JP2020522544A - Method of treating hyperlipidemia in diabetic patients by administration of a PCSK9 inhibitor - Google Patents
Method of treating hyperlipidemia in diabetic patients by administration of a PCSK9 inhibitor Download PDFInfo
- Publication number
- JP2020522544A JP2020522544A JP2019567608A JP2019567608A JP2020522544A JP 2020522544 A JP2020522544 A JP 2020522544A JP 2019567608 A JP2019567608 A JP 2019567608A JP 2019567608 A JP2019567608 A JP 2019567608A JP 2020522544 A JP2020522544 A JP 2020522544A
- Authority
- JP
- Japan
- Prior art keywords
- patient
- antibody
- antigen
- binding fragment
- insulin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 238000000034 method Methods 0.000 title claims abstract description 310
- 206010012601 diabetes mellitus Diseases 0.000 title claims description 66
- 229940122392 PCSK9 inhibitor Drugs 0.000 title description 43
- 208000031226 Hyperlipidaemia Diseases 0.000 title description 5
- 239000000427 antigen Substances 0.000 claims abstract description 295
- 108091007433 antigens Proteins 0.000 claims abstract description 295
- 102000036639 antigens Human genes 0.000 claims abstract description 295
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 claims abstract description 291
- 239000012634 fragment Substances 0.000 claims abstract description 277
- 238000002560 therapeutic procedure Methods 0.000 claims abstract description 213
- 229940125396 insulin Drugs 0.000 claims abstract description 148
- 102000004877 Insulin Human genes 0.000 claims abstract description 146
- 108090001061 Insulin Proteins 0.000 claims abstract description 146
- 208000001072 type 2 diabetes mellitus Diseases 0.000 claims abstract description 93
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 claims abstract description 74
- 208000035150 Hypercholesterolemia Diseases 0.000 claims abstract description 65
- 230000002526 effect on cardiovascular system Effects 0.000 claims abstract description 46
- 108010028554 LDL Cholesterol Proteins 0.000 claims description 158
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 claims description 136
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 106
- 239000003112 inhibitor Substances 0.000 claims description 78
- 239000000556 agonist Substances 0.000 claims description 57
- 101710180553 Proprotein convertase subtilisin/kexin type 9 Proteins 0.000 claims description 56
- 102100038955 Proprotein convertase subtilisin/kexin type 9 Human genes 0.000 claims description 56
- 201000001320 Atherosclerosis Diseases 0.000 claims description 44
- 108010023302 HDL Cholesterol Proteins 0.000 claims description 43
- 150000002632 lipids Chemical class 0.000 claims description 42
- 206010003210 Arteriosclerosis Diseases 0.000 claims description 40
- -1 fibrates Substances 0.000 claims description 40
- 208000029078 coronary artery disease Diseases 0.000 claims description 35
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 claims description 30
- 239000002245 particle Substances 0.000 claims description 28
- 101001098868 Homo sapiens Proprotein convertase subtilisin/kexin type 9 Proteins 0.000 claims description 27
- 239000003472 antidiabetic agent Substances 0.000 claims description 27
- 230000003178 anti-diabetic effect Effects 0.000 claims description 25
- 108010056301 Apolipoprotein C-III Proteins 0.000 claims description 24
- 102000004895 Lipoproteins Human genes 0.000 claims description 22
- 108090001030 Lipoproteins Proteins 0.000 claims description 22
- 102100030970 Apolipoprotein C-III Human genes 0.000 claims description 21
- 108010004460 Gastric Inhibitory Polypeptide Proteins 0.000 claims description 21
- 102100039994 Gastric inhibitory polypeptide Human genes 0.000 claims description 21
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 claims description 21
- 239000005557 antagonist Substances 0.000 claims description 21
- 102000002148 Diacylglycerol O-acyltransferase Human genes 0.000 claims description 20
- 108010001348 Diacylglycerol O-acyltransferase Proteins 0.000 claims description 20
- 102100033839 Glucose-dependent insulinotropic receptor Human genes 0.000 claims description 20
- 101710163236 Glucose-dependent insulinotropic receptor Proteins 0.000 claims description 20
- 108010041872 Islet Amyloid Polypeptide Proteins 0.000 claims description 20
- 102000036770 Islet Amyloid Polypeptide Human genes 0.000 claims description 20
- OLNTVTPDXPETLC-XPWALMASSA-N ezetimibe Chemical compound N1([C@@H]([C@H](C1=O)CC[C@H](O)C=1C=CC(F)=CC=1)C=1C=CC(O)=CC=1)C1=CC=C(F)C=C1 OLNTVTPDXPETLC-XPWALMASSA-N 0.000 claims description 20
- COCFEDIXXNGUNL-RFKWWTKHSA-N Insulin glargine Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@H]1CSSC[C@H]2C(=O)N[C@H](C(=O)N[C@@H](CO)C(=O)N[C@H](C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3C=CC(O)=CC=3)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3NC=NC=3)NC(=O)[C@H](CO)NC(=O)CNC1=O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(=O)NCC(O)=O)=O)CSSC[C@@H](C(N2)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](NC(=O)CN)[C@@H](C)CC)[C@@H](C)CC)[C@@H](C)O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)CC=1C=CC=CC=1)C(C)C)C1=CN=CN1 COCFEDIXXNGUNL-RFKWWTKHSA-N 0.000 claims description 19
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 claims description 19
- 239000008103 glucose Substances 0.000 claims description 19
- IDHVLSACPFUBDY-QCDLPZBNSA-N xenin Chemical compound C([C@@H](C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(O)=O)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CN=CN1 IDHVLSACPFUBDY-QCDLPZBNSA-N 0.000 claims description 19
- 108010057186 Insulin Glargine Proteins 0.000 claims description 18
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 claims description 18
- 230000001419 dependent effect Effects 0.000 claims description 18
- PBGKTOXHQIOBKM-FHFVDXKLSA-N insulin (human) Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@H]1CSSC[C@H]2C(=O)N[C@H](C(=O)N[C@@H](CO)C(=O)N[C@H](C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3C=CC(O)=CC=3)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3NC=NC=3)NC(=O)[C@H](CO)NC(=O)CNC1=O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O)=O)CSSC[C@@H](C(N2)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](NC(=O)CN)[C@@H](C)CC)[C@@H](C)CC)[C@@H](C)O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)CC=1C=CC=CC=1)C(C)C)C1=CN=CN1 PBGKTOXHQIOBKM-FHFVDXKLSA-N 0.000 claims description 18
- 229960002869 insulin glargine Drugs 0.000 claims description 17
- 229960003512 nicotinic acid Drugs 0.000 claims description 16
- 235000001968 nicotinic acid Nutrition 0.000 claims description 16
- 239000011664 nicotinic acid Substances 0.000 claims description 16
- 229940125753 fibrate Drugs 0.000 claims description 15
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 claims description 15
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 claims description 14
- 229960002027 evolocumab Drugs 0.000 claims description 14
- 229960000815 ezetimibe Drugs 0.000 claims description 14
- 238000012986 modification Methods 0.000 claims description 14
- 230000004048 modification Effects 0.000 claims description 14
- 208000010125 myocardial infarction Diseases 0.000 claims description 14
- 229960002855 simvastatin Drugs 0.000 claims description 14
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 claims description 13
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 claims description 13
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 claims description 13
- 229960005370 atorvastatin Drugs 0.000 claims description 13
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 claims description 13
- 230000009885 systemic effect Effects 0.000 claims description 13
- 102100025353 G-protein coupled bile acid receptor 1 Human genes 0.000 claims description 12
- 101000976075 Homo sapiens Insulin Proteins 0.000 claims description 12
- 108010065920 Insulin Lispro Proteins 0.000 claims description 12
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 claims description 12
- 239000012190 activator Substances 0.000 claims description 12
- SEERZIQQUAZTOL-ANMDKAQQSA-N cerivastatin Chemical compound COCC1=C(C(C)C)N=C(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC(O)=O)=C1C1=CC=C(F)C=C1 SEERZIQQUAZTOL-ANMDKAQQSA-N 0.000 claims description 12
- 229960005110 cerivastatin Drugs 0.000 claims description 12
- 229960003765 fluvastatin Drugs 0.000 claims description 12
- 229960004844 lovastatin Drugs 0.000 claims description 12
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 claims description 12
- VGYFMXBACGZSIL-MCBHFWOFSA-N pitavastatin Chemical compound OC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 VGYFMXBACGZSIL-MCBHFWOFSA-N 0.000 claims description 12
- 229960002797 pitavastatin Drugs 0.000 claims description 12
- 229960002965 pravastatin Drugs 0.000 claims description 12
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 claims description 12
- 229950009885 ralpancizumab Drugs 0.000 claims description 12
- 229940044601 receptor agonist Drugs 0.000 claims description 12
- 239000000018 receptor agonist Substances 0.000 claims description 12
- 229960000672 rosuvastatin Drugs 0.000 claims description 12
- BPRHUIZQVSMCRT-VEUZHWNKSA-N rosuvastatin Chemical compound CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC(O)=O BPRHUIZQVSMCRT-VEUZHWNKSA-N 0.000 claims description 12
- 229940122502 Cholesterol absorption inhibitor Drugs 0.000 claims description 11
- 229940123232 Glucagon receptor agonist Drugs 0.000 claims description 11
- 101000857733 Homo sapiens G-protein coupled bile acid receptor 1 Proteins 0.000 claims description 11
- 108010073961 Insulin Aspart Proteins 0.000 claims description 11
- 108010089308 Insulin Detemir Proteins 0.000 claims description 11
- RCHHVVGSTHAVPF-ZPHPLDECSA-N apidra Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@H]1CSSC[C@H]2C(=O)N[C@H](C(=O)N[C@@H](CO)C(=O)N[C@H](C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3C=CC(O)=CC=3)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3N=CNC=3)NC(=O)[C@H](CO)NC(=O)CNC1=O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O)=O)CSSC[C@@H](C(N2)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](NC(=O)CN)[C@@H](C)CC)[C@@H](C)CC)[C@@H](C)O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@@H](N)CC=1C=CC=CC=1)C(C)C)C1=CNC=N1 RCHHVVGSTHAVPF-ZPHPLDECSA-N 0.000 claims description 11
- 229960004717 insulin aspart Drugs 0.000 claims description 11
- 108700039926 insulin glulisine Proteins 0.000 claims description 11
- 229960002068 insulin lispro Drugs 0.000 claims description 11
- UGOZVNFCFYTPAZ-IOXYNQHNSA-N levemir Chemical compound CCCCCCCCCCCCCC(=O)NCCCC[C@@H](C(O)=O)NC(=O)[C@@H]1CCCN1C(=O)[C@H]([C@@H](C)O)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)CNC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H]1NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=2C=CC(O)=CC=2)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=2N=CNC=2)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=2N=CNC=2)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)CC=2C=CC=CC=2)C(C)C)CSSC[C@@H]2NC(=O)[C@@H](NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)CN)[C@@H](C)CC)C(C)C)CSSC[C@H](NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H](CO)NC(=O)[C@H]([C@@H](C)O)NC2=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N[C@@H](CSSC1)C(=O)N[C@@H](CC(N)=O)C(O)=O)CC1=CC=C(O)C=C1 UGOZVNFCFYTPAZ-IOXYNQHNSA-N 0.000 claims description 11
- 229940012843 omega-3 fatty acid Drugs 0.000 claims description 11
- 235000020660 omega-3 fatty acid Nutrition 0.000 claims description 11
- 239000006014 omega-3 oil Substances 0.000 claims description 11
- 239000003352 sequestering agent Substances 0.000 claims description 11
- IGRCWJPBLWGNPX-UHFFFAOYSA-N 3-(2-chlorophenyl)-n-(4-chlorophenyl)-n,5-dimethyl-1,2-oxazole-4-carboxamide Chemical compound C=1C=C(Cl)C=CC=1N(C)C(=O)C1=C(C)ON=C1C1=CC=CC=C1Cl IGRCWJPBLWGNPX-UHFFFAOYSA-N 0.000 claims description 10
- DGENZVKCTGIDRZ-UHFFFAOYSA-N 3-[4-[(3-phenoxyphenyl)methylamino]phenyl]propanoic acid Chemical compound C1=CC(CCC(=O)O)=CC=C1NCC1=CC=CC(OC=2C=CC=CC=2)=C1 DGENZVKCTGIDRZ-UHFFFAOYSA-N 0.000 claims description 10
- SWLAMJPTOQZTAE-UHFFFAOYSA-N 4-[2-[(5-chloro-2-methoxybenzoyl)amino]ethyl]benzoic acid Chemical compound COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(C(O)=O)C=C1 SWLAMJPTOQZTAE-UHFFFAOYSA-N 0.000 claims description 10
- 229940077274 Alpha glucosidase inhibitor Drugs 0.000 claims description 10
- 229940123208 Biguanide Drugs 0.000 claims description 10
- 102100040134 Free fatty acid receptor 4 Human genes 0.000 claims description 10
- 101800001586 Ghrelin Proteins 0.000 claims description 10
- 102400000442 Ghrelin-28 Human genes 0.000 claims description 10
- 229940122904 Glucagon receptor antagonist Drugs 0.000 claims description 10
- 102000030595 Glucokinase Human genes 0.000 claims description 10
- 108010021582 Glucokinase Proteins 0.000 claims description 10
- 102000001554 Hemoglobins Human genes 0.000 claims description 10
- 108010054147 Hemoglobins Proteins 0.000 claims description 10
- 101000890672 Homo sapiens Free fatty acid receptor 4 Proteins 0.000 claims description 10
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 claims description 10
- FYZPCMFQCNBYCY-WIWKJPBBSA-N Insulin degludec Chemical compound CC[C@H](C)[C@H](NC(=O)CN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H]1CSSC[C@@H]2NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CSSC[C@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](Cc3c[nH]cn3)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)Cc3ccccc3)C(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](Cc3c[nH]cn3)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](Cc3ccc(O)cc3)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](Cc3ccc(O)cc3)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](Cc3ccc(O)cc3)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC2=O)C(=O)N[C@@H](CC(N)=O)C(O)=O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](Cc2ccc(O)cc2)C(=O)N[C@@H]([C@@H](C)O)C(=O)N2CCC[C@H]2C(=O)N[C@@H](CCCCNC(=O)CC[C@H](NC(=O)CCCCCCCCCCCCCCC(O)=O)C(O)=O)C(O)=O)NC1=O)[C@@H](C)O)[C@@H](C)CC FYZPCMFQCNBYCY-WIWKJPBBSA-N 0.000 claims description 10
- 208000001145 Metabolic Syndrome Diseases 0.000 claims description 10
- 208000005764 Peripheral Arterial Disease Diseases 0.000 claims description 10
- 208000030831 Peripheral arterial occlusive disease Diseases 0.000 claims description 10
- 102000003728 Peroxisome Proliferator-Activated Receptors Human genes 0.000 claims description 10
- 108090000029 Peroxisome Proliferator-Activated Receptors Proteins 0.000 claims description 10
- 108091006299 SLC2A2 Proteins 0.000 claims description 10
- 108091006300 SLC2A4 Proteins 0.000 claims description 10
- 229940100389 Sulfonylurea Drugs 0.000 claims description 10
- 229940123464 Thiazolidinedione Drugs 0.000 claims description 10
- 102400000752 Xenin Human genes 0.000 claims description 10
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 claims description 10
- 239000003888 alpha glucosidase inhibitor Substances 0.000 claims description 10
- 210000000941 bile Anatomy 0.000 claims description 10
- 229950011350 bococizumab Drugs 0.000 claims description 10
- 239000003795 chemical substances by application Substances 0.000 claims description 10
- 230000001906 cholesterol absorption Effects 0.000 claims description 10
- 108010050259 insulin degludec Proteins 0.000 claims description 10
- 229960004225 insulin degludec Drugs 0.000 claims description 10
- 229960003948 insulin detemir Drugs 0.000 claims description 10
- 229960000696 insulin glulisine Drugs 0.000 claims description 10
- 229940125425 inverse agonist Drugs 0.000 claims description 10
- 229950004994 meglitinide Drugs 0.000 claims description 10
- GCYXWQUSHADNBF-AAEALURTSA-N preproglucagon 78-108 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 GCYXWQUSHADNBF-AAEALURTSA-N 0.000 claims description 10
- 239000011734 sodium Substances 0.000 claims description 10
- 229910052708 sodium Inorganic materials 0.000 claims description 10
- 229940083542 sodium Drugs 0.000 claims description 10
- 108010006643 xenin 25 Proteins 0.000 claims description 10
- ZOBPZXTWZATXDG-UHFFFAOYSA-N 1,3-thiazolidine-2,4-dione Chemical compound O=C1CSC(=O)N1 ZOBPZXTWZATXDG-UHFFFAOYSA-N 0.000 claims description 9
- 102100036506 11-beta-hydroxysteroid dehydrogenase 1 Human genes 0.000 claims description 9
- 101710186107 11-beta-hydroxysteroid dehydrogenase 1 Proteins 0.000 claims description 9
- 206010002388 Angina unstable Diseases 0.000 claims description 9
- XNCOSPRUTUOJCJ-UHFFFAOYSA-N Biguanide Chemical compound NC(N)=NC(N)=N XNCOSPRUTUOJCJ-UHFFFAOYSA-N 0.000 claims description 9
- 239000002126 C01EB10 - Adenosine Substances 0.000 claims description 9
- 229940125827 GPR40 agonist Drugs 0.000 claims description 9
- 102000001253 Protein Kinase Human genes 0.000 claims description 9
- 102000004117 Somatostatin receptor 3 Human genes 0.000 claims description 9
- 108090000674 Somatostatin receptor 3 Proteins 0.000 claims description 9
- 208000007814 Unstable Angina Diseases 0.000 claims description 9
- 229960005305 adenosine Drugs 0.000 claims description 9
- 239000002260 anti-inflammatory agent Substances 0.000 claims description 9
- 229940090124 dipeptidyl peptidase 4 (dpp-4) inhibitors for blood glucose lowering Drugs 0.000 claims description 9
- 239000003626 gastrointestinal polypeptide Substances 0.000 claims description 9
- 201000004332 intermediate coronary syndrome Diseases 0.000 claims description 9
- 108060006633 protein kinase Proteins 0.000 claims description 9
- 229940044551 receptor antagonist Drugs 0.000 claims description 9
- 239000002464 receptor antagonist Substances 0.000 claims description 9
- 239000000021 stimulant Substances 0.000 claims description 9
- YROXIXLRRCOBKF-UHFFFAOYSA-N sulfonylurea Chemical class OC(=N)N=S(=O)=O YROXIXLRRCOBKF-UHFFFAOYSA-N 0.000 claims description 9
- 208000032382 Ischaemic stroke Diseases 0.000 claims description 8
- 229940123483 GPR142 agonist Drugs 0.000 claims description 7
- 206010000891 acute myocardial infarction Diseases 0.000 claims description 7
- 239000002955 immunomodulating agent Substances 0.000 claims description 7
- 229940121363 anti-inflammatory agent Drugs 0.000 claims description 6
- 239000002253 acid Substances 0.000 claims description 5
- 239000003524 antilipemic agent Substances 0.000 claims description 4
- 230000005764 inhibitory process Effects 0.000 claims description 4
- VOMXSOIBEJBQNF-UTTRGDHVSA-N novorapid Chemical compound C([C@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CS)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H](CO)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CS)NC(=O)[C@H](CS)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](NC(=O)CN)[C@@H](C)CC)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(O)=O)C1=CC=C(O)C=C1.C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CS)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CS)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)CC=1C=CC=CC=1)C(C)C)C1=CN=CN1 VOMXSOIBEJBQNF-UTTRGDHVSA-N 0.000 claims description 4
- FJLGEFLZQAZZCD-MCBHFWOFSA-N (3R,5S)-fluvastatin Chemical compound C12=CC=CC=C2N(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC(O)=O)=C1C1=CC=C(F)C=C1 FJLGEFLZQAZZCD-MCBHFWOFSA-N 0.000 claims 3
- GNKDKYIHGQKHHM-RJKLHVOGSA-N ghrelin Chemical compound C([C@H](NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)CN)COC(=O)CCCCCCC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C1=CC=CC=C1 GNKDKYIHGQKHHM-RJKLHVOGSA-N 0.000 claims 3
- WNRQPCUGRUFHED-DETKDSODSA-N humalog Chemical compound C([C@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CS)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H](CO)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CS)NC(=O)[C@H](CS)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](NC(=O)CN)[C@@H](C)CC)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(O)=O)C1=CC=C(O)C=C1.C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CS)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CS)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)CC=1C=CC=CC=1)C(C)C)C1=CN=CN1 WNRQPCUGRUFHED-DETKDSODSA-N 0.000 claims 3
- 239000008194 pharmaceutical composition Substances 0.000 abstract description 23
- 238000011282 treatment Methods 0.000 description 120
- 239000000902 placebo Substances 0.000 description 61
- 229940068196 placebo Drugs 0.000 description 61
- 238000012216 screening Methods 0.000 description 55
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 54
- 238000002347 injection Methods 0.000 description 42
- 239000007924 injection Substances 0.000 description 42
- 230000008859 change Effects 0.000 description 39
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 32
- 239000003814 drug Substances 0.000 description 30
- 229940079593 drug Drugs 0.000 description 28
- 230000009467 reduction Effects 0.000 description 28
- 239000000203 mixture Substances 0.000 description 25
- 210000004602 germ cell Anatomy 0.000 description 24
- 238000004458 analytical method Methods 0.000 description 20
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 18
- UFTFJSFQGQCHQW-UHFFFAOYSA-N triformin Chemical compound O=COCC(OC=O)COC=O UFTFJSFQGQCHQW-UHFFFAOYSA-N 0.000 description 17
- 235000001014 amino acid Nutrition 0.000 description 16
- 235000012000 cholesterol Nutrition 0.000 description 16
- 238000006467 substitution reaction Methods 0.000 description 16
- DTHNMHAUYICORS-KTKZVXAJSA-N Glucagon-like peptide 1 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 DTHNMHAUYICORS-KTKZVXAJSA-N 0.000 description 15
- 238000012360 testing method Methods 0.000 description 15
- 108010007622 LDL Lipoproteins Proteins 0.000 description 14
- 102000007330 LDL Lipoproteins Human genes 0.000 description 14
- 230000002411 adverse Effects 0.000 description 14
- 210000004369 blood Anatomy 0.000 description 14
- 239000008280 blood Substances 0.000 description 14
- 238000009472 formulation Methods 0.000 description 14
- 235000018102 proteins Nutrition 0.000 description 14
- 102000004169 proteins and genes Human genes 0.000 description 14
- 108090000623 proteins and genes Proteins 0.000 description 14
- 101710095342 Apolipoprotein B Proteins 0.000 description 13
- 102100040202 Apolipoprotein B-100 Human genes 0.000 description 13
- 238000012384 transportation and delivery Methods 0.000 description 13
- 102100040214 Apolipoprotein(a) Human genes 0.000 description 12
- 101710115418 Apolipoprotein(a) Proteins 0.000 description 12
- AIRYAONNMGRCGJ-FHFVDXKLSA-N CC[C@H](C)[C@H](NC(=O)CN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H]1CSSC[C@@H]2NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CSSC[C@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](Cc3c[nH]cn3)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)Cc3ccccc3)C(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](Cc3c[nH]cn3)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](Cc3ccc(O)cc3)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](Cc3ccc(O)cc3)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](Cc3ccc(O)cc3)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC2=O)C(=O)N[C@@H](CC(N)=O)C(O)=O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](Cc2ccc(O)cc2)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N2CCC[C@H]2C(=O)N[C@@H]([C@@H](C)O)C(O)=O)NC1=O)[C@@H](C)O)[C@@H](C)CC Chemical compound CC[C@H](C)[C@H](NC(=O)CN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H]1CSSC[C@@H]2NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CSSC[C@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](Cc3c[nH]cn3)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)Cc3ccccc3)C(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](Cc3c[nH]cn3)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](Cc3ccc(O)cc3)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](Cc3ccc(O)cc3)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](Cc3ccc(O)cc3)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC2=O)C(=O)N[C@@H](CC(N)=O)C(O)=O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](Cc2ccc(O)cc2)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N2CCC[C@H]2C(=O)N[C@@H]([C@@H](C)O)C(O)=O)NC1=O)[C@@H](C)O)[C@@H](C)CC AIRYAONNMGRCGJ-FHFVDXKLSA-N 0.000 description 12
- 101800000224 Glucagon-like peptide 1 Proteins 0.000 description 12
- 102100040918 Pro-glucagon Human genes 0.000 description 12
- 150000001413 amino acids Chemical class 0.000 description 12
- 208000010668 atopic eczema Diseases 0.000 description 12
- 208000020832 chronic kidney disease Diseases 0.000 description 12
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 12
- 230000002829 reductive effect Effects 0.000 description 12
- 230000009977 dual effect Effects 0.000 description 11
- 125000000487 histidyl group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C([H])=N1 0.000 description 11
- 210000002966 serum Anatomy 0.000 description 11
- 230000001225 therapeutic effect Effects 0.000 description 11
- ZGGHKIMDNBDHJB-NRFPMOEYSA-M (3R,5S)-fluvastatin sodium Chemical compound [Na+].C12=CC=CC=C2N(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 ZGGHKIMDNBDHJB-NRFPMOEYSA-M 0.000 description 10
- 108020004414 DNA Proteins 0.000 description 10
- 210000004027 cell Anatomy 0.000 description 10
- DDRJAANPRJIHGJ-UHFFFAOYSA-N creatinine Chemical compound CN1CC(=O)NC1=N DDRJAANPRJIHGJ-UHFFFAOYSA-N 0.000 description 10
- 230000000694 effects Effects 0.000 description 10
- 108090000765 processed proteins & peptides Proteins 0.000 description 10
- 238000012549 training Methods 0.000 description 10
- WEDIKSVWBUKTRA-WTKGVUNUSA-N CC[C@H](C)[C@H](NC(=O)CN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H]1CSSC[C@@H]2NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CSSC[C@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](Cc3c[nH]cn3)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)Cc3ccccc3)C(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](Cc3c[nH]cn3)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](Cc3ccc(O)cc3)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](Cc3ccc(O)cc3)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](Cc3ccc(O)cc3)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC2=O)C(=O)N[C@@H](CC(N)=O)C(O)=O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](Cc2ccc(O)cc2)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)NC1=O)[C@@H](C)O)[C@@H](C)CC Chemical compound CC[C@H](C)[C@H](NC(=O)CN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H]1CSSC[C@@H]2NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CSSC[C@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](Cc3c[nH]cn3)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)Cc3ccccc3)C(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](Cc3c[nH]cn3)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](Cc3ccc(O)cc3)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](Cc3ccc(O)cc3)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](Cc3ccc(O)cc3)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC2=O)C(=O)N[C@@H](CC(N)=O)C(O)=O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](Cc2ccc(O)cc2)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)NC1=O)[C@@H](C)O)[C@@H](C)CC WEDIKSVWBUKTRA-WTKGVUNUSA-N 0.000 description 9
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 9
- 230000000172 allergic effect Effects 0.000 description 9
- 230000007423 decrease Effects 0.000 description 9
- 238000009826 distribution Methods 0.000 description 9
- 230000014509 gene expression Effects 0.000 description 9
- 230000035772 mutation Effects 0.000 description 9
- 230000035935 pregnancy Effects 0.000 description 9
- 238000011160 research Methods 0.000 description 9
- 238000007920 subcutaneous administration Methods 0.000 description 9
- 108010059886 Apolipoprotein A-I Proteins 0.000 description 8
- 102000005666 Apolipoprotein A-I Human genes 0.000 description 8
- 208000024172 Cardiovascular disease Diseases 0.000 description 8
- 241000699666 Mus <mouse, genus> Species 0.000 description 8
- 230000009102 absorption Effects 0.000 description 8
- 238000010521 absorption reaction Methods 0.000 description 8
- 102000053786 human PCSK9 Human genes 0.000 description 8
- 102000004420 Creatine Kinase Human genes 0.000 description 7
- 108010042126 Creatine kinase Proteins 0.000 description 7
- 102000025171 antigen binding proteins Human genes 0.000 description 7
- 108091000831 antigen binding proteins Proteins 0.000 description 7
- 230000036772 blood pressure Effects 0.000 description 7
- BGHSOEHUOOAYMY-JTZMCQEISA-N ghrelin Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)CN)C1=CC=CC=C1 BGHSOEHUOOAYMY-JTZMCQEISA-N 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- 102000004196 processed proteins & peptides Human genes 0.000 description 7
- BPYKTIZUTYGOLE-IFADSCNNSA-N Bilirubin Chemical compound N1C(=O)C(C)=C(C=C)\C1=C\C1=C(C)C(CCC(O)=O)=C(CC2=C(C(C)=C(\C=C/3C(=C(C=C)C(=O)N\3)C)N2)CCC(O)=O)N1 BPYKTIZUTYGOLE-IFADSCNNSA-N 0.000 description 6
- 206010020772 Hypertension Diseases 0.000 description 6
- 229940127355 PCSK9 Inhibitors Drugs 0.000 description 6
- 238000006243 chemical reaction Methods 0.000 description 6
- 239000003638 chemical reducing agent Substances 0.000 description 6
- 230000009850 completed effect Effects 0.000 description 6
- 201000010099 disease Diseases 0.000 description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 6
- 238000013146 percutaneous coronary intervention Methods 0.000 description 6
- 238000009597 pregnancy test Methods 0.000 description 6
- 230000004044 response Effects 0.000 description 6
- HSINOMROUCMIEA-FGVHQWLLSA-N (2s,4r)-4-[(3r,5s,6r,7r,8s,9s,10s,13r,14s,17r)-6-ethyl-3,7-dihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]-2-methylpentanoic acid Chemical compound C([C@@]12C)C[C@@H](O)C[C@H]1[C@@H](CC)[C@@H](O)[C@@H]1[C@@H]2CC[C@]2(C)[C@@H]([C@H](C)C[C@H](C)C(O)=O)CC[C@H]21 HSINOMROUCMIEA-FGVHQWLLSA-N 0.000 description 5
- 229940127291 Calcium channel antagonist Drugs 0.000 description 5
- 206010010904 Convulsion Diseases 0.000 description 5
- 206010012689 Diabetic retinopathy Diseases 0.000 description 5
- HTQBXNHDCUEHJF-XWLPCZSASA-N Exenatide Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 HTQBXNHDCUEHJF-XWLPCZSASA-N 0.000 description 5
- 108010011459 Exenatide Proteins 0.000 description 5
- 206010022095 Injection Site reaction Diseases 0.000 description 5
- XVVOERDUTLJJHN-UHFFFAOYSA-N Lixisenatide Chemical compound C=1NC2=CC=CC=C2C=1CC(C(=O)NC(CC(C)C)C(=O)NC(CCCCN)C(=O)NC(CC(N)=O)C(=O)NCC(=O)NCC(=O)N1C(CCC1)C(=O)NC(CO)C(=O)NC(CO)C(=O)NCC(=O)NC(C)C(=O)N1C(CCC1)C(=O)N1C(CCC1)C(=O)NC(CO)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)CC)NC(=O)C(NC(=O)C(CC(C)C)NC(=O)C(CCCNC(N)=N)NC(=O)C(NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(CCC(O)=O)NC(=O)C(CCC(O)=O)NC(=O)C(CCSC)NC(=O)C(CCC(N)=O)NC(=O)C(CCCCN)NC(=O)C(CO)NC(=O)C(CC(C)C)NC(=O)C(CC(O)=O)NC(=O)C(CO)NC(=O)C(NC(=O)C(CC=1C=CC=CC=1)NC(=O)C(NC(=O)CNC(=O)C(CCC(O)=O)NC(=O)CNC(=O)C(N)CC=1NC=NC=1)C(C)O)C(C)O)C(C)C)CC1=CC=CC=C1 XVVOERDUTLJJHN-UHFFFAOYSA-N 0.000 description 5
- 208000002193 Pain Diseases 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- 230000009471 action Effects 0.000 description 5
- 125000000539 amino acid group Chemical group 0.000 description 5
- 239000003613 bile acid Substances 0.000 description 5
- 238000013270 controlled release Methods 0.000 description 5
- 229940109239 creatinine Drugs 0.000 description 5
- 230000034994 death Effects 0.000 description 5
- 238000011156 evaluation Methods 0.000 description 5
- 230000007717 exclusion Effects 0.000 description 5
- YMTINGFKWWXKFG-UHFFFAOYSA-N fenofibrate Chemical compound C1=CC(OC(C)(C)C(=O)OC(C)C)=CC=C1C(=O)C1=CC=C(Cl)C=C1 YMTINGFKWWXKFG-UHFFFAOYSA-N 0.000 description 5
- 230000003993 interaction Effects 0.000 description 5
- 238000002372 labelling Methods 0.000 description 5
- 229960001093 lixisenatide Drugs 0.000 description 5
- 108010004367 lixisenatide Proteins 0.000 description 5
- 201000000083 maturity-onset diabetes of the young type 1 Diseases 0.000 description 5
- 230000007935 neutral effect Effects 0.000 description 5
- DHHVAGZRUROJKS-UHFFFAOYSA-N phentermine Chemical compound CC(C)(N)CC1=CC=CC=C1 DHHVAGZRUROJKS-UHFFFAOYSA-N 0.000 description 5
- 229920001223 polyethylene glycol Polymers 0.000 description 5
- 229920001184 polypeptide Polymers 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- 208000005176 Hepatitis C Diseases 0.000 description 4
- 108060003951 Immunoglobulin Proteins 0.000 description 4
- LTXREWYXXSTFRX-QGZVFWFLSA-N Linagliptin Chemical compound N=1C=2N(C)C(=O)N(CC=3N=C4C=CC=CC4=C(C)N=3)C(=O)C=2N(CC#CC)C=1N1CCC[C@@H](N)C1 LTXREWYXXSTFRX-QGZVFWFLSA-N 0.000 description 4
- 108010003723 Single-Domain Antibodies Proteins 0.000 description 4
- 210000001015 abdomen Anatomy 0.000 description 4
- 230000002378 acidificating effect Effects 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 229940124599 anti-inflammatory drug Drugs 0.000 description 4
- 229940090047 auto-injector Drugs 0.000 description 4
- 230000003111 delayed effect Effects 0.000 description 4
- 210000003743 erythrocyte Anatomy 0.000 description 4
- 102000018358 immunoglobulin Human genes 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 210000000265 leukocyte Anatomy 0.000 description 4
- 230000000926 neurological effect Effects 0.000 description 4
- 108010029667 pramlintide Proteins 0.000 description 4
- NRKVKVQDUCJPIZ-MKAGXXMWSA-N pramlintide acetate Chemical compound C([C@@H](C(=O)NCC(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CS)NC(=O)[C@@H](N)CCCCN)[C@@H](C)O)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 NRKVKVQDUCJPIZ-MKAGXXMWSA-N 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- 238000003860 storage Methods 0.000 description 4
- 238000011477 surgical intervention Methods 0.000 description 4
- 238000001356 surgical procedure Methods 0.000 description 4
- 238000012546 transfer Methods 0.000 description 4
- 238000011269 treatment regimen Methods 0.000 description 4
- 150000003626 triacylglycerols Chemical class 0.000 description 4
- 210000000689 upper leg Anatomy 0.000 description 4
- VOUAQYXWVJDEQY-QENPJCQMSA-N 33017-11-7 Chemical compound OC(=O)CC[C@H](N)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)NCC(=O)NCC(=O)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N1[C@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O)CCC1 VOUAQYXWVJDEQY-QENPJCQMSA-N 0.000 description 3
- 108010088751 Albumins Proteins 0.000 description 3
- 102000009027 Albumins Human genes 0.000 description 3
- 102400000345 Angiotensin-2 Human genes 0.000 description 3
- 101800000733 Angiotensin-2 Proteins 0.000 description 3
- 102000030169 Apolipoprotein C-III Human genes 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- 108010075254 C-Peptide Proteins 0.000 description 3
- 239000004072 C09CA03 - Valsartan Substances 0.000 description 3
- 101100112922 Candida albicans CDR3 gene Proteins 0.000 description 3
- 102000012336 Cholesterol Ester Transfer Proteins Human genes 0.000 description 3
- 108010061846 Cholesterol Ester Transfer Proteins Proteins 0.000 description 3
- 208000028698 Cognitive impairment Diseases 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 3
- 108060006698 EGF receptor Proteins 0.000 description 3
- 101000887427 Homo sapiens Probable G-protein coupled receptor 142 Proteins 0.000 description 3
- 206010020751 Hypersensitivity Diseases 0.000 description 3
- CZGUSIXMZVURDU-JZXHSEFVSA-N Ile(5)-angiotensin II Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC=1C=CC=CC=1)C([O-])=O)NC(=O)[C@@H](NC(=O)[C@H](CCCNC(N)=[NH2+])NC(=O)[C@@H]([NH3+])CC([O-])=O)C(C)C)C1=CC=C(O)C=C1 CZGUSIXMZVURDU-JZXHSEFVSA-N 0.000 description 3
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 3
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 3
- 108010033266 Lipoprotein(a) Proteins 0.000 description 3
- 102000057248 Lipoprotein(a) Human genes 0.000 description 3
- YSDQQAXHVYUZIW-QCIJIYAXSA-N Liraglutide Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCNC(=O)CC[C@H](NC(=O)CCCCCCCCCCCCCCC)C(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=C(O)C=C1 YSDQQAXHVYUZIW-QCIJIYAXSA-N 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- 206010028980 Neoplasm Diseases 0.000 description 3
- 102100039861 Probable G-protein coupled receptor 142 Human genes 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 3
- 238000008050 Total Bilirubin Reagent Methods 0.000 description 3
- 208000026935 allergic disease Diseases 0.000 description 3
- ZSBOMTDTBDDKMP-OAHLLOKOSA-N alogliptin Chemical compound C=1C=CC=C(C#N)C=1CN1C(=O)N(C)C(=O)C=C1N1CCC[C@@H](N)C1 ZSBOMTDTBDDKMP-OAHLLOKOSA-N 0.000 description 3
- 229950006323 angiotensin ii Drugs 0.000 description 3
- 239000000883 anti-obesity agent Substances 0.000 description 3
- 239000002220 antihypertensive agent Substances 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- SNPPWIUOZRMYNY-UHFFFAOYSA-N bupropion Chemical compound CC(C)(C)NC(C)C(=O)C1=CC=CC(Cl)=C1 SNPPWIUOZRMYNY-UHFFFAOYSA-N 0.000 description 3
- 208000010877 cognitive disease Diseases 0.000 description 3
- 230000036461 convulsion Effects 0.000 description 3
- 238000013461 design Methods 0.000 description 3
- 230000035487 diastolic blood pressure Effects 0.000 description 3
- 239000000539 dimer Substances 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 229960001519 exenatide Drugs 0.000 description 3
- 229960002297 fenofibrate Drugs 0.000 description 3
- 210000004408 hybridoma Anatomy 0.000 description 3
- 230000001024 immunotherapeutic effect Effects 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 239000004026 insulin derivative Substances 0.000 description 3
- 230000000302 ischemic effect Effects 0.000 description 3
- 210000004698 lymphocyte Anatomy 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 231100000682 maximum tolerated dose Toxicity 0.000 description 3
- 231100000350 mutagenesis Toxicity 0.000 description 3
- AHLBNYSZXLDEJQ-FWEHEUNISA-N orlistat Chemical compound CCCCCCCCCCC[C@H](OC(=O)[C@H](CC(C)C)NC=O)C[C@@H]1OC(=O)[C@H]1CCCCCC AHLBNYSZXLDEJQ-FWEHEUNISA-N 0.000 description 3
- 229920001983 poloxamer Polymers 0.000 description 3
- 230000002062 proliferating effect Effects 0.000 description 3
- 230000007115 recruitment Effects 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- 230000000250 revascularization Effects 0.000 description 3
- 238000002864 sequence alignment Methods 0.000 description 3
- MFFMDFFZMYYVKS-SECBINFHSA-N sitagliptin Chemical compound C([C@H](CC(=O)N1CC=2N(C(=NN=2)C(F)(F)F)CC1)N)C1=CC(F)=C(F)C=C1F MFFMDFFZMYYVKS-SECBINFHSA-N 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- RMMXLENWKUUMAY-UHFFFAOYSA-N telmisartan Chemical compound CCCC1=NC2=C(C)C=C(C=3N(C4=CC=CC=C4N=3)C)C=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C(O)=O RMMXLENWKUUMAY-UHFFFAOYSA-N 0.000 description 3
- 238000011285 therapeutic regimen Methods 0.000 description 3
- 238000004448 titration Methods 0.000 description 3
- 210000002700 urine Anatomy 0.000 description 3
- 229960005486 vaccine Drugs 0.000 description 3
- 230000004580 weight loss Effects 0.000 description 3
- XFJAMQQAAMJFGB-ZQGJOIPISA-N (2s,3r,4r,5s,6r)-2-[3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)-4-ethylphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3OCCOC3=CC=2)C(CC)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O XFJAMQQAAMJFGB-ZQGJOIPISA-N 0.000 description 2
- VTAKZNRDSPNOAU-UHFFFAOYSA-M 2-(chloromethyl)oxirane;hydron;prop-2-en-1-amine;n-prop-2-enyldecan-1-amine;trimethyl-[6-(prop-2-enylamino)hexyl]azanium;dichloride Chemical compound Cl.[Cl-].NCC=C.ClCC1CO1.CCCCCCCCCCNCC=C.C[N+](C)(C)CCCCCCNCC=C VTAKZNRDSPNOAU-UHFFFAOYSA-M 0.000 description 2
- PTIFVLOBVCIMKL-UHFFFAOYSA-N 2-[3-[1-(4-chlorophenyl)cyclopropyl]-[1,2,4]triazolo[4,3-a]pyridin-8-yl]propan-2-ol Chemical compound N=1N=C2C(C(C)(O)C)=CC=CN2C=1C1(C=2C=CC(Cl)=CC=2)CC1 PTIFVLOBVCIMKL-UHFFFAOYSA-N 0.000 description 2
- CZGVOBIGEBDYTP-VSGBNLITSA-N 3-[[(3r,5r)-3-butyl-3-ethyl-7-methoxy-1,1-dioxo-5-phenyl-4,5-dihydro-2h-1$l^{6},4-benzothiazepin-8-yl]methylamino]pentanedioic acid Chemical compound C1([C@@H]2C3=CC(OC)=C(CNC(CC(O)=O)CC(O)=O)C=C3S(=O)(=O)C[C@@](N2)(CC)CCCC)=CC=CC=C1 CZGVOBIGEBDYTP-VSGBNLITSA-N 0.000 description 2
- 102100029077 3-hydroxy-3-methylglutaryl-coenzyme A reductase Human genes 0.000 description 2
- 102100026105 3-ketoacyl-CoA thiolase, mitochondrial Human genes 0.000 description 2
- 108010006229 Acetyl-CoA C-acetyltransferase Proteins 0.000 description 2
- 108010005094 Advanced Glycation End Products Proteins 0.000 description 2
- 102000054930 Agouti-Related Human genes 0.000 description 2
- 101710127426 Agouti-related protein Proteins 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- 239000005485 Azilsartan Substances 0.000 description 2
- 239000002083 C09CA01 - Losartan Substances 0.000 description 2
- 239000002947 C09CA04 - Irbesartan Substances 0.000 description 2
- 239000005537 C09CA07 - Telmisartan Substances 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 102100033868 Cannabinoid receptor 1 Human genes 0.000 description 2
- 101710187010 Cannabinoid receptor 1 Proteins 0.000 description 2
- 229920001268 Cholestyramine Polymers 0.000 description 2
- 229920002905 Colesevelam Polymers 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- 101710088194 Dehydrogenase Proteins 0.000 description 2
- 201000004624 Dermatitis Diseases 0.000 description 2
- 206010012434 Dermatitis allergic Diseases 0.000 description 2
- 102100025012 Dipeptidyl peptidase 4 Human genes 0.000 description 2
- 208000032928 Dyslipidaemia Diseases 0.000 description 2
- 108030001679 Endothelin-converting enzyme 1 Proteins 0.000 description 2
- 102000048186 Endothelin-converting enzyme 1 Human genes 0.000 description 2
- 238000012276 Endovascular treatment Methods 0.000 description 2
- LCDDAGSJHKEABN-MLGOLLRUSA-N Evogliptin Chemical compound C1CNC(=O)[C@@H](COC(C)(C)C)N1C(=O)C[C@H](N)CC1=CC(F)=C(F)C=C1F LCDDAGSJHKEABN-MLGOLLRUSA-N 0.000 description 2
- 102100027844 Fibroblast growth factor receptor 4 Human genes 0.000 description 2
- 101710182387 Fibroblast growth factor receptor 4 Proteins 0.000 description 2
- 102000038624 GSKs Human genes 0.000 description 2
- 108091007911 GSKs Proteins 0.000 description 2
- 101000930822 Giardia intestinalis Dipeptidyl-peptidase 4 Proteins 0.000 description 2
- 102000017011 Glycated Hemoglobin A Human genes 0.000 description 2
- 108010014663 Glycated Hemoglobin A Proteins 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 108010090613 Human Regular Insulin Proteins 0.000 description 2
- 102000013266 Human Regular Insulin Human genes 0.000 description 2
- 208000013016 Hypoglycemia Diseases 0.000 description 2
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 2
- 206010022562 Intermittent claudication Diseases 0.000 description 2
- 101710172072 Kexin Proteins 0.000 description 2
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- 108010001831 LDL receptors Proteins 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 108010092277 Leptin Proteins 0.000 description 2
- 102000016267 Leptin Human genes 0.000 description 2
- 208000017170 Lipid metabolism disease Diseases 0.000 description 2
- 108010019598 Liraglutide Proteins 0.000 description 2
- 102100024640 Low-density lipoprotein receptor Human genes 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 206010027525 Microalbuminuria Diseases 0.000 description 2
- 208000000112 Myalgia Diseases 0.000 description 2
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 2
- 108091028043 Nucleic acid sequence Proteins 0.000 description 2
- 101800001672 Peptide YY(3-36) Proteins 0.000 description 2
- 208000018262 Peripheral vascular disease Diseases 0.000 description 2
- MFOCDFTXLCYLKU-CMPLNLGQSA-N Phendimetrazine Chemical compound O1CCN(C)[C@@H](C)[C@@H]1C1=CC=CC=C1 MFOCDFTXLCYLKU-CMPLNLGQSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 102100026918 Phospholipase A2 Human genes 0.000 description 2
- 108010058864 Phospholipases A2 Proteins 0.000 description 2
- 206010035664 Pneumonia Diseases 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 229920001213 Polysorbate 20 Polymers 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- AJLFOPYRIVGYMJ-UHFFFAOYSA-N SJ000287055 Natural products C12C(OC(=O)C(C)CC)CCC=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 AJLFOPYRIVGYMJ-UHFFFAOYSA-N 0.000 description 2
- 208000006011 Stroke Diseases 0.000 description 2
- 108090000787 Subtilisin Proteins 0.000 description 2
- 102000011923 Thyrotropin Human genes 0.000 description 2
- 108010061174 Thyrotropin Proteins 0.000 description 2
- KJADKKWYZYXHBB-XBWDGYHZSA-N Topiramic acid Chemical compound C1O[C@@]2(COS(N)(=O)=O)OC(C)(C)O[C@H]2[C@@H]2OC(C)(C)O[C@@H]21 KJADKKWYZYXHBB-XBWDGYHZSA-N 0.000 description 2
- 208000032109 Transient ischaemic attack Diseases 0.000 description 2
- 108010062497 VLDL Lipoproteins Proteins 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 230000005856 abnormality Effects 0.000 description 2
- 229940124308 alpha-adrenoreceptor antagonist Drugs 0.000 description 2
- 210000003484 anatomy Anatomy 0.000 description 2
- 229940125364 angiotensin receptor blocker Drugs 0.000 description 2
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 2
- 229940030600 antihypertensive agent Drugs 0.000 description 2
- 230000000923 atherogenic effect Effects 0.000 description 2
- 230000003143 atherosclerotic effect Effects 0.000 description 2
- 201000008937 atopic dermatitis Diseases 0.000 description 2
- 238000007681 bariatric surgery Methods 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- 239000002876 beta blocker Substances 0.000 description 2
- 229940097320 beta blocking agent Drugs 0.000 description 2
- IIBYAHWJQTYFKB-UHFFFAOYSA-N bezafibrate Chemical compound C1=CC(OC(C)(C)C(O)=O)=CC=C1CCNC(=O)C1=CC=C(Cl)C=C1 IIBYAHWJQTYFKB-UHFFFAOYSA-N 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 230000005540 biological transmission Effects 0.000 description 2
- 229960001058 bupropion Drugs 0.000 description 2
- 102220365045 c.89T>A Human genes 0.000 description 2
- 239000000480 calcium channel blocker Substances 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- HTQMVQVXFRQIKW-UHFFFAOYSA-N candesartan Chemical compound CCOC1=NC2=CC=CC(C(O)=O)=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C1=NN=NN1 HTQMVQVXFRQIKW-UHFFFAOYSA-N 0.000 description 2
- GPUADMRJQVPIAS-QCVDVZFFSA-M cerivastatin sodium Chemical compound [Na+].COCC1=C(C(C)C)N=C(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 GPUADMRJQVPIAS-QCVDVZFFSA-M 0.000 description 2
- MVCQKIKWYUURMU-UHFFFAOYSA-N cetilistat Chemical compound C1=C(C)C=C2C(=O)OC(OCCCCCCCCCCCCCCCC)=NC2=C1 MVCQKIKWYUURMU-UHFFFAOYSA-N 0.000 description 2
- 229960001152 colesevelam Drugs 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 238000002586 coronary angiography Methods 0.000 description 2
- 210000004351 coronary vessel Anatomy 0.000 description 2
- 230000003013 cytotoxicity Effects 0.000 description 2
- 231100000135 cytotoxicity Toxicity 0.000 description 2
- DBOBAIHRZONIPT-GHCHSQRSSA-N decanedioic acid;2,2-dimethyl-3-[3-[3-methyl-4-[[5-propan-2-yl-3-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-1h-pyrazol-4-yl]methyl]phenoxy]propylamino]propanamide Chemical compound OC(=O)CCCCCCCCC(O)=O.C=1C=C(OCCCNCC(C)(C)C(N)=O)C=C(C)C=1CC1=C(C(C)C)NN=C1O[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O.C=1C=C(OCCCNCC(C)(C)C(N)=O)C=C(C)C=1CC1=C(C(C)C)NN=C1O[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O DBOBAIHRZONIPT-GHCHSQRSSA-N 0.000 description 2
- 230000007547 defect Effects 0.000 description 2
- 230000007812 deficiency Effects 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- 235000005911 diet Nutrition 0.000 description 2
- 230000037213 diet Effects 0.000 description 2
- 229940074619 diovan Drugs 0.000 description 2
- 208000037765 diseases and disorders Diseases 0.000 description 2
- 239000002934 diuretic Substances 0.000 description 2
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 2
- 238000002592 echocardiography Methods 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 210000000981 epithelium Anatomy 0.000 description 2
- OROAFUQRIXKEMV-LDADJPATSA-N eprosartan Chemical compound C=1C=C(C(O)=O)C=CC=1CN1C(CCCC)=NC=C1\C=C(C(O)=O)/CC1=CC=CS1 OROAFUQRIXKEMV-LDADJPATSA-N 0.000 description 2
- IHIUGIVXARLYHP-YBXDKENTSA-N evacetrapib Chemical compound C1([C@@H](N(CC=2C=C(C=C(C=2)C(F)(F)F)C(F)(F)F)C2=NN(C)N=N2)CCC2)=CC(C)=CC(C)=C1N2C[C@H]1CC[C@H](C(O)=O)CC1 IHIUGIVXARLYHP-YBXDKENTSA-N 0.000 description 2
- 229950011259 evogliptin Drugs 0.000 description 2
- 235000019197 fats Nutrition 0.000 description 2
- 230000010030 glucose lowering effect Effects 0.000 description 2
- 208000002672 hepatitis B Diseases 0.000 description 2
- 238000002657 hormone replacement therapy Methods 0.000 description 2
- 229940038661 humalog Drugs 0.000 description 2
- 229940103471 humulin Drugs 0.000 description 2
- 201000001421 hyperglycemia Diseases 0.000 description 2
- 230000009610 hypersensitivity Effects 0.000 description 2
- 230000001631 hypertensive effect Effects 0.000 description 2
- 230000002218 hypoglycaemic effect Effects 0.000 description 2
- 239000012678 infectious agent Substances 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 208000014674 injury Diseases 0.000 description 2
- 238000003780 insertion Methods 0.000 description 2
- 230000037431 insertion Effects 0.000 description 2
- 208000021156 intermittent vascular claudication Diseases 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 229960002198 irbesartan Drugs 0.000 description 2
- YCPOHTHPUREGFM-UHFFFAOYSA-N irbesartan Chemical compound O=C1N(CC=2C=CC(=CC=2)C=2C(=CC=CC=2)C=2[N]N=NN=2)C(CCCC)=NC21CCCC2 YCPOHTHPUREGFM-UHFFFAOYSA-N 0.000 description 2
- 230000003907 kidney function Effects 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 210000002414 leg Anatomy 0.000 description 2
- 229940039781 leptin Drugs 0.000 description 2
- NRYBAZVQPHGZNS-ZSOCWYAHSA-N leptin Chemical compound O=C([C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CC(C)C)CCSC)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CS)C(O)=O NRYBAZVQPHGZNS-ZSOCWYAHSA-N 0.000 description 2
- 230000037356 lipid metabolism Effects 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- MBBCVAKAJPKAKM-UHFFFAOYSA-N lomitapide Chemical compound C12=CC=CC=C2C2=CC=CC=C2C1(C(=O)NCC(F)(F)F)CCCCN(CC1)CCC1NC(=O)C1=CC=CC=C1C1=CC=C(C(F)(F)F)C=C1 MBBCVAKAJPKAKM-UHFFFAOYSA-N 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 210000003141 lower extremity Anatomy 0.000 description 2
- RLSSMJSEOOYNOY-UHFFFAOYSA-N m-cresol Chemical compound CC1=CC=CC(O)=C1 RLSSMJSEOOYNOY-UHFFFAOYSA-N 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 238000002483 medication Methods 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 229960004452 methionine Drugs 0.000 description 2
- AJLFOPYRIVGYMJ-INTXDZFKSA-N mevastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=CCC[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 AJLFOPYRIVGYMJ-INTXDZFKSA-N 0.000 description 2
- BOZILQFLQYBIIY-UHFFFAOYSA-N mevastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CCC=C21 BOZILQFLQYBIIY-UHFFFAOYSA-N 0.000 description 2
- 238000012544 monitoring process Methods 0.000 description 2
- 210000003205 muscle Anatomy 0.000 description 2
- 238000002703 mutagenesis Methods 0.000 description 2
- DQCKKXVULJGBQN-XFWGSAIBSA-N naltrexone Chemical compound N1([C@@H]2CC3=CC=C(C=4O[C@@H]5[C@](C3=4)([C@]2(CCC5=O)O)CC1)O)CC1CC1 DQCKKXVULJGBQN-XFWGSAIBSA-N 0.000 description 2
- 229960003086 naltrexone Drugs 0.000 description 2
- 239000002547 new drug Substances 0.000 description 2
- 238000012633 nuclear imaging Methods 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- VTRAEEWXHOVJFV-UHFFFAOYSA-N olmesartan Chemical compound CCCC1=NC(C(C)(C)O)=C(C(O)=O)N1CC1=CC=C(C=2C(=CC=CC=2)C=2NN=NN=2)C=C1 VTRAEEWXHOVJFV-UHFFFAOYSA-N 0.000 description 2
- 229940125395 oral insulin Drugs 0.000 description 2
- 229960001243 orlistat Drugs 0.000 description 2
- 238000004806 packaging method and process Methods 0.000 description 2
- 239000004031 partial agonist Substances 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 229960003562 phentermine Drugs 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- RHGYHLPFVJEAOC-FFNUKLMVSA-L pitavastatin calcium Chemical group [Ca+2].[O-]C(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1.[O-]C(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 RHGYHLPFVJEAOC-FFNUKLMVSA-L 0.000 description 2
- 238000013439 planning Methods 0.000 description 2
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 2
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 2
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 2
- 229920000136 polysorbate Polymers 0.000 description 2
- 229940068977 polysorbate 20 Drugs 0.000 description 2
- 229920000053 polysorbate 80 Polymers 0.000 description 2
- 229940068968 polysorbate 80 Drugs 0.000 description 2
- 229960003611 pramlintide Drugs 0.000 description 2
- QGJUIPDUBHWZPV-SGTAVMJGSA-N saxagliptin Chemical compound C1C(C2)CC(C3)CC2(O)CC13[C@H](N)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 QGJUIPDUBHWZPV-SGTAVMJGSA-N 0.000 description 2
- 108010033693 saxagliptin Proteins 0.000 description 2
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 2
- VGKDLMBJGBXTGI-SJCJKPOMSA-N sertraline Chemical compound C1([C@@H]2CC[C@@H](C3=CC=CC=C32)NC)=CC=C(Cl)C(Cl)=C1 VGKDLMBJGBXTGI-SJCJKPOMSA-N 0.000 description 2
- UNAANXDKBXWMLN-UHFFFAOYSA-N sibutramine Chemical compound C=1C=C(Cl)C=CC=1C1(C(N(C)C)CC(C)C)CCC1 UNAANXDKBXWMLN-UHFFFAOYSA-N 0.000 description 2
- 210000002027 skeletal muscle Anatomy 0.000 description 2
- 230000000391 smoking effect Effects 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 230000006641 stabilisation Effects 0.000 description 2
- 238000011105 stabilization Methods 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- 238000013517 stratification Methods 0.000 description 2
- 238000009662 stress testing Methods 0.000 description 2
- 238000010254 subcutaneous injection Methods 0.000 description 2
- 239000007929 subcutaneous injection Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 208000011117 substance-related disease Diseases 0.000 description 2
- 150000005846 sugar alcohols Polymers 0.000 description 2
- 229940099093 symlin Drugs 0.000 description 2
- 230000035488 systolic blood pressure Effects 0.000 description 2
- 229950000034 teneligliptin Drugs 0.000 description 2
- WGRQANOPCQRCME-PMACEKPBSA-N teneligliptin Chemical compound O=C([C@H]1NC[C@H](C1)N1CCN(CC1)C1=CC(=NN1C=1C=CC=CC=1)C)N1CCSC1 WGRQANOPCQRCME-PMACEKPBSA-N 0.000 description 2
- VCVWXKKWDOJNIT-ZOMKSWQUSA-N tesofensine Chemical compound C1([C@H]2C[C@@H]3CC[C@@H](N3C)[C@@H]2COCC)=CC=C(Cl)C(Cl)=C1 VCVWXKKWDOJNIT-ZOMKSWQUSA-N 0.000 description 2
- 229950009970 tesofensine Drugs 0.000 description 2
- 229940127279 trajenta Drugs 0.000 description 2
- 230000009261 transgenic effect Effects 0.000 description 2
- 238000011830 transgenic mouse model Methods 0.000 description 2
- 201000010875 transient cerebral ischemia Diseases 0.000 description 2
- 230000032258 transport Effects 0.000 description 2
- 230000008733 trauma Effects 0.000 description 2
- 238000002562 urinalysis Methods 0.000 description 2
- OBHRVMZSZIDDEK-UHFFFAOYSA-N urobilinogen Chemical compound CCC1=C(C)C(=O)NC1CC1=C(C)C(CCC(O)=O)=C(CC2=C(C(C)=C(CC3C(=C(CC)C(=O)N3)C)N2)CCC(O)=O)N1 OBHRVMZSZIDDEK-UHFFFAOYSA-N 0.000 description 2
- ACWBQPMHZXGDFX-QFIPXVFZSA-N valsartan Chemical compound C1=CC(CN(C(=O)CCCC)[C@@H](C(C)C)C(O)=O)=CC=C1C1=CC=CC=C1C1=NN=NN1 ACWBQPMHZXGDFX-QFIPXVFZSA-N 0.000 description 2
- 229960001254 vildagliptin Drugs 0.000 description 2
- SYOKIDBDQMKNDQ-XWTIBIIYSA-N vildagliptin Chemical compound C1C(O)(C2)CC(C3)CC1CC32NCC(=O)N1CCC[C@H]1C#N SYOKIDBDQMKNDQ-XWTIBIIYSA-N 0.000 description 2
- 238000005303 weighing Methods 0.000 description 2
- 208000016261 weight loss Diseases 0.000 description 2
- UBQNRHZMVUUOMG-UHFFFAOYSA-N zonisamide Chemical compound C1=CC=C2C(CS(=O)(=O)N)=NOC2=C1 UBQNRHZMVUUOMG-UHFFFAOYSA-N 0.000 description 2
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 1
- QUKZWYNGKYRRKE-RIKNOMPASA-N (1r,3r)-2-[(2r)-2-amino-2-(3-hydroxy-1-adamantyl)acetyl]-2-azabicyclo[3.1.0]hexane-3-carbonitrile;3-(diaminomethylidene)-1,1-dimethylguanidine Chemical compound CN(C)C(=N)N=C(N)N.C1C(C2)CC(C3)CC2(O)CC13[C@@H](N)C(=O)N1[C@@H](C#N)CC2C[C@H]21 QUKZWYNGKYRRKE-RIKNOMPASA-N 0.000 description 1
- PCOMIRCNMMNOAP-CQSZACIVSA-N (1s)-1-[5-[[3-(2-methylpyridin-3-yl)oxy-5-pyridin-2-ylsulfanylpyridin-2-yl]amino]-1,2,4-thiadiazol-3-yl]ethane-1,2-diol Chemical compound CC1=NC=CC=C1OC1=CC(SC=2N=CC=CC=2)=CN=C1NC1=NC([C@H](O)CO)=NS1 PCOMIRCNMMNOAP-CQSZACIVSA-N 0.000 description 1
- XUFXOAAUWZOOIT-SXARVLRPSA-N (2R,3R,4R,5S,6R)-5-[[(2R,3R,4R,5S,6R)-5-[[(2R,3R,4S,5S,6R)-3,4-dihydroxy-6-methyl-5-[[(1S,4R,5S,6S)-4,5,6-trihydroxy-3-(hydroxymethyl)-1-cyclohex-2-enyl]amino]-2-oxanyl]oxy]-3,4-dihydroxy-6-(hydroxymethyl)-2-oxanyl]oxy]-6-(hydroxymethyl)oxane-2,3,4-triol Chemical compound O([C@H]1O[C@H](CO)[C@H]([C@@H]([C@H]1O)O)O[C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O)C(CO)=C1)O)C)[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O XUFXOAAUWZOOIT-SXARVLRPSA-N 0.000 description 1
- ZWASRJHIEFYJGL-BFUOFWGJSA-N (2r)-1,1,1-trifluoro-2-[3-[(2r)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2-methylpiperazin-1-yl]sulfonylphenyl]propan-2-ol Chemical compound C([C@H]1C)N(C=2C(=CC(F)=CC=2)C(F)(F)F)CCN1S(=O)(=O)C1=CC=CC([C@@](C)(O)C(F)(F)F)=C1 ZWASRJHIEFYJGL-BFUOFWGJSA-N 0.000 description 1
- MSFZPBXAGPYVFD-NFBCFJMWSA-N (2r)-2-amino-3-[1-[3-[2-[2-[2-[4-[[(5s)-5,6-diamino-6-oxohexyl]amino]butylamino]-2-oxoethoxy]ethoxy]ethylamino]-3-oxopropyl]-2,5-dioxopyrrolidin-3-yl]sulfanylpropanoic acid Chemical compound NC(=O)[C@@H](N)CCCCNCCCCNC(=O)COCCOCCNC(=O)CCN1C(=O)CC(SC[C@H](N)C(O)=O)C1=O MSFZPBXAGPYVFD-NFBCFJMWSA-N 0.000 description 1
- VEPOHXYIFQMVHW-PVJVQHJQSA-N (2r,3r)-2,3-dihydroxybutanedioic acid;(2s,3s)-3,4-dimethyl-2-phenylmorpholine Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O.O1CCN(C)[C@@H](C)[C@@H]1C1=CC=CC=C1 VEPOHXYIFQMVHW-PVJVQHJQSA-N 0.000 description 1
- LOGFVTREOLYCPF-KXNHARMFSA-N (2s,3r)-2-[[(2r)-1-[(2s)-2,6-diaminohexanoyl]pyrrolidine-2-carbonyl]amino]-3-hydroxybutanoic acid Chemical compound C[C@@H](O)[C@@H](C(O)=O)NC(=O)[C@H]1CCCN1C(=O)[C@@H](N)CCCCN LOGFVTREOLYCPF-KXNHARMFSA-N 0.000 description 1
- BTCRKOKVYTVOLU-SJSRKZJXSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[[4-(2-cyclopropyloxyethoxy)phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=CC(OCCOC3CC3)=CC=2)=C1 BTCRKOKVYTVOLU-SJSRKZJXSA-N 0.000 description 1
- NPBCMXATLRCCLF-IRRLEISYSA-N (2s,4r)-4-[(3r,5s,6r,7r,8r,9s,10s,12s,13r,14s,17r)-6-ethyl-3,7,12-trihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]-2-methylpentanoic acid Chemical compound C([C@@]12C)C[C@@H](O)C[C@H]1[C@@H](CC)[C@@H](O)[C@@H]1[C@@H]2C[C@H](O)[C@]2(C)[C@@H]([C@H](C)C[C@H](C)C(O)=O)CC[C@H]21 NPBCMXATLRCCLF-IRRLEISYSA-N 0.000 description 1
- GCERFBKFVDLDKD-NRYJBHHQSA-N (3r)-3-amino-1-[3-(trifluoromethyl)-6,8-dihydro-5h-[1,2,4]triazolo[4,3-a]pyrazin-7-yl]-4-(2,4,5-trifluorophenyl)butan-1-one;3-(diaminomethylidene)-1,1-dimethylguanidine;phosphoric acid;hydrochloride Chemical compound Cl.OP(O)(O)=O.CN(C)C(=N)N=C(N)N.C([C@H](CC(=O)N1CC=2N(C(=NN=2)C(F)(F)F)CC1)N)C1=CC(F)=C(F)C=C1F GCERFBKFVDLDKD-NRYJBHHQSA-N 0.000 description 1
- JJKOQZHWYLMASZ-FJWDNACWSA-N (3s,7r,8r,9s,10r,13s,14s,17r)-17-ethynyl-10,13-dimethyl-1,2,3,4,7,8,9,11,12,14,15,16-dodecahydrocyclopenta[a]phenanthrene-3,7,17-triol Chemical compound C1[C@@H](O)CC[C@]2(C)[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3[C@@H](O)C=C21 JJKOQZHWYLMASZ-FJWDNACWSA-N 0.000 description 1
- MZZLGJHLQGUVPN-PVPMGCCUSA-N (4R,5S)-5-[3,5-bis(trifluoromethyl)phenyl]-3-[[2-(4-fluoro-2-methoxy-5-propan-2-ylphenyl)-5-(trifluoromethyl)phenyl]methyl]-4-methyl-1,3-oxazolidin-2-one Chemical compound COC1=C(C=C(C(C)C)C(F)=C1)C1=C(CN2[C@H](C)[C@@H](OC2=O)C2=CC(=CC(=C2)C(F)(F)F)C(F)(F)F)C=C(C=C1)C(F)(F)F MZZLGJHLQGUVPN-PVPMGCCUSA-N 0.000 description 1
- QIQSYARFOIKJJR-LUTWCBITSA-N (4z,7z,10z,13z,16z,19z)-docosa-4,7,10,13,16,19-hexaenoic acid;(4z,7z,10z,13z,16z)-docosa-4,7,10,13,16-pentaenoic acid;(7z,10z,13z,16z,19z)-docosa-7,10,13,16,19-pentaenoic acid;(6z,9z,12z,15z,18z)-henicosa-6,9,12,15,18-pentaenoic acid;(5z,8z,11z,14z,17z)-i Chemical compound CCCCC\C=C/C\C=C/CCCCCCCC(O)=O.CCCCC\C=C/C\C=C/C\C=C/CCCCC(O)=O.CC\C=C/C\C=C/C\C=C/CCCCCCCC(O)=O.CC\C=C/C\C=C/C\C=C/C\C=C/CCCCCCC(O)=O.CC\C=C/C\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O.CC\C=C/C\C=C/C\C=C/C\C=C/C\C=C/CCCCC(O)=O.CCCCC\C=C/C\C=C/C\C=C/C\C=C/C\C=C/CCC(O)=O.CC\C=C/C\C=C/C\C=C/C\C=C/C\C=C/CCCCCC(O)=O.CC\C=C/C\C=C/C\C=C/C\C=C/C\C=C/C\C=C/CCC(O)=O QIQSYARFOIKJJR-LUTWCBITSA-N 0.000 description 1
- TXNPQZGSVXLGGP-MMTVBGGISA-N (6s)-6-(2-hydroxy-2-methylpropyl)-3-[(1s)-1-[4-(1-methyl-2-oxopyridin-4-yl)phenyl]ethyl]-6-phenyl-1,3-oxazinan-2-one Chemical compound C1([C@@]2(CC(C)(C)O)CCN(C(O2)=O)[C@@H](C)C=2C=CC(=CC=2)C2=CC(=O)N(C)C=C2)=CC=CC=C1 TXNPQZGSVXLGGP-MMTVBGGISA-N 0.000 description 1
- UCTWMZQNUQWSLP-VIFPVBQESA-N (R)-adrenaline Chemical compound CNC[C@H](O)C1=CC=C(O)C(O)=C1 UCTWMZQNUQWSLP-VIFPVBQESA-N 0.000 description 1
- RTHCYVBBDHJXIQ-MRXNPFEDSA-N (R)-fluoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-MRXNPFEDSA-N 0.000 description 1
- ZEUITGRIYCTCEM-KRWDZBQOSA-N (S)-duloxetine Chemical compound C1([C@@H](OC=2C3=CC=CC=C3C=CC=2)CCNC)=CC=CS1 ZEUITGRIYCTCEM-KRWDZBQOSA-N 0.000 description 1
- OAAZMUGLOXGVNH-UHFFFAOYSA-N 1-(3-hydroxyazetidin-1-yl)-2-(6-hydroxy-2-phenyl-2-adamantyl)ethanone Chemical compound C1C(O)CN1C(=O)CC1(C=2C=CC=CC=2)C2CC(C3O)CC1CC3C2 OAAZMUGLOXGVNH-UHFFFAOYSA-N 0.000 description 1
- VOGXDRFFBBLZBT-AAQCHOMXSA-N 2-[(5z,8z,11z,14z,17z)-icosa-5,8,11,14,17-pentaenoxy]butanoic acid Chemical compound CC\C=C/C\C=C/C\C=C/C\C=C/C\C=C/CCCCOC(CC)C(O)=O VOGXDRFFBBLZBT-AAQCHOMXSA-N 0.000 description 1
- NFTMKHWBOINJGM-UHFFFAOYSA-N 2-[1-(5-ethylpyrimidin-2-yl)piperidin-4-yl]-4-[[4-(tetrazol-1-yl)phenoxy]methyl]-1,3-thiazole Chemical compound N1=CC(CC)=CN=C1N1CCC(C=2SC=C(COC=3C=CC(=CC=3)N3N=NN=C3)N=2)CC1 NFTMKHWBOINJGM-UHFFFAOYSA-N 0.000 description 1
- AYKLXGCULGWUJX-UHFFFAOYSA-N 2-[5-chloro-2-[[1-[(3,4-difluorophenyl)methyl]-4-[(4-methylsulfonylphenyl)methyl]pyrrole-2-carbonyl]amino]-1,3-thiazol-4-yl]acetic acid Chemical compound C1=CC(S(=O)(=O)C)=CC=C1CC1=CN(CC=2C=C(F)C(F)=CC=2)C(C(=O)NC=2SC(Cl)=C(CC(O)=O)N=2)=C1 AYKLXGCULGWUJX-UHFFFAOYSA-N 0.000 description 1
- HPGJSAAUJGAMLV-UHFFFAOYSA-N 2-[[2-[[cyclohexyl-(4-propoxycyclohexyl)carbamoyl]amino]-1,3-thiazol-5-yl]sulfanyl]acetic acid Chemical compound C1CC(OCCC)CCC1N(C(=O)NC=1SC(SCC(O)=O)=CN=1)C1CCCCC1 HPGJSAAUJGAMLV-UHFFFAOYSA-N 0.000 description 1
- NZPSYYOURGWZCM-UHFFFAOYSA-N 2-butyl-3-[[4-[2-(2h-tetrazol-5-yl)phenyl]phenyl]methyl]-1,3-diazaspiro[4.4]non-1-en-4-one;6-chloro-1,1-dioxo-3,4-dihydro-2h-1$l^{6},2,4-benzothiadiazine-7-sulfonamide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O.O=C1N(CC=2C=CC(=CC=2)C=2C(=CC=CC=2)C2=NNN=N2)C(CCCC)=NC21CCCC2 NZPSYYOURGWZCM-UHFFFAOYSA-N 0.000 description 1
- RFVNOJDQRGSOEL-UHFFFAOYSA-N 2-hydroxyethyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCCO RFVNOJDQRGSOEL-UHFFFAOYSA-N 0.000 description 1
- RZPAXNJLEKLXNO-UKNNTIGFSA-N 22-Hydroxycholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)C(O)CCC(C)C)[C@@]1(C)CC2 RZPAXNJLEKLXNO-UKNNTIGFSA-N 0.000 description 1
- FODHWOBAQBTTFS-UHFFFAOYSA-N 3-[4-[2-(2-methylphenyl)ethynyl]phenyl]propanoic acid Chemical compound CC1=CC=CC=C1C#CC1=CC=C(CCC(O)=O)C=C1 FODHWOBAQBTTFS-UHFFFAOYSA-N 0.000 description 1
- OCNBSSLDAIWTKS-UHFFFAOYSA-N 3-[[[3,5-bis(trifluoromethyl)phenyl]methyl-(2-methyltetrazol-5-yl)amino]methyl]-n,n-bis(cyclopropylmethyl)-8-methylquinolin-2-amine Chemical compound C1CC1CN(CC1CC1)C=1N=C2C(C)=CC=CC2=CC=1CN(C1=NN(C)N=N1)CC1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 OCNBSSLDAIWTKS-UHFFFAOYSA-N 0.000 description 1
- 101710158485 3-hydroxy-3-methylglutaryl-coenzyme A reductase Proteins 0.000 description 1
- VUUUHLIHQHVLLE-UHFFFAOYSA-N 4-[(5-fluoropyridin-2-yl)methoxy]-1-(5-methyl-1,2,3,4-tetrahydropyrido[4,3-b]indol-7-yl)pyridin-2-one Chemical compound C1=C2N(C)C=3CCNCC=3C2=CC=C1N(C(C=1)=O)C=CC=1OCC1=CC=C(F)C=N1 VUUUHLIHQHVLLE-UHFFFAOYSA-N 0.000 description 1
- XWBXJBSVYVJAMZ-UHFFFAOYSA-N 4-[4-(2-adamantylcarbamoyl)-5-tert-butylpyrazol-1-yl]benzoic acid Chemical compound CC(C)(C)C1=C(C(=O)NC2C3CC4CC(C3)CC2C4)C=NN1C1=CC=C(C(O)=O)C=C1 XWBXJBSVYVJAMZ-UHFFFAOYSA-N 0.000 description 1
- LUUMLYXKTPBTQR-UHFFFAOYSA-N 4-chloro-n-[5-methyl-2-(7h-pyrrolo[2,3-d]pyrimidine-4-carbonyl)pyridin-3-yl]-3-(trifluoromethyl)benzenesulfonamide Chemical compound C=1C(C)=CN=C(C(=O)C=2C=3C=CNC=3N=CN=2)C=1NS(=O)(=O)C1=CC=C(Cl)C(C(F)(F)F)=C1 LUUMLYXKTPBTQR-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-M 4-hydroxybenzoate Chemical compound OC1=CC=C(C([O-])=O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-M 0.000 description 1
- NKOHRVBBQISBSB-UHFFFAOYSA-N 5-[(4-hydroxyphenyl)methyl]-1,3-thiazolidine-2,4-dione Chemical class C1=CC(O)=CC=C1CC1C(=O)NC(=O)S1 NKOHRVBBQISBSB-UHFFFAOYSA-N 0.000 description 1
- AYJRTVVIBJSSKN-UHFFFAOYSA-N 5-[4-[[6-(4-methylsulfonylphenyl)pyridin-3-yl]oxymethyl]piperidin-1-yl]-3-propan-2-yl-1,2,4-oxadiazole Chemical compound CC(C)C1=NOC(N2CCC(COC=3C=NC(=CC=3)C=3C=CC(=CC=3)S(C)(=O)=O)CC2)=N1 AYJRTVVIBJSSKN-UHFFFAOYSA-N 0.000 description 1
- 239000005541 ACE inhibitor Substances 0.000 description 1
- 239000008498 ALS-L1023 Substances 0.000 description 1
- 102000014156 AMP-Activated Protein Kinases Human genes 0.000 description 1
- 108010011376 AMP-Activated Protein Kinases Proteins 0.000 description 1
- 206010069754 Acquired gene mutation Diseases 0.000 description 1
- 208000020576 Adrenal disease Diseases 0.000 description 1
- 201000010000 Agranulocytosis Diseases 0.000 description 1
- 229940123338 Aldosterone synthase inhibitor Drugs 0.000 description 1
- 201000000736 Amenorrhea Diseases 0.000 description 1
- 206010001928 Amenorrhoea Diseases 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 208000000044 Amnesia Diseases 0.000 description 1
- 102000045205 Angiopoietin-Like Protein 4 Human genes 0.000 description 1
- 108700041144 Angiopoietin-Like Protein 8 Proteins 0.000 description 1
- 102100034604 Angiopoietin-like protein 8 Human genes 0.000 description 1
- 102100025668 Angiopoietin-related protein 3 Human genes 0.000 description 1
- 101710085848 Angiopoietin-related protein 3 Proteins 0.000 description 1
- 101710085845 Angiopoietin-related protein 4 Proteins 0.000 description 1
- VWVKUNOPTJGDOB-BDHVOXNPSA-N Anhydrous tofogliflozin Chemical compound C1=CC(CC)=CC=C1CC1=CC=C(CO[C@@]23[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O3)O)C2=C1 VWVKUNOPTJGDOB-BDHVOXNPSA-N 0.000 description 1
- 108010032595 Antibody Binding Sites Proteins 0.000 description 1
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 1
- 208000032467 Aplastic anaemia Diseases 0.000 description 1
- 108091023037 Aptamer Proteins 0.000 description 1
- 101100460776 Arabidopsis thaliana NPY2 gene Proteins 0.000 description 1
- 101100460782 Arabidopsis thaliana NPY4 gene Proteins 0.000 description 1
- 208000006820 Arthralgia Diseases 0.000 description 1
- 206010003591 Ataxia Diseases 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 102100034159 Beta-3 adrenergic receptor Human genes 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 206010006482 Bronchospasm Diseases 0.000 description 1
- HEYVINCGKDONRU-UHFFFAOYSA-N Bupropion hydrochloride Chemical compound Cl.CC(C)(C)NC(C)C(=O)C1=CC=CC(Cl)=C1 HEYVINCGKDONRU-UHFFFAOYSA-N 0.000 description 1
- 239000002080 C09CA02 - Eprosartan Substances 0.000 description 1
- 239000002081 C09CA05 - Tasosartan Substances 0.000 description 1
- 239000002053 C09CA06 - Candesartan Substances 0.000 description 1
- OBMZMSLWNNWEJA-XNCRXQDQSA-N C1=CC=2C(C[C@@H]3NC(=O)[C@@H](NC(=O)[C@H](NC(=O)N(CC#CCN(CCCC[C@H](NC(=O)[C@@H](CC4=CC=CC=C4)NC3=O)C(=O)N)CC=C)NC(=O)[C@@H](N)C)CC3=CNC4=C3C=CC=C4)C)=CNC=2C=C1 Chemical compound C1=CC=2C(C[C@@H]3NC(=O)[C@@H](NC(=O)[C@H](NC(=O)N(CC#CCN(CCCC[C@H](NC(=O)[C@@H](CC4=CC=CC=C4)NC3=O)C(=O)N)CC=C)NC(=O)[C@@H](N)C)CC3=CNC4=C3C=CC=C4)C)=CNC=2C=C1 OBMZMSLWNNWEJA-XNCRXQDQSA-N 0.000 description 1
- 108091008927 CC chemokine receptors Proteins 0.000 description 1
- PWDLDBWXTVILPC-WGAVTJJLSA-N CC(C)(N)CC1=CC=CC=C1.C1O[C@@]2(COS(N)(=O)=O)OC(C)(C)O[C@H]2[C@@H]2OC(C)(C)O[C@@H]21 Chemical compound CC(C)(N)CC1=CC=CC=C1.C1O[C@@]2(COS(N)(=O)=O)OC(C)(C)O[C@H]2[C@@H]2OC(C)(C)O[C@@H]21 PWDLDBWXTVILPC-WGAVTJJLSA-N 0.000 description 1
- 102000005674 CCR Receptors Human genes 0.000 description 1
- 108010002025 CER-001 Proteins 0.000 description 1
- 108010055448 CJC 1131 Proteins 0.000 description 1
- 108010007056 CKGGRAKDC-GG-D(KLAKLAK)2 Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- GHOSNRCGJFBJIB-UHFFFAOYSA-N Candesartan cilexetil Chemical compound C=12N(CC=3C=CC(=CC=3)C=3C(=CC=CC=3)C3=NNN=N3)C(OCC)=NC2=CC=CC=1C(=O)OC(C)OC(=O)OC1CCCCC1 GHOSNRCGJFBJIB-UHFFFAOYSA-N 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- 229940123239 Cholesterol synthesis inhibitor Drugs 0.000 description 1
- 241000251730 Chondrichthyes Species 0.000 description 1
- KPSRODZRAIWAKH-JTQLQIEISA-N Ciprofibrate Natural products C1=CC(OC(C)(C)C(O)=O)=CC=C1[C@H]1C(Cl)(Cl)C1 KPSRODZRAIWAKH-JTQLQIEISA-N 0.000 description 1
- BMOVQUBVGICXQN-UHFFFAOYSA-N Clinofibrate Chemical compound C1=CC(OC(C)(CC)C(O)=O)=CC=C1C1(C=2C=CC(OC(C)(CC)C(O)=O)=CC=2)CCCCC1 BMOVQUBVGICXQN-UHFFFAOYSA-N 0.000 description 1
- 108020004705 Codon Proteins 0.000 description 1
- 229920002911 Colestipol Polymers 0.000 description 1
- 208000031288 Combined hyperlipidaemia Diseases 0.000 description 1
- VGMFHMLQOYWYHN-UHFFFAOYSA-N Compactin Natural products OCC1OC(OC2C(O)C(O)C(CO)OC2Oc3cc(O)c4C(=O)C(=COc4c3)c5ccc(O)c(O)c5)C(O)C(O)C1O VGMFHMLQOYWYHN-UHFFFAOYSA-N 0.000 description 1
- 206010010144 Completed suicide Diseases 0.000 description 1
- 208000032170 Congenital Abnormalities Diseases 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 108091035707 Consensus sequence Proteins 0.000 description 1
- 206010011224 Cough Diseases 0.000 description 1
- FFEARJCKVFRZRR-SCSAIBSYSA-N D-methionine Chemical compound CSCC[C@@H](N)C(O)=O FFEARJCKVFRZRR-SCSAIBSYSA-N 0.000 description 1
- 229930182818 D-methionine Natural products 0.000 description 1
- XUIIKFGFIJCVMT-GFCCVEGCSA-N D-thyroxine Chemical compound IC1=CC(C[C@@H](N)C(O)=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-GFCCVEGCSA-N 0.000 description 1
- JVHXJTBJCFBINQ-ADAARDCZSA-N Dapagliflozin Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)=CC=C1Cl JVHXJTBJCFBINQ-ADAARDCZSA-N 0.000 description 1
- 208000019505 Deglutition disease Diseases 0.000 description 1
- 108700022150 Designed Ankyrin Repeat Proteins Proteins 0.000 description 1
- 208000001380 Diabetic Ketoacidosis Diseases 0.000 description 1
- 208000007342 Diabetic Nephropathies Diseases 0.000 description 1
- 208000032131 Diabetic Neuropathies Diseases 0.000 description 1
- 208000019872 Drug Eruptions Diseases 0.000 description 1
- 206010013700 Drug hypersensitivity Diseases 0.000 description 1
- DVJAMEIQRSHVKC-BDAKNGLRSA-N Dutogliptin Chemical compound OB(O)[C@@H]1CCCN1C(=O)CN[C@H]1CNCC1 DVJAMEIQRSHVKC-BDAKNGLRSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 208000017701 Endocrine disease Diseases 0.000 description 1
- 108050009340 Endothelin Proteins 0.000 description 1
- 102000002045 Endothelin Human genes 0.000 description 1
- 102000010180 Endothelin receptor Human genes 0.000 description 1
- 108050001739 Endothelin receptor Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 206010014950 Eosinophilia Diseases 0.000 description 1
- MCIACXAZCBVDEE-CUUWFGFTSA-N Ertugliflozin Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@@]23O[C@@](CO)(CO2)[C@@H](O)[C@H](O)[C@H]3O)=CC=C1Cl MCIACXAZCBVDEE-CUUWFGFTSA-N 0.000 description 1
- 206010015150 Erythema Diseases 0.000 description 1
- CTKXFMQHOOWWEB-UHFFFAOYSA-N Ethylene oxide/propylene oxide copolymer Chemical compound CCCOC(C)COCCO CTKXFMQHOOWWEB-UHFFFAOYSA-N 0.000 description 1
- 208000010201 Exanthema Diseases 0.000 description 1
- 108010043222 Exubera Proteins 0.000 description 1
- 102100037362 Fibronectin Human genes 0.000 description 1
- 108010067306 Fibronectins Proteins 0.000 description 1
- 102100026148 Free fatty acid receptor 1 Human genes 0.000 description 1
- 108010017464 Fructose-Bisphosphatase Proteins 0.000 description 1
- 102000027487 Fructose-Bisphosphatase Human genes 0.000 description 1
- 101710154531 G-protein coupled bile acid receptor 1 Proteins 0.000 description 1
- 102400000921 Gastrin Human genes 0.000 description 1
- 108010052343 Gastrins Proteins 0.000 description 1
- 208000018522 Gastrointestinal disease Diseases 0.000 description 1
- HEMJJKBWTPKOJG-UHFFFAOYSA-N Gemfibrozil Chemical compound CC1=CC=C(C)C(OCCCC(C)(C)C(O)=O)=C1 HEMJJKBWTPKOJG-UHFFFAOYSA-N 0.000 description 1
- ZWPRRQZNBDYKLH-VIFPVBQESA-N Gemigliptin Chemical compound C([C@@H](N)CC(=O)N1CC2=C(C(=NC(=N2)C(F)(F)F)C(F)(F)F)CC1)N1CC(F)(F)CCC1=O ZWPRRQZNBDYKLH-VIFPVBQESA-N 0.000 description 1
- 208000035451 General disorders and administration site conditions Diseases 0.000 description 1
- 206010071602 Genetic polymorphism Diseases 0.000 description 1
- 229940089838 Glucagon-like peptide 1 receptor agonist Drugs 0.000 description 1
- 102400000324 Glucagon-like peptide 1(7-37) Human genes 0.000 description 1
- 101800004266 Glucagon-like peptide 1(7-37) Proteins 0.000 description 1
- FAEKWTJYAYMJKF-QHCPKHFHSA-N GlucoNorm Chemical compound C1=C(C(O)=O)C(OCC)=CC(CC(=O)N[C@@H](CC(C)C)C=2C(=CC=CC=2)N2CCCCC2)=C1 FAEKWTJYAYMJKF-QHCPKHFHSA-N 0.000 description 1
- 108010086800 Glucose-6-Phosphatase Proteins 0.000 description 1
- 102000003638 Glucose-6-Phosphatase Human genes 0.000 description 1
- 108010058940 Glutamyl Aminopeptidase Proteins 0.000 description 1
- 102000006485 Glutamyl Aminopeptidase Human genes 0.000 description 1
- 102000007390 Glycogen Phosphorylase Human genes 0.000 description 1
- 108010046163 Glycogen Phosphorylase Proteins 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 206010019233 Headaches Diseases 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- 208000032843 Hemorrhage Diseases 0.000 description 1
- 208000016988 Hemorrhagic Stroke Diseases 0.000 description 1
- 101000912510 Homo sapiens Free fatty acid receptor 1 Proteins 0.000 description 1
- 208000030673 Homozygous familial hypercholesterolemia Diseases 0.000 description 1
- 102000005561 Human Isophane Insulin Human genes 0.000 description 1
- 108010084048 Human Isophane Insulin Proteins 0.000 description 1
- 102100021102 Hyaluronidase PH-20 Human genes 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- 102100030643 Hydroxycarboxylic acid receptor 2 Human genes 0.000 description 1
- 101710125793 Hydroxycarboxylic acid receptor 2 Proteins 0.000 description 1
- 108090000895 Hydroxymethylglutaryl CoA Reductases Proteins 0.000 description 1
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 1
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 1
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 1
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 1
- 102000003777 Interleukin-1 beta Human genes 0.000 description 1
- 108090000193 Interleukin-1 beta Proteins 0.000 description 1
- 229940122254 Intermediate acting insulin Drugs 0.000 description 1
- FFEARJCKVFRZRR-UHFFFAOYSA-N L-Methionine Natural products CSCCC(N)C(O)=O FFEARJCKVFRZRR-UHFFFAOYSA-N 0.000 description 1
- LEVWYRKDKASIDU-IMJSIDKUSA-N L-cystine Chemical compound [O-]C(=O)[C@@H]([NH3+])CSSC[C@H]([NH3+])C([O-])=O LEVWYRKDKASIDU-IMJSIDKUSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 229930195722 L-methionine Natural products 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 229940127470 Lipase Inhibitors Drugs 0.000 description 1
- 108010092217 Long-Acting Insulin Proteins 0.000 description 1
- 102000016261 Long-Acting Insulin Human genes 0.000 description 1
- 229940100066 Long-acting insulin Drugs 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 108010083928 MDCO-216 Proteins 0.000 description 1
- 206010054805 Macroangiopathy Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 102400001132 Melanin-concentrating hormone Human genes 0.000 description 1
- 101800002739 Melanin-concentrating hormone Proteins 0.000 description 1
- 208000026139 Memory disease Diseases 0.000 description 1
- FLOSMHQXBMRNHR-QPJJXVBHSA-N Methazolamide Chemical compound CC(=O)\N=C1\SC(S(N)(=O)=O)=NN1C FLOSMHQXBMRNHR-QPJJXVBHSA-N 0.000 description 1
- 102100023174 Methionine aminopeptidase 2 Human genes 0.000 description 1
- 108090000192 Methionyl aminopeptidases Proteins 0.000 description 1
- IBAQFPQHRJAVAV-ULAWRXDQSA-N Miglitol Chemical compound OCCN1C[C@H](O)[C@@H](O)[C@H](O)[C@H]1CO IBAQFPQHRJAVAV-ULAWRXDQSA-N 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 206010028391 Musculoskeletal Pain Diseases 0.000 description 1
- 208000029027 Musculoskeletal and connective tissue disease Diseases 0.000 description 1
- RTHCYVBBDHJXIQ-UHFFFAOYSA-N N-methyl-3-phenyl-3-[4-(trifluoromethyl)phenoxy]propan-1-amine Chemical compound C=1C=CC=CC=1C(CCNC)OC1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-UHFFFAOYSA-N 0.000 description 1
- 108010057466 NF-kappa B Proteins 0.000 description 1
- 102000003945 NF-kappa B Human genes 0.000 description 1
- 108091007491 NSP3 Papain-like protease domains Proteins 0.000 description 1
- 108090000028 Neprilysin Proteins 0.000 description 1
- 102000003729 Neprilysin Human genes 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- 102000003797 Neuropeptides Human genes 0.000 description 1
- 108090000189 Neuropeptides Proteins 0.000 description 1
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical class O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 1
- 108010036875 ORMD-0801 Proteins 0.000 description 1
- 206010030124 Oedema peripheral Diseases 0.000 description 1
- 239000005480 Olmesartan Substances 0.000 description 1
- UQGKUQLKSCSZGY-UHFFFAOYSA-N Olmesartan medoxomil Chemical compound C=1C=C(C=2C(=CC=CC=2)C2=NNN=N2)C=CC=1CN1C(CCC)=NC(C(C)(C)O)=C1C(=O)OCC=1OC(=O)OC=1C UQGKUQLKSCSZGY-UHFFFAOYSA-N 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- 102400000319 Oxyntomodulin Human genes 0.000 description 1
- 101800001388 Oxyntomodulin Proteins 0.000 description 1
- 108700005984 PP 1420 Proteins 0.000 description 1
- 108010016731 PPAR gamma Proteins 0.000 description 1
- 102000000536 PPAR gamma Human genes 0.000 description 1
- 102000018886 Pancreatic Polypeptide Human genes 0.000 description 1
- 108700020479 Pancreatic hormone Proteins 0.000 description 1
- 206010033661 Pancytopenia Diseases 0.000 description 1
- 101710176384 Peptide 1 Proteins 0.000 description 1
- 229940122054 Peroxisome proliferator-activated receptor delta agonist Drugs 0.000 description 1
- 206010034972 Photosensitivity reaction Diseases 0.000 description 1
- 208000014993 Pituitary disease Diseases 0.000 description 1
- 206010035226 Plasma cell myeloma Diseases 0.000 description 1
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical compound C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 208000034809 Product contamination Diseases 0.000 description 1
- 108010044159 Proprotein Convertases Proteins 0.000 description 1
- 102000006437 Proprotein Convertases Human genes 0.000 description 1
- 102000053067 Pyruvate Dehydrogenase Acetyl-Transferring Kinase Human genes 0.000 description 1
- GSINGUMRKGRYJP-VZWAGXQNSA-N Remogliflozin Chemical compound C1=CC(OC(C)C)=CC=C1CC1=C(C)N(C(C)C)N=C1O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 GSINGUMRKGRYJP-VZWAGXQNSA-N 0.000 description 1
- 102000013272 Renalase Human genes 0.000 description 1
- 108010090629 Renalase Proteins 0.000 description 1
- 108090000783 Renin Proteins 0.000 description 1
- 206010057190 Respiratory tract infections Diseases 0.000 description 1
- 208000017442 Retinal disease Diseases 0.000 description 1
- 206010038923 Retinopathy Diseases 0.000 description 1
- 101150055528 SPAM1 gene Proteins 0.000 description 1
- DLSWIYLPEUIQAV-UHFFFAOYSA-N Semaglutide Chemical compound CCC(C)C(NC(=O)C(Cc1ccccc1)NC(=O)C(CCC(O)=O)NC(=O)C(CCCCNC(=O)COCCOCCNC(=O)COCCOCCNC(=O)CCC(NC(=O)CCCCCCCCCCCCCCCCC(O)=O)C(O)=O)NC(=O)C(C)NC(=O)C(C)NC(=O)C(CCC(N)=O)NC(=O)CNC(=O)C(CCC(O)=O)NC(=O)C(CC(C)C)NC(=O)C(Cc1ccc(O)cc1)NC(=O)C(CO)NC(=O)C(CO)NC(=O)C(NC(=O)C(CC(O)=O)NC(=O)C(CO)NC(=O)C(NC(=O)C(Cc1ccccc1)NC(=O)C(NC(=O)CNC(=O)C(CCC(O)=O)NC(=O)C(C)(C)NC(=O)C(N)Cc1cnc[nH]1)C(C)O)C(C)O)C(C)C)C(=O)NC(C)C(=O)NC(Cc1c[nH]c2ccccc12)C(=O)NC(CC(C)C)C(=O)NC(C(C)C)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CCCNC(N)=N)C(=O)NCC(O)=O DLSWIYLPEUIQAV-UHFFFAOYSA-N 0.000 description 1
- 108010026951 Short-Acting Insulin Proteins 0.000 description 1
- 229940123958 Short-acting insulin Drugs 0.000 description 1
- 208000000453 Skin Neoplasms Diseases 0.000 description 1
- 208000019498 Skin and subcutaneous tissue disease Diseases 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- QNAZTOHXCZPOSA-UHFFFAOYSA-N Sobetirome Chemical compound C1=C(O)C(C(C)C)=CC(CC=2C(=CC(OCC(O)=O)=CC=2C)C)=C1 QNAZTOHXCZPOSA-UHFFFAOYSA-N 0.000 description 1
- 229940123451 Somatostatin receptor 3 agonist Drugs 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 206010042464 Suicide attempt Diseases 0.000 description 1
- 229940127103 TKS-1225 Drugs 0.000 description 1
- 229940127105 TT-401 Drugs 0.000 description 1
- 229920002359 Tetronic® Polymers 0.000 description 1
- 102100033451 Thyroid hormone receptor beta Human genes 0.000 description 1
- AUYYCJSJGJYCDS-LBPRGKRZSA-N Thyrolar Chemical class IC1=CC(C[C@H](N)C(O)=O)=CC(I)=C1OC1=CC=C(O)C(I)=C1 AUYYCJSJGJYCDS-LBPRGKRZSA-N 0.000 description 1
- JLRGJRBPOGGCBT-UHFFFAOYSA-N Tolbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=C(C)C=C1 JLRGJRBPOGGCBT-UHFFFAOYSA-N 0.000 description 1
- 102100033121 Transcription factor 21 Human genes 0.000 description 1
- 101710119687 Transcription factor 21 Proteins 0.000 description 1
- 241000224526 Trichomonas Species 0.000 description 1
- 102000008316 Type 4 Melanocortin Receptor Human genes 0.000 description 1
- 108010021436 Type 4 Melanocortin Receptor Proteins 0.000 description 1
- 206010045261 Type IIa hyperlipidaemia Diseases 0.000 description 1
- 101710204865 Tyrosine-protein phosphatase 1 Proteins 0.000 description 1
- 208000003443 Unconsciousness Diseases 0.000 description 1
- 206010046306 Upper respiratory tract infection Diseases 0.000 description 1
- LEHOTFFKMJEONL-UHFFFAOYSA-N Uric Acid Chemical compound N1C(=O)NC(=O)C2=C1NC(=O)N2 LEHOTFFKMJEONL-UHFFFAOYSA-N 0.000 description 1
- TVWHNULVHGKJHS-UHFFFAOYSA-N Uric acid Natural products N1C(=O)NC(=O)C2NC(=O)NC21 TVWHNULVHGKJHS-UHFFFAOYSA-N 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- 208000024248 Vascular System injury Diseases 0.000 description 1
- 208000012339 Vascular injury Diseases 0.000 description 1
- 206010069690 Vertebral foraminal stenosis Diseases 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- FZNCGRZWXLXZSZ-CIQUZCHMSA-N Voglibose Chemical compound OCC(CO)N[C@H]1C[C@](O)(CO)[C@@H](O)[C@H](O)[C@H]1O FZNCGRZWXLXZSZ-CIQUZCHMSA-N 0.000 description 1
- 229940127104 WYE-155189 Drugs 0.000 description 1
- YDTUJCNTIMWHPJ-NRFANRHFSA-N [(1r)-2-[4-[6-(4-chlorophenyl)-4-oxothieno[3,2-d]pyrimidin-3-yl]-2-methoxyphenoxy]-1-cyclopropylethyl] dihydrogen phosphate Chemical compound C1([C@@H](OP(O)(O)=O)COC2=CC=C(C=C2OC)N2C(C=3SC(=CC=3N=C2)C=2C=CC(Cl)=CC=2)=O)CC1 YDTUJCNTIMWHPJ-NRFANRHFSA-N 0.000 description 1
- YVPOVOVZCOOSBQ-AXHZAXLDSA-N [(1s,3r,7s,8s,8ar)-8-[2-[(2r,4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-3,7-dimethyl-1,2,3,7,8,8a-hexahydronaphthalen-1-yl] (2s)-2-methylbutanoate;pyridine-3-carboxylic acid Chemical compound OC(=O)C1=CC=CN=C1.C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 YVPOVOVZCOOSBQ-AXHZAXLDSA-N 0.000 description 1
- WNWXXAPGHTVCDL-OKDJMAGBSA-N [(1s,3r,7s,8s,8ar)-8-[2-[(2r,4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-3,7-dimethyl-1,2,3,7,8,8a-hexahydronaphthalen-1-yl] 2,2-dimethylbutanoate;pyridine-3-carboxylic acid Chemical compound OC(=O)C1=CC=CN=C1.C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 WNWXXAPGHTVCDL-OKDJMAGBSA-N 0.000 description 1
- 101710159466 [Pyruvate dehydrogenase (acetyl-transferring)] kinase, mitochondrial Proteins 0.000 description 1
- 229960002632 acarbose Drugs 0.000 description 1
- XUFXOAAUWZOOIT-UHFFFAOYSA-N acarviostatin I01 Natural products OC1C(O)C(NC2C(C(O)C(O)C(CO)=C2)O)C(C)OC1OC(C(C1O)O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O XUFXOAAUWZOOIT-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 229940023375 adipex-p Drugs 0.000 description 1
- 210000000577 adipose tissue Anatomy 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000000048 adrenergic agonist Substances 0.000 description 1
- 239000000674 adrenergic antagonist Substances 0.000 description 1
- 229940126157 adrenergic receptor agonist Drugs 0.000 description 1
- 229940034653 advicor Drugs 0.000 description 1
- 238000001261 affinity purification Methods 0.000 description 1
- 229940078883 afrezza Drugs 0.000 description 1
- 230000001270 agonistic effect Effects 0.000 description 1
- 229960004733 albiglutide Drugs 0.000 description 1
- OGWAVGNOAMXIIM-UHFFFAOYSA-N albiglutide Chemical compound O=C(O)C(NC(=O)CNC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)CNC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)CNC(=O)C(NC(=O)CNC(=O)C(N)CC=1(N=CNC=1))CCC(=O)O)C(O)C)CC2(=CC=CC=C2))C(O)C)CO)CC(=O)O)C(C)C)CO)CO)CC3(=CC=C(O)C=C3))CC(C)C)CCC(=O)O)CCC(=O)N)C)C)CCCCN)CCC(=O)O)CC4(=CC=CC=C4))C(CC)C)C)CC=6(C5(=C(C=CC=C5)NC=6)))CC(C)C)C(C)C)CCCCN)CCCNC(=N)N OGWAVGNOAMXIIM-UHFFFAOYSA-N 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 239000002170 aldosterone antagonist Substances 0.000 description 1
- 229940083712 aldosterone antagonist Drugs 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 229960001667 alogliptin Drugs 0.000 description 1
- 239000002160 alpha blocker Substances 0.000 description 1
- 102000030484 alpha-2 Adrenergic Receptor Human genes 0.000 description 1
- 108020004101 alpha-2 Adrenergic Receptor Proteins 0.000 description 1
- 229940000806 amaryl Drugs 0.000 description 1
- 231100000540 amenorrhea Toxicity 0.000 description 1
- HTIQEAQVCYTUBX-UHFFFAOYSA-N amlodipine Chemical compound CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1Cl HTIQEAQVCYTUBX-UHFFFAOYSA-N 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 229950009977 anagliptin Drugs 0.000 description 1
- LDXYBEHACFJIEL-HNNXBMFYSA-N anagliptin Chemical compound C=1N2N=C(C)C=C2N=CC=1C(=O)NCC(C)(C)NCC(=O)N1CCC[C@H]1C#N LDXYBEHACFJIEL-HNNXBMFYSA-N 0.000 description 1
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 1
- 239000004037 angiogenesis inhibitor Substances 0.000 description 1
- 239000002333 angiotensin II receptor antagonist Substances 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 230000003042 antagnostic effect Effects 0.000 description 1
- 229940074323 antara Drugs 0.000 description 1
- 229940127003 anti-diabetic drug Drugs 0.000 description 1
- 230000002058 anti-hyperglycaemic effect Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 230000009830 antibody antigen interaction Effects 0.000 description 1
- 230000000890 antigenic effect Effects 0.000 description 1
- 229940127088 antihypertensive drug Drugs 0.000 description 1
- 229940125710 antiobesity agent Drugs 0.000 description 1
- 229940127218 antiplatelet drug Drugs 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- NETXMUIMUZJUTB-UHFFFAOYSA-N apabetalone Chemical compound C=1C(OC)=CC(OC)=C(C(N2)=O)C=1N=C2C1=CC(C)=C(OCCO)C(C)=C1 NETXMUIMUZJUTB-UHFFFAOYSA-N 0.000 description 1
- 229940112930 apidra Drugs 0.000 description 1
- 239000002830 appetite depressant Substances 0.000 description 1
- 239000012062 aqueous buffer Substances 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 206010003119 arrhythmia Diseases 0.000 description 1
- 208000011775 arteriosclerosis disease Diseases 0.000 description 1
- 208000015337 arteriosclerotic cardiovascular disease Diseases 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 229940058087 atacand Drugs 0.000 description 1
- 229940034406 atorvastatin / ezetimibe Drugs 0.000 description 1
- FQCKMBLVYCEXJB-MNSAWQCASA-L atorvastatin calcium Chemical compound [Ca+2].C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC([O-])=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1.C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC([O-])=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 FQCKMBLVYCEXJB-MNSAWQCASA-L 0.000 description 1
- 229960002731 azilsartan Drugs 0.000 description 1
- KGSXMPPBFPAXLY-UHFFFAOYSA-N azilsartan Chemical compound CCOC1=NC2=CC=CC(C(O)=O)=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C1=NOC(=O)N1 KGSXMPPBFPAXLY-UHFFFAOYSA-N 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 235000004251 balanced diet Nutrition 0.000 description 1
- 210000000270 basal cell Anatomy 0.000 description 1
- 229940120049 belviq Drugs 0.000 description 1
- HYHMLYSLQUKXKP-UHFFFAOYSA-N bempedoic acid Chemical compound OC(=O)C(C)(C)CCCCCC(O)CCCCCC(C)(C)C(O)=O HYHMLYSLQUKXKP-UHFFFAOYSA-N 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- YXKTVDFXDRQTKV-HNNXBMFYSA-N benzphetamine Chemical compound C([C@H](C)N(C)CC=1C=CC=CC=1)C1=CC=CC=C1 YXKTVDFXDRQTKV-HNNXBMFYSA-N 0.000 description 1
- 229960002837 benzphetamine Drugs 0.000 description 1
- ANFSNXAXVLRZCG-RSAXXLAASA-N benzphetamine hydrochloride Chemical compound [Cl-].C([C@H](C)[NH+](C)CC=1C=CC=CC=1)C1=CC=CC=C1 ANFSNXAXVLRZCG-RSAXXLAASA-N 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 229940125388 beta agonist Drugs 0.000 description 1
- 108010014502 beta-3 Adrenergic Receptors Proteins 0.000 description 1
- 229960000516 bezafibrate Drugs 0.000 description 1
- 230000001588 bifunctional effect Effects 0.000 description 1
- 150000004283 biguanides Chemical class 0.000 description 1
- BPYKTIZUTYGOLE-UHFFFAOYSA-N billirubin-IXalpha Natural products N1C(=O)C(C)=C(C=C)C1=CC1=C(C)C(CCC(O)=O)=C(CC2=C(C(C)=C(C=C3C(=C(C=C)C(=O)N3)C)N2)CCC(O)=O)N1 BPYKTIZUTYGOLE-UHFFFAOYSA-N 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 230000007698 birth defect Effects 0.000 description 1
- 238000010241 blood sampling Methods 0.000 description 1
- 229940046049 bontril Drugs 0.000 description 1
- OZVBMTJYIDMWIL-AYFBDAFISA-N bromocriptine Chemical compound C1=CC(C=2[C@H](N(C)C[C@@H](C=2)C(=O)N[C@]2(C(=O)N3[C@H](C(N4CCC[C@H]4[C@]3(O)O2)=O)CC(C)C)C(C)C)C2)=C3C2=C(Br)NC3=C1 OZVBMTJYIDMWIL-AYFBDAFISA-N 0.000 description 1
- 229960002802 bromocriptine Drugs 0.000 description 1
- NOJMTMIRQRDZMT-GSPXQYRGSA-N bromocriptine methanesulfonate Chemical compound CS(O)(=O)=O.C1=CC(C=2[C@H](N(C)C[C@@H](C=2)C(=O)N[C@]2(C(=O)N3[C@H](C(N4CCC[C@H]4[C@]3(O)O2)=O)CC(C)C)C(C)C)C2)=C3C2=C(Br)NC3=C1 NOJMTMIRQRDZMT-GSPXQYRGSA-N 0.000 description 1
- 206010006451 bronchitis Diseases 0.000 description 1
- 229960004111 buformin Drugs 0.000 description 1
- XSEUMFJMFFMCIU-UHFFFAOYSA-N buformin Chemical compound CCCC\N=C(/N)N=C(N)N XSEUMFJMFFMCIU-UHFFFAOYSA-N 0.000 description 1
- 229940014641 bydureon Drugs 0.000 description 1
- 229940084891 byetta Drugs 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 229940022418 caduet Drugs 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- QXDMQSPYEZFLGF-UHFFFAOYSA-L calcium oxalate Chemical compound [Ca+2].[O-]C(=O)C([O-])=O QXDMQSPYEZFLGF-UHFFFAOYSA-L 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 229960001713 canagliflozin Drugs 0.000 description 1
- VHOFTEAWFCUTOS-TUGBYPPCSA-N canagliflozin hydrate Chemical compound O.CC1=CC=C([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)C=C1CC(S1)=CC=C1C1=CC=C(F)C=C1.CC1=CC=C([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)C=C1CC(S1)=CC=C1C1=CC=C(F)C=C1 VHOFTEAWFCUTOS-TUGBYPPCSA-N 0.000 description 1
- 229960000932 candesartan Drugs 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 230000007211 cardiovascular event Effects 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 230000001364 causal effect Effects 0.000 description 1
- 230000034303 cell budding Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 229950002397 cetilistat Drugs 0.000 description 1
- JUFFVKRROAPVBI-PVOYSMBESA-N chembl1210015 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(=O)N[C@H]1[C@@H]([C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO[C@]3(O[C@@H](C[C@H](O)[C@H](O)CO)[C@H](NC(C)=O)[C@@H](O)C3)C(O)=O)O2)O)[C@@H](CO)O1)NC(C)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 JUFFVKRROAPVBI-PVOYSMBESA-N 0.000 description 1
- AOXOCDRNSPFDPE-UKEONUMOSA-N chembl413654 Chemical compound C([C@H](C(=O)NCC(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](C)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)CNC(=O)[C@@H](N)CCC(O)=O)C1=CC=C(O)C=C1 AOXOCDRNSPFDPE-UKEONUMOSA-N 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 229960002174 ciprofibrate Drugs 0.000 description 1
- KPSRODZRAIWAKH-UHFFFAOYSA-N ciprofibrate Chemical compound C1=CC(OC(C)(C)C(O)=O)=CC=C1C1C(Cl)(Cl)C1 KPSRODZRAIWAKH-UHFFFAOYSA-N 0.000 description 1
- 229950003072 clinofibrate Drugs 0.000 description 1
- 229960005049 clofibride Drugs 0.000 description 1
- CXQGFLBVUNUQIA-UHFFFAOYSA-N clofibride Chemical compound CN(C)C(=O)CCCOC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 CXQGFLBVUNUQIA-UHFFFAOYSA-N 0.000 description 1
- ASARMUCNOOHMLO-WLORSUFZSA-L cobalt(2+);[(2r,3s,4r,5s)-5-(5,6-dimethylbenzimidazol-1-yl)-4-hydroxy-2-(hydroxymethyl)oxolan-3-yl] [(2s)-1-[3-[(1r,2r,3r,4z,7s,9z,12s,13s,14z,17s,18s,19r)-2,13,18-tris(2-amino-2-oxoethyl)-7,12,17-tris(3-amino-3-oxopropyl)-3,5,8,8,13,15,18,19-octamethyl-2 Chemical compound [Co+2].[N-]([C@@H]1[C@H](CC(N)=O)[C@@]2(C)CCC(=O)NC[C@H](C)OP([O-])(=O)O[C@H]3[C@H]([C@H](O[C@@H]3CO)N3C4=CC(C)=C(C)C=C4N=C3)O)\C2=C(C)/C([C@H](C\2(C)C)CCC(N)=O)=N/C/2=C\C([C@H]([C@@]/2(CC(N)=O)C)CCC(N)=O)=N\C\2=C(C)/C2=N[C@]1(C)[C@@](C)(CC(N)=O)[C@@H]2CCC(N)=O ASARMUCNOOHMLO-WLORSUFZSA-L 0.000 description 1
- 229960002604 colestipol Drugs 0.000 description 1
- GMRWGQCZJGVHKL-UHFFFAOYSA-N colestipol Chemical compound ClCC1CO1.NCCNCCNCCNCCN GMRWGQCZJGVHKL-UHFFFAOYSA-N 0.000 description 1
- 238000011284 combination treatment Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 238000002591 computed tomography Methods 0.000 description 1
- 238000004590 computer program Methods 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 239000003433 contraceptive agent Substances 0.000 description 1
- 230000002254 contraceptive effect Effects 0.000 description 1
- 229940012193 contrave Drugs 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 229960001334 corticosteroids Drugs 0.000 description 1
- 229940066901 crestor Drugs 0.000 description 1
- 230000009260 cross reactivity Effects 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 229940015838 cycloset Drugs 0.000 description 1
- 229940029644 cymbalta Drugs 0.000 description 1
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 1
- 229960003067 cystine Drugs 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 229960003834 dapagliflozin Drugs 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 208000033679 diabetic kidney disease Diseases 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- 229940120144 didrex Drugs 0.000 description 1
- 235000021045 dietary change Nutrition 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 235000015872 dietary supplement Nutrition 0.000 description 1
- XXEPPPIWZFICOJ-UHFFFAOYSA-N diethylpropion Chemical compound CCN(CC)C(C)C(=O)C1=CC=CC=C1 XXEPPPIWZFICOJ-UHFFFAOYSA-N 0.000 description 1
- 229960004890 diethylpropion Drugs 0.000 description 1
- ICFXZZFWRWNZMA-UHFFFAOYSA-N diethylpropion hydrochloride Chemical compound [Cl-].CC[NH+](CC)C(C)C(=O)C1=CC=CC=C1 ICFXZZFWRWNZMA-UHFFFAOYSA-N 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 1
- 229960004166 diltiazem Drugs 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 230000001882 diuretic effect Effects 0.000 description 1
- 229940030606 diuretics Drugs 0.000 description 1
- 208000002173 dizziness Diseases 0.000 description 1
- 229960003638 dopamine Drugs 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 206010013663 drug dependence Diseases 0.000 description 1
- 238000007905 drug manufacturing Methods 0.000 description 1
- 238000002651 drug therapy Methods 0.000 description 1
- 229960005175 dulaglutide Drugs 0.000 description 1
- 108010005794 dulaglutide Proteins 0.000 description 1
- 229950003693 dutogliptin Drugs 0.000 description 1
- 235000013601 eggs Nutrition 0.000 description 1
- 229960003345 empagliflozin Drugs 0.000 description 1
- OBWASQILIWPZMG-QZMOQZSNSA-N empagliflozin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=CC(O[C@@H]3COCC3)=CC=2)=C1 OBWASQILIWPZMG-QZMOQZSNSA-N 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- ZUBDGKVDJUIMQQ-UBFCDGJISA-N endothelin-1 Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(O)=O)NC(=O)[C@H]1NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@@H](CC=2C=CC(O)=CC=2)NC(=O)[C@H](C(C)C)NC(=O)[C@H]2CSSC[C@@H](C(N[C@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N2)=O)NC(=O)[C@@H](CO)NC(=O)[C@H](N)CSSC1)C1=CNC=N1 ZUBDGKVDJUIMQQ-UBFCDGJISA-N 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 239000002792 enkephalinase inhibitor Substances 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 229940035000 epanova Drugs 0.000 description 1
- 206010015037 epilepsy Diseases 0.000 description 1
- 229940015979 epipen Drugs 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 229960004563 eprosartan Drugs 0.000 description 1
- 229950011248 eprotirome Drugs 0.000 description 1
- 229950006535 ertugliflozin Drugs 0.000 description 1
- 231100000321 erythema Toxicity 0.000 description 1
- AKFNKZFJBFQFAA-DIOPXHOYSA-N ethyl 4-[[2-[(2s,4s)-2-cyano-4-fluoropyrrolidin-1-yl]-2-oxoethyl]amino]bicyclo[2.2.2]octane-1-carboxylate Chemical compound C1CC(C(=O)OCC)(CC2)CCC12NCC(=O)N1C[C@@H](F)C[C@H]1C#N AKFNKZFJBFQFAA-DIOPXHOYSA-N 0.000 description 1
- 229960003501 etofibrate Drugs 0.000 description 1
- XXRVYAFBUDSLJX-UHFFFAOYSA-N etofibrate Chemical compound C=1C=CN=CC=1C(=O)OCCOC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 XXRVYAFBUDSLJX-UHFFFAOYSA-N 0.000 description 1
- 229950000005 evacetrapib Drugs 0.000 description 1
- 229950004341 evinacumab Drugs 0.000 description 1
- 230000005713 exacerbation Effects 0.000 description 1
- 201000005884 exanthem Diseases 0.000 description 1
- 239000013604 expression vector Substances 0.000 description 1
- 210000003414 extremity Anatomy 0.000 description 1
- 229940012151 exubera Drugs 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 229960002464 fluoxetine Drugs 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- AIWAEWBZDJARBJ-PXUUZXDZSA-N fz7co35x2s Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CCCCNC(=O)COCCOCCNC(=O)CCN1C(C=CC1=O)=O)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 AIWAEWBZDJARBJ-PXUUZXDZSA-N 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 229960003627 gemfibrozil Drugs 0.000 description 1
- 229960002458 gemigliptin Drugs 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- 208000004104 gestational diabetes Diseases 0.000 description 1
- AFLFKFHDSCQHOL-IZZDOVSWSA-N gft505 Chemical compound C1=CC(SC)=CC=C1C(=O)\C=C\C1=CC(C)=C(OC(C)(C)C(O)=O)C(C)=C1 AFLFKFHDSCQHOL-IZZDOVSWSA-N 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 229960004580 glibenclamide Drugs 0.000 description 1
- WIGIZIANZCJQQY-RUCARUNLSA-N glimepiride Chemical compound O=C1C(CC)=C(C)CN1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)N[C@@H]2CC[C@@H](C)CC2)C=C1 WIGIZIANZCJQQY-RUCARUNLSA-N 0.000 description 1
- 229960004346 glimepiride Drugs 0.000 description 1
- 229960001381 glipizide Drugs 0.000 description 1
- ZJJXGWJIGJFDTL-UHFFFAOYSA-N glipizide Chemical compound C1=NC(C)=CN=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZJJXGWJIGJFDTL-UHFFFAOYSA-N 0.000 description 1
- 230000024924 glomerular filtration Effects 0.000 description 1
- 108010063245 glucagon-like peptide 1 (7-36)amide Proteins 0.000 description 1
- ZNNLBTZKUZBEKO-UHFFFAOYSA-N glyburide Chemical compound COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZNNLBTZKUZBEKO-UHFFFAOYSA-N 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000005484 gravity Effects 0.000 description 1
- 238000001631 haemodialysis Methods 0.000 description 1
- 231100000869 headache Toxicity 0.000 description 1
- 208000014951 hematologic disease Diseases 0.000 description 1
- 230000000322 hemodialysis Effects 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- 239000003395 histamine H3 receptor antagonist Substances 0.000 description 1
- 239000000710 homodimer Substances 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 230000002727 hyperosmolar Effects 0.000 description 1
- 208000006575 hypertriglyceridemia Diseases 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 210000004347 intestinal mucosa Anatomy 0.000 description 1
- 208000020658 intracerebral hemorrhage Diseases 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- 229950000991 ipragliflozin Drugs 0.000 description 1
- AHFWIQIYAXSLBA-RQXATKFSSA-N ipragliflozin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(F)C(CC=2SC3=CC=CC=C3C=2)=C1 AHFWIQIYAXSLBA-RQXATKFSSA-N 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- ZIPLFLRGHZAXSJ-UHFFFAOYSA-N isoquinoline-5-carboxylic acid Chemical compound N1=CC=C2C(C(=O)O)=CC=CC2=C1 ZIPLFLRGHZAXSJ-UHFFFAOYSA-N 0.000 description 1
- 239000000644 isotonic solution Substances 0.000 description 1
- 229940103445 janumet Drugs 0.000 description 1
- 229940090473 januvia Drugs 0.000 description 1
- 235000015110 jellies Nutrition 0.000 description 1
- 229940103513 kazano Drugs 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- 229940060975 lantus Drugs 0.000 description 1
- 230000002045 lasting effect Effects 0.000 description 1
- 229940095570 lescol Drugs 0.000 description 1
- 108010000849 leukocyte esterase Proteins 0.000 description 1
- 229940102988 levemir Drugs 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 201000002818 limb ischemia Diseases 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 229960002397 linagliptin Drugs 0.000 description 1
- 229940044185 lipofen Drugs 0.000 description 1
- 108010022197 lipoprotein cholesterol Proteins 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 229940034394 liptruzet Drugs 0.000 description 1
- 229960002701 liraglutide Drugs 0.000 description 1
- 229940092923 livalo Drugs 0.000 description 1
- 238000007449 liver function test Methods 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- CHHXEZSCHQVSRE-UHFFFAOYSA-N lobeglitazone Chemical compound C1=CC(OC)=CC=C1OC1=CC(N(C)CCOC=2C=CC(CC3C(NC(=O)S3)=O)=CC=2)=NC=N1 CHHXEZSCHQVSRE-UHFFFAOYSA-N 0.000 description 1
- 229950007685 lobeglitazone Drugs 0.000 description 1
- 229960003566 lomitapide Drugs 0.000 description 1
- WRZCAWKMTLRWPR-VSODYHHCSA-N lorcaserin hydrochloride hemihydrate Chemical compound O.Cl.Cl.C[C@H]1CNCCC2=CC=C(Cl)C=C12.C[C@H]1CNCCC2=CC=C(Cl)C=C12 WRZCAWKMTLRWPR-VSODYHHCSA-N 0.000 description 1
- 229960004773 losartan Drugs 0.000 description 1
- KJJZZJSZUJXYEA-UHFFFAOYSA-N losartan Chemical compound CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2[N]N=NN=2)C=C1 KJJZZJSZUJXYEA-UHFFFAOYSA-N 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 150000002681 magnesium compounds Chemical class 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 238000007726 management method Methods 0.000 description 1
- ORRDHOMWDPJSNL-UHFFFAOYSA-N melanin concentrating hormone Chemical compound N1C(=O)C(C(C)C)NC(=O)C(CCCNC(N)=N)NC(=O)CNC(=O)C(C(C)C)NC(=O)C(CCSC)NC(=O)C(NC(=O)C(CCCNC(N)=N)NC(=O)C(NC(=O)C(NC(=O)C(N)CC(O)=O)C(C)O)CCSC)CSSCC(C(=O)NC(CC=2C3=CC=CC=C3NC=2)C(=O)NC(CCC(O)=O)C(=O)NC(C(C)C)C(O)=O)NC(=O)C2CCCN2C(=O)C(CCCNC(N)=N)NC(=O)C1CC1=CC=C(O)C=C1 ORRDHOMWDPJSNL-UHFFFAOYSA-N 0.000 description 1
- 230000006984 memory degeneration Effects 0.000 description 1
- 206010027175 memory impairment Diseases 0.000 description 1
- 208000023060 memory loss Diseases 0.000 description 1
- 238000010197 meta-analysis Methods 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- OETHQSJEHLVLGH-UHFFFAOYSA-N metformin hydrochloride Chemical compound Cl.CN(C)C(=N)N=C(N)N OETHQSJEHLVLGH-UHFFFAOYSA-N 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 108700008455 metreleptin Proteins 0.000 description 1
- 229960000668 metreleptin Drugs 0.000 description 1
- 229940099246 mevacor Drugs 0.000 description 1
- 229950009116 mevastatin Drugs 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 239000011859 microparticle Substances 0.000 description 1
- 238000000386 microscopy Methods 0.000 description 1
- 230000003228 microsomal effect Effects 0.000 description 1
- 229960001110 miglitol Drugs 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 239000002395 mineralocorticoid Substances 0.000 description 1
- 229960003365 mitiglinide Drugs 0.000 description 1
- WPGGHFDDFPHPOB-BBWFWOEESA-N mitiglinide Chemical compound C([C@@H](CC(=O)N1C[C@@H]2CCCC[C@@H]2C1)C(=O)O)C1=CC=CC=C1 WPGGHFDDFPHPOB-BBWFWOEESA-N 0.000 description 1
- 238000001823 molecular biology technique Methods 0.000 description 1
- PXZWGQLGAKCNKD-DPNMSELWSA-N molport-023-276-326 Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(O)=O)[C@@H](C)O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 PXZWGQLGAKCNKD-DPNMSELWSA-N 0.000 description 1
- AGAHNABIDCTLHW-UHFFFAOYSA-N moperone Chemical compound C1=CC(C)=CC=C1C1(O)CCN(CCCC(=O)C=2C=CC(F)=CC=2)CC1 AGAHNABIDCTLHW-UHFFFAOYSA-N 0.000 description 1
- 210000002200 mouth mucosa Anatomy 0.000 description 1
- 210000003097 mucus Anatomy 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- 208000031225 myocardial ischemia Diseases 0.000 description 1
- 239000003887 narcotic antagonist Substances 0.000 description 1
- 229960000698 nateglinide Drugs 0.000 description 1
- OELFLUMRDSZNSF-BRWVUGGUSA-N nateglinide Chemical compound C1C[C@@H](C(C)C)CC[C@@H]1C(=O)N[C@@H](C(O)=O)CC1=CC=CC=C1 OELFLUMRDSZNSF-BRWVUGGUSA-N 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 229940117337 nesina Drugs 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- 230000007971 neurological deficit Effects 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 239000002840 nitric oxide donor Substances 0.000 description 1
- 230000000422 nocturnal effect Effects 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 239000000820 nonprescription drug Substances 0.000 description 1
- 229960002748 norepinephrine Drugs 0.000 description 1
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 1
- 229940103453 novolin Drugs 0.000 description 1
- 229940112879 novolog Drugs 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- NRWORBQAOQVYBJ-GJZUVCINSA-N obicetrapib Chemical compound N=1C=C(OCCCC(O)=O)C=NC=1N([C@H]1C[C@@H](CC)N(C2=CC=C(C=C21)C(F)(F)F)C(=O)OCC)CC1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 NRWORBQAOQVYBJ-GJZUVCINSA-N 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 229960005117 olmesartan Drugs 0.000 description 1
- 229960001199 olmesartan medoxomil Drugs 0.000 description 1
- MKMPWKUAHLTIBJ-ISTRZQFTSA-N omarigliptin Chemical compound C1([C@H]2OC[C@@H](C[C@@H]2N)N2CC3=CN(N=C3C2)S(=O)(=O)C)=CC(F)=CC=C1F MKMPWKUAHLTIBJ-ISTRZQFTSA-N 0.000 description 1
- 229940001450 onglyza Drugs 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 230000008816 organ damage Effects 0.000 description 1
- 206010033675 panniculitis Diseases 0.000 description 1
- 208000035824 paresthesia Diseases 0.000 description 1
- 229940000596 parlodel Drugs 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 229940021222 peritoneal dialysis isotonic solution Drugs 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000002974 pharmacogenomic effect Effects 0.000 description 1
- 229960000436 phendimetrazine Drugs 0.000 description 1
- ORMNNUPLFAPCFD-DVLYDCSHSA-M phenethicillin potassium Chemical compound [K+].N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C([O-])=O)=O)C(=O)C(C)OC1=CC=CC=C1 ORMNNUPLFAPCFD-DVLYDCSHSA-M 0.000 description 1
- 229960003243 phenformin Drugs 0.000 description 1
- ICFJFFQQTFMIBG-UHFFFAOYSA-N phenformin Chemical compound NC(=N)NC(=N)NCCC1=CC=CC=C1 ICFJFFQQTFMIBG-UHFFFAOYSA-N 0.000 description 1
- AZUIUVJESCFSLJ-UHFFFAOYSA-N phenyl 6-[[3-methoxy-2-[4-(trifluoromethyl)phenyl]benzoyl]amino]-3,4-dihydro-1h-isoquinoline-2-carboxylate Chemical compound C=1C=C(C(F)(F)F)C=CC=1C=1C(OC)=CC=CC=1C(=O)NC(C=C1CC2)=CC=C1CN2C(=O)OC1=CC=CC=C1 AZUIUVJESCFSLJ-UHFFFAOYSA-N 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 229930029653 phosphoenolpyruvate Natural products 0.000 description 1
- DTBNBXWJWCWCIK-UHFFFAOYSA-N phosphoenolpyruvic acid Chemical compound OC(=O)C(=C)OP(O)(O)=O DTBNBXWJWCWCIK-UHFFFAOYSA-N 0.000 description 1
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 1
- 208000007578 phototoxic dermatitis Diseases 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 229940068065 phytosterols Drugs 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 230000001817 pituitary effect Effects 0.000 description 1
- 208000017402 pituitary gland disease Diseases 0.000 description 1
- 239000000106 platelet aggregation inhibitor Substances 0.000 description 1
- 229960000502 poloxamer Drugs 0.000 description 1
- 229920001993 poloxamer 188 Polymers 0.000 description 1
- 229940044519 poloxamer 188 Drugs 0.000 description 1
- 239000008389 polyethoxylated castor oil Substances 0.000 description 1
- 108700002854 polyethylene glycol(B1)- insulin Proteins 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229950008882 polysorbate Drugs 0.000 description 1
- CJMKTEIIPMBTJB-DXFHJFHKSA-M potassium;[(2r,3r,4s,5r,6r)-6-[[3-[(3s,4r,5r)-3-butyl-7-(dimethylamino)-3-ethyl-4-hydroxy-1,1-dioxo-4,5-dihydro-2h-1$l^{6}-benzothiepin-5-yl]phenyl]carbamoylamino]-3,5-dihydroxy-4-phenylmethoxyoxan-2-yl]methyl sulfate Chemical compound [K+].O([C@H]1[C@H](O)[C@@H](COS([O-])(=O)=O)O[C@H]([C@@H]1O)NC(=O)NC=1C=CC=C(C=1)[C@@H]1C2=CC(=CC=C2S(=O)(=O)C[C@@]([C@@H]1O)(CC)CCCC)N(C)C)CC1=CC=CC=C1 CJMKTEIIPMBTJB-DXFHJFHKSA-M 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- VWBQYTRBTXKKOG-IYNICTALSA-M pravastatin sodium Chemical compound [Na+].C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC([O-])=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 VWBQYTRBTXKKOG-IYNICTALSA-M 0.000 description 1
- 230000002028 premature Effects 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000003405 preventing effect Effects 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- WPDCHTSXOPUOII-UHFFFAOYSA-N propan-2-yl 4-[5-methoxy-6-[(2-methyl-6-methylsulfonylpyridin-3-yl)amino]pyrimidin-4-yl]oxypiperidine-1-carboxylate Chemical compound N1=CN=C(OC2CCN(CC2)C(=O)OC(C)C)C(OC)=C1NC1=CC=C(S(C)(=O)=O)N=C1C WPDCHTSXOPUOII-UHFFFAOYSA-N 0.000 description 1
- 230000013777 protein digestion Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 229940103440 qsymia Drugs 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 108700027806 rGLP-1 Proteins 0.000 description 1
- 238000002708 random mutagenesis Methods 0.000 description 1
- 206010037844 rash Diseases 0.000 description 1
- 230000009103 reabsorption Effects 0.000 description 1
- 230000010837 receptor-mediated endocytosis Effects 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 238000003259 recombinant expression Methods 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 230000011514 reflex Effects 0.000 description 1
- 238000005057 refrigeration Methods 0.000 description 1
- 229940126844 remogliflozin Drugs 0.000 description 1
- 229950011516 remogliflozin etabonate Drugs 0.000 description 1
- UAOCLDQAQNNEAX-ABMICEGHSA-N remogliflozin etabonate Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](COC(=O)OCC)O[C@H]1OC1=NN(C(C)C)C(C)=C1CC1=CC=C(OC(C)C)C=C1 UAOCLDQAQNNEAX-ABMICEGHSA-N 0.000 description 1
- 238000012959 renal replacement therapy Methods 0.000 description 1
- 239000002461 renin inhibitor Substances 0.000 description 1
- 230000036454 renin-angiotensin system Effects 0.000 description 1
- 229940086526 renin-inhibitors Drugs 0.000 description 1
- 229960002354 repaglinide Drugs 0.000 description 1
- 238000009256 replacement therapy Methods 0.000 description 1
- FDBYIYFVSAHJLY-UHFFFAOYSA-N resmetirom Chemical compound N1C(=O)C(C(C)C)=CC(OC=2C(=CC(=CC=2Cl)N2C(NC(=O)C(C#N)=N2)=O)Cl)=N1 FDBYIYFVSAHJLY-UHFFFAOYSA-N 0.000 description 1
- 230000000284 resting effect Effects 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 229960000804 ronifibrate Drugs 0.000 description 1
- AYJVGKWCGIYEAK-UHFFFAOYSA-N ronifibrate Chemical compound C=1C=CN=CC=1C(=O)OCCCOC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 AYJVGKWCGIYEAK-UHFFFAOYSA-N 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- LALFOYNTGMUKGG-BGRFNVSISA-L rosuvastatin calcium Chemical compound [Ca+2].CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O.CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O LALFOYNTGMUKGG-BGRFNVSISA-L 0.000 description 1
- 231100000279 safety data Toxicity 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 229960004937 saxagliptin Drugs 0.000 description 1
- 229950011186 semaglutide Drugs 0.000 description 1
- 108010060325 semaglutide Proteins 0.000 description 1
- 238000010206 sensitivity analysis Methods 0.000 description 1
- 229940126842 sergliflozin Drugs 0.000 description 1
- HFLCZNNDZKKXCS-OUUBHVDSSA-N sergliflozin Chemical compound C1=CC(OC)=CC=C1CC1=CC=CC=C1O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HFLCZNNDZKKXCS-OUUBHVDSSA-N 0.000 description 1
- 239000003775 serotonin noradrenalin reuptake inhibitor Substances 0.000 description 1
- 230000000697 serotonin reuptake Effects 0.000 description 1
- 239000003772 serotonin uptake inhibitor Substances 0.000 description 1
- 229960002073 sertraline Drugs 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 230000001568 sexual effect Effects 0.000 description 1
- 229960004425 sibutramine Drugs 0.000 description 1
- 229940103449 simcor Drugs 0.000 description 1
- 229960004034 sitagliptin Drugs 0.000 description 1
- 238000002741 site-directed mutagenesis Methods 0.000 description 1
- 201000000849 skin cancer Diseases 0.000 description 1
- 208000017520 skin disease Diseases 0.000 description 1
- 231100000046 skin rash Toxicity 0.000 description 1
- 201000010106 skin squamous cell carcinoma Diseases 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- MUZRGKSNUTWRAF-UHFFFAOYSA-M sodium;2-[4-[4-[5-[[6-(trifluoromethyl)pyridin-3-yl]amino]pyridin-2-yl]phenyl]cyclohexyl]acetate Chemical compound [Na+].C1CC(CC(=O)[O-])CCC1C1=CC=C(C=2N=CC(NC=3C=NC(=CC=3)C(F)(F)F)=CC=2)C=C1 MUZRGKSNUTWRAF-UHFFFAOYSA-M 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 230000000392 somatic effect Effects 0.000 description 1
- 230000037439 somatic mutation Effects 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 208000023516 stroke disease Diseases 0.000 description 1
- 210000004304 subcutaneous tissue Anatomy 0.000 description 1
- 201000009032 substance abuse Diseases 0.000 description 1
- 231100000736 substance abuse Toxicity 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 238000002636 symptomatic treatment Methods 0.000 description 1
- 206010042772 syncope Diseases 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 229960000651 tasosartan Drugs 0.000 description 1
- ADXGNEYLLLSOAR-UHFFFAOYSA-N tasosartan Chemical compound C12=NC(C)=NC(C)=C2CCC(=O)N1CC(C=C1)=CC=C1C1=CC=CC=C1C=1N=NNN=1 ADXGNEYLLLSOAR-UHFFFAOYSA-N 0.000 description 1
- 108010048573 taspoglutide Proteins 0.000 description 1
- WRGVLTAWMNZWGT-VQSPYGJZSA-N taspoglutide Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NC(C)(C)C(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)C(C)(C)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 WRGVLTAWMNZWGT-VQSPYGJZSA-N 0.000 description 1
- 229950007151 taspoglutide Drugs 0.000 description 1
- 229960005187 telmisartan Drugs 0.000 description 1
- 230000002123 temporal effect Effects 0.000 description 1
- 229940034887 tenuate Drugs 0.000 description 1
- 229940078806 teveten Drugs 0.000 description 1
- 150000001467 thiazolidinediones Chemical class 0.000 description 1
- 230000002537 thrombolytic effect Effects 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 229940036555 thyroid hormone Drugs 0.000 description 1
- 239000005495 thyroid hormone Substances 0.000 description 1
- 108091008762 thyroid hormone receptors ß Proteins 0.000 description 1
- 229940034208 thyroxine Drugs 0.000 description 1
- XUIIKFGFIJCVMT-UHFFFAOYSA-N thyroxine-binding globulin Natural products IC1=CC(CC([NH3+])C([O-])=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-UHFFFAOYSA-N 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 229950006667 tofogliflozin Drugs 0.000 description 1
- 229960005371 tolbutamide Drugs 0.000 description 1
- 229940035305 topamax Drugs 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 229960004394 topiramate Drugs 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 239000001226 triphosphate Substances 0.000 description 1
- 235000011178 triphosphate Nutrition 0.000 description 1
- UNXRWKVEANCORM-UHFFFAOYSA-N triphosphoric acid Chemical compound OP(O)(=O)OP(O)(=O)OP(O)(O)=O UNXRWKVEANCORM-UHFFFAOYSA-N 0.000 description 1
- 239000003174 triple reuptake inhibitor Substances 0.000 description 1
- 210000005239 tubule Anatomy 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 229940116269 uric acid Drugs 0.000 description 1
- 208000019206 urinary tract infection Diseases 0.000 description 1
- 229960004699 valsartan Drugs 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 229940007428 victoza Drugs 0.000 description 1
- 230000001755 vocal effect Effects 0.000 description 1
- 229960001729 voglibose Drugs 0.000 description 1
- PNAMDJVUJCJOIX-XVZWKFLSSA-N vytorin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1.N1([C@@H]([C@H](C1=O)CC[C@@H](O)C=1C=CC(F)=CC=1)C=1C=CC(O)=CC=1)C1=CC=C(F)C=C1 PNAMDJVUJCJOIX-XVZWKFLSSA-N 0.000 description 1
- 229940009349 vytorin Drugs 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 229940002552 xenical Drugs 0.000 description 1
- 229940051223 zetia Drugs 0.000 description 1
- 229940072168 zocor Drugs 0.000 description 1
- 229940020965 zoloft Drugs 0.000 description 1
- 229940061639 zonegran Drugs 0.000 description 1
- 229960002911 zonisamide Drugs 0.000 description 1
- 229940018503 zyban Drugs 0.000 description 1
Images

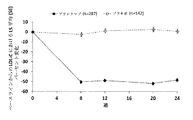
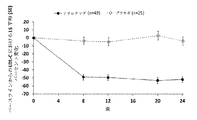


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/40—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/22—Hormones
- A61K38/28—Insulins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/54—Medicinal preparations containing antigens or antibodies characterised by the route of administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/545—Medicinal preparations containing antigens or antibodies characterised by the dose, timing or administration schedule
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Diabetes (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Endocrinology (AREA)
- Epidemiology (AREA)
- Emergency Medicine (AREA)
- Gastroenterology & Hepatology (AREA)
- Zoology (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Peptides Or Proteins (AREA)
Abstract
インスリン療法を受けている、高コレステロール血症および1型または2型真性糖尿病を有する心血管リスクの高い患者を治療する方法が提供される。これらの方法は、一般的に、インスリン療法と組み合わせて、hPCSK9抗体に特異的に結合する抗体またはその抗原結合断片を含む医薬組成物を患者に投与することを含む。Methods are provided for treating patients at high cardiovascular risk with hypercholesterolemia and type 1 or type 2 diabetes mellitus who are receiving insulin therapy. These methods generally involve administering to a patient a pharmaceutical composition comprising an antibody or antigen-binding fragment thereof that specifically binds to the hPCSK9 antibody in combination with insulin therapy.
Description
関連出願
この出願は、2017年6月9日に出願された米国仮特許出願第62/517,672号、2017年7月13日に出願された米国仮特許出願第62/532,162号、および2018年5月4日に出願された欧州特許出願第18305565.6号に対する優先権の利益を主張する。これらの関連出願のそれぞれの内容は、参照によりそれらの全体を本明細書に組み入れる。
Related Applications This application is related to US provisional patent application No. 62/517,672 filed on June 9, 2017, US provisional patent application No. 62/532,162 filed on July 13, 2017, And claim the benefit of priority to European Patent Application No. 183055565.6, filed May 4, 2018. The contents of each of these related applications are incorporated herein by reference in their entirety.
発明の分野
本発明は、脂質およびリポタンパク質のレベル上昇に関連付けられる疾患および障害の治療的処置の分野に関する。より具体的には、本発明は、高コレステロール血症を含む高脂血症を有する糖尿病患者を治療するためのPCSK9阻害剤の使用に関する。
FIELD OF THE INVENTION The present invention relates to the field of therapeutic treatment of diseases and disorders associated with elevated levels of lipids and lipoproteins. More specifically, the present invention relates to the use of PCSK9 inhibitors for treating diabetic patients with hyperlipidemia, including hypercholesterolemia.
高脂血症は、血中の脂質および/もしくはリポタンパク質のレベル上昇によって特徴付けられ、またはそれに関連付けられる疾患および障害を包含する一般的な用語である。高脂血症には、高コレステロール血症、高トリグリセリド血症、複合型高脂血症、および上昇したリポタンパク質a(Lp(a))が含まれる。多数の集団における高脂血症の特定の形態は、高コレステロール血症である。 Hyperlipidemia is a general term that encompasses diseases and disorders characterized by or associated with elevated levels of blood lipids and/or lipoproteins. Hyperlipidemia includes hypercholesterolemia, hypertriglyceridemia, combined hyperlipidemia, and elevated lipoprotein a (Lp(a)). A particular form of hyperlipidemia in many populations is hypercholesterolemia.
高コレステロール血症、特に低密度リポタンパク質(LDL)コレステロール(LDL−C)レベルの増加は、アテローム性動脈硬化症および冠状動脈性心疾患(CHD)の発症の主要なリスクを構成する(非特許文献1)。低密度リポタンパク質コレステロールは、コレステロール低下療法の主要な標的として特定されており、有効な代替治療評価項目として受け入れられている。多数の研究は、LDL−Cレベルの低下が、LDL−CレベルとCHD事象の間に強い直接的な関係があるCHDのリスクを減少させることを実証している;LDL−Cにおける1mmol/L(約40mg/dL)ごとの減少では、心血管疾患(CVD)の死亡率および罹患率が22%低下する。LDL−Cの大幅な減少は、CHD事象の大幅な減少をもたらし、集中的対標準的なスタチン治療の比較データは、LDL−Cレベルが低いほど、心血管(CV)リスクが非常に高い患者における利益がより大きいことを示唆する。 Hypercholesterolemia, especially increased low density lipoprotein (LDL) cholesterol (LDL-C) levels, constitutes a major risk of developing atherosclerosis and coronary heart disease (CHD). Reference 1). Low density lipoprotein cholesterol has been identified as a major target for cholesterol lowering therapy and has been accepted as an effective alternative therapeutic endpoint. Numerous studies have demonstrated that lowering LDL-C levels reduces the risk of CHD, which has a strong direct relationship between LDL-C levels and CHD events; 1 mmol/L in LDL-C. Every (about 40 mg/dL) reduction reduces cardiovascular disease (CVD) mortality and morbidity by 22%. A significant reduction in LDL-C results in a significant reduction in CHD events and comparative data from intensive versus standard statin treatments show that lower LDL-C levels are associated with a significantly higher cardiovascular (CV) risk in patients. Suggests that the profits in
心血管疾患(CVD)は、1型(T1)または2型(T2)真性糖尿病(DM)を有する患者の罹患率および死亡率の主要な原因であり、インスリン治療された糖尿病患者は、さらに高いCVリスクを有する。さらに、アテローム性動脈硬化症(ASCVD)を有する者の間に共存するDMが存在すると、CV事象のリスクが有意に増加する。一部の研究とメタ分析は、スタチンを使用したLDL−Cの低下は、DM患者におけるCV事象の有意な減少をもたらし、付随するエゼチミブを使用した追加のLDL−Cの低下と関連付けられたさらなるCVリスクが減少することを示している。しかしながら、現在利用可能な治療法を用いても、DMを有する多数の患者は、持続的な脂質異常を継続的に有し、したがって、CV事象の残ったリスクにさらされている。 Cardiovascular disease (CVD) is a major cause of morbidity and mortality in patients with type 1 (T1) or type 2 (T2) diabetes mellitus (DM), with insulin-treated diabetic patients being even higher. Have CV risk. Moreover, the presence of coexisting DM among persons with atherosclerosis (ASCVD) significantly increases the risk of CV events. Some studies and meta-analysis show that lowering LDL-C using statins resulted in a significant reduction in CV events in DM patients, and was associated with an additional lowering LDL-C using ezetimibe. It shows that the CV risk is reduced. However, even with the currently available treatments, many patients with DM continue to have persistent dyslipidemia and thus are at residual risk of CV events.
現在のLDL−C低下医薬には、抗PCSK9抗体などのプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)阻害剤が含まれる。抗PCSK9抗体は広範な臨床研究を受けているが、糖尿病患者におけるアリロクマブの有効性および安全性は十分に理解されていない。したがって、当該技術分野では、CVリスクが高いインスリン療法を受けている糖尿病患者における高コレステロール血症の治療に最適な有効性および安全性を提供する抗PCSK9抗体の治療レジメンを特定する必要がある。 Current LDL-C lowering drugs include proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors such as anti-PCSK9 antibodies. Although anti-PCSK9 antibodies have undergone extensive clinical research, the efficacy and safety of arilocumab in diabetic patients is not fully understood. Therefore, there is a need in the art to identify therapeutic regimens for anti-PCSK9 antibodies that provide optimal efficacy and safety for the treatment of hypercholesterolemia in diabetic patients undergoing insulin therapy at high CV risk.
本開示は、インスリン療法を受けている真性糖尿病(DM)患者における高コレステロール血症を治療する方法を提供する。特定の実施形態では、本方法は、ヒトPCSK9に特異的に結合する抗体またはその抗原結合断片の1回またはそれ以上の用量を、高コレステロール血症および糖尿病を有する患者に投与することを含む。特定の実施形態では、患者は心血管リスクが高い。特定の実施形態では、患者は、インスリン療法に加えて、付随する抗糖尿病療法を受ける。 The present disclosure provides a method of treating hypercholesterolemia in a diabetes mellitus (DM) patient undergoing insulin therapy. In certain embodiments, the method comprises administering one or more doses of an antibody or antigen-binding fragment thereof that specifically binds human PCSK9 to a patient with hypercholesterolemia and diabetes. In certain embodiments, the patient has an increased cardiovascular risk. In certain embodiments, the patient receives concomitant antidiabetic therapy in addition to insulin therapy.
一態様によれば、本方法は、1型真性糖尿病(T1DM)を有する患者における高コレステロール血症を治療する方法を含み、本方法は、
(a)(i)T1DM、および(ii)最大耐容性スタチン療法によって適切に制御されていない高コレステロール血症のインスリン療法を受けている心血管リスクの高い患者を選択すること;ならびに
(b)患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片の75mg、150mgもしくは300mgを投与すること
を含み、患者は付随するインスリン療法を受ける。
According to one aspect, the method comprises a method of treating hypercholesterolemia in a
(A) selecting (i) T1DM, and (ii) high cardiovascular risk patients receiving insulin therapy for hypercholesterolemia not adequately controlled by maximally tolerated statin therapy; and (b) The method comprises administering to a
特定の実施形態では、75mgの抗体または抗原結合断片が2週間ごとに患者に投与される。他の実施形態では、150mgの抗体または抗原結合断片が2週間ごとに患者に投与される。他の実施形態では、300mgの抗体または抗原結合断片が4週間ごとに患者に投与される。 In certain embodiments, 75 mg of antibody or antigen-binding fragment is administered to the patient every two weeks. In another embodiment, 150 mg of antibody or antigen-binding fragment is administered to the patient every two weeks. In another embodiment, 300 mg of antibody or antigen-binding fragment is administered to the patient every 4 weeks.
特定の実施形態では、抗体またはその抗原結合断片は、配列番号1/6を含むHCVR/LCVRアミノ酸配列対の重鎖および軽鎖CDRを含む。特定の実施形態では、抗体またはその抗原結合断片は、配列番号2、3、および4に記載の3つの重鎖CDR、ならびに配列番号7、8、および10に記載の3つの軽鎖CDRを含む。特定の実施形態では、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有する重鎖可変領域(HCVR)、および配列番号6のアミノ酸配列を有する軽鎖可変領域(LCVR)を含む。特定の実施形態では、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含む抗体またはその抗原結合断片との結合について競合する。特定の実施形態では、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含む抗体と同じ、PCSK9上のエピトープに結合する。特定の実施形態では、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含む抗体のエピトープと重複するPCSK9上のエピトープに結合する。 In certain embodiments, the antibody or antigen-binding fragment thereof comprises the heavy and light chain CDRs of the HCVR/LCVR amino acid sequence pair comprising SEQ ID NO: 1/6. In certain embodiments, the antibody or antigen-binding fragment thereof comprises the three heavy chain CDRs set forth in SEQ ID NOS: 2, 3 and 4 and the three light chain CDRs set forth in SEQ ID NOS: 7, 8 and 10. .. In certain embodiments, the antibody or antigen-binding fragment thereof comprises a heavy chain variable region (HCVR) having the amino acid sequence of SEQ ID NO:1 and a light chain variable region (LCVR) having the amino acid sequence of SEQ ID NO:6. In certain embodiments, the antibody or antigen-binding fragment thereof competes for binding with an antibody or antigen-binding fragment thereof that comprises HCVR having the amino acid sequence of SEQ ID NO:1 and LCVR having the amino acid sequence of SEQ ID NO:6. In a particular embodiment, the antibody or antigen-binding fragment thereof binds to the same epitope on PCSK9 as the antibody comprising HCVR having the amino acid sequence of SEQ ID NO:1 and LCVR having the amino acid sequence of SEQ ID NO:6. In certain embodiments, the antibody or antigen-binding fragment thereof binds to an epitope on PCSK9 that overlaps with an epitope of the antibody that includes HCVR having the amino acid sequence of SEQ ID NO:1 and LCVR having the amino acid sequence of SEQ ID NO:6.
特定の実施形態では、抗体またはその抗原結合断片は、配列番号86、87、88、90、91、および92を有する重鎖ならびに軽鎖CDRアミノ酸配列を含む。特定の実施形態では、抗体またはその抗原結合断片は、配列番号85に示されるアミノ酸配列と少なくとも90%、95%、もしくは99%同一であるアミノ酸配列を有するHCVR、および配列番号89に示されるアミノ酸配列と少なくとも90%、95%、もしくは99%同一であるアミノ酸配列を有するLCVRを含む。 In certain embodiments, the antibody or antigen-binding fragment thereof comprises heavy and light chain CDR amino acid sequences having SEQ ID NOs: 86, 87, 88, 90, 91, and 92. In certain embodiments, the antibody or antigen-binding fragment thereof comprises HCVR having an amino acid sequence that is at least 90%, 95%, or 99% identical to the amino acid sequence set forth in SEQ ID NO:85, and the amino acid set forth in SEQ ID NO:89. It includes an LCVR having an amino acid sequence that is at least 90%, 95%, or 99% identical to the sequence.
特定の実施形態では、抗体またはその抗原結合断片は、アリロクマブ、エボロクマブ、ボコシズマブ、ロデルシズマブ、ラルパンシズマブ、およびLY3015014からなる群から選択される。特定の実施形態では、抗体またはその抗原結合断片はアリロクマブである。 In certain embodiments, the antibody or antigen-binding fragment thereof is selected from the group consisting of arilocumab, evolocumab, bococizumab, rodercizumab, ralpancizumab, and LY301504. In certain embodiments, the antibody or antigen-binding fragment thereof is arilocumab.
特定の実施形態では、本明細書に開示される方法は、(c)例えば8週間後に、患者におけるLDL−Cレベルが閾値レベルより低い場合、約2週間ごとに1回またはそれ以上の次の用量の75mgの抗体またはその抗原結合断片を患者に投与するか、または、例えば8週間後に、患者におけるLDL−Cレベルが閾値レベル以上の場合、約2週間ごとに1回またはそれ以上の次の用量の150mgの抗体またはその抗原結合断片を投与することをさらに含む。特定の実施形態では、閾値レベルは70mg/dLである。 In certain embodiments, the methods disclosed herein include (c) about eight weeks later, if LDL-C levels in the patient are below a threshold level, about once every two weeks or more of the following: A dose of 75 mg of the antibody or antigen-binding fragment thereof is administered to the patient, or if LDL-C levels in the patient are above a threshold level, for example after 8 weeks, about once every 2 weeks or more of the following: It further comprises administering a dose of 150 mg of the antibody or antigen binding fragment thereof. In a particular embodiment, the threshold level is 70 mg/dL.
特定の実施形態では、本明細書に開示される方法は、(c)例えば8週間後に、患者におけるLDL−Cレベルが閾値レベルより低い場合、約4週間ごとに1回またはそれ以上の次の用量の300mgの抗体またはその抗原結合断片を患者に投与するか、または、例えば8週間後に、患者におけるLDL−Cレベルが閾値レベル以上の場合、約2週間ごとに1回またはそれ以上の次の用量の150mgの抗体またはその抗原結合断片を投与することをさらに含む。特定の実施形態では、閾値レベルは70mg/dLである。 In certain embodiments, the methods disclosed herein include (c) about every four weeks, if after 8 weeks LDL-C levels in the patient are below a threshold level, one or more of the following: A dose of 300 mg of the antibody or antigen-binding fragment thereof is administered to the patient, or if LDL-C levels in the patient are above a threshold level, for example after 8 weeks, about once every 2 weeks or more of the following: It further comprises administering a dose of 150 mg of the antibody or antigen binding fragment thereof. In a particular embodiment, the threshold level is 70 mg/dL.
特定の実施形態では、抗体またはその抗原結合断片は皮下投与される。 In certain embodiments, the antibody or antigen-binding fragment thereof is administered subcutaneously.
特定の実施形態では、患者は、付随する脂質修飾療法(LMT)をさらに受ける。特定の実施形態では、LMTは、スタチン、コレステロール吸収阻害剤、フィブラート、ナイアシン、オメガ−3脂肪酸、および胆汁酸封鎖剤からなる群から選択される。特定の実施形態では、LMTはスタチン療法である。特定の実施形態では、スタチンは、アトルバスタチン、ロスバスタチン、シンバスタチン、プラバスタチン、ロバスタチン、フルバスタチン、ピタバスタチン、およびセリバスタチンからなる群から選択される。特定の実施形態では、スタチン療法は、最大耐性量のスタチン療法である。特定の実施形態では、コレステロール吸収阻害剤はエゼチミブである。特定の実施形態では、患者は、スタチンに対して不耐性である。 In certain embodiments, the patient further receives concomitant lipid modification therapy (LMT). In certain embodiments, the LMT is selected from the group consisting of statins, cholesterol absorption inhibitors, fibrates, niacin, omega-3 fatty acids, and bile sequestrants. In certain embodiments, LMT is statin therapy. In certain embodiments, the statin is selected from the group consisting of atorvastatin, rosuvastatin, simvastatin, pravastatin, lovastatin, fluvastatin, pitavastatin, and cerivastatin. In certain embodiments, the statin therapy is a maximally resistant amount of statin therapy. In certain embodiments, the cholesterol absorption inhibitor is ezetimibe. In certain embodiments, the patient is intolerant to statins.
特定の実施形態では、インスリン療法は、ヒトインスリン、インスリングラルギン、インスリングルリジン、インスリンデテミル、インスリンリスプロ、インスリンデグルデック、インスリンアスパルト、および基礎インスリンからなる群から選択される。特定の実施形態では、患者は、インスリン療法に加えてさらなる付随する抗糖尿病療法を受ける。特定の実施形態では、追加の付随する抗糖尿病療法は、グルカゴン様ペプチド1(GLP−1)療法、胃腸ペプチド、グルカゴン受容体アゴニストまたはアンタゴニスト、グルコース依存性インスリン分泌性ポリペプチド(GIP)受容体アゴニストまたはアンタゴニストアンタゴニスト、グレリンアンタゴニストまたは逆アゴニスト、キセニン、キセニン類似体、ビグアナイド、スルホニル尿素、メグリチニド、チアゾリジンジオン、DPP−4阻害剤、アルファ−グルコシダーゼ阻害剤、ナトリウム依存性グルコース輸送体2(SGLT−2)阻害剤、SGLT−1阻害剤、ペルオキシソーム増殖因子活性化受容体(PPAR−)(アルファ、ガンマまたはアルファ/ガンマ)アゴニストまたはモジュレータ、アミリン、アミリンアナログ、Gタンパク質共役受容体119(GPR119)アゴニスト、GPR40アゴニスト、GPR120アゴニスト、GPR142アゴニスト、全身または低吸収性TGR5アゴニスト、糖尿病免疫療法薬、メタボリックシンドロームおよび糖尿病を治療するための抗炎症薬、アデノシンモノプリン酸活性化プロテインキナーゼ(AMPK)刺激薬、11−ベータ−ヒドロキシステロイドデヒドロゲナーゼ1の阻害剤、グルコキナーゼの活性化剤、ジアシルグリセロールO−アシルトランスフェラーゼ(DGAT)の阻害剤、グルコーストランスポータ−4のモジュレータ、ソマトスタチン受容体3アゴニスト、脂質減少剤、ならびにそれらの組み合わせからなる群から選択される。
In certain embodiments, the insulin therapy is selected from the group consisting of human insulin, insulin glargine, insulin glulisine, insulin detemir, insulin lispro, insulin degludec, insulin aspart, and basal insulin. In certain embodiments, the patient receives additional concomitant antidiabetic therapy in addition to insulin therapy. In certain embodiments, the additional concomitant anti-diabetic therapy is glucagon-like peptide 1 (GLP-1) therapy, gastrointestinal peptide, glucagon receptor agonist or antagonist, glucose-dependent insulinotropic polypeptide (GIP) receptor agonist. Or antagonist antagonist, ghrelin antagonist or inverse agonist, xenin, xenin analogue, biguanide, sulfonylurea, meglitinide, thiazolidinedione, DPP-4 inhibitor, alpha-glucosidase inhibitor, sodium-dependent glucose transporter 2 (SGLT-2) Inhibitor, SGLT-1 inhibitor, peroxisome proliferator-activated receptor (PPAR-) (alpha, gamma or alpha/gamma) agonist or modulator, amylin, amylin analog, G protein coupled receptor 119 (GPR119) agonist, GPR40 Agonists, GPR120 agonists, GPR142 agonists, systemic or low absorption TGR5 agonists, diabetic immunotherapeutic agents, anti-inflammatory agents for treating metabolic syndrome and diabetes, adenosine monopurine activated protein kinase (AMPK) stimulants, 11- Inhibitors of beta-
特定の実施形態では、抗体またはその抗原結合断片は、患者のLDL−Cレベルを、例えば、少なくとも30%、35%、40%、もしくは45%減少させる。特定の実施形態では、抗体またはその抗原結合断片は、患者の非HDL−Cレベルを、例えば、少なくとも25%、30%、35%、もしくは40減少させる。特定の実施形態では、抗体またはその抗原結合断片は、患者のアポリポタンパク質C3(ApoC3)レベルを(例えば、治療の12週間または24週間後に少なくとも約6.0%、約6.5%、約7.0%または約7.5%)減少させる。特定の実施形態では、抗体またはその抗原結合断片は、患者のリポタンパク質粒子の数を(例えば、治療の12週間または24週間後に少なくとも約20%、約30%、約40%または約50%)減少させる。他の実施形態では、抗体またはその抗原結合断片は、患者のリポタンパク質粒子のサイズを(例えば、治療の12週間または24週間後に少なくとも約1.5%、約2%、約2.5%、または約3%)減少させる。 In certain embodiments, the antibody or antigen-binding fragment thereof reduces LDL-C levels in a patient, eg, by at least 30%, 35%, 40%, or 45%. In certain embodiments, the antibody or antigen-binding fragment thereof reduces non-HDL-C levels in a patient, eg, by at least 25%, 30%, 35%, or 40. In certain embodiments, the antibody or antigen-binding fragment thereof increases apolipoprotein C3 (ApoC3) levels in a patient (eg, at least about 6.0%, about 6.5%, about 7 weeks or 12 weeks after treatment). 0.0% or about 7.5%). In certain embodiments, the antibody or antigen-binding fragment thereof increases the number of lipoprotein particles in a patient (eg, at least about 20%, about 30%, about 40% or about 50% after 12 or 24 weeks of treatment). Reduce. In other embodiments, the antibody or antigen-binding fragment thereof increases the size of the patient's lipoprotein particles (eg, at least about 1.5%, about 2%, about 2.5% after 12 or 24 weeks of treatment). Or about 3%).
特定の実施形態では、抗体またはその抗原結合断片は、(a)患者のヘモグロビンA1c(HbA1c)レベルに影響を及ぼさない;および/または(b)患者の空腹時血漿グルコース(FPG)レベルに影響を及ぼさない In certain embodiments, the antibody or antigen-binding fragment thereof does not (a) affect the patient's hemoglobin A1c (HbA1c) levels; and/or (b) affects the patient's fasting plasma glucose (FPG) levels. Does not reach
別の態様によれば、本方法は、1型真性糖尿病(T1DM)を有する患者における高コレステロール血症を治療する方法を含み、本方法は、
(a)(i)T1DM、および(ii)最大耐容スタチン療法によっては適切に制御されない高コレステロール血症を有する、インスリン療法を受けている心血管リスクの高い患者を選択すること;
(b)患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片を75mg、2週間ごとに投与すること;ならびに
(c)例えば8週間後に、患者におけるLDL−Cレベルが70mg/dLより低い場合、約2週間ごとに1回またはそれ以上の次の用量の75mgの抗体またはその抗原結合断片を患者に投与するか、または、例えば8週間後に、患者におけるLDL−Cレベルが70mg/dL以上の場合、約2週間ごとに1回またはそれ以上の次の用量の150mgの抗体またはその抗原結合断片を投与すること
を含み、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含み、患者は付随するインスリン療法を受ける。
According to another aspect, the method comprises a method of treating hypercholesterolemia in a patient having diabetes mellitus type 1 (T1DM), the method comprising:
Selecting (a) (i) T1DM, and (ii) patients with high cardiovascular risk receiving insulin therapy who have hypercholesterolemia that is not adequately controlled by maximally tolerated statin therapy;
(B) administering to the patient 75 mg of an antibody or an antigen-binding fragment thereof that specifically binds to human proprotein convertase subtilisin/kexin type 9 (PCSK9) every 2 weeks; and (c) after 8 weeks, for example. , If the LDL-C level in the patient is lower than 70 mg/dL, the patient is administered with one or more subsequent doses of 75 mg of the antibody or antigen-binding fragment thereof about once every two weeks, or, for example, for 8 weeks. Later, when the LDL-C level in the patient is 70 mg/dL or more, the method comprises administering 150 mg of the antibody or antigen-binding fragment thereof at one or more subsequent doses about every two weeks, wherein the antibody or antigen-binding fragment thereof is administered. The binding fragment comprises HCVR having the amino acid sequence of SEQ ID NO:1 and LCVR having the amino acid sequence of SEQ ID NO:6, and the patient undergoes concomitant insulin therapy.
別の態様によれば、本方法は、1型真性糖尿病(T1DM)を有する患者における高コレステロール血症を治療する方法を含み、本方法は、
(a)(i)T1DM、および(ii)最大耐容スタチン療法によっては適切に制御されない高コレステロール血症を有する、インスリン療法を受けている心血管リスクの高い患者を選択すること;
(b)患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片を300mg、4週間ごとに投与すること;ならびに
(c)8週間後に、患者におけるLDL−Cレベルが閾値レベルより高い場合、約4週間ごとに1回またはそれ以上の次の投薬量の300mgの抗体またはその抗原結合断片を患者に投与するか、または、8週間後に、患者におけるLDL−Cレベルが閾値レベル以下の場合、約2週間ごとに1回またはそれ以上の次の投薬量の150mgの抗体またはその抗原結合断片を投与すること
を含み、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含み、患者は付随するインスリン療法を受ける。一実施形態では、閾値レベルは15mg/dLである。別の実施形態では、閾値レベルは25mg/dLである。
According to another aspect, the method comprises a method of treating hypercholesterolemia in a patient having diabetes mellitus type 1 (T1DM), the method comprising:
Selecting (a) (i) T1DM, and (ii) patients with high cardiovascular risk receiving insulin therapy who have hypercholesterolemia that is not adequately controlled by maximally tolerated statin therapy;
(B) administering to a patient 300 mg of an antibody or an antigen-binding fragment thereof that specifically binds to human proprotein convertase subtilisin/kexin type 9 (PCSK9) every 4 weeks; and (c) after 8 weeks, If the LDL-C level in the patient is above the threshold level, the patient is administered one or more subsequent doses of 300 mg of the antibody or antigen binding fragment thereof about every four weeks, or after eight weeks, If the LDL-C level in the patient is below a threshold level, comprising administering 150 mg of the antibody or antigen-binding fragment thereof at one or more subsequent doses about every two weeks, the antibody or antigen-binding fragment thereof. Includes an HCVR having the amino acid sequence of SEQ ID NO:1, and an LCVR having the amino acid sequence of SEQ ID NO:6, wherein the patient receives concomitant insulin therapy. In one embodiment, the threshold level is 15 mg/dL. In another embodiment, the threshold level is 25 mg/dL.
別の態様によれば、本方法は、2型真性糖尿病(T2DM)を有する患者における高コレステロール血症を治療する方法を含み、本方法は、
(a)(i)T2DM、および(ii)最大耐容性スタチン療法によって適切に制御されていない高コレステロール血症のインスリン療法を受けている心血管リスクの高い患者を選択すること;ならびに
(b)患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片の75mg、150mgもしくは300mgを投与すること
を含み、患者は付随するインスリン療法を受ける。
According to another aspect, the method comprises a method of treating hypercholesterolemia in a patient having diabetes mellitus type 2 (T2DM), the method comprising:
(A) selecting (i) T2DM, and (ii) high cardiovascular risk patients receiving insulin therapy for hypercholesterolemia not adequately controlled by maximally tolerated statin therapy; and (b) Comprising administering to a patient 75 mg, 150 mg or 300 mg of an antibody or antigen-binding fragment thereof that specifically binds to the human proprotein convertase subtilisin/kexin type 9 (PCSK9), the patient undergoing concomitant insulin therapy.
特定の実施形態では、75mgの抗体または抗原結合断片が2週間ごとに患者に投与される。他の実施形態では、150mgの抗体または抗原結合断片が2週間ごとに患者に投与される。他の実施形態では、300mgの抗体または抗原結合断片が4週間ごとに患者に投与される。 In certain embodiments, 75 mg of antibody or antigen-binding fragment is administered to the patient every two weeks. In another embodiment, 150 mg of antibody or antigen-binding fragment is administered to the patient every two weeks. In another embodiment, 300 mg of antibody or antigen-binding fragment is administered to the patient every 4 weeks.
特定の実施形態では、抗体またはその抗原結合断片は、配列番号1/6を含むHCVR/LCVRアミノ酸配列対の重鎖および軽鎖CDRを含む。特定の実施形態では、抗体またはその抗原結合断片は、配列番号2、3、および4に記載の3つの重鎖CDR、ならびに配列番号7、8、および10に記載の3つの軽鎖CDRを含む。特定の実施形態では、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有する重鎖可変領域(HCVR)、および配列番号6のアミノ酸配列を有する軽鎖可変領域(LCVR)を含む。特定の実施形態では、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含む抗体またはその抗原結合断片との結合について競合する。特定の実施形態では、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含む抗体と同じ、PCK9上のエピトープに結合する。特定の実施形態では、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含む抗体のエピトープと重複するPCSK9上のエピトープに結合する。 In certain embodiments, the antibody or antigen-binding fragment thereof comprises the heavy and light chain CDRs of the HCVR/LCVR amino acid sequence pair comprising SEQ ID NO: 1/6. In certain embodiments, the antibody or antigen-binding fragment thereof comprises the three heavy chain CDRs set forth in SEQ ID NOS: 2, 3 and 4 and the three light chain CDRs set forth in SEQ ID NOS: 7, 8 and 10. .. In certain embodiments, the antibody or antigen-binding fragment thereof comprises a heavy chain variable region (HCVR) having the amino acid sequence of SEQ ID NO:1 and a light chain variable region (LCVR) having the amino acid sequence of SEQ ID NO:6. In certain embodiments, the antibody or antigen-binding fragment thereof competes for binding with an antibody or antigen-binding fragment thereof that comprises HCVR having the amino acid sequence of SEQ ID NO:1 and LCVR having the amino acid sequence of SEQ ID NO:6. In a particular embodiment, the antibody or antigen-binding fragment thereof binds to the same epitope on PCK9 as the antibody comprising HCVR having the amino acid sequence of SEQ ID NO:1 and LCVR having the amino acid sequence of SEQ ID NO:6. In certain embodiments, the antibody or antigen-binding fragment thereof binds to an epitope on PCSK9 that overlaps with an epitope of the antibody that includes HCVR having the amino acid sequence of SEQ ID NO:1 and LCVR having the amino acid sequence of SEQ ID NO:6.
特定の実施形態では、抗体またはその抗原結合断片は、それぞれ配列番号85および89で示されるアミノ酸配列を含む重鎖可変領域(HCVR)ならびに軽鎖可変領域(LCVR)の相補性決定領域(CDR)を含む。特定の実施形態では、抗体またはその抗原結合断片は、配列番号86、87、88、90、91、および92を有する重鎖ならびに軽鎖CDRアミノ酸配列を含む。特定の実施形態では、抗体またはその抗原結合断片は、配列番号85に示されるアミノ酸配列と少なくとも90%、95%、または99%同一であるアミノ酸配列を有するHCVR、および配列番号89に示されるアミノ酸配列と少なくとも90%、95%、または99%同一であるアミノ酸配列を有するLCVRを含む。 In certain embodiments, the antibody or antigen-binding fragment thereof has a complementarity determining region (CDR) of a heavy chain variable region (HCVR) and a light chain variable region (LCVR) comprising the amino acid sequences set forth in SEQ ID NOS:85 and 89, respectively. including. In certain embodiments, the antibody or antigen-binding fragment thereof comprises heavy and light chain CDR amino acid sequences having SEQ ID NOs: 86, 87, 88, 90, 91, and 92. In certain embodiments, the antibody or antigen-binding fragment thereof comprises HCVR having an amino acid sequence that is at least 90%, 95%, or 99% identical to the amino acid sequence set forth in SEQ ID NO:85, and the amino acid set forth in SEQ ID NO:89. It includes an LCVR having an amino acid sequence that is at least 90%, 95%, or 99% identical to the sequence.
特定の実施形態では、抗体またはその抗原結合断片は、アリロクマブ、エボロクマブ、ボコシズマブ、ロデルシズマブ、ラルパンシズマブ、およびLY3015014からなる群から選択される。特定の実施形態では、抗体またはその抗原結合断片はアリロクマブである。 In certain embodiments, the antibody or antigen-binding fragment thereof is selected from the group consisting of arilocumab, evolocumab, bococizumab, rodercizumab, ralpancizumab, and LY301504. In certain embodiments, the antibody or antigen-binding fragment thereof is arilocumab.
特定の実施形態では、本明細書に開示される方法は、(c)患者におけるLDL−Cレベルが閾値レベルより低い場合、約2週間ごとに1回またはそれ以上の次の用量の75mgの抗体もしくはその抗原結合断片を患者に投与するか、または患者におけるLDL−Cレベルが閾値レベル以上の場合、約2週間ごとに1回またはそれ以上の次の用量の150mgの抗体もしくはその抗原結合断片を投与することをさらに含む。特定の実施形態では、閾値レベルは70mg/dLである。 In certain embodiments, the methods disclosed herein include (c) if LDL-C levels in a patient are below a threshold level, about every two weeks at one or more subsequent doses of 75 mg of antibody. Alternatively, the antigen-binding fragment thereof is administered to the patient, or if the LDL-C level in the patient is above the threshold level, about every two weeks one or more subsequent doses of 150 mg antibody or antigen-binding fragment thereof are administered. Further comprising administering. In a particular embodiment, the threshold level is 70 mg/dL.
特定の実施形態では、本明細書に開示される方法は、(c)例えば8週間後に、患者におけるLDL−Cレベルが閾値レベルより低い場合、約4週間ごとに1回またはそれ以上の次の用量の300mgの抗体もしくはその抗原結合断片を患者に投与するか、または、例えば8週間後に、患者におけるLDL−Cレベルが閾値レベル以上の場合、約2週間ごとに1回またはそれ以上の次の用量の150mgの抗体もしくはその抗原結合断片を投与することをさらに含む。特定の実施形態では、閾値レベルは70mg/dLである。 In certain embodiments, the methods disclosed herein include (c) about every four weeks, if after 8 weeks LDL-C levels in the patient are below a threshold level, one or more of the following: A dose of 300 mg of the antibody or antigen-binding fragment thereof is administered to the patient, or if LDL-C levels in the patient are above a threshold level, for example after 8 weeks, about once every 2 weeks or more of the following: Further comprising administering a dose of 150 mg of the antibody or antigen binding fragment thereof. In a particular embodiment, the threshold level is 70 mg/dL.
特定の実施形態では、抗体またはその抗原結合断片は皮下投与される。 In certain embodiments, the antibody or antigen-binding fragment thereof is administered subcutaneously.
特定の実施形態では、患者は、付随する脂質修飾療法(LMT)をさらに受ける。特定の実施形態では、LMTは、スタチン、コレステロール吸収阻害剤、フィブラート、ナイアシン、オメガ−3脂肪酸、および胆汁酸封鎖剤からなる群から選択される。特定の実施形態では、LMTはスタチン療法である。特定の実施形態では、スタチンは、アトルバスタチン、ロスバスタチン、シンバスタチン、プラバスタチン、ロバスタチン、フルバスタチン、ピタバスタチン、およびセリバスタチンからなる群から選択される。特定の実施形態では、スタチン療法は、最大耐性量のスタチン療法である。特定の実施形態では、コレステロール吸収阻害剤はエゼチミブである。 In certain embodiments, the patient further receives concomitant lipid modification therapy (LMT). In certain embodiments, the LMT is selected from the group consisting of statins, cholesterol absorption inhibitors, fibrates, niacin, omega-3 fatty acids, and bile sequestrants. In certain embodiments, LMT is statin therapy. In certain embodiments, the statin is selected from the group consisting of atorvastatin, rosuvastatin, simvastatin, pravastatin, lovastatin, fluvastatin, pitavastatin, and cerivastatin. In certain embodiments, the statin therapy is a maximally resistant amount of statin therapy. In certain embodiments, the cholesterol absorption inhibitor is ezetimibe.
特定の実施形態では、患者は、スタチンにして不耐性である。 In certain embodiments, the patient is statin intolerant.
特定の実施形態では、インスリン療法は、ヒトインスリン、インスリングラルギン、インスリングルリジン、インスリンデテミル、インスリンリスプロ、インスリンデグルデック、インスリンアスパルト、および基礎インスリンからなる群から選択される。 In certain embodiments, the insulin therapy is selected from the group consisting of human insulin, insulin glargine, insulin glulisine, insulin detemir, insulin lispro, insulin degludec, insulin aspart, and basal insulin.
特定の実施形態では、患者は、インスリン療法に加えて付随する抗糖尿病療法をさらに受ける。特定の実施形態では、追加の付随する抗糖尿病療法は、グルカゴン様ペプチド1(GLP−1)療法、胃腸ペプチド、グルカゴン受容体アゴニストまたはアンタゴニスト、グルコース依存性インスリン分泌性ポリペプチド(GIP)受容体アゴニストまたはアンタゴニスト、グレリンアンタゴニストまたは逆アゴニスト、キセニン、キセニン類似体、ビグアナイド、スルホニル尿素、メグリチニド、チアゾリジンジオン、DPP−4阻害剤、アルファ−グルコシダーゼ阻害剤、ナトリウム依存性グルコース輸送体2(SGLT−2)阻害剤、SGLT−1阻害剤、ペルオキシソーム増殖因子活性化受容体(PPAR−)(アルファ、ガンマまたはアルファ/ガンマ)アゴニストまたはモジュレータ、アミリン、アミリンアナログ、Gタンパク質共役受容体119(GPR119)アゴニスト、GPR40アゴニスト、GPR120アゴニスト、GPR142アゴニスト、全身または低吸収性TGR5アゴニスト、糖尿病免疫療法薬、メタボリックシンドロームおよび糖尿病を治療するための抗炎症薬、アデノシンモノプリン酸活性化プロテインキナーゼ(AMPK)刺激薬、11−ベータ−ヒドロキシステロイドデヒドロゲナーゼ1の阻害剤、グルコキナーゼの活性化剤、ジアシルグリセロールO−アシルトランスフェラーゼ(DGAT)の阻害剤、グルコーストランスポータ−4のモジュレータ、ソマトスタチン受容体3アゴニスト、脂質減少剤、ならびにそれらの組み合わせからなる群から選択される。
In certain embodiments, the patient further receives concomitant antidiabetic therapy in addition to insulin therapy. In certain embodiments, the additional concomitant anti-diabetic therapy is glucagon-like peptide 1 (GLP-1) therapy, gastrointestinal peptide, glucagon receptor agonist or antagonist, glucose-dependent insulinotropic polypeptide (GIP) receptor agonist. Or antagonist, ghrelin antagonist or inverse agonist, xenin, xenin analogue, biguanide, sulfonylurea, meglitinide, thiazolidinedione, DPP-4 inhibitor, alpha-glucosidase inhibitor, sodium-dependent glucose transporter 2 (SGLT-2) inhibition Agent, SGLT-1 inhibitor, peroxisome proliferator-activated receptor (PPAR-) (alpha, gamma or alpha/gamma) agonist or modulator, amylin, amylin analog, G protein coupled receptor 119 (GPR119) agonist, GPR40 agonist , GPR120 agonists, GPR142 agonists, systemic or low absorption TGR5 agonists, diabetic immunotherapeutic agents, anti-inflammatory agents for treating metabolic syndrome and diabetes, adenosine monopurinate activated protein kinase (AMPK) stimulants, 11-beta -
特定の実施形態では、抗体またはその抗原結合断片は、患者のLDL−Cレベルを、例えば、少なくとも30%、35%、40%、もしくは45%減少させる。特定の実施形態では、抗体またはその抗原結合断片は、患者の非HDL−Cレベルを、例えば、少なくとも20%、25%、30%、もしくは35%減少させる。特定の実施形態では、抗体またはその抗原結合断片は、患者のアポリポタンパク質C3(ApoC3)レベルを(例えば、治療の12週間または24週間後に少なくとも約6.0%、約6.5%、約7.0%または約7.5%)減少させる。特定の実施形態では、抗体またはその抗原結合断片は、患者のリポタンパク質粒子の数を(例えば、治療の12週間または24週間後に少なくとも約20%、約30%、約40%または約50%)減少させる。他の実施形態では、抗体またはその抗原結合断片は、患者のリポタンパク質粒子のサイズを(例えば、治療の12週間または24週間後に少なくとも約1.5%、約2%、約2.5%、または約3%)減少させる。 In certain embodiments, the antibody or antigen-binding fragment thereof reduces LDL-C levels in a patient, eg, by at least 30%, 35%, 40%, or 45%. In certain embodiments, the antibody or antigen-binding fragment thereof reduces non-HDL-C levels in a patient by, for example, at least 20%, 25%, 30%, or 35%. In certain embodiments, the antibody or antigen-binding fragment thereof increases apolipoprotein C3 (ApoC3) levels in a patient (eg, at least about 6.0%, about 6.5%, about 7 weeks or 12 weeks after treatment). 0.0% or about 7.5%). In certain embodiments, the antibody or antigen-binding fragment thereof increases the number of lipoprotein particles in a patient (eg, at least about 20%, about 30%, about 40% or about 50% after 12 or 24 weeks of treatment). Reduce. In other embodiments, the antibody or antigen-binding fragment thereof increases the size of the patient's lipoprotein particles (eg, at least about 1.5%, about 2%, about 2.5% after 12 or 24 weeks of treatment). Or about 3%).
特定の実施形態では、抗体またはその抗原結合断片は、(a)患者のヘモグロビンA1c(HbA1c)レベルに影響を及ぼさない;および/または(b)患者の空腹時血漿グルコース(FPG)レベルに影響を及ぼさない。 In certain embodiments, the antibody or antigen-binding fragment thereof does not (a) affect the patient's hemoglobin A1c (HbA1c) levels; and/or (b) affects the patient's fasting plasma glucose (FPG) levels. Does not reach.
別の態様によれば、本方法は、2型真性糖尿病(T2DM)を有する患者における高コレステロール血症を治療する方法を含み、本方法は、
(a)(i)T2DM、および(ii)最大耐性量のスタチン療法によっては適切に制御されない高コレステロール血症を有する、インスリン療法を受けている心血管リスクの高い患者を選択すること;
(b)患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片を75mg、2週間ごとに投与すること;ならびに
(c)例えば8週間後に、患者におけるLDL−Cレベルが70mg/dLより低い場合、約2週間ごとに1回またはそれ以上の次の用量の75mgの抗体もしくはその抗原結合断片を患者に投与するか、または、例えば8週間後に、患者におけるLDL−Cレベルが70mg/dL以上の場合、約2週間ごとに1回またはそれ以上の次の用量の150mgの抗体もしくはその抗原結合断片を投与すること
を含み、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含み、患者は付随するインスリン療法を受ける。
According to another aspect, the method comprises a method of treating hypercholesterolemia in a patient having diabetes mellitus type 2 (T2DM), the method comprising:
(A) selecting patients with high cardiovascular risk receiving insulin therapy, who have hypercholesterolemia that is not adequately controlled by (i) T2DM, and (ii) maximally tolerated statin therapy;
(B) administering to the patient 75 mg of an antibody or an antigen-binding fragment thereof that specifically binds to human proprotein convertase subtilisin/kexin type 9 (PCSK9) every 2 weeks; and (c) after 8 weeks, for example. , If the LDL-C level in the patient is lower than 70 mg/dL, the patient is administered with one or more subsequent doses of 75 mg of the antibody or antigen-binding fragment thereof about once every two weeks or, for example, for 8 weeks. Later, if the LDL-C level in the patient is 70 mg/dL or more, comprising administering 150 mg of the antibody or antigen-binding fragment thereof at one or more subsequent doses about every two weeks, the antibody or antigen thereof. The binding fragment comprises HCVR having the amino acid sequence of SEQ ID NO:1 and LCVR having the amino acid sequence of SEQ ID NO:6, and the patient undergoes concomitant insulin therapy.
別の態様によれば、本方法は、2型真性糖尿病(T2DM)を有する患者における高コレステロール血症を治療する方法を含み、本方法は、
(a)(i)T2DM、および(ii)最大耐性量のスタチン療法によっては適切に制御されない高コレステロール血症を有する、インスリン療法を受けている心血管リスクの高い患者を選択すること;
(b)患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片を300mg、4週間ごとに投与すること;ならびに
(c)8週間後に、患者におけるLDL−Cレベルが閾値レベルより高い場合、約4週間ごとに1回またはそれ以上の次の投薬量の300mgの抗体もしくはその抗原結合断片を患者に投与するか、または、8週間後に、患者におけるLDL−Cレベルが閾値レベル以下の場合、約2週間ごとに1回またはそれ以上の次の投薬量の150mgの抗体もしくはその抗原結合断片を投与すること
を含み、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含み、患者は付随するインスリン療法を受ける。一実施形態では、閾値レベルは15mg/dLである。別の実施形態では、閾値レベルは25mg/dLである。
According to another aspect, the method comprises a method of treating hypercholesterolemia in a patient having diabetes mellitus type 2 (T2DM), the method comprising:
(A) selecting patients with high cardiovascular risk receiving insulin therapy, who have hypercholesterolemia that is not adequately controlled by (i) T2DM, and (ii) maximally tolerated statin therapy;
(B) administering to a patient 300 mg of an antibody or an antigen-binding fragment thereof that specifically binds to human proprotein convertase subtilisin/kexin type 9 (PCSK9) every 4 weeks; and (c) after 8 weeks, If the LDL-C level in the patient is above the threshold level, the patient is administered one or more subsequent doses of 300 mg of the antibody or antigen-binding fragment thereof about every four weeks, or after 8 weeks, If the LDL-C level in the patient is below the threshold level, the method comprises administering 150 mg of the antibody or antigen-binding fragment thereof at one or more subsequent doses about every two weeks, the antibody or antigen-binding fragment thereof. Includes an HCVR having the amino acid sequence of SEQ ID NO:1, and an LCVR having the amino acid sequence of SEQ ID NO:6, wherein the patient receives concomitant insulin therapy. In one embodiment, the threshold level is 15 mg/dL. In another embodiment, the threshold level is 25 mg/dL.
別の態様によれば、本方法は、T2DMおよびアテローム性動脈硬化性心血管疾患(ASCVD)を有する患者における高コレステロール血症を治療する方法を含み、本方法は、
(a)(i)T2DM、(ii)ASCVD、および(iii)最大耐性量のスタチン療法によって適切に制御されていない高コレステロール血症のインスリン療法を受けている心血管リスクの高い患者を選択すること;ならびに
(b)患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片の75mg、150mgもしくは300mgを投与すること
を含み、患者は付随するインスリン療法を受ける。
According to another aspect, the method comprises a method of treating hypercholesterolemia in a patient having T2DM and atherosclerotic cardiovascular disease (ASCVD), the method comprising:
Select patients at high cardiovascular risk who are receiving insulin therapy for hypercholesterolemia not adequately controlled by (a) (i) T2DM, (ii) ASCVD, and (iii) maximally tolerated statin therapy And (b) administering to the patient 75 mg, 150 mg or 300 mg of an antibody or antigen-binding fragment thereof that specifically binds to human proprotein convertase subtilisin/kexin type 9 (PCSK9), the patient being Receive insulin therapy.
特定の実施形態では、ASCVDは、冠状動脈性心疾患(CHD)、虚血性脳卒中、または末梢動脈疾患として定義される。特定の実施形態では、CHDは、急性心筋梗塞、無症候性心筋梗塞、および不安定狭心症を含む。 In certain embodiments, ASCVD is defined as coronary heart disease (CHD), ischemic stroke, or peripheral arterial disease. In certain embodiments, CHD comprises acute myocardial infarction, subclinical myocardial infarction, and unstable angina.
特定の実施形態では、75mgの抗体または抗原結合断片が2週間ごとに患者に投与される。特定の実施形態では、150mgの抗体または抗原結合断片が2週間ごとに患者に投与される。特定の実施形態では、300mgの抗体または抗原結合断片が4週間ごとに患者に投与される。 In certain embodiments, 75 mg of antibody or antigen-binding fragment is administered to the patient every two weeks. In certain embodiments, 150 mg of antibody or antigen-binding fragment is administered to the patient every two weeks. In certain embodiments, 300 mg of antibody or antigen-binding fragment is administered to the patient every 4 weeks.
特定の実施形態では、抗体またはその抗原結合断片は、配列番号1/6を含むHCVR/LCVRアミノ酸配列対の重鎖および軽鎖CDRを含む。特定の実施形態では、抗体またはその抗原結合断片は、配列番号2、3、および4に記載の3つの重鎖CDR、ならびに配列番号7、8、および10に記載の3つの軽鎖CDRを含む。特定の実施形態では、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有する重鎖可変領域(HCVR)、および配列番号6のアミノ酸配列を有する軽鎖可変領域(LCVR)を含む。特定の実施形態では、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含む抗体と同じ、PCSK9上のエピトープに結合する。特定の実施形態では、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含む抗体のエピトープと重複するPCSK9上のエピトープに結合する。 In certain embodiments, the antibody or antigen-binding fragment thereof comprises the heavy and light chain CDRs of the HCVR/LCVR amino acid sequence pair comprising SEQ ID NO: 1/6. In certain embodiments, the antibody or antigen-binding fragment thereof comprises the three heavy chain CDRs set forth in SEQ ID NOS: 2, 3 and 4 and the three light chain CDRs set forth in SEQ ID NOS: 7, 8 and 10. .. In certain embodiments, the antibody or antigen-binding fragment thereof comprises a heavy chain variable region (HCVR) having the amino acid sequence of SEQ ID NO:1 and a light chain variable region (LCVR) having the amino acid sequence of SEQ ID NO:6. In a particular embodiment, the antibody or antigen-binding fragment thereof binds to the same epitope on PCSK9 as the antibody comprising HCVR having the amino acid sequence of SEQ ID NO:1 and LCVR having the amino acid sequence of SEQ ID NO:6. In certain embodiments, the antibody or antigen-binding fragment thereof binds to an epitope on PCSK9 that overlaps with an epitope of the antibody that includes HCVR having the amino acid sequence of SEQ ID NO:1 and LCVR having the amino acid sequence of SEQ ID NO:6.
特定の実施形態では、抗体またはその抗原結合断片は、それぞれ配列番号85および89で示されるアミノ酸配列を含む重鎖可変領域(HCVR)ならびに軽鎖可変領域(LCVR)の相補性決定領域(CDR)を含む。特定の実施形態では、抗体またはその抗原結合断片は、配列番号86、87、88、90、91、および92を有する重鎖ならびに軽鎖CDRアミノ酸配列を含む。特定の実施形態では、抗体またはその抗原結合断片は、配列番号85に示されるアミノ酸配列と少なくとも90%、95%、または99%同一であるアミノ酸配列を有するHCVR、および配列番号89に示されるアミノ酸配列と少なくとも90%、95%、または99%同一であるアミノ酸配列を有するLCVRを含む。 In certain embodiments, the antibody or antigen-binding fragment thereof has a complementarity determining region (CDR) of a heavy chain variable region (HCVR) and a light chain variable region (LCVR) comprising the amino acid sequences set forth in SEQ ID NOS:85 and 89, respectively. including. In certain embodiments, the antibody or antigen-binding fragment thereof comprises heavy and light chain CDR amino acid sequences having SEQ ID NOs: 86, 87, 88, 90, 91, and 92. In certain embodiments, the antibody or antigen-binding fragment thereof comprises HCVR having an amino acid sequence that is at least 90%, 95%, or 99% identical to the amino acid sequence set forth in SEQ ID NO:85, and the amino acid set forth in SEQ ID NO:89. It includes an LCVR having an amino acid sequence that is at least 90%, 95%, or 99% identical to the sequence.
特定の実施形態では、抗体またはその抗原結合断片は、アリロクマブ、エボロクマブ、ボコシズマブ、ロデルシズマブ、ラルパンシズマブ、およびLY3015014からなる群から選択される。特定の実施形態では、抗体またはその抗原結合断片はアリロクマブである。 In certain embodiments, the antibody or antigen-binding fragment thereof is selected from the group consisting of arilocumab, evolocumab, bococizumab, rodercizumab, ralpancizumab, and LY301504. In certain embodiments, the antibody or antigen-binding fragment thereof is arilocumab.
特定の実施形態では、本方法は、
(c)患者におけるLDL−Cレベルが閾値レベルより低い場合、約2週間ごとに1回またはそれ以上の次の投薬量の75mgの抗体もしくはその抗原結合断片を患者に投与するか、または患者におけるLDL−Cレベルが閾値レベル以上の場合、約2週間ごとに1回またはそれ以上の次の投薬量の150mgの抗体もしくはその抗原結合断片を投与すること
をさらに含む。
In certain embodiments, the method comprises
(C) if the LDL-C level in the patient is below a threshold level, then administer to the patient one or more subsequent doses of 75 mg of the antibody or antigen-binding fragment thereof about once every two weeks, or in the patient If the LDL-C level is above the threshold level, it further comprises administering about one or more subsequent doses of 150 mg of the antibody or antigen-binding fragment thereof about once every two weeks.
特定の実施形態では、本方法は、
(c)患者におけるLDL−Cレベルが閾値レベルより低い場合、約4週間ごとに1回またはそれ以上の次の投薬量の300mgの抗体もしくはその抗原結合断片を患者に投与するか、または患者におけるLDL−Cレベルが閾値レベル以上の場合、約2週間ごとに1回またはそれ以上の次の投薬量の150mgの抗体もしくはその抗原結合断片を投与すること
をさらに含む。
In certain embodiments, the method comprises
(C) if the LDL-C level in the patient is below a threshold level, the patient is administered with one or more subsequent doses of 300 mg of the antibody or antigen-binding fragment thereof about once every four weeks, or in the patient If the LDL-C level is above the threshold level, it further comprises administering about once every two weeks or more of the next dose of 150 mg of the antibody or antigen-binding fragment thereof.
特定の実施形態では、閾値レベルは70mg/dLである。 In a particular embodiment, the threshold level is 70 mg/dL.
特定の実施形態では、抗体またはその抗原結合断片は皮下投与される。 In certain embodiments, the antibody or antigen-binding fragment thereof is administered subcutaneously.
特定の実施形態では、患者は、付随する脂質修飾療法(LMT)をさらに受ける。特定の実施形態では、LMTは、スタチン、コレステロール吸収阻害剤、フィブラート、ナイアシン、オメガ−3脂肪酸、および胆汁酸封鎖剤からなる群から選択される。特定の実施形態では、LMTはスタチン療法である。特定の実施形態では、スタチンは、アトルバスタチン、ロスバスタチン、シンバスタチン、プラバスタチン、ロバスタチン、フルバスタチン、ピタバスタチン、およびセリバスタチンからなる群から選択される。特定の実施形態では、スタチン療法は、最大耐性量のスタチン療法である。特定の実施形態では、コレステロール吸収阻害剤はエゼチミブである。 In certain embodiments, the patient further receives concomitant lipid modification therapy (LMT). In certain embodiments, the LMT is selected from the group consisting of statins, cholesterol absorption inhibitors, fibrates, niacin, omega-3 fatty acids, and bile sequestrants. In certain embodiments, LMT is statin therapy. In certain embodiments, the statin is selected from the group consisting of atorvastatin, rosuvastatin, simvastatin, pravastatin, lovastatin, fluvastatin, pitavastatin, and cerivastatin. In certain embodiments, the statin therapy is a maximally resistant amount of statin therapy. In certain embodiments, the cholesterol absorption inhibitor is ezetimibe.
特定の実施形態では、患者はスタチンに対して不耐性である。 In certain embodiments, the patient is intolerant to statins.
特定の実施形態では、インスリン療法は、ヒトインスリン、インスリングラルギン、インスリングルリジン、インスリンデテミル、インスリンリスプロ、インスリンデグルデック、インスリンアスパルト、および基礎インスリンからなる群から選択される。特定の実施形態では、患者は、インスリン療法に加えて付随する抗糖尿病療法を受ける。特定の実施形態では、追加の抗糖尿病療法は、グルカゴン様ペプチド1(GLP−1)療法、胃腸ペプチド、グルカゴン受容体アゴニストまたはアンタゴニスト、グルコース依存性インスリン分泌性ポリペプチド(GIP)受容体アゴニストまたはアンタゴニスト、グレリンアンタゴニストまたは逆アゴニスト、キセニン、キセニン類似体、ビグアナイド、スルホニル尿素、メグリチニド、チアゾリジンジオン、DPP−4阻害剤、アルファ−グルコシダーゼ阻害剤、ナトリウム依存性グルコース輸送体2(SGLT−2)阻害剤、SGLT−1阻害剤、ペルオキシソーム増殖因子活性化受容体(PPAR−)(アルファ、ガンマまたはアルファ/ガンマ)アゴニストまたはモジュレータ、アミリン、アミリンアナログ、Gタンパク質共役受容体119(GPR119)アゴニスト、GPR40アゴニスト、GPR120アゴニスト、GPR142アゴニスト、全身または低吸収性TGR5アゴニスト、糖尿病免疫療法薬、メタボリックシンドロームおよび糖尿病を治療するための抗炎症薬、アデノシンモノプリン酸活性化プロテインキナーゼ(AMPK)刺激薬、11−ベータ−ヒドロキシステロイドデヒドロゲナーゼ1の阻害剤、グルコキナーゼの活性化剤、ジアシルグリセロールO−アシルトランスフェラーゼ(DGAT)の阻害剤、グルコーストランスポータ−4のモジュレータ、ソマトスタチン受容体3アゴニスト、脂質減少剤、ならびにそれらの組み合わせからなる群から選択される。
In certain embodiments, the insulin therapy is selected from the group consisting of human insulin, insulin glargine, insulin glulisine, insulin detemir, insulin lispro, insulin degludec, insulin aspart, and basal insulin. In certain embodiments, the patient receives concomitant antidiabetic therapy in addition to insulin therapy. In certain embodiments, the additional anti-diabetic therapy is glucagon-like peptide 1 (GLP-1) therapy, gastrointestinal peptide, glucagon receptor agonist or antagonist, glucose-dependent insulinotropic polypeptide (GIP) receptor agonist or antagonist. , Ghrelin antagonist or inverse agonist, xenin, xenin analog, biguanide, sulfonylurea, meglitinide, thiazolidinedione, DPP-4 inhibitor, alpha-glucosidase inhibitor, sodium-dependent glucose transporter 2 (SGLT-2) inhibitor, SGLT-1 inhibitor, peroxisome proliferator-activated receptor (PPAR-) (alpha, gamma or alpha/gamma) agonist or modulator, amylin, amylin analog, G protein coupled receptor 119 (GPR119) agonist, GPR40 agonist, GPR120 Agonists, GPR142 Agonists, Systemic or Low Absorption TGR5 Agonists, Diabetic Immunotherapeutic Agents, Anti-inflammatory Drugs for Treating Metabolic Syndrome and Diabetes, Adenosine Monopurinate Activated Protein Kinase (AMPK) Stimulators, 11-beta-Hydroxy From inhibitors of
特定の実施形態では、抗体またはその抗原結合断片は、患者のLDL−Cレベルを、例えば、少なくとも30%、35%、40%、もしくは45%減少させる。特定の実施形態では、抗体またはその抗原結合断片は、患者の非HDL−Cレベルを、例えば、少なくとも20%、25%、30%、35%減少させる。特定の実施形態では、抗体またはその抗原結合断片は、患者のApoC3レベルを減少させる。特定の実施形態では、抗体またはその抗原結合断片は、患者のリポタンパク質粒子の数および/またはサイズを減少させる。特定の実施形態では、抗体またはその抗原結合断片は:
(a)患者のヘモグロビンA1c(HbA1c)レベルに影響を及ぼさず;および/または
(b)患者の空腹時血漿グルコース(FPG)レベルに影響を及ぼさない。
In certain embodiments, the antibody or antigen-binding fragment thereof reduces LDL-C levels in a patient, eg, by at least 30%, 35%, 40%, or 45%. In certain embodiments, the antibody or antigen-binding fragment thereof reduces non-HDL-C levels in a patient, eg, by at least 20%, 25%, 30%, 35%. In certain embodiments, the antibody or antigen-binding fragment thereof reduces ApoC3 levels in the patient. In certain embodiments, the antibody or antigen-binding fragment thereof reduces the number and/or size of lipoprotein particles in a patient. In a particular embodiment, the antibody or antigen-binding fragment thereof is:
(A) does not affect the patient's hemoglobin A1c (HbA1c) levels; and/or (b) does not affect the patient's fasting plasma glucose (FPG) levels.
別の態様によれば、本方法は、T2DMおよびASCVDを有する患者における高コレステロール血症を治療する方法を含み、本方法は、
(a)(i)T2DM、(ii)ASCVD、および(iii)最大耐性量のスタチン療法によって適切に制御されない高コレステロール血症を有する、インスリン療法を受けている心血管リスクの高い患者を選択すること;
(b)患者に2週間ごとに、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片を75mg投与すること;および
(c)患者におけるLDL−Cレベルが70mg/dLより低い場合、約2週間ごとに1回またはそれ以上の次の用量の75mgの抗体もしくはその抗原結合断片を患者に投与するか、または患者におけるLDL−Cレベルが70mg/dL以上の場合、約2週間ごとに1回またはそれ以上の次の用量の150mgの抗体もしくはその抗原結合断片を投与すること
を含み、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含み、患者は付随するインスリン療法を受ける。
According to another aspect, the method comprises a method of treating hypercholesterolemia in a patient having T2DM and ASCVD, the method comprising:
Select (a) high cardiovascular risk patients undergoing insulin therapy with hypercholesterolemia not adequately controlled by (i) T2DM, (ii) ASCVD, and (iii) maximally resistant statin therapy thing;
(B) administering to the patient every two
他の実施形態は、以下の詳細な説明の検討から明らかになるであろう。 Other embodiments will be apparent from a consideration of the following detailed description.
本方法は、記載される特定の方法および実験条件に限定されない。これは、このような方法および条件は変化する可能性があるためである。また、本方法の範囲は添付の特許請求の範囲によってのみ限定されるため、本明細書で使用される用語は、特定の実施形態を説明することのみを目的としており、限定することを意図していないこともまた理解されたい。 The method is not limited to the particular method and experimental conditions described. This is because such methods and conditions may change. Also, as the scope of the method is limited only by the scope of the appended claims, the terminology used herein is for the purpose of describing particular embodiments only, and is not intended to be limiting. It should also be understood that not.
別に定義されない限り、本明細書で使用されるすべての技術用語および科学用語は、当業者によって一般的に理解されるのと同じ意味を有する。本明細書で使用するとき、用語「約」は、特定の列挙された数値に関して使用される場合、その値が列挙された値から1%以下しか変化し得ないことを意味する。例えば、本明細書で使用するとき、「約100」という表現は、99および101、ならびにその間のすべての値(例えば、99.1、99.2、99.3、99.4など)を含む。 Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art. As used herein, the term "about," when used in reference to a particular recited numerical value, means that the value may vary from the recited value by no more than 1%. For example, as used herein, the expression "about 100" includes 99 and 101 and all values in between (eg, 99.1, 99.2, 99.3, 99.4, etc.). ..
本明細書に記載されるものと類似または同等の任意の方法および材料が使用されるが、好ましい方法および材料をここに記載する。本明細書で言及されるすべての刊行物は、それらの全体を記載するために参照により本明細書に組み入れる。 Although any methods and materials similar or equivalent to those described herein can be used, the preferred methods and materials are described herein. All publications mentioned herein are incorporated herein by reference to describe their entirety.
インスリン療法中の高コレステロール血症および糖尿病を有する患者を治療する方法
インスリン療法中の高コレステロール血症の糖尿病患者を治療するための方法および組成物が提供される。特定の実施形態によれば、これらの方法は、このような患者の血清中のリポタンパク質レベル(例えば、LDL−Cおよび/またはLp(a))の減少をもたらす。
Methods of Treating Patients with Hypercholesterolemia and Diabetes During Insulin Therapy Provided are methods and compositions for treating diabetic patients with hypercholesterolemia during insulin therapy. According to certain embodiments, these methods result in reduction of lipoprotein levels (eg, LDL-C and/or Lp(a)) in the serum of such patients.
本開示はまた、インスリン療法中に高コレステロール血症を有する糖尿病患者の治療に使用するための、PCSK9阻害剤(例えば、PCSK9(例えば、ヒトPCSK9)に特異的に結合する抗体またはその抗原結合断片)、またはPCSK9阻害剤を含む組成物を提供する。特定の実施形態では、PCSK9阻害剤または組成物は、このような患者の血清中のリポタンパク質(例えば、LDL−Cおよび/またはLp(a))のレベルを減少させるのに有用である。 The present disclosure also provides an antibody or antigen-binding fragment thereof that specifically binds to a PCSK9 inhibitor (eg, PCSK9 (eg, human PCSK9)) for use in the treatment of diabetic patients having hypercholesterolemia during insulin therapy. ), or a composition comprising a PCSK9 inhibitor. In certain embodiments, PCSK9 inhibitors or compositions are useful in reducing the levels of lipoproteins (eg, LDL-C and/or Lp(a)) in the serum of such patients.
本明細書で使用するとき、用語「リポタンパク質」とは、タンパク質と脂質の両方を含む生体分子粒子を意味する。リポタンパク質の例には、例えば、低密度リポタンパク質(LDL)、超低密度リポタンパク質(VLDL)、中密度リポタンパク質(IDL)、およびリポタンパク質(a)(Lp(a))が含まれる。 As used herein, the term "lipoprotein" means a biomolecule particle that includes both proteins and lipids. Examples of lipoproteins include, for example, low density lipoprotein (LDL), very low density lipoprotein (VLDL), medium density lipoprotein (IDL), and lipoprotein (a) (Lp(a)).
しばしば単に糖尿病と呼ばれる真性糖尿病は、身体が十分なインスリンを産生しないため、または産生されるインスリンに細胞が応答しないために、ヒトが高い血糖値を有する一群の代謝疾患である。最も一般的な糖尿病のタイプは次の通りである:(1)身体がインスリンを産生できない1型糖尿病;(2)経時的なインスリン欠乏症の増加を併発する、身体がインスリンを適切に使用できない2型糖尿病;および(3)女性が妊娠により糖尿病を発症する場合の妊娠糖尿病。糖尿病のすべての形態は、典型的には、多年後に発症する長期合併症のリスクを高める。これらの長期合併症のほとんどは、血管の損傷に基づいており、より大きな血管のアテローム性動脈硬化症から生じる「大血管」疾患と、小血管の損傷から生じる「微小血管」疾患の2つのカテゴリに分割される。大血管疾患の状態の例は、虚血性心疾患、心筋梗塞、脳卒中および末梢血管疾患である。微小血管疾患の例は、糖尿病性網膜症、糖尿病性腎症、および糖尿病性神経障害である。
Diabetes mellitus, often referred to simply as diabetes, is a group of metabolic disorders in which humans have high blood glucose levels, either because the body does not produce enough insulin or because cells do not respond to the insulin produced. The most common types of diabetes are: (1)
特定の実施形態によれば、治療される患者は、1型真性糖尿病(T1DM)または2型真性糖尿病(T2DM)を有し、インスリン療法を受けている。特定の実施形態では、患者は、少なくとも1年間、T1DMまたはT2DMと診断されている。特定の実施形態では、患者は、30歳前にT1DMと診断された。特定の実施形態では、T1DM患者は、0.2pmol/mLより低いCペプチドレベルを有する。特定の実施形態では、患者は、10%より低いグリコシル化ヘモグロビン(HbA1c)レベルを有する。
According to certain embodiments, the patient to be treated has
特定の実施形態によれば、治療される患者は、脂質修飾療法(LMT)によって適切に制御されない高コレステロール血症を有する。患者の血清LDL−C濃度が、認識された医学的に許容されるレベル、例えば、LMTで少なくとも4週間後、(患者の冠状動脈心疾患の相対的リスクを考慮して)70mg/dL未満に減少しない場合、高コレステロール血症はLMTによって適切に制御されないと見なされる。特定の実施形態では、LMTは最大限に許容されるスタチン療法である。本明細書で使用するとき、「最大耐性量のスタチン療法」とは、患者に許容できない有害な副作用を引き起こすことなく、患者に投与されるスタチンの最高用量を意味する。例えば、本明細書に開示される方法は、アトルバスタチン(アトルバスタチン+エゼチミブを含む)、ロスバスタチン、セリバスタチン、ピタバスタチン、フルバスタチン、ロバスタチン、シンバスタチン(シンバスタチン+エゼチミブを含む)、プラバスタチン、およびそれらの組み合わせからなる群から選択されるスタチンの1日用量によって適切に制御されない高コレステロール血症を有するT1DMまたはT2DMを有する患者を治療することを含む。特定の実施形態では、患者はこの療法にして不耐性である場合、患者は付随するスタチン療法を受けない。スタチン不耐症の患者は、例えば、疼痛、痛み、脱力感、またはけいれん、スタチン療法中に開始または増加し、スタチン療法を中止すると停止するものなどの緊張または外傷によるもの以外の骨格筋関連の症状があり得る。 According to certain embodiments, the patient to be treated has hypercholesterolemia that is not adequately controlled by lipid modification therapy (LMT). The patient's serum LDL-C concentration falls below 70 mg/dL (taking into account the patient's relative risk of coronary heart disease) after a recognized medically acceptable level, eg, LMT, for at least 4 weeks. If not reduced, hypercholesterolemia is considered not properly controlled by LMT. In certain embodiments, LMT is a maximally tolerated statin therapy. As used herein, "maximum tolerable dose of statin therapy" means the highest dose of statin administered to a patient without causing unacceptable adverse side effects in the patient. For example, the methods disclosed herein include the group consisting of atorvastatin (including atorvastatin + ezetimibe), rosuvastatin, cerivastatin, pitavastatin, fluvastatin, lovastatin, simvastatin (including simvastatin + ezetimibe), pravastatin, and combinations thereof. Comprising treating a patient with T1DM or T2DM having hypercholesterolemia that is not adequately controlled by the daily dose of statin selected from In certain embodiments, if a patient is intolerant to this therapy, the patient will not receive concomitant statin therapy. Patients with statin intolerance include, for example, pain, pain, weakness, or convulsions, skeletal muscle-related factors other than those due to tension or trauma, such as those that start or increase during statin therapy and stop when statin therapy is stopped. There may be symptoms.
患者の選択
本発明の方法および組成物は、高コレステロール血症および糖尿病を有し、インスリン療法を受けている患者の治療に有用である。治療される患者はまた、1つまたはそれ以上の追加の選択基準を示し得る。例えば、患者の計算されたLDL−Cレベルが70mg/dL、100mg/dL、または130mg/dL以上である場合、患者は治療のために選択される。患者は、場合により、少なくとも4週間、少なくとも1つの他の脂質修飾療法(LMT)と組み合わせて、最大耐性量のスタチンで治療され、または患者がスタチン不耐性の場合、患者は、少なくとも1つの非スタチンLMTの最大耐性量で、少なくとも4週間治療されている。スタチンの最大耐性量は、例えば、地域の慣行または地方のガイドラインに基づいて処方された用量、または小児患者の地方の処方情報で指定されている高用量への悪影響により最大耐性量のとして定義できます。スタチン不耐性は、例えば、少なくとも2つのスタチンに耐えられないこととして定義される:1つのスタチンは最低の1日の開始用量であり、もう1つは任意の用量であり、骨格筋関連の症状によるものである。疼痛、痛み、脱力感、またはけいれん、スタチン療法中に開始もしくは増加し、スタチン療法が中止される場合に停止したものなどの緊張または外傷によるものを除く。スタチンの毎日のレジメンを受けていない患者(例えば、週に1〜3回)はまた、毎日の用量に耐えられないとみなされる。
Patient Selection The methods and compositions of the invention are useful in treating patients with hypercholesterolemia and diabetes who are receiving insulin therapy. The patient to be treated may also exhibit one or more additional selection criteria. For example, if a patient's calculated LDL-C level is 70 mg/dL, 100 mg/dL, or 130 mg/dL or greater, the patient is selected for treatment. The patient is treated with a maximally tolerable amount of statin, optionally in combination with at least one other lipid modification therapy (LMT) for at least 4 weeks, or if the patient is statin intolerant, the patient is treated with at least one non- The maximum tolerated dose of statin LMT has been treated for at least 4 weeks. The maximum tolerated dose of a statin can be defined as the maximum tolerated dose due to adverse effects on, for example, doses prescribed according to local practice or local guidelines, or high doses specified in local prescribing information for pediatric patients. I will. Statin intolerance is defined as, for example, inability to tolerate at least two statins: one statin is the lowest daily starting dose, the other is any dose, and skeletal muscle-related symptoms It is due to. Except for pain, pain, weakness, or convulsions, tension or trauma, such as those that started or increased during statin therapy and stopped when statin therapy was discontinued. Patients who are not on a daily regimen of statins (eg, 1-3 times a week) are also considered tolerated daily doses.
さらに、患者が心血管(CV)リスクが高い場合、患者は治療のために選択される。特定の実施形態では、高CVリスク患者は、心血管疾患(CVD)および/または少なくとも1つのう追加のCVリスク因子の文書化された病歴を有する。CVDには、限定されないが、冠状動脈性心疾患(CHD)およびCHDのリスク同等物が含まれる。CHDには、限定されないが、急性心筋梗塞(MI)、サイレントMI、不安定狭心症、冠動脈血行再建術(例えば、経皮的冠動脈インターベンション(PCI)または冠動脈バイパス術(CABG))、および臨床的に重要なCHD(例えば、侵襲性診断または冠動脈造影、トレッドミルを使用したストレステスト、ストレス心エコー検査、または核イメージングなどの非侵襲的テスト)が含まれる。CHDリスク同等物には、限定されないが、末梢動脈疾患(例えば、実施例2の試験対象患者基準に記載されているもの)、およびアテローム血栓性起源の24時間を超えて持続する局所虚血性神経学的欠損を伴う以前の虚血性脳卒中が含まれる。CVリスク因子には、限定されないが、高血圧、現在の喫煙、男性で45歳以上、女性で55歳以上、ミクロ/マクロアルブミン尿症の病歴、糖尿病性網膜症の病歴、早発性CHDの家族歴(55歳未満の父親または兄弟;65歳未満の母親または姉妹)、低HDL−C(男性<40mg/dL[1.0mmol/L]および女性<50mg/dL[1.3mmol/L])、ならびに文書化された慢性腎疾患(CKD)(例えば、実施例2の試験対象患者基準で定義されているもの)が含まれる。 Further, if a patient is at high cardiovascular (CV) risk, the patient is selected for treatment. In certain embodiments, high CV risk patients have a documented history of cardiovascular disease (CVD) and/or at least one additional CV risk factor. CVD includes, but is not limited to, coronary heart disease (CHD) and risk equivalents of CHD. CHD includes, but is not limited to, acute myocardial infarction (MI), silent MI, unstable angina, coronary revascularization (eg, percutaneous coronary intervention (PCI) or coronary artery bypass surgery (CABG)), and Includes clinically important CHD (eg, non-invasive tests such as invasive diagnostics or coronary angiography, treadmill stress testing, stress echocardiography, or nuclear imaging). CHD risk equivalents include, but are not limited to, peripheral arterial disease (eg, those described in the study patient criteria of Example 2), and focal ischemic nerves of more than 24 hours of atherothrombotic origin. Includes a previous ischemic stroke with a biological defect. CV risk factors include, but are not limited to, hypertension, current smoking, males over 45 years old, females over 55 years old, history of micro/macroalbuminuria, history of diabetic retinopathy, family of early onset CHD. History (father or siblings under 55; mother or sister under 65), low HDL-C (male <40 mg/dL [1.0 mmol/L] and female <50 mg/dL [1.3 mmol/L]) , As well as the documented chronic kidney disease (CKD) (eg, as defined in the study patient criteria of Example 2).
特定の実施形態では、高CVリスク患者は、アテローム性動脈硬化性心血管疾患(ASCVD)を有する。特定の実施形態では、ASCVDは、冠状動脈性心疾患(CHD)、虚血性脳卒中、または末梢動脈疾患として定義される。特定の実施形態では、CHDは、急性心筋梗塞、無症候性心筋梗塞、および不安定狭心症を含む。特定の実施形態では、CHDは、急性心筋梗塞、無症候性心筋梗塞、または不安定狭心症として定義される。 In certain embodiments, the high CV risk patient has atherosclerotic cardiovascular disease (ASCVD). In certain embodiments, ASCVD is defined as coronary heart disease (CHD), ischemic stroke, or peripheral arterial disease. In certain embodiments, CHD comprises acute myocardial infarction, subclinical myocardial infarction, and unstable angina. In certain embodiments, CHD is defined as acute myocardial infarction, subclinical myocardial infarction, or unstable angina.
インスリン療法
本明細書に示されるように、本発明の方法による治療のために選択される糖尿病患者は、インスリンまたはその誘導体を含むインスリン療法を受けており、継続して受けている。市場にあるインスリンは、インスリンの起源(例えば、ウシ、ブタ、ヒトのインスリン)、およびさらにはそれらの組成が異なるため、作用のプロファイル(作用の開始および作用の持続時間)に影響を与える。異なるインスリン製品を組み合わせることにより、多種多様な作用プロファイルを取得し、可能な限り生理学に近い血糖値を設定することができる。例示的なインスリン療法には、ヒトインスリンなどの天然に存在するインスリン、ならびにインスリングラルギン(Gly(A21)−Arg(B31)−Arg(B32)ヒトインスリン、例えば、Lantus(登録商標))などの、作用持続時間が延長された修飾インスリンが含まれ得る。インスリングラルギンは、酸性の透明な溶液として注入され、皮下組織の生理的pH範囲における溶液特性のため、安定した六量体アソシエートとして沈殿される。インスリングラルギンは1日1回注射され、その平坦な血清プロファイル、および夜間低血糖リスクの関連した減少により、他の長時間活性インスリンよりも注目に値する(Schubert−Zsilaveczら、2巻:125〜130頁(2001年))。インスリングラルギンは、100U/mLより高い濃度、例えば、270〜330U/mLのインスリングラルギンまたは300U/mLのインスリングラルギン(欧州特許第2387989号に開示される)で投与される。他の例示的なインスリン療法には、インスリングルリジン(例えば、Apidra(登録商標))、インスリンデテミル(例えば、Levemir(登録商標))、インスリンリスプロ(例えば、Humalog(登録商標)、Liprolog(登録商標))、インスリンデグルデック(例えば、DegludecPlus(登録商標)、IdegLira(NN9068))、インスリンアスパルトおよびアスパルト製剤(例えば、NovoLog(登録商標))、基礎インスリンおよび類似体(例えばmLY2605541、LY2963016、NN1436)、PEG化インスリンリスプロ(例えば、LY−275585)、長時間作用型インスリン(例えば、NN1436、Insumera(PE0139)、AB−101、AB−102、Sensulin LLC)、中間作用型インスリン(例えば、Humulin(登録商標)N、Novolin(登録商標)N)、速効型および短時間作用型インスリン(例えば、Humulin(登録商標)R、Novolin(登録商標)R、Linjeta(登録商標)(VIAject(登録商標))、PH20インスリン、NN1218、HinsBet(登録商標))、予混合インスリン、SuliXen(登録商標)、NN1045、インスリン+Symlin(登録商標)、PE−0139、ACP−002ハイドロゲルインスリン、経口、吸入、経皮および口腔または舌下インスリン(例えば、Exubera(登録商標)、Nasulin(登録商標)、Afrezza(登録商標)、インスリントレゴピル、TPM−02インスリン、Capsulin(登録商標)、Oral−lyn(登録商標)、Cobalamin(登録商標)経口インスリン、ORMD−0801、Oshadi経口インスリン、NN1953、NN1954、NN1956、VIAtab(登録商標))が含まれる。また、二官能性リンカーによってアルブミンまたは別のタンパク質に結合しているインスリン誘導体も適している。
Insulin Therapy As shown herein, diabetic patients selected for treatment by the methods of the invention have been and continue to receive insulin therapy comprising insulin or a derivative thereof. The insulins on the market influence the profile of action (onset of action and duration of action) due to the different origins of insulin (eg, bovine, porcine, human insulin), and even their composition. By combining different insulin products, it is possible to obtain a wide variety of action profiles and set blood glucose levels as close to physiological as possible. Exemplary insulin therapies include naturally occurring insulin, such as human insulin, as well as insulin glargine (Gly(A21)-Arg(B31)-Arg(B32) human insulin, such as Lantus®). Modified insulins with extended duration of action may be included. Insulin glargine is infused as an acid clear solution and is precipitated as a stable hexameric associate due to its solution properties in the physiological pH range of subcutaneous tissue. Insulin glargine is injected once daily and is more noteworthy than other long-acting insulins due to its flattened serum profile and associated reduction in nocturnal hypoglycemia risk (Schubert-Zsilavecz et al., 2:125-130. Page (2001)). Insulin glargine is administered at a concentration higher than 100 U/mL, eg 270-330 U/mL insulin glargine or 300 U/mL insulin glargine (disclosed in EP 2387989). Other exemplary insulin therapies include insulin glulisine (eg, Apidra®), insulin detemir (eg, Levemir®), insulin lispro (eg, Humalog®, Liprolog®). )), insulin degludec (eg, DegludecPlus®, IdegLira (NN9068)), insulin aspart and aspart formulations (eg, NovoLog®), basal insulin and analogs (eg mLY2605541, LY2963016, NN1436). ), PEGylated insulin lispro (e.g. LY-275585), long-acting insulin (e.g. NN1436, Insumera (PE0139), AB-101, AB-102, Sensulin LLC), intermediate-acting insulin (e.g. Humulin (e.g. (Registered trademark) N, Novolin (registered trademark) N), fast-acting and short-acting insulin (eg, Humulin (registered trademark) R, Novolin (registered trademark) R, Linjeta (registered trademark) (VIAject (registered trademark)) , PH20 insulin, NN1218, HinsBet®), premixed insulin, SuliXen®, NN1045, insulin+Symlin®, PE-0139, ACP-002 hydrogel insulin, oral, inhalation, transdermal and Oral or sublingual insulin (eg, Exubera®, Nasulin®, Afrezza®, insulin tregopyr, TPM-02 insulin, Capsulin®, Oral-lyn®, Cobalamin® Oral Insulin, ORMD-0801, Oshadi Oral Insulin, NN1953, NN1954, NN1956, VIAtab®). Also suitable are insulin derivatives linked to albumin or another protein by a bifunctional linker.
PCSK9阻害剤
本方法は、PCSK9阻害剤を含む治療組成物を患者に投与することを含む。本明細書で使用するとき、「PCSK9阻害剤」は、ヒトPCSK9に結合または相互作用し、インビトロまたはインビボでPCSK9の正常な生物学的機能を阻害する任意の薬剤である。PCSK9阻害剤のカテゴリの非限定的な例には、小分子PCSK9アンタゴニスト、PCSK9発現または活性の核酸ベースの阻害剤(例えば、siRNAまたはアンチセンス)、PCSK9と特異的に相互作用するペプチドベースの分子(例えば、ペプチボディ)、PCSK9と特異的に相互作用する受容体分子、LDL受容体のリガンド結合部分を含むタンパク質、PCSK9結合足場分子(例、DARPins、HEATリピートタンパク質、ARMリピートタンパク質、テトラトリコペプチドリピートタンパク質、フィブロネクチンベースの足場構築物、および天然に存在するリピートタンパク質に基づく他の足場など[例えば、BoersmaおよびPluckthun、2011年、Curr.Opin.Biotechnol.22巻:849〜857頁、およびそこで引用された文献を参照])、および抗PCSK9アプタマーまたはその一部が含まれる。特定の実施形態によれば、本方法との関連で使用されるPCSK9阻害剤は、ヒトPCSK9に特異的に結合する抗PCSK9抗体または抗体の抗原結合断片である。
PCSK9 Inhibitor The method comprises administering to the patient a therapeutic composition comprising a PCSK9 inhibitor. As used herein, a "PCSK9 inhibitor" is any agent that binds or interacts with human PCSK9 and inhibits PCSK9's normal biological function in vitro or in vivo. Non-limiting examples of categories of PCSK9 inhibitors include small molecule PCSK9 antagonists, nucleic acid-based inhibitors of PCSK9 expression or activity (eg, siRNA or antisense), peptide-based molecules that specifically interact with PCSK9. (Eg, peptibodies), receptor molecules that specifically interact with PCSK9, proteins that include the ligand-binding portion of the LDL receptor, PCSK9-binding scaffold molecules (eg, DARPins, HEAT repeat proteins, ARM repeat proteins, tetratricopeptide repeats) Proteins, fibronectin-based scaffold constructs, and other scaffolds based on naturally occurring repeat proteins [eg Boersma and Pluckthun, 2011, Curr. Opin. Biotechnol. 22:849-857, and cited therein. Literature]), and anti-PCSK9 aptamers or parts thereof. According to a particular embodiment, the PCSK9 inhibitor used in the context of the method is an anti-PCSK9 antibody or an antigen-binding fragment of an antibody that specifically binds human PCSK9.
「ヒトプロタンパク質転換酵素サブチリシン/ケキシン9型」または「ヒトPCSK9」または「hPCSK9」という用語は、本明細書で使用するとき、配列番号197に示される核酸配列および配列番号198のアミノ酸配列を有するPCSK9、またはその生物学的に活性な断片を指す。 The term "human proprotein convertase subtilisin/kexin type 9" or "human PCSK9" or "hPCSK9", as used herein, has the nucleic acid sequence set forth in SEQ ID NO:197 and the amino acid sequence of SEQ ID NO:198. Refers to PCSK9, or a biologically active fragment thereof.
用語「抗体」とは、本明細書で使用するとき、ジスルフィド結合によって相互接続された4つのポリペプチド鎖、2つの重(H)鎖および2つの軽(L)鎖、ならびにその多量体(例、IgM)を含む免疫グロブリン分子を指すことが意図される。各重鎖は、重鎖可変領域(本明細書においてはHCVRまたはVHと略記される)および重鎖定常領域を含む。重鎖定常領域は、3つのドメイン、CH1、CH2およびCH3を含む。各軽鎖は、軽鎖可変領域(本明細書においてはLCVRまたはVLと略記される)および軽鎖定常領域を含む。軽鎖定常領域は、1つのドメイン(CL1)を含む。VHおよびVL領域は、フレームワーク領域(FR)と呼ばれるより保存された領域が散在する、相補性決定領域(CDR)と呼ばれる超可変性の領域にさらに細分化される。各VHおよびVLは、3つのCDRと4つのFRで構成され、アミノ末端からカルボキシ末端に向かって、次の順序:FR1、CDR1、FR2、CDR2、FR3、CDR3、FR4で配置される。異なる実施形態では、抗PCSK9抗体(またはその抗原結合部分)のFRは、ヒト生殖系列配列と同一であり、または天然もしくは人工的に改変される。アミノ酸コンセンサス配列は、2つ以上のCDRの並列分析に基づいて定義される。
The term "antibody," as used herein, includes four polypeptide chains, two heavy (H) chains and two light (L) chains interconnected by disulfide bonds, and multimers thereof (eg, , IgM). Each heavy chain comprises a heavy chain variable region (herein abbreviated as HCVR or V H) and a heavy chain constant region. The heavy chain constant region comprises three domains, a
用語「抗体」とは、本明細書で使用するとき、完全な抗体分子の抗原結合断片も含む。抗体の「抗原結合部分」、抗体の「抗原結合断片」等という用語は、本明細書で使用するとき、抗原に特異的に結合して複合体を形成する、任意の天然に存在する、酵素的に得られる、合成の、または遺伝子的に操作されたポリペプチドまたは糖タンパク質を含む。抗体の抗原結合断片は、例えば、タンパク質消化、または抗体可変ドメインおよび場合により定常ドメインをコードするDNAの操作および発現を伴う組換え遺伝子操作技法などの任意の適した標準技法を使用して完全抗体分子から誘導される。このようなDNAは公知であり、および/または、例えば、商業的供給源、DNAライブラリ(例えば、ファージ抗体ライブラリを含む)から容易に入手可能であるか、もしくは合成される。DNAを配列決定し、化学的にまたは分子生物学技術を使用することによって操作して、例えば、1つまたはそれ以上の可変ドメインおよび/もしくは定常ドメインを適切な構成に配置するか、またはコドンを導入する、システイン残基を作製する、アミノ酸を修飾、付加もしくは欠失させる等が可能である。 The term "antibody" as used herein also includes antigen binding fragments of whole antibody molecules. The term "antigen-binding portion" of an antibody, "antigen-binding fragment" of an antibody, etc., as used herein, refers to any naturally occurring enzyme that specifically binds an antigen to form a complex. Incorporated, synthetic, or genetically engineered polypeptides or glycoproteins. Antigen-binding fragments of antibodies may be prepared as a whole antibody using any suitable standard technique such as, for example, protein digestion, or recombinant genetic engineering techniques involving manipulation and expression of DNA encoding antibody variable domains and optionally constant domains. Derived from the molecule. Such DNAs are known and/or are readily available, eg, from commercial sources, DNA libraries (including, eg, phage antibody libraries), or synthesized. The DNA is sequenced and engineered chemically or by using molecular biology techniques to place, for example, one or more variable and/or constant domains in a suitable configuration, or codons. It is possible to introduce, create cysteine residues, modify, add or delete amino acids, etc.
「抗原結合断片」の非限定的な例としては、(i)Fab断片;(ii)F(ab’)2断片;(iii)Fd断片;(iv)Fv断片;(v)一本鎖Fv(scFv)分子;(vi)dAb断片;および(vii)抗体の超可変領域を模倣するアミノ酸残基(例えば、CDR3ペプチドなどの単離された相補性決定領域(CDR))、または制約させたFR3−CDR3−FR4ペプチドからなる最小認識単位が挙げられる。ドメイン特異的抗体、単一ドメイン抗体、ドメイン欠失した抗体、キメラ抗体、CDR移植抗体、ダイアボディ、トリアボディ、テトラボディ、ミニボディ、ナノボディ(例えば、一価ナノボディ、二価ナノボディ等)、小モジュラー免疫薬(SMIP)、およびサメ可変IgNARドメインなどの他の操作された分子もまた、本明細書で使用するとき、「抗原結合断片」という表現に包含される。 Non-limiting examples of "antigen-binding fragment" include (i) Fab fragment; (ii) F(ab')2 fragment; (iii) Fd fragment; (iv) Fv fragment; (v) single chain Fv. (ScFv) molecule; (vi) dAb fragment; and (vii) amino acid residues that mimic the hypervariable region of the antibody (eg, isolated complementarity determining regions (CDRs) such as CDR3 peptides), or constrained A minimal recognition unit consisting of the FR3-CDR3-FR4 peptide can be mentioned. Domain-specific antibody, single domain antibody, domain-deleted antibody, chimeric antibody, CDR-grafted antibody, diabodies, triabodies, tetrabodies, minibodies, Nanobodies (eg monovalent Nanobodies, divalent Nanobodies, etc.), small Modular immunodrugs (SMIPs) and other engineered molecules such as shark variable IgNAR domains are also encompassed by the expression "antigen-binding fragment" as used herein.
抗体の抗原結合断片は、典型的には、少なくとも1つの可変ドメインを含む。可変ドメインは、任意のサイズまたはアミノ酸組成であり、一般的に、1つまたはそれ以上のフレームワーク配列に隣接するか、またはそれを有するフレーム内に少なくとも1つのCDRを含む。VLと関連付けられたVHドメインを有する抗原結合断片において、VHおよびVLドメインは、任意の適切な配置において互いに相対的に位置される。例えば、可変領域は、二量体であってよく、VH−VH、VH−VL、またはVL−VL二量体を含有し得る。あるいは、抗体の抗原結合断片は、モノマーVHまたはVLドメインを含有し得る。 Antigen-binding fragments of antibodies typically include at least one variable domain. Variable domains are of any size or amino acid composition and generally include at least one CDR in frame with, or adjacent to, one or more framework sequences. In an antigen binding fragment that has a V H domain associated with a V L , the V H and V L domains are located relative to each other in any suitable arrangement. For example, the variable region may be dimeric and may contain VH - VH , VH - VL , or VL - VL dimers. Alternatively, the antigen-binding fragment of an antibody may contain monomeric VH or VL domains.
特定の実施形態では、抗体の抗原結合断片は、少なくとも1つの定常ドメインに共有結合された少なくとも1つの可変ドメインを含み得る。抗体の抗原結合断片内で見出される可変ドメインおよび定常ドメインの非限定的な例示的構成としては、(i)VH−CH1;(ii)VH−CH2;(iii)VH−CH3;(iv)VH−CH1−CH2;(v)VH−CH1−CH2−CH3;(vi)VH−CH2−CH3;(vii)VH−CL;(viii)VL−CH1;(ix)VL−CH2;(x)VL−CH3;(xi)VL−CH1−CH2;(xii)VL−CH1−CH2−CH3;(xiii)VL−CH2−CH3;および(xiv)VL−CLが挙げられる。上に列挙される例示的な構成のうちのいずれかを含む可変ドメインおよび定常ドメインのいずれの構成において、可変ドメインおよび定常ドメインは、互いに直接連結されか、または完全もしくは部分的ヒンジもしくはリンカー領域によって連結される。ヒンジ領域は、少なくとも2つ(例えば、5、10、15、20、40、60以上)のアミノ酸からなり得、これが単一ポリペプチド分子中の隣接する可変ドメインおよび/または定常ドメイン間の可撓性または半可撓性連結をもたらす。さらに、本発明の抗体の抗原結合断片は、互いにおよび/または1つもしくはそれ以上のモノマーVHもしくはVLドメインとの(例えば、ジスルフィド結合による)非共有会合で上に列挙される可変ドメイン構成および定常ドメイン構成のいずれかのホモ二量体またはヘテロ二量体(または他の多量体)を含み得る。
In certain embodiments, antigen-binding fragments of antibodies may comprise at least one variable domain covalently linked to at least one constant domain. Non-limiting exemplary configuration of the variable and constant domains are found within the antigen-binding fragment of an antibody, (i) V H -C H 1; (ii) V H -
完全抗体分子と同様に、抗原結合断片は、単一特異性または多重特異性(例えば、二重特異性)である。抗体の多重特異性抗原結合断片は、典型的には、少なくとも2つの異なる可変ドメインを含み、各可変ドメインは、別個の抗原または同じ抗原上の異なるエピトープに特異的に結合することができる。本明細書に開示される例示的な二重特異性抗体形式を含む任意の多重特異性抗体形式は、当該技術分野において利用可能なルーチン技法を使用して、本方法の抗体の抗原結合断片との関連での使用のために適合される。 Like whole antibody molecules, antigen-binding fragments are monospecific or multispecific (eg, bispecific). Multispecific antigen-binding fragments of antibodies typically contain at least two different variable domains, each variable domain capable of specifically binding to a distinct antigen or to different epitopes on the same antigen. Any multispecific antibody format, including the exemplary bispecific antibody formats disclosed herein, can be labeled with an antigen-binding fragment of an antibody of this method using routine techniques available in the art. Adapted for use in the context of.
抗体の定常領域は、補体を固定し、細胞依存性の細胞毒性を媒介する抗体の能力において重要である。したがって、抗体のアイソタイプは、抗体が細胞毒性を媒介することが望ましいかどうかに基づいて選択される。 The constant region of an antibody is important in the ability of the antibody to fix complement and mediate cell-dependent cytotoxicity. Thus, antibody isotypes are selected based on whether it is desirable for the antibody to mediate cytotoxicity.
用語「ヒト抗体」とは、本明細書で使用するとき、ヒト生殖細胞系免疫グロブリン配列に由来する可変領域および定常領域を有する抗体を含むことが意図される。ヒト抗体は、それにもかかわらず、例えば、CDRにおいて、および特定のCDR3において、ヒト生殖細胞系免疫グロブリン配列によってコードされていないアミノ酸残基(例えば、インビトロでのランダムもしくは部位特異的突然変異誘発によるか、またはインビボでの体細胞突然変異によって誘導された突然変異)を含み得る。しかしながら、用語「ヒト抗体」とは、本明細書で使用するとき、マウスなどの別の哺乳類動物の生殖細胞系に由来するCDR配列が、ヒトフレームワーク配列に移植されている抗体を含むことを意図しない。 The term "human antibody", as used herein, is intended to include antibodies having variable and constant regions derived from human germline immunoglobulin sequences. Human antibodies nevertheless have amino acid residues not encoded by human germline immunoglobulin sequences, eg, in CDRs, and in certain CDR3s (eg, by random or site-directed mutagenesis in vitro). Or mutations induced by somatic mutations in vivo). However, the term "human antibody" as used herein includes antibodies in which CDR sequences derived from the germ line of another mammal, such as a mouse, have been grafted into human framework sequences. Not intended.
用語「組換えヒト抗体」は、本明細書で使用するとき、宿主細胞に形質転換された組換え発現ベクターを使用して発現された抗体などの、組換え手段によって調製、発現、作製、または単離されたすべてのヒト抗体(以下にさらに記載される)、組換え、複合ヒト抗体ライブラリから単離された抗体(下でさらに記載される)、ヒト免疫グロブリン遺伝子にトランスジェニックである動物(例えば、マウス)から単離された抗体(例えば、Taylorら、(1992年)Nucl.Acids Res.20巻:6287〜6295頁を参照されたい)、またはヒト免疫グロブリン遺伝子配列の、他のDNA配列へのスプライシングを伴う任意の他の手段によって調製、発現、作製、または単離された抗体を含むことが意図される。このような組換えヒト抗体は、ヒト生殖細胞系免疫グロブリン配列に由来する可変領域および定常領域を有する。しかしながら、特定の実施形態では、このような組換えヒト抗体は、インビトロ突然変異誘発(または、ヒトIg配列にトランスジェニックな動物が使用される場合は、インビボでの体細胞突然変異誘発)に供され、したがって、組換え抗体のVHおよびVL領域のアミノ酸配列は、ヒト生殖細胞系VHおよびVL配列に由来する一方で、インビボではヒト抗体生殖細胞系レパートリー内に天然に存在しない配列であり得る。 The term "recombinant human antibody" as used herein is prepared, expressed, produced, or produced by recombinant means, such as an antibody expressed using a recombinant expression vector transformed into a host cell. All isolated human antibodies (further described below), antibodies isolated from recombinant, complex human antibody libraries (further described below), animals transgenic for human immunoglobulin genes ( For example, antibodies isolated from mice (see, eg, Taylor et al., (1992) Nucl. Acids Res. 20:6287-6295), or other DNA sequences of human immunoglobulin gene sequences. It is intended to include antibodies prepared, expressed, produced or isolated by any other means that involves splicing into. Such recombinant human antibodies have variable and constant regions derived from human germline immunoglobulin sequences. However, in certain embodiments, such recombinant human antibodies are subjected to in vitro mutagenesis (or in vivo somatic mutagenesis if animals transgenic for human Ig sequences are used). Thus, the amino acid sequences of the V H and V L regions of the recombinant antibody are derived from human germline V H and V L sequences while in vivo sequences that do not naturally occur within the human antibody germline repertoire. Can be
ヒト抗体は、ヒンジ不均一性に関連する2つの形態で存在することができる。一形態において、免疫グロブリン分子は、二量体が鎖間重鎖ジスルフィド結合によって一緒に保持されている約150〜160kDaの安定な4本鎖構築物を含む。第2の形態では、二量体は鎖間ジスルフィド結合を介して連結されておらず、共有結合した軽鎖と重鎖(半抗体)から構成される約75〜80kDaの分子が形成される。これらの形態は、アフィニティ精製後でさえも分離するのが非常に困難であった。 Human antibodies can exist in two forms associated with hinge heterogeneity. In one form, the immunoglobulin molecule comprises a stable four chain construct of about 150-160 kDa in which the dimers are held together by interchain heavy chain disulfide bonds. In the second form, the dimers are not linked via interchain disulfide bonds, forming a molecule of approximately 75-80 kDa composed of covalently bound light and heavy chains (half antibodies). These forms were very difficult to separate even after affinity purification.
様々な無傷のIgGアイソタイプにおける第2の形態の出現頻度は、限定されないが、抗体のヒンジ領域アイソタイプに関連する構造の違いによる。ヒトIgG4ヒンジのヒンジ領域の単一アミノ酸置換は、ヒトIgG1ヒンジを使用して通常観察されるレベルまで、第2の形態(Angalら、(1993年)Molecular Immunology 30巻:105頁)の出現を有意に減らすことができる。本方法は、所望の抗体形態の収率を改善するために、例えば、生産において望ましいヒンジ、CH2またはCH3領域に1つまたはそれ以上の突然変異を有する抗体を包含する。 The frequency of occurrence of the second form in various intact IgG isotypes is not limited, but is due to structural differences associated with the hinge region isotype of the antibody. Single amino acid substitutions in the hinge region of the human IgG4 hinge lead to the appearance of the second form (Angal et al., (1993) Molecular Immunology 30:105) to the level normally observed using the human IgG1 hinge. Can be significantly reduced. The method includes, in order to improve the yield of the desired antibody form, for example, preferably a hinge in the production, an antibody having one or more mutations in the C H 2 or C H 3 region.
「単離抗体」とは、本明細書で使用するとき、その天然環境の少なくとも1つの成分から同定および分離され、ならびに/または回収された抗体を意味する。例えば、生物の少なくとも1つの成分から、または抗体が天然に存在するか、もしくは天然に産生される組織または細胞から分離または除去された抗体は、本方法の目的で「単離された抗体」である。単離された抗体はまた、組換え細胞内のインサイチュの抗体も含む。単離された抗体は、少なくとも1つの精製または単離工程に供された抗体である。特定の実施形態によれば、単離された抗体は、他の細胞材料および/または化学物質を実質的に含まない。 "Isolated antibody", as used herein, means an antibody that has been identified and separated from and/or recovered from at least one component of its natural environment. For example, an antibody that has been separated or removed from at least one component of an organism or from a tissue or cell in which the antibody naturally occurs or is naturally produced is an "isolated antibody" for the purposes of this method. is there. Isolated antibody also includes the antibody in situ within recombinant cells. An isolated antibody is one that has been subjected to at least one purification or isolation step. According to certain embodiments, the isolated antibody is substantially free of other cellular material and/or chemicals.
「特異的に結合する」等の用語は、抗体またはその抗原結合断片が、生理的条件下で比較的安定している抗原と複合体を形成することを意味する。抗体が抗原に特異的に結合するかどうかを決定するための方法は、当該技術分野において周知であり、例えば、平衡透析、表面プラズモン共鳴等が挙げられる。例えば、本方法との関連で使用される場合、PCSK9に「特異的に結合する」抗体は、表面プラズモン共鳴アッセイにおいて測定した場合、約1000nM未満、約500nM未満、約300nM未満、約200nM未満、約100nM未満、約90nM未満、約80nM未満、約70nM未満、約60nM未満、約50nM未満、約40nM未満、約30nM未満、約20nM未満、約10nM未満、約5nM未満、約4nM未満、約3nM未満、約2nM未満、約1nM未満、もしくは約0.5nM未満のKDを有するPCSK9またはその一部分に結合する抗体を含む。しかしながら、ヒトPCSK9に特異的に結合する単離された抗体は、他(非ヒト)種由来のPCSK9分子などの他の抗原に対する交差反応性を有する。 By "specifically binds" and the like is meant that the antibody or antigen binding fragment thereof forms a complex with an antigen that is relatively stable under physiological conditions. Methods for determining whether an antibody specifically binds to an antigen are well known in the art and include, for example, equilibrium dialysis, surface plasmon resonance and the like. For example, an antibody that "specifically binds" to PCSK9 when used in the context of the present method, is less than about 1000 nM, less than about 500 nM, less than about 300 nM, less than about 200 nM, as measured in a surface plasmon resonance assay. <100 nM, <90 nM, <80 nM, <70 nM, <60 nM, <50 nM, <40 nM, <30 nM, <20 nM, <10 nM, <5 nM, <4 nM, <3 nM. comprising less than about 2 nM, less than about 1 nM, or antibodies that bind to PCSK9 or a portion thereof having a K D of less than about 0.5 nM. However, isolated antibodies that specifically bind to human PCSK9 have cross-reactivity to other antigens, such as PCSK9 molecules from other (non-human) species.
本方法に有用な抗PCSK9抗体は、抗体が由来した対応する生殖系列配列と比較して、重鎖および軽鎖可変ドメインのフレームワークならびに/またはCDR領域において1つまたはそれ以上のアミノ酸置換、挿入および/もしくは欠失を含み得る。このような突然変異は、本明細書に開示されるアミノ酸配列を、例えば、公開の抗体配列データベースから入手可能な生殖系列配列と比較することにより、容易に確認することができる。本方法は、本明細書に開示されるアミノ酸配列のいずれかに由来する抗体およびその抗原結合断片の使用を含み、1つまたはそれ以上のフレームワークおよび/もしくはCDR領域内の1つまたはそれ以上のアミノ酸が、抗体が由来する生殖系列配列の対応する残基に対して、または別のヒト生殖系列配列の対応する残基に対して、もしくは対応する生殖系列残基の保存的アミノ酸置換に対して突然変異される(このような配列変化は、本明細書において総称して「生殖細胞系列突然変異」と称される)。当業者は、本明細書に開示される重鎖および軽鎖可変領域配列から開始して、1つまたはそれ以上の個々の生殖系列突然変異またはそれらの組み合わせを含む多数の抗体および抗原結合断片を容易に産生することができる。特定の実施形態では、VHおよび/またはVLドメイン内のフレームワークならびに/またはCDR残基のすべては、抗体が由来する元の生殖系列配列に見られる残基に突然変異して戻される。他の実施形態では、特定の残基のみが、元の生殖系列配列に突然変異して戻され、例えば、FR1の最初の8アミノ酸内またはFR4の最後の8アミノ酸内に見られる突然変異残基のみ、またはCDR1、CDR2もしくはCDR3内に見られる変異残基のみである。他の実施形態では、1つまたはそれ以上のフレームワークおよび/またはCDR残基は、異なる生殖系列配列(すなわち、抗体が元々由来した生殖系列配列とは異なる生殖系列配列)の対応する残基に突然変異される。さらに、抗体は、フレームワークおよび/またはCDR領域内に2つ以上の生殖細胞系列変異の任意の組み合わせを含み得、例えば、特定の個々の残基が特定の生殖細胞系列配列の対応する残基に突然変異し、一方、元の生殖細胞系列とは異なる特定の他の残基が維持されるか、または異なる生殖系列配列の対応する残基に突然変異する。一度取得されると、1つまたはそれ以上の生殖細胞系突然変異を含む抗体および抗原結合断片は、結合特異性の改善、結合親和性の増加、拮抗性もしくは作動性の生物学的特性の改善または増大(場合に応じて)、免疫原性の減少などの1つまたはそれ以上の望ましい特性について簡単に試験することができる。この一般的な方法で得られた抗体および抗原結合断片の使用は、本方法に包含される。 Anti-PCSK9 antibodies useful in the present methods have one or more amino acid substitutions, insertions in the framework and/or CDR regions of the heavy and light chain variable domains as compared to the corresponding germline sequence from which the antibody was derived. And/or may include deletions. Such mutations can be readily identified by comparing the amino acid sequences disclosed herein with, for example, germline sequences available from public antibody sequence databases. The method comprises the use of antibodies and antigen-binding fragments thereof derived from any of the amino acid sequences disclosed herein and one or more within one or more framework and/or CDR regions. To the corresponding residue in the germline sequence from which the antibody is derived, or to the corresponding residue in another human germline sequence, or to a conservative amino acid substitution in the corresponding germline residue. (Such sequence changes are collectively referred to herein as "germline mutations"). One of skill in the art will begin with the heavy and light chain variable region sequences disclosed herein and will develop a large number of antibodies and antigen-binding fragments that include one or more individual germline mutations or combinations thereof. It can be easily produced. In certain embodiments, all of the framework and/or CDR residues within the VH and/or VL domain are mutated back to residues found in the original germline sequence from which the antibody was derived. In other embodiments, only certain residues are mutated back into the original germline sequence, eg, mutated residues found within the first 8 amino acids of FR1 or the last 8 amino acids of FR4. Only, or only the variant residues found within CDR1, CDR2 or CDR3. In other embodiments, one or more framework and/or CDR residues are at corresponding residues in a different germline sequence (ie, a germline sequence that differs from the germline sequence from which the antibody was originally derived). To be mutated. Further, the antibody may comprise any combination of two or more germline mutations within the framework and/or CDR regions, eg, where a particular individual residue corresponds to a particular germline sequence. , While maintaining certain other residues that differ from the original germline, or mutating to corresponding residues in different germline sequences. Once obtained, antibodies and antigen-binding fragments containing one or more germline mutations have improved binding specificity, increased binding affinity, improved antagonistic or agonistic biological properties. Alternatively, one can simply test for one or more desirable properties such as increase (as the case may be), decreased immunogenicity, and the like. The use of antibodies and antigen-binding fragments obtained by this general method is included in the method.
本方法は、1つまたはそれ以上の保存的置換を有する、本明細書に開示されるHCVR、LCVR、および/またはCDRアミノ酸配列のいずれかの変異体を含む抗PCSK9抗体の使用を含む。例えば、本方法は、本明細書に開示されるHCVR、LCVR、および/またはCDRアミノ酸配列のいずれかに対して、例えば、10以下、8以下、6以下、4以下などの保存的アミノ酸置換を有する、HCVR、LCVR、および/またはCDRアミノ酸配列を有する抗PCSK9抗体の使用を含む。 The method involves the use of an anti-PCSK9 antibody comprising a variant of any of the HCVR, LCVR, and/or CDR amino acid sequences disclosed herein with one or more conservative substitutions. For example, the method comprises conservative amino acid substitutions, eg, 10 or less, 8 or less, 6 or less, 4 or less, with any of the HCVR, LCVR, and/or CDR amino acid sequences disclosed herein. Comprising the use of an anti-PCSK9 antibody having an HCVR, LCVR, and/or CDR amino acid sequence.
用語「表面プラズモン共鳴」とは、本明細書で使用するとき、例えば、BIAcore(商標)システム(Biacore Life Sciences division of GE Healthcare、Piscataway、NJ)を使用して、バイオセンサーマトリックス内のタンパク質濃度の変化を検出することによって、リアルタイム相互作用の分析を可能にする光学現象を指す。 The term “surface plasmon resonance” as used herein, for example, uses the BIAcore™ system (Biacore Life Sciences division of GE Healthcare, Piscataway, NJ) to determine the concentration of proteins within a biosensor matrix. By detecting changes, it refers to an optical phenomenon that allows analysis of real-time interactions.
用語「KD」とは、本明細書で使用するとき、特定の抗体−抗原相互作用の平衡解離定数を指すことが意図される。 The term “K D ”, as used herein, is intended to refer to the equilibrium dissociation constant of a particular antibody-antigen interaction.
用語「エピトープ」とは、パラトープとして公知である抗体分子の可変領域における特定の抗原結合部位と相互作用する抗原決定基を指す。単一の抗原が1を超えるエピトープを有し得る。したがって、異なる抗体は抗原上の異なる領域に結合し得、異なる生物学的効果を有する場合がある。エピトープは、立体的または線状であり得る。立体構造エピトープは、線状ポリペプチド鎖の異なるセグメントからの空間的に並置されたアミノ酸によって生成される。線状エピトープは、ポリペプチド鎖の隣接するアミノ酸残基によって生成されるエピトープである。特定の状況において、エピトープは、抗原上の糖、ホスホリル基、またはスルホニル基の部分を含み得る。 The term "epitope" refers to an antigenic determinant that interacts with a particular antigen binding site in the variable region of an antibody molecule known as the paratope. A single antigen may have more than one epitope. Thus, different antibodies may bind to different regions on the antigen and may have different biological effects. Epitopes can be conformational or linear. Conformational epitopes are produced by spatially juxtaposed amino acids from different segments of a linear polypeptide chain. A linear epitope is an epitope produced by adjacent amino acid residues of a polypeptide chain. In certain circumstances, an epitope can include moieties of sugar, phosphoryl, or sulfonyl groups on the antigen.
特定の実施形態によれば、本方法において使用される抗PCSK9抗体は、pH依存性の結合特性を有する抗体である。本明細書で使用するとき、「pH依存性結合」という表現は、抗体またはその抗原結合断片が「中性pHと比較して酸性pHでPCSK9への結合の減少」を示すことを意味する(本開示の目的のために、両方の表現が互換的に使用される)。例えば、「pH依存性結合特性を有する抗体」には、酸性pHよりも中性pHで高い親和性でPCSK9に結合する抗体およびその抗原結合断片が含まれる。特定の実施形態では、抗体および抗原結合断片は、産生pHより中性pHで少なくとも3、5、10、15、20、25、30、35、40、45、50、55、60、65、70、75、80、85、90、95、100倍、またはそれ以上の親和性でPCSK9に結合する。 According to a particular embodiment, the anti-PCSK9 antibody used in the method is an antibody with pH-dependent binding properties. As used herein, the phrase "pH-dependent binding" means that the antibody or antigen-binding fragment thereof exhibits "reduced binding to PCSK9 at acidic pH compared to neutral pH" ( For the purposes of this disclosure, both expressions are used interchangeably). For example, "antibodies with pH-dependent binding properties" include antibodies and antigen-binding fragments thereof that bind PCSK9 with higher affinity at neutral pH than at acidic pH. In certain embodiments, the antibody and antigen-binding fragment are at least 3, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70 at a neutral pH above the production pH. , 75, 80, 85, 90, 95, 100, or 100-fold or more affinity binding to PCSK9.
この態様によれば、pH依存性結合特性を有する抗PCSK9抗体は、親の抗PCSK9抗体と比較して1つまたはそれ以上のアミノ酸変異を保有し得る。例えば、pH依存性結合特性を有する抗PCSK9抗体は、例えば、親抗PCSK9抗体の1つまたはそれ以上のCDRに1つまたはそれ以上のヒスチジン置換または挿入を含み得る。したがって、特定の実施形態によれば、親抗体の1つまたはそれ以上のCDRの1つまたはそれ以上のアミノ酸に対するヒスチジン残基の置換を除いて、親抗PCSK9抗体のCDRアミノ酸配列と同一であるCDRアミノ酸配列(例えば、重鎖および軽鎖CDR)を含む抗PCSK9抗体を投与することを含む方法が提供される。pH依存性結合を有する抗PCSK9抗体は、例えば、親抗体の単一のCDR、または親抗PCSK9抗体の複数の(例えば、2、3、4、5、または6個の)CDR全体に分布して、1、2、3、4、5、6、7、8、9個、またはそれ以上のヒスチジン置換を保有し得る。例えば、本方法は、親抗PCSK9抗体のHCDR1の1つもしくはそれ以上のヒスチジン置換、HCDR2の1つもしくはそれ以上のヒスチジン置換、HCDR3の1つもしくはそれ以上のヒスチジン置換、LCDR1の1つもしくはそれ以上のヒスチジン置換、LCDR2の1つもしくはそれ以上のヒスチジン置換、および/またはLCDR3の1つもしくはそれ以上のヒスチジン置換を含むpH依存性結合を有する抗PCSK9抗体の使用を含む。 According to this aspect, the anti-PCSK9 antibody with pH-dependent binding properties may carry one or more amino acid mutations as compared to the parent anti-PCSK9 antibody. For example, an anti-PCSK9 antibody with pH-dependent binding properties can include, for example, one or more histidine substitutions or insertions in one or more CDRs of the parent anti-PCSK9 antibody. Thus, according to a particular embodiment, it is identical to the CDR amino acid sequence of the parent anti-PCSK9 antibody, except for the substitution of a histidine residue for one or more amino acids of one or more CDRs of the parent antibody. Methods are provided that include administering an anti-PCSK9 antibody that comprises CDR amino acid sequences (eg, heavy and light chain CDRs). Anti-PCSK9 antibodies with pH-dependent binding are distributed, for example, in a single CDR of the parent antibody or in multiple CDRs (eg, 2, 3, 4, 5, or 6) of the parent anti-PCSK9 antibody. , May carry 1, 2, 3, 4, 5, 6, 7, 8, 9 or more histidine substitutions. For example, the method comprises one or more histidine substitutions of HCDR1 of the parent anti-PCSK9 antibody, one or more histidine substitutions of HCDR2, one or more histidine substitutions of HCDR3, one or more of LCDR1. Includes the use of anti-PCSK9 antibodies with pH-dependent binding, including histidine substitutions above, one or more histidine substitutions in LCDR2, and/or one or more histidine substitutions in LCDR3.
本明細書で使用するとき、「酸性pH」という表現は、6.0以下(例えば、約6.0未満、約5.5未満、約5.0未満など)のpHを意味する。「酸性pH」という表現には、約6.0、5.95、5.90、5.85、5.8、5.75、5.7、5.65、5.6、5.55、5.5、5.45、5.4、5.35、5.3、5.25、5.2、5.15、5.1、5.05、5.0、またはそれ以下のpH値が含まれる。本明細書で使用するとき、「中性pH」という表現は、約7.0から約7.4のpHを意味する。「中性pH」という表現には、約7.0、7.05、7.1、7.15、7.2、7.25、7.3、7.35、および7.4のpH値が含まれる。 As used herein, the expression "acidic pH" means a pH of 6.0 or less (eg, less than about 6.0, less than about 5.5, less than about 5.0, etc.). The expression "acidic pH" includes about 6.0, 5.95, 5.90, 5.85, 5.8, 5.75, 5.7, 5.65, 5.6, 5.55, PH values of 5.5, 5.45, 5.4, 5.35, 5.3, 5.25, 5.2, 5.15, 5.1, 5.05, 5.0 or less Is included. As used herein, the expression "neutral pH" means a pH of about 7.0 to about 7.4. The expression "neutral pH" refers to pH values of about 7.0, 7.05, 7.1, 7.15, 7.2, 7.25, 7.3, 7.35, and 7.4. Is included.
本方法との関連で使用される抗PCSK9抗体の非限定的な例には、例えば、アリロクマブ、エボロクマブ、ボコシズマブ、ロデルシズマブ、ラルパンシズマブ、LY3015014、または前述の抗体のいずれかの抗原結合部分が含まれる。 Non-limiting examples of anti-PCSK9 antibodies used in the context of the present method include, for example, allilocumab, evolocumab, bococizumab, rodercizumab, ralpancizumab, LY301504, or an antigen-binding portion of any of the foregoing antibodies.
ヒト抗体の調製
トランスジェニックマウスにおいてヒト抗体を生成する方法は、当該技術分野において公知である。このような既知の方法はいずれも、本方法との関連で使用して、ヒトPCSK9に特異的に結合するヒト抗体を作製する。
Preparation of Human Antibodies Methods for producing human antibodies in transgenic mice are known in the art. All such known methods are used in the context of this method to produce human antibodies that specifically bind human PCSK9.
VELOCIMMUNE(商標)技術(例えば、米国特許第6,596,541号、Regeneron Pharmaceuticalsを参照)またはモノクローナル抗体を生成する他の任意の既知の方法を使用して、ヒト可変領域およびマウス定常領域を有する、PCSK9に対する高親和性キメラ抗体が最初に単離される。VELOCIMMUNE(登録商標)技術は、内因性マウス定常領域遺伝子座に機能的に連結されたヒト重鎖および軽鎖可変領域を含むゲノムを有するトランスジェニックマウスの生成を含み、それにより、マウスは、抗原刺激に応答してヒト可変領域およびマウス定常領域を含む抗体を産生する。抗体の重鎖および軽鎖の可変領域をコードするDNAは単離され、ヒト重鎖および軽鎖定常領域をコードするDNAに機能的に連結される。次に、DNAは、完全なヒト抗体を発現することができる細胞において発現される。 Having human variable regions and mouse constant regions using VELOCIMMUNE™ technology (see, eg, US Pat. No. 6,596,541, Regeneron Pharmaceuticals) or any other known method of producing monoclonal antibodies. , A high affinity chimeric antibody to PCSK9 is first isolated. The VELOCIMMUNE® technology involves the generation of a transgenic mouse having a genome containing human heavy and light chain variable regions operably linked to an endogenous mouse constant region locus, whereby the mouse is In response to stimulation, it produces an antibody containing human variable regions and mouse constant regions. The DNA encoding the variable regions of the antibody heavy and light chains are isolated and operably linked to the DNA encoding the human heavy and light chain constant regions. The DNA is then expressed in cells capable of expressing fully human antibodies.
一般的に、VELOCIMMUNE(登録商標)マウスは、目的の抗原で攻撃され、抗体を発現するマウスからリンパ細胞(B細胞など)が回収される。リンパ細胞を骨髄腫細胞株と融合させて不死のハイブリドーマ細胞株を調製し、このようなハイブリドーマ細胞株をスクリーニングおよび選択して、目的の抗原に特異的な抗体を産生するハイブリドーマ細胞株を同定する。重鎖および軽鎖の可変領域をコードするDNAを単離し、重鎖および軽鎖の望ましいアイソタイプ定常領域に連結する。このような抗体タンパク質は、CHO細胞などの細胞で産生される。あるいは、抗原特異的キメラ抗体または軽鎖および重鎖の可変ドメインをコードするDNAは、抗原特異的リンパ球から直接単離される。 In general, VELOCIMMUNE (registered trademark) mice are challenged with an antigen of interest, and lymphocytes (B cells and the like) are recovered from mice expressing antibodies. Immortal hybridoma cell lines are prepared by fusing lymphocytes with myeloma cell lines, and such hybridoma cell lines are screened and selected to identify hybridoma cell lines that produce antibodies specific for the antigen of interest. .. DNA encoding the variable regions of the heavy and light chains are isolated and ligated to the desired isotype constant regions of the heavy and light chains. Such antibody proteins are produced in cells such as CHO cells. Alternatively, the DNA encoding the antigen-specific chimeric antibody or the variable domains of the light and heavy chains is isolated directly from the antigen-specific lymphocytes.
最初に、ヒト可変領域およびマウス定常領域を有する高親和性キメラ抗体が単離される。抗体は、当業者に公知である標準的な手順を使用して、親和性、選択性、エピトープなどを含む望ましい特性について特徴付けられ、選択される。マウス定常領域は、完全なヒト抗体、例えば、野生型もしくは修飾IgG1またはIgG4を生成するために、所望のヒト定常領域に置換される。選択された定常領域は特定の用途に応じて変化し得るが、高親和性抗原結合および標的特異性の特徴は可変領域にある。 First, a high affinity chimeric antibody having human variable regions and mouse constant regions is isolated. Antibodies are characterized and selected for desirable properties including affinity, selectivity, epitope, etc. using standard procedures known to those of skill in the art. The mouse constant region is replaced with the desired human constant region to produce fully human antibodies, eg, wild-type or modified IgG1 or IgG4. The constant region chosen may vary depending on the particular application, but features of high affinity antigen binding and target specificity lie in the variable region.
一般的に、固相上または溶液相中のいずれかに固定化された抗原に結合することにより測定される場合、使用される抗体は上記のように高親和性を保有する。マウス定常領域は、完全なヒト抗体を生成するために、所望のヒト定常領域に置換される。選択された定常領域は特定の用途に応じて変化し得るが、高親和性抗原結合および標的特異性の特徴は可変領域にある。 Generally, the antibody used possesses high affinity, as described above, as measured by binding to the antigen immobilized either on the solid phase or in solution phase. The mouse constant region is replaced with the desired human constant region in order to produce a fully human antibody. The constant region chosen may vary depending on the particular application, but features of high affinity antigen binding and target specificity lie in the variable region.
本方法との関連で使用されるPCSK9に特異的に結合するヒト抗体または抗体の抗原結合断片の具体例には、配列番号1および11からなる群から選択されるアミノ酸配列、または少なくとも90%、少なくとも95%、少なくとも98%もしくは少なくとも99%の配列同一性を有する実施的に類似したその配列を有する重鎖可変領域(HCVR)内に含まれる3つの重鎖CDR(HCDR1、HCDR2およびHCDR3)を含む任意の抗体または抗原結合断片が含まれる。あるいは、本方法との関連で使用されるPCSK9に特異的に結合するヒト抗体または抗体の抗原結合断片の具体例には、配列番号37、45、53、61、69、77、85、93、101、109、117、125、133、141、149、157、165、173、181、および189からなる群から選択されるアミノ酸配列、または少なくとも90%、少なくとも95%、少なくとも98%もしくは少なくとも99%の配列同一性を有する実施的に類似したその配列を有する重鎖可変領域(HCVR)内に含まれる3つの重鎖CDR(HCDR1、HCDR2およびHCDR3)を含む任意の抗体または抗原結合断片が含まれる。抗体または抗原結合断片は、配列番号6および15からなる群から選択されるアミノ酸配列、または少なくとも90%、少なくとも95%、少なくとも98%もしくは少なくとも99%の配列同一性を有する実施的に類似したその配列を有する軽鎖可変領域(LCVR)内に含まれる3つの軽鎖CDR(LCVR1、LCVR2、LCVR3)を含み得る。あるいは、抗体または抗原結合断片は、配列番号41、49、57、65、73、81、89、97、105、113、121、129、137、145、153、161、169、177、185、および193からなる群から選択されるアミノ酸配列、または少なくとも90%、少なくとも95%、少なくとも98%もしくは少なくとも99%の配列同一性を有する実施的に類似したその配列を有する軽鎖可変領域(LCVR)内に含まれる3つの軽鎖CDR(LCVR1、LCVR2、LCVR3)を含み得る。 Specific examples of human antibodies or antigen-binding fragments of antibodies that specifically bind to PCSK9 used in the context of this method include an amino acid sequence selected from the group consisting of SEQ ID NOs: 1 and 11, or at least 90%, Three heavy chain CDRs (HCDR1, HCDR2 and HCDR3) contained within a heavy chain variable region (HCVR) having practically similar sequences with at least 95%, at least 98% or at least 99% sequence identity. Included are any antibodies or antigen-binding fragments that include. Alternatively, specific examples of human antibodies or antigen-binding fragments of antibodies that specifically bind to PCSK9 used in the context of this method include SEQ ID NOs: 37, 45, 53, 61, 69, 77, 85, 93, An amino acid sequence selected from the group consisting of 101, 109, 117, 125, 133, 141, 149, 157, 165, 173, 181, and 189, or at least 90%, at least 95%, at least 98% or at least 99%. Any antibody or antigen-binding fragment comprising three heavy chain CDRs (HCDR1, HCDR2 and HCDR3) contained within a heavy chain variable region (HCVR) having practically similar sequences having the sequence identity of .. The antibody or antigen-binding fragment is an amino acid sequence selected from the group consisting of SEQ ID NOs: 6 and 15, or a substantially similar amino acid sequence thereof having at least 90%, at least 95%, at least 98% or at least 99% sequence identity. It may include three light chain CDRs (LCVR1, LCVR2, LCVR3) contained within a light chain variable region (LCVR) having a sequence. Alternatively, the antibody or antigen-binding fragment is SEQ ID NO: 41, 49, 57, 65, 73, 81, 89, 97, 105, 113, 121, 129, 137, 145, 153, 161, 169, 177, 185, and Within a light chain variable region (LCVR) having an amino acid sequence selected from the group consisting of 193, or an operatively similar sequence having at least 90%, at least 95%, at least 98% or at least 99% sequence identity. Can include the three light chain CDRs contained in (LCVR1, LCVR2, LCVR3).
2つのアミノ酸配列間の配列同一性は、参照アミノ酸配列、すなわち、配列番号で識別されるアミノ酸配列の全長にわたって、2つのアミノ酸配列間に最良の配列アライメントを使用して、および/または配列間の最良の配列アライメントの領域を用いて決定され、最良の配列アラインメントは、公知のツール、例えば、標準設定を使用するAlign、好ましくはEMBOSS::針、Matrix:Blosum62、Gap Open 10.0、Gap Extend 0.5を用いて得られる。 Sequence identity between two amino acid sequences may be determined using the best sequence alignment between the two amino acid sequences over the entire length of the reference amino acid sequence, ie the amino acid sequence identified by SEQ ID NO:, and/or between the sequences. Regions of best sequence alignments were determined and the best sequence alignments were determined using known tools, eg Align using standard settings, preferably EMBOSS::needle, Matrix:Blosum62, Gap Open 10.0, Gap Extend 10.0. Obtained using 0.5.
特定の実施形態において、抗体または抗原結合タンパク質は、配列番号1/6および11/15からなる群から選択される重鎖ならびに軽鎖可変領域アミノ酸配列対(HCVR/LCVR)からの6つのCDR(HCDR1、HCDR2、HCDR3、LCDR1、LCDR2およびLCDR3)を含む。あるいは、特定の実施形態では、抗体または抗原結合タンパク質は、配列番号37/41、45/49、53/57、61/65、69/73、77/81、85/89、93/97、101/105、109/113、117/121、125/129、133/137、141/145、149/153、157/161、165/169、173/177、181/185、and 189/193からなる群から選択される重鎖および軽鎖可変領域アミノ酸配列対(HCVR/LCVR)からの6つのCDR(HCDR1、HCDR2、HCDR3、LCDR1、LCDR2およびLCDR3)を含む。 In certain embodiments, the antibody or antigen-binding protein is a heavy chain and light chain variable region amino acid sequence pair selected from the group consisting of SEQ ID NOs: 1/6 and 11/15 and six CDRs (HCVR/LCVR) from the pair. HCDR1, HCDR2, HCDR3, LCDR1, LCDR2 and LCDR3). Alternatively, in certain embodiments, the antibody or antigen binding protein is SEQ ID NO: 37/41, 45/49, 53/57, 61/65, 69/73, 77/81, 85/89, 93/97, 101. /105, 109/113, 117/121, 125/129, 133/137, 141/145, 149/153, 157/161, 165/169, 173/177, 181/185, and 189/193 6 CDRs (HCDR1, HCDR2, HCDR3, LCDR1, LCDR2 and LCDR3) from the heavy and light chain variable region amino acid sequence pairs (HCVR/LCVR) selected from
特定の実施形態において、本方法において使用される抗PCSK9抗体または抗原結合タンパク質は、配列番号2/3/4/7/8/10(mAb316P[「REGN727」または「アリロクマブ」とも呼ばれる))および12/13/14/16/17/18(mAb300N)(米国特許出願公開番号2010/0166768を参照)および12/13/14/16/17/18から選択されるHCDR1/HCDR2/HCDR3/LCDR1/LCDR2/LCDR3アミノ酸配列を有し、配列番号16は、アミノ酸残基30でロイシンに対するヒスチジンの置換(L30H)を含む。 In certain embodiments, the anti-PCSK9 antibody or antigen binding protein used in the method is SEQ ID NOs: 2/3/4/7/8/10 (mAb316P [also referred to as "REGN727" or "arilocumab")) and 12 HCDR1/HCDR2/HCDR3/LCDR1/LCDR2 selected from /13/14/16/17/18 (mAb300N) (see U.S. Patent Application Publication No. 2010/0166768) and 12/13/14/16/17/18 /LCDR3 amino acid sequence, SEQ ID NO: 16 comprises a histidine substitution for leucine at amino acid residue 30 (L30H).
特定の実施形態では、抗体または抗原結合タンパク質は、配列番号1/6および11/15からなる群から選択されるHCVR/LCVRアミノ酸配列対を含む。特定の例示的な実施形態では、抗体または抗原結合タンパク質は、配列番号1のHCVRアミノ酸配列および配列番号6のLCVRアミノ酸配列を含む。特定の例示的な実施形態では、抗体または抗原結合タンパク質は、配列番号11のHCVRアミノ酸配列および配列番号15のLCVRアミノ酸配列を含む。特定の例示的な実施形態では、抗体または抗原結合タンパク質は、配列番号11のHCVRアミノ酸配列、およびアミノ酸残基30でロイシンに対するヒスチジンの置換を含む配列番号15のLCVRアミノ酸配列(L30H)を含む。
In certain embodiments, the antibody or antigen binding protein comprises an HCVR/LCVR amino acid sequence pair selected from the group consisting of SEQ ID NOs: 1/6 and 11/15. In certain exemplary embodiments, the antibody or antigen binding protein comprises the HCVR amino acid sequence of SEQ ID NO:1 and the LCVR amino acid sequence of SEQ ID NO:6. In certain exemplary embodiments, the antibody or antigen binding protein comprises the HCVR amino acid sequence of SEQ ID NO:11 and the LCVR amino acid sequence of SEQ ID NO:15. In certain exemplary embodiments, the antibody or antigen binding protein comprises the HCVR amino acid sequence of SEQ ID NO:11 and the LCVR amino acid sequence of SEQ ID NO:15 (L30H) that includes a histidine substitution for leucine at
医薬組成物および投与方法
本方法は、PCSK9阻害剤を患者に投与することを含み、PCSK9阻害剤は医薬組成物内に含まれる。医薬組成物は、適切な担体、賦形剤、および適切な移動、送達、耐性などを提供する他の薬剤とともに製剤化される。多数の適切な製剤は、すべての製薬化学者に公知である処方に見出される:Remington’s Pharmaceutical Sciences,Mack Publishing Company、Easton、PA。これらの製剤には、例えば、粉末、ペースト、軟膏、ゼリー、ワックス、油、脂質、脂質(カチオン性またはアニオン性)含有ベシクル(LIPOFECTIN(商標)など)、DNAコンジュゲート、無水吸収ペースト、水中油および油中水エマルジョン、エマルジョンカーボワックス(様々な分子量のポリエチレングリコール)、半固体ゲル、ならびにカーボワックスを含む半固体混合物が含まれる。Powellら、「Compendium of excipients for parenteral formulations」PDA(1998年)J Pharm Sci Technol 52巻:238〜311頁を参照されたい。
Pharmaceutical Compositions and Methods of Administration The methods include administering a PCSK9 inhibitor to a patient, the PCSK9 inhibitor being included within the pharmaceutical composition. The pharmaceutical composition is formulated with suitable carriers, excipients, and other agents that provide suitable transfer, delivery, resistance, etc. A number of suitable formulations are found in formulations known to all pharmaceutical chemists: Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA. These formulations include, for example, powders, pastes, ointments, jellies, waxes, oils, lipids, lipid (cationic or anionic) containing vesicles (such as LIPOFECTIN™), DNA conjugates, anhydrous absorption pastes, oil-in-water. And water-in-oil emulsions, emulsion carbowaxes (polyethylene glycols of various molecular weights), semisolid gels, and semisolid mixtures containing carbowaxes. See Powell et al., "Compendium of exclusives for parental formulations" PDA (1998) J Pharm Sci Technol 52:238-311.
本方法との関連で使用される抗PCSK9抗体を含む例示的な医薬製剤には、米国特許第8,795,669号(とりわけ、アリロクマブを含む例示的な製剤)または国際公開第2013/166448号または国際公開第2012/168491号に記載されている製剤のいずれかが含まれる。 Exemplary pharmaceutical formulations containing an anti-PCSK9 antibody for use in the context of the present method include US Pat. No. 8,795,669 (among others, an exemplary formulation comprising arilocumab) or WO 2013/166448. Also included are any of the formulations described in WO 2012/168491.
様々な送達システムが公知であり、医薬組成物を投与するために使用され、例えば、リポソーム、微粒子、マイクロカプセル、変異体ウイルスを発現できる組換え細胞、受容体媒介エンドサイトーシスへのカプセル化が挙げられる(例えば、Wuら、1987年、J.Biol.Chem.262巻:4429〜4432頁を参照されたい)。投与方法には、限定されないが、皮内、筋肉内、腹腔内、静脈内、皮下、鼻腔内、硬膜外、および経口経路が含まれる。組成物は、任意の便利な経路、例えば、注入またはボーラス注射、上皮もしくは粘膜の内層(例えば、口腔粘膜、直腸および腸粘膜など)を介した吸収により投与され、他の生物学的に活性な薬剤と一緒に投与される。 Various delivery systems are known and used to administer pharmaceutical compositions, including liposomes, microparticles, microcapsules, recombinant cells capable of expressing mutant virus, encapsulation into receptor-mediated endocytosis. (See, eg, Wu et al., 1987, J. Biol. Chem. 262:4429-4432). Modes of administration include, but are not limited to, intradermal, intramuscular, intraperitoneal, intravenous, subcutaneous, intranasal, epidural, and oral routes. The composition is administered by any convenient route, such as infusion or bolus injection, absorption through the epithelial or mucosal lining (eg, oral mucosa, rectal and intestinal mucosa), and other biologically active Administered with drug.
医薬組成物は、標準の針と注射器で皮下または静脈内に送達される。加えて、皮下送達に関して、ペン送達デバイスは、容易に医薬組成物を送達することにおいて用途を有する。このようなペン送達デバイスは、再利用可能または使い捨てであり得る。再利用可能なペン送達装置は、一般的に、医薬組成物を含む交換可能なカートリッジを利用する。カートリッジ内のすべての医薬組成物が投与され、カートリッジが空になったら、空のカートリッジを容易に廃棄し、医薬組成物を含む新しいカートリッジと交換する。次に、ペン配送デバイスを再利用する。使い捨てペンデリバリーデバイスには、交換可能なカートリッジはない。むしろ、使い捨てペン送達装置は、装置内のリザーバーに保持された医薬組成物で事前充填されている。リザーバーから医薬組成物が空になると、デバイス全体が廃棄される。 The pharmaceutical composition is delivered subcutaneously or intravenously with a standard needle and syringe. In addition, for subcutaneous delivery, pen delivery devices have application in delivering pharmaceutical compositions easily. Such pen delivery devices can be reusable or disposable. Reusable pen delivery devices generally utilize replaceable cartridges containing the pharmaceutical composition. When all the pharmaceutical composition in the cartridge has been dispensed and the cartridge is empty, the empty cartridge is easily discarded and replaced with a new cartridge containing the pharmaceutical composition. Then reuse the pen delivery device. Disposable pen delivery devices do not have replaceable cartridges. Rather, the disposable pen delivery device is prefilled with the pharmaceutical composition held in a reservoir within the device. When the pharmaceutical composition is emptied from the reservoir, the entire device is discarded.
多数の再利用可能なペンおよび自動注射器送達装置は、医薬組成物の皮下送達に用途がある。例としては、限定されないが、ほんの数例を挙げると、AUTOPEN(商標)(Owen Mumford,Inc.、Woodstock、UK)、DISETRONIC(商標)ペン(Disetronic Medical Systems、Bergdorf、Switzerland)、HUMALOG MIX 75/25(商標)ペン、HUMALOG(商標)ペン、HUMALIN 70/30(商標)ペン(Eli Lilly and Co.、Indianapolis、IN)、NOVOPEN(商標)I、IIおよびIII(Novo Nordisk、Copenhagen、Denmark)、NOVOPEN JUNIOR(商標)(Novo Nordisk、Copenhagen、Denmark)、BD(商標)ペン(Becton Dickinson、Franklin Lakes、NJ)、OPTIPEN(商標)、OPTIPEN PRO(商標)、OPTIPEN STARLET(商標)、ならびにOPTICLIK(商標)(sanofi−aventis、Frankfurt、Germany)が含まれる。本方法の医薬組成物の皮下送達における用途を有する使い捨てペン送達装置の例には、限定されないが、ほんの数例を挙げると、SOLOSTAR(商標)ペン(sanofi−aventis)、FLEXPEN(商標)(Novo Nordisk)、およびKWIKPEN(商標)(Eli Lilly)、SURECLICK(商標)オートインジェクタ(Amgen、Thousand Oaks、CA)、PENLET(商標)(Haselmeier、Stuttgart、Germany)、EPIPEN(Dey,L.P.)、およびHUMIRA(商標)ペン(Abbott Labs、Abbott Park I)が含まれる。
A number of reusable pens and auto-injector delivery devices find use in subcutaneous delivery of pharmaceutical compositions. Examples include, but are not limited to, AUTOPEN(TM) (Owen Mumford, Inc., Woodstock, UK), DISETRONIC(TM) pens (Distronic Medical Systems, Bergdorf, SwitzerlandMIUG/L). 25(TM) Pen, HUMALOG(TM) Pen,
特定の状況では、医薬組成物は、制御放出システムで送達される。一実施形態では、ポンプが使用される(Langer(前掲);Sefton、1987年、CRC Crit.Ref.Biomed.Eng.14巻:201頁を参照されたい)。別の実施形態では、ポリマー材料が使用される;Medical Applications of Controlled Release、LangerおよびWise(編集)、1974年、CRC Pres.、Boca Raton、Floridaを参照されたい。さらに別の実施形態では、制御放出システムは、組成物の標的の近くに配置され、したがって、全身用量の一部のみを必要とする(例えば、Goodson、1984年、Medical Applications of Controlled Release、前掲、2巻、115〜138頁を参照されたい)。他の制御放出システムについては、Langer、1990年、Science 249巻:1527〜1533頁で検討されている。 In certain situations, the pharmaceutical composition is delivered in a controlled release system. In one embodiment, a pump is used (see Langer (supra); Sefton, 1987, CRC Crit. Ref. Biomed. Eng. 14:201). In another embodiment, polymeric materials are used; Medical Applications of Controlled Release, Langer and Wise, 1974, CRC Pres. , Boca Raton, Florida. In yet another embodiment, the controlled release system is placed in proximity to the target of the composition and thus requires only a portion of the systemic dose (eg Goodson, 1984, Medical Applications of Controlled Release, supra, 2, pp. 115-138). Other controlled release systems are discussed in Langer, 1990, Science 249:1527-1533.
注射用製剤は、静脈内、皮下、皮内および筋肉内注射、点滴などのための剤形を含み得る。これらの注射用製剤は、公知の方法によって調製される。例えば、注射用製剤は、例えば、注射に従来使用されている滅菌水性媒体または油性媒体に上記の抗体またはその塩を溶解、懸濁もしくは乳化することにより調製される。注射用の水性媒体としては、例えば、生理食塩水、グルコースおよび他の補助剤を含む等張液などがあり、これらは、適切な可溶化剤、例えば、アルコール(例えば、エタノール)、多価アルコール(例えば、プロピレングリコール、ポリエチレングリコール)、非イオン性界面活性剤[例えば、ポリソルベート80、HCO−50(硬化ヒマシ油のポリオキシエチレン(50mol)付加物)]などと組み合わせて使用される。油性媒体として、例えば、ゴマ油、大豆油などがあり、これらは、可溶化剤、例えば、安息香酸ベンジル、ベンジルアルコールなどと組み合わせて使用される。こうして調製された注射液は、適切なアンプルに充填することが好ましい。
Injectable formulations may include dosage forms for intravenous, subcutaneous, intradermal and intramuscular injection, infusion and the like. These injectable preparations are prepared by known methods. For example, an injectable preparation is prepared, for example, by dissolving, suspending or emulsifying the above-mentioned antibody or a salt thereof in a sterile aqueous medium or oily medium conventionally used for injection. Aqueous vehicles for injection include, for example, isotonic solutions containing saline, glucose and other adjuvants, which are suitable solubilizers such as alcohols (eg ethanol), polyhydric alcohols. (Eg, propylene glycol, polyethylene glycol), a nonionic surfactant [eg,
有利には、上記の経口または非経口使用のための医薬組成物は、活性成分の用量に適合するのに適した単位用量の剤形に調製される。単位用量のこのような剤形には、例えば、錠剤、丸剤、カプセル剤、注射剤(アンプル)、坐剤などが含まれる。 Advantageously, the pharmaceutical compositions for oral or parenteral use described above are prepared into unit dose forms suitable for matching the dose of active ingredient. Such unit dose forms include, for example, tablets, pills, capsules, injections (ampoules), suppositories, and the like.
投薬量
患者に投与されるPCSK9阻害剤(例えば、抗PCSK9抗体)の量は、一般的に、治療有効量である。本明細書で使用するとき、「治療有効量」という語句は、LDL−C、ApoB、ApoB100、非HDL−C、総コレステロール、VLDL−C、トリグリセリド、ApoC3、TRL粒子、Lp(a)および残存コレステロールからなる群から選択される1つまたはそれ以上のパラメータにおいて、検出可能な減少(ベースラインから少なくとも約5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、またはそれ以上)をもたらすPCSK9阻害剤の用量を意味する。
Dosage The amount of PCSK9 inhibitor (eg, anti-PCSK9 antibody) administered to a patient is generally a therapeutically effective amount. As used herein, the phrase "therapeutically effective amount" refers to LDL-C, ApoB, ApoB100, non-HDL-C, total cholesterol, VLDL-C, triglycerides, ApoC3, TRL particles, Lp(a) and residual. Detectable reduction (at least about 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40% from baseline in one or more parameters selected from the group consisting of cholesterol , 45%, 50%, 55%, 60%, 65%, 70%, 75% or more) of the PCSK9 inhibitor.
抗PCSK9抗体の場合、治療有効量は、約0.05mgから約600mg、例えば、約0.05mg、約0.1mg、約1.0mg、約1.5mg、約2.0mg、約10mg、約20mg、約30mg、約40mg、約50mg、約60mg、約70mg、約75mg、約80mg、約90mg、約100mg、約110mg、約120mg、約130mg、約140mg、約160mg、約170mg、約180mg、約190mg、約200mg、約210mg、約220mg、約230mg、約240mg、約250mg、約260mg、約270mg、約280mg、約290mg、約300mg、約310mg、約320mg、約330mg、約340mg、約350mg、約360mg、約370mg、約380mg、約390mg、約400mg、約410mg、約420mg、約430mg、約440mg、約450mg、約460mg、約470mg、約480mg、約490mg、約500mg、約510mg、約520mg、約530mg、約540mg、約550mg、約560mg、約570mg、約580mg、約590mg、または約600mgの抗PCSK9抗体である。特定の例示的な実施形態によれば、抗PCSK9抗体の治療有効量は、30mg、40mgもしくは75mg(例えば、体重が50kg未満、および/または17歳以下の患者のアリロクマブの場合)、50mg、75mgもしくは150mg(例えば、体重が50kg以上、および/または17歳以下の患者のアリロクマブの場合)、または140mgもしくは420mg(例えば、エボロクマブの場合)である。PCSK9阻害剤の他の投薬量は、当業者には明らかである。 For anti-PCSK9 antibodies, a therapeutically effective amount is from about 0.05 mg to about 600 mg, such as about 0.05 mg, about 0.1 mg, about 1.0 mg, about 1.5 mg, about 2.0 mg, about 10 mg, about. 20 mg, about 30 mg, about 40 mg, about 50 mg, about 60 mg, about 70 mg, about 75 mg, about 80 mg, about 90 mg, about 100 mg, about 110 mg, about 120 mg, about 130 mg, about 140 mg, about 160 mg, about 170 mg, about 180 mg, About 190 mg, about 200 mg, about 210 mg, about 220 mg, about 230 mg, about 240 mg, about 250 mg, about 260 mg, about 270 mg, about 280 mg, about 290 mg, about 300 mg, about 310 mg, about 320 mg, about 330 mg, about 340 mg, about 350 mg. , About 360 mg, about 370 mg, about 380 mg, about 390 mg, about 400 mg, about 410 mg, about 420 mg, about 430 mg, about 440 mg, about 450 mg, about 460 mg, about 470 mg, about 480 mg, about 490 mg, about 500 mg, about 510 mg, about 520 mg, about 530 mg, about 540 mg, about 550 mg, about 560 mg, about 570 mg, about 580 mg, about 590 mg, or about 600 mg of anti-PCSK9 antibody. According to certain exemplary embodiments, a therapeutically effective amount of an anti-PCSK9 antibody is 30 mg, 40 mg or 75 mg (eg, for arilocumab in a patient weighing less than 50 kg and/or 17 years or younger), 50 mg, 75 mg. Or 150 mg (eg in the case of arilocumab in patients weighing 50 kg or more and/or 17 years or younger), or 140 mg or 420 mg (eg in the case of evolocumab). Other dosages for PCSK9 inhibitors will be apparent to those of skill in the art.
個々の用量内に含まれる抗PCSK9抗体の量は、患者の体重1キログラムあたりの抗体のミリグラム数(すなわち、mg/kg)で表される。例えば、抗PCSK9抗体は、体重1kgあたり約0.0001から約10mg/kgの用量で患者に投与される。 The amount of anti-PCSK9 antibody contained within an individual dose is expressed in milligrams of antibody per kilogram of patient body weight (ie, mg/kg). For example, the anti-PCSK9 antibody is administered to a patient at a dose of about 0.0001 to about 10 mg/kg body weight.
投与レジメン
特定の実施形態によれば、複数回用量のPCSK9阻害剤(すなわち、PCSK9阻害剤を含む医薬組成物)は、定義された時間経過にわたって(例えば、毎日の治療用スタチンレジメンまたは他のバックグランドLMTに加えて)対象に投与され得る。この態様による方法は、複数回用量のPCSK9阻害剤を対象に連続的に投与することを含む。本明細書で使用するとき、「連続的に投与する」とは、PCSK9阻害剤の各用量が、異なる時間点、例えば、所定の間隔(例えば、時間、日、週または月)で区切られた異なる日に対象に投与されることを意味する。本方法は、PCSK9阻害剤の単一の初期用量、続いてPCSK9阻害剤の1回またはそれ以上の二次用量、および場合によりPCSK9阻害剤の1回またはそれ以上の三次用量を患者に連続的に投与することを含む。
Dosing Regimens According to certain embodiments, multiple doses of a PCSK9 inhibitor (ie, a pharmaceutical composition comprising a PCSK9 inhibitor) are administered over a defined time course (eg, a daily therapeutic statin regimen or other dosage regimen). It can be administered to the subject (in addition to Grand LMT). The method according to this aspect comprises sequentially administering to the subject multiple doses of a PCSK9 inhibitor. As used herein, "consecutively administered" means that each dose of PCSK9 inhibitor is separated at different time points, eg, at predetermined intervals (eg, hours, days, weeks or months). Means being administered to a subject on a different day. The method provides a patient with a single initial dose of PCSK9 inhibitor, followed by one or more secondary doses of PCSK9 inhibitor, and optionally one or more tertiary doses of PCSK9 inhibitor. Administration to.
「一次投薬」、「二次投薬」、および「三次投薬」という用語は、PCSK9阻害剤を含む医薬組成物の個々の投薬の投与の時間的順序を指す。したがって、「初回投薬」は、治療レジメンの開始時に投与される投薬である(「ベースライン投薬」とも呼ばれる);「二次投薬」は、初回投薬後に投与される投薬である;「三次投薬」は、二次投薬後に投与される投薬である。初回、二次、および三次投薬は、すべて同じ量のPCSK9阻害剤を含み得るが、一般的には投与頻度に関連して互いに異なる可能性がある。しかしながら、特定の実施形態では、初回、二次、および/または三次投薬に含まれるPCSK9阻害剤の量は、治療の過程で互いに異なる(例えば、適宜、上下に調整される)。特定の実施形態では、2回以上(例えば、2、3、4、または5回)の用薬は、治療レジメンの開始時に「負荷用量」として投与され、その後、より低頻度基準(例えば、「維持用量」)で投与される後続の用量が投与される。 The terms "primary dose," "secondary dose," and "tertiary dose" refer to the temporal sequence of administration of individual doses of a pharmaceutical composition comprising a PCSK9 inhibitor. Thus, a "primary dose" is a dose administered at the beginning of a treatment regimen (also called a "baseline dose"); a "secondary dose" is a dose administered after the initial dose; a "tertiary dose". Is the medication administered after the second dose. The first, second, and third doses may all contain the same amount of PCSK9 inhibitor, but may differ from one another, generally with respect to frequency of administration. However, in certain embodiments, the amount of PCSK9 inhibitor included in the initial, secondary, and/or tertiary dosing will be different (eg, adjusted up or down as appropriate) over the course of treatment. In certain embodiments, two or more (eg, 2, 3, 4, or 5) doses of the drug are administered as a “loading dose” at the beginning of the treatment regimen, and then on a less frequent basis (eg, “ Subsequent doses administered in a maintenance dose") are administered.
例示的な実施形態によれば、各二次用量および/または三次用量は、直前の投薬から1〜26(例えば、1、1、1と1/2、2、2と1/2、3、3と1/2、4、4と1/2、5、5と1/2、6、6と1/2、7、7と1/2、8、8と1/2、9、9と1/2、10、10と1/2、11、11と1/2、12、12と1/2、13、13と1/2、14、14と1/2、15、15と1/2、16、16と1/2、17、17と1/2、18、18と1/2、19、19と1/2、20、20と1/2、21、21と1/2、22、22と1/2、23、23と1/2、24、24と1/2、25、25と1/2、26、26と1/2、またはそれ以上)週後に投与される。「直前の用量」という語句は、本明細書で使用するとき、一連の複数回投与において、用量を介入することなく、その次の用量を投与する前に、患者に投与される抗原結合分子の用量を意味する。
According to an exemplary embodiment, each secondary and/or tertiary dose is from 1 to 26 (
この態様による方法は、PCSK9阻害剤の任意の数の二次用量および/または三次用量を患者に投与することを含み得る。例えば、特定の実施形態では、単一の二次用量のみが患者に投与される。他の実施形態では、2つ以上(例えば、2、3、4、5、6、7、8、またはそれ以上)の二次用量が患者に投与される。同様に、特定の実施形態では、単一の三次用量のみが患者に投与される。他の実施形態では、2つ以上(例えば、2、3、4、5、6、7、8、またはそれ以上)の三次用量が患者に投与される。 The method according to this aspect can include administering to the patient any number of secondary and/or tertiary doses of the PCSK9 inhibitor. For example, in certain embodiments, only a single secondary dose is administered to the patient. In other embodiments, two or more (eg, 2, 3, 4, 5, 6, 7, 8 or more) secondary doses are administered to the patient. Similarly, in certain embodiments, only a single tertiary dose is administered to the patient. In other embodiments, two or more (eg, 2, 3, 4, 5, 6, 7, 8 or more) tertiary doses are administered to the patient.
複数の二次用量を伴う実施形態では、各二次用量は、他の二次用量と同じ頻度で投与される。例えば、各二次用量は、直前の用量の1〜2、4、6、8またはそれ以上の週の後に患者に投与される。同様に、複数の三次用量を伴う実施形態では、各三次用量は、他の三次用量と同じ頻度で投与される。例えば、各三次用量は、直前の用量の1〜2、4、6、8またはそれ以上の週の後に患者に投与される。あるいは、二次および/または三次用量が患者に投与される頻度は、治療レジメンの過程にわたって変更することができる。投与の頻度はまた、臨床検査後の個々の患者のニーズに応じて、医師が治療の過程で調整される。
In embodiments with multiple secondary doses, each secondary dose is administered at the same frequency as the other secondary doses. For example, each secondary dose is administered to the
本方法は、漸増オプション(本明細書では「用量変更」とも呼ばれる)を含む投与レジメンを含む。本明細書で使用するとき、「漸増オプション」とは、PCSK9阻害剤の特定数の投薬を受けた後、患者が1つまたはそれ以上の定義された治療パラメータの特定の減少を達成していない場合、PCSK9阻害剤の投薬量がその後増加する。例えば、2週間ごとに1回の頻度で患者に75mg用量の抗PCSK9抗体を投与することを含む治療レジメンの場合では、8週間後(すなわち、0週目、2週目、4週目、6週目、8週目で5回投薬が投与された)、患者が70mg/dL未満の血清LDL−C濃度に達していない場合、抗PCSK9抗体の用量は、その後、例えば、2週間ごとに1回投与される150mgに増加される(例えば、10週目または12週目以降に開始する)。
The method includes a dosing regimen that includes a titration option (also referred to herein as "dose modification"). As used herein, an "escalation option" is one in which a patient has not achieved a particular reduction in one or more defined therapeutic parameters after receiving a particular number of doses of a PCSK9 inhibitor. In some cases, the dosage of PCSK9 inhibitor is then increased. For example, in the case of a treatment regimen that involves administering to a patient a 75 mg dose of anti-PCSK9 antibody once every two weeks, 8 weeks later (ie, 0 weeks, 2 weeks, 4 weeks, 6 weeks). If the patient has not reached a serum LDL-C concentration of less than 70 mg/dL, then the dose of anti-PCSK9 antibody is then, for example, 1 every 2 weeks. The dose is increased to 150 mg (eg, starting at
特定の実施形態において、PCSK9に特異的に結合する抗体またはその抗原結合断片は、2週間ごとに1回の頻度で約75mgの用量で患者に投与される。特定の実施形態では、1回またはそれ以上、2回またはそれ以上、3回またはそれ以上、4回またはそれ以上、または5回またはそれ以上の投薬後に測定された患者のLDL−Cが<70mg/dLである場合、約75mgの用量が維持される。特定の実施形態では、1回またはそれ以上、2回またはそれ以上、3回またはそれ以上、4回またはそれ以上、または5回またはそれ以上の投薬後に測定された患者のLDL−Cが≧70mg/dLのままである場合、約75mgの用量は中止され、その後、PCSK9に特異的に結合する抗体またはその抗原結合断片は、2週間ごとに1回の頻度で約150mgの用量で患者に投与される。 In certain embodiments, the antibody or antigen-binding fragment thereof that specifically binds to PCSK9 is administered to the patient at a dose of about 75 mg, once every two weeks. In certain embodiments, a patient's LDL-C is <70 mg measured after one or more, two or more, three or more, four or more, or five or more doses. /DL, a dose of about 75 mg is maintained. In certain embodiments, a patient's LDL-C is ≧70 mg measured after one or more, two or more, three or more, four or more, or five or more doses. /DL remains, the dose of about 75 mg is discontinued and thereafter the antibody or antigen-binding fragment thereof that specifically binds to PCSK9 is administered to the patient at a dose of about 150 mg once every two weeks. To be done.
特定の実施形態では、PCSK9に特異的に結合する抗体またはその抗原結合断片は、4週間ごとに1回の頻度で約300mgの用量で患者に投与される。特定の実施形態では、1回またはそれ以上、2回またはそれ以上、3回またはそれ以上、4回またはそれ以上、または5回またはそれ以上の投薬後に測定された患者のLDL−Cが<70mg/dLである場合、約300mgの用量が維持される。特定の実施形態では、1回またはそれ以上、2回またはそれ以上、3回またはそれ以上、4回またはそれ以上、または5回またはそれ以上の投薬後に測定された患者のLDL−Cが≧70mg/dLのままである場合、約300mgの用量は中止され、その後、PCSK9に特異的に結合する抗体またはその抗原結合断片は、2週間ごとに1回の頻度で約150mgの用量で患者に投与される。 In certain embodiments, the antibody or antigen-binding fragment thereof that specifically binds to PCSK9 is administered to the patient at a dose of about 300 mg once every four weeks. In certain embodiments, a patient's LDL-C is <70 mg measured after one or more, two or more, three or more, four or more, or five or more doses. /DL, a dose of about 300 mg is maintained. In certain embodiments, a patient's LDL-C is ≧70 mg measured after one or more, two or more, three or more, four or more, or five or more doses. /DL, the dose of about 300 mg is discontinued, and then the antibody or antigen-binding fragment thereof that specifically binds to PCSK9 is administered to the patient at a dose of about 150 mg once every two weeks. To be done.
特定の実施形態では、PCSK9に特異的に結合する抗体またはその抗原結合断片は、2週間ごとに1回の頻度で約150mgの用量で患者に投与される。 In certain embodiments, the antibody or antigen-binding fragment thereof that specifically binds to PCSK9 is administered to the patient at a dose of about 150 mg once every two weeks.
特定の実施形態では、PCSK9に特異的に結合する抗体またはその抗原結合断片が、2週間ごとに1回の頻度で約150mgの用量で患者に投与される場合、約150mgの用量は、少なくとも1回の投薬または少なくとも2、3、4、もしくは5回の連続投薬後に測定された患者のLDL−Cが<10、15、20、または25mg/dLである場合に中断され、その後、PCSK9に特異的に結合する抗体またはその抗原結合断片は、2週間ごとに1回の頻度で約75mgの用量で患者に投与される。理論に拘束されることを望まないが、非常に低いLDL−Cレベル(例えば、<10、15、20、または25mg/dL)が糖尿病を悪化させるという仮説が立てられる。特定の実施形態では、約150mgの用量が一定用量として患者に投与される。特定の実施形態では、約150mgの用量は、本明細書に開示される用量調整後に患者に投与される(例えば、2週間ごとに約75mg、または4週間ごとに約300mg)。 In certain embodiments, when the antibody or antigen-binding fragment thereof that specifically binds to PCSK9 is administered to the patient at a dose of about 150 mg once every two weeks, the dose of about 150 mg is at least 1 Interrupted if the patient's LDL-C is <10, 15, 20, or 25 mg/dL measured after one dose or at least 2, 3, 4, or 5 consecutive doses, and then specific for PCSK9 The antibody or antigen-binding fragment thereof that specifically binds is administered to the patient at a dose of about 75 mg once every two weeks. Without wishing to be bound by theory, it is hypothesized that very low LDL-C levels (eg <10, 15, 20, or 25 mg/dL) exacerbate diabetes. In certain embodiments, a dose of about 150 mg is administered to a patient as a fixed dose. In certain embodiments, a dose of about 150 mg is administered to a patient after the dose adjustments disclosed herein (eg, about 75 mg every 2 weeks, or about 300 mg every 4 weeks).
併用療法
本明細書の他の箇所に記載されるように、本方法は、患者の以前に処方された脂質修飾療法(LMT)と組み合わせて(「に加えて」)PCSK9阻害剤を患者に投与することを含み得る。LMTには、限定されないが、スタチン、フィブラート、ナイアシン(例えば、ニコチン酸およびその誘導体)、胆汁酸封鎖剤、エゼチミブ、ロミタピド、フィトステロール、オルリスタットなどが含まれる。例えば、PCSK9阻害剤は、安定した毎日の治療用スタチンレジメンと組み合わせて患者に投与される。PCSK9阻害剤が本方法との関連で組み合わせて投与される例示的な毎日の治療用スタチンレジメンには、例えば、アトルバスタチン(毎日10、20、40または80mg)、(アトルバスタチン/エゼチミブ毎日10/10または40/10)、ロスバスタチン(毎日5、10または20mg)、セリバスタチン(毎日0.4または0.8mg)、ピタバスタチン(毎日1、2または4mg)、フルバスタチン(毎日20、40または80mg)、シンバスタチン(毎日5、10、20、40または80mg)、シンバスタチン/エゼチミブ(毎日10/10、20/10、40/10または80/10mg)、ロバスタチン(毎日10、20、40または80mg)、プラバスタチン(毎日10、20、40または80mg)、およびそれらの組み合わせが含まれる。特定の実施形態では、スタチン療法は、患者に対する最大耐性のスタチン療法である。PCSK9阻害剤が本方法との関連で組み合わせて投与される他のLMTには、例えば、(1)コレステロールの取り込みおよび/または胆汁酸の再吸収を阻害する薬剤(例えば、エゼチミブ);(2)リポタンパク質の異化を増加させる薬剤(例えば、ナイアシン);ならびに/または(3)22−ヒドロキシコレステロールなどのコレステロール除去に役割を果たすLXR転写因子のアクチベーターが含まれる。
Combination Therapy As described elsewhere herein, the method involves administering to a patient a PCSK9 inhibitor in combination (in addition to) a patient's previously prescribed lipid modification therapy (LMT). Can include doing. LMTs include, but are not limited to, statins, fibrates, niacin (eg, nicotinic acid and its derivatives), bile sequestrants, ezetimibe, romitapid, phytosterols, orlistat, and the like. For example, the PCSK9 inhibitor is administered to a patient in combination with a stable daily therapeutic statin regimen. Exemplary daily therapeutic statin regimens in which a PCSK9 inhibitor is administered in combination in the context of the present method include, for example, atorvastatin (10, 20, 40 or 80 mg daily), (atorvastatin/
特定の実施形態によれば、PCSK9阻害剤(例えば、抗PCSK9抗体、例えばアリロクマブ、エボロクマブ、ボコシズマブ、ロデルシズマブ、ラルパンシズマブまたはLY3015014)をアンジオポエチン様タンパク質3の阻害剤(例えば、抗ANGPTL3抗体、例えば、REGN1500)、アンジオポエチン様タンパク質4の阻害剤(例えば、抗ANGPTL4抗体、例えば、米国特許第9,120,851号において「H1H268P」または「H4H284P」と呼ばれる抗ANGPTL4抗体)、またはアンジオポエチン様タンパク質8の阻害剤(例えば、抗ANGPTL8抗体)と組み合わせて、患者に投与することを含む方法が提供される。
According to certain embodiments, a PCSK9 inhibitor (eg, an anti-PCSK9 antibody, such as allilocumab, evolocumab, bokocizumab, rodercizumab, ralpancizumab, or LY30105014) is an inhibitor of angiopoietin-like protein 3 (eg, an anti-ANGPTL3 antibody, eg, REGN1500). , An angiopoietin-like protein 4 inhibitor (eg, an anti-ANGPTL4 antibody, eg, an anti-ANGPTL4 antibody referred to as “H1H268P” or “H4H284P” in US Pat. No. 9,120,851), or an angiopoietin-
特定の実施形態によれば、インスリン療法に加えて、PCSK9阻害剤(例えば、抗PCSK9抗体、例えばアリロクマブ、エボロクマブ、ボコシズマブ、ロデルシズマブ、ラルパンシズマブまたはLY3015014)をさらなる抗糖尿病療法と組み合わせて患者に投与することを含む方法が提供される。例示的な追加の抗糖尿病療法には、限定されないが、以下が含まれる:
(a)Rote Liste 2016に記載されているすべての薬剤(例えば、Rote Liste 2014年、12章に記載されているすべての抗糖尿病薬)、Rote Liste 2016年、06章に記載されているすべての減量剤または食欲抑制薬、Rote Liste 2016年、58章に記載されているすべての脂質減少薬、Rote Liste 2016年、17章に記載されているすべての降圧薬、Rote Listeに記載されているすべての腎保護薬、またはRote Liste 2016年、36章に記載されているすべての利尿薬;
(b)GLP−1、GLP−1類似体、およびGLP−1受容体アゴニストを含むグルカゴン様ペプチド1(GLP−1)療法、例えば:GLP−1(7−37)、GLP−1(7−36)アミド、リキシセナチド(例えば、Lyxumia(登録商標))、エキセナチド(例えば、Exendin−4、rExendin−4、Byetta(登録商標)、Bydureon(登録商標)、エキセナチドNexP)、エキセナチド−LAR、リラグルチド(例えば、Victoza(登録商標))、セマグルチド、タスポグルチド、アルビグルチド、デュラグルチド、アルブゴン、オキシントモデュリン、ゲニプロシド、ACP−003、CJC−1131、CJC−1134−PC、GSK−2374697、PB−1023、TTP−054、ラングレナチド(HM−11260C)、CM−3、GLP−1 Eligen、AB−201、ORMD−0901、NN9924、NN9926、NN9927、Nodexen、Viador−GLP−1、CVX−096、ZYOG−1、ZYD−1、ZP−3022、CAM−2036、DA−3091、DA−15864、ARI−2651、ARI−2255、エキセナチド−XTEN(VRS−859)、エキセナチド−XTEN+グルカゴン−XTEN(VRS−859+AMX−808)ならびにポリマー結合GLP−1およびGLP−1類似体;
(c)デュアルGLP−1/GIPアゴニスト(例えば、RG−7697(MAR−701)、MAR−709、BHM081、BHM089、BHM098);デュアルGLP−1/グルカゴン受容体アゴニスト(例えば、BHM−034、OAP−189(PF−05212389、TKS−1225)、TT−401/402、ZP2929、LAPS−HMOXM25、MOD−6030);
(d)デュアルGLP−1/ガストリンアゴニスト(例えば、ZP−3022);
(e)胃腸ペプチド、例えば、ペプチドYY3−36(PYY3−36)またはその類似体および膵臓ポリペプチド(PP)またはその類似体;
(f)グルカゴン受容体アゴニストまたはアンタゴニスト、グルコース依存性インスリン分泌性ポリペプチド(GIP)受容体アゴニストまたはアンタゴニスト、グレリンアンタゴニストまたは逆アゴニスト、キセニンおよびその類似体。
(g)ジペプチジルペプチダーゼ−IV(DPP−4)阻害剤、例えば:アログリプチン(例えば、Nesina(登録商標)、Kazano(登録商標))、リナグリプチン(例えば、Ondero(登録商標)、Trajenta(登録商標)、Trajenta(登録商標)、Trayenta(登録商標))、サキサグリプチン(例えば、Onglyza(登録商標)、Komboglyze XR(登録商標))、シタグリプチン(例えば、Januvia(登録商標)、Xelevia(登録商標)、Tesavel(登録商標)、Janumet(登録商標)、Velmetia(登録商標)、Juvisync(登録商標)、JanumetXR(登録商標))、アナグリプチン、テネリグリプチン(例えば、Tenelia(登録商標))、トレラグリプチン、ビルダグリプチン(例えば、Galvus(登録商標)、Galvumet(登録商標))、ゲミグリプチン、オマリグリプチン、エボグリプチン、デュトグリプチン、DA−1229、MK−3102、KM−223、KRP−104、PBL−1427、塩酸ピノキサシン、およびAri−2243;
(h)ナトリウム依存性グルコース輸送体2(SGLT−2)阻害剤、例えば:カナグリフロジン、ダパグリフロジン、レモグリフロジン、レモグリフロジンエタボネート、セルグリフロジン、エンパグリフロジン、イプラグリフロジン、トフォグリフロジン、ルセオグリフロジン、エルツグリフロジン、EGT−0001442、LIK−066、SBM−TFC−039、およびKGA−3235(DSP−3235);
(i)SGLT−2およびSGLT−1の二重阻害剤(例えば、LX−4211、LIK066);
(j)SGLT−1阻害剤(例えば、LX−2761、KGA−3235など)または回腸胆汁酸転移(IBAT)阻害剤(例えば、GSK−1614235+GSK−2330672など)などの抗肥満薬と組み合わせたSGLT−1阻害剤;
(k)ビグアナイド(例えば、メトホルミン、ブホルミン、フェンホルミン);
(l)チアゾリジンジオン(例えば、ピオグリタゾン、ロシグリタゾン)、グリタゾン類似体(例えば、ロベグリタゾン);
(m)ペルオキシソーム増殖因子活性化受容体(PPAR−)(アルファ、ガンマまたはアルファ/ガンマ)アゴニストまたはモジュレータ(例えば、サログリタザール(例えば、リパグリン(登録商標))、GFT−505)、またはPPARガンマ部分アゴニスト(例えば、Int−131);
(n)スルホニル尿素(例えば、トルブタミド、グリベンクラミド、グリメピリド、アマリル(登録商標)、グリピジド)およびメグリチニド(例、ナテグリニド、レパグリニド、ミチグリニド);
(o)アルファ−グルコシダーゼ阻害剤(例えば、アカルボース、ミグリトール、ボグリボース);
(q)アミリンおよびアミリン類似体(例えば、プラムリンチド、Symlin(登録商標));
(p)Gタンパク質共役受容体119(GPR119)アゴニスト(例えば、GSK−1292263、PSN−821、MBX−2982、APD−597、ARRY−981、ZYG−19、DS−8500、HM−47000、YH−Chem1);
(q)GPR40アゴニスト(例えば、TUG−424、P−1736、P−11187、JTT−851、GW9508、CNX−011−67、AM−1638、AM−5262);
(r)GPR120アゴニストおよびGPR142アゴニスト;
(s)全身性または低吸収性TGR5(GPBAR1=Gタンパク質共役胆汁酸受容体1)アゴニスト(例えば、INT−777、XL−475、SB756050);
(t)糖尿病免疫療法薬、例えば:経口CCケモカイン受容体2型(CCR−2)アンタゴニスト(例えば、CCX−140、JNJ−41443532)、インターロイキン1ベータ(IL−1β)アンタゴニスト(例えば、AC−201)、または経口モノクローナル抗体(MoA)(例えば、メタロザミド、VVP808、PAZ−320、P−1736、PF−05175157、PF−04937319);
(v)メタボリックシンドロームおよび糖尿病の治療のための抗炎症剤、例えば:核因子カッパB阻害剤(例えば、Triolex(登録商標));
(w)アデノシン一リン酸活性化プロテインキナーゼ(AMPK)刺激剤、例えば:イメグリミン(PXL−008)、デビオ−0930(MT−63−78)、R−118;
(x)11−ベータ−ヒドロキシステロイドデヒドロゲナーゼ1(11−ベータ−HSD−1)の阻害剤(例えば、LY2523199、BMS770767、RG−4929、BMS816336、AZD−8329、HSD−016、BI−135585);
(y)グルコキナーゼの活性化剤(例えば、PF−04991532、TTP−399(GK1−399)、GKM−001(ADV−1002401)、ARRY−403(AMG−151)、TAK−329、TMG−123、ZYGK1);
(z)ジアシルグリセロールO−アシルトランスフェラーゼ(DGAT)の阻害剤(例えば、プラジガスタット(LCQ−908))、タンパク質チロシンホスファターゼ1の阻害剤(例えば、トロデュスクミン)、グルコース−6−ホスファターゼの阻害剤、フルクトース−1,6−ビスホスファターゼの阻害剤、グリコーゲンホスホリラーゼの阻害剤、ホスホエノールピルビン酸カルボキシキナーゼの阻害剤、グリコーゲンシンターゼキナーゼの阻害剤、ピルビン酸デヒドロゲナーゼキナーゼの阻害剤;
(aa)グルコーストランスポータ−4のモジュレータ、ソマトスタチン受容体3アゴニスト(例えば、MK−4256);
(bb)1つまたはそれ以上の脂質減少薬もまた組み合わせパートナーとして適している、例えば、3−ヒドロキシ−3−メチルグルタリル−コエンザイム−A−レダクターゼ(HMG−CoA−レダクターゼ)阻害剤、例えば、シンバスタチン(例えば、Zocor(登録商標)、Inegy(登録商標))、Simcor(登録商標))、アトルバスタチン(例えば、Sortis(登録商標)、Caduet(登録商標))、ロスバスタチン(例えば、Crestor(登録商標))、プラバスタチン(例えば、Lipostat(登録商標)、Selipran(登録商標))、フルバスタチン(例えば、Lescol(登録商標))、ピタバスタチン(例えば、Livazo(登録商標)、Livalo(登録商標))、ロバスタチン(例えば、Mevacor(登録商標)、Advicor(登録商標))、メバスタチン(例えば、Compactin(登録商標))、リバスタチン、セリバスタチン(Lipobay(登録商標))、フィブラート、例えば、ベザフィブラート(例えば、Cedur(登録商標)遅延)、シプロフィブラート(例えば、Hyperlipen(登録商標))、フェノフィブラート(例えば、Antara(登録商標)、Lipofen(登録商標)、リパンチル(登録商標))、ゲムフィブロジル(例えば、Lopid(登録商標)、Gevilon(登録商標))、エトフィブラート、シンフィブラート、ロニフィブラート、クリノフィブラート、クロフィブリド、ニコチン酸およびその誘導体(例えば、ナイアシンの徐放性製剤を含むナイアシンなど)、ニコチン酸受容体1アゴニスト(例えば、GSK−256073)、PPAR−deltaアゴニスト、アセチルCoA−アセチルトランスフェラーゼ(ACAT)阻害剤(例えば、avasimibe)、コレステロール吸収阻害剤(例えば、エゼチミブ、Ezetrol(登録商標)、Zetia(登録商標)、Liptruzet(登録商標)、Vytorin(登録商標)、S−556971)、胆汁酸結合物質(例えば、コレスチラミン、コレセベラム)、回腸胆汁酸輸送(IBAT)阻害剤(例えば、GSK−2330672、LUM−002)、ミクロソームトリグリセリド転移タンパク質(MTP)阻害剤(例えば、ロミタピド(AEGR−733)、SLx−4090、グラノタピド)、プロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)のモジュレータ(例えば、アリロクマブ(REGN727/SAR236553)、AMG−145、LGT−209、PF−04950615、MPSK3169A、LY3015014、ALD−306、ALN−PCS、BMS−962476、SPC5001、ISIS−394814、1B20、LGT−210、1D05、BMS−PCSK9Rx−2、SX−PCK9、RG7652)、LDL受容体アップレギュレーター、例えば、肝臓選択的甲状腺ホルモン受容体ベータアゴニスト(例えば、エピロチローム(KB−2115)、MB07811、ソベチロメム(QRX−431)、VIA−3196、ZYT1)、HDL上昇化合物タンパク質、例えば、コレステリルエステル転移タンパク質(CETP)阻害剤(例えば、アナセトラピブ(MK0859)、ダルセトラピブ、エバセトラピブ、JTT−302、DRL−17822、TA−8995、R−1658、LY−2484595、DS−1442)、またはデュアルCETP/PCSK9阻害剤(例えば、K−312)、ATP結合カセット(ABC1)レギュレーター、脂質代謝モジュレータ(例えば、BMS−823778、TAP−301、DRL−21994、DRL−21995)、ホスホリパーゼA2(PLA2)阻害剤(例えば、ダラプラディブ、Tyrisa(登録商標)、バレスプラディブ、リラプラディブ)、ApoA−Iエンハンサ(例えば、RVX−208、CER−001、MDCO−216、CSL−112)、コレステロール合成阻害剤(例えば、ETC−1002)、脂質代謝調節剤(例えば、BMS−823778、TAP−301、DRL−21994、DRL−21995)ならびにオメガ3脂肪酸およびその誘導体(例えば、イコサペントエチル(AMR101)、Epanova(登録商標)、AKR−063、NKPL−66、PRC−4016、CAT−2003);
(cc)ブロモクリプチン(例えば、Cycloset(登録商標)、Parlodel(登録商標))、フェンテルミンおよびフェンテルミン製剤または組み合わせ(例えば、Adipex−P、Ionamin、Qsymia(登録商標))、ベンズフェタミン(例えば、Didrex(登録商標))、ジエチルプロピオン(例えば、Tenuate(登録商標))、フェンジメトラジン(例えば、Adipost(登録商標))、Bontril(登録商標))、ブプロピオンおよび組み合わせ(例えば、Zyban(登録商標)、Wellbutrin XL(登録商標)、Contrave(登録商標)、Empatic(登録商標))、シブトラミン(例えば、Reductil(登録商標)、Meridia(登録商標))、トピラマート(例えば、Topamax(登録商標))、ゾニサミド(例えば、Zonegran(登録商標))、テソフェンシン、オピオイドアンタゴニスト、例えば、ナルトレキソン(例えば、Naltrexin(登録商標)、ナルトレキソン+ブプロピオン)、カンナビノイド受容体1(CB1)アンタゴニスト(例えば、TM−38837)、メラニン濃縮ホルモン(MCH−1)アンタゴニスト(例えば、BMS−830216、ALB−127158(a))、MC4受容体アゴニストおよび部分アゴニスト(例えば、AZD−2820、RM−493)、神経ペプチドY5(NPY5)またはNPY2アンタゴニスト(例えば、ベルネペリト、S−234462)、NPY4アゴニスト(例えば、PP−1420)、ベータ−3−アドレナリン受容体アゴニスト、レプチンまたはレプチン模倣薬、5−ヒドロキシトリプタミン2c(5HT2c)受容体のアゴニスト(例えば、ロカゼリン、Belviq(登録商標))、プラムリンチド/メトレレプチン、リパーゼ阻害剤、例えば、セチリスタット(例えば、Cametor(登録商標))、オルリスタット(例えば、Xenical(登録商標)、Calobalin(登録商標))、血管新生阻害剤(例えば、ALS−L1023)、ベタヒスチジンおよびヒスタミンH3アンタゴニスト(例えば、HPP−404)、AgRP(アグーチ関連タンパク質)阻害剤(例えば、TTP−435)、セロトニン再取り込み阻害剤、例えば、フルオキセチン(例えば、Fluctine(登録商標))、デュロキセチン(例えば、Cymbalta(登録商標))、デュアルまたはトリプルモノアミン取り込み阻害剤(ドーパミン、ノルエピネフリン、セロトニン再取り込み)、例えば、セルトラリン(例えば、Zoloft(登録商標))、テソフェンシン、メチオニンアミノペプチダーゼ2)(MetAP2)阻害剤(例えば、ベララニブ)、および線維芽細胞増殖因子受容体4(FGFR4)の産生に対するアンチセンスオリゴヌクレオチド(例えば、ISIS−FGFR4Rx)または禁止標的化ペプチド−1(例えば、Adipotide(登録商標));ならびに
(dd)酸化窒素ドナー、AT1アンタゴニストまたはアンギオテンシンII(AT2)受容体アンタゴニスト、例えば、テルミサルタン(例えば、Kinzal(登録商標)、Micardis(登録商標))、カンデサルタン(例えば、Atacand(登録商標)、Blopress(登録商標))、バルサルタン(例えば、Diovan(登録商標)、Co−Diovan(登録商標))、ロサルタン(例えば、Cosaar(登録商標))、エプロサルタン(例えば、Teveten(登録商標))、イルベサルタン(例えば、Aprovel(登録商標)、CoAprovel(登録商標))、オルメサルタン(例えば、Votum(登録商標)、Olmetec(登録商標))、タソサルタン、アジルサルタン(例えば、Edarbi(登録商標))、デュアルアンジオテンシン受容体遮断薬(デュアルARB)、アンジオテンシン変換酵素(ACE)阻害剤、ACE−2活性剤、レニン阻害剤、プロレニン阻害剤、エンドセリン変換酵素(ECE)阻害剤、エンドセリン受容体(ET1/ETA)遮断薬、エンドセリンアンタゴニスト、利尿薬、アルドステロンアンタゴニスト、アルドステロン合成酵素阻害薬、アルファ遮断薬、アルファ−2アドレナリン受容体のアンタゴニスト、ベータ遮断薬、混合アルファ/ベータ遮断薬、カルシウムアンタゴニスト、カルシウムチャネル遮断薬(CCB)、カルシウムチャネル遮断薬ジルチアゼムの鼻製剤(例えば、CP−404)、デュアルミネラルコルチコイド/CCB、中央作用性降圧剤、中性エンドペプチダーゼの阻害剤、アミノペプチダーゼ−A阻害剤、バソペプチド阻害剤、二重血管ペプチド阻害剤、例えばネプリライシン−ACE阻害剤またはネプリライシン−ECE阻害剤、二重作用性AT受容体−ネプリライシン阻害剤、二重AT1/ETAアンタゴニスト、高度糖化末端−製品(AGE)ブレーカー、組換えレナラーゼ、血圧ワクチン、例えば抗RAAS(レニン−アンジオテンシン−アルドステロン系)ワクチン、AT1−またはAT2−ワクチン、高血圧薬理ゲノミクスに基づく薬物、例えば高血圧反応を伴う遺伝的多型のモジュレータ、血小板凝集阻害剤、およびその他;ならびに
(ee)適切なそれらの組み合わせ。
According to certain embodiments, in addition to insulin therapy, administration of a PCSK9 inhibitor (eg, an anti-PCSK9 antibody, such as allilocumab, evolocumab, bococizumab, rodercizumab, ralpancizumab or LY301504) to the patient in combination with an additional antidiabetic therapy. A method including is provided. Exemplary additional antidiabetic therapies include, but are not limited to:
(A) All drugs listed in Rote Liste 2016 (eg, all anti-diabetic drugs listed in Chapter 12, Rote Liste 2014), all listed in Chapter 06, Rote Liste 2016 Weight loss agents or appetite suppressants, All Lipid-reducing drugs listed in Chapter 58, Rote Liste 2016, All antihypertensive agents listed in Chapter 17, Rote Liste 2016, All listed in Rote Liste Or a diuretic listed in Chapter 36, Rote Liste 2016;
(B) Glucagon-like peptide 1 (GLP-1) therapy including GLP-1, GLP-1 analogs, and GLP-1 receptor agonists, eg: GLP-1(7-37), GLP-1(7- 36) Amide, lixisenatide (eg Lyxumia®), exenatide (eg Exendin-4, rExendin-4, Byetta®, Bydureon®, exenatide NexP), exenatide-LAR, liraglutide. , Victoza (registered trademark), semaglutide, taspoglutide, albiglutide, dulaglutide, arbugon, oxyntomodulin, geniproside, ACP-003, CJC-1131, CJC-1134-PC, GSK-2374697, PB-1023, TTP-054. , Langlenatide (HM-11260C), CM-3, GLP-1 Eligen, AB-201, ORMD-0901, NN9924, NN9926, NN9927, Nodexen, Viador-GLP-1, CVX-096, ZYOG-1, ZYD-1. , ZP-3022, CAM-2036, DA-3091, DA-15864, ARI-2651, ARI-2255, Exenatide-XTEN(VRS-859), Exenatide-XTEN+Glucagon-XTEN(VRS-859+AMX-808) and polymer bond. GLP-1 and GLP-1 analogs;
(C) Dual GLP-1/GIP agonist (for example, RG-7697 (MAR-701), MAR-709, BHM081, BHM089, BHM098); Dual GLP-1/glucagon receptor agonist (for example, BHM-034, OAP -189 (PF-05212389, TKS-1225), TT-401/402, ZP2929, LAPS-HMOXM25, MOD-6030);
(D) dual GLP-1/gastrin agonist (eg ZP-3022);
(E) gastrointestinal peptides such as peptide YY3-36 (PYY3-36) or analogs thereof and pancreatic polypeptide (PP) or analogs thereof;
(F) Glucagon receptor agonist or antagonist, glucose-dependent insulinotropic polypeptide (GIP) receptor agonist or antagonist, ghrelin antagonist or inverse agonist, xenin and analogs thereof.
(G) dipeptidyl peptidase-IV (DPP-4) inhibitors, for example: alogliptin (eg Nesina (registered trademark), Kazano (registered trademark)), linagliptin (eg Ondero (registered trademark), Trajenta (registered trademark)). , Trajenta®, Trayenta®, saxagliptin (eg, Onglyza®, Komboglyze XR®), sitagliptin (eg, Januvia®, Xelevia®, Tesave). (Registered trademark), Janumet (registered trademark), Velmetia (registered trademark), Juvissync (registered trademark), JanumetXR (registered trademark), anagliptin, teneligliptin (for example, Tenelia (registered trademark)), trellagliptin, vildagliptin (for example, Galvus (registered trademark)). Registered trademark), Galvumet (registered trademark)), gemigliptin, omalipliptin, evogliptin, dutogliptin, DA-1229, MK-3102, KM-223, KRP-104, PBL-1427, pinoxacin hydrochloride, and Ari-2243;
(H) Sodium-dependent glucose transporter 2 (SGLT-2) inhibitors, for example: canagliflozin, dapagliflozin, remogliflozin, remogliflozin etabonate, sergliflozin, empagliflozin, ipragliflozin, tofogliflozin, Luceogliflozin, Ertugliflozin, EGT-0001442, LIK-066, SBM-TFC-039, and KGA-3235 (DSP-3235);
(I) dual inhibitors of SGLT-2 and SGLT-1 (eg LX-4211, LIK066);
(J) SGLT-in combination with an antiobesity agent such as an SGLT-1 inhibitor (eg, LX-2761, KGA-3235, etc.) or an ileal bile acid transfer (IBAT) inhibitor (eg, GSK-1614235+GSK-2330672). 1 inhibitor;
(K) biguanides (eg, metformin, buformin, phenformin);
(L) thiazolidinediones (eg, pioglitazone, rosiglitazone), glitazone analogs (eg, lobeglitazone);
(M) Peroxisome proliferator-activated receptor (PPAR-) (alpha, gamma or alpha/gamma) agonists or modulators (e.g. salogritazar (e.g. Lipagulin(R)), GFT-505), or PPAR gamma partial agonists. (For example, Int-131);
(N) Sulfonylureas (eg tolbutamide, glibenclamide, glimepiride, amaryl®, glipizide) and meglitinides (eg nateglinide, repaglinide, mitiglinide);
(O) alpha-glucosidase inhibitors (eg acarbose, miglitol, voglibose);
(Q) amylin and amylin analogues (eg pramlintide, Symlin®);
(P) G protein-coupled receptor 119 (GPR119) agonist (for example, GSK-1292263, PSN-821, MBX-2982, APD-597, ARRY-981, ZYG-19, DS-8500, HM-47000, YH-). Chem1);
(Q) GPR40 agonist (for example, TUG-424, P-1736, P-11187, JTT-851, GW9508, CNX-011-67, AM-1638, AM-5262);
(R) a GPR120 agonist and a GPR142 agonist;
(S) Systemic or low-absorbing TGR5 (GPBAR1=G protein-coupled bile acid receptor 1) agonist (eg, INT-777, XL-475, SB756050);
(T) Diabetic immunotherapeutic agents, for example: oral CC chemokine receptor type 2 (CCR-2) antagonist (for example CCX-140, JNJ-41434352),
(V) anti-inflammatory agents for the treatment of metabolic syndrome and diabetes, eg: nuclear factor kappa B inhibitors (eg Triolex®);
(W) adenosine monophosphate-activated protein kinase (AMPK) stimulants, for example: imeglymine (PXL-008), debio-0930 (MT-63-78), R-118;
(X) inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 (11-beta-HSD-1) (e.g., LY2523199, BMS770767, RG-4929, BMS816336, AZD-8329, HSD-016, BI-135585);
(Y) Activator of glucokinase (for example, PF-04991532, TTP-399 (GK1-399), GKM-001 (ADV-1002401), ARRY-403 (AMG-151), TAK-329, TMG-123. , ZYGK1);
(Z) an inhibitor of diacylglycerol O-acyltransferase (DGAT) (for example, prazistat (LCQ-908)), an inhibitor of protein tyrosine phosphatase 1 (for example, troduscumin), an inhibitor of glucose-6-phosphatase, An inhibitor of fructose-1,6-bisphosphatase, an inhibitor of glycogen phosphorylase, an inhibitor of phosphoenolpyruvate carboxykinase, an inhibitor of glycogen synthase kinase, an inhibitor of pyruvate dehydrogenase kinase;
(Aa) a modulator of glucose transporter-4, a
(Bb) one or more lipid-lowering agents are also suitable as combination partners, for example 3-hydroxy-3-methylglutaryl-coenzyme-A-reductase (HMG-CoA-reductase) inhibitors, for example: Simvastatin (eg, Zocor®, Inegy®), Simcor®), atorvastatin (eg Sortis®, Caduet®), rosuvastatin (eg Crestor®). ), pravastatin (eg, Lipostat®, Seliplan®), fluvastatin (eg, Lescol®), pitavastatin (eg, Livazo®, Livalo®), lovastatin (). For example, Mevacor®, Advicor®), mevastatin (eg Compactin®), rivastatin, cerivastatin (Lipobay®), fibrates, eg bezafibrate (eg Cedur®). Delay), ciprofibrate (e.g. Hyperlipen(R)), fenofibrate (e.g. Antara(R), Lipofen(R), Ripanchir(R)), gemfibrozil (e.g. Lipoid(R), Geviron. (Registered trademark), etofibrate, synfibrate, ronifibrate, clinofibrate, clofibride, nicotinic acid and its derivatives (such as niacin including a sustained-release preparation of niacin),
(Cc) Bromocriptine (eg Cycloset®, Parlodel®), phentermine and phentermine formulations or combinations (eg Adipex-P, Ionamine, Qsymia®), benzphetamine (eg Didrex(TM)). )), diethylpropion (eg Tenuate®), phendimetrazine (eg Adipost®), Bontril®), bupropion and combinations (eg Zyban®), Wellbutrin XL (registered trademark), Contrave (registered trademark), Empatic (registered trademark)), sibutramine (for example, Reductil (registered trademark), Merida (registered trademark)), topiramate (for example, Topamax (registered trademark)), zonisamide (registered trademark). For example, Zonegran®), tesofensine, opioid antagonists such as naltrexone (eg Naltrexin®, naltrexone+bupropion), cannabinoid receptor 1 (CB1) antagonists (eg TM-38837), melanin-concentrating hormone. (MCH-1) antagonists (eg BMS-830216, ALB-127158(a)), MC4 receptor agonists and partial agonists (eg AZD-2820, RM-493), neuropeptide Y5 (NPY5) or NPY2 antagonists (eg. For example, berneperito, S-234462), NPY4 agonists (eg PP-1420), beta-3-adrenergic receptor agonists, leptin or leptin mimetics, 5-hydroxytryptamine 2c (5HT2c) receptor agonists (eg locazelin). , Belviq®), pramlintide/metreleptin, lipase inhibitors such as cetilistat (eg Cametor®), orlistat (eg Xenical®, Calobalin®), angiogenesis inhibitors. (E.g. ALS-L1023), betahistidine and histamine H3 antagonists (e.g. HPP-404), AgRP (agouti-related protein) inhibitors (e.g. TTP-435), serotonin reuptake inhibitors e.g. fluoxetine (e.g. Fluctine (registered trademark), duloxet (Eg Cymbalta®), dual or triple monoamine uptake inhibitors (dopamine, norepinephrine, serotonin reuptake), eg sertraline (eg Zoloft®), tesofensine, methionine aminopeptidase 2) (MetAP2). ) Inhibitors (eg belaranib), and antisense oligonucleotides (eg ISIS-FGFR4Rx) or the production of fibroblast growth factor receptor 4 (FGFR4) or prohibited targeting peptide-1 (eg Adipotide®). ); and (dd) nitric oxide donors, AT1 antagonists or angiotensin II (AT2) receptor antagonists such as telmisartan (eg Kinzal®, Micardis®), candesartan (eg Atacand®). , Blopress(R), Valsartan (e.g. Diovan(R), Co-Diovan(R)), Losartan (e.g. Cosaar(R)), Eprosartan (e.g. Teveten(R)), Irbesartan (eg, Aprovel®, CoAprovel®), Olmesartan (eg, Votum®, Olmetec®), Tasosartan, Azilsartan (eg, Edarbi®), Dual Angiotensin Receptor blocker (dual ARB), angiotensin converting enzyme (ACE) inhibitor, ACE-2 activator, renin inhibitor, prorenin inhibitor, endothelin converting enzyme (ECE) inhibitor, endothelin receptor (ET1/ETA) blockade Drugs, endothelin antagonists, diuretics, aldosterone antagonists, aldosterone synthase inhibitors, alpha blockers, alpha-2 adrenergic receptor antagonists, beta blockers, mixed alpha/beta blockers, calcium antagonists, calcium channel blockers (CCB ), a nasal formulation of the calcium channel blocker diltiazem (eg CP-404), dual mineralocorticoid/CCB, centrally acting antihypertensive agents, inhibitors of neutral endopeptidases, aminopeptidase-A inhibitors, vasopeptide inhibitors, Heavy vascular peptide inhibitors such as neprilysin-ACE inhibitor or neprilysin-ECE inhibitor, dual action Sex AT receptor-neprilysin inhibitor, dual AT1/ETA antagonist, advanced glycation end-product (AGE) breaker, recombinant renalase, blood pressure vaccine, eg anti-RAAS (renin-angiotensin-aldosterone system) vaccine, AT1- or AT2 -Vaccines, drugs based on hypertensive pharmacogenomics, such as modulators of genetic polymorphisms associated with hypertensive responses, platelet aggregation inhibitors, and others; and (ee) suitable combinations thereof.
特定の実施形態では、追加の抗糖尿病療法は、GLP−1療法(例えば、リキシセナチド)である。特定の実施形態では、GLP−1療法は、メチオニン(例えば、L−メチオニンまたはD−メチオニン)とともに製剤化される。特定の実施形態では、ポリソルベート(例えば、ポリソルベート20、ポリソルベート80)、ポロキサマー(例えば、ポロキサマー188)、塩化ベンザルコニウム、ヒスチジン、リジン、および/またはEDTAは、GLP−1療法の製剤に存在しないか、または実質的に存在しない。特定の実施形態では、GLP−1療法の製剤は、界面活性剤、例えば、ポリオール(例えば、ポリプロピレングリコール、ポリエチレングリコール、ポロキサマー、プルロニック、テトロニクス)、部分的および脂肪酸エステルならびに多価アルコールのエステル、例えば、グリセロールおよびソルビトールのエステル(例えば、Span(登録商標)、Tween(登録商標)、Myrj(登録商標)、Brij(登録商標)、Cremophor(登録商標))を含まないか、または実質的に含まない。GLP−1療法の製剤は、適切な防腐剤(例えば、フェノール、m−クレゾール、ベンジルアルコール、および/またはp−ヒドロキシ安息香酸エステル)および適切な張度調整剤(例えば、グリセロール、デキストロース、ラクトース、ソルビトール、マンニトール、グルコース、NaCl、カルシウムまたはマグネシウム化合物、例えば、CaCl2)を含むことができる。グリセロール、デキストロース、ラクトース、ソルビトール、マンニトール、およびグルコースの濃度は通常、100〜250mMの範囲であり、NaClは最大150mMの濃度である。
In certain embodiments, the additional antidiabetic therapy is GLP-1 therapy (eg, lixisenatide). In certain embodiments, GLP-1 therapy is formulated with methionine (eg, L-methionine or D-methionine). In certain embodiments, is polysorbate (eg,
特定の実施形態では、患者が受けるインスリン療法は、追加の抗糖尿病療法(例えば、インスリン療法ではない前述の抗糖尿病療法のいずれか)と組み合わされる。例えば、特定の実施形態では、抗糖尿病療法は、インスリン療法(例えば、インスリングラルギン)とGLP−1療法(例えば、リキシセナチド)の組み合わせを含む。これらの療法は、個別に、または単一の医薬組成物で提供することができる。例えば、インスリングラルギンおよびリキシセナチドは、毎日の注射用に単一の医薬組成物(例えば、Soliqua(登録商標)100/33)に製剤化することができる。
In certain embodiments, the insulin therapy that the patient receives is combined with an additional anti-diabetic therapy (eg, any of the aforementioned anti-diabetic therapies that is not insulin therapy). For example, in certain embodiments, antidiabetic therapy comprises a combination of insulin therapy (eg, insulin glargine) and GLP-1 therapy (eg, lixisenatide). These therapies can be provided individually or in a single pharmaceutical composition. For example, insulin glargine and lixisenatide can be formulated into a single pharmaceutical composition (eg,
本方法との関連では、追加の治療活性成分、例えば、上記に列挙された薬剤またはその誘導体のいずれかは、PCSK9阻害剤の投与の直前、同時、または直後に投与される;(本開示の目的のために、このような投与レジメンは、追加の治療活性成分と「組み合わせた」PCSK9阻害剤の投与とみなされる)。本方法は、PCSK9阻害剤が本明細書の他の場所に記載される追加の治療活性成分の1つまたはそれ以上と同時に製剤化される医薬組成物およびその使用方法を含む。 In the context of this method, the additional therapeutically active ingredient, eg, any of the agents listed above or derivatives thereof, is administered immediately prior to, concurrently with, or shortly after administration of the PCSK9 inhibitor; For purposes of such administration, such a dosing regimen is considered as administration of a PCSK9 inhibitor "in combination with" an additional therapeutically active ingredient). The methods include pharmaceutical compositions and methods of use thereof in which a PCSK9 inhibitor is co-formulated with one or more of the additional therapeutically active ingredients described elsewhere herein.
アドオン療法としてのPCSK9阻害剤の投与
本治療方法は、高コレステロール血症および糖尿病を有する患者を、PCKS9に特異的に結合する抗体またはその抗原結合断片などのPCSK9阻害剤で治療することを含み、PCSK9阻害剤は、患者の既存の毎日の治療用インスリンおよび/またはスタチンレジメンへのアドオンなどの、患者の既存のインスリン療法および/またはLMT(該当する場合)へのアドオンとして投与することができる。
Administration of PCSK9 Inhibitors as Add-on Therapy The method of treatment comprises treating a patient with hypercholesterolemia and diabetes with a PCSK9 inhibitor, such as an antibody that specifically binds to PCKS9 or an antigen-binding fragment thereof, The PCSK9 inhibitor can be administered as an add-on to the patient's existing insulin therapy and/or LMT (if applicable), such as an add-on to the patient's existing daily therapeutic insulin and/or statin regimen.
例えば、本方法は、アドオン療法レジメンを含み、PCSK9阻害剤は、同じ安定した複数の毎日のインスリンレジメンおよび/または毎日の治療用スタチンレジメン(すなわち、スタチンと同じ投薬量)へのアドオン療法として投与され、患者はPCSK9阻害剤を受ける前であった。他の実施形態では、PCSK9阻害剤は、インスリンおよび/またはスタチンの投薬量より多いまたは少ない量のインスリンおよび/またはスタチンを含む治療用インスリンおよび/またはスタチンレジメンへの追加療法として投与され、患者はPCSK9阻害剤を投与する前であった。例えば、特定の投薬頻度および投薬量で投与されるPCSK9阻害剤を含む治療レジメンを開始した後、患者に投与または処方されるインスリンおよび/またはスタチンの1日用量は、PCSK9阻害剤治療レジメンを開始する前に患者が服用していた1日のスタチン用量と比較して、患者の治療ニーズに応じて、(a)同じままである、(b)増加する、または(c)減少する(例えば、漸増または漸減する)可能性がある。 For example, the method includes an add-on therapy regimen, wherein the PCSK9 inhibitor is administered as an add-on therapy to the same stable multiple daily insulin regimen and/or daily therapeutic statin regimen (ie, the same dosage as the statin). And the patient was prior to receiving the PCSK9 inhibitor. In other embodiments, the PCSK9 inhibitor is administered as an add-on therapy to a therapeutic insulin and/or statin regimen that includes an insulin and/or statin in an amount greater or less than the insulin and/or statin dosage. Before administration of the PCSK9 inhibitor. For example, after initiating a therapeutic regimen comprising a PCSK9 inhibitor administered at a particular dosing frequency and dosage, a daily dose of insulin and/or statin administered or prescribed to a patient initiates a PCSK9 inhibitor therapeutic regimen. (A) stays the same, (b) increases, or (c) decreases compared to the daily statin dose that the patient was taking prior to taking (eg, Gradually increase or decrease).
治療効果
本方法は、LDL−C、ApoB、ApoB100、非HDL−C、総コレステロール、VLDL−C、トリグリセリド、Lp(a)、HDL−C、LDL粒子数、LDL粒子サイズ、ApoC3、ApoA−1、トリグリセリドに富むリポタンパク質コレステロール(TRL−C)、および残留コレステロールからなる群から選択される1つまたはそれ以上の脂質成分の血清レベルの減少をもたらす。特定の実施形態によれば、PCSK9阻害剤を含む医薬組成物の患者への投与は、少なくとも約25%、30%、35%、40%、45%、50%、55%、60%、またはそれ以上の血清低密度リポタンパク質コレステロール(LDL−C)のベースラインからの平均パーセントの減少;少なくとも約25%、30%、35%、40%、45%、50%、55%、60%、またはそれ以上のApoBのベースラインからの平均パーセントの減少;少なくとも約25%、30%、35%、40%、45%、50%、55%、60%、またはそれ以上のApoB100のベースラインからの平均パーセントの減少;少なくとも約25%、30%、35%、40%、45%、50%、55%、60%、またはそれ以上の非HDL−Cのベースラインからの平均パーセントの減少;少なくとも約10%、15%、20%、25%、30%、35%、40%、またはそれ以上の総コレステロールのベースラインからの平均パーセントの減少;少なくとも約5%、10%、15%、20%、25%、30%、またはそれ以上のVLDL−Cのベースラインからの平均パーセントの減少;少なくとも約5%、10%、15%、20%、25%、30%、35%、またはそれ以上のトリグリセリドのベースラインからの平均パーセントの減少;少なくとも約20%、25%、30%、35%、40%、45%、50%、またはそれ以上のLDL粒子の数のベースラインからの平均パーセントの減少;少なくとも約1.5%、2%、2.5%、3%、3.5%もしくは4%、またはそれ以上のLDL粒子のサイズのベースラインからの平均パーセントの減少;少なくとも約5%、5.5%、6.0%、6.5%、7.0%、7.5%、8.0%、9.0%、10%、またはそれ以上のアポリポタンパク質C3(ApoC3)のベースラインからの平均パーセントの減少;少なくとも約1%、2%、3%、4%、5%、またはそれ以上のHDL−Cのベースラインからの平均パーセントの増加;少なくとも約1%、2%、3%、4%、5%、またはそれ以上のApoA−1のベースラインからの平均パーセントの増加;少なくとも約5%、10%、15%、20%、25%、30%、またはそれ以上のTRL−Cのベースラインからの平均パーセントの減少;および/または少なくとも約5%、10%、15%、20%、25%、またはそれ以上のLp(a)のベースラインからの平均パーセントの減少をもたらす。
Therapeutic effect The present method comprises LDL-C, ApoB, ApoB100, non-HDL-C, total cholesterol, VLDL-C, triglyceride, Lp(a), HDL-C, LDL particle number, LDL particle size, ApoC3, ApoA-1. , Lipoprotein cholesterol rich in triglycerides (TRL-C), and a decrease in serum levels of one or more lipid components selected from the group consisting of residual cholesterol. According to certain embodiments, administering to the patient a pharmaceutical composition comprising a PCSK9 inhibitor is at least about 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, or Further reduction in mean percentage of serum low density lipoprotein cholesterol (LDL-C) from baseline; at least about 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, Or more average percent reduction from baseline of ApoB; at least about 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, or more of baseline ApoB100 An average percent reduction from non-HDL-C baseline of at least about 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, or more; At least about 10%, 15%, 20%, 25%, 30%, 35%, 40%, or more, an average percent reduction in total cholesterol from baseline; at least about 5%, 10%, 15%, 20%, 25%, 30%, or more, an average percent reduction from baseline of VLDL-C; at least about 5%, 10%, 15%, 20%, 25%, 30%, 35%, or A greater average percent reduction in triglyceride from baseline; from a baseline number of LDL particles of at least about 20%, 25%, 30%, 35%, 40%, 45%, 50%, or more. Average percent reduction; at least about 1.5%, 2.5%, 2.5%, 3%, 3.5% or 4%, or more, average percent reduction from baseline in LDL particle size; at least About 5%, 5.5%, 6.0%, 6.5%, 7.0%, 7.5%, 8.0%, 9.0%, 10%, or more apolipoprotein C3( ApoC3) decrease in mean percent from baseline; increase in mean percent from baseline of HDL-C of at least about 1%, 2%, 3%, 4%, 5% or more; at least about 1% 2%, 3%, 4%, 5%, or more increase in mean percentage from baseline of ApoA-1; at least about 5%, 10%, 15%, 20%, 25%, 30%, Or more reduction in average percent of TRL-C from baseline; and/or at least about 5%, 10%, 15%, 20%, 25%, or more. Results in an average percent reduction from baseline of Lp(a) above.
本方法は、インスリン療法を受けている高コレステロール血症およびT1DMを有する患者を治療することを含み、本方法は、約75〜150mg/用量の投薬量、約2週間ごともしくは4週間ごとに約1回の投薬頻度、または本明細書に開示される漸増投薬レジメンに従った投薬レジメンで、患者に複数用量の抗PCSK9抗体またはその抗原結合断片を投与することを含む。抗PCSK9抗体による治療の約8、10、12、14、16、18、20、22、24週間以上の後、患者は、ベースラインから少なくとも35%、50%、または60%のLDL−Cレベルの減少を示し得る。特定の実施形態では、抗PCSK9抗体による1週間またはそれ以上の治療の後、患者は、ベースラインから約35%、50%、または60%以上のLDL−Cレベルの減少を示す。 The method comprises treating a patient with hypercholesterolemia and T1DM undergoing insulin therapy, the method comprising a dosage of about 75-150 mg/dose, about every 2 or 4 weeks. Administering multiple doses of anti-PCSK9 antibody or antigen-binding fragment thereof to a patient at a single dosing frequency, or in a dosing regimen according to the escalating dosing regimen disclosed herein. After about 8,10,12,14,16,18,20,22,24 or more weeks of treatment with anti-PCSK9 antibody, patients have at least 35%, 50%, or 60% LDL-C levels from baseline. Can show a decrease in In certain embodiments, after treatment with anti-PCSK9 antibody for one week or more, the patient exhibits a decrease in LDL-C levels of about 35%, 50%, or 60% or more from baseline.
本方法はまた、インスリン療法を受けている高コレステロール血症およびT2DMを有する患者を治療することを含み、本方法は、約75〜150mg/用量の投薬量、約2週間ごともしくは4週間ごとに約1回の投薬頻度、または本明細書に開示される漸増投薬レジメンに従った投薬レジメンで、患者に複数用量の抗PCSK9抗体またはその抗原結合断片を投与することを含む。抗PCSK9抗体による治療の約8、10、12、14、16、18、20、22、24週間以上の後、患者は、ベースラインから少なくとも40%、48%、または54%のLDL−Cレベルの減少を示し得る。特定の実施形態では、抗PCSK9抗体による1週間またはそれ以上の治療の後、患者は、ベースラインから約40%、48%、または54%以上のLDL−Cレベルの減少を示す。 The method also includes treating a patient with hypercholesterolemia and T2DM undergoing insulin therapy, the method comprising: a dosage of about 75-150 mg/dose, about every 2 or 4 weeks. Comprising administering to the patient multiple doses of an anti-PCSK9 antibody or antigen-binding fragment thereof, at a dosing frequency of about once, or a dosing regimen according to the escalating dosing regimen disclosed herein. After about 8, 10, 12, 14, 16, 18, 20, 22, 24 or more weeks of treatment with anti-PCSK9 antibody, patients have LDL-C levels of at least 40%, 48%, or 54% from baseline. Can show a decrease in In certain embodiments, after treatment with anti-PCSK9 antibody for one week or more, the patient exhibits a decrease in LDL-C levels of about 40%, 48%, or 54% or more from baseline.
本明細書に開示されるように、本方法は、患者の糖尿病パラメータを変更しない。例えば、特定の実施形態では、本方法は、患者のヘモグロビンA1c(HbA1c)レベルに影響を及ぼさない(例えば、1%、2%、3%、4%、5%、6%、7%、8%、9%、または10%を超えて変化しない)。特定の実施形態では、本方法は、患者の空腹時血漿グルコース(FPG)レベルに影響を及ぼさない(例えば、2%、4%、6%、8%、10%、12%、15%、18%、または20%を超えて変化しない)。 As disclosed herein, the method does not alter the patient's diabetes parameters. For example, in certain embodiments, the method does not affect the patient's hemoglobin A1c (HbA1c) levels (eg, 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8). %, 9%, or no more than 10%). In certain embodiments, the method does not affect fasting plasma glucose (FPG) levels in the patient (eg, 2%, 4%, 6%, 8%, 10%, 12%, 15%, 18). %, or does not change by more than 20%).
さらなる実施形態では、本発明は、1型真性糖尿病(T1DM)を有する患者の高コレステロール血症を治療するための、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片の使用に関する。
In a further embodiment, the invention provides an antibody that specifically binds to the human proprotein convertase subtilisin/kexin type 9 (PCSK9) for treating hypercholesterolemia in a patient with
なおさらなる実施形態では、本発明は、1型真性糖尿病(T1DM)を有する患者における高コレステロール血症を治療する方法に関する。
In yet a further embodiment, the invention relates to a method of treating hypercholesterolemia in a patient with
一実施形態では、上記使用および/または方法は、
(a)インスリン療法を受けている心血管リスクの高い患者であって、
(i)T1DM、および
(ii)最大耐性のスタチン療法によっては適切に制御されない高コレステロール血症
を有する患者を選択する工程;ならびに
(b)患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片を75mg、150mgもしくは300mg投与する工程
を含み、患者は付随するインスリン療法を受ける。
In one embodiment, the use and/or method comprises
(A) a patient at high cardiovascular risk receiving insulin therapy,
(I) selecting a patient with hypercholesterolemia that is not adequately controlled by T1DM, and (ii) maximally resistant statin therapy; Administering 75 mg, 150 mg or 300 mg of an antibody or antigen-binding fragment thereof that specifically binds to PCSK9), the patient undergoing concomitant insulin therapy.
上記使用および/または方法の一実施形態では、75mgの抗体または抗原結合断片は2週間ごとに患者に投与される。 In one embodiment of the above uses and/or methods, 75 mg of antibody or antigen binding fragment is administered to the patient every two weeks.
上記使用および/または方法の一実施形態では、150mgの抗体または抗原結合断片は2週間ごとに患者に投与される。 In one embodiment of the above uses and/or methods, 150 mg of antibody or antigen binding fragment is administered to the patient every two weeks.
上記使用および/または方法の一実施形態では、300mgの抗体または抗原結合断片は4週間ごとに患者に投与される。 In one embodiment of the above uses and/or methods, 300 mg of antibody or antigen binding fragment is administered to the patient every 4 weeks.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、配列番号2、3、および4に記載の3つの重鎖CDR、ならびに配列番号7、8、および10に記載の3つの軽鎖CDRを含む。 In one embodiment of the above uses and/or methods, the antibody or antigen-binding fragment thereof comprises the three heavy chain CDRs set forth in SEQ ID NOs:2, 3, and 4, and the 3 set forth in SEQ ID NOs:7, 8 and 10. Contains one light chain CDR.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有する重鎖可変領域(HCVR)および配列番号6のアミノ酸配列を有する軽鎖可変領域(LCVR)を含む。 In one embodiment of the above use and/or method, the antibody or antigen binding fragment thereof comprises a heavy chain variable region (HCVR) having the amino acid sequence of SEQ ID NO:1 and a light chain variable region (LCVR) having the amino acid sequence of SEQ ID NO:6. )including.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、アリロクマブ、エボロクマブ、ボコシズマブ、ロデルシズマブ、ラルパンシズマブおよびLY3015014からなる群から選択される In one embodiment of the use and/or method above, the antibody or antigen-binding fragment thereof is selected from the group consisting of allilocoumab, evolocumab, bococizumab, rodercizumab, ralpancizumab and LY301504.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片はアリロクマブである。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof is arilocumab.
一実施形態では、上記使用および/または方法は、
(c)患者におけるLDL−Cレベルが閾値レベルより低い場合、約2週間ごとに1回またはそれ以上の次の投薬量の75mgの抗体もしくはその抗原結合断片を患者に投与するか、または患者におけるLDL−Cレベルが閾値レベル以上の場合、約2週間ごとに1回またはそれ以上の次の投薬量の150mgの抗体もしくはその抗原結合断片を投与する工程
をさらに含む。
In one embodiment, the use and/or method comprises
(C) if the LDL-C level in the patient is below the threshold level, the patient is administered with one or more subsequent doses of 75 mg of the antibody or antigen-binding fragment thereof about once every two weeks, or in the patient If the LDL-C level is above the threshold level, the method further comprises the step of administering one or more subsequent doses of 150 mg of the antibody or antigen-binding fragment thereof about once every two weeks.
一実施形態では、上記使用および/または方法は、
(c)患者におけるLDL−Cレベルが閾値レベルより低い場合、約4週間ごとに1回またはそれ以上の次の投薬量の300mgの抗体もしくはその抗原結合断片を患者に投与するか、または患者におけるLDL−Cレベルが閾値レベル以上の場合、約2週間ごとに1回またはそれ以上の次の投薬量の150mgの抗体もしくはその抗原結合断片を投与する工程
をさらに含む。
In one embodiment, the use and/or method comprises
(C) if the LDL-C level in the patient is below a threshold level, the patient is administered with one or more subsequent doses of 300 mg of the antibody or antigen-binding fragment thereof about once every four weeks, or in the patient If the LDL-C level is above the threshold level, the method further comprises the step of administering one or more subsequent doses of 150 mg of the antibody or antigen-binding fragment thereof about once every two weeks.
上記使用および/または方法の一実施形態では、閾値レベルは70mg/dLである。 In one embodiment of the uses and/or methods above, the threshold level is 70 mg/dL.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は皮下投与される。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof is administered subcutaneously.
上記使用および/または方法の一実施形態では、患者は、付随する脂質修飾療法(LMT)をさらに受ける。 In one embodiment of the above uses and/or methods, the patient further undergoes concomitant lipid modification therapy (LMT).
上記使用および/または方法の一実施形態では、LMTは、スタチン、コレステロール吸収阻害剤、フィブラート、ナイアシン、オメガ−3脂肪酸、および胆汁酸封鎖剤からなる群から選択される。 In one embodiment of the use and/or method above, the LMT is selected from the group consisting of statins, cholesterol absorption inhibitors, fibrates, niacin, omega-3 fatty acids, and bile sequestrants.
上記使用および/または方法の一実施形態では、LMTはスタチン療法である。 In one embodiment of the above uses and/or methods, LMT is statin therapy.
上記使用および/または方法の一実施形態では、スタチンは、アトルバスタチン、ロスバスタチン、シンバスタチン、プラバスタチン、ロバスタチン、フルバスタチン、ピタバスタチン、およびセリバスタチンからなる群から選択される。 In one embodiment of the use and/or method above, the statin is selected from the group consisting of atorvastatin, rosuvastatin, simvastatin, pravastatin, lovastatin, fluvastatin, pitavastatin, and cerivastatin.
上記使用および/または方法の一実施形態では、スタチン療法が最大耐性のスタチン療法である。 In one embodiment of the above uses and/or methods, the statin therapy is the most resistant statin therapy.
上記使用および/または方法の一実施形態では、コレステロール吸収阻害剤はエゼチミブである。 In one embodiment of the above uses and/or methods, the cholesterol absorption inhibitor is ezetimibe.
上記使用および/または方法の一実施形態では、患者はスタチンにして不耐性である。 In one embodiment of the above uses and/or methods, the patient is statin intolerant.
上記使用および/または方法の一実施形態では、インスリン療法は、ヒトインスリン、インスリングラルギン、インスリングルリジン、インスリンデテミル、インスリンリスプロ、インスリンデグルデック、インスリンアスパルト、および基礎インスリンからなる群から選択される。 In one embodiment of the use and/or method above, the insulin therapy is selected from the group consisting of human insulin, insulin glargine, insulin glulisine, insulin detemir, insulin lispro, insulin degludec, insulin aspart, and basal insulin. It
上記使用および/または方法の一実施形態では、患者は、インスリン療法に加えて付随する抗糖尿病療法を受ける。 In one embodiment of the above uses and/or methods, the patient receives insulin therapy along with concomitant antidiabetic therapy.
上記使用および/または方法の一実施形態では、追加の付随する抗糖尿病療法は、グルカゴン様ペプチド1(GLP−1)療法、胃腸ペプチド、グルカゴン受容体アゴニストまたはアンタゴニスト、グルコース依存性インスリン分泌性ポリペプチド(GIP)受容体アゴニストまたはアンタゴニスト、グレリンアンタゴニストまたは逆アゴニスト、キセニン、キセニン類似体、ビグアナイド、スルホニル尿素、メグリチニド、チアゾリジンジオン、DPP−4阻害剤、アルファ−グルコシダーゼ阻害剤、ナトリウム依存性グルコース輸送体2(SGLT−2)阻害剤、SGLT−1阻害剤、ペルオキシソーム増殖因子活性化受容体(PPAR−)(アルファ、ガンマまたはアルファ/ガンマ)アゴニストまたはモジュレータ、アミリン、アミリンアナログ、Gタンパク質共役受容体119(GPR119)アゴニスト、GPR40アゴニスト、GPR120アゴニスト、GPR142アゴニスト、全身または低吸収性TGR5アゴニスト、糖尿病免疫療法薬、メタボリックシンドロームおよび糖尿病を治療するための抗炎症薬、アデノシンモノプリン酸活性化プロテインキナーゼ(AMPK)刺激薬、11−ベータ−ヒドロキシステロイドデヒドロゲナーゼ1の阻害剤、グルコキナーゼの活性化剤、ジアシルグリセロールO−アシルトランスフェラーゼ(DGAT)の阻害剤、グルコーストランスポータ−4のモジュレータ、ソマトスタチン受容体3アゴニスト、脂質減少剤、ならびにそれらの組み合わせからなる群から選択される。
In one embodiment of the above uses and/or methods, the additional concomitant antidiabetic therapy is glucagon-like peptide 1 (GLP-1) therapy, gastrointestinal peptide, glucagon receptor agonist or antagonist, glucose-dependent insulinotropic polypeptide. (GIP) receptor agonist or antagonist, ghrelin antagonist or inverse agonist, xenin, xenin analogue, biguanide, sulfonylurea, meglitinide, thiazolidinedione, DPP-4 inhibitor, alpha-glucosidase inhibitor, sodium-dependent glucose transporter 2 (SGLT-2) inhibitor, SGLT-1 inhibitor, peroxisome proliferator-activated receptor (PPAR-) (alpha, gamma or alpha/gamma) agonist or modulator, amylin, amylin analog, G protein coupled receptor 119 ( GPR119) Agonist, GPR40 Agonist, GPR120 Agonist, GPR142 Agonist, Systemic or Low Absorption TGR5 Agonist, Diabetic Immunotherapeutic, Anti-inflammatory Drug for Treating Metabolic Syndrome and Diabetes, Adenosine Monopurinate Activated Protein Kinase (AMPK) Stimulants, 11-beta-
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、患者のLDL−Cレベルを少なくとも30%、35%、40%、または45%減少させる。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof reduces LDL-C levels in the patient by at least 30%, 35%, 40%, or 45%.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、患者の非HDL−Cレベルを少なくとも25%、30%、35%、もしくは40%減少させる。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof reduces non-HDL-C levels in the patient by at least 25%, 30%, 35%, or 40%.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、患者のアポリポタンパク質C3(ApoC3)レベルを減少させる。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof reduces apolipoprotein C3 (ApoC3) levels in the patient.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、患者におけるリポタンパク質粒子の数および/またはサイズを減少させる。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof reduces the number and/or size of lipoprotein particles in the patient.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は:
(a)患者のヘモグロビンA1c(HbA1c)レベルに影響を及ぼさない;および/または
(b)患者の空腹時血漿グルコース(FPG)レベルに影響を及ぼさない。
In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof is:
(A) does not affect the patient's hemoglobin A1c (HbA1c) levels; and/or (b) does not affect the patient's fasting plasma glucose (FPG) levels.
さらなる実施形態では、本発明は、1型真性糖尿病(T1DM)を有する患者における高コレステロール血症を治療するための使用および/または方法に関し、本方法は、
(a)インスリン療法を受けている心血管リスクの高い患者であって、
(i)T1DM、および
(ii)最大耐性のスタチン療法によっては適切に制御されない高コレステロール血症
を有する患者を選択する工程;
(b)患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片を75mg、2週間ごとに投与する工程;ならびに
(c)患者におけるLDL−Cレベルが70mg/dLより低い場合、約2週間ごとに1回またはそれ以上の次の用量の75mgの抗体もしくはその抗原結合断片を患者に投与するか、または患者におけるLDL−Cレベルが70mg/dL以上の場合、約2週間ごとに1回またはそれ以上の次の用量の150mgの抗体もしくはその抗原結合断片を投与する工程
を含み、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含み、患者は付随するインスリン療法を受ける。
In a further embodiment, the invention relates to a use and/or method for treating hypercholesterolemia in a patient with
(A) a patient at high cardiovascular risk receiving insulin therapy,
(I) selecting T1DM, and (ii) patients with hypercholesterolemia not adequately controlled by maximally resistant statin therapy;
(B) administering to the patient 75 mg of an antibody or an antigen-binding fragment thereof that specifically binds to the human proprotein convertase subtilisin/kexin type 9 (PCSK9) every 2 weeks; and (c) LDL- in the patient. If the C level is less than 70 mg/dL, the patient is administered about one or more of the following doses of 75 mg of the antibody or antigen-binding fragment thereof about every two weeks, or the LDL-C level in the patient is 70 mg/dL. In the case of dL or more, it comprises the step of administering the following doses of about 150 mg of the antibody or its antigen-binding fragment once or more about every two weeks, wherein the antibody or its antigen-binding fragment has the amino acid sequence of SEQ ID NO: 1. Having HCVR, and an LCVR having the amino acid sequence of SEQ ID NO:6, the patient undergoes concomitant insulin therapy.
さらなる実施形態では、本発明は、2型真性糖尿病(T2DM)を有する患者における高コレステロール血症を治療するための使用および/または方法に関し、本方法は、
(a)インスリン療法を受けている心血管リスクの高い患者であって、
(i)T2DM、および
(ii)最大耐性のスタチン療法によって適切に制御されていない高コレステロール血症を有する患者を選択する工程;ならびに
(b)患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片の75mg、150mgもしくは300mgを投与する工程と
を含み、患者は付随するインスリン療法を受ける。
In a further embodiment, the invention relates to a use and/or method for treating hypercholesterolemia in a patient with diabetes mellitus type 2 (T2DM), the method comprising:
(A) a patient at high cardiovascular risk receiving insulin therapy,
(I) selecting a patient with hypercholesterolemia not adequately controlled by T2DM, and (ii) maximally resistant statin therapy; Administering 75 mg, 150 mg or 300 mg of an antibody or antigen-binding fragment thereof that specifically binds (PCSK9), and the patient receives concomitant insulin therapy.
上記使用および/または方法の一実施形態では、75mgの抗体または抗原結合断片は2週間ごとに患者に投与される。 In one embodiment of the above uses and/or methods, 75 mg of antibody or antigen binding fragment is administered to the patient every two weeks.
上記使用および/または方法の一実施形態では、150mgの抗体または抗原結合断片は2週間ごとに患者に投与される。 In one embodiment of the above uses and/or methods, 150 mg of antibody or antigen binding fragment is administered to the patient every two weeks.
上記使用および/または方法の一実施形態では、300mgの抗体または抗原結合断片は4週間ごとに患者に投与される。 In one embodiment of the above uses and/or methods, 300 mg of antibody or antigen binding fragment is administered to the patient every 4 weeks.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、配列番号2、3、および4に記載の3つの重鎖CDR、ならびに配列番号7、8、および10に記載の3つの軽鎖CDRを含む。 In one embodiment of the above uses and/or methods, the antibody or antigen-binding fragment thereof comprises the three heavy chain CDRs set forth in SEQ ID NOs:2, 3, and 4, and the 3 set forth in SEQ ID NOs:7, 8 and 10. Contains one light chain CDR.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有する重鎖可変領域(HCVR)、および配列番号6のアミノ酸配列を有する軽鎖可変領域(LCVR)を含む。 In one embodiment of the above use and/or method, the antibody or antigen binding fragment thereof comprises a heavy chain variable region (HCVR) having the amino acid sequence of SEQ ID NO:1 and a light chain variable region having the amino acid sequence of SEQ ID NO:6 ( LCVR).
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、アリロクマブ、エボロクマブ、ボコシズマブ、ロデルシズマブ、ラルパンシズマブ、およびLY3015014からなる群から選択される。 In one embodiment of the above uses and/or methods, the antibody or antigen-binding fragment thereof is selected from the group consisting of allilocoumab, evolocumab, bococizumab, rodercizumab, ralpancizumab, and LY301504.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片はアリロクマブである。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof is arilocumab.
上記使用および/または方法の一実施形態では、
(c)患者におけるLDL−Cレベルが閾値レベルより低い場合、約2週間ごとに1回またはそれ以上の次の投薬量の75mgの抗体もしくはその抗原結合断片を患者に投与するか、または患者におけるLDL−Cレベルが閾値レベル以上の場合、約2週間ごとに1回またはそれ以上の次の投薬量の150mgの抗体もしくはその抗原結合断片を投与する工程
をさらに含む。
In one embodiment of the above uses and/or methods,
(C) if the LDL-C level in the patient is below the threshold level, the patient is administered with one or more subsequent doses of 75 mg of the antibody or antigen-binding fragment thereof about once every two weeks, or in the patient If the LDL-C level is above the threshold level, the method further comprises the step of administering one or more subsequent doses of 150 mg of the antibody or antigen-binding fragment thereof about once every two weeks.
上記使用および/または方法の一実施形態では、
(c)患者におけるLDL−Cレベルが閾値レベルより低い場合、約4週間ごとに1回またはそれ以上の次の投薬量の300mgの抗体もしくはその抗原結合断片を患者に投与するか、または患者におけるLDL−Cレベルが閾値レベル以上の場合、約2週間ごとに1回またはそれ以上の次の投薬量の150mgの抗体もしくはその抗原結合断片を投与する工程
をさらに含む。
In one embodiment of the above uses and/or methods,
(C) if the LDL-C level in the patient is below a threshold level, the patient is administered with one or more subsequent doses of 300 mg of the antibody or antigen-binding fragment thereof about once every four weeks, or in the patient If the LDL-C level is above the threshold level, the method further comprises the step of administering one or more subsequent doses of 150 mg of the antibody or antigen-binding fragment thereof about once every two weeks.
上記使用および/または方法の一実施形態では、閾値レベルは70mg/dLである。 In one embodiment of the uses and/or methods above, the threshold level is 70 mg/dL.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は皮下投与される。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof is administered subcutaneously.
上記使用および/または方法の一実施形態では、患者は、付随する脂質修飾療法(LMT)をさらに受ける。 In one embodiment of the above uses and/or methods, the patient further undergoes concomitant lipid modification therapy (LMT).
上記使用および/または方法の一実施形態では、LMTは、スタチン、コレステロール吸収阻害剤、フィブラート、ナイアシン、オメガ−3脂肪酸、および胆汁酸封鎖剤からなる群から選択される。 In one embodiment of the use and/or method above, the LMT is selected from the group consisting of statins, cholesterol absorption inhibitors, fibrates, niacin, omega-3 fatty acids, and bile sequestrants.
上記使用および/または方法の一実施形態では、LMTはスタチン療法である。 In one embodiment of the above uses and/or methods, LMT is statin therapy.
上記使用および/または方法の一実施形態では、スタチンは、アトルバスタチン、ロスバスタチン、シンバスタチン、プラバスタチン、ロバスタチン、フルバスタチン、ピタバスタチン、およびセリバスタチンからなる群から選択される。 In one embodiment of the use and/or method above, the statin is selected from the group consisting of atorvastatin, rosuvastatin, simvastatin, pravastatin, lovastatin, fluvastatin, pitavastatin, and cerivastatin.
上記使用および/または方法の一実施形態では、スタチン療法は最大耐性量のスタチン療法である。 In one embodiment of the above uses and/or methods, the statin therapy is a maximally resistant amount of statin therapy.
上記使用および/または方法の一実施形態では、コレステロール吸収阻害剤はエゼチミブである。 In one embodiment of the above uses and/or methods, the cholesterol absorption inhibitor is ezetimibe.
上記使用および/または方法の一実施形態では、患者はスタチンにして不耐性である。 In one embodiment of the above uses and/or methods, the patient is statin intolerant.
上記使用および/または方法の一実施形態では、インスリン療法は、ヒトインスリン、インスリングラルギン、インスリングルリジン、インスリンデテミル、インスリンリスプロ、インスリンデグルデック、インスリンアスパルト、および基礎インスリンからなる群から選択される。 In one embodiment of the use and/or method above, the insulin therapy is selected from the group consisting of human insulin, insulin glargine, insulin glulisine, insulin detemir, insulin lispro, insulin degludec, insulin aspart, and basal insulin. It
上記使用および/または方法の一実施形態では、患者は、インスリン療法に加えて付随する抗糖尿病療法を受ける。 In one embodiment of the above uses and/or methods, the patient receives insulin therapy along with concomitant antidiabetic therapy.
上記使用および/または方法の一実施形態では、追加の付随する抗糖尿病療法は、グルカゴン様ペプチド1(GLP−1)療法、胃腸ペプチド、グルカゴン受容体アゴニストまたはアンタゴニスト、グルコース依存性インスリン分泌性ポリペプチド(GIP)受容体アゴニストまたはアンタゴニスト、グレリンアンタゴニストまたは逆アゴニスト、キセニン、キセニン類似体、ビグアナイド、スルホニル尿素、メグリチニド、チアゾリジンジオン、DPP−4阻害剤、アルファ−グルコシダーゼ阻害剤、ナトリウム依存性グルコース輸送体2(SGLT−2)阻害剤、SGLT−1阻害剤、ペルオキシソーム増殖因子活性化受容体(PPAR−)(アルファ、ガンマまたはアルファ/ガンマ)アゴニストまたはモジュレータ、アミリン、アミリンアナログ、Gタンパク質共役受容体119(GPR119)アゴニスト、GPR40アゴニスト、GPR120アゴニスト、GPR142アゴニスト、全身または低吸収性TGR5アゴニスト、糖尿病免疫療法薬、メタボリックシンドロームおよび糖尿病を治療するための抗炎症薬、アデノシンモノプリン酸活性化プロテインキナーゼ(AMPK)刺激薬、11−ベータ−ヒドロキシステロイドデヒドロゲナーゼ1の阻害剤、グルコキナーゼの活性化剤、ジアシルグリセロールO−アシルトランスフェラーゼ(DGAT)の阻害剤、グルコーストランスポータ−4のモジュレータ、ソマトスタチン受容体3アゴニスト、脂質減少剤、ならびにそれらの組み合わせからなる群から選択される。
In one embodiment of the above uses and/or methods, the additional concomitant antidiabetic therapy is glucagon-like peptide 1 (GLP-1) therapy, gastrointestinal peptide, glucagon receptor agonist or antagonist, glucose-dependent insulinotropic polypeptide. (GIP) receptor agonist or antagonist, ghrelin antagonist or inverse agonist, xenin, xenin analogue, biguanide, sulfonylurea, meglitinide, thiazolidinedione, DPP-4 inhibitor, alpha-glucosidase inhibitor, sodium-dependent glucose transporter 2 (SGLT-2) inhibitor, SGLT-1 inhibitor, peroxisome proliferator-activated receptor (PPAR-) (alpha, gamma or alpha/gamma) agonist or modulator, amylin, amylin analog, G protein coupled receptor 119 ( GPR119) Agonist, GPR40 Agonist, GPR120 Agonist, GPR142 Agonist, Systemic or Low Absorption TGR5 Agonist, Diabetic Immunotherapeutic, Anti-inflammatory Drug for Treating Metabolic Syndrome and Diabetes, Adenosine Monopurinate Activated Protein Kinase (AMPK) Stimulants, 11-beta-
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、患者のLDL−Cレベルを少なくとも30%、35%、40%、もしくは45%減少させる。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof reduces LDL-C levels in the patient by at least 30%, 35%, 40%, or 45%.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、患者の非HDL−Cレベルを少なくとも20%、25%、30%、もしくは35%減少させる。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof reduces non-HDL-C levels in the patient by at least 20%, 25%, 30%, or 35%.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、患者のApoC3レベルを減少させる。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof reduces ApoC3 levels in the patient.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、患者におけるリポタンパク質粒子の数および/またはサイズを減少させる。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof reduces the number and/or size of lipoprotein particles in the patient.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は:
(a)患者のヘモグロビンA1c(HbA1c)レベルに影響を及ぼさない;および/または
(b)患者の空腹時血漿グルコース(FPG)レベルに影響を及ぼさない。
In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof is:
(A) does not affect the patient's hemoglobin A1c (HbA1c) levels; and/or (b) does not affect the patient's fasting plasma glucose (FPG) levels.
さらなる実施形態では、本発明は、2型真性糖尿病(T2DM)を有する患者における高コレステロール血症を治療するための使用および/または方法に関し、本方法は、
(a)インスリン療法を受けている心血管リスクの高い患者であって、
(i)T2DM、および
(ii)最大耐性のスタチン療法によっては適切に制御されない高コレステロール血症
を有する患者を選択する工程;
(b)患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片を75mg、2週間ごとに投与する工程;ならびに
(c)患者におけるLDL−Cレベルが70mg/dLより低い場合、約2週間ごとに1回またはそれ以上の次の用量の75mgの抗体もしくはその抗原結合断片を患者に投与するか、または患者におけるLDL−Cレベルが70mg/dL以上の場合、約2週間ごとに1回またはそれ以上の次の用量の150mgの抗体もしくはその抗原結合断片を投与する工程
を含み、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含み、患者は付随するインスリン療法を受ける。
In a further embodiment, the invention relates to a use and/or method for treating hypercholesterolemia in a patient with diabetes mellitus type 2 (T2DM), the method comprising:
(A) a patient at high cardiovascular risk receiving insulin therapy,
Selecting (i) T2DM, and (ii) patients with hypercholesterolemia not adequately controlled by maximally tolerant statin therapy;
(B) administering to the patient 75 mg of an antibody or an antigen-binding fragment thereof that specifically binds to the human proprotein convertase subtilisin/kexin type 9 (PCSK9) every 2 weeks; and (c) LDL- in the patient. If the C level is less than 70 mg/dL, the patient is administered about one or more of the following doses of 75 mg of the antibody or antigen-binding fragment thereof about every two weeks, or the LDL-C level in the patient is 70 mg/dL. In the case of dL or more, it comprises the step of administering the following doses of about 150 mg of the antibody or its antigen-binding fragment once or more about every two weeks, wherein the antibody or its antigen-binding fragment has the amino acid sequence of SEQ ID NO: 1. Having HCVR, and an LCVR having the amino acid sequence of SEQ ID NO:6, the patient undergoes concomitant insulin therapy.
さらなる実施形態では、本発明は、2型真性糖尿病(T2DM)およびアテローム性動脈硬化性心血管疾患(ASCVD)を有する患者における高コレステロール血症を治療するための、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片の使用に関する。
In a further embodiment, the invention provides a human proprotein convertase subtilisin/kexin for treating hypercholesterolemia in patients with
なおさらなる実施形態では、本発明は、T2DMおよびASCVDを有する患者における高コレステロール血症を治療する方法に関する。 In yet a further embodiment, the invention relates to a method of treating hypercholesterolemia in a patient having T2DM and ASCVD.
一実施形態では、上記使用および/または方法は、
(a)インスリン療法を受けている心血管リスクの高い患者であって、
(i)T2DM、
(ii)ASCVD、および
(iii)最大耐性のスタチン療法によって適切に制御されていない高コレステロール血症を有する患者を選択する工程;ならびに
(b)患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片の75mg、150mgもしくは300mgを投与する工程
を含み、患者は付随するインスリン療法を受ける。
In one embodiment, the use and/or method comprises
(A) a patient at high cardiovascular risk receiving insulin therapy,
(I) T2DM,
(Ii) selecting a patient with hypercholesterolemia that is not adequately controlled by ASCVD, and (iii) maximally resistant statin therapy; Administering 75 mg, 150 mg or 300 mg of an antibody or antigen-binding fragment thereof that specifically binds (PCSK9), and the patient receives concomitant insulin therapy.
上記使用および/または方法の一実施形態では、ASCVDは、冠状動脈性心疾患(CHD)、虚血性脳卒中、または末梢動脈疾患として定義される。 In one embodiment of the above uses and/or methods, ASCVD is defined as coronary heart disease (CHD), ischemic stroke, or peripheral arterial disease.
上記使用および/または方法の一実施形態では、CHDは、急性心筋梗塞、無症候性心筋梗塞、および不安定狭心症を含む。 In one embodiment of the above uses and/or methods, CHD comprises acute myocardial infarction, subclinical myocardial infarction, and unstable angina.
上記使用および/または方法の一実施形態では、75mgの抗体または抗原結合断片は2週間ごとに患者に投与される。 In one embodiment of the above uses and/or methods, 75 mg of antibody or antigen binding fragment is administered to the patient every two weeks.
上記使用および/または方法の一実施形態では、150mgの抗体または抗原結合断片は2週間ごとに患者に投与される。 In one embodiment of the above uses and/or methods, 150 mg of antibody or antigen binding fragment is administered to the patient every two weeks.
上記使用および/または方法の一実施形態では、300mgの抗体または抗原結合断片は4週間ごとに患者に投与される。 In one embodiment of the above uses and/or methods, 300 mg of antibody or antigen binding fragment is administered to the patient every 4 weeks.
上記使用および/または方法の一実施形態では、配列番号2、3、および4に記載の3つの重鎖CDR、ならびに配列番号7、8、および10に記載の3つの軽鎖CDRを含む。 In one embodiment of the above uses and/or methods, it comprises the three heavy chain CDRs set forth in SEQ ID NOs: 2, 3 and 4 and the three light chain CDRs set forth in SEQ ID NOs: 7, 8 and 10.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有する重鎖可変領域(HCVR)、および配列番号6のアミノ酸配列を有する軽鎖可変領域(LCVR)を含む。 In one embodiment of the above use and/or method, the antibody or antigen binding fragment thereof comprises a heavy chain variable region (HCVR) having the amino acid sequence of SEQ ID NO:1 and a light chain variable region having the amino acid sequence of SEQ ID NO:6 ( LCVR).
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、アリロクマブ、エボロクマブ、ボコシズマブ、ロデルシズマブ、ラルパンシズマブ、およびLY3015014からなる群から選択される。 In one embodiment of the above uses and/or methods, the antibody or antigen-binding fragment thereof is selected from the group consisting of allilocoumab, evolocumab, bococizumab, rodercizumab, ralpancizumab, and LY301504.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片がアリロクマブである。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof is arilocumab.
一実施形態では、上記使用および/または方法は、
(c)患者におけるLDL−Cレベルが閾値レベルより低い場合、約2週間ごとに1回またはそれ以上の次の投薬量の75mgの抗体もしくはその抗原結合断片を患者に投与するか、または患者におけるLDL−Cレベルが閾値レベル以上の場合、約2週間ごとに1回またはそれ以上の次の投薬量の150mgの抗体もしくはその抗原結合断片を投与する工程
をさらに含む。
In one embodiment, the use and/or method comprises
(C) if the LDL-C level in the patient is below the threshold level, the patient is administered with one or more subsequent doses of 75 mg of the antibody or antigen-binding fragment thereof about once every two weeks, or in the patient If the LDL-C level is above the threshold level, the method further comprises the step of administering one or more subsequent doses of 150 mg of the antibody or antigen-binding fragment thereof about once every two weeks.
一実施形態では、上記使用および/または方法は、
(c)患者におけるLDL−Cレベルが閾値レベルより低い場合、約4週間ごとに1回またはそれ以上の次の投薬量の300mgの抗体またはその抗原結合断片を患者に投与するか、または患者におけるLDL−Cレベルが閾値レベル以上の場合、約2週間ごとに1回またはそれ以上の次の投薬量の150mgの抗体またはその抗原結合断片を投与する工程
を含む。
In one embodiment, the use and/or method comprises
(C) if the LDL-C level in the patient is below the threshold level, the patient is administered with one or more subsequent doses of 300 mg of the antibody or antigen-binding fragment thereof about once every four weeks, or in the patient If the LDL-C level is above the threshold level, it comprises the step of administering one or more subsequent doses of 150 mg of the antibody or antigen binding fragment thereof about once every two weeks.
上記使用および/または方法の一実施形態では、閾値レベルは70mg/dLである。 In one embodiment of the uses and/or methods above, the threshold level is 70 mg/dL.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は皮下投与される。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof is administered subcutaneously.
上記使用および/または方法の一実施形態では、患者は、付随する脂質修飾療法(LMT)をさらに受ける。 In one embodiment of the above uses and/or methods, the patient further undergoes concomitant lipid modification therapy (LMT).
上記使用および/または方法の一実施形態では、LMTは、スタチン、コレステロール吸収阻害剤、フィブラート、ナイアシン、オメガ−3脂肪酸、および胆汁酸封鎖剤からなる群から選択される。 In one embodiment of the use and/or method above, the LMT is selected from the group consisting of statins, cholesterol absorption inhibitors, fibrates, niacin, omega-3 fatty acids, and bile sequestrants.
上記使用および/または方法の一実施形態では、LMTはスタチン療法である。 In one embodiment of the above uses and/or methods, LMT is statin therapy.
上記使用および/または方法の一実施形態では、スタチンは、アトルバスタチン、ロスバスタチン、シンバスタチン、プラバスタチン、ロバスタチン、フルバスタチン、ピタバスタチン、およびセリバスタチンからなる群から選択される。 In one embodiment of the use and/or method above, the statin is selected from the group consisting of atorvastatin, rosuvastatin, simvastatin, pravastatin, lovastatin, fluvastatin, pitavastatin, and cerivastatin.
上記使用および/または方法の一実施形態では、スタチン療法が最大耐性量のスタチン療法である。 In one embodiment of the above uses and/or methods, the statin therapy is the most resistant amount of statin therapy.
上記使用および/または方法の一実施形態では、コレステロール吸収阻害剤はエゼチミブである。 In one embodiment of the above uses and/or methods, the cholesterol absorption inhibitor is ezetimibe.
上記使用および/または方法の一実施形態では、患者はスタチンにして不耐性である。 In one embodiment of the above uses and/or methods, the patient is statin intolerant.
上記使用および/または方法の一実施形態では、インスリン療法は、ヒトインスリン、インスリングラルギン、インスリングルリジン、インスリンデテミル、インスリンリスプロ、インスリンデグルデック、インスリンアスパルト、および基礎インスリンからなる群から選択される。 In one embodiment of the use and/or method above, the insulin therapy is selected from the group consisting of human insulin, insulin glargine, insulin glulisine, insulin detemir, insulin lispro, insulin degludec, insulin aspart, and basal insulin. It
上記使用および/または方法の一実施形態では、患者は、インスリン療法に加えて付随する抗糖尿病療法を受ける。 In one embodiment of the above uses and/or methods, the patient receives insulin therapy along with concomitant antidiabetic therapy.
上記使用および/または方法の一実施形態では、追加の付随する抗糖尿病療法は、グルカゴン様ペプチド1(GLP−1)療法、胃腸ペプチド、グルカゴン受容体アゴニストまたはアンタゴニスト、グルコース依存性インスリン分泌性ポリペプチド(GIP)受容体アゴニストまたはアンタゴニスト、グレリンアンタゴニストまたは逆アゴニスト、キセニン、キセニン類似体、ビグアナイド、スルホニル尿素、メグリチニド、チアゾリジンジオン、DPP−4阻害剤、アルファ−グルコシダーゼ阻害剤、ナトリウム依存性グルコース輸送体2(SGLT−2)阻害剤、SGLT−1阻害剤、ペルオキシソーム増殖因子活性化受容体(PPAR−)(アルファ、ガンマまたはアルファ/ガンマ)アゴニストまたはモジュレータ、アミリン、アミリンアナログ、Gタンパク質共役受容体119(GPR119)アゴニスト、GPR40アゴニスト、GPR120アゴニスト、GPR142アゴニスト、全身または低吸収性TGR5アゴニスト、糖尿病免疫療法薬、メタボリックシンドロームおよび糖尿病を治療するための抗炎症薬、アデノシンモノプリン酸活性化プロテインキナーゼ(AMPK)刺激薬、11−ベータ−ヒドロキシステロイドデヒドロゲナーゼ1の阻害剤、グルコキナーゼの活性化剤、ジアシルグリセロールO−アシルトランスフェラーゼ(DGAT)の阻害剤、グルコーストランスポータ−4のモジュレータ、ソマトスタチン受容体3アゴニスト、脂質減少剤、ならびにそれらの組み合わせからなる群から選択される。
In one embodiment of the above uses and/or methods, the additional concomitant antidiabetic therapy is glucagon-like peptide 1 (GLP-1) therapy, gastrointestinal peptide, glucagon receptor agonist or antagonist, glucose-dependent insulinotropic polypeptide. (GIP) receptor agonist or antagonist, ghrelin antagonist or inverse agonist, xenin, xenin analogue, biguanide, sulfonylurea, meglitinide, thiazolidinedione, DPP-4 inhibitor, alpha-glucosidase inhibitor, sodium-dependent glucose transporter 2 (SGLT-2) inhibitor, SGLT-1 inhibitor, peroxisome proliferator-activated receptor (PPAR-) (alpha, gamma or alpha/gamma) agonist or modulator, amylin, amylin analog, G protein coupled receptor 119 ( GPR119) Agonist, GPR40 Agonist, GPR120 Agonist, GPR142 Agonist, Systemic or Low Absorption TGR5 Agonist, Diabetic Immunotherapeutic, Anti-inflammatory Drug for Treating Metabolic Syndrome and Diabetes, Adenosine Monopurinate Activated Protein Kinase (AMPK) Stimulants, 11-beta-
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、患者のLDL−Cレベルを少なくとも30%、35%、40%、もしくは45%減少させる。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof reduces LDL-C levels in the patient by at least 30%, 35%, 40%, or 45%.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、患者の非HDL−Cレベルを少なくとも20%、25%、30%、もしくは35%減少させる。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof reduces non-HDL-C levels in the patient by at least 20%, 25%, 30%, or 35%.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、患者のApoC3レベルを減少させる。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof reduces ApoC3 levels in the patient.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は、患者におけるリポタンパク質粒子の数および/またはサイズを減少させる。 In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof reduces the number and/or size of lipoprotein particles in the patient.
上記使用および/または方法の一実施形態では、抗体またはその抗原結合断片は:
(a)患者のヘモグロビンA1c(HbA1c)レベルに影響を及ぼさない;および/または
(b)患者の空腹時血漿グルコース(FPG)レベルに影響を及ぼさない。
In one embodiment of the above uses and/or methods, the antibody or antigen binding fragment thereof is:
(A) does not affect the patient's hemoglobin A1c (HbA1c) levels; and/or (b) does not affect the patient's fasting plasma glucose (FPG) levels.
さらなる実施形態では、本発明は、T2DMおよびASCVDを有する患者における高コレステロール血症を治療するための使用および/または方法に関し、本方法は、
(a)インスリン療法を受けている心血管リスクの高い患者であって、
(i)T2DM、
(ii)ASCVD、および
(iii)最大耐性のスタチン療法によって適切に制御されない高コレステロール血症
を有する患者を選択すること;
(b)2週間ごとに患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する、75mgの抗体またはその抗原結合断片を投与すること;ならびに
(c)患者におけるLDL−Cレベルが70mg/dLより低い場合、約2週間ごとに1回またはそれ以上の次の用量の75mgの抗体もしくはその抗原結合断片を患者に投与するか、または患者におけるLDL−Cレベルが70mg/dL以上の場合、約2週間ごとに1回またはそれ以上の次の用量の150mgの抗体もしくはその抗原結合断片を投与すること
を含み、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含み、患者は付随するインスリン療法を受ける。
In a further embodiment, the invention relates to a use and/or method for treating hypercholesterolemia in a patient having T2DM and ASCVD, the method comprising:
(A) a patient at high cardiovascular risk receiving insulin therapy,
(I) T2DM,
Selecting patients with hypercholesterolemia not adequately controlled by (ii) ASCVD, and (iii) maximally resistant statin therapy;
(B) administering to the patient every two
以下の実施例は、本発明の方法および組成物を作製および使用する方法の完全な開示および説明を当業者に提供するために提示されており、発明者がそれらの発明としてみなす範囲を限定することを意図するものではない。使用された数値(例えば、量、温度など)に関して正確性を確保するための努力が行われたが、いくつかの実験誤差および偏差を考慮する必要がある。別段の指示がない限り、部は重量部であり、分子量は平均分子量であり、温度は摂氏温度であり、圧力は大気圧または大気圧付近である。 The following examples are presented to provide those of ordinary skill in the art with a complete disclosure and description of how to make and use the methods and compositions of the present invention, and limit the scope of what the inventors regard as their invention. It is not intended to be. Efforts have been made to ensure accuracy with respect to numbers used (eg amounts, temperature, etc.) but some experimental errors and deviations should be accounted for. Unless indicated otherwise, parts are parts by weight, molecular weight is average molecular weight, temperature is in degrees Celsius, and pressure is at or near atmospheric.
ヒトPCSK9に対するヒト抗体の生成
ヒト抗PCSK9抗体は、米国特許第8,062,640号に記載されているように生成された。以下の実施例で使用される例示的なPCSK9阻害剤は、「mAb316P」と呼ばれるヒト抗PCSK9抗体であり、「REGN727」または「アリロクマブ」としても知られている。mAb316Pは、以下のアミノ酸配列特性を有する:配列番号5を含む重鎖および配列番号9を含む軽鎖;配列番号1を含む重鎖可変領域(HCVR)および配列番号6を含む軽鎖可変ドメイン(LCVR);配列番号2を含む重鎖相補性決定領域1(HCDR1)、配列番号3を含むHCDR2、配列番号4を含むHCDR3、配列番号7を含む軽鎖相補性決定領域1(LCDR1)、配列番号8を含むLCDR2、および配列番号10を含むLCDR3。
Generation of Human Antibodies to Human PCSK9 Human anti-PCSK9 antibodies were generated as described in US Pat. No. 8,062,640. An exemplary PCSK9 inhibitor used in the examples below is the human anti-PCSK9 antibody called "mAb316P", also known as "REGN727" or "arilocumab". mAb316P has the following amino acid sequence characteristics: a heavy chain comprising SEQ ID NO:5 and a light chain comprising SEQ ID NO:9; a heavy chain variable region (HCVR) comprising SEQ ID NO:1 and a light chain variable domain comprising SEQ ID NO:6 ( LCVR); heavy chain complementarity determining region 1 (HCDR1) containing SEQ ID NO:2, HCDR2 containing SEQ ID NO:3, HCDR3 containing SEQ ID NO:4, light chain complementarity determining region 1 (LCDR1) containing SEQ ID NO:7, sequence
1型または2型糖尿病を有し、最大耐性のLDL−C減少療法で適切に制御されない心血管リスクが高い高コレステロール血症を有するインスリン治療患者におけるアリロクマブの有効性および安全性を評価するためのランダム化二重盲検プラセボ対照並行群間研究
導入
世界中で3億8千万人を超える人々が糖尿病を患っており、そのほとんどは心血管疾患(CVD)で死亡する。糖尿病のない人々と比較して、糖尿病を有する人々は、CVDを発症するリスクが高く、関連する臨床的合併症を早期の年齢で発症し、平均寿命が約6〜7年短くなっている。疾患の人件費が高いことに加えて、CVDはこれらの患者の全体的な医療費に大きく関わる。
To assess the efficacy and safety of arilocumab in insulin-treated patients with hypercholesterolemia with high cardiovascular risk who have
オデッセイDM−インスリンと名付けられたこの研究は、高心血管(CV)リスクの高コレステロール血症を伴うインスリン療法中の1型または2型真性糖尿病を有し、他の脂質修飾療法(LMT)の有無にかかわらず、スタチンの最大耐性量で十分に制御されていない成人患者を含んだ。
This study, termed Odyssey DM-Insulin, has
研究の目的
本研究の主要な目的は:(a)インスリンで治療された糖尿病を有し、最大耐性のLDL−C減少療法で適切に制御されない高コレステロール血症を有する、心血管リスクの高い患者における24週間の治療後の計算された低密度リポタンパク質コレステロール(LDL−C)の減少におけるアリロクマブの有効性をプラセボと比較して評価すること;ならびに(b)インスリンで治療された糖尿病を有する患者におけるアリロクマブの安全性および耐性を評価することである。
Objectives of the study The main objectives of the study were: (a) high cardiovascular risk patients with insulin-treated diabetes mellitus with hypercholesterolemia not adequately controlled by maximally tolerant LDL-C reduction therapy. Assessing the efficacy of alilocumab in comparison to placebo in reducing calculated low density lipoprotein cholesterol (LDL-C) after 24 weeks of treatment in; and (b) patients with diabetes treated with insulin. To assess the safety and resistance of alilocumab in.
研究の二次目的は、12週目および24週目での他の脂質パラメータ(例えば、測定されたLDL−C、非高密度リポタンパク質コレステロール(非HDL−C)、アポリポタンパク質B(Apo B)、総コレステロール(TC)、リポタンパク質a(Lp(a))、高密度リポタンパク質コレステロール(HDL−C)、トリグリセリド(TG)レベル、トリグリセリドに富んだリポタンパク質(TGRL)、アポリポタンパク質A−1(Apo A−1)、アポリポタンパク質C3(ApoC3)、およびLDL粒子の数およびサイズ)に対するプラセボと比較したアリロクマブの有効性を評価することであった。 Secondary objectives of the study were other lipid parameters at weeks 12 and 24 (eg measured LDL-C, non-high density lipoprotein cholesterol (non-HDL-C), apolipoprotein B (Apo B). , Total cholesterol (TC), lipoprotein a (Lp(a)), high density lipoprotein cholesterol (HDL-C), triglyceride (TG) levels, triglyceride-rich lipoprotein (TGRL), apolipoprotein A-1 ( It was to assess the efficacy of arilocumab compared to placebo on Apo A-1), apolipoprotein C3 (ApoC3), and LDL particle numbers).
研究設計
これは、1型または2型真性糖尿病のCVリスクが高く、最大耐性のLDL−C低下療法で高コレステロール血症が適切に制御されていない、インスリン治療患者に皮下(SC)注射により投与されるアリロクマブの有効性および安全性を評価するための第3b相のランダム化された二重盲検プラセボ対照多国籍および多施設研究であった。この研究は、最大3週間のスクリーニング期間、24週間の二重盲検治療期間、および二重盲検治療期間の終了後8週間の安全観察期間から構成されていた。
Study Design This is administered by subcutaneous (SC) injection to insulin-treated patients who have a high CV risk of
患者は、スタチン不耐性でない限り、他の脂質修飾療法(LMT)の有無にかかわらず、安定した最大耐性量のスタチン療法を受けていた。スタチン用量および投薬レジメン、ならびに他の脂質修飾治療の用量および投薬レジメン(該当する場合)は、スクリーニング期間前の4週間、スクリーニング期間中、およびスクリーニングからランダム化までを含む全研究期間を通じて安定であった。患者は、スクリーニングから24週目の来院までの全研究期間を通じて、グルコースおよび脂質管理のために安定した食事を摂っていた。患者は、地方/地域のケア基準に従って糖尿病の治療を受けていた。
Patients were receiving stable maximally tolerated doses of statin therapy with or without other lipid modification therapy (LMT), unless they were statin intolerant. Statin doses and dosing regimens, as well as doses and dosing regimens for other lipid-modifying therapies (if applicable), were stable for 4 weeks prior to the screening period, during the screening period, and throughout the study, including from screening to randomization. It was Patients had a stable diet for glucose and lipid control throughout the study, from screening to the
患者は、糖尿病の種類(すなわち、1型糖尿病対2型糖尿病)によって層別化された。約400人の患者がランダム化されたときに、2型糖尿病患者の募集が完了した。1型糖尿病患者の募集は、対象募集期間の終わりに完了した。
Patients were stratified by type of diabetes (ie,
アリロクマブは、75mg Q2Wの開始用量で12週間皮下投与され、8週目での来院時にLDL−Cが≧70mg/dL(1.81mmol/L)である場合、12週目にアリロクマブ150mg Q2Wへと盲検法で漸増させた。8週目の来院時にLDL−C<70mg/dL(1.81mmol/L)の患者は、治療期間が終了するまでアリロクマブ75mg Q2Wを継続した。
Arilocumab was administered subcutaneously at a starting dose of 75 mg Q2W for 12 weeks, and if LDL-C was ≧70 mg/dL (1.81 mmol/L) at the 8th week visit, then arilocumab 150 mg Q2W was administered at 12th week. Blinded titration. Patients with LDL-C <70 mg/dL (1.81 mmol/L) at
血液サンプルからの脂質パラメータに関するデータは、ランダム化後にマスクされた。治験責任医師または患者は、治験責任医師の判断による患者の安全性を除き、ランダム化後24週目の来院まで患者の脂質値を独立して評価する試みは行われなかった。 Data on lipid parameters from blood samples were masked after randomization. The investigator or patient did not attempt to independently evaluate the patient's lipid levels until the 24th visit after randomization, except for patient safety at the investigator's discretion.
患者は、−3、0、8、12、20、および24週目に研究現場を訪れ、各来院時に検査作業を行った。さらに、4週目と32週目に電話回答が行われた。 Patients visited the study site at -3, 0, 8, 12, 20, and 24 weeks and underwent laboratory work at each visit. In addition, telephone answers were given at the 4th and 32nd weeks.
調査医薬品(IMP)の最終投与から70日以内に発生した有害事象(AE)が文書化された。重篤な有害事象(SAE)または特に注目すべき有害事象(AESI)のある患者は、消散、安定化、または死亡するまで追跡された。 Adverse events (AEs) that occurred within 70 days of the last dose of study drug (IMP) were documented. Patients with Serious Adverse Events (SAE) or Adverse Events of Special Interest (AESI) were followed up until resolution, stabilization, or death.
患者の選択
本研究は、T1DMを有する76人の患者およびT2DMを有する441人の患者を含む合計517人の患者を登録した。
Patient Selection This study enrolled a total of 517 patients, including 76 patients with T1DM and 441 patients with T2DM.
試験対象患者基準
この研究に登録した患者は、以下の基準のすべてを満たした:
Patient inclusion criteria Patients enrolled in this study met all of the following criteria:
(1)インスリンで治療された1型または2型糖尿病を有し、LDL−Cレベルが70mg/dL(1.81mmol/L)以上であり、他のLMTの有無にかかわらず、スクリーニング来院前(−3週)の少なくとも4週間、患者に耐性であるスタチンの安定した最大用量/レジメンによって適切に制御されていない患者。患者が耐性であるスタチンの最大用量/レジメンは、治験責任医師の判断または懸念に基づいて患者が耐性である最高の登録用量/レジメンであった。スタチン投与量が少ない患者が許容できる理由のいくつかの例には、限定されないが、高用量への悪影響、高齢、低体格指数(BMI)、地域の慣習、地方の処方情報、または併用薬が含まれた。患者は、用量が一貫して摂取されている限り、スタチンの隔日投与を受けていた可能性があった(例えば、毎週月曜日、水曜日、金曜日など)。1種を超えるスタチンによる併用治療は許可されていなかった。治験責任医師が判断したように、スタチン不耐制を文書化した患者、および結果としてスタチン療法を中止した患者もまた研究の対象となった。スタチンの最大用量/レジメンではない理由(スタチン不耐性を含む)は、症例報告書に文書化された。
(1) Patients with
(2)スクリーニング来院時の18歳以上または成年の法定年齢のいずれかの患者。 (2) Patients who are 18 years of age or older at the screening visit or legal age of adulthood.
(3)スクリーニング来院前の少なくとも1年に1型または2型糖尿病と診断された患者(−3週)。1型糖尿病と診断された患者は、以下の基準をすべて満たす必要があった:
(a)30歳前に診断。
(b)診断後6か月以内に、毎日複数回の注射レジメン/基礎−食事インスリンレジメンまたはインスリンポンプレジメンで治療された;ならびに
(c)スクリーニング来院時にC−ペプチド<0.2pmol/mL。
(3) Patients with
(A) Diagnosed before the age of 30.
(B) Treated with multiple daily injection regimens/basal-dietary insulin regimens or insulin pump regimens within 6 months of diagnosis; and (c) C-peptide <0.2 pmol/mL at screening visit.
(4)スクリーニング来院時のグリコシル化ヘモグロビン(HbA1c)<10%(−3週)。治験責任医師の判断に基づいて、研究中にHbA1cの減少を標的とする計画がなかったという条件で、HbA1cが上昇した患者(最大10%)が適格であった。 (4) Glycosylated hemoglobin (HbA1c) at screening visit <10% (-3 weeks). Patients with elevated HbA1c (up to 10%) were eligible, based on the investigator's discretion, provided that there were no plans to target reduction of HbA1c during the study.
(5)CVDの文書化された病歴(CHDおよび/またはCHDリスク同等物を含む)ならびに/または少なくとも1つの追加のCVリスク因子を有する患者。 (5) Patients with a documented history of CVD (including CHD and/or CHD risk equivalents) and/or at least one additional CV risk factor.
CHDの病歴には、以下のうちの少なくとも1つが含まれた:
(a)急性心筋梗塞(MI);
(b)サイレントMI;
(c)不安定狭心症;
(d)冠動脈血行再建術(例えば、経皮的冠動脈インターベンション(PCI)または冠動脈バイパス移植手術(CABG));および
(e)侵襲的または非侵襲的検査(例えば、冠動脈造影、トレッドミルを使用したストレス検査、ストレス心エコー検査、または核イメージング)によって診断された臨床的に重要なCHD。
The history of CHD included at least one of the following:
(A) acute myocardial infarction (MI);
(B) Silent MI;
(C) unstable angina;
(D) coronary revascularization (eg, percutaneous coronary intervention (PCI) or coronary artery bypass graft surgery (CABG)); and (e) invasive or non-invasive examination (eg, using coronary angiography, treadmill). Clinically significant CHD diagnosed by stress testing, stress echocardiography, or nuclear imaging).
同等のCHDリスクには、次の少なくとも1つが含まれる:
(a)以下の基準の少なくとも1つを満たす末梢動脈疾患の文書化:
(i)安静時のいずれかの脚の足首上腕インデックス≦0.90とともに、推定されるアテローム性動脈硬化症の現在の間欠性跛行(再現性があり、運動によって生じ、10分以内に安静によって緩和される下肢の筋肉の不快感);
(ii)アテローム性動脈硬化症による片足または両足の血管内処置または外科的介入を伴う、間欠性跛行(再現性があり、運動によって生じ、10分以内に安静によって緩和される下肢の筋肉の不快感)の病歴;および
(iii)アテローム性動脈硬化症による血栓溶解、血管内処置または片足または両足の外科的介入を伴う重症肢虚血の病歴;ならびに
(b)アテローム血栓性の原因であると見なされる、24時間を超えて持続する局所虚血性神経学的欠損を伴う以前の虚血性脳卒中の文書化。出血および非虚血性神経疾患を除外するために、コンピューター断層撮影または磁気ラジオイメージングを行う必要がある。
Equivalent CHD risk includes at least one of the following:
(A) Documentation of peripheral arterial disease that meets at least one of the following criteria:
(I) Current intermittent claudication of recurrent probable atherosclerosis (reproducible, exercise-induced, with rest within 10 minutes, with resting ankle-brachial index ≤ 0.90 of either leg). Discomfort in the muscles of the lower limbs that is relieved);
(Ii) Intermittent claudication (reproducible, exercise-induced lower limb muscle deficiency relieved by rest within 10 minutes) with endovascular treatment or surgical intervention of one or both feet due to atherosclerosis. (Iii) a history of pleasing); and (iii) a history of severe limb ischemia with atherosclerotic thrombolysis, endovascular treatment or surgical intervention of one or both legs; and (b) atherosclerotic causes. Documented prior ischemic stroke with focal ischemic neurological deficits lasting for >24 hours. Computed tomography or magnetic radioimaging should be done to rule out hemorrhage and non-ischemic neurological disease.
心血管リスク因子には、以下の少なくとも1つが含まれた:
(a)高血圧(降圧薬で確立);
(b)現在の喫煙者;
(c)男性は45歳以上、女性は55歳以上;
(d)ミクロ/マクロアルブミン尿の病歴;
(e)糖尿病性網膜症の病歴(前増殖性または増殖性);
(f)未熟なCHDの家族歴(55歳未満の父親または兄弟;65歳未満の母親または姉妹);
(g)低HDL−C(男性<40mg/dL(1.0mmol/L)および女性<50mg/dL(1.3mmol/L));ならびに
(h)スクリーニング来院を含む、3か月以上、15≦eGFR<60mL/分/1.73m2によって定義される文書化された慢性腎疾患(CKD)。
Cardiovascular risk factors included at least one of the following:
(A) Hypertension (established with antihypertensive drug);
(B) current smokers;
(C) Men over 45 years old, women over 55 years old;
(D) History of micro/macroalbuminuria;
(E) History of diabetic retinopathy (pre-proliferative or proliferative);
(F) Family history of premature CHD (father or siblings under 55; mother or sister under 65);
(G) low HDL-C (male <40 mg/dL (1.0 mmol/L) and female <50 mg/dL (1.3 mmol/L)); and (h) 3 months or more, including screening visit, 15 Documented chronic kidney disease (CKD) defined by ≦eGFR<60 mL/min/1.73 m 2 .
(6)署名された書面によるインフォームドコンセント
除外基準
上記の試験対象患者基準をすべて満たした患者を以下の除外基準についてスクリーニングした:
(6) Signed Written Informed Consent Exclusion Criteria Patients who meet all of the above inclusion criteria are screened for the following exclusion criteria:
(1)研究方法論に関連する除外基準:
(a)研究中に新しいLMTを開始するか、または現在のLMTの用量を変更するように計画する;
(b)スタチン不耐性でない限り、スクリーニング来院前(−3週)またはスクリーニングからランダム化までの少なくとも4週間、LMT(スタチンまたは他のLMTを含む)の安定した用量ではなく、この症例では、スクリーニング来院前の4週間/スクリーニング期間中にスタチンはない;
(c)スクリーニング来院前(−3週)またはスクリーニングとランダム化来院の間の少なくとも4週間、安定した用量でなかった脂質に影響を与える可能性のある栄養補助食品または市販薬の使用;
(d)スクリーニング来院前(−3週)、またはスクリーニングとランダム化来院の間の4週間以内、酵母米製品の使用;
(e)ランダム化前の少なくとも6週間に安定したレジメンで下垂体/副腎疾患の代替療法として使用しない限り、全身性コルチコステロイドの使用。局所、関節内、鼻、吸入および眼のステロイド療法は「全身」とは見なされず、許可された;
(f)スクリーニング来院前の過去6週間(3週目)にレジメンが安定しており、研究中にレジメンを変更する計画がない場合を除き、継続的なホルモン補充療法の使用;
(g)最近(スクリーニング来院前の3か月以内(−3週)またはスクリーニングとランダム化来院の間)のMI、入院につながる不安定狭心症、制御不能な心不整脈、CABG、PCI、頸動脈手術またはステント留置、脳卒中、一過性虚血発作(TIA)、末梢血管疾患に対する血管内処置または外科的介入;
(h)研究中に予定されたPCI、CABG、頸動脈または末梢血行再建を受ける予定;
(i)過去12か月以内のニューヨーク心臓協会(NYHA)クラスIIIまたはIV心不全(表1を参照)の病歴;
(j)スクリーニング時またはランダム化来院時の収縮期血圧>180mmHgまたは拡張期血圧>110mmHg;
(k)スクリーニング来院前の2か月以内(−3週)、スクリーニングとランダム化の間に血漿失調症治療を受けた患者、またはそれを受ける計画がある患者。
(l)出血性脳卒中の病歴;
(m)PCSK9の機能喪失の病歴(すなわち、遺伝的突然変異または配列変異)またはホモ接合型家族性高コレステロール血症の病歴;
(n)適切に治療された基底細胞皮膚がん、扁平上皮細胞がん、もしくは上皮内子宮頸がんを除く、過去5年以内の新しいがんまたはがんの活発な進行;
(o)陽性HIV検査の病歴;
(p)1か月または5半減期のいずれか長い期間内に任意の有効な治験薬を服用した患者;
(q)スクリーニング来院前(−3週目)にコレステロール減少食を以前に指示されていない患者;
(r)スクリーニング中に同意を取り下げた患者(署名済みICFから開始);
(s)治験責任医師が判断した、スクリーニング来院前の2か月以内に5kgを超える変動として定義された不安定な体重;
(t)BMI>45kg/m2、または肥満手術、減量プログラム、または研究中に減量薬を開始する計画;
(u)減量薬の最近の開始(すなわち、スクリーニング来院前の3か月以内、もしくはスクリーニングとランダム化の間)または最近の肥満手術(過去6か月以内)および治験責任医師によって判断された積極的な減量段階にある;
(v)スクリーニング来院前の少なくとも6か月間にインスリンで治療されていない患者、またはスクリーニング来院前の少なくとも3か月間に、安定したインスリンレジメンで治療されていない患者(すなわち、インスリンの種類、注射の一般的な時期/頻度、基礎のみなどの投与モードもしくはパターン(2型糖尿病)、基礎の食事の変化など)、または研究期間中にインスリンの種類/頻度または注射方法の変更が必要になる可能性がある患者;
(w)スクリーニング前の少なくとも3か月間、安定したインスリン投薬量ではない(すなわち、治験責任医師が判断した場合、1日のインスリン投薬量の合計が30%を超える変動)、または治験責任医師が判断した、研究の過程でインスリン/抗高血糖レジメンの強化を必要とする可能性(例えば、新しい薬剤の追加、インスリン投薬量の漸増計画など);
(x)患者が摂取した他の血糖降下薬は、スクリーニング来院前の少なくとも3か月は安定していない;
(y)スクリーニング来院前の2か月以内の最近の糖尿病の代償不全の病歴(すなわち、糖尿病性ケトアシドーシスまたは高浸透圧性高血糖状態(HHS));
(z)研究中に腎代替療法を受けている、または受けようと計画している(例えば、血液透析、腎移植など);
(aa)血清脂質またはリポタンパク質に影響を与えることが知られている、臨床的に重要な未管理の内分泌疾患の存在。甲状腺置換療法を受けている患者は、サイロキシンの投薬量がスクリーニング前の少なくとも3か月間安定しており、患者の感受性甲状腺刺激ホルモン(s−TSH)レベルが、スクリーニング来院時に、検査室の正常範囲の±10%以内であった場合に含めることができる;
(bb)スクリーニング期間中の検査所見(妊娠検査を除き、ランダム化検査を含まない):
(i)血清TG>400mg/dL(4.52mmol/L)(1回の反復検査が許可される);
(ii)出産可能性のある女性の陽性の血清または尿妊娠検査;
(iii)B型肝炎表面抗原またはC型肝炎抗体の陽性検査;
(iv)腎疾患の食事における4変数の変更によるeGFR<15mL/分/1.73m2;
(MDRD)式;
(v)ALTまたはAST>3×ULN(1回の反復検査が許可される);もしくは
(vi)クレアチンホスホキナーゼ(CPK)>3×ULN(1回の反復検査が許可される);または
(cc)次のような条件/状況:
(i)平均余命の短い患者;
(ii)一次評価にバイアスをかける可能性のある併用治療の要件;
(iii)特定のプロトコル要件を満たすことが不可能(例えば、入院の必要性、研究への来院能力など);
(iv)患者は、治験責任医師または副治験責任医師、研究助手、薬剤師、治験コーディネーター、他のスタッフもしくはプロトコルの実施に直接関与したその血縁者であった;
(v)非協力的、または患者が研究手順に適合しない可能性がある状態;
(vi)研究において患者をランダム化することを不可能にする何らかの技術的/管理上の理由;または
(vii)治験責任医師または副治験責任医師の判断により、治験の安全な完了が妨げられたり、主要な全身疾患、平均余命の短い患者などの評価項目の評価が制限される、スクリーニング時に特定された臨床的に重大な異常。
(1) Exclusion criteria related to research methodology:
(A) Plan to start a new LMT during the study or change the current LMT dose;
(B) Stable doses of LMT (including statins or other LMTs) prior to screening visit (-3 weeks) or at least 4 weeks from screening to randomization, unless statin intolerant, and in this case screening 4 weeks prior to visit/no statins during screening;
(C) Use of dietary supplements or over-the-counter medications that may affect lipids that were not at stable doses for at least 4 weeks prior to screening visit (-3 weeks) or between screening and randomized visits;
(D) Pre-screening visit (-3 weeks) or within 4 weeks between screening and randomized visits, using yeast rice product;
(E) Use of systemic corticosteroids unless used as an alternative therapy for pituitary/adrenal disease in a stable regimen for at least 6 weeks prior to randomization. Topical, intraarticular, nasal, inhalation and ocular steroid therapy are not considered "whole body" and are permitted;
(F) Use of continuous hormone replacement therapy unless the regimen was stable during the last 6 weeks (third week) prior to the screening visit and there is no plan to change the regimen during the study;
(G) Recent MI (within 3 months prior to screening visit (-3 weeks) or between screening and randomized visit), unstable angina leading to hospitalization, uncontrolled cardiac arrhythmia, CABG, PCI, cervical Endovascular surgery or surgical intervention for arterial surgery or stent placement, stroke, transient ischemic attack (TIA), peripheral vascular disease;
(H) Will undergo PCI, CABG, carotid or peripheral revascularization scheduled during the study;
(I) History of New York Heart Association (NYHA) Class III or IV heart failure (see Table 1) within the last 12 months;
(J) systolic blood pressure >180 mmHg or diastolic blood pressure >110 mmHg at screening or randomized visit;
(K) Patients who have undergone or are planned to receive treatment for plasmatic ataxia within 2 months (-3 weeks) before screening visit, between screening and randomization.
(L) History of hemorrhagic stroke;
(M) History of PCSK9 loss of function (ie, genetic or sequence mutations) or homozygous familial hypercholesterolemia;
(N) New cancer or active progression of cancer within the last 5 years, excluding appropriately treated basal cell skin cancer, squamous cell carcinoma, or intraepithelial cervical cancer;
(O) History of positive HIV test;
(P) Patients taking any active study drug within one month or five half-lives, whichever is longer;
(Q) Patients who have not been previously instructed on a cholesterol-reduced diet prior to the screening visit (-3 weeks);
(R) Patients withdrawing consent during screening (starting with signed ICF);
(S) Unstable body weight, defined by the Investigator, defined as a variation of more than 5 kg within 2 months prior to the screening visit;
(T) BMI> 45kg / m 2, or bariatric surgery, weight loss program or plan to start a weight-loss drugs during the study;
(U) Recent onset of weight-loss drug (ie, within 3 months prior to screening visit, or between screening and randomization) or recent bariatric surgery (within the last 6 months) and aggressiveness determined by the investigator. In a general weight loss stage;
(V) Patients who have not been treated with insulin for at least 6 months prior to the screening visit, or who have not been treated with a stable insulin regimen for at least 3 months before the screening visit (ie, type of insulin, General timing/frequency, dosing mode or pattern such as basal only (
(W) It is not a stable insulin dosage for at least 3 months prior to screening (ie total daily insulin dosage varies by more than 30%, as judged by the investigator), or Possibly determined to require enhanced insulin/antihyperglycemic regimen in the course of the study (eg, addition of new drugs, insulin escalation regimen, etc.);
(X) Other hypoglycemic drugs taken by the patient are not stable for at least 3 months before the screening visit;
(Y) A recent history of decompensation of diabetes within the two months prior to the screening visit (ie diabetic ketoacidosis or hyperosmolar hyperglycemia (HHS));
(Z) undergoing or planning to undergo renal replacement therapy during the study (eg, hemodialysis, renal transplant, etc.);
(Aa) Presence of clinically significant uncontrolled endocrine disorders known to affect serum lipids or lipoproteins. Patients undergoing thyroid replacement therapy had thyroxine dosages stable for at least 3 months prior to screening and their sensitive thyroid stimulating hormone (s-TSH) levels at the screening visit normal range. Can be included if within ±10% of;
(Bb) Laboratory findings during the screening period (excluding pregnancy tests, not including randomized tests):
(I) Serum TG>400 mg/dL (4.52 mmol/L) (one repeat test allowed);
(Ii) Positive serum or urine pregnancy test for women of childbearing potential;
(Iii) hepatitis B surface antigen or hepatitis C antibody positive test;
(Iv) eGFR<15 mL/min/1.73 m 2 due to 4 variable changes in the diet for renal disease;
(MDRD) formula;
(V) ALT or AST>3xULN (one repeat test allowed); or (vi) creatine phosphokinase (CPK)>3xULN (one repeat test allowed); or ( cc) Conditions/situations such as:
(I) Patients with short life expectancy;
(Ii) requirements for combination treatments that may bias the primary assessment;
(Iii) unable to meet certain protocol requirements (eg need for hospitalization, ability to visit the study, etc.);
(Iv) Patient was the investigator or co-investigator, research assistant, pharmacist, study coordinator, other staff member or his relatives directly involved in the implementation of the protocol;
(V) non-cooperative or conditions in which the patient may not be compatible with the study procedure;
(Vi) some technical/administrative reason that makes it impossible to randomize patients in the study; or (vii) the investigator or co-investigator's decision may interfere with the safe completion of the trial. , Clinically significant abnormalities identified at screening that limit the evaluation of endpoints such as major systemic disease, and patients with short life expectancy.

(2)実対照薬および/または必須のバックグラウンド療法に関する除外基準:バックグラウンド療法に対するすべての禁忌またはそれぞれの国産品表示に示される使用(適切な場合)の警告/警戒 (2) Exclusion Criteria for Active Controls and/or Essential Background Therapies: All Contraindications to Background Therapies or Warnings/Warnings of Use (if Appropriate) Shown on Each Domestic Product Label
(3)アリロクマブの現在の知識に関連する除外基準:
(a)アリロクマブまたはアリロクマブのいずれかの成分に対する過敏症;
(b)妊娠中または授乳中の女性;
(c)避妊の非常に効果的な方法によって保護されていない出産の可能性のある女性(ICF、および/または特定の地方の要件の場合は地方のプロトコル補遺で定義される)および/または妊娠検査を受ける意思がないもしくは受けることができない女性。出産の可能性がある女性は、スクリーニングおよび組み入れ来院時に妊娠検査が陰性であることを確認する必要がある。彼女らは、研究治療の全期間を通して、IMPの最後の注射後少なくとも10週間、効果的な避妊法を使用しなければならない。適用された避妊法は、「人による使用のための医薬品の登録について技術的要件の調和に関する国際会議。M3(R2):人の臨床試験および医薬品の製造承認の実施のための非臨床安全性試験に関するガイダンス。ICH。2009年6月:1−25」による避妊の非常に効果的な方法のための基準を満たさなければならなかった。閉経後の女性は、少なくとも12か月間は無月経でなければならない。
(3) Exclusion criteria related to the current knowledge of Arilokumab:
(A) Hypersensitivity to Arilocumab or any component of Arilocumab;
(B) Pregnant or lactating women;
(C) Women of childbearing potential (as defined in the ICF and/or the local protocol addendum for certain local requirements) not protected by highly effective methods of contraception and/or pregnancy. Women who are unwilling or unable to undergo the test. Women of childbearing potential need to confirm a negative pregnancy test at screening and enrollment visits. They must use effective contraception throughout the study treatment for at least 10 weeks after the last injection of IMP. The contraceptive method applied is: “International Conference on Harmonization of Technical Requirements for Registration of Drugs for Human Use. M3 (R2): Nonclinical Safety for the Implementation of Human Clinical Trials and Drug Manufacturing Approval. Trial guidance. ICH. June 2009: 1-25” had to meet the criteria for a very effective method of contraception. Postmenopausal women must be amenorrhea for at least 12 months.
研究治療
調査医薬品
滅菌アリロクマブ製剤は、自動注射器(事前充填ペンとしても知られている)で、ショ糖、ヒスチジン、およびポリソルベート20をともに1mLの体積で含む水性緩衝液pH6.0中に75mg/mLおよび150mg/mLの濃度で供給された。アリロクマブの無菌プラセボは、患者が注射トレーニングを行うため、およびプラセボ治療群の患者のために、タンパク質を1mL容量の事前充填ペンとして添加することなく、アリロクマブと同じ製剤で調製された。スクリーニング期間中、患者(または別の指定された者)は、IMPの最初の投与前に、事前充填ペンを使用してプラセボ自己注射トレーニングを実行する必要があった。
Research Therapeutics Investigational Drugs A sterile Arilocumab formulation is an automatic syringe (also known as a prefilled pen), 75 mg/mL in an aqueous buffer pH 6.0 containing sucrose, histidine, and
アリロクマブにランダム化された患者の場合、初期用量は75mgであり、Q2Wに1回皮下投与された。8週目のLDL−C値が≧70mg/dL(1.81mmol/L)である場合、アリロクマブにランダム化された患者について、12週目で用量を盲検法で150mg Q2Wに増加させた。プラセボにランダム化された患者には、24週間の治療期間中、Q2Wで皮下注射が行われた。
For patients randomized to alilocumab, the initial dose was 75 mg, given subcutaneously once in Q2W. If the LDL-C value at
投与経路および投与方法
事前充填されたペントレーニングガイド(自動注射器トレーニングガイド)が現場に提供され、使用説明書(使用のための自動注射器)が患者に提供された。IMPの各投与は、腹部、大腿部、または上腕の外側領域(三角筋領域)への1mLの皮下注射からなった。別の併用薬がIMP注射と同じ部位に注射されている場合、患者はIMPの投与に別の部位を使用するように勧められた。
Route of Administration and Method of Administration A pre-filled pen training guide (auto-injector training guide) was provided to the scene and instructions for use (auto-injector for use) were provided to the patient. Each dose of IMP consisted of a 1 mL subcutaneous injection in the lateral region of the abdomen, thigh, or upper arm (the deltoid region). Patients were advised to use another site for the administration of IMP if another concomitant drug was injected at the same site as the IMP injection.
IMPは、自己注射によって、または別の指定された者(例えば、配偶者、血縁者など)によって投与することができる。研究中に指定された者がアリロクマブを患者に注射することになった場合、注射を投与する前にこの者が適切に訓練されていることが確認された。IMPの管理を計画している者は、研究スタッフによって訓練された。 IMP can be administered by self-injection or by another designated person (eg, spouse, relatives, etc.). If a person designated during the study decided to inject a patient with alilocumab, it was confirmed that this person was properly trained prior to administering the injection. Those planning to manage the IMP were trained by research staff.
訓練中および研究の過程で必要に応じて、患者(または注射を投与する別の指定された者(例えば、配偶者、血縁者など))に指示が与えられた。最初の来院時、および必要に応じて他の来院時に綿密な監督およびフィードバックが行われた。 Instructions were given to the patient (or another designated person (eg, spouse, relative, etc.) receiving the injection) during training and as needed during the course of the study. Close supervision and feedback were provided at the first visit and, if necessary, at other visits.
使用済みの事前充填ペンは、患者に提供された鋭利物容器に廃棄された。皮下IMP注射は、解剖学的領域(例えば、右大腿部、次に左大腿部または右腹部、次いで左腹部)内で回転させることが推奨された。また、患者には、研究中に別の解剖学的領域(例えば、大腿部、腹部、または上腕の外側領域など)に注射する選択肢があった。 The used prefilled pen was discarded in the sharps container provided to the patient. Subcutaneous IMP injections were recommended to be rotated within the anatomical region (eg, right thigh, then left thigh or right abdomen, then left abdomen). Patients also had the option of injecting into another anatomical region during the study, such as the thigh, abdomen, or lateral region of the upper arm.
患者はIMPを冷蔵庫に保存するように求められた。投与前に、IMPを室温の安全な場所に約30〜40分間置く。その後、IMPはできるだけ早く投与する必要がある。 The patient was asked to store the IMP in the refrigerator. Prior to administration, place IMP in a safe place at room temperature for about 30-40 minutes. Then IMP should be administered as soon as possible.
投与の時期
スクリーニング期間中、患者または指定された者は、最初のIMP注射前に、事前に充填されたペンを使用してプラセボ自己注射トレーニングを実施しなければならなかった。
Timing of Dosing During the screening period, patients or designated persons had to perform placebo self-injection training using pre-filled pens prior to the first IMP injection.
ランダム化来院時に、最初のIMP注射は、直接の現場スタッフの監督下で、患者または別の指定された物(例えば、配偶者、血縁者など)によって現場で行われた。この研究では、この最初の注射後少なくとも30分間、患者を調査現場で監視した。指定された者が研究の過程で変更した場合、新しい指定された者はプラセボで訓練された。 At the randomized visit, the first IMP injection was performed on-site by the patient or another designated entity (eg, spouse, relative, etc.) under the supervision of direct field staff. In this study, patients were monitored on-site for at least 30 minutes after this first injection. If the designated person changed during the course of the study, the new designated person was trained on placebo.
次に、IMP皮下注射は、Q2Wから最後の注射まで、臨床外で実施された。現場来院と同日に注射が予定されている場合は、採血が完了した後にIMPが投与された。例外的な症例では、患者が研究現場で注射を行うことを望み、その現場で注射の投与に対応する準備ができた場合、それも許可された。 Subcutaneous IMP injections were then performed off-clinically, from Q2W to the last injection. If injections were scheduled for the same day as the site visit, IMP was administered after blood collection was completed. In exceptional cases, if the patient wanted to make an injection at the study site and was ready to administer the injection at that site, it was also allowed.
IMPは、理想的にはほぼ同じ時刻に、Q2Wの皮下に投与されるべきである。しかしながら、ウィンドウ期間が±3日間であっても許可された。時刻は、患者の好みに基づいていた。 IMP should ideally be administered subcutaneously Q2W at about the same time. However, it was allowed even if the window period was ±3 days. The time of day was based on patient preference.
誤って、または他の状況により、注射を逃した日から7日を超えて遅れるか、または完全に逃した場合、患者は、遅れた注射を投与せずにIMP投与の元のスケジュールに戻るように要求された。誤って、または他の状況により、注射を逃した日から7日以下で遅れた場合、患者は遅れて注射を行い、次に、IMP投与の元のスケジュールを再開するように要求された。 Inadvertently or due to other circumstances, if the injection is delayed more than 7 days after the missed date or is missed altogether, the patient should return to the original schedule of IMP administration without giving the delayed injection. Requested to. Inadvertently or due to other circumstances, if the injection was delayed by less than 7 days from the day it was missed, the patient was required to make the delayed injection and then restart the original schedule of IMP administration.
非治験薬
薬物はバックグラウンド療法または潜在的な救助薬物のいずれかであるため、以下のクラスの薬物が非IMPとして特定された:
(a)スタチン;
(b)コレステロール吸収阻害剤(エゼチミブ);
(c)胆汁酸結合性金属イオン封鎖剤(例えば、コレスチラミン、コレスチポール、コレセベラム);
(d)ニコチン酸;
(e)フィブラート(例えば、フェノフィブラート);
(f)オメガ3脂肪酸(毎日≧1000mg);および
(g)インスリン。
Non-investigated Drugs The following classes of drugs have been identified as non-IMP because the drug is either a background therapy or a potential rescue drug:
(A) statin;
(B) cholesterol absorption inhibitor (ezetimibe);
(C) bile acid-binding sequestrants (eg cholestyramine, colestipol, colesevelam);
(D) nicotinic acid;
(E) fibrate (eg, fenofibrate);
(F) omega-3 fatty acids (>1000 mg daily); and (g) insulin.
スタチンを含むバックグラウンドLMTについて、現場は、患者の安全性監視および管理のための国産品ラベルに従った。患者は、研究中に他のLMTの有無にかかわらず、患者が耐性であるスタチン療法の安定した最大用量/レジメンを受けた。脂質プロファイル値は、ランダム化後に得られたサンプルから盲検化された。それでも、安全上の理由から、患者のバックグラウンドLMTを決定するために、現場はTG警告を認識した。 For background LMT containing statins, the site followed a domestic product label for patient safety monitoring and management. Patients received a stable maximal dose/regimen of statin therapy for which they were resistant, with or without other LMTs during the study. Lipid profile values were blinded from the samples obtained after randomization. Nevertheless, for safety reasons, the scene recognized the TG alert to determine the patient's background LMT.
スクリーニング来院(3週目)から24週目の来院まで、バックグラウンドLMTは変更されなかった。この期間中、治験責任医師の判断によると、重要な懸念(限定されないが、中央研究所によって投稿されたTG警告を含む)がそのような変更を保証した例外的な状況にない限り、他のスタチンもしくは他のLMTの用量調整、中止、または開始は行われなかった。反復テストによって確認されたTG警告について、治験責任医師は調査を行い、患者を管理し、医学的判断に従ってバックグラウンドLMTを変えた。
Background LMT was unchanged from the Screening Visit (Week 3) to
患者が薬物療法に耐性であり、安定した用量を維持した場合、すべてのフィブラートがエントリー時に許可された。患者が研究の過程でフィブラートの導入を必要とした場合(すなわち、TG警告に対応する救助治療として)、フェノフィブラートのみを追加することが許可された。バックグランドLMTおよびインスリンはスポンサーから提供された。患者は、地域の規制に準拠してこれらの薬を入手した。 All fibrates were allowed at entry if the patient was resistant to drug therapy and maintained a stable dose. If patients needed the introduction of fibrate during the course of the study (ie, as a rescue treatment in response to TG alerts), only fenofibrate was allowed to be added. Background LMT and insulin were provided by the sponsor. Patients obtained these medications in compliance with local regulations.
盲検化手順
アリロクマブおよびアリロクマブ用のプラセボは、同じように適合した事前充填ペンで提供され、同じように包装され、盲検者を保護するためのラベル付けが含まれた。各治療キットには、スポンサーからのコンピュータープログラムによって生成された番号でラベルされた。治療キットの番号は、患者のランダム化時と、1日24時間、週7日間利用可能なIVRS/IWRSを介してスケジュールされた患者の来院時に、治験責任医師によって取得された。
Blinding Procedures Arilocumab and placebo for Arilocumab were provided with similarly adapted prefilled pens, similarly packaged, and included labeling to protect the blind. Each treatment kit was labeled with a number generated by a computer program from the sponsor. The treatment kit number was obtained by the Investigator at the time of patient randomization and at the patient's visit scheduled via IVRS/IWRS available 24 hours a day, 7 days a week.
二重盲検設計によれば、研究患者、治験責任医師および研究現場の職員は、研究治療について盲検のままであり、以下に説明する状況を除き、ランダム化(治療コード)へのアクセスはなかった。 The double-blind design allows study patients, investigators, and staff at the study site to remain blind to study treatment and will not have access to randomization (treatment code) except in the circumstances described below. There wasn't.
有害事象
治療コードは、任意の疑わしい予期しない重篤な副作用(SUSAR)、すなわち、任意の予期しない(CIBの特定のセクションによる)、および治験責任医師および/またはスポンサーの判断に従ったIMPの使用に合理的に関連した、重大な有害事象を保健当局に報告するために医薬品安全性監視部門によって盲検化されなかった。
Adverse Events Treatment Code indicates any suspicious and unexpected serious side effects (SUSAR), ie any unexpected (according to a particular section of the CIB), and use of IMP at the discretion of the Investigator and/or Sponsor Was not blinded by the Drug Safety Oversight Department to report a serious adverse event reasonably related to
脂質パラメータ
中央研究所によって実施されたランダム化来院後に得られた血液サンプルからの脂質パラメータ値は、達成されたLDL−Cレベルに基づいて患者の治療群を推測することができなかったため、現場に伝達されなかった。スポンサーの運用チームは、最終的なデータベースロックが発生するまで、患者の識別に関連する脂質パラメータへのアクセスはなかった。安全の目的で、ランダム化が治験責任医師に送信された後はいつでも、TG値≧500mg/dLの場合にTGが警告を出す。
Lipid Parameters Lipid parameter values from blood samples obtained after randomized visits conducted by the Central Research Institute were unable to infer the treatment group of patients based on the achieved LDL-C levels, thus Not transmitted. The sponsor's operations team had no access to the lipid parameters associated with patient identification until the eventual database lock occurred. For safety purposes, the TG will alert if the TG value ≧500 mg/dL, whenever randomization is sent to the Investigator.
二重盲検治療期間の終わり(24週目の来院)に、治験責任医師は、標準的な慣行に従って患者の脂質を管理し続けた。ランダム化後の脂質値は、原資料で編集され、スポンサーと共有されなかった。
At the end of the double-blind treatment period (
抗アリロクマブ抗体
患者の抗アリロクマブ抗体の結果は、研究の進行中に現場に伝達されなかった。スポンサーの運用チームは、最終的なデータベースロックが発生するまで、患者識別番号に関連付けられた抗アリロクマブ抗体の結果へのアクセスはなかった。患者の抗アリロクマブ抗体価の決定に関与する検査技師は、運用チームから除外され、いずれもの潜在的な非盲検化を防ぐためのプロセスが設定された。
Anti-arilocumab antibody Patient anti-arilocumab antibody results were not transmitted to the site during the course of the study. The sponsor's operations team had no access to the anti-arilocumab antibody results associated with the patient identification number until the eventual database lock occurred. Laboratory technicians involved in determining the patient's anti-arilocumab titer were excluded from the operations team and a process in place to prevent any potential unblinding.
研究中のランダム化コードの破壊
AEの場合、患者を治療するためにIMPの知識が必要とされる状況でコードが破壊された。可能であれば、コードを破壊する前にモニタリングチーム/研究医との連絡が開始された。すべての呼び出しは、呼び出しの日時、監視チーム内で連絡した者の名前、患者ID、リクエストの文書、および非盲検化の有無の決定を含むように、監視チームによって適切に文書化された。
Randomized Code Breaking During the Study In the case of AE, the code was broken in situations where knowledge of IMP was required to treat patients. If possible, contact with the monitoring team/research physician was initiated before breaking the code. All calls were properly documented by the surveillance team, including the date and time of the call, the name of the person contacted within the surveillance team, the patient ID, the request documentation, and the unblinded decision.
現地でどのシステムが使用されたかに応じて、および/またはその目的のためにスポンサーから提供された任意の他の電話番号を呼び出すことにより、音声自動応答システム(IVRS)/ウェブ自動音声応答システム(IWRS)の適切なモジュールを使用することにより、いつでもコードブレイキングを実行することができる。しかしながら、症例の非盲検化をする前に、治験担当医師に連絡して症例について検討することが好ましかった。ブラインドが破壊された場合、治験責任医師は、日付、時刻、およびコードが破損した理由を文書化し、この情報をe−CRFの適切なページに報告するように要求された。非盲検化の理由を文書化する場合、治験責任医師は、IMPの性質に関するいずれの詳細も提供しなかった。治験責任医師は、データベースを閉鎖するまで、IMPの詳細をスポンサーの代表者または任意のスタッフメンバーに漏らさなかった。さらに、フォーム(例:AE、SAE)に記入する場合、フォームの研究治療は開示されなかった。 Depending on which system was used locally and/or by calling any other telephone number provided by the sponsor for that purpose, an interactive voice response system (IVRS)/web automated voice response system ( Code breaking can be performed at any time by using the appropriate modules of IWRS). However, it was preferable to contact the investigator to discuss the case before unblinding the case. If the blinds were destroyed, the investigator was required to document the date, time, and reason why the code was corrupted and report this information on the appropriate page of the e-CRF. When documenting the reasons for unblinding, the investigator did not provide any details regarding the nature of the IMP. The Investigator did not reveal IMP details to the sponsor's representative or any staff member until the database was closed. Further, when filling out forms (eg, AE, SAE), the form's study treatment was not disclosed.
暗号解読資料はまた、「24時間警報システム」を担当するエンティティにも保存されたが、このシステムは非常に例外的な場合にのみ使用する必要がある(すなわち、IVR/IWRシステムが利用できない、または治験責任医師および/もしくは現場スタッフに連絡できないなど)。しかしながら、IVRSを使用して非盲検化することが好ましい選択肢である。治験責任医師は、臨床監視チームから、地方の暗号解読資料の入手可能性について通知を受けた。関連する「24時間警報システム」電話番号を含む患者カードが、研究に参加するすべての患者に提供された。規制当局の報告要件に準拠するために、SAEの一部に対して(つまり、関連性があり予期しないSAEの一部に対して)スポンサーによる非盲検化も許可された。 The decryption material was also stored in the entity responsible for the "24 hour alarm system", but this system should only be used in very exceptional cases (ie IVR/IWR systems are not available, Or unable to contact the investigator and/or field staff). However, open-labeling using IVRS is the preferred option. The Investigator was notified by the clinical oversight team about the availability of local cryptanalysis material. Patient cards containing the associated "24-hour alert system" telephone numbers were provided to all patients participating in the study. In order to comply with regulatory reporting requirements, sponsor unblinding was also permitted for some SAEs (ie for some relevant and unexpected SAEs).
コードが破壊された場合、患者はIMP投与を永久に中止した。 If the code was destroyed, the patient permanently discontinued IMP administration.
治療群に患者を割り当てる方法
治療キット番号のランダム化リストは、スポンサーによって中央で生成された。IMP(アリロクマブ75または150mgキット、またはプラセボキット)は、このリストに従って包装された。
How to Assign Patients to Treatment Groups A randomized list of treatment kit numbers was centrally generated by the sponsor. IMP (
試験供給運用管理者は、治療キット番号のランダム化リストを提供し、研究生物統計学者は、集中治療配分システム提供者にランダム化スキームを提供した。次に、この集中治療配分システム提供者は、患者に治療を配分する患者ランダム化リストを生成した。 The trial supply operations manager provided a randomized list of treatment kit numbers, and the research biostatist provided a randomized scheme to the intensive care allocation system provider. The intensive care distribution system provider then generated a patient randomized list that allocated the treatments to the patients.
患者は、二重盲検治療期間中にプラセボまたはアリロクマブのいずれかを受けるようにランダム化された。ランダム化アリロクマブ:プラセボ比は2:1であった。ランダム化された各患者には、対応する治療キット番号(再補給来院)がいくつかあり、これらは集中治療配分システムによって配分された。ランダム化は、糖尿病の種類(つまり、1型対2型)によって層別化された。
Patients were randomized to receive either placebo or arilocumab during the double-blind treatment period. The randomized arilocumab:placebo ratio was 2:1. Each randomized patient had a corresponding treatment kit number (refill visit), which was distributed by the intensive care distribution system. Randomization was stratified by type of diabetes (ie,
治療キット番号は、ランダム化来院(1目日、0週目)時に集中治療配分システムを使用して配分され、次に、12週目に再補給来院として、必要に応じて予定外の来院時に配分された。 Treatment kit numbers will be distributed using the intensive care distribution system at randomized visits (1st day, 0th week) and then at 12th week as a restocking visit, as needed at unscheduled visits. Was distributed.
アリロクマブ治療群の患者については、12週目に配分された治療キットは、漸増規則に従う8週目のLDL−Cレベルに基づいていた。研究現場およびスポンサーに対して盲目的な方法で進めるために、中央検査室と集中治療配分システムの提供者との間で定期的なデータ転送が計画された。 For patients in the arilocumab treatment group, treatment kits distributed at 12 weeks were based on LDL-C levels at 8 weeks according to the titration rules. Periodic data transfer was planned between the central laboratory and the provider of the intensive care distribution system to proceed blindly to the research site and sponsors.
患者をランダム化する前に、治験責任医師または被指名人は、集中治療配分システムに連絡しなければならなかった。 The investigator or designee had to contact the intensive care distribution system before randomizing patients.
ランダム化された患者は、そのログファイルから文書化されるように、集中治療配分システムからの治療キット番号で登録および割り当てられた患者として定義された。この研究では、患者を複数回ランダム化することはできなかった。集中治療配分システムに連絡せずに治療が使用された場合、患者はランダム化されていないとみなされ、研究から外された。 Randomized patients were defined as patients enrolled and assigned a treatment kit number from the Intensive Care Distribution System, as documented from their log files. The study could not randomize patients multiple times. If treatment was used without contacting the intensive care allocation system, patients were considered non-randomized and removed from the study.
現場の選択に応じて、IVRSおよびIWRSという2種類の集中治療配分システムが使用された。 Two types of intensive care distribution systems were used, IVRS and IWRS, depending on site preference.
包装およびラベリング
二重盲検治療期間の間、アリロクマブまたはアリロクマブ用のプラセボのいずれかの二重盲検治療キットは、子供に耐性のあるパッケージに6つの事前充填ペンを含むように調製された。ブラインドを保護するために、注射用のすべての二重盲検治療キットの箱は同じ外観と感触を有していたため、二重盲検ラベルでラベリングされる。
Packaging and Labeling During the double-blind treatment period, a double-blind treatment kit, either arilocumab or a placebo for arilocumab, was prepared to contain 6 pre-filled pens in a child-resistant package. To protect the blinds, the boxes of all double-blind treatment kits for injection had the same look and feel and are therefore labeled with double-blind labels.
注射のための二重盲検治療キットに加えて、スクリーニングの来院でのランダム化前に行われる注射投与について患者に指示する目的で、アリロクマブ充填済みペン用のプラセボを1つ含むトレーニングキットを調製した(3週目、来院1)。必要と思われる場合は、ランダム化前に追加のトレーニングキットを使用して、アリロクマブのプラセボによる2回目の注射トレーニングを実施した。プラセボによる注射トレーニングが実施され、CRFに文書化された。これには、患者にIMPを投与した指定者が研究中に変更されたかどうかも含まれる。 In addition to a double-blind treatment kit for injections, a training kit with a placebo for an alilocumab-filled pen was prepared to instruct the patient about injections to be performed prior to randomization at screening visits Yes (Third week, Visit 1). If deemed necessary, a second injection training with placebo of alilocumab was performed using an additional training kit prior to randomization. Injection training with placebo was performed and documented on the CRF. This also includes whether the designated person who administered IMP to the patient changed during the study.
包装は投与スケジュールに従った。ラベリングの内容は、現地の規制仕様および要件に準拠していた。 Packaging followed the dosing schedule. The labeling content complied with local regulatory specifications and requirements.
保存条件および保存期間
治験責任医師または他の許可された人(例えば、薬剤師)は、現地の規制、ラベル付け仕様、ポリシー、および手順に従って、IMPを堅牢安全な場所に保存する責任があった。IMPの保存条件の調節、特に温度の調節(冷蔵保存など)および使用中の安定性に関する情報とIMPの取り扱いに関する指示は、スポンサーが提供する規則に従って管理された。
Storage Conditions and Storage Period The investigator or other authorized person (eg, pharmacist) was responsible for storing the IMP in a robust and secure location in accordance with local regulations, labeling specifications, policies, and procedures. Information on IMP storage conditions, in particular temperature control (such as refrigeration) and stability during use and instructions on handling the IMP were governed by rules provided by the sponsor.
IMPは、現場で+2℃から+8℃(36°Fから46°F)の冷蔵庫に保存された。現場の冷蔵庫の温度を毎日チェックし、ログシートに記録した。調査現場に保存されていたIMPは、ラベルに示された保存条件に従って、治験責任医師または被指名人または他の権限のある人の責任の下、適切な鍵のかかった部屋に保存された。 The IMP was stored in the refrigerator at +2°C to +8°C (36°F to 46°F) in the field. The temperature of the on-site refrigerator was checked daily and recorded on a log sheet. The IMP, which had been stored at the study site, was stored in an appropriate locked room under the responsibility of the investigator or designee or other authorized person, according to the storage conditions indicated on the label.
研究現場来院での患者へのIMPキットの提供後、研究現場から患者の冷蔵庫へのIMPキットの輸送のための適切な準備は整っていた。 After provision of the IMP kit to the patient at the study site visit, the appropriate preparations for transport of the IMP kit from the study site to the patient's refrigerator were in place.
研究の評価項目
ベースライン特性には、標準的な人口統計学(例えば、年齢、人種、体重、身長など)、病歴を含む疾患特性、および各患者の投薬歴が含まれた。
Study endpoints Baseline characteristics included standard demographics (eg age, race, weight, height, etc.), disease characteristics including medical history, and medication history for each patient.
主要な有効性評価項目
主要な有効性評価項目は、治療順守に関係なくすべてのLDL−C値(ITT推定値)を使用して、包括解析(ITT)集団のベースラインから24週までのLDL−Cの変化率であった。変化率は、100×(24週で計算されたLDL−C値−ベースラインで計算されたLDL−C値)/ベースラインで計算されたLDL−C値、として定義された。
Primary Efficacy Endpoints The primary efficacy endpoints are all LDL-C values (ITT estimates) regardless of treatment adherence, LDL from baseline to 24 weeks in the global analysis (ITT) population. It was the rate of change of -C. The rate of change was defined as 100 x (LDL-C value calculated at 24 weeks-LDL-C value calculated at baseline)/LDL-C value calculated at baseline.
ベースラインで計算されたLDL−C値は、最初の二重盲検IMP注射前に得られた最後のLDL−Cレベルであった。24週目に計算されたLDL−Cは、24週目の分析ウィンドウ内で得られたLDL−Cレベルであった。8〜24週のすべての計算されたLDL−C値(予定または予定外、絶食または絶食なし)は、上記の定義に従って、適切な場合、主要な有効性評価項目の値を提供するために使用することができた。 The LDL-C value calculated at baseline was the final LDL-C level obtained before the first double-blind IMP injection. The LDL-C calculated at 24 weeks was the LDL-C level obtained within the 24 week analysis window. All calculated LDL-C values from 8-24 weeks (scheduled or unscheduled, fasted or not fasted) are used to provide values for the primary efficacy endpoint, when appropriate, according to the definition above. We were able to.
主要な安全性評価項目
安全パラメータ(AE、検査室パラメータ、バイタルサイン)は、研究を通じて評価された。安全性データの観察は、次の通りであった:
(a)事前治療期間は、署名されたインフォームドコンセントから二重盲検IMP注射の最初の投与までとして定義された。
(b)治療緊急有害事象(TEAE)期間は、二重盲検IMP注射の最初の投薬からIMP注射の最終投与までの時間+70日(10週間)として定義された。これは、二重盲検IMPの停止後10週間まで、治療の残存効果が期待されるためである。
(c)治療後期間は、TEAE期間の終了日の翌日から、すべてのSAEおよびAESIのいずれか最後の来訪者の消散/安定化までの時間として定義された。
Main safety endpoints Safety parameters (AE, laboratory parameters, vital signs) were assessed throughout the study. Observations of safety data were as follows:
(A) Pretreatment period was defined as from signed informed consent to the first dose of double-blind IMP injection.
(B) The treatment emergency adverse event (TEAE) period was defined as the time from the first dose of double-blind IMP injection to the last dose of IMP injection + 70 days (10 weeks). This is because the residual effect of the treatment is expected up to 10 weeks after stopping the double-blind IMP.
(C) The post-treatment period was defined as the time from the day after the end of the TEAE period to the resolution/stabilization of all SAE and AESI last visitors.
AEは、医薬品を投与した患者または臨床調査患者での任意の不都合な医学的出来事であり、必ずしもこの治療と因果関係を有する必要はなかった。 An AE is any adverse medical event in a patient who received the drug or in a clinical study patient and did not necessarily have a causal relationship with this treatment.
SAEは、任意の用量で以下のような任意の不都合な医学的出来事であった:
(a)死に至った;
(b)生命を脅かすものであった。「重篤」の定義における「生命を脅かす」という用語は、患者が事象の時点で死のリスクにさらされている事象を指した;それがより深刻な場合に、仮説的に死を引き起こし得た事象については言及しなかった;
(c)入院を必要とするか、または既存の入院期間を延長する;
(d)永続的または重大な障害/無能力をもたらした;
(e)先天異常/出生時欠損であった;または
(f)医学的に重要な事象であった。
SAE was any adverse medical event at any dose, such as:
(A) died;
(B) It was life-threatening. The term "life-threatening" in the definition of "serious" refers to an event in which the patient is at risk of death at the time of the event; if it is more serious, it may hypothetically cause death. No mention was made of the events that occurred;
(C) requires hospitalization or extends existing hospital stay;
(D) resulted in permanent or significant disability/incapacity;
(E) Birth defects/natal defects; or (f) Medically significant events.
迅速な報告が他の状況、例えば、即座に命にかかわる重大な出来事ではなく、死亡または入院をもたらさないが、患者を危険にさらす可能性があり、または医療もしくは上記の定義にリストされている他の結果の1つを防ぐための外科的介入(具体的な手段または矯正治療)を必要とする重大な医学的事象において適切であるかどうかの決定において、医学的および科学的判断を用いるべきである。 Prompt reporting does not result in death or hospitalization in other situations, for example, not an immediate life-threatening event, but may endanger the patient, or is listed in the medical or definition above. Medical and scientific judgment should be used in determining whether it is appropriate in a serious medical event requiring surgical intervention (specific measures or orthodontic treatment) to prevent one of the other outcomes Is.
医学的に重要な事象の以下のリストは、医学的に重要な事象とみなすべき状態を決定するためのガイドラインとして役立つことが意図された。リストは、網羅的であることを意図していなかった:
(a)アレルギー性気管支けいれん、血液疾患(すなわち、無顆粒球症、再生不良性貧血、骨髄形成不全、骨髄異形成、汎血球減少症など)、またはけいれん(発作、てんかん、てんかん発作、欠勤など)の緊急治療室または自宅での集中治療;
(b)薬物依存または薬物乱用の発生;
(c)ALT>3×ULN+総ビリルビン>2×ULNまたは無症状のALT増加>10×ULN;
(d)自殺未遂または自殺を示唆する何らかの事象。
(e)失神、意識喪失(採血の結果として記録された場合を除く);
(f)水疱性皮膚発疹;
(g)研究中に診断されたまたは研究中に悪化したがん;
(h)慢性神経変性疾患(新たに診断された)または研究中に悪化;ならびに
(i)医薬品を介した感染性病原体の伝播の疑いがある場合(例えば、製品汚染)、感染性病原体の伝播が疑われる。
The following list of medically significant events was intended to serve as a guideline for determining which conditions should be considered medically significant events. The list was not intended to be exhaustive:
(A) Allergic bronchospasm, blood diseases (ie, agranulocytosis, aplastic anemia, myelodysplasia, myelodysplasia, pancytopenia, etc.) or convulsions (seizures, epilepsy, seizures, absences, etc.) ) Emergency room or home intensive care;
(B) occurrence of drug dependence or substance abuse;
(C) ALT>3×ULN+total bilirubin>2×ULN or asymptomatic ALT increase>10×ULN;
(D) Attempted suicide or any event suggesting suicide.
(E) Syncope, loss of consciousness (except when recorded as a result of blood sampling);
(F) Bullous skin rash;
(G) Cancers diagnosed during the study or aggravated during the study;
(H) Chronic neurodegenerative disease (newly diagnosed) or worse during the study; and (i) Transmission of the infectious agent if there is a suspicion of transmission of the infectious agent via a drug (eg, product contamination). Is suspected.
特に注目すべき有害事象(AESI)は、事前に指定された方法で監視、文書化、および管理する必要があるAE(重大または非重大)である。この研究では、AESIは、次の通りであった:
(a)ALTの増加:ALT≧3×ULN(ベースラインALT<ULNの場合)またはALT≧2倍のベースライン値(ベースラインALT≧ULNの場合);
(b)アレルギー事象:治験責任医師がアレルギーとみなす(またはアレルギー成分を含む)アレルギー性薬物反応および/または局所注射部位反応。これは、治験責任医師の医学的判断に従って、過敏症/アレルギーのさらなる評価のために他の医師と相談する必要があったAESIとして報告する必要がある;
(c)妊娠:治験中または治験薬の最終投薬後の70日以内に、女性患者または男性患者のパートナー(女性パートナーおよび現地の規制当局により許可されている場合)で発生する妊娠。妊娠は、すべてのケースでAESIとして記録された。妊娠は、1つまたはそれ以上のSAE基準を満たしている場合にのみ、SAEとして認定された。研究に含まれる女性患者の妊娠の場合、研究製品は中止された。妊娠の追跡調査は、結果が決定されるまで必須であった;
(d)IMPによる症候性の過剰投薬。過量投薬(偶発的または意図的)は、治験責任医師が疑う事象または患者が自発的に通知する事象(体系的な注射回数に基づくものではない)であり、意図された治療間隔内の意図された用量の少なくとも2回(つまり、2回以上の注射が7暦日未満に投与される)として定義され、「症候性過剰投薬(偶発的または意図的)」という用語を使用して報告され、これは、括弧内の状況(例えば、「症候性過剰投薬(偶発的)」または「症候性過剰投薬(意図的)」)を示した;患者を監視し、適切な対症療法を開始した。過剰投薬の状況は、個別のAE/SAEフォームに入力された逐語的および症状(ある場合)で明確に特定された。無症候性の過剰投薬は、標準的なAEとして報告するよう要求された;
(e)神経学的事象:追加の検査/手順および/または専門医への紹介を必要とする神経学的事象は、AESIとして報告するよう要求された。事象に追加の検査/手順および/または専門家への紹介が必要なかった場合、標準AEとして報告することが要求された;ならびに
(f)神経認知事象:すべての神経認知事象はAESIとみなされた。
Adverse events (AESI) of particular interest are AEs (significant or non-significant) that need to be monitored, documented, and managed in a predesignated manner. In this study, AESI was as follows:
(A) Increase in ALT: ALT≧3×ULN (if baseline ALT<ULN) or ALT≧2 times baseline value (if baseline ALT≧ULN);
(B) Allergic event: Allergic drug reaction and/or local injection site reaction that the investigator considers to be allergic (or includes allergic components). This should be reported as an AESI that, according to the investigator's medical judgment, had to consult with another physician for further evaluation of hypersensitivity/allergy;
(C) Pregnancy: Pregnancy that occurs in a female or male patient partner (if permitted by the female partner and local regulatory authority) during the study or within 70 days after the last dose of study drug. Pregnancy was recorded as AESI in all cases. Pregnancy was certified as SAE only if it met one or more SAE criteria. In the case of pregnancy of a female patient included in the study, the study product was discontinued. Follow-up of pregnancy was mandatory until outcome was determined;
(D) Symptomatic overdose with IMP. Overdose (accidental or intentional) is an event suspected by the investigator or voluntarily notified by the patient (not based on a systematic number of injections) and is intended within the intended treatment interval. Defined as at least two doses (that is, two or more injections administered in less than seven calendar days), reported using the term "symptomatic overdose (accidental or intentional)", This indicated the situation in brackets (eg, "symptomatic overdose (accidental)" or "symptomatic overdose (intentional)"; the patient was monitored and appropriate symptomatic treatment was initiated. Overdose status was clearly identified by verbal and symptom (if any) entered on individual AE/SAE forms. Asymptomatic overdose was required to report as standard AE;
(E) Neurological events: Neurological events requiring additional examination/procedure and/or referral to a specialist were required to be reported as AESI. If the event did not require any additional testing/procedures and/or referrals to experts, it was required to report as a standard AE; and (f) Neurocognitive events: All neurocognitive events were considered AESI. It was
二次的有効性評価項目
本研究の主要な二次的評価項目は、以下の通りであった:
(a)有効性治療期間中のすべてのLDL−C値を使用した、ベースラインから24週目までの計算されたLDL−Cの変化率(治療時推定値);
(b)ベースラインから24週目までの測定されたLDL−Cの変化率(ITT推定値);
(c)ベースラインから12週目までの計算されたLDL−Cの変化率(ITT推定値);
(d)ベースラインから12週目までの測定されたLDL−Cの変化率(ITT推定値);
(e)ベースラインから24週目までの非HDL−Cの変化率(ITT推定値);
(f)ベースラインから24週目までのApo Bの変化率(ITT推定値);
(g)ベースラインから24週目までの総コレステロールの変化率(ITT推定値);
(h)24週でLDL−C<70mg/dLに達した患者の割合(治療中の推定値);
(i)24週でLDL−C<50mg/dLに達した患者の割合(治療中の推定値);
(j)24週で非HDL−C<100mg/dLに達する患者の割合(治療時推定値);
(k)24週で非HDL−C<80mg/dLに達した患者の割合(治療時推定値);
(l)ベースラインから24週目までのLp(a)の変化率(ITT推定値);
(m)ベースラインから24週目までのHDL−Cの変化率(ITT推定値);
(n)ベースラインから24週目までのTGの変化率(ITT推定値);
(o)ベースラインから24週目までのLDL−C粒子数の変化率(ITT推定値);ならびに
(p)ベースラインから24週目までのLDL−C粒子サイズの変化率(ITT推定値)。
Secondary efficacy endpoints The primary secondary endpoints of this study were:
(A) Efficacy rate of change in calculated LDL-C from baseline to
(B) Change rate of LDL-C measured from baseline to week 24 (ITT estimated value);
(C) Calculated rate of change in LDL-C from baseline to Week 12 (ITT estimate);
(D) Rate of change in LDL-C measured from baseline to Week 12 (ITT estimate);
(E) Non-HDL-C change rate from baseline to week 24 (ITT estimate);
(F) Apo B change rate from baseline to 24th week (ITT estimated value);
(G) Rate of change in total cholesterol from baseline to week 24 (ITT estimate);
(H) Percentage of patients who reached LDL-C <70 mg/dL at 24 weeks (estimate during treatment);
(I) Percentage of patients who reached LDL-C <50 mg/dL at 24 weeks (estimate during treatment);
(J) Proportion of patients reaching non-HDL-C<100 mg/dL at 24 weeks (treatment estimate);
(K) Percentage of patients who reached non-HDL-C <80 mg/dL at 24 weeks (estimate at the time of treatment);
(L) Change rate of Lp(a) from baseline to 24th week (ITT estimated value);
(M) Change rate of HDL-C from baseline to 24th week (ITT estimated value);
(N) Change rate of TG from baseline to 24th week (ITT estimated value);
(O) Change rate of LDL-C particle number from baseline to 24th week (ITT estimated value); and (p) Change rate of LDL-C particle size from baseline to 24th week (ITT estimated value) ..
以下の糖尿病関連評価項目もまた研究で測定された:
(a)ベースラインから12週目および24週目までのHbA1cの絶対変化(ITTおよび治療中の推定値);
(b)ベースラインから12週目および24週目までのFPGの絶対変化(ITTおよび治療中の推定値);
(c)ベースラインから12週目および24週目までの1日のインスリン総投与量の絶対変化(ITTおよび治療中の推定値);ならびに
(d)ベースラインから12週目および24週目までのグルコース減少治療の数の絶対変化(ITTおよび治療推定値)。
The following diabetes-related endpoints were also measured in the study:
(A) Absolute change in HbA1c from baseline to weeks 12 and 24 (estimated during ITT and treatment);
(B) Absolute change in FPG from baseline to Week 12 and Week 24 (ITT and treatment estimates);
(C) Absolute change in total daily insulin dose from baseline to Weeks 12 and 24 (ITT and estimated during treatment); and (d) Baseline to
この研究の他の有効性評価項目には、以下が含まれる:
(a)ベースラインから12週目までの計算されたLDL−Cの変化率(治療時推定値);
(b)ベースラインから第12週および第24週までの測定されたLDL−Cの変化率(治療時推定値);
(c)ベースラインから12週目(ITTおよび治療中の推定量)および24週目(治療中の推定量)までの非HDL、Apo B、総コレステロール、Lp(a)、HDL−C、およびTGの変化率;
(d)12週目(ITTおよび治療中の推定値)および24週目(ITT推定値)で計算されたLDL−C<50およびさらに<70mg/dLに達する患者の割合;
(e)12週目および24週目に計算されたLDL−Cのベースラインから50%以上減少した患者の割合(ITT推定値);
(f)12週目(ITTおよび治療中の推定値)および24週目(ITTの推定値)で非HDL−C<80mg/dL、およびさらには<100mg/dLに達した者の割合;
(g)12週目および24週目でApo B<80mg/dLに達した患者の割合(ITTおよび治療中の推定値);
(h)ベースラインから12週(ITTおよび治療中の推定値)および24週(治療中の推定値)までのLDL−C粒子数およびサイズの変化率;
(i)ベースラインから12週目および24週目までのTGRL、Apo A−1、およびApo C−IIIの変化率(ITTおよび治療推定値);
(j)ベースラインから12週目および24週目までの比率Apo B/Apo A−1およびTC/HDL−Cの絶対変化(ITTおよび治療中の推定値);
(k)<8%または≧8%のベースラインA1cによると、12週目および24週目で計算されたLDL−C<70および<50mg/dLに達した患者の割合(ITTおよび治療中の推定値);ならびに
(l)<中央値A1cまたは≧中央値A1cのベースラインA1cに従って、12週目および24週目に計算されたLDL−C<70mg/dLおよび<50mg/dLに達した患者の割合(ITTおよび治療中の推定値)。
Other endpoints of this study's efficacy included:
(A) Calculated change rate of LDL-C from baseline to Week 12 (estimated value at treatment);
(B) Rate of change in LDL-C measured from baseline to week 12 and week 24 (estimate at treatment);
(C) Non-HDL, Apo B, total cholesterol, Lp(a), HDL-C, from baseline to Week 12 (ITT and estimated dose during treatment) and Week 24 (estimated amount during treatment), and TG change rate;
(D) Percentage of patients reaching LDL-C <50 and even <70 mg/dL calculated at Week 12 (ITT and treatment estimates) and Week 24 (ITT estimates);
(E) Percentage of patients with 50% or more reduction from baseline LDL-C calculated at weeks 12 and 24 (ITT estimate);
(F) Percentage of those who reached non-HDL-C <80 mg/dL, and even <100 mg/dL at Week 12 (ITT and treatment estimates) and Week 24 (ITT estimates);
(G) Percentage of patients who reached Apo B<80 mg/dL at 12 and 24 weeks (ITT and treatment estimates);
(H) Percent change in LDL-C particle number and size from baseline to week 12 (ITT and treatment estimates) and 24 weeks (treatment estimates);
(I) Percent change in TGRL, Apo A-1, and Apo C-III from baseline to Week 12 and Week 24 (ITT and treatment estimate);
(J) Absolute changes in the ratios Apo B/Apo A-1 and TC/HDL-C from baseline to Week 12 and Week 24 (ITT and estimated values during treatment);
(K) Percentage of patients who reached LDL-C <70 and <50 mg/dL calculated at
研究手順
週0のウィンドウ期間は+3日であった。8、12、および24週のウィンドウ期間は±3日であった。4、20、および32週のウィンドウ期間は±7日であった。1日目/組み入れ来院後のすべての来院について、1回の来院日が変更された場合、次の来院は図1に概説されている元のスケジュールに従って行われた。
Study Procedure The
採血
脂質パラメータ(例えば、TC、LDL−C、HDL−C、TG、非HDL−C、Apo A、Apo B、Apo C−III、Lp(a)、LDL粒子サイズおよび数)ならびにさらに血漿グルコース関する採血を含むすべての採血が、午前中、絶食状態(すなわち、一晩、少なくとも10から12時間速く喫煙を控える)で、研究を通じて、すべての現場来院のためのIMP注射の前に行われた。採血前の48時間以内のアルコール摂取および24時間以内の激しい運動は勧められなかった。患者が空腹状態ではなかった場合、血液サンプルは収集されず、翌日(またはこの日付にできるだけ近い日)に患者に新しい予約が、絶食の指示(上記の条件を参照)とともに与えられた。
Blood collection related to lipid parameters (eg TC, LDL-C, HDL-C, TG, non-HDL-C, Apo A, Apo B, Apo C-III, Lp(a), LDL particle size and number) and also plasma glucose. All blood draws, including blood draws, were done in the morning in the fasted state (ie, refrain from smoking fast for at least 10 to 12 hours overnight) prior to IMP injection for all field visits throughout the study. Alcohol consumption within 48 hours and vigorous exercise within 24 hours prior to blood collection were not recommended. If the patient was not hungry, no blood sample was collected and the next day (or as close as possible to this date) the patient was given a new appointment with a fasting instruction (see conditions above).
臨床検査
図1に概説された研究スケジュールおよび以下のガイドラインに従って、検査室データを収集した:
(a)血液学:4週目と20週目を除くすべての来院;必要に応じて、治験責任医師の臨床的判断に基づいて、0週目に実施することができる;
(b)化学:来院3および6を除くすべての来院;すべての患者に対して0週目に実施すべき血漿グルコースを除き、治験責任医師の臨床的判断に基づいて、必要に応じて、0週目に実施することができる;
(c)HbA1c:スクリーニング時ならびに0、12、および24週目;
(d)脂質パネル:スクリーニング時ならびに0、8、12、20、および24週目;
(e)ベータ定量化によるLDL−Cの測定:0、12、および24週目;
(f)他の脂質評価(Apo B、Apo A−1、Apo C−III、LDL粒子サイズと数、Lp[a]):0、12、および24週目;
(g)肝臓パネル:来院3および6を除くすべての来院;必要に応じて、治験責任医師の臨床的判断に基づいて、0週目に実施することができる。総ビリルビン値が正常範囲を超える場合、共役ビリルビンと非共役ビリルビンへの分化が自動的に行われる;
(h)クレアチンホスホキナーゼ(CPK):来院3および6を除くすべての来院;必要に応じて、治験責任医師の臨床的判断に基づいて、0週目に実施することができる;
(i)B型肝炎表面抗原:スクリーニングのみ;
(j)C型肝炎抗体:スクリーニング時および24週目;研究中にALTが増加した場合、C型肝炎抗体を測定する必要がある;研究中にC型肝炎抗体が陽性であった場合、反射テストが実施された;
(k)妊娠検査(妊娠の可能性がある女性のみ):スクリーニング時のみの血清妊娠検査、0週目と24週目の尿妊娠検査;
(l)甲状腺刺激ホルモン:甲状腺ホルモン補充療法を受けている患者のみのスクリーニング;
(m)C−ペプチド:スクリーニングのみ;
(n)0週目のみのPCSK9レベル;ならびに
(o)抗アリロクマブ抗体(0週目、12週目、および24週目)。
Laboratory Testing Laboratory data were collected according to the study schedule outlined in Figure 1 and the following guidelines:
(A) Hematology: All visits except 4th and 20th week; if necessary, it can be performed at 0th week based on the clinical judgment of the investigator;
(B) Chemistry: All visits except
(C) HbA1c: at screening and at
(D) Lipid panel: at screening and at 0, 8, 12, 20, and 24 weeks;
(E) Measurement of LDL-C by beta quantification:
(F) Other lipid evaluations (Apo B, Apo A-1, Apo C-III, LDL particle size and number, Lp[a]): 0, 12 and 24 weeks;
(G) Liver panel: All visits except
(H) Creatine Phosphokinase (CPK): All visits except
(I) Hepatitis B surface antigen: screening only;
(J) Hepatitis C antibody: at screening and at
(K) Pregnancy test (only for women who may be pregnant): serum pregnancy test only at screening, urine pregnancy test at 0 and 24 weeks;
(L) Thyroid stimulating hormone: screening only for patients undergoing thyroid hormone replacement therapy;
(M) C-peptide: screening only;
(N) PCSK9 levels only at
尿サンプリング
尿検査は、スクリーニングおよび24週目の来院時に実施された。ディップスティックを実施し、pH、比重、および血液、タンパク質、グルコース、ケトン、硝酸塩、白血球エステラーゼ、ウロビリノーゲン、およびビリルビンの存在を評価した。ディップスティックが異常な場合、標準顕微鏡検査が実施された。顕微鏡検査により、赤血球(RBC)、RBC塊、白血球(WBC)、WBC塊、上皮細胞(移行、腎尿細管、および扁平上皮)、円柱(硝子、上皮、WBC、RBC、顆粒、脂肪、細胞、幅広い、ろう状)、結晶(リン酸三、シュウ酸カルシウム、リン酸カルシウム、炭酸カルシウム、尿酸、無定形、ビウリン酸アンモニウム、ビリルビン、ロイシン、チロシン、シスチン)、細菌、酵母出芽、酵母菌糸、トリコモナス、卵型脂肪体、脂肪、粘液、および精子)の存在について評価した。
Urine sampling Urinalysis was performed at screening and at
スクリーニングおよび24週目の来院時に、アルブミン:クレアチニン比を計算するために、アルブミンおよびクレアチニンについてスポット尿検査を実施した。何らかの臨床的に関連する異常な検査値は、関係する患者に対して何らかの決定を下す前に確認のためにすぐに再チェックされた。
Spot urinalysis for albumin and creatinine was performed to calculate the albumin: creatinine ratio at screening and at
身体検査
一般的な身体検査が実施された。新しい臨床的に重大な異常またはベースラインからの悪化が組み入れ後に検出された場合、AEが報告され、患者はさらなる治験および/または治験責任医師の医学的判断に従って専門家の診察を考慮された。
Physical Examination A general physical examination was performed. If new clinically significant abnormalities or exacerbations from baseline were detected after enrollment, AEs were reported and patients were considered for further clinical trials and/or specialist consultation according to the investigator's medical judgment.
血圧と心拍数
血圧(BP)は、標準化された条件下で座位で、同日のおよそ同時間に、同じ腕で、同じ装置を用いて(患者が少なくとも5分間、座位で快適に休んだ後)測定された。値は、e−CRFに記録された;収縮期血圧と拡張期血圧の両方が記録された。最初のスクリーニング来院で、BPは両腕で測定された。この来院時に拡張期血圧が最も高い腕が決定され、研究中、この腕で血圧が測定された。この最高値はe−CRFに記録された。
Blood pressure and heart rate Blood pressure (BP) is measured in the sitting position under standardized conditions, approximately at the same time on the same day, on the same arm, using the same device (after the patient has been comfortably rested in the sitting position for at least 5 minutes). Was measured. Values were recorded on e-CRF; both systolic and diastolic blood pressures were recorded. At the first screening visit, BP was measured on both arms. The arm with the highest diastolic blood pressure was determined at this visit and blood pressure was measured on this arm during the study. This highest value was recorded on the e-CRF.
心拍数は、BPの測定時に測定された。 Heart rate was measured at the time of BP measurement.
体重および身長
患者は、下着または非常に軽い衣服を着用し、靴は着用せず、膀胱を空にして、体重を測定した。同じスケールが研究全体で使用された。
Weight and Height Patients wore underwear or very light clothing, no shoes, empty bladder and weighed. The same scale was used throughout the study.
自己報告された身長は許容できないため、身長を測定した。 Height was measured as self-reported height is unacceptable.
iTAQ問診
iTAQは、問診表を完了する前の4週間にわたる治療の受容性を評価するための患者報告結果(PRO)尺度であった。患者は8週目および24週目の来院時に完了するように要求された。
iTAQ Interview The iTAQ was a patient-reported outcome (PRO) measure to assess the acceptability of treatment for 4 weeks prior to completing the questionnaire. Patients were required to complete at the
インスリンログ
患者は、各来院の少なくとも7日前に(該当する場合、基礎インスリンおよび食事時インスリンについて)患者の毎日のインスリン用量を記録し、この情報を次の研究来院に持参すために、インスリンログを完了するように指示された。患者は、研究来院の7日よりも前に1日のインスリン用量を記録することができるが、しかしながら、各来院前の最後の7日間に収集された情報のみがCRFに入力された。
Insulin Log Patients should record their daily insulin doses (for basal and prandial insulin, if applicable) at least 7 days before each visit and use this information to bring this information to the next study visit. Was instructed to complete. Patients can record their daily insulin doses prior to 7 days prior to study visit, however, only the information collected during the last 7 days prior to each visit was entered into the CRF.
結果
計76人のT1DMを有する患者および441人のT2DMを有する患者が登録された。
Results A total of 76 patients with T1DM and 441 patients with T2DM were enrolled.
T1DMでランダム化された76人の患者全員が治療され、したがって、安全集団に含まれた。T1DMを有する2人のランダム化された患者(両方ともアリロクマブ群)は、包括解析(ITT)集団に含まれなかった。 All 76 patients randomized with T1DM were treated and therefore included in the safety population. Two randomized patients with T1DM, both in the arilocumab group, were not included in the global analysis (ITT) population.
441人のT2DMを有する患者のうち、3人は治療されず(1人はアリロクマブ群および2人はプラセボ群)、したがって、安全集団には含まれなかった。12人のランダム化されたT2DMを有する患者(7人はアリロクマブ群および5人はプラセボ群)はITT集団に含まれなかった。 Of the 441 patients with T2DM, 3 were untreated (1 in the arilocumab group and 2 in the placebo group) and were therefore not included in the safety population. Twelve patients with randomized T2DM (7 in the arilocumab group and 5 in the placebo group) were not included in the ITT population.
ベースラインまたは24週目までの分析ウィンドウのいずれかで利用可能な計算されたLDL−C値がない場合、患者はITT集団に含まれなかった。
Patients were not included in the ITT population if there were no calculated LDL-C values available at either baseline or the analysis window up to
研究患者
T1DMを有する6人(7.9%)の患者は、研究治療を早々に中止した(3人[5.9%]はアリロクマブ群(2人の患者はAEにより中止した)および3人[12.0%]はプラセボ群(2人の患者はAEにより中止した)。アリロクマブ群の3人の患者全員もまた研究期間を完了しなかったが、プラセボ群では2人の患者もまた研究期間を完了せず、1人の患者が研究期間の完了まで研究に残った。
Study Patients Six (7.9%) patients with T1DM discontinued study treatment prematurely (3 [5.9%] in the alilocumab group (2 patients discontinued due to AE) and 3). [12.0%] placebo (2 patients discontinued due to AE) All 3 patients in the arilocumab group also did not complete the study period, but 2 patients in the placebo group also studied The period was not completed and one patient remained in the study until the completion of the study period.
T2DMを有する39人(8.8%)の患者が研究治療を早期に中止した(アリロクマブ群で29人(9.9%)、プラセボ群で10人(6.8%))。アリロクマブ群の29人の患者のうち、18人の患者も研究を中止し、11人の患者が、研究期間が完了するまで研究に残った。プラセボ群では、10人の患者のうち、7人の患者も研究期間を完了せず、3人の患者が、研究期間が完了するまで残った。 Thirty-nine (8.8%) patients with T2DM discontinued study treatment prematurely (29 (9.9%) in the alilocumab group and 10 (6.8%) in the placebo group). Of the 29 patients in the arilocumab group, 18 also discontinued the study and 11 remained in the study until the study period was completed. Of the 10 patients in the placebo group, 7 patients did not complete the study period and 3 patients remained until the study period was completed.
人口統計およびベースライン特性
ベースライン特性は、一般的に、アリロクマブおよびプラセボ群で類似していた。T1DMを有するランダム化患者の60.5%は男性であり、T2DMを有する患者のランダム化患者の54.2%は男性であった。平均年齢が56.1(SD=9.5)であるT1DMを有する患者は、平均年齢が64.0(SD=9.1)であるT2DMを有する患者よりも若かった。T1DMを有する患者の平均BMIは30.0kg/m2(SD=5.9)であり、T2DMを有する患者の平均BMIは32.6kg/m2(SD=5.06)であった。
Demographic and Baseline Characteristics Baseline characteristics were generally similar for the arilocumab and placebo groups. 60.5% of the randomized patients with T1DM were male and 54.2% of the randomized patients with T2DM were male. Patients with T1DM with an average age of 56.1 (SD=9.5) were younger than those with T2DM with an average age of 64.0 (SD=9.1). Patients with T1DM had an average BMI of 30.0 kg/m 2 (SD=5.9) and patients with T2DM had an average BMI of 32.6 kg/m 2 (SD=5.06).
ベースラインで計算されたLDL−Cは、T2DMを有する患者(平均=110.4mg/dL、SD=37.3)よりもT1DMを有する患者(平均=121.0mg/dL、SD=51.2)で高かった。ベースライン時のトリグリセリドは、T1DM中央値(Q1:Q3)=102.0mg/dL(76.5:135.0)の患者では、T2DM中央値(Q1:Q3)=147.0mg/dL(105.0:212.0)の患者よりも低かった。 LDL-C calculated at baseline was higher in patients with T1DM (mean=121.0 mg/dL, SD=51.2) than in patients with T2DM (mean=110.4 mg/dL, SD=37.3). ) Was high. Triglyceride at baseline had median T1DM (Q1:Q3)=102.0 mg/dL (76.5:135.0) and median T2DM (Q1:Q3)=147.0 mg/dL(105 0.0:212.0).
他の脂質パラメータに関して、T1DM患者は、ベースラインからのLDL−C粒子数(LS平均)の減少率が12週目で40.7%、および24週目で44.4%であり、LDL−C粒子サイズの減少率が12週目で2.3%、および24週目で2.3%であった。ApoC3は、これらの患者では12週目で6.9%、および24週目で7.5%減少した。 For other lipid parameters, T1DM patients had a reduction in LDL-C particle count (LS mean) from baseline of 40.7% at 12 weeks and 44.4% at 24 weeks, indicating that LDL- The reduction rate of C particle size was 2.3% at the 12th week and 2.3% at the 24th week. ApoC3 was reduced in these patients by 6.9% at 12 weeks and 7.5% at 24 weeks.
他の脂質パラメータに関して、T2DM患者は12週で37.6%、24週で38.3%のLDL−C粒子数(LS平均)のベースラインからの減少率、ならびに12週で2.6%、および24週で2.8%のLDL−C粒子サイズの減少率を示した。これらの患者のApoC3は、12週で6.3%、24週で5.8%減少した。 For other lipid parameters, T2DM patients had a decrease in LDL-C particle count (LS mean) from baseline of 37.6% at 12 weeks, 38.3% at 24 weeks, and 2.6% at 12 weeks. , And the reduction rate of LDL-C particle size of 2.8% at 24 weeks. ApoC3 in these patients was reduced by 6.3% at 12 weeks and 5.8% at 24 weeks.
T1DMの患者では、糖尿病およびインスリン使用の平均期間は治療群間で類似していた。糖尿病の平均期間は34.92年(SD=12.67)であり、インスリン使用の平均期間は34.81年(SD=12.77)であった。T2DM患者では、治療群間で糖尿病およびインスリン使用の平均期間は類似していた。T2DMにおける糖尿病の平均期間は16.75年(SD=8.13)であり、インスリン使用の平均期間は8.01年(SD=6.90)であった。 In patients with T1DM, the mean duration of diabetes and insulin use was similar between treatment groups. The average duration of diabetes was 34.92 years (SD = 12.67) and the average duration of insulin use was 34.81 years (SD = 12.77). In T2DM patients, the mean duration of diabetes and insulin use was similar between treatment groups. The average duration of diabetes in T2DM was 16.75 years (SD=8.13) and the average duration of insulin use was 8.01 years (SD=6.90).
高コレステロール血症の期間は、一般的に、治療群間ならびにT1DMおよびT2DMの患者間で類似していた。 The duration of hypercholesterolemia was generally similar between treatment groups and between patients with T1DM and T2DM.
研究者によって報告されたように、スタチン不耐性の患者の割合は、T1DMの患者で31.6%、T2DMの患者で23.8%であった。 As reported by the investigators, the proportion of patients with statin intolerance was 31.6% in patients with T1DM and 23.8% in patients with T2DM.
ランダム化時にフィブラートを投与された患者の割合は、T1DMの患者で2.6%、T2DMの患者で8.8%であった。 The proportion of patients receiving fibrate at randomization was 2.6% in patients with T1DM and 8.8% in patients with T2DM.
ランダム化時のコレステロール吸収阻害剤(エゼチミブを含む)の患者の割合は、プラセボ群(7.6%)よりもアリロクマブ群(13.6%)、特にT2DMを有する患者において高く、患者45人(15.3%)対10人の患者(6.8%)であった。 The proportion of patients with cholesterol absorption inhibitors (including ezetimibe) at randomization was higher in the alilocumab group (13.6%) than in the placebo group (7.6%), especially in patients with T2DM, with 45 patients ( 15.3%) vs. 10 patients (6.8%).
心血管の病歴およびリスク因子は、一般的に治療群間で類似していた。T1DMとT2DMの患者の間で次の相違が観察された。
(1)ASCVDは、T2DM対T1DM患者において、冠動脈心疾患(34.7%対15.8%)および脳卒中(8.2%対2.6%)の頻度が高く、PAD(4.3%対9.2%)の頻度が低いT1DM患者よりもT2DM患者の方が頻度が高かった(40.1%対21.1%)。
(2)ASCVDのない患者のうち、T1DMを有する患者の56.7%が標的臓器損傷(微量アルブミン尿、マクロアルブミン尿)および/またはCKDおよび/または網膜症を有していたのに対し、T2DMを有する患者では39.4%であった。ASCVDのない患者の間でも、以下の追加の心血管リスク因子が観察された:
(a)T2DM患者よりもT1DMでより頻繁である:現在喫煙者(20.0%対14.0%)、前増殖性糖尿病性網膜症(36.7%対12.9%)および増殖性糖尿病性網膜症(20.0%対5.7%)。
(b)T1DMの頻度はT2DM患者よりも低い:高血圧(55.0%対84.8%)、微量アルブミン尿(10.0%対19.7%)、低HDL−C(16.7%対28.0%)
(3)ASCVDのない患者における3つ以上の追加のCVリスク因子の存在は、T1DM患者の45%およびT2DM患者の55.7%で観察された。
Cardiovascular history and risk factors were generally similar between treatment groups. The following differences were observed between patients with T1DM and T2DM.
(1) ASCVD has a high frequency of coronary heart disease (34.7% vs. 15.8%) and stroke (8.2% vs. 2.6%) in patients with T2DM vs. T1DM and PAD (4.3%). T2DM patients were more frequent (40.1% vs. 21.1%) than T1DM patients with a lower frequency (vs. 9.2%).
(2) Among patients without ASCVD, 56.7% of patients with T1DM had target organ damage (microalbuminuria, macroalbuminuria) and/or CKD and/or retinopathy, In patients with T2DM it was 39.4%. The following additional cardiovascular risk factors were also observed among patients without ASCVD:
(A) More frequent in T1DM than in T2DM patients: current smokers (20.0% vs 14.0%), preproliferative diabetic retinopathy (36.7% vs 12.9%) and proliferative. Diabetic retinopathy (20.0% vs 5.7%).
(B) T1DM frequency is lower than in T2DM patients: hypertension (55.0% vs 84.8%), microalbuminuria (10.0% vs 19.7%), low HDL-C (16.7%). 28.0%)
(3) The presence of three or more additional CV risk factors in patients without ASCVD was observed in 45% of T1DM patients and 55.7% of T2DM patients.
全体として、T1DMおよびT2DM患者は、両方の治療群で高強度および中程度の強度のスタチンで治療され、患者の割合が高いほど中強度のスタチンで治療された(58.9%)。全体として、T1DMおよびT2DM患者の59.0%はスタチンのみで治療された。 Overall, T1DM and T2DM patients were treated with high and moderate intensity statins in both treatment groups, with a higher proportion of patients treated with moderate intensity statins (58.9%). Overall, 59.0% of T1DM and T2DM patients were treated with statins alone.
調査医薬品への安全集団の曝露を表10に要約する。 The exposure of the safety population to the study drug is summarized in Table 10.
有効性
一次有効性評価項目
アリロクマブは、T1DMおよびT2DMを有する患者のITT集団において、ベースラインから24週目までの計算されたLDL−Cの変化のパーセンテージにおいてプラセボよりも優れていた(表11および12、ならびに図2および3に示す)。T1DM患者のうち、LDL−C<70mg/dL(<1.8mmol/L)を達成する個体の割合は、アリロクマブ群で70.2%、プラセボ群で5.1%(P<0.0001)であり、LDL−C<50mg/dL(1.3mmol/L)を達成した個体の割合は、アリロクマブ群で55.1%、プラセボ群で0%であった(P値は計算不能)。T2DM患者のうち、LDL−C<70mg/dL(<1.8mmol/L)を達成した個体の割合は、アリロクマブ群で76.4%、プラセボ群で7.4%(P<0.0001)であり、LDL−C<50mg/dL(1.3mmol/L)を達成した個体の割合は、アリロクマブ群で50.7%、プラセボ群で2.4%であった(P<0.0001)。有効性の主要評価項目の感度分析では、両方の集団で同様の結果が示された(データを示さず)。
Efficacy Primary Efficacy Endpoints Arilokumab was superior to placebo in the percentage of calculated LDL-C change from baseline to
二次有効性評価項目
アリロクマブは、非HDL−C、ApoB、総コレステロール、およびLp(a)のレベルのベースラインから24週目までの有意な減少(プラセボとの差)、ならびにT1DMおよびT2DMを有する患者のITT集団におけるHDL−Cの増加をもたらした(それぞれ、表13および14に示す)。
Secondary efficacy endpoints Arilocumab significantly reduced non-HDL-C, ApoB, total cholesterol, and Lp(a) levels from baseline to week 24 (difference from placebo), and T1DM and T2DM. Resulted in an increase in HDL-C in the ITT population of patients with (shown in Tables 13 and 14, respectively).
糖尿病関連の評価項目
全体として、FPGおよびHbA1c、およびグルコース減少治療は、両方の治療群のT1DMおよびT2DMを有する患者において経時的に安定したままであった。
Diabetes-related endpoints Overall, FPG and HbA1c, and glucose-lowering treatment remained stable over time in patients with T1DM and T2DM in both treatment groups.
HbA1cに関して、T1DMコホートでは、アリロクマブ群において、平均HbA1c%は、ベースラインで7.84%(SD=0.94)、平均絶対変化=−0.03%(0.6)であったが、一方、プラセボ群において、平均HbA1cは、ベースラインで7.68%(0.78)、平均絶対変化=−0.23%(0.36)であった。T2DMコホートでは、アリロクマブ群において、平均HbA1cは、ベースラインで7.52%(0.96)、平均絶対変化=0.18%(0.74)であったが、一方、プラセボ群において、平均HbA1cは、ベースラインで7.54%(1.02)、平均絶対変化=0.06%(0.66)であった。 Regarding HbA1c, in the T1DM cohort the mean HbA1c% was 7.84% (SD=0.94) at baseline and mean absolute change=−0.03% (0.6) in the allilocumab group. On the other hand, in the placebo group, the average HbA1c was 7.68% (0.78) at baseline and the average absolute change was −0.23% (0.36). In the T2DM cohort, mean HbA1c was 7.52% (0.96) at baseline and mean absolute change=0.18% (0.74) in the allilocumab group, while mean in placebo group. HbA1c had a baseline of 7.54% (1.02) with an average absolute change of 0.06% (0.66).
FPGに関して、T1DMコホートでは、アリロクマブ群において、平均FPGは、ベースラインで173mg/dL(SD=70.6)、平均絶対変化=−0.03mg/dL(0.6)であり、一方、プラセボ群において、平均FPGは、ベースラインで166.5mg/dL(75.6)、平均絶対変化=14.6mg/dL(75.9)であった。T2DMコホートでは、アリロクマブ群において、平均FPGは、ベースラインで154.1mg/dL(50.1)、平均絶対変化=9.5mg/dL(61.8)であり、一方、プラセボ群において、平均FPGは、ベースラインで153.5mg/dL(52.5)、平均絶対変化=10.0mg/dL(47.0)であった。 Regarding FPG, in the T1DM cohort, the mean FPG was 173 mg/dL (SD=70.6) at baseline, mean absolute change=−0.03 mg/dL (0.6) in the allilocumab group, while placebo. In the group, the mean FPG was 166.5 mg/dL (75.6) at baseline, mean absolute change=14.6 mg/dL (75.9). In the T2DM cohort, the mean FPG was 154.1 mg/dL (50.1) at baseline and mean absolute change = 9.5 mg/dL (61.8) at baseline in the arilocumab group, while mean in the placebo group. FPG was 153.5 mg/dL (52.5) at baseline, mean absolute change=10.0 mg/dL (47.0).
安全性
合計で344人の患者(51人のT1DMおよび293人のT2DM)がアリロクマブに曝露され、170人の患者(25人のT1DMおよび145人のT2DM)がプラセボに曝露された。
Safety A total of 344 patients (51 T1DM and 293 T2DM) were exposed to arilocumab and 170 patients (25 T1DM and 145 T2DM) were exposed to placebo.
全体として、T1DMまたはT2DMを有する患者の安全集団において、治療中に発生した有害事象(TEAE)を有する患者の割合は、治療群全体で類似していた(表15を参照されたい)。 Overall, in the safe population of patients with T1DM or T2DM, the proportion of patients with adverse events (TEAEs) that occurred during treatment was similar across treatment groups (see Table 15).
TEAEは、以下の器官分類(SOC)でより頻繁に(≧10%)報告された:
(a)感染症および侵入(アリロクマブで21.8%対プラセボで21.8%);
(b)胃腸障害(アリロクマブで13.1%対プラセボで12.4%);
(c)筋骨格および結合組織障害(アリロクマブで21.5%対プラセボで15.9%);ならびに
(d)一般的な障害および投与部位の状態(アリロクマブで11.0%対プラセボで8.8%)
TEAEs were reported more frequently (≧10%) with the following organ classifications (SOC):
(A) Infection and invasion (21.8% for allilocumab vs. 21.8% for placebo);
(B) Gastrointestinal disorders (13.1% for Arilokumab vs. 12.4% for placebo);
(C) Musculoskeletal and connective tissue disorders (21.5% for allilocumab vs. 15.9% for placebo); and (d) General disorders and administration site conditions (11.0% for allilocumab vs 8. for placebo). 8%)
PTレベルで、アリロクマブ群で最も頻繁に報告されたTEAE(≧2%)と、プラセボ群との差が≧0.5%であったのは、アリロクマブ群の頻度の減少によった:筋肉痛(4.4%対1.8%)、関節痛(2.9%対1.8%)、気管支炎(2.6%対0.6%)、めまい(2.6%対1.2%)、および末梢浮腫(2.0%対0.6%)。対照的に、プラセボ群で最も頻繁に報告されたTEAE(≧2%)と、アリロクマブ群との差が≧0.5%であったのは、インフルエンザ(2.3%対2.9%)、四肢の痛み(1.7%対2.9%)、低血糖(1.7%対2.4%)、咳(1.5%対2.9%)、筋骨格痛(1.2%対2.4%)、上気道感染(0.9%対2.4%)、高血糖(0.9%対2.4%)、および肺炎(0.6%対2.4%)であった。 Differences in PT levels between TEAEs (≥2%) most frequently reported in the alilocumab group and placebo groups were ≥0.5% due to the reduced frequency of the alilocumab group: myalgia (4.4% vs 1.8%), arthralgia (2.9% vs 1.8%), bronchitis (2.6% vs 0.6%), dizziness (2.6% vs 1.2). %), and peripheral edema (2.0% vs. 0.6%). In contrast, the most frequently reported TEAEs (≥2%) in the placebo group and ≥0.5% between the allilocumab group were flu (2.3% vs. 2.9%). , Limb pain (1.7% vs 2.9%), hypoglycemia (1.7% vs 2.4%), cough (1.5% vs 2.9%), musculoskeletal pain (1.2%). % Vs 2.4%), upper respiratory tract infection (0.9% vs 2.4%), hyperglycemia (0.9% vs 2.4%), and pneumonia (0.6% vs 2.4%). Met.
全体として、アリロクマブ群の25人の患者(7.3%)およびプラセボ群の14人の患者(8.2%)の患者で治療中に発生したSAEが報告された。いずれかの治療群の2人以上の患者で報告されたSAE(PTレベル)は、肺炎(アリロクマブ群の1人の患者(0.3%)対プラセボ群の2人の患者(1.2%))、椎骨孔狭窄(アリロクマブ群の2人の患者(0.6%)対プラセボ群の患者なし)、および尿路感染症(2人の患者(0.6%)対プラセボ群の患者なし)であった。最初のIMP投与(来院3)の1か月後、プラセボ群のT2DM患者で、心筋梗塞による1人の死亡が報告された。全体として、アリロクマブ群の17人の患者(4.9%)とプラセボ群の4人の患者(2.4%)がTEAEを経験し、永続的な治療中止をもたらした。PTレベルで、治療群の2人以上の患者で永続的な治療中止につながるTEAE患者の割合は次の通りであった:頭痛(アリロクマブ群で2人の患者(0.6%)対プラセボ群で患者なし)、認知障害(2人の患者(0.6%)対患者なし)、アレルギー性皮膚炎(2人の患者(0.6%)対患者なし)および筋肉痛(3人の患者(0.9%)対2人の患者(1.2%))。 Overall, SAEs that occurred during treatment were reported in 25 patients (7.3%) in the arilocumab group and 14 patients (8.2%) in the placebo group. SAE (PT levels) reported in 2 or more patients in either treatment group was pneumonia (1 patient in the allilocumab group (0.3%) vs. 2 patients in the placebo group (1.2%). )), vertebral foraminal stenosis (2 patients in the allilocumab group (0.6%) vs no placebo group), and urinary tract infections (2 patients (0.6%) vs no placebo group). )Met. One month after the first IMP administration (Visit 3), one T2DM patient in the placebo group reported one death due to myocardial infarction. Overall, 17 patients in the arilocumab group (4.9%) and 4 patients in the placebo group (2.4%) experienced TEAEs, resulting in permanent treatment discontinuation. At the PT level, the proportion of TEAE patients with permanent discontinuation of treatment in 2 or more patients in the treatment group was as follows: Headache (2 patients in the allilocumab group (0.6%) vs placebo group). No patient), cognitive impairment (2 patients (0.6%) vs no patient), allergic dermatitis (2 patients (0.6%) vs no patient) and myalgia (3 patients). (0.9%) vs. 2 patients (1.2%)).
特別な関心の有害事象(AESI)に関して、AESI基準を満たすALTの増加は、ALT≧3×ULN(ベースラインALT<ULNの場合)またはALT≧ベースライン値の2倍(ベースラインALT≧ULNの場合)として定義された。これらの事象は、アリロクマブ群の2人の患者(0.6%)とプラセボ群の1人の患者(0.6%)で報告された。 For adverse events of special interest (AESI), an increase in ALT that meets the AESI criteria is ALT≧3×ULN (if baseline ALT<ULN) or ALT≧twice the baseline value (baseline ALT≧ULN). If defined as). These events were reported in 2 patients (0.6%) in the arilocumab group and 1 patient (0.6%) in the placebo group.
AESI基準を満たすアレルギー薬物反応は、さらなる評価のために別の医師との協議を必要とするアレルギー事象として定義された。これらの事象は、アリロクマブ群の5人の患者(1.5%)に対して、プラセボ群の4人の患者(2.4%)で報告された。これらの反応は、主に皮膚および皮下組織の障害であり、アリロクマブ群の患者3人(0.9%)(アレルギー性皮膚炎1、湿疹1、光線過敏反応1)、およびプラセボ群の患者2人(1.2%)(皮膚炎1、薬疹1)で報告された。アリロクマブ群の他の2つのAESIアレルギー性薬物反応は、薬物過敏症と好酸球増加症であった。
An allergic drug response that meets the AESI criteria was defined as an allergic event that required consultation with another physician for further evaluation. These events were reported in 4 patients (2.4%) in the placebo group compared to 5 patients (1.5%) in the arilocumab group. These reactions were mainly skin and subcutaneous tissue disorders, with 3 patients (0.9%) in the allilocumab group (1 allergic dermatitis, 1 eczema, 1 photosensitivity reaction) and 2 patients in the placebo group. It was reported in humans (1.2%) (
AESI基準を満たす神経学的事象は、追加の検査/手順および/または専門家への紹介を必要とする神経学的事象として定義された。このような事象は、アリロクマブ群の1人の患者(0.3%)(感覚異常)とプラセボ群の1人の患者(0.6%)(嚥下障害)で報告された。両方の事象は、T2DM患者で報告された。 Neurological events that met the AESI criteria were defined as neurological events that required additional testing/procedures and/or referral to a specialist. Such events were reported in 1 patient (0.3%) (paresthesia) in the allilocumab group and 1 patient (0.6%) in the placebo group (dysphagia). Both events were reported in T2DM patients.
すべての神経認知事象はAESIとみなされた。スポンサーまたはFDA群ごとの神経認知事象が、アリロクマブ群の4人の患者(1.2%)とプラセボ群の患者なしで報告された。すべての事象はT2DM患者で報告された:認知障害は2人の患者(0.6%)で、記憶障害と記憶喪失はそれぞれ1人の患者(0.3%)で報告された。注目すべきは、2つの認知障害も永続的な治療中止をもたらした。 All neurocognitive events were considered AESI. Neurocognitive events by sponsor or FDA group were reported in 4 patients (1.2%) in the arilocumab group and no patients in the placebo group. All events were reported in T2DM patients: cognitive impairment was reported in 2 patients (0.6%) and memory impairment and memory loss were reported in 1 patient each (0.3%). Of note, two cognitive impairments also resulted in permanent treatment discontinuation.
AESI基準を満たす局所注射部位反応は、アレルギー性であり、別の医師との協議を必要とする局所注射部位反応、または臨床的に重要な非アレルギー性局所注射部位反応(例えば、径>2.5cmを有する腫れまたは紅斑の反応;活動を妨げる反応)のいずれかとして定義された。IMPに関連する研究者ごとに確認された(「eCRFあたり」)LISRは、アリロクマブ群の6人の患者(1.7%)対プラセボ群の8人の患者(4.7%)で報告された(アリロクマブに対してプラセボの注射部位反応)。さらなる評価のために他の医師と相談する必要がある反応として定義されたAESI基準を満たす局所注射部位反応(LISR)は報告されなかった。 Local injection site reactions that meet the AESI criteria are allergic and require local consultation with another physician, or clinically significant non-allergic local injection site reactions (eg, diameter >2. Swelling or erythema reaction with 5 cm; reaction preventing activity). Investigator-confirmed (“per eCRF”) LISR associated with IMP was reported in 6 patients (1.7%) in the arilocumab group versus 8 patients (4.7%) in the placebo group. (Placebo injection site reaction to arilocumab). No local injection site reaction (LISR) was reported that met the AESI criteria, which was defined as a reaction that needed to be consulted with another physician for further evaluation.
症候性の過剰投薬または妊娠の報告はなかった。 There were no reports of symptomatic overdose or pregnancy.
肝機能検査(ALT、AST、ALP、総ビリルビン)、CPKおよび腎機能検査(クレアチニン、eGFR、BUN)の分析は、研究されたパラメータのいずれかに対する経時変化の治療群間の差異を明らかにしなかった。PCSA分析では、研究中にどの治療群でもALT増加のPCSAは特定されなかった。ベースライン時のCPK値が正常な患者では、アリロクマブ群の患者7人(2.1%)に対してプラセボ群の患者1人(0.6%)で>3ULN(および≦10ULN)の増加が報告された。すべての患者はT2DMであった。10ULNを超えるCPKの増加は報告されなかった。 Analysis of liver function tests (ALT, AST, ALP, total bilirubin), CPK and renal function tests (creatinine, eGFR, BUN) did not reveal differences between treatment groups in time course for any of the parameters studied It was PCSA analysis did not identify ALT-increasing PCSA in any treatment group during the study. In patients with normal baseline CPK levels, there was a >3ULN (and ≤10ULN) increase in 1 placebo (0.6%) vs. 7 patients (2.1%) in the allilocumab group. Reported. All patients had T2DM. No increase in CPK above 10 ULN was reported.
ベースラインの状態に関係なく、治療期間中に糸球体濾過率(GFR)が軽度、中度、または重度に減少した患者の割合において、数値的に小さな差が観察された:アリロクマブ群においてそれぞれ49.7%、28.1%および3.8%の患者において、ならびにプラセボ群においてそれぞれ50.6%、24.4%と3.6%の患者において、GFRの軽度、中度、重度の減少。同様に、血中クレアチニンの増加(≧30%および<100%)は、アリロクマブ群の13人(3.8%)の患者とプラセボ群の5人の患者(3.0%)で測定された。血中クレアチニンが100%以上増加した患者はいなかった。腎機能に有意な差はなかった。 Numerically small differences were observed in the proportion of patients with mild, moderate, or severe reductions in glomerular filtration rate (GFR) during the treatment period regardless of baseline status: 49 each in the alilocumab group Mild, moderate, and severe reduction in GFR in 0.7%, 28.1%, and 3.8% of patients, and in 50.6%, 24.4%, and 3.6% of patients in the placebo group, respectively. .. Similarly, increases in blood creatinine (≧30% and <100%) were measured in 13 (3.8%) patients in the alilocumab group and 5 (3.0%) in the placebo group. .. None of the patients had blood creatinine increased by more than 100%. There was no significant difference in renal function.
バイタルサインについて有意な差は、治療群間で観察されなかった。 No significant difference in vital signs was observed between treatment groups.
2型真性糖尿病およびオデッセイDM−インスリン臨床試験からのASCVDを有する個体の分析
糖尿病を有する個体は、高レベルのアテローム生成リポタンパク質およびコレステロールを有し、これは、低密度リポタンパク質コレステロール(LDL−C)、非高密度リポタンパク質コレステロール(非HDL−C)、アポリポタンパク質B(ApoB)、および低密度リポタンパク質粒子数(LDL−PN)の上昇によって反映される。動脈硬化性心血管疾患(ASCVD)の存在は、将来の心血管事象のリスクを高める。
Analysis of Individuals With ASCVD from
この分析では、本発明者らは、T2DM、高LDL−C、または非HDL−Cを有する個体間でのアリロクマブの有効性および安全性を評価し、DM−インスリン研究において最大耐性のスタチンを受けるASCVDを確立した。ASCVDおよびT1DMのDM−インスリン研究参加者は、この群の個体数が少ないため、この分析には含まれなかった(アリロクマブ:n=11;プラセボ:n=5)。この実施例で使用されるように、ASCVDは冠状動脈性心疾患(CHD;急性および無症候性心筋梗塞(MI)、および不安定狭心症)、虚血性脳卒中、または末梢動脈疾患として定義された。 In this analysis, we evaluate the efficacy and safety of arilocumab among individuals with T2DM, high LDL-C, or non-HDL-C and receive the most resistant statins in the DM-insulin study. Established ASCVD. Participants in the DM-insulin study of ASCVD and T1DM were not included in this analysis due to the low population in this group (arilocumab: n=11; placebo: n=5). As used in this example, ASCVD is defined as coronary heart disease (CHD; acute and subclinical myocardial infarction (MI), and unstable angina), ischemic stroke, or peripheral arterial disease. It was
ベースラインおよび有効性データは、研究に従って分析された。有効性分析には、非HDL−C、LDL−C、ApoB、およびLDL−PNのベースラインからの24週目のパーセンテージ減少、および24週目で非HDL−C<100mg/dL(<2.59mmol/L)、LDL−C<70mg/dL(<1.81mmol/L)、およびApoB<80mg/dLを達成する個体のパーセンテージが含まれる。包括解析(ITT)の分析には、ベースラインLDL−C値および最大24週までの少なくとも1つのLDL−C値を有するランダム化された全個体を含んだ。
Baseline and efficacy data were analyzed according to the study. Efficacy analysis included a non-HDL-C, LDL-C, ApoB, and LDL-PN percentage reduction from baseline at
この分析は、確立されたASCVDおよびT2DMを有する177人のDM−インスリン個体を含んだ(表16)。 This analysis included 177 DM-insulin individuals with established ASCVD and T2DM (Table 16).
ASCVDに加えて、治療配分に関係なく、分析した個体の89.3%に高血圧の病歴があり、28.2%に慢性腎疾患(CKD)があった。合計で、20.3%が虚血性脳卒中の病歴を示し、10.7%が末梢動脈疾患(PAD)を患っていた。ベースラインでは、平均(標準偏差[SD])非HDL−Cレベルは144.2(46.2)mg/dL[3.73(1.20)mmol/L]であった;平均LDL−Cレベルは、治療配分に関係なく、108.7(39.1)mg/dL[2.82(1.01)mmol/L]であった。 In addition to ASCVD, regardless of treatment allocation, 89.3% of individuals analyzed had a history of hypertension and 28.2% had chronic kidney disease (CKD). In total, 20.3% had a history of ischemic stroke and 10.7% had peripheral arterial disease (PAD). At baseline, mean (standard deviation [SD]) non-HDL-C levels were 144.2 (46.2) mg/dL [3.73 (1.20) mmol/L]; mean LDL-C. The level was 108.7 (39.1) mg/dL [2.82 (1.01) mmol/L] regardless of treatment distribution.
効能
アリロクマブは、対照に対して、24週目でベースラインから非HDL−C、ApoB、LDL−PN、およびLDL−Cを減少させた(図4)。24週目で、対照に対して、非HDL−C<100mg/dL(<2.59mmol/L)、LDL−C<70mg/dL(<1.81mmol/L)、およびApoB<80mg/dLを達成した個体の割合が大幅に増加した(すべてP<0.0001;図5)。
Efficacy Arilocumab reduced non-HDL-C, ApoB, LDL-PN, and LDL-C from baseline at 24 weeks relative to controls (Figure 4). At
安全性
安全性分析は、T2DM、高LDL−Cまたは非HDL−C、DM−インスリン研究で最大耐性のスタチンを受けて確立されたASCVDを有する個体、ならびにT1DMまたはT2DM、高LDL−Cまたは非HDL−C、およびDM−脂質異常症研究で最大耐性のスタチンを受けて確立されたASCVDを有する個体、プールされた個体群で実施された(全体として参照により本明細書に組み入れる、Chan et al. (2017) Ann Transl Med. 5(23):477を参照されたい)。合計で、個体の66.4%(アリロクマブ)および67.0%(対照)が治療により発現した有害事象を報告した(TEAE;表17)。有害事象のパターンは、両方の群で類似していた。HbA1cの平均(SD)レベルは、ベースライン(アリロクマブ:7.3[0.9]%;対照:7.3[0.9]%)および24週目(アリロクマブ:7.6[1.2]%;対照:7.5[1.2]%;安全性分析)で各治療群において類似していた。FPGレベルはまた、ベースライン(アリロクマブ:154.2[47.9]mg/dL、8.6[2.7]mmol/L;対照:149.5[43.7]mg/dL、8.3[2.4]mmol/L)および24週目(アリロクマブ:164.7[54.9]mg/dL、9.1[3.0]mmol/L;対照:159.4[48.4]mg/dL、8.9[2.7]mmol/L;安全性分析)で治療配分に関係なく類似していた。
Safety Safety analysis was conducted for T2DM, high LDL-C or non-HDL-C, individuals with ASCVD established by receiving a maximally resistant statin in the DM-insulin study, as well as T1DM or T2DM, high LDL-C or non-high. HDL-C, and DM-Dyslipidemia studies were performed on individuals with ASCVD established and pooled populations who received the most resistant statins (Chan et al., incorporated herein by reference in its entirety). (2017) Ann Transl Med. 5(23):477). In total, 66.4% (arilocumab) and 67.0% (control) of individuals reported treatment-emergent adverse events (TEAE; Table 17). The pattern of adverse events was similar in both groups. Mean (SD) levels of HbA1c were baseline (arilocumab: 7.3 [0.9]%; control: 7.3 [0.9]%) and week 24 (arilocumab: 7.6 [1.2]. ]; control: 7.5 [1.2]%; safety analysis) were similar in each treatment group. FPG levels were also measured at baseline (arilocumab: 154.2 [47.9] mg/dL, 8.6 [2.7] mmol/L; control: 149.5 [43.7] mg/dL, 8. 3 [2.4] mmol/L) and week 24 (arilocumab: 164.7 [54.9] mg/dL, 9.1 [3.0] mmol/L; control: 159.4 [48.4]. ] Mg/dL, 8.9 [2.7]mmol/L; safety analysis) were similar regardless of treatment distribution.
結論
最大耐性のスタチンにもかかわらず高いLDL−Cレベルを有したT2DMおよびASCVDを有する個体において、アリロクマブは、アテローム性コレステロール含有量およびLDL−PNを対照に対して有意に減少させた。
Conclusions In individuals with T2DM and ASCVD who had high LDL-C levels despite maximally tolerant statins, alilocumab significantly reduced atherogenic cholesterol content and LDL-PN relative to controls.
Claims (86)
(a)(i)T1DM、および(ii)最大耐性のスタチン療法によっては適切に制御されない高コレステロール血症を有する、インスリン療法を受けている心血管リスクの高い患者を選択する工程;ならびに
(b)患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片を75mg、150mgもしくは300mg投与する工程
を含み、患者は付随するインスリン療法を受ける前記方法。 A method of treating hypercholesterolemia in a patient having type 1 diabetes mellitus (T1DM), the method comprising:
(A) selecting (i) T1DM, and (ii) patients with high cardiovascular risk receiving insulin therapy who have hypercholesterolemia not adequately controlled by maximally resistant statin therapy; and (b) ) Administering to a patient 75 mg, 150 mg or 300 mg of an antibody or antigen-binding fragment thereof that specifically binds to the human proprotein convertase subtilisin/kexin type 9 (PCSK9), wherein the patient receives the associated insulin therapy Method.
をさらに含む、請求項1〜8のいずれか一項に記載の方法。 (C) if the LDL-C level in the patient is below the threshold level, the patient is administered with one or more subsequent doses of 75 mg of the antibody or antigen-binding fragment thereof about once every two weeks, or in the patient 9. Any of claims 1-8, further comprising the step of administering one or more subsequent doses of 150 mg of antibody or antigen-binding fragment thereof about every two weeks if the LDL-C level is above the threshold level. The method described in paragraph 1.
をさらに含む、請求項1〜9のいずれか一項に記載の方法。 (C) if the LDL-C level in the patient is below a threshold level, the patient is administered with one or more subsequent doses of 300 mg of the antibody or antigen-binding fragment thereof about once every four weeks, or in the patient 10. Any of claims 1-9, further comprising the step of administering one or more subsequent doses of 150 mg of the antibody or antigen-binding fragment thereof about every two weeks if the LDL-C level is above the threshold level. The method described in paragraph 1.
(a)患者のヘモグロビンA1c(HbA1c)レベルに影響を及ぼさない;および/または
(b)患者の空腹時血漿グルコース(FPG)レベルに影響を及ぼさない
請求項1〜26のいずれか一項に記載の方法。 The antibody or antigen-binding fragment thereof is:
27. Any of claims 1-26, wherein (a) does not affect the patient's hemoglobin A1c (HbA1c) level; and/or (b) does not affect the patient's fasting plasma glucose (FPG) level. the method of.
(a)(i)T1DM、および(ii)最大耐性のスタチン療法によっては適切に制御されていない高コレステロール血症を有する、インスリン療法を受けている心血管リスクの高い患者を選択する工程;
(b)患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片を75mg、2週間ごとに投与する工程;ならびに
(c)患者におけるLDL−Cレベルが70mg/dLより低い場合、約2週間ごとに1回またはそれ以上の次の用量の75mgの抗体もしくはその抗原結合断片を患者に投与するか、または患者におけるLDL−Cレベルが70mg/dL以上の場合、約2週間ごとに1回またはそれ以上の次の用量の150mgの抗体もしくはその抗原結合断片を投与する工程
を含み、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含み、患者は付随するインスリン療法を受ける前記方法。 A method of treating hypercholesterolemia in a patient having type 1 diabetes mellitus (T1DM), the method comprising:
(A) selecting patients at high cardiovascular risk undergoing insulin therapy with hypercholesterolemia not adequately controlled by (i) T1DM, and (ii) maximally resistant statin therapy;
(B) administering to the patient 75 mg of an antibody or an antigen-binding fragment thereof that specifically binds to the human proprotein convertase subtilisin/kexin type 9 (PCSK9) every 2 weeks; and (c) LDL- in the patient. If the C level is less than 70 mg/dL, the patient is administered about one or more of the following doses of 75 mg of the antibody or antigen-binding fragment thereof about every two weeks, or the LDL-C level in the patient is 70 mg/dL. In the case of dL or more, it comprises the step of administering the following doses of about 150 mg of the antibody or its antigen-binding fragment once or more about every two weeks, wherein the antibody or its antigen-binding fragment has the amino acid sequence of SEQ ID NO: 1. And HCLC having the amino acid sequence of SEQ ID NO: 6, wherein the patient receives concomitant insulin therapy.
(a)(i)T2DM、および(ii)最大耐性のスタチン療法によって適切に制御されていない高コレステロール血症を有する、インスリン療法を受けている心血管リスクの高い患者を選択する工程;ならびに
(b)患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片の75mg、150mgもしくは300mgを投与する工程
を含み、患者は付随するインスリン療法を受ける前記方法。 A method of treating hypercholesterolemia in a patient with type 2 diabetes mellitus (T2DM), the method comprising:
(A) selecting (i) T2DM, and (ii) patients at high cardiovascular risk receiving insulin therapy who have hypercholesterolemia not adequately controlled by maximally resistant statin therapy; b) comprising the step of administering to the patient 75 mg, 150 mg or 300 mg of an antibody or an antigen-binding fragment thereof that specifically binds to the human proprotein convertase subtilisin/kexin type 9 (PCSK9), the patient receiving concomitant insulin therapy. The method of receiving.
をさらに含む、請求項29〜36のいずれか一項に記載の方法。 (C) if the LDL-C level in the patient is below the threshold level, the patient is administered with one or more subsequent doses of 75 mg of the antibody or antigen-binding fragment thereof about once every two weeks, or in the patient 37. Any of claims 29-36, further comprising the step of administering one or more subsequent doses of 150 mg of the antibody or antigen binding fragment thereof about once every two weeks if the LDL-C level is above the threshold level. The method described in paragraph 1.
をさらに含む、請求項29〜36のいずれか一項に記載の方法。 (C) if the LDL-C level in the patient is below the threshold level, administer to the patient one or more subsequent doses of 300 mg of the antibody or antigen-binding fragment thereof about once every four weeks, or in the patient 37. Any of claims 29-36, further comprising the step of administering one or more subsequent doses of 150 mg of the antibody or antigen binding fragment thereof about once every two weeks if the LDL-C level is above the threshold level. The method described in paragraph 1.
(a)患者のヘモグロビンA1c(HbA1c)レベルに影響を及ぼさない;および/または
(b)患者の空腹時血漿グルコース(FPG)レベルに影響を及ぼさない
請求項29〜54のいずれか一項に記載の方法。 The antibody or antigen-binding fragment thereof is:
55. Any of claims 29-54, wherein (a) does not affect the patient's hemoglobin A1c (HbA1c) level; and/or (b) does not affect the patient's fasting plasma glucose (FPG) level. the method of.
(a)(i)T2DM、および(ii)最大耐性のスタチン療法によっては適切に制御されない高コレステロール血症を有する、インスリン療法を受けている心血管リスクの高い患者を選択する工程;
(b)患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片を75mg、2週間ごとに投与する工程;ならびに
(c)患者におけるLDL−Cレベルが70mg/dLより低い場合、約2週間ごとに1回またはそれ以上の次の用量の75mgの抗体もしくはその抗原結合断片を患者に投与するか、または患者におけるLDL−Cレベルが70mg/dL以上の場合、約2週間ごとに1回またはそれ以上の次の用量の150mgの抗体もしくはその抗原結合断片を投与する工程
を含み、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含み、患者は付随するインスリン療法を受ける前記方法。 A method of treating hypercholesterolemia in a patient with type 2 diabetes mellitus (T2DM), the method comprising:
(A) selecting patients at high cardiovascular risk undergoing insulin therapy with hypercholesterolemia not adequately controlled by (i) T2DM and (ii) maximally resistant statin therapy;
(B) administering to the patient 75 mg of an antibody or an antigen-binding fragment thereof that specifically binds to the human proprotein convertase subtilisin/kexin type 9 (PCSK9) every 2 weeks; and (c) LDL- in the patient. If the C level is less than 70 mg/dL, the patient is administered about one or more of the following doses of 75 mg of the antibody or antigen-binding fragment thereof about every two weeks, or the LDL-C level in the patient is 70 mg/dL. In the case of dL or more, it comprises the step of administering the following doses of about 150 mg of the antibody or its antigen-binding fragment once or more about every two weeks, wherein the antibody or its antigen-binding fragment has the amino acid sequence of SEQ ID NO: 1. And HCLC having the amino acid sequence of SEQ ID NO: 6, wherein the patient receives concomitant insulin therapy.
(a)(i)T2DM、(ii)ASCVD、および(iii)最大耐性のスタチン療法によって適切に制御されていない高コレステロール血症のインスリン療法を受けている心血管リスクの高い患者を選択する工程;ならびに
(b)患者に、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片の75mg、150mgもしくは300mgを投与する工程
を含み、患者は付随するインスリン療法を受ける前記方法。 A method of treating hypercholesterolemia in a patient having T2DM and atherosclerotic cardiovascular disease (ASCVD), the method comprising:
Selecting patients at high cardiovascular risk undergoing insulin therapy for hypercholesterolemia not adequately controlled by (a) (i) T2DM, (ii) ASCVD, and (iii) maximally resistant statin therapy And (b) the step of administering to the patient 75 mg, 150 mg or 300 mg of an antibody or an antigen-binding fragment thereof that specifically binds to human proprotein convertase subtilisin/kexin type 9 (PCSK9), and the patient is incidental The above method of receiving insulin therapy.
をさらに含む、請求項57〜66のいずれか一項に記載の方法。 (C) if the LDL-C level in the patient is below the threshold level, the patient is administered with one or more subsequent doses of 75 mg of the antibody or antigen-binding fragment thereof about once every two weeks, or in the patient 67. Any of claims 57-66, further comprising the step of administering one or more subsequent doses of 150 mg of antibody or antigen binding fragment thereof about once every two weeks if the LDL-C level is above the threshold level. The method described in paragraph 1.
をさらに含む、請求項57〜66のいずれか一項に記載の方法。 (C) if the LDL-C level in the patient is below the threshold level, administer to the patient one or more subsequent doses of 300 mg of the antibody or antigen-binding fragment thereof about once every four weeks, or in the patient 67. Any of claims 57-66, further comprising the step of administering one or more subsequent doses of 150 mg of antibody or antigen binding fragment thereof about once every two weeks if the LDL-C level is above the threshold level. The method described in paragraph 1.
(a)患者のヘモグロビンA1c(HbA1c)レベルに影響を及ぼさない;および/または
(b)患者の空腹時血漿グルコース(FPG)レベルに影響を及ぼさない
請求項57〜84のいずれか一項に記載の方法。 The antibody or antigen-binding fragment thereof is:
85. Any of claims 57-84, wherein (a) does not affect the patient's hemoglobin A1c (HbA1c) level; and/or (b) does not affect the patient's fasting plasma glucose (FPG) level. the method of.
(a)(i)T2DM、(ii)ASCVD、および(iii)最大耐性のスタチン療法によって適切に制御されない高コレステロール血症を有する、インスリン療法を受けている心血管リスクの高い患者を選択する工程;
(b)患者に2週間ごとに、ヒトプロタンパク質転換酵素スブチリシン/ケキシン9型(PCSK9)に特異的に結合する抗体またはその抗原結合断片を75mg、2週間ごとに投与する工程;ならびに
(c)患者におけるLDL−Cレベルが70mg/dLより低い場合、約2週間ごとに1回またはそれ以上の次の用量の75mgの抗体もしくはその抗原結合断片を患者に投与するか、または患者におけるLDL−Cレベルが70mg/dL以上の場合、約2週間ごとに1回またはそれ以上の次の用量の150mgの抗体もしくはその抗原結合断片を投与する工程
を含み、抗体またはその抗原結合断片は、配列番号1のアミノ酸配列を有するHCVR、および配列番号6のアミノ酸配列を有するLCVRを含み、患者は付随するインスリン療法を受ける前記方法。 A method of treating hypercholesterolemia in a patient having T2DM and ASCVD, the method comprising:
(A) Selecting patients at high cardiovascular risk receiving insulin therapy with hypercholesterolemia not adequately controlled by (i) T2DM, (ii) ASCVD, and (iii) maximally resistant statin therapy ;
(B) a step of administering to the patient every 2 weeks, 75 mg of an antibody or an antigen-binding fragment thereof that specifically binds to human proprotein convertase subtilisin/kexin type 9 (PCSK9), every 2 weeks; and (c) If the LDL-C level in the patient is less than 70 mg/dL, the patient is administered with one or more subsequent doses of 75 mg antibody or antigen-binding fragment thereof about once every two weeks, or LDL-C in the patient. When the level is 70 mg/dL or higher, the method comprises the step of administering 150 mg of the antibody or antigen-binding fragment thereof at a dose of about once every two weeks or more, wherein the antibody or the antigen-binding fragment is SEQ ID NO: 1 A HCVR having the amino acid sequence of SEQ ID NO: 6 and an LCVR having the amino acid sequence of SEQ ID NO: 6, wherein the patient receives concomitant insulin therapy.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023111915A JP2023123842A (en) | 2017-06-09 | 2023-07-07 | Method for treating hyperlipemia in diabetes patient by administration of pcsk9 inhibitor |
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762517672P | 2017-06-09 | 2017-06-09 | |
| US62/517,672 | 2017-06-09 | ||
| US201762532162P | 2017-07-13 | 2017-07-13 | |
| US62/532,162 | 2017-07-13 | ||
| EP18305565 | 2018-05-04 | ||
| EP18305565.6 | 2018-05-04 | ||
| PCT/IB2018/054182 WO2018225041A1 (en) | 2017-06-09 | 2018-06-09 | Methods for treating hyperlipidemia in diabetic patients by administering a pcsk9 inhibitor |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2023111915A Division JP2023123842A (en) | 2017-06-09 | 2023-07-07 | Method for treating hyperlipemia in diabetes patient by administration of pcsk9 inhibitor |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2020522544A true JP2020522544A (en) | 2020-07-30 |
| JP2020522544A5 JP2020522544A5 (en) | 2021-07-26 |
Family
ID=62815099
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019567608A Pending JP2020522544A (en) | 2017-06-09 | 2018-06-09 | Method of treating hyperlipidemia in diabetic patients by administration of a PCSK9 inhibitor |
| JP2023111915A Pending JP2023123842A (en) | 2017-06-09 | 2023-07-07 | Method for treating hyperlipemia in diabetes patient by administration of pcsk9 inhibitor |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2023111915A Pending JP2023123842A (en) | 2017-06-09 | 2023-07-07 | Method for treating hyperlipemia in diabetes patient by administration of pcsk9 inhibitor |
Country Status (9)
| Country | Link |
|---|---|
| EP (1) | EP3634469A1 (en) |
| JP (2) | JP2020522544A (en) |
| KR (1) | KR20200026826A (en) |
| CN (1) | CN110913889A (en) |
| AU (1) | AU2018280567A1 (en) |
| CA (1) | CA3066317A1 (en) |
| IL (1) | IL271212A (en) |
| MX (1) | MX2019014831A (en) |
| TW (2) | TW201904608A (en) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2012210480B2 (en) | 2011-01-28 | 2017-05-18 | Sanofi Biotechnology | Human antibodies to PCSK9 for use in methods of treating particular groups of subjects |
| AR087305A1 (en) | 2011-07-28 | 2014-03-12 | Regeneron Pharma | STABILIZED FORMULATIONS CONTAINING ANTI-PCSK9 ANTIBODIES, PREPARATION METHOD AND KIT |
| JP2018523684A (en) | 2015-08-18 | 2018-08-23 | リジェネロン・ファーマシューティカルズ・インコーポレイテッドRegeneron Pharmaceuticals, Inc. | Anti-PCSK9 inhibitory antibody for treating hyperlipidemic patients undergoing lipoprotein apheresis |
| CN113876956B (en) * | 2020-07-01 | 2023-07-04 | 陈敏 | Application of PCSK9 inhibitor in preparation of product for promoting skin pigmentation |
| CN114525258A (en) * | 2020-10-30 | 2022-05-24 | 未来智人再生医学研究院(广州)有限公司 | Pluripotent stem cell expressing PCSK9 blocker or derivative thereof and application |
| WO2023051822A1 (en) * | 2021-09-30 | 2023-04-06 | 北京安龙生物医药有限公司 | Targeting oligonucleotide for treating diseases associated with pcsk9 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JO3672B1 (en) * | 2008-12-15 | 2020-08-27 | Regeneron Pharma | High Affinity Human Antibodies to PCSK9 |
| MX2017000628A (en) * | 2014-07-16 | 2017-04-27 | Sanofi Biotechnology | Methods for treating high cardiovascular risk patients with hypercholesterolemia. |
| RU2735521C2 (en) * | 2014-07-16 | 2020-11-03 | Санофи Байотекнолоджи | Methods of treating patients with heterozygous familial hypercholesterolemia (hefh) |
| JP2018523684A (en) * | 2015-08-18 | 2018-08-23 | リジェネロン・ファーマシューティカルズ・インコーポレイテッドRegeneron Pharmaceuticals, Inc. | Anti-PCSK9 inhibitory antibody for treating hyperlipidemic patients undergoing lipoprotein apheresis |
-
2018
- 2018-06-08 TW TW107119851A patent/TW201904608A/en unknown
- 2018-06-08 TW TW111142924A patent/TW202310872A/en unknown
- 2018-06-09 JP JP2019567608A patent/JP2020522544A/en active Pending
- 2018-06-09 EP EP18737034.1A patent/EP3634469A1/en not_active Withdrawn
- 2018-06-09 CA CA3066317A patent/CA3066317A1/en active Pending
- 2018-06-09 CN CN201880037888.2A patent/CN110913889A/en active Pending
- 2018-06-09 AU AU2018280567A patent/AU2018280567A1/en active Pending
- 2018-06-09 MX MX2019014831A patent/MX2019014831A/en unknown
- 2018-06-09 KR KR1020197038159A patent/KR20200026826A/en not_active Application Discontinuation
-
2019
- 2019-12-05 IL IL271212A patent/IL271212A/en unknown
-
2023
- 2023-07-07 JP JP2023111915A patent/JP2023123842A/en active Pending
Non-Patent Citations (3)
| Title |
|---|
| DIRK MULLERWIELAND, CARDIOVASC.DIABETOL, vol. VOL:16,NR:70, JPN5020007003, 25 May 2017 (2017-05-25), ISSN: 0004812357 * |
| HELEN M COLHOUN: "NO EFFECT OF PCSK9 INHIBITOR ALIROCUMAB ON THE INCIDENCE OF DIABETES IN A POOLED 以下備考", EUROPEAN HEART JOURNAL, vol. VOL:37, NR:39, JPN5020007004, 26 July 2016 (2016-07-26), pages 2981 - 2989, ISSN: 0004812358 * |
| LAWRENCE A LEITER: "LIPID-LOWERING EFFICACY AND SAFETY OF ALIROCUMAB IN PATIENTS WITH OR WITHOUT DIABETES: 以下備考", DIABETES, OBESITY AND METABOLISM, vol. VOL:19, NR:7, JPN5020007002, 16 February 2017 (2017-02-16), pages 989 - 996, ISSN: 0005005944 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2023123842A (en) | 2023-09-05 |
| AU2018280567A1 (en) | 2020-01-23 |
| CN110913889A (en) | 2020-03-24 |
| RU2019144346A3 (en) | 2021-10-13 |
| TW202310872A (en) | 2023-03-16 |
| MX2019014831A (en) | 2020-02-13 |
| CA3066317A1 (en) | 2018-12-13 |
| TW201904608A (en) | 2019-02-01 |
| IL271212A (en) | 2020-01-30 |
| EP3634469A1 (en) | 2020-04-15 |
| RU2019144346A (en) | 2021-07-09 |
| KR20200026826A (en) | 2020-03-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20190031774A1 (en) | Methods for treating hyperlipidemia in diabetic patients by administering a pcsk9 inhibitor | |
| JP6994484B2 (en) | Use of PCSK9 inhibitors to treat hyperlipidemia | |
| JP2020522544A (en) | Method of treating hyperlipidemia in diabetic patients by administration of a PCSK9 inhibitor | |
| EP3331908B1 (en) | Anti-angptl8 antibodies and uses thereof | |
| AU2014274077B2 (en) | Methods for treating autosomal dominant hypercholesterolemia associated with PCSK9 gain-of-function mutations | |
| JP2022169718A (en) | Methods for preventing or treating allergy by administering an il-4r antagonist | |
| KR102482375B1 (en) | Methods for treating high cardiovascular risk patients with hypercholesterolemia | |
| JP2017506626A (en) | Methods for treating patients with hypercholesterolemia that are not adequately managed with moderate dose statin therapy | |
| US20180134781A1 (en) | Methods of Treating Obesity with Anti-ANGPTL8 Antibodies | |
| US11779633B2 (en) | Glucagon-like peptide 1 receptor agonists and uses thereof | |
| RU2772712C2 (en) | Methods for treatment of hyperlipidemia in patients with diabetes by injection of pcsk9 inhibitor | |
| ES2779126T3 (en) | Using a PCSK9 Inhibitor to Treat Hyperlipidemia | |
| US20230272112A1 (en) | Use of a pcsk9 inhibitor to treat homozygous familial hypercholesterolemia | |
| RU2777328C2 (en) | Methods for preventing or treating allergies by introducing an il-4r antagonist | |
| EA045412B1 (en) | GLUCAGON-LIKE PEPTIDE 1 RECEPTOR AGONISTS AND THEIR APPLICATIONS |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210607 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20210607 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20220628 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20220926 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20221128 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20230307 |