JP5659030B2 - 高電圧機器用絶縁樹脂組成物の製造方法 - Google Patents
高電圧機器用絶縁樹脂組成物の製造方法 Download PDFInfo
- Publication number
- JP5659030B2 JP5659030B2 JP2011017149A JP2011017149A JP5659030B2 JP 5659030 B2 JP5659030 B2 JP 5659030B2 JP 2011017149 A JP2011017149 A JP 2011017149A JP 2011017149 A JP2011017149 A JP 2011017149A JP 5659030 B2 JP5659030 B2 JP 5659030B2
- Authority
- JP
- Japan
- Prior art keywords
- clay mineral
- layered clay
- epoxy resin
- weight
- silane coupling
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images


Landscapes
- Compositions Of Macromolecular Compounds (AREA)
- Organic Insulating Materials (AREA)
Description
(a)層状粘土鉱物を水、水系混合溶媒のうちいずれか一方で膨潤させて層状粘土鉱物分散液を得る工程と、
(b)前記層状粘土鉱物分散液にシランカップリング剤を加えて層状粘土鉱物表面を有機官能化し、有機官能化層状粘土鉱物分散液を得る工程と、
(c)前記有機官能化層状粘土鉱物分散液に、エポキシ樹脂およびエポキシ樹脂と相溶な有機溶媒を加えて混練する工程と
を含む。
工程(a)では、層状粘土鉱物を水、水系混合溶媒のうちいずれか一方で膨潤させ、層状粘土鉱物分散液を得る。
工程(a)で得られた層状粘土鉱物分散液にシランカップリング剤を加えて層状粘土鉱物表面を有機官能化し、有機官能化層状粘土鉱物分散液を得る。
式中、nは1〜3である。
Xは、加水分解性基または水酸基であり、例えば炭素数1〜8のアルコキシ基(例えばメトキシ基、エトキシ基、プロポキシ基、ブトキシ基など)、炭素数3〜8のアルケニルオキシ基(例えばイソプロペノキシ基、1−エチル−2−メチルビニルオキシム基など)、炭素数3〜8のケトオキシム基(ジメチルケトオキシム基、メチルエチルケトオキシム基など)、炭素数2〜8のアルキルオキシ基(アセトキシ基、プロピオノキシ基、ブチロイロキシ基、ベンォイルオキシム基など)、アミノ基(ジメチルアミノ基、ジエチルアミノ基など)、アミノキシ基(ジメチルアミノキシ基、ジエチルアミノキシ基など)、ハロゲン原子(塩素原子、臭素原子など)などである。これらの中で炭素数1〜4のアルコキシ基が反応性の点から好ましい。Xが複数個存在する場合、それらは互いに異なっていてもよい。
Yは、炭素数1〜25の炭化水素基であって置換基を有していても良い。当該置換基としては、エポキシ基、アミノ基、アミド基、カルボキシル基、メルカプト基、ビニル基、メタクリル基、スフィド基、水酸基、ハロゲン原子などである。Yが複数個存在する場合、それらは互いに異なっていてもよい。
工程(b)で得られた有機官能化層状粘土鉱物分散液に、エポキシ樹脂およびエポキシ樹脂と相溶な有機溶媒を加えて混練する。
ンジアミン、ジアミノジフェニルメタン、ジアミノジフェニルスルフォン、ジアミノジエ
チルジフェニルメタン、ジシアンジアミド、有機酸ジヒドラジドが挙げられる。
無水物、ポリアゼライン酸無水物、ポリセバシン酸無水物、ポリ(エチルオクタデカン二酸)無水物、ポリ(フェニルヘキサデカン二酸)無水物、メチルテトラヒドロ無水フタル
酸、メチルヘキサヒドロ無水フタル酸、無水メチルハイミック酸、ヘキサヒドロ無水フタ
ル酸、テトラヒドロ無水フタル酸、トリアルキルテトラヒドロ無水フタル酸、メチルシク
ロへキセンジカルボン酸無水物、無水フタル酸、無水トリメリット酸、無水ピロメリット
酸、ベンゾフェノンテトラカルボン酸、エチレングリコールビストリメリテート、グリセ
ロールトリストリメリテート、無水ヘット酸、テトラブロモ無水フタル酸、無水ナジック
酸、無水メチルナジック酸、無水ポリアゼライン酸が挙げられる。
−4−メチルイミダゾール、2−ヘプタデシルイミダゾールが挙げられる。また、ポリメ
ルカプタン系硬化剤の具体例としては、例えばポリサルファイド、チオエステルが挙げら
れる。
メタノール20重量%を含む水/メタノール混合溶媒に、層状粘土鉱物(クニミネ工業社製クレイ、クニピアF)を加えて攪拌し、層状粘土鉱物を膨潤および剥離させた濃度5重量%の層状粘土鉱物分散液を準備した。この層状粘土鉱物分散液に、シランカップリング剤であるZ−6040(東レ・ダウコーニング社製)をクレイの重量に対して3重量%加え、60℃で4時間加熱および混合した後、アセトンとトルエンをメタノールと同量加え、30分攪拌した。その後、ビスフェノールA型エポキシ樹脂(ジャパンエポキシレジン社製、エピコート828)100重量部を加えて60℃にて30分間攪拌した後、脱溶媒した。脱溶媒後の水分量は0.06ppmであった。次いで、これを3本ロールミル混合機(井上製作所社製、S−43/4×11)に5回以上通過させ、混練し、層状粘土鉱物分散樹脂を得た。
メタノール20重量%を含む水/メタノール混合溶媒に層状粘土鉱物(クニミネ工業社製クレイ、クニピアF)を加えて攪拌し、膨潤および剥離させた濃度5重量%の層状粘土鉱物分散液を得た。この層状粘土鉱物分散液に、シランカップリング剤であるZ−6040(東レ・ダウコーニング社製)をクレイの重量に対して3重量%加え、60℃で4時間加熱および混合した後、アセトンとトルエンをメタノールと同量加え、30分攪拌した。その後、ビスフェノールA型エポキシ樹脂(ジャパンエポキシレジン社製、エピコート828)100重量部を加えて60℃にて30分間攪拌した。その後、さらに無機フィラーとして粒子径約20μmのシリカ粒子を120重量部加えて攪拌しながら脱溶媒して層状粘土鉱物分散樹脂を得た。脱溶媒後の水分量は0.06ppmであった。
ビスフェノールA型エポキシ樹脂100重量部に、酸無水物系硬化剤86重量部とアミン系硬化促進剤1重量部からなる混合物を加え、混練した後、100℃、5mbarにて15分間真空混合を行った。実施例1と同様の条件で一次硬化および二次硬化を行い、同様の試験片を作製した。
エポキシ樹脂100重量部とシリカ粒子(粒子径約20μm)120重量部との混合物に、酸無水物系硬化剤86重量部とアミン系硬化促進剤1重量部と前記シリカ粒子230重量部との混合物を加えて混練した後、100℃、5mbarにて15分間真空混合を行った。実施例1と同様の条件で一次硬化および二次硬化を行い、同様の試験片を作製した。
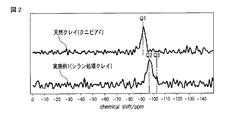
シランカップリング剤と層状粘土鉱物との反応を確認するため、日本電子社製、JMN−ECA400を用いてNMR測定を行った。実施例1に記載の条件でシランカップリング処理した後、エポキシ樹脂投入前に回収した層状粘土鉱物分散液をメタノール、アセトン、トルエンによりこの順番で洗浄し、遠心分離により沈降した層状粘土鉱物を回収し、十分に乾燥させたものをサンプルとして用いた。測定核は29Siとし、交差分極マジックスピニング法(CP/MAS:コンタクトタイム1 ms、90°パルス2.8 ms)により測定した。
各実施例において得られた65mm×3mm×1mmの各試験片について、オリエンテック社製、RHEOVIBRONを用いて動的粘弾測定を行った。測定周波数1Hz、昇温速度2℃/min、引っ張りモードにて測定した。実施例1および比較例1の試験片について得られた結果を図4に示す。
実施例1で得られた層状粘土鉱物樹脂の硬化物における層状粘土鉱物の分散状態を、透過型電子顕微鏡(TEM)(日立社製、H−7100:加速電圧100kV)を用いて観察した。結果を図5に示す。
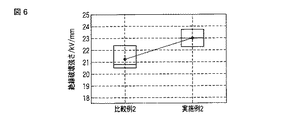
実施例2および比較例2で得られた層状粘土鉱物樹脂の硬化物の絶縁破壊強さを、球−平板電極を用いて昇圧速度0.6 kV/minで測定した。測定結果を図6に示す。
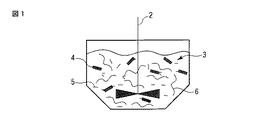
2…攪拌機
3…水または水系混合溶媒
4…層状粘土鉱物
5…シランカップリング剤
6…エポキシ樹脂
Claims (5)
- (a)層状粘土鉱物を水、水系混合溶媒のうちいずれか一方で膨潤させて層状粘土鉱物分散液を得る工程と、
(b)前記層状粘土鉱物分散液にシランカップリング剤を加えて層状粘土鉱物表面を有機官能化し、有機官能化層状粘土鉱物分散液を得る工程と、
(c)前記有機官能化層状粘土鉱物分散液に、エポキシ樹脂およびエポキシ樹脂と相溶な有機溶媒を加えて混練する工程と
を含むことを特徴とする高電圧機器用絶縁樹脂組成物の製造方法。 - 工程(c)においてさらに無機フィラーを加えることを特徴とする請求項1に記載の高電圧機器用絶縁樹脂組成物の製造方法。
- 前記シランカップリング剤が、前記層状粘土鉱物の重量に対して0.1〜10重量%用いられることを特徴とする請求項1または2に記載の製造方法。
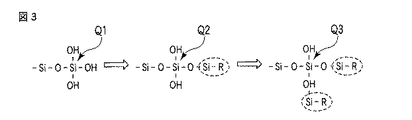
- 前記シランカップリング剤と前記層状粘土鉱物の表面の水酸基が化学結合を形成していることを特徴とする請求項1〜3のいずれか1項に記載の製造方法。
- 工程(c)で得られた混練物から溶媒を全て除去することを特徴とする請求項1〜4のいずれか1項に記載の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011017149A JP5659030B2 (ja) | 2011-01-28 | 2011-01-28 | 高電圧機器用絶縁樹脂組成物の製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011017149A JP5659030B2 (ja) | 2011-01-28 | 2011-01-28 | 高電圧機器用絶縁樹脂組成物の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2012158622A JP2012158622A (ja) | 2012-08-23 |
| JP5659030B2 true JP5659030B2 (ja) | 2015-01-28 |
Family
ID=46839431
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011017149A Active JP5659030B2 (ja) | 2011-01-28 | 2011-01-28 | 高電圧機器用絶縁樹脂組成物の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5659030B2 (ja) |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4476646B2 (ja) * | 2004-03-03 | 2010-06-09 | 株式会社東芝 | 高電圧機器用の絶縁樹脂組成物、絶縁材料とその製造方法、および絶縁構造体 |
| JP4969816B2 (ja) * | 2005-08-22 | 2012-07-04 | 株式会社東芝 | 樹脂組成物とその製造方法およびそれを用いた電気機器 |
-
2011
- 2011-01-28 JP JP2011017149A patent/JP5659030B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP2012158622A (ja) | 2012-08-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102725802B (zh) | 电绝缘体系 | |
| CN104177780B (zh) | 一种户外型电气绝缘改性环氧树脂组合物 | |
| JP5185890B2 (ja) | 高電圧電気機器用絶縁注型樹脂及びこれを用いた高電圧電気機器 | |
| CN102816411B (zh) | 电绝缘材料以及使用它的高电压设备 | |
| WO2008023692A1 (fr) | Formule de résine pour moulage, matériau isolant l'utilisant et structure isolante | |
| CN102388096B (zh) | 直接重叠注塑 | |
| JP2006057017A (ja) | 高電圧機器用耐部分放電性絶縁樹脂組成物、耐部分放電性絶縁材料及び絶縁構造体 | |
| JP5250003B2 (ja) | 樹脂材料及びこれを用いた高電圧機器 | |
| KR102021318B1 (ko) | 전력 전송 및 분배를 위한 절연 복합체 | |
| CN101816049A (zh) | 具有提高的电击穿强度的电绝缘体系 | |
| CN105255112A (zh) | 一种环氧树脂富勒烯复合材料及其制备方法 | |
| CN101506301A (zh) | 浇铸型树脂组合物及采用它的绝缘材料、绝缘结构体 | |
| Mo et al. | The influence of the interface between mica and epoxy matrix on properties of epoxy-based dielectric materials with high thermal conductivity and low dielectric loss | |
| JP4969816B2 (ja) | 樹脂組成物とその製造方法およびそれを用いた電気機器 | |
| WO2009104292A1 (ja) | 耐部分放電性樹脂組成物の製造方法、耐部分放電性樹脂組成物、および耐部分放電性絶縁材料 | |
| WO2013123648A1 (en) | Curable epoxy composition with milled glass fiber | |
| JP4476646B2 (ja) | 高電圧機器用の絶縁樹脂組成物、絶縁材料とその製造方法、および絶縁構造体 | |
| JP5659030B2 (ja) | 高電圧機器用絶縁樹脂組成物の製造方法 | |
| JP4314112B2 (ja) | 六フッ化硫黄ガス絶縁機器用繊維強化複合材料およびその製造方法 | |
| JP4387786B2 (ja) | エポキシ樹脂組成物および注型絶縁物 | |
| EP3070115A1 (en) | Epoxy resin composition with improved dielectric breakdown strength | |
| JP7533821B2 (ja) | グリシジルエーテル基含有化合物、硬化性樹脂組成物、硬化物及び積層体 | |
| JP7533820B2 (ja) | グリシジルエーテル基含有化合物、硬化性樹脂組成物、硬化物及び積層体 | |
| JP2018193517A (ja) | 複合絶縁樹脂組成物、並びにこれを用いた高電圧機器および遮断器 | |
| JP2014031391A (ja) | エポキシ樹脂およびそれを用いた電気機器 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20131114 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20131205 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20131212 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20131219 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20131226 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20140109 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20140116 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140318 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20140319 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140512 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20141104 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20141201 |
|
| R150 | Certificate of patent (=grant) or registration of utility model |
Ref document number: 5659030 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |