KR20120007331A - An improved process for the preparation of carbapenem compounds - Google Patents
An improved process for the preparation of carbapenem compounds Download PDFInfo
- Publication number
- KR20120007331A KR20120007331A KR1020100068045A KR20100068045A KR20120007331A KR 20120007331 A KR20120007331 A KR 20120007331A KR 1020100068045 A KR1020100068045 A KR 1020100068045A KR 20100068045 A KR20100068045 A KR 20100068045A KR 20120007331 A KR20120007331 A KR 20120007331A
- Authority
- KR
- South Korea
- Prior art keywords
- formula
- compound
- carbonyl
- mercapto
- thio
- Prior art date
Links
Images

Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D477/00—Heterocyclic compounds containing 1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula:, e.g. carbapenicillins, thienamycins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulphur-containing hetero ring
- C07D477/10—Heterocyclic compounds containing 1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula:, e.g. carbapenicillins, thienamycins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulphur-containing hetero ring with hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 4, and with a carbon atom having three bonds to hetero atoms with at the most one bond to halogen, e.g. an ester or nitrile radical, directly attached in position 2
- C07D477/12—Heterocyclic compounds containing 1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula:, e.g. carbapenicillins, thienamycins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulphur-containing hetero ring with hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 4, and with a carbon atom having three bonds to hetero atoms with at the most one bond to halogen, e.g. an ester or nitrile radical, directly attached in position 2 with hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, attached in position 6
- C07D477/14—Heterocyclic compounds containing 1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula:, e.g. carbapenicillins, thienamycins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulphur-containing hetero ring with hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 4, and with a carbon atom having three bonds to hetero atoms with at the most one bond to halogen, e.g. an ester or nitrile radical, directly attached in position 2 with hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, attached in position 6 with hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, attached in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Abstract
Description
본 발명은 엔올포스페이트화합물인 화학식III의 C-2위치에, 보호된 메르캅토화합물을 축합 반응시킨 후 물속에서 무정형의 보호된 카르바페넴계 화합물을 제조하고, 이를 건조하지 않으며 정제도 하지 않은채, 팔라듐히드록시드-카르본 금속촉매 하에 수소화 반응을 수행하여 탈보호화 함으로써 고수율 및 고순도로 2-(치환메르캅토)-1β-메틸-카르바페넴계 화합물을 제조하는 신규한 방법에 관한 것이다.
The present invention provides an amorphous protected carbapenem-based compound in water after condensation of a protected mercapto compound at the C-2 position of the general formula (III), which is an enol phosphate compound, without drying it and without purification. The present invention relates to a novel process for preparing 2- (substituted mercapto) -1β-methyl-carbapenem-based compounds in high yield and high purity by performing dehydrogenation under a palladium hydroxide-carbon metal catalyst.
1976년에 티에나마이신이 발견된 이래 카르바페넴계 항생물질 연구가 활발해져서 이미페넴을 비롯하여 많은 우수한 항균활성을 갖는 카르바페넴계 화합물이 개발되었다. 그러나 이들 카르바페넴계 화합물의 상당수는 데히드로펩타아제-1(DHP-1)에 의해 용이하게 대사되는 단점을 갖고 있었다.Since the discovery of thienamycin in 1976, carbapenem-based antibiotics have been actively researched, and carbapenem-based compounds having many excellent antimicrobial activities, including imipenem, have been developed. However, many of these carbapenem compounds have the disadvantage of being easily metabolized by dehydropeptase-1 (DHP-1).
그 때문에 DHP-1 에 대한 안정성 향상을 목표로 한 연구가 활발히 이루어져, 1984년에 머크사에 의해 우수한 항균 활성을 유지하면서도 화학적, 물리적으로 안정적이고 DHP-1에 대해서도 우수한 저항성을 갖는 1β-메틸-카르바페넴계 화합물(이미페넴)이 개발 되었다.As a result, research aimed at improving the stability of DHP-1 has been actively conducted, and in 11984, 1β-methyl- having chemical and physical stability and excellent resistance to DHP-1 while maintaining excellent antimicrobial activity by Merck. Carbapenem compounds (imipenem) have been developed.
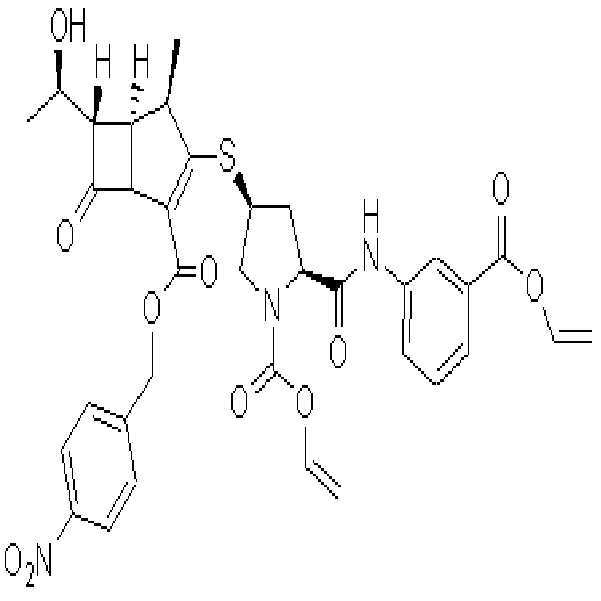
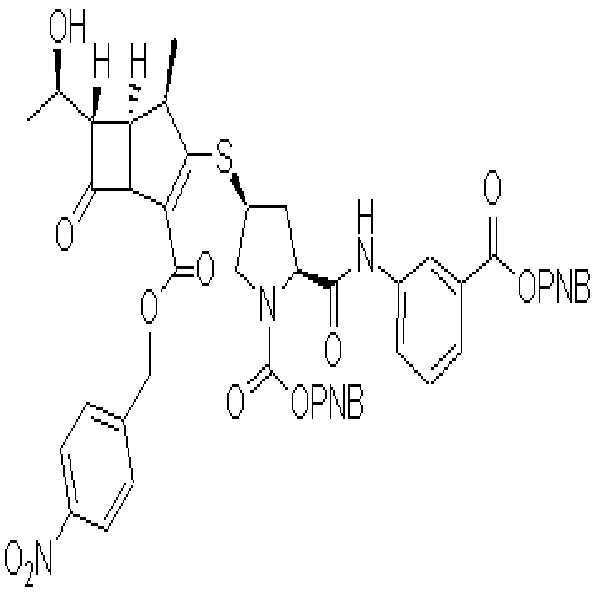
그 이후 여러 회사에서 엔올포스페이트 화합물인 화학식III에, 보호된 메르캅토화합물인 HR2를 축합 반응하여, 치환된 메르캅토화합물인 화학식II를 제조한 후 탈보호하여 2-(치환메르캅토)-1β-메틸-카르바페넴계 항생물질인 화학식I의 화합물들을 개발하여 상용화하고 있다.
Since then, several companies have condensed the protected mercapto compound HR2 with the enolphosphate compound of formula III to prepare a substituted mercapto compound of formula II, followed by deprotection to produce 2- (substituted mercapto) -1β-. Compounds of formula (I), a methyl-carbapenem antibiotic, have been developed and commercialized.
[화학식III][Formula III]

[화학식II][Formula II]

[화학식I]Formula I
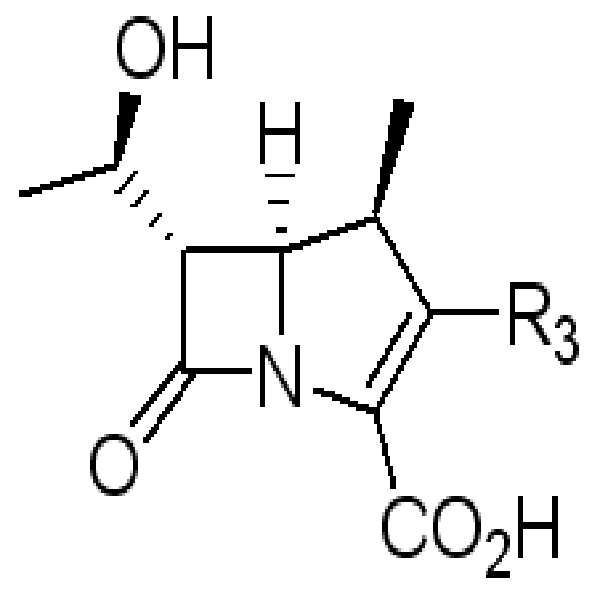
PNB는 파라니트로벤질기이다.PNB is a paranitrobenzyl group.
R2는,R 2 is
(2S,4S)-4-메르캅토-2-[[[3-(2-프로페닐옥시)카르보닐]페닐]아미노]카르보닐-1-피롤리딘카르복실산 2-프로페닐에스테르, (2S,4S)-4-메르캅토-2-[[[3-(p-니트로벤질옥시)카르보닐]페닐]아미노]카르보닐-1-피롤리딘카르복실산 p-니트로벤질에스테르, 6-티오-6,7-디히드로-5H-피라졸로[1,2-a][1,2,4]트리아졸트리플루오로아세트산염, 2-설파모일아미노메틸-1-p-니트로벤질옥시카보닐-4-메르캅토피롤리딘, 또는 4-니트로벤질(2S,4S)-2-[(디메틸아미노)카보닐]-4-메르캅토피롤리딘-1-카르복실레이트이다.(2S, 4S) -4-mercapto-2-[[[3- (2-propenyloxy) carbonyl] phenyl] amino] carbonyl-1-pyrrolidinecarboxylic acid 2-propenyl ester, ( 2S, 4S) -4-mercapto-2-[[[3- (p-nitrobenzyloxy) carbonyl] phenyl] amino] carbonyl-1-pyrrolidinecarboxylic acid p-nitrobenzyl ester, 6- Thio-6,7-dihydro-5H-pyrazolo [1,2-a] [1,2,4] triazoletrifluoroacetic acid salt, 2-sulfamoylaminomethyl-1-p-nitrobenzyloxycarbo Nyl-4-mercaptopyrrolidin, or 4-nitrobenzyl (2S, 4S) -2-[(dimethylamino) carbonyl] -4-mercaptopyrrolidine-1-carboxylate.
R3는,R 3 is
(2S,4S)-4-메르캅토-2-[[(3-카르복시페닐)아미노]카르보닐]-3-피롤리딘, 6-티오-6,7-디히드로-5H-피라졸로[1,2-a][1,2,4]트리아졸, 4-티오-2-설파모일아미노메틸피롤리딘, 또는 4-티오-(2S),(4S)-2-[(디메틸아미노)카보닐]피롤리딘이다.
(2S, 4S) -4-mercapto-2-[[(3-carboxyphenyl) amino] carbonyl] -3-pyrrolidine, 6-thio-6,7-dihydro-5H-pyrazolo [1 , 2-a] [1,2,4] triazole, 4-thio-2-sulfamoylaminomethylpyrrolidine, or 4-thio- (2S), (4S) -2-[(dimethylamino) carbo Nil] pyrrolidine.
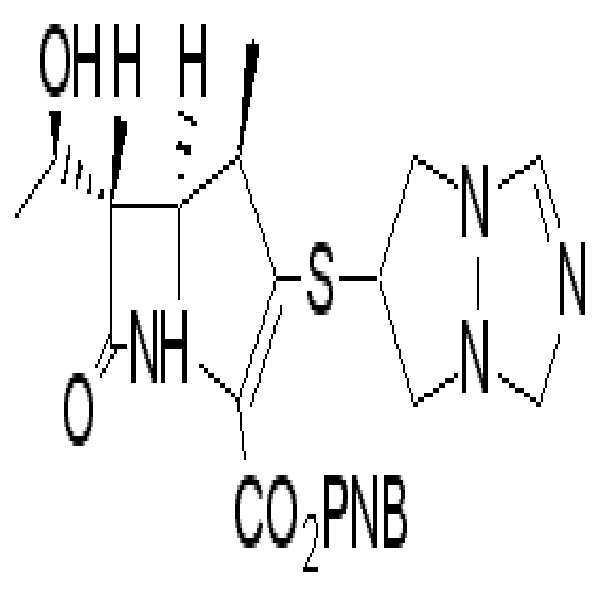
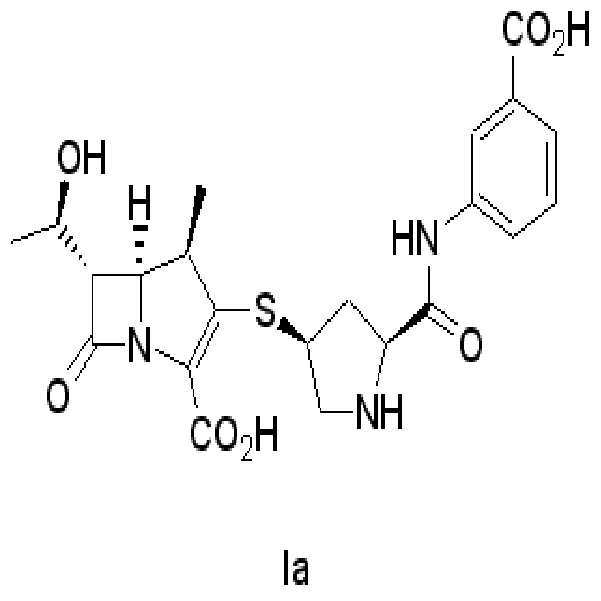
화학식I의 화합물로서, 하기 화학식Ia로 나타내는 에르타페넴,As the compound of formula (I), ertafenem represented by formula (Ia)

하기 화학식Ib로 나타내는 비아페넴,Viapenem represented by the following formula (Ib),

하기 화학식Ic로 나타내는 도리페넴,Doripenem, represented by Formula Ic,

하기 화학식Id로 나타내는 메로페넴 등이 개발되었다.
Meropenem and the like represented by the following formula Id have been developed.

이들 2-(치환메르캅토)-1β-메틸-카르바페넴계 화합물들의 제조방법을 살펴보면, 출발물질로 사용되는 엔올포스페이트 화합물인 화학식III은 Tetrahedron Letter 2293(1982); Tetrahedron 48, 55(1992);US 4,888,344(1987)에서의 반응식 1과 같은 방법으로 제조하여 사용하였다.
Looking at the preparation method of these 2- (substituted mercapto) -1β-methyl-carbapenem compounds, Formula III, which is an enol phosphate compound used as a starting material, is described in Tetrahedron Letter 2293 (1982); It was prepared and used in the same manner as in Scheme 1 in Tetrahedron 48, 55 (1992); US 4,888,344 (1987).
[반응식 1]Scheme 1
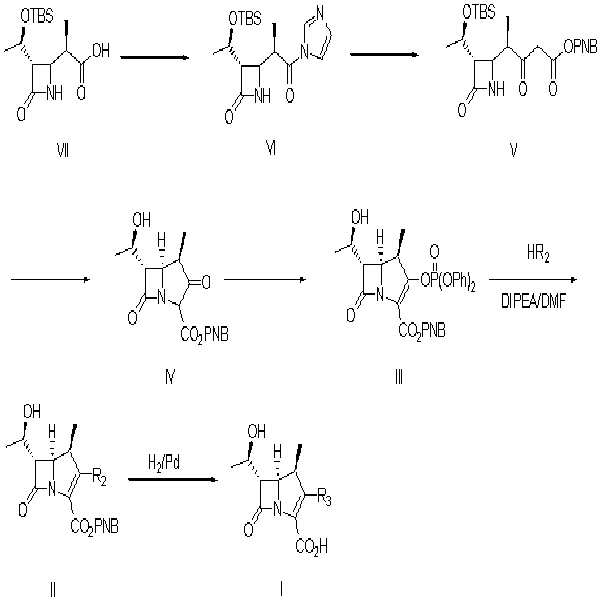
상기반응식 1에서 TBS는 t-부틸디메틸실릴기를 나타내고, PNB는 p-니트로벤질기를 나타낸다.In Scheme 1, TBS represents a t-butyldimethylsilyl group, and PNB represents a p-nitrobenzyl group.
R2는,R 2 is
(2S,4S)-4-메르캅토-2-[[[3-(2-프로페닐옥시)카르보닐]페닐]아미노]카르보닐-1-피롤리딘카르복실산 2-프로페닐에스테르, (2S,4S)-4-메르캅토-2-[[[3-(p-니트로벤질옥시)카르보닐]페닐]아미노]카르보닐-1-피롤리딘카르복실산 p-니트로벤질에스테르, 6-티오-6,7-디히드로-5H-피라졸로[1,2-a][1,2,4]트리아졸트리플루오로아세트산염, 2-설파모일아미노메틸-1-p-니트로벤질옥시카보닐-4-메르캅토피롤리딘, 또는 4-니트로벤질(2S,4S)-2-[(디메틸아미노)카보닐]-4-메르캅토피롤리딘-1-카르복실레이트이다.(2S, 4S) -4-mercapto-2-[[[3- (2-propenyloxy) carbonyl] phenyl] amino] carbonyl-1-pyrrolidinecarboxylic acid 2-propenyl ester, ( 2S, 4S) -4-mercapto-2-[[[3- (p-nitrobenzyloxy) carbonyl] phenyl] amino] carbonyl-1-pyrrolidinecarboxylic acid p-nitrobenzyl ester, 6- Thio-6,7-dihydro-5H-pyrazolo [1,2-a] [1,2,4] triazoletrifluoroacetic acid salt, 2-sulfamoylaminomethyl-1-p-nitrobenzyloxycarbo Nyl-4-mercaptopyrrolidin, or 4-nitrobenzyl (2S, 4S) -2-[(dimethylamino) carbonyl] -4-mercaptopyrrolidine-1-carboxylate.
R3는,R 3 is
(2S,4S)-4-메르캅토-2-[[(3-카르복시페닐)아미노]카르보닐]-3-피롤리딘, 6-티오-6,7-디히드로-5H-피라졸로[1,2-a][1,2,4]트리아졸, 4-티오-2-설파모일아미노메틸피롤리딘, 또는 4-티오-(2S),(4S)-2-[(디메틸아미노)카보닐]피롤리딘이다.(2S, 4S) -4-mercapto-2-[[(3-carboxyphenyl) amino] carbonyl] -3-pyrrolidine, 6-thio-6,7-dihydro-5H-pyrazolo [1 , 2-a] [1,2,4] triazole, 4-thio-2-sulfamoylaminomethylpyrrolidine, or 4-thio- (2S), (4S) -2-[(dimethylamino) carbo Nil] pyrrolidine.
상기 반응식 1에서, 종래의 엔올포스페이트화합물인 화학식III의 화합물과, 보호된 메르캅토화합물인 HR2화합물를 축합반응 하여, 보호된 2-(치환메르캅토)-1β-메틸-카르바페넴계 항생물질을 제조하는 방법을 살펴보면, US 5,478,820(1995), US 4,888344(1987), JP 35366(1992), EP480100(1992), JP35366(1992), JP272426(1996)에서는 축합반응 후 유기용매로 추출한 다음 컬럼크로마토그라피 방법으로, 보호된 2-(치환메르캅토)-1β-메틸-카르바페넴(화학식IIa, IIb, IIc, 및 IId)을 60%~80%의 낮은 수율로 분리하였다. In Scheme 1, a condensation reaction of a compound of formula III, which is a conventional enol phosphate compound, and HR 2 , which is a protected mercapto compound, is used to prepare a protected 2- (substituted mercapto) -1β-methyl-carbapenem antibiotic. In the manufacturing method, US 5,478,820 (1995), US 4,888344 (1987), JP 35366 (1992), EP480100 (1992), JP35366 (1992), and JP272426 (1996) were extracted with an organic solvent and then column By chromatographic method, protected 2- (substituted mercapto) -1β-methyl-carbapenem (formulas IIa, IIb, IIc, and IId) were isolated in low yields of 60% to 80%.
또한 US2007/0249827A1에서는 보호된 2-(치환메르캅토)-1β-메틸-카르바페넴인 화학식IId 화합물을 유기용매에서 결정화하여 87%의 수율로 수득하였고 결정형으로 등록하였다. Also in US2007 / 0249827A1, the compound of formula IId, which is protected 2- (substituted mercapto) -1β-methyl-carbapenem, was crystallized in an organic solvent, obtained in a yield of 87%, and registered as crystalline form.
두 번째 반응인 보호된 2-(치환메르캅토)-1β-메틸-카르바페넴의 탈보호화반응을 살펴보면, WO2006/035300(2005), EP0528678(2001), US 4,888344(1987), EP289801(1990)에서 금속 촉매로 고가의 5~10% 팔라듐-카르본을 사용하였으나 수율이 40~75%밖에 되지 않았다.Deprotection of the second reaction, protected 2- (substituted mercapto) -1β-methyl-carbapenem, is described in WO2006 / 035300 (2005), EP0528678 (2001), US 4,888344 (1987), EP289801 ( In 1990, an expensive 5-10% palladium-carbon was used as the metal catalyst, but the yield was only 40-75%.
그 원인은 팔라듐-카르본을 사용하여 탈보호화할 경우 불순물이 다량 생성되어 수율저하의 원인이 되었다.The reason for this is that when deprotection is carried out using palladium-carbon, a large amount of impurities are generated, which causes a decrease in yield.
이에 본 연구진은 이러한 문제점을 해결하기 위하여 카르바페넴계 항생제 제조에 범용적으로 적용가능하고, 수율 손실을 최소화하여 경제적이며, 불순물을 최소화하여 대량생산이 가능한 획기적인 방법을 개발하게 되었다.
In order to solve this problem, the researchers have developed a breakthrough method that can be widely applied to the production of carbapenem antibiotics, is economical by minimizing yield loss, and minimizes impurities.
종래의 카르바페넴계 항생제 제조과정의 문제점은,Problems of the conventional carbapenem antibiotic manufacturing process,
첫째, 엔올포스페이트화합물인 화학식III과 보호된 메르캅토화합물인 HR2을반응시킨 후, 화학식II와 같은 보호된 2-(치환메르캅토)-1β-메틸-카르바페넴 화합물들을 분리할 때, 비용이 고가이며 생산현장에서 사용하기 어려운 컬럼크로마토그래피 방법을 사용해야 하는 문제점이 있다. First, the cost of separating the protected 2- (substituted mercapto) -1β-methyl-carbapenem compounds such as formula (II) after reacting the enol phosphate compound (III) with the protected mercapto compound (HR 2 ) There is a problem in that the column chromatography method which is expensive and difficult to use at the production site should be used.
또 다른 방법인 유기용매에서 결정화하여 분리하는 방법을 살펴보면, 메로페넴의 전구체인 IId 화합물의 경우, 유기용매에서 결정화하기 때문에 10%이상의 수율 손실을 피할 수 없게 된다. 따라서, 수율을 극대화할 수 있는 경제적인 방법이 절실히 필요한 상태이다. In another method, crystallization and separation in an organic solvent, the IId compound, which is a precursor of meropenem, cannot be avoided due to crystallization in an organic solvent. Therefore, there is an urgent need for an economic method to maximize yield.
둘째, 보호된 2-(치환메르캅토)-1β-메틸-카르바페넴에서 탈보호하기위하여 고가인 금속촉매제인 팔라듐-카르본를 사용할 경우 불순물의 생성을 최소화할 수 없기 때문에 불순물을 최소화할 수 있는 다른 금속촉매를 사용하여 수율증가를 이루고, 제조공정을 단순화하여 산업적으로 적용하기에 적합한 2-(치환메르캅토)-1β-메틸-카르바페넴의 제조방법을 개발하는데 그 목적이 있다.
Second, the use of an expensive metal catalyst, palladium-carbon, for deprotection from protected 2- (substituted mercapto) -1β-methyl-carbapenem can minimize the formation of impurities, which can be minimized. The purpose is to develop a method for preparing 2- (substituted mercapto) -1β-methyl-carbapenem suitable for industrial application by increasing yield using other metal catalysts and simplifying the manufacturing process.
상기목적을 달성하기 위하여 2-(치환메르캅토)-1β-메틸-카르바페넴계 화합물인 화학식I의 개선된 제조방법은,In order to achieve the above object, an improved preparation method of formula (I), which is a 2- (substituted mercapto) -1β-methyl-carbapenem-based compound,
i) 하기 화학식III으로 표시되는 엔올포스페이트화합물과 보호된 메르캅토화합물인 HR2을 염기 존재 하에 축합 반응하여, 보호된 2-(치환메르캅토)-1β-메틸-카르바페넴 화합물인 화학식II의 화합물을 물속에서 무정형으로 고체화하여 분리하는 단계;i) Condensation reaction of an enol phosphate compound represented by the following general formula (III) with HR 2 , a protected mercapto compound, in the presence of a base to give a protected 2- (substituted mercapto) -1β-methyl-carbapenem compound of Separating the compound by solidifying it amorphously in water;
ii) 보호된 2-(치환메르캅토)-1β-메틸-카르바페넴화합물인 하기 화학식II의 화합물을, 촉매제로 팔라듐히드록시드-카르본의 존재하에 탈보호화함으로써 화학식I의 화합물을 제조하는 단계를 포함하는 것을 특징으로 한다.
ii) preparing a compound of formula (I) by deprotecting a compound of formula (II) which is a protected 2- (substituted mercapto) -1β-methyl-carbapenem compound in the presence of palladium hydroxide-carbon as a catalyst; Characterized in that it comprises a step.
[반응식2][Scheme 2]

상기 반응식 2에서, PNB은 p-니트로벤질기를 나타내고In Scheme 2, PNB represents a p-nitrobenzyl group
R2는,R 2 is
(2S,4S)-4-메르캅토-2-[[[3-(2-프로페닐옥시)카르보닐]페닐]아미노]카르보닐-1-피롤리딘카르복실산 2-프로페닐에스테르, (2S,4S)-4-메르캅토-2-[[[3-(p-니트로벤질옥시)카르보닐]페닐]아미노]카르보닐-1-피롤리딘카르복실산 p-니트로벤질에스테르, 6-티오-6,7-디히드로-5H-피라졸로[1,2-a][1,2,4]트리아졸트리플루오로아세트산염, 2-설파모일아미노메틸-1-p-니트로벤질옥시카보닐-4-메르캅토피롤리딘, 또는 4-니토로벤질(2S,4S)-2-[(디메틸아미노)카보닐]-4-메르캅토피롤리딘-1-카복실레이트이다.(2S, 4S) -4-mercapto-2-[[[3- (2-propenyloxy) carbonyl] phenyl] amino] carbonyl-1-pyrrolidinecarboxylic acid 2-propenyl ester, ( 2S, 4S) -4-mercapto-2-[[[3- (p-nitrobenzyloxy) carbonyl] phenyl] amino] carbonyl-1-pyrrolidinecarboxylic acid p-nitrobenzyl ester, 6- Thio-6,7-dihydro-5H-pyrazolo [1,2-a] [1,2,4] triazoletrifluoroacetic acid salt, 2-sulfamoylaminomethyl-1-p-nitrobenzyloxycarbo Nyl-4-mercaptopyrrolidine, or 4-nitorobenzyl (2S, 4S) -2-[(dimethylamino) carbonyl] -4-mercaptopyrrolidine-1-carboxylate.
R3는,R 3 is
(2S,4S)-4-메르캅토-2-[[(3-카르복시페닐)아미노]카르보닐]-3-피롤리딘, 6-티오-6,7-디히드로-5H-피라졸로[1,2-a][1,2,4]트리아졸, 4-티오-2-설파모일아미노메틸피롤리딘, 또는 4-티오-(2S),(4S)-2-[(디메틸아미노)카르보닐]피롤리딘이다.(2S, 4S) -4-mercapto-2-[[(3-carboxyphenyl) amino] carbonyl] -3-pyrrolidine, 6-thio-6,7-dihydro-5H-pyrazolo [1 , 2-a] [1,2,4] triazole, 4-thio-2-sulfamoylaminomethylpyrrolidine, or 4-thio- (2S), (4S) -2-[(dimethylamino) car Carbonyl] pyrrolidine.
본 발명의 일 태양에 따르면, i) 단계에서 제조된 무정형의 화학식 II의 화합물들은 더이상 정제 및 건조되지 않고 탈보호화될 수 있다.
According to one aspect of the invention, the amorphous compounds of formula II prepared in step i) can be deprotected without further purification and drying.
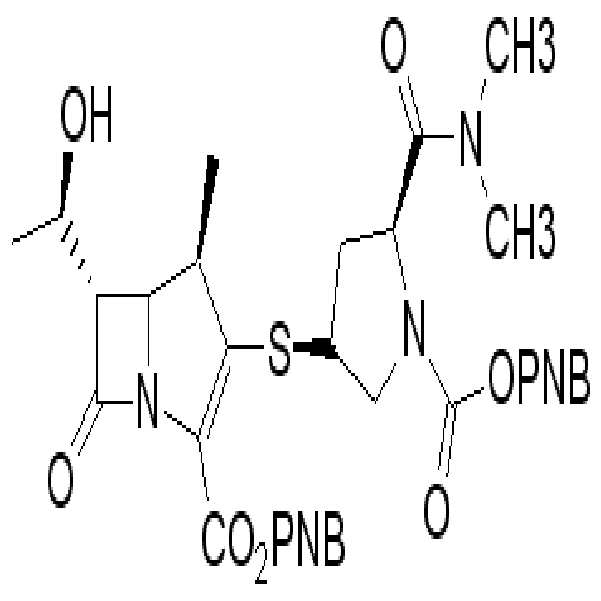
화학식 II의 화합물들은 하기 화학식 IIa, IIa-1, IIb, IIc, 및/또는 IId로 나타내는 보호된 2-(치환메르캅토)-1β-메틸-카르바페넴 유도체들이다.
Compounds of formula II are protected 2- (substituted mercapto) -1β-methyl-carbapenem derivatives represented by the formulas IIa, IIa-1, IIb, IIc, and / or IId.
[화학식 IIa][Formula IIa]

[화학식 IIa-1][Formula IIa-1]

[화학식 IIb][Formula IIb]
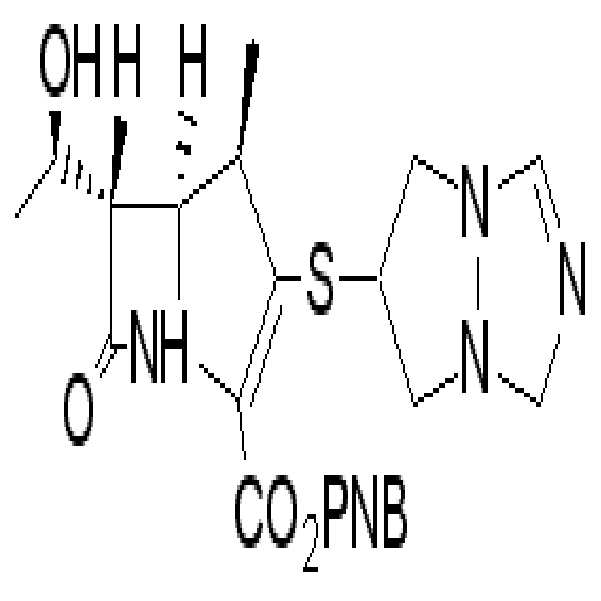
[화학식 IIc]Formula IIc]

[화학식 IId][Formula IId]
상기 반응식 2의 과정을 살펴보면,Looking at the process of Scheme 2,
i)단계에서 상기 화학식III으로 표시되는 엔올포스페이트화합물과 보호된메르캅토화합물인 HR2을 축합반응시킨 후 냉각수에 반응액을 가하여 무정형의 고체로 분리한 다음, ii)단계에 사용할 수 있는 방법이며, 상기 분리된 무정형의 고체는 더이상 정제 및 건조과정을 거치지 않을 수 있다.Condensation reaction of the enol phosphate compound represented by Chemical Formula III in step i) with HR 2 , which is a protected mercapto compound, and the reaction solution is added to cooling water to separate an amorphous solid, followed by step ii). In addition, the separated amorphous solid may no longer be subjected to purification and drying.
따라서 i)단계 제조수율이 98%이상으로 손실이 거의 발생하지 않고 효율적이면서도 99%의 고순도로 제조할 수 있는 경제적인 방법이다.
Therefore, step i) yield is more than 98%, almost no loss, and it is an economical method that can be manufactured with high purity of 99% with efficiency.
한편, 상기 반응식 2의 ii)단계는, 보호된 2-(치환메르캅토)-1β-메틸-카르바페넴 화합물인 화학식II을 탈보호화하는 방법으로서, 기존에 사용된 팔라듐-카르본대신 팔라듐하이드록시드-카르본을 사용함으로써 불순물을 최소화하여 고순도 및 고수율로 2-(치환메르캅토)-1β-메틸-카르바페넴 유도체인 화학식I을 제조할 수 있는, 경제적이며 대량생산이 가능한 효율적인 제조방법이다.
On the other hand, step ii) of Scheme 2 is a method of deprotecting the general formula (II), which is a protected 2- (substituted mercapto) -1β-methyl-carbapenem compound, and palladium hydride instead of palladium-carbon. Efficient preparation for economical and mass production to produce formula (I), 2- (substituted mercapto) -1β-methyl-carbapenem derivative, in high purity and high yield by minimizing impurities by using roxide-carbon It is a way.
본 발명에 따르면, 보호된 2-(치환메르캅토)-1β-메틸-카르바페넴 유도체들을 물속에서 결정화하기 때문에 수율이 98%이상으로 수율손실이 거의 없으며, 또한 습체의 수득물을 건조하지 않고 다음 단계인 탈보호반응을 수행할 수 있으므로 산업적으로 매우 효율적인 방법이라 할 수 있다.According to the present invention, since the protected 2- (substituted mercapto) -1β-methyl-carbapenem derivatives are crystallized in water, the yield is more than 98%, and there is almost no loss of yield, and without drying the obtained product of the wet body. The next step, deprotection, can be carried out, making it an industrially very efficient method.
또한, 탈보호 반응시 팔라듐-카르본 대신 팔라듐히드록시드-카르본을 사용함으로써 불순물의 생성을 최소화하여 고 순도 및 고 수율로 제조할 수 있는 매우 경제적인 방법을 개발하였다.
In addition, by using palladium hydroxide-carbon instead of palladium-carbon in the deprotection reaction, a very economical method for producing impurities with high purity and high yield was minimized.
도 1은 본 발명의 실시예에 따라 제조된 화학식IIc화합물에 대한 XRD 그래프로서, 무정형인 것을 나타내고 있다.1 is an XRD graph of a compound of formula IIc prepared according to an embodiment of the present invention, which shows that it is amorphous.
본 발명에 따른 2-(치환메르캅토)-1β-메틸-카르바페넴의 제조방법을 상기 반응식 2를 근거로 보다 구체적으로 설명하면 다음과 같다.A method for preparing 2- (substituted mercapto) -1β-methyl-carbapenem according to the present invention will be described in more detail based on Scheme 2 as follows.
첫 번째 반응인 i)단계의 반응 과정은 상기 화학식 III의 엔올포스페이트화합물을, 염기 존재 하에서 보호된 메르캅토화합물인 HR2와 축합 반응시킨 후, 냉각수에 가하여 화학식 IIa, IIa-1, IIb, IIc, 및/또는 IId 화합물들을 무정형으로 분리하는 단계이다. The reaction process of step i), the first reaction, is carried out by condensation reaction of the enol phosphate compound of formula III with HR 2 , a mercapto compound protected in the presence of a base, and then added to cooling water And / or amorphous separation of the IId compounds.
반응물인 HR2의 사용량은 엔올포스페이트화합물인 화학식 III에 대하여 1~1.2당량이고, 반응용매로는 극성용매인 아세토니트릴, 디메틸포름아미드, 디메틸아세트아미드 중에서 선택된 용매 또는 이들의 혼합용매를 사용할 수 있다. The reactant HR 2 is used in an amount of 1 to 1.2 equivalents based on the chemical formula III of the enol phosphate compound, and a reaction solvent selected from acetonitrile, dimethylformamide, and dimethylacetamide, which are polar solvents, or a mixed solvent thereof may be used. .
염기로는 3급 아민 화합물, 예를 들면, N,N-디메틸아미노피리딘의 방향족아민, 디이소프로필에틸아민, 트리에틸아민, 또는 트리메틸아민을 사용할 수 있으며, 바람직하게는 디이소프로필에틸아민을 사용한다. 염기의 사용량은 엔올포스페이트화합물인 화학식 III에 대하여 1~10당량이며, 바람직하게는 1~3당량이다. 반응은 25~-50℃, 바람직하게는 -15~-20℃에서 1~3시간 반응시킨다. 반응완료 후 결정화할 경우, 사용된 용매의 4배 내지 10배 부피의 냉각된 물에 반응액을 서서히 적가하여 무정형의 고체로 분리한다. 이때 반응액을 냉각수에 적가하면서 pH가 7이 넘지 않도록 1N HCl로 조절하면서 적가하여야 한다. 바람직하게는 pH 4~7 사이로 조절한다.As the base, a tertiary amine compound, for example, an aromatic amine of N, N-dimethylaminopyridine, diisopropylethylamine, triethylamine, or trimethylamine can be used, and preferably diisopropylethylamine is used. use. The use amount of base is 1-10 equivalents, preferably 1-3 equivalents to the general formula (III) which is an enol phosphate compound. The reaction is carried out at 25 to 50 ° C, preferably at -15 to 20 ° C for 1 to 3 hours. In the case of crystallization after completion of the reaction, the reaction solution is slowly added dropwise to the cooled water of 4 to 10 times the volume of the solvent used to separate it into an amorphous solid. At this time, the reaction solution was added dropwise to the cooling water while adjusting with 1N HCl so that the pH does not exceed 7. Preferably it is adjusted between pH 4-7.
상기에서 i)단계에서 얻은 무정형의 조생성물인 화학식 IIa, IIa-1, IIb, IIc, 및/또는 IId의 화합물들은 건조 및 더 이상의 정제과정 없이, 두 번째 반응인 탈보호화 반응에 사용할 수 있다.The compounds of formulas IIa, IIa-1, IIb, IIc, and / or IId, which are amorphous crude products obtained in step i), can be used in the second reaction deprotection reaction without drying and further purification.
두 번째 반응인 ii)단계의 반응 과정은 상기 무정형의 조생성물인 화학식IIa, IIa-1, IIb, IIc, 및/또는 IId의 화합물들을 수소화 반응시켜서 탈보호화 하여 본 발명이 목적하는 상기화학식 Ia, Ib, Ic, 및/또는 Id의 화합물들을 제조하는 단계이다.The reaction process of the second reaction step ii) is a deprotected by hydrogenation of the compounds of the amorphous crude product (IIa, IIa-1, IIb, IIc, and / or IId) of the general formula Ia, Preparing compounds of Ib, Ic, and / or Id.
상기한 탈보호화 반응은 조생성물인 화학식IIa, IIa-1, IIb, IIc, 및/또는 IId의 화합물에 대하여 0.2 내지 20몰%의 팔라듐하이드록시드-카르본 금속 촉매 하에 수행할 수 있다.The above deprotection reaction can be carried out under 0.2 to 20 mole% palladium hydroxide-carbon metal catalyst for the crude product of formulas IIa, IIa-1, IIb, IIc, and / or IId.
반응용매로는 정제수, 테트라히드로푸란, 메탄올, 에탄올, 프로판올,이소프로판올, 부탄올, 에틸아세테이트로 이루어진 그룹 중에서 선택된 단독용매 또는 이들의 혼합용매를 사용할 수 있다. As the reaction solvent, a single solvent selected from the group consisting of purified water, tetrahydrofuran, methanol, ethanol, propanol, isopropanol, butanol, and ethyl acetate or a mixed solvent thereof may be used.
반응온도는 0~25℃, 바람직하게는 15℃이고. 2~10kg/m2 압력 하에서 2 내지 8시간 반응한 다음 에틸아세테이트로 추출하여 불순물을 제거하고, 수층의 pH를 3~6으로 조절한다. 수층에 아세톤, 테트라히드로푸란, 메탄올, 프로판올, 또는 이소프로판올을 가하고 0~5℃에서 3~24시간 동안 결정화한 후 여과하고 건조하여, 두 단계 전체수율을 76~80%의 고 수율과 98%이상의 고 순도로 제조하였다.
Reaction temperature is 0-25 degreeC, Preferably it is 15 degreeC. After reacting for 2 to 8 hours at a pressure of 2 ~ 10kg / m 2 and then extracted with ethyl acetate to remove impurities, the pH of the aqueous layer is adjusted to 3-6. Acetone, tetrahydrofuran, methanol, propanol, or isopropanol was added to the aqueous layer and crystallized at 0-5 ° C. for 3 to 24 hours, followed by filtration and drying, yielding a high yield of 76-80% and a yield of at least 98%. Made to high purity.
본 발명은 하기의 실시예에 의하여 보다 더 잘 이해될 수 있으며, 하기의 실시예는 본 발명을 예시하기 위한 것이며 첨부된 특허 청구범위에 의하여 보호범위를 제한하고자 하는 것은 아니다.
The invention can be better understood by the following examples, which are intended to illustrate the invention and are not intended to limit the scope of the claims by the appended claims.
[실시예]
EXAMPLE
A. 화학식 III의 화합물로부터 화학식 II의 화합물의 조생성물 제조
A. Preparation of crude product of compound of formula II from compound of formula III
실시예 1Example 1
(4R,5S,6S)-p-니트로벤질-3-[[(3S,5S)-5-[(디메틸아미노)카르보닐]-1-[[4-니트로벤질)옥시]카르보닐]-3-피롤리디닐]티오-6-[(1R)-1-히드록시에틸]-4-메틸-7-옥소-1-아자바이시클로[3,2,0]헵트-2-엔-카르복실레이트(화학식 IId의 화합물)의 제조
(4R, 5S, 6S) -p-nitrobenzyl-3-[[(3S, 5S) -5-[(dimethylamino) carbonyl] -1-[[4-nitrobenzyl) oxy] carbonyl] -3 -Pyrrolidinyl] thio-6-[(1R) -1-hydroxyethyl] -4-methyl-7-oxo-1-azabicyclo [3,2,0] hept-2-ene-carboxylate Preparation of (Compound of Formula IId)
디메틸포름아미드 500ml에 화학식III의 화합물인 엔올포스페이트 화합물 120g(201.9mmol)과 4-니트로벤질-(2S,4S)-2-[(디메틸아미노)카르보닐]-4-메르캅토피롤리딘-1-카복실레이트 74g(209.4mmol)을 가하여 용해시킨 후 -20℃로 냉각하였다. 여기에 디이소프로필에틸아민 40ml를 서서히 적가한 후, 동 온도에서 1시간 교반하였다. 2L의 냉각수에 상기 반응액을 서서히 적가하여 결정화하였다. 이때 pH가 7이 넘지 않도록 1N 염산수용액으로 조절하며 적가하고 최종 pH를 6.5로 조절하였다.120 g (201.9 mmol) of the compound of formula III and 4-nitrobenzyl- (2S, 4S) -2-[(dimethylamino) carbonyl] -4-mercaptopyrrolidine-1 in 500 ml of dimethylformamide 74 g (209.4 mmol) of carboxylate was added to dissolve and cooled to -20 ° C. 40 ml of diisopropylethylamine was slowly added dropwise thereto, and then stirred at the same temperature for 1 hour. The reaction solution was slowly added dropwise to 2 L of cooling water to crystallize. At this time, the pH was adjusted to 1 N hydrochloric acid so as not to exceed 7 and the final pH was adjusted to 6.5.
0~5℃에서 1시간 교반 후 여과하여 화학식 IId의 화합물의 조생성물 450g(습체)을 얻었다.After stirring for 1 hour at 0-5 [deg.] C., filtration gave 450 g (wet product) of the crude product of the compound of formula IId.
건조수율: 138g(98%)Drying yield: 138g (98%)
순도(HPLC): 99.4%
Purity (HPLC): 99.4%
실시예 2Example 2
(1R,5S,6S)-2-[(3S,5S)-1-p-니트로벤질-5-(N-설파모일아미노)메틸피롤리딘-3-일]티오-6[(1R)-1-히드록시에틸-1-메틸-1-카르바-2-페넴-3-카르복실산 p-니트로벤질에스테르(화학식IIc의 화합물)의 제조
(1R, 5S, 6S) -2-[(3S, 5S) -1-p-nitrobenzyl-5- (N-sulfamoylamino) methylpyrrolidin-3-yl] thio-6 [(1R)- Preparation of 1-hydroxyethyl-1-methyl-1-carba-2-phenem-3-carboxylic acid p-nitrobenzyl ester (compound of formula IIc)
디메틸아세트아미드 50ml에 화학식III의 화합물인 엔올포스페이트 화합물 12g(20.2mmol)과 (2S,4S)-1-p-니트로벤질옥시카르보닐-2-(N-설파모일아미노)메틸-4-메르캅토피롤리딘 8.16g(20.9.mmol)을 가하여 용해시킨 후 -20℃로 냉각하였다. 여기에 디이소프로필에틸아민4ml(23mmol)를 서서히 적가한 후 동 온도에서 1시간 교반하였다.12 g (20.2 mmol) of the compound of formula III and (2S, 4S) -1-p-nitrobenzyloxycarbonyl-2- (N-sulfamoylamino) methyl-4-mercap in 50 ml of dimethylacetamide 8.16 g (20.9. Mmol) of torrolidine was added and dissolved, followed by cooling to -20 ° C. 4 ml (23 mmol) of diisopropylethylamines were slowly added dropwise thereto, followed by stirring at the same temperature for 1 hour.
300ml의 냉각수에 상기 반응액을 서서히 적가하여 결정화하였다. 이때 pH가 7이 넘지 않도록 1N HCl로 조절하며 적가하여 pH를 6.0로 조절하였다.The reaction solution was slowly added dropwise to 300 ml of cooling water to crystallize. At this time, the pH was adjusted to 6.0 by adding dropwise with 1N HCl so as not to exceed pH 7.
0~5℃에서 1시간 교반 후 여과하여 화학식IIc의 화합물의 조생성물 55g(습체)을 얻었다.After stirring for 1 hour at 0-5 ° C., filtration gave 55 g (wet product) of the crude product of the compound of formula IIc.
건조수율:14.57g(98.2%)Drying yield: 14.57 g (98.2%)
순도: 99.2%
Purity: 99.2%
실시예 3Example 3
6-[[2(4R,5S,6S)-카르복시-6-[(1R)-히드록시에틸]-4-메틸-7-옥소-1-아자비시클로[3,2,0]헵트-2-엔-3-일]티오]-6,7-디히드로-5H-피라졸로[1,2-a][1,2,4]트리아졸 p-니트로벤질에스테르(화학식 IIb의 화합물)의 제조
6-[[2 (4R, 5S, 6S) -carboxy-6-[(1R) -hydroxyethyl] -4-methyl-7-oxo-1-azabicyclo [3,2,0] hept-2- Preparation of En-3-yl] thio] -6,7-dihydro-5H-pyrazolo [1,2-a] [1,2,4] triazole p-nitrobenzylester (compound of Formula IIb)
아세토니트릴 50ml에 화학식III의 화합물인 엔올포스페이트 화합물 12g(20.2mmol)과 6-티오-6,7-디히드로-5H-피라졸로[1,2-a][1,2,4]트리아졸트리플루오로아세트산염 5.37g(20.9.mmol)을 가하여 용해시킨 후 -20℃로 냉각하였다. 여기에 디이소프로필에틸아민 5ml를 서서히 적가한 후, 동 온도에서 1시간 교반하였다.12 g (20.2 mmol) of the compound of formula III and 6-thio-6,7-dihydro-5H-pyrazolo [1,2-a] [1,2,4] triazoletri in 50 ml of acetonitrile 5.37 g (20.9. Mmol) of fluoroacetic acid was added to dissolve and cooled to -20 ° C. 5 ml of diisopropylethylamine was slowly added dropwise thereto, followed by stirring at the same temperature for 1 hour.
400ml의 냉각수에 상기 반응액을 서서히 적가하여 결정화하였다. 이때 pH가 7이 넘지 않도록 1N HCl로 조절하며 적가하여 pH를 6.8로 조절하였다.The reaction solution was slowly added dropwise to 400 ml of cooling water to crystallize. At this time, the pH was adjusted to 6.8 by dropwise addition with 1N HCl so as not to exceed pH 7.
0~5℃에서 1시간 교반 후 여과하여 화학식 IIb의 화합물의 조생성물 30g(습체)을 얻었다.After stirring for 1 hour at 0-5 ° C., filtration gave 30 g (wet product) of the crude product of the compound of formula IIb.
건조수율: 9.73g(99%)Drying yield: 9.73 g (99%)
순도: 99.1%
Purity: 99.1%
실시예 4Example 4
[4R-[3(3S*,5S*),4α,5β,6β(R*)]]-6-(1-히드록시에틸)-4-메틸-3-[[[1-[(2-프로페닐옥시)카르보닐]-5-[[3-[(2-프로페닐옥시)카르보닐]페닐]아미노]카르보닐-3-피롤리딘일]티오]-7-옥소-1-아자바이시클로[3,2,0]헵트-2-엔-2-카르복실산 p-니트로벤질에스테르(화학식 IIa의 화합물)의 제조
[4R- [3 (3S *, 5S *), 4α, 5β, 6β (R *)]]-6- (1-hydroxyethyl) -4-methyl-3-[[[1-[(2- Propenyloxy) carbonyl] -5-[[3-[(2-propenyloxy) carbonyl] phenyl] amino] carbonyl-3-pyrrolidinyl] thio] -7-oxo-1-azabicyclo Preparation of [3,2,0] hept-2-ene-2-carboxylic acid p-nitrobenzyl ester (compound of formula IIa)
아세토니트릴 40ml,디메틸포름아미드 10ml에 화학식III의 화합물인 엔올포스페이트 화합물 12g(20.2mmol)과 (2S,시스)-4-메르캅토-2-[[[3-(2-프로페닐옥시)카르보닐]페닐]아미노]카르보닐]-1-피롤리딘카르복실산 2-프로페닐에스테르 7.57g(20.9.mmol)을 가하여 용해시킨 후 -20℃로 냉각하였다. 여기에 디이소프로필에틸아민 10ml를 서서히 적가한 후 동 온도에서 1시간 교반하였다.12 g (20.2 mmol) of the compound of formula III and (2S, cis) -4-mercapto-2-[[[3- (2-propenyloxy) carbonyl in 40 ml of acetonitrile and 10 ml of dimethylformamide ] Phenyl] amino] carbonyl] -1-pyrrolidinecarboxylic acid 2-propenyl ester 7.57 g (20.9. Mmol) was added and dissolved, and it cooled to -20 degreeC. 10 ml of diisopropylethylamine was slowly added dropwise thereto, followed by stirring at the same temperature for 1 hour.
400ml의 냉각수에 상기 반응액을 서서히 적가하여 결정화하였다. 이때 pH가 7이 넘지 않도록 1N HCl로 조절하며 적가하여 pH를 6.8로 조절하였다.The reaction solution was slowly added dropwise to 400 ml of cooling water to crystallize. At this time, the pH was adjusted to 6.8 by dropwise addition with 1N HCl so as not to exceed pH 7.
0~5℃에서 1시간 교반 후 여과하여 화학식 IIa의 화합물의 조생성물 35g(습체)을 얻었다.After stirring for 1 hour at 0-5 ° C., filtration gave 35 g (wet product) of the crude product of the compound of formula IIa.
건조수율:14.34g(98.5%)Drying yield: 14.34 g (98.5%)
순도: 99.3%
Purity: 99.3%
실시예 5Example 5
[4R-[3(3S*,5S*),4α,5β,6β(R*)]]-6-(1-히드록시에틸)-4-메틸l-3-[[[1-[(2-p-니트로벤질옥시)카르보닐]페닐]아미노]카르보닐-3-피롤리딘닐]티오-7-옥소-1-아자바이시클로[3,2,0]헵트-2-엔-2-카르복실산 p-니트로벤질에스테르(화학식 IIa-1의 화합물)의 제조
[4R- [3 (3S *, 5S *), 4α, 5β, 6β (R *)]]-6- (1-hydroxyethyl) -4-methyll-3-[[[1-[(2 -p-nitrobenzyloxy) carbonyl] phenyl] amino] carbonyl-3-pyrrolidinyl] thio-7-oxo-1-azabicyclo [3,2,0] hept-2-ene-2-car Preparation of the acid p-nitrobenzyl ester (compound of formula IIa-1)
아세토니트릴 40ml, 디메틸아세트아미드 10ml에 화학식III의 화합물인 엔올포스페이트 화합물 12g(20.2mmol)과 (2S,시스)-4-메르캅토-2-[[[3-(2-p-니트로벤질옥시)카르보닐]페닐]아미노]카르보닐]-1-피롤리딘카르복실산 p-니트로벤질에스테르 12.13g(20.9.mmol)을 가하여 용해한 후 -20℃로 냉각하였다. 여기에 디이소프로필에틸아민 8ml를 서서히 적가한 후, 동 온도에서 1시간 교반하였다.12 g (20.2 mmol) of the compound of formula III and (2S, cis) -4-mercapto-2-[[[3- (2-p-nitrobenzyloxy) in 40 ml of acetonitrile and 10 ml of dimethylacetamide Carbonyl] phenyl] amino] carbonyl] -1-pyrrolidinecarboxylic acid p-nitrobenzyl ester 12.13g (20.9.mmol) was added, melt | dissolved, and it cooled to -20 degreeC. 8 ml of diisopropylethylamine was slowly added dropwise thereto, followed by stirring at the same temperature for 1 hour.
400ml의 냉각수에 상기 반응액을 서서히 적가하여 결정화하였다. 이때 pH가 7이 넘지 않도록 1N HCl로 조절하며 적가하여 pH를 6.8로 조절하였다.The reaction solution was slowly added dropwise to 400 ml of cooling water to crystallize. At this time, the pH was adjusted to 6.8 by dropwise addition with 1N HCl so as not to exceed pH 7.
0~5℃에서 1시간 교반 후 여과하여 화학식 IIa-1의 화합물의 조생성물 40g(습체)을 얻었다.After stirring for 1 hour at 0-5 ° C., filtration was performed to obtain 40 g (wet product) of the crude product of the compound of formula IIa-1.
건조수율 :18.31g(98%)Drying yield: 18.31 g (98%)
순도: 99.3%
Purity: 99.3%
B. 화학식 II의 화합물로부터 화학식 I의 화합물 제조
B. Preparation of Compounds of Formula (I) from Compounds of Formula (II)
실시예 6Example 6
(4R,5S,6S)-3-[[(3S,5S)-5-[(디메틸아미노)카르보닐]-3-피롤리디닐]티오]-6-[(1R)-1-히드록시에틸]-4-메틸-7-옥소-1-아자비시클로[3,2,0]헵트-2-엔-카르복실산(화학식Id의 화합물)의 제조
(4R, 5S, 6S) -3-[[(3S, 5S) -5-[(dimethylamino) carbonyl] -3-pyrrolidinyl] thio] -6-[(1R) -1-hydroxyethyl ] -4-Methyl-7-oxo-1-azabicyclo [3,2,0] hept-2-ene-carboxylic acid (compound of Formula Id)
실시예 1에서 얻은 화학식IId의 화합물인 조생성물 450g(습체)을 THF 1.4L 에 용해시킨 후 버퍼수용액 1.1L(N-몰포린 20ml를 정제수 1.08L에 가한 후 아세트산으로 pH7로 조절한 용액)를 가하고 5% 팔라듐히드록시드-카르본 28g을 가하고 실온에서 6시간 동안 수소를 가하여 5kg/m2의 압력 하에 탈보호반응을 하였다.After dissolving 450 g (wet product) of the compound of formula IId obtained in Example 1 in 1.4 L of THF, 1.1 L of buffered solution (20 ml of N-morpholine was added to 1.08 L of purified water and adjusted to pH 7 with acetic acid) 28 g of 5% palladium hydroxide-carbon was added thereto, followed by dehydrogenation at a pressure of 5 kg / m 2 by adding hydrogen at room temperature for 6 hours.
반응액을 여과하고 물 50ml로 세척한 후 에틸아세테이트 1L를 가하여 층 분리하였다. 수층을 0~5℃로 냉각하고 여기에 6L의 아세톤을 가하여 일야 교반 후 여과하였다. 고체를 냉각된 아세톤으로 세척하고 건조하여 화학식Id의 화합물 58.8g을 얻었다.The reaction solution was filtered, washed with 50 ml of water, and then separated into layers by adding 1 L of ethyl acetate. The aqueous layer was cooled to 0-5 [deg.] C., and 6L of acetone was added thereto, followed by overnight stirring and filtering. The solid was washed with cold acetone and dried to give 58.8 g of compound of formula Id.
두단계 총 수율:76%Two-step total yield: 76%
순도: 99.3%(HPLC)Purity: 99.3% (HPLC)
1H-NMR(D2O)δ 1.24(3H,d), 1.32(3H,d), 1.92(1H,m), 3.02(3H,s), 3.08(3H,s), 3.31~3.50(3H,m), 3.68(1H,m), 4.07(1H,m), 4.22~4.30(2H,m)
1 H-NMR (D 2 O) δ 1.24 (3H, d), 1.32 (3H, d), 1.92 (1H, m), 3.02 (3H, s), 3.08 (3H, s), 3.31 ~ 3.50 (3H , m), 3.68 (1H, m), 4.07 (1H, m), 4.22-4.30 (2H, m)
실시예 7Example 7
(1R,5S,6S)-6[(1R)-1-히드록시에틸]-2-[5-설프아미도메틸피롤리딘-3-일]티오-1-메틸-1-카르바-2-페넴-3-카르복실산(화합물Ic의 화합물)의 제조
(1R, 5S, 6S) -6 [(1R) -1-hydroxyethyl] -2- [5-sulamidomethylpyrrolidin-3-yl] thio-1-methyl-1-carba-2 Preparation of Penem-3-carboxylic Acid (Compound of Compound Ic)
실시예 2에서 얻은 화학식IIc의 화합물인 조생성물 55g(wet)을 THF 140ml 에 용해한 후 버퍼수용액 110ml(N-몰포린 2ml를 정제수 108ml에 가한 후 아세트산으로 pH7로 조절한 용액)를 가하고 5% 팔라듐히드록시드-카르본 5.6g을 가하고 실온에서 6시간 동안 수소를 가하여 5kg/m2의 압력 하에 탈보호반응을 하였다.After dissolving 55 g (wet) of the crude product of formula IIc obtained in Example 2 in 140 ml of THF, 110 ml of buffer solution (2 ml of N-morpholine was added to 108 ml of purified water and adjusted to pH 7 with acetic acid) and 5% palladium 5.6 g of hydroxide-carbon was added and hydrogen was added at room temperature for 6 hours to effect deprotection under a pressure of 5 kg / m 2 .
반응액을 여과하고 물 10ml로 세척한 후 에틸아세테이트 100ml를 가하고 30분 동안 교반 후 층 분리하였다. 수층을 0~5℃로 냉각하고 pH를 5.5로 조절하고 600ml의 아세톤을 가하여 일야 교반 후 여과하였다. 고체를 냉각된 아세톤으로 세척하고 건조하여 화합물Ic의 화합물 6.8g을 얻었다.The reaction solution was filtered, washed with 10 ml of water, 100 ml of ethyl acetate was added, the mixture was stirred for 30 minutes, and the layers were separated. The aqueous layer was cooled to 0-5 [deg.] C., the pH was adjusted to 5.5, and 600 ml of acetone was added, followed by overnight stirring and filtering. The solid was washed with cold acetone and dried to give 6.8 g of compound Ic.
두 단계 총 수율:80.3%Two stage total yield: 80.3%
순도: 99.1%(HPLC)Purity: 99.1% (HPLC)
1H-NMR(D2O)δ1.23(3H,d), 1.28(3H,d), 1.65~1.84(1H,m), 2.63~2.82(1H,m), 3.26~3.60( 5H,m), 3.64~3.77(1H,m), 3.85~4.12(2H,m), 4.16~4.30(2H,m)
1 H-NMR (D 2 O) δ1.23 (3H, d), 1.28 (3H, d), 1.65 ~ 1.84 (1H, m), 2.63 ~ 2.82 (1H, m), 3.26 ~ 3.60 (5H, m ), 3.64-3.77 (1H, m), 3.85-4.22 (2H, m), 4.16-4.30 (2H, m)
실시예8Example 8
(1R,5S,6S)-2-[6,7-디히드로-5H-피라졸로[1,2-a][1,2,4]트리아졸륨-6-일 ) ]티오-6-[(R)-1-히드록시에틸-1-메틸-카르바페넴-3-카르복실레이트(화학식Ib의 화합물)의 제조
(1R, 5S, 6S) -2- [6,7-dihydro-5H-pyrazolo [1,2-a] [1,2,4] triazolium-6-yl ) ] thio-6-[( R) -1-hydroxyethyl-1-methyl-carbapenem-3-carboxylate (compound of Formula Ib)
실시예 3에서 얻은 화학식IIb의 화합물인 조생성물 30g(wet)을 THF 140ml, 아세테이트버퍼수용액 110ml(N-몰포린 2ml를 정제수 108ml에 가한 후 아세트산으로 pH7로 조절한 용액)를 가하고 5% 팔라듐히드록시드-카르본 2.8g을 가하고 실온에서 2시간 동안 수소를 가하여 5kg/m2의 압력하에 탈보호반응을 하였다.30 g (wet) of the crude product, a compound of Formula IIb obtained in Example 3, was added with 140 ml of THF and 110 ml of aqueous acetate buffer solution (2 ml of N-morpholine was added to 108 ml of purified water and adjusted to pH 7 with acetic acid), followed by 5% palladium hydroxide. 2.8 g of loxide-carbon was added and hydrogen was added at room temperature for 2 hours to effect deprotection under a pressure of 5 kg / m 2 .
반응액을 여과하고 물 10ml로 세척한 후 에틸아세테이트 100ml를 가하고 30분 교반 후 층 분리하였다. 수층을 0~5℃로 냉각하고 pH를 5.5로 조절한 후 이소프로필알코올 600ml을 가하고 일야 교반한 다음 여과하였다. 고체를 냉각된 이소프로필알코올로 세척하고 건조하여 화학식Ib의 화합물 5.44g을 얻었다.The reaction solution was filtered, washed with 10 ml of water, 100 ml of ethyl acetate was added, the mixture was stirred for 30 minutes, and the layers were separated. The aqueous layer was cooled to 0˜5 ° C., the pH was adjusted to 5.5, and 600 ml of isopropyl alcohol was added, stirred overnight, and filtered. The solid was washed with cold isopropyl alcohol and dried to give 5.44 g of compound of formula Ib.
두 단계 총 수율:77%Two stage total yield: 77%
순도: 99.03%(HPLC)Purity: 99.03% (HPLC)
1H-NMR(D2O)δ: 1.28(3H,d), 1.32(3H,d), 3.45(1H,dd), 3.57(1H,dd), 4.32(1H,q), 4.36(1H,dd), 4.74~4.85(2H,m), 4.97~5.06(1H,m), 5.07~5.18(2H,m), 9.07(1H,s), 9.08(1H,s)
1 H-NMR (D 2 O) δ: 1.28 (3H, d), 1.32 (3H, d), 3.45 (1H, dd), 3.57 (1H, dd), 4.32 (1H, q), 4.36 (1H, dd), 4.74 to 4.85 (2H, m), 4.97 to 5.06 (1H, m), 5.07 to 5.18 (2H, m), 9.07 (1H, s), 9.08 (1H, s)
실시예9Example 9
(4R,5S,6S)-3-[[(3S,5S)-5-[[(3-카르복시페닐)아미노]카르보닐-3-피롤리디닐]티오-6-[(1R)-1-히드록시에틸]-4-메틸-7-옥소-1-아자바이시클로[3,2,0]헵트-2-엔-2-카르복실산(화학식Ia의 화합물)의 제조
(4R, 5S, 6S) -3-[[(3S, 5S) -5-[[(3-carboxyphenyl) amino] carbonyl-3-pyrrolidinyl] thio-6-[(1R) -1- Preparation of Hydroxyethyl] -4-methyl-7-oxo-1-azabicyclo [3,2,0] hept-2-ene-2-carboxylic acid (compound of Formula Ia)
실시예 5에서 얻은 화학식IIa-1의 화합물인 조생성물 40g(wet)을 THF 140ml,아세테이트버퍼수용액 110ml(N-몰포린 2ml를 정제수 108ml에 가한 후 아세트산으로 pH7로 조절한 용액)를 가하고 5% 팔라듐히드록시드-카르본 5.6g을 가하고 실온에서 6시간 동안 5kg/m2의 압력하에 수소화 반응하여 탈보호반응을 하였다.40 g (wet) of the crude product of Formula IIa-1 obtained in Example 5 was added to 140 ml of THF and 110 ml of an aqueous buffer buffer solution (2 ml of N-morpholine was added to 108 ml of purified water and adjusted to pH 7 with acetic acid), and 5% 5.6 g of palladium hydroxide-carbon was added and deprotected by hydrogenation under a pressure of 5 kg / m 2 for 6 hours at room temperature.
반응액을 여과하고 물 10ml로 세척한 후 에틸아세테이트 100ml를 가하고 30분 교반 후 층 분리하였다. 수층을 0~5℃로 냉각하고 pH를 4.5로 조절한 후 이소프로필알코올 600ml을 가하고 일야 교반 한 다음 여과하였다. 고체를 냉각된 이소프로필알코올로 세척하여 화학식Ia 화합물 7.5g을 얻었다.The reaction solution was filtered, washed with 10 ml of water, 100 ml of ethyl acetate was added, the mixture was stirred for 30 minutes, and the layers were separated. The aqueous layer was cooled to 0-5 ° C., the pH was adjusted to 4.5, and 600 ml of isopropyl alcohol was added, stirred overnight, and filtered. The solid was washed with cold isopropyl alcohol to give 7.5 g of compound of formula Ia.
두 단계 총 수율:78%Two stage total yield: 78%
순도: 98.5%(HPLC)Purity: 98.5% (HPLC)
1H-NMR(DMSO-d6)δ: 1.17(6H,d), 1.95(1H,m), 2.84(1H,m), 3.11(1H,dd), 3.23(1H,dd), 3.41(1H,q), 3.67(1H,dd), 3.90(1H,q), 3.40(1H,t), 4.22(1H,dd), 4.27(1H,t), 7.48(1H,t), 7.71(1H,d), 7.87(1H,d), 8.26(1H,s)
1 H-NMR (DMSO-d 6 ) δ: 1.17 (6H, d), 1.95 (1H, m), 2.84 (1H, m), 3.11 (1H, dd), 3.23 (1H, dd), 3.41 (1H , q), 3.67 (1H, dd), 3.90 (1H, q), 3.40 (1H, t), 4.22 (1H, dd), 4.27 (1H, t), 7.48 (1H, t), 7.71 (1H, d), 7.87 (1 H, d), 8.26 (1 H, s)
상기 실시예에서와 같이, 2-(치환메르캅토)-1β-메틸-카르바페넴 유도체들, 즉, 화학식Ia, Ib, Ic, 및/또는 Id를 제조하기 위한 두 단계 전체수율이 각각 78, 77. 80, 및 76%로서 수율이 높으며, 순도 또한 98%이상으로 제조할 수 있고, IIa, IIa-1, IIb, IIc, 및/또는 IId를 정제하지 않으며 건조하지 않고서 다음 탈보호화 반응을 할 수 있어 효율적이었다.
As in the above examples, the two-step overall yield for preparing 2- (substituted mercapto) -1β-methyl-carbapenem derivatives, i.e., Formulas Ia, Ib, Ic, and / or Id, respectively, was 78, 77. The yield is high as 80, and 76%, the purity can be prepared to 98% or more, and the next deprotection reaction can be carried out without purifying IIa, IIa-1, IIb, IIc, and / or IId without drying. It was able and was efficient.
Claims (7)
ii) 하기 화학식II로 나타내는 화합물을 탈보호화 하여 하기 화학식I로 나타내는 화합물을 제조하는 단계를 포함하는 것을 특징으로 하는 카르바페넴계 화합물의 제조방법:
[화학식III]

[화학식II]

[화학식I]

상기 식에서 PNB는 파라니트로벤질기이고,
R2는
(2S,4S)-4-메르캅토-2-[[[3-(2-프로페닐옥시)카르보닐]페닐]아미노]카르보닐-1-피롤리딘카르복실산 2-프로페닐에스테르, (2S,4S)-4-메르캅토-2-[[[3-(p-니트로벤질옥시)카르보닐]페닐]아미노]카르보닐-1-피롤리딘카르복실산 p-니트로벤질에스테르, 6-티오-6,7-디히드로-5H-피라졸로[1,2-a][1,2,4]트리아졸트리플루오로아세트산염, 2-설파모일아미노메틸-1-p-니트로벤질옥시카보닐-4-메르캅토피롤리딘, 또는 4-니트로벤질(2S,4S)-2-[(디메틸아미노)카보닐]-4-메르캅토피롤리딘-1-카르복실레이트이며,
R3는
(2S,4S)-4-메르캅토-2-[[(3-카르복시페닐)아미노]카르보닐]-3-피롤리딘, 6-티오-6,7-디히드로-5H-피라졸로[1,2-a][1,2,4]트리아졸, 4-티오-2-설파모일아미노메틸피롤리딘, 또는 4-티오-(2S),(4S)-2-[(디메틸아미노)카보닐]피롤리딘이다.
i) condensation of the compound represented by the following formula (III) with the protected mercapto compound HR 2 to form an amorphous separation of the compound represented by the following formula (II) in water; And
ii) a method for preparing a carbapenem-based compound, comprising the step of deprotecting the compound represented by the following formula (II) to produce a compound represented by the following formula (I):
[Formula III]
[Formula II]
Formula I
Wherein PNB is a paranitrobenzyl group,
R 2 is
(2S, 4S) -4-mercapto-2-[[[3- (2-propenyloxy) carbonyl] phenyl] amino] carbonyl-1-pyrrolidinecarboxylic acid 2-propenyl ester, ( 2S, 4S) -4-mercapto-2-[[[3- (p-nitrobenzyloxy) carbonyl] phenyl] amino] carbonyl-1-pyrrolidinecarboxylic acid p-nitrobenzyl ester, 6- Thio-6,7-dihydro-5H-pyrazolo [1,2-a] [1,2,4] triazoletrifluoroacetic acid salt, 2-sulfamoylaminomethyl-1-p-nitrobenzyloxycarbo Nyl-4-mercaptopyrrolidine, or 4-nitrobenzyl (2S, 4S) -2-[(dimethylamino) carbonyl] -4-mercaptopyrrolidine-1-carboxylate,
R 3 is
(2S, 4S) -4-mercapto-2-[[(3-carboxyphenyl) amino] carbonyl] -3-pyrrolidine, 6-thio-6,7-dihydro-5H-pyrazolo [1 , 2-a] [1,2,4] triazole, 4-thio-2-sulfamoylaminomethylpyrrolidine, or 4-thio- (2S), (4S) -2-[(dimethylamino) carbo Nil] pyrrolidine.
상기 화학식 II로 나타내는 화합물은 하기 화학식 IIa, IIa-1, IIb, IIc, 또는 IId의 화합물이고,
상기 화학식 I로 나타내는 화합물은 하기 화학식 Ia, Ib, Ic, 또는 Id의 화합물인 방법:
[화학식 IIa]
[화학식 IIa-1]
[화학식 IIb]
[화학식 IIc]

IIc
[화학식 IId]

[화학식Ia]
[화학식Ib]

[화학식Ic]

[화학식Id]

The compound represented by Formula II is a compound of Formula IIa, IIa-1, IIb, IIc, or IId,
Wherein the compound represented by Formula I is a compound of Formula Ia, Ib, Ic, or Id:
[Formula IIa]
[Formula IIa-1]
[Formula IIb]
Formula IIc]
IIc
[Formula IId]
Formula Ia
Formula Ib
Formula Ic
[Formula Id]
단계 i)의 반응용매로 디메틸포름아미드, 디메틸아세트아미드 및 아세토니트릴로 이루어진 그룹 중에서 선택된 용매 또는 이들의 혼합용매를 사용하는 방법.The method according to claim 1 or 2,
Method of using a solvent selected from the group consisting of dimethylformamide, dimethylacetamide and acetonitrile or a mixed solvent thereof as the reaction solvent of step i).
단계 i)의 분리단계는, 반응액을 냉각수에 가하여 pH 4~7사이에서 무정형의 화학식II의 화합물을 분리하는 것을 특징으로 하는 방법.The method according to claim 1 or 2,
The separation step of step i) is characterized in that the reaction solution is added to cooling water to separate the amorphous compound of formula II between pH 4-7.
상기 분리된 무정형의 화학식 II의 화합물을 더 이상 정제 및 건조하지 않고 탈보호화 반응을 수행하는 방법.The method of claim 4, wherein
A process for carrying out the deprotection reaction without further purification and drying of the isolated amorphous compound of formula II.
단계 ii)의 탈보호화 반응은 금속촉매로서 팔라듐하이드록사이드-카르본 존재하에 수행되는 것을 특징으로 하는 방법.The method according to claim 1 or 2,
The deprotection reaction of step ii) is carried out in the presence of palladium hydroxide-carbon as a metal catalyst.
단계 ii)의 반응용매로 정제수, 테트라히드로푸란, 메탄올, 에탄올, 프로판올, 이소프로판올, 부탄올 및 에틸아세테이트로 이루어진 그룹 중에서 선택된 단독용매 또는 이들의 혼합용매를 사용하는 방법.
The method according to claim 1 or 2,
Method of using a single solvent or a mixed solvent thereof selected from the group consisting of purified water, tetrahydrofuran, methanol, ethanol, propanol, isopropanol, butanol and ethyl acetate as the reaction solvent of step ii).
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020100068045A KR20120007331A (en) | 2010-07-14 | 2010-07-14 | An improved process for the preparation of carbapenem compounds |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020100068045A KR20120007331A (en) | 2010-07-14 | 2010-07-14 | An improved process for the preparation of carbapenem compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20120007331A true KR20120007331A (en) | 2012-01-20 |
Family
ID=45612705
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020100068045A KR20120007331A (en) | 2010-07-14 | 2010-07-14 | An improved process for the preparation of carbapenem compounds |
Country Status (1)
| Country | Link |
|---|---|
| KR (1) | KR20120007331A (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014057079A1 (en) * | 2012-10-12 | 2014-04-17 | Sandoz Ag | Preparation of ertapenem intermediates |
| CN110698480A (en) * | 2018-07-09 | 2020-01-17 | 武汉启瑞药业有限公司 | Synthesis and purification method of ertapenem intermediate |
-
2010
- 2010-07-14 KR KR1020100068045A patent/KR20120007331A/en not_active Application Discontinuation
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014057079A1 (en) * | 2012-10-12 | 2014-04-17 | Sandoz Ag | Preparation of ertapenem intermediates |
| JP2015533142A (en) * | 2012-10-12 | 2015-11-19 | サンド・アクチエンゲゼルシヤフト | Production of ertapenem intermediate |
| US9546171B2 (en) | 2012-10-12 | 2017-01-17 | Sandoz Ag | Preparation of ertapenem intermediates |
| CN110698480A (en) * | 2018-07-09 | 2020-01-17 | 武汉启瑞药业有限公司 | Synthesis and purification method of ertapenem intermediate |
| CN110698480B (en) * | 2018-07-09 | 2023-09-08 | 武汉启瑞药业有限公司 | Synthesis and purification method of ertapenem intermediate |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20090264643A1 (en) | Process for The Preparation of Beta-Lactam Antibiotic | |
| EP2388261B1 (en) | Improved process for the preparation of carbapenem using carbapenem intermediates and recovery of carbapenem | |
| US8097719B2 (en) | Meropenem intermediate in novel crystalline form and a method of manufacture of meropenem | |
| EP0182213A1 (en) | Carbapenem compounds and production thereof | |
| JP2008501657A (en) | Crystalline meropenem intermediate | |
| WO2011141847A1 (en) | An improved process for the preparation of meropenem | |
| WO2012038979A2 (en) | A process for preparation of ertapenem | |
| JP4100908B2 (en) | Production method of basic antibiotics and inorganic acid salts and oxalate intermediates | |
| KR20120007331A (en) | An improved process for the preparation of carbapenem compounds | |
| US9169258B2 (en) | Doripenem intermediate compound, preparation process therefor and use thereof, and preparation process for doripenem | |
| JP2010540433A (en) | Synthetic intermediate acid addition salts of carbapenem antibiotics and process for producing the same | |
| KR100781821B1 (en) | Process for preparing carbapenem compound | |
| US20110288289A1 (en) | Preparation of Carbapenem Intermediate and Their Use | |
| KR101573049B1 (en) | Crystalline form doripenem monohydrate and preparation method thereof | |
| JP2015533142A (en) | Production of ertapenem intermediate | |
| KR101050976B1 (en) | Acid addition salts of synthetic intermediates of carbapenem antibiotics and preparation methods thereof | |
| CN113929684B (en) | Meropenem intermediate and preparation method thereof | |
| KR20110062515A (en) | Process for preparing meropenem trihydrate | |
| KR102144777B1 (en) | A process for the preparation of crystalline Meropenem trihydrate | |
| CN106083859B (en) | Preparation method of imipenem monohydrate crystal | |
| KR100717131B1 (en) | 1beta;-METHYLCARBAPENEM DERIVATIVES HAVING PYRROLIDINE THIOL DERIVATIVES AND THE INTERMEDIATE FOR PREPARING THE SAME | |
| KR100473398B1 (en) | 1b-methylcarbapenem derivative having pyrrolidine derivative with oxime moiety and method for preparation of the same | |
| KR100473365B1 (en) | 1b-methylcarbapenem derivative having pyrrolidine derivative with oxadiazole moiety and method for preparation of the same | |
| KR100535253B1 (en) | 1b-methylcarbapenem derivatives having pyrrolidine with aminoethylcarbamoyl derivatives moiety and method for preparation of the same | |
| CN102617433B (en) | Method for preparing (2S, 4R)-1-paranitro carbobenzoxy-4-methylsulfonyl pyrrolidine-2-methanol |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WITN | Withdrawal due to no request for examination |