JP2004131701A - Polyetherester elastomer - Google Patents
Polyetherester elastomer Download PDFInfo
- Publication number
- JP2004131701A JP2004131701A JP2003203870A JP2003203870A JP2004131701A JP 2004131701 A JP2004131701 A JP 2004131701A JP 2003203870 A JP2003203870 A JP 2003203870A JP 2003203870 A JP2003203870 A JP 2003203870A JP 2004131701 A JP2004131701 A JP 2004131701A
- Authority
- JP
- Japan
- Prior art keywords
- polyetherester elastomer
- group
- methyl
- ether glycol
- oxetane
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images
Landscapes
- Polyesters Or Polycarbonates (AREA)
Abstract
Description
【0001】
【発明の属する技術分野】
本発明は高温から低温までの物性の変化が少なく、広い温度域でゴム状弾性を有するポリエーテルエステルエラストマーに関する。
【0002】
【従来の技術】
ポリエーテルエステルエラストマーは、主としてポリブチレンテレフタレートをハードセグメントとし、ポリエーテルグリコールをソフトセグメントとして共重合されており、ゴム状弾性を有し、機械強度、耐熱性、及び耐油性に優れることから、自動車部品、電気・電子部品、工業用部品、精密機械部品、生活用品等に広く利用されている。
【0003】
通常、ソフトセグメントとしてポリテトラメチレンエーテルグリコールが広く利用されているが、そのポリテトラメチレンエーテルグリコール成分が、低温領域において結晶化を起こし、使用条件によっては温度とともに物性が変化してしまい、例えば、低温領域においてゴム状弾性が発現しなくなるという問題があった。
【0004】
このような問題を解決する為に、原料のポリテトラメチレンエーテルグリコールの分子量分布(Mv/Mn)を狭く(Mv/Mn≦1.60)し、数平均分子量(Mn)を比較的小さくする試みがなされている(特公平3−40732号公報)。ここで、分子量分布(Mv/Mn)は、末端水酸基価より求めた数平均分子量(Mn)の、式Mv=antilog(0.4931ogη+3.0646)で規定される粘度平均分子量(Mv)に対する比の値である。但し、ηは40°Cの温度における溶融粘度をポアズで示した値である。
【0005】
また、ポリエーテルグリコールの結晶化を防ぐ目的で、側鎖にメチル基のついたもの、例えばポリ(2−メチル−1,3−プロピレンオキシ)グリコールをソフトセグメントとして使用すること(例えば、特許文献1参照)や、ポリエーテルグリコールの結晶性を防ぐ目的で、ネオペンチレンオキシドとテトラメチレンオキシドからなるポリエーテルグリコールをソフトセグメントして使用すること(例えば、特許文献2参照)が提案されている。
【0006】
【特許文献1】
特公平3−80170号公報
【特許文献2】
特許3117805号公報
【0007】
【発明が解決しようとする課題】
しかしながら、ポリテトラメチレンエーテルグリコールの分子量分布(Mv/Mn)を狭くしても、使用できるポリテトラメチレンエーテルグリコールの数平均分子量(Mn)は限定されており、特に融点を高く設定したいときは、耐熱性と低温特性、耐屈曲摩耗性及び弾性回復性等との物性バランスに劣る欠点を有する。
【0008】
また、ソフトセグメントとして側鎖にメチル基のついたポリエーテルグリコールを使用する場合には、比較的多量の側鎖にメチル基のついたポリオキシアルキレンユニットを有さないと効果が発現しないことや、原料の環状エーテル(例えばメチルテトラヒドロフラン、メチルオキセタン)が高価であることなどから、この方法は工業的には殆ど利用されていないのが現状である。
さらにネオペンチレンオキシドとテトラメチレンオキシドからなるポリエーテルグリコールをソフトセグメントとして使用しても、より低温度領域では物性変化が起こり、性能的にはまだ不十分である。
【0009】
本発明はこのような不具合を解決するためのものであり、ソフトセグメント成分となるポリエーテルグリコールを適切に選定することにより、高温から低温までの物性の変化を少なくし、ゴム状弾性を広い温度域で有する新規なポリエーテルエステルエラストマーを提供することを目的とする。
【0010】
【課題を解決するための手段】
本発明者等は、上記の問題点を解決するために鋭意研究を重ねた結果、ある特定の粘弾性挙動を示すポリエーテルエステルエラストマーが、低温においても良好なゴム状弾性を示すことを見出し、本発明に至った。
【0011】
すなわち、本発明の要旨は、縦振動での動的粘弾性測定の−50℃における貯蔵弾性率の常用対数の値(logE’−50)が、
【数2】
7.0≦logE’−50≦8.1
【0012】
であることを特徴とするポリエーテルエステルエラストマーに存している。
本発明の他の要旨は、DSCによる昇温スピード10℃/minでの測定において、−50℃〜100℃における吸熱熱量ΔHfが0mJ/mg〜8mJ/mg以下であることを特徴とするポリエーテルエステルエラストマーに存している。
本発明のもう一つの要旨は、―50℃〜100℃の間に融点を持たないことを特徴とするポリエーテルエステルエラストマーに存している。
【0013】
本発明の他のもう一つの要旨は、ポリアルキレンエーテルグリコールセグメントの数平均分子量が400〜10000であることを特徴とするポリエーテルエステルエラストマーに存している。
【0014】
【発明の実施の形態】
ポリエーテルエステルエラストマーとしては種々のものが知られているが、その代表的なものは、主として芳香族ポリエステルからなるハードセグメントと、主として長鎖ポリエーテルからなるソフトセグメントとを主な構成成分とするポリエーテルエステルエラストマーである。
【0015】
本発明に係るポリエーテルエステルエラストマーは縦振動での動的粘弾性測定の−50℃における貯蔵弾性率の常用対数の値(logE’−50)が、
【0016】
【数3】
7.0≦logE’−50≦8.1
【0017】
である。logE’−50が7.0より小さいと柔らかすぎて強度が出ず、8.1より大きいと硬くてもろくなる。好ましくは7.2〜8.0で、より好ましくは7.3〜7.9である。通常のポリエーテルエステルエラストマーは0℃以下の低温では硬くてもろくなる。特に−50℃以下では硬くなり過ぎてゴム状弾性が発現しない。しかし、本発明のポリエーテルエステルエラストマーは、縦振動による動的粘弾性の−50℃のlogE’の値が7.0〜8.1と低い値であり、柔軟性を保持している。
【0018】
さらに本発明に係るポリエーテルエステルエラストマーは、DSCによる昇温スピード10℃/minでの測定において、−50℃〜100℃における吸熱熱量ΔHfが0mJ/mg〜8mJ/mgであることが好ましい。より好ましくは0mJ/mg〜5mJ/mgであり、より好ましくは0mJ/mg〜3mJ/mgである。8mJ/mgを越える吸熱ピークがあると、常用温度域である−50℃〜100℃の間で機械強度、ゴム状弾性等が変化し、低温特性が低下する場合がある。
さらに本発明に係わるポリエーテルエステルエラストマーは、―50℃〜100℃の間に融点を持たないことがさらに好ましい。この間に融点があると、常用温度域であるー50℃〜100℃の間で機械強度、ゴム状弾性等が変化し、低温特性が低下することがある。
【0019】
本発明においてハードセグメントはジカルボン酸と短鎖のジオールとの組み合わせにより構成される。本発明のジカルボン酸成分をなす芳香族ジカルボン酸及びそのエステル形成性誘導体としては、ポリエステルの原料、特にポリエーテルエステル系エラストマーの原料として一般的に用いられているものが使用でき、例えばテレフタル酸、イソフタル酸、フタル酸、2,6−ナフタレンジカルボン酸等が挙げられる。なかでも、テレフタル酸、2,6−ナフタレンジカルボン酸が好ましく、特にテレフタル酸が好適である。これらの芳香族ジカルボン酸も所望ならば2種以上を併用してもよい。また、芳香族ジカルボン酸のアルキルエステルを用いる場合は、上記の芳香族ジカルボン酸のジメチルエステルやジエチルエステル等が用いられる。好ましいものは、ジメチルテレフタレート及び2,6−ジメチルナフタレンジカルボキシレートである。
【0020】
本発明における短鎖ジオール成分としては、ポリエステルの原料、特にポリエーテルエステル系エラストマーの原料として通常用いられるものが使用できる。例えば、エチレングリコール、1,2−プロピレングリコール、トリメチレングリコール、1,4−ブタンジオール、1,4−シクロヘキサンジオール、1,4−シクロヘキサンジメタノール等が挙げられ、中でも1,4−ブタンジオール、エチレングリコールが好ましく、特に1,4−ブタンジオールが好ましい。これらのジオールは、通常、単独で用いるが、所望ならば2種以上の混合物を使用することもできる。
また、上記のジオール及びジカルボン酸成分以外に3官能のアルコールやカルボン酸、又はそのエステルを少量共重合させてもよく、更にアジピン酸等の脂肪族ジカルボン酸、又はそのジアルキルエステルも少量ならば共重合成分として使用できる。
【0021】
本発明のポリエーテルエステルエラストマーにおけるソフトセグメント、即ち長鎖ポリエステルを構成するポリアルキレンエーテルグリコールとしては、数平均分子量が400〜10000であることが好ましい。600〜6000のものがより好ましく、特に1000〜5000のものが好適である。この数平均分子量が400未満になると、重合する最終ポリエーテルエステルのハード/ソフト比にもよるが、通常は短鎖ポリエステル(ハードセグメント)の平均連鎖長が小さくなり、融点降下が激しくなって耐熱性に劣るため、ポリエーテルエステルエラストマーとしてそのまま材料に使用する場合に好ましくない。また、10000を越えると、ポリアルキレンエーテルグリコールの粘度が上がり、系内での相分離が起きやすく、共重合等で得られるポリマーの物性が低下する傾向となる。なお、ここでいう「数平均分子量」とは、ゲル浸透クロマトグラフィー(GPC)で測定されたものである。GPCのキャリブレーションには、英国POLYMER LABORATORIES社のPOLYTETRAHYDROFURAN
キャリブレーションキットを使用すればよい。
【0022】
ポリアルキレンエーテルグリコールとしては、例えば、ポリエチレンエーテルグリコール、ポリ(1,2−プロピレンエーテル)グリコール、ポリ(1,3−プロピレンエーテル)グリコール、ポリ(テトラメチレンエーテル)グリコール、ポリ(ヘキサメチレンエーテル)グリコール、エチレンオキシドとプロピレンオキシドのブロック又はランダム共重合体、エチレンオキシドとTHFのブロック又はランダム共重合体、ポリ(プロピレンエーテル)ジイミドジ酸等が挙げられる。一般にポリアルキレンエーテルグリコールは結晶性であり、融点を持つ。そのため上記のポリアルキレンエーテルグリコールを用いてポリエーテルエステルエラストマーを重合すると、通常、−50℃〜100℃の低温域でポリアルキレンエーテルグリコール由来の融点(吸熱ピーク)が発現し、その融点を境に物性が大きく変化する。そこで、ポリアルキレンエーテルグリコールとしては、融点がないもの、融点が低いもの、融点があっても融解熱量(吸熱熱量)が低いものが好ましい。
上記のような性質を持つポリアルキレンエーテルグリコールのうち、下記式(1)の構造を有するものが好ましい。
【0023】
【化3】

式(1)中R1、R2は任意の置換基であり同一でも互いに異なっていてもよい。合成の容易さから、R1、R2は水素、アルキル基、ハロゲン原子、アルコキシ基、アリールオキシ基、アシルオキシ基、アシル基、アリールカルボキシル基、アリールカルボニル基、トリアルキルシリル基、トリアリールシリル基、トリアルキルシリルオキシ基、トリアリールシリルオキシ基、から選ばれることが好ましい。
【0025】
また、これらの基に含まれる炭化水素部分の水素は、ハロゲン原子、アルコキシ基、アリールオキシ基、アシルオキシ基、アシル基、アリールカルボキシル基、アリールカルボニル基、トリアルキルシリル基、トリアリールシリル基、トリアルキルシリルオキシ基、トリアリールシリルオキシ基、アルキル基、ビニル基、アルキルスルフォキシ基、アリールスルフォキシ基、シアノ基等で置換されていてもよい。ただし、R1、R2がともにメチル基であるものは所望の性能を示さないため、これを除く。また、R1、R2は炭素原子、酸素原子またはケイ素原子を介して互いに結合していてもよい。
ポリアルキレンエーテルグリコール中の式(1)の構造を有するユニットの含有量は、2モル%から100モル%が好ましい。含有量が少なすぎると所望の特性が得られないため、より好ましくは5モル%から100モル%である。
【0026】
さらに、これらの内でも、ポリアルキレンエーテルグリコールの側鎖に水素原子以外の原子やアルキル鎖等を導入したり、異なるアルキレンエーテルを共重合させたりして結晶性を下げ、−50℃〜100℃における吸熱熱量を下げることが好ましい。ポリアルキレンエーテルグリコールに側鎖を導入する方法としては過酸化物などを用いてグラフトさせたり、側鎖のついたアルキレンオキサイドユニットを共重合させる等の方法が挙げられる。特に側鎖のついたアルキレンオキサイドユニットを共重合させることが好ましい。重合性から考えると、オキセタン誘導体やテトラヒドロフラン誘導体等を開環させてアルキレンオキサイドユニットとし、他のポリアルキレンエーテルグリコールと共重合させることが好ましい。この共重合ポリアルキレンエーテルグリコールを用いることによって、ポリエーテルエステルエラストマーの低温における融点を飛躍的に低下、または消滅させることができる。さらに側鎖がメチル以上のアルキル鎖をもったオキセタン誘導体を用いると、少量で効果を十分に発現する。このようなオキセタン誘導体やテトラヒドロフラン誘導体等を開環させてアルキレンオキサイドユニットとし、他のポリアルキレンエーテルグリコールと共重合させる反応方法としては公知の方法が使える。
オキセタン誘導体とは式(2)で表されるものである。
【0027】
【化4】
式中、R3、R4は任意の置換基であり同一でも互いに異なっていてもよいが、ともに水素原子またはメチル基、あるいは一方が水素原子で他方がメチル基の場合を除く。また、R3、R4は炭素原子、酸素原子、ケイ素原子を介して結合していてもよい。
R3、R4がともに水素原子またはメチル基、あるいは一方が水素原子で他方がメチル基の場合、−50℃ではゴム状弾性が発現しないので好ましくない。
【0029】
合成の容易さから、R3、R4は、ハロゲン原子、アルコキシ基、アリールオキシ基、アシルオキシ基、アシル基、アリールカルボキシル基、アリールカルボニル基、トリアルキルシリル基、トリアリールシリル基、トリアルキルシリルオキシ基、トリアリールシリルオキシ基、アルキル基(メチル基は除く)から選ばれることが好ましい。これらの基に含まれる炭化水素部分の水素は、ハロゲン原子、アルコキシ基、アリールオキシ基、アシルオキシ基、アシル基、アリールカルボキシル基、アリールカルボニル基、トリアルキルシリル基、トリアリールシリル基、トリアルキルシリルオキシ基、トリアリールシリルオキシ基、アルキル基、ビニル基、アルキルスルフォキシ基、アリールスルフォキシ基、シアノ基で置換されていてもよい。
【0030】
また原料の入手の容易さから、R3、R4がハロゲン原子、炭素数2から10の直鎖または分岐鎖アルキル基、一つ以上のハロゲン原子で置換された炭素数2から10の直鎖または分岐鎖アルキル基、式(3)または式(4)で表されるものであることがより好ましい。
【0031】
【化5】
R5O−A− ・ ・ ・ 式(3)
R5(O−B)n−OA− ・ ・ ・ 式(4)
【0032】
式(3)、(4)において、nは1から10の整数、A、Bは炭素数1〜10のアルキレン基、R5は炭素数1〜20のアルキル基、アシル基、アシルアルキル基、アシルオキシアルキル基、トリアルキルシリル基、アルキルスルフォニル基、アリールスルフォニル基であり、R5、A、Bのいずれも直鎖でも分岐鎖でもよく、環構造を含んでもよい。またR5、A、Bが一つ以上のハロゲン原子、アルコキシ基、アルコキシアルキル基、アシルオキシアルキル基、アリールオキシ基、アリールオキシアルキル基、アリールカルボキシアルキル基、アルキルスルフォキシアルキル基、アリールスルフォキシアルキル基、シアノ基で置換されていてもよい。
【0033】
これらの中でも、得られる物性と原料の価格等のバランスから特に好ましいオキセタン誘導体としては、式(2)中のR3、R4がモノハロゲン化メチル基、又は、炭素数2から10の直鎖または分岐鎖のアルキル基で表される3,3−ジアルキルオキセタン、3,3−ビス(モノハロゲン化メチル)オキセタン、3−アルキル−3−モノハロゲン化メチルオキセタン、また式(2)中のR3が炭素数2から10の直鎖または分岐鎖アルキル基、R4が式(3)、(4)で表されるオキセタン誘導体があげられる。これらのオキセタン誘導体は、工業的に入手可能なネオペンチルグリコール等の2,2−ジアルキル−1,3−プロパンジオール、ペンタエリスリトール、トリメチロールエタンやトリメチロールプロパン等のトリメチロールアルカン類を原料にして、そのまま環化したり、水酸基をハロゲン化した後に環化したりすることによりオキセタン骨格をつくり、必要に応じて官能基を導入することにより製造される。
【0034】
オキセタン誘導体の具体例としては、オキセタン;3−エチルオキセタン等の3−アルキルオキセタン類;3,3−ジエチルオキセタン等の3,3−ジアルキルオキセタン類;3−エチル−3−クロロメチルオキセタン等の3−アルキル−3−ハロゲン化メチルオキセタン;3,3−ビス(クロロメチル)オキセタン、3,3−ビス(ブロモメチル)オキセタン、3,3−ビス(フルオロメチル)オキセタン等の3,3−ビス(ハロゲン化メチル)オキセタン類;3−メトキシ−3−メチルオキセタン、3−エトキシ−3−メチルオキセタン、3−プロポキシ−3−メチルオキセタン、3−ブトキシ−3−メチルオキセタン、3−メトキシ−3−エチルオキセタン、3−エトキシ−3−エチルオキセタン、3−プロポキシ−3−エチルオキセタン、3−ブトキシ−3−エチルオキセタン等の3−アルコキシ−3−アルキルオキセタン類;3−メトキシメチル−3−メチルオキセタン、3−エトキシメチル−3−メチルオキセタン、3−プロポキシメチル−3−メチルオキセタン、3−ブトキシメチル−3−メチルオキセタン、3−メトキシメチル−3−エチルオキセタン、3−エトキシメチル−3−エチルオキセタン、3−プロポキシメチル−3−エチルオキセタン、3−ブトキシメチル−3−エチルオキセタン等の3−アルコキシメチル−3−アルキルオキセタン類;3−(2−メトキシエトキシ)メチル−3−メチルオキセタン、3−(2−(2−メトキシエトキシ)エトキシ)メチル−3−メチルオキセタン、3−(2−(2−(2−メトキシエトキシ)エトキシ)エトキシ)メチル−3−メチルオキセタン、3−(2−エトキシエトキシ)メチル−3−メチルオキセタン、3−(2−(2−エトキシエトキシ)エトキシ)メチル−3−メチルオキセタン、3−(2−(2−(2−エトキシエトキシ)エトキシ)エトキシ)メチル−3−メチルオキセタン、3−(2−プロポキシエトキシ)メチル−3−メチルオキセタン、3−(2−ブトキシエトキシ)メチル−3−メチルオキセタン、3−(2−メトキシエトキシ)メチル−3−エチルオキセタン、3−(2−(2−メトキシエトキシ)エトキシ)メチル−3−エチルオキセタン、3−(2−(2−(2−メトキシエトキシ)エトキシ)エトキシ)メチル−3−エチルオキセタン、3−(2−エトキシエトキシ)メチル−3−エチルオキセタン、3−(2−(2−エトキシエトキシ)エトキシ)メチル−3−エチルオキセタン、3−(2−(2−(2−エトキシエトキシ)エトキシ)エトキシ)メチル−3−エチルオキセタン、3−(2−プロポキシエトキシ)メチル−3−エチルオキセタン、3−(2−ブトキシエトキシ)メチル−3−エチルオキセタン、3−(2−アセトキシエトキシ)メチル−3−メチルオキセタン、3−(2−アセトキシエトキシ)メチル−3−エチルオキセタン等に代表される、式(2)においてR3が炭素数1から10のアルキル基、R4が式(4)であらわされ、式(4)のnは1から10の整数、Aはメチレン基、Bはエチレン基、R5は炭素数1〜20のアルキル基、アシル基であらわされるオキセタン類;3,3−ビス(メトキシメチル)オキセタン、3,3−ビス(エトキシメチル)オキセタン、3,3−ビス(プロポキシメチル)オキセタン、3,3−ビス(ブトキシメチル)オキセタンのような3,3−ビス(アルコキシメチル)オキセタン類;3,3−ビス((2−メトキシエトキシ)メチル)オキセタン、3,3−ビス((2−(2−メトキシエトキシ)エトキシ)メチル)オキセタン、3,3−ビス((2−(2−(2−メトキシエトキシ)エトキシ)エトキシ)メチル)オキセタン、3,3−ビス((2−エトキシエトキシ)メチル)オキセタン、3,3−ビス((2−(2−エトキシエトキシ)エトキシ)メチル)オキセタン、3,3−ビス((2−(2−(2−エトキシエトキシ)エトキシ)エトキシ)メチル)オキセタン、3,3−ビス((2−プロポキシエトキシ)メチル)オキセタン、3,3−ビス((2−ブトキシエトキシ)メチル)オキセタン、3,3−ビス((2−アセトキシエトキシ)メチル)オキセタン等に代表される、式(2)においてR3、R4が式(4)であらわされ、式(4)のnは1から10の整数、Aはメチレン基、Bはエチレン基、R5は炭素数1〜20のアルキル基、アシル基であらわされるオキセタン類;3−(2−メトキシプロポキシ)メチル−3−エチルオキセタン、3−(3−n−ブトキシ−2−アセトキシプロポキシ)メチル−3−エチルオキセタン、3−(3−フェノキシ−2−アセトキシプロポキシ)メチル−3−エチルオキセタン、3−(2−トリメチルシリルオキシプロポキシ)メチル−3−エチルオキセタン等に代表される、式(2)においてR3が炭素数1から10のアルキル基、R4が式(4)であらわされ、式(4)のnは1から10の整数、Aはメチレン基、Bは置換基を有するエチレン基、R3はアルキル基、アシル基、トリアルキルシリル基であらわされるオキセタン類;3−トリメチルシリルオキシメチル−3−メチルオキセタン、3−トリメチルシリルオキシメチル−3−エチルオキセタンのようなトリアルキルシリルオキシ基を有するオキセタン類;などが挙げられる。
ただし、下記式(5)で表されるオキセタン誘導体は、酸触媒存在下で開環重合性の低いオルトエステルを形成することがあり、適さない。
【0035】
【化6】
(式(5)中、R6、R7は水素又は任意の置換基。)
本発明のポリエーテルエステルエラストマー全体に占める全ポリエーテルグリコールセグメント(ソフトセグメント)の量は10〜90重量%であり、好ましくは30〜75重量%、特に50〜70重量%であるのが好ましい。この値はポリエーテルエステルエラストマーを用いた目的とする最終成型品の要求物性による。ソフトセグメントの量が10重量%より小さいと軟質性、耐油性、耐熱性に劣り、エラストマーとしての満足のいく物性は期待できない。また、この量が90重量%を越えると軟質性は相当付与されるが、同時にハードセグメントの平均連鎖長が短くなり、物理的架橋点であるハードブロックが外力に対して抵抗できずに機械強度が著しく低下して、もはやエラストマー材料としては用途がなくなってしまう。また、融点も相当低下するため耐熱性にも劣り、好ましくない。より好ましいソフトセグメントの量は25〜75重量%である。
【0037】
本発明のポリエーテルエステルエラストマーがさらに他のポリエステル樹脂、ポリカーボネート樹脂、ポリアミド樹脂等の改質剤として使用される場合は、50重量%以上のソフトセグメント成分が含まれるポリエーテルエステルエラストマーを使用することが出来る。
【0038】
このような本発明のポリエーテルエステルエラストマーは、以下に示す公知の方法で製造できる。例えば、ジカルボン酸の低級アルコールジエステル、過剰量の低分子量グリコールおよびポリエーテルグリコールを触媒の存在下エステル交換反応させ、続いて得られる反応生成物を減圧下重縮合する方法、あるいはジカルボン酸とグリコール及びポリエーテルグリコールを触媒の存在下エステル化反応させ、ついで得られる生成物を重縮合する方法、また予め短鎖ポリエステル(例えばポリブチレンテレフタレート)を作っておき、これに他のジカルボン酸やジオールもしくはポリエーテルグリコールを加えたり、もしくは他の共重合ポリエステルを添加してエステル交換によりランダム化させる方法など何れの方法をとっても良い。
【0039】
エステル交換反応またはエステル化反応と重縮合反応に共通の触媒として、テトラ(イソプロポキシ)チタネート、テトラ(n−ブトキシ)チタネートに代表されるテトラアルキルチタネート、これらテトラアルキルチタネートとアルキレングリコールとの反応生成物、テトラアルキルチタネートの部分加水分解物、チキタニウムヘキサアルコキサイドの金属塩、チタニウムヘキサアルコキサイドの金属塩、チタンのカルボン酸塩、チタニル化合物等のTi系触媒が好ましい他、モノn−ブチルモノヒドロキシスズオキサイド、モノn−ブチルスズトリアセテート、モノn−ブチルスズモノオクチレート、モノn−ブチルスズモノアセテート等のモノアルキルスズ化合物、ジn−ブチルスズオキサイド、ジn−ブチルスズジアセテート、ジフェニルスズオキサイド、ジフェニルスズジアセテート、ジn−ブチルスズジオクチレート等のジアルキル(またはジアリール)スズ化合物等が挙げられる。この他Mg、Pb、Zr、Zn、Sb等の金属または金属酸化物触媒が有用である。これらの触媒は単独で、あるいは2種以上組み合わせて使用しても良い。特に単独で使用する場合には、テトラアルキルチタネート、または三酸化アンチモンが、組み合わせて使用する場合にはテトラアルキルチタネートと酢酸マグネシウムを用いることが好ましい。
【0040】
エステル化あるいは重縮合触媒の添加量は生成ポリマーに対して0.005〜0.5重量%が好ましく、特に0.03〜0.2重量%が好ましい。これら触媒はエステル交換またはエステル化反応開始時に添加した後、重縮合反応時に再び添加してもしなくても良い。また、ジカルボン酸やグリコールの一部としてポリカルボン酸や多官能ヒドロキシ化合物、オキシ酸等が共重合されていても良い。多官能成分は高粘度化成分として有効に作用し、その共重合体中の含有量は3モル%以下がよい。かかる多官能成分として用いることが出来るものにはトリメリット酸、トリメシン酸、ピロメリット酸、ベンゾフェノンテトラカルボン酸、ブタンテトラカルボン酸、グリセリン、ペンタエリスリトールおよびそれらのエステル、酸無水物等を挙げることができる。
【0041】
また、酸化防止剤をポリエーテルエステルエラストマーの製造中または製造後の任意の時期に加えることが出来るが、特にポリエーテルグリコールが高温に曝される時点、例えば重縮合反応に入る時点でポリエーテルグリコールの酸化劣化を防止する為重縮合反応を阻害せず、また触媒の機能を損なわない酸化防止剤を加えることが望ましい。これらの酸化防止剤としては燐酸、亜燐酸の脂肪族、芳香族又はアルキル基置換芳香族エステルや、次亜燐酸誘導体、フェニルホスホン酸、フェニルホスフィン酸、ジフェニルホスホン酸、ポリホスホネート、ジアルキルペンタエリスリトールジホスファイト、ジアルキルビスフェノールAジホスファイト等のリン化合物、フェノール系誘導体特にヒンダードフェノール化合物、チオエーテル系、ジチオ酸塩系、メルカプトベンズイミダゾール系、チオカルバニリド系、チオジプロピオン酸エステル等のイオウを含む化合物、スズマレート、ジブチルスズモノオキシド等のスズ系化合物を用いることができる。これらは単独で用いても2種以上組み合わせて用いても構わない。
【0042】
これら安定剤の添加量は、ポリエーテルエステルエラストマー100重量部に対し、0.01〜2重量部であることが望ましい。また、必要に応じて本発明におけるポリエーテルグリコールを、それ以外のポリエーテルグリコールで一部置換しても良い。かかる置換に用いられるポリエーテルグリコールとしてポリ(エチレンオキシ)グリコール、ポリ(プロピレンオキシ)グリコール、ポリ(テトラメチレンオキシ)グリコール、ポリ(1,2−プロピレンオキシ)グリコール、エチレンオキシドとプロピレンオキシドのブロック又はランダム共重合体、エチレンオキシドとTHFのブロック又はランダム共重合体、ポリ(2−メチル−1,3−プロピレンオキシ)グリコール、ポリ(プロピレンオキシ)ジイミドジ酸等が挙げられる。
【0043】
最終的には得られたポリエーテルエステルエラストマーに対し必要に応じて、本発明の目的、効果を損なわない範囲で、任意の成分を配合することができる。具体的には、酸化防止剤、カオリン、シリカ、マイカ、二酸化チタン、アルミナ、炭酸カルシウム、ケイ酸カルシウム、クレー、カオリン、ケイソウ土、アスベスト、硫酸バリウム、硫酸アルミニウム、硫酸カルシウム、塩基性炭酸マグネシウム、二硫化モリブデン、グラファイト、ガラス繊維、炭素繊維等の充填剤や補強材、ステアリン酸亜鉛やステアリン酸ビスアマイドのような滑剤ないしは離型剤、着色の為のカーボンブラック、群青、酸化チタン、亜鉛華、ぺんがら、紺青、アゾ顔料、ニトロ顔料、レーキ顔料、フタロシアニン顔料等の染顔料、オクタブロモジフェニル、テトラブロモビスフェノールポリカーボネート等の難燃化剤、ヒンダードアミン系光安定剤、紫外線吸収剤、発泡剤、エポキシ化合物やイソシアネート化合物等の増粘剤、シリコーンオイルやシリコーン樹脂等、公知の各種添加剤を用いることが出来る。
【0044】
【実施例】
以下、本発明を実施例により更に具体的に説明するが、本発明はその要旨を超えない限り以下の実施例に限定されるものではない。まず、実施例、比較例で用いた評価方法、材料を以下に記す。
<評価方法>
(1)引張強さ:JIS K6251(3号ダンベル)、試料は2mm厚のプレスシートを用いた。
(2)引っ張り伸び:JIS K6251(3号ダンベル)、試料は2mm厚のプレスシートを用いた。
(3)融点:SEIKO電子工業社製示差熱量計(RDC220)により、ポリエーテルエステルエラストマー、約6mgを用いて実施した。まず、30℃から290℃まで50℃/minの速度で昇温し、290℃で3分間保持したあと、10℃/minで−130℃まで降温し−130℃で10分間保持したあと、再度、昇温速度10℃/minで290℃まで昇温した。そのときの吸熱ピークを測定し、そのトップの温度を調べ、融点とした。
(4)ΔHf:SEIKO電子工業社製示差熱量計(RDC220)により、ポリエーテルエステルエラストマー、約5mgを用いて実施した。まず、30℃から290℃まで50℃/minの速度で昇温し、290℃で3分間保持したあと、3℃/minで−130℃まで降温し−130℃で10分間保持したあと、再度、昇温速度10℃/minで290℃まで昇温した。そのときの−50℃〜100℃における吸熱熱量をΔHfとして測定した。
【0045】
(5)logE‘:東洋精機(株)製レオグラフソリッドを用い、縦振動による動的粘弾性を測定した。昇温速度2℃/min、測定周波数10Hz、張力10g/mm2、歪み0.1%の条件で−130℃から150℃まで測定した。測定には、ポリエーテルエステルエラストマーをプレス(温度220℃、プレス時間5分)して厚み0.3mmのプレスシートを作成し、これを幅6mm、長さ3mmに切り出したものを測定サンプルとして用いた。動的貯蔵弾性率(E’)と動的損失弾性率(E”)とを測定した。
(6)アイゾット:JIS K7110に準拠してアイゾット衝撃強さを23℃と−50℃で測定した。破壊されなかった場合は表中に「NB」、破壊された場合には「Break」と記した。
【0046】
<材料>
実施例および比較例で使用された、ポリエーテルエステルエラストマーのソフトセグメントに用いるポリアルキレンエーテルグリコールとしては、以下のものを使用した。
(1)A1:3−ブトキシメチル−3−エチルオキセタン共重合ポリテトラメチレンエーテルグリコール(両末端が水酸基)(Mn=2119、Mw=3456Mw/Mn=1.63、3−ブトキシメチル−3−エチルオキセタン含率=10.1モル%)を下記製造例1に従って調製した。
【0047】
(製造例1)
9mol%のZrO2−SiO2複合酸化物触媒を窒素気流中500℃で1時間乾燥した。ガラス製容器に無水酢酸62.0g、テトラヒドロフラン400.5g、3−ブトキシメチル−3−エチルオキセタン7.7g(テトラヒドロフラン:3−ブトキシメチル−3−エチルオキセタン=89.9:10.1モル比)を入れ、窒素雰囲気下で攪拌しながら乾燥後の触媒11.2gを加え、水温を調節しておいた水浴へ入れ、ポンプを用いて3−ブトキシメチル−3−エチルオキセタンを0.18g/分で滴下しながら常圧、40℃で2時間反応を行った。(3−ブトキシメチル−3−エチルオキセタンを合計22.1g滴下した。)
【0048】
反応終了後、水12gを加え反応液を濾過し触媒を除去し、未反応物を蒸留により除去してポリエーテルポリオールジエステル105.4gを得た。得られたポリエーテルポリオールジエステル105.4g及びメタノール316.2g、水酸化ナトリウム0.5gを蒸留装置を備えた反応器に仕込み、メタノール/酢酸メチルの共沸混合物を留出させながら4時間エステル交換を行った。その後、未反応のメタノールを室温で減圧にて除去した後、メタノール158.1g、水158.1gをを加え、窒素下50℃にて30分加熱攪拌し、15分静置した後、上層(オリゴマーを多く含む)を除去した。このオリゴマー除去操作を2回行った後、下層をテトラヒドロフラン316.2gに溶解し、活性白土5.3gを加えて室温で2時間攪拌した後、濾過して活性白土を除去した。得られた濾液からテトラヒドロフランを留去し、3−ブトキシメチル−3−エチルオキセタン共重合ポリテトラメチレンエーテルグリコール99.1gを得た。
【0049】
(2)A2:3−オクトキシメチル−3−エチルオキセタン共重合ポリテトラメチレンエーテルグリコール(両末端が水酸基)(Mn=2023、Mw=3479 Mw/Mn=1.72、3−オクトキシメチル−3−エチルオキセタン含率= 9.1モル%)を、製造例1において、3−ブトキシメチル−3−エチルオキセタン7.7gを3−オクトキシメチル−3−エチルオキセタン10.2gに変更したこと以外は同様に調製した。
【0050】
(3)A3:3−メトキシメチル−3−エチルオキセタン共重合ポリテトラメチレンエーテルグリコール(両末端が水酸基)(Mn=2262、Mw=3774Mw/Mn=1.67、3−メトキシメチル−3−エチルオキセタン含率=モル10.2%)を、製造例1において、3−ブトキシメチル−3−エチルオキセタン7.7gを3−メトキシメチル−3−エチルオキセタン5.8gに変更したこと以外は同様に調製した。
(4)A4:ポリテトラメチレンエーテルグリコール:三菱化学(株)製、 Mn=2131、Mw=4349、Mw/Mn=2.04
(5)A5:PTG−L2000(保土谷化学工業株式会社製)、3−メチル−テトラヒドロフラン共重合ポリテトラメチレンエーテルグリコール(両末端が水酸基)(Mn=2141、Mw=4111、Mw/Mn=1.92、3−メチル−テトラヒドロフラン含率=16モル%)を用いた。
【0051】
<実施例及び比較例>
(実施例1)
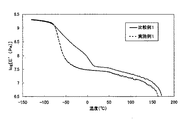
縮合用リアクターにジメチルテレフタレート(東京化成製)49g、1,4−ブタンジオール(東京化成製)38g、製造例1で製造した3−ブトキシメチル−3−エチルオキセタン共重合ポリテトラメチレンエーテルグリコール98gを仕込み、窒素置換後、窒素雰囲気下150℃で30分かけて加熱溶融した。次いでテトラブトキシチタネート(キシダ化学製)を1,4−ブタンジオールに6重量%にて溶解した溶液3.7gを添加し、回転数150rpmで撹拌しながら150℃で60分間保持した後、2時間かけて230°Cに昇温し、230℃で15分間保持して、エステル交換反応を行った。得られた溶融物中に、イルガノックス1330(チバスペシャリティーケミカル製)を1,4ブタンジオールに5重量%で分散したスラリー5.4mlを添加し、さらに45分間かけて245℃まで昇温させながら、85分間かけて1mmHgまで減圧し、その条件下で重縮合反応を行った。1mmHgの状態になってから230分後にトルクが2.0kgに達したので回転数を100rpmに落とし、さらに50分後にトルクが2.0kgに達した時点で回転数を50rpmまで落とし、さらにその55分後にトルクが2.0kgに達した時点で25rpmまで回転数を落として、最後にその40分後にトルクが1.7kgに達した時点で攪拌を止め、リアクター内を窒素で常圧に複圧し、重縮合反応を停止した。その後、リアクターの内容物をストランド状に取り出し、前述の評価方法に従って各評価を実施した。結果を表1に示す。また、動的貯蔵弾性率(E’)の温度依存性を解析した。その結果を図1に示す。
【0052】
(実施例2)
実施例1において、A1に代えて、製造例2で製造した3−オクトキシメチル−3−エチルオキセタン共重合ポリテトラメチレンエーテルグリコール(A2)を98g用いた以外は全て同様にして、エステル交換反応と縮合反応を行った。添加剤の種類及び調合比率も同様である。評価結果を表1に示す。
(実施例3)
実施例1において、A1に代えて、製造例3で製造した3−メトキシメチル−3−エチルオキセタン共重合ポリテトラメチレンエーテルグリコール(A3)を98g用いた以外は全て同様にして、エステル交換反応と縮合反応を行った。添加剤の種類及び調合比率も同様である。評価結果を表1に示す。
【0053】
(比較例1)
実施例1において、A1に代えてポリテトラメチレンエーテルグリコール(A4)98gを用いたこと以外は同様にして、エステル交換反応と縮合反応を行った。添加剤の種類及び調合比率も同様である。評価結果を表1に示す。また、動的貯蔵弾性率(E’)の温度依存性を解析した。その結果を図1に示す。
(比較例2)
実施例1において、A1に代えて、製造例5で製造した3−メチル−テトラヒドロフラン共重合ポリテトラメチレンエーテルグリコール(A5)を98g用いた以外は全て同様にして、エステル交換反応と縮合反応を行った。添加剤の種類及び調合比率も同様である。評価結果を表1に示す。
【0054】
【表1】
<結果の評価>
1)比較例1はlogE’−50の値が規定外の8.64であり、ΔHfも規定外の23.3mJ/mgであるため、―50℃でのアイゾット衝撃値が低い。
2)比較例2はlogE’−50の値が規定外の8.14であり、ΔHfも規定外の10.1mJ/mgであるため、―50℃でのアイゾット衝撃値が低い。
【0056】
【発明の効果】
本発明のポリエーテルエステルエラストマーは、従来のこの種のポリエステルエラストマーに比べて低温で硬化しないため、高温から低温までの物性の変化を少なく、ゴム状弾性を広い温度域で有する。そのために、耐寒・耐熱・耐油性が要求され、かつゴム状弾性の要求される熱可塑性エラストマーとして自動車部品(特にエンジン周り、内装)や、チューブ、ホース、ギア、電線被覆材等の工業用品、日用品、ポリエステルやポリカーボネート樹脂の耐衝撃性改良材として好適に使用できる。
【図面の簡単な説明】
【図1】実施例1のポリエーテルエステルエラストマーと比較例1のポリエーテルエステルエラストマーとにおける、動的貯蔵弾性率(E’)の温度変化を示すグラフである。[0001]
TECHNICAL FIELD OF THE INVENTION
The present invention relates to a polyetherester elastomer having little change in physical properties from high to low temperatures and having rubber-like elasticity in a wide temperature range.
[0002]
[Prior art]
Polyetherester elastomers are mainly copolymerized with polybutylene terephthalate as a hard segment and polyether glycol as a soft segment, have rubber-like elasticity, and are excellent in mechanical strength, heat resistance, and oil resistance. Widely used for parts, electric / electronic parts, industrial parts, precision machine parts, household goods, etc.
[0003]
Usually, polytetramethylene ether glycol is widely used as a soft segment, but the polytetramethylene ether glycol component causes crystallization in a low temperature region, and physical properties change with temperature depending on use conditions. There is a problem that rubber-like elasticity does not appear in a low temperature range.
[0004]
In order to solve such a problem, attempts have been made to narrow the molecular weight distribution (Mv / Mn) of the raw material polytetramethylene ether glycol (Mv / Mn ≦ 1.60) and to make the number average molecular weight (Mn) relatively small. (Japanese Patent Publication No. 3-40732). Here, the molecular weight distribution (Mv / Mn) is the ratio of the number average molecular weight (Mn) determined from the terminal hydroxyl value to the viscosity average molecular weight (Mv) defined by the formula Mv = antilog (0.4931 log η + 3.0646). Value. Here, η is a value in which the melt viscosity at a temperature of 40 ° C. is indicated by poise.
[0005]
Further, in order to prevent crystallization of polyether glycol, one having a methyl group on a side chain, for example, poly (2-methyl-1,3-propyleneoxy) glycol is used as a soft segment (for example, see Patent Document 1). For the purpose of preventing crystallinity of polyether glycol, use of polyether glycol composed of neopentylene oxide and tetramethylene oxide as a soft segment has been proposed (for example, see Patent Document 2). .
[0006]
[Patent Document 1]
Japanese Patent Publication No. 3-80170
[Patent Document 2]
Japanese Patent No. 3117805
[0007]
[Problems to be solved by the invention]
However, even if the molecular weight distribution (Mv / Mn) of polytetramethylene ether glycol is narrowed, the number average molecular weight (Mn) of polytetramethylene ether glycol that can be used is limited, and particularly when it is desired to set a high melting point, There is a disadvantage that the physical properties such as heat resistance and low temperature properties, flex wear resistance and elastic recovery are poor.
[0008]
In addition, when a polyether glycol having a methyl group on the side chain is used as the soft segment, the effect is not exhibited unless a relatively large amount of a polyoxyalkylene unit having a methyl group on the side chain is present. Since the starting material cyclic ethers (eg, methyltetrahydrofuran, methyloxetane) are expensive, this method is hardly used industrially at present.
Further, even if a polyether glycol composed of neopentylene oxide and tetramethylene oxide is used as a soft segment, physical properties change in a lower temperature range, and the performance is still insufficient.
[0009]
The present invention is intended to solve such a problem, and by appropriately selecting polyether glycol as a soft segment component, changes in physical properties from high to low temperatures are reduced, and rubber-like elasticity is increased over a wide temperature range. It is an object of the present invention to provide a novel polyetherester elastomer having a range.
[0010]
[Means for Solving the Problems]
The present inventors have conducted intensive studies to solve the above problems, and as a result, found that a polyetherester elastomer exhibiting a specific viscoelastic behavior exhibits good rubber-like elasticity even at a low temperature, The present invention has been reached.
[0011]
That is, the gist of the present invention is that a value of logarithmic common logarithm of storage elastic modulus at −50 ° C. (log E ′) in dynamic viscoelasticity measurement in longitudinal vibration -50 )But,
(Equation 2)
7.0 ≦ logE ′ -50 ≦ 8.1
[0012]
Wherein the polyetherester elastomer is characterized in that:
Another gist of the present invention is that the endothermic heat ΔH at −50 ° C. to 100 ° C. in measurement at a heating rate of 10 ° C./min by DSC. f Is 0 mJ / mg to 8 mJ / mg or less.
Another gist of the present invention resides in a polyetherester elastomer characterized by having no melting point between -50 ° C and 100 ° C.
[0013]
Another aspect of the present invention resides in a polyetherester elastomer, wherein the number average molecular weight of the polyalkylene ether glycol segment is from 400 to 10,000.
[0014]
BEST MODE FOR CARRYING OUT THE INVENTION
Various types of polyetherester elastomers are known, and typical ones are mainly composed of a hard segment mainly composed of an aromatic polyester and a soft segment mainly composed of a long-chain polyether. It is a polyetherester elastomer.
[0015]
The polyetherester elastomer according to the present invention has a common logarithmic value (logE ') of the storage modulus at -50 [deg.] C. in dynamic viscoelasticity measurement in longitudinal vibration. -50 )But,
[0016]
[Equation 3]
7.0 ≦ logE ′ -50 ≦ 8.1
[0017]
It is. logE ' -50 If it is less than 7.0, it is too soft to give strength, and if it is more than 8.1, it is hard and brittle. Preferably it is 7.2-8.0, More preferably, it is 7.3-7.9. Ordinary polyetherester elastomers are hard and brittle at low temperatures below 0 ° C. In particular, if the temperature is lower than -50 ° C, the rubber becomes too hard to exhibit rubbery elasticity. However, the polyetherester elastomer of the present invention has a low value of logE 'at -50C of dynamic viscoelasticity due to longitudinal vibration as low as 7.0 to 8.1, and retains flexibility.
[0018]
Furthermore, the polyetherester elastomer according to the present invention has an endothermic heat ΔH at −50 ° C. to 100 ° C. in measurement at a heating rate of 10 ° C./min by DSC. f Is preferably from 0 mJ / mg to 8 mJ / mg. It is more preferably 0 mJ / mg to 5 mJ / mg, and more preferably 0 mJ / mg to 3 mJ / mg. If there is an endothermic peak exceeding 8 mJ / mg, the mechanical strength, rubber-like elasticity, and the like may change between -50 ° C and 100 ° C, which is the normal temperature range, and the low-temperature characteristics may decrease.
Further, the polyetherester elastomer according to the present invention more preferably does not have a melting point between −50 ° C. and 100 ° C. If there is a melting point during this period, the mechanical strength, rubber-like elasticity, and the like change in the normal temperature range of −50 ° C. to 100 ° C., and the low-temperature characteristics may decrease.
[0019]
In the present invention, the hard segment is composed of a combination of a dicarboxylic acid and a short-chain diol. As the aromatic dicarboxylic acid and the ester-forming derivative thereof constituting the dicarboxylic acid component of the present invention, those commonly used as raw materials for polyesters, particularly, for polyetherester elastomers, for example, terephthalic acid, Isophthalic acid, phthalic acid, 2,6-naphthalenedicarboxylic acid, and the like. Of these, terephthalic acid and 2,6-naphthalenedicarboxylic acid are preferable, and terephthalic acid is particularly preferable. If desired, two or more of these aromatic dicarboxylic acids may be used in combination. When an alkyl ester of an aromatic dicarboxylic acid is used, the above-mentioned dimethyl ester or diethyl ester of the aromatic dicarboxylic acid is used. Preferred are dimethyl terephthalate and 2,6-dimethylnaphthalenedicarboxylate.
[0020]
As the short-chain diol component in the present invention, those usually used as a raw material for polyester, particularly a polyetherester-based elastomer can be used. For example, ethylene glycol, 1,2-propylene glycol, trimethylene glycol, 1,4-butanediol, 1,4-cyclohexanediol, 1,4-cyclohexanedimethanol, and the like, among which 1,4-butanediol, Ethylene glycol is preferred, and 1,4-butanediol is particularly preferred. These diols are usually used alone, but if desired, a mixture of two or more kinds can also be used.
In addition to the above-mentioned diol and dicarboxylic acid components, a small amount of a trifunctional alcohol or carboxylic acid, or an ester thereof may be copolymerized. Further, if a small amount of an aliphatic dicarboxylic acid such as adipic acid or a dialkyl ester thereof is used, the copolymer may be copolymerized. It can be used as a polymerization component.
[0021]
The soft segment in the polyetherester elastomer of the present invention, that is, the polyalkylene ether glycol constituting the long-chain polyester, preferably has a number average molecular weight of 400 to 10,000. Those having 600 to 6000 are more preferable, and those having 1000 to 5000 are particularly preferable. When the number average molecular weight is less than 400, the average chain length of the short-chain polyester (hard segment) usually decreases, depending on the hard / soft ratio of the final polyetherester to be polymerized, and the melting point drops sharply, resulting in heat resistance. It is not preferable when it is used as a material as a polyetherester elastomer as it is because of poor properties. On the other hand, if it exceeds 10,000, the viscosity of the polyalkylene ether glycol increases, phase separation easily occurs in the system, and the physical properties of the polymer obtained by copolymerization or the like tend to decrease. Here, the “number average molecular weight” is measured by gel permeation chromatography (GPC). The calibration of GPC requires POLYMER LABORATORIES UK's POLYTETRAHYDROFURAN.
What is necessary is just to use a calibration kit.
[0022]
Examples of the polyalkylene ether glycol include polyethylene ether glycol, poly (1,2-propylene ether) glycol, poly (1,3-propylene ether) glycol, poly (tetramethylene ether) glycol, poly (hexamethylene ether) glycol And a block or random copolymer of ethylene oxide and propylene oxide, a block or random copolymer of ethylene oxide and THF, and poly (propylene ether) diimide diacid. Generally, polyalkylene ether glycols are crystalline and have a melting point. Therefore, when a polyetherester elastomer is polymerized using the above-mentioned polyalkylene ether glycol, a melting point (endothermic peak) derived from the polyalkylene ether glycol usually appears in a low temperature range of −50 ° C. to 100 ° C. Physical properties change greatly. Therefore, it is preferable that the polyalkylene ether glycol has no melting point, has a low melting point, and has a low heat of fusion (endothermic heat) even if it has a melting point.
Among the polyalkylene ether glycols having the above properties, those having the structure of the following formula (1) are preferable.
[0023]
Embedded image
R in the formula (1) 1 , R 2 Is an arbitrary substituent and may be the same or different. Because of the ease of synthesis, R 1 , R 2 Is hydrogen, alkyl, halogen, alkoxy, aryloxy, acyloxy, acyl, arylcarboxyl, arylcarbonyl, trialkylsilyl, triarylsilyl, trialkylsilyloxy, triarylsilyloxy It is preferred to be selected from the group
[0025]
Further, the hydrogen of the hydrocarbon moiety contained in these groups may be a halogen atom, an alkoxy group, an aryloxy group, an acyloxy group, an acyl group, an arylcarboxyl group, an arylcarbonyl group, a trialkylsilyl group, a triarylsilyl group, It may be substituted with an alkylsilyloxy group, a triarylsilyloxy group, an alkyl group, a vinyl group, an alkylsulfoxy group, an arylsulfoxy group, a cyano group, or the like. Where R 1 , R 2 Are both methyl groups because they do not exhibit the desired performance. Also, R 1 , R 2 May be bonded to each other via a carbon atom, an oxygen atom or a silicon atom.
The content of the unit having the structure of the formula (1) in the polyalkylene ether glycol is preferably from 2 mol% to 100 mol%. If the content is too small, desired characteristics cannot be obtained, so that the content is more preferably 5 mol% to 100 mol%.
[0026]
Furthermore, among them, the atoms other than hydrogen atoms or alkyl chains are introduced into the side chains of the polyalkylene ether glycol, or different alkylene ethers are copolymerized to lower the crystallinity, and -50 ° C to 100 ° C. Is preferably reduced. Examples of a method of introducing a side chain into the polyalkylene ether glycol include a method of grafting using a peroxide or the like, and a method of copolymerizing an alkylene oxide unit having a side chain. In particular, it is preferable to copolymerize an alkylene oxide unit having a side chain. From the viewpoint of polymerizability, it is preferable to open a ring of an oxetane derivative, a tetrahydrofuran derivative, or the like to form an alkylene oxide unit, and copolymerize it with another polyalkylene ether glycol. By using this copolymerized polyalkylene ether glycol, the melting point of the polyetherester elastomer at a low temperature can be drastically reduced or eliminated. Further, when an oxetane derivative having an alkyl chain having a methyl or higher side chain is used, the effect is sufficiently exhibited with a small amount. A known method can be used as a reaction method of ring-opening such an oxetane derivative or tetrahydrofuran derivative to form an alkylene oxide unit and copolymerizing it with another polyalkylene ether glycol.
The oxetane derivative is represented by the formula (2).
[0027]
Embedded image
Where R 3 , R 4 Is an arbitrary substituent, which may be the same or different from each other, except that both are a hydrogen atom or a methyl group, or a case where one is a hydrogen atom and the other is a methyl group. Also, R 3 , R 4 May be bonded via a carbon atom, an oxygen atom or a silicon atom.
R 3 , R 4 Are both a hydrogen atom or a methyl group, or one of them is a hydrogen atom and the other is a methyl group.
[0029]
Because of the ease of synthesis, R 3 , R 4 Represents a halogen atom, an alkoxy group, an aryloxy group, an acyloxy group, an acyl group, an arylcarboxyl group, an arylcarbonyl group, a trialkylsilyl group, a triarylsilyl group, a trialkylsilyloxy group, a triarylsilyloxy group, or an alkyl group. (Excluding a methyl group). Hydrogen in the hydrocarbon moiety contained in these groups includes a halogen atom, an alkoxy group, an aryloxy group, an acyloxy group, an acyl group, an arylcarboxyl group, an arylcarbonyl group, a trialkylsilyl group, a triarylsilyl group, and a trialkylsilyl. It may be substituted with an oxy group, a triarylsilyloxy group, an alkyl group, a vinyl group, an alkylsulfoxy group, an arylsulfoxy group, or a cyano group.
[0030]
Also, due to the availability of raw materials, R 3 , R 4 Is a halogen atom, a straight-chain or branched-chain alkyl group having 2 to 10 carbon atoms, a straight-chain or branched-chain alkyl group having 2 to 10 carbon atoms substituted with one or more halogen atoms, formula (3) or (4) ) Is more preferable.
[0031]
Embedded image
R 5 O-A- ... Formula (3)
R 5 (OB) n-OA- formula (4)
[0032]
In the formulas (3) and (4), n is an integer of 1 to 10, A and B are an alkylene group having 1 to 10 carbon atoms, R 5 Is an alkyl group having 1 to 20 carbon atoms, an acyl group, an acylalkyl group, an acyloxyalkyl group, a trialkylsilyl group, an alkylsulfonyl group, an arylsulfonyl group; 5 , A, and B may be linear or branched, and may include a ring structure. Also R 5 , A and B are one or more halogen atoms, alkoxy groups, alkoxyalkyl groups, acyloxyalkyl groups, aryloxy groups, aryloxyalkyl groups, arylcarboxyalkyl groups, alkylsulfoxyalkyl groups, arylsulfoxyalkyl groups Or a cyano group.
[0033]
Of these, oxetane derivatives which are particularly preferred in view of the balance between the physical properties to be obtained and the price of the raw material are those represented by R in the formula (2). 3 , R 4 Is a monohalogenated methyl group or a linear or branched alkyl group having 2 to 10 carbon atoms, 3,3-dialkyloxetane, 3,3-bis (monohalogenated methyl) oxetane, 3-alkyl -3-monohalogenated methyloxetane, and R in the formula (2) 3 Is a linear or branched alkyl group having 2 to 10 carbon atoms, R 4 Are oxetane derivatives represented by the formulas (3) and (4). These oxetane derivatives are produced from industrially available 2,2-dialkyl-1,3-propanediol such as neopentyl glycol, pentaerythritol, and trimethylolalkanes such as trimethylolethane and trimethylolpropane. The oxetane skeleton is formed by cyclization as it is, or cyclization after halogenation of a hydroxyl group, and a functional group is introduced as necessary.
[0034]
Specific examples of the oxetane derivative include: oxetane; 3-alkyloxetanes such as 3-ethyloxetane; 3,3-dialkyloxetanes such as 3,3-diethyloxetane; and 3-alkyloxetanes such as 3-ethyl-3-chloromethyloxetane. -Alkyl-3-halogenated methyl oxetane; 3,3-bis (halogen) such as 3,3-bis (chloromethyl) oxetane, 3,3-bis (bromomethyl) oxetane, and 3,3-bis (fluoromethyl) oxetane Methyl) oxetanes; 3-methoxy-3-methyloxetane, 3-ethoxy-3-methyloxetane, 3-propoxy-3-methyloxetane, 3-butoxy-3-methyloxetane, 3-methoxy-3-ethyloxetane , 3-ethoxy-3-ethyloxetane, 3-propoxy-3-ethyloxe 3-alkoxy-3-alkyloxetanes such as 3-butoxy-3-ethyloxetane; 3-methoxymethyl-3-methyloxetane, 3-ethoxymethyl-3-methyloxetane, 3-propoxymethyl-3-methyl Oxetane, 3-butoxymethyl-3-methyloxetane, 3-methoxymethyl-3-ethyloxetane, 3-ethoxymethyl-3-ethyloxetane, 3-propoxymethyl-3-ethyloxetane, 3-butoxymethyl-3-ethyl 3-alkoxymethyl-3-alkyloxetanes such as oxetane; 3- (2-methoxyethoxy) methyl-3-methyloxetane; 3- (2- (2-methoxyethoxy) ethoxy) methyl-3-methyloxetane; -(2- (2- (2-methoxyethoxy) ethoxy) ethoxy Methyl-3-methyloxetane, 3- (2-ethoxyethoxy) methyl-3-methyloxetane, 3- (2- (2-ethoxyethoxy) ethoxy) methyl-3-methyloxetane, 3- (2- (2- (2-ethoxyethoxy) ethoxy) ethoxy) methyl-3-methyloxetane, 3- (2-propoxyethoxy) methyl-3-methyloxetane, 3- (2-butoxyethoxy) methyl-3-methyloxetane, 3- ( 2-methoxyethoxy) methyl-3-ethyloxetane, 3- (2- (2-methoxyethoxy) ethoxy) methyl-3-ethyloxetane, 3- (2- (2- (2-methoxyethoxy) ethoxy) ethoxy) Methyl-3-ethyloxetane, 3- (2-ethoxyethoxy) methyl-3-ethyloxetane, 3- (2- (2- Ethoxyethoxy) ethoxy) methyl-3-ethyloxetane, 3- (2- (2- (2-ethoxyethoxy) ethoxy) ethoxy) methyl-3-ethyloxetane, 3- (2-propoxyethoxy) methyl-3-ethyl Represented by oxetane, 3- (2-butoxyethoxy) methyl-3-ethyloxetane, 3- (2-acetoxyethoxy) methyl-3-methyloxetane, 3- (2-acetoxyethoxy) methyl-3-ethyloxetane and the like. In formula (2), R 3 Is an alkyl group having 1 to 10 carbon atoms, R 4 Is represented by the formula (4), wherein n in the formula (4) is an integer of 1 to 10, A is a methylene group, B is an ethylene group, R 5 Is an oxetane represented by an alkyl group or an acyl group having 1 to 20 carbon atoms; 3,3-bis (methoxymethyl) oxetane, 3,3-bis (ethoxymethyl) oxetane, 3,3-bis (propoxymethyl) oxetane 3,3-bis (alkoxymethyl) oxetanes such as 3,3-bis (butoxymethyl) oxetane; 3,3-bis ((2-methoxyethoxy) methyl) oxetane, 3,3-bis ((2 -(2-methoxyethoxy) ethoxy) methyl) oxetane, 3,3-bis ((2- (2- (2-methoxyethoxy) ethoxy) ethoxy) methyl) oxetane, 3,3-bis ((2-ethoxyethoxy) ) Methyl) oxetane, 3,3-bis ((2- (2-ethoxyethoxy) ethoxy) methyl) oxetane, 3,3-bis ((2- (2 (2-ethoxyethoxy) ethoxy) ethoxy) methyl) oxetane, 3,3-bis ((2-propoxyethoxy) methyl) oxetane, 3,3-bis ((2-butoxyethoxy) methyl) oxetane, 3,3- In the formula (2) represented by bis ((2-acetoxyethoxy) methyl) oxetane or the like, R 3 , R 4 Is represented by the formula (4), wherein n in the formula (4) is an integer of 1 to 10, A is a methylene group, B is an ethylene group, R 5 Is an oxetane represented by an alkyl group or an acyl group having 1 to 20 carbon atoms; 3- (2-methoxypropoxy) methyl-3-ethyloxetane, 3- (3-n-butoxy-2-acetoxypropoxy) methyl-3 In the formula (2) represented by -ethyloxetane, 3- (3-phenoxy-2-acetoxypropoxy) methyl-3-ethyloxetane, 3- (2-trimethylsilyloxypropoxy) methyl-3-ethyloxetane, etc. 3 Is an alkyl group having 1 to 10 carbon atoms, R 4 Is represented by the formula (4), n in the formula (4) is an integer of 1 to 10, A is a methylene group, B is an ethylene group having a substituent, R 3 Is an oxetane represented by an alkyl group, an acyl group or a trialkylsilyl group; an oxetane having a trialkylsilyloxy group such as 3-trimethylsilyloxymethyl-3-methyloxetane or 3-trimethylsilyloxymethyl-3-ethyloxetane And the like.
However, the oxetane derivative represented by the following formula (5) is not suitable because an orthoester having low ring-opening polymerizability may be formed in the presence of an acid catalyst.
[0035]
Embedded image
(In the formula (5), R 6 , R 7 Is hydrogen or an optional substituent. )
The total amount of the polyether glycol segment (soft segment) in the whole polyetherester elastomer of the present invention is 10 to 90% by weight, preferably 30 to 75% by weight, particularly preferably 50 to 70% by weight. This value depends on the required physical properties of the final molded article intended using the polyetherester elastomer. When the amount of the soft segment is less than 10% by weight, softness, oil resistance and heat resistance are inferior, and satisfactory physical properties as an elastomer cannot be expected. When the amount exceeds 90% by weight, the softness is considerably imparted, but at the same time, the average chain length of the hard segments is shortened, and the hard block, which is a physical cross-linking point, cannot resist external force and has a mechanical strength. Is remarkably reduced, and is no longer used as an elastomer material. In addition, the melting point is considerably lowered, so that the heat resistance is also poor, which is not preferable. A more preferred amount of the soft segment is 25-75% by weight.
[0037]
When the polyetherester elastomer of the present invention is used as a modifier for other polyester resins, polycarbonate resins, polyamide resins, etc., use a polyetherester elastomer containing a soft segment component of 50% by weight or more. Can be done.
[0038]
Such a polyetherester elastomer of the present invention can be produced by a known method described below. For example, a lower alcohol diester of dicarboxylic acid, a transesterification reaction of an excessive amount of low molecular weight glycol and polyether glycol in the presence of a catalyst, and then a polycondensation of the resulting reaction product under reduced pressure, or a dicarboxylic acid and glycol and A method in which polyether glycol is subjected to an esterification reaction in the presence of a catalyst, and then the obtained product is subjected to polycondensation. Alternatively, a short-chain polyester (for example, polybutylene terephthalate) is prepared in advance, and another dicarboxylic acid, diol or polyether is prepared. Any method such as a method in which ether glycol is added, or another copolymerized polyester is added and randomization is performed by transesterification may be employed.
[0039]
As a catalyst common to the transesterification reaction or the esterification reaction and the polycondensation reaction, a tetraalkyl titanate represented by tetra (isopropoxy) titanate and tetra (n-butoxy) titanate, and reaction formation between these tetraalkyl titanates and alkylene glycol Products, partial hydrolysates of tetraalkyl titanates, metal salts of titanium hexaalkoxide, metal salts of titanium hexaalkoxide, titanium carboxylate, titanyl compounds and other Ti-based catalysts, and mono-n- Monoalkyltin compounds such as butyl monohydroxytin oxide, mono-n-butyltin triacetate, mono-n-butyltin monooctylate, mono-n-butyltin monoacetate, di-n-butyltin oxide, di-n-butyltin diacetate, dif Nils dibutyltin oxide, diphenyltin diacetate, dialkyl di n- butyltin Geo lactylate etc. (or diaryl) tin compounds. In addition, a metal or metal oxide catalyst such as Mg, Pb, Zr, Zn, and Sb is useful. These catalysts may be used alone or in combination of two or more. In particular, when used alone, it is preferable to use tetraalkyl titanate or antimony trioxide, and when used in combination, it is preferable to use tetraalkyl titanate and magnesium acetate.
[0040]
The amount of the esterification or polycondensation catalyst to be added is preferably 0.005 to 0.5% by weight, particularly preferably 0.03 to 0.2% by weight, based on the produced polymer. These catalysts may be added at the start of the transesterification or esterification reaction, and may or may not be added again at the time of the polycondensation reaction. Further, a polycarboxylic acid, a polyfunctional hydroxy compound, an oxyacid, or the like may be copolymerized as a part of the dicarboxylic acid or the glycol. The polyfunctional component effectively acts as a high viscosity component, and its content in the copolymer is preferably 3 mol% or less. Those that can be used as such a polyfunctional component include trimellitic acid, trimesic acid, pyromellitic acid, benzophenonetetracarboxylic acid, butanetetracarboxylic acid, glycerin, pentaerythritol and their esters, acid anhydrides and the like. it can.
[0041]
The antioxidant can be added during the production of the polyetherester elastomer or at any time after the production, and especially when the polyetherglycol is exposed to a high temperature, for example, when it enters the polycondensation reaction. It is desirable to add an antioxidant which does not inhibit the polycondensation reaction and does not impair the function of the catalyst in order to prevent oxidative deterioration of the catalyst. Examples of these antioxidants include aliphatic, aromatic or alkyl group-substituted aromatic esters of phosphoric acid and phosphorous acid, hypophosphorous acid derivatives, phenylphosphonic acid, phenylphosphinic acid, diphenylphosphonic acid, polyphosphonates, dialkylpentaerythritol diacids. Phosphites, phosphorus compounds such as dialkylbisphenol A diphosphite, phenol derivatives, especially hindered phenol compounds, thioether compounds, dithio acid salts, mercaptobenzimidazole compounds, thiocarbanilide compounds, compounds containing sulfur such as thiodipropionate, tinmalate And tin-based compounds such as dibutyltin monoxide. These may be used alone or in combination of two or more.
[0042]
The addition amount of these stabilizers is desirably 0.01 to 2 parts by weight based on 100 parts by weight of the polyetherester elastomer. Further, if necessary, the polyether glycol in the present invention may be partially substituted with another polyether glycol. Poly (ethyleneoxy) glycol, poly (propyleneoxy) glycol, poly (tetramethyleneoxy) glycol, poly (1,2-propyleneoxy) glycol, block or random of ethylene oxide and propylene oxide are used as the polyether glycol used for such substitution. Copolymers, block or random copolymers of ethylene oxide and THF, poly (2-methyl-1,3-propyleneoxy) glycol, poly (propyleneoxy) diimidodiic acid, and the like.
[0043]
Finally, an optional component can be added to the finally obtained polyetherester elastomer, if necessary, as long as the object and effects of the present invention are not impaired. Specifically, antioxidants, kaolin, silica, mica, titanium dioxide, alumina, calcium carbonate, calcium silicate, clay, kaolin, diatomaceous earth, asbestos, barium sulfate, aluminum sulfate, calcium sulfate, basic magnesium carbonate, Fillers and reinforcing materials such as molybdenum disulfide, graphite, glass fiber, and carbon fiber, lubricants or release agents such as zinc stearate and bis amide stearate, carbon black for coloring, ultramarine, titanium oxide, zinc oxide, Pigments, navy blue, azo pigments, nitro pigments, lake pigments, dyes such as phthalocyanine pigments, flame retardants such as octabromodiphenyl, tetrabromobisphenol polycarbonate, hindered amine light stabilizers, ultraviolet absorbers, foaming agents, epoxy Compounds, isocyanate compounds, etc. Thickeners, silicone oils and silicone resins, can be used various known additives.
[0044]
【Example】
EXAMPLES Hereinafter, the present invention will be described more specifically with reference to examples, but the present invention is not limited to the following examples as long as the gist is not exceeded. First, evaluation methods and materials used in Examples and Comparative Examples are described below.
<Evaluation method>
(1) Tensile strength: JIS K6251 (No. 3 dumbbell), a 2 mm thick press sheet was used as a sample.
(2) Tensile elongation: JIS K6251 (No. 3 dumbbell), 2 mm thick pressed sheet was used as a sample.
(3) Melting point: Performed with a differential calorimeter (RDC220) manufactured by SEIKO Electronics Co., Ltd. using about 6 mg of a polyetherester elastomer. First, the temperature was raised from 30 ° C. to 290 ° C. at a rate of 50 ° C./min, kept at 290 ° C. for 3 minutes, cooled down to −130 ° C. at 10 ° C./min, kept at −130 ° C. for 10 minutes, and then again The temperature was raised to 290 ° C. at a rate of 10 ° C./min. The endothermic peak at that time was measured, the temperature at the top was determined, and it was defined as the melting point.
(4) ΔH f : Using a differential calorimeter (RDC220) manufactured by SEIKO Denshi Kogyo Co., Ltd., using about 5 mg of a polyetherester elastomer. First, the temperature was raised from 30 ° C. to 290 ° C. at a rate of 50 ° C./min, kept at 290 ° C. for 3 minutes, lowered to −130 ° C. at 3 ° C./min, kept at −130 ° C. for 10 minutes, and then again The temperature was raised to 290 ° C. at a rate of 10 ° C./min. The amount of heat absorbed at −50 ° C. to 100 ° C. is ΔH f Was measured.
[0045]
(5) log E ': Dynamic viscoelasticity due to longitudinal vibration was measured using Rheograph Solid manufactured by Toyo Seiki Co., Ltd. Heating rate 2 ° C / min, measurement frequency 10Hz, tension 10g / mm 2 The temperature was measured from -130 ° C to 150 ° C under the condition of 0.1% strain. For the measurement, a pressed sheet having a thickness of 0.3 mm was prepared by pressing a polyetherester elastomer (temperature: 220 ° C., pressing time: 5 minutes), and this was cut out to a width of 6 mm and a length of 3 mm and used as a measurement sample. Was. The dynamic storage modulus (E ') and the dynamic loss modulus (E ") were measured.
(6) Izod: Izod impact strength was measured at 23 ° C and -50 ° C in accordance with JIS K7110. In the table, “NB” was written in the table when the sample was not broken, and “Break” was written in the table when the sample was broken.
[0046]
<Material>
As the polyalkylene ether glycol used for the soft segment of the polyether ester elastomer used in Examples and Comparative Examples, the following was used.
(1) A1: 3-butoxymethyl-3-ethyloxetane copolymerized polytetramethylene ether glycol (both terminals are hydroxyl groups) (Mn = 2119, Mw = 3456 Mw / Mn = 1.63, 3-butoxymethyl-3-ethyl (Oxetane content = 10.1 mol%).
[0047]
(Production Example 1)
9 mol% ZrO 2 -SiO 2 The composite oxide catalyst was dried at 500 ° C. for 1 hour in a nitrogen stream. In a glass container, 62.0 g of acetic anhydride, 400.5 g of tetrahydrofuran, and 7.7 g of 3-butoxymethyl-3-ethyloxetane (tetrahydrofuran: 3-butoxymethyl-3-ethyloxetane = 89.9: 10.1 molar ratio). Was added thereto, and 11.2 g of the dried catalyst was added thereto while stirring under a nitrogen atmosphere. The mixture was placed in a water bath in which the water temperature was adjusted, and 3-butoxymethyl-3-ethyloxetane was added at 0.18 g / min using a pump. The reaction was carried out at normal pressure and 40 ° C. for 2 hours while the solution was dropped. (Total 22.1 g of 3-butoxymethyl-3-ethyloxetane was added dropwise.)
[0048]
After completion of the reaction, 12 g of water was added, the reaction solution was filtered to remove the catalyst, and unreacted substances were removed by distillation to obtain 105.4 g of polyether polyol diester. 105.4 g of the obtained polyether polyol diester, 316.2 g of methanol, and 0.5 g of sodium hydroxide were charged into a reactor equipped with a distillation apparatus, and transesterification was performed for 4 hours while distilling an azeotropic mixture of methanol / methyl acetate. Was done. Then, after removing unreacted methanol at room temperature under reduced pressure, 158.1 g of methanol and 158.1 g of water were added, and the mixture was heated and stirred at 50 ° C. for 30 minutes under nitrogen, allowed to stand for 15 minutes, and then stirred for 15 minutes. Oligomer-rich) was removed. After this oligomer removal operation was performed twice, the lower layer was dissolved in 316.2 g of tetrahydrofuran, 5.3 g of activated clay was added, the mixture was stirred at room temperature for 2 hours, and then filtered to remove the activated clay. Tetrahydrofuran was distilled off from the obtained filtrate to obtain 99.1 g of polybutamethylene ether glycol copolymerized with 3-butoxymethyl-3-ethyloxetane.
[0049]
(2) A2: 3-octoxymethyl-3-ethyloxetane copolymerized polytetramethylene ether glycol (both terminals are hydroxyl groups) (Mn = 2023, Mw = 3479 Mw / Mn = 1.72, 3-octoxymethyl- (3-ethyloxetane content = 9.1 mol%) was changed from 7.7 g of 3-butoxymethyl-3-ethyloxetane to 10.2 g of 3-octoxymethyl-3-ethyloxetane in Production Example 1. Other than that, it prepared similarly.
[0050]
(3) A3: 3-methoxymethyl-3-ethyloxetane copolymerized polytetramethylene ether glycol (both terminals are hydroxyl groups) (Mn = 2262, Mw = 3774 Mw / Mn = 1.67, 3-methoxymethyl-3-ethyl Oxetane content = mol 10.2%) in Preparation Example 1, except that 7.7 g of 3-butoxymethyl-3-ethyloxetane was changed to 5.8 g of 3-methoxymethyl-3-ethyloxetane. Prepared.
(4) A4: polytetramethylene ether glycol: manufactured by Mitsubishi Chemical Corporation, Mn = 2131, Mw = 4349, Mw / Mn = 2.04
(5) A5: PTG-L2000 (manufactured by Hodogaya Chemical Industry Co., Ltd.), 3-methyl-tetrahydrofuran copolymerized polytetramethylene ether glycol (both terminals are hydroxyl groups) (Mn = 2141, Mw = 4111, Mw / Mn = 1) .92, 3-methyl-tetrahydrofuran content = 16 mol%).
[0051]
<Examples and comparative examples>
(Example 1)
In a reactor for condensation, 49 g of dimethyl terephthalate (manufactured by Tokyo Kasei), 38 g of 1,4-butanediol (manufactured by Tokyo Kasei), and 98 g of copolymerized polytetramethylene ether glycol of 3-butoxymethyl-3-ethyloxetane manufactured in Production Example 1 were added. After charging and purging with nitrogen, the mixture was heated and melted at 150 ° C. for 30 minutes in a nitrogen atmosphere. Next, 3.7 g of a solution of tetrabutoxytitanate (manufactured by Kishida Chemical Co., Ltd.) dissolved in 1,4-butanediol at 6% by weight was added, and the mixture was kept at 150 ° C. for 60 minutes while stirring at 150 rpm for 2 hours. Then, the temperature was raised to 230 ° C., and the mixture was kept at 230 ° C. for 15 minutes to perform a transesterification reaction. To the obtained melt, 5.4 ml of a slurry obtained by dispersing Irganox 1330 (manufactured by Ciba Specialty Chemical) at 5% by weight in 1,4-butanediol was added, and the temperature was further raised to 245 ° C. over 45 minutes. Then, the pressure was reduced to 1 mmHg over 85 minutes, and a polycondensation reaction was performed under the reduced pressure. At 230 minutes after the state of 1 mmHg, the torque reached 2.0 kg, so the rotation speed was reduced to 100 rpm. After 50 minutes, when the torque reached 2.0 kg, the rotation speed was reduced to 50 rpm, and 55 When the torque reaches 2.0 kg later, the rotation speed is reduced to 25 rpm when the torque reaches 1.7 kg, and finally, when the torque reaches 1.7 kg after 40 minutes, the reactor is double-pressurized to normal pressure with nitrogen in the reactor. The polycondensation reaction was stopped. Thereafter, the contents of the reactor were taken out in a strand form, and each evaluation was performed according to the evaluation method described above. Table 1 shows the results. Further, the temperature dependence of the dynamic storage modulus (E ') was analyzed. The result is shown in FIG.
[0052]
(Example 2)
A transesterification reaction was performed in the same manner as in Example 1 except that 98 g of the 3-octoxymethyl-3-ethyloxetane copolymerized polytetramethylene ether glycol (A2) produced in Production Example 2 was used instead of A1. And a condensation reaction. The same applies to the type of additive and the mixing ratio. Table 1 shows the evaluation results.
(Example 3)
A transesterification reaction was performed in the same manner as in Example 1, except that 98 g of the 3-methoxymethyl-3-ethyloxetane copolymerized polytetramethylene ether glycol (A3) produced in Production Example 3 was used instead of A1. A condensation reaction was performed. The same applies to the type of additive and the mixing ratio. Table 1 shows the evaluation results.
[0053]
(Comparative Example 1)
A transesterification reaction and a condensation reaction were carried out in the same manner as in Example 1, except that 98 g of polytetramethylene ether glycol (A4) was used instead of A1. The same applies to the type of additive and the mixing ratio. Table 1 shows the evaluation results. Further, the temperature dependence of the dynamic storage modulus (E ') was analyzed. The result is shown in FIG.
(Comparative Example 2)
The transesterification reaction and the condensation reaction were carried out in the same manner as in Example 1 except that 98 g of the 3-methyl-tetrahydrofuran copolymerized polytetramethylene ether glycol (A5) produced in Production Example 5 was used instead of A1. Was. The same applies to the type of additive and the mixing ratio. Table 1 shows the evaluation results.
[0054]
[Table 1]
<Evaluation of results>
1) Comparative Example 1 is logE ' -50 Is an irregular value of 8.64 and ΔH f Is also 23.3 mJ / mg, which is out of the specified range, so that the Izod impact value at −50 ° C. is low.
2) Comparative Example 2 is logE ' -50 Is 8.14 out of the specified range, and ΔH f Is also 10.1 mJ / mg, which is out of the range, so that the Izod impact value at −50 ° C. is low.
[0056]
【The invention's effect】
Since the polyetherester elastomer of the present invention does not cure at a low temperature as compared with a conventional polyester elastomer of this type, the change in physical properties from a high temperature to a low temperature is small and the rubber-like elasticity has a wide temperature range. For this reason, automotive parts (particularly around engines, interiors), industrial products such as tubes, hoses, gears, electric wire covering materials, etc. It can be suitably used as an impact resistance improving material for daily necessities and polyester and polycarbonate resins.
[Brief description of the drawings]
FIG. 1 is a graph showing temperature changes in dynamic storage modulus (E ′) of a polyetherester elastomer of Example 1 and a polyetherester elastomer of Comparative Example 1.
Claims (8)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003203870A JP2004131701A (en) | 2002-08-14 | 2003-07-30 | Polyetherester elastomer |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002236476 | 2002-08-14 | ||
| JP2003203870A JP2004131701A (en) | 2002-08-14 | 2003-07-30 | Polyetherester elastomer |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2004131701A true JP2004131701A (en) | 2004-04-30 |
| JP2004131701A5 JP2004131701A5 (en) | 2006-05-11 |
Family
ID=32301045
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003203870A Pending JP2004131701A (en) | 2002-08-14 | 2003-07-30 | Polyetherester elastomer |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2004131701A (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018105914A3 (en) * | 2016-12-09 | 2018-08-09 | 주식회사 삼양사 | Thermoplastic polyether ester elastomer comprising anhydrosugar alcohol derivative and method for preparing same |
| US10995212B2 (en) | 2016-12-09 | 2021-05-04 | Samyang Corporation | Thermoplastic polyether ester elastomer comprising anhydrosugar alcohol derivative and method for preparing same |
-
2003
- 2003-07-30 JP JP2003203870A patent/JP2004131701A/en active Pending
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018105914A3 (en) * | 2016-12-09 | 2018-08-09 | 주식회사 삼양사 | Thermoplastic polyether ester elastomer comprising anhydrosugar alcohol derivative and method for preparing same |
| US10995212B2 (en) | 2016-12-09 | 2021-05-04 | Samyang Corporation | Thermoplastic polyether ester elastomer comprising anhydrosugar alcohol derivative and method for preparing same |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US3651014A (en) | Segmented thermoplastic copolyester elastomers | |
| KR100475758B1 (en) | Polyetherester elastomer | |
| JPS6259737B2 (en) | ||
| JPH0115532B2 (en) | ||
| US20120302722A1 (en) | Copolyether ester elastomer | |
| US20080207839A1 (en) | Block Copolyetherester Elastomer and Preparation Thereof | |
| US8378057B2 (en) | Polyether ester block copolymer | |
| US3891604A (en) | Segmented thermoplastic copolyester elastomer | |
| JP2006316262A (en) | Polyetherester block copolymer | |
| EP0608976B1 (en) | Esteramide copolymers and production thereof | |
| WO2009123287A1 (en) | Process for producing thermoplastic polyester elastomer and polycarbonate oligomer composition as starting material for the thermoplastic polyester elastomer | |
| JPH08511578A (en) | Polyetherester block copolymer elastomer | |
| JP2004131701A (en) | Polyetherester elastomer | |
| Aleksandrovic et al. | Poly (ether–ester) s modified with different amounts of fumaric moieties | |
| JP3117805B2 (en) | Polyetherester elastomer | |
| EP0275988A2 (en) | Polyamide copolymers | |
| KR100793195B1 (en) | Aromatic amide block copolymer and process of producing the same | |
| JPH07216072A (en) | Production of polyether ester block copolymer | |
| JP3117806B2 (en) | Method for producing polyetherester elastomer | |
| JP2005060645A (en) | Polyester-based block copolymer and method for producing the same | |
| JP4449375B2 (en) | Aromatic amide block copolymer and process for producing the same | |
| JP3418231B2 (en) | Polyetherester block copolymer and method for producing the same | |
| JP3625972B2 (en) | Method for producing polyester elastomer | |
| JPH03229724A (en) | Production of polyester elastomer copolymer | |
| JPH09268253A (en) | Polyester block copolymer composition and its production |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Written amendment |
Effective date: 20060322 Free format text: JAPANESE INTERMEDIATE CODE: A523 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20060322 |
|
| A977 | Report on retrieval |
Effective date: 20080331 Free format text: JAPANESE INTERMEDIATE CODE: A971007 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080924 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20090217 |