JP2017193564A - 無水形態のピリジン誘導体 - Google Patents
無水形態のピリジン誘導体 Download PDFInfo
- Publication number
- JP2017193564A JP2017193564A JP2017114602A JP2017114602A JP2017193564A JP 2017193564 A JP2017193564 A JP 2017193564A JP 2017114602 A JP2017114602 A JP 2017114602A JP 2017114602 A JP2017114602 A JP 2017114602A JP 2017193564 A JP2017193564 A JP 2017193564A
- Authority
- JP
- Japan
- Prior art keywords
- formula
- compound
- disorders
- crystalline form
- anhydrous crystalline
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- LNYOHOASVKBDQJ-IGSIKQDTSA-N CC(C)(C(N(C)c(c(-c(cc1)c(C)cc1F)c1)cnc1N(C1)[C@H](CO)CN2[C@@H]1COCC2)O)c1cc(C(F)(F)F)cc(C(F)(F)F)c1 Chemical compound CC(C)(C(N(C)c(c(-c(cc1)c(C)cc1F)c1)cnc1N(C1)[C@H](CO)CN2[C@@H]1COCC2)O)c1cc(C(F)(F)F)cc(C(F)(F)F)c1 LNYOHOASVKBDQJ-IGSIKQDTSA-N 0.000 description 1
- HVZDJZDLYLRRKC-MOJJIBGUSA-N CC(C)(C1OC1N(C)C1C=NC(N(C2)[C@H](CO)CN3[C@@H]2COCC3)=CC1c(cc1)c(C)cc1F)c1cc(C(F)(F)F)cc(C(F)(F)F)c1 Chemical compound CC(C)(C1OC1N(C)C1C=NC(N(C2)[C@H](CO)CN3[C@@H]2COCC3)=CC1c(cc1)c(C)cc1F)c1cc(C(F)(F)F)cc(C(F)(F)F)c1 HVZDJZDLYLRRKC-MOJJIBGUSA-N 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Psychiatry (AREA)
- Addiction (AREA)
- Anesthesiology (AREA)
- Pain & Pain Management (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
【課題】精神障害、うつ病、気分障害、不安症、睡眠障害および物質関連障害に有効な治療薬又は予防薬の提供。【解決手段】図1に示す粉末X線回折(XRPD)パターンによって特徴づけられる、式(I)で表される化合物の無水結晶形態1。及び1種類以上の医薬的に許容される担体又は希釈剤を含んでなる、医薬組成物。前記化合物を精神障害、うつ病、気分障害、不安症、睡眠障害及び物質関連障害の治療又は予防のために用いる方法。【選択図】図1
Description
本発明は、無水結晶形態の2−[3,5−ビス(トリフルオロメチル)フェニル]−N−{4−(4−フルオロ−2−メチルフェニル)−6−[(7S,9aS)−7−(ヒドロキシメチル)ヘキサヒドロピラジノ[2,1−c][1,4]オキサジン−8(1H)−イル]−3−ピリジニル}−N,2−ジメチルプロパンアミド、それを含んでなる医薬製剤、治療におけるそれらの使用およびそれらを調製するプロセスに関する。この化合物は、NK1およびNK3受容体のアンタゴニストであるので、精神障害、うつ病、気分障害、不安症、睡眠障害および物質関連障害の治療に使用することができる。
WO07/028654は、NK1およびNK3受容体のアンタゴニストであり、精神障害の治療に使用することができるいくつかのピリジン誘導体またはその医薬的に許容可能な塩について記載している。特に、化合物2−[3,5−ビス(トリフルオロメチル)フェニル]−N−{4−(4−フルオロ−2−メチルフェニル)−6−[(7S,9aS)−7−(ヒドロキシメチル)ヘキサヒドロピラジノ[2,1−c][1,4]オキサジン−8(1H)−イル]−3−ピリジニル}−N,2−ジメチルプロパンアミドまたはその医薬的に許容される塩について、WO07/028654に記載されている。
2−[3,5−ビス(トリフルオロメチル)フェニル]−N−{4−(4−フルオロ−2−メチルフェニル)−6−[(7S,9aS)−7−(ヒドロキシメチル)ヘキサヒドロピラジノ[2,1−c][1,4]オキサジン−8(1H)−イル]−3−ピリジニル}−N,2−ジメチルプロパンアミドの塩酸塩についてもWO07/028654に記載されている。
2−[3,5−ビス(トリフルオロメチル)フェニル]−N−{4−(4−フルオロ−2−メチルフェニル)−6−[(7S,9aS)−7−(ヒドロキシメチル)ヘキサヒドロピラジノ[2,1−c][1,4]オキサジン−8(1H)−イル]−3−ピリジニル}−N,2−ジメチルプロパンアミドの構造を、以下の式Iに示す。
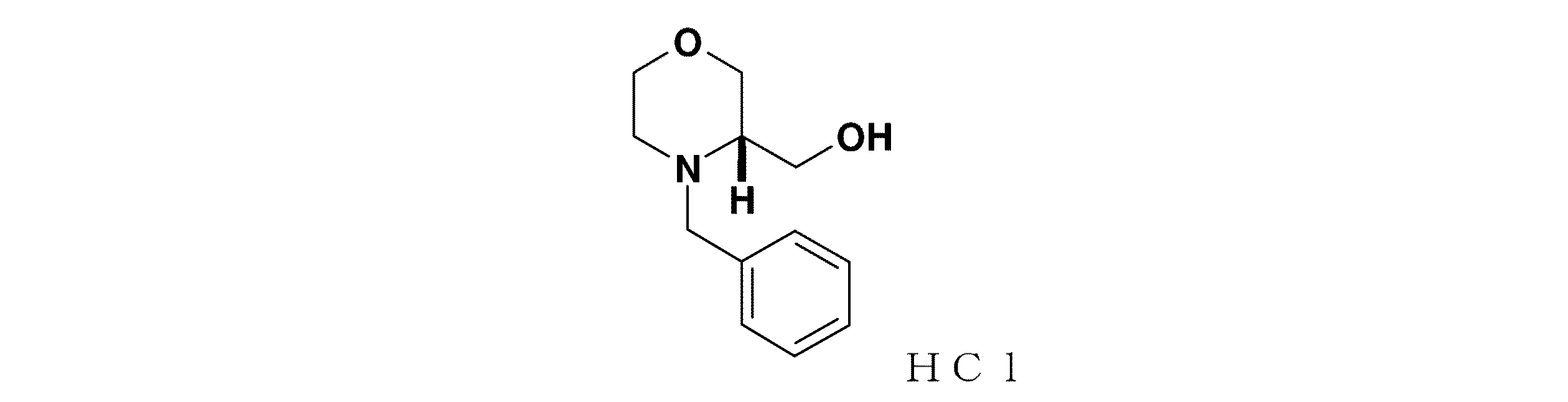
式(I)の化合物の医薬的に許容される塩には、塩酸、臭化水素酸、ヨウ化水素酸、リン酸、メタリン酸、硝酸および硫酸などの無機酸と形成された酸付加塩、ならびに酒石酸、酢酸、トリフルオロ酢酸、クエン酸、リンゴ酸、乳酸、フマル酸、安息香酸、ギ酸、プロピオン酸、グリコール酸、グルコン酸、マレイン酸、コハク酸、カンファースルホン酸、イソチオン酸、粘液酸、ゲンチシン酸、イソニコチン酸、サッカリン酸、グルクロン酸、フロン酸、グルタミン酸、アスコルビン酸、アンスラニル酸、サリチル酸、フェニル酢酸、マンデル酸、エンボン酸(パモ酸)、メタンスルホン酸、エタンスルホン酸、パントテン酸、ステアリン酸、スルファニル酸、アルギン酸、ガラクツロン酸、アリールスルホン酸、例えば、ベンゼンスルホン酸および4−メチルベンゼンスルホン酸などの有機酸と形成された酸付加塩が含まれる。
式(I)の化合物またはその塩酸塩は、WO07/028654に記載の手順に従って、部分的に非結晶または完全に非結晶な固体として得られ、これは吸湿性がある。非結晶固体および特に吸湿性の固体は、一般的には、低かさ高密度であり、かつ流動性が不十分であるので、製剤加工条件下で取り扱いが難しい。
したがって、製剤加工および医薬組成物において都合よく使用することができる優れた生理化学的特性を有する結晶形態の式(I)の化合物のニーズがある。
式(I)を有する無水結晶形態の2−[3,5−ビス(トリフルオロメチル)フェニル]−N−{4−(4−フルオロ−2−メチルフェニル)−6−[(7S,9aS)−7−(ヒドロキシメチル)ヘキサヒドロピラジノ[2,1−c][1,4]オキサジン−8(1H)−イル]−3−ピリジニル}−N,2−ジメチルプロパンアミドを今見出した。
本発明の第1の態様において、無水結晶形態の式(I)の化合物を提供する。
本発明の第2の態様において、無水結晶形態1(Form1)の式(I)の化合物を提供する。
本発明の第3の態様において、無水結晶形態1(Form1)の式(I)の化合物を提供し、このXRPDパターンは2θ角の単位で表され、銅KαX線照射を用いる回折計により得られる。
本発明の第4の態様において、図1と実質的に同じ粉末X線回折(XRPD)パターンによって特徴づけられる無水結晶形態1(Form1)の式(I)の化合物を提供し、このXRPDパターンは2θ角の単位で表され、銅KαX線照射を用いる回折計により得られ、このXRPDパターンは、本質的に以下の位置4.3±0.1、7.9±0.1、9.8±0.1、10.7±0.1、10.8±0.1、13.3±0.1、14.0±0.1、15.1±0.1°に2θ角ピークを含んでなり、これらの値は、それぞれ、20.4、11.1、9.0、8.3、8.2、6.6、6.3および5.9オングストローム(Å)の格子面間隔(d−spacing)に対応する。
本発明の第5の態様において、図1と実質的に同じ粉末X線回折(XRPD)パターンによって特徴づけられる無水結晶形態1(Form1)の式(I)の化合物を提供し、このXRPDパターンは2θ角の単位で表され、銅KαX線照射を用いる回折計により得られ、このXRPDパターンは、本質的に以下の位置4.3±0.1、7.9±0.1、9.8±0.1、10.7±0.1、10.8±0.1、13.1±0.1、13.2±0.1、13.3±0.1、14.0±0.1、14.4±0.1、15.0±0.1、15.1±0.1、15.7±0.1、15.9±0.1、16.3±0.1、16.5±0.1、16.8±0.1、17.0±0.1、17.4±0.1、17.5±0.1、18.1±0.1、18.2±0.1、18.7±0.1、19.4±0.1、19.7±0.1、20.0±0.1、20.1±0.1、20.2±0.1、20.5±0.1、20.7±0.1、21.5±0.1、21.8±0.1°に2θ角ピークを含んでなり、これらの値は、それぞれ、20.4、11.1、9.0、8.3、8.2、6.8、6.7、6.6、6.3、6.1、5.9、5.6、5.6、5.4、5.4、5.3、5.2、5.1、5.1、4.9、4.9、4.7、4.6、4.5、4.4、4.4、4.4、4.3、4.3、4.1および4.1オングストローム(Å)の格子面間隔(d−spacing)に対応する。
第6の態様として、本発明は、図3と実質的に同じ13C固体核磁気共鳴(SSNMR)スペクトルによって特徴づけられる無水結晶形態1(Form1)の式(I)の化合物を提供し、このSSNMRスペクトルは、13Cの観察のために100.56MHzの周波数で作動する分光計において、8kHzの回転周波数で、Bruker社製4mmの3重共鳴マジックアングルスピンプローブを用いる交差分極パルスシーケンスを用いて得られた。
第7の態様として、本発明は、13Cの観察のために100.56MHzの周波数で作動する分光計において、8kHzの回転周波数で、Bruker社製4mmの3重共鳴マジックアングルスピンプローブを用いる交差分極パルスシーケンスを用いて得られたSSNMRスペクトルによって特徴づけられる無水結晶形態1(Form1)の式(I)の化合物を提供し、このSSNMRは、175.03±0.2、163.30±0.2、158.66±0.2、156.50±0.2、149.79±0.2、148.17±0.2、146.96±0.2、139.56±0.2、133.19±0.2、132.32±0.2、129.76±0.2、126.5±0.2、124.15±0.2、120.37±0.2、119.20±0.2、118.16±0.2、112.83±0.2、70.62±0.2、67.61±0.2、64.96±0.2、59.89±0.2、57.08±0.2、55.50±0.2、52.41±0.2、48.35±0.2、40.79±0.2、32.75および21.12±0.2ppmの化学シフトを含んでなる。
別の態様として、本発明は、本発明による無水結晶形態の式(I)の化合物を含んでなる医薬組成物を提供する。この医薬組成物は、さらに、1種類以上の医薬的に許容される担体または希釈剤を含んでもよい。
別の態様として、本発明は、本発明による無水結晶形態の有効量の式(I)の化合物を哺乳類に投与することを含んでなる、精神障害、うつ病、気分障害、不安症、睡眠障害および物質関連障害の治療法または予防法を提供する。
別の態様として、本発明は、本発明による無水結晶形態の有効量の式(I)の化合物を哺乳類に投与することを含んでなる、統合失調症、うつ病およびアルコール依存症の治療法または予防法を提供する。
別の態様として、本発明は、治療に使用するための、本発明による無水結晶形態の式(I)の化合物を提供する。
別の態様として、本発明は、精神障害、うつ病、気分障害、不安症、睡眠障害および物質関連障害の治療または予防のための医薬の調製における、本発明による無水結晶形態の式(I)の化合物の使用を提供する。
別の態様として、本発明は、精神障害、うつ病、気分障害、不安症、睡眠障害および物質関連障害の治療または予防に使用するための、本発明による無水結晶形態の式(I)の化合物を提供する。
別の態様として、本発明は、統合失調症、うつ病およびアルコール依存症の治療または予防のための医薬の調製における、本発明による無水結晶形態の式(I)の化合物の使用を提供する。
別の態様として、本発明は、統合失調症、うつ病およびアルコール依存症の治療または予防に使用するための、本発明による無水結晶形態の式(I)の化合物を提供する。
本明細書で使用する「有効量」という用語は、例えば、研究者または臨床医が求めている組織、系、動物もしくはヒトの生物学的または医学的反応を引き起こす薬物または医薬品の量を意味する。さらに、「治療有効量」という用語は、かかる量を受け取らなかった対応する対象と比較して、疾患、障害、もしくは副作用の治療の改善、治癒、予防、または寛解をもたらすか、または疾患もしくは障害が発展する速度の減少をもたらす任意の量を意味する。この用語は、正常な生理的機能を増強するのに効果的な量も、その範囲内に含む。
本明細書で使用する「実質的に同じ粉末X線回折パターン」という用語は、本明細書で言及する回折パターン±0.1°内の2θ値の回折ピークを有するそれらの粉末X線回折パターンが、この言及する回折パターンの範囲内にあるという意味だと理解される。同様に、「少なくとも実質的に表1のピークを含んでなる」という用語は、対象となる表の±0.1°内の2θ値の回折ピークを有するそれらの粉末X線回折パターンが、表1で言及される回折パターンの範囲内にあるという意味だと理解される。
本発明の文脈内で、本明細書で使用する指標について説明する用語は、米国精神医学会(American Psychiatric Association)が出版した「精神障害の診断および統計学的マニュアル(the Diagnostic and Statistical Manual of Mental Disorders)」第4版(DSM−IV)および/または国際疾患分類(the International Classification of Diseases)第10版(ICD−10)の中で分類されている。本明細書で述べる障害の様々なサブタイプは本発明の一部と考えられる。以下に列挙した疾患の後の括弧内の数字は、DSM−IVの分類コードを表す。
本発明の文脈内で、「精神障害」という用語には、妄想型(295.30)、解体型(295.10)、緊張型(295.20)、未分化型(295.90)および残遺型(295.60)のサブタイプを含む統合失調症;統合失調症様障害(295.40);双極型およびうつ型のサブタイプを含む統合失調感情障害(295.70);恋愛(Erotomanic)型、誇大(Gradiose)型、嫉妬型、被害型、身体型、混合型および不特定型のサブタイプを含む妄想障害(297.1);短期精神障害(298.8);共通の精神障害(297.3);妄想および幻覚を伴うサブタイプを含む一般身体疾患による精神障害;妄想(293.81)および幻覚(293.82)を伴うサブタイプを含む物質誘発性精神障害;ならびに特定不能の精神障害(298.9)が含まれる。
うつ病および気分障害という用語には、大うつ病エピソード、躁病エピソード、混合エピソードおよび軽躁エピソード;大うつ病性障害、気分変調性障害(300.4)、特定不能のうつ病性障害(311)を含むうつ病性障害;I型双極性障害、II型双極性障害(軽躁エピソードを伴う再発性大うつ病エピソード)(296.89)、気分循環性障害(301.13)および特定不能の双極性障害(296.80)を含む双極性障害;うつ病の特徴、大うつ病様エピソード、躁病の特徴および混合型の特徴を伴うサブタイプを含む一般的身体疾患による気分障害(293.83)を含む他の気分障害、(うつ病の特徴、躁病の特徴および混合型の特徴を伴うサブタイプを含む)物質誘発性気分障害および特定不能の気分障害(296.90)が含まれる。
不安症という用語には、パニック発作;広場恐怖症を伴わないパニック障害(300.01)および広場恐怖症を伴うパニック障害(300.21)を含むパニック障害;広場恐怖症、パニック障害の既往歴のない広場恐怖症(300.22)、動物型、自然環境型、血液注入傷害(Blood−Injection−Injury)型、状況型および他の型のサブタイプを含む特定恐怖症(300.29、以前は単純恐怖症)、社会恐怖症(社会不安障害、300.23)、強迫性障害(300.3)、外傷後ストレス障害(309.81)、急性ストレス障害(308.3)、全般性不安障害(300.02)、一般身体疾患による不安障害(293.84)、物質誘発性不安障害、分離不安障害(309.21)、不安を伴う適応障害(309.24)および特定不能の不安障害(300.00)が含まれる。
睡眠障害という用語には、原発性不眠症(307.42)、原発性過眠症(307.44)、ナルコレプシー(347)、呼吸関連睡眠障害(780.59)、概日リズム睡眠障害(307.45)および特定不能の睡眠異常(307.47)などの睡眠異常などの原発性睡眠障害;悪夢障害(307.47)、睡眠時驚愕症(307.46)、睡眠時遊行症(307.46)および特定不能の睡眠時異常行動(307.47)などの睡眠時随伴症などの原発性睡眠障害;別の精神障害に関連した不眠症(307.42)および別の精神障害に関連した過眠症(307.44)などの別の精神障害に関連した睡眠障害;一般身体疾患による睡眠障害、特に、神経障害、神経因性疼痛、脚不穏症候群、心臓および肺の疾患などの疾患を伴う睡眠障害;不眠型、過眠型、睡眠時随伴型および混合型のサブタイプを含む物質誘発性睡眠障害;睡眠時無呼吸症候群ならびに時差ぼけ症候群が含まれる。
物質関連障害という用語には、物質依存、物質切望および物質濫用などの物質使用障害;物質中毒、物質離脱、物質誘発性せん妄、物質誘発性持続性認知症、物質誘発性持続性健忘障害、物質誘発性精神障害、物質誘発性気分障害、物質誘発性不安障害、物質誘発性の性機能不全、物質誘発性睡眠障害および幻覚剤持続性知覚障害(フラッシュバック)などの物質誘発性障害;アルコール依存(303.90)、アルコール濫用(305.00)、アルコール中毒(303.00)、アルコール離脱(291.81)、アルコール中毒せん妄、アルコール離脱せん妄、アルコール誘発性持続性認知症、アルコール誘発性持続性健忘障害、アルコール誘発性精神障害、アルコール誘発性気分障害、アルコール誘発性不安障害、アルコール誘発性の性的機能不全、アルコール誘発性睡眠障害および特定不能のアルコール関連障害(291.9)などのアルコール関連障害;アンフェタミン依存(304.40)、アンフェタミン濫用(305.70)、アンフェタミン中毒(292.89)、アンフェタミン離脱(292.0)、アンフェタミン中毒せん妄、アンフェタミン誘発性精神障害、アンフェタミン誘発性気分障害、アンフェタミン誘発性不安障害、アンフェタミン誘発性の性的機能不全、アンフェタミン誘発性睡眠障害および特定不能のアンフェタミン関連障害(292.9)などのアンフェタミン(またはアンフェタミン様)関連障害;カフェイン中毒(305.90)、カフェイン誘発性不安障害、カフェイン誘発性睡眠障害および特定不能のカフェイン関連障害(292.9)などのカフェイン関連障害;大麻依存(304.30)、大麻濫用(305.20)、大麻中毒(292.89)、大麻中毒せん妄、大麻誘発性精神障害、大麻誘発性不安障害および特定不能の大麻関連障害(292.9)などの大麻関連障害;コカイン依存(304.20)、コカイン濫用(305.60)、コカイン中毒(292.89)、コカイン離脱(292.0)、コカイン中毒せん妄、コカイン誘発性精神障害、コカイン誘発性気分障害、コカイン誘発性不安障害、コカイン誘発性の性的機能不全、コカイン誘発性睡眠障害および特定不能のコカイン関連障害(292.9)などのコカイン関連障害;幻覚剤依存(304.50)、幻覚剤濫用(305.30)、幻覚剤中毒(292.89)、幻覚剤持続性知覚障害(フラッシュバック)(292.89)、幻覚剤中毒せん妄、幻覚剤誘発性精神障害、幻覚剤誘発性気分障害、幻覚剤誘発性不安障害および特定不能の幻覚剤関連障害(292.9)などの幻覚剤関連障害;吸入剤依存(304.60)、吸入剤濫用(305.90)、吸入剤中毒(292.89)、吸入剤中毒せん妄、吸入剤誘発性持続性認知症、吸入剤誘発性精神障害、吸入剤誘発性気分障害、吸入剤誘発性不安障害および特定不能の吸入剤関連障害(292.9)などの吸入剤関連障害;ニコチン依存(305.1)、ニコチン離脱(292.0)および特定不能のニコチン関連障害(292.9)などのニコチン関連障害;オピオイド依存(304.00)、オピオイド濫用(305.50)、オピオイド中毒(292.89)、オピオイド離脱(292.0)、オピオイド中毒せん妄、オピオイド誘発性精神障害、オピオイド誘発性気分障害、オピオイド誘発性の性的機能不全、オピオイド誘発性睡眠障害および特定不能のオピオイド関連障害(292.9)などのオピオイド関連障害;フェンシクリジン依存(304.60)、フェンシクリジン濫用(305.90)、フェンシクリジン中毒(292.89)、フェンシクリジン中毒せん妄、フェンシクリジン誘発性精神障害、フェンシクリジン誘発性気分障害、フェンシクリジン誘発性不安障害および特定不能のフェンシクリジン関連障害(292.9)などのフェンシクリジン(またはフェンシクリジン様)関連障害;鎮静剤、睡眠薬または抗不安薬依存(304.10)、鎮静剤、睡眠薬または抗不安薬濫用(305.40)、鎮静剤、睡眠薬または抗不安薬中毒(292.89)、鎮静剤、睡眠薬または抗不安薬離脱(292.0)、鎮静剤、睡眠薬または抗不安薬中毒せん妄、鎮静剤、睡眠薬または抗不安薬離脱せん妄、鎮静剤、睡眠薬または抗不安薬持続性認知症、鎮静剤、睡眠薬または抗不安薬持続性健忘障害、鎮静剤、睡眠薬または抗不安薬誘発性精神障害、鎮静剤、睡眠薬または抗不安薬誘発性気分障害、鎮静剤、睡眠薬または抗不安薬誘発性不安障害、鎮静剤、睡眠薬または抗不安薬誘発性の性的機能不全、鎮静剤、睡眠薬または抗不安薬誘発性睡眠障害および特定不能の鎮静剤、睡眠薬または抗不安薬関連障害(292.9)などの鎮静剤、睡眠薬または抗不安薬関連障害;多物質依存(304.80)などの多物質関連障害;およびタンパク質同化ステロイド、硝酸塩吸入剤および亜酸化窒素などの他の(または未知の)物質関連障害が含まれる。
驚くことに、特に良好な医薬品特性を有する式(I)の化合物を、無水結晶形態で得ることができることを今見出した。
くさび型の結合は、この結合が紙面の上にあることを示す。点線の結合は、この結合が紙面の下にあることを示す。
式(I)の化合物の多形相は、粉末X線回折(XRPD)、示差走査熱量測定(DSC)および固体NMR(SSNMR)を含むが、これらに限定されないいくつかの従来の分析技術を用いて特徴づけ、かつ区別することができる。
多形は、2つ以上の別々の結晶相に結晶化する成分または化合物の能力と定義される。
したがって、多形は同じ分子式を共有する別々の固体であるが、全ての固体の特性はその構造によってきまるので、異なる多形は、異なる溶解度特性、異なる融点、異なる溶解特性、異なる熱安定性および/または光安定性、異なる有効期限、異なる懸濁特性および異なる生理学的吸収速度などの別々の物理的特性を示し得る。結晶性固体中に溶媒を含むと溶媒和物になり、溶媒が水の場合は水和物になる。
したがって、多形は同じ分子式を共有する別々の固体であるが、全ての固体の特性はその構造によってきまるので、異なる多形は、異なる溶解度特性、異なる融点、異なる溶解特性、異なる熱安定性および/または光安定性、異なる有効期限、異なる懸濁特性および異なる生理学的吸収速度などの別々の物理的特性を示し得る。結晶性固体中に溶媒を含むと溶媒和物になり、溶媒が水の場合は水和物になる。
したがって、本発明は、無水結晶形態の式(I)の化合物を提供する。
一実施形態において、式(I)の化合物の無水結晶形態は形態1である。
別の実施形態において、無水結晶形態1(Form1)の式(I)の化合物は、図1と実質的に同じ粉末X線回折(XRPD)パターンによって特徴づけられ、このXRPDパターンは2θ角の単位で表され、銅KαX線照射を用いる回折計により得られる。
この無水結晶形態1(Form1)の粉末X線回折(XRPD)パターンは、分析化学および物理的評価の技術者に公知の従来の技術および機器を用いて決定することができる。図1の回折パターンは、X’Celerator検出器を用いて、PANalytical社製X’Pert Pro粉末回折計、モデルPW3050/60で取得した。取得条件は、照射:Cu Kα、発生電圧:45kV、発生電流:40mA、ステップサイズ:0.008°2θ、ステップあたりの時間:575秒、発散スリットの種類:固定、発散スリットのサイズ:0.4354°、測定温度:20〜25℃の範囲、角度計の半径:240mmである。試料を0.7mmの毛細管に詰めることによって、試料を調製した。
特有のXRPD角および格子面間隔(d−spacing)を表1に示す。誤差範囲は、ピーク指定のそれぞれについて約±0.1°2θである。
特有のXRPD角および格子面間隔(d−spacing)を表1に示す。誤差範囲は、ピーク指定のそれぞれについて約±0.1°2θである。
実施例1の方法Bから得られた無水結晶形態1(Form1)の粉末試料を用いて、図1のXRPDパターンを得た。度(x軸)の2θ角を、秒あたりの計数率に関するピーク強度(y軸)に対してプロットした。このXRDパターンは特定形態に特有であり、2θ角(°)または格子面間隔(d−spacing)(Å)で表すことができる一連の特有の回折ピークを示す。
2θ回折角および対応する格子面間隔(d−spacing)値は、XRDパターンの様々なピークの位置を説明し、格子面間隔(d−spacing)値は、ブラッグ(Bragg)の式を用いて、観察される2θ角および銅Kα1波長を用いて算出される。使用した特定の回折計および分析者の試料調製技術に基づいて、観察された2θ角および格子面間隔(d−spacing)にわずかな変動が見込まれる。相対ピーク強度については、より大きな変動が見込まれる。相対ピーク強度の大きな変動は、結晶形態学の違いに起因する優先方位により観察され得る。それらの値を測定する温度によっても、観察される2θ角および格子面間隔(d−spacing)における変動が観察され得る。
化合物の正確な結晶形態の同定は、主に、観察される2θ角または格子面間隔(d−spacing)に基づくべきである。
無水結晶形態1(Form1)の式(I)の化合物を同定するために、特定の特徴的な2θ角が4.3±0.1、7.9±0.1、9.8±0.1、10.7±0.1、10.8±0.1、13.3±0.1、14.0±0.1、15.1±0.1°に生じ、これらの値は、それぞれ、20.4、11.1、9.0、8.3、8.2、6.6、6.3および5.9オングストローム(Å)の格子面間隔(d−spacing)に対応する。
当該技術者は、これらの特徴的な2θ角ピークまたは格子面間隔(d−spacing)から無水結晶形態1(Form1)を同定することができるが、状況によっては、無水結晶形態1(Form1)の同定のために、追加の2θ角または格子面間隔(d−spacing)を頼ることが望ましい場合もある。
したがって、無水結晶形態1(Form1)の式(I)の化合物は、典型的には、本質的に以下の位置4.3±0.1、7.9±0.1、9.8±0.1、10.7±0.1、10.8±0.1、13.1±0.1、13.2±0.1、13.3±0.1、14.0±0.1、14.4±0.1、15.0±0.1、15.1±0.1、15.7±0.1、15.9±0.1、16.3±0.1、16.5±0.1、16.8±0.1、17.0±0.1、17.4±0.1、17.5±0.1、18.1±0.1、18.2±0.1、18.7±0.1、19.4±0.1、19.7±0.1、20.0±0.1、20.1±0.1、20.2±0.1、20.5±0.1、20.7±0.1、21.5±0.1、21.8±0.1°に2θ角ピークを示し、これらの値は、それぞれ、20.4、11.1、9.0、8.3、8.2、6.8、6.7、6.6、6.3、6.1、5.9、5.9、5.6、5.6、5.4、5.4、5.3、5.2、5.1、5.1、4.9、4.9、4.7、4.6、4.5、4.4、4.4、4.4、4.3、4.3、4.1および4.1オングストローム(Å)の格子面間隔(d−spacing)に対応する。
上記に報告した2θ角の割り当ておよび格子面間隔(d−spacing)のそれぞれには多少の誤差範囲が存在する。格子面間隔(d−spacing)を決定する際の誤差は、回折走査角の増大または格子面間隔(d−spacing)の減少に伴って減少する。前述の2θ角の誤差範囲は、前述のピークの割り当てのそれぞれに対して約±0.1°である。
2θ角の割り当ておよび格子面間隔(d−spacing)に多少の誤差範囲があり得るので、無水結晶形態1(Form1)の式(I)の化合物の試料の特定形態を同定するために、XRPDパターンを比較する好ましい方法は、未知試料のXRPDパターンを既知形態のXRPDパターンの上に重ね合わせることである。例えば、当該技術者は、本明細書に記載の方法を用いて得た式(I)の化合物の未知試料のXRPDパターンを図1の上に重ね合わせることができ、当技術分野の専門知識と知見を用いて、この未知試料のXRPDパターンが、無水結晶形態1(Form1)の式(I)の化合物のXRPDパターンと実質的に同じであるかどうかを容易に判定することができる。
2θ角(°)および格子面間隔(d−spacing)(Å)を考慮すれば、無水結晶形態1(Form1)の式(I)の化合物は、以下のXRPDパターンの特徴を示す。
無水結晶形態1(Form1)の式(I)の化合物のXRPDパターンの前述の特性に基づき、当該技術者は、無水結晶形態1(Form1)の式(I)の化合物を容易に同定することができる。本明細書に記載の方法を用いて得られる無水結晶形態1(Form1)の式(I)の化合物の試料のXRPDパターンは追加のピークを示す可能性があることを、当該技術者は理解するであろう。前述の表は、その特定の結晶形態に特徴的な最も強いピークを示す。この表は、無水結晶形態1(Form1)の式(I)の化合物が示すピークの網羅的なリストではない。
実施例1の方法Aの粉末X線回折(XRPD)パターンは、図1に示したパターンと一致する。
固体核磁気共鳴(SSNMR)は、無水結晶形態1(Form1)の式(I)の化合物の物理的特徴を同定するための別の従来の分析技術である。無水結晶形態1(Form1)の式(I)の化合物のSSNMRスペクトルは独特である。本発明による、無水結晶形態1(Form1)の式(I)の化合物の固体NMRスペクトルは、分析化学および物理的評価の技術者に公知の従来の機器および技術を用いて測定される。
図3の13C固体NMRデータを、399.87MHzの1H周波数で作動するBruker社製Avance 400 triple−resonance spectrometerを用いて取得した。示した13CのSSNMRスペクトルを、8kHzの回転周波数で、Bruker社製4mmの3重共鳴マジックアングルスピンプローブによる交差分極パルスシーケンスを用いて得た。75〜90kHzの直線的な出力ランプ(linear power ramp)を、交差分極効率を高めるために1Hチャネルで用いた。5パルスの全サイドバンド抑圧パルスシーケンスにより、スピニングサイドバンド除去した。1Hデカップリングは、Spinal−64 sequenceを用いて得たが、19Fデカップリングは、回転周期あたり1πパルスを用いるπ−パルスデカップリングにより得た。特徴的な13C NMRピークの位置を、0ppm(100万分の1)のテトラメチルシランに対して記録し、機器の変動および較正により、精度は±0.2ppmである。
温度296K、回転速度8kHzでの13C観察について、100.56MHzの周波数で作動する分光計を用いて、無水結晶形態1(Form1)の式(I)の化合物の固体NMRスペクトルにおいて観察された特定の特徴的化学シフトには、以下のものが含まれる:175.03±0.2、163.30±0.2、158.66±0.2、156.50±0.2、149.79±0.2、148.17±0.2、146.96±0.2、139.56±0.2、133.19±0.2、132.32±0.2、129.76±0.2、126.5±0.2、124.15±0.2、120.37±0.2、119.20±0.2、118.16±0.2、112.83±0.2、70.62±0.2、67.61±0.2、64.96±0.2、59.89±0.2、57.08±0.2、55.50±0.2、52.41±0.2、48.35±0.2、40.79±0.2、32.75±0.2、および21.12±0.2ppm。
使用した特定の分光計および分析者の試料調製技術に基づいて、観察された化学シフトにわずかな変動が見込まれる。多少の誤差範囲が、上記で記録した化学シフトのそれぞれに存在する。前述の化学シフトの誤差範囲は約±0.2ppmである。
化学シフトの割り当てにおいて多少の誤差範囲が生じ得るので、式(I)の化合物の未知の形態が無水結晶形態1(Form1)であるかどうかを判定する好ましい方法は、この試料のSSNMRスペクトルを図3に示すSSNMRスペクトルの上に重ね合わせることである。当該技術者は、本明細書に記載の方法を用いて得られる式(I)の化合物の未知の形態のNMRスペクトルを図3の上に重ね合わせることができ、当技術分野の専門知識と知見を用いて、未知試料のNMRスペクトルが無水結晶形態1(Form1)の式(I)の化合物のNMRスペクトルと実質的に同じであるかどうかを容易に判定することができる。
具体的には、図3の13C固体NMRデータは、本特許出願の実施例1の方法Bの試料に対応する。
前述の分析技術はいずれも、無水結晶形態1(Form1)の式(I)の化合物を同定するために単独でまたは併用して用いることができる。さらに、物理的評価の他の方法を使用して、無水結晶形態1(Form1)の式(I)の化合物を同定し、特徴づけることもできる。無水結晶形態の物理的評価または同定に有用であることが当該技術者に公知である好適な技術の例として、示差走査熱量測定法(DSC)および赤外(IR)分光法が挙げられるが、これらに限定されない。これらの技術は、式(I)の化合物の未知形態の試料を特徴づけるために単独でまたは他の技術と併用して使用することができる。
無水結晶形態の式(I)の化合物およびこれを含んでなる医薬組成物は、動物、例えば、ヒトなどの哺乳類における治療、特に、精神障害、うつ病、気分障害、不安症、睡眠障害の治療または予防に有用である。PCT公開WO07/028654(この内容は参照によりその全体が本明細書に組込まれる)に開示される様々な治療上の使用は、無水結晶形態の式(I)の化合物に同様に適用できる。
別の態様において、本発明は、無水結晶形態の有効量の式(I)の化合物を含んでなる医薬組成物を提供する。
別の態様において、本発明は、本発明による無水結晶形態1(Form1)の有効量の式(I)の化合物を含んでなる医薬組成物を提供する。この医薬組成物は、さらに、1種類以上の医薬的に許容される担体または希釈剤を含んでもよい。
かかる医薬組成物は、1種類以上の医薬的に許容される担体または希釈剤を含んでもよい。適切な医薬組成物およびそれらの調製法の例は、PCT公開WO07/028654に開示されており、この内容は参照によりその全体が本明細書に組込まれる。都合のよいことに、適切な医薬組成物は、従来技術を用いて、ならびに、使用する時には、担体および希釈剤を用いて調製することができる。錠剤およびカプセル製剤などの経口投与用の医薬組成物が好ましい。
別の態様において、本発明は、治療に使用するための、本発明による無水結晶形態1(Form1)の式(I)の化合物を提供する。
別の態様において、本発明は、精神障害、うつ病、気分障害、不安症、睡眠障害および物質関連障害の治療および予防のための医薬の調製における、本発明による無水結晶形態1(Form1)の式(I)の化合物の使用を提供する。
別の態様において、本発明は、統合失調症、うつ病、気分障害およびアルコール依存症の治療または予防のための医薬の調製において使用するための、本発明による無水結晶形態1(Form1)の式(I)の化合物の使用を提供する。
別の態様において、本発明は、精神障害、うつ病、気分障害、不安症、睡眠障害および物質関連障害の治療および予防における、本発明による無水結晶形態1(Form1)の式(I)の化合物の使用を提供する。
別の態様において、本発明は、統合失調症、うつ病、気分障害およびアルコール依存症の治療または予防に使用するための、本発明による無水結晶形態1(Form1)の式(I)の化合物を提供する。
非晶形の式(I)の化合物は、PCT公開WO07/028654(この内容は、参照によりその全体が本明細書に組込まれる)に記載の方法に従って調製することができる。
特定の無水結晶形態の式(I)の化合物の特定の調製法を、以下の実施例に提供する。
本発明に使用する無水結晶形態の式(I)の化合物を、他の治療薬と併用して使用してもよい。同様に、本発明の医薬製剤は、1種類以上の追加の治療薬を含んでもよい。PCT公開WO07/028654(この内容は、参照によりその全体が本明細書に組込まれる)に開示され、式(I)の化合物またはその塩と併用してもよい様々な治療薬は、同様に、本発明による無水結晶形態の式(I)の化合物に適用できる。
したがって、本発明は、さらなる態様において、精神障害を治療または予防するために、追加の治療薬と共に無水結晶形態の式(I)の化合物を含んでなる併用法を提供する。
したがって、本発明は、さらなる態様において、精神障害を治療または予防するために、追加の治療薬と共に無水結晶形態1(Form1)の式(I)の化合物を含んでなる併用法を提供する。
無水結晶形態の式(I)の化合物を他の治療薬と併用して用いる時、これらの化合物を、任意の都合のよい経路によって、連続または同時のいずれか一方で投与してもよい。
同じ製剤に併用する時、これらの2つの化合物は安定していて、互いに相性がよく、かつこの製剤の他の成分と相性がよくなければならず、投与のために処方され得ることは理解されるであろう。別々に処方する時、これらを、当該技術分野においてかかる化合物について知られるような方法で、任意の都合のよい処方で提供することができる。
無水結晶形態の式(I)の化合物を第2の治療薬と併用して用いる時、各化合物の用量は、これらの化合物を単独で用いる時の用量と異なり得る。当該技術者は、適切な用量を容易に理解するであろう。
以下の実施例は説明の目的のみを意図しており、いかなる方法においても本発明の範囲を限定することを意図しない。
以下の手順において、各出発物質の後に、説明についての言及を、例によって提供する。これを、単に、熟練化学者への補助のために提供する。この出発物質は、必ずしも、言及されるバッチから調製されたわけではない。
本明細書で使用する、これらのプロセス、模式図および例において使用する記号および慣習は、現代の科学文献、例えば、the Journal of the American Chemical Societyまたはthe Journal of Biological Chemistryで用いられる記号および慣習と一致する。
具体的には、以下の略語が、実施例および本明細書全体において使用され得る。
g(グラム) mg(ミリグラム)
L(リットル) mL(ミリリットル)
μL(マイクロリットル) psi(ポンド・平方インチ)
M(モル) mM(ミリモル)
N(通常) kg(キログラム)
i.v.(静脈内) Hz(ヘルツ)
MHz(メガヘルツ) mol(モル)
MIBK(メチルイソブチルケトン) w/w(重量/重量)
mmol(ミリモル) RT(室温)
min(分) hまたはhrs(時間)
mp(融点) TLC(薄層クロマトグラフィー)
Tr(保管期間) RP(逆相)
THF(テトラヒドロフラン) DMSO(ジメチルスルホキシド)
EtOAc(酢酸エチル) DME(1,2−ジメトキシエタン)
DCM(ジクロロメタン) DCE(ジクロロエタン)
DMF(N,N−ジメチルホルムアミド) HOAc(酢酸)
Psig(重量ポンド毎平方インチゲージ)
MTBE(メチルtert−ブチルエーテル)
IPAc(酢酸イソプロピル) Et3N(トリエチルアミン)
wt/vol(重量/体積) IPA(イソプロピルアルコール)
乾燥減量(LOD) barg(棒ゲージ)
rpm(毎分回転数) q.s.(適量)
1−プロパンホスホン酸無水物(T3P)
BH3−THF(ボラン−テトラヒドロフラン錯体)
HPLC(高速液体クロマトグラフィー)
g(グラム) mg(ミリグラム)
L(リットル) mL(ミリリットル)
μL(マイクロリットル) psi(ポンド・平方インチ)
M(モル) mM(ミリモル)
N(通常) kg(キログラム)
i.v.(静脈内) Hz(ヘルツ)
MHz(メガヘルツ) mol(モル)
MIBK(メチルイソブチルケトン) w/w(重量/重量)
mmol(ミリモル) RT(室温)
min(分) hまたはhrs(時間)
mp(融点) TLC(薄層クロマトグラフィー)
Tr(保管期間) RP(逆相)
THF(テトラヒドロフラン) DMSO(ジメチルスルホキシド)
EtOAc(酢酸エチル) DME(1,2−ジメトキシエタン)
DCM(ジクロロメタン) DCE(ジクロロエタン)
DMF(N,N−ジメチルホルムアミド) HOAc(酢酸)
Psig(重量ポンド毎平方インチゲージ)
MTBE(メチルtert−ブチルエーテル)
IPAc(酢酸イソプロピル) Et3N(トリエチルアミン)
wt/vol(重量/体積) IPA(イソプロピルアルコール)
乾燥減量(LOD) barg(棒ゲージ)
rpm(毎分回転数) q.s.(適量)
1−プロパンホスホン酸無水物(T3P)
BH3−THF(ボラン−テトラヒドロフラン錯体)
HPLC(高速液体クロマトグラフィー)
特に示さない限り、全ての温度を℃(摂氏温度)で表す。特に言及しない限り、全ての反応を不活性雰囲気下、室温で行った。
特にはっきり記載しない限り、実施例において、陽子磁気共鳴(NMR)スペクトルを、Bruker社製装置で、400または700MHzにて記録し、化学シフトを、内部標準として残留溶媒線を用いてppm(δ)で報告する。分離パターンは、sを一重線、dを二重線、tを三重線、qを四重線、mを多重線、bを幅広と指定する。示差走査熱量測定(DSC)を、1分あたり10℃の走査速度にてTA Q1000熱量計で行った。
アルミニウムパンに1〜2mgの試料サイズを測り入れ、パンの蓋を上部に置き、このパンを密封することなく軽く圧着させた。
アルミニウムパンに1〜2mgの試料サイズを測り入れ、パンの蓋を上部に置き、このパンを密封することなく軽く圧着させた。
中間体1
(3R)−4−ベンジル−5−オキソモルホリン−3−カルボン酸
(3R)−4−ベンジル−5−オキソモルホリン−3−カルボン酸
300ガロンのハステロイ反応器に、N−ベンジル−D−セリン(50.0kg)を入れ、その後、テトラヒドロフラン(THF、271.2kg)を加えた。その後、この溶液を0℃まで冷却し、水中(152.5L)の炭酸カリウム(53.1kg)溶液を、−5℃〜5℃の間の温度を維持しながら一度に加えた。この温度を0℃まで再び調節した後、塩化クロロアセチル(40.2kg)を、4℃以下の温度を維持しながら、1時間かけて少量ずつ加えた。この混合物を、0〜4℃の間で30分間撹拌し、さらに別の塩化クロロアセチル(4.4kg)を一度に加えた。0〜4℃で、撹拌をさらに30分間続けた。
10℃以下の反応温度を維持しながら、50%水酸化ナトリウム水溶液(82.0kg)を50分かけて加えた。最終pH終点は13〜13.5になるべきである。添加を完了した後、この溶液を3〜5℃まで冷却し、4時間、この温度で撹拌した。反応が完了したことを(HPLCで)決定した後、これを20〜22℃まで温め、ヘプタン(75.0kg)を加えて、激しく撹拌した。塩基性の水層を収集し、ヘプタンを除去し、この水層を再び反応器に戻し入れた。この塩基性の水溶液をもう一度ヘプタン(107.8kg)で洗浄し、この反応器に戻し入れ、これを3℃まで冷却した後、温度を<6℃を維持しながら、1〜1.5時間かけて12N HCl(193.8kg)を少量ずつ加えることによって、pHを<2.0まで下げて調整した。酸添加を完了した後、この白色固体の懸濁液を3〜5℃まで冷却し、さらに2時間撹拌した後、濾過した。その後、このケーキを冷水(3〜5℃、50.0L)ですすぎ、乾燥させた後、LODが<0.6%になるまで55〜60℃の真空下に置き、白色固体として表題化合物を得た(54.9kg、91.2%収率)。1H NMR(DMSO−d6)δ7.37−7.24(m,5H),5.25(d,J=15.4Hz,1H),4.17(m,2H),4.12(m 1H),3.94(dd,J=5.6,3.2Hz,1H),3.92(dd,J=16.8,3.2Hz,1H),3.82(d,J=15.4Hz,1H)。
10℃以下の反応温度を維持しながら、50%水酸化ナトリウム水溶液(82.0kg)を50分かけて加えた。最終pH終点は13〜13.5になるべきである。添加を完了した後、この溶液を3〜5℃まで冷却し、4時間、この温度で撹拌した。反応が完了したことを(HPLCで)決定した後、これを20〜22℃まで温め、ヘプタン(75.0kg)を加えて、激しく撹拌した。塩基性の水層を収集し、ヘプタンを除去し、この水層を再び反応器に戻し入れた。この塩基性の水溶液をもう一度ヘプタン(107.8kg)で洗浄し、この反応器に戻し入れ、これを3℃まで冷却した後、温度を<6℃を維持しながら、1〜1.5時間かけて12N HCl(193.8kg)を少量ずつ加えることによって、pHを<2.0まで下げて調整した。酸添加を完了した後、この白色固体の懸濁液を3〜5℃まで冷却し、さらに2時間撹拌した後、濾過した。その後、このケーキを冷水(3〜5℃、50.0L)ですすぎ、乾燥させた後、LODが<0.6%になるまで55〜60℃の真空下に置き、白色固体として表題化合物を得た(54.9kg、91.2%収率)。1H NMR(DMSO−d6)δ7.37−7.24(m,5H),5.25(d,J=15.4Hz,1H),4.17(m,2H),4.12(m 1H),3.94(dd,J=5.6,3.2Hz,1H),3.92(dd,J=16.8,3.2Hz,1H),3.82(d,J=15.4Hz,1H)。
中間体2
[(3S)−4−ベンジルモルホリン−3−イル]メタノール塩酸塩
[(3S)−4−ベンジルモルホリン−3−イル]メタノール塩酸塩
500ガロンのグラスライニング製反応器に、テトラヒドロフラン(401.2kg)、続いて、ボロン−ジメチルスルフィド錯体(10M,93.1kg)を入れた。この溶液を35℃まで加熱した後、テトラヒドロフラン(253.4kg)に溶解した中間体1(47.5kg)を、40℃以下の温度を維持しながら、1時間かけて加えた。添加後、この混合物を35℃でさらに4時間撹拌した。この反応が完了した時(HPLC)、温度を35〜45℃の間に維持しながら、アセトン(178.5kg)を1時間かけて加えた。その後、この溶液を35℃でさらに1時間撹拌した後、40℃まで温め、一晩撹拌した。次の日、この溶液を35℃まで冷却し、水(213.7L)を、1時間かけて制御した方法で加えた。添加を完了した後、この溶液を真空下に置き、THFを蒸留して取り除き、最終体積は238Lに達した。水(83.1L)および酢酸エチル(471.3kg)を加え、この溶液を40℃まで加熱した後、水酸化ナトリウム(2M,462L)を加えた。この混合物を2分間撹拌した後、静置させた。水層を取り除き、酢酸エチル(471.3kg)で再び洗浄した。2回の酢酸エチル洗浄液(942.6kg)をこの反応器に戻し入れ、20%ブライン溶液(199.9L)で洗浄した。下部のブライン層を取り除いて、廃棄した。この酢酸エチル溶液を真空下に置き、蒸留して、最終体積を333Lにした。この溶液を20℃まで冷却し、30ミクロンのPallフィルターに通してきれいな反応器に移した。この反応器を酢酸エチル(85.7kg)ですすいだ後、この酢酸エチルを移送ラインおよびPallフィルターに通過させた。濾過した酢酸エチル溶液にメタノール(172.8kg)を加えた。この反応を10℃まで冷却した後、15℃以下の温度を維持しながら、クロロトリメチルシラン(22.2kg)を15分かけて加えた。
その後、この反応を5℃まで冷却し、少なくとも2時間撹拌した。スラリーをこの反応器から取り除き、濾過した。湿ったケーキを冷たい酢酸エチル(64.3kg)で洗浄し、乾燥させた後、50〜55℃で8時間、真空下に置き、白色固体として表題化合物を得た(43.1kg,87.6%収率)。1H NMR(DMSO−d6)δ10.88(br s,1H),7.62(m,2H),7.45(m,3H),5.72(br s,1H),4.79(dd,J=12.9,2.6Hz,1H),4.19(dd,J=12.9,7.5Hz,1H),3.98(ddd,J=15.4,12.2,3.2Hz,1H),3.94−3.81(m,3H),3.78−3.66(m,2H),3.39−3.27(m,1H),3.11−2.97(m,1H),2.89(d,J=10.8Hz,1H)。
その後、この反応を5℃まで冷却し、少なくとも2時間撹拌した。スラリーをこの反応器から取り除き、濾過した。湿ったケーキを冷たい酢酸エチル(64.3kg)で洗浄し、乾燥させた後、50〜55℃で8時間、真空下に置き、白色固体として表題化合物を得た(43.1kg,87.6%収率)。1H NMR(DMSO−d6)δ10.88(br s,1H),7.62(m,2H),7.45(m,3H),5.72(br s,1H),4.79(dd,J=12.9,2.6Hz,1H),4.19(dd,J=12.9,7.5Hz,1H),3.98(ddd,J=15.4,12.2,3.2Hz,1H),3.94−3.81(m,3H),3.78−3.66(m,2H),3.39−3.27(m,1H),3.11−2.97(m,1H),2.89(d,J=10.8Hz,1H)。
中間体3
(7S,9aS)−7−[(ベンジルオキシ)メチル]オクタヒドロピラジノ[2,1−c][1,4]オキサジン二シュウ酸塩
(7S,9aS)−7−[(ベンジルオキシ)メチル]オクタヒドロピラジノ[2,1−c][1,4]オキサジン二シュウ酸塩
ステージ3a
200ガロンの高圧ハステロイ反応器に、中間体2(42.7kg)、続いて、20%水酸化パラジウム/炭素(4.3kg、約50%含水)を入れた。この反応器を密封し、窒素をパージした。その後、エタノール(200proof,336.9kg)を入れた。撹拌器を許容可能な最大スピードに設定し、この反応を20〜30℃の間まで加熱し、この反応器を水素ガスで30psigまで加圧した。反応完了までの間(約3時間)、水素の取り込みを監視した。この反応が完了した後(HPLC)、このエタノール混合物を、セライトを詰めたPallフィルターに通過させた。その後、このフィルターを追加量のエタノール(101.1kg)ですすいだ。これらのエタノール溶液を合わせて、次の反応に直接用いた。このエタノール溶液を蒸発させ、乾燥させることによって分析試料(S)−1−モルホリン−3−イル−メタノール塩酸塩を得て、その後、これをNMRによって分析した。1H NMR(DMSO−d6)δ9.38(br s,1H),5.43(t,J=5.2Hz,1H),3.87(ddd,J=15.5,12.0,3.4Hz,2H),3.66(ddd,J=13.3,11.0,2.7Hz,1H),3.61−3.49(m,3H),3.27−3.17(m,1H),3.13(dt,J=13.0,2.5Hz,1H),3.02(ddd,J=14.4,10.7,3.7Hz,1H)。
200ガロンの高圧ハステロイ反応器に、中間体2(42.7kg)、続いて、20%水酸化パラジウム/炭素(4.3kg、約50%含水)を入れた。この反応器を密封し、窒素をパージした。その後、エタノール(200proof,336.9kg)を入れた。撹拌器を許容可能な最大スピードに設定し、この反応を20〜30℃の間まで加熱し、この反応器を水素ガスで30psigまで加圧した。反応完了までの間(約3時間)、水素の取り込みを監視した。この反応が完了した後(HPLC)、このエタノール混合物を、セライトを詰めたPallフィルターに通過させた。その後、このフィルターを追加量のエタノール(101.1kg)ですすいだ。これらのエタノール溶液を合わせて、次の反応に直接用いた。このエタノール溶液を蒸発させ、乾燥させることによって分析試料(S)−1−モルホリン−3−イル−メタノール塩酸塩を得て、その後、これをNMRによって分析した。1H NMR(DMSO−d6)δ9.38(br s,1H),5.43(t,J=5.2Hz,1H),3.87(ddd,J=15.5,12.0,3.4Hz,2H),3.66(ddd,J=13.3,11.0,2.7Hz,1H),3.61−3.49(m,3H),3.27−3.17(m,1H),3.13(dt,J=13.0,2.5Hz,1H),3.02(ddd,J=14.4,10.7,3.7Hz,1H)。
ステージ3b
300ガロンのグラスライニング製反応器に、(S)−1−モルホリン−3−イル−メタノール塩酸塩(ステージ3aにおける収率100%に基づいて579.5kg)を含むステージ3aのエタノール溶液を入れた。この溶液を真空下で蒸留して、339Lにした。温度を25℃に調節した後、この反応器に、N,N−ジイソプロピルエチルアミン(69.2kg)を入れ、10分間撹拌した。その後、エタノール(66.9kg)中N−Boc−O−ベンジル−D−セリン(42.4kg)溶液をこの反応器に入れ、25℃で1時間撹拌した。1時間撹拌した後、30℃以下の温度を維持しながら、1−T3P(酢酸エチル中約50%、114.8kg)を少なくとも30分かけて少量ずつ加えた。添加後、この反応を25℃で20分間撹拌した。この反応が完了した時(HPLC)、この反応器に水酸化ナトリウム(3N,229.4kg)を入れた。その後、この反応器を真空下に置き、エタノールを蒸留して取り除き、最終体積は339Lに達した。その後、MTBE(470.6kg)を加え、この混合物を10分間撹拌し、少なくとも15分間静置させた。下部の塩基性の水層を取り除いた後、このMTBEを、1N HCl(256.6L)、3N水酸化ナトリウム(256.6L)および20%ブライン溶液(213.5L)でそれぞれ洗浄した。このMTBE層を反応器から取り除き、次の反応に直接用いた。
300ガロンのグラスライニング製反応器に、(S)−1−モルホリン−3−イル−メタノール塩酸塩(ステージ3aにおける収率100%に基づいて579.5kg)を含むステージ3aのエタノール溶液を入れた。この溶液を真空下で蒸留して、339Lにした。温度を25℃に調節した後、この反応器に、N,N−ジイソプロピルエチルアミン(69.2kg)を入れ、10分間撹拌した。その後、エタノール(66.9kg)中N−Boc−O−ベンジル−D−セリン(42.4kg)溶液をこの反応器に入れ、25℃で1時間撹拌した。1時間撹拌した後、30℃以下の温度を維持しながら、1−T3P(酢酸エチル中約50%、114.8kg)を少なくとも30分かけて少量ずつ加えた。添加後、この反応を25℃で20分間撹拌した。この反応が完了した時(HPLC)、この反応器に水酸化ナトリウム(3N,229.4kg)を入れた。その後、この反応器を真空下に置き、エタノールを蒸留して取り除き、最終体積は339Lに達した。その後、MTBE(470.6kg)を加え、この混合物を10分間撹拌し、少なくとも15分間静置させた。下部の塩基性の水層を取り除いた後、このMTBEを、1N HCl(256.6L)、3N水酸化ナトリウム(256.6L)および20%ブライン溶液(213.5L)でそれぞれ洗浄した。このMTBE層を反応器から取り除き、次の反応に直接用いた。
ステージ4
300ガロンのグラスライニング製反応器に、[(R)−1−ベンジルオキシメチル−2−((S)−3−ヒドロキシメチル−モルホリン−4−イル)−2−オキソ−エチル]カルバミン酸tert−ブチルエステル(48.0kg)を含むステージ3bのMTBE溶液(503.6kg)を入れた。大気圧で、このMTBEを蒸留して取り除き、最終体積は144Lに達した。温度を25℃に調節した後、THF(426.7kg)を加えた。この溶液を大気圧で蒸留して取り除き、最終体積は192Lになった。再度、この溶液を25℃まで冷却し、THF(426.7kg)を加えた。この溶液を大気圧下で蒸留して144Lにし、25℃まで冷却し、さらにTHF(57.6kg)を加えて、最終溶液体積を193Lにした。カールフィッシャー分析を行い、溶液中に存在する水の量を決定した(追加のBH3−THFを加え、溶液中に検出される全ての水を消費させた)。別の(きれいな、THFですすいだ)550ガロンのグラスライニング製反応器に、BH3−THF(1.0M,327.4kg)を入れて、35℃まで加熱した。窒素圧下で、[(R)−1−ベンジルオキシメチル−2−((S)−3−ヒドロキシメチル−モルホリン−4−イル)−2−オキソ−エチル]カルバミン酸tert−ブチルエステルを含むこのTHF溶液を、45℃以下の温度を維持しながら、ゆっくり(少なくとも2時間)BH3−THF溶液に加えた。追加量のTHF(10.7kg)を用いて、これらを洗浄し、反応器に加えた。この反応を35℃で2.5時間維持し、試料を少しとって反応完了を確認した(HPLC)。この反応が完了した後、45℃以下の温度を維持しながら、アセトン(49.4kg)を1時間かけてゆっくり入れた。その後、この反応を少なくとも8時間撹拌した後、45℃以下の温度を維持しながら、メタノール(113.9kg)を1.5時間かけてゆっくり入れた。この溶液を35℃で3時間撹拌した後、真空下で、蒸留して216Lにした。温度を25℃に調節した後、MTBE(355.2kg)、続いて、15分間かけて水酸化ナトリウム(3N,259.6kg)を加えた。この溶液を10分間激しく撹拌した後、静置させた。塩基性の水層を取り除いた後、このMTBE層を20%ブライン溶液(240L)で洗浄した。このブライン溶液を取り除き、真空下で、このMTBEを蒸留して120Lにし、25℃まで冷却した後、酢酸イソプロピル(502.3kg)を反応器に入れた。真空下で、この溶液を蒸留して384Lにした後、この反応器から取り除き、10ミクロンのフィルターに通して、次の反応に直接用いた。
300ガロンのグラスライニング製反応器に、[(R)−1−ベンジルオキシメチル−2−((S)−3−ヒドロキシメチル−モルホリン−4−イル)−2−オキソ−エチル]カルバミン酸tert−ブチルエステル(48.0kg)を含むステージ3bのMTBE溶液(503.6kg)を入れた。大気圧で、このMTBEを蒸留して取り除き、最終体積は144Lに達した。温度を25℃に調節した後、THF(426.7kg)を加えた。この溶液を大気圧で蒸留して取り除き、最終体積は192Lになった。再度、この溶液を25℃まで冷却し、THF(426.7kg)を加えた。この溶液を大気圧下で蒸留して144Lにし、25℃まで冷却し、さらにTHF(57.6kg)を加えて、最終溶液体積を193Lにした。カールフィッシャー分析を行い、溶液中に存在する水の量を決定した(追加のBH3−THFを加え、溶液中に検出される全ての水を消費させた)。別の(きれいな、THFですすいだ)550ガロンのグラスライニング製反応器に、BH3−THF(1.0M,327.4kg)を入れて、35℃まで加熱した。窒素圧下で、[(R)−1−ベンジルオキシメチル−2−((S)−3−ヒドロキシメチル−モルホリン−4−イル)−2−オキソ−エチル]カルバミン酸tert−ブチルエステルを含むこのTHF溶液を、45℃以下の温度を維持しながら、ゆっくり(少なくとも2時間)BH3−THF溶液に加えた。追加量のTHF(10.7kg)を用いて、これらを洗浄し、反応器に加えた。この反応を35℃で2.5時間維持し、試料を少しとって反応完了を確認した(HPLC)。この反応が完了した後、45℃以下の温度を維持しながら、アセトン(49.4kg)を1時間かけてゆっくり入れた。その後、この反応を少なくとも8時間撹拌した後、45℃以下の温度を維持しながら、メタノール(113.9kg)を1.5時間かけてゆっくり入れた。この溶液を35℃で3時間撹拌した後、真空下で、蒸留して216Lにした。温度を25℃に調節した後、MTBE(355.2kg)、続いて、15分間かけて水酸化ナトリウム(3N,259.6kg)を加えた。この溶液を10分間激しく撹拌した後、静置させた。塩基性の水層を取り除いた後、このMTBE層を20%ブライン溶液(240L)で洗浄した。このブライン溶液を取り除き、真空下で、このMTBEを蒸留して120Lにし、25℃まで冷却した後、酢酸イソプロピル(502.3kg)を反応器に入れた。真空下で、この溶液を蒸留して384Lにした後、この反応器から取り除き、10ミクロンのフィルターに通して、次の反応に直接用いた。
ステージ5a
20Lのジャケット付きライニング製反応器に、ステージ4から得た[(S)−1−ベンジルオキシメチル−2−((S)−3−ヒドロキシメチル−モルホリン−4−イル)−エチル]カルバミン酸tert−ブチルエステル(ステージ4の理論的収率を100%と仮定して0.916kg)を含む酢酸イソプロピル(3.7L)溶液を入れた。この溶液にDCM(9.2L)およびトリエチルアミン(0.940L)を入れた。この溶液を5℃まで冷却し、塩化メタンスルホニル(0.690g)を、15℃以下の温度を保つ速度で加えた。添加後、この反応を20℃まで温め、HPLCにより完了が確認されるまで(約20時間)撹拌した。この反応を水(4.6L)で急冷し、10分間撹拌した後、静置した。この水層を廃棄した後、有機層を1N HCl(4.6L)および5%NaHCO3(4.6L)で連続して洗浄した。洗浄後、この有機層を真空下で蒸留して2.8Lにした。追加量の酢酸イソプロピル(1.8L)をこの反応器に入れ、この溶液を次の反応に直接用いた。
20Lのジャケット付きライニング製反応器に、ステージ4から得た[(S)−1−ベンジルオキシメチル−2−((S)−3−ヒドロキシメチル−モルホリン−4−イル)−エチル]カルバミン酸tert−ブチルエステル(ステージ4の理論的収率を100%と仮定して0.916kg)を含む酢酸イソプロピル(3.7L)溶液を入れた。この溶液にDCM(9.2L)およびトリエチルアミン(0.940L)を入れた。この溶液を5℃まで冷却し、塩化メタンスルホニル(0.690g)を、15℃以下の温度を保つ速度で加えた。添加後、この反応を20℃まで温め、HPLCにより完了が確認されるまで(約20時間)撹拌した。この反応を水(4.6L)で急冷し、10分間撹拌した後、静置した。この水層を廃棄した後、有機層を1N HCl(4.6L)および5%NaHCO3(4.6L)で連続して洗浄した。洗浄後、この有機層を真空下で蒸留して2.8Lにした。追加量の酢酸イソプロピル(1.8L)をこの反応器に入れ、この溶液を次の反応に直接用いた。
ステージ5b
20Lのジャケット付きライニング製反応器に6N HCl(4.58L)を入れ、10℃まで冷却した。この酸溶液に、20℃以下の温度を維持する速度で、ステージ5aの[(S)−1−ベンジルオキシメチル−2−((R)−3−クロロメチル−モルホリン−4−イル)−エチル]カルバミン酸tert−ブチルエステル溶液を加えた。この反応を25℃まで温め、HPLCにより完了が確認されるまで(1時間)撹拌した。この撹拌を停止し、2層に分けた。下部の酸性の層を取り除き、上部の有機層を廃棄した。この酸性の水層を反応器に戻し入れ、IPAc(4.6L)で洗浄した。再び、水層を取り除き、有機層を廃棄した。その後、この反応器にこの水層、続いて、IPAc(4.6L)を入れた。この混合物を2℃まで冷却し、20℃以下の温度を維持する速度で、50% NaOH(1.154kg)を加えた。この添加後、この混合物を5分間撹拌し、水層のpHを調べた(約7.0でなければならない)。層を分離し、下部の水層を取り除いて、廃棄した。その後、上部のIPAc層を、真空下の蒸留により、3.0L(3.27vol)まで減らした。アセトニトリル(10.0L)をこの反応器に入れ、この溶液を、真空下の蒸留により、3.0Lまで減らした。最後に、追加量のアセトニトリル(4.5L)を入れ、この溶液を次の反応に直接用いた。
20Lのジャケット付きライニング製反応器に6N HCl(4.58L)を入れ、10℃まで冷却した。この酸溶液に、20℃以下の温度を維持する速度で、ステージ5aの[(S)−1−ベンジルオキシメチル−2−((R)−3−クロロメチル−モルホリン−4−イル)−エチル]カルバミン酸tert−ブチルエステル溶液を加えた。この反応を25℃まで温め、HPLCにより完了が確認されるまで(1時間)撹拌した。この撹拌を停止し、2層に分けた。下部の酸性の層を取り除き、上部の有機層を廃棄した。この酸性の水層を反応器に戻し入れ、IPAc(4.6L)で洗浄した。再び、水層を取り除き、有機層を廃棄した。その後、この反応器にこの水層、続いて、IPAc(4.6L)を入れた。この混合物を2℃まで冷却し、20℃以下の温度を維持する速度で、50% NaOH(1.154kg)を加えた。この添加後、この混合物を5分間撹拌し、水層のpHを調べた(約7.0でなければならない)。層を分離し、下部の水層を取り除いて、廃棄した。その後、上部のIPAc層を、真空下の蒸留により、3.0L(3.27vol)まで減らした。アセトニトリル(10.0L)をこの反応器に入れ、この溶液を、真空下の蒸留により、3.0Lまで減らした。最後に、追加量のアセトニトリル(4.5L)を入れ、この溶液を次の反応に直接用いた。
ステージ5c
20Lのジャケット付きライニング製反応器にアセトニトリル(10.8L)およびEt3N(1.22kg)を入れ、その後、60℃まで加熱した。反応温度を約60℃に保ちながら、ステージ5bの[(S)−1−ベンジルオキシメチル−2−((R)−3−クロロメチル−モルホリン−4−イル)−エチルアミンアセトニトリル溶液を1時間かけてこの反応器に加えた。その後、HPLCにより反応の完了が確認されるまで、この混合物を60℃で撹拌した。その後、この溶液を、真空下で3.0Lまで濃縮した。この蒸留後、酢酸エチル(2.0L)を加え、スラリー(Et3N・HCl)を20℃まで冷却した。このスラリーをこの反応器から取り除き、濾過し、収集して、この生成溶液を保存した。Et3N・HClケーキを酢酸エチル(2.8L)で洗浄した。(7S,9aS)−7−[(ベンジルオキシ)メチル]オクタヒドロピラジノ[2,1−c][1,4]オキサジンを含む濾液を合わせて、ステージ5dに直接用いた。(7S,9aS)−7−[(ベンジルオキシ)メチル]オクタヒドロピラジノ[2,1−c][1,4]オキサジンの量を、重量/重量HPLCアッセイにより決定した。
20Lのジャケット付きライニング製反応器にアセトニトリル(10.8L)およびEt3N(1.22kg)を入れ、その後、60℃まで加熱した。反応温度を約60℃に保ちながら、ステージ5bの[(S)−1−ベンジルオキシメチル−2−((R)−3−クロロメチル−モルホリン−4−イル)−エチルアミンアセトニトリル溶液を1時間かけてこの反応器に加えた。その後、HPLCにより反応の完了が確認されるまで、この混合物を60℃で撹拌した。その後、この溶液を、真空下で3.0Lまで濃縮した。この蒸留後、酢酸エチル(2.0L)を加え、スラリー(Et3N・HCl)を20℃まで冷却した。このスラリーをこの反応器から取り除き、濾過し、収集して、この生成溶液を保存した。Et3N・HClケーキを酢酸エチル(2.8L)で洗浄した。(7S,9aS)−7−[(ベンジルオキシ)メチル]オクタヒドロピラジノ[2,1−c][1,4]オキサジンを含む濾液を合わせて、ステージ5dに直接用いた。(7S,9aS)−7−[(ベンジルオキシ)メチル]オクタヒドロピラジノ[2,1−c][1,4]オキサジンの量を、重量/重量HPLCアッセイにより決定した。
ステージ5d
20Lのジャケット付きライニング製反応器に、シュウ酸(ステージ5cの(7S,9aS)−7−[(ベンジルオキシ)メチル]オクタヒドロピラジノ[2,1−c][1,4]オキサジン溶液のwt/wt HPLCアッセイに基づいて0.304kg、1.0eq)、エタノール(200proof、2.75L)および酢酸エチル(6.4L)を入れた。この混合物を60℃まで加熱し、50℃を超える温度を維持する速度で、ステージ5cの(7S,9aS)−7−[(ベンジルオキシ)メチル]オクタヒドロピラジノ[2,1−c][1,4]オキサジンの粗酢酸エチル溶液をこの反応器に加えた。この溶液を60℃で30分間撹拌し、20℃まで冷却した後、20℃で一晩撹拌した。固体を取り除き、濾過した後、酢酸エチル:エタノール(6:1、1.8L)溶液で洗浄した。その後、この固体を真空下、50℃のオーブンに入れ、乾燥させ、ベージュ色の結晶性固体として表題化合物を得た(0.306kg)。この表題化合物の第2の収穫物を母液から得た(0.133kg)。1H NMR(DMSO−d6)δ9.5−8.5(br s,4H)7.41−7.36(m,3H),7.35−7.27(m,2H),4.57(d,J=2.0Hz,2H),3.94(t,J=8.3Hz,1H),3.76−3.61(m,4H),3.45(ddd,J=13.9,11.7,2.2Hz,1H),3.06(t,J=11.8Hz,1H),2.94(dd,J=12.7,2.9Hz,1H),2.79(d,J=12.4Hz,1H),2.67(t,J=12.5Hz,1H),2.56(d,J=11.4,1H),2.43(dd,J=12.7,3.4Hz,1H),2.39−2.29(m,1H),2.14(ddd,J=14.9,11.5,3.2Hz,1H)。
20Lのジャケット付きライニング製反応器に、シュウ酸(ステージ5cの(7S,9aS)−7−[(ベンジルオキシ)メチル]オクタヒドロピラジノ[2,1−c][1,4]オキサジン溶液のwt/wt HPLCアッセイに基づいて0.304kg、1.0eq)、エタノール(200proof、2.75L)および酢酸エチル(6.4L)を入れた。この混合物を60℃まで加熱し、50℃を超える温度を維持する速度で、ステージ5cの(7S,9aS)−7−[(ベンジルオキシ)メチル]オクタヒドロピラジノ[2,1−c][1,4]オキサジンの粗酢酸エチル溶液をこの反応器に加えた。この溶液を60℃で30分間撹拌し、20℃まで冷却した後、20℃で一晩撹拌した。固体を取り除き、濾過した後、酢酸エチル:エタノール(6:1、1.8L)溶液で洗浄した。その後、この固体を真空下、50℃のオーブンに入れ、乾燥させ、ベージュ色の結晶性固体として表題化合物を得た(0.306kg)。この表題化合物の第2の収穫物を母液から得た(0.133kg)。1H NMR(DMSO−d6)δ9.5−8.5(br s,4H)7.41−7.36(m,3H),7.35−7.27(m,2H),4.57(d,J=2.0Hz,2H),3.94(t,J=8.3Hz,1H),3.76−3.61(m,4H),3.45(ddd,J=13.9,11.7,2.2Hz,1H),3.06(t,J=11.8Hz,1H),2.94(dd,J=12.7,2.9Hz,1H),2.79(d,J=12.4Hz,1H),2.67(t,J=12.5Hz,1H),2.56(d,J=11.4,1H),2.43(dd,J=12.7,3.4Hz,1H),2.39−2.29(m,1H),2.14(ddd,J=14.9,11.5,3.2Hz,1H)。
中間体4
2−[3,5−ビス(トリフルオロメチル)フェニル]−N−{4−(4−フルオロ−2−メチルフェニル)−6−[(7S,9aS)−7−{[(フェニルメチル)オキシ]メチル}ヘキサヒドロピラジノ[2,1−c][1,4]オキサジン−8(1H)−イル]−3−ピリジニル}−N,2−ジメチルプロパンアミド二塩酸塩
2−[3,5−ビス(トリフルオロメチル)フェニル]−N−{4−(4−フルオロ−2−メチルフェニル)−6−[(7S,9aS)−7−{[(フェニルメチル)オキシ]メチル}ヘキサヒドロピラジノ[2,1−c][1,4]オキサジン−8(1H)−イル]−3−ピリジニル}−N,2−ジメチルプロパンアミド二塩酸塩
100ガロンのグラスライニング製反応器に、中間体3(10.5kg)、トルエン(173kg)および1N NaOH(200L)を入れた。この二相混合物を75℃まで加熱し、5分間撹拌した。撹拌を停止し、これらの層を30分間静置した。75℃で、下部の塩基性の水層を取り除いた。25℃まで冷却した後、水(100L)を加え、10分間撹拌した。この混合物を15分間静置し、下部の水層を取り除いた。トルエン層を真空下で40Lまで濃縮した後、新しいトルエン34.6kgを加えた。この溶液を、カールフィッシャー分析により、その水分含有量について調べた(<0.05wt/vol)。
この溶液を水含有量について調べた後、さらにトルエン(77.9kg)を加え、最終溶液の体積は180Lになった。別の100ガロンのグラスライニング製反応器に、((2−[3,5−ビス(トリフルオロメチル)フェニル]−N−[6−クロロ−4−(4−フルオロ−2−メチルフェニル)−3−ピリジニル]−N,2−ジメチルプロパンアミド(10.0kg)を入れ、続いてナトリウムt−ブトキシド(3.2kg)およびパラジウム(0)ビス−トリ−t−ブチルホスフィン(0.957kg)を入れた。その後、中間体3のトルエン溶液をこれらの固体に加え、85℃まで加熱した。この反応を、HPLCにより反応が完了するまで(約2〜4時間)、85℃で撹拌した。この溶液を25℃まで冷却した後、20%NaHSO3水溶液(100L)をこの反応器に入れた。この二相混合物を60℃まで加熱し、1時間撹拌し、25℃まで冷却して戻した。この二相混合物をこの反応器から取り除き、Celiteを詰めたPallフィルターでろ過した。この濾液をトルエン(8.7kg)ですすぎ、最初の濾液と合わせ、この反応器に戻し入れ、30分間静置した。静置後、下部の水層を取り除き、廃棄した。その後、この反応器に5%システイン水溶液(100L)を入れた。この混合物を60℃まで加熱し、1時間撹拌した後、25℃まで冷却した。再び、この二相の混合物をこの反応器から取り除き、Celiteを詰めたPallフィルターでろ過した。この濾液をトルエン(8.7kg)ですすぎ、合わせた濾液を反応器に戻し入れ、10%aq塩化ナトリウム(40kg)を加えた。この混合物を15分間撹拌し、30分間静置した。下の水層を取り除き、廃棄した。
その後、この反応器に、5%システイン水溶液(100L)を入れた。この混合物を60℃まで加熱し、1時間撹拌した後、25℃まで冷却した。この二相の混合物をこの反応器から取り除き、Celiteを詰めたPallフィルターでろ過した。この濾液をトルエン(8.7kg)ですすぎ、合わせた濾液を反応器に戻し入れ、10%aq塩化ナトリウム(40kg)を加えた。この混合物を15分間撹拌し、30分間静置した。下の水層を取り除き、廃棄した。5%重炭酸ナトリウム水溶液(70L)をこのトルエン層に加え、10分間撹拌した。これらの層を30分間静置し、下の重炭酸層を取り除き、廃棄した。
2%塩化ナトリウム水溶液(70L)を加え、15分間撹拌した。この混合物を30分間静置し、下の水層を取り除き、廃棄した。第2の2%塩化ナトリウム水溶液(70L)を加え、15分間撹拌した。この混合物を30分間静置し、下の水層を取り除き、廃棄した。上部のトルエン層を真空蒸留下で40Lまで濃縮した後、追加量のトルエン(60.6kg)を加えた。この溶液の水分含有量を、カールフィッシャー分析を用いて調べた(<1.0%wt/vol)。その後、この反応器に、ジオキサン中4N HCl(10.3kg)を入れ、25℃で30分間撹拌した。撹拌を終了した後、トルエン(77.9kg)を加え、溶液を真空下で蒸留し、最終体積は100Lになった。追加のトルエン(77.9kg)を入れ、再び、この溶液を真空下で減少させ、最終体積は100Lになった。
試料をガスクロマトグラフィー分析のために取り、溶液中の1,4−ジオキサンの含有量を決定した(<0.55%1,4−ジオキサン)。溶液の温度を25℃に調節し、n−ヘプタン(47.9kg)を、この反応器に少なくとも30分かけてゆっくり加えた。このスラリーを25℃で少なくとも4時間撹拌した。この固体をこの反応器から取り除き、濾過した。濾過ケーキを27.4kgのn−ヘプタンで洗浄した。その後、この固体を40℃の真空オーブンに一晩入れ、黄褐色固体として表題化合物(12.3kg,78.8%収率)を得た。1H NMR(DMSO−d6)δ8.02(s,1H),7.95(s,1H),7.73(br s,2H),7.27(m,5H),7.18(s,1H),7.16(s,1H),7.11(br s,1H),6.83(s,1H),5.12(br m,6H),4.64(d,J=12.5Hz,1H),4.50(d,J=11.7Hz,1H),4.41(m,1H),4.31−4.14(m,2H),4.05−3.76(m,4H),3.60(d,J=12.7Hz,1H),3.51−3.39(m,2H),3.35−3.12(m,2H),3.12−3.0(m,1H),2.55(m,1H),2.36−1.96(m,4H),1.56−1.15(m,4H)。
この溶液を水含有量について調べた後、さらにトルエン(77.9kg)を加え、最終溶液の体積は180Lになった。別の100ガロンのグラスライニング製反応器に、((2−[3,5−ビス(トリフルオロメチル)フェニル]−N−[6−クロロ−4−(4−フルオロ−2−メチルフェニル)−3−ピリジニル]−N,2−ジメチルプロパンアミド(10.0kg)を入れ、続いてナトリウムt−ブトキシド(3.2kg)およびパラジウム(0)ビス−トリ−t−ブチルホスフィン(0.957kg)を入れた。その後、中間体3のトルエン溶液をこれらの固体に加え、85℃まで加熱した。この反応を、HPLCにより反応が完了するまで(約2〜4時間)、85℃で撹拌した。この溶液を25℃まで冷却した後、20%NaHSO3水溶液(100L)をこの反応器に入れた。この二相混合物を60℃まで加熱し、1時間撹拌し、25℃まで冷却して戻した。この二相混合物をこの反応器から取り除き、Celiteを詰めたPallフィルターでろ過した。この濾液をトルエン(8.7kg)ですすぎ、最初の濾液と合わせ、この反応器に戻し入れ、30分間静置した。静置後、下部の水層を取り除き、廃棄した。その後、この反応器に5%システイン水溶液(100L)を入れた。この混合物を60℃まで加熱し、1時間撹拌した後、25℃まで冷却した。再び、この二相の混合物をこの反応器から取り除き、Celiteを詰めたPallフィルターでろ過した。この濾液をトルエン(8.7kg)ですすぎ、合わせた濾液を反応器に戻し入れ、10%aq塩化ナトリウム(40kg)を加えた。この混合物を15分間撹拌し、30分間静置した。下の水層を取り除き、廃棄した。
その後、この反応器に、5%システイン水溶液(100L)を入れた。この混合物を60℃まで加熱し、1時間撹拌した後、25℃まで冷却した。この二相の混合物をこの反応器から取り除き、Celiteを詰めたPallフィルターでろ過した。この濾液をトルエン(8.7kg)ですすぎ、合わせた濾液を反応器に戻し入れ、10%aq塩化ナトリウム(40kg)を加えた。この混合物を15分間撹拌し、30分間静置した。下の水層を取り除き、廃棄した。5%重炭酸ナトリウム水溶液(70L)をこのトルエン層に加え、10分間撹拌した。これらの層を30分間静置し、下の重炭酸層を取り除き、廃棄した。
2%塩化ナトリウム水溶液(70L)を加え、15分間撹拌した。この混合物を30分間静置し、下の水層を取り除き、廃棄した。第2の2%塩化ナトリウム水溶液(70L)を加え、15分間撹拌した。この混合物を30分間静置し、下の水層を取り除き、廃棄した。上部のトルエン層を真空蒸留下で40Lまで濃縮した後、追加量のトルエン(60.6kg)を加えた。この溶液の水分含有量を、カールフィッシャー分析を用いて調べた(<1.0%wt/vol)。その後、この反応器に、ジオキサン中4N HCl(10.3kg)を入れ、25℃で30分間撹拌した。撹拌を終了した後、トルエン(77.9kg)を加え、溶液を真空下で蒸留し、最終体積は100Lになった。追加のトルエン(77.9kg)を入れ、再び、この溶液を真空下で減少させ、最終体積は100Lになった。
試料をガスクロマトグラフィー分析のために取り、溶液中の1,4−ジオキサンの含有量を決定した(<0.55%1,4−ジオキサン)。溶液の温度を25℃に調節し、n−ヘプタン(47.9kg)を、この反応器に少なくとも30分かけてゆっくり加えた。このスラリーを25℃で少なくとも4時間撹拌した。この固体をこの反応器から取り除き、濾過した。濾過ケーキを27.4kgのn−ヘプタンで洗浄した。その後、この固体を40℃の真空オーブンに一晩入れ、黄褐色固体として表題化合物(12.3kg,78.8%収率)を得た。1H NMR(DMSO−d6)δ8.02(s,1H),7.95(s,1H),7.73(br s,2H),7.27(m,5H),7.18(s,1H),7.16(s,1H),7.11(br s,1H),6.83(s,1H),5.12(br m,6H),4.64(d,J=12.5Hz,1H),4.50(d,J=11.7Hz,1H),4.41(m,1H),4.31−4.14(m,2H),4.05−3.76(m,4H),3.60(d,J=12.7Hz,1H),3.51−3.39(m,2H),3.35−3.12(m,2H),3.12−3.0(m,1H),2.55(m,1H),2.36−1.96(m,4H),1.56−1.15(m,4H)。
中間体5
2−[3,5−ビス(トリフルオロメチル)フェニル]−N−{4−(4−フルオロ−2−メチルフェニル)−6−[(7S,9aS)−7−(ヒドロキシメチル)ヘキサヒドロピラジノ[2,1−c][1,4]オキサジン−8(1H)−イル]−3−ピリジニル}−N,2−ジメチルプロパンアミド二塩酸塩モノ−イソプロパノール溶媒和物
2−[3,5−ビス(トリフルオロメチル)フェニル]−N−{4−(4−フルオロ−2−メチルフェニル)−6−[(7S,9aS)−7−(ヒドロキシメチル)ヘキサヒドロピラジノ[2,1−c][1,4]オキサジン−8(1H)−イル]−3−ピリジニル}−N,2−ジメチルプロパンアミド二塩酸塩モノ−イソプロパノール溶媒和物
20Lのジャケット付きライニング製反応器に、中間体4(1.12kg)、10%パラジウム炭素(約50%含水、0.225kg)、イソプロパノール(10.08L)、水(1.12L)および濃塩酸(0.140kg)を入れた。この混合物を激しく撹拌し、50℃まで加熱し、HPLCにより反応の完了が確認されるまで(約3.5〜5時間)、0.25 bargの水素圧下に置いた。この反応が完了した後、この反応器に窒素をパージし、25℃まで冷却した。この反応物をこの反応器から取り出し、Celiteを詰めたPallフィルターでろ過し、パラジウム触媒を除去した。イソプロパノール(10.1L)および水(1.12L)を反応器に加えた後、Pallフィルターに通過させた。濾液を合わせ、イソプロパノール溶液(約24.0L)中に表題化合物を得た。当量を用いて別の反応を行い、(前のと)合わせて、イソプロパノール溶液(約48.0L)中に表題化合物(2.16kg)を得た。これらの合わせた溶液を、0.45ミクロンフィルターを通して50Lのジャケット付きライニング製反応器に入れた。この溶液を真空下で蒸留して8.0Lにした後、追加のIPA(22.4L)をこの反応器に入れた。再び、この溶液を真空下で蒸留して8.0Lにし、追加のIPA(22.4L)を加えた。
もう一度、この溶液を8.0Lまで減少させ、IPA(19.1L)を加えた。試料を取り、カールフィッシャー分析を用いて水分含有量を調べた(<0.4%wt/vol)。
この溶液を25℃に調節し、ジオキサン中4N HCl(1.35L)を加えた。その後、この反応を65℃まで加熱し、30分間撹拌した。この溶液を25℃まで冷却し、2,2,4−トリメチルペンタン(11.2L)を加え、スラリーを一晩撹拌した。固体をこの反応器から取り除き、濾過した。濾過ケーキを、IPAと2,2,4−トリメチルペンタンの1:1溶液(5.0L)で洗浄した。固体を乾かし、30℃の真空オーブンに一晩入れ、パールホワイトの固体として表題化合物(1.914kg,88.6%収率)を得た。1H NMR(DMSO−d6)δ11.42(br s,1H),8.01(s,1H),7.97(br s,1H),7.73(br s,2H),7.18(d,J=10.2Hz,1H),7.12(br m,2H),6.95(s,1H),5.78(br s,5H),4.77−4.65(m,1H),4.57−4.43(m,1H),4.21(dd,J=12.7,1.2Hz,1H),4.06−3.81(m,5H),3.77(sept,J=6.1Hz,IPA,1H),3.64(d,J=12.7Hz,1H),3.52−3.42(m,1H),3.42(d,J=11.7Hz,1H),3.29(dd,J=12.4,4.4Hz,1H),3.24−3.09(m,2H),2.65−2.54(m,1H),2.38−2.05(m,4H),1.56−1.11(m,4H),1.03(d,J=6.2Hz,IPA,6H)。
もう一度、この溶液を8.0Lまで減少させ、IPA(19.1L)を加えた。試料を取り、カールフィッシャー分析を用いて水分含有量を調べた(<0.4%wt/vol)。
この溶液を25℃に調節し、ジオキサン中4N HCl(1.35L)を加えた。その後、この反応を65℃まで加熱し、30分間撹拌した。この溶液を25℃まで冷却し、2,2,4−トリメチルペンタン(11.2L)を加え、スラリーを一晩撹拌した。固体をこの反応器から取り除き、濾過した。濾過ケーキを、IPAと2,2,4−トリメチルペンタンの1:1溶液(5.0L)で洗浄した。固体を乾かし、30℃の真空オーブンに一晩入れ、パールホワイトの固体として表題化合物(1.914kg,88.6%収率)を得た。1H NMR(DMSO−d6)δ11.42(br s,1H),8.01(s,1H),7.97(br s,1H),7.73(br s,2H),7.18(d,J=10.2Hz,1H),7.12(br m,2H),6.95(s,1H),5.78(br s,5H),4.77−4.65(m,1H),4.57−4.43(m,1H),4.21(dd,J=12.7,1.2Hz,1H),4.06−3.81(m,5H),3.77(sept,J=6.1Hz,IPA,1H),3.64(d,J=12.7Hz,1H),3.52−3.42(m,1H),3.42(d,J=11.7Hz,1H),3.29(dd,J=12.4,4.4Hz,1H),3.24−3.09(m,2H),2.65−2.54(m,1H),2.38−2.05(m,4H),1.56−1.11(m,4H),1.03(d,J=6.2Hz,IPA,6H)。
実施例1
無水結晶形態1(Form1)の2−[3,5−ビス(トリフルオロメチル)フェニル]−N−{4−(4−フルオロ−2−メチルフェニル)−6−[(7S,9aS)−7−(ヒドロキシメチル)ヘキサヒドロピラジノ[2,1−c][1,4]オキサジン−8(1H)−イル]−3−ピリジニル}−N,2−ジメチルプロパンアミドの調製
方法A
8gの非結晶遊離塩基の2−[3,5−ビス(トリフルオロメチル)フェニル]−N−{4−(4−フルオロ−2−メチルフェニル)−6−[(7S,9aS)−7−(ヒドロキシメチル)ヘキサヒドロピラジノ[2,1−c][1,4]オキサジン−8(1H)−イル]−3−ピリジニル}−N,2−ジメチルプロパンアミドを、ホイルで覆った500mL容器に分注した。この固体を、350rpmのオーバーヘッド撹拌により、ポリブロックの中で、120mlのイソオクタンでスラリーにした。このスラリーを、5℃/分の速度で70℃まで加熱した。この流動性のあるスラリーは、15分以内に淡黄色からクリーム色に変わった。70℃で一晩置いた後、このスラリーを1℃/分の速度で25℃まで冷却した。porosity3フィルターを用いて、真空下での濾過により固体を単離した。淡黄色の湿気のある粉末を乾燥濾過し、クリーム色の粉末を得た(固体1)。
無水結晶形態1(Form1)の2−[3,5−ビス(トリフルオロメチル)フェニル]−N−{4−(4−フルオロ−2−メチルフェニル)−6−[(7S,9aS)−7−(ヒドロキシメチル)ヘキサヒドロピラジノ[2,1−c][1,4]オキサジン−8(1H)−イル]−3−ピリジニル}−N,2−ジメチルプロパンアミドの調製
方法A
8gの非結晶遊離塩基の2−[3,5−ビス(トリフルオロメチル)フェニル]−N−{4−(4−フルオロ−2−メチルフェニル)−6−[(7S,9aS)−7−(ヒドロキシメチル)ヘキサヒドロピラジノ[2,1−c][1,4]オキサジン−8(1H)−イル]−3−ピリジニル}−N,2−ジメチルプロパンアミドを、ホイルで覆った500mL容器に分注した。この固体を、350rpmのオーバーヘッド撹拌により、ポリブロックの中で、120mlのイソオクタンでスラリーにした。このスラリーを、5℃/分の速度で70℃まで加熱した。この流動性のあるスラリーは、15分以内に淡黄色からクリーム色に変わった。70℃で一晩置いた後、このスラリーを1℃/分の速度で25℃まで冷却した。porosity3フィルターを用いて、真空下での濾過により固体を単離した。淡黄色の湿気のある粉末を乾燥濾過し、クリーム色の粉末を得た(固体1)。
この粉末はこの容器の中で丸い凝集体を形成したので、いくぶんかの物質は濾過できなかった。前に回収した濾液をこの容器に再び分注した。この凝集体物質を、70℃で7時間撹拌することによって、スラリーに分散させた。その後、この実験を1℃/分の速度で25℃まで冷却し、そのままで3夜置いた。濾過により、クリーム色の粉末を得た(固体2)。固体1および固体2を、40℃の真空下で19時間乾燥させ、表題化合物(7.087g)を得た。
方法B
100ガロンのグラスライニング製反応器に、中間体5(10.1kg)、続いて、MTBE(112.1kg)を入れた。この混合物に、2.5N NaOH(50.5L)を加えた。この反応を40℃に達するまで撹拌した後、40℃で15分間静置した。下部の水層を取り除き、廃棄した。その後、この反応器に、10%L−システイン水溶液(50.5L)を入れた。この二相混合物を40℃まで加熱し、1時間撹拌した。この混合物を15分間静置し、下部の水層を取り除いて、廃棄した。その後、この反応器に水(50.5L)を入れた。この混合物を40℃で15分間撹拌し、60分間静置し、下部の水層を取り除いて、廃棄した。再び、水(50.5L)を入れ、この反応を40℃で15分間撹拌した後、60分間静置した。下層を取り除いて、廃棄した。MTBE層を大気圧下で蒸留し、約25Lにした。このMTBE溶液を55℃まで温めた後、50〜55℃の間に温度を維持しながら、2,2,4−トリメチルペンタン(58.3kg)を1時間かけてゆっくり加えた。その後、この溶液を大気圧下で加熱し、最終体積は約40Lになった。
その後、この溶液を75℃まで冷却し、IPA(6.0kg)をこの反応器に加えた。この溶液を55℃まで冷却した後、カートリッジフィルターに通過させて、きれいな100ガロンのグラスライニング製反応器に入れた。カートリッジフィルターを通して、追加の2,2,4−トリメチルペンタン(31.1kg)をこの反応器に加えた。添加後、この溶液を70℃まで加熱し、30分間撹拌した後、50℃まで冷却して戻した。IPA(0.0657kg)および2,2,4−トリメチルペンタン(0.5222kg)中の無水結晶形態1(Form1)の2−[3,5−ビス(トリフルオロメチル)フェニル]−N−{4−(4−フルオロ−2−メチルフェニル)−6−[(7S,9aS)−7−(ヒドロキシメチル)ヘキサヒドロピラジノ[2,1−c][1,4]オキサジン−8(1H)−イル]−3−ピリジニル}−N,2−ジメチルプロパンアミドのシードスラリー(0.0838kg)を、カートリッジフィルターで濾過した溶媒を用いて調製した。その後、このシードスラリーを50℃の反応器に入れた。その後、このスラリーを、50℃で少なくとも3時間撹拌した。その後、2,2,4−トリメチルペンタン(29.2kg)を、定量ポンプにより、3時間かけて(カートリッジフィルターを通して)この反応器に加えた。この添加が完了した後、このスラリーを50℃で一晩置き、4.5時間かけて0℃まで冷却した後、0℃で少なくとも3時間置いた。その後、このスラリーを取り除いて、乾燥濾過機により濾過した。その後、この濾過ケーキを、カートリッジフィルターに通過させてこの反応器に入れた冷たい(0℃)2,2,4−トリメチルペンタン(2×23.3kg)で2回洗浄した。この固体を、真空下の50℃の乾燥濾過機の中で15時間乾燥させた(LOD<0.5%)。その後、この固体をこの乾燥機から取り出し、Quadro(登録商標)Comil(登録商標)、C−101によりふるいにかけ、ポリエチレンバッグで二重に裏打ちされたHDPE(高密度ポリエチレン)ドラムに回収し、白色固体として表題化合物(6.1kg、72.6%収率)を得た。DSCにより、分解を伴う開始融点=162℃であることが分かった。1H NMR(DMSO−d6)δ8.01(s,1H),7.85(s,1H),7.73(br s,2H),7.15(d,J=10.0Hz,1H),7.11(br m,2H),6.60(s,1H),4.68(dd,J=6.4,1.7Hz,1H),4.27−4.16(m,1H),4.16−4.0(m,1H),3.81−3.69(m,3H),3.55(dd,J=11.7,2.0Hz,1H),3.45−3.36(m,1H),3.15(t,J=10.5Hz,1H),3.02(d,J=10.7Hz,1H),2.64(d,J=11.8Hz,1H),2.58−2.53(m,2H),2.32−2.01(m,8H),1.57−1.12(m,6H)。
100ガロンのグラスライニング製反応器に、中間体5(10.1kg)、続いて、MTBE(112.1kg)を入れた。この混合物に、2.5N NaOH(50.5L)を加えた。この反応を40℃に達するまで撹拌した後、40℃で15分間静置した。下部の水層を取り除き、廃棄した。その後、この反応器に、10%L−システイン水溶液(50.5L)を入れた。この二相混合物を40℃まで加熱し、1時間撹拌した。この混合物を15分間静置し、下部の水層を取り除いて、廃棄した。その後、この反応器に水(50.5L)を入れた。この混合物を40℃で15分間撹拌し、60分間静置し、下部の水層を取り除いて、廃棄した。再び、水(50.5L)を入れ、この反応を40℃で15分間撹拌した後、60分間静置した。下層を取り除いて、廃棄した。MTBE層を大気圧下で蒸留し、約25Lにした。このMTBE溶液を55℃まで温めた後、50〜55℃の間に温度を維持しながら、2,2,4−トリメチルペンタン(58.3kg)を1時間かけてゆっくり加えた。その後、この溶液を大気圧下で加熱し、最終体積は約40Lになった。
その後、この溶液を75℃まで冷却し、IPA(6.0kg)をこの反応器に加えた。この溶液を55℃まで冷却した後、カートリッジフィルターに通過させて、きれいな100ガロンのグラスライニング製反応器に入れた。カートリッジフィルターを通して、追加の2,2,4−トリメチルペンタン(31.1kg)をこの反応器に加えた。添加後、この溶液を70℃まで加熱し、30分間撹拌した後、50℃まで冷却して戻した。IPA(0.0657kg)および2,2,4−トリメチルペンタン(0.5222kg)中の無水結晶形態1(Form1)の2−[3,5−ビス(トリフルオロメチル)フェニル]−N−{4−(4−フルオロ−2−メチルフェニル)−6−[(7S,9aS)−7−(ヒドロキシメチル)ヘキサヒドロピラジノ[2,1−c][1,4]オキサジン−8(1H)−イル]−3−ピリジニル}−N,2−ジメチルプロパンアミドのシードスラリー(0.0838kg)を、カートリッジフィルターで濾過した溶媒を用いて調製した。その後、このシードスラリーを50℃の反応器に入れた。その後、このスラリーを、50℃で少なくとも3時間撹拌した。その後、2,2,4−トリメチルペンタン(29.2kg)を、定量ポンプにより、3時間かけて(カートリッジフィルターを通して)この反応器に加えた。この添加が完了した後、このスラリーを50℃で一晩置き、4.5時間かけて0℃まで冷却した後、0℃で少なくとも3時間置いた。その後、このスラリーを取り除いて、乾燥濾過機により濾過した。その後、この濾過ケーキを、カートリッジフィルターに通過させてこの反応器に入れた冷たい(0℃)2,2,4−トリメチルペンタン(2×23.3kg)で2回洗浄した。この固体を、真空下の50℃の乾燥濾過機の中で15時間乾燥させた(LOD<0.5%)。その後、この固体をこの乾燥機から取り出し、Quadro(登録商標)Comil(登録商標)、C−101によりふるいにかけ、ポリエチレンバッグで二重に裏打ちされたHDPE(高密度ポリエチレン)ドラムに回収し、白色固体として表題化合物(6.1kg、72.6%収率)を得た。DSCにより、分解を伴う開始融点=162℃であることが分かった。1H NMR(DMSO−d6)δ8.01(s,1H),7.85(s,1H),7.73(br s,2H),7.15(d,J=10.0Hz,1H),7.11(br m,2H),6.60(s,1H),4.68(dd,J=6.4,1.7Hz,1H),4.27−4.16(m,1H),4.16−4.0(m,1H),3.81−3.69(m,3H),3.55(dd,J=11.7,2.0Hz,1H),3.45−3.36(m,1H),3.15(t,J=10.5Hz,1H),3.02(d,J=10.7Hz,1H),2.64(d,J=11.8Hz,1H),2.58−2.53(m,2H),2.32−2.01(m,8H),1.57−1.12(m,6H)。
粉末X線回折(XRPD)
XRPDパターンを、モノクロメータ装備のX’Celerator検出器を用いるPANalytical社製X’−Pert Pro粉末回折計モデルPW3050/60で、銅KαX線照射を用いて取得した。取得条件は、照射:Cu Kα、発生電圧:45kV、発生電流:40mA、ステップサイズ:0.008°2θ、ステップあたりの時間:575秒、発散スリットの種類:固定、発散スリットのサイズ:0.4354°、測定温度:20〜25℃の範囲、角度計の半径:240mmである。試料を、実施例1の方法B数ミリグラムを0.7mmの毛細管に詰めることによって調製する。パターンを表1に示す。
XRPDパターンを、モノクロメータ装備のX’Celerator検出器を用いるPANalytical社製X’−Pert Pro粉末回折計モデルPW3050/60で、銅KαX線照射を用いて取得した。取得条件は、照射:Cu Kα、発生電圧:45kV、発生電流:40mA、ステップサイズ:0.008°2θ、ステップあたりの時間:575秒、発散スリットの種類:固定、発散スリットのサイズ:0.4354°、測定温度:20〜25℃の範囲、角度計の半径:240mmである。試料を、実施例1の方法B数ミリグラムを0.7mmの毛細管に詰めることによって調製する。パターンを表1に示す。
実施例1の方法Aの粉末X線回折(XRPD)パターンは、図1に報告したパターンと一致する。
熱分析
示差走査熱量測定(DSC)を、TA Q1000熱量計で行った。実施例1の方法Bの試料をアルミニウムパンに測り入れ、パンの蓋を上部に置き、このパンを密封することなく軽く圧着させた。走査速度10℃/分。試料サイズ1〜2mg。無水結晶形態1(Form1)の式(I)の化合物のサーモグラムを図2に示す。
示差走査熱量測定(DSC)を、TA Q1000熱量計で行った。実施例1の方法Bの試料をアルミニウムパンに測り入れ、パンの蓋を上部に置き、このパンを密封することなく軽く圧着させた。走査速度10℃/分。試料サイズ1〜2mg。無水結晶形態1(Form1)の式(I)の化合物のサーモグラムを図2に示す。
実施例1方法Aの示差走査熱量測定(DSC)サーモグラムは、図2に報告するサーモグラムと一致する。
DSCデータを報告する時、事象の開始温度またはピーク温度が報告され得る。電流充電中は、開始温度のみ報告される。この開始温度は、主要な事象の接線とベースラインとの交差点である。
162℃の開始温度でのややシャープな非対称の融解級熱は分解を伴った。
融解が分解を伴う時は、同じ物質の異なるバッチにおいて開始融点に小さな変動が観察される場合があることを当該技術者は理解するであろう。
固体核磁気共鳴
実施例1の方法Bの13C固体NMRスペクトルを図3に示す。データを、399.87MHzの1H周波数で作動するBruker社製Avance 400 triple−resonance spectrometerを用いて取得した。示した13CのSSNMRスペクトルは、8kHzの回転周波数で、Bruker社製4mmの3重共鳴マジックアングルスピンプローブを用いる交差分極パルスシーケンスを用いて得られた。75〜90kHzの直線的な出力ランプ(linear power ramp)を、交差分極効率を高めるために1Hチャネル上で用いた。5パルスの全サイドバンド抑圧パルスシーケンスにより、スピニングサイドバンド除去した。1Hデカップリングは、Spinal−64 sequenceを用いて得たが、19Fデカップリングは、回転周期あたり1πパルスを用いるπ−パルスデカップリングにより得た。特徴的な13C NMRピーク位置を、0ppm(100万分の1)のテトラメチルシランに対して記録し、機器の変動および較正により、精度は±0.2ppmである。
実施例1の方法Bの13C固体NMRスペクトルを図3に示す。データを、399.87MHzの1H周波数で作動するBruker社製Avance 400 triple−resonance spectrometerを用いて取得した。示した13CのSSNMRスペクトルは、8kHzの回転周波数で、Bruker社製4mmの3重共鳴マジックアングルスピンプローブを用いる交差分極パルスシーケンスを用いて得られた。75〜90kHzの直線的な出力ランプ(linear power ramp)を、交差分極効率を高めるために1Hチャネル上で用いた。5パルスの全サイドバンド抑圧パルスシーケンスにより、スピニングサイドバンド除去した。1Hデカップリングは、Spinal−64 sequenceを用いて得たが、19Fデカップリングは、回転周期あたり1πパルスを用いるπ−パルスデカップリングにより得た。特徴的な13C NMRピーク位置を、0ppm(100万分の1)のテトラメチルシランに対して記録し、機器の変動および較正により、精度は±0.2ppmである。
医薬組成物
無水結晶形態1(Form1)の式(I)の化合物は、通常、患者に投与される前に医薬組成物に処方されるが、必ずしもそうであるわけではない。一態様において、本発明は、無水結晶形態1(Form1)の式(I)の化合物を含んでなる医薬組成物を対象にする。
無水結晶形態1(Form1)の式(I)の化合物は、通常、患者に投与される前に医薬組成物に処方されるが、必ずしもそうであるわけではない。一態様において、本発明は、無水結晶形態1(Form1)の式(I)の化合物を含んでなる医薬組成物を対象にする。
無水結晶形態1の式(I)の化合物の錠剤は、無水結晶形態1(Form1)の式(I)の化合物を20mg、25mg、45mg、100mgおよび200mg含んでなり、有効成分の即時放出をもたらす茶褐色のフィルムでコーティングされた丸い(または200mg長の楕円形)錠剤として経口投与用に処方されている。
賦形剤および錠剤の定量的組成のリストを以下の表2に報告する。
無水結晶形態1(Form1)の式(I)の化合物の錠剤20mg、25mg、45mg、100mgおよび200mgを、湿式造粒プロセス、乾式混合プロセス、打錠プロセスおよびフィルムコーティングプロセスを用いて製造した。
原体、ラクトース一水和物、結晶セルロースおよびクロスカルメロースナトリウムをふるいにかけ、約5分間、高剪断ミキサー造粒機の中で乾式混合した。この原体、ラクトース一水和物、結晶セルロースおよびクロスカルメロースナトリウムの乾燥混合物上に造粒水を噴霧した。湿った顆粒を、流動層乾燥機の中で、約40分間約65℃で乾燥させ(<2%LOD)、コニカルミル(ふるいサイズ813μm)を用いて粉砕し、ビン混合機の中でラクトース、結晶セルロースおよびクロスカルメロースナトリウムを約20分間混合した。ステアリン酸マグネシウムを潤滑のためにこのビン混合機に加え、この混合物を約3分間混合した。
この混合物を適切な別の(一打)打錠機を用いて圧縮し、コーティングしていない錠剤を得た。Opadry(登録商標)黄色03B22133を、精製水および調製したフィルムコーティング懸濁液と共に撹拌しながら混合容器の中に入れた。これらの錠剤を、適切なパンコーティング機に入れてフィルムコーティングした(約3%重量増加)。
本発明の第5の態様において、図1と実質的に同じ粉末X線回折(XRPD)パターンによって特徴づけられる無水結晶形態1(Form1)の式(I)の化合物を提供し、このXRPDパターンは2θ角の単位で表され、銅KαX線照射を用いる回折計により得られ、このXRPDパターンは、本質的に以下の位置4.3±0.1、7.9±0.1、9.8±0.1、10.7±0.1、10.8±0.1、13.1±0.1、13.2±0.1、13.3±0.1、14.0±0.1、14.4±0.1、15.0±0.1、15.1±0.1、15.7±0.1、15.9±0.1、16.3±0.1、16.5±0.1、16.8±0.1、17.0±0.1、17.4±0.1、17.5±0.1、18.1±0.1、18.2±0.1、18.7±0.1、19.4±0.1、19.7±0.1、20.0±0.1、20.1±0.1、20.2±0.1、20.5±0.1、20.7±0.1、21.5±0.1、21.8±0.1°に2θ角ピークを含んでなり、これらの値は、それぞれ、20.4、11.1、9.0、8.3、8.2、6.8、6.7、6.6、6.3、6.1、5.9、5.9、5.6、5.6、5.4、5.4、5.3、5.2、5.1、5.1、4.9、4.9、4.7、4.6、4.5、4.4、4.4、4.4、4.3、4.3、4.1および4.1オングストローム(Å)の格子面間隔(d−spacing)に対応する。
Claims (13)
- 無水結晶形態の式(I)の化合物:
- 前記無水結晶形態が形態1(Form 1)である、請求項1に記載の無水結晶形態の式(I)の化合物。
- 図1と実質的に同じ粉末X線回折(XRPD)パターンによって特徴づけられる、請求項2に記載の無水結晶形態1(Form 1)の式(I)の化合物。
- 前記XRPDパターンが、本質的に以下の位置:4.3±0.1、7.9±0.1、9.8±0.1、10.7±0.1、10.8±0.1、13.3±0.1、14.0±0.1、15.1±0.1°に2θ角ピークを含んでなり、これらの値が、それぞれ、20.4、11.1、9.0、8.3、8.2、6.6、6.3および5.9オングストローム(Å)の格子面間隔(d−spacing)に対応する、請求項2または3に記載の無水結晶形態1(Form 1)の式(I)の化合物。
- 前記XRPDパターンが、本質的に以下の位置:4.3±0.1、7.9±0.1、9.8±0.1、10.7±0.1、10.8±0.1、13.1±0.1、13.2±0.1、13.3±0.1、14.0±0.1、14.4±0.1、15.0±0.1、15.1±0.1、15.7±0.1、15.9±0.1、16.3±0.1、16.5±0.1、16.8±0.1、17.0±0.1、17.4±0.1、17.5±0.1、18.1±0.1、18.2±0.1、18.7±0.1、19.4±0.1、19.7±0.1、20.0±0.1、20.1±0.1、20.2±0.1、20.5±0.1、20.7±0.1、21.5±0.1、21.8±0.1°に2θ角ピークを含んでなり、これらの値が、それぞれ、20.4、11.1、9.0、8.3、8.2、6.8、6.7、6.6、6.3、6.1、5.9、5.9、5.6、5.6、5.4、5.4、5.3、5.2、5.1、5.1、4.9、4.9、4.7、4.6、4.5、4.4、4.4、4.4、4.3、4.3、4.1および4.1オングストローム(Å)の格子面間隔(d−spacing)に対応する、請求項2または3に記載の無水結晶形態1(Form 1)の式(I)の化合物。
- 図3と実質的に同じ13C固体核磁気共鳴(SSNMR)スペクトルによって特徴づけられ、前記固体SSNMRスペクトルが、13Cの観察のために100.56MHzの周波数で作動する分光計において、8kHzの回転周波数で、Bruker社製4mmの3重共鳴マジックアングルスピンプローブを用いる交差分極パルスシーケンスを用いて得られた、請求項2に記載の無水結晶形態1(Form 1)の式(I)の化合物。
- 前記SSNMRが、175.03±0.2、163.30±0.2、158.66±0.2、156.50±0.2、149.79±0.2、148.17±0.2、146.96±0.2、139.56±0.2、133.19±0.2、132.32±0.2、129.76±0.2、126.5±0.2、124.15±0.2、120.37±0.2、119.20±0.2、118.16±0.2、112.83±0.2、70.62±0.2、67.61±0.2、64.96±0.2、59.89±0.2、57.08±0.2、55.50±0.2、52.41±0.2、48.35±0.2、40.79±0.2、32.75および21.12±0.2ppmの化学シフトを含んでなる、請求項2または6に記載の無水結晶形態1(Form 1)の式(I)の化合物。
- 請求項1〜7のいずれか一項に記載の無水結晶形態の式(I)の化合物を含んでなる、医薬組成物。
- 1種類以上の医薬的に許容される担体または希釈剤をさらに含んでなる、請求項8に記載の医薬組成物。
- 治療に使用するための、請求項1〜7のいずれか一項に記載の無水結晶形態の式(I)の化合物。
- 精神障害、うつ病、気分障害、不安症、睡眠障害および物質関連障害の治療または予防のための医薬の調製における、請求項1〜7のいずれか一項に記載の無水結晶形態の式(I)の化合物の使用。
- 精神障害、うつ病、気分障害、不安症、睡眠障害および物質関連障害の治療または予防に使用するための、請求項1〜7のいずれか一項に記載の無水結晶形態の式(I)の化合物。
- 有効量の請求項1〜7のいずれか一項に記載の無水結晶形態の式(I)の化合物を哺乳類に投与することを含んでなる、精神障害、うつ病、気分障害、不安症、睡眠障害および物質関連障害の治療法または予防法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US23743509P | 2009-08-27 | 2009-08-27 | |
| US61/237,435 | 2009-08-27 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015139845A Division JP6404186B2 (ja) | 2009-08-27 | 2015-07-13 | 無水形態のピリジン誘導体 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2017193564A true JP2017193564A (ja) | 2017-10-26 |
Family
ID=42790943
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012526048A Pending JP2013503134A (ja) | 2009-08-27 | 2010-08-25 | 無水形態のピリジン誘導体 |
| JP2015139845A Active JP6404186B2 (ja) | 2009-08-27 | 2015-07-13 | 無水形態のピリジン誘導体 |
| JP2017114602A Pending JP2017193564A (ja) | 2009-08-27 | 2017-06-09 | 無水形態のピリジン誘導体 |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012526048A Pending JP2013503134A (ja) | 2009-08-27 | 2010-08-25 | 無水形態のピリジン誘導体 |
| JP2015139845A Active JP6404186B2 (ja) | 2009-08-27 | 2015-07-13 | 無水形態のピリジン誘導体 |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US8796269B2 (ja) |
| EP (1) | EP2470545B1 (ja) |
| JP (3) | JP2013503134A (ja) |
| KR (1) | KR20120056258A (ja) |
| CN (1) | CN102639536B (ja) |
| AU (1) | AU2010288502B2 (ja) |
| CA (1) | CA2772168C (ja) |
| DK (1) | DK2470545T3 (ja) |
| EA (1) | EA021411B1 (ja) |
| ES (1) | ES2440938T3 (ja) |
| IL (1) | IL217922A (ja) |
| IN (1) | IN2012DN01292A (ja) |
| MX (1) | MX2012002369A (ja) |
| PL (1) | PL2470545T3 (ja) |
| SG (1) | SG178231A1 (ja) |
| WO (1) | WO2011023733A1 (ja) |
| ZA (1) | ZA201200812B (ja) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013503134A (ja) * | 2009-08-27 | 2013-01-31 | グラクソスミスクライン・リミテッド・ライアビリティ・カンパニー | 無水形態のピリジン誘導体 |
| EP3915560A1 (en) | 2014-06-25 | 2021-12-01 | Emory University | Methods of managing conditioned fear with neurokinin receptor antagonists |
| JP6902682B2 (ja) * | 2015-05-18 | 2021-07-14 | カンディー セラピューティクス リミテッド | 性ホルモン依存性疾患を治療するためのnk−1/nk−3受容体デュアルアンタゴニスト |
| AU2017376900A1 (en) * | 2016-12-15 | 2019-05-23 | Glaxosmithkline Intellectual Property Development Limited | NRF2 compounds |
| EA202092131A1 (ru) | 2018-03-14 | 2020-11-27 | Кэнди Терапьютикс Лимитед | Новый фармацевтический препарат, содержащий двойные антагонисты рецептора nk-1/nk-3 |
| WO2021030335A1 (en) * | 2019-08-12 | 2021-02-18 | Millendo Therapeutics, Inc. | A stereoisomerically pure nk-3 receptor antagonist and crystalline forms thereof |
| AU2020381762B2 (en) | 2019-11-15 | 2026-01-22 | KaNDy Therapeutics Limited | New chemical process for making 6-chloro-4-(4-fluoro-2-methylphenyl)pyridin-3-amine a key intermediate of NT-814 |
| EP4431512A1 (en) | 2023-03-16 | 2024-09-18 | Bayer Consumer Care AG | Novel dual nk-1/nk-3 receptor antagonists |
| WO2025242584A1 (en) | 2024-05-24 | 2025-11-27 | Bayer Consumer Care Ag | A stable soft gelatin capsule formulation for elinzanetant |
| WO2025242585A1 (en) | 2024-05-24 | 2025-11-27 | Bayer Consumer Care Ag | Crystalline forms of salts of elinzanetant and compositions containing salts of elinzanetant |
| WO2025242583A1 (en) | 2024-05-24 | 2025-11-27 | Bayer Consumer Care Ag | Novel formulation comprising elinzanetant in a solid dispersion |
| WO2025261856A1 (en) * | 2024-06-18 | 2025-12-26 | Bayer Consumer Care Ag | Improved process for producing an elinzanetant precursor |
| US12533358B1 (en) | 2025-05-14 | 2026-01-27 | Bayer Consumer Care Ag | Methods of treatment with elinzanetant |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006514999A (ja) * | 2003-01-31 | 2006-05-18 | エフ.ホフマン−ラ ロシュ アーゲー | 2−(3,5−ビス−トリフルオロメチル−フェニル)−N−[6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イル)−4−(4−フルオロ−2−メチル−フェニル)−ピリジン−3−イル]−N−メチル−イソブチルアミドの新規な結晶変形 |
| JP2009507801A (ja) * | 2005-09-09 | 2009-02-26 | スミスクライン・ビーチャム・コーポレイション | ピリジン誘導体および精神異常の処置におけるそれらの使用 |
| WO2009087667A2 (en) * | 2007-12-11 | 2009-07-16 | Cipla Ltd. | A new method for preparation of zopiclone and its polymorphs |
| JP2015139845A (ja) * | 2014-01-29 | 2015-08-03 | 京セラ株式会社 | ドリルおよびそれを用いた切削加工物の製造方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE60045564D1 (de) * | 1999-02-24 | 2011-03-03 | Hoffmann La Roche | 4-Phenylpyridinderivate und deren Verwendung als NK-1 Rezeptorantagonisten |
| GB0203022D0 (en) * | 2002-02-08 | 2002-03-27 | Glaxo Group Ltd | Chemical compounds |
| JP4490421B2 (ja) * | 2003-07-03 | 2010-06-23 | エフ.ホフマン−ラ ロシュ アーゲー | 統合失調症を処置するデュアルnk1/nk3アンタゴニスト |
| GB0814340D0 (en) * | 2008-08-05 | 2008-09-10 | Smithkline Beecham Corp | Anhydrous crystol form fo a pyridine derivative |
| JP2013503134A (ja) * | 2009-08-27 | 2013-01-31 | グラクソスミスクライン・リミテッド・ライアビリティ・カンパニー | 無水形態のピリジン誘導体 |
-
2010
- 2010-08-25 JP JP2012526048A patent/JP2013503134A/ja active Pending
- 2010-08-25 WO PCT/EP2010/062420 patent/WO2011023733A1/en not_active Ceased
- 2010-08-25 AU AU2010288502A patent/AU2010288502B2/en active Active
- 2010-08-25 DK DK10747619.4T patent/DK2470545T3/da active
- 2010-08-25 CA CA2772168A patent/CA2772168C/en active Active
- 2010-08-25 MX MX2012002369A patent/MX2012002369A/es active IP Right Grant
- 2010-08-25 KR KR1020127004865A patent/KR20120056258A/ko not_active Ceased
- 2010-08-25 EA EA201270302A patent/EA021411B1/ru not_active IP Right Cessation
- 2010-08-25 PL PL10747619T patent/PL2470545T3/pl unknown
- 2010-08-25 SG SG2012007464A patent/SG178231A1/en unknown
- 2010-08-25 US US13/389,525 patent/US8796269B2/en active Active
- 2010-08-25 ES ES10747619.4T patent/ES2440938T3/es active Active
- 2010-08-25 EP EP10747619.4A patent/EP2470545B1/en active Active
- 2010-08-25 IN IN1292DEN2012 patent/IN2012DN01292A/en unknown
- 2010-08-25 CN CN201080037918.3A patent/CN102639536B/zh active Active
-
2012
- 2012-02-02 ZA ZA2012/00812A patent/ZA201200812B/en unknown
- 2012-02-02 IL IL217922A patent/IL217922A/en active IP Right Grant
-
2015
- 2015-07-13 JP JP2015139845A patent/JP6404186B2/ja active Active
-
2017
- 2017-06-09 JP JP2017114602A patent/JP2017193564A/ja active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006514999A (ja) * | 2003-01-31 | 2006-05-18 | エフ.ホフマン−ラ ロシュ アーゲー | 2−(3,5−ビス−トリフルオロメチル−フェニル)−N−[6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イル)−4−(4−フルオロ−2−メチル−フェニル)−ピリジン−3−イル]−N−メチル−イソブチルアミドの新規な結晶変形 |
| JP2009507801A (ja) * | 2005-09-09 | 2009-02-26 | スミスクライン・ビーチャム・コーポレイション | ピリジン誘導体および精神異常の処置におけるそれらの使用 |
| WO2009087667A2 (en) * | 2007-12-11 | 2009-07-16 | Cipla Ltd. | A new method for preparation of zopiclone and its polymorphs |
| JP2015139845A (ja) * | 2014-01-29 | 2015-08-03 | 京セラ株式会社 | ドリルおよびそれを用いた切削加工物の製造方法 |
Non-Patent Citations (4)
| Title |
|---|
| 厚生省医薬安全局審査管理課長: "医薬品の残留溶媒ガイドラインについて", 医薬審, vol. 第307号, JPN6014031669, 1998, pages 1 - 11, ISSN: 0003897904 * |
| 平山令明, 有機化合物結晶作製ハンドブック−原理とノウハウ−, JPN6014035600, 25 July 2008 (2008-07-25), pages 17 - 23, ISSN: 0003897901 * |
| 浅原 照三, 溶剤ハンドブック, JPN6011026738, 1985, pages 47 - 51, ISSN: 0003897903 * |
| 高田則幸: "創薬段階における原薬Formスクリーニングと選択", PHARM STAGE, vol. 6, no. 10, JPN6009053755, 15 January 2007 (2007-01-15), pages 20 - 25, ISSN: 0003897902 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2016000739A (ja) | 2016-01-07 |
| IL217922A0 (en) | 2012-03-29 |
| WO2011023733A1 (en) | 2011-03-03 |
| US8796269B2 (en) | 2014-08-05 |
| ES2440938T3 (es) | 2014-01-31 |
| BR112012003435A2 (pt) | 2016-02-23 |
| PL2470545T3 (pl) | 2014-03-31 |
| US20120157450A1 (en) | 2012-06-21 |
| EA201270302A1 (ru) | 2012-07-30 |
| CA2772168C (en) | 2019-01-08 |
| CN102639536B (zh) | 2015-03-18 |
| ZA201200812B (en) | 2013-03-27 |
| JP6404186B2 (ja) | 2018-10-10 |
| AU2010288502B2 (en) | 2013-12-12 |
| IN2012DN01292A (ja) | 2015-06-05 |
| EP2470545B1 (en) | 2013-10-09 |
| CN102639536A (zh) | 2012-08-15 |
| CA2772168A1 (en) | 2011-03-03 |
| JP2013503134A (ja) | 2013-01-31 |
| KR20120056258A (ko) | 2012-06-01 |
| MX2012002369A (es) | 2012-03-29 |
| SG178231A1 (en) | 2012-03-29 |
| AU2010288502A1 (en) | 2012-02-23 |
| IL217922A (en) | 2016-06-30 |
| DK2470545T3 (da) | 2014-01-13 |
| EA021411B1 (ru) | 2015-06-30 |
| EP2470545A1 (en) | 2012-07-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6404186B2 (ja) | 無水形態のピリジン誘導体 | |
| JP5701246B2 (ja) | 有機化合物 | |
| DK2767531T3 (en) | Cyclic n, n'-diarylthioureas and n, n'-diarylurines as androgen receptor antagonists, anti-cancer agent, method of preparation and use thereof | |
| US9365565B2 (en) | Salts of aza-bicyclic di-aryl ethers and pharmaceuticals thereof | |
| CA2721119A1 (en) | Anhydrous crystal form of orvepitant maleate | |
| KR20110052686A (ko) | 피리딘 유도체의 결정질 형태 | |
| CA3127469C (en) | Polymorphic forms of a substituted-quinoxaline-type bridged-piperidine compound | |
| US20180273507A1 (en) | Crystal forms of a m1 receptor positive allosteric modulator | |
| BR112012003435B1 (pt) | Composto na forma cristalina anidra, composição farmacêutica compreendendo dito composto e uso terapêutico do mesmo | |
| WO2025227089A1 (en) | Solid state forms of (2r,3s)-2-(4-(cyclopentylamino)phenyl)-1-(2-fluoro-6-methylbenzoyl)-n-(4-methyl-3-(trifluoromethyl)phenyl)piperidine-3-carboxamide | |
| CA3250447A1 (en) | MACROCYCLIC PYRIMIDINE DERIVATIVE, ITS PREPARATION PROCESS, AND PHARMACEUTICAL COMPOSITION INTENDED FOR THE PREVENTION OR TREATMENT OF A NEURODEGENERATIVE DISEASE AND CONTAINING THIS DERIVATIVE AS AN ACTIVE INGREDIENT | |
| NZ619574B2 (en) | Salts of aza-bicyclic di-aryl ethers and methods to make them or their precursors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20180223 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20180523 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20181016 |