JP4958905B2 - ペプチド鎖発現を増大させるための物質および方法 - Google Patents
ペプチド鎖発現を増大させるための物質および方法 Download PDFInfo
- Publication number
- JP4958905B2 JP4958905B2 JP2008526258A JP2008526258A JP4958905B2 JP 4958905 B2 JP4958905 B2 JP 4958905B2 JP 2008526258 A JP2008526258 A JP 2008526258A JP 2008526258 A JP2008526258 A JP 2008526258A JP 4958905 B2 JP4958905 B2 JP 4958905B2
- Authority
- JP
- Japan
- Prior art keywords
- dna
- seq
- cells
- expression
- peptide chain
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/24—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against cytokines, lymphokines or interferons
- C07K16/244—Interleukins [IL]
- C07K16/248—IL-6
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2812—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against CD4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/36—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against blood coagulation factors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/67—General methods for enhancing the expression
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Genetics & Genomics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Immunology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Zoology (AREA)
- Biomedical Technology (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Hematology (AREA)
- Microbiology (AREA)
- Plant Pathology (AREA)
- Physics & Mathematics (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Peptides Or Proteins (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Description
Furthら、Nucl.Acids.Res.、19、6205−6208(1991)
本発明の一局面は、配列番号1および配列番号2に示される配列を有する核酸を含んでなる単離されたDNAである。
本明細で引用される、限定されるものでないが特許および特許出願を挙げることができる全部の刊行物は、完全に示されるかのように引用することにより本明細書に組み込まれる。
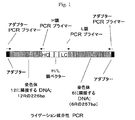
C128D細胞株は、ヒト/マウスキメラ抗体全体の発現を可能にするように2種の組換えDNA構築物で安定にトランスフェクトされているSp2/0ハツカネズミ(Mus musculus)系統BALB/c骨髄腫由来細胞株である。第一の構築物はヒトG1定常領域とともにマウス抗CD4 HC可変領域をコードした。第二の構築物はヒトκ定常領域とともにマウス抗CD4 LC可変領域をコードした。C128D細胞中で、HおよびL鎖ポリペプチドは極めて高レベル(消費済み(spent)振とうフラスコ中158mg/L;灌流バイオリアクター中1g/L/日)で発現された。加えて、最低1コピーのLC遺伝子および最低1コピーのHC遺伝子構築物が、C128D細胞株のゲノムDNA中に相互にすぐ隣接して組込んでいる。図1を参照されたい。
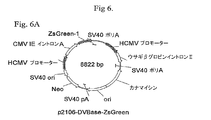
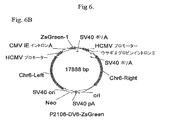
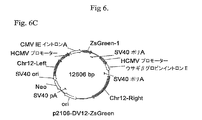
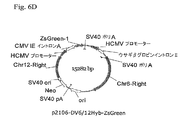
緑色蛍光タンパク質(GFP)レポーター遺伝子に連結された6R、6L、12Rおよび12L DNAを含有するベクター構築物を、組換え遺伝子発現を増大させる要素の同定を容易にするために作成した。全ベクターは標準的分子生物学的技術を使用して構築し、そしてプラスミド地図を全部図6に示す。
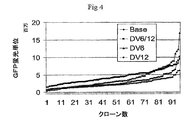
IL−6に対するヒトIgG1K抗体をコードするHCおよびLC遺伝子に連結した6Rおよび12R DNAを含有するベクター構築物を作成した。1構築物はp2106−DVBase−Antibody(DV−BASE−Ab)であった。DV−BASE−Abは、GFPレポーター遺伝子が抗体HC遺伝子で置換されかつLC遺伝子が該ベクターに存在するウサギβ−グロビンイントロンII要素に対し5’に挿入されたことを除き、p2106−DVBASE−ZsGreen(図6に示される)に本質的に同一である。第二の構築物はp2106−DV6/12Hyb−Antibody(DV6/12−Ab)であった。DV6/12−Abは、GFPレポーター遺伝子がHC遺伝子で置換されかつLC遺伝子が該ベクターに存在するウサギβグロビンイントロンII要素に対し5’に挿入されたことを除き、p2106−DV6/12Hyb−ZsGreen(図6に示される)に本質的に同一である。DV6/12−Ab中で、12R DNAはHC遺伝子の5’側に連結され、また、6R DNAはLC遺伝子の3’側に連結される。DV6/12は、C128D細胞中に見出される6Rおよび12R DNA要素の配置を表す。全ベクターは標準的分子生物学技術を使用して構築した。
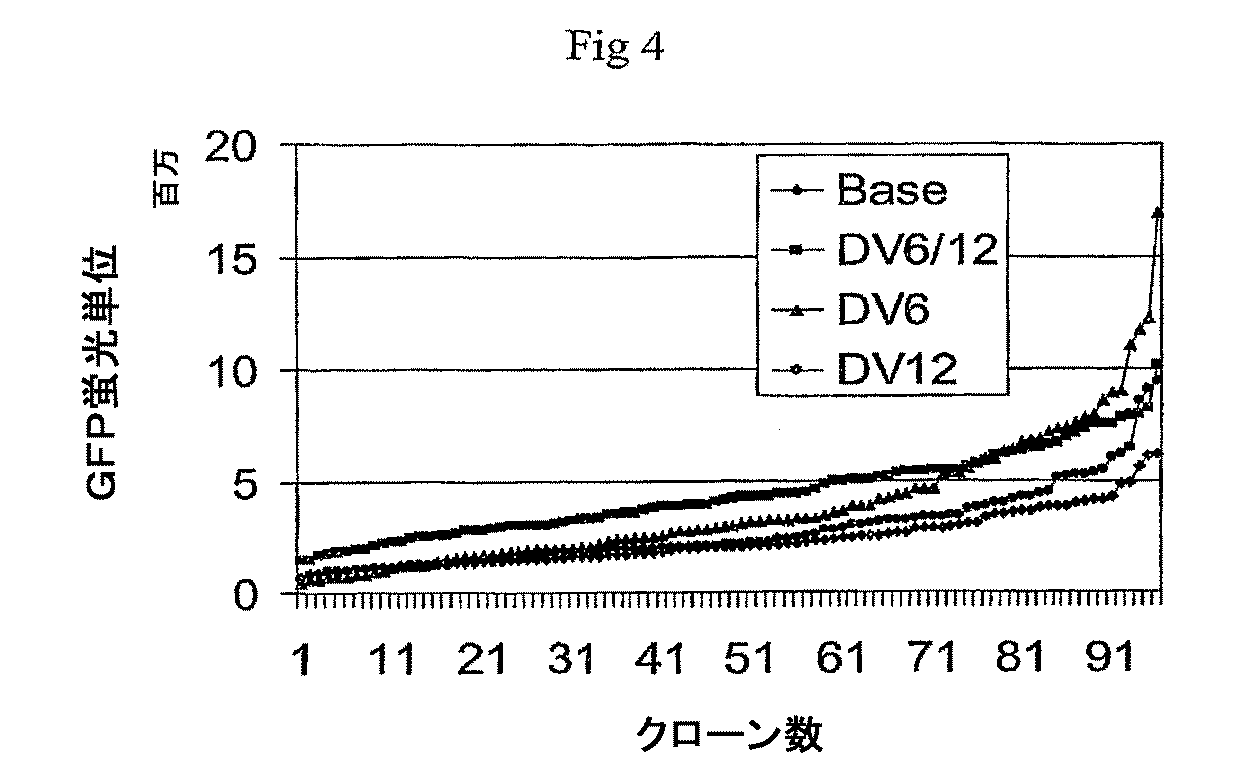
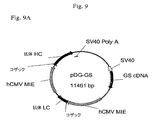
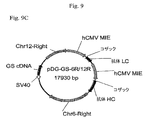
組織因子に対するヒト抗体をコードするHおよびL鎖遺伝子に連結された6Rおよび12R DNAを含有するベクター構築物を、示されるとおり作成した(プラスミド地図を図9に示す)。全ベクターは標準的分子生物学技術を使用して構築した。
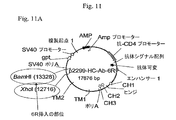
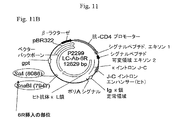
IL−6に対するマウス可変/ヒト定常キメラ抗体をコードするHCおよびLC遺伝子に連結した6R DNAを含有するベクター構築物を、示されるとおり作成した(プラスミド地図を図11に示す)。全ベクターは標準的分子生物学技術を使用して構築した。
図10に示されるデータは、12R DNAおよびそのフラグメントが核マトリックス結合活性を有することを示す。核マトリックス結合活性は、マトリックス付着領域(MAR)を含んでなるDNAと一般に関連する。MARは、転写エンハンサーおよび障壁型インスレーターDNA要素としばしば緊密に関連しかつ頻繁にそれらに隣接する。「障壁型インスレーター」は、隣接する凝縮クロマチンがそれ以外は転写的に活性の遺伝子の遺伝子座に侵食しかつ抑制することを予防する障壁として作用することにより、遺伝子発現を増大させ得る。
組換え遺伝子発現を増大させかつ6Rおよび12R DNA中に存在する中核DNA要素は容易に同定し得る。最初に、6Rおよび12R DNAのフラグメントを生成しかつサブクローニングすることができる。6Rおよび12R DNAフラグメントは制限酵素消化若しくはPCRのような技術を使用して生成し得る。こうしたフラグメントは、いずれかの大きさの5’、3’若しくは内部に配置された欠失をもつ6Rおよび12R DNAを包含しうる。フラグメントは、5’、3’若しくは内的に配置された挿入若しくは置換をもつ6Rおよび12R DNAもまた包含しうる。必要な場合は、当業者に公知の技術を使用して、制限フラグメントのようなフラグメントから5’若しくは3’DNAオーバーハングを排除し得る。
Claims (8)
- 配列番号1および配列番号2に示される配列を有する核酸を含んでなる単離されたDNA。
- 配列番号1に示される配列を有する核酸が、ペプチド鎖をコードする組換えDNAの3’側に作動可能に連結される、請求項1に記載のDNA。
- グルタミン合成酵素をコードするDNAをさらに含んでなる、請求項2に記載のDNA。
- ペプチド鎖をコードする組換えDNAに作動可能に連結される、配列番号1および配列番号2に示される配列を有する核酸を含んでなる単離されたDNA。
- 組換えDNAがcDNAである、請求項4に記載のDNA。
- 配列番号1に示される配列を有する核酸が、組換えDNAの3’側に作動可能に連結され、かつ、配列番号2に示される配列を有する核酸が、組換えDNAの5’側に作動可能に連結される、請求項4に記載のDNA。
- グルタミン合成酵素をコードするDNAをさらに含んでなる、請求項6に記載のDNA。
- ペプチド鎖が抗体フラグメントである、請求項7に記載のDNA。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US70756405P | 2005-08-11 | 2005-08-11 | |
| US60/707,564 | 2005-08-11 | ||
| PCT/US2006/031506 WO2007022009A2 (en) | 2005-08-11 | 2006-08-11 | Materials and methods to increase peptide chain expression |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2009504160A JP2009504160A (ja) | 2009-02-05 |
| JP4958905B2 true JP4958905B2 (ja) | 2012-06-20 |
Family
ID=37758241
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008526258A Expired - Fee Related JP4958905B2 (ja) | 2005-08-11 | 2006-08-11 | ペプチド鎖発現を増大させるための物質および方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US7498150B2 (ja) |
| EP (1) | EP1957660B1 (ja) |
| JP (1) | JP4958905B2 (ja) |
| AT (1) | ATE494386T1 (ja) |
| DE (1) | DE602006019486D1 (ja) |
| WO (1) | WO2007022009A2 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101790812B (zh) | 2007-08-20 | 2013-11-20 | myFC股份公司 | 具有反馈传感器的燃料电池组件 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK1395669T3 (da) | 2001-01-26 | 2009-11-16 | Selexis Sa | Matriks bindingsregioner og fremgangsmåder til anvendelse af disse |
-
2006
- 2006-08-11 JP JP2008526258A patent/JP4958905B2/ja not_active Expired - Fee Related
- 2006-08-11 DE DE602006019486T patent/DE602006019486D1/de active Active
- 2006-08-11 AT AT06789720T patent/ATE494386T1/de not_active IP Right Cessation
- 2006-08-11 EP EP06789720A patent/EP1957660B1/en not_active Not-in-force
- 2006-08-11 WO PCT/US2006/031506 patent/WO2007022009A2/en not_active Ceased
- 2006-08-11 US US11/503,103 patent/US7498150B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| US7498150B2 (en) | 2009-03-03 |
| EP1957660B1 (en) | 2011-01-05 |
| JP2009504160A (ja) | 2009-02-05 |
| EP1957660A4 (en) | 2009-06-17 |
| DE602006019486D1 (de) | 2011-02-17 |
| WO2007022009A2 (en) | 2007-02-22 |
| WO2007022009A3 (en) | 2007-11-22 |
| US20080213829A1 (en) | 2008-09-04 |
| EP1957660A2 (en) | 2008-08-20 |
| ATE494386T1 (de) | 2011-01-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12503520B2 (en) | Compositions and methods for making antibodies based on use of an expression-enhancing locus | |
| KR101183764B1 (ko) | 프로모터 | |
| PT939763E (pt) | Recombinação dirigida de adn medida por troca | |
| US20230062964A1 (en) | Modified mice that produce heavy chain only antibodies | |
| KR20110089846A (ko) | 키메라 항체의 제조를 위한 인간 이외의 포유동물 | |
| CN102165060A (zh) | 新的调控组件 | |
| EP2938728B1 (en) | Artificial introns | |
| US20220220509A1 (en) | Mammalian cell lines with sirt-1 gene knockout | |
| US20200087679A1 (en) | Expression cassette | |
| CN114008212A (zh) | 通过以限定的组织形式靶向整合多个表达盒来产生三价抗体表达细胞的方法 | |
| JP2013509188A (ja) | Sorf構築物および複数の遺伝子発現 | |
| CN114008081A (zh) | 通过以限定的组织形式靶向整合多个表达盒来产生二价双特异性抗体表达细胞的方法 | |
| JP4958905B2 (ja) | ペプチド鎖発現を増大させるための物質および方法 | |
| WO2014102101A1 (en) | Novel intron sequences | |
| EP2938726B1 (en) | Heterologous intron within a signal peptide | |
| CN120322561A (zh) | 新型转座酶系统 | |
| US20240076405A1 (en) | Expression vectors for immunoglobulins and applications thereof | |
| HK40061338A (en) | Mammalian cell lines with sirt-1 gene knockout | |
| HK40060850A (en) | Method for the generation of a bivalent, bispecific antibody expressing cell by targeted integration of multiple expression cassettes in a defined organization | |
| NZ626252B2 (en) | Expression cassette | |
| HK1232253B (zh) | 生产多肽的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20090710 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20111018 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20120118 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20120130 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120220 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120313 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120319 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150330 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 Ref document number: 4958905 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |