JP6541581B2 - Low concentration antibody preparation - Google Patents
Low concentration antibody preparation Download PDFInfo
- Publication number
- JP6541581B2 JP6541581B2 JP2015562515A JP2015562515A JP6541581B2 JP 6541581 B2 JP6541581 B2 JP 6541581B2 JP 2015562515 A JP2015562515 A JP 2015562515A JP 2015562515 A JP2015562515 A JP 2015562515A JP 6541581 B2 JP6541581 B2 JP 6541581B2
- Authority
- JP
- Japan
- Prior art keywords
- formulation
- polysorbate
- therapeutic protein
- surfactant
- protein
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images





Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39591—Stabilisation, fragmentation
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/16—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing nitrogen, e.g. nitro-, nitroso-, azo-compounds, nitriles, cyanates
- A61K47/18—Amines; Amides; Ureas; Quaternary ammonium compounds; Amino acids; Oligopeptides having up to five amino acids
- A61K47/183—Amino acids, e.g. glycine, EDTA or aspartame
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/20—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing sulfur, e.g. dimethyl sulfoxide [DMSO], docusate, sodium lauryl sulfate or aminosulfonic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2809—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against the T-cell receptor (TcR)-CD3 complex
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/40—Immunoglobulins specific features characterized by post-translational modification
- C07K2317/41—Glycosylation, sialylation, or fucosylation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/52—Constant or Fc region; Isotype
Description
本発明は、治療用タンパク質のための製剤の分野に関する。より具体的には、本発明は低濃度の治療用タンパク質のための製剤、および該製剤を製造する方法に関する。 The present invention relates to the field of formulations for therapeutic proteins. More specifically, the present invention relates to formulations for low concentrations of therapeutic proteins, and methods of manufacturing the formulations.
さらなる操作を行うことなく臨床投与を実施できる状態にある多数のバイオ医薬品が製剤化されかつ提供されているが、多くの製品は看護師、薬剤師、または医師による様々な程度の取扱いを必要とする。取扱いおよび投与の間は、タンパク質の物理的および化学的安定性が維持されていなければならない。安定性の喪失は、タンパク質製剤が静脈注射用(i.v.)溶液で低濃度まで希釈され、その結果賦形剤濃度が低下して本来の薬品製剤の組成および特性が変更された場合に生じる可能性がある。バイオ医薬製品をi.v.経路により送達する場合、タンパク質特性、製剤組成、送達すべき活性産物の濃度、希釈剤の選択、接触面、ならびに注入の時間および速度を含む幾つかの要素を考慮しなければならない。接触面は、タンパク質がそれらの両親媒性の性質により界面で吸着する傾向にあるため、特に興味深い。注射器ならびにi.v.注入容器およびラインへの多様なプラスチックポリマーの広範囲に及ぶ使用のため、吸着によるタンパク質損失のリスクが特に低濃度(<0.5 mg/mL)で大きい。従って、特に低用量製品のための薬品開発時には、フィルター、容器、注射器、およびチューブへの吸着に起因するタンパク質損失について調査し、かつこれに取り組まなければならない。 Although many biopharmaceuticals have been formulated and provided that are ready for clinical administration without further manipulation, many products require varying degrees of handling by a nurse, pharmacist, or physician . During handling and administration, the physical and chemical stability of the protein should be maintained. The loss of stability can occur when the protein preparation is diluted to a low concentration in an intravenous solution (iv), resulting in a reduction of the excipient concentration and a change in the composition and properties of the original drug preparation There is. When delivering a biopharmaceutical product by the iv route, several factors must be considered, including protein characteristics, formulation composition, concentration of active product to be delivered, choice of diluent, interface, and time and rate of injection. It does not. Contact surfaces are of particular interest because proteins tend to adsorb at the interface due to their amphiphilic nature. Due to the widespread use of various plastic polymers in syringes and iv injection containers and lines, the risk of protein loss due to adsorption is particularly large at low concentrations (<0.5 mg / mL). Thus, especially during drug development for low dose products, protein loss due to adsorption to filters, containers, syringes and tubes must be investigated and addressed.
本発明は、低濃度治療用タンパク質に適した製剤を提供するものである。 The present invention provides formulations suitable for low concentration therapeutic proteins.
発明の概要
ある態様においては、本発明は、以下a)およびb):a)治療用タンパク質;およびb)界面活性剤;ここで該治療用タンパク質に対する該界面活性剤のモル比は少なくとも100である、を含む該治療用タンパク質のための製剤を対象とする。
SUMMARY OF THE INVENTION In one aspect, the present invention provides a) a) and b): a therapeutic protein; and b) a surfactant; wherein the molar ratio of the surfactant to the therapeutic protein is at least 100. The present invention is directed to a formulation for said therapeutic protein, including
別の態様においては、本発明は、以下a)およびb):a)治療用タンパク質;およびb)抗酸化剤を含み、ここで該治療用タンパク質に対する該抗酸化剤のモル比は少なくとも750である、該治療用タンパク質のための製剤を対象とする。 In another aspect, the invention comprises the following a) and b): a) a therapeutic protein; and b) an antioxidant, wherein the molar ratio of said antioxidant to said therapeutic protein is at least 750 Certain formulations for the therapeutic protein are directed.
本発明は特定の方法、試薬、化合物、組成物、または生物系に限定されるものではなく、それらは当然変化しうることを理解されたい。同様に、本明細書中で使用した用語は単に特定の実施形態を説明するためのものであって、限定を意図したものではないことも理解されたい。本明細書および添付した特許請求の範囲において使用する場合、単数形「a」、「an」、および「the」は、文脈上明らかに他の意味を指している場合を除き、複数の指示対象を含む。従って、例えば、「糖(a sugar)」への言及は、2つ以上の糖の組み合わせなどを含む。 It is to be understood that the present invention is not limited to particular methods, reagents, compounds, compositions, or biological systems, which can, of course, vary. Similarly, it is also to be understood that the terminology used herein is for the purpose of describing particular embodiments only and is not intended to be limiting. As used in this specification and the appended claims, the singular forms "a," "an," and "the" are intended to include plural referents unless the context clearly indicates otherwise. including. Thus, for example, reference to "a sugar" includes combinations of two or more sugars and the like.
本明細書中で使用する「約」は、測定可能な値、例えば量、時間的期間などに言及している場合、指定値からの±20%または±10%、例えば±5%、±1%、および±0.1%の変動が開示方法を実施する上で適切であることから、かかる変動を包含することを意図している。 As used herein, “about” when referring to a measurable value, such as amount, time period, etc., ± 20% or ± 10%, eg ± 5%, ± 1 from the stated value Such variations are intended to be included, as variations of% and ± 0.1% are appropriate for carrying out the disclosed method.
他に定義しない限り、本明細書中で使用する全ての技術的および化学的用語は、本発明が属する分野の当業者に通常理解されているものと同じ意味を有する。本明細書中に記載されている方法および材料と類似した、または等価な方法および材料はいずれも本発明の試験のための実施に際して使用することができるが、好適な材料および方法は本明細書中に記載されている。本発明を説明しかつ権利を主張する際には、下記用語を使用する。 Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood to one of ordinary skill in the art to which this invention belongs. Although any methods and materials similar or equivalent to those described herein can be used in the practice of the present invention for testing, preferred materials and methods are described herein. It is described in the inside. In describing and claiming the present invention, the following terms will be used.
ある態様においては、本発明は、以下a)およびb):a)治療用タンパク質;およびb)界面活性剤;ここで該治療用タンパク質に対する該界面活性剤のモル比は少なくとも100である、を含む該治療用タンパク質のための製剤を対象とする。 In one embodiment, the present invention comprises the following a) and b): a) therapeutic protein; and b) surfactant; wherein the molar ratio of said surfactant to said therapeutic protein is at least 100: It is directed to a formulation for said therapeutic protein, including.
ある特定の実施形態においては、前記の治療用タンパク質に対する界面活性剤のモル比は、少なくとも150、少なくとも200、少なくとも250、少なくとも300、少なくとも400、および少なくとも500からなる群より選択される。ある実施形態においては、前記の治療用タンパク質に対する界面活性剤のモル比は約545である。 In certain embodiments, the molar ratio of surfactant to therapeutic protein is selected from the group consisting of at least 150, at least 200, at least 250, at least 300, at least 400, and at least 500. In one embodiment, the molar ratio of surfactant to therapeutic protein is about 545.
ある特定の実施形態においては、前記界面活性剤は、ポリソルベート-20、ポリソルベート-40、ポリソルベート-60、ポリソルベート-65、ポリソルベート-80、ポリソルベート-85、ポロキサマー188、およびそれらの混合物からなる群より選択される。ある実施形態においては、前記製剤は、約0.01%w/v〜約0.5%w/vの界面活性剤を含む。ある実施形態においては、約0.1%w/v、約0.2%w/v、約0.3%w/v、約0.4%w/v、または約0.5%w/vの界面活性剤である。ある実施形態においては、前記製剤は約0.1%w/vのポリソルベート80を含む。
In certain embodiments, the surfactant is selected from the group consisting of polysorbate-20, polysorbate-40, polysorbate-60, polysorbate-65, polysorbate-80, polysorbate-85, poloxamer 188, and mixtures thereof Be done. In one embodiment, the formulation comprises about 0.01% w / v to about 0.5% w / v surfactant. In certain embodiments, about 0.1% w / v, about 0.2% w / v, about 0.3% w / v, about 0.4% w / v, or about 0.5% w / v surfactant. In one embodiment, the formulation comprises about 0.1% w /
ある態様においては、本発明は、以下a)およびb):a)治療用タンパク質;およびb)抗酸化剤、ここで該治療用タンパク質に対する該抗酸化剤のモル比は少なくとも750である、を含む該治療用タンパク質のための製剤を対象とする。 In certain embodiments, the present invention comprises the following: a) and b): a therapeutic protein; and b) an antioxidant, wherein the molar ratio of the antioxidant to the therapeutic protein is at least 750. It is directed to a formulation for said therapeutic protein, including.
ある特定の実施形態においては、前記の治療用タンパク質に対する抗酸化剤のモル比は、少なくとも1000、少なくとも1250、少なくとも1500、少なくとも2000、少なくとも2500、少なくとも3000、少なくとも3500、少なくとも4000、少なくとも4500、少なくとも5000、少なくとも5500、少なくとも6000、少なくとも6500、および少なくとも7000からなる群より選択される。ある実施形態においては、前記の治療用タンパク質に対する抗酸化剤のモル比は約7143である。ある特定の実施形態においては、前記抗酸化剤は、メチオニン、システイン、グルタチオン、およびモノチオグリセロールからなる群より選択される。他の実施形態においては、前記抗酸化剤は金属キレート剤である。金属キレート剤としては、エチレンジアミン四酢酸塩(「EDTA」)、エチレングリコール四酢酸(「EGTA」)、チアミンテトラヒドロフルフリルジスルフィド(「TTFD」)、および2,3-ジメルカプトコハク酸(「DMSA」)が挙げられるが、これらに限定されない。ある特定の実施形態においては、前記製剤は約1 mM〜約50 mMの抗酸化剤を含む。ある実施形態においては、前記製剤は、約5 mM、約10 mM、約15 mM、約20 mM、約25 mM、約30 mM、約35 mM、約40 mM、または約45 mMの抗酸化剤を含む。ある実施形態においては、前記製剤は約10 mMのメチオニンを含む。 In certain embodiments, the molar ratio of antioxidant to therapeutic protein is at least 1000, at least 1250, at least 1500, at least 2000, at least 2500, at least 3000, at least 3500, at least 4000, at least 4500, at least It is selected from the group consisting of 5000, at least 5500, at least 6000, at least 6500, and at least 7000. In one embodiment, the molar ratio of antioxidant to therapeutic protein is about 7143. In certain embodiments, the antioxidant is selected from the group consisting of methionine, cysteine, glutathione, and monothioglycerol. In another embodiment, the antioxidant is a metal chelator. Metal chelating agents include ethylenediamine tetraacetate ("EDTA"), ethylene glycol tetraacetic acid ("EGTA"), thiamine tetrahydrofurfuryl disulfide ("TTFD"), and 2,3-dimercaptosuccinic acid ("DMSA") And the like, but is not limited thereto. In certain embodiments, the formulation comprises about 1 mM to about 50 mM of an antioxidant. In certain embodiments, the formulation is about 5 mM, about 10 mM, about 15 mM, about 20 mM, about 25 mM, about 30 mM, about 35 mM, about 40 mM, or about 45 mM of an antioxidant including. In one embodiment, the formulation comprises about 10 mM methionine.
ある特定の実施形態においては、前記製剤は緩衝液をさらに含み、この際、該製剤のpHは約4.0〜約8.0である。ある特定の実施形態においては、前記製剤のpHは、約4.0、約4.5、約5.0、約5.5、約6.0、約6.5、約7.0、約7.2、約7.5、または約8.0である。ある実施形態においては、該緩衝液は、ヒスチジン、酢酸塩、コハク酸塩、およびクエン酸塩からなる群より選択される。ある特定の実施形態においては、前記製剤は約1 mM〜約100 mMの緩衝液を含む。ある実施形態においては、前記製剤は、約10 mM、約20 mM、約30 mM、約40 mM、約50 mM、約60 mM、約70 mM、約80 mM、約90 mM、または約100 mMの緩衝液を含む。ある実施形態においては、前記製剤は約20 mMのヒスチジンをpH約6.5で含む。 In certain embodiments, the formulation further comprises a buffer, wherein the pH of the formulation is about 4.0 to about 8.0. In certain embodiments, the pH of the formulation is about 4.0, about 4.5, about 5.0, about 5.5, about 6.0, about 6.5, about 7.0, about 7.2, about 7.5, or about 8.0. In one embodiment, the buffer is selected from the group consisting of histidine, acetate, succinate, and citrate. In certain embodiments, the formulation comprises about 1 mM to about 100 mM buffer. In certain embodiments, the formulation is about 10 mM, about 20 mM, about 30 mM, about 40 mM, about 50 mM, about 60 mM, about 70 mM, about 80 mM, about 90 mM, or about 100 mM. Buffer. In one embodiment, the formulation comprises about 20 mM histidine at a pH of about 6.5.
ある特定の実施形態においては、前記治療用タンパク質は抗原結合性タンパク質である。ある実施形態においては、該抗原結合性タンパク質は抗体またはその断片である。ある実施形態においては、該抗原結合性タンパク質は免疫グロブリン単一可変ドメインである。ある実施形態においては、該抗原結合性タンパク質はヒトCD3に結合する。ある実施形態においては、この抗原結合性ポリペプチドは抗-CD3抗体である。ある実施形態においては、該抗-CD3抗体は、配列番号1を含む重鎖および配列番号2を含む軽鎖を含む。 In certain embodiments, the therapeutic protein is an antigen binding protein. In one embodiment, the antigen binding protein is an antibody or a fragment thereof. In one embodiment, the antigen binding protein is an immunoglobulin single variable domain. In one embodiment, the antigen binding protein binds human CD3. In one embodiment, the antigen binding polypeptide is an anti-CD3 antibody. In one embodiment, the anti-CD3 antibody comprises a heavy chain comprising SEQ ID NO: 1 and a light chain comprising SEQ ID NO: 2.
ある特定の実施形態においては、前記治療用タンパク質は、約0.01 mg/mL〜約1 mg/mLの濃度で存在する。ある実施形態においては、前記治療用タンパク質は、約0.1 mg/mL〜約0.5 mg/mLの濃度で存在する。ある実施形態においては、前記治療用タンパク質は約0.2 mg/mLの濃度で存在する。 In certain embodiments, the therapeutic protein is present at a concentration of about 0.01 mg / mL to about 1 mg / mL. In one embodiment, the therapeutic protein is present at a concentration of about 0.1 mg / mL to about 0.5 mg / mL. In one embodiment, the therapeutic protein is present at a concentration of about 0.2 mg / mL.
ある実施形態においては、前記製剤は再構成された製剤である。ある実施形態においては、前記製剤は液体医薬製剤である。ある実施形態においては、前記製剤は非経口投与に適したものである。 In one embodiment, the formulation is a reconstituted formulation. In one embodiment, the formulation is a liquid pharmaceutical formulation. In one embodiment, the formulation is suitable for parenteral administration.
ある態様においては、本発明は、治療用タンパク質のための製剤であって、以下a)〜d):a)治療用タンパク質;およびb)約0.01%w/v〜約0.5%w/vの界面活性剤、ここで該治療用タンパク質に対する該界面活性剤のモル比は少なくとも100である;c)約1 mM〜約50 mMの抗酸化剤、ここで治療用タンパク質に対する該抗酸化剤のモル比は少なくとも750である;およびd)約1 mM〜約100 mMの緩衝液を含み、ここで上記製剤のpHは約4.0〜約8.0である、上記製剤を対象とする。 In certain embodiments, the present invention is a formulation for a therapeutic protein, comprising a) to d): a) a therapeutic protein; and b) about 0.01% w / v to about 0.5% w / v Surfactant, wherein the molar ratio of said surfactant to said therapeutic protein is at least 100; c) from about 1 mM to about 50 mM of an antioxidant, wherein said molar ratio of said antioxidant to a therapeutic protein And d) comprising about 1 mM to about 100 mM of a buffer, and wherein the pH of the formulation is about 4.0 to about 8.0.
ある実施形態においては、前記製剤は約0.01 mM〜約1.0 mMのEDTAをさらに含む。ある特定の実施形態においては、前記製剤は、約0.05 mM、約0.1 mM、約0.15 mM、約0.2 mM、0.25 mM、約0.3 mM、約0.35 mM、約0.4 mM、0.45 mM、約0.5 mM、約0.55 mM、約0.6 mM、0.65 mM、約0.7 mM、約0.75 mM、約0.8 mM、約0.85 mM、約0.9 mM、約0.95 mM、または約1.0 mMのEDTAを含む。ある実施形態においては、前記製剤は約0.05 mMのEDTAを含む。 In one embodiment, the formulation further comprises about 0.01 mM to about 1.0 mM EDTA. In certain embodiments, the formulation is about 0.05 mM, about 0.1 mM, about 0.15 mM, about 0.2 mM, about 0.25 mM, about 0.3 mM, about 0.35 mM, about 0.4 mM, about 0.45 mM, about 0.5 mM, About 0.55 mM, about 0.6 mM, 0.65 mM, about 0.7 mM, about 0.75 mM, about 0.8 mM, about 0.85 mM, about 0.9 mM, about 0.95 mM, or about 1.0 mM EDTA. In one embodiment, the formulation comprises about 0.05 mM EDTA.
「ポリペプチド」、「ペプチド」及び「タンパク質」は本明細書において、アミノ酸残基のポリマーを言及するために、互換的に用いられる。ポリペプチドは、天然(組織由来の)起源、組換え又は原核若しくは真核細胞調製物由来の天然発現、又は合成法により化学的に生産されるものであり得る。該用語は、一以上のアミノ酸残基が、対応する天然のアミノ酸の人工的な化学模倣物であるアミノ酸ポリマー、並びに天然のアミノ酸ポリマー及び非天然のアミノ酸ポリマーに適用される。アミノ酸模倣物は、アミノ酸の一般的な化学構造とは異なる構造を有するが、天然のアミノ酸と同様の方法で機能する化学化合物を指す。非天然残基は、科学文献及び特許文献において十分に記載されている;天然アミノ酸残基の模倣物として有用なわずかな例示的な非天然成分及びガイドラインが以下に記載される。芳香族アミノ酸の模倣物は、例えば、D-又はL-ナフィルアラニン(naphylalanine);D-又はL-フェニルグリシン;D-又はL-2チエネイルアラニン(thieneylalanine);D-又はL-1,-2,3-又は4-ピレネイルアラニン(pyreneylalanine);D-又はL-3チエネイルアラニン(thieneylalanine);D-又はL-(2-ピリジニル)-アラニン;D-又はL-(3-ピリジニル)-アラニン;D-又はL-(2-ピラジニル)-アラニン;D-又はL-(4-イソプロピル)-フェニルグリシン;D-(トリフルオロメチル)-フェニルグリシン;D-(トリフルオロメチル)-フェニルアラニン;D-p-フルオロ-フェニルアラニン;D-又はL-p-ビフェニルフェニルアラニン;K-又はL-p-メトキシ-ビフェニルフェニルアラニン;D-又はL-2インドール(アルキル)アラニン;及びD-又はL-アルキルアラニン(alkylalanine)によって置換することによって作製され得、ここでアルキルは置換されたか又は未置換のメチル、エチル、プロピル、ヘキシル、ブチル、ペンチル、イソプロピル、イソ-ブチル、2級-イソチル(isotyl)、イソ-ペンチル、又は非酸性アミノ酸であり得る。非天然アミノ酸の芳香環として、例えば、チアゾイル、チオフェニル、ピラゾリル、ベンゾイミダゾリル、ナフチル、フラニル、ピロリル、及びピリジル芳香環が挙げられる。 "Polypeptide", "peptide" and "protein" are used interchangeably herein to refer to a polymer of amino acid residues. The polypeptide may be of natural (tissue-derived) origin, recombinant or naturally expressed from a prokaryotic or eukaryotic cell preparation, or chemically produced by synthetic methods. The terms apply to amino acid polymers in which one or more amino acid residues is an artificial chemical mimic of the corresponding naturally occurring amino acid, as well as to naturally occurring amino acid polymers and non-naturally occurring amino acid polymer. Amino acid mimetics refers to chemical compounds that have a structure that is different from the general chemical structure of an amino acid, but that functions in a manner similar to a naturally occurring amino acid. Non-naturally occurring residues are well described in the scientific and patent literature; few exemplary non-natural components and guidelines useful as mimetics of natural amino acid residues are described below. Mimetics of aromatic amino acids are, for example, D- or L-nafilalanine (naphylalanine); D- or L-phenylglycine; D- or L-2 thienailalalanine (thieneylalanine); D- or L-1,- 2,3- or 4-pyrenailalalanine; D- or L-3 thienailalalanine; D- or L- (2-pyridinyl) -alanine; D- or L- (3-pyridinyl) D- or L- (2-pyrazinyl) -alanine; D- or L- (4-isopropyl) -phenylglycine; D- (trifluoromethyl) -phenylglycine; D- (trifluoromethyl) -phenylalanine D-p-fluoro-phenylalanine; D- or L-p-biphenyl phenylalanine; K- or L-p-methoxy-biphenyl phenylalanine; D- or L-2 indole (alkyl) alanine; and D- or L-alkyl Substitute by alanine (alkylalanine) Can be prepared by reaction, where alkyl is substituted or unsubstituted methyl, ethyl, propyl, hexyl, butyl, pentyl, isopropyl, iso-butyl, secondary-isotyl, iso-pentyl or non- It may be an acidic amino acid. As an aromatic ring of a nonnatural amino acid, for example, thiazoyl, thiophenyl, pyrazolyl, benzimidazolyl, naphthyl, furanyl, pyrrolyl and pyridyl aromatic ring can be mentioned.
本明細書で用いられる「ペプチド」は、本明細書にて具体的に例示されるペプチドの保存的変異であるペプチドを含む。本明細書で用いられる「保存的変異」は、別の生物学的に類似する残基によるアミノ酸残基の置換を意味する。保存的変異の例として、限定されるものではないが、イソロイシン、バリン、ロイシン、アラニン、システイン、グリシン、フェニルアラニン、プロリン、トリプトファン、チロシン、ノルロイシン、又はメチオニン等の一つの疎水性残基を別の疎水性残基で置換すること、又はアルギニンのリジンでの置換、グルタミン酸のアスパラギン酸での置換、若しくはグルタミンのアスパラギンでの置換等の一つの極性残基を別の極性残基で置換すること等が挙げられる。互いに置換可能である中性親水性アミノ酸として、アスパラギン、グルタミン、セリン、及びスレオニンが挙げられる。「保存的変異」はまた、その置換ポリペプチドに対して産生された抗体が、非置換ポリペプチドとも免疫反応を起こす限り、非置換の親アミノ酸の代わりに置換アミノ酸を用いることも含む。そのような保存的置換は、本発明のペプチドのクラスの定義の範囲内である。本明細書で用いられる「カチオン性」とは、pH7.4にて正味の正電荷を有するいずれのペプチドをも意味する。ペプチドの生物活性は、当業者に公知であり、本明細書に記載される標準的な方法によって測定することができる。 As used herein, "peptide" includes peptides that are conservative variations of the peptides specifically exemplified herein. As used herein, "conservative mutation" refers to the replacement of an amino acid residue by another, biologically similar residue. Examples of conservative mutations include, but are not limited to, one hydrophobic residue such as, but not limited to isoleucine, valine, leucine, alanine, cysteine, glycine, phenylalanine, proline, tryptophan, tyrosine, norleucine, or methionine. Substitution of hydrophobic residues or substitution of one polar residue with another such as substitution of arginine with lysine, substitution of glutamic acid with aspartic acid, or substitution of glutamine with asparagine, etc. Can be mentioned. Neutral hydrophilic amino acids that can be substituted for one another include asparagine, glutamine, serine and threonine. "Conservative mutation" also includes using a substituted amino acid in place of a non-substituted parent amino acid, as long as the antibody raised against the substituted polypeptide also produces an immune response with the non-substituted polypeptide. Such conservative substitutions are within the definition of the class of peptides of the invention. As used herein, "cationic" means any peptide having a net positive charge at pH 7.4. The biological activity of the peptide is known to the person skilled in the art and can be measured by the standard methods described herein.
「組換え」とは、タンパク質に関連して用いられる場合、異種核酸若しくはタンパク質の導入によって、又は天然核酸若しくはタンパク質の変化によって修飾されたタンパク質を指す。 "Recombinant" when used in connection with a protein refers to a protein that has been modified by the introduction of heterologous nucleic acids or proteins or by changes in natural nucleic acids or proteins.
本明細書で用いられる「治療タンパク質」は、哺乳動物に投与されて、例えば研究者若しくは医師が求めている組織、系、動物、若しくはヒトの生物学的又は医学的応答を惹起することができるあらゆるタンパク質及び/又はポリペプチドを指す。治療タンパク質は、2つ以上の生物学的又は医学的応答を引き起こし得る。さらに、用語「治療有効量」は、そのような量を受けていない対応する被験者と比較して、限定されるものではないが、疾患、障害、若しくは副作用の治癒、予防、若しくは寛解、又は疾患若しくは障害の進行速度の低下をもたらす、いかなる量をも意味する。この用語はまた、正常な生理学的機能の向上に有効である量、ならびに第二の医薬剤の治療効果を向上又は補助する患者の生理学的機能を引き起こすのに有効である量もその範囲内に含む。 As used herein, a "therapeutic protein" can be administered to a mammal to elicit, for example, a biological or medical response in a tissue, system, animal, or human that a researcher or physician seeks Refers to any protein and / or polypeptide. Therapeutic proteins can cause more than one biological or medical response. Furthermore, the term "therapeutically effective amount" refers to, but is not limited to, the cure, prevention, or amelioration of the disease, disorder, or side effects, as compared to a corresponding subject who has not received such an amount. Or any amount that results in a reduction in the rate of progression of the disorder. Also within the scope of this term is an amount that is effective to improve normal physiological function, as well as an amount that is effective to trigger a patient's physiological function that enhances or supplements the therapeutic effect of the second pharmaceutical agent. Including.
本明細書において特定される全ての「アミノ酸」残基は、天然のL‐配置である。標準的なポリペプチド命名法に従って、アミノ酸残基に対する略語を以下に示す。

全てのアミノ酸残基配列は、本明細書において、その左から右への配向が慣例的なアミノ末端からカルボキシ末端への方向である式によって表されることには留意されたい。 It should be noted that all amino acid residue sequences are represented herein by a formula whose left to right orientation is the conventional amino terminus to carboxy terminus orientation.
他の実施形態では、ポリペプチドは、抗原結合ポリペプチドである。一実施形態では、抗原結合ポリペプチドは、可溶性受容体、抗体、抗体断片、免疫グロブリン単一可変ドメイン、Fab、F(ab')2、Fv、ジスルフィド結合Fv、scFv、閉鎖コンフォメーションの多重特異性抗体、ジスルフィド結合scFv、又は二重特異性抗体からなる群より選択される。 In another embodiment, the polypeptide is an antigen binding polypeptide. In one embodiment, the antigen binding polypeptide comprises a soluble receptor, antibody, antibody fragment, immunoglobulin single variable domain, Fab, F (ab ') 2, Fv, disulfide linked Fv, scFv, closed conformation multispecific It is selected from the group consisting of a sex antibody, a disulfide linked scFv, or a bispecific antibody.
本明細書で用いられる用語「抗原結合ポリペプチド」は、抗体、抗体断片、及び抗原に結合する能力を有するその他のタンパク質構築物を指す。 As used herein, the term "antigen binding polypeptide" refers to antibodies, antibody fragments, and other protein constructs that have the ability to bind antigen.
用語「Fv、Fc、Fd、Fab、又はF(ab)2」は、これらの標準的な意味で用いられる(例えば、Harlow et al., Antibodies A Laboratory Manual, Cold Spring Harbor Laboratory, (1988)参照)。 The terms "Fv, Fc, Fd, Fab or F (ab) 2" are used in their standard sense (see, for example, Harlow et al., Antibodies A Laboratory Manual, Cold Spring Harbor Laboratory (1988). ).
「キメラ抗体」は、ドナー抗体由来の天然の可変領域(軽鎖及び重鎖)を、アクセプター抗体由来の軽鎖及び重鎖定常領域と合わせて含む、遺伝子操作された抗体の種類を指す。 A "chimeric antibody" refers to a type of genetically engineered antibody that comprises naturally occurring variable regions (light and heavy chains) derived from a donor antibody in combination with light and heavy chain constant regions derived from an acceptor antibody.
「ヒト化抗体」は、非ヒトドナー免疫グロブリン由来のCDRを有し、分子の残りの免疫グロブリン由来部分は、一つ(以上)のヒト免疫グロブリン由来である遺伝子操作された抗体の種類を指す。加えて、フレームワーク支持残基(framework support residues)は、結合親和性を保存するために、改変されてよい(例えば、Queen et al., Proc. Natl. Acad Sci USA, 86:10029-10032 (1989)、Hodgson et al., Bio/Technology, 9:421 (1991)を参照)。適切なヒトアクセプター抗体は、従来のデータベース、例えば、KABAT.RTM.データベース、Los Alamosデータベース、及びSwiss Proteinデータベースより、ドナー抗体のヌクレオチド及びアミノ酸配列に対する相同性によって選択され得る。ドナー抗体のフレームワーク領域に対する相同性(アミノ酸に基づく)によって特徴付けられるヒト抗体は、ドナーCDRの挿入のための重鎖定常領域及び/又は重鎖可変フレームワーク領域を提供するのに適切であり得る。軽鎖定常又は可変フレームワーク領域を提供することができる適切なアクセプター抗体は、同様の方法で選択することができる。アクセプター抗体重鎖及び軽鎖が、同じアクセプター抗体に由来する必要はないことには留意されたい。先行文献には、そのようなヒト化抗体作製のためのいくつかの方法が記載されており、例えば、欧州特許第0239400A号及び欧州特許第054951A号を参照されたい。
A "humanized antibody" refers to a type of genetically engineered antibody that has CDRs from non-human donor immunoglobulins and the remaining immunoglobulin-derived portion of the molecule is derived from one (or more) human immunoglobulins. In addition, framework support residues may be modified to preserve binding affinity (eg, Queen et al., Proc. Natl. Acad Sci USA, 86: 1029-10032 ( 1989), Hodgson et al., Bio / Technology, 9: 421 (1991)). Suitable human acceptor antibodies can be selected by homology to the nucleotide and amino acid sequences of the donor antibody from conventional databases, such as the KABAT.RTM. Database, the Los Alamos database, and the Swiss Protein database. Human antibodies characterized by homology (based on amino acids) to the framework region of the donor antibody are suitable to provide a heavy chain constant region and / or a heavy chain variable framework region for insertion of donor CDRs. obtain. Suitable acceptor antibodies that can provide light chain constant or variable framework regions can be selected in a similar manner. It should be noted that the acceptor antibody heavy and light chains need not be from the same acceptor antibody. The prior art describes several methods for such humanized antibody production, see, for example,
用語「ドナー抗体」は、その可変領域、CDR、若しくはその他の機能性断片、又はそれらのアナログのアミノ酸配列を、第一の免疫グロブリンパートナーへ与え、それによって、改変免疫グロブリンコード領域を提供し、及びその結果として、ドナー抗体に特徴的である抗原特異性及び中和活性を有する改変抗体の発現を提供する抗体(モノクローナル及び/又は組換え)を指す。 The term "donor antibody" provides the amino acid sequence of its variable region, CDRs, or other functional fragments, or analogs thereof, to a first immunoglobulin partner, thereby providing a modified immunoglobulin coding region. And as a result, it refers to an antibody (monoclonal and / or recombinant) that provides for expression of a modified antibody having antigen specificity and neutralizing activity that is characteristic of the donor antibody.
用語「アクセプター抗体」は、ドナー抗体に対して異種であり、その重鎖及び/若しくは軽鎖フレームワーク領域、ならびに/又はその重鎖及び/若しくは軽鎖定常領域をコードするアミノ酸配列のすべて(又はいずれかの部分であるが、いくつかの実施形態ではすべて)を、第一の免疫グロブリンパートナーへ与える抗体(モノクローナル及び/又は組換え)を指す。特定の実施形態では、ヒト抗体は、アクセプター抗体である。 The term "acceptor antibody" is heterologous to the donor antibody and comprises all of the amino acid sequences encoding its heavy and / or light chain framework regions and / or its heavy and / or light chain constant regions (or Refers to an antibody (monoclonal and / or recombinant) that provides any portion, but in some embodiments, to a first immunoglobulin partner. In certain embodiments, the human antibody is an acceptor antibody.
「CDR」は、免疫グロブリン重鎖及び軽鎖の超可変領域である、抗体の相補性決定領域アミノ酸配列として定義される(例えば、Kabat et al., Sequences of Proteins of Immunological Interest, 4th Ed., U.S. Department of Health and Human Services, National Institutes of Health (1987)を参照されたい)。免疫グロブリンの可変部分には、3つの重鎖CDR及び3つの軽鎖CDR(又はCDR領域)が存在する。したがって、本明細書で用いられる場合、「CDR」は、3つの重鎖CDRすべて、又は3つの軽鎖CDRすべて(又は、適当な場合、すべての重鎖CDR及びすべての軽鎖CDRの両方)を意味する。抗体の構造及びタンパク質フォールディングは、他の残基が抗原結合領域の一部と見なされることを意味し得、当業者にはそのように理解されるであろう。例えば、Chothia et al., (1989) Conformations of immunoglobulin hypervariable regions、 Nature 342, p 877-883、を参照されたい。 "CDR" is defined as the complementarity determining region amino acid sequence of an antibody, which is the hypervariable region of an immunoglobulin heavy and light chain (e.g. Kabat et al., Sequences of Proteins of Immunological Interest, 4th Ed., See US Department of Health and Human Services, National Institutes of Health (1987)). There are three heavy chain CDRs and three light chain CDRs (or CDR regions) in the variable part of the immunoglobulin. Thus, as used herein, “CDR” refers to all three heavy chain CDRs, or all three light chain CDRs (or, where appropriate, both all heavy chain CDRs and all light chain CDRs) Means The structure of the antibody and protein folding can mean that other residues are considered to be part of the antigen binding region, and will be so understood by those skilled in the art. See, for example, Chothia et al., (1989) Conformations of immunoglobulin hypervariable regions, Nature 342, p 877-883.
本明細書で用いられる場合、用語「ドメイン」は、タンパク質の残りの部分とは独立した三次構造を有するフォールドされたタンパク質構造を指す。一般的に、ドメインは、タンパク質の別々の機能特性を担っており、多くの場合、タンパク質及び/又はそのドメインの残りの機能を喪失することなく、付加、除去、又は他のタンパク質へ転移され得る。「抗体単一可変ドメイン」は、抗体可変ドメインに特徴的な配列を含むフォールドされたポリペプチドドメインである。従って、それは、完全な抗体可変ドメイン、及び修飾可変ドメイン、例えば、一つ以上のループが抗体可変ドメインに特徴的ではない配列によって置き換えられたもの、又は、切断された、又はN末端若しくはC末端伸長を含む抗体可変ドメイン、ならびに少なくとも完全長ドメインの結合活性及び特異性を維持する可変ドメインのフォールドされた断片を含む。 As used herein, the term "domain" refers to a folded protein structure having a tertiary structure independent of the rest of the protein. In general, domains are responsible for the separate functional properties of a protein, and can often be added, removed or transferred to other proteins without loss of the protein and / or the remaining function of that domain . An "antibody single variable domain" is a folded polypeptide domain that comprises sequences characteristic of antibody variable domains. Thus, it may be a complete antibody variable domain, and a modified variable domain, eg, one or more of the loops replaced by sequences not characteristic of the antibody variable domain, or truncated, or N-terminal or C-terminal An antibody variable domain comprising an extension, and a folded fragment of the variable domain which maintains the binding activity and specificity of at least the full length domain.
語句「免疫グロブリン単一可変ドメイン」は、異なるV領域又はドメインとは独立して、抗原又はエピトープと特異的に結合する抗体可変ドメイン(VH、VHH、VL)を意味する。免疫グロブリン単一可変ドメインは、他の異なる可変領域又は可変ドメインとのフォーマット(例えば、ホモダイマー又はヘテロマルチマー)として存在し得、ここで、他の領域又はドメインは、単一免疫グロブリン可変ドメインによる抗原結合に必要ではない(すなわち、免疫グロブリン単一可変ドメインは、追加の可変ドメインとは独立して抗原と結合する)。「ドメイン抗体」又は「dAb」は、その用語が本明細書にて用いられる場合、抗原と結合することができる「免疫グロブリン単一可変ドメイン」と同じである。免疫グロブリン単一可変ドメインは、ヒト抗体可変ドメインであり得るが、(例えば、国際公開第00/29004号に開示される)げっ歯類、コモリザメ、及びラクダ科(Camelid)VHHdAb(ナノボディ)等、他の種由来の単一抗体可変ドメインも含む。ラクダ科VHHは、元々軽鎖のない重鎖抗体を産生するラクダ、ラマ、アルパカ、ヒトコブラクダ、及びグアナコを含む種由来である免疫グロブリン単一可変ドメインポリペプチドである。かかるVHHドメインは、本技術分野で利用可能である標準的な技術に従ってヒト化することができ、かかるドメインは、依然として、本発明において「ドメイン抗体」とみなされる。本明細書で用いられる場合、VHは、ラクダ科VHHドメインを含む。NARVは、コモリザメを含む軟骨魚類中にて同定された別の種類の免疫グロブリン単一可変ドメインである。これらのドメインは、新規抗原受容体可変領域(Novel Antigen Receptor variable region)(一般的に、V(NAR)又はNARVと略される)としても知られる。さらなる詳細については、Mol. Immunol. 44, 656-665 (2006)及び米国特許出願第20050043519A号を参照されたい。 The phrase "immunoglobulin single variable domain" means an antibody variable domain (V H , V HH , V L ) that specifically binds to an antigen or epitope independently of the different V regions or domains. The immunoglobulin single variable domain may be present as a format (e.g. homodimer or heteromultimer) with another different variable region or variable domain, wherein the other region or domain is an antigen according to a single immunoglobulin variable domain It is not necessary for binding (ie, the immunoglobulin single variable domain binds the antigen independently of the additional variable domain). A "domain antibody" or "dAb" as the term is used herein is the same as an "immunoglobulin single variable domain" capable of binding an antigen. The immunoglobulin single variable domain may be a human antibody variable domain, but is disclosed (for example, as disclosed in WO 00/29004) rodents, common sharks, and camelid (Chamelid) V HH dAbs (nanobodies) Also includes single antibody variable domains from other species, etc. Camelid V HH are originally immunoglobulin single variable domain polypeptide is a species derived from a light chain without camels which produce heavy chain antibodies, llama, alpaca, dromedary, and guanaco. Such V HH domains can be humanized according to standard techniques available in the art and such domains are still considered as "domain antibodies" in the present invention. As used herein, V H includes camelid V HH domains. NARV is another type of immunoglobulin single variable domain that has been identified in cartilaginous fish, including common sharks. These domains are also known as Novel Antigen Antigen Receptor variable regions (generally abbreviated as V (NAR) or NARV). For further details, see Mol. Immunol. 44, 656-665 (2006) and U.S. Patent Application No. 20050043519A.
用語「エピトープ結合ドメイン」は、異なるV領域又はドメインとは独立して、抗原又はエピトープと特異的に結合するドメインを指し、これは、ドメイン抗体(dAb)、例えばヒト、ラクダ科、又はサメ免疫グロブリン単一可変ドメインであってよい。 The term "epitope binding domain" refers to a domain that specifically binds to an antigen or epitope independently of different V regions or domains, which is a domain antibody (dAb) such as human, camelid or shark immunity It may be a globulin single variable domain.
本明細書で用いられる場合、用語「抗原結合部位」は、抗原と特異的に結合することができるタンパク質上の部位を意味し、これは、単一ドメイン、例えばエピトープ結合ドメインであり得、又はこれは、標準的な抗体上で見ることができる対となったVH/VLドメインであってもよい。本発明のいくつかの態様では、一本鎖Fv(ScFv)ドメインは、抗原結合部位を提供することができる。 As used herein, the term "antigen binding site" means a site on a protein capable of specifically binding to an antigen, which may be a single domain, such as an epitope binding domain, or This may be a paired V H / V L domain that can be found on standard antibodies. In some aspects of the invention, a single chain Fv (ScFv) domain can provide an antigen binding site.
用語「mAbdAb」及び「dAbmAb」は、本発明の抗原結合タンパク質を指すために本明細書で用いられる。これら2つの用語は、互換的に用いられ得、本明細書で用いられる場合、同じ意味を有することを意図している。 The terms "mAb dAb" and "dAb mAb" are used herein to refer to the antigen binding protein of the invention. These two terms may be used interchangeably and are intended to have the same meaning as used herein.
別の実施形態においては、前記製剤は追加の賦形剤をさらに含む。「賦形剤」としては、限定するものではないが、安定剤、例えば、ヒト血清アルブミン(hsa)、ウシ血清アルブミン(bsa)、α-カゼイン、グロブリン、α-ラクトアルブミン、LDH、リゾチーム、ミオグロビン、オボアルブミン、RNアーゼA;緩衝剤、例えば、クエン酸、HEPES、ヒスチジン、酢酸カリウム、クエン酸カリウム、リン酸カリウム(KH2PO4)、酢酸ナトリウム、炭酸水素ナトリウム、クエン酸ナトリウム、リン酸ナトリウム(NAH2PO4)、Tris塩基、およびTris-HCl;アミノ酸/代謝産物、例えば、グリシン、アラニン(α-アラニン、β-アラニン)、アルギニン、ベタイン、ロイシン、リジン、グルタミン酸、アスパラギン酸、ヒスチジン、プロリン、4-ヒドロキシプロリン、サルコシン、γ-アミノ酪酸(GABA)、オピン(アラノピン、オクトピン、ストロンビン)、およびトリメチルアミンN-オキシド(TMAO);界面活性剤、例えば、ポリソルベート20および80、ならびにポロキサマー407:脂質分子、例えば、ホスファチジルコリン、エタノールアミン、およびアセチルトリプトファナート:ポリマー、例えば、ポリエチレングリコール(PEG)、およびポリビニルピロリドン(PVP)10、24、40;低分子量の賦形剤、例えば、アラビノース、セロビオース、エチレングリコール、フルクトース、フコース、ガラクトース、グリセリン/グリセロール、グルコース、イノシトール、ラクトース、マルトース、マルトトリオース、マンノース、メリビオース、2-メチル-2,4-ペンタンジオール、オクツロース、プロピレングリコール、ラフィノース、リボース、スクロース、トレハロース、キシリトール、およびキシロース;ならびに高分子量の賦形剤、例えば、セルロース、β-シクロデキストリン、デキストラン(10 kd)、デキストラン(40 kd)、デキストラン(70 kd)、フィコール、ゼラチン、ヒドロキシプロピルメチル-セルロース、ヒドロキシエチルデンプン、マルトデキストリン、メトセル、peg (6 kd)、ポリデキストロース、ポリビニルピロリドン(PVP) k15(10 kd)、PVP(40 kd)、PVP k30(40 kd)、PVP k90(1000 kd)、セファデックスG 200、およびデンプン;抗酸化剤、例えば、アスコルビン酸、システインHCl、チオグリセロール、チオグリコール酸、チオソルビトール、およびグルタチオン;還元剤、例えば、システインHCl、ジチオトレイトール、および他のチオールまたはチオフェン;キレート剤、例えば、EDTA、EGTA、グルタミン酸、およびアスパラギン酸;無機塩/金属、例えば、Ca2+、Ni2+、Mg2+、Mn2+、Na2SO4、(NH4)2SO4、Na2HPO4/NaH2PO4、K2HPO4/KH2PO4、MgSO4、およびNaF;有機塩、例えば、酢酸Na、ポリエチレンNa、カプリン酸Na(オクタン酸Na)、プロピオン酸塩、乳酸塩、コハク酸塩、およびクエン酸塩;有機溶媒、例えば、アセトニトリル、ジメチルスルホキシド(dmso)、およびエタノールが挙げられる。 In another embodiment, the formulation further comprises an additional excipient. As the "excipient", but not limited to, stabilizers, such as human serum albumin (hsa), bovine serum albumin (bsa), α-casein, globulin, α-lactalbumin, LDH, lysozyme, myoglobin , Ovalbumin, RNase A; buffer, for example, citric acid, HEPES, histidine, potassium acetate, potassium citrate, potassium phosphate (KH 2 PO 4 ), sodium acetate, sodium hydrogencarbonate, sodium bicarbonate, sodium citrate, phosphoric acid Sodium (NAH 2 PO 4 ), Tris base, and Tris-HCl; amino acids / metabolites such as glycine, alanine (α-alanine, β-alanine), arginine, betaine, leucine, lysine, glutamic acid, aspartic acid, histidine , Proline, 4-hydroxyproline, sarcosine, γ-aminobutyric acid (GABA), opine (alanopine, octopine, stronbi ), And trimethylamine N-oxide (TMAO); surfactants such as polysorbate 20 and 80, and poloxamer 407: lipid molecules such as phosphatidylcholine, ethanolamine, and acetyltriptophanate: polymers such as polyethylene glycol (PEG) And polyvinylpyrrolidone (PVP) 10, 24, 40; low molecular weight excipients such as arabinose, cellobiose, ethylene glycol, fructose, fucose, galactose, glycerol / glycerol, glucose, inositol, lactose, maltose, maltotri Aus, mannose, melibiose, 2-methyl-2,4-pentanediol, octulose, propylene glycol, raffinose, ribose, sucrose, trehalose, xylitol, and xylose; High molecular weight excipients such as cellulose, β-cyclodextrin, dextran (10 kd), dextran (40 kd), dextran (70 kd), ficoll, gelatin, hydroxypropyl methyl-cellulose, hydroxyethyl starch, Maltodextrin, Methocel, peg (6 kd), polydextrose, polyvinyl pyrrolidone (PVP) k15 (10 kd), PVP (40 kd), PVP k30 (40 kd), PVP k90 (1000 kd), Sephadex G 200, And starches; antioxidants such as ascorbic acid, cysteine HCl, thioglycerol, thioglycollic acid, thiosorbitol, and glutathione; reducing agents such as cysteine HCl, dithiothreitol and other thiols or thiophenes; chelating agents, For example, EDTA, EGTA, glutamic acid and aspartic acid; inorganic salts / metals such as Ca 2+ , Ni 2+ , Mg 2+ , Mn 2+ , Na 2 SO 4 , (NH 4 ) 2 SO 4 , Na 2 HPO 4 / NaH 2 PO 4 , K 2 HPO 4 / KH 2 PO 4 , MgSO 4 , and NaF; Salts, such as sodium acetate, polyethylene Na, sodium caprate (sodium octanoate), propionate, lactate, succinate, and citrate; organic solvents such as, for example, acetonitrile, dimethyl sulfoxide (dmso), and ethanol Can be mentioned.
ある実施形態においては、前記製剤を約4.0〜約8.0のpHに製剤化する。ある実施形態においては、前記製剤を約6.5のpHに製剤化する。ある実施形態においては、前記製剤は約10 mM〜約50 mMのヒスチジンを含む。ある実施形態においては、前記製剤は約20 mMのヒスチジンを含む。 In one embodiment, the formulation is formulated to a pH of about 4.0 to about 8.0. In one embodiment, the formulation is formulated to a pH of about 6.5. In one embodiment, the formulation comprises about 10 mM to about 50 mM histidine. In one embodiment, the formulation comprises about 20 mM histidine.
前記治療用タンパク質を安定させるために使用する薬剤は、前記の製剤過程の任意の段階で添加することができる。例えば、前記の緩衝液、治療用タンパク質の前、後、もしくはそれらと同時に、または任意の賦形剤と共に添加することができる。 The agents used to stabilize the therapeutic protein can be added at any stage of the formulation process. For example, it can be added before, after, or at the same time as the aforementioned buffer, therapeutic protein, or together with an optional excipient.
本発明の製剤は、全身投与を含む任意の適切な投与経路により投与してもよい。全身投与としては、経口投与、非経口投与、経皮投与、直腸内投与、および吸入による投与が挙げられる。非経口投与とは経腸、経皮、または吸入によるもの以外の投与経路を指しており、また典型的には注射または注入によるものをいう。非経口投与としては、静脈内、筋肉内、および皮下への注射または注入が挙げられる。吸入とは、口から吸入されるか鼻腔から吸入されるかにかかわらず、患者の肺への投与を指す。 The formulations of the invention may be administered by any suitable route of administration, including systemic administration. Systemic administration includes oral, parenteral, transdermal, rectal, and by inhalation. Parenteral administration refers to routes of administration other than enteral, transdermal, or by inhalation, and typically refers to injection or infusion. Parenteral administration includes intravenous, intramuscular and subcutaneous injection or infusion. Inhalation refers to administration to the patient's lungs, whether inhaled through the mouth or through the nasal cavity.
本発明は、本発明の方法のいずれかにより製造された安定な製剤もまた対象とする。
(実施例)
The invention is also directed to stable formulations made by any of the methods of the invention.
(Example)
実施例1--オテリキシズマブは低濃度-低用量mAbである
オテリキシズマブは、そのFcドメイン内にグリコシル化部位を持たない、ヒトCD3に対するヒト化mAb(IgG1)である。臨床用量(1日当たり0.1〜0.5 mg)の正確な調製および投与を容易にするため、この薬品を、様々な1日量に対応する種々のバイアル単位表示を用いて0.2 mg/mLの濃度で作成した。該用量の投与には、等張0.9%食塩水(生理食塩水)を含有するi.v.バッグ中での前記mAbの希釈と、標準的な注入ポンプによる全バッグ内容物の送達が必要であった。この投薬方法により、i.v.バッグ中のタンパク質濃度は2μg/mLという低さになった。直接的な結果としては、有意なタンパク質損失のリスクが非常に高かった。こうした理由から、商業的に関連のあるi.v.投与セットを用いた短期間の安定性および適合性試験を実施することにより、既存製品の投与の実行可能性を評価するだけでなく、提案した希釈および投与設計の意図を支持するであろう代替製剤を開発した。
Example 1-Oteroxizumab is a Low Concentration-Low Dose mAb Oterixizumab is a humanized mAb (IgG1) to human CD3 that has no glycosylation site in its Fc domain. To facilitate accurate preparation and administration of clinical doses (0.1-0.5 mg per day), this drug is made at a concentration of 0.2 mg / mL using different vial unit indications corresponding to different daily doses did. Administration of the dose required dilution of the mAb in an iv bag containing isotonic 0.9% saline (saline) and delivery of the entire bag contents by a standard infusion pump. This dosing method resulted in protein concentrations in the iv bag as low as 2 μg / mL. As a direct consequence, the risk of significant protein loss was very high. For these reasons, the proposed dilutions and evaluations of the administration feasibility of existing products by performing short-term stability and compatibility studies with commercially relevant iv administration sets, as well as the proposed dilution and An alternative formulation was developed that would support the intent of dosing design.
実施例2--使用時安定性試験中の許容できないmAb損失の評価
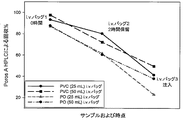
ヒスチジン緩衝液のみからなる前記の0.2 mg/mL製剤の投与の実行可能性を評価するために、市販のi.v.投与セットを使用して安定性試験を実行した。目標用量(0.1 mg)と同等の薬品量をi.v.生理食塩水バッグに添加してから、この希釈した製品の安定性を評価した。吸着による潜在的なタンパク質損失を最小限に抑えるために25または50 mLという比較的少ない生理食塩水量を選択したところ、i.v.バッグにおける活性濃度はそれぞれ4および2μg/mLとなった。さらに、2つの異なる種類の材料(ポリオレフィンPO、およびポリ塩化ビニルPVC)で構成されているi.v.バッグについて評価した。該バッグを研究室の作業台に設置し、周囲温度および照明条件に曝露した。各試験構成に対して3セットのバッグを調製した:バッグ1は初期条件の試験としての役割を果たし、バッグ2は保留(hold)を表し、またバッグ3は、インラインフィルターを備えたi.v.ライン(B. Braun Horizon Pump Filtered i.v.セット)を通じてのタンパク質溶液の臨床的調製および注入をシミュレートするために使用した。該バッグは二重に調製しかつ試験した。i.v.バッグ調製直後(i.v.バッグ中0時間保留)、およびi.v.バッグ中での2時間保留の後にサンプルを採集して試験を行った。追加のサンプルは、前記i.v.ラインを通じての2時間注入の後に採集した。ポリソルベート80(PS80)を全ての保持サンプルに0.001%の最終濃度で添加することにより、採集用チューブ内での吸着による損失を予防した。平均回収率(%)結果はPoros A-HPLC分析により測定した。回収は前記バッグ中の目的物濃度に基づいて算出し、また報告値は2つの異なる調製物の平均値とした。
Example 2-Evaluation of unacceptable mAb loss during in-use stability studies To evaluate the feasibility of administering the above 0.2 mg / mL formulation consisting of histidine buffer only, using a commercial iv dosing set The stability test was performed. An amount of drug equivalent to the target dose (0.1 mg) was added to the iv saline bag and the stability of this diluted product was assessed. When a relatively small saline volume of 25 or 50 mL was chosen to minimize potential protein loss due to adsorption, the active concentration in the iv bag was 4 and 2 μg / mL, respectively. In addition, an iv bag composed of two different types of materials (polyolefin PO and polyvinyl chloride PVC) was evaluated. The bag was placed on a laboratory bench and exposed to ambient temperature and lighting conditions. Three sets of bags were prepared for each test configuration: bag 1 serves as a test of initial conditions,
Poros A-HPLC分析により測定した平均回収率(%)結果を図1にプロットした。全てのPOバッグにおける回収(23%〜88%の範囲)は、PVCバッグにおける回収(41%〜98%の範囲)より低い傾向にあった。全体としては、2時間の室温保存後の全試験系における損失は10%を優に上回っていた。さらに、前記製品をi.v.ラインおよびフィルターを通じて注入した場合、前記タンパク質の50%超がバッグの材料に関係なく失われた。 The average recovery (%) results determined by Poros A-HPLC analysis are plotted in FIG. Recovery in all PO bags (range 23% to 88%) tended to be lower than recovery in PVC bags (range 41% to 98%). Overall, the loss in all test systems after 2 hours of room temperature storage was well over 10%. Furthermore, when the product was injected through an iv line and filter, more than 50% of the protein was lost regardless of the bag material.
実施例3--投与中の吸着による損失に対する安定性を改善するためのi.v.バッグ中の界面活性剤濃度の最適化
ヒスチジン緩衝液のみで製剤化されたmAbの投与時の許容できないタンパク質回収を理由に、界面活性剤ポリソルベート80(PS80)の前記製剤への組み入れがその用量の90%以上の送達を可能にしうるか否かを判定するために第2試験を実施した。従って、0.1 mgのタンパク質の調製および注入について、50 mLの生理食塩水を含有するPOバッグを、インラインフィルターを備えたi.v.ラインと組み合わせて使用して試験した(2μg/mL mAb)。初期の結果は、PVCバッグでの回収と比較した場合のPOバッグでのより低い回収を示しており、このことはPOバッグが許容しうるタンパク質回収を実証するためのより困難な障害でありうることを示しているので、このPOバッグのみを調べた。
Example 3-Optimization of surfactant concentration in iv bag to improve stability to loss due to adsorption during dosing due to unacceptable protein recovery upon administration of mAb formulated with histidine buffer alone Then, a second study was performed to determine if incorporation of surfactant polysorbate 80 (PS 80) into the formulation could allow for the delivery of 90% or more of the dose. Thus, for preparation and injection of 0.1 mg of protein, PO bags containing 50 mL of saline were tested using in combination with an iv line equipped with an in-line filter (2 μg / mL mAb). Initial results show lower recovery in PO bags as compared to recovery in PVC bags, which may be a more difficult obstacle for PO bags to demonstrate acceptable protein recovery As it shows, I examined only this PO bag.
同様の実験計画を、この第2試験で次の通りに使用した:0.2 mg/mLの濃度でありかつ様々な濃度のPS80(0.02、0.05、0.1、および0.3%w/v)を含有するmAbストック溶液を新たに調製し、さらに先に記載したようにi.v.注入バッグ(POのみ)に添加した。このi.v.生理食塩水バッグ中の最終mAb濃度は2μg/mLであり、また最終界面活性剤濃度は0.0002、0.0005、0.001、および0.003%であった。追加のPS80を(少なくとも0.001%の最終濃度まで)分析前に全サンプルに添加した。試験したi.v.ラインは、フィルターを備えたBaxter Continu-Flo Solution Setであった。mAb濃度は、初期、およびi.v.バッグ中での2時間保留または選択したi.v.ラインを通じての注入の後に測定した。回収結果はPoros A-HPLC分析により判定した。回収は前記バッグ中の目的物濃度に基づいて算出し、また報告値は2つの異なる調製物の平均値とした。 A similar experimental design was used in this second study as follows: mAbs at a concentration of 0.2 mg / mL and containing various concentrations of PS80 (0.02, 0.05, 0.1, and 0.3% w / v) Stock solutions were freshly prepared and added to iv infusion bags (PO only) as described above. The final mAb concentration in this iv saline bag was 2 μg / mL, and the final surfactant concentrations were 0.0002, 0.0005, 0.001, and 0.003%. Additional PS80 (to a final concentration of at least 0.001%) was added to all samples prior to analysis. The iv line tested was a Baxter Continu-Flo Solution Set equipped with a filter. The mAb concentration was measured initially and after infusion for two hours with a hold in an iv bag or through a selected iv line. Recovery results were determined by Poros A-HPLC analysis. The recovery was calculated based on the concentration of target in the bag and the reported value was the average of two different preparations.
Poros A-HPLC分析により判定した平均回収%を図2にプロットした。試験条件に応じて、その測定値は66%〜105%となった。全ての試験PS80濃度における約95%のタンパク質回収が、2時間保留中のi.v.バッグにおいて観察された。従って、バッグ中0.0002%という低さのPS80濃度(薬品製剤中0.02%に相当)は、mAbの吸着媒介性の損失を抑制するのに十分であった。しかし、図2に示すように、i.v.ラインおよびフィルター上でのさらなる有意な損失が認められる。界面活性剤濃度へのタンパク質損失全体の依存性は非線形であると考えられ、回収の90%近くが50 mL i.v.バッグ中0.001%(または製品中0.1%)以上のPS80濃度で達成された。 The average% recovery determined by Poros A-HPLC analysis is plotted in FIG. Depending on the test conditions, the measured value is 66% to 105%. Approximately 95% protein recovery at all tested PS80 concentrations was observed in the 2 hour pending iv bag. Thus, a PS80 concentration as low as 0.0002% in the bag (equivalent to 0.02% in the drug formulation) was sufficient to suppress the loss of adsorption mediatedness of the mAb. However, as shown in FIG. 2, additional significant losses on iv lines and filters are observed. The overall protein loss dependence on surfactant concentration was considered to be non-linear, with nearly 90% recovery achieved at PS80 concentrations of 0.001% (or 0.1% in product) in 50 mL iv bags.
実施例4--PS80-含有製品の安定性:酸化リスクの評価
加速条件下のタンパク質安定性は従来、有効期間の予測を与えるためだけでなく、長期保存用の最適な製剤を選択するためにも、バイオ医薬品の開発時に利用されている。ここで、PS80-含有製剤の安定性を評価するため、具体的にはPS80が長期保存中に酸化的分解によりmAbの安定性に悪影響を及ぼしうるリスクを評価するために、40℃で短期(2週間)加速安定性試験を行った。
Example 4-Stability of PS80-Containing Products: Assessment of Oxidation Risk Protein stability under accelerated conditions is traditionally not only to give a prediction of shelf life, but also to select the optimal formulation for long-term storage Is also used in the development of biopharmaceuticals. Here, in order to evaluate the stability of the PS80-containing preparation, specifically, to evaluate the risk that PS80 may adversely affect the stability of mAb by oxidative degradation during long-term storage, 2) Accelerated stability test was conducted.
ヒスチジン緩衝液で調製した様々なPS80ストック溶液を用いてmAb原薬を希釈することにより、目的の製剤を調製した。各バイアル中の最終mAb濃度は0.2 mg/mLであった。各試験条件を複数のガラスバイアルに等しく割り当て(aliquoted)、さらに該バイアルを40℃で2週間保存した。分析試験にはサイズ排除クロマトグラフィー(「SEC」)、キャピラリー等電点電気泳動(「cIEF」)、および質量分析法(「MS」)による分析を含めた。 The desired formulations were prepared by diluting the mAb drug substance with various PS80 stock solutions prepared in histidine buffer. The final mAb concentration in each vial was 0.2 mg / mL. Each test condition was equally assigned to multiple glass vials, and the vials were further stored at 40 ° C. for 2 weeks. Analytical tests included analysis by size exclusion chromatography ("SEC"), capillary isoelectric focusing ("cIEF"), and mass spectrometry ("MS").
メチオニン残基の酸化は、MSを利用して検出できる、メチオニンスルホキシドの形成および16 Daの質量シフトをもたらす。MSを使用することにより、酸化および非酸化ペプチドの相対量を比較した。MSの結果は表1に記載されている。mAb対照サンプル(賦形剤不含緩衝液中)に関しては、試験したメチオニン残基における測定酸化%は11%という高さであった。界面活性剤が製剤中に存在する場合、2つの最も曝露されたメチオニン残基における有意に上昇したレベルのmAb酸化(最大30%)が観察された。酸化はさらに凝集および他の二次分解プロセスを引き起こす可能性があることから、この酸化%の上昇は長期安定性低下の前兆となる。 Oxidation of methionine residues results in the formation of methionine sulfoxide and a mass shift of 16 Da which can be detected using MS. The relative amounts of oxidized and nonoxidized peptides were compared by using MS. The results of MS are described in Table 1. For the mAb control sample (in excipient free buffer), the measured% oxidation at the methionine residue tested was as high as 11%. When surfactant was present in the formulation, significantly elevated levels of mAb oxidation (up to 30%) at the two most exposed methionine residues were observed. This increase in oxidation is a precursor to long term stability loss as oxidation can further cause aggregation and other secondary degradation processes.
実施例5--選択した抗酸化剤の存在下でのPS80-含有製品の増大した安定性
提案した0.1%PS80含有製剤中のmAbの化学的安定性を改善するであろう賦形剤をスクリーニングするために、追加の研究をさらに行った。5つの異なる抗酸化剤(複数種のフリーラジカル捕捉剤および1種の金属キレート剤を含む)について試験した。
Example 5-Enhanced Stability of PS80-Containing Products in the Presence of Selected Antioxidants Screening of Excipients That Will Improve Chemical Stability of the mAb in the Proposed 0.1% PS80-Containing Formulation In order to do that, we did additional research. Five different antioxidants, including multiple free radical scavengers and one metal chelator, were tested.
目的の製剤は、ヒスチジン緩衝液で調製した、次の通りの様々な賦形剤ストック溶液:PS80(ポリソルベート80);Met(メチオニン);Cys(システイン);Glut(グルタチオン);MTG(モノチオグリセロール);EDTA(エデト酸二ナトリウム)を用いてmAb原薬を希釈することにより調製した。この製品バイアル中の最終抗酸化剤濃度は次の通り:5 mM Met、5 mM Cys、5 mM Glut、5 mM MTG、および1 mM EDTAであった。各バイアル中の最終mAb濃度は0.2 mg/mLであった。各試験条件を複数のガラスバイアルに等しく割り当て、さらに該バイアルを40℃で2週間保存した。分析試験にはcIEFおよびMSによる分析を含めた。 Formulations of interest were prepared in histidine buffer, various excipient stock solutions as follows: PS80 (polysorbate 80); Met (methionine); Cys (cysteine); Glut (glutathione); MTG (monothioglycerol) ); Prepared by diluting mAb drug substance with EDTA (disodium edetate). The final antioxidant concentrations in this product vial were as follows: 5 mM Met, 5 mM Cys, 5 mM Glut, 5 mM MTG, and 1 mM EDTA. The final mAb concentration in each vial was 0.2 mg / mL. Each test condition was equally assigned to a plurality of glass vials, and the vials were further stored at 40 ° C. for 2 weeks. Analytical tests included analysis by cIEF and MS.
表2に見られるように、抗酸化剤はいずれも前記界面活性剤の負の安定性影響を弱めることが可能であった。データは、メチオニンを含有する製剤において最も低い程度の酸化を示している。EDTAは、酸化を制御する点ではMetより有効性が低いと思われたが、それでもPS80-依存性酸化の程度を4倍近く有意に減少させた。 As seen in Table 2, all antioxidants were able to attenuate the negative stability impact of the surfactant. The data show the lowest degree of oxidation in formulations containing methionine. Although EDTA appeared to be less effective than Met in controlling oxidation, it still significantly reduced the degree of PS80-dependent oxidation by nearly 4-fold.
表2にはキャピラリー等電点電気泳動(cIEF)分析による電荷分析の結果も示されている。主%の最大値は、抗酸化剤を含有していない対照製剤に対して観察され、またMetまたはEDTAをさらに含むPS80-含有製剤に対しても観察された。cIEFを利用して評価したところ、最も安定性が低い製剤はCys、Glut、またはMTGを含有していた。
表2
0.1%PS80および様々な抗酸化剤を用いて製剤化した0.2 mg/mL mAbの安定性:バイアル中40℃で2週間保存した後のMSおよびcIEF分析の結果
Table 2 also shows the results of charge analysis by capillary isoelectric focusing (cIEF) analysis. Maximum% values were observed for the control formulations that did not contain antioxidants, and also for PS80-containing formulations that additionally contained Met or EDTA. The formulation with the least stability, as assessed using cIEF, contained Cys, Glut, or MTG.
Table 2
Stability of 0.2 mg / mL mAb formulated with 0.1% PS80 and various antioxidants: Results of MS and cIEF analysis after storage for 2 weeks at 40 ° C. in a vial
MS結果とcIEF結果との間の矛盾を説明する助けとするために、SDS-PAGE分析も実施した。Cys、Glut、またはMTGを含有する製剤に関する該SDS-PAGEの結果は、40℃で2週間保存したサンプルに関して、ジスルフィドシャッフリングによる化学的分解の兆候である、非還元ゲルにおけるバンドの有意なシフトを示した(図3)。SDS-PAGEにより検出されたCys、Glut、またはMTGの存在下でのmAbの化学的不安定性は、cIEFによる酸性ピークの増加とよく相関していた(表2)。従って、MetおよびEDTAは製品中のジスルフィド結合パターンの破壊を示さないたった2つの被験抗酸化剤であり、またこれらは最適な抗酸化活性を有することが確認された。抗酸化剤を含有する製剤の総合的な製品安定性は、以下:Met>EDTA>>Cys、Glut、MTGのようにランク付けすることができる。 SDS-PAGE analysis was also performed to help explain the discrepancy between MS and cIEF results. The SDS-PAGE results for formulations containing Cys, Glut, or MTG show a significant shift of bands in non-reduced gels that is indicative of chemical degradation by disulfide shuffling for samples stored for 2 weeks at 40 ° C. Shown (Figure 3). Chemical instability of the mAb in the presence of Cys, Glut, or MTG detected by SDS-PAGE correlated well with the increase of the acidic peak by cIEF (Table 2). Thus, Met and EDTA were the only two tested antioxidants that did not show disruption of the disulfide bond pattern in the product, and were also confirmed to have optimal antioxidant activity. The overall product stability of formulations containing antioxidants can be ranked as follows: Met> EDTA >> Cys, Glut, MTG.
実施例6--再製剤化製品の長期安定性および機能的抗酸化剤レベルの確認
PS80、Met、およびEDTAを含有する0.2 mg/mL mAb製剤の長期および加速安定性を評価するために、開発的安定性試験(development stability study)を実施した。0〜40 mMに及ぶ一連の6つのMet濃度を含めた幅広い条件について試験した。EDTAレベルは、mAbに関する以前の経験に基づいて0.05 mMに固定した。全部で8種の製剤を安定した状態で設置した。試験した製剤の概要を表3に示す。製剤A〜Fには、固定レベルのEDTA(0.05 mM)およびPS80(0.1%)と共に0、5、10、15、20、および40 mMのMetを組み入れた。製剤GおよびHは、それぞれ、PS80(単独)および緩衝液のみ(賦形剤無し)の対照としての役割を果たした。各製剤を複数のガラスバイアルに等分して入れ、さらに該バイアルを冷蔵(2〜8℃、一次)および25℃(加速)条件で倒立させて保存した。各バイアル中のmAb濃度は0.2 mg/mLであった。分析的データ取得(analytical pulls)を1、2、3、および6ヶ月間計画し、その結果を初期と比較した。
表3
製剤開発的安定性試験の実験構成。各処方コードに関係する製剤組成を詳述する
Example 6-Confirmation of long-term stability and functional antioxidant levels of the reformulated product
A development stability study was conducted to evaluate the long-term and accelerated stability of 0.2 mg / mL mAb formulations containing PS80, Met, and EDTA. A wide range of conditions were tested, including a series of 6 Met concentrations ranging from 0-40 mM. EDTA levels were fixed at 0.05 mM based on previous experience with mAbs. A total of eight preparations were placed in stable condition. A summary of the tested formulations is shown in Table 3. Formulations AF incorporated 0, 5, 10, 15, 20, and 40 mM Met with fixed levels of EDTA (0.05 mM) and PS80 (0.1%). Formulations G and H served as controls for PS80 (alone) and buffer only (no excipient), respectively. Each formulation was aliquoted into multiple glass vials, and the vials were stored inverted under refrigerated (2-8 ° C., primary) and 25 ° C. (accelerated) conditions. The mAb concentration in each vial was 0.2 mg / mL. Analytical pulls were planned for 1, 2, 3 and 6 months and the results were compared with the initial.
Table 3
Experimental configuration of formulation developmental stability test. Detailed formulation composition related to each prescription code
安定性に関する酸化レベルはMSを利用して測定した。表4は、タンパク質のトリプシン消化とその結果生じたペプチドのHPLC分離の後にMS定量化により決定したメチオニン残基の酸化レベルを列挙したものである。2〜8℃および25℃で6ヶ月間保存したサンプルC(PS80、Met、およびEDTA)、G(PS80単独)、ならびにH(賦形剤無し)の代表的な酸化傾向を図4にプロットした。酸化は、初期には全サンプルにおいて(理論上)最も曝露されたメチオニン残基254(M254)で観察された。興味深いことに、サンプル間の差異が初期分析の時点で存在し、サンプルGおよびHは第2曝露メチオニン残基430(M430)で〜3%の酸化を示した。サンプルA〜Fは少なくとも1種の抗酸化剤(Met)を含有していたが、一方のサンプルGおよびHは含有していなかった;従って、これらのデータは保存時の安定性を表している可能性があるだけでなく、サンプルの調製および分析中の分解を反映している可能性もある。EDTAとMetの複合添加の保護効果は、両抗酸化剤が存在するサンプルB〜Fにおいて明らかであった。具体的には、2〜8℃および25℃で6ヶ月という期間にわたり、M254での酸化は低いままであり(≦4.1%)、M430では酸化は殆どまたは全く観察されなかった。これらのデータは、抗酸化剤を組み合わせることの有効性を証明するものである。 Oxidation levels for stability were measured using MS. Table 4 lists the oxidation levels of methionine residues determined by MS quantification following tryptic digestion of proteins and HPLC separation of the resulting peptides. Representative oxidation trends of samples C (PS80, Met, and EDTA), G (PS80 alone), and H (without excipient) stored at 2-8 ° C. and 25 ° C. for 6 months are plotted in FIG. . Oxidation was initially observed at the most exposed (theoretically) most exposed methionine residue 254 (M254) in all samples. Interestingly, differences between samples were present at the time of initial analysis, and samples G and H showed ̃3% oxidation at the second exposed methionine residue 430 (M430). Samples A to F contained at least one antioxidant (Met) but not one of samples G and H; thus, these data represent storage stability Not only is it possible, but it may also reflect degradation during sample preparation and analysis. The protective effect of the combined addition of EDTA and Met was evident in samples B-F where both antioxidants were present. Specifically, oxidation at M254 remained low (≦ 4.1%) over a period of 6 months at 2-8 ° C. and 25 ° C., and little or no oxidation was observed at M430. These data demonstrate the effectiveness of combining antioxidants.
EDTAを含有しMetを含まないサンプルAのMS分析は、様々な酸化機構の識別に役立った。サンプルAにおける酸化レベルは対照サンプルH(緩衝液単独)における酸化レベルよりわずかに低いままであり、かつ対照サンプルG(PS80)における酸化レベルよりはるかに低いままであったが、それらはEDTAに加えてMetを含有するサンプルB〜Fで測定された酸化レベルより高かった。対応するMS結果から以前の3つの仮説:1.酸化はPS80の非存在下でさえも分解経路である、2.酸化は冷蔵保存時に生じ、かつ上昇した保存温度では有意に加速する、3.PS80の添加は恐らくは過酸化物-および金属-依存性経路の両方を介して酸化的分解経路を加速させる、が裏付けられたため、対照サンプルG(PS80)とH(緩衝液、賦形剤無し)との比較はさらに決定的に重要であった。25℃で6ヶ月間保存したサンプルに関して収集したデータにより、酸化は予想通り、PS80対照製剤(G)において最高レベル(100%)に達することが示された。加えて、製剤(H)において検出された酸化レベルは、抗酸化剤の組み合わせを含有するサンプル(A〜F)における酸化レベルより有意に高かった。これらのデータは、2種の抗酸化剤Met(フリーラジカル捕捉剤)とEDTA(金属キレート剤)との組み合わせが酸化を本質的に予防すること、ならびにそのクエンチング機構が温度依存性ではないことを示している。 MS analysis of EDTA-free, Met-free sample A helped to identify the various oxidation mechanisms. Although the oxidation level in sample A remained slightly lower than that in control sample H (buffer alone) and much lower than the oxidation level in control sample G (PS80), they added to EDTA Were higher than the oxidation levels measured in samples B-F containing Met. The previous three hypotheses from the corresponding MS results: 1. Oxidation is a degradation pathway even in the absence of PS80, 2. Oxidation occurs during cold storage, and accelerates significantly at elevated storage temperatures, 3. The addition of PS80 probably accelerated the oxidative degradation pathway via both peroxide- and metal-dependent pathways, so that control sample G (PS80) and H (buffer, no excipient) Comparison with was even more critical. Data collected on samples stored for 6 months at 25 ° C. indicated that oxidation reached the highest level (100%) in the PS80 control formulation (G), as expected. In addition, the oxidation level detected in formulation (H) was significantly higher than the oxidation level in samples (A-F) containing the combination of antioxidants. These data show that the combination of the two antioxidants Met (free radical scavenger) and EDTA (metal chelating agent) essentially prevents oxidation, and that its quenching mechanism is not temperature dependent. Is shown.
表4
異なるレベルの界面活性剤、抗酸化剤、および金属キレート剤を含有する、様々な保存温度での6ヶ月間の保存後のオテリキシズマブ安定性サンプルA〜H中の様々なメチオニン残基について報告された、MS分析によるペプチドマッピングの結果。各処方コードに関係する製剤組成は表3に詳細に記載されている。
Table 4
Reported for various methionine residues in otelixizumab stability samples A to H after storage for 6 months at different storage temperatures, containing different levels of surfactant, antioxidants and metal chelating agents , Results of peptide mapping by MS analysis. The formulation composition associated with each prescription code is described in detail in Table 3.
実施例7--低濃度/低用量オテリキシズマブ製剤の組成は、高界面活性剤レベルでの2種の抗酸化剤の独自の組み合わせである
下記表5は、低濃度オテリキシズマブ製剤(0.2 mg/mL)の組成と賦形剤機能の概要である。賦形剤のレベルおよび機能性は本製剤独自のものである。
Example 7-Composition of low concentration / low dose oteliximab formulation is a unique combination of two antioxidants at high surfactant level Table 5 below shows low concentration oteliximab formulation (0.2 mg / mL) It is a summary of the composition and excipient function of The level and functionality of the excipients are unique to this formulation.
実施例8--他の市販のmAb製剤との比較
他の市販のmAbと比較した場合、前記のオテリキシズマブの組成は高い賦形剤/mAb比(複数あり)を呈する。下記表6を参照されたい。
Example 8-Comparison with Other Commercially Available mAb Formulations When compared to other commercial mAbs, the composition of Oterliximab described above exhibits high excipient / mAb ratio (s). See Table 6 below.
界面活性剤-含有オテリキシズマブ製剤(0.2 mg/mL mAbおよび0.1%PS80)では、PS80/mAb比は約545である。比較すると、10 mg/mL mAbおよび0.22%PS80の濃度で製剤化されているエクリズマブ(ソリリス)は、その対応するPS80/mAb比は約25である。別のmAbであるリツキシマブでは、PS80濃度は0.07%である;従って、10 mg/mLの製品の場合、約8というPS80/mAb比を算出することができる。これらのケースのいずれにおいても、界面活性剤はモル過剰である;しかし、その過剰度は、オテリキシズマブに対して提案した比(545)よりもはるかに小さい。界面活性剤のかかる大モル過剰時には、該界面活性剤それ自体の純度および化学的安定性がmAb分子の安定性と相互に関連し合うため、それらを考慮する必要がある。 For surfactant-containing Oterliximab formulations (0.2 mg / mL mAb and 0.1% PS80), the PS80 / mAb ratio is about 545. By comparison, eculizumab (Solilith) formulated at a concentration of 10 mg / mL mAb and 0.22% PS80 has a corresponding PS80 / mAb ratio of about 25. For another mAb, rituximab, the PS80 concentration is 0.07%; therefore, for a 10 mg / mL product, a PS80 / mAb ratio of approximately 8 can be calculated. In any of these cases, the surfactant is in molar excess; however, the excess is much smaller than the proposed ratio for oteliximab (545). At such a large molar excess of surfactant, the purity and chemical stability of the surfactant itself must be taken into consideration as it correlates with the stability of the mAb molecule.
試験した抗酸化剤-含有オテリキシズマブ製剤(0.2 mg/mL mAbおよび5 mM Met)では、Met/mAb比は約3,750であった。文献には、5という最適比がHER2 mAbに関して報告されている。また、Met/mAb比が約100である他の非経口バイオ医薬製剤の実例も存在する(フォリトロピンαおよびβ)。本発明者らの比は幾つかの市販製品に見出される比をはるかに上回っているが、本発明者らの製剤におけるより高いMet濃度は、高いPS80濃度と直接相関している可能性があり、その上高いPS80/mAb比とも相関している可能性がある。
[1] 以下a)およびb):a)治療用タンパク質;およびb)界面活性剤;ここで該治療用タンパク質に対する該界面活性剤のモル比が少なくとも100である、を含む該治療用タンパク質のための製剤。
[2] 前記の治療用タンパク質に対する界面活性剤のモル比が、少なくとも150、少なくとも200、少なくとも250、少なくとも300、少なくとも400、および少なくとも500からなる群より選択される、1記載の製剤。
[3] 前記の治療用タンパク質に対する界面活性剤のモル比が約545である、1または2記載の製剤。
[4] 前記界面活性剤が、ポリソルベート-20、ポリソルベート-40、ポリソルベート-60、ポリソルベート-65、ポリソルベート-80、ポリソルベート-85、ポロキサマー88、およびそれらの混合物からなる群より選択される、1〜3のいずれか1項記載の製剤。
[5] 前記製剤がc)抗酸化剤をさらに含み、ここで前記治療用タンパク質に対する該抗酸化剤のモル比が少なくとも750である、1〜4のいずれか1項記載の製剤。
[6] 前記の治療用タンパク質に対する抗酸化剤のモル比が、少なくとも5500、少なくとも6000、少なくとも6500、および少なくとも7000からなる群より選択される、5記載の製剤。
[7] 前記の治療用タンパク質に対する抗酸化剤のモル比が約7143である、5または6記載の製剤。
[8] 前記抗酸化剤が、メチオニン、システイン、グルタチオン、およびモノチオグリセロールからなる群より選択される、5〜7のいずれか1項記載の製剤。
[9] 前記製剤がd)緩衝液をさらに含み、ここで前記製剤のpHが約4.0〜約8.0である、1〜8のいずれか1項記載の製剤。
[10] 前記緩衝液が、ヒスチジン、酢酸塩、クエン酸塩、およびコハク酸塩からなる群より選択される、9記載の製剤。
[11] 前記治療用タンパク質が抗原結合性タンパク質である、1〜10のいずれか1項記載の製剤。
[12] 前記抗原結合性タンパク質が抗体またはその断片である、11記載の製剤。
[13] 前記抗原結合性タンパク質が免疫グロブリン単一可変ドメインである、11記載の製剤。
[14] 前記抗原結合性タンパク質がヒトCD3に結合する、11記載の製剤。
[15] 前記の抗原結合性ポリペプチドが抗-CD3抗体である、14記載の製剤。
[16] 前記抗-CD3抗体が、配列番号1を含む重鎖および配列番号2を含む軽鎖を含む、15記載の製剤。
[17] 前記治療用タンパク質が約0.01 mg/mL〜約1 mg/mLの濃度で存在する、1〜16のいずれか1項記載の製剤。
[18] 前記治療用タンパク質が約0.1 mg/mL〜約0.5 mg/mLの濃度で存在する、1〜17のいずれか1項記載の製剤。
[19] 前記治療用タンパク質が約0.2 mg/mLの濃度で存在する、1〜18のいずれか1項記載の製剤。
[20] 前記製剤が再構成された製剤である、1〜19のいずれか1項記載の製剤。
[21] 前記製剤が液体医薬製剤である、1〜20のいずれか1項記載の製剤。
[22] 前記製剤が非経口投与に適したものである、1〜21のいずれか1項記載の製剤。
[23] 治療用タンパク質のための製剤であって、以下a)〜d):a)治療用タンパク質;およびb)約0.01%w/v〜約0.5%w/vの界面活性剤、ここで該治療用タンパク質に対する該界面活性剤のモル比が少なくとも100である;c)約1 mM〜約50 mMの抗酸化剤、ここで治療用タンパク質に対する該抗酸化剤のモル比が少なくとも750である;およびd)約1 mM〜約100 mMの緩衝液を含み、ここで上記製剤のpHが約4.0〜約8.0である、上記製剤。
[24] 前記治療用タンパク質が抗体であり、前記界面活性剤がポリソルベート80であり、前記抗酸化剤がメチオニンであり、かつ前記緩衝液がヒスチジンである、23記載の製剤。
[25] 前記抗体が約0.2 mg/mLの濃度で存在する、24記載の製剤。
[26] 前記製剤が約0.01 mM〜約1.0 mMのEDTAをさらに含む、1〜25のいずれか1項記載の製剤。
[27] 前記製剤が約0.01 mM〜約0.1 mMのEDTAを含む、26記載の製剤。
[28] 前記製剤が約0.05 mMのEDTAを含む、27記載の製剤。
For the antioxidant-containing oteliximab formulations tested (0.2 mg / mL mAb and 5 mM Met), the Met / mAb ratio was approximately 3,750. In the literature, an optimal ratio of 5 has been reported for the HER2 mAb. There are also examples of other parenteral biopharmaceutical formulations where the Met / mAb ratio is about 100 (follitropins alpha and beta). Although our ratio far exceeds that found in some commercial products, higher Met concentrations in our formulations may be directly correlated with higher PS80 concentrations. And may also be correlated with high PS80 / mAb ratios .
[1] A therapeutic protein of the following including a) and b): a) a therapeutic protein; and b) a surfactant, wherein the molar ratio of the surfactant to the therapeutic protein is at least 100. Formulation for.
[2] The preparation according to 1, wherein the molar ratio of surfactant to therapeutic protein is selected from the group consisting of at least 150, at least 200, at least 250, at least 300, at least 400, and at least 500.
[3] The preparation according to 1 or 2, wherein the molar ratio of the surfactant to the therapeutic protein is about 545.
[4] The surfactant is selected from the group consisting of polysorbate-20, polysorbate-40, polysorbate-60, polysorbate-65, polysorbate-80, polysorbate-85, poloxamer 88, and a mixture thereof, 1 to 4 The preparation according to any one of 3.
[5] The preparation according to any one of 1-4, wherein the preparation further comprises c) an antioxidant, wherein the molar ratio of the antioxidant to the therapeutic protein is at least 750.
[6] The preparation according to 5, wherein the molar ratio of the antioxidant to the therapeutic protein is selected from the group consisting of at least 5500, at least 6000, at least 6500, and at least 7000.
[7] The preparation according to 5 or 6, wherein the molar ratio of the antioxidant to the therapeutic protein is about 7143.
[8] The preparation according to any one of 5 to 7, wherein the antioxidant is selected from the group consisting of methionine, cysteine, glutathione and monothioglycerol.
[9] The preparation according to any one of 1-8, wherein the preparation further comprises d) a buffer, wherein the pH of the preparation is about 4.0 to about 8.0.
[10] The preparation according to 9, wherein the buffer is selected from the group consisting of histidine, acetate, citrate and succinate.
[11] The preparation according to any one of 1 to 10, wherein the therapeutic protein is an antigen binding protein.
[12] The preparation according to 11, wherein the antigen binding protein is an antibody or a fragment thereof.
[13] The preparation according to 11, wherein the antigen binding protein is an immunoglobulin single variable domain.
[14] The preparation according to 11, wherein the antigen binding protein binds to human CD3.
[15] The preparation according to 14, wherein the antigen-binding polypeptide is an anti-CD3 antibody.
[16] The preparation according to 15, wherein the anti-CD3 antibody comprises a heavy chain comprising SEQ ID NO: 1 and a light chain comprising SEQ ID NO: 2.
[17] The preparation according to any one of 1 to 16, wherein the therapeutic protein is present at a concentration of about 0.01 mg / mL to about 1 mg / mL.
[18] The preparation according to any one of 1 to 17, wherein the therapeutic protein is present at a concentration of about 0.1 mg / mL to about 0.5 mg / mL.
[19] The preparation according to any one of 1 to 18, wherein the therapeutic protein is present at a concentration of about 0.2 mg / mL.
[20] The preparation according to any one of 1 to 19, wherein the preparation is a reconstituted preparation.
[21] The preparation according to any one of 1 to 20, wherein the preparation is a liquid pharmaceutical preparation.
[22] The preparation according to any one of 1 to 21, wherein the preparation is suitable for parenteral administration.
[23] A formulation for a therapeutic protein, the following a) to d): a) therapeutic protein; and b) about 0.01% w / v to about 0.5% w / v of a surfactant, The molar ratio of the surfactant to the therapeutic protein is at least 100; c) an antioxidant of about 1 mM to about 50 mM, wherein the molar ratio of the antioxidant to the therapeutic protein is at least 750 And d) the above formulation, comprising about 1 mM to about 100 mM of buffer, wherein the pH of the formulation is about 4.0 to about 8.0.
[24] The formulation according to 23, wherein the therapeutic protein is an antibody, the surfactant is
[25] The preparation according to 24, wherein the antibody is present at a concentration of about 0.2 mg / mL.
[26] The preparation according to any one of 1 to 25, wherein the preparation further comprises about 0.01 mM to about 1.0 mM EDTA.
[27] The preparation according to 26, wherein the preparation comprises about 0.01 mM to about 0.1 mM EDTA.
[28] The preparation according to 27, wherein the preparation comprises about 0.05 mM EDTA.
Claims (12)
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361787709P | 2013-03-15 | 2013-03-15 | |
| US61/787,709 | 2013-03-15 | ||
| PCT/IB2014/059757 WO2014141152A2 (en) | 2013-03-15 | 2014-03-13 | Low concentration antibody formulations |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2016513635A JP2016513635A (en) | 2016-05-16 |
| JP6541581B2 true JP6541581B2 (en) | 2019-07-10 |
Family
ID=50397215
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015562515A Active JP6541581B2 (en) | 2013-03-15 | 2014-03-13 | Low concentration antibody preparation |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US10537638B2 (en) |
| EP (2) | EP2968535A2 (en) |
| JP (1) | JP6541581B2 (en) |
| KR (1) | KR20150132332A (en) |
| CN (1) | CN105073136A (en) |
| AU (1) | AU2014229282B2 (en) |
| BR (1) | BR112015023498A2 (en) |
| CA (1) | CA2902289A1 (en) |
| IL (1) | IL240754A0 (en) |
| RU (1) | RU2015137685A (en) |
| SG (1) | SG11201506499YA (en) |
| TW (1) | TW201501724A (en) |
| WO (1) | WO2014141152A2 (en) |
Families Citing this family (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101870555B1 (en) | 2011-08-23 | 2018-06-22 | 로슈 글리카트 아게 | Bispecific antibodies specific for t-cell activating antigens and a tumor antigen and methods of use |
| WO2014056783A1 (en) | 2012-10-08 | 2014-04-17 | Roche Glycart Ag | Fc-free antibodies comprising two fab-fragments and methods of use |
| RU2015140915A (en) | 2013-02-26 | 2017-04-03 | Роше Гликарт Аг | BSPECIFIC ANTI-BINDING MOLECULES ACTIVATING T-CELLS |
| MY192312A (en) | 2013-02-26 | 2022-08-17 | Roche Glycart Ag | Bispecific t cell activating antigen binding molecules |
| SG11201506499YA (en) | 2013-03-15 | 2015-09-29 | Glaxosmithkline Ip No 2 Ltd | Low concentration antibody formulations |
| DK3083689T3 (en) | 2013-12-17 | 2020-08-03 | Genentech Inc | Anti-CD3 antibodies and methods of use |
| PE20170263A1 (en) | 2014-08-04 | 2017-03-30 | Hoffmann La Roche | T-CELL ACTIVATING ANTIGEN-BINDING BI-SPECIFIC MOLECULES |
| US9751946B2 (en) | 2014-09-12 | 2017-09-05 | Genentech, Inc. | Anti-CLL-1 antibodies and immunoconjugates |
| BR112017010324A2 (en) | 2014-11-20 | 2018-05-15 | F. Hoffmann-La Roche Ag | method for treating or slowing cancer progression in an individual, molecules, methods for enhancing immune function in an individual and for selecting a patient for treatment, kits, pharmaceutical composition and uses of a combination of one molecule |
| TWI731861B (en) | 2015-06-16 | 2021-07-01 | 美商建南德克公司 | HUMANIZED AND AFFINITY MATURED ANTIBODIES TO FcRH5 AND METHODS OF USE |
| CN107847568B (en) | 2015-06-16 | 2022-12-20 | 豪夫迈·罗氏有限公司 | anti-CLL-1 antibodies and methods of use |
| WO2016204966A1 (en) | 2015-06-16 | 2016-12-22 | Genentech, Inc. | Anti-cd3 antibodies and methods of use |
| AR106188A1 (en) | 2015-10-01 | 2017-12-20 | Hoffmann La Roche | ANTI-CD19 HUMANIZED HUMAN ANTIBODIES AND METHODS OF USE |
| CN107849137B (en) | 2015-10-02 | 2021-11-26 | 豪夫迈·罗氏有限公司 | Bispecific anti-CEAXCD 3T cell activating antigen binding molecules |
| EP3371311B1 (en) | 2015-11-06 | 2021-07-21 | Orionis Biosciences BV | Bi-functional chimeric proteins and uses thereof |
| JP7325186B2 (en) | 2015-12-09 | 2023-08-14 | エフ・ホフマン-ラ・ロシュ・アクチェンゲゼルシャフト | Type II anti-CD20 antibody for reducing the formation of anti-drug antibodies |
| EP3397287A1 (en) | 2015-12-30 | 2018-11-07 | Genentech, Inc. | Formulations with reduced degradation of polysorbate |
| MX2018008347A (en) | 2016-01-08 | 2018-12-06 | Hoffmann La Roche | Methods of treating cea-positive cancers using pd-1 axis binding antagonists and anti-cea/anti-cd3 bispecific antibodies. |
| JP7166923B2 (en) | 2016-02-05 | 2022-11-08 | オリオニス バイオサイエンシズ ビーブイ | Targeted therapeutic agents and their uses |
| FI3433280T3 (en) | 2016-03-22 | 2023-06-06 | Hoffmann La Roche | Protease-activated t cell bispecific molecules |
| EP3435982A1 (en) | 2016-03-31 | 2019-02-06 | VHsquared Limited | Compositions |
| SE540001C2 (en) * | 2016-04-05 | 2018-02-20 | Kyttinge Invest Ab | Self-propelling trolley assembly |
| EP3519437B1 (en) | 2016-09-30 | 2021-09-08 | F. Hoffmann-La Roche AG | Bispecific antibodies against p95her2 |
| AR109621A1 (en) * | 2016-10-24 | 2018-12-26 | Janssen Pharmaceuticals Inc | FORMULATIONS OF VACCINES AGAINST GLUCOCONJUGADOS OF EXPEC |
| AU2017361081A1 (en) | 2016-11-15 | 2019-05-23 | Genentech, Inc. | Dosing for treatment with anti-CD20/anti-CD3 bispecific antibodies |
| WO2018144999A1 (en) | 2017-02-06 | 2018-08-09 | Orionis Biosciences, Inc. | Targeted engineered interferon and uses thereof |
| JP7263320B2 (en) | 2017-08-22 | 2023-04-24 | バイオジェン・エムエイ・インコーポレイテッド | Pharmaceutical composition containing anti-beta-amyloid antibody |
| AU2018335780A1 (en) * | 2017-09-22 | 2020-03-26 | Immunogen, Inc. | Methods of preventing methionine oxidation in immunoconjugates |
| EP3688033A4 (en) * | 2017-09-29 | 2021-06-23 | Janssen Biotech, Inc. | Novel formulations which stabilize low dose antibody compositions |
| AU2019218959A1 (en) | 2018-02-08 | 2020-09-03 | Genentech, Inc. | Bispecific antigen-binding molecules and methods of use |
| MA55033A (en) | 2019-02-18 | 2021-12-29 | Lilly Co Eli | THERAPEUTIC ANTIBODY FORMULATION |
| EP3986571A1 (en) | 2019-06-21 | 2022-04-27 | Sorriso Pharmaceuticals, Inc. | Polypeptides |
| CA3144566A1 (en) | 2019-06-21 | 2020-12-24 | Sorriso Pharmaceuticals, Inc. | Polypeptides |
| BR112022007776A2 (en) * | 2019-10-25 | 2022-07-05 | Amgen Inc | COMPOSITIONS AND METHODS TO MINIMIZE PROTEIN LOSS AT LOW PROTEIN CONCENTRATIONS |
| PE20221511A1 (en) | 2019-12-13 | 2022-10-04 | Genentech Inc | ANTI-LY6G6D ANTIBODIES AND METHODS OF USE |
| AU2021291011A1 (en) | 2020-06-19 | 2023-01-05 | F. Hoffmann-La Roche Ag | Antibodies binding to CD3 and CD19 |
| US20230406930A1 (en) * | 2022-04-13 | 2023-12-21 | Genentech, Inc. | Pharmaceutical compositions of therapeutic proteins and methods of use |
| US11958906B2 (en) * | 2022-04-13 | 2024-04-16 | Genentech, Inc. | Pharmaceutical compositions of mosunetuzumab and methods of use |
Family Cites Families (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS57106673A (en) | 1980-12-24 | 1982-07-02 | Chugai Pharmaceut Co Ltd | Dibenzo(b,f)(1,4)oxazepin derivative |
| GB8607679D0 (en) | 1986-03-27 | 1986-04-30 | Winter G P | Recombinant dna product |
| EP0689843B1 (en) * | 1993-12-17 | 2003-09-10 | Mochida Pharmaceutical Co., Ltd. | Composition containing soluble thrombomodulins |
| JP4454571B2 (en) * | 1998-03-06 | 2010-04-21 | 中外製薬株式会社 | Protein-free formulation |
| IL127127A0 (en) | 1998-11-18 | 1999-09-22 | Peptor Ltd | Small functional units of antibody heavy chain variable regions |
| DK1419179T3 (en) | 2001-08-10 | 2010-06-21 | Univ Aberdeen | Antigen binding domains from fish |
| US20030104996A1 (en) | 2001-08-30 | 2003-06-05 | Tiansheng Li | L-methionine as a stabilizer for NESP/EPO in HSA-free formulations |
| CA2463473A1 (en) | 2001-10-16 | 2003-04-24 | Symbiontics Inc. | Targeted therapeutic proteins |
| PT1441589E (en) | 2001-11-08 | 2012-08-13 | Abbott Biotherapeutics Corp | Stable liquid pharmaceutical formulation of igg antibodies |
| CA2600836A1 (en) * | 2005-03-08 | 2006-09-14 | Pharmacia & Upjohn Company Llc | Composition comprising an antibody against macrophage colony-stimulating factor (m-csf) and a chelating agent |
| US7989665B2 (en) | 2005-04-05 | 2011-08-02 | Firmenich Sa | Hydrogenation of esters with Ru/tetradentate ligands complexes |
| WO2007033230A2 (en) * | 2005-09-12 | 2007-03-22 | Novimmune S.A. | Anti-cd3 antibody formulations |
| JP5490714B2 (en) | 2007-11-28 | 2014-05-14 | メディミューン,エルエルシー | Protein preparation |
| JP2010241718A (en) * | 2009-04-03 | 2010-10-28 | Kyowa Hakko Kirin Co Ltd | Stable aqueous solution preparation of antibody |
| CA2765220A1 (en) * | 2009-07-14 | 2011-01-20 | Biogen Idec Ma Inc. | Methods for inhibiting yellow color and peroxide formation in a composition |
| US20120294866A1 (en) * | 2010-01-19 | 2012-11-22 | F. Hoffmann-La Roche Ag | Pharmaceutical formulation for proteins |
| WO2011163566A2 (en) * | 2010-06-25 | 2011-12-29 | Tolerx, Inc. | Methods of treating patients with immune-related diseases |
| US9045381B2 (en) | 2010-10-19 | 2015-06-02 | Yeda Research And Development Co. Ltd. | Ruthenium complexes and their uses in processes for formation and/or hydrogenation of esters, amides and derivatives thereof |
| WO2013003680A1 (en) | 2011-06-30 | 2013-01-03 | Genentech, Inc. | Anti-c-met antibody formulations |
| SG11201506499YA (en) | 2013-03-15 | 2015-09-29 | Glaxosmithkline Ip No 2 Ltd | Low concentration antibody formulations |
-
2014
- 2014-03-13 SG SG11201506499YA patent/SG11201506499YA/en unknown
- 2014-03-13 BR BR112015023498A patent/BR112015023498A2/en not_active Application Discontinuation
- 2014-03-13 WO PCT/IB2014/059757 patent/WO2014141152A2/en active Application Filing
- 2014-03-13 TW TW103108897A patent/TW201501724A/en unknown
- 2014-03-13 US US14/775,900 patent/US10537638B2/en active Active
- 2014-03-13 JP JP2015562515A patent/JP6541581B2/en active Active
- 2014-03-13 EP EP14714396.0A patent/EP2968535A2/en not_active Ceased
- 2014-03-13 KR KR1020157028852A patent/KR20150132332A/en not_active Application Discontinuation
- 2014-03-13 EP EP19198476.4A patent/EP3686217A1/en not_active Withdrawn
- 2014-03-13 CA CA2902289A patent/CA2902289A1/en not_active Abandoned
- 2014-03-13 CN CN201480016095.4A patent/CN105073136A/en active Pending
- 2014-03-13 AU AU2014229282A patent/AU2014229282B2/en not_active Ceased
- 2014-03-13 RU RU2015137685A patent/RU2015137685A/en not_active Application Discontinuation
-
2015
- 2015-08-23 IL IL240754A patent/IL240754A0/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| SG11201506499YA (en) | 2015-09-29 |
| US10537638B2 (en) | 2020-01-21 |
| KR20150132332A (en) | 2015-11-25 |
| TW201501724A (en) | 2015-01-16 |
| CN105073136A (en) | 2015-11-18 |
| BR112015023498A2 (en) | 2017-10-10 |
| WO2014141152A2 (en) | 2014-09-18 |
| EP2968535A2 (en) | 2016-01-20 |
| WO2014141152A3 (en) | 2014-12-04 |
| US20160000916A1 (en) | 2016-01-07 |
| AU2014229282A1 (en) | 2015-10-08 |
| IL240754A0 (en) | 2015-10-29 |
| RU2015137685A3 (en) | 2018-03-05 |
| RU2015137685A (en) | 2017-04-20 |
| CA2902289A1 (en) | 2014-09-18 |
| JP2016513635A (en) | 2016-05-16 |
| EP3686217A1 (en) | 2020-07-29 |
| AU2014229282B2 (en) | 2017-02-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6541581B2 (en) | Low concentration antibody preparation | |
| AU2012240050B2 (en) | Formulations with reduced viscosity | |
| AU2015260758B2 (en) | Antibody formulation | |
| US20140044727A1 (en) | Formulations with reduced viscosity | |
| US10933141B2 (en) | Formulations with reduced degradation of polysorbate | |
| TW201347791A (en) | Antibody formulation | |
| KR20140044305A (en) | Lyophilized formulations | |
| WO2014141149A1 (en) | Formulations with reduced viscosity | |
| KR20230009897A (en) | Formulations containing anti-IL-23p19 antibodies, methods for their preparation and uses |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20170217 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20171003 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20171219 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20180522 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20180821 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20181204 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190404 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20190418 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20190521 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20190611 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6541581 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |