되어 있다. 상기 시험에서, CPG2의 활성 부위 (SB43-gal)에 대해 지향된 갈락토실화된 항체는 순환 효소를 제거하고 불활성화시키기 위해 제공되었다. 추가 요법을 방해하는 인간 항-마우스 항체 반응 (HAMA)이 2주 후 모든 환자에서 발견되었다고 나피에르(Napier)는 언급하였다 (768페이지의 면역 반응이라는 표제하의 두번째 칼럼 참조). ADEPT 요법과 관련하여 상기 제거제의 사용을 촉진시키기 위해 면역억제제가 사용될 수 있다고 나피에르는 제안하였다.
WO 91/17761에는 ADEPT 기법과는 상이한 기법이 기재되어 있다. 이러한 기법에서는 세포독소가 치료학상 오직 이환된 세포만을 치료하는 데 사용될 수 있도록 길항제는 정상 세포에 표적화된다. 따라서, 상기 방법의 목표는 정상 세포를 보호하는 것이다.
EP0308208은 순환으로부터의 신속한 제거를 가능하게 하는 인간 간 아시알로당단백질 수용체에 결합하는 글리코시드 잔기에의 접합체 의해 변형된 항체 및 항체 접합체에 관한 것이다.
EP0733072 및 US 5,876,691에는 암배아 항원 (CEA)에 특이적인 항체가 기재되어 있다.
제거제로서 글리코실화된 제2 또는 추가 항체를 사용하는 것은 여러 가지 문제들에 관련되는데, 그 중 가장 중요한 것은 하기와 같다:
1) 제2 (제거) 항체는 필연적으로 면역원성이며, 이에 상기 항체는 반복하여 주기적으로 사용하는 것이 몇 회로 제한된다.
2) 효소에 대해 높은 친화도를 가지고 있는 바, 이를 조정하는 것이 곤란하다. 친화도가 높다는 것은 또한 표적 위치에 국소화된 효소에 매우 강력하게 결합함으로써 국소 약물 생성의 효능을 제한한다는 것을 의미한다.
3) 상기 항체가 고분자량이라는 것은 융합 단백질에 결합하여 그를 제거하기 위해서는 큰 질량의 당단백질이 접종되어야 한다는 것을 의미한다.
4) 제2 항체 성분의 비용, 생산 및 조절에 관한 문제가 비교적 곤란하다.
이러한 이유에서 새로운 종류의 제거제가 요구되고 있다. 따라서, 본 발명은 효소 및 표적 위치에서 특이적으로 결합하는 결합 분자의 접합체를 이 접합체에 대한 펩티드의 결합을 통해 대상체에서 비-표적 위치로부터 제거하는 펩티드 제거제를 제공하며, 여기서, 펩티드는 간 세포에 의해 발현되는 아시알로당단백질 수용체에 결합하는 것이다. 이를 통해 비-표적 위치에서 발견되는 접합체는 내재화된다. 따라서, 펩티드가 간 세포에 의해 발현되는 아시알로당단백질 수용체에 결합하게 되면 펩티드의 제거는 촉진된다. 특정 실시양태에서, 펩티드는 아시알로당단백질 수용체를 발현하는 간 세포에의 결합에 의해 이루어지는 간을 통한 제거가 촉진되도록 글리코실화된다. 더욱 구체적으로, 본 발명은 효소 및 표적 위치에 특이적으로 결합하는 결합 분자의 접합체를 상기 효소의 활성 부위에 대한 펩티드의 결합을 통해 대상체에서 비-표적 위치로부터 제거하는 펩티드 제거제를 제공하며, 여기서, 펩티드 (또한 제거제)는 간 세포에 의해 발현되는 아시알로당단백질 수용체에 결합하는 것이다. 이를 통해 비-표적 위치에서 발견되는 접합체는 내재화된다. 따라서, 펩티드가 간 세포에 의해 발현되는 아시알로당단백질 수용체에 결합하게 되면 펩티드의 제거는 촉진된다. 특정 실시양태에서, 펩티드는 아시알로당단백질 수용체를 발현하는 간 세포에의 결합에 의해 이루어지는 간을 통한 제거가 촉진되도록 글리코실화된다.
대상체는 전형적으로 인간 또는 동물이고, 바람직하게는 인간이다. 따라서, 제거는 일반적으로 간을 통해 순환으로부터 이루어진다. 본 발명의 펩티드 제거제는 간 세포에 의해 발현되는 아시알로당단백질 수용체에 결합하게 되고, 이를 통해 비-표적 위치에서 발견되는 접합체는 내재화된다.
하기와 같은 이유에서 본 발명의 제거제는 항체 기반 제거제와 관련된 문제들을 해소시켜 준다:
1) 본 발명의 제거제는 저분자량이기 때문에 비-면역원성이다.
2) 원하는 결합 친화도에 도달할 때까지 대체 아미노산을 이용한 위치 치환을 수행함으로써 효소 결합 부위에 대한 그의 친화도는 조정될 수 있다.
3) 저분자량이라는 것은 또한 융합 단백질보다 훨씬 더 작은 질량으로도 주어진 제거율을 달성할 수 있다는 것을 의미한다.
4) 본 펩티드 제거제는 공지된 화학적 방법에 의해 합성 화학물질로서 제조될 수 있는 바, 이에 비용, 생산 효율 및 조절 장벽에 관한 문제는 거의 없다.
상기 언급한 바와 같이, ADEPT 시스템은 대상체에서 표적 위치, 예를 들어, 종양 관련 항원에 결합하는 항체, 또는 다른 결합 분자를 통해 효소 활성을 국소화하는 것에 의존한다. ADEPT 시스템은, 대량이지만 고도로 국소화된 용량의 약물, 예를 들어, 세포독성제를 이환된 세포 (예를 들어, 암 세포), 또는 질병 유발 세포 (예를 들어, 기생충 세포)로 전달하기 위해 함께 협력하는 수개의 분자 모듈로 구성된다.
본 발명의 펩티드 제거제는 효소 및 결합 분자의 접합체, 더욱 구체적으로 효소 및 특히 효소의 활성 부위에 결합한다. 결합 분자는 표적 위치에 결합할 수 있고, 이로써 효소를 상기 특정 위치로 국소화시킬 수 있는 임의의 분자일 수 있다. 특정 실시양태에서, 효소는 표적 위치에서의 결합 분자의 특이적인 결합을 통해 표적 위치로 집중된다. 따라서, 결합 분자는 수용체, 항원, 또는 대상체의 세포에 의해 발현되는, 특히, 세포 표면 상에서 안정적으로 발현되는 다른 분자에 결합할 수 있다. 바람직하게는, 배타적인 것은 아니지만, 결합 분자는 항체 또는 그의 항원-결합 유도체를 포함하거나, 그로 이루어지거나, 또는 본질적으로 그로 이루어진다. IgG 이뮤노글로불린이 가장 전형적으로 사용되기는 하지만, 임의의 적합한 항체가 사용될 수 있다. 물론, 중쇄 항체 및 인간화된 형태의 비-인간 항체도 "항체"라는 용어의 범주내 포함된다. 일반적으로는 모노클로날 항체가 바람직하지만, 항체는 폴리클로날 항체일 수도 있다. 항원-결합 유도체는 관심 대상 표적에 특이적으로 결합할 수 있는 능력을 보유하는 항체의 모든 단편 및 다른 유도체를 포함한다. 대개 1가 단편이 사용된다. 그 예로는 Fab 단편, scFv, 단일 도메인 항체, 나노바디, 미니바디, 디아바디, 트리아바디 등이 포함된다.
결합 분자는 표적 위치에 특이적으로 결합함으로써 비-표적 위치, 예를 들어, 특정 항원을 발현하지 않는 세포에의 원치않는 결합을 피할 수 있다. 이는 또한 표적 위치에서 빠르게 포획될 수 있도록 하는 데 충분한 고도의 친화도를 가져야 한다. 그러나, 결합 친화도는 결합 분자가 표적 위치에 충분히 접근할 수 있을 정도, 예를 들면, 종양 관련 항원에 결합할 정도의 친화도를 의미하며, 결합 분자는 오직 임의의 종양 종괴 주변에만 결합할 수 있도록 매우 단단히 결합하지는 않아야 한다. 실제로, 임의의 선택적 결합능은 초점화된 치료학적 작용을 제공하기에 충분하며, 필요한 최소 효율은 적용마다 달라질 것이다. 바람직하게는, 결합 분자의 결합 친화도는 결합 분자가 결합한 표적 위치로부터 상기 분자의 누출을 최소화하는 낮은 해리 상수 (즉, 느린 오프 속도 (off-rate))와 관련이 있다. 따라서, 표적 위치에서 표적화된 유효 작용이 확실하게 이루어지도록, 가능한 한 결합 친화도 및 동력학적 성질은 균형을 이룬다.
효소는 효소가 그와 결합 분자와의 결합에 의해 표적 위치로 전달될 수 있도록 하는 방식으로 결합 분자와 접합체를 형성한다. 전형적으로, 효소는 결합 분자와 공유적으로 결합함으로써 어디서든 결합제 분자는 결합할 수 있고, 또한 적어도 하나의 효소 분자가 결합될 수 있다. 따라서, 상기에서 주시된 결합 특징들은 실제로 연속 아미노산 서열로서 결합 분자 및 효소 모두를 발현하는 단일 유전자로부터 유래되고, 적절한 작용성 도메인으로 폴딩된 결합 분자-효소 융합 단백질 또는 결합 분자-효소 화학적 접합체 형태의 하이브리드 분자에 적용된다. 효소는 표적 위치에서 전구약물을 활성 약물로 전환시키는 작용을 한다. 효소는 대상체에 천연적으로는 존재하지 않아서, 대상체에서 원치않는 활성 및 부작용을 막는 효소인 것인 바람직하다. 효소의 역할은 효소가 작용할 때까지는 비-독성인 전구약물 분자를 변형시켜 활성 약물, 일반적으로, 수명이 짧은 활성 약물을 유리시키는 것이다. 따라서, 결합 분자에 기반한 효소 활성의 국소화를 통해 전구약물은 오직 표적 위치에 바로 가까이에 위치할 때에만 확실하게 절단된다. 특정 실시양태에서, 전구약물은 효소에 의해 절단되었을 때 고도로 독성이지만 수명은 짧은 분자를 형성하는 물질로서, 이러한 특성을 통해 전구약물은 효능은 있지만, 오직 전구약물이 절단되는 위치 부근에 존재하는 세포만을 사멸시킬 수 있게 되는 것이 보장된다. 특정 실시양태에서, 알킬화제, 예를 들어, 벤조산 머스타드 유도체가 사용된다. 바람직하게는, 약물의 작용은 농도 의존적이다. 알킬화제는 그의 말단 글루타메이트를 통해 불활성화되고, 이로써, 상기 글루타메이트 잔기를 절단하는 작용을 하는 CPG2와 함께 사용하기에 이상적인 파트너가 된다. 표적 위치에서 생성된 고농도의 약물은 다시 혈액 내로 역누출되어 독성을 유발할 수 있는 바, 이에 짧은 반감기가 활성 약물의 중요한 속성이 된다.
표적 위치에서 불활성 전구약물을 활성 약물로 전환시킬 수 있는 임의의 적합한 효소가 사용될 수 있다. ADEPT 시스템과 관련하여 이미 사용되고 있는 적합한 효소로는 카르복시펩티다제 G2, 알칼리성 포스파타제, 베타-글루코로니다제, 페니실린-V-아미다제, 베타-락타마제, 베타-글루코시다제 및 니트로리덕타제가 포함된다. 본 발명의 펩티드 제거제를 생성하는 데 유용한, 잠재적으로 유용한 효소/전구약물 조합은 하기 표 1에 열거되어 있는 것과 같다.
<표 1>

본 발명과 관련하여 특히 바람직한 효소는 카르복시펩티다제 G2 (CPG2)이다. CPG2는 (슈도모나스(Psuedomonas)로부터 유래된) 세균 펩티다제이다. 이는 폴레이트 가수분해 효소로서, 엽산 및 그의 유사체, 예를 들어, 메토트렉세이트로부터 C-말단 글루타메이트 모이어티를 가수분해한다. 동종 CPG2의 분자 질량은 83,000이지만 (겔 여과), SDS PAGE 후에는 분자 질량이 41,400이 수득되는데, 이는 효소가 이량체라는 것을 나타낸다. 이는 각각 작용성인 무손상 이량체 중 2개의 활성 부위를 가지며, 이는 그의 구조와 작용에 있어 아연에 의존한다.
결합 분자와 효소의 접합체는 일반적으로 유전적 수단에 의해, 또는 다수의 공지된 화학적 가교 결합 방법들 중 하나를 사용하는 화학적 접합에 의해 제조된다. 유전적 수단이란 일반적으로는 효소 및 결합 분자 단백질, 둘 모두가 2가지 작용 모두를 가진 단일의 조합된 단백질 ("융합 단백질")로서 발현되도록 각 유전자를 함께 스플라이싱하는 것을 포함한다. 글리코실화된 융합 단백질은 제공되는 즉시 간을 통해 그 자신의 제거를 개시하여 결과적으로는 비-글리코실화된 형태보다도 더 소량의 효소가 종양으로 전달되는 바, 융합 단백질이 글리코실화되지 않도록 하는 것이 중요하다.
본 발명의 특정 실시양태에서, 펩티드 제거제는 효소에 결합시 효소 활성을 방지하거나 또는 억제한다. 따라서, 바람직하게는, 펩티드는 효소 활성에 의해 변형되지 않는다. "변형되지 않는다"라는 것은 효소가 제거될 수 있도록 펩티드가 효소에 대한 결합 친화도를 보유한다는 것을 의미하며; 단, 친화도를 상실하지 않는다면 변형이 이루어질 수도 있다. 펩티드가 효소 활성에 대해 저항성을 띠는 경우, 펩티드가 활성 부위에 결합하는 것이 보다 효과적인 제거 방식이다. 본 발명의 펩티드가 효소 활성에 대해 저항성을 띨 수 있게 하는 한가지 방식은 하나 이상의 D형 아미노산을 도입하는 것이다. 예를 들어, CPG2의 활성 부위에 결합하도록 디자인된 펩티드 제거제 중 D-아미노산 글루타메이트를 포함시키는 것은 상기 아미노산에서의 펩티드 결합의 가수분해를 방지하는 작용을 할 수 있다.
결합 분자-효소 접합체는 표적 위치에 결합한다. 이는 전형적으로는 관심 대상 세포 유형, 예를 들여, 종양 세포이다. 표적 위치에의 특이적인 결합은 관심 대상 세포 유형에 대해 특이적인 항원에의 결합에 기인하는 것일 수 있다. 특정 실시양태에서, 표적 위치는 종양 세포에 의해 발현되는 항원이다. 따라서, 예를 들어, 종양학적으로 적용하는 경우, 비-암성 숙주 세포를 공격할 위험을 회피하기 위해 항원 발현은 이상적으로는 암 세포에 대해 특이적이어야 한다. 항원은 종양 관련된 것일 수 있지만, 종양-특이성인 것이 바람직하다. 추가로, 항원은 관심 대상 세포의 표면 (막) 상에서 안정적으로 발현되어야 한다. 그러나, 본 발명과 관련하여 방출 또는 배출 이전에 일시적으로 발현되는 것도 가능하다. 표적 위치를 특징화하는 항원은 바람직하게는 자발적으로 또는 결합 분자가 결합한 경우에 신속하게 내재화되지 않는다. 그 이유는 전구약물이 전형적으로는 전신 투여되고, 세포내 효소는 순환 중 전구약물과 쉽게 상호작용하지 않기 때문이다. 표적 위치에 대한 구체적인 일례로는 암배아 항원 (CEA)으로서 공지된 항원을 발현하는 세포이다. 이러한 항원은 상당부의 결장직장 암종 (CRC)에 의해 발현되며, 이로써 CRC를 국소적으로 치료할 수 있다. CEA에 결합할 수 있는 항체 및 유도체는 당업계 공지되어 있으며, 이의 예로는, 본원에서 논의되는 ScFv 항-CEA가 있고, 이는 CPG2와의 융합 단백질로서 제조될 수 있거나, 또는 그렇지 않을 수도 있다.
본 발명의 펩티드 제거제는 간 세포에 의해 발현되는 아시알로당단백질 수용체에 결합한다. 이를 통해 비-표적 위치에서 발견되는 접합체는 내재화된다. 따라서, 펩티드가 간 세포에 의해 발현되는 아시알로당단백질 수용체에 결합하게 되면 펩티드의 제거는 촉진된다. 특정 실시양태에서, 펩티드는 아시알로당단백질 수용체를 발현하는 간 세포에의 결합에 의해 이루어지는 간을 통한 제거가 촉진되도록 글리코실화된다. 따라서, 펩티드는 간 세포에 도달하여 아시알로당단백질 수용체를 통해 이용될 수 있도록 하는 데 충분한 안정성을 가진, 펩티드 제거제와 결합 분자 및 효소의 접합체 사이의 복합체가 순환 중에 형성될 수 있도록 하는 결합 친화도로 효소에 결합하는 아미노산 서열로 구성된다. 그러나, 펩티드 제거제의 오프 속도는 표적 위치에 결합된 결합 분자 및 효소의 접합체로부터 전구약물의 투여 요법에 따른 기간 이내에 해리될 수 있도록 충분히 빨라야 하며, 그렇지 않을 경우, 전구약물의 활성 약물로의 전환은 방해받게 될 것이다. 따라서, 또한 한편에서는 간 세포 상에서 발현된 아시알로당단백질 수용체에 대한 친화도와, 다른 한편에서는 결합 분자 및 효소의 접합체에 대한 펩티드의 친화도 사이에는 균형이 이루어져야 한다. 특정 실시양태에서, 펩티드는 결합 분자 및 효소의 접합체에 대한, 및 특히 효소(의 활성 부위)에 대하여, 비-표적 위치로부터 효소를 제거할 수 있을 정도의 충분한 친화도를 갖는다. 특정 실시양태에서, 펩티드는 (또한) 표적 위치에서의 효소 활성에 유의하게 영향을 미치지 않을 정도로 충분히 빠른 해리 속도/높은 해리 상수를 가진다. 더욱 구체적으로, 결합 분자 및 효소의 접합체에 대한, 및 특히 효소(의 활성 부위)에 대한 펩티드의 친화도는 그의 표적 위치에 대한 결합 분자의 결합 친화도보다 약 10배 (예를 들어, 약 5 내지 약 15배 (상기 범위 사이의 모든 값 포함)) 더 낮을 수 있다. 따라서, 결합 분자 및 효소의 접합체의 표적 위치에의 결합이 제거에 비해 유리하지만, 결합 분자 및 효소의 접합체에 대한, 및 특히 효소(의 활성 부위)에 대한 펩티드의 친화도는, 아시알로당단백질 수용체에의 결합 후 내재화를 통해 이루어지는, 간을 통한 비-결합 접합체의 효율적인 제거가 확실하게 이루어질 수 있을 정도로 여전히 충분하다.
적절한 동력학 및 친화도 특성을 나타내는 본 발명의 펩티드 제거제는 본원에서 예시된다. 그러한 동력학 및 친화도 특성은 임의의 적합한 수단을 사용하여 측정될 수 있다. 동력학적 파라미터 및 친화도 파라미터는, 주지 기법인 (예를 들어, 지이 헬쓰케어(GE healthcare)로부터 상업적으로 입수가능한 것 - 비아코어 시스템(Biacore system)) 표면 플라스몬 공명 (SPR)을 사용하는 결합 연구에 기반하여 도출할 수 있다. 적합한 장치를 사용하여 수득한 데이터를 적절한 소프트웨어에 의해 분석함으로써 관련 파라미터를 자동으로 도출할 수 있다. 적절한 결합 모델을 소프트웨어에 적용시켜 곡선에의 피트가 확실하게 가능한 한 근접하도록 할 수 있다. 0.2 미만의 chi2 값을 허용되는 피트로 간주할 수 있다.
특정 실시양태에서, 1-상태 결합 모델 (1:1 랭뮤어(Langmuir) 결합 모델)을 사용하여 상업적으로 입수가능한 비아이벨류에이션(Biaevaluation) 소프트웨어를 사용함으로써 데이터를 수득할 수 있다. 따라서, 본 발명의 펩티드 제거제는 특정 실시양태에서 약 1 x 103 M-1s-1 내지 1 x 107 M-1s-1 사이의 임의의 값, 또는 약 1 x 104 M-1s-1 내지 1 x 106 M-1s-1 사이의 임의의 값, 또는 약 1 x 105 M-1s-1 내지 5 x 105 M-1s-1 사이의 임의의 값인 온 속도 (결합 속도 상수, Ka)를 나타낼 수 있다. 본 발명의 특정 펩티드에 대한 구체적인 Ka 값은 본원에서 기술된다 (2.26 x 105 M-1s-1의 Ka). 본 발명의 펩티드 제거제는 특정 실시양태에서 약 1 x 10-5 s-1 내지 1 x 10-1 s-1 사이의 임의의 값, 또는 약 1 x 10-4 s-1 내지 1 x 10-2 s-1 사이의 임의의 값, 또는 약 1 x 10-3 s-1 내지 5 x 10-3 s-1 사이의 임의의 값인 오프 속도 (해리 속도 상수, Kd)를 나타낼 수 있다. 본 발명의 특정 펩티드에 대한 구체적인 Kd 값은 본원에서 기술된다 (1.14 x 10-3 s-1의 Kd).
따라서, 본 발명의 펩티드 제거제는 1:1 랭뮤어 모델에 따라, 특정 실시양태에서 약 1 x 106 M-1 내지 1 x 1010 M-1 사이의 임의의 값, 또는 약 1 x 107 M-1 내지 1 x 109 M-1 사이의 임의의 값, 또는 약 1 x 108 M-1 내지 5 x 108 M-1 사이의 임의의 값인 유도 평형 결합 상수 (KA)를 나타낼 수 있다. 본 발명의 특정 펩티드에 대한 구체적인 KA 값은 본원에서 기술된다 (약 1.98 x 108 M-1). 이로써, 특정 실시양태에서 약 1 x 10-7 M 내지 1 x 10-11 M 사이의 임의의 값, 또는 약 1 x 10-8 M 내지 1 x 10-10 M 사이의 임의의 값, 또는 약 4 x 10-9 M 내지 6 x 10-9 M 사이의 임의의 값인 정도의, 본 발명의 펩티드 제거제에 대한 해리 상수를 얻을 수 있다. 본 발명의 특정 펩티드에 대한 구체적인 KD 값은 본원에서 기술된다 (약 5.04 x 10-9 M).
별법으로, 특정 실시양태에서, 2-상태 결합 모델을 사용하여 상업적으로 입수가능한 비아이벨류에이션 소프트웨어를 사용함으로써 데이터를 수득할 수 있다. 따라서, 본 발명의 펩티드 제거제는 특정 실시양태에서 약 1 x 103 M-1s-1 내지 1 x 107 M-1s-1 사이의 임의의 값, 또는 약 1 x 104 M-1s-1 내지 1 x 106 M-1s-1 사이의 임의의 값, 또는 1 x 105 M-1s-1 내지 5 x 105 M-1s-1 사이의 임의의 값인 1차 온 속도 (결합 속도 상수, Ka1)를 나타낼 수 있다. 본 발명의 특정 펩티드에 대한 구체적인 Ka1 값은 본원에서 기술된다 (2.84 x 105 M-1s-1의 Ka1). 따라서, 본 발명의 펩티드 제거제는 특정 실시양태에서 약 0.001 s-1 내지 1 s-1 사이의 임의의 값, 또는 약 0.01 s-1 내지 0.1 s-1 사이의 임의의 값, 또는 약 0.01 s-1 내지 0.05 s-1 사이의 임의의 값 또는 0.02 s-1인 2차 온 속도 (결합 속도 상수, Ka2)를 나타낼 수 있다. 본 발명의 특정 펩티드에 대한 구체적인 Ka2 값은 본원에서 기술된다 (0.014 s-1의 Ka2).
본 발명의 펩티드 제거제는 특정 실시양태에서 약 0.001 s-1 내지 1 s-1 사이의 임의의 값, 또는 약 0.01 s-1 내지 0.1 s-1 사이의 임의의 값, 또는 약 0.01 s-1 내지 0.05 s-1 사이의 임의의 값 또는 0.03 s-1인 1차 오프 속도 (해리 속도 상수, Kd1)를 나타낼 수 있다. 본 발명의 특정 펩티드에 대한 구체적인 Kd1 값은 본원에서 기술된다 (0.0258 s-1의 Kd1). 본 발명의 펩티드 제거제는 특정 실시양태에서 약 1 x 10-5 s-1 내지 1 x 10-1 s-1 사이의 임의의 값, 또는 약 1 x 10-4 s-1 내지 1 x 10-2 s-1 사이의 임의의 값, 또는 약 1 x 10-3 s-1 내지 5 x 10-3 s-1 사이의 임의의 값인 2차 오프 속도 (해리 속도 상수, Kd2)를 나타낼 수 있다. 본 발명의 특정 펩티드에 대한 구체적인 Kd2 값은 본원에서 기술된다 (1.58 x 10-3 s-1의 Kd2).
본 발명의 펩티드 제거제는 2-상태 결합 모델 피트에 기초하여, 특정 실시양태에서 약 1 x 105 M-1 내지 1 x 1010 M-1 사이의 임의의 값, 또는 약 1 x 107 M-1 내지 1 x 109 M-1 사이의 임의의 값, 또는 약 1 x 108 M-1 내지 2 x 108 M-1 사이의 임의의 값인 유도 평형 결합 상수 (K)를 나타낼 수 있다. 본 발명의 특정 펩티드에 대한 구체적인 K 값은 본원에서 기술된다 (약 1.09 x 108 M-1). 이로써, 특정 실시양태에서 약 1 x 10-5 M 내지 1 x 10-10 M 사이의 임의의 값, 또는 약 1 x 10-7 M 내지 1 x 10-9 M 사이의 임의의 값인 정도의, 본 발명의 펩티드 제거제에 대한 해리 상수를 얻을 수 있다. 본 발명의 특정 펩티드에 대한 평균화된 구체적인 KD 값은 2-상태 반응 피트에 기초하여 본원에서 기술된다 (약 9 nM).
본 발명의 펩티드 제거제는 또한 효소 활성을 억제시킬 수 있는 적합한 능력을 갖는 것으로 밝혀졌다. 따라서, 본 발명의 펩티드 제거제는 마이크로몰 크기의 최대 절반 억제 농도 (IC50)를 나타낼 수 있다. 적합한 펩티드는 약 10 내지 약 1,000 μM, 더욱 구체적으로, 약 20 내지 200 μM, 예를 들어, 약 50 내지 150 μM의 IC50을 나타낼 수 있다.
상기 논의된 바와 같이, 펩티드는 결합 분자 및 효소의 접합체에 결합하고, 특정 실시양태에서, 효소에, 특히, 효소의 활성 부위에 결합한다. 따라서, 특정 실시양태에서, 펩티드는 충분히 높은 친화도로 효소의 활성 부위에 결합함으로써 효소 활성을 불활성화시키거나 차단하는 수단으로서 작용할 뿐만 아니라, 효소를 간으로 유도하는 수단으로서 작용한다. 바람직한 실시양태에서, 펩티드는 비록 상기 활성 부위에는 맞기는 하지만, 효소 활성에 대해서는 저항성을 띤다. 효소에는 결합하지만, 효소 활성에 대한 기질로서는 작용하지 않는 적합한 펩티드가 본원에서 상세하게 기술된다. 따라서, 본 발명의 펩티드 제거제의 서열 및 구조는 전형적으로 관심 대상 효소의 활성 부위에 의해 결정된다. 특정 실시양태에서, 펩티드는 활성 부위에 결합하지만, 효소에 의해 전환되지는 않는 기질 유사체이다. 따라서, 펩티드에 대한 출발점은 기질 분자의 서열일 수 있으며, 이어서, 이는 펩티드 제거제가 제조되도록 적절히 변형될 수 있다.
예를 들어, CPG2의 천연 기질은 엽산이다. 엽산은 아미드 결합을 통해 글루탐산에 연결된 프테로산으로 이루어진 분자이다. CPG2는 글루타메이트-프테로산 아미드 결합을 가수분해한다. 따라서, 본 발명의 펩티드는 상기 구조를 기초로 한 것일 수 있다. 특정 실시양태에서, 펩티드는 글루타메이트 잔기를 포함하지만, (천연적으로 발생된 L형과 반대되는) D형이다. 본 발명의 펩티드는 서열 - 트립토판-페닐알라닌-글루타메이트 (WFE)를 포함할 수 있고, 임의로는 L-아미노산 대신 D-아미노산으로서의 글루타메이트를 도입할 수 있다. CPG2에 의한 글루타메이트-페닐알라닌 펩티드 결합이 용해되지 못하도록 하기 위해 D형 아미노산을 포함한다. 상기 서열은 가능하게는 아미노 말단에는 비시클릭 질소 치환된 환 구조, 중간 부분에는 6-원 방향족 환, 및 카르복시 말단에는 글루타메이트를 가진 트리펩티드를 가지는 바, 구조상 엽산과 가장 유사하다. 본 발명의 펩티드는 상기 기본 아미노산 서열에 대한 변형을 나타낼 수 있다. 당업자에게는 자명할 것인 바, 예를 들어, 아미노산을 전하 또는 소수성 또는 크기가 동일하거나 유사한 다른 아미노산으로 대체하는 등의 보존적 치환을 가할 수 있다. 따라서, 예를 들어, 각각 지방족 측쇄를 가진 글리신, 알라닌, 발린, 류신 및 이소류신 등의 아미노산은 특정 실시양태에서 서로서로 치환될 수 있다. 페닐알라닌, 티로신 및 트립토판 각각은 방향족 측쇄를 가지는 바, 따라서, 특정 실시양태에서 이들은 서로서로 치환될 수 있다. 시스테인 및 메티오닌 둘 모두는 황-함유 측쇄를 가지는 바, 따라서, 특정 실시양태에서 이들은 서로서로 치환될 수 있다. 세린 및 트레오닌은 지방족 히드록실 측쇄를 가지는 바, 따라서, 특정 실시양태에서 이들은 서로서로 치환될 수 있다. 리신, 아르기닌, 및 히스티딘은 염기성 측쇄를 가지는 바, 따라서, 특정 실시양태에서 이들은 서로서로 치환될 수 있다. 아스파르테이트 및 글루타메이트는 모두 산성이며, 특정 실시양태에서 이들은 서로서로 치환될 수 있다. 유사하게, 이들의 아미드 유도체, 아스파라긴 및 글루타민은 특정 실시양태에서 서로서로 치환될 수 있다. 비-천연 아미노산은 효소 (효소의 활성 부위)에의 결합을 개선시키기 위한 목적으로 도입될 수 있다. 분자 구조에 대하여 특징이 잘 규명되어 있는 비-천연 아미노산 다수가 당업계에 공지되어 있으며, 이들은 결합 특징을 개선시키기 위한 목적으로 합성된 펩티드 내로 쉽게 도입될 수 있다. 예로서 2-아미노아디프산 (Aad), 2-아미노아디프산 (bAad), 베타-알라닌, 베타-아미노프로피온산 (bAla), 2-아미노부티르산 (Abu), 4-아미노부티르산, 피페리딘산 (4Abu), 6-아미노카프로산 (Acp), 2-아미노헵탄산 (Ahe), 2-아미노이소부티르산 (Aib), 3-아미노이소부티르산 (bAib), 2-아미노피멜린산 (Apm), 2,4-디아미노부티르산 (Dbu), 데스모신 (Des), 2,2'-디아미노피멜린산 (Dpm), 2,3-디아미노프로피온산 (Dpr), N-에틸글리신 (EtGly), N-에틸아스파라긴 (EtAsn), 히드록시리신 (Hyl), 알로-히드록시리신 (aHyl), 3-히드록시프롤린 (3Hyp), 4-히드록시프롤린 (4Hyp), 이소데스모신 (Ide), 알로-이소류신 (aIle), N-메틸글리신, 사르코신 (MeGly), N-메틸이소류신 (MeIle), 6-N-메틸리신 (MeLys), N-메틸발린 (MeVal), 노르발린 (Nva), 노르류신 (Nle) 및 오르니틴 (Orn)을 들 수 있다.
CPG2에 결합하고 효소 활성을 효과적으로 억제시키는 본 발명의 구체적인 펩티드 제거제가 본원에서 기술된다. 그러한 펩티드는 하기 아미노산 서열:
아미노-나프토산 (ANA)-글루타메이트 (Glu)를 포함하거나, 본질적으로 그로 이루어지거나, 또는 그로 이루어질 수 있다.
본 발명의 펩티드는 하기 화학식 I의 디펩티드를 포함하거나, 본질적으로 그로 이루어지거나, 또는 그로 이루어질 수 있다.
<화학식 I>

본 발명의 제거제와 관련된 바람직한 특성을 나타내는 상기와 같은 디펩티드를 본원에서 제시한다. 상기 펩티드는 CPG2의 활성 부위에 대하여 나노몰 크기의 친화도를 가지지만, 기질로서의 작용을 하는 것은 아니다. IC50은 마이크로몰 크기로 50 내지 150 μM 사이, 더욱 구체적으로 약 88.5 μM이다. 본원에서 기술하는 바와 같이, 펩티드는 효소의 활성 부위에 결합하는 것 이외에도 또한 간 세포에 의해 발현되는 아시알로당단백질 수용체에 결합함으로써 간을 통한 제거가 촉진되어야 한다. 따라서, 상기와 같은 기본 디펩티드는 본원에서 논의되는 바와 같이, 제거를 허용하도록 확장될 수 있다. 그러한 펩티드는 3, 4, 5, 6, 7, 8, 9 또는 10개 이상의 아미노산 길이일 수 있고, 글리코실화될 수 있다.
본 발명의 특정 실시양태에서, 펩티드는 아미노산 서열:
세린 (Ser) - 알라닌 (Ala) - 아미노-나프토산 (ANA) - 글루타메이트 (Glu) (서열 1)를 포함하거나, 본질적으로 그로 이루어지거나, 또는 그로 이루어질 수 있다.
펩티드는 제거가 촉진되도록 변형될 수 있다. 펩티드는 아시알로당단백질 수용체를 발현하는 간 세포에 결합할 수 있도록 적합한 잔기에서 글리코실화될 수 있다. 특정 실시양태에서, 세린 잔기가 글리코실화된다. 펩티드는 본원에서 논의되는 바와 같이, 추가의 아미노산 잔기, 예를 들어, 총 길이가 4, 5, 6, 7, 8, 9 또는 10개의 아미노산을 포함하여, 적절하게는 최대 10, 20 또는 30개까지의 아미노산으로 될 수 있다. 본 발명의 펩티드는 하기 화학식 II의 펩티드를 포함하거나, 본질적으로 그로 이루어지거나, 또는 그로 이루어질 수 있다.
<화학식 II>

본 발명의 제거제와 관련된 바람직한 특성을 나타내는 상기와 같은 디펩티드를 본원에서 제시한다. 상기 펩티드는 CPG2의 활성 부위에 대하여 나노몰 크기의 친화도를 가지지만, 기질로서의 작용을 하는 것은 아니다. IC50은 마이크로몰 크기로 50 내지 150 μM 사이, 더욱 구체적으로 약 110 μM이다. 본원에서 논의되는 바와 같이, 본 발명의 펩티드는 천연적으로 발생되는 L형과 반대되는 D형 아미노산을 함유할 수 있다(상이한 광학 이성질체). 특히, 글루타메이트 잔기는 D형으로 제공될 수 있다. 또한, 본원에서 논의되는 바와 같이, 본 발명의 펩티드 제거제로서의 관능성이 유지되는 한, 펩티드 중의 아미노산 중 어느 것에 대하여 치환 및 특히 보존적 치환이 이루어질 수 있다.
본 발명의 펩티드는 최소의 면역원성을 띠는 바, 항체 기반 제거제에 비하여 이점을 제공한다. 이는 그 길이가 상대적으로 짧기 때문이다. 본 발명의 펩티드 제거제들은 각각 전형적으로 그 크기가 작음에도 불구하고, 뚜렷이 다른 2개의 도메인으로 구성된다. 제1 도메인은 표적 효소의 활성 부위에 결합은 하지만, 효소 작용에 의해 공유적으로 변경되지는 않는, 짧은 아미노산 서열을 포함한다 (상기 논의된 바와 같다). 상기 도메인은 비-천연 아미노산, 예를 들어, D형 아미노산을 포함할 수 있는데, 이러한 아미노산은 펩티드가 일단 활성 부위에 결합한 후에도 효소가 펩티드에 작용하지 못하도록 한다. 따라서, 특정 실시양태에서, 본 발명의 펩티드의 제1 도메인의 길이는 10, 15 또는 20개 이하의 아미노산이다. 더욱 특정 실시양태에서, 펩티드의 제1 도메인의 길이는 3 내지 10개의 아미노산, 예를 들어, 그 길이는 3, 4, 5, 6, 7, 8, 9 또는 10개의 아미노산이다. 특정 실시양태에서, 펩티드, 또는 그의 제1 도메인은 헵타펩티드이다.
제2 도메인은 간 세포에 의해 발현된 아시알로당단백질 수용체에 결합한다. 이를 통해 비-표적 위치에서 발견되는 접합체는 내재화된다. 따라서, 펩티드가 간 세포에 의해 발현되는 아시알로당단백질 수용체에 결합하게 되면 펩티드의 제거는 촉진된다. 특정 실시양태에서, 펩티드는 연속되는 아미노산 서열, 또는 대체 화학적 구조 부착물을 포함할 수 있다. 각각은, 아시알로당단백질 수용체를 가진 간 세포에 결합할 수 있고, 그에 의해 이용될 수 있는 기인, 하나 이상의 펜던트 당, 바람직하게는 갈락토스를 지닌다. 따라서, 특정 실시양태에서, 본 발명의 펩티드 제거제의 글리코실화는 하나 이상의 갈락토스 기를 커플링하는 것을 수반한다. 특정 실시양태에서, 제2 도메인은 하나 이상의 갈락토실 세린 아미노산 (즉, 세린 히드록실 기의 O 잔기에 공유적으로 연결된 갈락토스)를 포함함으로써 O-연결된 글리코실 펩티드를 생성한다. 상기 물질은 많은 공급업체로부터 상업적으로 입수가능하다. 필요에 따라, 임의 수의 상기 추가의 갈락토실 세린 기를 공지된 펩티드 화학 기법에 의해 첨가함으로써 간 세포 (간세포(hepatocyte))의 아시알로당단백질 수용체에의 글리코실 펩티드의 결합을 강화시킬 수 있다. 갈락토실 세린은 펩티드 (펩티드의 제1 도메인)에 직접 첨가될 수 있거나, 또는 링커 아미노산 또는 아미노산 스트레치를 통해 첨가될 수 있다. 따라서, 링커는 하나 이상의 아미노산, 특히, 글리신 잔기를 포함할 수 있다. 다수의 갈락토스 모이어티가 첨가될 수 있는데, 이들은 각각 하나 이상의 스페이서 아미노산에 의해 이격화될 수 있다. 따라서, 갈락토실 기의 배향 및 배치를 최적화시키기 위해 하나 이상의 추가의 아미노산에 의해 추가의 갈락토실 세린 잔기들 사이의 공간이 구축될 수 있다. 물론, 적절하다면, 갈락토실 세린 대신 대체 갈락토실 아미노산이 사용될 수 있다.
또한, 펩티드의 아미노산 서열이 (인간) 대상체의 임의의 활성 펩티드를 모방하지 않는 것이 중요한데; 그렇지 않을 경우, 펩티드는 바람직한 못한 생물학적 활성을 가질 수 있다. 추가로, 펩티드는 바람직한 못한 활성을 초래할 수 있는 임의의 내인성 (인간) 단백질(의 활성 부위), 특히, 효소 또는 수용체에 결합하지 않아야 한다. 아미노산 서열 데이터베이스 검색을 사용하여 후보물질을 확인한 후, 예를 들어, 적절한 시험관내 시험을 사용하여 시험할 수 있다.
전형적으로, 본 발명의 펩티드는 화학적으로 합성된다. 그러나, 원하는 경우, 재조합적으로 제조된 펩티드가 사용될 수 있다. 이어서, 재조합적으로 제조된 펩티드를 발현 후에 글리코실화시킬 수 있거나, 발현을 유도하는 적절한 세포 유형이 선택된 경우에는 세포에서 글리코실화시킬 수 있다. 본 발명의 펩티드 제거제를 제조하는 적합한 반응식은 실험 실시예를 참고로 하여 본원에서 보다 상세히 기재된다.
본 발명의 펩티드는 임의의 적합한 기법에 의해 제조될 수 있다. 이는 유전공학처리된 바이러스 또는 세포의 표면 상에 어레이된 랜덤 펩티드 라이브러리 (파지 디스플레이, 2-하이브리드 시스템 등)을 수단으로 제조될 수 있거나, 또는 합성되어 구조화된 표면 상에 어레이될 수 있다 (예를 들어, 펩스캔(PEPSCAN) 라이브러리). 펩스캔 프로토콜에 따라 수용액 중에서 표적 효소를 펩티드가 어레이된 상기 표면에 노출시킨 후, 이어서 프로세싱하여 디스플레이된 펩티드를 통해 효소의 상기 표면에의 결합을 검출할 것이다. 상기 절차를 1회 수행한 후에는 단지 약한 결합만을 얻을 수 있지만, 이어서 양성 결합 서열은 보다 장쇄의 펩티드를 제조하기 위한 확장을 비롯한 추가 변형을 위한 출발점으로서 사용될 수 있다. 이어서, 이러한 보다 장쇄의 펩티드를 스크리닝하고, 서열의 각 위치에 있는 아미노산의 체계적인 변경을 통해 개선된 결합제를 추가로 변형시킴으로써 최적화된 펩티드를 제조할 수 있다. 특정의 변경은 결합률을 약간 더 높이고, 이어서, 원하는 친화도 및 특이성을 가진 결합 서열이 출현할 때까지 상기와 같이 좀더 개선된 서열을 추가의 더 많은 회차의 체계적 치환에 대한 추가 출발점으로서 사용한다. 이어서, 구축물을 완성하기 위해 당을 가진 도메인을 첨가할 수 있다. 공지의 표준 펩티드 합성법 및 글리코실화 방법에 의해 이들 물질을 제조할 수 있다.
따라서, 본 발명은 또한
a. 임의로 효소 기질의 구조적 유사체인 출발 펩티드를 기초로 하여, 펩티드 어레이를 제조하는 단계,
b. 효소 및 표적 위치에 특이적으로 결합하는 결합 분자의 접합체에 대한 결합 친화도에 대해 펩티드 어레이를 스크리닝하고, 결합 친화도를 갖는 것을 선별하는 단계,
c. 임의로, 단계 b에서의 결합 친화도를 갖는 펩티드의 아미노산 서열을 변형시키고, 변형된 펩티드를 사용하여 단계 b를 반복함으로써 결합 친화도 개선에 대해 시험하는 단계,
d. 임의로, 단계 c에서 개선된 결합 친화도를 갖는 것으로 나타난 변형된 펩티드의 각 잔기에서 치환을 실시하고, 단계 b를 반복하여 치환된 펩티드 중 임의의 것이 추가로 개선된 결합 친화도를 갖는지 여부를 측정하는 단계,
e. 임의로, 단계 c 또는 d로부터 생성된 (개선된 결합 친화도를 갖는) 펩티드가 효소 활성을 방지하거나 또는 억제할 수 있는 능력을 갖는지 여부를 측정하는 단계,
f. 임의로, 대상체로부터 얻은 일련의 대조군 단백질을 사용하여 시험함으로써 효소에 대한 결합 특이성을 확인하는 단계,
g. 간 세포에 의해 발현된 아시알로당단백질 수용체에 결합함으로써 간을 통한 제거를 촉진하는 능력에 대해 펩티드를 시험하는 단계를 포함하며,
여기서, 단계 e 및 f가 실시되는 경우, 이들은 임의 순서로 수행될 수 있고, 단계 g는 단계 a에서 간 세포에 의해 발현된 아시알로당단백질 수용체에 결합함으로써 간을 통한 제거를 촉진할 수 있는 펩티드 어레이를 제공하는 것을 비롯한, 상기 방법 중 임의의 시점에서 수행될 수 있는 것인, (최적화된) 본 발명의 펩티드 제거제를 제조하는 방법을 제공한다.
단계 g의 펩티드는 확실하게 펩티드가 간 세포에 의해 발현된 아시알로당단백질 수용체에 결합함으로써 간을 통한 제거가 촉진될 수 있도록 하기 위해 변형될 수 있다. 따라서, 본 발명의 특정 실시양태에서, 단계 g는 펩티드 또는 펩티드들을 글리코실화하는 것을 포함한다. 따라서, 단계 a는 글리코실화된 펩티드 어레이를 제공하는 것을 포함할 수 있다.
따라서, 펩티드 어레이는 전형적으로 고체 표면 상에, 예를 들어, 펩스캔 유형의 실시양태에서는 다중-웰 플레이트의 웰 중에 고정화된다. 임의의 적합한 고체 표면이 사용될 수 있다. 고체 표면 상에서 펩티드를 합성할 수 있거나, 또는 펩티드를 합성한 후, 이어서 고정화시킬 수 있다. 별법으로, 파지 디스플레이 유형의 실시양태 또는 관련 기술에서, 효소 (또는 결합 분자 및 효소의 접합체)는 고정화되는 실체이며, 파지 디스플레이 라이브러리는 고정화된 효소에 대해 스크리닝된다.
펩티드 어레이는 펩티드 라이브러리를 포함할 수 있다. 파지 디스플레이 또는 관련 기술은 가장 강력한 결합제를 확인하기 위해 수회차에 걸쳐 디스플레이되고 효소에 대해 스크리닝된, 본질상 랜덤 펩티드 라이브러리에 의존할 수 있다. 특정 실시양태에서, 출발 펩티드는 결합 분자 및 효소의 접합체, 및 특정 실시양태에서는, 효소, 특히 효소의 활성 부위에 결합하도록 디자인된 짧은 펩티드이다. 펩티드의 출발 서열 및 구조는 관심 대상 효소의 활성 부위의 형상에 의해 결정될 수 있다. 특정 실시양태에서, 출발 펩티드는 활성 부위에 결합할 수는 있지만, 효소에 의해 전환되지는 않는 예측된 기질 유사체이다. 따라서, 펩티드에 대한 출발점은 기질 분자의 서열 또는 구조일 수 있으며, 이어서, 이는 펩티드 제거제를 제조하는 본 발명에 따라 변형될 수 있다. 특정 실시양태에서, 이는 트리펩티드일 수 있다. 예를 들어, 상기에서 논의된 바와 같이, CPG2의 천연 기질은 엽산이다. 따라서, 본 방법에 대한 출발점은 상기 구조를 기초로 할 수 있다. 특정 실시양태에서, 펩티드 출발점은 엽산에서 발견되는 글루타메이트 잔기를 포함하지만, (보통은 L형인 것과 반대되는) D형이다. 출발점 펩티드는 서열 - 트립토판-페닐알라닌-글루타메이트 (WFE)를 포함할 수 있고, 임의로는 L-아미노산 대신 D-아미노산으로서 글루타메이트를 도입할 수 있다. 별법으로, 출발점은 디펩티드 ANA-Glu이거나, 또는 서열 1의 펩티드 (Ser-Ala-ANA-Glu)일 수 있다. 이어서, 본 발명의 방법은 상기 출발 서열을 체계적으로 변형시킴으로써 원하는 특성을 가진 펩티드 제거제를 제조할 수 있다. 따라서, 펩티드 어레이 또는 라이브러리는 다양한 트리펩티드 유도체와 함께 트리펩티드 WFE를 포함할 수 있다. 유도체는 모든 아미노산이 체계적으로 치환된 펩티드를 포함할 수 있거나, 또는 예를 들어, (본원에 정의된 것과 같은) 오직 보존적으로 치환된 아미노산만을 포함할 수 있다.
단계 b는 표적 위치에 특이적으로 결합하는 결합 분자 및 효소의 접합체에 대한 결합 친화도에 대해 펩티드 어레이를 스크리닝하고, 결합 친화도를 갖는 것을 선별하는 것을 포함한다. 명시된 바와 같이, 결합에 대해 스크리닝하는 것은 전형적으로는 효소 활성 부위에의 결합에 대해 스크리닝하는 것이다. 임의의 적합한 스크린을 사용할 수 있다. 파지 디스플레이 유형의 실시양태에서, 스크린은 결합에 기반한다. 세척 단계에서는 임의의 비-결합 파지는 세척 제거된다. 결합 펩티드를 발현하는 파지가 풍부해질 수 있도록, 파지 용리 및 더 많은 파지 생산을 위한 추가 감염 후에 단계 b를 다수 회 반복할 수 있다. 이어서, 파지에 의해 발현되는 서열에 기초하여 효소에 결합할 수 있는 펩티드의 서열을 쉽게 결정할 수 있다. 이어서, 원하는 경우, 회수된 펩티드를 상기 방법의 추가 단계에 따라 변형시킬 수 있다.
펩스캔 유형의 실시양태에서, 효소 (또는 효소-결합 분자 접합체)를 라벨로 표지할 수 있다. 펩티드가 효소에 결합할 경우, 이는 고정화될 것이다. 세척 단계 후, 효소에 직접 결합하거나, 또는 효소가 표지된 경우에는 라벨에 결합하는 리포터를 첨가한다. 리포터는 그 자체가 효소, 예를 들어, 알칼리성 포스파타제일 수 있다. 리포터가 반드시 효소 또는 라벨 자체에 결합하는 것은 아니며, 이는 유사하게 라벨 또는 효소에 결합하는 결합 분자에 부착될 수 있다. 따라서, 특정 실시양태에서, 효소는 비오틴으로 표지된다. 리포터는 아비딘 분자, 예를 들어, 스트렙타비딘에 부착된다. 비오틴과 스트렙타비딘 사이의 상호작용을 통해 리포터 활성이 효소의 부위로 효과적으로 국소화되고, 이로써, (적절한 세척 단계 후) 성공적으로 효소에 결합한 펩티드의 위치가 밝혀질 수 있다. 본 방법의 구체적인 예는 실시예 1에 기재되어 있다.
임의적 단계 c는 전형적으로 결합 친화도를 개선시키고자 하는 시도로 펩티드 아미노산 서열을 확장하는 것을 수반한다. 그러나, 예를 들어, 기존 아미노산을 대체하기 위해 비-천연 아미노산을 사용하여 치환하는 것과 같은 다른 변형도 이루어질 수 있다. 특정 실시양태에서, 한번에 1, 2, 3, 4 또는 5개의 아미노산이 확장될 수 있다. 이어서, 그렇게 확장된 펩티드를 단계 b에 따라 다시 시험함으로써 어떤 변형이 결합 친화도의 개선을 초래하는지를 결정한다.
이어서, 단계 d는, 단계 c로부터 얻은, 개선된 결합 친화도를 나타내는 펩티드 또는 펩티드들에 대해 전체 위치 스캔하는 것이다. "전체 위치 스캔"은 각각의 개별 아미노산을 치환하여 일련의 펩티드를 제조함으로써 결합이 추가로 개선될 수 있는지 여부를 측정하는 것을 의미하는 것이다. 이를 통해 표적 효소에 대하여 최적의 결합 친화도를 갖는 펩티드 또는 펩티드들이 제조된다.
이어서, 추가 시험을 수행할 수 있다. 따라서, 접합체에 대해 개선된 결합 친화도를 갖는 펩티드가 또한 효소 활성을 방지하거나 또는 억제할 수 있는 능력을 갖는지 여부를 측정하기 위해 펩티드 또는 펩티드들을 시험할 수 있다. 상기 논의된 바와 같이, 펩티드가 활성 부위에 결합하도록 디자인된 경우, (결합 친화도에 대해 시험하는) 스크린은 효소에 의해 작용을 받는 펩티드가 양성 결과를 가져오지 못하도록 할 수 있기 때문에 상기 단계는 필요하지 않을 수도 있다.
상기 논의된 바와 같이, 펩티드의 아미노산 서열이 (인간) 대상체의 임의의 활성 펩티드를 모방하지 않는 것 또한 중요한데; 그렇지 않을 경우, 펩티드는 바람직한 못한 생물학적 활성을 가질 수 있다. 나아가, 펩티드는 임의의 내인성 (인간) 효소의 활성 부위에 결합하지 않아야 한다. 따라서, 본 발명의 방법은 임의로 대상체로부터 얻은 일련의 대조군 단백질을 사용하여 시험함으로써 효소에 대한 결합 특이성을 확인하는 것을 포함할 수 있다. 적합한 대조군은 관심 대상 효소에 기초하여 결정될 수 있다. 예를 들어, 아미노산 서열 또는 구조 데이터베이스 검색을 사용하여 유사한 활성 부위 및/또는 1차 아미노산 서열을 가진 단백질을 확인하고, 그에 따라 결합에 대해 시험할 수 있다. 추가로 또는 별법으로, 임의의 부작용이 있는지에 대해 측정하기 위해 적합한 동물 모델에서 펩티드를 시험할 수 있다. 특정 실시양태에서, 본 방법은 본 방법에 의해 제조된 펩티드의 아미노산 서열을 인간 아미노산 서열 데이터베이스와 비교하여 펩티드가 비바람직한 생물학적 활성을 가질 가능성이 낮음을 확인하는 단계를 추가로 포함한다.
펩티드 (또는 펩티드들)는 간 세포에 의해 발현되는 아시알로당단백질 수용체에 결합한다. 이를 통해 비-표적 위치에서 발견되는 접합체는 내재화된다. 따라서, 펩티드가 간 세포에 의해 발현되는 아시알로당단백질 수용체에 결합하게 되면 펩티드의 제거는 촉진된다. 특정 실시양태에서, 펩티드 또는 펩티드들은 본 발명에서 제거제로서 유용하도록 글리코실화된다. 글리코실화는 본 방법의 임의 시점에서 이루어질 수 있다. 전형적으로는, 이는 일단 하나 이상의 적합한 후보물질이 본 발명의 방법에 따라 제조된 후에 이루어진다. 이는 펩티드 또는 펩티드들의 추가 시험 이전에 이루어질 수 있다. 특별히 파지 디스플레이 유형의 실시양태에서, 글리코실화는 후보 결합 펩티드의 단리 이후에 이루어진다 - 파지에 의해 발현된 펩티드는 글리코실화되지 않을 것이다. 대안적 실시양태에서, 출발 펩티드는 글리코실화된 것일 수 있거나, 또는 펩티드는 단계 b 또는 d 이전에 글리코실화될 수 있다. 글리코실화는 임의의 적합한 수단을 통해 이루어질 수 있고, 그러한 수단의 일례는 본원에서 논의된다 (논의에서는 필요한 변경을 가하는 것을 적용한다).
본 발명의 펩티드 제거제는 특히 치료 방법, 예를 들어, 효소를 사용함으로써 결합 분자 및 효소의 접합체를 통해 효소를 국소화시켜 전구약물을 활성 약물로 전환시키는 ADEPT에 적용될 수 있다. 따라서, 본 발명은 또한 본 발명의 펩티드 제거제가 사용되는 치료 방법, 예를 들어, 효소를 사용함으로써 결합 분자 및 효소의 접합체를 통해 효소를 국소화시켜 전구약물을 활성 약물로 전환시키는 ADEPT를 제공한다. 유사하게, 본 발명은 효소를 사용함으로써 결합 분자 및 효소의 접합체를 통해 효소를 국소화시켜 전구약물을 활성 약물로 전환시키는 치료 방법에서, 특히 항체 지향 효소 전구약물 치료법 (ADEPT)에서 제거제로서의 본 발명의 펩티드의 용도를 제공한다. 이러한 측면은 또한 의학적 용도면에서 만들어질 수 있다. 따라서, 본 발명은 효소를 사용함으로써 결합 분자 및 효소의 접합체를 통해 효소를 국소화시켜 전구약물을 활성 약물로 전환시키는 치료 방법에서, 특히 항체 지향 효소 전구약물 치료법 (ADEPT)에서 제거제로서 사용하기 위한 본 발명의 펩티드를 제공한다. 유사하게, 본 발명은 효소를 사용함으로써 결합 분자 및 효소의 접합체를 통해 효소를 국소화시켜 전구약물을 활성 약물로 전환시키는 치료 방법에서, 특히 항체 지향 효소 전구약물 치료법 (ADEPT)에서 사용하기 위한 제거제의 제조에서의 본 발명의 펩티드의 용도를 제공한다.
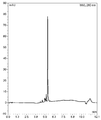
도 1은 CP014 갈락토스 모이어티가 아세틸화된 생성물의 HPLC 스펙트럼이다.
도 2는 CP014 갈락토스 모이어티가 탈아세틸화된 생성물의 HPLC 스펙트럼이다.
도 3은 CP014의 구조를 확인하는 ESI 데이터 (전기분무 질량 분광측정법)를 나타낸 것이다.
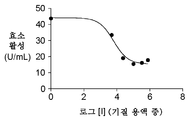
도 4는 CP014에 의한 ScFV 항-CEA-CPG2의 CPG2 효소 활성 억제를 보여주는 것으로서, CP014가 ScFV 항-CEA-CPG2의 억제제임을 제시한다. IC50 계산치는 110 μM이다.
도 5는 CP014가 ScFV 항-CEA-CPG2에 의해 전환되지 않는다는 것을 보여주는 앰플렉스 레드 검정의 결과를 나타낸 것이다. 앰플렉스 레드 검정에서 기질인 엽산 및 메토트렉세이트는 양성 대조군으로서 제시된다. CP014를 사용하였을 때, 형광 신호가 없다는 것은 CPG2가 기질로서 CP014를 사용할 수 없다는 것을 입증한다.
도 6a는 1:1 랭뮤어 결합 모델을 적용한 CP014와 ScFv 항-CEA-CPG2 융합 단백질의 상호작용을 제시하는 비아코어 데이터를 나타낸 것이다.
도 6b는 비아코어 데이터의 1:1 랭뮤어 결합 모델에의 피트를 제시하는 잔차 플롯을 나타낸 것이다.
도 6c는 2-상태 반응 피트를 적용한 CP014와 ScFv 항-CEA-CPG2 융합 단백질의 상호작용을 제시하는 비아코어 데이터를 나타낸 것이다.
도 6d는 비아코어 데이터의 2-상태 결합 모델에의 피트를 제시하는 잔차 플롯을 나타낸 것이다.
도 7은 주요 분자량 피크를 제시하는 CP006의 전기분무 질량 스펙트럼을 나타낸 것이다.
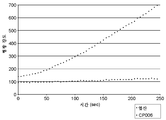
도 8은 억제 검정에서의 CP006에 의한 ScFv 항-CEA-CPG2 융합물의 억제를 나타낸 것이다. 스펙트럼 데이터는 미가공 데이터이다.
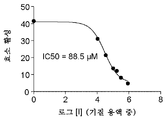
도 9는 억제 검정에서의 CP006에 의한 ScFv 항-CEA-CPG2 융합물의 억제를 나타낸 것이다. 억제 곡선은 IC50이 88.5 μM인 것을 제시한다.
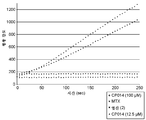
도 10은 CP006 및 MFECP에 대한 낮은 기질 활성을 제시하는 앰플렉스 레드 분석법의 결과를 나타낸 것이다. 기질 농도는 12.5 μM이었다.
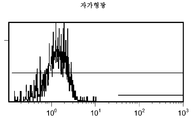
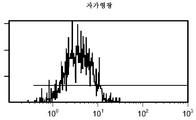
도 11a/b는 대조군 (CEA 표면 항원 음성) 세포주 (CRL 1573)를 사용하여 수행된, 자가형광에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
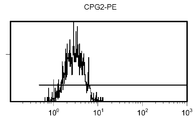
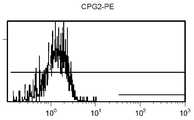
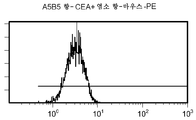
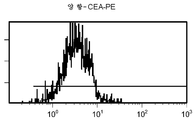
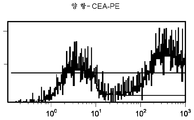
도 11 c/d는 대조군 (CEA 표면 항원 음성) 세포주 (CRL 1573)를 사용하여 수행된, CPG2-PE 결합에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
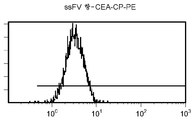
도 11e/f는 대조군 (CEA 표면 항원 음성) 세포주 (CRL 1573)를 사용하여 수행된, ssFV 항-CEA-CP-PE 결합에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
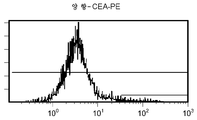
도 11g/h는 대조군 (CEA 표면 항원 음성) 세포주 (CRL 1573)를 사용하여 수행된, 양 항-CEA-PE 결합에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
도 11 i/j는 대조군 (CEA 표면 항원 음성) 세포주 (CRL 1573)를 사용하여 수행된, A5B5 항-CEA + 염소 항-마우스-PE 결합에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
도 12a/b는 CEA 표면 항원 양성 세포주 (CCI-229)를 사용하여 수행된, 자가형광에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
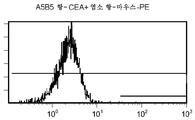
도 12c/d는 CEA 표면 항원 양성 세포주 (CCI-229)를 사용하여 수행된, CPG2-PE 결합에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
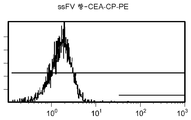
도 12e/f는 CEA 표면 항원 양성 세포주 (CCI-229)를 사용하여 수행된, ssFV 항-CEA-CP-PE 결합에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
도 12g/h는 CEA 표면 항원 양성 세포주 (CCI-229)를 사용하여 수행된, 양 항-CEA-PE 결합에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
도 12i/j는 CEA 표면 항원 양성 세포주 (CCI-229)를 사용하여 수행된, A5B5 항-CEA + 염소 항-마우스-PE 결합에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
도 2는 CP014 갈락토스 모이어티가 탈아세틸화된 생성물의 HPLC 스펙트럼이다.
도 3은 CP014의 구조를 확인하는 ESI 데이터 (전기분무 질량 분광측정법)를 나타낸 것이다.
도 4는 CP014에 의한 ScFV 항-CEA-CPG2의 CPG2 효소 활성 억제를 보여주는 것으로서, CP014가 ScFV 항-CEA-CPG2의 억제제임을 제시한다. IC50 계산치는 110 μM이다.
도 5는 CP014가 ScFV 항-CEA-CPG2에 의해 전환되지 않는다는 것을 보여주는 앰플렉스 레드 검정의 결과를 나타낸 것이다. 앰플렉스 레드 검정에서 기질인 엽산 및 메토트렉세이트는 양성 대조군으로서 제시된다. CP014를 사용하였을 때, 형광 신호가 없다는 것은 CPG2가 기질로서 CP014를 사용할 수 없다는 것을 입증한다.
도 6a는 1:1 랭뮤어 결합 모델을 적용한 CP014와 ScFv 항-CEA-CPG2 융합 단백질의 상호작용을 제시하는 비아코어 데이터를 나타낸 것이다.
도 6b는 비아코어 데이터의 1:1 랭뮤어 결합 모델에의 피트를 제시하는 잔차 플롯을 나타낸 것이다.
도 6c는 2-상태 반응 피트를 적용한 CP014와 ScFv 항-CEA-CPG2 융합 단백질의 상호작용을 제시하는 비아코어 데이터를 나타낸 것이다.
도 6d는 비아코어 데이터의 2-상태 결합 모델에의 피트를 제시하는 잔차 플롯을 나타낸 것이다.
도 7은 주요 분자량 피크를 제시하는 CP006의 전기분무 질량 스펙트럼을 나타낸 것이다.
도 8은 억제 검정에서의 CP006에 의한 ScFv 항-CEA-CPG2 융합물의 억제를 나타낸 것이다. 스펙트럼 데이터는 미가공 데이터이다.
도 9는 억제 검정에서의 CP006에 의한 ScFv 항-CEA-CPG2 융합물의 억제를 나타낸 것이다. 억제 곡선은 IC50이 88.5 μM인 것을 제시한다.
도 10은 CP006 및 MFECP에 대한 낮은 기질 활성을 제시하는 앰플렉스 레드 분석법의 결과를 나타낸 것이다. 기질 농도는 12.5 μM이었다.
도 11a/b는 대조군 (CEA 표면 항원 음성) 세포주 (CRL 1573)를 사용하여 수행된, 자가형광에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
도 11 c/d는 대조군 (CEA 표면 항원 음성) 세포주 (CRL 1573)를 사용하여 수행된, CPG2-PE 결합에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
도 11e/f는 대조군 (CEA 표면 항원 음성) 세포주 (CRL 1573)를 사용하여 수행된, ssFV 항-CEA-CP-PE 결합에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
도 11g/h는 대조군 (CEA 표면 항원 음성) 세포주 (CRL 1573)를 사용하여 수행된, 양 항-CEA-PE 결합에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
도 11 i/j는 대조군 (CEA 표면 항원 음성) 세포주 (CRL 1573)를 사용하여 수행된, A5B5 항-CEA + 염소 항-마우스-PE 결합에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
도 12a/b는 CEA 표면 항원 양성 세포주 (CCI-229)를 사용하여 수행된, 자가형광에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
도 12c/d는 CEA 표면 항원 양성 세포주 (CCI-229)를 사용하여 수행된, CPG2-PE 결합에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
도 12e/f는 CEA 표면 항원 양성 세포주 (CCI-229)를 사용하여 수행된, ssFV 항-CEA-CP-PE 결합에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
도 12g/h는 CEA 표면 항원 양성 세포주 (CCI-229)를 사용하여 수행된, 양 항-CEA-PE 결합에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
도 12i/j는 CEA 표면 항원 양성 세포주 (CCI-229)를 사용하여 수행된, A5B5 항-CEA + 염소 항-마우스-PE 결합에 대한 대조군의 유동 세포측정법 결과를 제시한 것이다.
본 발명은 하기의 비-제한적인 실시예를 참고로 하여 기술될 것이다:
실시예
1 - 효소
CPG2
에 대한
글리코실화된
결합제 펩티드를 포함하는 제거제 생성
펩스캔 기술 형태로 트리펩티드의 라이브러리를 합성하였다 (2 & 3). 플라스틱 웰 표면 안쪽에 부착된 로봇식 수단에 의해 펩티드를 합성하였다. 일련물에서 제1 펩티드는 CPG2에 대한 천연 기질인 엽산의 구조를 기초로 한 것이었다. 엽산은 아미드 결합을 통해 글루탐산에 연결된 프테로산으로 이루어진 분자이다. CPG2는 글루타메이트-프테로산 아미드 결합을 가수분해한다. 이 제1 트리펩티드는 하기 서열 - 트립토판-페닐알라닌-글루타메이트 (WFE)를 갖지만, 글루타메이트는 L-아미노산 대신 D-아미노산인데, 그 이유는 글루타메이트-페닐알라닌 펩티드 결합이 CPG2에 의한 용해의 대상이 되지 않도록 하기 위함이다. 가능하게는 아미노 말단에는 비시클릭 질소 치환된 환 구조, 중간 부분에는 6-원 방향족 환, 및 카르복시 말단에는 글루타메이트를 가진 트리펩티드를 통해 엽산과 가장 유사하게 되는 바, 상기 서열을 선택하였다. 트리펩티드 라이브러리의 나머지는 세 위치 각각에서의 모든 가능한 아미노산의 치환에 의해 상기 출발 구조물과는 체계적으로 상이한 펩티드로 이루어졌다.
트윈 20(Tween 20) (0.05% v/v) 및 소 혈청 알부민 (0.1% w/v)을 함유하는 포스페이트 완충 염수 (PBSTA) 중의 CPG2-비오틴 접합체 (0.1 ug/ml) 수용액을 각 웰에 분배하고, 60분 동안 인큐베이션한 후, 웰을 비우고 세정하였다. 이어서, PBSTA + 0.5 mM 염화마그네슘 중 스트렙타비딘-알칼리성 포스파타제 접합체 (1 ug/ml) 용액을 모든 웰에 제공하고, 추가로 60분 동안 다시 인큐베이션한 후, 웰을 다시 비우고 세정하였다. 다음 검정 단계에서는, 디에탄올아민 (600 ml의 정제수 중 105 ml) 중의 1 mg/ml의 알칼리성 포스파타제 (파라-니트로페놀 포스페이트)에 대한 기질 용액을 웰에 제공하고, 이를 추가의 60분 동안 다시 인큐베이션하거나, 또는 일부 색상이 발색될 때까지, 최대 48시간 동안 인큐베이션하였다. 마지막으로, 특수 판독기로 405 nm 파장에서 광학 밀도를 판독함으로써 상기 웰을 발색에 대해 평가하였다.
웰이 색상을 띨 경우, 이는 웰 표면에 부착된 펩티드가 가수분해되지 않고 비오틴화된 CPG2 효소에 결합하였다는 것을 나타내는 것이며, 색상의 강도는 결합한 CPG2의 양에 정비례하는데, 이는 결합 강도 (친화도)를 나타낸다.
CPG2에 결합한 서열을 기록하고, 순위매김하였다. 이어서, 제1 펩티드 어레이를 생성했던 것과 동일한 방식으로 디펩티드의 모든 가능한 변형이 결합 트리펩티드에 부착된 확장형 펩티드 어레이를 제조하기 위한 출발점으로서 가장 높은 결합을 나타내는 서열을 사용하였다. CPG2 결합의 검출에 대한 동일의 검정 절차를 상기 웰에도 다시 적용시키고, 가장 강력한 결합제를 다시 확인하고 순위매김하였다.
전체 절차를 다시 반복하여 추가로 2개의 아미노산 확장부를 첨가하여 가장 강력한 결합 헵타펩티드를 확인하였다. 이어서, 가장 강력한 결합제에 대해 전체 위치 스캔을 수행하여 최상의 결합 서열의 임의 변형이 여전히 보다 강력한 결합을 제공할 수 있는지 여부를 측정하였다.
이러한 서열에 대한 검정 단계 중 임의의 단계에서 두번째로 가장 강력한 결합제로 진행하여 상기 서열의 펩티드 변형물을 형성할 수 있는데, 이는 가장 강력한 후보물질로부터 유도된 라이브러리에서 보다 더 강력한 결합제가 검출되지 않을 경우에 따를 수 있는 절차이다.
결합 펩티드를 발견하기 위해서는 본원에 기술된 기본 펩티드 선별 기법 (펩스캔)이 다른 많은 방식으로 변형될 수 있다는 것을 인식할 것이다. 본원에 기술된 예시적 방법은 단지 그 방법이 사용될 수 있는 가능한 방식들 중 하나일 뿐이다.
일단 만족할만한 결합 서열을 발견하고 나면, 추가 실험 및 개발을 착수하는 데 충분한 양으로 유리 펩티드 샘플을 합성할 수 있다. 특히, 하나 이상의 갈락토실 세린 아미노산 (즉, 세린 히드록실 기의 O 잔기에 공유적으로 연결된 갈락토스 (이는 많은 공급업체로부터 상업적으로 입수가능하다))으로 이루어진 확장부를 양 말단에 가진 변이체를 제조함으로써 O-연결된 글리코실 펩티드를 생성할 수 있다. 필요에 따라, 임의 수의 상기 추가의 갈락토실 세린 기를 공지된 펩티드 화학 기법에 의해 첨가함으로써 간 세포 (간세포)의 아시알로당단백질 수용체에 대한 글리코실 펩티드의 결합을 강화시킬 수 있다. 이러한 결합은 단리된 C형 렉틴의 사용에 의해 또는 단리된 래트 간세포에 의해 시험관내에서 모델링될 수 있다.
갈락토실 세린은 헵타펩티드에 직접 첨가될 수 있거나, 또는 링커 아미노산 또는 아미노산 스트레치, 바람직하게는 (반드시 그러한 것은 아니지만) 하나 이상의 글리신 잔기로 구성된 것을 통해 첨가될 수 있다. 갈락토실 기의 배향 및 배치를 최적화시키기 위해 하나 이상의 추가의 아미노산을 이용하여 추가의 갈락토실 세린 잔기들 사이의 공간을 구축할 수 있다.
다른 갈락토실 아미노산도 쉽게 입수가능하고 펩티드 합성 화학법을 잘 따르다면, 갈락토실 세린 대신 사용될 수 있다.
이러한 방식으로 다수의 CPG2-결합 펩티드가 개발될 수 있고, 이로써 일련의 후보 분자를 형성할 수 있다. 이어서, 각 후보 분자를 간 세포에의 결합, CPG2에의 결합, 프로테아제에 대한 안정성, 보관시 안정성, 독성 부재 등에 대해 추가 회차로 특징화하고 선별할 수 있다. 암 항원에 결합하는 표적화 분자의 친화도에 부합하는 CPG2에 대한 친화도 (즉, 암 항원에 대하여 표적화 항체의 친화도보다 대략 10배 더 낮은 친화도)를 가진 펩티드를 선별하기 위해 상기와 같은 펩티드를 다수 생성한다.
실시예
2: 제거제 효소 결합제
CP014
CP014
로 지정된 화합물에 대한 화학적 설명 및
그래프식
표현

합성 및 정제 방법
CEM 리버티 마이크로웨이브 신시사이저(CEM Liberty Microwave Synthesiser)를 사용하고 Fmoc 화학법 (반응식 1 (여기서, R=수지))을 적용하여 고체상 펩티드 합성법 (SPPS)에 의해 펩티드 CP014를 합성하였다. 수지 Fmoc-Glu(OtBu)-NovaSynTGA (머크바이오사이언스(MerckBioscience))를 0.1 mmol의 크기로 사용하였다. 모든 후속 Fmoc-빌딩 블록은 NeoMPS, 바켐(Bachem) 및 머크바이오사이언스로부터 구입하였다. FMoc-세린 갈락토스 (아세틸로 보호된 것)는 덱스트라 랩스(Dextra Labs)로부터 입수하였다.
SPPS
단계 1

SPPS
단계 2

SPPS
단계
3

수지로부터의 펩티드 절단 및 정제
합성 완료시, 수지를 DCM (10 mL) 및 EtOH (10 mL)로 세척하고, 데시케이터에서 24 h 동안 건조시켰다. 24 h 후, 완성된 펩티드를 데시케이터로부터 제거하고, 3 h 동안 95% TFA, 2.5% d.H2O, 및 2.5% TIPS로 구성된 10 mL 용액에 가하였다. 상기 시간이 경과한 후, 감압하에 반응물을 증발시켜 무색 오일을 수득하였다. 저온 TBME 중에서 오일을 침전시켜 갈색의 조 고체를 수득하였다 (반응식 4). 조 생성물을 d.H2O 중 50% MeCN 500 ㎕에 용해시킨 후, 추가로 600 ㎕ 5% MeCN (0.1% TFA)로 희석시킨 후, 여과하고, 오닉스(Onyx) C18 칼럼 상에서 역상 프로그램을 사용하고 12분 동안에 걸쳐 5에서 100% MeCN (0.1% TFA)의 구배 용리를 적용하여 정제하였다. 생성물 수집을 위한 체류 시간은 280 nm에서 5.6분이었다 (도 2). 수집된 분획을 조합하고, 가압하에 환원시켜 갈색 오일을 수득하고, 이를 d.H2O (4 mL) 중에 용해시키고, 18 h 동안 0.2 mBar에서 냉동 건조시켜 갈색 고체를 수득하였다.

갈락토스 모이어티로부터의 아세틸 보호의 제거 및 정제
생성물을 최소량의 무수 MeOH 중에 용해시킨 후, 무수 MeOH 중 1 M의 NaOMe 용액을 pH 9-10이 될 때까지 적가함으로써 갈락토스 모이어티로부터 아세틸 보호를 제거하였다. 이어서, 반응물을 1 h 동안 교반시켰다. 상기 시간이 경과한 후, 아세트산을 pH 7이 될 때까지 적가하고, 감압하에 휘발물질을 제거하여 갈색 고체를 수득하였다 (반응식 5). 생성물을 d.H2O 중 50% MeCN 500 ㎕ 중에 용해시킨 후, 추가로 600 ㎕의 5% MeCN (0.1% TFA)으로 희석시키고, 여과하고, 오닉스 C18 칼럼 상에서 표준 역상 프로그램을 사용하고 12분 동안에 걸쳐 5에서 100% MeCN (0.1% TFA)의 구배 용리를 적용하여 정제하였다. 생성물 수집을 위한 체류 시간은 280 nm에서 4.7분이었다 (도 3). 수집된 분획을 조합하고, 가압하에 환원시켜 갈색 고체를 수득하고, 이를 d.H2O (4 mL) 중에 용해시키고, 18 h 동안 0.2 mBar에서 냉동 건조 조건에 적용하여 갈색 고체를 수득하였다.

사용된 분석 방법 및 데이터:
펩티드 CP014에 대하여 하기와 같은 HPLC 트레이스를 수득하였다 (도 1 및 2). 오닉스 C18 칼럼을 사용하고, 12분 동안에 걸친 5에서 100% MeCN (0.1% TFA)의 구배 용리를 적용하는 표준 역상 조건을 사용하였다. 용리 구배: 처음에 5% MeCN (0.1% TFA) → 제8분에 1.5분 동안 100% MeCN (0.1% TFA) → 다시 5% MeCN (0.1% TFA)으로 마지막까지.
LCMS 전기분무 (ESI) m/z (ESI) 637 (M+H+, 25%)를 사용하여 CP014의 구조를 확인하였다. 결과는 도 3에 제시되어 있다.
효소 억제 실험 및
IC50
계산
CPG2
활성의 억제제로서의
CP014
를 평가하는 데 사용된 효소 억제 검정 방법
분광광도 측정 검정을 사용하여 CP014가 CPG2 활성에 미치는 효과를 측정하였다. 농도 40 mM가 되도록 CP014를 검정 완충액 (0.2 mM 염화아연을 함유하는 0.1 M 트리스-HCl 완충액 (pH 7.3))에 용해시켰다. 상기 용액의 다양한 분취량을 큐벳에 첨가하고, 검정 완충액을 첨가하여 각 큐벳 중의 용액의 부피가 900 ㎕가 되도록 조정하였다. 검정 완충액 중 100 ㎕ 분취량의 0.6 mM 메토트렉세이트 용액을 각 큐벳에 첨가하여 0.06 mM (60 μM)의 메토트렉세이트 기질 농도를 얻었다. 큐벳 중 CP014의 농도 범위는 0 내지 400 μM이었다.
큐벳 중 기질 용액을 오븐에서 37℃로 가열하였다. 이어서, 큐벳을 차례로, 펠티에(Peltier)를 사용하여 37℃로 유지된 분광광도계에 위치시켰다. 10 ㎕ 분취량의 융합 단백질 (ScFV 항-CEA-CPG2) 용액 또는 CPG2 용액을 첨가한 후, 짧게 교반함으로써 검정을 개시하였다. 320 nm에서의 흡광도 감소를 측정함으로써 메토트렉세이트 기질의 가수분해에 대해 모니터링하였다.
37℃에서 1분당 1 μmol의 메토트렉세이트의 가수분해를 촉매하는 효소의 양을 CPG2 활성의 1 유닛 (1 U)으로 정의한다. 메토트렉세이트의 몰 흡광 계수는 8,300 L mol-1 cm-1인 것으로 측정되었다. 각 검정에 대한 흡광도 대 시간의 플롯을 작성하고, 0 내지 0.2분 사이의 데이터를 직선이 되도록 피팅하였다. 이어서, 직선 기울기에 -12 및 ScFV 항-CEA-CPG2/CPG2 용액의 희석 배율을 곱하여 효소 활성 (U/mL)을 얻었다.
한 변형된 검정에 따라 CPG2 검정 이전에 4℃에서 일정 기간 동안 다양한 양의 CP014와 ScFV 항-CEA-CPG2 또는 CPG를 인큐베이션하였다. 상기 인큐베이션 단계를 수행한 후, 37℃에서 검정 완충액 중 1 mL 분취량의 0.06 mM 메토트렉세이트를 함유하는 큐벳에 10 ㎕ 분취량의 각 혼합물을 첨가하였다. 큐벳의 내용물을 교반한 후, 상기 기술된 바와 같이 흡광도 감소에 대해 측정한 후, 이어서 효소 활성에 대해서 측정하였다.
CPG2 활성을 억제시킴에 있어서의 CP014의 유효성을 그의 IC50 값, 즉, 최대 절반 억제 농도로 표시하였다. 그래프패드 프리즘 5(GraphPad Prism 5) 소프트웨어를 사용하여, 비-선형 회귀를 효소 활성 대 CP014 농도 로그값의 플롯에 적용시킴으로써 IC50 값을 측정하였다.
잠재적인
CPG2
기질에 대해서 스크리닝하기 위한 비-증폭형
앰플렉스
레드 검정
분광광도 측정 검정을 사용함으로써 CP014가 CPG2 활성에 대한 잠재적인 억제제라는 것이 확립되었다. 상기 물질이 효소에 대한 기질이 아니라, 억제제라는 것을 확인하기 위해, 인비트로젠 인크.(Invitrogen Inc.)에 의해 개발된 변형된 버전의 검정을 수행하였다. 소위 앰플렉스 레드 검정이라는 것을 사용함으로써 식품 샘플 중의 L-글루타메이트를 측정하고, 연속하여 글루타메이트 옥시다제 활성을 모니터링할 수 있다. 검정 절차에서 증폭 단계를 제거함으로써 CPG2에 대한 기질로서의 화합물을 스크리닝할 수 있었다. 변형된 검정에서, CPG2의 기질로부터 절단된 L-글루탐산은 글루타메이트 옥시다제에 의해 산화되어 α-케토글루타레이트, 암모니아 및 과산화수소를 생성한다. 이어서, 양고추냉이 퍼옥시다제 (HRP)에 의해 촉매된 반응에서 과산화수소를 10-아세틸-3,7-디히드록시페녹사진 (앰플렉스 레드 시약)과 1:1의 화학량론으로 반응시켜 고도의 형광성을 띠는 생성물인 레소루핀(resorufin)을 수득하였다.
검정 완충액 (0.2 mM 염화아연을 함유하는 0.1 M 트리스-HCl 완충액 (pH 7.5)) 중의 25 μM 및 200 μM의 CP014 용액, 및 검정 완충액 중 25 μM의 엽산 용액을 제조하였다. DMSO 중 10 mM 앰플렉스 레드 시약 용액 2.5 ㎕, 검정 완충액 중 HRP 용액 5 ㎕ (100 U/mL), 검정 완충액 중 L-글루타메이트 옥시다제 용액 32 ㎕ (5 U/mL) 및 460.5 ㎕ 검정 완충 용액을 조합하여 앰플렉스 레드 시약 혼합물을 제조하였다. 검정 완충액을 사용하여 ScFV 항-CEA-CPG2 융합 단백질을 적절하게 희석하였다.
50 ㎕ 분취량의 CP014 용액 또는 25 μM 엽산 용액 및 50 ㎕ 분취량의 앰플렉스 레드 시약 혼합물을 흑색 마이크로타이터 플레이트의 웰에 첨가하였다. 플레이트를 덮개로 덮은 후, 10분 동안 37℃에서 인큐베이션하였다. 덮개를 제거하고, 1 ㎕ 분취량의 ScFV 항-CEA-CPG2를 첨가하였다. 짧게 교반한 후, 플레이트를 BMG 플루오스타 오메가(BMG Fluostar Omega) 플레이트 판독기에 두고, 5초 간격으로 245초 동안의 기간에 걸쳐 형광 강도를 모니터링하였다. 여기 필터 파장을 544 nm으로 설정하고, 방출 필터 파장은 590 nm으로 설정하였다. 물질이 엽산과 같은 CPG2에 대한 기질일 경우, 형광 강도 대 시간 (초)의 플롯은 초기 유도기 이후 선형 관계를 나타낸다. 플롯 중 직선 부분의 기울기는 CPG2 활성과 정비례하는 것으로 나타났다.
ScFV 항-CEA-CPG2를 대략 2 U/mL의 농도로 희석시켰을 때, 12.5 μM 엽산 대조군 검정의 경우에 계산된 기울기 값은 대략 4.0이었다. 플레이트 웰 중 12.5 μM 및 100 μM의 두 농도 모두에서 CP014는 음성 대조군 샘플 (즉, ScFV 항-CEA-CPG2 용액 대신 검정 완충액을 웰에 첨가한 것)의 것보다 크지 않은 기울기 값을 산출하였는데, 이는 상기 물질이 CPG2에 대한 기질이 아니라는 것을 나타내는 것이다.
IC50
평가 및
CP014
에 대한 기질 가능성
앞서 기술된 바와 같이 IC50을 측정하고, 도 4에 제시된 바와 같이 그래프패드 프리즘을 사용하여 데이터를 축소시켰다. 기질 가능성에 대하여 도해하는 앰플렉스 레드 검정 데이터는 도 5에 제시되어 있다.
데이터를 통해 CP014가 CPG2 활성을 억제하고 (도 4), 나아가 이는 기질로서 변경되거나 사용되지 않는다는 것 (도 5)이 입증되었다.
표면
플라스몬
공명 (
SPR
)을 사용한 결합 연구
제조사 (지이 헬쓰케어)가 권고한 바와 같은 EDC/NHS 화학법을 사용하여 펩티드를 CM5 칩에 커플링시켰다. 분석물을 HBS-P 완충액 (0.01 M HEPES (pH 7.4), 0.15 M NaCl, 0.005% v/v 계면활성제 P20) 중에서 구성하고, 5 ㎕/min으로 180초 동안 표면 상에 주입한 후, 300초 동안의 해리 시간을 가졌다. 재생 완충액 (10 mM CHAPS, 1 mM 구아니디늄 히드로클로라이드, 2 M NaCl)을 2회에 걸쳐 10초간 주입함으로써 잔류 결합을 제거하였다. 반응 신호 및 기준선이 실험 전반에 걸쳐 일관되는지에 대해 체크하고, 15 ㎕/min 및 75 ㎕/min의 유속을 시험하여 물질 수송에 기인한 최소의 효과를 확인하였다.
도 6a 및 6c는 각각 오버레이된 4개의 센서그램을 제시한다. 이는 고정화된 CP014 펩티드의 칩 표면 상에 12.5, 25, 50 및 100 nM ScFV 항-CEA-CPG2를 주입한 것으로부터 얻은, 블랭크가 차감된 실시간 반응이다. 비아코어 소프트웨어에서 공급받은 2개의 다른 알고리즘 ("비아이벨류에이션")을 사용하여 데이터를 피팅하였다. 도 6a에 제시되어 있는 첫번째 것은 1:1 결합 모델 (랭뮤어)이고, 도 6c에 제시되어 있는 두번째 것은 2-상태 반응 피트이다. 두 피트 모두 정규화된 센서그램에 매우 근접하게 정렬되어 있다. 이는 예상치로부터의 최소 편차를 나타내는 잔차 플롯으로부터 알 수 있다 (각각 도 6b 및 6d). 추가로, Chi2 값은 각각 0.154 및 0.0882이고, 이는 이러한 유형의 계산에 대한 허용 범위 (<0.2)내에 포함된다. 두 피트 모두 해리 상수 (결합 상수 KD)는 각각 5 nM 및 9 nM에서 매우 일치하였다.
도출된 동력학적 파라미터 및 친화도 파라미터는 각각 하기 표 1 및 2에 요약되어 있다:
<표 1>

<표 2>

실시예
3: 제거제 효소 결합제
CP006
CP006
으로 지정된 화합물에 대한 화학적 설명 및
그래프식
표현
CP006은 CP014의 전신이며, 그의 합성법 및 구조는 하기 기술된다.

CP006
의 합성
IIDQ 수지 (500 mg, 1.91 mmol/g)를 세정하고, 1시간 동안 10 ml 아세토니트릴 중에서 팽윤시키고, 진공하에서 용매를 배출시켰다. 이어서, Boc-아미노 나프토산 (폴리펩티드, 100 mg, 0.35 mmol) 및 H-Glu(OtBu)-OtBu (바켐, 108 mg, 0.37 mmol)를 5 ml 아세토니트릴 중에 용해시키고, 미리 팽윤시킨 수지에 첨가하고, RT에서 72시간 동안 완만하게 교반시켰다. 반응물을 깨끗한 플라스크 내로 배출시키고, 진공에서 용매를 제거하였다. 잔류물에 50 ml의 50% TFA/DCM을 첨가하고, 2시간 동안 격렬하게 교반시켰다. 용매를 제거한 후, 잔류물을 빙냉 tert-부틸 메틸에테르로 2회에 걸쳐 세정하고, 경사분리시켜 황색 고체만이 남아 있도록 하였다. 건조된 조 물질을 5% 아세토니트릴 (0.1% TFA)에 다시 용해시키고, 5-100% 아세토니트릴 0.1% TFA의 증가식 구배를 사용하여 HPLC에 의해 정제하였다. 풀링된 분획을 진공에서 환원시키고, 50% 아세토니트릴 중에서 희석시킨 후, 추가로 냉동 건조시켜 백색 분말을 수득하였다. ESMS -ve 질량 예측치: 316.11, 질량 측정치: 316.04.
효소 억제 및 CPG2에 대한 기질로서의 유용성에 대한 평가 방법은 앞서 기술된 방법을 사용하여 시험하였다.
CP006 은 ScFv 항- CEA - CPG2 에서 CPG2 활성의 억제제이며, 불량한 기질이다.
앞서 기술된 바와 같이 IC50을 측정하고, 도 9에 제시된 바와 같이 그래프패드 프리즘을 사용하여 데이터를 축소시켰다. 기질 가능성을 도해하는 앰플렉스 레드 검정 데이터는 도 10에 제시되어 있다.
실시예
4: 양 항-
CEA
및
ScFv
항-
CEA
-
CPG2
융합 단백질을 사용한 세포 결합 연구
유동 세포측정법을 사용한 분석법으로 양에서 생성된 CEA에 대한 항체, 및 ScFv 항-CEA-CPG2 융합 단백질을 검토하는 연구를 수행하였다.
CEA
발현 세포를 사용하여 수행된
유동 세포
측정법에 의한
ScFv
항-
CEA
-
CPG2
의
결합능
측정.
물질:
세포주 : CRL 1573 - 대조군 세포주, CCL 229 (LoVo) - CEA 발현 세포주
항체/접합체 : CPG2-PE (피코에리트린), CPG2 100 U/ml (100 U의 접합체의 1:100 희석액).
ScFv 항-CEA-CPG2-PE UCL, 40-50 U/ml, 0.71 mg/ml 단백질, BSA 함유 완충액 중 2.5 ㎍/ml.
양 항-CEA-PE, CF 1110-PE; 670 ㎍/ml의 스톡.
3.9 mg/ml의 마우스 항-인간 CEA UCL, BSA 함유 완충액 중 2.5 ㎍/ml.
0.25 mg/ml의 염소 항-마우스-PE 써던 바이오테크(Southern Biotech) 1050-09 L2806-XH69Z 카파 쇄 특이, 1 ㎍/ml로 사용됨.
4.5 ml 폴리스티렌 튜브, PBS 아지드, BSA, PBS+0.5% 포름알데히드 (폴리사이언시즈(Polysciences) #18814), 프로피듐 아이오딘 (오르페겐(Orpegen), 100 mg/ml)
250 G, 5 mins, 4℃로 설정된 원심분리 에펜도르프(Eppendorf) 5810R;
쿨터(Coulter) XL 유동 세포측정기.
방법 :
배양된 세포 둘 모두를 튜브당 2x105개의 세포로 사용하였고, 튜브 중의 세포는 하기를 함유하도록 제조되었다:
1. 어느 것도 첨가되지 않음, 자가형광 시험,
2. 프로피듐 아이오딘 (최종 농도 10 ㎍/ml),
3. CPG2-PE,
4. ScFv 항-CEA-CPG2-PE,
5. 양 항-CEA-PE,
6. 마우스 항-CEA/항-마우스-PE.
세포를 스핀 다운시키고, 100 ㎕의 항체 용액을 각 튜브에 첨가하였다. 100 ㎕의 완충액을 튜브 1 및 2에 첨가하고, 나중까지 냉장고에 남겨 두었다.
튜브를 4℃에서 1시간 동안 암실에서 인큐베이션하고, 1 ml PBSAB를 첨가하고, 상기와 같이 스핀시켰다. 상기 세척액 중에 튜브 1을 포함시켰다. 상청액을 제거하고, 100 ㎕ 접합체를 튜브 6에 첨가하고, 4℃에서 1 hr 동안 암실에서 추가로 인큐베이션하였다.
150 ㎕의 0.5% 포름알데히드를 튜브 1, 3, 4 및 5에 첨가하고, 측정할 때까지 (약 2시간) 냉장고에 보관하였다. 상기와 같이 튜브 6을 세척하고, 상기 튜브에 포름알데히드를 첨가하였다. 측정하기 30분 전에, 100 ㎕의 프로피듐 아이오딘을 튜브 2에 첨가하고, 측정하기 전, 1 ml의 PBS를 샘플 모두에 첨가하였다.
대조군 세포주는 어느 항-CEA 항체에 대해서도 어떤 특이적인 결합을 보이지 않았다 (도 11a 내지 j). 양 항체의 비-특이적인 결합은 약간 있었다 (도 11h). 도 11a 내지 11j (즉, 도 11a, 11c, 11e, 11g 및 11i) 각각의 첫번째 도면은 샘플간에 어떤 차이도 보이지 않는 대조군이다.
CCL 229 세포는 음성 대조군 세포주에 비하여 약간 더 큰 자가형광을 나타내었다 (도 12b). CPG2 대조군은 세포주에 결합하지는 않았지만 (도 12d), CEA 발현 세포주 중 2개의 세포 군집, 즉, 표면 단백질 CEA를 제시하는 세포 군집 및 그렇지 않은 세포 군집이 존재하였다. 이러한 현상은 3개의 항-CEA 항체 모두의 조합을 통해 나타났다 (도 12f, 12h 및 12j).
참고문헌

본 발명은 그 범주를 본원에 기술된 특정 실시양태로 제한하고자 하는 것은 아니다. 실제로, 본원에 기술된 것 이외에도, 본 발명의 다양한 변형이 상기 상세한 설명 및 첨부하는 도면으로부터 당업자에게 자명할 것이다. 그러한 변형 또한 첨부된 특허청구범위의 범주 내에 포함시키고자 한다. 또한, 본원에 기술된 모든 실시양태는 적절히 광범위하게 적용될 수 있고 임의의 및 모든 다른 일관된 실시양태와 조합될 수 있다고 간주된다.
본원에서 다양한 공개문헌이 인용되었으며, 이들의 개시내용은 그 전문이 참고로 포함된다.
SEQUENCE LISTING
<110> Mologic Limited
<120> PEPTIDE CLEARING AGENTS
<130> P109133WO00
<150> GB0916749.5
<151> 2009-09-23
<160> 1
<170> PatentIn version 3.5
<210> 1
<211> 4
<212> PRT
<213> Artificial sequence
<220>
<223> synthesised peptide
<220>
<221> S
<222> (1)..(1)
<223> S may be glycosylated
<220>
<221> X
<222> (3)..(3)
<223> X = amino-naphthoic acid
<400> 1
Ser Ala Xaa Glu
1
Claims (31)
- 대상체에서
a. 효소; 및
b. 표적 위치에 특이적으로 결합하는 결합 분자
의 접합체를 효소의 활성 부위에 대한 펩티드의 결합을 통해 비-표적 위치로부터 제거하는 펩티드 제거제이며, 여기서, 펩티드 제거제는 또한 간 세포에 의해 발현되는 아시알로당단백질 수용체에 결합함으로써 간을 통한 제거를 촉진하는 것인 펩티드 제거제. - 제1항에 있어서, 펩티드가 아시알로당단백질 수용체를 발현하는 간 세포에의 결합에 의해 이루어지는 간을 통한 제거가 촉진되도록 글리코실화된 것인 펩티드.
- 제1항 또는 제2항에 있어서, 펩티드가 효소에 결합시 효소 활성을 방지하거나 또는 억제하는 것인 펩티드.
- 제1항 내지 제3항 중 어느 한 항에 있어서, 펩티드가 효소 활성에 의해 변형되지 않는 것인 펩티드.
- 제1항 내지 제4항 중 어느 한 항에 있어서, 디펩티드 아미노-나프토산 (ANA)-글루타메이트 (Glu)를 포함하는 펩티드.
- 제1항 내지 제5항 중 어느 한 항에 있어서, 아미노산 서열 세린 (Ser)-알라닌 (Ala)-아미노-나프토산 (ANA)-글루타메이트 (Glu) (서열 1)를 포함하는 펩티드.
- 제6항에 있어서, 세린 잔기가 글리코실화된 것인 펩티드.
- 제1항 내지 제7항 중 어느 한 항에 있어서, 효소가 표적 위치에서의 결합 분자의 특이적인 결합을 통해 표적 위치로 집중되는 것인 펩티드.
- 제1항 내지 제8항 중 어느 한 항에 있어서, 결합 분자가 항체 또는 그의 항원-결합 유도체인 펩티드.
- 제1항 내지 제9항 중 어느 한 항에 있어서, 효소가 표적 위치에서 전구약물을 활성 약물로 전환시키는 작용을 하는 것인 펩티드.
- 제1항 내지 제10항 중 어느 한 항에 있어서, 펩티드가 효소에 대하여 비-표적 위치로부터의 효소의 제거를 가능하게 할 정도의 충분한 친화도를 갖는 것인 펩티드.
- 제1항 내지 제11항 중 어느 한 항에 있어서, 펩티드가 표적 위치에서의 효소 활성에 유의하게 영향을 미치지 않을 정도로 충분히 빠른 해리 속도 또는 높은 해리 상수를 갖는 것인 펩티드.
- 제1항 내지 제12항 중 어느 한 항에 있어서, 효소에 대한 펩티드의 친화도가 그의 표적 위치에 대한 결합 분자의 결합 친화도보다 약 10배 더 낮은 것인 펩티드.
- 제1항 내지 제13항 중 어느 한 항에 있어서, 표적 위치가 종양 세포에 의해 발현되는 항원인 펩티드.
- 제2항 내지 제14항 중 어느 한 항에 있어서, 글리코실화가 하나 이상의 갈락토스 기를 커플링하는 것을 수반하는 것인 펩티드.
- 제1항 내지 제15항 중 어느 한 항에 있어서, 펩티드가 10개 이하의 아미노산 길이인 펩티드.
- 제1항 내지 제16항 중 어느 한 항에 있어서, 펩티드가 최소의 면역원성인 펩티드.
- 제1항 내지 제17항 중 어느 한 항에 있어서, 펩티드가 화학적으로 합성된 것인 펩티드.
- 제1항 내지 제18항 중 어느 한 항에 있어서, 효소가 비-인간 기원인 펩티드.
- 제1항 내지 제19항 중 어느 한 항에 있어서, 효소가 카르복시펩티다제 G2, 알칼리성 포스파타제, 베타-글루코로니다제, 페니실린-V-아미다제, 베타-락타마제, 베타-글루코시다제 및 니트로리덕타제로부터 선택되는 것인 펩티드.
- 제18항에 있어서, 효소가 카르복시펩티다제 G2 (CPG2)인 펩티드.
- a. 바람직하게는 효소 기질의 구조적 유사체인 출발 펩티드를 기초로 하여, 펩티드 어레이를 제조하는 단계;
b. 효소 및 표적 위치에서 특이적으로 결합하는 결합 분자의 접합체에 대한 결합 친화도에 대해 펩티드 어레이를 스크리닝하고, 결합 친화도를 갖는 펩티드를 선별하는 단계;
c. 임의로, 단계 b의 결합 친화도를 갖는 펩티드의 아미노산 서열을 변형시키고, 변형된 펩티드를 사용하여 단계 b를 반복함으로써 결합 친화도 개선에 대해 시험하는 단계;
d. 임의로, 개선된 결합 친화도를 갖도록 단계 c에서 발견된 변형된 펩티드의 각 잔기에서 치환을 실시하고, 단계 b를 반복하여 치환된 펩티드 중 임의의 것이 추가로 개선된 결합 친화도를 갖는지 여부를 측정하는 단계;
e. 임의로, 단계 c 또는 d로부터 생성된 (개선된 결합 친화도를 갖는) 펩티드 또는 펩티드들이 효소 활성을 방지하거나 또는 억제할 수 있는 능력을 갖는지 여부를 측정하는 단계;
f. 임의로, 대상체로부터 얻은 일련의 대조군 단백질을 사용하여 시험함으로써 효소에 대한 결합 특이성을 확인하는 단계;
g. 간 세포에 의해 발현된 아시알로당단백질 수용체에 결합함으로써 간을 통한 제거를 촉진할 수 있는 능력에 대해 펩티드 또는 펩티드들을 시험하는 단계
를 포함하며; 여기서, 단계 e 및 f가 실시되는 경우, 이들은 임의 순서로 수행될 수 있고, 단계 g는 단계 a에서 간 세포에 의해 발현된 아시알로당단백질 수용체에 결합함으로써 간을 통한 제거를 촉진시킬 수 있는 펩티드 어레이를 제공하는 것을 비롯한, 상기 방법 중 임의의 시점에서 수행될 수 있는 것인, 제1항 내지 제21항 중 어느 한 항의 펩티드 제거제를 제조하는 방법. - 제22항에 있어서, 단계 g가 펩티드 또는 펩티드들을 글리코실화하는 것을 포함하고, 따라서, 단계 a는 글리코실화된 펩티드 어레이를 제공하는 것을 포함할 수 있는 것인 방법.
- 제22항 또는 제23항에 있어서, 단계 a가 펩티드 라이브러리를 사용하여 실시되는 것인 방법.
- 제22항 내지 제24항 중 어느 한 항에 있어서, 효소가 CPG2인 방법.
- 제22항 내지 제25항 중 어느 한 항에 있어서, 출발 펩티드가 트리펩티드 WFE인 방법.
- 제22항 내지 제26항 중 어느 한 항에 있어서, 상기 방법에 의해 제조된 펩티드의 아미노산 서열을 인간 아미노산 서열 데이터베이스와 비교하여 펩티드가 비바람직한 생물학적 활성을 가질 가능성이 낮음을 확인하는 단계를 추가로 포함하는 방법.
- 제1항 내지 제27항 중 어느 한 항의 펩티드를 제거제로 사용하는 것을 포함하는 항체 지향 효소 전구약물 치료법 (antibody directed enzyme pro-drug therapy; ADEPT) 방법.
- 항체 지향 효소 전구약물 치료법 (ADEPT)에서 제거제로서의 제1항 내지 제28항 중 어느 한 항의 펩티드의 용도.
- 제1항 내지 제29항 중 어느 한 항에 있어서, 항체 지향 효소 전구약물 치료법 (ADEPT)에서 제거제로서 사용하기 위한 펩티드.
- 항체 지향 효소 전구약물 치료법 (ADEPT)에서 사용하기 위한 제거제의 제조에서의 제1항 내지 제30항 중 어느 한 항의 펩티드의 용도.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB0916749.5A GB0916749D0 (en) | 2009-09-23 | 2009-09-23 | Peptide cleaning agents |
| GB0916749.5 | 2009-09-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20120084298A true KR20120084298A (ko) | 2012-07-27 |
Family
ID=41327500
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020127010173A KR20120084298A (ko) | 2009-09-23 | 2010-09-23 | 펩티드 제거제 |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US8846861B2 (ko) |
| EP (1) | EP2470216A1 (ko) |
| JP (1) | JP2013505287A (ko) |
| KR (1) | KR20120084298A (ko) |
| CN (1) | CN102665768B (ko) |
| AU (1) | AU2010299643B2 (ko) |
| BR (1) | BR112012006389A2 (ko) |
| CA (1) | CA2774081A1 (ko) |
| GB (1) | GB0916749D0 (ko) |
| IL (1) | IL218713A0 (ko) |
| IN (1) | IN2012DN02241A (ko) |
| MX (1) | MX2012003565A (ko) |
| NZ (1) | NZ598802A (ko) |
| SG (1) | SG179015A1 (ko) |
| WO (1) | WO2011036457A1 (ko) |
| ZA (1) | ZA201201527B (ko) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB2435510A (en) | 2006-02-23 | 2007-08-29 | Mologic Ltd | Enzyme detection product and methods |
| US9850296B2 (en) | 2010-08-10 | 2017-12-26 | Ecole Polytechnique Federale De Lausanne (Epfl) | Erythrocyte-binding therapeutics |
| US9517257B2 (en) | 2010-08-10 | 2016-12-13 | Ecole Polytechnique Federale De Lausanne (Epfl) | Erythrocyte-binding therapeutics |
| AU2011289579B2 (en) | 2010-08-10 | 2016-11-17 | Ecole Polytechnique Federale De Lausanne | Erythrocyte-binding therapeutics |
| US10946079B2 (en) | 2014-02-21 | 2021-03-16 | Ecole Polytechnique Federale De Lausanne | Glycotargeting therapeutics |
| US10953101B2 (en) | 2014-02-21 | 2021-03-23 | École Polytechnique Fédérale De Lausanne (Epfl) | Glycotargeting therapeutics |
| US10046056B2 (en) | 2014-02-21 | 2018-08-14 | École Polytechnique Fédérale De Lausanne (Epfl) | Glycotargeting therapeutics |
| AU2015233084B2 (en) | 2014-02-21 | 2020-12-17 | Anokion Sa | Glycotargeting therapeutics |
| US11253579B2 (en) | 2017-06-16 | 2022-02-22 | The University Of Chicago | Compositions and methods for inducing immune tolerance |
| CA3162687A1 (en) | 2020-01-31 | 2021-08-05 | Mark George Saulnier | Asgpr-binding compounds for the degradation of extracellular proteins |
| EP4334360A2 (en) | 2021-05-03 | 2024-03-13 | Avilar Therapeutics, Inc. | Potent asgpr-binding compounds for the degradation of immunoglobulins and other proteins |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6075010A (en) | 1992-06-09 | 2000-06-13 | Neorx Corporation | Small molecular weight ligand-hexose containing clearing agents |
| US4859449A (en) | 1987-09-14 | 1989-08-22 | Center For Molecular Medicine And Immunology | Modified antibodies for enhanced hepatocyte clearance |
| US20030068322A1 (en) | 1988-04-18 | 2003-04-10 | Immunomedics, Inc. | Methods of antibody-directed enzyme-prodrug therapy |
| AU652939B2 (en) | 1990-05-11 | 1994-09-15 | University Of Connecticut, The | Targeted protection from cytotoxins |
| GB9324807D0 (en) | 1993-12-03 | 1994-01-19 | Cancer Res Campaign Tech | Tumour antibody |
| US6172045B1 (en) * | 1994-12-07 | 2001-01-09 | Neorx Corporation | Cluster clearing agents |
| ATE458499T1 (de) | 1995-06-07 | 2010-03-15 | Immunomedics Inc | Verbesserte abgabe von diagnostischen und therapeutischen stoffen an einem zielort |
| GB9524942D0 (en) * | 1995-12-06 | 1996-02-07 | Aepact Ltd | Drug therapy |
| WO1998019162A1 (en) * | 1996-10-31 | 1998-05-07 | Novalon Pharmaceutical Corporation | Identification of drugs using complementary combinatorial libraries |
| GB9624993D0 (en) * | 1996-11-30 | 1997-01-15 | Aepact Ltd | Tumour therapy |
| US6962702B2 (en) * | 1998-06-22 | 2005-11-08 | Immunomedics Inc. | Production and use of novel peptide-based agents for use with bi-specific antibodies |
| US7138103B2 (en) * | 1998-06-22 | 2006-11-21 | Immunomedics, Inc. | Use of bi-specific antibodies for pre-targeting diagnosis and therapy |
| US6465612B1 (en) * | 1998-09-23 | 2002-10-15 | The Regents Of The University Of California | Synthetic peptides, conjugation reagents and methods |
| ES2662177T3 (es) * | 2001-04-20 | 2018-04-05 | University Of Georgia Research Foundation, Inc. | Inactivadores de sitio activo |
| CA2622441A1 (en) * | 2005-09-27 | 2007-04-05 | Amunix, Inc. | Proteinaceous pharmaceuticals and uses thereof |
| ATE514711T1 (de) | 2007-10-16 | 2011-07-15 | Toscana Biomarkers S R L | Galactosylierte peptide, deren zubereitung und verwendung zur diagnose von autoimmunerkrankungen |
-
2009
- 2009-09-23 GB GBGB0916749.5A patent/GB0916749D0/en not_active Ceased
-
2010
- 2010-09-23 IN IN2241DEN2012 patent/IN2012DN02241A/en unknown
- 2010-09-23 NZ NZ598802A patent/NZ598802A/xx not_active IP Right Cessation
- 2010-09-23 WO PCT/GB2010/001796 patent/WO2011036457A1/en active Application Filing
- 2010-09-23 CN CN201080042526.6A patent/CN102665768B/zh not_active Expired - Fee Related
- 2010-09-23 MX MX2012003565A patent/MX2012003565A/es not_active Application Discontinuation
- 2010-09-23 SG SG2012015806A patent/SG179015A1/en unknown
- 2010-09-23 JP JP2012530331A patent/JP2013505287A/ja active Pending
- 2010-09-23 EP EP10757623A patent/EP2470216A1/en not_active Withdrawn
- 2010-09-23 KR KR1020127010173A patent/KR20120084298A/ko not_active Application Discontinuation
- 2010-09-23 BR BR112012006389A patent/BR112012006389A2/pt not_active IP Right Cessation
- 2010-09-23 AU AU2010299643A patent/AU2010299643B2/en not_active Ceased
- 2010-09-23 CA CA2774081A patent/CA2774081A1/en not_active Abandoned
- 2010-09-23 US US13/497,967 patent/US8846861B2/en not_active Expired - Fee Related
-
2012
- 2012-02-29 ZA ZA2012/01527A patent/ZA201201527B/en unknown
- 2012-03-18 IL IL218713A patent/IL218713A0/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| AU2010299643B2 (en) | 2014-06-26 |
| JP2013505287A (ja) | 2013-02-14 |
| US20130053543A1 (en) | 2013-02-28 |
| CN102665768A (zh) | 2012-09-12 |
| ZA201201527B (en) | 2013-05-29 |
| US8846861B2 (en) | 2014-09-30 |
| AU2010299643A1 (en) | 2012-03-22 |
| SG179015A1 (en) | 2012-04-27 |
| WO2011036457A1 (en) | 2011-03-31 |
| EP2470216A1 (en) | 2012-07-04 |
| IL218713A0 (en) | 2012-05-31 |
| NZ598802A (en) | 2013-10-25 |
| BR112012006389A2 (pt) | 2016-11-22 |
| GB0916749D0 (en) | 2009-11-04 |
| CN102665768B (zh) | 2015-01-21 |
| CA2774081A1 (en) | 2011-03-31 |
| MX2012003565A (es) | 2012-08-03 |
| IN2012DN02241A (ko) | 2015-08-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR20120084298A (ko) | 펩티드 제거제 | |
| JP7417432B2 (ja) | 新規リンカー、その製造方法およびその応用 | |
| KR102746415B1 (ko) | Mt1-mmp에 특이적인 바이사이클릭 펩타이드 리간드 | |
| JP7291395B2 (ja) | IgG結合ペプチド、及び該ペプチドによる抗体の特異的修飾 | |
| AU784285B2 (en) | Methods and compositions for prolonging elimination half-times of bioactive compounds | |
| ES2855998T3 (es) | Etiqueta transglutamina para bioconjugación específica de sitio eficiente | |
| CN105979971B (zh) | 抗体-药物缀合物和免疫毒素 | |
| JP2019077731A (ja) | 改変した抗体組成物、それを作製および使用する方法 | |
| ES2864013T3 (es) | Conjugados de fármaco y anticuerpo activable anti-CD71 y métodos de uso de los mismos | |
| JP2012511033A5 (ko) | ||
| CN109689112A (zh) | 化学选择性的巯基与烯基或炔基磷酰胺的偶联 | |
| JP5677454B2 (ja) | 細胞内ターゲット結合用二座ペプチドバインダー | |
| US20180339043A1 (en) | Modified antibodies | |
| CA3033614A1 (en) | Tgf-.beta. antagonist conjugates | |
| Ucar et al. | A new radio-theranostic agent candidate: Synthesis and analysis of (ADH-1) c-EDTA conjugate | |
| WO2024150174A1 (en) | Conditionally activated immunocytokines and methods of use | |
| van Baal | Peptides and proteins in dendritic assemblies | |
| WO2011150418A2 (en) | Compositions and methods for treating pathologies |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PA0105 | International application |
Patent event date: 20120420 Patent event code: PA01051R01D Comment text: International Patent Application |
|
| PG1501 | Laying open of application | ||
| PC1203 | Withdrawal of no request for examination | ||
| WITN | Application deemed withdrawn, e.g. because no request for examination was filed or no examination fee was paid |