FR2796381A1 - Derives bicycliques de thioazepine ou de caprolactame, leur procede de preparation et les intermediaires de ce procede, leur application comme medicaments et les compositions pharmaceutiques les contenant - Google Patents
Derives bicycliques de thioazepine ou de caprolactame, leur procede de preparation et les intermediaires de ce procede, leur application comme medicaments et les compositions pharmaceutiques les contenant Download PDFInfo
- Publication number
- FR2796381A1 FR2796381A1 FR9909006A FR9909006A FR2796381A1 FR 2796381 A1 FR2796381 A1 FR 2796381A1 FR 9909006 A FR9909006 A FR 9909006A FR 9909006 A FR9909006 A FR 9909006A FR 2796381 A1 FR2796381 A1 FR 2796381A1
- Authority
- FR
- France
- Prior art keywords
- formula
- alkyl
- aryl
- group
- compounds
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 238000011282 treatment Methods 0.000 title claims description 22
- 206010020751 Hypersensitivity Diseases 0.000 title claims description 11
- 230000007815 allergy Effects 0.000 title claims description 11
- 208000001132 Osteoporosis Diseases 0.000 title claims description 7
- 206010028980 Neoplasm Diseases 0.000 title claims description 6
- 208000015181 infectious disease Diseases 0.000 title claims description 6
- JBKVHLHDHHXQEQ-UHFFFAOYSA-N Caprolactam Natural products O=C1CCCCCN1 JBKVHLHDHHXQEQ-UHFFFAOYSA-N 0.000 title abstract description 10
- 208000020084 Bone disease Diseases 0.000 title description 2
- -1 Rg = H Inorganic materials 0.000 claims abstract description 112
- 150000003839 salts Chemical class 0.000 claims abstract description 50
- 239000000651 prodrug Substances 0.000 claims abstract description 46
- 229940002612 prodrug Drugs 0.000 claims abstract description 46
- 239000000203 mixture Substances 0.000 claims abstract description 30
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims abstract description 29
- 125000003118 aryl group Chemical group 0.000 claims abstract description 25
- 229910052799 carbon Inorganic materials 0.000 claims abstract description 19
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 19
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 15
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 13
- 150000002367 halogens Chemical class 0.000 claims abstract description 13
- 125000000623 heterocyclic group Chemical group 0.000 claims abstract description 12
- 125000005842 heteroatom Chemical group 0.000 claims abstract description 11
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 10
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 9
- 229910006069 SO3H Inorganic materials 0.000 claims abstract description 7
- 125000000753 cycloalkyl group Chemical group 0.000 claims abstract description 7
- 150000003536 tetrazoles Chemical class 0.000 claims abstract description 6
- 229910006074 SO2NH2 Inorganic materials 0.000 claims abstract description 3
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 claims abstract description 3
- 125000004953 trihalomethyl group Chemical group 0.000 claims abstract description 3
- 150000001875 compounds Chemical class 0.000 claims description 157
- 238000002360 preparation method Methods 0.000 claims description 46
- 238000000034 method Methods 0.000 claims description 40
- 150000001412 amines Chemical class 0.000 claims description 29
- 239000003814 drug Substances 0.000 claims description 27
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 27
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 22
- 150000003254 radicals Chemical class 0.000 claims description 22
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 claims description 21
- 239000002253 acid Substances 0.000 claims description 17
- 230000009471 action Effects 0.000 claims description 16
- 125000000738 acetamido group Chemical group [H]C([H])([H])C(=O)N([H])[*] 0.000 claims description 15
- 230000008569 process Effects 0.000 claims description 14
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 14
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 13
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 12
- 230000024279 bone resorption Effects 0.000 claims description 12
- 238000006243 chemical reaction Methods 0.000 claims description 12
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims description 12
- 208000006386 Bone Resorption Diseases 0.000 claims description 11
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 11
- 125000000524 functional group Chemical group 0.000 claims description 11
- 201000010099 disease Diseases 0.000 claims description 10
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 10
- 230000002265 prevention Effects 0.000 claims description 10
- 238000010511 deprotection reaction Methods 0.000 claims description 9
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 9
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 8
- 239000003795 chemical substances by application Substances 0.000 claims description 8
- 230000002401 inhibitory effect Effects 0.000 claims description 8
- NYERMPLPURRVGM-UHFFFAOYSA-N thiazepine Chemical compound S1C=CC=CC=N1 NYERMPLPURRVGM-UHFFFAOYSA-N 0.000 claims description 8
- 229910018828 PO3H2 Inorganic materials 0.000 claims description 7
- 125000002619 bicyclic group Chemical group 0.000 claims description 7
- 150000001721 carbon Chemical group 0.000 claims description 7
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 7
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 claims description 7
- 239000008194 pharmaceutical composition Substances 0.000 claims description 7
- 239000002243 precursor Substances 0.000 claims description 7
- 208000023275 Autoimmune disease Diseases 0.000 claims description 6
- 239000000543 intermediate Substances 0.000 claims description 6
- 208000024172 Cardiovascular disease Diseases 0.000 claims description 5
- 206010061218 Inflammation Diseases 0.000 claims description 5
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical compound OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 claims description 5
- 208000026935 allergic disease Diseases 0.000 claims description 5
- 201000011510 cancer Diseases 0.000 claims description 5
- 230000006806 disease prevention Effects 0.000 claims description 5
- 230000036039 immunity Effects 0.000 claims description 5
- 230000004054 inflammatory process Effects 0.000 claims description 5
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 claims description 5
- 230000002062 proliferating effect Effects 0.000 claims description 5
- 208000037803 restenosis Diseases 0.000 claims description 5
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 claims description 4
- 239000000654 additive Substances 0.000 claims description 4
- 150000005840 aryl radicals Chemical class 0.000 claims description 4
- 125000004429 atom Chemical group 0.000 claims description 4
- 230000008878 coupling Effects 0.000 claims description 4
- 238000010168 coupling process Methods 0.000 claims description 4
- 238000005859 coupling reaction Methods 0.000 claims description 4
- 229910052739 hydrogen Inorganic materials 0.000 claims description 4
- 239000001257 hydrogen Substances 0.000 claims description 4
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 4
- 125000006239 protecting group Chemical group 0.000 claims description 4
- 125000004434 sulfur atom Chemical group 0.000 claims description 4
- 150000004912 thiazepines Chemical class 0.000 claims description 4
- 150000003852 triazoles Chemical class 0.000 claims description 4
- KJRKUBSCNFISGN-UHFFFAOYSA-N 1,4-thiazepine-2-carboxamide Chemical compound S1C(=CN=CC=C1)C(=O)N KJRKUBSCNFISGN-UHFFFAOYSA-N 0.000 claims description 3
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 3
- 125000002373 5 membered heterocyclic group Chemical group 0.000 claims description 2
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 claims description 2
- 150000001348 alkyl chlorides Chemical class 0.000 claims description 2
- SKOLWUPSYHWYAM-UHFFFAOYSA-N carbonodithioic O,S-acid Chemical class SC(S)=O SKOLWUPSYHWYAM-UHFFFAOYSA-N 0.000 claims description 2
- 238000009903 catalytic hydrogenation reaction Methods 0.000 claims description 2
- 230000000694 effects Effects 0.000 claims description 2
- 239000012634 fragment Substances 0.000 claims description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 claims description 2
- TUJKJAMUKRIRHC-UHFFFAOYSA-N hydroxyl Chemical compound [OH] TUJKJAMUKRIRHC-UHFFFAOYSA-N 0.000 claims description 2
- 239000012038 nucleophile Substances 0.000 claims description 2
- 230000036961 partial effect Effects 0.000 claims description 2
- 229910052705 radium Inorganic materials 0.000 claims description 2
- 229910052701 rubidium Inorganic materials 0.000 claims description 2
- 125000006652 (C3-C12) cycloalkyl group Chemical group 0.000 claims 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 claims 1
- 125000004430 oxygen atom Chemical group O* 0.000 claims 1
- 125000000217 alkyl group Chemical group 0.000 abstract description 15
- 229910052757 nitrogen Inorganic materials 0.000 abstract description 7
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 abstract description 3
- 125000004453 alkoxycarbonyl group Chemical group 0.000 abstract description 2
- 125000004104 aryloxy group Chemical group 0.000 abstract description 2
- 125000005530 alkylenedioxy group Chemical group 0.000 abstract 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 63
- 239000000047 product Substances 0.000 description 49
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 48
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 36
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 26
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 24
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 21
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 18
- 230000002829 reductive effect Effects 0.000 description 18
- 235000018102 proteins Nutrition 0.000 description 16
- 108090000623 proteins and genes Proteins 0.000 description 16
- 102000004169 proteins and genes Human genes 0.000 description 16
- 102000014400 SH2 domains Human genes 0.000 description 15
- 108050003452 SH2 domains Proteins 0.000 description 15
- 238000001819 mass spectrum Methods 0.000 description 14
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 13
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 12
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 238000004587 chromatography analysis Methods 0.000 description 11
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 9
- 230000005764 inhibitory process Effects 0.000 description 9
- 210000002997 osteoclast Anatomy 0.000 description 9
- 239000000243 solution Substances 0.000 description 9
- 239000007858 starting material Substances 0.000 description 9
- 238000003786 synthesis reaction Methods 0.000 description 9
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 8
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 8
- 239000000460 chlorine Substances 0.000 description 8
- VFRSADQPWYCXDG-LEUCUCNGSA-N ethyl (2s,5s)-5-methylpyrrolidine-2-carboxylate;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CCOC(=O)[C@@H]1CC[C@H](C)N1 VFRSADQPWYCXDG-LEUCUCNGSA-N 0.000 description 8
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 8
- 239000003446 ligand Substances 0.000 description 8
- 239000011734 sodium Substances 0.000 description 8
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 7
- 238000005481 NMR spectroscopy Methods 0.000 description 7
- 239000012074 organic phase Substances 0.000 description 7
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 7
- 229960005190 phenylalanine Drugs 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 7
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- TUCNEACPLKLKNU-UHFFFAOYSA-N acetyl Chemical compound C[C]=O TUCNEACPLKLKNU-UHFFFAOYSA-N 0.000 description 6
- 150000001408 amides Chemical class 0.000 description 6
- 239000002585 base Substances 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 6
- 230000003197 catalytic effect Effects 0.000 description 6
- 230000003993 interaction Effects 0.000 description 6
- 238000002821 scintillation proximity assay Methods 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 6
- PJUPKRYGDFTMTM-UHFFFAOYSA-N 1-hydroxybenzotriazole;hydrate Chemical compound O.C1=CC=C2N(O)N=NC2=C1 PJUPKRYGDFTMTM-UHFFFAOYSA-N 0.000 description 5
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 5
- 108060006706 SRC Proteins 0.000 description 5
- 229910052801 chlorine Inorganic materials 0.000 description 5
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 5
- 125000005328 phosphinyl group Chemical group [PH2](=O)* 0.000 description 5
- 125000001424 substituent group Chemical group 0.000 description 5
- 229960004441 tyrosine Drugs 0.000 description 5
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 4
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 4
- UVZMNGNFERVGRC-UHFFFAOYSA-N 4-cyclohexylbutanoic acid Chemical compound OC(=O)CCCC1CCCCC1 UVZMNGNFERVGRC-UHFFFAOYSA-N 0.000 description 4
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 4
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- 239000005557 antagonist Substances 0.000 description 4
- IYYIVELXUANFED-UHFFFAOYSA-N bromo(trimethyl)silane Chemical compound C[Si](C)(C)Br IYYIVELXUANFED-UHFFFAOYSA-N 0.000 description 4
- 210000004027 cell Anatomy 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 239000011737 fluorine Substances 0.000 description 4
- 229910052731 fluorine Inorganic materials 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 4
- 125000002950 monocyclic group Chemical group 0.000 description 4
- 210000002569 neuron Anatomy 0.000 description 4
- 239000000825 pharmaceutical preparation Substances 0.000 description 4
- 108090000765 processed proteins & peptides Proteins 0.000 description 4
- 108020003175 receptors Proteins 0.000 description 4
- 102000005962 receptors Human genes 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 4
- IYCLBDOEZOYCDB-UHFFFAOYSA-N 1,4-thiazepine-2-carboxylic acid Chemical compound S1C(=CN=CC=C1)C(=O)O IYCLBDOEZOYCDB-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 3
- 102000000395 SH3 domains Human genes 0.000 description 3
- 108050008861 SH3 domains Proteins 0.000 description 3
- 102000001332 SRC Human genes 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 125000002252 acyl group Chemical group 0.000 description 3
- 239000011324 bead Substances 0.000 description 3
- 210000000988 bone and bone Anatomy 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 3
- 238000001704 evaporation Methods 0.000 description 3
- 230000008020 evaporation Effects 0.000 description 3
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 3
- 238000005984 hydrogenation reaction Methods 0.000 description 3
- 230000003834 intracellular effect Effects 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 108091005981 phosphorylated proteins Proteins 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- DLNQPUNIZRZGQD-VBIHGRSYSA-N (2s)-2-[[(2s)-2-[[(2s,3s)-2-[[(2s)-1-[(2s,3s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-5-amino-2-[[(2s)-1-[(2s)-2-amino-4-carboxybutanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-3-(4-phosphonooxyphenyl)propanoyl]amino]-4-carboxybutanoyl]amino]-4-ca Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@H](C(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(O)=O)[C@@H](C)CC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CCC(O)=O)C1=CC=C(OP(O)(O)=O)C=C1 DLNQPUNIZRZGQD-VBIHGRSYSA-N 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical group NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 2
- 239000007993 MOPS buffer Substances 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 208000029725 Metabolic bone disease Diseases 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- DBTDEFJAFBUGPP-UHFFFAOYSA-N Methanethial Chemical compound S=C DBTDEFJAFBUGPP-UHFFFAOYSA-N 0.000 description 2
- 206010049088 Osteopenia Diseases 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- 230000005856 abnormality Effects 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 238000005804 alkylation reaction Methods 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 2
- 150000001602 bicycloalkyls Chemical group 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- MGNZXYYWBUKAII-UHFFFAOYSA-N cyclohexa-1,3-diene Chemical compound C1CC=CC=C1 MGNZXYYWBUKAII-UHFFFAOYSA-N 0.000 description 2
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 2
- 239000000975 dye Substances 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- AEOCXXJPGCBFJA-UHFFFAOYSA-N ethionamide Chemical compound CCC1=CC(C(N)=S)=CC=N1 AEOCXXJPGCBFJA-UHFFFAOYSA-N 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 239000007943 implant Substances 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- CFHGBZLNZZVTAY-UHFFFAOYSA-N lawesson's reagent Chemical compound C1=CC(OC)=CC=C1P1(=S)SP(=S)(C=2C=CC(OC)=CC=2)S1 CFHGBZLNZZVTAY-UHFFFAOYSA-N 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- 239000003094 microcapsule Substances 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- OKDQKPLMQBXTNH-UHFFFAOYSA-N n,n-dimethyl-2h-pyridin-1-amine Chemical compound CN(C)N1CC=CC=C1 OKDQKPLMQBXTNH-UHFFFAOYSA-N 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 229920005862 polyol Polymers 0.000 description 2
- 150000003077 polyols Chemical class 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- DTGKSKDOIYIVQL-WEDXCCLWSA-N (+)-borneol Chemical group C1C[C@@]2(C)[C@@H](O)C[C@@H]1C2(C)C DTGKSKDOIYIVQL-WEDXCCLWSA-N 0.000 description 1
- ATVFTGTXIUDKIZ-YFKPBYRVSA-N (2r)-2-[(2-methylpropan-2-yl)oxycarbonylamino]-3-sulfanylpropanoic acid Chemical compound CC(C)(C)OC(=O)N[C@@H](CS)C(O)=O ATVFTGTXIUDKIZ-YFKPBYRVSA-N 0.000 description 1
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 1
- 125000006545 (C1-C9) alkyl group Chemical group 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- QVCUKHQDEZNNOC-UHFFFAOYSA-N 1,2-diazabicyclo[2.2.2]octane Chemical compound C1CC2CCN1NC2 QVCUKHQDEZNNOC-UHFFFAOYSA-N 0.000 description 1
- 125000004815 1,2-dimethylethylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([*:2])C([H])([H])[H] 0.000 description 1
- QPPOMEOQNLTFRU-UHFFFAOYSA-N 1,4-thiazepine Chemical compound S1C=CC=NC=C1 QPPOMEOQNLTFRU-UHFFFAOYSA-N 0.000 description 1
- 125000004806 1-methylethylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- 125000000530 1-propynyl group Chemical group [H]C([H])([H])C#C* 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- PJBFHGHMMOGYNF-UHFFFAOYSA-N 2-amino-3-ethoxy-3-oxopropanoic acid;hydrochloride Chemical compound Cl.CCOC(=O)C(N)C(O)=O PJBFHGHMMOGYNF-UHFFFAOYSA-N 0.000 description 1
- VKPPFDPXZWFDFA-UHFFFAOYSA-N 2-chloroethanamine Chemical compound NCCCl VKPPFDPXZWFDFA-UHFFFAOYSA-N 0.000 description 1
- QQLSWNGYIUYOQV-UHFFFAOYSA-N 2-iodoethylcyclohexane Chemical compound ICCC1CCCCC1 QQLSWNGYIUYOQV-UHFFFAOYSA-N 0.000 description 1
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- MTJGVAJYTOXFJH-UHFFFAOYSA-N 3-aminonaphthalene-1,5-disulfonic acid Chemical compound C1=CC=C(S(O)(=O)=O)C2=CC(N)=CC(S(O)(=O)=O)=C21 MTJGVAJYTOXFJH-UHFFFAOYSA-N 0.000 description 1
- CDPQZLVBRUMMRQ-UHFFFAOYSA-N 3-cyclohexylbutanoic acid Chemical compound OC(=O)CC(C)C1CCCCC1 CDPQZLVBRUMMRQ-UHFFFAOYSA-N 0.000 description 1
- KIRFVQALDHTBPN-UHFFFAOYSA-N 3-cyclohexylpropanehydrazide Chemical compound NNC(=O)CCC1CCCCC1 KIRFVQALDHTBPN-UHFFFAOYSA-N 0.000 description 1
- 125000005917 3-methylpentyl group Chemical group 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- CMSJFUFHVIAXHE-UHFFFAOYSA-N 4-cyclohexylbutanehydrazide Chemical compound NNC(=O)CCCC1CCCCC1 CMSJFUFHVIAXHE-UHFFFAOYSA-N 0.000 description 1
- 229940090248 4-hydroxybenzoic acid Drugs 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 206010065687 Bone loss Diseases 0.000 description 1
- DZGPFLZCNYMBFC-UHFFFAOYSA-N C(C)OC(=O)N1SC=CC=CC1 Chemical compound C(C)OC(=O)N1SC=CC=CC1 DZGPFLZCNYMBFC-UHFFFAOYSA-N 0.000 description 1
- 0 CCCc1nnc2[n]1CCSC[C@@]2** Chemical compound CCCc1nnc2[n]1CCSC[C@@]2** 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- CCPHAMSKHBDMDS-UHFFFAOYSA-N Chetoseminudin B Natural products C=1NC2=CC=CC=C2C=1CC1(SC)NC(=O)C(CO)(SC)N(C)C1=O CCPHAMSKHBDMDS-UHFFFAOYSA-N 0.000 description 1
- 208000017667 Chronic Disease Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 208000018035 Dental disease Diseases 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 208000037147 Hypercalcaemia Diseases 0.000 description 1
- 201000002980 Hyperparathyroidism Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- BOWUOGIPSRVRSJ-YFKPBYRVSA-N L-2-aminohexano-6-lactam Chemical compound N[C@H]1CCCCNC1=O BOWUOGIPSRVRSJ-YFKPBYRVSA-N 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 108010058398 Macrophage Colony-Stimulating Factor Receptor Proteins 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 206010027452 Metastases to bone Diseases 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- LSDPWZHWYPCBBB-UHFFFAOYSA-N Methanethiol Chemical compound SC LSDPWZHWYPCBBB-UHFFFAOYSA-N 0.000 description 1
- 101100268066 Mus musculus Zap70 gene Proteins 0.000 description 1
- 102000004108 Neurotransmitter Receptors Human genes 0.000 description 1
- 108090000590 Neurotransmitter Receptors Proteins 0.000 description 1
- 238000010934 O-alkylation reaction Methods 0.000 description 1
- 208000010191 Osteitis Deformans Diseases 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 208000027868 Paget disease Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- ZHZZWIQPCAMTIM-UHFFFAOYSA-N [C]1=CC=CC2=CC=CC=C12 Chemical compound [C]1=CC=CC2=CC=CC=C12 ZHZZWIQPCAMTIM-UHFFFAOYSA-N 0.000 description 1
- FPQVGDGSRVMNMR-JCTPKUEWSA-N [[(z)-(1-cyano-2-ethoxy-2-oxoethylidene)amino]oxy-(dimethylamino)methylidene]-dimethylazanium;tetrafluoroborate Chemical compound F[B-](F)(F)F.CCOC(=O)C(\C#N)=N/OC(N(C)C)=[N+](C)C FPQVGDGSRVMNMR-JCTPKUEWSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 125000003670 adamantan-2-yl group Chemical group [H]C1([H])C(C2([H])[H])([H])C([H])([H])C3([H])C([*])([H])C1([H])C([H])([H])C2([H])C3([H])[H] 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 238000012382 advanced drug delivery Methods 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 125000004450 alkenylene group Chemical group 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 238000004873 anchoring Methods 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 238000005349 anion exchange Methods 0.000 description 1
- 125000005428 anthryl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C3C(*)=C([H])C([H])=C([H])C3=C([H])C2=C1[H] 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000000010 aprotic solvent Substances 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 229940053197 benzodiazepine derivative antiepileptics Drugs 0.000 description 1
- 125000003310 benzodiazepinyl group Chemical class N1N=C(C=CC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 125000006367 bivalent amino carbonyl group Chemical group [H]N([*:1])C([*:2])=O 0.000 description 1
- 210000001772 blood platelet Anatomy 0.000 description 1
- 208000019664 bone resorption disease Diseases 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 238000005341 cation exchange Methods 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 125000000068 chlorophenyl group Chemical group 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- HJZLEGIHUQOJBA-UHFFFAOYSA-N cyclohexane propionic acid Chemical compound OC(=O)CCC1CCCCC1 HJZLEGIHUQOJBA-UHFFFAOYSA-N 0.000 description 1
- 125000006547 cyclononyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- 239000012973 diazabicyclooctane Substances 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000003628 erosive effect Effects 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 239000007941 film coated tablet Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- HHLFWLYXYJOTON-UHFFFAOYSA-N glyoxylic acid Chemical compound OC(=O)C=O HHLFWLYXYJOTON-UHFFFAOYSA-N 0.000 description 1
- 239000003163 gonadal steroid hormone Substances 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000003394 haemopoietic effect Effects 0.000 description 1
- 210000000777 hematopoietic system Anatomy 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 108091008044 human SRC Proteins 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 230000000148 hypercalcaemia Effects 0.000 description 1
- 208000030915 hypercalcemia disease Diseases 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 208000027202 mammary Paget disease Diseases 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 210000000110 microvilli Anatomy 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- DOTMOQHOJINYBL-UHFFFAOYSA-N molecular nitrogen;molecular oxygen Chemical compound N#N.O=O DOTMOQHOJINYBL-UHFFFAOYSA-N 0.000 description 1
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 1
- FEKRFYZGYUTGRY-UHFFFAOYSA-N n'-ethylmethanediimine Chemical compound CCN=C=N FEKRFYZGYUTGRY-UHFFFAOYSA-N 0.000 description 1
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 210000004898 n-terminal fragment Anatomy 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 230000030991 negative regulation of bone resorption Effects 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 230000011164 ossification Effects 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- RQKYHDHLEMEVDR-UHFFFAOYSA-N oxo-bis(phenylmethoxy)phosphanium Chemical compound C=1C=CC=CC=1CO[P+](=O)OCC1=CC=CC=C1 RQKYHDHLEMEVDR-UHFFFAOYSA-N 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 238000010647 peptide synthesis reaction Methods 0.000 description 1
- 201000001245 periodontitis Diseases 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 125000001844 prenyl group Chemical group [H]C([*])([H])C([H])=C(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 239000002516 radical scavenger Substances 0.000 description 1
- 239000012429 reaction media Substances 0.000 description 1
- 230000022983 regulation of cell cycle Effects 0.000 description 1
- 230000000979 retarding effect Effects 0.000 description 1
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 238000010517 secondary reaction Methods 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 229940037128 systemic glucocorticoids Drugs 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- LFKDJXLFVYVEFG-UHFFFAOYSA-N tert-butyl carbamate Chemical compound CC(C)(C)OC(N)=O LFKDJXLFVYVEFG-UHFFFAOYSA-N 0.000 description 1
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- JKCJXAVMBWFTTC-UHFFFAOYSA-N thiazepine 1-oxide Chemical compound O=S1C=CC=CC=N1 JKCJXAVMBWFTTC-UHFFFAOYSA-N 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- IMNIMPAHZVJRPE-UHFFFAOYSA-N triethylenediamine Chemical compound C1CN2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 description 1
- 150000008648 triflates Chemical class 0.000 description 1
- AALUTIYNYXEFNT-UHFFFAOYSA-N trimethylsilane hydroiodide Chemical compound C[SiH](C)C.I AALUTIYNYXEFNT-UHFFFAOYSA-N 0.000 description 1
- CSRZQMIRAZTJOY-UHFFFAOYSA-N trimethylsilyl iodide Substances C[Si](C)(C)I CSRZQMIRAZTJOY-UHFFFAOYSA-N 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 229940005605 valeric acid Drugs 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 125000004933 β-carbolinyl group Chemical group C1(=NC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D281/00—Heterocyclic compounds containing rings of more than six members having one nitrogen atom and one sulfur atom as the only ring hetero atoms
- C07D281/02—Seven-membered rings
- C07D281/04—Seven-membered rings having the hetero atoms in positions 1 and 4
- C07D281/06—Seven-membered rings having the hetero atoms in positions 1 and 4 not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
L'invention a pour objet un dérivé bicyclique de thiazépine ou de caprolactame de formule (I) (CF DESSIN DANS BOPI) dans laquelle [A0 ], [A1 ], A2 , V, Y, W et R1 ont les significations indiquées dans la description, leurs sels et leurs promédicaments. L'invention de plus a pour objet les procédés de préparation des composés de formule (I), les intermédiaires de ce procédé, leur application à titre de médicaments, notamment comme inhibiteurs du domaine Src SH2 et inhibiteurs de la résorption osseuse médiée par les ostéoclastes, et les compositions pharmaceutiques les renfermant.
Description
<Desc/Clms Page number 1>
La présente invention a pour objet de nouveaux dérivés bicycliques de thiazépine ou de caprolactame, leur procédé de préparation et les intermédiaires de ce procédé, leur application comme médicaments et les compositions pharmaceutiques les contenant.
L'invention a pour objet un dérivé bicyclique de thiazépine ou de caprolactame de formule (I)

dans laquelle Het, [Ao] , [A1], A2, V, Y, W et R1 ont les significations indiquées plus bas, leurs sels physiologiquement acceptables et leurs promédicaments. Les composés de formule (I) sont des composés ayant une activité pharmacologique et sont donc utilisables à titre de médicaments. Ce sont notamment des antagonistes du domaine Src SH2 et ils inhibent la résorption osseuse médiée par les ostéoclastes. Ils sont donc utiles pour le traitement thérapeutique ou prophylactique de maladies qui sont causées au moins en partie par une augmentation non désirée de la résorption osseuse, par exemple l'ostéoporose. L'invention de plus a pour objet les procédés de préparation des composés de formule (I), les intermédiaires de ce procédé, leur application, en particulier à titre de médicament et les compositions pharmaceutiques les renfermant.
dans laquelle Het, [Ao] , [A1], A2, V, Y, W et R1 ont les significations indiquées plus bas, leurs sels physiologiquement acceptables et leurs promédicaments. Les composés de formule (I) sont des composés ayant une activité pharmacologique et sont donc utilisables à titre de médicaments. Ce sont notamment des antagonistes du domaine Src SH2 et ils inhibent la résorption osseuse médiée par les ostéoclastes. Ils sont donc utiles pour le traitement thérapeutique ou prophylactique de maladies qui sont causées au moins en partie par une augmentation non désirée de la résorption osseuse, par exemple l'ostéoporose. L'invention de plus a pour objet les procédés de préparation des composés de formule (I), les intermédiaires de ce procédé, leur application, en particulier à titre de médicament et les compositions pharmaceutiques les renfermant.
Domaine de l'invention a) L'activité catalytique Tyrosine kinase
L'activité catalytique tyrosine kinase est associée soit à des récepteurs possédant des domaines transmembranaires (EDF,DPGF,c-fms..) soit à des protéines intracellulaires (T.
L'activité catalytique tyrosine kinase est associée soit à des récepteurs possédant des domaines transmembranaires (EDF,DPGF,c-fms..) soit à des protéines intracellulaires (T.
<Desc/Clms Page number 2>
Hunter, Biochem Soc. Trans ; 307-327 (1996). Les tyrosines kinases intracellulaires ("non récepteur") sont réparties en 8 sous familles principalement selon la similarité de séquence de leur domaine catalytique et ont été appelées du nom de leur membre le plus connu (Src, Csk, Btk, Abl, Fak, Syk, Jak, Fsp). A l'intérieur de cette classe de tyrosines kynases intracellulaires, la famille Src contient 9 membres partageant entre eux 70% d'homologie de séquence (Src, Fyn, Yes, Frg, Lyn, Hck, Lck, Blk, Yrd chez les vertébrés). b)- Protéines de la famille Src
Les protéines de la famille Src partagent une structure commune qui est caractérisée par les éléments suivants : un petit fragment N-terminal impliqué dans l'ancrage à la membrane (appelé parfois aussi SH4), un domaine unique peu conservé (40-70 résidus), un domaine SH3 qui se lie à des séquences riches en prolines (50 résidus), un domaine SH2 impliqué dans la reconnaissance de séquence peptidique spécifiques phosphorylées (100 résidus), un domaine catalytique (tyrosine kinase, 250 résidus) et enfin un court domaine C-terminal impliqué dans la régulation. c)- Fonction des protéines tyrosines kinases de la famille c-Src
Les protéines tyrosine kinases de la famille c-Src ont des rôles multiples et se chevauchent au niveau de la transduction des signaux impliqués dans toute une série de situations biologiques différentes, notamment, la croissance et la différentiation cellulaire, le contrôle du cycle cellulaire, de la forme et de l'adhésion des cellules ainsi que la régulation des canaux ioniques et des récepteurs neurotransmetteurs.
Les protéines de la famille Src partagent une structure commune qui est caractérisée par les éléments suivants : un petit fragment N-terminal impliqué dans l'ancrage à la membrane (appelé parfois aussi SH4), un domaine unique peu conservé (40-70 résidus), un domaine SH3 qui se lie à des séquences riches en prolines (50 résidus), un domaine SH2 impliqué dans la reconnaissance de séquence peptidique spécifiques phosphorylées (100 résidus), un domaine catalytique (tyrosine kinase, 250 résidus) et enfin un court domaine C-terminal impliqué dans la régulation. c)- Fonction des protéines tyrosines kinases de la famille c-Src
Les protéines tyrosine kinases de la famille c-Src ont des rôles multiples et se chevauchent au niveau de la transduction des signaux impliqués dans toute une série de situations biologiques différentes, notamment, la croissance et la différentiation cellulaire, le contrôle du cycle cellulaire, de la forme et de l'adhésion des cellules ainsi que la régulation des canaux ioniques et des récepteurs neurotransmetteurs.
Tandis que l'expression de la plupart des membres de la famille de Src est limitée au système hématopoïétique, les protéines Src, Fyn et Yes sont exprimées dans un grand nombre de tissus et type cellulaires. Le gène c-Src en particulier
<Desc/Clms Page number 3>
est très largement exprimé dans les neurones, les plaquettes et les ostéoclastes. d) Résorption osseuse et c-Src
Les fonctions biologiques de plusieurs de ces protéines ont finalement commencé à être mieux comprises avec la réalisation des expériences de knock out. Ainsi Soriano et al (Cell 64,693-702 (1991) ) ont récemment généré une lignée de souris transgénique dont le gène de c-Src n'était pas fonctionnel. L'observation du phénotype des souris homozygotes pour cette mutation a fait apparaître plusieurs faits extrêmement intéressants : elles ne présentaient pas d'anormalité patente au niveau des neurones ou des plaquettes, par contre ces souris étaient toutes atteintes d'une résorption osseuse très diminuée qui donne lieu à des anormalités du squelette (S. N. Popoff et al ; 17 (5) 445 (1995)). Il a ensuite été démontré que ces souris possédaient des ostéoclastes en nombre normal, mais que ces derniers ne possédaient pas de bordure en brosse et n'étaient pas capables de résorption normale dans le test du pit (B.F.
Les fonctions biologiques de plusieurs de ces protéines ont finalement commencé à être mieux comprises avec la réalisation des expériences de knock out. Ainsi Soriano et al (Cell 64,693-702 (1991) ) ont récemment généré une lignée de souris transgénique dont le gène de c-Src n'était pas fonctionnel. L'observation du phénotype des souris homozygotes pour cette mutation a fait apparaître plusieurs faits extrêmement intéressants : elles ne présentaient pas d'anormalité patente au niveau des neurones ou des plaquettes, par contre ces souris étaient toutes atteintes d'une résorption osseuse très diminuée qui donne lieu à des anormalités du squelette (S. N. Popoff et al ; 17 (5) 445 (1995)). Il a ensuite été démontré que ces souris possédaient des ostéoclastes en nombre normal, mais que ces derniers ne possédaient pas de bordure en brosse et n'étaient pas capables de résorption normale dans le test du pit (B.F.
Boyce et al. J. Clin. Invest. 90,1622-27 (1992)).
Donc, seuls les ostéoclastes semblent affectés par la mutation. L'hypothèse est la suivante : dans les neurones, plaquettes et autres cellules dans lesquelles Src exerce une fonction, cette fonction pourrait être prise en charge par un membre très proche de la famille (fyn, lyn ou yes) ; revanche, cette substitution fonctionnelle n'aurait pas lieu dans les ostéoclastes. e)- Inhibition de la protéine Src
Dès lors que cette protéine semble essentiellement dans les processus de résorption osseuse et n'apparaît pas vitale pour d'autres fonctions, la demanderesse se propose de trouver de nouveaux composés l'inhibant pour enrayer la résorption osseuse.
Dès lors que cette protéine semble essentiellement dans les processus de résorption osseuse et n'apparaît pas vitale pour d'autres fonctions, la demanderesse se propose de trouver de nouveaux composés l'inhibant pour enrayer la résorption osseuse.
<Desc/Clms Page number 4>
Cette inhibition peut s'effectuer soit au niveau du domaine SH2, soit au niveau du domaine SH3 (impliqué dans la reconnaissance et l'interaction avec d'autres protéines), soit au niveau du domaine catalytique (tyrosine kinase).
Etat de la technique Les demandes de brevets suivantes décrivent des composés inhibant la liaison de protéines contenant un domaine SH2 avec une protéine phosphorylée.
WO 98/40093 décrit des dérivés hétérocycliques bicycliques, W097/12903 décrit des dérivés du phényle, W097/30079 et W096/23813 décrivent des dérivés peptidiques, 95/25118 décrit des dérivés de benzodiazépine, EP 727211 et WO 96/24343 décrivent l'utilisation de certains composés spécifiques du domaine SH2 pour traiter la résorption osseuse ainsi que les maladies autoimmunes.
D'autres investigations ont permis de montrer que les dérivés bicycliques thiazépine ou de caprolactame de formule (I) présentent une forte activité comme antagoniste du domaine humain Src SH2 et de la résorption osseuse médiée via les ostéoclastes.
L'invention a pour objet un composé de formule (I)

dans laquelle, - W est représente -CH2- ou -S- ; - Het représente un aryle hétérocyclique à 5 ou 6 chainons, accolé en position 3 et 4 à la thiazépine et renfermant de 1 à 3 hétéroatomes choisis parmi N, 0 ou S, dans lequel l'atome de soufre peut être substitué par 0,1 ou 2 atomes d'oxygènes,
dans laquelle, - W est représente -CH2- ou -S- ; - Het représente un aryle hétérocyclique à 5 ou 6 chainons, accolé en position 3 et 4 à la thiazépine et renfermant de 1 à 3 hétéroatomes choisis parmi N, 0 ou S, dans lequel l'atome de soufre peut être substitué par 0,1 ou 2 atomes d'oxygènes,
<Desc/Clms Page number 5>
- V, lié à un atome de carbone de l'hétérocycle, représente un groupement R3, OR3 ou SR3, - [Ao] représente un groupement choisi parmi - (C (Ra) (Rb) )n-CO-NRi- ou - (C (Ra) (Rb) ) n-NRi-CO-, n étant égal à 0,1,2 ou 3, - [Ai] représente un groupement choisi parmi

-CH (Z) - (C1-C9) -alkyle- (C5-Clq) -aryle-, -CH (Z) - (C5-Clq) -aryle-, - (C1-C4) -alkyle- (C5-Clq) -aryle-, et - (C5-C14)-aryle-, dans lesquels Z est un atome d'hydrogène, un tétrazole,
NRcR'c, NHCORc, CONHRc, NHC02Rc, NHCONHRc, NHSORc ou
NHS02Rc; les dits radicaux aryles étant substitués ou non substitués par R2 ou par R'2; - A2 représente un groupement choisi parmi -PO(ORd)(ORe), -OPO(ORd)(ORe), -B (ORd) (ORe) , -CRfRgPO(ORd)(ORe), -0-CRfRgPO (ORd) (ORe) , -CRfRg-C02Rd, -0-CRfRg-C02Rd, -N(CRfRg-C02Rd)2, -C02Rd, -CRfRg-CH2-
C02Rd, -CRfRgS03H, -O-CRfRgSO3H, -S03H, -S02NH2, -NHCO-
PO(ORd)(ORe) et -CO-PO(ORd)(ORe); - R3, Ra, Rb, Rc, Rd, Re ou Ri, indépendants les uns des autres, identiques ou différents, représentant un atome d' hydrogène, un radical (C1-C8)-alkyle-, (C3-C18)-
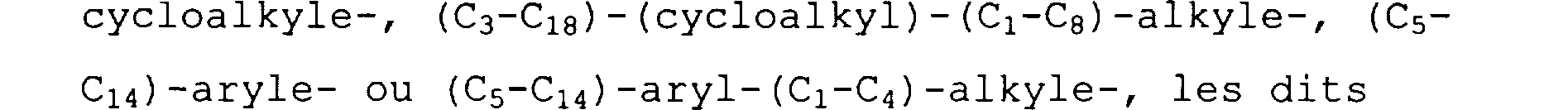
cycloalkyle-, (C3-Cla) - (cycloalkyl) - (C1-Ca) -alkyle-, (C,5C14) -aryle- ou (CS-C14) -aryl- (C1-C4) -alkyle-, les dits radicaux étant substitués ou non substitués par R2; - Rf et Rg identiques ou différents représentant un atome d'hydrogène, un halogène, ORc, NHRc, un radical C02Rd,
CH2CO2Rd, SO3H, PO(ORd)(ORe) ou tétrazole ; - éventuellement Ra et Rb forment ensemble avec l'atome de carbone qui les porte, un groupement cycloalkyle renfermant de 3 à 6 carbones, - Y, lié à un atome de carbone de l'hétérocycle, représente un atome C=O, C=S, C=NH ou S02;
-CH (Z) - (C1-C9) -alkyle- (C5-Clq) -aryle-, -CH (Z) - (C5-Clq) -aryle-, - (C1-C4) -alkyle- (C5-Clq) -aryle-, et - (C5-C14)-aryle-, dans lesquels Z est un atome d'hydrogène, un tétrazole,
NRcR'c, NHCORc, CONHRc, NHC02Rc, NHCONHRc, NHSORc ou
NHS02Rc; les dits radicaux aryles étant substitués ou non substitués par R2 ou par R'2; - A2 représente un groupement choisi parmi -PO(ORd)(ORe), -OPO(ORd)(ORe), -B (ORd) (ORe) , -CRfRgPO(ORd)(ORe), -0-CRfRgPO (ORd) (ORe) , -CRfRg-C02Rd, -0-CRfRg-C02Rd, -N(CRfRg-C02Rd)2, -C02Rd, -CRfRg-CH2-
C02Rd, -CRfRgS03H, -O-CRfRgSO3H, -S03H, -S02NH2, -NHCO-
PO(ORd)(ORe) et -CO-PO(ORd)(ORe); - R3, Ra, Rb, Rc, Rd, Re ou Ri, indépendants les uns des autres, identiques ou différents, représentant un atome d' hydrogène, un radical (C1-C8)-alkyle-, (C3-C18)-
cycloalkyle-, (C3-Cla) - (cycloalkyl) - (C1-Ca) -alkyle-, (C,5C14) -aryle- ou (CS-C14) -aryl- (C1-C4) -alkyle-, les dits radicaux étant substitués ou non substitués par R2; - Rf et Rg identiques ou différents représentant un atome d'hydrogène, un halogène, ORc, NHRc, un radical C02Rd,
CH2CO2Rd, SO3H, PO(ORd)(ORe) ou tétrazole ; - éventuellement Ra et Rb forment ensemble avec l'atome de carbone qui les porte, un groupement cycloalkyle renfermant de 3 à 6 carbones, - Y, lié à un atome de carbone de l'hétérocycle, représente un atome C=O, C=S, C=NH ou S02;
<Desc/Clms Page number 6>
- R1 représente un groupement-OH, -ORc, -NRcR'c, (C1-C4)alkyle- ou halogène;
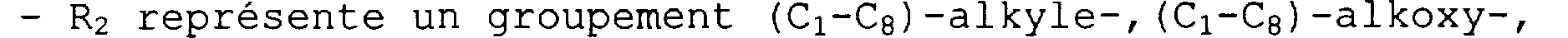
- R2 représente un groupement (C1-C8) -alkyle-, (C1-C8) -alkoxy-, hydroxy- (C1-C8) -alkyle-, (C1-C8) -alkoxy- (C1-C8) -alkyle-, (C1-C3) - alkylènedioxy-, (C5-C14)-aryle-, (C5-C14) -aryloxy-, (C5-C14) - aryl- (C1-C4) -alkyle-, (C5-Cl4) -aryl- (Cl-C4) -alkyloxy-, halogène, trihalogénométhyle-, hydroxyle, nitro, formyle, cyano, carboxy, -CONHz, (C1-C8)-alkyloxycarbonyle-, amino, - NH- ( (C1-C4) -alkyle) , -N ( (C1-C4) alkyle) 2, -NHCO- (C1-C4) -alkyle, -NHCO-(C5-Ci4)-aryle, -CONH-(C1-C4)-alkyle, -CONH- (C5-C14) -

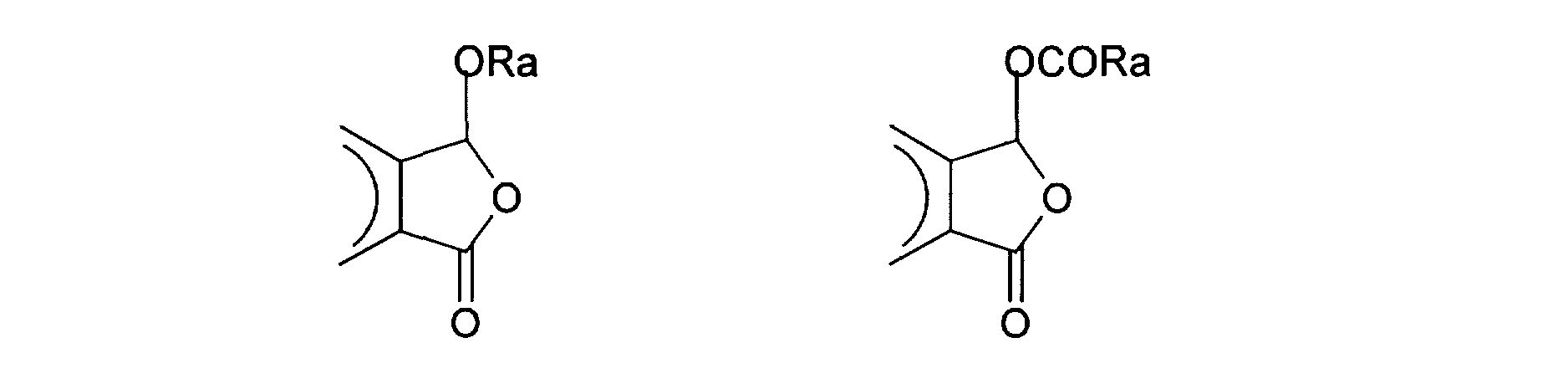
aryle, -CO- (C1-C4) -alkyle ou -CO- (C5-Clq) -aryle; - R'2a les mêmes valeurs que A2 ; - éventuellement A2 et R2 forment ensemble avec l'aryle qui les porte, l'un des cycles à 5 chaînons suivants :
le trait en pointillé indique que le groupement Y-Ri est optionnel, lesdits composés de formule (I) étant sous toutes leurs formes isomères possibles, seuls ou en mélange dans un rapport quelconque, ainsi que leurs sels physiologiquement acceptables et leurs promédicaments (prodrugs).
- R2 représente un groupement (C1-C8) -alkyle-, (C1-C8) -alkoxy-, hydroxy- (C1-C8) -alkyle-, (C1-C8) -alkoxy- (C1-C8) -alkyle-, (C1-C3) - alkylènedioxy-, (C5-C14)-aryle-, (C5-C14) -aryloxy-, (C5-C14) - aryl- (C1-C4) -alkyle-, (C5-Cl4) -aryl- (Cl-C4) -alkyloxy-, halogène, trihalogénométhyle-, hydroxyle, nitro, formyle, cyano, carboxy, -CONHz, (C1-C8)-alkyloxycarbonyle-, amino, - NH- ( (C1-C4) -alkyle) , -N ( (C1-C4) alkyle) 2, -NHCO- (C1-C4) -alkyle, -NHCO-(C5-Ci4)-aryle, -CONH-(C1-C4)-alkyle, -CONH- (C5-C14) -
aryle, -CO- (C1-C4) -alkyle ou -CO- (C5-Clq) -aryle; - R'2a les mêmes valeurs que A2 ; - éventuellement A2 et R2 forment ensemble avec l'aryle qui les porte, l'un des cycles à 5 chaînons suivants :
le trait en pointillé indique que le groupement Y-Ri est optionnel, lesdits composés de formule (I) étant sous toutes leurs formes isomères possibles, seuls ou en mélange dans un rapport quelconque, ainsi que leurs sels physiologiquement acceptables et leurs promédicaments (prodrugs).
Tous les radicaux qui peuvent se retrouver plusieurs fois dans les composés de formule (I), par exemple le radical R2, sont indépendants les uns des autres et peuvent être identiques ou différents.
Les radicaux alkyles peuvent être linéaires ou ramifiés, saturés ou mono- ou poly-insaturés. Ceci s'applique également lorsqu'ils portent un substituant ou lorsqu'ils sont inclus
<Desc/Clms Page number 7>
dans des groupements tels que par exemple alkoxy, alkoxycarbonyle ou aralkyle.
Par (C1-C8)-alkyle on entend notamment les radicaux méthyle, éthyle, propyle, butyle, pentyle, hexyle, heptyle, octyle, les n-isomères de ces radicaux, isopropyle, isobutyle, isopentyle, néopentyle, isohexyle, 3méthylpentyle, 2,3,4-triméthylhexyle, sec-butyle, tertbutyle, tert-pentyle. Parmi les radicaux préférés on peut citer méthyle, éthyle, n-propyle, isopropyle, n-butyle, isobutyle, et tert-butyle.
Les radicaux alkylènes bivalents correspondant aux radicaux monovalents cités plus haut sont par exemple les radicaux méthylène, éthylène, 1,3-propylène, 1,2-propylène (=l-méthyléthylène), 2,3-butylène (=1,2-diméthyléthylène), 1,4-butylène, ou 1,6-hexylène.
Les radicaux alkyles insaturés sont par exemple les radicaux alkényles tels que vinyle, 1-propényle, allyle, butényle, 3-méthyl-2-butényle ou les radicaux alkynyles tels que éthynyle, 1-propynyle ou propargyle.
Par radicaux alkylène bivalents insaturés on entend les radicaux alkénylène et alkynylène qui peuvent également être linéaires ou ramifiés. Il s'agit par exemple des radicaux vinylène, propénylène, éthynylène ou propynylène.
Les radicaux cycloalkyles peuvent être monocycliques, bicycliques ou tricycliques. Il s'agit par exemple des radicaux cyclopropyle, cyclobutyle, cyclopentyle, cyclohexyle, cycloheptyle, cyclooctyle, cyclononyle, cyclodécyle, cycloundécyle, cyclododécyle, cyclotétradécyle ou cyclooctadécyle qui le cas échéant peuvent être substitués par exemple par un alkyle renfermant de 1 à 4 atomes de carbone. Comme radicaux cycloalkyles substitués, on peut citer le 4 méthylcyclohexyle et le 2,3-diméthylcyclohexyle.
Les radicaux bicycloalkyles et tricycloalkyles peuvent être non substitués ou substitués en toute position, par
<Desc/Clms Page number 8>
exemple par un ou plusieurs groupes oxo et/ou 1 ou plusieurs groupes alkyles identiques ou différents tels que méthyle ou isopropyle et de préférence méthyle. La liaison de jonction du radical bi ou tricyclique peut se situer en toutes positions de la molécule. La liaison peut se situer au niveau de l'atome de carbone ponté ou d'un des autres atomes de carbone. Cette liaison peut présenter également toutes positions du point de vue de la stéréochimie, par exemple exo ou endo. Comme exemple de radicaux bicycloalkyles ou tricycloalkyles, on peut citer le camphanyle, le bornyle, l'adamantyle tel que le 1-adamantyle ou le 2-adamantyle, le caranyle, l'épiisobornyle, l'épibornyle, le norbornyle ou le norpinanyle.
Par halogène on entend fluor, chlore, brome ou iode.
Lorsque Rf et Rg représentent un halogène, il s'agit de préférence de fluor.
Par le terme (C5-C14) -aryle on entend - soit les radicaux (C5-C14) -aryle hétérocycliques (= (C5-C1)hétéroaryle), dans lesquels les atomes de carbone du cycle sont remplacés par un hétéroatome tel que azote, oxygène ou soufre, - soit les radicaux (C6-C14)-aryle carbocyclique.
Parmi les radicaux(C6-C14)-aryle carbocycliques, on peut citer le phényle, le naphtyle,le biphénylyle, l'anthryle ou le fluorényle et tout particulièrement le 1-naphtyle, le 2naphtyle et le phényle. Il s'agit de préférence du phényle.
Sauf indication contraire, les radicaux aryles, en particulier phényle, peuvent être non substitués ou substitués par un ou plusieurs radicaux identiques ou différents choisis parmi (C1-C8)-alkyle, en particulier

(Ci-C4) alkyle, (C1-C8) -alkoxy, formyle, halogène tel que fluor, chlore et brome, nitro, amino, trifluorométhyle, hydroxyle, -CH20H, méthylènedioxy, cyano, hydroxycarbonyle,
(Ci-C4) alkyle, (C1-C8) -alkoxy, formyle, halogène tel que fluor, chlore et brome, nitro, amino, trifluorométhyle, hydroxyle, -CH20H, méthylènedioxy, cyano, hydroxycarbonyle,
<Desc/Clms Page number 9>
aminocarbonyle, (C1-C4)-alkoxycarbonyle, phényle, phénoxy, benzyle et benzyloxy. En général, au plus 2 groupes nitro peuvent être utilisés dans les composés de formule (I) selon l'invention.
Dans le cas du phényle monosubstitué, le substituant peut être situé en position 2,3 ou 4, et de préférence en position 3 ou 4. Dans le cas où le phényle est disubstitué, les substituants peuvent être en position 2,3 ou 2,4 ou 2,5 ou 2,6 ou 3,4 ou 3,5. De préférence, dans les phényles disubstitués, les deux substituants sont en position 3,4.
Lorsque ce phényle est trisubstitué les positions sont les suivantes : 2,3,4 ou 2,3,5 ou 2,3,6 ou 2,4,5 ou 2,4,6 ou 3,4,5. De la même manière, les radicaux naphtyles ou d'autres radicaux aryles peuvent être substitués en toute position, par exemple le radical 1-naphtyle en position 2-, 3-, 4-, 5-, 6-, 7-, et 8 et le radical 2-naphtyle en position 1-, 3-, 4-, 5-, 6-, et 7.
Le groupement (CS-C14)-aryle peut représenter également un système aromatique monocyclique ou polycyclique dans lequel 1,2,3,4 ou 5 atomes de carbone du cycle sont remplacés par des hétéroatomes, en particulier identiques ou différents du groupe constitué de l'azote, l'oxygène et le soufre. Parmi les groupements (C5-C14)-aryle hétérocycliques (= (CS-C14) - hétéroaryle) on peut citer les groupements 2-pyridyle, 3pyridyle, 4-pyridyle, pyrrolyle, furyle, thiényle, imidazolyle, pyrazolyle, oxazolyle, isoxazolyle, thiazolyle, isothiazolyle, tétrazolyle, pyridyle, pyrazinyle, pyrimidinyle, indolyle, isoindolyle, indazolyle, phtalazinyle, quinolyle, isoquinolyle, quinoxalinyle, quinazolinyle, cinnolinyle, P-carbolinyle, ou encore des dérivés benzocondensés, cyclopenta-, cyclohexa-, ou cyclohepta- condensés de ces radicaux. Le système hétérocyclique peut être substitué par les mêmes substituants cités plus haut pour le système carbocyclique.
<Desc/Clms Page number 10>
Parmi les radicaux hétéroaryles, sont préférés les systèmes aromatiques monocycliques ou bicycliques ayant 1,2 ou 3 hétéroatomes, en particulier 1 ou 2 hétéroatomes, choisis parmi N, 0 ou S, et qui sont non substitués ou substitués par les groupes tels que (C1-C6)-alkyle, (Ci-Ce)alkoxy, fluor, chlore, nitro, amino, trifluorométhyle, hydroxyle, formyle, (C1-C4)-alkoxycarbonyle, phényle, phénoxy, benzyloxy, et benzyle.
Tout particulièrement, on peut citer les systèmes aromatiques monocycliques ou bicyclique de 5 à 10 chaînons renfermant de 1 à 3 hétéroatomes, en particulier 1 ou 2 hétéroatomes, choisis parmi N, 0 et S et qui peuvent être non substitués ou substitués par 1 ou 2 substituants tels que

(C1-C9)-alkyle, (C1-C9)-alkoxy, formyle, phényle, phénoxy, benzyle et benzyloxy.
(C1-C9)-alkyle, (C1-C9)-alkoxy, formyle, phényle, phénoxy, benzyle et benzyloxy.
Les atomes de carbone optiquement actifs contenus dans les composés de formule (I) peuvent indépendamment les uns des autres présenter la configuration R ou la configuration S.
Les composés de formule (I) peuvent être sous la forme d'énantiomères purs ou de diastéréoisomères purs ou sous la forme d'un mélange d'énantiomères, par exemple sous forme de racémates ou de mélanges de diastéréoisomères.
La présente invention a donc pour objet les énantiomères purs, les mélanges de ces énantiomères, les diastéréoisomères purs et les mélanges de ces diastéréoisomères.
L'invention comprend les mélanges de deux ou plus de deux stéréoisomères de formule (I) et tous les rapports de ces stéréoisomères au sein des dits mélanges.
Les composés de formule (I) peuvent le cas échéant être présent sous forme d'isomères E ou d'isomères Z. L'invention a donc pour objet les isomères E purs, les isomères Z purs et les mélanges E/Z selon un rapport quelconque.
<Desc/Clms Page number 11>
Enfin l'invention comprend les différents régioisomères liés à la position ortho, para ou méta de A2. De préférence A2 est en position para et R2 est en position méta.
Les diastéréoisomères, incluant les isomères E/Z peuvent être séparés en isomères individuels, par exemple par chromatographie. Les racémates peuvent être séparés en deux énantiomères par des méthodes courantes telles que la chromatographie en phase chirale ou par des méthodes de résolution.
Les sels physiologiquement acceptables des composés de formule (I) sont en particulier des sels pharmaceutiquement utilisables ou non toxiques ou physiologiquement utilisables.
Lorsque les composés de formule (I) renferment un groupe acide tel que l'acide carboxylique, il s'agit par exemple des sels de métaux alcalins ou alcalino terreux tels que les sels de sodium, de potassium, de magnésium, de calcium, et également les sels formés avec des ions ammonium quaternaires physiologiquement acceptables tel que l'ammoniac et des amines organiques physiologiquement acceptables telles que par exemple la triéthylamine, l'éthanolamine ou le tris-(2- hydroxyéthyle)amine.
Lorsque les composés de formule (I) contiennent un groupe basique, ils peuvent former un sel d'addition avec les acides par exemple avec les acides inorganiques tels que l'acide chlorhydrique, sulfurique, phosphorique ou avec les acides organiques carboxyliques tels que l'acide acétique, trifluoroacétique, citrique, benzoique, maléique, fumarique, tartrique, méthanesulfonique ou para toluène sulfonique.
Les composés de formule (I) qui comportent un groupe basique et un groupe acide peuvent être présents sous forme de Zwiterions (bétaînes), qui sont également inclus dans la présente invention.
Les sels des composés de formule (I) peuvent être obtenus par les méthodes ordinaires connues de l'homme du
<Desc/Clms Page number 12>
métier, par exemple en combinant un composé de formule (I) avec un acide organique ou inorganique ou une base dans un solvant ou un dispersant ou à partir d'un autre sel par échange de cation ou d'anion.
L'invention inclut également tous les sels des composés de formule (I) qui, à cause de leur faible acceptabilité physiologique, ne sont pas directement utilisable comme médicament, mais sont utilisables comme produits intermédiaires pour mettre en oeuvre des modifications chimiques ultérieures au niveau des composés de formule (I) ou comme produits de départ pour la préparation de sels physiologiquement acceptables.
La présente invention inclut également tous les solvates des composés de formule (I) par exemples les hydrates, les solvates formés avec les alcools, et tous les dérivés des composés de formule (I), par exemple les esters, prodrugs et autres dérivés physiologiquement acceptables, ainsi que les métabolites des composés de formule (I).
L'invention a plus particulièrement pour objet les prodrugs des composés de formule (I) qui peuvent être transformées en composés de formule (I) in vivo dans les conditions physiologiques. Les prodrugs des composés de formule (I), à savoir les dérivés chimiquement modifiés des composés de formule (I) afin d'obtenir des propriétés améliorées de manière désirée, sont connus de l'homme du métier.
Pour avoir plus d'information sur le type de prodrug envisagé dans la présente invention, on peut citer les ouvrages suivants : Fleicher et al., Advanced Drug Delivery Review 19 (1996) 115-130 ; of prodrugs, H. Bundgaard, Ed., Elsevier, 1985 ; H. Bungaard, Drugs of the Future 16 (1991) 443 ; et al. Bioorg. Med. Chem. Lett. 4 (1994) 1985 ; Safadi et al. Pharmaceutical Res. 10 (1993) 1350.
<Desc/Clms Page number 13>
Parmi les prodrugs appropriées des composés de formule (I) on peut citer de préférence : - les prodrugs sous forme d'esters du groupement phosphate ou phosphonate, - les prodrugs sous forme d'esters des groupes carboxyliques, en particulier du groupe COOH, - les prodrugs sous forme d'acyle et de carbamate pour les groupes contenant un azote acylable tel que les groupes amino.
Dans les prodrugs acylées ou sous forme de carbamate, une ou plusieurs fois, par exemple deux fois, un atome d'hydrogène situé sur l'atome d'azote est remplacé par un groupe acyle ou carbamate. Parmi les groupes acyles ou carbamates préférés, on peut citer les groupes RioCO-, R11OCO-, dans lesquels R10 est un hydrogène ou un radical (C1-C18)-alkyle, (C3-C18)-

cycloalkyle, (C3-Cl8) -cycloalkyl- (Cl-C8) -alkyle, (C5-C14) -aryle, dans lequel 1 à 5 atomes de carbone peuvent être remplacés par des hétéroatomes tels que N,O,S ou (C5-C14)-aryl-(C1- C8)alkyle, dans lequel 1 à 5 atomes de carbone dans la partie aryle peuvent être remplacés par des hétéroatomes tels que N,O,S et (R11 à les mêmes valeurs que R10 à l'exception d'hydrogène.
cycloalkyle, (C3-Cl8) -cycloalkyl- (Cl-C8) -alkyle, (C5-C14) -aryle, dans lequel 1 à 5 atomes de carbone peuvent être remplacés par des hétéroatomes tels que N,O,S ou (C5-C14)-aryl-(C1- C8)alkyle, dans lequel 1 à 5 atomes de carbone dans la partie aryle peuvent être remplacés par des hétéroatomes tels que N,O,S et (R11 à les mêmes valeurs que R10 à l'exception d'hydrogène.
Parmi les modes de réalisation préférés, l'invention a plus particulièrement pour objet les composés de formule (I) dans laquelle W représente un atome de soufre.
Parmi les mode de réalisation préférés, l'invention a plus particulièrement pour objet les composés de formule (I) dans laquelle Het représente un hétérocycle aromatique à 5 ou 6 chainons renfermant 2 ou 3 atomes d'azote et -[Ao]- représente le groupement-NHCO-.
Tout particulièrement, l'invention a pour objet un dérivé de thiazépine accolé en position 3 et 4 à un triazole de formule (la)
<Desc/Clms Page number 14>

dans laquelle V, A1 et A2 sont tels que définis précédemment.
Tout particulièrement, l'invention a pour objet un dérivé de thiazépine accolé en position 3 et 4 à un imidazole de formule (Ib)

dans laquelle V, Y, Ri, A1 et A2 sont tels que définis précédemment .
dans laquelle V, Y, Ri, A1 et A2 sont tels que définis précédemment .
L'invention a également plus particulièrement pour objet les composés de formules (I), (la) ou (Ib) dans lesquelles -A2 représente de préférence -0-P03H2, -CF2-PO3H2 ou -CH(C02H)2 et est notamment en position para.
L'invention a également plus particulièrement pour objet les composés de formules (I), (la) ou (Ib) dans lesquelles -

[api]- représente -CH (Z) - (C1-C4) -alkyl- (CS-C14) -aryle- ou (C5- C14)-aryle- le dit aryle étant éventuellement substitué par un groupement R2.ou R'2.
[api]- représente -CH (Z) - (C1-C4) -alkyl- (CS-C14) -aryle- ou (C5- C14)-aryle- le dit aryle étant éventuellement substitué par un groupement R2.ou R'2.
L'invention a également plus particulièrement pour objet les composés de formules (I), (la) ou (Ib) dans lesquelles Z représente de préférence NHCORc, NHCO2Rc, dans lesquels Rc représente un radical alkyle renfermant de 1 à 6 atomes de carbone, le carbone portant le groupement Z présentant la configuration S.
<Desc/Clms Page number 15>
L'invention a également plus particulièrement pour objet les composés de formules (I), (la) ou (Ib) dans lesquelles V représente de préférence OR3 ou R3 dans lesquels R3 représente un groupement (C1-C6)-alkyle-, (C3-C12) -cycloalkyle-, (C3-C12) -

cycloalkyl- (C1-C6) -alkyle- ou aryl-(CI-C6)-alkyle-
L'invention a également plus particulièrement pour objet les composés de formules (I) ou (Ib) dans lesquelles Y est un groupement C=0 et R1 est OH, NH2 ou un groupement alkyloxy renfermant de 1 à 6 atomes de carbone.
cycloalkyl- (C1-C6) -alkyle- ou aryl-(CI-C6)-alkyle-
L'invention a également plus particulièrement pour objet les composés de formules (I) ou (Ib) dans lesquelles Y est un groupement C=0 et R1 est OH, NH2 ou un groupement alkyloxy renfermant de 1 à 6 atomes de carbone.
Les composés préférés de formule (I), (la) ou (Ib) sont les composés dans lesquels un ou plusieurs radicaux ont les significations préférées citées plus haut.
L'invention a tout particulièrement pour objet les composés de formules (la) telle que définie précédemment dans laquelle

-[Ai]- représente -CH (Z) - (C1-C9) -alkyl-phényle- ou -phényle- ledit phényle étant éventuellement substitué en méta par un groupement R2 ou R'2 - A2 représente -O-PO3H2, -CF2-PO3H2 ou -CH(C02H)2 et est en position para, - Z représente NHCORc, NHCO2Rc, dans lesquels Rc représente un radical alkyle renfermant de 1 à 6 atomes de carbone, le carbone portant le groupement Z présentant la configuration S; - V représente un radical R3 représentant un groupement (C3-

C12) -cycloalkyl- (Cl-C6) -alkyle-, (Cl-C6)-alkyle ou aryl- (C1-
C6) -alkyle-, lesdits composés de formule (I) étant sous toutes leurs formes isomères possibles, seuls ou en mélange dans un rapport quelconque, ainsi que leurs sels physiologiquement acceptables et leurs promédicaments (prodrugs).
-[Ai]- représente -CH (Z) - (C1-C9) -alkyl-phényle- ou -phényle- ledit phényle étant éventuellement substitué en méta par un groupement R2 ou R'2 - A2 représente -O-PO3H2, -CF2-PO3H2 ou -CH(C02H)2 et est en position para, - Z représente NHCORc, NHCO2Rc, dans lesquels Rc représente un radical alkyle renfermant de 1 à 6 atomes de carbone, le carbone portant le groupement Z présentant la configuration S; - V représente un radical R3 représentant un groupement (C3-
C12) -cycloalkyl- (Cl-C6) -alkyle-, (Cl-C6)-alkyle ou aryl- (C1-
C6) -alkyle-, lesdits composés de formule (I) étant sous toutes leurs formes isomères possibles, seuls ou en mélange dans un rapport quelconque, ainsi que leurs sels physiologiquement acceptables et leurs promédicaments (prodrugs).
L'invention a tout particulièrement pour objet les composés de formules (Ib) telle que définie précédemment dans laquelle
<Desc/Clms Page number 16>
-[A,]- représente -CH(Z)-(Cl-C4)-alkyl-phényle- ou -phényle-, ledit phényle étant éventuellement substitué en méta par un groupement R2 ou R'2 - A2 représente -0-P03H2, -CF2-P03H2 ou -CH(CO2H)2et est en position para, - Z représente NHCORc, NHC02Rc, dans lesquels Rc représente un radical alkyle renfermant de 1 à 6 atomes de carbone, le carbone portant le groupement Z présentant la configuration S ; - V représente un radical OR3 dans lequel le radical R3 représentant un groupement (C3-C12)-cycloalkyl-(C1-C6)- alkyle-, (Ci-C6) -alkyle ou aryl-(C1-C6)-alkyle-, - Y est un groupement C=O, et - R1 est un groupement hydroxy, NH2, ou un groupement alkyloxy renfermant de 1 à 6 atomes de carbone, lesdits composés de formule (I) étant sous toutes leurs formes isomères possibles, seuls ou en mélange dans un rapport quelconque, ainsi que leurs sels physiologiquement acceptables et leurs promédicaments (prodrugs).
L'invention a également pour objet les composés de formule (I), (la ) ou (Ib) dont les noms suivent : . Acide [[4-((2S)-2-(acétylamino)-3-[[(9S)-3-[(3-
cyclohexyl)propyl]-5,6,8,9-tétrahydro-1,2,4-triazolo-[4,3- d][1,4]thiazépin-9-yl]amino]-3oxopropyl]phényl]difluorométhyl]-phosphonique; . Acide [[4-((2S)-2-(acétylamino)-3-[[(9S)-3-butyl-5,6,8,9-

tétrahydro-1,2,4-triazolo-[4,3-d][1,4]thiazépin-9-yl]amino]- 3-oxopropyl]phényl]difluorométhyl]-phosphonique; . Acide [[4-((2S)-2-(acétylamino)-3-[[(9S)-3-[(2- cyclohexyl)éthyl]-5,6,8,9-tétrahydro-1,2,4-triazolo-[4,3- d][1,4]thiazépin-9-yl]amino]-3oxopropyl]phényl]difluorométhyl]-phosphonique; . Acide [[4-((2S)-2-(acétylamino)-3-[[(9S)-3-[(3-(4- chlorophényl)propyl]-5,6,8,9-tétrahydro-1,2,4-triazolo-[4,3-
cyclohexyl)propyl]-5,6,8,9-tétrahydro-1,2,4-triazolo-[4,3- d][1,4]thiazépin-9-yl]amino]-3oxopropyl]phényl]difluorométhyl]-phosphonique; . Acide [[4-((2S)-2-(acétylamino)-3-[[(9S)-3-butyl-5,6,8,9-
tétrahydro-1,2,4-triazolo-[4,3-d][1,4]thiazépin-9-yl]amino]- 3-oxopropyl]phényl]difluorométhyl]-phosphonique; . Acide [[4-((2S)-2-(acétylamino)-3-[[(9S)-3-[(2- cyclohexyl)éthyl]-5,6,8,9-tétrahydro-1,2,4-triazolo-[4,3- d][1,4]thiazépin-9-yl]amino]-3oxopropyl]phényl]difluorométhyl]-phosphonique; . Acide [[4-((2S)-2-(acétylamino)-3-[[(9S)-3-[(3-(4- chlorophényl)propyl]-5,6,8,9-tétrahydro-1,2,4-triazolo-[4,3-
<Desc/Clms Page number 17>
d][1,4]thiazépin-9-yl]amino]-3oxopropyl]phényl]difluorométhyl]-phosphonique; . N2-acétyl-N-[(3-[(4-chlorophényl)propyl]-5,6,8,9- tétrahydro-1,2,4-triazolo-[4,3-d][1,4]thiazépin-9-yl]-4- (phosphonooxy)-L-Phénylalaninamide; . N-[(9S)-3-(2-cyclohexyléthyl)-5,6,8,9-tétrahydro-1,2,4-

triazolo[4,3-d][1,4]thiazépin-9-yl)-4-phosphonooxy)- Benzenamide ; . 9-(9S)-[[(2S)-2(acétylamino)-3-[4- (phosphonodifluorométhyl)phényl]-1-oxopropyl)amino]-3-(2- cyclohexyléthoxy)-5,6,8,9-tétrahydro-Imidazo[1,2d][1,4]thiazépine-2-carboxamide; . 9-(9S)-[[(2S)-2(acétylamino)-3-[4-(phosphonooxy)phényl]-1- oxopropyl)amino]-3-(2-cyclohexyléthoxy)-5,6,8,9-tétrahydroImidazo[1,2-d][1,4]thiazépine-2-carboxylate d'éthyle ; ainsi que leurs sels physiologiquement acceptables et leurs promédicaments (prodrugs).
triazolo[4,3-d][1,4]thiazépin-9-yl)-4-phosphonooxy)- Benzenamide ; . 9-(9S)-[[(2S)-2(acétylamino)-3-[4- (phosphonodifluorométhyl)phényl]-1-oxopropyl)amino]-3-(2- cyclohexyléthoxy)-5,6,8,9-tétrahydro-Imidazo[1,2d][1,4]thiazépine-2-carboxamide; . 9-(9S)-[[(2S)-2(acétylamino)-3-[4-(phosphonooxy)phényl]-1- oxopropyl)amino]-3-(2-cyclohexyléthoxy)-5,6,8,9-tétrahydroImidazo[1,2-d][1,4]thiazépine-2-carboxylate d'éthyle ; ainsi que leurs sels physiologiquement acceptables et leurs promédicaments (prodrugs).
La présente invention a également pour objet un procédé de préparation des composés de formule (I). Les composés peuvent généralement être préparés, par exemple au cours d'une synthèse convergente par couplage de deux ou plusieurs fragments qui peuvent être dérivées par rétrosynthèse des composés de formule (I). Afin d'éviter que les groupes fonctionnels puissent mener à des réactions indésirables ou secondaires au cours de chaque étape de synthèse, il peut être avantageux ou nécessaire au cours de la synthèse des composés de formule (I), d'introduire les groupes fonctionnels sous forme de précurseurs qui sont par la suite convertis en groupes fonctionnels désirés ou de bloquer temporairement ces groupes fonctionnels en mettant en oeuvre une stratégie de groupe protecteur appropriée à la synthèse qui est connue par l'homme de l'art (Greene, Wuts protective Group in Organic Synthesis, Wiley 1991)
<Desc/Clms Page number 18>
Ainsi les composés de formule (I) peuvent être préparés, par exemple, en couplant une amine de formule (II)

dans laquelle Het, Y, V, W et Ri sont tels que définis plus haut pour la formule (I) et où, le cas échéant, les groupes fonctionnels sont sous forme de précurseurs ou sous forme protégée, avec un acide ou un dérivé d'acide de formule (III)
X-C (=O)-[A1]-A'2 dans laquelle [Ai] est tel que défini plus haut dans la formule (I) et A'2 représentant, outre les valeurs de A2, le radical hydroxy, X est un groupe substituable par un nucléophile et où, le cas échéant, les groupes fonctionnels sont sous forme de précurseurs ou sous forme protégée, lesdits groupes fonctionnels éventuellement présents sous forme de précurseur ou sous forme protégée, étant par la suite convertis en groupes présents dans les composés de formule (I) .
dans laquelle Het, Y, V, W et Ri sont tels que définis plus haut pour la formule (I) et où, le cas échéant, les groupes fonctionnels sont sous forme de précurseurs ou sous forme protégée, avec un acide ou un dérivé d'acide de formule (III)
X-C (=O)-[A1]-A'2 dans laquelle [Ai] est tel que défini plus haut dans la formule (I) et A'2 représentant, outre les valeurs de A2, le radical hydroxy, X est un groupe substituable par un nucléophile et où, le cas échéant, les groupes fonctionnels sont sous forme de précurseurs ou sous forme protégée, lesdits groupes fonctionnels éventuellement présents sous forme de précurseur ou sous forme protégée, étant par la suite convertis en groupes présents dans les composés de formule (I) .
Certains composés de formule (I) peuvent si désiré ou si nécessaire, subir une ou plusieurs des réactions suivantes dans un ordre approprié : - action d'un agent de déprotection partielle, - action d'un agent de déprotection totale, - action de H-P(O)(ORd)(ORe) lorsque A'2 représente OH, - salification.
<Desc/Clms Page number 19>
Le groupement COX dans la formule (III) est de préférence le groupe acide carboxylique ou un dérivé activé de l'acide carboxylique. X, par exemple est hydroxyle ou halogène, en particulier, chlore ou brome, alkoxy, de préférence méthoxy ou éthoxy, aryloxy, par exemple phénoxy, pentafluorophényloxy, phénylthio, méthylthio, 2-pyridylthio ou un hétérocycle azoté lié en particulier via un azote tel que par exemple 1-imidazolyle. X peut être également par exemple (C1-C4)-alkyle-O-CO-O- ou tolylsulfonyloxy et le dérivé d'acide activé peut être un anhydride mixte.
Si X est un hydroxyle, donc si l'amine de formule (II) réagit avec un acide carboxylique de formule (III), alors l'acide carboxylique est d'abord activé. L'activation peut être effectuée par exemple avec le dicyclohexylcarbodiimide (DCCI) ou avec le 0-((cyano(éthoxycarbonyl)-métylène)amino)- 1,1,3,3-tétraméthyluronium tétrafluoroborate (TOTU; Kônig et al, Proc. 21st Europ. Peptide Symp. 1990 (Eds Giralt, Andreu), Escom, Leiden 1991, p.243) ou d'autres agents activants courants en synthèse peptidique.
Outre les amines libres de formule (II), les sels d'amines peuvent également être mis en oeuvre dans la réaction avec les composés de formule (III), les amines libres étant formés in-situ ou par une étape séparée au moyen d'une base.
La réaction d'un dérivé activé d'acide carboxylique de formule (III) avec l'amine (ou dérivé) de formule (II) est de préférence effectuée d'une manière connue en soi dans un solvant organique protique ou aprotique, mais inerte. Dans ce cas on utilise des solvants tels que le méthanol, l'isopropanol, le tert-butanol, le diméthylformamide ou le tétrahydrofurane à des températures allant de 0 C à la température de reflux de ces solvants, notamment lors de la réaction des esters méthyliques ou éthyliques (X est un méthoxy ou un éthoxy) avec les amines.
<Desc/Clms Page number 20>
Les réactions des acides de formule (III) (COX = COzH) avec les amines libres sont avantageusement mises en oeuvre dans un solvant aprotique inerte tel que le diméthylformamide, le tétrahydrofurane, le diméthoxyéthane, ou le dioxane, le cas échéant en additionnant une base telle que par exemple le tert-butoxide de potassium, le méthoxide de sodium ou une base organique telle que la N-méthylmorpholine. Cependant, l'eau peut également être utilisée comme solvant dans les réactions des composés de formule (II) avec les amines de formule (III), par exemple en utilisant une base telle que l'hydroxyde de sodium.
Si X est le chlore, la réaction sera de préférence menée par addition d'un piégeur d'acide, par exemple d'une base ou d'un excès d'amine (ou dérivé). Le mélange réactionnel est ensuite traité et si désiré le produit réactionnel est purifié selon les méthodes connues de l'homme du métier.
La réaction d'acylation mettant en oeuvre le composé de formule (III) [A'2]-[A1]-COOH, s'effectue de préférence en présence d'EDC (chlorhydrate de 1-(3-diméthylaminopropyl)-3- éthyl-carbodiimide) et de HOBt (1-hydroxybenzotriazole hydrate) dans un solvant dipolaire aprotique tel que le diméthylformamide.
Les groupes protecteurs éventuellement présents dans les composés obtenus à partir des composés de formule (II) et (III) sont ensuite éliminés par des procédés classiques ; par exemple, les groupes tertbutyl ester sont convertis en acide carboxylique par traitement avec de l'acide trifluoroacétique, les groupes benzyles sont éliminés par hydrogénation ou encore les groupes fluorénylméthoxycarbonyle sont éliminés en présence d'amine secondaire.
Lorsque l'amine est protégée (NHC02Rb), elle l'est notamment par un groupement Boc. Le retour à l'amine libre peut s'effectuer par action d'acide trifluoroacétique.
Lorsque cette amine est protégée par CO2CH2Ph, la
<Desc/Clms Page number 21>
déprotection s'effectue de préférence en présence Pd(OH)2, MgO dans le cyclohexadiene au reflux.

Pour obtenir une monodébenzylation([A2]=OP(0)(OH)(OBn)), on met en oeuvre en particulier l'iodure de sodium et la réaction s'effectue au reflux de l'acétone, ou le DABCO (1,4diazabicyclo[2.2.2]octane) dans le toluène.
Pour obtenir une monodébenzylation([A2]=OP(0)(OH)(OBn)), on met en oeuvre en particulier l'iodure de sodium et la réaction s'effectue au reflux de l'acétone, ou le DABCO (1,4diazabicyclo[2.2.2]octane) dans le toluène.
Pour obtenir une déprotection totale ([A2]=OP(O) (OH)2), on effectue de préférence une hydrogénation classique en présence de Pd/C à 10% ou on utilisera l'acide trifluoroacétique dans le dichlorométhane.
Lorsque [A'2] représente OH, la réaction de phosphorylation s'effectue avec HP(0)(ORd)(ORe) en présence de N'N- disopropyléthylamine (DIPEA) et de N,Ndiméthylaminopyridine (DMAP) dans l'acétonitrile en présence de tétrachlorure de carbone.
Si nécessaire, la conversion en des sels physiologiquement acceptables s'effectue par des procédés connus de l'homme du métier.
L'invention a plus particulièrement pour objet un procédé tel que décrit plus haut dans lequel le composé de formule (III) est choisi parmi N-Boc-Tyr-0-(P03Bn2), N-Ac-Tyr- 0-(P03Bn2), N-Boc-Tyr-CF2-(P03Et2) et N-Ac-Tyr-CF2-(PO3Et2).
L'invention a également pour objet un procédé de préparation de composés de formule (II) caractérisé en ce que l'on soumet un composé de formule (lia)

P étant un groupement protecteur de l'amine, à l'action d'un agent de formation de dérivés thiocétones correspondants tel que le P2S5, afin d'obtenir le composé de formule (IIb)
P étant un groupement protecteur de l'amine, à l'action d'un agent de formation de dérivés thiocétones correspondants tel que le P2S5, afin d'obtenir le composé de formule (IIb)
<Desc/Clms Page number 22>

que l'on soumet à l'action d'un iodure ou chlorure d'alkyle renfermant de 1 à 4 atomes de carbone en milieu basique, afin d'obtenir le composé de formule (IIc)

que l'on cyclise par -soit par action d'un dérivé de formule (IVa)
V-CO-NH-NH2, afin d'obtenir le dérivé bicyclique de formule (IIda)dans lequel l'hétérocycle accolé à la thiazépine est un triazole
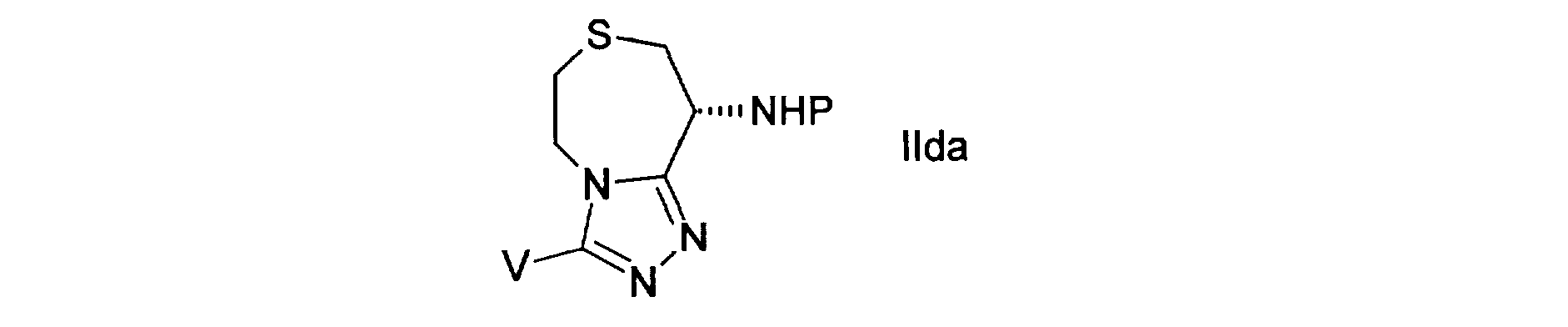
-soit par action d'un dérivé de formule (IVb) H2N-CH (C02Et) 2 (Y-Rl) puis, le cas échéant, transformation d'un groupement C02Et en YR1 désiré, afin d'obtenir le dérivé bicyclique de formule (IIdb)dans lequel l'hétérocycle accolé à la thiazépine est un imidazole
<Desc/Clms Page number 23>
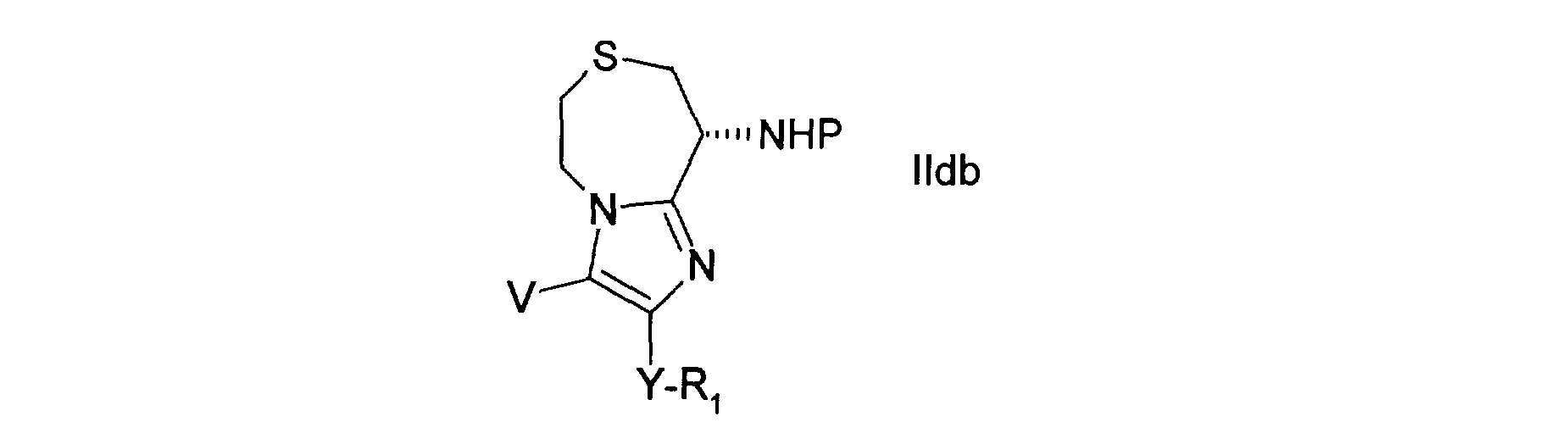
V représentant un groupement hydroxyle qui est ensuite alkylé, composés de formules (IIda) ou (IIdb) que l'on déprotège, notamment par hydrogénation catalytique, afin d'obtenir l'amine correspondante de formule (IIea) ou (IIeb).

L'invention a tout particulièrement pour objet le procédé de préparation, tel que défini plus haut, des composés de formule (la), telle que défini précédemment, caractérisé en ce que le produit de départ est un composé de formule (Ilea)
L'invention a tout particulièrement pour objet le procédé de préparation, tel que défini plus haut, des composés de formule (Ib), telle que défini précédemment, caractérisé en ce que le produit de départ est un composé de formule (IIeb)
La réaction de conversion de l'amide en thioamide s'effectue notamment en présence du réactif de Lawesson dans le toluène.
L'invention a tout particulièrement pour objet le procédé de préparation, tel que défini plus haut, des composés de formule (Ib), telle que défini précédemment, caractérisé en ce que le produit de départ est un composé de formule (IIeb)
La réaction de conversion de l'amide en thioamide s'effectue notamment en présence du réactif de Lawesson dans le toluène.
L'alkylation du thioamide s'effectue en particulier par action du iodure de méthyle en présence d'une base forte telle que l'hydrure de sodium.
<Desc/Clms Page number 24>
La réaction de cyclisation mettant en #uvre un composé de formule (IVa) ou (IVb) s'effectue de préférence dans un solvant polaire tel que l'éthanol.
L'invention a donc pour objet les procédés décrits plus haut et les nouveaux composés intermédiaires de formules (II), (IIb) (Ile), (IIda), (IIdb), (IIea) et (IIeb).
Les composés de départ de formule (III) qui sont ensuite liés avec les composés de formule (II) pour donner les composés de formule (I) sont commerciaux, peuvent être préparés selon des procédés décrits dans la littérature ou encore sont accessibles par analogie. A titre d'exemple on utilise de préférence, les produits de formule (III) suivants: - le 0-[bis(phénylméthoxy)phosphinyl]-N-[(1,1- diméthyléthoxy)carbonyl]-L-tyrosine : (N-Boc-Tyr-0-(P03Bn2), origine : Penissula Laboratories, Inc - Le N-acétyl-0-[bis-(diphénylméthoxy)phosphinyl]-L-tyrosine : N-Ac-Tyr-O-(PO3Bn2) - Le N-[(1,1-diméthyléthoxy)carbonyl]-4-[bis- éthoxyphosphinyl)difluorométhyl]-L-phénylalanine : N-BocTyr-CF2- (P03Et2) - Le N-acétyl-4-[bis-éthoxyphosphinyl)difluorométhyl]-L- phénylalanine : N-Ac-Tyr-CF2- (P03Et2)
La préparation des composés de formule (II) est illustrée dans les exemples décrits plus bas, étant entendu que la présente invention n'est pas restreinte à ces synthèses ou ces produits de départ. Il n'y a pas de difficulté majeure pour l'homme du métier de prévoir des modifications des synthèses décrites dans notre demande pour la préparation d'autres composés de formule (II) ou (III) selon l'invention.
La préparation des composés de formule (II) est illustrée dans les exemples décrits plus bas, étant entendu que la présente invention n'est pas restreinte à ces synthèses ou ces produits de départ. Il n'y a pas de difficulté majeure pour l'homme du métier de prévoir des modifications des synthèses décrites dans notre demande pour la préparation d'autres composés de formule (II) ou (III) selon l'invention.
Les composés de formule (lia) (série thiazépine) sont préparés à partir de la N-Boc-cystéine selon la méthode décrite dans FEBS Lett. (1984) 174(1), 76.
<Desc/Clms Page number 25>
Les composés de formule (II) avec Z représentant un groupement-CH2- (série caprolactame), sont préparés, par analogie, de façon équivalente au composé de formule (II) en série thiazépinone. Le composé de départ sera alors le L-alpha-amino-epsilon-caprolactame (Aldrich).
Les composés de formule (I) peuvent être utilisés pour inhiber la liaison d'une protéine Src à une protéine phosphorylée analogue.
Cette inhibition peut s'effectuer soit au niveau du domaine SH2, soit au niveau du domaine SH3 (impliqué dans la reconnaissance et l'interaction avec d'autres protéines), soit au niveau du domaine catalytique (tyrosine kinase).
Les composés selon l'invention ont tout particulièrement une affinité vis-à-vis du domaine SH2 de la protéine Src.
Les composés de formule (I) peuvent donc être utilisés pour inhiber la liaison d'une protéine Src contenant le domaine SH2, à une protéine phosphorylée analogue, la méthode consistant en l'administration au patient dont le traitement requiert l'inhibition du domaine SH2, une quantité inhibitrice du composé selon l'invention.
L'action des composés de formule (I) peut être démontrée par exemple dans un test dans lequel l'inhibition de la liaison du ligand EPQpYEEIPIYL radiomarqué à la protéine SH2 est déterminée par Scintillation proximity assay (SPA). Des précisions sur ce test sont données plus bas. Comme antagonistes du domaine Src SH2, les composés de formule (I) et leurs sels physiologiquement acceptables et leurs prodrugs conviennent en général pour le traitement ou la prévention de maladies liées aux interactions entre domaine SH2 et leur ligands ou qui peuvent être influencés par l'inhibition des interactions de ce type, pour soulager ou guérir lorsqu'une inhibition des interactions de ce type est désirée. Comme on l'a expliqué au début, une telle interaction joue un rôle important dans la résorption osseuse.
<Desc/Clms Page number 26>
Les composés de formule (I) sont tout particulièrement des antagonistes du récepteur humain Src SH2 et sont capables ainsi par exemple d'inhiber l'adhésion des ostéoclastes sur la surface de l'os et ainsi la résorption osseuse par les ostéoclastes.
Les maladies de l'os dont le traitement ou la prévention nécessitent l'emploi des composés de formule (I) ou de leurs prodrugs, sont notamment l'ostéoporose, l'hypercalcémie, l'ostéopénie, par exemple causée par les métastases osseuses, les désordres dentaires par exemple les parodontites, l'hyperparathyroïdisme, les érosions périarticulaires dans l'arthrite rhumatoïde, la maladie de Paget, l'ostéopénie induite par l'immobilisation. En outre les composés de formule (I) peuvent être utilisés pour soulager, empêcher ou traiter les désordres de l'os qui sont causés par les traitements, par les glucocorticoides, les thérapies liées à la prise de stéroides ou de corticostéroïdes ou par les déficiences d'hormones sexuelles mâles ou femelles.
Tous ces désordres sont caractérisés par une perte osseuse, qui est basée par un défaut d'équilibre entre la formation osseuse et la destruction osseuse et qui peut être influencé favorablement par l'inhibition de la résorption osseuse par les ostéoclastes.
Les composés selon l'invention, par leur affinité avec le domaine Src-SH2, peuvent également être utilisés dans d'autres applications thérapeutiques. Par exemple on sait que les plaquettes et les neurones sont des tissus qui expriment Src-SH2 également. En outre plusieurs protéines de cette famille étant majoritairement exprimées dans le système hémétopoiétique, de nombreuses applications dans le traitement de l'immunité, de l'infection, de l'allergie et des maladies autoimmunes sont envisageables.
Enfin, les composés de formule (I) peuvent également être utilisés pour inhiber la liaison d'une protéine
<Desc/Clms Page number 27>
contenant le domaine SH2, autre que Src, à une protéine phosphorylée analogue, la méthode consistant en l'administration au patient dont le traitement requiert l'inhibition du domaine SH2, une quantité inhibitrice du composé selon l'invention.
Les protéines contenant le domaine SH2 autres que Src sont choisies parmi Fyn, Lck, Yes, Blk, Lyn, Fgr, Hck, Yrk,Abl et Zap 70.
Les composés selon l'invention peuvent ainsi être utilisés dans le traitement de maladies telles que les maladies prolifératives, le cancer, la resténose, l'inflammation ; les allergies ou les maladies cardiovasculaires.
Parmi les médicaments de l'invention, on peut citer particulièrement les composés décrits dans la partie expérimentale.
Parmi ces produits, l'invention a plus particulièrement pour objet, à titre de médicaments, les composés de formule (I) listés précédemment.
La présente invention a donc pour objet un composé de formule (I), et/ou ses sels physiologiquement acceptables et/ou ses prodrugs, à titre de médicament.
La présente invention plus particulièrement pour objet un composé de formule (I), et/ou ses sels physiologiquement acceptables et/ou ses prodrugs, à titre de médicament ayant une activité inhibitrice du récepteur Src SH2.
La présente invention plus particulièrement pour objet un composé de formule (I), et/ou ses sels physiologiquement acceptables et/ou ses prodrugs, à titre de médicament ayant une activité inhibitrice de la résorption osseuse ou pour le traitement ou la prévention de l'ostéoporose.
La présente invention plus particulièrement pour objet un composé de formule (I), et/ou ses sels physiologiquement acceptables et/ou ses prodrugs, à titre de médicament pour le
<Desc/Clms Page number 28>
traitement ou la prévention de l'immunité, de l'infection, de l'allergie, et des maladies autoimmunes.
La présente invention plus particulièrement pour objet un composé de formule (I), et/ou ses sels physiologiquement acceptables et/ou ses prodrugs, à titre de médicament pour le traitement ou la prévention de maladies telles que les maladies prolifératives, le cancer, la resténose, l'inflammation, les allergies ou les maladies cardiovasculaires.
La présente invention a également pour objet l'utilisation des composés de formule (I) et/ou leurs sels physiologiquement acceptables et/ou leurs prodrugs pour la préparation de médicaments destinés à la prévention ou au traitement de l'ostéoporose.
La présente invention a également pour objet l'utilisation des composés de formule (I) et/ou leurs sels physiologiquement acceptables et/ou leurs prodrugs pour la préparation de médicaments destinés au traitement ou à la prévention de l'immunité, de l'infection, de l'allergie, et des maladies autoimmunes.
La présente invention a également pour objet l'utilisation des composés de formule (I) et/ou leurs sels physiologiquement acceptables et/ou leurs prodrugs pour la préparation de médicaments destinés au traitement ou à la prévention de maladies telles que les maladies prolifératives, le cancer, la resténose, l'inflammation, les allergies ou les maladies cardiovasculaires.
Les composés de formule (I) ainsi que leurs sels physiologiquement acceptables et leurs prodrugs peuvent être administrés aux animaux, de préférence aux mammifères et en particulier aux êtres humains comme médicaments à titre thérapeutique ou prophylactique.
Ils peuvent être administrés tels quels ou en mélange avec un ou plusieurs autres composés de formule (I) ou encore
<Desc/Clms Page number 29>
sous la forme d'une préparation pharmaceutique (composition pharmaceutique) qui permet une administration entérale ou parentérale et qui renferme à titre de composé actif une dose efficace d'au moins un composé de formule (I) et/ou ses sels physiologiquement acceptables et/ou ses prodrugs ainsi qu'un ou plusieurs excipients pharmaceutiquements acceptables et éventuellement un ou plusieurs additifs.
Les médicaments peuvent être administrés oralement, par exemple sous forme de pilule, de comprimés, de comprimés enrobés, de pelliculés, de granules, de gélules et capsules molles, de solutions, de sirops, d'émulsion, de suspension ou de mélange d'aérosol.
L'administration peut cependant être effectuée par voie rectale, par exemple sous forme de suppositoire ou par voie parentérale, par exemple sous forme de solutions injectables ou d'infusions, de microcapsules ou d'implants, ou par voie percutanée, par exemple sous la forme de pommade, de solutions, de pigments ou de colorants, par voie transdermique (patches) ou par d'autres voies telles que sous la forme d'aérosol ou de spray nasal.
Les compositions pharmaceutiques selon l'invention sont préparées selon des méthodes connues en soi, des supports organiques ou inorganiques, pharmaceutiquement inertes étant additionnés aux composés de formule (I) et/ou leurs sels physiologiquement acceptables et/ou leurs prodrugs.
Pour la production de pilule, de comprimés, de comprimés enrobés et de capsule en gélatine dure, il est possible d'utiliser par exemple, du lactose, de l'amidon de maïs ou ses dérivés, du talc, de l'acide stéarique ou ses sels, etc.
Les supports convenables pour des capsules en gélatine molle ou pour les suppositoires sont par exemple les graisses, les cires les polyols semi-solides ou liquides, les huiles natu relies ou modifiées etc. Les véhicules appropriés pour la préparation de solutions, par exemple les solutions injecta-
<Desc/Clms Page number 30>
bles, les émulsions ou les sirops sont par exemple l'eau, les alcools, le glycérol, les polyols, le sucrose, les sucres invertis, le glucose, les huiles végétales, etc. Les supports convenables pour les microcapsules ou les implants sont par exemple les copolymères d'acide glyoxilique et d'acide lactique. Les préparations pharmaceutiques contiennent normalement de 0,5% à 90% en poids de composés de formule (I) et/ou leurs sels physiologiquement acceptables.
En plus des principes actifs et des excipients, les préparations pharmaceutiques peuvent contenir des additifs tels que par exemple des diluants, des désintégrants, des liants, des lubrifiants, des agents mouillant, des stabilisants, des émulsifiants, des préservateurs, des agents sucrant, des colorants des agents de flaveurs ou des aromatisants, des épaississants, des agents tampons, et aussi des solvants ou des solubilisants ou des agents pour obtenir un effet retard et également des sels pour modifier la pression osmotique, des agents d'enrobage ou des antioxydants.
Elles peuvent également contenir deux ou plusieurs composés de formule (I) et/ou leurs sels physiologiquement acceptables et/ou leurs prodrugs. En outre, en plus d'au moins un ou plusieurs composés de formule (I) et/ou leurs sels physiologiquement acceptables et/ou leurs prodrugs, ils peuvent contenir au moins un ou plusieurs autres principes actifs utilisables à titre thérapeutique ou prophylactique.
La présente invention a donc pour objet les compositions pharmaceutiques qui permettent une administration entérale ou parentérale et qui, renferment à titre de composé actif une dose efficace d'au moins un composé de formule (I) et/ou ses sels physiologiquement acceptables et/ou ses prodrugs ainsi qu'un ou plusieurs excipients pharmaceutiquement acceptables, et éventuellement un ou plusieurs additifs usuels.
Lorsqu'on utilise les composés de formule (I), les doses peuvent varier à l'intérieur de limites larges et doivent
<Desc/Clms Page number 31>
être fixées en fonction de la personne à traiter. Ceci dépend par exemple du composé employé ou de la nature et de la sévérité de la maladie à traiter et si on se trouve dans des conditions graves ou chronique ou si on met en oeuvre un traitement prophylactique.
Les préparations pharmaceutiques (compositions pharmaceutiques) renferment normalement de 0,2 à 500 mg, et de préférence de 1 à 200 mg de composé de formule (I) et/ou leurs sels physiologiquement acceptables et/ou leurs prodrugs.
Dans le cas d'une administration par voie orale, la dose quotidienne varie en général de 0,01 à 100 mg/kg et de préférence de 0,1 à 50 mg/kg, en particulier de 0,1 à 5 mg/kg. Par exemple pour un adulte de 75kg on pourra envisager une dose quotidienne variant de 0,3 à 0,5 mg/kg.
Dans le cas d'une administration par voie intraveineuse, la dose quotidienne varie approximativement de 0,01 à 100 mg/kg et de préférence de 0,05 à 10 mg/kg.
La dose quotidienne peut être divisée, en particulier dans le cas de l'administration de grande quantité de principe actif, en plusieurs, par exemple 2,3 ou 4 parts. Le cas échéant, en fonction du comportement individuel, il peut être nécessaire d'administrer les différentes doses de manière croissante ou décroissante. Mise à part l'utilisation des composés de formule (I) comme médicaments, on peut également envisager leur utilisation comme véhicule ou support de composés actifs afin de transporter ces composés actifs de manière spécifique vers un site d'action (Drug targeting, voir Targeted Drug Delivery, R. C. Juliano, Handbook of Experimental Pharmacology, Vol 100, Ed. Born, G. V.R. et al, Springer Verlag). Les composés actifs qui peuvent être transportés sont en particulier ceux utilisés pour le traitement ou la prévention des maladies citées plus haut.
<Desc/Clms Page number 32>
Les composés de formule (I) et leurs sels peuvent également être employés comme agent de diagnostique, par exemple pour des méthodes in vitro ou comme auxiliaire dans des études biochimiques dans lesquelles on désire inhiber le domaine SRC SH2 humain. Ils peuvent en outre être utilisés comme intermédiaires pour la préparation d'autres composés, en particulier d'autres agents actifs, qui sont accessibles à partir des composés de formule (I), par exemple par modification ou introduction de radicaux ou de groupes fonctionnels.
Exemples
Les produits ont été identifiés par spectre de masse (SM), infrarouge (IR), spectre RMN et/ou leur Rf.
Les produits ont été identifiés par spectre de masse (SM), infrarouge (IR), spectre RMN et/ou leur Rf.
Les composés qui ont été purifiés par chromatographie en utilisant un éluant qui contient par exemple de l'acide acétique ou trifuoroacétique et qui ensuite sont séchés ou dans lesquels lors de la dernière étape de synthèse, par exemple de l'acide trifluoroacétique a été utilisé pour éliminer un groupe protecteur tert-butyle, contiennent parfois, en fonction de la manière dont le produit a été séché, l'acide provenant de l'éluant ou de la dernière étape de synthèse et donc se trouvent partiellement ou complètement sous la fortme du sel de l'acide utilisé, par exemple sous la forme d'un sel d'acide acétique ou trifluoroacétique. Ils peuvent également être plus ou moins hydratés.
Abrévations/noms chimiques éventuellement employés : AcOEt : acétate d'éthyle ; EDC : chlorhydrate de 1-(3diméthylaminopropyl)-3-éthyl-carbodiimide ; DMF : diméthylformamide ; HOBt : 1-hydroxybenzotriazole hydrate MeOH : méthanol ; TEA : triéthylamine ; TFA : acide trifluoroacétique ; THF : tétrahydrofurane ; MCPBA :acide méta-chloroperoxybenzoique ; DBU : 1,8diazabicyclo[5.4.0]undec-7-ene ; DPPA : diphénylphosphorylazide ; DMSO : diméthylsulfoxyde ; Pd/C
<Desc/Clms Page number 33>
Palladium sur charbon ; Boc : terbutoxycarbonyl ; CBz : benzyloxycarbonyl ; DCC 1,3-dicyclohexylcarbodiimide ; TMSBr : bromotriméthylsilane ; TMSI : iodure de triméthylsilane.
IR : Infrarouge ; RMN : Résonnance Magnétique Nucléaire ; SM : Spectre de Masse ; ep. : Épaulement ; F : fort ; s : singulet ; d : doublet ; t :triplet ; quad : quadruplet ; quint : quintuplet ; 1 : large ; m : multiplet ou massif; J : constante de couplage ; Rf : facteur de rétention (chromatographie).
Préparation P1 (9S)-3-(3-cyclohexylpropyl)-5,6,8,9-tétrahydro-1,2,4triazolo[4,3-d][1,4]thiazépine-9-amine Stade a : (hexahydro-5-thioxo-1,4-thiazépin-6-yl)-carbamate de (1,1diméthyléthyle)
On porte au reflux pendant 1 heure le mélange constitué de 2,86 g de (hexahydro-5-oxo-1,4-thiazépin-6-yl)-carbamate de (l,l-diméthyléthyle)(voir la préparation plus bas) dans 50 ml de toluène et 2,4 g de réactif de Lawesson puis laisse revenir à température ambiante, filtre et lave les cristaux obtenus. On obtient 2,82 g de produit attendu.
On porte au reflux pendant 1 heure le mélange constitué de 2,86 g de (hexahydro-5-oxo-1,4-thiazépin-6-yl)-carbamate de (l,l-diméthyléthyle)(voir la préparation plus bas) dans 50 ml de toluène et 2,4 g de réactif de Lawesson puis laisse revenir à température ambiante, filtre et lave les cristaux obtenus. On obtient 2,82 g de produit attendu.
IR (CHC13) 3383 cm-1=C-NH ; 1703 cm-1C=O ; 1747 cm-1 (ep.) ; 1516 cm-1 (Max) ; 1483 cm-1 Amide II, système conjugué ; 1369 cm-1Me du tertbutyle.
Stade b : (5-méthylthio-2,3,6,7-tétrahydro-1,4-thiazépin-6-yl)carbamate de (l,l-diméthyléthyle)
On ajoute, en 30 minutes à 0 C, 0,57 g d'hydrure de sodium à (50% huile)à 2,8 g du produit préparé au stade précédent dans 50 ml de tétrahydrofuranne, agite 15 minutes à cette température puis ajoute goutte à goutte 1,45 ml de iodure de méthyle. On agite 30 minutes à température ambiante puis verse dans de l'eau, extrait par 3 fois 50 ml d'acétate d'éthyle, sèche la phase organique et évapore sous pression
On ajoute, en 30 minutes à 0 C, 0,57 g d'hydrure de sodium à (50% huile)à 2,8 g du produit préparé au stade précédent dans 50 ml de tétrahydrofuranne, agite 15 minutes à cette température puis ajoute goutte à goutte 1,45 ml de iodure de méthyle. On agite 30 minutes à température ambiante puis verse dans de l'eau, extrait par 3 fois 50 ml d'acétate d'éthyle, sèche la phase organique et évapore sous pression
<Desc/Clms Page number 34>
réduite. L'extrait sec est recristallisé dans de l'éther éthylique. On obtient 1 g de produit attendu.
IR (CHC13) -NH : 3440 cm-1 ; C=O : 1714 cm-1 ; système conjugué + amide II : 1629, 1621, 1603 et 1486 cm-1 ; SM 277+ = MH+ ; 221+ = MH+ - tBu ; 173+ = 221+ - [CH3SH] Stade c : [(9S)-3-(3-cyclohexylpropyl)-5,6,8,9-tétrahydro-1,2,4- triazolo[4,3-d][1,4]thiazépin-9-yl]-carbamate de (1,1- diméthyléthyle)
On porte au reflux pendant 20 h le mélange constitué de 0,1 g du produit préparé au stade précédent dans 5 ml d'éthanol et 0,4 g d'hydrazide de l'acide 4-cyclohexyl- butanoïque sous la forme d'un sel d'acide trifluoroacétique, puis évapore sous pression réduite jusqu'à obtention d'un extrait sec que l'on purifie par chromatographie en éluant avec le mélange dichlorométhane/méthanol 95/5. On obtient 0,09g de produit cyclisé attendu Rf (CH2Cl2/MeOH 95/5) = 0,3 Préparation du (hexahydro-5-oxo-1,4-thiazépin-6-yl)-carbamate de (1,1-diméthyléthyle)
A une solution de 20 g de cystéine dans 250 ml de méthanol, on ajoute 71,2 ml de TEA puis 14,85 g de chloroéthylamine en 30 minutes. Le milieu réactionnel est agité 2 heures à température ambiante puis une nuit à reflux.
On porte au reflux pendant 20 h le mélange constitué de 0,1 g du produit préparé au stade précédent dans 5 ml d'éthanol et 0,4 g d'hydrazide de l'acide 4-cyclohexyl- butanoïque sous la forme d'un sel d'acide trifluoroacétique, puis évapore sous pression réduite jusqu'à obtention d'un extrait sec que l'on purifie par chromatographie en éluant avec le mélange dichlorométhane/méthanol 95/5. On obtient 0,09g de produit cyclisé attendu Rf (CH2Cl2/MeOH 95/5) = 0,3 Préparation du (hexahydro-5-oxo-1,4-thiazépin-6-yl)-carbamate de (1,1-diméthyléthyle)
A une solution de 20 g de cystéine dans 250 ml de méthanol, on ajoute 71,2 ml de TEA puis 14,85 g de chloroéthylamine en 30 minutes. Le milieu réactionnel est agité 2 heures à température ambiante puis une nuit à reflux.
Après évaporation, on coule dans l'eau, filtre, extrait à l'acétate d'éthyle, sèche sur MgS04, évapore puis recristallise dans l'acétate d'éthyle. On obtient 3,48 g de produit attendu.
Préparation de l'hydrazide de l'acide 4-cyclohexyl-butanoïque
L'hydrazide de l'acide 4-cyclohexyl-butanoïque est préparé selon la méthode suivante : On mélange 2 g d'acide cyclohexane butyrique (Aldrich) avec 12 ml de chlorure de
L'hydrazide de l'acide 4-cyclohexyl-butanoïque est préparé selon la méthode suivante : On mélange 2 g d'acide cyclohexane butyrique (Aldrich) avec 12 ml de chlorure de
<Desc/Clms Page number 35>
thionyle, porte au reflux pendant 1 heure 30 minutes, évapore sous pression réduite jusqu'à obtention d'un extrait sec et effectue un entraînement au toluène pour éliminer l'excès de chlorure de thionyle. Le chlorure d'acide ainsi préparé est mis en solution dans du dichlorométhane et on introduit goutte à goutte cette solution dans le mélange constitué de 1,55 g de Terbutylcarbazate dans 10 ml de dichlorométhane et 1,7 ml de triéthylamine. On agite 2 heures à température ambiante puis verse sur de l'eau et extrait par deux fois 50 ml de dichlorométhane. La phase organique est séchée puis évaporée sous pression réduite. On obtient 3,54 g d'hydrazide attendue.
La déprotection s'effectue en agitant pendant 6 heures à température ambiante 3,54 g de produit protégé préparé plus haut avec 14 ml d'acide trifluoroacétique dans 20 ml de dichlorométhane. Après évaporation sous pression réduite, on obtient 6g de produit déprotégé attendu.
SM 184+ = M+ ; 185+ = [M+H]+ ; 226+ = MH+ + CH3CN Stade d : (9S)-3-(3-cyclohexylpropyl)-5,6,8,9-tétrahydro-1,2,4- triazolo[4,3-d][1,4]thiazépine-9-amine (sel de trifluoroacétate)
A 90 mg de produit obtenu au stade précédent dans 5 ml de dichlorométhane, on ajoute 0,6 ml d'acide trifluoroacétique, agite trois heures à température ambiante puis évapore sous pression réduite jusqu'à obtention d'un extrait sec. On obtient 110 mg de produit attendu Préparation 2 (9S)-3-(3-propyl)-5,6,8,9-tétrahydro-1,2,4-triazolo[4,3d][1,4]thiazépine-9-amine (sel de trifluoroacétate)
On opère comme à la préparation 1 mais en utilisant lors du stade c 0,33 g de produit obtenu au stade b de la préparation 1 et 0,2 g d'hydrazide de l'acide valérique [CH3-
A 90 mg de produit obtenu au stade précédent dans 5 ml de dichlorométhane, on ajoute 0,6 ml d'acide trifluoroacétique, agite trois heures à température ambiante puis évapore sous pression réduite jusqu'à obtention d'un extrait sec. On obtient 110 mg de produit attendu Préparation 2 (9S)-3-(3-propyl)-5,6,8,9-tétrahydro-1,2,4-triazolo[4,3d][1,4]thiazépine-9-amine (sel de trifluoroacétate)
On opère comme à la préparation 1 mais en utilisant lors du stade c 0,33 g de produit obtenu au stade b de la préparation 1 et 0,2 g d'hydrazide de l'acide valérique [CH3-
<Desc/Clms Page number 36>
(CH2)3-CONH-NH2] (Commercial, Lancaster) . On obtient 320 mg d'amine déprotégée attendue.
Préparation 3 (9S)-3-(2-cyclohexyléthyl)-5,6,8,9-tétrahydro-1,2,4triazolo[4,3-d][1,4]thiazépine-9-amine (sel de trifluoroacétate)
On opère comme à la préparation 1 mais en utilisant lors du stade c, 1,1 g de produit obtenu au stade b de la préparation 1 et 1 g d'hydrazide de l'acide 3-cyclohexylpropanoique. On obtient 660 mg d'amine déprotégée attendue Préparation de l'hydrazide de l'acide 3-cyclohexylpropanoique l'hydrazide de l'acide 3-cyclohexyl-propanoique est préparé selon la méthode indiquée pour la préparation de l'hydrazide de l'acide 4-cyclohexyl-butanoïque mais à partir d'acide 3 cyclohexane propionique (Aldrich).
On opère comme à la préparation 1 mais en utilisant lors du stade c, 1,1 g de produit obtenu au stade b de la préparation 1 et 1 g d'hydrazide de l'acide 3-cyclohexylpropanoique. On obtient 660 mg d'amine déprotégée attendue Préparation de l'hydrazide de l'acide 3-cyclohexylpropanoique l'hydrazide de l'acide 3-cyclohexyl-propanoique est préparé selon la méthode indiquée pour la préparation de l'hydrazide de l'acide 4-cyclohexyl-butanoïque mais à partir d'acide 3 cyclohexane propionique (Aldrich).
Préparation 4 (9S)-3-[3-(4-chlorophényl)propyl]-5,6,8,9-tétrahydro-1,2,4-

triazolo[4,3-d][1,4]thiazépine-9-amine (sel de trifluoroacétate)
On opère comme à la préparation 1 mais en utilisant lors du stade c 0,2 g de produit obtenu au stade b de la préparation 1 et 0,2 g d'hydrazide de l'acide para-chloro-3phényl-butanoique. On obtient 260 mg d'amine déprotégée attendue Préparation de l'hydrazide de l'acide para-chloro-3-phénylbutanoique
L'hydrazide de l'acide 3-cyclohexyl-butanoique est préparé selon la méthode indiquée pour la préparation de l'hydrazide de l'acide 4-cyclohexyl-butanoîque mais à partir d'acide parachloro-3-phényl-butanoique (Aldrich)
triazolo[4,3-d][1,4]thiazépine-9-amine (sel de trifluoroacétate)
On opère comme à la préparation 1 mais en utilisant lors du stade c 0,2 g de produit obtenu au stade b de la préparation 1 et 0,2 g d'hydrazide de l'acide para-chloro-3phényl-butanoique. On obtient 260 mg d'amine déprotégée attendue Préparation de l'hydrazide de l'acide para-chloro-3-phénylbutanoique
L'hydrazide de l'acide 3-cyclohexyl-butanoique est préparé selon la méthode indiquée pour la préparation de l'hydrazide de l'acide 4-cyclohexyl-butanoîque mais à partir d'acide parachloro-3-phényl-butanoique (Aldrich)
<Desc/Clms Page number 37>
Préparation 5 9-(9S)-amino-3-(2-cyclohexyléthoxy)-5,6,8,9-tétrahydroimidazo[1,2-d[1,4]thiazépine-2-carboxylate d'éthyle (sel de trifluoroacétate) Stade a : 3-hydroxy-9-[(9S)-[[(2,2-diméthyl)éthoxy]carbonylamino]- 5,6,8,9-tétrahydro-imidazo[1,2-d[1,4]thiazépine-2-carboxylate d'éthyle
A une solution de 0,74 g de chlorhydrate d'aminomalonate d'éthyle dans 20 ml d'éthanol et 0,49 ml de triéthylamine on ajoute 0,85 mg du produit préparé au stade b de la préparation 1 et agite 24 h à température ambiante. On évapore ensuite sous pression réduite, reprend l'extrait sec obtenu par 50 ml d'acétate d'éthyle, lave, sèche et évapore de nouveau sous pression réduite. Les cristaux obtenus sont essorés, lavés à l'éther et séchés. On obtient 0,633 g de produit attendu.
A une solution de 0,74 g de chlorhydrate d'aminomalonate d'éthyle dans 20 ml d'éthanol et 0,49 ml de triéthylamine on ajoute 0,85 mg du produit préparé au stade b de la préparation 1 et agite 24 h à température ambiante. On évapore ensuite sous pression réduite, reprend l'extrait sec obtenu par 50 ml d'acétate d'éthyle, lave, sèche et évapore de nouveau sous pression réduite. Les cristaux obtenus sont essorés, lavés à l'éther et séchés. On obtient 0,633 g de produit attendu.
IR (CHC13) 3394 cm-1+ absorption générale OH/NH (NH) ; 1707 cm-1 (C=O) ; 1653 cm-1 région C=O et/ou C=N ; 1594,1533 cm-1 : système conjugué + amide II Stade b : 3-(2-cyclohexyléthoxy)-9-[(9S)-[[(2,2-diméthyl)éthoxy] carbonylamino]-5,6,8,9-tétrahydro-imidazo[1,2d[1,4]thiazépine-2-carboxylate d'éthyle
A 220 mg du produit obtenu au stade précédent, on ajoute 5 ml de diméthylformamide, 400 mg de carbonate de césium, 220 mg de iodure de cyclohexyléthyle et agite 4 h à température ambiante. On verse le mélange réactionnel dans de l'eau et extrait par deux fois 50 ml d'acétate d'éthyle. La phase organique est séchée puis évaporée sous pression réduite jusqu'à obtention d'un extrait sec que l'on purifie par chromatographie en éluant avec le mélange dichlorométhane/acétate d'éthyle 80/20 ; obtient ainsi 50
A 220 mg du produit obtenu au stade précédent, on ajoute 5 ml de diméthylformamide, 400 mg de carbonate de césium, 220 mg de iodure de cyclohexyléthyle et agite 4 h à température ambiante. On verse le mélange réactionnel dans de l'eau et extrait par deux fois 50 ml d'acétate d'éthyle. La phase organique est séchée puis évaporée sous pression réduite jusqu'à obtention d'un extrait sec que l'on purifie par chromatographie en éluant avec le mélange dichlorométhane/acétate d'éthyle 80/20 ; obtient ainsi 50
<Desc/Clms Page number 38>
mg de produit de 0-alkylation attendu (ainsi que 160 mg de produit de C-alkylation).
SM 490+ = MNa+ ; 434+ = MNa+ - tBu ; 390+ = MNa+ - Boc ; 957+ = [2M + Na]+ Stade c : 9-(9S)-amino-3-(2-cyclohexyléthoxy)-5,6,8,9-tétrahydroimidazo[1,2-d[1,4]thiazépine-2-carboxylate d'éthyle (sel de trifluoroacétate)
A 50 mg du produit obtenu au stade précédent dans 2 ml de dichlorométhane, on ajoute 0,3 ml d'acide trifluoroacétique, agite 2 h à température ambiante puis évapore sous pression réduite. On obtient 70 mg de produit attendu Préparation 6 9-(9S)-amino-3-(2-cyclohexyléthoxy)-5,6,8,9-tétrahydroimidazo[1,2-d[1,4]thiazépine-2-carboxamide (sel de trifluoroacétate) Stade a : Acide 3-(2-cyclohexyléthoxy)-9-[(9S)-[[(2,2-diméthyl)éthoxy] carbonylamino]-5,6,8,9-tétrahydro-imidazo[1,2- d[1,4]thiazépine-2-carboxylique
On agite 6 h à 45 C le mélange constitué de 0,36 g d'ester préparé au stade b de la préparation 5 dans 10 ml d'éthanol et 0,75 ml de soude 2N, verse le mélange réactionnel dans l'eau, lave avec 50 ml d'acétate d'éthyle, acidifie avec 0,1N d'acide chlorhydrique puis extrait par trois fois 20 ml d'acétate d'éthyle. La phase organique est séchée puis évaporée sous pression réduite. On obtient 120 mg de produit attendu.
A 50 mg du produit obtenu au stade précédent dans 2 ml de dichlorométhane, on ajoute 0,3 ml d'acide trifluoroacétique, agite 2 h à température ambiante puis évapore sous pression réduite. On obtient 70 mg de produit attendu Préparation 6 9-(9S)-amino-3-(2-cyclohexyléthoxy)-5,6,8,9-tétrahydroimidazo[1,2-d[1,4]thiazépine-2-carboxamide (sel de trifluoroacétate) Stade a : Acide 3-(2-cyclohexyléthoxy)-9-[(9S)-[[(2,2-diméthyl)éthoxy] carbonylamino]-5,6,8,9-tétrahydro-imidazo[1,2- d[1,4]thiazépine-2-carboxylique
On agite 6 h à 45 C le mélange constitué de 0,36 g d'ester préparé au stade b de la préparation 5 dans 10 ml d'éthanol et 0,75 ml de soude 2N, verse le mélange réactionnel dans l'eau, lave avec 50 ml d'acétate d'éthyle, acidifie avec 0,1N d'acide chlorhydrique puis extrait par trois fois 20 ml d'acétate d'éthyle. La phase organique est séchée puis évaporée sous pression réduite. On obtient 120 mg de produit attendu.
IR (CHC13) 3424 cm-1 -NH ; 1743 cm-1, 1709 cm-1 C=O ; 1582 cm-1, 1489 cm-1 système conjugué + amide II
<Desc/Clms Page number 39>
Stade b : [2-(2S)-(aminocarbonyl)-3-(2-cyclohexyléthoxy)-5,6,8,9- tétrahydro-Imidazo[1,2-d[1,4]thiazépine-6-yl]-Carbamate de (l,l-diméthyléthyle)
A 120 mg d'acide obtenu au stade précédent dans 5 ml de tétrahydrofuranne, on ajoute à 0 C, 77 mg dechlorhydrate de 1 éthyl 3-(3-diméthyl amino) propyl carbodiimide, 54 mg d'hydrate de 1-hydroxy benzotriazole et 38 l de N-méthy morpholine et agite 1 heure à 0 C. On ajoute ensuite 25 ul d'ammoniac aqueux 28%, agite de nouveau 1 heure à 0 C, laisse remonter la température à température ambiante, acidifie par de l'acide chlorhydrique 1N puis extrait par trois fois 20 ml d'acétate d'éthyle. La phase organique est séchée puis évaorée sous pression réduite. On obtient 45 mg de produit attendu.
A 120 mg d'acide obtenu au stade précédent dans 5 ml de tétrahydrofuranne, on ajoute à 0 C, 77 mg dechlorhydrate de 1 éthyl 3-(3-diméthyl amino) propyl carbodiimide, 54 mg d'hydrate de 1-hydroxy benzotriazole et 38 l de N-méthy morpholine et agite 1 heure à 0 C. On ajoute ensuite 25 ul d'ammoniac aqueux 28%, agite de nouveau 1 heure à 0 C, laisse remonter la température à température ambiante, acidifie par de l'acide chlorhydrique 1N puis extrait par trois fois 20 ml d'acétate d'éthyle. La phase organique est séchée puis évaorée sous pression réduite. On obtient 45 mg de produit attendu.
SM 461+ = [M + Na]+ ; 405+ = MNa+ - tBu ; 361+ = MNa+ - C02tBu 437- = [M-H]Stade c : 9-(9S)-amino-3-(2-cyclohexyléthoxy)-5,6,8,9-tétrahydroimidazo[1,2-d[1,4]thiazépine-2-carboxamide (sel de trifluoroacétate)
A 45 mg du produit obtenu au stade précédent dans 2 ml de dichlorométhane, on ajoute 0,3 ml d'acide trifluoroacétique, agite 2h à température ambiante puis évapore sous pression réduite. On obtient 45 mg de produit attendu.
A 45 mg du produit obtenu au stade précédent dans 2 ml de dichlorométhane, on ajoute 0,3 ml d'acide trifluoroacétique, agite 2h à température ambiante puis évapore sous pression réduite. On obtient 45 mg de produit attendu.
Exemple 1 Acide [[4- 2S)-2-(acétylamino)-3-[[(9S)-3-[(3- cyclohexyl)propyl]-5,6,8,9-tétrahydro-l,2,4-triazolo-[4,3- d][1,4]thiazépin-9-yl]amino]-3-oxopropyl]phényl] difluorométhyl]-phosphonique
<Desc/Clms Page number 40>
Stade a : [[4-((2S)-2-(acétylamino)-3-[[(9S)-3-[(3-cyclohexyl)propyl]- 5,6,8,9-tétrahydro-1,2,4-triazolo-[4,3-d][1,4]thiazépin-9- yl]amino]-3-oxopropyl]phényl]difluorométhyl]-phosphonate de diéthyle
A 100 mg de N-acétyl-4-[bis- éthoxyphosphinyl)difluorométhyl]-L-phénylalanine ( N-Ac-TyrCF2-PO3Et2 ) dans 8 ml de dichlorométhane et 2 ml de diméthylformamide, on ajoute, sous atmosphère d'azote et à 0 C, 46 mg de chlorhydrate de 1 éthyl 3-(3-diméthylamino) propyl carbodiimide et 33 mg d'hydrate de 1-hydroxy benzotriazole. On agite quinze minutes à 0 C puis ajoute une solution de l'amine préparée à la préparation P1 (0,2mmol) dans 2 ml de dichlorométhane et 30 l de triéthylamine. On agite une nuit à température ambiante, verse dans de l'eau, extrait par trois fois 50 ml d'acétate d'éthyle, sèche la phase organique et évapore sous pression réduite jusqu'à obtention d'un extrait sec que l'on purifie par chromatographie en éluant avec le mélange dichlorométhane/méthanol 95/5. On obtient 44 mg du produit attendu.
A 100 mg de N-acétyl-4-[bis- éthoxyphosphinyl)difluorométhyl]-L-phénylalanine ( N-Ac-TyrCF2-PO3Et2 ) dans 8 ml de dichlorométhane et 2 ml de diméthylformamide, on ajoute, sous atmosphère d'azote et à 0 C, 46 mg de chlorhydrate de 1 éthyl 3-(3-diméthylamino) propyl carbodiimide et 33 mg d'hydrate de 1-hydroxy benzotriazole. On agite quinze minutes à 0 C puis ajoute une solution de l'amine préparée à la préparation P1 (0,2mmol) dans 2 ml de dichlorométhane et 30 l de triéthylamine. On agite une nuit à température ambiante, verse dans de l'eau, extrait par trois fois 50 ml d'acétate d'éthyle, sèche la phase organique et évapore sous pression réduite jusqu'à obtention d'un extrait sec que l'on purifie par chromatographie en éluant avec le mélange dichlorométhane/méthanol 95/5. On obtient 44 mg du produit attendu.
SM 670+ = [M+H]+ ; 692+ = [M+Na]+ ; 1361+ = [2M + Na]+ ; 668- = [MH]- 704- = [M + Cl]' ; 640 - MH- - Et Stade b : Acide [[4-((2S)-2-(acétylamino)-3-[[(9S)-3-[(3- cyclohexyl)propyl]-5,6,8,9-tétrahydro-1,2,4-triazolo-[4,3d][1,4]thiazépin-9-yl]amino]-3-oxopropyl]phényl] difluorométhyl]-phosphonique
On porte au reflux pendant 2 heures le mélange constitué de 44mg de produit préparé au stade précédent dans 5 ml de dichlorométhane et 0,1 ml de bromotriméthylsilane, évapore ensuite sous pression réduite jusqu'à obtention d'un extrait sec et effectue trois entraînement au méthanol jusqu'à
On porte au reflux pendant 2 heures le mélange constitué de 44mg de produit préparé au stade précédent dans 5 ml de dichlorométhane et 0,1 ml de bromotriméthylsilane, évapore ensuite sous pression réduite jusqu'à obtention d'un extrait sec et effectue trois entraînement au méthanol jusqu'à
<Desc/Clms Page number 41>
élimination complète du réactif. On purifie l'extrait sec par chromatographie en éluant avec un mélange méthanol/eau 60/40 et lyophilise le produit. On obtient 17 mg de produit attendu.
SM MH+ = 614+ ; MH+ - CH2CH2CH2-cyclohexane = 490+ ; MNa+ = 636+ ; 2MH+ = 1227+ ; 2MNa+ = 1249+ ; (M-H)- - 612- RMN (DMSO) 0,84 (m, 2H) 1,02 à 1,30 (m, 7H) 1,63 (m, 7H) : chaîne et - (CH2)2-cyclohexane ; 1,77 (s) 1,81 (s) : CH3CO ; 2,60 à 3,20 (m, 8H) : CH2S et CH2-C= ; 4, 20 à 4, 55 (m, 3H) : CH2-CH2-N ; 4,66 (m, 1H) 5,35 (m, 1H) : CH2-CH-NHCO ; 7,23 à 7,48 (m, 4H) H aromatiques ; 8,28 (d) 8,76 (d dédoublé) : CO-NH-CH.
Exemple 2
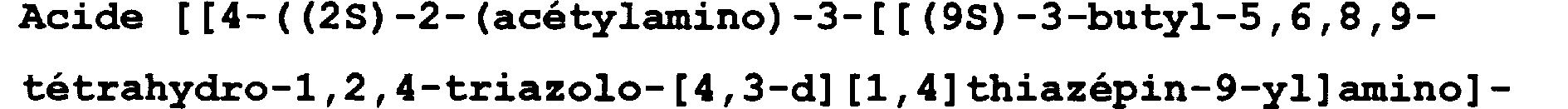
Acide [ [4- ( (2S) -2- (acétylamino) -3- [ [ (9S) -3-butyl-5, 6, 8, 9tétrahydro-1,2,4-triazolo-[4,3-d][1,4]thiazépin-9-yl]amino]- 3-oxopropyl]phényl]difluorométhyl]-phosphonique
On opère comme à l'exemple 1 mais avec 160 mg d'amine préparée à la préparation 2 et 129 mg de N-acétyl-4-[bis- éthoxyphosphinyl)difluorométhyl]-L-phénylalanine ( N-Ac-Tyr- CF2-PO3Et2 ). On obtient 65 mg de produit attendu.
Acide [ [4- ( (2S) -2- (acétylamino) -3- [ [ (9S) -3-butyl-5, 6, 8, 9tétrahydro-1,2,4-triazolo-[4,3-d][1,4]thiazépin-9-yl]amino]- 3-oxopropyl]phényl]difluorométhyl]-phosphonique
On opère comme à l'exemple 1 mais avec 160 mg d'amine préparée à la préparation 2 et 129 mg de N-acétyl-4-[bis- éthoxyphosphinyl)difluorométhyl]-L-phénylalanine ( N-Ac-Tyr- CF2-PO3Et2 ). On obtient 65 mg de produit attendu.
IR (CHC13) C=O 1648cm-1; Système conjugué, aromatique et amide II : 1540 cm-1 SM MH+ - 546+ ; MNa+ = 574+; (M-H)- - 544- RMN (DMSO) 0,92 (t, dédoublé) ; 1,38 (m) 1,63 (m) 2,87 (m) : chaîne nButyle; 1,79 (s) 1,82 (s) : CH3CO ; 2,80 à 3,10 (m) : CH2S et CH2-Ph ; 4,35 à 4,72 (m, 3H) : -CH2-N et CH ; (m) -CH-NHCO ; 7,30 à 7,48 (m, 4H) H aromatiques ; 8,28 (d) 8,88 (d) : 2 CH-NHCO Exemple 3 Acide [[4- 2S)-2-(acétylamino)-3-[[(9S)-3-[(2-
<Desc/Clms Page number 42>
cyclohexyl)éthyl]-5,6,8,9-tétrahydro-1,2,4-triazolo-[4,3- d][1,4]thiazépin-9-yl]amino]-3-oxopropyl]phényl] difluorométhyl]-phosphonique
On opère comme à l'exemple 1 mais avec 0,4g d'amine préparée à la préparation 3 et 160 mg de N-acétyl-4-[bis- éthoxyphosphinyl)difluorométhyl]-L-phénylalanine ( N-Ac-Tyr- CF2-P03Et2 ). On obtient 46 mg de produit attendu.
On opère comme à l'exemple 1 mais avec 0,4g d'amine préparée à la préparation 3 et 160 mg de N-acétyl-4-[bis- éthoxyphosphinyl)difluorométhyl]-L-phénylalanine ( N-Ac-Tyr- CF2-P03Et2 ). On obtient 46 mg de produit attendu.
SM MNa+ = 622+; (M-H) - = 598- RMN (DMSO) 0,92 (m) 1,10 à 1,80 (m) : chaîne alkyle ; (s) CH3CO 2,80 à 3,10 (m complexe) : CH2S et CH2-C= ; 4,40 à 4,70 (m) 5,43 (m) et 5,52 (m) : -CH-X ; 7,30 à 7,50 (m) H aromatiques ; 8,28 (d) 8,92 (dl) : 2 -CH-NHCO Exemple 4 Acide [[4- 2S)-2-(acétylamino)-3-[[(9S)-3-[(3-(4- chlorophényl)propyl]-5,6,8,9-tétrahydro-l,2,4-triazolo-[4,3- d][1,4]thiazépin-9-yl]amino]-3-oxopropyl]phényl] difluorométhyl]-phosphonique
On opère comme à l'exemple 1 mais avec l'amine préparée à la préparation 4 (0,2mmol) et 94 mg de N-acétyl-4-[bis- éthoxyphosphinyl)difluorométhyl]-L-phénylalanine ( N-Ac-Tyr- CF2-PO3Et2 ). On obtient 35 mg de produit attendu.
On opère comme à l'exemple 1 mais avec l'amine préparée à la préparation 4 (0,2mmol) et 94 mg de N-acétyl-4-[bis- éthoxyphosphinyl)difluorométhyl]-L-phénylalanine ( N-Ac-Tyr- CF2-PO3Et2 ). On obtient 35 mg de produit attendu.
SM (M-H)- = 640- RMN (DMSO) 1, 67 (s) 1, 71 (s) CH3CO ; 1, 81 (m, CH2 central) ; 2, 55 à 2, 85 (m, 10H) 2, 93 (m, 1H) : CH2S et CH2-C= ; 4,10 à 4, 65 (m, 3 à 4H) 5,25 (m, 1H) : -CH2-N-C= et CH-NHCO ; 7,10 à 7,42 (m, 8H) aromatiques ; 8,19 (d, 1H) 8,66 (d dédoublé 1H) : 2 -CH-NHCO Exemple 5 N2-acétyl-N-[(3-[(4-chlorophényl)propyl]-5,6,8,9-tétrahydro-

1,2,4-triazolo-[4,3-d][1,4]thiazépin-9-yl]-4-(phosphonooxy)- L-Phénylalaninamide
1,2,4-triazolo-[4,3-d][1,4]thiazépin-9-yl]-4-(phosphonooxy)- L-Phénylalaninamide
<Desc/Clms Page number 43>
Stade a : N2-acétyl-N-[(3-[(4-chlorophényl)propyl]-5,6,8,9-tétrahydro- 1,2,4-triazolo-[4,3-d][1,4]thiazépin-9-yl]-4-[bis- (diphénylméthoxy)phosphinyl]-L-Phénylalaninamide
On opère comme à l'exemple 1 stade a mais avec l'amine préparée à la préparation 1 (0,2mmol) et 120 mg de N-acétyl- 0-[bis-(diphénylméthoxy)phosphinyl]-L-tyrosine (N-Ac-Tyr-0- (P03Bn2)). On obtient 53 mg de produit attendu.
On opère comme à l'exemple 1 stade a mais avec l'amine préparée à la préparation 1 (0,2mmol) et 120 mg de N-acétyl- 0-[bis-(diphénylméthoxy)phosphinyl]-L-tyrosine (N-Ac-Tyr-0- (P03Bn2)). On obtient 53 mg de produit attendu.
SM (M-H)- = 786- ; [M+Na]+ - 810+ Stade b : N2-acétyl-N-[(3-[(4-chlorophényl)propyl]-5,6,8,9-tétrahydro- 1,2,4-triazolo-[4,3-d][1,4]thiazépin-9-yl]-4-(phosphonooxy)- L-Phénylalaninamide
On place sous atmosphère d'hydrogène pendant 20 heures à température ambiante, le mélange constitué de 50 mg du produit préparé au stade précédent dans 5 ml de méthanol et 35 mg de Palladium à 10% sur charbon. On obtient après filtration et évaporation sous pression réduite 32 mg de produit brut que l'on purifie par chromatographie en éluant avec le mélange méthanol/Eau 60/40. On obtient après lyophilisation 13 mg de produit pur attendu SM (M-H)- - 606RMN (DMSO) 1,76 (s) 1,78 (s) CH3CO ; 1,91 (m, CH2 central) ; 2,60 à 3,02 (m, 10H) : CH2S et CH2-C= ; 4,20 à 4,65 (m, 3H) 5,35 (m, 1H) : -CH2-N-C= et CH-NHCO ; 7,07 (m, 4H) 7,24 (dl, 2H) 7,34 (dl, 2H) aromatiques ; 8,20 (d, 1H) 8,61 (d) 8,68 (d, 1H) : 2-CHNHCO Exemple 6 N-[(9S)-3-(2-cyclohexyléthyl)-5,6,8,9-tétrahydro-1,2,4-
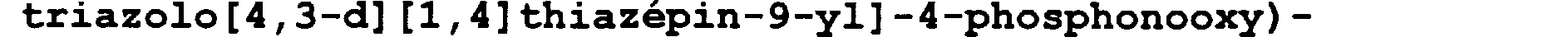
triazolo[4,3-d][l,4jthiazépin-9-yl]-4-phosphonooxy)- Benzenamide
On place sous atmosphère d'hydrogène pendant 20 heures à température ambiante, le mélange constitué de 50 mg du produit préparé au stade précédent dans 5 ml de méthanol et 35 mg de Palladium à 10% sur charbon. On obtient après filtration et évaporation sous pression réduite 32 mg de produit brut que l'on purifie par chromatographie en éluant avec le mélange méthanol/Eau 60/40. On obtient après lyophilisation 13 mg de produit pur attendu SM (M-H)- - 606RMN (DMSO) 1,76 (s) 1,78 (s) CH3CO ; 1,91 (m, CH2 central) ; 2,60 à 3,02 (m, 10H) : CH2S et CH2-C= ; 4,20 à 4,65 (m, 3H) 5,35 (m, 1H) : -CH2-N-C= et CH-NHCO ; 7,07 (m, 4H) 7,24 (dl, 2H) 7,34 (dl, 2H) aromatiques ; 8,20 (d, 1H) 8,61 (d) 8,68 (d, 1H) : 2-CHNHCO Exemple 6 N-[(9S)-3-(2-cyclohexyléthyl)-5,6,8,9-tétrahydro-1,2,4-
triazolo[4,3-d][l,4jthiazépin-9-yl]-4-phosphonooxy)- Benzenamide
<Desc/Clms Page number 44>
Stade a : N-[(9S)-3-(2-cyclohexyléthyl)-5,6,8,9-tétrahydro-1,2,4-

triazolo[4,3-d][1,4]thiazépin-9-yl]-4-hydroxy)-Benzenamide
On agite une heure, sous azote, le mélange, constitué de 56 mg d'acide 4-hydroxybenzoique dans 8 ml de dichlorométhane et 1 ml de diméthylformamide, 78 mg d'EDC.HCl et 75 mg d'HOBt puis on ajoute 112 mg d'amine préparée à la préparation 3 dans 5 ml de dichlorométhane. On agite 3 heures à température ambiante, verse dans de l'eau, extrait par trois fois 50 ml de dichlorométhane, sèche et évapore sous pression réduite.
triazolo[4,3-d][1,4]thiazépin-9-yl]-4-hydroxy)-Benzenamide
On agite une heure, sous azote, le mélange, constitué de 56 mg d'acide 4-hydroxybenzoique dans 8 ml de dichlorométhane et 1 ml de diméthylformamide, 78 mg d'EDC.HCl et 75 mg d'HOBt puis on ajoute 112 mg d'amine préparée à la préparation 3 dans 5 ml de dichlorométhane. On agite 3 heures à température ambiante, verse dans de l'eau, extrait par trois fois 50 ml de dichlorométhane, sèche et évapore sous pression réduite.
On obtient un extrait sec que l'on purifie par chromatographie en éluant avec le mélange dichlorométhane/Méthanol 95/5. On obtient ainsi 60 mg de produit attendu.
SM 401+ = [M + H]+ ; 423+ = [M + Na]+ ; 823+ = [2M + Na]+ ; 399- - [M-H]- ; 799- = [2M-H]Stade b : N-[(9S)-3-(2-cyclohexyléthyl)-5,6,8,9-tétrahydro-1,2,4- triazolo[4,3-d][1,4]thiazépin-9-yl]-4-[[bis(phénylméthoxy) phosphinyl]oxy]-Benzenamide
On mélange pendant 12 heures à température ambiante sous atmosphère d'azote le mélange constitué de 60 mg du produit préparé au stade précédent dans 4 ml d'acétonitrile, 2 ml de dioxanne 71 l de tétrachlorure de carbone,2 mg de DMAP, 50 l de dibenzylphosphite et 51 l d'éthyldiisopropylamine. On verse ensuite dans de l'eau, extrait par trois fois 20 ml de dichlorométhane, sèche la phase organique et évapore sous pression réduite jusqu'à obtention d'un extrait sec que l'on purifie par chromatographie en éluant avec le mélange dichlorométhane/méthanol 95/5. On obtient 30 mg de produit attendu.
On mélange pendant 12 heures à température ambiante sous atmosphère d'azote le mélange constitué de 60 mg du produit préparé au stade précédent dans 4 ml d'acétonitrile, 2 ml de dioxanne 71 l de tétrachlorure de carbone,2 mg de DMAP, 50 l de dibenzylphosphite et 51 l d'éthyldiisopropylamine. On verse ensuite dans de l'eau, extrait par trois fois 20 ml de dichlorométhane, sèche la phase organique et évapore sous pression réduite jusqu'à obtention d'un extrait sec que l'on purifie par chromatographie en éluant avec le mélange dichlorométhane/méthanol 95/5. On obtient 30 mg de produit attendu.
Rf (CHzClz/MeOH 90/10) : 0,45 Stade c :
<Desc/Clms Page number 45>
N-[(9S)-3-(2-cyclohexyléthyl)-5,6,8,9-tétrahydro-1,2,4- triazolo[4,3-d][1,4]thiazépin-9-yl]-4-phosphonooxy)- Benzenamide
On hydrogène pendant 20 heures à température ambiante 30 mg du produit préparé au stade précédent dans 5 ml de méthanol en présence de 30 mg de palladium à 10% sur charbon.
On hydrogène pendant 20 heures à température ambiante 30 mg du produit préparé au stade précédent dans 5 ml de méthanol en présence de 30 mg de palladium à 10% sur charbon.
Ensuite, on filtre, évapore sous pression réduite et purifie par chromatographie en phase inverse en éluant avec le mélange méthanol/eau 60/40 puis on lyophilise. On obtient 2 mg de produit attendu SM 479- - [M-H] - ; 399' = MH- - [-P (0) (OH)2] Exemple 7 9-(9S)-[[(2S)-2(acétylamino)-3-[4- (phosphonodifluorométhyl)phényl]-1-oxopropyl)amino]-3-(2cyclohexyléthoxy)-5,6,8,9-tétrahydro-Imidazo[1,2-d][1,4] thiazépine-2-carboxamide
On opère comme à l'exemple 1 stades a et b mais à partir de 45 mg d'amine préparée à la préparation 6 sous forme de sel de triflate et 50 mg de N-acétyl-4-[bis- éthoxyphosphinyl)difluorométhyl]-L-phénylalanine ( N-Ac-Tyr- CF2-P03Et2 ). On obtient après déprotection avec du bromotriméthylsilane 33 mg de produit attendu.
On opère comme à l'exemple 1 stades a et b mais à partir de 45 mg d'amine préparée à la préparation 6 sous forme de sel de triflate et 50 mg de N-acétyl-4-[bis- éthoxyphosphinyl)difluorométhyl]-L-phénylalanine ( N-Ac-Tyr- CF2-P03Et2 ). On obtient après déprotection avec du bromotriméthylsilane 33 mg de produit attendu.
SM 656- = [M-H]- Exemple 8 9-(9S)-[[(2S)-2(acétylamino)-3-[4-(phosphonooxy)phényl]-loxopropyl)amino]-3-(2-cyclohexyléthoxy)-5,6,8,9-tétrahydro- Imidazo[1,2-d][1,4]thiazépine-2-carboxylate d'éthyle
On opère comme à l'exemple 5 stades a et b mais à partir de l'amine préparée à la préparation 5 (0,107mmol) et 62 mg de N-acétyl-O-[bis-(diphénylméthoxy)phosphinyl]-L-tyrosine (N-Ac-Tyr-0-(P03Bn2)). On obtient après déprotection par hydrogénation (Pd 10%/charbon) 15 mg de produit attendu.
On opère comme à l'exemple 5 stades a et b mais à partir de l'amine préparée à la préparation 5 (0,107mmol) et 62 mg de N-acétyl-O-[bis-(diphénylméthoxy)phosphinyl]-L-tyrosine (N-Ac-Tyr-0-(P03Bn2)). On obtient après déprotection par hydrogénation (Pd 10%/charbon) 15 mg de produit attendu.
<Desc/Clms Page number 46>
SM 697 = [M-H]Na2+ ; 675 = MNa+ ; 653 = MH+ RMN (DMSO) 0,93 (m, 2H du cyclohexyle) ; 1,10 à 2,00 (m, 13H, chaîne aliphatique) ; 1,80 (s) 1,84 (s) CH3CO ; 2,60 à 3,10 (m, 10H) CH2S, Ph-CH2- ; 4,10 à 5,20 (m, 10H) CH2X et CHX ; 7,00 à 7,35 (m, 5H aromatiques) ; (m) 8,48 (d) 8,63 (d) 2H CO-NH-CH.
Etude pharmacologique des produits de l'invention . Mesure des IC50 par scintillation proximity essay (SPA).
La capacité des molécules à se lier à la protéine SrcSH2 est évaluée par compétition (mesure de l'ICso) dans un test appelé Scintillation proximity assay (SPA). Ce test utilise des billes PVT (Amersham) qui contiennent le scintillant. Ces billes coatées avec de la streptavidine sont incubées dans un tampon MOPS dans des plaques 99 puits avec 50nM de protéine SH2 préalablement biotinylée puis avec le ligand EPQpYEEIPIYL marqué à l'iode 125. En se liant à la protéine fixée sur la bille le ligand active le scintillant qui émet une lumière mesurée dans un compteur à scintillation Wallac micropeta. La valeur obtenue en absence de tout compétiteur correspond à la liaison maximale du ligand sur la protéine. Cette méthode ne nécessite pas de séparer le ligand libre du lié.
Les molécules à tester sont diluées dans le tampon MOPS contenant 2% DMSO et ajoutées à différentes concentrations de 0,lnM à 100 M dans les puits contenant le ligand, avant l'addition des billes coatées avec Src-SH2. Après 1 heure d'incubation une mesure de la liaison est effectuée au compteur à scintillation. L'inhibition de la liaison du ligand radiomarqué par les différentes concentrations de compétiteur permet d'évaluer les IC5o des molécules testées.
Résultat Résultats SPA : Peptide de référence
PYEEI - 0,2-0,4 umol
PYEEI - 0,2-0,4 umol
<Desc/Clms Page number 47>
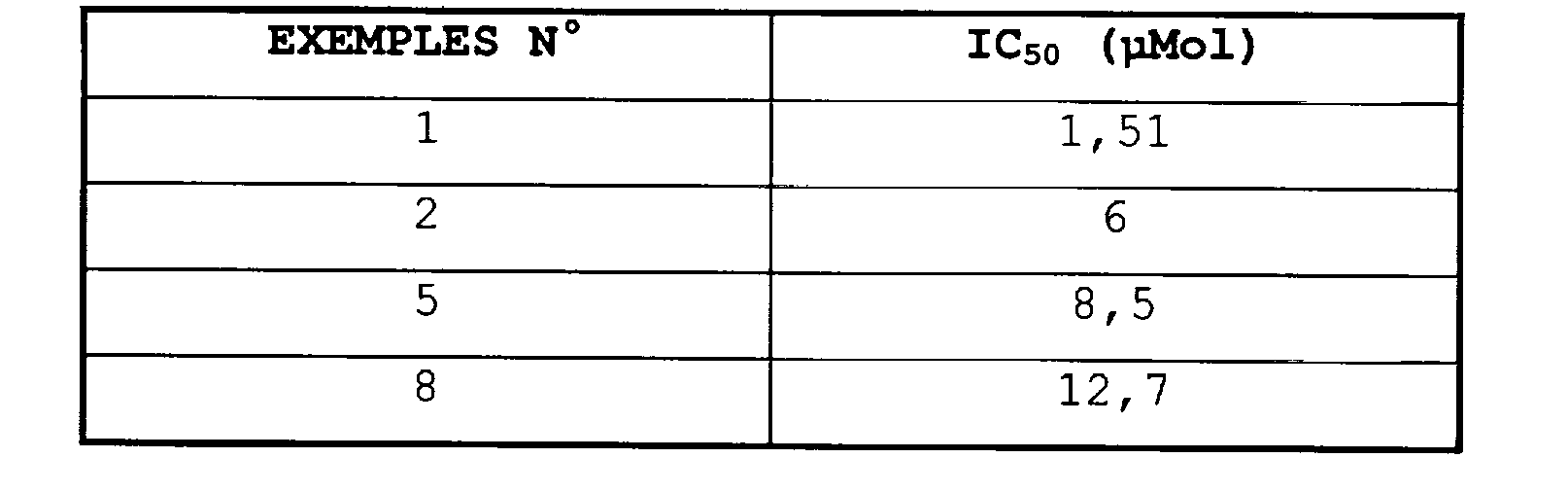
<tb>
<tb> EXEMPLES <SEP> ? <SEP> IC50 <SEP> (pMol)
<tb> 1 <SEP> 1,51 <SEP>
<tb> 2 <SEP> 6 <SEP>
<tb> 5 <SEP> 8,5
<tb> 8 <SEP> 12,7 <SEP>
<tb>
<tb> EXEMPLES <SEP> ? <SEP> IC50 <SEP> (pMol)
<tb> 1 <SEP> 1,51 <SEP>
<tb> 2 <SEP> 6 <SEP>
<tb> 5 <SEP> 8,5
<tb> 8 <SEP> 12,7 <SEP>
<tb>
Claims (29)
- -CH (Z ) - (C5-C14 ) -aryle-, - (C1-C4 ) -alkyle- (C5-C14 ) -aryle-, et - (C5-C14)-aryle-, dans lesquels Z est un atome d'hydrogène, un tétrazole, NRcR'c, NHCORc, CONHRc, NHC02Rc, NHCONHRc, NHSORc ou NHS02Rc; les dits radicaux aryles étant substitués ou non substitués par R2 ou par R'2; - A2 représente un groupement choisi parmi -PO(ORd)(ORe), -OPO(ORd)(ORe), -B (ORd) (ORe) , -CRfRgPO(ORd)(ORe), -0-CRfRgPO(ORd)(ORe), -CRfRg-C02Rd, -0- CRfRg-C02Rd, -N (CRfRg-C02Rd) 2, -C02Rd, -CRfRg-CH2-CO2Rd, - CRfRgS03H, -0-CRfRgS03H, -S03H, -S02NH2, -NHCO-PO (ORd) (ORe) et -CO-PO(ORd)(ORe);
 dans laquelle, - W représente un groupement -CH2- ou S ; - Het représente un aryle hétérocyclique à 5 ou 6 chainons, accolé en position 3 et 4 à la thiazépine et renfermant de 1 à 3 hétéroatomes choisis parmi N, 0 ou S, dans lequel l'atome de soufre peut être substitué par 0,1 ou 2 atomes d'oxygènes, - V, lié à un atome de carbone de l'hétérocycle, représente un groupement R3, OR3 ou SR3, - [Ao] représente un groupement choisi parmi -(C(Ra)(Rb))n-CO-NRi- ou -(C(Ra)(Rb))n-NRi-CO-, n étant égal à 0,1,2 ou 3, - [Ai] représente un groupement choisi parmi -CH (Z) - (Ci-C4) -alkyle- (C5-C14) -aryle-,
dans laquelle, - W représente un groupement -CH2- ou S ; - Het représente un aryle hétérocyclique à 5 ou 6 chainons, accolé en position 3 et 4 à la thiazépine et renfermant de 1 à 3 hétéroatomes choisis parmi N, 0 ou S, dans lequel l'atome de soufre peut être substitué par 0,1 ou 2 atomes d'oxygènes, - V, lié à un atome de carbone de l'hétérocycle, représente un groupement R3, OR3 ou SR3, - [Ao] représente un groupement choisi parmi -(C(Ra)(Rb))n-CO-NRi- ou -(C(Ra)(Rb))n-NRi-CO-, n étant égal à 0,1,2 ou 3, - [Ai] représente un groupement choisi parmi -CH (Z) - (Ci-C4) -alkyle- (C5-C14) -aryle-, REVENDICATIONS 1) Un composé de formule (I)<Desc/Clms Page number 49>
REVENDICATIONS 1) Un composé de formule (I)<Desc/Clms Page number 49> NH- ( (C1-C4) -alkyle) , -N ( (C1-C4) alkyle) z, -NHCO-(Cl-C4) -alkyle, -NHCO- (CS-C14) -aryle, -CONH- (Cl-C4) -alkyle, -CONH- (CS-C1q) aryle, -CO- (C1-Cq) -alkyle ou -C0- (C5-Ci4) -aryle; - R'2 a les mêmes valeurs que A2; - éventuellement A2 et R2 forment ensemble avec l'aryle qui les porte, l'un des cycles à 5 chaînons suivants :
NH- ( (C1-C4) -alkyle) , -N ( (C1-C4) alkyle) z, -NHCO-(Cl-C4) -alkyle, -NHCO- (CS-C14) -aryle, -CONH- (Cl-C4) -alkyle, -CONH- (CS-C1q) aryle, -CO- (C1-Cq) -alkyle ou -C0- (C5-Ci4) -aryle; - R'2 a les mêmes valeurs que A2; - éventuellement A2 et R2 forment ensemble avec l'aryle qui les porte, l'un des cycles à 5 chaînons suivants : C3) -alkylènedioxy-, {C5-Ci4) -aryle-, (C5-C14) -aryloxy-, (C5- C14) -aryl- (C1-C4) -alkyle-, (C5-C14) -aryl- (C1-C4) -alkyloxy-, halogène, trihalogénométhyle-, hydroxyle, nitro, formyle, cyano, carboxy, -CONH2, (C1-C8)-alkyloxycarbonyle-, amino, -
C3) -alkylènedioxy-, {C5-Ci4) -aryle-, (C5-C14) -aryloxy-, (C5- C14) -aryl- (C1-C4) -alkyle-, (C5-C14) -aryl- (C1-C4) -alkyloxy-, halogène, trihalogénométhyle-, hydroxyle, nitro, formyle, cyano, carboxy, -CONH2, (C1-C8)-alkyloxycarbonyle-, amino, - - R3, Ra, Rb, Rc, Rd, Re ou Ri, indépendants les uns des autres, identiques ou différents, représentant un atome d' hydrogène, un radical (C1-C8)-alkyle-, (C3-C18) -cycloalkyle- , (C3-C18)- (cycloalkyl) - (Cl-C8) -alkyle-, (C5-C14) -aryle- ou (C5-C14)-aryl-(C1-C4)-alkyle-, les dits radicaux étant substitués ou non substitués par R2; - Rf et Rg identiques ou différents représentant un atome d'hydrogène, un halogène, ORc, NHRc, un radical C02Rd, CH2C02Rd, SO3H, PO (ORd) (ORe) ou tétrazole; - éventuellement Ra et Rb forment ensemble avec l'atome de carbone qui les porte, un groupement cycloalkyle renfermant de 3 à 6 carbones, - Y, lié à un atome de carbone de l'hétérocycle, représente un atome C=0, C=S, C=NH ou SO2; - Ri représente un groupement-OH, -ORc,-NRcR'c, (C1-C4)alkyle- ou halogène; - R2 représente un groupement (C1-C8)-alkyle-, (C1-C8)-alkoxy-, hydroxy- (C1-C8) -alkyle-, (C1-C8) -alkoxy- (Cl-C8) -alkyle-, (CI-<Desc/Clms Page number 50>le trait en pointillé indique que le groupement Y-Ri est optionnel, lesdits composés de formule (I) étant sous toutes leurs formes isomères possibles, seuls ou en mélange dans un rapport quelconque, ainsi que leurs sels physiologiquement acceptables et leurs promédicaments (prodrugs).
- R3, Ra, Rb, Rc, Rd, Re ou Ri, indépendants les uns des autres, identiques ou différents, représentant un atome d' hydrogène, un radical (C1-C8)-alkyle-, (C3-C18) -cycloalkyle- , (C3-C18)- (cycloalkyl) - (Cl-C8) -alkyle-, (C5-C14) -aryle- ou (C5-C14)-aryl-(C1-C4)-alkyle-, les dits radicaux étant substitués ou non substitués par R2; - Rf et Rg identiques ou différents représentant un atome d'hydrogène, un halogène, ORc, NHRc, un radical C02Rd, CH2C02Rd, SO3H, PO (ORd) (ORe) ou tétrazole; - éventuellement Ra et Rb forment ensemble avec l'atome de carbone qui les porte, un groupement cycloalkyle renfermant de 3 à 6 carbones, - Y, lié à un atome de carbone de l'hétérocycle, représente un atome C=0, C=S, C=NH ou SO2; - Ri représente un groupement-OH, -ORc,-NRcR'c, (C1-C4)alkyle- ou halogène; - R2 représente un groupement (C1-C8)-alkyle-, (C1-C8)-alkoxy-, hydroxy- (C1-C8) -alkyle-, (C1-C8) -alkoxy- (Cl-C8) -alkyle-, (CI-<Desc/Clms Page number 50>le trait en pointillé indique que le groupement Y-Ri est optionnel, lesdits composés de formule (I) étant sous toutes leurs formes isomères possibles, seuls ou en mélange dans un rapport quelconque, ainsi que leurs sels physiologiquement acceptables et leurs promédicaments (prodrugs). - 2) Composés de formule (I) telle que définie à la revendication 1 dans laquelle W représente un atome de soufre.
- 3) Composés de formule (I) telle que définie à la revendication 1 ou 2 dans laquelle Het représente un hétérocycle aromatique à 5 ou 6 chaînons renfermant 2 ou 3 atomes d'azote et -[Ao]- représente le groupement -NHCO-.
- 4) Dérivés de thiazépine accolé en position 3 et 4 à un triazole tels que définis à l'une quelconque des revendications 1 à 3 répondant à la formule (la)
 V, Al et A2 étant tels que définis à la revendication 1.
V, Al et A2 étant tels que définis à la revendication 1. - 5) Dérivé de thiazépine accolé en position 3 et 4 à un imidazole tels que définis à l'une quelconque des revendications 1 à 3 répondant à la formule (Ib)
 V, Y, Ri, Ai et A2 étant tels que définis à la revendication 1.<Desc/Clms Page number 51>
V, Y, Ri, Ai et A2 étant tels que définis à la revendication 1.<Desc/Clms Page number 51> - 6) Composé de formules (I), (la) ou (Ib) telles que définies à l'une quelconque des revendications 1 à 5 dans lesquelles A2 représente -O-P03H2, -CF2-PO3H2 ou -CH(CO2H)2 et se trouve en position para.
- 7) Composé de formules (I), (la) ou (Ib) telles que définies à l'une quelconque des revendications 1 à 5 dans lesquelles -
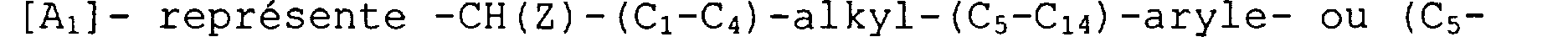 [A1J - représente -CH (Z) - (C1-C4) -alkyl- (C5-Clq) -aryle- ou (C5- C14)-aryle-,ledit aryle étant éventuellement substitué par un groupement R2 ou R'2, R2, R'2et Z étant tels que définis à la revendication 1.
[A1J - représente -CH (Z) - (C1-C4) -alkyl- (C5-Clq) -aryle- ou (C5- C14)-aryle-,ledit aryle étant éventuellement substitué par un groupement R2 ou R'2, R2, R'2et Z étant tels que définis à la revendication 1. - 8) Composé de formules (I), (la) ou (Ib) telles que définies à l'une quelconque des revendications 1 à 5 dans lesquelles Z représente de préférence NHCORc, NHC02Rc, dans lesquels Rc représente un radical alkyle renfermant de 1 à 6 atomes de carbone, le carbone portant le groupement Z présentant la configuration S
- 9) Composé de formules (I), (la) ou (Ib) telles que définies à l'une quelconque des revendications 1 à 5 dans V représente de préférence OR3 ou R3 dans lesquels R3 représente un groupement (C1-C6) -alkyle-, (C3-C12) -cycloalkyle-, (C3-C12) - cycloalkyl- (C1-C6) -alkyle- ou aryl- (C1-C6) -alkyle-
- 10) Composé de formules (I) ou (Ib) telles que définies à l'une quelconque des revendications 1 à 5 dans lesquelles Y est un groupement C=O et R1 est OH, NH2 ou un groupement alkyloxy renfermant de 1 à 6 atomes de carbone.
- 11) Un composé de formule (la) telle que définie à l'une quelconque des revendications 4 et 6 à 9 dans laquelle -[Ai]- représente -CH (Z) - (C1-C4) -alkyl-phényle- ou-phényle- éventuellement substitué en méta par un groupement R2 ou R'2 - A2 représente -O-PO3H2, -CF2-PO3H2 ou -CH(C02H)2 et est en position para, - Z représente NHCORc, NHC02Rc, dans lesquels Rc représente un radical alkyle renfermant de 1 à 6 atomes de carbone, le<Desc/Clms Page number 52>C1z) -cycloalkyl- (C1-C6) -alkyle-, (C1-C6) -alkyle ou aryl- (C1-C6) - alkyle-, lesdits composés de formule (I) étant sous toutes leurs formes isomères possibles, seuls ou en mélange dans un rapport quelconque, ainsi que leurs sels physiologiquement acceptables et leurs promédicaments (prodrugs).
 carbone portant le groupement Z présentant la configuration S; - V représente un radical R3 représentant un groupement (C3-
carbone portant le groupement Z présentant la configuration S; - V représente un radical R3 représentant un groupement (C3- - 12) Un composé de formule (Ib) telle que définie à l'une quelconque des revendications 5 à 9 dans laquelle -[Ai]- représente -CH(Z)-(C1-4)-alkyl-phényle- ou-phényle- éventuellement substitué en méta par un groupement R2 ou R'2, - A2 représente -O-PO3H2, -CF2-PO3H2 ou -CH(C02H)2 et est en position para, - Z représente NHCORc, NHC02Rc, dans lesquels Rc représente un radical alkyle renfermant de 1 à 6 atomes de carbone, le carbone portant le groupement Z présentant la configuration S; - V représente un radical OR3 dans lequel R3 représente un
 groupement un groupement (C3-C12) -cycloalkyl- (C1-C6) -alkyle- , (C1-C6) -alkyle ou aryl- (C1-C6) -alkyle-, - Y est un groupement C=0, et - R1 est un groupement hydroxy, NH2, ou un groupement alkyloxy renfermant de 1 à 6 atomes de carbone, lesdits composés de formule (I) étant sous toutes leurs formes isomères possibles, seuls ou en mélange dans un rapport quelconque, ainsi que leurs sels physiologiquement acceptables et leurs promédicaments (prodrugs).
groupement un groupement (C3-C12) -cycloalkyl- (C1-C6) -alkyle- , (C1-C6) -alkyle ou aryl- (C1-C6) -alkyle-, - Y est un groupement C=0, et - R1 est un groupement hydroxy, NH2, ou un groupement alkyloxy renfermant de 1 à 6 atomes de carbone, lesdits composés de formule (I) étant sous toutes leurs formes isomères possibles, seuls ou en mélange dans un rapport quelconque, ainsi que leurs sels physiologiquement acceptables et leurs promédicaments (prodrugs). - 13) les composés de formule (I) tels que définis à l'une quelconque des revendications 1 à 12 dont les noms suivent : . Acide [[4-((2S)-2-(acétylamino)-3-[[(9S)-3-[(3- cyclohexyl)propyl]-5,6,8,9-tétrahydro-1,2,4-triazolo-[4,3d][1,4]thiazépin-9-yl]amino]-3-<Desc/Clms Page number 53>Imidazo[1,2-dJ[1,4]thiazépine-2-carboxylate d'éthyle; ainsi que leurs sels physiologiquement acceptables et leurs promédicaments (prodrugs).
 triazolo[4,3-d][1,4]thiazépin-9-yl]-4-phosphonooxy)- Benzenamide; . 9-(9S)-[[(2S)-2(acétylamino)-3-[4- (phosphonodifluorométhyl)phényl]-1-oxopropyl)amino]-3-(2- cyclohexyléthoxy)-5,6,8,9-tétrahydro-Imidazo[1,2d][1,4]thiazépine-2-carboxamide; . 9-(9S)-[[(2S)-2(acétylamino)-3-[4-(phosphonooxy)phényl]-1- oxopropyl)amino]-3-(2-cyclohexyléthoxy)-5,6,8,9-tétrahydro-
triazolo[4,3-d][1,4]thiazépin-9-yl]-4-phosphonooxy)- Benzenamide; . 9-(9S)-[[(2S)-2(acétylamino)-3-[4- (phosphonodifluorométhyl)phényl]-1-oxopropyl)amino]-3-(2- cyclohexyléthoxy)-5,6,8,9-tétrahydro-Imidazo[1,2d][1,4]thiazépine-2-carboxamide; . 9-(9S)-[[(2S)-2(acétylamino)-3-[4-(phosphonooxy)phényl]-1- oxopropyl)amino]-3-(2-cyclohexyléthoxy)-5,6,8,9-tétrahydro-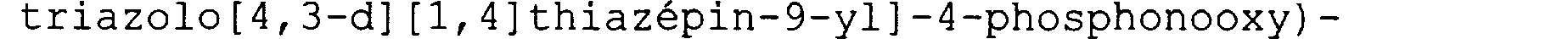 tétrahydro-1,2,4-triazolo-[4,3-d][1,4]thiazépin-9-yl]amino]- 3-oxopropyl]phényl]difluorométhyl]-phosphonique; . Acide [[4-((2S)-2-(acétylamino)-3-[[(9S)-3-[(2- cyclohexyl)éthyl]-5,6,8,9-tétrahydro-1,2,4-triazolo-[4,3d][1,4]thiazépin-9-yl]amino]-3oxopropyl]phényl]difluorométhyl]-phosphonique; . Acide [[4-((2S)-2-(acétylamino)-3-[[(9S)-3-[(3-(4- chlorophényl)propyl]-5,6,8,9-tétrahydro-1,2,4-triazolo-[4,3- d][1,4]thiazépin-9-yl]amino]-3- oxopropyl]phényl]difluorométhyl]-phosphonique; . N2-acétyl-N-[(3-[(4-chlorophényl)propyl]-5,6,8,9- tétrahydro-1,2,4-triazolo-[4,3-d][1,4]thiazépin-9-yl]-4- (phosphonooxy)-L-Phénylalaninamide; . N-[(9S)-3-(2-cyclohexyléthyl)-5,6,8,9-tétrahydro-1,2,4-
tétrahydro-1,2,4-triazolo-[4,3-d][1,4]thiazépin-9-yl]amino]- 3-oxopropyl]phényl]difluorométhyl]-phosphonique; . Acide [[4-((2S)-2-(acétylamino)-3-[[(9S)-3-[(2- cyclohexyl)éthyl]-5,6,8,9-tétrahydro-1,2,4-triazolo-[4,3d][1,4]thiazépin-9-yl]amino]-3oxopropyl]phényl]difluorométhyl]-phosphonique; . Acide [[4-((2S)-2-(acétylamino)-3-[[(9S)-3-[(3-(4- chlorophényl)propyl]-5,6,8,9-tétrahydro-1,2,4-triazolo-[4,3- d][1,4]thiazépin-9-yl]amino]-3- oxopropyl]phényl]difluorométhyl]-phosphonique; . N2-acétyl-N-[(3-[(4-chlorophényl)propyl]-5,6,8,9- tétrahydro-1,2,4-triazolo-[4,3-d][1,4]thiazépin-9-yl]-4- (phosphonooxy)-L-Phénylalaninamide; . N-[(9S)-3-(2-cyclohexyléthyl)-5,6,8,9-tétrahydro-1,2,4- oxopropyl]phényl]difluorométhyl]-phosphonique; . Acide [[4-((2S)-2-(acétylamino)-3-[[(9S)-3-butyl-5,6,8,9-
oxopropyl]phényl]difluorométhyl]-phosphonique; . Acide [[4-((2S)-2-(acétylamino)-3-[[(9S)-3-butyl-5,6,8,9- - 14) Un procédé de préparation des composés de formule (I) tels que définis à l'une quelconque des revendications 1 à 13, qui comprend le couplage de deux ou plusieurs fragments qui peuvent être dérivés par rétrosynthèse des composés de formule (I).
- 15) Un procédé selon la revendication 14 dans lequel on fait réagir une amine de formule (II)<Desc/Clms Page number 54>X-C (=O)-[A1]-A'2 dans laquelle [Ai] est tel que défini plus haut dans la formule (I) et A'2 représentant, outre les valeurs de A2, le radical hydroxy, X est un groupe substituable par un nucléophile et où, le cas échéant, les groupes fonctionnels sont sous forme de précurseurs ou sous forme protégée, lesdits groupes fonctionnels éventuellement présents sous forme de précurseur ou sous forme protégée, étant par la suite convertis en groupes présents dans les composés de formule (I),certains composés de formule (I) pouvant si désiré ou si nécessaire, subir une ou plusieurs des réactions suivantes dans un ordre approprié : - action d'un agent de déprotection partielle - action d'un agent de déprotection totale, - action de H-P(O)(ORd)(ORe) lorsque A'2 représente OH, - salification.dans laquelle Het, Y, V, w et Ri sont tels que définis à la revendication 1 pour la formule (I) et où, le cas échéant, les groupes fonctionnels sont sous forme de précurseurs ou sous forme protégée, avec un acide ou un dérivé d'acide de formule (III)

- 16) Procédé selon la revendication 15 caractérisé en ce que le composé de formule (III) est choisi parmi :
 N-Boc-Tyr-0- ( P03Bn2 ) , N-Ac-Tyr-0-(PO3Bn2), N-Boc-Tyr-CF2- (P03Et2) , N-Ac-Tyr-CF2-(PO3Et2).<Desc/Clms Page number 55>
N-Boc-Tyr-0- ( P03Bn2 ) , N-Ac-Tyr-0-(PO3Bn2), N-Boc-Tyr-CF2- (P03Et2) , N-Ac-Tyr-CF2-(PO3Et2).<Desc/Clms Page number 55> - 17) Procédé selon la revendication 15 de formation des composés de formule (la) telle que définie à la revendication 4 caractérisé en ce que le composé de formule (II)est le composé de formule (IIea) suivant :
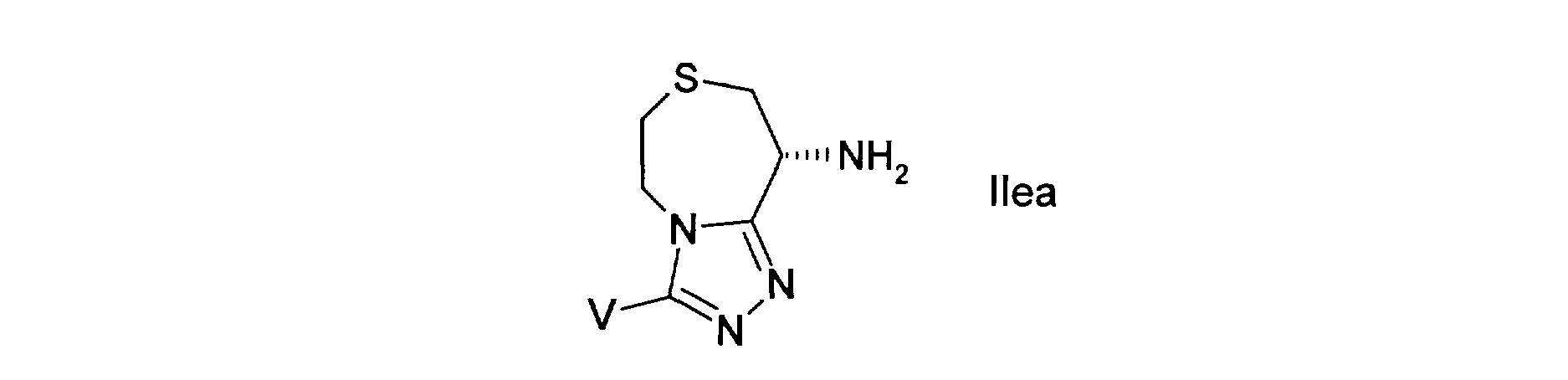 V étant tel que défini à la revendication 1.
V étant tel que défini à la revendication 1. - 18) Procédé selon la revendication 15 de formation des composés de formule (Ib) telle que définie à la revendication 5 caractérisé en ce que le composé de formule (II)est le composé de formule (IIeb) suivant :
 V, Y et RI étant tels que définis à la revendication 1.
V, Y et RI étant tels que définis à la revendication 1. - 19) Procédé de préparation des composés de formule (II) telle que définie à la revendication 16 caractérisé en ce que l'on soumet un composé de formule (lia)
 <Desc/Clms Page number 56>H2N-CH- (CO2Et)2 puis, le cas échéant, transformation d'un groupement C02Et en YR1 désiré, Y et Ri étant tels que définis à la revendication-soit par action d'un dérivé de formule (IVb)
<Desc/Clms Page number 56>H2N-CH- (CO2Et)2 puis, le cas échéant, transformation d'un groupement C02Et en YR1 désiré, Y et Ri étant tels que définis à la revendication-soit par action d'un dérivé de formule (IVb) V-CO-NH-NH2, V étant tel que défini à la revendication 1, afin d'obtenir le dérivé bicyclique de formule (IIda)dans lequel l'hétérocycle accolé à la thiazépine est un triazoleque l'on cyclise par -soit par action d'un dérivé de formule (IVa)
V-CO-NH-NH2, V étant tel que défini à la revendication 1, afin d'obtenir le dérivé bicyclique de formule (IIda)dans lequel l'hétérocycle accolé à la thiazépine est un triazoleque l'on cyclise par -soit par action d'un dérivé de formule (IVa) que l'on soumet à l'action d'un iodure ou chlorure d'alkyle renfermant de 1 à 4 atomes de carbone en milieu basique, afin d'obtenir le composé de formule (IIc)
que l'on soumet à l'action d'un iodure ou chlorure d'alkyle renfermant de 1 à 4 atomes de carbone en milieu basique, afin d'obtenir le composé de formule (IIc) P étant un groupement protecteur de l'amine, à l'action d'un agent de formation de dérivés thiocétones correspondants tel que le P2S5, afin d'obtenir le composé de formule (IIb)<Desc/Clms Page number 57>V représentant un groupement hydroxyle qui est ensuite alkylé, composés de formules (IIda) ou (IIdb) que l'on déprotège, notamment par hydrogénation catalytique, afin d'obtenir l'amine correspondante de formules (IIea) ou (IIeb) telles que définies aux revendications 17 et 18.
P étant un groupement protecteur de l'amine, à l'action d'un agent de formation de dérivés thiocétones correspondants tel que le P2S5, afin d'obtenir le composé de formule (IIb)<Desc/Clms Page number 57>V représentant un groupement hydroxyle qui est ensuite alkylé, composés de formules (IIda) ou (IIdb) que l'on déprotège, notamment par hydrogénation catalytique, afin d'obtenir l'amine correspondante de formules (IIea) ou (IIeb) telles que définies aux revendications 17 et 18. 1, afin d'obtenir le dérivé bicyclique de formule (IIdb)dans lequel l'hétérocycle accolé à la thiazépine est un imidazole
1, afin d'obtenir le dérivé bicyclique de formule (IIdb)dans lequel l'hétérocycle accolé à la thiazépine est un imidazole - 20) A titre de médicament, un composé de formule (I) et/ou ses sels physiologiquement acceptables et/ou ses prodrugs tel que défini à l'une quelconque des revendications 1 à 13.
- 21) Une composition pharmaceutique comprenant au moins un composé de formule (I) tel que défini à l'une quelconque des revendications 1 à 12 et/ou ses sels physiologiquement acceptables et/ou ses prodrugs ainsi qu'un ou plusieurs excipients pharmaceutiquement acceptables et éventuellement un ou plusieurs additifs.
- 22) A titre de médicament ayant une activité inhibitrice du récepteur Src SH2, un composé de formule (I) et/ou ses sels physiologiquement acceptables et/ou ses prodrugs tel que défini à l'une quelconque des revendications 1 à 13.<Desc/Clms Page number 58>
- 23) A titre de médicament ayant une activité inhibitrice de la résorption osseuse ou pour le traitement ou la prévention de l'ostéoporose, un composé de formule (I) et/ou ses sels physiologiquement acceptables et/ou ses prodrugs tel que défini à l'une quelconque des revendications 1 à 13.
- 24) A titre de médicament pour le traitement ou la prévention de l'immunité, de l'infection, de l'allergie, et des maladies autoimmunes, un composé de formule (I) et/ou ses sels physiologiquement acceptables et/ou ses prodrugs tel que défini à l'une quelconque des revendications 1 à 13.
- 25) A titre de médicament pour le traitement ou la prévention de maladies telles que les maladies prolifératives, le cancer, la resténose, l'inflammation, les allergies ou les maladies cardiovasculaires, un composé de formule (I) et/ou ses sels physiologiquement acceptables et/ou ses prodrugs tel que défini à l'une quelconque des revendications 1 à 13.
- 26) Utilisation des composés de formule (I) et/ou leurs sels physiologiquement acceptables et/ou leurs prodrugs tels que définis à l'une quelconque des revendications 1 à 13 pour la préparation de médicaments destinés à la prévention ou au traitement de l'ostéoporose.
- 27) Utilisation des composés de formule (I) et/ou leurs sels physiologiquement acceptables et/ou leurs prodrugs tels que définis à l'une quelconque des revendications 1 à 13 pour la préparation de médicaments destinés au traitement ou à la prévention de l'immunité, de l'infection, de l'allergie, et des maladies autoimmunes.
- 28) Utilisation des composés de formule (I) et/ou leurs sels physiologiquement acceptables et/ou leurs prodrugs tels que définis à l'une quelconque des revendications 1 à 13 pour la préparation de médicaments destinés au traitement ou à la prévention de maladies telles que les maladies prolifératives, le cancer, la resténose, l'inflammation, les allergies ou les maladies cardiovasculaires.<Desc/Clms Page number 59>
- 29) Intermédiaires de formules (II), (IIb), (Ile), (IIda), (IIdb), (IIea) et (IIeb) tels que définis aux revendications 15,17, 18 et 19.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9909006A FR2796381A1 (fr) | 1999-07-12 | 1999-07-12 | Derives bicycliques de thioazepine ou de caprolactame, leur procede de preparation et les intermediaires de ce procede, leur application comme medicaments et les compositions pharmaceutiques les contenant |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9909006A FR2796381A1 (fr) | 1999-07-12 | 1999-07-12 | Derives bicycliques de thioazepine ou de caprolactame, leur procede de preparation et les intermediaires de ce procede, leur application comme medicaments et les compositions pharmaceutiques les contenant |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| FR2796381A1 true FR2796381A1 (fr) | 2001-01-19 |
Family
ID=9547992
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| FR9909006A Pending FR2796381A1 (fr) | 1999-07-12 | 1999-07-12 | Derives bicycliques de thioazepine ou de caprolactame, leur procede de preparation et les intermediaires de ce procede, leur application comme medicaments et les compositions pharmaceutiques les contenant |
Country Status (1)
| Country | Link |
|---|---|
| FR (1) | FR2796381A1 (fr) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008078725A1 (fr) * | 2006-12-26 | 2008-07-03 | Daiichi Sankyo Company, Limited | Dérivé de thiazépine |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0665241A (ja) * | 1992-08-19 | 1994-03-08 | Takeda Chem Ind Ltd | 縮合イミダゾール化合物、その製造法及び剤 |
| WO1995025118A2 (fr) * | 1994-03-15 | 1995-09-21 | Trustees Of Tufts University | Inhibiteurs d'interactions des domaines sh2 |
| WO1997012903A1 (fr) * | 1995-10-04 | 1997-04-10 | Warner-Lambert Company | Composes, compositions et procedes permettant d'inhiber la liaison de proteines contenant un domaine sh2 avec des proteines phosphorylees de meme origine |
| WO1997030079A1 (fr) * | 1996-02-15 | 1997-08-21 | Pharmacia & Upjohn S.P.A. | Peptides antagonistes de la mitogenese et de la motogenese cellulaires et leur utilisation therapeutique |
| WO2000005246A1 (fr) * | 1998-07-21 | 2000-02-03 | Hoechst Marion Roussel | Derives de thioazepinone, procede de preparation et intermediaires de ce procede, application comme medicaments et compositions pharmaceutiques les contenant |
-
1999
- 1999-07-12 FR FR9909006A patent/FR2796381A1/fr active Pending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0665241A (ja) * | 1992-08-19 | 1994-03-08 | Takeda Chem Ind Ltd | 縮合イミダゾール化合物、その製造法及び剤 |
| WO1995025118A2 (fr) * | 1994-03-15 | 1995-09-21 | Trustees Of Tufts University | Inhibiteurs d'interactions des domaines sh2 |
| WO1997012903A1 (fr) * | 1995-10-04 | 1997-04-10 | Warner-Lambert Company | Composes, compositions et procedes permettant d'inhiber la liaison de proteines contenant un domaine sh2 avec des proteines phosphorylees de meme origine |
| WO1997030079A1 (fr) * | 1996-02-15 | 1997-08-21 | Pharmacia & Upjohn S.P.A. | Peptides antagonistes de la mitogenese et de la motogenese cellulaires et leur utilisation therapeutique |
| WO2000005246A1 (fr) * | 1998-07-21 | 2000-02-03 | Hoechst Marion Roussel | Derives de thioazepinone, procede de preparation et intermediaires de ce procede, application comme medicaments et compositions pharmaceutiques les contenant |
Non-Patent Citations (2)
| Title |
|---|
| CHEMICAL ABSTRACTS, vol. 121, no. 7, 15 August 1994, Columbus, Ohio, US; abstract no. 083336, AONO T ET AL: "Preparation of fused imidazole derivatives inhibiting formation of lipid peroxide" XP002137195 * |
| OSAMU UCHIKAWA: "Aminothiazole derivatives. I. A convenient synthesis of monocyclic and condensed 5-aminothiazole derivatives", JOURNAL OF HETEROCYCLIC CHEMISTRY., vol. 31, no. 4, 1994, HETEROCORPORATION. PROVO., US, pages 877 - 887, XP002137194, ISSN: 0022-152X * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008078725A1 (fr) * | 2006-12-26 | 2008-07-03 | Daiichi Sankyo Company, Limited | Dérivé de thiazépine |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2368891B1 (fr) | Dérivés de pyrimidines en tant qu' antagonistes du récepteur de la vitronectine | |
| CA3061611A1 (fr) | Modulateurs de l'interaction sestrine-gator2 et utilisations de ces derniers | |
| JP5976322B2 (ja) | テトラヒドロ−イミダゾ[1,5−a]ピラジン誘導体塩、その製造方法及び医薬用途 | |
| CZ356298A3 (cs) | Deriváty benzazepinon-N-octové kyseliny, substituované kyselinou fosfonovou, jakož i způsob jejich výroby a léčiva, obsahující tyto sloučeniny | |
| EP0532410A1 (fr) | Dérivés hétérocycliques N-substitués, comme inhibiteurs de l'angiotensive II | |
| EP1664047A1 (fr) | Derives d'indolizine 1,2,3,6,7,8 substituee, inhibiteurs des fgfs, leur procede de preparation et les compositions pharmaceutiques les contenant | |
| TW201902507A (zh) | 治療病毒感染之組合療法 | |
| EP1100801B1 (fr) | Utilisation de derives de la cysteine pour preparer un medicament destine a traiter les pathologies qui resultent de la formation de la proteine g het | |
| EP2070914B1 (fr) | Nouveaux dérivés antagonistes du récepteur de la vitronectine, leur procédé de préparation, leur application comme médicaments et les compositions pharmaceutiques les renfermant | |
| EP1257552B1 (fr) | Derives de xanthine, intermediaires et application au traitement de l'osteoporose | |
| WO2000005246A1 (fr) | Derives de thioazepinone, procede de preparation et intermediaires de ce procede, application comme medicaments et compositions pharmaceutiques les contenant | |
| FR2796381A1 (fr) | Derives bicycliques de thioazepine ou de caprolactame, leur procede de preparation et les intermediaires de ce procede, leur application comme medicaments et les compositions pharmaceutiques les contenant | |
| CA2896209A1 (fr) | 3,5-diaryl-azaindoles comme inhibiteurs de la proteine dyrk1a pour le traitement des deficiences cognitives liees au syndrome de down et a la maladie d'alzheimer | |
| EP1133478B1 (fr) | Nouveaux derives d'acylguanidines, leur procede de preparation, leur application comme medicaments et les compositions pharmaceutiques les renfermant | |
| EP1282621B1 (fr) | derives antagonistes du recepteur de la vitronectine | |
| EP1387841B1 (fr) | Nouveaux derives d'azole ou de triazole, leur procede de preparation et leur application comme fongicides | |
| FR2793796A1 (fr) | Nouveaux derives d'imidazoles, leur procede de preparation et les intermediaires de ce procede, leur application comme medicaments et les compositions pharmaceutiques les contenant | |
| EP1133491B1 (fr) | Derives du benzofurane, leur procede de preparation, leur application comme medicaments et les compositions pharmaceutiques les renfermant | |
| EP1409461B1 (fr) | Nouveaux derives d'azole ou de triazole, leur procede de preparation et leur application comme fongicides | |
| CA2968483A1 (fr) | Derives hydroxybisphosphoniques hydrosolubles de la doxorubicine | |
| EP1133476B1 (fr) | Derives d'iminoguanidines, leur procede de preparation, leur application comme medicaments |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| TP | Transmission of property |