JP5748351B2 - Hydrogenation catalyst for aromatic compounds and method for hydrogenating aromatic compounds using the catalyst - Google Patents
Hydrogenation catalyst for aromatic compounds and method for hydrogenating aromatic compounds using the catalyst Download PDFInfo
- Publication number
- JP5748351B2 JP5748351B2 JP2012071107A JP2012071107A JP5748351B2 JP 5748351 B2 JP5748351 B2 JP 5748351B2 JP 2012071107 A JP2012071107 A JP 2012071107A JP 2012071107 A JP2012071107 A JP 2012071107A JP 5748351 B2 JP5748351 B2 JP 5748351B2
- Authority
- JP
- Japan
- Prior art keywords
- catalyst
- raw material
- hydrogenation
- aqueous solution
- hydrogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000003054 catalyst Substances 0.000 title claims description 85
- 238000005984 hydrogenation reaction Methods 0.000 title claims description 74
- 238000000034 method Methods 0.000 title claims description 43
- 150000001491 aromatic compounds Chemical class 0.000 title claims description 36
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 claims description 75
- 239000002994 raw material Substances 0.000 claims description 70
- 239000001257 hydrogen Substances 0.000 claims description 39
- 229910052739 hydrogen Inorganic materials 0.000 claims description 39
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 37
- 229910052759 nickel Inorganic materials 0.000 claims description 36
- 229910052750 molybdenum Inorganic materials 0.000 claims description 30
- 229910052809 inorganic oxide Inorganic materials 0.000 claims description 27
- 229910052707 ruthenium Inorganic materials 0.000 claims description 26
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 claims description 24
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 claims description 24
- 238000006243 chemical reaction Methods 0.000 claims description 23
- 239000011733 molybdenum Substances 0.000 claims description 23
- 239000007788 liquid Substances 0.000 claims description 8
- 239000007864 aqueous solution Substances 0.000 description 57
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 39
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 34
- 230000000694 effects Effects 0.000 description 22
- 230000002378 acidificating effect Effects 0.000 description 19
- 239000000047 product Substances 0.000 description 18
- 230000009467 reduction Effects 0.000 description 16
- 239000000243 solution Substances 0.000 description 15
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 14
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 14
- 229910004298 SiO 2 Inorganic materials 0.000 description 11
- 229910052782 aluminium Inorganic materials 0.000 description 11
- 229910018072 Al 2 O 3 Inorganic materials 0.000 description 10
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 10
- 238000000975 co-precipitation Methods 0.000 description 10
- 150000007529 inorganic bases Chemical class 0.000 description 10
- 238000004519 manufacturing process Methods 0.000 description 10
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 9
- 238000001035 drying Methods 0.000 description 9
- 239000007789 gas Substances 0.000 description 9
- 238000005470 impregnation Methods 0.000 description 9
- 229910052751 metal Inorganic materials 0.000 description 9
- 239000002184 metal Substances 0.000 description 9
- 238000002360 preparation method Methods 0.000 description 9
- RWSOTUBLDIXVET-UHFFFAOYSA-N Dihydrogen sulfide Chemical compound S RWSOTUBLDIXVET-UHFFFAOYSA-N 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- 229910000037 hydrogen sulfide Inorganic materials 0.000 description 8
- 239000000203 mixture Substances 0.000 description 7
- 229910000029 sodium carbonate Inorganic materials 0.000 description 7
- 235000017550 sodium carbonate Nutrition 0.000 description 7
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 6
- 230000000052 comparative effect Effects 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- -1 hydrogen sulfide Chemical class 0.000 description 6
- UAEPNZWRGJTJPN-UHFFFAOYSA-N methylcyclohexane Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 239000002253 acid Substances 0.000 description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 4
- 229910018879 Pt—Pd Inorganic materials 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- 229910001593 boehmite Inorganic materials 0.000 description 4
- 239000008119 colloidal silica Substances 0.000 description 4
- 230000007423 decrease Effects 0.000 description 4
- 238000006477 desulfuration reaction Methods 0.000 description 4
- 230000023556 desulfurization Effects 0.000 description 4
- 229930195733 hydrocarbon Natural products 0.000 description 4
- 150000002430 hydrocarbons Chemical class 0.000 description 4
- FAHBNUUHRFUEAI-UHFFFAOYSA-M hydroxidooxidoaluminium Chemical compound O[Al]=O FAHBNUUHRFUEAI-UHFFFAOYSA-M 0.000 description 4
- 239000012535 impurity Substances 0.000 description 4
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 4
- 229910000510 noble metal Inorganic materials 0.000 description 4
- 239000000377 silicon dioxide Substances 0.000 description 4
- 150000003464 sulfur compounds Chemical class 0.000 description 4
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 3
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 3
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 3
- 102100025490 Slit homolog 1 protein Human genes 0.000 description 3
- 101710123186 Slit homolog 1 protein Proteins 0.000 description 3
- 238000002441 X-ray diffraction Methods 0.000 description 3
- 239000001099 ammonium carbonate Substances 0.000 description 3
- 235000012501 ammonium carbonate Nutrition 0.000 description 3
- 235000011114 ammonium hydroxide Nutrition 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- GYNNXHKOJHMOHS-UHFFFAOYSA-N methyl-cycloheptane Natural products CC1CCCCCC1 GYNNXHKOJHMOHS-UHFFFAOYSA-N 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 229910017604 nitric acid Inorganic materials 0.000 description 3
- 238000002161 passivation Methods 0.000 description 3
- 230000000704 physical effect Effects 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 238000010792 warming Methods 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- 229910021536 Zeolite Inorganic materials 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- MWPLVEDNUUSJAV-UHFFFAOYSA-N anthracene Chemical compound C1=CC=CC2=CC3=CC=CC=C3C=C21 MWPLVEDNUUSJAV-UHFFFAOYSA-N 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 239000008367 deionised water Substances 0.000 description 2
- 229910021641 deionized water Inorganic materials 0.000 description 2
- HNPSIPDUKPIQMN-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical compound O=[Si]=O.O=[Al]O[Al]=O HNPSIPDUKPIQMN-UHFFFAOYSA-N 0.000 description 2
- 238000010304 firing Methods 0.000 description 2
- 150000002431 hydrogen Chemical class 0.000 description 2
- 239000003350 kerosene Substances 0.000 description 2
- 239000011259 mixed solution Substances 0.000 description 2
- KBJMLQFLOWQJNF-UHFFFAOYSA-N nickel(ii) nitrate Chemical compound [Ni+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O KBJMLQFLOWQJNF-UHFFFAOYSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- YNPNZTXNASCQKK-UHFFFAOYSA-N phenanthrene Chemical compound C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 235000011118 potassium hydroxide Nutrition 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 229910001925 ruthenium oxide Inorganic materials 0.000 description 2
- YBCAZPLXEGKKFM-UHFFFAOYSA-K ruthenium(iii) chloride Chemical compound [Cl-].[Cl-].[Cl-].[Ru+3] YBCAZPLXEGKKFM-UHFFFAOYSA-K 0.000 description 2
- WOCIAKWEIIZHES-UHFFFAOYSA-N ruthenium(iv) oxide Chemical compound O=[Ru]=O WOCIAKWEIIZHES-UHFFFAOYSA-N 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 235000011121 sodium hydroxide Nutrition 0.000 description 2
- NTHWMYGWWRZVTN-UHFFFAOYSA-N sodium silicate Chemical compound [Na+].[Na+].[O-][Si]([O-])=O NTHWMYGWWRZVTN-UHFFFAOYSA-N 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 239000010457 zeolite Substances 0.000 description 2
- 229910000873 Beta-alumina solid electrolyte Inorganic materials 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- 229910021586 Nickel(II) chloride Inorganic materials 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 229910019891 RuCl3 Inorganic materials 0.000 description 1
- 239000004115 Sodium Silicate Substances 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 229910010413 TiO 2 Inorganic materials 0.000 description 1
- FMRLDPWIRHBCCC-UHFFFAOYSA-L Zinc carbonate Chemical compound [Zn+2].[O-]C([O-])=O FMRLDPWIRHBCCC-UHFFFAOYSA-L 0.000 description 1
- 238000005299 abrasion Methods 0.000 description 1
- MQRWBMAEBQOWAF-UHFFFAOYSA-N acetic acid;nickel Chemical compound [Ni].CC(O)=O.CC(O)=O MQRWBMAEBQOWAF-UHFFFAOYSA-N 0.000 description 1
- ZOIORXHNWRGPMV-UHFFFAOYSA-N acetic acid;zinc Chemical compound [Zn].CC(O)=O.CC(O)=O ZOIORXHNWRGPMV-UHFFFAOYSA-N 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 229910001854 alkali hydroxide Inorganic materials 0.000 description 1
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 1
- 150000008041 alkali metal carbonates Chemical class 0.000 description 1
- VXAUWWUXCIMFIM-UHFFFAOYSA-M aluminum;oxygen(2-);hydroxide Chemical compound [OH-].[O-2].[Al+3] VXAUWWUXCIMFIM-UHFFFAOYSA-M 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- 239000011609 ammonium molybdate Substances 0.000 description 1
- APUPEJJSWDHEBO-UHFFFAOYSA-P ammonium molybdate Chemical compound [NH4+].[NH4+].[O-][Mo]([O-])(=O)=O APUPEJJSWDHEBO-UHFFFAOYSA-P 0.000 description 1
- 235000018660 ammonium molybdate Nutrition 0.000 description 1
- 229940010552 ammonium molybdate Drugs 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 150000001448 anilines Chemical class 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- UBAZGMLMVVQSCD-UHFFFAOYSA-N carbon dioxide;molecular oxygen Chemical compound O=O.O=C=O UBAZGMLMVVQSCD-UHFFFAOYSA-N 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000000571 coke Substances 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 230000009849 deactivation Effects 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- 229910052734 helium Inorganic materials 0.000 description 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 1
- 150000004687 hexahydrates Chemical class 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 238000007654 immersion Methods 0.000 description 1
- 230000003100 immobilizing effect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 238000004898 kneading Methods 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 229910052748 manganese Inorganic materials 0.000 description 1
- WSFSSNUMVMOOMR-NJFSPNSNSA-N methanone Chemical compound O=[14CH2] WSFSSNUMVMOOMR-NJFSPNSNSA-N 0.000 description 1
- 229910000476 molybdenum oxide Inorganic materials 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229940078494 nickel acetate Drugs 0.000 description 1
- QMMRZOWCJAIUJA-UHFFFAOYSA-L nickel dichloride Chemical compound Cl[Ni]Cl QMMRZOWCJAIUJA-UHFFFAOYSA-L 0.000 description 1
- 229910000480 nickel oxide Inorganic materials 0.000 description 1
- LGQLOGILCSXPEA-UHFFFAOYSA-L nickel sulfate Chemical compound [Ni+2].[O-]S([O-])(=O)=O LGQLOGILCSXPEA-UHFFFAOYSA-L 0.000 description 1
- 229910000008 nickel(II) carbonate Inorganic materials 0.000 description 1
- 229910000363 nickel(II) sulfate Inorganic materials 0.000 description 1
- ZULUUIKRFGGGTL-UHFFFAOYSA-L nickel(ii) carbonate Chemical compound [Ni+2].[O-]C([O-])=O ZULUUIKRFGGGTL-UHFFFAOYSA-L 0.000 description 1
- PQQKPALAQIIWST-UHFFFAOYSA-N oxomolybdenum Chemical compound [Mo]=O PQQKPALAQIIWST-UHFFFAOYSA-N 0.000 description 1
- GNRSAWUEBMWBQH-UHFFFAOYSA-N oxonickel Chemical compound [Ni]=O GNRSAWUEBMWBQH-UHFFFAOYSA-N 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- 238000005504 petroleum refining Methods 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 239000002574 poison Substances 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 235000019353 potassium silicate Nutrition 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 239000010453 quartz Substances 0.000 description 1
- 238000011946 reduction process Methods 0.000 description 1
- GTCKPGDAPXUISX-UHFFFAOYSA-N ruthenium(3+);trinitrate Chemical compound [Ru+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O GTCKPGDAPXUISX-UHFFFAOYSA-N 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 238000007873 sieving Methods 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 238000005549 size reduction Methods 0.000 description 1
- 235000019795 sodium metasilicate Nutrition 0.000 description 1
- 229910052911 sodium silicate Inorganic materials 0.000 description 1
- 238000003980 solgel method Methods 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000002344 surface layer Substances 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 229910052723 transition metal Inorganic materials 0.000 description 1
- 150000003624 transition metals Chemical class 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- 239000004246 zinc acetate Substances 0.000 description 1
- 239000011667 zinc carbonate Substances 0.000 description 1
- 235000004416 zinc carbonate Nutrition 0.000 description 1
- 229910000010 zinc carbonate Inorganic materials 0.000 description 1
- 229910052726 zirconium Inorganic materials 0.000 description 1
Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/584—Recycling of catalysts
Description
本発明は、芳香族化合物の水素化触媒及び当該触媒を用いる水素化方法に関する。さらに詳しくは、ニッケル、モリブデン及びルテニウムを含有する芳香族化合物の水素化触媒及び当該触媒を用いる水素化方法に関する。 The present invention relates to an aromatic compound hydrogenation catalyst and a hydrogenation method using the catalyst. More specifically, the present invention relates to a hydrogenation catalyst for an aromatic compound containing nickel, molybdenum and ruthenium, and a hydrogenation method using the catalyst.
芳香族化合物の水素化反応は、石油精製における灯油留分や軽油留分の脱硫工程などで実際に進行している反応である。また、化成品や溶剤を製造する技術としても古くから行われている。 The hydrogenation reaction of an aromatic compound is a reaction that actually proceeds in the desulfurization process of a kerosene fraction and a light oil fraction in petroleum refining. Moreover, it has been performed for a long time as a technique for producing chemical products and solvents.
従来、芳香族化合物の水素化触媒としては、PtやPdなどの貴金属やNiやCuなどの遷移金属が活性金属種として用いられ、これらの金属種をアルミナやシリカ、ゼオライトといった各種担体に担持した触媒が主に使用されている。
これらの触媒を用いて、水素化反応を行う場合は、固定床反応器などが主に用いられ、その反応条件は、水素雰囲気下、反応温度100〜400℃、反応圧力0.1〜4MPaで行われる(例えば、特許文献1及び2を参照)。
Conventionally, as hydrogenation catalysts for aromatic compounds, noble metals such as Pt and Pd and transition metals such as Ni and Cu are used as active metal species, and these metal species are supported on various carriers such as alumina, silica, and zeolite. Catalysts are mainly used.
When performing a hydrogenation reaction using these catalysts, a fixed bed reactor or the like is mainly used. The reaction conditions are a hydrogen atmosphere, a reaction temperature of 100 to 400 ° C., and a reaction pressure of 0.1 to 4 MPa. (For example, see Patent Documents 1 and 2).
一方で、水素不存在下で、炭化水素中の硫黄分を効率よく除去し得る脱硫剤として、ニッケル、モリブデン及びルテニウムを含有する脱硫剤が報告されている(特許文献3及び4参照。)。 On the other hand, desulfurization agents containing nickel, molybdenum and ruthenium have been reported as desulfurization agents that can efficiently remove sulfur in hydrocarbons in the absence of hydrogen (see Patent Documents 3 and 4).
芳香族化合物の水素化反応をより効率的、低コストで行うために、触媒活性の向上(生産性向上、触媒コスト低減、装置サイズの縮小等)、反応条件の温和化(低温化、低圧化による装置コスト、運転動力低減)、安価な原料水素の利用が求められている。 In order to carry out the hydrogenation reaction of aromatic compounds more efficiently and at a low cost, the catalyst activity is improved (productivity improvement, catalyst cost reduction, equipment size reduction, etc.), and the reaction conditions are reduced (lower temperature, lower pressure). Equipment cost and operating power reduction), and the use of inexpensive raw material hydrogen is demanded.
この中で、安価な原料水素としては、例えば製油所水素化精製装置から排出される副生水素や、製鉄所のコークス炉排ガス中の水素が挙げられる。しかしながら、これらのガスは全圧自体が低く、かつ水素以外の低級炭化水素ガスなどを含む、いわゆる水素分圧が低いガスである。加えて、硫化水素などの硫黄化合物を含むことが多く、低品位な水素である。一般的に、水素分圧が低い場合には水素化反応の速度が低下する。また、硫化水素などの硫黄化合物は、水素化触媒を被毒し活性劣化の原因となる。
以上のことから、低温、低水素分圧の温和な条件であり、かつ硫黄化合物存在下においても高活性な水素化触媒が求められている。
Among these, inexpensive raw material hydrogen includes, for example, by-product hydrogen discharged from a refinery hydrotreating apparatus and hydrogen in a coke oven exhaust gas from an ironworks. However, these gases are gases having a low total pressure per se and a low so-called hydrogen partial pressure including lower hydrocarbon gases other than hydrogen. In addition, it often contains sulfur compounds such as hydrogen sulfide, and is low-grade hydrogen. Generally, when the hydrogen partial pressure is low, the speed of the hydrogenation reaction decreases. In addition, sulfur compounds such as hydrogen sulfide poison the hydrogenation catalyst and cause activity deterioration.
In view of the above, there is a demand for a hydrogenation catalyst that has mild conditions of low temperature and low hydrogen partial pressure and that is highly active even in the presence of a sulfur compound.
このような状況下、本発明らは鋭意検討した結果、低い反応温度、低い水素分圧、さらに硫化水素が共存する条件であっても、効率的に芳香族化合物を水素化することができる触媒を見出し、本発明を完成するに至った。 Under such circumstances, the present inventors have intensively studied, and as a result, a catalyst capable of efficiently hydrogenating an aromatic compound even under conditions where a low reaction temperature, a low hydrogen partial pressure, and hydrogen sulfide coexist. As a result, the present invention has been completed.
すなわち、本発明は、下記の芳香族化合物の水素化触媒及び芳香族化合物の水素化方法に関するものである。
(1) ニッケルを酸化物(NiO)換算で50〜95質量%、モリブデンを酸化物(MoO3)換算で0.5〜25質量%、ルテニウムを酸化物(RuO2)換算で0.1〜12質量%、及び無機酸化物を含有し、NiOの結晶子径が3nm以下であり、かつ比表面積が150〜600m2/gであることを特徴とする芳香族化合物の水素化触媒。
(2) 前記(1)の芳香族化合物の水素化触媒を用い、反応温度50〜400℃、水素分圧0.05〜3MPa、[水素(mol)]/[原料芳香族化合物(mol)]比1〜15、液空間速度0.01〜10h−1の条件下で、芳香族化合物を水素化することを特徴とする芳香族化合物の水素化方法。
That is, the present invention relates to the following aromatic compound hydrogenation catalyst and aromatic compound hydrogenation method.
(1) Nickel is 50 to 95 mass% in terms of oxide (NiO), molybdenum is 0.5 to 25 mass% in terms of oxide (MoO 3 ), and ruthenium is 0.1 to 0.1 in terms of oxide (RuO 2 ). A hydrogenation catalyst for an aromatic compound, comprising 12% by mass and an inorganic oxide, having a crystallite diameter of NiO of 3 nm or less and a specific surface area of 150 to 600 m 2 / g.
(2) Using the aromatic compound hydrogenation catalyst of (1) above, reaction temperature 50 to 400 ° C., hydrogen partial pressure 0.05 to 3 MPa, [hydrogen (mol)] / [raw material aromatic compound (mol)] A method for hydrogenating an aromatic compound, wherein the aromatic compound is hydrogenated under conditions of a ratio of 1 to 15 and a liquid space velocity of 0.01 to 10 h- 1 .
本発明の芳香族化合物の水素化触媒は、特定の組成や物性を有することにより、高純度の水素雰囲気下はもとより、水素分圧が低く、さらに硫化水素等の不純物が含まれている条件においても、低い温度で、ベンゼン、トルエン、ナフタレン等の芳香族化合物を効率的に水素化することができる。 The aromatic compound hydrogenation catalyst of the present invention has a specific composition and physical properties, so that the hydrogen partial pressure is low and the impurities such as hydrogen sulfide are contained in the high purity hydrogen atmosphere. However, aromatic compounds such as benzene, toluene, and naphthalene can be efficiently hydrogenated at a low temperature.
<芳香族化合物の水素化触媒の組成>
本発明の芳香族化合物の水素化触媒(以下、「本発明の水素化触媒」ということがある。)は、ニッケルを酸化物(NiO)換算で50〜95質量%、モリブデンを酸化物(MoO3)換算で0.5〜25質量%、ルテニウムを酸化物(RuO2)換算で0.1〜12質量%、及び無機酸化物を含有し、NiOの結晶子径が3nm以下であり、かつ比表面積が150〜600m2/gであることを特徴とする。本発明の水素化触媒は、ニッケル、モリブデン、及びルテニウムを特定の組成比で含有し、かつ特定の結晶子径や比表面積であることにより、硫化水素等の不純物が含まれている場合であっても、温和な条件で効率よく、芳香族化合物を水素化することができる。
<Composition of aromatic hydrogenation catalyst>
The aromatic compound hydrogenation catalyst of the present invention (hereinafter sometimes referred to as “the hydrogenation catalyst of the present invention”) is 50 to 95% by mass of nickel in terms of oxide (NiO), and molybdenum as an oxide (MoO). 3 ) 0.5 to 25% by mass in terms of conversion, 0.1 to 12% by mass of ruthenium in terms of oxide (RuO 2 ), and an inorganic oxide, the crystallite diameter of NiO being 3 nm or less, and The specific surface area is 150 to 600 m 2 / g. The hydrogenation catalyst of the present invention contains nickel, molybdenum, and ruthenium in a specific composition ratio, and has a specific crystallite diameter and specific surface area, thereby containing impurities such as hydrogen sulfide. However, the aromatic compound can be efficiently hydrogenated under mild conditions.
本発明の水素化触媒におけるニッケルの含有量は、酸化物(NiO)換算で50〜95質量%、好ましくは60〜90質量%、より好ましくは70〜90質量%である。ニッケル酸化物量が50質量%以上であれば所望の水素化活性が発現されるため好ましく、95質量%以下であれば、水素化活性が飽和せず、またNi同士の凝集による水素化活性の低下が生じにくいため好ましい。 Content of nickel in the hydrogenation catalyst of this invention is 50-95 mass% in conversion of an oxide (NiO), Preferably it is 60-90 mass%, More preferably, it is 70-90 mass%. If the amount of nickel oxide is 50% by mass or more, a desired hydrogenation activity is exhibited, and if it is 95% by mass or less, the hydrogenation activity is not saturated, and the hydrogenation activity is reduced due to aggregation of Ni. Is preferred because
本発明の水素化触媒におけるモリブデンの含有量は、酸化物(MoO3)換算で0.5〜25質量%、好ましくは0.5〜20質量%、より好ましくは1〜10質量%である。モリブデン酸化物量が0.5質量%以上であれば所望の水素化活性が発現されるため好ましく、25質量%以下であれば、水素化活性が飽和せず、また水素化活性の低下が生じにくいため好ましい The content of molybdenum in the hydrogenation catalyst of the present invention, 0.5 to 25 wt% of an oxide (MoO 3) in terms of, preferably 0.5 to 20 wt%, more preferably 1 to 10 mass%. If the amount of molybdenum oxide is 0.5% by mass or more, the desired hydrogenation activity is exhibited, which is preferable. If the amount is 25% by mass or less, the hydrogenation activity is not saturated and the reduction in hydrogenation activity is less likely to occur. Preferred
本発明の水素化触媒におけるルテニウムの含有量は、酸化物(RuO2)換算で0.1〜12質量%、好ましくは0.1〜10質量%である。ルテニウム酸化物量が0.1質量%以上であれば所望の水素化活性が発現されるため好ましく、12質量%以下であれば、水素化活性が飽和せず、また経済的にも望ましい。 The ruthenium content in the hydrogenation catalyst of the present invention is 0.1 to 12% by mass, preferably 0.1 to 10% by mass in terms of oxide (RuO 2 ). If the amount of ruthenium oxide is 0.1% by mass or more, a desired hydrogenation activity is exhibited, and if it is 12% by mass or less, the hydrogenation activity is not saturated and it is economically desirable.
本発明の水素化触媒においては、上記ニッケル、モリブデン及びルテニウムに加えてさらに、無機酸化物を含有する。無機酸化物を用いると、それに水素化活性金属が分散付着しその分散性が良くなり、水素化活性が向上し、触媒寿命の延長が期待される。また、触媒の成型性や強度も向上するため、無機酸化物を用いることは高活性かつ高耐久性の触媒を得る上で望ましい。 The hydrogenation catalyst of the present invention further contains an inorganic oxide in addition to the above nickel, molybdenum and ruthenium. When an inorganic oxide is used, the hydrogenation active metal is dispersed and attached to the oxide, so that the dispersibility is improved, the hydrogenation activity is improved, and the catalyst life is expected to be extended. Further, since the moldability and strength of the catalyst are also improved, it is desirable to use an inorganic oxide for obtaining a highly active and highly durable catalyst.
無機酸化物の種類は特に限定されないが、Si、Al、B、Mg、Ce、Zr、P、Ti、W、Mnからなる群から選ばれるいずれか1種の元素の酸化物もしくはこれらの混合物、又は2種以上の元素の複合酸化物が好ましい。これらの無機酸化物は結晶構造が無定形であっても結晶性であっても構わない。例えば、SiO2、Al2O3、TiO2、B2O3、MgO、SiO2−Al2O3、Al2O3−B2O3、SiO2−MgO、ゼオライトなどが挙げられる。 The kind of the inorganic oxide is not particularly limited, but an oxide of any one element selected from the group consisting of Si, Al, B, Mg, Ce, Zr, P, Ti, W, Mn, or a mixture thereof, Or the composite oxide of 2 or more types of elements is preferable. These inorganic oxides may have an amorphous or crystalline crystal structure. For example, SiO 2, Al 2 O 3 , TiO 2, B 2 O 3, MgO, SiO 2 -Al 2 O 3, Al 2 O 3 -B 2 O 3, SiO 2 -MgO, zeolite and the like.
各種無機酸化物の中でも、比較的高い比表面積を有し、成形性、圧壊強度、磨耗性に優れる、SiO2、Al2O3、及びSiO2−Al2O3が特に好ましい。このSiO2−Al2O3は、通常、後述する触媒の焼成工程においてSi原料及びAl原料の両者を含む混合物を焼成する過程で生成することができる。 Among various inorganic oxides, SiO 2 , Al 2 O 3 , and SiO 2 —Al 2 O 3 having a relatively high specific surface area and excellent in moldability, crushing strength, and abrasion properties are particularly preferable. This SiO 2 —Al 2 O 3 can usually be produced in the course of firing a mixture containing both the Si raw material and the Al raw material in the catalyst firing step described later.
無機酸化物成分含有量については、特に制限はなく、各種条件において適宜選定すればよいが、通常は触媒全体に対して好ましくは0.5〜49.4質量%、より好ましくは0.5〜40質量%、さらに好ましくは0.5〜30質量%の範囲であればよい。含有量が0.5質量%以上であれば、無機酸化物成分としての効果が十分に発揮され、また49.4質量%以下であれば、水素化活性成分の低下による脱硫性能の低下が防ぐことができ、好ましい。 The content of the inorganic oxide component is not particularly limited and may be appropriately selected under various conditions. Usually, it is preferably 0.5 to 49.4% by mass, more preferably 0.5 to 4% based on the entire catalyst. It may be in the range of 40% by mass, more preferably 0.5-30% by mass. When the content is 0.5% by mass or more, the effect as the inorganic oxide component is sufficiently exhibited, and when it is 49.4% by mass or less, the desulfurization performance is prevented from being lowered due to the reduction of the hydrogenation active component. Can be preferred.
本発明の水素化触媒の含有成分の一つであるニッケルの酸化物(NiO)状態における結晶子径は、3nm以下、好ましくは2.6nm以下、さらに好ましくは2.4nm以下である。一般に活性点の結晶子サイズが小さいほど表面積が増加し、活性が高くなる。NiOの結晶子径が3nm以下であれば、十分な水素化活性が発揮され好ましい。 The crystallite diameter in the oxide (NiO) state of nickel, which is one of the components of the hydrogenation catalyst of the present invention, is 3 nm or less, preferably 2.6 nm or less, more preferably 2.4 nm or less. Generally, the smaller the crystallite size at the active site, the greater the surface area and the higher the activity. If the crystallite diameter of NiO is 3 nm or less, sufficient hydrogenation activity is exhibited, which is preferable.
また、本発明の水素化触媒の比表面積は、還元処理前の状態で150〜600m2/gであり、180〜500m2/gであることが好ましい。比表面積が150m2/g以上であれば、水素化のための活性点の数が多くなり、十分な水素化活性が得られて好ましい。また、比表面積が600m2/g以下であれば、相対的に平均細孔径が大きくなり、十分な水素化活性が得られて好ましい。 Moreover, the specific surface area of the hydrogenation catalyst of this invention is 150-600 m < 2 > / g in the state before a reduction process, and it is preferable that it is 180-500 m < 2 > / g. A specific surface area of 150 m 2 / g or more is preferable because the number of active sites for hydrogenation increases and sufficient hydrogenation activity is obtained. Moreover, if a specific surface area is 600 m < 2 > / g or less, an average pore diameter becomes comparatively large and sufficient hydrogenation activity is obtained and it is preferable.
本発明の水素化触媒の形状については特に規定されず、成型体(押出し円柱、タブレット円柱、球など)、メッシュで篩い分けられた粒状体、粉末などいずれの状態でもかまわないが、取り扱いの簡便さを考えると、成型体又はメッシュで篩い分けられた粒状体が好ましい。触媒の形状を成型体あるいはメッシュで篩い分けられた粒状体にするためには、無機酸化物を用いることが望ましい。また、触媒の大きさは、成型体、メッシュで篩い分けられた粒状体に関らず特に限定されないが、通常直径、あるいは長さが0.1〜10mm、より好ましくは0.1〜5mmであることが好ましい The shape of the hydrogenation catalyst of the present invention is not particularly specified, and may be in any state such as a molded body (extruded cylinder, tablet cylinder, sphere, etc.), a granular body sieved with a mesh, or a powder. In view of the above, a granular body that is sieved with a molded body or a mesh is preferable. In order to make the shape of the catalyst into a granulated body or a granular body screened with a mesh, it is desirable to use an inorganic oxide. Further, the size of the catalyst is not particularly limited regardless of the size of the molded body or the granular material sieved with a mesh, but usually the diameter or length is 0.1 to 10 mm, more preferably 0.1 to 5 mm. Preferably
<芳香族化合物の水素化触媒の製造方法>
触媒の製造方法については特に規定されず、任意の方法で適宜調製することができる。例えば、無機酸化物を用いて、含浸法、混練法、共沈法、ゾルゲル法、平衡吸着法などにより製造することができ、ニッケル、モリブデン及びルテニウムを有効的に機能させるためには含浸法及び共沈法が好ましい。
ニッケル成分の添加には、1回の操作による担持量をより多くし得るため、含浸法よりも共沈法がより好ましい。ルテニウム成分の添加には、高い活性が得られることから含浸法がより好ましい。
<Method for Producing Aromatic Compound Hydrogenation Catalyst>
The method for producing the catalyst is not particularly defined and can be appropriately prepared by any method. For example, using an inorganic oxide, it can be produced by an impregnation method, a kneading method, a coprecipitation method, a sol-gel method, an equilibrium adsorption method, etc. In order to effectively function nickel, molybdenum and ruthenium, A coprecipitation method is preferred.
For the addition of the nickel component, the coprecipitation method is more preferable than the impregnation method because the loading amount by one operation can be increased. For the addition of the ruthenium component, an impregnation method is more preferable because high activity is obtained.
以下に本発明の水素化触媒の好適な製造方法について具体的に説明するが、本発明の水素化触媒の製造方法はこれに限定されるものではない Although the suitable manufacturing method of the hydrogenation catalyst of this invention is demonstrated concretely below, the manufacturing method of the hydrogenation catalyst of this invention is not limited to this.
〔Ni、Mo共沈+Ru含浸(1)〕
好適な触媒の製造方法の第一の方法について説明する。
この方法では、まず、ニッケル原料を含む酸性水溶液と、モリブデン原料を含む塩基性水溶液を別個に調製する。無機酸化物原料は、酸性水溶液又は塩基性水溶液のいずれにも添加することができる。2種以上の無機酸化物原料を使用する場合は、無機酸化物原料を両方の水溶液に添加してもよい。
[Ni, Mo coprecipitation + Ru impregnation (1)]
A first method for producing a suitable catalyst will be described.
In this method, first, an acidic aqueous solution containing a nickel raw material and a basic aqueous solution containing a molybdenum raw material are separately prepared. The inorganic oxide raw material can be added to either an acidic aqueous solution or a basic aqueous solution. When using 2 or more types of inorganic oxide raw materials, you may add an inorganic oxide raw material to both aqueous solution.
例えば、無機酸化物としてSiO2及びAl2O3を含む触媒を製造する場合、ニッケル原料及びアルミニウム原料を含む酸性水溶液と、モリブデン原料、Si原料及び無機塩基を含む塩基性水溶液をそれぞれ調製する。また、無機酸化物としてSiO2のみを含む触媒を製造する場合は、例えば、ニッケル原料を含む酸性水溶液と、モリブデン原料、Si原料及び無機塩基を含む塩基性水溶液をそれぞれ調製する。 For example, when producing a catalyst containing SiO 2 and Al 2 O 3 as inorganic oxides, an acidic aqueous solution containing a nickel raw material and an aluminum raw material and a basic aqueous solution containing a molybdenum raw material, a Si raw material and an inorganic base are prepared. In the production of the catalyst containing only SiO 2 as the inorganic oxide, for example, to prepare an acidic aqueous solution containing nickel material, molybdenum material, a basic aqueous solution containing a Si source and inorganic bases, respectively.
ニッケル原料としては特に限定されないが、硝酸ニッケル、硫酸ニッケル、塩化ニッケル、酢酸ニッケルなどの水溶性ニッケル金属塩及びその水和物が好適に使用できる。モリブデン原料としては特に限定されないが、モリブデン酸アンモニウム、モリブドリン酸などの水溶性モリブデン金属塩及びその水和物が好適に使用できる。これらのニッケル原料やモリブデン原料は、それぞれ単独で用いても、二種以上を組み合わせて用いてもよい。
また、アルミニウム原料としては、特に限定されないが、ベーマイト、擬ベーマイト、γアルミナ、βアルミナなどが好ましい。これらは粉体状、あるいはゾルの形態で用いることができ、一種用いてもよく、二種以上を組み合わせて用いてもよい。
上記ニッケル原料及びアルミニウム原料を含む酸性水溶液は、塩酸、硫酸、硝酸などの酸によって調製することが好ましい。
Although it does not specifically limit as a nickel raw material, Water-soluble nickel metal salts, such as nickel nitrate, nickel sulfate, nickel chloride, nickel acetate, and its hydrate can be used conveniently. Although it does not specifically limit as a molybdenum raw material, Water-soluble molybdenum metal salts, such as ammonium molybdate and molybdophosphoric acid, and its hydrate can be used conveniently. These nickel raw materials and molybdenum raw materials may be used alone or in combination of two or more.
Further, the aluminum raw material is not particularly limited, but boehmite, pseudoboehmite, γ alumina, β alumina and the like are preferable. These can be used in the form of powder or sol, and may be used alone or in combination of two or more.
The acidic aqueous solution containing the nickel raw material and the aluminum raw material is preferably prepared with an acid such as hydrochloric acid, sulfuric acid or nitric acid.
さらに、Si原料としては、特に限定されないが、シリカや水ガラス、メタケイ酸ソーダ、珪藻土、メソポーラスシリカ(MCM41)などが好ましい。
また、無機塩基としては、アルカリ金属の炭酸塩や水酸化物などが好ましく、例えば炭酸ナトリウム、炭酸水素ナトリウム、炭酸カリウム、水酸化ナトリウム、水酸化カリウムなどが挙げられ、特に炭酸ナトリウムが好適である。
この無機塩基の使用量は、次の工程において、酸性水溶液と塩基性水溶液との混合液が実質上中性から塩基性になるように選ぶのが有利である。Si原料及び無機塩基は、単独で用いてもよく、二種以上を組み合わせて用いてよい。
なお、アルミニウム原料やSi原料は、触媒に無機酸化物成分を加えるために用いるものである。これは、後記する第二、第三の方法でも同様である。
Furthermore, the Si raw material is not particularly limited, but silica, water glass, sodium metasilicate, diatomaceous earth, mesoporous silica (MCM41) and the like are preferable.
The inorganic base is preferably an alkali metal carbonate or hydroxide, such as sodium carbonate, sodium hydrogen carbonate, potassium carbonate, sodium hydroxide, potassium hydroxide, etc., with sodium carbonate being particularly preferred. .
The amount of the inorganic base used is advantageously selected so that the mixed solution of the acidic aqueous solution and the basic aqueous solution is substantially neutral to basic in the next step. Si raw material and inorganic base may be used independently and may be used in combination of 2 or more types.
The aluminum raw material and Si raw material are used for adding an inorganic oxide component to the catalyst. The same applies to the second and third methods described later.
次に、調製した各水溶液を、それぞれ25〜90℃に加温し、両者を混合する。そして、液温を25〜90℃に保持しながら0.5〜3時間程度撹拌し、反応を完結させる。酸性水溶液と塩基性水溶液の混合後のpHは6以上であることが好ましく、6〜11の範囲であることがより好ましく、6.5〜10の範囲であることがさらに好ましい。pHが6以上であれば、ニッケル、モリブデンが効率よく沈殿するため好ましい。また、pHが11以下であることが、無機塩基の使用量を節減することができて、製造コスト面から好ましい。 Next, each prepared aqueous solution is heated at 25-90 degreeC, respectively, and both are mixed. And it stirs about 0.5 to 3 hours, hold | maintaining liquid temperature at 25-90 degreeC, and completes reaction. The pH after mixing the acidic aqueous solution and the basic aqueous solution is preferably 6 or more, more preferably in the range of 6 to 11, and still more preferably in the range of 6.5 to 10. A pH of 6 or more is preferable because nickel and molybdenum precipitate efficiently. Moreover, it is preferable from the surface of manufacturing cost that the usage-amount of an inorganic base can be saved that pH is 11 or less.
反応させた水溶液の沈殿物をろ過、水洗後、固形物を公知の方法により50〜150℃程度の温度で乾燥処理する。このようにして得られた乾燥処理物を、好ましくは200〜450℃の範囲の温度において1〜5時間焼成する。 After the precipitate of the reacted aqueous solution is filtered and washed with water, the solid is dried at a temperature of about 50 to 150 ° C. by a known method. The dried product thus obtained is preferably fired at a temperature in the range of 200 to 450 ° C. for 1 to 5 hours.
そして、上記のようにして得られた焼成体に、ルテニウム原料をイオン交換水に溶解した水溶液を含浸担持し、乾燥後、アルカリ性水溶液でルテニウム成分を不溶・固定化し、ろ過・水洗・乾燥する。 Then, the fired body obtained as described above is impregnated and supported with an aqueous solution in which a ruthenium raw material is dissolved in ion-exchanged water.
ルテニウム原料としては、特に限定されないが、塩化ルテニウム、硝酸ルテニウムなどの水溶性ルテニウム金属塩及びその水和物が好適に使用できる。
また、ルテニウム成分を固定化するのに用いるアルカリ性水溶液としては、特に限定されないが、アンモニア水、炭酸アンモニウム、水酸化ナトリウム、水酸化カリウム、炭酸ナトリウム、炭酸水素ナトリウム等の水溶液を使用できる。
Although it does not specifically limit as a ruthenium raw material, Water-soluble ruthenium metal salts, such as ruthenium chloride and ruthenium nitrate, and its hydrate can be used conveniently.
Moreover, the alkaline aqueous solution used for immobilizing the ruthenium component is not particularly limited, but aqueous solutions of ammonia water, ammonium carbonate, sodium hydroxide, potassium hydroxide, sodium carbonate, sodium hydrogen carbonate and the like can be used.
ルテニウム成分の含浸、固定化物を乾燥させる際の温度としては120℃以下であることが好ましい。120℃以下であれば、酸化ルテニウムの生成を抑制でき、後の還元工程を効率化することができる。また、乾燥方法は特に限定されず、常圧での乾燥、減圧での乾燥、空気中での乾燥、不活性ガス雰囲気下での乾燥を任意に選ぶことができる。 The temperature at which the ruthenium component is impregnated and the immobilized product is dried is preferably 120 ° C. or lower. If it is 120 degrees C or less, the production | generation of ruthenium oxide can be suppressed and a subsequent reduction | restoration process can be made efficient. Further, the drying method is not particularly limited, and any of drying under normal pressure, drying under reduced pressure, drying in air, and drying under an inert gas atmosphere can be arbitrarily selected.
〔Ni、Mo共沈+Ru含浸(2)〕
次に、好適な触媒の製造方法の第二の方法について説明する。この方法では、まず、ニッケル原料及びモリブデン原料を含む酸性水溶液と、無機酸化物原料を含む塩基性水溶液を別個に調製する。2種以上の無機酸化物原料を使用する場合は、無機酸化物原料を酸性水溶液にも添加することができる。
[Ni, Mo coprecipitation + Ru impregnation (2)]
Next, a second method for producing a suitable catalyst will be described. In this method, first, an acidic aqueous solution containing a nickel raw material and a molybdenum raw material and a basic aqueous solution containing an inorganic oxide raw material are separately prepared. When using 2 or more types of inorganic oxide raw materials, an inorganic oxide raw material can also be added to acidic aqueous solution.
例えば、無機酸化物としてSiO2及びAl2O3を含む触媒を製造する場合は、ニッケル原料、モリブデン原料及びアルミニウム原料を含む酸性水溶液と、Si原料及び無機塩基を含む塩基性水溶液をそれぞれ調製する。 For example, when producing a catalyst containing SiO 2 and Al 2 O 3 as inorganic oxides, an acidic aqueous solution containing nickel raw material, molybdenum raw material and aluminum raw material, and a basic aqueous solution containing Si raw material and inorganic base are prepared. .
ニッケル原料、モリブデン原料、アルミニウム原料、Si原料、無機塩基としては、第一の方法と同様のものを用いることができる。また、ニッケル原料、モリブデン原料、及びアルミニウム原料を含む酸性水溶液は、塩酸、硫酸、硝酸などの酸によって調製することが好ましい。また、酸性水溶液と塩基性水溶液の混合後のpHは、第一の方法で述べたpHと同様の範囲とすることが好ましい。
調製した酸性水溶液と塩基性水溶液は、第一の方法と同様の条件で、混合して反応を完結させ、生成した沈殿物は、ろ過、水洗後、乾燥処理し、乾燥処理物を焼成する。
As the nickel raw material, the molybdenum raw material, the aluminum raw material, the Si raw material, and the inorganic base, those similar to the first method can be used. Moreover, it is preferable to prepare acidic aqueous solution containing nickel raw material, molybdenum raw material, and aluminum raw material with acids, such as hydrochloric acid, a sulfuric acid, and nitric acid. The pH after mixing the acidic aqueous solution and the basic aqueous solution is preferably in the same range as the pH described in the first method.
The prepared acidic aqueous solution and basic aqueous solution are mixed to complete the reaction under the same conditions as in the first method. The produced precipitate is filtered, washed with water, dried, and the dried product is fired.
さらに、こうして得られた焼成体に、第一の方法と同様に、ルテニウム原料を溶解した水溶液を含浸担持し、乾燥後、アルカリ性水溶液でルテニウム成分を不溶・固定化し、ろ過・水洗・乾燥する。この際、ルテニウム原料、アルカリ性水溶液としては、第一の方法と同様のものを用いることができる。次いで、得られたルテニウム成分の含浸、固定化物を、第一の方法と同様に、乾燥させる。 Further, the fired body thus obtained is impregnated and supported with an aqueous solution in which a ruthenium raw material is dissolved, and after drying, the ruthenium component is insoluble and fixed with an alkaline aqueous solution, followed by filtration, washing with water and drying. At this time, as the ruthenium raw material and the alkaline aqueous solution, those similar to the first method can be used. Next, the obtained impregnated and immobilized ruthenium component is dried in the same manner as in the first method.
〔Ni共沈+Mo、Ru含浸(3)〕
次に、好適な触媒の製造方法の第三の方法について説明する。この方法では、まず、ニッケル原料を含む酸性水溶液と、無機酸化物原料を含む塩基性水溶液を別個に調製する。2種以上の無機酸化物原料を使用する場合は、無機酸化物原料を酸性水溶液にも添加することができる。
[Ni coprecipitation + Mo, Ru impregnation (3)]
Next, the 3rd method of the manufacturing method of a suitable catalyst is demonstrated. In this method, first, an acidic aqueous solution containing a nickel raw material and a basic aqueous solution containing an inorganic oxide raw material are separately prepared. When using 2 or more types of inorganic oxide raw materials, an inorganic oxide raw material can also be added to acidic aqueous solution.
例えば、無機酸化物としてSiO2及びAl2O3を含む触媒を製造する場合は、ニッケル原料及びアルミニウム原料を含む酸性水溶液と、Si原料及び無機塩基を含む塩基性水溶液をそれぞれ調製する。 For example, when producing a catalyst containing SiO 2 and Al 2 O 3 as inorganic oxides, an acidic aqueous solution containing a nickel raw material and an aluminum raw material and a basic aqueous solution containing a Si raw material and an inorganic base are prepared.
ニッケル原料、アルミニウム原料、Si原料、無機塩基としては、第一の方法と同様のものを用いることができる。また、ニッケル原料及びアルミニウム原料を含む酸性水溶液は、塩酸、硫酸、硝酸などの酸によって調製することが好ましい。また、酸性水溶液と塩基性水溶液の混合後のpHは、第一の方法で述べたpHと同様の範囲とすることが好ましい。
調製した酸性水溶液と塩基性水溶液は、第一の方法と同様の条件で、混合して反応を完結させ、生成した沈殿物は、ろ過、水洗後、乾燥処理し、乾燥処理物を焼成する。
As the nickel raw material, the aluminum raw material, the Si raw material, and the inorganic base, those similar to the first method can be used. Moreover, it is preferable to prepare acidic aqueous solution containing a nickel raw material and an aluminum raw material with acids, such as hydrochloric acid, a sulfuric acid, and nitric acid. The pH after mixing the acidic aqueous solution and the basic aqueous solution is preferably in the same range as the pH described in the first method.
The prepared acidic aqueous solution and basic aqueous solution are mixed to complete the reaction under the same conditions as in the first method. The produced precipitate is filtered, washed with water, dried, and the dried product is fired.
得られた焼成物に、モリブデン原料をイオン交換水に溶解した水溶液を含浸担持させる。モリブデン原料がイオン交換水で溶解しない場合は少量のアンモニア水を加えても良い。
この際、モリブデン原料としては、第一の方法と同様のものを用いることができる。
得られたモリブデン原料水溶液の含浸物を、公知の方法により50〜150℃程度の温
度で乾燥処理し、その乾燥処理物を、好ましくは200〜450℃の範囲の温度において1〜5時間焼成する
The obtained fired product is impregnated with an aqueous solution obtained by dissolving a molybdenum raw material in ion-exchanged water. If the molybdenum raw material is not dissolved in ion-exchanged water, a small amount of ammonia water may be added.
At this time, as the molybdenum raw material, the same material as in the first method can be used.
The obtained impregnated product of molybdenum raw material aqueous solution is dried at a temperature of about 50 to 150 ° C. by a known method, and the dried product is preferably fired at a temperature in the range of 200 to 450 ° C. for 1 to 5 hours.
さらに、こうして得られた焼成体に、第一の方法と同様に、ルテニウム原料を溶解した水溶液を含浸担持し、乾燥後、アルカリ性水溶液でルテニウム成分を不溶・固定化し、ろ過・水洗・乾燥する。この際、ルテニウム原料、アルカリ性水溶液としては、第一の方法と同様のものを用いることができる。次いで、得られたルテニウム成分の含浸、固定化物を、第一の方法と同様に、乾燥させる。 Further, the fired body thus obtained is impregnated and supported with an aqueous solution in which a ruthenium raw material is dissolved, and after drying, the ruthenium component is insoluble and fixed with an alkaline aqueous solution, followed by filtration, washing with water and drying. At this time, as the ruthenium raw material and the alkaline aqueous solution, those similar to the first method can be used. Next, the obtained impregnated and immobilized ruthenium component is dried in the same manner as in the first method.
<芳香族化合物の水素化方法>
上記のようにして製造した本発明の水素化触媒は、水素化反応に供す前に、還元処理しておくことが好ましい。これにより、触媒の含有金属が活性化され、芳香族化合物を水素化しやすい状態となる。
<Method of hydrogenating aromatic compounds>
The hydrogenation catalyst of the present invention produced as described above is preferably subjected to a reduction treatment before being subjected to a hydrogenation reaction. Thereby, the metal contained in the catalyst is activated and the aromatic compound is easily hydrogenated.
還元方法は、水素、CO等による気相還元、ホルムアルデヒド、エタノール等を用いた液相還元等の公知の方法を用いることが可能であるが、気相による水素化還元が好ましく、この場合、水素雰囲気で200〜500℃で行うことが好ましく、300〜450℃の温度で行うことがより好ましい。 As the reduction method, a known method such as gas phase reduction using hydrogen, CO, etc., liquid phase reduction using formaldehyde, ethanol, or the like can be used, but hydrogen reduction by gas phase is preferable, and in this case, hydrogen reduction It is preferable to carry out at 200-500 degreeC by atmosphere, and it is more preferable to carry out at the temperature of 300-450 degreeC.
なお、水素化還元処理は、芳香族化合物の水素化反応の反応器内(オンサイト)で行ってもよく、事前の水素化還元処理装置(オフサイト)で行ってもかまわないが、水素化反応の温度よりも還元処理温度の方が高いため、使用する反応器の設計温度が高くなることなどを考慮すると、オフサイト還元が好ましい。さらにオフサイト水素化還元処理においては、還元処理後に触媒を空気中に取り出すと還元されたNi金属やRu金属の急激な酸化による発熱が起き、NiやRu、その他の含有成分が凝集し、表面積低下の恐れがある。よって、還元処理後、空気中に抜き出す前に、微量の酸素や二酸化炭素などを用いる金属表面の不動態化処理を施すことがさらに好ましい。上記不動態化処理では金属Niの表層の一部をNi酸化物(NiO)に酸化することで空気中に抜き出した場合でも、それ以上のNiの酸化が進まず安定に取り扱うことが可能となる。なお、このとき生成するNiOの粒子径は不動態化処理の条件によっては粒子径が多少変動する場合があるが、本発明において規定しているNiOの粒子径は還元、不動態化処理前の状態のものをいう。 The hydroreduction treatment may be performed in the reactor (on-site) of the hydrogenation reaction of the aromatic compound, or may be performed in a prior hydroreduction treatment apparatus (off-site). Since the reduction treatment temperature is higher than the reaction temperature, off-site reduction is preferable in consideration of the fact that the design temperature of the reactor used is increased. Further, in the off-site hydroreduction treatment, when the catalyst is taken out into the air after the reduction treatment, heat is generated due to rapid oxidation of the reduced Ni metal or Ru metal, Ni, Ru, and other components are aggregated, and the surface area There is a risk of decline. Therefore, it is more preferable to passivate the metal surface using a trace amount of oxygen, carbon dioxide, or the like after the reduction treatment and before extraction into the air. In the above passivation treatment, even when a part of the surface layer of metal Ni is oxidized to Ni oxide (NiO) and extracted into the air, further Ni oxidation does not proceed and it can be handled stably. . The particle diameter of NiO produced at this time may vary somewhat depending on the conditions of the passivation treatment, but the particle diameter of NiO defined in the present invention is the same as that before the reduction and passivation treatment. It means the state.
本発明の水素化触媒を用いた芳香族化合物の水素化反応では、通常、反応器に触媒を充填し、水素供給下で原料の芳香族化合物を触媒と接触させることにより水素化が行われる。
芳香族化合物と触媒とを接触させる方法としては、一般的には、固定床式反応器内に触媒層を形成し、原料の芳香族化合物及び水素を供給し行うことができる。
In the hydrogenation reaction of an aromatic compound using the hydrogenation catalyst of the present invention, the hydrogenation is usually carried out by filling the catalyst into a reactor and bringing the starting aromatic compound into contact with the catalyst under hydrogen supply.
As a method for bringing the aromatic compound and the catalyst into contact, generally, a catalyst layer is formed in a fixed bed reactor, and the raw material aromatic compound and hydrogen can be supplied.
水素化反応の条件としては、水素分圧は0.05〜3MPa、好ましくは、0.1〜1MPaが好ましい。一般に水素分圧が高いほど水素化反応には有利であるが、本発明では低い水素分圧においても効率的に水素化反応を進めることができるため、低品位な水素を有効に利用することができるとともに、昇圧器で全圧を高め水素分圧を高くする必要もなく、設備面や運転動力での投資を軽減することができる。 As conditions for the hydrogenation reaction, the hydrogen partial pressure is 0.05 to 3 MPa, preferably 0.1 to 1 MPa. In general, the higher the hydrogen partial pressure, the more advantageous for the hydrogenation reaction. However, in the present invention, the hydrogenation reaction can be efficiently carried out even at a low hydrogen partial pressure, so that low-grade hydrogen can be used effectively. In addition, it is not necessary to increase the total pressure with the booster to increase the hydrogen partial pressure, thereby reducing investment in facilities and operating power.
また、原料芳香族化合物に対する水素のモル比([水素(mol)]/[原料芳香族化合物(mol)]比)は、1〜15、好ましくは、3〜7である。一般に原料に対する水素の量が多いほど水素化反応には有利であるが、この範囲を保つことにより、効率的に水素化反応を進めることができる。 The molar ratio of hydrogen to the raw material aromatic compound ([hydrogen (mol)] / [raw material aromatic compound (mol)] ratio) is 1 to 15, preferably 3 to 7. Generally, the larger the amount of hydrogen relative to the raw material, the more advantageous for the hydrogenation reaction. However, maintaining this range allows the hydrogenation reaction to proceed efficiently.
用いる水素の純度は、不純物が含まれておらず、純度の高い水素を用いることができるのは言うまでもないが、本発明では、水素純度が低く、また、不純物として硫化水素などの硫黄化合物がある程度含まれていても、効率的に水素化を進めることができる。
水素純度に関しては、用いるガスの全圧にもよるが例えば、20vol%程度以上あれば好適に用いることができる。また、硫化水素は、100(容量)ppm程度までであれば、水素とともに存在していても、水素化活性の急激な失活を引き起こすことはない。
Needless to say, the purity of hydrogen used does not include impurities and high-purity hydrogen can be used. However, in the present invention, the hydrogen purity is low, and impurities such as sulfur compounds such as hydrogen sulfide are used to some extent. Even if it is contained, hydrogenation can proceed efficiently.
Regarding hydrogen purity, although depending on the total pressure of the gas used, for example, about 20 vol% or more, it can be suitably used. Further, hydrogen sulfide does not cause rapid deactivation of hydrogenation activity even if it is present with hydrogen as long as it is up to about 100 (volume) ppm.
また、反応温度は50〜400℃、好ましくは70〜300℃、より好ましくは70〜200℃、更に好ましくは80〜200℃、より更に好ましくは80〜150℃である。本発明では、低温でも水素化が進行するのが特徴であるが、50℃以下では、反応速度が低下し、また、逆に高温すぎる場合には、触媒中のニッケル成分が凝集して活性サイト数が減少し、水素化の性能が低下する恐れがある。
液空間速度(LHSV)は0.01〜10h−1、好ましくは0.1〜3h−1である。
Moreover, reaction temperature is 50-400 degreeC, Preferably it is 70-300 degreeC, More preferably, it is 70-200 degreeC, More preferably, it is 80-200 degreeC, More preferably, it is 80-150 degreeC. The present invention is characterized in that hydrogenation proceeds even at a low temperature. However, at a temperature of 50 ° C. or lower, the reaction rate decreases, and conversely, when the temperature is too high, the nickel component in the catalyst aggregates and becomes active sites. The number may decrease and the hydrogenation performance may be reduced.
Liquid hourly space velocity (LHSV) 0.01~10h -1, preferably 0.1~3h -1.
原料とする芳香族化合物としては、ベンゼン、トルエン、キシレン、フェノール類、チオフェノール類、アニリン類などの単環芳香族化合物やナフタレン、アントラセン、フェナントレンなどの多環芳香族化合物といった、いわゆるベンゼン核を有する炭化水素類やチオフェン、ピリジン、フランなどの複素環芳香族化合物などが挙げられるが、炭化水素類であれば特に限定されるものではなく、例えば、灯油や軽油を原料として、各留分中の芳香族化合物を水素化することも可能である。 The aromatic compounds used as raw materials include so-called benzene nuclei such as monocyclic aromatic compounds such as benzene, toluene, xylene, phenols, thiophenols and anilines, and polycyclic aromatic compounds such as naphthalene, anthracene and phenanthrene. Hydrocarbons and heterocyclic aromatic compounds such as thiophene, pyridine, furan, etc., but are not particularly limited as long as they are hydrocarbons. For example, kerosene or light oil is used as a raw material in each fraction. It is also possible to hydrogenate the aromatic compound.
次に、本発明を実施例により、さらに詳細に説明するが、本発明はこれらの例によって何ら限定されるものではない。
まず、実施例及び比較例における触媒の物性、及びトルエン転化率の機器分析方法を以下に示す。
EXAMPLES Next, although an Example demonstrates this invention further in detail, this invention is not limited at all by these examples.
First, the physical properties of the catalysts and the instrumental analysis methods for toluene conversion in Examples and Comparative Examples are shown below.
<触媒の比表面積測定>
BET(Braunauer−Emmett−Tailor specific surface area)比表面積の測定には、日本ベル社製表面積測定装置(Belsorp Mini)を用いた。試料約200〜300mgを精秤し、これを石英製の試料管に充填し、10-1〜10-3mmHg台に減圧しながら室温から400℃まで1時間かけて昇温し、減圧下、同温度で3時間保持して脱気処理を行った。その後、減圧しながら室温まで降温させ、高純度ヘリウムガスで置換し、脱気後の試料重量を精秤した。この後、液化窒素温度で窒素吸着を行い、比表面積を測定した。
<Measurement of specific surface area of catalyst>
A BET surface area measuring device (Belsorb Mini) was used for measurement of BET (Braunauer-Emmett-Tailor specific surface area) specific surface area. About 200 to 300 mg of the sample is precisely weighed, filled in a quartz sample tube, heated from room temperature to 400 ° C. over 1 hour while reducing the pressure to 10 −1 to 10 −3 mmHg level, The deaeration treatment was performed by maintaining at the same temperature for 3 hours. Thereafter, the temperature was lowered to room temperature while reducing the pressure, the gas was replaced with high purity helium gas, and the weight of the sample after deaeration was precisely weighed. Thereafter, nitrogen adsorption was performed at the liquefied nitrogen temperature, and the specific surface area was measured.
<触媒のX線回折分析方法>
株式会社リガク社製X線回折装置(RINT−2500V)を用いた。測定する試料を粉砕し、試料板に詰め、走査範囲5〜90°、試料回転速度20rpm、発散スリット1°、散乱スリット1°、受光スリット1°、スキャンスピード2°/minでX線回折(線源Cu−Kα線)測定を行った。
NiOの結晶子径はX線回折測定により2θ=62°付近にピークトップを有する回折ピークを検出し、Scherrerの式により算出した。
<X-ray diffraction analysis method of catalyst>
An X-ray diffractometer (RINT-2500V) manufactured by Rigaku Corporation was used. The sample to be measured is pulverized and packed in a sample plate. X-ray diffraction (scanning range 5 to 90 °, sample rotation speed 20 rpm, divergence slit 1 °, scattering slit 1 °, light receiving slit 1 °, scan speed 2 ° / min) (Source Cu—Kα ray) measurement was performed.
The crystallite diameter of NiO was calculated by the Scherrer equation by detecting a diffraction peak having a peak top in the vicinity of 2θ = 62 ° by X-ray diffraction measurement.
<トルエンの水素化反応>
内径15mmの反応管に触媒10mLを充填し、反応温度50〜350℃、LHSV=1.0h−1、水素/トルエン=5mol/mol、全圧0.6MPa、水素分圧0.12MPa、H2S=0〜50ppmの条件で反応を行った。また、生成油のFID−GC分析を行い、面積値よりトルエンの転化率を下記の式で求めた。なお、トルエン水素化生成物としてはメチルシクロヘキサンのみが確認され、それ以外の化合物は検出されなかった。
<Hydrogenation reaction of toluene>
A reaction tube with an inner diameter of 15 mm is filled with 10 mL of catalyst, reaction temperature 50 to 350 ° C., LHSV = 1.0 h −1 , hydrogen / toluene = 5 mol / mol, total pressure 0.6 MPa, hydrogen partial pressure 0.12 MPa, H 2. The reaction was performed under the condition of S = 0 to 50 ppm. Moreover, the FID-GC analysis of the product oil was performed, and the conversion rate of toluene was calculated | required by the following formula from the area value. In addition, only methylcyclohexane was confirmed as a toluene hydrogenation product, and other compounds were not detected.
式:トルエン転化率(%)=[メチルシクロヘキサンの面積値]/([トルエンの面積値]+[メチルシクロヘキサンの面積値])×100 Formula: Toluene conversion rate (%) = [Area value of methylcyclohexane] / ([Area value of toluene] + [Area value of methylcyclohexane]) × 100
[実施例1;Ni、Mo共沈+Ru含浸(1)(第一の方法)]
ベーマイトAP-3(触媒化成工業製)1.24g、1N−HNO3水溶液40mLをイオン交換水1Lに加え、80℃に加温後、Ni(NO3)2・6H2Oを149g加えて調製液Aを得た。別途用意したイオン交換水1LにコロイダルシリカスノーテックスXS(日産化学製)33.9g、炭酸ナトリウム99.4g、(NH4)6Mo7O24・5H2Oを3.0g加え、80℃に加温し、調製液Bを得た。調製液AとBを80℃に保持しながら、B液をA液に瞬時に加えて、1時間攪拌した。その後、イオン交換水を5L用いて洗浄し、ろ過後に空気中120℃で12時間乾燥させた後、400℃で1時間焼成し、得られた焼成物を破砕し、1.0mmと1.4mmの網目を有する篩で篩い分けた。次いで、RuCl3・nH2O(小島化学薬品製、Ru含有量:41mass%、n=1〜3)2.3gをイオン交換水11.4gに溶解させた水溶液に、上記メッシュ破砕したもの30gを1時間浸漬し、該メッシュ破砕したものに該水溶液を含浸担持させ、乾燥後、7N−NH3水150gに1時間漬け、イオン交換水2Lで洗浄、ろ過し、120℃で乾燥し、触媒1を得た。
[Example 1; Ni, Mo coprecipitation + Ru impregnation (1) (first method)]
Boehmite AP-3 (manufactured by Catalysts & Chemicals Industries) 1.24 g, a 1N-HNO 3 solution 40mL was added to ion-exchanged water 1L, after warming to 80 ° C., was added 149g of Ni (NO 3) 2 · 6H 2 O Preparation Liquid A was obtained. Separately prepared ion-exchanged water 1L colloidal silica Snowtex XS (produced by Nissan Chemical) 33.9 g, sodium carbonate 99.4g, (NH 4) 6 Mo 7 O 24 · 5H 2 O was added 3.0 g, in 80 ° C. It heated and the preparation liquid B was obtained. While maintaining the prepared solutions A and B at 80 ° C., the solution B was instantaneously added to the solution A and stirred for 1 hour. Thereafter, it was washed with 5 L of ion-exchanged water, and after filtration, dried in air at 120 ° C. for 12 hours, calcined at 400 ° C. for 1 hour, and the obtained calcined product was crushed, 1.0 mm and 1.4 mm The sieve was screened with a sieve having the following mesh. Next, 30 g of the above-mentioned mesh crushed in an aqueous solution in which 2.3 g of RuCl 3 .nH 2 O (manufactured by Kojima Chemical Co., Ru content: 41 mass%, n = 1 to 3) is dissolved in 11.4 g of ion-exchanged water. 1 hour, impregnating and supporting the aqueous solution on the mesh crushed, dried, soaked in 150 g of 7N-NH 3 water for 1 hour, washed with 2 L of ion-exchanged water, filtered, dried at 120 ° C., catalyst 1 was obtained.
[実施例2;Ni、Mo共沈+Ru含浸(1)(第一の方法)]
1N−HNO3水溶液40mLをイオン交換水1Lに加え、80℃に加温後、Ni(NO3)2・6H2Oを164g加え調製液Aを得た。別途用意したイオン交換水1LにコロイダルシリカスノーテックスXS(日産化学製)26.7g、炭酸ナトリウム99.4g、(NH4)6Mo7O24・5H2Oを1. 2g加え、80℃に加温し、調製液Bを得た。調製液AとBを80℃に保持しながら、B液をA液に瞬時に加えて、1時間攪拌した。その後、イオン交換水を5L用いて洗浄し、ろ過後に空気中120℃で12時間乾燥させた後、400℃で1時間焼成し、得られた焼成物を破砕し、1.0mmと1.4mmの網目を有する篩で篩い分けた。次いで、RuCl3・nH2O(小島化学薬品製、Ru含有量:41mass%、n=1〜3)0.6gをイオン交換水11.4gに溶解させた水溶液に、上記メッシュ破砕したもの30gを1時間浸漬し、該メッシュ破砕したものに該水溶液を含浸担持させ、乾燥後、7N−NH3水150gに1時間漬け、イオン交換水2Lで洗浄、ろ過し、120℃で乾燥し、触媒2を得た。
[Example 2; Ni, Mo coprecipitation + Ru impregnation (1) (first method)]
40 mL of 1N-HNO 3 aqueous solution was added to 1 L of ion exchange water, heated to 80 ° C., and then 164 g of Ni (NO 3 ) 2 .6H 2 O was added to obtain Preparation A. Colloidal silica SNOWTEX XS in deionized water 1L separately prepared (manufactured by Nissan Chemical Industries, Ltd.) 26.7 g, sodium carbonate 99.4 g, a 6 Mo 7 O 24 · 5H 2 O (NH 4) 1. 2g was added and it heated at 80 degreeC and the preparation liquid B was obtained. While maintaining the prepared solutions A and B at 80 ° C., the solution B was instantaneously added to the solution A and stirred for 1 hour. Thereafter, it was washed with 5 L of ion-exchanged water, and after filtration, dried in air at 120 ° C. for 12 hours, calcined at 400 ° C. for 1 hour, and the obtained calcined product was crushed, 1.0 mm and 1.4 mm The sieve was screened with a sieve having the following mesh. Next, 30 g of the above-mentioned mesh crushed in an aqueous solution in which 0.6 g of RuCl 3 .nH 2 O (manufactured by Kojima Chemical Co., Ru content: 41 mass%, n = 1 to 3) is dissolved in 11.4 g of ion-exchanged water. 1 hour, impregnating and supporting the aqueous solution on the mesh crushed, dried, soaked in 150 g of 7N-NH 3 water for 1 hour, washed with 2 L of ion-exchanged water, filtered, dried at 120 ° C., catalyst 2 was obtained.
[実施例3;Ni共沈+Mo、Ru含浸(第三の方法)]
ベーマイトAP-3(触媒化成工業製)1.24gをイオン交換水1Lに加え、80℃に加温後、Ni(NO3)2・6H2Oを149g加え調製液Aを得た。別途用意したイオン交換水1LにコロイダルシリカスノーテックスXS(日産化学製)33.9g、炭酸ナトリウム79.5gを加え、80℃に加温し、調製液Bを得た。調製液AとBを80℃に保持しながら、B液をA液に10分間で加えて、1時間攪拌した。その時の80℃におけるpHは7.9であった。その後、イオン交換水を5L用いて洗浄し、ろ過後に空気中120℃で12時間乾燥させた後、400℃で1時間焼成し、得られた成型物を破砕し、1.0mmと1.4mmの網目を有する篩で篩い分けし、成形体を得た。その内の20gに、(NH4)6Mo7O24・5H2O1.3gを水と7NNH3水を9:1の比で混合した水溶液8gに溶かした水溶液を含浸し、空気中120℃で12時間乾燥、400℃で1時間焼成した。その後、RuCl3・nH2O(小島化学薬品製、Ru含有量41mass% 、n=1〜3)1.5gをイオン交換水7.6gに溶解させた水溶液に、上記焼成品20gを1時間浸漬し、該メッシュ破砕したものに該水溶液を含浸担持させ、乾燥後、7N−NH3水100gに1時間漬け、イオン交換水2Lで洗浄、ろ過し、120℃で乾燥し、触媒3を得た。
[Example 3; Ni coprecipitation + Mo, Ru impregnation (third method)]
Boehmite AP-3 (manufactured by Catalysts & Chemicals Industries) 1.24 g was added to deionized water 1L, after warming to 80 ° C., to obtain a Ni (NO 3) 2 · 6H 2 O and 149g added preparation A. Colloidal silica snowtex XS (Nissan Chemical Co., Ltd.) 33.9 g and sodium carbonate 79.5 g were added to 1 L of ion-exchanged water separately prepared, and heated to 80 ° C. to obtain Preparation B. While maintaining the preparations A and B at 80 ° C., the solution B was added to the solution A over 10 minutes and stirred for 1 hour. At that time, the pH at 80 ° C. was 7.9. Thereafter, it was washed with 5 L of ion-exchanged water, and after filtration, dried in air at 120 ° C. for 12 hours, calcined at 400 ° C. for 1 hour, and the resulting molded product was crushed, 1.0 mm and 1.4 mm The resulting product was sieved with a sieve having a mesh of to obtain a molded product. To 20g of them, (NH 4) 6 Mo 7 O 24 · 5H 2 O1.3g the water and 7NNH 3 water 9: 1 of an aqueous solution dissolved in mixed aqueous 8g ratio impregnated in air 120 ° C. And dried at 400 ° C. for 1 hour. Thereafter, 20 g of the fired product was immersed in an aqueous solution in which 1.5 g of RuCl3 · nH2O (manufactured by Kojima Chemical Co., Ru content 41 mass%, n = 1 to 3) was dissolved in 7.6 g of ion-exchanged water for 1 hour. The aqueous solution was impregnated and supported on the mesh crushed, dried, soaked in 100 g of 7N-NH 3 water for 1 hour, washed with 2 L of ion-exchanged water, filtered, and dried at 120 ° C. to obtain Catalyst 3.
[参考例1;Ni、Mo共沈]
ベーマイトAP-3(触媒化成工業製)1.24g、1N−HNO3水溶液40mLをイオン交換水1Lに加え、80℃に加温後、Ni(NO3)2・6H2Oを149g加え調製液A を得た。別途用意したイオン交換水1LにコロイダルシリカスノーテックスXS(日産化学製)33.9g、炭酸ナトリウム99.4g、(NH4)6Mo7O24・5H2Oを3.0g加え、80℃に加温し、調製液Bを得た。調製液AとBを80℃に保持しながら、B液をA液に瞬時に加えて、1時間攪拌した。その後、イオン交換水を5L用いて洗浄し、ろ過後に空気中120℃で12時間乾燥させた後、400℃で1時間焼成し、得られた焼成物を破砕し、1.0mmと1.4mmの網目を有する篩で篩い分けし、触媒4を得た。
[Reference Example 1: Ni and Mo coprecipitation]
Boehmite AP-3 (Shokubai Kasei Kogyo) 1.24 g, was added a 1N-HNO 3 solution 40mL ion-exchanged water 1L, after warming to 80 ℃, Ni (NO 3) 2 · 6H 2 O and 149g added preparation A was obtained. Separately prepared ion-exchanged water 1L colloidal silica Snowtex XS (produced by Nissan Chemical) 33.9 g, sodium carbonate 99.4g, (NH 4) 6 Mo 7 O 24 · 5H 2 O was added 3.0 g, in 80 ° C. It heated and the preparation liquid B was obtained. While maintaining the prepared solutions A and B at 80 ° C., the solution B was instantaneously added to the solution A and stirred for 1 hour. Thereafter, it was washed with 5 L of ion-exchanged water, and after filtration, dried in air at 120 ° C. for 12 hours, calcined at 400 ° C. for 1 hour, and the obtained calcined product was crushed, 1.0 mm and 1.4 mm The catalyst 4 was obtained by sieving with a sieve having a mesh of.
[比較例1;Pt−Pd/SiO2−Al2O3]
シリカ−アルミナ(シリカ/アルミナ質量比=20/80、直径1/16インチの柱状成形物)37.7gを投入し、そこへ10%塩酸水溶液31.26gに塩化白金酸6水和物0.5005gと塩化パラジウム0.6278gを溶解させた溶液を、ピペットを用いて添加した。約25℃で2時間浸漬後、風乾し、マッフル炉で浸漬混合物の温度を120℃に上げ、約1時間乾燥させた。次いで、500℃で4時間焼成し、触媒5を得た。
[Comparative Example 1; Pt—Pd / SiO 2 —Al 2 O 3 ]
37.7 g of silica-alumina (silica / alumina mass ratio = 20/80, columnar molded product having a diameter of 1/16 inch) was added thereto, and 31.26 g of 10% hydrochloric acid aqueous solution was added to 31.26 g of chloroplatinic acid hexahydrate. A solution in which 5005 g and palladium chloride 0.6278 g were dissolved was added using a pipette. After being immersed at about 25 ° C. for 2 hours, it was air-dried, and the temperature of the immersion mixture was raised to 120 ° C. in a muffle furnace and dried for about 1 hour. Subsequently, it baked at 500 degreeC for 4 hours, and the catalyst 5 was obtained.
実施例1〜3、参考例1及び比較例1で得られた触媒1〜5の組成、物性及びトルエンの水素化反応を実施した結果を表1に示す。表1の結果より、本発明の水素化触媒は、0.12MPa程度の低い水素分圧において、Pt−Pd系の貴金属触媒よりも、より低温度で水素化反応を触媒することができ、かつトルエンの水素化活性が高いことが分かった。 Table 1 shows the compositions, physical properties, and results of the toluene hydrogenation reaction performed in Examples 1 to 3, Reference Example 1 and Comparative Example 1. From the results in Table 1, the hydrogenation catalyst of the present invention can catalyze the hydrogenation reaction at a lower temperature than the Pt—Pd noble metal catalyst at a hydrogen partial pressure as low as about 0.12 MPa, and It was found that toluene hydrogenation activity was high.
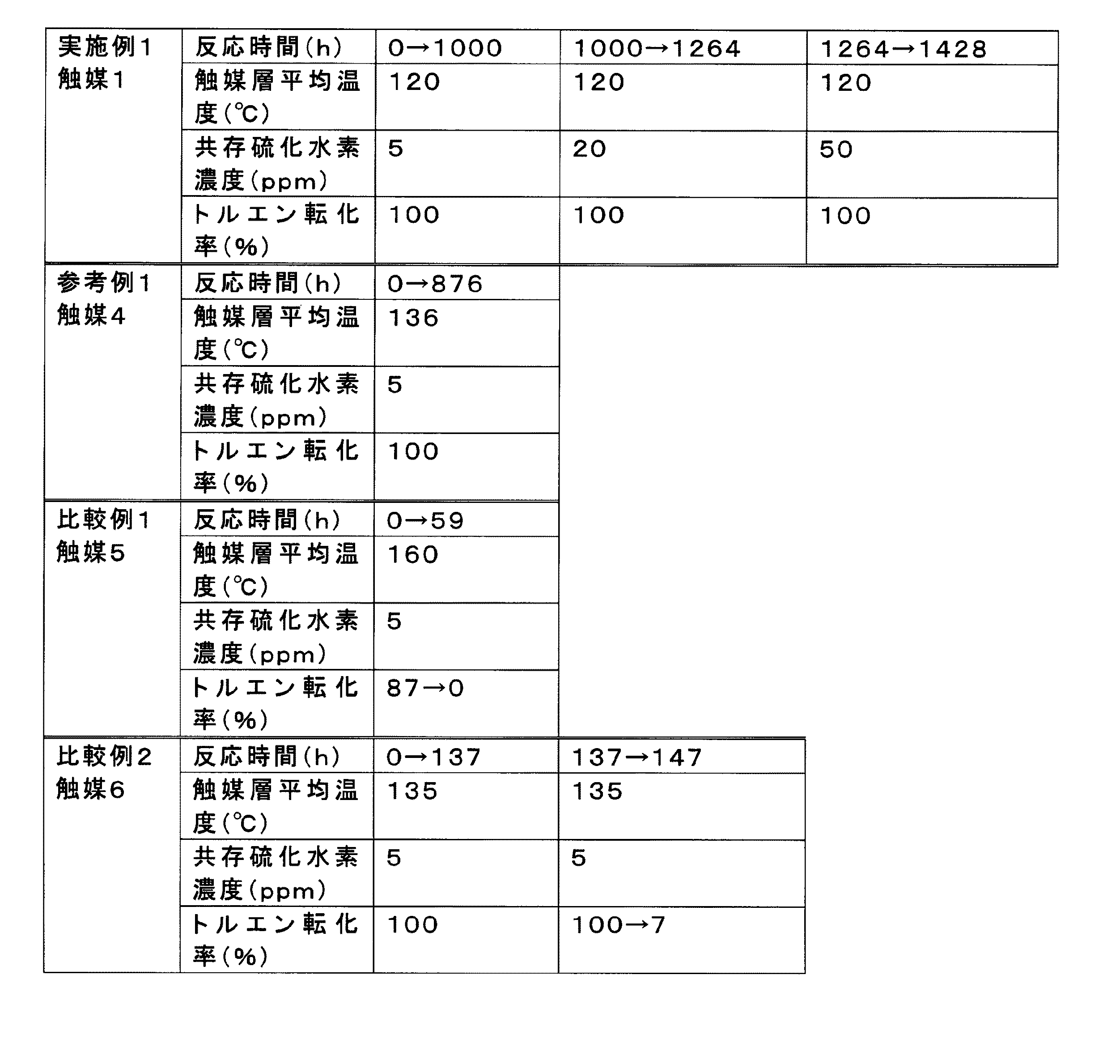
[比較例2;Ni/ZnO]
酢酸亜鉛94.5gと硝酸ニッケル77.8gを1200mLの水に溶解し両者の混合溶液を調製し、この溶液に、炭酸アンモニウム22gを200mLの水に溶解した炭酸アンモニウム水溶液と15%のアンモニア水を加えて、炭酸亜鉛と塩基性炭酸ニッケルの沈澱を得、12時間程放置した。この沈澱物を濾過、水洗後、120℃で12時間乾燥し、200℃で1時間、300℃で2時間、400℃で1時間、510℃で16時間焼成し、触媒6を得た。触媒6のNiO含有量は29%、比表面積は9m2/g、NiO結晶子径は5.5nmであった。
[Comparative Example 2; Ni / ZnO]
Dissolve 94.5 g of zinc acetate and 77.8 g of nickel nitrate in 1200 mL of water to prepare a mixed solution. To this solution, an aqueous solution of ammonium carbonate in which 22 g of ammonium carbonate was dissolved in 200 mL of water and 15% aqueous ammonia. In addition, a precipitate of zinc carbonate and basic nickel carbonate was obtained and left for about 12 hours. The precipitate was filtered, washed with water, dried at 120 ° C. for 12 hours, and calcined at 200 ° C. for 1 hour, 300 ° C. for 2 hours, 400 ° C. for 1 hour, and 510 ° C. for 16 hours to obtain Catalyst 6. The catalyst 6 had a NiO content of 29%, a specific surface area of 9 m 2 / g, and a NiO crystallite diameter of 5.5 nm.
実施例1の触媒1、比較例1の触媒5、比較例2の触媒6を用いて硫化水素共存下でトルエンの水素化反応を行い、活性への影響を調べた。結果を表2に示す。触媒1は、H2Sが共存する条件下であるにもかかわらず、1428時間という非常に長時間、高い水素化活性が維持されていた。これに対して、触媒5では反応時間59時間で失活してしまい、触媒6では反応時間147時間ではトルエン転化率はたった7%しかなかった。すなわち、表2の結果より、本発明の水素化触媒は、Pt−Pd系の貴金属触媒に比べて非常に耐H2Sに優れることが分かった。なお、ルテニウムを含有していない触媒4は、876時間もの長時間、高い水素化活性が維持されており、本発明の水素化触媒と同様にPt−Pd系の貴金属触媒に比べて非常に耐H2Sに優れることが分かった。 Hydrogenation reaction of toluene was conducted in the presence of hydrogen sulfide using the catalyst 1 of Example 1, the catalyst 5 of Comparative Example 1, and the catalyst 6 of Comparative Example 2, and the influence on the activity was examined. The results are shown in Table 2. The catalyst 1 maintained a high hydrogenation activity for a very long time of 1428 hours under the condition where H 2 S coexists. In contrast, catalyst 5 was deactivated at a reaction time of 59 hours, and catalyst 6 had a toluene conversion of only 7% at a reaction time of 147 hours. That is, from the results of Table 2, it was found that the hydrogenation catalyst of the present invention was extremely excellent in H 2 S resistance compared to Pt—Pd noble metal catalysts. The catalyst 4 containing no ruthenium maintains a high hydrogenation activity for a long time of 876 hours, and is much more resistant than the Pt—Pd noble metal catalyst as in the hydrogenation catalyst of the present invention. It was found to be excellent in H 2 S.
Claims (2)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012071107A JP5748351B2 (en) | 2012-03-27 | 2012-03-27 | Hydrogenation catalyst for aromatic compounds and method for hydrogenating aromatic compounds using the catalyst |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012071107A JP5748351B2 (en) | 2012-03-27 | 2012-03-27 | Hydrogenation catalyst for aromatic compounds and method for hydrogenating aromatic compounds using the catalyst |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2013202435A JP2013202435A (en) | 2013-10-07 |
| JP5748351B2 true JP5748351B2 (en) | 2015-07-15 |
Family
ID=49522144
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012071107A Active JP5748351B2 (en) | 2012-03-27 | 2012-03-27 | Hydrogenation catalyst for aromatic compounds and method for hydrogenating aromatic compounds using the catalyst |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5748351B2 (en) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR3025728B1 (en) * | 2014-09-11 | 2018-04-20 | IFP Energies Nouvelles | MESOPOROUS CATALYST BASED ON NICKEL AND ITS USE IN HYDROGENATION. |
| CN114453005B (en) * | 2020-10-21 | 2023-09-01 | 中国石油化工股份有限公司 | Hydrofining catalyst and preparation method and application thereof |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4896766B2 (en) * | 2006-02-24 | 2012-03-14 | コスモ石油株式会社 | Hydrocarbon desulfurization agent |
| JP4878868B2 (en) * | 2006-02-24 | 2012-02-15 | コスモ石油株式会社 | Hydrocarbon desulfurization agent |
| JP5261801B2 (en) * | 2006-09-20 | 2013-08-14 | 中国石油化工股▲分▼有限公司 | Nickel catalysts for selective hydrogenation |
-
2012
- 2012-03-27 JP JP2012071107A patent/JP5748351B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP2013202435A (en) | 2013-10-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5357170B2 (en) | Selective nickel-based hydrogenation catalyst and its production | |
| JP5815842B2 (en) | catalyst | |
| TWI617660B (en) | Hydrodesulfurization catalyst for light oil, and hydrotreating method of light oil | |
| JP4896766B2 (en) | Hydrocarbon desulfurization agent | |
| JP2016203074A (en) | Hydrotreating catalyst for hydrocarbon oil, production method of the same, and hydrotreating method | |
| JP5748351B2 (en) | Hydrogenation catalyst for aromatic compounds and method for hydrogenating aromatic compounds using the catalyst | |
| CN104971767B (en) | A kind of embedded catalyst for methanation in presence of sulfur and preparation method and application | |
| Guo et al. | Titanium silicalite-1 supported bimetallic catalysts for selective hydrogenolysis of 5-hydroxymethylfurfural to biofuel 2, 5-dimethylfuran | |
| JP5841481B2 (en) | Method for hydrotreating heavy residual oil | |
| JP5748352B2 (en) | Hydrogenation catalyst for aromatic compounds and method for hydrogenating aromatic compounds using the catalyst | |
| JP4773116B2 (en) | Method for producing catalyst for producing hydrocarbons from synthesis gas, and method for producing hydrocarbons from synthesis gas using the catalyst | |
| JP4922783B2 (en) | Hydrocarbon desulfurization agent | |
| Grimm et al. | Transition metal promoted combustion of rice husk and rice straw towards an energy optimized synthesis of biogenic silica | |
| JP2013027838A (en) | Method of regenerating hydrogenation catalyst | |
| CN113731427B (en) | Dual-function desulfurization catalyst, preparation method thereof and hydrocarbon oil desulfurization method | |
| JP5979443B2 (en) | Method for producing hydrogen | |
| JP4878868B2 (en) | Hydrocarbon desulfurization agent | |
| JP2007160250A (en) | Method of manufacturing catalyst, catalytic cracking catalyst and method of producing low-sulfur catalytically-cracked gasoline | |
| JPH0661464B2 (en) | Catalyst for hydrodesulfurization and denitrification of heavy hydrocarbon oils | |
| JP5948657B2 (en) | Hydrogen production method | |
| JP2009045526A (en) | Method for preparing nickel-ruthenium desulfurization agent and nickel-ruthenium catalyst | |
| JP5841480B2 (en) | Method for hydrotreating heavy residual oil | |
| JP4931099B2 (en) | Catalyst for producing cycloolefin and method for producing cycloolefin | |
| JP4955483B2 (en) | Method for producing hydrocarbon desulfurization agent | |
| JP5031790B2 (en) | Method for producing catalyst for hydrorefining of light oil and hydrorefining method of light oil |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20140307 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20150122 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20150127 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20150414 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20150511 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5748351 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |