JP4789271B2 - エラーが最小化された核酸分子の合成 - Google Patents
エラーが最小化された核酸分子の合成 Download PDFInfo
- Publication number
- JP4789271B2 JP4789271B2 JP2008543550A JP2008543550A JP4789271B2 JP 4789271 B2 JP4789271 B2 JP 4789271B2 JP 2008543550 A JP2008543550 A JP 2008543550A JP 2008543550 A JP2008543550 A JP 2008543550A JP 4789271 B2 JP4789271 B2 JP 4789271B2
- Authority
- JP
- Japan
- Prior art keywords
- molecules
- nucleotide sequence
- sequence
- endonuclease
- fragments
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000007523 nucleic acids Chemical class 0.000 title claims description 47
- 102000039446 nucleic acids Human genes 0.000 title claims description 45
- 108020004707 nucleic acids Proteins 0.000 title claims description 45
- 230000015572 biosynthetic process Effects 0.000 title claims description 32
- 238000003786 synthesis reaction Methods 0.000 title claims description 32
- 238000000034 method Methods 0.000 claims description 108
- 239000002773 nucleotide Substances 0.000 claims description 108
- 125000003729 nucleotide group Chemical group 0.000 claims description 108
- 239000012634 fragment Substances 0.000 claims description 76
- 108091034117 Oligonucleotide Proteins 0.000 claims description 73
- 102000004533 Endonucleases Human genes 0.000 claims description 37
- 108010042407 Endonucleases Proteins 0.000 claims description 37
- 108090000623 proteins and genes Proteins 0.000 claims description 33
- 238000006243 chemical reaction Methods 0.000 claims description 16
- 230000002194 synthesizing effect Effects 0.000 claims description 16
- 238000003752 polymerase chain reaction Methods 0.000 claims description 14
- 108091028043 Nucleic acid sequence Proteins 0.000 claims description 13
- 230000003321 amplification Effects 0.000 claims description 13
- 238000003199 nucleic acid amplification method Methods 0.000 claims description 13
- 238000000137 annealing Methods 0.000 claims description 12
- 102000007260 Deoxyribonuclease I Human genes 0.000 claims description 8
- 108010008532 Deoxyribonuclease I Proteins 0.000 claims description 8
- 241000588724 Escherichia coli Species 0.000 claims description 8
- 102000004099 Deoxyribonuclease (Pyrimidine Dimer) Human genes 0.000 claims description 6
- 108010082610 Deoxyribonuclease (Pyrimidine Dimer) Proteins 0.000 claims description 6
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 claims description 5
- 229910052748 manganese Inorganic materials 0.000 claims description 5
- 239000011572 manganese Substances 0.000 claims description 5
- 238000012163 sequencing technique Methods 0.000 claims description 5
- 238000010367 cloning Methods 0.000 claims description 4
- 239000013598 vector Substances 0.000 claims description 4
- 238000012545 processing Methods 0.000 claims description 3
- 230000008569 process Effects 0.000 description 14
- 108020004414 DNA Proteins 0.000 description 13
- 238000004925 denaturation Methods 0.000 description 9
- 230000036425 denaturation Effects 0.000 description 9
- 102000053602 DNA Human genes 0.000 description 6
- 238000003776 cleavage reaction Methods 0.000 description 6
- 230000007017 scission Effects 0.000 description 6
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 5
- 238000013459 approach Methods 0.000 description 5
- 238000007796 conventional method Methods 0.000 description 5
- 230000004927 fusion Effects 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 230000000295 complement effect Effects 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- 150000001413 amino acids Chemical group 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 235000018102 proteins Nutrition 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 108090000328 Arrestin Proteins 0.000 description 2
- 238000000018 DNA microarray Methods 0.000 description 2
- 108060002716 Exonuclease Proteins 0.000 description 2
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 229940024606 amino acid Drugs 0.000 description 2
- 235000001014 amino acid Nutrition 0.000 description 2
- 102000023732 binding proteins Human genes 0.000 description 2
- 108091008324 binding proteins Proteins 0.000 description 2
- 230000001276 controlling effect Effects 0.000 description 2
- 102000013165 exonuclease Human genes 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 229960000310 isoleucine Drugs 0.000 description 2
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical group CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 2
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- 229930024421 Adenine Natural products 0.000 description 1
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 1
- 102000003916 Arrestin Human genes 0.000 description 1
- 102000012410 DNA Ligases Human genes 0.000 description 1
- 108010061982 DNA Ligases Proteins 0.000 description 1
- 238000007702 DNA assembly Methods 0.000 description 1
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 1
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 1
- 102000003960 Ligases Human genes 0.000 description 1
- 108090000364 Ligases Proteins 0.000 description 1
- 229910021380 Manganese Chloride Inorganic materials 0.000 description 1
- GLFNIEUTAYBVOC-UHFFFAOYSA-L Manganese chloride Chemical compound Cl[Mn]Cl GLFNIEUTAYBVOC-UHFFFAOYSA-L 0.000 description 1
- 108010086093 Mung Bean Nuclease Proteins 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 108010010677 Phosphodiesterase I Proteins 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 108020004682 Single-Stranded DNA Proteins 0.000 description 1
- ZQTUNIWBUQUKAM-UHFFFAOYSA-N [3-[2-(1h-imidazol-1-ium-4-yl)ethylamino]-3-oxopropyl]azanium;dichloride Chemical compound Cl.Cl.NCCC(=O)NCCC1=CN=CN1 ZQTUNIWBUQUKAM-UHFFFAOYSA-N 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000013065 commercial product Substances 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 108010052305 exodeoxyribonuclease III Proteins 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 239000011565 manganese chloride Substances 0.000 description 1
- 229940099607 manganese chloride Drugs 0.000 description 1
- 235000002867 manganese chloride Nutrition 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000000203 mixture Substances 0.000 description 1
- 238000007857 nested PCR Methods 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 108091008146 restriction endonucleases Proteins 0.000 description 1
- 229920002477 rna polymer Polymers 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 108010068698 spleen exonuclease Proteins 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 229940113082 thymine Drugs 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1093—General methods of preparing gene libraries, not provided for in other subgroups
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/102—Mutagenizing nucleic acids
- C12N15/1027—Mutagenizing nucleic acids by DNA shuffling, e.g. RSR, STEP, RPR
Landscapes
- Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Zoology (AREA)
- Biomedical Technology (AREA)
- Wood Science & Technology (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Plant Pathology (AREA)
- Molecular Biology (AREA)
- Microbiology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Crystallography & Structural Chemistry (AREA)
- Bioinformatics & Computational Biology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Saccharide Compounds (AREA)
Description
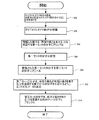
エラーコレクション(error correction)を備える核酸分子を合成するための方法が提供される。所望の完全長のヌクレオチド配列を有する分子の合成は、一般的に、所望の完全長のヌクレオチド配列の断片を有することを意図するオリゴヌクレオチドで開始し、任意に、他の所望のヌクレオチド、例えば、基質にオリゴヌクレオチドを結合するためのヌクレオチドを含む。オリゴヌクレオチドは、所望の完全長の配列の両鎖用に合成されてよく、合成におけるオリゴヌクレオチドの使用の効率を高め、したがって、そのコストをコントロールする。オリゴヌクレオチドは増幅されて、所望の完全長のヌクレオチド配列を有することを意図する、第一セットの分子にアセンブル(assembled)される。所望のヌクレオチド配列にアセンブルされる分子(複数)のフィデリティを改善するために、オリゴヌクレオチドが、そのヌクレオチド配列に従ってグループ化されることが確実にされ得る。第一セットでの分子は変性されて、所望の完全長のヌクレオチド配列を有することを意図する、第二セットの分子を形成するようにアニールされる。第二セットでの分子は、例えば、配列エラーが存する、第二セットの分子にブラントカット(blunt cut)を形成する、エンドヌクレアーゼを、分子(複数)に混合することによって、第二セットでの分子に沿ってランダムで、より小さいセグメントに切断される。該より小さいセグメントは、所望の完全長のヌクレオチド配列を有することを意図する、一セットの分子にアセンブルされる。核酸分子の合成プロセスの終りの近くで、この手法で分子切断を促すことによって、従来の方法で得ることができるよりも、ヌクレオチド配列のエラーがほとんど無い、一セットの完全長の分子を得ることができる。
なお、ここに、まとめとして、本発明の好ましい実施の形態(形態シリーズI及び形態シリーズII)を示す。
形態シリーズI
(形態1)
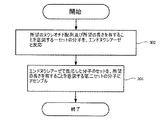
核酸分子を合成するための方法であって、該方法は、
オリゴヌクレオチド断片を、各断片が所望のヌクレオチド配列を有するよう、得るステップと、
該オリゴヌクレオチド断片を増幅するステップと、
該増幅したオリゴヌクレオチド断片を、所望の長さを有するよう、第一セットの分子にアセンブルするステップと、
該第一セットの分子を変性するステップと、
該変性された分子を第二セットの分子にアニーリングするステップと、
所望の長さよりも短い長さを有するべく第三セットの分子を産生するように、該第二セットの分子をエンドヌクレアーゼと反応させるステップと、
該第三セットの分子を該所望の長さの第四セットの分子にアセンブルするステップと
を含む、方法。
(形態2)
前記所望のヌクレオチド配列は、一以上の自然に生じる遺伝子配列を含む、形態1の方法。
(形態3)
前記所望のヌクレオチド配列は、一以上の合成のヌクレオチド配列を含む、形態1の方法。
(形態4)
前記所望のヌクレオチド配列は、一以上の自然に生じる遺伝子配列と、一以上の合成のヌクレオチド配列とのハイブリッドを含む、形態1の方法。
(形態5)
前記オリゴヌクレオチド断片を得るステップは、前記断片を合成するステップをさらに含む、形態1の方法。
(形態6)
前記オリゴヌクレオチド断片を得るステップは、マイクロチップ上で前記断片を合成するステップをさらに含む、形態5の方法。
(形態7)
前記オリゴヌクレオチド断片を得るステップは、前記所望のヌクレオチド配列の両鎖用のアダプタープライマーを用いて、マイクロチップ上に前記断片を合成するステップをさらに含む、形態6の方法。
(形態8)
前記オリゴヌクレオチド断片を得るステップは、前記所望のヌクレオチド配列の両鎖用の前記断片を合成するステップをさらに含む、形態5の方法。
(形態9)
さらに、前記断片の前記配列に従って、前記増幅されたオリゴヌクレオチド断片をグループ化するステップを含む、形態1の方法。
(形態10)
前記増幅されたオリゴヌクレオチド断片をグループ化するステップは、各別(separate)の容器に、固有(unique)の配列を共有する、断片の各グループを配置するステップをさらに含む、形態9の方法。
(形態11)
前記増幅されたオリゴヌクレオチド断片をグループ化するステップは、固有(unique)のアダプタープライマーを用いるステップをさらに含む、形態9の方法。
(形態12)
前記増幅したオリゴヌクレオチド断片を、所望の長さを有するよう第一セットの分子にアセンブルするステップは、一以上のオーバーラップ‐イクステンション(overlap-extension)ポリメラーゼ連鎖反応を用いることをさらに含む、形態1の方法。
(形態13)
前記第三セットの分子を前記所望の長さの第四セットの分子にアセンブルするステップは、一以上のオーバーラップ‐イクステンションポリメラーゼ連鎖反応を用いることをさらに含む、形態1の方法。
(形態14)
さらに、一以上のラウンドないし回数(round)の追加的なエラーコレクションによって、前記第四セットの分子を処理するステップを含む、形態1の方法。
(形態15)
さらに、一以上のポリメラーゼ連鎖反応によって前記第四セットの分子を増幅するステップを含む、形態1の方法。
(形態16)
さらに、前記第四セットの分子を、一以上のベクターにクローニングするステップを含む、形態1の方法。
(形態17)
さらに、前記第四セットの分子を、一以上のベクターにクローニングするステップを含む、形態15の方法。
(形態18)
さらに、前記第四セットの分子をシーケンシングするステップを含む、形態1の方法。
(形態19)
さらに、前記第四セットの分子をシーケンシングするステップを含む、形態1の方法。
(形態20)
エラーコレクションの方法であって、該方法は、
エンドヌクレアーゼで、所望のヌクレオチド配列を有するよう、かつ、所望の長さを有するよう一セットの分子を反応するステップと、
該エンドヌクレアーゼで反応した分子のセットを、該所望の長さを有するよう第二セットの分子にアセンブルするステップとを含む方法。
(形態21)
DNAアセンブリのための方法であって、該方法は、
オーバーラップを有する二本鎖DNA断片を合成するステップと、
エキソヌクレアーゼで該断片を反応させることによって、一本鎖のオーバーラップ(複数)を生成するステップと、
一本鎖結合タンパク質と反応させることによって該オーバーラップ(複数)をアニーリングするステップと、
DNAポリメラーゼでギャップを埋めるステップと、
DNAリガーゼでニックをシーリングするステップとを含む方法。
形態シリーズII
(形態1)
核酸分子の合成によるエラーコレクションのための方法であって、該方法は、
(a)オリゴヌクレオチド断片を、各断片が所望の長さの所望のヌクレオチド配列の一部を有するよう、得るステップと、
(b)該オリゴヌクレオチド断片を増幅するステップと、
(c)該増幅したオリゴヌクレオチド断片を、当該所望の長さを有するよう、第一セットの分子にアセンブルするステップと、
(d)該第一セットの分子を変性するステップと、
(e)該変性された分子を当該所望の長さを有するよう、第二セットの分子にアニーリングするステップと、
(f)該第二セットの分子を複数のエンドヌクレアーゼと反応させ、それによって、配列エラーの部位で該第二セットの分子にブラントカットを導入し、前記所望の長さよりも短い長さの断片を含む第三セットの分子を産生し、ここで、当該複数のエンドヌクレアーゼは任意の配列エラーの部位で切断するようにさせるステップと、
(g)断片を含む該第三セットの分子を該所望の長さの第四セットの分子にアセンブルし、それによって、当該第一セットの分子と比較して、第四セットの分子のエラー数が減少されるステップと
を含む、方法。
(形態2)
前記所望のヌクレオチド配列は、一以上の自然に生じる遺伝子配列を含む、形態1の方法。
(形態3)
前記所望のヌクレオチド配列は、一以上の合成のヌクレオチド配列を含む、形態1又は2の方法。
(形態4)
前記所望のヌクレオチド配列は、一以上の自然に生じる遺伝子配列と、一以上の合成のヌクレオチド配列とのハイブリッドを含む、形態1の方法。
(形態5)
前記オリゴヌクレオチド断片を得るステップは、前記断片を合成するステップを含む、形態1〜4のいずれかの方法。
(形態6)
前記オリゴヌクレオチド断片を得るステップは、マイクロチップ上で断片を合成するステップを含む、形態1〜5のいずれかの方法。
(形態7)
前記オリゴヌクレオチド断片を得るステップは、前記所望のヌクレオチド配列の両鎖の部分を有するように意図された一セットの断片を合成するステップを含む、形態1〜6のいずれかの方法。
(形態8)
さらに、アセンブルされる前に前記断片の前記配列に従って、前記オリゴヌクレオチド断片をグループ化するステップを含む、形態1〜7のいずれかの方法。
(形態9)
増幅に先立って前記オリゴヌクレオチドが得られ各別(separate)の容器に保持され、前記増幅されたオリゴヌクレオチド断片をグループ化するステップは、固有(unique)の配列を共有する、断片の各グループを各別(separate)の容器に保持されるステップを含む、形態1〜8のいずれかの方法。
(形態10)
前記増幅されたオリゴヌクレオチド断片をグループ化するステップは、固有(unique)のアダプタープライマーを用いるステップを含む、形態9の方法。
(形態11)
前記増幅した断片を第一セットの分子にアセンブルするか、又は前記第三セットの分子を第四セットの分子にアセンブルするステップは、オーバーラップ‐イクステンションポリメラーゼ連鎖反応を用いることを含む、形態1〜10のいずれかの方法。
(形態12)
さらに、一以上のラウンドないし回数(round)の追加的なエラーコレクションによって、前記第四セットの分子を処理するステップを含む、形態1〜11のいずれかの方法。
(形態13)
さらに、一以上のポリメラーゼ連鎖反応によって前記第四セットの分子を増幅するステップを含む、形態1〜12のいずれかの方法。
(形態14)
さらに、前記第四セットの分子を、一以上のベクターにクローニングするステップを含む、形態1〜13のいずれかの方法。
(形態15)
さらに、前記第四セットの分子をシーケンシングするステップを含む、形態1〜14のいずれかの方法。
(形態16)
前記複数のエンドヌクレアーゼはT7エンドヌクレアーゼIを含む、形態1〜15のいずれかの方法。
(形態17)
前記複数のエンドヌクレアーゼは3つのエンドヌクレアーゼを含む、形態1〜16のいずれかの方法。
(形態18)
前記複数のエンドヌクレアーゼはマングビーン(Mung Bean)エンドヌクレアーゼ、T7エンドヌクレアーゼI及びE.coliエンドヌクレアーゼVを含む、形態17の方法。
(形態19)
前記ステップ(f)の反応はマンガンの存在下で実行される、形態1〜18のいずれかの方法。
(形態20)
前記複数のエンドヌクレアーゼとの反応は、さらに、前記第二セットの分子をランダムにエラーがない(error-free)部位で切断する、形態1〜19のいずれかの方法。
所望の配列の範囲内のヌクレオチド配列で核酸分子を合成することは、分子生物学及びゲノム分野における断えざる挑戦である。過去数十年にわたって、多くの研究努力は、エラーが最小化された核酸分子を合成することに向けられてきた。ヌクレオチド配列エラーの著しい減少、及び/又は、合成効率の増大、若しくは、コストの減少を提供する方法は、基礎生医学及び生物工学研究の進展を可能にし、バイオテクノロジー産業の生産性を改善する。それらの問題に対する従来技術のアプローチは、様々な手段による、オリゴヌクレオチドの精製を含む。
104 オリゴ
106 第一セットの完全長の分子
108 一以上の分子(配列エラーを含む)
110 一以上の分子(配列エラーを含まない)
112 変性された、完全長の(一本鎖)分子
114 配列エラーを有しない、一以上の(一本鎖)分子
116 一以上の配列エラーを有する、一以上の分子
118 所望のヌクレオチド配列を有することを意図した、第二セットの完全長の分子
120 第二セットの完全長の分子(配列エラーを含む)
122 切断
124 第四セットの完全長の分子
Claims (20)
- 核酸分子の合成によるエラーコレクションのための方法であって、該方法は、
(a)オリゴヌクレオチド断片を、各断片が所望の長さの所望のヌクレオチド配列の一部を有するよう、得るステップと、
(b)該オリゴヌクレオチド断片を増幅するステップと、
(c)該増幅したオリゴヌクレオチド断片を、当該所望の長さを有するよう、第一セットの分子にアセンブルするステップと、
(d)該第一セットの分子を変性するステップと、
(e)該変性された分子を当該所望の長さを有するよう、第二セットの分子にアニーリングするステップと、
(f)該第二セットの分子を複数のエンドヌクレアーゼと反応させ、それによって、配列エラーの部位で該第二セットの分子にブラントカットを導入し、前記所望の長さよりも短い長さの断片を含む第三セットの分子を産生し、ここで、当該複数のエンドヌクレアーゼは任意の配列エラーの部位で切断するようにさせるステップと、
(g)断片を含む該第三セットの分子を該所望の長さの第四セットの分子にアセンブルし、それによって、当該第一セットの分子と比較して、第四セットの分子のエラー数が減少されるステップと
を含む、方法。 - 前記所望のヌクレオチド配列は、一以上の自然に生じる遺伝子配列を含む、請求項1の方法。
- 前記所望のヌクレオチド配列は、一以上の合成のヌクレオチド配列を含む、請求項1の方法。
- 前記所望のヌクレオチド配列は、一以上の自然に生じる遺伝子配列と、一以上の合成のヌクレオチド配列とのハイブリッドを含む、請求項1の方法。
- 前記オリゴヌクレオチド断片を得るステップは、前記断片を合成するステップを含む、請求項1の方法。
- 前記オリゴヌクレオチド断片を得るステップは、マイクロチップ上で断片を合成するステップを含む、請求項5の方法。
- 前記オリゴヌクレオチド断片を得るステップは、前記所望のヌクレオチド配列の両鎖の部分を有するように意図された一セットの断片を合成するステップを含む、請求項1の方法。
- さらに、アセンブルされる前に前記断片の前記配列に従って、前記ステップ(b)のオリゴヌクレオチド断片をグループ化するステップを含む、請求項1の方法。
- 増幅に先立って前記オリゴヌクレオチドが得られ各別(separate)の容器に保持され、固有(unique)の配列を共有する、断片の各グループが各別(separate)の容器に保持される、請求項1の方法。
- 前記増幅されたオリゴヌクレオチド断片をグループ化するステップは、固有(unique)のアダプタープライマーを用いるステップを含む、請求項8の方法。
- 前記増幅した断片を第一セットの分子にアセンブルするか、又は前記第三セットの分子を第四セットの分子にアセンブルするステップは、オーバーラップ‐イクステンションポリメラーゼ連鎖反応を用いることを含む、請求項1の方法。
- さらに、一以上のラウンドの追加的なエラーコレクションによって、前記第四セットの分子を処理するステップを含む、請求項1の方法。
- さらに、一以上のポリメラーゼ連鎖反応によって前記第四セットの分子を増幅するステップを含む、請求項1の方法。
- さらに、前記第四セットの分子を、一以上のベクターにクローニングするステップを含む、請求項1の方法。
- さらに、前記第四セットの分子をシーケンシングするステップを含む、請求項1の方法。
- 前記複数のエンドヌクレアーゼはT7エンドヌクレアーゼIを含む、請求項1の方法。
- 前記複数のエンドヌクレアーゼは3つのエンドヌクレアーゼを含む、請求項1の方法。
- 前記複数のエンドヌクレアーゼはマングビーン(Mung Bean)エンドヌクレアーゼ、T7エンドヌクレアーゼI及びE.coliエンドヌクレアーゼVを含む、請求項17の方法。
- 前記ステップ(f)の反応はマンガンの存在下で実行される、請求項1の方法。
- 前記複数のエンドヌクレアーゼとの反応は、前記第二セットの分子を配列エラーの部位およびランダムにエラーがない(error-free)部位の両部位で切断する、請求項1の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US74146905P | 2005-12-02 | 2005-12-02 | |
| US60/741,469 | 2005-12-02 | ||
| PCT/US2006/046457 WO2007065035A2 (en) | 2005-12-02 | 2006-12-04 | Synthesis of error-minimized nucleic acid molecules |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2009518013A JP2009518013A (ja) | 2009-05-07 |
| JP2009518013A5 JP2009518013A5 (ja) | 2011-04-07 |
| JP4789271B2 true JP4789271B2 (ja) | 2011-10-12 |
Family
ID=38092910
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008543550A Active JP4789271B2 (ja) | 2005-12-02 | 2006-12-04 | エラーが最小化された核酸分子の合成 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US7704690B2 (ja) |
| EP (1) | EP1963525B2 (ja) |
| JP (1) | JP4789271B2 (ja) |
| AU (1) | AU2006320275B2 (ja) |
| CA (1) | CA2642514C (ja) |
| WO (1) | WO2007065035A2 (ja) |
Families Citing this family (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MY143596A (en) * | 2005-08-11 | 2011-06-15 | Synthetic Genomics Inc | In vitro recombination method |
| WO2008115632A2 (en) * | 2007-02-09 | 2008-09-25 | The Regents Of The University Of California | Method for recombining dna sequences and compositions related thereto |
| DK2255013T3 (en) * | 2008-02-15 | 2016-09-12 | Synthetic Genomics Inc | Methods for in vitro joining and combinatorial assembly of nucleic acid molecules. |
| US10093552B2 (en) | 2008-02-22 | 2018-10-09 | James Weifu Lee | Photovoltaic panel-interfaced solar-greenhouse distillation systems |
| US9259662B2 (en) | 2008-02-22 | 2016-02-16 | James Weifu Lee | Photovoltaic panel-interfaced solar-greenhouse distillation systems |
| HUE031394T2 (en) | 2008-02-23 | 2017-07-28 | James Weifu Lee | Designer organisms for photobiological butanol production from carbon dioxide and water |
| US8986963B2 (en) * | 2008-02-23 | 2015-03-24 | James Weifu Lee | Designer calvin-cycle-channeled production of butanol and related higher alcohols |
| EP2664678B1 (en) * | 2008-10-24 | 2014-10-08 | Epicentre Technologies Corporation | Transposon end compositions and methods for modifying nucleic acids |
| US9080211B2 (en) | 2008-10-24 | 2015-07-14 | Epicentre Technologies Corporation | Transposon end compositions and methods for modifying nucleic acids |
| US10174368B2 (en) | 2009-09-10 | 2019-01-08 | Centrillion Technology Holdings Corporation | Methods and systems for sequencing long nucleic acids |
| CN102858995B (zh) | 2009-09-10 | 2016-10-26 | 森特瑞隆技术控股公司 | 靶向测序方法 |
| US20120252682A1 (en) * | 2011-04-01 | 2012-10-04 | Maples Corporate Services Limited | Methods and systems for sequencing nucleic acids |
| US20130109596A1 (en) | 2011-09-26 | 2013-05-02 | Life Technologies Corporation | High efficiency, small volume nucleic acid synthesis |
| WO2014106167A1 (en) * | 2012-12-31 | 2014-07-03 | Advanced Liquid Logic, Inc. | Digital microfluidic gene synthesis and error correction |
| US10563240B2 (en) | 2013-03-14 | 2020-02-18 | Life Technologies Corporation | High efficiency, small volume nucleic acid synthesis |
| EP3169781B1 (en) | 2014-07-15 | 2020-04-08 | Life Technologies Corporation | Compositions and methods for nucleic acid assembly |
| LT3557262T (lt) | 2014-12-09 | 2022-11-10 | Life Technologies Corporation | Didelio efektyvumo nukleorūgščių sintezė mažame tūryje |
| WO2017007925A1 (en) | 2015-07-07 | 2017-01-12 | Thermo Fisher Scientific Geneart Gmbh | Enzymatic synthesis of nucleic acid sequences |
| US20180291413A1 (en) | 2015-10-06 | 2018-10-11 | Thermo Fisher Scientific Geneart Gmbh | Devices and methods for producing nucleic acids and proteins |
| GB2566986A (en) | 2017-09-29 | 2019-04-03 | Evonetix Ltd | Error detection during hybridisation of target double-stranded nucleic acid |
| US20210163922A1 (en) * | 2018-03-19 | 2021-06-03 | Modernatx, Inc. | Assembly and error reduction of synthetic genes from oligonucleotides |
| US20210147830A1 (en) | 2018-06-29 | 2021-05-20 | Thermo Fisher Scientific Geneart Gmbh | High throughput assembly of nucleic acid molecules |
| GB201905303D0 (en) | 2019-04-15 | 2019-05-29 | Thermo Fisher Scient Geneart Gmbh | Multiplex assembly of nucleic acid molecules |
| EP4114972A1 (en) | 2020-03-06 | 2023-01-11 | Life Technologies Corporation | High sequence fidelity nucleic acid synthesis and assembly |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10500561A (ja) * | 1994-02-17 | 1998-01-20 | アフィマックス テクノロジーズ エヌ. ブイ. | ランダムフラグメント化および再組立によるdna変異誘発 |
| JP2004526423A (ja) * | 2000-12-01 | 2004-09-02 | コーネル リサーチ ファンデーション インコーポレーテッド | 結合エンドヌクレアーゼ開裂および連結反応を使用した核酸の相違の検出方法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6165793A (en) * | 1996-03-25 | 2000-12-26 | Maxygen, Inc. | Methods for generating polynucleotides having desired characteristics by iterative selection and recombination |
| US20040023327A1 (en) * | 1995-12-07 | 2004-02-05 | Short Jay M. | End selection in directed evolution |
| ES2464132T3 (es) * | 2000-04-07 | 2014-05-30 | Eiken Kagaku Kabushiki Kaisha | Procedimiento de amplificación de ácido nucleico utilizando como molde ácido nucleico bicatenario |
| US7879580B2 (en) * | 2002-12-10 | 2011-02-01 | Massachusetts Institute Of Technology | Methods for high fidelity production of long nucleic acid molecules |
| US20050106590A1 (en) * | 2003-05-22 | 2005-05-19 | Lathrop Richard H. | Method for producing a synthetic gene or other DNA sequence |
| WO2006026614A2 (en) * | 2004-08-27 | 2006-03-09 | Wisconsin Alumni Research Foundation | Method of error reduction in nucleic acid populations |
| US20070196834A1 (en) * | 2005-09-09 | 2007-08-23 | Francesco Cerrina | Method and system for the generation of large double stranded DNA fragments |
-
2006
- 2006-12-04 WO PCT/US2006/046457 patent/WO2007065035A2/en active Application Filing
- 2006-12-04 JP JP2008543550A patent/JP4789271B2/ja active Active
- 2006-12-04 EP EP06844856.2A patent/EP1963525B2/en active Active
- 2006-12-04 US US11/633,686 patent/US7704690B2/en active Active
- 2006-12-04 AU AU2006320275A patent/AU2006320275B2/en active Active
- 2006-12-04 CA CA2642514A patent/CA2642514C/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10500561A (ja) * | 1994-02-17 | 1998-01-20 | アフィマックス テクノロジーズ エヌ. ブイ. | ランダムフラグメント化および再組立によるdna変異誘発 |
| JP2004526423A (ja) * | 2000-12-01 | 2004-09-02 | コーネル リサーチ ファンデーション インコーポレーテッド | 結合エンドヌクレアーゼ開裂および連結反応を使用した核酸の相違の検出方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US7704690B2 (en) | 2010-04-27 |
| EP1963525B1 (en) | 2013-11-20 |
| EP1963525A2 (en) | 2008-09-03 |
| CA2642514C (en) | 2011-06-07 |
| CA2642514A1 (en) | 2007-06-07 |
| US20070128649A1 (en) | 2007-06-07 |
| WO2007065035A2 (en) | 2007-06-07 |
| WO2007065035A3 (en) | 2007-11-22 |
| EP1963525A4 (en) | 2009-03-18 |
| AU2006320275B2 (en) | 2012-06-07 |
| AU2006320275A1 (en) | 2007-06-07 |
| EP1963525B2 (en) | 2017-10-11 |
| JP2009518013A (ja) | 2009-05-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4789271B2 (ja) | エラーが最小化された核酸分子の合成 | |
| US11768200B2 (en) | Methods for maintaining the integrity and identification of a nucleic acid template in a multiplex sequencing reaction | |
| CN108060191B (zh) | 一种双链核酸片段加接头的方法、文库构建方法和试剂盒 | |
| EP2585593B1 (en) | Methods for polynucleotide library production, immortalization and region of interest extraction | |
| US9255291B2 (en) | Oligonucleotide ligation methods for improving data quality and throughput using massively parallel sequencing | |
| JP2009518013A5 (ja) | ||
| JP2005509433A (ja) | 多重pcr | |
| US20130123117A1 (en) | Capture probe and assay for analysis of fragmented nucleic acids | |
| US20230056763A1 (en) | Methods of targeted sequencing | |
| JP2022534625A (ja) | 標的ゲノム領域の捕捉と分析 | |
| CA3178211A1 (en) | Methods for ligation-coupled-pcr | |
| CA2962254C (en) | Composition and method for processing dna | |
| JP5129498B2 (ja) | 核酸クローニング法 | |
| WO2017183648A1 (ja) | 多項目増幅手法 | |
| EP3655572A1 (en) | Rapid library construction for high throughput sequencing | |
| WO2022239632A1 (ja) | 合成dna分子の製造方法 | |
| US20230122979A1 (en) | Methods of sample normalization | |
| Quan | Genetic Assembly, Error-Correction and a High-Throughput Screening Strategy for | |
| JP2011010591A (ja) | Dnaの増幅方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20091204 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20091204 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A711 Effective date: 20100122 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20100122 |
|
| RD03 | Notification of appointment of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7423 Effective date: 20101018 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20101021 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20101018 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110216 |
|
| A871 | Explanation of circumstances concerning accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A871 Effective date: 20110216 |
|
| A975 | Report on accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A971005 Effective date: 20110224 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110328 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20110713 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20110715 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140729 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4789271 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |