JP5437394B2 - 抗炎症性マクロライド - Google Patents
抗炎症性マクロライド Download PDFInfo
- Publication number
- JP5437394B2 JP5437394B2 JP2011546833A JP2011546833A JP5437394B2 JP 5437394 B2 JP5437394 B2 JP 5437394B2 JP 2011546833 A JP2011546833 A JP 2011546833A JP 2011546833 A JP2011546833 A JP 2011546833A JP 5437394 B2 JP5437394 B2 JP 5437394B2
- Authority
- JP
- Japan
- Prior art keywords
- formula
- compound
- methyl
- homoerythromycin
- aza
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 230000003110 anti-inflammatory effect Effects 0.000 title description 7
- 239000003120 macrolide antibiotic agent Substances 0.000 title description 6
- 150000001875 compounds Chemical class 0.000 claims description 168
- -1 2-diethylaminoethanoyl Chemical group 0.000 claims description 87
- 150000003839 salts Chemical class 0.000 claims description 71
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 41
- 210000000440 neutrophil Anatomy 0.000 claims description 37
- 201000003883 Cystic fibrosis Diseases 0.000 claims description 28
- 206010006448 Bronchiolitis Diseases 0.000 claims description 27
- 239000008194 pharmaceutical composition Substances 0.000 claims description 24
- 206010062952 diffuse panbronchiolitis Diseases 0.000 claims description 22
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 claims description 21
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 claims description 20
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 20
- 125000006239 protecting group Chemical group 0.000 claims description 20
- 208000006673 asthma Diseases 0.000 claims description 19
- 230000000414 obstructive effect Effects 0.000 claims description 19
- 206010006451 bronchitis Diseases 0.000 claims description 18
- 239000003814 drug Substances 0.000 claims description 18
- 201000010099 disease Diseases 0.000 claims description 17
- 150000003431 steroids Chemical class 0.000 claims description 17
- 208000013616 Respiratory Distress Syndrome Diseases 0.000 claims description 16
- 201000009267 bronchiectasis Diseases 0.000 claims description 16
- 238000006243 chemical reaction Methods 0.000 claims description 16
- 208000027157 chronic rhinosinusitis Diseases 0.000 claims description 16
- 208000027866 inflammatory disease Diseases 0.000 claims description 16
- 206010063837 Reperfusion injury Diseases 0.000 claims description 15
- 230000008595 infiltration Effects 0.000 claims description 15
- 238000001764 infiltration Methods 0.000 claims description 15
- 208000012947 ischemia reperfusion injury Diseases 0.000 claims description 15
- 201000000596 systemic lupus erythematosus Diseases 0.000 claims description 15
- 206010014561 Emphysema Diseases 0.000 claims description 14
- 201000000028 adult respiratory distress syndrome Diseases 0.000 claims description 14
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 14
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 13
- 230000004054 inflammatory process Effects 0.000 claims description 12
- 208000011580 syndromic disease Diseases 0.000 claims description 12
- 206010061218 Inflammation Diseases 0.000 claims description 11
- 230000002159 abnormal effect Effects 0.000 claims description 11
- 230000003915 cell function Effects 0.000 claims description 11
- 238000004519 manufacturing process Methods 0.000 claims description 11
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 10
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 10
- 206010018364 Glomerulonephritis Diseases 0.000 claims description 9
- 201000005569 Gout Diseases 0.000 claims description 9
- 206010018634 Gouty Arthritis Diseases 0.000 claims description 9
- 201000004681 Psoriasis Diseases 0.000 claims description 9
- 206010047115 Vasculitis Diseases 0.000 claims description 9
- 201000001320 Atherosclerosis Diseases 0.000 claims description 8
- 208000009693 Gingival Hyperplasia Diseases 0.000 claims description 8
- 208000022559 Inflammatory bowel disease Diseases 0.000 claims description 8
- 241001303601 Rosacea Species 0.000 claims description 8
- 206010040047 Sepsis Diseases 0.000 claims description 8
- 201000001245 periodontitis Diseases 0.000 claims description 8
- 201000007094 prostatitis Diseases 0.000 claims description 8
- 201000004700 rosacea Diseases 0.000 claims description 8
- 208000017520 skin disease Diseases 0.000 claims description 8
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims description 6
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 6
- 230000005855 radiation Effects 0.000 claims description 6
- 239000013543 active substance Substances 0.000 claims description 4
- 230000001684 chronic effect Effects 0.000 claims description 4
- 229940126214 compound 3 Drugs 0.000 claims description 2
- 239000003085 diluting agent Substances 0.000 claims description 2
- 238000013160 medical therapy Methods 0.000 claims description 2
- 230000009885 systemic effect Effects 0.000 claims description 2
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 claims 3
- 229910052802 copper Inorganic materials 0.000 claims 3
- 239000010949 copper Substances 0.000 claims 3
- 206010028980 Neoplasm Diseases 0.000 claims 1
- 201000009890 sinusitis Diseases 0.000 claims 1
- 239000000203 mixture Substances 0.000 description 68
- 239000000243 solution Substances 0.000 description 66
- 238000000034 method Methods 0.000 description 62
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 47
- 239000000047 product Substances 0.000 description 45
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 42
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 39
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 35
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 35
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 33
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 31
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 31
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 30
- 238000011282 treatment Methods 0.000 description 29
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 27
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 26
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 24
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 23
- 239000007787 solid Substances 0.000 description 21
- 239000002904 solvent Substances 0.000 description 21
- 238000001237 Raman spectrum Methods 0.000 description 20
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 19
- 239000000543 intermediate Substances 0.000 description 19
- 238000005481 NMR spectroscopy Methods 0.000 description 18
- 239000000872 buffer Substances 0.000 description 17
- 238000001914 filtration Methods 0.000 description 17
- 239000011541 reaction mixture Substances 0.000 description 17
- 239000000523 sample Substances 0.000 description 17
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 15
- 239000012044 organic layer Substances 0.000 description 15
- 239000002244 precipitate Substances 0.000 description 15
- 239000002002 slurry Substances 0.000 description 15
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 14
- 210000004027 cell Anatomy 0.000 description 14
- 235000019439 ethyl acetate Nutrition 0.000 description 14
- 239000000706 filtrate Substances 0.000 description 14
- 208000015768 polyposis Diseases 0.000 description 14
- 239000000725 suspension Substances 0.000 description 14
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 13
- 206010051379 Systemic Inflammatory Response Syndrome Diseases 0.000 description 13
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 13
- 229910052739 hydrogen Inorganic materials 0.000 description 13
- 239000010410 layer Substances 0.000 description 13
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 12
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 125000000217 alkyl group Chemical group 0.000 description 12
- 238000004458 analytical method Methods 0.000 description 12
- 210000004369 blood Anatomy 0.000 description 12
- 239000008280 blood Substances 0.000 description 12
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 12
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropyl acetate Chemical compound CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 12
- 239000000463 material Substances 0.000 description 12
- 238000012360 testing method Methods 0.000 description 12
- 238000002411 thermogravimetry Methods 0.000 description 12
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 11
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 11
- 229910052757 nitrogen Inorganic materials 0.000 description 11
- 210000002381 plasma Anatomy 0.000 description 11
- 238000003756 stirring Methods 0.000 description 11
- 238000001069 Raman spectroscopy Methods 0.000 description 10
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 10
- 238000000113 differential scanning calorimetry Methods 0.000 description 10
- 239000002158 endotoxin Substances 0.000 description 10
- 239000003921 oil Substances 0.000 description 10
- 235000019198 oils Nutrition 0.000 description 10
- 229920006395 saturated elastomer Polymers 0.000 description 10
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 9
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 9
- 241001465754 Metazoa Species 0.000 description 9
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 239000002253 acid Substances 0.000 description 9
- 239000001257 hydrogen Substances 0.000 description 9
- 238000000338 in vitro Methods 0.000 description 9
- 229920006008 lipopolysaccharide Polymers 0.000 description 9
- 239000000843 powder Substances 0.000 description 9
- 238000003556 assay Methods 0.000 description 8
- 239000002775 capsule Substances 0.000 description 8
- 239000000969 carrier Substances 0.000 description 8
- 239000003937 drug carrier Substances 0.000 description 8
- 239000002609 medium Substances 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 7
- 206010009900 Colitis ulcerative Diseases 0.000 description 7
- 208000011231 Crohn disease Diseases 0.000 description 7
- 229920002472 Starch Polymers 0.000 description 7
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 7
- 201000006704 Ulcerative Colitis Diseases 0.000 description 7
- 230000000844 anti-bacterial effect Effects 0.000 description 7
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 7
- 230000000694 effects Effects 0.000 description 7
- 229960001031 glucose Drugs 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- 239000008107 starch Substances 0.000 description 7
- 229940032147 starch Drugs 0.000 description 7
- 235000019698 starch Nutrition 0.000 description 7
- 239000003826 tablet Substances 0.000 description 7
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 6
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 6
- 241000699670 Mus sp. Species 0.000 description 6
- 239000002202 Polyethylene glycol Substances 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- 239000003570 air Substances 0.000 description 6
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 6
- 230000037396 body weight Effects 0.000 description 6
- 235000014113 dietary fatty acids Nutrition 0.000 description 6
- 239000000194 fatty acid Substances 0.000 description 6
- 229930195729 fatty acid Natural products 0.000 description 6
- 239000012530 fluid Substances 0.000 description 6
- 238000009472 formulation Methods 0.000 description 6
- 238000004128 high performance liquid chromatography Methods 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- 229920001223 polyethylene glycol Polymers 0.000 description 6
- 229920000642 polymer Polymers 0.000 description 6
- 239000011780 sodium chloride Substances 0.000 description 6
- 239000012453 solvate Substances 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 5
- ULGZDMOVFRHVEP-RWJQBGPGSA-N Erythromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 ULGZDMOVFRHVEP-RWJQBGPGSA-N 0.000 description 5
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- 230000001154 acute effect Effects 0.000 description 5
- 239000000654 additive Substances 0.000 description 5
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 5
- 239000012267 brine Substances 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 235000010980 cellulose Nutrition 0.000 description 5
- 229920002678 cellulose Polymers 0.000 description 5
- 210000000038 chest Anatomy 0.000 description 5
- 239000011248 coating agent Substances 0.000 description 5
- 238000000576 coating method Methods 0.000 description 5
- 230000003111 delayed effect Effects 0.000 description 5
- 238000000605 extraction Methods 0.000 description 5
- 235000011187 glycerol Nutrition 0.000 description 5
- 238000010438 heat treatment Methods 0.000 description 5
- 150000002431 hydrogen Chemical class 0.000 description 5
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 5
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 5
- 229920000609 methyl cellulose Polymers 0.000 description 5
- 235000010981 methylcellulose Nutrition 0.000 description 5
- 239000001923 methylcellulose Substances 0.000 description 5
- 229960002900 methylcellulose Drugs 0.000 description 5
- 239000003208 petroleum Substances 0.000 description 5
- 230000002829 reductive effect Effects 0.000 description 5
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 5
- 239000007921 spray Substances 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 4
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 4
- 241000894006 Bacteria Species 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 4
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 4
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 4
- 230000004913 activation Effects 0.000 description 4
- 235000010443 alginic acid Nutrition 0.000 description 4
- 229920000615 alginic acid Polymers 0.000 description 4
- APKFDSVGJQXUKY-INPOYWNPSA-N amphotericin B Chemical compound O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1/C=C/C=C/C=C/C=C/C=C/C=C/C=C/[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-INPOYWNPSA-N 0.000 description 4
- 239000003242 anti bacterial agent Substances 0.000 description 4
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 4
- 230000001580 bacterial effect Effects 0.000 description 4
- 239000011230 binding agent Substances 0.000 description 4
- 239000001768 carboxy methyl cellulose Substances 0.000 description 4
- 239000001913 cellulose Substances 0.000 description 4
- 239000007979 citrate buffer Substances 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 239000008121 dextrose Substances 0.000 description 4
- 238000001035 drying Methods 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 4
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 4
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 4
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 4
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 208000015181 infectious disease Diseases 0.000 description 4
- 230000000968 intestinal effect Effects 0.000 description 4
- 229940011051 isopropyl acetate Drugs 0.000 description 4
- 239000008101 lactose Substances 0.000 description 4
- 229960001375 lactose Drugs 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 229910052763 palladium Inorganic materials 0.000 description 4
- 239000002245 particle Substances 0.000 description 4
- 239000002953 phosphate buffered saline Substances 0.000 description 4
- 230000000291 postprandial effect Effects 0.000 description 4
- 239000003755 preservative agent Substances 0.000 description 4
- 239000012488 sample solution Substances 0.000 description 4
- 239000003381 stabilizer Substances 0.000 description 4
- 239000011550 stock solution Substances 0.000 description 4
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 4
- WMOVHXAZOJBABW-UHFFFAOYSA-N tert-butyl acetate Chemical compound CC(=O)OC(C)(C)C WMOVHXAZOJBABW-UHFFFAOYSA-N 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- 238000005160 1H NMR spectroscopy Methods 0.000 description 3
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 3
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 3
- 239000001856 Ethyl cellulose Substances 0.000 description 3
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 3
- FEWJPZIEWOKRBE-JCYAYHJZSA-L L-tartrate(2-) Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O FEWJPZIEWOKRBE-JCYAYHJZSA-L 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 229930195725 Mannitol Natural products 0.000 description 3
- 241000588655 Moraxella catarrhalis Species 0.000 description 3
- SGXDXUYKISDCAZ-UHFFFAOYSA-N N,N-diethylglycine Chemical compound CCN(CC)CC(O)=O SGXDXUYKISDCAZ-UHFFFAOYSA-N 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- 241000193998 Streptococcus pneumoniae Species 0.000 description 3
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 3
- 239000000443 aerosol Substances 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- 229940072056 alginate Drugs 0.000 description 3
- 235000019270 ammonium chloride Nutrition 0.000 description 3
- 239000000010 aprotic solvent Substances 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- FOCAUTSVDIKZOP-UHFFFAOYSA-N chloroacetic acid Chemical compound OC(=O)CCl FOCAUTSVDIKZOP-UHFFFAOYSA-N 0.000 description 3
- 229940106681 chloroacetic acid Drugs 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 230000000052 comparative effect Effects 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- 238000001938 differential scanning calorimetry curve Methods 0.000 description 3
- 239000007884 disintegrant Substances 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 229960003276 erythromycin Drugs 0.000 description 3
- 235000019325 ethyl cellulose Nutrition 0.000 description 3
- 229920001249 ethyl cellulose Polymers 0.000 description 3
- 239000012091 fetal bovine serum Substances 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 239000003205 fragrance Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 239000008103 glucose Substances 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 150000004820 halides Chemical class 0.000 description 3
- 150000003840 hydrochlorides Chemical class 0.000 description 3
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 3
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 3
- 210000004969 inflammatory cell Anatomy 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 210000004072 lung Anatomy 0.000 description 3
- 239000000594 mannitol Substances 0.000 description 3
- 235000010355 mannitol Nutrition 0.000 description 3
- 229960001855 mannitol Drugs 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 3
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 3
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 3
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 3
- 230000002685 pulmonary effect Effects 0.000 description 3
- 238000012216 screening Methods 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 239000011343 solid material Substances 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 229960002920 sorbitol Drugs 0.000 description 3
- 235000010356 sorbitol Nutrition 0.000 description 3
- 229940031000 streptococcus pneumoniae Drugs 0.000 description 3
- 238000013456 study Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- 238000011200 topical administration Methods 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- 239000006216 vaginal suppository Substances 0.000 description 3
- 239000012224 working solution Substances 0.000 description 3
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 2
- AXFJPPIWFDKUIH-UHFFFAOYSA-N 2-(diethylamino)acetic acid N-ethylethanamine Chemical compound C(C)NCC.C(C)N(CC)CC(=O)O AXFJPPIWFDKUIH-UHFFFAOYSA-N 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- GJCOSYZMQJWQCA-UHFFFAOYSA-N 9H-xanthene Chemical compound C1=CC=C2CC3=CC=CC=C3OC2=C1 GJCOSYZMQJWQCA-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- APKFDSVGJQXUKY-KKGHZKTASA-N Amphotericin-B Natural products O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1C=CC=CC=CC=CC=CC=CC=C[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-KKGHZKTASA-N 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 2
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 241000588724 Escherichia coli Species 0.000 description 2
- 241000606768 Haemophilus influenzae Species 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 2
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 2
- 206010020751 Hypersensitivity Diseases 0.000 description 2
- BAPJBEWLBFYGME-UHFFFAOYSA-N Methyl acrylate Chemical compound COC(=O)C=C BAPJBEWLBFYGME-UHFFFAOYSA-N 0.000 description 2
- 229920000881 Modified starch Polymers 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Chemical compound IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 2
- 206010029379 Neutrophilia Diseases 0.000 description 2
- 229930182555 Penicillin Natural products 0.000 description 2
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- 229920001214 Polysorbate 60 Polymers 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- WINXNKPZLFISPD-UHFFFAOYSA-M Saccharin sodium Chemical compound [Na+].C1=CC=C2C(=O)[N-]S(=O)(=O)C2=C1 WINXNKPZLFISPD-UHFFFAOYSA-M 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- 229920002125 Sokalan® Polymers 0.000 description 2
- 241000191967 Staphylococcus aureus Species 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 208000011341 adult acute respiratory distress syndrome Diseases 0.000 description 2
- 125000003545 alkoxy group Chemical group 0.000 description 2
- 150000005215 alkyl ethers Chemical class 0.000 description 2
- 230000007815 allergy Effects 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 229960003942 amphotericin b Drugs 0.000 description 2
- 229940121363 anti-inflammatory agent Drugs 0.000 description 2
- 239000002260 anti-inflammatory agent Substances 0.000 description 2
- 229940124599 anti-inflammatory drug Drugs 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 2
- 125000001589 carboacyl group Chemical group 0.000 description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 2
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 2
- 239000004359 castor oil Substances 0.000 description 2
- 235000019438 castor oil Nutrition 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 229960002626 clarithromycin Drugs 0.000 description 2
- AGOYDEPGAOXOCK-KCBOHYOISA-N clarithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@](C)([C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)OC)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 AGOYDEPGAOXOCK-KCBOHYOISA-N 0.000 description 2
- 210000001072 colon Anatomy 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- 230000002354 daily effect Effects 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- DOIRQSBPFJWKBE-UHFFFAOYSA-N dibutyl phthalate Chemical compound CCCCOC(=O)C1=CC=CC=C1C(=O)OCCCC DOIRQSBPFJWKBE-UHFFFAOYSA-N 0.000 description 2
- 238000007865 diluting Methods 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 230000009429 distress Effects 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 229940047650 haemophilus influenzae Drugs 0.000 description 2
- 229910052736 halogen Inorganic materials 0.000 description 2
- 125000001475 halogen functional group Chemical group 0.000 description 2
- 150000002367 halogens Chemical class 0.000 description 2
- 150000005826 halohydrocarbons Chemical class 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 2
- 229940071826 hydroxyethyl cellulose Drugs 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 239000012442 inert solvent Substances 0.000 description 2
- 230000028709 inflammatory response Effects 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 208000014674 injury Diseases 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 239000006199 nebulizer Substances 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 230000017095 negative regulation of cell growth Effects 0.000 description 2
- 230000003448 neutrophilic effect Effects 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 229940049954 penicillin Drugs 0.000 description 2
- 235000019271 petrolatum Nutrition 0.000 description 2
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 2
- 239000008363 phosphate buffer Substances 0.000 description 2
- 239000000049 pigment Substances 0.000 description 2
- 229920001983 poloxamer Polymers 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- 239000003380 propellant Substances 0.000 description 2
- 235000013772 propylene glycol Nutrition 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 238000010926 purge Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 239000011347 resin Substances 0.000 description 2
- 229920005989 resin Polymers 0.000 description 2
- 230000029058 respiratory gaseous exchange Effects 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- DAEPDZWVDSPTHF-UHFFFAOYSA-M sodium pyruvate Chemical compound [Na+].CC(=O)C([O-])=O DAEPDZWVDSPTHF-UHFFFAOYSA-M 0.000 description 2
- 239000012265 solid product Substances 0.000 description 2
- 239000003549 soybean oil Substances 0.000 description 2
- 235000012424 soybean oil Nutrition 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 229960005322 streptomycin Drugs 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 229960004793 sucrose Drugs 0.000 description 2
- 125000000542 sulfonic acid group Chemical group 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000002511 suppository base Substances 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 238000001757 thermogravimetry curve Methods 0.000 description 2
- 239000002562 thickening agent Substances 0.000 description 2
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 2
- 208000037816 tissue injury Diseases 0.000 description 2
- 235000010487 tragacanth Nutrition 0.000 description 2
- 239000000196 tragacanth Substances 0.000 description 2
- 229940116362 tragacanth Drugs 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- URAYPUMNDPQOKB-UHFFFAOYSA-N triacetin Chemical compound CC(=O)OCC(OC(C)=O)COC(C)=O URAYPUMNDPQOKB-UHFFFAOYSA-N 0.000 description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- 229920001285 xanthan gum Polymers 0.000 description 2
- 239000000811 xylitol Substances 0.000 description 2
- 235000010447 xylitol Nutrition 0.000 description 2
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 2
- 229960002675 xylitol Drugs 0.000 description 2
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- RXZBMPWDPOLZGW-XMRMVWPWSA-N (E)-roxithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=N/OCOCCOC)/[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 RXZBMPWDPOLZGW-XMRMVWPWSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- LVGUZGTVOIAKKC-UHFFFAOYSA-N 1,1,1,2-tetrafluoroethane Chemical compound FCC(F)(F)F LVGUZGTVOIAKKC-UHFFFAOYSA-N 0.000 description 1
- LDMOEFOXLIZJOW-UHFFFAOYSA-N 1-dodecanesulfonic acid Chemical compound CCCCCCCCCCCCS(O)(=O)=O LDMOEFOXLIZJOW-UHFFFAOYSA-N 0.000 description 1
- NOGFHTGYPKWWRX-UHFFFAOYSA-N 2,2,6,6-tetramethyloxan-4-one Chemical compound CC1(C)CC(=O)CC(C)(C)O1 NOGFHTGYPKWWRX-UHFFFAOYSA-N 0.000 description 1
- BDKLKNJTMLIAFE-UHFFFAOYSA-N 2-(3-fluorophenyl)-1,3-oxazole-4-carbaldehyde Chemical compound FC1=CC=CC(C=2OC=C(C=O)N=2)=C1 BDKLKNJTMLIAFE-UHFFFAOYSA-N 0.000 description 1
- IHCCLXNEEPMSIO-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperidin-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1CCN(CC1)CC(=O)N1CC2=C(CC1)NN=N2 IHCCLXNEEPMSIO-UHFFFAOYSA-N 0.000 description 1
- MFGOFGRYDNHJTA-UHFFFAOYSA-N 2-amino-1-(2-fluorophenyl)ethanol Chemical compound NCC(O)C1=CC=CC=C1F MFGOFGRYDNHJTA-UHFFFAOYSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical group CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- RVHOBHMAPRVOLO-UHFFFAOYSA-N 2-ethylbutanedioic acid Chemical compound CCC(C(O)=O)CC(O)=O RVHOBHMAPRVOLO-UHFFFAOYSA-N 0.000 description 1
- HDECRAPHCDXMIJ-UHFFFAOYSA-N 2-methylbenzenesulfonyl chloride Chemical compound CC1=CC=CC=C1S(Cl)(=O)=O HDECRAPHCDXMIJ-UHFFFAOYSA-N 0.000 description 1
- LEACJMVNYZDSKR-UHFFFAOYSA-N 2-octyldodecan-1-ol Chemical compound CCCCCCCCCCC(CO)CCCCCCCC LEACJMVNYZDSKR-UHFFFAOYSA-N 0.000 description 1
- AZKSAVLVSZKNRD-UHFFFAOYSA-M 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide Chemical compound [Br-].S1C(C)=C(C)N=C1[N+]1=NC(C=2C=CC=CC=2)=NN1C1=CC=CC=C1 AZKSAVLVSZKNRD-UHFFFAOYSA-M 0.000 description 1
- UOQHWNPVNXSDDO-UHFFFAOYSA-N 3-bromoimidazo[1,2-a]pyridine-6-carbonitrile Chemical compound C1=CC(C#N)=CN2C(Br)=CN=C21 UOQHWNPVNXSDDO-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- HIQIXEFWDLTDED-UHFFFAOYSA-N 4-hydroxy-1-piperidin-4-ylpyrrolidin-2-one Chemical compound O=C1CC(O)CN1C1CCNCC1 HIQIXEFWDLTDED-UHFFFAOYSA-N 0.000 description 1
- DFGKGUXTPFWHIX-UHFFFAOYSA-N 6-[2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]acetyl]-3H-1,3-benzoxazol-2-one Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)CC(=O)C1=CC2=C(NC(O2)=O)C=C1 DFGKGUXTPFWHIX-UHFFFAOYSA-N 0.000 description 1
- WSVLPVUVIUVCRA-KPKNDVKVSA-N Alpha-lactose monohydrate Chemical compound O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O WSVLPVUVIUVCRA-KPKNDVKVSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 244000144730 Amygdalus persica Species 0.000 description 1
- 108010011485 Aspartame Proteins 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 238000006237 Beckmann rearrangement reaction Methods 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 241000167854 Bourreria succulenta Species 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 125000002853 C1-C4 hydroxyalkyl group Chemical group 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- 229920000623 Cellulose acetate phthalate Polymers 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 229920001661 Chitosan Polymers 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 240000000560 Citrus x paradisi Species 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 1
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 229930006677 Erythromycin A Natural products 0.000 description 1
- 241001360526 Escherichia coli ATCC 25922 Species 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 238000012404 In vitro experiment Methods 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- 244000070406 Malus silvestris Species 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 240000008790 Musa x paradisiaca Species 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- NIPNSKYNPDTRPC-UHFFFAOYSA-N N-[2-oxo-2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 NIPNSKYNPDTRPC-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 208000000592 Nasal Polyps Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 208000031662 Noncommunicable disease Diseases 0.000 description 1
- 206010053159 Organ failure Diseases 0.000 description 1
- 235000019482 Palm oil Nutrition 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 239000004264 Petrolatum Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 241000384969 Priva Species 0.000 description 1
- 206010036790 Productive cough Diseases 0.000 description 1
- HDSBZMRLPLPFLQ-UHFFFAOYSA-N Propylene glycol alginate Chemical compound OC1C(O)C(OC)OC(C(O)=O)C1OC1C(O)C(O)C(C)C(C(=O)OCC(C)O)O1 HDSBZMRLPLPFLQ-UHFFFAOYSA-N 0.000 description 1
- 235000006040 Prunus persica var persica Nutrition 0.000 description 1
- 239000004373 Pullulan Substances 0.000 description 1
- 229920001218 Pullulan Polymers 0.000 description 1
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 235000019485 Safflower oil Nutrition 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- BCKXLBQYZLBQEK-KVVVOXFISA-M Sodium oleate Chemical compound [Na+].CCCCCCCC\C=C/CCCCCCCC([O-])=O BCKXLBQYZLBQEK-KVVVOXFISA-M 0.000 description 1
- HVUMOYIDDBPOLL-XWVZOOPGSA-N Sorbitan monostearate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O HVUMOYIDDBPOLL-XWVZOOPGSA-N 0.000 description 1
- 239000004147 Sorbitan trioleate Substances 0.000 description 1
- PRXRUNOAOLTIEF-ADSICKODSA-N Sorbitan trioleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCC\C=C/CCCCCCCC)[C@H]1OC[C@H](O)[C@H]1OC(=O)CCCCCCC\C=C/CCCCCCCC PRXRUNOAOLTIEF-ADSICKODSA-N 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 241000193996 Streptococcus pyogenes Species 0.000 description 1
- 241000320123 Streptococcus pyogenes M1 GAS Species 0.000 description 1
- 102000019259 Succinate Dehydrogenase Human genes 0.000 description 1
- 108010012901 Succinate Dehydrogenase Proteins 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- 244000172533 Viola sororia Species 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 229920000392 Zymosan Polymers 0.000 description 1
- YKTSYUJCYHOUJP-UHFFFAOYSA-N [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] Chemical compound [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] YKTSYUJCYHOUJP-UHFFFAOYSA-N 0.000 description 1
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 125000004466 alkoxycarbonylamino group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000005103 alkyl silyl group Chemical group 0.000 description 1
- OENHQHLEOONYIE-UKMVMLAPSA-N all-trans beta-carotene Natural products CC=1CCCC(C)(C)C=1/C=C/C(/C)=C/C=C/C(/C)=C/C=C/C=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C OENHQHLEOONYIE-UKMVMLAPSA-N 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- SNAAJJQQZSMGQD-UHFFFAOYSA-N aluminum magnesium Chemical compound [Mg].[Al] SNAAJJQQZSMGQD-UHFFFAOYSA-N 0.000 description 1
- 239000012080 ambient air Substances 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 239000012491 analyte Substances 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 230000000845 anti-microbial effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 229940127219 anticoagulant drug Drugs 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 235000021016 apples Nutrition 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 125000005228 aryl sulfonate group Chemical group 0.000 description 1
- 239000000605 aspartame Substances 0.000 description 1
- IAOZJIPTCAWIRG-QWRGUYRKSA-N aspartame Chemical compound OC(=O)C[C@H](N)C(=O)N[C@H](C(=O)OC)CC1=CC=CC=C1 IAOZJIPTCAWIRG-QWRGUYRKSA-N 0.000 description 1
- 235000010357 aspartame Nutrition 0.000 description 1
- 229960003438 aspartame Drugs 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 229960004099 azithromycin Drugs 0.000 description 1
- MQTOSJVFKKJCRP-BICOPXKESA-N azithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)N(C)C[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 MQTOSJVFKKJCRP-BICOPXKESA-N 0.000 description 1
- 235000021015 bananas Nutrition 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 239000011648 beta-carotene Substances 0.000 description 1
- 235000013734 beta-carotene Nutrition 0.000 description 1
- TUPZEYHYWIEDIH-WAIFQNFQSA-N beta-carotene Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1=C(C)CCCC1(C)C)C=CC=C(/C)C=CC2=CCCCC2(C)C TUPZEYHYWIEDIH-WAIFQNFQSA-N 0.000 description 1
- 229960002747 betacarotene Drugs 0.000 description 1
- 239000006177 biological buffer Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 210000001772 blood platelet Anatomy 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- CZBZUDVBLSSABA-UHFFFAOYSA-N butylated hydroxyanisole Chemical compound COC1=CC=C(O)C(C(C)(C)C)=C1.COC1=CC=C(O)C=C1C(C)(C)C CZBZUDVBLSSABA-UHFFFAOYSA-N 0.000 description 1
- HUCVOHYBFXVBRW-UHFFFAOYSA-M caesium hydroxide Inorganic materials [OH-].[Cs+] HUCVOHYBFXVBRW-UHFFFAOYSA-M 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 229940081734 cellulose acetate phthalate Drugs 0.000 description 1
- 229940081733 cetearyl alcohol Drugs 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 235000019693 cherries Nutrition 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 206010009887 colitis Diseases 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 229960001334 corticosteroids Drugs 0.000 description 1
- 239000006184 cosolvent Substances 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 229960001681 croscarmellose sodium Drugs 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 239000000625 cyclamic acid and its Na and Ca salt Substances 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 238000002784 cytotoxicity assay Methods 0.000 description 1
- 231100000263 cytotoxicity test Toxicity 0.000 description 1
- GVJHHUAWPYXKBD-UHFFFAOYSA-N d-alpha-tocopherol Natural products OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000007123 defense Effects 0.000 description 1
- 239000007857 degradation product Substances 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 238000010520 demethylation reaction Methods 0.000 description 1
- 230000001066 destructive effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 238000003113 dilution method Methods 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 229940088679 drug related substance Drugs 0.000 description 1
- 238000010410 dusting Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 239000003221 ear drop Substances 0.000 description 1
- 229940047652 ear drops Drugs 0.000 description 1
- 235000013399 edible fruits Nutrition 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003974 emollient agent Substances 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 239000003797 essential amino acid Substances 0.000 description 1
- 235000020776 essential amino acid Nutrition 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- MVPICKVDHDWCJQ-UHFFFAOYSA-N ethyl 3-pyrrolidin-1-ylpropanoate Chemical compound CCOC(=O)CCN1CCCC1 MVPICKVDHDWCJQ-UHFFFAOYSA-N 0.000 description 1
- 229960004667 ethyl cellulose Drugs 0.000 description 1
- 229960001617 ethyl hydroxybenzoate Drugs 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 239000004403 ethyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010228 ethyl p-hydroxybenzoate Nutrition 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- NUVBSKCKDOMJSU-UHFFFAOYSA-N ethylparaben Chemical compound CCOC(=O)C1=CC=C(O)C=C1 NUVBSKCKDOMJSU-UHFFFAOYSA-N 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 238000013100 final test Methods 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 229940044170 formate Drugs 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 125000005456 glyceride group Polymers 0.000 description 1
- 239000001087 glyceryl triacetate Substances 0.000 description 1
- 235000013773 glyceryl triacetate Nutrition 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 210000000224 granular leucocyte Anatomy 0.000 description 1
- 239000003979 granulating agent Substances 0.000 description 1
- 210000003714 granulocyte Anatomy 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- UBHWBODXJBSFLH-UHFFFAOYSA-N hexadecan-1-ol;octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO.CCCCCCCCCCCCCCCCCCO UBHWBODXJBSFLH-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 150000005828 hydrofluoroalkanes Chemical class 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 description 1
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 description 1
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 1
- 230000002519 immonomodulatory effect Effects 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000005462 in vivo assay Methods 0.000 description 1
- 238000012750 in vivo screening Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 230000004968 inflammatory condition Effects 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 208000030603 inherited susceptibility to asthma Diseases 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 208000028867 ischemia Diseases 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 125000005928 isopropyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(OC(*)=O)C([H])([H])[H] 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 238000012332 laboratory investigation Methods 0.000 description 1
- 150000003951 lactams Chemical class 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229940099563 lactobionic acid Drugs 0.000 description 1
- 229960001021 lactose monohydrate Drugs 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 230000004199 lung function Effects 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 229940041033 macrolides Drugs 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 229940037627 magnesium lauryl sulfate Drugs 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 1
- 229940057948 magnesium stearate Drugs 0.000 description 1
- HBNDBUATLJAUQM-UHFFFAOYSA-L magnesium;dodecyl sulfate Chemical compound [Mg+2].CCCCCCCCCCCCOS([O-])(=O)=O.CCCCCCCCCCCCOS([O-])(=O)=O HBNDBUATLJAUQM-UHFFFAOYSA-L 0.000 description 1
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 230000000873 masking effect Effects 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 210000003622 mature neutrocyte Anatomy 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 125000005395 methacrylic acid group Chemical group 0.000 description 1
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 244000000010 microbial pathogen Species 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 239000011859 microparticle Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- 239000007923 nasal drop Substances 0.000 description 1
- 229940100662 nasal drops Drugs 0.000 description 1
- 229920005615 natural polymer Polymers 0.000 description 1
- 230000007302 negative regulation of cytokine production Effects 0.000 description 1
- 208000004235 neutropenia Diseases 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- 239000012454 non-polar solvent Substances 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000008008 oral excipient Substances 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 229940116315 oxalic acid Drugs 0.000 description 1
- KHPXUQMNIQBQEV-UHFFFAOYSA-N oxaloacetic acid Chemical compound OC(=O)CC(=O)C(O)=O KHPXUQMNIQBQEV-UHFFFAOYSA-N 0.000 description 1
- 239000003346 palm kernel oil Substances 0.000 description 1
- 235000019865 palm kernel oil Nutrition 0.000 description 1
- 239000002540 palm oil Substances 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 229960000292 pectin Drugs 0.000 description 1
- 230000010412 perfusion Effects 0.000 description 1
- 206010034674 peritonitis Diseases 0.000 description 1
- 229940066842 petrolatum Drugs 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 239000008180 pharmaceutical surfactant Substances 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 210000004180 plasmocyte Anatomy 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 239000003880 polar aprotic solvent Substances 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 239000004584 polyacrylic acid Substances 0.000 description 1
- 229920000193 polymethacrylate Polymers 0.000 description 1
- 235000010989 polyoxyethylene sorbitan monostearate Nutrition 0.000 description 1
- 239000001818 polyoxyethylene sorbitan monostearate Substances 0.000 description 1
- 229920002503 polyoxyethylene-polyoxypropylene Polymers 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229950008882 polysorbate Drugs 0.000 description 1
- 229940113124 polysorbate 60 Drugs 0.000 description 1
- 229940100467 polyvinyl acetate phthalate Drugs 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 235000019422 polyvinyl alcohol Nutrition 0.000 description 1
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 1
- 229910000105 potassium hydride Inorganic materials 0.000 description 1
- 239000004540 pour-on Substances 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 235000010409 propane-1,2-diol alginate Nutrition 0.000 description 1
- 239000000770 propane-1,2-diol alginate Substances 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 229940095574 propionic acid Drugs 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 239000003586 protic polar solvent Substances 0.000 description 1
- 235000019423 pullulan Nutrition 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 229940076788 pyruvate Drugs 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 239000002516 radical scavenger Substances 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 238000005057 refrigeration Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- BOLDJAUMGUJJKM-LSDHHAIUSA-N renifolin D Natural products CC(=C)[C@@H]1Cc2c(O)c(O)ccc2[C@H]1CC(=O)c3ccc(O)cc3O BOLDJAUMGUJJKM-LSDHHAIUSA-N 0.000 description 1
- 238000001226 reprecipitation Methods 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 229960005224 roxithromycin Drugs 0.000 description 1
- 235000005713 safflower oil Nutrition 0.000 description 1
- 239000003813 safflower oil Substances 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 230000001953 sensory effect Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000002453 shampoo Substances 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 230000000391 smoking effect Effects 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 229940087562 sodium acetate trihydrate Drugs 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 229960001462 sodium cyclamate Drugs 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 229940054269 sodium pyruvate Drugs 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 229940045902 sodium stearyl fumarate Drugs 0.000 description 1
- 235000011076 sorbitan monostearate Nutrition 0.000 description 1
- 239000001587 sorbitan monostearate Substances 0.000 description 1
- 229940035048 sorbitan monostearate Drugs 0.000 description 1
- 235000019337 sorbitan trioleate Nutrition 0.000 description 1
- 229960000391 sorbitan trioleate Drugs 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 208000024794 sputum Diseases 0.000 description 1
- 210000003802 sputum Anatomy 0.000 description 1
- 239000012086 standard solution Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 229940086735 succinate Drugs 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 239000007916 tablet composition Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000005622 tetraalkylammonium hydroxides Chemical class 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 238000011287 therapeutic dose Methods 0.000 description 1
- 150000007970 thio esters Chemical class 0.000 description 1
- 239000003104 tissue culture media Substances 0.000 description 1
- 230000000451 tissue damage Effects 0.000 description 1
- 231100000827 tissue damage Toxicity 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- 229960001295 tocopherol Drugs 0.000 description 1
- 229930003799 tocopherol Natural products 0.000 description 1
- 235000010384 tocopherol Nutrition 0.000 description 1
- 239000011732 tocopherol Substances 0.000 description 1
- 210000003437 trachea Anatomy 0.000 description 1
- 238000005809 transesterification reaction Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
- 229960002622 triacetin Drugs 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- JOYRKODLDBILNP-UHFFFAOYSA-N urethane group Chemical group NC(=O)OCC JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 1
- 229940120293 vaginal suppository Drugs 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000004034 viscosity adjusting agent Substances 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 239000003871 white petrolatum Substances 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 1
- OENHQHLEOONYIE-JLTXGRSLSA-N β-Carotene Chemical compound CC=1CCCC(C)(C)C=1\C=C\C(\C)=C\C=C\C(\C)=C\C=C\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C OENHQHLEOONYIE-JLTXGRSLSA-N 0.000 description 1
Images

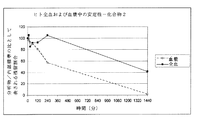

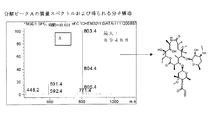














Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/04—Heterocyclic radicals containing only oxygen as ring hetero atoms
- C07H17/08—Hetero rings containing eight or more ring members, e.g. erythromycins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7048—Compounds having saccharide radicals and heterocyclic rings having oxygen as a ring hetero atom, e.g. leucoglucosan, hesperidin, erythromycin, nystatin, digitoxin or digoxin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Pulmonology (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Epidemiology (AREA)
- Urology & Nephrology (AREA)
- Rheumatology (AREA)
- Dermatology (AREA)
- Immunology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pain & Pain Management (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Description
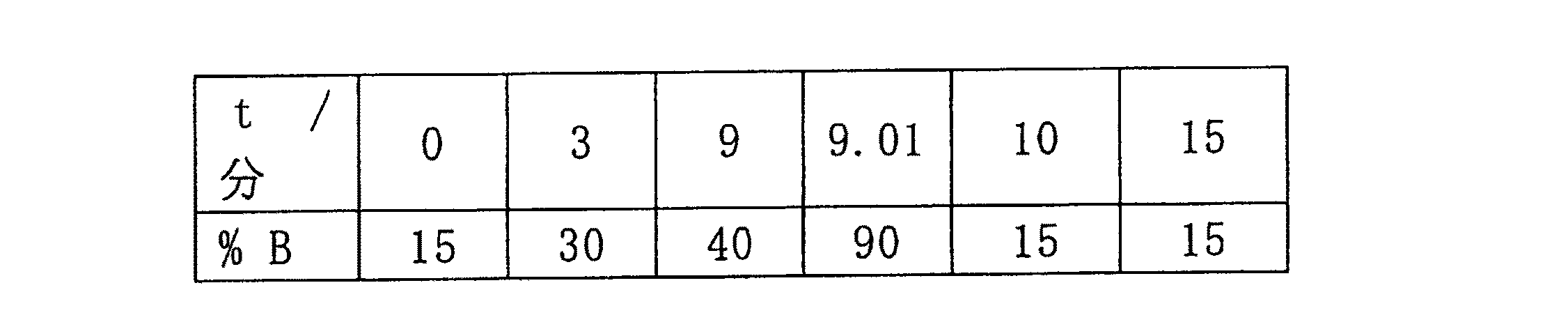
Aは、−C(O)−、−NHC(O)−、−C(O)NH−、−N(R7)CH2−、−CH2N(R7)−、−CH(OH)−および−C(=NOR7)−から選択される2価基であり;
R1は、−OC(O)(CH2)nNR8R9、−O−(CH2)nNR8R9、−OC(O)N(R7)(CH2)nNR8R9、
−O(CH2)nCN、−OC(O)(CH2)nN(CH2)nNR8R9または−OC(O)CH=CH2であり、ただし、R1が−OC(O)CH=CH2である場合、R3はメチルであってはならず;
R2は、水素またはヒドロキシル保護基であり;
R3は、水素、非置換C1〜4アルキル、または末端炭素原子がCNもしくはNH2基で置換されているC1〜4アルキル、またはC1〜5アルカノイルであり;
R4は、水素、C1〜4アルキルまたはC2〜6アルケニルまたはヒドロキシル保護基1であり;
R5は、ヒドロキシ、メトキシ基、−OC(O)(CH2)nNR8R9、−O−(CH2)nNR8R9または−O(CH2)nCNであり;
R6は、ヒドロキシであり;あるいは
R5およびR6は、介入原子と一緒になって以下の構造:
を有する環式基を形成し;
R7は、水素またはC1〜6アルキルであり;
R8およびR9は、それぞれ独立に、水素、C3〜7シクロアルキル、C1〜18アルキルであって、C1〜18アルキルは、
(i)中断されていない、または−O−、−S−および−N(R7)−から選択される1〜3個の2価基によって中断されており;および/または
(ii)非置換である、またはハロゲン、OH、NH2、N−(C1〜C6)アルキルアミノ(好ましくは、N−メチルアミノもしくはN−エチルアミノ)、N,N−ジ(C1〜C6−アルキル)アミノ(好ましくは、ジメチルアミノ、ジエチルアミノもしくはジイソプロピルアミノ)、CN、NO2、OCH3、飽和もしくは不飽和のC3〜8員非芳香環、酸素、硫黄および窒素から選択される1〜2個のヘテロ原子を含む飽和もしくは不飽和の2〜6個の炭素原子を含む非芳香複素環、アルキルカルボニルアルコキシ、ならびにアルコキシカルボニルアミノから選択される1〜3個の基によって置換されており;または
R8およびR9は、それらが結合する窒素と一緒になって、2〜6個の炭素原子を含む非芳香複素環を形成し、それは
i)酸素、硫黄および窒素から選択される0または1個の追加のヘテロ原子を含む、飽和または不飽和であり;および/または
ii)非置換である、またはC1〜5アルカノイルおよびC1〜6アルキルから選択される1〜2個の基によって置換されており、ここでC1〜6アルキルは、中断されていない、または−O−、−S−および−N(R7)−から選択される1〜3個の2価基によって中断されており、および/または、非置換である、またはOH、NH2、非置換である、もしくはC1〜4アルキル、ハロ、NH2、OH、SH、C1〜6アルコキシおよびC1〜4ヒドロキシアルキルから選択される基によって置換されている、2〜6個の炭素原子を含む非芳香複素環、非置換である、もしくはC1〜4アルキル、ハロ、NH2、OH、SH、C1〜6アルコキシおよびC1〜4ヒドロキシアルキルから選択される基によって置換されている、C3〜7シクロアルキルから選択される1〜2個の基によって置換されており;
nは1〜8の整数である;]
によって表されるマクロライド化合物、およびその薬学的に許容される誘導体を開示している。この文献は抗炎症活性を有しているとしてこれらの化合物を記載している。
1)図4と実質的に同じXRPDパターンによって特徴づけられる、式(I)によって表される結晶性化合物;
2)図5と実質的に同じDSCサーモグラムによって特徴づけられる、式(I)によって表される結晶性化合物;および/または
3)図6と実質的に同じTGA曲線によって特徴づけられる、式(I)によって表される結晶性化合物
を指す。
1)図7と実質的に同じXRPDパターンによって特徴づけられる、式(I)によって表される結晶性化合物;
2)図8と実質的に同じDSCサーモグラムによって特徴づけられる、式(I)によって表される結晶性化合物;および/または
3)図9と実質的に同じTGA曲線によって特徴づけられる、式(I)によって表される結晶性化合物
を指す。
1)図10と実質的に同じXRPDパターンによって特徴づけられる、式(I)によって表される結晶性化合物;
2)図11と実質的に同じラマンスペクトルによって特徴づけられる、式(I)によって表される化合物の結晶形態
を指す。
本発明の方法で使用するために、式(I)によって表される化合物またはその薬学的に許容される塩を原薬として投与することが可能であるが、有効成分を医薬製剤、例えば薬剤が投与の所期の経路および標準的な薬務に関して選択される少なくとも1つの薬学的に許容される担体との混合物中にあるものして、提供するのが好ましい。
式(I)によって表される化合物およびその塩は、以降で概説する一般的な方法によって調製され得、前記方法は本発明のさらなる態様を構成する。
HOC(O)CH2L (VI)
[式中、Lは、ハロゲン化物(例えば、塩化物、臭化物またはヨウ化物)、またはスルホン酸基(例えば、トシラート、メタンスルホナートまたはトリフラート)などの、上で定義した適当な脱離基である。]
によって表されるカルボン酸またはカルボン酸の適当な活性化誘導体との反応によって調製され得る。カルボキシル基の適当な活性化誘導体は、カルボン酸(III)について上で定義したものである。反応は、式(II)によって表される化合物と式(III)によって表されるカルボン酸との反応について上記の条件を使用して行われる。
本発明の化合物が、好中球浸潤から生じる好中球支配性炎症性疾患および/または好中球の異常細胞機能に関する疾患の治療における治療上の利点を提供する有利な特性を有する可能性は、例えば以下のアッセイを使用して説明され得る。
インビトロ血液および血漿安定性プロトコル
ヒト全血および血漿中のインビトロ安定性の調査を37℃で24時間にわたって行う。
24時間の、25℃、pH2〜pH8の緩衝液中、および37℃の生体関連培養液(SGF pH1.6、空腹時SIF pH6.5および食後SIF pH5.0)中の安定性を監視する。試料をLC/MSによって分析する。
pH2−リン酸緩衝液
pH3−クエン酸緩衝液
pH4−クエン酸緩衝液
pH5−クエン酸緩衝液
pH6−クエン酸緩衝液
pH7−リン酸緩衝液
pH8−TRIS緩衝液
SGF(人工胃液(Simulated Gastric Fluid))pH1.6
空腹時SIF(人工腸液(Simulated Intestinal Fluid))pH6.5
食後SIF(人工腸液)pH5.0
カラム:Waters XBridge C18、2.1×50mm、3.5μm
ガードカラム:Waters XBridge C18、2.1×10mm、3.5μm
移動相:A)0.1%HCOOH/H2O
B)0.1%HCOOH/CH3CN
勾配タイムテーブル:
カラム温度:25℃
試料温度:25℃(緩衝液中の安定性アッセイについて)
37℃(生体関連培養液中の安定性アッセイについて)
インジェクター容量:3μL(注入プログラムなし)
API−ES、陽イオンモード
質量範囲:150〜1500
フラグメンター:100
ゲイン:1.0
閾値:150
ステップサイズ:0.20
ガス温度:350℃
乾燥ガスフロー:13L/分
ネブライザー圧力:35psig
キャピラリー電圧:正 4000V
負 4000V
a)緩衝液中の安定性アッセイについて
化合物をCH3CNに溶解し、1mg/mLのストック溶液を作成する。ストック溶液を適当な緩衝液を用いて0.2mg/mLの濃度(4mL緩衝液中1mL溶液)に希釈し、使用溶液を作成する。
b1)pH1.6のSGF培養液の試験について
化合物をpH1.6のSGF培養液に溶解し、化合物1および2の場合には0.2mg/mL溶液、化合物3の場合には0.1mg/mL溶液を得る。次いで、溶液を37℃の水浴中に保管し、LC/MS分析のために適当な時点で取り出す。分析の前に、調製した試料溶液のpH値を確認し、必要に応じて調整する。
化合物をpH6.5の空腹時SIF培養液に溶解し、2mg/mL溶液を得る。次いで、溶液をバイアルに入れて、37℃の水浴中に保管し、LC/MS分析のために適当な時点で取り出し、CH3CNで10倍希釈する。分析の前に、調製した試料溶液のpH値を確認し、必要に応じて調整する。
化合物をpH5.0の食後SIF培養液に溶解し、2mg/mL溶液を得る。次いで、溶液をバイアルに入れて、37℃の水浴中に保管し、LC/MS分析のために適当な時点で取り出し、CH3CNで40倍希釈する。分析の前に、調製した試料溶液のpH値を確認し、必要に応じて調整する。
雄BALB/cJマウスにおける細菌性リポ多糖(LPS)によって誘発される肺好中球増加症
腹腔内投与(i.p.)のために、化合物を10mg/mLの最終濃度に溶解する。化合物の必要量を最初にジメチルスルホキシド(DMSO、シグマ社製)に溶解し、次いで、0.5%(w/v)メチルセルロースで希釈して、最終DMSO濃度が5%(v/v)となるようにする。得られた溶液を動物10g当たり0.2mLの用量容積で適用する。そのため、化合物用量は200mg/kgである。
抗菌性スクリーニング
臨床的に関連する細菌(Staphylococcus aureus ATCC13709、Streptococcus pneumoniae ATCC49619、Streptococcus pyogenes ATCC700294、Moraxella catarrhalis ATCC23246、Haemophilus influenzae ATCC49247およびEscherichia coli ATCC25922)に対する化合物の全細胞抗菌活性を、臨床検査標準(CLSI)推奨手順(Document M7〜A6A7、Methods for Dilution Susceptibility Tests for Bacteria that Grow Aerobically)を使用する微量液体希釈法によって測定する。化合物をDMSOに溶解し、50mMストック溶液を作成する。ストック溶液を適当な培養液(Haemophilus influenzaeにはヘモフィリステスト培地、Staphylococcus aureus、Moraxella catarrhalisおよびEscherichia coliにはミューラーヒントン培地、ならびにStreptococcus pyogenesおよびStreptococcus pneumoniaeには5%ウマ血清を補充したミューラーヒントン培地)中128μg/mLの濃度に希釈して使用溶液を作成する。Tecan Genesis150Workstation(Tecan Group Ltd.社製 Mannedorf、スイス)を使用して、U字型底96ウェルマイクロタイタープレート中に、使用溶液の2倍希釈系列(50μLアリコート)を調製する。化合物を希釈した後、50μLアリコートの試験単離物(約1×106cfu/ml)を、マイクロタイタープレートの各々のウェルに添加する。最終的な試験濃度は、0.125〜64μg/mLに及ぶ。接種されたプレートを周囲空気中、35℃で18〜24時間インキュベートする。最小発育阻止濃度(MIC)は、目に見える発育を阻害した最も低い化合物の濃度として決定する。
10%ウシ胎児血清(FBS;BioWest社製)、50U/mLペニシリン、50μg/mLストレプトマイシンおよび2.5μg/mLアムホテリシンB(ファンギゾン)(全てギブコ社製)を補充したRPMI1640培地(Institute of Immunology製、Zagreb)中でTHP−1細胞を培養する。
HepG2細胞は、10%FBS、1mMピルビン酸ナトリウム、0.1mM非必須アミノ酸、50U/mLペニシリン、50μg/mLストレプトマイシンおよび2.5μg/mLアムホテリシンB(ファンギゾン)(全てギブコ社製)を含むイーグル最小必須培地(MEM;ギブコ社製)中で維持する。
細胞発育の阻害%=OD492処理細胞/OD492未処理細胞×100
を使用して計算する。
粉末X線回折(XRPD)データは、X’Celerator検出器を使用して、PANalytical X’Pert Pro粉末回折装置、モデルPW3040/60で得た。収集条件は以下の通りであった。
a)放射線:Cu Kα、発生装置電圧:45kV、発生装置電流:40mA、開始角度:3.0°2θ、終了角度:40.0°2θ、ステップサイズ:0.0167°2θ、ステップ当たりの時間:104.7秒;または
b)放射線:Cu Kα、発生装置電圧:40kV、発生装置電流:45mA、開始角度:2.0°2θ、終了角度:40.0°2θ、ステップサイズ:0.0167°2θ、ステップ当たりの時間:31.75秒。
数ミリグラムの試料をSiウエハー(ゼロバックグラウンド)プレート上に載せ、分析のために粉末の薄層にすることによって、試料を調製した。
各々の2シータ角度割当ておよびd−格子面間隔にはいくらかの許容誤差が存在する。許容誤差は、値が測定される正確な温度を含むいくつかの因子に依存するだろう。前述の2シータ角度における許容誤差は、各々の前述のピーク割当てにつき約±0.2度である。2シータ角度割当ておよびd−格子面間隔においてはいくらかの許容誤差が可能であるので、式(I)によって表される化合物の試料の特定の形態を同定するためにXRPDパターンを比較する有用な方法は、式(I)によって表される化合物のある試料形態のXRPDパターンを、式(I)によって表される化合物の他の試料形態のXRPDパターンの上に重ねることである。
全ラマン分析は、自動XYZステージを備えるKaiser Optical Systems HoloWell分散型ラマン分光計で行った。785nmのダイオードレーザーによって励起をもたらした。約5〜20mgの試料をホウケイ酸塩フリットプレート上に、またはガラス試料バイアル中に置いた。観察されたラマンピークのわずかな変動は、使用した特定の分光計および分析者の試料調製技術に基づくと考えられる。前述のバンド位置の許容誤差は±2cm−1である。本明細書で提供されるラマンスペクトルは、x軸にcm−1の波数およびy軸に任意の単位の強度を示す。
冷凍冷却システムを備えるMettler Toledo DSC 822e熱量計でDSCを行った。試料をピンホールアルミニウム皿中で、10℃/分の加熱速度で25℃から300℃まで加熱した。試料に対して50mL/分の窒素パージを維持した。
観察された吸熱の有意な変動は、式(I)によって表される化合物の各々の形態のDSCサーモグラムに関しては、使用した特定の装置および皿の配置、分析者の試料調製技術ならびに試料の粒度および重量に基づくと考えられる。吸熱特性においては通常いくらかの許容誤差が存在する。すなわち、許容誤差は約±2.00℃程度である。
Mettler Toledo TGA/SDTA 851eシステムでTGAを行った。試料を予め風袋計量したアルミナるつぼに入れ、10℃/分の加熱速度で25℃から300℃まで加熱した。試料に対して50mL/分の窒素パージを維持した。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA
方法A
3’−N−デメチル−2’−O−3’−N−ジ−{[(フェニルメチル)オキシ]カルボニル}−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA
クロロギ酸ベンジル(30mL、209.7mmol)中のNaHCO3(13.2g、157.3mmol)の加熱した(60℃)懸濁液に、6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA(10g、13.1mmol)を2時間で徐々に添加した。反応混合物を60℃でさらに1時間攪拌した。次いで、反応混合物を室温に冷却し、ヘキサン(150mL)および水(150mL)を添加して、得られた粘着性沈殿を濾別し、EtOAc(150mL)に溶解して、水(2×100mL)で洗浄した。無水Na2SO4で乾燥後、有機層を蒸発させ、残渣にジエチルエーテル(80mL)を添加し、氷冷下でヘキサン(200mL)を添加することによって生成物を沈殿させた。得られた沈殿を濾過し、冷ヘキサン(50mL)で洗浄して標記中間体1(12.7g)、MS(ES+)m/z:1039.28[MH+Na]+を得た。
ジエチルアミノ酢酸
ジエチルアミン(230mL、2.2mol)中のクロロ酢酸(30g、0.32mol)の溶液を一晩攪拌した。得られた白色沈殿を濾過によって除去した。濾液を蒸発乾固し、黄色油を得て、それにEtOAc(300mL)を添加した。得られた沈殿を再度濾過によって除去した。濾液を蒸発させて黄色固体残渣(44g)を得、それをヘキサン(2×100mL)、EtOAc(2×50mL)および再度ヘキサン(50mL)で洗浄し、次いで真空中で乾燥させて標記中間体2(38g)を得た。
1H NMR(500MHz、CHLOROFORM−d)δppm8.85(1H、br.s)、3.56(2H、s)、3.29(4H、q、J=7.3Hz)、1.36(6H、t、J=7.3Hz)。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−2’−O−3’−N−ジ−{[(フェニルメチル)オキシ]カルボニル}−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA
DCM(35mL)中の中間体1(12.7g、12.5mmol)の溶液に、EDC×HCl(14.4g、74.9mmol)、DMAP(9.15g、74.9mmol)および中間体2(ジエチルアミノ酢酸)(9.83g、74.9mmol)を添加した。反応混合物を室温で一晩(約17時間)攪拌し、EtOAc(450mL)で希釈し、飽和NaHCO3(3×170mL)、飽和NH4Cl(3×170mL)および食塩水(350mL)で洗浄した。有機相を無水Na2SO4で乾燥させ、溶媒を蒸発させ、残渣を石油エーテル(250mL)に懸濁し、2時間攪拌し、濾過し、石油エーテル(30mL)で洗浄して標記中間体3(12.2g)、MS(ES+)m/z:1130.39[MH]+を得た。
方法1.EtOH(200mL)中の中間体3(12.2g、10.8mmol)の溶液に、10%Pd/C(1.22g)を添加し、反応混合物を5バールで一晩(約17時間)水素化(hydrogenolize)した。触媒を濾過によって除去し、濾液を蒸発させた。残渣にDCM(200mL)および水(200mL)を添加し、pHを4.7に調整した(1N HCl)。層を分離し、DCM(3×150mL)でpH4.7での抽出を繰り返した。水層に新しいDCM(150mL)を添加し、pHを5.7に調整した(1N NaOH)。層を分離し、DCM(2×20mL、7×150mL)でpH5.7での抽出を繰り返した。pH5.7の合わせた有機層を150mLの容積に濃縮し、水(150mL)を添加し、pHを9.5に調整した(水/NH4OH=1/1)。有機層を分離し、蒸発させて標記生成物実施例1(7.25g、MS(ES+)m/z:862.34[MH]+)を得た。
MS(ES+)m/z:862.11[MH]+。
1H NMR(500MHz、CHLOROFORM−d)δppm6.12(1H、d、J=8.2Hz)、4.88(1H、d、J=4.9Hz)、4.72(1H、d、J=9.8Hz)、4.68(1H、dd、J=10.7、1.5Hz)、4.55(1H、d、J=7.6Hz)、4.39(1H、m)、4.21(1H、dd、J=6.1、1.5Hz)、4.16(1H、m)、3.79(1H、m)、3.74(1H、d、J=6.4Hz)、3.43(1H、d、J=17.2Hz)、3.31(3H、s)、3.31(3H、s)、3.31(1H、d、J=17.1Hz)、3.28(1H、d、J=4.3Hz)、3.21(1H、br.s.)、3.14(1H、dd、J=9.8、7.3Hz)、2.83(1H、m)、2.69(4H、q、J=7.3Hz)、2.59(1H、ddd、J=11.5、9.8、4.6Hz)、2.43(3H、s)、2.37(1H、d、J=15.0Hz)、2.22(1H、dq、J=7.3、7.1Hz)、2.01(1H、dd、J=15.0、8.2Hz)、1.89(3H、m)、1.62(1H、dd、J=15.0、4.9Hz)、1.56(1H、m)、1.33(3H、s)、1.29(1H、d、J=13.7Hz)、1.24(3H、d、J=7.6Hz)、1.16(12H、m)、1.12(3H、s)、1.09(4H、d、J=7.1Hz)、1.07(6H、t、J=7.3Hz)、0.98(3H、d、J=7.6Hz)、0.90(3H、t、J=7.5Hz)。
中間体1:
3’−N−デメチル−2’−O−3’−N−ジ−{[(フェニルメチル)オキシ]カルボニル}−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA
工程a)6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA
6−O−メチルエリスロマイシンA 9(E)−オキシム(2.0kg)を50Lガラスライニング反応器に装入し、引き続いてアセトン(20L)を装入した。混合物を−10℃に冷却し、アセトン(5.5L)中のトルエン−2−スルホニルクロリド(775g、1.55eq)を20分間添加した。添加中、温度は−8℃に上がった。次いで、内部温度を0℃未満に維持しながら、水(25L)中のNaHCO3(460g)を1時間40分間滴加した。混合物を20℃に加温し、2時間攪拌して、次いで1M KOH(6L)を添加した。加熱マントルを70℃に設定し、反応物容積を真空中で約18Lに減少させた。混合物を10℃に冷却し、ヌッチェ(ザイツK200フィルター)で濾過し、濾過ケークを水(8L)で洗浄した。重い灰白色沈殿をフィルター上で一晩、次いで真空中32℃で風乾して標記生成物(1670g、Y=83.5%、LC−MS純度は参照に一致する)を得た。
バッチA) 6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA(1.01kg)を20L反応器に装入し、引き続いてジオキサン(4L)およびNaHCO3(1.49kg)を装入した。加熱マントル温度を90℃に設定した。内部温度が72℃に達したら、クロロギ酸ベンジル(800mL、4.4equiv.カルボベンゾキシクロリドまたはフェニルメチルクロロホルマートとしても知られている)の添加を開始し、4時間10分間続けた。30分後、余分のクロロギ酸ベンジル(30mL)を添加した。LC−MSによって完全な転換が確認されたので、混合物を20℃に冷却し、MTBE(6L)で希釈し、0.01M KOH(5L)でクエンチして、一晩攪拌した。水層を除去し、有機層を水(2×5L)および食塩水(2L)で洗浄した。有機層の容積は約7Lであり、この溶液を追加の漏斗に移した。ヘプタン(14L)を反応器に装入し、10℃に冷却した。次いで、追加の漏斗からのMTBE溶液を、攪拌ヘプタンに慎重に添加した。溶液の約2/3を添加した後に、懸濁粒子が凝塊を形成し始め、反応器の底に粘着性油が得られた。ほぼ全てのヘプタンを除去した後、全てが溶解するまでMTBE(1L)およびDCM(5L)を添加した。
得られた溶液をバッチBに添加した。
ジエチルアミノ酢酸
ジエチルアミン(9.5L)を、20Lガラスライニング反応器に装入し、3℃に冷却した。MTBE(1.8L)中のクロロ酢酸(1.21kg)を冷却しながら1時間にわたって添加した−添加中、温度は16℃に上がった。混合物を20℃に加温し、一晩攪拌し、ロータリーエバポレーターで濃縮して黄色油を得た。この油をiPrOAcとともに再濃縮し、iPrOAc(4.5L)と混合し、一晩攪拌した。灰白色吸湿性沈殿を濾過し、iPrOAc(1.5L)で洗浄し、直ちに乾燥キャビネットに入れて潮解を避けた。乾燥後、標記生成物が得られた(820g、Y=49%)。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−2’−O−3’−N−ジ−{[(フェニルメチル)オキシ]カルボニル}−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA
DCM(10L)を50Lガラスライニング反応器に装入し、内部温度を+5℃に調整した。次いで、全試薬を一度に以下の順で装入した:3’−N−デメチル−2’−O−3’−N−ジ−{[(フェニルメチル)オキシ]カルボニル}−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA(2800g、湿潤物質)、ジエチルアミノ酢酸(800g、2.2eq)、DMAP(745g、2.2eq)およびEDC×HCl(1170g、2.2eq)。これは+2℃への吸熱温度減少をもたらした。混合物を+20℃で一晩攪拌し、+10℃に冷却し、飽和NaHCO3水溶液(20L)でクエンチし、0.5時間攪拌した。EtOAcを添加し(25L)、水層を分離し捨てて、有機層を飽和NH4Cl水溶液(2×12L)および食塩水(10L)で洗浄した。容積を真空中で約8Lに減少させた。ヘプタン(45L)を第2反応器に装入し、8〜10℃に冷却した。DCM/EtOAc濃縮溶液を、よく攪拌しながら20分間にわたって、冷却したヘプタンに慎重に添加した。得られた白色非晶質沈殿をヌッチェ(ザイツK200フィルター)で濾過し、ヘプタン(8L)で洗浄し、一晩フィルター上で風乾して粗製標記生成物(2.5kg、Y=79%、LC−MSプロファイルは参照に一致する)を得た。
粗製固体(2.38kg)を50℃で還流MTBE(18.3L)に溶解し、内部温度を約50℃に維持しながら、ヘプタン(15L)を20分間にわたってゆっくり添加した。混合物を接種し、20℃に冷却させ、一晩攪拌した。白色沈殿を濾過し、MTBE/ヘプタン1:1(5L)、次いで純ヘプタン(3L)で洗浄し、24時間風乾して、標記生成物(1.9kg、Y=63%、LC−MSプロファイルは参照に一致する、LC純度98.5%)を得た。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−2’−O−3’−N−ジ−{[(フェニルメチル)オキシ]カルボニル}−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA(1.9kg)をMeOH(16L)に溶解し、得られた溶液を20Lオートクレーブに装入し、引き続いてMeOH(0.5L)中の10%Pd/C(150g、湿気60%H2O)を添加した。オートクレーブを排気し、水素を充満させた(全ての残存酸素を除去するために2度繰り返した)。オートクレーブを水素で再充満した後、混合物を1.5〜3バールで10時間水素化した。セライトを通してPd/Cを濾過し、ケークをMeOH(1L)で洗浄した。得られた溶液を50L反応器に装入し、Pdスカベンジャー(Biotage MP−TMT、50g)を添加し、混合物を室温で一晩攪拌した。次いで、混合物を濾過し、プラスチック容器に移して−10℃で保管した。その後、MeOH混合物を反応器に移し、ほぼ乾燥するまで濃縮した。次いで、EtOAcを装入し(12.5L)、引き続いて水(10L)および1M HCl溶液(3L)を装入した。有機相を分離し捨てた。水相に、固体NaHCO3(280g)およびEtOAc(10L)を添加し、食塩水(1L)を添加して分離を達成した。層を分離した後、生成物をEtOAc(2×7L)で抽出し、合わせた有機相を真空中でほぼ乾燥するまで濃縮し、ヘプタン(2L)で希釈して、再度濃縮した。次いで、ジエチルエーテル(7L)を添加し、混合物を3.5時間攪拌し、白色沈殿を濾過し、約2Lのジエチルエーテルで洗浄し、一晩風乾して標記生成物実施例1(946g、Y=65%、1H NMRプロファイルは参照に一致する、C/3’−NHCH3のまわりに位置するいくつかのプロトンのケミカルシフトにおいて差異が存在し、これは部分的なHCl塩形成を示唆している、HPLC純度97.8%)を得た。
相当量の沈殿が反応器中に残った。これをMeOHに溶解し、ロータリーエバポレーターで濃縮した。得られた油状残渣をジエチルエーテル(2L)中にスラリーにし、3時間攪拌し、濾過し、ジエチルエーテル(300mL)で洗浄し、一晩風乾して追加量の粗製標記生成物(250g、Y=17%)を得た。
アンモニア水または飽和NaHCO3溶液のいずれかを使用して、遊離塩基化(freebasing)を達成した:
生成物(20g、10gの部分的HCl塩+10gの粗製物)をDCM(150mL)に溶解し、アンモニア水溶液(12.5%、100mL)または飽和NaHCO3溶液(100mL)のいずれかと混合し、1.5時間攪拌した。層を分離し、水層を追加のDCM(75mL)で抽出した。合わせた有機層をほぼ乾燥するまで濃縮し、ジエチルエーテル(100mL)中にスラリーにし、容積をわずかに減少させ(約90mLまで)、3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA、形態1(XRPDパターンは、実施例11、形態1のXRPDパターンに一致した)のゆっくりした沈殿を得た。
中間体1:
3’−N−デメチル−2’−O−3’−N−ジ−{[(フェニルメチル)オキシ]カルボニル}−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA
工程a)6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA
2.5±2.5℃に冷却したアセトン(72.0L)中の6−O−メチルエリスロマイシンA 9(E)−オキシム(12.00kg)の溶液に、水(120.0L)中のNaHCO3(3.3kg)の溶液を2時間添加し、温度を2.5±2.5℃に維持した。アセトン(12.0L)中のp−トルエンスルホニルクロリド(4.79kg)の溶液を、冷却下15分以内でこの混合物に添加し、次いで、周囲温度に加温し(1時間15分)、2時間攪拌した。反応混合物を2等分し(約102.0L)、その各々を新しい反応器に移し、各々半分に水(96.0L)を添加し、反応混合物を約1.5時間で7.5±2.5℃に冷却した。次いで、各々半分を、10.0Lの予め調製したNaOH(36.0Lの水中の1.80kg)水溶液を約1時間添加することによって、pHをpH9.0〜11.5に調整した。得られた懸濁液を7.5±2.5℃で2時間熟成させ、遠心濾過によって単離し、水で3度洗浄して(24.0L、48.0Lおよび24.0L)、湿潤生成物(13.50kg)を得た。湿潤生成物を真空下40±5℃で10時間乾燥させて、標記生成物(10.82kg、1H NMRプロファイルは参照に一致する)を得た。
バッチA) ジオキサン(40.0L)を反応器に装入し、温度を30±5℃に設定し、6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA(8.00kg)、引き続いてNaHCO3(10.56kg)を添加した。30±5℃で30分間、この混合物に、クロロギ酸ベンジル(9.00kg)をゆっくり添加した。添加後、混合物を60±5℃に加熱し、2.5時間維持した。反応混合物を新しい反応器に移し、7.5±7.5℃に冷却し、MTBE(80.0L)で希釈し、次いで7.5±7.5℃で、トリエチルアミン(8.0L)でゆっくり処理した。予め冷却した0.01N水酸化カリウム溶液(60.0Lの水中の4.035kg)を、7.5±7.5℃の上記反応混合物に添加し、12.5±2.5℃で10分間攪拌した。層を分離し、温度を12.5±5℃に維持しながら、上層の有機層を水で2度(2×40.0L)、次いで塩化ナトリウム水溶液(40.0L中8.00kg)で洗浄した。
有機層を、留出物が観察されなくなるまで、高真空(NLT650mm/Hg)下60℃未満で濃縮した。MTBE(16.0L)を残渣に添加し、32.5±7.5℃で攪拌して、溶液を得た。得られた溶液を追加の容器に移し、30±5℃で1時間にわたってヘキサン(160.0L)にゆっくり添加し、MTBE(4.0L)で追加の容器をすすいだ。混合物を30±5℃で45分間攪拌し、2.5±2.5℃に冷却し、90分間熟成させ、遠心濾過によって単離した。ケークをヘキサン(16.0L)で洗浄し、真空下30±5℃で5時間乾燥させて粗製固体生成物を得た。
粗製固体を、MTBE(16.0L)およびトリエチルアミン(8.0L)の混合物中に懸濁し、65±10℃で12時間加熱した後、周囲温度に冷却し、さらなるMTBE(16.0L)で希釈した。スラリーを周囲温度でさらに12時間熟成させた後、遠心濾過によって単離した。ケークをMTBE(16.0L)で洗浄し、真空下30±5℃で10時間乾燥させて、標記生成物(6.95kg)を得た。
有機層を、留出物が観察されなくなるまで、高真空(NLT650mm/Hg)下60℃未満で濃縮した。MTBE(20.0L)を残渣に添加し、32.5±7.5℃で攪拌して、溶液を得た。得られた溶液を追加の容器に移し、30±5℃で1時間20分間にわたってヘキサン(200.0L)にゆっくり添加し、MTBE(5.0L)で追加の容器をすすいだ。混合物を30±5℃で30分間攪拌し、2.5±2.5℃に冷却し、90分間熟成させ、遠心濾過によって単離した。ケークをヘキサン(20.0L)で洗浄し、真空下30±5℃で5時間乾燥させて粗製固体生成物を得た。
粗製固体を、MTBE(20.0L)およびトリエチルアミン(10.0L)の混合物中に懸濁し、65±10℃で9時間加熱した後、周囲温度に冷却し、さらなるMTBE(20.0L)で希釈した。スラリーを周囲温度でさらに10時間熟成させた後、遠心濾過によって単離した。ケークをMTBE(15.0L)で洗浄し、真空下65±10℃で8時間乾燥させて、標記生成物(8.75kg)を得た。
ジエチルアミノ酢酸
ジエチルアミン(112.0L)を反応器に装入し、2.5±2.5℃に冷却した。温度を5±5℃に維持しながら、クロロ酢酸(14.00kg)を30分間少しずつ添加した。混合物を1時間、30±5℃に加温し、次いで、同温度で一晩(約17時間)攪拌した。反応混合物を、ヌッチェフィルターを通して濾過し、濾液を乾燥容器に回収した。ケークを酢酸エチル(28.0L)で洗浄し、濾液を乾燥容器に回収した。
酢酸エチル(70.0L)を反応器に装入し、湿潤ケークを添加し、30±5℃で20分間スラリー化した。ヌッチェフィルターを通してスラリーを濾過し、濾液を乾燥容器に回収した。ケークを酢酸エチル(28.0L)で洗浄し、濾液を乾燥容器に回収した。
合わせた回収濾液を新しい反応器に装入し、留出物が観察されなくなるまで、真空下60℃未満で溶媒を蒸留して取り除いた。残渣を30±5℃に冷却し、アセトニトリル(14.0L)およびアセトン(28.0L)の混合物を添加し、25±2℃で11時間攪拌した。得られた吸湿性沈殿を窒素フロー下で濾過した。湿潤ケークをアセトン(20.0L)で洗浄し、真空下(650mmHg)45±5℃で約1.5日間乾燥させて標記生成物(4.32kg)を得た。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−2’−O−3’−N−ジ{[(フェニルメチル)オキシ]カルボニル}−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA
DCM(40.0L)を反応器に装入し、温度を30±5℃に調整し、3’−N−デメチル−2’−O−3’−N−ジ−{[(フェニルメチル)オキシ]カルボニル}−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA(8.0kg)を添加して、透明な溶液が観察されるまで攪拌した。次いで、攪拌し、温度を30±5℃に維持している状態で、ジエチルアミノ酢酸(1.55kg)、引き続いてDMAP(0.96kg)を添加した。反応混合物を2.5±2.5℃に冷却した。EDC×HCl(1.50kg)をこの温度で添加して透明な溶液を得た。反応混合物を25±5℃にゆっくり加温し、25±5℃で5時間攪拌した。さらなるDMAP(1.20kg)を添加し、反応物を再度2.5±2.5℃に冷却した。さらなるEDC×HCl(1.88kg)をこの温度で添加して透明な溶液を得た。反応混合物を25±5℃にゆっくり加温し、25±5℃で11時間攪拌した。
次いで、留出物が観察されなくなるまで、混合物を真空下30℃未満で濃縮し、残渣をトルエン(80.0L)に溶解し、混合物を15±5℃に冷却した。有機溶液を炭酸水素ナトリウム水溶液(40.0Lの水中の4.00kg)で2度洗浄し、各々の抽出後の水層を15±5℃に維持した。上層の有機層を塩化アンモニウム水溶液(40.0Lの水中の12.00kg)で2度洗浄し、各々の抽出後の水層を10〜15℃に維持した。合わせた水性洗浄液(炭酸水素ナトリウムおよび塩化アンモニウム)をトルエン(40.0L)で逆抽出し、有機層を炭酸水素ナトリウム水溶液(40.0Lの水中の4.00kg)および塩化アンモニウム水溶液(40.0Lの水中の12.00kg)で連続的に洗浄した。
合わせた有機相を塩化ナトリウム水溶液(40.0Lの水中の12.00kg)で洗浄し、有機層を無水硫酸ナトリウムで乾燥させ、ヌッチェフィルターを通して濾過し、トルエン(16.0L)で洗浄した。約5.0〜5.5容積が反応器内部に残るまで、合わせた濾液および洗浄液を真空下50℃未満で濃縮した。ヘプタン(80.0L)を35±5℃で30分間にわたってゆっくり添加し、得られた懸濁液を30±5℃で13時間攪拌した。遠心濾過によって生成物を単離し、ヘプタン(16.0L)で洗浄した。生成物を高真空下(NLT650mm/Hg)40±5℃で11時間乾燥させて標記生成物(7.30kg)を得た。
反応器を排気し、窒素で洗浄し(2度繰り返した)、窒素フロー下でi−PrOH(36.0L)および3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−2’−O−3’−N−ジ{[(フェニルメチル)オキシ]カルボニル}−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA(4.5kg)を添加し、溶液を10分間攪拌した。次いで、10%Pd/C(0.225kg)、引き続いてトリエチルアミン(1.80L)を添加し、窒素フローを止め、反応器を排気し、20psi圧力が得られるまで水素を充満させた(2度繰り返した)。反応器を水素で再充満した後、混合物を20psi、30±5℃で2時間水素化した。ヌッチェフィルターのHyflowベッドを使用してパラジウム炭素を除去し、i−PrOH(18.0L)で洗浄し、50℃未満で留出物がなくなるまで、合わせた濾液を高真空下(NLT650mm/Hg)で濃縮した。新しいi−PrOH(9.0L)を残渣に添加し、50℃未満で留出物がなくなるまで、混合物を高真空下(NLT650mm/Hg)で濃縮した。
濃縮物質をIPA(45.0L)に溶解し、きれいな反応器に移し、7.5±2.5℃に冷却し、チオシリカ(0.18kg)で処理し、7.5±2.5℃で3時間攪拌した。ヌッチェフィルターのHyflowベッドを通した濾過によってチオシリカを除去し、i−PrOH(18.0L)で洗浄し、50℃未満で留出物がなくなるまで、合わせた濾液を高真空下(NLT650mm/Hg)で濃縮した。
ヘプタン(13.5L)を上記混合物に添加し、50℃未満で留出物がなくなるまで、高真空下(NLT650mm/Hg)で濃縮した。さらなるヘプタン(13.5L)を上記混合物に添加し、50℃未満で留出物がなくなるまで高真空下(NLT650mm/Hg)で濃縮した。ヘプタン(22.5L)を上記混合物に添加し、得られた懸濁液を20±5℃で90分間攪拌した。生成物を遠心濾過によって単離し、ヘプタン(9.0L)で洗浄した。湿潤ケークを高真空下(NLT650mm/Hg)30±5℃で10時間乾燥させて粗製標記生成物(2.9kg)を得た。
27.5±2.5℃の酢酸イソプロピル(33.6L)中の粗製3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA(4.2kg)の溶液を10分間攪拌し、次いで、透明な溶液が観察されるまで、追加量の酢酸イソプロピル(12.6L)を添加した。Micronフィルターを通して溶液を濾過し、酢酸イソプロピル(2.1L)で洗浄し、透明な反応器に移して、3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA、形態1(0.01kg)を接種した。得られた混合物を27.5±2.5℃で30分間攪拌し、次いで、ヘプタン(96.6L)を27.5±2.5℃で2時間ゆっくり添加した。懸濁液を27.5±2.5℃で12時間10分間攪拌し、2.5±2.5℃に冷却し、この温度で2.5時間攪拌した。生成物を濾過によって単離し、冷ヘプタン:酢酸イソプロピル混合物(2:1 5.65L/2.74L)および最終的にヘプタン(8.4L)で洗浄した。生成物を高真空下(NLT650mm/Hg)40±5℃で一定重量まで乾燥させて、3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA、形態1(3.46kg)(1H NMRプロファイルは参照に一致する、HPLC純度98.0%)を得た。
3’−N−デメチル−4”−O−(3−ジエチルアミノプロピオニル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA
MSデータ(MS(ES+)m/z:876.7[MH]+)はWO2006/087644中に述べられているものに一致する。
4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA
MSデータ(MS(ES+)m/z:877.09[MH]+)はWO2006/087644中に述べられているものに一致する。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA酢酸塩
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA(100mg、0.116mmol)をi−PrOH(0.7mL)に溶解し、酢酸(13.9μL、0.244mmol)を添加した。混合物を0℃で冷却し、n−ヘキサン(10mL)およびジイソプロピルエーテル(3mL)を滴加した。次いで、溶媒を蒸発させ、乾燥後に標記化合物(103mg)を白色固体として得た。
MS(ES+)m/z:862.8(95.3%)
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンAコハク酸塩
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA(75mg)、コハク酸(20mg)および酢酸t−ブチル(300μL)をバイアル中に添加し、攪拌下で2日間にわたって温度を0〜40℃でサイクルさせた。スラリーを単離し、得られた固体を真空下40℃で60時間乾燥させて標記生成物を得た。
1H NMR(400MHz、CHLOROFORM−d)δppm7.61、6.46、6.44、6.12、6.10、5.21、5.08、5.06、4.93、4.92、4.89、4.88、4.75、4.72、4.69、4.67、4.60、4.58、4.54、4.53、4.51、4.49、4.37、4.36、4.34、4.33、4.32、4.21、4.19、4.17、4.15、4.08、4.06、3.82、3.77、3.74、3.72、3.65、3.64、3.61、3.55、3.51、3.48、3.44、3.31、3.31、3.28、3.22、3.06、3.05、3.03、3.01、2.85、2.84、2.82、2.73、2.71、2.69、2.67、2.58、2.56、2.54、2.52、2.41、2.37、2.31、2.24、2.22、2.10、2.07、2.04、1.99、1.97、1.92、1.90、1.88、1.86、1.79、1.66、1.64、1.61、1.59、1.57、1.56、1.52、1.51、1.50、1.48、1.47、1.45、1.43、1.42、1.40、1.32、1.29、1.28、1.26、1.23、1.22、1.18、1.17、1.16、1.14、1.13、1.12、1.10、1.08、1.06、0.99、0.97、0.92、0.90、0.88。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA安息香酸塩
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA(75mg)、安息香酸(10.6mg)および酢酸t−ブチル(300μL)をバイアル中に添加し、攪拌下で2日間にわたって温度を0〜40℃でサイクルさせた。スラリーを単離し、得られた固体を真空下40℃で60時間乾燥させて標記生成物を得た。
1H NMR(400MHz、CHLOROFORM−d)δppm8.08、8.06、7.52、7.51、7.49、7.44、7.42、7.40、7.27、6.11、6.09、4.89、4.88、4.74、4.71、4.70、4.67、4.60、4.58、4.39、4.38、4.37、4.35、4.34、4.21、4.20、4.18、4.16、3.75、3.74、3.49、3.44、3.40、3.38、3.36、3.35、3.32、3.31、3.22、2.85、2.83、2.82、2.80、2.74、2.72、2.70、2.68、2.59、2.40、2.37、2.24、2.22、2.20、2.05、2.05、2.02、2.01、1.99、1.97、1.88、1.86、1.65、1.64、1.61、1.60、1.57、1.55、1.45、1.41、1.38、1.33、1.30、1.25、1.23、1.22、1.20、1.19、1.17、1.15、1.13、1.11、1.10、1.08、1.06、0.99、0.97、0.92、0.90、0.89。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA L−酒石酸塩
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA(75mg)、L−酒石酸(13.5mg)および酢酸t−ブチル(300μL)をバイアル中に添加し、攪拌下で2日間にわたって温度を0〜40℃でサイクルさせた。スラリーを単離し、得られた固体を真空下40℃で60時間乾燥させて標記生成物を得た。
1H NMR(400MHz、CHLOROFORM−d)δppm7.19、7.17、7.10、7.08、6.14、6.12、4.81、4.80、4.66、4.63、4.61、4.59、4.49、4.48、4.32、4.30、4.29、4.28、4.23、4.12、4.11、4.09、4.07、3.72、3.71、3.70、3.67、3.66、3.42、3.38、3.27、3.24、3.12、2.78、2.76、2.75、2.67、2.65、2.63、2.61、2.51、2.32、2.28、2.17、2.15、2.13、1.95、1.93、1.90、1.89、1.82、1.81、1.79、1.61、1.57、1.56、1.53、1.52、1.37、1.25、1.22、1.18、1.16、1.14、1.12、1.10、1.09、1.07、1.05、1.03、1.01、0.99、0.94、0.92、0.85、0.83、0.81.
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA二塩酸塩
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA(100mg)およびアセトニトリル(200μL)をバイアル中に添加した。得られたスラリーを攪拌し、70℃に加熱した。追加量のアセトニトリル(300μL)を添加し、引き続いて塩酸(19μL、水中37%w/w)を添加したところ、固体が形成し、攪拌困難となった。したがって、さらなる量のアセトニトリル(200μL)を添加し、反応混合物を0℃に冷却し、その温度で15時間維持した。スラリーを濾過し、得られた固体を真空下40℃で24時間乾燥させて標記生成物を得た。
1H NMR(400MHz、CHLOROFORM−d)δppm7.57、7.55、5.10、5.09、4.99、4.98、4.96、4.96、4.93、4.87、4.85、4.83、4.53、4.52、4.49、4.48、4.46、4.45、4.44、4.42、4.14、4.12、4.11、3.82、3.81、3.71、3.69、3.63、3.48、3.48、3.47、3.38、3.36、3.34、3.32、3.31、3.31、3.31、3.30、3.30、3.28、3.25、3.21、3.12、3.11、3.09、3.07、3.06、2.81、2.79、2.78、2.77、2.76、2.72、2.69、2.51、2.47、2.40、2.38、2.38、2.36、2.34、2.26、2.23、2.22、2.20、2.03、2.02、2.00、1.99、1.89、1.88、1.87、1.86、1.85、1.85、1.83、1.83、1.78、1.77、1.75、1.73、1.56、1.55、1.54、1.53、1.52、1.51、1.50、1.48、1.43、1.40、1.37、1.35、1.33、1.31、1.29、1.24、1.23、1.22、1.21、1.19、1.18、1.17、1.15、1.14、1.12、1.06、1.04、1.03、1.01、0.92、0.90、0.88。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンAリン酸塩
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA(75mg)、リン酸(6μL、水中85%w/w)および酢酸t−ブチル(300μL)をバイアル中に添加し、攪拌下で2日間にわたって温度を0〜40℃でサイクルさせた。スラリーを単離し、得られた固体を真空下40℃で60時間乾燥させて標記生成物を得た。
1H NMR(400MHz、CHLOROFORM−d)δppm7.19、7.17、7.10、7.08、6.07、4.81、4.80、4.65、4.62、4.59、4.48、4.46、4.30、4.13、4.11、4.09、4.07、3.65、3.64、3.58、3.54、3.49、3.25、3.23、3.14、2.77、2.75、2.72、2.70、2.51、2.32、2.28、2.16、2.15、2.13、1.96、1.94、1.92、1.89、1.84、1.82、1.80、1.78、1.77、1.61、1.57、1.55、1.53、1.52、1.47、1.45、1.38、1.35、1.25、1.19、1.17、1.16、1.13、1.12、1.10、1.09、1.07、1.05、1.04、1.03、1.02、1.02、0.92、0.90、0.84、0.83、0.81。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA非晶質形態
上記実施例1、方法A、方法1と同様の手順に従って得た3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA(1.33g)をアセトンに溶解し、次いで、石油エーテルの添加によって沈殿させ、標記生成物(1.2g)を、図3のXRPDパターンによって特徴づけられる非晶質形態として得た。
MS(ES+)m/z:862.47[MH]+。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA−形態1
実施例10の非晶質3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA(500mg)を、攪拌により室温でアセトニトリル(2.5mL)に溶解した。透明な溶液が得られて1〜2分以内に、物質が沈殿し始めた。懸濁液を室温で1.5時間攪拌した。白色沈殿を真空下で濾過し、アセトニトリル(3×0.5mL)で洗浄し、室温で1時間乾燥させて標記生成物(307mg)形態1を得た。単離した固体物質をXRPD、DSCおよびTGAによって分析した。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA−形態2
実施例10の非晶質3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA(100mg)を脱塩水(1.0mL)に添加した。得られた懸濁液に、アセトン(1mL)を添加したところ、ゴム様物質が得られた。追加量のアセトン(0.4mL)を添加し、わずかに加熱してゴム様物質を溶解した。さらなる脱塩水(0.4mL)を添加した。約90%の透明な溶液を新しいバイアルに移し、次いでそれをパラフィルムで被覆し、室温で維持して結晶化させた。15日後、濾過によって、得られた結晶(単一結晶、小六角形)を単離して標記生成物(23mg)形態2を得た。単離した結晶を大気中、周囲温度で2時間維持し、XRPD、DSCおよびTGAによって分析した。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA−形態3
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA、形態1(700mg)を、メタノール:水50%(3mL)とともにSyn10チューブに入れた。スラリーを攪拌下で4日間にわたって0〜40℃で温度サイクルさせた。固体を濾別し、真空下40℃で一晩乾燥させて標記生成物形態3を得た。単離した固体物質を、1H NMR、XRPDおよびラマンによって分析した。
1H NMR(400MHz、DMSO−D6)は、溶媒ピークの存在を示さなかった。
1H NMR(400MHz、DMSO−D6)δppm0.81(t、J=7.3Hz、3H)、0.89(d、J=6.8Hz、4H)、0.92〜0.99(m、9H)、1.00〜1.05(m、9H)、1.06〜1.13(m、9H)、1.22(s、3H)、1.38(s、1H)、1.66〜1.75(m、1H)、1.75〜1.87(m、2H)、1.91(s、1H)、2.13(d、J=13.9Hz、1H)、2.25(s、3H)、2.31(d、J=15.4Hz、2H)、2.53〜2.62(m、4H)、2.80〜2.87(m、1H)、3.13(s、2H)、3.14〜3.20(m、1H)、3.24(s、3H)、3.27(s、4H)、3.36〜3.44(m、1H)、3.54(d、J=6.6Hz、1H)、3.70(s、1H)、3.85(s、1H)、3.94(d、J=7.3Hz、1H)、4.30〜4.39(m、3H)、4.56(d、J=9.8Hz、1H)、4.77(d、J=4.6Hz、1H)、4.89(dd、J=9.8、2.0Hz、1H)、5.00(d、J=4.9Hz、1H)、7.17(d、J=8.3Hz、1H)。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA−形態4
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA、形態1(100mg)およびIPA:水=1:1 v:v(0.5mL)をバイアル中に添加し、攪拌下で5日間にわたって0〜40℃で温度サイクルさせた。得られた溶液を室温で蒸発させて標記生成物形態4を得た。単離した物質を1H NMRおよびラマンによって分析した。
1H NMR(400MHz、DMSO−D6)δppm7.27、6.09、4.89、4.88、4.74、4.71、4.69、4.67、4.58、4.56、4.40、4.23、4.21、4.07、4.05、4.04、4.02、4.00、3.82、3.81、3.80、3.78、3.77、3.75、3.46、3.42、3.34、3.32、3.30、3.25、3.23、3.20、3.17、3.15、2.87、2.86、2.84、2.82、2.80、2.72、2.71、2.69、2.67、2.65、2.62、2.60、2.59、2.46、2.40、2.36、2.26、2.24、2.22、2.20、2.19、2.05、2.03、2.01、1.99、1.95、1.92、1.90、1.88、1.86、1.84、1.65、1.60、1.34、1.32、1.26、1.24、1.23、1.21、1.19、1.17、1.16、1.13、1.12、1.10、1.08、1.06、1.00、0.98、0.92、0.90、0.89、0.01。
形態4のラマンスペクトルを図12に示す。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA−形態5
方法A:3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA、形態1(25mg)をイソプロピルエーテル(500μL)にスラリー化し、振盪下で24時間にわたって0〜40℃で温度サイクルさせた。次いで、温度を20℃に設定し、反応混合物を2日間振盪した。固体を濾別し、真空下で1時間乾燥させて標記生成物形態5を得た。単離した固体物質をラマンによって分析した。形態5のラマンスペクトルを図13に示す。
方法B:3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA、形態1(25mg)をt−ブチルメチルエーテル(500μL)にスラリー化し、振盪下で24時間にわたって0〜40℃で温度サイクルさせた。次いで、温度を20℃に設定し、追加量の3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA、形態1(約25mg)をスパチュラで添加して濃厚なスラリーを得、これを20℃で2日間振盪した。固体を濾別し、真空下で1時間乾燥させて、標記生成物形態5を得た。これは実施例15、方法Aで得られた物質と一致するラマンスペクトルに従っていた。1H NMR(400MHz、DMSO−D6)は、溶媒ピークの存在を示さなかった。
1H NMR(400MHz、DMSO−D6)δppm0.81(t、J=7.5Hz、3H)、0.89(d、J=6.8Hz、4H)、0.92〜0.99(m、9H)、1.00〜1.05(m、9H)、1.06〜1.14(m、9H)、1.22(s、3H)、1.31〜1.46(m、1H)、1.63〜1.73(m、1H)、1.75〜1.86(m、2H)、1.87〜1.97(m、1H)、2.11(s、1H)、2.25(s、3H)、2.31(d、J=15.4Hz、2H)、2.59(qd、J=7.2、1.6Hz、4H)、2.84(d、J=6.1Hz、1H)、3.13(s、2H)、3.15〜3.19(m、1H)、3.24(s、3H)、3.25〜3.28(m、4H)、3.35〜3.45(m、1H)、3.54(d、J=6.6Hz、1H)、3.70(s、1H)、3.85(s、1H)、3.96(d、J=7.8Hz、1H)、4.30〜4.40(m、3H)、4.56(d、J=10.0Hz、1H)、4.77(d、J=4.6Hz、1H)、4.89(dd、J=9.9、2.1Hz、1H)、5.00(d、J=4.6Hz、1H)、7.17(d、J=8.1Hz、1H)。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA−形態6
酢酸メチル(500μL)中の3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA、形態1(50mg)の溶液を振盪下で24時間にわたって0〜40℃で温度サイクルさせた。次いで、温度を20℃に設定し、追加量の3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA、形態1(約50mg)をスパチュラで添加して濃厚なスラリーを得、これを20℃で2日間振盪した。スラリーを濾別し、300μLの飽和濾液を、キャップをして約2℃の冷蔵庫中に1ヶ月間置いて標記生成物形態6を得、これを溶液の慎重なピペット操作によって単離した。ラマンによる分析前に固体を窒素フロー下で短時間乾燥させた。1H NMR(DMSO−d6中)は、溶媒の存在を示さなかった。形態6のベースラインを補正したラマンスペクトルを図14に示す。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA−形態7
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA、形態1(75mg)をHPLCバイアルインサートに入れ、テトラヒドロフラン(1.5mL)を含むHPLCバイアルに入れ、キャップを締め、周囲温度に5日間置いたところ、固体の潮解が引き起こされた。得られた溶液の1/3を新しいHPLCバイアルインサートに入れ、それをn−ヘプタン(1.5mL)を含むHPLCバイアルに入れ、周囲温度に2週間にわたって置いて標記生成物形態7を得、これを溶液の慎重なピペット操作によって単離した。ラマンによる分析前に固体を窒素フロー下で短時間乾燥させた。1H NMR(DMSO−d6中)は、THF(<0.25eq)の存在を示した。形態7のラマンスペクトルを図15に示す。
3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA−形態8
メタノール(500μL)中の3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA、形態1(200mg)の溶液を振盪下で24時間にわたって0〜40℃で温度サイクルさせた。次いで、温度を20℃に設定し、追加量の3’−N−デメチル−4”−O−(2−ジエチルアミノエタノイル)−6−O−メチル−9a−アザ−9a−ホモエリスロマイシンA、形態1(約175mg)をスパーテルで滴加して濃厚なスラリーを得、これを20℃で2日間振盪した。スラリーを濾別し、200μLの濾液をメタノール(200μL)で希釈した。200μLのアリコートを蒸発皿に入れ、蒸発乾固させて標記生成物形態6を得た。単離した物質をラマンによって分析した。形態8のラマンスペクトルを図16に示す。
安定性
インビトロ血液および血漿安定性
インビトロ血液および血漿安定性プロトコルに記載の方法論を使用して、実施例1〜3の化合物のインビトロ安定性を測定した。実施例1および3は、24時間にわたって全血中および血漿中の両方において安定であることを示した。24時間後、実施例2は不安定性を示し、約41%の化合物が全血中に残っており、2%未満が血漿中に残っていた(図1A〜C参照)。
緩衝液中および生体関連培養液中の安定性プロトコルに記載の方法論を使用して、25℃、pH2〜pH8の緩衝液中および37℃の生体関連培養液(SGF pH1.6、空腹時SIF pH6.5および食後SIF pH5.0)中の実施例1〜3の化合物の安定性を24時間測定した。
25℃、pH7の緩衝液中で24時間後、実施例2の78.4%が残っていた。
25℃、pH8の緩衝液中で24時間後、実施例2の44.6%が残っていた。
37℃、pH1.6のSGF中で24時間後、実施例2の79.0%が残っていた。
37℃、pH6.5の空腹時SIF中で24時間後、実施例2の63.7%が残っていた。
インビトロプロトコルに記載の方法論を使用して、実施例1〜3の化合物の抗菌剤としてのインビトロ活性を測定した。実施例1および2は、全6菌株に対して測定可能な活性を有さないことが分かった(各々の場合につきMIC>64μg/mL)。実施例3は、抗菌活性を示すことが分かり、Moraxella catarrhalis ATCC23246およびStreptococcus pneumoniae ATCC49619に対して8μg/mLの最小発育阻止濃度(MIC)を有するが、他の4菌株に対しては測定可能な活性を示さなかった(各々の場合につきMIC>64μg/mL)。
雄BALB/cJマウスにおける細菌性リポ多糖によって誘発される肺好中球増加症についてのインビボプロトコル(ここおよび前に記載)および/または国際公開WO2006/087644に記載の雄BALB/cJマウスにおける細菌性リポ多糖によって誘発される肺好中球増加症についてのインビボプロトコル中に記載の方法論を使用して、実施例1〜3の化合物の抗炎症剤としてのインビボ活性を測定した。実施例1、2および3は、総細胞数の平均90%を超える阻害を示した。平均して、実施例1は89%超、実施例2は70%超、および実施例3は79%超の、試験化合物200mg/kgの単一用量を腹腔内投与(i.p.)された処理動物のBALF中の好中球数の阻害を示した。
実施例1の化合物は、抗炎症性スクリーニングにおいて活性を示し、有意な抗菌活性を有さず、かつ血漿、全血、pH2〜pH8の緩衝液および生体関連培養液中で安定であるという点で、有利な特性を有しており、それがこの化合物を抗炎症剤としての開発に非常に適したものとしている。
Claims (12)
- 式(I):
- 請求項1に記載の式(I)によって表される化合物、またはその薬学的に許容される塩。
- 式(I):
- 2シータ角度の点から表現され、かつ、銅Kα放射線を使用する回折計によって得られるXRPDパターンによって特徴づけられる結晶形態1である、請求項1に記載の式(I)によって表される化合物であって、前記XRPDパターンは、それぞれ19.1、15.9、11.5、9.2、8.4、8.0、7.7、7.5、7.2、6.3、5.6、4.8、4.7、4.3、4.2および4.0オングストローム(Å)のd−格子面間隔に対応する、4.6、5.6、7.7、9.6、10.6、11.0、11.6、11.8、12.2、14.1、15.9、18.4、19.0、20.5、21.1および22.0度からなる群から選択される少なくとも5つの位置の2シータ角度(2θ/°)を含んでなる化合物。
- 2シータ角度の点から表現され、かつ、銅Kα放射線を使用する回折計によって得られるXRPDパターンによって特徴づけられる結晶形態2である、請求項1に記載の式(I)によって表される化合物であって、前記XRPDパターンは、それぞれ9.7、9.5、8.5、8.1、7.7、7.4、6.8、6.6、6.0、5.8、5.6、5.4、5.3、4.7、4.6、4.6、4.5および4.3オングストローム(Å)のd−格子面間隔に対応する、9.1、9.4、10.4、10.9、11.5、12.0、13.0、13.4、14.7、15.4、15.7、16.5、16.8、18.7、19.2、19.5、19.8および20.8度からなる群から選択される少なくとも5つの位置の2シータ角度(2θ/°)を含んでなる化合物。
- 2シータ角度の点から表現され、かつ、銅Kα放射線を使用する回折計によって得られるXRPDパターンによって特徴づけられる結晶形態3である、請求項1に記載の式(I)によって表される化合物であって、前記XRPDパターンは、それぞれ16.0、12.1、11.3、9.7、9.4、8.9、8.4、8.1、7.7、7.4、7.0、6.0、5.7、5.6、5.3、5.2、4.9、4.7および4.6オングストローム(Å)のd−格子面間隔に対応する、5.5、7.3、7.9、9.1、9.5、10.0、10.5、11.0、11.5、12.0、12.6、14.8、15.5、15.8、16.6、16.9、18.2、18.9および19.5度からなる群から選択される少なくとも5つの位置の2シータ角度(2θ/°)を含んでなる化合物。
- a)式(II):
HOC(O)CH2NR3R4 (III)
(式中、R3およびR4は各々エチルである、またはそれぞれ独立に、エチルに転換できる基である)によって表されるカルボン酸またはカルボン酸の適当な活性化誘導体とを反応させ、引き続いてヒドロキシル保護基R1、アミノ保護基R2をその後に除去し、必要な場合には、−NR3R4基を−N(CH2CH3)2へ転換すること;または
b)式(IV):
NR3R4 (V)
(式中、R3およびR4は各々エチルである、またはそれぞれ独立に、エチルに転換できる基である)によって表されるアミンとを反応させ、引き続いてヒドロキシル保護基R1、アミノ保護基R2をその後に除去し、必要な場合には、−NR3R4基を−N(CH2CH3)2へ転換すること
を含んでなる、請求項1に記載の化合物を調製する方法。 - 少なくとも1種の薬学的に許容される賦形剤、希釈剤および/または担体と共に、請求項1から6のいずれか一項に記載の化合物、またはその薬学的に許容される塩を含んでなる、医薬組成物。
- 医学療法に使用するための、請求項1から6のいずれか一項に記載の化合物。
- 慢性閉塞性肺疾患、嚢胞性線維症、びまん性汎細気管支炎、閉塞性細気管支炎、気管支炎、気管支拡張症、急性呼吸窮迫症候群、重症のまたはステロイド抵抗性の喘息、気腫、慢性鼻副鼻腔炎、関節リウマチ、痛風性関節炎、炎症性腸疾患、糸球体腎炎、虚血再潅流による傷害、アテローム性動脈硬化症、乾癬および脈管炎などの皮膚病、全身性エリテマトーデス、全身性炎症反応症候群、敗血症、虚血再潅流障害、酒さ、歯周炎、歯肉過形成および前立腺炎症候群から選択される、好中球浸潤から生じる好中球支配性炎症性疾患および/または好中球の異常細胞機能に関する疾患を治療するための薬剤の製造における、請求項1から6のいずれか一項に記載の化合物の使用。
- 前記疾患が、慢性閉塞性肺疾患、嚢胞性線維症、びまん性汎細気管支炎、閉塞性細気管支炎、気管支炎、気管支拡張症、急性呼吸窮迫症候群、重症のまたはステロイド抵抗性の喘息、気腫および慢性鼻副鼻腔炎から選択される、請求項10に記載の使用。
- 1つまたは複数のさらなる治療上活性な薬剤を含んでなる、請求項8に記載の医薬組成物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14860909P | 2009-01-30 | 2009-01-30 | |
| US61/148,609 | 2009-01-30 | ||
| PCT/EP2010/050967 WO2010086349A1 (en) | 2009-01-30 | 2010-01-28 | Anti-inflammatory macrolide |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2012516304A JP2012516304A (ja) | 2012-07-19 |
| JP5437394B2 true JP5437394B2 (ja) | 2014-03-12 |
Family
ID=41698466
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011546833A Expired - Fee Related JP5437394B2 (ja) | 2009-01-30 | 2010-01-28 | 抗炎症性マクロライド |
Country Status (23)
| Country | Link |
|---|---|
| US (1) | US8227428B2 (ja) |
| EP (1) | EP2384333B1 (ja) |
| JP (1) | JP5437394B2 (ja) |
| KR (1) | KR20110099791A (ja) |
| CN (1) | CN102378763A (ja) |
| AR (1) | AR075340A1 (ja) |
| AU (1) | AU2010209732B2 (ja) |
| BR (1) | BRPI1006114A2 (ja) |
| CA (1) | CA2748847A1 (ja) |
| CL (1) | CL2011001829A1 (ja) |
| CO (1) | CO6400226A2 (ja) |
| DO (1) | DOP2011000242A (ja) |
| EA (1) | EA201101139A1 (ja) |
| ES (1) | ES2412059T3 (ja) |
| IL (1) | IL213871A0 (ja) |
| MA (1) | MA32997B1 (ja) |
| MX (1) | MX2011008074A (ja) |
| PE (1) | PE20120220A1 (ja) |
| SG (1) | SG172782A1 (ja) |
| TW (1) | TW201038277A (ja) |
| UY (1) | UY32408A (ja) |
| WO (1) | WO2010086349A1 (ja) |
| ZA (1) | ZA201104653B (ja) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5036558B2 (ja) | 2005-01-13 | 2012-09-26 | グラクソ グループ リミテッド | 抗炎症活性を有するマクロライド |
| AR075340A1 (es) | 2009-01-30 | 2011-03-23 | Glaxosmithkline Zagreb | 3'-n-desmetil-4''-o-(2-dietilaminoetanoil)-6-o-metil-9a-aza-9a-homoeritromicina a,formas cristalinas 1, 2 y 3, proceso para su preparacion, composicion farmaceutica que lo comprende y su uso para la preparacion de un medicamento util para el tratamiento de enfermedades inflamatorias |
| US20130045939A1 (en) * | 2010-03-19 | 2013-02-21 | Catabasis Pharmaceuticals, Inc. | Fatty acid macrolide derivatives and their uses |
| WO2011131749A1 (en) * | 2010-04-23 | 2011-10-27 | Glaxo Group Limited | New 14 and 15 membered macrolides for the treatment of neutrophil dominated inflammatory diseases |
| JP6116206B2 (ja) * | 2012-11-26 | 2017-04-19 | クリニプロ株式会社 | 吸入用パウダーの製造方法 |
| WO2018131724A1 (en) * | 2017-01-13 | 2018-07-19 | Tobishi Pharmaceutical Co., Ltd. | Neutrophil activation regulator |
| AU2021232959B2 (en) | 2020-03-12 | 2023-09-07 | Zoetis Services Llc | Immunomodulating urea azalides |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US525051A (en) | 1894-08-28 | Adjustable chair | ||
| US3725385A (en) | 1970-11-02 | 1973-04-03 | Abbott Lab | Process for the demethylation of 3-amino macrolides |
| EP0126344A2 (en) | 1983-05-20 | 1984-11-28 | Abbott Laboratories | Tripeptide esters of therapeutic agents |
| NZ224501A (en) * | 1987-05-06 | 1990-01-29 | Adir | Tylosin and carbomycin oxime derivatives and pharmaceutical compositions |
| SI9011409A (en) | 1990-07-18 | 1995-10-31 | Pliva Pharm & Chem Works | O-methyl azitromycin derivates, methods and intermediates for their preparation and methods for preparation of pharmaceuticals products which comprise them |
| EP0895999A1 (en) | 1997-08-06 | 1999-02-10 | Pfizer Products Inc. | C-4" substituted macrolide antibiotics |
| HRP980189B1 (en) | 1998-04-06 | 2004-04-30 | Pliva Pharm & Chem Works | Novel 15-membered lactams ketolides |
| PL353316A1 (en) * | 1999-05-18 | 2003-11-17 | Pfizer Products Inc. | Novel crystalline forms of a macrolide antibiotic |
| HRP20010301A2 (en) | 2001-04-27 | 2001-12-31 | Pliva D D | New therapeutic indication for azithromycin in the treatment of non-infective inflammatory diseases |
| CN100577681C (zh) | 2002-07-08 | 2010-01-06 | 葛兰素伊斯特拉齐瓦基森塔萨格勒布公司 | 用于治疗炎性疾病和病症的新化合物、组合物和方法 |
| ITMI20021726A1 (it) | 2002-08-01 | 2004-02-02 | Zambon Spa | Macrolidi ad attivita' antiinfiammatoria. |
| GB0310986D0 (en) | 2003-05-13 | 2003-06-18 | Glaxo Group Ltd | Novel compounds |
| EP1841776B1 (en) | 2005-01-13 | 2008-12-31 | GlaxoSmithKline istrazivacki centar Zagreb d.o.o. | Decladinosyl-macrolides with anti-inflammatory activity |
| JP5036558B2 (ja) * | 2005-01-13 | 2012-09-26 | グラクソ グループ リミテッド | 抗炎症活性を有するマクロライド |
| WO2006106440A1 (en) | 2005-01-14 | 2006-10-12 | Glaxosmithkline Istrazivacki Centar Zagreb D.O.O. | Macrolide compounds containing biotin and photo-affinity group for macrolide target identification |
| AR075340A1 (es) | 2009-01-30 | 2011-03-23 | Glaxosmithkline Zagreb | 3'-n-desmetil-4''-o-(2-dietilaminoetanoil)-6-o-metil-9a-aza-9a-homoeritromicina a,formas cristalinas 1, 2 y 3, proceso para su preparacion, composicion farmaceutica que lo comprende y su uso para la preparacion de un medicamento util para el tratamiento de enfermedades inflamatorias |
-
2010
- 2010-01-28 AR ARP100100227A patent/AR075340A1/es not_active Application Discontinuation
- 2010-01-28 US US12/695,272 patent/US8227428B2/en not_active Expired - Fee Related
- 2010-01-28 SG SG2011046216A patent/SG172782A1/en unknown
- 2010-01-28 BR BRPI1006114A patent/BRPI1006114A2/pt not_active IP Right Cessation
- 2010-01-28 MA MA34057A patent/MA32997B1/fr unknown
- 2010-01-28 PE PE2011001406A patent/PE20120220A1/es not_active Application Discontinuation
- 2010-01-28 CA CA2748847A patent/CA2748847A1/en not_active Abandoned
- 2010-01-28 MX MX2011008074A patent/MX2011008074A/es active IP Right Grant
- 2010-01-28 ES ES10701369T patent/ES2412059T3/es active Active
- 2010-01-28 AU AU2010209732A patent/AU2010209732B2/en not_active Expired - Fee Related
- 2010-01-28 EP EP10701369.0A patent/EP2384333B1/en active Active
- 2010-01-28 WO PCT/EP2010/050967 patent/WO2010086349A1/en active Application Filing
- 2010-01-28 EA EA201101139A patent/EA201101139A1/ru unknown
- 2010-01-28 UY UY0001032408A patent/UY32408A/es unknown
- 2010-01-28 TW TW099102336A patent/TW201038277A/zh unknown
- 2010-01-28 KR KR1020117017844A patent/KR20110099791A/ko not_active Ceased
- 2010-01-28 CN CN2010800149961A patent/CN102378763A/zh active Pending
- 2010-01-28 JP JP2011546833A patent/JP5437394B2/ja not_active Expired - Fee Related
-
2011
- 2011-06-23 ZA ZA2011/04653A patent/ZA201104653B/en unknown
- 2011-06-30 IL IL213871A patent/IL213871A0/en unknown
- 2011-07-26 DO DO2011000242A patent/DOP2011000242A/es unknown
- 2011-07-28 CL CL2011001829A patent/CL2011001829A1/es unknown
- 2011-07-29 CO CO11095916A patent/CO6400226A2/es not_active Application Discontinuation
Also Published As
| Publication number | Publication date |
|---|---|
| MX2011008074A (es) | 2011-08-17 |
| IL213871A0 (en) | 2011-07-31 |
| CO6400226A2 (es) | 2012-03-15 |
| DOP2011000242A (es) | 2011-09-15 |
| US20100197623A1 (en) | 2010-08-05 |
| TW201038277A (en) | 2010-11-01 |
| EA201101139A1 (ru) | 2012-03-30 |
| PE20120220A1 (es) | 2012-04-04 |
| SG172782A1 (en) | 2011-08-29 |
| CN102378763A (zh) | 2012-03-14 |
| CL2011001829A1 (es) | 2011-10-14 |
| EP2384333B1 (en) | 2013-04-17 |
| EP2384333A1 (en) | 2011-11-09 |
| US8227428B2 (en) | 2012-07-24 |
| AU2010209732B2 (en) | 2012-12-20 |
| JP2012516304A (ja) | 2012-07-19 |
| AU2010209732A1 (en) | 2011-07-21 |
| MA32997B1 (fr) | 2012-01-02 |
| KR20110099791A (ko) | 2011-09-08 |
| ES2412059T3 (es) | 2013-07-10 |
| WO2010086349A1 (en) | 2010-08-05 |
| CA2748847A1 (en) | 2010-08-05 |
| UY32408A (es) | 2010-07-30 |
| BRPI1006114A2 (pt) | 2018-02-06 |
| ZA201104653B (en) | 2012-02-29 |
| AR075340A1 (es) | 2011-03-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5437394B2 (ja) | 抗炎症性マクロライド | |
| AU2006215315B2 (en) | Macrolides with anti-inflammatory activity | |
| JP3666788B2 (ja) | 3,6−ケタール及びエノールエーテルマクロライド抗生物質 | |
| WO2011131749A1 (en) | New 14 and 15 membered macrolides for the treatment of neutrophil dominated inflammatory diseases | |
| CN107129514B (zh) | 红霉素a酮内酯类抗生素衍生物、其制备方法及应用 | |
| ES2317527T3 (es) | Decladinosil-macrolidos que presentan actividad anti-inflamatoria. | |
| EP0771564A1 (en) | Interleukin-5 production inhibitor | |
| US20110039795A1 (en) | 2'-o,3'-n-bridged macrolides | |
| JP2012516305A (ja) | 好中球優位の炎症性疾患の治療のための9−デオキソ−9a−メチル−9a−アザ−9a−ホモエリスロマイシンa誘導体 | |
| JP2006528947A (ja) | 4”−置換マクロライド | |
| JP2023071705A (ja) | グラナチシンbの多形体 | |
| CN105524132A (zh) | 含有喹啉取代基的红霉素a酮内酯类抗生素衍生物、其制备方法及应用 | |
| CN111662351B (zh) | 新奥克梯隆型皂苷元衍生物及制备抗耐药菌药物应用 | |
| JP7309608B2 (ja) | 抗がん活性を有するステロイドサポニン | |
| JPH0853355A (ja) | Il−5産生抑制剤 | |
| HK1112920B (en) | Macrolides with anti-inflammatory activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130423 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130718 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130725 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130815 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130822 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130924 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20131001 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20131017 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20131112 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20131211 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |