FR2674853A1 - Derives de piperidinylguanidine, leur preparation et leur application en therapeutique. - Google Patents
Derives de piperidinylguanidine, leur preparation et leur application en therapeutique. Download PDFInfo
- Publication number
- FR2674853A1 FR2674853A1 FR9104010A FR9104010A FR2674853A1 FR 2674853 A1 FR2674853 A1 FR 2674853A1 FR 9104010 A FR9104010 A FR 9104010A FR 9104010 A FR9104010 A FR 9104010A FR 2674853 A1 FR2674853 A1 FR 2674853A1
- Authority
- FR
- France
- Prior art keywords
- sep
- general formula
- group
- mmol
- mixture
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 0 *N(*)C(CC1)CCN1C(*1)=NC(CC2)C1CCC2(*1*CCCC1)I Chemical compound *N(*)C(CC1)CCN1C(*1)=NC(CC2)C1CCC2(*1*CCCC1)I 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
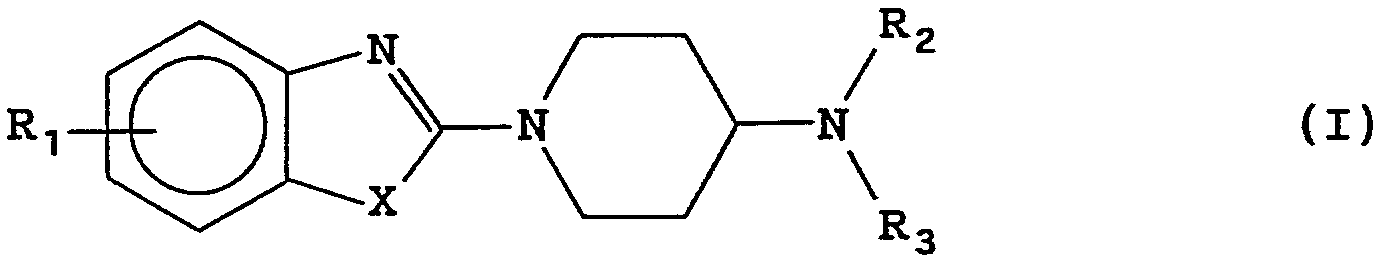
Composés répondant à la formule générale (I) (CF DESSIN DANS BOPI) dans laquelle R1 représente un atome d'hydrogène ou d'halogène ou un groupe (C1 -C4 )alkyle, trifluorométhyle, nitro, amino, uréido, N'-(C1 -C4 )alkyluréido ou N'-phényluréido, R2 représente un atome d'hydrogène ou un groupe (C1 -C4 )alkyle ou benzyle, R3 représente soit un atome d'hydrogène, soit un groupe amidino de formule C(-NH2 )=N-H, et X représente soit un atome d'oxygène ou de soufre, soit un groupe imino de formule N-R4 dans laquelle R4 représente un atome d'hydrogène ou un groupe (C1 -C7 )alkyle, méthoxyéthyle, éthoxycarbonylméthyle, carbamoylméthyle, phényle ou 4-chlorophényle, à l'état de bases libres ou de sels d'addition à des acides. Application en thérapeutique.
Description
La présente invention a pour objet des dérivés de pipéridinylguanidine, leur préparation et leur application en thérapeutique.
Les composés de l'invention répondent à la formule générale (I) dans laquelle

R1 représente un atome d'hydrogène ou d'halogène ou un
groupe (C1-C4)alkyle, trifluorométhyle, nitro, amino,
uréido, N'-(C1-C4)alkyluréido ou N'-phényluréido,
R2 représente un atome d'hydrogène ou un groupe (C1-C4)
alkyle ou benzyle,
R3 représente soit un atome d'hydrogène, soit un groupe
amidino de formule C(-NH2)=N-H, et
X représente soit un atome d'oxygène ou de soufre, soit
un groupe imino de formule N-R4 dans laquelle R4 repré
sente un atome d'hydrogène ou un groupe (C1-C7)alkyle,
méthoxyéthyle, éthoxycarbonylméthyle, carbamoylméthyle,
phényle ou 4-chlorophényle.
groupe (C1-C4)alkyle, trifluorométhyle, nitro, amino,
uréido, N'-(C1-C4)alkyluréido ou N'-phényluréido,
R2 représente un atome d'hydrogène ou un groupe (C1-C4)
alkyle ou benzyle,
R3 représente soit un atome d'hydrogène, soit un groupe
amidino de formule C(-NH2)=N-H, et
X représente soit un atome d'oxygène ou de soufre, soit
un groupe imino de formule N-R4 dans laquelle R4 repré
sente un atome d'hydrogène ou un groupe (C1-C7)alkyle,
méthoxyéthyle, éthoxycarbonylméthyle, carbamoylméthyle,
phényle ou 4-chlorophényle.
Les composés de l'invention peuvent exister à l'étant de bases libres ou de sels d'addition à des acides.
Conformément à l'invention, on peut les préparer selon un procédé illustré par le schéma de la page suivante.
On fait d'abord réagir un dérivé chloré de formule générale (II), dans laquelle X et R1 sont tels que définis ci-dessus, avec le 8-aza-1,4-dioxaspiroC4.5ldécane de formule (III), dans les conditions de réaction habituelles entre un dérivé halogéné et une amine secondaire, c'est-à-dire par chauffage dans un solvant organique tel que le diméthylsulfoxyde ou le 3-méthylbutan-2-ol.On obtient l'intermédiaire de formule générale (IV) qui, lorsque X représente N-H, peut ensuite, si on le désire, être alkylé de manière connue à l'aide d'un halogénoalcane en Cl-c7 par exemple d'un iodoalcane en pré-
Schéma

Schéma
<tb> <SEP> N <SEP> O
<tb> (il) <SEP> R1 <SEP> e <SEP> \ <SEP> Cl <SEP> + <SEP> HN <SEP> 9 <SEP> X <SEP> (III)
<tb> <SEP> x <SEP> O
<tb> <SEP> N <SEP> O
<tb> <SEP> R1 <SEP> \\tN <SEP> (Iv)
<tb> <SEP> x <SEP> O
<tb> <SEP> M <SEP> kylaton
<tb> <SEP> optionnelle <SEP> ç
<tb> <SEP> lorsque <SEP> X=NH <SEP> :<SEP> H
<tb> <SEP> N-H <SEP> 4 <SEP> N-R5
<tb> <SEP> R,-Tp=i <SEP> O <SEP> (v)
<tb> <SEP> i) <SEP> H2N-R2
<tb> <SEP> ii) <SEP> H2/Pd
<tb> <SEP> N <SEP> > <SEP> R2
<tb> <SEP> 1 <SEP> iH <SEP> (la)
<tb> <SEP> H2NCN
<tb> <SEP> N <SEP> R2
<tb> <SEP> R1 <SEP> x\NN <SEP> (lob)
<tb> <SEP> HN
<tb> <SEP> HN
<tb> sence d'hydrure de sodium, dans un solvant organique tel que le diméthylformamide. On hydrolyse ensuite la fonction acétal en fonction cétone, en milieu acide, pour obtenir la cétone de formule générale (V), puis on fait réagir cette dernière avec une amine de formule générale R2-NH2, dans laquelle R2 est tel que défini ci-dessus, ce qui, après hydrogénation catalytique, donne un composé de formule générale (Ia), qui correspond à la formule générale (I) lorsque R3 représente l'hydrogène.
<tb> (il) <SEP> R1 <SEP> e <SEP> \ <SEP> Cl <SEP> + <SEP> HN <SEP> 9 <SEP> X <SEP> (III)
<tb> <SEP> x <SEP> O
<tb> <SEP> N <SEP> O
<tb> <SEP> R1 <SEP> \\tN <SEP> (Iv)
<tb> <SEP> x <SEP> O
<tb> <SEP> M <SEP> kylaton
<tb> <SEP> optionnelle <SEP> ç
<tb> <SEP> lorsque <SEP> X=NH <SEP> :<SEP> H
<tb> <SEP> N-H <SEP> 4 <SEP> N-R5
<tb> <SEP> R,-Tp=i <SEP> O <SEP> (v)
<tb> <SEP> i) <SEP> H2N-R2
<tb> <SEP> ii) <SEP> H2/Pd
<tb> <SEP> N <SEP> > <SEP> R2
<tb> <SEP> 1 <SEP> iH <SEP> (la)
<tb> <SEP> H2NCN
<tb> <SEP> N <SEP> R2
<tb> <SEP> R1 <SEP> x\NN <SEP> (lob)
<tb> <SEP> HN
<tb> <SEP> HN
<tb> sence d'hydrure de sodium, dans un solvant organique tel que le diméthylformamide. On hydrolyse ensuite la fonction acétal en fonction cétone, en milieu acide, pour obtenir la cétone de formule générale (V), puis on fait réagir cette dernière avec une amine de formule générale R2-NH2, dans laquelle R2 est tel que défini ci-dessus, ce qui, après hydrogénation catalytique, donne un composé de formule générale (Ia), qui correspond à la formule générale (I) lorsque R3 représente l'hydrogène.
Une autre variante de procédé, non illustrée par un schéma, consiste à préparer un composé de formule générale (Ia) à partir d'un composé de formule générale (II) et d'une pipéridine déjà substituée en position 4 par un groupe NH-R2, en présence d'une base.
Si l'on désire un composé de formule générale (I) dans laquelle R3 représente un groupe amidino, on fait finalement réagir un composé de formule générale (Ia) avec le cyanamide pour obtenir une guanidine de formule générale (Ib).
Il va de soi qu'un composé de formule générale (I) peut être transformé en un autre composé de formule générale (1) par toute méthode connue.
Ainsi on peut intervenir par exemple au stade (Ib, R1=H) pour introduire un substituant R1 autre que l'hydrogène, tel que, par exemple, un groupe nitro au moyen d'acide nitrique, ledit groupe nitro pouvant ensuite, si on le désire, être réduit en groupe amino puis transformé en groupe uréido.
Les composés de départ sont décrits dans la littérature ou bien peuvent être préparés selon des méthodes qui y sont décrites ou connues de l'homme du métier.
Ainsi, par exemple, des benzoxazoles de formule générale (II,
X=O) sont décrits dans J. Med. Chem., 1988, 31, 1719-1728.
X=O) sont décrits dans J. Med. Chem., 1988, 31, 1719-1728.
Ceux dans la formule desquels X représente un groupe phénylimino ou (4-chlorophényl)imino sont accessibles à partir de la N-phénylbenzène-1,2-diamine ou de la N-(4-chlorophényl)benzène-1,2-diamine, respectivement, d'abord par réaction avec l'urée pour former la 1-phénylbenzimidazolone ou la 1-(4-chlorophényl)benzimidazolone, respectivement, puis par réaction avec le chlorure de phosphoryle.
Des benzimidazoles analogues aux composés de formule générale (Ia) sont, par ailleurs, décrits dans la demande de brevet européen N00217700.
La pipéridine de formule (III) est disponible dans le commerce.
Les exemples qui suivent illustrent en détail la préparation de quelques composés selon l'invention. Les microanalyses élémentaires et les spectres IR et RMN confirment les structures des produits obtenus. Les numéros indiqués entre parenthèses dans les titres des exemples correspondent à ceux du tableau donné plus loin.
Exemple 1 (Composé N05).
N-[1-[1-(1-Méthyléthyl)-1H-benzimidazol-2-ylApipéridin-4-yl]- méthanamine, fumarate.
1.1 8-(1H-Benzimidazol-2-yl)-8-aza-1,4-dioxaspiro[4.5]dé-
cane.
cane.
On chauffe à 1250C pendant 5h un mélange de 20 g (131 mmoles) de 2-chloro-1H-benzimidazole et 20,53 g (144 mmoles) de 8-aza-1,4-dioxaspiro[4.5]décane dans 100 ml de 3-méthylbutanol. On évapore le solvant et on recristallise le résidu dans un mélange de dichlorométhane et d'éther diéthylique. On obtient 33 g de chlorhydrate dont on libère la base par extraction avec le dichlorométhane en milieu basique.
Point de fusion : 3010C.
1.2 8-[1-(1-Méthyléthyl)-1H-benzimidazol-2-yl]-8-aza
1,4-dioxaspiro[4.5]décane.
1,4-dioxaspiro[4.5]décane.
A 1 g (250 mmoles) d'hydrure de sodium à 60% dans l'huile et 25 ml de diméthylformamide placés sous argon on ajoute, goutte à goutte, 5,5 g (21,12 mmoles) de 8-(1H-benzimida zol-2-yl)-8-aza-1,4-dioxaspiro[4.5]décane en solution dans 30 ml de diméthylformamide. On agite le mélange à 0 C pendant 30mn puis on ajoute 10 ml, soit 17 g (100 mmoles) de 2-iodopropane en solution dans 10 ml de diméthylformamide, et on poursuit l'agitation à 0 C pendant 2h. On évapore le solvant sous pression réduite, on reprend le résidu avec de l'eau et on l'extrait deux fois avec de l'acétate d'éthyle. Après lavage, séchage et évaporation de la phase organique on obtient 3,2 g de produit.
Point de fusion : 1210C.
1.3 1-[1-(1-Méthyléthyl)-1H-benzimidazol-2-yl]pipéridin
4-one.
4-one.
On chauffe au reflux pendant 1h 3,4 g (13,1 mmoles) de 8-[1-(1-méthyléthyl)-1H-benzimidazol-2-yl]-8-aza-1,4-dioxa spiro(4.5]décane dans 50 ml d'acide chlorhydrique à 10% dans l'acide acétique, on évapore le mélange, on reprend le résidu avec de l'eau et on l'extrait deux fois avec du dichlorométhane. Après lavage, séchage et évaporation de la phase organique on obtient 2,35 g de produit.
Point de fusion : 1610C.
1.4 N-[1-[1-(1-Méthyléthyl)-1H-benzimidazol-2-yl]pipéridin-
4-yl]méthanamine, fumarate.
4-yl]méthanamine, fumarate.
A une solution de 0,2 g (6,45 mmoles) de méthanamine dans 30 ml de méthanol on ajoute 1 g (3,9 mmoles) de 1-[1-(1-mé- thyléthyl)-1H-benzimidazol-2-yl]pipéridin-4-one et on effectue une hydrogénation en présence de 0,1 g de charbon palladié à 10%, à température ambiante, pendant 6h. On filtre le catalyseur, on évapore le filtrat, et on purifie le résidu d'évaporation par chromatographie sur colonne de gel de silice en éluant avec un mélange de dichlorométhane/méthanol/ammoniaque 9/8/2. On obtient 1 g de produit (point de fusion : 870C) dont on prépare le fumarate avec un équivalent d' acide fumarique.
Point de fusion : 1900C.
ExemPle 2 (Composé N06).
N-[1-(Benzothiazol-2-yl)pipériodin-4-yl]-N-méthylguanidine, dichlorhydrate.
A un mélange de 1,4 g (5,7 mmoles) de N-[(benzothiazol-2-yl]- pipéridin-4-yl]méthanamine et 244 mg (5,8 mmoles) de cyanamide finement broyé on ajoute 0,4 ml d'acide acétique et on chauffe le tout à 1400C. Après 5mn le mélange se prend en masse, on ajoute du méthanol et on chauffe au reflux jusqu'à dissolution totale du solide. On évapore le méthanol, on lave le résidu avec du chloroforme, on l'essore, on le reprend avec du méthanol acidifié par de l'éther diéthylique saturé d'acide chlorhydrique, et on évapore les solvants. On recristallise le résidu dans un mélange de méthanol et d'éther diéthylique, et finalement on isole 1,1 g de poudre blanche.
Point de fusion : 283-286 C.
ExemPle 3 (Composé N010).
N-Méthyl-N-[1-[1-(1-méthyléthyl)-1H-benzimidazol-2-yl]pipéri din-4-yl J guanidine, acétate.
On mélange, au bain d'huile à 800C, 10,5 g (38,5 mmoles) de
N-[1-[1-(1-méthyléthyl)-1H-benzimidazol-2-yl]pipéridin-4-yl]méthanamine, on ajoute 2,3 ml d'acide acétique, on agite le mélange pour l'homogénéiser, puis on ajoute 1,8 g (42 mmoles) de cyanamide et on chauffe le mélange à 110 C pendant 30mn.
N-[1-[1-(1-méthyléthyl)-1H-benzimidazol-2-yl]pipéridin-4-yl]méthanamine, on ajoute 2,3 ml d'acide acétique, on agite le mélange pour l'homogénéiser, puis on ajoute 1,8 g (42 mmoles) de cyanamide et on chauffe le mélange à 110 C pendant 30mn.
On ajoute 50 ml de chloroforme pour fluidifier le mélange, on chauffe au reflux pendant quelques minutes, on filtre le mélange chaud, on lave le solide avec du chloroforme et on le sèche. On obtient 11,2 g de produit.
Point de fusion : 2470C.
Exemple 4 (Composé N014)
N-t1-(1-Éthoxycarbonylméthyl-1H-benzimidazol-2-yl)pipéridin- 4-yl J -N-méthylguanidine, acétate.
N-t1-(1-Éthoxycarbonylméthyl-1H-benzimidazol-2-yl)pipéridin- 4-yl J -N-méthylguanidine, acétate.
4.1 2-(8-Aza-1,4-dioxaspiro[4.5]décan-8-yl)-1H-benzimida
zole-1 -acétate d'éthyle.
zole-1 -acétate d'éthyle.
Dans un ballon tricol de 250 ml on introduit 2,4 g (60 mmoles) d'hydrure de sodium à 60% dans de l'huile minérale, puis 50 ml de diméthylformamide, puis 12,9 g de 8-aza-8-(1-H benzimidazol-2-yl-1,4-dioxaspiro[4.5]décane. On attend la fin du dégagement d'hydrogène, on agite le mélange encore pendant 15mn, on le place dans un bain de glace et on ajoute, goutte à goutte, 10 g, soit 6,7 ml (60 mmoles), de bromoacétate d'éthyle.
On agite le mélange pendant 1h à température ambiante, puis on le verse dans de l'eau glacée contenant de l'éther diéthylique, on sépare le précipité par filtration, on le lave à l'eau et à 1'l'éther diéthylique, et on le sèche sous pression réduite. On obtient 12 g de produit.
Point de fusion : 1200C.
4.2 2-(4-Oxopipéridin-1-yl)-1 H-benzimidazole-1-acétate
d'éthyle.
d'éthyle.
On chauffe à 800C pendant 2h un mélange de 1l g (31,8 mmoles) de 2-(8-aza-1,4-dioxaspiro[4.5]décan-8-yl)-1H-benzimidazole- 1-acétate d'éthyle et 110 ml d'acide formique à 98%. On laisse refroidir, on évapore l'acide, on ajoute du carbonate de sodium, et on extrait le mélange trois fois avec de l'éther diéthylique. On lave la phase organique avec une solution aqueuse saturée de chlorure de sodium, on la sèche sur sulfate de magnésium et on évapore le solvant. On obtient 9,5 g de produit.
4.3 2-E 4- (Méthylamino)pipérin-1 -yl J -1 H-benzimidazole-1 -ace-
tate d'éthyle.
tate d'éthyle.
On introduit dans un flacon de Parr 4 g (120 mmoles) de méthylamine et 100 ml d'éthanol, on refroidit le mélange dans un bain de glace, on ajoute 30 g de charbon palladié à 5%, puis 13,8 g (45 mmoles) de 2-(4-oxopipéridin-1-yl)-1H-benzi- midazole-l -acétate a éthyle.
On effectue une hydrogénation pendant 30mn, on filtre le catalyseur, on évapore l'éthanol à température ambiante, on reprend le résidu avec du toluène, qu'on évapore à 500C, on fait cristalliser le résidu dans de l'éther diéthylique et on le recristallise dans un mélange d'alcool isopropylique et d'éther diisopropylique. On obtient 8,5 g de produit.
Point de fusion : 1050C.
4.4 N-[1-(1-nthoxycarbonylméthyl-1H-benzimidazol-2-yl)pipé-
ridin-4-yll-N-méthylguanidine, acétate.
ridin-4-yll-N-méthylguanidine, acétate.
On chauffe à 1100C un mélange de 3,16 g (10 mmoles) de 2-[4-(méthylamino)pipérin-1-yl]-1H-benzimidazole-1-acétate d'éthyle, 0,6 ml d'acide acétique et 0,46 g (11 mmoles) de cyanamide. Le mélange se prend en masse au bout de 30mn et on le triture avec du chloroforme. On le chauffe au reflux pendant 1h, on le filtre, on lave le solide au chloroforme puis à l'éther diéthylique, et on le recristallise dans un mélange d'éthanol et d'éther diéthylique. On obtient 1,5 de produit.
Point de fusion : 204 C.
ExemPle 5 (Composé N015).
N-[1-(1-Carbamoylméthyl-1H-benzimidazol-2-yl)pipéridin-4-yl]
N-méthylguanidine, fumarate.
N-méthylguanidine, fumarate.
On introduit dans un autoclave de 40 ml 0,6 g (1,43 mmoles) d'ac@tate de N-[1-(1-éthoxycarbonylméthyl-1H-benzimidazol2-yl)pipéridin-4-yl]-N-méthylguanidine et 20 ml d'éthanol refroidi et saturé d'ammoniac, et on chauffe le mélange au bain d'huile à 1000C pendant 6h. On évapore l'éthanol, on reprend le résidu avec du méthanol, et on ajoute un équivalent d'acide fumarique et un peu d'éther diéthylique.
On obtient 0,5 g de solide qu'on recristallise dans un mélange de méthanol et d'éther diéthylique, ce qui laisse finalement 0,3 g de produit pur.
Point de fusion : 2560C.
Exemple 6 (Composé N 16).
N-Méthyl-N-[1-(1-phényl-1H-benzimidazol-2-yl)pipéridin-4-yl]guanidine, acétate.
6.1 I -Phényl-1 -H-benzimidazolone.
On chauffe à 1900C , au bain d'huile, un mélange de 18,4 g (100 mmoles) de N-phénylbenzène-1,2-diamine et 7,5 g (125 mmoles) d'urée, pendant 2h30.
En fin de réaction le mélange est solide. On le reprend avec de l'eau et de l'éther diéthylique, on essore le solide, on le lave avec de l'eau et de l'éther diéthylique et on le sèche. On obtient 19,4 g de produit.
Point de fusion : 2040C.
6.2 2-Chloro-1-phénjyl-1-H-benzimidazole.
On chauffe au reflux pendant 3h un mélange de 18,5 g (88 mmoles) de 1-phényl-1-H-benzimidazolone et 130 ml de chlorure de phosphoryle. On évapore le "solvant", on ajoute avec précaution de l'eau au résidu et on le chauffe à 500C jusqu'à destruction du complexe. On ajoute de l'ammoniaque jusqu'à pH=8, on extrait le mélange avec de l'éther diéthylique, on lave la phase organique à l'eau, on évapore le solvant et on purifie le résidu par chromatographie sur colonne de gel de silice en éluant avec du dichlorométhane. On obtient 15 g de produit.
Point de fusion : 700C.
6.3 8-(1-Phényl-1H-benzimidazol-2-yl)-8-aza-1,4-dioxaspi ro(4.SJdêcane.
On chauffe à 130 C , au bain d'huile, un mélange de 6 g (42 mmoles) de 8-aza-1,4-dioxaspiro[4.5]décane et 8 g (35 mmoles) de 2-chloro-1-phényl-1-H-benzimidazole, pendant 2h sous atmosphère d'argon.
On reprende le mélange avec de l'eau et de la soude, on l'extrait avec du dichlorométhane, on sèche la phase aqueuse, on évapore le solvant et on purifie le résidu huileux par chromatographie sur colonne de gel de silice en éluant avec un mélange 97,5/2,5 de dichlorométhane/méthanol. Après recristallisation dans l'éther diéthylique on obtient 7,75 g de produit.
Point de fusion : 1470C.
6.4 1-(1-Phényl-1H-benzimidazol-2-yl)pipéridin-4-one.
On chauffe à 600C pendant 3h un mélange de 11,8 g (35,2 mmoles) de 8(1 -phényl-1 H-benzimidazol-2-yl) -8-aza-1 , 4-dioxa- spiro(4.53décane, 150 ml d'acide acétique et 16 ml d'acide chlorhydrique concentré.
On évapore le mélange sous pression réduite, on ajoute de la soude 2N et de l'éther diéthylique, on filtre le précipité, on le lave à l'eau et à l'éther diéthylique et on le sèche.
On obtient 10,2 g de produit.
Point de fusion : 1570C.
6.5 N-[1-(1-Phényl-1H-benzimidazol-2-yl)pipéridin-4-yl]mé
thanamine.
thanamine.
On introduit dans un flacon de Parr 10 g (34 mmoles) de 1-(1-phényl-1H-benzimidazol-2-yl)pipéridin-4-one, 110 ml de méthanol et 9 g (~300 mmoles) de méthylamine, et on effectue une hydrogénation en présence de 0,5 g de charbon palladié à 10% sous environ 0,28 MPa pendant 3h.
On filtre le catalyseur, on évapore le filtrat et on reprend le résidu avec de l'hexane bouillant. On essore le produit qui cristallise et on le lave à l'hexane. On obtient 9,5 g de produit.
Point de fusion : 1130C.
6.6 N-Méthyl-N-[1-(1-phényl-1H-benzimidazol-2-yl)pipéridin
4-yl Jguanidine, acétate.
4-yl Jguanidine, acétate.
On chauffe à 1300C, au bain d'huile, un mélange de 3,06 g (10 mmoles) de N-[1-(1-phényl-1H-benzimidazol-2-yl)pipéridin 4-yl]méthanamine et 0,6 ml (10 mmoles) d'acide acétique et on ajoute 0,46 g (11 mmoles) de cyanamide. On observe une fusion progressive du mélange et, après 20mn, un produit cristallise. On maintient le chauffage à 1300C pendant 30mn.
On reprend les cristaux avec du chloroforme bouillant pendant 2h, on les essore et on les lave avec du chloroforme. Après séchage on obtient 3,5 g de solide qu'on recristallise dans l'alcool isopropylique. On obtient 2,58 g de produit.
Point de fusion : 2450C.
Exemple 7 (Composé N021).
NS (5-Chlorobenzoxazol-2-yl)pipéridin-4-yl]méthanamine, fumarate.
7.1 5-Chlorobenzoxazole-2-thiol.
On chauffe au reflux pendant 60h un mélange de 50 g (348 mmoles) de 2-amino-4-chlorophénol, 300 ml d'éthanol et 80 ml, soit 100 g (1,32 mole) de disulfure de carbone. On filtre le précipité verdâtre, on le sèche en présence d'anhydride phosphorique, on le recristallise dans 600 ml d'acide acétique, on le lave à l'éther diéthylique et on le sèche. On obtient 29 g de composé.
Point de fusion : 2760C.
7.2 2,5-Dichlorobenzoxazole.
On agite à température ambiante un mélange de 28,14 g (152 mmoles) de 5-chlorobenzoxazole-2-thiol et 250 ml de toluène, et on ajoute, par petites portions, 92,2 g (188 mmoles) de pentachlorure de phosphore. On maintient l'agitation à température ambiante pendant 20mn, puis on chauffe au reflux pendant 2h45. On refroidit le mélange, et on évapore le toluène sous pression réduite. On obtient 31 g de produit qu'on utilise tel quel dans l'étape suivante.
7.3 N-El - ( 5-Chlorobenzoxazol-2-yl ) pipéridin-4-yl Jméthan-
amine, fumarate.
amine, fumarate.
On mélange 2,5 g (13,3 mmoles) de 2,5-dichlorobenzoxazole, 4,5 g (16 mmoles) de dibromhydrate de N-(pipéridin-4-yl)méthanamine et 6,7 g (53,2 mmoles) de carbonate de potassium dans 25 ml de diméthylsulfoxyde, et on chauffe lentement jusqu'à 900C. On laisse refroidir le mélange, devenu noir, on le jette sur de la glace, et on l'extrait trois fois avec de l'éther diéthylique. On lave la phase organique à l'eau, on la sèche sur sulfate de magnésium et on évapore le solvant sous pression réduite. On obtient 2,35 g de résidu cristallisé qu'on purifie par chromatographie sur colonne de gel de silice en éluant avec un mélange de dichlorométhane/ammoniaque 99/1 ; on obtient finalement 1,88 g de solide cristallin.
On prépare le fumarate avec 1,73 g (6,51 mmole) de cette base et 0,756 g (6,51 mmole) d'acide fumarique dans environ 15 ml d'alcool isopropylique et on isole finalement 2,00 g de produit.
Point de fusion : 213,5-215,50C.
Exemple 8 (Composé N027).
N-Méthyl-N-t1-[1-(1-méthyléthyl)-6-nitro-1H-benzimidazol- 2-yl]pipéridin-4-yl]guanidine, dichlorhydrate.
A un mélange de 3,74 g (10 mmoles) d'acétate de N-méthyl
N-[1-[1-(1-méthyléthyl)-1H-benzimidazol-2-yl]pipéridin-4-yl]guanidine et 30 ml d'acide trifluoroacétique on ajoute 10 ml (20 mmoles) d'une solution 2N d'acide nitrique dans l'acide trifluoroacétique, et on chauffe au reflux pendant 4h. On évapore l'acide trifluoroacétique, on reprend le résidu avec de l'éther diéthylique, on le filtre, on lave le solide avec de l'éther diéthylique pour éliminer toute trace d'acide trifluoroacétique, on le reprend avec de l'eau et on prépare le dichlorhydrate en faisant passer la solution sur résine échangeuse d'ions (Cl-). On évapore l'éluat, on recristallise le résidu une fois dans l'éthanol, puis trois fois dans l'eau, et on obtient finalement 0,8 g de dichlorhydrate trihydraté.
N-[1-[1-(1-méthyléthyl)-1H-benzimidazol-2-yl]pipéridin-4-yl]guanidine et 30 ml d'acide trifluoroacétique on ajoute 10 ml (20 mmoles) d'une solution 2N d'acide nitrique dans l'acide trifluoroacétique, et on chauffe au reflux pendant 4h. On évapore l'acide trifluoroacétique, on reprend le résidu avec de l'éther diéthylique, on le filtre, on lave le solide avec de l'éther diéthylique pour éliminer toute trace d'acide trifluoroacétique, on le reprend avec de l'eau et on prépare le dichlorhydrate en faisant passer la solution sur résine échangeuse d'ions (Cl-). On évapore l'éluat, on recristallise le résidu une fois dans l'éthanol, puis trois fois dans l'eau, et on obtient finalement 0,8 g de dichlorhydrate trihydraté.
Point de fusion : 2150C (décomposition).
Exemple 9 (Composé N029).
N-Méthyl-N-[1-[6-amino-1-(1-méthyléthyl)-1H-benzimidazol2-yl]pipéridin-4-yl]guanidine, dichlorhydrate.
On soumet 0,35 g (0,72 mmole) de dichlorhydrate trihydraté de
N-méthyl-N-[1-[1-(1-méthyléthyl)-6-nitro-1H-benzimidazol 2-yllpipéridin-4-yllguanidine à une hydrogénation en présence d'oxyde de platine sous environ 0,35 MPa pendant 1h.
N-méthyl-N-[1-[1-(1-méthyléthyl)-6-nitro-1H-benzimidazol 2-yllpipéridin-4-yllguanidine à une hydrogénation en présence d'oxyde de platine sous environ 0,35 MPa pendant 1h.
On filtre le catalyseur, on évapore le filtrat, et on recristallise le résidu d'abord dans un mélange d'alcool isopropylique et d'éthanol, puis dans l'eau. On obtient 0,12 g de produit.
Point de fusion : 2120C (décomposition).
ExemPle 10 (Composé N031).
N-Méthyl-N-[1-[6-(N'-butyluréido)-1-(1-méthyléthyl)-1H-benzi midazol-2-yl]pipéridin-4-yllguanidine, acétate.
A une solution de 0,39 g (1 mmole) d'acétate de N-méthyl
N-[1-[6-amino-1-(1-méthyléthyl)-1H-benzimidazol-2-yl]pipéridin-4-yl]guanidine dans 4 ml d'eau, à température ambiante et au bain à ultra sons, on ajoute 0,13 ml (1,2 mmole) d'isocyanate de butyle. On laisse réagire le mélange pendant 15mn, puis on évapore le solvant sous pression réduite et on recristallise le résidu dans un mélange d'alcool isopropylique et d'éther diisopropylique. On obtient 0,35 g de produit.
N-[1-[6-amino-1-(1-méthyléthyl)-1H-benzimidazol-2-yl]pipéridin-4-yl]guanidine dans 4 ml d'eau, à température ambiante et au bain à ultra sons, on ajoute 0,13 ml (1,2 mmole) d'isocyanate de butyle. On laisse réagire le mélange pendant 15mn, puis on évapore le solvant sous pression réduite et on recristallise le résidu dans un mélange d'alcool isopropylique et d'éther diisopropylique. On obtient 0,35 g de produit.
Point de fusion : 2340C (décomposition).
Le tableau qui suit illustre les structures chimiques et les propriétés physiques de quelques composés selon l'invention. Tableau


N <SEP> R1 <SEP> R2 <SEP> R3 <SEP> X <SEP> Sel <SEP> ou <SEP> base <SEP> F <SEP> ( C)
<tb> <SEP> 1 <SEP> H <SEP> H <SEP> H <SEP> N-CH(CH3)2 <SEP> 3/2 <SEP> fum <SEP> 196-197
<tb> <SEP> 2 <SEP> H <SEP> H <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 1 <SEP> AcOH <SEP> 217
<tb> <SEP> 3 <SEP> H <SEP> CH3 <SEP> H <SEP> S <SEP> base <SEP> 87
<tb> <SEP> 4 <SEP> H <SEP> CH3 <SEP> H <SEP> N-CH3 <SEP> 1 <SEP> fum <SEP> 194
<tb> <SEP> 5 <SEP> H <SEP> CH3 <SEP> H <SEP> N-CH(CH3)2 <SEP> 1 <SEP> fum <SEP> 190
<tb> <SEP> 6 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> S <SEP> 2 <SEP> HCl <SEP> 283-286
<tb> <SEP> 7 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-H <SEP> 2 <SEP> HCl <SEP> > 320
<tb> <SEP> 8 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH3 <SEP> 1 <SEP> AcOH <SEP> 278
<tb> <SEP> 9 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH2CH3 <SEP> 1 <SEP> AcOH <SEP> 247-249
<tb> 10 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 1 <SEP> AcOH <SEP> 247
<tb> Tableau (suite)

<tb> <SEP> 1 <SEP> H <SEP> H <SEP> H <SEP> N-CH(CH3)2 <SEP> 3/2 <SEP> fum <SEP> 196-197
<tb> <SEP> 2 <SEP> H <SEP> H <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 1 <SEP> AcOH <SEP> 217
<tb> <SEP> 3 <SEP> H <SEP> CH3 <SEP> H <SEP> S <SEP> base <SEP> 87
<tb> <SEP> 4 <SEP> H <SEP> CH3 <SEP> H <SEP> N-CH3 <SEP> 1 <SEP> fum <SEP> 194
<tb> <SEP> 5 <SEP> H <SEP> CH3 <SEP> H <SEP> N-CH(CH3)2 <SEP> 1 <SEP> fum <SEP> 190
<tb> <SEP> 6 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> S <SEP> 2 <SEP> HCl <SEP> 283-286
<tb> <SEP> 7 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-H <SEP> 2 <SEP> HCl <SEP> > 320
<tb> <SEP> 8 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH3 <SEP> 1 <SEP> AcOH <SEP> 278
<tb> <SEP> 9 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH2CH3 <SEP> 1 <SEP> AcOH <SEP> 247-249
<tb> 10 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 1 <SEP> AcOH <SEP> 247
<tb> Tableau (suite)
N <SEP> R1 <SEP> R2 <SEP> R3 <SEP> X <SEP> Sel <SEP> ou <SEP> base <SEP> F <SEP> ( C)
<tb> 11 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH2-CH(CH3)2 <SEP> 2 <SEP> HCl <SEP> 285-290 <SEP> (d)
<tb> 12 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH2-cC6H11 <SEP> 1 <SEP> AcOH <SEP> 242
<tb> 13 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH2CH2-O-CH3 <SEP> 1 <SEP> AcOH <SEP> 218
<tb> 14 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH2-COO-CH2CH3 <SEP> 1 <SEP> AcOH <SEP> 204
<tb> 15 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH2-CO-NH2 <SEP> 1/2 <SEP> fum <SEP> 256
<tb> 16 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-C6H5 <SEP> 1 <SEP> AcOH <SEP> 245
<tb> 17 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-C6H4-4-Cl <SEP> 1 <SEP> AcOH <SEP> 260
<tb> 18 <SEP> H <SEP> nC4H9 <SEP> H <SEP> N-CH(CH3)2 <SEP> 1 <SEP> fum <SEP> 218-220
<tb> 19 <SEP> H <SEP> CH2C6H5 <SEP> H <SEP> N-CH(CH3)2 <SEP> 1 <SEP> fum <SEP> 216-217
<tb> 20 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> O <SEP> 1 <SEP> mal <SEP> 204-206
<tb> 21 <SEP> 5-Cl <SEP> CH3 <SEP> N <SEP> O <SEP> 1 <SEP> fum <SEP> 213,5-215,5
<tb> 22 <SEP> 6-Cl <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 2 <SEP> HCl <SEP> 266 <SEP> (d)
<tb> 23 <SEP> 5-CH3 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 1 <SEP> AcOH <SEP> 231
<tb> 24 <SEP> 6-CH3 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 2 <SEP> HCl <SEP> 267 <SEP> (d)
<tb> 25 <SEP> 5-CF3 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> base <SEP> 270 <SEP> (d)
<tb> 26 <SEP> 5-NO2 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 2 <SEP> HCl, <SEP> 1 <SEP> H2O <SEP> 245 <SEP> (d)
<tb> 27 <SEP> 6-NO2 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 2 <SEP> HCl <SEP> 215 <SEP> (d)
<tb> 28 <SEP> 5-NH2 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 1 <SEP> AcOH <SEP> 226-230 <SEP> (d)
<tb> Tableau (suite et fin)

<tb> 11 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH2-CH(CH3)2 <SEP> 2 <SEP> HCl <SEP> 285-290 <SEP> (d)
<tb> 12 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH2-cC6H11 <SEP> 1 <SEP> AcOH <SEP> 242
<tb> 13 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH2CH2-O-CH3 <SEP> 1 <SEP> AcOH <SEP> 218
<tb> 14 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH2-COO-CH2CH3 <SEP> 1 <SEP> AcOH <SEP> 204
<tb> 15 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH2-CO-NH2 <SEP> 1/2 <SEP> fum <SEP> 256
<tb> 16 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-C6H5 <SEP> 1 <SEP> AcOH <SEP> 245
<tb> 17 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-C6H4-4-Cl <SEP> 1 <SEP> AcOH <SEP> 260
<tb> 18 <SEP> H <SEP> nC4H9 <SEP> H <SEP> N-CH(CH3)2 <SEP> 1 <SEP> fum <SEP> 218-220
<tb> 19 <SEP> H <SEP> CH2C6H5 <SEP> H <SEP> N-CH(CH3)2 <SEP> 1 <SEP> fum <SEP> 216-217
<tb> 20 <SEP> H <SEP> CH3 <SEP> C(-NH2)=NH <SEP> O <SEP> 1 <SEP> mal <SEP> 204-206
<tb> 21 <SEP> 5-Cl <SEP> CH3 <SEP> N <SEP> O <SEP> 1 <SEP> fum <SEP> 213,5-215,5
<tb> 22 <SEP> 6-Cl <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 2 <SEP> HCl <SEP> 266 <SEP> (d)
<tb> 23 <SEP> 5-CH3 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 1 <SEP> AcOH <SEP> 231
<tb> 24 <SEP> 6-CH3 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 2 <SEP> HCl <SEP> 267 <SEP> (d)
<tb> 25 <SEP> 5-CF3 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> base <SEP> 270 <SEP> (d)
<tb> 26 <SEP> 5-NO2 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 2 <SEP> HCl, <SEP> 1 <SEP> H2O <SEP> 245 <SEP> (d)
<tb> 27 <SEP> 6-NO2 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 2 <SEP> HCl <SEP> 215 <SEP> (d)
<tb> 28 <SEP> 5-NH2 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 1 <SEP> AcOH <SEP> 226-230 <SEP> (d)
<tb> Tableau (suite et fin)
N <SEP> R1 <SEP> R2 <SEP> R3 <SEP> X <SEP> Sel <SEP> ou <SEP> base <SEP> F <SEP> ( C)
<tb> 29 <SEP> 6-NH2 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 2 <SEP> HCl <SEP> 212 <SEP> (d)
<tb> 30 <SEP> 6-NHCONH2 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 1 <SEP> AcOH <SEP> 240 <SEP> (d)
<tb> 31 <SEP> 6-NHCONH(CH2)3CH3 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 1 <SEP> AcOH <SEP> 234 <SEP> (d)
<tb> 32 <SEP> 6-NHCONHC6H5 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 1 <SEP> AcOH <SEP> 237 <SEP> (d)
<tb> Légende
Dans la colonne "R1", le nombre placé devant le symbole d'un substituant autre que H indique sa position sur l'hétérocycle.
<tb> 29 <SEP> 6-NH2 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 2 <SEP> HCl <SEP> 212 <SEP> (d)
<tb> 30 <SEP> 6-NHCONH2 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 1 <SEP> AcOH <SEP> 240 <SEP> (d)
<tb> 31 <SEP> 6-NHCONH(CH2)3CH3 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 1 <SEP> AcOH <SEP> 234 <SEP> (d)
<tb> 32 <SEP> 6-NHCONHC6H5 <SEP> CH3 <SEP> C(-NH2)=NH <SEP> N-CH(CH3)2 <SEP> 1 <SEP> AcOH <SEP> 237 <SEP> (d)
<tb> Légende
Dans la colonne "R1", le nombre placé devant le symbole d'un substituant autre que H indique sa position sur l'hétérocycle.
Dans la colonne "R2", nC4H9 désigne un groupe n-butyle et CH2C6H5 désigne un groupe benzyle.
Dans la colonne "X", cC6H11 désigne un groupe cyclohexyle, et C6H5 et C6H4-4-Cl désignent respectivement un groupe phényle et 4-chlorophényle.
Dans la colonne "Sel ou base", "fum" désigne un fumarate, "mal" désigne un maléate, "HCl" désigne un chlorhydrate, "AcOH" désigne un acétate et "ox" désigne un oxalate ; le nombre qui précède chacune de ces abréviations, dans le cas des sels, indique le le nombre de moles d'acide (ou d'eau) pour une mole de base.
Dans la colonne "F( C)", "(d)" indique un point de fusion avec décomposition.
Les composés de l'invention ont fait l'objet d'essais pharmacologiques qui ont montré leur intérêt comme substances actives de médicaments.
Ainsi ils ont été testés quant à leurs effets inhibiteurs de la liaison de la [ [ H]quipazine avec les récepteurs sérotoninergiques de type 5-HT3 présents dans le-cortex cérébral du rat, selon une variante de la méthode décrite par Milburn et
Peroutka (J. Neurochem., 52, 1787-1792, 1989).
Peroutka (J. Neurochem., 52, 1787-1792, 1989).
On utilise des rats mâles Sprague-Dawley, de 150 à 200 g, dans tous les essais. On en prélève le cortex cérébral et on l'homogénéise dans 20 volumes (poids/volume) de tampon Hepes 25 mM ou de tampon Hepes 25 mM contenant du chlorure de sodium (180 mM), du chlorure de calcium (2,5 mM), du chlorure de potassium (5 mM) et du chlorure de magnésium (1,2 mM) (pH=7,4), à l'aide d'un broyeur Polytron@@. Après centrifugation de la suspension pendant 10mn à 45000xg, on remet le culot en suspens ion dans le volume initial de tampon, contenant éventuellement 0,05% de Triton X-100", et on effectue une première incubation de 30mn à 370C. On effectue ensuite encore deux centrifugations comme précédemment décrit, et on reprend le culot final dans 11,7 volumes de tampon Hepes 25 mM à pH=7,4.
On détermine la liaison de la E3HJquipazine (51,6-69,8 Ci/mmole, New'England Nuclear, Boston, Ma, USA) en faisant incuber 150 ul de la suspension membranaire avec le radioligand (0,8 nM) dans un volume final de 1 ml, pendant 30mn à 250C, en l'absence ou en la présence du composé à étudier. L'incubation a lieu en présence de 0,1 WM de paroxétine et de 1 uM de kétansérine. La liaison non spécifique est déterminée en présence de 1 WM d'ondansetron.Après l'incubation, on dilue le mélange testé avec 5 ml de tampon Tris-HCl glacé 50 mM (pH=7,4 à OOC). On collecte les membranes par filtration sur des filtres Whatman GF/Bt prétraités avec 0,05% de polyéthylèneimine, et on les lave avec trois volumes de 5 ml de tampon Tris-HCl glacé 50 mM.
La radioactivité retenue sur les filtres est mesurée par spectrométrie de scintillation liquide à une efficacité de 50 à 60%.
Les résultats s'expriment par la concentration (CI50) de composé étudié qui inhibe 50% de la liaison de la E 3HJquipa- zine, déterminée par une méthode graphique ou mathématique.
Les composés de l'invention les plus actifs dans cet essai se caractérisent par des CI, inférieures à 1 nM (1
Les composés de l'invention ont été également testés quant à leur effet sur le réflexe de Bezold-Jarisch, c'est-à-dire une intense bradycardie, provoqué par injection intraveineuse de sérotonine. Ce reflexe met en jeu la stimulation des récepteurs spécifiques 5-HT3 du nerf vague, ce qui provoque une dépolarisation et donc une sécrétion d'acétylcholine,-qui est le neurotransmetteur vagal naturel.
Les composés de l'invention ont été également testés quant à leur effet sur le réflexe de Bezold-Jarisch, c'est-à-dire une intense bradycardie, provoqué par injection intraveineuse de sérotonine. Ce reflexe met en jeu la stimulation des récepteurs spécifiques 5-HT3 du nerf vague, ce qui provoque une dépolarisation et donc une sécrétion d'acétylcholine,-qui est le neurotransmetteur vagal naturel.
On anesthésie des rats mâles Sprague-Dawley à l'uréthane (1 à 25 g/kg par voie intrapéritonéale), on mesure la pression sanguine grâce à un cathéter placé dans l'artère carotide, et on se sert des impulsions de pression pour activer un cardiotachymètre. Des canules sont placées dans les deux veines fémorales pour faciliter l'administration intraveineuse des produits.
On trace les courbes doses/réponses de la bradycardie provoquée par injection de doses de 30 pg/kg de sérotonine, avant et après l'injection des composés à étudier.
Les composés de l'invention les plus actifs dans cet essai inhibent la bradycardie provoquée par la sérotonine d'au moins 50% à une dose de 10 pg/kg administrée par voie intraveineuse.
Un autre essai in vivo est celui de la vidange gastrique chez le rat, selon un protocole décrit par Scarpignato (J. Pharmacol., (Paris) 14(2), 261-268, 1983).
Les animaux sont des rats CD mâles de 180 à 200 g, à jeun depuis 24h. Les composés à étudier sont administrés par voie intrapéritonéale ou orale, 15 ou 30mn avant l'absorption d'un repas liquide non digestible (méthylcellulose et rouge de phénol). Les animaux sont sacrifiés îOmn après l'administration du repas, et la quantité de rouge de phénol restant dans l'estomac est dosée par spectrophotométrie, un groupe de rats témoins étant sacrifié immédiatement après le repas.
Les composés de l'invention les plus actifs dans cet essai augmentent la vidange gastrique après une dose administrée de 1 mg/kg par voie intrapéritonéale ou orale.
Les composés de l'invention ont également été étudiés quant à leurs effets sur l'émèse chez le furet (mâle, 1 à 1,4 kg), selon une méthode décrite par Costall et al. (Neuropharma cology 25(8), 959-961, 1986).
Les composés à étudier, ou du sérum physiologique, sont administrés par voie intraveineuse (veine jugulaire), sous anesthésie à l'halothane, immédiatement avant une perfusion intraveineuse de cisplatine (10 mg/kg en 1Omn).
On observe ensuite les animaux pendant 3h, en notant le nombre de crises émétiques, le nombre total de spasmes et de vomissements, ainsi que le délai d'apparition de la première crise.
Les composés de l'invention les plus actifs dans cet essai montrent un effet anti-émétique après administration d'une dose inférieure à 5 mg/kg par voie intraveineuse.
Enfin les composés de l'invention ont été étudiés quant à leurs effets sur les récepteurs 5-HT atypiques (5-HT4) dans l'iléon de cobaye, d'après Craig et Clarke, J. Pharm. Exp.
Ther., 252 (3), 1378-1386, (1990).
Des cobayes tricolores Jegard mâles de 300 à 400 g sont assommés et saignés. On prélève rapidement un fragment d'iléon de 3 cm environ, au niveau de la jonction iléocoecale, et on le lave avec 10 ml de tampon Krebs tiédi (composition en mM : NaCl=118 ; CaCl2=2,6 ; KCl=4,9
NaH2PO4=1 ; MgSO4=1,2 ; NaHCO3=25 ; Glucose=11,7). L'iléon est monté sur une pipette de 2 ml et le muscle longitudinal est séparé soigneusement avec un coton dentaire imbibé de tampon
Krebs. L'organe est connecté à un transducteur isométrique sous une tension basale de 0,5 g et maintenu dans un bain de
Krebs à 370C aéré par un courant carbogène. Après un repos d'environ 30mn on applique une stimulation électrique (0,2 Hz ; 1,5 ms ; tension supramaximale s45 V) grâce à des électrodes de champ H.Sachs, modèle F2H reliées à un stimulateur Grass S88n', jusqu'à stabilisation des contrac tions (ou "twitches"). On ajoute alors dans le bain 3-10 M de phénoxybenzamine, ce qui diminue l'amplitude des contractions jusqu'à environ 50% (130mon). L'organe est ensuite lavé six fois à 5mn d'intervalle. Avant le quatrième lavage on ajoute de la sérotonine (3-10 7M). Si nécessaire on diminue l'amplitude des contractions à 50% de l'amplitude supramaximale par diminution de la tension électrique après le sixième lavage. On construit une courbe concentration-effet par additions cumulatives, à lmn d'intervalle, du composé à étudier.
NaH2PO4=1 ; MgSO4=1,2 ; NaHCO3=25 ; Glucose=11,7). L'iléon est monté sur une pipette de 2 ml et le muscle longitudinal est séparé soigneusement avec un coton dentaire imbibé de tampon
Krebs. L'organe est connecté à un transducteur isométrique sous une tension basale de 0,5 g et maintenu dans un bain de
Krebs à 370C aéré par un courant carbogène. Après un repos d'environ 30mn on applique une stimulation électrique (0,2 Hz ; 1,5 ms ; tension supramaximale s45 V) grâce à des électrodes de champ H.Sachs, modèle F2H reliées à un stimulateur Grass S88n', jusqu'à stabilisation des contrac tions (ou "twitches"). On ajoute alors dans le bain 3-10 M de phénoxybenzamine, ce qui diminue l'amplitude des contractions jusqu'à environ 50% (130mon). L'organe est ensuite lavé six fois à 5mn d'intervalle. Avant le quatrième lavage on ajoute de la sérotonine (3-10 7M). Si nécessaire on diminue l'amplitude des contractions à 50% de l'amplitude supramaximale par diminution de la tension électrique après le sixième lavage. On construit une courbe concentration-effet par additions cumulatives, à lmn d'intervalle, du composé à étudier.
Les réponses sont mesurées en termes de capacité à restaurer l'amplitude des contractions au niveau de celle obtenue grâce à la tension supramaximale et après traitement avec la phénoxybenzamine.
Les composés de l'invention se comportent comme des agonistes, agonistes partiels ou antagonistes desdits récepteurs, certains d'entre eux étant actifs à des concentrations inférieures à 10 nM.
Les résultats des essais biologiques montrent que les composés de l'invention sont des ligands des récepteurs de type 5-HT. Comme montré ci-dessus, ils ont en particulier une interaction avec les récepteurs de types 5-HT3 et 5-HT4.
Ils peuvent donc être utilisés pour le traitement et la prévention des désordres dans lesquels les récepteurs 5-HT sont impliqués, tels que nausées et vomissements, par exemple consécutifs à un traitement antitumoral ou à l'administration d'un anesthésique ; troubles du système nerveux central tels que la schizophrénie, la manie, l'anxiété et la dépression troubles de la cognition tels que la démence sénile ou pré- sénile d'Alzheimer, dyskinésie, douleurs, migraines et maux de tête ; troubles de la dépendance ou du sevrage d'alcool ou de drogues ; troubles de la fonction gastrointestinale tels que dyspepsie, ulcère peptique, aigreurs d'estomac, flatulences ; troubles du système cardiovasculaire et troubles respiratoires.
A cet effet ils peuvent être présentés sous toutes formes appropriées à l'administration orale ou parentérale, telles que comprimés, dragées, gélules, capsules, suspensions ou solutions buvables ou injectables, etc, en association avec des excipients convenables, et dosées pour permettre une administration de 0,005 à 5 mg/kg de 1 à 4 fois par jour.
Claims (6)
1. Composés répondant à la formule générale (I) dans laquelle
phényle ou 4-chlorophényle, ainsi que leurs sels d'addition à des acides acceptables en pharmacologie.
méthoxyéthyle, éthoxycarbonylméthyle, carbamoylméthyle,
sente un atome d'hydrogène ou un groupe (C1-C7)alkyle,
un groupe imino de formule N-R4 dans laquelle R4 repré
X représente soit un atome d'oxygène ou de soufre, soit
amidino de formule C(-NH2)=N-H, et
R3 représente soit un atome d'hydrogène, soit un groupe
alkyle ou benzyle,
uréido, N'-(C1-C4)alkyluréido ou N'-phényluréido, Rye. représente un atome d'hydrogène ou un groupe (C1-C4)
groupe (C1-C4)alkyle, trifluorométhyle, nitro, amino,
R1 représente un atome d'hydrogène ou d'halogène ou un
2. Composés selon la revendication 1, caractérisés en ce que
R2 représente un groupe méthyle.
3. Composés selon l'une des revendications 1 et 2, caractérisés en ce que X représente un groupe imino de formule N- (C1-C7) alkyle.
4. Procédé de préparation d'un composé selon la revendication 1, caractérisé en ce qu'on fait réagir un dérivé chloré de formule générale (II) (dans laquelle X et R1 sont tels que définis dans la revendication 1) avec le 8-aza-1,4-dioxa spiro[4.5]décane de formule (III)

pour obtenir l'intermédiaire de formule générale (IV)

qui, lorsque X représente N-H, peut ensuite, si on le désire, être alkylé de manière connue, puis on hydrolyse la fonction acétal en fonction cétone, pour obtenir la cétone de formule générale (v)

puis on fait réagir cette dernière avec une amine de formule générale R2-NH2 (dans laquelle R2 est tel que défini dans la revendication 1) ce qui, après hydrogénation catalytique, donne un composé de formule générale (Ia)

qui correspond à la formule générale (I) lorsque R3 représente l'hydrogène, puis, si l'on désire un composé de formule générale (I) dans laquelle R3 représente un groupe amidino, on fait réagir le composé de formule générale (Ia) avec le cyanamide pour obtenir une guanidine de formule générale (Ib)

5. Médicament caractérisé en ce qu'il contient un composé selon l'un des revendications 1 à 3.
6. Composition pharmaceutique caracérisée en ce qu'elle contient un composé selon l'une des revendications 1 à 3.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9104010A FR2674853B1 (fr) | 1991-04-03 | 1991-04-03 | Derives de piperidinylguanidine, leur preparation et leur application en therapeutique. |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9104010A FR2674853B1 (fr) | 1991-04-03 | 1991-04-03 | Derives de piperidinylguanidine, leur preparation et leur application en therapeutique. |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| FR2674853A1 true FR2674853A1 (fr) | 1992-10-09 |
| FR2674853B1 FR2674853B1 (fr) | 1995-01-20 |
Family
ID=9411396
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| FR9104010A Expired - Fee Related FR2674853B1 (fr) | 1991-04-03 | 1991-04-03 | Derives de piperidinylguanidine, leur preparation et leur application en therapeutique. |
Country Status (1)
| Country | Link |
|---|---|
| FR (1) | FR2674853B1 (fr) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993024117A2 (fr) * | 1992-05-23 | 1993-12-09 | Smithkline Beecham Plc | Medicaments pour le traitement de l'anxiete |
| WO1994000113A2 (fr) * | 1992-06-27 | 1994-01-06 | Smithkline Beecham Plc | Medicaments renfermant des antagonistes du recepteur de 5-ht¿4? |
| WO1997010823A1 (fr) * | 1995-09-18 | 1997-03-27 | Glaxo Group Limited | Antagonistes du recepteur de 5-ht3 contre la dyskinesie |
| FR2773800A1 (fr) * | 1998-01-20 | 1999-07-23 | Synthelabo | Derives de benzimidazole, leur procede de preparation et leur application en therapeutique |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0304353A1 (fr) * | 1987-07-23 | 1989-02-22 | Synthelabo | Dérivés de benzimidazole et leur préparation |
| EP0186087B1 (fr) * | 1984-12-22 | 1989-08-23 | Dr. Karl Thomae GmbH | Tétrahydro-benzothiazoles, leur procédé de préparation et leur utilisation comme intermédiaires ou médicaments |
| EP0356234A2 (fr) * | 1988-08-25 | 1990-02-28 | Fujisawa Pharmaceutical Co., Ltd. | Benzazoles, procédés de préparation et compositions pharmaceutiques les contenant |
| EP0409692A2 (fr) * | 1989-07-13 | 1991-01-23 | Rhone-Poulenc Sante | Dérivés d'imino-2 hétérocyclylalkyl-3 benzothiazoline, leur préparation et les médicaments les contenant |
| US4988699A (en) * | 1989-03-14 | 1991-01-29 | Warner-Lambert Company | Substituted tetrahydrobenzothiazoles as dopaminergic agents |
| EP0445026A1 (fr) * | 1990-02-27 | 1991-09-04 | Adir Et Compagnie | Dérivés d'aminométhyl-pipéridine, leur procÀ©dé de préparation et les compositions pharmaceutiques qui les contiennent |
-
1991
- 1991-04-03 FR FR9104010A patent/FR2674853B1/fr not_active Expired - Fee Related
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0186087B1 (fr) * | 1984-12-22 | 1989-08-23 | Dr. Karl Thomae GmbH | Tétrahydro-benzothiazoles, leur procédé de préparation et leur utilisation comme intermédiaires ou médicaments |
| EP0304353A1 (fr) * | 1987-07-23 | 1989-02-22 | Synthelabo | Dérivés de benzimidazole et leur préparation |
| EP0356234A2 (fr) * | 1988-08-25 | 1990-02-28 | Fujisawa Pharmaceutical Co., Ltd. | Benzazoles, procédés de préparation et compositions pharmaceutiques les contenant |
| US4988699A (en) * | 1989-03-14 | 1991-01-29 | Warner-Lambert Company | Substituted tetrahydrobenzothiazoles as dopaminergic agents |
| EP0409692A2 (fr) * | 1989-07-13 | 1991-01-23 | Rhone-Poulenc Sante | Dérivés d'imino-2 hétérocyclylalkyl-3 benzothiazoline, leur préparation et les médicaments les contenant |
| EP0445026A1 (fr) * | 1990-02-27 | 1991-09-04 | Adir Et Compagnie | Dérivés d'aminométhyl-pipéridine, leur procÀ©dé de préparation et les compositions pharmaceutiques qui les contiennent |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993024117A2 (fr) * | 1992-05-23 | 1993-12-09 | Smithkline Beecham Plc | Medicaments pour le traitement de l'anxiete |
| WO1993024117A3 (fr) * | 1992-05-23 | 1994-04-14 | Smithkline Beecham Plc | Medicaments pour le traitement de l'anxiete |
| US5763459A (en) * | 1992-05-23 | 1998-06-09 | Smithkline Beecham P.L.C. | Medicaments for the treatment of anxiety |
| WO1994000113A2 (fr) * | 1992-06-27 | 1994-01-06 | Smithkline Beecham Plc | Medicaments renfermant des antagonistes du recepteur de 5-ht¿4? |
| WO1994000113A3 (fr) * | 1992-06-27 | 1994-04-14 | Smithkline Beecham Plc | Medicaments renfermant des antagonistes du recepteur de 5-ht¿4? |
| WO1997010823A1 (fr) * | 1995-09-18 | 1997-03-27 | Glaxo Group Limited | Antagonistes du recepteur de 5-ht3 contre la dyskinesie |
| FR2773800A1 (fr) * | 1998-01-20 | 1999-07-23 | Synthelabo | Derives de benzimidazole, leur procede de preparation et leur application en therapeutique |
Also Published As
| Publication number | Publication date |
|---|---|
| FR2674853B1 (fr) | 1995-01-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US4710500A (en) | 1-(4'-fluorophenyl)-3,5-substituted indoles useful in the treatment of psychic disorders and pharmaceutical compositions thereof | |
| US5418241A (en) | Piperidine derivatives, their preparation and their application in therapeutics | |
| EP0507650B1 (fr) | Dérivés de pipéridine, leur préparation et leur application en thérapeutique | |
| KR100787649B1 (ko) | 1-[(벤조이미다졸-1-일)퀴놀린-8-일]피페리딘-4-일아민유도체의 제조 방법 | |
| JPH04506222A (ja) | N−置換複素環誘導体およびその製法 | |
| CZ398191A3 (en) | Novel 4-amino alkyl-2(3h)-indolones and pharmaceutical compositions containing them | |
| DE69028934T2 (de) | Tetrahydrobenzimidazol-Derivate | |
| EP0302788A1 (fr) | Dérivés de [(pipéridinyl-4)méthyl]-2 tétrahydro-1,2,3,4 9H-pyrido[3,4-b]indole, leur préparation et leur application en thérapeutique | |
| EP0306375A1 (fr) | Dérivés de[(pipéridinyl-4)méthyl]-2 tétrahydro-1,2,3,4 isoquinoléine, leur préparation et leur application en thérapeutique | |
| KR20090116716A (ko) | 칸나비노이드-cb1 길항작용과 아세틸콜린에스테라제 억제가 조합된 화합물 | |
| CA2547391C (fr) | Nouveaux derives de benzimidazole et d'imidazo-pyridine et leur utilisation en tant que medicament | |
| CH625518A5 (fr) | ||
| US4977175A (en) | 4,5,6,7-tetrahydrobenzimidazole derivatives as 5HT3 -antagonists | |
| DE69713110T2 (de) | 4-aminoethoxy-indolon-derivate | |
| US6391896B1 (en) | 3-Tetrahydropyridin-4-yl indoles for treatment of psychotic disorders | |
| FR2674853A1 (fr) | Derives de piperidinylguanidine, leur preparation et leur application en therapeutique. | |
| US5096900A (en) | (4-piperidyl)methyl-2,3-dihydro-1h-isoindole and -2,3,4,5-tetrahydro-1h-benzazepine derivatives, their preparation and their application in therapy | |
| CA2317515A1 (fr) | Derives d'oxazol utilises comme agonistes du recepteur 1a de la serotonine | |
| JP3588363B2 (ja) | キノキサリンジオン類 | |
| FR2618436A1 (fr) | Derives de piperidine, leur preparation et leur application en therapeutique | |
| US5081128A (en) | 2,3-dihydro-1h-isoindole derivatives and their application in therapy | |
| FR2499991A1 (fr) | Nouveaux 2,1,3-benzothiadiazoles et 2,1,3-benzoxadiazoles, leur preparation et leur application comme medicaments | |
| EA001658B1 (ru) | Хиноксалиндионы | |
| AU722616B2 (en) | 5-aminoalkoxy-1,4-dihydroquinoxaline-2,3-diones being dopamine agonists | |
| FR2773800A1 (fr) | Derives de benzimidazole, leur procede de preparation et leur application en therapeutique |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ST | Notification of lapse |