本開示がより容易に理解され得ることを保証するべく、特定の用語をまず定義する。追加の定義は、詳細な説明全体を通して記載される。
「CD40」という用語は腫瘍壊死因子受容体スーパーファミリーメンバ5を指す。「CD40」という用語は、バリアント、アイソフォーム、ホモログ、オーソログ、およびパラログを含む。例えば、ヒトCD40タンパク質に特異的な抗体は、特定の場合において、サルなど、ヒト以外の種のCD40タンパク質と交差反応し得る。他の実施形態において、ヒトCD40タンパク質に特異的な抗体は、ヒトCD40タンパク質に完全に特異的であり得、他の種または他のタイプに対して交差反応性を示さないことがあり得、または、すべての他の種ではないが特定の他の種のCD40と交差反応し得る。
「ヒトCD40」という用語は、Genbankアクセッション番号NP_001241.1(SEQ ID NO:68)を有するヒトCD40のアミノ酸配列など、ヒトのアミノ酸配列を有するCD40タンパク質を指す。「サルまたはアカゲザルCD40」および「マウスCD40」という用語はそれぞれ、例えば、Genbankアクセッション番号NP_001252791.1(SEQ ID NO.:70)およびNP_035741.2(SEQ ID NO.:72)をそれぞれ有するアミノ酸配列を有するものなどのサルおよびマウスCD40配列を指す。
本明細書において言及される「抗体」という用語は、全抗体および任意の抗原結合断片(すなわち、「抗原結合部分」)またはその一本鎖を含む。全抗体は、ジスルフィド結合によって相互接続された少なくとも2つの重(H)鎖および2つの軽鎖(L)を含む糖タンパク質である。各重鎖は、重鎖可変領域(本明細書においてVHと略記される)および重鎖定常領域を含む。重鎖定常領域は、3つのドメイン、すなわち、CH1、CH2、およびCH3を含む。各軽鎖は、軽鎖可変領域(本明細書においてVLと略記される)および軽鎖定常領域を含む。軽鎖定常領域は1つのドメインCLを含む。VHおよびVL領域は更に、相補性決定領域(CDR)と称される超可変性の領域に細分化でき、フレームワーク領域(FR)と称される、より保存的な領域が点在する。各VHおよびVLは、3つのCDRおよび4つのFRから構成され、アミノ末端からカルボキシ末端まで、FR1、CDR1、FR2、CDR2、FR3、CDR3、FR4の順に配置される。重鎖および軽鎖の可変領域は、抗原と相互作用する結合ドメインを含む。抗体の定常領域は、免疫系の様々な細胞(例えば、エフェクター細胞)、および、古典的補体経路の第1成分(C1q)を含む、宿主の組織または因子に対する免疫グロブリンの結合を媒介できる。
本明細書において使用される場合、抗体の「抗原結合部分」(または単に「抗体部分」)という用語は、抗原(例えばCD40タンパク質)に特異的に結合する能力を保持する抗体の1または複数の断片を指す。抗体の抗原結合機能は、完全長抗体の断片によって実行され得ることが示された。抗体の「抗原結合部分」という用語に包含される結合断片の例は、(i)VL、VH、CLおよびCH1ドメインから成る一価断片であるFab断片、(ii)ヒンジ領域においてジスルフィド架橋によって連結された2つのFab断片を含む二価断片であるF(ab')2断片、(iii)VHおよびCH1ドメインから成るFd断片、(iv)抗体の単一アームのVLおよびVHドメインから成るFv断片、(v)VHドメインから成るdAb断片(Ward et al., (1989) Nature 341:544-546)、(vi)隔離された相補性決定領域(CDR)、および、(viii)単一の可変ドメインおよび2つの定常ドメインを含む重鎖可変領域であるナノボディを含む。更に、Fv断片の2つのドメインであるVLおよびVHは、別の遺伝子によってコードされるが、組み換えの方法を使用することにより、合成リンカによって連結することができ、VLおよびVH領域が対合して一価分子(一本鎖Fv(scFv)として知られ、例えば、Bird et al., (1988) Science 242:423-426; and Huston et al., (1988) Proc. Natl. Acad. Sci. USA 85:5879-5883)を参照されたい)を形成する単一タンパク質鎖となることが可能となる。そのような一本鎖抗体はまた、抗体の「抗原結合部分」という用語に包含されることが意図される。これらの抗体断片は、当業者に知られている従来の技法を使用して取得され、断片は、完全な抗体と同じ方式で、使用性に関してスクリーニングされる。
本明細書において使用される場合、「単離抗体」とは、異なる抗原特異性を有する他の抗体を実質的に含まない抗体を指すことが意図される(例えば、CD40タンパク質に特異的に結合する単離抗体は、CD40タンパク質以外の抗原に特異的に結合する抗体を実質的に含まない)。しかしながら、ヒトCD40タンパク質に特異的に結合する単離抗体は他の種のCD40タンパク質など、他の抗原に対する交差反応性を有し得る。更に、単離抗体は、他の細胞材料および/または化学物質を実質的に含まないことがあり得る。
本明細書において使用される場合、「モノクローナル抗体」または「モノクローナル抗体組成物」という用語は、単一分子組成物の抗体分子の調製物を指す。モノクローナル抗体組成物は、特定のエピトープに対して、単一結合特異性および親和性を示す。
本明細書において使用される場合、「マウス抗体」という用語は、フレームワークおよびCDR領域の両方がマウス生殖細胞系免疫グロブリン配列に由来する可変領域を有する抗体を含むことが意図される。更に、抗体が定常領域を含む場合、定常領域も、マウス生殖細胞系免疫グロブリン配列に由来する。開示のマウス抗体は、マウス生殖細胞系免疫グロブリン配列によってエンコードされないアミノ酸残基(例えば、in vitroのランダムまたは部位特異的変異誘発、または、in vivoの体細胞変異によって導入される変異)を含み得る。しかしながら、本明細書において使用される場合、「マウス抗体」という用語は、別の哺乳類種の生殖系列に由来するCDR配列がマウスのフレームワーク配列に移植された抗体を含むことが意図されない。
「キメラ抗体」という用語は、非ヒトの供給元からの遺伝物質をヒトの遺伝物質と組み合わせることによって作られる抗体を指す。または、より一般に、キメラ抗体は、別の種の遺伝物質を有する特定の種の遺伝物質を有する抗体である。
本明細書において使用される場合、「ヒト化抗体」という用語は、ヒトにおいて天然に産生される抗体バリアントとの類似性を増加させるように改変されたタンパク質配列を有する非ヒト種の抗体を指す。
「抗原を認識する抗体」および「抗原に特異的な抗体」という語句は、本明細書において、「抗原に特異的に結合する抗体」という用語と交換可能に使用される。
本明細書において使用される場合、「ヒトCD40に特異的に結合する抗体」とは、ヒトCD40タンパク質(および、場合によっては、1または複数の非ヒト種からのCD40タンパク質)に結合するが、非CD40タンパク質に実質的に結合しない抗体を指すことが意図される。好ましくは、抗体は、「高親和性」で、すなわち、KD5.0×10-8M以下、より好ましくは、1.0×10-8M以下、より好ましくは、5.0×10-9以下でヒトCD40タンパク質に結合する。
本明細書において使用される場合、タンパク質または細胞に「実質的に結合しない」という用語は、タンパク質または細胞に結合しない、または、高親和性で結合しない、すなわち、1.0×10-6M以上、より好ましくは1.0×10-5M以上、より好ましくは1.0×10-4M以上、より好ましくは1.0×10-3M以上、更により好ましくは1.0×10-2M以上のKDでタンパク質または細胞に結合することを意味する。
IgG抗体に対する「高親和性」という用語は、標的抗原に対して、1.0×10-6M以下、より好ましくは5.0×10-8M以下、更により好ましくは1.0×10-8M以下、更により好ましくは5.0×10-9M以下、更により好ましくは1.0×10-9M以下のKDを有する抗体を指す。しかしながら、「高親和性」結合は、他の抗体アイソタイプに対して変動し得る。例えば、IgMアイソタイプに対する「高親和性」結合は、10-6M以下、より好ましくは10-7M以下、更により好ましくは10-8M以下のKDを有する抗体を指す。
本明細書において使用される場合、「Kassoc」または「Ka」という用語は、特定の抗体‐抗原相互作用の会合速度を指すことを意図し、本明細書において使用される場合、「Kdis」または「Kd」という用語は、特定の抗体‐抗原相互作用の解離速度を指すことを意図する。本明細書において使用される場合、「KD」という用語は、Kaに対するKdの比(すなわち、Kd/Ka)から取得される解離定数を指すことを意図し、モル濃度(M)で表現される。抗体のKD値は、本技術分野において十分に確立された方法を使用して決定され得る。抗体のKDを決定するための好ましい方法は、表面プラズモン共鳴を使用する、好ましくは、Biacore(登録商標)システムなどのバイオセンサシステムを使用することによるものである。
半数効果濃度としても知られる「EC50」という用語は、指定された曝露時間後のベースラインと最大値との間の中間の応答を誘導する抗体の濃度を指す。
「対象」という用語は、任意のヒトまたは非ヒト動物を含む。「非ヒト動物」という用語は、すべての脊椎動物、例えば、非ヒト霊長類、ヒツジ、イヌ、ネコ、ウシ、ウマ、ニワトリ、両生類、および爬虫類など、哺乳類および非哺乳類を含むが、非ヒト霊長類、ヒツジ、イヌ、ネコ、ウシ、ウマなどの哺乳類が好ましい。
「アゴニストCD40抗体」または「アゴニスト抗CD40抗体」という用語は、CD40に結合してCD40シグナル伝達を活性化または誘導し、免疫細胞活性化および増殖、ならびにサイトカインおよびケモカイン産生を促進する抗CD40抗体を指す。「アンタゴニストCD40抗体」という用語は、CD40Lの係合によって誘導され得るCD40シグナル伝達をブロックまたは阻害する抗CD40抗体を指す。
「治療有効量」という用語は、疾患または病状(癌など)に関連する症状を予防または緩和する、および/または、疾患または病状の重篤度を低減するのに十分な本開示の抗体の量を意味する。治療有効量は、治療される病状の文脈に沿うものと理解され、実際の有効量は、当業者によって容易に認識される。
開示の様々な態様が、以下のサブセクションで更に詳細に説明される。
ヒトCD40に対する結合特異性を有する抗CD40抗体、および、その有利な機能特性
開示の例示的な抗体またはその抗原結合部分は、高親和性、例えば、1×10-8M以下のKDでヒトCD40に特異的に結合する。抗体はまた、サルCD40との交差反応性を有するが、マウスCD40とは結合しない。
開示の例示的な抗体は、CD40シグナル伝達を活性化または誘導するアゴニストCD40抗体であり、従って、免疫細胞活性化および増殖、ならびにサイトカインおよびケモカイン産生に関与する。
開示の例示的な抗体またはその抗原結合部分は、従来技術のアゴニスト抗CD40抗体と同等か、それ以上のin vivo抗腫瘍作用を有し、毒性はそれと同じか、またはより低い。抗体投与が停止した後でも、腫瘍は増殖しないか、または、更には完全に消失する。
好ましくは、開示の抗体はモノクローナル抗体である。追加的または代替的に、抗体は例えば、マウス、キメラ、または、ヒト化モノクローナル抗体であり得る。
モノクローナル抗CD40抗体
開示の例示的な抗体は、下および以下の例において説明されるような構造的および化学的な特徴を有するモノクローナル抗体である。例示的な抗CD40抗体のVHアミノ酸配列は、SEQ ID NO:37、38、39、40、41、42、43、44、45、46、47、48、49または50に記載される。例示的な抗CD40抗体のVLアミノ酸配列は、SEQ ID NO:51、52、53、54、55、56、57、58、59、60、または61に示される。例示的な抗体の重鎖/軽鎖可変領域のアミノ酸配列は、下の表1にまとめられている。いくつかのクローンは同じVHまたはVLを共有する。これらの例示的な抗体は、SEQ ID NO:62、63または64に記載される重鎖定常領域、および、SEQ ID NO:65または66に記載される軽鎖定常領域を含み得、重鎖可変領域のC末端は重鎖定常領域のN末端に連結され得、軽鎖可変領域のC末端は、軽鎖定常領域のN末端に連結され得る。
表1における重鎖可変領域CDRおよび軽鎖可変領域CDRは、Kabatナンバリングシステムによって定義された。しかしながら、本技術分野においてよく知られるように、CDR領域は、重鎖/軽鎖可変領域配列に基づいて、Chothia、IMGT、AbM、またはContactナンバリングシステム/方法など、他のシステムによっても決定できる。
ヒトCD40に結合する他の抗CD40抗体のVHおよびVL配列(またはCDR配列)は、本開示の抗CD40抗体のVHおよびVL配列(またはCDR配列)と組み合わされ得る。好ましくは、VHおよびVL鎖(またはそのような鎖の中のCDR)が組み合わされるとき、特定のVH/VLペアからのVH配列は、構造的に同様のVH配列と置き換えられる。同様に、好ましくは、特定のVH/VLペアからのVL配列は、構造的に同様のVL配列と置き換えられる。
従って、一実施形態において、開示の抗体またはその抗原結合部分は、以下を含む。
(a)表1において上に列挙されたアミノ酸配列を含む重鎖可変領域、および、
(b)表1において上に列挙されたアミノ酸配列を含む軽鎖可変領域、または、別の抗CD40抗体のVL(抗体はヒトCD40に特異的に結合する)
別の実施形態において、開示の抗体またはその抗原結合部分は、以下を含む。
(a)表1において上に列挙された重鎖可変領域のCDR1、CDR2、およびCDR3領域、ならびに、
(b)表1において上に列挙された軽鎖可変領域のCDR1、CDR2、およびCDR3領域、または、別の抗CD40抗体のCDR(抗体はヒトCD40に特異的に結合する)
更に別の実施形態において、抗体またはその抗原結合部分は、ヒトCD40に結合する他の抗体のCDR、例えば、重鎖可変領域からのCDR1および/またはCDR3、および/または、異なる抗CD40抗体の軽鎖可変領域からのCDR1、CDR2、および/またはCDR3と組み合わされた抗CD40抗体の重鎖可変CDR2領域を含む。
表1
重鎖/軽鎖可変領域のアミノ酸配列
加えて、本技術分野において、CDR1および/またはCDR2ドメインから独立のCDR3ドメインは単独で、類似の抗原に対する抗体の結合特異性を決定できること、ならびに、共通のCDR3配列に基づいて同じ結合特異性を有する複数の抗体を予測的に生成できることが知られている。例えば、Klimka et al.,, British J. of Cancer 83(2):252-260 (2000); Beiboer et al.,, J. Mol. Biol. 296:833-849 (2000); Rader et al.,, Proc. Natl. Acad. Sci. U.S.A. 95:8910-8915 (1998); Barbas et al.,, J. Am. Chem. Soc. 116:2161-2162 (1994); Barbas et al.,, Proc. Natl. Acad. Sci. U.S.A. 92:2529-2533 (1995); Ditzel et al.,, J. Immunol. 157:739-749 (1996); Berezov et al.,, BIAjournal 8: Scientific Review 8 (2001); Igarashi et al.,, J. Biochem (Tokyo) 117:452-7 (1995); Bourgeois et al.,, J. Virol 72:807-10 (1998); Levi et al.,, Proc. Natl. Acad. Sci. U.S.A. 90:4374-8 (1993); Polymenis and Stoller, J. Immunol. 152:5218-5329 (1994) and Xu and Davis, Immunity 13:37-45 (2000)を参照されたい。また、米国特許第6,951,646号;第6,914,128号;第6,090,382号;第6,818,216号;第6,156,313号;第6,827,925号;第5,833,943号;第5,762,905号および第5,760,185号を参照されたい。これらの参照の各々は、全体が参照によって本明細書に組み込まれる。
従って、別の実施形態において、開示の抗体は、抗CD40抗体の重鎖可変領域のCDR2、および、抗CD40抗体の重鎖および/または軽鎖可変領域の少なくともCDR3、または、別の抗CD40抗体の重鎖および/または軽鎖可変領域のCDR3を含み、抗体はヒトCD40に特異的に結合可能である。これらの抗体は好ましくは、(a)CD40に結合するために競合し、(b)機能的特性を保持し、(c)同じエピトープに結合し、および/または、(d)本開示の抗CD40抗体と同様の結合親和性を有する。更に別の実施形態において、抗体は更に、抗CD40抗体の軽鎖可変領域のCDR2、または、別の抗CD40抗体の軽鎖可変領域のCDR2を含み得、抗体はヒトCD40に特異的に結合可能である。別の実施形態において、開示の抗体は、抗CD40抗体の重鎖および/または軽鎖可変領域のCDR1、または、別の抗CD40抗体の重鎖および/または軽鎖可変領域のCDR1を含み得、抗体はヒトCD40に特異的に結合可能である。
保存的改変
別の実施形態において、開示の抗体は、1または複数の保存的改変によって、本開示の抗CD40抗体とは異なる、CDR1、CDR2、およびCDR3配列の重鎖および/または軽鎖可変領域配列を含む。本技術分野において、抗原結合を排除しない特定の保存的配列改変が行われ得ることが理解される。例えば、Brummell et al., (1993) Biochem 32:1180-8; de Wildt et al., (1997) Prot. Eng. 10:835-41; Komissarov et al., (1997) J. Biol. Chem. 272:26864-26870; Hall et al., (1992) J. Immunol. 149:1605-12; Kelley and O'Connell (1993) Biochem.32:6862-35; Adib-Conquy et al., (1998) Int. Immunol.10:341-6 and Beers et al., (2000) Clin. Can. Res. 6:2835-43を参照されたい。
従って、一実施形態において、抗体は、CDR1、CDR2、およびCDR3配列を含む重鎖可変領域、および/または、CDR1、CDR2、およびCDR3配列を含む軽鎖可変領域を備え、(a)重鎖可変領域CDR1配列は、上の表1に列挙される配列、および/または、その保存的改変を含み、および/または、(b)重鎖可変領域CDR2配列は、上の表1に列挙される配列、および/または、その保存的改変を含み、(c)重鎖可変領域CDR3配列は、上の表1に列挙された配列、および、その保存的改変を含み、および/または、(d)軽鎖可変領域CDR1および/またはCDR2および/またはCDR3配列は、上の表1に列挙された配列、および/または、その保存的改変を含み、(e)抗体はヒトCD40に特異的に結合する。
本開示の抗体は、ヒトCD40に対する高親和性結合など、上に記載れた以下の機能特性の1または複数を保有する。
様々な実施形態において、抗体は例えば、マウス、ヒト、ヒト化、またはキメラ抗体であり得る。
本明細書において使用される場合、「保存的配列改変」という用語は、アミノ酸配列を含む抗体の結合特性に著しく影響しない、または、それを変更しないアミノ酸改変を指すことを意図する。そのような保存的改変はアミノ酸置換、追加および欠失を含む。部位特異的変異誘発およびPCR媒介変異誘発など、本技術分野において知られている標準的な技法によって、開示の抗体に改変が導入され得る。保存的アミノ酸置換とは、アミノ酸残基が、同様の側鎖を有するアミノ酸残基に置き換えられることである。同様の側鎖を有するアミノ酸残基のファミリが本技術分野において定義されている。これらのファミリは、塩基性側鎖(例えば、リシン、アルギニン、ヒスチジン)、酸性側鎖(例えば、アスパラギン酸、グルタミン酸)、極性無電荷側鎖(例えば、グリシン、アスパラギン、グルタミン、セリン、スレオニン、チロシン、システイン、トリプトファン)、非極性側鎖(例えば、アラニン、バリン、ロイシン、イソロイシン、プロリン、フェニルアラニン、メチオニン)、ベータ分岐側鎖(例えば、スレオニン、バリン、イソロイシン)および芳香族側鎖(例えば、チロシン、フェニルアラニン、トリプトファン、ヒスチジン)を有するアミノ酸を含む。したがって、開示の抗体のCDR領域における1または複数のアミノ酸残基は、同じ側鎖ファミリからの他のアミノ酸残基に置き換えられ得、変更された抗体は、本明細書において記載された機能的アッセイを使用して、保持された機能(すなわち、上に記載された機能)について試験され得る。
操作および改変された抗体
開示の抗体は、改変された抗体を操作するために、本開示の抗CD40抗体のVH/VL配列の1または複数を有する抗体を出発材料として使用して調製され得る。抗体は、一方または両方の可変領域(すなわち、VHおよび/またはVL)、例えば、1または複数のCDR領域、および/または、1または複数のフレームワーク領域における1または複数の残基を改変することによって操作され得る。追加的または代替的に、例えば抗体のエフェクター機能を変更するために、抗体は、定常領域内の残基を改変することによって操作され得る。
特定の実施形態において、抗体の可変領域を操作するために、CDR移植が使用され得る。抗体は、主に6の重鎖および軽鎖相補性決定領域(CDR)に位置するアミノ酸残基を通じて標的抗原と相互作用する。この理由から、CDR内のアミノ酸配列は、CDRの外部の配列より、個別の抗体間の多様性が高い。CDR配列は、大部分の抗体抗原相互作用を担うので、異なる特性を有する異なる抗体からのフレームワーク配列に移植された、特定の天然に発生する抗体からのCDR配列を含む発現ベクターを構築することによって、特定の天然に発生する抗体の特性を模倣する組み換え抗体を発現させることが可能である(例えば、Riechmann et al., (1998) Nature 332:323-327; Jones et al., (1986) Nature 321:522-525; Queen et al., (1989) Proc. Natl. Acad.を参照されたい、また、U.S.A.86:10029-10033、米国特許第5,225,539号;第5,530,101号;第5,585,089号;第5,693,762号および第6,180,370号を参照されたい)。
従って、開示の別の実施形態は、上に記載された本開示の配列を含むCDR1、CDR2、およびCDR3配列を含む重鎖可変領域、および/または、上に記載された本開示の配列を含むCDR1、CDR2、およびCDR3配列を含む軽鎖可変領域を備える単離モノクローナル抗体、または、その抗原結合部分に関連する。これらの抗体は本開示のモノクローナル抗体のVHおよびVL CDR配列を含むが、異なるフレームワーク配列を含み得る。
そのようなフレームワーク配列は、公共のDNAデータベース、または、生殖系列抗体遺伝子配列を含む公開されている参考文献から取得できる。例えば、ヒト重鎖および軽鎖可変領域遺伝子の生殖系列DNA配列は、「VBase」ヒト生殖系列配列データベース(インターネットで入手可能www.mrc-cpe.cam.ac.uk/vbase)、ならびに、上記で引用されるKabat et al., (1991);Tomlinson et al., (1992) J. Mol. Biol. 227:776-798;およびCox et al., (1994) Eur. J. Immunol. 24:827-836で見つけることができる。これらの各々の内容は、参照によって本明細書に明示的に組み込まれる。別の例として、ヒト重鎖および軽鎖可変領域遺伝子の生殖系列DNA配列はGenbankデータベースで見ることができる。例えば、HCo7 HuMAbマウスにおいて見られる以下の重鎖生殖系列配列は、添付のGenbankアクセッション番号1-69(NG--0010109、NT--024637&BC070333)、3-33(NG--0010109&NT--024637)および3-7(NG--0010109&NT--024637)において入手可能である。別の例として、HCo12 HuMAbマウスにおいて見られる以下の重鎖生殖系列配列は、添付のGenbankアクセッション番号1‐69(NG--0010109、NT--024637&BC070333)、5-51(NG--0010109 &NT--024637)、4-34(NG--0010109&NT--024637)、3-30.3(CAJ556644)&3-23(AJ406678)において利用可能である。
抗体タンパク質配列は、当業者に知られている、Gapped BLAST (上記のAltschul et al.,(1997))と呼ばれる配列類似性検索方法の1つを使用して、まとめられたタンパク質配列データベースと比較される。
開示の抗体において使用される好ましいフレームワーク配列は、開示の抗体によって使用されるフレームワーク配列と同様の構造である。VH CDR1、CDR2、およびCDR3配列は、生殖系列免疫グロブリン遺伝子において見られるものと同一の配列を有するフレームワーク領域に移植され得るか(フレームワーク配列は生殖系列免疫グロブリン遺伝子に由来する)、または、CDR配列は、生殖系列配列と比較して1または複数の変異を含むフレームワーク領域に移植され得る。例えば、特定の場合において、抗体の抗原結合能力を維持または強化するために、フレームワーク領域内の残基を変異させることが有益であることが分かった(例えば、米国特許第5,530,101号;第5,585,089号;第5,693,762号および第6,180,370号を参照されたい)。
別のタイプの可変領域改変は、VHおよび/またはVL CDR1、CDR2、および/またはCDR3領域におけるアミノ酸残基を変異させ、これによって、目的の抗体の1または複数の結合特性(例えば親和性)を改善することである。変異を導入するために、部位特異的変異誘発またはPCR媒介変異誘発が実行され得、抗体結合に対する影響、または、他の目的の機能特性が、本技術分野において知られているように、in vitroまたはin vivoアッセイで評価され得る。好ましくは、保存的改変(本技術分野において知られている)が導入される。変異は、アミノ酸置換、追加、または欠失であり得るが、好ましくは置換である。更に、典型的には、CDR領域内の1、2、3、4または5以下の残基が変更される。
従って、別の実施形態において、開示は、(a)本開示の配列、または、1、2、3、4もしくは5つのアミノ酸置換、欠失、もしくは追加を有するアミノ酸配列を含むVH CDR1領域、(b)本開示の配列、または、1、2、3、4、もしくは5つのアミノ酸置換、欠失、もしくは追加を有するアミノ酸配列を含むVH CDR2領域、(c)本開示の配列、または、1、2、3、4、もしくは5つのアミノ酸置換、欠失、もしくは追加を有するアミノ酸配列を含むVH CDR3領域、(d)本開示の配列、または、1、2、3、4、もしくは5つのアミノ酸置換、欠失、もしくは追加を有するアミノ酸配列を含むVL CDR1領域、(e)本開示の配列、または、1、2、3、4、もしくは5つのアミノ酸置換、欠失、もしくは追加を有するアミノ酸配列を含むVL CDR2領域、および(f)本開示の配列、または、1、2、3、4、もしくは5つのアミノ酸置換、欠失、もしくは追加を有するアミノ酸配列を含むVL CDR3領域を備える重鎖可変領域を備える、単離された抗CD40モノクローナル抗体またはその抗原結合部分を提供する。
操作された開示の抗体は、例えば、抗体の特性を改善するために、VHおよび/またはVL内のフレームワーク残基に改変が行われたものを含む。典型的には、そのようなフレームワーク改変は、抗体の免疫原性を減少させるために行われる。例えば、1つのアプローチは、1または複数のフレームワーク残基を、対応する生殖系列配列に「復帰変異」させることである。より具体的には、体細胞変異を経験した抗体は、抗体の由来元である生殖系列配列とは異なるフレームワーク残基を含み得る。そのような残基は、抗体フレームワーク配列を、抗体の由来元である生殖系列配列と比較することによって識別され得る。
別のタイプのフレームワーク改変は、T細胞エピトープを除去するために、フレームワーク領域内、または、更には1または複数のCDR領域内の1または複数の残基を変異させ、これによって、抗体の潜在的な免疫原性を低減することを伴う。このアプローチは、「脱免疫化」とも呼ばれ、米国特許出願公開第20030153043号に更に詳細に記載されている。
フレームワークまたはCDR領域内で行われる改変に加えて、または替えて、開示の抗体は、典型的には、血中半減期、補体結合、Fc受容体結合、および/または、抗原依存的細胞毒性など、抗体の1または複数の機能的特性を変更するために、Fc領域内の改変を含むように操作され得る。更に、開示の抗体は、化学的に改変され得る(例えば、1または複数の化学的部分が抗体に取り付けられ得る)か、または、そのグリコシル化を変更するために改変され得、これにより、同様に抗体の1または複数の機能的特性が変更される。
一実施形態において、CH1のヒンジ領域は、ヒンジ領域におけるシステイン残基の数が変更されるように、例えば、増加または減少するように改変される。このアプローチは米国特許第5,677,425号に更に記載される。CH1のヒンジ領域におけるシステイン残基の数は、例えば、軽鎖および重鎖の組み立てを促進するために、または、抗体の安定性を増加または減少させるために変更される。
別の実施形態において、抗体のFcヒンジ領域は、抗体の生物学的半減期を減少させるために、変異される。より具体的には、ネイティブのFcヒンジドメインのスタフィロコッカスタンパク質A(SpA)結合と比べて弱いSpA結合を抗体が有するように、1または複数のアミノ酸変異が、Fcヒンジ断片のCH2‐CH3ドメインインタフェース領域に導入される。このアプローチは、米国特許第6,165,745号に更に詳細に記載されている。
更に別の実施形態において、抗体のグリコシル化は改変される。例えば、グリコシル化された抗体が作られ得る(すなわち、抗体はグリコシル化を有しない)。グリコシル化は、例えば、抗原に対する抗体の親和性を増加させるために変更され得る。そのような炭化水素改変は、例えば、抗体配列内のグリコシル化の1または複数の部位を変更することによって達成され得る。例えば、1または複数の可変領域フレームワークグリコシル化部位の除去をもたらす1または複数のアミノ酸置換が行われ得、これによって、当該部位のグリコシル化を除去する。そのようなアグリコシル 化により、抗原に対する抗体の親和性が増加し得る。例えば、米国特許第5,714,350号および第6,350,861号を参照されたい。
追加的または代替的に、フコシル残基の量が低減された低フコシル化抗体、または、二分GlcNac構造を増加させた抗体など、グリコシル化のタイプが変更された抗体が作られ得る。そのような変更されたグリコシル化パターンは、抗体のADCC能力を増加させることが実証された。そのような炭化水素改変は、例えば、グリコシル化機構を変更した宿主細胞において、抗体を発現させることによって達成され得る。グリコシル化機構を変更した細胞が本技術分野において説明され、開示の組み換え抗体を発現させるための宿主細胞として使用され得、これによって、グリコシル化が変更された抗体が産生される。例えば、細胞株Ms704、Ms705、およびMs709は、Ms704、Ms705、およびMs709細胞株において発現する抗体がそれらの炭化水素上にフコースを有しないように、フコシルトランスフェラーゼ遺伝子FUT8(α(1,6)‐フコシルトランスフェラーゼ)を有しない。Ms704、Ms705、およびMs709 FUT8‐/‐細胞株が、2つの置換ベクターを使用して、CHO/DG44細胞におけるFUT8遺伝子の標的化された破壊によって作成された(米国特許出願公開第20040110704号およびYamane-Ohnuki et al., (2004) Biotechnol Bioeng 87:614-22を参照されたい)。別の例として、EP1,176,195は、フコシルトランスフェラーゼをエンコードする、機能的に破壊されたFUT8遺伝子を有する細胞株を記載し、そのような細胞株において発現する抗体は、α-1,6結合関連酵素を低減または除去することによって低フコシル化を示す。EP1,176,195はまた、抗体のFc領域に結合するN‐セチルグルコサミンにフコースを追加するための酵素活性が低い、または、酵素活性を有さない細胞株、例えば、ラット骨髄腫細胞株YB2/0 (ATCC CRL 1662)を記載する。PCT公開WO03/035835は、フコースをAsn(297)連結炭化水素に取り付ける能力が低減したバリアントCHO細胞株、Lec13細胞を記載する。この結果、その宿主細胞において発現する抗体の低フコシル化も生じる(Shields et al., (2002) J. Biol. Chem. 277:26733-26740も参照されたい)。改変されたグリコシル化プロファイルを有する抗体はまた、PCT公開WO06/089231に記載されるように、ニワトリの卵において産生され得る。代替的に、改変されたグリコシル化プロファイルを有する抗体が、アオウキクサなどの植物細胞において産生さ得る。植物系における抗体産生のための方法が、2006年8月11日に出願されたAlston & Bird LLP attorney docket No. 040989/314911に対応する米国特許出願において開示されている。PCT公開WO99/54342は、グリコシルトランスフェラーゼ(例えば、β(1,4)‐N‐アセチルグルコサミントランスフェラーゼIII(GnTIII))を改変する糖タンパク質を発現するように操作された細胞株を記載する。それにより、操作された細胞株において発現する抗体は、抗体のADCC活性の増加をもたらす二分GlcNac構造の増加を示す(Umana et al., (1999) Nat. Biotech. 17:176-180も参照されたい)。代替的に、抗体のフコース残基は、フコシダーゼ酵素を使用して切断され得る。例えば、フコシダーゼα‐L‐フコシダーゼは、フコシル残基を抗体から除去する(Tarentino et al., (1975) Biochem. 14:5516-23)。
本開示によって想定される本明細書の抗体の別の改変はPEG化である。抗体は、例えば、抗体の生物学的(例えば、血中)半減期を増加させるためにPEG化され得る。抗体をPEG化するために、抗体またはその断片は典型的には、1または複数のPEG基が抗体または抗体断片に取り付けられる条件下において、PEGの反応性エステルまたはアルデヒド誘導体などのポリエチレングリコール(PEG)と反応される。好ましくは、PEG化は、反応性PEG分子(または、類似の反応性水溶性ポリマー)とのアシル化反応またはアルキル化反応を介して実行される。本明細書において使用される場合、「ポリエチレングリコール」という用語は、モノ(C1‐C10)アルコキシまたはアリールオキシ‐ポリエチレングリコールまたはポリエチレングリコール‐マレイミドなど、他のタンパク質を誘導体化するために使用されるPEGの任意の形態を包含することが意図される。特定の実施形態において、PEG化される抗体は、アグリコシル化抗体である。タンパク質をPEG化するための方法が本技術分野において知られ、開示の抗体に適用され得る。例えば、EPO 154 316およびEP 0 401 384を参照されたい。
抗体の物理的特性
開示の抗体は、その異なるクラスを検出および/または区別するための様々な物理的特性を特徴とし得る。
例えば、抗体は、軽鎖または重鎖可変領域のいずれにおける1または複数のグリコシル化部位を含み得る。そのようなグリコシル化部位は、変更された抗原結合に起因して、抗体の免疫原性の増加、または、抗体のpKの変更をもたらし得る(Marshall et al (1972) Annu Rev Biochem 41:673-702; Gala and Morrison (2004) J Immunol 172:5489-94; Wallick et al (1988) J Exp Med 168:1099-109; Spiro (2002) Glycobiology 12:43R-56R; Parekh et al (1985) Nature 316:452-7; Mimura et al., (2000) Mol Immunol 37:697-706)。グリコシル化は、N‐X‐S/T配列を含むモチーフにおいて発生することが知られている。いくつかの場合において、可変領域グリコシル化を含まない抗CD40抗体を有することが好ましい。これは、可変領域においてグリコシル化モチーフを含まない抗体を選択すること、または、グリコシル化領域内の残基を変異させることのいずれによって達成され得る。
好ましい実施形態において、抗体はアスパラギン異性部位を含まない。アスパラギンの脱アミド化は、N‐GまたはD‐G配列で発生し、ポリペプチド鎖にねじれを導入して安定性を減少させる(イソアスパラギン酸作用)イソアスパラギン酸残基の作成をもたらし得る。
各抗体は、一般に6~9.5のpH範囲に収まる固有の等電点(pI)を有する。IgG1抗体のpIは典型的には、7~9.5のpH範囲に収まり、IgG4抗体のpIは、典型的には、6~8のpH範囲に収まる。正常範囲外のpIを有する抗体は、in vivo条件下において、いくらかのアンフォールディングおよび不安定性を有し得ると考えられる。したがって、正常範囲に収まるpI値を含む抗CD40抗体を有することが好ましい。これは、正常範囲におけるpIを有する抗体を選択すること、または、荷電表面残基を変異させることのいずれかによって達成できる。
開示の抗体をエンコードする核酸分子
別の態様において、開示は、開示の抗体の重鎖および/または軽鎖可変領域またはCDRをエンコードする核酸分子を提供する。核酸は、全細胞、細胞溶解液において、または、部分的に精製された、もしくは実質的に純粋な形態で存在し得る。核酸は、標準的な技法によって、他の細胞成分または他の混入物質、例えば、他の細胞核酸またはタンパク質などから精製されるとき、「単離」される、または、「実質的に純粋」になる。開示の核酸は、例えば、DNAまたはRNAであり得、イントロン配列を含んでも含まなくてもよい。好ましい実施形態において、核酸はcDNA分子である。
開示の核酸は、標準的な分子生物学の技法を使用して取得され得る。ハイブリドーマ(例えば、下で更に説明される、ヒト免疫グロブリン遺伝子を保有する遺伝子組み換えマウスから調製されたハイブリドーマ)によって発現される抗体については、ハイブリドーマによって作られる抗体の軽鎖および重鎖をエンコードするcDNAは、標準的なPCR増幅またはcDNAクローン技法によって取得され得る。免疫グロブリン遺伝子ライブラリから(例えば、ファージディスプレイ技法を使用して)取得される抗体については、そのような抗体をエンコードする核酸は、遺伝子ライブラリから回収され得る。
好ましくは、開示の核酸分子は、CD40モノクローナル抗体のVHおよびVL配列またはCDRをエンコードするものを含む。VHおよびVLセグメントをエンコードするDNA断片が取得されると、これらのDNA断片は、例えば、可変領域遺伝子を完全長抗体鎖遺伝子、Fab断片遺伝子、またはscFv遺伝子に変換するために、標準的な組み換えDNA技法によって更に操作され得る。これらの操作において、VLまたはVHをエンコードするDNA断片は、抗体定常領域またはフレキシブルリンカなどの、別のタンパク質をエンコードする別のDNA断片に作動可能に連結される。この文脈において使用される場合、「作動可能に連結される」という用語は、2つのDNA断片によってエンコードされるアミノ酸配列がインフレームに留まるように、2つのDNA断片が結合されることを意味するように意図される。
VH領域をエンコードする単離DNAは、VHをエンコードするDNAを、重鎖定常領域(CH1、CH2、およびCH3)をエンコードする別のDNA分子に作動可能に連結することによって、完全長重鎖遺伝子に変換され得る。ヒト重鎖定常領域遺伝子の配列は、本技術分野において知られ、これらの領域を包含するDNA断片は標準的なPCR増幅によって取得され得る。重鎖定常領域は、IgG1、IgG2、IgG3、IgG4、IgA、IgE、IgM、またはIgD定常領域であり得るが、もっとも好ましくは、IgG1またはIgG4定常領域である。Fab断片重鎖遺伝子については、VHをエンコードするDNAは、重鎖CH1定常領域のみをエンコードする別のDNA分子に作動可能に連結され得る。
VL領域をエンコードする単離DNAは、VLをエンコードするDNAを、軽鎖定常領域CLをエンコードする別のDNA分子に作動可能に連結することによって、完全長軽鎖遺伝子(ならびにFab軽鎖遺伝子)に変換され得る。ヒト軽鎖定常領域遺伝子の配列は、本技術分野において知られ、これらの領域を包含するDNA断片は標準的なPCR増幅によって取得され得る。好ましい実施形態において、軽鎖定常領域は、カッパまたはラムダ定常領域であり得る。
scFv遺伝子を作成するために、VHおよびVLをエンコードするDNA断片が、フレキシブルリンカをエンコードする、例えば、アミノ酸配列(Gly4-Ser)3をエンコードする別の断片に作動可能に連結される。その結果、VHおよびVL配列は、VLおよびVH領域がフレキシブルリンカによって結合された、連続する一本鎖タンパク質として発現され得る(例えば、Bird et al., (1988) Science 242:423-426; Huston et al., (1988) Proc. Natl. Acad. Sci. USA 85:5879-5883; McCafferty et al.,, (1990) Nature 348:552-554を参照されたい)。
開示のモノクローナル抗体の産生
本開示のモノクローナル抗体(mAb)は、Kohler and Milstein (1975) Nature 256: 495の周知の体細胞ハイブリダイゼーション(ハイブリドーマ)技法を使用して産生され得る。モノクローナル抗体を産生するための他の実施形態は、Bリンパ球のウイルスまたは発癌性形質転換、および、ファージディスプレイ技法を含む。キメラまたはヒト化抗体も本技術分野において周知である。例えば、米国特許第4,816,567号;第5,225,539号;第5,530,101号;第5,585,089号;第5,693,762号および第6,180,370号を参照されたい。これらの内容は、全体として参照によって本明細書に具体的に組み込まれる。
開示のモノクローナル抗体を生成するトランスフェクトーマの産生
開示の抗体はまた、本技術分野において周知のように、例えば、組み換えDNA技法および遺伝子トランスフェクション方法(例えば、Morrison, S. (1985) Science 229:1202)の組み合わせを使用して、宿主細胞トランスフェクトーマによって産生され得る。一実施形態において、標準的な分子生物学的技法によって取得される部分的または完全長軽鎖および重鎖をエンコードするDNAは、1または複数の発現ベクターに挿入される。その結果、遺伝子は、転写および翻訳制御配列に作動可能に連結される。この文脈において、「作動可能に連結される」という用語は、ベクター内の転写および翻訳制御配列が、抗体遺伝子の転写および翻訳を制御する意図される機能を担うように、抗体遺伝子がベクターにライゲーションされることを意味するように意図される。
「制御配列」という用語は、プロモータ、エンハンサ、および、抗体遺伝子の転写または翻訳を制御する他の発現制御要素(例えば、ポリアデニル化シグナル)を含むことが意図される。そのような制御配列は、例えば、Goeddelに記載される(Gene Expression Technology. Methods in Enzymology 185, Academic Press, San Diego, Calif. (1990))。哺乳類宿主細胞発現のための好ましい制御配列は、サイトメガロウイルス(CMV)、シミアンウイルス40(SV40)、アデノウィルスに由来するプロモータおよび/またはエンハンサ、例えば、アデノウイルス主要後期プロモータ(AdMLP)およびポリオーマなど、哺乳類細胞において高レベルのタンパク質発現を指令するウイルス要素を含む。代替的に、ユビキチンプロモータまたはβグロブリンプロモータなど、非ウイルス性制御配列が使用され得る。なお更に、ヒトT細胞白血病ウイルス1型のSV40初期プロモータおよび長い末端反復からの配列を含むSRαプロモータシステムなどの異なる由来の配列から構成される制御要素(Takebe et al., (1988) Mol. Cell. Biol. 8:466-472)も使用され得る。発現ベクターおよび発現対照配列は、使用される発現宿主細胞に適合するように選択される。
抗体軽鎖遺伝子および抗体重鎖遺伝子は、同じ、または別個の発現ベクターに挿入され得る。好ましい実施形態において、任意の抗体アイソタイプの完全長抗体遺伝子を作成するために可変領域が使用される。それらは、所望のアイソタイプの重鎖定常領域および軽鎖定常領域を既にエンコードしている発現ベクターに挿入される。その結果、VHセグメントは、ベクター内のCHセグメントに作動可能に連結され、VLセグメントは、ベクター内のCLセグメントに作動可能に連結される。追加的または代替的に、組み換え発現ベクタは、宿主細胞からの抗体鎖の分泌を促進するシグナルペプチドをエンコードし得る。抗体鎖遺伝子は、シグナルペプチドが抗体鎖遺伝子のアミノ末端にインフレームに連結されるように、ベクターにクローニングされ得る。シグナルペプチドは、免疫グロブリンシグナルペプチド、または、異種混合のシグナルペプチド(すなわち、非免疫グロブリンタンパク質からのシグナルペプチド)であり得る。
抗体鎖遺伝子および制御配列に加えて、開示の組み換え発現ベクタは、宿主細胞におけるベクターの複製を制御する配列(例えば、複製起点)および選択可能マーカ遺伝子など、追加の配列を保有し得る。選択可能マーカ遺伝子は、ベクターが導入された宿主細胞の選択を容易にする(例えば、米国特許第4,399,216号、第4,634,665号および第5,179,017号を参照)。例えば、選択可能マーカ遺伝子は典型的には、ベクターが導入された宿主細胞に対して、G418、ハイグロマイシン、またはメトトレキサートなどの薬剤に対する抵抗性を付与する。好ましい選択可能マーカ遺伝子は、ジヒドロ葉酸還元酵素(DHFR)遺伝子(メトトレキサート選択/増幅と共に、dhfr宿主細胞において使用)およびneo遺伝子(G418選択用)を含む。
軽鎖および重鎖の発現のために、重鎖および軽鎖をエンコードする発現ベクターが、標準的な技法によって宿主細胞にトランスフェクトされる。「トランスフェクション」という用語の様々な形態は、外来性DNAを原核生物または真核生物の宿主細胞に導入するために一般に使用される幅広い様々な技法、例えば、電気穿孔法、りん酸カルシウム沈殿、DEAEデキストラントランスフェクションなどを包含することが意図される。開示の抗体を原核生物または真核生物のいずれの宿主細胞において発現することも理論的に可能であるが、真核細胞(哺乳類宿主細胞がもっとも好ましい)における抗体の発現がもっとも好ましい。なぜなら、そのような真核細胞、特に哺乳類細胞は、原核生物の細胞と比較して、適切に折り畳まれた、免疫活性抗体を組み立てて分泌する可能性が高いからである。
好ましくは、開示の組み換え抗体を発現するための哺乳類宿主細胞は、チャイニーズハムスター卵巣細胞(CHO細胞)(Urlaub and Chasin, (1980) Proc. Natl. Acad. Sci. USA 77:4216-4220に記載されるdhfr-CHO細胞を含み、例えばR. J. Kaufman and P. A. Sharp (1982) J. Mol. Biol. 159:601-621に記載されるようにDHFR選択可能マーカーと共に使用される)、NSO骨髄腫細胞、COS細胞およびSP2細胞を含む。特に、NSO骨髄腫細胞と共に使用するための別の好ましい発現系は、WO87/04462、WO89/01036およびEP338,841において開示されるGS遺伝子発現系である。抗体遺伝子をエンコードする組み換え発現ベクタが哺乳類宿主細胞に導入されるとき、抗体は、宿主細胞における抗体の発現、または、より好ましくは、宿主細胞が増殖する培養培地への抗体の分泌を可能にするのに十分な期間にわたって宿主細胞を培養することによって産生される。抗体は、標準的なタンパク質精製方法を使用して培養培地から回収され得る。
二重特異性分子
別の態様において、本開示は、少なくとも2つの異なる結合部位または標的分子に結合する二重特異性分子を生成するために、少なくとも1つの他の機能性分子、例えば、別のペプチドまたはタンパク質(例えば、受容体に対する別の抗体またはリガンド)に連結された1または複数の開示の抗体を含む二重特異性分子を特徴とする。したがって、本明細書において使用される場合、「二重特異性分子」は、3以上の特異性を有する分子を含む。
実施形態において、二重特異性分子は、抗Fc結合特異性および抗CD40結合特異性に加えて、第3の特異性を有する。第3の特異性は、抗増強因子(EF)、例えば、細胞毒性活性に関与する表面タンパク質に結合する分子に対するものであり得、これによって標的細胞に対する免疫反応を増加させる。例えば、抗増強因子は、(例えば、CD2、CD3、CD8、CD28、CD4、CD40、またはICAM‐1を介して)細胞毒性T細胞または他の免疫細胞に結合し得、標的細胞に対する免疫反応の増加をもたらす。
二重特異性分子には多くの異なるフォーマットおよびサイズがあり得る。サイズの範囲の一端では、二重特異性分子は、従来の抗体フォーマットを保持するが、同一の特異性の2つの結合アームを有する代わりに、異なる特異性を各々が有する2つの結合アームを有する。他端は、ペプチド鎖によって連結される2つの一本鎖抗体断片(scFv)から成る二重特異性分子、いわゆるBs(scFv)2コンストラクトである。中サイズの二重特異性分子は、ペプチジルリンカによって連結される2つの異なるF(ab)断片を含む。これらのフォーマットおよび他のフォーマットの二重特異性分子は、遺伝子操作、体細胞ハイブリダイゼーション、または化学的方法によって調製され得る。例えば、上記で引用されるKufer et al; Cao and Suresh, Bioconjugate Chemistry, 9 (6), 635-644 (1998); and van Spriel et al.,, Immunology Today, 21 (8), 391-397 (2000)およびそこで挙げられる参考文献を参照されたい。
医薬組成物
別の態様において、本開示は、薬学的に許容される担体と共に製剤化される本開示の1または複数の抗体を含む医薬組成物を提供する。組成物は任意で、別の抗体、または、抗VISTA抗体などの薬剤など、1または複数の追加の薬学的な活性成分を含み得る。開示の医薬組成物はまた、例えば、別の抗癌剤、別の抗炎症剤または抗微生物剤などとの併用療法において投与され得る。
医薬組成物は任意の数の賦形剤を含み得る。使用できる賦形剤として、担体、表面活性剤、増粘剤、乳化剤、固体結合剤、分散助剤、懸濁助剤、可溶化剤、着色剤、着香剤、コーティング、崩壊剤、潤滑剤、甘味料、保存料、等張剤、およびそれらの組み合わせが挙げられる。好適な賦形剤の選択および使用は、Gennaro, ed., Remington: The Science and Practice of Pharmacy, 20th Ed. (Lippincott Williams & Wilkins 2003)において教示され、その開示は、参照によって本明細書に組み込まれる。
医薬組成物は、好ましくは(例えば注射または注入による)静脈内、筋肉内、皮下、非経口、脊髄または表皮投与に好適である。投与経路に応じて、活性成分を材料でコーティングして、酸の作用およびそれを不活性化し得る他の自然条件から保護することができる。本明細書において使用される「非経口投与」という語句は、通常は注射による、経腸および局所投与以外の投与様式を意味し、静脈内、筋肉内、動脈内、髄腔内、被膜内、眼窩内、心臓内、皮内、腹腔内、気管内、皮下、表皮下、関節内、被膜下、くも膜下、脊髄内、硬膜外および胸骨内の注射および注入を含むが、これらに限定されない。代替的に、本開示の抗体は、局所、表皮または粘膜の投与経路、例えば、鼻腔内、経口、膣、直腸、舌下または局所などの非経口経路を介して投与され得る。
医薬組成物は、無菌水溶液または分散系の形態であり得る。また、それらはマイクロエマルション、リポソーム、または、高い薬剤濃度に好適な他の規則構造で製剤化され得る。
単回投与形態を生成するために担体物質と組み合わせることができる活性成分の量は、治療される対象、および、特定の投与方法に応じて変動し、一般的に、治療効果を生じさせる組成物の量である。一般に、この量の範囲は、薬学的に許容される担体と組み合わされた、100%中、約0.01%から約99%の活性成分、好ましくは、約0.1%から約70%、もっとも好ましくは、約1%から約30%の活性成分である。
最適な所望の反応(例えば治療反応)を提供するように投与計画が調整される。例えば、単回ボーラスを投与でき、いくつかの分割された用量を経時的に投与でき、または、治療状況の緊急事態による指示に応じて用量を比例的に低減または増加させることができる。投与を容易にするために、および、投与量を統一するために、単位用量形態で非経口組成物を製剤化することは特に有利である。本明細書において使用される単位用量形態とは、治療される対象のための単位投与量として好適な物理的に別個の単位を指す。各単位は、必要な医薬担体と組み合わせて所望の治療効果を生じさせるために算出された活性成分の予め定められた量を含む。抗体は代替的に、徐放性製剤として投与できる。この場合、必要な投与頻度が少ない。
抗体の投与については、投与量は、宿主の体重の約0.0001~100mg/kg、より一般的には、0.01~5mg/kgの範囲であり得る。例えば、投与量は、0.3mg/kg体重、1mg/kg体重、3mg/kg体重、5mg/kg体重、または10mg/kg体重、または、1~10mg/kgの範囲であり得る。例示的な治療計画は、1週間に1回、2週間に1回、3週間に1回、4週間に1回、1か月に1回、3か月に1回、または、3~6か月に1回の投与を伴う。開示の抗CD40抗体の好ましい投与計画は、経静脈投与を介して1mg/kg体重、または、3mg/kg体重を含み、抗体は、以下の投与スケジュール、すなわち、(i)6回の投与は4週間ごと、その後3か月ごと、(ii)3週間ごと、(iii)3mg/kg体重を1回、その後、1mg/kg体重を3週間ごとのうち1つを使用して提供される。いくつかの方法において、約1~1000μg/ml(いくつかの方法において、約25~300μg/ml)の血漿抗体濃度を達成するように、投与量が調整される。
開示の抗CD40抗体の「治療有効投与量」は、好ましくは、疾患症状の重症度の減少、無疾患症状期間の頻度および期間の増加、または、疾患の苦痛に起因する機能障害または能力障害の予防を引き起こす。例えば、担癌の対象を治療する場合、「治療有効投与量」は好ましくは、未治療の対象と比較して、腫瘍増殖を少なくとも約20%、より好ましくは少なくとも約40%、さらに好ましくは少なくとも約60%、なお更に好ましくは少なくとも約80%阻害する。治療抗体の治療有効量は、腫瘍サイズを低減でき、または、そうでなければ、典型的にはヒトである、または別の哺乳類であり得る対象における症状を緩和する。
医薬組成物は、インプラント、経皮パッチ、およびマイクロカプセル化送達システムを含む、放出制御製剤であり得る。エチレン酢酸ビニル、ポリ酸無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、ポリ乳酸など、生分解性、生体適合性ポリマーを使用できる。例えば、Sustained and Controlled Release Drug Delivery Systems, J. R. Robinson, ed., Marcel Dekker, Inc., New York, 1978を参照されたい。
治療用組成物は、(1)無針皮下注射装置(例えば、米国特許第5,399,163; 5,383,851; 5,312,335; 5,064,413; 4,941,880; 4,790,824;および4,596,556号)、(2)マイクロ注入ポンプ(米国特許第4,487,603号)、(3)経皮装置(米国特許第4,486,194号)、(4)注入機器(米国特許第4,447,233および4,447,224号)、(5)浸透圧装置(米国特許第4,439,196および4,475,196号)などの医療装置を介して投与され得る。これらの開示は、参照によって本明細書に組み込まれる。
特定の実施形態において、開示のモノクローナル抗体は、in vivoの適切な分散を確実にするために製剤化され得る。例えば、開示の治療抗体が血液脳関門を超えることを確実にするために、それらは、特定の細胞または臓器への選択的輸送を強化する標的化部分を追加的に含み得るリポソームにおいて製剤化され得る。例えば、米国特許第4,522,811号;第5,374,548号;第5,416,016号;第5,399,331号;V. V. Ranade (1989) J. Clin.Pharmacol.29:685; Umezawa et al.,, (1988) Biochem. Biophys. Res. Commun. 153:1038; Bloeman et al., (1995) FEBS Lett.357:140; M. Owais et al., (1995) Antimicrob. Agents Chemother. 39:180; Briscoe et al., (1995) Am. J. Physiol. 1233:134; Schreier et al., (1994) J. Biol. Chem. 269:9090; Keinanen and Laukkanen (1994) FEBS Lett. 346:123;およびKillion and Fidler (1994) Immunomethods 4:273を参照されたい。
開示の使用および方法
本開示の抗体またはその抗原結合部分(組成物、二重特異性)は、例えば、癌、炎症性疾患または感染症の治療および/または防止に関与する、多くのin vitroおよびin vivoの用途を有する。抗体は、腫瘍増殖を阻害するために、例えばin vivoでヒトの対象に投与され得る。
癌細胞の増殖および生存を阻害する、開示の抗CD40抗体の能力を考慮すると、開示は、腫瘍の増殖が対象において阻害されるように、開示の抗体を対象に投与する段階を含む、対象における腫瘍細胞の増殖を阻害するための方法を提供する。開示の抗体によって治療することができる腫瘍の非限定的な例には、原発性および/または転移性のB細胞リンパ腫、慢性リンパ球性白血病、多発性骨髄腫、黒色腫、結腸腺癌、膵臓癌、大腸癌、胃腸癌、前立腺癌、膀胱癌、腎臓癌、卵巣癌、子宮頸癌、乳癌、肺癌、および鼻咽頭癌が含まれるが、これらに限定されない。追加的に、難治性または再発性の悪性腫瘍の増殖が、開示の抗体を使用して阻害され得る。
別の態様において、開示は、治療有効量の開示の抗体またはその抗原結合部分を対象に投与する段階を含む、対象における炎症性疾患、感染症、アテローム血栓症、または呼吸器疾患を治療するための方法を提供する。追加の抗炎症剤、抗微生物剤、または他の治療剤が、開示の抗体、または、その抗原結合部分と共に投与され得る。
一般に、開示の抗体は、対象における免疫反応を強化するために使用され得る。
開示のこれらの方法および他の方法は下で更に詳細に説明される。
併用療法
別の態様において、開示は、本開示の抗CD40抗体(またはその抗原結合部分)が、対象における腫瘍増殖の阻害において有効である1または複数の追加の抗体と共に同時投与される併用療法の方法を提供する。一実施形態において、開示は、抗CD40抗体、ならびに、抗VISTA抗体、抗LAG‐3抗体、抗PD‐L1抗体、および抗PD‐1抗体、および/または抗CTLA-4抗体などの1または複数の追加の抗体を対象に投与する段階を含む、対象における腫瘍増殖を阻害するための方法を提供する。特定の実施形態において、対象はヒトである。
CD40シグナル伝達活性化は更に、標準的な癌治療とも組み合わされ得る。例えば、CD40シグナル伝達活性化が、CTLA‐4および/またはLAG‐3および/またはPD‐1ブロック、また、化学療法計画と組み合わされ得る。例えば、化学療法剤が、細胞毒性剤であり得る抗CD40抗体と共に投与され得る。例えば、エピルビシン、オキサリプラチン、5‐FUが、抗CD40療法を受ける患者に投与される。
任意で、抗CD40および1または複数の追加の抗体(例えば、抗CTLA‐4および/または抗LAG‐3および/または抗PD‐1抗体)の組み合わせが更に、癌細胞、精製された腫瘍抗原(組換えタンパク質、ペプチド、および炭化水素分子を含む)、および、免疫刺激サイトカインをエンコードする遺伝子でトランスフェクトされた細胞などを含む免疫原と組み合わされ得る(He et al., (2004) J. Immunol. 173:4919-28)。使用され得る腫瘍ワクチンの非限定的な例は、gp100、MAGE抗原、Trp-2、MART1および/またはチロシナーゼのペプチドなど、黒色腫抗原のペプチド、または、サイトカインGM-CSFを発現するようにトランスフェクトされた腫瘍細胞を含む。
抗CD40抗体と組み合わされ得る他の療法は、インターロイキン2(IL‐2)投与、放射線、手術、またはホルモン抑制を含むが、これらに限定されない。
本明細書において説明される治療剤の組み合わせは、薬学的に許容される担体における単一の組成物として同時に投与され得るか、または、薬学的に許容される担体における各薬剤と共に別個の組成物として同時に投与され得る。別の実施形態において、治療剤の組み合わせは、順次に投与され得る。
更に、併用療法の1より多くの用量が順次に投与される場合、順次投与の順序は、投与の各時点において、逆になり得、または、同じ順序に維持され得、順次の投与は、同時の投与、または、それらの任意の組み合わせと組み合わされ得る。
本開示は以下の例において更に図示されるが、これは、更なる限定として解釈されるべきでない。本出願全体において引用される、すべての図面およびすべての参考文献、Genbank配列、特許および特許出願公開の内容は、参照によって本明細書に明示的に組み込まれる。
例
例1 ヒト、アカゲザルまたはマウスCD40を安定的に発現するHEK293A細胞株の構築
ヒト、アカゲザルまたはマウスCD40を過剰発現する安定的な細胞株は、HEK293A細胞(Cobioer、NJ、中国)を使用して構築されたい。簡潔に説明すると、ヒト、アカゲザル、またはマウスCD40のcDNA配列(SEQ ID NO:68、70、72にそれぞれ記載されているアミノ酸配列をエンコードするSEQ ID NO:67、69、71)が合成され、次に、EcoRI 部位とBamHI 部位との間で、pLV-EGFP(2A)-Puroベクターにサブクローニングされた。レンチウイルスが、Lipofectamine 3000キット(Thermo Fisher Scientific、米国)の指示に従って、pLV-EGFP(2A)-Puro-CD40、psPAXおよびpMD2.Gプラスミドの共トランスフェクションによってHEK-293T細胞(Cobioer、NJ、中国)において生成された。共トランスフェクションの3日後、レンチウイルスは、それぞれのHEK-293T細胞の細胞培養培地(10%FBS(Cat#:FND500、Excell)を有するDMEM培地(Cat#:SH30022.01、Gibco))から採取された。最後に、HEK293A細胞にレンチウイルスを感染させ、ヒト、アカゲザル、またはマウスCD40を安定的に発現するHEK293A細胞株、すなわち、HEK293A/ヒトCD40、HEK293A/アカゲザルCD40またはHEK293A/マウスCD40細胞を生成した。次に、トランスフェクトされたHEK293A細胞を、0.2μg/mlのピューロマイシン(Cat #:A11138-03、Gibco)を含む培地(DMEM+10%FBS)において7日間培養した。ヒトCD40およびアカゲザルCD40の発現は、市販の抗ヒトCD40抗体(PE‐抗ヒトCD40、Biolegend、米国、Cat#:313006)を使用して、FACSによって確認された。同様に、マウスCD40の発現は、市販の抗マウスCD40抗体(PE‐抗マウスCD40、Biolegend、米国、Cat#:124609)を使用して、FACSによって確認された。
例2:ヒトCD40に対するモノクローナルマウス抗体を生成する例示的なハイブリドーマ細胞株の産生
いくつかの改変を有する従来のハイブリドーマ融合技術を使用して、ネズミ抗ヒトCD40モノクローナル抗体(mAb)が生成された。
免疫化
下の表2におけるスキームに従って、13匹のBALB/cマウス(Beijing Vital River Laboratory Animal Technology Co., Ltd,、北京、中国)に、組み換えヒトCD40 (ECD)-his (Sino Biological,中国,Cat#:10774-H08H)および/または組み換えアカゲザルCD40(ECD)-hFc (Sino Biological,中国,Cat#:90097-C02H)を注射した。ヒトCD40(ECD)‐hisおよびアカゲザルCD40(ECD)‐hFcは、等しい体積の完全フロイントアジュバント(SIGMA、米国、Cat#:F5881-10*10ML)、不完全フロイントアジュバント(SIGMA、米国、Cat#:F5506-6*10ML)、またはPBSを用いて、超音波処理によって乳化された。
表2
免疫化スキーム
各追加免疫の1週間後、組み換えヒトCD40(ECD)‐hFc(Sino Biological、中国、Cat#:10774-H02H)およびアカゲザルCD40(ECD)‐hFc(Sino Biological、中国、Cat#:90097-C02H)を使用して、ELISAによる力価測定のために、50μlのネズミ血清が各マウスから採取された。力価測定はまた、例1において調製された、HEK293A過剰発現ヒトCD40、アカゲザルCD40、またはマウスCD40を使用して、FACSによって行われた。
最終追加免疫後のELISAおよびFACS分析結果に基づいて、最高の血清力価を有する7匹のマウスがハイブリドーマ細胞株生成のために選択された。
ハイブリドーマ細胞株の生成
小さい改変を有する従来のハイブリドーマ融合技術を使用して、ハイブリドーマ細胞株が生成された。
最終追加免疫の4日後、マウスを犠牲にし、脾臓を採取して、PBSにおいて単一の細胞懸濁液として調製した。DMEM培地(Hyclone、Cat#:SH30243.01B)を用いて、脾臓細胞を3回洗浄した。対数期における生存骨髄腫細胞SP2/0 (ATCC、CRL-1581)を、ネズミ脾臓細胞と1:4の比で混合した。次に細胞を2回洗浄し、次に、PEG(Sigma、Cat#:P7181)との細胞融合を実行した。DMEM培地を用いて、融合後の細胞を3回洗浄し、10%FBSおよび1X HAT(Sigma、H0262)が添加された細胞増殖培地(RPMI培地1640(Gibco、Cat#:C22400500CP))に懸濁した。細胞懸濁液を96ウェル細胞培養プレートにおいて、ウェルあたり200μl(5×104細胞/ウェル)で平板培養し、37℃の加湿した5%CO2インキュベータにおいて7日間にわたってインキュベートした。次に、増殖培地を、10%FBS+1X HT(Sigma、H0137)が添加された新鮮な増殖培地で置き換えた。2~3日後、ハイブリドーマ細胞をELISAおよびFACSによってスクリーニングした。
ELISAによるハイブリドーマ細胞株のスクリーニング
最初にハイスループットELISA結合アッセイを使用して、ヒトCD40に結合するモノクローナル抗体を産生するハイブリドーマクローンについてスクリーニングした。ヒトCD40に結合するモノクローナル抗体を産生するハイブリドーマクローンを、アカゲザルまたはマウスCD40と交差反応する能力について、更に試験した。
ELISAアッセイのために、96ウェルELISAプレートを100μl/ウェルのヒトCD40(ECD)‐his(0.5μg/ml、Sino Biological、中国、Cat#:10774-H08H)、アカゲザルCD40(ECD)‐hFc(0.5μg/ml、Sino Biological、中国、Cat#:90097-C02H)、または、ネズミCD40‐His(0.5μg/ml、Sino Biological、中国、Cat#50324-M03H)で、室温において一晩コーティングした。プレートをPBST緩衝液(PBS+0.05% Tween20)で3回洗浄し、200μlのブロック緩衝液(1%BSA、1%ヤギ血清、および、0.05% Tween20を含むPBS)で、室温において2時間ブロックし、PBSTで3回洗浄した。次に、ハイブリドーマ細胞培養液の上清を、希釈緩衝液(1%BSA、1%ヤギ血清、および、0.01% Tween20を含むPBS)で10倍希釈し、ウェルあたり100μlとなるようにプレートに添加した。室温で1時間インキュベートした後、プレートをPBSTで3回洗浄し、次に、100μlのヤギ抗マウスFc‐HRP(1:5000、Sigma、米国、Cat#:A9309-1ml)を各ウェルに添加した。室温で1時間インキュベートした後に、プレートをPBSTで3回洗浄し、次に、80μlのTMBを各ウェルに添加した。5~10分後、80μlの0.16M硫酸を各ウェルに添加し、次に、SpectraMaxR i3X(Molecular Devies、米国)でOD450を読んだ。
ELISAアッセイによって、234のハイブリドーマクローンがヒトおよびアカゲザルCD40の両方に特異的に結合することが識別された。
FACSによるハイブリドーマ細胞株のスクリーニング
234のハイブリドーマクローンが、HEK293A細胞上で発現するヒト、アカゲザル、またはマウスCD40に対する結合能力について更にスクリーニングされた。簡潔に説明すると、例1において調製された100,000個のHEK293AヒトCD40細胞、HEK293A/アカゲザルCD40細胞、または、HEK293A/マウスCD40細胞が、96ウェルプレートの各ウェルに播種され、希釈緩衝液(PBS+1%BSA、1%ヤギ血清、および0.01% Tween20)で10倍に希釈されたハイブリドーマ細胞培養上清をプレートに添加した(100μl/ウェル)。4℃で1時間インキュベートした後に、プレートをPBSTで3回洗浄した。次に、500倍に希釈されたAPCヤギ抗マウスIgG(BioLegen、米国、Cat#:405308)と共に、細胞がプレートに添加された。4℃で1時間インキュベートした後に、プレートをPBSで3回洗浄し、次に、FACSマシン(BD)を使用して細胞蛍光をモニタリングした。
FACSスクリーニングに基づいて、HEK293A/ヒトCD40およびHEK293A/アカゲザルCD40細胞の両方に対する高い結合能力を示した162の陽性クローンが取得された。
抗CD40抗体を産生するハイブリドーマクローンのサブクローニング
162のハイブリドーマクローンが2回のサブクローニングを受けた。サブクローニングの間、各親クローンからの複数のサブクローン(n>3)が選択され、上に記載されるように、ELISAおよびFACSアッセイによって確認された。このプロセスを通じて選択されたサブクローンは、モノクローナル抗体を産生するハイブリドーマ細胞として定義された。最後に、ヒトおよびサルCD40の両方に対する高い結合能力を有する108のサブクローン(各親クローンから1サブクローン)が取得された。
HEK Blue活性アッセイによるハイブリドーマ細胞株のスクリーニング
108のサブクローンを96ウェルプレートに拡げ、次に、5日間培養した。ヒトCD40に対するアゴニスト活性を有するCD40抗体を識別するためのHEK-Blue活性アッセイのために上清を採取した。
簡潔に説明すると、ヒトCD40(SEQ ID NO:68)を発現する安定的なHEK-Blueレポータ細胞株(HEK-Blue/CD40と呼ばれる)が、HEK-Blueヌル1_v細胞(Invivogen、サンディエゴ、カリフォルニア州)にCD40発現レンチウイルス(例1において生成)を感染させることによって確立され、その後、10μg/mlのピューロマイシンで選択された。
HEK-Blueレポータアッセイのために、200μlの培養培地(DMEM培地(Hyclone、米国、Cat#:SH30243.01)+10%FBS(Excell、中国、Cat#:FND500)+10μg/mlピューロマイシン(GIBCO、米国、Cat#:A11138-03)+100μg/mlノルモイシン(登録商標)(Invivogen,米国,Cat#:ant-nr-2)、100μg/mlゼオシン(Invivogen,米国,Cat#:ant-Zn-5))に再懸濁された40000のHEK-Blue/CD40細胞を96ウェルプレートにおいて平板培養し、37℃で一晩培養した。2日目に、200μlのDMEM培地を各ウェルに添加し、培養培地と置き換えた。7時間後、ウェルにおけるDMEM培地は、100μl/ウェルのHEK Blue Detection緩衝液(Invitrogen;米国;Cat#:hb-det3)および100μl/ウェルのハイブリドーマ細胞培養上清で置き換えた。結果として生じた混合物を、適切な青色が現れるまで、5%のCO2の存在下で、37℃でインキュベートした。SpectraMaxマイクロプレートリーダ(Molecular Devices、米国、SpectraMaxR i3X)を使用してOD630が測定された。抗HEL抗体(LifeTein, LLC、米国、Cat.#:LT12031)をネガティブコントロールとして使用し、RO7009789(ヒトIgG2/カッパ定常領域を有する、US7338660B2において開示されたアミノ酸配列を使用して調製されたアゴニスト抗体)、および、CD40の天然のリガンドおよび活性化因子であるCD40L(Sino Biological、中国、Cat:10239-H08E)をポジティブコントロールとして使用した。
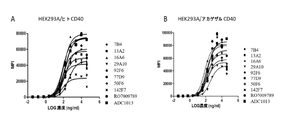
図1に示されるように、38のクローンは、異なるレベルのCD40アゴニスト活性を示し、他はアゴニスト活性を示さなかった。
例3 マウス抗CD40モノクローナル抗体の精製
上述のHEK-Blueアッセイに基づいて、高いHEK-Blue活性を有する20のクローン(下の表3を参照されたい)を、更なる特性評価について選択した。20の選択されたクローンからのモノクローナルマウス抗体を精製した。簡潔に説明すると、1%HT(Gibco、Cat#:11067-030)が添加された100mlの新鮮な無血清培地(Gibco、米国、Cat#:12045-076)を各々が有するT175細胞培養フラスコにおいて各サブクローンのハイブリドーマ細胞を増殖させた。37℃の5%CO2を有するインキュベータに細胞培養液を10日間置いた。細胞培養液を採取後、3500rpm、5分間の遠心分離を行い、次に、0.22μmカプセルを使用して濾過を行い、細胞片を除去した。次に、前平衡タンパク質Aアフィニティカラム(GE、米国、Cat#:17040501)を使用してモノクローナルマウス抗体を精製し、溶出緩衝液(20mMクエン酸、pH3.0~pH3.5)で溶出した。次に、抗体をPBS緩衝液(pH7.0)で維持し、NanoDrop計器を使用して、その濃度を決定した。
精製された各抗体のアイソタイプは、製造者のマニュアルに従って、Rapid Isotyping Kit with Kappa and Lambda-Mouse(Thermal、米国、Cat#:26179)およびMouse Monoclonal Antibody Isotyping Reagents(Sigma、米国、Cat#:IS02-1KT)を使用することによって決定された。アイソタイプ決定の結果および選択された上位20クローンの発現力価を表3にまとめた。
表3
抗OD40抗体クローンのアイソタイプおよび発現力価
例4 ヒトおよびサルCD40に結合された精製マウス抗CD40モノクローナル抗体
精製マウス抗CD40モノクローナル抗体はまず、ELISAアッセイによって特性評価され、組み換えヒト、サル、またはマウスCD40タンパク質に対する結合親和性を決定した。
ELISAプレートを、500ng/mlヒトCD40(ECD)‐his(Sino Biological、中国、Cat#:10774-H08H)でコーティングし、4℃で一晩放置した。200μlのブロック緩衝液(1%BSA、1%ヤギ血清および0.05% Tween20を含むPBS)でウェルをブロックし、室温で2時間放置し、次に、100μlの連続希釈した抗CD40抗体(40000ng/mlから開始)を各ウェルに添加して、室温で1時間インキュベートした。プレートをPBST(PBS+0.05% Tween 20)で3回洗浄し、5000倍希釈したヤギ抗マウスIgG‐HRP(Simga、米国、Cat#:A9309-1ml)を添加し、室温で1時間にわたってインキュベートした。新しく調製したUltra-TMB(BD、米国、Cat#:555214)を用いて、プレートを室温で5分間現像した。SpectraMax(登録商標) i3X(Molecular Devies、米国)で450nmの吸光度を読んだ。
サルまたはマウスCD40に対する20のCD40 mAbの種交差反応性を、ダイレクトELISAによって更に評価した。簡潔に説明すると、500ng/mlサルCD40(ECD)‐hFc(Sino Biological、中国、Cat#:90097-C02H)またはマウスCD40‐hFc(Sino Biological、中国、Cat#:50324-M03H)を96ウェルELISAプレート上にコーティングし、その後、連続希釈された100μlの抗CD40抗体(40000ng/mlから開始)と共にインキュベートした。その後、HRP(Sigma、米国、Cat#:A9309-1ml)と結合されたヤギ抗マウスIgGを使用した。抗CD40抗体RO7009789およびADC1013(ヒトIgG1/カッパ定常領域を有する、US2016/0311916A1において開示されたアミノ酸配列を使用して調製した)を参照として使用した。
これらの結合試験のEC
50値を表4にまとめる。51F7を除く、20の抗体すべては、明らかにサルCD40と交差反応したが、マウスCD40とは交差反応しなかったことが分かる。
表4
ヒト、サル、またはマウスCD40に対する20のマウス抗CD40mAbの結合能力
例5 HEK293A細胞で発現した、ヒトおよびアカゲザルCD40に結合された例示的なマウス抗CD40モノクローナル抗体
抗CD40抗体が、HEK293A細胞で発現するヒト、サル、またはマウスCD40に結合するかどうかを更に決定するために、例1において生成されたヒト、サル、またはマウスCD40をそれぞれ安定的に過剰発現するHEK293A細胞を使用して、FACSによる細胞ベース結合アッセイを実行した。簡潔に説明すると、105のHEK293A細胞を96ウェルプレートの各ウェルに播種し、連続希釈された抗CD40抗体をプレートに添加した。4℃で1時間インキュベートした後に、プレートをPBSTで3回洗浄した。次に、500倍希釈されたAPC結合ヤギ抗マウスIgG(BioLegen、米国、Cat#:405308)をプレートに添加した。4℃で1時間インキュベートした後に、プレートをPBSで3回洗浄し、次に、FACSマシン(BD)を使用して細胞蛍光をモニタリングした。
下の表5に示されるように、マウス抗CD40モノクローナル抗体はすべて、ヒトおよびアカゲザルCD40の両方に対して高い結合能力を示したが、マウスCD40には結合しなかった(データは示していない)。
表5
ヒトおよびサルCD40に対するマウス抗OX‐40抗体の結合親和性
例6 マウス抗CD40抗体がヒトCD40‐CD40L相互作用を阻害または促進した
ヒトCD40に対するヒトCD40Lの結合をブロックまたは促進する能力について、精製された抗CD40抗体を更に解析した。簡潔に説明すると、500ng/mlのヒトCD40L(Sino Biological、中国、Cat#10239-H08E)で、96ウェルのELISAプレートを4℃で一晩コーティングした。200μlのブロック緩衝液(PBS+2%BSA)を用いて、プレートを室温で2時間ブロックした。次に、連続希釈された抗CD40抗体(40μg/mlから開始)を、2μg/mlのヒトCD40‐hFc(Sino biological, Cat#:10774-H02H)と混合し、37℃で1時間インキュベートし、次に、この混合物をウェルに添加し、室温で1時間インキュベートした。PBST(PBS+0.05% Tween20)を用いてプレートを3回洗浄し、抗ヒトIgG FC‐HRP(1:5000、Sigma、米国、Cat#:A0170-1ML)を添加し、次に、室温で1時間インキュベートした。プレートをPBSTで3回洗浄し、次に、80μlのTMBを各ウェルに添加した。5~10分後、80μlの0.16M硫酸を各ウェルに添加し、次に、SpectraMaxR i3X(Molecular Devies、米国)でOD450を測定した。
興味深いことに、データは、6のマウス抗体(13A2、16A6、7B4、50F6、142F7および101C12)がヒトCD40‐CD40L相互作用を促進し、別の3の抗体(23B8、92F6、82D3)がCD40‐CD40L相互作用をブロックしたことを示し、残りはCD40‐CD40L相互作用に対する明確な影響を示さなかった。4の代表的な抗体の結果を図2に示す。
例7 例示的なマウス抗CD40抗体のアゴニスト活性の決定
選択されたマウス抗CD40抗体がアゴニスト活性を有するかどうかを決定するために、HEK-Blue活性アッセイが実行された。簡潔に説明すると、例2において生成されたHEK-Blue/CD40細胞をDMEM培地(Hyclone、米国、Cat#:SH30243.01)+10%FBS(Excell、中国、Cat#:FND500)+10μg/mlピューロマイシン(GIBCO、米国、Cat#:A11138-03)+100μg/mlノルモシン(Invivogen、米国、Cat#:ant-nr-2)+100μg/mlゼオシン(Invivogen、米国、Cat#:ant-Zn-5)においてインキュベートした。200μlの培養培地における40,000のHEK-Blue/CD40細胞を、96ウェルアッセイプレートの各ウェルに分注し、37℃で培養した。一晩(約12時間)インキュベートした後に、200μlの新鮮なDMEM培地を使用して培養培地を置き換えた。7時間後、各ウェルにおけるDMEM培地を、様々な濃度(100μg/ml~0.01ng/ml)で抗CD40抗体を含む100μL/ウェルのHEK Blue Detection緩衝液(Invivogen;米国;Cat#:hb-det3)で置き換えた。適切な青色が現れるまで、細胞を37℃でインキュベートした。SpectraMaxマイクロプレートリーダ(Molecular Devices、米国、SpectraMaxR i3X)を使用して、630nmにおける吸光度を測定した。
EC
50値を下の表6にまとめた。代表的な曲線を図3に示した。見て分かるように、20のマウス抗体はすべて、HEK-Blueアッセイにおいて異なるアゴニスト活性を示し、CD40下流シグナル伝達を刺激する能力を示唆した。
表6
抗CD40抗体のアゴニスト活性
例8 エピトープビニング
エピトープビニングのために、競合ELISAアッセイを実行した。簡潔に説明すると、5μg/mlのR07009789またはADC1013を用いて、96ウェルプレートを4℃で一晩コーティングした。200μlのブロック緩衝液(1%BSA、1%ヤギ血清および0.05% Tween20を含むPBS)を用いて、ウェルを室温で2時間ブロックした。ヒトCD40(ECD)‐His(Sino Biological、中国、Cat#:10774-H08H)を0.5μg/mlに希釈し、プレートに添加し、次に室温で1時間インキュベートした。ELISAプレートをPBSTで3回洗浄し、次に、精製された抗体を1μg/mlに希釈し、各ウェルに添加し、室温で1時間インキュベートした。ELISAプレートをPBSTで3回洗浄し、次に、1:20000で希釈された抗マウスFc‐HRP(Sigma、米国、Cat#:A9309-1MC)を各ウェルに添加し、室温で1時間インキュベートした。新しく調製したUltra‐TMB(Huzhou Yingchuang、中国、Cat#: TMB-S-003)を用いて、プレートを室温で5分間現像し、SpectraMaxマイクロプレートリーダ(Molecular Devices;米国;SpectraMaxR i3X)上で、450nmの吸光度を測定した(OD450)。
結果を表7にまとめた。7のマウス抗体(101C12、142F7、89D11、13A2、16A6、7B4および50F6)が両方の参照抗体と競合し、抗体9A7、92F6、19H4、16F4および51F7は、いずれの参照抗体とも競合を示さなかった。残りの抗体は、2つの参照抗体のいずれかと競合した。
表7
競合ELISAによるエピトープビニング
例9 例示的なアゴニスト抗CD40抗体が樹状細胞成熟を促進した
マウス抗CD40抗体のアゴニスト活性を更に決定するために樹状細胞成熟アッセイを実行した。簡潔に説明すると、1人の健康なヒトのドナーの血液試料からのPBMCを、密度勾配遠心分離によって採取し、次に、RPMI1640培地で再懸濁した。PBMCを37℃のインキュベータで2時間培養し、容器の壁に付着した細胞を単離単球として採取した。24ウェルプレートにおける、10%FBSが添加されたRPMI1640培地において、100ng/mlの組み換えヒトGM‐CSF(R&D、米国、Cat#:7954-GM)および100ng/mlの組み換えヒトIL‐4(R&D、米国、Cat#:6507‐IL)と共に単球を培養した。3日後、培地の半分を新鮮な培地で置き換えた。培養の6日目に、抗CD40抗体(10μg/mlまたは1μg/ml)、または、対照の抗体(RO7009789、ADC1013、およびHel(LifeTein、米国、Cat#:LT12031))を細胞に添加し、プレートを更に48時間培養した。FITCマウス抗ヒトCD83(BD、米国、Cat#:556910)、PEマウス抗ヒトCD86(BD、Cat#:555658)およびBV650マウス抗ヒトCD80(BD、米国、Cat#:564158)を、FACSによるDC活性化マーカーの染色のために使用した。
代表的な抗体の結果を図4に示した。マウス抗CD40抗体16A6、29A10、7B4および13A2は、抗Helアイソタイプの対照と比較して、成熟した樹状細胞のバイオマーカーであるCD86の発現を増加させ、抗体16A6、29A10、7B4および13A2は、両方とも共刺激分子であるCD80およびCD83の発現を著しくアップレギュレートした。
例10 例示的なキメラ抗CD40抗体の発現および精製
8の抗体(13A2、16A6、7B4、29A10、92F6、77D9、50F6および142F7)が更なる試験のために選択された。文献(Juste et al., (2006), Anal Biochem. 1;349(1):159-61)に記載された変性プライマーのセットと共に、標準的なPCR方法を使用して、選択された8の候補の抗体の可変領域配列をハイブリドーマ細胞からクローニングした。重鎖可変領域+ヒトIgG2定常領域をエンコードする配列、または、軽鎖可変領域+ヒトカッパ定常領域をエンコードする配列(それぞれ、SEQ ID NO:63、65において記載される重鎖定常領域および軽鎖定常領域のアミノ酸配列)をpCDNA3.1(Invitrogen、Carlsbad、米国)のXhoI/BamHI制限部位に挿入することによって発現ベクターを構築した。重鎖可変領域のC末端は、ヒトIgG2定常領域のN末端に連結され、軽鎖可変領域のC末端は、ヒトカッパ定常領域のN末端に連結された。可変領域のアミノ酸SEQ ID番号を上の表1にまとめた。
発現ベクターは、HEK-293F細胞(Cobioer、NJ、中国)にトランスフェクトされたPEIである。特に、HEK-293F細胞は、Free Style(登録商標)293発現培地(Gibco、Cat#:12338-018)において培養され、1:3のDNA:PEI比でポリエチレンイミン(PEI)を使用して発現ベクターでトランスフェクトされた(DNAは細胞培地ミリリットルあたり1.5μg)。トランスフェクトされたHEK-293F細胞を、120RPMで振とうしながら、5%CO2下で、37℃でインキュベータにおいて培養した。10~12日後、上清を採取し、モノクローナル抗体を例3において説明されたように精製した。
例11 HEK293A細胞で発現されたヒトまたはアカゲザルCD40に結合された例示的なキメラ抗CD40モノクローナル抗体
例5のプロトコルに従って、例1において生成されたHEK293A/ヒトCD40細胞、HEK293A/アカゲザルCD40細胞、およびHEK293A/マウスCD40細胞に結合する能力について、キメラ抗CD40抗体を更に特性評価した。図5に示されるように、キメラ抗体は、ヒトおよびサルCD40の両方に対して高い結合親和性を有した。これらのキメラ抗体は、マウスCD40に結合しなかった(データは示されていない)。
例12 例示的なキメラ抗CD40モノクローナル抗体はアゴニスト活性を有し、樹状細胞成熟を促進した。
例7および例9において説明されるプロトコルに従って、HEK-Blueアッセイおよび樹状細胞成熟アッセイによって、CD40シグナル伝達活性化に対する作用について、キメラ抗体を評価した。RO7009789、APX005(ヒトIgG1/カッパ定常領域を有するWO2014/070934A1において開示されるアミノ酸配列を使用して調製された)および/またはADC1013を参照として使用した。
図6において示される8のキメラ抗体は、親モノクローナル抗体と同様の機能活性を示した。図7は、すべての試験されたキメラ抗体が、成熟した樹状細胞のバイオマークであるCD86のアップレギュレーションによって示唆されるように、樹状細胞の成熟を促進したことを示した。
例13 例示的な抗CD40抗体のヒト化
上で説明された特性評価およびアッセイに基づいて、2つの候補抗体7B4および13A2が、ヒト化および更なる調査のために選択された。ネズミ抗体のヒト化は、下で詳細に説明されるような、よく確立されたCDR移植方法(参照によって全体が本明細書に組み込まれる米国特許第5,225,539号)を使用して実行された。
ネズミ抗体7B4および13A2のヒト化についてのアクセプタフレームワークを選択するために、7B4および13A2の軽鎖および重鎖可変領域配列をNCBIのウェブサイト(http:// www.ncbi.nlm.nih. gov/igblast)におけるヒト免疫グロブリン遺伝子データベースに対してBLASTを行った。7B4および13A2に対して最高の相同性を有するヒト生殖系列IGVHおよびIGVKをヒト化のアクセプタとして選択した。抗体7B4および13A2については、選択されたヒト重鎖アクセプタはIGHV4-28*06であり、選択されたヒト軽鎖アクセプタは、下の表8に列挙されたIGKV2-30*02である。
CDRループ構造の支持において重要な役割を担い得る主なフレームワーク残基を識別するべく、7B4および13A2の可変ドメインについて、3次元構造をシミュレートし、それにより、ヒト化抗体における復帰変異を設計した。選択された構造テンプレートは、L-CDR1、L-CDR2、L-CDR3、H-CDR1、H-CDR2およびH-CDR3において、それぞれ7B4および13A2と同じクラスのカノニカルループ構造を有した。選択された構造テンプレートを使用して、ネズミのフレームワークを重鎖および軽鎖についてのヒトのアクセプタのフレームワークと置き換えることによって構造モデルを構築した。次に、3次元構造モデリングシミュレーションを実行し、CDRループ構造または重鎖および軽鎖のインタフェースを支持するのに重要であり得る主なフレームワーク残基を識別した。ネズミ抗体およびヒトアクセプタがフレームワークにおける特定の部位において同一の残基を共有するとき、ヒト生殖系列残基は維持された。他方、ネズミ抗体およびヒト生殖系列アクセプタがフレームワークにおける特定の部位において異なる残基を有するとき、この残基の重要性は、構造モデリングによって評価された。ネズミ抗体のフレームワークにおける残基がCDR残基と相互作用し、それに影響することが分かった場合、この残基は、ネズミ残基に復帰変異された。
表8
抗体構造シミュレーションにおいて使用された構造テンプレート
上に説明された構造モデリングに基づいて、重鎖について5つの潜在的な復帰変異(I49M、V68I、M70I、K44N、G45K)および13A2の軽鎖について5つの潜在的な復帰変異(M4L、R51L、F76L、Y92F、Q105S)が識別された。7B4については、重鎖について5つの潜在的な復帰変異(I49M、V68I、M70I、K44N、G45K)が識別され、軽鎖について4つの復帰変異(M4L、R51L、Y92F、Q105S)が識別された。
表1にまとめられているように、合計5つのヒト化抗体を用いて、7B4および13A2の両方については、3つのヒト化重鎖可変領域および3つのヒト化軽鎖可変領域が設計された。
ヒト化重鎖可変領域+ヒトIgG2定常領域をエンコードする配列、および、軽鎖可変領域+ヒトカッパ定常領域をエンコードする配列(それぞれ、SEQ ID NO:63および65に記載された重鎖定常領域および軽鎖定常領域のアミノ酸配列)を化学的に合成し、次に、それぞれBamH IおよびXho I制限部位を使用して、pCDNA3.1(+)ベースの発現ベクター(Invitrogen、米国)にサブクローニングした。重鎖可変領域のC末端をヒトIgG2定常領域のN末端に連結し、軽鎖可変領域のC末端をヒトカッパ定常領域のN末端に連結した。すべての発現コンストラクトをDNA配列によって確認した。HEK293F発現系(Invitrogen、米国)を重鎖および軽鎖発現ベクターでトランスフェクトし、例10に説明されたプロトコルに従って、10のヒト化抗CD40抗体(13A2が5、7B4が5)を一時的に発現させた。ヒト化抗体を例3に説明されたように精製した。
例14 例示的なキメラおよびヒト化抗CD40抗体の特性評価
例5に説明されたプロトコルに従って、HEK293A/ヒトCD40細胞およびHEK293AアカゲザルCD40細胞への結合の能力について、キメラおよびヒト化抗CD40抗体の特性評価を更に行った。また、例7および例9におけるプロトコルに従って、HEK-BlueアッセイにおいてCD40シグナル伝達を活性化する能力、および、樹状細胞成熟を促進する能力についてそれぞれ試験し、樹状細胞は3名の健康なヒトドナーから採取された。製造者の指示に従って、ヒトIL12(p40)ELISAキット(BD、米国、Cat#:551116)を使用することによって、樹状細胞によるIL‐12(p40)分泌を測定した。
図8、図9、図10(ドナー1)、図11(ドナー2)、および図12(ドナー3)に示されるように、ヒト化抗CD40抗体13A2-VH3VL2、13A2-VH3VL3および7B4VH2VL2は、最高の結合、アゴニスト、および機能活性を示した。
例15 ヒトCD40に対する例示的なキメラまたはヒト化抗CD40抗体の結合親和性
BIAcore(登録商標)8K計器(GE Life Sciences)に対するヒトCD40に対するキメラまたはヒト化抗CD40抗体の結合親和性を決定するためにSPRアッセイを実行した。簡潔に説明すると、ヒトCD40(ECD)‐hisタンパク質(Sino Biological、中国、Cat#:10774-H08H)の100~200の応答単位(RU)がCM5バイオセンサチップ(Cat#:BR-1005-30、GE Life Sciences)に結合され、その後、1Mエタノールアミンを用いて、未反応の群のブロックを行った。0.3μM~10μMの濃度範囲の連続希釈された抗体をSPR泳動緩衝液(HBS-EP緩衝液、pH7.4、GE Life Sciences;米国;Cat#:BR-1006-69)に30μl/分で注入した。ブランク対照のRUを減算して結合能力を計算した。1対1のラングミュア結合モデル(BIA Evaluation Software、GE Life Sciences)を使用して会合速度(k
a)および解離速度(k
d)を計算した。平衡解離定数K
Dをk
d/k
a比として計算した。抗体のSPRで決定された結合曲線を図13に示した。キメラまたはヒト化抗体の結合親和性を表9に列挙した。
表9
ヒトCD40に対する抗CD40抗体の結合親和性
例16 例示的なキメラまたはヒト化抗CD40抗体のエピトープマッピング
ELISAによって、結合エピトープについてキメラおよびヒト化抗CD40抗体を試験した。
CD40細胞外ドメイン(ECD)には4つの個別のシステインリッチドメイン(CRD)、すなわち、CRD1、CRD2、CRD3およびCRD4がある。CD40 ECDの構造に基づいて、1つの完全長CD40 ECD、5つのCD40 ECD欠失体、および、4つのCD40 ECD変異体を生成した。その情報は下の表10において見ることができる。これらの組換えタンパク質は、タンパク質分泌のためにN末端においてシグナルペプチド(SEQ ID NO:83)に連結され、ELISAアッセイのためにC末端においてmFcタグ(SEQ ID NO:84)に連結される。これらの組換えタンパク質をエンコードするDNA配列を合成し、pCDNA3.1ベクターにサブクローニングした。例10におけるプロトコルに従って、組換えタンパク質の発現および精製を実行した。例2におけるプロトコルに従って、組み換えCD40タンパク質に対するmAbの結合能力を評価するためにELISAアッセイを実行した。
図14のパネルAに示されるように、すべての抗体は完全長CD40 ECDに結合したが、欠失体には結合しなかった。これは、CD40のCRD1ドメインが抗体の結合に関与し、重要であることを示す。図14のパネルBは、キメラ13A2抗体、3つのヒト化抗体、およびADC1013が、CD40変異体2-4に結合できないことを示した。このことは、5つの抗体が同一または同様の結合エピトープを共有することを示す。RO7009789は変異体1、2および4に結合できず、APX005は変異体2および4に結合できない。
表10
CD40 ECD欠失体および変異体
注:CRDΔ1:欠失したCDR1ドメイン
例17 ヒトCD40に特異的に結合する例示的なヒト化抗CD40抗体
相同性アミノ酸配列を有する他のヒトTNFRSFメンバーと比較してヒトCD40に対する抗CD40抗体の結合特異性を決定するために、例2に説明されるプロトコルに従ってELISAアッセイを実行した。
ヒトCD40(ECD)‐his(TNFRSF5、Sino Biological、中国、Cat#:10774-H08H)、ヒトOX40-his(TNFRSF4、Sino Biological、中国、Cat#:10481-H08H)、ヒトHVEM-mFc(TNFRSF14、ACRO、中国、Cat#:HVM-H5255)、ヒト4-1BB(TNFRSF9、ACRO、中国、Cat#:41B-H522a)、ヒトNGFR(TNFRSF16、Sino Biological、中国、Cat#:13184-H08H)、ヒトDR6(TNFRSF21、Sino Biological、中国、Cat#:10175-H08H)、ヒトRANK(TNFRSF11、ACRO、中国、Cat#:RAL-H5240)に対する抗CD40抗体の結合親和性を試験した。
図15に示すように、13A2VH3VL3および7B4VH2VL2のいずれも、組み換えヒトOX40(TNFRSF4)、HVEM(TNFRSF14)、4‐1BB(TNFRSF9)、NGFR(TNFRSF16)、DR6(TNFRSF21)、またはRANK(TNFRSF11)に対する結合を示さなかった。このことは、13A2VH3VL3および7B4VH2VL2がヒトCD40に特異的に結合したことを示唆する。
例18 操作された抗CD40抗体はより良いアゴニスト活性を有した。
アゴニスト抗CD40抗体の最適な生物学的抗腫瘍作用は、Fc受容体(FcR)の共係合(coengagement)を必要とすることが示唆されている(Richman and Vonderheide, (2014) Cancer Immunol Res 2(1): 19-26)。したがって、13A2VH3VL3または7B4VH2VL2の重鎖/軽鎖可変領域+ヒトIgG1/カッパ定常領域を有する抗CD40抗体を調製した。ヒトIgG1定常領域は、267EおよびL328F変異(SEQ ID NO:64において記載された、変異したIgG1定常領域のアミノ酸配列)を有するように操作した。取得された抗体をそれぞれ、13A2-VH3VL3-IgG1(SE/LF)および7B4-VH2VL2-IgG1(SE/LF)と名付け、例9におけるプロトコルに従って、樹状細胞成熟を促進する能力について更に特性評価を行った。RO7009789、ADC1013、およびAPX005をポジティブコントロールとして使用し、Helをネガティブコントロールとして使用した。
図16に示すように、13A2-VH3VL3-IgG1(SE/LF)および7B4-VH2VL2-IgG1(SE/LF)は両方とも、親の抗体と比べて、樹状細胞成熟の促進において、著しく高いアゴニスト活性を示し、13A2-VH3VL3-IgG1(SE/LF)は、すべての試験された抗体の中で最高の活性を有する。
例19 例示的なヒト化抗CD40抗体がin vivo抗腫瘍作用を有した。
13A2-VH3VL3または7B4-VH2VL2の重鎖/軽鎖可変領域およびマウスIgG1/カッパ定常領域(それぞれ、SEQ ID NO:62、66に記載されたマウスIgG1/カッパ定常領域のアミノ酸配列)を有する抗CD40抗体のin vivo抗腫瘍活性を、ヒトCD40を有する遺伝子組み換えマウス(GemPharmatech Co. Ltd、中国)にMC38ネズミ結腸腺癌を移植することによって確立された動物モデルにおいて試験した。マウスモデルにおけるFc機能を強化するためにマウスIgG1/カッパ定常領域を使用した。0日目に各マウスの片方の脇腹に1×106のMC38細胞を皮下注射した。腫瘍が約80mm3に増殖したとき、動物を5群に無作為に割り付けた(8マウス/群)。次に、13A2-VH3VL3、7B4-VH2VL2、RO7009789、APX005またはPBSを10mg/kg/日の用量で、4日目、7日目、11日目、14日目、18日目、21日目に動物に腹腔内投与した。RO7009789およびAPX005は両方とも、マウスIgG1/カッパ定常領域(それぞれ、SEQ ID NO:62および66に記載されたマウスIgG1/カッパ定常領域のアミノ酸配列)を有するように操作された。
腫瘍サイズおよびマウス体重を経時的に追跡した。腫瘍の寸法(幅および長さ)をノギスで測定し、腫瘍体積を公式TV=(長さ×幅2)/2で計算した。抗体投与群における腫瘍体積が3.5cm3に到達する前に実験を終了した。腫瘍体積の差を識別するために一元配置分散分析を使用した。
25日目に、各群からのより大きい腫瘍を有する4匹のマウスをT細胞分析のために選択した。マウスを犠牲にした直後に腫瘍を採取し、コラゲナーゼを有するHanks緩衝液に配置した。次に、腫瘍を小片に切断し、コラゲナーゼを有するHanks緩衝液において、穏やかに振とうしながら37℃で30分間インキュベートした。その後、コラゲナーゼを不活性化して免疫細胞の生存を維持するために10mlのRPMI1640+10%FBSを各試料に添加した。試料を70μmの細胞フィルタ膜(Corning、Cat#:352350)に通し、新しい管に配置した。試料をペレット化して、1×107細胞/mlの密度でPBSF緩衝液(PBS+2%FBS)に再懸濁した。試料をPBSF緩衝液で2回洗浄した。各試料を2つの部分に分割した。一方には、抗CD45(Brilliant Violet 785(登録商標)抗マウスCD45抗体;Biolegend;米国;Cat#:103149)、抗CD8(APC抗マウスCD8a抗体;Biolegend;米国;Cat#:100712)、抗CD3(FITC抗マウスCD3抗体;Biolegend;米国;Cat#:100203)および抗CD4(PerCP抗マウスCD4抗体;Biolegend;米国;Cat#:100432)蛍光抗体混合物を添加し、他方には、抗CD11c(APC抗マウスCD8a抗体;Biolegend;米国;Cat#:100712)、抗CD80(APC抗マウスCD11c抗体;Biolegend;米国;Cat#:117310)、抗CD83(PE/Cy7抗マウスCD83抗体;Biolegend;米国;Cat#:121518)および抗CD86(FITC抗マウスCD86抗体;Biolegend;米国;Cat#:105005)蛍光抗体混合物を添加した。結果として生じる混合物を4℃で30分インキュベートした。細胞をPBSF緩衝液によって2回洗浄し、FACSマシン(BD)上で解析した。
図17に示されるように、抗CD40抗体を用いた処置により、ネガティブコントロール群と比較して、腫瘍増殖が著しく低減または阻害された。ただし、反応は個体により異なる。腫瘍増殖の阻害はすべてのマウス(7B4VH2VL2またはAPX005投与群)または大部分のマウス(13A2VH3VL3投与群では8のうち6、RO7009789投与群では8のうち7)で観察された。28日目に、7B4VH2VL2投与群における残り4匹のマウスすべてにおいて腫瘍が消失し、APX005投与群における残り4匹のマウスのうち2匹においては腫瘍がまったく無かった。
抗CD40抗体を用いた治療により、抗体の毒性に起因して、マウス体重の低減が生じ得る。図18および下の表11に示されるように、25日目の13A2VH3VL3群におけるマウスの体重が、腫瘍重量を考慮しても(25日目の腫瘍は、サイズが約1000mm
3、重量が1.2g)初期の重量と比較してやや増加した。このことは、抗体13A2VH3VL3の毒性が低いことを示している。換言すると、13A2VH3VL3を用いた治療は、RO7009789群において観察されたような著しい体重の低減を生じさせず、7B4VH2VL2またはAPX005群と比較して、マウス体重に対する影響が小さかった。
表11
5群における経時的なマウス体重(平均±標準誤差(g)、群あたり8マウス)
図19は、抗体7B4VH2VL2によって、CD45+細胞におけるCD45+CD3+CD8+細胞およびCD45+CD3+CD4+細胞の両方の割合が明らかに増加したことを示す。13A2VH3VL3で治療したマウスにおいてもCD45+CD3+CD8+細胞の割合が増加した。加えて、図20は、7B4VH2VL2処置によって、腫瘍浸潤樹状細胞(CD45+CD11c+細胞)におけるCD80およびCD83発現が著しく増加したことを示す。このことは、樹状細胞成熟の促進における強いアゴニスト活性を示す。
いくつかの例示的な抗体重鎖/軽鎖可変領域のアミノ酸配列を下にまとめた。
開示は1または複数の実施形態に関連して上で説明されたが、開示はこれらの実施形態に限定されず、説明は添付の請求項の思想および範囲内に含まれ得るすべての代替形態、修正形態、および同等物を包含することが意図されていることが理解されるべきである。本明細書において参照される引用はすべて、参照によって全体的に更に組み込まれる。
[考えられる他の項目]
(項目1)
CDR1領域、CDR2領域、およびCDR3領域を含む重鎖可変領域、ならびに、CDR1領域、CDR2領域、およびCDR3領域を含む軽鎖可変領域を備え、
上記重鎖CDR1領域、CDR2領域、およびCDR3領域、ならびに、上記軽鎖CDR1領域、CDR2領域、およびCDR3領域は、(1)それぞれSEQ ID NO:1、8、15、21、27および31;(2)それぞれSEQ ID NO:1、9、15、21、27および31;(3)それぞれ2、9、15、21、27および31;(4)それぞれSEQ ID NO:3、10、16、22、28および32;(5)それぞれSEQ ID NO:4、11、17、23、29および33;(6)それぞれSEQ ID NO:5、12、18、24、27および34;(7)それぞれSEQ ID NO:6、13、19、25、27および35;または(8)それぞれSEQ ID NO:7、14、20、26、30および36のアミノ酸配列を含む、
腫瘍壊死因子受容体CD40に結合する単離モノクローナル抗体またはその抗原結合部分。
(項目2)
上記重鎖可変領域は、SEQ ID NO:37、38、39、40、41、42、43、44、45、46、47、48、49または50に対して少なくとも80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%または100%の同一性を有するアミノ酸配列を含む、項目1に記載の抗体またはその抗原結合部分。
(項目3)
上記軽鎖可変領域は、SEQ ID NO:51、52、53、54、55、56、57、58、59、60または61に対して少なくとも80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%または100%の同一性を有するアミノ酸配列を含む、項目1に記載の抗体またはその抗原結合部分。
(項目4)
上記重鎖可変領域および上記軽鎖可変領域は、(1)それぞれSEQ ID NO:37および51:(2)それぞれSEQ ID NO:38および52:(3)それぞれSEQ ID NO:39および53:(4)それぞれSEQ ID NO:40および54:(5)それぞれSEQ ID NO:41および55:(6)それぞれSEQ ID NO:44および58:(7)それぞれSEQ ID NO:45および59:(8)それぞれSEQ ID NO:46および60:(9)それぞれSEQ ID NO:46および61:(10)それぞれSEQ ID NO:47および60:(11)それぞれSEQ ID NO:47および61:(12)それぞれSEQ ID NO:48および59:(13)それぞれSEQ ID NO:49および60:(14)それぞれSEQ ID NO:49および61:(15)それぞれSEQ ID NO:50および60:または(16)それぞれSEQ ID NO:50および61に対して少なくとも80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%または100%の同一性を有するアミノ酸配列を含む、項目1に記載の抗体またはその抗原結合部分。
(項目5)
SEQ ID NO:62、63、または64に対して少なくとも80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%または100%の同一性を有するアミノ酸配列を有する重鎖可変領域に連結された重鎖定常領域、および/または、SEQ ID NO:65または66に対して少なくとも80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94% 95%、96%、97%、98%、99%または100%の同一性を有するアミノ酸配列を有する軽鎖可変領域に連結された軽鎖定常領域を備える、項目1に記載の抗体またはその抗原結合部分。
(項目6)
(a)ヒトまたはサルCD40に結合し、(b)マウスCD40に結合せず、(c)ヒトCD40‐ヒトCD40L相互作用をブロックまたは促進し、(d)CD40シグナル伝達を活性化し、(e)DC細胞成熟を促進し、(f)CD4+および/またはCD8+T細胞増殖を促進する、項目1に記載の抗体またはその抗原結合部分。
(項目7)
マウス、ヒト、キメラ、またはヒト化抗体である、項目1に記載の抗体またはその抗原結合部分。
(項目8)
IgG1、IgG2、またはIgG4アイソタイプである項目1に記載の抗体またはその抗原結合部分。
(項目9)
項目1に記載の抗体またはその抗原結合部分と、薬学的に許容される担体とを備える医薬組成物。
(項目10)
抗腫瘍剤および/またはサイトカインを更に含える、項目10に記載の医薬組成物。
(項目11)
癌疾患の治療に使用するための、項目1に記載の抗体またはその抗原結合部分。
(項目12)
上記癌疾患は固形または非固形腫瘍である、項目11に記載の使用するための抗体またはその抗原結合部分。
(項目13)
上記癌疾患は、B細胞リンパ腫、慢性リンパ球性白血病、多発性骨髄腫、黒色腫、結腸腺癌、膵臓癌、大腸癌、胃腸癌、前立腺癌、膀胱癌、腎臓癌、卵巣癌、子宮頸癌、乳癌、肺癌、鼻咽頭癌から成る群から選択される、項目12に記載の使用するための抗体またはその抗原結合部分。
(項目14)
項目1に記載の抗体またはその抗原結合部分が、免疫刺激抗体、共刺激抗体、化学療法剤、および/またはサイトカインと共に投与される、項目12に記載の使用するための抗体またはその抗原結合部分。
(項目15)
上記免疫刺激抗体は、抗VISTA抗体、抗PD‐1抗体、抗PD‐L1抗体、抗LAG‐3抗体、抗TIM‐3抗体、抗STAT3抗体、および抗ROR1抗体から成る群から選択される、項目14に記載の使用するための抗体またはその抗原結合部分。
(項目16)
上記共刺激抗体は、抗CD137抗体または抗GITR抗体である、項目14に記載の使用するための抗体またはその抗原結合部分。
(項目17)
上記化学療法剤はエピルビシン、オキサリプラチン、および/または、5‐フルオロウラシルである、項目14に記載の使用するための抗体またはその抗原結合部分。
(項目18)
上記サイトカインはGM‐CSFおよび/またはIL‐4である、項目14に記載の使用するための抗体またはその抗原結合部分。
(項目19)
項目1に記載の抗体またはその抗原結合断片を備える二重特異性分子。
(項目20)
項目1に記載の抗体またはその抗原結合部分をエンコードするポリヌクレオチドを備える宿主細胞。