JP2005296867A - Hydrocarbon hydrotreating catalyst composition and hydrocarbon oil hydrotreating method using the catalyst composition. - Google Patents
Hydrocarbon hydrotreating catalyst composition and hydrocarbon oil hydrotreating method using the catalyst composition. Download PDFInfo
- Publication number
- JP2005296867A JP2005296867A JP2004118974A JP2004118974A JP2005296867A JP 2005296867 A JP2005296867 A JP 2005296867A JP 2004118974 A JP2004118974 A JP 2004118974A JP 2004118974 A JP2004118974 A JP 2004118974A JP 2005296867 A JP2005296867 A JP 2005296867A
- Authority
- JP
- Japan
- Prior art keywords
- catalyst composition
- catalyst
- hydrotreating
- silica
- alumina
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 239000003054 catalyst Substances 0.000 title claims abstract description 149
- 239000000203 mixture Substances 0.000 title claims abstract description 74
- 239000004215 Carbon black (E152) Substances 0.000 title claims abstract description 35
- 229930195733 hydrocarbon Natural products 0.000 title claims abstract description 35
- 150000002430 hydrocarbons Chemical class 0.000 title claims abstract description 34
- 238000000034 method Methods 0.000 title claims abstract description 19
- 150000002697 manganese compounds Chemical class 0.000 claims abstract description 35
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 84
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 claims description 44
- 239000002245 particle Substances 0.000 claims description 43
- 239000000377 silicon dioxide Substances 0.000 claims description 42
- 239000011572 manganese Substances 0.000 claims description 38
- 229910052751 metal Inorganic materials 0.000 claims description 36
- 239000002184 metal Substances 0.000 claims description 36
- MCMNRKCIXSYSNV-UHFFFAOYSA-N ZrO2 Inorganic materials O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 claims description 26
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N titanium dioxide Inorganic materials O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 claims description 26
- 238000005984 hydrogenation reaction Methods 0.000 claims description 9
- 229910052809 inorganic oxide Inorganic materials 0.000 claims description 6
- 230000000737 periodic effect Effects 0.000 claims description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 4
- 229910052739 hydrogen Inorganic materials 0.000 claims description 4
- 239000001257 hydrogen Substances 0.000 claims description 4
- 238000003763 carbonization Methods 0.000 claims 1
- 125000001183 hydrocarbyl group Chemical group 0.000 claims 1
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 abstract description 57
- 230000000694 effects Effects 0.000 abstract description 34
- 229910052759 nickel Inorganic materials 0.000 abstract description 28
- 238000006477 desulfuration reaction Methods 0.000 abstract description 22
- 230000023556 desulfurization Effects 0.000 abstract description 22
- 229910052720 vanadium Inorganic materials 0.000 abstract description 11
- GPPXJZIENCGNKB-UHFFFAOYSA-N vanadium Chemical compound [V]#[V] GPPXJZIENCGNKB-UHFFFAOYSA-N 0.000 abstract description 11
- 150000001875 compounds Chemical class 0.000 abstract description 4
- GEYXPJBPASPPLI-UHFFFAOYSA-N manganese(iii) oxide Chemical compound O=[Mn]O[Mn]=O GEYXPJBPASPPLI-UHFFFAOYSA-N 0.000 description 58
- 239000003921 oil Substances 0.000 description 31
- JKQOBWVOAYFWKG-UHFFFAOYSA-N molybdenum trioxide Chemical compound O=[Mo](=O)=O JKQOBWVOAYFWKG-UHFFFAOYSA-N 0.000 description 26
- 239000002002 slurry Substances 0.000 description 26
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 24
- 229910018072 Al 2 O 3 Inorganic materials 0.000 description 18
- 229910052750 molybdenum Inorganic materials 0.000 description 18
- 239000000243 solution Substances 0.000 description 18
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 17
- 239000011733 molybdenum Substances 0.000 description 17
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 16
- 229910052698 phosphorus Inorganic materials 0.000 description 16
- 239000011574 phosphorus Substances 0.000 description 16
- 239000011148 porous material Substances 0.000 description 15
- 230000000052 comparative effect Effects 0.000 description 14
- 229910004298 SiO 2 Inorganic materials 0.000 description 13
- 239000007864 aqueous solution Substances 0.000 description 13
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 12
- 229910052748 manganese Inorganic materials 0.000 description 12
- 150000002736 metal compounds Chemical class 0.000 description 12
- 229910000008 nickel(II) carbonate Inorganic materials 0.000 description 12
- ZULUUIKRFGGGTL-UHFFFAOYSA-L nickel(ii) carbonate Chemical compound [Ni+2].[O-]C([O-])=O ZULUUIKRFGGGTL-UHFFFAOYSA-L 0.000 description 12
- 238000006243 chemical reaction Methods 0.000 description 11
- VASIZKWUTCETSD-UHFFFAOYSA-N manganese(II) oxide Inorganic materials [Mn]=O VASIZKWUTCETSD-UHFFFAOYSA-N 0.000 description 10
- 238000007324 demetalation reaction Methods 0.000 description 9
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 8
- 239000002994 raw material Substances 0.000 description 7
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 6
- MIVBAHRSNUNMPP-UHFFFAOYSA-N manganese(2+);dinitrate Chemical compound [Mn+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O MIVBAHRSNUNMPP-UHFFFAOYSA-N 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 6
- 229910052753 mercury Inorganic materials 0.000 description 6
- 239000011734 sodium Substances 0.000 description 6
- 229910052708 sodium Inorganic materials 0.000 description 6
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 description 5
- 230000032683 aging Effects 0.000 description 5
- DIZPMCHEQGEION-UHFFFAOYSA-H aluminium sulfate (anhydrous) Chemical compound [Al+3].[Al+3].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O DIZPMCHEQGEION-UHFFFAOYSA-H 0.000 description 5
- ANBBXQWFNXMHLD-UHFFFAOYSA-N aluminum;sodium;oxygen(2-) Chemical compound [O-2].[O-2].[Na+].[Al+3] ANBBXQWFNXMHLD-UHFFFAOYSA-N 0.000 description 5
- 238000001704 evaporation Methods 0.000 description 5
- 238000004231 fluid catalytic cracking Methods 0.000 description 5
- 229910001388 sodium aluminate Inorganic materials 0.000 description 5
- 229910052717 sulfur Inorganic materials 0.000 description 5
- 239000011593 sulfur Substances 0.000 description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 4
- 229910010413 TiO 2 Inorganic materials 0.000 description 4
- 239000000295 fuel oil Substances 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 239000000017 hydrogel Substances 0.000 description 4
- 235000019353 potassium silicate Nutrition 0.000 description 4
- NTHWMYGWWRZVTN-UHFFFAOYSA-N sodium silicate Chemical compound [Na+].[Na+].[O-][Si]([O-])=O NTHWMYGWWRZVTN-UHFFFAOYSA-N 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 4
- 238000009826 distribution Methods 0.000 description 3
- 238000001125 extrusion Methods 0.000 description 3
- 238000005470 impregnation Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 229910052721 tungsten Inorganic materials 0.000 description 3
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical group CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 238000005299 abrasion Methods 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- 229910017052 cobalt Inorganic materials 0.000 description 2
- 239000010941 cobalt Substances 0.000 description 2
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 238000000227 grinding Methods 0.000 description 2
- 238000004898 kneading Methods 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 231100000572 poisoning Toxicity 0.000 description 2
- 230000000607 poisoning effect Effects 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 238000004062 sedimentation Methods 0.000 description 2
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 2
- 239000010937 tungsten Substances 0.000 description 2
- -1 vanadium and nickel Chemical class 0.000 description 2
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- YGTJVXYTFSYHIO-UHFFFAOYSA-L C([O-])([O-])=O.[Ni+2].P(O)(O)(O)=O Chemical compound C([O-])([O-])=O.[Ni+2].P(O)(O)(O)=O YGTJVXYTFSYHIO-UHFFFAOYSA-L 0.000 description 1
- 229910017028 MnSi Inorganic materials 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- 238000004523 catalytic cracking Methods 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- LRCFXGAMWKDGLA-UHFFFAOYSA-N dioxosilane;hydrate Chemical compound O.O=[Si]=O LRCFXGAMWKDGLA-UHFFFAOYSA-N 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 238000010304 firing Methods 0.000 description 1
- 230000005484 gravity Effects 0.000 description 1
- 238000009776 industrial production Methods 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 229910052976 metal sulfide Inorganic materials 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 229910000510 noble metal Inorganic materials 0.000 description 1
- DCKVFVYPWDKYDN-UHFFFAOYSA-L oxygen(2-);titanium(4+);sulfate Chemical compound [O-2].[Ti+4].[O-]S([O-])(=O)=O DCKVFVYPWDKYDN-UHFFFAOYSA-L 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000002574 poison Substances 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 238000002459 porosimetry Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 229910052702 rhenium Inorganic materials 0.000 description 1
- WUAPFZMCVAUBPE-UHFFFAOYSA-N rhenium atom Chemical group [Re] WUAPFZMCVAUBPE-UHFFFAOYSA-N 0.000 description 1
- 230000005070 ripening Effects 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 229910000348 titanium sulfate Inorganic materials 0.000 description 1
- 238000002834 transmittance Methods 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- ZXAUZSQITFJWPS-UHFFFAOYSA-J zirconium(4+);disulfate Chemical compound [Zr+4].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O ZXAUZSQITFJWPS-UHFFFAOYSA-J 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G45/00—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds
- C10G45/02—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds to eliminate hetero atoms without changing the skeleton of the hydrocarbon involved and without cracking into lower boiling hydrocarbons; Hydrofinishing
- C10G45/04—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds to eliminate hetero atoms without changing the skeleton of the hydrocarbon involved and without cracking into lower boiling hydrocarbons; Hydrofinishing characterised by the catalyst used
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/32—Manganese, technetium or rhenium
- B01J23/34—Manganese
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G45/00—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds
- C10G45/02—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds to eliminate hetero atoms without changing the skeleton of the hydrocarbon involved and without cracking into lower boiling hydrocarbons; Hydrofinishing
- C10G45/04—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds to eliminate hetero atoms without changing the skeleton of the hydrocarbon involved and without cracking into lower boiling hydrocarbons; Hydrofinishing characterised by the catalyst used
- C10G45/12—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds to eliminate hetero atoms without changing the skeleton of the hydrocarbon involved and without cracking into lower boiling hydrocarbons; Hydrofinishing characterised by the catalyst used containing crystalline alumino-silicates, e.g. molecular sieves
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/70—Catalyst aspects
Landscapes
- Chemical & Material Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Materials Engineering (AREA)
- Crystallography & Structural Chemistry (AREA)
- Catalysts (AREA)
- Production Of Liquid Hydrocarbon Mixture For Refining Petroleum (AREA)
Abstract
Description
本発明は、炭化水素油の水素化処理触媒組成物に関し、さらに詳しくはバナジウムやニッケルなどの金属化合物を多量に含む炭化水素油の水素化処理に使用して、脱硫活性が低下することなく高い脱メタル活性を示す炭化水素油の水素化処理触媒組成物に関する。 The present invention relates to a hydrotreating catalyst composition for hydrocarbon oils, and more specifically, it is used for hydrotreating hydrocarbon oils containing a large amount of metal compounds such as vanadium and nickel, and the desulfurization activity is high without decreasing. The present invention relates to a hydroprocessing catalyst composition for hydrocarbon oils that exhibits demetalization activity.
従来の水素化処理触媒は、実装置においてバナジウムやニッケルなどの金属化合物を多量に含む炭化水素油(以下、重質油ということがある)の水素化処理に工業的に使用した場合に、金属化合物などによる触媒細孔の閉塞が生じて脱硫活性、脱メタル活性が失活するため長時間の運転ができないという問題があった。
また、水素化処理装置の下流に流動接触分解(FCC)装置を有する製油所では、水素化処理して得られた生成油を接触分解の原料油として使用するため、水素化処理装置で金属化合物の除去が十分に行われないと、生成油中に含まれるバナジウムやニッケルなどの金属化合物がFCC触媒を被毒して失活させるためFCC触媒の使用量が増加するという問題が起こる。このため、水素化処理装置において重質油を水素化処理する際、より効果的に脱硫、脱メタルを行う水素化処理触媒組成物が望まれていた。
A conventional hydrotreating catalyst is a metal that is used in industrial equipment for hydrotreating hydrocarbon oils (hereinafter sometimes referred to as heavy oil) containing a large amount of metal compounds such as vanadium and nickel. The catalyst pores are blocked by a compound or the like, and the desulfurization activity and demetallization activity are deactivated, so that there is a problem that the operation cannot be performed for a long time.
Also, in refineries that have fluid catalytic cracking (FCC) equipment downstream of the hydrotreating equipment, the product oil obtained by hydrotreating is used as the feedstock for catalytic cracking. If the removal of the catalyst is not performed sufficiently, a metal compound such as vanadium or nickel contained in the product oil poisons the FCC catalyst and deactivates the FCC catalyst, resulting in an increase in the amount of FCC catalyst used. For this reason, there has been a demand for a hydrotreating catalyst composition that performs desulfurization and demetalization more effectively when hydrotreating heavy oil in a hydrotreating apparatus.
前述の要望に応じて種々の水素化処理触媒組成物に関する提案がなされている。例えば、特許文献1には、炭化水素供給原料の選択的水添脱窒素法で、鉄とMo、W及びこれらの混合物より成る群から選ばれた少なくとも一つの金属との無定形硫化物を含む触媒を用いることが記載されており、該触媒にNi、Co、Zn、Mn、Cuおよびこれらの混合物より成る群から選ばれた助触媒金属の少なくとも一つの金属硫化物を含めると、炭化水素供給原料から窒素の選択的除去に優れた効果を有することが開示されている。 Various hydroprocessing catalyst compositions have been proposed in response to the aforementioned needs. For example, Patent Document 1 includes an amorphous sulfide of iron and at least one metal selected from the group consisting of Mo, W, and mixtures thereof in a selective hydrodenitrogenation process of a hydrocarbon feedstock. The use of a catalyst is described, wherein the catalyst includes at least one metal sulfide of a promoter metal selected from the group consisting of Ni, Co, Zn, Mn, Cu, and mixtures thereof, providing a hydrocarbon feed It is disclosed that it has an excellent effect on selective removal of nitrogen from a raw material.
特許文献2には、第VIIB族の少なくとも1つの元素を含む触媒および水素化処理におけるその使用法の発明で、少なくとも1つのマトリックスと、第VIIB族の少なくとも1つの金属と、第VIII族の少なくとも1つの非貴金属と、モリブデンおよびタングステンよりなる群の中から選ばれる第VIB族の少なくとも1つの金属と、場合によっては第VIIA族の少なくとも1つの元素と、リン、ホウ素またはケイ素よりなる群の中から選ばれかつ触媒の活性を改善する追加元素とからなる触媒が記載されており、第VIIB族の少なくとも1つの金属はレニウムまたはマンガンが好ましいことが開示されている。 In US Pat. No. 5,689,059, a catalyst comprising at least one element of Group VIIB and its use in hydroprocessing, it comprises at least one matrix, at least one metal of Group VIIB, and at least of Group VIII. In a group consisting of one non-noble metal, at least one Group VIB metal selected from the group consisting of molybdenum and tungsten, and optionally at least one element from Group VIIA, and phosphorus, boron or silicon And a catalyst consisting of an additional element that improves the activity of the catalyst is described, wherein at least one metal of Group VIIB is preferably rhenium or manganese.
従来、バナジウムやニッケルなどの金属化合物を多量に含む炭化水素油の水素化処理では、脱メタル活性の高い脱メタル触媒でバナジウムやニッケルなどの金属化合物を除去し、次いで、脱硫活性の高い水素化処理触媒で硫黄や窒素を除去する方法が行われている。そのため、従来の脱メタル触媒は、脱メタル活性は高くても脱硫活性が低く、また、脱メタル触媒の後段で使用される水素化処理触媒は高い脱硫活性を有しても脱メタル活性が低かった。
本発明の目的は、バナジウムやニッケルなどの金属化合物を多量に含む炭化水素油の水素化処理に使用して、高い脱メタル活性を有し、しかも高い脱硫活性を有する炭化水素油の水素化処理触媒組成物および該触媒組成物を使用した炭化水素油の水素化処理方法を提供することにある。
Conventionally, in the hydrotreatment of hydrocarbon oils containing a large amount of metal compounds such as vanadium and nickel, metal compounds such as vanadium and nickel are removed with a demetallation catalyst having high demetallation activity, and then hydrogenation with high desulfurization activity is performed. A method of removing sulfur and nitrogen with a treatment catalyst has been performed. Therefore, even if the conventional demetallation catalyst has a high demetallation activity, the desulfurization activity is low, and the hydrotreating catalyst used in the subsequent stage of the demetallation catalyst has a low demetallation activity even if it has a high desulfurization activity. It was.
An object of the present invention is to use a hydrocarbon oil hydrotreating a high amount of metal compounds such as vanadium and nickel to have a high demetallation activity and a high desulfurization activity. It is an object of the present invention to provide a catalyst composition and a method for hydrotreating a hydrocarbon oil using the catalyst composition.
本発明者は、前述の目的を達成するために鋭意研究を重ねた結果、水素化処理触媒組成物中にマンガン化合物が微粒子の状態で存在している触媒組成物は高い脱メタル活性を示すにも係わらず脱硫活性が低下しないことを見出し本発明を完成するに至った。
即ち、本発明の第1は、担体に水素化活性金属成分を担持してなる水素化処理触媒組成物中に粒子状のマンガン化合物を含有することを特徴とする炭化水素油の水素化処理触媒組成物に関する。
本発明の第2は、前記粒子状のマンガン化合物の平均粒子径が0.5〜20μmの範囲にあることを特徴とする請求項1記載の炭化水素油の水素化処理触媒組成物に関する。
本発明の第3は、水素化処理触媒組成物中における前記粒子状のマンガン化合物の含有量が0.5〜60重量%の範囲であることを特徴とする請求項1または2記載の炭化水素油の水素化処理触媒組成物に関する。
本発明の第4は、前記粒子状のマンガン化合物が、Mn2O3であることを特徴とする請求項1〜3のいずれかに記載の炭化水素油の水素化処理触媒組成物に関する。
本発明の第5は、前記担体がアルミナ、シリカ−アルミナ、シリカ、シリカ−チタニア、シリカ−ジルコニアから選ばれた多孔性無機酸化物であることを特徴とする請求項1〜4のいずれかに記載の炭化水素油の水素化処理触媒組成物に関する。
本発明の第6は、前記水素化活性金属成分が周期律表の第VIB族金属、第VIII族金属から選ばれた少なくとも1種の金属成分を含有することを特徴とする請求項1〜5のいずれかに記載の炭化水素油の水素化処理触媒組成物に関する。
本発明の第7は、触媒として請求項1〜6のいずれかに記載の水素化処理触媒組成物を使用することを特徴とする炭化水素油の水素化処理方法に関する。
As a result of intensive studies to achieve the above-mentioned object, the present inventor has found that a catalyst composition in which a manganese compound is present in a fine particle state in a hydroprocessing catalyst composition exhibits high demetallation activity. Nevertheless, the present inventors have found that the desulfurization activity does not decrease and have completed the present invention.
That is, the first aspect of the present invention is a hydrotreating catalyst for hydrocarbon oil, characterized in that the hydrotreating catalyst composition comprising a carrier supporting a hydrotreating metal component contains a particulate manganese compound. Relates to the composition.
The second aspect of the present invention relates to the hydrotreating catalyst composition for hydrocarbon oil according to claim 1, wherein the average particle size of the particulate manganese compound is in the range of 0.5 to 20 μm.
A third aspect of the present invention is the hydrocarbon according to claim 1 or 2, wherein the content of the particulate manganese compound in the hydrotreating catalyst composition is in the range of 0.5 to 60 wt%. The present invention relates to an oil hydrotreating catalyst composition.
A fourth aspect of the present invention relates to the hydrocarbon oil hydrotreating catalyst composition according to any one of claims 1 to 3, wherein the particulate manganese compound is Mn 2 O 3 .
According to a fifth aspect of the present invention, the carrier is a porous inorganic oxide selected from alumina, silica-alumina, silica, silica-titania, and silica-zirconia. The hydrocarbon oil hydrotreating catalyst composition described.
According to a sixth aspect of the present invention, the hydrogenation active metal component contains at least one metal component selected from Group VIB metals and Group VIII metals of the Periodic Table. The hydrocarbon oil hydrotreating catalyst composition according to any one of the above.
A seventh aspect of the present invention relates to a hydrocarbon oil hydrotreating method, wherein the hydrotreating catalyst composition according to any one of claims 1 to 6 is used as a catalyst.
本発明の炭化水素油の水素化処理触媒組成物は、担体に水素化活性金属成分を担持してなる水素化処理触媒組成物中に粒子状のマンガン化合物を含有することを特徴とする。
本発明での粒子状のマンガン化合物とは、水素化処理触媒組成物中にマンガン化合物が均一に存在するのではなく、粒子状態のマンガン化合物が不均一に存在することを意味する。前記粒子状のマンガン化合物は、その平均粒子径が0.5〜20μmの範囲にあることが好ましい。該粒子状のマンガン化合物の平均粒子径が0.5μmよりも小さい場合には、バナジウムやニッケルなどの金属化合物の捕捉能が弱くなることがあり、所望の脱メタル活性や脱硫活性が得られないことがある。また、該平均粒子径が20μmよりも大きい場合には、得られる水素化処理触媒組成物の強度が工業触媒用としては弱くなることがある。該粒子状のマンガン化合物の平均粒子径は、更に好ましくは1〜10μmの範囲にあることが望ましい。
なお、水素化処理触媒組成物中の前記粒子状のマンガン化合物の平均粒子径は、走査型電子顕微鏡(SEM;Scanning Electron Microscope)による反射電子像の少なくとも5カ所の画像から、マンガン化合物の粒子の最大径を500個以上測定して求めた平均値である。
The hydrotreating catalyst composition for hydrocarbon oil of the present invention is characterized in that the hydrotreating catalyst composition obtained by supporting a hydrotreating metal component on a carrier contains a particulate manganese compound.
The particulate manganese compound in the present invention means that the manganese compound is not uniformly present in the hydrotreating catalyst composition, but the manganese compound in the particle state is unevenly present. The particulate manganese compound preferably has an average particle size in the range of 0.5 to 20 μm. When the average particle size of the particulate manganese compound is smaller than 0.5 μm, the ability to capture metal compounds such as vanadium and nickel may be weak, and the desired demetalization activity and desulfurization activity cannot be obtained. Sometimes. Moreover, when this average particle diameter is larger than 20 micrometers, the intensity | strength of the hydrotreating catalyst composition obtained may become weak for an industrial catalyst use. The average particle diameter of the particulate manganese compound is more preferably in the range of 1 to 10 μm.
The average particle diameter of the particulate manganese compound in the hydrotreating catalyst composition is determined based on the image of the manganese compound particles from at least five images of the reflected electron images obtained by a scanning electron microscope (SEM; Scanning Electron Microscope). An average value obtained by measuring 500 or more maximum diameters.
本発明の水素化処理触媒組成物は、前記粒子状のマンガン化合物の含有量が0.5〜60重量%の範囲にあることが好ましい。該含有量が0.5重量%より少ない場合には、所望の脱メタル活性が得られないことがあり、また、該含有量が60重量%より多い場合には、脱硫活性が低下することがある。前記粒子状のマンガン化合物の含有量は、更に好ましくは、1〜40重量%の範囲にあることが望ましい。 In the hydrotreating catalyst composition of the present invention, the content of the particulate manganese compound is preferably in the range of 0.5 to 60% by weight. If the content is less than 0.5% by weight, the desired demetalization activity may not be obtained. If the content is more than 60% by weight, the desulfurization activity may be reduced. is there. The content of the particulate manganese compound is more preferably in the range of 1 to 40% by weight.
前記粒子状のマンガン化合物原料としては、水素化処理触媒組成物中で粒子状態ができる化合物に転移できる化合物であれば使用可能で、例えば、MnO、MnO2、Mn2O3、Mn3O4、MnSO4、MnP、MnSiO3、ZnMn2O4、ZnMn2O3、Zn2MnO4、ZnMn3O7、Zn2Mn3O8、ZnMn(SO4)2、ZnMn(PO4)2、Zn2MnSi2O7、LiZnMn3O8などが例示され、MnO、MnO2、Mn2O3、Mn3O4、ZnMn2O4、ZnMn2O3、Zn2MnO4、ZnMn3O7、Zn2Mn3O8、などの酸化物は、工業生産を考えると形態的に安定でかつ比較的安価で入手しやすいので好適である。
前記粒子状マンガン化合物は、特に、Mn2O3であることが好ましい。Mn2O3は、特にバナジウムやニッケルとの親和性が強いため、これらの金属化合物はMn2O3粒子に選択的に捕捉されるので金属化合物による活性点の被毒や、また、金属化合物の堆積による細孔閉塞が起こりにくい。そのため、水素化処理触媒組成物は脱硫活性が低下することなく高い脱メタル活性を有する。
As the particulate manganese compound raw material, any compound that can be transferred to a compound capable of forming a particle state in the hydrotreating catalyst composition can be used. For example, MnO, MnO 2 , Mn 2 O 3 , Mn 3 O 4 , MnSO 4 , MnP, MnSiO 3 , ZnMn 2 O 4 , ZnMn 2 O 3 , Zn 2 MnO 4 , ZnMn 3 O 7 , Zn 2 Mn 3 O 8 , ZnMn (SO 4 ) 2 , ZnMn (PO 4 ) 2 , Examples include Zn 2 MnSi 2 O 7 , LiZnMn 3 O 8 , MnO, MnO 2 , Mn 2 O 3 , Mn 3 O 4 , ZnMn 2 O 4 , ZnMn 2 O 3 , Zn 2 MnO 4 , ZnMn 3 O 7 Oxides such as Zn 2 Mn 3 O 8 are suitable because they are morphologically stable and relatively inexpensive and easily available in view of industrial production.
The particulate manganese compound is particularly preferably Mn 2 O 3 . Since Mn 2 O 3 has a particularly strong affinity with vanadium and nickel, these metal compounds are selectively captured by Mn 2 O 3 particles, so that poisoning of active sites by metal compounds, and metal compounds Pore clogging due to the deposition of the is difficult to occur. Therefore, the hydrotreating catalyst composition has high demetalization activity without desulfurization activity decreasing.
本発明において、水素化活性成分を担持する担体としては、通常の水素化処理触媒組成物に使用される多孔性無機酸化物が使用可能であり、例えば、アルミナ、シリカ、チタニア、ジルコニア、シリカ−アルミナ、チタニア−アルミナ、ジルコニア−アルミナ、リン−アルミナ、ボリア−アルミナ、シリカ−ボリア−アルミナ、シリカ−チタニア、シリカ−チタニア−アルミナ、シリカ−ジルコニア、シリカ−ジルコニア−アルミナなどが例示される。特に、アルミナ、シリカ−アルミナ、シリカ、シリカ−チタニア、シリカ−ジルコニアから選ばれた多孔性無機酸化物は、成形性がよく、細孔径や細孔容積などの物性コントロールが比較的容易にできるため好ましい。 In the present invention, a porous inorganic oxide used in a conventional hydroprocessing catalyst composition can be used as a carrier supporting a hydrogenation active component. For example, alumina, silica, titania, zirconia, silica- Examples include alumina, titania-alumina, zirconia-alumina, phosphorus-alumina, boria-alumina, silica-boria-alumina, silica-titania, silica-titania-alumina, silica-zirconia, silica-zirconia-alumina, and the like. In particular, porous inorganic oxides selected from alumina, silica-alumina, silica, silica-titania, and silica-zirconia have good moldability and can easily control physical properties such as pore diameter and pore volume. preferable.
また、本発明における水素化活性金属成分としては、周期律表の第VIB族金属、第VIII族金属から選ばれた少なくとも1種の金属成分であり、具体的には、第VIB族金属としては特にモリブデンやタングステンが好ましく、第VIII族金属としては特にニッケルやコバルトが好ましい。該水素化活性金属成分は、必要に応じて、さらにリンなどの他の成分を含むものであってもよい。特に、該水素化活性金属成分は、モリブデンとニッケルおよび/またはコバルトの組み合わせが、脱硫活性が高いことから好適である。
本発明の水素化処理触媒組成物は、前述の周期律表の第VIB族金属成分を酸化物として1〜20重量%、周期律表の第VIII族金属成分を酸化物として0.5〜8重量%の範囲で含有することが好ましい。
The hydrogenation active metal component in the present invention is at least one metal component selected from Group VIB metal and Group VIII metal of the Periodic Table, and specifically, as Group VIB metal, In particular, molybdenum and tungsten are preferable, and nickel and cobalt are particularly preferable as the Group VIII metal. The hydrogenation active metal component may further contain other components such as phosphorus, if necessary. In particular, the hydrogenation active metal component is preferably a combination of molybdenum and nickel and / or cobalt because of its high desulfurization activity.
The hydrotreating catalyst composition of the present invention comprises 1 to 20% by weight of the Group VIB metal component of the periodic table as an oxide, and 0.5 to 8 of the Group VIII metal component of the periodic table as an oxide. It is preferable to contain in the range of weight%.
本発明の炭化水素油の水素化処理触媒組成物は、例えば、前述の多孔性無機酸化物の前駆物質と所望の平均粒子径を有する前述のマンガン化合物粒子を所望の割合で混合、混練して押出成型に適した捏和物を調製し、そして、通常の方法で該捏和物を所望の形状に押出成型した後、乾燥、焼成して担体を調製し、該担体に前述の水素化活性金属成分を通常の方法で担持して製造される。また、該水素化処理触媒組成物は、前述の多孔性無機酸化物の前駆物質と所望の平均粒子径を有する前述のマンガン化合物粒子と共に前述の水素化活性金属成分を含む水溶液を混合、混練して押出成型に適した捏和物を調製した後、所望の形状に押出成型し、乾燥、焼成して製造することもできる。 The hydrocarbon oil hydrotreating catalyst composition of the present invention comprises, for example, mixing and kneading the aforementioned porous inorganic oxide precursor and the aforementioned manganese compound particles having a desired average particle diameter in a desired ratio. A kneaded product suitable for extrusion molding is prepared, and the kneaded product is extruded into a desired shape by a usual method, then dried and calcined to prepare a carrier, and the above-mentioned hydrogenation activity is added to the carrier. It is produced by supporting a metal component by an ordinary method. The hydrotreating catalyst composition is prepared by mixing and kneading an aqueous solution containing the aforementioned hydrogenation active metal component together with the aforementioned porous inorganic oxide precursor and the aforementioned manganese compound particles having a desired average particle size. After preparing a kneaded product suitable for extrusion molding, it can be produced by extrusion molding into a desired shape, drying and firing.
本発明の炭化水素油の水素化処理触媒組成物は、表面積(SA)が100〜350m2/g、水銀圧入法による細孔容積(PV)が0.40〜1.10ml/g、水銀圧入法で測定した細孔分布での平均細孔直径が8〜30nmの範囲にあることが望ましい。
なお、水銀圧入法で測定した細孔分布での平均細孔直径は、接触角150度、表面張力480dyn/cmの値を使用して測定した細孔直径4.2nm(水銀圧入圧力400MPaに相当)以上の細孔容積の1/2に相当する細孔直径である。
特に、水素化処理反応塔の前段で使用される脱メタルを主目的とする水素化処理触媒組成物である場合は、好ましくは、表面積(SA)が100〜250m2/g、水銀圧入法による細孔容積(PV)が0.50〜1.10ml/g、水銀圧入法で測定した細孔分布での平均細孔直径が10〜30nmの範囲にあることが望ましい。
The hydrocarbon oil hydrotreating catalyst composition of the present invention has a surface area (SA) of 100 to 350 m 2 / g, a pore volume (PV) by mercury intrusion method of 0.40 to 1.10 ml / g, and mercury intrusion. The average pore diameter in the pore distribution measured by the method is desirably in the range of 8 to 30 nm.
The average pore diameter in the pore distribution measured by the mercury intrusion method is 4.2 nm (corresponding to a mercury intrusion pressure of 400 MPa) measured using a contact angle of 150 degrees and a surface tension of 480 dyn / cm. ) The pore diameter corresponding to 1/2 of the above pore volume.
In particular, in the case of a hydrotreating catalyst composition mainly intended for demetalization used in the preceding stage of the hydrotreating reaction tower, the surface area (SA) is preferably 100 to 250 m 2 / g, by a mercury intrusion method. The pore volume (PV) is preferably 0.50 to 1.10 ml / g, and the average pore diameter in the pore distribution measured by mercury porosimetry is preferably in the range of 10 to 30 nm.
本発明の炭化水素油の水素化処理触媒組成物は、通常の重質油の水素化処理方法で使用可能であり、また、通常の水素化処理条件が採用可能で、例えば、反応温度は300〜450℃の範囲、水素分圧は5〜25MPaの範囲、液空間速度(LHSV)は0.1〜5.0hr−1の範囲が例示される。 The hydrocarbon oil hydrotreating catalyst composition of the present invention can be used in an ordinary heavy oil hydrotreating method, and can employ ordinary hydrotreating conditions. For example, the reaction temperature is 300. A range of ˜450 ° C., a hydrogen partial pressure of 5 to 25 MPa, and a liquid space velocity (LHSV) of 0.1 to 5.0 hr −1 are exemplified.
本発明の炭化水素油の水素化処理触媒組成物は、触媒組成物中に粒子状のマンガン化合物が存在するので、炭化水素油中に含まれるバナジウムやニッケルなどの金属化合物は水素化処理の際に該粒子状のマンガン化合物に選択的に捕捉されてバナジウムやニッケルなどの金属化合物による触媒組成物の活性点の被毒が緩和されるため、また、触媒組成物表面での金属化合物の堆積が少なくなるので触媒組成物の細孔閉塞が起こりにくい。そのため、本発明の水素化処理触媒組成物は、重質油の水素化処理に使用して脱硫活性が低下することなく高い脱メタル活性を有する。 The hydrocarbon oil hydrotreating catalyst composition of the present invention contains particulate manganese compounds in the catalyst composition, so that metal compounds such as vanadium and nickel contained in the hydrocarbon oil are subjected to hydrotreating. In addition, it is selectively trapped by the particulate manganese compound and the poisoning of the active site of the catalyst composition by the metal compound such as vanadium or nickel is alleviated. Therefore, the pores of the catalyst composition are hardly blocked. Therefore, the hydrotreating catalyst composition of the present invention is used for heavy oil hydrotreating and has a high demetallizing activity without a decrease in desulfurization activity.
以下に実施例を示して本発明をさらに具体的に説明するが、本発明はこれにより何ら限定されるものではない。 The present invention will be described more specifically with reference to the following examples, but the present invention is not limited thereto.
実施例1
本出願人に係わる再公表WO95/15920号公報に記載のアルミナの製造装置を使用してアルミナ水和物を調製した。即ち、薬液添加口2箇所を持つ循環ラインを設けたタンクに純水719kgを張り込み、これにイソブチレンと無水マレイン酸の共重合物0.5kgを添加し、約2時間高速攪拌し完全に溶解させた。この水溶液にアルミン酸ナトリウム水溶液(Al2O3として濃度22重量%)2kgを攪拌しながら添加し、60℃に加温し循環させた。次いで硫酸アルミニウム水溶液(Al2O3として濃度7重量%)を添加して種子アルミナ水和物スラリーを調製した。このスラリーに、アルミン酸ナトリウム水溶液(Al2O3として濃度22重量%)107.1kgと硫酸アルミニウム水溶液(Al2O3として濃度7重量%)168.3kgを各々35.7kg/hrと56.1kg/hrの添加速度で、温度60℃、pH7.9〜8.1を保ち、攪拌および循環させながら、3時間かけて添加してアルミナ水和物を調合した。得られたアルミナ水和物調合スラリーを洗浄してナトリウムおよび硫酸根を除去したアルミナ水和物スラリーを得た。このアルミナ水和物のスラリーに純水を加えて、Al2O3濃度10重量%のスラリーに調製し、15重量%アンモニア水にて該スラリーpHを11に調製した後、還流器のついた熟成タンクにて95℃で8時間熟成した。熟成終了後、このスラリー30kg(Al2O3として3kg)をスチームジャケット付き双腕型ニーダーにより蒸発濃縮しながら捏和し、可塑性のある捏和物(V)とした。この捏和物(V)に平均粒子径0.2μmの三酸化二マンガン92.8g(Mn2O3として)を加えて、双腕型ニーダーにて20分間練った。この三酸化二マンガン入りのアルミナ捏和物をオーガー式押し出し機で、1.8mmの四つ葉型の柱状に押し出し成形した。得られたアルミナ成形品は、110℃で16時間乾燥した後、さらに650℃で2時間焼成して粒子状Mn2O3を3.0重量%(担体基準)含有するアルミナ担体を得た。
該担体にモリブデンとニッケルとリンを酸化物として触媒組成物基準で5.9重量%、 1.45重量%、1.5重量%となるように三酸化モリブデンと炭酸ニッケルのリン酸水溶液を周知の方法で含浸した後、回転式乾燥機を用いて室温から250℃まで昇温乾燥した。さらにこの乾燥品は、550℃で1時間空気中にて焼成して触媒Aを調製した。
なお、触媒Aの走査型電子顕微鏡(SEM;Scanning Electron Microscope)による反射電子像の5カ所の画像から、三酸化二マンガン粒子の最大径を1カ所画像当たり100個測定して求めた(以下の実施例についても同様)平均粒子径は0.2μmであった。
触媒Aの性状を表1に示す。
Example 1
Alumina hydrate was prepared using the alumina production apparatus described in the republished WO95 / 15920 related to the present applicant. That is, 719 kg of pure water was put in a tank provided with a circulation line having two chemical solution addition ports, 0.5 kg of a copolymer of isobutylene and maleic anhydride was added thereto, and the mixture was stirred at high speed for about 2 hours to completely dissolve. It was. To this aqueous solution, 2 kg of an aqueous sodium aluminate solution (concentration 22% by weight as Al 2 O 3 ) was added with stirring, heated to 60 ° C. and circulated. Next, an aqueous aluminum sulfate solution (concentration 7% by weight as Al 2 O 3 ) was added to prepare a seed alumina hydrate slurry. To this slurry, 107.1 kg of sodium aluminate aqueous solution (concentration 22 wt% as Al 2 O 3 ) and 168.3 kg of aqueous aluminum sulfate solution (concentration 7 wt% as Al 2 O 3 ) were respectively 35.7 kg / hr and 56. With the addition rate of 1 kg / hr, the temperature was kept at 60 ° C. and pH 7.9 to 8.1, and the mixture was added over 3 hours while stirring and circulating to prepare alumina hydrate. The obtained alumina hydrate preparation slurry was washed to obtain an alumina hydrate slurry from which sodium and sulfate radicals were removed. Pure water was added to the alumina hydrate slurry to prepare a slurry with an Al 2 O 3 concentration of 10% by weight, and the slurry pH was adjusted to 11 with 15% by weight ammonia water, followed by a reflux. Aging was performed at 95 ° C. for 8 hours in an aging tank. After completion of aging, 30 kg of this slurry ( 3 kg as Al 2 O 3) was kneaded while evaporating and concentrating with a double-arm kneader equipped with a steam jacket to obtain a flexible kneaded product (V). To this kneaded product (V), 92.8 g of dimanganese trioxide having an average particle diameter of 0.2 μm (as Mn 2 O 3 ) was added, and kneaded with a double-arm kneader for 20 minutes. The alumina kneaded product containing dimanganese trioxide was extruded into an 1.8 mm four-leaf type column by an auger type extruder. The obtained alumina molded article was dried at 110 ° C. for 16 hours, and further calcined at 650 ° C. for 2 hours to obtain an alumina carrier containing 3.0% by weight (based on the carrier) of particulate Mn 2 O 3 .
An aqueous phosphoric acid solution of molybdenum trioxide and nickel carbonate is well known so that the support contains molybdenum, nickel, and phosphorus as oxides to 5.9 wt%, 1.45 wt%, and 1.5 wt% based on the catalyst composition. After the impregnation by the above method, the temperature was dried from room temperature to 250 ° C. using a rotary dryer. Further, this dried product was calcined in air at 550 ° C. for 1 hour to prepare Catalyst A.
The maximum diameter of the dimanganese trioxide particles was determined by measuring 100 particles per image from the five images of the reflected electron image of the catalyst A using a scanning electron microscope (SEM). The same applies to the examples) The average particle size was 0.2 μm.
Properties of catalyst A are shown in Table 1.
実施例2
実施例1において、平均粒子径0.5μmの三酸化二マンガンを使用した以外は実施例1と同様にして、触媒Bを調製した。
触媒Bの性状を表1に示す。
Example 2
In Example 1, Catalyst B was prepared in the same manner as in Example 1, except that dimanganese trioxide having an average particle size of 0.5 μm was used.
Properties of catalyst B are shown in Table 1.
実施例3
実施例1において、平均粒子径2.1μmの三酸化二マンガンを使用し、三酸化二マンガンの添加量を15.1g(Mn2O3として)とした以外は実施例1と同様にして、触媒Cを調製した。
触媒Cの性状を表1に示す。
Example 3
In Example 1, except that dimanganese trioxide having an average particle diameter of 2.1 μm was used and the addition amount of dimanganese trioxide was 15.1 g (as Mn 2 O 3 ), Catalyst C was prepared.
The properties of catalyst C are shown in Table 1.
実施例4
実施例1において、平均粒子径2.1μmの三酸化二マンガンを使用した以外は実施例1と同様にして、触媒Dを調製した。
触媒Dの性状を表1に示す。
Example 4
A catalyst D was prepared in the same manner as in Example 1 except that dimanganese trioxide having an average particle diameter of 2.1 μm was used in Example 1.
Properties of catalyst D are shown in Table 1.
実施例5
実施例1において、平均粒子径2.2μmの一酸化マンガンを使用し、一酸化マンガンの添加量を92.8g(Mn2O3として)とした以外は実施例1と同様にして、触媒Eを調製した。
触媒Eの性状を表1に示す。
Example 5
In Example 1, except that manganese monoxide having an average particle diameter of 2.2 μm was used, and the amount of manganese monoxide added was 92.8 g (as Mn 2 O 3 ), the same as in Example 1, Catalyst E Was prepared.
Properties of catalyst E are shown in Table 1.
実施例6
実施例1において、平均粒子径10.4μmの三酸化二マンガンを使用した以外は実施例1と同様にして、触媒Fを調製した。
触媒Fの性状を表1に示す。
Example 6
In Example 1, Catalyst F was prepared in the same manner as in Example 1, except that dimanganese trioxide having an average particle size of 10.4 μm was used.
Properties of catalyst F are shown in Table 1.
実施例7
実施例1において、平均粒子径2.1μmの三酸化二マンガンを使用し、三酸化二マンガンの添加量を333.3g(Mn2O3として)とした以外は実施例1と同様にして、触媒Gを調製した。
触媒Gの性状を表1に示す。
Example 7
In Example 1, except that dimanganese trioxide having an average particle diameter of 2.1 μm was used, and the addition amount of dimanganese trioxide was 333.3 g (as Mn 2 O 3 ), the same as in Example 1, Catalyst G was prepared.
Properties of catalyst G are shown in Table 1.
実施例8
実施例1において、平均粒子径2.1μmの三酸化二マンガンを使用し、三酸化二マンガンの添加量を4500g(Mn2O3として)とした以外は実施例1と同様にして、触媒Hを調製した。
触媒Hの性状を表1に示す。
Example 8
In Example 1, except that dimanganese trioxide having an average particle diameter of 2.1 μm was used and the addition amount of dimanganese trioxide was changed to 4500 g (as Mn 2 O 3 ), the catalyst H Was prepared.
Properties of catalyst H are shown in Table 1.
実施例9
実施例1において、平均粒子径20.3μmの三酸化二マンガンを使用した以外は実施例1と同様にして、触媒Iを調製した。
触媒Iの性状を表1に示す。
Example 9
In Example 1, Catalyst I was prepared in the same manner as Example 1 except that dimanganese trioxide having an average particle size of 20.3 μm was used.
Properties of catalyst I are shown in Table 1.
実施例10
実施例1において、平均粒子径23.4μmの三酸化二マンガンを使用した以外は実施例1と同様にして、触媒Jを調製した。
触媒Jの性状を表1に示す。
Example 10
A catalyst J was prepared in the same manner as in Example 1 except that dimanganese trioxide having an average particle size of 23.4 μm was used.
Properties of catalyst J are shown in Table 1.
実施例11
実施例1と同様にして調製した捏和物(V)に平均粒子径2.1μmの三酸化二マンガン92.8g(Mn2O3として)を加えて、更に、モリブデンとニッケルとリンを酸化物として触媒組成物基準でそれぞれ5.9重量%、1.45重量%、1.5重量%となるように三酸化モリブデンと炭酸ニッケルのリン酸水溶液を加えて、双腕型ニーダーにて20分間混練した。この三酸化二マンガンとモリブデンとニッケルとリンが入ったアルミナ捏和物をオーガー式押し出し機で、1.8mmの四つ葉型の柱状に押し出し成形した。得られたアルミナ成形品は、110℃で16時間乾燥した後、さらに680℃で2時間焼成して触媒Kを得た。
触媒Kの性状を表1に示す。
Example 11
92.8 g of dimanganese trioxide (as Mn 2 O 3 ) having an average particle size of 2.1 μm was added to the kneaded material (V) prepared in the same manner as in Example 1, and further molybdenum, nickel and phosphorus were oxidized. As a product, a phosphoric acid aqueous solution of molybdenum trioxide and nickel carbonate was added so as to be 5.9 wt%, 1.45 wt%, and 1.5 wt% on the basis of the catalyst composition, respectively. Kneaded for a minute. The alumina kneaded material containing dimanganese trioxide, molybdenum, nickel and phosphorus was extruded into a 1.8 mm four-leaf type column by an auger type extruder. The obtained alumina molded article was dried at 110 ° C. for 16 hours and then calcined at 680 ° C. for 2 hours to obtain catalyst K.
Properties of catalyst K are shown in Table 1.
実施例12
水硝子に硫酸を加えて調製したシリカヒドロゲルを熟成した後、これにアルミン酸ナトリウム水溶液と硫酸アルミニウム溶液を加えて、SiO2/Al2O3重量比70/30のシリカアルミナ水和物を調製した。このシリカアルミナ水和物のスラリーを洗浄してナトリウムおよび硫酸根を除去したシリカアルミナ水和物スラリーを得た。
このスラリー30kg(SiO2−Al2O3として3kg)をスチームジャケット付き双腕型ニーダーにより蒸発濃縮しながら捏和、可塑性のある捏和物(W)とした。
この捏和物に平均粒子径2.1μmの三酸化二マンガン92.8g(Mn2O3として)を加えて、双腕型ニーダーにて20分間混練した。この三酸化二マンガン入りのシリカアルミナ捏和物をオーガー式押し出し機で、1.8mmの四つ葉型の柱状に押し出し成形した。得られたシリカアルミナ成形品は、110℃で16時間乾燥した後、さらに680℃で2時間焼成してマンガン粒子を3重量%含有するシリカアルミナ(SiO2−Al2O3)担体を得た。
該シリカアルミナ担体にモリブデンとニッケルとリンを酸化物として触媒組成物基準でそれぞれ5.9重量%、1.45重量%、1.5重量%となるように三酸化モリブデンと炭酸ニッケルのリン酸水溶液を含浸した後、回転式乾燥機を用いて室温から250℃まで昇温乾燥した。さらに得られた乾燥品は、550℃で1時間空気中にて焼成し、触媒Lを調製した。
触媒Lの性状を表1に示す。
Example 12
After aging a silica hydrogel prepared by adding sulfuric acid to water glass, a sodium aluminate aqueous solution and an aluminum sulfate solution are added thereto to prepare a silica alumina hydrate having a SiO 2 / Al 2 O 3 weight ratio of 70/30. did. This silica alumina hydrate slurry was washed to obtain a silica alumina hydrate slurry from which sodium and sulfate radicals had been removed.
30 kg of this slurry ( 3 kg as SiO 2 -Al 2 O 3) was kneaded and evaporated to a flexible kneaded product (W) while evaporating and concentrating with a double-arm kneader with a steam jacket.
To this kneaded product, 92.8 g of dimanganese trioxide having an average particle diameter of 2.1 μm (as Mn 2 O 3 ) was added and kneaded for 20 minutes with a double-arm kneader. This silica-alumina kneaded product containing dimanganese trioxide was extruded into an 1.8 mm four-leaf column by an auger type extruder. The obtained silica alumina molded article was dried at 110 ° C. for 16 hours, and further calcined at 680 ° C. for 2 hours to obtain a silica alumina (SiO 2 —Al 2 O 3 ) support containing 3% by weight of manganese particles. .
Molybdenum trioxide and nickel carbonate phosphoric acid with molybdenum, nickel and phosphorus as oxides on the silica-alumina support so as to be 5.9 wt%, 1.45 wt% and 1.5 wt%, respectively, based on the catalyst composition After impregnating with the aqueous solution, it was dried by heating from room temperature to 250 ° C. using a rotary dryer. Further, the obtained dried product was calcined in the air at 550 ° C. for 1 hour to prepare catalyst L.
Properties of the catalyst L are shown in Table 1.
実施例13
水硝子に硫酸を加えて調製したシリカヒドロゲルを熟成した後、これを洗浄してナトリウムおよび硫酸根を除去したシリカ水和物スラリーを得た。
このスラリー30kg(SiO2として3kg)をスチームジャケット付き双腕型ニーダーにより蒸発濃縮しながら捏和して、可塑性のある捏和物(X)とした。
この捏和物に平均粒子径2.1μmの三酸化二マンガン92.8g(Mn2O3として)を加えて、双腕型ニーダーにて20分間混練した。この三酸化二マンガン入りのシリカ捏和物をオーガー式押し出し機で、1.8mmの四つ葉型の柱状に押し出し成形した。
得られたシリカ成形品は、110℃で16時間乾燥した後、さらに680℃で2時間焼成してマンガン粒子を3重量%含有するシリカ担体を得た。
該シリカ担体にモリブデンとニッケルとリンを酸化物として触媒組成物基準で5.9重量%、1.45重量%、1.5重量%となるように三酸化モリブデンと炭酸ニッケルのリン酸水溶液を含浸した後、回転式乾燥機を用いて室温から250℃まで昇温乾燥した。
さらに得られた乾燥品は、550℃で1時間空気中にて焼成し、触媒Mを調製した。
触媒Mの性状を表1に示す。
Example 13
A silica hydrogel prepared by adding sulfuric acid to water glass was aged and then washed to obtain a silica hydrate slurry from which sodium and sulfate radicals had been removed.
30 kg of this slurry (3 kg as SiO 2 ) was kneaded while evaporating and concentrating with a double-arm kneader equipped with a steam jacket, to obtain a plastic kneaded product (X).
To this kneaded product, 92.8 g of dimanganese trioxide having an average particle size of 2.1 μm (as Mn 2 O 3 ) was added and kneaded for 20 minutes with a double-arm kneader. This silica kneaded material containing dimanganese trioxide was extruded into an 1.8 mm four-leaf type column by an auger type extruder.
The obtained silica molded article was dried at 110 ° C. for 16 hours, and further fired at 680 ° C. for 2 hours to obtain a silica carrier containing 3% by weight of manganese particles.
An aqueous phosphoric acid solution of molybdenum trioxide and nickel carbonate is added to the silica support so that it becomes 5.9 wt%, 1.45 wt% and 1.5 wt% based on the catalyst composition using molybdenum, nickel and phosphorus as oxides. After impregnation, the temperature was dried from room temperature to 250 ° C. using a rotary dryer.
Furthermore, the obtained dried product was calcined in the air at 550 ° C. for 1 hour to prepare Catalyst M.
The properties of the catalyst M are shown in Table 1.
実施例14
水硝子に硫酸を加えて調製したシリカヒドロゲルを熟成した後、これに硫酸チタン溶液を加えてSiO2/TiO2重量比90/10のシリカチタニア水和物を調製した。このシリカチタニア水和物のスラリーを洗浄してナトリウムおよび硫酸根を除去したシリカチタニア水和物スラリーを得た。
このスラリー30kg(SiO2−TiO2として3kg)をスチームジャケット付き双腕型ニーダーにより蒸発濃縮しながら捏和して、可塑性のある捏和物(Y)とした。
この捏和物に平均粒子径2.1μmの三酸化二マンガン92.8g(Mn2O3として)を加えて、双腕型ニーダーにて20分間混練した。この三酸化二マンガン入りのシリカチタニア捏和物をオーガー式押し出し機で、1.8mmの四つ葉型の柱状に押し出し成形した。得られたシリカチタニア成形品は、110℃で16時間乾燥した後、さらに680℃で2時間焼成してマンガン粒子を3重量%含有するシリカチタニア(SiO2−TiO2)担体を得た。
該シリカチタニア担体にモリブデンとニッケルとリンを酸化物として触媒組成物基準で5.9重量%、1.45重量%、1.5重量%となるように三酸化モリブデンと炭酸ニッケルのリン酸水溶液を含浸した後、回転式乾燥機を用いて室温から250℃まで昇温乾燥した。さらにこの乾燥品は、550℃で1時間空気中にて焼成し、触媒Nを調製した。
触媒Nの性状を表1に示す。
Example 14
A silica hydrogel prepared by adding sulfuric acid to water glass was aged, and then a titanium sulfate solution was added thereto to prepare silica titania hydrate having a SiO 2 / TiO 2 weight ratio of 90/10. The silica titania hydrate slurry was washed to obtain a silica titania hydrate slurry from which sodium and sulfate radicals were removed.
30 kg of this slurry (3 kg as SiO 2 —TiO 2 ) was kneaded while evaporating and concentrating with a double-arm kneader with a steam jacket to obtain a plastic kneaded product (Y).
To this kneaded product, 92.8 g of dimanganese trioxide having an average particle size of 2.1 μm (as Mn 2 O 3 ) was added and kneaded for 20 minutes with a double-arm kneader. This silica titania kneaded material containing dimanganese trioxide was extruded into an 1.8 mm four-leaf column by an auger type extruder. The obtained silica titania molded article was dried at 110 ° C. for 16 hours, and further fired at 680 ° C. for 2 hours to obtain a silica titania (SiO 2 —TiO 2 ) support containing 3% by weight of manganese particles.
An aqueous phosphoric acid solution of molybdenum trioxide and nickel carbonate so that the silica titania carrier has molybdenum, nickel, and phosphorus as oxides to 5.9 wt%, 1.45 wt%, and 1.5 wt% based on the catalyst composition. After being impregnated, it was dried at an elevated temperature from room temperature to 250 ° C. using a rotary dryer. Further, this dried product was calcined in the air at 550 ° C. for 1 hour to prepare catalyst N.
Properties of catalyst N are shown in Table 1.
実施例15
水硝子に硫酸を加えて調製したシリカヒドロゲルを熟成した後、これに硫酸ジルコニウム溶液を加えてSiO2/ZrO2重量比80/20のシリカジルコニア水和物を調製した。このシリカジルコニア水和物のスラリーを洗浄してナトリウムおよび硫酸根を除去したシリカジルコニア水和物スラリーを得た。
このスラリー30kg(SiO2−ZrO2として3kg)をスチームジャケット付き双腕型ニーダーにより蒸発濃縮しながら捏和して、可塑性のある捏和物(Z)とした。
この捏和物に平均粒子径2.1μmの三酸化二マンガン92.8g(Mn2O3として)を加えて、双腕型ニーダーにて20分間混練した。この三酸化二マンガン入りのシリカジルコニア捏和物をオーガー式押し出し機で、1.8mmの四つ葉型の柱状に押し出し成形した。得られたシリカジルコニア成形品は、110℃で16時間乾燥した後、さらに680℃で2時間焼成してマンガン粒子を3重量%含有するシリカジルコニア(SiO2−ZrO2)担体を得た。
該シリカジルコニア担体にモリブデンとニッケルとリンを酸化物として触媒組成物基準で5.9重量%、1.45重量%、1.5重量%となるように三酸化モリブデンと炭酸ニッケルのリン酸水溶液を含浸した後、回転式乾燥機を用いて室温から250℃まで昇温乾燥した。さらにこの乾燥品は、550℃で1時間空気中にて焼成し、触媒Oを調製した。
触媒Oの性状を表1に示す。
Example 15
A silica hydrogel prepared by adding sulfuric acid to water glass was aged, and then a zirconium sulfate solution was added thereto to prepare a silica zirconia hydrate having a SiO 2 / ZrO 2 weight ratio of 80/20. This silica zirconia hydrate slurry was washed to obtain a silica zirconia hydrate slurry from which sodium and sulfate radicals had been removed.
30 kg of this slurry (3 kg as SiO 2 —ZrO 2 ) was kneaded while evaporating and concentrating with a double-arm kneader equipped with a steam jacket to give a plastic kneaded product (Z).
To this kneaded product, 92.8 g of dimanganese trioxide having an average particle size of 2.1 μm (as Mn 2 O 3 ) was added and kneaded for 20 minutes with a double-arm kneader. The silica zirconia kneaded material containing dimanganese trioxide was extruded and formed into a 1.8 mm four-leaf column by an auger type extruder. The obtained silica zirconia molded article was dried at 110 ° C. for 16 hours, and further calcined at 680 ° C. for 2 hours to obtain a silica zirconia (SiO 2 —ZrO 2 ) support containing 3% by weight of manganese particles.
An aqueous phosphoric acid solution of molybdenum trioxide and nickel carbonate so that the silica zirconia support has 5.9 wt%, 1.45 wt% and 1.5 wt% based on the catalyst composition using molybdenum, nickel and phosphorus as oxides. After being impregnated, it was dried at an elevated temperature from room temperature to 250 ° C. using a rotary dryer. Further, this dried product was calcined in the air at 550 ° C. for 1 hour to prepare Catalyst O.
Properties of catalyst O are shown in Table 1.
比較例1
実施例1と同様にして調製した捏和物(V)をオーガー式押し出し機で、1.8mmの四つ葉型の柱状に押し出し成形した。得られたアルミナ成形品は、110℃で16時間乾燥した後、さらに680℃で2時間焼成してアルミナ担体を得た。
該アルミナ担体にモリブデンとニッケルとリンを酸化物として触媒組成物基準で5.9重量%、1.45重量%、1.5重量%となるように三酸化モリブデンと炭酸ニッケルのリン酸水溶液を含浸した後、回転式乾燥機を用いて室温から250℃まで昇温乾燥した。さらにこの乾燥品は、550℃で1時間空気中にて焼成し、触媒Pを調製した。
触媒Pの性状を表2に示す。
Comparative Example 1
The kneaded product (V) prepared in the same manner as in Example 1 was extruded into a 1.8 mm four-leaf type column using an auger type extruder. The obtained alumina molded article was dried at 110 ° C. for 16 hours and then calcined at 680 ° C. for 2 hours to obtain an alumina carrier.
An aqueous phosphoric acid solution of molybdenum trioxide and nickel carbonate is added to the alumina support so that it becomes 5.9 wt%, 1.45 wt% and 1.5 wt% based on the catalyst composition using molybdenum, nickel and phosphorus as oxides. After impregnation, the temperature was dried from room temperature to 250 ° C. using a rotary dryer. Further, this dried product was calcined in the air at 550 ° C. for 1 hour to prepare catalyst P.
Properties of the catalyst P are shown in Table 2.
比較例2
実施例1と同様にして調製した捏和物(V)をオーガー式押し出し機で、1.8mmの四つ葉型の柱状に押し出し成形した。得られたアルミナ成形品は、110℃で16時間乾燥した後、さらに680℃で2時間焼成してアルミナ担体を得た。
該アルミナ担体に、三酸化二マンガンとして担体基準で3重量%となるように硝酸マンガンおよびモリブデンとニッケルとリンを酸化物として触媒組成物基準で5.9重量%、1.45重量%、1.5重量%となるように三酸化モリブデンと炭酸ニッケルとリン酸との水溶液を含浸した後、回転式乾燥機を用いて室温から250℃まで昇温乾燥した。さらにこの乾燥品は、550℃で1時間空気中にて焼成し、触媒Qを調製した。
触媒Qの性状を表2に示す。
Comparative Example 2
The kneaded product (V) prepared in the same manner as in Example 1 was extruded into an 1.8 mm four-leaf type column using an auger type extruder. The obtained alumina molded article was dried at 110 ° C. for 16 hours and then calcined at 680 ° C. for 2 hours to obtain an alumina carrier.
To the alumina support, 5.9% by weight, 1.45% by weight, and 1.45% by weight based on the catalyst composition using manganese nitrate, molybdenum, nickel and phosphorus as oxides so as to be 3% by weight as dimanganese trioxide. After impregnating with an aqueous solution of molybdenum trioxide, nickel carbonate and phosphoric acid so as to be 5% by weight, it was dried by heating from room temperature to 250 ° C. using a rotary dryer. Further, this dried product was calcined in the air at 550 ° C. for 1 hour to prepare Catalyst Q.
Properties of catalyst Q are shown in Table 2.
比較例3
実施例1と同様にして、循環ライン及び薬液添加口2箇所を持つタンクに純水719kgを張り込み、これにイソブチレンと無水マレイン酸の共重合物0.5kgを添加し、約2時間高速攪拌し完全に溶解させた。
この溶液にアルミン酸ナトリウム水溶液(Al2O3として濃度22重量%)2kgを攪拌しながら添加し、60℃に加温し循環させた。次いで硫酸アルミニウム溶液(Al2O3として濃度7重量%)を添加して種子アルミナスラリーを調製した。
このスラリーに、アルミン酸ナトリウム水溶液(Al2O3として濃度22重量%)107.1kgと硫酸アルミニウム溶液(Al2O3として濃度7重量%)168.3kgを各々35.7kg/hrと56.1kg/hrの添加速度で、温度60℃下pH7.9〜8.1を保ち、攪拌および循環させながら、3時間かけて添加した。また、同じタイミングで、このタンクに別にとりつけた薬液添加口から硝酸マンガン水溶液(Mn2O3として1重量%)110.7kgを36.9kg/hrの添加速度で、3時間かけて添加した。
このアルミナ−マンガン(Al2O3−Mn2O3)水和物調合スラリーを洗浄してナトリウムおよび硫酸根、硝酸根を除去したアルミナ−マンガン水和物スラリーを得た。
このアルミナ−マンガン水和物のスラリーに純水を加えて、Al2O3−Mn2O3濃度10重量%のスラリーに調製し、15重量%アンモニア水にてpH11に調製した後還流器のついた熟成タンクにて95℃で8時間熟成した。
熟成終了後、このスラリー30kg(Al2O3+Mn2O3として3kg)をスチームジャケット付き双腕型ニーダーにより蒸発濃縮しながら捏和して、可塑性のある捏和物とした。このアルミナ−マンガン捏和物をオーガー式押し出し機で、1.8mmの四つ葉型の柱状に押し出し成形した。得られたアルミナ−マンガン成形品は、110℃で16時間乾燥した後、さらに680℃で2時間焼成して97重量%Al2O3−3重量%Mn2O3担体(アルミナ−マンガン担体)を得た。
該アルミナ−マンガン担体にモリブデンとニッケルとリンを酸化物として触媒組成物基準で5.9重量%、1.45重量%、1.5重量%となるように三酸化モリブデンと炭酸ニッケルのリン酸水溶液を含浸した後、回転式乾燥機を用いて室温から250℃まで昇温乾燥した。さらにこの乾燥品は、550℃で1時間空気中にて焼成し、触媒Rを調製した。
触媒Rの性状を表2に示す。
Comparative Example 3
In the same manner as in Example 1, 719 kg of pure water was put into a tank having a circulation line and two chemical addition ports, 0.5 kg of a copolymer of isobutylene and maleic anhydride was added thereto, and the mixture was stirred at high speed for about 2 hours. It was completely dissolved.
To this solution, 2 kg of an aqueous sodium aluminate solution (concentration: 22% by weight as Al 2 O 3 ) was added with stirring, heated to 60 ° C. and circulated. Next, an aluminum sulfate solution (7% by weight as Al 2 O 3 ) was added to prepare a seed alumina slurry.
To this slurry, 107.1 kg of an aqueous solution of sodium aluminate (concentration 22% by weight as Al 2 O 3 ) and 168.3 kg of aluminum sulfate solution (concentration 7% by weight as Al 2 O 3 ) were respectively 35.7 kg / hr and 56. It was added over 3 hours at a rate of 1 kg / hr while maintaining a pH of 7.9 to 8.1 at a temperature of 60 ° C. while stirring and circulating. Further, at the same timing, 110.7 kg of an aqueous manganese nitrate solution (1 wt% as Mn 2 O 3 ) was added at a rate of 36.9 kg / hr over 3 hours from a chemical solution addition port separately attached to this tank.
This alumina-manganese (Al 2 O 3 —Mn 2 O 3 ) hydrate preparation slurry was washed to obtain an alumina-manganese hydrate slurry from which sodium, sulfate radicals and nitrate radicals were removed.
Pure water is added to the alumina-manganese hydrate slurry to prepare a slurry having an Al 2 O 3 —Mn 2 O 3 concentration of 10% by weight, adjusted to pH 11 with 15% by weight ammonia water, It was aged at 95 ° C. for 8 hours in the attached aging tank.
After the ripening, the (as Al 2 O 3 + Mn 2 O 3 3kg) slurry 30kg was kneaded while evaporated by a twin-arm kneader with a steam jacket was kneaded product with a plastic. This alumina-manganese kneaded product was extruded into an 1.8 mm four-leaf type column by an auger type extruder. The obtained alumina-manganese molded article was dried at 110 ° C. for 16 hours, and further calcined at 680 ° C. for 2 hours to be 97 wt% Al 2 O 3 -3 wt% Mn 2 O 3 support (alumina-manganese support) Got.
Phosphoric acid of molybdenum trioxide and nickel carbonate so that the alumina-manganese support has molybdenum, nickel, and phosphorus as oxides, and becomes 5.9 wt%, 1.45 wt%, and 1.5 wt% based on the catalyst composition. After impregnating with the aqueous solution, it was dried by heating from room temperature to 250 ° C. using a rotary dryer. Further, this dried product was calcined in the air at 550 ° C. for 1 hour to prepare Catalyst R.
Table 2 shows the properties of the catalyst R.
比較例4
実施例12と同様にして調製した捏和物(W)をオーガー式押し出し機で、1.8mmの四つ葉型の柱状に押し出し成形した。得られたシリカアルミナ成形品は、110℃で16時間乾燥した後、さらに680℃で2時間焼成してシリカアルミナ(SiO2−Al2O3)担体を得た。
該シリカアルミナ担体に、三酸化二マンガンとして担体基準で3重量%となるように硝酸マンガンおよびモリブデンとニッケルとリンを酸化物として触媒組成物基準で5.9重量%、1.45重量%、1.5重量%となるように三酸化モリブデンと炭酸ニッケルとリン酸との水溶液を含浸した後、回転式乾燥機を用いて室温から250℃まで昇温乾燥した。さらにこの乾燥品は、550℃で1時間空気中にて焼成し、触媒Sを調製した。
触媒Sの性状を表2に示す。
Comparative Example 4
The kneaded product (W) prepared in the same manner as in Example 12 was extruded into a 1.8 mm four-leaf type column with an auger type extruder. The obtained silica alumina molded article was dried at 110 ° C. for 16 hours, and further calcined at 680 ° C. for 2 hours to obtain a silica alumina (SiO 2 —Al 2 O 3 ) support.
To the silica-alumina support, 5.9% by weight, 1.45% by weight, based on the catalyst composition, using manganese nitrate and molybdenum, nickel, and phosphorus as oxides so as to be 3% by weight as dimanganese trioxide. After impregnating with an aqueous solution of molybdenum trioxide, nickel carbonate and phosphoric acid so as to be 1.5% by weight, the mixture was dried at an elevated temperature from room temperature to 250 ° C. using a rotary dryer. Further, this dried product was calcined in the air at 550 ° C. for 1 hour to prepare Catalyst S.
Table 2 shows the properties of the catalyst S.
比較例5
実施例13と同様にして調製した捏和物(X)をオーガー式押し出し機で、1.8mmの四つ葉型の柱状に押し出し成形した。得られたシリカ成形品は、110℃で16時間乾燥した後、さらに680℃で2時間焼成してシリカ担体を得た。
該シリカ担体に、三酸化二マンガンとして担体基準で3重量%となるように硝酸マンガンおよびモリブデンとニッケルとリンを酸化物として触媒組成物基準で5.9重量%、1.45重量%、1.5重量%となるように三酸化モリブデンと炭酸ニッケルとリン酸との水溶液を含浸した後、回転式乾燥機を用いて室温から250℃まで昇温乾燥した。さらにこの乾燥品は、550℃で1時間空気中にて焼成し、触媒Tを調製した。
触媒Tの性状を表2に示す。
Comparative Example 5
The kneaded material (X) prepared in the same manner as in Example 13 was extruded into an 1.8 mm four-leaf type column using an auger type extruder. The obtained silica molded article was dried at 110 ° C. for 16 hours, and further calcined at 680 ° C. for 2 hours to obtain a silica carrier.
To the silica support, 5.9% by weight, 1.45% by weight, and 1.45% by weight based on the catalyst composition using manganese nitrate, molybdenum, nickel, and phosphorus as oxides so as to be 3% by weight as dimanganese trioxide. After impregnating with an aqueous solution of molybdenum trioxide, nickel carbonate and phosphoric acid so as to be 5% by weight, it was dried by heating from room temperature to 250 ° C. using a rotary dryer. Further, this dried product was calcined in the air at 550 ° C. for 1 hour to prepare Catalyst T.
Table 2 shows the properties of the catalyst T.
比較例6
実施例14と同様にして調製した捏和物(Y)をオーガー式押し出し機で、1.8mmの四つ葉型の柱状に押し出し成形した。得られたシリカチタニア成形品は、110℃で16時間乾燥した後、さらに680℃で2時間焼成してシリカチタニア(SiO2−TiO2)担体を得た。
該シリカチタニア担体に、三酸化二マンガンとして担体基準で3重量%となるように硝酸マンガンおよびモリブデンとニッケルとリンを酸化物として触媒組成物基準で5.9重量%、1.45重量%,1.5重量%となるように三酸化モリブデンと炭酸ニッケルとリン酸との水溶液を含浸した後、回転式乾燥機を用いて室温から250℃まで昇温乾燥した。さらにこの乾燥品は、550℃で1時間空気中にて焼成し、触媒Uを調製した。
触媒Uの性状を表2に示す。
Comparative Example 6
The kneaded product (Y) prepared in the same manner as in Example 14 was extruded into a 1.8 mm four-leaf type column using an auger type extruder. The obtained silica titania molded article was dried at 110 ° C. for 16 hours, and further calcined at 680 ° C. for 2 hours to obtain a silica titania (SiO 2 —TiO 2 ) support.
To the silica titania support, 5.9% by weight, 1.45% by weight, based on the catalyst composition, using manganese nitrate and molybdenum, nickel, and phosphorus as oxides so as to be 3% by weight as the support based on dimanganese trioxide, After impregnating with an aqueous solution of molybdenum trioxide, nickel carbonate and phosphoric acid so as to be 1.5% by weight, the mixture was dried at an elevated temperature from room temperature to 250 ° C. using a rotary dryer. Furthermore, this dried product was calcined in the air at 550 ° C. for 1 hour to prepare Catalyst U.
Table 2 shows the properties of the catalyst U.
比較例7
実施例15と同様にして調製した捏和物(Z)をオーガー式押し出し機で、1.8mmの四つ葉型の柱状に押し出し成形した。得られたシリカジルコニア成形品は、110℃で16時間乾燥した後、さらに680℃で2時間焼成してシリカジルコニア(SiO2−ZrO2)担体を得た。
該シリカジルコニア担体に、三酸化二マンガンとして担体基準で3重量%となるように硝酸マンガンおよびモリブデンとニッケルとリンを酸化物として触媒組成物基準で5.9重量%、1.45重量%、1.5重量%となるように三酸化モリブデンと炭酸ニッケルとリン酸との水溶液を含浸した後、回転式乾燥機を用いて室温から250℃まで昇温乾燥した。さらにこの乾燥品は、550℃で1時間空気中にて焼成し、触媒Vを調製した。
触媒Vの性状を表2に示す。
Comparative Example 7
The kneaded product (Z) prepared in the same manner as in Example 15 was extruded into a 1.8 mm four-leaf type column using an auger type extruder. The obtained molded product of silica zirconia was dried at 110 ° C. for 16 hours, and further calcined at 680 ° C. for 2 hours to obtain a silica zirconia (SiO 2 —ZrO 2 ) support.
To the silica zirconia support, 5.9% by weight, 1.45% by weight, based on the catalyst composition, using manganese nitrate and molybdenum, nickel, and phosphorus as oxides so as to be 3% by weight as dimanganese trioxide. After impregnating with an aqueous solution of molybdenum trioxide, nickel carbonate and phosphoric acid so as to be 1.5% by weight, the mixture was dried at an elevated temperature from room temperature to 250 ° C. using a rotary dryer. Further, this dried product was calcined in the air at 550 ° C. for 1 hour to prepare Catalyst V.
Properties of catalyst V are shown in Table 2.
実施例16 活性評価試験
実施例1〜15で調製した触媒A〜Oおよび比較例1〜7で調製した触媒P〜Vについて、固定床式のマイクロリアクターを用いて次に示す条件で反応温度を変えて活性評価試験を行った。
反応条件; 触媒充填量 200 ml
反応圧力 15 MPa
液空間速度(LHSV) 0.45 hr−1
水素/油比(H2/HC) 800 Nm3/kl
反応温度 370℃、380℃、390℃
また、原料油には下記性状の常圧残渣油を使用した。
原料油性状; 比重(15/4℃) 0.9831g/cm3
残炭 11.7 重量%
アスファルテン分 5.2 重量%
イオウ分 4.236 重量%
メタル(Ni+V)量 85.3 重量%
活性評価は、上記反応条件下で反応温度380℃一定のもと原料油を通油し、定期的に測定した生成油中のメタル(Ni+V)量と通油量から触媒に堆積したメタル量(MOC:Metal on Catalyst)が15重量%になった時に、目標反応温度を370、380、390℃と変えて反応させ、実際の反応温度に対して脱メタル率及び脱硫率をプロットして引いた直線から、反応温度380℃に対応する脱メタル率及び脱硫率を求めた。
その値を表1及び2に示す。
なお、脱メタル率および脱硫率は次式により求めた。
脱メタル率={(原料油中のメタル濃度−水素化処理生成油中のメタル濃度)/原料油中のメタル濃度}×100
脱硫率={(原料油中の硫黄濃度−水素化処理生成油中の硫黄濃度)/原料油中の硫黄濃度}×100
実施例、比較例中、原料のマンガン化合物の平均粒子径は、沈降速度による光透過性や沈降法により測定した値を示す。
表1及び2における、触媒中のマンガン化合物の平均粒子径(μm)は、触媒のある断面を走査型電子顕微鏡で写真を撮り、写真からマンガン化合物の粒子の大きさを測定した値を示す。
Example 16 Activity Evaluation Test Regarding the catalysts A to O prepared in Examples 1 to 15 and the catalysts P to V prepared in Comparative Examples 1 to 7, the reaction temperature was changed under the following conditions using a fixed bed type microreactor. The activity evaluation test was conducted by changing.
Reaction conditions; catalyst loading 200 ml
Reaction pressure 15 MPa
Liquid space velocity (LHSV) 0.45 hr −1
Hydrogen / oil ratio (H 2 / HC) 800 Nm 3 / kl
Reaction temperature 370 ° C, 380 ° C, 390 ° C
Moreover, the normal pressure residual oil of the following property was used for raw material oil.
Raw material oil properties; specific gravity (15/4 ° C.) 0.9831 g / cm 3
Remaining charcoal 11.7% by weight
Asphaltene content 5.2% by weight
Sulfur content 4.236% by weight
Metal (Ni + V) amount 85.3 wt%
The activity was evaluated by passing the feedstock oil under a constant reaction temperature of 380 ° C. under the above reaction conditions, and periodically measuring the amount of metal (Ni + V) in the produced oil and the amount of metal deposited on the catalyst ( When the MOC (Metal on Catalyst) reached 15% by weight, the target reaction temperature was changed to 370, 380, and 390 ° C., and the demetalization rate and desulfurization rate were plotted and drawn against the actual reaction temperature. From the straight line, the demetallation rate and desulfurization rate corresponding to the reaction temperature of 380 ° C. were determined.
The values are shown in Tables 1 and 2.
The demetalization rate and desulfurization rate were determined by the following formulas.
Demetalization rate = {(metal concentration in raw material oil−metal concentration in hydrotreated product oil) / metal concentration in raw material oil} × 100
Desulfurization rate = {(sulfur concentration in feedstock-sulfur concentration in hydrotreated oil) / sulfur concentration in feedstock} × 100
In the examples and comparative examples, the average particle diameter of the raw material manganese compound indicates the value measured by the light transmittance by the sedimentation rate or the sedimentation method.
In Tables 1 and 2, the average particle size (μm) of the manganese compound in the catalyst is a value obtained by measuring the size of the manganese compound particles from the photograph taken with a scanning electron microscope.
表1及び2の結果から、本発明における実施例1〜11の触媒A〜Kは、触媒中に粒子状マンガン化合物が存在している触媒で、これらの触媒はマンガン化合物を含有していない比較例1の触媒Pやマンガン化合物が粒子として存在していない比較例2、3の触媒Q、Rよりも脱硫率および脱メタル率の値が大きく、脱硫活性および脱メタル活性に優れていることが判る。
実施例12に示す触媒Lと比較例4に示す触媒S、実施例13に示す触媒Mと比較例5に示す触媒T、実施例14に示す触媒Nと比較例6に示す触媒U、実施例15に示す触媒Oと比較例7に示す触媒Vは、各々担体成分は同じで、マンガン化合物が粒子として存在するものとしないものとを比較したものである。この結果から、先と同様にマンガン化合物粒子が存在している触媒の方が脱硫率、脱メタル率の値が大きく、脱硫活性および脱メタル活性に優れていることが判る。
From the results of Tables 1 and 2, Catalysts A to K of Examples 1 to 11 in the present invention are catalysts in which particulate manganese compounds are present in the catalyst, and these catalysts do not contain manganese compounds. The catalyst P and manganese compound of Example 1 are not present as particles, and the desulfurization rate and demetalization rate are larger than those of Catalysts Q and R of Comparative Examples 2 and 3, and are excellent in desulfurization activity and demetalization activity. I understand.
Catalyst L shown in Example 12 and Catalyst S shown in Comparative Example 4, Catalyst M shown in Example 13 and Catalyst T shown in Comparative Example 5, Catalyst N shown in Example 14 and Catalyst U shown in Comparative Example 6, Example The catalyst O shown in 15 and the catalyst V shown in Comparative Example 7 have the same carrier component and are compared with those in which the manganese compound is not present as particles. From this result, it can be seen that the catalyst in which manganese compound particles are present as in the previous case has higher desulfurization rate and demetalization value, and is superior in desulfurization activity and demetalization activity.
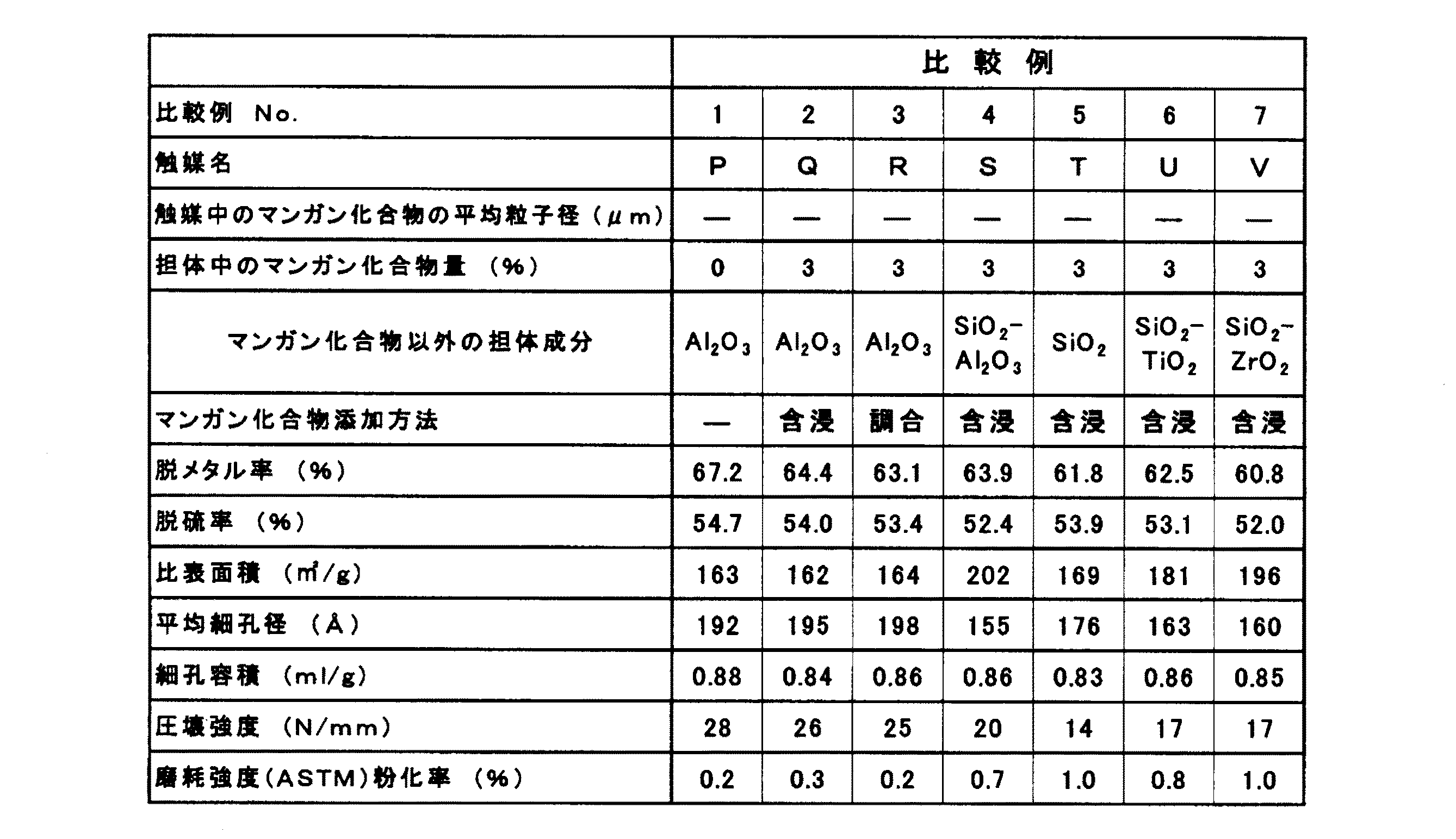
表1、表2において、
(注) 1)圧壊強度は、前処理として、試料を500℃で1時間焼成したものを室温までデシケーターにて冷却したものから長さ4mm以上の試料40個以上を木屋式硬度計(圧縮子3.18mm)を用いて圧縮し、破砕された時の荷重を求めて次式により算出した。
圧壊強度(N/mm)=S×9.807/L×n
ここで、Sは加圧荷重の総和(kg)、Lは圧縮子の径(3.18mm)、nは測定個数を表す。
2)磨耗強度粉化率は、ASTM法D 4058−96に基づいて求めた。
具体的には、前処理として、850μmの篩で静かに篩い分けた篩上の試料を500℃で1時間焼成した後室温までデシケーターにて冷却し、この中から試料を100g秤量し、ASTM法D 4058−96に定められたドラム内に入れ、60±5R.P.Mの速度で30分間回転させた。この試料を全量回収し、850μmの篩で静かに篩い分け、その篩上の試料を500℃で1時間焼成した後室温までデシケーターにて冷却し、これを秤量して次式により求めた。
磨耗強度粉化率(%)=(W0−W)/W0 ×100
ここで、W0は試料重量(g)、Wは測定後の850μm篩上焼成試料の重量(g)を表す。
In Table 1 and Table 2,
(Note) 1) The crushing strength was measured as a pretreatment of 40 or more samples of a length of 4 mm or more from a sample fired at 500 ° C. for 1 hour and cooled with a desiccator to a room temperature. 3.18 mm), and the load when crushed was obtained and calculated by the following formula.
Crushing strength (N / mm) = S × 9.807 / L × n
Here, S is the total pressure load (kg), L is the diameter of the compressor (3.18 mm), and n is the number of measurements.
2) The abrasion strength powdering rate was determined based on ASTM method D 4058-96.
Specifically, as a pretreatment, a sample on a sieve gently sieved with an 850 μm sieve was baked at 500 ° C. for 1 hour, cooled to room temperature with a desiccator, 100 g of the sample was weighed, and ASTM method D 4058-96, put in the drum specified 60 ± 5R. P. Rotated at M speed for 30 minutes. The whole amount of this sample was collected and gently sieved with a 850 μm sieve, the sample on the sieve was baked at 500 ° C. for 1 hour, cooled to room temperature with a desiccator, weighed, and determined by the following formula.
Abrasion strength powdering rate (%) = (W 0 −W) / W 0 × 100
Here, W 0 represents the sample weight (g), and W represents the weight (g) of the baked sample on the 850 μm sieve after the measurement.
Claims (7)
A hydrotreating method for hydrocarbon oil, wherein the hydrotreating catalyst composition according to any one of claims 1 to 6 is used as a catalyst.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004118974A JP4471717B2 (en) | 2004-04-14 | 2004-04-14 | Hydrocarbon hydrotreating catalyst composition and hydrocarbon oil hydrotreating method using the catalyst composition. |
| KR1020050030691A KR101158924B1 (en) | 2004-04-14 | 2005-04-13 | Catalyst composition for hydrogenating hydrocarbon oil and hydrotreatment of hydrocarbon oil by using the same |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004118974A JP4471717B2 (en) | 2004-04-14 | 2004-04-14 | Hydrocarbon hydrotreating catalyst composition and hydrocarbon oil hydrotreating method using the catalyst composition. |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2005296867A true JP2005296867A (en) | 2005-10-27 |
| JP4471717B2 JP4471717B2 (en) | 2010-06-02 |
Family
ID=35329058
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004118974A Expired - Lifetime JP4471717B2 (en) | 2004-04-14 | 2004-04-14 | Hydrocarbon hydrotreating catalyst composition and hydrocarbon oil hydrotreating method using the catalyst composition. |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP4471717B2 (en) |
| KR (1) | KR101158924B1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012139617A (en) * | 2010-12-28 | 2012-07-26 | Jgc Catalysts & Chemicals Ltd | Hydrogenation treatment catalyst for hydrocarbon oil and hydrogen treatment method using the same |
| CN117943077A (en) * | 2022-10-31 | 2024-04-30 | 中国石油化工股份有限公司 | A catalyst for light distillate oil hydrodenitrogenation and its preparation method and application |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH07316565A (en) * | 1994-05-23 | 1995-12-05 | Nippon Oil Co Ltd | Hydroprocessing method for heavy oil |
| EP0946294B1 (en) * | 1996-11-07 | 2002-09-25 | Institut Français du Pétrole | Catalyst having at least one element of group viib and its use in hydro-treating |
| JP2001104790A (en) * | 1999-10-07 | 2001-04-17 | Tonengeneral Sekiyu Kk | Hydrotreating catalyst and method for hydrotreating hydrocarbon oil using the same |
| US7105140B2 (en) * | 2002-03-04 | 2006-09-12 | Conocophillips Company | Desulfurization compositions |
-
2004
- 2004-04-14 JP JP2004118974A patent/JP4471717B2/en not_active Expired - Lifetime
-
2005
- 2005-04-13 KR KR1020050030691A patent/KR101158924B1/en not_active Expired - Fee Related
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012139617A (en) * | 2010-12-28 | 2012-07-26 | Jgc Catalysts & Chemicals Ltd | Hydrogenation treatment catalyst for hydrocarbon oil and hydrogen treatment method using the same |
| CN117943077A (en) * | 2022-10-31 | 2024-04-30 | 中国石油化工股份有限公司 | A catalyst for light distillate oil hydrodenitrogenation and its preparation method and application |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20060045658A (en) | 2006-05-17 |
| KR101158924B1 (en) | 2012-06-21 |
| JP4471717B2 (en) | 2010-06-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5545602A (en) | Catalyst with specified pore size distribution | |
| EP1773969B1 (en) | Two-step hydroprocessing method for heavy hydrocarbon oil | |
| JP4638610B2 (en) | Hydrotreating catalyst and hydrotreating method | |
| CA2429944C (en) | Alumina having novel pore structure, method of making and catalysts made therefrom | |
| EP2424663B1 (en) | Method for making a hydroconversion multi-metallic catalyst | |
| JP6506430B2 (en) | Improved resid hydroprocessing catalyst containing titania | |
| JP3821483B2 (en) | Catalyst for hydrotreatment and its utilization | |
| TWI611836B (en) | Catalyst support and preparation method thereof | |
| EP2772308B1 (en) | Hydrogenation catalyst and method for producing same | |
| JP4818163B2 (en) | Alumina support, hydrodemetallation catalyst using the same, and production method thereof | |
| CA2360121C (en) | Hydroprocessing catalyst and use thereof | |
| JPH06500728A (en) | hydrogenation catalyst | |
| JP4369871B2 (en) | Heavy material HPC process using a mixture of catalysts | |
| JP2000000470A (en) | Hydrotreating catalyst and method for hydrotreating heavy oil | |
| KR20170003593A (en) | A catalyst and its use for the selective hydrodesulfurization of an olefin containing hydrocarbon feedstock | |
| JP2002363575A (en) | Method for two-stage hydrotreatment of heavy hydrocarbon oil | |
| CN109475845A (en) | Hydroprocessing catalyst and method for preparing the same | |
| JP4471717B2 (en) | Hydrocarbon hydrotreating catalyst composition and hydrocarbon oil hydrotreating method using the catalyst composition. | |
| JP5645652B2 (en) | Hydrocarbon hydrotreating catalyst and hydrotreating method using the same | |
| JP4800637B2 (en) | Hydrocarbon oil hydrotreating catalyst composition and hydrocarbon oil hydrotreating method using the catalyst composition | |
| JP2010201281A (en) | Hydrogen demetalization catalyst for hydrocarbon oil and hydrogen treating method using the catalyst | |
| JP5334632B2 (en) | Hydrocarbon hydrotreating catalyst and hydrotreating method using the same | |
| JP3949375B2 (en) | Hydrocarbon hydrotreating catalyst composition and hydrotreating method using the same | |
| JP3333231B2 (en) | Hydrodesulfurization catalyst and method for producing the same | |
| JPH06134312A (en) | Hydrogenation treating catalyt composition |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20070412 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20091109 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20091117 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100115 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100216 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20100302 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130312 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4471717 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130312 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140312 Year of fee payment: 4 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |