JP5349323B2 - 修飾ヌクレオチドを含む転写物を生成させるための材料および方法 - Google Patents
修飾ヌクレオチドを含む転写物を生成させるための材料および方法 Download PDFInfo
- Publication number
- JP5349323B2 JP5349323B2 JP2009542267A JP2009542267A JP5349323B2 JP 5349323 B2 JP5349323 B2 JP 5349323B2 JP 2009542267 A JP2009542267 A JP 2009542267A JP 2009542267 A JP2009542267 A JP 2009542267A JP 5349323 B2 JP5349323 B2 JP 5349323B2
- Authority
- JP
- Japan
- Prior art keywords
- ome
- modified
- transcription
- nucleic acid
- triphosphate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims description 245
- 125000003729 nucleotide group Chemical group 0.000 title claims description 156
- 239000000463 material Substances 0.000 title description 11
- 108091023037 Aptamer Proteins 0.000 claims description 362
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 336
- 238000013518 transcription Methods 0.000 claims description 306
- 230000035897 transcription Effects 0.000 claims description 306
- 150000007523 nucleic acids Chemical class 0.000 claims description 210
- 102000039446 nucleic acids Human genes 0.000 claims description 190
- 108020004707 nucleic acids Proteins 0.000 claims description 190
- 239000000203 mixture Substances 0.000 claims description 164
- 239000002773 nucleotide Substances 0.000 claims description 141
- 101710137500 T7 RNA polymerase Proteins 0.000 claims description 140
- XKMLYUALXHKNFT-UUOKFMHZSA-N Guanosine-5'-triphosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O XKMLYUALXHKNFT-UUOKFMHZSA-N 0.000 claims description 109
- 239000011541 reaction mixture Substances 0.000 claims description 93
- 235000001014 amino acid Nutrition 0.000 claims description 87
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 claims description 82
- 229920001223 polyethylene glycol Polymers 0.000 claims description 76
- 239000001226 triphosphate Substances 0.000 claims description 76
- 235000011178 triphosphate Nutrition 0.000 claims description 76
- 238000012986 modification Methods 0.000 claims description 72
- 230000004048 modification Effects 0.000 claims description 66
- NYHBQMYGNKIUIF-UUOKFMHZSA-N Guanosine Chemical class C1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O NYHBQMYGNKIUIF-UUOKFMHZSA-N 0.000 claims description 64
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 62
- PGAVKCOVUIYSFO-XVFCMESISA-N UTP Chemical compound O[C@@H]1[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O[C@H]1N1C(=O)NC(=O)C=C1 PGAVKCOVUIYSFO-XVFCMESISA-N 0.000 claims description 57
- 229910001425 magnesium ion Inorganic materials 0.000 claims description 57
- 229910001437 manganese ion Inorganic materials 0.000 claims description 57
- 229950010342 uridine triphosphate Drugs 0.000 claims description 53
- -1 -modified nucleotide triphosphates Chemical class 0.000 claims description 52
- PGAVKCOVUIYSFO-UHFFFAOYSA-N uridine-triphosphate Natural products OC1C(O)C(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)OC1N1C(=O)NC(=O)C=C1 PGAVKCOVUIYSFO-UHFFFAOYSA-N 0.000 claims description 49
- JLVVSXFLKOJNIY-UHFFFAOYSA-N Magnesium ion Chemical compound [Mg+2] JLVVSXFLKOJNIY-UHFFFAOYSA-N 0.000 claims description 37
- UNXRWKVEANCORM-UHFFFAOYSA-N triphosphoric acid Chemical compound OP(O)(=O)OP(O)(=O)OP(O)(O)=O UNXRWKVEANCORM-UHFFFAOYSA-N 0.000 claims description 37
- 102000009617 Inorganic Pyrophosphatase Human genes 0.000 claims description 35
- 108010009595 Inorganic Pyrophosphatase Proteins 0.000 claims description 35
- MIKUYHXYGGJMLM-GIMIYPNGSA-N Crotonoside Natural products C1=NC2=C(N)NC(=O)N=C2N1[C@H]1O[C@@H](CO)[C@H](O)[C@@H]1O MIKUYHXYGGJMLM-GIMIYPNGSA-N 0.000 claims description 30
- 150000001413 amino acids Chemical class 0.000 claims description 30
- NYHBQMYGNKIUIF-UHFFFAOYSA-N D-guanosine Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(CO)C(O)C1O NYHBQMYGNKIUIF-UHFFFAOYSA-N 0.000 claims description 29
- 229940029575 guanosine Drugs 0.000 claims description 29
- 239000011777 magnesium Substances 0.000 claims description 27
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 claims description 22
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical class O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 claims description 22
- 235000004279 alanine Nutrition 0.000 claims description 22
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 claims description 21
- DRTQHJPVMGBUCF-XVFCMESISA-N Uridine Chemical group O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-XVFCMESISA-N 0.000 claims description 20
- 230000002209 hydrophobic effect Effects 0.000 claims description 20
- ZKHQWZAMYRWXGA-KQYNXXCUSA-J ATP(4-) Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-J 0.000 claims description 18
- 150000001412 amines Chemical class 0.000 claims description 18
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 claims description 17
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 claims description 17
- 150000002357 guanidines Chemical class 0.000 claims description 14
- 239000002202 Polyethylene glycol Substances 0.000 claims description 13
- KITLPLLNIZOYIJ-UUOKFMHZSA-N [[(2r,3s,4r,5r)-5-(2-amino-6-oxo-7,8-dihydro-3h-purin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] phosphono hydrogen phosphate Chemical group C1=2NC(N)=NC(=O)C=2NCN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O KITLPLLNIZOYIJ-UUOKFMHZSA-N 0.000 claims description 13
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 claims description 12
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 claims description 10
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 claims description 10
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 claims description 9
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 claims description 9
- 239000002777 nucleoside Substances 0.000 claims description 9
- 229940104230 thymidine Drugs 0.000 claims description 9
- RGWHQCVHVJXOKC-SHYZEUOFSA-N dCTP Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](CO[P@](O)(=O)O[P@](O)(=O)OP(O)(O)=O)[C@@H](O)C1 RGWHQCVHVJXOKC-SHYZEUOFSA-N 0.000 claims description 8
- 239000002126 C01EB10 - Adenosine Substances 0.000 claims description 7
- 229960005305 adenosine Drugs 0.000 claims description 7
- DRTQHJPVMGBUCF-PSQAKQOGSA-N beta-L-uridine Natural products O[C@H]1[C@@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-PSQAKQOGSA-N 0.000 claims description 7
- DRTQHJPVMGBUCF-UHFFFAOYSA-N uracil arabinoside Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UHFFFAOYSA-N 0.000 claims description 7
- 229940045145 uridine Drugs 0.000 claims description 7
- UHDGCWIWMRVCDJ-UHFFFAOYSA-N 1-beta-D-Xylofuranosyl-NH-Cytosine Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(CO)O1 UHDGCWIWMRVCDJ-UHFFFAOYSA-N 0.000 claims description 5
- UHDGCWIWMRVCDJ-PSQAKQOGSA-N Cytidine Natural products O=C1N=C(N)C=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-PSQAKQOGSA-N 0.000 claims description 5
- 125000003118 aryl group Chemical group 0.000 claims description 5
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 5
- UHDGCWIWMRVCDJ-ZAKLUEHWSA-N cytidine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-ZAKLUEHWSA-N 0.000 claims description 5
- 125000001041 indolyl group Chemical group 0.000 claims description 5
- SXUXMRMBWZCMEN-UHFFFAOYSA-N 2'-O-methyl uridine Natural products COC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 SXUXMRMBWZCMEN-UHFFFAOYSA-N 0.000 claims description 4
- XKMLYUALXHKNFT-UHFFFAOYSA-N rGTP Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O XKMLYUALXHKNFT-UHFFFAOYSA-N 0.000 description 96
- 230000027455 binding Effects 0.000 description 85
- 238000009739 binding Methods 0.000 description 85
- 108091034117 Oligonucleotide Proteins 0.000 description 82
- 238000006243 chemical reaction Methods 0.000 description 76
- 102100029880 Glycodelin Human genes 0.000 description 64
- 101000585553 Homo sapiens Glycodelin Proteins 0.000 description 64
- ZKHQWZAMYRWXGA-KQYNXXCUSA-N Adenosine triphosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-N 0.000 description 57
- PCDQPRRSZKQHHS-XVFCMESISA-N CTP Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 PCDQPRRSZKQHHS-XVFCMESISA-N 0.000 description 54
- ATHGHQPFGPMSJY-UHFFFAOYSA-N spermidine Chemical compound NCCCCNCCCN ATHGHQPFGPMSJY-UHFFFAOYSA-N 0.000 description 50
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 46
- 108020004414 DNA Proteins 0.000 description 44
- 239000003446 ligand Substances 0.000 description 41
- 230000008569 process Effects 0.000 description 41
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 38
- 239000000872 buffer Substances 0.000 description 38
- RZCIEJXAILMSQK-JXOAFFINSA-N TTP Chemical compound O=C1NC(=O)C(C)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 RZCIEJXAILMSQK-JXOAFFINSA-N 0.000 description 37
- 108090000623 proteins and genes Proteins 0.000 description 37
- 102000004169 proteins and genes Human genes 0.000 description 36
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 32
- 235000018102 proteins Nutrition 0.000 description 30
- 235000013928 guanylic acid Nutrition 0.000 description 28
- 239000003814 drug Substances 0.000 description 27
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 26
- 239000007995 HEPES buffer Substances 0.000 description 25
- RQFCJASXJCIDSX-UUOKFMHZSA-N guanosine 5'-monophosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H]1O RQFCJASXJCIDSX-UUOKFMHZSA-N 0.000 description 25
- 229940063673 spermidine Drugs 0.000 description 25
- 229920002594 Polyethylene Glycol 8000 Polymers 0.000 description 23
- 239000013504 Triton X-100 Substances 0.000 description 23
- 229920004890 Triton X-100 Polymers 0.000 description 23
- 238000006467 substitution reaction Methods 0.000 description 23
- 210000004027 cell Anatomy 0.000 description 22
- 239000000499 gel Substances 0.000 description 22
- 230000001225 therapeutic effect Effects 0.000 description 22
- 230000000694 effects Effects 0.000 description 21
- 239000000243 solution Substances 0.000 description 21
- UMCBIDVJVPVDLH-UHFFFAOYSA-N diphosphono hydrogen phosphate;guanidine Chemical class NC(N)=N.OP(O)(=O)OP(O)(=O)OP(O)(O)=O UMCBIDVJVPVDLH-UHFFFAOYSA-N 0.000 description 20
- 230000002441 reversible effect Effects 0.000 description 20
- 125000001424 substituent group Chemical group 0.000 description 20
- 238000003786 synthesis reaction Methods 0.000 description 20
- 238000004458 analytical method Methods 0.000 description 19
- 238000000338 in vitro Methods 0.000 description 19
- 229920000642 polymer Polymers 0.000 description 19
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 18
- 230000035772 mutation Effects 0.000 description 18
- 230000015572 biosynthetic process Effects 0.000 description 17
- 239000013615 primer Substances 0.000 description 17
- 108090000765 processed proteins & peptides Proteins 0.000 description 17
- 102000004196 processed proteins & peptides Human genes 0.000 description 16
- 230000003321 amplification Effects 0.000 description 15
- 238000003199 nucleic acid amplification method Methods 0.000 description 15
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 14
- 229920001515 polyalkylene glycol Polymers 0.000 description 14
- 102100034608 Angiopoietin-2 Human genes 0.000 description 13
- 101000924533 Homo sapiens Angiopoietin-2 Proteins 0.000 description 13
- 230000028993 immune response Effects 0.000 description 13
- 238000001727 in vivo Methods 0.000 description 13
- 125000002264 triphosphate group Chemical group [H]OP(=O)(O[H])OP(=O)(O[H])OP(=O)(O[H])O* 0.000 description 13
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 12
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 12
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 12
- 239000000020 Nitrocellulose Substances 0.000 description 12
- 108091028043 Nucleic acid sequence Proteins 0.000 description 12
- 238000002474 experimental method Methods 0.000 description 12
- 230000006870 function Effects 0.000 description 12
- 230000003308 immunostimulating effect Effects 0.000 description 12
- 229920001220 nitrocellulos Polymers 0.000 description 12
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 12
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 12
- PCDQPRRSZKQHHS-CCXZUQQUSA-N Cytarabine Triphosphate Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 PCDQPRRSZKQHHS-CCXZUQQUSA-N 0.000 description 11
- 229910052799 carbon Inorganic materials 0.000 description 11
- WAEMQWOKJMHJLA-UHFFFAOYSA-N Manganese(2+) Chemical compound [Mn+2] WAEMQWOKJMHJLA-UHFFFAOYSA-N 0.000 description 10
- 101710163270 Nuclease Proteins 0.000 description 10
- 239000004677 Nylon Substances 0.000 description 10
- 125000003295 alanine group Chemical group N[C@@H](C)C(=O)* 0.000 description 10
- 230000000903 blocking effect Effects 0.000 description 10
- 125000005647 linker group Chemical group 0.000 description 10
- 229920001778 nylon Polymers 0.000 description 10
- 150000003230 pyrimidines Chemical class 0.000 description 10
- 239000000126 substance Substances 0.000 description 10
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 10
- 101000957776 Escherichia coli O157:H7 Mannose-1-phosphate guanylyltransferase 1 Proteins 0.000 description 9
- 108010006785 Taq Polymerase Proteins 0.000 description 9
- 239000002299 complementary DNA Substances 0.000 description 9
- 239000013604 expression vector Substances 0.000 description 9
- 125000001909 leucine group Chemical group [H]N(*)C(C(*)=O)C([H])([H])C(C([H])([H])[H])C([H])([H])[H] 0.000 description 9
- 238000010839 reverse transcription Methods 0.000 description 9
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical group [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 9
- 238000012546 transfer Methods 0.000 description 9
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 8
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 8
- 238000003556 assay Methods 0.000 description 8
- 238000010494 dissociation reaction Methods 0.000 description 8
- 230000005593 dissociations Effects 0.000 description 8
- 238000009826 distribution Methods 0.000 description 8
- 125000000487 histidyl group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C([H])=N1 0.000 description 8
- 239000012160 loading buffer Substances 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 210000002966 serum Anatomy 0.000 description 8
- 230000000087 stabilizing effect Effects 0.000 description 8
- 238000012360 testing method Methods 0.000 description 8
- 239000004475 Arginine Substances 0.000 description 7
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 7
- 230000004913 activation Effects 0.000 description 7
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 7
- 150000001875 compounds Chemical class 0.000 description 7
- 230000003993 interaction Effects 0.000 description 7
- 239000013642 negative control Substances 0.000 description 7
- 230000006320 pegylation Effects 0.000 description 7
- 229920001184 polypeptide Polymers 0.000 description 7
- 239000002213 purine nucleotide Substances 0.000 description 7
- 239000002719 pyrimidine nucleotide Substances 0.000 description 7
- 238000011160 research Methods 0.000 description 7
- 239000011780 sodium chloride Substances 0.000 description 7
- 235000000346 sugar Nutrition 0.000 description 7
- 229940124597 therapeutic agent Drugs 0.000 description 7
- 102000004190 Enzymes Human genes 0.000 description 6
- 108090000790 Enzymes Proteins 0.000 description 6
- QGWNDRXFNXRZMB-UUOKFMHZSA-N GDP Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O QGWNDRXFNXRZMB-UUOKFMHZSA-N 0.000 description 6
- 101001124867 Homo sapiens Peroxiredoxin-1 Proteins 0.000 description 6
- 229910021380 Manganese Chloride Inorganic materials 0.000 description 6
- GLFNIEUTAYBVOC-UHFFFAOYSA-L Manganese chloride Chemical compound Cl[Mn]Cl GLFNIEUTAYBVOC-UHFFFAOYSA-L 0.000 description 6
- 102100026066 Phosphoprotein associated with glycosphingolipid-enriched microdomains 1 Human genes 0.000 description 6
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 6
- 239000006180 TBST buffer Substances 0.000 description 6
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical group O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 6
- 230000015556 catabolic process Effects 0.000 description 6
- 230000004087 circulation Effects 0.000 description 6
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 6
- 238000006731 degradation reaction Methods 0.000 description 6
- 238000012217 deletion Methods 0.000 description 6
- 230000037430 deletion Effects 0.000 description 6
- 238000013461 design Methods 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- QGWNDRXFNXRZMB-UHFFFAOYSA-N guanidine diphosphate Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(COP(O)(=O)OP(O)(O)=O)C(O)C1O QGWNDRXFNXRZMB-UHFFFAOYSA-N 0.000 description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 6
- 238000011534 incubation Methods 0.000 description 6
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 6
- 229910001629 magnesium chloride Inorganic materials 0.000 description 6
- 239000011565 manganese chloride Substances 0.000 description 6
- 229940099607 manganese chloride Drugs 0.000 description 6
- 235000002867 manganese chloride Nutrition 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- 102000044158 nucleic acid binding protein Human genes 0.000 description 6
- 108700020942 nucleic acid binding protein Proteins 0.000 description 6
- 238000000159 protein binding assay Methods 0.000 description 6
- 230000004044 response Effects 0.000 description 6
- 239000000758 substrate Substances 0.000 description 6
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 6
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 5
- 241000282693 Cercopithecidae Species 0.000 description 5
- 101000782195 Homo sapiens von Willebrand factor Proteins 0.000 description 5
- 102000008235 Toll-Like Receptor 9 Human genes 0.000 description 5
- 108010060818 Toll-Like Receptor 9 Proteins 0.000 description 5
- SSQWUPFNONDRPX-AEHJODJJSA-N [(2R,3R,4R,5R)-4-amino-5-(2-amino-6-oxo-1H-purin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphono hydrogen phosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@@]1(N)O SSQWUPFNONDRPX-AEHJODJJSA-N 0.000 description 5
- BCOONYUGFLPFLH-AEHJODJJSA-N [(2R,3R,4S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-4-fluoro-3,4-dihydroxyoxolan-2-yl]methyl dihydrogen phosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@]1(O)F BCOONYUGFLPFLH-AEHJODJJSA-N 0.000 description 5
- GYJOUNAUSNJSIO-AEHJODJJSA-N [(2r,3r,4r,5r)-4-amino-5-(2-amino-6-oxo-3h-purin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl dihydrogen phosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@]1(N)O GYJOUNAUSNJSIO-AEHJODJJSA-N 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 230000000295 complement effect Effects 0.000 description 5
- 238000000502 dialysis Methods 0.000 description 5
- 150000002009 diols Chemical class 0.000 description 5
- 238000012869 ethanol precipitation Methods 0.000 description 5
- 239000012528 membrane Substances 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 238000011002 quantification Methods 0.000 description 5
- 238000003757 reverse transcription PCR Methods 0.000 description 5
- 238000000926 separation method Methods 0.000 description 5
- 241000894007 species Species 0.000 description 5
- 238000010561 standard procedure Methods 0.000 description 5
- 239000006228 supernatant Substances 0.000 description 5
- 101100076239 Drosophila melanogaster Mctp gene Proteins 0.000 description 4
- 229910019142 PO4 Inorganic materials 0.000 description 4
- 102000012753 TIE-2 Receptor Human genes 0.000 description 4
- 108010090091 TIE-2 Receptor Proteins 0.000 description 4
- JUOREOKPMJLFRU-LIQNOFJUSA-N [(2r,3s)-5-(2-amino-6-oxo-3h-purin-9-yl)-3-hydroxyoxolan-2-yl]methyl dihydrogen phosphate;azane Chemical compound N.C1=2NC(N)=NC(=O)C=2N=CN1C1C[C@H](O)[C@@H](COP(O)(O)=O)O1 JUOREOKPMJLFRU-LIQNOFJUSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 4
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 4
- 125000000637 arginyl group Chemical group N[C@@H](CCCNC(N)=N)C(=O)* 0.000 description 4
- 230000008827 biological function Effects 0.000 description 4
- 238000010367 cloning Methods 0.000 description 4
- 108091036078 conserved sequence Proteins 0.000 description 4
- CIKGWCTVFSRMJU-KVQBGUIXSA-N dGDP Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(O)=O)O1 CIKGWCTVFSRMJU-KVQBGUIXSA-N 0.000 description 4
- 150000002148 esters Chemical group 0.000 description 4
- 125000003827 glycol group Chemical group 0.000 description 4
- 230000003100 immobilizing effect Effects 0.000 description 4
- 230000002163 immunogen Effects 0.000 description 4
- 230000006872 improvement Effects 0.000 description 4
- 150000002611 lead compounds Chemical class 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- 210000000056 organ Anatomy 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 4
- 239000010452 phosphate Substances 0.000 description 4
- 230000013878 renal filtration Effects 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- 150000003384 small molecules Chemical class 0.000 description 4
- 238000010532 solid phase synthesis reaction Methods 0.000 description 4
- 238000004448 titration Methods 0.000 description 4
- 102000016911 Deoxyribonucleases Human genes 0.000 description 3
- 108010053770 Deoxyribonucleases Proteins 0.000 description 3
- AHCYMLUZIRLXAA-SHYZEUOFSA-N Deoxyuridine 5'-triphosphate Chemical compound O1[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)C[C@@H]1N1C(=O)NC(=O)C=C1 AHCYMLUZIRLXAA-SHYZEUOFSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 108060002716 Exonuclease Proteins 0.000 description 3
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 3
- 108091008103 RNA aptamers Proteins 0.000 description 3
- 230000004570 RNA-binding Effects 0.000 description 3
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 3
- BTVMPTGYVOHMLG-AEHJODJJSA-N [(2R,3R,4S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-4-fluoro-3,4-dihydroxyoxolan-2-yl]methyl phosphono hydrogen phosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@]1(O)F BTVMPTGYVOHMLG-AEHJODJJSA-N 0.000 description 3
- 125000004429 atom Chemical group 0.000 description 3
- 230000009286 beneficial effect Effects 0.000 description 3
- 230000001588 bifunctional effect Effects 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 230000021615 conjugation Effects 0.000 description 3
- 229940104302 cytosine Drugs 0.000 description 3
- 238000001212 derivatisation Methods 0.000 description 3
- 238000011033 desalting Methods 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 102000013165 exonuclease Human genes 0.000 description 3
- 230000005714 functional activity Effects 0.000 description 3
- 238000001502 gel electrophoresis Methods 0.000 description 3
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000003780 insertion Methods 0.000 description 3
- 230000037431 insertion Effects 0.000 description 3
- 238000002703 mutagenesis Methods 0.000 description 3
- 231100000350 mutagenesis Toxicity 0.000 description 3
- 230000005257 nucleotidylation Effects 0.000 description 3
- 229940046166 oligodeoxynucleotide Drugs 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 150000008300 phosphoramidites Chemical class 0.000 description 3
- 230000036470 plasma concentration Effects 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 150000003212 purines Chemical class 0.000 description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 238000012163 sequencing technique Methods 0.000 description 3
- 238000002741 site-directed mutagenesis Methods 0.000 description 3
- 238000005556 structure-activity relationship Methods 0.000 description 3
- 150000008163 sugars Chemical class 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- 229940035893 uracil Drugs 0.000 description 3
- DCESWPKOLYIMNH-UHFFFAOYSA-N 2-(2,5-dioxopyrrolidin-1-yl)propanoic acid Chemical compound OC(=O)C(C)N1C(=O)CCC1=O DCESWPKOLYIMNH-UHFFFAOYSA-N 0.000 description 2
- PMUNIMVZCACZBB-UHFFFAOYSA-N 2-hydroxyethylazanium;chloride Chemical compound Cl.NCCO PMUNIMVZCACZBB-UHFFFAOYSA-N 0.000 description 2
- 108020000946 Bacterial DNA Proteins 0.000 description 2
- 102000004127 Cytokines Human genes 0.000 description 2
- 108090000695 Cytokines Proteins 0.000 description 2
- 102000053602 DNA Human genes 0.000 description 2
- 108091008102 DNA aptamers Proteins 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- 102100027286 Fanconi anemia group C protein Human genes 0.000 description 2
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 2
- 101000955962 Homo sapiens Vacuolar protein sorting-associated protein 51 homolog Proteins 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 108091093037 Peptide nucleic acid Proteins 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- 101500027983 Rattus norvegicus Octadecaneuropeptide Proteins 0.000 description 2
- 108091028664 Ribonucleotide Proteins 0.000 description 2
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- 108010090804 Streptavidin Proteins 0.000 description 2
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical class OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 2
- 102000002689 Toll-like receptor Human genes 0.000 description 2
- 108020000411 Toll-like receptor Proteins 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 208000006673 asthma Diseases 0.000 description 2
- 229960000074 biopharmaceutical Drugs 0.000 description 2
- 229920001400 block copolymer Polymers 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 230000009918 complex formation Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 230000009260 cross reactivity Effects 0.000 description 2
- NHVNXKFIZYSCEB-XLPZGREQSA-N dTTP Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)C1 NHVNXKFIZYSCEB-XLPZGREQSA-N 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 239000005547 deoxyribonucleotide Substances 0.000 description 2
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 2
- 239000000032 diagnostic agent Substances 0.000 description 2
- 229940039227 diagnostic agent Drugs 0.000 description 2
- 239000001177 diphosphate Substances 0.000 description 2
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical group [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 2
- 235000011180 diphosphates Nutrition 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 229940073579 ethanolamine hydrochloride Drugs 0.000 description 2
- 230000002349 favourable effect Effects 0.000 description 2
- 238000000684 flow cytometry Methods 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 125000003843 furanosyl group Chemical group 0.000 description 2
- 229920001519 homopolymer Polymers 0.000 description 2
- 102000052216 human VPS51 Human genes 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 230000005847 immunogenicity Effects 0.000 description 2
- 238000010348 incorporation Methods 0.000 description 2
- 208000015181 infectious disease Diseases 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- 238000004811 liquid chromatography Methods 0.000 description 2
- 239000012139 lysis buffer Substances 0.000 description 2
- 229920002521 macromolecule Polymers 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 239000003607 modifier Substances 0.000 description 2
- 238000000329 molecular dynamics simulation Methods 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N monoethanolamine hydrochloride Natural products NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- 239000000178 monomer Substances 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 230000009871 nonspecific binding Effects 0.000 description 2
- 238000002515 oligonucleotide synthesis Methods 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- 230000003285 pharmacodynamic effect Effects 0.000 description 2
- 150000004713 phosphodiesters Chemical class 0.000 description 2
- 229920002401 polyacrylamide Polymers 0.000 description 2
- 238000006116 polymerization reaction Methods 0.000 description 2
- 150000003141 primary amines Chemical class 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 125000001500 prolyl group Chemical group [H]N1C([H])(C(=O)[*])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 2
- 229920005604 random copolymer Polymers 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000010076 replication Effects 0.000 description 2
- 239000002336 ribonucleotide Substances 0.000 description 2
- 125000002652 ribonucleotide group Chemical group 0.000 description 2
- 125000000548 ribosyl group Chemical group C1([C@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 2
- 238000005070 sampling Methods 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 230000009870 specific binding Effects 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- PFNFFQXMRSDOHW-UHFFFAOYSA-N spermine Chemical compound NCCCNCCCCNCCCN PFNFFQXMRSDOHW-UHFFFAOYSA-N 0.000 description 2
- 230000002269 spontaneous effect Effects 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- ZFXYFBGIUFBOJW-UHFFFAOYSA-N theophylline Chemical compound O=C1N(C)C(=O)N(C)C2=C1NC=N2 ZFXYFBGIUFBOJW-UHFFFAOYSA-N 0.000 description 2
- 230000014616 translation Effects 0.000 description 2
- 238000000870 ultraviolet spectroscopy Methods 0.000 description 2
- 230000003827 upregulation Effects 0.000 description 2
- 229960005486 vaccine Drugs 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- QDZOEBFLNHCSSF-PFFBOGFISA-N (2S)-2-[[(2R)-2-[[(2S)-1-[(2S)-6-amino-2-[[(2S)-1-[(2R)-2-amino-5-carbamimidamidopentanoyl]pyrrolidine-2-carbonyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-N-[(2R)-1-[[(2S)-1-[[(2R)-1-[[(2S)-1-[[(2S)-1-amino-4-methyl-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]pentanediamide Chemical compound C([C@@H](C(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(N)=O)NC(=O)[C@@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](N)CCCNC(N)=N)C1=CC=CC=C1 QDZOEBFLNHCSSF-PFFBOGFISA-N 0.000 description 1
- BJFCLFGYYYYQJY-YFHUEUNASA-N 1-[(2R,3R,4R,5R)-4-hydroxy-5-(hydroxymethyl)-3-methoxyoxolan-2-yl]-5-(1H-indol-2-ylmethylidene)-1,3-diazinane-2,4-dione Chemical compound CO[C@@H]1[C@@H]([C@H](O[C@H]1N2CC(=CC3=CC4=CC=CC=C4N3)C(=O)NC2=O)CO)O BJFCLFGYYYYQJY-YFHUEUNASA-N 0.000 description 1
- YKBGVTZYEHREMT-KVQBGUIXSA-N 2'-deoxyguanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 YKBGVTZYEHREMT-KVQBGUIXSA-N 0.000 description 1
- YKBGVTZYEHREMT-UHFFFAOYSA-N 2'-deoxyguanosine Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1CC(O)C(CO)O1 YKBGVTZYEHREMT-UHFFFAOYSA-N 0.000 description 1
- RGNOTKMIMZMNRX-XVFCMESISA-N 2-amino-1-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-4-one Chemical compound NC1=NC(=O)C=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 RGNOTKMIMZMNRX-XVFCMESISA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- BGTXMQUSDNMLDW-AEHJODJJSA-N 2-amino-9-[(2r,3s,4r,5r)-3-fluoro-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-3h-purin-6-one Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](CO)[C@@H](O)[C@]1(O)F BGTXMQUSDNMLDW-AEHJODJJSA-N 0.000 description 1
- ZLOIGESWDJYCTF-UHFFFAOYSA-N 4-Thiouridine Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=S)C=C1 ZLOIGESWDJYCTF-UHFFFAOYSA-N 0.000 description 1
- ZLOIGESWDJYCTF-XVFCMESISA-N 4-thiouridine Chemical class O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=S)C=C1 ZLOIGESWDJYCTF-XVFCMESISA-N 0.000 description 1
- OCKGFTQIICXDQW-ZEQRLZLVSA-N 5-[(1r)-1-hydroxy-2-[4-[(2r)-2-hydroxy-2-(4-methyl-1-oxo-3h-2-benzofuran-5-yl)ethyl]piperazin-1-yl]ethyl]-4-methyl-3h-2-benzofuran-1-one Chemical compound C1=C2C(=O)OCC2=C(C)C([C@@H](O)CN2CCN(CC2)C[C@H](O)C2=CC=C3C(=O)OCC3=C2C)=C1 OCKGFTQIICXDQW-ZEQRLZLVSA-N 0.000 description 1
- LQLQRFGHAALLLE-UHFFFAOYSA-N 5-bromouracil Chemical class BrC1=CNC(=O)NC1=O LQLQRFGHAALLLE-UHFFFAOYSA-N 0.000 description 1
- KSNXJLQDQOIRIP-UHFFFAOYSA-N 5-iodouracil Chemical class IC1=CNC(=O)NC1=O KSNXJLQDQOIRIP-UHFFFAOYSA-N 0.000 description 1
- LRSASMSXMSNRBT-UHFFFAOYSA-N 5-methylcytosine Chemical compound CC1=CNC(=O)N=C1N LRSASMSXMSNRBT-UHFFFAOYSA-N 0.000 description 1
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 108700031308 Antennapedia Homeodomain Proteins 0.000 description 1
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 1
- 108010000241 Arthropod Proteins Proteins 0.000 description 1
- 238000009010 Bradford assay Methods 0.000 description 1
- FGUUSXIOTUKUDN-IBGZPJMESA-N C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 Chemical compound C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 FGUUSXIOTUKUDN-IBGZPJMESA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 102000020313 Cell-Penetrating Peptides Human genes 0.000 description 1
- 108010051109 Cell-Penetrating Peptides Proteins 0.000 description 1
- 206010057250 Cell-mediated cytotoxicity Diseases 0.000 description 1
- 108091026890 Coding region Proteins 0.000 description 1
- MIKUYHXYGGJMLM-UUOKFMHZSA-N Crotonoside Chemical compound C1=NC2=C(N)NC(=O)N=C2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O MIKUYHXYGGJMLM-UUOKFMHZSA-N 0.000 description 1
- 108020001019 DNA Primers Proteins 0.000 description 1
- 239000003155 DNA primer Substances 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- 101100481408 Danio rerio tie2 gene Proteins 0.000 description 1
- 241000255581 Drosophila <fruit fly, genus> Species 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 238000012286 ELISA Assay Methods 0.000 description 1
- 102000004533 Endonucleases Human genes 0.000 description 1
- 108010042407 Endonucleases Proteins 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- 206010015866 Extravasation Diseases 0.000 description 1
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 1
- 108010070875 Human Immunodeficiency Virus tat Gene Products Proteins 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 229930010555 Inosine Natural products 0.000 description 1
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 description 1
- 108010074328 Interferon-gamma Proteins 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- 102000043129 MHC class I family Human genes 0.000 description 1
- 108091054437 MHC class I family Proteins 0.000 description 1
- 102000043131 MHC class II family Human genes 0.000 description 1
- 108091054438 MHC class II family Proteins 0.000 description 1
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 description 1
- 101100481410 Mus musculus Tek gene Proteins 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- 108091005461 Nucleic proteins Proteins 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical compound CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 description 1
- 101710149951 Protein Tat Proteins 0.000 description 1
- 108091030071 RNAI Proteins 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- 102400000096 Substance P Human genes 0.000 description 1
- 101800003906 Substance P Proteins 0.000 description 1
- 230000005867 T cell response Effects 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 1
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 231100000230 acceptable toxicity Toxicity 0.000 description 1
- 230000033289 adaptive immune response Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 238000001261 affinity purification Methods 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229940059260 amidate Drugs 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 229940125644 antibody drug Drugs 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 210000000612 antigen-presenting cell Anatomy 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 239000012830 cancer therapeutic Substances 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000005779 cell damage Effects 0.000 description 1
- 208000037887 cell injury Diseases 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 238000002144 chemical decomposition reaction Methods 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 230000000139 costimulatory effect Effects 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 230000008260 defense mechanism Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- VGONTNSXDCQUGY-UHFFFAOYSA-N desoxyinosine Natural products C1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 VGONTNSXDCQUGY-UHFFFAOYSA-N 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- 239000012470 diluted sample Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000006471 dimerization reaction Methods 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 230000009881 electrostatic interaction Effects 0.000 description 1
- 239000012149 elution buffer Substances 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- DNJIEGIFACGWOD-UHFFFAOYSA-N ethyl mercaptane Natural products CCS DNJIEGIFACGWOD-UHFFFAOYSA-N 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- NPUKDXXFDDZOKR-LLVKDONJSA-N etomidate Chemical compound CCOC(=O)C1=CN=CN1[C@H](C)C1=CC=CC=C1 NPUKDXXFDDZOKR-LLVKDONJSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 230000036251 extravasation Effects 0.000 description 1
- 235000013861 fat-free Nutrition 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 1
- 230000009368 gene silencing by RNA Effects 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 229920000140 heteropolymer Polymers 0.000 description 1
- 229920006158 high molecular weight polymer Polymers 0.000 description 1
- 229940034998 human von willebrand factor Drugs 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 230000015788 innate immune response Effects 0.000 description 1
- 229960003786 inosine Drugs 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- BPHPUYQFMNQIOC-NXRLNHOXSA-N isopropyl beta-D-thiogalactopyranoside Chemical compound CC(C)S[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O BPHPUYQFMNQIOC-NXRLNHOXSA-N 0.000 description 1
- 210000003292 kidney cell Anatomy 0.000 description 1
- 150000002605 large molecules Chemical class 0.000 description 1
- 150000002634 lipophilic molecules Chemical class 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- 229920001427 mPEG Polymers 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 238000009115 maintenance therapy Methods 0.000 description 1
- 229910052748 manganese Inorganic materials 0.000 description 1
- 239000011572 manganese Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- YACKEPLHDIMKIO-UHFFFAOYSA-N methylphosphonic acid Chemical compound CP(O)(O)=O YACKEPLHDIMKIO-UHFFFAOYSA-N 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- SIUPPTRKFWMXIE-UHFFFAOYSA-N n-ethyl-n'-propylmethanediimine Chemical compound CCCN=C=NCC SIUPPTRKFWMXIE-UHFFFAOYSA-N 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 108091008104 nucleic acid aptamers Proteins 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 125000003835 nucleoside group Chemical group 0.000 description 1
- 229920002113 octoxynol Polymers 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 230000010412 perfusion Effects 0.000 description 1
- 238000002823 phage display Methods 0.000 description 1
- SXADIBFZNXBEGI-UHFFFAOYSA-N phosphoramidous acid Chemical group NP(O)O SXADIBFZNXBEGI-UHFFFAOYSA-N 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 229920000724 poly(L-arginine) polymer Polymers 0.000 description 1
- 108010011110 polyarginine Proteins 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 238000011533 pre-incubation Methods 0.000 description 1
- 238000011165 process development Methods 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 238000001742 protein purification Methods 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 238000004064 recycling Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000012772 sequence design Methods 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 230000037432 silent mutation Effects 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 229940126586 small molecule drug Drugs 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 229940063675 spermine Drugs 0.000 description 1
- 238000009987 spinning Methods 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 230000002966 stenotic effect Effects 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 230000001839 systemic circulation Effects 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229960000278 theophylline Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 229940021747 therapeutic vaccine Drugs 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 150000005691 triesters Chemical class 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 210000003606 umbilical vein Anatomy 0.000 description 1
- 238000003828 vacuum filtration Methods 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 239000013598 vector Substances 0.000 description 1
- 238000012800 visualization Methods 0.000 description 1
- 108010047303 von Willebrand Factor Proteins 0.000 description 1
- 102100036537 von Willebrand factor Human genes 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
Images














Landscapes
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Description
この本特許出願は、米国特許法§119(e)の下、以下の仮出願への利益を主張する:2006年12月22日に出願された米国仮特許出願第60/876,780号および2007年1月10日に出願された米国仮特許出願第60/879,830号。これらの各々は、それらの全体が参考として本明細書に援用される。
本発明は、核酸を転写するための材料および方法、特に、テンプレート指向性重合において修飾酵素を用いて核酸、特にアプタマーへの修飾ヌクレオチドの導入を増加させるための、修飾酵素および材料および方法に関する。
本発明はまた、以下の項目を提供する。
(項目1)
核酸を転写する方法であり、
a)(i)2’―OMe ATP、2’―OMe GTP、2’―OMe CTP、2’―OMe TTPおよび2’―OMe UTPより選択される2’―OMe修飾ヌクレオチド三リン酸(2’―OMe NTP)のいずれかを導入することが可能な修飾T7RNAポリメラーゼであり、上記修飾RNAポリメラーゼが、対応する非修飾RNAポリメラーゼの2’―修飾NTPを導入する能力と比較して、上記2’―修飾ヌクレオチド三リン酸(2’―修飾NTP)を導入する能力が増加している、修飾T7RNAポリメラーゼと、(ii)少なくとも一つの核酸転写テンプレートと、(iii)ヌクレオチド三リン酸であり、上記ヌクレオチド三リン酸がグアニジン三リン酸を含み、上記グアニジン三リン酸が全て2’―OMe修飾されている、ヌクレオチド三リン酸とを含む、転写反応混合物を調製するステップと;
b)一本鎖核酸が生じる条件下で、上記転写反応混合物を転写するステップであり、上記生じる一本鎖核酸の、最初のグアノシンヌクレオチドの後の全てのグアニジンヌクレオチドが、2’―OMe修飾されている、ステップと
を含む、方法。
(項目2)
上記転写反応混合物が、マグネシウムイオン、マンガンイオン、またはマグネシウムおよびマンガンイオンの両方をさらに含む、項目1に記載の方法。
(項目3)
上記核酸転写テンプレートが、少なくとも部分的に二本鎖である、項目1に記載の方法。
(項目4)
上記核酸転写テンプレートが、完全二本鎖である、項目1に記載の方法。
(項目5)
上記生じた一本鎖核酸における最初のグアノシヌクレオチドが、2’―OMe修飾されている、項目1に記載の方法。
(項目6)
上記修飾T7RNAポリメラーゼが、野生型T7ポリメラーゼに対して639位および784位で改変されたアミノ酸を含み、上記784位の改変アミノ酸がアラニンであるときには、上記639位の改変アミノ酸がフェニルアラニンでない、項目1に記載の方法。
(項目7)
上記修飾T7RNAポリメラーゼが、639位にロイシン、784位にアラニンを含む、項目6に記載の方法。
(項目8)
上記二本鎖オリゴヌクレオチド転写テンプレートが、上記オリゴヌクレオチド転写テンプレートの5’末端の固定領域に導入されたリーダー配列をさらに含む、項目3に記載の方法。
(項目9)
上記リーダー配列が、全てプリンのリーダー配列である、項目8に記載の方法。
(項目10)
上記全てプリンのリーダー配列が、少なくとも8ヌクレオチド長、少なくとも10ヌクレオチド長;少なくとも12ヌクレオチド長;および少なくとも14ヌクレオチド長より選択される長さを有する、項目9に記載の方法。
(項目11)
上記反応転写条件が、マグネシウムおよびマンガンイオンの両方を含み、マグネシウムイオンの濃度が、マンガンイオンの濃度より3.0〜3.5倍高い、項目2に記載の方法。
(項目12)
各NTP、2’―修飾NTPまたは2’―OMe NTPが、約0.5mMの濃度で存在し、上記反応転写条件が、マグネシウムおよびマンガンイオンの両方を含み、マグネシウムイオンの濃度が約5.0mMであり、マンガンイオンの濃度が約1.5mMである、項目2に記載の方法。
(項目13)
各NTP、2’―修飾NTPまたは2’―OMe NTPが、約1.0mMの濃度で存在し、上記反応転写条件が、マグネシウムおよびマンガンイオンの両方を含み、マグネシウムイオンの濃度が約6.5mMであり、マンガンイオンの濃度が約2.0mMである、項目2に記載の方法。
(項目14)
各NTP、2’―修飾NTPまたは2’―OMe NTPが、約2.0mMの濃度で存在し、上記反応転写条件が、マグネシウムおよびマンガンイオンの両方を含み、マグネシウムイオンの濃度が約9.6mMであり、マンガンイオンの濃度が約2.9mMである、項目2に記載の方法。
(項目15)
上記転写反応混合物が、置換グアノシンまたはグアノシンをさらに含む、項目1に記載の方法。
(項目16)
上記置換グアノシンまたはグアノシンが、GMPである、項目15に記載の方法。
(項目17)
上記転写反応混合物が、ポリアルキレングリコールをさらに含む、項目1に記載の方法。
(項目18)
上記ポリアルキレングリコールが、ポリエチレングリコールである、項目17に記載の方法。
(項目19)
上記転写反応混合物が、無機ピロホスファターゼをさらに含む、項目1に記載の方法。
(項目20)
上記転写反応混合物が、2’―デオキシシチジン三リン酸(2’―デオキシCTP)、2’―O―Meアデノシン三リン酸(2’―OMe ATP)、2’―OMeグアノシン三リン酸GTP(2’―OMe GTP)、および2’―OMeウリジン三リン酸(2’―OMe UTP)または2’―OMeチミジンヌクレオチド三リン酸(2’―OMe TTP)の混合物をさらに含む、項目1に記載の方法。
(項目21)
上記転写反応混合物が、2’―デオキシチミジン三リン酸(2’―デオキシTTP)または2’―デオキシウリジン三リン酸(2’―デオキシUTP)および2’―O―Meアデノシン三リン酸(2’―OMe ATP)、2’―OMeグアノシン三リン酸GTP(2’―OMe GTP)、および2’―OMeシチジン三リン酸(2’―OMe CTP)の混合物をさらに含む、項目1に記載の方法。
(項目22)
上記転写反応混合物が、2’―OHチミジン三リン酸(2’―OH TTP)または2’―OHウリジン三リン酸(2’―OH UTP)および2’―O―Meアデノシン三リン酸(2’―OMe ATP)、2’―OMeグアノシン三リン酸GTP(2’―OMe
GTP)、および2’―OMeシチジン三リン酸(2’―OMe CTP)の混合物をさらに含む、項目1に記載の方法。
(項目23)
上記転写反応混合物が、側鎖で修飾された核酸塩基をさらに含む、項目1に記載の方法。
(項目24)
上記核酸塩基が、ウリジンの5―および6―位、チミジンの5―および6―位、シチジンの5および6―位および環外アミン、アデノシンの2―、7―および8―位および環外アミン、グアノシンの7―および8―位および環外アミンからなる群より選択される位置で修飾される、項目23に記載の方法。
(項目25)
修飾核酸塩基が、ウラシル、シトシンまたはチミジンである、項目24に記載の方法。
(項目26)
上記側鎖が、疎水性芳香族側鎖である、項目23に記載の方法。
(項目27)
上記側鎖が、ベンジルまたはインドリルである、項目26に記載の方法。
(項目28)
アプタマーを同定する方法であり、上記アプタマーが2’―OMe修飾ヌクレオチドを含む、方法であり、
a)(i)2’―OMe ATP、2’―OMe GTP、2’―OMe CTP、2’―OMe TTPおよび2’―OMe UTPより選択される2’―OMe修飾ヌクレオチド三リン酸(2’―OMe NTP)のいずれかを導入することが可能な修飾T7RNAポリメラーゼであり、上記修飾RNAポリメラーゼが、対応する非修飾RNAポリメラーゼの2’―修飾NTPを導入する能力と比較して、上記2’―修飾ヌクレオチド三リン酸(2’―修飾NTP)を導入する能力が増加している、修飾T7RNAポリメラーゼと、(ii)少なくとも一つの核酸転写テンプレートと、(iii)ヌクレオチド三リン酸であり、上記ヌクレオチド三リン酸がグアニジン三リン酸を含み、上記グアニジン三リン酸が全て2’―OMe修飾されている、ヌクレオチド三リン酸とを含む、転写反応混合物を調製するステップと;
b)上記修飾T7RNAポリメラーゼにより、一本鎖核酸の候補混合物の上記核酸分子に、少なくとも一つの修飾2’―OMeヌクレオチド三リン酸(2’―OMe NTP)が導入される条件下で、上記転写反応混合物を転写することにより、上記一本鎖核酸の候補混合物を調製するステップであり、上記候補混合物の上記一本鎖核酸分子の、最初のグアノシンヌクレオチドの後の全てのグアニジンヌクレオチドが、2’―OMe修飾されている、ステップと;
c)上記候補混合物を上記標的分子と接触させるステップと、
d)上記候補混合物の親和性と比較して、標的分子に対する親和性が増加した上記核酸を、上記候補混合物から分離するステップと、
e)アプタマーが濃縮された混合物を生じるために、上記親和性が増加した核酸を増幅するステップを含み、それによって、アプタマーの上記グアニジンヌクレオチドの一つを選択的に除いて、全て2’―OMe修飾されたグアニジンヌクレオチドを含む、上記標的分子に対する上記アプタマーが、同定される、
方法。
(項目29)
上記転写反応混合物が、マグネシウムイオン、マンガンイオンまたはマグネシウムおよびマンガンイオンの両方をさらに含む、項目28に記載の方法。
(項目30)
上記核酸転写テンプレートが、少なくとも部分的に二本鎖である、項目28に記載の方法。
(項目31)
上記核酸転写テンプレートが、完全二本鎖である、項目28に記載の方法。
(項目32)
上記生じた一本鎖核酸における最初のグアノシヌクレオチドが、2’―OMe修飾されている、項目28に記載の方法。
(項目33)
上記修飾T7RNAポリメラーゼが、野生型T7ポリメラーゼに対して639位および784位で改変されたアミノ酸を含み、上記784位の改変アミノ酸がアラニンであるときには、上記639位の改変アミノ酸がフェニルアラニンでない、項目28に記載の方法。
(項目34)
上記修飾T7RNAポリメラーゼが、639位にロイシン、784位にアラニンを含む、項目33に記載の方法。
(項目35)
上記二本鎖オリゴヌクレオチド転写テンプレートが、上記オリゴヌクレオチド転写テンプレートの5’末端の固定領域に導入されたリーダー配列をさらに含む、項目30に記載の方法。
(項目36)
上記リーダー配列が、全てプリンのリーダー配列である、項目35に記載の方法。
(項目37)
上記全てプリンのリーダー配列が、少なくとも8ヌクレオチド長、少なくとも10ヌクレオチド長;少なくとも12ヌクレオチド長;および少なくとも14ヌクレオチド長より選択される長さを有する、項目36に記載の方法。
(項目38)
上記反応転写条件が、マグネシウムおよびマンガンイオンの両方を含み、マグネシウムイオンの濃度が、マンガンイオンの濃度より3.0〜3.5倍高い、項目29に記載の方法。
(項目39)
各NTP、2’―修飾NTPまたは2’―OMe NTPが、約0.5mMの濃度で存在し、上記反応転写条件が、マグネシウムおよびマンガンイオンの両方を含み、マグネシウムイオンの濃度が約5.0mMであり、マンガンイオンの濃度が約1.5mMである、項目29に記載の方法。
(項目40)
各NTP、2’―修飾NTPまたは2’―OMe NTPが、約1.0mMの濃度で存在し、上記反応転写条件が、マグネシウムおよびマンガンイオンの両方を含み、マグネシウムイオンの濃度が約6.5mMであり、マンガンイオンの濃度が約2.0mMである、項目29に記載の方法。
(項目41)
各NTP、2’―修飾NTPまたは2’―OMe NTPが、約2.0mMの濃度で存在し、上記反応転写条件が、マグネシウムおよびマンガンイオンの両方を含み、マグネシウムイオンの濃度が約9.6mMであり、マンガンイオンの濃度が約2.9mMである、項目29に記載の方法。
(項目42)
上記転写反応混合物が、置換グアノシンまたはグアノシンをさらに含む、項目28に記載の方法。
(項目43)
上記置換グアノシンまたはグアノシンが、GMPである、項目42に記載の方法。
(項目44)
上記転写反応混合物が、ポリアルキレングリコールをさらに含む、項目28に記載の方法。
(項目45)
上記ポリアルキレングリコールが、ポリエチレングリコールである、項目44に記載の方法。
(項目46)
上記転写反応混合物が、無機ピロホスファターゼをさらに含む、項目28に記載の方法。
(項目47)
上記転写反応混合物が、2’―デオキシシチジン三リン酸(2’―デオキシCTP)、2’―O―Meアデノシン三リン酸(2’―OMe ATP)、2’―OMeグアノシン三リン酸GTP(2’―OMe GTP)、および2’―OMeウリジン三リン酸(2’―OMe UTP)または2’―OMeチミジンヌクレオチド三リン酸(2’―OMe TTP)の混合物をさらに含む、項目28に記載の方法。
(項目48)
上記転写反応混合物が、2’―デオキシチミジン三リン酸(2’―デオキシTTP)または2’―デオキシウリジン三リン酸(2’―デオキシUTP)および2’―O―Meアデノシン三リン酸(2’―OMe ATP)、2’―OMeグアノシン三リン酸GTP(2’―OMe GTP)、および2’―OMeシチジン三リン酸(2’―OMe CTP)の混合物をさらに含む、項目28に記載の方法。
(項目49)
上記転写反応混合物が、2’―OHチミジン三リン酸(2’―OH TTP)または2’―OHウリジン三リン酸(2’―OH UTP)および2’―O―Meアデノシン三リン酸(2’―OMe ATP)、2’―OMeグアノシン三リン酸GTP(2’―OMe
GTP)、および2’―OMeシチジン三リン酸(2’―OMe CTP)の混合物をさらに含む、項目28に記載の方法。
(項目50)
上記転写反応混合物が、側鎖で修飾された核酸塩基をさらに含む、項目28に記載の方法。
(項目51)
上記核酸塩基が、ウリジンの5―および6―位、チミジンの5―および6―位、シチジンの5および6―位および環外アミン、アデノシンの2―、7―および8―位および環外アミン、グアノシンの7―および8―位および環外アミンからなる群より選択される位置で修飾される、項目50に記載の方法。
(項目52)
修飾核酸塩基が、ウラシル、シトシンまたはチミジンである、項目51に記載の方法。
(項目53)
上記側鎖が、疎水性芳香族側鎖である、項目50に記載の方法。
(項目54)
上記側鎖が、ベンジルまたはインドリルである、項目53に記載の方法。
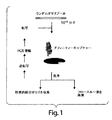
アプタマーを生成する好ましい方法は、概して図1に示され、in vitro選択と呼もばれる、“Systematic Evolution of Ligands by Exponential Enrichment(試験管内人工進化法)”(「SELEX(商標)」)と称されるプロセスによる。SELEX(商標)プロセスは、標的分子に対する高特異性結合をもつ核酸分子のin vitro進化のための方法であり、例えば、1990年6月11日出願の、現在は放棄されている米国特許出願第07/536428号、“Nucleic Acid Ligands”という題の米国特許第5,475,096号、および“Nucleic Acid Ligands”という題の米国特許第5,270,163号(WO91/19813も参照)に記載されている。SELEX(商標)は、選択および増幅の反復的サイクルを実行することにより、任意の所望のレベルの標的結合親和性をもつ、本明細書において「核酸リガンド」とも称されるアプタマーを得るために使用できる。
一実施形態においては、P(O)O基がP(O)S(「チオエート」)、P(S)S(「ジチオエート」)、P(O)NR2(「アミデート」)、P(O)R、P(O)OR’、COまたはCH2(「ホルムアセタール」)、または3’―アミン(―NH―CH2―CH2―)により置換されたオリゴヌクレオチドが提供され、各RまたはR’は、独立してHであるか置換もしくは非置換アルキルである。―O―、―N―、または―S―結合を介して、隣接するヌクレオチドに結合基が結合され得る。オリゴヌクレオチドにおける全ての結合が同一である必要はない。
アプタマーが、治療薬および/または特定の種類の診断薬としての使用に適するためには、合成が安価であり、in vivoで安全および安定していることが好ましい。野生型RNAおよびDNAアプタマーは通常、ヌクレアーゼにより分解されやすいため、in vivoで安定してない。2’―位における修飾基の導入により、ヌクレアーゼ分解に対する抵抗が大きく高められうる。
本発明の2’修飾アプタマーは、野生型ポリメラーゼ、例えば野生型T7ポリメラーゼより高い、フラノース2’位に大きな置換基を有する修飾ヌクレオチドの導入率を有する修飾ポリメラーゼ、例えば変異T7ポリメラーゼを用いてつくられる。例えば、639位のチロシン残基がフェニルアラニンに変えられている(Y639F)変異T7ポリメラーゼは、2’デオキシ、2’アミノ―、および2’フルオロ―ヌクレオチド三リン酸(NTPs)を容易に基質として利用し、様々な用途のために修飾RNAsを合成するために広く使われている。しかし、この変異T7ポリメラーゼは、2’―OMeまたは2’―アジド(2’―N3)置換基等の大きな2’―置換基を伴うNTPsを容易に利用(すなわち導入)できないことが報告されている。大きな2’置換基の導入のために、Y639F変異に加えて、784位のヒスチジンがアラニン残基に変えられた変異T7ポリメラーゼが、記載されており(Y639F/H784A)、修飾ピリミジンNTPsを導入するために、限られた状況において使われている。Padilla,R.およびSousa,R.,Nucleic Acids Res.,2002,30(24):138を参照。639位のチロシン残基がフェニルアラニンに変えられ、784位のヒスチジン残基がアラニンに変えられ、378位のリジン残基がアルギニンに変えられている変異T7RNAポリメラーゼ(Y639F/H784A/K378R)が、修飾プリンおよびピリミジンNTPs、例えば2’―OMe NTPsを導入するために、限られた状況において使用されているが、転写のための2’―OH GTPのスパイクを含む。Burmeister等、(2005)Chemistry and Biology,12:25―33を参照。転写のための2’―OH GTPスパイクの包含により、完全に2’―OMeではなく2’―OH GTPの存在に依存しうるアプタマーが生じうる。

2’―修飾SELEX(商標)法の転写条件に、多数の因子が重要であると決定されている。2’修飾、特に2’―OMe修飾ヌクレオチドを含む転写混合物による転写条件のために記載される因子は、2’修飾および側鎖核酸塩基修飾ヌクレオチド三リン酸を含む転写混合物による本発明の転写方法と関連しても用いられる。本発明に用いられる側鎖修飾ヌクレオチド三リン酸が、説明されている。例えば、各々が参照により全体として本明細書に組み込まれる、Vaught JD,Dewey T,Eaton BE.J Am Chem Soc.2004;126:11231―11237;Nieuwlandt D,West M,Cheng X,Kirshenheuter G,Eaton BE.Chembiochem.2003,4,651―654;Ito T,Ueno Y,Komatsu Y,Matsuda A.Nucleic Acids Res.2003;31,2514―2523;Dewey,TM,Mundt,AA,Crouch,GJ,Zyzniewyski,MC,Eaton,BE.J Am Chem Soc 1995,117,8474―8475;Jager S,Rasched G,Kornreich―Leshem H,Engeser M,Thum O,Famulok M.J Am Chem Soc.2005,127,15071―15082;Tarasow TM,Tarasow SL,Eaton BE.Nature.1997,389,54―57;Liu D,Gugliotti LA,Wu T,Dolska M,Tkachenko AG,Shipton MK,Eaton BE,Feldheim DL.Langmuir.2006,22,5862―5866;Gugliotti LA,Feldheim DL,Eaton BE.J Am Chem Soc.2005,127,17814―17818;Gugliotti LA,Feldheim DL,Eaton BE.Science.2004,304,850―852;Kuwahara M,Hanawa K,Ohsawa K,Kitagata R,Ozaki H,Sawai H.Bioorg Med Chem.2006,14,2518―2526;Saitoh H,Nakamura A,Kuwahara M,Ozaki H,Sawai H.Nucleic Acids Res Suppl.2002,2,215―216;Kuwahara M,Ohbayashi T,Hanawa K,Shoji A,Ozaki AN,Ozaki H,Sawai H.Nucleic Acids Res Suppl.2002,2,83―84.Mehedi Masud M,Ozaki―Nakamura A,Kuwahara M,Ozaki H,Sawai H Chembiochem.2003,4,584―548;Masud MM,Ozaki―Nakamura A,Satou F,Ohbayashi T,Ozaki H,Sawai H.Nucleic Acids Res Suppl.2001;(l):21―2;Obayashi T,Masud MM,Ozaki AN,Ozaki H,Kuwahara M,Sawai H.Bioorg Med Chem Lett.2002,12,1167―70;Sawai H,Ozaki―Nakamura A,Mine M,Ozaki H..Bioconjug Chem.2002,2,309―316;Ohbayashi T,Kuwahara M,Hasegawa M,Kasamatsu T,Tamura T,Sawai H Org Biomol Chem.2005,3,2463―2468;Shoji A,Hasegawa T,Kuwahara M,Ozaki H,Sawai H.Bioorg Med Chem Lett.2006 Oct 28;[Epub ahead of print]Kuwahara M,Nagashima J,Hasegawa M,Tamura T,Kitagata R,Hanawa K,Hososhima S,Kasamatsu T,Ozaki H,Sawai H.Nucleic Acids Res.2006,34,5383―5394;Ito Y.Nucleic Acids Symp Ser.997,37,259―260;Schoetzau T,Langner J,Moyroud E,Roehl I,Vonhoff S,Klussmann S.Bioconjug Chem.2003,5,919―926;Mehedi Masud M,Ozaki―Nakamura A,Kuwahara M,Ozaki H,Sawai H.Chembiochem.2003,4,584―588;およびEbara Y,Kaihatsu K,Ueji S.Nucleic Acids Symp Ser.1999,42,175―176を参照。
所望の標的に結合するアプタマーが同定されたら、同定されたアプタマー配列の結合および/または機能的特性をさらに増加させるために、いくつかの技術が選択的に実行されうる。SELEX(商標)プロセス(例えば2’―修飾SELEX(商標))により同定される所望の標的に結合するアプタマー、例えばMNA、dCmD、dTmV、rUmV、dUmVアプタマーが、所望の結合および/または機能的特性を有する最小のアプタマー配列(本明細書において「最小化コンストラクト」とも称される)を得るために、選択的に切断されうる。これを達成する一つの方法は、最小化コンストラクトのデザインを知るための、フォールディングプログラムおよび配列分析(例えば保存モチーフおよび/または共変動を探すための、選択から得られるクローン配列のアライニング)の使用による。最小化コンストラクトのデザインを知るために、アプタマー配列の5’および3’境界を決定するために、生化学的プロービング実験も行われうる。その後、最小化コンストラクトが化学合成され、それらが得られた非最小化配列と比較して、結合および機能的特性につき試験されうる。一連の5’、3’および/または内部欠損を含むアプタマー配列の変異体も、直接化学合成され、それらが得られた非最小化アプタマー配列と比較して、結合および/または機能的特性につき試験されうる。
(1)すでに体内に存在する置換基、例えば2’―デオキシ、2’―リボ、2’―O―メチルプリンまたはピリミジンまたは5―メチルシトシン。
脊椎動物の免疫系による細菌DNAの認識は、特定の配列コンテキストにおける非メチル化CGジヌクレオチドの認識に基づく(「CpGモチーフ」)。このようなモチーフを認識する一つのレセプターは、異なる微生物成分を認識することにより先天性の免疫反応に関与するToll―likeレセプター(〜10メンバー)のファミリーのメンバー、Toll―likeレセプター9(「TLR9」)である。TLR9は、非メチル化オリゴデオキシヌクレオチド(「ODN」)CpG配列を配列特異的様式で結合する。CpGのモチーフの認識が防御機構を誘発し、先天性および最終的に後天性の免疫反応につながる。例えばマウスにおけるTLR9の活性化は、抗原提示細胞の活性化、MHCクラスIおよびII分子のアップレギュレーション、および重要な共刺激分子およびIL―12およびIL―23を含むサイトカインの発現を誘導する。この活性化は、TH1サイトカインIFN―ガンマの強いアップレギュレーションを含めて、直接および間接的にBおよびT細胞反応を増強する。CpG配列に対する反応は、集合的に、感染症からの保護、ワクチンに対する免疫反応の改善、喘息に対する有効な反応、および抗体依存―細胞媒介細胞傷害の改善につながる。したがって、CpG ODNsは、感染症からの保護を提供し、免疫アジュバントまたは癌治療薬(単独療法またはmAbや他の療法と組み合わせて)として機能し、喘息およびアレルギー反応を減少させうる。
アプタマーを含む全てのオリゴヌクレオチドベースの治療薬の薬物動態学的特性が、所望の薬学的用途に適合するように調整されることが重要である。細胞外標的に対するアプタマーには(アンチセンスおよびRNAiベースの治療薬の場合のように)細胞内送達と関連した問題がないが、このようなアプタマーはなお、標的器官および組織に分布され、所望の薬剤投与計画と整合した一定期間体内にとどまることが可能でなければならない。
上述の通り、高分子量の非免疫原性ポリマーによる核酸の誘導体化は、核酸の薬物動態学的および薬力学的特性を変更し、それらをより有効な治療剤とする可能性を有する。活性の好ましい変化には、ヌクレアーゼによる分解に対する抵抗の増加、腎臓による濾過の減少、免疫系に対する曝露の減少、および治療薬の体内分布の変更を含みうる。
高分子量PAG―核酸―PAG結合体は、二つ以上の反応部位を含む核酸と単官能性活性化PEGの反応により調製されうる。一実施形態においては、核酸は二反応性または二活性化され、二つの反応部位:従来のホスホラミダイト合成によりオリゴヌクレオチドに導入された5’―アミノ基および3’―アミノ基、例えば図2に示されるような3’―5’―ジ―PEG化を含む。代替的実施形態では、例えばピリミジンの5―位、プリンの8―位またはリボースの2’―位を一級アミンの付着のための部位として用いて、内部の位置に反応部位が導入されうる。このような実施形態においては、核酸がいくつかの活性化または反応性部位を有することができ、多重活性化されていると言われる。合成および精製の後、修飾オリゴヌクレオチドは、自発的加水分解を最小化しながら、オリゴヌクレオチド反応部位との選択反応を促進する条件下で、単活性化PEGと組み合わせられる。好ましい実施形態においては、モノメトキシ―PEGがスクシンイミジルプロピオン酸で活性化され、pH8.3で共役反応が行われる。二置換PEGの合成を誘発するために、オリゴヌクレオチドに対して化学量論的過剰のPEGが提供される。反応の後、完全、部分的、および非結合種を分離するために、PEG―核酸結合体がゲル電気泳動または液体クロマトグラフィにより精製される。
本発明の方法に用いられる変異T7RNAポリメラーゼは、以下のように調製されうる。T7RNAポリメラーゼ(図3Aおよび3Bにそれぞれ示され、Bull,J.J等、J.Mol.Evol.,57(3),241―248(2003)に記載される核酸およびアミノ酸配列を変異させて、LA変異体(Y639L/H784A)、LAR変異体(Y639L/H784A/K378R)、LLA変異体(P266L/Y639L/H784A)またはLLAR変異体P266L/Y639L/H784A/K378R)を生じうる。T7RNAポリメラーゼが、発現ベクター(T7RNAポリメラーゼ発現ベクターの例が、参照により全体として本明細書組み込まれるに米国特許出願第5,869,320号に記載される)に含まれ、または変異誘発後に発現ベクターに挿入されうる。変異T7RNAポリメラーゼは、タンパク質精製の間の容易性のためにHis―tagを選択的に含むために、操作されうる。
変異T7ポリメラーゼ核酸配列を含む発現ベクターは、BL21(DE3)コンピテント細胞(Stratagene、カリフォルニア州)に形質転換され、20分間氷上でインキュベートされる。チューブを2分間42℃に入れることにより、熱ショックが行われる。チューブを1分間氷に置いた後、1mlのLブロス(「LB」)が加えられ、37℃のシェーカで45分間インキュベートされる。100ulの培養液が、LB+Amp寒天プレートにプレートされ、一晩37℃でインキュベートされる。
実施例2A:2’―OH GTPを伴わない2’―O―メチル転写
2’―OH GTPの滴定を用いて、2’―OH GTPの濃度に対するY639L/H784A/K378R変異ポリメラーゼの感受性をテストするために、実験が行われた。
Y639L/H784A/K378R変異T7RNAポリメラーゼを用い、2’―OH GTPを用いないMNA転写の忠実度およびバイアスを評価するために、追加的な実験が行われた。忠実度をテストするために、単一のクローン配列がPCRにより増幅され、Y639L/H784A/K378Rポリメラーゼを用いた2’―OH GTPを伴わないMNA転写をプログラムするために使用され、PAGEにより精製され、RQl DNアーゼを用いて残りのDNAテンプレートが消化され(そしてDNAテンプレートの非存在がPCRによりアッセイされ)、転写物が逆転写され(Thermoscript、Invitrogen、カリフォルニア州カールズバッド)、PCRにより増幅された。このPCR産物が配列決定された後、欠失、挿入および置換の数が計算された。この実験で配列決定された1300の塩基のうちに、欠失および挿入は観察されず、三つの置換が観察された(図7を参照)。これらの数は、30―ヌクレオチド縮重領域内にコードされる配列情報がSELEX(商標)の次のラウンドに忠実に伝達されるチャンスが、93%であることを示唆し、この数は非常に高いため、野生型RNAにつき測定される数を上回る。
二本鎖転写テンプレートを生成するために、ポリメラーゼ連鎖反応をプログラムするために以下のDNAテンプレートおよびプライマが用いられた。Nは、ATGCの各々である確率がほぼ等しい縮重位置を示し、全ての配列が3’から5’方向にリストされる:PCRテンプレート(ARC2118)
in vitro転写を用いて2’―O―メチルヌクレオチドおよび2’―デオキシヌクレオチドの両者を導入するY639L/H784A/K378R変異ポリメラーゼの能力をテストするために実験が行われた。
2’―OMeプリンおよびピリミジンヌクレオチド(以下に「MNA」)からなるプールを用いて、ヒトAng2(以下に「h―Ang2」)に対するアプタマーを同定するために、選択が実行された。選択戦略は、h―Ang2に特異的な高親和性アプタマーをもたらした。
プール調製
配列
100μLの最終的体積の選択バッファー(1X DulbeccoのPBS(DPBS))において100pmolのタンパク質と330pmol(2×1014分子)のMNA ARC2118プールを1時間室温でインキュベートすることにより、選択が開始された。RNA―タンパク質複合体および未結合RNA分子が、0.45ミクロンニトロセルローススピンカラム(Schleicher and Schuell、ニューハンプシャー州キーン)を用いて分離された。カラムはKOHで前処置され(1mL 0.5M KOH中にカラムフィルタ浸漬、15分RT;スピン。1mL dH2O中にフィルタ浸漬 5分RT;スピン)、1mL 1X PBSで二回洗浄されてから、プール:Ang2複合体を含む溶液がカラムに加えられ、2分間1500xgで遠心分離された。フィルタは、非特異的結合物を除去するために、500μL DPBSにより二回洗浄された。2×100μL溶離バッファー(7M尿素、100mM酢酸ナトリウム、3mMEDTA、95℃に予熱)の添加によりRNAが溶出されてから、エタノールにより沈殿させられた。プライマ配列番号27を用いて、製造者の指示に従って、ThermoScript RT―PCR(商標)システム(Invitrogen、カリフォルニア州カールズバッド)により、RNAが逆転写された。配列番号26および配列番号27を用いて、製造者の指示に従って、Taqポリメラーゼ(New England Biolabs、マサチューセッツ州ベヴァリー)によるPCRにより、cDNAが増幅された。プール調製のために上述のようにテンプレートが転写され、変性ポリアクリルアミドゲルで精製された。
プールのタンパク質結合親和性をモニタするために、選択の全体にわたりドットブロット結合アッセイが行われた。トレース32P―エンド標識したプールRNAが、h―Ang2と組み合わされ、DPBSバッファー中で30μLの最終的体積で、30分間室温でインキュベートされた。混合物がドットブロット装置に適用され(Minifold―1Dot Blot、Acrylic、Schleicher and Schuell、ニューハンプシャー州キーン)、(上から下へ)ニトロセルロース、ナイロン、およびゲルブロット膜とアセンブルされた。タンパク質に結合されたRNAはニトロセルロースフィルタに捕らえられ;非タンパク結合RNAはナイロンフィルタに捕らえられる。ラウンド9でh―Ang2結合の濃縮の開始が見られた。ラウンド9、10および12のプールテンプレートが、TOPO TAクローニングキット(Invitrogen、カリフォルニア州カールズバッド)を使用して製造者の指示に従ってクローニングされ、化学合成のために26のユニーククローンが選択され、解離定数(KD)が決定された。簡単にいうと、合成RNAsがγ―32P ATPで5’末端標識され、ドットブロットアッセイおよび1X DPBS(w/Ca2+およびMg2+)(Gibco、Catalog#14040、Invitrogen、カリフォルニア州カールズバッド)のバッファー条件を用いてKD値が決定された。式:RNA結合画分=振幅*(((AptConc+[h―Ang2]+KD)−SQRT((AptConc+[h―Ang2]+KD)2−4(AptConc*[h―Ang2])))/(2*AptConc))+バックグラウンドにデータをあてはめてKDsが推定された。結果は、下表5にレポートされる。
Elisaアッセイ
いくつかアプタマーが、Tie2レセプターに対するAng2結合を妨げる能力を測定するために準備されたELISAアッセイにおいてテストされた。Tie2レセプターを捕捉するために、100μLのPBS(pH7.4)中の150ngのTie2―Fc(R&D systems 313―TI―100―CF、ミネソタ州ミネアポリス)が、96ウェルMaxisorbプレート(NUNC#446612、ニューヨーク州ロチェスター)に置かれ、4℃で一晩インキュベートされた。捕捉の間、様々な濃度の合成RNA50μLが、50μLの3.6nM Ang2(200ng/mL)(R&D systems、623―AN―025/CF、ミネソタ州ミネアポリス)(0.2% BSAを伴うPBS中)と、0.1% BSAを伴うPBS中1.8nM(100ng/mL)の最終Ang2濃度で混合され、1時間室温でインキュベートされた。一晩のインキュベーション後に捕捉溶液が除去され、200μLのTBST(25mM Tris―HCl pH7.5、150mM NaClおよび0.01% Tween20)によりプレートが三回洗浄された。そして、プレートが、5%脱脂粉ミルクを含む200μL TBSTにより室温で30分間ブロックされた。ブロッキングの後、プレートが200μLのTBSTで再び室温で三回洗浄され、合成RNA:Ang2混合物がプレートに加えられ、1時間室温でインキュベートされた。そして、プレートが200μLのTBSTで三回洗浄され、100μLのビオチン化ヤギ抗Ang2抗体(1:1000;R&D Systems BAF623、ミネソタ州ミネアポリス)が加えられ、室温で1時間インキュベートされた。200μLのTBSTにより三回洗浄したあと、100μLのHRP連結ストレプトアビジン(1:200;R&D systems#DY998、ミネソタ州ミネアポリス)が加えられ、0.5時間室温でインキュベートされた。そして、プレートが再び200μLのTBSTにより三回洗浄され、100μLのTMP溶液(Pierce、#34028)が加えられ、暗所で、室温で5分間インキュベートされた。2N H2SO4を含む100μLの溶液が加えられて反応が停止され、プレートがSpectroMaxで、450nmで読み取られた。結果は、下表5の最終的欄に与えられる。
ヒト臍静脈内皮細胞(「HUVEC」)(ATCC)およびK293細胞、ヒトTie2レセプターを過剰発現する細胞系統を用いて、細胞膜上のTie2レセプターに対するAng2の結合を阻害する特異的MNA Ang2アプタマーのIC50が決定された。簡単にいうと、組換え哺乳類発現ベクターpCDNA3.1―Tie2が293細胞(ATCC、ヴァージニア州マナサス)にトランスフェクションされ、G418(Invitrogen、カリフォルニア州カールズバッド)による選択の後、安定したクローンが得られた。フローサイトメトリは、HUVECおよびK293細胞の両方のTie2タンパク質の発現を示した。Ang2滴定アッセイにより、HUVECおよびK293細胞に対する細胞アプタマー阻害アッセイのAng2(R&D Systems、ミネソタ州ミネアポリス)の量がさらに決定され、それぞれ1および0.1μg/mLであった。
2’―OMeプリンおよびピリミジンヌクレオチドからなるプール(以下に「mRmY」)を用いて、ヒトIgE(以下に「h―IgE」)に対するアプタマーを同定するために、選択が行われた。選択戦略は、h―IgEに特異的な高親和性アプタマーをもたらした。
プール調製
配列
330pmol(2×1014分子)のMNA ARC2118プールを、BSAブロックされた疎水性プレート(Maxisorpプレート、Nunc、ニューヨーク州ロチェスター)に結合した24pmolのタンパク質とともに100μLの最終体積の選択バッファー(1X DulbeccoのPBS(DPBS))中で、室温で1時間インキュベートすることにより選択が開始されれた。ウェルは、非特異的結合物を除去するために、120μL DPBSにより四回洗浄された。RNAが溶出され、プライマ配列番号27を用いて、ThermoScript RT―PCR(商標)システム(Invitrogen、カリフォルニア州カールズバッド)により、製造者の指示に従って逆転写された。配列番号26および配列番号27を用いて、製造者の指示に従って、Taqポリメラーゼ(New England Biolabs、マサチューセッツ州ベヴァリー)を使用したPCRによりcDNAが増幅された。プール調製で上述したようにテンプレートが転写され、変性ポリアクリルアミドゲルで精製された。
プールのタンパク質結合親和性をモニタするために、選択の全体にわたりドットブロット結合アッセイが行われた。トレース32P―エンド標識したプールRNAが、h―IgEと組み合わされ、DPBSバッファー中で30μLの最終体積で、30分間室温でインキュベートされた。混合物がドットブロット装置に適用され(Minifold―1 Dot Blot、Acrylic、Schleicher and Schuell、ニューハンプシャー州キーン)、(上から下へ)ニトロセルロース、ナイロン、およびゲルブロット膜とアセンブルされた。タンパク質に結合されたRNAは、ニトロセルロースフィルタに捕らえられ;非タンパク結合RNAは、ナイロンフィルタに捕らえられる。ラウンド8でh―IgE結合の濃縮の開始が見られた。ラウンド5、8および12のプールテンプレートが、TOPO TAクローニングキット(Invitrogen、カリフォルニア州カールズバッド)を使用して製造者の指示に従ってクローニングされた。配列決定データから、ラウンド8のプールが全配列の59%を含む単一の主要クローンに収束したことが明らかになった。この主要クローンと三つの可能性のあるミニマーが化学合成のために選択され、解離定数(KD)が決定された。簡単にいうと、合成RNAsがγ―32P ATPで5’末端標識され、ドットブロットアッセイおよび1X DPBS(w/Ca2+およびMg2+)(Gibco、Catalog#14040、Invitrogen、カリフォルニア州カールズバッド)のバッファー条件を用いてKD値が決定された。式:RNA結合画分=振幅*(((AptConc+[h―IgE]+KD)−SQRT((AptConc+[h―IgE]+KD)2−4(AptConc*[h―IgE])))/(2*AptConc))+バックグラウンドにデータをあてはめてKDsが推定された。主要クローンは、約800pMのKDを有した。最善の結合のミニマーの、サルIgE(m―IgE)に対する結合もテストされたが、サルIgEタンパク質に対する交差反応結合は示されなかった。この交差反応性の欠如は、ELISAによっても確認された。3’末端に逆方向dTを伴うミニマーが、医薬品化学プロセスの親分子として用いられた。
IgE特異的MNAミニマーの一つの化学組成(図9)が、化合物の血漿安定性を維持しながら親和性および能力を改善するために変更された。プロセスは、最小化IgEアプタマーの一連の誘導体の設計、合成および評価を含み、一連の各誘導体は、いずれの残基が置換に耐えるかを決定するため、各所定のヌクレオチドで単一の修飾を含んだ。第一修飾組は、各ユニーク2’―OMeヌクレオチドへのデオキシヌクレオチドの置換であった。別個の修飾ラウンドにおいては、各誘導体が異なるヌクレオチド間結合位置で単一のホスホロチオエート修飾を含む一連の誘導体が合成された。これらの修飾の最初の段階において生成されたデータを用いて、最小化アプタマーの構造活性相関(SAR)が確立された。後の修飾段階においては、最初のSARデータに基づいて設計された複合置換組でアプタマーが合成され、テストされた。複合置換パネルから、2’―OMeから2’―デオキシへの置換がその組成に導入された、39ヌクレオチドの長さのアプタマーが同定された。さらに、2’―OMeから2’―デオキシへの一つの置換およびリン酸塩からホスホロチオエートへの四つの置換がその組成に導入された、39ヌクレオチドの長さの、得られた最小化修飾アプタマーが同定された。図10に示すように、このデオキシ/ホスホロチオエート修飾アプタマーは、最小化非修飾親アプタマーならびに二つの2’―OMeからデオキシへの置換を有する最小化親アプタマーの両方と比較して、結合親和性の増加を示す。
最小化非修飾親アプタマーおよびデオキシ/ホスホロチオエート修飾アプタマーが、ヒト、ラットおよびサル血清における安定性を決定するためにアッセイされた。各アプタマーが、90%血清中5μMの最終濃度になるように、1mlのプール血清に加えられた。アプタマーは振盪を伴って37℃でインキュベートされ、0、0.5、1、4、24、48、72、および98時間でタイムポイントがとられた。各タイムポイントで、インキュベート試料からの90μlのストックが、10μlの0.5M EDTAに加えられ、BIACORE 2000システムを用いた後の安定性分析のために−20℃で冷凍された。
2’―OMe ATP、GTP、およびUTPヌクレオチド、および2’―デオキシCTPヌクレオチド(以下に「dCmD」)からなるプールを用いて、ヒトフォンビルブラント因子(以下に「h―vWF」)に対するアプタマーを同定するために、選択が行われた。選択戦略から、h―vWFに特異的な高親和性アプタマーが生じた。
プール調製
配列
100μLの最終体積の選択バッファー(1X DulbeccoのPBS(DPBS)において、HSAブロックされた疎水性プレート(Maxisorpプレート、Nunc、ニューヨーク州ロチェスター)に結合された20pmolのタンパク質とともに330pmol(2×1014分子)のdCmD ARC4218プールを1時間室温でインキュベートすることにより、選択が開始された。非特異的結合物を除去するために、200μL DPBSによりウェルが五回洗浄された。RNAが溶出され、プライマARC4208(配列番号30)を用いて、製造者の指示に従ってThermoScript RT―PCR(商標)システム(Invitrogen、カリフォルニア州カールズバッド)により逆転写された。プライマARC4219(配列番号29)およびARC4208(配列番号30)を用いて、製造者の指示に従って、Taqポリメラーゼ(New England Biolabs、マサチューセッツ州ベヴァリー)によるPCRによりcDNAが増幅された。プール調製につき上述したようにテンプレートが転写された。エタノール沈殿によりテンプレートが精製され、その後Micro Bio―spinカラム(Cat.#732―6250、Bio−Rad Laboratories、カリフォルニア州ハーキュリーズ)を用いた脱塩ステップが行われた。
プールのタンパク質結合親和性をモニタするために、選択の全体にわたりドットブロット結合アッセイが行われた。トレース32P―エンド標識したプールRNAが、h―vWFと組み合わされ、DPBSバッファー中で30μLの最終体積で、室温で30分間インキュベートされた。混合物がドットブロット装置に適用され(Minifold―1 Dot Blot、Acrylic、Schleicher and Schuell、ニューハンプシャー州キーン)、(上から下へ)ニトロセルロース、ナイロン、およびゲルブロット膜とアセンブルされた。タンパク質に結合されたRNAは、ニトロセルロースフィルタに捕らえられ;非タンパク結合RNAは、ナイロンフィルタに捕らえられる。ラウンド5でh―vWF結合の濃縮の開始が見られた。ラウンド5およびラウンド6プールの解離定数(KD)が決定された。簡単にいうと、プールRNAsがγ―32P ATPで5’末端標識され、ドットブロットアッセイおよび1X DPBS(w/Ca2+およびMg2+)(Gibco、Catalog#14040、Invitrogen、カリフォルニア州カールズバッド)のバッファー条件を用いてKD値が決定された。式:RNA結合画分=振幅*(((AptConc+[h―vWF]+KD)−SQRT((AptConc+[h―vWF]+KD)2−4(AptConc*[h―vWF])))/(2*AptConc))+バックグラウンドにデータをあてはめてKDsが推定された。ラウンド6プールは、約2nMのKDを有した。プールは、Alドメインに対する結合についてもテストされ、約4nMのKDを有した。
2’―OMe ATP、GTP、およびUTPヌクレオチド、および2’―デオキシCTPヌクレオチド(以下に「dCmD」)からなるプールを用いて、ヒトIgE(以下に「h―IgE」)に対するアプタマーを同定するために選択が行われた。選択戦略から、h―IgEに特異的な高親和性アプタマーが生じた。
プール調製
配列
330pmol(2×1014分子)のdCmD ARC4218プールを、HSAブロックされた疎水性プレート(Maxisorpプレート、Nunc、ニューヨーク州ロチェスター)に結合した20pmolのタンパク質とともに100μLの最終体積の選択バッファー(1X DulbeccoのPBS(DPBS))中で、室温で1時間インキュベートすることにより選択が開始されれた。ウェルは、非特異的結合物を除去するために、200μL DPBSにより五回洗浄された。RNAが溶出され、プライマARC4208(配列番号30)を用いて、ThermoScript RT―PCR(商標)システム(Invitrogen、カリフォルニア州カールズバッド)により、製造者の指示に従って逆転写された。プライマARC4219(配列番号29)およびARC4208(配列番号30)を用いて、製造者の指示に従って、Taqポリメラーゼ(New England Biolabs、マサチューセッツ州ベヴァリー)を用いたPCRにより、cDNAが増幅された。プール調製につき上述したように、テンプレートが転写された。エタノール沈殿によりテンプレートが精製された後、Micro Bio―spinカラム(Cat.#732―6250、Bio―Rad Laboratories、カリフォルニア州ハーキュリーズ)を用いて脱塩ステップが行われた。
プールのタンパク質結合親和性をモニタするために、選択の全体にわたりドットブロット結合アッセイが行われた。トレース32P―エンド標識したプールRNAが、h―IgEと組み合わされ、DPBSバッファー中で30μLの最終体積で、室温で30分間インキュベートされた。混合物がドットブロット装置に適用され(Minifold―1 Dot Blot、Acrylic、Schleicher and Schuell、ニューハンプシャー州キーン)、(上から下へ)ニトロセルロース、ナイロン、およびゲルブロット膜とアセンブルされた。タンパク質に結合されたRNAはニトロセルロースフィルタに捕らえられ;非タンパク結合RNAはナイロンフィルタに捕らえられる。ラウンド5でh―IgE結合の濃縮の開始が見られた。ラウンド5および6プールの解離定数(KD)が決定された。簡単にいうと、プールRNAsがγ―32P ATPで5’末端標識され、ドットブロットアッセイおよび1X DPBS(w/Ca2+およびMg2+)(Gibco、Catalog#14040、Invitrogen、カリフォルニア州カールズバッド)のバッファー条件を用いてKD値が決定された。式:RNA結合画分=振幅*(((AptConc+[h―IgE]+KD)−SQRT((AptConc+[h―IgE]+KD)2―4(AptConc*[h―IgE])))/(2*AptConc))+バックグラウンドにデータをあてはめてKDsが推定された。ラウンド6プールは、約80nMのKDを有した。
Y639L/H784A/K378R変異ポリメラーゼが、in vitro転写を用いて2’―O―メチルA、C、Gおよび2’―O―メチル―5インドリルメチレンUを転写産物に導入するために用いられる。修飾ウリジンが、以下にヌクレオシドとして示される:
治療目的のタンパク質標的に対するSELEXが、複数の縮重位置および上の実施例7に記載のMNA―I転写条件を含む転写テンプレートを用いて実行される。
200nMの40ヌクレオチドの内側の連続縮重領域を伴う二本鎖転写テンプレート、200mM HEPES、40mM DTT、2mMスペルミジン、0.01%TritonX―100、各1.5mMの2’―OMe TTP、2’―OMe ATP、2’―OMe CTP、2’―OMe GTP、8mM MgCl2、2.5mM MnCl2、1mM GMP、10%PEG 8000、2.5単位/ml無機ピロホスファターゼ、pH7.5およびK378R/Y639F/H784A変異T7ポリメラーゼにより、400ul転写が準備された。溶液が37℃で一晩インキュベートされ、沈殿され、変性ローディングバッファーに溶解され、変性PAGEを用いて分析された後、UVシャドウイングを用いて蛍光板上で可視化された。隣接するウェルにロードされた対応するMNA転写産物と共移動したバンドが観察された。2’―OMe TTPを伴わずにインキュベートされた陰性対照転写物は、観察可能なバンドを生成しなかった。
200nMの二本鎖転写テンプレートARC3205、200mM HEPES、40mM DTT、2mMスペルミジン、0.01%TritonX―100、各1.5mMの2’―デオキシTTP、2’―OMe ATP、2’―OMe CTP、2’―OMe GTP、8mM MgCl2、2.5mM MnCl2、1mM GMP、10%PEG8000、2.5単位/ml無機ピロホスファターゼ、pH7.5およびK378R/Y639F/H784A変異T7ポリメラーゼにより、400ul転写が準備された。溶液が37℃で一晩インキュベートされ、沈殿され、変性ローディングバッファーに溶解され、変性PAGEを用いて分析された後、UVシャドウイングを用いて蛍光板上で可視化された。隣接するウェルにロードされた対応するMNA転写産物と共移動したバンドが観察された。2’―デオキシTTPを伴わずにインキュベートされた陰性対照転写物は、観察可能なバンドを生成しなかった。
200nMの40ヌクレオチドの内側の連続縮重領域を伴う二本鎖転写テンプレート、200mM HEPES、40mM DTT、2mMスペルミジン、0.01%TritonX―100、各1.5mMの2’―OH TTP、2’―OMe ATP、2’―OMe CTP、2’―OMe GTP、8mM MgCl2、2.5mM MnCl2、1mM GMP、10%PEG8000、無機ピロホスファターゼ2.5単位/ml、pH7.5およびK378R/Y639F/H784A変異T7ポリメラーゼにより、400ul転写が準備された。溶液が37℃で一晩インキュベートされ、沈殿され、変性ローディングバッファーに溶解され、変性PAGEを用いて分析された後、UVシャドウイングを用いて蛍光板上で可視化された。隣接するウェルにロードされた対応するMNA転写産物と共移動したバンドが観察された。2’―OH TTPを伴わずにインキュベートされた陰性対照転写物は、観察可能なバンドを生成しなかった。
200nMの二本鎖転写テンプレートARC3205、200mM HEPES、40mM DTT、2mMスペルミジン、0.01%TritonX―100、各1.5mMの2’―デオキシUTP、2’―OMe ATP、2’―OMe CTP、2’―OMe GTP、8mM MgCl2、2.5mM MnCl2、1mM GMP、10%PEG8000、無機ピロホスファターゼ2.5単位/ml、pH7.5およびK378R/Y639F/H784A変異T7ポリメラーゼにより、400ul転写が準備された。溶液が37℃で一晩インキュベートされ、沈殿され、変性ローディングバッファーに溶解され、変性PAGEを用いて分析された後、UVシャドウイングを用いて蛍光板上で可視化された。隣接するウェルにロードされた対応するMNA転写産物と共移動したバンドが観察された。2’―デオキシUTPを伴わずにインキュベートされた陰性対照転写物は、観察可能なバンドを生成しなかった。
200nMの二本鎖転写テンプレートARC3205、200mM HEPES、40mM DTT、2mMスペルミジン、0.01%TritonX―100、各1.5mMの2’―OH UTP、2’―OMe ATP、2’―OMe CTP、2’―OMe GTP、8mM MgCl2、2.5mM MnCl2、1mM GMP、10%PEG8000、無機ピロホスファターゼ2.5単位/ml、pH7.5およびK378R/Y639F/H784A変異T7ポリメラーゼにより、400ul転写が準備された。溶液が37℃で一晩インキュベートされ、沈殿され、変性ローディングバッファーに溶解され、変性PAGEを用いて分析された後、UVシャドウイングを用いて蛍光板上で可視化された。隣接するウェルにロードされた対応するMNA転写産物と共移動したバンドが観察された。2’―OH UTPを伴わずにインキュベートされた陰性対照転写物は、観察可能なバンドを生成しなかった。
200nMの二本鎖転写テンプレートARC3205、200mM HEPES、40mM DTT、2mMスペルミジン、0.01%TritonX―100、各1.5mMの2’―デオキシTTP、2’―OMe ATP、2’―OMe CTP、2’―OH GTP、8mM MgCl2、2.5mM MnCl2、1mM GMP、10%PEG8000、無機ピロホスファターゼ2.5単位/ml、pH7.5およびK378R/Y639F/H784A変異T7ポリメラーゼにより、400ul転写が準備された。溶液が37℃で一晩インキュベートされ、沈殿され、変性ローディングバッファーに溶解され、変性PAGEを用いて分析された後、UVシャドウイングを用いて蛍光板上で可視化された。隣接するウェルにロードされた対応するMNA転写産物と共移動したバンドが観察された。2’―デオキシTTPを伴わずにインキュベートされた陰性対照転写は、観察可能なバンドを生成しなかった。
200nMの40ヌクレオチドの内側の連続縮重領域を伴う二本鎖転写テンプレート、200mM HEPES、40mM DTT、2mMスペルミジン、0.01%TritonX―100、各1.5mMの2’―OMe QTP、2’―OMe ATP、2’―OMe CTP、2’―OH GTP、8mM MgCl2、2.5mM MnCl2、1mM GMP、10%PEG8000、無機ピロホスファターゼ2.5単位/ml、pH7.5およびK378R/Y639F/H784A変異T7ポリメラーゼにより、400ul転写が準備された。2’―OMe QTP(mQ)が下に示される:
50nMの30ヌクレオチドの内側の連続縮重領域を伴う二本鎖転写テンプレート、200mM HEPES、40mM DTT、2mMスペルミジン、0.01%TritonX―100、各1.5mMの2’―OMe UTP、2’―OMe ATP、2’―OMe CTPおよび2’―OMe GTP、または各1.5mMの2’―OH UTP、2’―OH ATP、2’―OH CTPおよび2’―OH GTP、8mM MgCl2、2.5mM MnCl2、1mM GMP、10%PEG8000、無機ピロホスファターゼ2.5単位/ml、pH7.5および以下に示される変異または野生型T7ポリメラーゼ(WTがK378R、FがK378R/Y639F、LがK378R/Y639L、FAがK378R/Y639F/H784A、LAがK378R/Y639L/H784A)により、400ulの転写が準備された。溶液が37℃で一晩インキュベートされ、沈殿され、変性ローディングバッファーに溶解され、変性PAGEを用いて分析された後、UVシャドウイングを用いて蛍光板上で可視化された。図11に示されるように、異なる転写の収率が観察された。
Claims (16)
- 核酸を転写する方法であり、
a)転写反応混合物を調製するステップであって、前記転写反応混合物は、
(i)2’―OMe ATP、2’―OMe GTP、2’―OMe CTP、2’―OMe TTPおよび2’―OMe UTPより選択される2’―OMe修飾ヌクレオチド三リン酸(2’―OMe NTP)のいずれかを導入することが可能な修飾T7 RNAポリメラーゼであり、前記修飾T7 RNAポリメラーゼが、対応する非修飾RNAポリメラーゼの2’―修飾ヌクレオチド三リン酸(2’―修飾NTP)を導入する能力と比較して、前記2’―修飾ヌクレオチド三リン酸(2’―修飾NTP)を導入する能力が増加しており、前記修飾T7 RNAポリメラーゼは、野生型T7 RNAポリメラーゼに対してアミノ酸639位および784位において変異を含み、前記639位におけるアミノ酸はロイシンに変化し、前記784位におけるアミノ酸はアラニンに変化している、修飾T7 RNAポリメラーゼと、
(ii)少なくとも一つの核酸転写テンプレートと、
(iii)ヌクレオチド三リン酸の混合物であり、前記ヌクレオチド三リン酸がグアニジン三リン酸を含み、前記グアニジン三リン酸が全て2’―OMe修飾されており、前記ヌクレオシド三リン酸の混合物は、2’−デオキシシチジン三リン酸(2’−デオキシCTP)、2’−OMeアデノシン三リン酸(2’−OMe ATP)、2’−OMeグアノシン三リン酸GTP(2’−OMe GTP)、および2’−OMeウリジン三リン酸(2’−OMe UTP)または2’−OMeチミジンヌクレオチド三リン酸(2’−OMe TTP)の混合物である、ヌクレオチド三リン酸の混合物と
を含む、転写反応混合物を調製するステップと;
b)一本鎖核酸が生じる条件下で、前記転写反応混合物を転写するステップであり、前記生じる一本鎖核酸の、最初のグアノシンヌクレオチドの後の全てのグアニジンヌクレオチドが、2’―OMe修飾されている、ステップと
を含む、方法。 - 前記転写反応混合物が、側鎖で修飾された核酸塩基をさらに含む、請求項1に記載の方法。
- 前記核酸塩基が、ウリジンの5―および6―位、チミジンの5―および6―位、シチジンの5および6―位および環外アミン、アデノシンの2―、7―および8―位および環外アミン、グアノシンの7―および8―位および環外アミンからなる群より選択される位置で修飾される、請求項2に記載の方法。
- 前記側鎖が、疎水性芳香族側鎖である、請求項2に記載の方法。
- 前記側鎖が、ベンジルまたはインドリルである、請求項4に記載の方法。
- 前記転写反応混合物が、マグネシウムイオン、マンガンイオン、マグネシウムおよびマンガンイオンの両方、GMP、ポリエチレングリコール、または無機ピロホスファターゼの1または複数をさらに含む、請求項1に記載の方法。
- 前記核酸転写テンプレートが、少なくとも部分的に二本鎖である、請求項1に記載の方法。
- 前記少なくとも部分的に二本鎖の核酸転写テンプレートが、前記核酸転写テンプレートの5’末端の固定領域に導入されたリーダー配列をさらに含む、請求項7に記載の方法。
- アプタマーを同定する方法であり、前記アプタマーが2’―OMe修飾ヌクレオチドを含む、方法であり、
a)転写反応混合物を調製するステップであって、前記転写反応混合物は、
(i)2’―OMe ATP、2’―OMe GTP、2’―OMe CTP、2’―OMe TTPおよび2’―OMe UTPより選択される2’―OMe修飾ヌクレオチド三リン酸(2’―OMe NTP)のいずれかを導入することが可能な修飾T7 RNAポリメラーゼであり、前記修飾T7 RNAポリメラーゼが、対応する非修飾RNAポリメラーゼの2’―修飾ヌクレオチド三リン酸(2’―修飾NTP)を導入する能力と比較して、前記2’―修飾ヌクレオチド三リン酸(2’―修飾NTP)を導入する能力が増加しており、前記T7 RNAポリメラーゼは、野生型T7 RNAポリメラーゼに対してアミノ酸639位および784位において変異を含み、前記639位におけるアミノ酸はロイシンに変化し、前記784位におけるアミノ酸はアラニンに変化している、修飾T7RNAポリメラーゼと、
(ii)少なくとも一つの核酸転写テンプレートと、
(iii)ヌクレオチド三リン酸の混合物であり、前記ヌクレオチド三リン酸がグアニジン三リン酸を含み、前記グアニジン三リン酸が全て2’―OMe修飾されており、前記ヌクレオシド三リン酸の混合物は、2’−デオキシシチジン三リン酸(2’−デオキシCTP)、2’−OMeアデノシン三リン酸(2’−OMe ATP)、2’−OMeグアノシン三リン酸GTP(2’−OMe GTP)、および2’−OMeウリジン三リン酸(2’−OMe UTP)または2’−OMeチミジンヌクレオチド三リン酸(2’−OMe TTP)の混合物である、ヌクレオチド三リン酸の混合物と
を含む、転写反応混合物を調製するステップと;
b)前記修飾T7RNAポリメラーゼにより、一本鎖核酸の候補混合物の前記核酸分子に、少なくとも一つの修飾2’―OMeヌクレオチド三リン酸(2’―OMe NTP)が導入される条件下で、前記転写反応混合物を転写することにより、前記一本鎖核酸の候補混合物を調製するステップであり、前記候補混合物の前記一本鎖核酸分子の、最初のグアノシンヌクレオチドの後の全てのグアニジンヌクレオチドが、2’―OMe修飾されている、ステップと;
c)前記候補混合物を前記標的分子と接触させるステップと、
d)前記候補混合物の親和性と比較して、標的分子に対する親和性が増加した前記核酸を、前記候補混合物から分離するステップと、
e)アプタマーが濃縮された混合物を生じるために、前記親和性が増加した核酸を増幅するステップを含み、それによって、アプタマーの前記グアニジンヌクレオチドの一つを選択的に除いて、全て2’―OMe修飾されたグアニジンヌクレオチドを含む、前記標的分子に対する前記アプタマーが、同定される、
方法。 - 前記転写反応混合物が、側鎖で修飾された核酸塩基をさらに含む、請求項9に記載の方法。
- 前記核酸塩基が、ウリジンの5―および6―位、チミジンの5―および6―位、シチジンの5および6―位および環外アミン、アデノシンの2―、7―および8―位および環外アミン、グアノシンの7―および8―位および環外アミンからなる群より選択される位置で修飾される、請求項10に記載の方法。
- 前記側鎖が、疎水性芳香族側鎖である、請求項10に記載の方法。
- 前記側鎖が、ベンジルまたはインドリルである、請求項12に記載の方法。
- 前記転写反応混合物が、マグネシウムイオン、マンガンイオン、マグネシウムおよびマンガンイオンの両方、GMP、ポリエチレングリコール、または無機ピロホスファターゼの1または複数をさらに含む、請求項9に記載の方法。
- 前記核酸転写テンプレートが、少なくとも部分的に二本鎖である、請求項9に記載の方法。
- 前記少なくとも部分的に二本鎖の核酸転写テンプレートが、前記核酸転写テンプレートの5’末端の固定領域に導入されたリーダー配列をさらに含む、請求項15に記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US87983007P | 2007-01-10 | 2007-01-10 | |
| US60/879,830 | 2007-01-10 | ||
| PCT/IB2007/004146 WO2008078180A2 (en) | 2006-12-22 | 2007-12-28 | Materials and methods for the generation of transcripts comprising modified nucleotides |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2011504357A JP2011504357A (ja) | 2011-02-10 |
| JP5349323B2 true JP5349323B2 (ja) | 2013-11-20 |
Family
ID=43638583
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009542267A Active JP5349323B2 (ja) | 2007-01-10 | 2007-12-28 | 修飾ヌクレオチドを含む転写物を生成させるための材料および方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5349323B2 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7016511B2 (ja) * | 2017-06-20 | 2022-02-07 | 学校法人甲南学園 | 核酸合成法 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2506748A1 (en) * | 2002-12-03 | 2004-06-17 | Archemix Corporation | Method for in vitro selection of 2'-substituted nucleic acids |
| US20050037394A1 (en) * | 2002-12-03 | 2005-02-17 | Keefe Anthony D. | Method for in vitro selection of 2'-substituted nucleic acids |
| AU2005287273B2 (en) * | 2004-09-07 | 2011-05-12 | Archemix Corp. | Aptamers to von Willebrand Factor and their use as thrombotic disease therapeutics |
| EP1907590B1 (en) * | 2005-06-30 | 2012-09-19 | Archemix LLC | T7 rna polymerase and methods for the generation of fully 2'-modified nucleic acid transcripts |
-
2007
- 2007-12-28 JP JP2009542267A patent/JP5349323B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP2011504357A (ja) | 2011-02-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8101385B2 (en) | Materials and methods for the generation of transcripts comprising modified nucleotides | |
| EP2207891B1 (en) | Materials and methods for the generation of transcripts comprising modified nucleotides | |
| JP5015923B2 (ja) | 完全な2’改変核酸転写物を生成するための材料および方法 | |
| EP1737879B1 (en) | Aptamer-mediated intracellular delivery of therapeutic oligonucleotides | |
| JP4176466B2 (ja) | 前立腺特異的膜抗原に対する核酸リガンド | |
| US20060030535A1 (en) | Controlled modulation of the pharmacokinetics and biodistribution of aptamer therapeutics | |
| JP2006508688A (ja) | 2’−置換核酸のインビトロ選択のための方法 | |
| JP2007527246A (ja) | ヒトil−12サイトカインファミリーに対するアプタマーおよび自己免疫疾患の治療剤としてのそれらの使用 | |
| US20160266133A1 (en) | Nucleic acid-scaffolded small molecule libraries | |
| US20080214489A1 (en) | Aptamer-mediated intracellular delivery of oligonucleotides | |
| US20090081679A1 (en) | Compositions and methods for in vivo SELEX | |
| US7579450B2 (en) | Nucleic acid ligands specific to immunoglobulin E and their use as atopic disease therapeutics | |
| WO2005052121A2 (en) | Multivalent aptamers | |
| JP5349323B2 (ja) | 修飾ヌクレオチドを含む転写物を生成させるための材料および方法 | |
| WO2009126632A1 (en) | Compositions and methods for the use of mutant t3 rna polymerases in the synthesis of modified nucleic acid transcripts | |
| KR20070044813A (ko) | 이뮤노글로불린 e에 대한 핵산 리간드 및 아토피성 질환치료제로서의 이의 용도 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20101221 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20111130 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20121228 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130327 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130403 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130426 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130508 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130527 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130603 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130604 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20130725 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20130820 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5349323 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |