JP2010065080A - 共重合体の製造方法、共重合体、および該共重合体を用いた電解質 - Google Patents
共重合体の製造方法、共重合体、および該共重合体を用いた電解質 Download PDFInfo
- Publication number
- JP2010065080A JP2010065080A JP2008230138A JP2008230138A JP2010065080A JP 2010065080 A JP2010065080 A JP 2010065080A JP 2008230138 A JP2008230138 A JP 2008230138A JP 2008230138 A JP2008230138 A JP 2008230138A JP 2010065080 A JP2010065080 A JP 2010065080A
- Authority
- JP
- Japan
- Prior art keywords
- formula
- copolymer
- group
- integer
- represented
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- UHOVQNZJYSORNB-UHFFFAOYSA-N c1ccccc1 Chemical compound c1ccccc1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 2
- HKKGIHCPNBYMFD-UHFFFAOYSA-N C1=CC=[I]C=C1 Chemical compound C1=CC=[I]C=C1 HKKGIHCPNBYMFD-UHFFFAOYSA-N 0.000 description 1
- HHLMWQDRYZAENA-UHFFFAOYSA-N Nc(cc1)ccc1Oc1ccc(C(C(F)(F)F)(C(F)(F)F)c(cc2)ccc2Oc(cc2)ccc2N)cc1 Chemical compound Nc(cc1)ccc1Oc1ccc(C(C(F)(F)F)(C(F)(F)F)c(cc2)ccc2Oc(cc2)ccc2N)cc1 HHLMWQDRYZAENA-UHFFFAOYSA-N 0.000 description 1
- BEKFRNOZJSYWKZ-UHFFFAOYSA-N Nc1ccc(C(C(F)(F)F)(C(F)(F)F)c(cc2)ccc2N)cc1 Chemical compound Nc1ccc(C(C(F)(F)F)(C(F)(F)F)c(cc2)ccc2N)cc1 BEKFRNOZJSYWKZ-UHFFFAOYSA-N 0.000 description 1
- UVUCUHVQYAPMEU-UHFFFAOYSA-N Nc1cccc(C(C(F)(F)F)(C(F)(F)F)c2cccc(N)c2)c1 Chemical compound Nc1cccc(C(C(F)(F)F)(C(F)(F)F)c2cccc(N)c2)c1 UVUCUHVQYAPMEU-UHFFFAOYSA-N 0.000 description 1
Images




Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/30—Hydrogen technology
- Y02E60/50—Fuel cells
Landscapes
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
- Conductive Materials (AREA)
- Fuel Cell (AREA)
- Polyoxymethylene Polymers And Polymers With Carbon-To-Carbon Bonds (AREA)
Abstract
【解決手段】一般式(1)で表される化合物(A)と、一般式(2)で表される化合物(B)とを、触媒(C)の存在下で重合させることを特徴とする共重合体の製造方法。
得られた共重合体の35℃における還元粘度(0.5質量%濃度のm-クレゾール溶解溶液)が0.55〜1.5dL/gであり、GPCによる重量平均分子量が30000以上である。nは、3〜50である。
【選択図】図4
Description
ルホフェニル基を有する2価の芳香族ジアミンが使用されている。
アミン)を使用しているので、かかるジアミンからのイミド環の加水分解耐久性に問題があり、高温高加湿下で高分子鎖の切断が生じ、耐水性や強度などの機械的特性が低下してしまうことを見出した。スルホン酸基を含まない疎水性のポリイミドは、高温耐水性に優れ、また、製膜性、化学的耐久性、機械的特性に優れる。一方、スルホン化ポリアリーレンは、高分子鎖の加水分解耐久性に優れるが、製膜性や機械的特性に劣る傾向がある。そこで、スルホン化ポリアリーレンオリゴマーと疎水性ポリイミドオリゴマーからなるブロ
ック共重合体は、両成分の特徴を備えた優れた固体高分子電解質型燃料電池用の電解質膜材料として期待できることを見出した。
[1]一般式(1)で表される化合物(A)と、一般式(2)で表される化合物(B)とを
、触媒(C)の存在下で重合させることを特徴とする共重合体の製造方法。
は、3〜50の整数を示す。
、Z’は、直接結合、−O−、−S−、−CO−、−SO2−、−SO−、-C(CF3)2-のい
ずれかを示す。
子、アルキル基、環状アルキル基、芳香族基を示す。式(d)中、mは0〜4の整数を示す
。a、bは、0〜4の整数を示す。なお+はXへの結合端、*は窒素原子への結合端を示す。]
[2]得られた共重合体のゲルパーミエーションクロマトグラフィー法で測定したポリスチ
レン換算重量平均分子量が15,000〜1,000,000である[1]の共重合体の製
造方法。
[3]前記式(1)において、繰り返し単位数を示すnが5〜30である、[1]の共重合体の製造方法。
[4]化合物(A)と化合物(B)の混合物と触媒(C)とを混合する工程、およびその後に
再度触媒(C)を添加する工程を有する[1]〜[3]の共重合体の製造方法。
[5]化合物(B)に触媒(C)を添加する工程、およびその後に化合物(A)を添加する工
程を有する[1]〜[3]の共重合体の製造方法。
[6]一般式(1')で表される構造単位(A')と、一般式(2')で表される構造を有する構造単位(B')とを含む共重合体。
、3〜50の整数を示す。
Zは、−O−、−S−、直接結合、−CO−、−SO2−、−SO−、-C(CF3)2-、のいずれかを示す。
x1およびx2は0〜4の整数を示す(ただし、yが0である場合にはx1は1〜4であり
、yが1である場合にはx1とx2のいずれか一方は1以上である)。yは0〜1の整数、zは0〜3の整数を示す。]
れかを示す。
子、アルキル基、環状アルキル基、芳香族基を示す。式(d)中、mは0〜4の整数を示す
。a、bは、0〜4の整数を示す。なお+は式(1’)の左端又は右端に相当する結合端、*は窒素原子への結合端を示す。]
[7]構造単位(B’)が下記式(3’)で表されることを特徴とする請求項6に記載の共
重合体。
[8]得られた共重合体のゲルパーミエーションクロマトグラフィー法で測定したポリスチ
レン換算重量平均分子量が15,000〜1,000,000である[6]又は[7]の共重合体。
[9]一般式(1)において、繰り返し単位数を示すnが、5〜30である[6]〜[8]の共重
合体。
[10]前記[6]〜8のいずれかに記載の共重合体を含んでなる、固体高分子型燃料電池用の
電解質。
[11]前記[10]の電解質を用いて得られる、固体高分子型燃料電池用の電解質膜。
本発明では、下記化合物(A)と(B)とを触媒(C)存在下に反応させることで、共重
合体を製造する。
化合物(A)
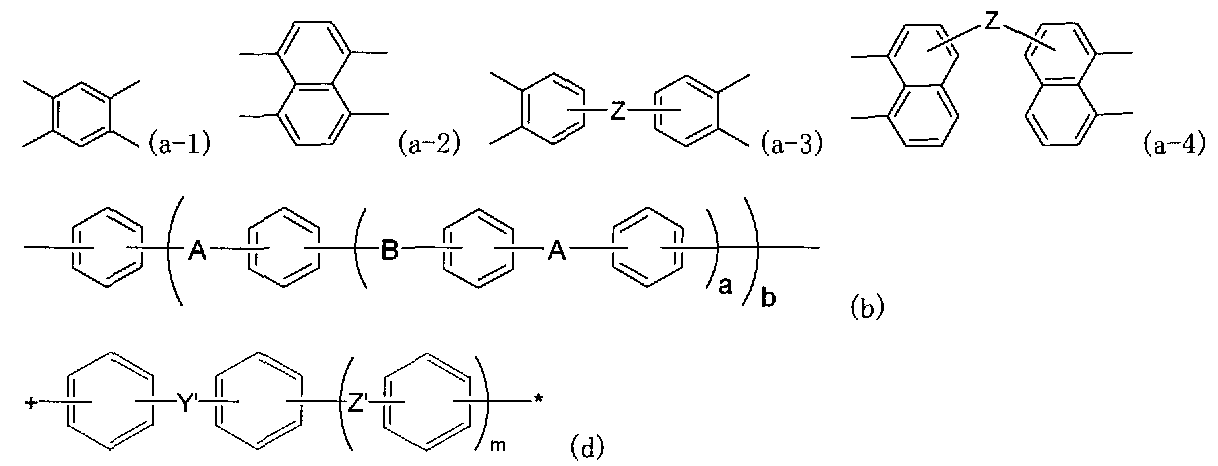
化合物(A)は一般式(1)で表される。
nは、3〜50の整数を示し、好ましくは5〜30である。このような繰り返し数であれば、高分子量の共重合体を得ることができる。
ホニル基、ベンゼンスルホニル基のいずれかを示す。
、Z’は、直接結合、−O−、−S−、−CO−、−SO2−、−SO−、-C(CF3)2-のい
ずれかを示す。
原子、フッ素原子、アルキル基、環状アルキル基、芳香族基を示す。a、bは、0〜4の整数を示す。
示す。
このような化合物として、特に本発明では下記式(1A)で表される化合物が好ましい。
Ar1は下記式(a-2)または(a-5)のいずれかで表される基であり、Ar2-1は下記式(b-1
)または(b-2)で表される基であり、Ar2-2は下記式(c-1)〜(c-3)のいずれかで表され
る基である。なお、式(1A)における、Ar2-1およびAr2-2は、いずれも式(1)のAr2の
定義に含まれる。
は窒素原子への結合端を示す。
を含むユニットの分率を示し、aおよびbは、ポリイミドオリゴマーに重合溶媒に対する溶解性および、オリゴマー鎖の疎水性に応じて、0〜1そして1〜0の範囲で決める。
上記化合物(A)は、次の2段階の反応で合成できる。まず式(1-a)で表される芳香族テ
トラカルボン酸類(好ましくは、テトラカルボン酸二無水物)と、下記式(1-b)で表され
る芳香族ジアミン化合物とを所定のn数になるように芳香族テトラカルボン酸類を所定量過剰に用いて反応させることにより、酸無水物末端のポリイミドオリゴマーを合成する。必要によりトリアミンや芳香族トリカルボン酸無水物等を併用することもできる。次に、酸無水物末端のポリイミドオリゴマーに、式(1-d)で表される芳香族モノアミン化合物を
反応させる。
NH2−Ar2−NH2 …(1-b)
X−Ar4−NH2 …(1-d)
(式中、X、Ar1〜Ar4は前記式(1)と同様である)
式(1-a)で表される芳香族テトラカルボン酸類としては、特に限定されるものではない
が、例えば、3,3',4,4'−ビフェニルテトラカルボン酸、2,3',3,4'−ビフェニルテトラカルボン酸、3,3',4,4'−ベンゾフェノンテトラカルボン酸、3,3',4,4'−ジフェニルエーテルテトラカルボン酸、ビス(3,4−ジカルボキシフェニル)メタン、2,2−ビス(3,4−ジ
カルボキシフェニル)プロパン、ピロメリット酸、1,4,5,8−ナフタレンテトラカルボン
酸、4,4'-ビナフチル-1.1',8,8'-テトラカルボン酸、3,4,9,10−ペリレンテトラカルボン酸、4,4'−(ヘキサフルオロイソプロピリデン)ジフタル酸、m−(ターフェニル)3,4,3",4"−テトラカルボン酸又はそれらの酸二無水物やエステル化物を挙げることができる
。このうち、1,4,5,8−ナフタレンテトラカルボン酸、4,4'-ビナフチル-1.1',8,8'-テト
ラカルボン酸が好ましい。
としては、4−ビス(4−アミノフェノキシ)ベンゼン、4−ビス(3−アミノフェノキシ)フェニルスルホン、2,2-[4−ビス(4−アミノフェノキシ)フェニル]ヘキサフロロプロパン、2,2-ジ(4−アミノフェノキシ)ヘキサフロロプロパン、2,2-ジ(3−アミノフェノキシ)ヘキサフロロプロパンなどを挙げることができる。
フェニル]ヘキサフロロプロパン、2,2-ジ(4−アミノフェノキシ)ヘキサフロロプロパン、2,2-ジ(3−アミノフェノキシ)ヘキサフロロプロパン(上記式(1A)で、Ar2-2を誘導す
るもの)を組合わせることも好ましい。
できる。
また、本発明では、ジアミンの他に3官能アミン(トリアミン)を使用することもできる。トリアミンを使用することで、分子中に分岐架橋構造を有するスルホン化芳香族ポリイミドとすることができる。トリアミンとしては、化合物としては、例えばトリメチルアミン、トリエチルアミンなど脂肪族アミンを使用してもよいが、ポリイミドの耐熱性の観点から、1,3,5‐トリス(4‐アミノフェノキシ)ベンゼン(TAPB)などの芳香族ア
ミンが好ましい。
水物も含む)を含んでいてもよい。
化合物(A)は公知のポリイミドの合成方法を適用することによって得ることができる。
例えば、クレゾールなどの極性溶媒中で、芳香族ジアミン(1-b)および(1-c)と芳香族テトラカルボン類(1-a)とを、必要に応じて、共沸溶媒としてトルエン又はキシレンなどを添
加し、50〜220℃に加熱し生成した水を除去しながら0.5〜100時間縮重合反応させる。また必要に応じて、安息香酸、イソキノリンなどを触媒として添加しても良い。
酸無水物末端のポリイミドオリゴマーに式(1-d)で表される芳香族モノアミン化合物を
添加し反応させる時、この芳香族モノアミンの添加量は酸無水物末端量の10モル%程度過剰が好ましい。
ゾフェノン、4-クロロ-4'-[ジトリフルオロメチル-(4-アミノフェニル)]メチルベンゾ
フェノン、4-クロロ-4'-[ジトリフルオロメチル-(4-アミノフェノキシ)フェニル]メチ
ルベンゾフェノンなどが例示される。
化合物(B)
本発明で使用される化合物(B)は、下記一般式(2)で表される芳香族スルホン酸系化
合物である。
ホニル基、ベンゼンスルホニル基のいずれかを示す。Yは、−CO−、−SO2−、−S
O−、―C(CF3)2−のいずれかを示す。Zは、直接結合、−O−、−S−、−CO−、−SO2−、−SO−、-C(CF3)2-のいずれかを示す。
ルカリ金属原子としては、リチウム、ナトリウム、カリウムなどが上げられる。また、脂肪族炭化水素基としては、炭素原子数4〜20のものが例示され、具体的にはtert−ブチル基、iso−ブチル基、n−ブチル基、sec−ブチル基、ネオペンチル基、シクロペンチル基、ヘキシル基、シクロヘキシル基、シクロペンチルメチル基、シクロヘキシルメチル基、アダマンチル基、アダマンチルメチル基、2−エチルヘキシル基、ビシクロ[2.2.1]ヘプチル基、ビシクロ[2.2.1]ヘプチルメチル基、テトラヒドロフルフリル基、2−メチルブチル基、3,3−ジメチル−2,4−ジオキソランメチル基、シクロヘキシルメチル基、アダマンチルメチル基、ビシクロ[2.2.1]ヘプチルメチル基などの直鎖状炭化水素基、分岐状炭化水素基、脂環式炭化水素基などが挙げられる。これらのうちn−ブチル基、ネオペンチル基、テトラヒドロフルフリル基、シクロペンチル基、シクロヘキシル基、シクロヘキシルメチル基、アダマンチルメチル基、ビシクロ[2.2.1]ヘプチルメチル基が好ましく、さらにはネオペンチル基が好ましい。
り、yが1である場合にはx1とx2のいずれか一方は1以上である)。
yは0〜1の整数、zは0〜3の整数を示す。
ェノンをスルホン酸化すれば、2,5−ジクロロ−(4’−スルホフェノキシ)ベンゾフェノンが得られる。
触媒(C)
本発明で使用される触媒としては、特に限定されないが、遷移金属化合物を含む触媒系が好ましい。この触媒系としては、遷移金属塩および配位子、または配位子が配位された遷移金属若しくはその塩(以下、「遷移金属(塩)」と記す。)、ならびに還元剤を必須成分とする。また、遷移金属塩以外の塩を添加することにより、重合速度を上げることができる。ここで、遷移金属塩としては、塩化ニッケル、臭化ニッケル、ヨウ化ニッケル、ニッケルアセチルアセトナートなどのニッケル化合物、塩化パラジウム、臭化パラジウム、ヨウ化パラジウムなどのパラジウム化合物、塩化鉄、臭化鉄、ヨウ化鉄などの鉄化合物、塩化コバルト、臭化コバルト、ヨウ化コバルトなどのコバルト化合物などが挙げられる。これらのうち特に、塩化ニッケル、臭化ニッケルなどが好ましい。また、配位子としては、トリフェニルホスフィン、2,2′−ビピリジン、1,5−シクロオクタジエン、1,3−ビス(ジフェニルホスフィノ)プロパンなどが挙げられるが、トリフェニルホスフィン、2,2′−ビピリジンが好ましい。上記配位子は、1種単独で、あるいは2種以上を併用することができる。
塩)が、上記モノマーの総計1モルに対し、通常、0.0001〜10モル、好ましくは0.01〜0.5モルである。0.0001モル未満では、重合反応が充分に進行せず、一方、10モルを超えると、分子量が低下するという問題がある。触媒系において、遷移金属塩および配位子を用いる場合、この配位子の使用割合は、遷移金属塩1モルに対し、通常、0.1〜100モル、好ましくは1〜10モルである。0.1モル未満では、触媒活性が不充分となり、一方、100モルを超えると、分子量が低下するという問題がある。また、触媒系における還元剤の使用割合は、モノマーの総計1モルに対し、通常、0.1〜100モル、好ましくは1〜10モルである。0.1モル未満では、重合が充分進行せず、一方、100モルを超えると、得られる重合体の精製が困難になるという問題がある。
触媒は、複数回に分けて添加してもよく、具体的には、予め調製した触媒に、化合物(A)および(B)を添加して反応させたのち、再度触媒を添加して、さらに重合をすすめてもよい。このように複数段にわけて触媒を添加すれば、重合の制御が容易となり、重合時間を短くできたり、共重合体の分子量を均一にできたり、また高分子量のものを得ることも可能となる。
重合条件
本発明では、前記化合物(A)と(B)とを触媒(C)存在下に反応させる。好ましくは
、化合物(A)及び化合物(B)の混合物と触媒(C)を混合することにより反応させる。また、化合物(A)と化合物(B)の混合物と触媒(C)を混合する工程およびその後に再度触媒(C)を添加する工程を有する重合方法を採用してもよいし、化合物(B)に触媒
(C)を添加する工程およびその後に化合物(A)を添加する工程を有する重合方法を採
用してもよい。
ことが望ましいので、通常、化合物(A)/(B)モル比が、1/5〜1/500の範囲であり、好ましくは1/10〜1/200の範囲にあることが望ましい。
反応濃度としては、各成分(A)および(B)の合計量が、3〜40 g/dlの範囲、好ましくは、15〜35 g/dlの範囲にあることが望ましい。
重合反応開始時の反応系では、化合物(B)のモル濃度が化合物Aのモル濃度より非常に大きく(10倍以上)、また化合物Bの反応性が高いので、まず化合物B同士が重合してBのオリ
ゴマーが生成し、それが化合物Aと順次反応していく。従って、得られた共重合体は、化
合物(A)および(B)のオリゴマーのブロック共重合体であり、化合物(B)のオリゴマーに由
来する構造単位が親水性ドメインをそしてポリイミドオリゴマーが疎水性ドメインを形成し、ミクロ相分離構造をとりやすいので、耐水性が高く、機械的特性、物理的耐久性も高く、かつプロトン伝導性も高い。
の芳香族プロトンのピークから、その構造を確認することができる。
[共重合体]
本発明にかかる共重合体は、下記一般式(1')で表される構造単位(A')と、一般式(2')で表される構造を有する構造単位(B')とを含み、下記式(C')で表されることを特徴とする。かかる共重合体は、上記製造方法で調製される。
物(B)から誘導される。
さらに、構造単位(B’)は、下記構造を有することが好ましい。
x1,x2,yおよびzは、式(2’)と同様である。
かかる共重合体は下記式(C')で表される。
k、Y、Z、R21,x1,x2,yおよびzは、式(2’)と同様である。cおよびdは、各構造単位のモル比を表す。]
構造単位(A')と(B')との組成比(すなわち式(C')中、cとdの比率)は、所望のプロトン伝導度やイオン交換容量によるが、通常、化合物(A)/(B)モル比が、1/5〜1/500の範囲であり、好ましくは1/10〜1/200の範囲にあることが望ましい。
なお、本発明では、前記構造単位(A')および(B')のほかに、本発明の効果を阻害しない範囲で少量の他の構造単位を含んでもよい。たとえば、特開2004-346163号公報に示さ
れるようなポリアリーレン構造単位を含んでいてもよい。このようなその他の構造単位は、全構造単位の10モル%以下で含まれていることが望ましい。
本発明の高分子固体電解質は、例えば一次電池用電解質、二次電池用電解質、燃料電池用プロトン伝導膜、表示素子、各種センサー、信号伝達媒体、固体コンデンサー、イオン交換膜などに利用可能である。
[プロトン伝導膜]
本発明のプロトン伝導膜は、上記スルホン酸基を有するポリイミド共重合体からなり、
かかるプロトン伝導膜を調製する際には、上記共重合体以外に、硫酸、リン酸などの無機酸、およびその塩、カルボン酸を含む有機酸、適量の水などを併用してもよい。
このような方法により得られるプロトン伝導膜は、その乾燥膜厚が、通常10〜100μm、好ましくは20〜80μmである。
本実施例で得られた共重合体は以下のようにして評価した。
評価方法
[分子量の測定]
共重合体の数平均分子量(Mn),重量平均分子量(Mw)は、溶媒にNMP緩衝溶液を用い、ゲルパーミエーションクロマトグラフィー(GPC:(東ソー(株)HCL-8220製))によって、ポ
リスチレン換算の分子量を求めた。NMP緩衝溶液は、NMP(3L)/リン酸(3.3mL)/臭化リチウム(7.83g)の比率で調製した。
[還元粘度]
還元粘度ηS P/cは、各実施例で得られた重合体を0.5wt%の濃度になるように1g/dLのLiCl含有NMPに溶解して、オストワルド粘度計を用いて35℃で測定した。
[イオン交換容量]
サンプルシートを15wt%食塩水に30℃で72時間浸漬し、溶出したプロトンをフェノールフ
タレイン指示薬を用いて、0.05MNaOH水溶液で滴定して求めた。
[1H-NMR]
溶媒として重水素化ジメチルスルホキシド(DMSO‐d6)を用いて、日本電子JEOL EX-270により測定した。
[吸水率]
膜サンプル約100mgを乾燥して乾燥重量Wdを測定した後、25℃で24時間水に浸
漬した。膜サンプルを水から取り出し、手早く表面に付着した水をティシュペーパーでふき取り、膨潤時の膜重量Wsを測定した。吸水率(Water uptake;WU)を次式から求めた。
WU(%)=(Ws‐Wd)/Wd×100
[サイズ変化]
直径2cmの円形サンプルシートを70%RH雰囲気に置き平衡後の厚み tsと直径lsを測る。つぎに、同じサンプルを25℃の水に5時間浸漬し、水中での厚み tと直径lを測る
。膜厚方向と膜面方向のサイズ変化、それぞれΔtc、Δlcを次式から求めた。
( Δtc = (t - ts) / ts
( Δlc = (l - ls) / ls
[耐水性試験]
・機械強度
膜厚30〜40μmの膜サンプルを130℃、飽和水蒸気中に100時間暴露した後、膜形状・強度の観点から、次の5段階で評価した。なおII〜Vで用いたフイルム片は、暴露
処理後、風乾し幅5mm長さ20mmの形状としたものである。
I: 膜形状を保持していない。膜が多くの小片に破れている。
II: フイルム片の両端をつかんで(つかみ代が5mm)、折り曲げると膜が破断。
III: 折り目の角度が0°となるようにフィルム片を、折り目をつけて曲げると破断。
IV: 折り目を付けて曲げても破断しないが、もとに曲げ戻すと破断。
V: 折り目を付けて曲げても、さらに曲げ戻しても破断せず。
・プロトン伝導度
また、飽和水蒸気暴露処理した膜を風乾後、60℃、100〜50%RHでプロトン伝導度を測定し、プロトン伝導度の観点から、次の3段階で評価した。
a:処理によりプロトン伝導度は20%以上低下
b:5〜20%低下
c:実験誤差(±5%)範囲内(変化なし)
[プロトン伝導度]
プロトン伝導度測定セルに膜シート(1.0cm×0.5cm)と4枚の白金黒電極板をとりつけ、温度制御した水中又は温度・湿度制御したチャンバー内にセットし、日置電気(株)製のLCRメーター(HIOKI3552-80)を用いて、100Hzから100kHzの周波数範囲で複素インピーダンス法により電気抵抗Rを測定し、プロトン伝導度σを次式から計算した。なお、表1での温度は60℃とした。
σ=d/(ts ws R)
ここで、dは2電極間距離(0.5cm)、tsとwsは、室温で70%RHにおける膜シートの厚さと幅である。水中でのプロトン伝導度の計算には、水中でのtsとws値を用いた。
[合成例1]
ポリイミドオリゴマー(A−1)の合成
窒素気流下で、乾燥した三口フラスコ中でビス[4-(3−アミノフェノキシ)]フェニルスルホン(BAPPS)8.650g(20.0mmol)と安息香酸5.13(42.0mmol)をm−クレゾール(125
ml)に溶解させ、次いでナフタレン−1,4,5,8−テトラカルボン酸二無水物(NTDA)8.050g(30.0mmol)を加え、80℃で4時間攪拌する。その後、イソキノリン5.42g(42.0mmol)を加え、さらに180℃で18時間反応し、酸無水物末端のポリイミドオリゴ
マー溶液を得た。この溶液に4−クロロ−4’−(4−アミノフェノキシ)ベンゾフェノン(CAPBP)7.12g(22.0mmol)を加え、80℃で4時間、さらに180℃で18時間反応させ、重合反応液を冷却後、アセトン中に加えた。得られた個体をアセトン洗浄後
、N-メチルピロリドン(NMP)に溶かし、アセトン中に加え、再沈精製することにより、
式(A−1)で表されるポリイミドオリゴマーを得た。nは約3であった。
ポリイミドオリゴマー(A−2)の合成
NTDA2.145g(8.0mmol)、BAPPS3.027g(7.0mmol)、CAPBP 0.712g(2.2mmol)、安息香酸1.37g(11.2mmol)、イソキノリン1.45g(11.2mmol)とm−クレゾール(33ml)を用いた以外は、合成例1と同様にして、ポリイミドオリゴマー(A−2)を得た。nは約8であった。
ポリイミドオリゴマー(A−3)の合成
NTDA10.995g(41.0mmol)、BAPPS16.867g(39.0mmol)、CAPBP 1.439g(4.4mmol)、安
息香酸7.01g(57.4mmol)、イソキノリン7.41g(57.4)とm−クレゾール(160ml)を用
いた以外は、合成例1と同様にして、ポリイミドオリゴマー(A−3)を得た。nは約20であった。
[合成例4]
スルホン化モノマー(B−1)の合成
2,5−ジクロロ安息香酸10.000g(52.4mmol)を塩化チオニル30 ml中で、80℃、6
時間還流し反応した後、真空蒸留精製により2,5−ジクロロ安息香酸クロライド9.210
g(44.0mmol)を得た。
冷却し、2,5−ジクロロ安息香酸クロライド 2.090g(10.0mmol)とベンゼン20mlの溶液を滴下し、1.5時間攪拌した。さらに25℃で10h反応した後、反応液を希塩酸水溶液に加え、有機相を分離した。有機相を食塩水溶液で洗浄後、溶媒を留去して得られた固体をエタノールから再結晶して、2,5−ジクロロベンゾフェノン 2.080(8.30mol)g
(収率83%)を得た。
2,5−ジクロロベンゾフェノン10.000g( 40.0mmol)を、濃硫酸(98%)15mlに溶かす。これに発煙硫酸(60%) 15mlをゆっくり加えて0℃で5時間、さらに60℃で12時
間攪拌した。反応液を冷却後、氷水300gに加え、食塩約50gを加え、塩析した。析
出した固体を水300gに溶かし、10%NaOH水溶液で中和後、食塩約50gを加え、塩析した。析出した固体を熱水から2回再結晶することにより3−(2,5−ジクロロベン
ゾイル)ベンゼンスルホン酸ナトリウム9.420g(26.7mmol)(収率67%)を得た。
スルホン化ポリフェレンオリゴマー(B−2)の合成
窒素置換したグローブボックス中で反応フラスコにジメチルアセトアミド(DMAc) 40ml、臭化ニッケル(II) 0.173g(0.8mmol)、トリフェニルホスフィン 1.560g(6.0mmol)、亜鉛 3.111g(47.6mmol)を加え、80℃で10分攪拌し、触媒を調整した。これに、3−(2,5−ジクロロベンゼゾイル)ベンゼンスルホン酸ナトリウム 4.000g(11.3mmol)を加え、80℃で6時間攪拌した。重合反応溶液を10wt%塩酸水溶液200mlに加え、一晩攪拌後、ろ過し、固体を得た。得られた固体をアセトンで洗浄し、側鎖型スルホン化ポリフェレンオリゴマー(B−2)を得た。GPC分析の結果、mは62であった。
スルホン化モノマー(B−3)の合成
丸底フラスコにジフェニルエーテル 17.87g(105mmol)と1,2-ジクロロエタン180m
lを加え溶かし、氷浴で冷却する。これに、2,5−ジクロロ安息香酸クロライド 20.90g(100mmol)と塩化アルミニウム 14.67g(110mmol)を1,2-ジクロロエタン210mlに溶
かした溶液を滴下し、0℃で1.5時間攪拌した。さらに25℃で10h反応した後、反応液を希塩酸溶液に加え、有機相を分離した。有機相を食塩水溶液で洗浄後、溶媒を留去して得られた固体をエタノールから再結晶を行うことにより2,5−ジクロロ−(4‘−フェノキシ)ベンゾフェノン 29.77g(87.0mmol)(収率87%)を得た。
時間攪拌した。反応溶液を氷水に加え、水酸化ナトリウムで中和した。析出した固体をろ過し、水から再結晶することにより2,5−ジクロロ−(4’−スルホフェノキシ)ベンゾフェノン(8.07g(18.0mmol)(収率62%)を得た。
スルホン化ポリフェレンオリゴマー(B−4)の合成
窒素置換したグローブボックス中で反応フラスコにDMAc20ml、臭化ニッケル(II) 0.069g(0.3mmol)、トリフェニルホスフィン0.618g(2.4mmol)、亜鉛1.232g(18.8mmol)
を加え、80℃で10分攪拌し、触媒を調整した。これに、2,5−ジクロロ−(4’−
スルホフェノキシ)ベンゾフェノン2.000g(4.5mmol)を加え、80℃で6時間攪拌した。重合反応溶液を10wt%塩酸水溶液200mlに加え、一晩攪拌後、ろ過し、固体を得た。得られた固体をアセトンで洗浄し、側鎖型スルホン化ポリフェレンオリゴマー(B−4)を得た。GPC分析結果からm=約62と見積もった。得られた側鎖型スルホン化ポリフェレンオリゴマー(B−4)のFT−IRスペクトルの結果を図3に示す。
窒素置換したグローブボックス中で反応フラスコにNMP5ml、臭化ニッケル(II)0.128g(0.60mmol)、トリフェニルホスフィン1.152g(4.4mmol)、亜鉛2.300g(35mmol)を加え、
80℃で10分攪拌し、触媒を調整した。これに、スルホン化モノマー(B−1)1.340g(3.80mmol)とポリイミドオリゴマー(A−1)1.000g(0.41mmol)を18mlのNMPに溶かした溶液を加え、80℃で39時間攪拌した。重合反応溶液を10wt%塩酸水溶液200ml
に加え、一晩攪拌後、ろ過し、固体を得た。得られた固体をアセトンで洗浄し乾燥させた。これをNMPに再溶解し、アセトンで再沈殿させた後、熱エタノール(78℃)で抽出し
、可溶部分(スルホン化モノマーB-1のオリゴマー)を除去することにより、スルホン化
ポリフェレンオリゴマーとポリイミドオリゴマーとの共重合ポリマー(C−1)を得た。
窒素置換したグローブボックス中で反応フラスコにNMP5ml、臭化ニッケル(II)0.083g(0.38mmol)、トリフェニルホスフィン0.700g(2.7mmol)、亜鉛1.410g(21.5mmol)を加え
、80℃で10分攪拌し、触媒を調製した。これに、スルホン化モノマー(B−1)1.734g(4.91mmol)とポリイミドオリゴマー(A−1)1.275g(0.52mmol)を10mlのNMPに溶かした溶液を加え、80℃で3時間攪拌した。さらに、7mol%の触媒(臭化ニッケル(II)0.083g(0.38mmol)、トリフェニルホスフィン0.700g(2.7mmol)、亜鉛1.410g(21.5mmol)、NMP5ml)を追加添加し、12時間更に重合させた。重合反応溶液を10wt%塩酸水溶液200mlに加え、一晩攪拌後、ろ過し、固体を得た。得られた固体をアセトンで洗浄し乾燥
させた。これを熱エタノールで抽出し、可溶部分(スルホン化モノマー(B-1)のオリゴ
マー)を除去することにより、スルホン化ポリフェレンオリゴマーとポリイミドオリゴマーとの共重合ポリマー(C−2)を得た。
[実施例3]
表2に従って反応条件を変更した以外は、実施例1と同様にして共重合体(C−3)を得た。
[実施例4]
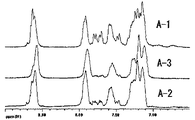
表2に従って反応条件を変更した以外は、実施例1と同様にして共重合体(C−4)を得た。得られた共重合体(C−4)の1H-NMRスペクトルを図4に示す。
表2に従って反応条件を変更した以外は、実施例2と同様にして共重合体(C−5)を得た。
表2に従って反応条件を変更した以外は、実施例7および10は実施例1と、実施例6および8、9は実施例2と同様にして共重合体(C−6〜C−10)を得た。実施例10で得られた共重合体(C−10)の1H-NMRスペクトルを図5に示す。
[実施例11]
窒素置換したグローブボックス中で反応フラスコにNMP2ml、臭化ニッケル(II)0.094g(0.43mmol)、トリフェニルホスフィン0.789g(3.0mmol)、亜鉛1.591g(24.3mmol)を加え
、80℃で10分攪拌し、触媒を調整した。これに、スルホン化モノマー(B−1)2.150g(6.09mmol)を5mlのNMPに溶かした溶液を加え、80℃で20分攪拌し反応させた後、
ポリイミドオリゴマー(A−3)0.850g(0.060mmol)をNMP3mlに溶かした溶液を加え2時間反応させた。さらに、7mol%の触媒(NMP5ml、臭化ニッケル(II)0.094g(0.43mmol)、トリフェニルホスフィン0.789g(3.0mmol)、亜鉛1.591g(24.3mmol)、NMP 2ml)を追加
添加し、5時間重合させた。重合反応溶液を10wt%塩酸水溶液300mlに加え、一晩
攪拌後、ろ過し、固体を得た。得られた固体をアセトンで洗浄し乾燥させた。これを熱水でソクスレー抽出し、可溶部分(スルホン化モノマーB-1のオリゴマー)を除去すること
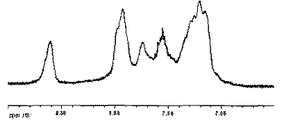
により、スルホン化ポリフェレンオリゴマーとポリイミドオリゴマーとの共重合ポリマー(C−11)を得た。得られた共重合体C−11の1HNMRスペクトルを図6に示す。
表2に従って反応条件を変更した以外は、実施例11と同様にして共重合体(C−12〜C−13)を得た
[比較例1]
窒素置換したグローブボックス中で反応フラスコにNMP5ml、臭化ニッケル(II)0.033g(0.15mmol)、トリフェニルホスフィン0.275g(1.1mmol)、亜鉛0.589g(9.0mmol)を加え、80℃で10分攪拌し、触媒を調整した。これに、スルホン化ポリフェレンオリゴマー(
B−4)1.800g(0.11mmol)とポリイミドオリゴマー(A−1)1.800g(0.7mmol)をNMP13mlに溶かした溶液を加え、80℃で30時間攪拌し重合させた。重合反応溶液を10wt%塩酸水溶液200mlに加え、一晩攪拌後、ろ過し、固体を得た。得られた固体をアセトン
で洗浄し乾燥させた。熱エタノールで抽出し、可溶部分(スルホン化モノマーB-2および
そのポリマー)を除去することにより、スルホン化ポリフェレンとポリイミドオリゴマーとの共重合ポリマー(C−14)を得た。
[比較例2]
窒素置換したグローブボックス中で反応フラスコにNMP5ml、臭化ニッケル(II)0.030g(0.14mmol)、トリフェニルホスフィン0.280g(1.1mmol)、亜鉛0.550g(8.4mmol)を加え、80℃で10分攪拌し、触媒を調整した。これに、スルホン化ポリフェレンオリゴマー(
B−4)1.360g(0.06mmol)とポリイミドオリゴマー(A−1)1.800g(0.7mmol)をNMP26mlに溶かした溶液を加え、80℃で72時間攪拌し重合させた。重合反応溶液を10wt%塩酸水溶液200mlに加え、一晩攪拌後、ろ過し、固体を得た。得られた固体をアセト
ンで洗浄し乾燥させた。熱エタノールで抽出し、可溶部分(スルホン化モノマーB-4およびそのポリマー)を除去することにより、スルホン化ポリフェレンオリゴマーとポリイミドオリゴマーとの共重合ポリマー(C−15)を得た。実施例をおよび比較例で得られた共重合体のイオン交換容量、還元粘度および分子量を表2に示す。
分子量の共重合体を得ることが可能である。すなわち、比較例1、2に示される合成方法では、スルホン化オリゴマー、すなわち、(B−2)あるいは(B−4)が、ポリイミドユニット、すなわち(A−1)と共重合が十分に起こっておらず、(B-2)或いは(B-4
)が単独重合したスルホン化ポリアリーレンが生成しており、それが、ポリマーの精製の過程で溶媒に溶出し設定通りのIECを有するポリマーが得られていない。一方、一般式(2)に示されるモノマーと一般式(1)に示されるポリイミドオリゴマーを用いてブロック共重合体を得る製造方法では、設定したIECと近いポリマーが得られており、これは、共重合が十分に起こっていることを示唆している。そのため、高分子量体が得られ還元粘度が高いポリマーを得ることが出来る。したがって、一般式(2)に示されるモノマーと一般式(1)に示されるポリイミドオリゴマーを用いてブロック共重合体を得る製造方法は、設定通りのイオン交換容量を有し、かつ、より高分子量の共重合体を得る製造方
法として非常に有効な方法である。
[評価試料の調製]
上記各実施例・比較例で得られた共重合体を電解質として、NMPに10wt%で溶解し、
ガラスシャーレ上に塗布し、120℃で16時間乾燥させることにより電解質膜を得た。作成
した電解質膜の水浸漬による吸水率およびサイズ変化プロトン伝導度および130℃飽和水
蒸気に100時間暴露処理による高温耐水性試験結果を表3に示す。
。
実施例4および実施例13において、イオン交換容量が1.9meq/g程度で、吸水率が70
%で比較的低いサイズ変化を示す膜が得られた。この膜は、60℃で50%RHの低湿度で13 mS/cm程度の高いプロトン伝導性を示し、これは、スルホン化芳香族炭化水素系高分
子電解質膜に比べて非常に高く、ナフィオン膜に匹敵する。また、水中での膜膨潤に伴うサイズ変化は、膜厚方向に比べて、膜面方向に小さい異方性を示し、膜電極接合体の接合安定性の点からも好ましい。
のGPC分析による分子量変化から調べた。実施例1および13において、処理前の表2に
示されているMnとMwの値は、暴露処理後、実施例1では、それぞれ14900、43200に、実施例11では、22000,55000になり、ほとんど低下しなかった。一方、スルホン化ジアミンから合成する通常のスルホン化ポリイミド膜では、イミド環の加水分解のため、高分子鎖の切断が起こり、分子量が大きく低下することが報告されている(例えば、非特許文献:Makuromolecules, 39巻3号1189-1198頁、2006年)。
飽和水蒸気の100h暴露処理によっても分子量の低下がほとんど生じないものと考えられる。また、表3に示すように、機械的強度およびプロトン伝導性の免からも優れた本発明で
得られる共重合体は、耐水性を有することが確認できた。
Claims (11)
- 一般式(1)で表される化合物(A)と、一般式(2)で表される化合物(B)とを、触媒(C)の存在下で重合させることを特徴とする共重合体の製造方法。
3〜50の整数を示す。
式(1)および(2)中、Xは、塩素、臭素、ヨウ素、メタンスルホニル基、トリフルオロメタンスルホニル基、ベンゼンスルホニル基のいずれかを示す。式(2)中、Yは、−CO
−、−SO2−、−SO−、-C(CF3)2-のいずれかを示し、Zは、直接結合、−O−、−S−、−CO−、−SO2−、−SO−、-C(CF3)2-、のいずれかを示す。
また、式(2)中、R21は、水素原子、アルカリ金属原子、脂肪族炭化水素基を示す。x1およびx2は0〜4の整数を示す(ただし、yが0である場合にはx1は1〜4であり、yが1である場合にはx1とx2のいずれか一方は1以上である)。yは0〜1の整数、zは0〜3の整数を示す。]
、Z’は、直接結合、−O−、−S−、−CO−、−SO2−、−SO−、-C(CF3)2-のい
ずれかを示す。
式(b)中、A,Bは、直接結合、−O−、−S−、−C(R’)2−、―C(CF3)2−、−CO−、−SO2−、−SO−のいずれかを示す。ただしR’は水素原子、フッ素原
子、アルキル基、環状アルキル基、芳香族基を示す。式(d)中、mは0〜4の整数を示す
。a、bは、0〜4の整数を示す。なお+はXへの結合端、*は窒素原子への結合端を示す。] - 得られた共重合体のゲルパーミエーションクロマトグラフィー法で測定したポリスチレン換算重量平均分子量が15,000〜1,000,000であることを特徴とする請求
項1に記載の共重合体の製造方法。 - 前記式(1)において、nが5〜30である、請求項1に記載の共重合体の製造方法。
- 化合物(A)と化合物(B)の混合物と触媒(C)とを混合する工程、およびその後に再度触媒(C)を添加する工程を有することを特徴とする、請求項1〜3のいずれかに記載の共重合体の製造方法。
- 化合物(B)に触媒(C)を添加する工程、およびその後に化合物(A)を添加する工程を有することを特徴とする、請求項1〜3のいずれかに記載の共重合体の製造方法。
- 一般式(1')で表される構造単位(A')と、一般式(2')で表される構造を有する
構造単位(B')とを含むことを特徴とする共重合体。
、3〜50の整数を示す。
式(2’)中、Yは、−CO−、−SO2−、−SO−、-C(CF3)2-のいずれかを示し、
Zは、−O−、−S−、直接結合、−CO−、−SO2−、−SO−、-C(CF3)2-、のいずれかを示す。
また、式(2’)中、R21は、水素原子、アルカリ金属原子、脂肪族炭化水素基を示す。
x1およびx2は0〜4の整数を示す(ただし、yが0である場合にはx1は1〜4であり
、yが1である場合にはx1とx2のいずれか一方は1以上である)。yは0〜1の整数、zは0〜3の整数を示す。]
れかを示す。
式(b)中、A,Bは、−O−、−S−、直接結合、−C(R’)2−、―C(CF3)2−、−CO−、−SO2−、−SO−のいずれかを示す。ただしR’は水素原子、フッ素原
子、アルキル基、環状アルキル基、芳香族基を示す。式(d)中、mは0〜4の整数を示す
。a、bは、0〜4の整数を示す。なお+は式(1’)の左端又は右端に相当する結合端、*は窒素原子への結合端を示す。] - 構造単位(B’)が下記式(3’)で表されることを特徴とする請求項6に記載の共重合体。
は、式(2’)と同様である。] - 得られた共重合体のゲルパーミエーションクロマトグラフィー法で測定したポリスチレン換算重量平均分子量が15,000〜1,000,000であることを特徴とする請求項6又は7に記載の共重合体。
- 一般式(1)において、繰り返し単位数を示すnが、5〜30である、請求項6〜8のいずれか一に記載の共重合体。
- 請求項6〜8のいずれかに記載の共重合体を含んでなる、固体高分子型燃料電池用の電解質。
- 請求項10に記載の電解質を用いて得られる、固体高分子型燃料電池用の電解質膜。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008230138A JP5421564B2 (ja) | 2008-09-08 | 2008-09-08 | 共重合体の製造方法、共重合体、および該共重合体を用いた電解質 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008230138A JP5421564B2 (ja) | 2008-09-08 | 2008-09-08 | 共重合体の製造方法、共重合体、および該共重合体を用いた電解質 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010065080A true JP2010065080A (ja) | 2010-03-25 |
| JP5421564B2 JP5421564B2 (ja) | 2014-02-19 |
Family
ID=42190944
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008230138A Expired - Fee Related JP5421564B2 (ja) | 2008-09-08 | 2008-09-08 | 共重合体の製造方法、共重合体、および該共重合体を用いた電解質 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5421564B2 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011089036A (ja) * | 2009-10-22 | 2011-05-06 | Jsr Corp | 新規な芳香族化合物および側鎖にスルホン酸基を含む芳香環を有するポリアリーレン系共重合体 |
| JP2012016247A (ja) * | 2010-07-05 | 2012-01-19 | Kansai Electric Power Co Inc:The | ポリイミド系高分子アクチュエータ、及びその製造方法 |
| JP2013231127A (ja) * | 2012-04-27 | 2013-11-14 | Kaneka Corp | 高分子電解質およびその利用 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004285116A (ja) * | 2003-03-19 | 2004-10-14 | Honda Motor Co Ltd | 高分子電解質およびプロトン伝導膜 |
| JP2004285283A (ja) * | 2003-03-25 | 2004-10-14 | Honda Motor Co Ltd | 高分子電解質およびプロトン伝導膜 |
| JP2005154578A (ja) * | 2003-11-26 | 2005-06-16 | Jsr Corp | 架橋型高分子電解質およびプロトン伝導膜 |
| JP2005336310A (ja) * | 2004-05-26 | 2005-12-08 | Jsr Corp | プロトン酸基を有する重合体、高分子固体電解質およびプロトン伝導膜 |
-
2008
- 2008-09-08 JP JP2008230138A patent/JP5421564B2/ja not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004285116A (ja) * | 2003-03-19 | 2004-10-14 | Honda Motor Co Ltd | 高分子電解質およびプロトン伝導膜 |
| JP2004285283A (ja) * | 2003-03-25 | 2004-10-14 | Honda Motor Co Ltd | 高分子電解質およびプロトン伝導膜 |
| JP2005154578A (ja) * | 2003-11-26 | 2005-06-16 | Jsr Corp | 架橋型高分子電解質およびプロトン伝導膜 |
| JP2005336310A (ja) * | 2004-05-26 | 2005-12-08 | Jsr Corp | プロトン酸基を有する重合体、高分子固体電解質およびプロトン伝導膜 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011089036A (ja) * | 2009-10-22 | 2011-05-06 | Jsr Corp | 新規な芳香族化合物および側鎖にスルホン酸基を含む芳香環を有するポリアリーレン系共重合体 |
| JP2012016247A (ja) * | 2010-07-05 | 2012-01-19 | Kansai Electric Power Co Inc:The | ポリイミド系高分子アクチュエータ、及びその製造方法 |
| JP2013231127A (ja) * | 2012-04-27 | 2013-11-14 | Kaneka Corp | 高分子電解質およびその利用 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5421564B2 (ja) | 2014-02-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI236486B (en) | Crosslinkable aromatic resin having protonic acid group, and ion conductive polymer membrane, binder and fuel cell using the resin | |
| Chen et al. | Synthesis and properties of novel side-chain-sulfonated polyimides from bis [4-(4-aminophenoxy)-2-(3-sulfobenzoyl)] phenyl sulfone | |
| Li et al. | Sulfonated polyimides bearing benzimidazole groups for proton exchange membranes | |
| Wang et al. | Sulfonated aromatic polyamides containing nitrile groups as proton exchange fuel cell membranes | |
| JP5642453B2 (ja) | リン酸ドープ電解質膜およびその製造方法並びにそれを含む燃料電池 | |
| US8742031B2 (en) | Solvent-soluble 6,6-polyimide copolymers and processes for preparing them | |
| Chen et al. | Synthesis and properties of sulfonated polyimides from homologous sulfonated diamines bearing bis (aminophenoxyphenyl) sulfone | |
| Mandal et al. | Sulfonated polyimides containing triphenylphosphine oxide for proton exchange membranes | |
| Chen et al. | Synthesis and properties of novel sulfonated polyimides bearing sulfophenyl pendant groups for fuel cell application | |
| Li et al. | Acid doped polybenzimidazoles containing 4-phenyl phthalazinone moieties for high-temperature PEMFC | |
| Mandal et al. | Sulfonated copolyimides containing trifluoromethyl and phosphine oxide moieties: synergistic effect towards proton exchange membrane properties | |
| KR20080028489A (ko) | 질소 함유 방향족 화합물 및 그의 제조 방법, 중합체 및양성자 전도막 | |
| Li et al. | Functionalized 4-phenyl phthalazinone-based polybenzimidazoles for high-temperature PEMFC | |
| JP5421564B2 (ja) | 共重合体の製造方法、共重合体、および該共重合体を用いた電解質 | |
| Guo et al. | Synthesis and properties of novel multiblock copolyimides consisting of benzimidazole-groups-containing sulfonated polyimide hydrophilic blocks and non-sulfonated polyimide hydrophobic blocks as proton exchange membranes | |
| CN101591436B (zh) | 含二氮杂萘酮联苯结构聚苯并咪唑及其制备方法 | |
| Li et al. | Synthesis and characterization of novel sulfonated polyimides from 1, 4-bis (4-aminophenoxy)-naphthyl-2, 7-disulfonic acid | |
| Hu et al. | Synthesis and characterization of sulfonated polyimides derived from 2, 2′-bis (4-sulfophenyl)-4, 4′-oxydianiline as polymer electrolyte membranes for fuel cell applications | |
| Liu et al. | Sulfonated naphthalenic polyimides containing ether and ketone linkages as polymer electrolyte membranes | |
| Zhang et al. | Novel aromatic proton-exchange polyelectrolytes via polyacylation of pre-sulfonated monomers | |
| Akbarian-Feizi et al. | Investigation on the preparation of new sulfonated polyimide fuel cell membranes in organic and ionic liquid media | |
| Guo et al. | Synthesis and properties of novel sulfonated polyimides from 4, 4′-(biphenyl-4, 4′-diyldi (oxo)) bis (1, 8-naphthalic anhydride) | |
| Akbarian‐Feizi et al. | Synthesis of new sulfonated copolyimides in organic and ionic liquid media for fuel cell application | |
| JP2002358978A (ja) | 高分子電解質膜及びその製造方法 | |
| JP2006152009A (ja) | スルホン化芳香族ポリイミド及び該ポリイミドよりなる電解質膜 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20110907 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110907 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20111028 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20120220 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20120220 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20130208 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130219 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130411 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20131112 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20131122 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5421564 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |