定義
特定の官能基及び化学用語の定義を、より詳細に以下に記載する。本開示の目的のために、化学元素を、元素周期表CAS方式、Handbook of Chemistry and Physics,75th Ed.の内部カバーに従って識別し、特定の官能基は、そこに記載されているように一般的に定義するものとする。さらに、有機化学の一般的な原理、及び特定の機能的部分及び反応性は、Organic Chemistry,Thomas Sorrell,University Science Books,Sausalito:1999に記載されており、その全内容は参照により本明細書に援用する。
本明細書及び特許請求の範囲で使用する単数形「a」、「an」、及び「the」は、文脈によってそうでないことが明確に示されない限り、複数の参照を含むものとする。したがって、例えば、「化合物」への言及には、複数のそのような化合物が包まれる。
約:用語「約」とは、値に関して本明細書中で使用する場合、言及する値に関する類似の値を指す。一般的に、内容に精通している当業者であれば、その内容における「約」に包含される関連する分散の程度を理解するであろう。例えば、いくつかの実施形態では、用語「約」は、言及する値の25%、20%、19%、18%、17%、16%、15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4%、3%、2%、1%以内、またはそれ未満の値の範囲を包含し得る。
投与:本明細書中で使用する場合、用語「投与」とは、一般的には、被験体または系への組成物の投与を指す。当業者であれば、適切な状況において、例えばヒトなどの被験体への投与に利用できる様々な経路を知っているであろう。例えば、いくつかの実施形態では、投与は、眼、経口、非経口、局所などであってもよい。いくつかの特定の実施形態では、投与は、気管支(例えば、気管支点滴による)、頬、皮膚(例えば、真皮、皮内、皮間、経皮などの1つ以上の局所であるか、またはそれらを含む場合があり)、経腸、動脈内、皮内、胃内、髄内、筋肉内、鼻腔内、腹腔内、くも膜下腔内、静脈内、脳室内、特定の器官内(例えば、肝臓内)、粘膜、経鼻、経口、直腸、皮下、舌下、局所、気管(例えば、気管内投与による)、経膣、硝子体などであってもよい。いくつかの実施形態では、投与は、間欠的(例えば、時間的に分離された複数回の投与)及び/または定期的(例えば、共通の期間によって分離された個々の投与)投与である投与を含み得る。いくつかの実施形態では、投与は、少なくとも選択された期間の連続投与(例えば、灌流)を含み得る。
アルツハイマー病:本明細書中で使用する場合、用語「アルツハイマー病」または「AD」とは、ヒト中枢神経系の進行性疾患を指す。脳のインスリンシグナル伝達は学習と記憶に重要であり、脳のインスリン抵抗性はADの主要な危険因子である。インスリンシグナル伝達の回復は、ADの潜在的な治療法として注目されている(White MF,Science 2003;302:1710-1;De Felice DG et al,Alzheimer’s & Dementia 2014;10:S26-S32)。特定の実施形態では、それは、高齢者に典型的な認知症によって、見当識障害、記憶喪失、言語障害、計算障害、または視空間失認によって、及び精神医学的徴候によって顕在化する。特定の実施形態では、それは、脳のいくつかの領域の変性ニューロンに関連している。本明細書中で使用する用語「認知症」とは、精神医学的症状を伴うまたは伴わないアルツハイマー型認知症が含まれるが、これらに限定されない。特定の実施形態では、本明細書中で提供する治療方法は、被験体の軽度、中程度及び重度のアルツハイマー病の治療に有効である。アルツハイマー病の段階には、さらに、「中程度または中期アルツハイマー病」とも呼ばれる「中程度に重度の認知機能低下」、「中程度に重度または中期アルツハイマー病」とも呼ばれる「重度の認知機能低下」、及び「重度または末期アルツハイマー病」とも呼ばれる「極めて重度の認知機能低下」が含まれる。中程度に重度の認知機能低下は、記憶の大きな欠落及び認知機能不全の表出が特徴である。この段階では、日々の活動に対する何らかの支援が不可欠となる。重度の認知機能低下では、記憶障害が悪化し続け、重大な性格の変化が現れる場合があり、影響を受けた個体は、日常的な日常活動で広範な支援を必要とする。末期アルツハイマー病または非常に重度の認知機能低下は、個体が環境に反応する能力、会話能力、そして最終的には運動を制御する能力を失う、この疾患の最終段階である。
生物学的活性:本明細書中で使用する場合、用語「生物学的活性」とは、目的の薬剤または実体によって達成される、観察可能な生物学的効果または結果を指す。例えば、いくつかの実施形態では、特異的結合相互作用は生物活性である。いくつかの実施形態では、生物学的経路または事象の調節(例えば、誘導、増強、または阻害)は生物学的活性である。いくつかの実施形態では、生物学的活性の存在または程度を、目的の生物学的経路または事象によって産生される直接的または間接的な産物の検出を通じて評価する。
併用療法:本明細書中で使用する場合、用語「併用療法」とは、被験体を、2つ以上の治療レジメン(例えば、2つ以上の治療薬)に同時に曝露させる状況を指す。いくつかの実施形態では、2つ以上のレジメンを同時に投与してもよく;いくつかの実施形態では、そのようなレジメンを連続して投与してもよい(例えば、第1のレジメンのすべての「用量」を、任意の用量の第2のレジメンの投与の前に投与する)。いくつかの実施形態では、そのような薬剤を重複した投薬レジメンで投与する。いくつかの実施形態では、併用療法の「投与」は、他の薬剤または治療法を受ける被験体に対する1つ以上の薬剤または治療法を組み合わせた投与を含み得る。明確にするために、併用療法では、個々の薬剤を単一の組成物で(または必然的に同時に)投与する必要はないが、いくつかの実施形態では、2つ以上の薬剤またはその活性部分を、組み合わせ組成物、または組み合わせ化合物(例えば、単一の化学複合体または共有結合体の一部として)で、一緒に投与してもよい。
比較可能:本明細書中で使用する場合、用語「比較可能」とは、互いに同一ではないが、それらの間の比較を可能にするのに十分類似している2つ以上の薬剤、実体、状況、条件セットなどを指し、それにより、当業者は、観察された相違点または類似点に基づいて結論を合理的に引き出し得ることを理解するであろう。いくつかの実施形態では、条件、状況、個体、または集団の比較可能なセットは、複数の実質的に同一の特徴及び1つまたは少数の多様な特徴によって特徴付けられる。当業者であれば、文脈において、比較可能とみなされる2つ以上のそのような薬剤、実体、状況、条件のセットなどについて、所与の状況においてどの程度の同一性が必要であるかを理解するであろう。例えば、当業者であれば、異なる状況、個体、または集団のもとで、またはそれを用いて得られる結果または観察される現象における相違が、様々なそれらの特徴の変化によって引き起こされるか、またはそれらの特徴の変化を示すという合理的な結論を保証するのに十分な数及び種類の実質的に同一の特徴によって特徴付けられる場合、状況、個体、または集団のセットは、互いに比較可能であることを理解するであろう。
糖尿病:糖尿病の中心的な特徴は、β細胞機能の障害である。I型及びII型糖尿病の両方で疾患の進行の初期に発生する1つの異常は、摂食誘発性の急速なインスリン反応の喪失である。その結果、肝臓はグルコースを生成し続け、それが食事の基本的な成分から摂取され吸収されるグルコースに追加される。
II型糖尿病:II型糖尿病の1つの特徴は、インスリン抵抗性と呼ばれるインスリン作用の障害である。インスリン抵抗性は、最大グルコース除去率(GERmax)の低下及びGERmaxを達成するために必要なインスリン濃度の増加の両方として現れる。したがって、所定のグルコース負荷を処理するには、より多くのインスリンが必要であり、そのインスリン濃度の増加をより長期間維持する必要がある。その結果、糖尿病患者は長時間にわたってグルコース濃度の上昇にさらされ、インスリン抵抗性がさらに悪化する。さらに、長期にわたる血糖値の上昇は、それ自体がβ細胞に対して有毒である。II型糖尿病の別の特徴は、血糖値の上昇に対する反応の遅延である。正常な個体は、通常、食物摂取後2~3分以内にインスリンを放出し始めるが、II型糖尿病の場合、血糖が上昇し始めるまで内在性インスリンを分泌することができず、その後、長いプラトーへの緩やかな濃度上昇である第二相動態を伴う。結果として、内在性グルコース産生は遮断されず、摂取後も継続し、患者は高血糖症(血糖値の上昇)を経験する。II型糖尿病は、様々な、及びあまりよく理解されていない状況から発生する。早期インスリン放出の早期の喪失、及び結果としての継続的なグルコース放出は、グルコース濃度の上昇に寄与する。高グルコースレベルはインスリン抵抗性を促進し、インスリン抵抗性は血清グルコース濃度の長期にわたる上昇を引き起こす。この状況は、自己増幅サイクルにつながる可能性があり、これにより、インスリン濃度が高くなると血糖値を制御する効果が低下する。さらに、上記のように、グルコースレベルの上昇はβ細胞に対して毒性を有し、機能的なβ細胞の数を低下させる。膵島に栄養を与える微小血管の成長または維持を損なう遺伝的欠損も、その劣化に役割を果たし得る(Glee,S.M.,et al. Nature Genetics 38:688-693,2006)。最終的に、膵臓は不全となり、個体は、I型糖尿病の人々と同様のインスリン欠乏症の発症へと進行する。
I型糖尿病:I型糖尿病は、身体の固有の免疫系による膵臓のインスリン産生細胞(β細胞)の破壊の結果として生じる。これは最終的に完全なインスリンホルモン欠乏症をもたらす。
剤形または単位剤形:本明細書中で使用する場合、用語「剤形または単位剤形」とは、被験体への投与のための活性薬剤(例えば、治療剤または診断剤)の物理的に別個の単位を指す。いくつかの実施形態では、そのような各単位は、所定量の活性薬剤を含む。いくつかの実施形態では、そのような量は、関連する集団に投与する場合に所望のまたは有益な結果と相関すると判断された投薬レジメンに従った(すなわち、治療的投薬レジメンを用いた)投与に適した単位投与量(またはその全体の割合)である。当業者であれば、特定の被験体に投与する治療用組成物または薬剤の総量は、1人以上の担当医によって決定され、また、複数の剤形の投与を含み得ることを理解している。
投薬レジメン:本明細書中で使用する場合、用語「投薬レジメン」とは、一般的には期間によって分けられ、被験体に個別に投与する単位用量のセット(一般的には複数)を指す。いくつかの実施形態では、所定の治療薬は、推奨される投薬レジメンを有し、それは1つ以上の用量を含み得る。いくつかの実施形態では、投薬レジメンは、それぞれが他の用量から時間的に分離された複数の投与を含む。いくつかの実施形態では、個々の投与は、同じ長さの期間だけ互いに分離され;いくつかの実施形態では、投薬レジメンは、複数の投与と、個々の投与を分離する少なくとも2つの異なる期間を含む。いくつかの実施形態では、投薬レジメン内のすべての投与は同じ単位投与量である。いくつかの実施形態では、投薬レジメン内の異なる投与は異なる量である。いくつかの実施形態では、投薬レジメンは、第1の用量での第1の投与と、それに続く第1の用量とは異なる第2の用量での1回以上の追加投与とを含む。いくつかの実施形態では、投薬レジメンは、第1の用量での第1の投与と、それに続く第1の用量と同じ第2の用量での1回以上の追加投与を含む。いくつかの実施形態では、投薬レジメンは、関連する集団に投与する場合、所望の、または有益な結果と関連付けられる(すなわち、治療的投薬レジメンである)。
賦形剤:本明細書中で使用する場合、用語「賦形剤」とは、例えば、所望の粘稠性または安定化効果を提供するか、またはそれらに寄与するために、医薬組成物に含まれ得る非治療薬を指す。いくつかの実施形態では、適切な薬学的賦形剤として、例えば、デンプン、グルコース、ラクトース、スクロース、ゼラチン、麦芽、コメ、小麦粉、チョーク、シリカゲル、ステアリン酸ナトリウム、モノステアリン酸グリセロール、タルク、塩化ナトリウム、乾燥スキムミルク、グリセロール、プロピレン、グリコール、水、エタノールなどが挙げられ得る。
ハネムーン期:本明細書中で使用する場合、1型糖尿病の用語「ハネムーン期」とは、早期インスリン放出の喪失を特徴とする疾患の初期段階を指し、残されたβ細胞機能がいくらかのインスリンを産生し、それは第二相動態により放出される。
高血糖症:本明細書中で使用する場合、用語「高血糖症」とは、通常より高い空腹時血糖濃度を特徴とする疾患、障害、または病態を指す。いくつかの実施形態では、高血糖症は、126mg/dL以上の血糖濃度を特徴とする。いくつかの実施形態では、高血糖症は、280mg/dL(15.6mM)以上の血糖濃度を特徴とする。
低血糖症:本明細書中で使用する場合、用語「低血糖症」とは、通常よりも低い血糖濃度を特徴とする疾患、障害、または病態を指す。いくつかの実施形態では、低血糖症は、63mg/dL(3.5mM)以下の血糖濃度を特徴とする。いくつかの実施形態では、低血糖症は、低血糖症の症状として認識される、認知障害、行動変化、蒼白、発汗低血圧症、紅潮及び衰弱などの症状を引き起こし、適切なカロリー摂取により解消する。いくつかの実施形態では、低血糖症は重度であり、それにより、グルカゴン注射、グルコース注入、または別の関係者による支援が必要である。
改善、増加、阻害または減少:本明細書中で使用する場合、用語「改善」、「増加」、「阻害」、及び「減少」、またはそれらの文法的均等物は、ベースラインまたは他の基準測定値に対する値を示す。いくつかの実施形態では、適切な基準測定値は、特定の薬剤または処置が存在せず(例えば、前及び/または後に)、その他の点では比較可能な条件下での、または適切な比較可能な基準薬剤の存在下での、特定の系における(例えば、単一の個体における)測定値であり得るか、または測定値を含み得る。いくつかの実施形態では、適切な基準測定値は、関連する薬剤または処置の存在下で、特定の方法で応答することが知られているか、または予想される、比較可能な系における測定値であり得るか、または測定値を含み得る。
インスリン関連障害:本明細書中で使用する場合、用語「インスリン関連障害」とは、哺乳類におけるインスリンの産生、調節、代謝、及び作用に関する障害を指す。インスリン関連障害として、糖尿病前症、I型糖尿病、II型糖尿病、低血糖症、高血糖症、インスリン抵抗性、分泌機能障害、筋肉減少症、膵臓β細胞機能の喪失、及び膵臓β細胞の喪失が挙げられるが、これらに限定されない。
インスリン関連障害を有する非インスリン依存性患者:本明細書中で使用する場合、用語「インスリン関連障害を有する非インスリン依存性患者」とは、外因的に提供するインスリンによる治療が、診断時の現在の標準的な治療ではない障害を有する患者を指す。外因的に投与するインスリンで治療しないインスリン関連障害を有するインスリン非依存患者には、早期II型糖尿病、ハネムーン期I型糖尿病、糖尿病前症及びインスリン産生細胞移植レシピエントが含まれる。
インスリン抵抗性:本明細書中で使用する場合、用語「インスリン抵抗性」とは、患者の細胞がインスリンに適切にまたは効率的に応答できないことを指す。膵臓は、より多くのインスリンを産生することにより、細胞レベルでこの問題に応答する。最終的に、インスリンに対する身体のニーズに膵臓が追いつくことができず、血流中に過剰なグルコースが蓄積する。インスリン抵抗性の患者は、多くの場合、高レベルの血糖値と血中を循環する高レベルのインスリンを同時に有する。
インスリン抵抗性障害:本明細書中で使用する場合、用語「インスリン抵抗性障害」とは、インスリン抵抗性によって引き起こされるか、またはインスリン抵抗性が寄与する任意の疾患または病態を指す。例として:糖尿病、肥満、メタボリックシンドローム、インスリン抵抗性症候群、シンドロームX、インスリン抵抗性、血圧上昇、高血圧、高血中コレステロール、脂質異常症、高脂血症、脂質異常症、脳卒中、冠動脈疾患または心筋梗塞を含む動脈硬化性疾患、高血糖症、高インスリン血症及び/または高プロインスリン血症、耐糖能障害、インスリン放出遅延、冠動脈心疾患を含む糖尿病合併症、狭心症、うっ血性心不全、脳卒中、認知症の認知機能、網膜症、神経障害、腎症、糸球体腎炎、糸球体硬化症、ネフローゼ症候群、高血圧性腎硬化症、いくつかの種類のがん(例えば、子宮内膜、乳房、前立腺、及び結腸)、妊娠の合併症、女性の生殖機能の低下(例えば、月経不順、不妊、排卵不順、多嚢胞性卵巣症候群(PCOS))、脂肪異栄養症、コレステロール関連障害、例えば、胆石、胆嚢炎及び胆石症、痛風、閉塞性睡眠時無呼吸及び呼吸器系の問題、骨関節炎、ならびに例えば骨粗鬆症などの骨量減少の予防及び治療が挙げられる。
異性体:当技術分野で知られているように、多くの化学物質(特に多くの有機分子及び/または多くの小分子)は、様々な構造(例えば、幾何、立体配座、同位体)及び/または光学異性体で存在することができる。例えば、任意のキラル中心がR及びS配置で存在することができ、二重結合はZ及びE立体配座異性体で存在することができ、特定の構造要素は2つ以上の互変異性体を採用することができ、特定の構造は1つ以上の同位体濃縮原子で置換することができる(例えば、水素の場合は重水素またはトリチウム、13Cの場合は12Cまたは14C、129Iの場合は131Iなど)。いくつかの実施形態では、文脈から当業者には明らかであるように、本明細書中の特定の化合物構造の描写または参照は、そのすべての構造異性体及び/または光学異性体を代表し得る。いくつかの実施形態では、文脈から当業者には明らかであるように、本明細書中の特定の化合物構造の描写または参照は、描写または参照する異性体形態のみを包含することを意図している。いくつかの実施形態では、様々な異性体形態で存在することができる化学物質を含む組成物は、複数のそのような形態を含み;いくつかの実施形態では、そのような組成物は単一の形態のみを含む。例えば、いくつかの実施形態では、様々な光学異性体(例えば、立体異性体、ジアステレオマーなど)として存在することができる化学物質を含む組成物には、そのような光学異性体のラセミ集団が含まれ;いくつかの実施形態では、そのような組成物は、単一の光学異性体のみを含み、及び/または一緒に光学活性を保持する複数の光学異性体を含む。
非経口:本明細書中で使用する場合、用語「非経口投与」及び「非経口で投与する」とは、通常は注射による、経腸及び局所投与以外の投与様式を指す。いくつかの実施形態では、非経口投与は、静脈内、筋肉内、動脈内、くも膜下腔内、嚢内、眼窩内、心臓内、皮内、腹腔内、経気管、皮下、表皮下、関節内、嚢下、くも膜下、脊髄内及び胸骨内注射及び/または注入であり得るか、またはそれらを含み得る。
部分的に不飽和:本明細書中で使用する場合、用語「部分的に不飽和」とは、少なくとも1つの二重結合または三重結合を含む環部分を指す。用語「部分的に不飽和」は、複数の不飽和部位を有する環を包含することを意図しているが、本明細書中で定義するように、アリールまたはヘテロアリール部分を含むことを意図していない。
医薬組成物:本明細書中で使用する場合、用語「医薬組成物」とは、活性薬剤を1つ以上の薬学的に許容される担体とともに製剤化した組成物を指す。いくつかの実施形態では、活性薬剤は、関連のある集団に投与する場合に所定の治療効果を達成する統計的に有意な確率を示す治療レジメンでの投与に適した単位用量で存在する。いくつかの実施形態では、医薬組成物を、以下に適合するものを含む固体または液体形態での投与のために特別に製剤化してもよい:経口投与、例えば、水薬(水性または非水性の溶液または懸濁液)、錠剤、例えば、頬、舌下、及び全身吸収を標的とするもの、ボーラス、散剤、顆粒剤、舌に塗布するためのペースト;非経口投与、例えば、皮下、筋肉内、静脈内または硬膜外注射による、例えば、滅菌溶液もしくは懸濁液、または徐放性製剤として;局所塗布、例えば、皮膚、肺、または口腔に塗布する、クリーム、軟膏、または徐放性パッチもしくはスプレーとして;膣内または直腸内、例えば、ペッサリー、クリーム、または発泡体として;舌下;眼内;経皮;または経鼻、肺内、及び他の粘膜表面へ。当業者であれば、一般的に、いくつかの実施形態では、ヒトまたは動物被験体への投与のために製剤化する任意の組成物は、その投与が処方箋を必要とするかどうかにかかわらず、医薬組成物と見なされ得ることを理解するであろう。したがって、例えば、いくつかの実施形態では、食物または食物サプリメント組成物(例えば、シェイクまたはスポーツドリンクまたは栄養サプリメント粉末などの液体または固体の消費可能組成物)は、医薬組成物とみなされ得る。代替的または追加的に、いくつかの実施形態では、医薬組成物は、例えば米国の食品医薬品局などの適切な政府機関による関連する被験体への投与のために特に調節及び承認される製剤であってもよい。いくつかの実施形態では、医薬組成物は、免許を有する医師の処方箋なしでは法的に投与できない組成物である。
薬学的に許容される:本明細書中で使用する場合、本明細書において開示する組成物を製剤化するために使用する担体、希釈剤、または賦形剤に対して適用する用語「薬学的に許容される」とは、その担体、希釈剤、または賦形剤が組成物の他の成分と適合しなければならず、また、そのレシピエントに有害であってはならないことを意味する。
薬学的に許容される担体:本明細書中で使用する用語「薬学的に許容される担体」とは、薬学的に許容される物質、組成物またはビヒクル、例えば、対象化合物を、ある器官、または身体の一部から、別の器官または身体の一部へ運搬または輸送することに関与する、物質をカプセル化する液体または固体の充填剤、希釈剤、賦形剤、または溶媒を指す。各担体は、製剤の他の成分と適合性があり、患者に有害でないという意味で「許容可能」でなければならない。薬学的に許容される担体として機能することができる物質のいくつかの例として:糖質、例えば、ラクトース、グルコース及びスクロース:デンプン類、例えば、コーンスターチ及びジャガイモデンプン;セルロース、及びその誘導体、例えば、カルボキシメチルセルロースナトリウム、エチルセルロース及び酢酸セルロース;粉末トラガカント;麦芽;ゼラチン;タルク;賦形剤、例えば、カカオバター及び坐薬ワックス;油類、例えば、落花生油、綿実油、ベニバナ油、ゴマ油、オリーブ油、コーン油及びダイズ油;グリコール類、例えば、プロピレングリコール;ポリオール類、例えば、グリセリン、ソルビトール、マンニトール及びポリエチレングリコール;エステル類、例えば、オレイン酸エチル及びラウリン酸エチル;寒天;緩衝剤、例えば、水酸化マグネシウム及び水酸化アルミニウム;アルギン酸;パイロジェンフリー水;等張食塩水;リンガー液;エチルアルコール;pH緩衝液;ポリエステル類、ポリカーボネート類及び/またはポリ無水物;ならびに医薬製剤に使用される他の非毒性適合物質が挙げられる。
糖尿病前症:本明細書中で使用する場合、用語「糖尿病前症」とは、患者が空腹時高血糖及び/または耐糖能障害を有する疾患、障害、または病態を指す。いくつかの実施形態では、糖尿病前症患者の空腹時血糖値は、100mg/dL(5.5mmol/L)~126mg/dL(7.0mmol/L)である。いくつかの実施形態では、糖尿病前症患者は、食後2時間の血糖値が、140mg/dL(7.8mmol/L)~200mg/dL(11.1mmol/L)である。
プロドラッグ:本明細書中で使用する場合、用語「プロドラッグ」とは、代謝変換により薬理学的に活性な薬剤に変換される、薬理学的に活性な、またはより一般的には不活性な化合物を指す。本明細書に記載の式のいずれかの化合物のプロドラッグは、式のいずれかの化合物に存在する官能基を、in vivoで切断して親化合物を放出し得るように修飾することにより調製される。in vivoでは、プロドラッグは生理学的条件下で化学変化を容易に受け(例えば、天然酵素(複数可)により加水分解されるか、または作用を受け)、薬理学的に活性な薬剤の遊離をもたらす。プロドラッグとして、本明細書に記載の式の化合物が挙げられ、その化合物においては、ヒドロキシル基、アミノ基、またはカルボキシル基が、in vivoで切断されて遊離ヒドロキシル基、アミノ基、またはカルボキシル基をそれぞれ再生し得る基に結合している。プロドラッグの例として、本明細書に記載の式のいずれかの化合物のエステル類(例えば、酢酸塩、ギ酸塩、及び安息香酸塩誘導体)、または生理学的pHに至らせるか酵素作用により活性な親薬物に変換される他の誘導体が挙げられるが、これらに限定されない。適切なプロドラッグ誘導体の選択及び調製の従来の手順は、当技術分野で説明されている(例えば、Bundgaard. Design of Prodrugs. Elsevier,1985参照)。
増殖性病態:本明細書中で使用する場合、用語「増殖性病態」とは、細胞増殖に関連する疾患または障害を指す。いくつかの実施形態では、増殖性疾患または障害は、がんであるか、がんを含む。いくつかの実施形態では、増殖性疾患または障害は、炎症性疾患または障害である。いくつかの実施形態では、増殖性疾患または障害は、自己免疫疾患または障害である。いくつかの実施形態では、増殖性疾患または障害は、微生物感染症(例えば、細菌感染症)である。
参照:本明細書中で使用する場合、用語「参照」とは、比較を実行する基準または対照を指す。例えば、いくつかの実施形態では、目的の薬剤、動物、個体、集団、試料、配列または値を、参照または対照の薬剤、動物、個体、集団、試料、配列または値と比較する。いくつかの実施形態では、参照または対照を、目的の試験または測定と実質的に同時に試験及び/または測定する。いくつかの実施形態では、参照または対照は、場合により有形の媒体で具現化される履歴参照または対照である。一般的には、当業者であれば理解するように、参照または対照は、評価中のものと比較可能な条件または状況の下で測定または特徴付けられる。当業者であれば、特定の可能な参照または対照への依存及び/または比較を正当化するのに十分な類似性が存在する場合を認識するであろう。
リスク:本明細書中で使用する場合、疾患、障害、及び/または病態の「リスク」とは、特定の個体が、疾患、障害、及び/または病態を発症する可能性を指す。いくつかの実施形態では、リスクをパーセンテージとして表す。いくつかの実施形態では、リスクは、0、1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90~最大100%までである。いくつかの実施形態では、リスクは、参照試料または参照試料群に関連するリスクに相対するリスクとして表される。いくつかの実施形態では、参照試料または参照試料群は、疾患、障害、病態、及び/または事象の既知のリスクを有する。いくつかの実施形態では、参照試料または参照試料群は、特定の個体と比較可能な個体に由来する試料である。いくつかの実施形態では、相対リスクは、0、1、2、3、4、5、6、7、8、9、10、またはそれ以上である。
固体形態:当技術分野で公知のように、多くの化学物質(特に多くの有機分子及び/または多くの小分子)は、例えば、アモルファス形態及び/または結晶形態などの様々な異なる固体形態をとることができる(例えば、多形体、水和物、溶媒和物など)。いくつかの実施形態では、そのような物質を、そのような単一の形態として(例えば、単一の多形体の純粋な調製物として)利用してもよい。いくつかの実施形態では、そのような物質を、そのような形態の混合物として利用してもよい。
被験体:本明細書中で使用する場合、用語「被験体」とは、生物、一般的には哺乳類(例えば、ヒト、いくつかの実施形態では出生前のヒトの形態を含む)を指す。いくつかの実施形態では、被験体は、関連する疾患、障害または病態に罹患している。いくつかの実施形態では、被験体は、疾患、障害、または病態に高感受性である。いくつかの実施形態では、被験体は、疾患、障害または病態の1つ以上の症状または特徴を示す。いくつかの実施形態では、被験体は、疾患、障害、または病態の症状または特徴をまったく示さない。いくつかの実施形態では、被験体は、疾患、障害、または病態に対する感受性またはリスクに特徴的な1つ以上の特徴を有する被験体である。いくつかの実施形態では、被験体は患者である。いくつかの実施形態では、被験体は、診断及び/または療法を施す及び/または施している個体である。
高感受性:疾患、障害、または病態(例えば、インフルエンザ)に「高感受性」である個体は、疾患、障害、または病態を発症するリスクを有する。いくつかの実施形態では、疾患、障害、または病態に高感受性の個体は、疾患、障害、または病態の症状をまったく示さない。いくつかの実施形態では、疾患、障害、または病態に高感受性の個体は、疾患、障害、及び/または病態を有すると診断されていない。いくつかの実施形態では、疾患、障害、または病態に高感受性の個体は、疾患、障害、または病態の発症に関連する条件にさらされている個体である。いくつかの実施形態では、疾患、障害、及び/または病態を発症するリスクは、集団ベースのリスクである(例えば、疾患、障害、または病態に罹患している個体の家族)。
治療有効量:本明細書中で使用する場合、用語「治療有効量」とは、それを投与する対象に所望の効果を生じさせる量を指す。いくつかの実施形態では、この用語は、治療的投薬レジメンに従って、疾患、障害、及び/または病態に罹患しているかまたは罹患しやすい集団に投与する場合に、疾患、障害、及び/または病態を治療するのに十分な量を指す。いくつかの実施形態では、治療有効量は、疾患、障害、及び/または病態の1つ以上の症状の発症率及び/または重症度を低下させ、及び/または発症を遅らせる量である。当業者であれば、用語「治療有効量」が、実際に特定の個体において治療の成功を達成することが必ずしも必要でないことを理解するであろう。むしろ、治療有効量は、そのような治療を必要とする患者に投与する場合に、有意な数の被験体において特定の望ましい薬理学的応答を提供する量であり得る。いくつかの実施形態では、治療有効量への言及は、1つ以上の特定の組織(例えば、疾患、障害または病態に影響を受ける組織)または体液(例えば、血液、唾液、血清、汗、涙、尿など)において測定されるような量への言及であり得る。当業者であれば、いくつかの実施形態において、治療有効量の特定の薬剤または療法を単回用量で製剤化及び/または投与することができることを理解するであろう。いくつかの実施形態では、治療的に有効な薬剤を、例えば投与レジメンの一部として、複数の用量で製剤化及び/または投与してもよい。
治療:本明細書中で使用する場合、用語「治療」(同様に「治療する」または「処置」)とは、特定の疾患、障害、及び/または病態の1つ以上の症状、特徴、及び/または原因を、部分的にまたは完全に、軽減、寛解、緩和、阻害、発症遅延させ(例えば、確立された開始時間または期間に関して)、重症度を低下させ、及び/または発生率を低下させる治療の投与を指す。いくつかの実施形態では、治療は、関連する疾患、障害、及び/または病態の徴候または症状を示さない被験体の、及び/または関連する疾患、障害、及び/または病態に罹患していると診断されていない被験体の、及び/または疾患、障害、及び/または病態の初期徴候のみを示す被験体の治療であってもよい。代替的または追加的に、いくつかの実施形態では、治療は、関連する疾患、障害及び/または病態の1つ以上の確立された徴候を示す被験体に対する治療であってもよい。いくつかの実施形態では、治療は、関連する疾患、障害、及び/または病態に罹患していると診断された被験体の治療であってもよい。いくつかの実施形態では、治療は、関連する疾患、障害、及び/または病態の発症リスクの増加と統計的に相関する1つ以上の感受性因子を有することが知られている被験体に対する治療であってもよい。いくつかの実施形態では、そのような治療は、疾患、障害、及び/または病態を発症するリスクを減らすこと、及び/または疾患、障害、または病態の1つ以上の特徴または症状の発症を遅らせることを指す。いくつかの実施形態では、治療は、関連する集団または系(例えば、モデル系)に適用する場合の統計的有意性とともに、適切な結果を達成すること(例えば、特定の疾患、障害、及び/または病態の1つ以上の症状、特徴、及び/または原因を、部分的にまたは完全に、軽減、寛解、緩和、阻害、発症遅延、重症度の低減、及び/または発生率の低減をすること)が実証されたレジメンによる治療の投与である。いくつかの実施形態では、1つ以上の症状の診断及び/または発症後に施す処置は「治療的」処置とみなされ、一方、診断及び/または症状の発症前に施す処置は「予防的」処置とみなされる。
単位用量:本明細書中で使用する場合、用語「単位用量」とは、医薬組成物の単回用量として及び/または物理的に別個の単位で投与する量を指す。多くの実施形態では、単位用量は所定量の活性薬剤を含む。いくつかの実施形態では、単位用量には、薬剤の単回用量の全体が含まれる。いくつかの実施形態では、全体の単回用量を達成するために、複数の単位用量を投与する。いくつかの実施形態では、意図する効果を達成するために、複数の単位用量の投与が必要であるか、または必要であることが予想される。単位用量は、例えば、所定量の1つ以上の治療薬を含む液体(例えば、許容される担体)の量、所定量の1つ以上の固体形態の治療薬、所定量の1つ以上の治療薬を含む徐放性の製剤または薬物送達装置などであってもよい。治療薬(複数可)に加えて様々な成分のいずれかを含む製剤中に単位用量が存在し得ることが理解されよう。以下に記載するように、例えば、許容される担体(例えば、薬学的に許容される担体)、希釈剤、安定剤、緩衝剤、防腐剤などを含ませてもよい。多くの実施形態では、特定の治療薬の適切な合計日用量に、一部または複数の単位用量を含ませてもよく、例えば担当医が健全な医学的判断の範囲内でそれを決定してもよいことは、当業者であれば理解するであろう。いくつかの実施形態では、任意の特定の被験体または生物に対する具体的な有効量レベルは、治療する障害及び障害の重症度;使用する特定の活性化合物の活性;使用する特定の組成物;被験体の年齢、体重、一般的な健康状態、性別及び食事;投与時間、及び使用する特定の活性化合物の排出速度;治療期間;使用する特定の化合物(複数可)と組み合わせてまたは同時に使用する薬物及び/または追加の療法、ならびに医学分野で周知の同様の要因を含む、様々な要因に依存し得る。
アルキル:本明細書中で使用する場合、用語「アルキル」とは、直鎖または分枝アルキル基を指す。例示的なC1-6アルキル基として、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、tert-ブチル、ペンチル、イソペンチル、ネオペンチル、及びヘキシルが挙げられるが、これらに限定されない。
ヘテロ原子:本明細書中で使用する場合、用語「ヘテロ原子」とは、酸素、硫黄、窒素、リン、またはケイ素(窒素、硫黄、リン、もしくはケイ素の任意の酸化形態;任意の塩基性窒素の四級化形態または;複素環の置換可能な窒素、例えばN(3,4-ジヒドロ-2H-ピロリルなど)、NH(ピロリジニルなど)もしくはNR+(N置換ピロリジニルなど)を含む)の1つ以上を含む。
炭素環式:本明細書中で使用する場合、用語「炭素環式」とは、完全に飽和しているか、または1つ以上の不飽和単位を含むが芳香族ではなく分子の残りの部分に単一の結合点を有する、単環式炭化水素を指す。
複素環式:本明細書中で使用する場合、用語「複素環式」とは、上記で定義したように、飽和または部分不飽和であり、炭素原子に加えて、1個以上、好ましくは1~4個のヘテロ原子を有する安定な単環式複素環部分を指す。複素環の環原子に関して使用する場合、用語「窒素」には置換窒素が含まれる。例として、酸素、硫黄、または窒素から選択される0~3個のヘテロ原子を含む飽和または部分不飽和環において、窒素は、N(3,4-ジヒドロ-2H-ピロリルなど)、NH(ピロリジニルなど)、または+NR(N置換ピロリジニルなど)であってもよい。複素環式環を、任意のヘテロ原子または炭素原子においてそのペンダント基に結合することができ、それにより安定な構造がもたらされ、場合により、環原子のいずれかを置換することができる。3~8員複素環の例として、テトラヒドロフラニル、テトラヒドロチオフェニル、ピロリジニル、ピペリジニル、ピロリニル、オキサゾリジニル、ピペラジニル、ジオキサニル、ジオキソラニル、ジアゼピニル、オキサゼピニル、チアゼピニル、及びモルホリニルが挙げられる。
ハロゲン:本明細書中で使用する場合、用語「ハロゲン」及び「ハロ」とは、F、Cl、Br、またはIを指す。
化合物C:本明細書中で使用する場合、用語「化合物C」とは、5’-メチルセレノアデノシンを指し;これは、(2R,4S,5S)-2-(6-アミノ-9H-プリン-9-イル)-5-((メチルセラニル)メチル)テトラヒドロフラン-3,4-ジオール、CAS登録番号5135-40-0としても知られており、その薬学的に許容される塩が含まれる。
化合物D:本明細書中で使用する場合、用語「化合物D」とは、5’-セレノアデノシルホモシステインを指し;これは、(2R)-2-アミノ-4-((((2S,3S,5R)-5-(6-アミノ-9H-プリン-9-イル)-3,4-ジヒドロキシテトラヒドロフラン-2-イル)メチル)セラニル)ブタン酸、CAS登録番号4053-91-2としても知られており、その薬学的に許容される塩が含まれる。
化合物E:本明細書中で使用する場合、用語「化合物E」とは、γ-グルタミル-メチルセレノ-システインまたはγ-L-グルタミル-Se-メチル-L-システインを指し;これは、N5-(1-カルボキシ-2-(メチルセラニル)エチル)-L-グルタミン、またはその薬学的に許容される塩としても知られる。
化合物CDE:本明細書中で使用する場合、用語「化合物CDE」とは、化合物C、化合物D、及び化合物Eの混合物、またはその薬学的に許容される塩を指す。
化合物
本開示は、単一化合物の処置後、糖尿病マウス、培養した肝細胞及び/または骨格筋細胞において、グルコースレベルを低下させ、耐糖能を向上させ、インスリン受容体機能及びその下流のシグナル伝達を回復または活性化し、グルコース取り込みのための細胞質小胞から細胞膜へのグルコース輸送体タンパク質(GLUT)移行のためのAS160リン酸化を増強し、グルコース取り込みを刺激し、腎機能障害及び/または肝臓損傷を伴わずに高インスリン血症を軽減することができる多くの例示的な化合物を提供する。例えば、実施例2は、化合物43、50、53、69、及び70のそれぞれがHepG2細胞のグルコース産生を低下させたことを示している。特に、化合物43は化合物CDEよりも高い効力を示した。HepG2及びラットH4IIE細胞の抗糖尿病薬メトホルミンと比較した場合、化合物43はより高い効力とより低い細胞毒性を示した。また、化合物#43は、糖尿病マウスの肝臓におけるG6pc(肝グルコース産生の重要な酵素遺伝子)の発現の阻害において化合物50よりも強力であり、マウス肝臓AML-12細胞において化合物CDE、化合物C、化合物D及び化合物50よりも強力であることも示した(実施例4)。さらに、本開示は、化合物43が、糖尿病マウスの高血糖症に対して化合物C、化合物50、化合物69及び化合物70よりも強力であること(実施例3);化合物43が耐糖能を有意に向上させ(実施例3)、インスリン抵抗性糖尿病Leprdb/dbマウスの肝臓及び骨格筋におけるインスリン受容体機能及びその下流シグナル伝達を増強/回復すること(実施例5及び7~8);ならびに、化合物43が、野生型マウスの応答に類似した糖尿病マウスのグルコースチャレンジに対する応答を誘発すること(実施例3)を示す。さらに、本開示は、肝臓及び骨格筋細胞の両方でのグルコース取り込みについて、化合物#43処置がAS160のリン酸化を促進し(細胞内小胞から細胞膜へのGLUTの移行を促進するために)(実施例8)、糖尿病マウスの肝臓及び培養マウス肝細胞においてGLUT4発現を刺激し(実施例6)、肝細胞及び骨格筋細胞においてインスリン作用を促進及び/または増強してグルコース取り込みを刺激し(実施例6~7)、インスリン抵抗性糖尿病マウスにおいて腎機能障害及び/または肝臓損傷を伴わずに高インスリン血症を軽減する(実施例9)ことができることを示す。
本開示はまた、その活性に寄与し得るセレン化合物の特徴も提供する。例えば、セレン化合物#43はグルコース産生の阻害において高い効力を有するが、その硫黄類似体化合物68は効力が低いこと(実施例2);ならびに化合物#43は、インスリン抵抗性糖尿病マウスの血糖値及びHbA1cレベルを低下させ、耐糖能を向上させる際に、化合物#68よりも強力であること(実施例3)が示された。また、2’及び3’位にジアセチルエステル(#43)、環状炭酸塩(化合物#50)、モルホリノカルボキシレート(#53)、ジプロパノイルエステル(#69)、またはジブタノイルエステル(#70)を含むセレン化合物の処置後にグルコース産生の阻害が観察され、化合物43の処置後に最大の効力が観察されたことが示された(実施例2)。さらに、5’メチルセレノ基及び5’セレノホモシステイン基を含む化合物は、HepG2細胞におけるグルコース産生の阻害において同様の効力を有することが示された(実施例2の化合物C対化合物D)。また、in vivo研究により、インスリン抵抗性糖尿病マウスの血糖値を低下させ、耐糖能を向上させる際に、化合物#43が化合物#69及び#70に比べてより強力であることを明らかにした(実施例3)。
いくつかの実施形態では、本開示は、式(1)の化合物:
またはその薬学的に許容される塩、プロドラッグ、もしくは異性体に関し、式中、
R
2及びR
3のそれぞれは、独立して、Hまたは-C(O)-Rであり、各Rは、独立して、C
1-6アルキルまたは3~8員の炭素環式または複素環式であり、R
2及びR
3は、両方ともHであってはならず;
あるいは、R
2とR
3は一緒になって-(CH
2)
n-C(O)-(CH
2)
m-を形成し、式中、n及びmのそれぞれは、独立して0~3であり、n+m≦3であり;
R
5は、-C
1-6アルキルまたは-C
1-6アルキル-CH(NH
2)COOHであり;
R
8は、Hまたはハロゲンであり;ならびに
Xは、Hまたはハロゲンであり;
式中、炭素環、複素環、-(CH
2)
n-、及び-(CH
2)
m-部分のそれぞれは、独立して、場合により、-OH、ハロゲン、NH
2、CN、またはC
1-6アルキルで1~3回置換されていてもよく;ならびに
各C
1-6アルキル部分は、独立して、場合により、-OH、ハロゲン、NH
2、またはCNで1~3回置換されていてもよい。
式(1)のいくつかの実施形態では、R8はHである。式(1)のいくつかの実施形態では、式(1)のR8はハロゲンである。式(1)のいくつかの実施形態では、式(1)のR8はFである。
式(1)のいくつかの実施形態では、XはHである。式(1)のいくつかの実施形態では、Xはハロゲンである。式(1)のいくつかの実施形態では、XはFである。
式(1)のいくつかの実施形態では、R5は-C1-6アルキルであり、これは、場合により、-OH、ハロゲン、NH2、またはCNにより1~3回置換されていてもよい。式(1)のいくつかの実施形態では、R5は-C1-6アルキルであり、場合により、ハロゲンにより1~3回置換されていてもよい。式(1)のいくつかの実施形態では、R5は非置換-C1-6アルキルである。式(1)のいくつかの実施形態では、R5は非置換の直鎖-C1-6アルキルである。式(1)のいくつかの実施形態では、R5はメチルである。式(1)のいくつかの実施形態では、R5はエチルである。式(1)のいくつかの実施形態では、R5はプロピルである。
式(1)のいくつかの実施形態では、R5は-C1-6アルキル-CH(NH2)COOHであり、式中、C1-6アルキルは、場合により、-OH、ハロゲン、NH2、またはCNにより1~3回置換されていてもよい。式(1)のいくつかの実施形態では、R5は-C1-6アルキル-CH(NH2)COOHであり、式中、C1-6アルキルは、場合により、ハロゲンにより1~3回置換されていてもよい。式(1)のいくつかの実施形態では、R5は-C1-6アルキル-CH(NH2)COOHであり、式中、C1-6アルキルは非置換である。式(1)のいくつかの実施形態では、R5は-CH2CH2-CH(NH2)COOHである。式(1)のいくつかの実施形態では、R5は-CH2-CH(NH2)COOHである。式(1)のいくつかの実施形態では、R5は-CH2CH2CH2-CH(NH2)COOHである。
式(1)のいくつかの実施形態では、R2はHであり、R3は-C(O)-Rであり、式中、Rは、C1-6アルキルまたは3~8員の炭素環式または複素環式であり、炭素環式部分及び複素環式部分のそれぞれは、独立して、場合により、-OH、ハロゲン、NH2、CN、またはC1-6アルキルで1~3回置換されていてもよく;各C1-6アルキルは、独立して、場合により、-OH、ハロゲン、NH2、またはCNで1~3回置換されていてもよい。
式(1)のいくつかの実施形態では、R3はHであり、式(1)のR2は-C(O)-Rであり、式中、Rは、C1-6アルキルまたは3~8員の炭素環式または複素環式であり、炭素環式部分及び複素環式部分は、独立して、場合により、-OH、ハロゲン、NH2、CN、またはC1-6アルキルで1~3回置換されていてもよく;各C1-6アルキルは、独立して、場合により、-OH、ハロゲン、NH2、またはCNで1~3回置換されていてもよい。
式(1)のいくつかの実施形態では、R2及びR3のそれぞれは、独立して、C(O)-Rであり、式中、各Rは、独立してC1-6アルキルまたは3~8員の炭素環式または複素環式であり、炭素環式部分及び複素環式部分のそれぞれは、独立して、場合により、-OH、ハロゲン、NH2、CN、またはC1-6アルキルで1~3回置換されていてもよく;各C1-6アルキルは、独立して、場合により、-OH、ハロゲン、NH2、またはCNで1~3回置換されていてもよい。
式(1)のいくつかの実施形態では、各Rは、独立して、3~8員の炭素環式または複素環式であり、炭素環式部分及び複素環式部分のそれぞれは、独立して、場合により、-OH、ハロゲン、NH
2、CN、またはC
1-6アルキルにより1~3回置換されていてもよく;各C
1-6アルキルは、独立して、場合により、-OH、ハロゲン、NH
2、またはCNで1~3回置換されていてもよい。式(1)のいくつかの実施形態では、各Rは、独立して、3~8員の炭素環式または複素環式であり、炭素環式部分及び複素環式部分のそれぞれは、独立して、場合により、ハロゲンで1~3回置換されていてもよい。式(1)のいくつかの実施形態では、各Rは、独立して、3~8員の炭素環式または複素環式であり、炭素環式部分及び複素環式部分のそれぞれは、独立して、場合により、ハロゲンで1~3回置換されていてもよい。式(1)のいくつかの実施形態では、各Rは、独立して、3~8員の非置換炭素環式または非置換複素環式である。式(1)のいくつかの実施形態では、各Rは、独立して、6員の非置換炭素環式または非置換複素環式である。式(1)のいくつかの実施形態では、各Rは、独立して非置換複素環である。式(1)のいくつかの実施形態では、Rは
である。
式(1)のいくつかの実施形態では、各Rは、独立してC1-6アルキルであり、各C1-6アルキルは、独立して、場合により、-OH、ハロゲン、NH2、またはCNにより1~3回置換されていてもよい。式(1)のいくつかの実施形態では、各Rは、独立してC1-6アルキルであり、各C1-6アルキルは、独立して、場合によりハロゲンにより1~3回置換されていてもよい。式(1)のいくつかの実施形態では、各Rは、独立して非置換C1-6アルキルである。式(1)のいくつかの実施形態では、各Rは、独立して非置換直鎖C1-6アルキルである。式(1)のいくつかの実施形態では、各Rは、独立して、メチル、エチル、またはプロピルである。式(1)のいくつかの実施形態では、RはCH3である。
式(1)のいくつかの実施形態では、R2とR3は一緒になって-(CH2)nC(O)-(CH2)m-を形成し、式中、n及びmのそれぞれは、独立して0~3であり、n+m≦3であり、-(CH2)n-及び-(CH2)m-部分のそれぞれは、独立して、場合により、-OH、ハロゲン、NH2、CN、またはC1-6アルキルで1~3回置換されていてもよく;各C1-6アルキルは、独立して、場合により、-OH、ハロゲン、NH2、またはCNで1~3回置換されていてもよい。式(1)のいくつかの実施形態では、-(CH2)n-及び-(CH2)m-部分のそれぞれは、独立して、場合によりハロゲンで1~3回置換されていてもよい。式(1)のいくつかの実施形態では、-(CH2)n-及び-(CH2)m-部分は非置換である。いくつかの実施形態では、n=m=0である。
いくつかの実施形態では、本開示は、式(2)の化合物:
またはその薬学的に許容される塩、プロドラッグ、もしくは異性体に関し、
式中、R
8は、Hまたはハロゲンであり;
Xは、Hまたはハロゲンであり;
各R
5’は、独立して、Hまたはハロゲンであり;ならびに
各Rは、独立してC
1-6アルキルであり、そのそれぞれは、独立して、場合によりハロゲンで1~3回置換されていてもよい。
式(2)のいくつかの実施形態では、C(R5’)3は、CF3、CHF2、CH2F、またはCH3である。
式(2)のいくつかの実施形態では、化合物は式(2’)の化合物である:
式(2)または(2’)のいくつかの実施形態では、R8はHである。式(2)または(2’)のいくつかの実施形態では、R8はハロゲンである。式(2)または(2’)のいくつかの実施形態では、R8はFである。
式(2)または(2’)のいくつかの実施形態では、XはHである。式(2)または(2’)のいくつかの実施形態では、Xはハロゲンである。式(2)または(2’)のいくつかの実施形態では、XはFである。
式(2)または(2’)のいくつかの実施形態では、Rは、それぞれ独立して非置換C1-6アルキルである。式(2)または(2’)のいくつかの実施形態では、Rは、それぞれ独立して、C1-3アルキルであり、そのそれぞれは、独立して、場合によりハロゲンにより1~3回置換されていてもよい。式(2)または(2’)のいくつかの実施形態では、Rは、それぞれ独立して、非置換C1-3アルキルである。式(2)または(2’)のいくつかの実施形態では、Rは、それぞれ独立して、-CH3、-CH2CH3、または-CH2CH2CH3である。
いくつかの実施形態では、本開示は、式(3)の化合物:
またはその薬学的に許容される塩、プロドラッグ、もしくは異性体に関し、
式中、R
8は、Hまたはハロゲンであり;
Xは、Hまたはハロゲンであり;ならびに
各R’は、独立してHまたはハロゲンである。
式(3)のいくつかの実施形態では、-Se-C(R’)3は、-Se-CH3、-Se-CHF2、-Se-CH2F、または-Se-CF3である。
式(3)のいくつかの実施形態では、化合物は式(3’)の化合物である:
式(3)または(3’)のいくつかの実施形態では、R8はHである。式(3)または(3’)のいくつかの実施形態では、R8はハロゲンである。式(3)または(3’)のいくつかの実施形態では、R8はFである。
式(3)または(3’)のいくつかの実施形態では、XはHである。式(3)または(3’)のいくつかの実施形態では、Xはハロゲンである。式(3)または(3’)のいくつかの実施形態では、XはFである。
式(3)または(3’)のいくつかの実施形態では、各C(R’)3は、独立して、CF3、CHF2、またはCH2F、またはCH3である。式(3)または(3’)のいくつかの実施形態では、各C(R’)3は、独立して、CH2FまたはCH3である。式(3)または(3’)のいくつかの実施形態では、各C(R’)3は、CH3である。
いくつかの実施形態では、本開示は、式:
の化合物、
またはその薬学的に許容される塩、プロドラッグ、もしくは異性体に関する。
製剤
いくつかの実施形態では、本開示は、式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくはその異性体を含み、及び/または送達する(すなわち、系または被験体への投与時に)組成物を提供する。いくつかの実施形態では、本開示は、式(1)~(3)のいずれか1つの単一の化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体のみを含む組成物を提供する。いくつかの実施形態では、本開示は、1つ以上の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体、及び本開示による、ヒトまたは動物被験体への投与に適した1つ以上の担体または賦形剤を含む組成物を提供する。
いくつかの実施形態では、本開示は、式(1)~(3)のいずれか1つの化合物の活性部分を送達する組成物を提供する。いくつかの実施形態では、組成物は、式(1)~(3)のいずれか1つの化合物の活性代謝物を含む。いくつかの実施形態では、組成物は、前記組成物の投与時に式(1)~(3)のいずれかの化合物の代謝産物を形成する化合物を含み、その代謝産物は関連する生物活性を維持している。
いくつかの実施形態では、本開示は、活性医薬成分(API)及び1つ以上の薬学的に許容される担体または賦形剤を含むという点で医薬組成物である組成物を提供する。いくつかの実施形態では、APIは、式(1)~(3)のいずれか1つの化合物であるか、またはそれを含む。いくつかの実施形態では、APIは、式(1)~(3)のいずれか1つの化合物からなる。いくつかの実施形態では、APIは、式(1)~(3)のいずれか1つの単一化合物からなる。
いくつかの実施形態では、本開示は、例えば、1つ以上の適切な(すなわち、薬学的に許容される)担体または賦形剤を式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体と組み合わせることによる、提供する組成物の製造方法を提供する。いくつかの実施形態では、1つ以上の薬学的に許容される担体または賦形剤は経口投与に適しており、混合物を経口製剤に製剤化する。いくつかの実施形態では、医薬組成物は固体剤形である。いくつかの実施形態では、固体剤形は、錠剤、カプセル剤、またはロゼンジである。いくつかの実施形態では、医薬組成物は、液体剤形(例えば、飲料)である。
Remington’s Pharmaceutical Sciences,Sixteenth Edition,E.W.Martin(Mack Publishing Co.,Easton,Pa.,1980)は、医薬組成物の製剤化に使用する様々な担体及びその調製のための公知の技術を開示している。いくつかの実施形態では、本開示は、薬学的に許容される量の本明細書に記載の化合物を含む医薬組成物を提供する。いくつかの実施形態では、単一剤形を生成するために担体物質と組み合わせることができる有効成分の量は、治療する宿主、及び/または特定の投与様式に応じて異なり得る。単一の剤形を生成するために担体物質と組み合わせることができる有効成分の量は、通常、治療効果を生成する化合物の量となる。一般的に、この量は、有効成分の約1%~約99%、約5%~約70%、または約10%~約30%の範囲である。
いくつかの実施形態では、湿潤剤、乳化剤、及び潤滑剤、例えば、ラウリル硫酸ナトリウム及びステアリン酸マグネシウム、ならびに着色剤、剥離剤、コーティング剤、甘味料、香味料及び芳香剤、防腐剤及び抗酸化剤も、組成物中に存在させてよい。
薬学的に許容される抗酸化剤の例として:水溶性抗酸化剤、例えば、アスコルビン酸、システイン塩酸塩、重硫酸ナトリウム、二亜硫酸ナトリウム、亜硫酸ナトリウムなど;油溶性抗酸化剤、例えば、パルミチン酸アスコルビル、ブチルヒドロキシアニソール(BHA)、ブチルヒドロキシトルエン(BHT)、レシチン、没食子酸プロピル、α-トコフェロールなど;及び金属キレート剤、例えば、クエン酸、エチレンジアミン四酢酸(EDTA)、ソルビトール、酒石酸、リン酸などが挙げられる。
いくつかの実施形態では、本発明の製剤には、経口、経鼻、局所(頬及び舌下を含む)、直腸内、膣内及び/または非経口投与に適した製剤が含まれる。いくつかの実施形態では、製剤を、単位剤形で簡便に提示してもよく、薬学の技術分野で周知の任意の方法によって調製してもよい。いくつかの実施形態では、本明細書に記載の製剤は、シクロデキストリン、リポソーム、ミセル形成剤、例えば胆汁酸、ならびにポリマー担体、例えばポリエステル及びポリ無水物からなる群から選択される賦形剤;ならびに本明細書に記載の化合物を含む。いくつかの実施形態では、本明細書に記載の製剤は、本明細書に記載の化合物を、経口で生体利用可能にする。
いくつかの実施形態では、そのような製剤の調製方法は、本明細書に記載の化合物を、1つ以上の薬学的に許容される担体または賦形剤、及び場合により1つ以上の補助成分と会合させる工程を含み得る。いくつかの実施形態では、製剤は、本明細書に記載の化合物を、液体担体、もしくは微粉化固体担体、またはその両方と均一かつ密接に会合させ、次いで必要に応じて製品を成形することにより調製する。
いくつかの実施形態では、経口投与に適した本明細書に記載の製剤は、それぞれ有効成分として所定量の本明細書に記載の化合物を含む、カプセル、カシェ、丸薬、錠剤、ロゼンジ(フレーバーベース、通常、スクロース及びアカシアまたはトラガカントを使用)、散剤、顆粒の形態、または水性もしくは非水性液体中の溶液もしくは懸濁液として、または水中油型もしくは油中水型液体エマルジョンとして、またはエリキシル剤またはシロップ剤として、またはトローチとして(ゼラチン及びグリセリン、またはスクロース及びアカシアなどの不活性基剤を使用して)及び/または口内洗浄剤、飲料などとしてもよい。いくつかの実施形態では、本明細書に記載の化合物を、代替的または追加的に、ボーラス、舐剤またはペーストとして投与してもよい。
いくつかの実施形態では、経口投与用の本明細書に記載の固体剤形(カプセル、錠剤、丸薬、糖衣錠、散剤、顆粒など)では、有効成分を、ナトリウムなどの1つ以上の薬学的に許容される担体、例えば、クエン酸塩またはリン酸二カルシウム、及び/または以下のいずれかと混合する:充填剤または増量剤、例えば、デンプン、ラクトース、スクロース、グルコース、マンニトール、及び/またはケイ酸;結合剤、例えば、カルボキシメチルセルロース、アルギン酸塩、ゼラチン、ポリビニルピロリドン、スクロース及び/またはアカシア;保湿剤、例えばグリセロール;崩壊剤、例えば、寒天、炭酸カルシウム、ジャガイモまたはタピオカデンプン、アルギン酸、特定のケイ酸塩、及び炭酸ナトリウム;溶解遅延剤、例えば、パラフィン;吸収促進剤、例えば、四級アンモニウム化合物;湿潤剤、例えば、セチルアルコール、グリセロールモノステアレート、及び非イオン性界面活性剤;吸収剤、例えば、カオリン及びベントナイト粘土;潤滑剤、例えば、タルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポリエチレングリコール、ラウリル硫酸ナトリウム、及びそれらの混合物;ならびに着色剤。いくつかの実施形態では、カプセル、錠剤、及び丸薬の場合、医薬組成物は緩衝剤も含み得る。また、いくつかの実施形態では、同様のタイプの固体組成物を、ラクトースまたは乳糖、ならびに高分子量ポリエチレングリコールなどの担体を使用して、軟質及び硬質ゼラチンカプセルにおいて充填剤として使用してもよい。
いくつかの実施形態では、錠剤を、場合により1つ以上の補助成分とともに、圧縮または成形によって作製してもよい。いくつかの実施形態では、圧縮錠剤を、結合剤(例えば、ゼラチンまたはヒドロキシプロピルメチルセルロース)、潤滑剤、不活性希釈剤、防腐剤、崩壊剤(例えば、デンプングリコール酸ナトリウムまたは架橋カルボキシメチルセルロースナトリウム)、界面活性剤または分散剤を使用して調製してもよい。いくつかの実施形態では、成形錠剤を、粉末化合物の混合物を不活性液体希釈剤で湿らせる適切な機械において作製してもよい。
いくつかの実施形態では、錠剤、及び本明細書に記載の医薬組成物の他の固体剤形、例えば、糖衣錠、カプセル、丸薬及び顆粒は、場合により、コーティング及びシェル、例えば、腸溶コーティング及び医薬製剤分野で周知の他のコーティングで刻み目を付けるかまたは調製してもよい。いくつかの実施形態では、それらを、例えば、所望の放出特性を提供するようにヒドロキシプロピルメチルセルロースを様々な割合で、他のポリマーマトリックス、リポソーム及び/またはミクロスフェアを使用して、内部の有効成分の徐放または制御放出を提供するように製剤化してもよい。いくつかの実施形態では、それらを、急速放出用に、例えば凍結乾燥により製剤化してもよい。いくつかの実施形態では、それらを、例えば、細菌保持フィルターによる濾過により、または滅菌水に溶解可能な滅菌固体組成物の形態の滅菌剤、もしくは使用直前に他の滅菌注射用媒体を組み込むことにより滅菌してもよい。いくつかの実施形態では、これらの組成物に場合により乳白剤を含ませてもよく、これらの組成物は、有効成分(複数可)を、胃腸管の特定の部分でのみ、またはそこにおいて優先的に、場合により遅延様式で放出する組成物であってもよい。使用できる包埋組成物の例として、高分子物質及びワックスが挙げられる。いくつかの実施形態では、有効成分は、適切な場合、上記の賦形剤の1つ以上を有するマイクロカプセル化形態であってもよい。
いくつかの実施形態では、本明細書に記載の化合物の経口投与用の液体剤形には、薬学的に許容されるエマルジョン、マイクロエマルジョン、溶液、懸濁液、シロップ及びエリキシル剤が含まれる。いくつかの実施形態では、有効成分に加えて、液体剤形は、当技術分野で一般的に使用される不活性希釈剤、例えば、水または他の溶媒、可溶化剤及び乳化剤、例えばエチルアルコール、イソプロピルアルコール、エチルカーボネート、酢酸エチル、ベンジルアルコール、安息香酸ベンジル、プロピレングリコール、1,3-ブチレングリコール、油(特に、綿実、落花生、トウモロコシ、胚芽、オリーブ、ヒマシ、及びゴマ油)、グリセロール、テトラヒドロフリルアルコール、ポリエチレングリコール及びソルビタンの脂肪酸エステル、ならびにそれらの混合物を含み得る。
いくつかの実施形態では、不活性希釈剤に加えて、経口組成物に、湿潤剤、乳化剤及び懸濁剤、甘味料、香味料、着色剤、芳香剤ならびに防腐剤などのアジュバントを含ませることができる。
いくつかの実施形態では、活性化合物に加えて、懸濁液には、例えば、エトキシ化イソステアリルアルコール、ポリオキシエチレンソルビトール及びソルビタンエステル、微結晶性セルロース、メタ水酸化アルミニウム、ベントナイト、寒天及びトラガカントなどの懸濁剤、ならびにそれらの混合物を含ませてもよい。
いくつかの実施形態では、直腸内または膣内投与用の本明細書に記載の製剤を坐薬として提示してもよく、本明細書に記載の1つ以上の化合物を、1つ以上の適切な非刺激性賦形剤または担体、例えば、カカオバター、ポリエチレングリコール、坐薬ワックスまたはサリチル酸塩と混合することによりこれを調製してもよく、この坐薬は、室温で固体であるが、体温では液体であり、それゆえに、直腸または膣腔内で溶けて活性化合物を放出する。
いくつかの実施形態では、膣内投与に適した本明細書に記載の製剤には、適切であることが当技術分野で知られている担体を含む、ペッサリー、タンポン、クリーム、ゲル、ペースト、フォームまたはスプレー製剤が含まれる。
いくつかの実施形態では、本明細書に記載の化合物の局所または経皮投与用の剤形には、散剤、スプレー、軟膏、ペースト、クリーム、ローション、ゲル、溶液、パッチ及び吸入剤が含まれる。いくつかの実施形態では、活性化合物を、無菌条件下で、薬学的に許容される担体、及び必要とされ得る保存剤、緩衝液、または噴射剤と混合してもよい。
いくつかの実施形態では、軟膏、ペースト、クリーム、及びゲルは、本明細書に記載の化合物に加えて、賦形剤、例えば、動物性及び植物性脂肪、油類、ワックス、パラフィン、デンプン、トラガカント、セルロース誘導体、ポリエチレングリコール、シリコーン、ベントナイト、ケイ酸、タルク及び酸化亜鉛、またはそれらの混合物を含み得る。
いくつかの実施形態では、散剤及びスプレーに、本明細書に記載の化合物に加えて、賦形剤、例えば、ラクトース、タルク、ケイ酸、水酸化アルミニウム、ケイ酸カルシウム及びポリアミド粉末、またはこれらの物質の混合物を含ませることができる。いくつかの実施形態では、スプレーに、慣習的な噴射剤、例えば、クロロフルオロ炭化水素、ならびに揮発性非置換炭化水素、例えば、ブタン及びプロパンをさらに含ませることができる。
いくつかの実施形態では、経皮パッチは、本明細書に記載の化合物の身体への制御された送達を提供するという追加の利点を有する。いくつかの実施形態では、化合物を適切な媒体に溶解または分散させることにより、そのような剤形を作ることができる。いくつかの実施形態では、吸収促進剤を使用して、皮膚を通過する化合物のフラックスを増加させることができる。いくつかの実施形態では、速度制御膜を提供するか、化合物をポリマーマトリックスまたはゲルに分散させることにより、そのようなフラックスの速度を制御することができる。
いくつかの実施形態では、本開示は、眼科用製剤、眼軟膏、散剤、溶液などを提供する。
いくつかの実施形態では、非経口投与に適した本明細書に記載の医薬組成物は、本明細書に記載の1つ以上の化合物と、1つ以上の薬学的に許容される無菌の等張水溶液または非水溶液、分散液、懸濁液またはエマルジョン、または無菌粉末とを組み合わせて含み、これを、使用直前に滅菌注射用溶液または分散液に再構成してもよく、これには、目的のレシピエントの血液または懸濁剤または増粘剤の存在下で、製剤を等張にする糖、アルコール、抗酸化剤、緩衝剤、静菌剤、溶質が含まれ得る。
本明細書に記載の医薬組成物に使用し得る適切な水性及び非水性担体の例として、水、エタノール、ポリオール(グリセロール、プロピレングリコール、ポリエチレングリコールなど)、及びそれらの適切な混合物、オリーブ油などの植物性油、ならびにオレイン酸エチルなどの注射可能な有機エステルが挙げられる。いくつかの実施形態では、例えば、レシチンなどのコーティング物質の使用により、分散液の場合には必要な粒子サイズの維持により、及び界面活性剤の使用により、適切な流動性を維持することができる。
いくつかの実施形態では、本明細書に記載の組成物に、防腐剤、湿潤剤、乳化剤、及び分散剤などのアジュバントを含ませてもよい。いくつかの実施形態では、例えば、パラベン、クロロブタノール、フェノールソルビン酸などの様々な抗菌剤及び抗真菌剤を含めることにより、対象化合物に対する微生物の作用の予防を確保し得る。いくつかの実施形態では、糖、塩化ナトリウムなどの等張剤を組成物に含めることが望ましい場合がある。いくつかの実施形態では、モノステアリン酸アルミニウム及びゼラチンなどの吸収を遅延させる薬剤を含めることにより、注射可能な医薬形態の長期吸収をもたらし得る。
いくつかの実施形態では、例えば、薬物の効果を長引かせるために、皮下または筋肉内注射からの薬物の吸収を遅延させることが望ましい。いくつかの実施形態では、これは、水への溶解度が低い結晶性または非晶質物質の液体懸濁液の使用により達成され得る。いくつかの実施形態では、薬物の吸収速度はその溶解速度に依存し、溶解速度は結晶サイズ及び結晶形態に依存し得る。いくつかの実施形態では、非経口投与する薬物形態の遅延吸収は、薬物を油性ビヒクルに溶解または懸濁することにより達成される。
いくつかの実施形態では、ポリラクチド-ポリグリコリドなどの生分解性ポリマー中に対象化合物のマイクロカプセルマトリックスを形成することにより、注射可能なデポー形態を作製する。いくつかの実施形態では、薬物とポリマーの比、及び使用する特定のポリマーの性質に応じて、薬物放出の速度を制御することができる。他の生分解性ポリマーの例として、ポリ(オルトエステル)及びポリ(無水物)が挙げられる。いくつかの実施形態では、体組織に適合するリポソームまたはマイクロエマルジョンに薬物を閉じ込めることにより、デポー注射製剤を調製する。
いくつかの実施形態では、薬物溶出形態には、コーティングまたは薬物添加ステント及び埋込装置が含まれる。いくつかの実施形態では、薬物溶出ステント及び他の装置に、化合物または医薬製剤をコーティングしてもよく、さらに徐放用に設計されたポリマーを含めてもよい。
いくつかの実施形態では、化合物または医薬製剤を経口投与する。いくつかの実施形態では、化合物または医薬製剤を静脈内投与する。いくつかの実施形態では、化合物を、切断可能なリンカーを介して、カテーテルとともに投与する固体支持体に付着させる。いくつかの実施形態では、投与経路には、舌下、筋肉内、及び経皮投与が含まれる。
いくつかの実施形態では、本明細書に記載の化合物を、医薬品としてヒト及び動物に投与し、それらは、それ自体で、または例えば0.1%~99.5%、もしくは0.5%~90%の有効成分を含む医薬組成物として薬学的に許容される担体と組み合わせて、投与することができる。
いくつかの実施形態では、本明細書に記載の化合物を、経口、非経口、局所、または直腸内投与してもよい。いくつかの実施形態では、それらを、当然ながら各投与経路に適した形で与える。いくつかの実施形態では、それらを、注射、吸入、目薬、軟膏、坐薬などにより錠剤またはカプセルの形態で、注射、注入または吸入による投与で;ローションまたは軟膏により局所に;及び坐薬により直腸内に投与する。
いくつかの実施形態では、本明細書に記載の化合物を、例えばエアロゾル、スプレーによって経口、経鼻で、散剤、軟膏または点滴によって直腸内、膣内、非経口、大槽内及び局所的に、ならびに頬側及び舌下を含む、任意の適切な投与経路による治療のために、ヒト及び他の動物に投与してもよい。
いくつかの実施形態では、適切な水和形態で使用し得る本明細書に記載の化合物、及び/または本明細書に記載の医薬組成物を、当業者に公知の従来の方法により薬学的に許容可能な剤形に製剤化する。
いくつかの実施形態では、本明細書に記載の医薬組成物中の有効成分の実際の用量レベルは、患者に毒性を与えずに、特定の患者、組成物、及び投与様式に対して所望の治療応答を達成するのに有効な量の有効成分を得るために、様々に異なっていてもよい。
いくつかの実施形態では、選択する用量レベルは、本明細書に記載の特定の化合物の活性、投与経路、投与時間、使用する特定の化合物の排出速度または代謝速度、治療期間、使用する特定の化合物と組み合わせて使用する他の薬物、化合物及び/または物質、治療中の患者の年齢、性別、体重、病態、健康全般及び以前の病歴、ならびに医学分野で周知の類似の要因、を含む様々な要因に依存する。
当技術分野の通常の技術を有する医師または獣医であれば、必要な医薬組成物の有効量を決定及び処方することができる。いくつかの実施形態では、医師または獣医は、医薬組成物中の本明細書に記載の化合物の投与を、所望の治療効果を達成するのに必要なレベルよりも低いレベルで開始し、次いで所望の効果が達成されるまで用量を徐々に増加させることができる。
いくつかの実施形態では、本明細書に記載の化合物または医薬組成物を、被験体に慢性的に提供する。いくつかの実施形態では、慢性処置には、1か月以上、1か月~1年、1年以上、またはそれ以上の繰り返し投与など、長期間にわたる任意の形態の繰り返し投与が含まれる。いくつかの実施形態では、慢性処置には、本明細書に記載の化合物または医薬組成物を、被験体の生涯にわたって繰り返し投与することが含まれる。いくつかの実施形態では、慢性処置には、例えば、1日1回以上、1週間に1回以上、または1か月に1回以上の定期的な投与が含まれる。いくつかの実施形態では、本明細書に記載の化合物の日用量などの適切な用量は、治療効果を生じさせるのに有効な最低用量となる化合物の量である。そのような有効量は一般的に、本明細書に記載の要因に依存する。いくつかの実施形態では、患者に対する本明細書に記載の化合物の用量は、示す効果のために使用する場合、1日あたり体重1kgあたり約0.0001~約100mgの範囲である。いくつかの実施形態では、日用量は、体重1kgあたり化合物0.001~50mgの範囲である。いくつかの実施形態では、日用量は、体重1kgあたり化合物0.01~10mgの範囲である。しかしながら、より低いまたはより高い用量を使用することができる。いくつかの実施形態では、年齢、疾患の進行、体重、または他の要因によって被験体の生理機能が変化するにつれて、被験体に投与する用量を修正してもよい。
いくつかの実施形態では、活性化合物の有効な日用量を、1日の間に、場合により単位投与形態で、適切な間隔で別々に投与する、2、3、4、5、6回、またはそれ以上の部分用量として投与してもよい。
いくつかの実施形態では、本明細書に記載の化合物を単独で投与する。いくつかの実施形態では、本明細書に記載の化合物を、本明細書に記載の医薬製剤(組成物)として投与する。
いくつかの実施形態では、本明細書に記載の化合物を、他の医薬品と同様に、ヒトまたは獣医学で使用するための任意の簡便な方法で投与するために製剤化してもよい。
化合物及び/または組成物の調製
いくつかの実施形態では、式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を、全体的または部分的に化学合成により調製してもよく;いくつかの実施形態では、部分的に化学合成によって調製する式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を、半合成的方法を用いて調製する。いくつかの実施形態では、式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を、単離により調製してもよい。
いくつかの実施形態では、本開示は、1つ以上の試料をアッセイして、例えば試料中の生物活性を検出することを含む、本明細書に記載の化合物及び/または組成物の調製方法を提供する。いくつかの実施形態では、1つ以上の試料は、式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を含む。いくつかの実施形態では、本明細書において提供する方法は、1つ以上の試料中の検出可能な生物活性の存在を検出及び/または確認する工程を含む。いくつかの実施形態では、本明細書において提供する方法は、1つ以上の試料に検出可能な生物活性が存在しないことを確認する工程を含む。
いくつかの実施形態では、生物活性はグルコース産生の阻害である。いくつかの実施形態では、生物活性をHepG2細胞で試験する。いくつかの実施形態では、生物活性をH4IIE細胞で試験する。
いくつかの実施形態では、生物活性は、血清HbA1cレベルの低下である。いくつかの実施形態では、生物活性を、インスリン抵抗性糖尿病マウスで試験する。
いくつかの実施形態では、生物活性は耐糖能の増強である。いくつかの実施形態では、生物活性を、インスリン抵抗性糖尿病マウスで試験する。
いくつかの実施形態では、生物活性はG6pc発現の阻害である。いくつかの実施形態では、生物活性を、AML-12細胞で試験する。いくつかの実施形態では、生物活性を、糖尿病刺激で刺激したAML-12細胞で試験する。いくつかの実施形態では、生物活性を、ヒトHepG2細胞で試験する。いくつかの実施形態では、生物活性を、インスリン抵抗性糖尿病マウスの肝臓で試験する。
いくつかの実施形態では、生物活性は、Pdk1、Akt、Foxo1及びAS160のリン酸化の増強である。いくつかの実施形態では、生物活性を肝臓で試験する。いくつかの実施形態では、生物活性を骨格筋で試験する。
いくつかの実施形態では、生物活性はGlut4発現の増強である。いくつかの実施形態では、生物活性を、マウス肝臓AML-12細胞で試験する。いくつかの実施形態では、生物活性を、インスリン抵抗性糖尿病マウスの肝臓で試験する。
いくつかの実施形態では、生物活性は、インスリン抵抗性状態の被験体におけるインスリンシグナル伝達の活性化及び/または回復である。いくつかの実施形態では、被験体は、有意なレベルの循環インスリンを特徴とする。いくつかの実施形態では、インスリン抵抗性状態は、被験体におけるリン酸化インスリン受容体のレベル及び/または活性の低下を特徴とする。いくつかの実施形態では、被験体は、糖尿病、及び/または糖尿病関連の疾患、障害、または病態を有する。
いくつかの実施形態では、生物活性はグルコース取り込みの増強である。いくつかの実施形態では、細胞は肝細胞及び骨格筋細胞である。いくつかの実施形態では、生物活性は血清インスリンレベルの低下である。いくつかの実施形態では、生物活性を、インスリン抵抗性糖尿病マウスで試験する。
化合物及び/または組成物の同定及び/または特徴決定
いくつかの実施形態では、本開示は、本明細書に記載の化合物及び/または組成物の同定及び/または特徴決定の方法を提供する。いくつかの実施形態では、そのような方法は、式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体をそれぞれ含む複数の試料を、その試料中の生物活性について試験し;及び、1つ以上のそのような試料中の前記生物活性の存在及び/またはレベルを決定する工程を含む。いくつかの実施形態では、提供する方法は、式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体の存在及び/またはレベルに関連する前記生物活性を検出することを含む。いくつかの実施形態では、提供する方法は、式(1)~(3)のいずれか1つの特定の化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を、その化合物の前記生物活性を検出することにより、同定及び/または特徴決定する工程を含む。
いくつかの実施形態では、生物活性はグルコース産生の阻害である。いくつかの実施形態では、生物活性をHepG2細胞で試験する。いくつかの実施形態では、生物活性をH4IIE細胞で試験する。
いくつかの実施形態では、生物活性は、血清HbA1cレベルの低下である。いくつかの実施形態では、生物活性を、インスリン抵抗性糖尿病マウスで試験する。
いくつかの実施形態では、生物活性は耐糖能の向上である。いくつかの実施形態では、生物活性を、インスリン抵抗性糖尿病マウスで試験する。
いくつかの実施形態では、生物活性はG6pc発現の阻害である。いくつかの実施形態では、生物活性を、AML-12細胞で試験する。いくつかの実施形態では、生物活性を、糖尿病刺激で刺激したAML-12細胞で試験する。いくつかの実施形態では、生物活性をヒトHepG2細胞で試験する。いくつかの実施形態では、生物活性を、インスリン抵抗性糖尿病マウスの肝臓で試験する。
いくつかの実施形態では、生物活性は、Pdk1、Akt、Foxo1、及びAS160のリン酸化の増強である。いくつかの実施形態では、生物活性を、肝臓で試験する。いくつかの実施形態では、生物活性を、骨格筋で試験する。
いくつかの実施形態では、生物活性はGlut4発現の増強である。いくつかの実施形態では、生物活性を、マウス肝臓AML-12細胞で試験する。いくつかの実施形態では、生物活性を、インスリン抵抗性糖尿病マウスの肝臓で試験する。
いくつかの実施形態では、生物活性は、インスリン抵抗性状態の被験体におけるインスリンシグナル伝達の活性化及び/または回復である。いくつかの実施形態では、被験体は、有意なレベルの循環インスリンを特徴とする。いくつかの実施形態では、インスリン抵抗性状態は、被験体におけるリン酸化インスリン受容体のレベル及び/または活性の低下を特徴とする。いくつかの実施形態では、被験体は、糖尿病、及び/または糖尿病関連の疾患、障害、または病態を有する。
いくつかの実施形態では、生物活性は、被験体の細胞へのグルコース取り込みの増強である。いくつかの実施形態では、生物活性は、肝細胞及び骨格筋細胞へのグルコース取り込みの増強である。いくつかの実施形態では、生物活性は、血清インスリンレベルの低下である。いくつかの実施形態では、生物活性を、インスリン抵抗性糖尿病マウスで試験する。
使用
本開示は、本明細書に記載の化合物、例えば化合物43が、インスリンを模倣して、グルコース産生を阻害し(例えば、実施例2参照);インスリン抵抗性糖尿病の被験体の血糖値及びHbA1cレベルを低下させ、高血糖症の発症を軽減し、耐糖能を向上させ(例えば、実施例3参照);インスリン抵抗性糖尿病の被験体の肝臓でのG6pc発現を抑制し、インスリンを模倣するが迂回して、培養マウス及びヒト肝細胞でのG6pc発現を抑制し、インスリン作用を増強し(例えば、実施例4参照);インスリンを模倣するが迂回して、Pdk1及びAktを活性化し、肝臓でのFoxo1のリン酸化を増強し(例えば、実施例5参照);インスリン抵抗性糖尿病の被験体の肝臓でのGlut4発現を増強し、インスリンを模倣するが迂回して、マウス肝細胞でのGlut4発現を増強し(例えば、実施例6参照)、ヒト肝細胞でのAS160のリン酸化(グルコース取り込みを増強するための、細胞質小胞から細胞膜へのGLUT4輸送への重要な事象)(例えば、実施例8参照)、及びマウス肝細胞でのグルコース取り込み(例えば、実施例6参照);インスリン抵抗性糖尿病の被験体の骨格筋のインスリンシグナル伝達Pdk1/Akt/Foxo1を増強及び/または回復させ(例えば、実施例7参照)、インスリンを模倣するが迂回して、Aktを活性化し、骨格筋細胞でのAS160のリン酸化を増強し(細胞質小胞から細胞膜へのGLUT4輸送への重要な事象)(例えば、実施例8参照)、及びインスリン作用を増強して骨格筋細胞でのグルコース取り込みを刺激し(例えば、実施例7参照);インスリン抵抗性糖尿病の被験体の骨格筋及び肝臓、ならびに培養マウス骨格筋細胞及びヒト肝細胞のインスリン受容体(Insr)機能を活性化及び/または回復させ(例えば、実施例8参照);ならびに腎機能を損なうことなく、及び/または肝臓損傷を引き起こすことなく、高インスリン血症を軽減する(実施例9)ことができることを提供する。したがって、本開示は、本明細書に記載の化合物が、グルコース代謝を調節し、本明細書に記載のインスリン関連障害を治療することに有用であることを明確に示す。
いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、グルコース代謝の調節方法及び/またはグルコース代謝障害の治療方法を提供する。いくつかの実施形態では、グルコース代謝障害は、正常範囲内にない血糖値を伴う。いくつかの実施形態では、グルコース代謝障害は、グルコースの取り込み及び/または輸送の欠陥に関連する。いくつかの実施形態では、グルコース代謝障害は、真性糖尿病、グリセルアルデヒド-3-リン酸デヒドロゲナーゼ欠損症、糖尿症、高血糖症、高インスリン症、または低血糖症である。
いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、グルコース輸送障害の治療方法を提供する。いくつかの実施形態では、グルコース輸送障害は、グルコース-ガラクトース吸収不良症、ファンコニー-ビッケル症候群、またはDe Vivo病(GLUT1欠損症候群(GLUT1DS))である。
いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、グルコース取り込みのための細胞質小胞から細胞膜へのグルコース輸送体タンパク質(GLUT)移行のためのAS160リン酸化の増強方法を提供する。
いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、肝臓及び骨格筋の両方におけるグルコース取り込みの増強方法を提供する。
いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、インスリン関連障害の治療方法を提供する。いくつかの実施形態では、インスリン関連障害は、糖尿病前症、I型糖尿病、II型糖尿病、低血糖症、高血糖症、インスリン抵抗性、分泌機能不全、膵臓β細胞機能の喪失、及び膵臓β細胞の喪失からなる群から選択される。いくつかの実施形態では、インスリン関連障害の患者は、インスリン関連障害を有するインスリン非依存患者である。
いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、インスリン抵抗性障害の治療方法を提供する。インスリン抵抗性障害の特定の例は上記に記載されている。いくつかの実施形態では、提供する方法は、II型糖尿病、高インスリン血症、高プロインスリン血症、網膜症、神経障害、または腎障害を治療するための方法である。いくつかの実施形態では、提供する方法は、腎機能を損なうことなく、及び/または肝臓損傷をもたらすことなく、高インスリン血症を軽減する。
いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、肥満の治療方法を提供する。
いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、糖尿病の治療方法を提供する。いくつかの実施形態では、糖尿病はI型糖尿病である。いくつかの実施形態では、糖尿病はII型糖尿病である。
いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、高血糖症の治療方法を提供する。
いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、グルコース産生の阻害方法を提供する。
いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、血清HbA1cレベルの低下方法を提供する。
いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、耐糖能の増加方法を提供する。
いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、G6pc発現の抑制方法を提供する。
いくつかの実施形態では、本開示は、式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、肝臓及び/または骨格筋におけるPdk1、Akt、及びFoxo1のリン酸化の増強方法を提供する。
いくつかの実施形態では、本開示は、式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、Glut4発現の増加方法を提供する。
いくつかの実施形態では、本開示は、式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、インスリン抵抗性状態の被験体におけるインスリンシグナル伝達の活性化及び/または回復方法を提供する。いくつかの実施形態では、被験体は、有意なレベルの循環インスリンを特徴とする。いくつかの実施形態では、インスリン抵抗性状態は、被験体におけるリン酸化インスリン受容体のレベル及び/または活性の低下を特徴とする。いくつかの実施形態では、被験体は、糖尿病、及び/または糖尿病関連疾患、障害、または病態を有する。いくつかの実施形態では、被験体はII型糖尿病を患っている。
いくつかの実施形態では、本開示は、式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、被験体の細胞へのグルコース取り込みの増強方法を提供する。いくつかの実施形態では、細胞は骨格筋細胞である。
化合物#43は、インスリンを模倣するが迂回して、肝臓及び骨格筋のインスリン受容体シグナル伝達を活性化し、インスリン抵抗性糖尿病の被験体においてもインスリン受容体機能を回復させることができることが示されている。これらの所見は、化合物#43が多嚢胞性卵巣症候群(PCOS)、アルツハイマー病(AD)、筋肉減少症などのインスリンシグナル伝達の欠陥を特徴とする疾患または症候群の治療に使用することができることを示唆している。
いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、多嚢胞性卵巣症候群(PCOS)の治療方法を提供する。PCOSはホルモンの不均衡であり、不規則な期間、望ましくない発毛、ざ瘡を引き起こす可能性がある。PCOSを患っている若い女性では、インスリンレベルが上昇していることが多く、これにより卵巣がより多くのアンドロゲンホルモンを産生し、不規則な期間またはわずかな期間、体毛、ざ瘡が増加する。PCOSを患うと、インスリン抵抗性及び2型糖尿病の発症を引き起こし得る。メトホルミンは、インスリン感受性を改善し、2型糖尿病の発症を予防するために、PCOSを患っている女性に処方されることが多い薬剤である。結果は、化合物#43が培養肝細胞のグルコース産生の阻害においてメトホルミンよりも強力であり、インスリン抵抗性糖尿病マウスのインスリン受容体機能を回復させることができることを示している。したがって、化合物#43はPCOSの治療に潜在的に有用であり得る。
いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、アルツハイマー病(AD)の治療方法を提供する。脳のインスリンシグナル伝達は学習と記憶に重要であり、脳のインスリン抵抗性はADの主要な危険因子である。インスリンシグナル伝達の回復は、ADの潜在的な治療法として注目されている(White MF,Science 2003;302:1710-1;De Felice DG et al,Alzheimer’s & Dementia 2014;10:S26-S32)。化合物#43がインスリン様活性を示し、インスリン抵抗性の被験体のインスリン受容体機能を回復させることができたことを示す上記の結果を考慮すると、化合物#43はADの治療に潜在的に有用であり得る。
いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、筋肉減少症の治療方法を提供する。筋肉減少症は、年齢の増加に伴う骨格筋量の進行性の喪失を特徴とし、筋力の低下、可動性及び機能の低下、疲労の増加、転倒に関連する傷害のリスクの増加、そして多くの場合虚弱(Candow and Chilibeck,2005;Sakuma and Yamaguchi,2012)をもたらす。筋肉生物学の最近の進歩により、インスリンシグナル伝達(Insr/PI3K/Akt)が筋肉タンパク質の合成、及び筋肉タンパク質分解の阻害(Akt/Foxo1を介した2つの萎縮症遺伝子Fbxo32及びTrim63の発現の阻害を介して)に重要であることが明らかとなっている。最適なインスリンシグナル伝達は、筋肉減少症を含む筋消耗プロセスを軽減する(Glass and Roubenoff,2010;Ryall et al.,2008;Sakuma and Yamaguchi,2012)。そのため、筋肉減少症の治療のための新規戦略として、インスリンシグナル伝達の刺激剤、アミノ酸サプリメント(タンパク質合成を向上させるための)及びプロテアソームタンパク質分解の阻害剤が注目されている。本明細書に記載するように、インスリン抵抗性糖尿病マウスにおいてさえ、骨格筋におけるInsr/Pdk1/Akt/Foxo1シグナル伝達を活性化する、化合物#43のインスリン様活性は、筋肉の萎縮状態を治療するためのこの化合物の潜在的な用途を示唆している。
本開示はまた、提供する化合物がミトコンドリア機能を増強する可能性が高く、したがって、ミトコンドリア疾患及び/または機能不全の治療に有用であることを示す。いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)の化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、ミトコンドリア関連疾患(例えば、機能不全ミトコンドリアによって引き起こされる)の治療方法を提供する。いくつかの実施形態では、ミトコンドリア関連疾患は、変性疾患(例えば、がん、心血管疾患及び心不全、2型糖尿病、アルツハイマー病及びパーキンソン病、脂肪肝疾患、白内障、骨粗鬆症、筋消耗、睡眠障害及び炎症性疾患、例えば、乾癬、関節炎及び大腸炎)であり得る。いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、ミトコンドリア機能の増強方法を提供する。
いくつかの実施形態では、本開示は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む、脳の糖新生の増強方法を提供する。いくつかの実施形態では、提供する方法は、脳内のグルコース取り込みを増加させる。いくつかの実施形態では、提供する方法は、記憶及び学習を含む脳機能を維持または回復させるための方法である。
併用療法
いくつかの実施形態では、本開示は、本明細書に記載の疾患、障害、または病態の併用療法のための本明細書に記載の化合物及び/または組成物の使用を提供する。いくつかの実施形態では、本開示は、前記疾患、障害、または病態の1つ以上の異なる治療を受けた、受けている、または受けるであろう疾患、障害、または病態の患者の治療方法を提供し、この方法は、治療有効量の式(1)~(3)のいずれか1つの化合物、またはその薬学的に許容される塩、プロドラッグ、もしくは異性体を投与することを含む。
いくつかの実施形態では、インスリン関連障害を有する患者のための1つ以上の療法は、例えば、I型糖尿病の場合、インスリン療法;例えば、初発性II型糖尿病の場合、食事及び運動;例えば、早期II型糖尿病の場合、経口抗糖尿病薬;メトホルミン;インスリン分泌促進薬、例えば、スルホニル尿素;グリタゾン;長時間作用型基礎インスリン;中間作用インスリン;及び短期(急速)作用インスリンからなる群から選択される。いくつかの実施形態では、インスリン療法は、皮下(SC)、静脈内、及び/または吸入による投与を伴う。いくつかの実施形態では、インスリン関連障害を有する患者のための1つ以上の療法は、現在開発中の療法、例えばインスリン-フマリルジケトピペラジン(FDKP)であってもよい。
実施例1:化合物#43、#50、#53、#69、#70、及び#68の合成
1a.アデノシン,5’-Se-メチル-5’-セリノ-,2’,3’-ジアセテート(化合物#43)の合成
スキーム:
合成手順:滴下漏斗、不活性ガス入口/出口及び温度計を備え、オーブン乾燥した50ml三口フラスコに、5’-Se-メチル-5’-セレノ-アデノシン(1.0グラム、0.0029モル、1.0モル当量)及び無水ピリジン(10ml)を入れた。反応セットを氷/塩浴に入れ、撹拌を開始した。溶液の温度が0℃に下がった時点で、無水酢酸(10ml、0.105モル、36.47モル当量)を15分間滴下し、無水酢酸の添加中、反応混合物の温度を5℃以下に維持した。反応混合物を5~10℃で6時間撹拌した。氷冷水(100ml)を加えて過剰の無水酢酸をクエンチし、10wt%のNaHCO
3水溶液を加えてpHを7に調整した。水性混合物を酢酸エチル(2×100ml)で抽出した。合わせた酢酸エチル抽出物を無水Na
2SO
4(1グラム)で乾燥させ、250mlの丸底フラスコに濾過する。濾液を減圧下、35~40℃で乾燥するまで濃縮し、粗生成物を淡黄色のシロップ状の液体として得た後、酢酸エチルとヘキサンの混合物(1:3 v/v)を使用してシリカゲルカラムに通すことによって、高純度の生成物をオフホワイトの結晶として得た(1.12グラム、収率:90.3%、HPLCによる純度:>99%)。
1b.アデノシン,5’-S-メチル-5’-チオ-,2’,3’-ジアセテート(化合物#68)の合成:
スキーム:
合成手順:滴下漏斗、不活性ガス入口/出口及び温度計を備え、オーブン乾燥した50ml三口フラスコに、5’-S-メチル-5’-チオ-アデノシン(1.0グラム、0.0033モル、1.0モル当量)及び無水ピリジン(10ml)を入れた。反応セットを氷/塩浴に入れ、撹拌を開始した。溶液の温度が0℃に下がった時点で、無水酢酸(10ml、0.105モル、31.8モル当量)を15分間滴下し、無水酢酸の添加中、反応混合物の温度を5℃以下に維持した。反応混合物を5~10℃で6時間撹拌した。氷冷水(100ml)を加えて過剰の無水酢酸をクエンチし、10wt%のNaHCO
3水溶液を加えてpHを7に調整した。水性混合物を酢酸エチル(2×100ml)で抽出した。合わせた酢酸エチル抽出物を無水Na
2SO
4(1グラム)で乾燥させ、250mlの丸底フラスコに濾過する。濾液を減圧下、35~40℃で乾燥するまで濃縮して、粗生成物を淡黄色のシロップ状の液体として得た後、酢酸エチルとヘキサンの混合物(1:3 v/v)を使用してシリカゲルカラムに通すことによって、高純度の生成物をオフホワイトの結晶として得た(1.08グラム、収率:87%、HPLCによる純度:>99%)。
1c.アデノシン,5’-Se-メチル-5’-セリノ-,2’,3’-ジプロピオネート(化合物#69)の合成:
スキーム:
合成手順:滴下漏斗、不活性ガス入口/出口及び温度計を備え、オーブン乾燥した50mlの三口フラスコに、5’-Se-メチル-5’-セレノ-アデノシン(1.0グラム、0.0029モル、1.0モル当量)及び無水ピリジン(10ml)を入れた。反応セットを氷/塩浴に入れ、撹拌を開始した。溶液の温度が0℃に下がった時点で、プロピオン酸無水物(10ml、0.078モル、27.0モル当量)を15分間滴下し、プロピオン酸無水物の添加中、反応混合物の温度を5℃以下に維持した。反応混合物を5~10℃で6時間撹拌した。氷冷水(100ml)を加えて過剰のプロピオン酸無水物をクエンチした後、10wt%のNaHCO
3水溶液を加えてpHを7に調整した。水性混合物を酢酸エチル(2×100ml)で抽出した。合わせた酢酸エチル抽出物を無水Na
2SO
4(1グラム)で乾燥させ、250mlの丸底フラスコに濾過する。濾液を減圧下、35~40℃で乾燥するまで濃縮して、粗生成物を淡黄色のシロップ状の液体として得た後、酢酸エチルとヘキサンの混合物(1:3 v/v)を使用してシリカゲルカラムに通すことによって、高純度の生成物をオフホワイトの結晶として得た(1.18グラム、収率:89.3%、HPLCによる純度:>99%)。
1d.アデノシン,5’-Se-メチル-5’-セリノ-,2’,3’-ジブタノエート(化合物#70)の合成:
スキーム:
合成手順:滴下漏斗、不活性ガス入口/出口及び温度計を備え、オーブン乾燥した50mlの三口フラスコに、5’-Se-メチル-5’-セレノ-アデノシン(1.0グラム、0.0029モル、1.0モル当量)及び無水ピリジン(10ml)を入れた。反応セットを氷/塩浴に入れ、撹拌を開始した。溶液の温度が0℃に下がった時点で、無水酪酸(10ml、0.078モル、27.0モル当量)を15分間滴下し、無水酪酸の添加中、反応混合物の温度を5℃未満に維持した。反応混合物を5~10℃で6時間撹拌した。氷冷水(100ml)を加えて過剰の無水酪酸をクエンチし、10wt%のNaHCO
3水溶液を加えてpHを7に調整した。水性混合物を酢酸エチル(2×100ml)で抽出した。合わせた酢酸エチル抽出物を無水Na
2SO
4(1グラム)で乾燥させ、250mlの丸底フラスコに濾過する。濾液を減圧下、35~40℃で乾燥するまで濃縮し、粗生成物を淡黄色のシロップ状の液体として得た後、酢酸エチルとヘキサンの混合物(1:3 v/v)を使用してシリカゲルカラムに通すことによって、高純度の生成物をオフホワイトの結晶として得た(1.20グラム、収率:85.7%、HPLCによる純度:>99%)。
1e.アデノシン,5’-Se-メチル-5’-セレノ-,環状2’,3’-カーボネート(化合物#50)の合成:
スキーム:
合成手順:滴下漏斗、不活性ガス入口/出口及び温度計を備え、オーブン乾燥した50ml三口フラスコに、5’-Se-メチル-5’-セレノ-アデノシン(1.0グラム、0.0029モル、1.0モル当量)及び無水ジメチルホルムアミド(20ml)を入れた。反応セットを氷/塩浴に入れ、撹拌を開始した。溶液の温度が0℃に下がった時点で、カルボニルジイミダゾール(CDI、0.57グラム、0.0035モル、1.21モル当量)を5℃未満で加えた。反応混合物をゆっくりと室温まで温め、次いでアルゴンガス雰囲気下、同じ温度で反応混合物を4時間撹拌した。溶媒を減圧下で除去して残渣を取得し、それをクロロホルム(5ml)とエタノール(数滴)の混合物に溶解して透明な溶液を取得し、有機層を1%の酢酸水溶液(2×1ml)で洗浄し、無水Na
2SO
4(1グラム)で乾燥させ、250ml丸底フラスコに濾過した。濾液を25~30℃の減圧下で乾燥するまで濃縮し、粗生成物を淡黄色のシロップ状の液体として得た。粗生成物を、エタノール/水混合物(1:1 v/v)の混合物に溶かし、減圧下、45~50℃で乾燥するまで濃縮して残渣を取得し、ヘキサン(25ml)を加え、10分間撹拌し、その後、減圧下、30~35℃で乾燥するまで濃縮して、所望の生成物をオフホワイトの結晶として得た(1.02グラム、収率:95.3%、HPLCによる純度:>99%)。
1f.アデノシン,5’-Se-メチル-5’-セレノ-,2’-モルホリノカルバメート及びアデノシン,5’-Se-メチル-5’-セレノ-,3’-モルホリノカルバメートの領域異性体混合物(化合物#53)の合成:
スキーム:
合成手順:滴下漏斗、不活性ガス入口/出口、及び温度計を備え、オーブン乾燥した50mlの三口フラスコに、アデノシン,5’-Se-メチル-5’-セレノ-,環状2’,3’-カルボネート(1.0グラム、0.0027モル、1.0モル当量)及び無水ジメチルホルムアミド(10ml)を入れた。モルホリン(0.26グラム、0.0029モル、1.1モル当量)を20~25℃で添加した。反応混合物を室温で1時間撹拌し、次いで45~50℃で減圧下、乾燥するまで濃縮して残渣を取得し、ヘキサン(25ml)を加え、10分間撹拌して所望の領域異性体混合生成物をオフホワイトの固体として沈殿させた(1.12グラム、収率:91%、HPLCによる純度:>99%)。
実施例2.
表1に挙げる合成化合物を、本明細書に記載の実施例におけるグルコース産生及び細胞生存率に対する効果について細胞培養(in vitro)で試験した。特に、試験した細胞は、ヒト肝細胞癌HepG2及びラット肝臓H4IIE肝細胞であった。
材料及び方法
細胞株及び化合物
ヒト肝癌HepG2及びラット肝癌H4IIE細胞は、アメリカ合衆国培養細胞系統保存機関(ATCC,Manassas,Virginia)から購入した。HepG2細胞及びH4IIE細胞は、10%FBSを添加したイーグル最小必須培地(EMEM)で培養した。
化合物#43及び表1に挙げた他の化合物は、合成するか、市販品供給元から入手した(入手可能な場合)。すべての被験化合物の純度は、HPLCで測定して≧99%であることを確認した。これらすべての化合物をDMSOまたは水に溶解して、実験用に126.7または12.67mMのストック溶液を得た。メトホルミン及びインスリンはシグマから購入した。
グルコース産生アッセイ
等しい数のヒトHepG2細胞またはラットH4IIE細胞(1~1.5×105細胞/ウェル)を、24時間、10%FBS含有培地中の96ウェルプレートに播種した。その後、細胞をPBSで2回洗浄し、100μlのグルコース産生培地(20mM乳酸ナトリウム、2mMピルビン酸ナトリウム及び5mM HEPESを補充したグルコースフリー、フェノールレッドフリーDMEM培地中の様々な濃度の化合物、メトホルミンまたはインスリンで、37℃、24時間(H4IIE細胞のみ)または48時間(HepG2細胞のみ)処置した。また、細胞を0.24%DMSO(実験で使用したDMSO溶媒の最大量)存在下でインキュベートした。
上記の処置の後、50μlの培養培地を回収し、製造元のプロトコールに従ってMolecular Probes Amplex Redグルコースアッセイキット(カタログ番号A22189)を使用してグルコース分析に供した。上記の化合物処置後の培養プレートの細胞数または生存率は、製造元のプロトコール及び指示に従って、PromegaのCellTiter96(登録商標)AQueous One Solution細胞増殖アッセイキットを使用して測定した。簡潔に述べると、グルコースアッセイのために50μlの培地を除去した後、細胞をAQueous One溶液(25μlのストック溶液及び50μlの予熱PBS/ウェルあたり)で37℃、1時間インキュベートし、各試料におけるOD490nmの吸光度をBio-Tekマイクロプレートリーダーで測定した。培養ウェルの細胞生存率を、培養細胞のOD490nmからプレーン培養培地(細胞の播種なし)のOD490nmを差し引くことにより測定した。培養細胞におけるグルコース産生は、培養培地のグルコース濃度を各ウェルの細胞生存率で正規化することにより取得した。上記の分析のために、各処置あたり少なくとも3つの試料を調べた。データは、これらの試料の平均値±SEMとして提示する。実験は少なくとも2回繰り返した。
グルコース産生の阻害に関する化合物#43またはメトホルミンの半数最大阻害濃度(IC50値)は、ED50Plus v1.0オンラインソフトウェアを使用して測定した。
結果及び考察
1.HepG2細胞のグルコース産生に対するインスリン及び化合物溶媒DMSOの効果
図1の左パネルに示すように、それぞれ10nM及び100nMのインスリン処置により、HepG2細胞におけるグルコース産生が20%及び30%減少したが、一方、最大体積を使用した化合物溶媒DMSOは、培養HepG2細胞のグルコースレベルに影響を及ぼさなかった。これらの結果は、HepG2細胞がグルコース産生の阻害においてインスリンに応答し、グルコース産生に対する被験化合物の観察された効果がDMSOの潜在的な効果によるものではないことを示唆している。これらの結果は、インスリン様活性を有し、肝細胞においてグルコース産生を阻害することができる化合物のスクリーニングのための適切な細胞系をHepG2細胞が構成することを確立する。
2.化合物#43はインスリンを模倣して、HepG2細胞のグルコース産生を阻害することができる
図1に示すように、無血清条件下、試験した用量の化合物#43の存在下でのHepG2細胞のインキュベーションにより、培地中のグルコースレベルが低下した。化合物#43(3.8μM)で処置した後に観察されたHepG2細胞によるグルコース産生の減少は、インスリン(100nM)を使用した場合に達成されたものと比較可能であったが、より高い用量の化合物#43(7.6、15.2及び30.4μM)は、100nMインスリンよりもさらに強力であった。さらに、試験したすべての用量での化合物#43処置後、HepG2細胞において細胞生存率の有意な減少は観察されなかった(データは示さず)。これらの結果は、化合物#43がHepG2細胞のグルコース産生を阻害することができるインスリン模倣薬であることを示唆している。
3.化合物#43自体は、HepG2細胞でのグルコース産生の阻害において、3つの化合物の組み合わせであるCDEよりも高いエンドポイント効力を示した
試験したすべての用量での化合物CDEによるHepG2細胞の処置(1:1:1のC/D/E比)によってもグルコース産生は阻害され、それらは100nMインスリンと比較可能な効力を有していた。3.8μMの用量の化合物#43は、HepG2細胞でのグルコース産生の阻害において、CDEの組み合わせ製品(3.8μMの各個々の化合物を含む)と同じくらい強力であった。しかしながら、7.6μM以上の用量の化合物#43は、HepG2細胞でのグルコース産生の阻害においてCDEよりも強力であった。これらの結果は、HepG2細胞でのグルコース産生の阻害において、化合物#43自体が3つの化合物の組み合わせであるCDEよりも高いエンドポイント効力を示したことを示した。化合物CDEにおけるセレン濃度は各投与点で化合物#43の3倍であることに留意すると、これらの結果は、セレン分子に加えて化合物#43の1つ以上の構造的特徴が、少なくとも特定の状況において、その活性に寄与し得ることを示唆している可能性がある。
4.化合物#43のセレン分子の代わりに硫黄を使用すると、HepG2でのグルコース産生の阻害が抑止される
図1の右側のパネルに示すように、硫黄含有化合物である化合物#68(化合物#43の硫黄類似体)または化合物#64(SAM)、ならびにリストに挙げた他の非セレン化合物による細胞の処置は、HepG2細胞でのグルコース産生の低下にほとんど影響を与えなかった。グルコース産生の阻害における化合物#43と#68の顕著な違いは、HepG2細胞でのグルコース産生の阻害において、化合物#43のセレン分子がその機能に寄与していることを示している。
5.HepG2細胞におけるグルコース産生の阻害に対するセレン化合物の異なる効果
上記のように、セレン含有化合物C、D、及びEの等モル混合物(CDE)は、示した用量でヒトHepG2細胞のグルコース産生を阻害した。図1のX軸に示す化合物の濃度は、混合物中の各Se含有化合物のSe濃度を指しており、合計Se濃度は、実際にはX軸上に示す濃度の3倍であることに留意されたい。これは、これらの実験で試験した単一分子候補のそれぞれと混合物成分の直接比較を容易にするために行った。
グルコース産生の阻害がセレンのみによるものである場合、CDEは化合物#43よりも3倍多いセレンを含むため、後者よりもよりロバストな応答を生じるであろうと予想され得る。その点については、明らかにそうではない。
図1(左側のパネル)は、化合物C、D、43、50、53、69及び70のそれぞれがグルコース産生を阻害し、その中で化合物43が最も強力であったことを示している。さらに、化合物EはHepG2細胞のグルコース産生を刺激することがわかった。
まとめると、これらの結果は以下のことを示唆している:
(i)化合物中のセレン分子は、グルコース産生の効果的な阻害に必要である(図1の左パネルの化合物#43を、その正確な硫黄類似体である右パネルの化合物#68と比較されたい)。
(ii)しかしながら、セレン分子だけではグルコース産生の阻害には不十分な場合がある(いくつかの場合では、セレンを含有する構造的に類似した化合物は、より低いまたは反対の効果を示した)。
(iii)セレン分子の濃度だけを高めても効力は増強されない場合がある(CDEのセレン濃度は化合物#43のセレン濃度の3倍であるが、それほど強力ではない)。
(iv)試験した化合物の中で、化合物#43は、肝細胞のグルコース産生の阻害において最も高い効力を示した。
6.リストに挙げたセレン化合物の構造的特徴の分析により、それらの活性に寄与し得る化学基を決定する
化合物CとDとの比較は、5’メチルセレノ基と5’セレノホモシステインが同様のグルコース産生阻害を提供し得ることを示している。化合物43、50、53、69、及び70の比較は、2’及び3’位のジアセチルエステル(#43)が環状カルボネート(化合物#50)、モルホリノカルボキシレート(#53)、ジプロパノイルエステル(#69)、及びジブタノイルエステル(#70)に比べて、より高いグルコース産生阻害を提供することを示している。アデニン、アデノシン及びアデノシンのいくつかの化学的バリアントは、HepG2細胞におけるグルコース産生を阻害しなかった。さらに、2つのアデニリルシクラーゼ阻害剤(#59及び61)もグルコース産生を阻害しなかったが、このことは、グルコース産生の阻害における化合物#43の作用が、細胞のAMPレベルの低下によるものではないと思われることを示している(Hardie DG. Cell Metabolism 2013:17(3):313-314)。同時に、この結果は、アデニンとセレン分子に加えて1つ以上の特徴が、少なくとも特定の状況において活性に寄与し得ることを示唆している。
7.HepG2及びラットH4IIE細胞でのグルコース産生の阻害における化合物#43とメトホルミン(周知の抗糖尿病薬)の比較研究
上記のように、HepG2細胞におけるロバストなグルコース産生阻害が、化合物#43処置後に観察された。この化合物の効果を、周知の抗糖尿病薬メトホルミンと比較した。上記及び図2の上部パネルに示すように、化合物#43処置後のHepG2細胞におけるグルコースレベルの用量依存的な低下が観察され、IC50(半数最大阻害濃度)値は15.5μMであった。図からわかるように、500μMの用量のメトホルミンは、HepG2細胞のグルコース産生を阻害しなかったが、代わりにいくつかの刺激効果を示した。高用量のメトホルミン(0.5~4mM)は、これらの細胞に対する細胞毒性を示した(データは示さず)。これらの結果は、インスリンと同様に、化合物#43がHepG2細胞のグルコース産生を阻害することができることを示している。
ラットH4IIE肝細胞は、メトホルミンに応答してグルコース産生を低下させることが報告されている。したがって、このラット肝細胞株を使用して、グルコース産生に対する化合物#43の阻害活性をさらに確認し、化合物#43の効力をメトホルミンと比較した。図2に示すように、それぞれ化合物#43及びメトホルミンで処置すると、無血清条件下でH4IIE細胞のグルコース産生が用量依存的に減少した。これらの化合物の毒性効果は、試験した用量において細胞生存率に対して観察されなかった(データは示さず)。化合物#43のIC50は17.8μMであり、これはHepG2細胞におけるIC50とほぼ同一である。対照的に、36.25μMの用量のメトホルミンは阻害活性をほとんどまたはまったく示さず、この実験におけるメトホルミンのIC50は275μMであった。これらの結果は、培養ラット肝細胞におけるグルコース産生の阻害において、化合物#43がメトホルミンよりも強力(少なくとも15倍強力)であることを示唆している。
まとめると、化合物#43、50、53、69及び70はすべて、培養肝細胞におけるグルコース産生阻害において活性を示した。しかしながら、化合物#43は、グルコース産生に対する阻害剤活性を試験した中で、はるかに最も強力な単一化合物であり、その活性は、HepG2細胞における高インスリン用量(100nM)の活性を上回るものであった。
さらに、化合物#43は、試験した両方の肝細胞株において、ビグアナイド薬であるメトホルミンよりもはるかに強力であることが示された。メトホルミンは現在、2型糖尿病の治療に使用される第一選択薬である。
実施例3:インスリン抵抗性及び糖尿病性レプチン受容体(Lepr)偶発ヌル変異マウスの血流中のグルコース及び/またはHbA1cレベルの調節ならびに耐糖能の向上における、化合物#43及び密接に関連するセレノ有機化合物(#C、#50、#69及び#70)、ならびに化合物#43の硫黄類似体(化合物#68)を用いた研究
材料及び方法
化合物
化合物#43、#C、#50、#68、#69及び#70を、Chemistry Laboratory of Alltech,Incにおいて合成した。試験したすべての化合物の純度は、HPLCによる測定で、≧99%であることが確認された。
動物
5~10週齢の雄の糖尿病性偶発変異(レプチン受容体変異)Leprdb/dbマウス(C57BL/6J系統)は、Jackson Laboratory(Bar Harbor,Maine)から購入し、病原体フリーの飼育室に収容し、餌と水を自由に摂取させた。
化合物#43及びその他の化合物による慢性処置
38日齢の雄のLeprdb/dbマウスに、0.2%DMSO、化合物#C、化合物#50、化合物#68、及び/または化合物#43(体重1キログラムあたり25μgセレンまたは硫黄当量の各化合物、滅菌生理食塩水で希釈)を含有する生理食塩水(0.09%NaCl)を、43~90日間の範囲の期間、毎日腹腔内(ip)注射した。上記の処置マウスの体重を、天秤を使用して毎日記録し、目に見える異常な動物の肉眼的形態及び歩行行動を毎日モニタリングした。処置後、動物を一晩絶食させ、その後、血糖値アッセイまたはHbA1cアッセイ、糖負荷試験または組織採取に供した。
化合物#43、#69及び#70の潜在的な抗糖尿病効果の比較研究のために、41日齢の雄のLeprdb/dbマウスに、0.2%DMSO、化合物#43、化合物#69、または化合物#70(体重1キログラムあたり25μgセレンの各化合物、滅菌生理食塩水で希釈)を含有する生理食塩水(0.09%NaCl)を、毎日腹腔内(ip)注射した。43日間の処置後、動物を一晩絶食させ、その後、血糖値試験及び糖負荷試験に供した。これらの化合物を90日間毎日処置した後、これらの動物の血清を収集し、次いで血中HbA1cアッセイに供した。
化合物#43の急性処置
一晩の絶食後、8~10週齢のLeprdb/dbマウスに、0.2%DMSOまたは化合物#43(体重1キログラムあたり、0.0054、0.054、0.54、または5.4mgの化合物#43(化合物のストックを滅菌生理食塩水中に希釈した)を含有する生理食塩水(0.09%NaCl)を腹腔内注射した。生理食塩水または化合物の注射後、マウスをケージに戻し、1、2、3、5、及び8時間にわたって、水を自由に摂取させたが、餌は与えなかった。注射後の各時点で、グルコースアッセイのために各マウスの尾から少量の血液を採取した。
非絶食条件下の追加の6週齢Leprdb/dbマウスに、0.2%DMSOを含有する生理食塩水(0.09%NaCl)、または化合物#43の単回投与(5.4mg化合物/kg体重)を腹腔内注射した。注射後、動物をケージに戻し、水と餌を自由に摂取させた。24時間後、グルコースアッセイのためにマウスの尾から少量の血液を採取した。
血糖値アッセイ
生理食塩水または化合物で処置した後、またはグルコースのボーラスを注射した後、マウスの尾の先端を切り取ることにより、各マウス由来の少量の血液を採取した。血糖値は、600mg/dLのグルコース測定の最大能力を有する血糖値測定器を使用して測定した。
糖負荷試験
糖負荷試験は、以前に記載されたように実施した(Li et al,Int J Biol Sci 2008;4:29-36)。簡潔に述べると、生理食塩水または化合物で処置した後、一晩絶食させたLeprdb/dbマウスに2グラム/kg体重の20%D-グルコースを腹腔内注射した。最大グルコース測定能力600mg/dLの血糖値測定器を使用して、グルコースを注射してから時間0(グルコース注射の直前)、0.25、0.5、1、及び2時間後の血糖値を測定した。このため、データ分析では、600mg/dLを超える血糖値は600mg/dLとしてカウントした。
血中及び血清HbA1cアッセイ
生理食塩水または化合物での処置後、マウス尾からの少量の血液をEDTAコーティングしたエッペンドルフチューブ(血液凝固を防ぐため)(Fisher Scientific)に回収し、次いでCrystal ChemまたはKamiyaのマウス糖化ヘモグロビンA1c ELISAキットを使用して、製造元のプロトコールに従ってHbA1cアッセイに供した。また、最終処置後、Kamiya Biomeidcal CompanyのマウスHbA1cキットを使用して、製造元のプロトコールに従ってマウス血清を回収した。
統計解析
該当する場合、スチューデントt検定を使用して、生理食塩水処置群と化合物処置群との差異の統計的有意性を判定し、0.05未満のP値を有意とみなした。データは、図に示すマウスの数の平均値±SEMとして提示する。
結果と考察
Leprdb/dbマウスは、レプチン受容体遺伝子(Lepr)のすべての既知のアイソフォームを欠損している。このホモ接合性マウスモデルは、耐糖能障害、インスリン感受性の低下、高血糖症、及び高インスリン血症を有する高悪性度II型糖尿病マウスモデルである。これらのマウスは、約3~4週齢で肉眼的肥満、10~14日で始まる血漿インスリンの上昇、及び約4~8週齢で高血糖症(すなわち、高血糖値)を発症する(Coleman DL. 1978 Diabetologia 14:141-8)。
in vitro研究により、化合物#43が、インスリンを模倣するが迂回して、化合物#Cまたは#50などの密接に関連する化合物よりもはるかに大きな効力でグルコース産生を阻害することが示された。さらに、化合物#43の硫黄含有類似体(化合物#68)は、HepG2細胞におけるグルコース産生に対してほとんどまたは全く阻害効果を有していなかった(図1~2)。したがって、インスリン抵抗性Leprdb/dbマウスは、血流中のグルコースを潜在的に低下させ、重度の糖尿病的バックグラウンドに対するインスリン感受性及び耐糖能を改善するうえでの実験化合物の使用を調査する理想的なin vivoモデル系である。
1.慢性処置後のLeprdb/dbマウスの高血糖症に対して試験した3つのセレノ有機化合物(#C、#50、#43)の中で最も強力な化合物である化合物#43
Leprdb/dbマウスで示すように、化合物の投与レジメンを採用して、高血糖症の治療における試験化合物の潜在的な役割を調査した。高血糖症の発症(生後約4~8週間で発症)前後に化合物を腹腔内注射することにより、マウスに毎日処置を施した。3つの化合物(すなわち、化合物#43、化合物#C及び化合物#50、同一濃度のセレンを送達)を43~52日間毎日注射して、これらのセレノ有機化合物がインスリン抵抗性のマウスの高血糖症に対して測定可能な効果を有するかどうかを調べた。
試験したすべての化合物による処置は、これらの変異マウスの体重増加に影響を与えなかったことがわかり(データは示さず)、このことは、試験したこれらの化合物が、Leprdb/dbマウスにおいて、表示した食物の消費に対する食欲の異常な増加の阻害効果をほとんどまたは全く有さない可能性が高いことを示している。また、処置期間中に生理食塩水で処置した(対照)マウスと、化合物で処置したLeprdb/dbマウスの間で動物の肉眼的形態及び歩行行動に目に見える差異はなかった(データは示さず)。これらの結果は、試験した用量でのこれらの化合物が、動物の行動または活動に対して毒性作用を及ぼさないことを示している。
化合物#C、#50、及び#43のうち、化合物#43で処置したLeprdb/dbマウスの血糖値は、同年齢の通常の野生型マウス(約100mg/dL、データは示さず)に比べてなお高かったにもかかわらず、化合物#43での処置は、Leprdb/dbマウスの血流中のグルコースレベルの最も有意な減少、すなわち対照に比べて約45%の減少をもたらした(図3左パネル参照)。さらに、これらの結果は、化合物#43がこの重度のII型糖尿病マウスモデルの血流中のグルコースレベルを有意に低下させることができることを明らかに示しており、この化合物の高血糖症予防の可能性を示している。さらに、これらの結果は、高血糖症に対するセレノ有機化合物の示差的効果の良いエビデンスを提供しており、化合物#43が高血糖症の治療に対する最も強力な化合物(これら3つの試験した化合物の中で)であることを示している。
さらに、化合物#C、#50及び#43で処置したLeprdb/dbマウスから血清を採取し、HbA1c試験に供した。HbA1cレベルは、過去2~3か月にわたる血糖濃度の長期的な指標であり、糖尿病患者の血糖値の履歴をモニタリングするために臨床医学で広く使用されている。図3に示すように、化合物#C、#50及び#43の中で、化合物#43による処置でのみ、生理食塩水処置群と比較した場合の血清HbA1cレベルの有意な減少(約20%減少)が生じた。これらの結果は、マウスの空腹時血糖値(図3の左パネル)及びin vitroでのグルコース産生(HepG2細胞、図1)に対するこれら3つの化合物の上記の観察された示差的効果と符合する。
したがって、これらの結果は、高血糖症に対するセレノ有機化合物の示差的効果が存在し、化合物#43が、これら3つのセレン含有化合物(#C、#50、及び#43)の中で最も強力な化合物であり、インスリン抵抗性の被験体における高血糖症の治療に対して明らかな潜在能力を有することへのin vivoでのエビデンスを与えるものとなっている。
2.化合物#43のセレン分子を試験用量の硫黄分子で置換すると、慢性処置後のLeprdb/dbマウスの高血糖症に対して有意な効果はない
in vitroでの研究では、化合物#68ではなく化合物#43がHepG2細胞におけるグルコース産生をロバストに阻害することが示された(図1)。上記の研究は、化合物#43がLeprdb/dbマウスのグルコース出力及びHbA1cレベルを低下させることができることを示している(図3)。Leprdb/dbマウスにおける化合物#43の抗糖尿病効果をさらに確認し、化合物#43のセレン分子がこの抗高血糖症効果に必要かどうかを調べるために、38日齢の雄のLeprdb/dbマウスに、等量のセレンまたは硫黄を含有する化合物#43及びその直接的な硫黄含有類似体である化合物#68を、腹腔内注射により、3か月間、毎日処置した。
ここでも、3か月間化合物で処置した後、Leprdb/dbマウスに目に見える形態学的異常、歩行行動異常または体重変化はなかったが、このことは、試験した用量の化合物#43または#68が、これらのマウスに対する明白な毒性効果を有していなかったことを示している。しかしながら、化合物#43を3か月間注射すると空腹時血糖値が統計的に有意に減少したが、一方、化合物#68は血糖値を有意に低下させなかった(図4、左パネル)。さらに劇的に、3か月間の化合物#43処置後の空腹時血糖値は、約135mg/dLに低下したが、これは、正常な非糖尿病/肥満マウスで報告されている血糖値(約100mg/dL)に近いものである。これらの血糖値の低下と一致して、3か月間の化合物#43処置後のLeprdb/dbマウス由来の血液試料中のHbA1cレベルも、対照(生理食塩水処置)群に比べて有意に減少した(約30%減少)。しかしながら、化合物#68処置は、Leprdb/dbマウスの血中HbA1cレベルに影響を及ぼさなかった。化合物#43によるこの90日間の処置後のLeprdb/dbマウスのHbA1cレベルの低下の程度(図4)は、42日間の化合物#43処置後のLeprdb/dbマウスに比べて、より顕著であると考えられる(図3)。
総合すると、これらのin vivoでの結果は、化合物#43が高悪性度II型糖尿病のマウスモデルにおいて血糖レベル及びHbA1cレベルを有意に低下させることができるという発見をさらに確証付けるものであり、糖尿病患者の高血糖症の治療における化合物#43の潜在能力を示している。さらに、結果は、化合物#43のセレン原子を硫黄原子で置換した後、化合物#43の抗糖尿病性が失われることも確認した。
3.化合物#43は、Leprdb/dbマウスにおいて、慢性処置後、化合物#69及び#70に比べて、より高い抗高血糖症性を示した
in vitroでの研究では、化合物#43の2’,3’位のジアセチル基をジプロパノイル基(化合物#69)またはブタノイル基(化合物#70)で置換すると、HepG2細胞におけるグルコース産生の阻害における化合物#43の活性が減弱することが示された(図2)。上記の研究は、化合物#43がLeprdb/dbマウスのグルコース出力及びHbA1cレベルを有意に低下させることができることを示している(図3~図4)。Leprdb/dbマウスにおける化合物#43の抗糖尿病効果をさらに確認し、この抗高血糖症効果に対する化合物#43のジアセチル基の寄与を調べるために、41日齢の雄のLeprdb/dbマウスに、等量のセレンを含有する化合物#43、#69及び#70を、腹腔内注射により、43日間及び90日間、毎日処置した。
ここでも、これらの3つの化合物で90日間処置した後、Leprdb/dbマウスに目に見える形態学的異常、歩行行動異常または体重変化はなかったが、このことは、試験した用量の化合物#43、#69または#70が、これらのマウスに対する明白な毒性効果を有していなかったことを示している。化合物#43を43日間注射すると、空腹時血糖値が統計的に有意に減少することが明らかとなった(生理食塩水群での約290mg/dLから188mg/dLへの約35%の減少)(図5左パネル)。化合物#43のジアセチル基をジブタノイル基(化合物#70)で置換した場合も、Leprdb/dbマウスの空腹時血糖値は低下したが、化合物#43に比べると有意ではなかった(図5左パネル)。化合物#43のジアセチル基をジプロパノイル基(化合物#69)に置換すると、Leprdb/dbマウスの空腹時血糖値がわずかに増加した(図5左パネル)。上記の観察結果をさらに検証するために、これらのLeprdb/dbマウスに、腹腔内注射により、等量のセレンを含有する化合物#43、#69及び#70を、さらに47日間毎日投与し続け(毎日の化合物処置を合計90日間)、血清を回収し、血中HbA1cアッセイに供した。図5の右パネルに示すように、化合物#43処置により、HbA1cレベルが有意に減少した(図5、右パネル)。最初の評価では、化合物#70処置でもHbA1cレベルが約50%減少することが示され(図5、右パネル)、一方、化合物#69処置ではHbA1cレベルの有意な減少は生じず(図5、右パネル);これらのデータをさらに検討した結果、化合物#69と化合物#70のいずれにも有意な効果は認められなかった(図5、右パネル)。総合すると、これらの結果は、化合物#43がインスリン抵抗性の被験体の高血糖症の治療に大きな可能性を秘めていることを示唆している。さらに、結果は、化合物#43の2’,3’位のジアセチル基がin vivoでの抗高血糖症機能に寄与しており、特定の状況では、1つまたは2つの炭素原子によるジアセチル基の延長がグルコース恒常性に対して悪影響を与えることを示している。
4.化合物#43の急性処置により、Leprdb/dbマウスの血糖値が用量依存的に低下した
上記の研究は、化合物#43での慢性処置により、血糖値レベル及びHbA1cレベルを有意に低下させることができることを示した。有効量の範囲を決定し、化合物#43に対する応答期間を確立するために、8~10週齢のLeprdb/db雄マウスを一晩絶食させ、次いで生理食塩水(化合物#43ストック溶媒の最大注入量である0.2%DMSOを含有する)、0.0054、0.054、0.54及び5.4mg/kg体重の化合物#43(ストック化合物を生理食塩水で希釈して作成)を腹腔内注射した。1、2、3、5及び8時間目の注射の直前と直後のLeprdb/dbマウス(空腹条件ではあるが水は自由に摂取できる)の血糖値を調べ、結果の血糖値を、各時点での個々の動物のそれぞれについてプロットした。
上記の用量の化合物#43の急性単回注射は、肉眼的形態及び歩行行動に対して目に見える毒性効果を引き起こさなかった。生理食塩水で処置したLeprdb/dbマウスでは、8時間の試験期間中に血糖値のわずかな低下(約50~70mg/dL)しかなく(図6)、これは、これらのLeprdb/dbマウスがグルコース除去能力に欠陥を示すという事実と合致している。しかしながら、試験したすべての用量での化合物#43処置は、注射後2時間または3時間の最低用量の化合物#43処置(この場合のグルコースレベルの減少は統計的に有意となる間際である)を除いては、血糖値が有意に減少した(各時点での生理食塩水群に比べて)(図6)。結果は、試験したすべての用量の化合物#43が、単回注射の1時間後に血糖値を低下させるのに有効であることを示した。これは、化合物#43が、グルコース低下効果を引き出すために、短期間(すなわち1時間)以内にin vivoで関連する標的組織に到達することができることを示している。絶食条件下での血糖値の低下における化合物#43の活性は、投与後2~5時間でピークに達し、この効果は少なくともさらに3時間維持された。試験した動物(一晩絶食させ、試験した8時間の間絶食させ続けた)をさらに絶食させることができなかったため、化合物#43の作用の最大有効期間はこれらの実験からは決定されなかった。
これらの結果は、1000倍の濃度範囲にわたる化合物#43による急性処置が、インスリン抵抗性及び2型糖尿病の広く使用されている動物モデルにおいて血糖値を有意に低下させることを示している。化合物#43は速効性(治療後≦1時間)であり、少なくとも8時間は活性を維持する。
5.化合物#43の急性処置は、より若いLeprdb/dbマウスの高血糖症への進行を減弱させる
上述のように、Leprdb/dbマウスは、10~14日齢から始まる血漿インスリンの上昇及びおよそ4~8週齢での高血糖症(すなわち、高血糖値)を示す(Coleman DL. 1978 Diabetologia 14:141-8)。化合物#43が高血糖症の発症を減弱させる潜在能力を有するかどうかを試験するために、より若いマウスに化合物#43の単回用量を急性注射で投与した。簡潔に述べると、通常の摂食条件下の6週齢のLeprdb/db雄マウスに、0.2%DMSOまたは化合物#43を含有する生理食塩水を5.4mg/kg体重の用量で1回腹腔内注射した。処置の24時間後、これらのマウスに食物及び水を自由に摂取させている間に、これらのマウスの血糖値を測定した。
図7に示すように、生理食塩水処置の24時間後のこれらの若いLeprdb/dbマウスの血糖値は有意に増加したが(約20%増加)、これは、これらのマウスが依然として高血糖症を発症するプロセス内にあることを示している。対照的に、化合物#43を処置すると、Leprdb/dbマウスの血糖値は有意に減少した(約20%)(図7)。
これらの結果は、化合物#43が高血糖症の発症を減弱させる潜在能力を有することを示唆している。さらに、これらの研究は、グルコース出力の低下における化合物#43の有効性が、摂食条件下のこれらの糖尿病マウスにおいて、少なくとも24時間継続する可能性が高いことも示している。
6.化合物#43投与後の糖尿病Leprdb/dbマウスの耐糖能の増強
糖負荷試験は、血糖が高く急速に上昇した後(例えば、通常は食事後)に身体がグルコースを処理する方法における異常を同定する。インスリンは、肝臓におけるグルコース産生の阻害だけでなく、筋肉、肝臓、脂肪細胞でのグルコースの取り込み、貯蔵、代謝でも重要な役割を果たし、血流中のグルコースレベルを低下させる。
糖尿病患者は、インスリンを産生できないか、またはインスリンに効率的に応答できず、グルコース恒常性を維持できないため、非常に低い耐糖能を有する。本明細書に記載のin vitroでの研究は、化合物#43がインスリンを模倣できるだけでなく、インスリンを迂回してグルコース産生を阻害することができることを示している(図1~図2)。Leprdb/dbマウスは、耐糖能障害及びインスリン抵抗性がこれらの変異マウスにおいて提示されるという事実を考慮すると、グルコース恒常性の維持における化合物#43の役割を調べる理想的なマウスII型糖尿病モデルである。したがって、Leprdb/dbマウスへのそれぞれの化合物の腹腔内注射後のマウスの耐糖能の向上に対する化合物#43及び他の構造的に類似した関連するセレン及び硫黄化合物の効果を調べた。
これらの雄マウスに、38日齢で、生理食塩水(0.2%DMSOを含む)、化合物#C、#43、または#50(体重1kgあたり25μgセレンの各化合物)を、43日間腹腔内注射した。処置の最後に、これらのマウスを一晩絶食させ、グルコース(2g/kg体重)を注射し、グルコース注射後0.25時間(15分)、0.5時間(30分)、1時間(60分)及び2時間(120分)時点で、血糖値を測定した。グルコース注射の直前(ゼロ時点と呼ばれる)の血糖値も記録した。
図8Aに示すように、グルコース注射後、生理食塩水で処置したLeprdb/dbマウスでは、以下の試験したすべての時点で0.25時間から始まる血糖値の有意な増加が観察された。本明細書に記載するように、これらの分析に使用した血糖値測定器のグルコース測定限界は600mg/dLであった。したがって、この限界を超えるグルコースレベルは600mg/dLとして記録した。したがって、特に生理食塩水で処置した動物の場合、グルコース注射後の試験した時点での特定の測定値は、真の血糖濃度の過小評価を十分に表している場合がある。
化合物#Cで処置したマウスでは、生理食塩水処置群と同様に、グルコース注射後の試験したすべての時点での血糖値は非常に高いままであった(図8A)。これらの結果は、試験した用量の化合物#Cがこれらのインスリン抵抗性糖尿病マウスの耐糖能を向上させなかったことを示唆している。
化合物#50の処置では、生理食塩水処置群と比較した場合、図8Aにおけるグルコース注射後0.25、0.5または1時間において明らかな減少はなかったものの、グルコース注射後2時間時点でグルコースレベルが有意に減少した。これらの結果は、化合物#50がこれらの糖尿病マウスの耐糖能を向上させるうえで何らかの効果を有する可能性が高いことを示唆している。
対照的に、化合物#43をで処置したLeprdb/dbマウスでは、グルコース注射後0.25、0.5及び1時間での血糖値は、生理食塩水、化合物#Cまたは#50で処置したマウスよりも明らかに低かった(図8A)。血糖値測定器の測定限界により、グルコース注射後のこれらの時点での、他の処置と比較した化合物#43処置マウスにおけるグルコースレベルの低下の程度は、図8Aに示すものよりもはるかに劇的であったと思われる。グルコース注射の2時間後、化合物#43処置Leprdb/dbマウスの血糖値は、生理食塩水、化合物#Cまたは化合物#50で処置した同腹仔よりもはるかに低く(図8A参照)、注射前の血糖値までほぼ完全に回復していた。グルコース注射後2時間での化合物#43処置Leprdb/dbマウスのグルコースレベルの低下は、生理食塩水または化合物#C処置マウスと比較した場合、有意に異なっていた(P<0.001)。これらの結果は、試験した用量での化合物#Cではなく化合物#43が、これらのインスリン抵抗性糖尿病マウスのインスリン作用を、耐糖能の向上によって評価されるように、ほぼ完全に回復させることができ、化合物#50もまた、グルコースクリアランスの向上においていくつかの有益な効果を有し得ることを示唆している。
Leprdb/dbマウスのグルコースクリアランスを向上させる化合物#43の高い潜在能力をさらに確認し、化合物#43のセレン原子がこの効果に必要かどうかを調べるために、38日齢の雄Leprdb/dbマウスに、等量のセレンまたは硫黄をそれぞれ含有する化合物#43または#68を2か月間、腹腔内注射することにより、毎日処置した。処置の終わりに、これらのマウスを一晩絶食させ、上記のように糖負荷試験に供した。
図8Bに示すように、化合物#68で処置したLeprdb/dbマウスにおいて、グルコースの注射後0.25時間で始まる以下の試験したすべての時点における血糖値の有意な増加が観察された。グルコース注射後2時間の時点で血糖値の明らかな低下はなかったが(0.25、0.5、1時間のレベルと比較した場合)、このことは、試験した用量の化合物#68が、これらのインスリン抵抗性糖尿病マウスにおける耐糖能の向上効果をほとんど有していないか、またはまったく有していないことを示している。
化合物#43で処置したLeprdb/dbマウスでは、グルコースチャレンジ注射前の血糖値は、化合物#68で処置したマウスよりも有意に低かった(P<0.05)。これらの結果は、この糖尿病マウスモデルにおける空腹時血糖値の低下において、化合物#43が化合物#68よりも強力であるという上記の観察結果と一致した(図4)。グルコース注射の0.25、0.5及び1時間後の化合物#43処置Leprdb/dbマウスの血糖値は、化合物#68で処置したマウスよりも低かった。グルコース注射の2時間後、化合物#43処置Leprdb/dbマウスの血糖値は、化合物#68で処置した同腹仔よりも有意に低かった(図8B参照、P<0.001)。この実験における化合物#43処置Leprdb/dbマウスのグルコースクリアランス曲線(図8B)は、上記の最初の糖負荷試験で観察された曲線(図8A)とほぼ同一であった。ここでも、血糖値測定器の測定限界により、グルコース注射後のこれらの時点での化合物#68処置マウスと比較した化合物#43処置マウスのグルコースレベルの減少は、図8Bに示すものよりもはるかに劇的であった。いずれにせよ、上記の結果は、試験した用量の化合物#43が耐糖能を劇的に向上させ、化合物#43のセレン原子を硫黄に置換すると、これらのインスリン抵抗性糖尿病Leprdb/dbマウスにおけるグルコースクリアランスの促進能力がほぼ完全に破壊されることをさらに確認するものである。
最後に、化合物#43のリボース基の2’及び3’位の両方のアセチル基をプロパノイルまたはブタノイル基で置換することにより、Leprdb/dbマウスのグルコースクリアランスを向上させることができるかどうかを調べた。41日齢の雄のLeprdb/dbマウスに、等量のセレンを含有する化合物#43、#69または#70を、それぞれ43日間、腹腔内注射によって毎日処置した。処置の終わりに、これらのマウスを一晩絶食させ、上記のように糖負荷試験に供した。
図8Cに示すように、生理食塩水で処置したLeprdb/dbマウスにおいて、グルコースの注射後0.25時間から開始する以下の試験したすべての時点における血糖値の有意な増加が観察された。グルコース注射後1時間の時点の前には血糖値の明らかな減少はなかったが(0.25及び0.5時間のグルコースレベルと比較した場合)、これらのインスリン抵抗性糖尿病マウスでは、グルコース注射後2時間においてグルコースレベルがわずかに減少していた。
化合物#69処置は、生理食塩水処置群と比較した場合、図8Cのグルコース注射後0.25、0.5または1時間における血糖値の明らかな低下はなかったものの、グルコース注射後2時間の時点でグルコースレベルのわずかであるが有意でない低下をもたらした。これらの結果は、化合物#69がこれらの糖尿病マウスの耐糖能を向上させるうえで、ある程度の効果を有し得ることを示唆している。
化合物#70処置マウスでは、グルコース注射前の空腹時血糖値は、生理食塩水処置群よりも低かった(図8C)。グルコース注入後、特に2時間の時点で、生理食塩水処置マウスと比較した場合、化合物#70処置マウスでは血糖値にわずかではあるが有意ではない減少があった。これらの結果は、化合物#69と同様に、試験した用量の化合物#70も、これらのインスリン抵抗性糖尿病マウスの耐糖能を向上させるうえで何らかの効果を有する可能性が高いことを示唆している。
対照的に、化合物#43処置Leprdb/dbマウスにおけるグルコース注射の0.25及び0.5時間後の血糖値は、生理食塩水、化合物#69または#70で処置したマウスよりも明らかに低かった(図8C)。グルコース注射の1時間後、化合物#43処置Leprdb/dbマウスの血糖値は、生理食塩水、化合物#69または化合物#70で処置した同腹仔よりも有意に低かった(図8C)。グルコース注射の2時間後でも、化合物#43処置Leprdb/dbマウスの血糖値は、生理食塩水、化合物#69または化合物#70で処置したマウスよりもはるかに低かった。グルコース注射の2時間後の化合物#43処置Leprdb/dbマウスのグルコースレベルの低下は、生理食塩水処置マウスと比較した場合に有意に異なっていた(P<0.05)。ここでも、血糖値測定器の測定限界のため、グルコース注射後の各時点での、他の処置に比べての化合物#43処置マウスにおけるグルコースレベルの減少の程度は、図8Cに示すものよりもはるかに劇的である可能性が高かった。いずれにせよ、これらの結果は、試験した用量での化合物#43がこれらのインスリン抵抗性糖尿病マウスの耐糖能を劇的に向上させることができることをさらに示している。また、これらの結果は、化合物#69及び#70がグルコースクリアランスの向上に有益な効果をもたらし得ることを示唆している。化合物#43の化学構造を化合物#69及び#70と比較すると、化合物#43のリボース基の2’及び3’位の両方のアセチル基が最適なグルコースクリアランス活性に不可欠であり、化合物#43のこれらのジアセチル基をジプロパノイルまたはジブタノイル基に置換すると、これらのインスリン抵抗性糖尿病Leprdb/dbマウスのグルコースクリアランスを促進する能力が有意に減弱することが明らかである。
まとめると、上記の研究は、試験した用量の化合物#43がLeprdb/dbマウスの耐糖能を有意に向上させることができることを示している。このプロセスでの化合物#43の作用は、骨格筋、肝臓、及び脂肪組織におけるグルコースのクリアランスにおけるインスリン感受性の向上により媒介される可能性が高い。さらに、セレンは化合物#43の作用に不可欠であるが、その存在自体のみでは、糖尿病の被験体にグルコースクリアランス能力を付与するのに十分ではない。セレン原子は、極めて特定の化学形態で提示しなければならない。これは、化合物#50の活性が低いことと、化合物Cの活性がないことから明らかであり;それらのどちらも化合物#43と構造的には非常に類似している。さらに、化合物#43のリボース基の2’及び3’位のアセチル基も、グルコースクリアランスにおける活性を維持するために必要である。
実施例4:糖尿病レプチン受容体(Lepr)偶発ヌル変異マウスの肝臓及び化合物#43処置後の培養肝細胞における糖新生酵素遺伝子G6pcの発現の阻害、ならびに培養肝細胞でのG6pc発現の阻害におけるインスリン作用の増強
肝臓は、グルコースを産生して血流中の正常なグルコースレベルを維持するための主要な器官である。グルコース-6-ホスファターゼ触媒サブユニット(G6pc)は、肝臓の糖新生に不可欠な酵素である。G6pc発現の調節に対する化合物#43の効果を、in vivo及びin vitroの両方で研究した。
材料及び方法
化合物
化合物#43、#C、#D、#E、及び#50は、Alltech,Inc.の化学実験室で合成した。試験したすべての化合物の純度は、HPLCによる測定で≧99%であることが確認された。
Leprdb/dbマウスにおける化合物#43及び#50のin vivo処置
38日齢の雄のLeprdb/dbマウス(C57BL/6J系統、Jackson Laboratoryから購入)に、0.2%DMSO、化合物#50、または化合物#43を含有する生理食塩水(0.09%NaCl)を毎日腹腔内注射した。52日間(体重1キログラムあたり25μgセレン当量の各化合物、滅菌生理食塩水で希釈)。処置後、肝臓を採取し、RNA分析を行った。
細胞株及び細胞増殖
ヒト肝癌HepG2及びマウス肝臓AML-12細胞は、アメリカ合衆国培養細胞系統保存機関(ATCC,Manassas,Virginia)から購入した。HepG2細胞は、10%FBSを添加したイーグル最小必須培地(EMEM)で増殖させた。AML-12細胞は、10%ウシ胎仔血清(FBS)、40ng/mlデキサメタゾン(Dex,Sigma)及び1×ITS(0.01mg/mlウシインスリン、0.0055mg/mlヒトトランスフェリン、5ng/ml亜セレン酸ナトリウムを含む)溶液(Sigma)を添加したダルベッコ改良イーグル培地及びハムF12(DMEM/F12)培地で増殖させた。
RNA分析のための細胞処置
基底G6pc発現(糖尿病刺激:8-CPT/Dexの非存在下)のRNA分析のために、増殖させたAML-12細胞及びHepG2細胞を、それぞれ10%FBS ITSフリー及びDexフリーのDMEM/F12培地及び10%FBS EMEM培地中、24ウェルプレート(0.5~2×105細胞/ウェル)上で一晩培養した。これらの細胞をPBSで2回すすぎ、残留血清を除去した。次いで、PBSで洗浄したHepG2細胞を、無血清EMEM培地中のインスリンまたは化合物#43の非存在下または存在下で40時間処置した。いくつかの実験では、PBSで洗浄したAML-12細胞を、無血清DMEM/F12培地中、化合物#43または他のセレン化合物の非存在下または存在下で、24時間インキュベートした。他の実験では、増殖させたAML-12細胞を、10%FBSを含むがITS/DexフリーのDMEM/F12培地で24時間、化合物#43(150または300ppb)の非存在下または存在下で前処置した。24時間処置後、AML-12細胞をPBSで2回洗浄し(培養液中の残留血清を除去するため)、次いで無血清DMEM/Dex培地中、インスリン、化合物#43またはその両方で6時間処置した。
糖尿病刺激誘発G6pc発現のRNA分析のために、AML-12細胞を、10%FBSを含むがITS/DexフリーのDMEM/F12培地中の化合物#43(150または300ppb)の非存在下または存在下で24時間前処置した。次いで、これらの細胞をPBSで2回洗浄して残留血清を除去し、無血清の通常のDMEM/F12培地中、インスリン(10または100nM)、または0.1mM 8-CPT(Sigma)及び0.5μM Dexの存在下または非存在下で化合物#43(150または300ppb)とともに、さらに6時間インキュベートした。
RNAの単離及びリアルタイムPCR分析
生理食塩水またはセレン化合物で処置したLeprdb/dbマウス由来の全RNAを、製造元のプロトコールに従ってQiagen RNAeasy RNA単離キットを使用して単離した。培養細胞由来の全RNAを、Trizol(Invitrogen)を使用して製造元のプロトコールに従って分離し、DNase I存在下でインキュベートして潜在的な混入ゲノムDNAを除去した。以前に記載されたように(Lan et al EMBO J 2003)、Applied-BioscienceのRTキットと事前設計したTaqmanプローブ(Invitrogen)を使用して、RNA試料をリアルタイムPCR(QRT-PCR)分析に供した。データは、各試料のアクチンB(Actb)mRNAレベルによって正規化し、3~5個の試料の平均値±SEMとして提示する。
統計解析
該当する場合、スチューデントt検定を使用して、P値が0.05未満である処置群間の統計的有意性を、統計的に有意であると判定した。
結果:
1.Leprdb/dbマウスの肝臓におけるG6pc mRNA発現の分析
以前の実験は、Leprdb/dbマウスの血糖値及びHbA1cレベルの阻害における化合物#43及び#50の異なる効果を示した(図3)。特定の仮説に拘泥することを望むものではないが、そのような異なる効果は、少なくとも部分的には、in vivoでの糖新生G6pc遺伝子の発現に対するこれらの化合物の潜在的な示差的作用によるものである可能性がある。したがって、Leprdb/dbマウスでのG6pc mRNA発現を、これら2つの化合物の慢性処置後に測定した。
図9に示すように、肝臓のG6pc mRNAレベルは、化合物#50の処置後、Leprdb/dbマウスでわずかに減少したが、有意ではなかった。しかしながら、化合物#43の処置により、生理食塩水で処置した対照と比較した場合、Leprdb/dbマウスの肝臓のG6pc mRNAレベルが劇的に減少(約56%減少)した(図9)。
総合すると、これらの結果は、G6pc発現の阻害においてこれらのセレン化合物の示差的効果があり得ること、及び化合物#43がこれらの重度のII型糖尿病マウスの肝臓におけるG6pc発現の強力な阻害剤であることのin vivoでのエビデンスを提供する。これらの結果は、化合物#43処置後のLeprdb/dbマウスの血糖値とHbA1cレベルの低下(図3)が、少なくとも部分的にはG6pc mRNA発現の減弱によるものであることを示唆している。さらに、G6pcの発現はインスリンシグナル伝達に応じて調節され、Leprdb/dbマウスがインスリン抵抗性であるため、これらの結果は、化合物#43がインスリンを迂回するか、インスリン作用を回復させてこれらの糖尿病マウスのG6pc発現を調節することができることを示唆している。
2.化合物#43処置後のマウス及びヒト肝細胞におけるG6pc mRNA発現の阻害ならびにG6pc発現の阻害におけるインスリン作用の増強
上記の研究は、化合物#43がインスリン抵抗性条件下で糖尿病マウスのG6pc発現を有意に阻害することができることを明らかにした。培養肝細胞を使用して、(a)G6pc発現に対するセレン化合物の示差的な効果があるかどうか、(b)肝臓のG6pc発現に化合物#43が直接影響を及ぼすかどうか、及び(c)G6pc発現の調節において化合物#43がインスリン作用を向上させることができるかどうかを調べた。化合物の効果を調べるために、2つの治療レジメン(無血清条件下での肝細胞の直接的な化合物処置、及び血清含有培地中の化合物による肝細胞の前処置とそれに続く無血清条件下での化合物の再処置)を実施して、これらの肝細胞におけるG6pc発現に対する化合物#43の効果を試験した。
最初に、マウス肝臓AML-12細胞を、無血清、インスリン-トランスフェリン-ナトリウム亜セレン酸サプリメント(ITS)及びデキサメタゾン(Dex)フリーの培地中、化合物なし(対照)、化合物CDEの組み合わせ、化合物#C、化合物#D、化合物#50及び化合物#43を、300十億分率(300ppb)のセレン用量(各化合物3.8μMに相当)で24時間処置し、G6pc発現に対するこれらのセレン化合物の示差的な効果があるかどうかを調べた。図10Aに示すように、化合物CDE(300ppbの各化合物)は、G6pc mRNA発現の有意な減少をもたらした。しかしながら、試験した用量での化合物#C、#D、または#50は、AML-12細胞におけるG6pc発現を有意に阻害しなかった。対照的に、同じセレン用量の化合物#43での処置は、AML-12細胞におけるG6pc発現のロバストな減少(対照群と比較した場合、約60%の減少)をもたらした(図10A)。化合物#43処置後のG6pc発現の減少の程度(60%)は、化合物CDE併用処置に比べてより顕著であった(約40%減少)。これらの結果は、これらのセレノ有機化合物がin vitroでのG6pc発現の阻害に示差的な影響を与える場合があり、化合物#43がプロセス中の試験したすべての化合物の中で最も強力な化合物であることを示唆している。これは、上記のin vivoマウスでの研究と一致している(図9)。この実験はAML-12細胞において完全に無血清の条件下(すなわち、インスリンまたは任意の他の増殖因子の非存在下)で行われたため、これらの結果は、化合物#43がインスリンを模倣するが迂回して、化合物CDEの組み合わせよりも高い効力で、AML-12細胞のG6pc発現を直接阻害することができることを示唆している。
次いで、別の肝細胞株であるヒトHepG2細胞を、無血清培地中、100nMのインスリンまたは600ppbの化合物#43の存在下で40時間インキュベートして、G6PC発現に対する化合物#43の直接的な阻害効果をさらに検証した。図10Bに示すように、インスリン処置はG6PC発現の有意な減少をもたらしたが、このことは、インスリンシグナル伝達がHepG2細胞で機能していることを示している。さらに、完全に無血清の条件下で化合物#43で処置した後のHepG2細胞のG6PC mRNAレベルは、対照群と比較すると有意に減弱していた(図10B)。化合物#43処置後のHepG2細胞のG6PC発現の減少は、図1で観察されたグルコース産生の減少と合致する。したがって、これらの結果は、化合物#43がインスリンを模倣するが、迂回して、G6PC発現を直接下方制御し、それによりHepG2細胞のグルコース産生を阻害することをさらに示唆している。
最後に、AML-12細胞を、血清を含むがITS/Dexフリーの培地中、化合物#43で24時間前処置し、続いて、FBS/ITS/Dexフリーの培地中、インスリンの存在下または非存在下でこの化合物を6時間再処置して、化合物#43がG6pc発現を阻害することができるかどうか、及びG6pc発現の下方制御においてインスリンと化合物#43の間に相加効果または相乗効果があるかどうかをさらに調べた。図10Cに示すように、10nMのインスリン処置により、対照群(図10Cの第1のバー)と比較した場合、G6pc mRNAレベルの有意な減少(約65%)が生じた。インスリンと同様に、化合物#43の処置(150ppbと300ppbの両方で)も、10nMインスリンと比較可能な減少レベルでG6pc発現の有意な減少をもたらした。さらに、G6pc mRNAレベルの減少は、化合物#43またはインスリン単独を用いた処置に比べて、化合物#43での前処置とそれに続く化合物#43及びインスリンの両方の共処置後のAML-12においてより顕著であった。これらの結果は、化合物#43がインスリンを模倣するが迂回して、AML-12細胞のG6pc発現を阻害することができるという上記の観察結果をさらに裏付けている。これらの結果は、化合物#43がAML-12細胞のG6pc発現の下方制御においてインスリン作用を増強することができることも示唆している。
3.擬似糖尿病条件下で培養したAML-12細胞での化合物#43処置後のG6pc発現の調節におけるG6pc発現の阻害及びインスリン作用の向上(8-CPT及びDexの両方により刺激)
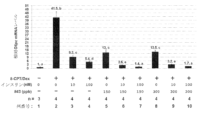
サイクリックAMP(8-CPT)及びDexは、肝臓におけるG6pc発現及びグルコース産生の周知の刺激剤であり、in vivoでの糖尿病状態を模倣する。G6pc発現に対する化合物#43の効果をさらに調べるために、細胞透過性8-(4-クロロフェニルチオ)cAMP(8-CPT)及びデキサメタゾン(Dex)で共処置したAML-12細胞でのG6pc mRNA発現を調べた。簡潔に述べると、AML-12肝細胞を、10%FBSを含むがITS/DexフリーのDMEM/F12培地で150ppbまたは300ppbの化合物#43の非存在下または存在下で24時間前処置した。PBSで2回洗浄した後、これらの細胞を、無血清培地中、10nMまたは100nMインスリン、0.1mM 8-CPT、及び0.5μM Dexの存在下または非存在下、これらのセレン化合物で6時間再処置した。これらの処置後、細胞を回収し、QRT-PCR分析に供した。
図11に示すように、8-CPT/Dexで処置したAML-12肝細胞では、G6pc mRNAの発現が41.5倍増加した(列#1対#2)。両方の用量のインスリン処置は、8-CPT/Dex群と比較した場合、AML-12細胞における8-CPT/Dex誘発G6pc発現を有意に減少させた(図11の列#3~4対列#2)。さらに、150及び300ppbの用量での化合物#43もまた、8-CPT/Dex誘導G6pc発現を有意に減弱させ(列#2の41.5から列#5の13へ、及び列#8の13.5へ減少、図11)、10nMインスリンと比較可能な効力を有していた(列#3、図11)。これらの研究は、インスリンと同様に、試験した用量での化合物#43単独で8-CPT/Dex誘発G6pc発現を阻害することができることを示した(8-CPT/Dex群と比較した場合、図11の列#5及び#8対#2、約68%の減少)。
さらに、化合物#43とインスリンとの併用処置(図11の列#6~7及び列#9~10)は、インスリン/化合物#43処置なし(列#2)、インスリンのみ(列#3~4)または化合物#43のみ(図11の列#5及び#8)と比較した場合、AML-12細胞における8-CPT/Dex誘発G6pc発現をさらに阻害した。より劇的に、150ppbの化合物#43と100nMのインスリン及び8-CPT/Dexの併用処置(列#7)、ならびに300ppbの化合物#43と100nMのインスリン及び8-CPT/Dexの併用処置(列#10)におけるG6pc mRNAレベルは、8-CPT/Dex処置単独(列#2)のレベルから、8-CPT/Dexなしの対照群(図11の列#1)に近いレベル(列#7、#10)まで、ロバストに減少した。言い換えれば、化合物#43(150または300ppb)と100nMのインスリンの共処置は、AML-12細胞における8-CPT/Dex誘導G6pc発現をほぼ完全に消失させた。
総合すると、これらの結果は、インスリンと同様に、試験した用量において化合物#43単独で8-CPT/Dex誘導G6pc発現を阻害することができ、また、AML-12細胞における8-CPT/Dex処置によるG6pcの発現増加を阻害するうえで、インスリンと化合物#43の両方の併用が、インスリン単独または化合物#43単独に比べてより効果的であることを示している。
上記のセレン化合物に応答したG6pc発現低下の効果は、試験した用量でのこれらの化合物が同じ実験条件下でAML-12細胞またはHepG2細胞の生存率に影響を及ぼさなかったことから、細胞生存に及ぼすこれらのセレン化合物の潜在的な毒性効果によるものではなかった(データは示さず)。
まとめると、これらの結果は、肝臓におけるG6pc発現の阻害に対する化合物#43及び#50の示差的効果が存在することを示した。少なくとも、データは、化合物#43がin vivo及びin vitroの両方で肝臓のG6pc発現を阻害する強力な化合物であることを示している。研究はさらに、通常の条件及び糖尿病をシミュレートする条件(すなわち、8-CPT/Dexによる細胞処置)のいずれで培養したマウス肝細胞及びヒト肝細胞においても、化合物#43がインスリンを模倣するが迂回して、G6pc発現を直接阻害することができることを明らかにした。さらに、化合物#43は、インスリン作用を向上させて、通常の条件及び擬似糖尿病条件のいずれで培養したAML-12細胞においても、G6pc発現を阻害することができる。総合すると、これらの結果は、化合物#43が、in vivo及びin vitroの両方で肝臓におけるG6pc発現を阻害することができ、したがってI型及びII型糖尿病の貴重な治療薬となりうる分子的エビデンスを提供する。
実施例5:化合物#43はインスリンを模倣するが迂回して、ホスホイノシチド依存性プロテインキナーゼ1(PDK1)及びプロテインキナーゼB(AKT)シグナル伝達を活性化し、in vivo及びin vitroで肝臓のフォークヘッドボックスタンパク質O1(FOXO1)のリン酸化を増強する
フォークヘッド転写因子FOXO1は、肝臓内の代謝、糖新生及びインスリン感受性において重要な役割を果たす。FOXO1の細胞内活性は、翻訳後修飾によって緊密に調節される。特に、FOXO1のリン酸化はFOXO1を核から排除し、それにより肝臓内のグルコース産生のためのG6pcなどの標的遺伝子へのアクセスを遮断する。インスリン抵抗性または糖尿病の個体では、核からFOXO1を排除するシグナルがないため、核内に存在し続け、G6pcの転写を刺激する。G6pcの発現増加は糖新生を促進し、高血糖症を引き起こす。
上記のように、in vivo及びin vitroの結果は、化合物#43がインスリン作用を模倣するが迂回してG6pc発現を阻害することができ、そのプロセスにおいてインスリン作用を向上させることができることを示した。FOXO1が肝臓の糖新生及びインスリン感受性の主要なシグナル伝達分子であり、PDK1とAKTがFOXO1の上流の2つの主要な中間シグナル伝達分子であることから、Leprdb/dbマウス、ヒト肝臓HepG2細胞及びマウス肝臓AML-12細胞において、化合物#43がインスリンと同様にFOXO1とその上流のシグナル伝達分子であるPDK1及びAKTを標的とするかどうかという疑問について調べた。
材料及び方法
化合物
化合物#43は、Alltech,Incの化学実験室で合成した。試験したこの化合物の純度は、HPLCによる測定で≧99%であることが確認された。
Leprdb/dbマウスにおける化合物#43のin vivo処置及び肝臓タンパク質の調製
出生後38日目の雄のLeprdb/dbマウス(C57BL/6J系統、Jackson Laboratoryから購入)に、0.2%DMSO、化合物#43(体重1キログラムあたり25μgセレンまたは硫黄当量の各化合物、滅菌生理食塩水で希釈)を含有する生理食塩水(0.09%NaCl)を52日間、毎日腹腔内注射した。処置後、肝臓を採取し、-80℃で保存した。
凍結肝組織を、完全プロテイナーゼ及びホスファターゼ阻害剤(Thermo-Fisher Scientific,Waltham,MA)を含有する無菌の氷冷PBS中で細かく刻み、組織ホモジナイザー(Thermo-Fisher Scientific,Waltham,MA)を用いて均質化した。これらの組織ホモジネートを、タンパク質を抽出するために、完全プロテイナーゼ/ホスファターゼ阻害剤を含有するThemo-Fisherの既製RIPA緩衝液(ホモジネート1部/RIPA緩衝液2部)で希釈した。ホモジネート中のタンパク質を、RIPA緩衝液で4℃、一晩抽出した。これらの一晩抽出したタンパク質溶解物を12000×gで4℃、30分間遠心分離し、これらの組織溶解物の上清中のタンパク質レベルを、Pierce Micro-BCAタンパク質アッセイキット(Thermo Scientific-Piece Biotechnology,Rockford,IL)を使用して製造元のプロトコールに従って測定した。
細胞培養
ヒト肝癌HepG2細胞及びマウス肝臓AML-12細胞株は、アメリカ合衆国培養細胞系統保存機関(ATCC,Manassas,Virginia)から購入した。HepG2細胞は、10%FBSを添加したイーグル最小必須培地(EMEM)で培養した。AML-12細胞は、10%ウシ胎仔血清(FBS)、40ng/mlデキサメタゾン(Dex,Sigma)及び1×ITS(0.01mg/mlウシインスリン、0.0055mg/mlヒトトランスフェリン、5ng/mlセレンナトリウムを含有する)溶液(Sigma)を添加したダルベッコ改良イーグル培地及びハムF12(DMEM/F12)培地で増殖させた。
タンパク質分析のための細胞処置
HepG2細胞を6ウェルプレートに播種し(7×105細胞/ウェル)、10%FBS EMEM培地で30時間培養した。次いで、これらの細胞をPBSで2回洗浄して残留血清を除去し、通常のEMEM培地中で一晩血清飢餓状態にした。これらの血清飢餓状態のHepG2細胞を、化合物#43(600ppb)の非存在下または存在下で、0分(処置直前)、30分、60分、90分、24時間、30時間及び40時間処置した。
AML-12細胞を使用して、糖尿病刺激剤による誘導後に化合物#43が肝細胞内のPdk1/Akt/Foxo1シグナル伝達分子を調節することができるかどうかを調べた。増殖させたAML-12細胞を6ウェル(1×106細胞/ウェル)プレートに播種し、10%FBSを含むがITS/DexフリーのDMEM/F12培地中で24時間培養した。次いで、これらの細胞をPBSで2回洗浄して残留血清を除去し、通常のDMEM/F12培地中で一晩血清飢餓状態にした。これらの血清飢餓状態のAML12細胞を、無血清の通常のDMEM/F12培地中、10nMインスリンもしくは化合物#43(300ppb)の非存在下(対照群)または存在下、糖尿病刺激剤8-CPT(0.1mM)及びDex(0.5μM)の組み合わせを用いて、それぞれ、60分間、90分間、6時間処置した。
上記の処置の後、培養したHepG2細胞及びAML-12細胞を氷冷PBSで2回すすぎ、完全プロテイナーゼ及びホスファターゼ阻害剤(Thermo-Fisher Scientific,Waltham,MA)を含有する氷冷RIPA緩衝液に氷上で30分間溶解させた。セルスクレーパーとホールピペットを使用して細胞溶解液を回収し、4℃、12000×gで30分間遠心分離してDNAペレットを除去し、タンパク質抽出物を取得した。これらの細胞溶解物の上清中のタンパク質レベルを、Pierce Micro-BCAタンパク質アッセイキット(Thermo Scientific-Piece Biotechnology,Rockford,IL)を使用して、製造元のプロトコールに従って測定した。
ウエスタンブロット分析
前述のように、肝組織タンパク質100マイクログラム、または対照及び化合物(複数可)処置したHepG2細胞もしくはAML-12細胞由来の全タンパク質5マイクログラムをSDS-PAGEゲル分離に供し、次いでPVDF膜に転写した。(Reddy et al. 2008 Science)。5%(w/v)のウシ血清アルブミン(Sigma,St.Louis,MO)を含有するリン酸緩衝生理食塩水(PBS)中で膜をブロックし、特異的な一次抗体の存在下でインキュベートした後、HRP結合抗マウスまたは抗ウサギ二次抗体(1:5000希釈、Cell Signaling Inc.)の存在下でインキュベートした。Gapdh(Li-COR,Lincoln,Nebraska)以外のすべての一次抗体はCell Signaling Incから購入した。膜ブロットの陽性シグナルは、Amershamのenhanced chemiluminescence Western Blotting Prime Detection試薬(GE Healthcare Lifescience,Pittsburgh,PA)を使用して検出した。膜ブロット上のこれらの発光シグナルの画像を、LI-COR Odyssey Fc Image system(Lincoln,Nebraska)を使用してキャプチャした。GE WB ECL-prime-detectionプロトコール(GE Healthcare Lifescience,Pittsburgh,PA)に記載されているように、同じ膜ブロットを除去し、別の抗体で再ブロットした。NIH ImageJソフトウェアを使用してウエスタンブロットのタンパク質バンド濃度を測定し、各試料のGapdhまたはActb/ACTBレベルで正規化した。データは、群ごとに3つの試料の平均値±SEMとして提示する。
統計解析
該当する場合、スチューデントt検定を実行して、2つの群間の統計的差異を決定した。0.05未満のP値を有意とみなした。
結果:
1.化合物#43による慢性処置後のインスリン抵抗性Leprdb/dbマウスの肝臓におけるPdk1、Akt、及びFoxo1のリン酸化の増強
動物試験により、化合物#43が、血糖値及びHbA1cレベルを低下させ、Leprdb/dbマウスにおける肝臓G6pc発現を阻害することができることが明らかとなった(図3~図7、図9)。空腹時血糖値及び血中HbA1cレベルの低下は、少なくとも部分的には、Leprdb/dbマウスの肝臓における糖新生G6pc遺伝子発現の減弱に起因する。肝臓でのG6pc発現は、インスリンシグナル伝達Pdk1/Akt/Foxo1カスケードによって制御される。したがって、このアプリケーションでは、これらのインスリン抵抗性Leprdb/dbマウスの肝臓で、化合物#43の慢性処置が、少なくともある程度、インスリンシグナル伝達を回復させる(すなわち、Pdk1/Akt/Foxoのリン酸化を増強する)ことができるかどうかを調べた。
出生後38日目のLeprdb/dbマウスに、生理食塩水または化合物#43を体重1kgあたり25μgセレンの用量で52日間、毎日腹腔内注射した。上記の処置の後、肝臓組織を採取し、インスリンシグナル伝達分子に対する特異的抗体を使用したウエスタンブロット分析に供した。図12Aに示すように、化合物#43処置マウスの肝臓におけるリン酸化Pdk1、トレオニン308でのリン酸化Akt、及びセリン256でのリン酸化Foxo1のタンパク質シグナルは、生理食塩水処置マウスのタンパク質シグナルよりも明らかに豊富あった。これらのウエスタンブロットの定量分析は、化合物#43での処置後、Leprdb/dbマウスの肝臓でpPdk1、pAkt及びpFoxo1のタンパク質レベルが有意に上昇したことを示した(図12B)。対照的に、化合物#43処置マウスでは全Aktレベルに有意な変化はなかった。Pdk1、Akt、及びFoxo1のリン酸化の増加は、Leprdb/dbマウスがインスリンに応答できないことが知られているにもかかわらず、化合物#43の慢性処置後のLeprdb/dbマウスの肝臓においてインスリン下流シグナル伝達カスケードが活性であることを強く示唆している。言い換えれば、これらの結果は、化合物#43がインスリン作用を少なくとも部分的に回復させるか、インスリンを迂回するか、またはその両方をして、Pdk1/Akt/Foxo1のリン酸化を刺激し、これらのインスリン抵抗性糖尿病マウスの肝臓におけるG6pc発現及びグルコース産生の減弱をもたらすことができることを示している。
2.化合物#43はインスリンを模倣するが迂回して、一時的にPDK1/AKTを活性化し、その後、無血清培地中で培養したヒトHepG2細胞のFOXO1を不活性化する
肝臓内でPDK1、AKT及びFOXO1のリン酸化を調節するための化合物#43のインスリン非依存性であるがインスリン様の効果があるかどうかを調べるために、血清飢餓状態のヒトHepG2細胞を、無血清培地中、対照及び600ppbの化合物#43で30分~48時間の様々な時間処置した。処置した細胞をウエスタンブロット分析に供した。
図13Aに示すように、30、60及び90分間の化合物#43処置後、HepG2細胞におけるリン酸化PDK1のタンパク質シグナルが明らかに増加したが、より長い処置時点(24時間処置後)ではそれがなかった。定量的研究により、化合物#43処置後、HepG2細胞におけるpPDK1の有意かつ一過性の増加が示され、ピーク増加は化合物処置後約60~90分であった(図13B)。同様に、T308でのリン酸化AKTの有意かつ一過性の増加は、30、60、90分間及び24時間の化合物#43処置後のHepG2細胞でも観察され、ピーク増加は60及び90分であった(図13A、C)。対照的に、試験したすべての時点での全AKTタンパク質レベルは、化合物#43処置後のHepG2細胞において有意に変化していなかった(図13A、D)。
T24でのリン酸化FOXO1のタンパク質レベルは、90分以上化合物#43で処置した後、HepG2細胞において有意に増加していた(図13A、E)。FOXO1のリン酸化の増加は、pPDK1及びpAKTの増加事象よりも後に観察された(図13A、13E対13B~C)。24時間未満の化合物#43処置後では、HepG2細胞の全FOXO1タンパク質レベルに有意な変化はなかった(図13A、13F)。しかしながら、化合物#43の長期処置(30時間または48時間)により、HepG2細胞の全FOXO1タンパク質がわずかではあるが統計的に有意に減少しており(図13A、F)、これは、HepG2細胞のリン酸化FOXO1の継続的な上昇から生じるFOXO1のプロテアソームタンパク質分解の潜在的な増加が原因であり得る。リン酸化FOXO1は核から排除され、これは直接的な結果として核FOXO1及びG6pc発現の減少を意味する。また、化合物#43処置したHepG2細胞ではグルコース産生の有意な減少(図1)及びG6PC発現(図10B)が、ならびに化合物#43処置したLeprdb/dbマウスでは、高血糖症の軽減及びG6pc発現の低下が観察された(図3~図7、図9)。
総合すると、上記の結果は、化合物#43がインスリンを模倣するが迂回して、PDK1及びAKTを一時的に活性化し、次いでヒト肝臓HepG2細胞のFOXO1を不活性化することができることを示した。
3.化合物#43はインスリンを模倣するが迂回して、Pdk1/Aktを一時的に活性化し、その後、擬似糖尿病条件下で培養したAML-12細胞においてFoxo1を不活性化する(8-CPTとDexの両方で刺激)
前述の実施例に記載したように、化合物#43は、インスリンを模倣するが迂回して、AML-12細胞における8-CPT/Dex誘導G6pc発現を阻害することができる(図11)。この効果は、これらのマウス肝細胞においてFoxo1を不活性化する化合物#43の潜在的なインスリン様活性によるものであり得る。したがって、このアプリケーションでは、擬似糖尿病条件下で培養したAML-12細胞でのインスリンシグナル伝達分子のタンパク質発現を調べた(8-CPT及びDexで刺激)。
図14Aに示すように、インスリン処置は、60分及び90分においてPdk1、Akt及びFoxo1のリン酸化を増強したが、このことは、擬似糖尿病条件下で培養したAML-12細胞がインスリンに応答性であることを示した。インスリンと同様に、化合物#43は、60分間の化合物処置後、これらの8-CPT/Dex処置AML-12細胞においてPdk1、Akt及びFoxo1のリン酸化も有意に誘導した(図14A~図14B)。化合物処置の90分後に、これらのAML-12細胞でpFoxo1タンパク質レベルの有意な増加が観察されたが、pPdk1、pAkt、Akt、及びFoxo1を含む他の試験したすべての分子のタンパク質レベルは、化合物#43処置後に有意に変化していなかった(図14A、C)。化合物#43処置の6時間では、依然としてpFoxo1レベルの有意な増加、及び全Foxo1タンパク質レベルのわずかではあるが有意な減少があった(図14A、D)。6時間の処置でのpFoxo1の増加及び全Foxo1タンパク質レベルのわずかな減少は、10nMのインスリン処置後のAML-12細胞でも観察された(図14A、E)。ここでも、6時間の処置での全Foxo1のわずかな減少は、糖尿病刺激を受けたこれらのAML-12細胞の細胞質ゾルでのFoxo1の連続的なリン酸化に起因するFoxo1タンパク質の標的化プロテアソームタンパク質分解によるものであり得る。化合物#43処置後のFoxo1のリン酸化の増強は、図11で観察される核Foxo1の減少及び8-CPT/Dex誘導G6pc発現の減少をもたらし得る。総合すると、これらの結果は、化合物#43は、インスリンと同様に、擬似糖尿病条件下で培養したAML-12細胞において、Pdk1及びAktのリン酸化を一時的に誘導し、次いでFoxo1のリン酸化を誘導することができる。
結論として、上記のin vitro及びin vivoでの研究により、化合物#43はインスリンを模倣するが迂回して、肝臓で一時的にPdk/Aktを活性化し、次いでFoxo1を不活性化することができることが示された。
実施例6:Leprdb/dbマウスの肝臓及び培養肝細胞におけるGlut4(SLC2A4)発現の増強、ならびに化合物#43処置後の培養肝細胞におけるグルコース取り込みの増強
in vivoでの研究により、化合物#43は、インスリン抵抗性Leprdb/dbマウスの血糖値を低下させ、グルコースクリアランスを向上させることができることが明らかになった(図3~図8)。これは、部分的には肝臓でのグルコース産生の減弱によるものであるが、血流から肝臓を含む末梢組織へのグルコース取り込みの増加によるものでもあり得る。Glut4は、重要なグルコース輸送体であり、全身のインスリン刺激に応答した、肝臓でのグルコース取り込みの間接的なFoxo1標的遺伝子である。したがって、Leprdb/dbマウス及び培養マウス肝臓AML-12細胞を化合物#43で処置して、肝臓でのGlut4発現及びグルコース取り込みに対する潜在的な効果を試験した。
材料及び方法
化合物
化合物#43及び化合物#50は、Alltech,Incの化学実験室で合成した。これらすべての化合物の純度は、HPLCによる測定で≧99%であることが確認された。
Leprdb/dbマウスにおける化合物#43及び#50のin vivo処置
出生後38日目の雄のLeprdb/dbマウス(C57BL/6J系統、Jackson Laboratoryから購入)に、0.2%DMSO、化合物#43、または化合物#50を含有する生理食塩水(0.09%NaCl)を52日間、毎日腹腔内注射した(体重1キログラムあたり25μgセレンの各化合物、滅菌生理食塩水で希釈)。処置後、肝臓を採取し、RNA分析に供した。
細胞培養
マウス肝臓AML-12細胞は、アメリカ合衆国培養細胞系統保存機関(ATCC,Manassas,Virginia)から購入した。これらの細胞を、10%ウシ胎仔血清(FBS)、40ng/mlデキサメタゾン(Dex,Sigma)及び1×ITS(0.01mg/mlウシインスリン、0.0055mg/mlヒトトランスフェリン、5ng/mlセレンナトリウム)溶液(Sigma)を添加したダルベッコ改変イーグル培地及びハムF12(DMEM/F12)培地で増殖させた。
RNA分析のための細胞処置
基底Glut4(Slc2a4)発現(糖尿病刺激剤8-CPT/Dexの非存在下)のRNA分析のために、増幅させたAML-12細胞を、10%FBSを含むがITS及びDexフリーのDMEM/F12培地中、24ウェル(1×105細胞/ウェル)プレート上で一晩培養した。これらの細胞をPBSで2回洗浄して残留血清を除去した後、無血清DMEM/F12培地中、ビヒクル(0.024%DMSO)または化合物#43(300ppb)の存在下で24時間インキュベートした。
擬似糖尿病条件下で培養したAML-12細胞におけるGlut4発現のRNA分析のために、増殖させたAML-12細胞を、10%FBSを含むがITS及びDexフリーのDMEM/F12培地中、24ウェル(2×105細胞/ウェル)プレート上で一晩培養した。次いで、これらの細胞をPBSで2回洗浄して潜在的な残留血清を除去し、通常のDMEM/F12培地で一晩血清飢餓状態にした。次いで、これらの血清飢餓状態のAML-12細胞を、無血清の通常のDMEM/F12培地中、糖尿病刺激剤である0.1mM 8-CPT(Sigma)及び0.5μM Dexの存在下で、ビヒクル(0.024%DMSO)または化合物#43(300ppb)とともに6時間及び24時間インキュベートした。
RNAの単離及びリアルタイムPCR分析
生理食塩水または化合物#43で処置したLeprdb/dbマウス由来の全肝臓RNAを、Qiagen RNAeasy RNA単離キットを使用して製造元のプロトコールに従って単離した。培養細胞由来の全RNAは、Trizol(Invitrogen)を使用して製造元のプロトコールに従って分離し、DNase Iの存在下でインキュベートして潜在的な混入ゲノムDNAを除去した。以前に説明したように(Lan et al EMBO J 2003)、Applied-BioscienceのRTキット及び事前設計したTaqmanプローブ(Invitrogen)を使用して、RNA試料をリアルタイムPCR分析に供した。データは、各試料のアクチンB(Actb)mRNAレベルによって正規化し、3~5試料の平均値±SEMとして提示する。
グルコース取り込みアッセイ
増殖させた同数のAML-12細胞を96ウェルプレートに播種し(1.5×105/ウェル)、10%FBSを含むがITS/DexフリーのDMEM/F12培地中で一晩培養した。次いで、これらの細胞をPBSで2回洗浄し(潜在的な残留血清を除去するため)、通常のDMEM/F12培地中、一晩血清飢餓状態にした。これらの血清飢餓状態のAML-12細胞を、グルコース/フェノールレッドフリーのDMEM培地中、処置なし(基底)、インスリン(10及び100nM)、または化合物#43(150、300及び600ppb)で37℃、1.5時間処置した。処置後、培地を除去し、細胞をPBSで1回洗浄し、1mMの2-デオキシグルコース(2DG)の存在下で室温、30分間インキュベートした。2DG処置した細胞を、次いで、PromegaのGlucose Uptake-Glo Assayキットを使用して製造元のプロトコールに従って、グルコース取り込み測定に供した。Bio-Tekルミノメーターを使用して、発光シグナルを記録した。
統計解析
該当する場合、スチューデントt検定を使用して、異なる処置群間の差異の統計的有意性を判定し、0.05未満のP値を有意な結果を表すものとみなした。
結果:
1.化合物#43及び化合物#50での慢性処置後のLeprdb/dbマウスの肝臓におけるGlut4 mRNA発現の分析
出生後38日目の雄のLeprdb/dbマウスに、0.2%DMSO、化合物#43、または化合物#50(体重1kgあたり25μgセレン当量の各化合物、滅菌生理食塩水で希釈)を含有する生理食塩水(0.09%NaCl)を52日間、毎日腹腔内注射した。これらの化合物処置後、肝臓を採取し、Glut4及びActb mRNAのRNA分析に供した。
図15に示すように、化合物#50処置は、Leprdb/dbマウスの肝臓におけるGlut4 mRNAレベルを数値的に増加させた(生理食塩水処置群と比較した場合)。しかしながら、化合物#43処置は、Leprdb/dbマウスの肝臓におけるGlut4 mRNA発現レベルの大幅で有意な増加(5倍;P=0.048)を誘発した(図15)。これらの結果は、肝臓におけるGlut4発現の刺激においてこれら2つのセレン化合物間に明確な差異があり、化合物#43がこの器官におけるGlut4発現の強力なエンハンサーであることを示唆している。これらの結果は、化合物#43で処置したLeprdb/dbマウスで観察された血糖値の低下と耐糖能の向上(図3~図8)が、糖尿病の被験体の血流から肝臓へのグルコース取り込みの増加をもたらすGlut4発現の増強に部分的に起因し得るものであったことも示唆している。
2.糖尿病刺激剤(8-CPT/Dex)の刺激の存在下または存在下で、マウス肝臓AML-12細胞における化合物#43処置後のGlut4 mRNA発現の増強
上記のin vivo研究は、化合物#43がインスリン抵抗性Leprdb/dbマウスの肝臓におけるGlut4発現を有意に刺激することができることを明らかにした。これは、肝臓組織に対する化合物#43の潜在的な全身効果または化合物#43の潜在的な直接効果によるものであり得る。後者のシナリオを試験するために、培養肝細胞を使用して、化合物#43が肝臓のGlut4発現を直接調節することができるかどうかを調べた。
化合物#43が正常なAML-12細胞(糖尿病刺激剤の投与なし)で基底Glut4発現を調節することができるかどうかを調べた。簡潔に述べると、AML-12細胞を、無血清及びITS/Dexフリーの培地中、ビヒクル(0.024%DMSO)及び化合物#43(300ppb)で24時間処置し、Glut4及びActb発現のRNA分析に供した。図16Aに示すように、化合物#43(300ppb)の処置により、AML-12細胞におけるGlut4 mRNA発現の有意な増加(約2.1倍の増加)がもたらされた。この実験が完全な無血清条件下で培養したAML-12細胞で行われたことを考慮すると、AML-12細胞での化合物#43処置後のGlut4発現の増強は、インスリン、血清、または増殖因子に依存しないことが示唆された。
化合物#43がGlut4発現を調節できるかどうかをさらに試験するために、糖尿病刺激剤にさらしたAML-12細胞を使用した。簡潔に述べると、AML-12細胞を一晩血清飢餓状態にし、その後、無血清の通常のDMEM/F12培地中、糖尿病刺激剤である0.1mM 8-CPT(Sigma)及び0.5μM Dexの存在下、ビヒクル(0.024%DMSO)または化合物#43(300ppb)とともに6時間及び24時間インキュベートした。図16Bに示すように、6時間及び24時間の両方での化合物#43の処置は、糖尿病刺激剤8-CPT及びDexで共処置したこれらのAML-12細胞におけるGlut4 mRNA発現の有意な増加をもたらした。したがって、これらの結果は、化合物#43が、擬似糖尿病条件下で培養したAML-12細胞のGlut4発現を直接調節することができることを示している。
3.化合物#43はインスリンを模倣するが迂回して、マウス肝臓AML-12細胞のグルコース取り込みを促進する
Glut4発現の増強は、化合物#43がインスリンを模倣するが迂回して、肝臓におけるグルコース取り込みを増強する可能性が高いことを示唆している。これを試験するために、マウス肝臓AML-12細胞でグルコース取り込み実験を実施した。簡潔に述べると、同数のAML-12細胞を96ウェルプレートに播種し、10%FBSを含むがITS/DexフリーのDMEM/F12培地中で24時間培養し、通常のDMEM/F12培地中で一晩血清飢餓状態にした。これらの血清飢餓状態のAML-12細胞を、グルコース/フェノールレッド/無血清DMEM培地中、インスリン(10及び100nM)または化合物#43(150、300及び600ppb)で37℃、1.5時間処置した。処置後、細胞を1mMの2-デオキシグルコース(2DG)の存在下で室温、30分間インキュベートした後、PromegaのGlucose Uptake-Gloアッセイキットを使用して発光分析に供した。検出した発光シグナルは、培養細胞へのグルコース取り込みを表す。
図17に示すように、10nMのインスリン処置は、グルコース取り込みを増強しなかった。しかしながら、100nMのインスリン処置により、AML-12細胞のグルコース取り込みが有意に増加した。さらに、試験した3つの用量すべてでの化合物#43処置により、グルコース取り込みが有意に増加し、600ppbの化合物#43で処置したAML-12細胞のグルコース取り込みの増加の程度は、100nMのインスリンと比較可能なものであった(図17)。AML-12細胞を無血清条件下で化合物#43処置したため、これらの結果は、インスリンと同様に、化合物#43が迅速に(1.5時間未満)作用して肝細胞のグルコース取り込みを直接刺激することができることを示している。肝臓でのグルコース取り込みの増加は、化合物#43がインスリン抵抗性Leprdb/dbマウスの血糖値を低下させ、グルコースクリアランスを向上させることができる理由の1つであり得る(図3~8)。
実施例7:インスリン抵抗性Leprdb/dbマウスの骨格筋におけるインスリンシグナル伝達の重要な下流分子であるリン酸化Pdk1、Akt及びFoxo1の発現の増強、ならびに分化したC2C12(骨格筋)細胞へのグルコース取り込みの刺激におけるインスリン及び化合物#43の両方の協同作用
材料及び方法
化合物
化合物#43は、Alltech,Incの化学実験室で合成した。この試験化合物の純度は、HPLCによる測定で≧99%であることが確認された。
Leprdb/dbマウスにおける化合物#43のin vivo処置
38日齢の雄のLeprdb/dbマウス(C57BL/6J系統、Jackson Laboratoryから購入)に、化合物#43(0.2%DMSOまたは体重1キログラムあたり0.136mg、滅菌生理食塩水で希釈)を含有する生理食塩水(0.09%NaCl)を52日間、毎日腹腔内注射した。処置後、腓腹筋の骨格筋試料を採取し、-80℃で保存した。
骨格筋タンパク質の調製とウエスタンブロット分析
凍結させた骨格筋を、完全プロテイナーゼ及びホスファターゼ阻害剤(Thermo-Fisher Scientific,Waltham,MA)を含有する無菌の氷冷PBS中で細かく刻み、組織ホモジナイザー(Thermo-Fisher Scientific,Waltham,MA)を使用して均質化した。これらの組織ホモジネートを、タンパク質を抽出するために、完全プロテイナーゼ/ホスファターゼ阻害剤を含有するThemo-FisherのRIPA緩衝液(ホモジネート1部/RIPA緩衝液2部)で希釈した。ホモジネート中のタンパク質を4℃のRIPA緩衝液で一晩抽出した。一晩抽出したこれらのタンパク質/組織溶解物を、12000×gで4℃、30分間遠心分離し、これらの組織溶解物の上清中のタンパク質レベルを、Pierce Micro-BCAタンパク質アッセイキット(Thermo Scientific-Piece Biotechnology,Rockford,IL)を使用して製造元のプロトコールに従って測定した。
100マイクログラムの骨格筋タンパク質をSDS-PAGEゲル分離に供し、次いで、以前に記載されたようにPVDF膜に転写した(Reddy et al. 2008 Science)。5%(w/v)のウシ血清アルブミン(Sigma,St.Louis,MO)を含有するリン酸緩衝生理食塩水(PBS)で膜をブロックし、特異的な一次抗体の存在下でインキュベートした後、HRP結合抗マウスまたは抗ウサギ二次抗体(1:5000希釈、Cell Signaling Inc.)の存在下でインキュベートした。β-チューブリンに対する抗体(LI-COR bioscience)以外のすべての一次抗体はCell Signaling Incから購入した。アマシャムのenhanced chemiluminescence Western Blotting Prime Detection試薬(GE Healthcare Lifescience,Pittsburgh,PA)を使用して、膜ブロットの陽性シグナルを検出した。LI-COR Odyssey Fc Image system(Lincoln,Nebraska)を使用して、膜ブロット上のこれらの発光シグナルの画像をキャプチャした。GE WB ECL-prime-detectionプロトコール(GE Healthcare Lifescience,Pittsburgh,PA)に記載されているように、同じ膜ブロットを除去し、別の抗体で再ブロットした。NIH ImageJソフトウェアを使用してウエスタンブロットのタンパク質バンド濃度を測定し、各試料のβ-チューブリンレベルで正規化した。データは、5つの動物試料の平均値±SEMとして提示する。
C2C12細胞培養、C2C12細胞の分化、及びグルコース取り込み分析
マウス筋芽細胞C2C12細胞は、アメリカ合衆国培養細胞系統保存機関(ATCC,Manassas,Virginia)から購入した。これらの細胞を、10%FBSを添加したDMEM培地で増殖させた。次いで、同数のC2C12細胞を96ウェルプレートに播種し(5000細胞/ウェル)、10%FBS DMEM培地で37℃、5日間培養した。前述のように、培養5日目に0.5%ウマ血清(Sigma)を含有するDMEM培地(分化培地)を使用してC2C12細胞を分化させ、これを7日間、毎日細胞に新鮮な10%FBS DMEM培地を補充して継続した(Misu et al,Cell Metabolism 12,483-495,2010)。分化後7日目に、分化したC2C12細胞をPBSで2回すすぎ、無血清のグルコースフリーDMEM培地中、0.006%DMSO(化合物#43溶媒)または化合物#43(300または600ppb)の存在下または非存在下で一晩前処置した。次いで、これらの細胞をPBSで1回洗浄し、その後、グルコース/フェノールレッドフリーのDMEM培地中、処置なし(基底)またはインスリン(200nM)、化合物#43(300及び600ppb)、またはインスリンと化合物#43の両方で、37℃、1.5時間処置した。処置後、培地を除去し、細胞をPBSで1回洗浄し、1mM 2-デオキシグルコース(2DG)の存在下で室温、30分間インキュベートした。2DG処置した細胞を、次いで、PromegaのGlucose Uptake-Glo Assayキットを使用して、製造元のプロトコールに従って、グルコース取り込みに供した。Bio-Tekルミノメーターを使用して、発光シグナルを記録した。
統計解析
該当する場合、スチューデントt検定を使用して、P値が0.05未満の異なる処置群間の差異の統計的有意性を判定した。
結果:
1.化合物#43の慢性処置後のインスリン抵抗性Leprdb/dbマウスの骨格筋におけるインスリン下流シグナル伝達分子Pdk1、Akt及びFoxo1のリン酸化の増加
肝臓に加えて、骨格筋は、全身性インスリンに応答したグルコース恒常性にとって重要な他の主要器官である。骨格筋におけるグルコース取り込みは、血流中の正常なグルコースレベルを維持する上で重要な役割を果たす。動物実験により、化合物#43が血糖値及びHbA1cレベルを低下させ、インスリン抵抗性Leprdb/dbマウスの耐糖能を有意に向上させることができることが明らかになった(図3図8)。これらの効果は、グルコースの取り込みを刺激するための骨格筋におけるインスリンシグナル伝達(すなわちPdk1/Akt)の回復によるものであり得る。したがって、化合物#43の慢性処置が、これらのインスリン抵抗性Leprdb/dbマウスの骨格筋における損傷したインスリンシグナル伝達を部分的に修復することができるかどうかを調べた。
出生後38日目のLeprdb/dbマウスに、生理食塩水(0.2%DMSOを含有する)または化合物#43を、体重1kgあたり0.136mgの化合物#43の用量で52日間、毎日腹腔内注射した。上記の処置後、骨格筋を採取し、それらのインスリンシグナル伝達分子に対する特異的抗体を使用してウエスタンブロット分析に供した。
図18Aに示すように、リン酸化Pdk1、トレオニン308のAkt、及びセリン256のFoxo1のタンパク質バンド濃度は、生理食塩水処置マウスに比べて化合物#43処置マウスの骨格筋で明らかに高かったが、全AktまたはFoxo1ではそうではなかった。これらのウエスタンブロットの定量分析により、生理食塩水で処置したマウスと比較した場合、化合物#43処置後、Leprdb/dbマウスの骨格筋においてpPdk1タンパク質レベルが増加したことが示された(図18B)。化合物#43処置マウスの骨格筋におけるpPdk1のレベルの増加は、統計的有意に近いものであった(P=0.11)。さらに、2つのインスリン/Pdk1下流シグナル伝達分子pAkt及びpFoxo1のタンパク質レベルは、化合物#43処置後、Leprdb/dbマウスの骨格筋において有意に上昇した(図18B)。対照的に、化合物#43処置マウスの骨格筋の全Akt及びFoxo1タンパク質レベルの有意な変化はなかった。Pdk1のリン酸化の増加とリン酸化Akt及びリン酸化Foxo1の有意な増加は、Leprdb/dbマウスがインスリン不応性であることが知られているにもかかわらず、化合物#43の慢性処置後、Leprdb/dbマウスの骨格筋においてインスリン下流シグナル伝達カスケードが活性化していることを示唆している。言い換えれば、これらの結果は、化合物#43がインスリン作用を回復させるか、インスリンを迂回するか、またはその両方でPI3Kを活性化して骨格筋におけるPdk1/Aktのリン酸化を誘導し、これらの重度のインスリン抵抗性のII型糖尿病マウスにおけるグルコース恒常性の調節において重要な機能を果たすことができることを示している。
2.インスリンと化合物#43の両方の処置後の分化マウスC2C12(骨格筋)細胞におけるグルコース取り込みの増強
ヒト及び他の哺乳類では、骨格筋は、通常、全身のインスリン刺激性グルコース取り込みの75%を占める。骨格筋のインスリンへの応答能力の低下は、全身のグルコース恒常性を著しく破壊する。骨格筋におけるグルコース取り込みのプロセスは、主にインスリンに応答するPI3K/Pdk1/Aktシグナル伝達カスケードを介して媒介されることが十分に実証されている。インスリン抵抗性Leprdb/dbマウスの骨格筋におけるPI3K/Pdk1/Aktシグナル伝達分子の活性化(図18)は、化合物#43が骨格筋のグルコース取り込みを直接調節し、そのプロセス中にインスリン作用を増強し得る可能性が高いことを示唆している。これらの可能性を試験するために、分化したマウスC2C12(骨格筋)細胞でグルコース取り込み実験を実施した。
簡潔に述べると、同数のC2C12細胞を96ウェルプレートに播種し(5000細胞/ウェル)、10%FBS DMEM培地中で37℃、5日間培養した。前述のように、これらの細胞を、骨格筋細胞になるように、0.5%ウマ血清(Sigma)を含有するDMEM培地を使用して7日間分化させた(Misu et al,Cell Metabolism 12,483-495,2010)。完全に分化したC2C12細胞を、無血清グルコースフリーDMEM培地中で0.006%DMSO(化合物#43溶媒)または化合物#43(300または600ppb)の存在下または非存在下で、一晩前処置した。次いで、これらの細胞を、グルコース/フェノールレッドフリーのDMEM培地中、処置なし(基底)またはインスリン(200nM)、化合物#43(300及び600ppb)、もしくはインスリンと化合物#43の両方の存在下で、37℃、1.5時間インキュベートした。処置後、細胞を1mM 2-デオキシグルコース(2DG)の存在下で室温、30分間インキュベートし、PromegaのGlucose Uptake-Gloアッセイキットを使用して発光分析に供した。検出した発光シグナルは、培養した分化C2C12細胞へのグルコースの取り込みを表す。
図19に示すように、200nMのインスリン単独または600ppbの化合物#43での処置により、これらの培養骨格筋細胞においてグルコース取り込みが16~19%増加した(ただし統計的に有意ではない、基底群と比較した場合)が、300ppbの化合物#43では増加しなかった。200nMのインスリンと同様の効力を有する高用量(600ppb)の化合物#43で処置した後、これらの培養骨格筋細胞のグルコース取り込みが増加する傾向があったことから、600ppbを上回る用量での化合物#43により、これらの分化した筋肉細胞におけるグルコース取り込みを直接かつ有意に高めることができる可能性がある。いずれにせよ、これらの結果から、試験した用量600ppbの化合物#43が200nMインスリンと比較可能な効力でグルコース取り込みを刺激することができることが示された。
インスリン(200nM)及び化合物#43(300ppb)の両方の共処置は、分化したC2C12細胞によるグルコース取り込みのロバストかつ有意な増加(63%)をもたらした(図19)。200nMのインスリン及び600ppbの化合物#43の共処置後、これらの分化した細胞で、より顕著なグルコース取り込みの増加(79%)が観察された。インスリンと化合物#43の両方の共処置後の分化したC2C12細胞におけるグルコース取り込みの増加の程度は、インスリンまたは化合物#43単独よりもはるかに高かったが、このことは、インスリンと化合物#43の間に相乗作用があったことを示している。したがって、上記の研究は、化合物#43がインスリンと協働して、分化した骨格筋細胞のグルコース取り込みを有意に増強することができることを示している。
総合すると、上記の研究は、化合物#43がインスリン抵抗性糖尿病Leprdb/dbマウスの骨格筋におけるインスリンシグナル伝達を活性化または回復させ(Pdk1/Akt/Foxo1のリン酸化の増強によって示される)、インスリン作用を増強して培養骨格筋(分化したC2C12)細胞のグルコース取り込みを増強することができることを示唆している。これらの結果は、化合物#43が血糖値を低下させ、インスリン抵抗性Leprdb/dbマウスの耐糖能を向上させることができるというさらなる分子的エビデンスを提供する(図3~図8)。
実施例8:化合物#43処置後のインスリン抵抗性Leprdb/dbマウスの骨格筋及び肝臓、ならびに培養したマウス骨格筋細胞及びヒト肝細胞におけるインスリン受容体(Insr)シグナル伝達の活性化、ならびに分化マウスC2C12(骨格筋)細胞及びヒト肝臓HepG2細胞におけるグルコース取り込みのための細胞内小胞から細胞膜へのGLUT4移行のためのAS160-keyのリン酸化に対する化合物#43のインスリン様効果
肝臓と骨格筋において、インスリンがInsrのαサブユニットに結合した後、Insrβは、Y1146で始まり、次いでY1150/51において、チロシン自己リン酸化を受け、その後、PI3K/Pdk1を活性化してAktのリン酸化を誘導することが十分に実証されている。TBC1ドメインファミリーメンバー4(TBC1D4)としても知られるAS160は、GLUT4タンパク質を細胞質小胞に保持する上で重要な役割を果たすAkt基質である。インスリンに応答したInsr/PI3k/Pdk1/Aktシグナル伝達の活性化はAS160をリン酸化し、GLUT4タンパク質の細胞質小胞から細胞膜への移行を促進して、骨格筋細胞及び肝細胞へのグルコース取り込みを促進する。したがって、化合物#43がInsrβのチロシンリン酸化を引き起こす可能性について、インスリン抵抗性Leprdb/dbマウスの骨格筋及び肝臓、ならびに分化したマウスC2C12(骨格筋)細胞及びヒト肝臓HepG2細胞で調べた。さらに、化合物#43がインスリンを模倣するが迂回して、分化したマウスC2C12細胞のインスリン受容体の下流シグナル伝達分子であるAktを活性化することができるかどうかを調べた。さらに、Akt標的基質であるAS160のリン酸化に対する化合物#43の効果を、分化したマウスC2C12細胞とヒト肝臓HepG2細胞の両方で調べた。
材料及び方法
化合物
化合物#43は、Alltech,Incの化学実験室で合成した。試験したこの化合物の純度は、HPLCによる測定で≧99%であることが確認された。
Leprdb/dbマウスにおける化合物#43のin vivo処置
出生後38日目の雄のLeprdb/dbマウス(C57BL/6J系統、Jackson Laboratoryから購入)に、化合物#43(0.2%DMSOまたは体重1キログラムあたり0.136mg、滅菌生理食塩水で希釈)を含有する生理食塩水(0.09%NaCl)を52日間、毎日腹腔内注射した。処置後、腓腹筋の骨格筋及び肝臓の試料を採取し、-80℃で保存した。
骨格筋及び肝臓タンパク質の調製
凍結した骨格筋及び肝臓組織を、完全プロテイナーゼ及びホスファターゼ阻害剤(Thermo-Fisher Scientific,Waltham,MA)を含有する無菌の氷冷PBS中で細かく刻み、組織ホモジナイザー(Thermo-Fisher Scientific,Waltham,MA)を使用して均質化した。これらの組織ホモジネートを、タンパク質を抽出するために完全プロテイナーゼ/ホスファターゼ阻害剤を含有するThemo-Fisherの既製RIPA緩衝液(ホモジネート1部/RIPA緩衝液2部)で希釈した。ホモジネート中のタンパク質をRIPA緩衝液中で4℃、一晩抽出した。一晩抽出したこれらのタンパク質/組織溶解物を12000×gで4℃、30分間遠心分離し、これらの組織溶解物の上清中のタンパク質レベルを、Pierce Micro-BCAタンパク質アッセイキット(Thermo Scientific-Piece Biotechnology,Rockford,IL)を使用して、製造元のプロトコールに従って測定した。
HepG2細胞及びC2C12細胞の細胞培養、C2C12細胞の分化、細胞処置ならびに培養細胞タンパク質抽出物の調製
ヒト肝癌HepG2細胞及びマウス筋芽細胞C2C12細胞株は、アメリカ合衆国培養細胞系統保存機関(ATCC,Manassas,Virginia)から購入した。HepG2細胞は10%FBSを添加したイーグル最小必須培地(EMEM)中で培養し、C2C12細胞は10%FBSを添加したDMEM培地中で増殖させた。
HepG2細胞を6ウェルプレートに播種し(7×105細胞/ウェル)、10%FBS EMEM培地中で30時間培養した。次いで、これらの細胞をPBSで2回洗浄して残留血清を除去し、通常のEMEM培地中で一晩血清飢餓状態にした。これらの血清飢餓状態のHepG2細胞を、無血清培地中、化合物の非存在下、または化合物#43(600ppb)で、0分間(処置の直前)、30分間及び60分間、処置した。
C2C12細胞を分化させるために、同数のこれらの筋芽細胞を最初に12ウェルプレートに播種し(60,000細胞/ウェル)、10%FBS DMEM培地中で37℃、5日間培養した。これらの細胞には、新鮮な10%FBS DMEM培地を毎日補充した。前述と同様に、培養後5日目に、0.5%ウマ血清(Sigma)を含有するDMEM培地(分化培地)を使用してC2C12細胞を7日間分化させた(Misu et al,Cell Metabolism 12,483-495,2010)。分化後7日目に、分化したC2C12細胞をPBSで2回すすぎ、無血清DMEM培地中で一晩インキュベートした。次いで、これらの血清飢餓状態の細胞を、無血清DMEM培地中、処置なし(基底)またはインスリン(200nM)、化合物#43(600ppb)、またはインスリンと化合物#43の両方で、37℃、5分間及び60分間処置した。
上記の処置の後、培養したHepG2細胞または分化したC2C12細胞を氷冷PBSで2回すすぎ、氷上で完全プロテイナーゼ及びホスファターゼ阻害剤(Thermo-Fisher Scientific,Waltham,MA)を含有する氷冷RIPA緩衝液に氷上で30分間溶解させた。細胞溶解物を、セルスクレーパー及びホールピペットを使用して回収し、次いで12000×gで4℃、30分間遠心分離してDNAペレットを除去し、タンパク質抽出物を取得した。これらの細胞溶解物の上清中のタンパク質レベルは、Pierce Micro-BCAタンパク質アッセイキット(Thermo Scientific-Piece Biotechnology,Rockford,IL)を使用して製造元のプロトコールに従って測定した。
ウエスタンブロット分析
前述のように、100マイクログラムの骨格筋または肝臓組織タンパク質、5マイクログラムのHepG2細胞タンパク質抽出物、または8マイクログラムの分化したC2C12細胞タンパク質抽出物をSDS-PAGEゲル分離に供し、次いでPVDF膜に転写した(Reddy et al. 2008 Science)。5%(w/v)のウシ血清アルブミン(Sigma,St.Louis,MO)を含有するリン酸緩衝生理食塩水(PBS)中で膜をブロックし、特異的な一次抗体の存在下でインキュベートした後、HRP結合抗マウスまたは抗ウサギ二次抗体(1:5000希釈、Cell Signaling Inc.)の存在下でインキュベートした。β-チューブリンに対する抗体(LI-COR bioscience)以外のすべての一次抗体はCell Signaling Incから購入した。アマシャムのenhanced chemiluminescence Western Blotting Prime Detection試薬(GE Healthcare Lifescience,Pittsburgh,PA)を使用して、膜ブロット上の陽性シグナルを検出した。LI-COR Odyssey Fc Image system(Lincoln,Nebraska)を使用して、膜ブロット上のこれらの発光シグナルの画像をキャプチャした。GE WB ECL-prime-detectionプロトコール(GE Healthcare Lifescience,Pittsburgh,PA)に記載されているように、同じ膜ブロットを除去し、別の抗体で再ブロットした。NIH ImageJソフトウェアを使用してウエスタンブロットのタンパク質バンド濃度を測定し、各試料のβ-チューブリンまたはACTBタンパク質レベルで正規化した。データは、群ごとに3~5個の試料の平均値±SEMとして提示する。
Y1146またはY1150/51のホスホInsrβの酵素免疫測定法(ELISA)
生理食塩水または化合物#43で処置したLeprdb/dbマウス由来の肝臓タンパク質試料を、PathScanホスホインスリン受容体β(Tyr1146またはTyr1150/1151)Sandwish ELISAキット(Cell Signaling Technology,Danvers,MA)を用いたELISAアッセイに供し、その際、タンパク質抽出物を捕捉するpInsrβY1146またはpInsrβY1150/1151抗体の存在下で4℃、一晩インキュベートする点(プロトコールに記載されているように37℃で2時間インキュベートする代わりに)以外は製造元のプロトコールに従った。Y1146での蛍光Insrβの検出には400マイクログラムの肝臓タンパク質抽出物を使用し、Y1150/1151での蛍光Insrβの検出には600マイクログラムの肝臓タンパク質抽出物を使用した。試験した試料の450nmでの吸光度(OD450)を、Bio-tekマイクロプレートリーダーを使用して記録した。各試料中の内部タンパク質対照であるβ-チューブリンのレベルを、100μgのタンパク質抽出物と特異的なβ-チューブリンモノクローナル抗体を使用したウエスタンブロット分析によって測定し、続いてNIH Image Jソフトウェアを使用して、ウエスタンブロットにおけるβ-チューブリンタンパク質のバンド濃度の定量分析を行った。次いで、試験した各試料のOD450をそのβ-チューブリンタンパク質レベルで正規化し、Y1146またはY1150/1151でのホスホInsrβのレベルを取得した。
統計解析
適用可能な場合、スチューデントt検定を使用して、処置群間の差異の統計的有意性を判定し、0.05未満のP値を有意とみなした。
結果:
1.化合物#43による慢性処置後のインスリン抵抗性Leprdb/dbマウスの骨格筋におけるInsrβのチロシンリン酸化の増強
上述のように、骨格筋は、全身性インスリンに応答するグルコース恒常性にとって絶対に不可欠である。動物実験では、化合物#43が、血糖値及びHbA1cレベルを低下させ、インスリン抵抗性Leprdb/dbマウスの耐糖能を向上させることができることが明らかになった(図3~図8)。これらの効果は、骨格筋で機能するインスリン受容体の潜在的な回復によるものである可能性があり、それにより、これらのインスリン抵抗性Leprdb/dbマウスにおいてグルコース取り込みを生じさせることができる。化合物#43で処置したインスリン抵抗性Leprdb/dbマウスの骨格筋でのPdk1及びAktのリン酸化の増強(図18)は、骨格筋において、化合物#43がインスリンを迂回するが模倣するか、またはインスリン作用を回復させて、Pdk1/Aktカスケードの上流のインスリンシグナル伝達カスケード分子を活性化することを示唆している。Y1146でのInsrβのチロシンリン酸化は、インスリンのInsrαへの結合後の活性化インスリン受容体シグナル伝達の最初の工程を反映しており、これは骨格筋におけるPI3K/Pdk1/Aktシグナル伝達の上流の重要な事象である。したがって、化合物#43による慢性処置が、これらのインスリン抵抗性Leprdb/dbマウスの骨格筋におけるInsrβのリン酸化を調節することができるかどうかを調べた。
出生後38日目のLeprdb/dbマウスに、生理食塩水(0.2%の化合物溶媒DMSOを含有)または化合物#43を、体重1kgあたり0.136mgの化合物#43の用量で52日間、毎日腹腔内注射した。上記の処置の後、骨格筋試料を回収し、前述のインスリンシグナル伝達分子に対する特異的抗体を使用したウエスタンブロット分析に供した。
図20Aに示すように、チロシン1146でのリン酸化Insrβのタンパク質バンド濃度は、化合物#43処置Leprdb/dbマウスの骨格筋において生理食塩水処置マウスよりもはるかに高かったが、全Insrβではそうではなかった。これらのウエスタンブロットの定量分析により、生理食塩水で処置したマウスと比べた場合、化合物#43で処置した後のLeprdb/dbマウスの骨格筋において、リン酸化Insrβタンパク質レベルがロバストに増加した(約2.5倍増加)ことが示された(図20B)。対照的に、化合物#43処置マウスの骨格筋の全Insrβタンパク質レベルの有意な変化はなかった(図20C)。これらの結果は、化合物#43で処置したLeprdb/dbマウスの骨格筋におけるInsrの重要なインスリンシグナル伝達分子であるPdk1及びAktのリン酸化の増加の観察と一致している(図18)。総合すると、これらの結果は、Leprdb/dbマウスがインスリンに応答できないように設計されているにもかかわらず、化合物#43での慢性処置後、Leprdb/dbマウスの骨格筋においてインスリン受容体が活性化されることを明確に示している。言い換えれば、これらの結果は、これらの重度のII型インスリン抵抗性糖尿病マウスの骨格筋において、化合物#43がインスリン作用を回復させるか、インスリンを迂回するか、またはその両方により、Insrβのチロシンリン酸化を刺激し、次いでPI3K/Pdk1/Aktシグナル伝達を活性化することができることを示唆している。
これらの知見は、血流からのグルコースクリアランスの増強、空腹時血糖値の低下、HbA1Cレベルの低下、及びインスリンとの相乗作用による骨格筋のグルコース取り込みの増強の観察とともに、化合物#43が、I型及びII型糖尿病のいずれに対しても効果的な治療薬であり得ることを強く示している。
2.化合物#43は、インスリンを模倣するが迂回して、分化したマウスC2C12(骨格筋)細胞のY1146、Pdk1、Akt、及びAS160のInsrβにおけるリン酸化を刺激する
化合物#43が骨格筋におけるインスリン受容体シグナル伝達を直接活性化することができるかどうかをさらに調べるため、血清飢餓状態の分化マウスC2C12(骨格筋)細胞を、無血清及びグルコースフリーのDMEM培地中、化合物#43(600ppb)、インスリン(200nM、陽性対照)、またはその両方の非存在下または存在下で、非常に短い期間(すなわち、5分間)及び60分間、インキュベートした。次いで、ウエスタンブロット分析を実行して、これらの分化した骨格筋細胞における活性化Insr(すなわち、Y1146のpInsr)ならびにリン酸化Pdk1、Akt及びAs160を含むその下流シグナル伝達分子のタンパク質発現レベルを調べた。
これらの培養骨格筋細胞における活性化Insrのタンパク質発現を、インスリンまたは化合物#43処置後に測定した。予想通り、分化したC2C12細胞において、5分間のインスリンでの処置により、Y1146でpInsrβが有意に増加したが、全Insrβはそうではなかった(図21A~図21B)。さらに、600ppbの化合物#43処置でも、これらの培養骨格筋細胞においてY1146のpInsrβが有意に増加したが、全Insrβはそうではなかった(図21A~図21B)。インスリン及び化合物#43の5分間の共処置では、これらの分化したC2C12細胞のpInsrβタンパク質レベルが増加する傾向があったが(図21A~図21B)、このことは、化合物#43またはインスリン処置後のInsrβの刺激は一過性の事象であることを示している。実際、インスリン、化合物#43またはその両方による長期処置(60分)では、pInsrβレベルは低下したが(図21C~図21D)、このことは、骨格筋細胞において、インスリンまたは化合物#43処置後のInsrの活性化が実際に一過性の事象であり、Insrβのチロシンリン酸化を調節する負のフィードバックが存在することをさらに示している。いずれにせよ、化合物#43の短時間(5分間)の処置後にこれらの培養骨格筋細胞で観察されたY1146でのInsrβのチロシンリン酸化の増強は、上記のインスリン抵抗性のLeprdb/dbマウスにおけるInsrβの活性化と一致している(図20)。これらの分化したC2C12細胞を、完全に無血清の条件下で、血清飢餓状態にし、培養し、化合物#43で処置したことから、これらの結果は、これらの分化した骨格筋細胞において、化合物#43が、インスリンを模倣するが迂回して、Insrβを直接かつ迅速に活性化することができることを示唆している。
Insrβのチロシンリン酸化は、インスリンに応答してPI3K/Pdk1シグナル伝達を活性化し、Aktのリン酸化を増強するため、これらの分化したC2C12骨格筋細胞において、化合物#43が、インスリンを模倣してAktリン酸化を調節することができるかどうかを調べた。図21Aに示すように、5分間のインスリン処置により、リン酸化Aktタンパク質レベルの有意な増加がもたらされたが、全Aktタンパク質レベルは有意に増加しなかった。しかしながら、このAktのリン酸化の増加は、インスリン処置の60分後のこれらの分化したC2C12細胞では観察されなかった(図21C~図21D)。対照的に、化合物#43の5分間の処置では、これらのC2C12細胞においてリン酸化Aktタンパク質レベルの有意な増加は引き起こされなかったが、化合物#43の60分間の処置後、これらの細胞においてリン酸化Aktタンパク質レベルのロバストな増加が観察された(図21C~図21D)。インスリンと化合物#43の両方の共処置により、試験した両方の時点(6分及び60分、図21)でpAktタンパク質レベルが有意に増加した。インスリンと化合物#43の共処置群におけるAktのリン酸化の増加レベルは、5分ではインスリン単独と比較可能であったが、一方、インスリンと化合物#43の両方の共処置後60分でのリン酸化Aktのタンパク質レベルは、化合物#43単独処置後のレベルとほぼ同一であった。これらの結果は、これらの骨格筋細胞でインスリンがInsr下流シグナル伝達分子Aktを一時的に活性化することができることを示唆している。さらに、これらの結果は、化合物#43が、インスリンほど速くはないものの、インスリンを模倣するが迂回して、これらの細胞のAktを活性化することができることを明らかにした。さらに、これらの結果は、これらの分化した骨格筋細胞におけるAktリン酸化の刺激においてインスリンと化合物#43の間に相乗作用がなかったことも示している。しかしながら、これらの結果は、化合物#43とインスリンの両方の共処置により、活性化されたAktシグナル伝達の期間を引き延ばすことができることを明らかにした。
上述のように、AS160はAkt標的基質であり、骨格筋細胞内の小胞におけるGLUT4タンパク質の保持に必要である。インスリン/Insrβ/PI3K/Aktシグナル伝達に応答したS588でのAS160のリン酸化により、細胞質小胞から細胞膜へのGLUT4タンパク質の移行が引き起こされ、グルコースの取り込みが促進される。in vivoでの研究により、化合物#43が、インスリン抵抗性Leprdb/dbマウスである糖尿病マウスにおける血糖値を低下させ、耐糖能を向上させることができることが明らかになった(図3~図8)。さらに、培養骨格筋細胞では、化合物#43処置後、特にインスリンと化合物#43の両方での共処置後に、グルコース取り込みの増加が観察された(図19)。したがって、化合物#43は、AS160のリン酸化によるグルコース取り込みのために骨格筋細胞のGLUT4移行を調節することができる可能性がある。したがって、無血清培地中、インスリン、化合物#43、またはその両方の5分間及び60分間の処置後の分化C2C12(骨格筋)細胞において、S588でのリン酸化AS160のタンパク質レベルを測定した。
図21A~図21Bに示すように、5分間のインスリン処置は、これらの分化したC2C12細胞において、AS160のタンパク質発現に影響を及ぼさなかったが、S588でのAS160のリン酸化の有意な増加(約2倍)をもたらした。しかしながら、60分間のインスリン処置後、リン酸化AS160の明らかな増加は観察されなかった(図21C~図21D)。これらの結果は、インスリンが骨格筋細胞におけるGLUT4移行のためにAS160のリン酸化を一時的に増強することができることを示唆している。
対照的に、化合物#43単独での5分間の処置は、これらの分化したC2C12細胞におけるリン酸化AS160及び全AS160のタンパク質レベルに影響を及ぼさなかった(図21A~図21B)。しかしながら、化合物#43で60分間処置した後の骨格筋細胞において、S588でのリン酸化AS160のレベルのロバストな増加(約6倍)が観察された(図21C~図21D)。これらの結果は、これらの培養骨格筋細胞において、化合物#43が、インスリンほど速くはないものの、インスリンを模倣して、GLUT4移行のためのAS160のリン酸化を誘導することができることを示唆している。
試験した両方の時点(5分及び60分)でのインスリン及び化合物#43の両方での共処置は、リン酸化AS160タンパク質レベルの有意な増加をもたらした(図21)。5分での共処置群におけるAS160のリン酸化の増加のレベルはインスリン単独と比較可能であったが(図21A~図21B)、このことは、共処置群で観察されたAS160リン酸化が主にこの時点でのインスリンの効果によるものであることを示している。同様に、処置後60分で、共処置群のリン酸化AS160のタンパク質レベルは、化合物#43単独処置後のレベルとほぼ同一であったが(図21C~図21D)、このことは、観察されたこの時点での共処置群のAS160のリン酸化が、主に化合物#43単独の効果によるものであることを示している。これらの結果は、これらの分化したインスリン応答性細胞において、化合物#43とインスリンの両方の共処置がAS160のリン酸化の持続時間を増加させ、グルコース取り込みのためのGLUT4移行を促進することができることを示唆している。
総合すると、これらの結果は、化合物#43が、分化したC2C12細胞において、インスリンを模倣するが迂回して、Insr(Insrβのチロシンリン酸化によって示される)を直接かつ迅速に活性化することができることを示している。これは、インスリン抵抗性Leprdb/db糖尿病マウスの骨格筋で観察されたInsrβの活性化と一致する(図20)。活性化されたInsrは、次いで、骨格筋のPI3K/Pdk1/Aktシグナル伝達を活性化して(図18に示すin vivo研究でさらに裏付けられる)、その後、AS160(公知のAkt標的基質)をリン酸化し、グルコース取り込みのための細胞質小胞から細胞膜へのGLUT4移行を増強することができる。実際、化合物#43の処置後、特にインスリンと化合物#43の両方の共処置後に、培養骨格筋細胞においてグルコース取り込みの増加が観察された(図19)。化合物#43処置後の骨格筋のグルコース取り込みの増強は、糖尿病マウスの血糖値を低下させ、耐糖能を向上させることができる(図3~図8)。要するに、これらの結果は、化合物#43が、骨格筋細胞において、インスリンを模倣するが迂回して、Insrβ/PI3K/Pdk1/Aktシグナルを直接活性化してAS160をリン酸化し、細胞質小胞から細胞膜へのGLUT4移行を増強して骨格筋へのグルコース取り込みを促進し、それにより、1型及び2型糖尿病の両方の主要な特徴及び病理に対抗することができることを示している。
3.化合物#43での慢性処置後のインスリン抵抗性Leprdb/dbマウスの肝臓におけるInsrβのチロシンリン酸化の増強
実施例6に記載の研究は、化合物#43がin vivio及びin vitroの両方で肝臓のPdk1/Akt/Foxo1のリン酸化を増強することができることを明らかにした(図12~図14)。これらすべての効果は、上記の骨格筋細胞と同様に、化合物#43処置後の肝細胞における上流シグナル伝達分子Insrの活性化に起因し得る。前述のように、Y1146及びその後のY1150/1151でのInsrβのチロシンリン酸化は、インスリンのInsrαへの結合後の活性化インスリン受容体シグナル伝達の最初の数ステップを反映しており、肝臓におけるPI3K/Pdk1/Akt/Foxo1シグナル伝達の上流の重要な事象である。したがって、化合物#43による慢性処置により、これらのインスリン抵抗性Leprdb/dbマウスの肝臓におけるInsrβのチロシンリン酸化を調節することができるかどうかを調べた。
出生後38日目のLeprdb/dbマウスに、生理食塩水(0.2%化合物溶媒DMSOを含有する)または化合物#43を、体重1kgあたり0.136mgの化合物#43の用量で52日間、毎日腹腔内注射した。上記の処置後、肝臓試料を採取し、Y1146の蛍光Insrβ及びY1150/1151の蛍光Insrβ(OD450を取得するために)のELISAアッセイ及び内部対照であるβ-チューブリンのウエスタンブロット分析に供した。各試料のOD450をそのβ-チューブリンタンパク質レベルで正規化した後、各試料のY1146またはY1150/1151の蛍光Insrβのレベルを取得した。
図22Aに示すように、生理食塩水処置マウスと比較した場合、化合物#43での処置後、Leprdb/dbマウスの肝臓において、チロシン1146のリン酸化Insrβのタンパク質レベルが有意に増加した(約2.9倍増加)。同様に、チロシン1150/1151のリン酸化Insrβのタンパク質レベルも、Leprdb/dbマウスの肝臓において、生理食塩水処置マウスと比較した場合、化合物#43での慢性処置後、有意に増加した(約2.95倍増加)(図22B)。これらの結果は、化合物#43で処置したLeprdb/dbマウスの肝臓におけるInsrの重要なインスリンシグナル伝達分子であるPdk1及びAktのリン酸化の増加の観察と一致する(図12)。総合すると、これらの結果は、Leprdb/dbマウスがインスリンに応答できないように設計されているにもかかわらず、化合物#43での慢性処置後にLeprdb/dbマウスの肝臓においてインスリン受容体が活性化されることを明確に示している。言い換えれば、これらの結果は、化合物#43が、これらの重度II型インスリン抵抗性糖尿病マウスの肝臓において、インスリン作用を回復させるか、インスリンを迂回するか、またはその両方で、Insrβのチロシンリン酸化を刺激し、その後、PI3K/Pdk1/Aktシグナル伝達を活性化することができることを示唆している。
4.化合物#43は、インスリンを模倣するが迂回して、ヒト肝臓HepG2細胞のY1146でのInsrβ及びS588でのAS160のリン酸化を刺激する
化合物#43が、インスリンを模倣するが迂回して、肝細胞のインスリン受容体を直接活性化することができるかどうかをさらに調べるために、ヒト肝臓HepG2細胞を一晩血清飢餓状態にし、次いで無血清及びグルコースフリーのDMEM培地中、化合物#43(600ppb)の存在下で30分間及び60分間インキュベートした。ウエスタンブロット分析を実施して、これらのヒト肝細胞における活性化INSR(すなわち、Y1146のpINSRβ)のタンパク質発現レベルを調べた。図23に示すように、600ppbの化合物#43の30分及び60分の処置の両方で、これらの培養肝細胞においてY1146のリン酸化INSRβが有意に増加したが、全INSRβは増加しなかった。これらの結果は、化合物#43を同じ期間処置した後のHepG2細胞におけるINSR下流シグナル伝達分子PDK1及びAKTのリン酸化の増加と一致する(図13)。これらの肝細胞は血清飢餓状態にあり、化合物#43の処置は完全に無血清の条件下で行われたことから、これらの結果は、化合物#43が、肝細胞において、グルコース産生のためのG6PC発現の阻害、及びグルコース取り込みのためのGLUT4発現の刺激のために、インスリンを模倣するが迂回して、INSRβのチロシンリン酸化を直接刺激し、PI3K/PI3K/AKTの活性化をもたらし、FOXO1を不活性化することができることを示唆している。
FOXO1に加えて、AS160は、脂肪細胞及び骨格筋などのインスリン標的組織における細胞内小胞から細胞膜へのグルコース移行に重要な役割を果たす別のAKT標的基質である。in vivo及びin vitroでの研究により、化合物#43が、糖尿病マウスの血糖値を低下させ、耐糖能を向上させることができ(図3~図8)、培養AML-12肝細胞のグルコース取り込みを増強することができる(図17)ことが示されたが、このことは、肝臓におけるグルコース取り込みの増加が、I型及びII型糖尿病に対抗して血糖値を下げ、耐糖能を向上させるうえでの化合物#43のメカニズムの1つであり得ることを示唆している。化合物#43処置後に誘発されるグルコース取り込みの増強は、肝細胞へのグルコース取り込みを刺激するための、GLUT4発現の増強(図15~図16で示す)、潜在的なGLUT4移行の増強、またはその両方に起因し得る。後者のシナリオに対処するために、化合物#43で処置したHepG2細胞のリン酸化AS160(AKT標的基質)タンパク質レベルを測定した。
図23に示すように、これらの培養ヒト肝細胞において、600ppbの化合物#43による30分及び60分の処置の両方で、S588でのリン酸化AS160が有意に増加したが、全AS160タンパク質レベルは増加しなかった。これらの結果は、化合物#43で同じ期間処置した後のHepG2細胞におけるAS160の上流の2つの重要なシグナル伝達分子であるPDK1及びAKTのリン酸化の増加と一致する(図13)。AS160のリン酸化は、グルコース取り込みのために細胞内小胞から細胞膜へのGLUT4移行を増強できることから、これらの結果は、化合物#43が、ヒト肝細胞において、インスリンを模倣するが迂回して、グルコース取り込みのための細胞質小胞から細胞膜へのGLUT4移行を刺激することができることを示唆している。これを裏付けるものとして、培養AML-12肝細胞において、化合物#43を1.5時間処置した後、グルコース取り込みの増強が観察されたことが想起されるであろう(図17)。したがって、肝細胞では、化合物#43は、Insr/PI3k/Pdk1/Akt/Foxo1シグナル伝達を介してGLUT4発現を刺激するだけでなく(図12~図17に示す)、Insr/PI3K/Pdk1/Akt/AS160を介して細胞質小胞から細胞膜へのGLUT4移行も増強し(図12~図17、図23で示す)、したがってグルコースの取り込みを増強するであろう。
まとめると、上記の研究はすべて、化合物#43が、インスリン抵抗性糖尿病Leprdb/dbマウスの骨格筋及び肝臓の両方でインスリン受容体機能を回復させることができる(Insrβのチロシンリン酸化の増強によって示される)ことを明らかにしている。in vitroでの研究は、化合物#43が、インスリンを厳密に模倣して、骨格筋細胞及びヒト肝細胞の両方でインスリン受容体を活性化することができることをさらに示している。さらに、これらの結果は、インスリンと同様に、化合物#43が、Pdk1/Aktシグナル伝達を活性化して、培養骨格筋細胞とヒト肝細胞の両方でAS160(AKT標的基質)のリン酸化を誘導することができることを示している。AS160のリン酸化の増強により、肝細胞及び骨格筋細胞の両方において、細胞質小胞から細胞膜へのGLUT4移行が促進され、グルコースの取り込みが促進され、糖尿病の状況において血糖値が低下し、耐糖能が向上する。
これら2つの組織、すなわち肝臓及び骨格筋は、II型糖尿病の発症及び病因においてはるかに最も重要であることを強調すべきである。化合物#43は、これらの組織におけるインスリンシグナル伝達カスケードを回復させる能力により、II型糖尿病の治療において非常に大きな治療的価値を有し得る。
さらに、化合物#43は、インスリンが添加されていないインスリン応答性細胞においてインスリン模倣薬として機能することができるため、I型糖尿病の有効な治療薬にもなり得るという強力な可能性が存在する。
実施例9:化合物#43の慢性処置は、インスリン抵抗性糖尿病のdb/dbマウスにおいて血清インスリン及びアラニンアミノトランスフェラーゼ(ALT)レベルの低下をもたらしたが、血清クレアチニンレベルの低下をもたらさなかった
材料及び方法
化合物
化合物#43は、Alltech,Incの化学実験室で合成した。試験したすべての化合物の純度は、HPLCによる測定で≧99%であることが確認された。
動物
5週齢の雄の糖尿病偶発変異(レプチン受容体変異)Leprdb/dbマウス(C57BL/6J系統)は、Jackson Laboratory(Bar Harbor,Maine)から購入し、病原体フリーの飼育室に収容し、餌と水を自由に摂取させた。3か月齢の野生型(非糖尿病)C57マウスもJackson Laboratoryから購入した。
化合物#43による慢性処置
38日齢の雄のLeprdb/dbマウスに、0.2%DMSOを含有する生理食塩水(0.09%NaCl)及び化合物#43(体重1キログラムあたり0.136mg、無菌生理食塩水で希釈)を52日間、毎日腹腔内(ip)注射した。3か月齢の野生型(非糖尿病)C57マウスの血清も採取した。処置後、これらの血清試料を採取し、インスリン、ALT及びクレアチニンアッセイに供した。
血清インスリン、ALT及びクレアチニンアッセイ
インスリン、ALT及びクレアチニンの血清中レベルを、それぞれ、Themo-Fisher ScientificのInsulin Mouse ELISAキット(カタログ番号EMINS)、SigmaのALT Activity Assayキット(カタログ番号MAK052)、及びAbcamのCreatinine Assayキット(カタログ番号Ab65340)を使用して、製造元のプロトコールに従って測定した。
統計解析
適用可能な場合、スチューデントt検定を使用して、生理食塩水処置群と化合物処置群との差異の統計的有意性を判定し、0.05未満のP値を有意とみなした。データは、図中に示すマウスの数の平均値±SEMとして提示する。
結果及び考察
図24Aに示すように、Leprdb/dbマウスは、インスリンレベルが約2586μIU/ml(非糖尿病の野生型マウスよりも約100倍高い)の高インスリン血症を示した。しかしながら、化合物#43処置により、そのレベルは非糖尿病の野生型マウスよりも依然として高いものの、血清インスリンレベルが劇的に減少(約80%の減少)した(図24A)。これらの結果は、化合物#43が糖尿病患者の高インスリン血症を治療する可能性を有していることを示唆している。
アラニンアミノトランスフェラーゼ(ALT)試験は、一般的には肝臓損傷を検出するために使用される。図24Bに示すように、生理食塩水で処置したLeprdb/dbマウスの血清ALTレベルは、非糖尿病マウスよりも有意に高かった。しかしながら、化合物#43処置により、血清ALTレベルが有意に低下した。これらの結果は、化合物#43の慢性処置(52日間毎日処置)が、肝臓毒性を示さず;むしろ、肝臓損傷に対する保護効果を有していたことを示唆している。
血中クレアチニン試験は、腎機能を評価するために広く使用されており、クレアチニンレベルの上昇は、腎機能障害または腎疾患を意味する。図24Cに示すように、化合物#43処置後、Leprdb/dbマウスの血清クレアチニンレベルに有意な変化はなかった。これらの結果は、化合物#43の慢性処置(52日間毎日処置)が腎機能に毒性効果を及ぼす可能性が低いことを示唆している。
総合すると、上記の結果は、糖尿病の被験体の高インスリン血症に対する化合物#43の潜在的な使用を示唆している。さらに、上記の結果は、化合物#43が肝臓及び腎臓の機能にほとんどまたはまったく毒性効果を及ぼさないことも示唆している。むしろ、化合物#43は肝臓の損傷に対する保護効果を示し得る。
結論(作用機序;図25)
本開示における実験結果を、実線の赤い矢印で示し、一方、予想される結果(公開された文献に基づく)を破線の矢印で示す。化合物#43は、インスリンを模倣するが迂回して、細胞膜の内表面上でインスリン受容体βサブユニットのチロシンリン酸化を迅速に誘導することができるため、インスリンが細胞表面でインスリン受容体αに結合して活性化する必要性はなくなる。これにより、肝臓と骨格筋の両方でPI3K/PDK1/AKTシグナル伝達カスケードが活性化される。言い換えると、インスリンまたは活性な細胞表面インスリン受容体を存在させる必要なく、正常なインスリンシグナル伝達を回復させることができる。肝細胞では、AKTの活性化によりFOXO1のリン酸化のロバストな増加が生じ、FOXO1の直接標的遺伝子G6PCの発現が有意に低下し;これは、糖尿病の被験体の肝細胞におけるグルコース産生の阻害につながる。さらに、FOXO1間接標的遺伝子GLUT4の発現、及び細胞質小胞から細胞膜へのGLUT4移行の重要な因子であるAKT標的基質AS160(TBC1D4)のリン酸化は、化合物#43処置後の肝細胞において増強され、その結果、肝細胞膜のGLUT4輸送タンパク質が増加し、血流からのグルコース取り込みが向上する。これらはすべて、I型及びII型糖尿病の被験体の耐糖能の向上を示す。骨格筋細胞では、化合物#43は、インスリンを模倣するが迂回して、INSRβ/PDK1/AKTシグナル伝達を活性化し、AS160(TBC1D4)のリン酸化をもたらすことができる。ここでも、これによって、細胞質小胞から細胞膜へのGLUT4移行が増強され、骨格筋細胞へのグルコース取り込みが促進され、最終的に、I型及びII型糖尿病の被験体の両方で血糖値の有意な低下と耐糖能の劇的な向上がもたらされる。示すように、化合物#43は、肝細胞におけるG6PC発現の阻害によりインスリン作用を増強することができる。G6PC発現によって駆動されるプロセスである肝臓による制御不能のグルコース産生は、2型糖尿病の重要な特徴であり、重要な問題でもある。糖尿病の肝臓におけるグルコース産生の抑制は、最も広く使用されている抗糖尿病薬のクラスであるビグアナイド、例えばメトホルミンの重要な作用機序である。化合物#43は、このプロセスを遮断する能力を有しているため、2型糖尿病の治療において潜在的に非常に価値が高い。さらに、化合物#43は、インスリン抵抗性糖尿病マウスの骨格筋のインスリン受容体機能を回復させ、インスリン作用を増強して、培養骨格筋細胞のグルコース取り込みを刺激することができる。総合すると、これらの結果は、ヒトのI型及びII型糖尿病に対する化合物#43の使用に関する大きな可能性を示している。
実施例10:無細胞系における化合物#43によるインスリン受容体タンパク質の直接活性化
材料及び方法
化合物
化合物#43及びその硫黄類似体化合物#68を、Alltech,Incの化学実験室で合成した。試験したこれらの化合物の純度は、HPLCによる測定で≧99%であることが確認された。
インスリン受容体(Insr)のin vitroリン酸化、及び活性化Insr(Insrβサブユニットの1146、1150、及び1151のリン酸化チロシン残基によって示される)のウエスタンブロット分析による検出
Insrのin vitroリン酸化を、以下の修正を加えたSigmaのプロトコールに従って実施した。簡潔に述べると、0.8μlのネイティブINSRストック溶液の原液を含有する10μlのネイティブインスリン受容体溶液(Sigma、カタログ番号I9266;50mM HEPES、pH7.6、150mM NaCl及び0.1%TritonX-100を含有する酵素希釈緩衝液で希釈)を、インスリン(Sigma)、DMSO(化合物#43溶媒)、化合物#43または化合物#68(50mM HEPES、pH7.6及び100μg/mlウシ血清アルブミンで希釈)を含有する同体積の溶液の存在下で氷上、30分間インキュベートした。次いで、0.2mM ATP、50mM HEPES、pH7.6、50mM MgCl2及び4mM MnCl2を含有する20μlの2×キナーゼ緩衝液を上記の反応液に加え、混合し、氷上で45分間インキュベートした。
5マイクロリットルの上記反応物を、直ちに、Insrβタンパク質(Cell Signaling Inc.)の1146、1150及び1151のリン酸化チロシン残基に対する特異的抗体を使用したウエスタンブロット分析に供した。NIH Image Jソフトウェアを使用して、タンパク質バンド濃度を測定した。
統計解析
該当する場合、スチューデントt検定を使用して、処置群間の差異の統計的有意性を判定し、0.05未満のP値を有意とみなした。
結果:
1.ラット肝臓組織から精製した天然のインスリン受容体タンパク質は、化合物#43とインスリンによってin vitro無細胞系で活性化された
培養肝細胞及び分化骨格筋細胞、ならびにT2D糖尿病マウスにおける研究により、化合物#43がin vitro及びin vivoの両方でインスリン受容体を活性化することができることが明らかになった。化合物#43がInsrの活性化に直接影響を及ぼすかどうかを調べるため、ネイティブインスリン受容体タンパク質(ラット肝組織から精製)のin vitro無細胞リン酸化アッセイを実施した。チロシン残基1146、1150及び1151においてリン酸化Insrβに対する特異的抗体を使用して、活性化Insrを検出した。予想通り、インスリン処置はこの無細胞in vitro系においてY1146/1150/1151においてInsrβのリン酸化を誘導することができた(図26)。重要なことに、化合物#43は、1.9及び3.8μMの用量で、Y1146/1150/1151においてInsrβのリン酸化を有意に増強することができ、リン酸化レベルの上昇は0.5μMインスリンと直接比較可能であった(図26)。これらの結果は、化合物#43がインスリンを忠実に模倣してインスリン受容体を直接活性化することができることを裏付けるものであり、化合物#43がI型糖尿病を含む糖尿病に対してインスリンを置き換えるものとなる可能性を有するというさらなる分子的エビデンスを提供している。
2.化合物#43の硫黄類似体である化合物#68は、無細胞系でのInsrの活性化において化合物#43よりもはるかに効果が低い
培養肝細胞及び2型糖尿病(T2D)Leprdb/dbマウスでの研究により、化合物#43の硫黄類似体である化合物#68は、in vitroでのグルコース産生の阻害(図1)及びT2Dマウスにおける高血糖症の減弱(図4)において化合物#43よりも効果が低いことが示された。無細胞系でのInsrの活性化において化合物#43と化合物#68の間に示差的な効果があるかどうかを調べるために、同量のネイティブインスリン受容体タンパク質を同じモル濃度(3.8μM)の化合物#43または化合物#68の存在下でインキュベートし、in vitroリン酸化アッセイに供した。図27に示すように、試験した用量で化合物#68ではなく化合物#43がInsrを活性化することができた。これらの研究は、Insrの活性化において化合物#68が試験した用量で化合物#43よりも効果が低いことを示唆しており、これは肝臓グルコース産生の阻害(図1)及びT2Dマウスにおける高血糖症(図4)に対する化合物#68の低い効力の説明となり得る。
実施例11:化合物#43の急性処置後のストレプトゾトシン(STZ)誘発1型糖尿病(T1D)マウスの血糖値の低下
材料及び方法
化合物
ストレプトゾトシン(STZ)はSigmaから購入した。化合物#43は、Alltech,IncのChemistry Laboratoryで合成した。化合物#43の純度は、HPLCによる測定で≧99%であることが確認された。
1型糖尿病(T1D)マウスモデル及びこれらのT1Dマウスの血糖値に対する化合物#43の効果
5週齢のC57/BL6雄マウスに、ストレプトゾトシン(STZ、55mg/kgマウス体重)を5日間、毎日腹腔内注射し、その後、回復のためにさらに14日間飼育室に収容した。これらのマウスの血糖値を、グルコメーターを使用して測定した。血糖値が500mg/dLを上回る動物を、I型糖尿病(T1D)とみなした。500~550mg/dLの非絶食時血糖値を有するこれらのT1Dマウスを一晩絶食させ、5.4mg/kg体重の化合物#43または2%DMSOを含有する生理食塩水(化合物#43ストック溶媒)を腹腔内注射した。注射後1、2及び3時間において、グルコメーターを使用してこれらのマウスの血糖値を測定した。
統計解析
スチューデントt検定を使用して、各時点での対照(DMSO)と化合物#43群との差異の統計的有意性を判定した。0.05未満のP値は、これら2つの群間の統計的に有意な差を示す。
結果
T1Dマウスの高血糖症に対する化合物#43の潜在能力を調べるために、500~550mg/dLの血糖値を有するSTZ誘発T1Dマウスを一晩絶食させ、DMSOまたは化合物#43(5.4mg/kg体重)を含有する生理食塩水を、1、2及び3時間、腹腔内注射し、血糖値の測定に供した。図28に示すように、3時間の期間中、対照マウス(DMSO含有生理食塩水を注射した)の血糖値に明白な変化はなかった。しかしながら、化合物#43で1時間処置した後、これらのT1Dマウスでは血糖値の低下傾向が観察された(図28)。さらに重要なことに、2時間及び3時間の化合物#43処置後のいずれにおいても、これらのT1Dマウスで血糖値の有意な低下が観察された(図28)。総合すると、これらの結果は、化合物#43がT1Dマウスの高血糖症を軽減することを裏付けるものである。
実施例においていくつかの実施形態を示しているが、本開示の他の実施形態を提供するためにそれらを変更し得ることは明らかである。したがって、本発明の範囲は、単に例示として示す特定の実施形態によってではなく、添付の特許請求の範囲によって定義されることが理解されるであろう。