JP2021532106A - 細菌由来ミニ細胞を含む組成物およびそれを使用する方法 - Google Patents
細菌由来ミニ細胞を含む組成物およびそれを使用する方法 Download PDFInfo
- Publication number
- JP2021532106A JP2021532106A JP2021503095A JP2021503095A JP2021532106A JP 2021532106 A JP2021532106 A JP 2021532106A JP 2021503095 A JP2021503095 A JP 2021503095A JP 2021503095 A JP2021503095 A JP 2021503095A JP 2021532106 A JP2021532106 A JP 2021532106A
- Authority
- JP
- Japan
- Prior art keywords
- cancer
- tumor
- cell
- agonist
- cells
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 239000000203 mixture Substances 0.000 title claims abstract description 125
- 238000000034 method Methods 0.000 title claims abstract description 46
- 230000001580 bacterial effect Effects 0.000 title claims description 111
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 353
- 239000000556 agonist Substances 0.000 claims abstract description 202
- 239000002246 antineoplastic agent Substances 0.000 claims abstract description 178
- 201000011510 cancer Diseases 0.000 claims abstract description 93
- 108010074328 Interferon-gamma Proteins 0.000 claims abstract description 89
- 102000008070 Interferon-gamma Human genes 0.000 claims abstract description 86
- 102000002227 Interferon Type I Human genes 0.000 claims abstract description 82
- 108010014726 Interferon Type I Proteins 0.000 claims abstract description 82
- 229940028894 interferon type ii Drugs 0.000 claims abstract description 54
- 210000004027 cell Anatomy 0.000 claims description 242
- 108010050904 Interferons Proteins 0.000 claims description 93
- 102000014150 Interferons Human genes 0.000 claims description 92
- 229940079322 interferon Drugs 0.000 claims description 90
- 108020004414 DNA Proteins 0.000 claims description 74
- 210000004881 tumor cell Anatomy 0.000 claims description 71
- 229940127089 cytotoxic agent Drugs 0.000 claims description 64
- 108090000623 proteins and genes Proteins 0.000 claims description 64
- 239000003446 ligand Substances 0.000 claims description 51
- 230000014509 gene expression Effects 0.000 claims description 45
- 102000039446 nucleic acids Human genes 0.000 claims description 44
- 108020004707 nucleic acids Proteins 0.000 claims description 44
- 150000007523 nucleic acids Chemical class 0.000 claims description 44
- 108020004459 Small interfering RNA Proteins 0.000 claims description 41
- SLURUCSFDHKXFR-WWMWMSKMSA-N (7s,9s)-7-[[(1s,3r,4as,9s,9ar,10as)-9-methoxy-1-methyl-3,4,4a,6,7,9,9a,10a-octahydro-1h-pyrano[1,2][1,3]oxazolo[3,4-b][1,4]oxazin-3-yl]oxy]-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione Chemical compound O=C1C2=CC=CC(OC)=C2C(=O)C(C(O)=C23)=C1C(O)=C3C[C@@](O)(C(=O)CO)C[C@@H]2O[C@H]1C[C@@H]2N3CCO[C@H](OC)[C@H]3O[C@@H]2[C@H](C)O1 SLURUCSFDHKXFR-WWMWMSKMSA-N 0.000 claims description 35
- 239000002924 silencing RNA Substances 0.000 claims description 32
- 108091032973 (ribonucleotides)n+m Proteins 0.000 claims description 31
- 239000002773 nucleotide Substances 0.000 claims description 26
- 125000003729 nucleotide group Chemical group 0.000 claims description 26
- 108010001857 Cell Surface Receptors Proteins 0.000 claims description 24
- 208000003174 Brain Neoplasms Diseases 0.000 claims description 22
- 239000003112 inhibitor Substances 0.000 claims description 18
- 102000040650 (ribonucleotides)n+m Human genes 0.000 claims description 17
- 241000699666 Mus <mouse, genus> Species 0.000 claims description 17
- 108091034117 Oligonucleotide Proteins 0.000 claims description 17
- 206010006187 Breast cancer Diseases 0.000 claims description 16
- 208000026310 Breast neoplasm Diseases 0.000 claims description 16
- 230000006907 apoptotic process Effects 0.000 claims description 16
- 239000002679 microRNA Substances 0.000 claims description 15
- 229940045799 anthracyclines and related substance Drugs 0.000 claims description 14
- 239000002158 endotoxin Substances 0.000 claims description 14
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 13
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 13
- 210000004962 mammalian cell Anatomy 0.000 claims description 13
- 201000002528 pancreatic cancer Diseases 0.000 claims description 13
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 13
- 201000000053 blastoma Diseases 0.000 claims description 12
- 210000003169 central nervous system Anatomy 0.000 claims description 12
- 201000008184 embryoma Diseases 0.000 claims description 12
- 238000001914 filtration Methods 0.000 claims description 12
- 229920001282 polysaccharide Polymers 0.000 claims description 12
- 239000005017 polysaccharide Substances 0.000 claims description 12
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 11
- 210000001519 tissue Anatomy 0.000 claims description 11
- 208000006468 Adrenal Cortex Neoplasms Diseases 0.000 claims description 10
- 201000005202 lung cancer Diseases 0.000 claims description 10
- 208000020816 lung neoplasm Diseases 0.000 claims description 10
- 108010056274 polo-like kinase 1 Proteins 0.000 claims description 10
- 230000004663 cell proliferation Effects 0.000 claims description 9
- 229920006008 lipopolysaccharide Polymers 0.000 claims description 9
- 229940046168 CpG oligodeoxynucleotide Drugs 0.000 claims description 8
- 238000002512 chemotherapy Methods 0.000 claims description 8
- 230000010837 receptor-mediated endocytosis Effects 0.000 claims description 8
- 206010009944 Colon cancer Diseases 0.000 claims description 7
- 101000669447 Homo sapiens Toll-like receptor 4 Proteins 0.000 claims description 7
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 7
- 102100039360 Toll-like receptor 4 Human genes 0.000 claims description 7
- RFCBNSCSPXMEBK-INFSMZHSSA-N c-GMP-AMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]3[C@@H](O)[C@H](N4C5=NC=NC(N)=C5N=C4)O[C@@H]3COP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 RFCBNSCSPXMEBK-INFSMZHSSA-N 0.000 claims description 7
- 229940106189 ceramide Drugs 0.000 claims description 7
- 239000003534 dna topoisomerase inhibitor Substances 0.000 claims description 7
- 230000002496 gastric effect Effects 0.000 claims description 7
- 201000007270 liver cancer Diseases 0.000 claims description 7
- 208000014018 liver neoplasm Diseases 0.000 claims description 7
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical class ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 claims description 7
- 229960004961 mechlorethamine Drugs 0.000 claims description 7
- 125000002757 morpholinyl group Chemical group 0.000 claims description 7
- 102000040430 polynucleotide Human genes 0.000 claims description 7
- 108091033319 polynucleotide Proteins 0.000 claims description 7
- 239000002157 polynucleotide Substances 0.000 claims description 7
- 229940044693 topoisomerase inhibitor Drugs 0.000 claims description 7
- 206010005003 Bladder cancer Diseases 0.000 claims description 6
- 201000009030 Carcinoma Diseases 0.000 claims description 6
- 102000008394 Immunoglobulin Fragments Human genes 0.000 claims description 6
- 108010021625 Immunoglobulin Fragments Proteins 0.000 claims description 6
- 208000006265 Renal cell carcinoma Diseases 0.000 claims description 6
- 229940123237 Taxane Drugs 0.000 claims description 6
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 6
- 150000002224 folic acids Chemical class 0.000 claims description 6
- 201000001441 melanoma Diseases 0.000 claims description 6
- 230000000242 pagocytic effect Effects 0.000 claims description 6
- 210000002784 stomach Anatomy 0.000 claims description 6
- 208000011580 syndromic disease Diseases 0.000 claims description 6
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 6
- YDNKGFDKKRUKPY-JHOUSYSJSA-N C16 ceramide Natural products CCCCCCCCCCCCCCCC(=O)N[C@@H](CO)[C@H](O)C=CCCCCCCCCCCCCC YDNKGFDKKRUKPY-JHOUSYSJSA-N 0.000 claims description 5
- 206010008342 Cervix carcinoma Diseases 0.000 claims description 5
- 206010014967 Ependymoma Diseases 0.000 claims description 5
- 101000669402 Homo sapiens Toll-like receptor 7 Proteins 0.000 claims description 5
- 206010025323 Lymphomas Diseases 0.000 claims description 5
- 208000003445 Mouth Neoplasms Diseases 0.000 claims description 5
- CRJGESKKUOMBCT-VQTJNVASSA-N N-acetylsphinganine Chemical compound CCCCCCCCCCCCCCC[C@@H](O)[C@H](CO)NC(C)=O CRJGESKKUOMBCT-VQTJNVASSA-N 0.000 claims description 5
- JMENXJYBCQFIRK-KRJDXUSZSA-N N-hexacosanoylisoglobotriaosyl ceramide Chemical compound O[C@@H]1[C@@H](O)[C@H](OC[C@H](NC(=O)CCCCCCCCCCCCCCCCCCCCCCCCC)[C@H](O)\C=C\CCCCCCCCCCCCC)O[C@H](CO)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O2)O)[C@@H](O)[C@@H](CO)O1 JMENXJYBCQFIRK-KRJDXUSZSA-N 0.000 claims description 5
- 206010060862 Prostate cancer Diseases 0.000 claims description 5
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 5
- 108010041388 Ribonucleotide Reductases Proteins 0.000 claims description 5
- 102000000505 Ribonucleotide Reductases Human genes 0.000 claims description 5
- 229940044665 STING agonist Drugs 0.000 claims description 5
- 206010039491 Sarcoma Diseases 0.000 claims description 5
- 102100031463 Serine/threonine-protein kinase PLK1 Human genes 0.000 claims description 5
- 102100039390 Toll-like receptor 7 Human genes 0.000 claims description 5
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 claims description 5
- 208000009956 adenocarcinoma Diseases 0.000 claims description 5
- 229930013930 alkaloid Natural products 0.000 claims description 5
- 230000033115 angiogenesis Effects 0.000 claims description 5
- 230000000340 anti-metabolite Effects 0.000 claims description 5
- 229940100197 antimetabolite Drugs 0.000 claims description 5
- 239000002256 antimetabolite Substances 0.000 claims description 5
- 210000004556 brain Anatomy 0.000 claims description 5
- ZVEQCJWYRWKARO-UHFFFAOYSA-N ceramide Natural products CCCCCCCCCCCCCCC(O)C(=O)NC(CO)C(O)C=CCCC=C(C)CCCCCCCCC ZVEQCJWYRWKARO-UHFFFAOYSA-N 0.000 claims description 5
- 201000010881 cervical cancer Diseases 0.000 claims description 5
- 208000029742 colonic neoplasm Diseases 0.000 claims description 5
- 230000004069 differentiation Effects 0.000 claims description 5
- 239000003937 drug carrier Substances 0.000 claims description 5
- 229940125697 hormonal agent Drugs 0.000 claims description 5
- 208000012987 lip and oral cavity carcinoma Diseases 0.000 claims description 5
- 230000036244 malformation Effects 0.000 claims description 5
- 230000001394 metastastic effect Effects 0.000 claims description 5
- 206010061289 metastatic neoplasm Diseases 0.000 claims description 5
- DDOVBCWVTOHGCU-QMXMISKISA-N n-[(e,2s,3r)-3-hydroxy-1-[(2r,3r,4s,5r,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxynonadec-4-en-2-yl]octadecanamide Chemical compound CCCCCCCCCCCCCCCCCC(=O)N[C@H]([C@H](O)\C=C\CCCCCCCCCCCCCC)CO[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O DDOVBCWVTOHGCU-QMXMISKISA-N 0.000 claims description 5
- VVGIYYKRAMHVLU-UHFFFAOYSA-N newbouldiamide Natural products CCCCCCCCCCCCCCCCCCCC(O)C(O)C(O)C(CO)NC(=O)CCCCCCCCCCCCCCCCC VVGIYYKRAMHVLU-UHFFFAOYSA-N 0.000 claims description 5
- 108700002563 poly ICLC Proteins 0.000 claims description 5
- 150000003230 pyrimidines Chemical class 0.000 claims description 5
- HVCOBJNICQPDBP-UHFFFAOYSA-N 3-[3-[3,5-dihydroxy-6-methyl-4-(3,4,5-trihydroxy-6-methyloxan-2-yl)oxyoxan-2-yl]oxydecanoyloxy]decanoic acid;hydrate Chemical compound O.OC1C(OC(CC(=O)OC(CCCCCCC)CC(O)=O)CCCCCCC)OC(C)C(O)C1OC1C(O)C(O)C(O)C(C)O1 HVCOBJNICQPDBP-UHFFFAOYSA-N 0.000 claims description 4
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 claims description 4
- 206010006143 Brain stem glioma Diseases 0.000 claims description 4
- 108090000994 Catalytic RNA Proteins 0.000 claims description 4
- 102000053642 Catalytic RNA Human genes 0.000 claims description 4
- 239000012623 DNA damaging agent Substances 0.000 claims description 4
- 229930186217 Glycolipid Natural products 0.000 claims description 4
- 101000800483 Homo sapiens Toll-like receptor 8 Proteins 0.000 claims description 4
- 208000009164 Islet Cell Adenoma Diseases 0.000 claims description 4
- 208000000172 Medulloblastoma Diseases 0.000 claims description 4
- 208000002030 Merkel cell carcinoma Diseases 0.000 claims description 4
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 claims description 4
- 239000012648 POLY-ICLC Substances 0.000 claims description 4
- 208000009565 Pharyngeal Neoplasms Diseases 0.000 claims description 4
- 206010034811 Pharyngeal cancer Diseases 0.000 claims description 4
- 208000007913 Pituitary Neoplasms Diseases 0.000 claims description 4
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 4
- 201000008183 Pulmonary blastoma Diseases 0.000 claims description 4
- 229940124614 TLR 8 agonist Drugs 0.000 claims description 4
- 102100033110 Toll-like receptor 8 Human genes 0.000 claims description 4
- 108091027569 Z-DNA Proteins 0.000 claims description 4
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 claims description 4
- 108010044540 auristatin Proteins 0.000 claims description 4
- PKFDLKSEZWEFGL-MHARETSRSA-N c-di-GMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]3[C@@H](O)[C@H](N4C5=C(C(NC(N)=N5)=O)N=C4)O[C@@H]3COP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 PKFDLKSEZWEFGL-MHARETSRSA-N 0.000 claims description 4
- 230000025084 cell cycle arrest Effects 0.000 claims description 4
- 208000017763 cutaneous neuroendocrine carcinoma Diseases 0.000 claims description 4
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 claims description 4
- 229960000975 daunorubicin Drugs 0.000 claims description 4
- 150000004676 glycans Chemical class 0.000 claims description 4
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 4
- 229960000908 idarubicin Drugs 0.000 claims description 4
- 208000010626 plasma cell neoplasm Diseases 0.000 claims description 4
- 229940115270 poly iclc Drugs 0.000 claims description 4
- 229940115272 polyinosinic:polycytidylic acid Drugs 0.000 claims description 4
- 150000003212 purines Chemical class 0.000 claims description 4
- 230000002207 retinal effect Effects 0.000 claims description 4
- 108091092562 ribozyme Proteins 0.000 claims description 4
- 210000004500 stellate cell Anatomy 0.000 claims description 4
- 210000000115 thoracic cavity Anatomy 0.000 claims description 4
- 229940044616 toll-like receptor 7 agonist Drugs 0.000 claims description 4
- 229940044655 toll-like receptor 9 agonist Drugs 0.000 claims description 4
- 210000001635 urinary tract Anatomy 0.000 claims description 4
- IRPOZWRRAFKYMQ-LMIAXWKISA-N 1-O-(alpha-D-galactopyranuronosyl)-N-tetradecanoyldihydrosphingosine Chemical compound CCCCCCCCCCCCCCC[C@@H](O)[C@@H](NC(=O)CCCCCCCCCCCCC)CO[C@H]1O[C@H](C(O)=O)[C@H](O)[C@H](O)[C@H]1O IRPOZWRRAFKYMQ-LMIAXWKISA-N 0.000 claims description 3
- 208000030507 AIDS Diseases 0.000 claims description 3
- 208000002008 AIDS-Related Lymphoma Diseases 0.000 claims description 3
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 claims description 3
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 claims description 3
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 3
- 108020005544 Antisense RNA Proteins 0.000 claims description 3
- YPMUZVRAVKLKGC-UHFFFAOYSA-N BBGL-II Natural products OC1C(O)C(O)C(COC(=O)CCCCCCCCCCCCCCC)OC1OC1CC2=CCC3C4CCC(C(C)CCCC(C)C)C4(C)CCC3C2(C)CC1 YPMUZVRAVKLKGC-UHFFFAOYSA-N 0.000 claims description 3
- 206010004593 Bile duct cancer Diseases 0.000 claims description 3
- 241000283690 Bos taurus Species 0.000 claims description 3
- 150000000700 C-glycosides Chemical class 0.000 claims description 3
- 241000283073 Equus caballus Species 0.000 claims description 3
- 241000282326 Felis catus Species 0.000 claims description 3
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 claims description 3
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 claims description 3
- 108091007460 Long intergenic noncoding RNA Proteins 0.000 claims description 3
- 206010025312 Lymphoma AIDS related Diseases 0.000 claims description 3
- 208000034578 Multiple myelomas Diseases 0.000 claims description 3
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims description 3
- 206010029260 Neuroblastoma Diseases 0.000 claims description 3
- 241000283973 Oryctolagus cuniculus Species 0.000 claims description 3
- 241001494479 Pecora Species 0.000 claims description 3
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 claims description 3
- 108010009460 RNA Polymerase II Proteins 0.000 claims description 3
- 102000009572 RNA Polymerase II Human genes 0.000 claims description 3
- 241000700159 Rattus Species 0.000 claims description 3
- 206010041067 Small cell lung cancer Diseases 0.000 claims description 3
- 108091027967 Small hairpin RNA Proteins 0.000 claims description 3
- 206010042971 T-cell lymphoma Diseases 0.000 claims description 3
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 claims description 3
- 201000002454 adrenal cortex cancer Diseases 0.000 claims description 3
- 229940045687 antimetabolites folic acid analogs Drugs 0.000 claims description 3
- 229910052799 carbon Inorganic materials 0.000 claims description 3
- 239000003184 complementary RNA Substances 0.000 claims description 3
- 238000011109 contamination Methods 0.000 claims description 3
- 150000001982 diacylglycerols Chemical class 0.000 claims description 3
- 230000002124 endocrine Effects 0.000 claims description 3
- 230000003211 malignant effect Effects 0.000 claims description 3
- 208000025854 malignant tumor of adrenal cortex Diseases 0.000 claims description 3
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims description 3
- 239000004055 small Interfering RNA Substances 0.000 claims description 3
- 208000000587 small cell lung carcinoma Diseases 0.000 claims description 3
- 206010062261 spinal cord neoplasm Diseases 0.000 claims description 3
- 210000002536 stromal cell Anatomy 0.000 claims description 3
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims description 3
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 claims description 2
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 claims description 2
- 229930182476 C-glycoside Natural products 0.000 claims description 2
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 claims description 2
- 208000001976 Endocrine Gland Neoplasms Diseases 0.000 claims description 2
- 208000000461 Esophageal Neoplasms Diseases 0.000 claims description 2
- 208000006168 Ewing Sarcoma Diseases 0.000 claims description 2
- 208000017604 Hodgkin disease Diseases 0.000 claims description 2
- 208000021519 Hodgkin lymphoma Diseases 0.000 claims description 2
- 208000010747 Hodgkins lymphoma Diseases 0.000 claims description 2
- 206010021042 Hypopharyngeal cancer Diseases 0.000 claims description 2
- 206010056305 Hypopharyngeal neoplasm Diseases 0.000 claims description 2
- 208000005016 Intestinal Neoplasms Diseases 0.000 claims description 2
- 206010061252 Intraocular melanoma Diseases 0.000 claims description 2
- 208000007766 Kaposi sarcoma Diseases 0.000 claims description 2
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 2
- 206010023825 Laryngeal cancer Diseases 0.000 claims description 2
- 206010062038 Lip neoplasm Diseases 0.000 claims description 2
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 claims description 2
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 claims description 2
- 201000007224 Myeloproliferative neoplasm Diseases 0.000 claims description 2
- 208000001894 Nasopharyngeal Neoplasms Diseases 0.000 claims description 2
- 206010061306 Nasopharyngeal cancer Diseases 0.000 claims description 2
- 206010030155 Oesophageal carcinoma Diseases 0.000 claims description 2
- 208000000821 Parathyroid Neoplasms Diseases 0.000 claims description 2
- 208000002471 Penile Neoplasms Diseases 0.000 claims description 2
- 206010034299 Penile cancer Diseases 0.000 claims description 2
- 208000033014 Plasma cell tumor Diseases 0.000 claims description 2
- 208000007452 Plasmacytoma Diseases 0.000 claims description 2
- 206010038389 Renal cancer Diseases 0.000 claims description 2
- 208000021712 Soft tissue sarcoma Diseases 0.000 claims description 2
- 208000024313 Testicular Neoplasms Diseases 0.000 claims description 2
- 206010057644 Testis cancer Diseases 0.000 claims description 2
- 208000024770 Thyroid neoplasm Diseases 0.000 claims description 2
- 208000002495 Uterine Neoplasms Diseases 0.000 claims description 2
- 201000005969 Uveal melanoma Diseases 0.000 claims description 2
- 206010047741 Vulval cancer Diseases 0.000 claims description 2
- 208000004354 Vulvar Neoplasms Diseases 0.000 claims description 2
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 claims description 2
- 208000008383 Wilms tumor Diseases 0.000 claims description 2
- 150000001783 ceramides Chemical class 0.000 claims description 2
- 208000011654 childhood malignant neoplasm Diseases 0.000 claims description 2
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 claims description 2
- 210000002308 embryonic cell Anatomy 0.000 claims description 2
- 201000011523 endocrine gland cancer Diseases 0.000 claims description 2
- 201000004101 esophageal cancer Diseases 0.000 claims description 2
- 201000008819 extrahepatic bile duct carcinoma Diseases 0.000 claims description 2
- 201000010536 head and neck cancer Diseases 0.000 claims description 2
- 208000024348 heart neoplasm Diseases 0.000 claims description 2
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 2
- 231100000844 hepatocellular carcinoma Toxicity 0.000 claims description 2
- 201000006866 hypopharynx cancer Diseases 0.000 claims description 2
- 201000002313 intestinal cancer Diseases 0.000 claims description 2
- 201000002529 islet cell tumor Diseases 0.000 claims description 2
- 201000010982 kidney cancer Diseases 0.000 claims description 2
- 210000000244 kidney pelvis Anatomy 0.000 claims description 2
- 206010023841 laryngeal neoplasm Diseases 0.000 claims description 2
- 201000006721 lip cancer Diseases 0.000 claims description 2
- 206010024627 liposarcoma Diseases 0.000 claims description 2
- 201000000564 macroglobulinemia Diseases 0.000 claims description 2
- 208000026037 malignant tumor of neck Diseases 0.000 claims description 2
- 208000026045 malignant tumor of parathyroid gland Diseases 0.000 claims description 2
- 201000008026 nephroblastoma Diseases 0.000 claims description 2
- 208000029974 neurofibrosarcoma Diseases 0.000 claims description 2
- 201000002575 ocular melanoma Diseases 0.000 claims description 2
- 201000008968 osteosarcoma Diseases 0.000 claims description 2
- 208000021284 ovarian germ cell tumor Diseases 0.000 claims description 2
- 208000022102 pancreatic neuroendocrine neoplasm Diseases 0.000 claims description 2
- 208000003154 papilloma Diseases 0.000 claims description 2
- 208000029211 papillomatosis Diseases 0.000 claims description 2
- 210000004738 parenchymal cell Anatomy 0.000 claims description 2
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 claims description 2
- 208000010916 pituitary tumor Diseases 0.000 claims description 2
- 208000029340 primitive neuroectodermal tumor Diseases 0.000 claims description 2
- 230000001953 sensory effect Effects 0.000 claims description 2
- 201000008261 skin carcinoma Diseases 0.000 claims description 2
- 201000003120 testicular cancer Diseases 0.000 claims description 2
- 201000002510 thyroid cancer Diseases 0.000 claims description 2
- 206010046766 uterine cancer Diseases 0.000 claims description 2
- 208000037965 uterine sarcoma Diseases 0.000 claims description 2
- 206010046885 vaginal cancer Diseases 0.000 claims description 2
- 208000013139 vaginal neoplasm Diseases 0.000 claims description 2
- 201000005102 vulva cancer Diseases 0.000 claims description 2
- VQFKFAKEUMHBLV-BYSUZVQFSA-N 1-O-(alpha-D-galactosyl)-N-hexacosanoylphytosphingosine Chemical compound CCCCCCCCCCCCCCCCCCCCCCCCCC(=O)N[C@H]([C@H](O)[C@H](O)CCCCCCCCCCCCCC)CO[C@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O VQFKFAKEUMHBLV-BYSUZVQFSA-N 0.000 claims 6
- 102000006240 membrane receptors Human genes 0.000 claims 6
- 108091070501 miRNA Proteins 0.000 claims 3
- 241000234671 Ananas Species 0.000 claims 2
- 235000007119 Ananas comosus Nutrition 0.000 claims 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 claims 2
- 208000006593 Urologic Neoplasms Diseases 0.000 claims 2
- 210000005036 nerve Anatomy 0.000 claims 2
- FJAAFGBZVPKOCR-HBHGLPGMSA-N 1-palmitoyl-2-cis-vaccenoyl-3-alpha-D-glucosyl-sn-glycerol Chemical compound CCCCCC\C=C/CCCCCCCCCC(=O)O[C@H](COC(=O)CCCCCCCCCCCCCCC)CO[C@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O FJAAFGBZVPKOCR-HBHGLPGMSA-N 0.000 claims 1
- SVUOLADPCWQTTE-UHFFFAOYSA-N 1h-1,2-benzodiazepine Chemical compound N1N=CC=CC2=CC=CC=C12 SVUOLADPCWQTTE-UHFFFAOYSA-N 0.000 claims 1
- 208000003200 Adenoma Diseases 0.000 claims 1
- 101800002638 Alpha-amanitin Proteins 0.000 claims 1
- 208000005440 Basal Cell Neoplasms Diseases 0.000 claims 1
- 206010004146 Basal cell carcinoma Diseases 0.000 claims 1
- 206010005949 Bone cancer Diseases 0.000 claims 1
- 208000018084 Bone neoplasm Diseases 0.000 claims 1
- 101000831496 Homo sapiens Toll-like receptor 3 Proteins 0.000 claims 1
- 208000010505 Nose Neoplasms Diseases 0.000 claims 1
- 206010061336 Pelvic neoplasm Diseases 0.000 claims 1
- 241000009328 Perro Species 0.000 claims 1
- 206010057846 Primitive neuroectodermal tumour Diseases 0.000 claims 1
- 208000015634 Rectal Neoplasms Diseases 0.000 claims 1
- RXGJTYFDKOHJHK-UHFFFAOYSA-N S-deoxo-amaninamide Natural products CCC(C)C1NC(=O)CNC(=O)C2Cc3c(SCC(NC(=O)CNC1=O)C(=O)NC(CC(=O)N)C(=O)N4CC(O)CC4C(=O)NC(C(C)C(O)CO)C(=O)N2)[nH]c5ccccc35 RXGJTYFDKOHJHK-UHFFFAOYSA-N 0.000 claims 1
- 208000031673 T-Cell Cutaneous Lymphoma Diseases 0.000 claims 1
- 102100024324 Toll-like receptor 3 Human genes 0.000 claims 1
- 239000004007 alpha amanitin Substances 0.000 claims 1
- MYYARUCLXARAEE-ZATZPJRKSA-N alpha-C-GalCer Chemical compound CCCCCCCCCCCCCCCCCCCCCCCCCCC(=O)N[C@@H](CC[C@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O)[C@H](O)[C@H](O)CCCCCCCCCCCCCCCCC MYYARUCLXARAEE-ZATZPJRKSA-N 0.000 claims 1
- CIORWBWIBBPXCG-SXZCQOKQSA-N alpha-amanitin Chemical compound O=C1N[C@@H](CC(N)=O)C(=O)N2C[C@H](O)C[C@H]2C(=O)N[C@@H]([C@@H](C)[C@@H](O)CO)C(=O)N[C@@H](C2)C(=O)NCC(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@H]1C[S@@](=O)C1=C2C2=CC=C(O)C=C2N1 CIORWBWIBBPXCG-SXZCQOKQSA-N 0.000 claims 1
- CIORWBWIBBPXCG-UHFFFAOYSA-N alpha-amanitin Natural products O=C1NC(CC(N)=O)C(=O)N2CC(O)CC2C(=O)NC(C(C)C(O)CO)C(=O)NC(C2)C(=O)NCC(=O)NC(C(C)CC)C(=O)NCC(=O)NC1CS(=O)C1=C2C2=CC=C(O)C=C2N1 CIORWBWIBBPXCG-UHFFFAOYSA-N 0.000 claims 1
- 229940049706 benzodiazepine Drugs 0.000 claims 1
- POQRWMRXUOPCLD-HIIAJUEOSA-N beta-D-galactosyl-N-(tetracosanoyl)sphingosine Chemical compound CCCCCCCCCCCCCCCCCCCCCCCC(=O)N[C@H]([C@H](O)\C=C\CCCCCCCCCCCCC)CO[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O POQRWMRXUOPCLD-HIIAJUEOSA-N 0.000 claims 1
- YIGARKIIFOHVPF-CNUVFPMCSA-N beta-D-glucosyl-N-(docosanoyl)sphingosine Chemical compound CCCCCCCCCCCCCCCCCCCCCC(=O)N[C@H]([C@H](O)\C=C\CCCCCCCCCCCCC)CO[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O YIGARKIIFOHVPF-CNUVFPMCSA-N 0.000 claims 1
- 230000000112 colonic effect Effects 0.000 claims 1
- 201000007241 cutaneous T cell lymphoma Diseases 0.000 claims 1
- 208000010932 epithelial neoplasm Diseases 0.000 claims 1
- 201000009277 hairy cell leukemia Diseases 0.000 claims 1
- 210000001821 langerhans cell Anatomy 0.000 claims 1
- 210000000214 mouth Anatomy 0.000 claims 1
- 201000005962 mycosis fungoides Diseases 0.000 claims 1
- 208000037830 nasal cancer Diseases 0.000 claims 1
- 208000023974 pharynx neoplasm Diseases 0.000 claims 1
- 201000004119 pineal parenchymal tumor of intermediate differentiation Diseases 0.000 claims 1
- 208000025638 primary cutaneous T-cell non-Hodgkin lymphoma Diseases 0.000 claims 1
- 206010038038 rectal cancer Diseases 0.000 claims 1
- 201000001275 rectum cancer Diseases 0.000 claims 1
- 208000015347 renal cell adenocarcinoma Diseases 0.000 claims 1
- 210000005132 reproductive cell Anatomy 0.000 claims 1
- 210000000278 spinal cord Anatomy 0.000 claims 1
- 210000003741 urothelium Anatomy 0.000 claims 1
- 229960005502 α-amanitin Drugs 0.000 claims 1
- 238000010586 diagram Methods 0.000 abstract description 4
- 239000003814 drug Substances 0.000 description 89
- 229940079593 drug Drugs 0.000 description 88
- 238000011282 treatment Methods 0.000 description 72
- 102000001301 EGF receptor Human genes 0.000 description 56
- 108060006698 EGF receptor Proteins 0.000 description 56
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 54
- 241000699670 Mus sp. Species 0.000 description 53
- -1 anthracycline Chemical class 0.000 description 48
- 210000004443 dendritic cell Anatomy 0.000 description 43
- 210000001744 T-lymphocyte Anatomy 0.000 description 41
- 210000000822 natural killer cell Anatomy 0.000 description 40
- 102000053602 DNA Human genes 0.000 description 37
- 102000004169 proteins and genes Human genes 0.000 description 35
- 230000000694 effects Effects 0.000 description 34
- 230000004044 response Effects 0.000 description 34
- 102000005962 receptors Human genes 0.000 description 31
- 108020003175 receptors Proteins 0.000 description 31
- 238000012384 transportation and delivery Methods 0.000 description 31
- 235000018102 proteins Nutrition 0.000 description 30
- 239000003795 chemical substances by application Substances 0.000 description 28
- 230000000259 anti-tumor effect Effects 0.000 description 26
- 241000894006 Bacteria Species 0.000 description 25
- 108090000765 processed proteins & peptides Proteins 0.000 description 25
- 102000004127 Cytokines Human genes 0.000 description 23
- 108090000695 Cytokines Proteins 0.000 description 23
- 230000004913 activation Effects 0.000 description 23
- 108700011259 MicroRNAs Proteins 0.000 description 22
- 230000008685 targeting Effects 0.000 description 22
- 230000027455 binding Effects 0.000 description 21
- 229960004679 doxorubicin Drugs 0.000 description 21
- 238000009472 formulation Methods 0.000 description 21
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 20
- 239000012528 membrane Substances 0.000 description 20
- 210000002865 immune cell Anatomy 0.000 description 19
- 210000002540 macrophage Anatomy 0.000 description 19
- 230000001225 therapeutic effect Effects 0.000 description 19
- 102000000844 Cell Surface Receptors Human genes 0.000 description 18
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 17
- 239000011780 sodium chloride Substances 0.000 description 17
- 239000000427 antigen Substances 0.000 description 16
- 108091007433 antigens Proteins 0.000 description 16
- 102000036639 antigens Human genes 0.000 description 16
- 229940126586 small molecule drug Drugs 0.000 description 16
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 16
- 238000004519 manufacturing process Methods 0.000 description 15
- 230000036457 multidrug resistance Effects 0.000 description 15
- 231100000331 toxic Toxicity 0.000 description 15
- 230000002588 toxic effect Effects 0.000 description 15
- 206010036790 Productive cough Diseases 0.000 description 14
- 238000009169 immunotherapy Methods 0.000 description 14
- 230000001965 increasing effect Effects 0.000 description 14
- 210000003802 sputum Anatomy 0.000 description 14
- 208000024794 sputum Diseases 0.000 description 14
- 230000004614 tumor growth Effects 0.000 description 14
- 229940034982 antineoplastic agent Drugs 0.000 description 13
- 238000003501 co-culture Methods 0.000 description 13
- 230000006870 function Effects 0.000 description 13
- 230000001404 mediated effect Effects 0.000 description 13
- 102100031256 Cyclic GMP-AMP synthase Human genes 0.000 description 12
- 108030002637 Cyclic GMP-AMP synthases Proteins 0.000 description 12
- 206010059866 Drug resistance Diseases 0.000 description 12
- 101001046686 Homo sapiens Integrin alpha-M Proteins 0.000 description 12
- 102100022338 Integrin alpha-M Human genes 0.000 description 12
- 229930012538 Paclitaxel Natural products 0.000 description 12
- 101710196623 Stimulator of interferon genes protein Proteins 0.000 description 12
- 239000012190 activator Substances 0.000 description 12
- WPIHMWBQRSAMDE-YCZTVTEBSA-N beta-D-galactosyl-(1->4)-beta-D-galactosyl-N-(pentacosanoyl)sphingosine Chemical compound CCCCCCCCCCCCCCCCCCCCCCCCC(=O)N[C@@H](CO[C@@H]1O[C@H](CO)[C@H](O[C@@H]2O[C@H](CO)[C@H](O)[C@H](O)[C@H]2O)[C@H](O)[C@H]1O)[C@H](O)\C=C\CCCCCCCCCCCCC WPIHMWBQRSAMDE-YCZTVTEBSA-N 0.000 description 12
- 238000003384 imaging method Methods 0.000 description 12
- 230000037361 pathway Effects 0.000 description 12
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 11
- 108010092160 Dactinomycin Proteins 0.000 description 11
- 206010033799 Paralysis Diseases 0.000 description 11
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 11
- 238000001727 in vivo Methods 0.000 description 11
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 11
- 229960001592 paclitaxel Drugs 0.000 description 11
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 11
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 10
- 102100040247 Tumor necrosis factor Human genes 0.000 description 10
- 210000000805 cytoplasm Anatomy 0.000 description 10
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 10
- 230000007246 mechanism Effects 0.000 description 10
- 239000002504 physiological saline solution Substances 0.000 description 10
- 102000004196 processed proteins & peptides Human genes 0.000 description 10
- 210000000952 spleen Anatomy 0.000 description 10
- 108010051479 Bombesin Proteins 0.000 description 9
- 102000013585 Bombesin Human genes 0.000 description 9
- 102000004889 Interleukin-6 Human genes 0.000 description 9
- 108090001005 Interleukin-6 Proteins 0.000 description 9
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 9
- 206010027406 Mesothelioma Diseases 0.000 description 9
- 108010056088 Somatostatin Proteins 0.000 description 9
- DNDCVAGJPBKION-DOPDSADYSA-N bombesin Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(N)=O)NC(=O)CNC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC=1NC2=CC=CC=C2C=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1NC(=O)CC1)C(C)C)C1=CN=CN1 DNDCVAGJPBKION-DOPDSADYSA-N 0.000 description 9
- 230000001472 cytotoxic effect Effects 0.000 description 9
- 229960000640 dactinomycin Drugs 0.000 description 9
- 210000000987 immune system Anatomy 0.000 description 9
- 230000003993 interaction Effects 0.000 description 9
- 238000011068 loading method Methods 0.000 description 9
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 9
- 238000011002 quantification Methods 0.000 description 9
- 210000004981 tumor-associated macrophage Anatomy 0.000 description 9
- 108700028369 Alleles Proteins 0.000 description 8
- 101001011382 Homo sapiens Interferon regulatory factor 3 Proteins 0.000 description 8
- 102100029843 Interferon regulatory factor 3 Human genes 0.000 description 8
- 108090000189 Neuropeptides Proteins 0.000 description 8
- 102000005157 Somatostatin Human genes 0.000 description 8
- 238000010162 Tukey test Methods 0.000 description 8
- 102000055135 Vasoactive Intestinal Peptide Human genes 0.000 description 8
- 108010003205 Vasoactive Intestinal Peptide Proteins 0.000 description 8
- 229940024606 amino acid Drugs 0.000 description 8
- 230000030741 antigen processing and presentation Effects 0.000 description 8
- 238000013459 approach Methods 0.000 description 8
- 230000032823 cell division Effects 0.000 description 8
- 230000001086 cytosolic effect Effects 0.000 description 8
- 231100000433 cytotoxic Toxicity 0.000 description 8
- 230000003013 cytotoxicity Effects 0.000 description 8
- 231100000135 cytotoxicity Toxicity 0.000 description 8
- 238000011161 development Methods 0.000 description 8
- 210000001163 endosome Anatomy 0.000 description 8
- VBUWHHLIZKOSMS-RIWXPGAOSA-N invicorp Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)C(C)C)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=C(O)C=C1 VBUWHHLIZKOSMS-RIWXPGAOSA-N 0.000 description 8
- 210000001165 lymph node Anatomy 0.000 description 8
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 8
- 229960000485 methotrexate Drugs 0.000 description 8
- 230000035772 mutation Effects 0.000 description 8
- 239000013642 negative control Substances 0.000 description 8
- 238000001543 one-way ANOVA Methods 0.000 description 8
- 230000003389 potentiating effect Effects 0.000 description 8
- 230000001105 regulatory effect Effects 0.000 description 8
- NHXLMOGPVYXJNR-ATOGVRKGSA-N somatostatin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N1)[C@@H](C)O)NC(=O)CNC(=O)[C@H](C)N)C(O)=O)=O)[C@H](O)C)C1=CC=CC=C1 NHXLMOGPVYXJNR-ATOGVRKGSA-N 0.000 description 8
- 229960000553 somatostatin Drugs 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- 229960003087 tioguanine Drugs 0.000 description 8
- 239000003981 vehicle Substances 0.000 description 8
- 241000282472 Canis lupus familiaris Species 0.000 description 7
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 7
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 7
- 108090001053 Gastrin releasing peptide Proteins 0.000 description 7
- 102000004862 Gastrin releasing peptide Human genes 0.000 description 7
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 description 7
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 description 7
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 7
- 102000043131 MHC class II family Human genes 0.000 description 7
- 108091054438 MHC class II family Proteins 0.000 description 7
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 7
- 102000003797 Neuropeptides Human genes 0.000 description 7
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 7
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 150000001875 compounds Chemical class 0.000 description 7
- 230000018109 developmental process Effects 0.000 description 7
- 230000028993 immune response Effects 0.000 description 7
- 230000002163 immunogen Effects 0.000 description 7
- 230000003308 immunostimulating effect Effects 0.000 description 7
- 238000001990 intravenous administration Methods 0.000 description 7
- 230000035800 maturation Effects 0.000 description 7
- 210000000581 natural killer T-cell Anatomy 0.000 description 7
- 239000002245 particle Substances 0.000 description 7
- 239000013612 plasmid Substances 0.000 description 7
- 230000028327 secretion Effects 0.000 description 7
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 7
- 229960000303 topotecan Drugs 0.000 description 7
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 7
- 230000001988 toxicity Effects 0.000 description 7
- 231100000419 toxicity Toxicity 0.000 description 7
- 210000005166 vasculature Anatomy 0.000 description 7
- 108010024976 Asparaginase Proteins 0.000 description 6
- 102000015790 Asparaginase Human genes 0.000 description 6
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 description 6
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 description 6
- 102000019034 Chemokines Human genes 0.000 description 6
- 108010012236 Chemokines Proteins 0.000 description 6
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 6
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 6
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 6
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 6
- 102100038192 Serine/threonine-protein kinase TBK1 Human genes 0.000 description 6
- 235000001014 amino acid Nutrition 0.000 description 6
- 150000001413 amino acids Chemical class 0.000 description 6
- 230000001093 anti-cancer Effects 0.000 description 6
- 229960003272 asparaginase Drugs 0.000 description 6
- DCXYFEDJOCDNAF-UHFFFAOYSA-M asparaginate Chemical compound [O-]C(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-M 0.000 description 6
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 6
- 210000000988 bone and bone Anatomy 0.000 description 6
- 229960004562 carboplatin Drugs 0.000 description 6
- 229940044683 chemotherapy drug Drugs 0.000 description 6
- 239000000356 contaminant Substances 0.000 description 6
- 230000034994 death Effects 0.000 description 6
- 231100000517 death Toxicity 0.000 description 6
- 238000013461 design Methods 0.000 description 6
- 229960005420 etoposide Drugs 0.000 description 6
- 229960002949 fluorouracil Drugs 0.000 description 6
- PUBCCFNQJQKCNC-XKNFJVFFSA-N gastrin-releasingpeptide Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(N)=O)NC(=O)CNC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)CNC(=O)[C@H](C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(C)C)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)C(C)C)[C@@H](C)O)C(C)C)[C@@H](C)O)C(C)C)C1=CNC=N1 PUBCCFNQJQKCNC-XKNFJVFFSA-N 0.000 description 6
- 208000005017 glioblastoma Diseases 0.000 description 6
- YLMAHDNUQAMNNX-UHFFFAOYSA-N imatinib methanesulfonate Chemical compound CS(O)(=O)=O.C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 YLMAHDNUQAMNNX-UHFFFAOYSA-N 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 229960004768 irinotecan Drugs 0.000 description 6
- 229960004857 mitomycin Drugs 0.000 description 6
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 6
- 238000004806 packaging method and process Methods 0.000 description 6
- 230000003285 pharmacodynamic effect Effects 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- 230000002829 reductive effect Effects 0.000 description 6
- 239000000523 sample Substances 0.000 description 6
- 229960001278 teniposide Drugs 0.000 description 6
- 238000002560 therapeutic procedure Methods 0.000 description 6
- JXLYSJRDGCGARV-CFWMRBGOSA-N vinblastine Chemical compound C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-CFWMRBGOSA-N 0.000 description 6
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical class C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 6
- 102100033350 ATP-dependent translocase ABCB1 Human genes 0.000 description 5
- IYMAXBFPHPZYIK-BQBZGAKWSA-N Arg-Gly-Asp Chemical compound NC(N)=NCCC[C@H](N)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(O)=O IYMAXBFPHPZYIK-BQBZGAKWSA-N 0.000 description 5
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 5
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 5
- 102100032367 C-C motif chemokine 5 Human genes 0.000 description 5
- 206010057248 Cell death Diseases 0.000 description 5
- 108010055166 Chemokine CCL5 Proteins 0.000 description 5
- 101800001982 Cholecystokinin Proteins 0.000 description 5
- 102100025841 Cholecystokinin Human genes 0.000 description 5
- 102100021906 Cyclin-O Human genes 0.000 description 5
- 101000897441 Homo sapiens Cyclin-O Proteins 0.000 description 5
- 101000665442 Homo sapiens Serine/threonine-protein kinase TBK1 Proteins 0.000 description 5
- 101000643024 Homo sapiens Stimulator of interferon genes protein Proteins 0.000 description 5
- 102000013462 Interleukin-12 Human genes 0.000 description 5
- 108010065805 Interleukin-12 Proteins 0.000 description 5
- 206010059282 Metastases to central nervous system Diseases 0.000 description 5
- 229930192392 Mitomycin Natural products 0.000 description 5
- 206010033128 Ovarian cancer Diseases 0.000 description 5
- 238000012228 RNA interference-mediated gene silencing Methods 0.000 description 5
- 102100028748 Transportin-1 Human genes 0.000 description 5
- 230000033289 adaptive immune response Effects 0.000 description 5
- 229940098174 alkeran Drugs 0.000 description 5
- 230000001640 apoptogenic effect Effects 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 210000004204 blood vessel Anatomy 0.000 description 5
- 229960002092 busulfan Drugs 0.000 description 5
- 238000002619 cancer immunotherapy Methods 0.000 description 5
- 229960005243 carmustine Drugs 0.000 description 5
- 210000000170 cell membrane Anatomy 0.000 description 5
- 239000002738 chelating agent Substances 0.000 description 5
- 229940107137 cholecystokinin Drugs 0.000 description 5
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 5
- 229960004316 cisplatin Drugs 0.000 description 5
- 125000004122 cyclic group Chemical group 0.000 description 5
- 229960004397 cyclophosphamide Drugs 0.000 description 5
- 230000009089 cytolysis Effects 0.000 description 5
- 210000000172 cytosol Anatomy 0.000 description 5
- 229960003901 dacarbazine Drugs 0.000 description 5
- 230000007423 decrease Effects 0.000 description 5
- 229960005501 duocarmycin Drugs 0.000 description 5
- VQNATVDKACXKTF-XELLLNAOSA-N duocarmycin Chemical compound COC1=C(OC)C(OC)=C2NC(C(=O)N3C4=CC(=O)C5=C([C@@]64C[C@@H]6C3)C=C(N5)C(=O)OC)=CC2=C1 VQNATVDKACXKTF-XELLLNAOSA-N 0.000 description 5
- 229930184221 duocarmycin Natural products 0.000 description 5
- LIQODXNTTZAGID-OCBXBXKTSA-N etoposide phosphate Chemical compound COC1=C(OP(O)(O)=O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 LIQODXNTTZAGID-OCBXBXKTSA-N 0.000 description 5
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 5
- 229960005277 gemcitabine Drugs 0.000 description 5
- 230000009368 gene silencing by RNA Effects 0.000 description 5
- 230000002209 hydrophobic effect Effects 0.000 description 5
- 229960002411 imatinib Drugs 0.000 description 5
- 230000002519 immonomodulatory effect Effects 0.000 description 5
- 230000006872 improvement Effects 0.000 description 5
- 230000001939 inductive effect Effects 0.000 description 5
- 229940100601 interleukin-6 Drugs 0.000 description 5
- 230000003834 intracellular effect Effects 0.000 description 5
- 229960001428 mercaptopurine Drugs 0.000 description 5
- 229960001156 mitoxantrone Drugs 0.000 description 5
- 210000001616 monocyte Anatomy 0.000 description 5
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 5
- 210000001539 phagocyte Anatomy 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 230000005855 radiation Effects 0.000 description 5
- IZTQOLKUZKXIRV-YRVFCXMDSA-N sincalide Chemical compound C([C@@H](C(=O)N[C@@H](CCSC)C(=O)NCC(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](N)CC(O)=O)C1=CC=C(OS(O)(=O)=O)C=C1 IZTQOLKUZKXIRV-YRVFCXMDSA-N 0.000 description 5
- 239000000243 solution Substances 0.000 description 5
- 239000006228 supernatant Substances 0.000 description 5
- 230000009885 systemic effect Effects 0.000 description 5
- 238000011269 treatment regimen Methods 0.000 description 5
- 229960003048 vinblastine Drugs 0.000 description 5
- 229960004528 vincristine Drugs 0.000 description 5
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 5
- 229960004276 zoledronic acid Drugs 0.000 description 5
- PNDPGZBMCMUPRI-HVTJNCQCSA-N 10043-66-0 Chemical compound [131I][131I] PNDPGZBMCMUPRI-HVTJNCQCSA-N 0.000 description 4
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 4
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 4
- 238000011729 BALB/c nude mouse Methods 0.000 description 4
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 4
- 229940122361 Bisphosphonate Drugs 0.000 description 4
- 108010089448 Cholecystokinin B Receptor Proteins 0.000 description 4
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 4
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 4
- 241000588724 Escherichia coli Species 0.000 description 4
- VJLLLMIZEJJZTE-VNQXHBPZSA-N HexCer(d18:1/16:0) Chemical compound CCCCCCCCCCCCCCCC(=O)N[C@H]([C@H](O)\C=C\CCCCCCCCCCCCC)COC1OC(CO)C(O)C(O)C1O VJLLLMIZEJJZTE-VNQXHBPZSA-N 0.000 description 4
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 4
- 101000852870 Homo sapiens Interferon alpha/beta receptor 1 Proteins 0.000 description 4
- 101000917858 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-A Proteins 0.000 description 4
- 101000917839 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-B Proteins 0.000 description 4
- 101001109501 Homo sapiens NKG2-D type II integral membrane protein Proteins 0.000 description 4
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 description 4
- 102100036714 Interferon alpha/beta receptor 1 Human genes 0.000 description 4
- 102000003814 Interleukin-10 Human genes 0.000 description 4
- 108090000174 Interleukin-10 Proteins 0.000 description 4
- 108010002350 Interleukin-2 Proteins 0.000 description 4
- 102000015696 Interleukins Human genes 0.000 description 4
- 108010063738 Interleukins Proteins 0.000 description 4
- ZCYVEMRRCGMTRW-AHCXROLUSA-N Iodine-123 Chemical compound [123I] ZCYVEMRRCGMTRW-AHCXROLUSA-N 0.000 description 4
- 102100029185 Low affinity immunoglobulin gamma Fc region receptor III-B Human genes 0.000 description 4
- 102100034216 Melanocyte-stimulating hormone receptor Human genes 0.000 description 4
- 206010027476 Metastases Diseases 0.000 description 4
- 102100022680 NKG2-D type II integral membrane protein Human genes 0.000 description 4
- 102000002002 Neurokinin-1 Receptors Human genes 0.000 description 4
- 108010040718 Neurokinin-1 Receptors Proteins 0.000 description 4
- 108091008606 PDGF receptors Proteins 0.000 description 4
- 102000011653 Platelet-Derived Growth Factor Receptors Human genes 0.000 description 4
- 102100040678 Programmed cell death protein 1 Human genes 0.000 description 4
- 108010078067 RNA Polymerase III Proteins 0.000 description 4
- 102000014450 RNA Polymerase III Human genes 0.000 description 4
- 108091028664 Ribonucleotide Proteins 0.000 description 4
- 108050001286 Somatostatin Receptor Proteins 0.000 description 4
- 102000011096 Somatostatin receptor Human genes 0.000 description 4
- DPOPAJRDYZGTIR-UHFFFAOYSA-N Tetrazine Chemical compound C1=CN=NN=N1 DPOPAJRDYZGTIR-UHFFFAOYSA-N 0.000 description 4
- 108010021428 Type 1 Melanocortin Receptor Proteins 0.000 description 4
- 108091008605 VEGF receptors Proteins 0.000 description 4
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 description 4
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical class OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 229960002932 anastrozole Drugs 0.000 description 4
- 230000005975 antitumor immune response Effects 0.000 description 4
- 229940108502 bicnu Drugs 0.000 description 4
- 230000033228 biological regulation Effects 0.000 description 4
- 150000004663 bisphosphonates Chemical class 0.000 description 4
- 208000002352 blister Diseases 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 230000021164 cell adhesion Effects 0.000 description 4
- 238000002659 cell therapy Methods 0.000 description 4
- 230000003833 cell viability Effects 0.000 description 4
- 229960002436 cladribine Drugs 0.000 description 4
- 230000000295 complement effect Effects 0.000 description 4
- 230000016396 cytokine production Effects 0.000 description 4
- 230000001419 dependent effect Effects 0.000 description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 4
- 238000002296 dynamic light scattering Methods 0.000 description 4
- 230000012202 endocytosis Effects 0.000 description 4
- 229960001904 epirubicin Drugs 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 4
- 229930004094 glycosylphosphatidylinositol Natural products 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 239000003102 growth factor Substances 0.000 description 4
- 102000050022 human STING1 Human genes 0.000 description 4
- 229960001101 ifosfamide Drugs 0.000 description 4
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 4
- 230000005934 immune activation Effects 0.000 description 4
- 230000005931 immune cell recruitment Effects 0.000 description 4
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 210000005007 innate immune system Anatomy 0.000 description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 4
- 102000006495 integrins Human genes 0.000 description 4
- 108010044426 integrins Proteins 0.000 description 4
- 229940047122 interleukins Drugs 0.000 description 4
- 238000002372 labelling Methods 0.000 description 4
- 210000000265 leukocyte Anatomy 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- OHSVLFRHMCKCQY-NJFSPNSNSA-N lutetium-177 Chemical compound [177Lu] OHSVLFRHMCKCQY-NJFSPNSNSA-N 0.000 description 4
- 210000003712 lysosome Anatomy 0.000 description 4
- 230000001868 lysosomic effect Effects 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 229960001924 melphalan Drugs 0.000 description 4
- 108020004999 messenger RNA Proteins 0.000 description 4
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 4
- 201000011519 neuroendocrine tumor Diseases 0.000 description 4
- 210000004940 nucleus Anatomy 0.000 description 4
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 4
- 229960001756 oxaliplatin Drugs 0.000 description 4
- 230000035515 penetration Effects 0.000 description 4
- 229960003171 plicamycin Drugs 0.000 description 4
- 230000010287 polarization Effects 0.000 description 4
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 4
- 229960000624 procarbazine Drugs 0.000 description 4
- XJMOSONTPMZWPB-UHFFFAOYSA-M propidium iodide Chemical compound [I-].[I-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CCC[N+](C)(CC)CC)=C1C1=CC=CC=C1 XJMOSONTPMZWPB-UHFFFAOYSA-M 0.000 description 4
- 230000002285 radioactive effect Effects 0.000 description 4
- 230000008707 rearrangement Effects 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 239000002336 ribonucleotide Substances 0.000 description 4
- 125000002652 ribonucleotide group Chemical group 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- 238000002603 single-photon emission computed tomography Methods 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- 229940124676 vascular endothelial growth factor receptor Drugs 0.000 description 4
- 230000035899 viability Effects 0.000 description 4
- JBHPLHATEXGMQR-LFWIOBPJSA-N vipivotide tetraxetan Chemical compound OC(=O)CC[C@H](NC(=O)N[C@@H](CCCCNC(=O)[C@H](CC1=CC=C2C=CC=CC2=C1)NC(=O)[C@H]1CC[C@H](CNC(=O)CN2CCN(CC(O)=O)CCN(CC(O)=O)CCN(CC(O)=O)CC2)CC1)C(O)=O)C(O)=O JBHPLHATEXGMQR-LFWIOBPJSA-N 0.000 description 4
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 3
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 3
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 description 3
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 3
- 206010002329 Aneurysm Diseases 0.000 description 3
- 108010006654 Bleomycin Proteins 0.000 description 3
- 108010073466 Bombesin Receptors Proteins 0.000 description 3
- 102100031650 C-X-C chemokine receptor type 4 Human genes 0.000 description 3
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 3
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 3
- 206010010356 Congenital anomaly Diseases 0.000 description 3
- 230000004543 DNA replication Effects 0.000 description 3
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 3
- 238000002965 ELISA Methods 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 102400000921 Gastrin Human genes 0.000 description 3
- 108010052343 Gastrins Proteins 0.000 description 3
- 102000005720 Glutathione transferase Human genes 0.000 description 3
- 108010070675 Glutathione transferase Proteins 0.000 description 3
- 101000922348 Homo sapiens C-X-C chemokine receptor type 4 Proteins 0.000 description 3
- 101001034652 Homo sapiens Insulin-like growth factor 1 receptor Proteins 0.000 description 3
- 101000959820 Homo sapiens Interferon alpha-1/13 Proteins 0.000 description 3
- 101001032342 Homo sapiens Interferon regulatory factor 7 Proteins 0.000 description 3
- 101000815628 Homo sapiens Regulatory-associated protein of mTOR Proteins 0.000 description 3
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 3
- 101000652747 Homo sapiens Target of rapamycin complex 2 subunit MAPKAP1 Proteins 0.000 description 3
- 101000648491 Homo sapiens Transportin-1 Proteins 0.000 description 3
- 101000956263 Homo sapiens Uncharacterized protein C19orf48 Proteins 0.000 description 3
- 102100039688 Insulin-like growth factor 1 receptor Human genes 0.000 description 3
- 102100040019 Interferon alpha-1/13 Human genes 0.000 description 3
- 102100037850 Interferon gamma Human genes 0.000 description 3
- 102100038070 Interferon regulatory factor 7 Human genes 0.000 description 3
- 102100020793 Interleukin-13 receptor subunit alpha-2 Human genes 0.000 description 3
- 108090000978 Interleukin-4 Proteins 0.000 description 3
- 102000004388 Interleukin-4 Human genes 0.000 description 3
- 231100000111 LD50 Toxicity 0.000 description 3
- 108010047230 Member 1 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 3
- 206010027452 Metastases to bone Diseases 0.000 description 3
- 102000010168 Myeloid Differentiation Factor 88 Human genes 0.000 description 3
- 108010077432 Myeloid Differentiation Factor 88 Proteins 0.000 description 3
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 3
- 108010037516 PSMA-617 Proteins 0.000 description 3
- 108091000080 Phosphotransferase Proteins 0.000 description 3
- 101710179684 Poly [ADP-ribose] polymerase Proteins 0.000 description 3
- 101710128017 Polyribonucleotide nucleotidyltransferase 1, mitochondrial Proteins 0.000 description 3
- 102100034410 Polyribonucleotide nucleotidyltransferase 1, mitochondrial Human genes 0.000 description 3
- 238000000692 Student's t-test Methods 0.000 description 3
- 230000005867 T cell response Effects 0.000 description 3
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 3
- GKLVYJBZJHMRIY-OUBTZVSYSA-N Technetium-99 Chemical compound [99Tc] GKLVYJBZJHMRIY-OUBTZVSYSA-N 0.000 description 3
- 108091046869 Telomeric non-coding RNA Proteins 0.000 description 3
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 3
- 229910052771 Terbium Inorganic materials 0.000 description 3
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 3
- 102000002689 Toll-like receptor Human genes 0.000 description 3
- 108020000411 Toll-like receptor Proteins 0.000 description 3
- 102100038573 Uncharacterized protein C19orf48 Human genes 0.000 description 3
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 3
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 3
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 3
- 229940122803 Vinca alkaloid Drugs 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 239000013543 active substance Substances 0.000 description 3
- 230000003044 adaptive effect Effects 0.000 description 3
- 229940009456 adriamycin Drugs 0.000 description 3
- 229960000473 altretamine Drugs 0.000 description 3
- 230000005809 anti-tumor immunity Effects 0.000 description 3
- 210000000612 antigen-presenting cell Anatomy 0.000 description 3
- 229940041181 antineoplastic drug Drugs 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 229960002756 azacitidine Drugs 0.000 description 3
- 210000003719 b-lymphocyte Anatomy 0.000 description 3
- 229960001561 bleomycin Drugs 0.000 description 3
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 3
- 230000008499 blood brain barrier function Effects 0.000 description 3
- 210000001218 blood-brain barrier Anatomy 0.000 description 3
- 210000001185 bone marrow Anatomy 0.000 description 3
- 229960000455 brentuximab vedotin Drugs 0.000 description 3
- 230000030833 cell death Effects 0.000 description 3
- 230000010261 cell growth Effects 0.000 description 3
- 230000036755 cellular response Effects 0.000 description 3
- 238000005119 centrifugation Methods 0.000 description 3
- 229960005395 cetuximab Drugs 0.000 description 3
- 210000003690 classically activated macrophage Anatomy 0.000 description 3
- 238000011284 combination treatment Methods 0.000 description 3
- 238000009295 crossflow filtration Methods 0.000 description 3
- 229960000684 cytarabine Drugs 0.000 description 3
- 230000001461 cytolytic effect Effects 0.000 description 3
- 239000002254 cytotoxic agent Substances 0.000 description 3
- 230000004041 dendritic cell maturation Effects 0.000 description 3
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 230000009977 dual effect Effects 0.000 description 3
- 239000012636 effector Substances 0.000 description 3
- INVTYAOGFAGBOE-UHFFFAOYSA-N entinostat Chemical compound NC1=CC=CC=C1NC(=O)C(C=C1)=CC=C1CNC(=O)OCC1=CC=CN=C1 INVTYAOGFAGBOE-UHFFFAOYSA-N 0.000 description 3
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 3
- 238000000684 flow cytometry Methods 0.000 description 3
- 229960002074 flutamide Drugs 0.000 description 3
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 3
- 150000002270 gangliosides Chemical class 0.000 description 3
- 102000006602 glyceraldehyde-3-phosphate dehydrogenase Human genes 0.000 description 3
- 108020004445 glyceraldehyde-3-phosphate dehydrogenase Proteins 0.000 description 3
- VKYKSIONXSXAKP-UHFFFAOYSA-N hexamethylenetetramine Chemical compound C1N(C2)CN3CN1CN2C3 VKYKSIONXSXAKP-UHFFFAOYSA-N 0.000 description 3
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 3
- 239000002955 immunomodulating agent Substances 0.000 description 3
- 230000001506 immunosuppresive effect Effects 0.000 description 3
- 230000001976 improved effect Effects 0.000 description 3
- APFVFJFRJDLVQX-AHCXROLUSA-N indium-111 Chemical compound [111In] APFVFJFRJDLVQX-AHCXROLUSA-N 0.000 description 3
- 208000015181 infectious disease Diseases 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 230000015788 innate immune response Effects 0.000 description 3
- 230000017306 interleukin-6 production Effects 0.000 description 3
- 230000002601 intratumoral effect Effects 0.000 description 3
- 229960005108 mepolizumab Drugs 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 230000002018 overexpression Effects 0.000 description 3
- 229960002340 pentostatin Drugs 0.000 description 3
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 239000002953 phosphate buffered saline Substances 0.000 description 3
- 102000020233 phosphotransferase Human genes 0.000 description 3
- 230000001766 physiological effect Effects 0.000 description 3
- 229920001184 polypeptide Polymers 0.000 description 3
- 238000002600 positron emission tomography Methods 0.000 description 3
- 230000035755 proliferation Effects 0.000 description 3
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 3
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 3
- 229960004641 rituximab Drugs 0.000 description 3
- 229960004540 secukinumab Drugs 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 230000011664 signaling Effects 0.000 description 3
- 230000002195 synergetic effect Effects 0.000 description 3
- 238000012385 systemic delivery Methods 0.000 description 3
- 238000012353 t test Methods 0.000 description 3
- GZCRRIHWUXGPOV-UHFFFAOYSA-N terbium atom Chemical compound [Tb] GZCRRIHWUXGPOV-UHFFFAOYSA-N 0.000 description 3
- 229960005267 tositumomab Drugs 0.000 description 3
- 238000013518 transcription Methods 0.000 description 3
- 229960000575 trastuzumab Drugs 0.000 description 3
- 229960001099 trimetrexate Drugs 0.000 description 3
- NOYPYLRCIDNJJB-UHFFFAOYSA-N trimetrexate Chemical compound COC1=C(OC)C(OC)=CC(NCC=2C(=C3C(N)=NC(N)=NC3=CC=2)C)=C1 NOYPYLRCIDNJJB-UHFFFAOYSA-N 0.000 description 3
- 230000014567 type I interferon production Effects 0.000 description 3
- 230000003827 upregulation Effects 0.000 description 3
- 239000013598 vector Substances 0.000 description 3
- XRASPMIURGNCCH-UHFFFAOYSA-N zoledronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CN1C=CN=C1 XRASPMIURGNCCH-UHFFFAOYSA-N 0.000 description 3
- ARLKVQYMFRECLV-JSGCOSHPSA-N (2s)-2-[[(2s)-2-amino-3-(1h-indol-3-yl)propanoyl]amino]-4-methylsulfanylbutanamide Chemical compound C1=CC=C2C(C[C@H](N)C(=O)N[C@@H](CCSC)C(N)=O)=CNC2=C1 ARLKVQYMFRECLV-JSGCOSHPSA-N 0.000 description 2
- RQFCJASXJCIDSX-UHFFFAOYSA-N 14C-Guanosin-5'-monophosphat Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(COP(O)(O)=O)C(O)C1O RQFCJASXJCIDSX-UHFFFAOYSA-N 0.000 description 2
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 2
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 2
- RXACEEPNTRHYBQ-UHFFFAOYSA-N 2-[[2-[[2-[(2-sulfanylacetyl)amino]acetyl]amino]acetyl]amino]acetic acid Chemical compound OC(=O)CNC(=O)CNC(=O)CNC(=O)CS RXACEEPNTRHYBQ-UHFFFAOYSA-N 0.000 description 2
- RGHYDLZMTYDBDT-UHFFFAOYSA-N 2-amino-8-ethyl-4-methyl-6-(1H-pyrazol-5-yl)-7-pyrido[2,3-d]pyrimidinone Chemical compound O=C1N(CC)C2=NC(N)=NC(C)=C2C=C1C=1C=CNN=1 RGHYDLZMTYDBDT-UHFFFAOYSA-N 0.000 description 2
- IGRZXNLKVUEFDM-UHFFFAOYSA-N 2-phenyl-N-(5-propan-2-yl-2-thiazolyl)acetamide Chemical compound S1C(C(C)C)=CN=C1NC(=O)CC1=CC=CC=C1 IGRZXNLKVUEFDM-UHFFFAOYSA-N 0.000 description 2
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 2
- MDOJTZQKHMAPBK-UHFFFAOYSA-N 4-iodo-3-nitrobenzamide Chemical compound NC(=O)C1=CC=C(I)C([N+]([O-])=O)=C1 MDOJTZQKHMAPBK-UHFFFAOYSA-N 0.000 description 2
- 108060000255 AIM2 Proteins 0.000 description 2
- MNFPZBOQEWMBOK-UHFFFAOYSA-N AS-I-145 Chemical compound C1=CC=CC2=C(CCCl)C(NC(=O)C3=CC=4C=C(C(=C(OC)C=4N3)OC)OC)=CC(N)=C21 MNFPZBOQEWMBOK-UHFFFAOYSA-N 0.000 description 2
- 102100030088 ATP-dependent RNA helicase A Human genes 0.000 description 2
- 108010027164 Amanitins Proteins 0.000 description 2
- 206010061424 Anal cancer Diseases 0.000 description 2
- 208000007860 Anus Neoplasms Diseases 0.000 description 2
- 206010003571 Astrocytoma Diseases 0.000 description 2
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 2
- 101710089792 BAG family molecular chaperone regulator 1 Proteins 0.000 description 2
- 102100037152 BAG family molecular chaperone regulator 1 Human genes 0.000 description 2
- 244000063299 Bacillus subtilis Species 0.000 description 2
- 235000014469 Bacillus subtilis Nutrition 0.000 description 2
- 102100035875 C-C chemokine receptor type 5 Human genes 0.000 description 2
- 101710149870 C-C chemokine receptor type 5 Proteins 0.000 description 2
- OBMZMSLWNNWEJA-XNCRXQDQSA-N C1=CC=2C(C[C@@H]3NC(=O)[C@@H](NC(=O)[C@H](NC(=O)N(CC#CCN(CCCC[C@H](NC(=O)[C@@H](CC4=CC=CC=C4)NC3=O)C(=O)N)CC=C)NC(=O)[C@@H](N)C)CC3=CNC4=C3C=CC=C4)C)=CNC=2C=C1 Chemical compound C1=CC=2C(C[C@@H]3NC(=O)[C@@H](NC(=O)[C@H](NC(=O)N(CC#CCN(CCCC[C@H](NC(=O)[C@@H](CC4=CC=CC=C4)NC3=O)C(=O)N)CC=C)NC(=O)[C@@H](N)C)CC3=CNC4=C3C=CC=C4)C)=CNC=2C=C1 OBMZMSLWNNWEJA-XNCRXQDQSA-N 0.000 description 2
- 102000008203 CTLA-4 Antigen Human genes 0.000 description 2
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 2
- 208000005623 Carcinogenesis Diseases 0.000 description 2
- 102000014914 Carrier Proteins Human genes 0.000 description 2
- 102000016362 Catenins Human genes 0.000 description 2
- 108010067316 Catenins Proteins 0.000 description 2
- 108090000266 Cyclin-dependent kinases Proteins 0.000 description 2
- 102000003903 Cyclin-dependent kinases Human genes 0.000 description 2
- 108010037462 Cyclooxygenase 2 Proteins 0.000 description 2
- 101710192476 Cytoplasmic polyadenylation element-binding protein Proteins 0.000 description 2
- 102100035186 DNA excision repair protein ERCC-1 Human genes 0.000 description 2
- 241000194033 Enterococcus Species 0.000 description 2
- 206010015866 Extravasation Diseases 0.000 description 2
- 108010029961 Filgrastim Proteins 0.000 description 2
- GYHNNYVSQQEPJS-OIOBTWANSA-N Gallium-67 Chemical compound [67Ga] GYHNNYVSQQEPJS-OIOBTWANSA-N 0.000 description 2
- 208000031448 Genomic Instability Diseases 0.000 description 2
- 206010018338 Glioma Diseases 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 239000000579 Gonadotropin-Releasing Hormone Substances 0.000 description 2
- 102100039619 Granulocyte colony-stimulating factor Human genes 0.000 description 2
- 102000009465 Growth Factor Receptors Human genes 0.000 description 2
- 108010009202 Growth Factor Receptors Proteins 0.000 description 2
- 102000006354 HLA-DR Antigens Human genes 0.000 description 2
- 108010058597 HLA-DR Antigens Proteins 0.000 description 2
- 102000002812 Heat-Shock Proteins Human genes 0.000 description 2
- 108010004889 Heat-Shock Proteins Proteins 0.000 description 2
- 101000864670 Homo sapiens ATP-dependent RNA helicase A Proteins 0.000 description 2
- 101001017818 Homo sapiens ATP-dependent translocase ABCB1 Proteins 0.000 description 2
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 2
- 101000876529 Homo sapiens DNA excision repair protein ERCC-1 Proteins 0.000 description 2
- 101000598002 Homo sapiens Interferon regulatory factor 1 Proteins 0.000 description 2
- 101001139134 Homo sapiens Krueppel-like factor 4 Proteins 0.000 description 2
- 101001017828 Homo sapiens Leucine-rich repeat flightless-interacting protein 1 Proteins 0.000 description 2
- 101001030069 Homo sapiens Major vault protein Proteins 0.000 description 2
- 101000972282 Homo sapiens Mucin-5AC Proteins 0.000 description 2
- 101000972276 Homo sapiens Mucin-5B Proteins 0.000 description 2
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 description 2
- 101000874165 Homo sapiens Probable ATP-dependent RNA helicase DDX41 Proteins 0.000 description 2
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 2
- 101000661459 Homo sapiens Tyrosine-protein kinase STYK1 Proteins 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- VSNHCAURESNICA-UHFFFAOYSA-N Hydroxyurea Chemical compound NC(=O)NO VSNHCAURESNICA-UHFFFAOYSA-N 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- 108010034143 Inflammasomes Proteins 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 108090001061 Insulin Proteins 0.000 description 2
- 102000004877 Insulin Human genes 0.000 description 2
- 102100036981 Interferon regulatory factor 1 Human genes 0.000 description 2
- 108010047761 Interferon-alpha Proteins 0.000 description 2
- 102000006992 Interferon-alpha Human genes 0.000 description 2
- 102100024064 Interferon-inducible protein AIM2 Human genes 0.000 description 2
- 108090000176 Interleukin-13 Proteins 0.000 description 2
- 102000003816 Interleukin-13 Human genes 0.000 description 2
- 101710112634 Interleukin-13 receptor subunit alpha-2 Proteins 0.000 description 2
- 102100020677 Krueppel-like factor 4 Human genes 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 2
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 2
- 102100033303 Leucine-rich repeat flightless-interacting protein 1 Human genes 0.000 description 2
- 102100038884 Major vault protein Human genes 0.000 description 2
- 239000000637 Melanocyte-Stimulating Hormone Substances 0.000 description 2
- 108010007013 Melanocyte-Stimulating Hormones Proteins 0.000 description 2
- 101710151321 Melanostatin Proteins 0.000 description 2
- 108010052285 Membrane Proteins Proteins 0.000 description 2
- 102000018697 Membrane Proteins Human genes 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 102100023727 Mitochondrial antiviral-signaling protein Human genes 0.000 description 2
- 101710142315 Mitochondrial antiviral-signaling protein Proteins 0.000 description 2
- 102100026888 Mitogen-activated protein kinase kinase kinase 7 Human genes 0.000 description 2
- 102100022494 Mucin-5B Human genes 0.000 description 2
- 101100335081 Mus musculus Flt3 gene Proteins 0.000 description 2
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 description 2
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 2
- 230000006051 NK cell activation Effects 0.000 description 2
- 206010061309 Neoplasm progression Diseases 0.000 description 2
- 102100037283 Neuromedin-B receptor Human genes 0.000 description 2
- 102400000064 Neuropeptide Y Human genes 0.000 description 2
- 102000007399 Nuclear hormone receptor Human genes 0.000 description 2
- 102000004316 Oxidoreductases Human genes 0.000 description 2
- 108090000854 Oxidoreductases Proteins 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 101710176384 Peptide 1 Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 235000008331 Pinus X rigitaeda Nutrition 0.000 description 2
- 235000011613 Pinus brutia Nutrition 0.000 description 2
- 241000018646 Pinus brutia Species 0.000 description 2
- 102100035727 Probable ATP-dependent RNA helicase DDX41 Human genes 0.000 description 2
- 101710089372 Programmed cell death protein 1 Proteins 0.000 description 2
- 102100038280 Prostaglandin G/H synthase 2 Human genes 0.000 description 2
- 101710120463 Prostate stem cell antigen Proteins 0.000 description 2
- 102100036735 Prostate stem cell antigen Human genes 0.000 description 2
- 102000000574 RNA-Induced Silencing Complex Human genes 0.000 description 2
- 108010016790 RNA-Induced Silencing Complex Proteins 0.000 description 2
- 101100247004 Rattus norvegicus Qsox1 gene Proteins 0.000 description 2
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 2
- 101710100969 Receptor tyrosine-protein kinase erbB-3 Proteins 0.000 description 2
- 102100029986 Receptor tyrosine-protein kinase erbB-3 Human genes 0.000 description 2
- 108020004511 Recombinant DNA Proteins 0.000 description 2
- IGLNJRXAVVLDKE-OIOBTWANSA-N Rubidium-82 Chemical compound [82Rb] IGLNJRXAVVLDKE-OIOBTWANSA-N 0.000 description 2
- 101000857870 Squalus acanthias Gonadoliberin Proteins 0.000 description 2
- 108010091105 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 2
- 102000018075 Subfamily B ATP Binding Cassette Transporter Human genes 0.000 description 2
- 108010025037 T140 peptide Proteins 0.000 description 2
- 229940124613 TLR 7/8 agonist Drugs 0.000 description 2
- 101150011375 Tab2 gene Proteins 0.000 description 2
- 210000000447 Th1 cell Anatomy 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- 102000008235 Toll-Like Receptor 9 Human genes 0.000 description 2
- 108010060818 Toll-Like Receptor 9 Proteins 0.000 description 2
- 102000040945 Transcription factor Human genes 0.000 description 2
- 108091023040 Transcription factor Proteins 0.000 description 2
- 102000004243 Tubulin Human genes 0.000 description 2
- 108090000704 Tubulin Proteins 0.000 description 2
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 2
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 2
- 102100037781 Tyrosine-protein kinase STYK1 Human genes 0.000 description 2
- 102100036976 X-ray repair cross-complementing protein 6 Human genes 0.000 description 2
- 101710124907 X-ray repair cross-complementing protein 6 Proteins 0.000 description 2
- VWQVUPCCIRVNHF-OUBTZVSYSA-N Yttrium-90 Chemical compound [90Y] VWQVUPCCIRVNHF-OUBTZVSYSA-N 0.000 description 2
- QVXFGVVYTKZLJN-KHPPLWFESA-N [(z)-hexadec-7-enyl] acetate Chemical compound CCCCCCCC\C=C/CCCCCCOC(C)=O QVXFGVVYTKZLJN-KHPPLWFESA-N 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 230000008649 adaptation response Effects 0.000 description 2
- 208000037844 advanced solid tumor Diseases 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 150000003797 alkaloid derivatives Chemical class 0.000 description 2
- 230000000735 allogeneic effect Effects 0.000 description 2
- CIORWBWIBBPXCG-JZTFPUPKSA-N amanitin Chemical compound O=C1N[C@@H](CC(N)=O)C(=O)N2CC(O)C[C@H]2C(=O)N[C@@H](C(C)[C@@H](O)CO)C(=O)N[C@@H](C2)C(=O)NCC(=O)N[C@@H](C(C)CC)C(=O)NCC(=O)N[C@H]1CS(=O)C1=C2C2=CC=C(O)C=C2N1 CIORWBWIBBPXCG-JZTFPUPKSA-N 0.000 description 2
- 229960003437 aminoglutethimide Drugs 0.000 description 2
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 description 2
- 239000003098 androgen Substances 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 230000000692 anti-sense effect Effects 0.000 description 2
- 229940045985 antineoplastic platinum compound Drugs 0.000 description 2
- 239000000074 antisense oligonucleotide Substances 0.000 description 2
- 238000012230 antisense oligonucleotides Methods 0.000 description 2
- 201000011165 anus cancer Diseases 0.000 description 2
- 150000008209 arabinosides Chemical class 0.000 description 2
- 229940078010 arimidex Drugs 0.000 description 2
- 108091008324 binding proteins Proteins 0.000 description 2
- 230000031018 biological processes and functions Effects 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 229940088954 camptosar Drugs 0.000 description 2
- 230000036952 cancer formation Effects 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 231100000504 carcinogenesis Toxicity 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 230000020411 cell activation Effects 0.000 description 2
- 230000003915 cell function Effects 0.000 description 2
- 210000002421 cell wall Anatomy 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- AOXOCDRNSPFDPE-UKEONUMOSA-N chembl413654 Chemical compound C([C@H](C(=O)NCC(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](C)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)CNC(=O)[C@@H](N)CCC(O)=O)C1=CC=C(O)C=C1 AOXOCDRNSPFDPE-UKEONUMOSA-N 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 230000014564 chemokine production Effects 0.000 description 2
- 230000002759 chromosomal effect Effects 0.000 description 2
- 210000000349 chromosome Anatomy 0.000 description 2
- 230000024321 chromosome segregation Effects 0.000 description 2
- 229950009003 cilengitide Drugs 0.000 description 2
- AMLYAMJWYAIXIA-VWNVYAMZSA-N cilengitide Chemical compound N1C(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](C(C)C)N(C)C(=O)[C@H]1CC1=CC=CC=C1 AMLYAMJWYAIXIA-VWNVYAMZSA-N 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 230000021615 conjugation Effects 0.000 description 2
- 210000002808 connective tissue Anatomy 0.000 description 2
- 239000002872 contrast media Substances 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 229940088547 cosmegen Drugs 0.000 description 2
- 102000003675 cytokine receptors Human genes 0.000 description 2
- 108010057085 cytokine receptors Proteins 0.000 description 2
- 108091007930 cytoplasmic receptors Proteins 0.000 description 2
- 229960002448 dasatinib Drugs 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 238000002716 delivery method Methods 0.000 description 2
- 229960001251 denosumab Drugs 0.000 description 2
- 238000000432 density-gradient centrifugation Methods 0.000 description 2
- NIJJYAXOARWZEE-UHFFFAOYSA-N di-n-propyl-acetic acid Natural products CCCC(C(O)=O)CCC NIJJYAXOARWZEE-UHFFFAOYSA-N 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- 238000001085 differential centrifugation Methods 0.000 description 2
- 238000009792 diffusion process Methods 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 229960003668 docetaxel Drugs 0.000 description 2
- 229940115080 doxil Drugs 0.000 description 2
- 239000000890 drug combination Substances 0.000 description 2
- 238000012377 drug delivery Methods 0.000 description 2
- 238000009510 drug design Methods 0.000 description 2
- 235000013399 edible fruits Nutrition 0.000 description 2
- 238000001493 electron microscopy Methods 0.000 description 2
- 229950005837 entinostat Drugs 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 2
- 229940011871 estrogen Drugs 0.000 description 2
- 239000000262 estrogen Substances 0.000 description 2
- 230000029142 excretion Effects 0.000 description 2
- 230000036251 extravasation Effects 0.000 description 2
- 229950008085 figitumumab Drugs 0.000 description 2
- 229960004177 filgrastim Drugs 0.000 description 2
- 102000006815 folate receptor Human genes 0.000 description 2
- 108020005243 folate receptor Proteins 0.000 description 2
- 229960000304 folic acid Drugs 0.000 description 2
- 235000019152 folic acid Nutrition 0.000 description 2
- 239000011724 folic acid Substances 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 229940006110 gallium-67 Drugs 0.000 description 2
- 201000011243 gastrointestinal stromal tumor Diseases 0.000 description 2
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 2
- 230000030279 gene silencing Effects 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- XLXSAKCOAKORKW-AQJXLSMYSA-N gonadorelin Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 XLXSAKCOAKORKW-AQJXLSMYSA-N 0.000 description 2
- 229940035638 gonadotropin-releasing hormone Drugs 0.000 description 2
- RQFCJASXJCIDSX-UUOKFMHZSA-N guanosine 5'-monophosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H]1O RQFCJASXJCIDSX-UUOKFMHZSA-N 0.000 description 2
- 210000002443 helper t lymphocyte Anatomy 0.000 description 2
- 229940022353 herceptin Drugs 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 229960001330 hydroxycarbamide Drugs 0.000 description 2
- 229960002751 imiquimod Drugs 0.000 description 2
- DOUYETYNHWVLEO-UHFFFAOYSA-N imiquimod Chemical compound C1=CC=CC2=C3N(CC(C)C)C=NC3=C(N)N=C21 DOUYETYNHWVLEO-UHFFFAOYSA-N 0.000 description 2
- 230000001024 immunotherapeutic effect Effects 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 229940055742 indium-111 Drugs 0.000 description 2
- 239000000411 inducer Substances 0.000 description 2
- 230000008595 infiltration Effects 0.000 description 2
- 238000001764 infiltration Methods 0.000 description 2
- 230000002757 inflammatory effect Effects 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 229950002133 iniparib Drugs 0.000 description 2
- 229940125396 insulin Drugs 0.000 description 2
- 229940076144 interleukin-10 Drugs 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000010255 intramuscular injection Methods 0.000 description 2
- 239000007927 intramuscular injection Substances 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 238000010253 intravenous injection Methods 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 230000002147 killing effect Effects 0.000 description 2
- 208000032839 leukemia Diseases 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 210000004698 lymphocyte Anatomy 0.000 description 2
- 229920002521 macromolecule Polymers 0.000 description 2
- 230000036210 malignancy Effects 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 230000009401 metastasis Effects 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- 238000013508 migration Methods 0.000 description 2
- ZAHQPTJLOCWVPG-UHFFFAOYSA-N mitoxantrone dihydrochloride Chemical compound Cl.Cl.O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO ZAHQPTJLOCWVPG-UHFFFAOYSA-N 0.000 description 2
- 108090000743 multidrug resistance protein 3 Proteins 0.000 description 2
- 102000004233 multidrug resistance protein 3 Human genes 0.000 description 2
- 229960003816 muromonab-cd3 Drugs 0.000 description 2
- OHDXDNUPVVYWOV-UHFFFAOYSA-N n-methyl-1-(2-naphthalen-1-ylsulfanylphenyl)methanamine Chemical compound CNCC1=CC=CC=C1SC1=CC=CC2=CC=CC=C12 OHDXDNUPVVYWOV-UHFFFAOYSA-N 0.000 description 2
- 229930014626 natural product Natural products 0.000 description 2
- CTMCWCONSULRHO-UHQPFXKFSA-N nemorubicin Chemical compound C1CO[C@H](OC)CN1[C@@H]1[C@H](O)[C@H](C)O[C@@H](O[C@@H]2C3=C(O)C=4C(=O)C5=C(OC)C=CC=C5C(=O)C=4C(O)=C3C[C@](O)(C2)C(=O)CO)C1 CTMCWCONSULRHO-UHQPFXKFSA-N 0.000 description 2
- BPGXUIVWLQTVLZ-OFGSCBOVSA-N neuropeptide y(npy) Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(O)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 BPGXUIVWLQTVLZ-OFGSCBOVSA-N 0.000 description 2
- 210000000440 neutrophil Anatomy 0.000 description 2
- 239000002547 new drug Substances 0.000 description 2
- 229960002450 ofatumumab Drugs 0.000 description 2
- 229960000572 olaparib Drugs 0.000 description 2
- FAQDUNYVKQKNLD-UHFFFAOYSA-N olaparib Chemical compound FC1=CC=C(CC2=C3[CH]C=CC=C3C(=O)N=N2)C=C1C(=O)N(CC1)CCN1C(=O)C1CC1 FAQDUNYVKQKNLD-UHFFFAOYSA-N 0.000 description 2
- 229920001542 oligosaccharide Polymers 0.000 description 2
- 150000002482 oligosaccharides Chemical class 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 229960004390 palbociclib Drugs 0.000 description 2
- AHJRHEGDXFFMBM-UHFFFAOYSA-N palbociclib Chemical compound N1=C2N(C3CCCC3)C(=O)C(C(=O)C)=C(C)C2=CN=C1NC(N=C1)=CC=C1N1CCNCC1 AHJRHEGDXFFMBM-UHFFFAOYSA-N 0.000 description 2
- WRUUGTRCQOWXEG-UHFFFAOYSA-N pamidronate Chemical compound NCCC(O)(P(O)(O)=O)P(O)(O)=O WRUUGTRCQOWXEG-UHFFFAOYSA-N 0.000 description 2
- 229940046231 pamidronate Drugs 0.000 description 2
- 201000008129 pancreatic ductal adenocarcinoma Diseases 0.000 description 2
- FPOHNWQLNRZRFC-ZHACJKMWSA-N panobinostat Chemical compound CC=1NC2=CC=CC=C2C=1CCNCC1=CC=C(\C=C\C(=O)NO)C=C1 FPOHNWQLNRZRFC-ZHACJKMWSA-N 0.000 description 2
- 244000052769 pathogen Species 0.000 description 2
- 230000001717 pathogenic effect Effects 0.000 description 2
- 210000000680 phagosome Anatomy 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 229940127557 pharmaceutical product Drugs 0.000 description 2
- 238000009520 phase I clinical trial Methods 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- IJAPPYDYQCXOEF-UHFFFAOYSA-N phthalazin-1(2H)-one Chemical compound C1=CC=C2C(=O)NN=CC2=C1 IJAPPYDYQCXOEF-UHFFFAOYSA-N 0.000 description 2
- 229940063179 platinol Drugs 0.000 description 2
- 150000003058 platinum compounds Chemical class 0.000 description 2
- 238000010837 poor prognosis Methods 0.000 description 2
- 230000037452 priming Effects 0.000 description 2
- 230000000770 proinflammatory effect Effects 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- PMXCMJLOPOFPBT-HNNXBMFYSA-N purvalanol A Chemical compound C=12N=CN(C(C)C)C2=NC(N[C@@H](CO)C(C)C)=NC=1NC1=CC=CC(Cl)=C1 PMXCMJLOPOFPBT-HNNXBMFYSA-N 0.000 description 2
- 239000003790 pyrimidine antagonist Substances 0.000 description 2
- 238000000163 radioactive labelling Methods 0.000 description 2
- 239000012217 radiopharmaceutical Substances 0.000 description 2
- 229940121896 radiopharmaceutical Drugs 0.000 description 2
- 230000002799 radiopharmaceutical effect Effects 0.000 description 2
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 2
- 230000007115 recruitment Effects 0.000 description 2
- 230000008439 repair process Effects 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- SUFUKZSWUHZXAV-BTJKTKAUSA-N rosiglitazone maleate Chemical compound [H+].[H+].[O-]C(=O)\C=C/C([O-])=O.C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O SUFUKZSWUHZXAV-BTJKTKAUSA-N 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 231100000004 severe toxicity Toxicity 0.000 description 2
- 230000001568 sexual effect Effects 0.000 description 2
- 229960002930 sirolimus Drugs 0.000 description 2
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 210000004988 splenocyte Anatomy 0.000 description 2
- 230000006641 stabilisation Effects 0.000 description 2
- 238000011105 stabilization Methods 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- 230000004936 stimulating effect Effects 0.000 description 2
- 230000035882 stress Effects 0.000 description 2
- QNUKRWAIZMBVCU-WCIBSUBMSA-N su9516 Chemical compound C12=CC(OC)=CC=C2NC(=O)\C1=C/C1=CN=CN1 QNUKRWAIZMBVCU-WCIBSUBMSA-N 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- DKPFODGZWDEEBT-QFIAKTPHSA-N taxane Chemical class C([C@]1(C)CCC[C@@H](C)[C@H]1C1)C[C@H]2[C@H](C)CC[C@@H]1C2(C)C DKPFODGZWDEEBT-QFIAKTPHSA-N 0.000 description 2
- 229940056501 technetium 99m Drugs 0.000 description 2
- 229940061353 temodar Drugs 0.000 description 2
- 108091008743 testicular receptors 4 Proteins 0.000 description 2
- 229960001196 thiotepa Drugs 0.000 description 2
- 230000035897 transcription Effects 0.000 description 2
- 230000007704 transition Effects 0.000 description 2
- 238000013519 translation Methods 0.000 description 2
- 230000005945 translocation Effects 0.000 description 2
- 229960001727 tretinoin Drugs 0.000 description 2
- APJYDQYYACXCRM-UHFFFAOYSA-N tryptamine Chemical compound C1=CC=C2C(CCN)=CNC2=C1 APJYDQYYACXCRM-UHFFFAOYSA-N 0.000 description 2
- 230000005747 tumor angiogenesis Effects 0.000 description 2
- 239000000439 tumor marker Substances 0.000 description 2
- 230000005751 tumor progression Effects 0.000 description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 2
- MSRILKIQRXUYCT-UHFFFAOYSA-M valproate semisodium Chemical compound [Na+].CCCC(C(O)=O)CCC.CCCC(C([O-])=O)CCC MSRILKIQRXUYCT-UHFFFAOYSA-M 0.000 description 2
- 229960000604 valproic acid Drugs 0.000 description 2
- 229960004854 viral vaccine Drugs 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- WWUZIQQURGPMPG-UHFFFAOYSA-N (-)-D-erythro-Sphingosine Natural products CCCCCCCCCCCCCC=CC(O)C(N)CO WWUZIQQURGPMPG-UHFFFAOYSA-N 0.000 description 1
- QDZOEBFLNHCSSF-PFFBOGFISA-N (2S)-2-[[(2R)-2-[[(2S)-1-[(2S)-6-amino-2-[[(2S)-1-[(2R)-2-amino-5-carbamimidamidopentanoyl]pyrrolidine-2-carbonyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-N-[(2R)-1-[[(2S)-1-[[(2R)-1-[[(2S)-1-[[(2S)-1-amino-4-methyl-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]pentanediamide Chemical compound C([C@@H](C(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(N)=O)NC(=O)[C@@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](N)CCCNC(N)=N)C1=CC=CC=C1 QDZOEBFLNHCSSF-PFFBOGFISA-N 0.000 description 1
- BRVFNEZMTRVUGW-QFIPXVFZSA-N (2s)-2-[6-[(3-methyl-1-oxo-4h-benzo[f]quinazolin-9-yl)methylamino]-3-oxo-1h-isoindol-2-yl]pentanedioic acid Chemical compound C1=C2C(=O)N([C@@H](CCC(O)=O)C(O)=O)CC2=CC(NCC2=CC=C3C=CC4=C(C3=C2)C(=O)N=C(N4)C)=C1 BRVFNEZMTRVUGW-QFIPXVFZSA-N 0.000 description 1
- LGNCNVVZCUVPOT-FUVGGWJZSA-N (2s)-2-[[(2r,3r)-3-[(2s)-1-[(3r,4s,5s)-4-[[(2s)-2-[[(2s)-2-(dimethylamino)-3-methylbutanoyl]amino]-3-methylbutanoyl]-methylamino]-3-methoxy-5-methylheptanoyl]pyrrolidin-2-yl]-3-methoxy-2-methylpropanoyl]amino]-3-phenylpropanoic acid Chemical compound CC(C)[C@H](N(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 LGNCNVVZCUVPOT-FUVGGWJZSA-N 0.000 description 1
- WOWDZACBATWTAU-FEFUEGSOSA-N (2s)-2-[[(2s)-2-(dimethylamino)-3-methylbutanoyl]amino]-n-[(3r,4s,5s)-1-[(2s)-2-[(1r,2r)-3-[[(1s,2r)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-oxopropyl]pyrrolidin-1-yl]-3-methoxy-5-methyl-1-oxoheptan-4-yl]-n,3-dimethylbutanamide Chemical compound CC(C)[C@H](N(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)C1=CC=CC=C1 WOWDZACBATWTAU-FEFUEGSOSA-N 0.000 description 1
- IDXCXSCCZNCXCL-XMADEQCMSA-N (2s)-2-[[(2s)-2-[[(2s)-1-[(2s)-2-[[(2s)-2-[[2-[[(2s,4r)-1-[(2s)-1-[(2s)-2-amino-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]-4-hydroxypyrrolidine-2-carbonyl]amino]acetyl]amino]-3-thiophen-2-ylpropanoyl]amino]-3-hydroxypropanoyl]pyrrolidine Chemical compound C1=CC(OC)=CC=C1C[C@@H](CN[C@@H](CCCN=C(N)N)C(O)=O)NC(=O)[C@H]1N(C(=O)[C@H](CO)NC(=O)[C@H](CC=2SC=CC=2)NC(=O)CNC(=O)[C@H]2N(C[C@H](O)C2)C(=O)[C@H]2N(CCC2)C(=O)[C@@H](N)CCCN=C(N)N)CCC1 IDXCXSCCZNCXCL-XMADEQCMSA-N 0.000 description 1
- QNRATNLHPGXHMA-XZHTYLCXSA-N (r)-(6-ethoxyquinolin-4-yl)-[(2s,4s,5r)-5-ethyl-1-azabicyclo[2.2.2]octan-2-yl]methanol;hydrochloride Chemical compound Cl.C([C@H]([C@H](C1)CC)C2)CN1[C@@H]2[C@H](O)C1=CC=NC2=CC=C(OCC)C=C21 QNRATNLHPGXHMA-XZHTYLCXSA-N 0.000 description 1
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical compound C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 1
- FBFJOZZTIXSPPR-UHFFFAOYSA-N 1-(4-aminobutyl)-2-(ethoxymethyl)imidazo[4,5-c]quinolin-4-amine Chemical compound C1=CC=CC2=C(N(C(COCC)=N3)CCCCN)C3=C(N)N=C21 FBFJOZZTIXSPPR-UHFFFAOYSA-N 0.000 description 1
- WUAPFZMCVAUBPE-NJFSPNSNSA-N 188Re Chemical compound [188Re] WUAPFZMCVAUBPE-NJFSPNSNSA-N 0.000 description 1
- CHTLVASIXCFOHC-UHFFFAOYSA-N 2,7-phenanthroline Chemical compound C1=NC=C2C3=CC=CN=C3C=CC2=C1 CHTLVASIXCFOHC-UHFFFAOYSA-N 0.000 description 1
- OOVTUOCTLAERQD-OJMBIDBESA-N 2-(2-chlorophenyl)-5,7-dihydroxy-8-[(2r,3s)-2-(hydroxymethyl)-1-methylpyrrolidin-3-yl]chromen-4-one;hydrochloride Chemical compound Cl.OC[C@@H]1N(C)CC[C@H]1C1=C(O)C=C(O)C2=C1OC(C=1C(=CC=CC=1)Cl)=CC2=O OOVTUOCTLAERQD-OJMBIDBESA-N 0.000 description 1
- YZBAXVICWUUHGG-UHFFFAOYSA-N 2-[[4-[2-[dimethyl(oxido)azaniumyl]ethylamino]-5,8-dihydroxy-9,10-dioxoanthracen-1-yl]amino]-n,n-dimethylethanamine oxide Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCC[N+](C)(C)[O-])=CC=C2NCC[N+](C)([O-])C YZBAXVICWUUHGG-UHFFFAOYSA-N 0.000 description 1
- DJQYYYCQOZMCRC-UHFFFAOYSA-N 2-aminopropane-1,3-dithiol Chemical compound SCC(N)CS DJQYYYCQOZMCRC-UHFFFAOYSA-N 0.000 description 1
- VQMWCYHBHMGBDP-UHFFFAOYSA-N 2h-1,2-benzodiazepine-3,4-dione Chemical compound N1C(=O)C(=O)C=C2C=CC=CC2=N1 VQMWCYHBHMGBDP-UHFFFAOYSA-N 0.000 description 1
- ZOOGRGPOEVQQDX-UUOKFMHZSA-N 3',5'-cyclic GMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-UUOKFMHZSA-N 0.000 description 1
- 125000003974 3-carbamimidamidopropyl group Chemical group C(N)(=N)NCCC* 0.000 description 1
- 125000004042 4-aminobutyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])N([H])[H] 0.000 description 1
- MSTNYGQPCMXVAQ-KIYNQFGBSA-N 5,6,7,8-tetrahydrofolic acid Chemical compound N1C=2C(=O)NC(N)=NC=2NCC1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 MSTNYGQPCMXVAQ-KIYNQFGBSA-N 0.000 description 1
- KDOPAZIWBAHVJB-UHFFFAOYSA-N 5h-pyrrolo[3,2-d]pyrimidine Chemical compound C1=NC=C2NC=CC2=N1 KDOPAZIWBAHVJB-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HFDKKNHCYWNNNQ-YOGANYHLSA-N 75976-10-2 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@@H](NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)N)C(C)C)[C@@H](C)O)C1=CC=C(O)C=C1 HFDKKNHCYWNNNQ-YOGANYHLSA-N 0.000 description 1
- OONFNUWBHFSNBT-HXUWFJFHSA-N AEE788 Chemical compound C1CN(CC)CCN1CC1=CC=C(C=2NC3=NC=NC(N[C@H](C)C=4C=CC=CC=4)=C3C=2)C=C1 OONFNUWBHFSNBT-HXUWFJFHSA-N 0.000 description 1
- 102000005416 ATP-Binding Cassette Transporters Human genes 0.000 description 1
- 108010006533 ATP-Binding Cassette Transporters Proteins 0.000 description 1
- 102100022410 ATP-dependent DNA/RNA helicase DHX36 Human genes 0.000 description 1
- 102100038266 ATP-dependent RNA helicase DDX54 Human genes 0.000 description 1
- 241000580482 Acidobacteria Species 0.000 description 1
- 241001156739 Actinobacteria <phylum> Species 0.000 description 1
- 241000186046 Actinomyces Species 0.000 description 1
- 241000203809 Actinomycetales Species 0.000 description 1
- 206010052747 Adenocarcinoma pancreas Diseases 0.000 description 1
- 241000204018 Anaeroplasma Species 0.000 description 1
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 1
- 101100043388 Arabidopsis thaliana SRK2D gene Proteins 0.000 description 1
- 241000203069 Archaea Species 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 208000004736 B-Cell Leukemia Diseases 0.000 description 1
- 208000003950 B-cell lymphoma Diseases 0.000 description 1
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 1
- QULDDKSCVCJTPV-UHFFFAOYSA-N BIIB021 Chemical compound COC1=C(C)C=NC(CN2C3=NC(N)=NC(Cl)=C3N=C2)=C1C QULDDKSCVCJTPV-UHFFFAOYSA-N 0.000 description 1
- 241000304886 Bacilli Species 0.000 description 1
- 108020000946 Bacterial DNA Proteins 0.000 description 1
- 241000606125 Bacteroides Species 0.000 description 1
- 101150017888 Bcl2 gene Proteins 0.000 description 1
- 206010061692 Benign muscle neoplasm Diseases 0.000 description 1
- 241001655328 Bifidobacteriales Species 0.000 description 1
- 241001474374 Blennius Species 0.000 description 1
- 208000031648 Body Weight Changes Diseases 0.000 description 1
- 102100037086 Bone marrow stromal antigen 2 Human genes 0.000 description 1
- 241000186146 Brevibacterium Species 0.000 description 1
- 102100022595 Broad substrate specificity ATP-binding cassette transporter ABCG2 Human genes 0.000 description 1
- 241000195940 Bryophyta Species 0.000 description 1
- 102100031172 C-C chemokine receptor type 1 Human genes 0.000 description 1
- 101710149814 C-C chemokine receptor type 1 Proteins 0.000 description 1
- 102100036305 C-C chemokine receptor type 8 Human genes 0.000 description 1
- 102100021942 C-C motif chemokine 28 Human genes 0.000 description 1
- 108010074051 C-Reactive Protein Proteins 0.000 description 1
- 102100032752 C-reactive protein Human genes 0.000 description 1
- 108700012439 CA9 Proteins 0.000 description 1
- 108091005932 CCKBR Proteins 0.000 description 1
- 108700012434 CCL3 Proteins 0.000 description 1
- 108010058905 CD44v6 antigen Proteins 0.000 description 1
- 102100025222 CD63 antigen Human genes 0.000 description 1
- 210000005236 CD8+ effector T cell Anatomy 0.000 description 1
- 101150066398 CXCR4 gene Proteins 0.000 description 1
- 206010006895 Cachexia Diseases 0.000 description 1
- 208000031968 Cadaver Diseases 0.000 description 1
- 102000000905 Cadherin Human genes 0.000 description 1
- 108050007957 Cadherin Proteins 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 1
- 102100024423 Carbonic anhydrase 9 Human genes 0.000 description 1
- 244000132059 Carica parviflora Species 0.000 description 1
- 235000014653 Carica parviflora Nutrition 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 108090000426 Caspase-1 Proteins 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 102000000013 Chemokine CCL3 Human genes 0.000 description 1
- 108010019670 Chimeric Antigen Receptors Proteins 0.000 description 1
- 241000606161 Chlamydia Species 0.000 description 1
- 101100009647 Chlamydomonas reinhardtii DIP13 gene Proteins 0.000 description 1
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 1
- 241000191368 Chlorobi Species 0.000 description 1
- 241001142109 Chloroflexi Species 0.000 description 1
- 108010009685 Cholinergic Receptors Proteins 0.000 description 1
- 108010059480 Chondroitin Sulfate Proteoglycans Proteins 0.000 description 1
- 102000005598 Chondroitin Sulfate Proteoglycans Human genes 0.000 description 1
- 241000238855 Chrysiogenes Species 0.000 description 1
- 241001112696 Clostridia Species 0.000 description 1
- UDMBCSSLTHHNCD-UHFFFAOYSA-N Coenzym Q(11) Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(O)=O)C(O)C1O UDMBCSSLTHHNCD-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 241000186216 Corynebacterium Species 0.000 description 1
- 208000009798 Craniopharyngioma Diseases 0.000 description 1
- 241001464430 Cyanobacterium Species 0.000 description 1
- 108010069514 Cyclic Peptides Proteins 0.000 description 1
- 102000001189 Cyclic Peptides Human genes 0.000 description 1
- 102100036279 DNA (cytosine-5)-methyltransferase 1 Human genes 0.000 description 1
- 108010041052 DNA Topoisomerase IV Proteins 0.000 description 1
- 229940123780 DNA topoisomerase I inhibitor Drugs 0.000 description 1
- 229940124087 DNA topoisomerase II inhibitor Drugs 0.000 description 1
- 241000450599 DNA viruses Species 0.000 description 1
- 230000004568 DNA-binding Effects 0.000 description 1
- 101100481404 Danio rerio tie1 gene Proteins 0.000 description 1
- KWHWFTSHDPJOTG-UHFFFAOYSA-N Deazaflavin Chemical compound C1=CC=C2C=C(C(=O)NC(=O)N3)C3=NC2=C1 KWHWFTSHDPJOTG-UHFFFAOYSA-N 0.000 description 1
- 241000192095 Deinococcus-Thermus Species 0.000 description 1
- 101000582926 Dictyostelium discoideum Probable serine/threonine-protein kinase PLK Proteins 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 108010066687 Epithelial Cell Adhesion Molecule Proteins 0.000 description 1
- 102000018651 Epithelial Cell Adhesion Molecule Human genes 0.000 description 1
- 241000402754 Erythranthe moschata Species 0.000 description 1
- DBVJJBKOTRCVKF-UHFFFAOYSA-N Etidronic acid Chemical compound OP(=O)(O)C(O)(C)P(O)(O)=O DBVJJBKOTRCVKF-UHFFFAOYSA-N 0.000 description 1
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 1
- 208000017259 Extragonadal germ cell tumor Diseases 0.000 description 1
- 241000605898 Fibrobacter Species 0.000 description 1
- 102100023600 Fibroblast growth factor receptor 2 Human genes 0.000 description 1
- 101710182389 Fibroblast growth factor receptor 2 Proteins 0.000 description 1
- 102100027842 Fibroblast growth factor receptor 3 Human genes 0.000 description 1
- 101710182396 Fibroblast growth factor receptor 3 Proteins 0.000 description 1
- 102100027844 Fibroblast growth factor receptor 4 Human genes 0.000 description 1
- 241000192125 Firmicutes Species 0.000 description 1
- 206010016717 Fistula Diseases 0.000 description 1
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 1
- 241001453172 Fusobacteria Species 0.000 description 1
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 1
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 1
- GYHNNYVSQQEPJS-YPZZEJLDSA-N Gallium-68 Chemical compound [68Ga] GYHNNYVSQQEPJS-YPZZEJLDSA-N 0.000 description 1
- 102000052874 Gastrin receptors Human genes 0.000 description 1
- 208000034534 Gastroenteropancreatic neuroendocrine neoplasm Diseases 0.000 description 1
- 206010051066 Gastrointestinal stromal tumour Diseases 0.000 description 1
- JRZJKWGQFNTSRN-UHFFFAOYSA-N Geldanamycin Natural products C1C(C)CC(OC)C(O)C(C)C=C(C)C(OC(N)=O)C(OC)CCC=C(C)C(=O)NC2=CC(=O)C(OC)=C1C2=O JRZJKWGQFNTSRN-UHFFFAOYSA-N 0.000 description 1
- 208000032612 Glial tumor Diseases 0.000 description 1
- 102000051325 Glucagon Human genes 0.000 description 1
- 108060003199 Glucagon Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical group NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 1
- NYHBQMYGNKIUIF-UUOKFMHZSA-N Guanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O NYHBQMYGNKIUIF-UUOKFMHZSA-N 0.000 description 1
- 102000003693 Hedgehog Proteins Human genes 0.000 description 1
- 108090000031 Hedgehog Proteins Proteins 0.000 description 1
- 241000167880 Hirundinidae Species 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000901942 Homo sapiens ATP-dependent DNA/RNA helicase DHX36 Proteins 0.000 description 1
- 101000883804 Homo sapiens ATP-dependent RNA helicase DDX54 Proteins 0.000 description 1
- 101000740785 Homo sapiens Bone marrow stromal antigen 2 Proteins 0.000 description 1
- 101000823298 Homo sapiens Broad substrate specificity ATP-binding cassette transporter ABCG2 Proteins 0.000 description 1
- 101000716063 Homo sapiens C-C chemokine receptor type 8 Proteins 0.000 description 1
- 101000897477 Homo sapiens C-C motif chemokine 28 Proteins 0.000 description 1
- 101000934368 Homo sapiens CD63 antigen Proteins 0.000 description 1
- 101000914324 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 5 Proteins 0.000 description 1
- 101000914321 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 7 Proteins 0.000 description 1
- 101000916489 Homo sapiens Chondroitin sulfate proteoglycan 4 Proteins 0.000 description 1
- 101000931098 Homo sapiens DNA (cytosine-5)-methyltransferase 1 Proteins 0.000 description 1
- 101000650600 Homo sapiens DNA-directed RNA polymerase I subunit RPA2 Proteins 0.000 description 1
- 101000851181 Homo sapiens Epidermal growth factor receptor Proteins 0.000 description 1
- 101000917134 Homo sapiens Fibroblast growth factor receptor 4 Proteins 0.000 description 1
- 101000882127 Homo sapiens Histone-lysine N-methyltransferase EZH2 Proteins 0.000 description 1
- 101100232357 Homo sapiens IL13RA1 gene Proteins 0.000 description 1
- 101100232360 Homo sapiens IL13RA2 gene Proteins 0.000 description 1
- 101000606465 Homo sapiens Inactive tyrosine-protein kinase 7 Proteins 0.000 description 1
- 101001001420 Homo sapiens Interferon gamma receptor 1 Proteins 0.000 description 1
- 101001032345 Homo sapiens Interferon regulatory factor 8 Proteins 0.000 description 1
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 1
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 description 1
- 101001033312 Homo sapiens Interleukin-4 receptor subunit alpha Proteins 0.000 description 1
- 101000916644 Homo sapiens Macrophage colony-stimulating factor 1 receptor Proteins 0.000 description 1
- 101000628547 Homo sapiens Metalloreductase STEAP1 Proteins 0.000 description 1
- 101001133081 Homo sapiens Mucin-2 Proteins 0.000 description 1
- 101000972284 Homo sapiens Mucin-3A Proteins 0.000 description 1
- 101000972286 Homo sapiens Mucin-4 Proteins 0.000 description 1
- 101000972273 Homo sapiens Mucin-7 Proteins 0.000 description 1
- 101000581981 Homo sapiens Neural cell adhesion molecule 1 Proteins 0.000 description 1
- 101000829725 Homo sapiens Phospholipid hydroperoxide glutathione peroxidase Proteins 0.000 description 1
- 101001066878 Homo sapiens Polyribonucleotide nucleotidyltransferase 1, mitochondrial Proteins 0.000 description 1
- 101000617725 Homo sapiens Pregnancy-specific beta-1-glycoprotein 2 Proteins 0.000 description 1
- 101000709305 Homo sapiens Replication protein A 14 kDa subunit Proteins 0.000 description 1
- 101001092206 Homo sapiens Replication protein A 32 kDa subunit Proteins 0.000 description 1
- 101001104307 Homo sapiens Ribonuclease 7 Proteins 0.000 description 1
- 101000575639 Homo sapiens Ribonucleoside-diphosphate reductase subunit M2 Proteins 0.000 description 1
- 101001038337 Homo sapiens Serine/threonine-protein kinase LMTK1 Proteins 0.000 description 1
- 101001038335 Homo sapiens Serine/threonine-protein kinase LMTK2 Proteins 0.000 description 1
- 101001038341 Homo sapiens Serine/threonine-protein kinase LMTK3 Proteins 0.000 description 1
- 101000829153 Homo sapiens Somatostatin receptor type 5 Proteins 0.000 description 1
- 101001056234 Homo sapiens Sperm mitochondrial-associated cysteine-rich protein Proteins 0.000 description 1
- 101000595548 Homo sapiens TIR domain-containing adapter molecule 1 Proteins 0.000 description 1
- 101000653679 Homo sapiens Translationally-controlled tumor protein Proteins 0.000 description 1
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 description 1
- 101000666868 Homo sapiens Vasoactive intestinal polypeptide receptor 2 Proteins 0.000 description 1
- 101000620751 Homo sapiens mRNA export factor RAE1 Proteins 0.000 description 1
- 206010020400 Hostility Diseases 0.000 description 1
- 241000701806 Human papillomavirus Species 0.000 description 1
- 101000776197 Human papillomavirus type 16 Probable protein E5 Proteins 0.000 description 1
- 101000767631 Human papillomavirus type 16 Protein E7 Proteins 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- 101150103227 IFN gene Proteins 0.000 description 1
- 101150026109 INSR gene Proteins 0.000 description 1
- 101150068197 INSRR gene Proteins 0.000 description 1
- 102000043138 IRF family Human genes 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 238000012404 In vitro experiment Methods 0.000 description 1
- 102100039813 Inactive tyrosine-protein kinase 7 Human genes 0.000 description 1
- 101710090028 Inositol-3-phosphate synthase 1 Proteins 0.000 description 1
- 102100022297 Integrin alpha-X Human genes 0.000 description 1
- 108010054267 Interferon Receptors Proteins 0.000 description 1
- 102000001617 Interferon Receptors Human genes 0.000 description 1
- 102100026720 Interferon beta Human genes 0.000 description 1
- 102100026688 Interferon epsilon Human genes 0.000 description 1
- 101710147309 Interferon epsilon Proteins 0.000 description 1
- 102100035678 Interferon gamma receptor 1 Human genes 0.000 description 1
- 102100036157 Interferon gamma receptor 2 Human genes 0.000 description 1
- 102100022469 Interferon kappa Human genes 0.000 description 1
- 102100038069 Interferon regulatory factor 8 Human genes 0.000 description 1
- 108090000467 Interferon-beta Proteins 0.000 description 1
- 102100027268 Interferon-stimulated gene 20 kDa protein Human genes 0.000 description 1
- 102100039078 Interleukin-4 receptor subunit alpha Human genes 0.000 description 1
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 1
- SHGAZHPCJJPHSC-NUEINMDLSA-N Isotretinoin Chemical compound OC(=O)C=C(C)/C=C/C=C(C)C=CC1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-NUEINMDLSA-N 0.000 description 1
- UETNIIAIRMUTSM-UHFFFAOYSA-N Jacareubin Natural products CC1(C)OC2=CC3Oc4c(O)c(O)ccc4C(=O)C3C(=C2C=C1)O UETNIIAIRMUTSM-UHFFFAOYSA-N 0.000 description 1
- 102000010638 Kinesin Human genes 0.000 description 1
- 108010063296 Kinesin Proteins 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 1
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 1
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 1
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 1
- 239000002176 L01XE26 - Cabozantinib Substances 0.000 description 1
- 201000005099 Langerhans cell histiocytosis Diseases 0.000 description 1
- 108010000817 Leuprolide Proteins 0.000 description 1
- 241000186781 Listeria Species 0.000 description 1
- 229910052765 Lutetium Inorganic materials 0.000 description 1
- 210000004322 M2 macrophage Anatomy 0.000 description 1
- 102000043129 MHC class I family Human genes 0.000 description 1
- 108091054437 MHC class I family Proteins 0.000 description 1
- 102100028198 Macrophage colony-stimulating factor 1 receptor Human genes 0.000 description 1
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 102100027754 Mast/stem cell growth factor receptor Kit Human genes 0.000 description 1
- 101710087603 Mast/stem cell growth factor receptor Kit Proteins 0.000 description 1
- 102400000740 Melanocyte-stimulating hormone alpha Human genes 0.000 description 1
- 101710200814 Melanotropin alpha Proteins 0.000 description 1
- SGDBTWWWUNNDEQ-UHFFFAOYSA-N Merphalan Chemical compound OC(=O)C(N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-UHFFFAOYSA-N 0.000 description 1
- 102000003735 Mesothelin Human genes 0.000 description 1
- 108090000015 Mesothelin Proteins 0.000 description 1
- 102100026712 Metalloreductase STEAP1 Human genes 0.000 description 1
- 108091030146 MiRBase Proteins 0.000 description 1
- 241001430197 Mollicutes Species 0.000 description 1
- 102100034256 Mucin-1 Human genes 0.000 description 1
- 108010008707 Mucin-1 Proteins 0.000 description 1
- 102100034263 Mucin-2 Human genes 0.000 description 1
- 102100022497 Mucin-3A Human genes 0.000 description 1
- 102100022693 Mucin-4 Human genes 0.000 description 1
- 102100022492 Mucin-7 Human genes 0.000 description 1
- 101100452374 Mus musculus Ikbke gene Proteins 0.000 description 1
- 101100043703 Mus musculus Sting1 gene Proteins 0.000 description 1
- 101100481406 Mus musculus Tie1 gene Proteins 0.000 description 1
- 108010021466 Mutant Proteins Proteins 0.000 description 1
- 102000008300 Mutant Proteins Human genes 0.000 description 1
- 241000204031 Mycoplasma Species 0.000 description 1
- 201000004458 Myoma Diseases 0.000 description 1
- QPCDCPDFJACHGM-UHFFFAOYSA-N N,N-bis{2-[bis(carboxymethyl)amino]ethyl}glycine Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(=O)O)CCN(CC(O)=O)CC(O)=O QPCDCPDFJACHGM-UHFFFAOYSA-N 0.000 description 1
- HRNLUBSXIHFDHP-UHFFFAOYSA-N N-(2-aminophenyl)-4-[[[4-(3-pyridinyl)-2-pyrimidinyl]amino]methyl]benzamide Chemical compound NC1=CC=CC=C1NC(=O)C(C=C1)=CC=C1CNC1=NC=CC(C=2C=NC=CC=2)=N1 HRNLUBSXIHFDHP-UHFFFAOYSA-N 0.000 description 1
- 108010001657 NK Cell Lectin-Like Receptor Subfamily K Proteins 0.000 description 1
- 102000000812 NK Cell Lectin-Like Receptor Subfamily K Human genes 0.000 description 1
- 101150117329 NTRK3 gene Proteins 0.000 description 1
- 206010028729 Nasal cavity cancer Diseases 0.000 description 1
- 108010004217 Natural Cytotoxicity Triggering Receptor 1 Proteins 0.000 description 1
- 102100032870 Natural cytotoxicity triggering receptor 1 Human genes 0.000 description 1
- 208000003788 Neoplasm Micrometastasis Diseases 0.000 description 1
- 102100027347 Neural cell adhesion molecule 1 Human genes 0.000 description 1
- 208000009277 Neuroectodermal Tumors Diseases 0.000 description 1
- 206010073103 Neuroendocrine breast tumour Diseases 0.000 description 1
- 101000871378 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) ATP-dependent RNA helicase dbp2 Proteins 0.000 description 1
- 102000017922 Neurotensin receptor Human genes 0.000 description 1
- 108060003370 Neurotensin receptor Proteins 0.000 description 1
- 101150056950 Ntrk2 gene Proteins 0.000 description 1
- 108091005461 Nucleic proteins Proteins 0.000 description 1
- KUIFHYPNNRVEKZ-VIJRYAKMSA-N O-(N-acetyl-alpha-D-galactosaminyl)-L-threonine Chemical compound OC(=O)[C@@H](N)[C@@H](C)O[C@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1NC(C)=O KUIFHYPNNRVEKZ-VIJRYAKMSA-N 0.000 description 1
- 229960005557 OSI-7904 Drugs 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- 238000012879 PET imaging Methods 0.000 description 1
- 102000038030 PI3Ks Human genes 0.000 description 1
- 108091007960 PI3Ks Proteins 0.000 description 1
- 102000018886 Pancreatic Polypeptide Human genes 0.000 description 1
- JNTOCHDNEULJHD-UHFFFAOYSA-N Penciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(CCC(CO)CO)C=N2 JNTOCHDNEULJHD-UHFFFAOYSA-N 0.000 description 1
- 108010067902 Peptide Library Proteins 0.000 description 1
- KHGNFPUMBJSZSM-UHFFFAOYSA-N Perforine Natural products COC1=C2CCC(O)C(CCC(C)(C)O)(OC)C2=NC2=C1C=CO2 KHGNFPUMBJSZSM-UHFFFAOYSA-N 0.000 description 1
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 1
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical class OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- 208000012641 Pigmentation disease Diseases 0.000 description 1
- 206010050487 Pinealoblastoma Diseases 0.000 description 1
- 208000007641 Pinealoma Diseases 0.000 description 1
- 102000001393 Platelet-Derived Growth Factor alpha Receptor Human genes 0.000 description 1
- 108010068588 Platelet-Derived Growth Factor alpha Receptor Proteins 0.000 description 1
- 102100024588 Podocalyxin-like protein 2 Human genes 0.000 description 1
- 108091010857 Podocalyxin-like protein 2 Proteins 0.000 description 1
- 102000002681 Polyribonucleotide nucleotidyltransferase Human genes 0.000 description 1
- 102100022019 Pregnancy-specific beta-1-glycoprotein 2 Human genes 0.000 description 1
- 102100027467 Pro-opiomelanocortin Human genes 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 108090000315 Protein Kinase C Proteins 0.000 description 1
- 102000003923 Protein Kinase C Human genes 0.000 description 1
- 101710150114 Protein rep Proteins 0.000 description 1
- 241000192142 Proteobacteria Species 0.000 description 1
- 102000052575 Proto-Oncogene Human genes 0.000 description 1
- 108700020978 Proto-Oncogene Proteins 0.000 description 1
- 101710141955 RAF proto-oncogene serine/threonine-protein kinase Proteins 0.000 description 1
- 108091005685 RIG-I-like receptors Proteins 0.000 description 1
- 108700031422 RMP 7 Proteins 0.000 description 1
- 108090000944 RNA Helicases Proteins 0.000 description 1
- 102000004409 RNA Helicases Human genes 0.000 description 1
- 229940122277 RNA polymerase inhibitor Drugs 0.000 description 1
- 101150066681 Rae1 gene Proteins 0.000 description 1
- 101001039269 Rattus norvegicus Glycine N-methyltransferase Proteins 0.000 description 1
- 102100029981 Receptor tyrosine-protein kinase erbB-4 Human genes 0.000 description 1
- 101710100963 Receptor tyrosine-protein kinase erbB-4 Proteins 0.000 description 1
- 101710152114 Replication protein Proteins 0.000 description 1
- 102100034372 Replication protein A 14 kDa subunit Human genes 0.000 description 1
- 102100035525 Replication protein A 32 kDa subunit Human genes 0.000 description 1
- 102100026006 Ribonucleoside-diphosphate reductase subunit M2 Human genes 0.000 description 1
- 229940127395 Ribonucleotide Reductase Inhibitors Drugs 0.000 description 1
- 101100355601 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) RAD53 gene Proteins 0.000 description 1
- 102100040293 Serine/threonine-protein kinase LMTK1 Human genes 0.000 description 1
- 102100040292 Serine/threonine-protein kinase LMTK2 Human genes 0.000 description 1
- 102100040291 Serine/threonine-protein kinase LMTK3 Human genes 0.000 description 1
- 101710106944 Serine/threonine-protein kinase TBK1 Proteins 0.000 description 1
- 102100023085 Serine/threonine-protein kinase mTOR Human genes 0.000 description 1
- 208000000453 Skin Neoplasms Diseases 0.000 description 1
- 108010017622 Somatomedin Receptors Proteins 0.000 description 1
- 102000004584 Somatomedin Receptors Human genes 0.000 description 1
- 102100029329 Somatostatin receptor type 1 Human genes 0.000 description 1
- 102100023806 Somatostatin receptor type 5 Human genes 0.000 description 1
- 102100026503 Sperm mitochondrial-associated cysteine-rich protein Human genes 0.000 description 1
- 241000230565 Sphingobacteriia Species 0.000 description 1
- 241001180364 Spirochaetes Species 0.000 description 1
- 241000187747 Streptomyces Species 0.000 description 1
- 102400000096 Substance P Human genes 0.000 description 1
- 101800003906 Substance P Proteins 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 101000983124 Sus scrofa Pancreatic prohormone precursor Proteins 0.000 description 1
- 230000006044 T cell activation Effects 0.000 description 1
- 210000000662 T-lymphocyte subset Anatomy 0.000 description 1
- 238000003917 TEM image Methods 0.000 description 1
- 102100036073 TIR domain-containing adapter molecule 1 Human genes 0.000 description 1
- 108010065917 TOR Serine-Threonine Kinases Proteins 0.000 description 1
- 102000003141 Tachykinin Human genes 0.000 description 1
- NAVMQTYZDKMPEU-UHFFFAOYSA-N Targretin Chemical compound CC1=CC(C(CCC2(C)C)(C)C)=C2C=C1C(=C)C1=CC=C(C(O)=O)C=C1 NAVMQTYZDKMPEU-UHFFFAOYSA-N 0.000 description 1
- CBPNZQVSJQDFBE-FUXHJELOSA-N Temsirolimus Chemical compound C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-FUXHJELOSA-N 0.000 description 1
- 241000131694 Tenericutes Species 0.000 description 1
- WDLRUFUQRNWCPK-UHFFFAOYSA-N Tetraxetan Chemical compound OC(=O)CN1CCN(CC(O)=O)CCN(CC(O)=O)CCN(CC(O)=O)CC1 WDLRUFUQRNWCPK-UHFFFAOYSA-N 0.000 description 1
- 241000186339 Thermoanaerobacter Species 0.000 description 1
- 241001143310 Thermotogae <phylum> Species 0.000 description 1
- 239000000365 Topoisomerase I Inhibitor Substances 0.000 description 1
- 239000000317 Topoisomerase II Inhibitor Substances 0.000 description 1
- 102100036502 Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase Human genes 0.000 description 1
- 102000009618 Transforming Growth Factors Human genes 0.000 description 1
- 108010009583 Transforming Growth Factors Proteins 0.000 description 1
- 102100029887 Translationally-controlled tumor protein Human genes 0.000 description 1
- 102000001742 Tumor Suppressor Proteins Human genes 0.000 description 1
- 108010040002 Tumor Suppressor Proteins Proteins 0.000 description 1
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 description 1
- 101150098329 Tyro3 gene Proteins 0.000 description 1
- 108010075974 Vasoactive Intestinal Peptide Receptors Proteins 0.000 description 1
- 102000012088 Vasoactive Intestinal Peptide Receptors Human genes 0.000 description 1
- 102100038388 Vasoactive intestinal polypeptide receptor 1 Human genes 0.000 description 1
- 101710137655 Vasoactive intestinal polypeptide receptor 1 Proteins 0.000 description 1
- 102100038286 Vasoactive intestinal polypeptide receptor 2 Human genes 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 108020000999 Viral RNA Proteins 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- VWQVUPCCIRVNHF-OIOBTWANSA-N Yttrium-86 Chemical compound [86Y] VWQVUPCCIRVNHF-OIOBTWANSA-N 0.000 description 1
- PNDPGZBMCMUPRI-XXSWNUTMSA-N [125I][125I] Chemical compound [125I][125I] PNDPGZBMCMUPRI-XXSWNUTMSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 102000034337 acetylcholine receptors Human genes 0.000 description 1
- 210000005006 adaptive immune system Anatomy 0.000 description 1
- UDMBCSSLTHHNCD-KQYNXXCUSA-N adenosine 5'-monophosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H]1O UDMBCSSLTHHNCD-KQYNXXCUSA-N 0.000 description 1
- 229950006790 adenosine phosphate Drugs 0.000 description 1
- 102000019997 adhesion receptor Human genes 0.000 description 1
- 108010013985 adhesion receptor Proteins 0.000 description 1
- 238000012382 advanced drug delivery Methods 0.000 description 1
- 238000005273 aeration Methods 0.000 description 1
- 108010081667 aflibercept Proteins 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- SRHNADOZAAWYLV-XLMUYGLTSA-N alpha-L-Fucp-(1->2)-beta-D-Galp-(1->4)-[alpha-L-Fucp-(1->3)]-beta-D-GlcpNAc Chemical compound O[C@H]1[C@H](O)[C@H](O)[C@H](C)O[C@H]1O[C@H]1[C@H](O[C@H]2[C@@H]([C@@H](NC(C)=O)[C@H](O)O[C@@H]2CO)O[C@H]2[C@H]([C@H](O)[C@H](O)[C@H](C)O2)O)O[C@H](CO)[C@H](O)[C@@H]1O SRHNADOZAAWYLV-XLMUYGLTSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 150000003927 aminopyridines Chemical class 0.000 description 1
- 238000000540 analysis of variance Methods 0.000 description 1
- 230000031016 anaphase Effects 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000002424 anti-apoptotic effect Effects 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 238000009175 antibody therapy Methods 0.000 description 1
- 230000000890 antigenic effect Effects 0.000 description 1
- 230000009876 antimalignant effect Effects 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 108010072041 arginyl-glycyl-aspartic acid Proteins 0.000 description 1
- 239000003886 aromatase inhibitor Substances 0.000 description 1
- 229940046844 aromatase inhibitors Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 230000017047 asymmetric cell division Effects 0.000 description 1
- 229940062310 avandia Drugs 0.000 description 1
- 229940120638 avastin Drugs 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 102000055104 bcl-X Human genes 0.000 description 1
- 108700000711 bcl-X Proteins 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 230000005266 beta plus decay Effects 0.000 description 1
- 230000005250 beta ray Effects 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 229960002938 bexarotene Drugs 0.000 description 1
- 210000000941 bile Anatomy 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 229940088623 biologically active substance Drugs 0.000 description 1
- QKSKPIVNLNLAAV-UHFFFAOYSA-N bis(2-chloroethyl) sulfide Chemical class ClCCSCCCl QKSKPIVNLNLAAV-UHFFFAOYSA-N 0.000 description 1
- 230000004579 body weight change Effects 0.000 description 1
- 230000004097 bone metabolism Effects 0.000 description 1
- 238000007469 bone scintigraphy Methods 0.000 description 1
- 210000004958 brain cell Anatomy 0.000 description 1
- 201000008211 brain sarcoma Diseases 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 229960002874 briakinumab Drugs 0.000 description 1
- HFCFMRYTXDINDK-WNQIDUERSA-N cabozantinib malate Chemical compound OC(=O)[C@@H](O)CC(O)=O.C=12C=C(OC)C(OC)=CC2=NC=CC=1OC(C=C1)=CC=C1NC(=O)C1(C(=O)NC=2C=CC(F)=CC=2)CC1 HFCFMRYTXDINDK-WNQIDUERSA-N 0.000 description 1
- 229940127093 camptothecin Drugs 0.000 description 1
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 1
- 230000004712 cancer cell adhesion Effects 0.000 description 1
- 230000009702 cancer cell proliferation Effects 0.000 description 1
- 230000000711 cancerogenic effect Effects 0.000 description 1
- 229960004117 capecitabine Drugs 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 150000001720 carbohydrates Chemical group 0.000 description 1
- 231100000315 carcinogenic Toxicity 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 210000004534 cecum Anatomy 0.000 description 1
- 210000005056 cell body Anatomy 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000011712 cell development Effects 0.000 description 1
- 230000022534 cell killing Effects 0.000 description 1
- 230000011748 cell maturation Effects 0.000 description 1
- 230000012292 cell migration Effects 0.000 description 1
- 230000010094 cellular senescence Effects 0.000 description 1
- 210000003850 cellular structure Anatomy 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000013522 chelant Substances 0.000 description 1
- ZWDPMBMJMPIWMJ-MZXODVADSA-N chembl430974 Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC(F)=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 ZWDPMBMJMPIWMJ-MZXODVADSA-N 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- WHTVZRBIWZFKQO-UHFFFAOYSA-N chloroquine Chemical compound ClC1=CC=C2C(NC(C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-UHFFFAOYSA-N 0.000 description 1
- ZYVSOIYQKUDENJ-WKSBCEQHSA-N chromomycin A3 Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@@H]1OC(C)=O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@@H](O)[C@H](O[C@@H]3O[C@@H](C)[C@H](OC(C)=O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@@H]1C[C@@H](O)[C@@H](OC)[C@@H](C)O1 ZYVSOIYQKUDENJ-WKSBCEQHSA-N 0.000 description 1
- 230000007012 clinical effect Effects 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 229940046044 combinations of antineoplastic agent Drugs 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- RYGMFSIKBFXOCR-IGMARMGPSA-N copper-64 Chemical compound [64Cu] RYGMFSIKBFXOCR-IGMARMGPSA-N 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 150000003983 crown ethers Chemical class 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- ZOOGRGPOEVQQDX-UHFFFAOYSA-N cyclic GMP Natural products O1C2COP(O)(=O)OC2C(O)C1N1C=NC2=C1NC(N)=NC2=O ZOOGRGPOEVQQDX-UHFFFAOYSA-N 0.000 description 1
- 108091008878 cytoplasmic PRRs Proteins 0.000 description 1
- 231100000599 cytotoxic agent Toxicity 0.000 description 1
- 229940043239 cytotoxic antineoplastic drug Drugs 0.000 description 1
- 210000004544 dc2 Anatomy 0.000 description 1
- 229960003603 decitabine Drugs 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000007123 defense Effects 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 239000005549 deoxyribonucleoside Substances 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 238000002059 diagnostic imaging Methods 0.000 description 1
- 239000012954 diazonium Substances 0.000 description 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-O diazynium Chemical compound [NH+]#N IJGRMHOSHXDMSA-UHFFFAOYSA-O 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 108010017219 dihydrodiol dehydrogenases Proteins 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XQRLCLUYWUNEEH-UHFFFAOYSA-L diphosphonate(2-) Chemical compound [O-]P(=O)OP([O-])=O XQRLCLUYWUNEEH-UHFFFAOYSA-L 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 125000004119 disulfanediyl group Chemical group *SS* 0.000 description 1
- AUZONCFQVSMFAP-UHFFFAOYSA-N disulfiram Chemical compound CCN(CC)C(=S)SSC(=S)N(CC)CC AUZONCFQVSMFAP-UHFFFAOYSA-N 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- 125000002185 docetaxel anhydrous group Chemical group 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 238000002651 drug therapy Methods 0.000 description 1
- 230000008482 dysregulation Effects 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 210000003890 endocrine cell Anatomy 0.000 description 1
- 230000002121 endocytic effect Effects 0.000 description 1
- 239000006274 endogenous ligand Substances 0.000 description 1
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 230000010502 episomal replication Effects 0.000 description 1
- 230000008472 epithelial growth Effects 0.000 description 1
- QTTMOCOWZLSYSV-QWAPEVOJSA-M equilin sodium sulfate Chemical compound [Na+].[O-]S(=O)(=O)OC1=CC=C2[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4C3=CCC2=C1 QTTMOCOWZLSYSV-QWAPEVOJSA-M 0.000 description 1
- 229960001433 erlotinib Drugs 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- 229960000752 etoposide phosphate Drugs 0.000 description 1
- 229960005167 everolimus Drugs 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 239000013613 expression plasmid Substances 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 238000011354 first-line chemotherapy Methods 0.000 description 1
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 1
- 229960000961 floxuridine Drugs 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 239000004052 folic acid antagonist Substances 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 125000002446 fucosyl group Chemical group C1([C@@H](O)[C@H](O)[C@H](O)[C@@H](O1)C)* 0.000 description 1
- 150000002254 galactosylceramides Chemical class 0.000 description 1
- 229950001109 galiximab Drugs 0.000 description 1
- 229910052733 gallium Inorganic materials 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- QTQAWLPCGQOSGP-GBTDJJJQSA-N geldanamycin Chemical compound N1C(=O)\C(C)=C/C=C\[C@@H](OC)[C@H](OC(N)=O)\C(C)=C/[C@@H](C)[C@@H](O)[C@H](OC)C[C@@H](C)CC2=C(OC)C(=O)C=C1C2=O QTQAWLPCGQOSGP-GBTDJJJQSA-N 0.000 description 1
- 229960000578 gemtuzumab Drugs 0.000 description 1
- 230000037440 gene silencing effect Effects 0.000 description 1
- 238000012226 gene silencing method Methods 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- MASNOZXLGMXCHN-ZLPAWPGGSA-N glucagon Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 MASNOZXLGMXCHN-ZLPAWPGGSA-N 0.000 description 1
- 229960004666 glucagon Drugs 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- PEDCQBHIVMGVHV-UHFFFAOYSA-N glycerol Substances OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 1
- 230000006867 granzyme B production Effects 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 210000002768 hair cell Anatomy 0.000 description 1
- 210000003128 head Anatomy 0.000 description 1
- 230000002489 hematologic effect Effects 0.000 description 1
- 239000000710 homodimer Substances 0.000 description 1
- 230000005745 host immune response Effects 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 229940075628 hypomethylating agent Drugs 0.000 description 1
- 210000003405 ileum Anatomy 0.000 description 1
- 239000012216 imaging agent Substances 0.000 description 1
- 229960003685 imatinib mesylate Drugs 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 230000008073 immune recognition Effects 0.000 description 1
- 230000006058 immune tolerance Effects 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 230000009851 immunogenic response Effects 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 238000013394 immunophenotyping Methods 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 230000000415 inactivating effect Effects 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- VVVPGLRKXQSQSZ-UHFFFAOYSA-N indolo[3,2-c]carbazole Chemical compound C1=CC=CC2=NC3=C4C5=CC=CC=C5N=C4C=CC3=C21 VVVPGLRKXQSQSZ-UHFFFAOYSA-N 0.000 description 1
- 229960005544 indolocarbazole Drugs 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 206010022000 influenza Diseases 0.000 description 1
- 229960003971 influenza vaccine Drugs 0.000 description 1
- 230000004941 influx Effects 0.000 description 1
- 238000011221 initial treatment Methods 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 108010085650 interferon gamma receptor Proteins 0.000 description 1
- 108010080375 interferon kappa Proteins 0.000 description 1
- 230000010468 interferon response Effects 0.000 description 1
- 108700027921 interferon tau Proteins 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 229940125425 inverse agonist Drugs 0.000 description 1
- XMBWDFGMSWQBCA-OIOBTWANSA-N iodane Chemical compound [124IH] XMBWDFGMSWQBCA-OIOBTWANSA-N 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229940044173 iodine-125 Drugs 0.000 description 1
- 229940084651 iressa Drugs 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- PXZQEOJJUGGUIB-UHFFFAOYSA-N isoindolin-1-one Chemical compound C1=CC=C2C(=O)NCC2=C1 PXZQEOJJUGGUIB-UHFFFAOYSA-N 0.000 description 1
- 239000003394 isomerase inhibitor Substances 0.000 description 1
- VDBNYAPERZTOOF-UHFFFAOYSA-N isoquinolin-1(2H)-one Chemical compound C1=CC=C2C(=O)NC=CC2=C1 VDBNYAPERZTOOF-UHFFFAOYSA-N 0.000 description 1
- 229960005280 isotretinoin Drugs 0.000 description 1
- 210000001630 jejunum Anatomy 0.000 description 1
- 230000003907 kidney function Effects 0.000 description 1
- VHOGYURTWQBHIL-UHFFFAOYSA-N leflunomide Chemical compound O1N=CC(C(=O)NC=2C=CC(=CC=2)C(F)(F)F)=C1C VHOGYURTWQBHIL-UHFFFAOYSA-N 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 108091023663 let-7 stem-loop Proteins 0.000 description 1
- 108091063478 let-7-1 stem-loop Proteins 0.000 description 1
- 108091049777 let-7-2 stem-loop Proteins 0.000 description 1
- 231100000636 lethal dose Toxicity 0.000 description 1
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 1
- 229960004338 leuprorelin Drugs 0.000 description 1
- 108010052322 limitin Proteins 0.000 description 1
- GZQKNULLWNGMCW-PWQABINMSA-N lipid A (E. coli) Chemical compound O1[C@H](CO)[C@@H](OP(O)(O)=O)[C@H](OC(=O)C[C@@H](CCCCCCCCCCC)OC(=O)CCCCCCCCCCCCC)[C@@H](NC(=O)C[C@@H](CCCCCCCCCCC)OC(=O)CCCCCCCCCCC)[C@@H]1OC[C@@H]1[C@@H](O)[C@H](OC(=O)C[C@H](O)CCCCCCCCCCC)[C@@H](NC(=O)C[C@H](O)CCCCCCCCCCC)[C@@H](OP(O)(O)=O)O1 GZQKNULLWNGMCW-PWQABINMSA-N 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000003908 liver function Effects 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 201000005296 lung carcinoma Diseases 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- 230000002934 lysing effect Effects 0.000 description 1
- 102100022885 mRNA export factor RAE1 Human genes 0.000 description 1
- 229940124302 mTOR inhibitor Drugs 0.000 description 1
- 230000034701 macropinocytosis Effects 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 239000003628 mammalian target of rapamycin inhibitor Substances 0.000 description 1
- 230000000873 masking effect Effects 0.000 description 1
- 239000012092 media component Substances 0.000 description 1
- 208000023356 medullary thyroid gland carcinoma Diseases 0.000 description 1
- 230000034217 membrane fusion Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 208000037819 metastatic cancer Diseases 0.000 description 1
- 208000011575 metastatic malignant neoplasm Diseases 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- CWWARWOPSKGELM-SARDKLJWSA-N methyl (2s)-2-[[(2s)-2-[[2-[[(2s)-2-[[(2s)-2-[[(2s)-5-amino-2-[[(2s)-5-amino-2-[[(2s)-1-[(2s)-6-amino-2-[[(2s)-1-[(2s)-2-amino-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-5 Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)OC)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CCCN=C(N)N)C1=CC=CC=C1 CWWARWOPSKGELM-SARDKLJWSA-N 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 108091074487 miR-34 stem-loop Proteins 0.000 description 1
- 108091092493 miR-34-1 stem-loop Proteins 0.000 description 1
- 108091059780 miR-34-2 stem-loop Proteins 0.000 description 1
- 108091089612 miR-352 stem-loop Proteins 0.000 description 1
- 244000000010 microbial pathogen Species 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 101150110189 minE gene Proteins 0.000 description 1
- 230000025608 mitochondrion localization Effects 0.000 description 1
- 230000011278 mitosis Effects 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- 230000001483 mobilizing effect Effects 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- VYGYNVZNSSTDLJ-HKCOAVLJSA-N monorden Natural products CC1CC2OC2C=C/C=C/C(=O)CC3C(C(=CC(=C3Cl)O)O)C(=O)O1 VYGYNVZNSSTDLJ-HKCOAVLJSA-N 0.000 description 1
- 230000000877 morphologic effect Effects 0.000 description 1
- 230000008450 motivation Effects 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 235000011929 mousse Nutrition 0.000 description 1
- 101150035528 mukB gene Proteins 0.000 description 1
- 231100000219 mutagenic Toxicity 0.000 description 1
- 230000003505 mutagenic effect Effects 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- FTTNHUULIMOCKS-WAYWQWQTSA-N n-[(z)-3-acetamidobut-2-en-2-yl]acetamide Chemical compound CC(=O)N\C(C)=C(\C)NC(C)=O FTTNHUULIMOCKS-WAYWQWQTSA-N 0.000 description 1
- LBWFXVZLPYTWQI-IPOVEDGCSA-N n-[2-(diethylamino)ethyl]-5-[(z)-(5-fluoro-2-oxo-1h-indol-3-ylidene)methyl]-2,4-dimethyl-1h-pyrrole-3-carboxamide;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C LBWFXVZLPYTWQI-IPOVEDGCSA-N 0.000 description 1
- RRTPWQXEERTRRK-UHFFFAOYSA-N n-[4-(4-amino-2-butylimidazo[4,5-c]quinolin-1-yl)oxybutyl]octadecanamide Chemical compound C1=CC=CC2=C3N(OCCCCNC(=O)CCCCCCCCCCCCCCCCC)C(CCCC)=NC3=C(N)N=C21 RRTPWQXEERTRRK-UHFFFAOYSA-N 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- OSTGTTZJOCZWJG-UHFFFAOYSA-N nitrosourea Chemical compound NC(=O)N=NO OSTGTTZJOCZWJG-UHFFFAOYSA-N 0.000 description 1
- 108091027963 non-coding RNA Proteins 0.000 description 1
- 102000042567 non-coding RNA Human genes 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 238000012633 nuclear imaging Methods 0.000 description 1
- 238000009206 nuclear medicine Methods 0.000 description 1
- URPYMXQQVHTUDU-OFGSCBOVSA-N nucleopeptide y Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 URPYMXQQVHTUDU-OFGSCBOVSA-N 0.000 description 1
- 229950005751 ocrelizumab Drugs 0.000 description 1
- 229950008516 olaratumab Drugs 0.000 description 1
- 229940046166 oligodeoxynucleotide Drugs 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000001582 osteoblastic effect Effects 0.000 description 1
- 229940045681 other alkylating agent in atc Drugs 0.000 description 1
- 229940127084 other anti-cancer agent Drugs 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 101150032584 oxy-4 gene Proteins 0.000 description 1
- 229950007318 ozogamicin Drugs 0.000 description 1
- 238000009116 palliative therapy Methods 0.000 description 1
- 208000021090 palsy Diseases 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 201000002094 pancreatic adenocarcinoma Diseases 0.000 description 1
- 208000021010 pancreatic neuroendocrine tumor Diseases 0.000 description 1
- 229960001972 panitumumab Drugs 0.000 description 1
- 230000001769 paralizing effect Effects 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 238000005192 partition Methods 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 210000004197 pelvis Anatomy 0.000 description 1
- 229960005079 pemetrexed Drugs 0.000 description 1
- QOFFJEBXNKRSPX-ZDUSSCGKSA-N pemetrexed Chemical compound C1=N[C]2NC(N)=NC(=O)C2=C1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 QOFFJEBXNKRSPX-ZDUSSCGKSA-N 0.000 description 1
- 102000014187 peptide receptors Human genes 0.000 description 1
- 108010011903 peptide receptors Proteins 0.000 description 1
- 229930192851 perforin Natural products 0.000 description 1
- 210000004976 peripheral blood cell Anatomy 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 230000002688 persistence Effects 0.000 description 1
- 229960002087 pertuzumab Drugs 0.000 description 1
- 238000002823 phage display Methods 0.000 description 1
- 230000002399 phagocytotic effect Effects 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- 208000001297 phlebitis Diseases 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 208000020943 pineal parenchymal cell neoplasm Diseases 0.000 description 1
- 201000002511 pituitary cancer Diseases 0.000 description 1
- 210000004180 plasmocyte Anatomy 0.000 description 1
- 229940031999 pneumococcal conjugate vaccine Drugs 0.000 description 1
- 239000008389 polyethoxylated castor oil Substances 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 231100000683 possible toxicity Toxicity 0.000 description 1
- 230000032361 posttranscriptional gene silencing Effects 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 208000037821 progressive disease Diseases 0.000 description 1
- 210000001236 prokaryotic cell Anatomy 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 208000023958 prostate neoplasm Diseases 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 230000000722 protumoral effect Effects 0.000 description 1
- 239000000649 purine antagonist Substances 0.000 description 1
- YAAWASYJIRZXSZ-UHFFFAOYSA-N pyrimidine-2,4-diamine Chemical compound NC1=CC=NC(N)=N1 YAAWASYJIRZXSZ-UHFFFAOYSA-N 0.000 description 1
- YUOCYTRGANSSRY-UHFFFAOYSA-N pyrrolo[2,3-i][1,2]benzodiazepine Chemical compound C1=CN=NC2=C3C=CN=C3C=CC2=C1 YUOCYTRGANSSRY-UHFFFAOYSA-N 0.000 description 1
- QNMMYUBZGLXCCK-UHFFFAOYSA-N pyrrolo[3,2-a]carbazole Chemical compound N1=C2C=CC=CC2=C2C1=C1C=CN=C1C=C2 QNMMYUBZGLXCCK-UHFFFAOYSA-N 0.000 description 1
- GZTPJDLYPMPRDF-UHFFFAOYSA-N pyrrolo[3,2-c]pyrazole Chemical compound N1=NC2=CC=NC2=C1 GZTPJDLYPMPRDF-UHFFFAOYSA-N 0.000 description 1
- 239000002096 quantum dot Substances 0.000 description 1
- AECPBJMOGBFQDN-YMYQVXQQSA-N radicicol Chemical compound C1CCCC(=O)C[C@H]2[C@H](Cl)C(=O)CC(=O)[C@H]2C(=O)O[C@H](C)C[C@H]2O[C@@H]21 AECPBJMOGBFQDN-YMYQVXQQSA-N 0.000 description 1
- 229930192524 radicicol Natural products 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 229960002633 ramucirumab Drugs 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 208000016691 refractory malignant neoplasm Diseases 0.000 description 1
- 230000012121 regulation of immune response Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229930002330 retinoic acid Natural products 0.000 description 1
- WUAPFZMCVAUBPE-IGMARMGPSA-N rhenium-186 Chemical compound [186Re] WUAPFZMCVAUBPE-IGMARMGPSA-N 0.000 description 1
- 239000002342 ribonucleoside Substances 0.000 description 1
- 229960003271 rosiglitazone maleate Drugs 0.000 description 1
- 102220210442 rs1057523485 Human genes 0.000 description 1
- HNMATTJJEPZZMM-BPKVFSPJSA-N s-[(2r,3s,4s,6s)-6-[[(2r,3s,4s,5r,6r)-5-[(2s,4s,5s)-5-[acetyl(ethyl)amino]-4-methoxyoxan-2-yl]oxy-6-[[(2s,5z,9r,13e)-13-[2-[[4-[(2e)-2-[1-[4-(4-amino-4-oxobutoxy)phenyl]ethylidene]hydrazinyl]-2-methyl-4-oxobutan-2-yl]disulfanyl]ethylidene]-9-hydroxy-12-(m Chemical compound C1[C@H](OC)[C@@H](N(CC)C(C)=O)CO[C@H]1O[C@H]1[C@H](O[C@@H]2C\3=C(NC(=O)OC)C(=O)C[C@@](C/3=C/CSSC(C)(C)CC(=O)N\N=C(/C)C=3C=CC(OCCCC(N)=O)=CC=3)(O)C#C\C=C/C#C2)O[C@H](C)[C@@H](NO[C@@H]2O[C@H](C)[C@@H](SC(=O)C=3C(=C(OC)C(O[C@H]4[C@@H]([C@H](OC)[C@@H](O)[C@H](C)O4)O)=C(I)C=3C)OC)[C@@H](O)C2)[C@@H]1O HNMATTJJEPZZMM-BPKVFSPJSA-N 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 229910052594 sapphire Inorganic materials 0.000 description 1
- 239000010980 sapphire Substances 0.000 description 1
- SIXSYDAISGFNSX-OUBTZVSYSA-N scandium-46 Chemical compound [46Sc] SIXSYDAISGFNSX-OUBTZVSYSA-N 0.000 description 1
- 238000004062 sedimentation Methods 0.000 description 1
- WUWDLXZGHZSWQZ-WQLSENKSSA-N semaxanib Chemical compound N1C(C)=CC(C)=C1\C=C/1C2=CC=CC=C2NC\1=O WUWDLXZGHZSWQZ-WQLSENKSSA-N 0.000 description 1
- 229950003647 semaxanib Drugs 0.000 description 1
- 238000009958 sewing Methods 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- 208000037968 sinus cancer Diseases 0.000 description 1
- 238000004513 sizing Methods 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 201000000849 skin cancer Diseases 0.000 description 1
- 230000004877 small RNA loading onto RISC Effects 0.000 description 1
- PUZPDOWCWNUUKD-UHFFFAOYSA-M sodium fluoride Chemical compound [F-].[Na+] PUZPDOWCWNUUKD-UHFFFAOYSA-M 0.000 description 1
- PUZPDOWCWNUUKD-ULWFUOSBSA-M sodium;fluorine-18(1-) Chemical compound [18F-].[Na+] PUZPDOWCWNUUKD-ULWFUOSBSA-M 0.000 description 1
- 210000004872 soft tissue Anatomy 0.000 description 1
- 229950007874 solanezumab Drugs 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 108090000586 somatostatin receptor 2 Proteins 0.000 description 1
- 102000004052 somatostatin receptor 2 Human genes 0.000 description 1
- 108010082379 somatostatin receptor type 1 Proteins 0.000 description 1
- 229960003787 sorafenib Drugs 0.000 description 1
- 230000004991 spatiotemporal regulation Effects 0.000 description 1
- WWUZIQQURGPMPG-KRWOKUGFSA-N sphingosine Chemical compound CCCCCCCCCCCCC\C=C\[C@@H](O)[C@@H](N)CO WWUZIQQURGPMPG-KRWOKUGFSA-N 0.000 description 1
- 101150087667 spk1 gene Proteins 0.000 description 1
- 229940068117 sprycel Drugs 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- ADNPLDHMAVUMIW-CUZNLEPHSA-N substance P Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(N)=O)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CCCN=C(N)N)C1=CC=CC=C1 ADNPLDHMAVUMIW-CUZNLEPHSA-N 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- NCEXYHBECQHGNR-QZQOTICOSA-N sulfasalazine Chemical compound C1=C(O)C(C(=O)O)=CC(\N=N\C=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-QZQOTICOSA-N 0.000 description 1
- NCEXYHBECQHGNR-UHFFFAOYSA-N sulfasalazine Natural products C1=C(O)C(C(=O)O)=CC(N=NC=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-UHFFFAOYSA-N 0.000 description 1
- 229960001940 sulfasalazine Drugs 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 229940034785 sutent Drugs 0.000 description 1
- 230000009747 swallowing Effects 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- 229940037128 systemic glucocorticoids Drugs 0.000 description 1
- 231100000057 systemic toxicity Toxicity 0.000 description 1
- 108060008037 tachykinin Proteins 0.000 description 1
- 101150047061 tag-72 gene Proteins 0.000 description 1
- AYUNIORJHRXIBJ-TXHRRWQRSA-N tanespimycin Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(NCC=C)C(=O)C=C1C2=O AYUNIORJHRXIBJ-TXHRRWQRSA-N 0.000 description 1
- 229950007866 tanespimycin Drugs 0.000 description 1
- 229950008160 tanezumab Drugs 0.000 description 1
- 229940120982 tarceva Drugs 0.000 description 1
- 150000004579 taxol derivatives Chemical class 0.000 description 1
- 229940063683 taxotere Drugs 0.000 description 1
- 229960004964 temozolomide Drugs 0.000 description 1
- 230000028016 temperature homeostasis Effects 0.000 description 1
- 229960000235 temsirolimus Drugs 0.000 description 1
- GZCRRIHWUXGPOV-CBESVEIWSA-N terbium-149 Chemical compound [149Tb] GZCRRIHWUXGPOV-CBESVEIWSA-N 0.000 description 1
- GZCRRIHWUXGPOV-AHCXROLUSA-N terbium-155 Chemical compound [155Tb] GZCRRIHWUXGPOV-AHCXROLUSA-N 0.000 description 1
- GZCRRIHWUXGPOV-NJFSPNSNSA-N terbium-161 Chemical compound [161Tb] GZCRRIHWUXGPOV-NJFSPNSNSA-N 0.000 description 1
- 150000003505 terpenes Chemical class 0.000 description 1
- RGYLYUZOGHTBRF-BIHRQFPBSA-N tetragastrin Chemical compound C([C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)CC=1C2=CC=CC=C2NC=1)CCSC)C(N)=O)C1=CC=CC=C1 RGYLYUZOGHTBRF-BIHRQFPBSA-N 0.000 description 1
- BKVIYDNLLOSFOA-OIOBTWANSA-N thallium-201 Chemical compound [201Tl] BKVIYDNLLOSFOA-OIOBTWANSA-N 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 229940127044 therapeutic radiopharmaceutical Drugs 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- PLHJCIYEEKOWNM-HHHXNRCGSA-N tipifarnib Chemical compound CN1C=NC=C1[C@](N)(C=1C=C2C(C=3C=C(Cl)C=CC=3)=CC(=O)N(C)C2=CC=1)C1=CC=C(Cl)C=C1 PLHJCIYEEKOWNM-HHHXNRCGSA-N 0.000 description 1
- 229950009158 tipifarnib Drugs 0.000 description 1
- 230000000451 tissue damage Effects 0.000 description 1
- 231100000827 tissue damage Toxicity 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 238000003146 transient transfection Methods 0.000 description 1
- 102000035160 transmembrane proteins Human genes 0.000 description 1
- 108091005703 transmembrane proteins Proteins 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 239000013638 trimer Substances 0.000 description 1
- 239000001226 triphosphate Substances 0.000 description 1
- 230000004565 tumor cell growth Effects 0.000 description 1
- 230000005851 tumor immunogenicity Effects 0.000 description 1
- 239000000225 tumor suppressor protein Substances 0.000 description 1
- 230000010472 type I IFN response Effects 0.000 description 1
- 231100000402 unacceptable toxicity Toxicity 0.000 description 1
- 210000003932 urinary bladder Anatomy 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 230000024883 vasodilation Effects 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 229960004449 vismodegib Drugs 0.000 description 1
- BPQMGSKTAYIVFO-UHFFFAOYSA-N vismodegib Chemical compound ClC1=CC(S(=O)(=O)C)=CC=C1C(=O)NC1=CC=C(Cl)C(C=2N=CC=CC=2)=C1 BPQMGSKTAYIVFO-UHFFFAOYSA-N 0.000 description 1
- 238000012800 visualization Methods 0.000 description 1
- WAEXFXRVDQXREF-UHFFFAOYSA-N vorinostat Chemical compound ONC(=O)CCCCCCC(=O)NC1=CC=CC=C1 WAEXFXRVDQXREF-UHFFFAOYSA-N 0.000 description 1
- 229960000237 vorinostat Drugs 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 230000004584 weight gain Effects 0.000 description 1
- 235000019786 weight gain Nutrition 0.000 description 1
- 208000016261 weight loss Diseases 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 229940053867 xeloda Drugs 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical compound [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 description 1
- 229940072168 zocor Drugs 0.000 description 1
- WHNFPRLDDSXQCL-UAZQEYIDSA-N α-msh Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(N)=O)NC(=O)[C@H](CO)NC(C)=O)C1=CC=C(O)C=C1 WHNFPRLDDSXQCL-UAZQEYIDSA-N 0.000 description 1
Images


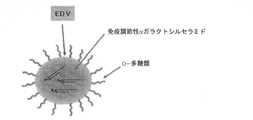






































































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/66—Microorganisms or materials therefrom
- A61K35/74—Bacteria
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7034—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin
- A61K31/704—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin attached to a condensed carbocyclic ring system, e.g. sennosides, thiocolchicosides, escin, daunorubicin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/337—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having four-membered rings, e.g. taxol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5383—1,4-Oxazines, e.g. morpholine ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
- A61K38/21—Interferons [IFN]
- A61K38/217—IFN-gamma
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/6807—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug or compound being a sugar, nucleoside, nucleotide, nucleic acid, e.g. RNA antisense
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6875—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody being a hybrid immunoglobulin
- A61K47/6879—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody being a hybrid immunoglobulin the immunoglobulin having two or more different antigen-binding sites, e.g. bispecific or multispecific immunoglobulin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5005—Wall or coating material
- A61K9/5063—Compounds of unknown constitution, e.g. material from plants or animals
- A61K9/5068—Cell membranes or bacterial membranes enclosing drugs
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/111—General methods applicable to biologically active non-coding nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
- C12N15/1137—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing against enzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/14—Type of nucleic acid interfering N.A.
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/14—Type of nucleic acid interfering N.A.
- C12N2310/141—MicroRNAs, miRNAs
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/17—Immunomodulatory nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2320/00—Applications; Uses
- C12N2320/30—Special therapeutic applications
- C12N2320/32—Special delivery means, e.g. tissue-specific
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y207/00—Transferases transferring phosphorus-containing groups (2.7)
- C12Y207/11—Protein-serine/threonine kinases (2.7.11)
- C12Y207/11001—Non-specific serine/threonine protein kinase (2.7.11.1), i.e. casein kinase or checkpoint kinase
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Genetics & Genomics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Zoology (AREA)
- Microbiology (AREA)
- General Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Wood Science & Technology (AREA)
- Mycology (AREA)
- Immunology (AREA)
- Biophysics (AREA)
- Gastroenterology & Hepatology (AREA)
- Plant Pathology (AREA)
- Biochemistry (AREA)
- Physics & Mathematics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Medicinal Preparation (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description
本発明の一実施形態は必要とする被験体を治療する方法に関し、この方法は、有効量の本明細書に開示される組成物を被験体に投与することを含む。一実施形態では、対象がヒト、非ヒト霊長類、イヌ、ネコ、ウシ、ヒツジ、ウマ、ウサギ、マウス、またはラットである。
一実施形態では、対象はヒトである。
一実施形態では、対象はがんに罹患している。1つの実施形態において、がんは、肺がん、乳がん、脳がん、肝臓がん、結腸がん、肛門がん、膵臓がん、および膀胱がんからなる群より選択される。一実施態様では、がんは急性リンパ芽球性白血病、急性骨髄性白血病、副腎皮質がん、AIDS関連がん、AIDS関連リンパ腫、虫垂がん、星状細胞腫、非定型奇形腫/ラブドイド腫瘍、基底細胞がん、膀胱がん、脳幹グリオーマ、脳腫瘍、乳がん、気管支腫瘍、バーキットリンパ腫、原発部位不明がん、カルチノイド腫瘍、主要サイト不明がん、中枢神経系非定型奇形腫様/ラブドイド腫瘍、中枢神経系胚芽腫、子宮頸がん、小児がん、脊索腫、慢性リンパ性白血病、慢性骨髄性白血病、慢性骨髄増殖性疾患、大腸がん、結腸直腸がん、頭蓋咽頭腫、皮膚T細胞リンパ腫、内分泌膵島細胞腫瘍、子宮体がん、上衣芽細胞腫、上衣芽腫、上衣腫、食道がん、感覚神経肉腫、ユーイング肉腫、頭蓋外胚細胞腫瘍、性腺外生殖細胞腫瘍、肝外胆管がん、胆がん、胃(胃)がん、胃腸カルチノイド腫瘍、消化管間質細胞腫瘍、消化管間質腫瘍(GIST)、妊娠性絨毛腫瘍 、神経膠腫、有毛細胞白血病、 心臓腫瘍、頭頸部がん、ホジキンリンパ腫、下咽頭がん、眼内黒色腫、小島細胞腫瘍、カポジ肉腫、腎臓ガン、ランゲルハンス細胞組織球症、喉頭ガン、唇ガン、肝臓がん、悪性繊維性組織サイト骨ガン、脂肪肉腫、髄芽腫、黒色腫、メルケル細胞がん、メルケル細胞がん、中皮腫、原発不明の転移性扁平上皮首がん、口腔がん、多発性内分泌腫瘍症候群、多発性骨髄腫/形質細胞腫瘍、菌状息肉症、骨髄異形成症候群、鼻腔がん、鼻咽頭がん、神経芽腫、非ホジキンリンパ腫、非黒色腫皮膚がん、非小細胞肺がん、口腔がん、口腔癌、中咽頭がん、骨肉腫、その他の脳および脊髄腫瘍、卵巣胚細胞腫瘍、卵巣低悪性度腫瘍、膵がん、乳頭腫症、副鼻腔がん、副甲状腺がん、骨盤がん、ペニスのがん、咽頭がん、中間的な分化を示す松果体実質細胞腫瘍、松果体芽腫、下垂体腫瘍、形質細胞新生物/多発性骨髄腫、胸膜肺芽腫、 原発性中枢神経系(CNS)リンパ腫、原発性肝細胞腫瘍、胸膜肺芽腫、原発性肝細胞がん、前立腺がん、腎細胞がん、腎細胞がん、腎細胞がん、網膜芽細胞腫、網膜芽細胞腫、横紋筋肉腫、セザリー症候群、唾液腺がん、小細胞肺がん、小腸がん、軟部組織肉腫、扁平上皮がん、頸部がん、胃(胃)がん、テント上原始神経外胚葉性腫瘍、T細胞リンパ腫、精巣がん、咽頭がん、胸腺がん、胸腺腫、甲状腺がん、移行上皮がん、腎盂および尿管の移行上皮がん、絨毛腫瘍、尿管がん、尿道がん、子宮がん、子宮肉腫、膣がん、外陰がん、ワルデンシュトレームマクログロブリン血症、およびウィルムス腫瘍からなる群より選択される。
一実施形態において、脳がんまたは脳腫瘍は、脳幹グリオーマ、中枢神経系非定型奇形腫様/ラブドイド腫瘍、中枢神経系胚芽腫、星細胞腫、頭蓋咽頭腫、上衣芽腫、上衣腫、髄芽腫、髄上皮腫、中間的分化の松果体実質腫瘍、テント上原始神経外胚葉性腫瘍および松果体芽腫からなる群より選択される。
一実施形態では、組成物が数週間にわたって少なくとも1週間に1回投与される。一実施形態において、組成物は、数週間〜数ヶ月にわたって週に少なくとも1回投与される。
一実施形態では、組成物が週に少なくとも1回、約2、約3、約4、約5、約6、約7、約8、約9、約10、約11、約12、約13、約14、約15、約16、約17、約18、約19または約20週間以上投与される。一実施形態では、組成物が毎週約2回投与される。1つの実施形態において、組成物は、約2、約3、約4、約5、約6、約7、約8、約9、約10、約11、約12、約13、約14、約15、約16、約17、約18、約19または約20週間以上、週2回投与される。
上記の概要および図面の以下の説明ならびに詳細な説明は、例示的かつ説明的なものである。それらは本発明のさらなる詳細を提供することを意図しているが、限定として解釈されるべきではない。他の目的、利点、および新規な特徴は、本発明の以下の詳細な説明から当業者には容易に明らかになるのであろう。
本発明は、(i)抗腫瘍剤およびI型インターフェロンアゴニスト;(ii)抗腫瘍剤およびII型インターフェロンアゴニスト;または(iii)抗腫瘍剤、I型インターフェロンアゴニスト、およびII型インターフェロンアゴニストの組み合わせを含む組成物であって、少なくとも抗腫瘍剤が無傷の細菌由来ミニ細胞内にパッケージされている組成物が、がん治療戦略を相乗的に改善することができるという発見に基づく。
A.細菌のミニ細胞デリバリー法の概要
がん細胞に化学療法剤を送達するための細菌ミニ細胞の使用は、以前に記載されている。がんを治療するためのこの送達方法は、毒性化学療法剤もしくは薬物、または機能性核酸を、典型的には直径約400nmである細菌由来ミニ細胞にパッケージ化する。典型的には、ミニ細胞が特異的がん細胞を標的とする抗体を有する。この抗体はがん細胞の表面に結合し、ミニ細胞はがん細胞によって細胞内に取り込まれる。このようにして、毒性化学療法剤は全身に広く分布せず、したがって、毒性薬物または化合物ががん細胞内に送達されるにつれて、副作用および不耐性の機会を減少させる。毒性化学療法剤の送達ビヒクルとして抗体標的化ミニ細胞を用いると、がん細胞を死滅させるのに必要な薬物がはるかに少なくなり、したがって治療指数が改善される。
(i) ミニ細胞中にパッケージ化化された抗悪性腫瘍剤とミニ細胞パッケージ化I型インターフェロンアゴニストとの併用
第1の実施形態では、細菌ミニ細胞にパッケージされたI型インターフェロンアゴニストと組み合わせた細菌ミニ細胞パッケージされた抗腫瘍剤の組み合わせに関する組成物および方法が記載される。

第2の実施形態は、ミニ細胞パッケージ化I型インターフェロンアゴニストまたはミニ細胞パッケージ化抗悪性腫瘍剤と、ミニ細胞パッケージ化I型インターフェロンアゴニストおよびII型インターフェロンアゴニスト(細菌ミニ細胞を含まない)とを組み合わせたミニ細胞パッケージ化防新生物剤を使用する方法および組成物を対象とする。
さらに、他のすべての治療選択肢を使い果たしたステージ4の膵臓がん患者で得られた結果は顕著であった。患者の腫瘍マーカー(CA 19−9)の量は最初の3回の投与後に90%以上低下し、これはわずか10日間の治療に相当した。10回投与後、これはさらに低下し、腫瘍マーカー値はほぼ95%低下した。この患者はまた、IV期膵がんを呈するほとんどの患者が経験する悪液質状態とは対照的に、有意な体重増加を示し、生活の質の顕著な改善を報告した。これらの結果は、特に進行膵がんに伴う予後不良を考えると劇的である。
要約すると、5例がEGFR(V)EDVPNU/DoxまたはEGFR(V)EDVPNU+EDV40mer/60mer(I型IFNアゴニスト)アImukin(II型IFNアゴニスト)の計69回を受けた。治療は忍容性が良好であり、免疫調節免疫賦活剤の添加は、単剤負荷EDVおよび標的EDVの安全性プロファイルを変化させないようであった。
実施例13は、ミニ細胞にパッケージされた抗新生物剤をII型インターフェロンアゴニスト、例えばIFN−繧ニ組み合わせることの有効性を評価するために実施された種々の研究の結果を記載している。結果は、II型インターフェロンアゴニストの追加が肺がんおよび乳がんを含む種々のがんの異種移植モデルにおいてミニ細胞にパッケージされた抗新生物剤の抗がん効果を増強または増強することを示す。さらに、実施例13に記載され、以下の表4に抜粋されるデータは、抗腫瘍剤単独に通常耐性である腫瘍の処置において、ミニ細胞中にパッケージされた抗腫瘍剤を含む組成物へのII型インターフェロンアゴニストの添加が腫瘍安定化を達成するために必須であることを実証する。したがって、ミニ細胞パッケージ化抗腫瘍剤をII型インターフェロンアゴニストと組み合わせることにより、薬物耐性を克服することができる。
本発明者らはまた、ミニ細胞パッケージ化抗腫瘍剤、ミニ細胞パッケージ化I型インターフェロンアゴニスト、およびII型インターフェロンアゴニスト(単独またはミニ細胞パッケージ化のいずれか)の三重組み合わせが、劇的な抗がん効果を生じ得ることを発見した。具体的には、実施例14が後期内因性腫瘍(脳がん、肉腫、またはメラノーマ)を有するイヌの処置を、ミニ細胞パッケージ化抗腫瘍剤、ミニ細胞パッケージ化I型インターフェロンアゴニスト、およびII型インターフェロンアゴニストの組合せで詳述する。結果は、組み合わせ組成物が十分に耐性であったことを示す。さらに、7匹の評価可能な動物のうち6匹(85.7%)では疾患は安定化したが、1匹のイヌはほぼ部分奏効(腫瘍サイズの29.8%の縮小)を達成した。
別の実施形態では、本発明がミニ細胞パッケージ化抗腫瘍薬と、ミニ細胞パッケージ化II型インターフェロンアゴニスト、例えば瘁|ガラクトシルセラミド(瘁|GC)との組み合わせを含み、I型インターフェロンアゴニストの非存在下で、驚くべき抗がん効果を示す発見に基づく組成物に関する。
実施例16は、腫瘍細胞表面受容体に標的化されたミニ細胞パッケージ化抗腫瘍剤が、がん免疫療法、例えば、細胞−免疫療法として機能することを実証するデータを詳細に記載する。特に、実施例は、マクロファージ、NK細胞および樹状細胞を含む自然免疫系の細胞を活性化する細菌ミニ細胞の能力を例示する。これに続いて樹状細胞の成熟と抗原提示が起こり、腫瘍特異的細胞傷害性T細胞が産生され、さらなる免疫細胞の腫瘍微小環境への動員をもたらす適応T細胞応答が起こる。このアプローチは免疫原性腫瘍微小環境を作り出し、複数の免疫細胞サブセットにも作用することで、患者に生じる可能性のある原発性および/または適応抵抗性を回避することによって、免疫療法による現在の落とし穴の一部を回避するものである。
実施例17は化合物に関連する重篤な毒性のために従来の手段を使用して送達することができない、超毒性抗腫瘍剤、例えばPNU159682の有効な送達を実証するデータを詳述する。具体的には実施例17がpM範囲の薬物耐性がん細胞であっても、PNU159682がIC50を有する超細胞毒素である方法を詳述し(Quintieriら、2005)、これは重篤な全身毒性のために化合物を臨床的に使用することができないことを意味する(StaudacherおよびBrown、2017)。しかし、細菌ミニ細胞中にカプセル化される場合、PNU159682のようなスーパー細胞毒素は、ほとんど副作用を伴わずに腫瘍に効果的に送達され得る。
上記のように、本発明の組成物は、少なくとも2つの異なる活性剤、抗腫瘍剤およびI型インターフェロンアゴニスト、II型インターフェロンアゴニスト、またはI型インターフェロンアゴニストおよびII型インターフェロンアゴニストと抗腫瘍剤との両方を含む。3つの異なる活性剤は、1つ、2つ、または3つの異なるミニ細胞にパッケージ化することができる。II型インターフェロンアゴニストはまた、ミニ細胞にパッケージされることなく、本発明の方法および組成物に含まれ得る。
「抗悪性腫瘍剤」とは、化学的、生物学的なものであるかにかかわらず、腫瘍細胞の増殖、発生、成熟又は拡散を阻止又は阻害する医薬品をいう。「抗悪性腫瘍剤」とは「抗悪性腫瘍剤」及び「化学療法剤」と互換的に使用されるものである
本開示の文脈において、所定の脳腫瘍患者を治療するための抗腫瘍剤を選択することは、従来の医療行為と一致して、いくつかの因子に依存する。これらの因子には患者の年齢、カルノフスキー・スコア、および患者が受けた可能性のあるあらゆる治療歴が含まれるが、これらに限定されない。一般に、神経腫瘍学の原則と実践、MMehta(Demos Medical Publishing 2011)、および神経腫瘍学の原則、DSchiffおよびPO’NeILl、edsを参照のこと。(McGraw−HILl 2005)。
(10) ヒストンデアセチラーゼ(HDAC)阻害薬、例えば、ボリノスタット、エンチノスタット(Sndx−275)、モセチノスタット(MGCD103)、パノビノスタット(LBH589)、ロミデプシン、バルプロ酸[0049]、サイクリン依存性キナーゼ(CDK)阻害剤、例えば、フラボピリドール、オロモウシン、ロスコビチン、ケンパウロン、AG−024322(ファイザー)、ファスカプリシン、リウビジン、プルバラノールA、NU2058、BML−259、SU 9516、PD−0332991、P276‐00[0050]、熱ショックタンパク質(HSP90)阻害剤、例えば、ゲルダナマイシン、タネスピマイシン、アルベスピマイシン、ラジシコール、 デグリン、およびBIIB021;(11) マウス二重微小染色体2(MDM2)阻害剤、例えば、Cis−イミダゾリン、ベンゾジアゼピンジオン、スピロ−オキシインドール、イソキノリノン、チオフェン、5−デアザフラビン、トリプタミン;(12) 未分化リンパ腫キナーゼ(ALK)阻害薬、例えば、アミノピリジン、ジアミノピリミジン、ピリドイソキノリン、ピロロピラゾール、インドロカルバゾール、ピロロピリミジン、ジアニリノピリミジン;(13) ベンズアミド、フタラジノン、トリサイクリックインドール、ベンズイミダゾール、インダゾール、ピロロカルバゾール、フタラジノン、イソインドリノンによって例示されるポリ[ADPリボース]ポリメラーゼ(PARP)阻害薬;および(14) アンサクリン、アスパラギナーゼ(El−spar)、ヒドロキシ尿素、ミトキサントロン(ノバントロン)、ミトタン(ライタントロン)、メイタンシノイド、レチノイン酸誘導体、骨髄増殖因子(サルグラモスチムおよびフィルグラスチム)、アミホスチン、葉酸代謝を破壊する薬剤、例えばペメトレキセド、リボヌクレオチドレダクターゼ阻害剤(ヒドロキシ尿素)、副腎皮質ステロイド阻害剤(ミトタン)、酵素(アスパラギナーゼおよびペグアスパラガーゼ)、抗微小管剤(エストラムスチン)、およびレチノイド(ベキサロテン、イソトレチノイン、トレチノイン(ATRA))によって例示される、その他の抗がん剤。
本開示に従って使用可能な活性薬剤は、上記に列挙した薬物種類または特定の薬剤に限定されない。異なる発見舞台は、がん細胞のユニークな分子特徴に向けられる新しい薬剤を産生し続ける;実際、数千のそのような化学的および生物学的薬剤が発見されており、そのうちのいくつかのみをここに列挙する。しかし、親水性または疎水性の多様な活性薬剤のパッケージ化に適応する、無傷の細菌由来ミニ細胞および死滅細菌細胞の驚くべき能力は本開示の発見に従って、ミニ細胞にパッケージ化場合に、本質的に任意のそのような薬剤ががんを治療する可能性を有することを意味する。
「放射性核種」とは、不安定な核をもつ原子、すなわち、核内で新たに生成された放射線粒子または原子電子のいずれかに付与されるのに利用可能な過剰なエネルギーを特徴とする原子である。本明細書では放射性核種を「放射性同位元素」、「放射性イメージング剤」、または「放射性標識」と呼ぶこともある。放射性核種はイメージングおよび/または治療目的で使用することができる。それらは、ミニ細胞内に含有され得るか、またはミニ細胞外表面上のリガンド、ペプチド、または糖脂質に付着され得る。結合は、直接またはリンカーを介して、メルカプトアセチルトリグリシン(MAG3)、DOTA、EDTA、HYNIC、DTPA、またはクラウンエーテルなどのキレート剤を含むキレート化部分を含有するリンカーを使用することができる。キレート剤は、ミニ細胞表面成分に直接付着させるか、またはリンカーを介してミニ細胞に付着させることができる。多くの放射性核種が当技術分野で知られており、イットリウム−90、テクネチウム−99m、ヨウ素−123、ヨウ素−124、ヨウ素−125、ヨウ素−131、ルビジウム−82、タリウム−201、ガリウム−67、フッ素−18、キセノン−133、およびインジウム−111など、多くの放射性核種が医療用途に適していることが知られている。
本開示において使用される抗腫瘍剤はまた、化学療法薬であり得る。本明細書において、「化学療法剤」、「化学療法剤」、および「化学療法剤」は、腫瘍細胞を殺すかまたは破壊する能力を有する薬剤を意味するために互換的に使用される。化学療法剤は以下にさらに詳述されるように、小分子薬物または生物学的薬物であり得る。
化学療法を目的として設計されたある種の分子は、許容できない毒性のために前臨床または臨床試験中に失敗する。本発明者らは高毒性または「超毒性」の化学療法薬をミニ細胞にパッケージ化し、続いて腫瘍患者に全身的に送達すると、薬剤が腫瘍細胞に送達されることを示した。さらに、腫瘍細胞が破壊され、薬剤含有細胞質が近傍の正常組織に放出された後でも、結果は正常組織に対する毒性ではない。これは薬剤が既にDNAなどの腫瘍細胞構造に結合しており、もはや正常細胞を攻撃できないためである。したがって、本発明はがん患者への高毒性(「超毒性」)化学療法薬の送達に特に有用である。
別の局面において、ミニ細胞は、生物学的化学療法薬を含み得る。このような薬物の実施例としては、アスパラギナーゼ、AIN―457、バピネオズマブ、ベリムマブ、ブレンツキシマブ、ブリアキンマブ、カナキヌマブ、セツキシマブ、ダロツズマブ、デノスマブ、エプラツズマブ、エストラフェナトクス、ファレツヅマブ、フィギツムマブ、ガリキシマブ、ゲムツズマブ、ギレンツキシマブ(WX-G250)、イプリツモマブ、イノツズマブ、イピリズマブ、メポリズマブ、ムロモナブ−CD3、ナプツモマブ、ネシツムマブ、ニモツムマブ、オクレリズマブ、オファツムマブ、、オテリズマブ、オゾガミシン、パジバキシマブ、パニツムマブ、ペルツヅマブ、ラムシルマブ、レスリズマブ、リツキシマブ、REGN88、ソラネズマブテプリズマブ、タネズマブ、チウキセタン、トシツモマブ、トラスツズマブ(ハーセプチン(登録商標))、トレメリムマブ、ベドリズマブ、ザルツムマブ、およびザノリムマブが挙げられるが、これらに限定されない。
「機能性核酸」とは、宿主細胞への導入の際に、タンパク質の発現を特異的に妨害する核酸分子をいい、がんの治療に関して、本開示によれば、無傷の細菌由来ミニ細胞を介してがん細胞に送達される機能性核酸ペイロードは腫瘍細胞増殖、血管新生または化学療法に対する抵抗性を促進する、および/またはアポトーシスまたは細胞周期停止を阻害する遺伝子、すなわち「がん促進遺伝子」を阻害することが好ましい。
本発明の組成物はI型インターフェロンアゴニスト、すなわち、I型インターフェロンのレベル(例えば、活性または発現レベル)を増加させる薬剤を含み得る。ヒトI型インターフェロン(IFN)は、免疫系の活性の調節を助けるインターフェロンタンパク質の大きなサブグループである。インターフェロンはインターフェロン受容体に結合する。I型IFNはすべて、IFNAR1鎖とIFNAR2鎖からなるIFN−α(IFNAR)として知られる特異的な細胞表面受容体複合体に結合する。IFNARはIFN−α(アルファ)、IFN−β(ベータ)、IFN−κ(カッパー)、IFN−δ(デルタ)、IFN−ε(イプシロン)、IFN−τ(タウ)、IFN−ω(オメガ)、IFN−ζ(ゼータ、リミチンとしても知られる)と呼ばれる哺乳類のI型IFNである。
図2は、免疫調節性の60mer二本鎖DNAを含むミニ細胞の例示的な実施形態のグラフ描写を示す。本発明者らは、EGFR標的ミニ細胞を有する二本鎖DNAなどのI型インターフェロンアゴニストの送達が細胞傷害性薬物を負荷されたミニ細胞の免疫賦活剤(すなわち、抗腫瘍効果を増強する)として作用することを発見した。実施例11を参照されたい。したがって、超毒性薬剤PNU−159682とパッケージされたミニ細胞を組み合わせることにより、抗腫瘍効果が増強され、この治療は後期膵臓がん患者によって十分に忍容性であった。実施例12を参照されたい。
二本鎖RNAはI型IFNの誘導因子である。RNAヘリカーゼであるレチノイン酸誘導遺伝子I(RIG―I)とメラノーマ分化関連遺伝シ5(MDA5)は、I型IFN分泌を誘発する細胞質受容体である。これらの受容体(RIG−I様受容体)はミトコンドリア局在アダプター分子IPS−1あるいはMAVSとキナーゼTBK1、IKKiを介してシグナルを伝達し、IRF3を活性化し、I型IFN易伝子の転写を誘導する(河合・秋良、2010)。RIG−IおよびMDA5は、それらの5’末端でトリリン酸化されたウイルスRNAに応答する(LeungおよびAmarasinghe、2016; Lu ら、2010; Marq ら、2011; Wang ら、2010)。
RNAポリメラーゼIIIは、ポリ(dA:dT)DNAのための細胞質ゾルDNAセンサーである(Ablasser ら、2009)。細胞質ゾルでは、RNAポリメラーゼIIIがポリ(dA:dT)を5’トリリン酸化を伴うRNAに変換する。変換された5’-ppp RNAは、次にRIG−I−MAVS経路とNF鵤活性化を開始させて、I型IFN分泌を誘発する。
サイトゾルDNAセンサー、IRFのDNA依存性アクチベーター(DAI)またはZ-DNA結合タンパク質1は、TBK1及びIRF3を介する機構において、右巻きのdsDNA高次構造(B-DNA)に応答してI型IFNを誘導することが知られている(Kawai and Akira、2010)。RNAポリメラーゼIIIもB−DNAを5’-ppp RNAに転写し、その後RIG−Iを介してI型IFN転写を活性化する(Chiuら、2009)。これらの転写因子がリン酸化されると、I型IFNファミリーのすべての遺伝子の発現を促すのに役立ち、それによってI型IFN産生が増幅される。多くのサイトゾルDNAセンサーが細胞内病原性DNAを認識することが報告されている。例えば、Xiaら、"DNA sensor cGAS-mediated immune recognition"、Protein Cell、7(11): 777−791 (2016)から抜粋された図26を参照されたい。
cGASは、細胞質DNAを認識するDNAセンサーである(Ablasser ら、2013a; Ablasser et al、2013b; Gao et al、2013a; Li et al、2013b; Schoggins ら、2014; Sun ら、2013; Wu ら、2013)。36bpより長い二本鎖DNA(dsDNA)がcGAS活性化に最適である(Gaoら、2013b)。ポストDNA結合、cGASはATPおよびGTPが触媒ポケットに入ることを可能にする立体配座変化を受け、STING−TBK1軸の強い活性剤であるcGAMPの合成をもたらす(CivrILら、2013; Gaoら、2013b; Kranzuschら、2013; Wuら、2013; Zhangら、2014)。cGASはdsDNAおよびDNA−RNAハイブリッドによって活性化され得る(MaNKanら、2014)。
細菌のセカンドメッセンジャーであるcyclic-di-GMPはDAIまたは他の既知の細胞質受容体とは無関係であるが、TBK1及びIRF3を必要とする機構を介して、I型IFNを強力に誘導する(McWhirterら、2009)。
一部の細胞種、例えばマクロファージやDCでは、I型IFNがそれぞれdsRNAやリポ多糖による膜貫通受容体トル様受容体3(TLR3)やTLR4のトリガーに応答して産生される。TLR3とTLR4はアダプター分子TRIFを介してシグナルを伝達し、TBK1と会合してIRF3を活性化する(川井・秋良、2010)。
環状ジヌクレオチド(CDN)[サイクリックジ(グアノシン5’一りん酸)、環状ジ(アデノシン5’一りん酸)、及び環状GMPは、インターフェロン遺伝子の細胞質パターン認識受容体刺激剤(STING)を介してTBK1/IRF3/1型インターフェロンシグナル伝達軸を活性化する病原体関連分子パターン分子(PAMP)のクラスである。
本発明の組成物および方法はII型IFNアゴニスト、すなわち、II型インターフェロンのレベル(例えば、活性または発現レベル)を増加させる薬剤を含み得る。型IIインターフェロン(IFN)のクラスは、現在、IFN縺iガンマ)と呼ばれるメンバーを含む。成熟IFN−繧ヘ抗平行ホモ二量体であり、IFN−γ受領隊(IFNGR)複合体に結合して標的細胞内でシグナルを誘発する。IFNGRはIFNGR1とIFNGR2と名づけられた各分子の2つのサブユニットからできている。IFN−繧ヘ、免疫応答および炎症応答の調節に関与している;ヒトではインターフェロン−繧ヘ1種類のみである。活性化T細胞やナチュラルキラー細胞で産生される。IFN−繧ヘI型IFNの作用を増強する。Th1細胞から放出されたIFN−繧ヘ感染部位に白血球を動員し、炎症を亢進させる。また、マクロファージを刺激して、飲み込まれた細菌を殺す。Th1細胞から放出されるIFN−繧ヘ、Th2応答の調節にも重要である。IFN−繧ヘ免疫応答の調節に重要な役割を果たしており、その産生は自己免疫疾患を引き起こす可能性がある。
「ミニ細胞」という用語はここでは染色体を欠き(「無染色体」)、二分裂の際に、細胞分裂とDNA分離との協調の乱れによって生じた細菌細胞の誘導体を意味するために用いられる。ミニ細胞はいわゆる「膜ブレブ」(大きさが約0.2・以下)のような他の小さな小胞とは異なっている。この小胞が特定の状況下で自然に生成・放出されるが、特定の遺伝子再編成やエピソーム遺伝子発現によるものではない。同じトークンによって、無傷のミニ細胞は同様ゴーストとは区別される。同様ゴーストは特異的な遺伝子再編成やエピソーム遺伝子発現のために生成されない。本開示で使用される細菌由来のミニ細胞は完全に無傷であり、したがって、破壊または分解され、除去さえされる外膜または画定膜によって特徴付けられる、他の無染色体形態の細菌細胞誘導体とは区別される。米国特許を参照されたい。第7,183,105号、第111欄、第54行以降。本開示のミニ細胞を特徴付ける無傷の膜は、腫瘍発明内で、取り込み後にペイロードが放出されるまで、ミニ細胞内での治療用ペイロードの保持を可能にする。
活性剤または抗腫瘍剤(例えば、小分子薬物、タンパク質および機能性核酸)は、複数の無傷のミニ細胞を活性剤と一緒に緩衝液中で共インキュベートすることによって、ミニ細胞中に直接パッケージ化され得る。緩衝液の組成は、この分野でよく知られている条件に応じて変化させ、無傷のミニ細胞における活性剤の負荷を最適化することができる。緩衝液はまた、薬剤に依存して(例えば、核酸ペイロードの場合、ヌクレオチド配列またはミニ細胞にロードされる核酸の長さに依存して)変化され得る。ローディングに適した例示的な緩衝液にはリン酸緩衝生理食塩水(PBS)が含まれるが、これに限定されない。一旦パッケージ化されると、活性剤はミニ細胞内に残り、分解から保護される。無菌生理食塩水中でインキュベートしたsiRNAパッケージ化ミニ細胞を用いた長期インキュベーション研究は例えば、siRNAの漏出がないことを示した。
本発明者らは腫瘍細胞周囲の血管が完全性の喪失を示すことを発見した。すなわち、血管は血液脳関門(BBB)環境においても、大きな開窓を有し、「漏れやすい」状態である。がん細胞が定着すると、新たな血管の形成を促進する物質−血管新生と呼ばれるプロセス−を分泌する。これらの血管は速やかに成長し、正常な血管とは異なり、50nmから1.2・(透過性亢進血管系)の「穴」(開窓部)で漏れやすい。リポソームなどの薬物送達粒子は、現在、腫瘍微小環境を支持する漏出性血管系からの血管外遊出を含む受動的方法によって腫瘍標的化をもたらすと考えられている。Hobbsら、1998。異常な腫瘍微小環境は間質性高血圧を特徴とし、この現象が抗がん抗体治療薬のアクセスを制限し得ることが示されているが、これは免疫リポソーム(Nielsenら、2002)およびQuantum Dotsに結合体化された抗体(Gaoら、2004)によって例示されるように、絶対的な障壁であるとは思われない。この現象は、特異的に指向された腫瘍抗体を保有するという追加の利点を有するEDVにも当てはまる。静注後、EDVは腫瘍の微小環境に漏出し、その後、がん細胞表面受容体との結合およびエンドサイトーシスを介して能動的なターゲティングが行われる。したがって、従来の理解とは対照的に、ミニ細胞と同じ大きさ、すなわち、BBBの上記のコンセンサス孔径限界よりもはるかに大きい粒子は、それにもかかわらず、漏出性血管壁の開窓よりも小さく、したがって、これらの開窓を通って腫瘍微小環境に受動的に溢出することができる。
正常細胞ではなく腫瘍細胞に過剰発現している受容体は、in vivo腫瘍イメージングの優れた候補である。今日まで、多くの腫瘍標的化ペプチドおよびそれらのアナログが、以下に記載されるように同定されている。
本発明はその範囲内に、(1)抗腫瘍剤、(2)I型IFNアゴニスト、および/または(3)II型IFNアゴニストの1つ以上の組み合わせをペイロードとして有するミニ細胞を含む組成物または製剤を含む。3つの成分すべてを含む組成物において、抗腫瘍剤、I型IFNアゴニスト、およびII型IFNアゴニストは、1つ以上のミニ細胞に含まれ得る。例えば:(a)抗腫瘍剤、I型IFNアゴニスト、およびII型IFNアゴニストは同じミニ細胞内に含まれ得る;(b)抗腫瘍剤およびI型IFNアゴニストが第1のミニ細胞内に含まれ得、そしてII型IFNアゴニストは第2のミニ細胞内に含まれ得る;(c)抗腫瘍剤およびII型IFNアゴニストが第1のミニ細胞内に含まれ得、そしてI型IFNアゴニストは第2のミニ細胞内に含まれ得る;または(d)抗腫瘍剤が第1のミニ細胞内に含まれ得、そしてII型IFNアゴニストは第2のミニ細胞内に含まれ得、そしてII型IFNアゴニストは第2のミニ細胞内に含まれ得る;または(e)抗腫瘍剤が第1のミニ細胞内に含まれ得、I型IFNアゴニストは第2のミニ細胞内に含まれ得、そしてII型IFNアゴニストは第3のミニ細胞内に含まれ得る。
本発明の製剤は局所的または全身的のいずれかで、所望の治療効果を達成するために、種々の経路を介して、および哺乳動物体内の種々の部位に投与され得る。送達は例えば、経口投与によって、体腔への製剤の適用によって、吸入または通気によって、または非経口、筋肉内、静脈内、門脈内、肝臓内、腹腔内、皮下、腫瘍内、または皮内投与によって達成され得る。投与の様式および部位は、標的細胞の位置に依存する。例えば、腫瘍転移は、標的ミニ細胞の静脈内送達を介してより効率的に処置され得る。原発性卵巣がんは、標的ミニ細胞の腹腔内送達を介して治療され得る。経路の組み合わせもまた、使用され得る。例えば、転移性膀胱がんにおいて、細胞傷害性薬物負荷および受容標的化ミニ細胞は膀胱内および静脈内に投与され得、免疫賦活剤パッケージ化(受容体標的化または非標的化)ミニ細胞は標的化薬物パッケージ化ミニ細胞と共に静脈内に投与され得る。標的化された、薬物パッケージ化ミニ細胞のin situ投与は膀胱表面露出腫瘍を標的とし得るが、静脈内投与されたミニ細胞の完全な組合せは組織局在腫瘍を標的とし得、また抗腫瘍免疫応答を誘発し得る。
本発明のミニ細胞は、汚染親細菌細胞を実質的に含まない。したがって、ミニ細胞含有製剤は、好ましくは107ミニ細胞当たり約1未満の汚染親細菌細胞、108ミニ細胞当たり約1未満の汚染親細菌細胞、109ミニ細胞当たり約1未満の汚染親細菌細胞、1010ミニ細胞当たり約1未満の汚染親細菌細胞、または1011ミニ細胞当たり約1未満の汚染親細菌細胞を含む。
本明細書中に記載される組成物は、癌に罹患している対象を処置するために使用され得る。本明細書に開示される方法は、少なくとも1つの抗腫瘍剤、インターフェロンI型アゴニスト、インターフェロンII型アゴニスト、またはインターフェロンI型アゴニストとインターフェロンII型アゴニストとの組み合わせを含む、本発明による組成物の有効量を対象に投与することを含む。抗新生物剤、インターフェロンI型アゴニスト、インターフェロンII型アゴニスト、またはインターフェロンI型アゴニストとインターフェロンII型アゴニストの組合せは、1つ以上のミニ細胞で構成される。
本明細書で使用される技術用語および科学用語は別段の定義がない限り、本発明が関係する当業者によって一般に理解される意味を有する。以下の説明および実施例において参照される材料、試薬などは、特に断らない限り、商業的供給源から入手可能である。
この実施例はミニ細胞(EDV)が化学療法薬の効率的な送達を提供し、マウス異種移植モデルにおいて腫瘍増殖を阻害することを示した。EDV標的化技術は、結腸癌、乳癌、卵巣癌、白血病、肺癌、中皮腫、および子宮癌を含む様々な癌のマウス異種移植モデルで試験されている。さらに、EDVがEGFR、ヒト上皮成長因子受容体2(HER2)、メソテリン(MSLN)、およびCD33を含む腫瘍細胞表面受容体に標的化されたので、種々の標的化部分が利用された。最後に、試験した標的EDVは、ドキソルビシン、パクリタキセル、モナストロール、イリノテカン、PNU159682のような超細胞傷害性薬剤、および新規チミジル酸シンターゼ阻害剤(OSI)を含む多種多様な細胞傷害性薬剤を含んでいた。PNU−159682は、ドキソルビシンよりも数千時間細胞傷害性であるアントラサイクリン類似体である。OSI7904は抗腫瘍活性を有するベンゾキナゾリン葉酸類似体である。チミジル酸シンターゼ阻害薬として、OSI−7904はチミジル酸シンターゼに非競合的に結合し、その結果、チミンヌクレオチド合成およびDNA複製が阻害される。
この実施例は、薬物を負荷したミニ細胞(EDV)がイヌに安全に投与され得ることを示した。
この実施例は、ミニ細胞(EDV)技術がサルによって十分に許容されることを示した。
3件のアカゲザル試験を実施し、空のEDV(1用量あたり2ラ1010まで)、EGFR標的ドキソルビシン負荷EDV(1用量あたり2×1010まで)、およびEGFR標的パクリタキセル負荷EDV(1用量あたり1×1011まで)の毒性を評価した。サルにEDVを週1回、5週間投与した(35日間反復投与試験)。
この実施例から、前臨床マウス試験(実施例1)、前臨床イヌ試験(実施例2)および前臨床サル試験(実施例3)では、わずかな炎症反応しか観察されなかったことが示された。これらの反応は、投与4時間後と同様に速やかに消失した。その他、血液学的及び生化学的パラメータに有意な変化は認められなかった。投与期間中、外観および行動は健康なままであった。
・ EDV表面露出免疫優性抗原に対する血清抗体反応は、LPSのO−多糖成分(IgGまたはIgM反応)である。抗O多糖抗体応答はT細胞非依存性であり、記憶応答を示さない。
・ 腫瘍細胞表面受容体(例えば、EGFR)にEDVを標的化するためのBsAbの構築に使用されるマウスIgGモノクローナル抗体に対する血清抗体応答。
本実施例は、進行固形腫瘍にパクリタキセル(Taxol(登録商標4 )ョ)を送達するためにミニ細胞(EDV)を使用する有望な結果を示した。
この実施例は、EGFRを標的としたドキソルビシンパッケージ化ミニ細胞(EDV)による治療が再発性膠芽腫に罹患した患者において忍容性が良好であったことを示している。患者の50%が疾患の安定化を示したが、部分奏効または完全奏効を経験した患者はいなかった。Whittle ら、J. Clin. Neurosci., 22(12):1889−1894(2015)。
この実施例では、マイクロRNA-16でパッケージされたEGFR標的EDVが有効性を検査した16人の患者のうち1人の中皮腫患者で部分奏効を示したのに対し、患者の62.5%は疾患が安定化し、患者の31.3%は疾患が進行したことが示された。試験データは、van Zandwijk ら、Lancet Oncol., 18(10):1386−1396 (2017)およびKaoら、Am.J. Respir. Crit. Care Med., 191(12): 1467-1469 (2015) に公開されている。この治療戦略は概して良好であった。
この実施例は、超毒性細胞傷害性化学療法剤であるPNU−159682が広範囲の他の化学療法薬よりも強力な癌細胞増殖阻害剤であることを示した。
この実施例はPNU−159682などの超毒性化学療法剤を送達するためにミニ細胞(EDV)を使用することが、肺癌異種移植モデルにおける腫瘍増殖を効果的に阻害することを示した。
この実施例はポロ様キナーゼ1(Plk1)、リボヌクレオチドレダクターゼ酵素1(RRM1)を標的とするsiRNA、またはmiRNA16aを標的とするsiRNAを送達するためにミニ細胞(EDV)を使用することが、癌細胞の増殖を効果的に阻害し得ることを示した。
この実施例は、インターフェロンI型アゴニストと併用される化学療法剤を送達するためにミニ細胞(EDV)を使用することが、肺癌異種移植片などの癌を治療するための有効な戦略であることを示した。インターフェロンI型アゴニストは、化学療法剤と同じまたは異なるミニ細胞中に存在同様。本実施例において、化学療法剤は超毒性薬物PNU−159682であり、インターフェロンI型アゴニストは、40mer二本鎖DNAである。
本実施例は、I型およびII型IFNアゴニストが進行固形腫瘍に罹患しているヒト患者におけるEGFR EDVPNU-159682 の抗癌作用を増大させることを示した。著しいことに、進行期膵臓癌患者においてさえ、この治療は、わずか3回の投与後に腫瘍マーカーレベルの90%の低下を生じ、患者は著しく改善された生活の質を示した。
薬剤および1型IFNアゴニストを含むミニ細胞:補助EDV40merを伴うEGFR(V) EDVPNU(1型IFNアゴニスト)を、進行固形腫瘍を有する被験者を対象とした非盲検単一施設探索的第1相試験(デザイナーEDV試験)において、3人の患者で評価した。適格とするために、標的化を容易にするために、患者は、腫瘍組織におけるEGFR発現のエビデンスを有する進行固形腫瘍の組織学的または細胞学的記録を得ることとした。患者は、標準的な1次、2次または3次治療レジメンの投与中または投与後に疾患の進行を示していなければならない。試験の主要なエンドポイントは、型EDV40mer(第1種IFNアゴニスト)とのEGFR(V)EDVPNU-159682の安全性と耐性を確立することであった。
シドニーのローヤルノースショア病院で最初の思いやりのある使用ケース研究が行われた。患者は68歳の女性で、ステージ IVの膵癌と診断された。Whipple手術(膵頭十二指腸切除術)を受け、第一選択治療としてゲムシタビンが投与された。FOLFIRINOX治療レジメンも受けたが、転移性肝疾患を発症した。彼女の腫瘍細胞をin vitroで試験し、PNU−159682に感受性であることが見出された。
この実施例はドキソルビシンをIFN−繧ニ組み合わせて送達するためにミニ細胞(EDV)を使用することが、マウス異種移植モデルにおいて改善された抗腫瘍効果を提供することを示した。
EGFREDVDox で処置したマウスは、A549肺癌異種移植片の腫瘍安定化を達成した(図20、黒四角)。対照的に、EGFREDVDoxおよびIFN−繧ナ処置したマウスは、全6回の投与後43日目までに非常に有意な腫瘍退縮を示した(図20、黒丸)。IFN−繧フみで処理したマウスは、抗腫瘍効果を示さず(図20、黒三角)、腫瘍は生理食塩水処理グループと同様に成長した(図20、白菱形)。従って、EGFREDVDoxとIFN−縺iII型IFN)とを組み合わせると、以下の表14に要約されるように、肺癌のマウス異種移植片モデルにおいて腫瘍退縮が生じた。
この実施例は、イヌの研究において、ミニ細胞技術および追加的にインターフェロン縺iイムキン)(II型IFN)を有する超毒性薬物PNU-159682およびI型IFNアゴニスト(40mer二本鎖DNA)を送達することが、良好な忍容性を示すことを示した。
この実施例から、パクリタキセルまたはドキソルビシンをパッケージ化したEDVはトル様受容体活性化と一致するサイトカイン反応をヒトで誘導するが、このEDV処理はインターフェロンII型反応を誘導しないことが示された。
この実施例は、腫瘍細胞表面受容体を標的としたEDVナノ細胞が自然免疫系および適応免疫系の活性化と併せて、スーパーサイトトキシンPNU−159682(682)を送達することによって、腫瘍に対する二重攻撃が可能な癌免疫療法として機能することを実証する。
682はpM範囲の薬物耐性癌細胞でさえIC50を有する超細胞毒素であるため(Quintieriら、2005)、このような化合物に関連する重篤な全身毒性のために、臨床的に使用することができない(StaudacherおよびBrown、2017)。しかし、EDVに封入した場合、682のようなスーパーサイトトキシンを、最小限の体重減少(%)、被毛の波打ちがほとんどないかまたは全くないこと、および様々な治療的ペイロードを担うEDVで処理されたマウスを含む以前の研究と相関するEp−EDV−682処理マウスにおける嗜眠または円背姿勢の出現がないことから明らかなように、副作用がほとんどなく腫瘍に効果的に送達することができる(MaCDiarmidら、2009; MaCDiarmidら、2007a; MaCDiarmidら、2007b; Sagnellaら、2018)(図35A−35D)。
本実施例の目的は、抗腫瘍剤を含む細菌ミニ細胞のNK細胞機能に対する影響を決定することであった。
本実施例の目的は、EDV処置後の腫瘍微小環境内のサイトカイン環境を調査することであった。
最初のin vitro実験では、EDV処理が効果的な化学療法薬を負荷した標的EDVに応答して、直接的相互作用または細胞死の結果のいずれかを介して樹状細胞の成熟をもたらしうることが示された。従って、この試験は、この結果がインビボでのDC成熟および抗原提示に翻訳され得、腫瘍特異的CD8+ 細胞傷害性T細胞の産生を生じるかどうかを試験することを目的とした。
この実験は、ステージIVの膵管腺癌(PDAC)患者(CEB)におけるEGFREDV682治療の使用の思いやりのある症例の臨床観察に関連する。
EnGeneICDream Vector (EDV): EDVは先に記載したように Salmonella enterica serovar Typhimur IUm(STyphimurium)minCDEstrainから産生され、精製された(MacDiarmidら、2007b)。薬物負荷、抗体標的化、凍結乾燥、および用量調製は以前に記載されている(MacDiarmidら、2007b; Sagnellaら、2018)。EDV調製物は、Zetasizer NanoシリーズおよびNanoSight LM20(Malvern Instrument)を用いた動的光散乱を用いてEDVサイズおよび数を評価する厳密な品質管理の対象とした。エンドトキシンレベルを、Endosafeポータブル試験システム(Charles River)を用いて評価した。薬物をEDVTM調製物から抽出し、先に記載したように高速液体クロマトグラフィーによって定量した(MacDiarmidら、2007b)。
この実施例は、癌細胞に対する瓱Cおよび遊離瓱CのEDV送達を対比する。
この実施例は、腫瘍に対するミニ細胞含有治療薬およびミニ細胞含有インターフェロンII型アゴニストの効力を例示する。この結果は、インターフェロンI型アゴニストを欠く組成物が腫瘍を効果的に処置するために使用され得ることを実証する。
Ablasserら、Nat., Immunol., 10(10):1065−72(2009)。
Ablasser ら、Nature., 498:380-384 (2013a)。
Ablasserら、Nature., 503:530−534(2013b)。
Adamusら、Contemp. Oncol (Ponzn), 22(1A):56−60(2018年3月)。
Aduro Biotech Inc. (2016),Novartis Pharmaceuticals. 進行/転移性固形腫瘍またはリンパ腫患者におけるMIW815(ADU−S100)の安全性および有効性の研究。臨床試験gov [インターネット]、ベセスダ(MD):米国国立医学図書館(US)、識別子: NCT02675439。https://ClinicalTrials.gov/show/NCT02675439から入手可能、(2016年7月01日引用)。
Ahmadzadehfar ら、「イットリウム-90微小球体による肝腫瘍のラジオ塞栓形成,」 Semin Nucl Med. 2010;40(2):105-121。
Ahmadzadehfarら、「去勢抵抗性転移性前立腺癌の177Lu−PSMA−DKFZ−617を用いた反復放射性リガンド療法の治療反応性および副作用」、Oncotarget.2016;7(11):12477-12488。
Alexopoulou ら、Nature., 413:732−738(2001)。
Alzahrani AS、AlShaikh O、Tuli M、Al-Sugair A、Alamawi R、Al-Rasheed MM. 「遺伝子組換えヒトチロトロピンの診断価値は、差別化された甲状がん患者のフォローアップにおいて、123I全身断層撮影を刺激した」Clin Nucl Med. 2012;37(3):229-234。
Andersson L、Blomberg L、Flegel M、Lepsa L、Nilsson B、Verlander M、「ペプチドの大規模な合成」、 Pept Sci. 2000; 55:227-250。
Anguilleら、Pharmacological Reviews 67., 731-753(2015)。
Barberら、Curr. Opin. Immunol., 23(1):10−20(2011)。
Belardelli ら、TRENDS in Immunology 23, 201−208 (2002)。
Bernardiniら、Frontiers in immunology 7, 402(2016)。
Birkholz ら(2015)、J Biol Chem. 「T細胞免疫機能の・ガラクトシルセラミドのT細胞免疫機能のアルファとオメガ」. NIH.gov [Internet]. Bethesda(MD):United States National Library of Medicine(米国)、識別子: PMC4505449、https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4505449/から利用可能。
Bobanga ら、Oncoimmunology 7, e1393598。
Bredel、Brain Res.Rev., 35:161(2001)。
Britton ら、Genes Dev., 12:1254-9 (1998)。
Brody ら、J. Clin. Oncol., 28:4324−4332(2010)。
Burckstummerら、Nat. Immuno., 10:266−272(2009)。
Burger M、Hartmann T、Krome M、Rawluk J、Tamamura H、Fujii NKipps TJ、Burger JA. 「CXCR4ケモカイン受容体(CD184)の低分子ペプチド阻害剤は、慢性リンパ性白血病B細胞におけるCXCL12の活性化、遊走、および抗アポトーシス反応に拮抗する」、Blood. 2005; 106:1824-1830。
Caplen.N.J., Expert Opin. Biol. Ther., 3; 575−86(2003)。
Caplen and Mousses、Ann. NY Acad. Sci., 1002:56−62(2003)。
Caravella and Lugovskoy, Curr. Opin. Chem. Biol., 14: 520−28(2010)。
Carrenoら、Clin Transl. Immunology, 5(4): e69(2016)。
Caskeyら、J.Exp. Med., 208:23572366(2011)。
Cauwelsら、Cancer research 78, 463−474 (2018)。
Chatalic KLS、Kwekkeboom DJ、de Jong M、「イメージングと治療のためのラジオペプチド:輝いている未来」、J Nucl Med. 2015; 56:1809-1812。
Chen ら、Int. J. Cancer, 93: 107(2001)。
Chikuma ら、Cancer Sci, 108: 574−580 (2017)。
Chiuら、Cell, 138:576−591(2009)。
Chuら、PLoS Biology, 4: 1122−36(2006)。
Civrilら、Nature, 498:332−337(2013)。
Clark-Curtiss and Curtiss、Methods Enzymol、101: 347−362 (1983)。
Colonnaら、Nat. Immunol., 5:1219−1226(2004)。
Corrales らl、Cell Rep., 11:1018−1030 (2015)。
Coryら、Cancer Commun.,3(7):207−12(1991)。
D'Aloia ら、Cell death & disease 9、282 (2018)。
D'Angiolella ら、Cell, 149:1023−34 (2012)。
Da SILvaら、Breast Cancer Res.,12: R46(1−13)(2010)。
Debinskiら、J. Neurooncol., 48: 103−11(2000)。
Debinski and Gibo、Mol. Med., 6: 440−49(2000)。
de Boer ら、J. Bacteriol., 174(1): 63−70 (1992)。
Deutscher SL、「癌の分子イメージングおよび診断におけるファージディスプレイ」、Chem Rev., 2010; 110:3196−3211。
Dine ら、Asia-Pacific journal of oncology nursing 4, 127−135(2017)。
Dobbsら、Cell Host Microbe, 18(2):15−24(2015)。
Dong ら、International journal of molecular sciences 17, 320(2016)。
Dowlingら、PLoS One, 8:e58164(2013)。
Dredge ら、Cancer immunology、immunotherapy: CII 51、521-531 (2002)。
Duanら、Mol., Cancer Ther, 3:833−8(2004)。
Duxburyら、Ann. Surg., 240:667−74(2004)。
Dynavax Technologies Corporation (2016)、「未治療の低悪性度B細胞リンパ腫患者を対象とした限局性低線量放射線療法との併用におけるSD-101の試験。2016. 臨床試験gov [インターネット]。Bethesda(MD):米国国立医学図書館(US)、識別子、 NCT02266147。https://ClinicalTrials.gov/show/NCT02266147から入手可能、(2016年7月01日引用)。
Emens ら、European journal of cancer 81, 116−129 (2017)。
Erathodiyil N、Ying JY. 「バイオイメージング用途のための無機ナノ粒子の官能化」、Acc Chem Res. 2011; 44:925-935. [PubMed: 21648430]。
Fang ら、Seminars in immunology 31, 37−54 (2017)。
Farkonaら、BMC medicine 14,73(2016)。
Ferlazzoら、The Journal of Immunology 172, 1333−1339 (2004)。
Fernandes-Alnemriら、Nature, 458:509-513 (2009)。
Field ら、Proc., Natl Acad. Sc IUSA,58:1004−1010(1967)。
Fitzgerald-Bocarsly ら、Biochimie 89, 843−855 (2007)。
Fuら、Sci. Transl. Med., 7(283):283ra252(2015)。
Fukuda、Curr. Protocols Molec. Biol.(Suppl. 26)、17.5.1−17.5.8 (1994)。
Gaoら、Nat Biotechnol., 22(8):969−976(2004)。
Gaoら、Science, 341:903−906(2013a)。
Gaoら、Cell, 153:1094−1107(2013b)。
Gerard SK、Cavalieri RR、 「画像診断によるI−123甲状腺腫瘍全身スキャン 6、24、48時間後」、Clin Nucl Med. 2002;27(1):1-8。
Ghosh A、Heston WDW、「 前立腺癌における腫瘍標的前立腺特異膜抗原(PSMA)とその制御」、J Cell Biochem. 2004;91(3):528-539。
Gitlinら、Proc. Natl Acad. Sci. USA, 103: 8459−8464(2006)。
Goh and Sorkin, Cold Spring Harb. Perspect. Biol., 5: a017459 (2013)。
Gray BP, Brown KC、「ペプチド組合わせ図書館:細胞結合ペプチドについての採鉱」、Chem Rev. 2013; 114: 1020−1081。
Gregoryら、Methods in Molecular Biology, 342: 33−47(2006)。
Gupta ら、「摘要 CT091: MEDI9197、アゴニスト、 TLR 7/8 アゴニスト の安全性と薬力学的活動」、 Cancer Research、AACR Annual Meeting 2017; April 1−5, 2017(2017年7月発行)。
Hansenら、EMBO J., 33(15):1654−66(2014)。
Harry E.J., Mol. Microbiol.,40(4):795-803(2001)。
Hershey, J. Allergy Clin. Immunol., 111: 677-90(2003)。
Hiragaら、J. Bacteriol., 171: 1496−1505 (1989)。
Hobbsら、Proc. Natl. Acad. Sci. USA, 95(8):4607−4612(1998)。
Holman BL、Tumeh SS. 単光子放出計算トモグラフィー(SPECT):応用とポテンシャル、JAMA.1990;263(4):561-564。
Hornungら、Nature., 458:514−518(2009)。
Hu & Lutkenhaus、Mol., Microbio., 34(1):82−90(1999)。
Iftodeら、Crit. Rev., Biochem., Mol. Biol., 34: 141−80(1999)。
Igarashi H、Fujimori N、Ito T、Nakamura T、Oono T、Nakamura K、Suzuki K、Jensen RT、Takayanagi R、血管作動性腸ペプチド(VIP)とVIPレセプター-治療的応用のための構造と機能の説明、 Int J Clin Med. 2011; 2:500-508。
Immune Design (2016)、Merck Sharp & Dohme Corp. 濾胞性非ホジキンリンパ腫患者におけるペムブロリズマブ併用または非併用の腫瘍内G100の研究、2017. 臨床試験gov [インターネット]、Bethesda(MD):米国国立医学図書館(US)、識別子: NCT02501473、https:// ClinicalTrials.gov/show/NCT02501473から入手可能(2016年7月01日引用)。
Jarboeら、Cancer Res.,67: 7983−86(2007)。
JeNKinsら、British journal of cancer 118、9−16 (2018)。
Jung ら、Translational oncology 11、686−690 (2018)。
Kaoら、Am. J., 191(12):1467−1469(2015)。
Kao et al、American Journal of Respiratory and Critical Care Medicine 191, 1467−1469 (2015)。
Kawai and Akira、Nat. Immunol., 11:373−384(2010)。
KhalILら、Proc Nat'l Acad. USA, 106: 11667−72(2009)。
Kimら、Proc. Natl. Acad. Sci. USA, 107:15181−15186 (2010)。
Kotaら、Cell, 137: 1005−17(2009)。
Kramer-Marek G、Capala J、現代の癌治療における核医学の役割、Tumour Biol. 2012;33(3):629-640。
Kranzuschら、Cell REp.,3:1362−1368(2013)。
Kriegら、Nature, 374:546−549(1995)。
Kwekkeboom DJ, de Herder WW, Kam BLら、放射性標識ソマトスタチン類似体[177 Lu−DOTA 0、Tyr3]オクトレオテートによる治療:毒性、有効性、および生存、J Clin Oncol. 2008;26(13):2124-2130。
Landskron ら、Journal of immunology research 2014, 149185 (2014)。
Leeら、Cancers 3, 3856−3893 (2011)。
Lemmon and Schlessinger、Cell, 141(7): 1117−134 (2010)。
Leung and Amarasinghe、Curr. Opin. Struct. Biol., 36:133−141(2016)。
Liら、Science,341:1390−1394(2013b)。
Liuら、Science,347(6227):aaa2630(2015)。
Luら、Structure,18:1032−1043(2010)。
Maら、Mol. Microbiol., 54: 99−108(2004)。
MacDiarmidら、PLoS One, 11(4)(2016)。
MacDiarmid ら、Nature biotechnology 27, 643−651 (2009)。
MacDiarmid ら、Cell cycle 6, 2099−2105 (2007a)。
MacDiarmid ら、Cancer cell 11, 431−445 (2007b)。
Majkowskaら、「低エネルギー・ベータ放射体47Scの複合体と骨の痛み治療のためのゾレドロン酸による177Lu」、Appl Radiat Isot. 2009;67(1):11-13。
Mankanら、EMBO J., 33:2937−2946(2014)。
McWhirterら、J. Exp. Med., 206:1899−1911(2009)。
Marqら、J Biol. Chem., 286:6108−6116(2011)。
Matsuno M、Matsui T、Iwasaki A、Arakawa Y、.ガストリン分泌におけるアセチルコリンとガストリン放出ペプチド(GRP)の役割、J Gastroenterol、1997; 32:579-586。
MedImmune LLC (2016)、固形癌患者に投与したMEDI9197の検討、2018. 臨床試験gov [インターネット]。Bethesda(MD):米国国立医学図書館(US)、識別子: NCT02556463、https://ClinicalTrials.gov/show/NCT02556463から入手可能、(2016年7月01日引用)。Mellman ら、Nature 480、480−489(2011)。
Merrifield R、固相ペプチド合成、Adv Enzymol Relat Areas Mol Biol. 2006; 32:221-296。
Meulen and Brady、Hum. Vaccin. Immunother., 13(1):15−16(2017)。
Mhawech-Fauceglia P、Zhang S、Terracciano L、ら、 正常および腫瘍組織における前立腺特異的膜抗原(PSMA)蛋白質発現と前立腺腺癌におけるその感受性および特異性:多重腫瘍組織マイクロアレイ法を用いた免疫組織化学的研究、Histopathology. 2007;50(4):472-483。
Morvan ら、Nature reviews Cancer 16, 7-19(2016)。
M・lerら、Frontiers in immunology 8, 304(2017)。
Muller C, Zhernosekov K, Koster U ら、PETおよびSPECT、ならびにα及びβ放射性核種療法のためのユニークな四組適合テルビウム放射性同位元素:新しい受容体標的葉酸誘導体を用いたin vivo概念実証研究。J Nucl Med. 2012;53(12):1951-1959。
シドニー・オーストラリア大学NHMRC臨床試験センター:シドニー(NSW):(2017)識別子ACTRN12617000037303 抗ヒトEGFR(ベクティビックス配列)標的EDVと免疫調節補助剤(EDV−40mer)を担持する同時非標的EDVを担持する細胞毒性薬物PNU−159682(EGFR(V)−EDV−PNU)の第1相試験2017年1月10日; https://www.anzctr.org.au/ACTRN12617000037303.aspx。
Nielsenら、Biochim. Biophys. Acta,1591(1−3), 109−118(2002)。
Niethら、FEBS Lett、545: 144−50 (2003)。
Oh and Park、Advanced Drug Delivery Rev, 61: 850−62 (2009)。
Ohki-Hamazaki H、Iwabuchi M、Maekawa F、ボムシン様ペプチドおよびそのレセプターの発展と機能、Int J Dev Biol. 2005; 49:293-300。
Oiseth ら、Journal of Cancer Metastasis and Treatment 3、250 (2017)。
Okada ら、J. Bacteriol., 176:917−22 (1994)。
Okanoら、J. Am. Chem. Soc., 128:7136−37(2006)。
Oncovir Inc(2016)、National Institutes of Health、Icahn School of Medicine at Mount Sinai、Bay Hematology Oncology、Emory University、University of Pittsburgh、National Cancer Institute、腫瘍内ヒルトノール(登録商標)による固形癌に対するIn Situ.自家治療ワクチン接種、2018臨床試験gov [インターネット]、Bethesda(MD):米国国立医学図書館(US)、識別子: NCT02423863。https:// ClinicalTrials.gov/show/NCT02423863から入手可能、(2016年7月01日引用)。
Oritz-Zapater ら、Nature Medicine, 18(1):83−90 (2011)。
Orzalliら、Proc. Natl. Acad. Sci. USA, 109: E3008−E3017(2012)。
Parkら、Breast Cancer Res.,4(3):95−99(2002)。
Palmedo H、骨転移の放射性核種療法、In: Clinical Nuclear Medicine. Berlin, Heidelberg: Springer Berlin Heidelberg; 2007:433-442。
Pillai ら、「放射性核種治療のための177Luの生産ロジスティックス」 Appl Radiat Isot. 2003; 59(2-3):109-118。
Quintieriら、Clinical Cancer Research 11、1608−1617 (2005)。
Raskin & de Boer、J.Bacteriol., 181: 6419−6424 (1999)。
ReeveおよびCornett、J. Virol., 15: 1308−16(1975)。
Reidら、Annals of oncology : official journal of European Society for Medical Oncology 24, 3128−3135 (2013)。
Rezvani ら、分子治療法 、the journal of the American Society of Gene Therapy 25, 1769−1781 (2017)。
Riceら、Semin. Nucl. Med., 41: 265−282(2011)。
Ruoslahti E.、 RGDおよびインテグリンに対する他の認識配列、Annu Rev Cell Dev Biol. 1996; 12:697-715。
Sagnellaら、Molecular cancer therapeutics 17, 1012−1023(2018)。
Santoni M、Scarpelli M、Mazzucchelli R. ら. 前立腺癌における個別化治療のための前立腺特異的膜抗原を標的とする:形態学的および分子的背景と将来の見込み、J Biol Regul Homeost Agents. 2014;28(4):555-563。
Sawa-Wejksza ら、Archivum immunologiae et therapiae experimentalis 66、97−111 (2018)。
Sazar、「自然宿主防御の起動 : ヒルトノール(ポリICLC)と悪性脳腫瘍」、Oncovir、Inc.、www.oncovir.com/id2 (2018年7月11日アクセス)。
Sharma ら、 Cell 168, 707−723 (2017)。
Sharpe、Immunological reviews 276、5−8 (2017)。
Showalter、Cytokine 97, 123−132 (2017)。
Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C、正常および悪性ヒト組織における前立腺に特有の膜抗原表現、 Clin Cancer Res. 1997;3(1):81-85。
Simmons ら、The Journal of Immunology 188, 3116−3126 (2012)。
Singh M、Mukhopadhyay KAlpha-melanocyte stimulating hormone: 新興の抗炎症性抗菌ペプチド、Biomed Res Int. 2014; 2014:874610。
Sioud, M., Trends Pharmacol. Sci., 25: 22−8(2004)。
Schogginsら、Nature,505:691−695(2014)。
Solomonら、PLos One,10: 1−17(2015)。
Staudacher ら、British journal of cancer 117, 1736−1742 (2017)。
Strand、FL.神経ペプチド:生理学的過程の調節因子、MIT press; 1999。
Stewart and D'Ari、J Bacteriol., 174:4513−6 (1992)。
Sunら、Science,339(6121):786−791(2013)。
Sunら、Biochem., Biophys. Res. Commun., 280:788(2001)。
Sun X、Li Y、L IU T、Li Z、Zhang X、Chen X、がん検診のためのペプチドベースの画像助剤、 Adv Drug Deliv Rev. 110−111: 38−51 (2017)。
Szkandera ら、British journal of cancer 110, 183−188 (2014)。
Takahashi H、Emoto K、Dubey M、Castner DG、Grainger DW. イメージング表面固定化化学:非接着性ヒドロゲル薄膜上の細胞パターン化との相関、Adv Funct Mater. 2008; 18:2079-2088。
Takaokaら、Nature, 448:501−505(2007)。
Takeshitaら、Molec. Ther., 18: 181−87(2010)。
Tanpureら、Bioorg. Med. Chem., 21: 8019−32(2013)。
Tatemoto, K、神経ペプチドY: 神経ペプチドYおよび関連ペプチドの歴史と概要、Springer; 2004、p1−21。
Teunissenら、「消化管の内分泌腫瘍ペプチド受容体放射性核種療法」、Best Pract Res Clin Gastroenterol, 2005;19(4):595-616。
Tyler-McMahon BM、Boules M、Richelson ENeurotensin: peptide for next millenn IUm. Regul Pept. 2000; 93:125-136。
Unterholznerら、Nat. Immunol., 11:997−1004(2010)。
Unterholznerら、Immunobiology, 128(11):1312−21(2013)。
van Zandwijk ら、Lancet Oncol、18(10): 1386−1396 (2017)。
van Zandwijk ら、The Lancet Oncology 18、1386−1396 (2017)。
Ventola、Pharmacy and Therapeutics 42、452−463 (2017)。
Wallace ら、Springer seminars in immunopathology 27, 49−64 (2005)。
Walrand S、Hesse M、Renaud L、Jamar FX線塞栓術後のPET/CT 90 Y線量測定に対する画像再構成の偏りの影響、J Nucl Med, 2015;56(3):494-495。
Wangら、Nat. Struct. Mol. Biol., 17:781−787(2010)。
Wangら、Immunity, 41(6):919−33(2014)。
Weckbecker G、Lewis I、Albert R、Schmid HA、Hoyer D、Bruns C、ソマトスタチン研究の機会:生物学的、化学的、治療的な側面、Nat Rev Drug Discov. 2003; 2:999-1017。
White & McCubrey、Leukemia、15: 1011−1021 (2001)。
Whittle ら、J Clin. Neurosci., 22(12):1889−1894(2015)。
Whittleら、Journal of clinical neuroscience: official journal of Neurosurgical Society of Australasia 22、1889−1894(2015)。
Wuら、Science, 339:826−830(2013)。
Wykoskyら、Clin Cancer Res., 14: 199−208(2008)。
Xiaら、Nat. Immunol., 16:366−375(2015)。
Yagueら、Gene Ther., 11: 1170−74(2004)。
Yang W、Luo D、Wang S、Wang R、Chen R、Liu Y、Zhu T、Ma X、Liu R、Xu G、TMTP1、転移を特異的に標的化する新規な腫瘍帰巣性ペプチド、Clin Cancer Res. 2008; 14:5494-5502。
Yangら、Nat. Immunol., 11:487−494(2010)。
Yiら、PLoS One, 8(10):e77846(2013)。
Yuanら、Scientific reports 5, 14273(2015)。
Zhangら、J Immunol., 186:4541−4545(2011a)。
Zhangら、Nat. Immunol., 12:959−965 (2011b)。
Zhangら、Cell REp., 6:421−430(2014)。
Zibert ら、Human Gene Therapy 15, 21−34 (2004)。
Ziegler-Heitbrock ら、Frontiers in immunology 4、23 (2013)。
Zitvogelら、Nature reviews Immunology 15、405−414 (2015)。
米国特許第8,591,862号。
米国特許第7,183,105号。
U.S. 2008/0051469。
WO 2000/067776。
WO 2003/033519。
WO 2004/113507。
WO 2005/056749。
WO 2005/079854。
WO 2009/027830。
月4日に出願された米国仮出願62/788,265に対し、35USC§119に基づく優先権利益を主
張し、その全内容が参照により本明細書に組み込まれる。
の全身デリバリーはがん治療において極めて重要な役割を果たすが、それはまた深刻な問
題を引き起こす。例えば、投与された薬物に対する正常な組織/器官の全身曝露は、重篤
な毒性を引き起こし得る。このことは、全身投与されるがん化学療法薬が薬剤の生物学的
利用能の低さおよび患者内の分布容積の大さを克服するために、しばしば非常に高い用量
で送達されなければならないという事実によって悪化する。また、全身的な薬剤投与は、
しばしば主要な血管内での安全なカテーテルの使用を必要とするので、侵襲的であり得る
。薬物の全身投与は末梢または中枢のいずれかの静脈の使用を必要とすることが多いため
、静脈炎などの局所合併症を引き起こす可能性がある。薬物の血管外遊出はまた、ビンカ
アルカロイドおよびアントラサイクリンの投与時に一般に見られるような、局所投与部位
での水泡/組織損傷をもたらし得る。
である。一次化学療法に反応しない腫瘍では、診断時に内因性耐性が存在する。後天性耐
性は初回治療によく反応する可能性があるが、再発時に耐性表現型を示す腫瘍で生じる。
このような腫瘍は、以前に使用された薬と、構造や作用機序が異なる薬を含む新しい薬の
両方に耐性を獲得する。MDR (多剤耐性)という用語は1種類の薬にさらされた後に、腫瘍細胞が構造的に無関係ないくつかの薬に対して交差耐性を示すようになる現象をいう。多剤耐性の機序は複雑で多因子性であるが、これは主としてがん細胞におけるゲノム不安定性および突然変異のレベルが高いためである。例示的な機構は、薬物不活性化、細胞膜ポンプによる薬物の排出、薬物流入の減少、薬物標的の突然変異およびアポトーシスの開始不全である。Bredel、2001;Chenら、2001; Sunら、2001; and White & McCubrey
、2001。
換した細胞を検出し、拒絶することができないと、がんの発生につながる可能性がある。
腫瘍は、免疫介在性拒絶反応から逃れるために複数の機構を用いる。これらのメカニズム
の多くは、現在、細胞レベルおよび分子レベルで知られている。このような知見にもかか
わらず、がん免疫療法は臨床において未だ確立された治療法ではない。
でき、同時にこれらの薬物を全身的に送達することに関連する問題を回避することができ
る、薬物の標的送達を提供することができる送達システムに対する大きな必要性が依然と
して存在する。本発明はこれらの必要性をみたすものである。
治療有効用量、および(b)インターフェロンI型アゴニストを含む精製された無傷の細菌由来ミニ細胞の治療有効用量を含む。別の実施形態では、組成物が(a)抗腫瘍剤を含む精製された無傷の細菌由来ミニ細胞の治療有効量、および(b)インターフェロンII型アゴニストを含む精製された無傷の細菌由来ミニ細胞の治療有効量を含む。なおさらなる実施形態において、組成物は、(a)抗腫瘍剤を含む、精製された無傷の細菌由来ミニ細胞
の治療有効用量;(b)インターフェロンI型アゴニストを含む、精製された無傷の細菌由来
ミニ細胞の治療有効用量;および(c)インターフェロンII型アゴニストを含む、精製さ
れた無傷の細菌由来ミニ細胞の治療有効用量を含む。
型アゴニスト、またはインターフェロンI型アゴニストおよびインターフェロンII型アゴ
ニストの組み合わせは2つ以上の精製された無傷の細菌由来ミニ細胞内にパッケージされ
る。一実施形態では抗腫瘍剤およびインターフェロンI型アゴニスト、インターフェロンII型アゴニスト、またはインターフェロンI型アゴニストおよびインターフェロンII型アゴニストの組み合わせは精製された無傷の細菌由来ミニ細胞の3つの別個の集団内にパッケージ化される。
フェロンII型アゴニストを含み、(a)抗腫瘍剤、インターフェロンI型アゴニスト、およびインターフェロンII型アゴニストは同じミニ細胞内に含まれ、(b)抗腫瘍剤およびインターフェロンII型アゴニストは第2のミニ細胞内に含まれ、(c)抗腫瘍剤およびインターフェロンII型アゴニストは第2のミニ細胞内に含まれ、(d)抗腫瘍剤は第1のミニ細胞内に含まれ、または(e)抗腫瘍剤は第2のミニ細胞内に含まれる。II型インターフェロンアゴニストが第3のミニ細胞内に含まれる。
を転写することができるポリヌクレオチドからなる群から選択される。一実施形態では、
抗腫瘍剤が超毒性化学療法薬である。一実施形態では、超毒性化学療法薬がモルホリニル
アントラサイクリン、メイタンシノイド、デュカルマイシン、アウリスタチン、カリケア
マイシン(DNA損傷剤)、α−アマニチン(RNAポリメラーゼII阻害剤)、センタマイシン(LiNK)、ピロロベンゾジアゼピン(LiNK)、ストレプトニグチン、窒素マスタード、ニトロソルエア、アルカンスルホネート、ピリミジン類似体、プリン類似体、代謝拮抗剤、葉酸類似体、アントラサイクリン、タキサン、ビンカアルカロイド、トポイソメラーゼ阻害剤、ホルモン剤、およびそれらの組合せからなる群より選択される。一実施形態では、モルホリニルアントラサイクリンがネモルビシン、PNU159682、イダルビシン、ダウノルビシン、カミノマイシン、およびオキソルビシンからなる群より選択される。一実施形態では、超毒性化学療法薬はPNU159682である。
、アンチセンスRNA、およびリボザイムからなる群より選択される。一実施形態では、
機能性核酸が腫瘍細胞増殖、血管新生、または化学療法に対する抵抗性を促進する、およ
び/またはアポトーシスもしくは細胞周期停止を阻害する遺伝子を阻害する。いくつかの
実施形態において、siRNAは、リボヌクレオチドレダクターゼM1(RRM1)発現
を阻害する。いくつかの実施形態において、siRNAは、ポロ様キナーゼ1(Plk1
)発現を阻害する。いくつかの実施形態では、miRNAはmiRNA16aである。
またはインターフェロンI型アゴニストとインターフェロンII型アゴニストの組み合わせ
はオリゴヌクレオチドである。一実施形態では、オリゴヌクレオチドが少なくとも約40
のヌクレオチド、少なくとも約50のヌクレオチド、または少なくとも約60のヌクレオ
チドの配列を含む。いくつかの実施形態では、オリゴヌクレオチドがPNPase1、ポ
リ(I:C)、ポリICLC、イミキモド、イミダゾキオリネレスキモド、cGAMPま
たはCpGオリゴデオキシヌクレオチドのポリヌクレオチド産物である。
さらに含む。一実施形態では、組成物がI型インターフェロンアゴニストを含むミニ細胞
に結合した二重特異性リガンドをさらに含む。一実施形態では、組成物がII型インターフ
ェロンアゴニストを含むミニ細胞に結合した二重特異性リガンドをさらに含む。
のアームと、非食作用性(non-phagocytotic)哺乳動物細胞表面受容体に対する特異性を有
する第2のアームとを含む。一実施形態では、ミニ細胞表面構造がミニ細胞表面上のリポ
多糖のO多糖成分である。1つの実施形態において、非貪食哺乳動物細胞表面受容体は、
ミニ細胞の受容体媒介エンドサイトーシスを活性化することができる。
形態では抗体または抗体断片が細菌由来のミニ細胞表面構造に対する特異性を有する第1
の多価アームと、がん細胞表面受容体に対する特異性を有する第2の多価アームとを含み
、がん細胞表面受容体はミニ細胞の受容体媒介エンドサイトーシスを活性化することがで
きる。
108ミニ細胞当たり約1未満の汚染親細菌細胞、109ミニ細胞当たり約1未満の汚染親
細菌細胞、1010ミニ細胞当たり約1未満の汚染親細菌細胞、または1011ミニ細胞当た
り約1未満の汚染親細菌細胞を含む。
ニ細胞が直径が約400nmである。一実施形態では、組成物が200nm濾過によって除去
可能な親細菌細胞汚染を含まない。
:(a)少なくとも約109;(b)少なくとも約1×109;(c)少なくとも約2×109
;(d)少なくとも約5×109;(e)少なくとも8×109;(f)約1011以下;(g)約1×1011以下;(h)約9×1010以下;または(i)約8×1010以下。
本発明の一実施形態は必要とする被験体を治療する方法に関し、この方法は、有効量の本明細書に開示される組成物を被験体に投与することを含む。一実施形態では、対象がヒト、非ヒト霊長類、イヌ、ネコ、ウシ、ヒツジ、ウマ、ウサギ、マウス、またはラットである。
一実施形態では、対象はヒトである。
一実施形態では、対象はがんに罹患している。1つの実施形態において、がんは、肺がん、乳がん、脳がん、肝臓がん、結腸がん、肛門がん、膵臓がん、および膀胱がんからなる群より選択される。一実施態様では、がんは急性リンパ芽球性白血病、急性骨髄性白血病、副腎皮質がん、AIDS関連がん、AIDS関連リンパ腫、虫垂がん、星状細胞腫、非定型奇形腫/ラブドイド腫瘍、基底細胞がん、膀胱がん、脳幹グリオーマ、脳腫瘍、乳がん、気管支腫瘍、バーキットリンパ腫、原発部位不明がん、カルチノイド腫瘍、主要サイト不明がん、中枢神経系非定型奇形腫様/ラブドイド腫瘍、中枢神経系胚芽腫、子宮頸がん、小児がん、脊索腫、慢性リンパ性白血病、慢性骨髄性白血病、慢性骨髄増殖性疾患、大腸がん、結腸直腸がん、頭蓋咽頭腫、皮膚T細胞リンパ腫、内分泌膵島細胞腫瘍、子宮体がん、上衣芽細胞腫、上衣芽腫、上衣腫、食道がん、感覚神経肉腫、ユーイング肉腫、頭蓋外胚細胞腫瘍、性腺外生殖細胞腫瘍、肝外胆管がん、胆がん、胃(胃)がん、胃腸カルチノイド腫瘍、消化管間質細胞腫瘍、消化管間質腫瘍(GIST)、妊娠性絨毛腫瘍 、神経膠腫、有毛細胞白血病、 心臓腫瘍、頭頸部がん、ホジキンリンパ腫、下咽頭がん、眼内黒色腫、小島細胞腫瘍、カポジ肉腫、腎臓ガン、ランゲルハンス細胞組織球症、喉頭ガン、唇ガン、肝臓がん、悪性繊維性組織サイト骨ガン、脂肪肉腫、髄芽腫、黒色腫、メルケル細胞がん、メルケル細胞がん、中皮腫、原発不明の転移性扁平上皮首がん、口腔がん、多発性内分泌腫瘍症候群、多発性骨髄腫/形質細胞腫瘍、菌状息肉症、骨髄異形成症候群、鼻腔がん、鼻咽頭がん、神経芽腫、非ホジキンリンパ腫、非黒色腫皮膚がん、非小細胞肺がん、口腔がん、口腔癌、中咽頭がん、骨肉腫、その他の脳および脊髄腫瘍、卵巣胚細胞腫瘍、卵巣低悪性度腫瘍、膵がん、乳頭腫症、副鼻腔がん、副甲状腺がん、骨盤がん、ペニスのがん、咽頭がん、中間的な分化を示す松果体実質細胞腫瘍、松果体芽腫、下垂体腫瘍、形質細胞新生物/多発性骨髄腫、胸膜肺芽腫、 原発性中枢神経系(CNS)リンパ腫、原発性肝細胞腫瘍、胸膜肺芽腫、原発性肝細胞がん、前立腺がん、腎細胞がん、腎細胞がん、腎細胞がん、網膜芽細胞腫、網膜芽細胞腫、横紋筋肉腫、セザリー症候群、唾液腺がん、小細胞肺がん、小腸がん、軟部組織肉腫、扁平上皮がん、頸部がん、胃(胃)がん、テント上原始神経外胚葉性腫瘍、T細胞リンパ腫、精巣がん、咽頭がん、胸腺がん、胸腺腫、甲状腺がん、移行上皮がん、腎盂および尿管の移行上皮がん、絨毛腫瘍、尿管がん、尿道がん、子宮がん、子宮肉腫、膣がん、外陰がん、ワルデンシュトレームマクログロブリン血症、およびウィルムス腫瘍からなる群より選択される。
一実施形態において、脳がんまたは脳腫瘍は、脳幹グリオーマ、中枢神経系非定型奇形腫様/ラブドイド腫瘍、中枢神経系胚芽腫、星細胞腫、頭蓋咽頭腫、上衣芽腫、上衣腫、髄芽腫、髄上皮腫、中間的分化の松果体実質腫瘍、テント上原始神経外胚葉性腫瘍および松果体芽腫からなる群より選択される。
一実施形態では、組成物が数週間にわたって少なくとも1週間に1回投与される。一実施形態において、組成物は、数週間〜数ヶ月にわたって週に少なくとも1回投与される。
一実施形態では、組成物が週に少なくとも1回、約2、約3、約4、約5、約6、約7、約8、約9、約10、約11、約12、約13、約14、約15、約16、約17、約18、約19または約20週間以上投与される。一実施形態では、組成物が毎週約2回投与される。1つの実施形態において、組成物は、約2、約3、約4、約5、約6、約7、約8、約9、約10、約11、約12、約13、約14、約15、約16、約17、約18、約19または約20週間以上、週2回投与される。
上記の概要および図面の以下の説明ならびに詳細な説明は、例示的かつ説明的なものである。それらは本発明のさらなる詳細を提供することを意図しているが、限定として解釈されるべきではない。他の目的、利点、および新規な特徴は、本発明の以下の詳細な説明から当業者には容易に明らかになるのであろう。
本発明は、(i)抗腫瘍剤およびI型インターフェロンアゴニスト;(ii)抗腫瘍剤およびI
I型インターフェロンアゴニスト;または(iii)抗腫瘍剤、I型インターフェロンアゴニ
スト、およびII型インターフェロンアゴニストの組み合わせを含む組成物であって、少な
くとも抗腫瘍剤が無傷の細菌由来ミニ細胞内にパッケージされている組成物が、がん治療
戦略を相乗的に改善することができるという発見に基づく。
トが無傷の細菌由来ミニ細胞内にパッケージされる、活性剤と免疫調節剤との組み合わせ
は、がん性細胞に対する劇的な効力、ならびに対象における薬物耐性発達の驚くべき欠如
を生じる。記載された組成物は、I型および/またはII型インターフェロンアゴニストの
ような免疫調節薬と組み合わせた抗腫瘍薬の全身送達に関連する毒性を回避して、相乗的
に改善されたがん治療戦略を提供する。
応答をもたらした(Emensら、2017; Farkonaら、2016; OisethおよびAziz、2017; Sharma
ら、2017; Ventola、2017)。しかし、現在の免疫療法戦略は様々な腫瘍型にわたって限
定された成功率をもたらし、最初に時間腫瘍退縮再発を促進することを実証する患者のか
なりの割合をもたらした(Emensら、2017; Mellmanら、2011; OisethおよびAziz、2017;
Sharmaら、2017; Ventola、2017)。
の機構を解明し、ここでは、免疫系の複数のアームの係合と組み合わせた超細胞毒素の送
達を介して腫瘍に対する二重攻撃を開始する。このアプローチは免疫原性腫瘍微小環境を
作り出し、それにより患者に生じるかもしれない一次耐性および/または適応耐性を回避
する複数の免疫細胞サブセットにも作用することにより、免疫療法による現在の落とし穴
のいくつかを回避する。
腫瘍免疫原性を欠き、したがって、利用可能な現在の戦略に対して初期応答を示さない(
Emensら、2017; OisethおよびAziz、2017; Sharmaら、2017)。このように、新規で頑健
な免疫療法アプローチの同定は、臨床転帰の有意な改善をもたらす可能性があり、依然と
して優先権の高い領域である。
かで達成されなければならない。第1に、腫瘍細胞死を介してインサイチュで誘導され得
るか、または外因的に送達され得る腫瘍細胞抗原は樹状細胞(DC)によって取り込まれ
なければならない(Anguilleら、2015; Emensら、2017; Jungら、2018; Mellmanら、2011)。抗原取り込みと併せて、DCは抗原の分化および増強されたプロセシングおよび提示を促す適切な成熟シグナルを受け取る必要があり、その結果、耐性とは反対の抗腫瘍機能が促進される(Anguilleら、2015; Emensら、2017; Jungら、2018; Mellmanら、2011; Simmonsら、2012)。これらの成熟した腫瘍抗原負荷DCはその後、腫瘍特異的細胞傷害性T細胞の産生、NKおよび/またはNKT細胞応答のトリガー、およびT−ヘルパー1型応答の増強を介して起こり得る抗腫瘍T細胞応答を効果的に生成しなければならない(Emensら、2017; Fangら、2017; Mellmanら、2011; Sharmaら、2017; Zitvogelら、2015)。抗腫瘍T細胞は免疫抑制シグナルが存在し得る腫瘍微小環境に最終的に入らなければならず、それらの抗腫瘍機能を有効に実施しなければならない(Emensら、2017; Mellmanら、2011)。これらの工程のいずれかで生じる問題は、免疫療法の有効性を妨げ、治療の全失敗をもたらし得る(Emensら、2017; Mellmanら、2011; Sharmaら、2017)。
ヒビターおよびキメラ抗原受容体T細胞療法(CAR)が含まれる(Emensら、2017; Mellma
nら、2011; OisethおよびAziz、2017; Sharmaら、2017; Ventola、2017)。細胞傷害性T
リンパ球抗原4(CTLA4)およびプログラム細胞死1/プログラム細胞死1リガンド(PD/PDL)などのチェックポイント阻害剤は免疫抑制シグナルの伝達を遮断し、腫瘍微小環境内で細胞傷害性Tリンパ球を活性化するように刺激することによって機能する(Dineら、2017; Jenkinsら、2018; Sharpe、2017)。これらの経路の阻害剤は特定のがんにおいて劇的な臨床結果を示しているが、異なるがんにわたる全体的な応答率は依然として低く(〜15−25%)、これらの治療に関連する免疫関連毒性は高い可能性がある(Dineら、2017; Emens ら、2017; Jenkins ら、2018; Sharpe、2017; Ventola、2017)。新たなチェックポイントが潜在的免疫ターゲットとして継続的に発見されていることにより、免疫抑制経路の精巧で多様なセットを利用することが可能であることは明らかである(Dineら、2017; Emensら、2017; Farkonaら、2016; Jenkinsら、2018; Sharpe、2017)。したがって、チェックポイント阻害剤に対する耐性の開発はハードルであり続けており、これらの問題を克服するために複数のチェックポイント阻害剤の組み合わせを利用する試みがなされているが、これはしばしば関連する毒性を悪化させる(Dineら、2017; Jenkinsら、2018; Sharmaら、2017;Ventola、2017)。
傷を開始するために強力なT細胞活性化を誘発することができる膜融合受容体を発現する
ための患者のT細胞の遺伝子操作を伴うCART細胞治療である(D’Aloiaら、2018’; Farkonaら、2016; Mellmanら、2011; Sharmaら、2017)。この治療アプローチは「液体」血液がんの治療において、前例のない臨床結果をもたらしたが、現在まで、良好な特異的抗原標的の欠如、腫瘍へのホーミング不良、腫瘍への溢出不良、および敵対的腫瘍微小環境内での持続性の欠如に関連する限界に起因して、固形悪性腫瘍を標的とする際に同等の応答をもたらさなかった(D’Aloiaら、2018’; Sharmaら、2017)。重度に前処置された患者からのリンパ球の利用可能性および長い製造時間に関連する実用的な限界も存在し、急速に進行する疾患を有する患者にとって実行可能な治療選択肢ではない(OisethおよびAziz、2017; Rezvaniら、2017)。
とによって生成される、直径400±20nmの非生存ナノ細胞からなる細菌由来の送達系
である(MacDiarmidら、2007b)。これらのナノ細胞は細胞傷害性薬物、siRNA、ま
たはmiRNAでパッケージ化され得、ナノ細胞の表面多糖への二重特異性抗体の付着を
介して腫瘍細胞表面受容体に特異的に標的化され得ることが実証されている(MacDiarmid
ら、2009; MacDiarmidら、2007b; Reidら、2013)。マウスおよびイヌの研究における静
脈内投与後はそれらのサイズのために血管系に保持されているが、その後、腫瘍関連漏出
性血管系を介して腫瘍に急速に溢出していることが実証されている(MacDiarmidら、2007
b; Sagnellaら、2018)。関連する二重特異性抗体を介する腫瘍細胞表面受容体の結合は
エンドソームへのマクロピノサイトーシス、およびリソソームにおける細胞内の変性赤血
球を介するペイロードの放出をもたらす(MacDiarmidら、2009; MacDiarmidら、2007b; S
agnellaら、2018)。これらのナノ細胞治療の安全性は3つの第I相臨床試験で実証されて
おり、様々な末期がん患者には1000回を超える用量が投与され、682の負荷EDV
が現在第I相試験で患者に送達されており、これまでに有望な安全性プロフィールが示さ
れている(2017; Kaoら、2015; Solomonら、2015; van Zandwijkら、2017; Whittleら、2
015)。
A. 細菌のミニ細胞デリバリー法の概要
がん細胞に化学療法剤を送達するための細菌ミニ細胞の使用は、以前に記載されている
。がんを治療するためのこの送達方法は、毒性化学療法剤もしくは薬物、または機能性核
酸を、典型的には直径約400nmである細菌由来ミニ細胞にパッケージ化する。典型的に
は、ミニ細胞が特異的がん細胞を標的とする抗体を有する。この抗体はがん細胞の表面に
結合し、ミニ細胞はがん細胞によって細胞内に取り込まれる。このようにして、毒性化学
療法剤は全身に広く分布せず、したがって、毒性薬物または化合物ががん細胞内に送達さ
れるにつれて、副作用および不耐性の機会を減少させる。毒性化学療法剤の送達ビヒクル
として抗体標的化ミニ細胞を用いると、がん細胞を死滅させるのに必要な薬物がはるかに
少なくなり、したがって治療指数が改善される。
ス(実施例1)、イヌ(実施例2)、およびサル(実施例3)における異種移植腫瘍に、
パクリタキセルまたはドキソルビシンなどの化学療法薬を送達し得ることを示した。標的
送達はがん細胞が化学療法剤の大部分を受け取り、低レベルの毒性を生じることを確実に
する。実施例1〜3を参照されたい; MacDiarmidら、2007b;MacDiarmidら、2007a; MacD
iarmidら、2009;およびMacDiarmidら、2016も参照されたい。さらに、ミニ細胞は、異種
移植モデルにおいて有意な免疫応答を誘導せず、ミニ細胞は忍容性が良好である(実施例
4)。したがって、無傷の細菌由来ミニ細胞は、進行固形腫瘍を標的としたドキソルビシ
ン(実施例5)、膠芽腫を標的としたドキソルビシン(実施例6)、および中皮腫を標的
としたMicroRNA−16a(実施例7)を含む例を有する、患者に抗がん剤を送達するための十分に許容されるビヒクルである。
は治癒をもたらさなかった。したがって、がん治療療法の改善が必要とされている。本発
明者らは、3つの異なるタイプのペイロードを有するミニ細胞の組み合わせを使用すると
、驚くほど劇的かつ有効な臨床効果が得られることを発見した。
ロンII型アゴニストを含むミニ細胞と組み合わせた化学療法剤(例えば、以下の実施例に
おいて、薬剤はPNU159682、超毒性化学療法剤である)を含むミニ細胞が相乗的
な抗腫瘍効果をもたらし、後期膵臓がんに罹患している患者によって十分に耐えられるこ
とを発見した。実施例12を参照されたい。実際、後期膵臓がん患者はこの治療後に著し
く改善された生活の質を示し、これは、その段階の患者にとって顕著である。このトリプ
ル・オブ・デュル・コンビネーション戦略は、がんの治療を相乗的に改善するものである
。本発明者らはまた、インターフェロンII型アゴニストを含むミニ細胞と組み合わせた化
学療法剤を含むミニ細胞が、相乗的な抗腫瘍効果を生じることを発見した。
フェロンアゴニストがない場合に、ミニ細胞パッケージ化抗腫瘍薬の2剤併用により、大
きなサイズの腫瘍に対して劇的な有効性もたらされることが発見された。このような結果
は、以前には記載されていない。一部の患者では、I型インターフェロンアゴニストとII
型インターフェロンアゴニストを併用すると、2種類のインターフェロンアゴニストが相
乗的に作用するのではなく競合的に作用する可能性があるため、逆効果となる可能性があ
ると仮定されている。このデータについては、以下で詳しく説明する。
の実施形態、方法論、プロトコル、または試薬に限定するものではない。同様に、ここで
使用される用語は特定の実施形態のみを記載し、本発明の範囲を限定するものではない。
(i) ミニ細胞中にパッケージ化化された抗悪性腫瘍剤とミニ細胞パッケージ化I型インターフェロンアゴニストとの併用
第1の実施形態では、細菌ミニ細胞にパッケージされたI型インターフェロンアゴニス
トと組み合わせた細菌ミニ細胞パッケージされた抗腫瘍剤の組み合わせに関する組成物お
よび方法が記載される。
成物で処置されたマウスにおける肺がん異種移植片モデルにおける結果を示すデータを記
載する。グループ1および5の動物に、無傷の細菌由来ミニ細胞中にパッケージされた化
学療法剤(PNU159682)と、無傷の細菌由来ミニ細胞中にパッケージされたI型
インターフェロンアゴニスト(40mer二本鎖DNAまたは50mer二本鎖DNA)との組
み合わせを投与した。全てのミニ細胞組成物は、腫瘍増殖の安定化をもたらした。しかし
ながら、最も劇的な結果は、実験のパート1における生理食塩水処理から生じた大きな腫
瘍サイズを処理した後に得られた。その後、生理食塩水処理対照群を、ミニ細胞パッケー
ジ化抗腫瘍剤とミニ細胞パッケージ化I型インターフェロンアゴニストとの組み合わせを
含む組成物で実験の第2部において処理した場合、腫瘍サイズは、5日間にわたって62
%減少した。
トの追加が劇的な腫瘍サイズ減少をもたらし、これはミニ細胞にパッケージされた抗腫瘍
剤をI型インターフェロンアゴニスト免疫賦活剤の非存在下で使用した場合には見られな
かった。結果を以下の表にまとめる。これらの結果は、ミニ細胞パッケージ化I型インタ
ーフェロンアゴニストの添加によるミニ細胞パッケージ化抗腫瘍剤に対する免疫賦活剤効
果を明らかに実証する。
I型インターフェロン作動薬(ミニ細胞パッケージではない)とミニ細胞パッケージ化抗
悪性腫瘍剤との併用
第2の実施形態は、ミニ細胞パッケージ化I型インターフェロンアゴニストまたはミニ
細胞パッケージ化抗悪性腫瘍剤と、ミニ細胞パッケージ化I型インターフェロンアゴニス
トおよびII型インターフェロンアゴニスト(細菌ミニ細胞を含まない)とを組み合わせた
ミニ細胞パッケージ化防新生物剤を使用する方法および組成物を対象とする。
床結果に反映される。具体的には、実施例12が(1)ミニ細胞パッケージ化抗腫瘍剤と
ミニ細胞パッケージ化I型インターフェロンアゴニストとの組み合わせ;および(2)ミニ細胞パッケージ化抗腫瘍剤、ミニ細胞パッケージ化I型インターフェロンアゴニスト、およびII型インターフェロンアゴニストの組み合わせを含む組成物で進行固形腫瘍を患う患者を処置した場合に起こったことに関する。
ジ化抗腫瘍剤の免疫賦活剤として使用されるI型およびII型IFNアゴニストの安全性プ
ロフィールを実証する。無傷の細菌由来ミニ細胞にパッケージ化されたI型インターフェ
ロンアゴニストが40mer二本鎖DNA(EDV40mer)または60mer二本鎖DNA(E
DV60mer)であった以下の表3のデータを参照されたい。
さらに、他のすべての治療選択肢を使い果たしたステージ4の膵臓がん患者で得られた結果は顕著であった。患者の腫瘍マーカー(CA 19−9)の量は最初の3回の投与後に90%以上低下し、これはわずか10日間の治療に相当した。10回投与後、これはさらに低下し、腫瘍マーカー値はほぼ95%低下した。この患者はまた、IV期膵がんを呈するほとんどの患者が経験する悪液質状態とは対照的に、有意な体重増加を示し、生活の質の顕著な改善を報告した。これらの結果は、特に進行膵がんに伴う予後不良を考えると劇的である。
要約すると、5例がEGFR(5)EDVPNU/DoxまたはEGFR(V)EDVPNU+EDV40mer/60mer(I型IFNアゴニスト)±Imukin(II型IFNアゴニスト)の計69回を受けた。治療は忍容性が良好であり、免疫調節免疫賦活剤の添加は、単剤負荷EDVおよび標的EDVの安全性プロファイルを変化させないようであった。
実施例13は、ミニ細胞にパッケージされた抗新生物剤をII型インターフェロンアゴニス
ト、例えばIFN−γと組み合わせることの有効性を評価するために実施された種々の研
究の結果を記載している。結果は、II型インターフェロンアゴニストの追加が肺がんおよ
び乳がんを含む種々のがんの異種移植モデルにおいてミニ細胞にパッケージされた抗新生
物剤の抗がん効果を増強または増強することを示す。さらに、実施例13に記載され、以
下の表4に抜粋されるデータは、抗腫瘍剤単独に通常耐性である腫瘍の処置において、ミ
ニ細胞中にパッケージされた抗腫瘍剤を含む組成物へのII型インターフェロンアゴニスト
の添加が腫瘍安定化を達成するために必須であることを実証する。したがって、ミニ細胞
パッケージ化抗腫瘍剤をII型インターフェロンアゴニストと組み合わせることにより、薬
物耐性を克服することができる。
ンおよびII型インターフェロンアゴニスト(単独またはミニ細胞パッケージ化のいずれか)
の三剤併用
本発明者らはまた、ミニ細胞パッケージ化抗腫瘍剤、ミニ細胞パッケージ化I型インタ
ーフェロンアゴニスト、およびII型インターフェロンアゴニスト(単独またはミニ細胞パ
ッケージ化のいずれか)の三重組み合わせが、劇的な抗がん効果を生じ得ることを発見し
た。具体的には、実施例14が後期内因性腫瘍(脳がん、肉腫、またはメラノーマ)を有
するイヌの処置を、ミニ細胞パッケージ化抗腫瘍剤、ミニ細胞パッケージ化I型インター
フェロンアゴニスト、およびII型インターフェロンアゴニストの組合せで詳述する。結果
は、組み合わせ組成物が十分に耐性であったことを示す。さらに、7匹の評価可能な動物
のうち6匹(85.7%)では疾患は安定化したが、1匹のイヌはほぼ部分奏効(腫瘍サ
イズの29.8%の縮小)を達成した。
胞パッケージ化II型インターフェロンアゴニストの二剤併用
別の実施形態では、本発明がミニ細胞パッケージ化抗腫瘍薬と、ミニ細胞パッケージ化
II型インターフェロンアゴニスト、例えばα−ガラクトシルセラミド(α−GC)との組み合わせを含み、I型インターフェロンアゴニストの非存在下で、驚くべき抗がん効果を示す発見に基づく組成物に関する。
ェロンII型アゴニストの二重組合せの効力を示すデータを記載する。この結果は、インタ
ーフェロンI型アゴニストを欠く組成物が腫瘍を効果的に処置するために使用され得るこ
とを実証する。図40および図42も参照されたい。実験結果は、生理食塩水およびEpミニ細胞Dox処置と比較して、Epミニ細胞Dox+ミニ細胞 α-GC (インターフェロンII型アゴニスト)を受けた併用処置群について、腫瘍進行の顕著な停止を示した。この結果はEpミニ細胞Doxに、ミニ細胞 α-GC 処理を追加することにより、免疫賦活剤効果の理論を支持する。
ジ化抗腫瘍剤をミニ細胞パッケージ化インターフェロンII型アゴニストと組み合わせたも
の)は、腫瘍のサイズ(大きな腫瘍のサイズを含む)を、約5%、約10%、約15%、
約20%、約25%、約30%、約35%、約40%、約45%、約50%、約55%、
約60%、約65%、約70%、約75%、約80%、約85%、約90%、約95%、
または約100%減少させ得る。約3日、約5日、約1週間、約2週間、約3週間、約1
、約2、約3、約4、約5、約6、約7、約8、約9、約10、約11、約12カ月、約
1.5年、約2年以上のような、任意の適切な時間にわたって、細胞サイズの減少を測定
することができる。
実施例16は、腫瘍細胞表面受容体に標的化されたミニ細胞パッケージ化抗腫瘍剤が、
がん免疫療法、例えば、細胞−免疫療法として機能することを実証するデータを詳細に記
載する。特に、実施例は、マクロファージ、NK細胞および樹状細胞を含む自然免疫系の
細胞を活性化する細菌ミニ細胞の能力を例示する。これに続いて樹状細胞の成熟と抗原提
示が起こり、腫瘍特異的細胞傷害性T細胞が産生され、さらなる免疫細胞の腫瘍微小環境
への動員をもたらす適応T細胞応答が起こる。このアプローチは免疫原性腫瘍微小環境を
作り出し、複数の免疫細胞サブセットにも作用することで、患者に生じる可能性のある原
発性および/または適応抵抗性を回避することによって、免疫療法による現在の落とし穴
の一部を回避するものである。
腫瘍を特異的に標的とする自然免疫応答および適応免疫応答を誘発する能力を示す。
的ミニ細胞での処置に続いてインビボで抗腫瘍表現型を採用することを示すデータを記載
する。これは、NK細胞が先天性免疫系の主要なエフェクター細胞であり、活性化シグナ
ルと阻害シグナルとのバランスによって厳密に調節されるので、有意である(Morvan and
Lanier、2016; Wallace and Smyth、2005)。NK細胞機能の障害は腫瘍発生、増殖、お
よび転移の増加と関連しており、したがって、抗腫瘍免疫応答に寄与することにおけるそ
の重要性は十分に実証されている(Fangら、2017; MorvanおよびLanier、2016; Rezvani
ら、2017; WallaceおよびSmyth、2005)。
の処置が腫瘍特異的CD8+ T細胞の産生を生じることを示すデータを詳述する。最初の
インビトロ実験は、EDV処理が効果的な化学療法剤を負荷された標的化EDVに応答し
て、直接的相互作用または細胞死の結果のいずれかを介して樹状細胞成熟をもたらし得る
ことを示した。従って、この試験は、この結果がインビボでのDC成熟および抗原提示に
翻訳され得、腫瘍特異的CD8+ 細胞傷害性T細胞の産生を生じるかどうかを試験するこ
とを目的とした。結果として得られたデーターは、ミニ細胞処理が腫瘍特異的CD8+ T
細胞の産生を首尾よく誘発したことを実証した。さらに、全T細胞数(CD3+)の有意
な増加、ならびにCD4+およびCD8+ T細胞数の両方の有意な増加がミニ細胞カプセ
ル化抗腫瘍剤(例えば、PNU159682)で処置されたマウスのリンパ節において見
られた(図31G)。処理されたマウスのリンパ節における成熟樹状細胞の有意な増加も
検出され(図31H)、4T1細胞で処理されたマウスから単離されたCD8+ T細胞間
の相互作用の可視化は、これらのT細胞が腫瘍細胞にペルフォリン(緑色)を付着させ、
そこに排出することができることを示している(図31I)。
的化細菌ミニ細胞が、この薬物を腫瘍部位に効果的に送達するだけでなく、多数の免疫細
胞サブセットを刺激することによって免疫療法剤として挙動する能力を実証する。実施例
は、マクロファージ、NK細胞およびCD8+T細胞を含む免疫細胞サブセットを、腫瘍
細胞を効果的に排除することができる抗腫瘍表現型に向かって押すための、ミニ細胞カプ
セル化抗腫瘍剤処理の能力を実証する。抗腫瘍薬の有効性と組み合わせると、腫瘍に対す
る二重の攻撃をもたらす。
って実現され始めたのは最近のことである(Farkonaら、2016; Ventola、2017)。細菌性
ミニ細胞は腫瘍への直接的な細胞毒性剤の送達を介して最初に免疫原性腫瘍微小環境を作
り出し、そこで抗腫瘍表現型に向けて直接的または間接的に自然免疫系を刺激する、独特
の複合細胞免疫療法である。次いで、この先天性免疫活性化は、腫瘍特異的細胞傷害性T
細胞が生じる適応応答を誘発する(図33)。
の毒性ペイロードを有する標的化ミニ細胞の投与用量の30%以上が2時間以内に腫瘍微
小環境に直接沈着する(MacDiarmidら、2007b)。標的細菌ミニ細胞は、腫瘍細胞上の受
容体に結合し(実施例21の場合、4T1およびCT26Ep12.1)、次いで、それらの負荷(抗腫瘍剤)を腫瘍細胞内に直接的に効果的に送達する。PNU159682は、腫瘍細胞に送達されてから24時間以内に迅速なアポトーシスを生じる非常に強力な超細胞毒である(図33A)。次いで、細菌ミニ細胞(例えば、Ep−EDV−682)処理によって産生されるアポトーシス細胞およびDAMPシグナルは腫瘍関連マクロファージ(TAM)などの自然免疫細胞と相互作用し、CD86のアップレギュレーションおよびTNFαおよびIL-6などのTh1炎症誘発性サイトカインの産生を刺激することができる(図33B)。これらの変化は腫瘍細胞を溶解し、サイトカインを放出して他の免疫細胞サブセットの活性化をシグナル伝達することができるマクロファージのM1分極の典型であり、したがって抗腫瘍特性を有することが示されている(Sawa−WejkszaおよびKandefer−Szerszen、2018; Yuan ら、2015)。
とができるが、これは現在の系において非常に低いレベルで起こることが予想される。T
AMは一般に、腫瘍微小環境において最も豊富な免疫細胞であり、TAMの数の増加は、
予後不良および腫瘍増殖の増加と関連することが実証されている(Sawa−WejkszaおよびK
andefer−Szerszen、2018)。これはTAMの大部分が腫瘍促進特性を有することが示されている抗炎症性M2マクロファージからなるが、炎症性M1マクロファージは抗腫瘍特性を示すという事実によるものである(Sawa−WejkszaおよびKandefer−Szerszen、2018; Yuan ら、2015)。実施例21は、4つの異なる腫瘍モデルにおいて、細菌ミニ細胞処理が腫瘍微小環境内のM1:M2バランスをシフトさせる能力を示す。異なる腫瘍モデルにおけるこのシフトの程度の差にもかかわらず、M1極性化の増加は、細菌ミニ細胞で処理されたマウスの腫瘍から単離されたTAMによる腫瘍細胞溶解の増加に翻訳されることが示された。M1への表現型シフトに加えて、細菌ミニ細胞処置マウスの腫瘍からのTAMはまた、免疫細胞動員、特にNK細胞、CD4+T細胞およびCD8+T細胞による腫瘍浸潤を促進するのに関与することが確立されているケモカインであるMIP−1αの増加量を分泌した(図33C)(Allenら、2018)。
よびプライミングを含むが、この戦略の成功は免疫寛容の発生、不十分な数のCD8+細
胞傷害性T細胞(CTL)の誘導、または不十分な抗腫瘍効力を有するもの、および腫瘍
微小環境の抑制性質を含む様々な因子のために限定されている(Anguilleら、2015; Jungら、2018; Landskronら、2014; OisethおよびAziz、2017)。細菌ミニ細胞処理は死にかけている腫瘍細胞(図33D)に応答して、腫瘍微小環境内のDCのインビボでのプライミングおよび成熟を可能にし、未成熟DCは、標的化され、薬物負荷された細菌ミニ細胞に応答して産生されるDAMPおよび/またはアポトーシス性腫瘍細胞体を取り込むことができる。次に、これらのDAMPおよび死につつある腫瘍細胞は、MHCクラスIおよびII分子を介してDC表面上の抗原提示のために処理され、同時にDC成熟が起こる。DC成熟プロセスのマーカーとして同定された共刺激分子CD86、CD80およびMHC
クラスIIのアップレギュレーションは細菌ミニ細胞処置マウスの腫瘍排液リンパ節におい
て検出される成熟DCのパーセンテージの増加と共に、細菌ミニ細胞処置腫瘍細胞と共培
養されたDCにおいて生じることが示された(Anguilleら、2015; Cauwelsら、2018; Simmonsら、2012)。成熟過程の間、DCはT細胞への抗原提示のために腫瘍排出リンパ節に移動し、それによって、CD4+Tヘルパー細胞および腫瘍に対する適応免疫反応を開始する腫瘍特異的CD8+CTLの産生を増加させる(図33E)。続いて、細菌ミニ細胞処理腫瘍細胞と共培養したDCによるIFNα/β、TNFα、IL−12p40、およびIL−6の産生の増加が検出され、さらに4T1及びCT26Ep12.1腫瘍モデルの両方で観察された腫瘍微小環境におけるIFNα濃度の有意な増加が認められた(実施例21)。腫瘍微小環境内の1型IFN(IFNα/βおよびIFN刺激遺伝子の発現レベルは良好な疾患転帰と相関することが示されており、実際には、免疫療法を含むがん治療の成功に必要である可能性さえある(Cauwelsら、2018年; Fitzgerald-BocarslyおよびFeng、2007年; Zitvogelら、2015年)。1型IFNの抗腫瘍活性はDC、TおよびBリンパ球、NK細胞、およびマクロファージの免疫細胞活性化を介して間接的に生じる(Cauwelsら、2018; Fitzgerald-BocarslyおよびFeng、2007; Showalterら、2017; Zitvogelら、2015)。
ニ細胞での処置は、NK細胞活性化を誘発し得、増加した細胞毒性をもたらす(図33F
)。NK細胞は抗原非方法悪性細胞を溶解する固有の能力を有するため、宿主への有害な
影響の可能性を避けるために、その活性化および機能状態を厳密に制御しなければならな
い。NK細胞を誘引し、腫瘍微小環境内でNK細胞を活性化する能力は、その抗腫瘍機能
を発揮する能力に不可欠である。Ep−EDV−682処理腫瘍の微小環境において有意に増加するIL−2、IFNγ、およびIFNαを含むサイトカインはサイトカイン産生の増加および細胞溶解機能の増強の両方に向かってNK細胞を活性化することが知られている(Fangら、2017年; FerlazzoおよびMunz,2004年; LeeおよびMargolin,2011年; MorvanおよびLanier,2016年; Rezvaniら、2017年)。実際、証拠は1型IFNがNK細胞毒性の活性化に必要であることを示す(FerlazzoおよびMunz、2004; Mullerら、2017)。さらに、1型IFNは細胞老化を誘導し、続いて腫瘍細胞におけるNKG2Dリガンド発現のアップレギュレーションを誘導し、それによってNK細胞によるそれらの排除を促進することができる(Mullerら、2017)。NKG2D受容体のアップレギュレーションはEp−EDV−682で処理したマウスの腫瘍内のNK細胞で観察され、この受容体はEp−EDV−682処理マウスから単離したNK細胞の細胞溶解能力に有意に寄与することが実証された。さらに、未成熟、中間および成熟マウスのNK細胞はケモカインMIP1αおよびRANTESに結合できるCCR1およびCCR5ケモカイン受容体の両方を発現しており、いずれもEp−EDV−682を投与した腫瘍のほか、Ep−EDV−682を投与したマウスのマクロファージおよびNK細胞によってアップレギュレートされている(BeRNArdiniら、2016年)。
瘍免疫応答を誘発することが可能な独特のがん治療戦略を表す。腫瘍に対する二重の攻撃
が起こり、まず、送達された治療に応答して細胞死を経て、続いて適応免疫応答を導く自
然免疫細胞活性化が起こる。この種の治療法には、免疫細胞の活性化がインビボでも主に
腫瘍部位で起こるという点で、現在の免疫療法戦略よりも一定の利点があり、これは急速
に変化する動的環境である。さらに、免疫原性腫瘍環境を作り出し、それらの腫瘍に対す
る免疫応答または単一の免疫細胞サブセットのみを標的とする治療への適応をほとんど示
さない患者に関連する問題を回避して、複数の免疫細胞サブセットに対する効果を誘発す
る。実施例21に記載の研究は新規のがん免疫治療薬としての細菌ミニ細胞の可能性を強
調し、将来の細菌ミニ細胞製剤はペイロードおよび標的化能力の両方に関してこの技術の
汎用性を考慮して、その固有の免疫原性をさらに活用することができる(MacDiarmidら、
2007a)。
実施例17は化合物に関連する重篤な毒性のために従来の手段を使用して送達すること
ができない、超毒性抗腫瘍剤、例えばPNU159682の有効な送達を実証するデータ
を詳述する。具体的には実施例17がpM範囲の薬物耐性がん細胞であっても、PNU1
59682がIC50を有する超細胞毒素である方法を詳述し(Quintieriら、2005)、
これは重篤な全身毒性のために化合物を臨床的に使用することができないことを意味する
(StaudacherおよびBrown、2017)。しかし、細菌ミニ細胞中にカプセル化される場合、
PNU159682のようなスーパー細胞毒素は、ほとんど副作用を伴わずに腫瘍に効果
的に送達され得る。
上記のように、本発明の組成物は、少なくとも2つの異なる活性剤、抗腫瘍剤およびI
型インターフェロンアゴニスト、II型インターフェロンアゴニスト、またはI型インター
フェロンアゴニストおよびII型インターフェロンアゴニストと抗腫瘍剤との両方を含む。
3つの異なる活性剤は、1つ、2つ、または3つの異なるミニ細胞にパッケージ化するこ
とができる。II型インターフェロンアゴニストはまた、ミニ細胞にパッケージされること
なく、本発明の方法および組成物に含まれ得る。
「抗悪性腫瘍剤」とは、化学的、生物学的なものであるかにかかわらず、腫瘍細胞の増
殖、発生、成熟又は拡散を阻止又は阻害する医薬品をいう。「抗悪性腫瘍剤」とは「抗悪
性腫瘍剤」及び「化学療法剤」と互換的に使用されるものである。
本開示の文脈において、所定の脳腫瘍患者を治療するための抗腫瘍剤を選択することは
、従来の医療行為と一致して、いくつかの因子に依存する。これらの因子には患者の年齢
、カルノフスキー・スコア、および患者が受けた可能性のあるあらゆる治療歴が含まれる
が、これらに限定されない。一般に、神経腫瘍学の原則と実践、M.Mehta(Demos Medical Publishing 2011)、および神経腫瘍学の原則、DSchiffおよびPO’Neill, edsを参照の
こと。(McGraw−Hill 2005)。
、化学療法薬の量が多くとも約750μg、約500μg、約250μg、約100μg、約50μg、約10μg、約5μg、約1μg、約0.5μg、または約0.1μgであり得る。別の態様では、組成物がミニ細胞にパッケージ化されずに使用される場合、薬物の治療有効量の約1/1,000未満、あるいは約1/2,000、1/5,000、1/10,000、1/20,000、1/50,000、1/100,000、1/200,000または1/500,000未満の量を有する化学療法薬物を含む。本開示のさらに別の態様によれば、組成物は、少なくとも約1nmolの化学療法薬を含むことができる。したがって、本開示はまた、化学療法薬の量が、それぞれ、少なくとも約2nmol、約3nmol、約4nmol、約5nmol、約10nmol、約20nmol、約50nmol、約100nmol、または約800nmolである実施形態を包含する。
子に依存する。これらの因子には患者の年齢、腫瘍の病期、および何であれ患者が受けた
可能性のある過去の治療法が含まれるが、これらに限定されない。
述されるクラスのうちの1つから選択され得る。これらの薬物はまた、薬物設計および発
明努力から設計された合成類似物でもあり得る。任意の公知の化学療法剤が、本発明の組
成物において利用され得る。公知の化学療法剤の例にはこれらに限定されないが、以下の
ものが含まれる:(1) マスタードガス誘導体(メクロレタミン、シクロホスファミド(シ
トキサン)、クロラムブシル(ロイケラン)、メルファラン、イホスファミドなど)、エ
チレンイミン(チオテパ(チオプレックス)およびヘキサメチルメラミン)、アルキルス
ルホネート(ブスルファン(ミレラン))、ヒドラジンおよびトリアジン(アルトレタミ
ン(ヘキサレン)、プロカルバジン(マチュラン)、ダカルバジン(DTIC)およびテモゾロミド)、ニトロソ尿素(カルムスチン、ロムスチンおよびストレプトゾシン)、金属塩(カルボプラチン、シスプラチン(プラチノール)およびオキサリプラチン)、メクロレタミン、メルファラン(アルケラン)などのアルキル化剤;(2) 植物アルカロイド、テルペノイドおよびトポイソメラーゼ阻害薬、例えばビンカアルカロイド(ビンクリスチン(オンコビン)、ビンブラスチン(ヴェルバン)、ビンデシン、およびビノレルビン)、タキサン(パクリタキセル(タキソール)およびドセタキセル(タキソテール))、ポドフィロトキシン(エトポシドおよびテニソピド)、およびカンプトテカン類似体(イリノテカンおよびトポテカン);(3) アントラサイクリン系(ドキソルビシン(アドリアマイシン、ルベックス、ドキシル)、ダウノルビシン、エピルビシン、ミトキサントロン、イダルビシン、デュオカルマイシンおよびダクチノマイシン(コスメゲン))、クロモマイシン(ダクチノマイシンおよびプリカマイシン(ミトラマイシン))、およびその他(ミトマイシンおよびブレオマイシン(ブレノキサン))などの抗腫瘍薬;(4) 葉酸拮抗薬(メトトレキサート)、ピリミジン拮抗薬(メトトレキサート)、ピリミジン拮抗薬(5−フルオロウラシル、フォクスウリジン、シタラビン、フルロウラシル(5−FU)、カペシタビンおよびゲムシタビン)、プリン拮抗薬(6−メルカプトプリン(プリンエトール)および6−チオグアニン)、6−チオプリン、およびアデノシンデアミナーゼ阻害薬(クラドリビン(ロイスタチン)、フルダラビン、ネララビンおよびペントスタチン)、アザシチジン、チオグアニン、およびシタラビン(ara−C)などの代謝拮抗薬;(5) トポイソメラーゼI阻害薬(イロノテカン、トポテカン)、トポイソメラーゼII阻害薬(アムサクリン、エトポシド、エトポシドリン酸、テニポシド)などのトポイソメラーゼ阻害薬;(6)エストロゲンおよびアンドロゲン阻害薬(タモキシフェンおよびフルタミド)、ゴナドトロピン放出ホルモンアゴニスト(リュープロリドおよびゴセレリン(ゾラデックス))、アロマターゼ阻害薬(アミノグルテチミドおよびアナストロゾール(アリミデックス))によって例示されるホルモン剤;(7) DNA低メチル化剤、例えばアザシチジン、デシタビン;(8) イニパリブ、オラパリブ、ベリパリブなどのポリ(アデノシン二リン酸[ADP]−リボース)ポリメラーゼ(PARP)経路阻害薬;(9) PI3K/Akt/mTOR経路阻害薬、例:エベロリムス;(10) ヒストンデアセチラーゼ(HDAC)阻害薬、例えば、ボリノスタット、エンチノスタット(Sndx−275)、モセチノスタット(MGCD103)、パノビノスタット(LBH589)、ロミデプシン、バルプロ酸[0049]、サイクリン依存性キナーゼ(CDK)阻害剤、例えば、フラボピリドール、オロモウシン、ロスコビチン、ケンパウロン、AG−024322(ファイザー)、ファスカプリシン、リウビジン、プルバラノールA、NU2058、BML−259、SU 9516、PD−0332991、P276‐00[0050]、熱ショックタンパク質(HSP90)阻害剤、例えば、ゲルダナマイシン、タネスピマイシン、アルベスピマイシン、ラジシコール、デグリン、およびBIIB021;(11) マウス二重微小染色体2(MDM2)阻害剤、例えば、Cis−イミダゾリン、ベンゾジアゼピンジオン、スピロ−オキシインドール、イソキノリノン、チオフェン、5−デアザフラビン、トリプタミン;(12) 未分化リンパ腫キナーゼ(ALK)阻害薬、例えば、アミノピリジン、ジアミノピリミジン、ピリドイソキノリン、ピロロピラゾール、インドロカルバゾール、ピロロピリミジン、ジアニリノピリミジン;(13) ベンズアミド、フタラジノン、トリサイクリックインドール、ベンズイミダゾール、インダゾール、ピロロカルバゾール、フタラジノン、イソインドリノンによって例示されるポリ[ADPリボース]ポリメラーゼ(PARP)阻害薬;および(14) アンサクリン、アスパラギナーゼ(El−spar)、ヒドロキシ尿素、ミトキサントロン(ノバントロン)、ミトタン(ライタントロン)、メイタンシノイド、レチノイン酸誘導体、骨髄増殖因子(サルグラモスチムおよびフィルグラスチム)、アミホスチン、葉酸代謝を破壊する薬剤、例えばペメトレキセド、リボヌクレオチドレダクターゼ阻害剤(ヒドロキシ尿素)、副腎皮質ステロイド阻害剤(ミトタン)、酵素(アスパラギナーゼおよびペグアスパラガーゼ)、抗微小管剤(エストラムスチン)、およびレチノイド(ベキサロテン、イソトレチノイン、トレチノイン(ATRA))によって例示されるその他の抗がん剤。
ラン、Ara-C、 アナストロゾール、 BiCNU、ビカルタミド、ブレオマイシン、
ブスルファン、カルボプラチン、カペシタビン、カルボムスチン、CCNU、クロラムブ
シル、シスプラチン、クラドリビン、CPT-11、シクロホスファミド、シタラビン、
シトキサン、ダカルバジン、ダクチノマイシン、ダウノルビシン、ドセタキセル、ドキソ
ルビシン、DTIC、エピルビシン、エトポシド、フロウリジン、フルダラビン、フルタミド、ホテムスチン、ゲムシタビン、ヘキサメチルアミン、ヒドロキシ尿素、イダルビシン、イホスファミド、イリノテカン、ロムスチン、メクロレサミン、メルファラン、メルカプトプリン、メトトレキサート、マイトマイシン、ミトタン、ミトキサントロン、オキサリプラチン、パクリタクセル、パミドロネート、ペントスタチン、プリカマイシン、プロカルバジン、ステロイド、ストレプトゾシン、STI―571、ストレプトゾシン、タモキシフェン、テモゾロミド、テニポシド、テトラジン、チオグアニン、チオテバ、チオグアニン、トムデックス、トポテカン、トレオスルファン、トリメトレキサート、ビンバラスティン、ビンクリスチン、ビンデシン、ビノレルビン、VP-16、ゼローダがある。
であり、強力な細胞毒性を有する。ヒト患者の使用には安全でないと考えられるが、毒性
の懸念のために、メイタンシノイドは本発明に従って、ミニ細胞を介した脳腫瘍患者への
送達に適している。
最初に単離された一連の関連する天然産物である。それらはまた、強力な細胞毒性を有す
るが、ヒトでの使用には安全でないと考えられる。メイタンシノイドと同様に、デュオカ
ルマイシンは、本発明における使用のための適切な化学療法薬である。
ーゼ、AIN―457、バピネオズマブ、べリムナブ、ブレンツキシマブ、ブリアキヌマブ、カナキヌマブ、セツキシマブ、ダロツズマブ、デンソスマブ、エプラツズマブ、エスタフェナトクス、ファルレツズマブ、フィギツムマブ、ガリキシマブ、ゲムツズマブ、ギレンツキシマブ(WX−G250)、ハーセプチン、イブリツモマブ、イノツズマブ、イポリズマブ、メポリズマブ、ムロモナブ−CD3、ナプツモマブ、ネシツツムマブ、ニモツムマブ、オクレリズマブ、オファツムマブ、オテリキシズマブ、オゾガマイシン、パギマクシブマブ、パニツムマブ、ラムシルマブ、ペルツズマブ、レスリズマブ、リツキシマブ、REGN88、ソラネズマブ、テプリズマブ、チウキセタン、トシツモマブ、トラスツズマブ、トレメリムマブ、ベドリズマブ、ザルツムマブおよびザノリムマブがある。
ara-C、アナストロゾール、BiCNU、ビカルタミド、ブレオマイシン、ブスルフ
ァン、カペシタビン、カルボプラチン、カルボプラチン、カルムスチン、CCNU、クロ
ラムブシル、シスプラチン、クラドリビン、CPT−11、シクロホスファミド、シタラビン、シトキシンアラビノシド、シトキサン、ダカルバジン、ダクチノマイシン、ダウノルビシン、デキソラゾキサン、ドセタクセル、ドキソルビシン、DTIC、エピルビシン、エチレンイミン、エトポシド、フロクスウリジン、フルオロウラシル、フルタミド、ホテムスチン、ゲムシタビン、ヘキサメチルアミン、ヒドロキシウレア、イホスファミド、イリノテカン、ロムスチン、メクロレタミン、メルファラン、メルカプトプリン、メトトレキサート、ミトマイシン、ミトタン、ミトキサントロン、オキサリプラチン、パクリタクセル、パミドタミン、ペントシタチン、プリカマイシン、プロカルバジン、ステロイド、ストレプトゾシン、STI-571、タモキシフェン、テモゾロミド、テニポシデ、テトラジン、チオグアニン、チオテパ、トムデックス、トモテカン、トレオスルファン、トリメトレキサート、ビンブラスチン、ビンクリスティン、ビンデシン、ビノレルビン、VP-16、キセロダ、アスパラギナーゼ、AIN-457、バピニュウツマブ、ベリムマブ、ブレンツキシマブ、ブリアキヌマブ、カナキヌマブ、セツキシマブ、ダロツズマブ、デノスマブ 、エプラツズマブ、エスタフェナトクス、ファレツツマブ、フィギツムマブ、ガリキシマブ,ゲムツツマブ、ギレンツキシマブ(WX―G250)、ヘルセプチン、イブリツモマブ、イントツツマブ、イプリズマブ、メポリズマブ、ムロモナブ−CD3、ナプツモマブ、ネシツモマブ、ニモツツマブ、オクレリズマブ、オファツムマブ、オテリズマブ、オゾガミシン、パジバキシマブ、パニツムマブ、パーツツマブ、ラムシルマブ、レスリツマブ、リツキシマブ、REGN88、ソラネズマブ、タネツマブ、テプリズマブ、ティウクセタン、トラスツツマブ、トシツモマブ、トレメリムマブ、ベドリズマブ、ザルツムマブ、ザノリムパブ、5FC 、アキュータンホフマンラロッシェ、AEE788ノバルティス、AMG-102、アンチネオプラストン、AQ4N(バノキサントロン)、AVANDIA(ロシグリタゾンマレイン酸塩)、アバスチン(ベバシズマブ)ジェネテック、BCNU、biCNUカルムスチン、CCI−779、CCNU、CCNUロムスチン、セレコキシブ(システミック)、クロロキン、シレンギチド(EMD121974)、CPT−11(CAMPTOSAR、イリノテカン)、ダサチニブ(BMS−354825、スプリセル)、樹状細胞療法、エトポシド(エポシン、エトポホス、ベペシド)、GDC−0449、グレベック(メシル酸イマチニブ)、グリアデルウエファー、ヒドロキシクロロキン、IL−13、IMC−3G3、免疫療法 、イレッサ(ZD-1839)、ラバチニブ(GW572016)、がん用メトトレキサート(システミック )、ノボキュア、OSI-774、 PCV、RAD001 ノバ―ティス (mTOR阻害剤)、ラパマイシン(ラパムネ、、シロリムス)、RMP−7、シンバスタチン、シロリムス、ソラフェニブ、SU−101、SU5416 スゲン、スルファサラジン(アズルフィデン)、スーテント(ファイザー)、TARCEVA(エロトニブHCl)、タキソール、TEMODAR schering-plough、TGF-Bアンチセンス、サロミド(サリドマイド)、トポテカン(システィミック)、VEGFトラップ、VEGF−trap、ボリノスタット(SAHA)、XL765、XL184、XL765、 ザルネストラ(tipifarnib)、ZOCOR(シムバスチン)、シクロホスファミド(シトキサン)、(Alkeran)、クロラムブシル(リューケラン)、チオペタ(チオプレックス)、ブスルファン(ミレラン)、プロカルバジン(マツラン)、ダカルバジン(DTIC)、アルトレタミン(ヘキサレン)、クロラムブシル、シスプラチン(プラチノール)、イホスファミド、メトトレキサート(MTX)、6-チオプリンス(メルカトプリン[6-MP]、チオグアニン[6-TG])、メルカプトプリン(プリンネトール)、リン酸フルダラシン、(ルイスタチン)、フルロラシル(5-FU)、シタラビン(ara−C)、アザシチジン、ビンブラスチン(Velban)、ビンクリスチン(オンコビン)、ポドフィロトキシン(エトポシド{VM−16}およびテニポシドVM{VM−26})、カンプトテシン(トポテカンおよびイリノテカン(タキソール)、パクリタクセル(タクソール)およびドセタクセル(タクソテレ)のようなタキサン、(アドリアマイシン、ルベックス、ドキシル)、ダクチノマイシン(コスメゲン)、プリカマイシン(ミズラマイシン)、マイトマイシン(ムタマイシン)、ブレオマイシン(ブレノキサン)、エストロゲンおよびアンドロゲン阻害薬(タモキシフェン)、ゴナドトロピン放出ホルモンアゴニスト(リュープロリドおよびゴセレリン(ゾラデックス)、アナストロマターゼ阻害薬(アミノグルテチミドおよびアナストロゾール(アリミデックス))、アムサクリン、アスパラギナーゼ(El-spar)、ミトキサントロン(ノバントロン)、ミトタン(ロイソドレン)、レチノイン酸誘導体、骨髄増殖因子(サーグラモスチムおよびフィルグラスチム)、アミホスチン、デシタビン、イニパリブ、オラパリブ、ベリパリブ、エベロリムス、ボリノスタット、エンチノスタット(SNDX−275)、モセチノスタット(MGCD0103)、パノビノスタット(LBH589)、ロミデプシン、バルプロ酸、フラボピリドール、オロモウシン、ロスコビチン、ケンパウロン、AG−024322(ファイザー)、ファスカプリジン、リュービディン、プルバラノールA、NU2058、BML−259、SU9516、PD−0332991、P276-00、ゲルダナマイシン、タネスピマイシン、アルベスピマイシン、ラディシコール、デグエリン、BIIB021、cis−イミダゾリン、ベンゾジアゼピンジオン、スピロ−オキシインドール、イソキノリノン、チオフェン、5−デアザフラビン、トリプタミン、アミノピリミジン、ジアミノピリミジン、ピリドイソキノリン、ピロロビラゾール、インドロカルバゾール、ピロロピリミジン、ジアニリノピリミジン、ベンズアミド、フタラジノン、トリサイクリックインドール、ベンズイミダゾール、インダゾール、ピロロカルバゾール、イソインドリノン モルホリニルアントラサイクリン、マイタンシノイド、デュサルマイシン、カリケアマイシン(DNA損傷剤)、α−アマニチン(RNAポリメラーゼ阻害剤)、センタナマイシン、ピロロベンゾジアゼピン、ニトロゲンムスタード、ストレプトニグチン、ニトロソウアート、アルカンスルホネート、ピリミジン類似体、プリン類似体、代謝拮抗剤、葉酸類似体、アントラサイクリン、タキサン、ビンカアルカロイド、トポイソメラーゼ阻害剤、ホルモン剤、およびこれらの任意の組合せからなる群より選択される化合物を含む。
本開示に従って使用可能な活性薬剤は、上記に列挙した薬物種類または特定の薬剤に限定されない。異なる発見舞台は、がん細胞のユニークな分子特徴に向けられる新しい薬剤を産生し続ける;実際、数千のそのような化学的および生物学的薬剤が発見されており、そのうちのいくつかのみをここに列挙する。しかし、親水性または疎水性の多様な活性薬剤のパッケージ化に適応する、無傷の細菌由来ミニ細胞および死滅細菌細胞の驚くべき能力は本開示の発見に従って、ミニ細胞にパッケージ化場合に、本質的に任意のそのような薬剤ががんを治療する可能性を有することを意味する。
限定されない機能性核酸である。種類の構成員は、以下でさらに議論される。
「放射性核種」とは、不安定な核をもつ原子、すなわち、核内で新たに生成された放射
線粒子または原子電子のいずれかに付与されるのに利用可能な過剰なエネルギーを特徴と
する原子である。本明細書では放射性核種を「放射性同位元素」、「放射性イメージング
剤」、または「放射性標識」と呼ぶこともある。放射性核種はイメージングおよび/また
は治療目的で使用することができる。それらは、ミニ細胞内に含有され得るか、またはミ
ニ細胞外表面上のリガンド、ペプチド、または糖脂質に付着され得る。結合は、直接また
はリンカーを介して、メルカプトアセチルトリグリシン(MAG3)、DOTA、EDT
A、HYNIC、DTPA、またはクラウンエーテルなどのキレート剤を含むキレート化
部分を含有するリンカーを使用することができる。キレート剤は、ミニ細胞表面成分に直
接付着させるか、またはリンカーを介してミニ細胞に付着させることができる。多くの放
射性核種が当技術分野で知られており、イットリウム−90、テクネチウム−99m、ヨ
ウ素−123、ヨウ素−124、ヨウ素−125、ヨウ素−131、ルビジウム−82、
タリウム−201、ガリウム−67、フッ素−18、キセノン−133、およびインジウ
ム−111など、多くの放射性核種が医療用途に適していることが知られている。
ム86、テルビウム152、テルビウム155、テルビウム149、テルビウム161、
テクネチウム99m、ヨウ素123、ヨウ素131、ルビジウム82、タリウム201、
ガリウム67、フッ素18、銅64、ガリウム68、キセノン133、インジウム111
、ルテチウム177、およびそれらの任意の組合せからなる群から選択される放射性同位
体を含む。
素には、例えば、ガンマおよびベータ放射体であるヨウ素-131およびルテチウム-17
7が含まれる。従って、これらの薬剤は、画像化および治療の両方のために使用され得る
。
(ガンマおよびベータ放射体)もまた、画像化および治療目的の両方のために使用され得
る(GerardおよびCavalieri、2002; Alzahraniら、2012)。
):152Tb(ベータプラス放射体)、155Tb(ガンマ放射体)、149Tb(アルファ放射体)、および161Tb(beta-particle)(Mullerら、2012; Walrandら、2015)である。
−99m(99mTc)またはヨウ素−123(123I)などのガンマエミッタは、ガンマカメラ(平面イメージング)またはSPECT(単一光子放出コンピュータ断層撮影)(Holman and Tumeh、1990)を用いて配置することができる。
して、広範な使用を見出している。いくつかの実施形態において、放射性核種は、抗腫瘍
剤として適切に使用される。
る。したがって、タンパク質または他のミニ細胞表面部分(下記参照)は、Pierce Biote
chnology Inc(Rockford、Ill.)から市販され、Rice ら、Semin. Nucl. Med.,41,265(2011)に詳述されている、Pierceヨウ素標識試薬の使用などの市販の標識手段を使用して、放射性核種で標識することができる。あるいは、放射性核種をミニ細胞内のタンパク質に組み込むことができる。
NAで形質転換する。非対称的な細胞分裂の際にミニ細胞が形成されると、プラスミドD
NAのいくつかのコピーがミニ細胞の細胞質に分離する。得られた組換えミニ細胞を、プ
ラスミドDNAからミニ細胞内で発現された外来タンパク質が放射性核種運搬アミノ酸に
組み込まれるような条件下で、放射性標識アミノ酸の存在下でインキュベートする。例え
ば、Clark-Curtiss anDCurtiss、Methods Enzymol、101:347−362(1983)のプロト
コールに従って、組換えミニ細胞を、35Sメチオニンを含む最小増殖培地中でインキュベ
ートし、それにより、新たに発現されたプラスミドコードタンパク質が35Sメチオニンを
組み込む。同様の手法を用いて、組換えミニ細胞を所望に応じて他の放射性標識でパッケ
ージ化することができる。
uppl. 26), 17.5.1-17.5.8 (1994)に記載された十分に確立されたプロトコールを使用し
て放射性標識され得る。ミニ細胞に固有のこのようなオリゴ糖の例として、グラム陰性菌
由来のミニ細胞の表面に見られるリポ多糖(LPS)のO−多糖成分が挙げられる(後述)。
腫瘍標的化リガンドとして使用される二重特異性抗体を放射性標識することである。米国
特許公開2007/0237744を参照されたく、その内容は参照により本明細書に組み込まれる
。すなわち、ミニ細胞上に「コーティングされた」二重特異性抗体は、放射性標識のため
にかなりの量のさらなる表面タンパク質を暴露する。したがって、抗体でコーティングさ
れたミニ細胞に関連する放射標識のより高い比活性を達成することが可能である。対照的
に、コーティングされていないミニ細胞の放射性標識、すなわち、放射性核種が風土病非
のみを標識する場合、より弱い標識(より低い比活性)を生じ得る。一実施形態では、こ
のより弱い標識がグラム陰性細菌に由来するミニ細胞の外膜関連タンパク質が以下でさら
に論じるように、ミニ細胞表面を覆うO-多糖の長鎖を含むLPSによってマスクされるために起こると考えられる。
を少なくとも減少させるのに十分な腫瘍内照射のレベルを与える用量または複数の用量で
送達される。治療の進行は、この線に沿って、ケースバイケースで監視することができる
。しかしながら、一般的には組成物中にパッケージ化される放射能の量が典型的には約3
0〜約50Gyのオーダーであるが、本発明はまた、約30Gy〜約200Gyの全範囲を与え
る、例えば約50〜約200Gyのような、より多量の放射能を意図する。
考慮すると、組成物中にパッケージされる放射能の量は上記よりもさらに低くてもよい。
したがって、一態様では、組成物が約20〜約40Gy、または約10〜約30Gy、または
約1〜約20Gy、または約10Gy未満を含む。
放射線を送達する機能を有する放射性同位元素を含み得る。いくつかの実施形態において
、リガンドは、Arg-Gly―Asp(RGD)ペプチド、ボンベシン(BBN)/ガス
トリン放出ペプチド(GRP)、コレシストキニン(CCK)/ガストリンペプチド、α−メラノサイト刺激ホルモン(α−MSH)、ニューロペプチド(NT)、[68Ga]Ga−PSMA−HBEDCC([68Ga]Ga−PSMA−11[PET])、[177Lu]Y−J591、[123I]I−MIP−1072、[131I]I−MIP−1095、68Gaまたは177Lu標識PSMA−I&T、68GaまたはLu標識DKFZ−PSMA−617(PSMA−617)、ソマトスタチン(SST)ペプチド、サブスタンスP、T140を含む腫瘍分子標的ペプチド1(TMTP1)、血管作動性腸ペプチド(VIP)、またはそれらの任意の組合せ。
くつかの実施形態では、結合はリンカーを介する。いくつかの実施形態では、腫瘍標的化
リガンドが放射性同位体または放射性同位体をキレート化するキレート剤部分の結合のた
めの官能基を含むペプチドを含む。結合に利用可能なペプチドの官能基にはリジン側鎖上
のε−アミノ基、アルギニン側鎖上のグアニジニウム基、アスパラギン酸またはグルタミン酸上のカルボキシル基、システインチオール、およびチロシン上のフェノールが含まれるが、これらに限定されない。最も一般的な共役反応は、カルボジイミド/Nヒドロキシスクシンイミジル(EDC/NHS)媒介カルボキシルおよびアミンカップリング、チオール基へのマレイミド共役、およびチロシン上のフェノールのジアゾニウム修飾である。ペプチドを画像化部分と結合させるための代表的な化学は多くの総説において見出すことができる(ErathodiyilおよびYing,2011; Takahashiら、2008)。
放射性同位元素はSPECT画像化のための99mTc、123I、および111In、ならびにPET画像化のための18F、64Cu、および68Gaを含むペプチド標識のために使用されている(Chatalicら、2015)。一般に、これらの放射性同位体は、キレート剤を介してペプチドに結合される。いくつかの広く使用されているキレート剤は、(Sunら、2017)に記載されている。ほとんどの治療用放射性医薬品は、β放出同位体(β−)標識されている。
射性同位元素から標的放射線を送達する。いくつかの実施形態では放射性同位元素が治療
用放射線放出剤として機能し、放射性同位元素によって提供される放射線の量は腫瘍に対
する治療効果を提供するのに十分である。いくつかの実施形態において、治療効果は、腫
瘍サイズの減少である。腫瘍は、約100%、約90%、約80%、約70%、約60%
、約50%、約40%、約30%、約20%、約10%、または約5%だけサイズが縮小
され得る。
めに使用され得る。骨代謝の程度に応じて、トレーサーは、骨、好ましくは骨芽細胞性骨
転移への接着を介して蓄積する。治療計画には、テクネチウム−99m−ヒドロキシエチ
リデンジホスホネート(HEDP)による骨シンチグラフィーを行い、代謝および転移病
変の範囲を推定する必要がある。ビスホスホネートHEDPは、レニウム186(βエミ
ッター、半減期:89時間、1.1MeV最大エネルギー、最大範囲:4.6mm)またはレ
ニウム188(βエミッター[85%、2.1MeV]およびγエミッター[15%、155 keV]、半減期:16.8時間、軟組織における最大範囲:10mm)のいずれかでの治療のために標識することができる(Palmedo、2007)。骨緩和療法のための新しい有望な放射性医薬品には、ゾレドロン酸の放射性標識複合体がある。ゾレドロン酸は環状側鎖をもつ新しい、最も強力なビスホスホネートの世代に属する。スカンジウム−46またはルテチウム−177で標識したゾレドロン酸の骨親和性は優れた吸収([177Lu]Lu−ゾレドロン酸に対して98%、[46Sc]Sc−ゾレドロン酸に対して82%)を示し、これは、サマリウム−153で標識したビスホスホネートよりもはるかに高い(最大:67%)(Majkowskaら、2009)。これらのビスホスホネートは骨転移の診断または治療として使用するために、無傷のミニ細胞に結合させることができる。
本開示において使用される抗腫瘍剤はまた、化学療法薬であり得る。本明細書において
、「化学療法剤」、「化学療法剤」、および「化学療法剤」は、腫瘍細胞を殺すかまたは
破壊する能力を有する薬剤を意味するために互換的に使用される。化学療法剤は以下にさ
らに詳述されるように、小分子薬物または生物学的薬物であり得る。
タンパク質またはポリマー高分子と比較して低分子量を有することを特徴とする化合物を
包含する。小分子薬物は典型的には約800ダルトン以下であり、Temodar(登録商標)
(テモゾロミド)によって例示されるように、約194ダルトンで、約150ダルトンの
下限を有し、これは神経膠芽腫および他のタイプの脳がんを処置するために使用される。
この文脈において、「約」は、適格な分子量値が測定精度の変動および数ダルトンまたは
数十ダルトンのオーダーの実験誤差を受けることを示す。したがって、小分子薬物は、約
900ダルトン以下、約800ダルトン以下、約700ダルトン以下、約600ダルトン
以下、約500ダルトン以下、または約400ダルトン以下、例えば約150〜約400
ダルトンの範囲の分子量を有することができる。より具体的には、小分子薬物が約400
ダルトン以上、約450ダルトン以上、約500ダルトン以上、約550ダルトン以上、
約600ダルトン以上、約650ダルトン以上、約700ダルトン以上、または約750
ダルトン以上の分子量を有することができる。別の実施形態では、ミニ細胞にパッケージ
された小分子薬物が約400〜約900ダルトン、約450〜約900ダルトン、約45
0〜約850ダルトン、約450〜約800ダルトン、約500〜約800ダルトン、ま
たは約550〜約750ダルトンの分子量を有する。
ミン、アルカンスルホネート、テトラジン、白金化合物、ピリミジン類似体、プリン類似
体、代謝拮抗物質、葉酸類似体、アントラサイクリン、タキサン、ビンカアルカロイド、
およびトポイソメラーゼ阻害剤などの上記に列挙したものが含まれるが、これらに限定さ
れない。従って、本発明において使用するための小分子薬物は特に、以下のいずれかの中
から選択され得る:エネジイン(例えば、ダインマイシンA、ユニラマイシン、カリケア
マイシンγ1およびカリケアマイシンθ1);メアマイシン、FR901464の合成類似体;ベンゾスベレン誘導体(例えば、Tanpureら、Bioorg Med. Chem., 21: 8019−32 (2013)に記載される);オーリスタチン(例えば、オーリスタチンE、モノメチルオーリスタチンE(MMAE))およびオーリスタチンF(これらは、ドラスタチンの合成類似体である);デュオカルマイシン(例えばデュオカルマイシンSAおよびCC−1065);メイタンシンおよびその誘導体(メイタンシノイド)(例えば、DM1およびDM4);イリノテカン(Camptosar(登録商標))ならびに他のトポイソメラーゼ阻害剤(例えば、トポテカン、エトポシド、ミトキサントロンおよびテニポシド);ならびにヤタケマイシン(その合成はOkanoら、2006によって詳述される)。
発明での使用に適したもの例証である:アクチノマイシンD、アルケラン、アラビアC、
アナストロゾール、BiCNU、ビカルタミド、ビサントレン、ブレオマイシン、ブスル
ファン、カペシタビン(Xeloda(登録商標))、カルボプラチン、カルボプラチナム、カルムスチン、CCNU、クロラムブシル、シスプラチン、クラドリビン、CPT11、シクロホスファミド、シタラビン、シトシンアラビノシド、シトキサン、ダカルバジン、ダクチノマイシン、ダクノルビシン、デキサラゾキサン、ドキソルビシン、DTIC、エピルビシン、エチレンイミン、エトポシド、フロキシウルジン、フルダラビン、フルオロウラシル、フルタミド、ホテムスチン、ゲムシタビン、ヘキサメチルアミン、ヒドロキシ尿素、イダルビシン、イホスファミドイリノテカン、イリノテカン、ロムスチン、メクロルエタミン、メルファラン、メルカプトプリン、メトテレキサエート、ミトマイシン、ミトタン、ミトキサントロン、オキサリプラチン、パクリタキセル、パミドロネート、ペントスタチン、プリカマイシン、プロカルバジン、ストレプトゾシン、STI−571、タモキシフェン、テモゾロミド、テニポシド、テトラジン、チオグアニン、チオテパ、トポテカン、トムデックス、トレオスルファン、トリメトレキサート、ビンブラスチン、ビンクリスチン、ビンデシン、ビンオレルビンおよびVP-16。
酸」を除く生物学的プロセスによって作製することができる任意の生物学的に活性な高分
子を意味すると定義され、サイズによってそのポリペプチドは上記に定義されるように小
分子薬物として適格である。したがって、「生物学的薬物」サブカテゴリーは、小分子薬
物および機能的核酸サブカテゴリーを除外し、それらと重複しない。生物学的薬物の例は
、例えば、医薬化学および薬物設計のツールを使用して、天然であろうと組換えであろう
と合成であろうと、治療用タンパク質および抗体である。
化学療法を目的として設計されたある種の分子は、許容できない毒性のために前臨床ま
たは臨床試験中に失敗する。本発明者らは高毒性または「超毒性」の化学療法薬をミニ細
胞にパッケージ化し、続いて腫瘍患者に全身的に送達すると、薬剤が腫瘍細胞に送達され
ることを示した。さらに、腫瘍細胞が破壊され、薬剤含有細胞質が近傍の正常組織に放出
された後でも、結果は正常組織に対する毒性ではない。これは薬剤が既にDNAなどの腫
瘍細胞構造に結合しており、もはや正常細胞を攻撃できないためである。したがって、本
発明はがん患者への高毒性(「超毒性」)化学療法薬の送達に特に有用である。
度の耐性を伴うかなりの不均一性の段階に達した可能性が高い。本記述の「毒性の強い化
学療法剤」または「超毒性の化学療法剤」とは、がん細胞に対する有効用量と比較して、
正常細胞に対する致死量が比較的低いため、従来の薬剤に対する耐性を克服できる化学療
法剤を指す。
ン、モルホリニルアントラサイクリン、およびそれらの誘導体が含まれるが、これらに限
定されない。メイタンシノイド(分子量:約738ダルトン)はメイタンシンの化学誘導
体の群であり、強力な細胞毒性を有する。ヒト患者の使用には安全ではないと考えられる
が、毒性の懸念のために、メイタンシノイドは本発明に従って、ミニ細胞を介した腫瘍患
者への送達に適している。デュオカルマイシン(分子量:約588ダルトン)は、ストレ
プトマイセス細菌から最初に単離された一連の関連する天然産物である。それらはまた、
強力な細胞毒性を有するが、ヒトでの使用には安全でないと考えられる。メイタンシノイ
ドと同様に、デュオカルマイシンは、本発明における使用のための適切な化学療法薬であ
る。
リン誘導体のクラスの化合物が例示されている。そのような誘導体の中には、ネモルビシ
ン(3’−デアミノ−3’−[2(S)−メトキシ−4−モルホリニル]ドキソルビシン)(MMDX)、およびその主要代謝産物であるPNU−159682(3’−デアミノ−3’−アンヒドロ−[2’’(S)−メトキシ−3’’(R)−ヒドロキシ−4’’−モルホリニル]ドキソルビシン)、ならびに米国特許第8,470,948号(その内容は参照により本明細書に組み込まれる)に記載されている他のそのような4つの誘導体がある:3’−デアミノ−3’’−4’−アンヒドロ−[2’’(S)−メトキシ−3’’(R)−ヒドロキシ−4’’−モルホリニル]−イダルビシン;3’−デアミノ−3’’−4’−アンヒドロ−[2’’(S)−メトキシ−3’’(R)−ヒドロキシ−4’’−モルホリニル]−ダウリルビシン;3’−デアミノ−3’’−4’ −アンヒドロ−2’’ (S)−メトキシ−3’’(R)−ヒドロキシ−4’’−モルホリニル]−カミノマイシン;および3’−デアミノ−3’’−4’−アンヒドロ−[2’’(S)−エトキシ−3’’(R)−ヒドロキシ−4’’−モルホリニル]d−オキソルビシン。
’アンヒドロ[2’’(S)メトキシ3’’(R)オキシ4’’モルホリニル]ドキソル
ビシン(PNU159682)を含む。本発明者らは、PNU−159682が多くの異
なる腫瘍細胞株における薬物耐性を克服すると思われる強力な薬物であり、多くの異なる
腫瘍細胞株に対する細胞毒性分析において、ある範囲の従来の化学療法薬よりもはるかに
強力であることを発見した。実施例8および9を参照されたい。さらに、ドキソルビシン
に耐性のヒト腫瘍異種移植片が、EGFR標的およびPNU−159682負荷EDVの
IV投与によって効果的に治療され得ることが、インビボマウス異種移植実験において示された。実施例11を参照。注目すべきことに、PNU−159682負荷EDVとI型インターフェロンアゴニストを併用すると、忍容性が良好であり、後期膵がん患者において相乗的かつ抗がん効果の改善が得られることがわかった。実施例12を参照されたい。したがって、本発明の一実施形態では、組成物が活性抗がん剤としてPNU159682を含むEGFR標的ミニ細胞を含む。
ン、カリケアマイシン(DNA損傷剤)、α−アマニチン(RNAポリメラーゼII阻害剤
)、センタナマイシン、ゲルダナマイシン、ピロロベンゾジアゼピン、ストレプトニグチ
ン、窒素マスタード、ニトロソルエース、エチレンイミン、アルカンスルホネート、テト
ラジン、白金化合物、ピリミジン類似体、プリン類似体、代謝拮抗剤、葉酸類似体、アン
トラサイクリン、タキサン、ビンカアルカロイド、トポイソメラーゼ阻害剤、およびホル
モン剤が含まれる。
別の局面において、ミニ細胞は、生物学的化学療法薬を含み得る。このような薬物の実
施例としては、アスパラギナーゼ、AIN―457、バピネオズマブ、ベリムマブ、ブレ
ンツキシマブ、ブリアキンマブ、カナキヌマブ、セツキシマブ、ダロツズマブ、デノスマ
ブ、エプラツズマブ、エストラフェナトクス、ファレツヅマブ、フィギツムマブ、ガリキ
シマブ、ゲムツズマブ、ギレンツキシマブ(WX-G250)、イプリツモマブ、イノツ
ズマブ、イピリズマブ、メポリズマブ、ムロモナブ−CD3、ナプツモマブ、ネシツムマブ、ニモツムマブ、オクレリズマブ、オファツムマブ、オテリズマブ、オゾガミシン、パ
ジバキシマブ、パニツムマブ、ペルツヅマブ、ラムシルマブ、レスリズマブ、リツキシマ
ブ、REGN88、ソラネズマブテプリズマブ、タネズマブ、チウキセタン、トシツモマ
ブ、トラスツズマブ(ハーセプチン(登録商標))、トレメリムマブ、ベドリズマブ、ザ
ルツムマブ、およびザノリムマブが挙げられるが、これらに限定されない。
「機能性核酸」とは、宿主細胞への導入の際に、タンパク質の発現を特異的に妨害する
核酸分子をいい、がんの治療に関して、本開示によれば、無傷の細菌由来ミニ細胞を介し
てがん細胞に送達される機能性核酸ペイロードは腫瘍細胞増殖、血管新生または化学療法
に対する抵抗性を促進する、および/またはアポトーシスまたは細胞周期停止を阻害する
遺伝子、すなわち「がん促進遺伝子」を阻害することが好ましい。
とによってタンパク質の発現を減少させる能力を有する。本開示のためのミニ細胞ペイロ
ードのこのカテゴリーには、とりわけ、siRNA、shRNA、ショートRNA(典型
的には400塩基長未満)、マイクロRNA(miRNA)、リボザイムおよびデコイR
NA、アンチセンス核酸、およびLincRNAなどの調節RNAが含まれる。「リボザイム
」とは他のRNA分子を塩基配列特異的に繰り返し切断できる酵素活性を有するRNA分
子をいい、「アンチセンスオリゴヌクレオチド」とは、特定の遺伝子転写産物の一部に相
補的な核酸分子であって、転写産物とハイブリダイズして翻訳を阻害することができるも
のをいい、アンチセンスオリゴヌクレオチドはRNAまたはDNAを含むことができ、「
LincRNA」または「長鎖遺伝子間非コードRNA」とは、200ヌクレオチドより長い非タンパク質コード転写産物を包含する。Khalilら、2009によって論じられているように、LincRNAは遺伝子の転写、スプライシング、および/または翻訳を調節することができる。
、本開示による使用に適した機能的核酸分子の供給源であり得る。したがって、本開示の
一実施形態では、無傷のミニ細胞が腫瘍促進遺伝子を標的とするために利用され得る転写
後遺伝子サイレンシングRNA干渉(RNAi)機構を媒介するsiRNA分子を保有す
る。例えば、MacDiarmidら、2009(抗体提示ミニ細胞は、化学療法薬、薬剤に対する耐性
を発現するのに対抗するsiRNA)、およびOhおよびPark、Advanced Drug Delivery R
ev., 61:850−62(2009)(それぞれ、乳がん、卵巣がん、子宮頸がん、肝臓がん、肺が
ん、および前立腺がんを治療する治療的なsiRNAsの配達)を参照されたい。
名された約10〜約30ヌクレオチド長の二本鎖RNA分子を指す。好ましくは、siR
NA分子は約12〜約28ヌクレオチド長であり、より好ましくは約15〜約25ヌクレ
オチド長であり、さらに好ましくは約19〜約23ヌクレオチド長であり、最も好ましく
は約21〜約23ヌクレオチド長である。したがって、siRNA分子は例えば、約12、約13、約14、約15、約16、約17、約18、約19、約20、約21、約22、約23、約24、約25、約26、約27、約28、または約29ヌクレオチドの長さであり得る。
21量体)とよばれるsiRNAは、19個の連続した塩基対合のためにアニーリングす
る2本の対向するRNA鎖を含むことができる。各鎖上の残りの2つのリボヌクレオチド
は「オーバーハング」を形成し、siRNAが異なる長さの2本の鎖を含む場合、鎖の長
い方がsiRNAの長さを示す。たとえば、21ヌクレオチド長の1本鎖と20ヌクレオ
チド長の2本鎖をもつdsRNAは21量体を構成する。
手可能である。例えば、コンピュータベースのsiRNA設計ツールは、www.dharmacon
.comのインターネット上で利用可能である。
遺伝子サイレンシングRNA干渉(RNAi)機構を媒介することができるmiRNAを
保有する。また、siRNAと同様に、miRNAによって媒介される遺伝子サイレンシ
ング効果を利用して、腫瘍促進遺伝子を標的とすることができる。例えば、Kotaら、2009
(トランスフェクションを介するmiRNAの送達はマウス肝臓がんモデルにおいて、が
ん細胞増殖の阻害、腫瘍特異的アポトーシスおよび疾患進行からの劇的な防御をもたらす
)、およびTakeshitaら、2010(一過性トランスフェクションを介する合成miRNAの
送達は、骨組織上の転移性前立腺腫瘍細胞の増殖を阻害した)を参照のこと。
いて、「miRNA」は一般に、(siRNAの場合のように二本鎖ではなく)約17〜
約27ヌクレオチド一本鎖RNA分子のクラスを意味する。したがって、miRNA分子
は例えば、約17、約18、約19、約20、約21、約22、約23、約24、約25
、約26、または約27ヌクレオチドの長さであり得る。好ましくは、miRNA分子が
約21〜約25ヌクレオチド長である。
補完しないことである。対照的に、siRNAはmRNA標的に対して完全に相補的でな
ければならない。したがって、siRNAは一般的に単一の、特異的な標的のサイレンシ
ングをもたらすが、miRNAは乱雑である。
または化学療法に対する抵抗性を促進する遺伝子を阻害することができる。阻害された遺
伝子自身も、アポトーシスや細胞周期停止を阻害することができる。機能的核酸によって
標的化され得る遺伝子の例は、以下に提供される。
瘍表現型を促進するタンパク質の遺伝子または転写物を標的とする。これらの文脈におけ
る機能性核酸戦略の成功裏の適用は当該技術分野において達成されているが、ミニ細胞ベ
クターの利点はない。例えば、Sioud, Trends Pharmacol. Sci., 2004;、Caplen、Exper
t Opin.Biol.Ther., 2003; Nieth ら、2003; Caplen and Mousses、2003; Duxburyら、2
004; Yagueら、2004;およびDuanら、2004を参照のこと。
白質は、獲得薬物耐性または内因性薬物耐性に寄与する可能性がある。腫瘍細胞などの罹
患細胞が、最初は薬物に反応するが、その後の治療サイクルで不応性となると、耐性表現
型が獲得される。獲得薬物耐性に関与する有用な標的には、P−糖タンパク質(P−gp、P−170、PGY1、MDR1、ABCB1、MDR関連タンパク質、多剤耐性タンパク質1)、MDR−2およびMDR−3などのATP結合カセットトランスポーターが含まれる。MRP2(多剤耐性関連タンパク質)、BCR−ABL(切断点クラスター領域)Abelsonプロトオンコジーン)、STI−571耐性関連タンパク質、肺耐性関連タンパク質、シクロオキシゲナーゼ−2、核因子κ、XRCC1(X線交差相補性グループ1)、ERCC1(切り出し交差相補性遺伝子)、GSTP1(グルタチオンS−トランスフェラーゼ)、変異β−チューブリン、およびIL−6などの増殖因子は、獲得薬物耐性に関与するさらなる標的である。
BCRP、APT11a、およびLRPなどのATP結合カセットトランスポーターが含
まれる。有用な標的には、アポトーシス抵抗性を促進するタンパク質も含まれる。これら
には、Bcl2(B細胞白血病/リンパ腫)、Bcl-XL、A1/Bfl1、局所接着
キナーゼ、ジヒドロジオールデヒドロゲナーゼ、およびp53変異蛋白質が含まれる。
らの例示は、β−カテニン、PKC−α(タンパク質キナーゼC)、C−RAF、DP97 Dead box RNAヘリカーゼ1、FLIP(DNAメチルトランスフェラーゼ1)、C−Sfc、53BPI、Polycomb群タンパク質EZH2(zeste相同体のエンハンサー)、ErbB1、HPV-16 E5およびE7(ヒト乳頭腫ウイルス初期5および初期7)、FortiLin& MCI1P(骨髄性細胞白血病1タンパク質)、DIP13α(DDC相互作用タンパク質13a)、MBD2(メチルCpG結合ドメイン)、p21、KLF4(Kruppel様因子4)、tpt/TCTP(翻訳制御腫瘍タンパク質)、SPK1およびSPK2(スフィンゴシンキナーゼ)、P300、PLK1(ポロ様キナーゼ−1)、Trp53、ErbB1である。VEGF(Vascular endothelial growth factor)、BAG−1(BCL2-associated athanogene 1)、MRP2、BCR−ABL、STI−571 resistance-associated protein、シクロオキシゲナーゼ−2、核因子κ、XRCC1、ERCC1、GSTP1、突然変異体−β−チューブリン、および増殖因子。
PB)によって例示されるグローバル調節エレメントである。例えば、CEPB4は膠芽
腫および膵臓がんにおいて過剰発現され、そこでは、このタンパク質が腫瘍増殖に関連す
る数百の遺伝子を活性化し、健康な細胞においては検出されない(Oritz−Zapaterら、20
11年)。したがって、本明細書に従って、神経膠芽腫の処置はCEPB4の過剰発現に対
抗する薬剤(例えば、siRNAまたは腫瘍細胞によるCEPB4発現を破壊する他の機
能的核酸分子)を含む、無傷の、細菌由来のミニ細胞を含む組成物の投与を介して達成さ
れ得る。
A1)、32kDa(RPA2)、および14kDa(RPA3)サブユニットから構成
される三量体複合体が含まれ、これはすべての生物におけるDNA複製に必須である。If
todeら、1999。
は、ポロ様キナーゼ(PLK1)があり、広範囲のがん細胞で過剰発現していることがわ
かった。実施例3、図12を参照されたい。本開示の発明者らはまた、Plk1(siP
lk1)発現を阻害するsiRNAが、中皮腫および副腎皮質がん細胞の増殖を阻害する
ことを見出した。実施例10を参照。したがって、いくつかの実施形態では、本開示のミ
ニ細胞はPlk1を含む。
レオシド5’−二リン酸から、DNAの複製や修復に必要な対応する2’−デオキシリボ
ヌクレオシド5’−三リン酸への転化を触媒するので、がんの治療標的となりうるリボヌ
クレオチドレダクターゼ(RR)などがある。D’Angiolellaら、2012を参照のこと。ヒ
トRRは2つのサブユニット、RRM1およびRRM2を含み、両方のサブユニットを標
的とする機能的核酸は、本発明において有用である。本開示の発明者らは、RRM1(s
iRRM1)を標的とするsiRNAがミニ細胞を送達すると、中皮腫および副腎皮質が
ん細胞増殖を強力に阻害することを示した。実施例10を参照。したがって、いくつかの
実施形態では、ミニ細胞がリボヌクレオチドレダクターゼM1(RRM1)発現を阻害す
るsiRNAを含む。
本発明の組成物はI型インターフェロンアゴニスト、すなわち、I型インターフェロンのレベル(例えば、活性または発現レベル)を増加させる薬剤を含み得る。ヒトI型インターフェロン(IFN)は、免疫系の活性の調節を助けるインターフェロンタンパク質の大きなサブグループである。インターフェロンはインターフェロン受容体に結合する。I型IFNはすべて、IFNAR1鎖とIFNAR2鎖からなるIFN−α(IFNAR)として知られる特異的な細胞表面受容体複合体に結合する。IFNARはIFN−α(アルファ)、IFN−β(ベータ)、IFN−κ(カッパー)、IFN−δ(デルタ)、IFN−ε(イプシロン)、IFN−τ(タウ)、IFN−ω(オメガ)、IFN−ζ(ゼータ、リミチンとしても知られる)と呼ばれる哺乳類のI型IFNである。
図2は、免疫調節性の60mer二本鎖DNAを含むミニ細胞の例示的な実施形態のグラ
フ描写を示す。本発明者らは、EGFR標的ミニ細胞を有する二本鎖DNAなどのI型イ
ンターフェロンアゴニストの送達が細胞傷害性薬物を負荷されたミニ細胞の免疫賦活剤(
すなわち、抗腫瘍効果を増強する)として作用することを発見した。実施例11を参照さ
れたい。したがって、超毒性薬剤PNU−159682とパッケージされたミニ細胞を組
み合わせることにより、抗腫瘍効果が増強され、この治療は後期膵臓がん患者によって十
分に忍容性であった。実施例12を参照されたい。
よって誘導することができる。具体的には、微生物病原体由来のサイトゾルDNAによる
自然免疫活性化がI型IFNおよびcGAMP、サイクリックGMP−AMP合成酵素(c
GAS)およびIFNγ誘導因子16(IFI16)などのサイトゾルDNAセンサーに
よって媒介される炎症誘発性サイトカインの強力な誘因である。例えば、Hansenら、2014
;およびUnterholznerら、2013を参照のこと。cGASは二本鎖DNAへのポスト結合、c
GASはIFN遺伝子の小胞体結合タンパク質刺激因子(STING)上にドッキングす
るセカンドメッセンジャーサイクリックGMP−AMPを産生する酵素能力を有する。Ba
rberら、2011。これにより、STINGがホモ二量体化し、ERから遊走し(Dobbsら、2015)、STINGをリン酸化するTANK結合キナーゼ1を動員することで、IFNの発現を開始させる転写因子IFN調節因子3が生じる。Dobbsら、2015; Wangら、2014;およびLiuら、2015を参照のこと。したがって、I型IFNの発現は上記および引用された参考文献に記載されるように、細胞質DNAセンサーによって認識され得る標的細胞に二本鎖DNAを送達することによって誘導され得る。
トを含む無傷のミニ細胞を含む。いくつかの実施形態において、I型IFNアゴニストは
本明細書中に記載されるように、I型IFNのDNAセンサ媒介誘導に適切なオリゴヌク
レオチドである。いくつかの実施形態では、オリゴヌクレオチドが少なくとも約10、少
なくとも約20、少なくとも約30、少なくとも約40、少なくとも約50、少なくとも
約60、少なくとも約70、少なくとも約80、少なくとも約90、少なくとも約100
、少なくとも約110、少なくとも約120、少なくとも約130、少なくとも約140
、少なくとも約150、少なくとも約160、少なくとも約170、少なくとも約180
、少なくとも約190、または少なくとも約200ヌクレオチドの配列を含む。別の実施
形態では、オリゴヌクレオチドが約10から約200までのヌクレオチドの配列、または
これらの2つの値の間の任意の量を含む。ある実施形態において、オリゴヌクレオチドは
、少なくとも約40ヌクレオチド、少なくとも約50ヌクレオチド、または少なくとも約
60ヌクレオチドの配列を含む。
得る。たとえば、TLR9活性化合成CpGオリゴデオキシ核酸(CpG−ODN)は、ヒトDNAとは対照的に非メタル化CpGモチフに富んでいるという、菌DNAの免疫刺激特性に基づいて設計された。Kriegら、1995。配列特徴および骨格修飾の最適化は、B細胞またはpDCのいずれかを優先的に活性化するCpGODNサブタイプをもたらした。したがって、本明細書では、CpG−ODNはメチル化されていてもメチル化されていなくてもよく、または両方の組み合わせであってもよいと考えられる。
子はそれらのアゴニストと共に、ミニ細胞を介して送達してI型IFN分泌を誘発するの
に適している。これらの分子には限定されるわけではないが、二本鎖RNA(dsRNA)、ポリ(dA:dT)DNA、二本鎖Z−DNAおよびB−DNA、36bpおよびDNA−RNAハイブリッドより長いDNA(dsDNA)、細菌のセカンドメッセンジャーであるサイクリックジGMP、TLR3、TLR4、TLR7、TLR8およびTLR9アゴニスト、ならびに以下により十分に説明されるSTINGアゴニストが含まれる。
二本鎖RNAはI型IFNの誘導因子である。RNAヘリカーゼであるレチノイン酸誘
導遺伝子I(RIG―I)とメラノーマ分化関連遺伝子5(MDA5)は、I型IFN分
泌を誘発する細胞質受容体である。これらの受容体(RIG−I様受容体)はミトコンドリア局在アダプター分子IPS−1あるいはMAVSとキナーゼTBK1、IKKiを介してシグナルを伝達し、IRF3を活性化し、I型IFN易伝子の転写を誘導する(河合・秋良、2010)。RIG−IおよびMDA5は、それらの5’末端でトリリン酸化されたウイルスRNAに応答する(LeungおよびAmarasinghe、2016; Lu ら、2010; Marq ら、2011; Wang ら、2010)。
RNAポリメラーゼIIIは、ポリ(dA:dT)DNAのための細胞質ゾルDNAセンサーである(Ablasser ら、2009)。細胞質ゾルでは、RNAポリメラーゼIIIがポリ(dA:dT)を5’トリリン酸化を伴うRNAに変換する。変換された5’-pppRNAは、次にRIG−I−MAVS経路とNFκB活性化を開始させて、I型IFN分泌を誘発する。
サイトゾルDNAセンサー、IRFのDNA依存性アクチベーター(DAI)またはZ-DNA結合タンパク質1は、TBK1及びIRF3を介する機構において、右巻きのdsDNA高次構造(B-DNA)に応答してI型IFNを誘導することが知られている(Kawai and Akira、2010)。RNAポリメラーゼIIIもB−DNAを5’−pppRNAに転写し、その後RIG−Iを介してI型IFN転写を活性化する(Chiuら、2009)。これらの転写因子がリン酸化されると、I型IFNファミリーのすべての遺伝子の発現を促すのに役立ち、それによってI型IFN産生が増幅される。多くのサイトゾルDNAセンサーが細胞内病原性DNAを認識することが報告されている。例えば、Xiaら、"DNA sensor cGAS-mediated immune recognition"、Protein Cell、7(11): 777−791 (2016)から抜粋された図26を参照されたい。
rら、2010)およびDAI(Takaokaら、2007)は二本鎖DNA(dsDNA)を検出し、STINGTBK1IRF3経路を活性化する。LRRFIP1はdsDNAと結合し、β−カテニンを介してIRF3の活性化を引き起こす(Yangら、2010)。DHX9およびDHX36はdsDNAと会合し、MyD88を介してNFκB活性化をもたらす(Kimら、2010)。Ku70はdsDNAと結合して、IRF1およびASCRF7の活性化を介してI型インターフェロン(IFN)を誘導する(Zhangら、2011a)。AIM2はASCおよびプロ−カスパーゼ−1を動員することにより、dsDNAと相互作用し、インフラマソームを活性化する(Burckstummerら、2009年; Fernandes-Alnemriら、2009年; Hornungら、2009年)。注目すべきことに、Sox2は好中球のサイトゾルで発現し、dsDNAに結合するとTab2/TAK1複合体を配列依存的に活性化する(Xiaら、2015)。
cGASは、細胞質DNAを認識するDNAセンサーである(Ablasserら、2013a; Ablasserら、2013b; Gaoら、2013a; Liら、2013b; Schogginsら、2014; Sunら、2013; Wuら、2013)。36bpより長い二本鎖DNA(dsDNA)がcGAS活性化に最適である(Gaoら、2013b)。ポストDNA結合、cGASはATPおよびGTPが触媒ポケットに入ることを可能にする立体配座変化を受け、STING−TBK1軸の強い活性剤であるCGAMPの合成をもたらす(Civrilら、2013; Gaoら、2013b; Kranzuschら、2013; Wuら、2013; Zhangら、2014)。cGASはdsDNAおよびDNA−RNAハイブリッドによって活性化され得る(Mankanら、2014)。
細菌のセカンドメッセンジャーであるcyclic-di-GMPはDAIまたは他の既
知の細胞質受容体とは無関係であるが、TBK1及びIRF3を必要とする機構を介して
、I型IFNを強力に誘導する(McWhirterら、2009)。
一部の細胞種、例えばマクロファージやDCでは、I型IFNがそれぞれdsRNAやリポ多糖による膜貫通受容体トル様受容体3(TLR3)やTLR4のトリガーに応答して産生される。TLR3とTLR4はアダプター分子TRIFを介してシグナルを伝達し、TBK1と会合してIRF3を活性化する(川井・秋良、2010)。
PDmAbと組み合わせて試験されている[Immune Design 2016](J.MeulenおよびS.Brady、「Immune Design」、Hum.Vaccin.Immunother.,13(1):15(2017))。TLR3アゴニストPolyiltonol(商標)およびTLR7/8アゴニストMEDI9197もまた、進行した到達可能な固形腫瘍を有する患者において試験されている(MedImmune 2016;Oncovir 201)。("自然宿主防御を起動させること; Hiltonol(ポリICLC)および悪性腫瘍、ASalzar、Oncovir、Inc.、www.oncovir.com/id2(accessed July 11、2018); およびGuptaら、"Abstract CT091: MEDI9197の安全性と薬力学的活動、 TLR 7/8 アゴニストは固形腫瘍と共に被検体内で管理される " Cancer Research、AACR Annual Meeting 2017; April 1-5、2017(published July 2017))。低線量放射線療法とともにCpG富化オリゴデオキシヌクレオチド(CpGODN、PF−3512676)などのTLRアゴニストの腫瘍内注射は、第I/II相臨床試験で進行期非ホジキンリンパ腫患者において臨床反応を示している[Dynavax 2016](Adamusら、2018)。
環状ジヌクレオチド(CDN)[サイクリックジ(グアノシン5’一りん酸)、環状ジ(アデノシン5’一りん酸)、及び環状GMPは、インターフェロン遺伝子の細胞質パターン認識受容体刺激剤(STING)を介してTBK1/IRF3/1型インターフェロンシグナル伝達軸を活性化する病原体関連分子パターン分子(PAMP)のクラスである。
ら、WTアレル、レファレンス(REF)アレル(R232H)、HAQアレル(R71H、G230A、R293Q)、AQアレル(G230A、R293Q)およびQアレル(R293Q)を含む5つのヒトSTING変種を識別した(Yiら、2013)。
る消化に抵抗性であり、培養ヒト細胞においてIFN−βのより高い発現を刺激し、ジチオ修飾を含まないCDNと比較してより強力な抗腫瘍免疫を誘導した(Corralesら、2015; Fuら、2015)。ヒトSTINGに対する親和性を増加させるために、MLRRS2 CDAは、1つの2’5’および1つの3’5’混合ホスホジエステル結合(2’,3’CDN)を有するリン酸架橋によって規定される非正準構造を含む。2’,3’混合結合構造は増大したSTING結合親和性を付与し(Gaoら、2013b)、真核生物cGASによって産生される内因性cGAMPにおいても見出される。MLRR―S2CDAは菌体3’,3’CDN(Corralesら、2015; Fuら、2015)によって誘導されたシグナル伝達に耐性であることが知られているREFアレルに対するドナーホモジゴースを含む、異なるSTING型を有する複数のドナーから単離されたヒト周辺血球(PBMC)中のIFN−βのHEK293T細胞STINGシグナリングアッセイおよび誘導されたIFN−βの誘導ドーズ依存性発現において、広範囲に既知のヒトSTINGアレルを活性化することが示された。
本発明の組成物および方法はII型IFNアゴニスト、すなわち、II型インターフェロン
のレベル(例えば、活性または発現レベル)を増加させる薬剤を含み得る。型IIインター
フェロン(IFN)のクラスは、現在、IFN−γ(ガンマ)と呼ばれるメンバーを含む。成熟IFN−γは抗平行ホモ二量体であり、IFN−γ受容体(IFNGR)複合体に結合して標的細胞内でシグナルを誘発する。IFNGRはIFNGR1とIFNGR2と名づけられた各分子の2つのサブユニットからできている。IFN−γは、免疫応答および炎症応答の調節に関与している;ヒトではインターフェロン−γは1種類のみである。活性化T細胞やナチュラルキラー細胞で産生される。IFN−γはI型IFNの作用を増強する。Th1細胞から放出されたIFN−γは感染部位に白血球を動員し、炎症を亢進させる。また、マクロファージを刺激して、飲み込まれた細菌を殺す。Th1細胞から放出されるIFN−γは、Th2応答の調節にも重要である。IFN−γは免疫応答の調節に重要な役割を果たしており、その産生は自己免疫疾患を引き起こす可能性がある。
物を包含する。ミニ細胞は細菌に由来するが、ミニ細胞自体はヒト患者においてII型イン
ターフェロン応答を活性化しない。実施例15を参照。本発明者らは、IFNγの添加が
ドキソルビシンを負荷したEGFR標的EDVの抗腫瘍効力を増大させ、異種移植モデル
において腫瘍退縮を引き起こすことを発見した。実施例13を参照されたい。さらに、(
i)超毒性化学療法薬PNU159682を負荷したEFGR標的化ミニ細胞、(ii)20ヌクレオチドを含む二本鎖DNAを負荷した非標的化ミニ細胞、および(iii)IFNγ生成物Imukinを含むミニ細胞を含む組成物は後期内因性腫瘍に罹患しているイヌにおいて十分に耐性があり、抗がん効果を誘導した。実施例14を参照。
す。例えば、Chikumaら、2017を参照のこと。IFNγサイトカインは自然抗原の結合によりナチュラルキラー細胞から放出されるが、スフィンゴ糖脂質化合物は自然免疫応答と獲得免疫応答の両方の強力な活性化因子として機能することができる。本発明者らは、スフィンゴ糖脂質への曝露がII型インターフェロン、IFN−γ、および多数のインターロイキン(Th1−、Th2−、および/またはTh17−型サイトカイン)を含む自然ナチュラルキラーT(iNKT)細胞による強力なサイトカイン応答を誘導することを発見した。例えば、Carrenoら、2016を参照のこと。次いで、iNKT細胞はDC成熟を誘導し、細胞傷害性T細胞応答の発生をもたらすT細胞ヘルパー様機能を示す。
されており、C−グリコシド形態のα−ガラクトシルセラミド(α−C−GalCer)、αガラクタトシルセラミド (α−GalCer)、12炭素アシル形態のガラクトシルセラミド(β−GalCer)、β−D−グルコピラノシルセラミド(β−GlcCer)、1,2−ジアシル−3−0−ガラクトシル−sn−グリセロール(BbGL−II)、ジアシルグリセロール含有糖脂質(Glc−DAG−s2)、ガングリオシド(GD3)、ガングリオトリアセラミド(Gg3Cer)、グリコシルホスファチジルイノシトール(GPI)、α−グルクロノシルセラミド(GSL−1またはGSL−4)、イソグロボトリヘキシルセラミド(iGb3)、リポホスホグリカン(LPG)、α−ガラクトシルセラミド類似体(OCH)、およびトレイトルセラミド。特定の実施形態において、本明細書中に開示されるミニ細胞は、II型IFNアゴニストとしてα−ガラクトシルセラミド(α−GalCer)を含む。
増強する観点から、II型IFNアゴニストを免疫系の細胞に直接送達することができる。
あるいは、標的でないEDVは免疫系の貪食細胞に取り込まれ、そこでエンドソームで分
解され、αGCは免疫活性化のためにiNKT細胞に提示される。したがって、いくつかの実施形態では、ミニ細胞がII型インターフェロンアゴニストの標的送達を提供する。他の実施形態では、本明細書に開示される組成物がII型インターフェロンアゴニストを含む非標的ミニ細胞を含む。
びグルココルチコイドがある。これらの因子を阻害するタンパク質や核酸は、IFN−γの産生を刺激することができる。
たは分泌を活性化する遺伝子もこの文脈での使用に適している。
の種類の有害作用を引き起こすことなくIFN−緕Y生を誘導できる多くのウイルスワク
チンが利用可能である。この種のウイルスワクチンの代表的なものは、インフルエンザ(
インフルエンザ)ワクチンである。
γの血清濃度が患者が薬物負荷、二重特異的抗体標的化ミニ細胞または死滅細菌細胞の投
与も受けた場合に低いことを示す。従って、1つの局面において、本発明の方法は、約3
0,000pg/mL以下の血清IFNγ濃度の増加を生じる。別の局面において、血清IF
N−γ濃度は、約5000pg/mL、1000pg/mL、900pg/mL、800pg/mL、70
0pg/mL、600pg/mL、500pg/mL、400pg/mL、300pg/mL、200pg/mL、
または100pg/mLより高くないまで増加する。さらなる局面において、得られる血清I
FNγ濃度は、少なくとも約10pg/mL、または少なくとも約20pg/mL、30pg/mL、
40pg/mL、50pg/mL、60pg/mL、70pg/mL、80pg/mL、90pg/mL、100pg
/mL、150pg/mL、200pg/mL、300pg/mL、400pg/mLまたは500pg/mLで
ある。
もしくはアナログである。いくつかの局面において、投与は、宿主血液のml当たり約0.
02ng〜1マイクログラムのIFN−γを達成する。一態様では、宿主血液中で達成されるIFNγ濃度が約0.1ng/ml〜約500ng/ml、約0.2ng/ml〜約200ng/ml、約0.5ng/ml〜約100ng/ml、約1ng/ml〜約50ng/ml、または約2ng/ml〜約20ng/mlである。
「ミニ細胞」という用語はここでは染色体を欠き(「無染色体」)、二分裂の際に、細
胞分裂とDNA分離との協調の乱れによって生じた細菌細胞の誘導体を意味するために用
いられる。ミニ細胞はいわゆる「膜ブレブ」(大きさが約0.2μm以下)のような他の小さな小胞とは異なっている。この小胞が特定の状況下で自然に生成・放出されるが、特定の遺伝子再編成やエピソーム遺伝子発現によるものではない。同じトークンによって、無傷のミニ細胞は同様ゴーストとは区別される。同様ゴーストは特異的な遺伝子再編成やエピソーム遺伝子発現のために生成されない。本開示で使用される細菌由来のミニ細胞は完全に無傷であり、したがって、破壊または分解され、除去さえされる外膜または画定膜によって特徴付けられる、他の無染色体形態の細菌細胞誘導体とは区別される。米国特許を参照されたい。第7,183,105号、第111欄、第54行以降。本開示のミニ細胞を特徴付ける無傷の膜は、腫瘍発明内で、取り込み後にペイロードが放出されるまで、ミニ細胞内での治療用ペイロードの保持を可能にする。
よって細胞の極性部位を脱抑制する結果として産生される無核の非生存ナノ粒子である。
Maら、2004。抑制解除は細菌が中心および極で分裂することを意味し、本開示の発明者ら
が示したミニ細胞をもたらす極性分裂は、様々な化学療法薬の効率的なパッケージ化を可
能にする漏出耐性のマイクロリザーバ担体として機能することができる。さらに、例えば
、粒子当たり約14,000分子しかパッケージ化できない現在のステルスリポソーム薬物キャリア(リポソームドキソルビシン)とは対照的に(Parkら、Breast Cancer Res、4(3):95−99(2002))、または5個未満の薬物分子を運ぶことができる「武装抗体」、EDVは、100万個までの薬物分子のペイロードを容易に収容することができる。さらに、EDVは二重特異性抗体を使用してがん細胞の表面上の過剰発現受容体を標的とすることができ(下記の部Dを参照)、これはインビトロおよびインビボの両方で、非常に有意な腫瘍増殖阻害および/または退行を可能にする。
菌細胞から調製することができる。原核生物の染色体複製は、中細胞隔形成を含む、正常
な二分裂に連結されている。たとえば大腸菌では、minCDのようなmin遺伝子の変
異によって、細胞分裂の際に細胞極での隔壁形成の阻害を取り除くことができ、その結果
、正常な娘細胞と染色体のないミニ細胞が産生される。de Boer ら、J.Bacteriol.,174:
63−70 (1992);Raskin & de Boer、J.Bacteriol.,181:6419−s24 (1999); Hu & Lutkenhaus、Mol.Microbio.,34:82−90(1999); Harry, Mol. Microbiol.,40:795(20
01)を参照されたい。
に影響を及ぼす一連の他の遺伝子再編成または突然変異に続いて、染色体のないミニ細胞
も生成される。ReeveおよびCornett、JVirol., 15:1308(1975)を参照のこと。ミニ細
胞はまた、細胞分裂/染色体分離に関与するタンパク質の遺伝子発現レベルの撹乱に続い
て形成され得る。例えば、minEの過剰発現は、極性分裂およびミニ細胞の産生を導く
。同様に、染色体のないミニ細胞は染色体分離における欠損、例えば、Bacilus subtilisにおけるsmc突然変異(Brittonら、Genes Dev.,12: 1254(1998))、BsubtilisにおけるspoOJ欠失(Iretonら、JBacteriol., 176: 532029(1994))、EcoliにおけるmukB突然変異(Hiragaら、JBacteriol., 171:1496(1989))、およびEcoliにおけるparC突然変異(StewartおよびD’Ari、JBacteriol., 174:4513(1992))に起因し得る。さらに、CafAは細胞分裂の速度を増強し得、そして/または複製後の染色体分配を阻害し得(Okadaら、JBacteriol., 176:917(1994))、連鎖細胞および染色体不含ミニ細胞の形成を生じる。
れは、これらの細菌における細菌細胞分裂の保存された性質によるグラム陽性またはグラ
ム陰性起源である。さらに、本開示において使用されるミニ細胞は上記のように、無傷の
細胞壁(すなわち、「無傷のミニ細胞」)を有するべきであり、そして特定の遺伝的再配
置またはエピソーム遺伝子発現に起因しない、他の小胞(例えば、膜ブレブ)と区別され
、そしてそれらから分離されるべきである。
またはグラム陰性であり得る。一態様では、親細菌がTerra-/Glidobacteria(BV1)、Prot
eobacteria(BV2)、Spirochaetes、Sphingobacteria、およびPlanctobacteriaを含む
BV4から1つ以上選択される。別の局面に追及すると、細菌は、Bacilli、Clostridia
またはTenericutes/MollicutesなどのFirmicutes(BV3)、またはActinomycetalesま
たはBifidobacterialesなどのActinobacteria(BV5)から選択される1つ以上である。
に定義されているような、細菌、シアノバテリア、真正細菌および古細菌の非生存原核細
胞である。このような細胞はそれらが無傷の細胞壁および/または細胞膜を有し、細菌種
に内因性である遺伝物質(核酸)を含む場合、「無傷」であると見なされる。死滅した細
菌細胞を調製する方法は例えば、米国特許出願公開第2008/0038296号に記載されており
、その内容は、参照により本明細書に組み込まれる。
ス−サームス)、シアノバクテリア、サーモフルバクテリア、好熱菌(アキフィカ、サー
モトガエ)、アルファ、ベータ、ガンマ(腸内細菌科)デルタまたはエプシロンプロテオ
バクテリア、スピロバクテリア、スピロヘータ、フィブロバクター、クロロビ/バクテロ
イデス、クラミジア/ベルコメクロビア、プラントミケス、アシドバクテリア、クリシオ
ゲネス、デフェリバクテリア、フソバクテリア、ゲムアチモナデテス、ニトロスピラ、シ
ネルギステス、ディクチオグロムス、レンティスファエラバシラス、バシラス、リステリ
ア科、ブドウ球菌、腸球菌科、エンテロコッカス、ラクトバシラセエ、ロイコノストック
、クロストリジウム、サーモアナエロバクター、マイコプラズマ目、アナエロプラズマ目
、アコレプラズマ目、アクチオマイシネ、アクチノマイセス、コリネバクテリウム、コリ
ネバクテリウム、フランキナーゼ、ミクロコッシネ、ブレビバクテリウム、およびビフィ
ズス菌から1種以上が選択される。
能な限り完全に単離されたミニ細胞または死滅した細菌細胞を含むべきである。遊離エン
ドトキシンおよび親細菌細胞を除去するために細菌由来のミニ細胞を精製するための方法
論は、例えば、その全体が参照により本明細書に組み込まれるWO 2004/113507に記載さ
れている。簡単には、(a)一般に0.2μmより小さい膜ブレブなどのより小さい小胞、(b)細胞膜から放出される遊離エンドトキシン、および(c)生きているか死んでいるかにかかわらず親細菌、およびそれらの破片(これも遊離エンドトキシンの供給源である)の除去を達成する。このような除去は、とりわけ、より小さい小胞および細胞残屑を除去するための0.2μm濾過、親細胞を誘導してフィラメントを形成した後に親細胞を除去するための0.45μm濾過、生きた細菌細胞を殺すための抗生物質、および遊離エンドトキシンに対する抗体を用いて実施することができる。
約400nmのサイズであり、すなわち、膜小胞および他のより小さい小胞より大きく、そ
して親細菌より小さいという本発明者らによる発見である。ミニ細胞のサイズ決定は、電
子顕微鏡法などの固体状態を使用することによって、または液体ベースの技術、例えば動
的光散乱によって達成することができる。そのような各技法によってもたらされるサイズ
値は誤差範囲を有する可能性があり、その値は、技法間でいくらか異なる可能性がある。
したがって、乾燥状態のミニ細胞のサイズは、電子顕微鏡法によって約400nm±50nm
と測定することができる。動的光散乱は、同じミニ細胞を約500nm±50nmのサイズで
あると測定することができる。また、薬物パッケージ化リガンド標的ミニ細胞は、やはり
動的光散乱を使用して、約400nm〜600nm±50nmであると測定することができる。
らミニ細胞を単離する目的のために、実際に容易に適応される。すなわち、無傷の細菌由
来ミニ細胞は、ミニ細胞に堅い球状構造を与える堅い膜によって囲まれた細胞質を特徴と
する。この構造は透過型電子顕微鏡写真で明らかであり、ミニ細胞の直径は硬い膜の外側
の境界の間でミニ細胞を横切って測定される。この測定により、上述した400nm±50
nmのサイズ値が得られた。
ポ多糖(LPS)のO-多糖成分であり、これはリピドAアンカーを介して外膜に埋め込
まれている。この成分は、鎖の反復単位当たり4〜5個の糖の70〜100個もの反復単
位を有する反復炭水化物残基単位の鎖である。これらの鎖はin vivoのように液体環境で
は剛性ではないので、サンゴ海環境で海藻の一般的な外観を与える波状の柔軟な構造をと
ることができる;すなわち、鎖は、ミニ細胞膜に固定されたままで液体と共に移動する。
キシンを含む。これに関して、約250EU以下、約200EU以下、約150EU以下、約1
00EU以下、約90EU以下、約80EU以下、約70EU以下、約60EU以下、約50EU以下
、約40EU以下、約30EU以下、約20EU以下、約15EU以下、約10EU以下、約9EU以
下、約8EU以下、約7EU以下、約6EU以下、約5EU以下、約4EU以下、約3EU以下、約2
EU以下、約1EU以下、約0.9EU以下、約0.8EU以下、約0.7EU以下、約0.6EU以
下、約0.5EU以下、約0.4EU以下、約0.4EU以下、約0.3EU以下、約0.2EU以
下、約0.1EU以下、約0.05EU以下、または約0.01EU以下。
ば、少なくとも約1×109、少なくとも約2×109、少なくとも約5×109、または
少なくとも8×109含むことができる。いくつかの実施形態において、組成物は約101
1個以下のミニ細胞または死滅したバクテリアセル、例えば約1×1011以下、または約
9×1010以下、または約8×1010以下を含む。
活性剤または抗腫瘍剤(例えば、小分子薬物、タンパク質および機能性核酸)は、複数
の無傷のミニ細胞を活性剤と一緒に緩衝液中で共インキュベートすることによって、ミニ
細胞中に直接パッケージ化され得る。緩衝液の組成は、この分野でよく知られている条件
に応じて変化させ、無傷のミニ細胞における活性剤の負荷を最適化することができる。緩
衝液はまた、薬剤に依存して(例えば、核酸ペイロードの場合、ヌクレオチド配列または
ミニ細胞にロードされる核酸の長さに依存して)変化され得る。ローディングに適した例
示的な緩衝液にはリン酸緩衝生理食塩水(PBS)が含まれるが、これに限定されない。
一旦パッケージ化されると、活性剤はミニ細胞内に残り、分解から保護される。無菌生理
食塩水中でインキュベートしたsiRNAパッケージ化ミニ細胞を用いた長期インキュベ
ーション研究は例えば、siRNAの漏出がないことを示した。
コードするプラスミドのようなベクターを親細菌細胞に形質転換することによって、ミニ
細胞に導入され得る。ミニ細胞が親細菌細胞から形成される場合、ミニ細胞は、プラスミ
ドおよび/または発現産物、抗腫瘍剤の特定のコピーを保持する。ミニ細胞へのパッケー
ジ化および発現産物のさらなる詳細は国際公開第03/033519号パンフレットにより提供さ
れ、その内容は参照によりその全体が本発明に組み込まれる。
する組換えミニ細胞が、食細胞および非食細胞に送達され得ることを実証した。国際公開
第03/033519号パンフレットはまた、エピソーム複製プラスミドDNA上に担持された異
種核酸を用いたミニ細胞産生親細菌株の遺伝的形質転換を記載した。親生物とミニ細胞を
分離すると、エピソームDNAの一部はミニ細胞に分離される。得られた組換えミニ細胞
は哺乳類食細胞に容易に飲み込まれ、細胞内ファゴリソソーム内で分解されるようになっ
た。さらに、組換えDNAの一部はファゴリソソーム膜を逃れ、哺乳類細胞核に輸送され
、そこで組換え遺伝子が発現した。
ッケージ化することができる。このようなアプローチは、薬物耐性およびアポトーシス耐
性と戦うために使用され得る。例えば、がん患者は日常的に化学療法薬に対する耐性を示
す。このような耐性は、とりわけ、多剤耐性(MDR)ポンプおよび抗アポトーシス遺伝
子のような遺伝子の過剰発現によって媒介され得る。この耐性に対抗するために、ミニ細
胞は、MDR席連遺伝子に対する治療的に有意な濃度の機能性核酸と共にパッケージされ得、そして化学療法の前に患者に投与され得る。さらに、異なるmRNA標的に向けられた同じミニ細胞複数の機能性核酸へのパッケージ化は、ほとんどの分子標的が突然変異を受け、複数の対立遺伝子を有するため、治療成功を高めることができる。ミニ細胞への核酸の直接パッケージ化のさらなる詳細は国際公開第2009/027830号パンフレットに提供されており、その内容は、参照によりその全体が本願に組み込まれる。
細胞細胞質との間に薬物の濃度勾配を作り出すことによってミニ細胞中にパッケージ化す
ることができる。細胞外培地がミニ細胞の細胞質よりも高い薬物濃度を含む場合、薬物は
、この濃度勾配を下ってミニ細胞の細胞質に自然に移動する。しかし、濃度勾配が逆にな
ると、薬物はミニ細胞から外に移動しない。例えば、米国特許出願公開第2008/0051469
号(その内容は参照により特に組み込まれる)に、薬物ローディングプロセスおよびその
驚くべき性質のさらなる詳細が見出される。
に溶解され得る。例えば、パクリタキセルはエタノールとクレモフォアEL(ポリエトキシル化ヒマシ油)との1:1ブレンドに溶解し、続いてPBS中で希釈して、水性媒体中で部分的に希釈され、薬物が溶液中に残ることを確実にするために最小量の有機溶媒を担持するパクリタキセルの溶液を達成することができる。ミニ細胞は、薬物負荷のためにこの最終培地中でインキュベートすることができる。従って、本発明者らは疎水性薬物でさえ、ミニ細胞の細胞質または膜中に拡散して、高い、そして治療的に有意な細胞質薬物負荷を達成し得ることを発見した。これは、ミニ細胞膜が疎水性リン脂質二重層から構成されており、疎水性分子の細胞質への拡散を防止することが期待されるため、予想外である。代表的な低分子薬物の多様性のミニ細胞へのローディングが示されており、様々なサイズおよび化学的特性が示されている:ドキソルビシン、パクリタキセル、フルオロ−パクリタキセル、シスプラチン、ビンブラスチン、モンサトロール、チミジル酸シンターゼ(TS)阻害剤OSI−7904、イリノテカン、5−フルオロウラシル、ゲムシタビン、およびカルボプラチン。さらに、ボードを横切って、得られた小分子薬物パッケージ化ミニ細胞は、インビトロおよびインビボで、有意な抗腫瘍効力を示す。
本発明者らは腫瘍細胞周囲の血管が完全性の喪失を示すことを発見した。すなわち、血
管は血液脳関門(BBB)環境においても、大きな開窓を有し、「漏れやすい」状態であ
る。がん細胞が定着すると、新たな血管の形成を促進する物質−血管新生と呼ばれるプロ
セス−を分泌する。これらの血管は速やかに成長し、正常な血管とは異なり、50nmから
1.2μm(透過性亢進血管系)の「穴」(開窓部)で漏れやすい。リポソームなどの薬物送達粒子は、現在、腫瘍微小環境を支持する漏出性血管系からの血管外遊出を含む受動的方法によって腫瘍標的化をもたらすと考えられている。Hobbsら、1998。異常な腫瘍微小環境は間質性高血圧を特徴とし、この現象が抗がん抗体治療薬のアクセスを制限し得ることが示されているが、これは免疫リポソーム(Nielsenら、2002)およびQuantum Dotsに結合体化された抗体(Gaoら、2004)によって例示されるように、絶対的な障壁であるとは思われない。この現象は、特異的に指向された腫瘍抗体を保有するという追加の利点を有するEDVにも当てはまる。静注後、EDVは腫瘍の微小環境に漏出し、その後、がん細胞表面受容体との結合およびエンドサイトーシスを介して能動的なターゲティングが行われる。したがって、従来の理解とは対照的に、ミニ細胞と同じ大きさ、すなわち、BBBの上記のコンセンサス孔径限界よりもはるかに大きい粒子は、それにもかかわらず、漏出性血管壁の開窓よりも小さく、したがって、これらの開窓を通って腫瘍微小環境に受動的に溢出することができる。
、それらに取り込まれることができる。したがって、抗腫瘍剤でパッケージ化されたミニ
細胞は腫瘍細胞の細胞質中に薬剤を放出し、それを殺す。
リガンドを介して標的哺乳動物腫瘍細胞に向けられる。いくつかの実施形態では、リガン
ドは「二重特異性」ですなわち、リガンドはミニ細胞および哺乳動物(腫瘍)細胞成分の
両方に対して特異性を示し、その結果、所与の小胞を標的細胞に結合させ、それによって
、後者が前者を飲み込む。ミニ細胞を腫瘍細胞に標的化するための二重特異性リガンドの
使用は国際公開第05/056749号パンフレットおよび国際公開第05/079854号パンフレット
にさらに記載されており、死滅した細菌細胞を腫瘍細胞に標的化するための二重特異性リ
ガンドの使用は、米国特許第8,591,862号にさらに記載されており、これらの記載内容は
、その全部を参照してここに盛り込んでいる。このようなリガンドが小胞に付着すると、
リガンドの非占有特異性(「単一特異性」)は、それが標的(腫瘍)哺乳動物細胞と相互
作用するまで関係する。多数の腫瘍標的化リガンドが当該分野で公知である(Hongら、20
11; Hoelderら、2012; Galluzziら、2013)。ソマトスタチン(SST)ペプチド、血管作動性腸ペプチド(VIP)、Arg(RGD)ペプチド、およびボンベシン/ガストリン放出ペプチド(BBN/GRP)などのいくつかのペプチドは腫瘍受容体イメージングについて首尾よく特徴付けられている(De Jongら、2009; Tweedle、2009; SchotteliusおよびWester、2009; Igarashiら、2011; Lavermanら、2012)。
正常細胞ではなく腫瘍細胞に過剰発現している受容体は、in vivo腫瘍イメージングの優れた候補である。今日まで、多くの腫瘍標的化ペプチドおよびそれらのアナログが、以下に記載されるように同定されている。
プチドは、エキソクリン、内分裂、サーモレギュレーション、スクロース規制、ならびに
細胞成長などの種々の生理的効果を示すニューロペプチドファミリーから成る(Ohki−Ha
mazaki他、2005)。ボンベシン様ペプチド受容体には、ニューロメジンB受容体、ボンベ
シン3受容体、GRP受容体、ボンベシン4受容体の4つのサブタイプがある。これらの受容体は、乳がん、卵巣がんおよび消化管間質腫瘍などの多くの腫瘍において過剰発現される。
α−MSHとその類似体はヒトメラノーマ転移の80%以上に発現するメラノコルチン−1受容体(MC―1r)に結合親和性を示し、メラノーマ標的イメージングと放射線療法の媒体として広く使用されている。
ドファミリーに属する(Tatemoto,2004)。NPY受容体は、神経芽細胞腫、肉腫、およ
び乳がんを含む様々な腫瘍において過剰発現される。
甲状腺髄様がんなど様々な腫瘍で同定されているNT受容体を標的としている(Tyler-McMahonら、2000)。したがって、それはがんイメージングのための魅力的な候補である。
ら、2014)。[68Ga]GaPSMA−HPEDCC([68Ga]GaPSMA−11[PET]としても知られる)、モノクローナル抗体(mAb)[177 Lu]Lu/[90 Y]y−J591(療法)、[123I]I−MIP−1072(平面/SPECT)、[131I]I−MIP−1095(療法)、およびPETのための68Gaまたは治療のための177Luで標識されたセラノスティック剤PSMA−I&TおよびDKFZ−PSMA−617(PSMA−617)を含む、PSMAを標的とするいくつかの利用可能な放射性医薬品がある。
る天然に存在するシクロペプチドホルモンである(Weckbeckerら、2003)。インスリン、
グルカゴン、その他のホルモンの分泌を阻害することができる。ソマトスタチン受容体(
SSTR;5つのサブタイプSSTR1〜SSTR5)は、神経膠腫、神経内分泌腫瘍お
よび乳房腫瘍を含む多くの腫瘍において過剰発現される。GEP系の神経内分泌腫瘍(N
EN)は、膵臓、空腸、回腸、盲腸、直腸、虫垂、結腸に由来することが最も多い。すべ
てのGEP−NENに共通する特徴は、内分泌細胞と神経細胞の複合的な特徴である。高
分化型NENはソマトスタチン受容体(SSTR)、特にSSTR―2サブタイプを過剰発現する。
モカイン受容体4型(CXCR4)の逆アゴニストである(Burgerら、2005)。その誘導
体はCXCR4造影剤として広く使用されている。
肝微小転移由来のがん細胞に特異的に結合することが見出されている5アミノ酸ペプチド
である(Yangら、2008)。
の成分との間の相互作用によって小胞の細胞膜に付着させることができる。発現されたリ
ガンドは、リガンドの腫瘍表面成分結合部分が露出されるように小胞の表面上に固定され
、その結果、小胞と哺乳動物腫瘍細胞とが接触すると、その部分が標的哺乳動物細胞表面
受容体に結合することができる。
て、あるいは殺された細胞になる前に細菌細胞によって発現され、提示される。この場合
、このリガンドは、小胞に対する特異性を必要とせず、哺乳動物細胞に特徴的な成分に対
してのみ特異性を提示する。すなわち、このような成分は腫瘍細胞がその表面に存在する
限り、腫瘍細胞自体に、または治療中の特定の種類の腫瘍細胞にさえも特異的である必要
はない。
関数として生じるこの蓄積は小胞パッケージ化治療ペイロードの腫瘍細胞への送達をもた
らし、次いで、これはパッケージ化小胞を内在化する。
ーシスに対して抵抗性である細胞を含む、一連の哺乳動物腫瘍細胞に適用可能であること
を見出した。例えば、抗HER2レセプターまたは抗EGFレセプターに向けられる抗体を含むリガンドは、肺、卵巣、脳、乳房、前立腺、および皮膚がん細胞のような一連の標的非貪食細胞上のそれぞれのレセプターにミニ細胞を結合することができる。
行する。すなわち、本発明の文脈において、適切な標的細胞は細胞表面受容体を提示し、
その結合は、小胞上のリガンドによって、その小胞のエンドサイトーシスを誘発する。
哺乳動物細胞表面受容体との間の相互作用がここでは「受容体媒介エンドサイトーシス」
(rME)経路と呼ばれる取り込み経路を、腫瘍細胞などの標的宿主細胞の後期エンドソーム/リソソームコンパートメントに活性化することができることを発見した。このrME経路によって、本発明者らは、細菌由来の小胞が初期エンドソーム、後期エンドソームおよびリソソームを通してプロセシングされ、哺乳動物宿主細胞の細胞質へのそれらのペイロードの放出を生じることを見出した。さらに、核酸であるペイロードは、後期エンドソーム/リソソームコンパートメントにおいて完全な分解を逃れるだけでなく、宿主細胞によっても発現される。
び標的細胞上の表面成分にそれぞれ結合し、後者の成分とのその相互作用がrME経路への小胞の取り込みを導くので、上記のように「二重特異性」であり得る。いずれにしても、本発明によれば、成分との相互作用が標的細胞表面からの細胞質内在化を伴うエンドサイトーシス経路に実質的にアクセスする場合、所与の標的細胞表面レセプターは、リガンドによる結合の候補であり得る。このような候補は候補成分をその表面上に提示する細胞型が候補を結合するリガンドを担持するミニ細胞とインビトロで共インキュベートされ、そしてまた、例えば、共焦点顕微鏡を介して視覚的に、検出に従う蛍光色素または他のマーカーに結合されるアッセイを介して、本発明における適合性について容易に評価される。この種のインビトロアッセイはMacDiarmid ら、2007bにより、図3の436頁の凡例に記載されている)したがって、観察されたマーカーの内部移行は、試験された標的細胞表面受容体が本発明に適しているというようなアッセイによる陽性指標を構成する。
たは多糖であり得る。好ましいリガンドは抗体である。本使用において、用語「抗体」は
免疫原性応答のインビトロまたはインビボ生成によって得られる免疫グロブリン分子を包
含し、従って、「抗体」カテゴリーはモノクローナル抗体およびヒト化抗体(例えば、一
本鎖抗体フラグメント(scFv)、二重特異性抗体など)を包含する。Caravella and
Lugovskoy(Curr. Opin. Chem.Biol., 14:520−28 (2010))の総説(ここではその全体
を参照として組み込む)によって証明されるように、多数の異なる二重特異性タンパク質
および抗体ベースのリガンドが知られている。本開示に従って有用な抗体は、公知の組換
えDNA技術によって得ることができる。
体を使用して、治療される腫瘍中の細胞にミニ細胞を標的化することができる。この点に
おける例示的な細胞表面受容体にはRTKである上皮成長因子受容体(EGFR)、血管
内皮成長因子受容体(VEGFR)、血小板由来成長因子受容体(PDGFR)およびイ
ンスリン様成長因子受容体(IGFR)のいずれかが含まれ、これらのそれぞれは脳腫瘍
を含むいくつかの固形腫瘍において高度に発現され、葉酸受容体はいくつかの下垂体腺腫
において過剰発現される。このような二重特異性リガンドは突然変異受容体または改変受
容体(例えば、ヒト神経膠芽腫多形腫瘍の50%〜80%において発現されるIL13R
α2受容体)に標的化され得る(Wykoskyら、2008; Jarboeら、2007;Debinskiら、2000
;およびOkadaら、1994を参照のこと)が、正常組織において発現されるその生理学的対
応物IL4R/IL13Rとは異なる。Hershey、2003を参照されたい。したがって、I
L13Rα2は正常な脳細胞には実質的に存在しない。Debinski and Gibo、2000を参照
。さらに、脳に転移する腫瘍はある種の受容体を過剰発現する可能性があり、これもまた
適切な標的となり得る。例えば、Da Silvaら、2010は、乳がんの脳転移がRTKのHERファミリーの全てのメンバーを発現することを示した。脳転移巣の20%でHER2が増幅・過剰発現、脳転移巣の21%でEGFRが過剰発現、脳転移巣の60%でHER3が過剰発現、脳転移巣の22%でHER4が過剰発現していた。興味深いことに、HER3発現は脳に存在する乳がん細胞で増加していた。
のメンバーである。RKTは膜貫通タンパク質のファミリーであり、他の膜内在性タンパ
ク質と同様の速度で構成的内部移行(エンドサイトーシス)を行う。Goh and Sorkin、20
13を参照のこと。RKTのファミリーは、Lemmon and Schlessingerが、Cell, 141(7)
:1117−134 (2010)に記載さしている。例示的なRTKは、ErbB EGFR、 Er
bB2、ErbB3、ErbB4 Ins InsR、IGF1R、InsRR PDGF PDGFRα、PDGFRβ、CSF1R/Fms、Kit/SCFR、Fit3/Flk2 VEGF VEGFR1/Fit1、VEGFR2/KDR、VEGFR3/Fit4 FGF FGFR1、FGFR2、FGFR3、FGFR4 PTK7 PTK7/Cck4 Trk TrkA、TrkB、TrkC Ror Ror1、Ror2 MuSK MET、Ron AXL、Mer、Tyro3 Tie Tie1、Tie2 Eph EphA1‐8、EPHA10、EphB1‐4、EphB6 Ret Ryk DDR DDR1、 DDR2 Ros LMR LMR1、LMR2、LMR3 ALK、LTK STYK1 SuRTK106/STYK1である。
(葉酸受容体)のファミリーであり、これは葉酸および還元型葉酸誘導体と結合し、テト
ラヒドロ葉酸の細胞内部への送達を媒介する;IL13などの同族サイトカインの内部移
行に役割を果たす膜結合サイトカイン受容体のファミリー;特定のがん細胞上に発現し、
同族モノクローナル抗体、例えばCD20の例でリツキシマブの内部移行を媒介するCD
20、CD33、メソテリンおよびHM1.24などの表面抗原;およびエンドソーム経
路を通って輸送され、がん細胞接着の主要橋渡し役である接着受容体ファミリー(インテ
グリン)である。本発明の1つの実施形態において、腫瘍細胞表面受容体は、インテグリ
ン、ニューロメジンB受容体、ボンベシン3受容体、GRP受容体、ボンベシン4受容体
、CCK2/ガストリン、メラノコルチン−1受容体(MC−1r)、ニューロペプチド
Y(NPY)受容体、ニューロテンシン(NT)受容体、前立腺特異的膜抗原(PSMA
)、ソマトスタチン(SST)受容体、ニューロキニン1受容体(NK1R)、ケモカイ
ン受容体型4(CXCR4)、血管作動性腸ペプチド(VIP)、上皮成長因子受容体(
EGFR)、血管内皮成長因子受容体(VEGFR)、血小板由来成長因子受容体(PD
GFR)、インスリンのような成長因子受容体(IGFR)およびこれらの組み合わせがあ
る。
独特に発現されるが、発現されないままであるか、低いレベルで発現されるか、または健
康な状態において接近非ないかのいずれかである抗原である。本発明の標的化リガンドに
よって特異的に結合され得るそのような標的抗原の例は、有利にはEpCAM、CCR5
、CD19、HER−2neu、HER−3、HER−4、EGFR、PSMA、CEA、MUC−1(ムチン)、MUC2、MUC3、MUC4、MUC5、MUC5、MUC7、BhcG、Lewis−Yから選択され得る。CD20、CD33、CD30、ガングリオシドGD3、9-O-アセチルGD3、GM1、フコシルSA、GD2、カルボアンヒドラーゼIX(MN/CA IX)、CD44v6、ソニックヘッジホッグ(Shh)、Wue-1、形質細胞抗原、(膜結合)IgE、コンドロイチン硫酸プロテオグリカン(MCSP)、CCR8、TNF-α前駆体、STEAP、前立腺幹細胞抗原(PSCA)、Ly-6、デスモグレイン4、E−カドヘリンネオエピトープ、胎児アセチルコリン受容体、CA19-9マーカーおよびミュラー管阻害物質(MIS)受容体タイプIIsTn(シアリル化Tn抗原; TAG-72)、FAP(線維芽細胞活性化抗原)、エンドシアリン、EGFRVIIILG、SASおよびCD63。
本発明はその範囲内に、(1)抗腫瘍剤、(2)I型IFNアゴニスト、および/または
(3)II型IFNアゴニストの1つ以上の組み合わせをペイロードとして有するミニ細胞
を含む組成物または製剤を含む。3つの成分すべてを含む組成物において、抗腫瘍剤、I
型IFNアゴニスト、およびII型IFNアゴニストは、1つ以上のミニ細胞に含まれ得る
。例えば:(a)抗腫瘍剤、I型IFNアゴニスト、およびII型IFNアゴニストは同じミニ
細胞内に含まれ得る;(b)抗腫瘍剤およびI型IFNアゴニストが第1のミニ細胞内に含ま
れ得、そしてII型IFNアゴニストは第2のミニ細胞内に含まれ得る;(c)抗腫瘍剤および
II型IFNアゴニストが第1のミニ細胞内に含まれ得、そしてI型IFNアゴニストは第
2のミニ細胞内に含まれ得る;または(d)抗腫瘍剤が第1のミニ細胞内に含まれ得、そ
してII型IFNアゴニストは第2のミニ細胞内に含まれ得、そしてII型IFNアゴニスト
は第2のミニ細胞内に含まれ得る;または(e)抗腫瘍剤が第1のミニ細胞内に含まれ得
、I型IFNアゴニストは第2のミニ細胞内に含まれ得、そしてII型IFNアゴニストは
第3のミニ細胞内に含まれ得る。
FNアゴニストの組み合わせをペイロードとして有するミニ細胞を含む組成物または製剤
を含む。いくつかの実施形態では、抗腫瘍剤およびI型IFNアゴニストまたはINFIIアゴニストが1つ以上のミニ細胞に含まれ得る。例えば、(a)抗腫瘍剤およびI型IFNアゴニストは同じミニ細胞内に含まれ得る;(b)抗腫瘍剤が第1のミニ細胞内に含まれ得、I型IFNアゴニストは第2のミニ細胞内に含まれ得る;(c)抗腫瘍剤およびII型IFNアゴニストが同じミニ細胞内に含まれ得る;または(d)抗腫瘍剤が第1のミニ細胞内に含まれ得、II型IFNアゴニストは第2のミニ細胞内に含まれ得る。
インターフェロンI型アゴニスト60mer二本鎖DNA、および/またはインターフェロンII型アゴニストα−ガラクトシルセラミドを含み、ここで、siPlk1、60mer二本鎖DNA、およびα−ガラクトシルセラミドは、1つ以上のミニ細胞内に含まれる。
フェロンI型アゴニスト60mer二本鎖DNA、および/またはインターフェロンII型アゴニストα−ガラクトシルセラミドを含み、siRRM1、60mer二本鎖DNA、およびα−ガラクトシルセラミドは1つ以上のミニ細胞内に含まれる。
インターフェロンI型アゴニスト60mer二本鎖DNA、および/またはインターフェロンII型アゴニストαガラクトシルセラミドを含み、PNU159682、60mer二本鎖DNA、および/またはαガラクトシルセラミドは1つ以上のミニ細胞内に含まれる。
。ミニ細胞およびリガンドは、本明細書に記載されるものいずれであってもよい。したが
って、本発明の二重特異性リガンドは、ミニ細胞の表面成分および標的哺乳動物細胞の表
面成分に結合することができる。
、組成物の薬物または薬物送達品質を過度に妨害しない他の成分を有するそのようなミニ
細胞、薬物、およびリガンドを含む製剤)は、1つまたは複数の薬学的に許容される担体
または賦形剤を使用して、従来の様式で製剤化することができる。
たは複数用量容器で、添加された防腐剤の有無にかかわらず、提示することができる。処
方物は油性または水性ビヒクル中の溶液、懸濁液、またはエマルジョンであり得、そして
処方剤(例えば、懸濁剤、安定剤および/または分散剤)を含み得る。適切な溶液はレシ
ピエントの血液と等張であり、生理食塩水、リンゲル液、およびデキストロース溶液によ
って例示される。あるいは、製剤が適切なビヒクル、例えば、無菌の発熱物質を含まない
水または生理食塩水で再構成するために、凍結乾燥粉末形態であり得る。製剤はまた、デ
ポー製剤の形態であってもよい。このような長時間作用性処方物は移植(例えば、皮下ま
たは筋肉内)によって、または筋肉内注射によって投与され得る。いくつかの実施形態で
は、投与することは経腸または非経口投与を含む。いくつかの実施形態では、投与するこ
とは経口、頬側、舌下、鼻腔内、直腸、膣、静脈内、筋肉内、および皮下注射から選択さ
れる投与を含む。
抗腫瘍剤の「治療有効」量は、本開示に従って、対象に投与された場合に薬理学的応答を
引き起こす、問題の薬剤、例えばsiRNAまたは超細胞傷害性薬剤の投薬量である。
イロードを保有するミニ細胞が投与される場合、動物モデルまたはヒト被験体のいずれか
において、腫瘍または腫瘍の症状の予防または改善を参照することによって測定され得る
。所定の例(例えば、特定の被験体)における「治療有効量」を証明する量は、そのよう
な投薬量が熟練した実施者によって「治療有効量」と見なされるとしても、腫瘍について
同様に処置される被験体の100%に対して有効ではないかもしれない。この点に関する
適切な用量はまた、例えば、腫瘍の型、段階、および重症度の関数として変化する。
その数は、どの抗腫瘍剤がミニ細胞中にパッケージ化されるか、および腫瘍を処置する際
のその薬剤の効力に基づいて確認され得る。この点において、治療効果は、腫瘍塊などの
臨床的または病理学的パラメーターを用いて測定することができる。従って、腫瘍質量の
減少または減少した増加は、治療効果を測定するために使用され得る。
本発明の製剤は局所的または全身的のいずれかで、所望の治療効果を達成するために、
種々の経路を介して、および哺乳動物体内の種々の部位に投与され得る。送達は例えば、
経口投与によって、体腔への製剤の適用によって、吸入または通気によって、または非経
口、筋肉内、静脈内、門脈内、肝臓内、腹腔内、皮下、腫瘍内、または皮内投与によって
達成され得る。投与の様式および部位は、標的細胞の位置に依存する。例えば、腫瘍転移
は、標的ミニ細胞の静脈内送達を介してより効率的に処置され得る。原発性卵巣がんは、
標的ミニ細胞の腹腔内送達を介して治療され得る。経路の組み合わせもまた、使用され得
る。例えば、転移性膀胱がんにおいて、細胞傷害性薬物負荷および受容標的化ミニ細胞は
膀胱内および静脈内に投与され得、免疫賦活剤パッケージ化(受容体標的化または非標的
化)ミニ細胞は標的化薬物パッケージ化ミニ細胞と共に静脈内に投与され得る。標的化さ
れた、薬物パッケージ化ミニ細胞のin situ投与は膀胱表面露出腫瘍を標的とし得るが、
静脈内投与されたミニ細胞の完全な組合せは組織局在腫瘍を標的とし得、また抗腫瘍免疫
応答を誘発し得る。
本発明のミニ細胞は、汚染親細菌細胞を実質的に含まない。したがって、ミニ細胞含有
製剤は、好ましくは107ミニ細胞当たり約1未満の汚染親細菌細胞、108ミニ細胞当た
り約1未満の汚染親細菌細胞、109ミニ細胞当たり約1未満の汚染親細菌細胞、1010
ミニ細胞当たり約1未満の汚染親細菌細胞、または1011ミニ細胞当たり約1未満の汚染
親細菌細胞を含む。
る。1つのこのような方法は、クロスフロー濾過(供給流量は膜表面に平行である; Forb
es,1987)およびデッドエンド濾過(供給流量は膜表面に垂直である)を組み合わせる。
必要に応じて、濾過の組み合わせの前に、細菌細胞のいくらかの部分を除去し、それによ
ってミニ細胞について上清を濃縮するために、低遠心力での差動遠心分離を行うことがで
きる。
、ミニ細胞バンドを勾配から収集し、任意に、ミニ細胞をさらなるラウンドの密度勾配遠
心分離に供して、純度を最大にする。本方法は、ミニ細胞含有試料に対して分画遠心分離
を実施する予備工程をさらに含むことができる。低遠心力で実施される場合、示差遠心分
離は親細菌細胞のいくらかの部分を除去し、それによって、ミニ細胞について上清を濃縮
する。
る。従って、ミニ細胞精製方法は、(a)ミニ細胞を含有する試料を、親細菌細胞が糸状
形態をとるように誘導する条件に供する工程、続いて(b)試料を濾過して精製ミニ細胞
調製物を得る工程を包含し得る。
み合わせは、以下の通りである:
工程A:ミニ細胞産生細菌細胞培養物の分化遠心分離。この工程は2,000gで約20分間行うことができ、上清中にミニ細胞を残しながら、ほとんどの親細菌細胞を除去する;
工程B:等張および非毒性密度勾配媒体を使用する密度勾配遠心分離。この工程はミニ細胞の損失を最小限に抑えながら、親細菌細胞を含む多くの汚染物質からミニ細胞を分離する。好ましくは、この工程は精製方法内で繰り返される;
工程C:親細菌細胞汚染をさらに減少させるために、0.45μmフィルターを通したクロスフロー濾過;
工程D:残存親細菌細胞のストレス誘導性フィラメント形成。これは、ミニ細胞懸濁液をいくつかのストレス誘導環境条件のいずれかに曝すことによって達成され得る;
工程E:親細菌細胞を死滅させるための抗生物質処理;
工程F:膜ブレブ、膜断片、細菌残屑、核酸、培地成分などの小さな汚染物質を除去し、ミニ細胞を濃縮するためのクロスフロー濾過。ミニ細胞を小さな汚染物質から分離するために、0.2μm濾過を使用することができ、ミニ細胞を濃縮するために、0.1μm濾過を使用することができる;
工程G:糸状の死んだ細菌細胞を除去するためのデッドエンド濾過。この
工程のために0.45μm濾過を使用することができる;
工程H:ミニ細胞調製物からのエンドトキシンの除去。抗脂質A被覆磁性ビーズをこの工程に用いることができる。
な生理学的効果を得るために、日常的なテストによって規定される適切な用量で使用され
得る。投与法は、患者の年齢、体重、性別、医学的状態;治療される状態の重症度、投与
経路、および患者の腎機能および肝機能を含む様々な因子に応じて選択され得る。
最適な精度は、ミニ細胞の動力学および標的部位および標的細胞に対する薬物の利用可能
性に基づくレジメンを必要とし得る。治療レジメンの至適濃度を決定する際には、ミニ細
胞または薬物の分布、平衡、および排泄を考慮してもよい。ミニ細胞および薬物の用量は
、組み合わせて使用される場合、所望の効果を達成するために調節され得る。
化することができる。例えば、1つ以上の投薬レジメンが選択され得、そして薬物動態学
的/薬力学的モデルが、1つ以上の投薬レジメンの薬物動態学的/薬力学的プロフィール
を決定するために使用され得る。次に、特定の薬物動態学的/薬力学的プロフィールに基
づいて所望の薬物動態学的/薬力学的応答を達成する投与のための投薬レジメンの1つが
選択され得る。例えば、WO 00/67776を参照のこと。
一実施形態では、製剤が数週間〜数ヶ月にわたって週に少なくとも1回投与される。
約8、約9、約10、約11、約12、約13、約14、約15、約16、約17、約1
8、約19、約20、約21、約22、約23、約24、約25、約26、約27、約2
8、約29、約30、または約31日間投与することができる。あるいは、製剤が約一日
に約1回、約2、約3、約4、約5、約6、約7、約8、約9、約10、約11、約12
、約13、約14、約15、約16、約17、約18、約19、約20、約21、約22
、約23、約24、約25、約26、約27、約28、約29、約30または約31日ま
たはそれ以上毎に投与され得る。
9、約10、約11、約12、約13、約14、約15、約16、約17、約18、約1
9または約20週間またはそれ以上毎に約1回投与されてもよい。あるいは、製剤が約2
、約3、約4、約5、約6、約7、約8、約9、約10、約11、約12、約13、約1
4、約15、約16、約17、約18、約19または約20週間以上、少なくとも週1回
投与され得る。
約10、約11、約12、約13、約14、約15、約16、約17、約18、約19ま
たは約20週間またはそれ以上毎約2回投与され得る。あるいは、製剤が約2、約3、約
4、約5、約6、約7、約8、約9、約10、約11、約12、約13、約14、約15
、約16、約17、約18、約19または約20週間以上、少なくとも週1回投与され得
る。
、約10、約11または約12カ月またはそれ以上ごとに約1回投与することができる。
与する。
間後の任意の時点で起こり得る。あるいは、薬物が数時間〜数日、おそらく数週間〜数ヶ
月後の任意の時点で、ミニ細胞の後に投与され得る。
約7、約8、約9、約10、約11、約12、約13、約14、約15、約16、約17
、約18、約19、約20、約21、約22、約23または約24時間前に投与され得る
。さらに、ミニ細胞は、薬物の投与の少なくとも約1、約2、約3、約4、約5、約6、
約7、約8、約9、約10、約11、約12、約13、約14、約15、約16、約17
、約18、約19、約20、約21、約22、約23、約24、約25、約26、約27
、約28、約29、約30または約31日前に投与され得る。さらに別の実施形態におい
て、ミニ細胞は、薬物の少なくとも約1、約2、約3、約4、約5、約6、約7、約8、
約9、約10、約11、約12、約13、約14、約15、約16、約17、約18、約
19または約20週間以上前に投与され得る。さらなる実施形態において、ミニ細胞は、
薬物の少なくとも約1、約2、約3、約4、約5、約6、約7、約8、約9、約10、約
11または約12ヶ月前に投与され得る。
の投与後、数分から数時間の任意の時点で起こり得る。あるいは、ミニ細胞が薬物の数時
間〜数日、おそらく数週間〜数ヶ月後の任意の時点で投与され得る。
本明細書中に記載される組成物は、癌に罹患している対象を処置するために使用され得
る。本明細書に開示される方法は、少なくとも1つの抗腫瘍剤、インターフェロンI型ア
ゴニスト、インターフェロンII型アゴニスト、またはインターフェロンI型アゴニストと
インターフェロンII型アゴニストとの組み合わせを含む、本発明による組成物の有効量を
対象に投与することを含む。抗新生物剤、インターフェロンI型アゴニスト、インターフ
ェロンII型アゴニスト、またはインターフェロンI型アゴニストとインターフェロンII型
アゴニストの組合せは、1つ以上のミニ細胞で構成される。
学的に受容可能なキャリアをさらに含む。
ために有用であり、ここで、被験体は、ヒト、非ヒト霊長類、イヌ、ネコ、ウシ、ヒツジ
、ウマ、ウサギ、マウス、またはラットである。
る。いくつかの実施形態では癌は肺癌、乳癌、脳癌、肝臓癌、結腸癌、膵臓癌、または膀
胱癌を含む。
、副腎皮質がん、 エイズ関連のガン、エイズ関連リンパ腫、肛門がん、虫垂ガン;星細胞
腫、非定型奇形腫様/ラブドイド腫瘍、基底細胞癌、膀胱がん、脳幹神経膠腫、脳腫瘍(
脳幹グリオーマ、中枢神経系非定型奇形腫様/ラブドイド腫瘍、中枢神経系胚芽腫、星細
胞腫、頭蓋咽頭腫、上衣芽腫、上衣腫、髄芽腫、髄上皮腫、松果体実質腫瘍、テント上の
原始神経外胚葉性腫瘍および松果体芽腫、乳癌、気管支腫瘍、バーキットリンパ腫、原発
部位不明の癌、カルチノイド腫瘍;未知の主要なサイトのがん、中枢神経系非定型奇形腫
/ラブドイド腫瘍、子宮頸癌、小児がん、脊索腫、慢性リンパ性白血病、慢性骨髄性白血
病、慢性脊髄増殖性障害、大腸がん、結腸癌、頭蓋咽頭腫、皮膚T細胞リンパ腫、内分泌
膵島細胞腫瘍、子宮内膜癌、上衣芽腫、上衣腫、食道癌、感覚神経芽腫、ユーイング肉腫
;頭蓋外胚、性腺外生殖細胞腫瘍、 肝外胚細胞腫瘍、胃(胃)がん、消化管カルチノイド
腫瘍、消化管間質腫瘍、グリオーマ、有毛細胞白血病、頭頸部がん、ホジキンリンパ腫、
下咽頭がん、眼内黒色腫、小島細胞腫瘍;カポジ肉腫、腎臓がん、ランゲルハンス細胞組
織球症、喉頭ガン;唇のガン、肝臓がん、悪性線維性組織球腫骨がん、髄芽腫、髄上皮腫;
黒色腫、メルケル細胞がん、メルケル細胞がん、中皮腫、原発不明の転移性扁平上皮性頸
部がん、口ガン、多発性内分泌腫瘍症候群、多発性骨髄腫/形質細胞腫瘍、菌状息肉腫、
骨髄異形成症候群、脊髄増殖性新生物;、鼻腔がん、鼻咽頭ガン、神経芽細胞腫、非ホジ
キンリンパ腫、非黒色腫皮膚がん、非小細胞肺がん、口腔がん、中咽頭がん、骨肉腫、そ
の他の脳腫瘍および脊髄腫瘍、卵巣がん、卵巣上皮がん、卵巣胚細胞 卵巣低悪性度腫瘍
、膵臓癌、乳頭腫症;副鼻腔ガン;副甲状腺癌、骨盤部癌、陰茎がん、咽頭がん、松果体腫
瘍、松果体芽腫、下垂体性腫瘍、形質細胞新生物/多発性骨髄腫、胸膜肺芽腫、一次性中
枢神経系(CNS)リンパ腫、原発性肝細胞癌、前立腺癌、直腸ガン、腎細胞(腎臓)癌
、腎細胞癌、気道癌、横紋筋肉腫、唾液腺癌、セザリー症候群、小さな細胞肺がん、小腸
癌、軟部組織肉腫、扁平上皮細胞癌、扁平上皮首ガン、胃(胃)癌、テント上原始神経外
胚葉性腫瘍、T細胞癌、精巣癌、咽頭癌、胸腺癌、胸腺腫、甲状腺癌、移行性細胞ガン、
腎盂と尿管の移行上皮がん、栄養芽層の腫瘍、尿道がん、子宮がん、子宮肉腫、膣がん、
外陰がん、ワルデンシュトレームマクログロブリン血症、またはウィルムス腫瘍が含まれ
る。
型奇形腫様/ラブドイド腫瘍、中枢神経系胚芽腫、星細胞腫、頭蓋咽頭腫、上衣芽腫、上
衣腫、髄芽腫、髄上皮腫、中間的分化の松果体実質腫瘍、テント上未分化原始神経外胚葉
性腫瘍および松果体芽腫からなる群より選択される。
本明細書で使用される技術用語および科学用語は別段の定義がない限り、本発明が関係
する当業者によって一般に理解される意味を有する。以下の説明および実施例において参
照される材料、試薬などは、特に断らない限り、商業的供給源から入手可能である。
び語句の意味は、以下に提供される。他の用語および語句は、本明細書全体を通して定義
される。
数の引用を含む。
範囲から逸脱せずに、列挙された数の実質的に周囲の数を指すことが意図されることを意
味する。本明細書で使用される場合、「約」は当業者によって理解され、それが使用され
る文脈である程度変化するのであろう。それが使用される文脈で与えられる当業者に明ら
かでない用語の使用がある場合、「約」は特定の用語の±10%までを意味するであろう
。
断、処置または治療が所望される任意の哺乳動物対象をいう。1つの好ましい実施形態に
おいて、個体、対象、宿主、または患者はヒトである。他の対象としてはウシ、ウマ、イ
ヌ、ネコ、モルモット、ウサギ、ラット、霊長類、およびマウスが挙げられるが、これら
に限定されない。
「癌腫」は、細胞増殖の制御の有意な喪失によって特徴付けられる異常な成長表現型を示
す細胞または組織を指す。がんには主にいくつかの種類がある。がんとは、皮膚内または
内臓を覆っている組織から発生するがんのことである。肉腫は、骨、軟骨、脂肪、筋肉、
血管、その他の結合組織や支持組織から発生するがんである。白血病は、骨髄などの造血
組織から発生するがんで、異常な血液細胞が大量に産生されて血液中に入るようになる。
リンパ腫と多発性骨髄腫は、免疫系の細胞から発生するがんである。中枢神経系がんは、
脳と脊髄の組織から発生するがんである。本発明の方法および組成物は特に、前癌性、悪
性、前転移性、転移性、および非転移性細胞に適用される。
および/または生理学的効果を得ることを指す。この効果は、腫瘍またはその症状を完全
にまたは部分的に予防するという点で予防的であり得、および/または腫瘍および/また
は腫瘍に起因する有害な効果について部分的または完全な安定化または治癒という点で治
療的であり得る。治療は、哺乳動物、特にヒトにおける腫瘍の任意の治療を包含する。所
望の効果は特に、腫瘍塊の減少または腫瘍塊増加の阻害として測定され得る腫瘍応答であ
る。腫瘍応答に加えて、全生存の増加、無増悪生存、または腫瘍再発までの時間、または
有害作用の減少もまた、所望の処置効果として臨床的に使用され得る。
己投与すること、および本明細書中に開示されるような薬剤の投与を処方または指示する
ことを含む。
のような処置を必要とする被験体において、活性剤が投与される特定の薬理学的効果を提
供する、被験体における活性剤投薬量または血漿濃度を意味する。有効量の活性剤は、た
とえそのような投薬量が当業者によって有効量であると見なされるとしても、本明細書中
に記載される状態/疾患を処置する際に常に有効であるとは限らないことが強調される。
性核酸をコードする任意の小分子薬物、タンパク質、機能性核酸、または多核酸である。
活性剤は、本明細書に記載される抗腫瘍薬、機能性酸、インターフェロンI型アゴニスト
またはII型アゴニストのいずれかであり得る。
見合った、過度の毒性、刺激、アレルギー反応、または他の問題または合併症を伴わずに
インビボで使用するのに適した、健全な医学的判断の範囲内にある化合物、材料、組成物
、および/または剤形を指す。
(2a)受容体結合を必要としないマクロピノサイトーシス、ならびに(2b)クラスリン介
在性エンドサイトーシス、(2c)カベオラ介在性エンドサイトーシスおよび(2d)クラス
リン/カベオラ非依存性エンドサイトーシスを含むカテゴリーを包含し、これらは全て後
期エンドソーム/リソソーム経路にアクセスする傾向がある。本発明者らはミニ細胞上の
リガンドと哺乳類細胞表面受容体との相互作用により、特定のエンドサイトーシス経路が
活性化され、後期エンドソーム/リソソームコンパートメントへの受容体媒介エンドサイ
トーシス(rME)が関与することを発見した。このようなエンドサイトーシス経路のお
かげで、本発明者らはさらに、ミニ細胞が標的哺乳動物細胞の細胞質中にそれらのペイロ
ードを放出することができることを発見した。ペイロードがコード核酸である場合、核酸
は、後期エンドソーム/リソソーム区画において完全に分解されるだけでなく、標的哺乳
動物細胞においても発現される。
理解を提供するものである。実施例は、薬剤耐性腫瘍細胞が(1)薬剤耐性をコードする
遺伝子の発現を減少または排除するように設計されたRNAi配列を担持する標的組換え
ミニ細胞の投与、および(2)癌細胞が感受性にされる薬剤を担持する標的化された、薬
剤パッケージ化ミニ細胞の投与によって、インビボで効果的に治療され得ることを実証す
る。
施例に記載された特定の条件または詳細に限定されるべきではないことを理解されたい。
明細書全体を通して、米国特許を含む、公に利用可能な文書へのあらゆる参照は、参照に
より特に援用される。
この実施例はミニ細胞(EDV)が化学療法薬の効率的な送達を提供し、マウス異種移
植モデルにおいて腫瘍増殖を阻害することを示した。EDV標的化技術は、結腸癌、乳癌
、卵巣癌、白血病、肺癌、中皮腫、および子宮癌を含む様々な癌のマウス異種移植モデル
で試験されている。さらに、EDVがEGFR、ヒト上皮成長因子受容体2(HER2)
、メソテリン(MSLN)、およびCD33を含む腫瘍細胞表面受容体に標的化されたの
で、種々の標的化部分が利用された。最後に、試験した標的EDVは、ドキソルビシン、
パクリタキセル、モナストロール、イリノテカン、PNU159682のような超細胞傷
害性薬剤、および新規チミジル酸シンターゼ阻害剤(OSI)を含む多種多様な細胞傷害
性薬剤を含んでいた。PNU−159682は、ドキソルビシンよりも数千時間細胞傷害
性であるアントラサイクリン類似体である。OSI7904は抗腫瘍活性を有するベンゾ
キナゾリン葉酸類似体である。チミジル酸シンターゼ阻害薬として、OSI−7904は
チミジル酸シンターゼに非競合的に結合し、その結果、チミンヌクレオチド合成およびD
NA複製が阻害される。
薬物パッケージされたEDVを使用した場合、大きな(>1000 mm3)腫瘍についても
観察された(表5を参照のこと)。遊離薬物(EDVに詰め込まれていない)で処理した
対照マウスは静脈炎の予想毒性を示し、最終的に体重が減少し、死亡した。これらの毒性
はEDV包装薬剤の反復投与では認められなかった。
000倍も低かったが、EDVによって送達された細胞傷害性薬物濃度は抗腫瘍効力を最
大にするのに十分であった。
この実施例は、薬物を負荷したミニ細胞(EDV)がイヌに安全に投与され得ることを
示した。
および薬剤包装EDVを最大98回まで、2年以上の経過で1匹の動物に安全に投与でき
ることが示された。一部のイヌでは投与後にインターロイキン−6(IL−6)、インタ
ーロイキン−10(IL-10)および腫瘍壊死因子α(TNF−α)の上昇を伴う軽度の温度上昇(最大1の上昇)が認められたが、重大な有害事象との関連は認められなかった。
が腫瘍増殖を阻害できることが示された。進行した非ホジキンリンパ腫の2匹のイヌを、
CD3標的、ドキソルビシンパッケージ化EDVで処理し、両者はリンパ節サイズの非常
に有意な減少により明らかなように、顕著な腫瘍退縮を示した。MacDiarmid ら、2007b。血管肉腫のイヌの60%以上が、CD33標的EDVおよびドキソルビシンパッケージ
化EDVで治療した場合、腫瘍の安定化または退縮を示した。
EGFR標的EDVで動物を治療した。MacDiarmid ら、2016。1匹のイヌ(20回を超
える用量を投与された11匹のイヌ)に98回までの反復用量を1×1010 の EGFR ミニ細胞Doxで投与したが、毒性の徴候は観察されなかった。客観的奏効率は23.53%(イヌ17匹中4匹;95%信頼区間、6.8−49.8%)であった。腫瘍反応を評価した15匹のイヌのうち、2匹は治療に対して完全反応(CR)を示し、2匹は治療に対して部分反応(PR)を示し(腫瘍容積の90〜98.95%の減少)、10匹は安定疾患(SD)、1匹は進行性疾患(PD)を示した。
布試験では、標的EDVが脳腫瘍に局在することが示され、EDVが血液脳関門を回避し
て腫瘍周辺に入ることができることが示唆された。消化管に局在することから、糞便を介
した排泄が示唆される。
この実施例は、ミニ細胞(EDV)技術がサルによって十分に許容されることを示した
。3件のアカゲザル試験を実施し、空のEDV(1用量あたり2×1010まで)、EGFR標的ドキソルビシン負荷EDV(1用量あたり2×1010まで)、およびEGFR標的パクリタキセル負荷EDV(1用量あたり1×1011まで)の毒性を評価した。サルにEDVを週1回、5週間投与した(35日間反復投与試験)。
後のIL−6の上昇が認められた。炎症マーカーC反応性蛋白もこれらの時期に増加した
が、有意な毒性や有害事象は観察されなかった。EGFRを標的としたドキソルビシンを
負荷したEDVのみによる治療経過中に、TNF−αの軽度の上昇が認められた。合計7
2匹のサルにEDV技術を安全に投与した。
この実施例から、前臨床マウス試験(実施例1)、前臨床イヌ試験(実施例2)および
前臨床サル試験(実施例3)では、わずかな炎症反応しか観察されなかったことが示され
た。これらの反応は、投与4時間後と同様に速やかに消失した。その他、血液学的及び生
化学的パラメータに有意な変化は認められなかった。投与期間中、外観および行動は健康
なままであった。
考慮された免疫応答は以下のとおりであった:
・ EDV表面露出免疫優性抗原に対する血清抗体反応は、LPSのO−多糖成分(IgGまたはIgM反応)である。抗O-多糖抗体応答はT細胞非依存性であり、記憶応答を示さない。
・ 腫瘍細胞表面受容体(例えば、EGFR)にEDVを標的化するためのBsAbの構
築に使用されるマウスIgGモノクローナル抗体に対する血清抗体応答。
びドキソルビシンパッケージ化EDVの投与3−4までに平均約10,000まで上昇し
た。その後の投与では、それ以上の力価の上昇はみられなかった。サルを用いた3試験(
健常動物)における抗LPS IgG力価は、プラトーに達する前の最初の2〜3回の投
与で概して軽度の上昇を示した。グラム陰性菌に対するワクチンで予想される抗O−多糖抗体力価は一般に数百万であるため、反応は主に用量依存的であり、最高用量レベルのEGFREDVDox では100を丁度超える最高力価まで上昇した。
でも測定されており、EGFR標的EDV(マウスIgG)を投与したサルでは、EGF
R抗体に応答して軽度の力価上昇が観察された。これらの結果から、BsAbを標的とし
た薬物パッケージ化EDVの投与は、その後の投与の有効性を妨げうる有意な抗LPS免
疫応答を誘発しない可能性があることが示唆される。これは、反復投与が実行可能な治療
選択肢である可能性が高いことを示唆しているため、免疫系が損なわれている可能性が高
い癌患者に特に関連がある。
(EGFR(Erb)EDVPac)を評価する第1相臨床試験
本実施例は、進行固形腫瘍にパクリタキセル(Taxol(登録商標))を送達するためにミニ細胞(EDV)を使用する有望な結果を示した。
化EDVのヒト患者における忍容性は良好であることが示されたが、治験において有害事
象または用量制限毒性が認められたため、かなりの数の患者が試験を終了しなければなら
なかった。さらに、この治療戦略は疾患の安定化を達成したが、この研究の患者のいずれ
も治療に対して部分的または完全奏効を示さなかった。この試験データの結果はSolomon
ら、2015年に公表されている。
アニュージーランド臨床試験登録(番号ACTRN12609000672257)に登録され、最終的な試験
報告書が入手可能であり、本試験は2015年のSolomonらに発表されている。
であった。
1010、1.5×1010、2×1010、および5×1010 のEGFR(Erb)ターゲットEDVの7段階で計236回の投与が提供された。28例中22例が少なくとも1サイクルの治療(週5回投与)を完了し、1例は9サイクル(約14カ月)にわたり45回の投与を受けた。投与に関連した死亡は認められなかった。最高耐量は1×1010 EGFR(Erb)EDVPacと同定され、特に長時間の発熱および肝機能検査(LFT)の一過性上昇の形態で、この用量以上で有意な毒性が観察された。本治療の忍容性は概して良好であり、示さた集団において許容可能な安全性所見が認められた。
発熱)および悪寒(悪寒)であり、患者の最大60%で認められた(グレード1〜2の重
症度)。ほとんどの患者で、投与4時間後にサイトカインIL-6、IL-8およびIL−
10の軽度の一過性の上昇が認められた。投与後24時間以内に投与前のレベルに回復し
た。これは、治療に対する軽微な炎症反応と一致する。
を必要とする有害事象が認められた。これらの事象を表7に要約し、以下の説明に記載す
る。
の忍容性は良好であり、意図した対象集団に対する安全性に特段の懸念はない。
は、全例で陰性であった。1例を除くすべての患者は、EGFR(Erb)EDVPacによる治療後に陽性のサルモネラ菌抗体価を発現した(27/28=96%)。抗LPS抗体価は投与3(Day 15)までにピークに達し、反復投与にもかかわらずそのレベルに維持された。アービタックス抗体価陽性を示した患者はいなかった。
反応は、病勢安定(SD)であった(RECIST規準に従って部分奏効または完全奏効
を達成した患者はいなかった)。第1サイクル終了時には22例中10例(45.5%)
に安定が認められ、22例中12例(55.5%)に病勢進行(PD)が確認された。用
量レベル1の患者1例は9サイクルの完全な治療を完了し、4サイクル目の終了時から疾
患は安定し、進行した状態が交互に認められた。彼女は197日というPDの発症までの
最も長い時間であった。
DVの忍容性は良好であり、患者の45.5%が疾患の安定化を示すことが実証された。
この実施例はまた、生存および疾患応答を改善するために癌治療戦略を改善することが望
ましいことを実証する。
GFR(V)EDVDox)を評価する第1相臨床試験
この実施例は、EGFRを標的としたドキソルビシンパッケージ化ミニ細胞(EDV)
による治療が再発性膠芽腫に罹患した患者において忍容性が良好であったことを示してい
る。患者の50%が疾患の安定化を示したが、部分奏効または完全奏効を経験した患者は
いなかった。Whittle ら、J. Clin. Neurosci., 22(12):1889−1894(2015)。
リア、シドニーおよびメルボルンの4カ所の腫瘍クリニックで実施され、オーストラリア
ニュージーランド臨床試験登録 (番号ACTRN12613000297729)に登録されており、最終臨
床試験報告書が入手可能である。この研究は、Whittleら、JClinのリスト草案に基づいて
発表されている。Neurosci.,22(12):1889−1894(2015)。
ドIVの膠芽腫と確定診断され、標準治療(最大限の安全な外科的切除、標準補助放射線/テモゾロミド、維持テモゾロミド治療を含む)を受けた後に疾患の再発または進行を経験した患者であった。
%までの患者で認められた軽度の発熱(発熱)、悪心および硬直(悪寒)であった(一般
的にグレード1〜2の重症度)。ほとんどの患者で、投与3時間後にサイトカインIL-
6、IL-8、IL-10、TNF−痰フ軽度の一過性の上昇が認められた。投与後24時
間以内に投与前のレベルに回復した。これは、治療に対する軽微な炎症反応と一致する。
上の治療関連有害事象を5例に認めた。これらの事象は、以下の表9に要約され、以下の
説明に記載される。
例はグレード1の発熱を伴うグレード3の体力低下の重篤な有害事象のために、サイクル
1の投与2の後の夕方に入院した。患者は試験登録時に抗産物抗体(EDVのサルモネラ
菌LPS成分に対する抗体)が陽性であったため、これは用量制限毒性とは考えられなか
った。この患者はさらに2回の治療を受けたが、事象の再発は認められなかった。
低血圧の重篤な有害事象が発現し、静脈内水分補給入院となった。これは、患者が治療を
受けたEColi尿路感染症に起因する可能性が高いため、DLTとはみなされなかった
。その後、治験薬を3回追加投与された。
、一部の投与後にグレード 3の肝酵素上昇が認められた。これらは、無症候性で一過性
であり、用量間でベースラインに戻ったため、用量制限毒性または重篤な有害事象とはみ
なされなかった。2例でグレード3の低リン酸血症が認められ、1例ではグレード4のリ
ンパ球減少症が認められた。これらはまた、一過性であり、治療的介入を必要としなかっ
たため、用量制限毒性や重篤な有害事象とも考えられなかった。
の懸念はない。
る抗体を評価した。スクリーニング時に14例中13例(93%)がサルモネラ菌抗体陰
性であった。用量レベル2に割り付けられた1例はスクリーニング時に陽性であった。3
回目の投与までに、全例で最初に抗体価の上昇が認められた。計47回の投与を受けた患
者が1例いたにもかかわらず、その後の投与量を超えて増強することなく力価が維持され
た。ベクチビックスに対する抗体を発現した患者はいなかった。
。完全寛解または部分寛解を経験した患者はいなかったが、50%が疾患の安定を示し、
疾患の進行を経験した患者はいなかった。1例の患者は試験の全期間にわたって疾患が安
定しており、ほぼ6サイクル(約12ヵ月)の治療を受けていたと報告された。
例全例が背景生存期間中央値5〜7カ月を超えて生存し、OS中央値は15.1カ月(範
囲9.1〜>18.4)であった。4人の被験者は最後の接触時に生存しており、この時
点で打ち切られた。少なくとも1サイクルの治療を完了したこれらのうちの2人(14.
3%)は、>18ヶ月間生存した。
事象がほとんどなく、患者の50%が疾患の安定化を経験した有望な結果を示した。しか
しながら、改善された治療戦略が必要とされている。
この実施例では、マイクロRNA-16でパッケージされたEGFR標的EDVが有効
性を検査した16人の患者のうち1人の中皮腫患者で部分奏効を示したのに対し、患者の
62.5%は疾患が安定化し、患者の31.3%は疾患が進行したことが示された。試験
データは、van Zandwijkら、Lancet Oncol., 18(10):1386−1396(2017)およびKaoら、Am.J. Respir. Crit. Care Med.,191(12): 1467-1469 (2015) に公開されている。この治療戦略は概して良好であった。
NA16aを評価した。試験の主要エンドポイントはVEDVmiRNA16aの最高耐量およびDLT
を確立し、反復投与の影響を評価し、VEDVmiRNA16aによる有効性の早期徴候を検出す
ることであった。EDVをEGFRに標的化するために使用した抗体は、現在のプロトコ
ール(ベクチビックスベースの配列)の抗体と同じであったことに留意されたい。試験の
第2のエンドポイントはVEDVmiRNA16aを投与された患者のQOLを評価し、治療中の
東部協力腫瘍学グループ(ECOG)の全身状態および肺機能パラメータの変化をモニタリングすることであった。探索的評価項目は、治療中の免疫マーカーおよびサイトカインマーカーの変化を評価した。
学的または細胞学的記録を受けていなければならない。患者は、ECOGのパーフォーマ
ンスステータスが0または1で、平均余命が3カ月以上の18歳以上の男女を対象とした
。患者は、標準的な1または2番目のライン治療レジメンの投与中または投与後に疾病の
進行を示さなければならず、十分な骨髄、肝臓および腎機能を有することが必要であった
。
され、2016年11月24日に終了した。本試験は、オーストラリアニュージーランド臨床試験
登録(番号ACTRN 12614001248651)および臨床試験gov(番号NCT02369198)に登録された
。合計27人の患者が5つの集団を超えて動員され、26人の患者が合計316用量のV
EDVmiRNA16aを受けた(1人の被験者はいずれかの治療を受ける前に死亡し、さらなる
解析から除外された)。この研究は、Kao ら、2015およびvan Zandwijk ら、2017により
発表されている。
サイトカイン反応の増加を避けるために、用量を1×109からフェーズ1当量用量に漸
増させる用量適応も評価した。デキサメタゾン(dex)適応も評価し、その後の用量の
前投薬のためにdexを徐々に減少させた。MTDは週1回5×109 VEDVmiR16aと
同定された。本治療は概して良好であり、示された集団において許容可能な安全性所見が
認められた。
与を受けた(治療のフルサイクルが5回以上)。観察された最良効果は1例の部分奏効で
あった。この症例は以下に記載するように、VEDVmiR16aによる治療に反応してほぼ完
全な寛解を示した。10人の患者(62.5%)は疾患の安定化を示し、5人の患者(3
1.3%)は疾患の進行を示した。生存期間中央値は36.5週、すなわち8.4か月(
範囲9.3〜>119.6週)であり、9例(60.0%)が6か月間生存し、このうち
5例は治療開始から12か月以上経過した時点で良好に生存していた。図4を参照された
い。
に劇的な臨床応答を示した(Kaoら、Am. J. Respir. Crit. Care Med., 191(12):46
7−1469(2015)を参照のこと)。8週間の期間の終了時、PET-CTスキャンで「完全
である」代謝反応が明らかになり、胸部CTスキャンで部分奏効が認められ、4週間後に
確認された。客観的画像反応は呼吸機能検査パラメータの顕著な改善を伴っていた。
盗汗もこのカテゴリーに含まれる)などの注入に伴う反応(96.2%)であった。これ
らの大部分の重症度は軽度から中等度であった。腫瘍部位の非心臓性胸痛も、注入後に1
4例の患者(53.8%)が経験した。これらの反応は治験実施計画書の改訂により対処
された。治験実施計画書の改訂版では、第1サイクルでは全被験者に用量漸増スケジュー
ルの修正を行ったため、注入に伴う反応が少なく、重症度も低かった。臨床検査では、炎
症性サイトカインおよび好中球の一過性の上昇、ならびに大多数の患者におけるVEDVm
iRNA16a注入直後のリンパ球の一過性の低下が明らかにされ、軽度の炎症反応と一致した
。
もしれないとされた。非心臓性胸痛及び注入に伴う反応は、いずれも2例に発現した。3
例で用量制限毒性が発現し、さらに2例で用量制限毒性と考えられる毒性が発現したが、
用量制限毒性の枠外で発現したため、用量制限毒性の基準には適合しなかった。投与に関
連した死亡は認められなかった。これらの事象は、以下の表11に要約され、以下の説明
に記載される。
の非心臓性胸痛の用量制限毒性が認められた。この被験者は減量された用量に進み、その
後、完全な強度に段階的に戻った。この集団を拡大して計6例の被験者を登録したが、そ
れ以上のDLTはなかった。
られた。第1は、用量制限毒性として分類された注入関連反応であった。この被験者は減
量された用量に進み、その後、完全な強度に段階的に戻った。2番目の事象は冠動脈虚血
を併発した持続的な心電図変化であり、第4週の投与で起こったため、用量制限毒性とし
て分類されなかった(以下でさらに考察する)。この被験者は試験から除外され、この集
団(最大投与量)にそれ以上の被験者は組み入れられなかった。
は、すべての被験者が減量を開始し、その後、治験薬に対する注入反応を最小限に抑える
ために、十分な強さまで漸増した。この集団で用量制限毒性を経験した被験者はいなかっ
た。
は経験されなかった。しかし、週2回投与に伴う臨床的負担が大きいため、この集団への
募集を中止することとした。
物の試験ではこれまで観察されていなかった。2例で重篤な有害事象として、減量又は中
止に至った心イベント(虚血及びたこつぼ型心筋症)が認められた。当該事象は投与7日
後に発現し、冠動脈疾患の既往歴があったことから、虚血は治験薬投与に起因する可能性
は低いと判断された。この被験者では、より早期の投与中に心電図変化が認められた。心
筋症事象に先立って急性輸液反応が発現したが、これはおそらくdex漸減の結果であると考えられた。この被験者には冠動脈疾患の既往もあった。さらに3例で投与後に一過性の心電図変化(T波異常)が認められたが、重篤とは分類されなかった。これらは、トロポニンの上昇、虚血または駆出率の変化とは関連していなかった。すべての事象が消失し、被験者は追加投与を受けたが、それ以上の心電図異常は認められなかった。それにもかかわらず、悪性胸膜中皮腫患者の高齢、および本疾患に関連する罹病率を考慮すると、これらの観察結果から、安全性委員会は、より厳格な心臓除外基準と、残りの試験および今後のあらゆる試験のための追加的な心臓モニタリングを推奨するに至った。
えられたが、有意な懸念はなかった。
するEDVを用いることによって、中皮腫を治療する上で有望な結果を示した。しかしな
がら、実施例の結果はまた、中皮腫のための改善された治療戦略の必要性を実証する。
療法剤よりも大きく腫瘍細胞系の増殖を阻害することを明らかにした
この実施例は、超毒性細胞傷害性化学療法剤であるPNU−159682が広範囲の他の
化学療法薬よりも強力な癌細胞増殖阻害剤であることを示した。
物であり、その親化合物(MMDXとドキソルビシン)の3,000倍以上の細胞毒性を
有することから、PNU―159682は、“超毒性”化学療法薬と考えられる。このよ
うな薬物の使用は、超毒性薬物の毒性レベルが死亡を含む重度の有害事象をもたらすため
、一般的に典型的な化学療法治療では不可能である。
に、図6は、PNU−159682およびデュオカルマイシンがドキソルビシン、ミトタ
ン、パクリタキセル、オキサリプラチン、およびミトキサントロンに対して高度に耐性で
あったステージIV患者に由来する2つのAdreno−cortical癌細胞株に対して強力な細胞
毒性効果を示したことを示す。さらに、図7は、PNU−159682がドキソルビシン
、パクリタキセルおよびドセタキセルに耐性であったヒト乳癌細胞株MDA-MB-468
の増殖を阻害したことを示す。図8は、PNU−159682がドキソルビシン、シスプ
ラチンに耐性であるヒト結腸直腸癌細胞株Caco-2(図8A)およびHCT116(図8B)の増殖を阻害したことを示す。図9は、PNU−159682がドキソルビシンおよびパクリタキセルIVAXに耐性であった神経膠芽腫細胞株U87−MGの増殖を阻害し得ることを示す。図10Aは、これらの細胞がドキソルビシン、ゲムザール、マトルトキサート、オキサリプラチン、イリノテカン、および5−フルオロウラシル(5−FU)に耐性であるにもかかわらず、PNU−159682がゲミシタビン感受性であるヒト膵臓細胞株(MiaPaca-2細胞、図10A)の増殖を阻害することができることを示す。図10Bは、PNU−159682がゲミシタビン耐性細胞(MiaPaca−2 GemR細胞、図10B)であるヒト膵臓細胞株の増殖を、これらの細胞がgemzarに耐性であったとしても阻害し得ることを示す。
とが非常に望ましいことを示しており、それは、薬剤が従来の非超毒性化学療法剤に対す
る耐性を示す癌の治療に有用であり得るからである。
植モデルにおいてヒト肺癌細胞の薬剤耐性を克服することができる
この実施例はPNU−159682などの超毒性化学療法剤を送達するためにミニ細胞
(EDV)を使用することが、肺癌異種移植モデルにおける腫瘍増殖を効果的に阻害する
ことを示した。
ローンを選択することによりドキソルビシン耐性とした。次に、これらの細胞を異種移植
片としてBalb/c nu/nuマウスに移植した。腫瘍体積が〜150mm3に達したとき
、(1群あたりn=7)マウスの4つの異なった群を、(i)生理食塩水、(ii)ドキソ
ルビシン(EGFREDVTM Dox)を負荷したEDVを標的とする上皮増殖因子受容体(EGFR)、(iii)PNU−159682(EGFREDVTM 682)を負荷したEDVを標的とするEGFR、および(iv)図11に実線矢印で示す時点でPNU−159682(EDVTM 682)を負荷した非標的EDVで静脈内(IV)処置した。EGFRを標的とし、PNU−159682を負荷したEDVの組成を図1にグラフで示す。
この実施例はポロ様キナーゼ1(Plk1)、リボヌクレオチドレダクターゼ酵素1(R
RM1)を標的とするsiRNA、またはmiRNA16aを標的とするsiRNAを送
達するためにミニ細胞(EDV)を使用することが、癌細胞の増殖を効果的に阻害し得る
ことを示した。
549、A549MDR(ドックス耐性A549セルライン、多薬剤耐性膜ポンプ、MDRを過剰表現する)、H2122、H358およびH441を含むいくつかの非小細胞肺がん(NSCLC)セルラインにおいて過剰に表されることが示された。図12はGAPDH(g)、KSP(K)、Plk1(P)、およびRRM1(R)の発現を示し、この発現は、示されたNSCLC細胞株におけるGAPDH発現と比較して示される。
、RRM1(siRRM1)およびPlk1(siPlk1)を標的とする阻害非コード
低分子干渉RNA「siRNA」を合成し、がん細胞株への送達のためにEDV中にパッ
ケージ化した。
とがわかった。EGFR標的、siRRM1パッケージEDVをMSTO(メソヘリオマ
細胞系)またはH295R(アドレノ皮質細胞系)にトランスフェクトした。トランスフ
ェクションの5日後、細胞増殖を測定し、その結果を図13に示し、非標的化siRRM
1パッケージ化EDVまたはEGFR標的化siNonsenseパッケージ化EDVを
用いた対照トランスフェクションと比較して、細胞増殖の非常に有意な阻害を示した。
EGFR導的化siRRM1パッケージ化EDVでの静脈内(IV)処置は図14に示さ
れるように、生理食塩水またはEGFR標的化siScrambledパッケージ化ED
Vと比較して、非常に有意な抗腫瘍効力を示した。siRRM1パッケージ化EDVを受
けたマウスから単離された腫瘍は、陰性対照を受けたマウスからの腫瘍よりも有意に小さ
かった。図15。
種移植モデルにおいて細胞毒性薬を負荷したEDVの抗腫瘍効果を増大させる
この実施例は、インターフェロンI型アゴニストと併用される化学療法剤を送達するた
めにミニ細胞(EDV)を使用することが、肺癌異種移植片などの癌を治療するための有
効な戦略であることを示した。インターフェロンI型アゴニストは、化学療法剤と同じま
たは異なるミニ細胞中に存在同様。本実施例において、化学療法剤は超毒性薬物PNU−
159682であり、インターフェロンI型アゴニストは、40mer二本鎖DNAである。
36、および38日目に、マウスをこれらのEDVの組み合わせで処置した。図18に示
すように、試験したEDVの全ての組み合わせは、腫瘍増殖の安定化をもたらした。対照
的に、生理食塩水処理対照群は〜650mm3の体積まで主要増殖を示した。第36日と3
8日では、腫瘍体積が〜650mm3の食塩水群マウスが、図18の下矢印で示されるよう
にEGFREDVPNU-159682 +EDV50merで処理された。
FREDVPNU-159682)を含むミニ細胞(EDV)と組合せて40mer二本鎖DNA(EDV40mer)を含むEDVは、肺癌のマウス異種移植片モデルにおいてEGFREDVPNU-159682単独で処置された腫瘍細胞と比較して、腫瘍のより有意な退縮を誘導した。図19において、Balb/C nu/nuマウスを、(i)黒丸=EGFREDVPNU-159682、(ii)黒三角=EGFREDVPNU-159682+EDV40mer、または(iii)黒四角=生理食塩水で処置した。結果を以下の表にまとめる。
Nアゴニスト( イムキン)を用いたEDVPNU-159682 の臨床的評価
本実施例は、I型およびII型IFNアゴニストが進行固形腫瘍に罹患しているヒト患者
におけるEGFREDVPNU-159682の抗癌作用を増大させることを示した。著しいことに、進行期膵臓癌患者においてさえ、この治療は、わずか3回の投与後に腫瘍マーカーレベルの90%の低下を生じ、患者は著しく改善された生活の質を示した。
:本発明者らは、対象が1型IFNアゴニスト40mer二本鎖DNA(EDV40mer)(1型IFNアゴニスト)または60mer二本鎖DNA(EDV60mer)(1型IFNアゴニスト)を負荷したミニ細胞と組合せて標的化および負荷EDVを受けた臨床事例研究を行った。
として、EGFR(V)EDVPNU-159682と補助EDV40mer(1型IFNアゴニスト)の併用療法を行った。2人の追加の患者は、緊急使用法のもとで、EDV60mer(1型IFNアゴニスト)と併用して負荷および標的EDVを受けた。IV期膵がんと診断された1人の緊急使用患者はEGFR(V)EDVPNU-159682およびEDV40mer(1型IFNアゴニスト)またはEDV60mer(1型IFNアゴニスト)による治療を受け、再発副腎皮質がんと診断された2人目の緊急使用患者はEGFR(V)EDVPNUおよびEDV60mer+イムキン(2型IFN)を受けた。
薬剤および1型IFNアゴニストを含むミニ細胞:補助EDV40merを伴うEGFR(V)EDVPNU(1型IFNアゴニスト)を、進行固形腫瘍を有する被験者を対象とした非盲検単一施設探索的第1相試験(デザイナーEDV試験)において、3人の患者で評価した。適格とするために、標的化を容易にするために、患者は、腫瘍組織におけるEGFR発現のエビデンスを有する進行固形腫瘍の組織学的または細胞学的記録を得ることとした。患者は、標準的な1次、2次または3次治療レジメンの投与中または投与後に疾患の進行を示していなければならない。試験の主要なエンドポイントは、型EDV40mer(第1種IFNアゴニスト)とのEGFR(V)EDVPNU-159682の安全性と耐性を確立することであった。
ニュージーランド臨床試験登録(番号ACTRN 12617000037303)に登録された。
09で計13回のEGFR(V)EDVPNU-159682を受けた。治療は、8週間の治療からなるサイクルにおいて、20分間のIV注入として週1回投与した。各サイクルの終了時に、患者は腫瘍の放射線学的評価を受けた。
は概して良好であった。他のEDV生成物の投与でみられるように、患者は一般的に炎症
性サイトカインであるIL−6、IL−8およびTNF−αの一時的な増加を経験し、これは治療用量間でベースラインに戻った。観察された血液学的パラメータの変化はEDV治療薬を用いた過去の試験でみられた変化を大きく反映しており、白血球(WBC)の軽度の自己限定性上昇、投与3時間後の好中球の上昇、およびそれに伴うリンパ球および単球の減少などであった。パラメータは、次の投与前の次の時点で正常に戻った。一部の被験者で血清リン酸塩濃度の軽度低下が認められたが、介入を必要とせず、用量間でベースライン値に回復した。
発熱が認められた。これらの患者は観察のために一晩入院し、事象は翌日までに消失した
。1例が用量制限毒性のため試験を中止した。2人目の患者は試験を継続し、追加投与を
受けた。
シドニーのローヤルノースショア病院で最初の思いやりのある使用ケース研究が行われ
た。患者は68歳の女性で、ステージIVの膵癌と診断された。Whipple手術(膵頭十二指腸切除術)を受け、第一選択治療としてゲムシタビンが投与された。FOLFIRINOX治療レジメンも受けたが、転移性肝疾患を発症した。彼女の腫瘍細胞をin vitroで試験し、PNU−159682に感受性であることが見出された。
R標的EDVを含むEDV生成物の2週間毎の用量を受けた。これらを、20mLの注入と
してIV送達した。患者は本プロトコールでの使用を意図したように、EGFR(V)標
的EDVおよびPNUを含むITG(609)標的EDVの両方、ならびにEDV40mer
(1型IFNアゴニスト)またはEDV60mer(1型IFNアゴニスト)を受けた。全部
で45回のEGFR(V)EDVPNU-159682+EDV40mer(または関連する商品EDV60mer)を受けた。EGFR(V)EDVPNU-159682とITG(609)EDVPNU-159682の量はそれぞれ2×109と4×109まで増量し、EDV40mer/60mer は5×108の一連の量で与えた。
なかった。予備的な結果は炎症性サイトカインであるIL-6、IL-8およびTNF−αの投与後の一過性の増加を示しており、他のEDV生成物の投与で見られたものと同様で
ある。これらの反応は一般に、その後の投与量にわたって低下した。抗炎症性サイトカイ
ンIL−10も投与後に一過性に増加した。興味深いことに、IFN−αは研究全体を通
して様々な時点で同様に増加し、これは、EDV40mer/60merによる刺激の結果である可能性が高い。投与2時間後にIFN−γの上昇は検出されなかった。
間に相当する最初の3回の投与後に90%以上低下した。10回投与後、これはさらに低
下し、腫瘍マーカー値がほぼ95%低下した。また、IV期膵がん患者のほとんどが経験
する悪液質状態とは対照的に、有意な体重増加を示し、生活の質の著しい改善を報告した
。このように、本症例研究の予備的な安全性および有効性の結果は、特に進行膵癌に伴う
予後不良を考慮すると、極めて有望である。
常に重い末期副腎皮質癌患者を対象とした2回目のコンパッショネートユースケーススタ
ディは、Royal North Shore Hospital(シドニー)で治療された。
。これは、EDV製品の投与で予想される。残念ながら、患者は重篤な免疫系抑制因子で
あることが知られている高レベルのコルチゾールを含む非常に高い疾患負荷を有し、第7
週のCTスキャンは進行性疾患を示したため、患者は試験から脱落した。
上皮成長因子受容体標的EDVの抗腫瘍効果を増強し、様々な癌の異種移植モデルにおい
て腫瘍退縮を引き起こす
この実施例はドキソルビシンをIFN−γと組み合わせて送達するためにミニ細胞(E
DV)を使用することが、マウス異種移植モデルにおいて改善された抗腫瘍効果を提供す
ることを示した。
りEGFREDVDoxおよびIFN−γ(0.5×104IU)を投与された(図23、実線)。図23に示すように、グループ1、2、および3は1週間あたり2回、示された投薬量であり(図23のx軸上の黒三角によって示される);一方、グループ4のマウスは1週間あたり3回、示された投薬量を投与された(図23のx軸上の白三角によって示される)。EGFREDVDoxおよびIFN−γ(0.5または0.75×104IU)で週2回または週3回処置したマウスは、耐性A549肺癌異種移植片の腫瘍安定化を達成した(図23)。対照的に、EGFREDVDoxで処置したマウスは抗腫瘍効力を示さず、腫瘍は生理食塩水で処置したグループと同様に増殖した。この結果はIFN−γ(0.5または0.75×104IU)を、通常は腫瘍単独に抵抗性である腫瘍の処置におけるEGFREDVDoxと共に含めることが、腫瘍安定化を達成するために必須であることを示唆する。したがって、EGFREDVDoxとIFN−γを組み合わせることにより、肺癌異種移植モデルにおける薬剤耐性を克服することができる(以下の表16に要約)。
この実施例は、イヌの研究において、ミニ細胞技術および追加的にインターフェロンγ(イムキン)(II型IFN)を有する超毒性薬物PNU-159682およびI型IFNアゴニスト(40mer二本鎖DNA)を送達することが、良好な忍容性を示すことを示した。
オン動物であった。各ペットの飼い主からインフォームドコンセントを得た。
merおよび/またはイムキン)と併用して、EGFRまたはITG(インテグリン)を標的とするPNU負荷EDVで治療した。脳腫瘍または肉腫のイヌをEGFR標的EDVで治療し(n=9)、黒色腫のイヌをITG導的EDVで治療した(n=4)。合計185週1回または隔週1回の用量を、アジュバントの有無にかかわらず、異なる組み合わせで投与し、最大73回の用量を単一のイヌに与えた。最大5×109のPNUロードEDVおよび標的EDV、最大2×109EDV40mer/50mer、イムキン、25μg/m2/用量を用いた。いずれの併用も全般的に忍容性は良好であり、最も多く認められた有害事象は他の
EDV生成物の投与時に認められた有害事象(軽度の嗜眠、発熱、悪心、嘔吐)と同様で
あった。免疫調節アジュバントの添加はEDV投与で見られる一般スペクトルを変化させ
たり、有害事象の頻度を増加させたりしないようであった。
(WBC、好中球、リンパ球、単球および好酸球)の軽度の一過性の減少が認められた。
同様の変化が、免疫調節アジュバントの添加の有無にかかわらず見られた。注目すべきは
、他のイヌおよびヒトの臨床試験では好中球は一般に投与後3時間で減少するのではなく
増加したことである。この軽度の好中球減少は、PNU負荷EDVによるイヌの治療に特
異的であると思われる。
NF−αの一過性の上昇が認められた。一般に、免疫調節アジュバントを含む用量は、PNU負荷EDV単独を含む用量よりも、これらのサイトカインのわずかに高い誘導をもたらした。また、投与後のIL−2量が低下した。これらのデータは、抗腫瘍負荷EDVおよび標的EDVと組み合わせて使用される免疫調節EDVアジュバントの安全性および忍容性を支持する。
たが、1匹のイヌではほぼ部分奏効(腫瘍サイズの29.8%の縮小)が得られた。1匹
のイヌは、11ヵ月以上の治療期間にわたって73回の投与を受け、試験期間中安定した
疾患を示した。このイヌは視神経を圧迫する腫瘍塊による視力喪失を示した。しかし、治
療の過程で視力は回復し、臨床症状の改善が示された。
この実施例から、パクリタキセルまたはドキソルビシンをパッケージ化したEDVはト
ル様受容体活性化と一致するサイトカイン反応をヒトで誘導するが、このEDV処理はイ
ンターフェロンII型反応を誘導しないことが示された。
に、サイトカインのパネルを、ヒト初回投与試験 (上記実施例5)および再発性膠芽腫ヒ
ト臨床試験(上記実施例6)において評価した。
)の患者3/22例、および再発性膠芽腫研究(図25参照)の患者1/14例において
、研究期間を通じてランダムな段階で上昇した。IFN−αの誘導は投与と一致しなかっ
た。インターフェロン経路は一般に、TLR経路のような細菌刺激よりもむしろウイルス
刺激によって活性化されるため、これらの選択された患者は試験中にウイルス感染が同時
に報告されていなかった可能性がある(例えば、風邪)。しかし、IFN−γを含む試験
された他のすべてのサイトカインは、標的EDVおよび負荷EDVの投与によって影響さ
れなかった。このことから、II型IFN−αの活性化(イムキンの添加を介して)は、異
なる抗腫瘍経路を刺激することによって標的EDVおよび負荷EDVの有効性を高めるた
めの実行可能なアプローチである可能性が示唆される。
された細菌由来ナノ細胞によって誘発される新規経路
この実施例は、腫瘍細胞表面受容体を標的としたEDVナノ細胞が自然免疫系および適
応免疫系の活性化と併せて、スーパーサイトトキシンPNU−159682(682)を
送達することによって、腫瘍に対する二重攻撃が可能な癌免疫療法として機能することを
実証する。
ことができる682活性化M1マクロファージおよびNK細胞を送達する標的EDVナノ
細胞を示す。これに続いて、樹状細胞の成熟が起こり、抗原提示および腫瘍特異的CD8
+ T細胞の生成が生じる。スーパーサイトトキシン送達と、先天性および適応性抗腫瘍免
疫応答の両方の活性化の組み合わせは、臨床腫瘍学において可能性を有する強力な癌細胞
免疫療法をもたらす。
は、腫瘍細胞内で細胞傷害性薬物を送達することができるだけでなく、同時に腫瘍を特異
的に標的とする自然免疫応答および適応免疫応答を誘発することができる。臨床的に、思
いやりのある使用症例患者では、682の負荷EDVが適応免疫を刺激しながら薬剤耐性
を克服することが示されている。前臨床試験は、EDV治療に起因する免疫療法経路が免
疫系による効果的な抗腫瘍応答を開始するのに必要な全ての工程に対処するアプローチを
包含することを実証した。この例は、マクロファージ、NK細胞および樹状細胞を含む自
然免疫系の細胞を活性化するEDVの能力を例示している。これに続いて樹状細胞の成熟
と抗原提示が起こり、腫瘍特異的細胞傷害性T細胞が産生され、さらなる免疫細胞の腫瘍
微小環境への動員をもたらす適応T細胞応答が起こる。このアプローチは免疫原性腫瘍微
小環境を作り出し、それにより患者に生じるかもしれない一次耐性および/または適応耐
性を回避する複数の免疫細胞サブセットにも作用することにより、免疫療法による現在の
ピットフォールのいくつかを回避する。
在に応答して、CD86発現量の有意な増加を誘発することができたが、682単独では
同じ応答を誘発しなかった(図27A)。さらに、EDV処理RAW細胞はTh1マクロ
ファージ分極の原因であると同定されている炎症誘発性サイトカインTNF−αおよびI
L−6の有意な増加を示した(Yuanら、2015)(図35Aおよび35B)。興味深いこと
に、Ep−EDV−682で処理し、続いてRAW264.7細胞と共培養したマウス腫
瘍細胞(4T1およびCT26Ep12.1)はまた、RAW264.7細胞上でのCD
86発現の有意な増加(図27B)ならびに炎症誘発性サイトカインTNF−αおよびIL−6の産生の有意な増加を生じさせることができた。(図27C−27D)。しかし、Ep−EDVまたは682処理のみではCD86発現に有意な変化を生じることはできず、682処理ではTh1サイトカインの産生の増加は認められなかったことから、EDV負荷682に応答した細胞死がその後のM1マクロファージの極性化を完全に誘導するために必要であることが示された。
した腫瘍細胞死の効果も試験した(図27E〜27I)。骨髄由来樹状細胞(BMDC)
を処理した腫瘍細胞(4T1及びCT26Ep12.1)と48時間共インキュベートし
た後、1型インターフェロンIFNαおよびIFNβの産生を評価した。樹状細胞による
1型インターフェロン産生の増加は樹状細胞の成熟および抗原提示の増強の機序として十
分に確立されており、NK細胞およびT細胞を含む他の免疫細胞サブセットとの相互作用
に不可欠である(Fitzgerald-Bocarsly and Feng、2007; Simmons ら、 2012)。Ep−E
DV−682処理4T1細胞とのBMDC共培養は、IFNα mRNA産生の約100倍の増加およびIFNβmRNA産生の約70倍の増加を伴う1型インターフェロンの両方の有意な増加を示した(図27E)。同様に、Ep−EDV−682処理CT26Ep12.1とのBMDC共培養は、IFNαmRNA産生の約300倍の増加およびIFNβmRNA産生の約60倍の増加を示した(図27F)。
及ぼしたかどうかを評価するために、CT26Ep12.1をEp−EDV−Doxで処
理したところ、IFNβmRNA産生の有意な〜20倍の増加が認められたが、IFNα
mRNA産生のわずかで有意ではない増加が認められたのみであった。Ep−EDVおよ
び682処理は、いずれの細胞株との共培養においても、1型インターフェロンmRNA
産生の増加を誘発することができなかった。しかしながら、CT26Ep12.1のドキ
ソルビシン(Dox)処理は、実際、IFNβmRNA産生の有意な〜5倍の増加を示し
た(図27F)。腫瘍細胞のドキソルビシン処理は免疫原性細胞死をもたらすことが知ら
れており、したがって、樹状細胞の成熟を促すことができる。しかしながら、IC50を
はるかに超える薬物用量は一般に、薬物単独でこのタイプの死が生じるために必要である
(Showalterら、2017)。BMDCをEDVと薬物で処理した4T1およびCT26Ep
12.1細胞と共インキュベートすると、CD86、MHCクラスII、およびCD80
が24時間以内にアップレギュレーションされ(図27G〜27I)、マウスJAWSI
I細胞で見られる同様の結果が得られた(図35C〜35E)EDVで処理した腫瘍細胞
と共培養したBMDCも、TNFアルファ、IL−12p40、およびIL−6の産生の
顕著かつ有意な増加を示した(図27J〜27L)。これらの結果は682を負荷したE
DVがマクロファージをM1抗腫瘍表現型に偏向させ、樹状細胞成熟を促進することがで
き、682単独では同様の応答を誘発することができないことを示す。
682はpM範囲の薬物耐性癌細胞でさえIC50を有する超細胞毒素であるため(Qu
intieriら、2005)、このような化合物に関連する重篤な全身毒性のために、臨床的に使
用することができない(StaudacherおよびBrown、2017)。しかし、EDVに封入した場
合、682のようなスーパーサイトトキシンを、最小限の体重減少(%)、被毛の波打ち
がほとんどないかまたは全くないこと、および様々な治療的ペイロードを担うEDVで処
理されたマウスを含む以前の研究と相関するEp−EDV−682処理マウスにおける嗜
眠または円背姿勢の出現がないことから明らかなように、副作用がほとんどなく腫瘍に効
果的に送達することができる(MacDiarmidら、2009; MacDiarmidら、2007a; MacDiarmidら、2007b; Sagnellaら、2018)(図35A−35D)。
かを有するEp−EDV−682処置BALB/cマウスにおいて、有意な腫瘍退縮が見られた(図28A〜28B)。Ep−EDV−682はまた、T84ヒト異種移植片および薬物耐性A549/MDR異種移植片を有する無胸腺BALB/cヌードマウスにおいて有意な腫瘍還元を促進することができ、ならびに大きな(〜600mm3)A549/MDR腫瘍において有意な腫瘍還元を誘発することができた(図28C〜28D)。
るが、最初のインビトロ実験はEDVがM1マクロファージ分極を駆動することを含むが
、これに限定されない多くの方法で免疫療法剤として挙動することもできることを示した
。これらの結果が腫瘍微小環境内の先天性免疫系のインビボ刺激にまで拡張されたかどう
かを確立するために、CD11b+免疫細胞を、生理食塩水、Ep−EDV、またはEp
−EDV−682で処理した4T1またはCT26Ep12.1マウス腫瘍から単離し、
xCELLigenceリアルタイムセルアナライザー(RTCA)において、それらの
対応する腫瘍細胞と5:1(エフェクター:標的)の比率でエクスビボで共培養した(図
28E〜28G)。接着性4T1細胞と共培養したCD11b+細胞は最初の接着および
沈降相を示し、その結果細胞指数が増加し、続いて活性相を示し、CD11b+ 細胞が接
着性腫瘍細胞を殺すのに効果的である場合には細胞指数が着実に減少し、腫瘍細胞が効果
的に溶解されず増殖し続けた場合には細胞指数が増加した。
1b+細胞は4T1細胞を殺すのに非常に効果的であったが、Ep−EDVまたは生理食
塩水で処置した腫瘍から単離したものは4T1腫瘍細胞を殺さなかった(図28E)。4T1腫瘍内のマクロファージの表現型決定(CD45+ CD11b+ Ly6G- Ly6C+)がCD86:CD206発現マクロファージの比の増加によって証明されるように、M1/M2比の増加を示した(図28F)。更に、Ep−EDV−682 4T1系細胞担持マウスからのCD1b+は4T1細胞(図36I)とex vivo共培養した場合、マクロファージ炎症蛋白質1α(CCL3/MIPα)の産生において、塩水およびEpEDV処理マウスと比較して2倍の増加を示した。MIP−1αの局所的産生は免疫エフェクター細胞浸透物を細胞微小環境中に誘引し、エフェクター細胞媒介抗菌反応を潜在化する主要因子として意味されていた(Allenら、2018; Zibertら、2004)。
DV−682で処置されたマウスの腫瘍から単離されたCD11b+細胞の増強された細
胞溶解活性を示した(図28G)。全般的に、CT26Ep12.1腫瘍由来のCD11b+細胞は4T1腫瘍由来のものよりも活性が高かったが、これは3つの処置群すべてについてのxCELLigence RTCAにおける細胞指数の着実な低下によって示される。しかしながら、細胞溶解はより顕著であり、Ep−EDV−682処置群におけるCD11b+ 細胞添加後1時間以内に始まり、CD11b+ 細胞添加後10時間で42%の生存率に低下した。対照的に、Ep−EDV処理サンプルでのサイトリズムの開始には5時間近くを要し、サリン処理群では7時間、CD11b+ 細胞追加後10時間での生存率はそれぞれ76%および86%であった。CT26Ep12.1種瘍から単離されたCD11b+細胞のフローサイトメトリー解析は、Ep−EDV−682処置腫瘍においてCD86:CD206比(M1/M2比)の有意な増大を示した(図28H)。
ウスにおける薬物耐性ヒト肺癌異種移植片(A549/MDR)においても見られ、EG
FR−EDV−682処置マウスの脾臓におけるM1/M2比の約2〜3倍の増加が生理
食塩水処置マウスまたは腫瘍増殖の減少に効果のないEDVで処置したマウスと比較して
観察された(図35E)。さらに、EGFR−EDV−682処置T84腫瘍において、
M1/M2比のわずかであるが有意な増加も検出された(図35F)。両方の免疫コンピ
テントマウス癌モデルと同様に、EGFR−EDV−682で処置されたA549/MD
R腫瘍を有するヌードマウスの腫瘍から単離されたCD11b+細胞は生理食塩水で処置
されたものと比較して、xCELLigence RTCAによって決定されるように、
A549/MDR腫瘍細胞へのCD11b+ 細胞付加の6.5時間後に28%の細胞溶解
を伴う優れた腫瘍細胞溶解を示した(図35Gおよび35H)。
とる
本実施例の目的は、抗腫瘍剤を含む細菌ミニ細胞のNK細胞機能に対する影響を決定す
ることであった。
T1またはCT26Ep12.1腫瘍を有する対照BALB/cマウスの脾臓からEp−EDV−682、EpEDVまたは生理食塩水による2週間処理後にNK細胞を単離した。脾臓NK細胞を、それらの対応する腫瘍細胞と20:1のE:T比でxCELLigenceRTCA中で共培養し、腫瘍細胞死を3〜4日間にわたって分析した(図29A〜29D)。
強力な細胞溶解を介して抗腫瘍特性を示したが、生理食塩水またはEp−EDVで処理し
たマウスはその標的腫瘍細胞に対してほとんどまたは全く細胞溶解能を示さなかった。C
T26Ep12.1を有するマウス由来のNK細胞は標的CT26Ep12.1細胞の迅
速な細胞溶解を示し、同時培養の最初の数時間以内にほぼ60%の標的細胞生存率に低下
し、50時間後にわずか18%の標的細胞生存率を維持した。Ep−EDV処理CT26
Ep12.1同様のNK細胞は50時間後に70〜80%の標的細胞生存率を有する低レ
ベルの細胞溶解能力を有し、一方、生理食塩水処理マウス同様のNK細胞は、同じ期間に
〜82%の標的細胞生存率を示した(図29D)。4T1腫瘍を有するEp−EDV−6
82処理マウス由来のNK細胞は依然として高度に活性であるが、70時間後に〜36%
の標的細胞生存率まで低下する、より緩やかな細胞溶解プロファイルを示し、一方、生理
食塩水またはEp−EDV処理マウス由来のNK細胞は90%の標的細胞生存率を維持し
た(図29Aおよび29B)。さらに、Ep−EDV−682で処理したマウスから単離
したNK細胞とのNK/4T1共培養は、生理食塩水で処理したマウスからのものと比較
して、TNFαおよびRANTESの両方の産生の2倍以上の増加を示した(図29Fおよび29G)。
は、免疫応答性マウス腫瘍モデル(図36Aおよび36C)と同様の細胞溶解プロファイ
ルを示した。EGFR−EDV−682処理マウスのNK細胞はその標的腫瘍細胞と共培
養すると、A549/MDRおよびT84腫瘍のいずれにおいても標的細胞の生存率が4
0%未満となったが、生理食塩水対照ではいずれも標的細胞の生存率が70%を維持した
。T84/NK細胞共培養物中のグランザイムB産生の試験は、Ep−EDV−682処
置マウス由来のNK細胞を含む共培養物中のグランザイムBの有意に高いレベルを明らか
にした(図36B)。
−682処理腫瘍のNK細胞(CD45+、CD11b+、DX5+)は、腫瘍免疫監視
において重要であることが知られているNK活性化受容体であるNKG2D発現を有意に増加させた(図29E)。腫瘍細胞表面上のNKG2DリガンドのアップレギュレーションはNK阻害シグナルを無効にするのに十分であり、したがって腫瘍細胞の細胞溶解を可能にする(Morvan and Lanier、2016)。NKG2D結合NKリガンドRAE-1、H60a、およびMULT−1についてのマウス腫瘍細胞株(CT26Ep12.1および4T1を含む)のスクリーニングにより、4T1およびCT26Ep12.1のいずれもスクリーニングした4つの細胞株の60aの発現レベルが最も高く、これがこれら2つの細胞株において最も高い全体的発現を有するリガンドであることが示された。さらに、マウス乳癌細胞系4T1およびEMT-6細胞はスクリーニングした全ての細胞系においてRAE-1の最高の発現を示したが、H60aよりはるかに低いレベルであり、一方、CT26Ep12.1はこのリガンドのレベルが非常に低かっただけであった。最後に、4T1細胞を除いて、全ての他の細胞系はMULT−1の発現を示さなかった(図29H)。
をさらに調べるために、NK細胞を4T1腫瘍を有するEp−EDV−682処理マウス
の脾臓から単離した。NK細胞を、xCELLigence RTCAシステムにおいて4T1細胞と共培養する前に、NKG2D受容体のその特定のリガンドへの結合をブロックするように設計された抗体と共にインキュベートした(図29I)。RAE−1に対する抗体は4T1細胞のNK細胞溶解の〜13%阻害をもたらしたが、H60aに対する抗体は〜21%阻害をもたらし、2つの抗体の組み合わせはNK細胞の細胞溶解能力の〜25%を阻害した(図29J)。
本実施例の目的は、EDV処置後の腫瘍微小環境内のサイトカイン環境を調査すること
であった。
酵素的方法で穏やかに解離させて、細胞の溶解がないことを確実にし、その結果、間質性
腫瘍サイトカインレベルを評価し得た(図30A〜30B)。腫瘍サイズがかなり違うた
めに、腫瘍を秤量し、測定し、そしてサイトカインレベルを、これらのサイズ差を説明す
るために、組織1グラムあたりに計算した。いずれの腫瘍も、Ep−EDV−682処理
に反応して腫瘍微小環境内のTNFαの有意な増加を示したが、この増加は生理食塩水処理と比較して、Ep−EDV−682処理後に10倍の増加を伴うCT26Ep12.1腫瘍においてより顕著であった。同様に、Ep−EDV−682処理は、4T1腫瘍におけるIFNαレベルの約2倍の増加、およびCT26Ep12.1腫瘍における15倍の増加を伴う、両腫瘍における間質IFNα濃度の有意な増加をもたらした。
12.1腫瘍で見られ、そこではIFNγレベルの5百倍の増加が起こったが、4T1腫瘍ではIFNγレベルのわずかではあるが有意な2倍の増加が起こった。Ep−EDV−682処理の結果としてのIL−1βレべルは4T1腫瘍で有意に減少したが、CT26Ep12.1腫瘍では有意ではないが増加を示した。IL2(〜4倍)およびIL4(〜3倍)の有意な増加が4T1腫瘍で生じたが、Ep−EDV−682処理後のIL6レベルに有意な変化はなかった。Ep−EDV−682処理マウスのCT26Ep12.1腫瘍ではIL−2量は150倍以上有意に増加したが、IL−6量は有意に3倍増加し、IL−4は2倍有意ではない増加が観察された。抗原提示細胞、NK細胞、およびT細胞などの免疫細胞による浸潤の主要な決定因子であることが示されているMIP−1α(CCL3)およびRANTES(CCL5)も試験した(Allenら、2018)。4T1およびCT26Ep12.1の両方はEp−EDV−682処理腫瘍における2つのケモカインのレベルの増加を示し、CT26Ep12.1腫瘍におけるMIP−1αレベルおよびEp−EDV−682で処理された4T1腫瘍におけるRANTESレベルの約3倍の有意な増加を示した。さらに、RANTESとMIP−1αレベルの2〜2.5倍の増加は有意ではなかったが、それぞれCT26Ep12.1と4T1 Ep−EDV処理の両方で起こった。一般に、Ep−EDV処置は、生理食塩水処置群と同様の間質腫瘍サイトカインおよびケモカインレベルをもたらした。
生(TNFα、IFNγ、IL−1β、IL−2、およびIL−10)を評価した。脾細胞を単独で培養するか、またはそれらの対応するマウス由来の分散した腫瘍細胞と共培養した。生理食塩水、Ep−EDV、およびEp−EDV−682での全身処置は、4T1またはCT26Ep12.1腫瘍モデルのいずれかからの脾細胞によるサイトカイン産生に対して有意な効果を有さなかった(図30C〜30G)。しかし、脾細胞を対応する処理腫瘍とともに培養すると、これもはやそうではなかった。TNF瘤Y生は、脾細胞のみと比較して、Ep−EDV−682で処理したマウスからの共培養、ならびに両腫瘍モデルからの生理食塩水およびEp−EDVで処理したマウスからの共培養において有意に増加した(図30C)。4T1モデルにおいて、IL−2産生は、Ep−EDV−682処置マウスからの共培養物において、生理食塩水処置マウスと比較して有意に増加した(図30D)。さらに、生理食塩水処理マウスの脾細胞を対応する腫瘍細胞と共培養した場合、IL−2産生の有意な減少が認められた。IL−2 CT26Ep12.1腫瘍を有するマウスから単離した脾細胞単独と比較して、全ての共培養において産生が有意に増加した。IL−1β産生における唯一の有意な変化は、生理食塩水処置マウスからの試料がEp−EDV−682処置マウスと比較して、インビボで見られる差に対応する有意な増加を示した4T1モデルからの共培養物において生じた(図30E)。IFNγ産生は脾細胞単独と、4T1腫瘍を有する生理食塩水処理マウスから分離した共培養の間で有意に減少し、Ep−EDV−682処理脾細胞/腫瘍細胞共培養の方が、生理食塩水またはEp−EDV処理4T1腫瘍担癌マウスから分離した共培養よりも有意に大きかった(図30F)。CT26Ep12.1腫瘍モデルにおいて、IFNγ産生は生理食塩水およびEp−EDV共培養と比較して、Ep−EDV−682処理脾細胞/腫瘍細胞共培養で減少したが、これはEp−EDVに対してのみ有意であった。最後に、IL−10産生は、CT26Ep12.1処置マウスからの生理食塩水処置脾細胞/腫瘍細胞共培養物において、脾細胞単独およびEDV処置群と比較して有意に増加した(図30G)。
最初のin vitro実験では、EDV処理が効果的な化学療法薬を負荷した標的EDVに応
答して、直接的相互作用または細胞死の結果のいずれかを介して樹状細胞の成熟をもたら
しうることが示された。従って、この試験は、この結果がインビボでのDC成熟および抗
原提示に翻訳され得、腫瘍特異的CD8+ 細胞傷害性T細胞の産生を生じるかどうかを試
験することを目的とした。
4T1またはCT26Ep12.1腫瘍を有するマウスから脾臓を取り出し、CD8+ T
細胞を単離した。次いで、CD8+ T細胞を対応する腫瘍細胞に添加し、xCELLig
ence RTCAを用いてこれらの細胞を特異的に認識し、殺すそれらの能について試
験した(図31Aおよび31C)。4T1を保有するマウスから単離され、生理食塩水ま
たはEp−EDVで処理されたCD8+ T細胞は4T1細胞に対して細胞毒性を示さな
かったが、Ep−EDV−682で処理されたマウスから単離されたCD8+ T細胞は30時間後に標的細胞の50%の細胞溶解を誘導した(図31B)。
たCD8+ T細胞は標的CT26Ep12.1細胞を殺すのに非常に効果的であり、エフ
ェクタ細胞を標的細胞に加えた20時間後に81%の細胞毒性が見られた(図31D)。
興味深いことに、Ep−EDV処置でさえ、CT26Ep12.1モデルにおいて腫瘍特
異的CD8+ T細胞の産生を誘発することができ、20時間後に40%の細胞毒性が明ら
かであったが、生理食塩水処置マウス由来のCD8+ T細胞はCT26Ep12.1細胞
に対して特異性を示さなかった。4T1腫瘍内のCD8+ T細胞の流動解析は、Ep−E
DV−682処置マウスの腫瘍内のCD8+ T細胞(CD45+、CD3+、CD8+)の
割合のわずかながら有意な増大を示した(図31E)。さらに、Ep−EDV−682処
置マウスの腫瘍内の調節性T細胞(CD45+、CD3+、CD4、CD25+)の割合が
有意に2倍低下した(図31F)。4T1腫瘍担癌マウスの腫瘍流量リンパ節におけるT
細胞数も流れを介して調べた。生理食塩水およびEp−EDV処理マウスの両方と比較し
て、Ep−EDV−682処理マウスのリンパ節において、全体的なT細胞数(CD3+
)の有意な増加、ならびにCD4+およびCD8+ T細胞数の両方の有意な増加が見られ
た(図31G)。4T1腫瘍を有するEp−EDV−682処置マウスのリンパ節におけ
る成熟樹状細胞の有意な増大も検出された(図31H)。4T1細胞で処置したEp−E
DV−682マウス由来の単離されたCD8+ T細胞間の相互作用の可視化はこれらのT
細胞が腫瘍細胞に付着し、パーフォリン(緑色)を腫瘍細胞に放出することができること
を示す(図31I)。
この実験は、ステージIVの膵管腺癌(PDAC)患者(CEB)におけるEGFREDV682治療の使用の思いやりのある症例の臨床観察に関連する。
含み、多発性低密度肝病変を明らかにした。腫瘍は陽電子放出断層撮影(PET)では活
発ではなかった。標準的な生化学検査および血液学的検査は、概して特記すべきものはな
かった。血清CA19―9およびC反応性蛋白質(CRP)も評価した。一部の消化器悪
性腫瘍、特に膵癌に発現する糖鎖抗原であるCA 19−9の血清中低濃度は、全生存率
および治療に対する反応の予後指標となることが示されている。同様に、上昇したCRP
レベルもまた、不良な臨床転帰と有意に関連することが示されている(Szkanderaら、201
4)。ゲムシタビンとFOLFIRINOXの後でさえ、CEBはCA19−9量が120,000kU/Lを超え、通常の3,000倍であり、かなり上昇したCRP量が64mg/Lであった。
性を調べた。頭部および尾部PDAC細胞の両方は低感度を示し、第1および第2の系統の薬物に対して部分的に応答しなかった(図37A)。対照的に、両者は682に対して極端な感度を示し、IC50はpM温度範囲であった(図37A)。さらに、上皮増殖因子受容体(EGFR)は、フローサイトメトリー(図37)によって、細胞あたり>200,000コピーで過剰発現されることが見出され、したがって、治療のために682を負荷したEDVを標的化するための理想的な受容体であることが見出された。
現型検査により、好ましい抗腫瘍反応を裏付ける可能性のある複数の細胞型内の変化が明
らかになった(Gating戦略−図38)。マクロファージおよび樹状細胞の前駆体である全
CD14+単球は、中間体(CD14+CD16++)単球サブセット(69%増加;図
32D)を含むスクリーニング用量(D1)(図32C)と比較した場合、D12で15
.28%から31.39%に増加した(105%増加)。中間の単球はT細胞に抗原を提
示する能力が最も高く、IL−12およびIFN−繧フ抗原特異的誘導が優れている(Zie
gler-Heitbrock and Hofer、2013)。
減少も観察され(図32F)、CD8+エフェクターT応答を駆動する原因であるCle
c9A+骨髄性樹状細胞(mDC)もまた増加した(D12で98%)(図32E)。こ
の増加は、D12におけるエフェクターCD8+ T細胞(50%増加)を含む全細胞傷害
性CD8+ T細胞(60%)の増加と一致していた(図32G)。エフェクターCD8+
T細胞プールは単球、マクロファージまたは樹状細胞によって提示された抗原と最近相互
作用し、腫瘍および/またはEDV抗原特異的T細胞を含むCD8+ T細胞である。消耗
したプログラム死−1(PD−1+)表現型を発現する細胞傷害性CD8+ T細胞は、P
D−L1を発現する腫瘍細胞による長期の細胞活性化とPD連結への感受性を示す、D1と比較してD12で17%減少した。この方法は腫瘍および腫瘍排出リンパ節内でよく起こる。このケーススタディで観察された結果は、前臨床マウスモデルで観察された結果と同様の傾向をたどる。
瘍部位に効果的に送達するだけでなく、複数の免疫細胞サブセットを刺激することによっ
て免疫療法として挙動する能力を示している。Ep−EDV−682処理が、マクロファ
ージ、NK細胞およびCD8+ T細胞を含む免疫細胞サブセットを、腫瘍細胞を効果的に
排除することができる抗腫瘍表現型に向かって押すことができることが実証されている。
この薬物の有効性と組み合わせると、腫瘍に二重の攻撃をもたらす。
性ペイロードを有する標的EDVの投与用量の30%以上が2時間以内に腫瘍微小環境に
直接沈着する(MacDiarmidら、2007b)。EpCAM標的化EDVは腫瘍細胞上の表面発現EpCAM(この場合、4T1およびCT26Ep12.1)に結合し、次いで、それらのペイロード(682)を腫瘍細胞内に直接的に有効に送達する。682は、腫瘍細胞に送達されてから24時間以内に迅速なアポトーシスを生じる非常に強力な超細胞毒である(図33A)。次に、Ep−EDV−682処理によって産生されたアポトーシス細胞およびDAMPシグナルは腫瘍関連マクロファージ(TAM)などの自然免疫細胞と相互作用し、CD86のアップレギュレーションおよびTNFアルファおよびIL−6などのTh1炎症誘発性サイトカインの産生を刺激することができる(図33B)。
胞活性化を誘発し得、増加した細胞毒性を導く(図33F)。NKG2D受容体のアップ
レギュレーションはEp−EDV−682で処理したマウスの腫瘍内のNK細胞上で観察
され、この受容体はEp−EDV−682処理マウスから単離したNK細胞の細胞溶解能
に有意に寄与することが実証された。さらに、未成熟、中間および成熟マウスNK細胞は
ケモカインMIP1αおよびRANTESに結合することができるCCR1およびCCR5ケモカイン受容体の両方を発現し、それらの両方はEp−EDV−682処理腫瘍ならびにEp−EDV−682処理マウス由来のマクロファージおよびNK細胞によって増加される(Bernardiniら、2016年)。
EnGeneIC Dream Vector (EDV):EDVは先に記載したように Salmonella enterica serovar Typhimurium(STyphimurium)minCDE-strainから産生され、精製された(MacDiarmidら、2007b)。薬物負荷、抗体標的化、凍結乾燥、および用量調製は以前に記載されている(MacDiarmidら、2007b; Sagnellaら、2018)。EDV調製物は、Zetasizer NanoシリーズおよびNanoSight LM20(Malvern Instrument)を用いた動的光散乱を用いてEDVサイズおよび数を評価する厳密な品質管理の対象とした。エンドトキシンレベルを、Endosafeポータブル試験システム(Charles River)を用いて評価した。薬物をEDVTM調製物から抽出し、先に記載したように高速液体クロマトグラフィーによって定量した(MacDiarmidら、2007b)。
い、Kaluzaソフトウェア(Beckman Coulter)を用いて分析した。
コ改変イーグル培地(DMEM)(Sigma)中で約70%コンフルエンスまで増殖させ、細胞
スクレーパーを用いて継代培養した。モウスがん細胞系(4T1およびCT26)は10
% FCSを含んでいるRPMI−1640培地(Sigma)の単層で成長させ、リン酸塩緩
衝塩素(PBS)/Trypsin EDTAを用いて週2〜3回継代培養した。全ての細胞を、5
% CO2を含む加湿大気中で37の培養物中で維持し、そして日常的にスクリーニングし
、そしてマイコプラズマを含まないことを確認した。マウス細胞株におけるEpCAM発現および受容体数を、Quantum Simply Cellular anti−Rat IgGミクロスフェア(Bangs Laboratory)を使用して、APC抗マウスCD326(Biolegend)を用いたフローサイトメトリーを使用して定量した。CT26はEpCAMについて陰性であることが示されたので、細胞をLipofectamine 2000(Thermo Fisher)を用いてマウスEpCAM ORFクローン(NM_008532.2)(Genescript)を含むDYKpCDNA3.1+Cで形質移入した。G418選択を使用して、CT26クローンを発現するEpCAMの純粋な集団を得、そして細胞をEpCAM発現について上記のようにスクリーニングした。クローンについて、増殖速度、薬剤感受性およびin vivo腫瘍原性を調べ、上記3つのパラメータが親CT26細胞株と類似している高いEpCAM発現を有するものを、その後のすべての研究(CT26Ep12.1)のために選択した。
×105細胞で6ウェルプレートに播種し、一晩インキュベートした。次いで、培地を、
以下の1μg/mL LPS(Sigma);100pmol PNU−159682(Najing Levena);Ep−EDV−682(500:1および1000:1-EDV:細胞)、Ep−EDV(5000:1-EDV:細胞)のうちの1つを含む新鮮な培地で置き換えるか、または未処理のままにした。細胞を、処理の6時間後および24時間後に、細胞スクレーパーを使用して回収し、そして試料を、DAPI(Sigma)、抗CD45 Brilliant Violet 510(BioLegend)、抗CD86 APC−Cy7(BioLegend)、および抗CD206 AF488(R&D Systems)で染色し、そしてフローサイトメトリーによって評価した。
をVersene(Gibco)で回収し、細胞を1mLのエッペンドルフ管に回収した。細胞を、Ep−
EDV(1000:1および5000:1−EDV:細胞);Ep−EDV−682(5
00:1および1000:1−EDV:細胞);Ep−EDV−Dox(10,000:
1−EDV:細胞)、100pmol PNU−159682、5μMドキソルビシン、または培地単独を含む10%FBS(Bovogen)を補充した1mLのDMEM(Sigma)に再懸濁した。薬物およびEDV量は選択された濃度が処置後最初の24時間以内に細胞死の開始を生じるように、MTSおよびXCELLigenceリアルタイム実験を介して確立した。次いで、細胞をPBSで十分に洗浄して、任意の非接着性EDVまたは過剰の薬物を除去した。処理した腫瘍細胞を、RAW264.7またはBMDCのいずれかで、腫瘍細胞: RAW264.7/BMDC/JUT IIの1:1の比率で一夜培養した。上清をELISA分析のために回収した。RAW264.7/腫瘍細胞共培養物を、細胞スクレーパーを使用して収集し、そして試料を、DAPI(Sigma)、抗CD45 Brilliant Violet 510(BioLegend)、抗CD86 APC−Cy7、および抗CD206 AF488で染色し、そしてフローサイトメトリーによって評価した。JAWSII/腫瘍細胞およびBMDC/腫瘍細胞共培養物をバーゼンで収集し、DAPI(Sigma)、CD11bAF488(Abcam)、CD11c PE(Molecular Probes)、抗CD45Brilliant Violet 510またはPECy5(BioLegend)、anti-CD86 APC−Cy7、MHCクラスII PECy5(Thermo Fisher)、MHCクラスII Brilliant Violet421(BioLegend)、7−AAD(BioLegend)、および/またはCD80PE(Thermo Fisher)で染色し、フローサイトメトリーによって評価した。RNAeasy Plus Mini Kit (Qiagen)を製造者のプロトコールに従って使用して、BMDC/腫瘍細胞共培養物からRNAを抽出した。簡単に述べると、細胞を溶解し、RLT緩衝液中でホモジナイズし、gDNAエリミネータースピンカラムに通した。70%エタノールをフロースルーに添加し、次いで試料をRNeasyスピンカラムに通し、洗浄し、RNaseを含まない水中で溶出した。RNA濃度はエッペンドルフ生物光度計プラスで測定した。RNAを使用して、製造者のプロトコールに従ってSuperScriptTMVILOTMCDNAシンセシスキット(サーモフィッシャー)を使用してCDNAを逆転写した。転写されたCDNAをqPCRのために1:2に希釈した。各qPCR反応は、5μL TaqMan fast advancedmaster mix(Thermo Fisher),0.5μL 20X Taqmanプライマー/プローブ混合物(IFNα Mm03030145_gH、IFNb1 Mm00439552_s1、GAPDH Mm99999915_g1; Thermo Fisher)および2.5μLの水を含んでいた。8μLの混合物および2μLのCDNAを96ウェルプレートに添加した。qPCRは、Applied Biosystems Real-TimEp CR Systemを用いて行った。データをエクセルにエクスポートし、ΔΔCtから相対定量を計算した。
従って、AEC 1/2016、AEC 14/2016、AEC 15/2011、およ
びAEC 11/2017の下で実施した。4T1およびCT26Ep12.1モデルに
ついては、6〜8週齢の雌BALB/cマウスをAnimal Resources Centreから得た。T84およびA549/MDRモデルについては、5〜7週齢のBALB/c Fox1nu/ARCを動物資源センターから入手した。少なくとも1週間の観察後に、50μLのPBS当たり5×10 4 の4T1細胞を右側の第3の乳房脂肪パッドに、または100μLのPBS当たり2×10 5 のCT26Ep12.1をBALB/cマウスの右側腹部に皮下注射した。ヒト異種移植片については、100μLのPBS/Matrigel(シグマ)あたり5×106 A549/MDRまたは1×107T84を右側腹部に河下注射した。4T1モデルに対する腫瘍誘発後7日目、平均腫瘍径が約90mm3であった場合には治療を開始し、CT26Ep12.1モデルについては9日目に、平均腫瘍径が約125mm3 のマウスを、生理食塩水、1×109 EpCAM標的EDV(Ep−EDV)、またはPNU−159682(Ep−EDV−682)を負荷した1×109 EpCAM標的EDVのうちの1つを用いて、週3回、2週間、腹腔内静脈注射により治療した。腫瘍を週3回測定し、腫瘍体積をπ/6(長さ×幅×高さ)として計算した。2週間の期間の終わりに、マウスを人道的に安楽死させ、ex vivo分析のために腫瘍および脾臓を採取した。A549/MDRおよびT84腫瘍の治療は、腫瘍がそれぞれ100〜120mm3および120〜150mm3に達したときに開始した。マウスを、生理食塩水、ドキソルビシン(EGFR−EDV−Dox)を負荷した1×109 EGFR標的EDV、PNU−159682(EGFR−EDV−682)を負荷した1x109 EGFR標的EDV、またはPNU−159682(EDV−682)を負荷した1×109 非標的EDVで処理した。
、秤量し、そしてgentleMACS(商標)Dissociatorを使用して、製造業者の指示に従って
37で組織解離キット(Miltenyi Biotec)を使用して酵素的に消化した。解離後、赤血球溶解緩衝液(Sigma)を用いて赤血球を除去した。洗浄後、細胞を70μm細胞ストレーナーに通して、凝集塊を除去した。CD11b+細胞を、MACS分離器(Miltenyi Biotec)上のLSカラム上のCD11b MACSビーズ(Miltenyi Biotec)を使用する陽性選択によって精製した。単離されたCD11b+細胞集団の純度を、APC抗マウスCD11b(Biolegend)を用いたフローサイトメトリーによって評価し、約80%純粋であることが示された(図39A)。
ナイズし、70μmメッシュストレーナーを通して濾過して、単細胞懸濁液を得、続いて、RBC溶解緩衝液を使用して赤血球溶解した。次いで、脾細胞を洗浄し、細胞数を実施してから、NKまたはCD8+ T細胞単離に進行した。NK細胞およびCD8+ T細胞を、NK細胞単離IIキット(Miltenyi Biotec)またはCD8a+ T細胞単離キット(Miltenyi Biotec)のいずれかを使用して、製造業者の説明書に従って、ネガティブ選択によって解離脾臓細胞から単離した。MACSセパレーター(Miltenyi Biotec)上のLSカラムを用いて細胞を分離した。NK細胞およびCD8+ T細胞調製物をフローサイトメトリーによって評価し、NK細胞純度は一貫して90%よりも高かったが(図39B)、CD8+ T細胞純度は一貫して86%よりも高かった(図39C)。NK細胞を、NK細胞媒介細胞溶解アッセイの前に、37℃で10% FBSを補充したRPMI−1640倍地中で一晩休止させた。CD8+ T細胞を、単離直後に腫瘍細胞に加え、CD8+ T細胞溶解を評価した。
溶解を監視した:細胞増殖特性および腫瘍細胞死を、xCELLigence DPシステムによりリアルタイムで監視した。そうするために、組織培養ウェルの基部を覆う円形電極は電気インピーダンスの変化を検出し、インピーダンス値を細胞指数(CI)に変換する。細胞指数測定値は、細胞接着の強度および細胞数に直接対応する。標的細胞(4T1、CT26Ep12.1、A549/MDR、またはT84)をEプレート16に播種した。細胞を付着させ、それらが対数増殖期に達するまで増殖させた。エフェクター細胞(CD11b+細胞、NK細胞又はCD8+ T細胞)は、5:1(CD11b+:腫瘍細胞)、20:1(NK細胞:マウス腫瘍細胞)、10:1(NK:ヒト腫瘍細胞)及び30:1(CD8+ T細胞:腫瘍細胞)のエフェクター/標的細胞比で標的細胞に添加された。エフェクター細胞を加えた後、このシステムは3〜4日間、定期的に測定し(5分または15分毎)、免疫細胞が仲介する腫瘍細胞の殺傷をモニターした。
溶解後、細胞をMACS緩衝液(Miltenyi Biotec)中で1:10のFcブロックと共に10
分間インキュベートした。10分間インキュベートした後、細胞を1回洗浄し、MACS緩衝液中の一次抗体パネルと共に、暗所で氷上で15分間インキュベートした。細胞を2回洗浄し、次いでフローサイトメトリー分析のために緩衝液に再懸濁した。T細胞、NK
細胞およびマクロファージ染色パネルでは以下の抗体が用いられた。:anti-CD45 P
ECy7 (BioLegend)、anti-CD45 BV510 (Biolegend)、anti-CD3e APC
−eFluor780 (eBioscience)、 anti-CD3 APC (Molecular Probes)、anti-CD
4 PE―TR (Abcam)、anti-CD8a FITC (eBioscience)、anti-CD8 BV5
10 (BioLegend)、anti-CD25 PE (Abcam)、anti-CD314 (NKG2D)、PE−e
Fluor610 (eBioscience)、anti-CD335 (NKp46)PECy7 (BioLegend)、a
nti-CD27 BV421 (BioLegend)、ant-CD183 (CXCR3) BV510 (BioLegend
)、anti−NKG2A/C/E FITC (eBioscience)、anti-CD11b APC (BD Pha
rmingen)、anti-Ly6C FITC(BioLegend)、 anti-Ly6G BV510 (BioLege
nd)、anti-F4/80 PE Dazzle594 (BioLegend)、anti-CD206 PEC
y7 (BioLegend)およびanti-CD86 APC―Cy7 (BioLegend)。単一染色対照およ
び/またはversacomp(Beckman Coulter)ビーズを蛍光補償のために使用した。DAPI (
Sigma)、ヨウ化プロピジウム(Sigma)、DRAQ5(Thermo Fisher)、またはlive/Dead Yellow(Thermo Fisher)を生細胞検出に使用した。未染色およびアイソタイプ対照を用いて、自己蛍光量を決定し、抗体特異性を確認した。
イトカインおよびケモカインレベルを測定するために、腫瘍を全ての皮膚を除去して注意
深く解剖し、無血清培地に入れ、重量を測定した。次いで、腫瘍を、エッペンドルフ mic
ropestles(Sigma)を使用して穏やかに破壊し、大きな断片が見えないことを確実にした。
細胞懸濁液を遠心分離し、上清を収集し、分析まで−80で保存した。脾細胞/腫瘍細胞
共培養のために、脾臓を解離させ、腫瘍を以前に記載されたように酵素的に消化した。次
いで、同じマウス同様の脾細胞および腫瘍を、10:1の比率で組織培養プレートに入れ
(脾細胞:腫瘍細胞)、72時間まで培養した。上清を24、48および72時間で回収
し、分析まで−80で保存した。マウスIL−1β、TNF−α、IL−2、IL−4、
IL−6、IFNα、IFNβ、RANTESおよびMIP−1αについて、製造業者の指示に従って腫瘍および脾細胞上清を分析した。IFNαキットはPBL Assay Scienceから入手し、IL−β、ATNF−α、IL−2、IL−4、IL−6、IFNγ、ARANTES(CCL5)およびMIP−1α(CCL3)デュオセットキットはR&Dシステムから入手した。各ELISAは、3,3’,5,5’−テトラメチルベンジジン(Sigma)基質を用いて開発した。マイクロウェルプレートを、Biotek uQuantプレートリーダーにおいて、参照波長として540nmを用いて450nmで読み取った。KCジュニアソフトウェアを使用して、4つのパラメータロジスティック曲線を標準に適合させ、試料を補間した。各アッセイの最小検出可能濃度(MDC)は、応答のs.d.に10を掛け、変曲点における標準曲線の勾配で割ることによって計算した。
Bは、以前に膵管腺癌(PDAC)に対して膵臓、胆、十二指腸、脾臓および胃の一部と
周囲リンパ節を除去するための完全なWhipple法を受けていた。膵頭部と膵尾部に腫瘍を
認め、ステージ IVと診断した。他の施設でゲムシタビンとその後のFOLFIRINOXによる治療を受けたが、治療中に肝臓に広範な転移性疾患を発症していた。彼女の化学療法の終了時に、ホイップル処置の16ヶ月後に、彼女は全ての治療選択肢を使い果たし、彼女の体重は62kgから45kgに減少し、彼女はオーストラリア治療薬局(TGA)コンパシオン酸塩使用スキームの下で投与することができる実験的EDV治療を求め、中皮腫(van Zandwijkら、2017)および再発性神経膠芽腫(Whittleら、2015)についての第I相試験において以前に試験されていた。シドニーのRoyal North Shore Hospitalの腫瘍科病棟で、CEBを第1サイクルとして週2回、7週間投与した。しかしながら、彼女の弱体化した状態のために、そしてEDVに固有のリポ多糖に対する耐性を潜在的に構築するために、用量は、サイクル内でゆっくりと増加した(以下の表17)。
前投薬が与えられた(van Zandwijkら、2017)。血清生化学的検査、血液学的検査および
サイトカイン発現を、各投与の前と3時間後に評価した。CA19−9およびC反応性タ
ンパク質量を少なくとも週2回モニターした。末梢血単核細胞(PBMC)を、抗腫瘍免
疫細胞数の変化について、投与前およびサイクル終了時にフローサイトメトリーにより調
べた。最初の外科的切除から腫瘍組織を得て、PDAC細胞を培養し、薬物感受性および
表面受容体発現について試験した。
データは、平均±標準偏差(SD)または平均の標準誤差(SEM)として表され。2
群間の統計的有意性は、スチューデントのt検定によって決定した。3群以上の群間の統
計学的有意差は、一元配置分散分析、続いてTukeyの多重比較検定により判定した。腫瘍
退縮研究の意義は、二元配置ANOVAに続いてTukeyの多重比較検定により決定した。すべての検定について、p値は以下のとおりであった:* p0.05、** p0.01、*** p0.001、および**** p0.0001。
介するαGCの表層提示
この実施例は、癌細胞に対するαGCおよび遊離αGCのEDV送達を対比する。
)。
側トレイリザーバを、P1000ピペットを使用して溶融1%アガロースで満たした(1
gのアガロースを100mlの水に溶解し、マイクロ波で溶解し、〜50に冷却させた)。
プレートを乾燥させ、室温で少なくとも30分間沈降させた。次いで、懸滴プレートの外
側ウェルを、50μLの無菌細胞培養培地(細胞なし)/ウェルで満たした。
αGC(陽性対照);未処理細胞と比較した空のミニ細胞およびミニ細胞 αGC で処理し、処理後8時間、16時間、24時間および48時間で収集した(図46A〜46D)。
25またはT75フラスコ中で増殖させた。ピペット助剤を用いてピペッティングするこ
とにより、培養培地を滅菌50ml管に注意深く収集し、フラスコの培養表面を5mlの滅菌
PBSで2回洗浄し、各洗浄後に同じ滅菌50ml管に収集した。接着細胞を、5mlの0.2
5%トリプシン/EDTAの添加によって収集し、そして37で3分間、または全ての細
胞がフラスコの表面から持ち上げられるまでインキュベートした。持ち上げた細胞を、ピ
ペット補助を用いて穏やかにピペッティングすることによって、注意深く単一細胞に分け
、前の工程で使用した試料無菌50ml管に移した。次いで、細胞懸濁液を300gで7分
間遠心分離し、上清を注意深くデカントした。細胞ペレットを、チューブの底を指でフリ
ックすることによって解離させ、5mlの予め温めたJAWSII培養培地に再懸濁した。
細胞懸濁液を、ピペット補助を用いて注意深くピペッティングすることによって、単一細
胞にさらに解離させた。細胞数を決定するために、10μLの細胞懸濁液を10μLのトリパンブルー溶液と混合し、EVE自動細胞カウンターを用いて分析した。
た。5×104JAWSII細胞および5×108ミニ細胞(ミニ細胞対細胞の比率1:1
000)をそれぞれの処理試料に使用し、JAWSII細胞培養培地中で総量50μLで培養した。アイソタイプ対照のために、余分な未処理試料を調製した。適量のミニ細胞を7分で12,000gで遠心法によりペレット化し、ピペットで上清を注意深く除去した。適切な量の生きたJAWS細胞(前のセクションからの細胞数に基づく)を、ペレット化されたミニ細胞に添加した。次いで、穏やかなピペッティングによって、ミニ細胞をシングルミニ細胞−細胞懸濁液に解離させた。次いで、滅菌培養培地を添加することによって、各サンプルの最終容量を作製した。未処理試料およびαGC処理試料については、5×104のJAWSII細胞を各サンプル使用し、総体積50μLのJAWSII細胞培養培地中で培養した。適切な量の生きたJAWSII細胞をエッペンドルフチューブに移した。次いで、滅菌培養培地を添加することによって、各サンプルの最終容量を作製した。1000ng/mLのαGCを、1000ng/mLのαGC(陽性対照)処理群で処理したJAWSII細胞のための細胞懸濁液に直接添加した。次いで、試料を、50μLの処理懸濁液/ウェルで懸滴プレートの各々のウェルに注意深く播種し、そして収集まで5% CO2で37℃でインキュベートした。
この実施例は、腫瘍に対するミニ細胞含有治療薬およびミニ細胞含有インターフェロン
II型アゴニストの効力を例示する。この結果は、インターフェロンI型アゴニストを欠く
組成物が腫瘍を効果的に処置するために使用され得ることを実証する。
0μLのPBS当たり2×105細胞を皮下注射することによって確立した。腫瘍は移植後
8日以内に約125mm3の開始体積まで増殖した。マウスを各投与群につき8匹のマウスで無作為に群分けした。Epミニ細胞Dox、ミニ細胞α-GC、およびEpミニ細胞+ミニ細胞α-GC組合せ)を用いて、生理食塩水処理のみと比較した。
〜800mm3腫瘍については16時間および24時間後に屠殺した(図44)。
組み合わせが腫瘍に対する有効性を実証することを示している。この結果は、インターフ
ェロンI型アゴニストを欠く組成物が、腫瘍を効果的に処置するために使用され得ること
を実証する。
物に様々な修正および変形を行うことができることが明らかであろう。したがって、本発
明は添付の特許請求の範囲およびその均等の範囲内に入る限り、本発明の修正および変形
を包含することが意図される。
Ablasserら、Nat., Immunol., 10(10):1065−72(2009)。
Ablasser ら、Nature., 498:380-384 (2013a)。
Ablasserら、Nature., 503:530−534(2013b)。
Adamusら、Contemp. Oncol (Ponzn), 22(1A):56−60(2018年3月)。
Aduro Biotech Inc.(2016),Novartis Pharmaceuticals. 進行/転移性固形腫瘍または
リンパ腫患者におけるMIW815(ADU−S100)の安全性および有効性の研究。臨床試験gov
[インターネット]、Bethesda(MD:米国国立医学図書館(US)、識別子: NCT02675
439。https://ClinicalTrials.gov/show/NCT02675439から入手可能、(2016年7月01日引
用)。
Ahmadzadehfar ら、「イットリウム-90微小球体による肝腫瘍のラジオ塞栓形成」 Sem
in Nucl Med. 2010;40(2):105-121。
Ahmadzadehfarら、「去勢抵抗性転移性前立腺癌の177Lu−PSMA−DKFZ−617を用いた反復
放射性リガンド療法の治療反応性および副作用」、Oncotarget.2016;7(11):12477-12488
。
Alexopoulou ら、Nature., 413:732−738(2001)。
Alzahrani AS、AlShaikh O、Tuli M、Al-Sugair A、Alamawi R、Al-Rasheed MM. 「遺伝
子組換えヒトチロトロピンの診断価値は、差別化された甲状がん患者のフォローアップに
おいて、123I全身断層撮影を刺激した」Clin Nucl Med. 2012;37(3):229-234。
Andersson L、Blomberg L、Flegel M、Lepsa L、Nilsson B、Verlander M、「ペプチド
の大規模な合成」、 Pept Sci. 2000; 55:227-250。
Anguilleら、Pharmacological Reviews 67., 731-753(2015)。
Barberら、Curr. Opin. Immunol., 23(1):10−20(2011)。
Belardelliら、TRENDS in Immunology 23, 201−208 (2002)。
Bernardiniら、Frontiers in immunology 7, 402(2016)。
Birkholzら(2015)、J Biol Chem.「T細胞免疫機能のガラクトシルセラミドのT細胞免疫機能のアルファとオメガ」. NIH.gov [Internet]. Bethesda(MD):Uni
ted States National Library of Medicine(米国)、識別子: PMC4505449、https://www.
ncbi.nlm.nih.gov/pmc/articles/PMC4505449/から利用可能。
Bobangaら、Oncoimmunology 7, e1393598。
Bredel、Brain Res.Rev., 35:161(2001)。
Brittonら、Genes Dev., 12:1254-9 (1998)。
Brodyら、J. Clin. Oncol., 28:4324−4332(2010)。
Burckstummerら、Nat. Immuno., 10:266−272(2009)。
Burger M、Hartmann T、Krome M、Rawluk J、Tamamura H、Fujii NKipps TJ、Burger JA.
「CXCR4ケモカイン受容体(CD184)の低分子ペプチド阻害剤は、慢性リンパ性白血病B細胞におけるCXCL12の活性化、遊走、および抗アポトーシス反応に拮抗する」、Blood.2005; 106:1824-1830。
Caplen.N.J., Expert Opin. Biol. Ther., 3; 575−86(2003)。
Caplen and Mousses、Ann. NY Acad. Sci., 1002:56−62(2003)。
Caravella and Lugovskoy, Curr. Opin. Chem. Biol., 14: 520−28(2010)。
Carrenoら、Clin Transl. Immunology, 5(4): e69(2016)。
Caskeyら、J.Exp. Med., 208:23572366(2011)。
Cauwelsら、Cancer research 78, 463−474 (2018)。
Chatalic KLS、Kwekkeboom DJ、de Jong M、「イメージングと治療のためのラジオペプチ
ド:輝いている未来」、J Nucl Med. 2015; 56:1809-1812。
Chenら、Int. J. Cancer, 93: 107(2001)。
Chikumaら、Cancer Sci, 108: 574−580 (2017)。
Chiuら、Cell, 138:576−591(2009)。
Chuら、PLoS Biology, 4: 1122−36(2006)。
Civrilら、Nature, 498:332−337(2013)。
Clark-Curtiss and Curtiss、Methods Enzymol、101: 347−362 (1983)。
Colonnaら、Nat. Immunol., 5:1219−1226(2004)。
Corralesら、Cell Rep., 11:1018−1030 (2015)。
Coryら、Cancer Commun.,3(7):207−12(1991)。
D'Aloiaら、Cell death & disease 9、282 (2018)。
D'Angiolellaら、Cell, 149:1023−34 (2012)。
Da Silvaら、Breast Cancer Res.,12: R46(1−13)(2010)。
Debinskiら、J. Neurooncol., 48: 103−11(2000)。
Debinski and Gibo、Mol. Med., 6: 440−49(2000)。
de Boerら、J. Bacteriol., 174(1): 63−70 (1992)。
Deutscher SL、「癌の分子イメージングおよび診断におけるファージディスプレイ」、Ch
em Rev., 2010; 110:3196−3211。
Dineら、Asia-Pacific journal of oncology nursing 4, 127−135(2017)。
Dobbsら、Cell Host Microbe, 18(2):15−24(2015)。
Dongら、International journal of molecular sciences 17, 320(2016)。
Dowlingら、PLoS One, 8:e58164(2013)。
Dredgeら、Cancer immunology、immunotherapy: CII 51、521-531 (2002)。
Duanら、Mol., Cancer Ther, 3:833−8(2004)。
Duxburyら、Ann. Surg., 240:667−74(2004)。
Dynavax Technologies Corporation (2016)、「未治療の低悪性度B細胞リンパ腫患者を
対象とした限局性低線量放射線療法との併用におけるSD-101の試験。2016. 臨床試験g
ov [インターネット]。Bethesda(MD):米国国立医学図書館(US)、識別子、 NCT022
66147。https://ClinicalTrials.gov/show/NCT02266147から入手可能、(2016年7月01日
引用)。
Emensら、European journal of cancer 81, 116−129 (2017)。
Erathodiyil N、Ying JY. 「バイオイメージング用途のための無機ナノ粒子の官能化」、
Acc Chem Res. 2011; 44:925-935. [PubMed: 21648430]。
Fangら、Seminars in immunology 31, 37−54 (2017)。
Farkonaら、BMC medicine 14,73(2016)。
Ferlazzoら、The Journal of Immunology 172, 1333−1339 (2004)。
Fernandes-Alnemriら、Nature, 458:509-513 (2009)。
Fieldら、Proc., Natl Acad. Sc IUSA,58:1004−1010(1967)。
Fitzgerald-Bocarsly ら、Biochimie 89, 843−855 (2007)。
Fuら、Sci. Transl. Med., 7(283):283ra252(2015)。
Fukuda、Curr. Protocols Molec. Biol.(Suppl. 26)、17.5.1−17.5.8(1994)。
Gaoら、Nat Biotechnol., 22(8):969−976(2004)。
Gaoら、Science, 341:903−906(2013a)。
Gaoら、Cell, 153:1094−1107(2013b)。
Gerard SK、Cavalieri RR、 「画像診断によるI−123甲状腺腫瘍全身スキャン 6、24
、48時間後」、Clin Nucl Med. 2002;27(1):1-8。
Ghosh A、Heston WDW、「 前立腺癌における腫瘍標的前立腺特異膜抗原(PSMA)とその制御」、J Cell Biochem. 2004;91(3):528-539。
Gitlinら、Proc. Natl Acad. Sci. USA, 103: 8459−8464(2006)。
Goh and Sorkin, Cold Spring Harb. Perspect. Biol., 5: a017459 (2013)。
Gray BP, Brown KC、「ペプチド組合わせ図書館:細胞結合ペプチドについての採鉱」、C
hem Rev. 2013; 114: 1020−1081。
Gregoryら、Methods in Molecular Biology, 342: 33−47(2006)。
Guptaら、「摘要 CT091: MEDI9197、アゴニスト、 TLR 7/8 アゴニスト の安全
性と薬力学的活動」、 Cancer Research、AACR Annual Meeting 2017; April 1−5, 2017
(2017年7月発行)。
Hansenら、EMBO J., 33(15):1654−66(2014)。
Harry E.J., Mol. Microbiol.,40(4):795-803(2001)。
Hershey, J. Allergy Clin. Immunol., 111: 677-90(2003)。
Hiragaら、J. Bacteriol., 171: 1496−1505 (1989)。
Hobbsら、Proc. Natl. Acad. Sci. USA, 95(8):4607−4612(1998)。
Holman BL、Tumeh SS. 単光子放出計算トモグラフィー(SPECT):応用とポテンシャル、JA
MA.1990;263(4):561-564。
Hornungら、Nature., 458:514−518(2009)。
Hu & Lutkenhaus、Mol., Microbio., 34(1):82−90(1999)。
Iftodeら、Crit. Rev., Biochem., Mol. Biol., 34: 141−80(1999)。
Igarashi H、Fujimori N、Ito T、Nakamura T、Oono T、Nakamura K、Suzuki K、Jensen
RT、Takayanagi R、血管作動性腸ペプチド(VIP)とVIPレセプター-治療的応用のための構造と機能の説明、 Int J Clin Med. 2011; 2:500-508。
Immune Design (2016)、Merck Sharp & Dohme Corp. 濾胞性非ホジキンリンパ腫患者に
おけるペムブロリズマブ併用または非併用の腫瘍内G100の研究、2017. 臨床試験gov [インターネット]、Bethesda(MD):米国国立医学図書館(US)、識別子: NCT02501473、https:// ClinicalTrials.gov/show/NCT02501473から入手可能(2016年7月01日引用)。
Jarboeら、Cancer Res.,67: 7983−86(2007)。
Jenkinsら、British journal of cancer 118、9−16 (2018)。
Jungら、Translational oncology 11、686−690 (2018)。
Kaoら、Am. J., 191(12):1467−1469(2015)。
Kaoら、American Journal of Respiratory and Critical Care Medicine 191, 1467
−1469 (2015)。
Kawai and Akira、Nat. Immunol., 11:373−384(2010)。
Khalilら、Proc Nat'l Acad. USA, 106: 11667−72(2009)。
Kimら、Proc. Natl. Acad. Sci. USA, 107:15181−15186 (2010)。
Kotaら、Cell, 137: 1005−17(2009)。
Kramer-Marek G、Capala J、現代の癌治療における核医学の役割、Tumour Biol. 2012;33
(3):629-640。
Kranzuschら、Cell REp.,3:1362−1368(2013)。
Kriegら、Nature, 374:546−549(1995)。
Kwekkeboom DJ, de Herder WW, Kam BLら、放射性標識ソマトスタチン類似体[177 Lu−D
OTA 0、Tyr3]オクトレオテートによる治療:毒性、有効性、および生存、J Clin Oncol.
2008;26(13):2124-2130。
Landskronら、Journal of immunology research 2014, 149185 (2014)。
Leeら、Cancers 3, 3856−3893 (2011)。
Lemmon and Schlessinger、Cell, 141(7): 1117−134 (2010)。
Leung and Amarasinghe、Curr. Opin. Struct. Biol., 36:133−141(2016)。
Liら、Science,341:1390−1394(2013b)。
Liuら、Science,347(6227):aaa2630(2015)。
Luら、Structure,18:1032−1043(2010)。
Maら、Mol. Microbiol., 54: 99−108(2004)。
MacDiarmidら、PLoS One, 11(4)(2016)。
MacDiarmidら、Nature biotechnology 27, 643−651 (2009)。
MacDiarmidら、Cell cycle 6, 2099−2105 (2007a)。
MacDiarmidら、Cancer cell 11, 431−445 (2007b)。
Majkowskaら、「低エネルギー・ベータ放射体47Scの複合体と骨の痛み治療のためのゾレドロン酸による177Lu」、Appl Radiat Isot. 2009;67(1):11-13。
Mankanら、EMBO J., 33:2937−2946(2014)。
McWhirterら、J. Exp. Med., 206:1899−1911(2009)。
Marqら、J Biol. Chem., 286:6108−6116(2011)。
Matsuno M、Matsui T、Iwasaki A、Arakawa Y、.ガストリン分泌におけるアセチルコリン
とガストリン放出ペプチド(GRP)の役割、J Gastroenterol、1997; 32:579-586。
MedImmune LLC (2016)、固形癌患者に投与したMEDI9197の検討、2018. 臨床試験gov [
インターネット]。Bethesda(MD):米国国立医学図書館(US)、識別子: NCT02556463
、https://ClinicalTrials.gov/show/NCT02556463から入手可能、(2016年7月01日引用)
。Mellman ら、Nature 480、480−489(2011)。
Merrifield R、固相ペプチド合成、Adv Enzymol Relat Areas Mol Biol.2006; 32:221-296。
Meulen and Brady、Hum. Vaccin. Immunother., 13(1):15−16(2017)。
Mhawech-Fauceglia P、Zhang S、Terracciano L、ら、 正常および腫瘍組織における前立
腺特異的膜抗原(PSMA)蛋白質発現と前立腺腺癌におけるその感受性および特異性:多重
腫瘍組織マイクロアレイ法を用いた免疫組織化学的研究、Histopathology. 2007;50(4):4
72-483。
Morvan ら、Nature reviews Cancer 16, 7-19(2016)。
Mullerら、Frontiers in immunology 8, 304(2017)。
Muller C, Zhernosekov K, Koster U ら、PETおよびSPECT、ならびにα及びβ放
射性核種療法のためのユニークな四組適合テルビウム放射性同位元素:新しい受容体標的
葉酸誘導体を用いたin vivo概念実証研究。J Nucl Med. 2012;53(12):1951-1959。
シドニー・オーストラリア大学NHMRC臨床試験センター:シドニー(NSW):(2017)識別子ACTRN12617000037303 抗ヒトEGFR(ベクティビックス配列)標的EDVと免疫調節補助剤(EDV−40mer)を担持する同時非標的EDVを担持する細胞毒性薬物PNU−159682(EGFR(V)−EDV−PNU)の第1相試験2017年1月10日; https://www.anzctr.org.au/ACTRN12617000037303.aspx。
Nielsenら、Biochim. Biophys. Acta,1591(1−3), 109−118(2002)。
Niethら、FEBS Lett、545: 144−50 (2003)。
Oh and Park、Advanced Drug Delivery Rev, 61: 850−62 (2009)。
Ohki-Hamazaki H、Iwabuchi M、Maekawa F、ボムシン様ペプチドおよびそのレセプターの
発展と機能、Int J Dev Biol. 2005; 49:293-300。
Oisethら、Journal of Cancer Metastasis and Treatment 3、250 (2017)。
Okadaら、J. Bacteriol., 176:917−22 (1994)。
Okanoら、J. Am. Chem. Soc., 128:7136−37(2006)。
Oncovir Inc(2016)、National Institutes of Health、Icahn School of Medicine at
Mount Sinai、Bay Hematology Oncology、Emory University、University of Pittsburgh
、National Cancer Institute、腫瘍内ヒルトノール(登録商標)による固形癌に対するIn
Situ.自家治療ワクチン接種、2018臨床試験gov [インターネット]、Bethesda(MD):
米国国立医学図書館(US)、識別子: NCT02423863。https:// ClinicalTrials.gov/show/
NCT02423863から入手可能、(2016年7月01日引用)。
Oritz-Zapater ら、Nature Medicine, 18(1):83−90 (2011)。
Orzalliら、Proc. Natl. Acad. Sci. USA, 109: E3008−E3017(2012)。
Parkら、Breast Cancer Res.,4(3):95−99(2002)。
Palmedo H、骨転移の放射性核種療法、In: Clinical Nuclear Medicine. Berlin, Heidel
berg: Springer Berlin Heidelberg; 2007:433-442。
Pillaiら、「放射性核種治療のための177Luの生産ロジスティックス」 Appl Radiat Iso
t. 2003; 59(2-3):109-118。
Quintieriら、Clinical Cancer Research 11、1608−1617 (2005)。
Raskin & de Boer、J.Bacteriol., 181: 6419−6424 (1999)。
ReeveおよびCornett、J. Virol., 15: 1308−16(1975)。
Reidら、Annals of oncology : official journal of European Society for Medical On
cology 24, 3128−3135 (2013)。
Rezvaniら、分子治療法 、the journal of the American Society of Gene Therapy 25,
1769−1781 (2017)。
Riceら、Semin. Nucl. Med., 41: 265−282(2011)。
Ruoslahti E.、 RGDおよびインテグリンに対する他の認識配列、Annu Rev Cell Dev Biol
. 1996; 12:697-715。
Sagnellaら、Molecular cancer therapeutics 17, 1012−1023(2018)。
Santoni M、Scarpelli M、Mazzucchelli R. ら. 前立腺癌における個別化治療のための前
立腺特異的膜抗原を標的とする:形態学的および分子的背景と将来の見込み、J Biol Reg
ul Homeost Agents. 2014;28(4):555-563。
Sawa-Wejksza ら、Archivum immunologiae et therapiae experimentalis 66、97−111
(2018)。
Sazar、「自然宿主防御の起動 : ヒルトノール(ポリICLC)と悪性脳腫瘍」、Oncovir、
Inc.、www.oncovir.com/id2 (2018年7月11日アクセス)。
Sharmaら、 Cell 168, 707−723 (2017)。
Sharpe、Immunological reviews 276、5−8 (2017)。
Showalter、Cytokine 97, 123−132 (2017)。
Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C、正常および悪性ヒト組
織における前立腺に特有の膜抗原表現、 Clin Cancer Res. 1997;3(1):81-85。
Simmonsら、The Journal of Immunology 188, 3116−3126 (2012)。
Singh M、Mukhopadhyay KAlpha-melanocyte stimulating hormone: 新興の抗炎症性抗菌
ペプチド、Biomed Res Int. 2014; 2014:874610。
Sioud, M., Trends Pharmacol. Sci., 25: 22−8(2004)。
Schogginsら、Nature,505:691−695(2014)。
Solomonら、PLos One,10: 1−17(2015)。
Staudacher ら、British journal of cancer 117, 1736−1742 (2017)。
Strand、FL.神経ペプチド:生理学的過程の調節因子、MIT press; 1999。
Stewart and D'Ari、J Bacteriol., 174:4513−6 (1992)。
Sunら、Science,339(6121):786−791(2013)。
Sunら、Biochem., Biophys. Res. Commun., 280:788(2001)。
Sun X、Li Y、Liu T、Li Z、Zhang X、Chen X、がん検診のためのペプチドベースの画像助剤、 Adv Drug Deliv Rev. 110−111: 38−51 (2017)。
Szkanderaら、British journal of cancer 110, 183−188 (2014)。
Takahashi H、Emoto K、Dubey M、Castner DG、Grainger DW. イメージング表面固定化化
学:非接着性ヒドロゲル薄膜上の細胞パターン化との相関、Adv Funct Mater. 2008; 18:
2079-2088。
Takaokaら、Nature, 448:501−505(2007)。
Takeshitaら、Molec. Ther., 18: 181−87(2010)。
Tanpureら、Bioorg. Med. Chem., 21: 8019−32(2013)。
Tatemoto, K、神経ペプチドY: 神経ペプチドYおよび関連ペプチドの歴史と概要、Sprin
ger; 2004、p1−21。
Teunissenら、「消化管の内分泌腫瘍ペプチド受容体放射性核種療法」、Best Pract Res
Clin Gastroenterol, 2005;19(4):595-616。
Tyler-McMahon BM、Boules M、Richelson ENeurotensin: peptide for next millenn I
Um. Regul Pept. 2000; 93:125-136。
Unterholznerら、Nat. Immunol., 11:997−1004(2010)。
Unterholznerら、Immunobiology, 128(11):1312−21(2013)。
van Zandwijkら、Lancet Oncol、18(10): 1386−1396 (2017)。
van Zandwijkら、The Lancet Oncology 18、1386−1396 (2017)。
Ventola、Pharmacy and Therapeutics 42、452−463 (2017)。
Wallace ら、Springer seminars in immunopathology 27, 49−64 (2005)。
Walrand S、Hesse M、Renaud L、Jamar FX線塞栓術後のPET/CT 90Y線量測定に対する画像再構成の偏りの影響、J Nucl Med, 2015;56(3):494-495。
Wangら、Nat. Struct. Mol. Biol., 17:781−787(2010)。
Wangら、Immunity, 41(6):919−33(2014)。
Weckbecker G、Lewis I、Albert R、Schmid HA、Hoyer D、Bruns C、ソマトスタチン研究
の機会:生物学的、化学的、治療的な側面、Nat Rev Drug Discov. 2003; 2:999-1017。
White & McCubrey、Leukemia、15: 1011−1021 (2001)。
Whittleら、J Clin. Neurosci., 22(12):1889−1894(2015)。
Whittleら、Journal of clinical neuroscience: official journal of Neurosurgical S
ociety of Australasia 22、1889−1894(2015)。
Wuら、Science, 339:826−830(2013)。
Wykoskyら、Clin Cancer Res., 14: 199−208(2008)。
Xiaら、Nat. Immunol., 16:366−375(2015)。
Yagueら、Gene Ther., 11: 1170−74(2004)。
Yang W、Luo D、Wang S、Wang R、Chen R、Liu Y、Zhu T、Ma X、Liu R、Xu G、TMTP1、
転移を特異的に標的化する新規な腫瘍帰巣性ペプチド、Clin Cancer Res. 2008; 14:5494
-5502。
Yangら、Nat. Immunol., 11:487−494(2010)。
Yiら、PLoS One, 8(10):e77846(2013)。
Yuanら、Scientific reports 5, 14273(2015)。
Zhangら、J Immunol., 186:4541−4545(2011a)。
Zhangら、Nat. Immunol., 12:959−965 (2011b)。
Zhangら、Cell REp., 6:421−430(2014)。
Zibertら、Human Gene Therapy 15, 21−34 (2004)。
Ziegler-Heitbrock ら、Frontiers in immunology 4、23 (2013)。
Zitvogelら、Nature reviews Immunology 15、405−414 (2015)。
米国特許第8,591,862号。
米国特許第7,183,105号。
U.S. 2008/0051469。
WO 2000/067776。
WO 2003/033519。
WO 2004/113507。
WO 2005/056749。
WO 2005/079854。
WO 2009/027830。
Claims (20)
- (a)少なくとも1つの抗腫瘍剤を含む、精製された無傷の細菌由来ミニ細胞の治療有効量、および
(b)インターフェロンI型アゴニスト、インターフェロンII型アゴニスト、またはインターフェロンI型アゴニストおよびインターフェロンII型アゴニストの組合せ、
を含む組成物。 - 前記組成物の要素(b)が、
(i)インターフェロンI型アゴニストを含む、精製された無傷の細菌由来ミニ細胞の治療有効量、または
(ii)インターフェロンII型アゴニストを含む、精製された無傷の細菌由来ミニ細胞の治療有効量、または
(iii)以下の組合せ:
(1)インターフェロンI型アゴニストを含む精製された無傷の細菌由来ミニ細胞の治療有効量、および
(2)インターフェロンII型アゴニストを含む精製された無傷の細菌由来ミニ細胞の治療有効量、
を含む、請求項1に記載の組成物。 - (a)前記抗腫瘍剤、およびインターフェロンI型アゴニスト、インターフェロンII型アゴニスト、またはインターフェロンI型アゴニストおよびインターフェロンII型アゴニストの組合せは、2つ以上の精製された無傷の細菌由来ミニ細胞内にパッケージ化され、または
(b)前記抗腫瘍剤、およびインターフェロンI型アゴニスト、インターフェロンII型アゴニスト、またはインターフェロンI型アゴニストおよびインターフェロンII型アゴニストの組合わせは、精製された無傷の細菌由来ミニ細胞の3つの別々の集団内にパッケージ化される、
請求項1または2に記載の組成物。 - 前記抗腫瘍剤、インターフェロンI型アゴニスト、およびインターフェロンII型アゴニストを含み、ここで、
(a)前記抗腫瘍剤、インターフェロンI型アゴニスト、およびインターフェロンII型アゴニストは、同じミニ細胞内に含まれ、
(b)前記抗腫瘍剤およびインターフェロンI型アゴニストは第1のミニ細胞内に含まれ、インターフェロンII型アゴニストは第2のミニ細胞内に含まれ、
(c)前記抗腫瘍剤およびインターフェロンII型アゴニストは第1のミニ細胞内に含まれ、インターフェロンI型アゴニストは第2のミニ細胞内に含まれ、
(d)前記抗腫瘍剤は第1のミニ細胞内に含まれ、インターフェロンI型アゴニストおよびインターフェロンII型アゴニストは第2のミニ細胞内に含まれ、または
(e)前記抗腫瘍剤は第1のミニ細胞内に含まれ、インターフェロンI型アゴニストは第2のミニ細胞内に含まれ、およびインターフェロンII型アゴニストは第3のミニ細胞内に含まれる、
請求項1〜3のいずれか1項に記載の組成物。 - 前記組成物が、インターフェロンI型アゴニストを含まない、請求項1〜4のいずれか1項に記載の組成物。
- (a)前記抗腫瘍剤は、放射性核種、化学療法薬、機能性核酸、および機能性核酸を転写することができるポリヌクレオチドからなる群から選択され、および/または
(b)前記抗腫瘍剤は、超毒性化学療法剤であり、および/または
(c)前記抗腫瘍剤は、モルホリニルアントラサイクリン、メイタンシノイド、デュカルマイシン、アウリスタチン、カリケアマイシン(DNA損傷剤)、α−アマニチン(RNAポリメラーゼII阻害剤)、センタマイシン、ピロロベンゾジアゼピン、ストレプトニグチン、窒素マスタード、ニトロソルエア、アルカンスルホネート、ピリミジン類似体、プリン類似体、代謝拮抗剤、葉酸類似体、アントラサイクリン、タキサン、ビンカアルカロイド、トポイソメラーゼ阻害剤、ホルモン剤、およびこれらの組合せからなる群より選択される超毒性化学療法剤であり、および/または
(d)前記抗腫瘍剤は、ネモルビシン、PNU-159682、イダルビシン、ダウノルビシン、カミノマイシン、およびオキソルビシンからなる群より選択されるモルホリニルアントラサイクリンである超毒性化学療法剤であり、および/または
(e)前記抗腫瘍剤は、PNU-159682である超毒性化学療法剤であり、および/または
(f)前記抗腫瘍剤は、siRNA、miRNA、shRNA、lincRNA、アンチセンスRNA、およびリボザイムからなる群より選択される機能性核酸であり、および/または
(g)前記抗腫瘍剤は、siRNA、miRNA、shRNA、lincRNA、アンチセンスRNA、およびリボザイムからなる群より選択される機能性核酸であり、ここで、機能性核酸は腫瘍細胞増殖、血管新生または化学療法に対する抵抗性を促進し、および/またはアポトーシスまたは細胞周期停止を阻害する遺伝子を阻害し、そして任意に、(i)siRNAは、リボヌクレオチドレダクターゼM1(RRM1)発現を阻害し、(ii)siRNAがポロ様キナーゼ1(Plk1)発現を阻害し、または(iii)miRNAがmiRNA16aである、
請求項1〜5のいずれか1項に記載の組成物。 - (a)インターフェロンI型アゴニスト、インターフェロンII型アゴニスト、またはインターフェロンI型アゴニストとインターフェロンII型アゴニストの組合せはオリゴヌクレオチドであり、および/または
(b)インターフェロンI型アゴニスト、インターフェロンII型アゴニスト、またはインターフェロンアI型ゴニストとインターフェロンII型アゴニストとの組み合わせはオリゴヌクレオチドであり、オリゴヌクレオチドは少なくとも約40ヌクレオチド、少なくとも約50ヌクレオチド、または少なくとも約60ヌクレオチドの配列を含み、および/または
(c)インターフェロンI型アゴニスト、インターフェロンII型アゴニスト、またはインターフェロンI型アゴニストとインターフェロンII型アゴニストの組み合わせはオリゴヌクレオチドであり、オリゴヌクレオチドはPNPase1、ポリ(I:C)、ポリ−ICLC、イミキモド、イミダゾキノリンエスキモド、cGAMPまたはCpG−オリゴデオキシヌクレオチドのポリヌクレオチド産物である、
請求項1〜6のいずれか1項に記載の組成物。 - インターフェロンI型アゴニストが、二本鎖RNA(dsRNA)、ポリ(dA:dT)DNA、二本鎖Z−DNAおよびB−DNA、36bpおよびDNA−RNAハイブリッドよりも長いDNA(dsDNA)、細菌セカンドメッセンジャーサイクリックジ−GMP、TLR3、TLR4、TLR7、TLR8およびTLR9アゴニスト、STINGアゴニスト、ならびにそれらの組み合わせからなる群より選択される請求項1〜7のいずれか一項記載の組成物。
- (a)インターフェロンII型アゴニストが、C−グリコシド型のα−ガラクトシルセラミド(α−C−GalCer)、α−ガラクトシルセラミド(α−GalCer)、12炭素アシル型ガラクトシルセラミド(β−GalCer)、β−D−グルコピラノシルセラミド(β−GlcCer)、1,2−ジアシル−3−ガラクトシル−sn−グリセロール(BbGL−II)、糖脂質を含むジアシルグリセロール(Glc−DAG−s2)、ガングリオキシド(GD3)、ガングリオトリアセラミド(Gg3Cer)、グリコシルホスファチジルイノシトール(GPI)、α−グルクロノシルセラミド(GSL−1またはGSL−4)、イソグロボトリヘキソシルセラミド(iGb3)、リポホスホグリカン(LPG)、ホスファチジルコリン(LPC)、α−ガラクトシルセラミド類似体(OCH)、スレイトールセラミドおよびそれらの組合わせからなる群より選択され、および/または
(b)インターフェロンII型作動薬はα−ガラクトシルセラミド(α−GalCer)である、
請求項1〜8のいずれか1項に記載の組成物。 - (a)抗腫瘍剤を含むミニ細胞に結合した二重特異性リガンド、および/または
(b)インターフェロンI型アゴニストを含むミニ細胞に結合した二重特異性リガンド、および/または
(c)インターフェロンII型アゴニストを含むミニ細胞に結合した二重特異性リガンド、を更に含む請求項1〜9のいずれか1項に記載の組成物。 - 前記二重特異性リガンドが、
(a) ミニ細胞表面構造に対する特異性を有する第1のアームと、非食作用性哺乳動物細胞表面受容体に対する特異性を有する第2のアームを含み、および/または
(b)ミニ細胞表面構造に対する特異性を有する第1のアームと、非食作用性哺乳動物細胞表面受容体に対する特異性を有し、ミニ細胞表面構造がミニ細胞表面上のリポ多糖のO−多糖成分である第2のアームとを含み、および/または
(c)ミニ細胞表面構造に対する特異性を有する第1のアームおよび非食作用性哺乳動物細胞表面受容体に対する特異性を有する第2のアームを含み、ここで、非貪食性哺乳動物細胞表面受容体は、ミニ細胞の受容体媒介エンドサイトーシスを活性化することができ、および/または
(d)二重特異性抗体または抗体断片を含み、および/または
(e)ここで、抗体または抗体断片は、細菌由来のミニ細胞表面構造に対する特異性を有する第1の多価アームと、癌細胞表面受容体に対する特異性を有する第2の多価アームとを含み、癌細胞表面受容体は、ミニ細胞の受容体媒介エンドサイトーシスを活性化することができる、
請求項10に記載の組成物。 - 前記組成物が、107ミニ細胞当たり約1未満の汚染親細菌細胞、108ミニ細胞当たり約1未満の汚染親細菌細胞、109ミニ細胞当たり約1未満の汚染親細菌細胞、1010ミニ細胞当たり約1未満の汚染親細菌細胞、または1011ミニ細胞当たり約1未満の汚染親細菌細胞を含む、請求項1〜11のいずれか1項に記載の組成物。
- 薬学的に受容可能なキャリアをさらに含む、請求項1〜12のいずれか1項に記載の組成物。
- 前記ミニ細胞が、直径約400nmである、請求項1〜13のいずれか1項に記載の組成物。
- 前記組成物が200nm濾過によって除去可能な親細菌細胞汚染を含まない、請求項1〜14のいずれか1項に記載の組成物。
- 前記組成物が、
(a)少なくとも約109、
(b)少なくとも約1×109、
(c)少なくとも約2×109、
(d)少なくとも約5×109、
(e)少なくとも8×109、
(f)約1011以下、
(g)約1×1011以下、
(h)約9×1010以下、または
(i)約8×1010以下
のミニ細胞または死滅した細菌細胞を含む、請求項1〜15のいずれか1項に記載の組成物。 - 有効量の前記組成物を被験体に投与することを含む、必要とする被験体を治療する方法において使用するための、請求項1〜16のいずれか1項に記載の組成物。
- 前記被検体が、ヒト、非ヒト霊長類、イヌ、ネコ、ウシ、ヒツジ、ウマ、ウサギ、マウス、またはラットである請求項17に記載の組成物。
- (a)前記被検体は癌に罹患しており、および/または
(b)前記被験体が癌に罹患しており、該癌が肺癌、乳癌、脳癌、肝臓癌、結腸癌、膵臓癌、および膀胱癌からなる群より選択され、および/または
(c)前記被験体が癌に罹患しており、該癌が急性リンパ芽球性白血病、急性骨髄性白血病、副腎皮質がん、エイズ関連がん、エイズ関連リンパ腫、虫垂がん、星状細胞腫、非定型奇形腫/ラブドイド腫瘍、基底細胞がん、膀胱がん、脳幹グリオーマ、脳腫瘍、乳がん、気管支腫瘍、バーキットリンパ腫、原発部位不明がん、カルチノイド腫瘍、主要サイト不明がん、中枢神経系非定型奇形腫様/ラブドイド腫瘍、中枢神経系胚芽腫、子宮頸がん、小児がん、脊索腫、慢性リンパ性白血病、慢性骨髄性白血病、慢性骨髄増殖性疾患、大腸がん、結腸直腸がん、頭蓋咽頭腫、皮膚T細胞リンパ腫、内分泌膵島細胞腫瘍、子宮体がん、上衣芽細胞腫、上衣芽腫、上衣腫、食道がん、感覚神経肉腫、ユーイング肉腫、頭蓋外胚細胞腫瘍、性腺外生殖細胞腫瘍、肝外胆管がん、胆がん、胃(胃)がん、胃腸カルチノイド腫瘍、消化管間質細胞腫瘍、消化管間質腫瘍(GIST)、妊娠性絨毛腫瘍 、神経膠腫、有毛細胞白血病、 心臓腫瘍、頭頸部がん、ホジキンリンパ腫、下咽頭がん、眼内黒色腫、小島細胞腫瘍、カポジ肉腫、腎臓ガン、ランゲルハンス細胞組織球症、喉頭ガン、唇ガン、肝臓がん、悪性繊維性組織サイト骨ガン、脂肪肉腫、髄芽腫、黒色腫、メルケル細胞がん、メルケル細胞がん、中皮腫、原発不明の転移性扁平上皮首がん、口腔がん、多発性内分泌腫瘍症候群、多発性骨髄腫/形質細胞腫瘍、菌状息肉症、骨髄異形成症候群、鼻腔がん、鼻咽頭がん、神経芽腫、非ホジキンリンパ腫、非黒色腫皮膚がん、非小細胞肺がん、口腔がん、口腔癌、中咽頭がん、骨肉腫、その他の脳および脊髄腫瘍、卵巣胚細胞腫瘍、卵巣低悪性度腫瘍、膵がん、乳頭腫症、副鼻腔がん、副甲状腺がん、骨盤がん、ペニスのがん、咽頭がん、中間的な分化を示す松果体実質細胞腫瘍、松果体芽腫、下垂体腫瘍、形質細胞新生物/多発性骨髄腫、胸膜肺芽腫、 原発性中枢神経系(CNS)リンパ腫、原発性肝細胞腫瘍、胸膜肺芽腫、原発性肝細胞がん、前立腺がん、腎細胞がん、腎細胞がん、腎細胞がん、網膜芽細胞腫、網膜芽細胞腫、横紋筋肉腫、セザリー症候群、唾液腺がん、小細胞肺がん、小腸がん、軟部組織肉腫、扁平上皮がん、頸部がん、胃(胃)がん、テント上原始神経外胚葉性腫瘍、T細胞リンパ腫、精巣がん、咽頭がん、胸腺がん、胸腺腫、甲状腺がん、移行上皮がん、腎盂および尿管の移行上皮がん、絨毛腫瘍、尿管がん、尿道がん、子宮がん、子宮肉腫、膣がん、外陰がん、ワルデンシュトレームマクログロブリン血症、およびウィルムス腫瘍および/または
(d)前記被験体は、脳幹グリオーマ、中枢神経系非定型奇形腫様/ラブドイド腫瘍、中枢神経系胚芽腫、星状細胞腫、頭蓋咽頭腫、上衣芽腫、上衣腫、髄芽腫、髄上皮腫、中間的分化の松果体実質腫瘍、テント上原始神経外胚葉性腫瘍および松果体芽腫からなる群より選択される脳腫瘍または脳腫瘍を罹患している、
請求項17または18に記載の組成物。 - 前記組成物が、
(a)少なくとも週に1回、数週間にわたり、および/または
(b)1週間に少なくとも1回、数週間から数ヶ月にわたり、および/または
(c)1週間に少なくとも1回、約2、約3、約4、約5、約6、約7、約8、約9、約10、約11、約12、約13、約14、約15、約16、約17、約18、約19または約20週間またはそれ以上、および/またはおよび/または
(d)毎週約2回、および/または
(e)約2、約3、約4、約5、約6、約7、約8、約9、約10、約11、約12、約13、約14、約15、約16、約17、約18、約19または約20週間、またはそれ以上にわたり、週に約2回、
投与される、請求項17〜19のいずれか1項に記載の組成物。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023222336A JP2024038199A (ja) | 2018-07-23 | 2023-12-28 | 細菌由来ミニ細胞を含む組成物およびそれを使用する方法 |
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862702172P | 2018-07-23 | 2018-07-23 | |
| US62/702,172 | 2018-07-23 | ||
| US201962788265P | 2019-01-04 | 2019-01-04 | |
| US62/788,265 | 2019-01-04 | ||
| PCT/IB2019/056259 WO2020021437A1 (en) | 2018-07-23 | 2019-07-22 | Compositions comprising bacterially derived minicells and methods of using the same |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2023222336A Division JP2024038199A (ja) | 2018-07-23 | 2023-12-28 | 細菌由来ミニ細胞を含む組成物およびそれを使用する方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2021532106A true JP2021532106A (ja) | 2021-11-25 |
| JPWO2020021437A5 JPWO2020021437A5 (ja) | 2022-06-28 |
Family
ID=69181384
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021503095A Pending JP2021532106A (ja) | 2018-07-23 | 2019-07-22 | 細菌由来ミニ細胞を含む組成物およびそれを使用する方法 |
| JP2023222336A Pending JP2024038199A (ja) | 2018-07-23 | 2023-12-28 | 細菌由来ミニ細胞を含む組成物およびそれを使用する方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2023222336A Pending JP2024038199A (ja) | 2018-07-23 | 2023-12-28 | 細菌由来ミニ細胞を含む組成物およびそれを使用する方法 |
Country Status (8)
| Country | Link |
|---|---|
| US (2) | US11504402B2 (ja) |
| EP (1) | EP3826654A4 (ja) |
| JP (2) | JP2021532106A (ja) |
| CN (1) | CN112689511A (ja) |
| AU (1) | AU2019311380A1 (ja) |
| CA (1) | CA3107095A1 (ja) |
| SG (1) | SG11202100507PA (ja) |
| WO (1) | WO2020021437A1 (ja) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9844598B2 (en) | 2011-12-13 | 2017-12-19 | Engeneic Molecular Delivery Pty Ltd | Bacterially derived, intact minicells for delivery of therapeutic agents to brain tumors |
| CN106999444B (zh) * | 2014-10-03 | 2023-12-12 | 安吉尼科分子传输公司 | 完整的细菌衍生的囊泡对小分子化合物的增强装载 |
| CA3073449A1 (en) | 2017-09-25 | 2019-03-28 | Agrospheres, Inc. | Compositions and methods for scalable production and delivery of biologicals |
| SG11202107306WA (en) * | 2019-01-04 | 2021-08-30 | Engeneic Molecular Delivery Pty Ltd | Encapsulated glycolipid antigens for treatment of neoplastic diseases |
| WO2021191796A1 (en) * | 2020-03-24 | 2021-09-30 | Engeneic Molecular Delivery Pty Ltd | Compositions and vaccines for treating and/or preventing viral infections, including coronavirus infections, and methods of using the same |
| EP4236996A4 (en) * | 2020-10-27 | 2024-11-13 | Rutgers, the State University of New Jersey | REPLICATION-FAILED VACCINES AND THEIR USES |
| CN114569627A (zh) * | 2020-12-02 | 2022-06-03 | 杭州星鳌生物科技有限公司 | 新型sting激动剂复合物的制备、组成及其在抗肿瘤药物中的应用 |
| EP4373520A1 (en) * | 2021-07-22 | 2024-05-29 | EnGeneIC Molecular Delivery Pty Ltd. | Compositions and vaccines for treating and/or preventing coronavirus variant infections and methods of using the same |
| WO2024218655A1 (en) * | 2023-04-17 | 2024-10-24 | Engeneic Molecular Delivery Pty Ltd | Compositions and methodology for delivery of alpha-galactosylceramide compounds |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016532639A (ja) * | 2013-10-04 | 2016-10-20 | エンジーンアイシー モレキュラー デリバリー ピーティーワイ リミテッド | 薬物を負荷した二重特異性リガンド標的化ミニ細胞およびインターフェロン−γを用いた併用腫瘍治療 |
| US20180100159A1 (en) * | 2016-10-06 | 2018-04-12 | Engenelc Molecular Delivery Pty. Ltd. | Bacterial minicells for delivering nucleic acid adjuvants and methods of using the same |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BR0010316A (pt) | 1999-05-11 | 2004-04-27 | Ortho Mcnell Pharmaceutical In | Método e sistema para obtenção de regimes de dosagem otimizados de epo para uma resposta farmacodinâmica/farmacocinética desejada em um paciente |
| US20030194798A1 (en) | 2001-05-24 | 2003-10-16 | Surber Mark W. | Minicell compositions and methods |
| US20050222057A1 (en) | 2001-10-15 | 2005-10-06 | Himanshu Brahmbhatt | Intact minicells as vectors for dna transfer and gene therapy in vitro and in vivo |
| US7611885B2 (en) | 2003-06-24 | 2009-11-03 | Engeneic Molecular Delivery Pty, Ltd. | Pharmaceutically compatible method for purifying intact bacterial minicells |
| CN101954095B (zh) | 2003-12-09 | 2013-03-13 | 基因分子传递股份有限公司 | 通过源自细菌的完整小细胞将靶基因递送至非吞噬哺乳动物细胞 |
| US8772013B2 (en) | 2004-02-02 | 2014-07-08 | Engeneic Molecular Delivery Pty Ltd | Methods for targeted in vitro and in vivo drug delivery to mammalian cells via bacterially derived intact minicells |
| SG135174A1 (en) | 2004-02-02 | 2007-09-28 | Engeneic Molecular Delivery Pty Ltd | Compositions and methods for targeted in vitro and in vivo drug delivery to mammalian cells via bacterially derived intact minicells |
| CN102921019B (zh) * | 2004-08-26 | 2015-04-01 | 恩杰内克分子递送有限公司 | 通过细菌性来源的完整小细胞向哺乳动物细胞递送功能性核酸 |
| PL2712618T3 (pl) | 2006-06-23 | 2017-07-31 | Engeneic Molecular Delivery Pty Ltd. | Ukierunkowane dostarczanie leków, terapeutycznych kwasów nukleinowych i funkcjonalnych kwasów nukleinowych do komórek ssaczych przez nienaruszone, uśmiercone komórki bakteryjne |
| SI2865755T1 (sl) | 2007-03-30 | 2017-06-30 | Engeneic Molecular Delivery Pty Ltd. | Intaktne minicelice, ki so izvedene iz bakterij in vključujejo regulatorno RNA |
| EP3750562A1 (en) * | 2012-10-02 | 2020-12-16 | Vaxiion Therapeutics, LLC | Immunomodulatory minicells and methods of use |
| US9006200B2 (en) * | 2013-03-13 | 2015-04-14 | Asbestos Diseases Research Foundation | MicroRNA-based approach to treating malignant pleural mesothelioma |
| CN106170299A (zh) * | 2014-01-22 | 2016-11-30 | 小利兰斯坦福大学托管委员会 | 用于抗体和抗体负载树突状细胞介导治疗的方法和组合物 |
-
2019
- 2019-07-22 CN CN201980060185.6A patent/CN112689511A/zh active Pending
- 2019-07-22 AU AU2019311380A patent/AU2019311380A1/en active Pending
- 2019-07-22 EP EP19839892.7A patent/EP3826654A4/en active Pending
- 2019-07-22 JP JP2021503095A patent/JP2021532106A/ja active Pending
- 2019-07-22 SG SG11202100507PA patent/SG11202100507PA/en unknown
- 2019-07-22 US US16/518,833 patent/US11504402B2/en active Active
- 2019-07-22 CA CA3107095A patent/CA3107095A1/en active Pending
- 2019-07-22 WO PCT/IB2019/056259 patent/WO2020021437A1/en unknown
-
2022
- 2022-11-18 US US17/990,402 patent/US20230079745A1/en active Pending
-
2023
- 2023-12-28 JP JP2023222336A patent/JP2024038199A/ja active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016532639A (ja) * | 2013-10-04 | 2016-10-20 | エンジーンアイシー モレキュラー デリバリー ピーティーワイ リミテッド | 薬物を負荷した二重特異性リガンド標的化ミニ細胞およびインターフェロン−γを用いた併用腫瘍治療 |
| US20180100159A1 (en) * | 2016-10-06 | 2018-04-12 | Engenelc Molecular Delivery Pty. Ltd. | Bacterial minicells for delivering nucleic acid adjuvants and methods of using the same |
Also Published As
| Publication number | Publication date |
|---|---|
| US11504402B2 (en) | 2022-11-22 |
| AU2019311380A1 (en) | 2021-02-11 |
| EP3826654A1 (en) | 2021-06-02 |
| JP2024038199A (ja) | 2024-03-19 |
| SG11202100507PA (en) | 2021-02-25 |
| US20200054689A1 (en) | 2020-02-20 |
| EP3826654A4 (en) | 2022-07-06 |
| WO2020021437A1 (en) | 2020-01-30 |
| CA3107095A1 (en) | 2020-01-30 |
| CN112689511A (zh) | 2021-04-20 |
| US20230079745A1 (en) | 2023-03-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20230079745A1 (en) | Compositions comprising bacterially derived minicells and methods of using the same | |
| US10357514B2 (en) | Treatment of cancer using anti-CD19 Chimeric Antigen Receptor | |
| WO2021053667A2 (en) | Combination cancer therapy and cytokine control therapy for cancer treatment | |
| TW202428301A (zh) | 治療性rna | |
| JP7330994B2 (ja) | 治療用ナノ生物学的組成物による訓練免疫の促進 | |
| KR102433719B1 (ko) | 약물 로딩된, 이중특이성 리간드-표적화된 미니세포 및 인터페론 감마에 의한 복합 종양 치료 | |
| JP7548911B2 (ja) | 腫瘍性疾患の治療のためのカプセル化グリコリピド抗原 | |
| EP4031655A2 (en) | Combination cancer therapy and cytokine control therapy for cancer treatment | |
| CN114729314A (zh) | 用于癌症治疗的组合癌症疗法和细胞因子控制疗法 | |
| TWI837437B (zh) | 嵌合抗原受體t細胞療法 | |
| US20230416746A1 (en) | Immuno-oncology targets to improve t-cell metabolic response | |
| BR122023023020A2 (pt) | Uso de células t para tratar linfoma de células do manto ou leucemia linfoblástica aguda, métodos de previsão e para melhorar a eficácia do tratamento com células t car | |
| Lee | Cell Surface Engineering and Its Applications in Cancer Therapy |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210423 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220620 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20220620 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20230516 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20230808 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20231003 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20240507 |