JP2017196544A - Separation material and column - Google Patents
Separation material and column Download PDFInfo
- Publication number
- JP2017196544A JP2017196544A JP2016086980A JP2016086980A JP2017196544A JP 2017196544 A JP2017196544 A JP 2017196544A JP 2016086980 A JP2016086980 A JP 2016086980A JP 2016086980 A JP2016086980 A JP 2016086980A JP 2017196544 A JP2017196544 A JP 2017196544A
- Authority
- JP
- Japan
- Prior art keywords
- group
- functional group
- separation material
- hydrophobic polymer
- polymer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Abstract
Description
本発明は、分離材及びカラムに関する。 The present invention relates to a separation material and a column.
従来、タンパク質に代表される生体高分子を分離精製する場合、一般的には、多孔質型の合成高分子を母体とするイオン交換体、親水性天然高分子の架橋ゲルを母体とするイオン交換体等が用いられている。上記の多孔質型の合成高分子を母体とするイオン交換体の場合、塩濃度による体積変化が小さいため、カラムに充填してクロマトグラフィーで用いた場合、通液時の耐圧性に優れる傾向がある。しかし、このイオン交換体は、タンパク質等の分離に用いた場合、疎水的相互作用に基づく不可逆吸着等の非特異吸着が起こるため、ピークの非対称化が発生する、又は該疎水的相互作用でイオン交換体に吸着されたタンパク質が吸着されたまま回収できないという問題点がある。 Conventionally, when separating and purifying biopolymers typified by proteins, generally, ion exchangers based on porous synthetic polymers and ion exchanges based on cross-linked gels of hydrophilic natural polymers are generally used. The body is used. In the case of an ion exchanger based on the above porous synthetic polymer, the volume change due to the salt concentration is small, so when packed in a column and used for chromatography, the pressure resistance during liquid passage tends to be excellent. is there. However, when this ion exchanger is used for separation of proteins, etc., nonspecific adsorption such as irreversible adsorption based on hydrophobic interaction occurs, so that peak asymmetries occur or ions are generated by the hydrophobic interaction. There is a problem that the protein adsorbed on the exchanger cannot be recovered while adsorbed.
一方、デキストラン、アガロース等の多糖に代表される親水性天然高分子の架橋ゲルを母体とするイオン交換体の場合、タンパク質の非特異吸着が殆どないという利点がある。ところが、このイオン交換体は、水溶液中で著しく膨潤し、溶液のイオン強度による体積変化、及び遊離酸形と負荷形との体積変化が大きく、機械的強度も十分ではないという欠点を有する。特に、架橋ゲルをクロマトグラフィーで使用する場合、通液時の圧力損失が大きく、通液によりゲルが圧密化するといった欠点がある。 On the other hand, an ion exchanger based on a cross-linked gel of a hydrophilic natural polymer typified by polysaccharides such as dextran and agarose has an advantage that there is almost no non-specific adsorption of protein. However, this ion exchanger swells remarkably in an aqueous solution, and has the disadvantage that the volume change due to the ionic strength of the solution, the volume change between the free acid form and the loaded form is large, and the mechanical strength is not sufficient. In particular, when a cross-linked gel is used in chromatography, there is a disadvantage that the pressure loss during liquid passage is large and the gel is consolidated by liquid passage.
親水性天然高分子の架橋ゲルが持つ欠点を克服するため、これをいわば「骨格」となる剛直な物質と組み合わせる試みがこれまでになされている(例えば、特許文献1〜7)。 In order to overcome the disadvantages of the hydrophilic natural polymer cross-linked gel, attempts have been made so far to combine it with a rigid substance that becomes a “skeleton” (for example, Patent Documents 1 to 7).
しかしながら、従来の分離材は、タンパク質の非特異吸着が多く、かつカラムに充填したときの通液性に劣るという問題がある。 However, the conventional separation material has many problems such as non-specific adsorption of proteins and poor liquid permeability when packed in a column.
そこで、本発明は、タンパク質の非特異吸着が低減され、カラムに充填したときの通液性に優れる分離材を提供することを目的とする。 Therefore, an object of the present invention is to provide a separation material that reduces nonspecific adsorption of proteins and has excellent liquid permeability when packed in a column.
本発明の具体的態様を以下に示す。
[1] 疎水性高分子粒子と、該疎水性高分子粒子の表面の少なくとも一部を被覆する被覆層と、を備え、上記被覆層が、水酸基及び水酸基以外の官能基を有する官能基含有高分子を含む、分離材。
[2] 上記水酸基以外の官能基が、カルボキシ基、エポキシ基、又はアミノ基である、[1]に記載の分離材。
[3] 水中での5%圧縮変形弾性率が70MPa以上である、[1]又は[2]に記載の分離材。
[4] カラムに充填した場合、カラム圧0.3MPaのときに通液速度が500cm/h以上である、[1]〜[3]のいずれかに記載の分離材。
[5] 上記疎水性高分子粒子が、スチレン系モノマに由来する構造単位を有する疎水性高分子を含む、[1]〜[4]のいずれかに記載の分離材。
[6] 上記官能基含有高分子が、多糖類又はその変性体に由来する構造を含む、[1]〜[5]のいずれかに記載の分離材。
[7] 上記官能基含有高分子が、アガロース又はその変性体に由来する構造を含む、[1]〜[6]のいずれかに記載の分離材。
[8] 上記官能基含有高分子が、架橋されている、[1]〜[7]のいずれかに記載の分離材。
[9] 上記疎水性高分子粒子の平均粒径が10〜500μmである、[1]〜[8]のいずれかに記載の分離材。
[10] 上記疎水性高分子粒子が多孔構造を有する、[1]〜[9]のいずれかに記載の分離材。
[11] [1]〜[10]のいずれかに記載の分離材を備えるカラム。
Specific embodiments of the present invention are shown below.
[1] A hydrophobic polymer particle and a coating layer that covers at least a part of the surface of the hydrophobic polymer particle, wherein the coating layer has a functional group-containing polymer having a hydroxyl group and a functional group other than a hydroxyl group. Separation material containing molecules.
[2] The separating material according to [1], wherein the functional group other than the hydroxyl group is a carboxy group, an epoxy group, or an amino group.
[3] The separation material according to [1] or [2], wherein a 5% compressive deformation elastic modulus in water is 70 MPa or more.
[4] The separation material according to any one of [1] to [3], wherein when the column is packed, the liquid flow rate is 500 cm / h or more when the column pressure is 0.3 MPa.
[5] The separating material according to any one of [1] to [4], wherein the hydrophobic polymer particles include a hydrophobic polymer having a structural unit derived from a styrene monomer.
[6] The separating material according to any one of [1] to [5], wherein the functional group-containing polymer includes a structure derived from a polysaccharide or a modified product thereof.
[7] The separating material according to any one of [1] to [6], wherein the functional group-containing polymer includes a structure derived from agarose or a modified product thereof.
[8] The separating material according to any one of [1] to [7], wherein the functional group-containing polymer is crosslinked.
[9] The separation material according to any one of [1] to [8], wherein the hydrophobic polymer particles have an average particle size of 10 to 500 μm.
[10] The separation material according to any one of [1] to [9], wherein the hydrophobic polymer particles have a porous structure.
[11] A column comprising the separation material according to any one of [1] to [10].
本発明によれば、タンパク質の非特異吸着が低減され、カラムに充填したときの通液性に優れる分離材及び当該分離材を備えるカラムを提供することができる。 ADVANTAGE OF THE INVENTION According to this invention, the nonspecific adsorption | suction of protein is reduced and a column provided with the separation material which is excellent in liquid permeability when it fills a column, and the said separation material can be provided.
以下、本発明の好適な実施形態について説明をするが、本発明はこれらの実施形態に何ら限定されるものではない。 Hereinafter, preferred embodiments of the present invention will be described, but the present invention is not limited to these embodiments.
<分離材>
本実施形態の分離材は、疎水性高分子粒子と、該疎水性高分子粒子の表面の少なくとも一部を被覆する被覆層と、を備え、上記被覆層が、水酸基及び水酸基以外の官能基を有する官能基含有高分子を含む。なお、例えば、疎水性高分子粒子が多孔構造を有するような態様である場合において、「疎水性高分子粒子の表面」とは、疎水性高分子粒子の外側の表面のみでなく、疎水性高分子粒子の内部における細孔の表面を含むものとする。本実施形態の分離材によれば、タンパク質の非特異吸着が低減されると共に、カラムに充填したときに優れた通液性を有する。また、本実施形態の分離材は、耐久性、耐アルカリ性及び耐圧性にも優れ、カラムに充填したときの吸着量(動的吸着量)も実用上充分に高いと考えられる。
<Separation material>
The separation material of this embodiment includes hydrophobic polymer particles and a coating layer that covers at least a part of the surface of the hydrophobic polymer particles, and the coating layer has hydroxyl groups and functional groups other than hydroxyl groups. A functional group-containing polymer. For example, in the case where the hydrophobic polymer particles have a porous structure, the “surface of the hydrophobic polymer particles” means not only the outer surface of the hydrophobic polymer particles but also the hydrophobic polymer particles. It shall include the surface of the pores inside the molecular particle. According to the separation material of the present embodiment, non-specific adsorption of proteins is reduced, and excellent liquid permeability is obtained when packed in a column. In addition, the separation material of this embodiment is excellent in durability, alkali resistance, and pressure resistance, and the adsorption amount (dynamic adsorption amount) when packed in a column is considered to be sufficiently high in practice.
本実施形態の分離材が上述の効果を発揮する理由を、本発明者らは、以下のように推測している。 The present inventors presume the reason why the separating material of the present embodiment exhibits the above-described effects as follows.
本実施形態の分離材は、上記疎水性高分子粒子と、上記被覆層とを備えることにより、疎水性高分子粒子及び上記官能基含有高分子の双方の利点を有すると考えられる。例えば、本実施形態の分離材は、上記疎水性高分子粒子を備えることにより、耐久性、耐アルカリ性及び耐圧性に優れ、かつカラムに充填したときに優れた通液性を発揮すると考えられる。また、本実施形態の分離材は、上記被覆層を備えることにより、タンパク質の非特異吸着が低減されると共に、タンパク質等の吸脱着が起こり易く、同一流速下でのタンパク質等の吸着量(動的吸着量)が大きいと考えられる。 The separation material of this embodiment is considered to have the advantages of both the hydrophobic polymer particles and the functional group-containing polymer by including the hydrophobic polymer particles and the coating layer. For example, it is considered that the separation material of this embodiment is excellent in durability, alkali resistance and pressure resistance by providing the hydrophobic polymer particles, and exhibits excellent liquid permeability when packed in a column. In addition, since the separation material of the present embodiment includes the coating layer, nonspecific adsorption of proteins is reduced, and adsorption and desorption of proteins and the like are likely to occur. It is considered that the amount of adsorption is large.
(疎水性高分子粒子)
本実施形態に係る疎水性高分子粒子は、疎水性を有する高分子(以下、場合により「疎水性高分子」という)を含む粒子である。疎水性高分子粒子の製造方法に特に制限はないが、例えば、疎水性を有する高分子を形成可能なモノマを重合させる方法が挙げられる。モノマとしては、疎水性を有する高分子を形成可能なものであれば、特に限定されないが、例えば、スチレン系モノマが挙げられる。すなわち、上記疎水性高分子粒子は、例えば、スチレン系モノマに由来する構造単位を有する疎水性高分子を含んでいてもよい。
(Hydrophobic polymer particles)
The hydrophobic polymer particles according to the present embodiment are particles containing a polymer having hydrophobicity (hereinafter sometimes referred to as “hydrophobic polymer”). Although there is no restriction | limiting in particular in the manufacturing method of hydrophobic polymer particle, For example, the method of polymerizing the monomer which can form the polymer which has hydrophobicity is mentioned. The monomer is not particularly limited as long as it can form a hydrophobic polymer, and examples thereof include a styrene monomer. That is, the hydrophobic polymer particles may include, for example, a hydrophobic polymer having a structural unit derived from a styrene monomer.
本実施形態に係る疎水性高分子粒子は、多孔構造を有していてもよい。すなわち、本実施形態に係る疎水性高分子粒子は、例えば、疎水性の多孔質高分子粒子であってもよい。多孔質高分子粒子は、例えば、多孔質化剤の存在下でモノマを重合させて得られるポリマを含む粒子であり、従来の懸濁重合、乳化重合等により合成することができる。 The hydrophobic polymer particles according to the present embodiment may have a porous structure. That is, the hydrophobic polymer particles according to the present embodiment may be, for example, hydrophobic porous polymer particles. The porous polymer particle is, for example, a particle containing a polymer obtained by polymerizing a monomer in the presence of a porosifying agent, and can be synthesized by conventional suspension polymerization, emulsion polymerization, or the like.
スチレン系モノマとしては、以下のような多官能性モノマ、単官能性モノマ等が挙げられる。 Examples of the styrenic monomer include the following polyfunctional monomers and monofunctional monomers.
多官能性モノマとしては、例えば、ジビニルベンゼン、ジビニルビフェニル、ジビニルナフタレン、ジビニルフェナントレン等のジビニル化合物が挙げられる。これらの多官能性モノマは、1種を単独で又は2種以上を組み合わせて用いることができる。上記の中でも耐久性、耐酸性、耐アルカリ性に優れる観点から、ジビニルベンゼンが好ましい。 Examples of the polyfunctional monomer include divinyl compounds such as divinylbenzene, divinylbiphenyl, divinylnaphthalene, and divinylphenanthrene. These polyfunctional monomers can be used alone or in combination of two or more. Among these, divinylbenzene is preferable from the viewpoint of excellent durability, acid resistance, and alkali resistance.
単官能性モノマとしては、例えば、スチレン、o−メチルスチレン、m−メチルスチレン、p−メチルスチレン、α−メチルスチレン、o−エチルスチレン、m−エチルスチレン、p−エチルスチレン、2,4−ジメチルスチレン、p−n−ブチルスチレン、p−t−ブチルスチレン、p−n−ヘキシルスチレン、p−n−オクチルスチレン、p−n−ノニルスチレン、p−n−デシルスチレン、p−n−ドデシルスチレン、p−メトキシスチレン、p−フェニルスチレン、p−クロロスチレン、3,4−ジクロロスチレン等のスチレン及びその誘導体が挙げられる。これらの単官能性モノマは、1種を単独で又は2種以上を組み合わせて用いることができる。上記の中でも、耐酸性及び耐アルカリ性に優れる観点から、スチレンが好ましい。モノマは、カルボキシ基、アミノ基、水酸基、アルデヒド基等の官能基を有するスチレン誘導体であってもよい。 Examples of the monofunctional monomer include styrene, o-methylstyrene, m-methylstyrene, p-methylstyrene, α-methylstyrene, o-ethylstyrene, m-ethylstyrene, p-ethylstyrene, 2,4- Dimethyl styrene, pn-butyl styrene, pt-butyl styrene, pn-hexyl styrene, pn-octyl styrene, pn-nonyl styrene, pn-decyl styrene, pn-dodecyl Examples thereof include styrene such as styrene, p-methoxystyrene, p-phenylstyrene, p-chlorostyrene, and 3,4-dichlorostyrene and derivatives thereof. These monofunctional monomers can be used alone or in combination of two or more. Among these, styrene is preferable from the viewpoint of excellent acid resistance and alkali resistance. The monomer may be a styrene derivative having a functional group such as a carboxy group, an amino group, a hydroxyl group, and an aldehyde group.
多孔質化剤としては、重合時に相分離を促し、粒子の多孔質化を促進する有機溶媒である脂肪族又は芳香族の炭化水素類、エステル類、ケトン類、エーテル類、アルコール類等が挙げられる。多孔質化剤の具体例は、トルエン、キシレン、シクロヘキサン、オクタン、酢酸ブチル、フタル酸ジブチル、メチルエチルケトン、ジブチルエーテル、1−ヘキサノール、2−オクタノール、デカノール、ラウリルアルコール、シクロヘキサノール、ジエチルベンゼン等を含む。これらの多孔質化材は、1種を単独で又は2種以上を組み合わせて用いることができる。 Examples of the porosifying agent include aliphatic or aromatic hydrocarbons, esters, ketones, ethers, alcohols, and the like, which are organic solvents that promote phase separation at the time of polymerization and promote pore formation of particles. It is done. Specific examples of the porosifying agent include toluene, xylene, cyclohexane, octane, butyl acetate, dibutyl phthalate, methyl ethyl ketone, dibutyl ether, 1-hexanol, 2-octanol, decanol, lauryl alcohol, cyclohexanol, diethylbenzene and the like. These porous materials can be used alone or in combination of two or more.
上記多孔質化剤の配合量は、モノマ全質量に対して、例えば、0〜200質量%であってもよい。多孔質化剤の量によって、疎水性高分子粒子の空隙率をコントロールできる。さらに、多孔質化剤の種類によって、疎水性高分子粒子の細孔の大きさ及び形状をコントロールすることができる。 The amount of the porosifying agent may be, for example, 0 to 200% by mass with respect to the total mass of the monomer. The porosity of the hydrophobic polymer particles can be controlled by the amount of the porous agent. Furthermore, the size and shape of the pores of the hydrophobic polymer particles can be controlled by the type of the porous agent.
溶媒として使用する水を多孔質化剤とすることもできる。水を多孔質化剤とする場合は、モノマに油溶性界面活性剤を溶解させ、水を吸収することによって、粒子を多孔質化することが可能である。 Water used as a solvent can also be used as a porosifying agent. When water is used as the porosifying agent, it is possible to make the particles porous by dissolving the oil-soluble surfactant in the monomer and absorbing water.
多孔質化に使用される油溶性界面活性剤としては、分岐C16〜C24脂肪酸、鎖状不飽和C16〜C22脂肪酸又は鎖状飽和C12〜C14脂肪酸のソルビタンモノエステル、例えば、ソルビタンモノラウレート、ソルビタンモノオレエート、ソルビタンモノミリステート又はヤシ脂肪酸から誘導されるソルビタンモノエステル;分岐C16〜C24脂肪酸、鎖状不飽和C16〜C22脂肪酸又は鎖状飽和C12〜C14脂肪酸のジグリセロールモノエステル、例えば、ジグリセロールモノオレエート(例えば、C18:1(炭素数18個、二重結合数1個)脂肪酸のジグリセロールモノエステル)、ジグリセロールモノミリステート、ジグリセロールモノイソステアレート又はヤシ脂肪酸のジグリセロールモノエステル;分岐C16〜C24アルコール(例えば、ゲルベアルコール)、鎖状不飽和C16〜C22アルコール又は鎖状飽和C12〜C14アルコール(例えば、ヤシ脂肪アルコール)のジグリセロールモノ脂肪族エーテル;及びこれらの混合物が挙げられる。 Examples of the oil-soluble surfactant used for the porous formation include sorbitan monoesters of branched C16 to C24 fatty acids, chain unsaturated C16 to C22 fatty acids or chain saturated C12 to C14 fatty acids, such as sorbitan monolaurate, sorbitan Sorbitan monoesters derived from monooleate, sorbitan monomyristate or coconut fatty acid; diglycerol monoesters of branched C16-C24 fatty acids, chain unsaturated C16-C22 fatty acids or chain saturated C12-C14 fatty acids, for example di- Glycerol monooleate (for example, diglycerol monoester of C18: 1 (18 carbon atoms, 1 double bond) fatty acid), diglycerol monomyristate, diglycerol monoisostearate or diglycerol monoester of coconut fatty acid Ester; Branch C16-C 4 alcohols (e.g., Guerbet alcohols), linear unsaturated C16~C22 alcohol or chain saturated C12~C14 alcohols (e.g., coconut fatty alcohols) diglycerol mono-fatty ethers; and mixtures thereof.
これらのうち、ソルビタンモノラウレート(例えば、SPAN(スパン、登録商標)20、好ましくは純度約40%を超える、より好ましくは純度約50%を超える、更に好ましくは純度約70%を超えるソルビタンモノラウレート);ソルビタンモノオレエート(例えば、SPAN(スパン、登録商標)80、好ましくは純度約40%、より好ましくは純度約50%、更に好ましくは純度約70%を超えるソルビタンモノオレエート);ジグリセロールモノオレエート(例えば、純度約40%を超える、より好ましくは純度約50%を超える、更に好ましくは純度約70%を超えるジグリセロールモノオレエート);ジグリセロールモノイソステアレート(例えば、好ましくは純度約40%を超える、より好ましくは純度約50%を超える、更に好ましくは純度約70%を超えるジグリセロールモノイソステアレート);ジグリセロールモノミリステート(好ましくは純度約40%を超える、より好ましくは純度約50%を超える、更に好ましくは純度約70%を超えるソルビタンモノミリステート);ジグリセロールのココイル(例えば、ラウリル基、ミリストイル基等)エーテル;及びこれらの混合物が好ましい。 Of these, sorbitan monolaurate (eg, SPAN 20), preferably greater than about 40% purity, more preferably greater than about 50% purity, and even more preferably greater than about 70% purity. Sorbitan monooleate (eg, SPAN 80, preferably about 40% purity, more preferably about 50% purity, more preferably more than about 70% purity sorbitan monooleate); Diglycerol monooleate (eg, greater than about 40% purity, more preferably greater than about 50% purity, more preferably greater than about 70% purity); diglycerol monoisostearate (eg, Preferably greater than about 40% purity, more preferably greater than about 50% purity More preferably a diglycerol monoisostearate of greater than about 70% purity); diglycerol monomyristate (preferably greater than about 40% purity, more preferably greater than about 50% purity, even more preferably about 70% purity). More preferred sorbitan monomyristate); cocoyl (eg, lauryl, myristoyl, etc.) ethers of diglycerol; and mixtures thereof.
これらの油溶性界面活性剤は、モノマ全質量に対して、5〜80質量%の範囲で用いることが好ましい。油溶性界面活性剤の含有量が5質量%以上であると、水滴の安定性が充分となることから、大きな単一孔を形成し易くなる。また、油溶性界面活性剤の含有量が80質量%以下であると、重合後に疎水性高分子粒子が形状をより保持し易くなる。 These oil-soluble surfactants are preferably used in the range of 5 to 80% by mass with respect to the total mass of the monomer. When the content of the oil-soluble surfactant is 5% by mass or more, the stability of the water droplets becomes sufficient, so that a large single hole is easily formed. Further, when the content of the oil-soluble surfactant is 80% by mass or less, it becomes easier for the hydrophobic polymer particles to retain the shape after polymerization.
上記重合反応には、水性媒体を用いることもできる。上記水性媒体としては、水、水と水溶性溶媒(例えば、低級アルコール)との混合媒体等が挙げられる。水性媒体には、界面活性剤が含まれていてもよい。界面活性剤としては、アニオン系、カチオン系、ノニオン系及び両性イオン系の界面活性剤のうち、いずれも用いることができる。 An aqueous medium can also be used for the polymerization reaction. Examples of the aqueous medium include water, a mixed medium of water and a water-soluble solvent (for example, lower alcohol), and the like. The aqueous medium may contain a surfactant. As the surfactant, any of anionic, cationic, nonionic and zwitterionic surfactants can be used.
アニオン系界面活性剤としては、例えば、オレイン酸ナトリウム、ヒマシ油カリ等の脂肪酸油、ラウリル硫酸ナトリウム、ラウリル硫酸アンモニウム等のアルキル硫酸エステル塩、ドデシルベンゼンスルホン酸ナトリウム等のアルキルベンゼンスルホン酸塩、アルキルナフタレンスルホン酸塩、アルカンスルホン酸塩、ジオクチルスルホコハク酸ナトリウム等のジアルキルスルホコハク酸塩、アルケルニルコハク酸塩(ジカリウム塩)、アルキルリン酸エステル塩、ナフタレンスルホン酸ホルマリン縮合物、ポリオキシエチレンアルキルフェニルエーテル硫酸エステル塩、ポリオキシエチレンラウリルエーテル硫酸ナトリウム等のポリオキシエチレンアルキルエーテル硫酸塩及びポリオキシエチレンアルキル硫酸エステル塩が挙げられる。 Examples of the anionic surfactant include fatty acid oils such as sodium oleate and castor oil potassium, alkyl sulfate salts such as sodium lauryl sulfate and ammonium lauryl sulfate, alkylbenzene sulfonates such as sodium dodecylbenzenesulfonate, and alkylnaphthalene sulfone. Acid salts, alkane sulfonates, dialkyl sulfosuccinates such as sodium dioctyl sulfosuccinate, alkenyl succinates (dipotassium salts), alkyl phosphate esters, naphthalene sulfonate formalin condensates, polyoxyethylene alkylphenyl ether sulfates Salts, polyoxyethylene alkyl ether sulfates such as sodium polyoxyethylene lauryl ether sulfate, and polyoxyethylene alkyl sulfate salts.
カチオン系界面活性剤としては、例えば、ラウリルアミンアセテート、ステアリルアミンアセテート等のアルキルアミン塩、及びラウリルトリメチルアンモニウムクロライド等の第四級アンモニウム塩が挙げられる。 Examples of the cationic surfactant include alkylamine salts such as laurylamine acetate and stearylamine acetate, and quaternary ammonium salts such as lauryltrimethylammonium chloride.
ノニオン系界面活性剤としては、例えば、ポリエチレングリコールアルキルエーテル類、ポリエチレングリコールアルキルアリールエーテル類、ポリエチレングリコールエステル類、ポリエチレングリコールソルビタンエステル類、ポリアルキレングリコールアルキルアミン又はアミド類等の炭化水素系ノニオン界面活性剤、シリコンのポリエチレンオキサイド付加物類、ポリプロピレンオキサイド付加物類等のポリエーテル変性シリコン系ノニオン界面活性剤、及びパーフルオロアルキルグリコール類等のフッ素系ノニオン界面活性剤が挙げられる。 Nonionic surfactants include, for example, hydrocarbon nonionic surfactants such as polyethylene glycol alkyl ethers, polyethylene glycol alkyl aryl ethers, polyethylene glycol esters, polyethylene glycol sorbitan esters, polyalkylene glycol alkylamines, or amides. Agents, polyether-modified silicon nonionic surfactants such as silicon polyethylene oxide adducts and polypropylene oxide adducts, and fluorine nonionic surfactants such as perfluoroalkyl glycols.
両性イオン系界面活性剤としては、例えば、ラウリルジメチルアミンオキサイド等の炭化水素界面活性剤、リン酸エステル系界面活性剤、及び亜リン酸エステル系界面活性剤が挙げられる。 Examples of zwitterionic surfactants include hydrocarbon surfactants such as lauryl dimethylamine oxide, phosphate ester surfactants, and phosphite ester surfactants.
界面活性剤は、1種を単独で又は2種以上を組み合わせて用いてもよい。上記界面活性剤の中でも、モノマ重合時の分散安定性の観点から、アニオン系界面活性剤が好ましい。 One surfactant may be used alone, or two or more surfactants may be used in combination. Among the surfactants, anionic surfactants are preferable from the viewpoint of dispersion stability during monomer polymerization.
上記重合反応においては、必要に応じて、重合開始剤を添加することもできる。上記重合開始剤としては、例えば、過酸化ベンゾイル、過酸化ラウロイル、オルソクロロ過酸化ベンゾイル、オルソメトキシ過酸化ベンゾイル、3,5,5−トリメチルヘキサノイルパーオキサイド、tert−ブチルパーオキシ−2−エチルヘキサノエート、ジ−tert−ブチルパーオキサイド等の有機過酸化物;及び2,2’−アゾビスイソブチロニトリル、1,1’−アゾビスシクロヘキサンカルボニトリル、2,2’−アゾビス(2,4−ジメチルバレロニトリル)等のアゾ系化合物が挙げられる。重合開始剤は、モノマ100質量部に対して、例えば、0.1〜7.0質量部の範囲で使用することができる。 In the said polymerization reaction, a polymerization initiator can also be added as needed. Examples of the polymerization initiator include benzoyl peroxide, lauroyl peroxide, orthochlorobenzoyl peroxide, orthomethoxybenzoyl peroxide, 3,5,5-trimethylhexanoyl peroxide, tert-butylperoxy-2-ethylhexa Organic peroxides such as noate and di-tert-butyl peroxide; and 2,2′-azobisisobutyronitrile, 1,1′-azobiscyclohexanecarbonitrile, 2,2′-azobis (2, Azo compounds such as 4-dimethylvaleronitrile). A polymerization initiator can be used in 0.1-7.0 mass parts with respect to 100 mass parts of monomers, for example.
重合温度は、モノマ及び重合開始剤の種類に応じて、適宜選択することができる。重合温度は、25〜110℃が好ましく、50〜100℃がより好ましい。 The polymerization temperature can be appropriately selected according to the type of monomer and polymerization initiator. The polymerization temperature is preferably 25 to 110 ° C, more preferably 50 to 100 ° C.
疎水性高分子粒子の合成において、粒子の分散安定性を向上させるために、高分子分散安定剤を用いてもよい。 In the synthesis of hydrophobic polymer particles, a polymer dispersion stabilizer may be used in order to improve the dispersion stability of the particles.
高分子分散安定剤としては、例えば、ポリビニルアルコール、ポリカルボン酸、セルロース類(ヒドロキシエチルセルロース、カルボキシメチルセルロース、メチルセルロース等)、及びポリビニルピロリドンが挙げられ、トリポリリン酸ナトリウム等の無機系水溶性高分子化合物も併用することができる。これらのうち、ポリビニルアルコール又はポリビニルピロリドンが好ましい。高分子分散安定剤の添加量は、モノマ100質量部に対して1〜10質量部が好ましい。 Examples of the polymer dispersion stabilizer include polyvinyl alcohol, polycarboxylic acid, celluloses (hydroxyethyl cellulose, carboxymethyl cellulose, methyl cellulose, etc.), and polyvinyl pyrrolidone, and inorganic water-soluble polymer compounds such as sodium tripolyphosphate are also included. Can be used together. Of these, polyvinyl alcohol or polyvinyl pyrrolidone is preferred. The addition amount of the polymer dispersion stabilizer is preferably 1 to 10 parts by mass with respect to 100 parts by mass of the monomer.
モノマが単独で重合することを抑えるために、亜硝酸塩類、亜硫酸塩類、ハイドロキノン類、アスコルビン酸類、水溶性ビタミンB類、クエン酸、ポリフェノール類等の水溶性の重合禁止剤を用いてもよい。 In order to prevent the monomer from polymerizing alone, water-soluble polymerization inhibitors such as nitrites, sulfites, hydroquinones, ascorbic acids, water-soluble vitamin Bs, citric acid, and polyphenols may be used.
上記疎水性高分子粒子の平均粒径は、通液性が更に向上する観点から、10μm以上が好ましい。上記疎水性高分子粒子の平均粒径は、カラムの分離性の観点から、500μm以下が好ましい。これらの観点から、上記疎水性高分子粒子の平均粒径は、例えば、10〜500μmであってもよい。 The average particle diameter of the hydrophobic polymer particles is preferably 10 μm or more from the viewpoint of further improving liquid permeability. The average particle diameter of the hydrophobic polymer particles is preferably 500 μm or less from the viewpoint of column separability. From these viewpoints, the hydrophobic polymer particles may have an average particle size of, for example, 10 to 500 μm.
疎水性高分子粒子の粒径の変動係数(C.V.)は、通液性が更に向上する観点から、3〜15%であることが好ましく、5〜15%であることがより好ましく、5〜10%であることが更に好ましい。C.V.を低減する方法としては、マイクロプロセスサーバー(株式会社日立製作所製)等の乳化装置により単分散化することが挙げられる。 The coefficient of variation (CV) of the particle size of the hydrophobic polymer particles is preferably 3 to 15%, more preferably 5 to 15%, from the viewpoint of further improving the liquid permeability. More preferably, it is 5 to 10%. C. V. As a method for reducing the above, monodispersion by an emulsification apparatus such as a microprocess server (manufactured by Hitachi, Ltd.) is exemplified.
疎水性高分子粒子又は分離材の平均粒径及び粒径のC.V.(変動係数)は、以下の測定法により求めることができる。
1)粒子又は分離材を、超音波分散装置を使用して水(界面活性剤等の分散剤を含む)に分散させ、1質量%の粒子又は分離材を含む分散液を調製する。
2)粒度分布計(シスメックスフロー、シスメックス株式会社製)を用いて、上記分散液中の粒子又は分離材約1万個の画像により平均粒径及び粒径のC.V.(変動係数)を測定する。
C. of average particle diameter and particle diameter of hydrophobic polymer particles or separation material V. (Coefficient of variation) can be determined by the following measurement method.
1) Disperse particles or a separating material in water (including a dispersing agent such as a surfactant) using an ultrasonic dispersion device to prepare a dispersion liquid containing 1% by mass of the particles or the separating material.
2) Using a particle size distribution meter (Sysmex Flow, manufactured by Sysmex Corporation), the average particle size and particle size of C.I. V. (Coefficient of variation) is measured.
疎水性高分子粒子が、多孔構造を有する場合、当該粒子の空隙率(細孔容積)は、疎水性高分子粒子の全体積基準で30体積%以上70体積%以下であることが好ましく、40体積%以上70体積%以下であることがより好ましい。また、疎水性高分子粒子は、細孔径分布におけるモード径(細孔径分布の最頻値、最大頻度細孔径)が0.1μm以上0.5μm未満である細孔、すなわちマクロポアー(マクロ孔)を有することが好ましい。疎水性高分子粒子の細孔径分布におけるモード径として、より好ましくは、0.2μm以上0.5μm未満である。細孔径分布におけるモード径が0.1μm以上であると、細孔内に物質が入り易くなる傾向にあり、細孔径分布におけるモード径が0.5μm未満であると、比表面積が充分なものになる。これらは上述の多孔質化剤により調整可能である。 When the hydrophobic polymer particles have a porous structure, the porosity (pore volume) of the particles is preferably 30% by volume or more and 70% by volume or less based on the total volume of the hydrophobic polymer particles. It is more preferable that the volume be not less than 70% by volume. In addition, the hydrophobic polymer particles have pores having a mode diameter (mode of pore diameter distribution, maximum frequency pore diameter) of 0.1 μm or more and less than 0.5 μm, that is, macropores. It is preferable to have. The mode diameter in the pore size distribution of the hydrophobic polymer particles is more preferably 0.2 μm or more and less than 0.5 μm. If the mode diameter in the pore size distribution is 0.1 μm or more, the substance tends to easily enter the pores. If the mode diameter in the pore diameter distribution is less than 0.5 μm, the specific surface area is sufficient. Become. These can be adjusted by the above-mentioned porous agent.
疎水性高分子粒子の比表面積は、30m2/g以上であることが好ましい。より高い実用性の観点から、比表面積は35m2/g以上であることがより好ましく、40m2/g以上であることが更に好ましい。比表面積が30m2/g以上であると、分離する物質の吸着量が大きくなる傾向にある。 The specific surface area of the hydrophobic polymer particles is preferably 30 m 2 / g or more. From the viewpoint of higher practicality, the specific surface area is more preferably 35 m 2 / g or more, and further preferably 40 m 2 / g or more. When the specific surface area is 30 m 2 / g or more, the adsorption amount of the substance to be separated tends to increase.
本実施形態に係る疎水性高分子粒子又は分離材の細孔径分布におけるモード径、比表面積及び空隙率は、水銀圧入測定装置(オートポア:株式会社島津製作所製)にて測定した値であり、以下のようにして測定する。試料約0.05gを、標準5mL粉体用セル(ステム容積0.4mL)に加え、初期圧21kPa(約3psia、細孔直径約60μm相当)の条件で測定する。水銀パラメータは、装置デフォルトの水銀接触角130°、水銀表面張力485dynes/cmに設定する。また、細孔径0.1〜3μmの範囲に限定してそれぞれの値を算出する。 The mode diameter, specific surface area and porosity in the pore size distribution of the hydrophobic polymer particles or separation material according to the present embodiment are values measured with a mercury intrusion measuring apparatus (Autopore: manufactured by Shimadzu Corporation), and the following Measure as follows. About 0.05 g of a sample is added to a standard 5 mL powder cell (stem volume 0.4 mL), and measurement is performed under an initial pressure of 21 kPa (about 3 psia, corresponding to a pore diameter of about 60 μm). Mercury parameters are set to a device default mercury contact angle of 130 ° and a mercury surface tension of 485 dynes / cm. Moreover, it limits to the range of 0.1-3 micrometers of pore diameters, and calculates each value.
(被覆層)
本実施形態に係る被覆層は、水酸基及び水酸基以外の官能基を有する官能基含有高分子を含む。なお、上記官能基含有高分子において、上記官能基は化学結合により高分子に結合している。被覆層がこのようなものであると、イオン交換性又はリガンドとの結合性が付与される。
(Coating layer)
The coating layer according to the present embodiment includes a functional group-containing polymer having a hydroxyl group and a functional group other than the hydroxyl group. In the functional group-containing polymer, the functional group is bonded to the polymer by a chemical bond. When the coating layer is such, ion exchange property or binding property with a ligand is imparted.
上記官能基としては、例えば、カルボキシ基、エポキシ基、アミノ基、4級アンモニウム基、スルホン酸基等が挙げられる。上記官能基は、カルボキシ基、エポキシ基、又はアミノ基であることが好ましい。アミノ基の具体例は、ジエチルアミノエチル(DEAE)基を含む。 Examples of the functional group include a carboxy group, an epoxy group, an amino group, a quaternary ammonium group, and a sulfonic acid group. The functional group is preferably a carboxy group, an epoxy group, or an amino group. Specific examples of amino groups include diethylaminoethyl (DEAE) groups.
上記官能基含有高分子は、例えば、下記式(a)又は(b)で表される基を含んでいてもよい。 The functional group-containing polymer may contain, for example, a group represented by the following formula (a) or (b).
式(a)中、Xは、カルボキシ基を含む基を表す。Xの具体例は、カルボキシ基で置換されたアルキル基を含む。 In formula (a), X represents a group containing a carboxy group. Specific examples of X include an alkyl group substituted with a carboxy group.
![]()
![]()
式(b)中、Yは、アミノ基を含む基、又はエポキシ基を含む基を示す。Yの具体例は、グリシジル基、及びジエチルアミノエチル基を含む。 In formula (b), Y represents a group containing an amino group or a group containing an epoxy group. Specific examples of Y include a glycidyl group and a diethylaminoethyl group.
上記官能基含有高分子は、親水性高分子であってもよい。 The functional group-containing polymer may be a hydrophilic polymer.
上記官能基含有高分子は、親水性が向上する観点から、水酸基を有する高分子又はその変性体に由来する構造を含んでいてもよい。例えば、上記官能基含有高分子は、水酸基を有する高分子又はその変性体から1つ以上の水酸基を除いた構造を有するものであってもよい。水酸基を有する高分子は、1分子中に2個以上の水酸基を有することが好ましく、親水性高分子であることがより好ましい。水酸基を有する高分子としては、例えば、多糖類及びポリビニルアルコールが挙げられる。多糖類としては、例えば、アガロース、デキストラン、セルロース、及びキトサンが挙げられる。すなわち、上記官能基含有高分子は、多糖類又はその変性体に由来する構造を含んでいてもよく、アガロース又はその変性体に由来する構造を含んでいてもよい。 From the viewpoint of improving hydrophilicity, the functional group-containing polymer may include a structure derived from a polymer having a hydroxyl group or a modified product thereof. For example, the functional group-containing polymer may have a structure in which one or more hydroxyl groups are removed from a polymer having a hydroxyl group or a modified product thereof. The polymer having a hydroxyl group preferably has two or more hydroxyl groups in one molecule, and more preferably a hydrophilic polymer. Examples of the polymer having a hydroxyl group include polysaccharides and polyvinyl alcohol. Examples of polysaccharides include agarose, dextran, cellulose, and chitosan. That is, the functional group-containing polymer may include a structure derived from a polysaccharide or a modified product thereof, or may include a structure derived from agarose or a modified product thereof.
水酸基を有する高分子の重量平均分子量(Mw)は、例えば、10000〜200000程度であってもよい。 The weight average molecular weight (Mw) of the polymer having a hydroxyl group may be, for example, about 10,000 to 200,000.
水酸基を有する高分子の変性体としては、例えば、水酸基を有する高分子を疎水基で変性したもの(以下、「疎水基変性体」ともいう)が挙げられる。上記官能基含有高分子が、このような変性体に由来する構造を含むと、界面吸着能が向上する傾向にある。疎水基としては、例えば、炭素数1〜6のアルキル基、炭素数6〜10のアリール基等が挙げられる。炭素数1〜6のアルキル基としては、例えば、メチル基、エチル基、及びプロピル基が挙げられる。炭素数6〜10のアリール基としては、例えば、フェニル基、及びナフチル基が挙げられる。疎水基は、例えば、水酸基と反応する官能基(例えば、エポキシ基)及び疎水基を有する化合物(例えば、グリシジルフェニルエーテル)を、水酸基を有する高分子と従来公知の方法で反応させることにより、導入することができる。 Examples of the modified polymer having a hydroxyl group include those obtained by modifying a polymer having a hydroxyl group with a hydrophobic group (hereinafter also referred to as “hydrophobic group-modified product”). When the functional group-containing polymer contains a structure derived from such a modified product, the interfacial adsorption ability tends to be improved. Examples of the hydrophobic group include an alkyl group having 1 to 6 carbon atoms and an aryl group having 6 to 10 carbon atoms. Examples of the alkyl group having 1 to 6 carbon atoms include a methyl group, an ethyl group, and a propyl group. Examples of the aryl group having 6 to 10 carbon atoms include a phenyl group and a naphthyl group. The hydrophobic group is introduced, for example, by reacting a functional group that reacts with a hydroxyl group (for example, an epoxy group) and a compound having a hydrophobic group (for example, glycidyl phenyl ether) with a polymer having a hydroxyl group by a conventionally known method. can do.
疎水基変性体において、疎水基変性体を構成する全構成単位の総モル量(疎水基を含有する構成単位及び疎水基を含有しない構成単位のモル量の総和)に対する、疎水基を含有する構成単位の含有割合は、官能基含有高分子を疎水性高分子粒子の表面に吸着させるための疎水的相互作用力の保持と、タンパク質の非特異吸着の抑制とのバランスの観点から、5〜30%であることが好ましく、10〜20%であることがより好ましく、12〜17%であることが更に好ましい。 In the hydrophobic group-modified product, a structure containing a hydrophobic group relative to the total molar amount of all the structural units constituting the hydrophobic group-modified product (sum of the molar amounts of the structural unit containing the hydrophobic group and the structural unit not containing the hydrophobic group). The content ratio of the unit is 5 to 30 from the viewpoint of balance between retention of the hydrophobic interaction force for adsorbing the functional group-containing polymer to the surface of the hydrophobic polymer particle and suppression of nonspecific adsorption of the protein. %, More preferably 10 to 20%, and still more preferably 12 to 17%.
カラム圧の上昇がより抑制される観点から、上記官能基含有高分子は架橋されていてもよい。 From the viewpoint of further suppressing the increase in column pressure, the functional group-containing polymer may be crosslinked.
(被覆層の形成方法)
本実施形態に係る被覆層は、例えば、疎水性高分子粒子表面に、水酸基を有する高分子又はその変性体を吸着させる工程(吸着処理工程)と、上記水酸基を有する高分子又はその変性体を架橋する工程(架橋処理工程)と、架橋された水酸基を有する高分子又はその変性体に官能基を導入する工程(官能基導入工程)と、を備える方法により形成できる。以下、これらの工程について、具体的に説明する。
(Formation method of coating layer)
The coating layer according to the present embodiment includes, for example, a step of adsorbing a polymer having a hydroxyl group or a modified product thereof on the surface of the hydrophobic polymer particles (adsorption treatment step), and a polymer having the hydroxyl group or a modified product thereof. It can be formed by a method comprising a step of crosslinking (crosslinking treatment step) and a step of introducing a functional group into the polymer having a crosslinked hydroxyl group or a modified product thereof (functional group introduction step). Hereinafter, these steps will be specifically described.
(吸着処理工程)
吸着処理工程においては、例えば、水酸基を有する高分子又はその変性体の溶液に、疎水性高分子粒子を含浸させることにより、疎水性高分子粒子表面に、水酸基を有する高分子又はその変性体を吸着させる。これにより、疎水性高分子粒子表面に、水酸基を有する高分子又はその変性体が固定化される。水酸基を有する高分子又はその変性体の溶液の溶媒としては、水酸基を有する高分子又はその変性体を溶解することのできるものであれば、特に限定されないが、水が最も一般的である。溶媒に溶解させる高分子の濃度は、5〜20(mg/mL)が好ましい。
(Adsorption treatment process)
In the adsorption treatment step, for example, a polymer having a hydroxyl group or a modified product thereof is formed on the surface of the hydrophobic polymer particle by impregnating the polymer solution having a hydroxyl group or a modified product thereof with the hydrophobic polymer particle. Adsorb. Thereby, the polymer which has a hydroxyl group, or its modified body is fix | immobilized on the hydrophobic polymer particle surface. The solvent for the polymer having a hydroxyl group or a modified product thereof is not particularly limited as long as it can dissolve the polymer having a hydroxyl group or a modified product thereof, but water is most commonly used. The concentration of the polymer dissolved in the solvent is preferably 5 to 20 (mg / mL).
含浸方法としては、例えば、水酸基を有する高分子又はその変性体の溶液に疎水性高分子粒子を加えて一定時間放置する方法が挙げられる。含浸時間は疎水性高分子粒子の表面状態によっても変わるが、例えば、疎水性高分子粒子が多孔構造を有する場合でも、通常一昼夜含浸すれば、高分子濃度が多孔質体の内部で外部濃度と平衡状態となる。 Examples of the impregnation method include a method in which hydrophobic polymer particles are added to a solution of a polymer having a hydroxyl group or a modified product thereof and left for a predetermined time. Although the impregnation time varies depending on the surface state of the hydrophobic polymer particles, for example, even when the hydrophobic polymer particles have a porous structure, the polymer concentration is usually equal to the external concentration inside the porous body if it is impregnated all day and night. It becomes an equilibrium state.
次いで、水、アルコール等の溶媒で洗浄し、未吸着分の水酸基を有する高分子又はその変性体を除去する。 Next, the polymer is washed with a solvent such as water or alcohol to remove the polymer having an unadsorbed hydroxyl group or a modified product thereof.
(架橋処理工程)
架橋処理工程においては、例えば、架橋剤を用いて、疎水性高分子粒子表面に吸着された水酸基を有する高分子又はその変性体を架橋反応させて、これらの架橋体を形成する。当該架橋体は、例えば、水酸基を有する3次元架橋網目構造を有する。
(Crosslinking process)
In the crosslinking treatment step, for example, using a crosslinking agent, a polymer having a hydroxyl group adsorbed on the surface of the hydrophobic polymer particles or a modified product thereof is subjected to a crosslinking reaction to form these crosslinked products. The crosslinked body has, for example, a three-dimensional crosslinked network structure having a hydroxyl group.
架橋剤としては、例えば、エピクロルヒドリン等のエピハロヒドリン、グルタルアルデヒド等のジアルデヒド化合物、メチレンジイソシアネート等のジイソシアネート化合物、及びエチレングリコールジグリシジルエーテル等のグリシジル化合物などのような水酸基に活性な官能基を2個以上有する化合物が挙げられる。また、水酸基を有する高分子又はその変性体が、例えば、アミノ基を有する化合物(キトサン又はその変性体等)である場合には、ジクロロオクタンのようなジハライド化合物も架橋剤として使用できる。 Examples of the crosslinking agent include two functional groups active on hydroxyl such as epihalohydrins such as epichlorohydrin, dialdehyde compounds such as glutaraldehyde, diisocyanate compounds such as methylene diisocyanate, and glycidyl compounds such as ethylene glycol diglycidyl ether. The compound which has the above is mentioned. Further, when the polymer having a hydroxyl group or a modified product thereof is, for example, a compound having an amino group (such as chitosan or a modified product thereof), a dihalide compound such as dichlorooctane can also be used as a crosslinking agent.
この架橋反応には通常触媒が用いられる。該触媒は架橋剤の種類に合わせて適宜従来公知のものを用いることができるが、例えば、架橋剤がエピクロルヒドリン等の場合には水酸化ナトリウム等のアルカリが有効であり、ジアルデヒド化合物の場合には塩酸等の鉱酸が有効である。 A catalyst is usually used for this crosslinking reaction. As the catalyst, a conventionally known catalyst can be appropriately used according to the type of the crosslinking agent. For example, when the crosslinking agent is epichlorohydrin or the like, an alkali such as sodium hydroxide is effective, and in the case of a dialdehyde compound. Mineral acids such as hydrochloric acid are effective.
架橋剤による架橋反応は、通常、水酸基を有する高分子又はその変性体が吸着された疎水性高分子粒子を、適当な媒体中に分散、懸濁させた系に架橋剤を添加することによって行われる。架橋剤の添加量は、水酸基を有する高分子として多糖類を使用した場合、単糖類の1単位を1モルとすると、それに対して、例えば、0.1〜100モル倍の範囲内で、分離材の性能に応じて選定することができる。架橋剤の添加量が、上記下限値以上であると、被覆層が疎水性高分子粒子上に良好に保持される傾向にある。また、架橋剤の添加量が上記上限値以下であれば、水酸基を有する高分子又はその変性体の反応率が高い場合でも、水酸基を有する高分子又はその変性体の特性が損なわれ難い。 The crosslinking reaction with a crosslinking agent is usually performed by adding a crosslinking agent to a system in which hydrophobic polymer particles adsorbed with a polymer having a hydroxyl group or a modified product thereof are dispersed and suspended in an appropriate medium. Is called. When the polysaccharide is used as the polymer having a hydroxyl group, the addition amount of the cross-linking agent is, for example, within a range of 0.1 to 100 mol times with respect to one unit of the monosaccharide as 1 mol. It can be selected according to the performance of the material. When the addition amount of the crosslinking agent is not less than the above lower limit value, the coating layer tends to be favorably retained on the hydrophobic polymer particles. Moreover, if the addition amount of a crosslinking agent is below the said upper limit, even if the reaction rate of the polymer which has a hydroxyl group or its modified body is high, the characteristic of the polymer which has a hydroxyl group or its modified body is hard to be impaired.
触媒の使用量としては、架橋剤の種類により異なるが、通常、水酸基を有する高分子として多糖類を使用する場合に、多糖類を形成する単糖類の1単位を1モルとすると、これに対して、例えば、0.01〜10モル倍の範囲、好ましくは0.1〜5モル倍の範囲で使用される。 The amount of the catalyst used varies depending on the type of the crosslinking agent. Usually, when a polysaccharide is used as the polymer having a hydroxyl group, if one unit of the monosaccharide forming the polysaccharide is 1 mole, For example, it is used in the range of 0.01 to 10 mole times, preferably in the range of 0.1 to 5 mole times.
例えば、架橋反応条件を温度条件とした場合、反応系の温度を上げ、その温度が反応温度に達すれば架橋反応が生起する。 For example, when the cross-linking reaction condition is a temperature condition, the cross-linking reaction occurs when the temperature of the reaction system is raised and the temperature reaches the reaction temperature.
水酸基を有する高分子又はその変性体が吸着された疎水性高分子粒子を分散、懸濁させる媒体は、例えば、高分子成分、架橋剤等を抽出してしまうことなく、かつ、架橋反応に不活性なものであることができる。上記媒体の具体例としては、水、トルエン、ジクロルベンゼン、ニトロメタン等が挙げられる。 A medium in which hydrophobic polymer particles to which a polymer having a hydroxyl group or a modified product thereof has been adsorbed is dispersed or suspended does not extract a polymer component, a crosslinking agent, etc. It can be active. Specific examples of the medium include water, toluene, dichlorobenzene, nitromethane and the like.
架橋反応は、例えば、1〜90℃の範囲の温度で行ってもよい。また、反応時間は、例えば、1〜36時間であってもよく、1〜24時間であってもよく、1〜10時間であってもよい。 The crosslinking reaction may be performed at a temperature in the range of 1 to 90 ° C, for example. The reaction time may be, for example, 1 to 36 hours, 1 to 24 hours, or 1 to 10 hours.
架橋度を調整する場合、架橋反応を段階的に行うことができる。例えば、一度架橋反応を行った後、再度架橋反応させることによって、架橋の進行度を調整できる。 When adjusting the degree of crosslinking, the crosslinking reaction can be carried out stepwise. For example, the degree of progress of crosslinking can be adjusted by performing the crosslinking reaction once and then performing the crosslinking reaction again.
架橋度は、熱分解による5%重量減少温度を測定し、その値から評価することができる。架橋度が高い場合、重量減少開始温度は高くなり、架橋度が低い場合、重量減少開始温度も低くなる。熱分解による5%重量減少温度は、例えば、示差熱熱重量測定装置を用いて測定できる。 The degree of crosslinking can be evaluated from a value obtained by measuring a 5% weight loss temperature due to thermal decomposition. When the degree of crosslinking is high, the weight reduction start temperature is high, and when the degree of crosslinking is low, the weight reduction start temperature is also low. The 5% weight loss temperature due to thermal decomposition can be measured using, for example, a differential thermothermal gravimetric apparatus.
架橋反応終了後、得られた粒子をろ別し、次いで、水、メタノール、エタノール等の親水性有機溶媒で洗浄し、未反応の高分子、懸濁用媒体等を除去する。これにより、疎水性高分子粒子の表面の少なくとも一部に、水酸基を有する高分子又はその変性体の架橋体を含む被覆層が形成された、架橋体被覆粒子を作製できる。 After completion of the crosslinking reaction, the obtained particles are filtered off and then washed with a hydrophilic organic solvent such as water, methanol, ethanol, etc. to remove unreacted polymer, suspending medium and the like. Thereby, the crosslinked body covering particle | grains by which the coating layer containing the bridge | crosslinking body of the polymer which has a hydroxyl group, or its modified body was formed in at least one part of the surface of hydrophobic polymer particle can be produced.
(官能基導入工程)
官能基導入工程は、上記架橋体に官能基を導入する工程である。上記架橋体に官能基を導入する方法に特に制限はなく、任意の方法を選択できる。上記架橋体に官能基を導入する方法としては、例えば、上記架橋体に官能基を直接導入する方法、及び上記架橋体が有する水酸基を介して官能基を導入する方法が挙げられる。官能基の導入方法に特に制限はなく、官能基の種類等により適宜選択できる。水酸基を介して官能基を導入する方法としては、例えば、ハロゲン化アルキル化合物を用いる方法が挙げられる。なお、水酸基を介して官能基を導入する場合、官能基は、架橋体が有する水酸基のうちの少なくとも一部に導入されていればよい。
(Functional group introduction process)
The functional group introduction step is a step of introducing a functional group into the crosslinked product. There is no restriction | limiting in particular in the method of introduce | transducing a functional group into the said crosslinked body, Arbitrary methods can be selected. Examples of the method for introducing a functional group into the crosslinked body include a method for directly introducing a functional group into the crosslinked body, and a method for introducing a functional group via a hydroxyl group of the crosslinked body. There is no restriction | limiting in particular in the introduction method of a functional group, According to the kind etc. of functional group, it can select suitably. Examples of the method for introducing a functional group through a hydroxyl group include a method using a halogenated alkyl compound. In addition, when introduce | transducing a functional group through a hydroxyl group, the functional group should just be introduce | transduced into at least one part of the hydroxyl group which a crosslinked body has.
ハロゲン化アルキル化合物としては、モノハロゲノ酢酸、モノハロゲノプロピオン酸等のモノハロゲノカルボン酸及びそのナトリウム塩、ジエチルアミノエチルクロライド等のハロゲン化アルキル基を少なくとも1つ有する1級、2級又は3級アミン等が挙げられる。ハロゲン化アルキル化合物は、硫黄化合物、臭化物又は塩化物であってもよい。 Examples of the halogenated alkyl compounds include monohalogenocarboxylic acids such as monohalogenoacetic acid and monohalogenopropionic acid and sodium salts thereof, primary, secondary or tertiary amines having at least one halogenated alkyl group such as diethylaminoethyl chloride. Can be mentioned. The alkyl halide compound may be a sulfur compound, bromide or chloride.
官能基として、弱酸性基であるカルボキシ基を導入する方法としては、例えば、上記ハロゲン化アルキル化合物として、モノハロゲノ酢酸、モノハロゲノプロピオン酸等のモノハロゲノカルボン酸又はそのナトリウム塩を反応させる方法が挙げられる。これらハロゲン化アルキル化合物の使用量は、架橋体被覆粒子の全質量に対して、0.2質量%以上であることが好ましい。 Examples of the method for introducing a carboxy group, which is a weakly acidic group, as the functional group include a method in which a monohalogenocarboxylic acid such as monohalogenoacetic acid or monohalogenopropionic acid or a sodium salt thereof is reacted as the halogenated alkyl compound. It is done. The amount of these halogenated alkyl compounds used is preferably 0.2% by mass or more based on the total mass of the crosslinked-coated particles.
官能基としてカルボキシ基を導入する方法は、例えば、上記架橋体が有する水酸基に、エピクロルヒドリン等のハロゲン基含有グリシジル化合物を反応させ、エポキシ基を導入した後、当該エポキシ基に、カルボン酸又はその塩を反応させる方法であってもよい。カルボン酸又はその塩としては、例えば、メルカプトプロピオン酸、及びメルカプト酢酸ナトリウムが挙げられる。 The method for introducing a carboxy group as a functional group is, for example, by reacting a halogen group-containing glycidyl compound such as epichlorohydrin with the hydroxyl group of the crosslinked product, introducing an epoxy group, and then introducing the carboxylic acid or a salt thereof into the epoxy group. May be a method of reacting. Examples of the carboxylic acid or a salt thereof include mercaptopropionic acid and sodium mercaptoacetate.
官能基として、エポキシ基を導入する方法としては、例えば、上記架橋体に、エピハロヒドリン、エチレングリコールジグリシジルエーテル、1,2,3,4−ジエポキシブタン等のエポキシ基含有化合物を反応させる方法が挙げられる。エピハロヒドリンとして好ましいものとしては、エピクロロヒドリン、エピブロモヒドリン、エピヨードヒドリン、β−メチルエピクロロヒドリン、α−メチルエピクロロヒドリン、γ−メチルエピクロロヒドリン等が挙げられる。これらのエポキシ基含有化合物の使用量は、架橋体被覆粒子の全質量に対して0.2質量%以上であることが好ましい。反応条件は、40〜90℃で0.5〜12時間であることが好ましい。 As a method for introducing an epoxy group as a functional group, for example, a method of reacting an epoxy group-containing compound such as epihalohydrin, ethylene glycol diglycidyl ether, 1,2,3,4-diepoxybutane, or the like with the above crosslinked product. Can be mentioned. Preferable examples of the epihalohydrin include epichlorohydrin, epibromohydrin, epiiodohydrin, β-methylepichlorohydrin, α-methylepichlorohydrin, γ-methylepichlorohydrin, and the like. It is preferable that the usage-amount of these epoxy group containing compounds is 0.2 mass% or more with respect to the total mass of bridge | crosslinking body covering particle | grains. The reaction conditions are preferably 40 to 90 ° C. and 0.5 to 12 hours.
官能基としてエポキシ基を導入する方法は、例えば、上記架橋体が有する水酸基に、エピクロルヒドリン等のハロゲン基含有グリシジル化合物を反応させる方法であってもよい。 The method for introducing an epoxy group as a functional group may be, for example, a method in which a halogen group-containing glycidyl compound such as epichlorohydrin is reacted with the hydroxyl group of the crosslinked product.
官能基として、アミノ基を導入する方法としては、例えば、上記ハロゲン化アルキル化合物のうち、水素原子の一部が塩素原子に置換されたアルキル基を少なくとも1つ有する、モノ−、ジ−又はトリ−アルキルアミン、モノ−、ジ−又はトリ−アルカノールアミン、モノ−アルキル−モノ−アルカノールアミン、ジ−アルキル−モノ−アルカノールアミン、モノ−アルキル−ジ−アルカノールアミン等を反応させる方法が挙げられる。これらのハロゲン化アルキル化合物の使用量としては、架橋体被覆粒子の全質量に対して0.2質量%以上であることが好ましい。反応条件は、40〜90℃で、0.5〜12時間であることが好ましい。当該方法に用いられるハロゲン化アルキル化合物の具体例は、ジエチルアミノエチルクロライド塩酸塩を含む。 As a method for introducing an amino group as a functional group, for example, among the above-mentioned halogenated alkyl compounds, mono-, di- or tri-containing compounds having at least one alkyl group in which a part of hydrogen atoms are substituted with chlorine atoms. -Alkylamine, mono-, di- or tri-alkanolamine, mono-alkyl-mono-alkanolamine, di-alkyl-mono-alkanolamine, mono-alkyl-di-alkanolamine and the like can be mentioned. The amount of these halogenated alkyl compounds used is preferably 0.2% by mass or more based on the total mass of the crosslinked product-coated particles. The reaction conditions are preferably 40 to 90 ° C. and 0.5 to 12 hours. Specific examples of the alkyl halide compound used in the method include diethylaminoethyl chloride hydrochloride.
官能基として、強酸性基であるスルホン酸基の導入方法としては、上記架橋体に対してエピクロロヒドリン等のグリシジル化合物を反応させ、亜硫酸ナトリウム、重亜硫酸ナトリウム等の亜硫酸塩又は重亜硫酸塩の飽和水溶液に架橋体被覆粒子を添加する方法等が挙げられる。反応条件は、30〜90℃で1〜10時間であることが好ましい。 As a method for introducing a sulfonic acid group that is a strongly acidic group as a functional group, a glycidyl compound such as epichlorohydrin is reacted with the cross-linked product, and a sulfite or bisulfite such as sodium sulfite or sodium bisulfite. And the like, and the like. The reaction conditions are preferably 30 to 90 ° C. and 1 to 10 hours.
また、アルカリ性雰囲気下で、上記架橋体に1,3−プロパンスルトンを反応させる方法によっても、上記架橋体に官能基を導入し得る。1,3−プロパンスルトンは、架橋体被覆粒子の全質量に対して0.4質量%以上使用することが好ましい。反応条件は、0〜90℃で0.5〜12時間であることが好ましい。 Also, a functional group can be introduced into the crosslinked product by a method of reacting the crosslinked product with 1,3-propane sultone in an alkaline atmosphere. 1,3-propane sultone is preferably used in an amount of 0.4% by mass or more based on the total mass of the crosslinked product-coated particles. The reaction conditions are preferably 0 to 90 ° C. and 0.5 to 12 hours.
本実施形態の分離材において、乾燥状態の分離材1g当たりの官能基のモル量(mmol)は、吸着させるタンパク質のサイズ、分離材の表面積、比重等に応じて適宜調整することが好ましい。 In the separation material of this embodiment, the molar amount (mmol) of the functional group per gram of the separation material in a dry state is preferably adjusted as appropriate according to the size of the protein to be adsorbed, the surface area of the separation material, the specific gravity and the like.
本実施形態の分離材は、イオン交換担体として好適に使用し得る。また、本実施形態の分離材は、アフィニティ精製、タンパク質の静電的相互作用による分離等にも好適に使用し得る。例えば、タンパク質を含む混合溶液の中に本実施形態の分離材を添加し、静電的相互作用によりタンパク質だけを分離材に吸着させた後、該分離材を溶液からろ別し、塩濃度の高い水溶液中に添加すれば、分離材に吸着しているタンパク質を容易に脱離、回収できる。また、本実施形態の分離材は、カラムクロマトグラフィー(例えば、液体クロマトグラフィー)において、使用することも可能である。より具体的には、本実施形態の分離材は、例えば、アフィニティークロマトグラフィー分野(特に抗体医薬品精製の分野)にも好適に利用できる。 The separation material of this embodiment can be suitably used as an ion exchange carrier. Moreover, the separation material of this embodiment can be suitably used for affinity purification, separation by electrostatic interaction of proteins, and the like. For example, after adding the separation material of the present embodiment to a mixed solution containing protein, adsorbing only the protein to the separation material by electrostatic interaction, the separation material is filtered from the solution, and the salt concentration If added to a high aqueous solution, the protein adsorbed on the separation material can be easily desorbed and recovered. Moreover, the separation material of this embodiment can also be used in column chromatography (for example, liquid chromatography). More specifically, the separation material of the present embodiment can be suitably used, for example, in the affinity chromatography field (particularly in the field of antibody drug purification).
本実施形態の分離材を用いて分離できる生体高分子としては、水溶性物質が好ましい。具体的には、血清アルブミン、免疫グロブリン等の血液タンパク質などのタンパク質、生体中に存在する酵素、バイオテクノロジーにより生産されるタンパク質生理活性物質、DNA、生理活性をするペプチド等の生体高分子などであり、好ましくは分子量が200万以下、より好ましくは50万以下である。また、公知の方法に従い、タンパク質の等電点、イオン化状態等によって、分離材の性質、条件等を選ぶ必要がある。公知の方法としては、例えば、特開昭60−169427号公報等に記載の方法が挙げられる。 The biopolymer that can be separated using the separation material of the present embodiment is preferably a water-soluble substance. Specific examples include proteins such as blood proteins such as serum albumin and immunoglobulin, enzymes present in the living body, protein bioactive substances produced by biotechnology, DNA, biopolymers such as bioactive peptides, etc. Yes, preferably the molecular weight is 2 million or less, more preferably 500,000 or less. In addition, according to a known method, it is necessary to select properties, conditions, etc. of the separation material depending on the isoelectric point, ionization state, etc. of the protein. Examples of known methods include the methods described in JP-A-60-169427.
本実施形態の分離材は、疎水性高分子粒子1g当たり30〜600mgの被覆層を備えると好ましい。被覆層の量は熱分解の重量減少等で測定することができる。 The separating material of the present embodiment preferably includes a coating layer of 30 to 600 mg per 1 g of the hydrophobic polymer particles. The amount of the coating layer can be measured by reducing the weight of pyrolysis.
本実施形態の分離材の水中での5%圧縮変形弾性率(湿潤状態での5%圧縮変形弾性率)は、例えば、70MPa以上であってもよく、75MPa以上であってもよく、80MPa以上であってもよい。 The 5% compressive deformation modulus in water (5% compressive deformation modulus in a wet state) of the separation material of the present embodiment may be, for example, 70 MPa or more, 75 MPa or more, or 80 MPa or more. It may be.
本明細書において、分離材の水中での5%圧縮変形弾性率(分離材が5%圧縮変形したときの圧縮弾性率、5%K値ともいう)は、以下の方法により算出された値をいう。
微小圧縮試験機(Fisher製)を用いて、室温(20〜25℃)条件にて荷重負荷速度1mN/秒で、四角柱の平滑な端面(50μm×50μm)により、予め水中に浸漬させた分離材を50mNまで圧縮したときの荷重及び圧縮変位を測定する。得られた測定値から、分離材が5%圧縮変形したときの圧縮弾性率(5%K値)を下記式により求める。また、上記測定中の変位量が最も大きく変化する点の荷重を破壊強度(mN)とする。
5%K値(MPa)=(3/21/2)・F・S−3/2・R−1/2
F:分離材が5%圧縮変形したときの荷重(mN)
S:分離材が5%圧縮変形したときの圧縮変位(mm)
R:分離材の半径(mm)
In this specification, the 5% compressive deformation modulus of the separating material in water (also referred to as compressive modulus and 5% K value when the separating material undergoes 5% compressive deformation) is a value calculated by the following method. Say.
Separation using a micro compression tester (manufactured by Fisher) at room temperature (20 to 25 ° C.) at a load rate of 1 mN / sec. The load and compression displacement when the material is compressed to 50 mN are measured. From the obtained measured value, the compression elastic modulus (5% K value) when the separating material is 5% compressively deformed is obtained by the following formula. Further, the load at the point at which the displacement during the measurement changes most greatly is defined as the breaking strength (mN).
5% K value (MPa) = (3/2 1/2 ) · F · S −3 / 2 · R −1/2
F: Load (mN) when the separating material is 5% compressively deformed
S: Compression displacement (mm) when the separating material is 5% compressively deformed
R: radius of separating material (mm)
本実施形態の分離材は、カラムに充填した場合、カラム圧0.3MPaのときに通液速度が500cm/h以上であることが好ましい。カラムクロマトグラフィーでタンパク質の分離を行う場合、タンパク質溶液等の通液速度としては、一般に400cm/h以下の範囲であるが、本実施形態の分離材を使用した場合は、通常のタンパク質分離用の分離材よりも速い通液速度500cm/h以上で使用することができる。なお、本明細書における通液速度とは、φ7.8×300mmのステンレスカラムに本実施形態の分離材を充填し、液を通した際の通液速度を表す。 When the separation material of this embodiment is packed in a column, it is preferable that the liquid passing speed is 500 cm / h or more when the column pressure is 0.3 MPa. When separating proteins by column chromatography, the flow rate of protein solution or the like is generally in the range of 400 cm / h or less. However, when the separation material of the present embodiment is used, the separation rate for normal protein separation is as follows. It can be used at a liquid passing speed of 500 cm / h or more faster than the separating material. In addition, the liquid flow rate in this specification represents the liquid flow rate when the separation material of this embodiment is filled in a stainless steel column of φ7.8 × 300 mm and the liquid is passed.
分離材の、平均細孔径、比表面積等は、疎水性高分子粒子の原料、多孔質化剤、水酸基を有する高分子等を適宜選択することによって、調整することができる。 The average pore diameter, specific surface area, etc. of the separating material can be adjusted by appropriately selecting the raw material of the hydrophobic polymer particles, the porous agent, the polymer having a hydroxyl group, and the like.
本実施形態の分離材には、上記官能基を介して又は上記官能基とは別に、例えば、リガンド(例えば、プロテインA)又はイオン交換基を導入することもできる。リガンド又はイオン交換基が更に導入された分離材は、例えば、アフィニティ精製、イオン交換精製等の用途に好適に使用し得る。 For example, a ligand (for example, protein A) or an ion exchange group can be introduced into the separation material of the present embodiment via the functional group or separately from the functional group. A separation material into which a ligand or ion exchange group is further introduced can be suitably used for applications such as affinity purification and ion exchange purification.
本実施形態のカラムは、本実施形態の分離材を備えるものである。本実施形態のカラムは、例えば、カラムに本実施形態の分離材を充填することで製造できる。カラムに本実施形態の分離材を充填する方法は特に制限されるものではなく、例えば、公知の方法を採用することができる。 The column of the present embodiment includes the separation material of the present embodiment. The column of this embodiment can be manufactured, for example, by filling the column with the separation material of this embodiment. The method for filling the column with the separation material of the present embodiment is not particularly limited, and for example, a known method can be employed.
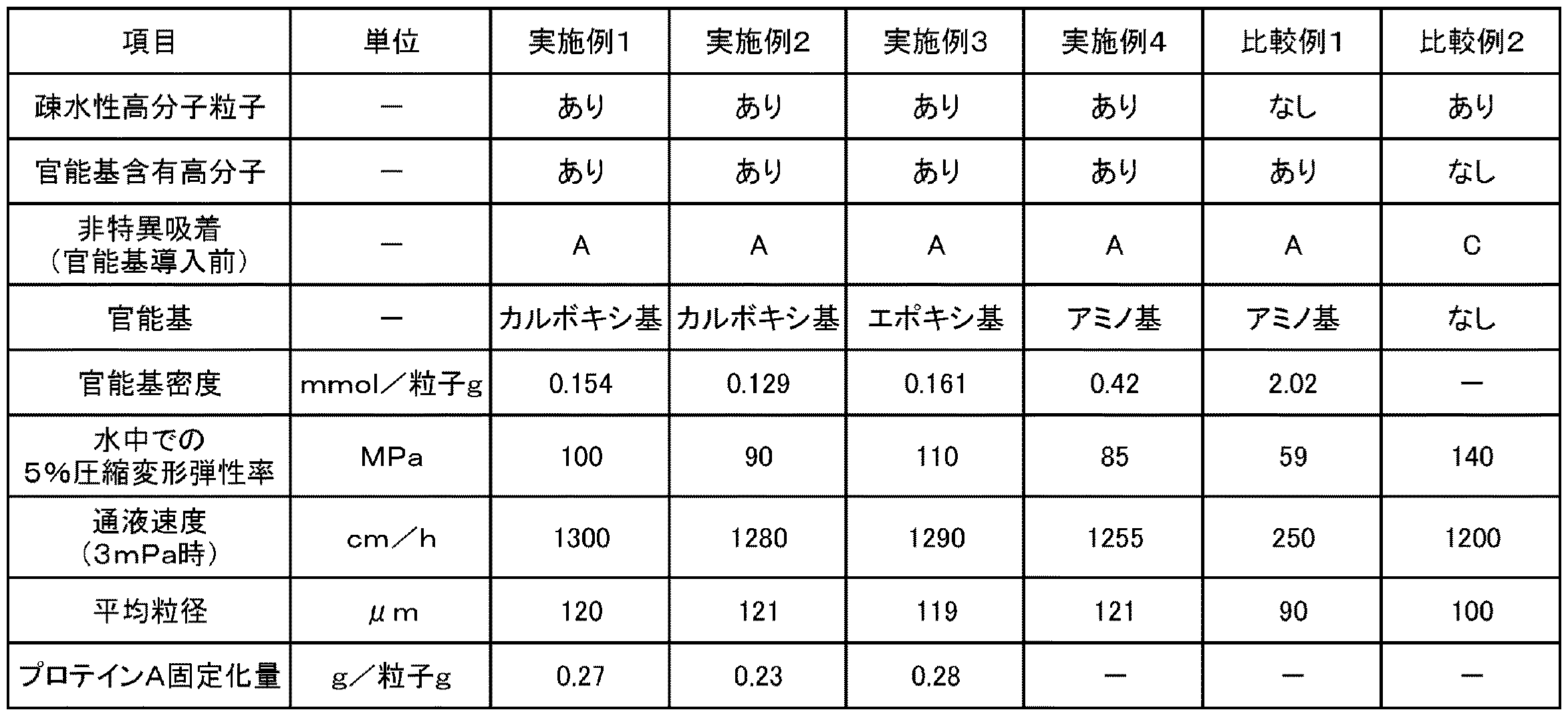
以下、本発明を実施例により説明するが、本発明はこれら実施例に限定されるものではない。 EXAMPLES Hereinafter, although an Example demonstrates this invention, this invention is not limited to these Examples.
(実施例1)
(疎水性の多孔質高分子粒子の合成)
500mLの三口フラスコに、モノマとしての純度96%のジビニルベンゼン(新日鉄住金株式会社製、商品名:DVB960)16g、多孔質化剤としてのヘキサノール16g及びジエチルベンゼン16g、並びに開始剤としての過酸化ベンゾイル0.64gをポリビニルアルコール(0.5質量%)分散剤水溶液に混合して、混合液を得た。得られた混合液を、マイクロプロセスサーバーを使用して乳化した後、得られた乳化液をフラスコに移し、80℃のウォーターバスで加熱しながら、撹拌機を用いて約8時間撹拌して粒子を得た。得られた粒子をろ別して、更にアセトンで洗浄し、疎水性の多孔質高分子粒子1を得た。得られた多孔質高分子粒子1の粒径をフロー型粒径測定装置で測定し、平均粒径を算出した。結果を表1に示す。
Example 1
(Synthesis of hydrophobic porous polymer particles)
In a 500 mL three-necked flask, 16 g of divinylbenzene having a purity of 96% as a monomer (manufactured by Nippon Steel & Sumikin Co., Ltd., trade name: DVB960), 16 g of hexanol and 16 g of diethylbenzene as a porous agent, and benzoyl peroxide 0 as an initiator .64 g was mixed with an aqueous polyvinyl alcohol (0.5% by mass) dispersant solution to obtain a mixed solution. The resulting mixture is emulsified using a microprocess server, and then the resulting emulsion is transferred to a flask and stirred with a stirrer for about 8 hours while heating in a water bath at 80 ° C. Got. The obtained particles were collected by filtration and further washed with acetone to obtain hydrophobic porous polymer particles 1. The particle size of the obtained porous polymer particles 1 was measured with a flow type particle size measuring device, and the average particle size was calculated. The results are shown in Table 1.
(水酸基を有する高分子への疎水基の導入)
アガロース水溶液(濃度2質量%)480mLに水酸化ナトリウム0.98g、及びグリシジルフェニルエーテル4.90gを投入して60℃で6時間反応させ、アガロースにフェニル基を導入した。得られた変性アガロースをイソプロピルアルコールで再沈殿させ、洗浄した。変性アガロースの疎水基含有量を下記方法により算出したところ、14.2%であった。
(Introduction of a hydrophobic group into a polymer having a hydroxyl group)
Sodium hydroxide 0.98 g and glycidyl phenyl ether 4.90 g were added to 480 mL of an agarose aqueous solution (concentration 2% by mass) and reacted at 60 ° C. for 6 hours to introduce a phenyl group into the agarose. The obtained modified agarose was reprecipitated with isopropyl alcohol and washed. The hydrophobic group content of the modified agarose was calculated by the following method and found to be 14.2%.
(変性アガロースの疎水基含有量の評価)
乾燥状態の粉末アガロース(変性されていないアガロース)と、揮発分0.1質量%未満まで乾燥させた変性アガロースとをそれぞれ70℃の純水に溶解させ、0.05質量%の水溶液サンプルを調製した。
(Evaluation of hydrophobic group content of modified agarose)
Dry powder agarose (unmodified agarose) and denatured agarose dried to a volatile content of less than 0.1% by mass are each dissolved in pure water at 70 ° C. to prepare a 0.05% by mass aqueous solution sample. did.
分光光度計により各水溶液の269nmの吸光度を測定して濃度を求め、下記式より疎水基含有量を算出した。
・疎水基含有量(%)=(CAG/(CHAG+CAG))×100
・CAG:変性されているアガロース構成単位の濃度(mmol/L)
=A/εGPE×1000
・CHAG:変性されていないアガロース構成単位の濃度(mmol/L)
=(変性されてないアガロース構成単位の濃度(g/L)/アガロース構成単位(306g/mol))×1000
・A:疎水基導入アガロースの真の吸光度
=疎水基を導入したアガロースの吸光度−変性されていないアガロースの吸収
・εGPE:グリシジルフェニルエーテルの吸光係数
=1372(L/(mol・cm))
・変性されていないアガロースの吸収=変性されてないアガロースの吸光度×(変性アガロースのサンプル濃度(mmol/L)/変性されてないアガロースのサンプル濃度(mmol/L))
・変性されてないアガロース構成単位の濃度(g/L)=変性アガロースのサンプル濃度(質量%)×10−変性されているアガロース構成単位の濃度(g/L)
・変性されているアガロース構成単位の濃度(g/L)=(CAG×変性されているアガロース構成単位(456g/mol))/1000
The absorbance at 269 nm of each aqueous solution was measured with a spectrophotometer to determine the concentration, and the hydrophobic group content was calculated from the following formula.
Hydrophobic group content (%) = (C AG / (C HAG + C AG )) × 100
C AG : concentration of denatured agarose constituent unit (mmol / L)
= A / ε GPE × 1000
C HAG : Concentration of unmodified agarose constituent unit (mmol / L)
= (Concentration of unmodified agarose constituent unit (g / L) / Agarose constituent unit (306 g / mol)) × 1000
A: True absorbance of hydrophobic group-introduced agarose = Absorbance of agarose with hydrophobic group introduced-Absorption of unmodified agarose • ε GPE : Absorption coefficient of glycidyl phenyl ether = 1372 (L / (mol · cm))
Absorption of undenatured agarose = absorbance of undenatured agarose × (sample concentration of denatured agarose (mmol / L) / sample concentration of unmodified agarose (mmol / L))
Non-denatured agarose constituent unit concentration (g / L) = denatured agarose sample concentration (mass%) × 10—denatured agarose constituent unit concentration (g / L)
Denatured agarose constituent unit concentration (g / L) = (C AG × modified agarose constituent unit (456 g / mol)) / 1000
(水酸基を有する高分子の変性体の粒子へのコーティング)
20mg/mLの変性アガロース水溶液に多孔質高分子粒子1を70mL/粒子gの濃度で投入し、55℃で24h撹拌して、多孔質高分子粒子1に変性アガロースを吸着させた。次いで、変性アガロースを吸着させた多孔質高分子粒子1をろ別して、更に熱水で洗浄した。
(Coating on particles of modified polymer having a hydroxyl group)
Porous polymer particles 1 were put into a 20 mg / mL modified agarose aqueous solution at a concentration of 70 mL / particle g and stirred at 55 ° C. for 24 hours to adsorb the modified agarose to the porous polymer particles 1. Subsequently, the porous polymer particles 1 on which the modified agarose was adsorbed were separated by filtration and further washed with hot water.
(コーティングした水酸基を有する高分子の変性体の架橋)
多孔質高分子粒子1の表面及び細孔内部に吸着した変性アガロースを次のようにして架橋した。変性アガロースが吸着した粒子10gを0.4M水酸化ナトリウム水溶液に分散させ、0.02Mエピクロロヒドリンを添加し、24時間室温にて撹拌した。その後、2質量%の熱ドデシル硫酸ナトリウム水溶液で洗浄後、純水で更に洗浄し、乾燥させることで、変性アガロースの架橋体を被覆層として有する架橋体被覆粒子を得た。
(Crosslinking of a modified polymer having a coated hydroxyl group)
The modified agarose adsorbed on the surface of the porous polymer particles 1 and inside the pores was crosslinked as follows. 10 g of particles adsorbed with modified agarose were dispersed in a 0.4 M aqueous sodium hydroxide solution, 0.02 M epichlorohydrin was added, and the mixture was stirred at room temperature for 24 hours. Then, after washing with a 2% by mass of hot sodium dodecyl sulfate aqueous solution, further washing with pure water and drying were performed to obtain crosslinked-coated particles having a crosslinked product of modified agarose as a coating layer.
(タンパク質の非特異吸着能の評価)
得られた架橋体被覆粒子0.2gを、BSA(Bovine Serum Albumin)濃度24mg/mLのTris−塩酸緩衝液(pH8.0)20mLに加え、24時間室温で撹拌した。その後、遠心分離で上澄み液を採取し、分光光度計で上澄み液の280nmの吸光度を測定することによって求めた上澄み液のBSA濃度より、粒子に吸着したBSA量を算出した。粒子1mLあたりのBSA量吸着量が、1mg以下である場合を「A」、1mg超5mg未満である場合を「B」、5mg以上である場合を「C」として評価した。結果を表1に示す。
(Evaluation of non-specific adsorption ability of protein)
0.2 g of the obtained crosslinked product-coated particles was added to 20 mL of Tris-hydrochloric acid buffer (pH 8.0) having a BSA (Bovine Serum Albumin) concentration of 24 mg / mL, and stirred at room temperature for 24 hours. Thereafter, the supernatant was collected by centrifugation, and the amount of BSA adsorbed on the particles was calculated from the BSA concentration of the supernatant obtained by measuring the absorbance at 280 nm of the supernatant with a spectrophotometer. The case where the amount of adsorbed BSA per mL of particles was 1 mg or less was evaluated as “A”, the case where it was more than 1 mg and less than 5 mg as “B”, and the case where it was 5 mg or more as “C”. The results are shown in Table 1.
(カルボキシ基の導入)
得られた架橋体被覆粒子(乾燥質量1g)を、0.4Mの水酸化ナトリウム水溶液10mLに加えた後、エピクロロヒドリン0.6gを更に加え、室温で撹拌しながら3時間反応させた。反応終了後、粒子をろ別し、500mLの水で洗浄して、架橋体にエポキシ基を導入した。次いで、粒子に、水10mL、及び1Mのメルカプト酢酸ナトリウム水溶液10mLを加え、室温で24時間撹拌し、反応させた。反応終了後、粒子をろ別し、1000mLの水で洗浄して、架橋体にカルボキシ基を導入した。これにより本実施形態の分離材を得た。なお、本実施例における官能基導入剤は、メルカプト酢酸ナトリウムである。
(Introduction of carboxy group)
The obtained crosslinked product-coated particles (dry mass 1 g) were added to 10 mL of a 0.4 M aqueous sodium hydroxide solution, 0.6 g of epichlorohydrin was further added, and the mixture was reacted at room temperature for 3 hours with stirring. After completion of the reaction, the particles were filtered off and washed with 500 mL of water to introduce epoxy groups into the crosslinked product. Next, 10 mL of water and 10 mL of 1M aqueous solution of mercaptoacetate were added to the particles, and the mixture was stirred at room temperature for 24 hours to be reacted. After completion of the reaction, the particles were filtered off and washed with 1000 mL of water to introduce a carboxy group into the crosslinked product. Thereby, the separation material of this embodiment was obtained. In addition, the functional group introducing agent in this example is sodium mercaptoacetate.
(カルボキシ基量の測定)
得られた分離材の官能基量を以下のように測定した。湿潤状態の分離材0.5gに、0.1Nの塩酸水溶液20gを加え、室温で30分撹拌した。撹拌後の分離材を吸引してろ別し、さらに、水500mLで洗浄し、洗浄液のpHが5以上であることを確認した。洗浄後の分離材に0.5Mの塩化ナトリウム水溶液10gを加え、室温で30分撹拌した。撹拌後の分離材の懸濁液を0.01N水酸化ナトリウム水溶液でpH7を終点として滴定することにより、乾燥状態の分離材1g当たりのカルボキシ基のモル量(mmol)を測定した。この結果を官能基密度(mmol/粒子g)として、表1に示す。
(Measurement of carboxy group amount)
The functional group amount of the obtained separating material was measured as follows. 20 g of 0.1N aqueous hydrochloric acid solution was added to 0.5 g of the wet separating material, and the mixture was stirred at room temperature for 30 minutes. The separation material after stirring was filtered by suction, and further washed with 500 mL of water, and it was confirmed that the pH of the cleaning solution was 5 or more. 10 g of a 0.5 M aqueous sodium chloride solution was added to the separation material after washing, and the mixture was stirred at room temperature for 30 minutes. By titrating the suspension of the separated material after stirring with a 0.01N sodium hydroxide aqueous solution at an end point of pH 7, the molar amount (mmol) of carboxy groups per 1 g of the separated material in a dry state was measured. This result is shown in Table 1 as the functional group density (mmol / particle g).
(リガンドの固定化)
得られた分離材0.09gに、プロテインAを0.02g、WSC(1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩)を0.02g、及びpH5.8に調整した50mMのPBSを2ml加えたのち、室温で24時間撹拌した。その後、粒子をろ別及び洗浄することによりリガンド固定化粒子を得た。
(Immobilization of ligand)
To 0.09 g of the obtained separating material, 0.02 g of protein A, 0.02 g of WSC (1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride), and 50 mM adjusted to pH 5.8 After adding 2 ml of PBS, the mixture was stirred at room temperature for 24 hours. Thereafter, the particles were filtered and washed to obtain ligand-immobilized particles.
(リガンド固定化量の算出)
24時間撹拌後の反応溶液から粒子をろ別した後、所定濃度の粒子を含む試料を調製した。当該試料の吸光度(280nm)を測定し、予め作成した検量線により、リガンド固定化量を算出した。結果を表1に示す。
(Calculation of ligand immobilization amount)
After filtering the particles from the reaction solution after stirring for 24 hours, a sample containing particles having a predetermined concentration was prepared. The absorbance (280 nm) of the sample was measured, and the amount of immobilized ligand was calculated using a calibration curve prepared in advance. The results are shown in Table 1.
(水中での5%圧縮変形弾性率の算出)
得られた分離材の水中での5%圧縮変形弾性率を以下のようにして算出した。結果を表1に示す。
(Calculation of 5% compressive deformation modulus in water)
The 5% compressive deformation elastic modulus of the obtained separating material in water was calculated as follows. The results are shown in Table 1.
微小圧縮試験機(Fisher製)を用いて、室温(20〜25℃)条件にて荷重負荷速度1mN/秒で、四角柱の平滑な端面(50μm×50μm)により、予め水中に浸漬させた分離材を50mNまで圧縮したときの荷重及び圧縮変位を測定する。得られた測定値から、分離材が5%圧縮変形したときの圧縮弾性率(5%K値)を下記式により求めることができる。また、上記測定中の変位量が最も大きく変化する点の荷重を破壊強度(mN)とする。 Separation using a micro compression tester (manufactured by Fisher) at room temperature (20 to 25 ° C.) at a load load rate of 1 mN / sec. And a smooth end face (50 μm × 50 μm) of a quadrangular column in advance. The load and compression displacement when the material is compressed to 50 mN are measured. From the measured value obtained, the compression modulus (5% K value) when the separating material is 5% compressively deformed can be obtained by the following equation. Further, the load at the point at which the displacement during the measurement changes most greatly is defined as the breaking strength (mN).
5%K値(MPa)=(3/21/2)・F・S−3/2・R−1/2
F:分離材が5%圧縮変形したときの荷重(mN)
S:分離材が5%圧縮変形したときの圧縮変位(mm)
R:分離材の半径(mm)
5% K value (MPa) = (3/2 1/2 ) · F · S −3 / 2 · R −1/2
F: Load (mN) when the separating material is 5% compressively deformed
S: Compression displacement (mm) when the separating material is 5% compressively deformed
R: radius of separating material (mm)
(カラム特性評価)
得られた分離材を、濃度30質量%のスラリー(溶媒:メタノール)として、φ7.8×300mmのステンレスカラムに、15分充填した。その後、カラムに流速を変えながら水を通し、流速とカラム圧との関係を測定し、0.3MPa時の通液速度(線流速)を測定した。結果を表1に示す。
(Column characteristic evaluation)
The obtained separating material was packed in a φ7.8 × 300 mm stainless steel column as a slurry (solvent: methanol) with a concentration of 30% by mass for 15 minutes. Thereafter, water was passed through the column while changing the flow rate, the relationship between the flow rate and the column pressure was measured, and the liquid flow rate (linear flow rate) at 0.3 MPa was measured. The results are shown in Table 1.
(実施例2)
実施例1と同様にして、架橋体被覆粒子を作製した。
(Example 2)
In the same manner as in Example 1, crosslinked product-coated particles were produced.
(カルボキシ基の導入)
得られた架橋体被覆粒子(乾燥質量1g)を、0.4Mの水酸化ナトリウム水溶液10mLに加えた後、エピクロロヒドリン0.6gを更に加え、室温で撹拌しながら3時間反応させた。反応終了後、粒子をろ別し、500mLの水で洗浄して、架橋体にエポキシ基を導入した。次いで、粒子に、水酸化ナトリウム5gを水15mLに溶解させた水酸化ナトリウム水溶液15mLと、メルカプトプロピオン酸5gとを加え、室温で24時間撹拌し、反応させた。反応終了後、粒子をろ別し、1000mLの水で洗浄して、架橋体にカルボキシ基を導入した。これにより本実施形態の分離材を得た。なお、本実施例における官能基導入剤は、メルカプトプロピオン酸である。
(Introduction of carboxy group)
The obtained crosslinked product-coated particles (dry mass 1 g) were added to 10 mL of a 0.4 M aqueous sodium hydroxide solution, 0.6 g of epichlorohydrin was further added, and the mixture was reacted at room temperature for 3 hours with stirring. After completion of the reaction, the particles were filtered off and washed with 500 mL of water to introduce epoxy groups into the crosslinked product. Next, 15 mL of an aqueous sodium hydroxide solution in which 5 g of sodium hydroxide was dissolved in 15 mL of water and 5 g of mercaptopropionic acid were added to the particles, and the mixture was stirred at room temperature for 24 hours to be reacted. After completion of the reaction, the particles were filtered off and washed with 1000 mL of water to introduce a carboxy group into the crosslinked product. Thereby, the separation material of this embodiment was obtained. The functional group introducing agent in this example is mercaptopropionic acid.
また、実施例1と同様にして、平均粒径、タンパク質の非特異吸着能、カルボキシ基量(官能基密度)、水中での5%圧縮変形弾性率、及びカラム特性(通液速度)を評価した。また、実施例1と同様にして、リガンドの固定化を行い、リガンド固定化量を算出した。結果を表1に示す。 Further, in the same manner as in Example 1, the average particle size, the nonspecific adsorption ability of the protein, the amount of carboxy groups (functional group density), the 5% compressive deformation elastic modulus in water, and the column characteristics (liquid flow rate) were evaluated. did. Further, in the same manner as in Example 1, the ligand was immobilized, and the amount of immobilized ligand was calculated. The results are shown in Table 1.
(実施例3)
実施例1と同様にして、架橋体被覆粒子を作製した。
(Example 3)
In the same manner as in Example 1, crosslinked product-coated particles were produced.
(エポキシ基の導入)
得られた架橋体被覆粒子(乾燥質量1g)を、0.4Mの水酸化ナトリウム水溶液10mLに加えた後、エピクロロヒドリン0.6gを更に加え、室温で撹拌しながら3時間反応させた。反応終了後、粒子をろ別し、500mLの水で洗浄し、架橋体にエポキシ基を導入した。これにより本実施形態の分離材を得た。
(Introduction of epoxy group)
The obtained crosslinked product-coated particles (dry mass 1 g) were added to 10 mL of a 0.4 M aqueous sodium hydroxide solution, 0.6 g of epichlorohydrin was further added, and the mixture was reacted at room temperature for 3 hours with stirring. After completion of the reaction, the particles were filtered off and washed with 500 mL of water to introduce epoxy groups into the crosslinked product. Thereby, the separation material of this embodiment was obtained.
(エポキシ基量の測定)
得られた分離材の官能基量を以下のように測定した。湿潤状態の分離材0.5gに水10g,1Nの塩酸水溶液1g加え、70℃で1時間撹拌し、反応させた。反応後、0.1N水酸化ナトリウムで中和滴定することにより、乾燥状態の分離材1g当たりのエポキシ基のモル量(mmol)を測定した。この結果を官能基密度(mmol/粒子g)として、表1に示す。
(Measurement of epoxy group content)
The functional group amount of the obtained separating material was measured as follows. 10 g of water and 1 g of a 1N aqueous hydrochloric acid solution were added to 0.5 g of a wet separating material, and the mixture was stirred at 70 ° C. for 1 hour to be reacted. After the reaction, neutralization titration with 0.1N sodium hydroxide was performed to measure the molar amount (mmol) of the epoxy group per 1 g of the separating material in a dry state. This result is shown in Table 1 as the functional group density (mmol / particle g).
(リガンドの固定化)
得られた分離材0.09gに、プロテインAを0.02g、及びpH10の0.5M炭酸バッファー(和光純薬工業社製の炭酸水素ナトリウム及び炭酸ナトリウムと、RO水とで調製)を2mL加え、30℃で24時間撹拌した。その後、粒子をろ別及び洗浄することによりリガンド固定化粒子を得た。
(Immobilization of ligand)
To 0.09 g of the obtained separation material, 0.02 g of protein A, and 2 mL of 0.5 M carbonate buffer (prepared with sodium hydrogen carbonate and sodium carbonate manufactured by Wako Pure Chemical Industries, Ltd. and RO water) at pH 10 are added. , And stirred at 30 ° C. for 24 hours. Thereafter, the particles were filtered and washed to obtain ligand-immobilized particles.
(リガンド固定化量の算出)
24時間撹拌後の反応溶液から粒子をろ別した後、所定濃度の粒子を含む試料を調製した。当該試料の吸光度(280nm)を測定し、予め作成した検量線により、リガンド固定化量を算出した。結果を表1に示す。
(Calculation of ligand immobilization amount)
After filtering the particles from the reaction solution after stirring for 24 hours, a sample containing particles having a predetermined concentration was prepared. The absorbance (280 nm) of the sample was measured, and the amount of immobilized ligand was calculated using a calibration curve prepared in advance. The results are shown in Table 1.
また、実施例1と同様にして、平均粒径、タンパク質の非特異吸着能、水中での5%圧縮変形弾性率、及びカラム特性(通液速度)を評価した。結果を表1に示す。 Further, in the same manner as in Example 1, the average particle size, the nonspecific adsorption ability of the protein, the 5% compressive deformation modulus in water, and the column characteristics (liquid passing speed) were evaluated. The results are shown in Table 1.
(実施例4)
実施例1と同様にして、架橋体被覆粒子を作製した。
Example 4
In the same manner as in Example 1, crosslinked product-coated particles were produced.
(アミノ基の導入)
得られた架橋体被覆粒子(乾燥質量20g)を、5Mの水酸化ナトリウム水溶液200mLに加え、室温で1時間放置した。別途、ジエチルアミノエチルクロライド塩酸塩の所定量(10g)を200mL添加し、水溶液の温度を70℃まで上げ、撹拌しながら2時間反応させた。反応終了後、ろ過、水/エタノール(体積比5/1)で3回洗浄し、架橋体にジエチルアミノエチル(DEAE)基を導入した。これにより本実施形態の分離材を得た。
(Introduction of amino group)
The obtained crosslinked product-coated particles (dry mass 20 g) were added to 200 mL of a 5 M aqueous sodium hydroxide solution and left at room temperature for 1 hour. Separately, 200 mL of a predetermined amount (10 g) of diethylaminoethyl chloride hydrochloride was added, the temperature of the aqueous solution was raised to 70 ° C., and the mixture was reacted for 2 hours with stirring. After completion of the reaction, the mixture was filtered and washed three times with water / ethanol (volume ratio 5/1) to introduce a diethylaminoethyl (DEAE) group into the crosslinked product. Thereby, the separation material of this embodiment was obtained.
(アミノ基量の測定)
得られた分離材の官能基量を以下のように測定した。湿潤状態の分離材0.5gを0.1N水酸化ナトリウム20mLに1時間浸漬し、室温で撹拌した。その後、溶液のpHが7以下となるように水で洗浄を行った。洗浄後の分離材を0.1N塩酸20mLに浸漬し、1時間撹拌させた。撹拌後の分離材をろ別後、塩酸水溶液を中和滴定することにより、乾燥状態の分離材1g当たりのアミノ基のモル量(mmol)を測定した。この結果を官能基密度(mmol/粒子g)として、表1に示す。
(Measurement of amino group content)
The functional group amount of the obtained separating material was measured as follows. 0.5 g of the wet separating material was immersed in 20 mL of 0.1N sodium hydroxide for 1 hour and stirred at room temperature. Thereafter, the solution was washed with water so that the pH of the solution was 7 or less. The separation material after washing was immersed in 20 mL of 0.1N hydrochloric acid and stirred for 1 hour. The separation material after stirring was filtered off, and the hydrochloric acid aqueous solution was neutralized and titrated to measure the molar amount (mmol) of amino groups per 1 g of the separation material in a dry state. This result is shown in Table 1 as the functional group density (mmol / particle g).
また、実施例1と同様にして、平均粒径、タンパク質の非特異吸着能、水中での5%圧縮変形弾性率、及びカラム特性(通液速度)を評価した。結果を表1に示す。 Further, in the same manner as in Example 1, the average particle size, the nonspecific adsorption ability of the protein, the 5% compressive deformation modulus in water, and the column characteristics (liquid passing speed) were evaluated. The results are shown in Table 1.
(比較例1)
市販のアガロース粒子(Capto DEAE(GEヘルスケア製、商品名))をそのまま分離材として用い、実施例1と同様に、平均粒径、タンパク質の非特異吸着能、水中での5%圧縮変形弾性率、及びカラム特性(通液速度)を評価した。結果を表1に示す。
(Comparative Example 1)
Commercially available agarose particles (Capto DEAE (manufactured by GE Healthcare, trade name)) were used as they were as separation materials, and in the same manner as in Example 1, average particle size, nonspecific adsorption ability of proteins, 5% compression deformation elasticity in water The rate and column characteristics (liquid flow rate) were evaluated. The results are shown in Table 1.
(比較例2)
多孔質高分子粒子1をそのまま分離材として用い、実施例1と同様に、平均粒径、タンパク質の非特異吸着能、水中での5%圧縮変形弾性率、及びカラム特性(通液速度)を評価した。結果を表1に示す。
(Comparative Example 2)
Using the porous polymer particles 1 as a separating material as they were, the same as in Example 1, the average particle size, the nonspecific adsorption ability of the protein, the 5% compressive deformation elastic modulus in water, and the column characteristics (liquid flow rate) evaluated. The results are shown in Table 1.
表1の結果から、実施例1〜4の分離材は、タンパク質の非特異吸着が低減され、カラムに充填したときの通液性に優れていることがわかる。また、実施例1〜4の分離材は、水中での5%圧縮変形弾性率も高いことがわかる。 From the results in Table 1, it can be seen that the separation materials of Examples 1 to 4 have reduced nonspecific adsorption of proteins and are excellent in liquid permeability when packed in a column. Moreover, it turns out that the separating material of Examples 1-4 has a high 5% compressive deformation elastic modulus in water.
Claims (11)
該疎水性高分子粒子の表面の少なくとも一部を被覆する被覆層と、を備え、
前記被覆層が、水酸基及び水酸基以外の官能基を有する官能基含有高分子を含む、分離材。 Hydrophobic polymer particles,
A coating layer covering at least a part of the surface of the hydrophobic polymer particles,
The separating material, wherein the coating layer includes a functional group-containing polymer having a hydroxyl group and a functional group other than a hydroxyl group.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016086980A JP6759679B2 (en) | 2016-04-25 | 2016-04-25 | Separator and column |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016086980A JP6759679B2 (en) | 2016-04-25 | 2016-04-25 | Separator and column |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017196544A true JP2017196544A (en) | 2017-11-02 |
| JP6759679B2 JP6759679B2 (en) | 2020-09-23 |
Family
ID=60237002
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016086980A Active JP6759679B2 (en) | 2016-04-25 | 2016-04-25 | Separator and column |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP6759679B2 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020036151A1 (en) * | 2018-08-14 | 2020-02-20 | 日立化成株式会社 | Separating material, method of manufacturing separating material, and column |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6463858A (en) * | 1987-04-24 | 1989-03-09 | Unilever Nv | Substrate and manufacture thereof |
| JPH01254247A (en) * | 1988-04-01 | 1989-10-11 | Mitsubishi Kasei Corp | Complex separation agent and manufacture thereof |
| US5030352A (en) * | 1990-01-25 | 1991-07-09 | Purdue Research Foundation | Coated media for chromatography |
| JPH055731A (en) * | 1991-06-28 | 1993-01-14 | Sekisui Chem Co Ltd | Carrier for affinity chromatography |
| JP2002507934A (en) * | 1997-06-25 | 2002-03-12 | アメルシャム・ファルマシア・バイオテック・アクチボラグ | Coated polymer articles and uses thereof |
| JP2008232764A (en) * | 2007-03-19 | 2008-10-02 | Tosoh Corp | Novel filler for filled bed and use therefor |
| WO2011125673A1 (en) * | 2010-03-31 | 2011-10-13 | Jsr株式会社 | Filler for affinity chromatography |
| JP2013512078A (en) * | 2009-12-01 | 2013-04-11 | エクステラ・メディカル・エルエルシー | Method for removing cytokines from blood using surface-immobilized polysaccharides |
-
2016
- 2016-04-25 JP JP2016086980A patent/JP6759679B2/en active Active
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6463858A (en) * | 1987-04-24 | 1989-03-09 | Unilever Nv | Substrate and manufacture thereof |
| JPH01254247A (en) * | 1988-04-01 | 1989-10-11 | Mitsubishi Kasei Corp | Complex separation agent and manufacture thereof |
| US5030352A (en) * | 1990-01-25 | 1991-07-09 | Purdue Research Foundation | Coated media for chromatography |
| JPH04505968A (en) * | 1990-01-25 | 1992-10-15 | パーデュー・リサーチ・ファウンデーション | Coating media for chromatography |
| JPH055731A (en) * | 1991-06-28 | 1993-01-14 | Sekisui Chem Co Ltd | Carrier for affinity chromatography |
| JP2002507934A (en) * | 1997-06-25 | 2002-03-12 | アメルシャム・ファルマシア・バイオテック・アクチボラグ | Coated polymer articles and uses thereof |
| JP2008232764A (en) * | 2007-03-19 | 2008-10-02 | Tosoh Corp | Novel filler for filled bed and use therefor |
| JP2013512078A (en) * | 2009-12-01 | 2013-04-11 | エクステラ・メディカル・エルエルシー | Method for removing cytokines from blood using surface-immobilized polysaccharides |
| WO2011125673A1 (en) * | 2010-03-31 | 2011-10-13 | Jsr株式会社 | Filler for affinity chromatography |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020036151A1 (en) * | 2018-08-14 | 2020-02-20 | 日立化成株式会社 | Separating material, method of manufacturing separating material, and column |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6759679B2 (en) | 2020-09-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6790834B2 (en) | Separator | |
| JP6848203B2 (en) | Separator and column | |
| WO2016117574A1 (en) | Separation material | |
| JPWO2018181738A1 (en) | Separation material | |
| JP6911464B2 (en) | Separator and column | |
| JP6778381B2 (en) | Separator | |
| JP6972606B2 (en) | Separation material, column, and method for manufacturing the separation material | |
| JP2021007915A (en) | Production method for coated particle and column production method | |
| JP6759679B2 (en) | Separator and column | |
| JP2017125815A (en) | Separation material and column | |
| WO2020036151A1 (en) | Separating material, method of manufacturing separating material, and column | |
| JP6746919B2 (en) | Separation material and column | |
| JP6676975B2 (en) | Separation materials and columns | |
| JP6874276B2 (en) | Separator and column | |
| JP6939021B2 (en) | Separator and column | |
| JP6926573B2 (en) | Separator and column | |
| WO2018174022A1 (en) | Separation material | |
| JP6852259B2 (en) | Separator and column | |
| JP6834132B2 (en) | Separator and column | |
| JP6676976B2 (en) | Separation materials and columns | |
| JP6778378B2 (en) | Separator and column | |
| JP2020015809A (en) | Porous polymer particle, separation material, column and method for producing porous polymer particle | |
| WO2018181925A1 (en) | Separating material for gel filtration and method for purifying water-soluble polymer substance | |
| JP2020018989A (en) | Separation material | |
| JP6834130B2 (en) | Separator and column |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20190325 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20200115 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200121 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200319 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20200804 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20200817 |
|
| R151 | Written notification of patent or utility model registration |
Ref document number: 6759679 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R151 |