JP2020507312A - 複数の宿主用の複数のdnaコンストラクトのアセンブリ及び編集のためのモジュラーユニバーサルプラスミド設計戦略 - Google Patents
複数の宿主用の複数のdnaコンストラクトのアセンブリ及び編集のためのモジュラーユニバーサルプラスミド設計戦略 Download PDFInfo
- Publication number
- JP2020507312A JP2020507312A JP2019539756A JP2019539756A JP2020507312A JP 2020507312 A JP2020507312 A JP 2020507312A JP 2019539756 A JP2019539756 A JP 2019539756A JP 2019539756 A JP2019539756 A JP 2019539756A JP 2020507312 A JP2020507312 A JP 2020507312A
- Authority
- JP
- Japan
- Prior art keywords
- dna
- crispr
- ctag
- sequence
- modular
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Images



















Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1003—Extracting or separating nucleic acids from biological samples, e.g. pure separation or isolation methods; Conditions, buffers or apparatuses therefor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/102—Mutagenizing nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/64—General methods for preparing the vector, for introducing it into the cell or for selecting the vector-containing host
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/66—General methods for inserting a gene into a vector to form a recombinant vector using cleavage and ligation; Use of non-functional linkers or adaptors, e.g. linkers containing the sequence for a restriction endonuclease
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPRs]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/35—Nature of the modification
- C12N2310/351—Conjugate
- C12N2310/3517—Marker; Tag
Abstract
本開示は、高スループットDNAアセンブリのインビトロ反応のための方法、組成物、及びキットを記載する。本開示はさらに、予め確認されたCRISPRプロトスペーサ/プロトスペーサ隣接モチーフ配列組合せを含むクローニングタグセグメントが側面に位置するモジュラー挿入DNA部分を含むモジュラーCRISPR DNAコンストラクトを記載する。
Description
関連出願の相互参照
[0001] 本出願は、2017年2月10日出願の米国仮特許出願第62/457,493号の優先権の利益を主張する。この出願は、全ての目的について、その全体が参照によって本明細書に組み込まれる。
[0001] 本出願は、2017年2月10日出願の米国仮特許出願第62/457,493号の優先権の利益を主張する。この出願は、全ての目的について、その全体が参照によって本明細書に組み込まれる。
分野
[0002] 本開示は、インビトロ誘導性遺伝子配列編集に用いられる系、方法、及び組成物に関する。本開示は、とりわけ、DNAクローニング、オリゴヌクレオチドのアセンブリの向上のための、そして微生物の向上のための誘導性配列複合体編集を用いる方法を記載する。
[0002] 本開示は、インビトロ誘導性遺伝子配列編集に用いられる系、方法、及び組成物に関する。本開示は、とりわけ、DNAクローニング、オリゴヌクレオチドのアセンブリの向上のための、そして微生物の向上のための誘導性配列複合体編集を用いる方法を記載する。
電子的に提出されたテキストファイルの説明
[0003] 本出願と共に電子的に提出されたテキストファイルの内容は、その全体が参照によって本明細書に組み込まれる:配列表のコンピュータ読取り可能なフォーマットコピー(ファイル名:ZYMR_010_01WO_SeqList_ST25.txt、記録日:2018年1月30日;ファイルサイズ:ほぼ800キロバイト)。
[0003] 本出願と共に電子的に提出されたテキストファイルの内容は、その全体が参照によって本明細書に組み込まれる:配列表のコンピュータ読取り可能なフォーマットコピー(ファイル名:ZYMR_010_01WO_SeqList_ST25.txt、記録日:2018年1月30日;ファイルサイズ:ほぼ800キロバイト)。
政府ライセンス権
[0004] 本発明は、DARPAによる、契約第HR0011-15-9-0014号の政府支援を受けてなされた。政府は、本発明において、一定の権利を有する。
[0004] 本発明は、DARPAによる、契約第HR0011-15-9-0014号の政府支援を受けてなされた。政府は、本発明において、一定の権利を有する。
背景
[0005] 生物学における主要な関心領域は、遺伝子配列のインビトロでの、そしてインビボでの標的修飾である。実際に、学術的な、そして商業的な遺伝的研究の最も大きな障壁の1つは、新規の遺伝的コンストラクトが、試験に先立って生成され得、又は後に修飾され得る速度であった。
[0005] 生物学における主要な関心領域は、遺伝子配列のインビトロでの、そしてインビボでの標的修飾である。実際に、学術的な、そして商業的な遺伝的研究の最も大きな障壁の1つは、新規の遺伝的コンストラクトが、試験に先立って生成され得、又は後に修飾され得る速度であった。
[0006] 制限部位認識又はDNAのハイブリダイゼーション及び増幅に頼る現在利用可能なクローニング技術は、後の修飾にとって、遅く、信頼できず、且つ扱いにくいことが判明した。クラスター化した規則的な配置の短い回文配列反復(Clustered Regularly Interspaced Short Palindromic Repeats(CRISPR))遺伝子編集系の発見は、研究者に、遺伝的修飾の更なるアベニューを提供した。しかしながら、この新しいアプローチさえ、高スループットモジュラークローニング用途にとって、非実用的なままである。
[0007] CRISPR編集位置は、例えば、多くの場合、プロトスペーサ隣接モチーフ(PAM)の位置によって制限される。新規のCRISPR誘導RNA設計及び遺伝子ターゲティングは、時間がかかって高価となり得、そしてまた低い効率の影響を受け易く、且つオフターゲット突然変異の可能性がある。
[0008] ゆえに、遺伝子配列の標的変更のための組成物及び方法の向上が必要とされている。
開示の概要
[0009] 一部の実施形態において、本開示は、モジュラーCRISPR DNAコンストラクトを利用する、高スループットDNAアセンブリのインビボ及びインビトロ反応のための方法、組成物、及びキットを教示する。
[0009] 一部の実施形態において、本開示は、モジュラーCRISPR DNAコンストラクトを利用する、高スループットDNAアセンブリのインビボ及びインビトロ反応のための方法、組成物、及びキットを教示する。
[0010] ゆえに、一部の実施形態において、本開示は、予め確認されたCRISPRプロトスペーサ/プロトスペーサ隣接モチーフ(PAM)配列組合せを含むクローニングタグセグメントが側面に位置するモジュラー挿入DNA部分を含むCRISPR DNAコンストラクトを教示する。一部の実施形態において、本開示は、CRISPRエンドヌクレアーゼでDNAを消化することを教示する。一部の実施形態において、本開示は、II型−クラス2 CRISPRエンドヌクレアーゼ(例えばCas9)でDNAを消化することを教示する。一部の実施形態において、本開示は、V型−クラス2 CRISPRエンドヌクレアーゼでDNAを消化することを教示する。一部の実施形態において、本開示は、DNAをCpf1エンドヌクレアーゼで消化することを教示する。
[0011] 一部の実施形態において、本開示は、CRISPRマルチクローン部位を含む組換えモジュラーCRISPR DNAコンストラクトを教示し、前記マルチクローン部位は:a)少なくとも2つの互いに異なるクローニングタグ(cTAG)であって、各cTAGが:i)1つ以上の確認されたCRISPRランディング部位であって、それぞれ、プロトスペーサ隣接モチーフ(PAM)に作動可能に連結されたプロトスペーサ配列を含み;前記確認されたCRISPRランディング部位の少なくとも1つが、モジュラーCRISPR DNAコンストラクト内で固有である、1つ以上の確認されたCRISPRランディング部位を含む、cTAGと;b)1つ以上のDNA挿入配列とを含み;i)前記cTAGのそれぞれが、1つ以上のDNA挿入配列のそれぞれの周りのフランキング位置内に分布しており;且つii)前記DNA挿入配列の少なくとも1つが、選択マーカーを含む。
[0012] 一部の実施形態において、本開示は、組換えモジュラーCRISPR DNAコンストラクトを教示し、前記モジュラーCRISPR DNAコンストラクトは、環状である。
[0013] 一部の実施形態において、本開示は、組換えモジュラーCRISPR DNAコンストラクトを教示し、前記モジュラーCRISPR DNAコンストラクトは、直鎖状である。
[0014] 一部の実施形態において、本開示は、組換えモジュラーCRISPR DNAコンストラクトを教示し、前記モジュラーCRISPR DNAコンストラクトは、生物のゲノム中に組み込まれる。
[0015] 一部の実施形態において、本開示は、組換えモジュラーCRISPR DNAコンストラクトを教示し、前記互いに異なるcTAGの少なくとも1つが、少なくとも2つの確認されたCRISPRランディング部位を含む。
[0016] 一部の実施形態において、本開示は、組換えモジュラーCRISPR DNAコンストラクトを教示し、CRISPRランディング部位の少なくとも1つが、Cas9エンドヌクレアーゼ用である。
[0017] 一部の実施形態において、本開示は、組換えモジュラーCRISPR DNAコンストラクトを教示し、CRISPRランディング部位の少なくとも1つが、Cpf1エンドヌクレアーゼ用である。
[0018] 一部の実施形態において、本開示は、組換えモジュラーCRISPR DNAコンストラクトを教示し、前記互いに異なるcTAGの少なくとも1つが、稀な(≧8塩基長)制限酵素部位を含む。
[0019] 一部の態様において、本開示は、組換えモジュラーCRISPR DNAコンストラクトを「MegaModular」コンストラクトと呼ぶ。
[0020] 一部の実施形態において、本開示は、組換え核酸分子を調製する方法を教示し、前記方法は:a):i)複数のDNA挿入部分であって、各DNA挿入部分の側面に、2つのクローニングタグ(cTAG)が位置しており、各cTAGが:1)1つ以上の確認されたCRISPRランディング部位であって、それぞれ、プロトスペーサ隣接モチーフ(PAM)に作動可能に連結されたプロトスペーサ配列を含む、1つ以上の確認されたCRISPRランディング部位を含む、複数のDNA挿入部分と;ii)複数のDNA挿入部分の少なくとも2つにおいて存在する前記cTAGの少なくとも1つを標的とする1つ以上のCRISPR複合体であって、各CRISPR複合体が;1)CRISPRエンドヌクレアーゼ、及び2)前記標的とされるcTAGの1つに前記CRISPRエンドヌクレアーゼを動員することができるガイドRNAを含む、1つ以上のCRISPR複合体とを含む混合物を形成することであって;前記混合物は、複数のDNA挿入部分の少なくとも2つにおける標的とされるcTAGの消化を可能にして、突出末端を生じさせる条件下でインキュベートされる、混合物を形成することと、b)(a)において生じた消化生成物を、適合する突出末端のハイブリダイゼーション、及びハイブリダイズされた末端の共有結合を可能にする条件でインキュベートすることと;を含み、結果として生じた組換え核酸分子は、前記方法においてライゲーションされる元の挿入部分の完全なcTAG配列を含む。
[0021] 一部の実施形態において、本開示は、組換え核酸分子を調製する方法を教示し、CRISPRエンドヌクレアーゼは、Cpf1である。
[0022] 一部の実施形態において、本開示は、組換え核酸分子を調製する方法を教示し、CRISPRエンドヌクレアーゼは、Cas9である。
[0023] 一部の実施形態において、本開示は、組換え核酸分子を調製する方法を教示し、前記方法は:i)ライゲーションに先立って、CRISPR複合体から、消化されたcTAG配列を分離する工程、又はii)ライゲーションに先立って、CRISPR複合体を不活化する工程を含む。
[0024] 一部の実施形態において、本開示は、組換え核酸分子を調製する方法を教示し、分離工程は、DNA精製工程を含む。
[0025] 一部の実施形態において、本開示は、組換え核酸分子を調製する方法を教示し、不活化工程は、前記CRISPR複合体の熱又は化学物質による不活化を含む。
[0026] 一部の実施形態において、本開示は、組換え核酸分子を調製する方法を教示し、複数のDNA挿入部分のそれぞれの2つのcTAGは、cTAG対を形成し、前記cTAG対は、前記方法においてライゲーションされるDNA挿入部分の他の全てのcTAG対に対して固有である。
[0027] 一部の実施形態において、本開示は、組換え核酸分子を調製する方法を教示し、各cTAG対におけるcTAGの少なくとも1つが、異なるcTAG対における少なくとも1つの他のcTAGと同じである。
[0028] 一部の実施形態において、本開示は、DNA配列編集方法を教示し、前記方法は:a)反応に:i)本開示のモジュラーCRISPR DNAコンストラクトと:ii)置換DNA挿入部分であって、前記置換DNA挿入部分は、第1及び第2の挿入cTAGが側面に位置しており;1)第1の挿入cTAGは、モジュラーCRISPR DNAコンストラクトの互いに異なるcTAGの1つの確認されたCRISPRランディング部位を含み、第2の挿入cTAGは、モジュラーCRISPR DNAコンストラクトの別の互いに異なるcTAGの確認されたCRISPRランディング部位を含む、置換DNA挿入部分と;iii)第1及び第2の挿入cTAGをそれぞれ標的とする第1及び第2のCRISPR複合体とを導入することであって、各CRISPR複合体は:1)CRISPRエンドヌクレアーゼ、及び2)前記標的とされる挿入cTAGの1つに前記CRISPRエンドヌクレアーゼを動員することができるガイドRNAを含み;第1及び第2のCRISPR複合体は、第1及び第2の挿入cTAG、並びにそれらの対応する互いに異なるcTAGを切断して、突出末端を生じさせる、導入することと、b)置換DNA挿入部分及びcTAGが消化された生じたモジュラーCRISPR DNAコンストラクトを:(a)適合する突出末端のハイブリダイゼーション、及びハイブリダイズされた末端の共有結合を可能にする条件下でインキュベートすることとを含み;結果として生じた編集されたモジュラーCRISPR DNAコンストラクトは、当該方法によってライゲーションされる元の挿入部分の完全なcTAG配列を含む。
[0029] 一部の実施形態において、本開示は、DNA配列編集方法を教示し、工程(b)の反応は、機能的リガーゼを含む。
[0030] 一部の実施形態において、本開示は、DNA配列編集方法を教示し、前記方法は:a)反応に:i)本開示のモジュラーCRISPR DNAコンストラクトと;ii)モジュラーCRISPR DNAコンストラクト内の2つの互いに異なるcTAGを標的とする少なくとも2つのCRISPR複合体であって、各CRISPR複合体は;1)CRISPRエンドヌクレアーゼ、及び2)前記標的とされる互いに異なるcTAGの1つに前記CRISPRエンドヌクレアーゼを動員することができるガイドRNAを含み;第1及び第2のCRISPR複合体は、モジュラーCRISPR DNAコンストラクト内の2つの互いに異なるcTAGを切断し、結果として生じた互いに異なるcTAGは、突出末端を含む、少なくとも2つのCRISPR複合体とを導入することと、b)第2の反応に:i)(a)においてcTAGが消化された生じたモジュラーCRISPR DNAコンストラクトと;ii)置換DNA挿入部分であって、前記置換DNA挿入部分は、第1及び第2の挿入cTAGが側面に位置しており;1)第1の挿入cTAGは、(a)において切断される、未消化の互いに異なるcTAGの1つのポリヌクレオチド配列を含み、そして第2の挿入cTAGは、(a)において切断される別の未消化の異なるcTAGのポリヌクレオチド配列を含み;そして2)第1及び第2の挿入cTAGは、(a)由来の互いに異なるcTAGの突出末端と適合する突出末端を含む、置換DNA挿入部分とを;適合する突出末端のハイブリダイゼーション、及びハイブリダイズされた末端の共有結合を可能にする条件下で導入することとを含み;結果として生じた編集されたモジュラーCRISPR DNAコンストラクトは、(a)において標的とされる元の未消化の互いに異なるcTAGの完全な配列を含む。
[0031] 一部の実施形態において、本開示は、DNA配列編集方法を教示し、工程(b)の反応は、機能的リガーゼを含む。
[0032] 一部の実施形態において、本開示は、DNA配列編集方法を教示し、CRISPRエンドヌクレアーゼは、Cpf1である。
[0033] 一部の実施形態において、本開示は、DNA配列編集方法を教示し、工程(a)はさらに、2つの切断された互いに異なるcTAGを一本鎖エキソヌクレアーゼで消化することによって、突出末端を有する互いに異なるcTAGを生成することを含む。一部の態様において、エキソヌクレアーゼ工程後に、リガーゼ及びポリメラーゼを加えて、ポリメラーゼ及びリガーゼにより接合部を修復することができる。一部の態様において、当該反応はまた、Cas9消化による平滑末端切断によりなされてもよい。
[0034] 一部の実施形態において、本開示は、DNA配列編集方法を教示し、CRISPRエンドヌクレアーゼは、Cas9である。
[0035] 一部の実施形態において、本開示は、CRISPRマルチクローン部位を含む組換えモジュラーCRISPR DNAコンストラクトを含む宿主細胞ゲノムを提供し、前記マルチクローン部位は:a)少なくとも2つの互いに異なるクローニングタグ(cTAG)であって、各cTAGは:i)1つ以上の確認されたCRISPRランディング部位であって、それぞれ、プロトスペーサ隣接モチーフ(PAM)に作動可能に連結されたプロトスペーサ配列を含み;前記確認されたCRISPRランディング部位の少なくとも1つが、モジュラーCRISPR DNAコンストラクト内で固有である、CRISPRランディング部位を含む、cTAGと;b)1つ以上のDNA挿入部分とを含み;i)前記互いに異なるcTAGのそれぞれが、1つ以上のDNA挿入部分のそれぞれの周りのフランキング位置内に分布している。
[0036] 一部の実施形態において、本開示は、組換え核酸分子を調製する方法を提供し、前記方法は:a)i)2つのクローニングタグ(cTAG)が側面に位置する複数のDNA挿入部分であって、各cTAGは:1)1つ以上の確認されたCRISPRランディング部位であって、それぞれ、プロトスペーサ隣接モチーフ(PAM)に作動可能に連結されたプロトスペーサ配列を含む、CRISPRランディング部位;及び2)稀な(≧8塩基)制限酵素認識部位を含み;少なくとも2つの挿入部分のcTAGの少なくとも1つが、同じ制限酵素部位を含む、複数のDNA挿入部分と;ii)複数のDNA挿入部分の少なくとも2つにおける稀な制限酵素部位を標的とする1つ以上の制限酵素と:を含む混合物を;複数のDNA挿入部分の少なくとも2つにおける、1つ以上の制限酵素による標的とされるcTAGの消化を可能にして、DNA末端が消化された挿入部分を生じさせる条件下でインキュベートすることと;b)工程(a)において生じたDNA末端が消化されたDNA挿入部分を、消化されたDNA末端の共有結合を可能にする条件下でインキュベートすることとを含み;結果として生じた組換え核酸分子は、前記方法において共有結合される元の挿入部分の完全なcTAG配列を含む。
[0037] 一部の実施形態において、本開示は、DNA配列編集方法を提供し、前記方法は:a)i)請求項1のモジュラーCRISPR DNAコンストラクトであって、互いに異なるcTAGの少なくとも2つが、稀な(≧8塩基)制限酵素認識部位を含む、モジュラーCRISPR DNAコンストラクトと;ii)置換DNA挿入部分であって、前記置換DNA挿入部分は、第1及び第2の挿入cTAGが側面に位置しており;1)第1の挿入cTAGは、モジュラーCRISPR DNAコンストラクトの互いに異なるcTAGの1つの稀な制限酵素認識部位を含み、そして第2の挿入cTAGは、モジュラーCRISPR DNAコンストラクトの別の異なるcTAGの稀な制限酵素認識部位を含む、置換DNA挿入部分と;iii)第1及び第2の挿入cTAG内の稀な制限酵素部位を標的とする1つ以上の制限酵素とを用意することであって;部分(i)及び(ii)がそれぞれ、部分(iii)と、単一又は別個の反応でインキュベートされ;1つ以上の制限酵素は、第1及び第2の挿入cTAG、並びにそれらの対応する互いに異なるcTAGの稀な制限酵素認識部位を切断して、消化されたDNA末端を生じさせる、用意することと:b)置換DNA挿入部分、及び工程(a)において生じたDNA末端が消化されたモジュラーCRISPR DNAコンストラクトを、消化されたDNA末端の共有結合を可能にする条件下でインキュベートすることとを含み;結果として生じた編集されたモジュラーCRISPR DNAコンストラクトは、前記方法によって共有結合される元の挿入部分の完全なcTAG配列を含む。
[0038] 一部の実施形態において、本開示は、組換え核酸分子を調製する方法を提供し、前記方法は:a)i)複数のDNA挿入部分であって、各DNA挿入部分は、2つのクローニングタグ(cTAG)が側面に位置しており、各cTAGは:1)1つ以上の確認されたCRISPRランディング部位であって、それぞれ、プロトスペーサ隣接モチーフ(PAM)に作動可能に連結されたプロトスペーサ配列を含み;DNA挿入部分の少なくとも2つが、同じcTAGを共有する、CRISPRランディング部位を含む、複数のDNA挿入部分;ii)一本鎖DNA(ssDNA)エキソヌクレアーゼ:を含む混合物を;少なくとも2つのDNA挿入部分における共有されるcTAGの消化を可能にすることによって、少なくとも2つのDNA挿入部分内に適合する突出DNA末端を生じさせる条件下でインキュベートすることと、b)(a)において生じたcTAGが消化されたDNA挿入部分を、少なくとも2つのDNA挿入部分の適合する突出DNA末端のハイブリダイゼーション及び共有結合を可能にする条件下でインキュベートすることとを含み;結果として生じた組換え核酸分子は、消化前の共有されるcTAGの完全なcTAG配列を含む。この反応は、ポリメラーゼで行われてもリガーゼで行われてもよく、これらは接合部を固定するのに用いられる。さらにこれは、予め消化されたベクターに実行され得る。
図面の簡単な説明
[0039]図1A−Cは、本開示のCRISPR/Cas9系及びCRISPR/Cpf1系の比較を示す。図1A Cas9エンドヌクレアーゼは、tracrRNA及びcrRNA複合体によってdsDNAを標的とするように動員される。図1B Cas9エンドヌクレアーゼはまた、単一のガイドRNA(sgRNA)として知られている人工的に融合されたtracrRNA配列及びcrRNA配列によってdsDNAを標的とするように動員され得る。Cas9エンドヌクレアーゼは、平滑末端を生じさせる。
図1Cは、本開示のCRISPR/Cas9系及びCRISPR/Cpf1系の比較を示す。Cpf1エンドヌクレアーゼは、crRNAガイドポリリボヌクレオチドしか必要としない。Cpf1エンドヌクレアーゼ切断は、5’突出部を有する二本鎖切断を生じさせる。
[0040]図2A−Cは、本開示のモジュラーCRISPRコンストラクトを利用する本クローニング方法の実施形態を示す。図2Aは、本開示に従って、Cas9又はCpf1ヌクレアーゼで容易に変更され得るモジュラーCRISPRプラスミドを図解する。前述のように、本開示のモジュラーCRISPRコンストラクトは、「MegaModular」コンストラクトと呼ばれ得る。数字によって表される互換可能なpart(部分)は、文字によって表される不変のcTAG配列が側面に位置する。partは、予めアセンブルされてあってもよいし、cTAG配列同一性に基づいてインビトロでアセンブルされてもよい。例となる挿入partを、図2Aの右側に示す。
図2Bは、本開示のモジュラーCRISPRコンストラクトを利用する本クローニング方法の実施形態を示す。いくつかの戦略、例えばCas9、Cpf1、又は制限酵素によるcTAGでの切断を用いて、プラスミド全体を再びアセンブルする必要なく個々の部分を置換することができる。cTAG配列は、以下に限定されないが、Cas9、Cpf1、制限、及び/又は組換え部位が挙げられる1つ以上のクローニング部位を含んでよい。図2Cは、本開示のモジュラーCRISPRコンストラクトを利用する本クローニング方法の実施形態を示す。生物のゲノム中に組み込まれると、cTAGは、予め確認された直交するgRNA配列を有する、ゲノム組み込まれたDNAの置換、挿入、又は除去を可能とする予め確認されたCas9ランディング部位又はCpf1ランディング部位として機能し続け得る。
[0041]図3A−Dは、本開示のモジュラーCRISPRコンストラクトを利用する本クローニング方法の実施形態を示す。図3Aは、本開示に従って、Cas9又はCpf1ヌクレアーゼで容易に変更され得るモジュラーCRISPRプラスミドを図解する。数字によって表される互換可能なpartは、文字によって表される不変のcTAG配列が側面に位置する。partは、予めアセンブルされてあってもよいし、cTAG配列同一性に基づいてインビボ又はインビトロでアセンブルされてもよい。例となる挿入partを、図3Aの右側に示す。
図3Bは、本開示のモジュラーCRISPRコンストラクトを利用する本クローニング方法の実施形態を示す。いくつかの戦略、例えばCas9、Cpf1、又は制限酵素によるcTAGでの切断を用いて、プラスミド全体を再びアセンブルする必要なく個々の部分を置換することができる。cTAG配列は、以下に限定されないが、Cas9、Cpf1、制限、及び/又は組換え部位が挙げられる1つ以上のクローニング部位を含んでよい。
[0041]図3Cは、本開示のモジュラーCRISPRコンストラクトを利用する本クローニング方法の実施形態を示す。図3Cは、挿入部分を除去する、又は既存のモジュラープラスミドからスタッファ配列を加える本開示の方法を示す。
[0041]図3Dは、本開示のモジュラーCRISPRコンストラクトを利用する本クローニング方法の実施形態を示す。本開示のモジュラープラスミドの挿入部分は、モジュラーCRISPRベクターの一部又は全体の、宿主細胞のゲノム中へのゲノム組み込み用の配列として機能し得る。
[099]図4は、実施例1のワンポットインビトロモジュラーCRISPRクローニングを示す。具体的には、あるプラスミドから別のものへのワンポット反応での挿入物の移入によるプラスミド13001009086の生成を示す。この反応の詳細を、実施例1で明らかにする。
[0042]図5は、実施例2のインビトロモジュラーCRISPRクローニング方法の実施形態を示す。各パネルは、実施例2に記載する実験設計の実例を示す。クロラムフェニコール耐性遺伝子を、カナマイシン耐性骨格プラスミド中にクローニングして、デュアル耐性プラスミドを生じさせた。次に、デュアル耐性プラスミドを細菌に形質転換してから、カナマイシン及びクロラムフェニコール抗生物質を補った培地中で当該細菌を培養した。耐性コロニーは、Cpf1クローニングアセンブリの成功を示した。
[0043]図6は、実施例2のインビトロモジュラーCRISPRクローニング方法の結果を示す。y軸は、カナマイシン及びクロラムフェニコールを補った培地中で増殖した、回収したコロニーの数を表す。耐性コロニーは、Cpf1クローニングアセンブリの成功を示す。結果は、デュアル耐性プラスミドのリガーゼ依存性アセンブリを示した。
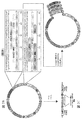
[0044]図7は、pJDI427のベクターマップを示す。Cpf1アセンブリに用いたCRISPRランディング部位を、cTAG M及びcTAG Nとしてラベル付けしている。関連する配列情報は、配列番号102に見出すことができる。
[0045]図8は、pJDI429のベクターマップを示す。Cpf1アセンブリに用いたCRISPRランディング部位を、cTAG N及びcTAG Oとしてラベル付けしている。関連する配列情報は、配列番号103に見出すことができる。
[0046]図9は、pJDI430のベクターマップを示す。Cpf1アセンブリに用いたCRISPRランディング部位を、cTAG P及びcTAG Nとしてラベル付けしている。関連する配列情報は、配列番号104に見出すことができる。
[0047]図10は、pJDI431のベクターマップを示す。Cpf1アセンブリに用いたCRISPRランディング部位を、cTAG P及びcTAG Oとしてラベル付けしている。関連する配列情報は、配列番号105に見出すことができる。
[0048]図11は、pJDI432のベクターマップを示す。Cpf1アセンブリに用いたCRISPRランディング部位を、cTAG M及びcTAG Nとしてラベル付けしている。関連する配列情報は、配列番号106に見出すことができる。
[0049]図12は、pJDI434のベクターマップを示す。Cpf1Cアセンブリに用いたCRISPRランディング部位を、cTAG N及びcTAG Oとしてラベル付けしている。関連する配列情報は、配列番号107に見出すことができる。
[0050]図13は、pJDI435のベクターマップを示す。Cpf1アセンブリに用いたCRISPRランディング部位を、cTAG P及びcTAG Nとしてラベル付けしている。関連する配列情報は、配列番号108に見出すことができる。
[0051]図14は、pJDI436のベクターマップを示す。Cpf1アセンブリに用いたCRISPRランディング部位を、cTAG P及びcTAG Oとしてラベル付けしている。関連する配列情報は、配列番号109に見出すことができる。
[0052]図15は、本開示の方法に従う、モジュラーCRISPRコンストラクトの例となる遺伝子編集を示す。具体的には、図15は、実施例3のmegamodular設計を用いる制限酵素消化及びライゲーションによるプラスミドアセンブリを示す。図15は、モジュラーCRISPRプラスミド骨格p1300283391及び適合するGFP含有挿入DNA部分を、ApaI及びPvuI制限酵素でそれぞれ消化して、適合するクローニングタグ末端が生じることを示す。消化した骨格及び挿入物を、インビトロでライゲーションして、新しいモジュラーCRISPRコンストラクトが生じる。
詳細な説明
定義
[0053] 以下の用語は、当業者によってよく理解されると考えられるが、以下の定義は、本開示の主題の説明を容易にするために示される。
定義
[0053] 以下の用語は、当業者によってよく理解されると考えられるが、以下の定義は、本開示の主題の説明を容易にするために示される。
[0054] 用語「a」又は「an」は、その実体の1つ以上を指す、すなわち、複数の指示対象を指し得る。したがって、用語「a」又は「an」、「1つ以上」、及び「少なくとも1つ」は、本明細書中で互換的に用いられる。加えて、不定冠詞「a」又は「an」による「要素」への言及は、要素の1つが存在すること、そして要素の1つしか存在しないことを文脈が明らかに要求しない限り、1つを超える要素が存在する可能性を除外しない。
[0055] 用語「原核生物」は、当該技術で認識されており、核を含有しない細胞を指す。原核生物は通常、2つのドメイン、細菌(Bacteria)及び古細菌(Archaea)の1つに分類される。古細菌ドメインと細菌ドメインの決定的な生物間差異は、16SリボソームRNA内のヌクレオチド塩基配列の根本的差異に基づく。
[0056] 「真核生物」は、細胞が、膜内に囲まれた核及び他の細胞小器官を含有するあらゆる生物である。真核生物は、分類群真核生物(Eukarya又はEukaryota)に属する。原核生物細胞(上述の細菌及び古細菌)とは別に真核細胞を設定する顕著な特徴は、真核細胞が、膜結合細胞小器官、とりわけ核を有することであり、核は、遺伝的材料を含有し、且つ核エンベロープによって囲まれている。
[0057] 用語「古細菌」は、メンドシクテス(Mendosicutes)門の生物のカテゴリ化を指し、これは典型的に、特殊な環境において見出され、且ついくつかの基準(リボソームタンパク質の数、及び細胞壁中のムラミン酸の欠乏が挙げられる)によって残りの原核生物から区別される。ssrRNA分析に基づいて、古細菌は、2つの系統学的に異なる群:クレン古細菌(Crenarchaeota)及びユーリ古細菌(Euryarchaeota)からなる。その生理学に基づいて、古細菌は、3つのタイプ:メタン生成菌(メタンを生成する原核生物);極端な好塩菌(非常に高い塩(NaCl)濃度にて生きる原核生物);及び極端な(ハイパー)サーモフィルス(非常に高い温度にて生きる原核生物)に系統化され得る。細菌から識別される一体化した古細菌の特徴(すなわち、細胞壁中にムレインなし、エステル結合膜脂質その他)の他に、当該原核生物は、特定の生息地に適合する特有の構造的属性又は生化学的属性を示す。クレン古細菌は主に、超好熱性の硫黄依存性原核生物からなり、ユーリ古細菌は、メタン生成菌及び極端な好塩菌を含有する。
[0058] 「細菌」又は「真正細菌」は、原核生物のドメインを指す。細菌は、以下の少なくとも11の異なる群を含む:(1)グラム陽性(gram+)菌(2つの主要な亜門がある):(1)高G+C群(アクチノミセス(Actinomycetes)、マイコバクテリウム(Mycobacteria)、ミクロコッカス(Micrococcus)その他);(2)低G+C群(バチルス(Bacillus)、クロストリジウム(Clostridia)、ラクトバチルス(Lactobacillus)、ブドウ球菌(Staphylococci)、連鎖球菌(Streptococci)、マイコプラズマ(Mycoplasmas));(2)プロテオバクテリア(Proteobacteria)、例えば、紅色光合成+非光合成グラム陰性菌(最も「一般的な」グラム陰性菌が挙げられる);(3)シアノバクテリア、例えば酸素発生型光合成生物;(4)スピロヘータ及び近縁種;(5)プランクトミセス(Planctomyces);(6)バクテロイデス(Bacteroides)、フラボバクテリア(Flavobacteria);(7)クラミジア(Chlamydia);(8)緑色硫黄細菌;(9)緑色非硫黄細菌(嫌気性光合成生物);(10)放射線抵抗性の球状細菌及び近縁生物;(11)サーモトーガ(Thermotoga)及びサーモシフォ・サーモフィルス(Thermosipho thermophiles)。
[0059] 用語「遺伝的に修飾された宿主細胞」、「組換え宿主細胞」、及び「組換え株」は、本明細書中で互換的に用いられており、本開示のクローニング方法及び形質転換方法によって遺伝的に修飾された宿主細胞を指す。ゆえに、当該用語は、由来した天然に存在する微生物と比較して、(例えば、遺伝的修飾が、微生物のコード核酸配列に影響を及ぼす場合に、)変更された、修飾された、又は異なる遺伝子型及び/又は表現型を示すように、遺伝的に変更され、修飾され、又は操作された宿主細胞(例えば、細菌、酵母細胞、真菌細胞、CHO、ヒト細胞その他)を含む。当該用語は、問題の特定の組換え微生物だけでなく、そのような微生物の後代又は潜在的な後代をも指すことが理解される。
[0060] 用語「遺伝的に操作された」は、宿主細胞のゲノムのあらゆる操作(例えば、核酸の挿入又は欠失による)を指し得る。
[0061] 本明細書中で用いられる「選択マーカー」は、多くの場合、特定の条件下で、これを含有する分子(例えばレプリコン)又は細胞を選択させることができる核酸セグメントである。当該マーカーは、以下に限定されないが、RNA、ペプチド、又はタンパク質の生成等の活性をコードすることもできるし、RNA、ペプチド、タンパク質、無機化合物又は無機組成物及び有機化合物又は有機組成物等のための結合部位を提供することもできる。選択マーカーの例として、限定されないが、以下が挙げられる:(1)普通であれば有毒な化合物に対して耐性をもたらす生成物(例えば抗生物質)をコードする核酸セグメント;(2)レシピエント細胞内で普通であれば欠如している生成物(例えば、tRNA遺伝子、栄養要求性マーカー)をコードする核酸セグメント;(3)遺伝子生成物の活性を抑制する生成物をコードする核酸セグメント;(4)容易に同定され得る生成物(例えば、表現型マーカー、例えばβ−ガラクトシダーゼ、緑色蛍光タンパク質(GFP)、黄色蛍光タンパク質(YFP)、シアン蛍光タンパク質(CFP)、及び細胞表面タンパク質)をコードする核酸セグメント;(5)細胞の生存及び/又は機能に対して普通であれば有害である他の生成物に結合する生成物をコードする核酸セグメント;(6)核酸セグメントのいずれかの活性を別のやり方で阻害して、可視の、又は選択可能な表現型をもたらす核酸(例えばアンチセンスオリゴヌクレオチド)をコードする核酸セグメント;(7)基質を修飾する他の生成物(例えば制限酵素)に結合する生成物をコードする核酸セグメント;(8)所望の分子を単離又は同定するのに用いられ得る核酸セグメント(例えば特定のタンパク質結合部位);(9)普通であれば機能し得ない特定のヌクレオチド配列を(例えば、分子の亜集団のPCR増幅用に)コードする核酸セグメント;並びに(10)不在である場合に、特定の化合物に耐性又は感受性を直接的又は間接的に付与する核酸セグメント。
[0062] 本明細書中で用いられる「対抗選択マーカー」(counterselectable marker又はcounterselection marker)は、選択直後に宿主生物の成長を排除する、又は阻害する核酸セグメントである。一部の実施形態において、本開示の対抗選択マーカーは、細胞を、化学物質/増殖条件/遺伝的背景の1つ以上に対して感受性にさせる。一部の実施形態において、本開示の対抗選択マーカーは、毒性遺伝子である。一部の実施形態において、対抗選択マーカーは、誘導プロモータによって発現される。
[0063] 本明細書中で用いられる用語「核酸」は、あらゆる長さのヌクレオチドのポリマー形態(リボヌクレオチド若しくはデオキシリボヌクレオチド、又はその類似体)を指す。当該用語は、分子の一次構造を指すので、二本鎖DNA及び一本鎖DNA、並びに二本鎖RNA及び一本鎖RNAを含む。また、修飾された核酸、例えばメチル化且つ/又はキャッピングされた核酸、修飾された塩基、骨格修飾等を含有する核酸も含む。用語「核酸」及び「ヌクレオチド配列」は、互換的に用いられる。
[0064] 本明細書中で用いられる用語「遺伝子」は、生体機能と関連するDNAのあらゆるセグメントを指す。ゆえに、遺伝子は、以下に限定されないが、コード配列及び/又はその発現に必要とされる調節配列を含む。遺伝子はまた、例えば、他のタンパク質のための認識配列を形成する発現されないDNAセグメントを含んでもよい。遺伝子は、注目のソースからのクローニング、又は知られている、若しくは予測される配列情報からの合成が挙げられる種々のソースから得られ得、所望のパラメータを有するように設計された配列を含んでよい。
[0065] 本明細書中で用いられる用語「相同」、「ホモログ」、又は「オルソログ」は、当該技術において知られており、そして、共通の祖先又はファミリーメンバーを共有し、且つ配列同一性の程度に基づいて決定される関連した配列を指す。用語「相同性」、「相同」、「実質的に類似」、及び「実質的に対応」は、本明細書中で互換的に用いられる。これらは、1つ以上のヌクレオチド塩基の変化が、遺伝子発現を媒介する、又は特定の表現型をもたらす核酸フラグメントの能力に影響を及ぼさない核酸フラグメントに言及する。当該用語はまた、最初の無修飾フラグメントに対する、結果として生じる核酸フラグメントの機能特性を実質的に変更しない1つ以上のヌクレオチドの欠失又は挿入等の、本開示の核酸フラグメントの修飾にも言及する。したがって、当業者が理解するように、本開示は、特定の例示的な配列を超えて包含することが理解される。当該用語は、1つの種、亜種、変種、品種、又は株に見出される遺伝子と、別の種、亜種、種類、品種、又は株の対応する、又は相当する遺伝子との関係を説明する。本開示の目的上、相同配列同士が比較される。「相同配列」、「ホモログ」、又は「オルソログ」は、機能的に関連していると思われ、考えられ、又はそうであることが知られている。機能的関係は、限定されないが以下が挙げられるいくつかの方法のいずれか1つで示され得る:(a)配列同一性の程度、及び/又は(b)同じ若しくは類似の生体機能。好ましくは、(a)及び(b)の双方が示される。相同性は、当該技術において容易に入手可能なソフトウェアプログラムを用いて判定され得、例えば、Current Protocols in Molecular Biology (F.M. Ausubel et al., eds., 1987) Supplement 30, section 7.718, Table 7.71において考察されるものがある。一部のアラインメントプログラムは、MacVector(Oxford Molecular Ltd, Oxford, U.K.)、ALIGN Plus(Scientific and Educational Software, Pennsylvania)、及びAlignX(Vector NTI, Invitrogen, Carlsbad, CA)である。別のアラインメントプログラムは、デフォルトパラメータを用いるSequencher(Gene Codes, Ann Arbor, Michigan)である。
[0066] 本明細書中で用いられる用語「ヌクレオチド変化」は、例えば、当該技術において十分に理解されるように、ヌクレオチド置換、欠失、及び/又は挿入を指す。例えば、突然変異は、サイレント置換、付加、又は欠失をもたらす変更を含有するが、コードされるタンパク質の特性若しくは活性、又はタンパク質が形成される方法を変更しない。
[0067] 本明細書中で用いられる用語「タンパク質修飾」は、例えば、当該技術において十分に理解されるように、アミノ酸置換、アミノ酸修飾、欠失、及び/又は挿入を指す。
[0068] 本明細書中で用いられる、核酸又はポリペプチドの用語「少なくとも一部」又は「フラグメント」は、そのような配列の最小のサイズ特性を有する部分、又は完全長分子のより大きなあらゆるフラグメント(最大、完全長分子を含む)を意味する。本開示のポリヌクレオチドのフラグメントは、遺伝的調節要素の生物学的に活性な部分をコードし得る。遺伝的調節要素の生物学的に活性な部分は、本明細書中で記載されるように、遺伝的調節要素を含む本開示のポリヌクレオチドの1つの部分を単離して、活性を評価することによって、調製され得る。同様に、ポリペプチドの部分は、4アミノ酸、5アミノ酸、6アミノ酸、7アミノ酸等であってよく、最大で完全長のポリペプチドに至ってよい。用いられることとなる部分の長さは、特定の用途によって決まることとなる。ハイブリダイゼーションプローブとして有用な核酸の部分は、12ヌクレオチドの短さであってもよい;一部の実施形態において、これは20ヌクレオチドである。エピトープとして有用なポリペプチドの部分は、4アミノ酸の短さであってもよい。全長ポリペプチドの機能を実行するポリペプチドの部分は通常、4アミノ酸よりも長くなるであろう。
[0069] 本明細書中で開示されるポリヌクレオチドのPCR増幅のために、オリゴヌクレオチドプライマーが、注目のあらゆる生物から抽出されたcDNA又はゲノムDNA由来の対応するDNA配列を増幅するためのPCR反応に用いられるように設計され得る。PCRプライマーを設計する方法及びPCRクローニング方法が、当該技術において通常知られており、Sambrook et al. (2001) Molecular Cloning: A Laboratory Manual (3rd ed., Cold Spring Harbor Laboratory Press, Plainview, New York)に開示されている。また、Innis et al., eds. (1990) PCR Protocols: A Guide to Methods and Applications (Academic Press, New York);Innis and Gelfand, eds. (1995) PCR Strategies (Academic Press, New York);及びInnis and Gelfand, eds. (1999) PCR Methods Manual (Academic Press, New York)も参照。PCRの既知の方法として、以下に限定されないが、対形成されたプライマー、ネステッドプライマー、単一の特異的なプライマー、縮重プライマー、遺伝子特異的プライマー、ベクター特異的プライマー、部分的にミスマッチされるプライマー等を用いる方法が挙げられる。
[0070] 本明細書中で用いられる用語「プライマー」は、増幅標的にアニールして、DNAポリメラーゼを付着させることによって、プライマー伸長生成物の合成が誘導される条件下、すなわち、重合のためのヌクレオチド及び剤、例えばDNAポリメラーゼの存在下、そして適切な温度及びpHに置かれたときに、DNA合成の開始点として機能することができるオリゴヌクレオチドを指す。(増幅)プライマーは好ましくは、増幅の効率を最大にするために、一本鎖である。好ましくは、プライマーは、オリゴデオキシリボヌクレオチドである。プライマーは、重合のための剤の存在下で、伸長生成物の合成を開始させるほど十分に長くなければならない。プライマーの正確な長さは、温度及びプライマーの組成(A/T対G/C含量)が挙げられる多く因子によって決まることとなる。PCR増幅等のDNA増幅の当該技術において一般的に用いられるように、一対の双方向プライマーが、1つのフォワードプライマー及び1つのリバースプライマーからなる。
[0071] 用語「ストリンジェンシー」又は「ストリンジェントハイブリダイゼーション条件」は、ハイブリッドの安定性に影響を及ぼすハイブリダイゼーション条件、例えば、温度、塩濃度、pH、ホルムアミド濃度等を指す。これらの条件は、プライマー又はプローブの、その標的核酸配列への特異的結合を最大にし、且つ非特異的結合を最小にするように、実験に基づいて最適化される。使用される用語は、プローブ又はプライマーが、その標的配列に、他の配列よりも検出可能となるように(例えばバックグラウンドに対して少なくとも2倍)ハイブリダイズすることとなる条件への言及を含む。ストリンジェント条件は、配列依存的であり、状況が違えば異なることとなる。配列が長くなるほど、より高い温度にて特異的にハイブリダイズする。通常、ストリンジェント条件は、定義されたイオン強度及びpHにて、特定の配列についての熱融解点(Tm)よりも約5℃低くなるように選択される。Tmは、相補的な標的配列の50%が、完全にマッチするプローブ又はプライマーにハイブリダイズする(定義されたイオン強度及びpH下の)温度である。典型的には、ストリンジェント条件は、pH7.0〜8.3にて、塩濃度が約1.0M Na+イオン未満、典型的には約0.01〜1.0M Na+イオン濃度(又は他の塩)であり、そして温度は、短いプローブ又はプライマー(例えば10〜50ヌクレオチド)について少なくとも約30℃であり、長いプローブ又はプライマー(例えば50ヌクレオチド超)について少なくとも約60℃である。ストリンジェント条件はまた、ホルムアミド等の脱安定化剤(destabilizing agent)の付加により達成されてもよい。例示的な低ストリンジェント条件又は「ストリンジェンシー引下げ条件」として、30%ホルムアミド、1M NaCl、1%SDSのバッファ溶液による37℃でのハイブリダイゼーション、及び2×SSC中での40℃での洗浄が挙げられる。例示的な高ストリンジェンシー条件として、50%ホルムアミド、1M NaCl、1%SDS中での37℃でのハイブリダイゼーション、及び0.1×SSC中での60℃での洗浄が挙げられる。ハイブリダイゼーション手順は、当該技術において周知であり、例えばAusubel et al., 1998及びSambrook et al., 2001に記載されている。一部の実施形態において、ストリンジェント条件は、1mM Na2EDTA、0.5〜20%ドデシル硫酸ナトリウム(例えば、0.5%、1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%又は20%)を含有する0.25M Na2HPO4バッファ(pH7.2)中での45℃でのハイブリダイゼーションに続く、0.1%(w/v)ドデシル硫酸ナトリウムを含有する5×SSC中での55℃〜65℃での洗浄である。
[0072] 本明細書中で用いられる用語「実質的に同じ」は、1、2、3、4、5、6、又は7以下のヌクレオチドが異なる2つのポリヌクレオチド配列を指す。cTAGの文脈で用いられる場合、用語「実質的に同じ」は、少なくとも1つのCRISPRランディング部位においてCRISPR切断を排除するように設計されたcTAGの1つのPAM又はプロトスペーサ領域中の突然変異を除いて、同じであろう2つのcTAGを表す。用語「実質的に同じ」が、用語「部分的な」配列又はcTAGと共に用いられる場合、当該組合せは、先に述べたような、2つの実質的に同じcTAG間の比較を指し、ここでcTAGの1つは、CRISPRエンドヌクレアーゼによって消化されている。ゆえに、当該用語は、PAM又はプロトスペーサ領域内の突然変異を除いて、記載されているcTAGが、第2のcTAGと(その未消化の形態で)同じであることを示すのに用いられることとなる。
[0073] 本明細書中で用いられる用語「プロモータ」は、コード配列又は機能性RNAの発現を制御することができるDNA配列を指す。プロモータ配列は、近位の上流要素及びより遠位の上流要素からなり得、後者の要素は多くの場合、エンハンサと呼ばれる。したがって、「エンハンサ」は、プロモータ活性を刺激することができるDNA配列であり、プロモータの固有の要素であっても、プロモータのレベル又は組織特異性を増強するために挿入された異種要素であってもよい。
[0074] 本明細書中で用いられる用語「異種」は、特定の生物において天然に見出されない核酸配列を指す。
[0075] 本明細書中で用いられる用語「内因性」、「内因性遺伝子」は、遺伝子の天然に存在するコピーを指す。
[0076] 本明細書中で用いられる用語「天然に存在する」は、天然に存在するソースに由来する遺伝子又は配列を指す。一部の実施形態において、天然に存在する遺伝子は、野生型遺伝子(非導入遺伝子)を指し、ソース生物内でのその内因性セッティング(endogenous setting)内に位置決めされているかも、異なる生物中に導入された場合に「異種」セッティング内に配置されるかも問わない。ゆえに、本開示の目的上、「天然に存在しない」配列は、合成された、突然変異させた、又はそれ以外では、既知の天然の配列とは異なる配列を有するように修飾された配列である。一部の実施形態において、修飾は、タンパク質レベルのもの(例えばアミノ酸置換)であってよい。他の実施形態において、修飾は、タンパク質配列に何ら影響を及ぼさないDNAレベルのもの(例えばコドン最適化)であってよい。一部の実施形態において、天然に存在しない配列は、コンストラクトであってよい。
[0077] 本明細書中で用いられる用語「外因性」は、用語「異種」と互換的に用いられており、その天然のソース以外のいくつかのソースに由来する物質を指す。例えば、用語「外因性タンパク質」又は「外因性遺伝子」は、非天然のソース又は位置由来の、生体系に人工的に供給されたタンパク質又は遺伝子を指す。内因性の遺伝子の、人工的に突然変異させた変異体は、本開示の目的上、「外因性」とみなす。
[0078] 本明細書中で用いられるフレーズ「組換えコンストラクト」、「発現コンストラクト」、「キメラコンストラクト」、「コンストラクト」、及び「組換えDNAコンストラクト」は、本明細書中で互換的に用いられる。組換えコンストラクトは、核酸フラグメントの人工的な組合せ、例えば、自然界で一緒に見出されない調節配列及びコード配列を含む。例えば、キメラコンストラクトは、異なるソースに由来する調節配列及びコード配列を含んでもよいし、同じソースに由来するが、自然界で見出されるものとは異なる様式で配置される調節配列及びコード配列を含んでもよい。そのようなコンストラクトは、それ自体で用いられてもよいし、ベクターと共に用いられてもよい。ベクターが用いられるならば、ベクターの選択は、当業者にとって周知の、宿主細胞を形質転換するのに用いられることとなる方法次第である。例えば、プラスミドベクターが用いられ得る。当業者であれば、本開示の単離された核酸フラグメントのいずれかを含む宿主細胞を首尾よく形質転換し、選択し、且つ増殖させるのに、ベクター上に存在しなければならない遺伝的要素を十分知っている。また、当業者であれば、独立した様々な形質転換事象が、発現の様々なレベル及びパターンをもたらすこととなること(Jones et al., (1985) EMBO J. 4:2411-2418;De Almeida et al., (1989) Mol. Gen. Genetics 218:78-86)、ゆえに複数の事象が、所望の発現レベル及びパターンを提示する系統を得るのにスクリーニングされなければならないことを認識するであろう。そのようなスクリーニングは、とりわけ、DNAのサザン分析、mRNA発現のノーザン分析、タンパク質発現のイムノブロッティング分析、又は表現型分析によって達成され得る。ベクターは、プラスミド、ウイルス、バクテリオファージ、プロウイルス、ファージミド、トランスポゾン、人工染色体等であってよく、これらは自律的に複製し、又は宿主細胞の染色体中に組み込むことができる。ベクターはまた、裸のRNAポリヌクレオチド、裸のDNAポリヌクレオチド、同じ鎖内でDNA及びRNAの双方で構成されるポリヌクレオチド、ポリ−リジン結合DNA又はRNA、ペプチド結合DNA又はRNA、リポソーム結合DNA等であってもよく、これらは自律的に複製しない。本明細書中で用いられる用語「発現」は、機能的最終生成物、例えば、mRNA又はタンパク質(前駆体又は成熟)の生成を指す。
[0079] 用語「作動可能に連結された」は、文脈において、本開示に従うプロモータポリヌクレオチドの、更なるオリゴヌクレオチド又はポリヌクレオチドとの、前記更なるポリヌクレオチドの転写をもたらす連続的な配置を意味する。一部の実施形態において、本開示のプロモータ配列は、遺伝子の5’UTR、又はオープンリーディングフレームの直前に挿入される。他の実施形態において、本開示の作動可能に連結されたプロモータ配列及び遺伝子配列は、1つ以上のリンカーヌクレオチドによって分離されている。CRISPRプロトスペーサ及びプロスペーサ(prospacer)隣接モチーフ(PAM)の文脈における用語「作動可能に連結された」は、CRISPRエンドヌクレアーゼ複合体によって高い効率にて切断され得る、近接して配置されたプロトスペーサ/PAM組合せ配列を指す。
[0080] 用語「CRISPR RNA」又は「crRNA」は、標的DNA配列とのハイブリダイズ、及びCRISPRエンドヌクレアーゼの動員を担うRNA鎖を指す。crRNAは、天然に存在し得、又はRNAを生成する既知のあらゆる方法に従って合成され得る。
[0081] 用語「ガイド配列」又は「スペーサ」は、標的DNAとのハイブリダイズを担うcrRNA又はガイドRNA(gRNA)の部分を指す。
[0082] 用語「プロトスペーサ」は、crRNA又はガイド鎖によって標的とされるDNA配列を指す。一部の実施形態において、プロトスペーサ配列は、CRISPR複合体のcrRNAガイド配列とハイブリダイズする。
[0083] 用語「シード領域」は、DNA配列CRISPRリボ核タンパク質複合体間の最初の複合体形成を担うリボ核配列(ribonucleic sequence)を指す。シード領域と標的DNA配列間のミスマッチは、crRNA/sgRNA配列の残部よりも強い作用を、標的部位の認識及び切断に及ぼす。一部の実施形態において、crRNA/gRNAのシード領域における単一のミスマッチが、CRISPR複合体をその結合部位にて不活性にすることができる。一部の実施形態において、Cas9エンドヌクレアーゼ用のシード領域は、ガイド配列の3’部分の最後の約12ntに沿って位置決めされ、これは、PAMに隣接するプロトスペーサ標的配列の部分に対応する(ハイブリダイズする)。一部の実施形態において、Cpf1エンドヌクレアーゼ用のシード領域は、ガイド配列の5’部分の最初の約5ntに沿って位置決めされ、これは、PAMに隣接するプロトスペーサ標的配列の部分に対応する(ハイブリダイズする)。
[0084] 用語「tracrRNA」は、トランスコードされた(trans-encoded)小さなRNAを指す。TracrRNAは、crRNAと相補的であり、且つcrRNAと塩基対形成して、crRNA/tracrRNAハイブリッドを形成し、これは、CRISPRエンドヌクレアーゼを標的配列に動員することができる。
[0085] 本明細書中で用いられる用語「ガイドRNA」又は「gRNA」は、CRISPRエンドヌクレアーゼを標的配列に動員することができるRNAの配列又は配列の組合せを指す。ゆえに、本明細書中で用いられるガイドRNAは、天然若しくは合成のcrRNA(例えば、Cpf1について)、天然若しくは合成のcrRNA/tracrRNAハイブリッド(例えば、Cas9について)、又は単一のガイドRNA(sgRNA)であり得る。
[0086] 本明細書中で用いられる用語「CRISPRランディング部位」は、CRISPR複合体によって標的とされ得るDNA配列を指す。ゆえに、一部の実施形態において、CRISPRランディング部位は、CRISPRエンドヌクレアーゼ複合体で切断され得る、近接して配置されたプロトスペーサ/プロトスペーサ隣接モチーフの組合せ配列を含む。用語「確認されたCRISPRランディング部位」は、前記配列の高い効率の切断を誘導することができるガイドRNAが存在するCRISPRランディング部位を指す。ゆえに、用語「確認された」は、配列が、CRISPR複合体によって切断可能であることが以前に示されていることを意味すると解釈されるべきである。各「確認されたCRISPRランディング部位」は、定義上、確認を伴う、試験されたガイドRNAの存在を裏付けることとなる。
[0087] 用語「付着末端(sticky end)」は、配列突出部を含む二本鎖ポリヌクレオチド分子の末端を指す。一部の実施形態において、付着末端は、5’又は3’の配列突出部を有するdsDNA分子の末端であり得る。一部の実施形態において、本開示の付着末端は、同じ、又は他の分子の適合する付着末端とハイブリダイズすることができる。ゆえに、一実施形態において、第1のDNAフラグメントの3’上の付着末端は、第2のDNAフラグメント上の適合する付着末端とハイブリダイズし得る。一部の実施形態において、これらのハイブリダイズされた付着末端は、リガーゼによって縫い合わされ(sewn together)得る。他の実施形態において、付着末端は、ライゲーションに先立って、dsDNA分子を完成させるのに突出部の伸長を必要とする場合がある。用語「遺伝的スカー」は、DNA操作方法によって核酸配列中に導入された、不所望のあらゆる配列を指す。例えば、一部の実施形態において、本開示は、制限酵素結合部位、配列アダプター、又はクローニングに対処するためのスペーサ、TA−部位、NHEJから残されたスカー等の遺伝的スカーを教示する。一部の実施形態において、本開示は、スカーレスクローニング方法及び遺伝子編集方法を教示する。
[0088] 本明細書中で用いられる用語「標的とされる」は、1つの部材又は分子が、別の部材又は分子と、非標的部材又は分子を除外するような特異性の程度で相互作用するであろうという予想を指す。例えば、本開示に従う第2のポリヌクレオチドを標的とする第1のポリヌクレオチドは、第2のポリヌクレオチドと配列特異的に(例えば、ワトソン−クリック型塩基対形成を介して)ハイブリダイズするように設計されている。一部の実施形態において、ハイブリダイゼーションの選択された領域は、ハイブリダイゼーションを、1つ又は複数の標的領域に対して固有になるように設計される。第2のポリヌクレオチドは、その標的配列(ハイブリダイゼーションの領域)が突然変異し、又はそれ以外では第2のポリヌクレオチドから除去/分離されれば、第1のターゲティングポリヌクレオチドの標的であることが終わり得る。
[0089] 本開示は、「MegaModular」コンストラクト又は設計として教示且つ記載されるユニバーサルモジュラーCRISPR DNAコンストラクト又は設計を指す。
DNAヌクレアーゼ
[0090] 一部の実施形態において、本開示は、DNAヌクレアーゼを利用する遺伝子編集/クローニングのための方法及び組成物を教示する。CRISPR複合体、転写活性化因子様エフェクタヌクレアーゼ(TALEN)、亜鉛フィンガーヌクレアーゼ(ZFN)、及びFokI制限酵素が、遺伝子編集ツールとして用いられている配列特異的ヌクレアーゼの一部である。これらの酵素は、そのヌクレアーゼ活性を、注目の配列を認識するように操作されたガイド領域との相互作用を通して、所望の標的遺伝子座に標的化することができる。一部の実施形態において、本開示は、CRISPRベースの遺伝子編集方法を教示する。
[0090] 一部の実施形態において、本開示は、DNAヌクレアーゼを利用する遺伝子編集/クローニングのための方法及び組成物を教示する。CRISPR複合体、転写活性化因子様エフェクタヌクレアーゼ(TALEN)、亜鉛フィンガーヌクレアーゼ(ZFN)、及びFokI制限酵素が、遺伝子編集ツールとして用いられている配列特異的ヌクレアーゼの一部である。これらの酵素は、そのヌクレアーゼ活性を、注目の配列を認識するように操作されたガイド領域との相互作用を通して、所望の標的遺伝子座に標的化することができる。一部の実施形態において、本開示は、CRISPRベースの遺伝子編集方法を教示する。
[0091] インビボCRISPRベースの編集の原理は、大部分は、天然の細胞のDNA修復系に依存している。ヌクレアーゼによって導入される二本鎖dsDNA切断は、非相同末端結合(NHEJ)若しくは相同指向性修復(HDR)、一本鎖アニーリング(SSA)、又はマイクロホモロジ末端結合(MMEJ)によって修復される。
[0092] HDRは、DNA切断の標的部位を囲む領域と相同な配列を含有する鋳型DNAに依存する。細胞の修復タンパク質が、外来から供給された、又は内因性のDNA配列と、DNA切断を囲む部位との相同性を用いて、切断を鋳型DNA上の配列と置換することでdsDNA切断を修復する。しかしながら、鋳型DNAを組み込めないと、結果としてNHEJ、MMEJ、又はSSAとなり得る。NHEJ、MMEJ、及びSSAは、標的部位でのヌクレオチドの挿入又は欠失(インデル)を多くの場合伴って、フレームシフト突然変異、又は未成熟終止コドンの挿入に起因するゲノムの標的領域の遺伝的ノックアウト(サイレンシング)をもたらす、エラーを起こしやすいプロセスである。また、Cpf1媒介編集が、エンドヌクレアーゼによって生じる突出部の従来のハイブリダイゼーションに続くライゲーションを介して機能し得る。
[0093] CRISPRエンドヌクレアーゼはまた、本開示の後の節で考察されるように、インビトロDNA操作に有用である。
CRISPR系
[0094] CRISPR(クラスター化した規則的な配置の短い回文配列反復:Clustered Regularly Interspaced Short Palindromic Repeats)及びCRISPR関連(cas)エンドヌクレアーゼは、元々、ウイルス及びプラスミドの侵入からの保護のために細菌及び古細菌によって進化した適応免疫系として発見された。細菌において天然に存在するCRISPR/Cas系は、以前に直面したウイルス及びプラスミドから獲得されたゲノム−ターゲティング配列(スペーサと呼ばれる)によって分離される塩基配列の短い回文反復からなる1つ以上のCas遺伝子及び1つ以上のCRISPRアレイで構成される(Wiedenheft, B., et. al. Nature. 2012; 482:331;Bhaya, D., et. al., Annu. Rev. Genet. 2011; 45:231;及びTerms, M.P. et. al., Curr. Opin. Microbiol. 2011; 14:321)。1つ以上のCRISPR遺伝子座を有する細菌及び古細菌は、CRISPRアレイの近位末端にて宿主染色体中に外来配列の短いフラグメント(プロトスペーサ)を組み込むことによって、ウイルス又はプラスミドのチャレンジに応答する。CRISPR遺伝子座の転写は、以前に直面した侵入核酸と相補的な配列を含有するCRISPR由来RNA(crRNA)のライブラリを生成する(Haurwitz, R.E., et. al., Science. 2012:329; 1355;Gesner, E.M., et. al., Nat. Struct. Mol. Biol. 2001:18; 688;Jinek, M., et. al., Science. 2012:337; 816-21)。crRNAによる標的認識は、標的DNAとの相補的塩基対形成を通して起こり、これは、Casタンパク質による外来配列の切断を導く(Jinek et. al. 2012“A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity.”Science. 2012:337; 816-821)。
[0094] CRISPR(クラスター化した規則的な配置の短い回文配列反復:Clustered Regularly Interspaced Short Palindromic Repeats)及びCRISPR関連(cas)エンドヌクレアーゼは、元々、ウイルス及びプラスミドの侵入からの保護のために細菌及び古細菌によって進化した適応免疫系として発見された。細菌において天然に存在するCRISPR/Cas系は、以前に直面したウイルス及びプラスミドから獲得されたゲノム−ターゲティング配列(スペーサと呼ばれる)によって分離される塩基配列の短い回文反復からなる1つ以上のCas遺伝子及び1つ以上のCRISPRアレイで構成される(Wiedenheft, B., et. al. Nature. 2012; 482:331;Bhaya, D., et. al., Annu. Rev. Genet. 2011; 45:231;及びTerms, M.P. et. al., Curr. Opin. Microbiol. 2011; 14:321)。1つ以上のCRISPR遺伝子座を有する細菌及び古細菌は、CRISPRアレイの近位末端にて宿主染色体中に外来配列の短いフラグメント(プロトスペーサ)を組み込むことによって、ウイルス又はプラスミドのチャレンジに応答する。CRISPR遺伝子座の転写は、以前に直面した侵入核酸と相補的な配列を含有するCRISPR由来RNA(crRNA)のライブラリを生成する(Haurwitz, R.E., et. al., Science. 2012:329; 1355;Gesner, E.M., et. al., Nat. Struct. Mol. Biol. 2001:18; 688;Jinek, M., et. al., Science. 2012:337; 816-21)。crRNAによる標的認識は、標的DNAとの相補的塩基対形成を通して起こり、これは、Casタンパク質による外来配列の切断を導く(Jinek et. al. 2012“A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity.”Science. 2012:337; 816-821)。
[0095] 少なくとも5つの主要なCRISPR系型(I、II、III、IV、及びV型)及び少なくとも16個の異なったサブタイプが存在する(Makarova, K.S., et al., Nat Rev Microbiol. 2015. Nat. Rev. Microbiol. 13, 722-736)。CRISPR系はまた、そのエフェクタタンパク質に基づいて分類される。クラス1系は、多サブユニットcrRNA−エフェクタ複合体を有するが、クラス2系では、エフェクタ複合体の全ての機能が、単一のタンパク質(例えば、Cas9又はCpf1)によって実行される。一部の実施形態において、本開示は、II型及び/又はV型の単一サブユニットエフェクタ系の使用を教示する。ゆえに、一部の実施形態において、本開示は、クラス2 CRISPR系の使用を教示する。
CRISPR/Cas9
[0096] 一部の実施形態において、本開示は、II型CRISPR系を用いた遺伝子編集方法を教示する。一部の実施形態において、II型CRISPR系は、Cas9酵素を用いる。II型系は、i)単一エンドヌクレアーゼタンパク質、ii)トランス活性化crRNA(tracrRNA)、及びiii)crRNAの5’末端の約20ヌクレオチド(nt)部分が標的核酸と相補的であるcrRNAに依存している。CRISPR crRNA鎖の、その標的DNAプロトスペーサと相補的である領域は、本明細書でガイド配列と呼ぶ。
[0096] 一部の実施形態において、本開示は、II型CRISPR系を用いた遺伝子編集方法を教示する。一部の実施形態において、II型CRISPR系は、Cas9酵素を用いる。II型系は、i)単一エンドヌクレアーゼタンパク質、ii)トランス活性化crRNA(tracrRNA)、及びiii)crRNAの5’末端の約20ヌクレオチド(nt)部分が標的核酸と相補的であるcrRNAに依存している。CRISPR crRNA鎖の、その標的DNAプロトスペーサと相補的である領域は、本明細書でガイド配列と呼ぶ。
[0097] 一部の実施形態において、II型系のtracrRNA及びcrRNA成分は、単一ガイドRNA(sgRNA)によって置換され得る。sgRNAは、例えば、標的DNA配列(ガイド配列)と相補的な少なくとも12〜20ヌクレオチド配列を含むヌクレオチド配列を含んでよく、そしてその3’末端に共通のスカフォールドRNA配列を含んでよい。本明細書中で用いられる「共通のスカフォールドRNA」は、tracrRNA配列を模倣するあらゆるRNA配列、又はtracrRNAとして機能するあらゆるRNA配列を指す。
[0098] Cas9エンドヌクレアーゼは、平滑末端DNA切断を生じさせ、そしてcrRNAオリゴ及びtracrRNAオリゴの組合せによって標的DNAに動員され、これによりエンドヌクレアーゼは、RNA CRISPR複合体の相補的ハイブリダイゼーションを介して、つなぎ留められる(tether)(図1Aの塗り潰した三角矢印参照)。
[0099] 一部の実施形態において、crRNA/エンドヌクレアーゼ複合体によるDNA認識は、標的プロトスペーサから下流側の、標的DNAの3’部分に位置決めされたプロトスペーサ隣接モチーフ(PAM)(例えば、5’−NGG−3’)との更なる相補的塩基対形成を必要とする(Jinek, M., et. al., Science. 2012:337; 816-821)。一部の実施形態において、Cas9によって認識されるPAMモチーフは、様々なCas9タンパク質について、変動する。
[0100] 一部の実施形態において、当業者であれば、本明細書中に開示されるCas9が、あらゆるソースに由来する、又はあらゆるソースから単離されるあらゆる変異体であってよいことを理解し得る。例えば、一部の実施形態において、本開示のCas9ペプチドとして、配列番号1、配列番号2、配列番号3、配列番号4、配列番号5、及び配列番号6から選択される配列番号の1つ以上が挙げられ得る。他の実施形態において、本開示のCas9ペプチドは、文献中に記載される突然変異の1つ以上を含んでよく、限定されないが、以下に記載される機能突然変異が挙げられる:Fonfara et al. Nucleic Acids Res. 2014 Feb; 42(4): 2577-90;Nishimasu H. et al. Cell. 2014 Feb 27; 156(5): 935-49;Jinek M. et al. Science. 2012 337:816-21;及びJinek M. et al. Science. 2014 Mar 14; 343(6176);また、米国特許出願第13/842,859号、2013年3月15日出願(この出願は、参照によって本明細書に組み込まれる)参照;さらに、米国特許第8,697,359号;米国特許第8,771,945号;米国特許第8,795,965号;米国特許第8,865,406号;米国特許第8,871,445号;米国特許第8,889,356号;米国特許第8,895,308号;米国特許第8,906,616号;米国特許第8,932,814号;米国特許第8,945,839号;米国特許第8,993,233号;及び米国特許第8,999,641号(これらの特許は全て、参照によって本明細書に組み込まれる)参照。ゆえに、一部の実施形態において、本明細書中で開示される系及び方法は、二本鎖ヌクレアーゼ活性を有する野生型Cas9タンパク質、一本鎖ニッカーゼとして機能するCas9突然変異体、又はヌクレアーゼ活性が修飾された他の突然変異体により用いられ得る。
CRISPR/Cpf1
[0101] 他の実施形態において、本開示は、V型CRISPR系を用いた遺伝子編集方法を教示する。一部の実施形態において、本開示は、プレボテラ(Prevotella)属及びフランキセラ(Francisella)属由来のCRISPR 1(Cpf1)を用いる方法を教示する。
[0101] 他の実施形態において、本開示は、V型CRISPR系を用いた遺伝子編集方法を教示する。一部の実施形態において、本開示は、プレボテラ(Prevotella)属及びフランキセラ(Francisella)属由来のCRISPR 1(Cpf1)を用いる方法を教示する。
[0102] 本開示のCpf1 CRISPR系は、i)単一のエンドヌクレアーゼタンパク質、及びii)crRNAであって、3’末端の一部が標的核酸と相補的であるガイド配列を含むcrRNAを含む。当該系において、Cpf1ヌクレアーゼは、crRNAによって、標的DNAに直接動員される(図1Bの塗り潰した三角矢印参照)。一部の実施形態において、Cpf1用のガイド配列は、検出可能なDNA切断を達成するのに少なくとも12nt、13nt、14nt、15nt、又は16ntでなければならず、そして効率的なDNA切断を達成するのに少なくとも14nt、15nt、16nt、17nt、又は18ntでなければならない。
[0103] 本開示のCpf1系は、Cas9と様々な形で異なる。第一に、Cas9とは異なり、Cpf1は、切断用の別個のtracrRNAを必要としない。一部の実施形態において、Cpf1 crRNAは、約42〜44塩基長の短さであり得る(その23〜25ntはガイド配列であり、19ntは構成的な直接反復配列である)。それに対して、組み合わされたCas9 tracrRNA及びcrRNA合成配列は、約100塩基長であり得る。一部の実施形態において、本開示は、Cpf1用のcrRNAを「ガイドRNA」と呼ぶこととする。
[0104] 第二に、Cpf1は、標的の5’上流側に位置決めされる「TTN」PAMモチーフを選ぶ。これは、Cas9系についての標的DNAの3’上に位置決めされる「NGG」PAMモチーフと対照的である。一部の実施形態において、ガイド配列に直ぐ先行するウラシル塩基は、置換され得ない(Zetsche, B. et al. 2015.“Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System”Cell 163, 759-771、この文献は、全ての目的について、その全体が参照によって本明細書に組み込まれる)。
[0105] 第三に、Cpf1について切られる部位は、約3〜5塩基単位で互い違いにされて(staggered)、これが「付着末端」を生じさせる(Kim et al., 2016.“Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells”、2016年6月6日にオンラインで公開)。約3〜5ntの突出部を有する当該付着末端は、NHEJ媒介ライゲーションを促進して、末端がマッチするDNAフラグメントの遺伝子編集を向上させると思われる。切られる部位は、標的DNAの3’末端にあり、PAMがある5’末端に対して遠位である。切られる位置は通常、crRNAにハイブリダイズされない鎖上の第18塩基、及びハイブリダイズされる相補鎖上の対応する第23塩基の後にくる(図1B)。
[0106] 第四に、Cpf1複合体において、「シード」領域は、ガイド配列の最初の5nt以内に位置決めされる。Cpf1 crRNAシード領域は、突然変異に高度に敏感であり、この領域中の単一の塩基置換でさえ、切断活性を大幅に引き下げ得る(Zetsche B. et al. 2015“Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System”Cell 163, 759-771参照)。批評的に、Cas9 CRISPR標的とは異なり、Cpf1系の切断部位とシード領域は、重ならない。Cpf1 crRNAターゲティングオリゴの設計に関する更なるガイダンスが、Zetsche B. et al. 2015.“Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System”Cell 163, 759-771に掲載されている。
[0107] 当業者であれば、本明細書中で開示されるCpf1が、あらゆるソースに由来する、又はあらゆるソースから単離されるあらゆる変異体であってよいことが理解されよう。例えば、一部の実施形態において、本開示のCpf1ペプチドとして、配列番号7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64から選択される配列番号の1つ以上、又はそれらのあらゆる変異体が挙げられ得る。
リガーゼ
[0108] 一部の実施形態において、本開示は、標的とされるCpf1複合体を介して標的DNAを切断してから、生じた付着末端をDNA挿入物とライゲーションする方法を教示する。一部の実施形態において、本開示は、標的DNAを切断するためのCpf1複合体、及びDNAを「縫い」合わせ返すためのリガーゼを用意する方法を教示する。他の実施形態において、本開示は、つなぎ留められるリガーゼ酵素を含む、修飾されたCpf1複合体を教示する。
[0108] 一部の実施形態において、本開示は、標的とされるCpf1複合体を介して標的DNAを切断してから、生じた付着末端をDNA挿入物とライゲーションする方法を教示する。一部の実施形態において、本開示は、標的DNAを切断するためのCpf1複合体、及びDNAを「縫い」合わせ返すためのリガーゼを用意する方法を教示する。他の実施形態において、本開示は、つなぎ留められるリガーゼ酵素を含む、修飾されたCpf1複合体を教示する。
[0109] 本明細書中で用いられる用語「リガーゼ」は、任意の数の酵素的試薬又は非酵素的試薬を含んでよい。例えば、リガーゼは、適切な条件下で、DNA分子、RNA分子、又はハイブリッド中の隣接するヌクレオチドの3’−OHと5’−リン酸との間でリン酸ジエステル結合を形成する、酵素的ライゲーション試薬又は触媒である。
[0110] 一部の実施形態において、本開示は、酵素リガーゼの使用を教示する。適合する温度感受性酵素リガーゼとして、以下に限定されないが、バクテリオファージT4リガーゼ及び大腸菌(E. coli)リガーゼが挙げられる。熱安定リガーゼとして、以下に限定されないが、Afuリガーゼ、Taqリガーゼ、Tflリガーゼ、Tthリガーゼ、Tth HB8リガーゼ、サームス(Thermus)属種AK16Dリガーゼ、及びpfuリガーゼが挙げられる(例えば、国際公開第2000/026381号、Wu et al., Gene, 76(2): 245-254, (1989)、及びLuo et al., Nucleic Acids Research, 24(15): 3071-3078 (1996)参照)。当業者であれば、任意の数の熱安定リガーゼが、好熱性の生物又は超好熱性の生物、例えば、真正細菌及び古細菌の特定の種から得られ得ること;そしてそのようなリガーゼは、開示される方法及びキットに使用され得ることが理解されよう。一部の実施形態において、可逆的に不活化された酵素(例えば米国特許第5,773,258号参照)が、本教示の一部の実施形態に使用されてよい。
[0111] 他の実施形態において、本開示は、化学ライゲーション剤の使用を教示する。化学ライゲーション剤として、以下に限定されないが、活性化剤、濃縮剤、及び還元剤、例えばカルボジイミド、臭化シアン(BrCN)、N−シアノイミダゾール、イミダゾール、1−メチルイミダゾール/カルボジイミド/シスタミン、ジチオスレイトール(DTT)、及び紫外線光が挙げられる。自動ライゲーション、すなわち、ライゲーティング剤の不在下での自発的ライゲーションもまた、本明細書中で本教示の範囲内である。化学ライゲーション方法の詳細なプロトコル及び適切な反応基の説明が、特に、Xu et al., Nucleic Acid Res., 27:875-81 (1999);Gryaznov and Letsinger, Nucleic Acid Res. 21: 1403-08 (1993);Gryaznov et al., Nucleic Acid Res. 22: 2366-69 (1994);Kanaya and Yanagawa, Biochemistry 25:7423-30 (1986);Luebke and Dervan, Nucleic Acids Res. 20: 3005-09 (1992);Sievers and von Kiedrowski, Nature 369:221-24 (1994);Liu and Taylor, Nucleic Acids Res. 26: 3300-04 (1999);Wang and Kool, Nucleic Acids Res. 22: 2326-33 (1994);Purmal et al., Nucleic Acids Res. 20: 3713-19 (1992);Ashley and Kushlan, Biochemistry 30:2927-33 (1991);Chu and Orgel, Nucleic Acids Res. 16: 3671-91 (1988);Sokolova et al., FEBS Letters 232:153-55 (1988);Naylor and Gilham, Biochemistry 5:2722-28 (1966);及び米国特許第5,476,930号において見出され得る。
[0112] 一部の実施形態において、本開示の方法、キット、及び組成物は、フォトライゲーション反応とも適合する。ライゲーション剤として適切な波長の光を用いるフォトライゲーションもまた、本教示の範囲内である。一部の実施形態において、フォトライゲーションは、以下に限定されないが、4−チオチミジン、5−ビニルウラシル及びその派生物、又はそれらの組合せが挙げられるヌクレオチド類似体を含むプローブを含む。一部の実施形態において、ライゲーション剤は:(a)UV−A範囲(約320nm〜約400nm)、UV−B範囲(約290nm〜約320nm)、又はそれらの組合せ内の光、(b)約300nm〜約375nmの波長の光、(c)約360nm〜約370nmの波長の光;(d)約364nm〜約368nmの波長の光、又は(e)約366nmの波長の光を含む。一部の実施形態において、フォトライゲーションは、可逆的である。フォトライゲーションの説明は、特に、Fujimoto et al., Nucl. Acid Symp. Ser. 42: 39-40 (1999);Fujimoto et al., Nucl. Acid Res. Suppl. 1:185-86 (2001);Fujimoto et al., Nucl. Acid Suppl., 2:155-56 (2002);Liu and Taylor, Nucl. Acid Res. 26: 3300-04 (1998)中で、そしてワールド・ワイド・ウェブ:sbchem.kyoto-u.ac.jp/saito-lab上で見出され得る。
ユニバーサルモジュラーCRISPR DNAコンストラクト及びその使用
[0113] 一部の実施形態において、本発明は、DNAコンストラクトのモジュラーアセンブリの戦略を説明する。一部の実施形態において、本開示のDNAアセンブリ方法は、プラスミド、小さな直鎖状DNA、及び形質転換された染色体遺伝子座が挙げられるあらゆるコンストラクトに適用可能である。
[0113] 一部の実施形態において、本発明は、DNAコンストラクトのモジュラーアセンブリの戦略を説明する。一部の実施形態において、本開示のDNAアセンブリ方法は、プラスミド、小さな直鎖状DNA、及び形質転換された染色体遺伝子座が挙げられるあらゆるコンストラクトに適用可能である。
[0114] 態様において、発明者らは、「MegaModular」設計のようなユニバーサルモジュラーCRISPR DNAコンストラクトに言及する。
従来のDNA編集及びアセンブリ技術の欠点
[0115] 従来の多成分DNAクローニング戦略は、配列が複雑な多成分DNAコンストラクトを効果的にアセンブルして修飾する能力に限界がある。例えば、制限酵素クローニングが、各DNA挿入物のクローニング接合部にて適切に位置決めされている固有の制限酵素認識部位、及びその、最終のベクター内でのデスティネーション部位のアベイラビリティによって、制限される。ゲートウェイクローニング技術が、同様に、多成分アセンブリに対応する比較的少ない数の固有の組換え部位によって制限される。
[0115] 従来の多成分DNAクローニング戦略は、配列が複雑な多成分DNAコンストラクトを効果的にアセンブルして修飾する能力に限界がある。例えば、制限酵素クローニングが、各DNA挿入物のクローニング接合部にて適切に位置決めされている固有の制限酵素認識部位、及びその、最終のベクター内でのデスティネーション部位のアベイラビリティによって、制限される。ゲートウェイクローニング技術が、同様に、多成分アセンブリに対応する比較的少ない数の固有の組換え部位によって制限される。
[0116] 従来のDNAアセンブリ技術の別のマイナス面は、配列を編集する能力が、多くの場合、構築の時間に制限されることである。例えば、効率的なアセンブリ戦略、例えばリガーゼサイクリング反応(LCR)の生成物は、初期アセンブリが完了すると、容易に修飾されない(Kok, S, et al., 2014“Rapid and Reliable DNA assembly via Ligase Cycling Reaction”ACS Synth. Biol., 3 (2): 97-106)。類似の懸念が、従来の制限酵素クローニングで生じ、その共通の制限認識部位は、制限部位を含有するポリヌクレオチドが、アセンブルされることとなるコンストラクト中に挿入されると、又は前記コンストラクトが、前記部位で満ちている染色体中に組み込まれた場合に、固有のクローニング点として機能しなくなる。ゆえに、シーケンシャルな制限クローニングを通して生じたベクターは、クローニングプロセスが順調に進行中であると、配列を固定又は更新するための提供される選択肢が、非常に少なくなる。
[0117] より新しい技術、例えば従来のCRISPR DNAアセンブリ技術ですら、類似の複雑性、以前のアセンブルされたコンストラクト/ベクター設計を繰り返す容易性、及び速度限界に直面し続けている(Wang, JW. et al., 2015“CRISPR/Cas9 nuclease combined with Gibson assembly for seamless cloning”BioTechniques, Vol 58, No. 4: 161-170)。CRISPRクローニングは、適合するプロトスペーサ隣接モチーフ(PAM)に隣接する、標的とされる機能ガイドRNAの設計を必要とする。標的部位内での適切なPAM配列のアベイラビリティが、ゲノム又はコンストラクト内の可能性があるDNA挿入位置の数に対する設計上の大きな制限となる。
[0118] さらに、ガイドRNA配列の設計及び試験は、多成分アセンブリについて大きな技術的課題を課す。当業者であれば、例えば、全てのgRNA配列が機能的であるわけではないこと、そしてCRISPR DNAアセンブリの有効な実施が時折、複数のgRNA配列変異体の設計及び確認を必要とする場合があることを認識するであろう。これらの制限は、特に、多成分アセンブリにおいて扱い難く、所望の修飾を首尾よくもたらす単一のgRNA配列の失敗が、元のクローニングプランにもはや納まらない以降のアセンブリ成分を再設計する必要を引き起こす虞がある。ゆえに、多成分アセンブリの接合部毎に複数のカスタムガイドRNAを必要とする技術を用いることもまた、非常に高価で、扱い難く、且つ非実用的となる虞がある。
モジュラーCRISPRタグアセンブリベクター及びそれを用いる方法
[0119] 一部の実施形態において、本開示は、先で記載される上述の従来技術と関連する多くの制限を克服するDNAアセンブリ方法を教示する。一部の実施形態において、本開示はまた、本発明の方法に用いられるモジュラーCRISPRアセンブリコンストラクト、組成物、及びキットを教示する。
[0119] 一部の実施形態において、本開示は、先で記載される上述の従来技術と関連する多くの制限を克服するDNAアセンブリ方法を教示する。一部の実施形態において、本開示はまた、本発明の方法に用いられるモジュラーCRISPRアセンブリコンストラクト、組成物、及びキットを教示する。
[0120] 一部の実施形態において、本開示は、1つ以上のCRISPRマルチクローナル部位(cMCS)を含むDNAコンストラクトを教示する。一部の実施形態において、本開示のcMCSは、記載されるDNAコンストラクトの一部のみを表す(すなわち、コンストラクトの一部のみが、本開示の方法に従って容易に編集可能である)。他の実施形態において、本開示のcMCSは、DNAコンストラクト全体が容易に編集可能であるように、コンストラクト全体の内部の重要な位置上に位置決めされている。ゆえに、一部の実施形態において、モジュラーcTAGベクターの全ての機能部分(例えば、アセンブリに必要とされる全ての起源、マーカー、カーゴ、要素)が、挿入DNA部分の内部に含まれており、そして本開示の遺伝子編集方法を介して容易に交換され得る。
[0121] 一部の実施形態において、本開示のcMCSは、1つ以上のクローニングタグ(cTAG)を含み、これらはそれぞれ、少なくとも1つの確認されたCRISPRターゲティング部位を含む。一部の実施形態において、本開示のcMCSはさらに、DNA挿入部分を含み、各DNA挿入部分には、一対のcTAGが側面に位置しており、1つ以上のcTAGを標的とする1つ以上のCRISPRエンドヌクレアーゼによるcMCSの消化が、前記側面に位置する挿入部分を解放して、適合するドナーDNA部分の挿入を可能とすることとなる。
[0122] 本明細書の図2及び図3は、本開示の方法に従う、モジュラーCRISPRアセンブリプラスミドコンストラクトの実施形態を示す。開示される例示のプラスミドは、一連のDNA挿入物(図2AのPart1〜8)を含有し、これらにはそれぞれ、図2Aにおいて、一対のcTAG(タグA〜H)が側面に位置する。適切なCRISPR/ガイド配列複合体によるこの例のcTAG A及びBの消化が、プラスミドのPart2を解放して、所望の特性を有する置換Part2挿入物の挿入を可能とすることとなる。
[0123] 当業者であれば、インビボでの、そしてインビトロでのベクターの配列特異的モジュラークローニング/編集を可能とする、本記載のベクター系の利点を直ちに認識するであろう。以下の節では、開示されるモジュラークローニングベクターの種々の態様、並びにそれらの、分子生物学、遺伝子治療、及び遺伝子編集への種々の用途を概説することとする。
モジュラーCRISPRベクター挿入部分
[0124] 一部の実施形態において、本開示の挿入部分は、CRISPR消化後の相同組換え挿入用のドナーDNA配列である。ゆえに、一部の実施形態において、本開示の挿入部分配列は、消化されるモジュラーCRISPRコンストラクトの末端に対する相同性が、注目する挿入配列であって、当該配列の相同組換え、ハイブリダイゼーション、及び挿入をトリガーするのに十分である配列が側面に位置する挿入配列を含む。
[0124] 一部の実施形態において、本開示の挿入部分は、CRISPR消化後の相同組換え挿入用のドナーDNA配列である。ゆえに、一部の実施形態において、本開示の挿入部分配列は、消化されるモジュラーCRISPRコンストラクトの末端に対する相同性が、注目する挿入配列であって、当該配列の相同組換え、ハイブリダイゼーション、及び挿入をトリガーするのに十分である配列が側面に位置する挿入配列を含む。
[0125] 他の実施形態において、本開示の挿入部分は、付着末端を介したハイブリダイゼーション及びライゲーション(例えば、Cpf1消化、制限酵素消化、Gibsonアセンブリ、又は他のハイブリダイゼーションベースのアセンブリの後に、LCRを含む)が可能なドナーDNA配列である。ゆえに、一部の実施形態において、本開示の挿入部分配列は、注目する挿入配列であって、消化されるモジュラーCRISPRコンストラクトの末端に対する相同性が、付着末端のハイブリダイゼーションを可能にするのに十分である配列が側面に位置する挿入配列を含む。
[0126] 他の更なる実施形態において、本開示の挿入部分は、平滑末端ライゲーション用のドナーDNA配列である。
[0127] 一部の実施形態において、本開示のモジュラーCRISPR DNAコンストラクトは、あらゆる挿入部分配列と適合する。ゆえに、本ベクターの部分は、以下に限定されないが、選択マーカー、複製起点、プロモータ、ターミネータ配列、他の調節配列、バーコード、組換え部位、又は使用者が注目する他の配列を含んでよい。一部の実施形態において、本開示の挿入部分は、1つ以上の遺伝子座中への相同組換え及び挿入をトリガーするための相同配列を含んでよい。一部の実施形態において、前記相同組換え挿入部分は、同じく組換え事象を介してゲノム中に挿入されることとなる他の挿入部分に前後することとなる。
[0128] 一部の実施形態において、本開示は、各挿入部分が、単一の配列(例えば、注目するプロモータのみ又は遺伝子のみ、図2A、Part8参照)を含むことを教示する。他の実施形態において、本開示は、1つ以上の挿入部分が、複数の要素、例えばプロモータ−注目する遺伝子(GOI)の組合せ、マルチサブユニットキメラタンパク質融合を、又はコンストラクト全体(プロモータ−GOI−ターミネータの組合せを含む、図2A、Part5参照)さえも含有してよいことを教示する。
[0129] 一部の実施形態において、本開示は、結合してない個々の挿入部分を教示する。すなわち、一部の実施形態において、本開示は、1つ又は複数の連結されていない挿入部分(図2A、結合してない挿入部分のリストを示す右側参照)を教示する。一部の実施形態において、本開示は、前記複数の部分を、1つ以上のモジュラーCRISPRコンストラクトにアセンブルする方法を教示する。一部の態様において、本開示は、MegaModularコンストラクトをアセンブルするためのキットを教示する。
[0130] 他の実施形態において、本開示は、部分的に、又は完全にアセンブルされたモジュラーCRISPR DNAコンストラクトを教示する。例えば、一部の実施形態において、本開示は、1、2、3、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100個、又はそれ以上、及びそれらの間のあらゆる範囲のアセンブルされた挿入部分を含むモジュラーCRISPR DNAコンストラクトを教示する。本開示はまた、前記挿入部分を含むキットを教示する。
[0131] 一部の実施形態において、前記アセンブルされた、又は部分的にアセンブルされたモジュラーCRISPR DNAコンストラクトは、直鎖状である。一部の実施形態において、前記アセンブルされた、又は部分的にアセンブルされたモジュラーCRISPR DNAコンストラクトは、環状(例えばプラスミド)である。一部の実施形態において、前記アセンブルされた、又は部分的にアセンブルされたモジュラーCRISPR DNAコンストラクトは、ゲノムDNA中に組み込まれる。
[0132] 一部の実施形態において、本開示のコンストラクトは最初に、更なるクローニング用のプレースホルダとして、短いスペーサ配列のみを含有することとなる(図3Cの「スタッファ」配列参照)。一部の実施形態において、挿入部分プレースホルダは、小さな、ランダム化された配列である。他の実施形態において、本開示のベクターは最初に、1つ以上のプレ選択された挿入DNA部分を含むこととなる。例えば、一部の実施形態において、モジュラーCRISPRコンストラクトは最初に、少なくとも1つの選択マーカー及び/又は少なくとも1つの複製起点を含むこととなる。
[0133] 適切な選択マーカーとして、以下に限定されないが、抗生物質耐性を付与する遺伝子、蛍光タンパク質をコードする遺伝子、tRNA遺伝子、栄養要求性マーカー、毒性遺伝子、表現型マーカー、アンチセンスオリゴヌクレオチド、制限酵素、制限酵素切断部位、酵素切断部位、タンパク質結合部位、及びPCRプライマー配列と相補的な配列が挙げられる。
[0134] 適切な抗生物質耐性遺伝子として、以下に限定されないが、クロラムフェニコール耐性遺伝子、アンピシリン耐性遺伝子、テトラサイクリン耐性遺伝子、ゼオシン耐性遺伝子、スペクチノマイシン耐性遺伝子、及びカナマイシン耐性遺伝子が挙げられる。
[0135] 本発明の特定の実施形態において、対抗選択マーカーは、毒性遺伝子である。適切な毒性遺伝子として、以下に限定されないが、ccdB遺伝子、1つ又は複数のter部位に結合するtusタンパク質をコードする遺伝子、kicB遺伝子、sacB遺伝子、ASK1遺伝子、ΦX174E遺伝子、及びDpnI遺伝子が挙げられる。一部の実施形態において、毒性選択マーカーの存在は、挿入が行われなかった、又は不成功であったことの指標として機能する。毒性選択マーカーはまた、毒性遺伝子により未だ適所で修飾されていないベクターを有する細胞に死を引き起こすことによって、陽性細胞の非修飾親ベクターのバックグラウンドを低下させるように機能し得る。
[0136] 本発明の方法の更なる実施形態において、モジュラーCRISPRコンストラクトは、1つ以上の毒性遺伝子及び1つ以上の抗生物質耐性遺伝子の双方を含んでよい。
[0137] 一部の実施形態において、モジュラーCRISPRコンストラクトは最初に、少なくとも1つの調節配列を含むこととなる。一部の実施形態において、本開示は、以下に限定されないが、マトリックス付着領域、発現インシュレータ配列、発現エンハンサ配列、プロモータ、5’UTR、3’UTR、ターミネータ配列、終止コドン、開始コドンその他を含むベクターを教示する。一部の実施形態において、モジュラーCRISPRコンストラクトは最初に、前記コンストラクトの染色体挿入を促進する配列(例えば、t−DNAボーダー、Cre/Lox、又は染色体配列に対して相同性の末端)を含むこととなる。一部の実施形態において、染色体挿入用の配列は、生物のゲノム中にモジュラーCRISPRコンストラクト全体を挿入するように配置される。他の実施形態において、染色体挿入用の配列は、モジュラーCRISPRコンストラクトの一部のみを挿入するように配置される(図3D参照)。
[0138] 一部の実施形態において、本開示の挿入部分は、更なるcTAGを含むことさえできる。挿入部分を通したcTAGの付加は、利用可能なクローニングスキームの複雑性を増大させることができ、そしてまた、置換され得る利用可能な挿入部分の数を拡大することによって、コンストラクトのサイズを拡大することもできる。
[0139] 一部の実施形態において、本開示の挿入部分は、従来のクローニング部位を含み得る。例えば、一部の実施形態において、本開示は、ゲートウェイ組換え部位、制限部位、Cre/Lox部位、又は他の従来のクローニング部位を含む挿入部分を教示する。
[0140] 一部の実施形態において、本開示は、従来のDNAコンストラクトから挿入部分を生じさせる方法を教示する。すなわち、一部の実施形態において、本開示は、cTAGを従来のDNAコンストラクトに(例えば、オリゴ、PCRフラグメント、プラスミド、又は他の利用可能なDNAセグメントに)加える方法を教示する。一部の実施形態において、本開示は、cTAGを、単一の成分、例えば、注目する遺伝子(GOI)、プロモータに加える方法を教示する。他の実施形態において、本開示は、cTAGを多要素コンストラクトに加える方法を教示する。
[0141] 当業者であれば、挿入部分を構築する方法を認識するであろう。例えば、一部の実施形態において、cTAGは、前記cTAGを含むプライマーによるPCR増幅を介してDNA分子中に組み込まれ得る。他の実施形態において、cTAGは、従来のクローン技術(例えば、制限酵素、Gibson、又は他のアセンブリ方法)を介して組み込まれ得る。さらに他の実施形態において、cTAGは、平滑末端ライゲーションを介して組み込まれ得る。
[0142] 一部の実施形態において、本開示の挿入部分は、広い種適合スペクトルを有し得る(例えば、マーカーが、原核生物及び真核生物の双方の発現配列を含有して、複数の生物において効力を発揮し得る)。他の実施形態において、本開示の挿入部分は、単一の種/属/科/目/綱/門/界又はドメイン内の生物への適用性が制限されるように設計される。一部の実施形態において、例えば、複製起点部分が、単一の種内でのみ、又は種の一群内でのみプラスミドを維持することができるようにすることができる。他の実施形態において、蛍光マーカーは、原核生物ドメイン及び真核生物ドメインの双方にわたって機能するようにコドン最適化され得る。
[0143] 一部の実施形態において、Cas9エンドヌクレアーゼは、標的配列のPAMから3〜4ヌクレオチド上流を切断する。ゆえに、Cas9複合体によるcTAG消化は、PAM配列、又は標的のプロトスペーサ配列の喪失を通して、cTAG機能の喪失をもたらし得る。一部の実施形態において、本開示は、ドナー挿入配列を、挿入直後にcTAG配列を(例えば、以前に失われたPAM又はプロトスペーサ配列の挿入を通して)再構成するように設計することによって、前記cTAG配列の機能を維持する方法を教示する。類似の対策が、Cpf1エンドヌクレアーゼを通して切断される配列について想定される。
[0144] 図2Bは、本開示のcTAG修復の概念を示す。Cas9エンドヌクレアーゼによる挿入part2の切断もまた、cTAG A及びBの一部の喪失をもたらす。相同組換えを介した挿入part2a〜2dのいずれか1つの以降の挿入により、全cTAG配列の回復がもたらされる。
[0145] 当業者であれば、挿入部分についてほぼ無限の選択肢を認識するであろう。挿入の上述のリストは、説明のためのものであることが意図されており、本開示の方法、キット、及びコンストラクトの適用性を制限するものとして決して解釈されるべきでない。
モジュラーCRISPRクローニングタグ
[0146] 一部の実施形態において、本開示のモジュラーCRISPRコンストラクトは、1つ以上のクローニングタグ(cTAG)を含む。一部の実施形態において、本開示のモジュラーCRISPRコンストラクトは、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100個、又はそれ以上のcTAGを含む。
[0146] 一部の実施形態において、本開示のモジュラーCRISPRコンストラクトは、1つ以上のクローニングタグ(cTAG)を含む。一部の実施形態において、本開示のモジュラーCRISPRコンストラクトは、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100個、又はそれ以上のcTAGを含む。
[0147] 一部の実施形態において、本開示は、各cTAGが、少なくとも1つの確認されたCRISPRプロトスペーサ/PAMの組合せ配列(「CRISPRランディング部位」)を含むことを教示する。すなわち、一部の実施形態において、cTAGは、少なくとも1つの実験的に確認された、高い効率のCRISPRランディング部位を含む。一部の実施形態において、本開示のcTAGは、ウェットベンチ実験(例えば、前記CRISPRランディング部位を標的とするCRISPR複合体によるcTAG配列のインビトロ切断)によって確認され得る。他の実施形態において、cTAG確認は、査読を受けた雑誌における切断の報告から想定されてもよい。
[0148] 一部の実施形態において、本開示のcTAGは、1、2、3、4、5、6、7、8、9、10個、又はそれ以上のCRISPRランディング部位を含む。一部の実施形態において、CRISPRランディング部位は、互いに重なる。他の実施形態において、CRISPRランディング部位は、cTAG内の互いに異なる、重ならない領域を占有する。一部の実施形態において、CRISPRランディング部位は、Cas9エンドヌクレアーゼ切断又はCpf1エンドヌクレアーゼ切断のいずれかに特異的であり得る。一部の実施形態において、CRISPRランディング部位は、他のあらゆる現行の、又は今のところ発見されているCRISPRエンドヌクレアーゼに特異的であり得る。
[0149] 他の実施形態において、本開示は、単一のcTAG内のマルチクローニング部位が、様々な生物全体にわたって機能するように設計され得ることを教示する。ゆえに、一部の実施形態において、cTAG Cpf1ランディング部位は、HR機構を欠く、又は下方制御する生物において好まれ得る。他の実施形態において、cTAGの制限部位が、最初のインビトロクローニングに好まれ得る一方、Cas9又はCpf1ランディング部位が、選択された真核生物においてインビボで起こるより複雑な編集に好まれ得る。
[0150] 一部の実施形態において、本開示は、cTAGが、1つ以上の非CRISPRクローニング配列を含み得ることを教示する。例えば、一部の実施形態において、本開示のcTAGは、制限酵素部位、組換え部位、トポイソメラーゼ部位、スプライシング部位、及びCre−Lox部位からなる群から選択される1つ以上の要素を含んでよい。
[0151] 一部の実施形態において、適切な制限酵素部位として、以下に限定されないが、AaII、AarI、AasI、AatII、Acc65I、AccB7I、AccI、AccIII、AciI、AclI、AcuI、AdeI、AfeI、AflII、AflIII、AgeI、AhdI、AleI、AloI、AluI、Alw21I、Alw26I、Alw44I、AlwI、AlwNI、ApaI、ApaLI、ApeKI、ApoI、AscI、AseI、AsiSI、AvaI、AvaII、AvrII、BaeI、BalI、BamHI、BanI、BanII、BbsI、BbuI、BbvCI、BbvI、BccI、BceAI、BcgI、BciVI、BclI、BcnI、BcuI、BfaI、BfiI、BfmI、BfrBI、BfuAI、BfuCI、BfuI、BglI、BglII、BlpI、Bme1390I、Bme1580I、BmgBI、BmrI、BmtI、BoxI、BpiI、BplI、BpmI、Bpu10I、Bpu1102I、BpuEI、BsaAI、BsaBI、BsaHI、BsaI、BsaJI、BsaMI、BsaWI、BsaXI、BseDI、BseGI、BseJI、BseLI、BseMI、BseMII、BseNI、BseRI、BseSI、BseXI、BseYI、BsgI、Bsh1236I、Bsh1285I、BshNI、BshTI、BsiEI、BsiHKAI、BsiWI、BslI、BsmAI、BsmBI、BsmFI、BsmI、BsoBI、Bsp19I、Bsp120I、Bsp1286I、Bsp1407I、Bsp143I、Bsp143II、Bsp68I、BspCNI、BspDI、BspEI、BspHI、BspLI、BspMI、BspPI、BspQI、BspTI、BsrBI、BsrDI、BsrFI、BsrGI、BsrI、BsrSI、BssHII、BssKI、BssSI、Bst1107I、Bst98I、BstAPI、BstBI、BstEII、BstF5I、BstNI、BstOI、BstUI、BstXI、BstYI、BstZI、BstZ17I、Bsu15I、Bsu36I、BsuRI、BtgI、BtgZI、BtsCI、BtsI、BveI、Cac8I、CaiI、CfoI、Cfr10I、Cfr13I、Cfr42I、Cfr9I、CfrI、ClaI、CpoI、Csp45I、Csp6I、CspI、CspCI、CviaII、CviKI−1、CviQI、DdeI、DpnI、DpnII、DraI、DraIII、DrdI、EaeI、EagI、Eam1104I、Eam1105I、EarI、EciI、Ecl136II、EclHKI、Eco105I、Eco130I、Eco147I、Eco24I、Eco31I、Eco32I、Eco47I、Eco47III、Eco52I、Eco57I、Eco57MI、Eco72I、Eco81I、Eco88I、Eco91I、EcolCRI、EcoNI、EcoO109I、EcoP15I、EcoRI、EcoRV、EheI、Esp3I、FatI、FauI、Fnu4HI、FokI、FseI、FspI、FspAI、GsuI、HaeII、HaeIII、HgaI、HhaI、Hin1I、Hin4I、Hin6I、HincII、HindIII、HinfI、HinP1I、HpaI、HpaII、HphI、Hpy166II、Hpy188I、Hpy188III、Hpy8I、Hpy99I、HpyAV、HpyCH4III、HpyCH4IV、HpyCH4V、HpyF10VI、Hsp92I、Hsp92II、I−PpoI、KasI、Kpn2I、KpnI、KspAI、LweI、MbiI、MboI、MboII、MfeI、MisI、MluI、MlyI、MmeI、MnlI、Mph1103I、MscI、MseI、MslI、MspA1I、MspI、MssI、MunI、Mva1269I、MvaI、MwoI、NaeI、NarI、NciI、NcoI、NdeI、NdeII、NgoMIV、NheI、NheI−HF、NlaIII、NlaIV、NmeAIII、NmuCI、NotI、NruI、NsbI、NsiI、NspI、OliI、PacI、PaeI、PaeR7I、PagI、PauI、PciI、PdiI、PdmI、Pfl23II、PflFI、PflMI、PfoI、PhoI、PleI、PmeI、PmlI、PpiI、PpuMI、PshAI、PsiI、Psp1406I、Psp5II、PspGI、PspOMI、PspXI、PstI、PsuI、PsyI、PvuI、PvuII、PvuII−HF、RsaI、RsrII、SacI、SacII、SalI、SalI−HF、SapI、SatI、Sau3AI、Sau96I、SbfI、ScaI、ScaI−HF、SchI、ScrFI、SdaI、SduI、SexAI、SfaNI、SfcI、SfiI、SfoI、SgfI、SgrAI、SinI、SmaI、SmiI、SmlI、SmuI、SnaBI、SpeI、SphI、SphI−HF、SspI、StuI、StyD4I、StyI、SwaI、TaaI、TaiI、TaqαI、TaqI、TasI、TatI、TauI、TfiI、TliI、Tru1I、Tru91、TseI、Tsp45I、Tsp509I、TspMI、TspRI、Tth111I、TurboNaeI、TurboNarI、Van91I、VspI、XagI、XapI、XbaI、XceI、XcmI、XhoI、XhoII、XmaI、XmaJI、XmiI、XmnI、及びZraIからなる群から選択される制限酵素によって認識される部位が挙げられる。また、態様として、ホーミングエンドヌクレアーゼ、例えば:I−SceI、I−CeuI、及びPI−PspIが挙げられる。これらの酵素についての対応する切断部位は、当該技術において知られている。
[0152] 一部の実施形態において、本開示は、8ヌクレオチド長以上の部位を認識する稀な制限酵素(≧8制限酵素)の使用を教示する。一部の実施形態において、本開示は、各cTAGにおける単一の稀な制限部位の使用を教示する。他の実施形態において、本開示のcTAGは、2つ以上の制限部位を含んでよい。以下の表1は、本発明に従うcTAGのリストを記載しており、それぞれ、稀な制限酵素部位を太字で示す。

[0153] 一部の実施形態において、本発明に用いられる適切な組換え部位として、限定されないが、以下が挙げられる:attB部位、attP部位、attL部位、attR部位、lox部位、psi部位、tnpI部位、dif部位、cer部位、frt部位、並びにそれらの突然変異体、変異体、及び派生物。本発明の特定の実施形態において、トポイソメラーゼ認識部位は、存在するならば、I型トポイソメラーゼによって認識されて結合され、これは、IB型トポイソメラーゼであってよい。IB型トポイソメラーゼの適切なタイプとして、以下に限定されないが、真核生物核I型トポイソメラーゼ及びポックスウイルストポイソメラーゼが挙げられる。一部の実施形態において、ポックスウイルストポイソメラーゼの適切なタイプとして、以下に限定されないが、ワクシニアウイルス、ショープ線維腫ウイルス、ORFウイルス、鶏痘ウイルス、伝染性軟属腫ウイルス、及びアムサクタ・モーリエ・エントモポックスウイルス(Amsacta morreientomopoxvirus)等のウイルスによって生成される、又はこれらから単離されるポックスウイルストポイソメラーゼが挙げられる。
[0154] 一部の実施形態において、CRISPRクローニング部位及び非CRISPRクローニング部位のcTAG配置は、使用者の選択に従ってオーダーされ得る。一部の実施形態において、本開示は、CRISPR結合部位が、挿入部分から最も遠く離れるようにオーダーされるべきであることを教示する。実例となる一実施形態において、cTAGは、以下のように5’〜3’に配置され得る:(Part I)[R1-A1-C1-C2]-(Part II)、式中、R=制限部位、A=リコンビナーゼ部位、及びC=CRISPRランディング部位。一部の実施形態において、Cは、複数の重複する、又は連続するCRISPR及び/又は制限ランディング部位を含んでよい。一部の実施形態において、本開示のcTAG上のクローニング部位の配置は、対称形となる(すなわち、クローニング部位のタイプの対称的順序を提供する)こととなる。
[0155] 他の実施形態において、本開示のcTAG上のクローニング部位の配置は、非対称的であってもよい。例えば、実例となる別の実施形態において、cTAGは、以下のように5’〜3’に配置され得る:(Part I)-[R1-A1-C1-C2]-(Part II)、式中、R=制限部位、A=リコンビナーゼ部位、及びC1〜2=CRISPRランディング部位。他の更なる実施形態において、cTAGは、以下のように5’〜3’に配置され得る:i)(Part I)-[R1-C1-C2]-(Part II)、ii)(Part I)-[R1-C1]-(Part II)、iii)(Part I)-[C1-C2]-(Part II)、又はそれらの逆順、式中、R=制限部位、A=リコンビナーゼ部位、及びC1〜2=CRISPRランディング部位。
[0156] 当業者であれば、種々のcTAG配置の利点及び用途を認識するであろう。例えば、単一タグの実施形態において、モジュラーコンストラクトは、単一のCRISPRエンドヌクレアーゼの消化による挿入を可能にするであろうが、第2のフランキングcTAG部位の欠如に起因して、前記挿入の除去又は置換を(より多くの、例えば更なるcTAGの更なる消化なしに)可能にしないであろう。一部の実施形態において、本開示は、挿入された部分が、それ自体で、更なるcTAGを含有して、cMCS内の可能性がある挿入部分位置の数を拡大することができることを教示する。
[0157] 他の実施形態において、本開示は、モジュラーCRISPRコンストラクトから1つ以上の挿入部分を除去する方法を教示する。一部の実施形態において、モジュラーCRISPRコンストラクトのcTAGの2つ以上が、適合する末端を生じさせることができる制限酵素結合部位を含む。一部の実施形態において、制限酵素部位は、互いに同じである。他の実施形態において、制限酵素部位は、互いに異なるが、前記部位の結果として生じる消化は、ハイブリダイゼーション及びライゲーションに適合する末端を生じさせる。一部の実施形態において、モジュラーCRISPRコンストラクトの部分の削除用の制限部位は、2つ以上のcTAGの他の末端に、結果として生じるライゲーション済みコンストラクトが、挿入部分の、cTAGに対する同じ比率をなお維持することとなるように配置される。
[0158] 一部の実施形態において、本開示は、本開示のモジュラーCRISPRコンストラクト内の削除に用いられる制限酵素部位が、適合する末端をもたらすあらゆる制限酵素であってよいことを教示する。他の実施形態において、本開示は、本開示のモジュラーCRISPRコンストラクト内の削除に用いられる制限酵素部位が、適合する末端をもたらすあらゆる稀な8≧塩基制限酵素であってよいことを教示する。選択された実施形態において、本開示は、本開示のモジュラーCRISPRコンストラクト内の削除に用いられる制限酵素部位が、I−SceI及びPI−PspIであってよいことを教示する。
[0159] 一部の実施形態において、本開示は、cTAG対を生じさせるように2つのcTAGが各挿入部分の側面に位置するモジュラーCRISPRコンストラクトを教示する。一部の実施形態において、上述のcTAG対は、挿入部分の選択的切断/置換を可能にする。例えば、図2Bに示されるように、cTAG A及びBを標的とするエンドヌクレアーゼによるモジュラーCRISPRプラスミドの消化は、挿入part2を特異的に除去することとなる。
[0160] 先で考察されるように、本開示の選択された実施形態は、エンドヌクレアーゼ切断後にcTAG機能を回復する置換挿入部分を提供する。ゆえに、図2Bに示されるように、置換挿入part2a〜2dは、モジュラーCRISPRプラスミド中への挿入後直ぐにcTAG A及びBの機能を回復することとなる配列を含む。
[0161] 一部の実施形態において、本開示は、cTAGが挿入部分の指向性をも制御し得ることを教示する。挿入部分中のcTAG末端とモジュラーCRISPRコンストラクト中の切断されたcTAGとの配列相同性が、相同組換え又はハイブリダイゼーション(例えば、Gibsonアプローチによる)のいずれかを通して、Cas9切断配列についての挿入指向性を決定することとなる。Cpf1配列中の挿入指向性はまた、いずれかのcTAG上のCpf1付着末端のワトソン−クリック型ハイブリダイゼーションを介しても制御され得る。
[0162] 一部の実施形態において、本開示はまた、代替のcTAG配置を提供する。例えば、一部の実施形態において、本開示のモジュラーCRISPRコンストラクトは、例えば、ネストcTAGに用いられる機能を提供するように設計されてよい。
[0163] 一部の実施形態において、本開示は、インビトロ及びインビボでの多成分アセンブリを可能にする、共有された重複「タグ」領域に基づく成分ベースのCRISPRアセンブリを教示する。一部の実施形態において、本開示のタグは、DNAコンストラクトからの未来のクローニング又はインビトロDNAアセンブリを促進するためのCRISPRランディング部位を含む。DNAコンストラクトが宿主生物のゲノム中に組み込まれるならば、予め選択されたCas9又はCpf1ランディング部位が、容易な遺伝的変更を促進し得る。一組の実験において、アセンブリ戦略は、複数の数及びタイプのDNA成分を含有する、複数の生物に用いられ得るDNAプラスミドの構築を可能にする。
[0164] 一部の実施形態において、このアセンブリ戦略は、所望のあらゆるDNA成分のセットをコードするプラスミドをアセンブル且つ急速に再アセンブルするのに用いられ得、代謝経路が挙げられる。他の実施形態において、組込みプラスミドへのcTAGの設計もまた、DNA成分を、宿主生物のゲノム内に、そしてそこから直接的にスワップするのに用いられ得、未来のプラスミドをクローニングする必要を回避する。
cTAG配列設計アルゴリズム
[0165] 一部の実施形態において、本開示は、cTAG内のCRISPRランディング部位を促進するように設計されたアルゴリズムを教示する。一部の実施形態において、CRISPRランディング部位は、既存の配列から同定される配列である。ゆえに、一部の実施形態において、本開示は、ソフトウェアプログラムの使用が、所望のガイド配列長に基づくインプットDNA配列、及び指定されたCRISPR酵素用のCRISPRモチーフ配列(PAM、プロトスペーサ隣接モチーフ)の双方の鎖上に、候補CRISPR標的配列を同定するように設計されることを教示する。例えば、PAM配列TTNを有する、フランシセラ・ノビシダ(Francisella novicida)U112由来のCpf1用の標的部位が、インプット配列上、そしてインプットの逆相補体上の双方で、5’−TTN−3’を検索することによって同定され得る。PAM配列TTTNを有する、ラクノスピラ科の細菌(Lachnospiraceae bacterium)及びアシダミノコッカス(Acidaminococcus)属種由来のCpf1用の標的部位が、インプット配列上、そしてインプットの逆相補体上の双方で、5’−TTTN−3’を検索することによって同定され得る。同様に、PAM配列NNAGAAWを有する、S.サーモフィルス(S. thermophilus)CRISPR1のCas9用の標的部位が、インプット配列上、そしてインプットの逆相補体上の双方で、5’−Nx−NNAGAAW−3’を検索することによって同定され得る。化膿連鎖球菌(S. pyogenes)のCas9用のPAM配列は、5’−NGG−3’である。
[0165] 一部の実施形態において、本開示は、cTAG内のCRISPRランディング部位を促進するように設計されたアルゴリズムを教示する。一部の実施形態において、CRISPRランディング部位は、既存の配列から同定される配列である。ゆえに、一部の実施形態において、本開示は、ソフトウェアプログラムの使用が、所望のガイド配列長に基づくインプットDNA配列、及び指定されたCRISPR酵素用のCRISPRモチーフ配列(PAM、プロトスペーサ隣接モチーフ)の双方の鎖上に、候補CRISPR標的配列を同定するように設計されることを教示する。例えば、PAM配列TTNを有する、フランシセラ・ノビシダ(Francisella novicida)U112由来のCpf1用の標的部位が、インプット配列上、そしてインプットの逆相補体上の双方で、5’−TTN−3’を検索することによって同定され得る。PAM配列TTTNを有する、ラクノスピラ科の細菌(Lachnospiraceae bacterium)及びアシダミノコッカス(Acidaminococcus)属種由来のCpf1用の標的部位が、インプット配列上、そしてインプットの逆相補体上の双方で、5’−TTTN−3’を検索することによって同定され得る。同様に、PAM配列NNAGAAWを有する、S.サーモフィルス(S. thermophilus)CRISPR1のCas9用の標的部位が、インプット配列上、そしてインプットの逆相補体上の双方で、5’−Nx−NNAGAAW−3’を検索することによって同定され得る。化膿連鎖球菌(S. pyogenes)のCas9用のPAM配列は、5’−NGG−3’である。
[0166] 同様に、PAM配列NGGNGを有する、S.サーモフィルス(S. thermophilus)CRISPRのCas9用の標的部位が、インプット配列上、そしてインプットの逆相補体上の双方で、5’−N,−NGGNG−3’を検索することによって同定され得る。
[0167] 他の実施形態において、本開示は、無からCRISPRランディング部位を設計する方法を教示する。当業者であれば、CRISPRランディング部位を本開示のガイドRNAと共に設計することが容易にできよう。そこで生じたプロトスペーサ配列は、先に述べたような所望のCRISPRエンドヌクレアーゼに適したPAMのモチーフと組み合わされる。
[0168] 一部の実施形態において、本開示は:配列番号65、66、67、68、69、70、71、72、73、74、78、79、80、81、及びそれらの組合せからなる群から選択される配列を含むcTAGを教示する。
[0169] DNA標的部位のゲノム中での複数の出現が、全ての潜在的部位を同定した後に、非特異的ゲノム編集に至り得るので、本開示は、一部の実施形態において、関連する参照ゲノム又はモジュラーCRISPRコンストラクト中で現れる回数に基づいて、配列をフィルタリングすることを教示する。配列特異性が「シード」配列(例えば、Cpf1媒介切断のためのガイド配列の最初の5nt)によって決定されるCRISPR酵素について、フィルタリング工程はまた、シードが同じ様々な配列をフィルタリングし得る。
[0170] 一部の実施形態において、アルゴリズムのツールは、特定のガイド配列について、潜在的なオフ標的部位を同定することもできる。例えば、一部の実施形態において、Cas−Offinderが、Cpf1について潜在的なオフ標的部位を同定するのに用いられ得る(Kim et al., 2016.“Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells”(2016年6月6日にオンラインで公開)参照)。他の公に利用可能なあらゆるCRISPR設計/同定ツールが用いられてもよく、例えば、Zhang lab’s crispr.mit.edu tool(Hsu, et al. 2013“DNA targeting specificity of RNA_guided Cas9 nucleases”Nature Biotech 31, 827-832参照)が挙げられる。
[0171] 一部の実施形態において、使用者は、シード配列の長さを選択することができてよい。使用者はまた、フィルタを通すためにゲノム中のシード:PAM配列の出現数を指定することができてよい。デフォルトは、ユニーク配列をスクリーニングすることである。フィルタリングレベルは、シード配列の長さ及びゲノム中での配列の出現数の双方を変えることによって変更される。プログラムは、加えて、又は代わりに、同定された標的配列の逆の相補体を提供することによって、報告される標的配列と相補的なガイド配列の配列を提供し得る。
モジュラーCRISPR DNAコンストラクトクローニング
[0172] 一部の実施形態において、本開示は、本開示のモジュラーCRISPR DNAコンストラクトを用いて、新しい組換え核酸分子を調製する方法を教示する。一部の実施形態において、本開示は、DNA部分アセンブリ方法を教示する。各方法の説明が、以下に記載される。
[0172] 一部の実施形態において、本開示は、本開示のモジュラーCRISPR DNAコンストラクトを用いて、新しい組換え核酸分子を調製する方法を教示する。一部の実施形態において、本開示は、DNA部分アセンブリ方法を教示する。各方法の説明が、以下に記載される。
DNAアセンブリ方法
[0173] 一部の実施形態において、本開示は、DNA部分のモジュラーアセンブリ方法を教示する。一部の実施形態において、本開示のDNAアセンブリ方法は、インビトロで行われる。ゆえに、一部の実施形態において、本開示は、i)少なくとも2つの挿入部分DNAを、少なくとも1つのCRISPR複合体と一緒に含む混合物を形成する工程と、ii)前記混合物を、挿入DNAのCRISPR消化用の条件でインキュベートする工程と、iii)続いて、2つの挿入部分DNAのそれぞれの消化由来の、適合する付着末端をハイブリダイズさせる工程と、iv)前記互いにハイブリダイズした末端をライゲーションして、新しい組換え核酸を生じさせる工程とを教示する。ゆえに、一部の実施形態において、本開示の挿入部分DNAは、一緒に消化される。他の実施形態において、本開示は、各挿入部分DNAを個々に、同じ又は異なるCRISPR複合体で消化する方法を教示する。一部の実施形態において、少なくとも1つの挿入部分が、CRISPR複合体によって消化されない。一部の実施形態において、本開示は、エキソヌクレアーゼ処理が、(後の節に記載されるデュアルCRISPR消化のために)工程iii)のハイブリダイゼーションに先立って行われることを教示する。
[0173] 一部の実施形態において、本開示は、DNA部分のモジュラーアセンブリ方法を教示する。一部の実施形態において、本開示のDNAアセンブリ方法は、インビトロで行われる。ゆえに、一部の実施形態において、本開示は、i)少なくとも2つの挿入部分DNAを、少なくとも1つのCRISPR複合体と一緒に含む混合物を形成する工程と、ii)前記混合物を、挿入DNAのCRISPR消化用の条件でインキュベートする工程と、iii)続いて、2つの挿入部分DNAのそれぞれの消化由来の、適合する付着末端をハイブリダイズさせる工程と、iv)前記互いにハイブリダイズした末端をライゲーションして、新しい組換え核酸を生じさせる工程とを教示する。ゆえに、一部の実施形態において、本開示の挿入部分DNAは、一緒に消化される。他の実施形態において、本開示は、各挿入部分DNAを個々に、同じ又は異なるCRISPR複合体で消化する方法を教示する。一部の実施形態において、少なくとも1つの挿入部分が、CRISPR複合体によって消化されない。一部の実施形態において、本開示は、エキソヌクレアーゼ処理が、(後の節に記載されるデュアルCRISPR消化のために)工程iii)のハイブリダイゼーションに先立って行われることを教示する。
[0174] 他の更なる実施形態において、本開示は、挿入部分末端をssDNAエキソヌクレアーゼに曝すこと、そして、生じた付着末端のハイブリダイゼーションに続く、ポリメラーゼによる任意選択の充填、及びライゲーションによる、挿入部分のGibson様結合を教示する。一部の実施形態において、1つ以上の挿入部分が、ssDNAエキソヌクレアーゼ処理に先立って、dsDNAエキソヌクレアーゼに曝される。一部の実施形態において、本開示は、1つ以上のCRISPRエンドヌクレアーゼ(例えば、後の節に記載されるデュアルCRISPR消化)によって消化された挿入部分又はモジュラーCRISPRベクターのGibson様結合を教示する。
[0175] 以下の節は、挿入部分及び本開示のモジュラーCRISPRコンストラクトがアセンブルされて編集され得る種々の方法を確認する一連の実例を提供する。以下に記載される技術のリストは、本開示の配列の実用性を強調する一連の実例を提供するが、限定となることは意図されていない。当業者であれば、本開示に従う挿入部分のアセンブリ及び編集を可能にする他の技術を認識するであろう。
[0176] 一部の実施形態において、本開示は、Cpf1及び/又はCas9 CRISPRエンドヌクレアーゼを包含する方法を記載する。これらの具体的なCRISPRエンドヌクレアーゼへの言及は、特許請求の範囲において明記されない限り、実例であり、限定となることは意図されていない。当業者であれば、他の既存の、又はこれまで発見されていないCRISPRエンドヌクレアーゼの、本開示のコンストラクト及び方法への利用を直ちに認識するであろう。Cpf1への言及は、スタガードDNA切断を触媒して付着DNA末端を生じさせることができる、現在知られている、又は発見されていないあらゆるCRISPRエンドヌクレアーゼの使用を包含するものと解釈され得る。Cas9への言及も同様に、dsDNAの平滑末端切断を触媒することができる、現在知られている、又は発見されていないあらゆるCRISPRエンドヌクレアーゼの使用を包含するものと解釈され得る。
インビトロCpf1
[0177] 一部の実施形態において、本開示のインビトロDNAアセンブリは、下記のようにCpf1 CRISPR複合体により行われる。第一に、2つ以上の挿入部分が、少なくとも2つの挿入部分間で共通するcTAGを標的とするCpf1 CRISPR複合体とインキュベートされる。一部の実施形態において、挿入部分は、単一の混合物中で一緒にインキュベートされる。他の実施形態において、挿入部分は、異なる混合物中でインキュベートされる。
[0177] 一部の実施形態において、本開示のインビトロDNAアセンブリは、下記のようにCpf1 CRISPR複合体により行われる。第一に、2つ以上の挿入部分が、少なくとも2つの挿入部分間で共通するcTAGを標的とするCpf1 CRISPR複合体とインキュベートされる。一部の実施形態において、挿入部分は、単一の混合物中で一緒にインキュベートされる。他の実施形態において、挿入部分は、異なる混合物中でインキュベートされる。
[0178] 第二に、一部の実施形態において、消化された生成物は精製されて、活性CRISPRヌクレアーゼが除去される。一部の実施形態において、精製は、消化された挿入部分からの活性Cpf1複合体の分離を包含する。一部の実施形態において、これは、DNA精製、例えばゲル精製又はカラム精製を通して達成され得る。他の実施形態において、精製は、Cpf1不活化によって、例えば熱又は化学物質による不活化を通して達成され得る。
[0179] 第三に、消化された挿入部分は、Cpf1複合体によって生じた適合する付着末端のハイブリダイゼーションに適した条件でインキュベートされる。次に、ハイブリダイズされた末端が、本開示の以前の部分において記載されたものが挙げられる、既知のあらゆるライゲーション方法に従ってライゲーションされる。
インビトロCas9
[0180] 他の実施形態において、本開示のインビトロDNAアセンブリは、下記のようにCas9 CRISPR複合体により行われる。第一に、2つ以上の挿入部分が、少なくとも2つの挿入部分間で共通するcTAGを標的とするCas9 CRISPR複合体とインキュベートされる。一部の実施形態において、挿入部分は、単一の混合物中で一緒にインキュベートされる。他の実施形態において、挿入部分は、様々な混合物中でインキュベートされる。
[0180] 他の実施形態において、本開示のインビトロDNAアセンブリは、下記のようにCas9 CRISPR複合体により行われる。第一に、2つ以上の挿入部分が、少なくとも2つの挿入部分間で共通するcTAGを標的とするCas9 CRISPR複合体とインキュベートされる。一部の実施形態において、挿入部分は、単一の混合物中で一緒にインキュベートされる。他の実施形態において、挿入部分は、様々な混合物中でインキュベートされる。
[0181] 第二に、一部の実施形態において、消化された生成物は精製されて、活性CRISPRヌクレアーゼが除去される。一部の実施形態において、精製は、消化された挿入部分からの活性Cas9複合体の分離を包含する。一部の実施形態において、これは、DNA精製、例えばゲル精製又はカラム精製を通して達成され得る。他の実施形態において、精製は、Cas9不活化によって、例えば熱又は化学物質による不活化を通して達成され得る。
[0182] 一部の実施形態において、Cas9消化された生成物についての第3の工程は、平滑末端−ライゲーションに適した条件で挿入部分をインキュベートするものである。
デュアルCRISPRアセンブリ
[0183] 他の実施形態において、本開示はまた、CRISPR消化された挿入部分のピースを、少なくとも1つの共有されるcTAG配列とアセンブルするGibsonアセンブリ型の方法(例えば、異なるCRISPRランディング部位にて消化された、適合するcTAGのアセンブリ)を教示する。ゆえに、一部の実施形態において、本開示は、下記のように、デュアルCRISPR消化アセンブリを教示する。
[0183] 他の実施形態において、本開示はまた、CRISPR消化された挿入部分のピースを、少なくとも1つの共有されるcTAG配列とアセンブルするGibsonアセンブリ型の方法(例えば、異なるCRISPRランディング部位にて消化された、適合するcTAGのアセンブリ)を教示する。ゆえに、一部の実施形態において、本開示は、下記のように、デュアルCRISPR消化アセンブリを教示する。
[0184] 第一に、2つ以上の挿入部分が、少なくとも2つの挿入部分間で共通する上述のcTAG内の、各部分の側面に位置する2つの異なるCRISPRランディング部位を標的とする2つのCRISPR複合体とインキュベートされる。
[0185] 一部の実施形態において、2つの異なるCRISPRランディング部位は、一緒に消化される。他の実施形態において、一方の挿入部分DNAが、あるCRISPRランディング部位を標的とするCRISPR複合体で消化され、そして他方の挿入部分DNAが、第2のCRISPRランディング標的部位を標的とする異なるCRISPR複合体で、別個の容器内で消化される。それぞれの場合において、これらの消化の結果は、2つの挿入DNA cTAGのそれぞれにおいて共有されるcTAGが、少なくとも1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、又は20bpの、互いとの配列重なりを含むこととなるということであろう。
[0186] 例えば、実例となる実施形態において、2つの挿入DNA部分間で共有されるcTAGは、以下のように5’〜3’に配置されることとなる:(Part I)-[R1-C1-C2]-(Part II)、式中、R=制限部位、C1=第1のCRISPRランディング部位、及びC2=第2のCRISPRランディング部位。この実例となる実施形態において、3’に共有されるcTAGを有する第1の挿入DNA部分は、C2を標的とするCRISPR複合体で消化されることとなり、そして5’に共有されるcTAGを有する第2の挿入DNA部分は、C1を標的とするCRISPR複合体で消化されることとなる。これにより、重なる配列がC1〜C2にまたがる2つのDNA挿入部分が生じることとなる。
[0187] 第二に、一部の実施形態において、消化された生成物は精製されて、活性CRISPRヌクレアーゼが除去される。一部の実施形態において、精製は、消化された挿入部分からの活性Cas9複合体の分離を包含する。一部の実施形態において、これは、DNA精製、例えばゲル精製又はカラム精製を通して達成され得る。他の実施形態において、精製は、CRISPR不活化によって、例えば熱又は化学物質による不活化を通して達成され得る。
[0188] 第三に、一部の実施形態において、CRISPR消化された挿入部分は、ssDNAエキソヌクレアーゼとインキュベートされて、2つの挿入DNA部分間で重なる付着末端が生じる。
[0189] 第四に、消化された挿入部分は、CRISPR複合体/エキソヌクレアーゼ消化によって生じた適合する付着末端のハイブリダイゼーションに適した条件でインキュベートされる。次に、ハイブリダイズされた末端が、本開示の以前の部分で記載されたものが挙げられる、既知のあらゆるライゲーション方法に従ってライゲーションされる。一部の実施形態において、ハイブリダイズされた部分は、ライゲーションに先立って、ポリメラーゼとインキュベートされて、欠けているあらゆる配列ギャップが満たされる。
架橋アセンブリ
[0190] 他の実施形態において、本開示は、挿入部分の消化されたcTAG配列と重なる架橋配列を含む第3のDNA配列の付加を通した、Cas9消化部分のGibsonアセンブリを教示する。
[0190] 他の実施形態において、本開示は、挿入部分の消化されたcTAG配列と重なる架橋配列を含む第3のDNA配列の付加を通した、Cas9消化部分のGibsonアセンブリを教示する。
[0191] この実例において、双方の挿入部分が、同じCRISPRランディング部位を標的とする同じCas9 CRISPR複合体で消化される。この実施形態において、消化されて生じたcTAGは、配列重なりを有することはない。ゆえに、一部の実施形態において、第3の工程は、Cas9消化された挿入部分が、ssDNAエキソヌクレアーゼでさらに消化されて、3’突出部又は5’突出部のいずれかを生じさせるものである。次に、エキソヌクレアーゼ消化された挿入部分は、CRISPR複合体及びエキソヌクレアーゼ消化の組合せによって生じた適合する付着末端の、架橋配列とのハイブリダイゼーションに適した条件でインキュベートされる。次に、ハイブリダイズされた末端が、本開示の以前の部分で記載されたものが挙げられる、既知のあらゆるライゲーション方法に従ってライゲーションされる。一部の実施形態において、本開示のエキソヌクレアーゼ消化は、第2の工程の前に行われる。
インビトロHDR
[0192] 他の実施形態において、本開示は、Cas9又はCpf1エンドヌクレアーゼによって消化された挿入部分DNAの末端をHDR複合体とアセンブルすることによって、前記消化された挿入部分の組換えをトリガーするインビトロ方法を教示する。
[0192] 他の実施形態において、本開示は、Cas9又はCpf1エンドヌクレアーゼによって消化された挿入部分DNAの末端をHDR複合体とアセンブルすることによって、前記消化された挿入部分の組換えをトリガーするインビトロ方法を教示する。
インビトロ相同組換え
[0193] 一部の実施形態において、本開示のインビボDNAアセンブリは、下記のようにCpf1又はCas9 CRISPR複合体により行われる。一実施形態において、少なくとも1つのcTAGを共有する2つ以上の挿入部分が、宿主細胞中に導入される。一部の実施形態において、相同なcTAG配列を共有するDNA挿入部分の存在は、相同組換えアセンブリ(例えば、酵母の相同組換え)をトリガーするのに十分であろう。
[0193] 一部の実施形態において、本開示のインビボDNAアセンブリは、下記のようにCpf1又はCas9 CRISPR複合体により行われる。一実施形態において、少なくとも1つのcTAGを共有する2つ以上の挿入部分が、宿主細胞中に導入される。一部の実施形態において、相同なcTAG配列を共有するDNA挿入部分の存在は、相同組換えアセンブリ(例えば、酵母の相同組換え)をトリガーするのに十分であろう。
[0194] 例えば、一部の実施形態において、2つの挿入DNA部分間の少なくとも1つの共有されるcTAG配列は、アセンブルされて直鎖状のコンストラクトを生じさせ得る。また、この実例となる実施形態において、2つの残りの外側のcTAGは、細胞内の別のベクターのcTAGと組み換えられる(例えば、既存のプラスミド又は染色体中に挿入される)ように設計され得る。他の実施形態において、2つの部分はさらに、2つの挿入DNA部分間での、第2の共有されるcTAGの組換えを通して、環状のコンストラクト中にアセンブルされ得る。アセンブルされたコンストラクトは、アセンブリに用いられた生物に用いられてもよいし、一部の実施形態において、精製されて第2の生物に形質転換されてもよい(例えば、酵母中でのアセンブリ、及び以降の細菌への形質転換)。
[0195] 他の実施形態において、cTAGが共有される1つ以上の挿入部分が、宿主細胞中への導入に先立って消化されてよい。ゆえに、一部の実施形態において、本開示は、解放される部分のインビボアセンブリに先立つ、より大きなベクターから挿入部分を解放するCRISPR消化を教示する。一部の実施形態において、消化はCas9により実行される。他の実施形態において、消化はCpf1により実行される。他の実施形態において、消化は制限酵素により実行される。一部の実施形態において、挿入部分のCRISPR消化は、インビトロで行われる。一部の実施形態において、消化された生成物は、アセンブリ宿主細胞中への挿入部分の形質転換に先立って、活性CRISPRエンドヌクレアーゼを除去するように精製される。
[0196] 一部の実施形態において、精製工程は、DNA精製、例えばゲル精製又はカラム精製を通して達成され得る。他の実施形態において、精製は、CRISPR不活化によって、例えば熱又は化学物質による不活化を通して達成され得る。
インビトロライゲーション
[0197] 一部の実施形態において、本開示は、CRISPRエンドヌクレアーゼによる再切断から挿入部分を保護する方法を教示する。一部の実施形態において、本開示の挿入部分は、DNA配列の化学修飾を介して、エンドヌクレアーゼ切断から保護され得る。例えば、一部の実施形態において、本開示は、ホスホロチオエートオリゴヌクレオチドを教示する。
[0197] 一部の実施形態において、本開示は、CRISPRエンドヌクレアーゼによる再切断から挿入部分を保護する方法を教示する。一部の実施形態において、本開示の挿入部分は、DNA配列の化学修飾を介して、エンドヌクレアーゼ切断から保護され得る。例えば、一部の実施形態において、本開示は、ホスホロチオエートオリゴヌクレオチドを教示する。
[0198] 一部の実施形態において、本開示の方法はとりわけ、マルチ部分DNAアセンブリに有用である。
[0199] 本明細書の図2Aは、本開示の方法に従う、マルチ部分DNAアセンブリの実例を示す。この例において、一連の8つのDNA部分(part1〜8)(それぞれ2つのcTAG(タグA〜H)を有する)が、インビトロで組み合わされてから、(インビボでの相同組換えを介して、又は先に述べるライゲーションを介して)セルフアセンブルすることができる。
DNA編集方法
[0200] 一部の実施形態において、本開示は、モジュラーCRISPR DNAコンストラクトの編集方法を教示する。一部の実施形態において、本開示のDNA編集方法は、先に記載されるDNAアセンブリ方法の同じ原理を使用するが、1つ以上の既存のモジュラーCRISPR DNAコンストラクトを編集する目的でそうする。
[0200] 一部の実施形態において、本開示は、モジュラーCRISPR DNAコンストラクトの編集方法を教示する。一部の実施形態において、本開示のDNA編集方法は、先に記載されるDNAアセンブリ方法の同じ原理を使用するが、1つ以上の既存のモジュラーCRISPR DNAコンストラクトを編集する目的でそうする。
[0201] 一部の実施形態において、本開示のDNA編集方法は、インビトロで行われる。ゆえに、一部の実施形態において、本開示は、i)モジュラーCRISPR DNAコンストラクト、及び少なくとも1つの挿入DNA部分を、少なくとも1つのCRISPR複合体と一緒に含む混合物を形成する工程と、ii)前記混合物を、挿入DNAのcTAG、及びその対応するモジュラーCRISPR DNAコンストラクトcTAGのCRISPR消化用の条件でインキュベートする工程と、続いてiii)上述の各cTAGの消化によって生じた適合する付着末端(Cpf1の場合)をハイブリダイズさせる工程と、iv)前記ハイブリダイズされた末端(又はCas9が用いられるならば、平滑末端)を互いにライゲーションして、新しい組換え核酸を生じさせる工程を教示する。一部の実施形態において、エキソヌクレアーゼ処理が、(後の節に記載されるデュアルCRISPR消化のために)工程iii)のハイブリダイゼーションに先立って行われる。一部の実施形態において、本開示の消化は、挿入部分DNA及びモジュラーCRISPR DNAコンストラクトについて、別々に行われる。一部の実施形態において、モジュラーCRISPR DNAコンストラクトのみが、CRISPR複合体で消化される。
インビトロCpf1
[0202] 一部の実施形態において、本開示のインビトロDNA編集方法は、下記のようにCpf1 CRISPR複合体により行われる。第一に、モジュラーCRISPR DNAコンストラクト及び少なくとも1つの挿入DNA部分が、挿入部分のcTAG、及びそれらの、モジュラーCRISPR DNAコンストラクト内の対応するタグを標的とするCpf1 CRISPR複合体とインキュベートされる。一部の実施形態において、モジュラーCRISPR DNA及び挿入部分DNAの消化は、別個の反応で行われる。
[0202] 一部の実施形態において、本開示のインビトロDNA編集方法は、下記のようにCpf1 CRISPR複合体により行われる。第一に、モジュラーCRISPR DNAコンストラクト及び少なくとも1つの挿入DNA部分が、挿入部分のcTAG、及びそれらの、モジュラーCRISPR DNAコンストラクト内の対応するタグを標的とするCpf1 CRISPR複合体とインキュベートされる。一部の実施形態において、モジュラーCRISPR DNA及び挿入部分DNAの消化は、別個の反応で行われる。
[0203] 第二に、一部の実施形態において、消化された生成物は精製されて、活性CRISPRヌクレアーゼが除去される。一部の実施形態において、精製は、消化されたヌクレオチドからの活性Cpf1複合体の分離を包含する。一部の実施形態において、これは、DNA精製、例えばゲル精製又はカラム精製を通して達成され得る。他の実施形態において、精製は、Cpf1不活化によって、例えば熱又は化学物質による不活化を通して達成され得る。
[0204] 第三に、消化されたモジュラーCRISPR DNAコンストラクト及び挿入部分は、Cpf1複合体によって生じた適合する付着末端のハイブリダイゼーションに適した条件でインキュベートされる。次に、ハイブリダイズされた末端が、本開示の以前の部分で記載されたものが挙げられる、既知のあらゆるライゲーション方法に従ってライゲーションされる。
インビトロCas9
[0205] 他の実施形態において、本開示のインビトロDNA編集方法は、下記のようにCas9 CRISPR複合体により行われる。第一に、モジュラーCRISPR DNAコンストラクト及び少なくとも1つの挿入DNA部分が、挿入部分のcTAG、及びその、モジュラーCRISPR DNAコンストラクト内の対応するタグを標的とするCas9 CRISPR複合体とインキュベートされる。一部の実施形態において、モジュラーCRISPR DNA及び挿入部分DNAの消化は、別個の反応で行われる。
[0205] 他の実施形態において、本開示のインビトロDNA編集方法は、下記のようにCas9 CRISPR複合体により行われる。第一に、モジュラーCRISPR DNAコンストラクト及び少なくとも1つの挿入DNA部分が、挿入部分のcTAG、及びその、モジュラーCRISPR DNAコンストラクト内の対応するタグを標的とするCas9 CRISPR複合体とインキュベートされる。一部の実施形態において、モジュラーCRISPR DNA及び挿入部分DNAの消化は、別個の反応で行われる。
[0206] 第二に、一部の実施形態において、消化された生成物は精製されて、活性CRISPRヌクレアーゼが除去される。一部の実施形態において、精製は、消化されたヌクレオチドからの活性Cas9複合体の分離を包含する。一部の実施形態において、これは、DNA精製、例えばゲル精製又はカラム精製を通して達成され得る。他の実施形態において、精製は、Cas9不活化によって、例えば熱又は化学物質による不活化を通して達成され得る。
[0207] 一部の実施形態において、Cas9消化された生成物についての第3の工程は、平滑末端ライゲーションに適した条件で挿入部分をインキュベートするものである。
Gibson編集
[0208] 他の実施形態において、本開示はまた、CRISPR消化されたコンストラクトの配列及び/又は無傷の重なるcTAG配列を含有する未消化挿入部分を編集するGibsonアセンブリ型の方法を教示する。ゆえに、一部の実施形態において、第3の工程は、Cas9消化されたモジュラーCRISPR DNAコンストラクト及び挿入部分が、ssDNAエキソヌクレアーゼでさらに消化されて、3’突出部又は5’突出部のいずれかを生じさせるものである。一部の実施形態において、本開示は、ssDNA消化に先立つ、非CRISPR消化挿入部分を短くするためのdsDNAエキソヌクレアーゼ消化を教示する。
[0208] 他の実施形態において、本開示はまた、CRISPR消化されたコンストラクトの配列及び/又は無傷の重なるcTAG配列を含有する未消化挿入部分を編集するGibsonアセンブリ型の方法を教示する。ゆえに、一部の実施形態において、第3の工程は、Cas9消化されたモジュラーCRISPR DNAコンストラクト及び挿入部分が、ssDNAエキソヌクレアーゼでさらに消化されて、3’突出部又は5’突出部のいずれかを生じさせるものである。一部の実施形態において、本開示は、ssDNA消化に先立つ、非CRISPR消化挿入部分を短くするためのdsDNAエキソヌクレアーゼ消化を教示する。
[0209] 次に、エキソヌクレアーゼ消化されたDNA部分は、CRISPR複合体及びエキソヌクレアーゼ消化の組合せによって生じた適合する付着末端のハイブリダイゼーションに適した条件下でインキュベートされる。次に、ハイブリダイズされた末端が、本開示の以前の部分で記載されたものが挙げられる、既知のあらゆるライゲーション方法に従ってライゲーションされる。一部の実施形態において、ハイブリダイズされたDNAは、ライゲーションに先立って、ポリメラーゼとインキュベートされて、欠けているDNA部分が満たされる。一部の実施形態において、本開示のエキソヌクレアーゼ消化は、CRISPR不活化工程の前に行われる。
[0210] 一部の実施形態において、消化された配列のライゲーションは、インビトロで起こってよい。
[0211] 他の実施形態において、本開示は、Cas9又はCpf1エンドヌクレアーゼによって消化されたモジュラーCRISPR DNAコンストラクト、及び少なくとも1つの未消化挿入部の末端を、HDR複合体とアセンブルすることによって、前記消化されたモジュラーCRISPR DNAコンストラクト及び少なくとも1つの挿入DNA部分の組換えをトリガーするインビトロ方法を教示する。
[0212] 本開示のDNA編集方法の一部の実施形態において、DNA挿入部分は、第2のモジュラーCRISPR DNAコンストラクト内に含まれる。ゆえに、一部の実施形態において、本開示のDNA編集方法は、あるモジュラーCRISPR DNAコンストラクトから別のものへのDNA挿入部分の移入を含む。
発現、精製、及び送達
[0213] 一部の実施形態において、本開示は、CRISPR複合体をコードするベクター、コンストラクト、及び核酸配列の方法及び組成物を教示する。一部の実施形態において、本開示は、Cas9又はCpf1タンパク質のトランスジェニック発現又は一過性発現用のプラスミドを教示する。一部の実施形態において、本開示は、以下に限定されないが、リガーゼ、リンカー、及びNLSが挙げられる、本明細書中に記載される他のポリペプチドの1つ以上のタンパク質融合のためのインフレーム配列を含むキメラCas9又はCpf1タンパク質をコードするプラスミドを教示する。
[0213] 一部の実施形態において、本開示は、CRISPR複合体をコードするベクター、コンストラクト、及び核酸配列の方法及び組成物を教示する。一部の実施形態において、本開示は、Cas9又はCpf1タンパク質のトランスジェニック発現又は一過性発現用のプラスミドを教示する。一部の実施形態において、本開示は、以下に限定されないが、リガーゼ、リンカー、及びNLSが挙げられる、本明細書中に記載される他のポリペプチドの1つ以上のタンパク質融合のためのインフレーム配列を含むキメラCas9又はCpf1タンパク質をコードするプラスミドを教示する。
[0214] 一部の実施形態において、本開示のプラスミド及びベクターは、Cas9/Cpf1タンパク質をコードし、そしてまたcrRNA/tracrRNA/sgRNA、及び/又は本開示のドナー挿入配列をコードすることとなる。他の実施形態において、操作された複合体の異なる成分は、1つ以上の異なるプラスミド中にコードされてよい。
[0215] 一部の実施形態において、本開示のプラスミドは、複数の種にわたって用いられ得る。他の実施形態において、本開示のプラスミドは、形質転換されることとなる生物に合わせて調整される。一部の実施形態において、本開示の配列は、遺伝子が編集されることとなる生物において発現するようにコドン最適化されることとなる。当業者であれば、遺伝子編集に十分な発現をもたらすプロモータを用いることの重要性を認識するであろう。一部の実施形態において、異なる種用のプラスミドは、異なるプロモータを必要とすることとなる。
[0216] 一部の実施形態において、本開示のプラスミド及びベクターは、注目する細胞において選択的に発現される。ゆえに、一部の実施形態において、本出願は、異所性プロモータ(ectopic promoter)、組織特異的プロモータ、発生調節プロモータ(developmentally-regulated promoter)、又は誘導プロモータの使用を教示する。一部の実施形態において、本開示はまた、ターミネータ配列の使用を教示する。
[0217] 一部の実施形態において、本開示はまた、Cpf1及び/又はCas9エンドヌクレアーゼタンパク質を発現させて精製する方法を教示する。一部の実施形態において、本開示は、本開示のタンパク質が、市販のいかなるタンパク質生成精製キット又はサービスによって生成されてもよいことを教示する。例えば、一部の実施形態において、本開示は、ポリヒスチジン(His)、グルタチオンs−トランスフェラーゼ(GST)、又は他の精製タグキメラ融合によるベクター中へのCas9及び/又はCpf1のクローニング方法を教示する。一部の実施形態において、本開示は、種々の原核生物及び真核生物、並びに無細胞のタンパク質生成系を教示する。例えば、一部の実施形態において、本開示は、大腸菌(E. coli)BL21中でのタンパク質発現プラスミドの発現を教示する。一部の実施形態において、タンパク質生成系は、タンパク質毒性の影響を和らげるように誘導可能であろう。例えば、一部の実施形態において、本開示は、IPTG又はアラビノース誘導系を用いる方法を教示する。
[0218] 一部の実施形態において、本開示はまた、親和性タグ(His−ニッケル、GST−グルタチオンその他)を含む、種々のタンパク質精製スキームを教示する。一部の実施形態において、本開示は、タンパク質精製のための未変性条件及び変性条件の双方を教示する。
[0219] 他の実施形態において、本開示は、以下に限定されないが、GenScript(登録商標)、ThermoFisher(登録商標)、及びNovoProtein(登録商標)が挙げられる1つ以上のタンパク質生成サービスを介した、Cas9及び/又はCpf1の生成を教示する。
形質転換
[0220] 一部の実施形態において、本開示は、本明細書中で開示されるプラスミド及びベクターの形質転換の使用を教示する。当業者であれば、本開示のプラスミドが、本明細書の他の部分において記載される既知のあらゆる系を通して細胞に形質転換され得ると認識するであろう。例えば、一部の実施形態において、本開示は、粒子砲撃による形質転換、化学的形質転換、アグロバクテリウム形質転換、ナノスパイク形質転換、エレクトロポレーション、及びウイルス形質転換を教示する。
[0220] 一部の実施形態において、本開示は、本明細書中で開示されるプラスミド及びベクターの形質転換の使用を教示する。当業者であれば、本開示のプラスミドが、本明細書の他の部分において記載される既知のあらゆる系を通して細胞に形質転換され得ると認識するであろう。例えば、一部の実施形態において、本開示は、粒子砲撃による形質転換、化学的形質転換、アグロバクテリウム形質転換、ナノスパイク形質転換、エレクトロポレーション、及びウイルス形質転換を教示する。
[0221] 一部の実施形態において、本開示のベクターは、形質転換、形質移入、形質導入、ウイルス感染、遺伝子銃、又はTi媒介遺伝子移入が挙げられる種々の技術のいずれを用いて宿主細胞中に導入されてもよい。特定の方法として、リン酸カルシウム形質移入、DEAEデキストラン媒介形質移入、リポフェクション、又はエレクトロポレーションが挙げられる(Davis, L., Dibner, M., Battey, I., 1986“Basic Methods in Molecular Biology”)。形質転換の他の方法として、例えば、酢酸リチウム形質転換及びエレクトロポレーションが挙げられる(例えば、Gietz et al., Nucleic Acids Res. 27: 69-74 (1992);Ito et al., J. Bacterol. 153:163-168 (1983);及びBecker and Guarente, Methods in Enzymology 194:182-187 (1991)参照)。一部の実施形態において、形質転換された宿主細胞は、組換え宿主株と呼ばれる。
[0222] 一部の実施形態において、本開示は、本開示の96ウェルプレートロボット工学プラットフォーム及び液体ハンドリングマシンを用いる細胞の高スループット形質転換を教示する。
[0223] 一部の実施形態において、本開示は、外因性タンパク質(Cpf1/Cas9及びDNAリガーゼ)、RNA(crRNA/tracRNA/GuideRNA)、及びDNA(挿入DNA部分又はモジュラーCRISPRコンストラクト)を細胞中に入れる方法を教示する。これを達成する種々の方法が、以前に記載されており、タンパク質/RNA/DNAの直接的な形質移入又はDNA形質転換に続くRNA及びタンパク質の細胞内発現が挙げられる(Dicarlo, J. E. et al.“Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems.”Nucleic Acids Res (2013). doi: 10.1093/nar/gkt135;Ren, Z. J., Baumann, R. G. & Black, L. W.“Cloning of linear DNAs in vivo by overexpressed T4 DNA ligase: construction of a T4 phage hoc gene display vector.”Gene 195, 303-311 (1997);Lin, S., Staahl, B. T., Alla, R. K. & Doudna, J. A.“Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery.”Elife 3, e04766 (2014))。
[0224] 一部の実施形態において、本開示は、先に記載される1つ以上の選択マーカーによる形質転換細胞のスクリーニングを教示する。そのような一実施形態において、カナマイシン耐性マーカー(KanR)を含むベクターで形質転換された細胞が、有効な量のカナマイシン抗生物質を含有する培地上にプレーティングされる。カナマイシン入りの培地上で認識できるコロニー形成単位により、ベクターカセットのゲノム中への組込みが推定される。所望の配列の挿入が、関連する挿入部位のPCR、制限酵素分析、及び/又は配列決定を介して裏付けられ得る。
[0225] 他の実施形態において、本開示の複合体の一部又は全体が、細胞に直接送達され得る。ゆえに、一部の実施形態において、本開示は、本開示のポリペプチド及び核酸の発現及び精製を教示する。当業者であれば、タンパク質及び核酸を精製するための多くの方法を認識するであろう。一部の実施形態において、ポリペプチドは、誘導可能なタンパク質生成系又は構成的タンパク質生成系、例えば細菌系、酵母系、植物細胞系、又は動物細胞系を介して発現され得る。一部の実施形態において、本開示はまた、親和性タグを介したタンパク質及び/若しくはポリペプチドの精製、又はカスタム抗体精製を教示する。他の実施形態において、本開示はまた、ポリヌクレオチドの化学合成方法を教示する。
[0226] 一部の実施形態において、当業者であれば、遺伝子発現用のウイルスベクター又はプラスミドが、本明細書中で開示される複合体を送達するのに用いられ得ることを認識するであろう。ウイルス様粒子(VLP)が、リボ核タンパク質複合体をカプセルに入れるのに用いられ得る。そして、本明細書中で開示される精製されたリボ核タンパク複合体が精製されて、エレクトロポレーション又はエレクトロインジェクションを介して細胞に送達され得る。
キット
[0227] 一部の実施形態において、本開示は、上述の方法及び組成物に開示される要素のいずれか1つ以上を含有するキットを提供する。一部の実施形態において、キットは、モジュラーCRISPR DNAコンストラクト、並びにキット及び必須のあらゆる試薬又は反応物質を用いるための使用説明書を含む。一部の実施形態において、ベクター系は、(a)モジュラーCRISPR DNAコンストラクト、(b)CRISPRエンドヌクレアーゼタンパク質及び必須の標的ガイドRNA(又は前記部材をコードする配列)を含むCRISPR複合体、並びに、場合によっては(c)本明細書中に記載される前掲の挿入DNA部分を含む。
[0227] 一部の実施形態において、本開示は、上述の方法及び組成物に開示される要素のいずれか1つ以上を含有するキットを提供する。一部の実施形態において、キットは、モジュラーCRISPR DNAコンストラクト、並びにキット及び必須のあらゆる試薬又は反応物質を用いるための使用説明書を含む。一部の実施形態において、ベクター系は、(a)モジュラーCRISPR DNAコンストラクト、(b)CRISPRエンドヌクレアーゼタンパク質及び必須の標的ガイドRNA(又は前記部材をコードする配列)を含むCRISPR複合体、並びに、場合によっては(c)本明細書中に記載される前掲の挿入DNA部分を含む。
[0228] 要素は、適切なあらゆるコンテナ、例えばバイアル、ボトル、若しくはチューブ、又は宿主細胞若しくはプラスミド中に、個々に提供されても組み合わせて提供されてもよい。一部の実施形態において、キットは、1つ以上の言語、例えば複数の言語の使用説明書を含む。
[0229] 一部の実施形態において、キットは、本明細書中に記載される要素(例えば、精製されたCpf1エンドヌクレアーゼ)の1つ以上を利用するプロセスに用いられる1つ以上の試薬を含む。試薬は、適切ないかなるコンテナ内に提供されてもよい。例えば、キットは、1つ以上の反応バッファ又は貯蔵バッファを提供してよい。試薬は、特定のアッセイに使用可能である形態で提供されてもよいし、使用前に1つ以上の他の成分の付加を必要とする形態(例えば、濃縮した形態又は凍結乾燥された形態)で提供されてもよい。バッファは、いかなるバッファであってもよく、以下に限定されないが、炭酸ナトリウムバッファ、重炭酸ナトリウムバッファ、ホウ酸バッファ、トリスバッファ、MOPSバッファ、HEPESバッファ、及びそれらの組合せが挙げられる。一部の実施形態において、バッファはアルカリ性である。一部の実施形態において、バッファは、pHが約7から約10である。一部の実施形態において、キットは、crRNA配列及び調節要素を作動可能に連結するような、ベクター中への挿入用のcrRNA配列に対応する1つ以上のオリゴヌクレオチドを含む。
実施例
[0230] 以下の実施例は、本開示の種々の実施形態を具体的に説明する目的で与えられており、いかなる形であれ本開示を限定することを意味しない。本開示中での変更、及び特許請求の範囲によって定義される本開示の精神に包含される他の用途が、当業者に思い浮かぶであろう。
[0230] 以下の実施例は、本開示の種々の実施形態を具体的に説明する目的で与えられており、いかなる形であれ本開示を限定することを意味しない。本開示中での変更、及び特許請求の範囲によって定義される本開示の精神に包含される他の用途が、当業者に思い浮かぶであろう。
実施例1:ワンポットインビトロモジュラーCRISPRクローニング
[0231] 本実施例は、ワンポット反応における、あるプラスミドから別のものへの挿入物の移入による、プラスミド13001009086(配列番号82)の生成を記載する。図4参照。
[0231] 本実施例は、ワンポット反応における、あるプラスミドから別のものへの挿入物の移入による、プラスミド13001009086(配列番号82)の生成を記載する。図4参照。
[0232] 双方のプラスミドが、注目する領域の側面に位置するクローニングタグ(cTAG K[配列番号78]/cTAG L[配列番号79]及びcTAG K’[配列番号80]/cTAG L’[配列番号81])を有する。編集されたプラスミドに向けてクローニング反応を駆動するために、Cpf1スペーサは、レシピエントプラスミド及びドナープラスミド(それぞれK/K’及びL/L’)上で相対する向きにある。このインサイドアウト/アウトサイドイン消化により、最終生成物中のCpf1スペーサを除去して、所望の生成物の再切断を除外する(図4中の、‘485プラスミドにおけるインサイドアウト消化及び‘784プラスミドにおけるアウトサイドイン消化を示す湾曲した矢印参照)。Cas9スペーサは残って、この部位での反復編集(iterative editing)が可能となる。ゆえに、MegaModularコンストラクトは、反復編集を可能にする迅速な単一ポット反応スキームを可能にする。
[0233] Cpf1タンパク質をGenscriptによって、そしてcrRNAをSynthegoによって合成した。ワンポット切断/ライゲーション反応のために、crRNA(crRNA1及びcrRNA3)と複合体形成したCpf1タンパク質を、ATPを含有するバッファ中のプラスミド(13000789485−配列番号83及び13000823784−配列番号84)及びDNAリガーゼに加えた。これらの成分を、切断及びライゲーション用に最適化した温度にて循環させた。
[0234] 反応物を、大腸菌(E. coli)に形質転換して、陽性クローンを配列決定して、新しい挿入物の挿入及びCpf1スペーサの喪失を裏付けた。
[0235] 欠失のために、用いたクローニングタグ内のCpf1部位は、プラスミドクロージャを可能にするような適合する突出部を生成しなければならない。cTAG L’を、2つのCpf1スペーサ(1つは、突出部がcTAG K’と適合しない挿入用の、そして第2のものは、突出部がcTAG K’と適合する欠失用のものである)を含有するように設計した。
実施例2:インビトロモジュラーCRISPRクローニング
[0236] 本実施例を、CRISPRクローニングの柔軟性を確認するために設計した。最初の工程として、カナマイシン耐性遺伝子又はクロラムフェニコール耐性遺伝子をコードするいくつかの耐性プラスミドを、ソースベクターpzHR039(配列番号100)及び13000223370(配列番号101)からそれぞれ作出した。カナマイシン耐性プラスミドをそれぞれ、GFP遺伝子の側面に位置する種々のCpf1ランディング部位を含むように設計した(消化したときに、当該プラスミドは「カナマイシン耐性プラスミド骨格」を生成する)。クロラムフェニコール耐性プラスミドをそれぞれ、クロラムフェニコール耐性遺伝子の側面に位置する種々のCpf1ランディング部位を含むように設計した(消化したときに、当該プラスミドは「クロラムフェニコール耐性挿入物」を生成する)。本実施例で用いた各プラスミドについての配列及びベクターマップを、表2に開示する。
[0236] 本実施例を、CRISPRクローニングの柔軟性を確認するために設計した。最初の工程として、カナマイシン耐性遺伝子又はクロラムフェニコール耐性遺伝子をコードするいくつかの耐性プラスミドを、ソースベクターpzHR039(配列番号100)及び13000223370(配列番号101)からそれぞれ作出した。カナマイシン耐性プラスミドをそれぞれ、GFP遺伝子の側面に位置する種々のCpf1ランディング部位を含むように設計した(消化したときに、当該プラスミドは「カナマイシン耐性プラスミド骨格」を生成する)。クロラムフェニコール耐性プラスミドをそれぞれ、クロラムフェニコール耐性遺伝子の側面に位置する種々のCpf1ランディング部位を含むように設計した(消化したときに、当該プラスミドは「クロラムフェニコール耐性挿入物」を生成する)。本実施例で用いた各プラスミドについての配列及びベクターマップを、表2に開示する。
[0237] 各カナマイシン耐性プラスミド及びクロラムフェニコール耐性プラスミドを、最初に、II型制限酵素KpnI−HF及びPvuI−HF(双方ともNEBから市販されている)でそれぞれ線状化した。各プラスミド上のKpnI制限部位及びPvuI制限部位の位置を、図7〜図14に記載するベクターマップ中に示す。線状化の後、耐性プラスミドはもはや、細菌宿主系において自己複製できなかった。
[0238] 次に、線状化した耐性プラスミドを、15ug(1.58uMの最終濃度)のCpf1酵素及び2uLの以下に記載する5uM各ガイドRNA(0.167uMの最終濃度)のプレインキュベートした混合物と60uLの反応で混合して、活性CRISPR複合体を形成した。
[0239] 本実施例で用いたCpf1酵素は、IDTから商業的に得た。Cpf1は、アシダミノコッカス(Acidaminococcus)属種Cpf1(AsCpf1)から得た。酵素をさらに、1つのN末端核局在配列(NLS)及び1つのC末端NLS、並びに3つのN末端FLAGタグ及び1つのC末端6−Hisタグを含むように修飾した。
[0240] 本実施例で用いたガイドRNAは、IDTから特注した。各ガイドRNAを、線状化された耐性プラスミド内に位置決めした様々なCRISPRランディング部位を標的とするように設計した。本実施例において、骨格プラスミドのCpf1ランディング部位を切り出したが、挿入物のライゲーション直後に回復させた。表2は、用いた各ガイドRNAのガイド配列部分を示す。このように、GFP遺伝子を各カナマイシン耐性プラスミドから切り出して、カナマイシン耐性プラスミド骨格を生成するように、混合物中のCRISPR複合体を設計した(図5、第2のパネル参照)。また、クロラムフェニコール耐性遺伝子をクロラムフェニコール耐性プラスミドから切り出して、クロラムフェニコール耐性挿入物を生成するように、混合物中のCRISPR複合体を設計した(図5、第2のパネル参照)。同様に、末端のハイブリダイゼーションが生じて、カナマイシン及びクロラムフェニコール「デュアル耐性」プラスミドを生成することとなる適合する突出部が生じるように、各反応のカナマイシン耐性プラスミド骨格及びクロラムフェニコール耐性挿入物を設計した。
[0241] Cpf1及びガイドRNAを含む線状化耐性プラスミド混合物を、メーカーの推奨するCpf1バッファ中で摂氏37度にて3時間インキュベートした。選択した反応物をアガロースゲル上でランさせて、生じたフラグメントを、標準的なDNA抽出キット(Zymo Researchキット、メーカーの使用説明書に従って用いた)を用いて精製した。精製(対照)及び未精製(試験)。
[0242] カナマイシン耐性プラスミド骨格及びクロラムフェニコール耐性挿入物(それぞれ2つの適合するCpf1付着末端を含む)を含むDNAフラグメントを、T4 DNAリガーゼ(NEBから市販されている)を有する、又は有していない新しい反応内で組み合わせて、NEB10−B細胞(NEBから市販されている)に形質転換した。形質転換細胞を、機能耐性プラスミドを含有しないあらゆる細胞の増殖を妨げるように設計した、カナマイシン及びクロラムフェニコールの双方を補った培地上にプレーティングした。
[0243] 個々のコロニーを、配列決定のために送って、Cpf1クローニングの接合部を裏付けた。また、回収したコロニーを、表2に記載するプライマーを用いるPCRを介して確認した。図5は、先で記載した一般的な実験設計を示すが、プラスミドを、先で記載したように、Cpf1消化に先立って線状化した。
[0244] 本実験の結果を、表3及び図6に示す。各形質転換についての反応番号を、最上部の行に沿って示しており、用いたガイドRNAを、表3の左側の欄に沿って一覧にしている。リガーゼを有する、又は有していない同じCpf1反応の比較は、リガーゼ酵素の存在下で、形質転換体において9.9倍の増大を示した。このことは、コロニー成長が、Cpf1消化後のカナマイシン及びクロラムフェニコールの二重耐性プラスミドの形成に起因したことを示す。リガーゼなし反応は、反応が特異的であることを確立するために設計した対応対照であり、未消化の耐性プラスミドの汚染レベルの存在に単純に起因しなかった。
[0245] 個々の16コロニーをサンガー配列決定して、上流側及び下流側のクローニング接合部の双方を確認した。7つの上流側で配列決定した接合部のうち7つ、そして9つの下流側の接合部のうち8つにおいて、T4 DNAリガーゼによる反応由来のCpf1媒介クローンは、正確な消化及びライゲーションを示した。
[0246] 反応物71及び72を、DNAゲル精製工程にかけなかったCpf1消化プラスミドで形質転換した。しかしながら、Cpf1酵素を、T4 DNAリガーゼの付加前に、供給者の使用説明書に従って熱不活化した(反応72)。反応71及び72は、同じリガーゼ依存性を示した。
[0247] PCT/US2017/042245(国際公開第2018/013990号、米国仮特許出願第62/362,909号の優先権を主張)の開示は、その全体が本明細書に組み込まれる。
実施例3:MegaModular設計を用いる、制限酵素消化及びライゲーションによるプラスミドアセンブリ
[0248] 本実施例は、本開示の方法に従う、モジュラーCRISPRベクターの遺伝的編集を記載する。図15は、本実施例に記載するモジュラーCRISPRプラスミド13000444591の遺伝的編集を示す。第一に、以前に構築したプラスミドから「スタッファ」挿入DNA部分を除去することによって、プラスミド骨格を調製した。スタッファ部分の側面に位置するクローニングタグ(cTAG)D(配列番号68)及びE(配列番号69)を制限酵素ApaI及びPvuIで消化することによって、スタッファ挿入DNA部分を除去した。生じたフラグメントを、ゲル電気泳動を介して分離して、プラスミド骨格に対応する所望の8.3kbのフラグメントをゲルから切り出して、標準的なシリカ膜カラムを用いて抽出した。
[0248] 本実施例は、本開示の方法に従う、モジュラーCRISPRベクターの遺伝的編集を記載する。図15は、本実施例に記載するモジュラーCRISPRプラスミド13000444591の遺伝的編集を示す。第一に、以前に構築したプラスミドから「スタッファ」挿入DNA部分を除去することによって、プラスミド骨格を調製した。スタッファ部分の側面に位置するクローニングタグ(cTAG)D(配列番号68)及びE(配列番号69)を制限酵素ApaI及びPvuIで消化することによって、スタッファ挿入DNA部分を除去した。生じたフラグメントを、ゲル電気泳動を介して分離して、プラスミド骨格に対応する所望の8.3kbのフラグメントをゲルから切り出して、標準的なシリカ膜カラムを用いて抽出した。
[0249] モジュラーCRISPRベクター用の新しい挿入部を生成するために、cTAG D及びcTAG Eが側面に位置する所望の挿入DNA部分を、ユニバーサルcTAGオリゴtagD_FWD(配列番号75)及びtagE_REV(配列番号76)を用いてPCR増幅した。生じた挿入物は、cTAG D及びcTAG Eが側面に位置するGFPマーカー遺伝子を含有した。生じたPCRフラグメントを、cTAG D内を切るApaI酵素及びcTAG E配列内を切るPvuI酵素で消化した。消化した挿入DNA部分を、標準的なシリカ膜カラムを用いて精製した。
[0250] 精製したモジュラーCRISPRベクター骨格及び挿入DNA部分を、リガーゼにより単一の反応物に組み合わせて、環状プラスミドを生成した。生じた編集GFP含有プラスミド13000444591の配列を、配列番号77に記載する。
実施例4:MegaModular設計を用いる、酵母相同組換えによるプラスミドアセンブリ
[0251] MegaModularタグが側面に位置するPCRフラグメントの酵母相同組換えによって、プラスミド13000283399(配列番号85)をアセンブルした。アセンブリに所望されるコンストラクトを、特異的なMegaModularタグが側面に位置するように、PCRによって増幅した。当該タグにより、出芽酵母(Saccharomyces cerevisiae)におけるフラグメントの指向性アセンブリが可能となった。というのも、タグそれ自体が、相同組換え用の重なる相同領域として機能したからである。具体的には、5つのフラグメントを、以下のように、MegaModularタグが側面に位置するPCRを介して増幅した:タグA−フラグメント1−タグB;タグB−フラグメント2−タグC;タグC−フラグメント3−タグD;タグD−フラグメント4−タグE;及びタグE−フラグメント5−タグF。これらのフラグメントを、酵母の複製起点及びTRP栄養要求性選択マーカーを含有する線状化アセンブリベクター、並びに一方の端部のタグA及び他方のタグFと共に、出芽酵母(S. cerevisiae)に形質転換した。アセンブルした環状プラスミドを、トリプトファンを欠く培地中での出芽酵母(S. cerevisiae)増殖によって選択した。当該プラスミドを回収して、大腸菌中で増幅させて、正確な高次構造を配列決定によって裏付けた。
[0251] MegaModularタグが側面に位置するPCRフラグメントの酵母相同組換えによって、プラスミド13000283399(配列番号85)をアセンブルした。アセンブリに所望されるコンストラクトを、特異的なMegaModularタグが側面に位置するように、PCRによって増幅した。当該タグにより、出芽酵母(Saccharomyces cerevisiae)におけるフラグメントの指向性アセンブリが可能となった。というのも、タグそれ自体が、相同組換え用の重なる相同領域として機能したからである。具体的には、5つのフラグメントを、以下のように、MegaModularタグが側面に位置するPCRを介して増幅した:タグA−フラグメント1−タグB;タグB−フラグメント2−タグC;タグC−フラグメント3−タグD;タグD−フラグメント4−タグE;及びタグE−フラグメント5−タグF。これらのフラグメントを、酵母の複製起点及びTRP栄養要求性選択マーカーを含有する線状化アセンブリベクター、並びに一方の端部のタグA及び他方のタグFと共に、出芽酵母(S. cerevisiae)に形質転換した。アセンブルした環状プラスミドを、トリプトファンを欠く培地中での出芽酵母(S. cerevisiae)増殖によって選択した。当該プラスミドを回収して、大腸菌中で増幅させて、正確な高次構造を配列決定によって裏付けた。
参照による組込み
[0252] 本明細書中で引用される全ての参考文献、記事、刊行物、特許、特許公報、及び特許出願は、全ての目的について、その全体が参照によって組み込まれる。しかしながら、本明細書中で引用されるいずれの参考文献、記事、刊行物、特許、特許公報、及び特許出願への言及も、これらが有効な先行技術を構成するという示唆の容認でもいかなる形態でもないし、且つそうとられるべきでないし、世界中のいずれの国においても共通の一般的知識の一部を形成しないし、且つそうするべきでない。
[0252] 本明細書中で引用される全ての参考文献、記事、刊行物、特許、特許公報、及び特許出願は、全ての目的について、その全体が参照によって組み込まれる。しかしながら、本明細書中で引用されるいずれの参考文献、記事、刊行物、特許、特許公報、及び特許出願への言及も、これらが有効な先行技術を構成するという示唆の容認でもいかなる形態でもないし、且つそうとられるべきでないし、世界中のいずれの国においても共通の一般的知識の一部を形成しないし、且つそうするべきでない。
Claims (49)
- CRISPRマルチクローン部位を含む組換えモジュラーCRISPR DNAコンストラクトであって、前記マルチクローン部位は:
a)少なくとも2つの互いに異なるクローニングタグ(cTAG)であって、それぞれが:
i)1つ以上の確認されたCRISPRランディング部位であって、それぞれ、プロトスペーサ隣接モチーフ(PAM)に作動可能に連結されたプロトスペーサ配列を含み;前記確認されたCRISPRランディング部位の少なくとも1つが、前記モジュラーCRISPR DNAコンストラクト内で固有である、1つ以上の確認されたCRISPRランディング部位を含む、cTAGと;
b)1つ以上のDNA挿入部分であって、
i)前記互いに異なるcTAGのそれぞれが、前記1つ以上のDNA挿入部分のそれぞれの周りのフランキング位置内に分布している、1つ以上のDNA挿入部分
を含む、組換えモジュラーCRISPR DNAコンストラクト。 - 前記モジュラーCRISPR DNAコンストラクトは、環状である、請求項1に記載のモジュラーCRISPR DNAコンストラクト。
- 前記モジュラーCRISPR DNAコンストラクトは、直鎖状である、請求項1に記載のモジュラーCRISPR DNAコンストラクト。
- 前記モジュラーCRISPR DNAコンストラクトは、生物のゲノム中に組み込まれる、請求項1に記載のモジュラーCRISPR DNAコンストラクト。
- 前記互いに異なるcTAGの少なくとも1つが、少なくとも2つの確認されたCRISPRランディング部位を含む、請求項1〜4のいずれか一項に記載のモジュラーCRISPR DNAコンストラクト。
- 前記CRISPRランディング部位の少なくとも1つが、Cas9エンドヌクレアーゼ用である、請求項5に記載のモジュラーCRISPR DNAコンストラクト。
- 前記CRISPRランディング部位の少なくとも1つが、Cpf1エンドヌクレアーゼ用である、請求項6に記載のモジュラーCRISPR DNAコンストラクト。
- 前記互いに異なるcTAGの少なくとも1つが、稀な(≧8塩基長)制限酵素部位を含む、請求項1に記載のモジュラーCRISPR DNAコンストラクト。
- 組換え核酸分子を調製する方法であって:
a):
i)複数のDNA挿入部分であって、各DNA挿入部分の側面に、2つのクローニングタグ(cTAG)が位置しており、各cTAGが:
1)1つ以上の確認されたCRISPRランディング部位であって、それぞれ、プロトスペーサ隣接モチーフ(PAM)に作動可能に連結されたプロトスペーサ配列を含む、1つ以上の確認されたCRISPRランディング部位を含む、複数のDNA挿入部分と;
ii)複数のDNA挿入部分の少なくとも2つにおいて存在する前記cTAGの少なくとも1つを標的とする1つ以上のCRISPR複合体であって、各CRISPR複合体が:
1)CRISPRエンドヌクレアーゼ、及び
2)前記標的とされるcTAGの1つに前記CRISPRエンドヌクレアーゼを動員することができるガイドRNA
を含む、1つ以上のCRISPR複合体と;
を含む混合物を、
前記複数のDNA挿入部分の少なくとも2つにおける前記標的とされるcTAGの消化を可能にして、消化されたDNA末端を生じさせる条件下で
インキュベートすることと、
b)(a)において生じたcTAGが消化された前記DNA挿入部分を、前記消化されたDNA末端の共有結合を可能にする条件下でインキュベートすることと;
を含み、結果として生じた組換え核酸分子は、前記方法においてライゲーションされる元の挿入部分の完全なcTAG配列を含む、方法。 - 前記消化されたDNA末端は、前記消化された末端の共有結合に先立って互いにハイブリダイズすることができる突出配列を有する付着末端である、請求項9に記載の方法。
- 前記CRISPRエンドヌクレアーゼは、Cpf1である、請求項10に記載の方法。
- 前記消化されたDNA末端は、平滑末端である、請求項9に記載の方法。
- 前記平滑末端はさらに、ssDNAエキソヌクレアーゼで消化されて、突出配列を有する付着末端が生じ、工程b)はさらに、前記消化された末端の共有結合に先立って前記突出配列にハイブリダイズすることができる架橋DNA配列を加えることを含む、請求項12に記載の方法。
- 前記CRISPRエンドヌクレアーゼは、Cas9である、請求項9、12、又は13に記載の方法。
- 前記方法はさらに:
i)工程b)に先立って、前記CRISPR複合体から、前記消化されたcTAG配列を分離する工程、又は
ii)工程b)に先立って、前記CRISPR複合体を不活化する工程
を含む、請求項9〜13のいずれか一項に記載の方法。 - 前記分離工程は、DNA精製工程を含む、請求項15に記載の方法。
- 前記不活化工程は、前記CRISPR複合体の熱又は化学物質による不活化を含む、請求項15に記載の方法。
- 前記複数のDNA挿入部分のそれぞれの前記2つのcTAGは、cTAG対を形成し、前記cTAG対は、前記方法においてライゲーションされる前記DNA挿入部分の他の全てのcTAG対に対して固有である、請求項9〜13のいずれか一項に記載の方法。
- 各cTAG対における前記cTAGの少なくとも1つが、異なるcTAG対における少なくとも1つの他のcTAGと同じである、請求項18に記載の方法。
- DNA配列編集方法であって、前記方法は:
a)用意することであって、
i)請求項1に記載のモジュラーCRISPR DNAコンストラクトと;
ii)置換DNA挿入部分であって、前記置換DNA挿入部分は、第1及び第2の挿入cTAGが側面に位置しており;
1)前記第1の挿入cTAGは、前記モジュラーCRISPR DNAコンストラクトの前記互いに異なるcTAGの1つの前記確認されたCRISPRランディング部位を含み、そして前記第2の挿入cTAGは、前記モジュラーCRISPR DNAコンストラクトの別の異なるcTAGの前記確認されたCRISPRランディング部位を含む、置換DNA挿入部分と;
iii)前記第1及び第2の挿入cTAGをそれぞれ標的とする第1及び第2のCRISPR複合体と
を用意し、各CRISPR複合体が:
1)CRISPRエンドヌクレアーゼ、及び
2)前記標的とされる挿入cTAGの1つに前記CRISPRエンドヌクレアーゼを動員することができるガイドRNA;
を含み、
部分(i)及び(ii)がそれぞれ、部分(iii)と、単一又は別個の反応でインキュベートされ;前記第1及び第2のCRISPR複合体は、前記第1及び第2の挿入cTAG、並びにそれらの対応する互いに異なるcTAGを切断して、消化されたDNA末端を生じさせる、用意することと:
b)前記置換DNA挿入部分、及び工程(a)において生じたDNA末端が消化されたモジュラーCRISPR DNAコンストラクトを、前記消化されたDNA末端の共有結合を可能にする条件下でインキュベートすることと;を含み、結果として生じた編集されたモジュラーCRISPR DNAコンストラクトは、前記方法によって共有結合される元の挿入部分の完全なcTAG配列を含む、DNA配列編集方法。 - 工程(b)の前記反応は、機能的リガーゼを含む、請求項20に記載のDNA配列編集方法。
- 前記方法はさらに:
i)工程(b)に先立って、前記切断された第1及び第2の挿入cTAG、並びにそれらの対応する互いに異なるcTAGを前記CRISPR複合体から分離する工程、又は
ii)工程(b)に先立って前記CRISPR複合体を不活化する工程
を含む、請求項20に記載の方法。 - 前記分離工程は、DNA精製工程を含む、請求項22に記載の方法。
- 前記不活化工程は、前記CRISPR複合体の熱又は化学物質による不活化を含む、請求項22に記載の方法。
- 前記消化されたDNA末端は、前記消化された末端の共有結合に先立って互いにハイブリダイズすることができる突出配列を有する付着末端である、請求項20に記載の方法。
- 前記CRISPRエンドヌクレアーゼは、Cpf1である、請求項20に記載の方法。
- 前記消化されたDNA末端は、平滑末端である、請求項20に記載の方法。
- 前記平滑末端はさらに、ssDNAエキソヌクレアーゼで消化されて、突出配列を有する付着末端が生じ、工程b)はさらに、前記消化された末端の共有結合に先立って前記突出配列にハイブリダイズすることができる架橋DNA配列を加えることを含む、請求項27に記載の方法。
- 前記CRISPRエンドヌクレアーゼは、Cas9である、請求項20、27、又は28に記載の方法。
- DNA配列編集方法であって、前記方法は:
a)第1の反応用に:
i)請求項1に記載のモジュラーCRISPR DNAコンストラクト:
ii)前記モジュラーCRISPR DNAコンストラクト内の互いに異なるcTAGの少なくとも1つをそれぞれ標的とする少なくとも2つのCRISPR複合体
を用意することであって、各CRISPR複合体は:
1)CRISPRエンドヌクレアーゼ、及び
2)前記標的とされる互いに異なるcTAGの1つに前記CRISPRエンドヌクレアーゼを動員することができるガイドRNA;
を含み;
前記第1及び第2のCRISPR複合体は、前記モジュラーCRISPR DNAコンストラクト内の前記標的とされる互いに異なるcTAGを切断することによって、消化されたcTAGを生じさせる、用意することと、
b)第2の反応用に:
i)工程(a)において生じたcTAGが消化された前記モジュラーCRISPR DNAコンストラクト;及び
ii)置換DNA挿入部分であって、前記置換DNA挿入部分は、第1及び第2の挿入cTAGが側面に位置している、置換DNA挿入部分;
を用意することであって、
1)前記第1の挿入cTAGは、工程(a)において切断される前記未消化の互いに異なるcTAGの1つと同じ、又は実質的に同じである配列を含み、前記第2の挿入cTAGは、工程(a)において切断される前記未消化の互いに異なるcTAGのもう1つと同じ、又は実質的に同じである配列を含む;用意することとを
部分(i)の前記モジュラーCRISPR DNAコンストラクト及び部分(ii)の前記置換DNA挿入部分の前記結合を可能にして、編集されたモジュラーCRISPR DNAコンストラクトを形成させる条件下で行い;結果として生じた編集されたモジュラーCRISPR DNAコンストラクトは、工程(a)において標的とされる前記未消化の互いに異なるcTAGと同じ、又は実質的に同じである再構成されたcTAGを含む、方法。 - 工程(b)の前記結合は、ライゲーションであり、前記置換DNA挿入部分はまた、ライゲーションに先立って前記CRISPR複合体によって消化される、請求項30に記載のDNA配列編集方法。
- 前記CRISPRエンドヌクレアーゼは、Cpf1である、請求項30に記載のDNA配列編集方法。
- 工程(b)の前記結合は、相同組換えである、請求項30に記載のDNA配列編集方法。
- 前記CRISPRエンドヌクレアーゼは、Cas9である、請求項30又は33に記載のDNA配列編集方法。
- 前記第1及び第2の反応は、別々でない、請求項30に記載のDNA配列編集方法。
- 前記DNA挿入部分は、前記CRISPRエンドヌクレアーゼによって消化されない、請求項33又は35に記載のDNA配列編集方法。
- cTAGが消化された前記モジュラーCRISPR DNAコンストラクト及び前記未消化の置換DNA挿入部分は双方とも、一本鎖DNA(ssDNA)エキソヌクレアーゼで消化されることによって、互いにハイブリダイズすることができる適合する突出DNA末端を形成する、請求項33又は35に記載のDNA配列編集方法。
- 前記第2の反応はさらに、前記適合する突出DNA末端の共有結合に先立って、あらゆる配列ギャップ内でファイルすることができるポリメラーゼ又はリガーゼを含む、請求項37に記載のDNA配列編集方法。
- 前記挿入部分は、CRISPR消化を妨げるように化学的に修飾されている、請求項36に記載のDNA配列編集方法。
- 前記第1及び第2の挿入cTAGは、それらの確認されたCRISPRランディング部位内に、突然変異したPAM又は突然変異したプロトスペーサを含む、請求項36に記載のDNA配列編集方法。
- CRISPRマルチクローン部位を含む組換えモジュラーCRISPR DNAコンストラクトを含む宿主細胞ゲノムであって、前記マルチクローン部位は:
a)少なくとも2つの互いに異なるクローニングタグ(cTAG)であって、各cTAGは:
i)1つ以上の確認されたCRISPRランディング部位であって、それぞれ、プロトスペーサ隣接モチーフ(PAM)に作動可能に連結されたプロトスペーサ配列を含み;前記確認されたCRISPRランディング部位の少なくとも1つが、前記モジュラーCRISPR DNAコンストラクト内で固有である、CRISPRランディング部位を含む、cTAGと;
b)1つ以上のDNA挿入部分であって、
i)前記互いに異なるcTAGのそれぞれが、前記1つ以上のDNA挿入部分のそれぞれの周りのフランキング位置内に分布している、1つ以上のDNA挿入部分
を含む宿主細胞ゲノム。 - 組換え核酸分子を調製する方法であって、
a)インキュベートすることであって、
i)2つのクローニングタグ(cTAG)が側面に位置する複数のDNA挿入部分であって、各cTAGは:
1)1つ以上の確認されたCRISPRランディング部位であって、それぞれ、プロトスペーサ隣接モチーフ(PAM)に作動可能に連結されたプロトスペーサ配列を含む、CRISPRランディング部位;及び
2)稀な(≧8塩基)制限酵素認識部位であって、少なくとも2つの挿入部分の前記cTAGの少なくとも1つが、同じ制限酵素部位を含む稀な(≧8塩基)制限酵素認識部位を含む、複数のDNA挿入部分と;
ii)前記複数のDNA挿入部分の少なくとも2つにおける前記稀な制限酵素部位を標的とする1つ以上の制限酵素と:
を含む混合物を;
前記複数のDNA挿入部分の少なくとも2つにおける、前記1つ以上の制限酵素による前記標的とされるcTAGの消化を可能にして、DNA末端が消化された挿入部分を生じさせる条件下でインキュベートすることと;
b)工程(a)において生じたDNA末端が消化された前記DNA挿入部分を、前記消化されたDNA末端の共有結合を可能にする条件下でインキュベートすることであって、結果として生じた組換え核酸分子は、前記方法において共有結合される元の挿入部分の完全なcTAG配列を含む、インキュベートすることを含む方法。 - DNA配列編集方法であって、
a)用意することであって、
i)請求項1に記載のモジュラーCRISPR DNAコンストラクトであって、前記互いに異なるcTAGの少なくとも2つが、稀な(≧8塩基)制限酵素認識部位を含む、モジュラーCRISPR DNAコンストラクトと;
ii)置換DNA挿入部分であって、前記置換DNA挿入部分は、第1及び第2の挿入cTAGが側面に位置しており;
1)前記第1の挿入cTAGは、前記モジュラーCRISPR DNAコンストラクトの前記互いに異なるcTAGの1つの前記稀な制限酵素認識部位を含み、そして前記第2の挿入cTAGは、前記モジュラーCRISPR DNAコンストラクトの別の異なるcTAGの前記稀な制限酵素認識部位を含む、置換DNA挿入部分と;
iii)前記第1及び第2の挿入cTAG内の前記稀な制限酵素部位を標的とする1つ以上の制限酵素と
を用意し;
部分(i)及び(ii)がそれぞれ、部分(iii)と、単一又は別個の反応でインキュベートされ;前記1つ以上の制限酵素は、第1及び第2の挿入cTAG、並びにそれらの対応する互いに異なるcTAGの前記稀な制限酵素認識部位を切断して、消化されたDNA末端を生じさせる、用意することと:
b)前記置換DNA挿入部分、及び工程(a)において生じたDNA末端が消化された前記モジュラーCRISPR DNAコンストラクトを、前記消化されたDNA末端の共有結合を可能にする条件下でインキュベートすることであって、結果として生じた編集されたモジュラーCRISPR DNAコンストラクトは、前記方法によって共有結合される元の挿入部分の完全なcTAG配列を含む、インキュベートすることを含むDNA配列編集方法。 - 前記第1及び第2の挿入cTAGは、同じ稀な制限酵素部位を含む、請求項43に記載のDNA配列編集方法。
- 組換え核酸分子を調製する方法であって、
a)インキュベートすることであって、
i)複数のDNA挿入部分であって、各DNA挿入部分は、2つのクローニングタグ(cTAG)が側面に位置しており、各cTAGは:
1)1つ以上の確認されたCRISPRランディング部位であって、それぞれ、プロトスペーサ隣接モチーフ(PAM)に作動可能に連結されたプロトスペーサ配列を含み;前記DNA挿入部分の少なくとも2つが、同じcTAGを共有する、CRISPRランディング部位を含む、複数のDNA挿入部分;
ii)一本鎖DNA(ssDNA)エキソヌクレアーゼ:
を含む混合物を;
前記少なくとも2つのDNA挿入部分における前記共有されるcTAGの消化を可能にすることによって、前記少なくとも2つのDNA挿入部分内に適合する突出DNA末端を生じさせる条件下でインキュベートすることと、
b)(a)において生じたcTAGが消化された前記DNA挿入部分を、前記少なくとも2つのDNA挿入部分の前記適合する突出DNA末端のハイブリダイゼーション及び共有結合を可能にする条件下でインキュベートすることであって、結果として生じた組換え核酸分子は、消化前の前記共有されるcTAGの完全なcTAG配列を含む、インキュベートすることを含む組換え核酸分子を調製する方法。 - 工程(a)における前記混合物はさらに、前記適合する突出DNA末端の共有結合に先立って、あらゆる配列ギャップ内でファイルすることができるポリメラーゼを含む、請求項45に記載の組換え核酸分子を調製する方法。
- 前記複数のDNA挿入部分は、工程(a)の前に、二本鎖DNA(dsDNA)エキソヌクレアーゼで消化される、請求項45に記載の組換え核酸分子を調製する方法。
- 前記複数のDNA挿入部分の1つ以上が、工程(a)におけるインキュベーションに先立ってCRISPRエンドヌクレアーゼで切断された、請求項45に記載の組換え核酸分子を調製する方法。
- 前記cTAGの1つ以上が、配列番号65〜74、78〜81、及びそれらの組合せからなる群から選択される、請求項1〜49のいずれか一項に記載の挿入部分。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762457493P | 2017-02-10 | 2017-02-10 | |
| US62/457,493 | 2017-02-10 | ||
| PCT/US2018/017573 WO2018148511A1 (en) | 2017-02-10 | 2018-02-09 | A modular universal plasmid design strategy for the assembly and editing of multiple dna constructs for multiple hosts |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2020507312A true JP2020507312A (ja) | 2020-03-12 |
| JP2020507312A5 JP2020507312A5 (ja) | 2021-03-25 |
Family
ID=63107838
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019539756A Withdrawn JP2020507312A (ja) | 2017-02-10 | 2018-02-09 | 複数の宿主用の複数のdnaコンストラクトのアセンブリ及び編集のためのモジュラーユニバーサルプラスミド設計戦略 |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US11098305B2 (ja) |
| EP (1) | EP3580337A4 (ja) |
| JP (1) | JP2020507312A (ja) |
| KR (1) | KR20190116282A (ja) |
| CN (1) | CN110312797A (ja) |
| CA (1) | CA3049989A1 (ja) |
| WO (1) | WO2018148511A1 (ja) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA3049989A1 (en) | 2017-02-10 | 2018-08-16 | Zymergen Inc. | A modular universal plasmid design strategy for the assembly and editing of multiple dna constructs for multiple hosts |
| CA3107002A1 (en) * | 2018-08-15 | 2020-04-30 | Zymergen Inc. | Applications of crispri in high throughput metabolic engineering |
| KR102487901B1 (ko) | 2019-04-04 | 2023-01-12 | 리제너론 파마슈티칼스 인코포레이티드 | 표적화된 변형의 표적화 벡터로의 무흔적 도입을 위한 방법 |
| CN114555803A (zh) * | 2019-10-14 | 2022-05-27 | 延世大学校产学协力团 | 新的前间区序列邻近基序序列以及利用其修饰细胞基因组中靶核酸的方法 |
| KR102178813B1 (ko) | 2019-10-18 | 2020-11-13 | 주식회사 다우진유전자연구소 | Dna 구조체를 적용한 간이 키트 |
| BR112022009584A2 (pt) * | 2019-11-18 | 2022-10-04 | Shanghai Bluecross Medical Science Inst | Sistema de edição de genes derivado de flavobacterium |
| US20230357796A1 (en) * | 2019-11-27 | 2023-11-09 | Danmarks Tekniske Universitet | Constructs, compositions and methods thereof having improved genome editing efficiency and specificity |
| CN113373130B (zh) * | 2021-05-31 | 2023-12-22 | 复旦大学 | Cas12蛋白、含有Cas12蛋白的基因编辑系统及应用 |
| GB202117455D0 (en) * | 2021-12-02 | 2022-01-19 | Academisch Ziekenhuis Leiden | Method of editing nucleic acid |
| WO2023194537A1 (en) * | 2022-04-08 | 2023-10-12 | Glaxosmithkline Intellectual Property Development Limited | Novel processes for the production of polynucleotides including oligonucleotides |
Family Cites Families (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE69433010T2 (de) | 1993-04-12 | 2004-06-09 | Northwestern University, Evanston | Verfahren zur darstellung von oligonukleotiden |
| US5773258A (en) | 1995-08-25 | 1998-06-30 | Roche Molecular Systems, Inc. | Nucleic acid amplification using a reversibly inactivated thermostable enzyme |
| AU776820B2 (en) | 1998-10-30 | 2004-09-23 | Cornell Research Foundation Inc. | High fidelity thermostable ligase and uses thereof |
| US20120149115A1 (en) * | 2009-06-11 | 2012-06-14 | Snu R&Db Foundation | Targeted genomic rearrangements using site-specific nucleases |
| GB2481425A (en) * | 2010-06-23 | 2011-12-28 | Iti Scotland Ltd | Method and device for assembling polynucleic acid sequences |
| PE20190844A1 (es) | 2012-05-25 | 2019-06-17 | Emmanuelle Charpentier | Modulacion de transcripcion con arn de direccion a adn generico |
| EP3252160B1 (en) | 2012-12-12 | 2020-10-28 | The Broad Institute, Inc. | Crispr-cas component systems, methods and compositions for sequence manipulation |
| SG10201801969TA (en) | 2012-12-12 | 2018-04-27 | Broad Inst Inc | Engineering and Optimization of Improved Systems, Methods and Enzyme Compositions for Sequence Manipulation |
| US20140310830A1 (en) | 2012-12-12 | 2014-10-16 | Feng Zhang | CRISPR-Cas Nickase Systems, Methods And Compositions For Sequence Manipulation in Eukaryotes |
| US8993233B2 (en) | 2012-12-12 | 2015-03-31 | The Broad Institute Inc. | Engineering and optimization of systems, methods and compositions for sequence manipulation with functional domains |
| US20140242664A1 (en) | 2012-12-12 | 2014-08-28 | The Broad Institute, Inc. | Engineering of systems, methods and optimized guide compositions for sequence manipulation |
| US8697359B1 (en) | 2012-12-12 | 2014-04-15 | The Broad Institute, Inc. | CRISPR-Cas systems and methods for altering expression of gene products |
| JP6980380B2 (ja) * | 2013-03-15 | 2021-12-15 | ザ ジェネラル ホスピタル コーポレイション | 短縮ガイドRNA(tru−gRNA)を用いたRNA誘導型ゲノム編集の特異性の増大 |
| US9873907B2 (en) | 2013-05-29 | 2018-01-23 | Agilent Technologies, Inc. | Method for fragmenting genomic DNA using CAS9 |
| EP3417880A1 (en) | 2013-06-05 | 2018-12-26 | Duke University | Rna-guided gene editing and gene regulation |
| DK3011029T3 (da) * | 2013-06-17 | 2020-03-16 | Broad Inst Inc | Administration, modificering og optimering af tandem-guidesystemer, fremgangsmåder og sammensætninger til sekvensmanipulering |
| BR112015031608A2 (pt) * | 2013-06-17 | 2017-08-22 | Massachusetts Inst Technology | Aplicação e uso dos sistemas crispr-cas, vetores e composições para direcionamento e terapia hepáticos |
| US10287590B2 (en) | 2014-02-12 | 2019-05-14 | Dna2.0, Inc. | Methods for generating libraries with co-varying regions of polynuleotides for genome modification |
| HUE049405T2 (hu) * | 2014-06-23 | 2020-09-28 | Regeneron Pharma | Nukleáz-közvetített DNS-összeállítás |
| US20180002706A1 (en) * | 2014-12-30 | 2018-01-04 | University Of South Florida | Methods and compositions for cloning into large vectors |
| US9790490B2 (en) * | 2015-06-18 | 2017-10-17 | The Broad Institute Inc. | CRISPR enzymes and systems |
| WO2018013990A1 (en) | 2016-07-15 | 2018-01-18 | Zymergen Inc. | Scarless dna assembly and genome editing using crispr/cpf1 and dna ligase |
| WO2017037304A2 (en) * | 2016-07-28 | 2017-03-09 | Dsm Ip Assets B.V. | An assembly system for a eukaryotic cell |
| CA3049989A1 (en) | 2017-02-10 | 2018-08-16 | Zymergen Inc. | A modular universal plasmid design strategy for the assembly and editing of multiple dna constructs for multiple hosts |
| CA3107002A1 (en) | 2018-08-15 | 2020-04-30 | Zymergen Inc. | Applications of crispri in high throughput metabolic engineering |
-
2018
- 2018-02-09 CA CA3049989A patent/CA3049989A1/en active Pending
- 2018-02-09 KR KR1020197022014A patent/KR20190116282A/ko active Search and Examination
- 2018-02-09 CN CN201880012880.0A patent/CN110312797A/zh active Pending
- 2018-02-09 JP JP2019539756A patent/JP2020507312A/ja not_active Withdrawn
- 2018-02-09 EP EP18751852.7A patent/EP3580337A4/en not_active Withdrawn
- 2018-02-09 WO PCT/US2018/017573 patent/WO2018148511A1/en unknown
-
2019
- 2019-08-08 US US16/535,245 patent/US11098305B2/en active Active
-
2021
- 2021-08-03 US US17/392,366 patent/US20210371856A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| US11098305B2 (en) | 2021-08-24 |
| KR20190116282A (ko) | 2019-10-14 |
| EP3580337A1 (en) | 2019-12-18 |
| CA3049989A1 (en) | 2018-08-16 |
| WO2018148511A1 (en) | 2018-08-16 |
| EP3580337A4 (en) | 2020-12-02 |
| US20210371856A1 (en) | 2021-12-02 |
| US20200123540A1 (en) | 2020-04-23 |
| CN110312797A (zh) | 2019-10-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11130955B2 (en) | Applications of CRISPRi in high throughput metabolic engineering | |
| US11098305B2 (en) | Modular universal plasmid design strategy for the assembly and editing of multiple DNA constructs for multiple hosts | |
| US20190330659A1 (en) | Scarless dna assembly and genome editing using crispr/cpf1 and dna ligase | |
| US20230272394A1 (en) | RNA-DIRECTED DNA CLEAVAGE BY THE Cas9-crRNA COMPLEX | |
| US20210355465A1 (en) | Engineered CRISPR-Cas9 Nucleases | |
| US10633642B2 (en) | Engineered CRISPR-Cas9 nucleases | |
| JP2022122919A (ja) | プログラム可能cas9-リコンビナーゼ融合タンパク質およびその使用 | |
| US20230074594A1 (en) | Genome editing using crispr in corynebacterium | |
| EP3778899A1 (en) | Rna-directed dna cleavage and gene editing by cas9 enzyme from neisseria meningitidis | |
| CN108064279B (zh) | 噬菌体λ整合酶的突变体 | |
| CA3192224A1 (en) | Base editing enzymes | |
| AU2022342157A1 (en) | Class ii, type v crispr systems | |
| US20230220424A1 (en) | Rapid removal of a self-replicating fungal plasmid for efficient marker cycling | |
| EP2848691B1 (en) | Broad host range expression vector for diverse prokaryotes | |
| AU2021333586A9 (en) | Systems and methods for transposing cargo nucleotide sequences | |
| AU2002325588B2 (en) | A composition comprising nucleic acids | |
| Belle | Marker exclusion by bacteriophage T4 is mediated by site-specific non-intron encoded homing endonucleases | |
| AU2007202518A1 (en) | Methods and compositions utilizing nucleic acids |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210208 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20210208 |
|
| A761 | Written withdrawal of application |
Free format text: JAPANESE INTERMEDIATE CODE: A761 Effective date: 20211101 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20211101 |