JP2013155358A - 乳化重合法及びヒドロゲルの合成方法 - Google Patents
乳化重合法及びヒドロゲルの合成方法 Download PDFInfo
- Publication number
- JP2013155358A JP2013155358A JP2012019218A JP2012019218A JP2013155358A JP 2013155358 A JP2013155358 A JP 2013155358A JP 2012019218 A JP2012019218 A JP 2012019218A JP 2012019218 A JP2012019218 A JP 2012019218A JP 2013155358 A JP2013155358 A JP 2013155358A
- Authority
- JP
- Japan
- Prior art keywords
- condensing agent
- dehydrating condensing
- group
- agent precursor
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images













Landscapes
- Polyamides (AREA)
Abstract
【課題】 反応開始の制御や反応速度の制御を行うことができる新規な乳化重合法及びヒドロゲルの合成方法を提供する
【解決手段】 水溶性の脱水縮合剤前駆体と脂溶性の脱水縮合剤前駆体とが結合することにより脱水縮合剤として機能する脱水縮合剤を用いて乳化重合を行う。この際、原料化合物を含む水相に水溶性の脱水縮合剤前駆体を添加しておき、乳化分散後、有機相に脂溶性の脱水縮合剤前駆体を加えることで、重合反応を進行させる。
【選択図】 図1
【解決手段】 水溶性の脱水縮合剤前駆体と脂溶性の脱水縮合剤前駆体とが結合することにより脱水縮合剤として機能する脱水縮合剤を用いて乳化重合を行う。この際、原料化合物を含む水相に水溶性の脱水縮合剤前駆体を添加しておき、乳化分散後、有機相に脂溶性の脱水縮合剤前駆体を加えることで、重合反応を進行させる。
【選択図】 図1
Description
本発明は、脱水縮合剤の水溶性部分と脂溶性部分とを分離して反応系に添加する新規な乳化重合法に関するものであり、さらには、これを応用したヒドロゲルの合成方法に関する。
ヒドロゲルは、三次元構造(ネットワーク)を有し、このネットワークの中に多量の水を媒体として含んだものであり、架橋点での結合様式の違いにより、共有結合からなる化学ゲルと、イオン結合や水素結合等からなる物理ゲルに分類される。例えば、ポリアクリルアミドゲルは、アクリルアミドがラジカル重合した化学ゲルの代表例である。寒天やゼラチンは、多糖が絡み合った物理ゲルの代表例である。
前述のヒドロゲルは、医療、食品、各種材料分野等、様々な分野において使用されており、特にナノサイズ、あるいはマイクロサイズの微粒子ヒドロゲルは、非常に有用な機能性材料として、各方面から注目を集めている。
前記微粒子ヒドロゲルのうち、化学ゲルを利用した微粒子ヒドロゲルは、例えば乳化重合による合成が一般的であり、原料化合物を含む水相を有機相中に乳化分散し、乳化分散の後、水相に含まれる原料化合物をラジカル共重合等による乳化重合させることにより合成することができる。
ここで、多様なヒドロゲルの合成を考えた場合、前記ラジカル共重合の他、合成法として脱水縮合による乳化重合等を採用することも考えられるが、この時、脱水縮合反応に供する原料化合物は、縮合するカルボキシ基やアミノ基の他、水酸基や硫酸基等、多くの親水性官能基を有するため、これらが可溶な水中で利用できる脱水縮合剤を使用しなくては合成できない、という大きな問題がある。水中で利用できるという要件を満たす縮合剤としては、僅かに水溶性カルボジイミド(EDC)等が知られているに過ぎず、その性能は必ずしも十分とは言えない。
このような状況から、水中で使用できる脱水縮合剤として、トリアジン化合物と第三級アミンとを反応させることにより得られる四級アンモニウム塩が開発されている(例えば、特許文献1や特許文献2等を参照)。これら四級アンモニウム塩は、トリアジン化合物と第三級アミンとを混合することで、室温下、数分で定量的(収率は概ね100%)に生成し、水中で脱水縮合剤として使用することができる。
例えば、特許文献1には、分子内に特定のトリアジン環を有する四級アンモニウム塩からなる縮合剤が開示されている。同様に、特許文献2には、トリアジン化合物と3級アミンとを反応させた四級アンモニウム塩からなり、水界面への集積性を有する脱水縮合剤が開示されている。これら特許文献に記載される脱水縮合剤は、多くの場合、前述の水溶性カルボジイミドよりも優れた性能を示す。
しかしながら、これまで、これら脱水縮合剤を用いて微粒子状のヒドロゲルを合成した例はなく、仮に、各特許文献に記載される脱水縮合剤を用いて公知の方法で微粒子状のヒドロゲルを合成しようとすると、多くの困難を伴うことが予想される。
例えば、原料化合物を含む水溶液に前述の脱水縮合剤を加え、脱水縮合反応が進行する前に乳化分散させる方法を採用した場合、乳化分散開始時から反応が進行してしまうので、水溶液の粘度が高く分散に時間を要する場合や、水溶液の濃度が高く縮合反応速度が速い場合等には、分散が間に合わず、目的が達せられないおそれがある。
また、脱水縮合剤水溶液の添加に伴い、水相の希釈が避けられないという問題もある。脱水縮合剤水溶液を添加すると、添加した水分量の分だけ反応物の濃度が下がることになる。合成されるヒドロゲルの架橋度や構造は、反応物の初期濃度に依存すると考えられ、濃度変化は品質保持の点等から、できるだけ避けることが望ましい。
さらに、脱水縮合剤は、乳化分散開始前に一度に加えるしかなく、反応速度を制御することはできない。すなわち、脱水縮合剤の添加速度による反応速度制御は不可能である。
一方、原料化合物を含む水相を有機相と乳化分散した後に脱水縮合剤を添加する方法を採用した場合、前述の脱水縮合剤が四級アンモニウム塩であり水溶性の塩であるので、低極性有機溶媒には溶解せず、有機相への添加は不可能である。
したがって、脱水縮合剤を水溶液として添加せざるを得ないが、この場合には、乳化分散した乳濁液の分散状態が変化してしまうという問題があり、また、水相における水の量が増えるので、乳化分散前に添加する場合と同様の問題が生ずる。
乳化重合を採用した場合の前記課題を解消するために、例えば原料化合物であるカルボン酸とアミンを含む水溶液中に脱水縮合剤を加え、水中での脱水縮合反応によりアミド化を行ってヒドロゲルを合成した後、固まり状態のゲルを粉砕し、篩にかけて粒度を揃えることも考えられるが、柔らかいゲルを粉砕することや篩にかけることは難しいのが実情である。また、粉砕や篩による分級が可能であるとしても、理想的な形状(球形)に成形することが難しいという大きな問題もある。
本発明は、このような従来技術が抱える課題を解消することを目的に提案されたものである。すなわち、本発明は、反応開始のタイミングの制御や反応速度の制御を行うことができ、脱水縮合剤を用いた乳化重合における前記課題を悉く解消し得る全く新しい乳化重合法及びヒドロゲルの合成方法を提供することを目的とする。
前述の目的を達成するために、本発明の乳化重合法は、水溶性の脱水縮合剤前駆体と脂溶性の脱水縮合剤前駆体とが結合することにより脱水縮合剤として機能する化合物を用いて乳化重合を行うに際し、原料化合物を含む水相に前記水溶性の脱水縮合剤前駆体を添加しておき、乳化分散後、有機相に前記脂溶性の脱水縮合剤前駆体を加えることで、重合反応を進行させることを特徴とする。
また、本発明のヒドロゲルの合成方法は、乳化分散により有機相中に水相を分散させ、水相に含まれる原料化合物を、水溶性の脱水縮合剤前駆体と脂溶性の脱水縮合剤前駆体とが結合することにより脱水縮合剤として機能する化合物を用いて乳化重合するヒドロゲルの合成方法であって、原料化合物を含む水相に前記水溶性の脱水縮合剤前駆体を添加しておき、乳化分散後、有機相に前記脂溶性の脱水縮合剤前駆体を加えることで、重合反応を進行させることを特徴とする。
前記水溶性の脱水縮合剤前駆体や脂溶性の脱水縮合剤前駆体は、いずれか一方のみでは脱水縮合剤として機能せず、脱水縮合反応は進行しない。本発明においては、乳化重合時には、水相に水溶性の脱水縮合剤前駆体が添加されるのみであるので、重合反応は進行しない。乳化分散後、有機相に脂溶性の脱水縮合剤前駆体を加えることで重合反応が開始される。
すなわち、本発明においては、脂溶性の脱水縮合剤前駆体の添加によって反応開始が制御され、水溶液の粘度や濃度等に依存することはない。また、本発明では、反応速度の制御も可能であり、脂溶性の脱水縮合剤前駆体の添加量や添加速度によって、重合速度が制御される。あるいは、水相中の脱水縮合剤濃度が水溶性の脱水縮合剤前駆体の初期濃度によって制御され、当該初期濃度を薄くすればゆっくり反応が進み、濃くすれば速く反応が進行する。
本発明によれば、反応開始の制御や反応速度の制御を行うことが可能な、新規な乳化重合法を提供することが可能である。これにより、反応液の粘度や濃度等に依存せず、反応液の初期濃度変化を引き起こすことなく重合反応を進行させることが可能な乳化重合法を実現することができる。
また、本発明によれば、前記乳化重合法をヒドロゲルの合成方法に適用することで、球形で粒度の均一性に優れたヒドロゲルを容易に合成することが可能である。さらに、得られるヒドロゲルは、反応後の表面修飾が容易であり、様々な分野への応用が期待できる。
以下、本発明を適用した乳化重合法及びヒドロゲルの合成方法の実施形態について、図面を参照して詳細に説明する。
先ず、本発明の乳化重合法、ヒドロゲルの合成方法では、水溶性の脱水縮合剤前駆体と脂溶性の脱水縮合剤前駆体とが結合することにより縮合剤として機能する脱水縮合剤を用いて乳化重合を行う。使用する脱水縮合剤は、前記要件を満たすものであれば、その種類は問わない。
使用する脱水縮合剤の具体例としては、例えば第三級アミン系化合物とトリアジン系化合物とを反応させて得られる脱水縮合剤を挙げることができ、この場合、前記第三級アミン系化合物が水溶性の脱水縮合剤前駆体、前記トリアジン系化合物が脂溶性の脱水縮合剤前駆体ということになる。
第三級アミン系化合物とトリアジン系化合物とを反応させて得られる脱水縮合剤としては、例えば、水溶性の脱水縮合剤前駆体が式(1)で表される第三級アミン化合物であり、前記脂溶性の脱水縮合剤前駆体が式(2)で表されるトリアジン化合物であり、脱水縮合剤が式(3)で表される化合物である。

なお、式(1)〜式(3)に共通している「置換されていてもよい」とは、1個以上の置換基を有していてもよいことを意味し、置換基としては、ハロゲン、ニトロ、シアノ、C1−6アルキル、C3−8シクロアルキル、C2−6アルケニル、C2−6アルキニル、C1−6アルコキシ、C1−6アルキレンジオキシ、C6−10アリール、C7−14アラルキル、C1−6アルコキシ−カルボニル、C7−14アラルキルオキシ−カルボニル、C1−6アルキル−カルボニル、C6−10アリール−カルボニル、C6−10アリールオキシ−カルボニル、C1−6アルキルスルホニル、C6−10アリールスルホニル、ホルミル、アジド、C1−6アルキルチオ、C6−C10アリールチオ、C1−6アルキル基で置換されていてもよいカルバモイル、トリC1−6アルキルシリル基、保護されたアミノ基等が挙げられる。中でも、ハロゲン、ニトロ、シアノ、C1−6アルキル、C1−6アルコキシ、メチレンジオキシ、C1−6アルコキシ−カルボニル、アセチル、ベンゾイル、ホルミル、カルバモイル、アジド、トリメチルシリル、トリエチルシリル、トリイソプロピルシリル、tert−ブチルジメチルシリル、ジメチルアミノ、アセチルアミノ、ベンジルオキシカルボニルアミノ、t−ブトキシカルボニルアミノが好ましい。これらが単一、もしくは複数置換されていてもよい
前記水溶性の脱水縮合剤前駆体は、トリアジン化合物と反応して脱水縮合能を有するトリアジニルアンモニウム塩を形成するものであればよく、具体的には、例えば、N−メチルモルホリン、メチルピペリジン、ジエチルメチルアミン、トリメチルアミン、ジメチルエチルアミン、キヌクリジン、メチルピロリジン等が挙げられ、中でもN−メチルモルホリンが特に好ましい。
最も好ましいのは、水溶性の脱水縮合剤前駆体が式(4)で表されるモルホリン化合物(以下、NMMと称する。)であり、前記脂溶性の脱水縮合剤前駆体が式(5)で表されるトリアジン化合物(以下、CDMTと称する。)であり、脱水縮合剤が式(6)で表される化合物(以下、DMT−MMと称する。)である。3級アミンにモルホリン骨格を導入することで、水溶性が向上する。
原料化合物は、前記脱水縮合剤により脱水縮合(重合)可能であるものであれば、やはりその種類は問わない。重合により水(分散媒)に高分子が網目状に分散してゲルを形成するものを原料化合物として選択すれば、乳化重合によりヒドロゲルを合成することが可能になる。例えば、原料化合物としてポリカルボン酸とポリアミンを選択すれば、前記脱水縮合剤による重合反応としてアミド化反応が進行し、ポリアミドゲルが合成される。その他、ヒドロゲルの合成を考えた場合、アミノ基やカルボキシ基等、縮合に関与する官能基を有する多糖類等も有用な原料化合物である。
次に、脱水縮合剤として前記DMT−MMを使用する場合を例にして、本発明の乳化重合法、ヒドロゲルの合成方法の概要について説明する。
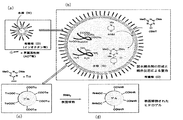
図1は、DMT−MMを脱水縮合剤として使用する乳化重合法、ヒドロゲルの合成方法の概要を示すものである。
乳化重合を行う場合、先ず、図1(a)に示すように、水相Wと有機相Oを乳化分散する必要がある。本例の場合、水相Wが分散相、有機相Oが連続相となるように乳化分散を行う。なお、乳化分散に際しては、乳化剤を使用することも可能である。乳化剤としては、公知のものがいずれも使用可能であるが、W/Oエマルション系においては、ジ(2−エチルヘキシル)スルホコハク酸ナトリウム(AOT)等が好適である。有機層Oに用いる有機溶媒は、水と混ざらず乳化懸濁液を形成しうる有機溶媒であれば如何なるものであってもよく、例えば、塩化メチレン、クロロホルム、ジククロエタン、酢酸メチル、酢酸エチル、酢酸プロピル、酢酸イソプロピル、酢酸ブチル、酢酸イソブチル、酢酸tert-ブチル、酢酸ベンジル、ジエチルエーテル、tert-ブチルメチルエーテル、ジフェニルエーテル、ジベンジルエーテル、パラフィン、ペンタン、ヘキサン、ヘプタン、オクタン、イオソオクタン、ニトロメタン、ニトロエタン、ニトロプロパン、ベンゼン、トルエン、キシレン、クロロベンゼン、ジクロロベンゼン、フルオロベンゼン、トリフルオロベンゼン、2H,3H-デカフルオロペンタン、エイコサフルオロノナン、ヘプタコサフルオロトリブチルアミン、ヘプタデカフルオロ-n-オクチルブロミド、ヘキサデカフルオロ(1,3-ジメチルシクロヘキサン)、ヘキサデカフルオロヘプタン、ヘキサフルオロベンゼン、オクタデカフルオロデカヒドロナフタレン、オクタデカフルオロオクタン、オクタフルオロシクロペンテン、オクタフルオロトルエン、ペンタデカフルオロトリエチルアミン、ペルフルオロ(2-ブチルテトラヒドロフラン)、ペルフルオロトリアミルアミン、テトラデカフルオロヘキサン、テトラデカフルオロメチルシクロヘキサン、テトラデカフルオロ-2-メチルペンタンヘキサン等を例示することができる。なお、有機溶媒の粘度は乳濁液の状態(安定性や粒径)に影響することから、粘性の液体状パラフィン類(流動パラフィン)などの有機溶媒を選択するか、もしくはこれらを混ぜて粘度を適性に調整すると良いこともある。
ヒドロゲルの合成においては、この乳化分散後の水相Wの大きさや均一さ等により合成される微粒子ヒドロゲルの大きさや均一さ等が決まるため、所望の微粒子の大きさに応じて乳化分散条件を決定すればよい。
前記乳化分散に際しては、予め水相Wに原料化合物(例えばジカルボン酸またはそれ以上のカルボキシ基を含むポリカルボン酸、ジアミンまたはそれ以上のアミノ基を含むポリアミン)及びNMMを溶解しておき、これらを含んだ水相Wを乳化分散する。NMM単独では脱水縮合剤として機能せず、この段落では何の反応も起こらないので、水相Wの粘度や濃度に依存することなく、必要なW/Oエマルションを形成させることが可能である。
前述の乳化分散の後、有機相OにCDMTを加え、反応を開始する。CDMTは、有機相Oに添加するので、有機溶媒(例えばトルエン)に溶解して添加することが好ましい。ここで用いる有機溶媒は、CDMTが溶解するものであれば有機相と同じ溶媒でも良いし異なる溶媒でも良い。乳化剤の希釈を避けるためには、乳化剤を添加した有機溶媒が好ましい。
図1(b)に示すように、脂溶性のCDMTは、有機相Oを移動し、やがて油水界面において水相W中のNMMと反応し、水溶性のDMT−MMとなる。このDMT−MMは、有機相Oに不溶であるので水相W内にとどまり、原料化合物(ポリカルボン酸とポリアミン)の縮合反応(重合反応)を引き起こす。本例の場合、ポリカルボン酸とポリアミンはアミド化反応により重合し、水(分散媒)に高分子が網目状に分散して形成されたヒドロゲルが合成される。前記アミド化反応後は、NMMは再生され、次のCDMTと反応してDMT−MMが生じ、再びアミド化反応に寄与する。
本発明では、DMT−MMを固体状態や溶液状態等で直接反応系に加えるのではなく、DMT−MMがCDMTとNMMとから定量的に生成することを利用して、W/Oエマルションの水相W内でDMT−MMを発生させて重合反応に利用することが大きな特徴事項である。
前記特徴を有する本発明の乳化重合法及びヒドロゲルの合成方法は、反応開始の制御や反応速度の制御といった点で、様々な利点を有する。先ず、反応開始の制御に関して、乳化分散時に重合は進行せず、乳化分散後にCDMTを加えて重合反応を開始するので、乳化分散液が適切な状態になった後、任意のタイミングで反応を開始できる。このとき、水相の初期の粘度や濃度、あるいは分散した水相の大きさ(乳濁した水粒子のサイズ)を変化させることなく反応を開始させることが可能である。また、NMMが触媒として作用するので、CDMTが過剰であってもすべて利用することが可能である。
反応速度の制御に関して言えば、CDMTの添加量と添加速度によって重合速度を制御することができ、CDMTがNMMより過剰であっても全て縮合剤に変換することができる。また、水相中の縮合剤濃度をNMMの初期濃度で制御することができ、NMMの初期濃度を薄くすれば反応がゆっくりと進行し、NMMの初期濃度を濃くすれば反応が速く進行する。
以上が本発明の乳化重合法、ヒドロゲルの合成方法の概要であるが、本発明で合成されるヒドロゲルは、重合反応後の表面修飾が可能であり、様々な分野への応用が期待できるという特徴も有する。
ヒドロゲルの表面修飾について説明すると、例えば、前記ポリカルボン酸とポリアミンを原料化合物とするヒドロゲルの合成において、ポリカルボン酸よりポリアミンの量を少なくすると、図1(c)に示すように、最終的に未反応のカルボキシ基がトリアジニルエステルのまま残ることになる。図1(c)において、ゲル表面のCOOTrzがトリアジニルエステル部分である。
前記トリアジニルエステルは、一定時間水中で安定であり、また脂溶性であるために水相Wから有機相Oに突き出した状態で存在し、加水分解を受け難くなる。そのため、重合反応後に例えばアミン等を加えれば、図1(d)に示すように、ゲル表面に化学修飾を施すことが可能になる。化学修飾としては、蛍光色素の導入や、電荷の導入、疎水性基の導入、その他、様々な機能性官能基の導入等を挙げることができる。
以上のように、本発明の乳化重合法やヒドロゲルの合成方法によれば、反応開始の制御や反応速度の制御を行うことが可能であり、水相の粘度や濃度、あるいは分散した水相の大きさ(乳濁した水粒子のサイズ)を初期条件のまま変化させることなく重合反応を開始させることが可能な乳化重合法を実現することができる。また、本発明によれば、前記乳化重合法をヒドロゲルの合成方法に適用することで、球形で粒度の均一性に優れた微粒子状のヒドロゲルを容易に合成することが可能である。さらに、得られるヒドロゲルは、反応後の表面修飾が容易であり、様々な分野への応用が期待できる。
なお、前記実施形態の説明は、ヒドロゲルの合成を中心に行ったが、本発明の乳化重合法はヒドロゲルの合成以外にも、種々の脱水縮合反応、重合反応に適用することが可能である。
本発明は、例えば脱水縮合するカルボン酸とアミンを含む水相にNMMを加え、有機相にCDMTを加えると、NMMとCDMTが界面で反応し、生じた水溶性のDMT−MMがそのまま有機相によって閉鎖された水相にとどまることを利用した方法であり、反応剤の特徴を全て理解して、幅広い学術分野にまたがる上記全ての事実を同時に理解した上でなければできない発想であり、これらの分野全てに通じることは極めてまれであることに鑑み、極めて斬新な技術ということができる。また、反応後に更に表面を修飾するという方法についても、官能基の性質を知っていないと発想できないメカニズムであり、粒子への付加価値や機能の導入が可能となり、新しいゲルの調製技術となり得るものである。
以下、本発明を適用した具体的な実施例について、実験結果を基に説明する。
実施例1
アルギン酸ナトリウム(50mg,0.25mmol)、エチレンジアミン二塩酸塩(3.3mg, 0.025mmol)、N−メチルモルホリン(27.5μL,0.25mmol)を蒸留水に溶かし、5.0mLの水溶液Aとした。別の試験管にAOT(200mM)を含むイソオクタン溶液(4mL)を入れ、ここに800rpmで撹拌しながら、水溶液A(255μL,カルボン酸残基0.013mmol)をゆっくりと加えてエマルションを調製した。
アルギン酸ナトリウム(50mg,0.25mmol)、エチレンジアミン二塩酸塩(3.3mg, 0.025mmol)、N−メチルモルホリン(27.5μL,0.25mmol)を蒸留水に溶かし、5.0mLの水溶液Aとした。別の試験管にAOT(200mM)を含むイソオクタン溶液(4mL)を入れ、ここに800rpmで撹拌しながら、水溶液A(255μL,カルボン酸残基0.013mmol)をゆっくりと加えてエマルションを調製した。
次に、このエマルションに、200mMのAOTを含むトルエン溶液(2.5mL)にCDMT(43.9mg,0.25mmol)を溶かして調製した溶液(100mM CDMT,129μL,0.013mmol)を加えて反応を開始した。15時間後、DMT−MM水溶液(361.1mM,36μL,0.013mmol)とCBA水溶液(19.4mM,664μL,0.013mmol)を加えて2時間反応させた。撹拌を止めて、静置させた後、有機相(上層)をパスツールピペットで取り除き、得られたゲルをイソオクタン、ジエチルエーテルで洗浄後、窒素気流により有機溶媒を蒸発させて微粒子ヒドロゲルを得た。
AOT:ジ(2‐エチルヘキシル)スルホコハク酸ナトリウム
CDMT:2−クロロ−4,6−ジメトキシ−1,3,5−トリアジン
DMT−MM:4−(4,6−ジメトキシ−1,3,5−トリアジン−2−イル)−4−メチルモルホリニウムクロリド
CBA:カスケードブルーエチレンジアミン三ナトリウム塩
AOT:ジ(2‐エチルヘキシル)スルホコハク酸ナトリウム
CDMT:2−クロロ−4,6−ジメトキシ−1,3,5−トリアジン
DMT−MM:4−(4,6−ジメトキシ−1,3,5−トリアジン−2−イル)−4−メチルモルホリニウムクロリド
CBA:カスケードブルーエチレンジアミン三ナトリウム塩
図2は、実施例1で作製した微粒子ヒドロゲルの位相差顕微鏡写真であり、図3は、実施例1で作製した微粒子ヒドロゲルのDLS(動的光散乱法)による粒度分布を示す図である。約3μm及び約30μmを中心粒径とする微粒子状のヒドロゲルが得られたことがわかる。
図4は、実施例1で作製し蛍光色素により表面修飾した微粒子ヒドロゲルの顕微鏡写真である。使用した蛍光色素は、化5に示すピラニン誘導体(CBA)であり、最大励起波長374〜378nm、399〜403nm、最大蛍光波長422〜430nmである。観察で用いたのは励起波長320〜400nm、蛍光波長410〜510nmである。なお、ピラニン誘導体(CBA)による表面修飾の反応式を化6に示す。
実施例2
アルギン酸ナトリウム(70mg,0.35mmol)、エチレンジアミン二塩酸塩(4.7mg, 0.035mmol)、N−メチルモルホリン(38.9μL,0.35mmol)を蒸留水に溶かし、2.0mLの水溶液Bとした。別の試験管にAOT(200mM)を含むイソオクタン溶液(2mL)を入れ、ここに撹拌しながら水溶液B(222μL,カルボン酸残基0.039mmol)をゆっくりと加えてエマルションを調製した。
アルギン酸ナトリウム(70mg,0.35mmol)、エチレンジアミン二塩酸塩(4.7mg, 0.035mmol)、N−メチルモルホリン(38.9μL,0.35mmol)を蒸留水に溶かし、2.0mLの水溶液Bとした。別の試験管にAOT(200mM)を含むイソオクタン溶液(2mL)を入れ、ここに撹拌しながら水溶液B(222μL,カルボン酸残基0.039mmol)をゆっくりと加えてエマルションを調製した。
次に、このエマルションに、200mMのAOTを含むトルエン溶液(1.5mL)にCDMT(105.8mg,0.60mmol)を溶かして調製した溶液(400mM CDMT,194μL, 0.078mmol)を加えて反応を開始した。6時間後、CBA水溶液(48.6mM,200μL,0.0097mmol)を加えて1時間反応させた。撹拌を止めて、静置させた後、有機相(上層)をパスツールピペットで取り除いた。得られたゲルを、イソオクタン及びジエチルエーテルで洗浄後、窒素気流により有機溶媒を蒸発させた。このゲルに蒸留水を加えたのち、2.0mLの容器に移して遠心(1分,13.4×103rpm)した後、水相を取り除き再度蒸留水を加えた。これを6回繰り返し、未反応のCBAを取り除き、CBA修飾された微粒子ヒドロゲルを得た。
図5は、実施例2で作製し蛍光色素により表面修飾した微粒子ヒドロゲルの顕微鏡写真である。使用した蛍光色素は、実施例1で使用したものと同様のピラニン誘導体であり、最大励起波長374〜378nm、399〜403nm、最大蛍光波長422〜430nmである。観察で用いたのは励起波長320〜400nm、蛍光波長410〜510nmである。本実施例においては、数百μmレベルの粒径を有する微粒子ヒドロゲルが合成された。
実施例3
アルギン酸ナトリウム(60mg,0.3mmol)、350mMエチレンジアミン二塩酸塩水溶液(85.7μL,0.03mmol)、N−メチルモルホリン(33μL,0.3mmol)を蒸留水に溶かし、6.0mLの水溶液Cとした。別の試験管にAOT(200mM)を含むイソオクタン溶液(6mL)を入れ、ここに撹拌しながら、水溶液C(666μL,カルボン酸残基0.034mmol)をゆっくりと加えてエマルションを調製した。
アルギン酸ナトリウム(60mg,0.3mmol)、350mMエチレンジアミン二塩酸塩水溶液(85.7μL,0.03mmol)、N−メチルモルホリン(33μL,0.3mmol)を蒸留水に溶かし、6.0mLの水溶液Cとした。別の試験管にAOT(200mM)を含むイソオクタン溶液(6mL)を入れ、ここに撹拌しながら、水溶液C(666μL,カルボン酸残基0.034mmol)をゆっくりと加えてエマルションを調製した。
次に、このエマルションに、200mMのAOTを含むトルエン溶液(7.0mL)にCDMT(491.5mg,2.8mmol)を溶かして調製した溶液(400mM CDMT,750μL,0.3mmol)を加えて反応を開始した。12時間後、撹拌を止めて、静置させた後、有機相(上層)をパスツールピペットで取り除いた。得られたゲルを、イソオクタン及びジエチルエーテルで洗浄後、窒素気流により有機溶媒を蒸発させた。このゲルに蒸留水を2.0mL加えて粒子を均一に分散させた後、L−リシン水溶液(666mM,1.0mL,0.67mmol)を加えて3時間撹拌した。次に、そのゲルを25℃で一晩透析(排除分子量:12,000〜16,000)し、リシン修飾された微粒子ゲルを得た。
図6は、実施例3で作製した微粒子ヒドロゲルのDLS(動的光散乱法)による粒度分布を示す図である。本実施例では、粒径1〜2μmの微粒子状のヒドロゲルが得られたことがわかる。
実施例4
アルギン酸ナトリウム(60mg,0.3mmol)、350mMエチレンジアミン二塩酸塩水溶液(85.7μL,0.03mmol)、N−メチルモルホリン(33μL,0.3mmol)を蒸留水に溶かし、6.0mLの水溶液Cとした。別の試験管にAOT(200mM)を含むイソオクタン溶液(6mL)を入れ、ここに撹拌しながら、水溶液C(666μL,カルボン酸残基0.034mmol)をゆっくりと加えてエマルションを調製した。
アルギン酸ナトリウム(60mg,0.3mmol)、350mMエチレンジアミン二塩酸塩水溶液(85.7μL,0.03mmol)、N−メチルモルホリン(33μL,0.3mmol)を蒸留水に溶かし、6.0mLの水溶液Cとした。別の試験管にAOT(200mM)を含むイソオクタン溶液(6mL)を入れ、ここに撹拌しながら、水溶液C(666μL,カルボン酸残基0.034mmol)をゆっくりと加えてエマルションを調製した。
次に、このエマルションに、200mMのAOTを含むトルエン溶液(4.0mL)にCDMT(280.9mg,1.6mmol)を溶かして調製した溶液(400mM CDMT,250μL,0.1mmol)を加えて反応を開始した。12時間後、撹拌を止めて、静置させた後、有機相(上層)をパスツールピペットで取り除いた。得られたゲルをイソオクタン、ジエチルエーテルで洗浄後、窒素気流により有機溶媒を蒸発させた。このゲルにL−リシン水溶液(68mM,500μL,0.034 mmol)、蒸留水500μL、DMT−MM水溶液(68mM,500μL,0.034mmol)を加えて粒子を均一に分散させ、微粒子ヒドロゲル溶液を得た。
図7は、実施例4で作製したリシン修飾微粒子ヒドロゲルのDLS(動的光散乱法)による粒度分布を示す図である。粒径350μmの微粒子状のヒドロゲルが得られている。
この実施例に倣って、最後に加える表面修飾剤をリシンの代わりにモノエタノールアミン、タウリン、N,N−ジメチルエチレンジアミンとし、これらで表面修飾された微粒子ヒドロゲルを合成した。各表面修飾における反応を化7に示す。
得られた微粒子ヒドロゲルのDLS(動的光散乱法)による粒度分布を図8〜図10に示す。表面修飾の相違により、得られる微粒子ヒドロゲルの平均粒径には、有意な変化は見られなかった。
また、モノエタノールアミン、タウリン、N,N−ジメチルエチレンジアミンで表面修飾した粒子のゼータ電位を測定した。結果を図11〜図13に示す。また、図14にゼータ電位散布図を示す。このゼータ電位散布図において、縦軸には、表面修飾後に負電荷を持つタウリン、中性で電荷を持たないエタノールアミン、正電荷をもつジメチルエチレンジアミンをそれぞれ修飾部位として導入したつ粒子のゼータ電位をとった。横軸には、それぞれの官能基の電荷数をとった。これらの図より、表面の電荷が反映されていると考えられる。
実施例5
コンドロイチン硫酸Cナトリウム(300mg,0.66mmol)、エチレンジアミン二塩酸塩(8.8mg,0.066mmol)、N−メチルモルホリン(73μL,0.66mmol)を蒸留水に溶かし、1.0mLの水溶液Dとした。別の試験管にAOT(200mM)を含むイソオクタン溶液(2mL)を入れ、ここに撹拌しながら、水溶液D(222μL,カルボン酸残基0.15mmol)をゆっくりと加えてエマルションを調製した。
コンドロイチン硫酸Cナトリウム(300mg,0.66mmol)、エチレンジアミン二塩酸塩(8.8mg,0.066mmol)、N−メチルモルホリン(73μL,0.66mmol)を蒸留水に溶かし、1.0mLの水溶液Dとした。別の試験管にAOT(200mM)を含むイソオクタン溶液(2mL)を入れ、ここに撹拌しながら、水溶液D(222μL,カルボン酸残基0.15mmol)をゆっくりと加えてエマルションを調製した。
次に、このエマルションに、200mMのAOTを含むトルエン溶液(2.0mL)にCDMT(176.0mg,1.0mmol)を溶かして調製した溶液(500mM CDMT,300μL,0.15mmol)を加えて反応を開始した。一晩反応させた後、撹拌を止めて、静置させた後、有機相(上層)をパスツールピペットで取り除いた。得られたゲルをイソオクタン、ジエチルエーテルで洗浄後、窒素気流により有機溶媒を蒸発させた。このゲルに蒸留水を1.8mL加えて粒子を均一に分散させ、微粒子ヒドロゲル溶液を得た。実施例2と同様な400μm前後の微粒子ヒドロゲルが得られた。
Claims (11)
- 水溶性の脱水縮合剤前駆体と脂溶性の脱水縮合剤前駆体とが結合することにより脱水縮合剤として機能する化合物を用いて乳化重合を行うに際し、
原料化合物を含む水相に前記水溶性の脱水縮合剤前駆体を添加しておき、乳化分散後、有機相に前記脂溶性の脱水縮合剤前駆体を加えることで、重合反応を進行させることを特徴とする乳化重合法。 - 前記水溶性の脱水縮合剤前駆体がアミン系化合物であり、前記脂溶性の脱水縮合剤前駆体がトリアジン系化合物であることを特徴とする請求項2記載の乳化重合法。
- 前記水溶性の脱水縮合剤前駆体が式(1)で表されるアミン化合物であり、前記脂溶性の脱水縮合剤前駆体が式(2)で表されるトリアジン化合物であり、脱水縮合剤が式(3)で表される化合物であることを特徴とする請求項2記載の乳化重合法。
- 前記重合反応がアミド化反応であることを特徴とする請求項3記載の乳化重合法。
- 前記水相が分散相であり、前記有機相が連続相であることを特徴とする請求項1から4のいずれか1項記載の乳化重合法。
- 乳化分散により有機相中に水相を分散させ、水相に含まれる原料化合物を、水溶性の脱水縮合剤前駆体と脂溶性の脱水縮合剤前駆体とが結合することにより脱水縮合剤として機能する化合物を用いて乳化重合するヒドロゲルの合成方法であって、
原料化合物を含む水相に前記水溶性の脱水縮合剤前駆体を添加しておき、乳化分散後、有機相に前記脂溶性の脱水縮合剤前駆体を加えることで、重合反応を進行させることを特徴とするヒドロゲルの合成方法。 - 前記水溶性の脱水縮合剤前駆体がアミン系化合物であり、前記脂溶性の脱水縮合剤前駆体がトリアジン系化合物であることを特徴とする請求項6記載のヒドロゲルの合成方法。
- 前記水溶性の脱水縮合剤前駆体が式(1)で表されるアミン化合物であり、前記脂溶性の脱水縮合剤前駆体が式(2)で表されるトリアジン化合物であり、脱水縮合剤が式(3)で表される化合物であることを特徴とする請求項7記載のヒドロゲルの合成方法。
- 前記重合反応がアミド化反応であることを特徴とする請求項8記載のヒドロゲルの合成方法。
- 前記重合反応の終了後、ヒドロゲル表面を化学修飾することを特徴とする請求項6から9のいずれか1項記載のヒドロゲルの合成方法。
- アミン水溶液を加えることで前記化学修飾を行うことを特徴とする請求項10記載のヒドロゲルの合成方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012019218A JP2013155358A (ja) | 2012-01-31 | 2012-01-31 | 乳化重合法及びヒドロゲルの合成方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012019218A JP2013155358A (ja) | 2012-01-31 | 2012-01-31 | 乳化重合法及びヒドロゲルの合成方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2013155358A true JP2013155358A (ja) | 2013-08-15 |
Family
ID=49050867
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012019218A Pending JP2013155358A (ja) | 2012-01-31 | 2012-01-31 | 乳化重合法及びヒドロゲルの合成方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2013155358A (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018511060A (ja) * | 2015-02-09 | 2018-04-19 | スリングショット バイオサイエンシーズ, インコーポレイテッド | 調整可能な光学特性を有するヒドロゲル粒子およびその使用の方法 |
| US10942109B2 (en) | 2012-04-06 | 2021-03-09 | Slingshot Biosciences, Inc. | Hydrogel particles with tunable optical properties |
| US11313782B2 (en) | 2020-01-24 | 2022-04-26 | Slingshot Biosciences, Inc. | Compositions and methods for cell-like calibration particles |
| US11598768B2 (en) | 2020-05-04 | 2023-03-07 | Slingshot Biosciences, Inc. | Compositions and methods for passive optical barcoding for multiplexed assays |
-
2012
- 2012-01-31 JP JP2012019218A patent/JP2013155358A/ja active Pending
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10942109B2 (en) | 2012-04-06 | 2021-03-09 | Slingshot Biosciences, Inc. | Hydrogel particles with tunable optical properties |
| JP2018511060A (ja) * | 2015-02-09 | 2018-04-19 | スリングショット バイオサイエンシーズ, インコーポレイテッド | 調整可能な光学特性を有するヒドロゲル粒子およびその使用の方法 |
| US11686661B2 (en) | 2015-02-09 | 2023-06-27 | Slingshot Biosciences, Inc. | Cytometric device hematology reference composition |
| US11747261B2 (en) | 2015-02-09 | 2023-09-05 | Slingshot Biosciences, Inc. | Hydrogel particles with tunable optical properties and methods for using the same |
| US11761877B2 (en) | 2015-02-09 | 2023-09-19 | Slingshot Biosciences, Inc. | Hydrogel particles with tunable optical properties and methods for using the same |
| US11927519B2 (en) | 2015-02-09 | 2024-03-12 | Slingshot Biosciences, Inc. | Synthetic human cell mimic hydrogel particle for cytometric or coulter device |
| US11313782B2 (en) | 2020-01-24 | 2022-04-26 | Slingshot Biosciences, Inc. | Compositions and methods for cell-like calibration particles |
| US11726023B2 (en) | 2020-01-24 | 2023-08-15 | Slingshot Biosciences, Inc. | Compositions and methods for cell-like calibration particles |
| US11598768B2 (en) | 2020-05-04 | 2023-03-07 | Slingshot Biosciences, Inc. | Compositions and methods for passive optical barcoding for multiplexed assays |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Zheng et al. | CO2 stimuli-responsive, injectable block copolymer hydrogels cross-linked by discrete organoplatinum (II) metallacycles via stepwise post-assembly polymerization | |
| Thompson et al. | Covalently cross-linked colloidosomes | |
| EP3436450B9 (en) | Reaction medium containing water-surfactant mixture | |
| Zhang et al. | Bioconjugated Janus particles prepared by in situ click chemistry | |
| US20070060658A1 (en) | Stabilization of organogels and hydrogels by azide-alkyne [3+2] cycloaddition | |
| Zhu et al. | Hydrophilic porous polymers based on high internal phase emulsions solely stabilized by poly (urethane urea) nanoparticles | |
| JP2013155358A (ja) | 乳化重合法及びヒドロゲルの合成方法 | |
| Liu et al. | Facile synthesis and chiral recognition of block and star copolymers containing stereoregular helical poly (phenyl isocyanide) and polyethylene glycol blocks | |
| JP2013531691A5 (ja) | ||
| JP4992090B2 (ja) | 静電結合型高分子ベシクル | |
| CN1802150A (zh) | 载体颗粒 | |
| Xiang et al. | One-pot synthesis for biocompatible amphiphilic hyperbranched polyurea micelles | |
| CN104519990A (zh) | 微囊的制造方法及微囊 | |
| CN111514107B (zh) | 一种活性氧响应型囊泡及其制备方法 | |
| Ren et al. | Multistimuli-responsive Pickering emulsion stabilized by Se-containing surfactant-modified chitosan | |
| Wu et al. | Aqueous-core capsules via interfacial free radical alternating copolymerization | |
| Plunkett et al. | Light-regulated electrostatic interactions in colloidal suspensions | |
| CN110028062A (zh) | 一种表面修饰油溶性氧化石墨烯的制备方法 | |
| Gao et al. | Janus-like single-chain polymer nanoparticles as two-in-one emulsifiers for aqueous and nonaqueous Pickering emulsions | |
| Wang et al. | Emulsion templating cyclic polymers as microscopic particles with tunable porous morphology | |
| Hwang et al. | A family of photolabile nitroveratryl-based surfactants that self-assemble into photodegradable supramolecular structures | |
| Cao et al. | Self-assembled nanostructures from amphiphilic globular protein–polymer hybrids | |
| JP5376290B2 (ja) | 尿素化合物、尿素化合物の自己集合体及び自己集合体を含有するオルガノゲル並びにオルガノゲルの製造方法 | |
| Sun et al. | Formation of n-Hexane-in-DMF Nonaqueous Pickering Emulsions: ABC Triblock Worms versus AB Diblock Worms | |
| JP7178340B2 (ja) | 水/界面活性剤混合物中における化学反応のための触媒 |