JP2010142232A - 加ピロリン酸分解活性化重合(pap) - Google Patents
加ピロリン酸分解活性化重合(pap) Download PDFInfo
- Publication number
- JP2010142232A JP2010142232A JP2009297339A JP2009297339A JP2010142232A JP 2010142232 A JP2010142232 A JP 2010142232A JP 2009297339 A JP2009297339 A JP 2009297339A JP 2009297339 A JP2009297339 A JP 2009297339A JP 2010142232 A JP2010142232 A JP 2010142232A
- Authority
- JP
- Japan
- Prior art keywords
- oligonucleotide
- pap
- nucleic acid
- template
- nucleotide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/6846—Common amplification features
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6813—Hybridisation assays
- C12Q1/6827—Hybridisation assays for detection of mutation or polymorphism
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/6858—Allele-specific amplification
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Microbiology (AREA)
- Biophysics (AREA)
- Analytical Chemistry (AREA)
- Physics & Mathematics (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Enzymes And Modification Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Saccharide Compounds (AREA)
Abstract
【解決手段】相補的な活性化可能オリゴヌクレオチドP*を、テンプレート鎖にアニーリングさせる工程。アニーリングした活性化可能オリゴヌクレオチドP*を加ピロリン酸分解する工程。活性化されたオリゴヌクレオチドP*を伸長することによって重合させ、所望の核酸鎖を合成する工程。テンプレート鎖から所望の核酸鎖を分離する工程。所望の核酸鎖の所望の増幅レベルが達成されるまで、前記工程を反復する工程。
【選択図】なし
Description
[0001]本発明は、核酸重合および増幅に関する。特に、本発明は、加ピロリン酸分解(pyrophosphorolysis)および重合を連続してカップリングする、核酸増幅の新規でそして一般的な方法に関する。該方法はアレル特異的増幅に適応されてきており、そして野生型アレルの存在下で、非常に稀なアレルを検出する特異性を非常に増加させることも可能である。我々は該方法を加ピロリン酸分解活性化重合(PAP)と称する。
[0011]本発明は、核酸テンプレート鎖上で、所望の核酸鎖を合成する、加ピロリン酸分解活性化重合(PAP)法である。該方法は、連続的に行われる以下の工程を含んでなる。
[0016](d)テンプレート鎖から工程(c)の所望の核酸鎖を分離する工程、および
[0017](e)所望の核酸鎖の所望の増幅レベルが達成されるまで、工程(a)〜(d)を反復する工程。
[0028](e)突然変異アレルの所望の増幅レベルが達成されるまで、工程(a)〜(d)を反復する工程。
[0038](e)所望の増幅レベルが達成されるまで、工程(a)〜(d)を反復する工程、および
[0039](f)増幅を生じるオリゴヌクレオチドP*の重複を解析することによって、核酸配列を順序正しく配置する工程。
[0041](a)多数の活性化可能オリゴヌクレオチドP*と、核酸のテンプレート鎖を、ハイブリダイゼーション条件下で混合する工程。すべてのオリゴヌクレオチドP*は、テンプレートと同じ数nのヌクレオチドを有し、そして集合的に、nヌクレオチドを有するすべてのありうる配列を構成する。すべてのオリゴヌクレオチドP*は、伸長不能3'末端を有する。伸長不能3'末端は、例えば伸長不能3'-デオキシヌクレオチドまたはアシクロヌクレオチドであることも可能である。十分に相補的なオリゴヌクレオチドP*は、いずれも、テンプレート鎖にハイブリダイズするであろう。伸長不能3'末端は、テンプレート鎖が3'末端に対応する位で相補的である場合にのみ、テンプレート鎖にハイブリダイズするであろう。
[0045](e)所望の増幅レベルが達成されるまで、工程(a)〜(d)を反復する工程、および
[0046](f)増幅を生じるオリゴヌクレオチドP*の配列を決定し、次いでこれらのオリゴヌクレオチドの重複を解析することによって、核酸配列を順序正しく配置する工程。
[0052](iii)リンカー連結工程には、合成された核酸鎖の3'末端へのリンカーの連結が含まれる。テンプレート非依存性重合であり、そして合成された核酸鎖の3'末端に余分な核酸配列が付加されるターミナルトランスフェラーゼ伸長を、リンカー連結工程の代わりに行うことも可能である。
[0058](a)核酸含有試料にオリゴヌクレオチドP*を添加する工程、ここでオリゴヌクレオチドP*は伸長不能3'末端を有し、オリゴヌクレオチドP*の3'末端残基は加ピロリン酸分解によって除去可能であり、そしてオリゴヌクレオチドP*は、試料に存在する標的核酸の実質的な相補鎖にアニーリングする;
[0059](b)標的核酸の実質的な相補鎖にアニーリングしたオリゴヌクレオチドP*の3'伸長不能末端を、加ピロリン酸分解によって除去して、オリゴヌクレオチドP*を非ブロッキング化し、ブロッキングされていないオリゴヌクレオチドを産生する工程;および
[0060](c)標的核酸の存在を検出する工程、ここで標的核酸の配列は、オリゴヌクレオチドP*の配列と実質的に相補的である。
[0063](a)核酸含有試料に2つのオリゴヌクレオチドP*を添加する工程、ここで各オリゴヌクレオチドP*は伸長不能3'末端を有し、オリゴヌクレオチドP*の3'末端残基は加ピロリン酸分解によって除去可能であり、一方のオリゴヌクレオチドP*ともう一方のオリゴヌクレオチドP*は、それぞれの3'端で、少なくとも1ヌクレオチド重複し、そして一方のオリゴヌクレオチドP*は、試料に存在する標的核酸の実質的な相補鎖にアニーリングし、そしてもう一方のオリゴヌクレオチドP*は、標的核酸の実質的な相補鎖の相補体にアニーリングする;
[0064](b)標的核酸にアニーリングしたオリゴヌクレオチドP*の3'伸長不能末端を、加ピロリン酸分解によって除去して、オリゴヌクレオチドP*を非ブロッキング化し、ブロッキングされていないオリゴヌクレオチドを産生する工程;および
[0065](c)標的核酸の存在を検出する工程、ここで標的核酸の配列は、オリゴヌクレオチドP*の配列に実質的に相補的である。
[0070]・活性化可能オリゴヌクレオチドP*は、3'末端に加えて、他の位でも、ブロッキングされたヌクレオチドを含有可能である。
[0075]・活性化は、3'エキソヌクレアーゼなどの別の機構によって起こることも可能である。3'エキソヌクレアーゼは、3'端にミスマッチがあるかどうかを弁別するため、マッチしたプライマーまたはミスマッチしたプライマーに特異性を有する可能性もある。3'エキソヌクレアーゼはどちらの方式でも使用可能である。該酵素がミスマッチを好む場合、上述のように使用可能であるが、一般的でない突然変異を検出する能力は、活性化に関するある程度の特異性に依存するだろうし、とはいえ、特異性は、部分的に、内部のミスマッチに由来する可能性もある。
[0111]本明細書において、以下の用語を用いる。
[0112]加ピロリン酸分解:ピロホスフェート(PPi)の存在下で、DNAポリメラーゼによって、ヌクレオチド鎖から3'ヌクレオチドを除去して、ヌクレオチド三リン酸を生成すること。これは重合反応の逆である。
[0115]PAP-A:稀な突然変異の検出に使用可能な、PAPに基づくアレル特異的増幅(図1)。
[0117]PAP-R:既知の配列内の未知の突然変異の検出のための、PAPに基づく再配列決定(図3および4)。
[0120]GVまたはAVアレル:本明細書において、モデル系として用いた、ドーパミンD1受容体遺伝子の一般的な多型のアレル(本明細書において、G0またはA0アレルとも称する)。
[0122]指数関数的PAP:指数関数的産物集積のためであり、そして少なくとも1つがP*である、2つの相対するオリゴヌクレオチドを用いたPAP。
[0124]PASA:特異的アレルのPCR増幅(アレル特異的PCRまたはARMSとしても知られる)。
[0126]突然変異負荷:組織内の体細胞突然変異の頻度およびパターン。
[0128]伸長不能3'末端(または端):伸長不能であるが、加ピロリン酸分解によって活性化可能である、オリゴヌクレオチドP*の3'末端(または端)のヌクレオチドまたはヌクレオチド類似体。伸長不能3'末端(または端)の例には、限定されるわけではないが、2'3'-ジデオキシヌクレオチド、アシクロヌクレオチド、3'-デオキシアデノシン(コルジセピン)、3'-アジド-3'-デオキシチミジン(AZT)、2',3'-ジデオキシイノシン(ddI)、2',3'-ジデオキシ-3'-チアシチジン(3TC)および2',3'-ジデヒドロ-2',3'-ジデオキシチミジン(d4T)が含まれる。
[0130]多重PAP:1つの反応試験管中または固体支持体、例えばマイクロアレイ上の、1より多いPAP(PAP、Bi-PAP、マッチまたはミスマッチPAP等)。
[0132]ミスマッチPAP:P*およびテンプレート間にミスマッチを有するPAP。
[0133]入れ子(nested)PAP:テンプレート核酸上で、1つの対が第二の対の内部に位置する、2以上の対のP*を用いるPAP。
[0137]核酸増幅に必須のDNAポリメラーゼは、以下の反応のいくつかまたはすべてを触媒する:i)デオキシヌクレオチド三リン酸またはその類似体の重合;ii)ピロホスフェート(PP)の存在下での二重鎖DNAの加ピロリン酸分解、[dNMP]n+x[PPi]...[dNMP]n-x+x[dNTP];iii)3'-5'エキソヌクレアーゼ活性(PPiを必要としない)、およびiv)5'-3'エキソヌクレアーゼ活性(DuetcherおよびKornberg、1969;KornbergおよびBaker、1992)。TaqおよびTfl DNAポリメラーゼに関しては、重合活性および5'-3'エキソヌクレアーゼ活性が報告されている(Chienら、1976;Kaledinら、1981;Longleyら、1990)。T7 SequenaseTMおよびThermoSequenaseTMDNAポリメラーゼに関しては、加ピロリン酸分解は、サンガー配列決定反応において、特定のジデオキシヌクレオチド末端セグメントの分解を導く可能性もある(TaborおよびRichardson、1990;Vander Hornら、1997)。
[0146]テンプレート鎖に、実質的に相補的な活性化可能オリゴヌクレオチドP*をアニーリングさせる工程。この活性化可能オリゴヌクレオチドP*は、3'末端に、伸長不能ヌクレオチドまたはその類似体を有する。
[0150](d)テンプレート鎖から工程(C)の所望の核酸鎖を分離する工程、および
[0151](e)所望の核酸鎖の所望の増幅レベルが達成されるまで、工程(A)〜(D)を反復する工程。
[0156]LM-PAPは工程(i)、(ii)、(iii)、(iv)および(v)によって、工程(i)、(ii)、(iii)および(vi)によって、工程(ii)、(iii)、(iv)および(v)によって、または工程(ii)、(iii)および(vi)によって、実行可能であり、工程は以下のとおりである。
[0158](ii)プライマーP1は遺伝子特異的であり、そしてその伸長には:1)テンプレート鎖に実質的に相補的なプライマーをアニーリングさせ;2)ヌクレオシド三リン酸またはその類似体および核酸ポリメラーゼの存在下、テンプレート鎖上でプライマーを伸長し、この伸長がテンプレート鎖上の切断部位で「脱線する」ことが含まれる。工程1)および2)を反復することも可能である。
[0160](iii)リンカー連結工程には、合成された核酸鎖の3'末端へのリンカーの連結が含まれる。テンプレート非依存性重合であり、そして合成された核酸鎖の3'末端に余分な核酸配列が付加されるターミナルトランスフェラーゼ伸長を、リンカー連結工程の代わりに行うことも可能である。
[0187]マイクロアレイ上のBi-PAP新規DNA配列決定を図25に示す。簡潔には、該方法はまず、P*対を用いたBi-PAP増幅物をすべて収集し、そして次いで、対の特異的下位配列を順序付けることによって、この収集物から未知のDNA配列を再構築する。
[0194]本発明は、PAPを用いてヒトD1ドーパミン受容体遺伝子内の多型部位における既知の突然変異を同定可能であることを例示する、以下の実施例から理解されるであろう。ジデオキシオリゴヌクレオチド配列、アシクロヌクレオチド配列、DNAポリメラーゼ、PPi濃度、アレル特異的テンプレート、pH、およびdNTP濃度の影響を調べた。原理を立証するため、実施例に報告する実験を行った。以下の実施例は、例示のため提供され、そしていかなる方式でも本発明を限定することを意図しない。当該技術分野に周知の標準技術またはそれらの文書に具体的に記載される技術を利用した。
PCRによるテンプレートの調製
[0195]2つのプライマー(T=5'GAC CTG CAG CAA GGG AGT CAG AAG3'(配列番号1)およびU=5'TCA TAC CGG AAA GGG CTG GAG ATA3'(配列番号2))を用いたPCRによって、ヒトD1ドーパミン受容体遺伝子の640 bp領域を増幅した(図6A)。TU:UT二重鎖産物はGenBank X55760のヌクレオチド33〜672に渡り、そしてG+C含量は55.3%である。よく生じるAからGへの多型は、ヌクレオチド229に位置し、G/G、A/AおよびG/Aの3つの遺伝子型を生じる(Liuら、1995)。PCR混合物は、50μlの体積を含有する:50 mM KCl、10 mM Tris/HCl、pH8.3、1.5 mM MgCl2、各200μMの4種のdNTP(Boehringer Mannheim)、各0.1μMのプライマー、2%DMSO、1 UのTaq DNAポリメラーゼ(Boehringer Mannheim)およびG/Gホモ接合体、A/Aホモ接合体またはG/Aヘテロ接合体由来の250 ngのゲノムDNA。周期条件には:95℃15秒間の変性、55℃30秒間のアニーリング、および72℃1分間の伸長で、全部で35周期が含まれた(Perkin-Elmer GeneAmp PCR系9600)。Centricon(登録商標)100微量濃縮装置(Amicon)上で3回保持することによって、プライマーおよび他の小分子から、PCR産物をおよそ10,000倍精製した。260 nmのUV吸光度によって、回収されたPCR産物の量を測定した。
[0196]ホープ市DNA/RNA化学研究室において、Perseptive Biosystems 8909合成装置(Framinsham)によってデオキシヌクレオチドオリゴヌクレオチドを合成し、そしてoligopureカートリッジ(Hamilton)によって精製した。ターミナルトランスフェラーゼによって3'末端ジデオキシヌクレオチドを付加した。混合物は、40μlの総体積を含有した:200 mMカコジル酸カリウム、25 mM Tris/HCl(25℃でpH6.6)、2.5 mM CoCl2、0.25 mg/mlのBSA、4000 pMのオリゴヌクレオチド、2.5 mM 2'3'-ddNTP(ddNTPに対する3'-OH末端のモル比は1:25であった)(Boehringer Mannheim)、125 Uのターミナルトランスフェラーゼ(Boehringer Mannheim)。反応を37℃で1時間インキュベーションし、そして次いで、最終濃度5 mMでEDTAを添加することによって停止した。ブタノールを用いることによって脱塩した後、TBE緩衝液(90 mM Tris/ホウ酸、1 mM EDTA、pH8.3)中の分離用7 M尿素/20%ポリアクリルアミドゲル電気泳動によってジデオキシオリゴヌクレオチドを精製した(Maniatisら、1982)。260 nmのUV吸光度によって、回収されたP*の量を測定した。
[0198]オリゴヌクレオチドP*およびUを用いるPAP、または1つのP*のみを用いるPAPによって、TU:UT二重鎖テンプレート内の469 bp領域を増幅した(表1および図6A)。PU:UP二重鎖産物は、GenBank X55760のヌクレオチド204〜672に対応し、そしてG+C含量は55.6%である。言及しない限り、PAP反応混合物は、Tfl DNAポリメラーゼでは、25μlの総体積を含有した:75 mM KCl、20 mM Tris/HCl(pH7.4)、1.5 mM MgCl2、各40μMの4種のdNTP(dATP、dTTP、dGTPおよびdCTP)、0.2μM P*、0.05μM Uオリゴヌクレオチド、300μM Na4PPi(20 MMストック溶液をHClでpH8.0に調整した)、1μCiの[α-32P]-dCTP(3000 Ci/nmol、Amersham)、1 UのTfl DNAポリメラーゼ(Promega)および2 ngのTU:UT。Taq DNAポリメラーゼでは、50 mM KCl、10 mM Tris/HCl(pH7.4)、2.0 mM MgCl2および1 UのTaq DNAポリメラーゼ(Boehringer Mannheim)を除いて、反応混合物は同じであった。PCRおよび他の対照の混合物は、添加するプライマーを除いて、同じであった。周期条件には:94℃15秒間、55℃1分間、および1分間で傾斜をつけて72℃に上昇させ、そして72℃2分間、全部で15周期が含まれた。
PAPに用いたオリゴヌクレオチド

bTは3'末端がTデオキシヌクレオチドであることを意味し、そしてG*は3'末端がGジデオキシヌクレオチドであることを意味する。太い大文字GおよびAは、それぞれ、GアレルおよびAアレルに対応するG塩基およびA塩基である。5'末端の第一の塩基は、GenBank X55760のヌクレオチド208に対応する。
fUおよび1つのP*を用いた増幅または1つのP*のみを用いた増幅。
[0200]3つの制限エンドヌクレアーゼ、AciI(5'C▼CGC3'/3'GGC▲G5')、EaeI(5'Py▼GGCCPu3'/3'PuCCGG▲Py5')およびEco0109I(5'PuG▼GNCCPy3'/3'PyCCNG▲GPu5')は各々、PU:UP二重鎖内に制限部位を有する。D5G*およびUを用いたPAPによって、G/Gアレルを増幅し;D1およびUを用いたPCR増幅を対照として用いた。40μlのPAP反応および2μlのPCR反応をCentricon(登録商標)100微量濃縮装置で精製し、そして濃縮し、そして制限エンドヌクレアーゼ:1×NE緩衝液3中、2.5 UのAciI;または1×NE緩衝液1中、3 UのEaeI;またはBSAを含むNE緩衝液4中の30 UのEco0109I(上記の酵素および緩衝液はすべてNew England BioLabsからのもの)によって産物を消化した。10μlの反応物を、37℃で2時間インキュベーションした。消化反応物を、上述のように、標準的2%アガロースゲルを通じて電気泳動した。
[0201]TflおよびTaq DNAポリメラーゼは、加ピロリン酸分解活性を含有することが示された。Tfl DNAポリメラーゼを利用して、D1ドーパミン受容体遺伝子のヌクレオチド229でGアレルを検出した(Liuら、1995)(図6A)。ddGまたはddAいずれかを用いて、3'末端でP*を合成した(表1を参照されたい)。3'末端ジデオキシヌクレオチドは、重合による直接伸長を阻害するが、P*がGアレルの相補鎖と特異的にハイブリダイズする場合、ピロホスフェート(PPi)の存在下で、加ピロリン酸分解によって除去可能である。分解されたオリゴヌクレオチドは、5'-3'方向の重合によって伸長可能である(図6Bおよび6C)。
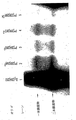
[0203]2つのオリゴヌクレオチド(P*およびU)、Tfl DNAポリメラーゼ、並びにG/GおよびA/AアレルのDNAテンプレートを用いてPAPを行った。多数のP*を試験した(表1)。D5G*(アレル特異的ヌクレオチドおよびジデオキシヌクレオチドが3'末端に同時局在する)およびD3G*(アレル特異的ヌクレオチドは、3'末端から2塩基である)は、PPiの存在下で、Gアレルを特異的に増幅した(図8A)。PPiを添加しないと、D5G*で特異的な産物はまったく観察されず、添加したPPiがPAPに必須の構成要素であることが示された(図8B、レーン6および15)。D3G*を用いるレーン4中、およびD4G*を用いるレーン5中に、かすかな産物が観察された(図8B)(以下を参照されたい)。
[0204]上記パラメータ各々を調べた。PAPは、7.4〜7.7の間のpH、200μM〜400μMの間の[PPi]、および25μM〜50μMの間の[dNTP]で最も効率的であった(表2)。Taq DNAポリメラーゼをTflの代わりに用いることが可能であり、効率は同様であった(表2)。
PAPに影響を及ぼすパラメータ
bPAP効率は:-、特異的産物(単数または複数)なし;±、非常に弱い特異的産物(単数または複数);+、弱い特異的産物(単数または複数);++、中程度の特異的産物(単数または複数);+++、強い特異的産物(単数または複数);++++、非常に強い特異的産物(単数または複数)と示される。
特異的産物の同一性
[0205]特異的産物の同一性を確認するため、制限エンドヌクレアーゼ消化を行った(図9)。3つの制限エンドヌクレアーゼAciI、EaeIおよびEco0109は各々、PU:UP二重鎖に制限部位を有する。期待される制限断片が見出された。D3G*およびUを用いても、同様の結果が観察された。
[0207]PPiの存在下、G/GおよびA/Aアレルから、1つのP*のみを用いた線形増幅のため、PAPを行った。D3G*およびD5G*で、PAPの特異的産物が得られたが、他のP*では得られなかった(図10、レーン4および6)。P*の効率は、オリゴヌクレオチドのサイズ、3'末端ジデオキシヌクレオチド、およびアレル特異的ヌクレオチドの位置によって影響を受けた。
[0213]実施例1は、加ピロリン酸分解後の重合を用いて、PASAの特異性を増加させうる証拠を提供する。有意な非特異的増幅は、2種類のエラーの連続したカップリングを必要とする(図7)。3'末端でミスマッチデオキシヌクレオチドを除去する、ミスマッチ加ピロリン酸分解の率は、正しいdNMPに対する誤ったdNMPの除去率として表すと、T7 DNAポリメラーゼでは10-5未満であると報告された(KornbergおよびBaker、1992;Wongら、1991)。重合によって置換突然変異を生成する、誤った取り込みの率は、正しいdNMPに対する誤ったdNMPの取り込み率として表すと、T7 DNAポリメラーゼでは10-5、そして大腸菌DNAポリメラーゼIでは10-4であると報告された(KornbergおよびBaker、1992;Wongら、1991;Bebenekら、1990)。Taq DNAポリメラーゼ、およびT7 DNAポリメラーゼの3'-5'エキソヌクレアーゼ欠損突然変異体、および大腸菌DNAポリメラーゼIでも、同様の結果が報告された(KornbergおよびBaker、1992;Wongら、1991;EckertおよびKunkel、1990)。(i)D5G*を用いたPAPにおける非特異的増幅による特異性は、ddNMPのミスマッチ加ピロリン酸分解の率がdNMPと同じであるならば、周期あたり10-5と概算される。(ii)非特異的増幅による特異性は、ミスマッチ加ピロリン酸分解および誤った取り込みが連続してカップリングされている場合、3.3×10-11と概算される。
[0214]TflまたはTaq DNAポリメラーゼを利用してG/GおよびA/Aアレルを増幅することによって、各P*を試験した。特異的増幅には、PPiおよびアレル特異的テンプレートの存在が必要である。さらに、増幅効率は、オリゴヌクレオチドのサイズ、3'末端ジデオキシヌクレオチド、P*の3'末端に比較したアレル特異的ヌクレオチドの位置に影響を受ける。
[0217]in vitroで酵素的に核酸を増幅するいくつかの方法が開発されてきており、そして既知の配列変異体を検出するのに適応させることも可能である。これらには、ポリメラーゼ連鎖反応(PCR)(Saikiら、1985;Saikiら、1988)、リガーゼ連鎖反応(LCR)(Landegrenら、1988;Barany、1991)および回転周期増幅(RCA)(Lizardiら、1998;Banerら、1998)が含まれる。PAPは多くの点で異なる:i)各増幅で、加ピロリン酸分解および重合を連続してカップリングし、ii)PAPのためのジデオキシオリゴヌクレオチドが少なくとも1つある。アシクロヌクレオチドなど、3'末端で3'-ヒドロキシル基を欠く、他の化学的に修飾されたヌクレオチドが同じ機能を果たしうる(以下の実施例12を参照されたい)、iii)一方の形式は線形増幅用であり、そして他方は指数関数増幅用であり、iv)増幅にはPPiが必要であり、v)有意な非特異的増幅は、ミスマッチ加ピロリン酸分解および誤った取り込み両方を必要とし、vi)PAPは既知の点突然変異を検出可能であり、そして野生型アレルから、非常に稀な突然変異アレルを検出する特異性を、非常に増加させることが可能である。
PCRによるテンプレートの調製
[0227]2つのプライマー(T=5'GAC CTG CAG CAA GGG AGT CAG AAG3'(配列番号1)およびU=5'TCA TAC CGG AAA GGG CTG GAG ATA3'(配列番号2))を用いたPCRによって、ヒトD1ドーパミン受容体遺伝子の640 bp領域を増幅した。TU:UT二重鎖産物はGenBank X55760のヌクレオチド33〜672に渡り、そして産物のG+C含量は55%である。よく生じるAからGの多型は、ヌクレオチド229に位置し、G/G、A/AおよびG/Aの3つの遺伝子型を生じる。PCR体積は50μlである:50 mM KCl、10 mM Tris/HCl、pH8.3、1.5 mM MgCl2、各200μMの4種のdNTP、各0.1μMのプライマー、2%DMSO、1 UのTaq DNAポリメラーゼ(Boehringer Mannheim)およびG/Gホモ接合体、A/Aホモ接合体またはG/Aヘテロ接合体由来の250 ngのゲノムDNA。周期条件には:GeneAmp PCR系9600(Perkin-Elmer Applied Biosystems)を用いた、94℃15秒間の変性、55℃30秒間のアニーリング、および72℃1分間の伸長で、全部で35周期が含まれた。Centricons 100微量濃縮装置(Amicon)上で3回保持することによって、プライマーおよび他の小分子から、PCR産物をおよそ10,000倍精製した。260 nmのUV吸光度によって、回収されたPCR産物の量を測定した。
[0228]ホープ市DNA/RNA化学研究室において、Perseptive Biosystems 8909合成装置(Framinsham)によってデオキシヌクレオチドオリゴヌクレオチドを合成し、そしてoligopureカートリッジ(Hamilton)によって精製した。ターミナルトランスフェラーゼによって3'末端ジデオキシヌクレオチドを付加した。混合物は、30μlの総体積を含有した:100 mMカコジル酸カリウム(pH7.2)、2.0 mM CoCl2、0.2 mM DTT、2500 pMのオリゴヌクレオチド、2 mM 2',3'-ddNTP(ddNTPに対する3'-OH末端のモル比は1:24であった)(Boehringer Mannheim)、100 Uのターミナルトランスフェラーゼ(GIBCO BRL)。反応を37℃で4時間インキュベーションし、そして次いで、最終濃度5 mMでEDTAを添加することによって停止した。Centri-spinTMカラム(Princeton Separations)を用いて脱塩した後、TBE緩衝液(90 mM Tris/ホウ酸、1 mM EDTA、pH8.3)中の分離用7 M尿素/20%ポリアクリルアミドゲル電気泳動によってP*を精製した(Maniatisら、1982)。260 nmのUV吸光度によって、回収されたP*の量を測定した。
[0230]オリゴヌクレオチドP*およびUを用いるPAP、またはP*のみを用いるPAPによって、TU:UT二重鎖テンプレート内の445〜469 bpの領域を増幅した。PU:UP二重鎖産物は、GenBank X55760のヌクレオチド204〜228から672に対応し、そしてG+C含量は56%である。PAP反応混合物は、25μlの総体積を含有した:50 mM KCl、10 mM Tris/HCl(pH7.6)、1.5 mM MgCl2、各100μMの4種のdNTP(dATP、dTTP、dGTPおよびdCTP)、0.1μM P*、0.1μM Uオリゴヌクレオチド(TCATACCGGAAAGGGCTGGAGATA(配列番号2))、300μM Na4PPi、2%DMSO、1μCiの[α-32P]dCTP(3000 Ci/mmol、Amersham)、1 UのAmpliTaqFS DNAポリメラーゼ(PE Applied Biosystems)または各0.5 UのAmpliTaqFSおよびTaq DNAポリメラーゼ、および10 ngのTU:UT。8 U ThermoSequenase(Amersham Pharmacia)または4 U ThermoSequenaseに、0.5 U Taqおよび2.5 mM MgCl2を加えた以外は同一の条件下で、ThermoSequenaseもまた試験した。周期条件には:94℃10秒間の変性、60℃1分間(ThermoSequenaseでは55℃)のアニーリング、および72℃2分間の伸長、全部で15周期が含まれた。
[0232]実施例1において、天然TflまたはTaq DNAポリメラーゼを用いて、3'末端にddGを持つP*のみが増幅された。AmpliTaqFSおよびThermoSequenase DNAポリメラーゼは、P*の3'末端のいかなる種類のジデオキシヌクレオチド(ddAMP、ddTMP、ddGMPまたはddCMP)もはるかに少なくしか区別せず、はるかにより高いPAP効率を達成することが見出された。例えば、ドーパミンD1受容体遺伝子の18量体であるが、3'末端にddGMPおよびddAMPを有する、P*(212)18G0およびP*(212)18A0(表3)は、それぞれ、GアレルおよびAアレルを特異的に増幅した。その収量比は1.4(図11Bのレーン9と11を比較されたい)であり、そしてしたがって、P*(212)18G0は、P*(212)18A0より、周期あたり4%、より効率的であると概算される。別のP*(228)26A-24=5'TAGGAACTTGGGGGGTGTCAGAGCCC*3'(配列番号12)は、3'末端にddCMPを持つ26量体であるが、3'末端にddCMPを持たないプライマーと同程度に効率的に増幅され、そして収量は、TflまたはTaqを用いたものと比較して、1,000倍増加すると概算された。さらに、PAPは、ヒトゲノムDNAから直接、セグメントを増幅した。
P * の長さおよびミスマッチに影響を受けるPAP特異性
cアレル特異的塩基までの3'末端からの距離:「0」=3'末端、-3=3'末端から3塩基。
ePAPのノイズ比(%)は、同一のP*による特異的アレル産物に対する非特異的アレル産物の相対収量、または同一テンプレートを用いることによる、天然型に対する、指定された突然変異P*の相対収量と定義される。特異的シグナルを<10%ノイズ比とする。
[0234]AmpliTaqFSを用いて、異なる長さおよびミスマッチを持つ、多様なP*を調べた(表3)。PAP効率に対する長さおよびミスマッチの影響を、同一テンプレート由来の異なる長さの2つのP*間の相対収量(%)として表し(図12)、これは、長さが各々2〜4塩基少ないと、0.0%〜201.5%で多様であった。PAPの特異性もまた、P*の長さおよびミスマッチに影響を受ける(表3)。ノイズ比(%)は、マッチ産物に対するミスマッチ産物の相対収量と定義され、そして特異的シグナルは、<10%ノイズ比でスコアされる。P*のアレル特異的塩基が3'末端にある場合、特異的アレルのみが増幅され、そして特異性はP*の長さには関連しなかった(図12A)。アレル特異的塩基がP*の3'末端にない場合、特異性はP*の長さに関連した。3'末端から15塩基までの、18量体P*における非3'末端ミスマッチはいずれも、増幅を引き起こさなかった(図12B〜12E)が、26量体P*における2つのこうしたミスマッチであっても、非特異的増幅を引き起こした。
異なって配置されたP * のPAP特異性
異なってミスマッチするP * を用いたPAPの特異性
bノイズ比(%)は、ミスマッチP*およびP*(212)18G0とGアレル特異的テンプレートの間の相対収量である。
[0241]P*の3'特異的下位配列の特性を適用して、平行した方式で、未知の配列変異体のスキャンまたはあらかじめ決定された配列の再配列決定を行うことも可能である。あらかじめ決定された配列の相補鎖上の各ヌクレオチドに、18量体などの4つの下流P*によってクエリーを行い(図11)、この下流P*は、3'末端で、ddAMP、ddTMP、ddGMPまたはddCMPいずれかが野生型配列および3つのありうる一塩基置換に対応することを除いて、同一の配列を有する。X塩基の相補鎖をスキャンするP*の数は、4およびXの乗算であり、これは指数関数的PAPまたは線形PAPいずれにも適している。4つの下流P*は、3'末端のddAMP、ddTMP、ddGMPおよびddCMPが、4つの蛍光色素によるなど、区別のため異なって標識されている場合、単一の点上に固定することさえ可能である。こうして、増幅シグナルは、加ピロリン酸分解によって、ddNMPがP*から除去される際、各色素の強度減少によって表されうる。線形PAPの1つの利点は、区別のため異なる色素で標識された4種のddNTPを一塩基伸長の基質として使用可能であることである。
[0244]PAPによる新規DNA配列決定の概念は、P*のすべてのありうる3'特異的下位配列を用いて、新規配列の3'特異的下位配列の存在を同定することである。P*の3'特異的下位配列の完全なセットは4nである。3'特異的下位配列は各々、4mの5'エンハンサー下位配列の完全なサブセットを有する。例えば、3'特異的下位配列としての16量体および5'エンハンサー下位配列としての2量体の完全なセットは、(A、T、G、C)(A、T、G、C)N16=418と示すことも可能である。
[0248]同一かまたは異なっているかを調べるため、2つのDNA配列を比較するフィンガープリンティングでは、3'特異的下位配列の不完全なセットを用いることによってP*の数を減少させる、単純な方法がある。これらを特定の順序に配置することによって、染色体位置とともに配列を同定することが可能である。ヒトゲノムには3×109 bp DNAがあることを考慮すると、特異性を増加させるために、1つのP*のみを用いるPAPより、2つのオリゴヌクレオチドを用いるPAPが好ましい。
[0250]オリゴヌクレオチドを用いることによるSBHにおいて、ハイブリダイゼーション、および重複部分を通じて陽性にハイブリダイズするプローブのアセンブリによってDNA配列を決定する。固定試料上の単一オリゴヌクレオチドハイブリダイゼーションは、最適ハイブリダイゼーションおよび洗浄条件下で、非常に特異的になりうることがずっと以前から知られており(Wallaceら、1979)、したがって、単一内部ミスマッチを含有するものから、完全なハイブリッドを区別することが可能である。アレイ中のオリゴヌクレオチドは、長さ11〜20ヌクレオチドであり、そして中央に7〜9塩基の特異的領域を有し、ミスマッチハイブリダイゼーションによって、非特異的シグナルが生成される。標準的ハイブリダイゼーションおよび洗浄条件下で、マッチおよびミスマッチ間の二重鎖安定性はまた、末端ミスマッチおよび隣接配列によって影響を受ける(Drmanacら、1989;Khrapkoら、1989;Ginot、1997)。
ゲノムDNAからのPAP増幅
[0256]本実施例は、ゲノムDNAからのPAP直接増幅を例示する。本実施例に用いるオリゴヌクレオチドを以下に列挙する。レーン番号は、図15のレーンを指す。
[0258]0.1μM濃度中の相対する上流オリゴヌクレオチドは:D1(420)24 U 5'ACGGCAGCACAGACCAGCGTGTTC3'(配列番号48)であり、各下流オリゴヌクレオチドと対形成した。詳細に関しては、表3の脚注を参照されたい。
LM-PCRおよびLM-PAPの特異性の比較
[0261]LM-PCRプロトコルには、ヒト・ドーパミンD1受容体遺伝子モデル系における、プライマー伸長、リンカー連結、PCR増幅、および指示された標識が含まれる(図16)。プライマー伸長(P1プライマー=5'TTGCCACTCAAGCGGTCCTCTCAT3'(配列番号49))の最初の10周期でVentR(エキソ-)DNAポリメラーゼを用いたことを除いて、本質的に記載されるように(Pfeiferら、1999)、UV処理したゲノムDNA試料に対して、ターミナルデオキシヌクレオチジルトランスフェラーゼ(TdT)を添加して、LM-PCRを行った(このプロトコルは、TD-PCRとして知られる)。温度周期は、95℃1分間、63℃3分間、および72℃3分間であった。シグナルを増進するため、ターミナルトランスフェラーゼをプロトコルに添加し、そしてLM-PCRのこの変型をTD-PCRと呼ぶ。ターミナルデオキシヌクレオチジルトランスフェラーゼ(TdT)テール付加の前に、Dynabeadを用いて、標的DNA分子を濃縮した。製造者に記載されるように、Expand Long Template PCR系3(BMB)を用いて、PCRを行った(P2プライマー=5'GAAGCAATCTGGCTGTGCAAAGTC3'(配列番号50))。直接標識を行う前に、QIAquick PCR精製キット(QIAGEN)を用いて、PCR産物を精製した。32P標識プライマー:
[0262]PAPによる直接標識工程をのぞいて、アレル特異的PCRとして、LM-PAPを行った(図16A)。32P標識プライマー:
10 4 〜10 5 テンプレート中の1つの突然変異を検出するためのPAP-Aの最適化
[265]1μgのラムダファージDNAは、2×1010コピーのテンプレートを含有する。1部分の突然変異体lacIテンプレートを104〜105部分の対照DNAテンプレート、例えば野生型lacIと混合することによって、PAPの特異性を決定する。PAP-Aの特異性は、ポリメラーゼのエラー率、P*の純度(現在の精製プロトコルでは<2×10-4)および抽出プロセスにおけるDNAテンプレート損傷の可能性の関数である。酵素種および濃度、並びにdNTP、PPi、Mg++またはMn++などの他の構成要素の濃度を試験することによって、PAPの収量および特異性を最適化する。DNAポリメラーゼなどの抗体が活性化する酵素を用いた、室温でのホットスタートPAPを用いて、誤った増幅を排除することも可能である。
PAP-Rの最適化
[0269]モデル系において、P*に沿ってミスマッチがあると活性化が阻害され、ミスマッチが5'端から2ヌクレオチドであった場合でさえも、活性化が阻害された(図14)。5'末端が2、6、9、および12ヌクレオチド下流に移動している、P*の18量体のさらなるセットもまた、活性化の阻害を示した(図13)。さらに、20量体および22量体もまた、一ヌクレオチドミスマッチでの阻害を示す(図12)。これらの知見を拡大適用し、そして再配列決定の堅固な方法の基礎を築くため、一塩基ミスマッチの位置およびP*の活性化の関係をさらに解析する。
LM-PAPの最適化
[0274]ヒト・ドーパミンD1受容体遺伝子およびマウスPgkl遺伝子をモデル系として用いて、LM-PAPまたはLM-PCRを利用する場合のクロマチン構造の解析を比較する。ドーパミンD1受容体遺伝子は上述されている。X染色体不活性化は、初期胚発生段階で起こる。雌性細胞中の2つのアレルが異なる発現状態を維持するため、これは、遺伝子制御の研究に好適な系である。Pgklは、ホスホグリセリン酸キナーゼ(PGK)をコードするX染色体連鎖ハウスキーピング遺伝子である。PGKは、解糖に重要な酵素であり、そして該遺伝子は、雌性体細胞の不活性X染色体(Xi)および雄性生殖細胞における以外は、いつでも活性であると期待される。
アレル特異的LM-PAPの最適化
[0276]コード領域および非コード領域両方のpgklaおよび1b遺伝子の多型部位が報告されている(Boerら、1990)。これらを用いて、アレル特異的P*を設計する。1つのアレル特異的オリゴヌクレオチドを、Pgk1遺伝子から前向きに選択し、そして1つを、ドーパミンD1受容体遺伝子から前向きに選択する。同一配列のブロッキングされたオリゴヌクレオチドおよびブロッキングされていないオリゴヌクレオチドを合成し、そしてそれぞれ、アレル特異的LM-PAPおよびLM-PCRを行う。シグナル対ノイズ比を定量化し、そして比較する。
マイクロアレイ上のPAP-R
[0277]最初の実験は、上述のように、エクソンBおよびHの2つの20ヌクレオチド領域に重点を置くであろう。PAP-Rの実験設計は、例えばGeniom(登録商標)装置を用いたマイクロアレイ上のP*オリゴヌクレオチドのデジタル光指示合成を用いたことを除いて、上述の実験と類似である。因子IX遺伝子のエクソンBおよびHの20 bp領域の野生型に、そしてすべての一塩基ミスマッチに相補的な、総数160のオリゴヌクレオチドを合成する。陽性対照として、因子IX遺伝子の隣接する160 bp領域に正確にマッチする、登録されたものから各々1ヌクレオチドずれていく160オリゴヌクレオチドを合成する。野生型および突然変異体試料から、ゲノムDNAを増幅し、オリゴヌクレオチドにアニーリングさせ、そして蛍光ジデオキシターミネーターを用いて、プライマー伸長を行う。固体支持体に関してプロトコルを最適化する。因子IXの2つの20 bpヌクレオチド領域にミスマッチするオリゴヌクレオチドの、すべてでないとしてもほとんどが、あるとしてもわずかなシグナルしか生じず、一方、160の対照オリゴヌクレオチドの大部分が強いシグナルを生じるように、プライマーの長さ、利用する酵素および反応条件の調整を行う。
ヒトおよびマウスゲノムDNAからのPAP直接増幅
[0281]2つのP*、P1*(配列番号45、Gアレル特異的)またはP2*(配列番号47、Aアレル特異的)および上流のブロッキングされていないプライマー(U;配列番号48)各々を用いてPAPを行い、D1ドーパミン遺伝子の180 bpセグメントを増幅した。P*は3'末端にddCおよびddGを含む26量体である。100 ngのヒトゲノムDNAを35周期増幅し、その後、2%ゲル電気泳動した。PAP反応混合物は、総体積25μlを含有した:50 mM KCl、20 mM HEPES/NaOH(25℃でpH6.9)、10 mM (NH4)2SO4、1.5 mM MgCl2、各40μMの4種のdNTP(dATP、dTTP、dGTPおよびdCTP)、0.1μM U、150μM Na4PPi、2%DMSO、0.5 UのAmpliTaqFSポリメラーゼ(PE Applied Biosystems)、0.5 UのTaqポリメラーゼ、および100 ngのヒトゲノムDNA。周期条件は、94℃15秒間、65℃30秒間、および72℃1分間であった。図17Aは、D1ドーパミン遺伝子のPAP増幅の結果を示す。レーン2および5において、P1*は、3'末端から24ヌクレオチドでAアレルテンプレートに特異的であり、したがってG/GおよびA/A遺伝子型間にはほとんどまたはまったく区別がない。レーン3および6において、P2*は、3'末端から2ヌクレオチドでAアレルテンプレートに特異的であり、したがって、A/A遺伝子型の特異的増幅がある。レーン1および5はPCR対照である。レーン4および8は、P*を含まない陰性対照である。レーンMはcpx DNA/HAEIVマーカー120 ngである。
アシクロヌクレオチドおよび多様なポリメラーゼを用いたPAP
λファージDNAテンプレート
[0285]挿入された大腸菌の野生型lacI遺伝子を含有する野生型λファージDNAテンプレート(Kohlerら、1991)をStratageneから購入した。Maniatisら(1982)にしたがって、SCS-8大腸菌細胞に形質転換したλファージプラークから、突然変異体λファージDNAテンプレートを調製した。該テンプレートは、lacI遺伝子のヌクレオチド369にTからGの突然変異を含有した。260 nmのUV吸光度によって、λファージDNAの量を測定した。
[0286]ターミナルトランスフェラーゼによって、3'末端アシクロヌクレオチドまたは3'末端ジデオキシヌクレオチドをデオキシヌクレオチドオリゴヌクレオチドに付加した。混合物は、総体積25μlを含有した:100 mMカコジル酸カリウム(pH7.2)、2.0 mM CoCl2、0.2 mM DTT、2 nMのオリゴヌクレオチド、2.4 mMアシクロNTP(アシクロNTPに対する3'-OH末端のモル比は1:30であった)(New England BioLabs)、または2.4 mM 2',3'-ddNTP(ddNTPに対する3'-OH末端のモル比は1:30であった)(Roche)、100 Uのターミナルトランスフェラーゼ(Invitrogen)。反応を37℃で6時間インキュベーションし、そして次いで、最終濃度5 mMでEDTAを添加することによって停止した。Centri-spin-20カラム(Princeton Separations)を用いて脱塩した後、30 mMトリエタノールアミン/トリシン緩衝液(25℃でpH7.9)を用いて分離用7 M尿素/18%ポリアクリルアミドゲル電気泳動によってP*を精製した(Maniatisら、1982;Liuら、1999b)。260 nmのUV吸光度によって、回収されたP*の量を測定した。
[0288]それぞれ、P*1およびO1,P*2およびO2、並びにP*1およびP2*を用いたPAPを調べた(図18Aおよび表6)。P*は長さ30または35ヌクレオチドであり、そして3'末端にアシクロヌクレオチドまたはジデオキシヌクレオチドを含有した。
オリゴヌクレオチドのリスト
[0292]上述のように、遺伝子操作されたDNAポリメラーゼであるTaqFS(InnisおよびGelfand、1999)は、PAPの効率を非常に改善した。3'末端ジデオキシヌクレオチドでブロッキングされたP*は、ピロホスフェート(PPi)およびアレルテンプレートの相補鎖の存在下で、3'末端ジデオキシヌクレオチドを除去する加ピロリン酸分解によって活性化可能である。次いで、活性化されたP*をDNA重合によって伸長可能である。
[0296]これらの結果は、サンガー配列決定で用いる2つのターミネーターをPAPのブロッカーとして使用可能であることを立証する。ターミネーターはまた、AIDSなどのウイルス疾病の療法、および癌療法としても記載されてきており、例えば3'-デオキシアデノシン(コルジセピン)、3'-アジド-3'-デオキシチミジン(AZT)、2',3'-ジデオキシイノシン(ddI)、2',3'-ジデオキシ-3'-チアシチジン(3TC)および2',3'-ジデヒドロ-2',3'-ジデオキシチミジン(d4T)がある。DNAポリメラーゼは、その三リン酸型を合成鎖に取り込むことが可能であり、そして取り込みは伸長の終結を引き起こす(GardnerおよびJack、1999;Chengら、1987;St.Clairら、1987;UenoおよびMitsuya、1997)。3'-アジド-3'-デオキシチミジン(AZT)、2',3'-ジデオキシ-3'-チアシチジン(3TC)および2',3'-ジデヒドロ-2',3'-ジデオキシチミジン(d4T)の一リン酸ヌクレオチドは、オリゴヌクレオチドの3'末端に位置した場合、HIV逆転写酵素またはその変異体による加ピロリン酸分解によって、除去可能である(Arionら、1998;Gotteら、2000;Meyerら、2000;Urbanら、2001)。これらの結果は、多様な種類のブロッカーに対する、そしてRNAテンプレートに対するPAPの適用を示す。
Bi-PAPによる非常に稀なアレルの検出
λファージDNAテンプレート
[0298]挿入された大腸菌の野生型lacI遺伝子を含有する野生型λファージDNAテンプレート(Kohlerら、1991)をStratageneから購入した。Maniatisら(1982)にしたがって、SCS-8大腸菌細胞に形質転換したλファージプラークから、3つの突然変異λファージDNAテンプレートを調製した。これらは、それぞれ、lacI遺伝子中、ヌクレオチド190位でAからTの突然変異、ヌクレオチド369でTからGの突然変異、そしてヌクレオチド369でTからCの突然変異を含有する。260 nmのUV吸光度によって、λファージDNAの量を測定した。
[0299]ターミナルトランスフェラーゼによって、オリゴデオキシヌクレオチドに3'末端ジデオキシヌクレオチドを付加した。混合物は、25μlの総体積を含有した:100 mMカコジル酸カリウム(pH7.2)、2.0 mM CoCl2、0.2 mM DTT、2 nMのオリゴヌクレオチド、2.4 mM 2',3'-ddNTP(ddNTPに対する3'-OH末端のモル比は1:30であった)(Roche)、100 Uのターミナルトランスフェラーゼ(Invitrogen)。反応を37℃で6時間インキュベーションし、そして次いで、最終濃度5 mMでEDTAを添加することによって停止した。Centri-spin-20カラム(Princeton Separations)を用いて脱塩した後、30 mMトリエタノールアミン/トリシン緩衝液(25℃でpH7.9)を用いて分離用7 M尿素/16%ポリアクリルアミドゲル電気泳動によってP*を精製した(Maniatisら、1982、Liuら、1999b)。260 nmのUV吸光度によって、回収されたP*の量を測定した。
[0301]lacI遺伝子のヌクレオチド190およびヌクレオチド369のBi-PAPアッセイを調べた。369位の上流のP*が42ヌクレオチドであることを除いて、P*は40ヌクレオチドであった。各P*は、3'末端に配列特異的ヌクレオチドを含有した。PAP反応混合物は、25μlの総体積を含有した:50 mM KCl、20 mM HEPES/NaOH(25℃でpH6.9)、10 mM (NH4)2SO4、1.5 mM MgCl2、各40μMの4種のdNTP(dATP、dTTP、dGTPおよびdCTP)、各0.1μMのP*、150μM Na4PPi、4%DMSO、1μCiの[α-32P]-dCTP(3000 Ci/mmol、Amersham)、1 UのAmpliTaqFS DNAポリメラーゼ(PE Applied Biosystems)、2000コピーのλファージDNAテンプレート、または別の箇所に記載したもの。周期条件は、92℃6秒間、68℃20秒間、および72℃20秒間で、全部で35周期であった。第一の周期前に、92℃1分間の変性工程を追加した。
[0303]突然変異産物を、同じサイズの野生型産物から区別するため、非変性SSCPゲル電気泳動を行った(Oritaら、1989)。2倍体積の装填緩衝液(7 M尿素および50%ホルムアミド)と反応物を混合し、煮沸し、そして氷上で迅速に冷却した。混合反応物10μl中の産物を、30 mMエタノールアミン/Capsco緩衝液(pH9.6)(Liuら、1999b)を用い、8%非変性PAGE-PLUS(Amresco)ゲルを通じて4℃で電気泳動した。ゲルを乾燥させ、そしてオートラジオグラフィーのため、Kodak X-OMATTM ARフィルムに曝露した。各増幅産物から3つまたは4つのバンドがゲル上に見られた。上の1つまたは2つのバンドは、かなりの量の増幅産物が存在した結果、電気泳動中、変性一本鎖セグメントがハイブリダイゼーションしたために二重染色されたDNAであった。増幅産物の濃度が増加すると、上部バンドの強度がさらに増加する。
[0304]遺伝子操作されたDNAポリメラーゼ、TaqFSは、PAPの効率を非常に改善した。劇的により高い効率のため、PAPの条件をさらに最適化し、数コピーのλファージDNAまたはヒトゲノムDNAテンプレートから、PAPが直接増幅することを可能にした。反応構成要素および熱周期措置を最適化し、最適化には、:i)dNTPに対するPPi比を本質的に一定に維持しながら、PPi濃度を減少させること、ii)低pH HEPES緩衝液(25℃でpH6.9)の使用、iii)(NH4)2SO3の添加、iv)TaqFS量の増加、およびv)より高いアニーリング温度が含まれる。
[0305]PAPは3.3×1011:1の選択度を有しうる(図19)。この潜在能力に近づくには、混乱させるエラー原因を排除する設計が必要である。λDNAのlacI遺伝子のA190T突然変異をモデル系として用いる。1つの下流P*および1つのブロッキングされていない上流オリゴヌクレオチドを伴うPAPにおいて、ブロッキングされていない上流オリゴヌクレオチドからの伸長エラーは、目的の稀な突然変異を生成する可能性があり、したがって、選択度を減少させる可能性がある。TaqFSの誤った取り込みの率が、取り込まれるヌクレオチドあたり10-4であり、そしてありうる3つの誤った取り込みのうち1つが、新たに合成される上流鎖上にA→Tの突然変異を生成するならば、副次的な悪影響のため、選択度が、3.3×10-5に減少する。この限界を取り除くため、Bi-PAPが開発された(図20A)。Bi-PAPでは、下流および上流オリゴヌクレオチドがどちらも、3'末端で目的のヌクレオチドに特異的なP*である。P*は3'末端で1ヌクレオチド重複する。
[0307]Bi-PAPが非常に高い選択度を持つことを立証するため、Bi-PAP反応に、1010コピーより多いDNAテンプレートを用いた。λ DNA 1μgは2×1010ベクターゲノムを含有するが、ヒトゲノムDNA 1μgは3.3×105ゲノムしか含有しないため、大腸菌のlacI遺伝子を含有するλ DNAをモデル系として選択した。この実験室において、野生型λ DNAが混入している可能性を回避するため、突然変異P*を用いた突然変異特異的Bi-PAPアッセイを選択して野生型λ DNAを増幅した。λファージプラークに感染した大腸菌を調べることによって、野生型λ DNAにおけるlacI遺伝子の自発的突然変異の相対頻度は、10-9未満と概算される。
3つの突然変異特異的Bi-PAPアッセイの要約 a
blacI遺伝子において転写物の第一のヌクレオチドの位をヌクレオチド1位と指定する(Farabaugh、1978)。P*の3'ヌクレオチドは、示される位に位置し、そして対応する突然変異に相補的である。
Bi-PAPによるマウス組織における突然変異負荷の測定
材料および方法
[0313]10日齢〜25ヶ月齢のマウスの肝臓、心臓、脂肪組織、大脳および小脳を迅速凍結し、そして使用するまで液体窒素下に保存した。Big Blueプロトコル(Stratagene取り扱い説明書マニュアル)にしたがって、DNAを抽出した。簡潔には、組織をホモジナイズし、そしてプロテイナーゼKで消化した。ゲノムDNAをフェノール/クロロホルムで抽出し、そしてエタノール沈殿した。DNAをTE緩衝液(10 mM Tris/HCl、1 mM EDTA、pH8.0)に溶解し、そして4℃で保存した。260 nmのUV吸光度によって、マウスゲノムDNAの量を測定した。
[0316]トランスジェニックマウス突然変異検出系によって、in vivoで自発的突然変異または誘導された突然変異の頻度およびパターンを決定することが可能である。Big Blue(登録商標)系は、突然変異標的として、大腸菌lacI遺伝子を含有する、染色体に組み込まれたλファージDNAを宿するトランスジェニックマウスを用いる(GrossenおよびVijg、1993;Gossenら、1989;Kohlerら、1990)。lacI遺伝子は、40のタンデム反復されたλ DNA中、各マウス二倍体ゲノム内に組み込まれている。
Bi-PAPによって測定された体細胞突然変異体頻度
b反応総数に比較したT369G突然変異の陽性シグナル数の比。
c突然変異体がポワソン分布にしたがって反応物中に分布し、そして1以上の突然変異体が反応物中にある場合、増幅が陽性であり、そして突然変異体が反応物中にない場合、陰性であると仮定する式(反応あたりの突然変異体がゼロである頻度=e-x、xは反応あたりの突然変異体の平均数)を用いて、反応あたりのT369G突然変異体の平均数を概算する。
核酸を検出する方法であって:
(a)核酸に第一のオリゴヌクレオチドP*をアニーリングさせ、ここで第一のオリゴヌクレオチドP*は伸長不能3'端を有し、第一のオリゴヌクレオチドP*の3'伸長不能末端は加ピロリン酸分解によって除去可能である;
(b)加ピロリン酸分解によって第一のオリゴヌクレオチドP*の3'伸長不能末端を除去して、ブロッキングされていない第一のオリゴヌクレオチドを産生し;
(c)ブロッキングされていない第一のオリゴヌクレオチドを伸長し;
(d)伸長された第一のオリゴヌクレオチドを検出することによって、核酸の存在を検出する
ことを含んでなる、前記方法。
核酸ポリメラーゼを用いて加ピロリン酸分解を行う、態様1の方法。
態様3:
核酸ポリメラーゼを用いて伸長を行う、態様1の方法。
伸長工程で存在するヌクレオチドまたはヌクレオチド類似体が標識を含有し、そして伸長された第一のオリゴヌクレオチド中の標識の存在が検出される、態様1の方法。
工程(a)、(b)および(c)を反復する、態様1の方法。
態様6:
第一のオリゴヌクレオチドP*の3'伸長不能末端が、核酸ポリメラーゼによって伸長不能であるが、加ピロリン酸分解によって除去可能である、ヌクレオチドまたはヌクレオチド類似体である、態様1の方法。
ヌクレオチドまたはヌクレオチド類似体が、3'デオキシヌクレオチド、2',3'-ジデオキシヌクレオチド、アシクロヌクレオチド、3'-デオキシアデノシン(コルジセピン)、3'-アジド-3'-デオキシチミジン(AZT)、2',3'-ジデオキシイノシン(ddI)、2',3'-ジデオキシ-3'-チアシチジン(3TC)および2',3'-ジデヒドロ-2',3'-ジデオキシチミジン(d4T)からなる群より選択される、態様6の方法。
第二のオリゴヌクレオチドを核酸の相補鎖にアニーリングさせる、態様1の方法。
態様9:
第二のオリゴヌクレオチドが、ブロッキングされていない第二のオリゴヌクレオチドである、態様8の方法。
第二のオリゴヌクレオチドを工程(c)で伸長する、態様9の方法。
態様11:
第二のオリゴヌクレオチドが伸長不能3'端を有する第二のオリゴヌクレオチドP*であり、第二のオリゴヌクレオチドP*の3'伸長不能末端が加ピロリン酸分解によって除去可能である、態様9の方法。
第二のオリゴヌクレオチドP*の3'伸長不能末端を工程(b)の加ピロリン酸分解によって除去して、ブロッキングされていない第二のオリゴヌクレオチドを産生し、そしてブロッキングされていない第二のオリゴヌクレオチドを工程(c)で伸長する、態様11の方法。
第二のオリゴヌクレオチドP*の3'伸長不能末端が、核酸ポリメラーゼによって伸長不能であるが、加ピロリン酸分解によって除去可能である、ヌクレオチドまたはヌクレオチド類似体である、態様11の方法。
ヌクレオチドまたはヌクレオチド類似体が、3'デオキシヌクレオチド、2',3'-ジデオキシヌクレオチド、アシクロヌクレオチド、3'-デオキシアデノシン(コルジセピン)、3'-アジド-3'-デオキシチミジン(AZT)、2',3'-ジデオキシイノシン(ddI)、2',3'-ジデオキシ-3'-チアシチジン(3TC)および2',3'-ジデヒドロ-2',3'-ジデオキシチミジン(d4T)からなる群より選択される、態様13の方法。
第一のオリゴヌクレオチドP*の3'伸長不能末端が、核酸ポリメラーゼによって伸長不能であるが、加ピロリン酸分解によって除去可能である、ヌクレオチドまたはヌクレオチド類似体であり、そして第二のオリゴヌクレオチドP*の3'伸長不能末端が、核酸ポリメラーゼによって伸長不能であるが、加ピロリン酸分解によって除去可能である、ヌクレオチドまたはヌクレオチド類似体である、態様11の方法。
ヌクレオチドまたはヌクレオチド類似体が、3'デオキシヌクレオチド、2',3'-ジデオキシヌクレオチド、アシクロヌクレオチド、3'-デオキシアデノシン(コルジセピン)、3'-アジド-3'-デオキシチミジン(AZT)、2',3'-ジデオキシイノシン(ddI)、2',3'-ジデオキシ-3'-チアシチジン(3TC)および2',3'-ジデヒドロ-2',3'-ジデオキシチミジン(d4T)からなる群より選択される、態様15の方法。
核酸を合成する方法であって:
(a)核酸に、伸長不能3'端を有する第一のオリゴヌクレオチドP*をアニーリングさせ、ここで第一のオリゴヌクレオチドP*の3'伸長不能末端は加ピロリン酸分解によって除去可能である;
(b)加ピロリン酸分解によって、核酸にアニーリングした第一のオリゴヌクレオチドP*の3'伸長不能末端を除去し;そして
(c)核酸ポリメラーゼを用いて、ブロッキングされていない第一のオリゴヌクレオチドを伸長する
ことを含んでなる、前記方法。
工程(a)〜(c)を反復する、態様17の方法。
態様19:
核酸ポリメラーゼを用いて加ピロリン酸分解および伸長を行う、態様17の方法。
第二のオリゴヌクレオチドを、前記の第一の鎖の相補体である核酸鎖にアニーリングさせる、態様17の方法。
第二のオリゴヌクレオチドが、ブロッキングされていない第二のオリゴヌクレオチドである、態様20の方法。
第二のオリゴヌクレオチドを工程(c)で伸長する、態様21の方法。
態様23:
工程(a)〜(c)を反復する、態様22の方法。
第二のオリゴヌクレオチドが伸長不能3'端を有する第二のオリゴヌクレオチドP*であり、第二のオリゴヌクレオチドP*の3'伸長不能末端が加ピロリン酸分解によって除去可能である、態様20の方法。
工程(a)〜(c)を反復する、態様24の方法。
態様26:
第一のオリゴヌクレオチドP*の3'伸長不能末端が、核酸ポリメラーゼによって伸長不能であるが、加ピロリン酸分解によって除去可能である、ヌクレオチドまたはヌクレオチド類似体である、態様17の方法。
ヌクレオチドまたはヌクレオチド類似体が、3'デオキシヌクレオチド、2',3'-ジデオキシヌクレオチド、アシクロヌクレオチド、3'-デオキシアデノシン(コルジセピン)、3'-アジド-3'-デオキシチミジン(AZT)、2',3'-ジデオキシイノシン(ddI)、2',3'-ジデオキシ-3'-チアシチジン(3TC)および2',3'-ジデヒドロ-2',3'-ジデオキシチミジン(d4T)からなる群より選択される、態様26の方法。
第二のオリゴヌクレオチドP*の3'伸長不能末端が、核酸ポリメラーゼによって伸長不能であるが、加ピロリン酸分解によって除去可能である、ヌクレオチドまたはヌクレオチド類似体である、態様24の方法。
ヌクレオチドまたはヌクレオチド類似体が、3'デオキシヌクレオチド、2',3'-ジデオキシヌクレオチド、アシクロヌクレオチド、3'-デオキシアデノシン(コルジセピン)、3'-アジド-3'-デオキシチミジン(AZT)、2',3'-ジデオキシイノシン(ddI)、2',3'-ジデオキシ-3'-チアシチジン(3TC)および2',3'-ジデヒドロ-2',3'-ジデオキシチミジン(d4T)からなる群より選択される、態様28の方法。
第一のオリゴヌクレオチドP*の3'伸長不能末端が、核酸ポリメラーゼによって伸長不能であるが、加ピロリン酸分解によって除去可能である、ヌクレオチドまたはヌクレオチド類似体であり、そして第二のオリゴヌクレオチドP*の3'伸長不能末端が、核酸ポリメラーゼによって伸長不能であるが、加ピロリン酸分解によって除去可能である、ヌクレオチドまたはヌクレオチド類似体である、態様24の方法。
ヌクレオチドまたはヌクレオチド類似体が、3'デオキシヌクレオチド、2',3'-ジデオキシヌクレオチド、アシクロヌクレオチド、3'-デオキシアデノシン(コルジセピン)、3'-アジド-3'-デオキシチミジン(AZT)、2',3'-ジデオキシイノシン(ddI)、2',3'-ジデオキシ-3'-チアシチジン(3TC)および2',3'-ジデヒドロ-2',3'-ジデオキシチミジン(d4T)からなる群より選択される、態様30の方法。
核酸を検出する方法であって:
(a)核酸に2つのオリゴヌクレオチドP*をアニーリングさせ、ここで各オリゴヌクレオチドP*は伸長不能3'端を有し、各オリゴヌクレオチドP*の3'伸長不能末端は加ピロリン酸分解によって除去可能であり、一方のオリゴヌクレオチドP*ともう一方のオリゴヌクレオチドP*は、それぞれの3'端で、少なくとも1ヌクレオチド重複し、そして一方のオリゴヌクレオチドP*は第一の核酸鎖にアニーリングし、そしてもう一方のオリゴヌクレオチドP*は、第一の核酸鎖の相補体である核酸鎖にアニーリングする;
(b)加ピロリン酸分解によって、アニーリングした第一および第二のオリゴヌクレオチドP*の3'伸長不能末端を除去して、ブロッキングされていないオリゴヌクレオチドを産生し;
(c)ブロッキングされていないオリゴヌクレオチドを伸長し;そして
(d)伸長されたオリゴヌクレオチドを検出する
ことを含んでなる、前記方法。
核酸ポリメラーゼを用いて加ピロリン酸分解を行う、態様32の方法。
態様34:
核酸ポリメラーゼを用いて伸長を行う、態様32の方法。
核酸ポリメラーゼを用いて加ピロリン酸分解および伸長を行う、態様32の方法。
態様36:
伸長工程で存在するヌクレオチドまたはヌクレオチド類似体が標識を含有し、そして伸長されたオリゴヌクレオチド中の標識の存在が検出される、態様32の方法。
工程(a)、(b)および(c)を反復する、態様32の方法。
態様38:
各オリゴヌクレオチドP*の3'伸長不能末端が、核酸ポリメラーゼによって伸長不能であるが、加ピロリン酸分解によって除去可能である、ヌクレオチドまたはヌクレオチド類似体である、態様32の方法。
ヌクレオチドまたはヌクレオチド類似体が、3'デオキシヌクレオチド、2',3'-ジデオキシヌクレオチド、アシクロヌクレオチド、3'-デオキシアデノシン(コルジセピン)、3'-アジド-3'-デオキシチミジン(AZT)、2',3'-ジデオキシイノシン(ddI)、2',3'-ジデオキシ-3'-チアシチジン(3TC)および2',3'-ジデヒドロ-2',3'-ジデオキシチミジン(d4T)からなる群より選択される、態様38の方法。
核酸検出法であって:
(a)核酸からテンプレート核酸を合成し;
(b)テンプレート核酸に、伸長不能3'端を有する第一のオリゴヌクレオチドP*をアニーリングさせ、ここで第一のオリゴヌクレオチドP*の3'伸長不能末端は加ピロリン酸分解によって除去可能である;
(c)加ピロリン酸分解によって、テンプレート核酸にアニーリングした第一のオリゴヌクレオチドP*の3'伸長不能末端を除去し;そして
(d)ブロッキングされていないオリゴヌクレオチドを伸長する
ことを含んでなる、前記方法。
(i)1つの核酸鎖に、配列特異的である第一の相補オリゴヌクレオチドをアニーリングさせ、そして核酸ポリメラーゼを用いて、該核酸鎖上で、前記の第一の相補オリゴヌクレオチドを伸長して、伸長された第一のオリゴヌクレオチドを産生し、そして(ii)伸長された第一のオリゴヌクレオチドの3'末端に第二のオリゴヌクレオチドを付加して、テンプレート核酸を産生する、態様40の方法。
連結によって、第二のオリゴヌクレオチドを、伸長された第一の相補オリゴヌクレオチドに付加する、態様41の方法。
ターミナルトランスフェラーゼ伸長によって、第二の相補オリゴヌクレオチドを、伸長された第一の相補オリゴヌクレオチドに付加する、態様41の方法。
テンプレート核酸を増幅する工程(a1)をさらに含んでなる、態様41の方法。
態様45:
増幅がポリメラーゼ連鎖反応である、態様44の方法。
増幅が加ピロリン酸分解活性化重合による、態様44の方法。
態様47:
核酸コピーを合成する反応によって、テンプレート核酸を合成する、態様40の方法。
増幅反応が重合連鎖反応である、態様47の方法。
態様49:
増幅反応が加ピロリン酸分解活性化重合反応である、態様47の方法。
核酸ポリメラーゼを用いて加ピロリン酸分解を行う、態様40の方法。
態様51:
核酸ポリメラーゼを用いて伸長を行う、態様40の方法。
核酸ポリメラーゼを用いて加ピロリン酸分解および伸長を行う、態様40の方法。
態様53:
伸長工程で存在するヌクレオチドまたはヌクレオチド類似体が標識を含有し、そして伸長されたオリゴヌクレオチド中の標識の存在が検出される、態様52の方法。
工程(a)〜(d)を反復する、態様40の方法。
態様55:
オリゴヌクレオチドP*の3'伸長不能末端が、核酸ポリメラーゼによって伸長不能であるが、加ピロリン酸分解によって除去可能である、ヌクレオチドまたはヌクレオチド類似体である、態様40の方法。
ヌクレオチドまたはヌクレオチド類似体が、3'デオキシヌクレオチド、2',3'-ジデオキシヌクレオチド、アシクロヌクレオチド、3'-デオキシアデノシン(コルジセピン)、3'-アジド-3'-デオキシチミジン(AZT)、2',3'-ジデオキシイノシン(ddI)、2',3'-ジデオキシ-3'-チアシチジン(3TC)および2',3'-ジデヒドロ-2',3'-ジデオキシチミジン(d4T)からなる群より選択される、態様55の方法。
工程(b)で、第二のオリゴヌクレオチドをテンプレート核酸にアニーリングさせる、態様40の方法。
第二のオリゴヌクレオチドが、工程(c)で伸長される、ブロッキングされていない第二のオリゴヌクレオチドである、態様57の方法。
第二のオリゴヌクレオチドが伸長不能3'端を有する第二のオリゴヌクレオチドP*であり、第二のオリゴヌクレオチドP*の3'伸長不能末端が、工程(c)の加ピロリン酸分解によって除去され、ブロッキングされていない第二のオリゴヌクレオチドが産生され、そしてブロッキングされていない第二のオリゴヌクレオチドが工程(c1)で伸長される、態様57の方法。
第二のオリゴヌクレオチドP*の3'伸長不能末端が、核酸ポリメラーゼによって伸長不能であるが、加ピロリン酸分解によって除去可能である、ヌクレオチドまたはヌクレオチド類似体である、態様59の方法。
ヌクレオチドまたはヌクレオチド類似体が、3'デオキシヌクレオチド、2',3'-ジデオキシヌクレオチド、アシクロヌクレオチド、3'-デオキシアデノシン(コルジセピン)、3'-アジド-3'-デオキシチミジン(AZT)、2',3'-ジデオキシイノシン(ddI)、2',3'-ジデオキシ-3'-チアシチジン(3TC)および2',3'-ジデヒドロ-2',3'-ジデオキシチミジン(d4T)からなる群より選択される、態様60の方法。
第二のオリゴヌクレオチドP*の3'伸長不能末端が、核酸ポリメラーゼによって伸長不能であるが、加ピロリン酸分解によって除去可能である、ヌクレオチドまたはヌクレオチド類似体である、態様59の方法。
ヌクレオチドまたはヌクレオチド類似体が、3'デオキシヌクレオチド、2',3'-ジデオキシヌクレオチド、アシクロヌクレオチド、3'-デオキシアデノシン(コルジセピン)、3'-アジド-3'-デオキシチミジン(AZT)、2',3'-ジデオキシイノシン(ddI)、2',3'-ジデオキシ-3'-チアシチジン(3TC)および2',3'-ジデヒドロ-2',3'-ジデオキシチミジン(d4T)からなる群より選択される、態様62の方法。
第一のオリゴヌクレオチドP*の3'伸長不能末端が、核酸ポリメラーゼによって伸長不能であるが、加ピロリン酸分解によって除去可能である、ヌクレオチドまたはヌクレオチド類似体である、態様59の方法。
ヌクレオチドまたはヌクレオチド類似体が、3'デオキシヌクレオチド、2',3'-ジデオキシヌクレオチド、アシクロヌクレオチド、3'-デオキシアデノシン(コルジセピン)、3'-アジド-3'-デオキシチミジン(AZT)、2',3'-ジデオキシイノシン(ddI)、2',3'-ジデオキシ-3'-チアシチジン(3TC)および2',3'-ジデヒドロ-2',3'-ジデオキシチミジン(d4T)からなる群より選択される、態様64の方法。
核酸が、切断されている核酸である、態様40の方法。
態様67:
核酸が損傷またはニックを有する二本鎖核酸である、態様40の方法。
核酸テンプレート鎖上で、所望の核酸鎖を合成する、加ピロリン酸分解活性化重合法であって
(a)3'末端に伸長不能3'-デオキシヌクレオチドを有し、そしてテンプレート鎖上の対応するヌクレオチドに関してミスマッチを有する、活性化可能オリゴヌクレオチドP*を、テンプレート鎖にアニーリングさせ、
(b)ピロホスフェート、および加ピロリン酸分解活性を有し、そして末端3'-デオキシヌクレオチドの除去によってオリゴヌクレオチドP*を活性化する酵素を用いて、生じた二重鎖を加ピロリン酸分解し、そして
(c)4種のヌクレオシド三リン酸および核酸ポリメラーゼの存在下で、テンプレート鎖上、活性化されたオリゴヌクレオチドP*を伸長することによって重合させ、所望の核酸鎖を合成する
ことを連続的に含んでなる、前記方法。
(d)テンプレート鎖から工程(c)の所望の核酸鎖を分離し、そして
(e)所望の核酸鎖の所望の増幅レベルが達成されるまで、工程(a)〜(d)を反復する
ことによる、所望の核酸鎖の増幅を含む、態様68の加ピロリン酸分解活性化重合法。
第二のオリゴヌクレオチドの存在下で行う、態様69の加ピロリン酸分解活性化重合法であって、工程(a)において、第二のオリゴヌクレオチドが、工程(d)の分離された所望の核酸鎖産物にアニーリングし、そして工程(c)には、所望の核酸鎖上で第二のオリゴヌクレオチドを伸長することによって重合させて、核酸テンプレート鎖のコピーを合成することが含まれ、そして工程(d)には、増幅が指数関数的であるように、所望の核酸鎖から、合成された核酸テンプレート鎖を分離することが含まれる、前記方法。
活性化可能オリゴヌクレオチドP*およびテンプレート鎖間のミスマッチが、テンプレート上の対応するヌクレオチドに関して生じる、態様70の加ピロリン酸分解活性化重合法。
工程(a)〜(c)を、サーモサイクラー上、2以上の温度段階として、連続して行う、態様69の加ピロリン酸分解活性化重合法。
Claims (24)
- 核酸検出法であって:
(a)核酸からテンプレート核酸を合成し、ここで前記テンプレート核酸は以下の:
(i)1つの核酸鎖に、配列特異的である第一の相補オリゴヌクレオチドをアニーリングさせ、そして核酸ポリメラーゼを用いて、該核酸鎖上で、前記の第一の相補オリゴヌクレオチドを伸長して、伸長された第一のオリゴヌクレオチドを産生する;そして
(ii)伸長された第一の相補オリゴヌクレオチドの3'末端に第二のオリゴヌクレオチドを付加してテンプレート核酸を産生する;
ことにより合成する;
(b)テンプレート核酸に、伸長不能3'端を有する第一のオリゴヌクレオチドP*および第二のオリゴヌクレオチドP*をアニーリングさせ、ここでそれぞれの3'伸長不能末端は加ピロリン酸分解によって除去可能である;
(c)加ピロリン酸分解によって、テンプレート核酸にアニーリングした第一のオリゴヌクレオチドP*および第二のオリゴヌクレオチドP*のそれぞれの3'伸長不能末端を除去して、ブロッキングされていない第一のオリゴヌクレオチドおよびブロッキングされていない第二のオリゴヌクレオチドを産生し;そして
(d)ブロッキングされていない第一のオリゴヌクレオチドおよびブロッキングされていない第二のオリゴヌクレオチドを伸長する;
ことを含んでなる、前記方法。 - 連結によって、伸長された第一の相補オリゴヌクレオチドに第二のオリゴヌクレオチドを付加する、請求項1に記載の方法。
- ターミナルトランスフェラーゼ伸長によって、伸長された第一の相補オリゴヌクレオチドに第二のオリゴヌクレオチドを付加する、請求項1に記載の方法。
- テンプレート核酸を増幅する工程(a1)をさらに含んでなる、請求項1〜3のいずれか1項記載の方法。
- 増幅がポリメラーゼ連鎖反応である、請求項1〜3の記載の方法。
- 増幅が加ピロリン酸分解活性化重合による、請求項4に記載の方法。
- テンプレート核酸が、核酸のコピーを合成する反応により合成される、請求項1に記載の方法。
- 核酸ポリメラーゼを用いて加ピロリン酸分解を行う、請求項1〜7のいずれか1項に記載の方法。
- 核酸ポリメラーゼを用いて伸長を行う、請求項1〜7のいずれか1項に記載の方法。
- 伸長工程で存在するヌクレオチド又はヌクレオチド類似体が標識を含有し、そして伸長されたオリゴヌクレオチド中の標識の存在が検出される、請求項1に記載の方法。
- 工程(a)〜(d)を反復する、請求項1に記載の方法。
- 核酸が、切断されている核酸である、請求項1〜11のいずれか1項に記載の方法。
- 核酸が損傷又はニックを有する二本鎖核酸である、請求項1〜11のいずれか1項に記載の方法。
- それぞれのオリゴヌクレオチドP*の3'伸長不能末端が、核酸ポリメラーゼによって伸長不能であるが、加ピロリン酸分解によって除去可能である、ヌクレオチド又はヌクレオチド類似体である、請求項1記載の方法。
- ヌクレオチド又はヌクレオチド類似体が、3'デオキシヌクレオチド、2',3'-ジデオキシヌクレオチド、アシクロヌクレオチド、3'-デオキシアデノシン(コルジセピン)、3'-アジド-3'-デオキシチミジン(AZT)、2',3'-ジデオキシイノシン(ddI)、2',3'-ジデオキシ-3'-チアシチジン(3TC)及び2',3'-ジデヒドロ-2',3'-ジデオキシチミジン(d4T)からなる群より選択される、請求項14に記載の方法。
- 核酸を検出する方法であって:
(a)核酸に2つのオリゴヌクレオチドP*をアニーリングさせ、ここで各オリゴヌクレオチドP*は伸長不能3'端を有し、各オリゴヌクレオチドP*の3'伸長不能末端は加ピロリン酸分解によって除去可能であり、一方のオリゴヌクレオチドP*ともう一方のオリゴヌクレオチドP*は、それぞれの3'端で、少なくとも1ヌクレオチド重複し、そして一方のオリゴヌクレオチドP*は第一の核酸鎖にアニーリングし、そしてもう一方のオリゴヌクレオチドP*は、第一の核酸鎖の相補体である核酸鎖にアニーリングする;
(b)加ピロリン酸分解によって、アニーリングした第一及び第二のオリゴヌクレオチドP*の3'伸長不能末端を除去して、ブロッキングされていないオリゴヌクレオチドを産生し;そして
(c)オリゴヌクレオチドP*の3'伸長不能末端の除去を検出する;
ことを含んでなる、前記方法。 - オリゴヌクレオチドP*の3'伸長不能末端を標識し、そしてオリゴヌクレオチドP*の3'伸長不能末端の除去の検出を、オリゴヌクレオチドP*の標識化の減少を検出することにより行う、請求項16に記載の方法。
- (a)1またはそれ以上のヌクレオチドおよび核酸ハイブリッド中へのヌクレオチドの導入を触媒する酵素を使用して、ブロッキングされていないオリゴヌクレオチドを伸長する工程;そして(b)伸長されたオリゴヌクレオチドを検出することにより核酸の存在を検出する工程;により、オリゴヌクレオチドP*の3'伸長不能末端の除去の検出を行う、請求項16に記載の方法。
- 2つの反応、すなわち、活性化されていない場合にはオリゴヌクレオチドが核酸テンプレート上で伸長することを阻害する不活性オリゴヌクレオチドを活性化する反応である第一の反応、そして活性化オリゴヌクレオチドを核酸テンプレート上で伸長する反応である第二の反応、を連続的に組み合わせて含むプロセスであって、ここで不活性オリゴヌクレオチドは、核酸テンプレートと3'側適合を有し、そして不活性オリゴヌクレオチドが核酸代謝酵素により活性化される、前記プロセス。
- 不活性オリゴヌクレオチドが、3'末端ブロッキングされている、請求項19に記載のプロセス。
- 核酸代謝酵素が、トポイソメラーゼ、ヘリカーゼ、RNase H、テロメラーゼ、または逆転写酵素である、請求項19に記載のプロセス。
- 4種のヌクレオチド三リン酸および核酸ポリメラーゼの存在下にて、伸長を行う、請求項19に記載のプロセス。
- オリゴヌクレオチドが、第一の反応の前およびその間にテンプレートに対して少なくとも部分的にハイブリダイズする、請求項19に記載のプロセス。
- 伸長が、核酸テンプレートの増幅の一部である、請求項19に記載のプロセス。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US37909202P | 2002-05-10 | 2002-05-10 | |
| US60/379,092 | 2002-05-10 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004503654A Division JP4500673B2 (ja) | 2002-05-10 | 2003-05-09 | 加ピロリン酸分解活性化重合(pap) |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2010142232A true JP2010142232A (ja) | 2010-07-01 |
| JP2010142232A5 JP2010142232A5 (ja) | 2010-09-09 |
| JP5133969B2 JP5133969B2 (ja) | 2013-01-30 |
Family
ID=29420484
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004503654A Expired - Fee Related JP4500673B2 (ja) | 2002-05-10 | 2003-05-09 | 加ピロリン酸分解活性化重合(pap) |
| JP2009297339A Expired - Fee Related JP5133969B2 (ja) | 2002-05-10 | 2009-12-28 | 加ピロリン酸分解活性化重合(pap) |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004503654A Expired - Fee Related JP4500673B2 (ja) | 2002-05-10 | 2003-05-09 | 加ピロリン酸分解活性化重合(pap) |
Country Status (6)
| Country | Link |
|---|---|
| EP (2) | EP1509619B1 (ja) |
| JP (2) | JP4500673B2 (ja) |
| AT (1) | ATE552349T1 (ja) |
| AU (1) | AU2003241401B8 (ja) |
| CA (2) | CA2805053A1 (ja) |
| WO (1) | WO2003095664A2 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015519084A (ja) * | 2012-06-15 | 2015-07-09 | イラミーナ インコーポレーテッド | 核酸ライブラリーの動力学排除増幅 |
| JP2021524287A (ja) * | 2018-07-19 | 2021-09-13 | バイオフィデリティ・リミテッド | 改善されたポリヌクレオチド配列検出法 |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0406863D0 (en) * | 2004-03-26 | 2004-04-28 | Qiagen As | Nucleic acid sequencing |
| GB0406864D0 (en) * | 2004-03-26 | 2004-04-28 | Qiagen As | Nucleic acid sequencing |
| US7745125B2 (en) * | 2004-06-28 | 2010-06-29 | Roche Molecular Systems, Inc. | 2′-terminator related pyrophosphorolysis activated polymerization |
| CA2706999C (en) * | 2007-11-28 | 2013-01-22 | F. Hoffmann-La Roche Ag | Mutant dna polymerases with improved pyrophosphorolysis activated polymerization (pap) ability |
| CN101381766B (zh) * | 2008-06-11 | 2012-04-18 | 厦门大学 | 一种基因稀有突变的定量检测方法 |
| WO2012082753A2 (en) | 2010-12-13 | 2012-06-21 | Life Technologies Corporation | Polymerization of nucleic acids using activation by polyphosphorolysis (app) reactions |
| WO2012151328A2 (en) | 2011-05-02 | 2012-11-08 | President And Fellows Of Harvard College | Spatial sequestration of dynamic nucleic acid circuits |
| WO2013119888A1 (en) | 2012-02-09 | 2013-08-15 | President And Fellows Of Harvard College | Selective nucleic acid amplification from nucleic acid pools |
| US11261494B2 (en) * | 2012-06-21 | 2022-03-01 | The Chinese University Of Hong Kong | Method of measuring a fractional concentration of tumor DNA |
| CN103266103B (zh) * | 2012-11-08 | 2016-08-31 | 天津精耐特基因生物技术有限公司 | 用于扩增核糖核酸的方法及分析该方法中rna或dna模板的方法 |
| WO2015010020A1 (en) | 2013-07-18 | 2015-01-22 | President And Fellows Of Harvard College | Specific nucleic acid amplification with compounded selectivity |
| CN108410980B (zh) * | 2018-01-22 | 2022-02-01 | 华大数极生物科技(深圳)有限公司 | 筛选甲基化pcr检测的目标区域的方法、试剂盒及应用 |
| US11268140B2 (en) * | 2018-06-12 | 2022-03-08 | Shaofeng Ding | Delayed pyrophosphorolysis activated polymerization |
| GB201919186D0 (en) * | 2019-12-23 | 2020-02-05 | Biofidelity Ltd | Simplified polynucleotide sequence detection method |
| US11634754B2 (en) | 2021-04-15 | 2023-04-25 | Biofidelity Ltd. | Nucleic acid enrichment and detection |
| CN115308079B (zh) * | 2022-08-26 | 2024-03-29 | 攀钢集团重庆钒钛科技有限公司 | 一种实验室钛精矿酸解率的表征方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001062975A2 (en) * | 2000-02-23 | 2001-08-30 | City Of Hope | Pyrophosphorolysis activated polymerization (pap): application to allele-specific amplification and nucleic acid sequence determination |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA1340806C (en) | 1986-07-02 | 1999-11-02 | James Merrill Prober | Method, system and reagents for dna sequencing |
| US6268146B1 (en) * | 1998-03-13 | 2001-07-31 | Promega Corporation | Analytical methods and materials for nucleic acid detection |
| US20020048760A1 (en) * | 1999-12-10 | 2002-04-25 | Hyseq, Inc. | Use of mismatch cleavage to detect complementary probes |
| US20010049102A1 (en) * | 2000-02-24 | 2001-12-06 | Huang Xiaohua C. | Methods for determining single nucleotide variations |
-
2003
- 2003-05-09 EP EP03731135A patent/EP1509619B1/en not_active Expired - Lifetime
- 2003-05-09 CA CA2805053A patent/CA2805053A1/en not_active Abandoned
- 2003-05-09 CA CA2485596A patent/CA2485596C/en not_active Expired - Fee Related
- 2003-05-09 AU AU2003241401A patent/AU2003241401B8/en not_active Ceased
- 2003-05-09 JP JP2004503654A patent/JP4500673B2/ja not_active Expired - Fee Related
- 2003-05-09 AT AT03731135T patent/ATE552349T1/de active
- 2003-05-09 WO PCT/US2003/014556 patent/WO2003095664A2/en not_active Ceased
- 2003-05-09 EP EP10183576.7A patent/EP2292787B1/en not_active Expired - Lifetime
-
2009
- 2009-12-28 JP JP2009297339A patent/JP5133969B2/ja not_active Expired - Fee Related
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001062975A2 (en) * | 2000-02-23 | 2001-08-30 | City Of Hope | Pyrophosphorolysis activated polymerization (pap): application to allele-specific amplification and nucleic acid sequence determination |
Non-Patent Citations (2)
| Title |
|---|
| JPN6009008066; Nucleic Acids Research Vol.26,No.7, 1998, p.1807-1811 * |
| JPN6009008067; Nucleic Acids Research Vol.30,No.2, 200201, p.598-604 * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015519084A (ja) * | 2012-06-15 | 2015-07-09 | イラミーナ インコーポレーテッド | 核酸ライブラリーの動力学排除増幅 |
| JP2017060465A (ja) * | 2012-06-15 | 2017-03-30 | イラミーナ インコーポレーテッド | 核酸ライブラリーの動力学排除増幅 |
| JP2018139607A (ja) * | 2012-06-15 | 2018-09-13 | イラミーナ インコーポレーテッド | 核酸ライブラリーの動力学排除増幅 |
| US11254976B2 (en) | 2012-06-15 | 2022-02-22 | Illumina, Inc. | Kinetic exclusion amplification of nucleic acid libraries |
| JP2021524287A (ja) * | 2018-07-19 | 2021-09-13 | バイオフィデリティ・リミテッド | 改善されたポリヌクレオチド配列検出法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2485596C (en) | 2013-04-16 |
| AU2003241401B8 (en) | 2009-08-06 |
| EP2292787B1 (en) | 2017-03-22 |
| WO2003095664A3 (en) | 2004-04-08 |
| AU2003241401B2 (en) | 2007-11-08 |
| CA2805053A1 (en) | 2003-11-20 |
| AU2003241401A1 (en) | 2003-11-11 |
| ATE552349T1 (de) | 2012-04-15 |
| WO2003095664A2 (en) | 2003-11-20 |
| JP5133969B2 (ja) | 2013-01-30 |
| JP4500673B2 (ja) | 2010-07-14 |
| EP1509619A2 (en) | 2005-03-02 |
| EP2292787A1 (en) | 2011-03-09 |
| EP1509619B1 (en) | 2012-04-04 |
| EP1509619A4 (en) | 2007-07-11 |
| JP2005525121A (ja) | 2005-08-25 |
| CA2485596A1 (en) | 2003-11-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5133969B2 (ja) | 加ピロリン酸分解活性化重合(pap) | |
| US7919253B2 (en) | Serial coupling of nucleic acid metabolism and extension for nucleic acid amplification | |
| EP2112233B1 (en) | Pyrophosphorolysis activated polymerization (PAP): application to allele-specific amplification and nucleic acid sequence determination | |
| AU2001241621A1 (en) | Pyrophosphorolysis activated polymerization (PAP): application to allele-specific amplification and nucleic acid sequence determination | |
| Liu et al. | PAP: detection of ultra rare mutations depends on P* oligonucleotides:“sleeping beauties” awakened by the kiss of pyrophosphorolysis | |
| JP2007530026A (ja) | 核酸配列決定 | |
| Carvalho et al. | Optimization of a multiplex minisequencing protocol for population studies and medical genetics | |
| CA2911712C (en) | Pyrophosphorolysis activated polymerization (pap) | |
| US20030228596A1 (en) | Template-driven nucleic acid amplifications | |
| Lowe | ACID REPEAT SEQUENCES BY $8 $8 2i DISCONTINUOUS PRIMER EXTENSION |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100723 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20111104 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20120206 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20120209 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120213 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20120213 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20121016 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20121108 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20151116 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |