ES2947741T3 - Preparación y uso de un barredor de especies reactivas de oxígeno - Google Patents
Preparación y uso de un barredor de especies reactivas de oxígeno Download PDFInfo
- Publication number
- ES2947741T3 ES2947741T3 ES21212401T ES21212401T ES2947741T3 ES 2947741 T3 ES2947741 T3 ES 2947741T3 ES 21212401 T ES21212401 T ES 21212401T ES 21212401 T ES21212401 T ES 21212401T ES 2947741 T3 ES2947741 T3 ES 2947741T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- mmol
- amino
- benzyl
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000002360 preparation method Methods 0.000 title description 16
- 239000003642 reactive oxygen metabolite Substances 0.000 title description 11
- 239000002516 radical scavenger Substances 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 444
- 238000000034 method Methods 0.000 claims abstract description 55
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 54
- 150000003839 salts Chemical class 0.000 claims abstract description 51
- 201000010099 disease Diseases 0.000 claims abstract description 35
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 claims description 205
- -1 methyl (2S)-2-{[(2S)-1-[(2S,3E,5S)-2-benzyl-5-{[ (tert-butoxy)carbonyl]amino}-7-methyloct-3-enoyl]pyrrolidin-2-yl]formamido}-5-({[(1-oxyl-2,2,6,6-tetramethylpiperidin-4-yl )oxy]carbonyl}amino)pentanoate Chemical compound 0.000 claims description 138
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 112
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 69
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 claims description 48
- 125000004485 2-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])([H])C1([H])* 0.000 claims description 46
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 38
- 230000004806 ferroptosis Effects 0.000 claims description 29
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 25
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 23
- RUZLIIJDZBWWSA-INIZCTEOSA-N methyl 2-[[(1s)-1-(7-methyl-2-morpholin-4-yl-4-oxopyrido[1,2-a]pyrimidin-9-yl)ethyl]amino]benzoate Chemical group COC(=O)C1=CC=CC=C1N[C@@H](C)C1=CC(C)=CN2C(=O)C=C(N3CCOCC3)N=C12 RUZLIIJDZBWWSA-INIZCTEOSA-N 0.000 claims description 21
- 229910052799 carbon Inorganic materials 0.000 claims description 18
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 18
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 17
- KAESVJOAVNADME-UHFFFAOYSA-N 1H-pyrrole Natural products C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 claims description 16
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 claims description 16
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 claims description 13
- 230000002401 inhibitory effect Effects 0.000 claims description 5
- 230000005764 inhibitory process Effects 0.000 claims description 5
- WWZKQHOCKIZLMA-UHFFFAOYSA-N Caprylic acid Natural products CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 claims description 3
- GONOPSZTUGRENK-UHFFFAOYSA-N benzyl(trichloro)silane Chemical compound Cl[Si](Cl)(Cl)CC1=CC=CC=C1 GONOPSZTUGRENK-UHFFFAOYSA-N 0.000 claims description 3
- HAYGHUSWXCEQRP-MPPAPVJUSA-N (2S)-2-[[(2S)-1-[(E,2S,5S)-2-benzyl-7-methyl-5-[(2-methylpropan-2-yl)oxycarbonylamino]oct-3-enoyl]pyrrolidine-2-carbonyl]amino]-5-[(1-hydroxy-2,2,6,6-tetramethylpiperidin-4-yl)oxycarbonylamino]pentanoic acid Chemical compound CC(C)C[C@@H](/C=C/[C@H](CC1=CC=CC=C1)C(=O)N2CCC[C@H]2C(=O)N[C@@H](CCCNC(=O)OC3CC(N(C(C3)(C)C)O)(C)C)C(=O)O)NC(=O)OC(C)(C)C HAYGHUSWXCEQRP-MPPAPVJUSA-N 0.000 claims 2
- 101100063435 Caenorhabditis elegans din-1 gene Proteins 0.000 claims 1
- 239000000203 mixture Substances 0.000 abstract description 137
- 238000011282 treatment Methods 0.000 abstract description 20
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 abstract description 16
- 208000024891 symptom Diseases 0.000 abstract description 13
- 230000036542 oxidative stress Effects 0.000 abstract description 4
- 206010012559 Developmental delay Diseases 0.000 abstract description 2
- 230000001629 suppression Effects 0.000 abstract 1
- 239000000243 solution Substances 0.000 description 330
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 273
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 219
- 238000003786 synthesis reaction Methods 0.000 description 198
- 230000015572 biosynthetic process Effects 0.000 description 195
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 153
- 235000019439 ethyl acetate Nutrition 0.000 description 136
- 239000011541 reaction mixture Substances 0.000 description 125
- 239000000047 product Substances 0.000 description 110
- 239000002904 solvent Substances 0.000 description 101
- 239000007787 solid Substances 0.000 description 100
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 97
- 229920006395 saturated elastomer Polymers 0.000 description 95
- 239000011734 sodium Substances 0.000 description 74
- 239000012044 organic layer Substances 0.000 description 72
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 64
- 239000012074 organic phase Substances 0.000 description 62
- 239000012267 brine Substances 0.000 description 61
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 61
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 53
- 239000008346 aqueous phase Substances 0.000 description 45
- 238000002953 preparative HPLC Methods 0.000 description 45
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 43
- 239000000460 chlorine Substances 0.000 description 41
- 239000008194 pharmaceutical composition Substances 0.000 description 41
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 39
- 239000006260 foam Substances 0.000 description 39
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 38
- 238000006243 chemical reaction Methods 0.000 description 34
- 239000003480 eluent Substances 0.000 description 32
- 239000003208 petroleum Substances 0.000 description 32
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 31
- 239000000546 pharmaceutical excipient Substances 0.000 description 29
- 238000003756 stirring Methods 0.000 description 27
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 23
- 210000004027 cell Anatomy 0.000 description 23
- 239000012043 crude product Substances 0.000 description 23
- 239000000126 substance Substances 0.000 description 22
- 238000010898 silica gel chromatography Methods 0.000 description 21
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 20
- 239000004480 active ingredient Substances 0.000 description 20
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 20
- 239000007832 Na2SO4 Substances 0.000 description 19
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 19
- 235000019270 ammonium chloride Nutrition 0.000 description 19
- 208000035475 disorder Diseases 0.000 description 19
- 229910052938 sodium sulfate Inorganic materials 0.000 description 19
- 235000011152 sodium sulphate Nutrition 0.000 description 19
- 125000003118 aryl group Chemical group 0.000 description 18
- 239000002552 dosage form Substances 0.000 description 18
- 239000000843 powder Substances 0.000 description 18
- WKTRRGAEDXTUJJ-UHFFFAOYSA-N (1,3-dioxoisoindol-2-yl) (1-hydroxy-2,2,6,6-tetramethylpiperidin-4-yl) carbonate Chemical compound C(ON1C(C2=CC=CC=C2C1=O)=O)(OC1CC(N(C(C1)(C)C)O)(C)C)=O WKTRRGAEDXTUJJ-UHFFFAOYSA-N 0.000 description 17
- 125000000217 alkyl group Chemical group 0.000 description 17
- 239000007864 aqueous solution Substances 0.000 description 17
- 239000007821 HATU Substances 0.000 description 16
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 16
- 238000005160 1H NMR spectroscopy Methods 0.000 description 15
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 15
- 125000000753 cycloalkyl group Chemical group 0.000 description 15
- 239000000741 silica gel Substances 0.000 description 15
- 229910002027 silica gel Inorganic materials 0.000 description 15
- 239000012453 solvate Substances 0.000 description 15
- 239000002253 acid Substances 0.000 description 14
- 125000001072 heteroaryl group Chemical group 0.000 description 14
- 239000010410 layer Substances 0.000 description 14
- 238000000746 purification Methods 0.000 description 14
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 13
- 238000004440 column chromatography Methods 0.000 description 13
- 239000003814 drug Substances 0.000 description 13
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 13
- 239000003921 oil Substances 0.000 description 13
- 235000019198 oils Nutrition 0.000 description 13
- 230000002829 reductive effect Effects 0.000 description 13
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 12
- 239000003153 chemical reaction reagent Substances 0.000 description 12
- 125000001424 substituent group Chemical group 0.000 description 12
- 239000000725 suspension Substances 0.000 description 12
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 11
- 125000004429 atom Chemical group 0.000 description 11
- 238000005859 coupling reaction Methods 0.000 description 11
- 238000009472 formulation Methods 0.000 description 11
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 11
- 239000004615 ingredient Substances 0.000 description 11
- 239000003826 tablet Substances 0.000 description 11
- GCTFTMWXZFLTRR-GFCCVEGCSA-N (2r)-2-amino-n-[3-(difluoromethoxy)-4-(1,3-oxazol-5-yl)phenyl]-4-methylpentanamide Chemical compound FC(F)OC1=CC(NC(=O)[C@H](N)CC(C)C)=CC=C1C1=CN=CO1 GCTFTMWXZFLTRR-GFCCVEGCSA-N 0.000 description 10
- XEVOHWHMATVWAF-LLSOJIOMSA-N (e,2s,5s)-2-benzyl-7-methyl-5-[(2-methylpropan-2-yl)oxycarbonylamino]oct-3-enoic acid Chemical compound CC(C)(C)OC(=O)N[C@@H](CC(C)C)\C=C\[C@@H](C(O)=O)CC1=CC=CC=C1 XEVOHWHMATVWAF-LLSOJIOMSA-N 0.000 description 10
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 10
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 10
- UKAWXALBBYVEGN-UHFFFAOYSA-N carbamic acid;dihydrochloride Chemical compound Cl.Cl.NC(O)=O UKAWXALBBYVEGN-UHFFFAOYSA-N 0.000 description 10
- 239000003795 chemical substances by application Substances 0.000 description 10
- 229940125900 compound 59 Drugs 0.000 description 10
- 239000013058 crude material Substances 0.000 description 10
- 238000004128 high performance liquid chromatography Methods 0.000 description 10
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 239000002585 base Substances 0.000 description 9
- 229920001577 copolymer Polymers 0.000 description 9
- 239000007788 liquid Substances 0.000 description 9
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 9
- 239000003755 preservative agent Substances 0.000 description 9
- 238000012360 testing method Methods 0.000 description 9
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 9
- OSFQDWVAUXECFW-PJNFCNLCSA-N (1-hydroxy-2,2,6,6-tetramethylpiperidin-4-yl) (2S)-2-[[(2S)-1-[(E,2S,5S)-2-benzyl-7-methyl-5-[(2-methylpropan-2-yl)oxycarbonylamino]oct-3-enoyl]pyrrolidine-2-carbonyl]amino]-5-(phenylmethoxycarbonylamino)pentanoate Chemical compound C(C1=CC=CC=C1)[C@H](C(=O)N1[C@@H](CCC1)C(=O)N[C@H](C(=O)OC1CC(N(C(C1)(C)C)O)(C)C)CCCNC(=O)OCC1=CC=CC=C1)\C=C\[C@H](CC(C)C)NC(=O)OC(C)(C)C OSFQDWVAUXECFW-PJNFCNLCSA-N 0.000 description 8
- 208000009304 Acute Kidney Injury Diseases 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 241000124008 Mammalia Species 0.000 description 8
- 238000005481 NMR spectroscopy Methods 0.000 description 8
- 208000033626 Renal failure acute Diseases 0.000 description 8
- 201000011040 acute kidney failure Diseases 0.000 description 8
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 8
- 229940079593 drug Drugs 0.000 description 8
- 230000000694 effects Effects 0.000 description 8
- 229910052757 nitrogen Inorganic materials 0.000 description 8
- 229910052708 sodium Inorganic materials 0.000 description 8
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 7
- JVVRCYWZTJLJSG-UHFFFAOYSA-N 4-dimethylaminophenol Chemical compound CN(C)C1=CC=C(O)C=C1 JVVRCYWZTJLJSG-UHFFFAOYSA-N 0.000 description 7
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-dimethylaminopyridine Substances CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 7
- XLCDKRLVOCOUMK-NRFANRHFSA-N 9H-fluoren-9-ylmethyl N-[(4S)-4-amino-5-oxo-5-(propan-2-ylamino)pentyl]carbamate Chemical compound N[C@@H](CCCNC(OCC1C2=CC=CC=C2C=2C=CC=CC1=2)=O)C(=O)NC(C)C XLCDKRLVOCOUMK-NRFANRHFSA-N 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 7
- 230000000670 limiting effect Effects 0.000 description 7
- 238000007911 parenteral administration Methods 0.000 description 7
- 230000001225 therapeutic effect Effects 0.000 description 7
- 239000003981 vehicle Substances 0.000 description 7
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 6
- 239000004698 Polyethylene Substances 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 6
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 6
- 239000000443 aerosol Substances 0.000 description 6
- 150000001412 amines Chemical class 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 239000002775 capsule Substances 0.000 description 6
- 239000003995 emulsifying agent Substances 0.000 description 6
- 239000000839 emulsion Substances 0.000 description 6
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 238000012986 modification Methods 0.000 description 6
- 230000004048 modification Effects 0.000 description 6
- 229910052760 oxygen Inorganic materials 0.000 description 6
- SXXCSLRNTOLYJK-AJYKVAJBSA-N propan-2-yl (2S)-2-[[(2S)-1-[(E,2S,5S)-2-benzyl-7-methyl-5-[(2-methylpropan-2-yl)oxycarbonylamino]oct-3-enoyl]pyrrolidine-2-carbonyl]amino]-5-(9H-fluoren-9-ylmethoxycarbonylamino)pentanoate Chemical compound C1=CC=CC=2C3=CC=CC=C3C(C1=2)COC(=O)NCCC[C@@H](C(=O)OC(C)C)NC(=O)[C@H]1N(CCC1)C([C@H](\C=C\[C@H](CC(C)C)NC(=O)OC(C)(C)C)CC1=CC=CC=C1)=O SXXCSLRNTOLYJK-AJYKVAJBSA-N 0.000 description 6
- 235000017550 sodium carbonate Nutrition 0.000 description 6
- 229910000029 sodium carbonate Inorganic materials 0.000 description 6
- 239000000375 suspending agent Substances 0.000 description 6
- BHHMFVASLNUQAJ-FAJYYVQYSA-N (2S)-1-[(E,2S,5S)-5-amino-2-benzyl-7-methyloct-3-enoyl]-N-[(2S)-5-(diaminomethylideneamino)-1-[(1-hydroxy-2,2,6,6-tetramethylpiperidin-4-yl)amino]-1-oxopentan-2-yl]pyrrolidine-2-carboxamide Chemical compound CC(C)C[C@H](N)\C=C\[C@H](CC1=CC=CC=C1)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCCNC(N)=N)C(=O)NC1CC(C)(C)N(O)C(C)(C)C1 BHHMFVASLNUQAJ-FAJYYVQYSA-N 0.000 description 5
- ZQEBQGAAWMOMAI-ZETCQYMHSA-N (2s)-1-[(2-methylpropan-2-yl)oxycarbonyl]pyrrolidine-2-carboxylic acid Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(O)=O ZQEBQGAAWMOMAI-ZETCQYMHSA-N 0.000 description 5
- 229920000858 Cyclodextrin Polymers 0.000 description 5
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 5
- 239000005977 Ethylene Substances 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 239000003937 drug carrier Substances 0.000 description 5
- 239000012156 elution solvent Substances 0.000 description 5
- 239000000796 flavoring agent Substances 0.000 description 5
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 5
- 229910052739 hydrogen Inorganic materials 0.000 description 5
- 239000000543 intermediate Substances 0.000 description 5
- 208000028867 ischemia Diseases 0.000 description 5
- 239000008297 liquid dosage form Substances 0.000 description 5
- 238000004519 manufacturing process Methods 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 230000002503 metabolic effect Effects 0.000 description 5
- 239000002207 metabolite Substances 0.000 description 5
- 239000002674 ointment Substances 0.000 description 5
- 229920000573 polyethylene Polymers 0.000 description 5
- 229920001223 polyethylene glycol Polymers 0.000 description 5
- 125000001500 prolyl group Chemical group [H]N1C([H])(C(=O)[*])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- 241000894007 species Species 0.000 description 5
- 238000004809 thin layer chromatography Methods 0.000 description 5
- KVCNMRFEXCVPHA-SFHVURJKSA-N 9H-fluoren-9-ylmethyl N-[(4S)-4,5-diamino-5-oxopentyl]carbamate Chemical compound N[C@@H](CCCNC(OCC1C2=CC=CC=C2C=2C=CC=CC1=2)=O)C(=O)N KVCNMRFEXCVPHA-SFHVURJKSA-N 0.000 description 4
- YJEMZAJZUDAHDC-QFIPXVFZSA-N 9H-fluoren-9-ylmethyl N-[(4S)-4-amino-5-(diethylamino)-5-oxopentyl]carbamate Chemical compound N[C@@H](CCCNC(OCC1C2=CC=CC=C2C=2C=CC=CC1=2)=O)C(=O)N(CC)CC YJEMZAJZUDAHDC-QFIPXVFZSA-N 0.000 description 4
- SUOYTCYRJFVEEK-VWLOTQADSA-N 9H-fluoren-9-ylmethyl N-[(4S)-4-amino-5-[(1-hydroxy-2,2,6,6-tetramethylpiperidin-4-yl)amino]-5-oxopentyl]carbamate Chemical compound N[C@@H](CCCNC(OCC1C2=CC=CC=C2C=2C=CC=CC1=2)=O)C(=O)NC1CC(N(C(C1)(C)C)O)(C)C SUOYTCYRJFVEEK-VWLOTQADSA-N 0.000 description 4
- WRRFBYDIVATCNS-UIOOFZCWSA-N 9H-fluoren-9-ylmethyl N-[(4S)-5-(diethylamino)-5-oxo-4-[[(2S)-pyrrolidine-2-carbonyl]amino]pentyl]carbamate Chemical compound C(C)N(C([C@H](CCCNC(OCC1C2=CC=CC=C2C=2C=CC=CC1=2)=O)NC(=O)[C@H]1NCCC1)=O)CC WRRFBYDIVATCNS-UIOOFZCWSA-N 0.000 description 4
- KOAFUIIQDBGLSM-VMPREFPWSA-N 9H-fluoren-9-ylmethyl N-[(4S)-5-[(1-hydroxy-2,2,6,6-tetramethylpiperidin-4-yl)amino]-5-oxo-4-[[(2S)-pyrrolidine-2-carbonyl]amino]pentyl]carbamate Chemical compound ON1C(CC(CC1(C)C)NC([C@H](CCCNC(OCC1C2=CC=CC=C2C=2C=CC=CC1=2)=O)NC(=O)[C@H]1NCCC1)=O)(C)C KOAFUIIQDBGLSM-VMPREFPWSA-N 0.000 description 4
- JIRBSFRNHRJLPI-QFIPXVFZSA-N 9H-fluoren-9-ylmethyl N-[(5S)-5-amino-6-oxo-6-(propan-2-ylamino)hexyl]carbamate Chemical compound N[C@@H](CCCCNC(OCC1C2=CC=CC=C2C=2C=CC=CC1=2)=O)C(=O)NC(C)C JIRBSFRNHRJLPI-QFIPXVFZSA-N 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 4
- 229910019142 PO4 Inorganic materials 0.000 description 4
- 239000002202 Polyethylene glycol Substances 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 4
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 125000002877 alkyl aryl group Chemical group 0.000 description 4
- 125000005119 alkyl cycloalkyl group Chemical group 0.000 description 4
- 125000005213 alkyl heteroaryl group Chemical group 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 239000003963 antioxidant agent Substances 0.000 description 4
- 235000006708 antioxidants Nutrition 0.000 description 4
- 235000010323 ascorbic acid Nutrition 0.000 description 4
- 229910052801 chlorine Inorganic materials 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- SWFKHPJXIUXVIL-VWLOTQADSA-N cyclohexyl (2S)-2-amino-6-(9H-fluoren-9-ylmethoxycarbonylamino)hexanoate Chemical compound C1=CC=CC=2C3=CC=CC=C3C(C1=2)COC(=O)NCCCC[C@@H](C(=O)OC1CCCCC1)N SWFKHPJXIUXVIL-VWLOTQADSA-N 0.000 description 4
- ZCNIGBRPLUULRN-NDEPHWFRSA-N cyclohexyl (2S)-6-(9H-fluoren-9-ylmethoxycarbonylamino)-2-[(2-methylpropan-2-yl)oxycarbonylamino]hexanoate Chemical compound CC(C)(C)OC(=O)N[C@@H](CCCCNC(=O)OCC1C2=CC=CC=C2C2=C1C=CC=C2)C(=O)OC1CCCCC1 ZCNIGBRPLUULRN-NDEPHWFRSA-N 0.000 description 4
- DPPGGXZJRQERHQ-WDYNHAJCSA-N cyclohexyl (2S)-6-(9H-fluoren-9-ylmethoxycarbonylamino)-2-[[(2R)-pyrrolidine-2-carbonyl]amino]hexanoate Chemical compound C1=CC=CC=2C3=CC=CC=C3C(C1=2)COC(=O)NCCCC[C@@H](C(=O)OC1CCCCC1)NC(=O)[C@@H]1NCCC1 DPPGGXZJRQERHQ-WDYNHAJCSA-N 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 239000002270 dispersing agent Substances 0.000 description 4
- 206010015037 epilepsy Diseases 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 125000001153 fluoro group Chemical group F* 0.000 description 4
- 229930195712 glutamate Natural products 0.000 description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 4
- 210000003734 kidney Anatomy 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- DBXHGXUJUYEEFZ-GOTSBHOMSA-N methyl (2S)-2-[[(2S)-2-amino-3-methylbutanoyl]amino]-5-(9H-fluoren-9-ylmethoxycarbonylamino)pentanoate Chemical compound COC(=O)[C@H](CCCNC(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2)NC(=O)[C@@H](N)C(C)C DBXHGXUJUYEEFZ-GOTSBHOMSA-N 0.000 description 4
- HCYJCOUPSQCMJK-UIOOFZCWSA-N methyl (2S)-5-(9H-fluoren-9-ylmethoxycarbonylamino)-2-[[(2S)-3-methyl-2-[(2-methylpropan-2-yl)oxycarbonylamino]butanoyl]amino]pentanoate Chemical compound C1=CC=CC=2C3=CC=CC=C3C(C1=2)COC(=O)NCCC[C@@H](C(=O)OC)NC([C@H](C(C)C)NC(=O)OC(C)(C)C)=O HCYJCOUPSQCMJK-UIOOFZCWSA-N 0.000 description 4
- RQDDIHNPUSEQFX-ZDUSSCGKSA-N methyl (2s)-2-amino-6-(phenylmethoxycarbonylamino)hexanoate Chemical compound COC(=O)[C@@H](N)CCCCNC(=O)OCC1=CC=CC=C1 RQDDIHNPUSEQFX-ZDUSSCGKSA-N 0.000 description 4
- 239000001301 oxygen Substances 0.000 description 4
- 235000021317 phosphate Nutrition 0.000 description 4
- 229920000139 polyethylene terephthalate Polymers 0.000 description 4
- 239000005020 polyethylene terephthalate Substances 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- 230000002335 preservative effect Effects 0.000 description 4
- 108090000765 processed proteins & peptides Proteins 0.000 description 4
- MPFIJCLCSOHUIB-NRFANRHFSA-N propan-2-yl (2S)-2-amino-5-(9H-fluoren-9-ylmethoxycarbonylamino)pentanoate Chemical compound C1=CC=CC=2C3=CC=CC=C3C(C1=2)COC(=O)NCCC[C@@H](C(=O)OC(C)C)N MPFIJCLCSOHUIB-NRFANRHFSA-N 0.000 description 4
- GQVWALDVBZTWEX-DQEYMECFSA-N propan-2-yl (2S)-5-(9H-fluoren-9-ylmethoxycarbonylamino)-2-[[(2S)-pyrrolidine-2-carbonyl]amino]pentanoate Chemical compound C1=CC=CC=2C3=CC=CC=C3C(C1=2)COC(=O)NCCC[C@@H](C(=O)OC(C)C)NC(=O)[C@H]1NCCC1 GQVWALDVBZTWEX-DQEYMECFSA-N 0.000 description 4
- 125000006239 protecting group Chemical group 0.000 description 4
- 230000010410 reperfusion Effects 0.000 description 4
- 239000008299 semisolid dosage form Substances 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 229910052717 sulfur Inorganic materials 0.000 description 4
- 239000006188 syrup Substances 0.000 description 4
- 235000020357 syrup Nutrition 0.000 description 4
- HNTZJMNMSUVVPQ-NSOVKSMOSA-N tert-butyl (2S)-2-[[(2S)-5-(9H-fluoren-9-ylmethoxycarbonylamino)-1-oxo-1-(propan-2-ylamino)pentan-2-yl]carbamoyl]pyrrolidine-1-carboxylate Chemical compound C1=CC=CC=2C3=CC=CC=C3C(C1=2)COC(=O)NCCC[C@@H](C(=O)NC(C)C)NC(=O)[C@H]1N(CCC1)C(=O)OC(C)(C)C HNTZJMNMSUVVPQ-NSOVKSMOSA-N 0.000 description 4
- MIOIBDAQYVAFLO-DEOSSOPVSA-N tert-butyl N-[(2S)-5-(9H-fluoren-9-ylmethoxycarbonylamino)-1-oxo-1-(propan-2-ylamino)pentan-2-yl]carbamate Chemical compound CC(C)NC(=O)[C@H](CCCNC(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2)NC(=O)OC(C)(C)C MIOIBDAQYVAFLO-DEOSSOPVSA-N 0.000 description 4
- 239000002562 thickening agent Substances 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- 238000012546 transfer Methods 0.000 description 4
- 239000004474 valine Substances 0.000 description 4
- 235000014393 valine Nutrition 0.000 description 4
- 239000004475 Arginine Substances 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 3
- 108010092160 Dactinomycin Proteins 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 3
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 3
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 3
- 239000004472 Lysine Substances 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- 108010033024 Phospholipid Hydroperoxide Glutathione Peroxidase Proteins 0.000 description 3
- 102100023410 Phospholipid hydroperoxide glutathione peroxidase Human genes 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- VDQKIDYOPUMJGQ-VQPCLXHQSA-N XJB-5-131 Chemical compound C([C@@H](/C=C/[C@@H](NC(=O)OC(C)(C)C)CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(=O)OCC=1C=CC=CC=1)C(=O)NC1CC(C)(C)N([O])C(C)(C)C1)C1=CC=CC=C1 VDQKIDYOPUMJGQ-VQPCLXHQSA-N 0.000 description 3
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 3
- 239000002671 adjuvant Substances 0.000 description 3
- 125000005466 alkylenyl group Chemical group 0.000 description 3
- 239000004599 antimicrobial Substances 0.000 description 3
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 3
- 125000003710 aryl alkyl group Chemical group 0.000 description 3
- 239000011668 ascorbic acid Substances 0.000 description 3
- 230000004071 biological effect Effects 0.000 description 3
- 239000012472 biological sample Substances 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 239000006172 buffering agent Substances 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 229920002678 cellulose Polymers 0.000 description 3
- 239000001913 cellulose Substances 0.000 description 3
- 239000002738 chelating agent Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000003086 colorant Substances 0.000 description 3
- 230000002301 combined effect Effects 0.000 description 3
- 239000006071 cream Substances 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 3
- 229960000640 dactinomycin Drugs 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- BFXLJWUGRPGMFU-UHFFFAOYSA-N dipropoxyphosphinothioyl n,n-diethylcarbamodithioate;sulfane Chemical compound S.CCCOP(=S)(OCCC)SC(=S)N(CC)CC BFXLJWUGRPGMFU-UHFFFAOYSA-N 0.000 description 3
- 239000008298 dragée Substances 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 230000008030 elimination Effects 0.000 description 3
- 238000003379 elimination reaction Methods 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- BKQFRNYHFIQEKN-UHFFFAOYSA-N erastin Chemical compound CCOC1=CC=CC=C1N1C(=O)C2=CC=CC=C2N=C1C(C)N1CCN(C(=O)COC=2C=CC(Cl)=CC=2)CC1 BKQFRNYHFIQEKN-UHFFFAOYSA-N 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 239000007941 film coated tablet Substances 0.000 description 3
- 235000013355 food flavoring agent Nutrition 0.000 description 3
- 235000003599 food sweetener Nutrition 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 229960003180 glutathione Drugs 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 125000001188 haloalkyl group Chemical group 0.000 description 3
- 150000004677 hydrates Chemical class 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 3
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 230000001965 increasing effect Effects 0.000 description 3
- 238000001802 infusion Methods 0.000 description 3
- 239000003112 inhibitor Substances 0.000 description 3
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 3
- 239000006210 lotion Substances 0.000 description 3
- 239000007937 lozenge Substances 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- QXVPHKSOBCWCBY-DTWKUNHWSA-N methyl (2R)-3-methyl-2-[[(2S)-pyrrolidine-2-carbonyl]amino]butanoate Chemical compound COC(=O)[C@H](NC(=O)[C@@H]1CCCN1)C(C)C QXVPHKSOBCWCBY-DTWKUNHWSA-N 0.000 description 3
- REYNUXDAVXHOHP-UHFFFAOYSA-N methyl 2-amino-5-(9H-fluoren-9-ylmethoxycarbonylamino)pentanoate Chemical compound COC(C(CCCNC(=O)OCC1C2=CC=CC=C2C2=CC=CC=C12)N)=O REYNUXDAVXHOHP-UHFFFAOYSA-N 0.000 description 3
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 3
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 125000003367 polycyclic group Chemical group 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 239000000651 prodrug Substances 0.000 description 3
- 229940002612 prodrug Drugs 0.000 description 3
- 239000003380 propellant Substances 0.000 description 3
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 238000012216 screening Methods 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 239000003381 stabilizer Substances 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 239000003765 sweetening agent Substances 0.000 description 3
- 238000007910 systemic administration Methods 0.000 description 3
- WRRRFHUTOXKMRF-AMVUTOCUSA-N tert-butyl (2S)-2-[[5-(9H-fluoren-9-ylmethoxycarbonylamino)-1-methoxy-1-oxopentan-2-yl]carbamoyl]pyrrolidine-1-carboxylate Chemical compound COC(=O)C(CCCNC(=O)OCC1C2=CC=CC=C2C2=C1C=CC=C2)NC(=O)[C@@H]1CCCN1C(=O)OC(C)(C)C WRRRFHUTOXKMRF-AMVUTOCUSA-N 0.000 description 3
- PYLXNPWBGFPCAN-VWLOTQADSA-N tert-butyl N-[(2S)-6-(9H-fluoren-9-ylmethoxycarbonylamino)-1-oxo-1-(propan-2-ylamino)hexan-2-yl]carbamate Chemical compound C(C)(C)NC([C@H](CCCCNC(OCC1C2=CC=CC=C2C=2C=CC=CC1=2)=O)NC(OC(C)(C)C)=O)=O PYLXNPWBGFPCAN-VWLOTQADSA-N 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- 238000011200 topical administration Methods 0.000 description 3
- 239000000080 wetting agent Substances 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 2
- 125000000027 (C1-C10) alkoxy group Chemical group 0.000 description 2
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 2
- 125000006727 (C1-C6) alkenyl group Chemical group 0.000 description 2
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 2
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 description 2
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 2
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 2
- LVGUZGTVOIAKKC-UHFFFAOYSA-N 1,1,1,2-tetrafluoroethane Chemical compound FCC(F)(F)F LVGUZGTVOIAKKC-UHFFFAOYSA-N 0.000 description 2
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 2
- KKHFRAFPESRGGD-UHFFFAOYSA-N 1,3-dimethyl-7-[3-(n-methylanilino)propyl]purine-2,6-dione Chemical compound C1=NC=2N(C)C(=O)N(C)C(=O)C=2N1CCCN(C)C1=CC=CC=C1 KKHFRAFPESRGGD-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- DFRAKBCRUYUFNT-UHFFFAOYSA-N 3,8-dicyclohexyl-2,4,7,9-tetrahydro-[1,3]oxazino[5,6-h][1,3]benzoxazine Chemical compound C1CCCCC1N1CC(C=CC2=C3OCN(C2)C2CCCCC2)=C3OC1 DFRAKBCRUYUFNT-UHFFFAOYSA-N 0.000 description 2
- GSDQYSSLIKJJOG-UHFFFAOYSA-N 4-chloro-2-(3-chloroanilino)benzoic acid Chemical compound OC(=O)C1=CC=C(Cl)C=C1NC1=CC=CC(Cl)=C1 GSDQYSSLIKJJOG-UHFFFAOYSA-N 0.000 description 2
- UZFMOKQJFYMBGY-UHFFFAOYSA-N 4-hydroxy-TEMPO Chemical compound CC1(C)CC(O)CC(C)(C)N1[O] UZFMOKQJFYMBGY-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 2
- JQUCWIWWWKZNCS-LESHARBVSA-N C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F Chemical compound C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F JQUCWIWWWKZNCS-LESHARBVSA-N 0.000 description 2
- SCJNYBYSTCRPAO-LXBQGUBHSA-N CN(C)C\C=C\C(=O)NC1=CC=C(N=C1)C(=O)N[C@@]1(C)CCC[C@H](C1)NC1=NC(C2=CNC3=CC=CC=C23)=C(Cl)C=N1 Chemical compound CN(C)C\C=C\C(=O)NC1=CC=C(N=C1)C(=O)N[C@@]1(C)CCC[C@H](C1)NC1=NC(C2=CNC3=CC=CC=C23)=C(Cl)C=N1 SCJNYBYSTCRPAO-LXBQGUBHSA-N 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- 241000282693 Cercopithecidae Species 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 2
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 2
- 238000004566 IR spectroscopy Methods 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 2
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 2
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 2
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 2
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 2
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 2
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 2
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 2
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 2
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- 235000019483 Peanut oil Nutrition 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- ZTHYODDOHIVTJV-UHFFFAOYSA-N Propyl gallate Chemical compound CCCOC(=O)C1=CC(O)=C(O)C(O)=C1 ZTHYODDOHIVTJV-UHFFFAOYSA-N 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 229910006124 SOCl2 Inorganic materials 0.000 description 2
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 2
- 229920001800 Shellac Polymers 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 2
- 239000004473 Threonine Substances 0.000 description 2
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 2
- XTXRWKRVRITETP-UHFFFAOYSA-N Vinyl acetate Chemical compound CC(=O)OC=C XTXRWKRVRITETP-UHFFFAOYSA-N 0.000 description 2
- 150000001241 acetals Chemical class 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 235000004279 alanine Nutrition 0.000 description 2
- 150000001447 alkali salts Chemical class 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 125000004103 aminoalkyl group Chemical group 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- MWPLVEDNUUSJAV-UHFFFAOYSA-N anthracene Chemical compound C1=CC=CC2=CC3=CC=CC=C3C=C21 MWPLVEDNUUSJAV-UHFFFAOYSA-N 0.000 description 2
- 238000003782 apoptosis assay Methods 0.000 description 2
- 230000006907 apoptotic process Effects 0.000 description 2
- 239000008135 aqueous vehicle Substances 0.000 description 2
- 229960005070 ascorbic acid Drugs 0.000 description 2
- 235000009582 asparagine Nutrition 0.000 description 2
- 229960001230 asparagine Drugs 0.000 description 2
- 235000003704 aspartic acid Nutrition 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- OTKPPUXRIADSGD-PPRNARJGSA-N avoparcina Chemical compound O([C@@H]1C2=CC=C(C(=C2)Cl)OC=2C=C3C=C(C=2O[C@H]2C([C@@H](O)[C@H](O)[C@@H](CO)O2)O[C@@H]2O[C@@H](C)[C@H](O)[C@H](N)C2)OC2=CC=C(C=C2)[C@@H](O)[C@H](C(N[C@H](C(=O)N[C@H]3C(=O)N[C@H]2C(=O)N[C@@H]1C(N[C@@H](C1=CC(O)=CC(O)=C1C=1C(O)=CC=C2C=1)C(O)=O)=O)C=1C=CC(O)=CC=1)=O)NC(=O)[C@H](NC)C=1C=CC(O[C@H]2[C@@H]([C@H](O)[C@@H](O)[C@H](C)O2)O)=CC=1)[C@H]1C[C@@H](N)[C@@H](O)[C@H](C)O1 OTKPPUXRIADSGD-PPRNARJGSA-N 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000005540 biological transmission Effects 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- 150000001649 bromium compounds Chemical class 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 239000004359 castor oil Substances 0.000 description 2
- 235000019438 castor oil Nutrition 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 229910052729 chemical element Inorganic materials 0.000 description 2
- RNFNDJAIBTYOQL-UHFFFAOYSA-N chloral hydrate Chemical compound OC(O)C(Cl)(Cl)Cl RNFNDJAIBTYOQL-UHFFFAOYSA-N 0.000 description 2
- 229960002327 chloral hydrate Drugs 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 239000008139 complexing agent Substances 0.000 description 2
- 230000008094 contradictory effect Effects 0.000 description 2
- 238000013270 controlled release Methods 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 230000000875 corresponding effect Effects 0.000 description 2
- 239000006184 cosolvent Substances 0.000 description 2
- DDRJAANPRJIHGJ-UHFFFAOYSA-N creatinine Chemical compound CN1CC(=O)NC1=N DDRJAANPRJIHGJ-UHFFFAOYSA-N 0.000 description 2
- 235000018417 cysteine Nutrition 0.000 description 2
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 229910052805 deuterium Inorganic materials 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- RWRIWBAIICGTTQ-UHFFFAOYSA-N difluoromethane Chemical compound FCF RWRIWBAIICGTTQ-UHFFFAOYSA-N 0.000 description 2
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 235000013870 dimethyl polysiloxane Nutrition 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 208000011325 dry age related macular degeneration Diseases 0.000 description 2
- 239000002662 enteric coated tablet Substances 0.000 description 2
- UJHBVMHOBZBWMX-UHFFFAOYSA-N ferrostatin-1 Chemical compound NC1=CC(C(=O)OCC)=CC=C1NC1CCCCC1 UJHBVMHOBZBWMX-UHFFFAOYSA-N 0.000 description 2
- 239000007888 film coating Substances 0.000 description 2
- 238000009501 film coating Methods 0.000 description 2
- 235000019634 flavors Nutrition 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 230000004907 flux Effects 0.000 description 2
- 238000001640 fractional crystallisation Methods 0.000 description 2
- 239000003349 gelling agent Substances 0.000 description 2
- 235000013922 glutamic acid Nutrition 0.000 description 2
- 239000004220 glutamic acid Substances 0.000 description 2
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 2
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 125000004438 haloalkoxy group Chemical group 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 2
- 150000002430 hydrocarbons Chemical group 0.000 description 2
- 239000000017 hydrogel Substances 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 150000004694 iodide salts Chemical class 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- NNPPMTNAJDCUHE-UHFFFAOYSA-N isobutane Chemical compound CC(C)C NNPPMTNAJDCUHE-UHFFFAOYSA-N 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 2
- 229960000310 isoleucine Drugs 0.000 description 2
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 2
- 239000007951 isotonicity adjuster Substances 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000007942 layered tablet Substances 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 239000003589 local anesthetic agent Substances 0.000 description 2
- 229960005015 local anesthetics Drugs 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 230000000873 masking effect Effects 0.000 description 2
- 238000004949 mass spectrometry Methods 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- AFCCDDWKHLHPDF-UHFFFAOYSA-M metam-sodium Chemical compound [Na+].CNC([S-])=S AFCCDDWKHLHPDF-UHFFFAOYSA-M 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- 239000000693 micelle Substances 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 239000004005 microsphere Substances 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- 230000006654 negative regulation of apoptotic process Effects 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 150000002894 organic compounds Chemical class 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- 239000000312 peanut oil Substances 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- ZQBAKBUEJOMQEX-UHFFFAOYSA-N phenyl salicylate Chemical compound OC1=CC=CC=C1C(=O)OC1=CC=CC=C1 ZQBAKBUEJOMQEX-UHFFFAOYSA-N 0.000 description 2
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 2
- 239000008055 phosphate buffer solution Substances 0.000 description 2
- 229920000435 poly(dimethylsiloxane) Polymers 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 230000003449 preventive effect Effects 0.000 description 2
- 235000018102 proteins Nutrition 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000003352 sequestering agent Substances 0.000 description 2
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 2
- 239000004208 shellac Substances 0.000 description 2
- 229940113147 shellac Drugs 0.000 description 2
- 235000013874 shellac Nutrition 0.000 description 2
- 229920002379 silicone rubber Polymers 0.000 description 2
- 238000002603 single-photon emission computed tomography Methods 0.000 description 2
- 210000003491 skin Anatomy 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 229940001584 sodium metabisulfite Drugs 0.000 description 2
- 235000010262 sodium metabisulphite Nutrition 0.000 description 2
- 239000008259 solid foam Substances 0.000 description 2
- 239000012265 solid product Substances 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 235000010356 sorbitol Nutrition 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 229960005322 streptomycin Drugs 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- 239000007940 sugar coated tablet Substances 0.000 description 2
- 238000009495 sugar coating Methods 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 238000001356 surgical procedure Methods 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- 230000008719 thickening Effects 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 125000001425 triazolyl group Chemical group 0.000 description 2
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical class CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- DVGQZTCCDCQQDH-UHFFFAOYSA-N (1-hydroxy-2,2,6,6-tetramethylpiperidin-4-yl) carbonochloridate Chemical compound C(OC1CC(N(C(C1)(C)C)O)(C)C)(=O)Cl DVGQZTCCDCQQDH-UHFFFAOYSA-N 0.000 description 1
- ZQEBQGAAWMOMAI-SSDOTTSWSA-N (2r)-1-[(2-methylpropan-2-yl)oxycarbonyl]pyrrolidine-2-carboxylic acid Chemical compound CC(C)(C)OC(=O)N1CCC[C@@H]1C(O)=O ZQEBQGAAWMOMAI-SSDOTTSWSA-N 0.000 description 1
- VJOBYERMPPUUOX-QMMMGPOBSA-N (2s)-2-(butoxycarbonylamino)-3-methylbutanoic acid Chemical compound CCCCOC(=O)N[C@@H](C(C)C)C(O)=O VJOBYERMPPUUOX-QMMMGPOBSA-N 0.000 description 1
- AOUOVFRSCMDPFA-QSDJMHMYSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-amino-3-carboxypropanoyl]amino]-4-carboxybutanoyl]amino]-3-methylbutanoyl]amino]butanedioic acid Chemical group OC(=O)C[C@@H](C(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](N)CC(O)=O AOUOVFRSCMDPFA-QSDJMHMYSA-N 0.000 description 1
- STPKWKPURVSAJF-LJEWAXOPSA-N (4r,5r)-5-[4-[[4-(1-aza-4-azoniabicyclo[2.2.2]octan-4-ylmethyl)phenyl]methoxy]phenyl]-3,3-dibutyl-7-(dimethylamino)-1,1-dioxo-4,5-dihydro-2h-1$l^{6}-benzothiepin-4-ol Chemical compound O[C@H]1C(CCCC)(CCCC)CS(=O)(=O)C2=CC=C(N(C)C)C=C2[C@H]1C(C=C1)=CC=C1OCC(C=C1)=CC=C1C[N+]1(CC2)CCN2CC1 STPKWKPURVSAJF-LJEWAXOPSA-N 0.000 description 1
- VUEGYUOUAAVYAS-JGGQBBKZSA-N (6ar,9s,10ar)-9-(dimethylsulfamoylamino)-7-methyl-6,6a,8,9,10,10a-hexahydro-4h-indolo[4,3-fg]quinoline Chemical compound C1=CC([C@H]2C[C@@H](CN(C)[C@@H]2C2)NS(=O)(=O)N(C)C)=C3C2=CNC3=C1 VUEGYUOUAAVYAS-JGGQBBKZSA-N 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- ICLYJLBTOGPLMC-KVVVOXFISA-N (z)-octadec-9-enoate;tris(2-hydroxyethyl)azanium Chemical compound OCCN(CCO)CCO.CCCCCCCC\C=C/CCCCCCCC(O)=O ICLYJLBTOGPLMC-KVVVOXFISA-N 0.000 description 1
- NPNPZTNLOVBDOC-UHFFFAOYSA-N 1,1-difluoroethane Chemical compound CC(F)F NPNPZTNLOVBDOC-UHFFFAOYSA-N 0.000 description 1
- QXOGPTXQGKQSJT-UHFFFAOYSA-N 1-amino-4-[4-(3,4-dimethylphenyl)sulfanylanilino]-9,10-dioxoanthracene-2-sulfonic acid Chemical compound Cc1ccc(Sc2ccc(Nc3cc(c(N)c4C(=O)c5ccccc5C(=O)c34)S(O)(=O)=O)cc2)cc1C QXOGPTXQGKQSJT-UHFFFAOYSA-N 0.000 description 1
- VUQPJRPDRDVQMN-UHFFFAOYSA-N 1-chlorooctadecane Chemical class CCCCCCCCCCCCCCCCCCCl VUQPJRPDRDVQMN-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- 125000004793 2,2,2-trifluoroethoxy group Chemical group FC(CO*)(F)F 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 description 1
- OEPOKWHJYJXUGD-UHFFFAOYSA-N 2-(3-phenylmethoxyphenyl)-1,3-thiazole-4-carbaldehyde Chemical compound O=CC1=CSC(C=2C=C(OCC=3C=CC=CC=3)C=CC=2)=N1 OEPOKWHJYJXUGD-UHFFFAOYSA-N 0.000 description 1
- QTMAZYGAVHCKKX-UHFFFAOYSA-N 2-[(4-amino-5-bromopyrrolo[2,3-d]pyrimidin-7-yl)methoxy]propane-1,3-diol Chemical compound NC1=NC=NC2=C1C(Br)=CN2COC(CO)CO QTMAZYGAVHCKKX-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- MDFXJBQEWLCGHP-MFOYZWKCSA-N 2-[2-[(z)-(pyridine-4-carbonylhydrazinylidene)methyl]phenoxy]acetic acid Chemical compound OC(=O)COC1=CC=CC=C1\C=N/NC(=O)C1=CC=NC=C1 MDFXJBQEWLCGHP-MFOYZWKCSA-N 0.000 description 1
- PAYROHWFGZADBR-UHFFFAOYSA-N 2-[[4-amino-5-(5-iodo-4-methoxy-2-propan-2-ylphenoxy)pyrimidin-2-yl]amino]propane-1,3-diol Chemical compound C1=C(I)C(OC)=CC(C(C)C)=C1OC1=CN=C(NC(CO)CO)N=C1N PAYROHWFGZADBR-UHFFFAOYSA-N 0.000 description 1
- 125000000022 2-aminoethyl group Chemical group [H]C([*])([H])C([H])([H])N([H])[H] 0.000 description 1
- CFWRDBDJAOHXSH-SECBINFHSA-N 2-azaniumylethyl [(2r)-2,3-diacetyloxypropyl] phosphate Chemical compound CC(=O)OC[C@@H](OC(C)=O)COP(O)(=O)OCCN CFWRDBDJAOHXSH-SECBINFHSA-N 0.000 description 1
- 125000004777 2-fluoroethyl group Chemical group [H]C([H])(F)C([H])([H])* 0.000 description 1
- 125000004918 2-methyl-2-pentyl group Chemical group CC(C)(CCC)* 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- WMPPDTMATNBGJN-UHFFFAOYSA-N 2-phenylethylbromide Chemical class BrCCC1=CC=CC=C1 WMPPDTMATNBGJN-UHFFFAOYSA-N 0.000 description 1
- ODJQKYXPKWQWNK-UHFFFAOYSA-N 3,3'-Thiobispropanoic acid Chemical compound OC(=O)CCSCCC(O)=O ODJQKYXPKWQWNK-UHFFFAOYSA-N 0.000 description 1
- TZZDVPMABRWKIZ-MFTLXVFQSA-N 3-[6-[4-[[1-[4-[(1R,2S)-6-hydroxy-2-phenyl-1,2,3,4-tetrahydronaphthalen-1-yl]phenyl]piperidin-4-yl]methyl]piperazin-1-yl]-3-oxo-1H-isoindol-2-yl]piperidine-2,6-dione Chemical compound OC=1C=C2CC[C@@H]([C@@H](C2=CC=1)C1=CC=C(C=C1)N1CCC(CC1)CN1CCN(CC1)C=1C=C2CN(C(C2=CC=1)=O)C1C(NC(CC1)=O)=O)C1=CC=CC=C1 TZZDVPMABRWKIZ-MFTLXVFQSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- MJKVTPMWOKAVMS-UHFFFAOYSA-N 3-hydroxy-1-benzopyran-2-one Chemical class C1=CC=C2OC(=O)C(O)=CC2=C1 MJKVTPMWOKAVMS-UHFFFAOYSA-N 0.000 description 1
- 125000004919 3-methyl-2-pentyl group Chemical group CC(C(C)*)CC 0.000 description 1
- NZAQRZWBQUIBSF-UHFFFAOYSA-N 4-(4-sulfobutoxy)butane-1-sulfonic acid Chemical compound OS(=O)(=O)CCCCOCCCCS(O)(=O)=O NZAQRZWBQUIBSF-UHFFFAOYSA-N 0.000 description 1
- MPMKMQHJHDHPBE-RUZDIDTESA-N 4-[[(2r)-1-(1-benzothiophene-3-carbonyl)-2-methylazetidine-2-carbonyl]-[(3-chlorophenyl)methyl]amino]butanoic acid Chemical compound O=C([C@@]1(N(CC1)C(=O)C=1C2=CC=CC=C2SC=1)C)N(CCCC(O)=O)CC1=CC=CC(Cl)=C1 MPMKMQHJHDHPBE-RUZDIDTESA-N 0.000 description 1
- XUXUHDYTLNCYQQ-UHFFFAOYSA-N 4-amino-TEMPO Chemical compound CC1(C)CC(N)CC(C)(C)N1[O] XUXUHDYTLNCYQQ-UHFFFAOYSA-N 0.000 description 1
- 125000004920 4-methyl-2-pentyl group Chemical group CC(CC(C)*)C 0.000 description 1
- 125000006163 5-membered heteroaryl group Chemical group 0.000 description 1
- GDUANFXPOZTYKS-UHFFFAOYSA-N 6-bromo-8-[(2,6-difluoro-4-methoxybenzoyl)amino]-4-oxochromene-2-carboxylic acid Chemical compound FC1=CC(OC)=CC(F)=C1C(=O)NC1=CC(Br)=CC2=C1OC(C(O)=O)=CC2=O GDUANFXPOZTYKS-UHFFFAOYSA-N 0.000 description 1
- IRBAWVGZNJIROV-SFHVURJKSA-N 9-(2-cyclopropylethynyl)-2-[[(2s)-1,4-dioxan-2-yl]methoxy]-6,7-dihydropyrimido[6,1-a]isoquinolin-4-one Chemical compound C1=C2C3=CC=C(C#CC4CC4)C=C3CCN2C(=O)N=C1OC[C@@H]1COCCO1 IRBAWVGZNJIROV-SFHVURJKSA-N 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- 229920001450 Alpha-Cyclodextrin Polymers 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 102000003669 Antiporters Human genes 0.000 description 1
- 108090000084 Antiporters Proteins 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- IYHHRZBKXXKDDY-UHFFFAOYSA-N BI-605906 Chemical compound N=1C=2SC(C(N)=O)=C(N)C=2C(C(F)(F)CC)=CC=1N1CCC(S(C)(=O)=O)CC1 IYHHRZBKXXKDDY-UHFFFAOYSA-N 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- PKMUHQIDVVOXHQ-HXUWFJFHSA-N C[C@H](C1=CC(C2=CC=C(CNC3CCCC3)S2)=CC=C1)NC(C1=C(C)C=CC(NC2CNC2)=C1)=O Chemical compound C[C@H](C1=CC(C2=CC=C(CNC3CCCC3)S2)=CC=C1)NC(C1=C(C)C=CC(NC2CNC2)=C1)=O PKMUHQIDVVOXHQ-HXUWFJFHSA-N 0.000 description 1
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 206010007559 Cardiac failure congestive Diseases 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 102000003952 Caspase 3 Human genes 0.000 description 1
- 108090000397 Caspase 3 Proteins 0.000 description 1
- 102000004041 Caspase 7 Human genes 0.000 description 1
- 108090000567 Caspase 7 Proteins 0.000 description 1
- 102000011727 Caspases Human genes 0.000 description 1
- 108010076667 Caspases Proteins 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 238000003734 CellTiter-Glo Luminescent Cell Viability Assay Methods 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 239000004709 Chlorinated polyethylene Substances 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- DSLZVSRJTYRBFB-LLEIAEIESA-N D-glucaric acid Chemical compound OC(=O)[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O DSLZVSRJTYRBFB-LLEIAEIESA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical class C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- 239000001692 EU approved anti-caking agent Substances 0.000 description 1
- 239000006145 Eagle's minimal essential medium Substances 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- IMROMDMJAWUWLK-UHFFFAOYSA-N Ethenol Chemical compound OC=C IMROMDMJAWUWLK-UHFFFAOYSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 108010024636 Glutathione Proteins 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- 206010019196 Head injury Diseases 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- 244000043261 Hevea brasiliensis Species 0.000 description 1
- 241001272567 Hominoidea Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical class Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 208000031226 Hyperlipidaemia Diseases 0.000 description 1
- 206010058222 Hypertensive cardiomyopathy Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- 150000000994 L-ascorbates Chemical class 0.000 description 1
- LEVWYRKDKASIDU-IMJSIDKUSA-N L-cystine Chemical compound [O-]C(=O)[C@@H]([NH3+])CSSC[C@H]([NH3+])C([O-])=O LEVWYRKDKASIDU-IMJSIDKUSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 239000004166 Lanolin Chemical class 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 241000238367 Mya arenaria Species 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- LVDRREOUMKACNJ-BKMJKUGQSA-N N-[(2R,3S)-2-(4-chlorophenyl)-1-(1,4-dimethyl-2-oxoquinolin-7-yl)-6-oxopiperidin-3-yl]-2-methylpropane-1-sulfonamide Chemical compound CC(C)CS(=O)(=O)N[C@H]1CCC(=O)N([C@@H]1c1ccc(Cl)cc1)c1ccc2c(C)cc(=O)n(C)c2c1 LVDRREOUMKACNJ-BKMJKUGQSA-N 0.000 description 1
- AVYVHIKSFXVDBG-UHFFFAOYSA-N N-benzyl-N-hydroxy-2,2-dimethylbutanamide Chemical compound C(C1=CC=CC=C1)N(C(C(CC)(C)C)=O)O AVYVHIKSFXVDBG-UHFFFAOYSA-N 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- QOVYHDHLFPKQQG-NDEPHWFRSA-N N[C@@H](CCC(=O)N1CCC(CC1)NC1=C2C=CC=CC2=NC(NCC2=CN(CCCNCCCNC3CCCCC3)N=N2)=N1)C(O)=O Chemical compound N[C@@H](CCC(=O)N1CCC(CC1)NC1=C2C=CC=CC2=NC(NCC2=CN(CCCNCCCNC3CCCCC3)N=N2)=N1)C(O)=O QOVYHDHLFPKQQG-NDEPHWFRSA-N 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 206010033546 Pallor Diseases 0.000 description 1
- 241000282579 Pan Species 0.000 description 1
- 239000005662 Paraffin oil Substances 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 229920012485 Plasticized Polyvinyl chloride Polymers 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000005062 Polybutadiene Substances 0.000 description 1
- 229920002367 Polyisobutene Polymers 0.000 description 1
- 108010039918 Polylysine Proteins 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- HCBIBCJNVBAKAB-UHFFFAOYSA-N Procaine hydrochloride Chemical compound Cl.CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 HCBIBCJNVBAKAB-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- 208000019155 Radiation injury Diseases 0.000 description 1
- 206010063897 Renal ischaemia Diseases 0.000 description 1
- 206010039020 Rhabdomyolysis Diseases 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 235000019485 Safflower oil Nutrition 0.000 description 1
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- 239000003490 Thiodipropionic acid Substances 0.000 description 1
- BZHJMEDXRYGGRV-UHFFFAOYSA-N Vinyl chloride Chemical compound ClC=C BZHJMEDXRYGGRV-UHFFFAOYSA-N 0.000 description 1
- 229930003427 Vitamin E Natural products 0.000 description 1
- DRBWRJPFNOBNIO-KOLCDFICSA-N [(2r)-1-[(2r)-2-(pyridine-4-carbonylamino)propanoyl]pyrrolidin-2-yl]boronic acid Chemical compound N([C@H](C)C(=O)N1[C@@H](CCC1)B(O)O)C(=O)C1=CC=NC=C1 DRBWRJPFNOBNIO-KOLCDFICSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- SPEUIVXLLWOEMJ-UHFFFAOYSA-N acetaldehyde dimethyl acetal Natural products COC(C)OC SPEUIVXLLWOEMJ-UHFFFAOYSA-N 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 150000008043 acidic salts Chemical class 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- HFHDHCJBZVLPGP-RWMJIURBSA-N alpha-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO HFHDHCJBZVLPGP-RWMJIURBSA-N 0.000 description 1
- 229940043377 alpha-cyclodextrin Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical class [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 1
- 230000000843 anti-fungal effect Effects 0.000 description 1
- 239000000729 antidote Substances 0.000 description 1
- 229940075522 antidotes Drugs 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 125000002102 aryl alkyloxo group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 230000003385 bacteriostatic effect Effects 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Chemical class 0.000 description 1
- 229940092738 beeswax Drugs 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical class OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229960001950 benzethonium chloride Drugs 0.000 description 1
- UREZNYTWGJKWBI-UHFFFAOYSA-M benzethonium chloride Chemical compound [Cl-].C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OCCOCC[N+](C)(C)CC1=CC=CC=C1 UREZNYTWGJKWBI-UHFFFAOYSA-M 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004601 benzofurazanyl group Chemical group N1=C2C(=NO1)C(=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- WHGYBXFWUBPSRW-FOUAGVGXSA-N beta-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO WHGYBXFWUBPSRW-FOUAGVGXSA-N 0.000 description 1
- 229960004853 betadex Drugs 0.000 description 1
- 230000002146 bilateral effect Effects 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- PFYXSUNOLOJMDX-UHFFFAOYSA-N bis(2,5-dioxopyrrolidin-1-yl) carbonate Chemical compound O=C1CCC(=O)N1OC(=O)ON1C(=O)CCC1=O PFYXSUNOLOJMDX-UHFFFAOYSA-N 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 150000001642 boronic acid derivatives Chemical class 0.000 description 1
- 125000005998 bromoethyl group Chemical group 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- 229940057971 butane Drugs 0.000 description 1
- 150000004648 butanoic acid derivatives Chemical class 0.000 description 1
- 125000004106 butoxy group Chemical group [*]OC([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 229920005549 butyl rubber Polymers 0.000 description 1
- 235000019282 butylated hydroxyanisole Nutrition 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 235000010410 calcium alginate Nutrition 0.000 description 1
- 239000000648 calcium alginate Substances 0.000 description 1
- 229960002681 calcium alginate Drugs 0.000 description 1
- OKHHGHGGPDJQHR-YMOPUZKJSA-L calcium;(2s,3s,4s,5s,6r)-6-[(2r,3s,4r,5s,6r)-2-carboxy-6-[(2r,3s,4r,5s,6r)-2-carboxylato-4,5,6-trihydroxyoxan-3-yl]oxy-4,5-dihydroxyoxan-3-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylate Chemical compound [Ca+2].O[C@@H]1[C@H](O)[C@H](O)O[C@@H](C([O-])=O)[C@H]1O[C@H]1[C@@H](O)[C@@H](O)[C@H](O[C@H]2[C@H]([C@@H](O)[C@H](O)[C@H](O2)C([O-])=O)O)[C@H](C(O)=O)O1 OKHHGHGGPDJQHR-YMOPUZKJSA-L 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical class C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- XNRHTMDHGDWBGP-UHFFFAOYSA-N carbamic acid;hydrochloride Chemical compound Cl.NC(O)=O XNRHTMDHGDWBGP-UHFFFAOYSA-N 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 150000001722 carbon compounds Chemical class 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 229940096529 carboxypolymethylene Drugs 0.000 description 1
- 238000003570 cell viability assay Methods 0.000 description 1
- 238000012054 celltiter-glo Methods 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 229940082500 cetostearyl alcohol Drugs 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 239000007910 chewable tablet Substances 0.000 description 1
- 238000004296 chiral HPLC Methods 0.000 description 1
- 239000012069 chiral reagent Substances 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- HRYZWHHZPQKTII-UHFFFAOYSA-N chloroethane Chemical class CCCl HRYZWHHZPQKTII-UHFFFAOYSA-N 0.000 description 1
- 125000002603 chloroethyl group Chemical group [H]C([*])([H])C([H])([H])Cl 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 208000010877 cognitive disease Diseases 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 230000009918 complex formation Effects 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 229940126179 compound 72 Drugs 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 229940109239 creatinine Drugs 0.000 description 1
- 150000001896 cresols Chemical class 0.000 description 1
- 239000002577 cryoprotective agent Substances 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000000000 cycloalkoxy group Chemical group 0.000 description 1
- 229940097362 cyclodextrins Drugs 0.000 description 1
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 1
- 229960003067 cystine Drugs 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000011033 desalting Methods 0.000 description 1
- 239000008355 dextrose injection Substances 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 150000008050 dialkyl sulfates Chemical class 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- UMNKXPULIDJLSU-UHFFFAOYSA-N dichlorofluoromethane Chemical compound FC(Cl)Cl UMNKXPULIDJLSU-UHFFFAOYSA-N 0.000 description 1
- 229940099364 dichlorofluoromethane Drugs 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- DENRZWYUOJLTMF-UHFFFAOYSA-N diethyl sulfate Chemical class CCOS(=O)(=O)OCC DENRZWYUOJLTMF-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- WBZKQQHYRPRKNJ-UHFFFAOYSA-L disulfite Chemical compound [O-]S(=O)S([O-])(=O)=O WBZKQQHYRPRKNJ-UHFFFAOYSA-L 0.000 description 1
- 239000012990 dithiocarbamate Substances 0.000 description 1
- 150000004659 dithiocarbamates Chemical class 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 238000001493 electron microscopy Methods 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 238000003821 enantio-separation Methods 0.000 description 1
- JPGQOUSTVILISH-UHFFFAOYSA-N enflurane Chemical compound FC(F)OC(F)(F)C(F)Cl JPGQOUSTVILISH-UHFFFAOYSA-N 0.000 description 1
- BJXYHBKEQFQVES-NWDGAFQWSA-N enpatoran Chemical compound N[C@H]1CN(C[C@H](C1)C(F)(F)F)C1=C2C=CC=NC2=C(C=C1)C#N BJXYHBKEQFQVES-NWDGAFQWSA-N 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 239000003248 enzyme activator Substances 0.000 description 1
- 229920005558 epichlorohydrin rubber Polymers 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- HQQADJVZYDDRJT-UHFFFAOYSA-N ethene;prop-1-ene Chemical group C=C.CC=C HQQADJVZYDDRJT-UHFFFAOYSA-N 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- RFHAOTPXVQNOHP-UHFFFAOYSA-N fluconazole Chemical compound C1=NC=NN1CC(C=1C(=CC(F)=CC=1)F)(O)CN1C=NC=N1 RFHAOTPXVQNOHP-UHFFFAOYSA-N 0.000 description 1
- 229960004884 fluconazole Drugs 0.000 description 1
- 125000003709 fluoroalkyl group Chemical group 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical class [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 230000001408 fungistatic effect Effects 0.000 description 1
- 125000003838 furazanyl group Chemical group 0.000 description 1
- YRTCKZIKGWZNCU-UHFFFAOYSA-N furo[3,2-b]pyridine Chemical compound C1=CC=C2OC=CC2=N1 YRTCKZIKGWZNCU-UHFFFAOYSA-N 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 210000004211 gastric acid Anatomy 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 150000002306 glutamic acid derivatives Chemical class 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 239000003979 granulating agent Substances 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 238000001631 haemodialysis Methods 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 230000000322 hemodialysis Effects 0.000 description 1
- UKACHOXRXFQJFN-UHFFFAOYSA-N heptafluoropropane Chemical compound FC(F)C(F)(F)C(F)(F)F UKACHOXRXFQJFN-UHFFFAOYSA-N 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 239000008173 hydrogenated soybean oil Substances 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- 229960004337 hydroquinone Drugs 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 239000007946 hypodermic tablet Substances 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 125000004857 imidazopyridinyl group Chemical group N1C(=NC2=C1C=CC=N2)* 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000005462 in vivo assay Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 238000011221 initial treatment Methods 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 229910003480 inorganic solid Inorganic materials 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007919 intrasynovial administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000007915 intraurethral administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 229920000554 ionomer Polymers 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 230000000302 ischemic effect Effects 0.000 description 1
- 239000001282 iso-butane Substances 0.000 description 1
- 229940035415 isobutane Drugs 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- TWBYWOBDOCUKOW-UHFFFAOYSA-M isonicotinate Chemical compound [O-]C(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-M 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 150000003893 lactate salts Chemical class 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 229940039717 lanolin Drugs 0.000 description 1
- 235000019388 lanolin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 231100001231 less toxic Toxicity 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 230000003859 lipid peroxidation Effects 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- 208000002780 macular degeneration Diseases 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 150000002688 maleic acid derivatives Chemical class 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 229940057917 medium chain triglycerides Drugs 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000001525 mentha piperita l. herb oil Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229940071648 metered dose inhaler Drugs 0.000 description 1
- 125000005397 methacrylic acid ester group Chemical group 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-M methanesulfonate group Chemical class CS(=O)(=O)[O-] AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 1
- NJTCEXRGYVYYIX-AABGKKOBSA-N methyl (2S)-2-[[(2S)-3-methyl-2-[[(2R)-pyrrolidine-2-carbonyl]amino]butanoyl]amino]-5-(phenylmethoxycarbonylamino)pentanoate Chemical compound C(C1=CC=CC=C1)OC(=O)NCCC[C@@H](C(=O)OC)NC([C@H](C(C)C)NC(=O)[C@@H]1NCCC1)=O NJTCEXRGYVYYIX-AABGKKOBSA-N 0.000 description 1
- REYNUXDAVXHOHP-IBGZPJMESA-N methyl (2S)-2-amino-5-(9H-fluoren-9-ylmethoxycarbonylamino)pentanoate Chemical compound C1=CC=CC=2C3=CC=CC=C3C(C1=2)COC(=O)NCCC[C@@H](C(=O)OC)N REYNUXDAVXHOHP-IBGZPJMESA-N 0.000 description 1
- BJNJTVSXZUTHRA-LBPRGKRZSA-N methyl (2s)-2-amino-5-(phenylmethoxycarbonylamino)pentanoate Chemical compound COC(=O)[C@@H](N)CCCNC(=O)OCC1=CC=CC=C1 BJNJTVSXZUTHRA-LBPRGKRZSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- GDOPTJXRTPNYNR-UHFFFAOYSA-N methyl-cyclopentane Natural products CC1CCCC1 GDOPTJXRTPNYNR-UHFFFAOYSA-N 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- GVZFUVXPTPGOQT-UHFFFAOYSA-M mitoq Chemical compound CS([O-])(=O)=O.O=C1C(OC)=C(OC)C(=O)C(CCCCCCCCCC[P+](C=2C=CC=CC=2)(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1C GVZFUVXPTPGOQT-UHFFFAOYSA-M 0.000 description 1
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 1
- 125000001620 monocyclic carbocycle group Chemical group 0.000 description 1
- 230000001095 motoneuron effect Effects 0.000 description 1
- 201000000585 muscular atrophy Diseases 0.000 description 1
- YGBMCLDVRUGXOV-UHFFFAOYSA-N n-[6-[6-chloro-5-[(4-fluorophenyl)sulfonylamino]pyridin-3-yl]-1,3-benzothiazol-2-yl]acetamide Chemical compound C1=C2SC(NC(=O)C)=NC2=CC=C1C(C=1)=CN=C(Cl)C=1NS(=O)(=O)C1=CC=C(F)C=C1 YGBMCLDVRUGXOV-UHFFFAOYSA-N 0.000 description 1
- LPOIGVZLNWEGJG-UHFFFAOYSA-N n-benzyl-5-(4-methylpiperazin-1-yl)-2-nitroaniline Chemical compound C1CN(C)CCN1C1=CC=C([N+]([O-])=O)C(NCC=2C=CC=CC=2)=C1 LPOIGVZLNWEGJG-UHFFFAOYSA-N 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical class C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 229920003052 natural elastomer Polymers 0.000 description 1
- 229920001194 natural rubber Polymers 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 201000001119 neuropathy Diseases 0.000 description 1
- 230000007823 neuropathy Effects 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 150000002829 nitrogen Chemical class 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 230000006610 nonapoptotic cell death Effects 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 239000000346 nonvolatile oil Substances 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- GLDOVTGHNKAZLK-UHFFFAOYSA-N octadecan-1-ol Chemical class CCCCCCCCCCCCCCCCCCO GLDOVTGHNKAZLK-UHFFFAOYSA-N 0.000 description 1
- QYSGYZVSCZSLHT-UHFFFAOYSA-N octafluoropropane Chemical compound FC(F)(F)C(F)(F)C(F)(F)F QYSGYZVSCZSLHT-UHFFFAOYSA-N 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000019645 odor Nutrition 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 239000003346 palm kernel oil Substances 0.000 description 1
- 235000019865 palm kernel oil Nutrition 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- GTLACDSXYULKMZ-UHFFFAOYSA-N pentafluoroethane Chemical compound FC(F)C(F)(F)F GTLACDSXYULKMZ-UHFFFAOYSA-N 0.000 description 1
- AEABQBMUYZBBCW-UHFFFAOYSA-N pentanamide Chemical compound CC[CH]CC(N)=O AEABQBMUYZBBCW-UHFFFAOYSA-N 0.000 description 1
- 235000019477 peppermint oil Nutrition 0.000 description 1
- 229960004692 perflenapent Drugs 0.000 description 1
- KAVGMUDTWQVPDF-UHFFFAOYSA-N perflubutane Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(F)(F)F KAVGMUDTWQVPDF-UHFFFAOYSA-N 0.000 description 1
- 229950003332 perflubutane Drugs 0.000 description 1
- NJCBUSHGCBERSK-UHFFFAOYSA-N perfluoropentane Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F NJCBUSHGCBERSK-UHFFFAOYSA-N 0.000 description 1
- 229960004065 perflutren Drugs 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 208000033808 peripheral neuropathy Diseases 0.000 description 1
- 238000005502 peroxidation Methods 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 229960000969 phenyl salicylate Drugs 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 235000011007 phosphoric acid Nutrition 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- PDDXOPNEMCREGN-UHFFFAOYSA-N phosphoric acid;trioxomolybdenum;hydrate Chemical compound O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.OP(O)(O)=O PDDXOPNEMCREGN-UHFFFAOYSA-N 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229940068196 placebo Drugs 0.000 description 1
- 239000000902 placebo Substances 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229920001490 poly(butyl methacrylate) polymer Polymers 0.000 description 1
- 229920001084 poly(chloroprene) Polymers 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 1
- 229920001515 polyalkylene glycol Polymers 0.000 description 1
- 229920002857 polybutadiene Polymers 0.000 description 1
- 229920001610 polycaprolactone Polymers 0.000 description 1
- 229920002721 polycyanoacrylate Polymers 0.000 description 1
- 229940068886 polyethylene glycol 300 Drugs 0.000 description 1
- 229940068918 polyethylene glycol 400 Drugs 0.000 description 1
- 229940057838 polyethylene glycol 4000 Drugs 0.000 description 1
- 229940068917 polyethylene glycols Drugs 0.000 description 1
- 229920001195 polyisoprene Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 239000004926 polymethyl methacrylate Substances 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920006324 polyoxymethylene Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229920002689 polyvinyl acetate Polymers 0.000 description 1
- 239000011118 polyvinyl acetate Substances 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 238000002600 positron emission tomography Methods 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000012746 preparative thin layer chromatography Methods 0.000 description 1
- 229960001309 procaine hydrochloride Drugs 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 235000010388 propyl gallate Nutrition 0.000 description 1
- 239000000473 propyl gallate Substances 0.000 description 1
- 229940075579 propyl gallate Drugs 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- RUOJZAUFBMNUDX-UHFFFAOYSA-N propylene carbonate Chemical compound CC1COC(=O)O1 RUOJZAUFBMNUDX-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical class CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 230000000541 pulsatile effect Effects 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- WVIICGIFSIBFOG-UHFFFAOYSA-N pyrylium Chemical compound C1=CC=[O+]C=C1 WVIICGIFSIBFOG-UHFFFAOYSA-N 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 150000004053 quinones Chemical class 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 235000005713 safflower oil Nutrition 0.000 description 1
- 239000003813 safflower oil Substances 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 150000003902 salicylic acid esters Chemical class 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 229910052711 selenium Inorganic materials 0.000 description 1
- 239000011669 selenium Substances 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 239000008354 sodium chloride injection Substances 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 239000008137 solubility enhancer Substances 0.000 description 1
- 239000002195 soluble material Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 238000002798 spectrophotometry method Methods 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 150000003890 succinate salts Chemical class 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003456 sulfonamides Chemical class 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 150000003892 tartrate salts Chemical class 0.000 description 1
- 235000019640 taste Nutrition 0.000 description 1
- 229920001897 terpolymer Polymers 0.000 description 1
- 150000003942 tert-butylamines Chemical class 0.000 description 1
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 1
- ZUHZGEOKBKGPSW-UHFFFAOYSA-N tetraglyme Chemical compound COCCOCCOCCOCCOC ZUHZGEOKBKGPSW-UHFFFAOYSA-N 0.000 description 1
- OULAJFUGPPVRBK-UHFFFAOYSA-N tetratriacontyl alcohol Chemical class CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCO OULAJFUGPPVRBK-UHFFFAOYSA-N 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- RTKIYNMVFMVABJ-UHFFFAOYSA-L thimerosal Chemical compound [Na+].CC[Hg]SC1=CC=CC=C1C([O-])=O RTKIYNMVFMVABJ-UHFFFAOYSA-L 0.000 description 1
- 229940033663 thimerosal Drugs 0.000 description 1
- 150000003567 thiocyanates Chemical class 0.000 description 1
- 235000019303 thiodipropionic acid Nutrition 0.000 description 1
- 230000009974 thixotropic effect Effects 0.000 description 1
- 238000003354 tissue distribution assay Methods 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M toluenesulfonate group Chemical group C=1(C(=CC=CC1)S(=O)(=O)[O-])C LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- 125000005490 tosylate group Chemical group 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
- 230000000472 traumatic effect Effects 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- 229940117013 triethanolamine oleate Drugs 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- YFNKIDBQEZZDLK-UHFFFAOYSA-N triglyme Chemical compound COCCOCCOCCOC YFNKIDBQEZZDLK-UHFFFAOYSA-N 0.000 description 1
- 229920011532 unplasticized polyvinyl chloride Polymers 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 230000002485 urinary effect Effects 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 235000019165 vitamin E Nutrition 0.000 description 1
- 239000011709 vitamin E Substances 0.000 description 1
- 229940046009 vitamin E Drugs 0.000 description 1
- 235000012431 wafers Nutrition 0.000 description 1
- 239000008136 water-miscible vehicle Substances 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/06—Tripeptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0821—Tripeptides with the first amino acid being heterocyclic, e.g. His, Pro, Trp
- C07K5/0823—Tripeptides with the first amino acid being heterocyclic, e.g. His, Pro, Trp and Pro-amino acid; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/07—Tetrapeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06139—Dipeptides with the first amino acid being heterocyclic
- C07K5/06165—Dipeptides with the first amino acid being heterocyclic and Pro-amino acid; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1024—Tetrapeptides with the first amino acid being heterocyclic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Cardiology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Heart & Thoracic Surgery (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Epidemiology (AREA)
- Psychology (AREA)
- Urology & Nephrology (AREA)
- Immunology (AREA)
- Hospice & Palliative Care (AREA)
- Gastroenterology & Hepatology (AREA)
- Psychiatry (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Vascular Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Hydrogenated Pyridines (AREA)
- Food Preservation Except Freezing, Refrigeration, And Drying (AREA)
Abstract
Un compuesto de fórmula (I), sus sales farmacéuticamente aceptables y sus enantiómeros o diastereómeros individuales. Composiciones y métodos útiles para el tratamiento o supresión de enfermedades, retrasos en el desarrollo y síntomas relacionados con el estrés oxidativo. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Preparación y uso de un barredor de especies reactivas de oxígeno
CAMPO DE LA INVENCIÓN
[0001] La presente solicitud es una solicitud divisional de EP17923551.0. La presente invención proporciona compuestos para su uso en métodos de tratamiento o erradicación de enfermedades, retrasos en el desarrollo y síntomas relacionados con el estrés oxidativo. Más concretamente, los métodos comprenden administrar una cantidad eficaz de un compuesto, que es un modulador de apoptosis y ferroptosis.
ANTECEDENTES DE LA INVENCIÓN
[0002] Mientras que las ERO (especies reactivas de oxígeno) localizadas son fundamentales para el desarrollo y metabolismo biológicos de la célula eucariota, el flujo excesivo de ERO sin restricciones conduce a disfunciones y/o muerte celulares (p. ej., artículos: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3484374/pdf/nihms412463.pdf y http://www.sciencedirect.com/science/article/pii/S0006291X16317715, acceso el 7 de julio de 2017).
[0003] Desde su hallazgo en 2012, la ferroptosis ha surgido como una vía mecanística fundamental para el exceso de flujo de ERO (especialmente en la mitocondria), que en última instancia conduce a la muerte celular no apoptótica (p. ej., artículos: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4764384/ y https://www.ncbi.nlm.nih gov/pmc/articles/PMC5072448/. acceso el 7 de julio de 2017). Como el elemento fundamental de la ferroptosis, la pérdida de actividad de la glutatión peroxidasa 4 (GPx4) posibilita un exceso mortal de ERO, que después conduce a la destrucción celular por estrés oxidativo (p. ej., peroxidación de los fosfolípidos poliinsaturados de la membrana) (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5003261/, acceso el 7 de julio de 2017). Dado que la GPx4 usa la glutatión (GSH) como cofactor fundamental para captar las ERO, el déficit de producción de GSH puede surgir a partir de la inducción bloqueada (p. ej., por medio de pequeños compuestos conocidos, como erastina y exceso de glutamatos) de su sistema Xc- 'contracorriente' (un antitransportador glutamato/cistina). Por otro lado, inducir el exceso de flujo de ERO por medio de ferroptosis puede destruir ciertas células cancerígenas (p. ej., artículo: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5345622/pdf/JCMM-21-648.pdf, acceso el 7 de julio de 2017).
[0004] Según los artículos anteriores, así como las referencias sobre otros inhibidores atribuibles a sus mecanismos de acción antiferróptoticos (p. ej., ferrostatinas: solicitudes de patente US 2016/0297748 y WO 2013/152039, quinonas, como MitoQ: http://www.discoverymedicine.com/Robin-A-J-Smith/2011/02/07/mitochondria-targeted-antioxidants-astherapies/), pequeños péptidos, como SS-31: (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4267688/, acceso el 7 de julio de 2017, y la publicación de patente WO2004070054 (A2)); nitróxidos, como XJB-5-131 y análogos de JP4-039: (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5043442/pdf/oc6b00199.pdf, acceso 7 de julio de 2017, y publicación de patente WO2012112851 (A2)). También se ha empleado XJB-5-131 en la práctica de la inhibición de la ferroptosis, donde desempeña un papel fundamental en la peroxidación lipídica intramitocondrial de la ferroptosis. Asimismo, el compuesto de referencia ferrostatina-1 y XJB-5-131 puede compartir mecanismos de acción similares o que ambos compuestos actúen en la misma vía de señalización. La ferroptosis se puede vincular a una variedad de enfermedades vastamente conocidas que se agrupan en términos generales entorno al sistema nervioso periférico (SNP) o central (SNC).
[0005] SNP: enfermedades cardiovasculares (p. ej., aterosclerosis, miocardiopatía hipertensiva, insuficiencia cardíaca congestiva, accidente cerebrovascular, etc.), metabólicas (p. ej., diabetes y sus complicaciones relacionadas como neuropatía, hiperlipidemia, etc.), urinarias (p. ej., lesión renal aguda (LRA)), relacionadas con la edad (p. ej., atrofia muscular esquelética, y degeneración macular seca asociada a la edad (DMAE seca, https://en.rn.wikipedia.org/wiki/Macular degeneration, acceso el 17 de julio de 2017) y traumatismo (p. ej., lesión por radiación, rabdomiolisis inducida, traumatismo craneoencefálico, cirugía mayor, etc.). En el caso de la LRA (https://en m.wikipedia.org/wiki/Acute kidney injury, acceso el 7 de julio de 2017), la FDA de EE. UU. hoy en día aún tiene pendiente aprobar un fármaco para curar específicamente esta enfermedad, que afecta especialmente al 30 % aprox. de los pacientes de EE. UU. hospitalizados en unidades de cuidados intensivos. Cuando la LRA progresa por desgracia a los estadios tardíos/crónicos, la hemodiálisis y, en última instancia el remplazo renal, se convierten en las principales opciones de tratamiento restantes.
[0006] SNC: enfermedades neuromotoras (p. ej., de Parkinson, de Huntington, esclerosis lateral amiotrófica, epilepsia, etc.) y cognitivas (p. ej., de Alzheimer (p. ej., https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4679930/pdf/fnins-09-00469.pdf, acceso el 7 de julio de 2017)). En el caso de la epilepsia, que afecta a más de 50 millones de personas en todo el mundo, una minoría significativa (20-30 %) de los pacientes son/se vuelven resistentes a más de 20 de los fármacos aprobados actualmente (p. ej., https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5068473/pdf7ndt-12-2605.pdf, acceso el 7 de julio de 2017). Sin lugar a duda, la investigación en curso probablemente vinculará más enfermedades
debidas a la ferroptosis y más inhibidores de esta desde su hallazgo en 2012. Por consiguiente, como ha quedado ejemplificado por la LRA y la epilepsia, también se necesitan mejores fármacos (probablemente novedosos) urgentemente para otras enfermedades que se vincularán a la ferroptosis en el futuro.
SUMARIO DE LA INVENCIÓN
[0007] La presente invención proporciona compuestos que modulan la ferroptosis en un sujeto; incluye ciertos derivados de TEMPO, y su preparación. Los compuestos de la presente invención se pueden usar para prevenir, tratar, aliviar un cierto trastorno o una enfermedad en un paciente.
[0008] La presente invención se refiere a un compuesto seleccionado de entre Compuesto (III), Compuesto (XXVIII), Compuesto (XXIV), Compuesto (XXX), Compuesto (Xx XI), Compuesto (XXXII), Compuesto (Xx X iII), Compuesto (XXXIV), Compuesto (XXXV), Compuesto (XXXVI), Compuesto (x Xx VII), Compuesto (Xx XVIII), Compuesto (XXXIX), Compuesto (XXXXIV), Compuesto (XXXXV), Compuesto (XXXXVI), y Compuesto (XXXx V iI), o una sal farmacéuticamente aceptable de cualquiera de los anteriores, para su uso en un método de inhibición de la ferroptosis en un paciente:
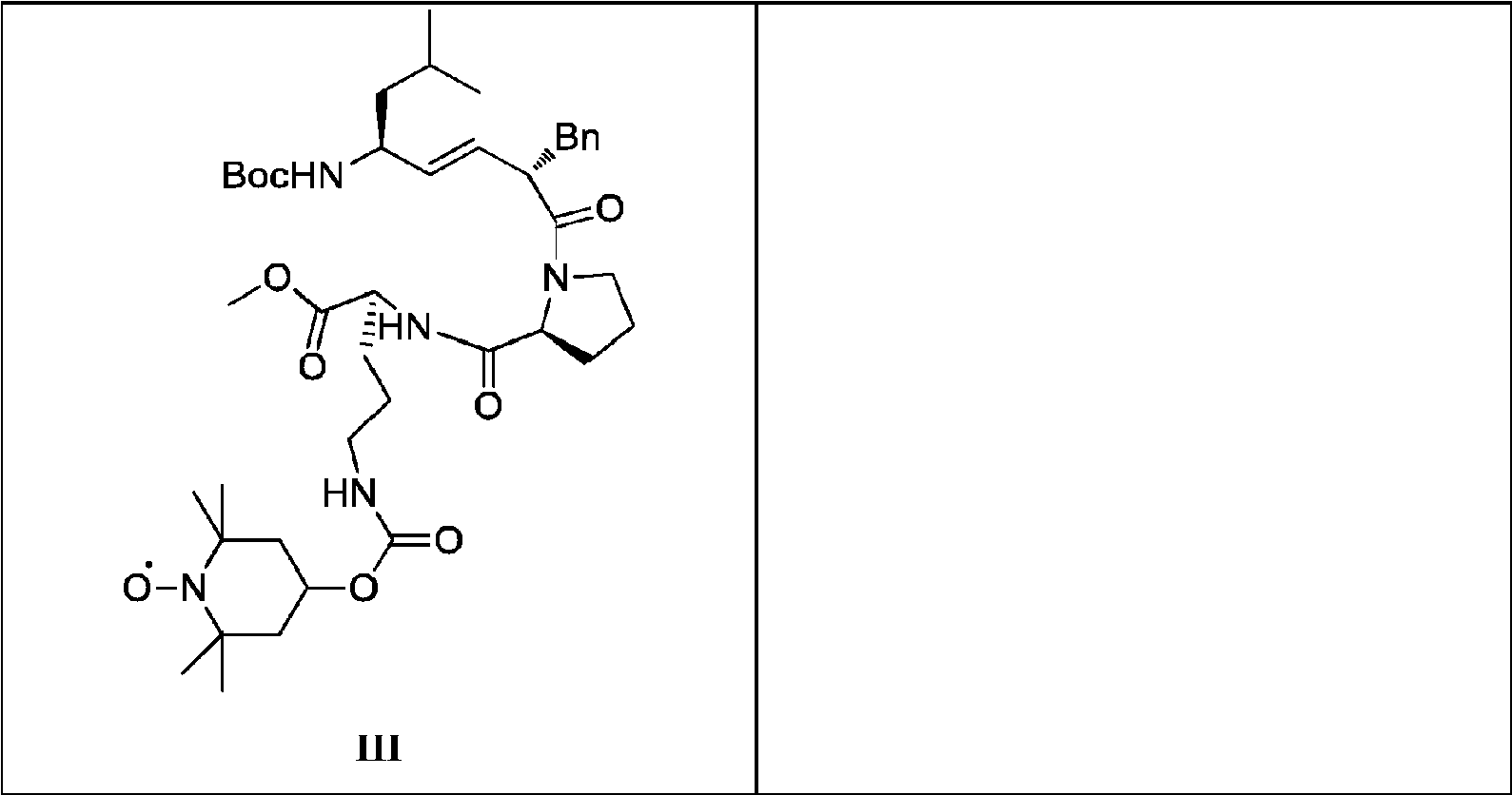




[0009] La presente divulgación se refiere a compuestos de fórmula (I), sales farmacéuticamente aceptables de este, y enantiómeros o diastereómeros individuales de este

donde
x es 0 o 1;
G está ausente, O, NH o CH2;
W es:

Donde Ri, R2, R3, o R4 es alquilo C1-C6, R5 es OH, u O,
n es 0,1,2 o 3,
L está ausente, o se selecciona de entre un grupo que consiste en: alquilo C1-C10, alcoxilo C1-C10, cicloalquilo, alquilcicloalquilo C1-C10, heterocicloalquilo, alquilheterocicloalquilo C1-C10, arilo, alquilarilo C1-C10, heteroarilo, y alquilheteroarilo C1-C10, donde dicho cicloalquilo o heterocicloalquilo tiene entre aprox. 3 y aprox. 7 carbonos en el anillo, y dicho arilo o heteroarilo tiene entre aprox. 5 y aprox. 10 carbonos en el anillo;
PEP es una fracción peptídica que tiene la siguiente estructura:

donde un símbolo * denota un centro quiral en una configuración (S) o (R);
B está ausente, N, NH, u O;
Xi se selecciona de entre un grupo que consiste en H, -(C = O)-O-Rm, -(SO)-O-Rm, -(SOz)-O-Rm,-(SO2)-N-(Rm)2 y -(C = O)-N-(Rm)2, donde Rm es alquilo C1-C6, alcoxilo C1-C6, alquenilo C1-C6, haloalquilo C1-C6, cicloalquilo, alquilcicloalquilo C1-C6, heterocicloalquilo, alquilheterocicloalquilo C1-C6, arilo, alquilarilo C1-C6, heteroarilo, o alquilheteroarilo C1-C6, donde dicho cicloalquilo o heterocicloalquilo tiene entre aprox. 3 y aprox. 7 carbonos en el anillo, y dicho arilo o heteroarilo tiene entre aprox. 5 y aprox.10 carbonos en el anillo;
Rn se selecciona de entre -O-X3, -S-X3, -NH-X3, o ~N-X4a(X4b);
X2 o X3 se selecciona de entre un grupo que consiste en H, alquilo C1-C10, alquenilo C1-C10, arilo, alquilarilo C1-C6, heteroarilo, y alquilheteroarilo C1-C6, donde dicho arilo o heteroarilo tiene entre 5 y 6 carbonos en el anillo;
X4a y X4b se seleccionan por separado de entre alquilo C1-C6, alquilo C1-C6 sustituido; X4a y X4b pueden, opcionalmente, formar juntos heterocicloalquilo C3-C8, heterocicloalquilo C3-C8 sustituido.
A está ausente, o se selecciona de entre un grupo que consiste en: alanina, leucina, isoleucina, fenilalanina, metionina, prolina, glicina, triptófano, serina, treonina, cisteína, tirosina, asparagina, glutamina, histidina, lisina, arginina, ácido aspártico, ácido glutámico, y valina, cada uno de los cuales puede estar en una configuración D o L; el residuo de amino está desprotegido o protegido por un grupo protector seleccionado de entre un grupo que consiste en Cbz y Fmoc;
con la condición de que PEP no sea uno de los siguientes:


[0010] Los compuestos de la presente invención o una sal farmacéuticamente aceptable de un compuesto de la presente invención, y enantiómeros y diastereómeros individuales de estos, son útiles como moduladores de ferroptosis. Por consiguiente, la invención se dirige a un compuesto de la presente invención o una sal farmacéuticamente aceptable de un compuesto de la presente invención, y enantiómeros y diastereómeros individuales de este para su uso en la reducción de las especies reactivas de oxígeno (ERO) en una célula, que comprende que una célula entre en contacto con una cantidad eficaz de dicho compuesto como un modulador de ferroptosis.
[0011] Los compuestos de la presente invención son útiles en métodos para modular la ferroptosis en un sujeto, que comprende exponer al sujeto a una cantidad eficaz de al menos un compuesto de la presente invención, o una sal farmacéuticamente aceptable de un compuesto de la presente invención, y enantiómeros y diastereómeros individuales de este, y al menos un excipiente farmacéuticamente aceptable, un portador, un adyuvante, un disolvente, una sustancia de soporte o una combinación de estos, y cantidades terapéuticamente eficaces adicionales de uno o más ingredientes activos auxiliares opcionales.
[0012] Cualquier modo de realización divulgado en la presente memoria se puede combinar con otros modos de realización siempre que no sean contradictorios entre sí, aunque los modos de realización se describan según diferentes aspectos de la invención. Además, cualquier rasgo técnico de un modo de realización se puede aplicar al rasgo técnico correspondiente en otros modos de realización siempre que no sean contradictorios entre sí, aunque los modos de realización se describan según diferentes aspectos de la invención.
[0013] Lo anterior tan solo resume ciertos aspectos divulgados en la presente memoria y no pretende ser de naturaleza restrictiva. Estos aspectos y otros aspectos y modos de realización, rasgos y beneficios adicionales de la invención quedarán patentes a partir de la siguiente descripción detallada y mediante la práctica de la invención.
DESCRIPCIÓN DETALLADA Y MODOS DE REALIZACIÓN CONCRETOS
[0014] La mayoría de las denominaciones químicas en la presente memoria se generaron por medio de la nomenclatura IUPAC. Algunas denominaciones químicas se generaron por medio de diferentes nomenclaturas o denominaciones comerciales o alternativas conocidas en la materia. En caso de conflicto entre denominaciones y estructuras, prevalecerán las estructuras.
DEFINICIONES Y TERMINOLOGÍA GENERAL
[0015] Ahora se hará referencia en detalle a ciertos modos de realización de la invención, ejemplos que se ilustran en las estructuras y fórmulas que acompañan. La invención pretende abarcar todas las alternativas, modificaciones y
equivalentes que se pueden incluir dentro del alcance de la presente invención, tal como definen las reivindicaciones. Un experto en la materia reconocerá muchos métodos y materiales similares o equivalentes a los descritos en la presente memoria que se podrían emplear en la práctica de la presente invención.
[0016] Se aprecia además que ciertos rasgos de la invención que, por claridad, se describen en el contexto de modos de realización independientes, también se pueden proporcionar combinados en un único modo de realización. En cambio, varios rasgos de la invención que, por brevedad, se describen en el contexto de un solo modo de realización, también se pueden proporcionar por separado o en cualquier subcombinación adecuada.
[0017] A no ser que se definan de otra manera, todos los términos técnicos y científicos usados en la presente memoria tienen el mismo significado que el comúnmente entendido por un experto en la materia a la que pertenece esta invención.
[0018] Tal como se usa en la presente memoria, las siguientes definiciones serán de aplicación a no ser que se indique lo contrario. A efectos de esta invención, se identifican los elementos químicos de acuerdo con la tabla periódica de los elementos químicos, versión CAS, y el Handbook of Chemistry and Physics, 75th Ed. 1994. Asimismo, los ingredientes generales de química orgánica se describen en Organic Chemistry, Thomas Sorrell, University Science Books, Sausalito: 1999, y March's Advanced Organic Chemistry by Michael B. Smith and Jerry March, John Wiley & Sons, New York: 2007.
[0019] Tal como se usa anteriormente, y a lo largo de esta divulgación, se entenderá que los siguientes términos, a no ser que se indique lo contrario, tienen los siguientes significados. Si falta alguna definición, prevalecerá la definición convencional conocida por el experto en la materia. Si alguna definición proporcionada en la presente memoria entra en conflicto con una definición proporcionada en cualquier publicación citada o es diferente a esta, prevalecerá la definición proporcionada en la presente memoria.
[0020] Tal como se usa en la presente memoria, los términos «incluyendo», «que contiene» y «comprendiendo/que comprende» se usan en su sentido abierto no limitativo.
[0021] Tal como se usa en la presente memoria, las formas singulares «un/una», «el/la» incluyen referentes plurales a no ser que el contexto claramente indique lo contrario.
[0022] Para proporcionar una descripción más concisa, algunas de las expresiones cuantitativas dadas en la presente memoria no se califican con el término «aprox.». Se entiende que, tanto si el término «aprox.» se usa explícitamente o no, cualquier cantidad dada en la presente memoria se refiere al valor real dado, y también se refiere a la aproximación a dicho valor dado que se deduciría razonablemente según los conocimientos básicos de la materia, incluyendo equivalentes y aproximaciones debido a las condiciones experimentales y/o de medición para dicho valor dado. Cada vez que un rendimiento se de en forma de un porcentaje, este rendimiento se refiere a una masa de la entidad para la que se indica el rendimiento con respecto a la cantidad máxima de la misma entidad que se podría obtener en las condiciones estequiométricas concretas. Las concentraciones que se dan como porcentajes se refieren a las relaciones de masa, a menos que se indique lo contrario.
Definiciones químicas
[0023] Tal como se usa en la presente memoria, «alquilo» se refiere a un grupo de hidrocarburo saturado de cadena lineal o ramificada que tiene entre 1 y 12 átomos de carbono. Los grupos de alquilo representativos incluyen, entre otros, metilo, etilo, n-propil, isopropilo, 2-metil-1 -propil, 2-metil-2-propil, 2-metil-1 -butil, 3-metil-1-butil, 2-metil-3-butil, 2,2-dimetil-1-propil, 2-metil-1 -pentil, 3-metil-1-pentil, 4-metil-1 -pentil, 2-metil-2-pentil, 3-metil-2-pentil, 4-metil-2-pentil, 2,2-dimetil-1-butil, 3,3-dimetil-1 -butil, 2-etil-1-butil, butilo, isobutilo, t-butil, n-pentil, isopentilo, neopentilo, n-hexil, y similares, y grupos alquilo más largos, como heptilo, octilo, y similares. Tal como se usa en la presente memoria, «alquilo inferior» se refiere a un alquilo que tiene entre 1 y 6 átomos de carbono.
[0024] El término «alquilamino», tal como se usa en la presente memoria, denota un grupo amino como se define en la presente memoria donde se reemplaza un átomo de hidrógeno del grupo amino por un grupo alquilo como se define en la presente memoria. Los grupos aminoalquilo se pueden definir mediante la siguiente fórmula general -NH-alquil. Esta fórmula general incluye grupos de las siguientes fórmulas generales: -NH-C1-C10 alquil y -NH-C1-C6 alquil Los ejemplos de grupos aminoalquilo incluyen, entre otros, aminometilo, aminoetilo, aminopropilo, aminobutilo.
[0025] El término «alcoxilo», tal como se usa en la presente memoria, incluye -O-(alquil), donde el alquilo se ha definido anteriormente.
[0026] Tal como se usa en la presente memoria, «alcoxialquilo» significa -(alquilenil)-O-(alquil), donde cada «alquilo» es independientemente un grupo alquilo definido anteriormente.
[0027] El término «amina», tal como se usa en la presente memoria se refiere a un grupo -NH2.
[0028] «Arilo» se refiere a un grupo aromático mono-, bi- o tricíclico, donde todos los anillos del grupo son aromáticos. Para los sistemas bi- o tricíclicos, los anillos aromáticos individuales se fusionan entre sí. Los grupos arilo ejemplares incluyen, entre otros, fenilo, naftaleno y antraceno.
[0029] «Ariloxilo», tal como se usa en la presente memoria, se refiere a un grupo -O-(aril) donde el arilo se define como anteriormente.
[0030] «Arilalquilo», tal como se usa en la presente memoria se refiere a un grupo -(alquilenil)-(aril), donde el alquilenilo y el arilo se definen como anteriormente. Los ejemplos no limitativos de arilalquilos comprenden un grupo alquilo inferior. Los ejemplos no limitativos de grupos arilalquilo adecuados incluyen bencilo, 2-fenetil y naftalenilmetilo.
[0031] «Arilalcoxi», tal como se usa en la presente memoria, se refiere a un grupo -(alquilenil)-aril, donde el alquilenilo y el arilo se definen como anteriormente.
[0032] Los términos «deuterio», «deuterado», tal como se usan en la presente memoria, se refieren a que son, o que están sustituidos por, un isótopo estable de hidrógeno que tiene un protón y un neutrón.
[0033] El término «halógeno», tal como se usa en la presente memoria, se refiere a flúor, cloro, bromo o yodo. El término «halo» representa cloro, fluoro, bromo o yodo.
[0034] El término «haloalquilo» denota un grupo alquilo como se ha definido anteriormente donde uno o más, por ejemplo, uno, dos o tres de los átomos de hidrógeno del grupo alquilo se reemplazan con un átomo de halógeno, por ejemplo, fluoro, bromo o cloro, en concreto, fluoro. Los ejemplos de haloalquilo incluyen, entre otros, monofluoro-, difluoro-, o trifluoro-metil, -etil o -propil, por ejemplo, 3,3,3-trifluoropropil, 2-fluoroetil, 2,2,2-trifluoroetil, fluorometilo, difluorometilo, o trifluorometilo, o bromoetilo o cloroetilo. De manera similar, el término «fluoroalquilo» se refiere a un grupo alquilo como se ha definido anteriormente sustituido por uno o más, por ejemplo, uno, dos o tres átomos de flúor.
[0035] El término «haloalcoxilo», tal como se usa en la presente memoria, se refiere a un grupo -O-(haloalquil) donde el haloalquilo se define como anteriormente. Los grupos haloalcoxilo ejemplares son bromoetoxilo, cloroetoxilo, trifluorometoxilo y 2,2,2-trifluoroetoxi.
[0036] El término «hidroxilo» se refiere a un grupo -OH.
[0037] El término «hidroxialquilo» denota un grupo alquilo que está sustituido por al menos un grupo hidroxilo, por ejemplo, uno, dos o tres grupo(s) hidroxilo. La porción de alquilo del grupo hidroxialquilo proporciona el punto de conexión al resto de la molécula. Los ejemplos de grupos hidroxialquilo incluyen, entre otros, hidroximetilo, hidroxietilo, 1-hidroxipropil, 2-hidroxiisopropil, 1,4-dihidroxibutil, y similares.
[0038] El término «oxo» significa un grupo = O y puede estar unido a un átomo de carbono o a un átomo de azufre. El término «N-óxido» se refiere a la forma oxidada de un átomo de nitrógeno.
[0039] Tal como se usa en la presente memoria, el término «cicloalquilo» se refiere a un carbociclo saturado o parcialmente saturado, monocíclico, policíclico fusionado, policíclico con puente o espiro policíclico que tiene entre 3 y 12 átomos de carbono en el anillo. Una categoría no limitativa de los grupos cicloalquilo son carbociclos monocíclicos saturados o parcialmente saturados que tienen entre 3 y 6 átomos de carbono. Los ejemplos ilustrativos de grupos cicloalquilo incluyen, entre otros, las siguientes fracciones:

[0040] El término «cicloalcoxi» se refiere a un grupo -O-(cicloalquil).
[0041] El término «heterocicloalquilo», tal como se usa en la presente memoria, se refiere al cicloalquilo, tal como se define en esta aplicación, siempre que uno o más de los carbonos en el anillo indicados estén reemplazados por una fracción seleccionada de entre -O-, -N = , -C(O)-, -S-.
[0042] Tal como se usa en la presente memoria, el término «heteroarilo» se refiere a un heterociclo aromático monocíclico, o policíclico fusionado, que tiene entre tres y 15 átomos en el anillo que se seleccionan de entre carbono, oxígeno, nitrógeno, selenio y azufre. Los grupos heteroarilo adecuados no incluyen sistemas de anillo que se deban cargar para ser aromáticos, como el pirilio. Algunos anillos heteroarilo adecuados de 5 miembros (como un heteroarilo monocíclico o como parte de un heteroarilo policíclico) tienen un átomo de oxígeno, azufre o nitrógeno, o uno de nitrógeno más uno de oxígeno o de azufre, o 2, 3, o 4 átomos de nitrógeno. Algunos anillos heteroarilo adecuados de 6 miembros (como un heteroarilo monocíclico o como parte de un heteroarilo policíclico) tienen 1, 2, o 3 átomos de nitrógeno. Los ejemplos de grupos heteroarilo incluyen, entre otros, piridinilo, imidazolilo, imidazopiridinilo, pirimidinilo, pirazolilo, triazolilo, pirazinilo, tetrazolilo, furilo, tienilo, isoxazolilo, tiazolilo, oxazolilo, isotiazolilo, pirrolilo, quinolinilo, isoquinolinilo, indolilo, bencimidazolilo, benzofuranilo, cinnolinilo, indazolilo, indolizinilo, ftalazinilo, piridazinilo, triazinilo, isoindolilo, pteridinilo, purinilo, oxadiazolilo, triazolilo, tiadiazolilo, furazanilo, benzofurazanilo, benzotiofenilo, benzotiazolilo, benzoxazolilo, quinazolinilo, quinoxalinilo, naftiridinilo y furopiridinilo.
[0043] Los expertos en la materia reconocerán que las especies de heteroarilo y los grupos cicloalquilo enumerados o ilustrados anteriormente no son exhaustivos, y que también se pueden seleccionar especies adicionales en el alcance de estos términos definidos.
[0044] Tal como se describen en la presente memoria, los compuestos divulgados en la presente memoria se pueden sustituir opcionalmente por uno o más sustituyentes, o como ejemplifican las clases, subclases y especies concretas de la invención.
[0045] Tal como se usa en la presente memoria, el término «sustituido» significa que el grupo o la fracción especificada lleva uno o más sustituyentes adecuados. Tal como se usa en la presente memoria, el término «no sustituido» significa que el grupo especificado no lleva sustituyentes. Tal como se usa en la presente memoria, el término «sustituido opcionalmente» significa que el grupo especificado no está sustituido o está sustituido por el número especificado de sustituyentes. Cuando el término «sustituido» se usa para describir un sistema estructural, se pretende que la sustitución se produzca en cualquier posición de valencia permitida en el sistema.
[0046] Tal como se usa en la presente memoria, «uno o más sustituyentes» denota de una al máximo número posible de sustituciones que se pueden producir en cualquier posición de valencia permitida en el sistema. En un cierto modo de realización, uno o más sustituyentes significa 1, 2, 3, 4 o 5 sustituyentes. En otro modo de realización, uno o más sustituyentes significa 1, 2 o 3 sustituyentes.
[0047] Se asume que cualquier átomo representado en la presente memoria con una valencia insatisfecha tiene el número suficiente de átomos de hidrógeno para satisfacer la valencia del átomo.
[0048] Cuando cualquier variable (p. ej., alquilo, alquilenilo, heteroarilo, R1, R2, o Ra) aparece en más de un lugar en cualquier fórmula o descripción proporcionada en la presente memoria, la definición de esa variable en cada caso es independiente de su definición en cualquier otro caso.
[0049] Los intervalos numéricos, tal como se usan en la presente memoria, pretenden incluir números enteros consecutivos. Por ejemplo, un intervalo expresado como «entre 0 y 4» o «0-4» incluye 0, 1, 2, 3 y 4, mientras que un intervalo expresado como el «10-20 %» incluye el 10 %, 11 %, 12 %, 13 %, 14 %, 15 %, 16 %, 17 %, 18 %, 19 % y el 20 %. De manera similar, los intervalos numéricos también pretenden incluir números enteros fraccionarios consecutivos. Por ejemplo, un intervalo expresado como el «1-2 %» incluiría el 1.0 %, 1.1 %, 1.2 %, 1.3 %, 1.4 %, 1.5 %, 1.6 %, 1.7 %, 1.8 %, 1.9 % y el 2.0 %.
[0050] Cuando se muestra una fracción multifuncional, el punto de unión con el núcleo se indica mediante una línea o guion. Por ejemplo, ariloxi- se refiere a una fracción en la que un átomo de oxígeno es el punto de unión con la molécula central mientras el arilo se une al átomo de oxígeno.
Definiciones adicionales
[0051] Tal como se usa en la presente memoria, el término «sujeto» engloba mamíferos y no mamíferos. Los ejemplos de mamíferos incluyen, entre otros, cualquier miembro del grupo de los mamíferos: seres humanos; primates no humanos,
como chimpancés y otras especies de monos y simios; animales de granja, como ganado bovino, equino, ovino, caprino, porcino; animales domésticos, como conejos, perros y gatos; y animales de laboratorio, incluyendo roedores, como ratas, ratones, cobayas y similares. Los ejemplos de no mamíferos incluyen, entre otros, aves, peces y similares. En un modo de realización de la presente invención, el mamífero es un ser humano.
[0052] «Paciente» incluye tanto seres humanos como animales.
[0053] El término «inhibidor» se refiere a una molécula, como un compuesto, un fármaco o un activador enzimático, o una hormona que bloquea o interfiere de otro modo con una actividad biológica concreta.
[0054] El término «modulador» se refiere a una molécula, como un compuesto de la presente invención, que aumenta o reduce, o que afecta de otro modo a la actividad de una proteína, un receptor y/o unos canales iónicos dados.
[0055] Los términos «cantidad eficaz» o «cantidad terapéuticamente eficaz» se refieren a una cantidad suficiente del agente para proporcionar el resultado biológico deseado. Ese resultado puede ser la reducción y/o alivio de las manifestaciones clínicas, síntomas o causas de una enfermedad o afección médica, o cualquier otra alteración deseada de un sistema biológico. Por ejemplo, una «cantidad eficaz» para uso terapéutico es la cantidad de un compuesto, o de una composición que comprende el compuesto, que es necesaria para proporcionar un cambio clínicamente relevante en un estado de enfermedad, síntoma o afección médica. Una persona con conocimientos básicos en la materia puede determinar una cantidad «eficaz» en cualquier caso concreto mediante la experimentación rutinaria. Por consiguiente, la expresión «cantidad eficaz» generalmente se refiere a la cantidad en la que la sustancia activa tiene un efecto terapéuticamente deseado.
[0056] Tal como se usa en la presente memoria, los términos «tratar» y «tratamiento» abarcan tanto el tratamiento «preventivo» como «curativo». Con tratamiento «preventivo» se pretende indicar un aplazamiento en el desarrollo de la enfermedad, de un síntoma de una enfermedad, o de una afección médica, contener los síntomas que puedan aparecer o reducir del riesgo de desarrollo o recurrencia de una enfermedad o síntoma. El tratamiento «curativo» incluye reducir la gravedad o contener el agravamiento de una enfermedad, un síntoma o una afección existentes. Por consiguiente, el tratamiento incluye aliviar o prevenir el agravamiento de los síntomas existentes de la enfermedad, prevenir que aparezcan síntomas, aliviar o prevenir las causas metabólicas subyacentes de los síntomas, inhibir el trastorno o la enfermedad, p. ej., detener el desarrollo del trastorno o la enfermedad, mitigar el trastorno o la enfermedad, provocar la regresión del trastorno o la enfermedad; mitigar una afección provocada por la enfermedad o el trastorno, o frenar los síntomas de la enfermedad o el trastorno.
[0057] Tal como se usa en la presente memoria, se debe entender que los términos «administración de un» y «administrar un» compuesto significan proporcionar un compuesto de la invención, composición farmacéutica que comprende un compuesto o un profármaco de un compuesto de la invención a un individuo que lo necesite. Se reconoce que un experto en la materia no limitativa puede tratar a un paciente que actualmente padece enfermedades o trastornos relacionados, o proporcionar un tratamiento profiláctico a un paciente que padece las enfermedades o los trastornos por medio de una cantidad efectiva del compuesto de la presente invención.
[0058] El término «composición», como se usa en la presente memoria, pretende abarcar un producto que comprende los ingredientes especificados en las cantidades especificadas, así como cualquier producto que derive, directa o indirectamente, de las combinaciones de los ingredientes especificados en las cantidades especificadas. Se pretende que este término, con respecto a la composición farmacéutica, abarque un producto que comprenda el/los ingrediente(s) activo(s) y los ingrediente(s) inerte(s) que componen el portador, así como cualquier otro producto que derive, directa o indirectamente, de una combinación, formación compleja o agregación de dos o más ingredientes cualquiera, o de los otros tipos de reacciones o efectos combinados para provocar la disociación de uno o más de los ingredientes. Por lo tanto, las composiciones farmacéuticas divulgadas en la presente memoria abarcan cualquier composición elaborada al mezclar un compuesto de la presente invención y un portador farmacéuticamente aceptable.
Descripciones químicas adicionales
[0059] Cualquier fórmula dada en la pretende memoria pretende representar compuestos que tienen estructuras representadas mediante la fórmula estructural, así como ciertas modificaciones o formas. Por ejemplo, los compuestos de cualquier fórmula dada en la presente memoria pueden tener centros asimétricos o quirales y, por lo tanto, existir en diferentes formas estereoisómeras. Se considera que todos los estereoisómeros, incluyendo los isómeros ópticos, enantiómeros y diastereómeros, de los compuestos de la fórmula general, y mezclas de estos, se encuentran dentro del alcance de la fórmula. Además, ciertas estructuras pueden existir como isómeros geométricos (es decir, isómeros cis y trans), como tautómeros o como atropisómeros. Se contemplan en la presente memoria todas estas formas isoméricas, y mezclas de estas, como parte de la presente invención. Por consiguiente, cualquier fórmula dada en la presente memoria
pretende representar un racemato, una o más formas enantioméricas, una o más formas diastereoméricas, una o más formas tautoméricas o atropisoméricas, y mezclas de estas.
[0060] «Estereoisómero» se refiere a compuestos que tienen una composición química idéntica, pero que difieren respecto de la disposición de los átomos o grupos en el espacio. Los estereoisómeros incluyen enantiómero, diastereómeros, confórmero (rotámero), isómero geométrico (cis/trans), atropisómero, etc.
[0061] «Quiral» se refiere a moléculas que tienen la propiedad no superponible con su imagen especular, mientras que el término «aquiral» se refiere a las moléculas que son superponibles con su imagen especular.
[0062] «Enantiómeros» se refiere a dos estereoisómeros de un compuesto que son imágenes especulares no superponibles entre sí.
[0063] «Diastereómeros» se refiere a un estereoisómero con dos o más centros de quiralidad y cuyas moléculas no son imágenes especulares entre sí Los diastereómeros tienen propiedades físicas diferentes, p. ej., puntos de fusión, puntos de ebullición, propiedades espectrales o actividades biológicas. Se puede separar una mezcla de diastereómeros por medio de procedimientos analíticos de alta resolución como electroforesis y cromatografía, como CLAR.
[0064] Las definiciones y convenciones estereoquímicas usadas en la presente memoria provienen por lo general de S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, New York; y Eliel, E. and Wilen, S., Stereochemistry of Organic Compounds, John Wiley & Sons, Inc., New York, 1994.
[0065] Muchos compuestos orgánicos existen en formas ópticamente activas, es decir, tienen la capacidad de rotar el plano de luz polarizada. Al describir un compuesto ópticamente activo, los prefijos D y L, o R y S, se usan para denotar la configuración absoluta de la molécula sobre su(s) centro(s) quiral(es). Los prefijos d o l o (+) y (-) se emplean para designar el signo de rotación del plano de luz polarizada mediante el compuesto, donde (-) o l significan que el compuesto es levorrotatorio. Un compuesto con el prefijo (+) o d es dextrorrotatorio. Se puede hacer referencia a un estereoisómero específico como un enantiómero, y una mezcla de estos estereoisómeros se denomina mezcla enantiomérica. Se hace referencia a una mezcla 50 : 50 de enantiómeros como una mezcla racémica o un racemato, que se puede producir cuando no ha habido esteroselección o estereoespecificidad en una reacción o un proceso químico.
[0066] Cualquier átomo asimétrico (p. ej., de carbono o similares) del/de los compuesto(s) divulgados en la presente memoria puede estar en configuración racémica o enriquecida enantioméricamente, por ejemplo, (R)-, (S)- o (R, S)-. En ciertos modos de realización, cada átomo asimétrico tiene al menos un 50 % de exceso enantiomérico, al menos un 60 % de exceso enantiomérico, al menos un 70 % de exceso enantiomérico, al menos un 80 % de exceso enantiomérico, al menos un 90 % de exceso enantiomérico, al menos un 95 % de exceso enantiomérico, o al menos un 99 % de exceso enantiomérico en la configuración (R)- o (S)-.
[0067] Dependiendo de la elección de materiales y procedimientos de partida, los compuestos pueden estar presentes en forma de uno de los estereoisómeros posibles o como mezclas de estos, como mezclas de racematos y diastereoisómeros, dependiendo del número de átomos de carbono asimétricos. Los isómeros ópticamente activos (R)- y (S)- se pueden preparar mediante sintones quirales o reactivos quirales, o separarse por medio de técnicas convencionales. Si el compuesto contiene un doble enlace, el sustituyente puede estar en configuración E o Z. Si el compuesto contiene un cicloalquilo disustituido, un sustituyente cicloalquilo puede tener una configuración cis- o trans- en comparación con otro sustituyente de la misma estructura de cicloalquilo.
[0068] Se puede separar cualquier mezcla resultante de estereoisómeros según las diferencias fisicoquímicas de los constituyentes, en los isómeros geométricos puros o esencialmente puros, enantiómeros, diastereómeros, por ejemplo, mediante cromatografía y/o cristalización fraccionada. Cualquier racemato resultante de productos finales o intermedios se puede separar en antípodas ópticas mediante métodos conocidos por los expertos en la materia, p. ej., mediante la separación de las sales diastereoméricas de estos. Los productos racémicos también se pueden separar mediante cromatografía quiral, p. ej., cromatografía líquida de alta resolución (CLAR) con un adsorbente quiral. También se pueden preparar los enantiómeros preferidos mediante síntesis asimétricas. Véanse, por ejemplo, Jacques, et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Principles of Asymmetric Synthesis (2nd Ed. Robert E. Gawley, Jeffrey Aubé, Elsevier, Oxford, UK, 2012); Eliel, E.L. Stereochemistry of Carbon Compounds (McGraw-Hill, NY, 1962); Wilen, S.H. Tables of Resolving Agents and Optical Resolutions p. 268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN 1972); Chiral Separation Techniques: A Practical Approach (Subramanian, G. Ed., Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007).
[0069] Se pueden separar las mezclas diastereoméricas en sus diastereómeros individuales según sus diferencias físicas y químicas mediante métodos conocidos por los expertos en la materia, como, por ejemplo, mediante cromatografía
y/o cristalización fraccionada. Los enantiómeros se pueden separar al convertir la mezcla enantiomérica en una mezcla diastereomérica mediante la reacción con un compuesto ópticamente activo adecuado (p. ej., un auxiliar quiral, como un alcohol quiral o cloruro de ácido de Mosher, o la formación de una mezcla de sales diastereoméricas), la separación de los diastereómeros y la trasformación (p. ej., hidrolización o desalación) de los diastereómeros individuales en los enantiómeros puros correspondientes. Los enantiómeros también se pueden separar por medio de la columna quiral de CLAR.
[0070] Los compuestos para uso según la invención pueden formar sales farmacéuticamente aceptables, que también están dentro del alcance de esta invención. Una «sal farmacéuticamente aceptable» se refiere a una sal de un ácido libre o a una base de un compuesto de fórmula (I) que no es tóxica, es tolerable fisiológicamente, es compatible con la composición farmacéutica en la que se formula, y es, por lo demás, adecuada para formulación y/o administración a un sujeto. Se entiende que la referencia a un compuesto en la presente memoria incluye la referencia a una sal farmacéuticamente aceptable de dicho compuesto a no ser que se indique lo contrario.
[0071] Las sales compuestas incluyen sales ácidas formadas con ácidos inorgánicos y/u orgánicos, así como sales básicas formadas con bases inorgánicas y/u orgánicas. Además, cuando un compuesto dado contiene tanto una fracción básica, como, entre otros, una piridina o un imidazol y una fracción ácida, como, entre otros, un ácido carboxílico, un experto en la materia reconocerá que el compuesto puede existir como un ion dipolar («sal interna»); estas sales se incluyen en el término «sal» tal como se usa en la presente memoria. Las sales de los compuestos para uso según la invención se pueden preparar, por ejemplo, al hacer reaccionar un compuesto con una cantidad de un ácido o una base adecuada, como una cantidad equivalente, en un medio como uno en que la sal precipita o en un medio acuoso seguido de liofilización.
[0072] Las sales ejemplares incluyen, entre otras, sales de sulfato, citrato, acetato, oxalato, cloruro, bromuro, yoduro, nitrato, bisulfato, fosfato, fosfato de ácido, isonicotinato, lactato, salicilato, ácido citrato, tartrato, oleato, tanato, pantotenato, bitartrato, ascorbato, succinato, maleato, gentisinato, fumarato, gluconato, glucuronato, sacarato, formiato, benzoato, glutamato, metanosulfonato («mesilato»), etanosulfonato, bencenosulfonato, p-toluenosulfonato, y de pamoato (es decir, 1,1'-metileno-bis(2-hidroxi-3-naftoato)). Una sal farmacéuticamente aceptable puede implicar la inclusión de otra molécula como un ion acetato, un ion succinato u otro contraión. El contraión puede ser cualquier fracción orgánica o inorgánica que estabilice la carga del compuesto original. Además, una sal farmacéuticamente aceptable puede tener más de un átomo con carga en su estructura. Los casos en que varios átomos con carga son parte de una sal farmacéuticamente aceptable pueden tener varios contraiones. De ahí que una sal farmacéuticamente aceptable pueda tener uno o más átomos con carga y/o uno o más contraiones.
[0073] Las sales de adición de ácido ejemplares incluyen acetatos, ascorbatos, benzoatos, bencenosulfonatos, bisulfatos, boratos, butiratos, citratos, alcanforatos, alcanforsulfonatos, fumaratos, clorhidratos, bromhidratos, yodhidratos, lactatos, maleatos, metanosulfonatos, naftalenosulfonatos, nitratos, oxalatos, fosfatos, propionatos, salicilatos, succinatos, sulfatos, tartratos, tiocianatos, toluenosulfonatos (también conocidos como tosilatos) y similares.
[0074] Las sales básicas ejemplares incluyen sales de amonio, sales de metales alcalinos, como sales de sodio, de litio y de potasio, sales de metales alcalinotérreos como sales de calcio y de magnesio, sales con bases orgánicas (por ejemplo, aminas orgánicas), como diciclohexilaminas, terc-butil aminas, y sales con aminoácidos, como arginina, lisina y similares. Los grupos básicos que contienen nitrógeno se pueden cuartenizar con agentes, como haluros de alquilo inferior (p. ej., cloruros, bromuros y yoduros de butilo, metilo y etilo), sulfatos de dialquilo (p. ej., sulfatos de dimetilo, dietilo y dibutilo), haluros de cadena larga (p. ej., cloruros, bromuros y yoduros de decilo, laurilo y estearilo), haluros de aralquilo (p. ej., bromuros de bencilo y fenetilo) y otros.
[0075] Asimismo, los ácidos y las bases considerados, en general, adecuados para la formación de sales farmacéuticamente útiles a partir de compuestos farmacéuticos se tratan, por ejemplo, en P. Stahl et al, Camille G. (eds.) Handbook of Pharmaceutical Salts. Properties, Selection and Use. (2002) Zurich: Wiley-VCH; S. Berge et al, Journal of Pharmaceutical Sciences (1977) 66(1) 1-19; P. Gould, International J. of Pharmaceutics (1986) 33201-217; Anderson et al, The Practice of Medicinal Chemistry (1996), Academic Press, New York; y en The Orange Book (Food & Drug Administration, MD, disponible en la FDA).
[0076] Asimismo, cualquier compuesto descrito en la presente memoria pretende referirse también a cualquier forma no disuelta, o a un hidrato, solvato o polimorfo de este compuesto, y mezclas de este, incluso si estas formas no se enumeran explícitamente. «Solvato» significa una asociación física de un compuesto de la invención con una o más moléculas de disolvente. Esta asociación física implica diversos grados de enlace iónico y covalente, incluyendo el enlace de hidrógeno. En ciertos casos, el solvato será capaz de aislarse, por ejemplo, cuando una o más moléculas de disolvente se incorporen en la red cristalina de un sólido cristalino. «Solvato» abarca tanto la fase de disolución como los solvatos aislables. Los solvatos adecuados incluyen los formados con disolventes farmacéuticamente aceptables como agua, etanol y similares. En algunos modos de realización, el disolvente es agua y los solvatos son hidratos.
[0077] Uno o más compuestos para uso según la invención se pueden transformar opcionalmente en un solvato. Los métodos de preparación de solvatos son conocidos, en general. Así, por ejemplo, M. Caira et al., J. Pharmaceutical Sci.,
93(3), 601-611 (2004), describe la preparación de los solvatos del fluconazol antifúngico en acetato de etilo, así como del agua. Se describen preparaciones similares de solvatos, hemisolvato, hidratos y similares en E. C. van Tonder et al, AAPS PharmSciTech., 5(1), article 12 (2004); y A. L. Bingham et al, Chem. Commun., 603-604 (2001). Un proceso representativo no limitativo implica disolver el compuesto de la invención en una cantidad adecuada de disolvente (disolvente orgánico o agua o una mezcla de estos) a una temperatura más alta que la ambiente, y enfriar la solución a una velocidad suficiente para formar cristales, que luego se aíslan mediante métodos estándar. Las técnicas analíticas como, por ejemplo, espectroscopia infrarroja, muestran la presencia del disolvente (o agua) en los cristales en forma de solvato (o hidrato).
[0078] La presente divulgación también se refiere a metabolitos farmacéuticamente activos de los compuestos de fórmula (I)a o (I)b, y usa estos metabolitos en los métodos de la invención. Un «metabolito farmacéuticamente activo» significa un producto farmacológicamente activo del metabolismo en el cuerpo de un compuesto de fórmula (I)a o (I)b o una sal de este. Se pueden determinar los metabolitos activos de un compuesto por medio de técnicas habituales conocidas o disponibles en la materia. Véanse, p. ej., Bertolini et al., J. Med. Chem. 1997, 40, 2011-2016; Shan et al., J.
Pharm. Sci. 1997, 86 (7), 765-767; Bagshawe, Drug Dev. Res. 1995, 34, 220-230; Bodor, Adv. Drug Res. 1984, 13, 255
331; Bundgaard, Design of Prodrugs (Elsevier Press, 1985); and Larsen, Design and Application of Prodrugs, Drug Design and Development (Krogsgaard-Larsen et al., eds., Harwood Academic Publishers, 1991).
[0079] Cualquier fórmula dada en la presente memoria también pretende representar formas no marcadas, así como formas marcadas isotópicamente de los compuestos. Los compuestos marcados isotópicamente tienen estructuras representadas mediante las fórmulas dadas en la presente memoria a excepción de que uno o más átomos se reemplazan con un átomo que tiene una masa atómica o un número másico seleccionados. Los ejemplos de isótopos que se pueden incorporar a los compuestos de la invención incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, flúor, cloro y yodo, como 2H, 3H, 11C, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F, 36Cl, y 125I, respectivamente. marcados isotópicamente son útiles en los estudios metabólicos (por ejemplo, con 14C), estudios cinéticos de reacción
(con, por ejemplo, 2H o 3H), técnicas de detección o diagnóstico por imagen [como la tomografía de emisión de positrones
(PET, por sus siglas en inglés) o tomografía computarizada por emisión de fotón único (SPECT, por sus siglas en inglés) incluyendo ensayos de distribución tisular de fármaco o sustrato, o en el tratamiento radiactivo de pacientes. En concreto, un compuesto marcado 18F o 11C puede ser especialmente adecuado para estudios de TEP o SPECT. Además, la sustitución con isótopos más pesados como el deuterio (es decir, 2H) puede plantear ciertas ventajas terapéuticas derivadas de una mayor estabilidad metabólica, por ejemplo, vida media in vivo aumentada o requisitos de posología reducidos. Los compuestos marcados isotópicamente de esta invención se pueden preparar, en general, al llevar a cabo los procedimientos divulgados en los esquemas o en los ejemplos y las preparaciones descritas a continuación al sustituir un reactivo marcado isotópicamente disponible fácilmente por un reactivo no marcado isotópicamente.
[0080] Se pretende que el uso de los términos «sal», «solvato», «polimorfo» y similares, con respecto a los compuestos descritos en la presente memoria, se apliquen por igual a la sal, el solvato y las formas polimorfas de enantiómeros, estereoisómeros, rotámeros, tautómeros, atropisómeros y racematos de los compuestos de la invención.
Abreviatura:


DESCRIPCIÓN DE LOS COMPUESTOS DE LA INVENCIÓN
[0081] La presente invención se refiere a un compuesto seleccionado de entre Compuesto (III), Compuesto (XXVIII), Compuesto (XXIV), Compuesto (XXX), Compuesto (Xx XI), Compuesto (XXXII), Compuesto (Xx X iII), Compuesto (XXXIV), Compuesto (XXXV), Compuesto (Xx Xv I), Compuesto (x Xx VII), Compuesto (Xx XVIII), Compuesto (XXXIX), Compuesto (XXXXIV), Compuesto (XXXXV), Compuesto (XXXXVI), y Compuesto (XXXx V iI), o una sal farmacéuticamente aceptable de cualquiera de los anteriores, para su uso en un método de inhibición de la ferroptosis en un paciente. Un aspecto de esta divulgación consiste en los compuestos, las composiciones, los kits y los antídotos para modular la transmisión de glutamato en mamíferos que tienen un compuesto de fórmula (I), sales farmacéuticamente aceptables de estos y enantiómeros o diastereómeros individuales de estos

donde
x es 0 o 1;
G está ausente, O, NH o CH2;
W es:

Donde R1, R2, R3, o R4 es alquilo C1-C6, R5 es OH, u O,
n es 0,1,2 o 3,
L está ausente, o se selecciona de entre un grupo que consiste en alquilo C1-C10, alcoxilo C1-C10, cicloalquilo, alquilcicloalquilo C1-C10, heterocicloalquilo, alquilheterocicloalquilo C1-C10, arilo, alquilarilo C1-C10, heteroarilo, y alquilheteroarilo C1-C10, donde dicho cicloalquilo o heterocicloalquilo tiene entre aprox. 3 y aprox. 7 carbonos en el anillo, y dicho arilo o heteroarilo tiene entre aprox. 5 y aprox. 10 carbonos en el anillo;
PEP es una fracción peptídica que tiene la siguiente estructura:

donde un símbolo * denota un centro quiral en una configuración (S) o (R);
Bestá ausente, N, NH, o O;
X1 se selecciona de entre un grupo que consiste en H, -(C = O)-O-Rm, -(SO)-O-Rm, -(SOz)-O-Rm, -(SO2)-N-(Rm)2 y -(C = O)-N-(Rm)2, donde Rm es alquilo C1-C6, alcoxilo C1-C6, alquenilo C1-C6, haloalquilo C1-C6, cicloalquilo, alquilcicloalquilo C1-C6, heterocicloalquilo, alquilheterocicloalquilo C1-C6, arilo, alquilarilo C1-C6, heteroarilo, o alquilheteroarilo C1-C6,
donde dicho cicloalquilo o heterocicloalquilo tiene entre aprox. 3 y aprox. 7 carbonos en el anillo, y dicho arilo o heteroarilo tiene entre aprox. 5 y aprox.10 carbonos en el anillo;
Rn se selecciona de entre -O-X3, -S-X3, -NH-X3, o ~N-X4a(X4b);
X2 o X3 se selecciona de entre un grupo que consiste en H, alquilo C1-C10, alquenilo C1-C10, arilo, alquilarilo C1-C6, heteroarilo, y alquilheteroarilo C1-C6, donde dicho arilo o heteroarilo tiene entre 5 y 6 carbonos en el anillo; X4a y X4b se seleccionan por separado de entre alquilo C1-C6, alquilo C1-C6 sustituido; X4a y X4b pueden, opcionalmente, formar juntos heterocicloalquilo C3-C8, heterocicloalquilo C3-C8 sustituido.
A está ausente, o se selecciona de entre un grupo que consiste en: alanina, leucina, isoleucina, fenilalanina, metionina, prolina, glicina, triptófano, serina, treonina, cisteína, tirosina, asparagina, glutamina, histidina, lisina, arginina, ácido aspártico, ácido glutámico, y valina, cada uno de los cuales puede estar en una configuración D o L; el residuo de amino está desprotegido, o protegido por un grupo protector seleccionado de entre un grupo que consiste en Cbz y Fmoc;
con la condición de que PEP no sea uno de los siguientes:



[0082] El compuesto de fórmula (I) se ilustra además mediante el siguiente grupo compuesto que consiste en:

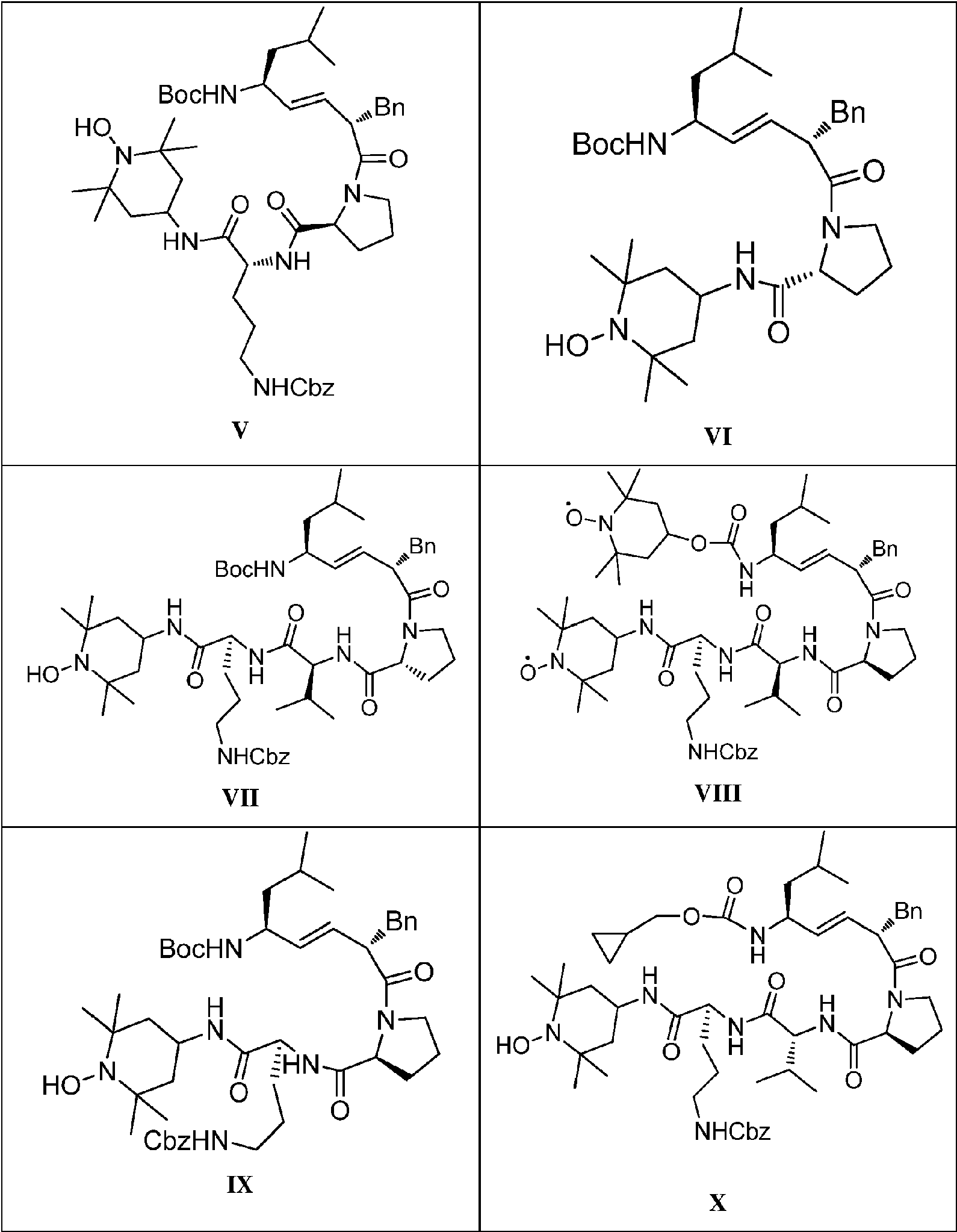







y sales farmacéuticamente aceptables de este.
[0083] La presente invención se refiere a un compuesto seleccionado de entre Compuesto (III), Compuesto (XXVIII), Compuesto (XXIV), Compuesto (XXX), Compuesto (Xx XI), Compuesto (XXXII), Compuesto (XXXIII), Compuesto (XXXIV), Compuesto (XXXV), Compuesto (XXXVI), Compuesto (XXXVII), Compuesto (XXXVIII), Compuesto (XXXIX), Compuesto (XXXXIV), Compuesto (x Xx XV), Compuesto (XXXXVI), y Compuesto (XXXx V iI), o una sal farmacéuticamente aceptable de cualquiera de los anteriores, para su uso en un método de inhibición de la ferroptosis en un paciente.
[0084] Un aspecto de la presente invención está relacionado con compuestos de la presente invención para su uso en el tratamiento, prevención, inhibición o eliminación de un trastorno o una enfermedad relacionada con el estrés oxidativo en un paciente.
[0085] Un aspecto de la presente invención está relacionado con los compuestos de la presente invención para su uso como un modulador de ferroptosis y para su uso en el tratamiento, la prevención, la inhibición o la eliminación de un trastorno o una enfermedad o una afección médica en un paciente al modular la ferroptosis en dicho paciente.
COMPOSICIÓN FARMACÉUTICA DE LOS COMPUESTOS DIVULGADOS EN LA PRESENTE MEMORIA Y PREPARACIÓN Y ADMINISTRACIÓN
[0086] La presente divulgación proporciona una composición farmacéutica que comprende compuestos de la presente divulgación, p. ej., compuestos de ejemplo. De acuerdo con los ejemplos específicos, la composición farmacéutica puede además comprender un excipiente, portador, adyuvante, disolvente farmacéuticamente aceptables y una combinación de estos.
[0087] La presente divulgación proporciona un método para tratar, prevenir o aliviar una enfermedad o un trastorno, que comprende administrar una cantidad segura y eficaz de una composición farmacéutica que contiene compuestos divulgados en la presente memoria con, opcionalmente, uno o más agentes activos terapéuticos auxiliares. La cantidad del compuesto de la composición farmacéutica divulgada en la presente memoria se refiere a una cantidad que puede ser efectivamente detectada para modular la ferroptosis en muestras biológicas y en un paciente. Se puede administrar el ingrediente activo a sujetos que necesiten este tratamiento en dosis que proporcionarán la eficacia farmacéutica ideal, que no se limita a los efectos terapéuticos deseados, en la vía de administración y en la duración del tratamiento. La posología variará de un paciente a otro dependiendo de la naturaleza y gravedad de la enfermedad, del peso del paciente, la dieta en particular que siga el paciente, el tratamiento concomitante y otros factores que reconocerán los expertos en la materia.
[0088] La posología real empleada puede variar dependiendo de las necesidades del paciente y la gravedad de la afección tratada. La determinación de la pauta posológica adecuada para una situación concreta no excede las habilidades en la materia. Por comodidad, la dosis diaria total se puede dividir y administrar en porciones durante el día según sea necesario. La cantidad y frecuencia de administración de los compuestos divulgados en la presente memoria y/o de sus sales farmacéuticamente aceptables se regulará de acuerdo con el criterio del médico tratante en función de factores como la edad, la afección y el tamaño del paciente, así como la gravedad de los síntomas tratados.
[0089] También se apreciará que ciertos compuestos divulgados en la presente memoria pueden existir de forma libre para el tratamiento, o si procede, como un derivado farmacéuticamente aceptable o un profármaco de estos. Un derivado farmacéuticamente aceptable incluye sales, ésteres, sales de estos ésteres farmacéuticamente aceptables o cualquier otro aducto o derivado que, al administrarse a un paciente que lo necesite, proporciona, directa o indirectamente, un compuesto como se describe en la presente memoria o un metabolito o residuo terapéuticamente eficaz de este.
[0090] Las composiciones farmacéuticas se pueden preparar y empaquetar a granel donde una cantidad segura y eficaz de un compuesto de fórmula (I) divulgado en la presente memoria se puede extraer y luego administrar al paciente, como con polvos o jarabes. En general, se administran al paciente niveles de dosificación de entre 0.0001 y 10 mg/kg de peso corporal al día para lograr una modulación eficaz de la transmisión disfuncional de glutamato. Alternativamente, las composiciones farmacéuticas se pueden preparar y empaquetar en forma de dosis unitarias donde cada unidad físicamente independiente contiene una cantidad segura y eficaz de un compuesto de fórmula (I) divulgado en la presente memoria.
[0091] Cuando las composiciones farmacéuticas de la presente divulgación también contienen uno o más ingredientes activos, además de un compuesto de la presente divulgación, la relación en peso del compuesto de la presente divulgación con el segundo ingrediente activo puede variar y depender de la dosis efectiva de cada ingrediente. Así, por ejemplo, cuando un compuesto de la presente divulgación se combina con otro agente, la relación en peso del compuesto de la
presente divulgación con el otro agente oscilará en general entre aprox. 1000 : 1 y aprox. 1 : 1000, como entre aprox. 200 : 1 y 1 : 200. Las combinaciones de un compuesto de la presente divulgación y otros ingredientes activos estarán, en general, dentro del intervalo anteriormente mencionado, pero se debería usar una dosis efectiva de cada ingrediente activo en la combinación en cada caso.
[0092] «Excipiente farmacéuticamente aceptable», tal como se usa en la presente memoria se refiere a un material, una composición o un vehículo farmacéuticamente aceptables implicados en dar forma o consistencia a la composición farmacéutica. Cada excipiente debe ser compatible con los demás ingredientes de la composición farmacéutica cuando se entremezclan, de manera que los efectos combinados que reducirían esencialmente la eficacia del compuesto divulgado en la presente memoria cuando se administra a un paciente y que darían lugar a composiciones farmacéuticamente inaceptables. Además, cada excipiente debe ser de una pureza suficientemente alta para que sea farmacéuticamente aceptable.
[0093] Los excipientes farmacéuticamente aceptables variarán dependiendo de la forma concreta de dosificación elegida. Además, los excipientes farmacéuticamente aceptables se pueden elegir para que tengan una función concreta en la composición. Por ejemplo, se pueden elegir ciertos excipientes farmacéuticamente aceptables por su capacidad para facilitar la producción de formas de dosificación uniformes. Ciertos excipientes farmacéuticamente aceptables se pueden elegir por su capacidad para facilitar la producción de formas de dosificación estables. Ciertos excipientes farmacéuticamente aceptables se pueden elegir por su capacidad para facilitar el porte o transporte de los compuestos divulgados en la presente memoria de un órgano o parte del cuerpo a otro órgano o parte del cuerpo una vez administrado al paciente. Ciertos excipientes farmacéuticamente aceptables se pueden elegir por su capacidad para mejorar la adhesión del paciente al tratamiento.
[0094] Los excipientes farmacéuticamente aceptables adecuados incluyen los siguientes tipos de excipientes: diluyentes, rellenos, aglutinantes, disgregantes, lubrificantes, deslizantes, agentes de granulación, agentes de recubrimiento, agentes humectantes, disolventes, codisolventes, agentes de suspensión, emulsionantes, edulcorantes, agentes saborizantes, agentes enmascaradores del sabor, agentes colorantes, agentes antiaglomerantes, humectantes, agentes quelantes, plastificantes, agentes para aumentar la viscosidad, antioxidantes, conservantes, estabilizadores, tensoactivos y agentes amortiguadores. El experto en la materia apreciará que ciertos excipientes farmacéuticamente aceptables pueden tener más de una función o pueden tener funciones alternativas dependiendo de la cantidad de excipiente presente en la formulación y de qué otros ingredientes están presentes en la formulación.
[0095] Los expertos en la materia poseen el conocimiento y la habilidad en la materia para poder seleccionar excipientes farmacéuticamente aceptables en cantidades adecuadas. Además, existen recursos disponibles para los expertos en la materia que describen excipientes farmacéuticamente aceptables y que pueden ser útiles para seleccionar excipientes farmacéuticamente aceptables adecuados. Los ejemplos incluyen Remington's Pharmaceutical Sciences (Mack Publishing Company), The Handbook of Pharmaceutical Additives (Gower Publishing Limited), y The Handbook of Pharmaceutical Excipients (the American Pharmaceutical Association and the Pharmaceutical Press).
[0096] En Remington: The Science and Practice of Pharmacy, 21st edition, 2005, ed. D.B. Troy, Lippincott Williams & Wilkins, Philadelphia, y Encyclopedia of Pharmaceutical Technology, eds. J. Swarbrick and J. C. Boylan, 1988-1999, Marcel Dekker, New York, se divulgan varios portadores usados en la formulación de composiciones farmacéuticamente aceptables y técnicas conocidas para la preparación de estas. Excepto en la medida en que cualquier medio portador sea incompatible con los compuestos divulgados en la presente memoria, como al producir cualquier efecto biológico no deseado o al tener un efecto combinado nocivo con cualquier/cualesquiera otro(s) compuesto(s) de la composición farmacéuticamente aceptable, cuyo uso se contempla en el alcance de esta divulgación.
[0097] Las composiciones farmacéuticas divulgadas en la presente memoria se preparan por medio de técnicas y métodos conocidos para los expertos en la materia. Se describen algunos de los métodos usados normalmente en la materia en Remington's Pharmaceutical Sciences (Mack Publishing Company).
[0098] Por lo tanto, otro aspecto de la presente divulgación está relacionado con un método para preparar una composición farmacéutica. La composición farmacéutica contiene el compuesto divulgado en la presente memoria y un excipiente farmacéuticamente aceptable, portador, adyuvante, vehículo o una combinación de estos, el método comprende mezclar varios ingredientes. La composición farmacéutica que contiene el compuesto divulgado en la presente memoria se puede preparar, por ejemplo, a una temperatura ambiente y presión normales.
[0099] El compuesto de la divulgación se formulará, por lo general, en una forma de dosificación adaptada para administración al paciente mediante la vía de administración deseada. Por ejemplo, las formas de dosificación incluyen las adaptadas para (1) administración por vía oral, como comprimidos, cápsulas, pastillas, píldoras, grageas, polvos, jarabes, elixires, suspensiones, soluciones, emulsiones, sobres y obleas; (2) administración por vía parenteral, como soluciones, suspensiones y polvos estériles para reconstitución; (3) administración por vía transdérmica, como parches
transdérmicos; (4) administración por vía rectal, como supositorios; (5) inhalación, como aerosoles, soluciones y polvos secos; y (6) administración por vía tópica, como cremas, pomadas, lociones, soluciones, ungüentos, pulverizadores, espumas y geles.
[0100] Las composiciones farmacéuticas proporcionadas en la presente memoria se pueden proporcionar como comprimidos de capa, comprimidos triturados, pastillas para chupar masticables, comprimidos de disolución rápida, comprimidos multicapa, o comprimidos con recubrimiento entérico, comprimidos recubiertos de azúcar o con cubierta pelicular. Los comprimidos con recubrimiento entérico son comprimidos de capa recubiertos con sustancias que resisten la acción del ácido gástrico, pero se disuelven o desintegran en el intestino, que por consiguiente protege los ingredientes activos del ambiente ácido del estómago. Los recubrimientos entéricos incluyen, entre otros, ácidos grasos, grasas, salicilato de fenilo, ceras, goma laca, goma laca amónica y acetoftalatos de celulosa. Los comprimidos recubiertos de azúcar son comprimidos de capa con un recubrimiento de azúcar alrededor, que puede ser beneficioso para enmascarar sabores u olores desagradables y proteger los comprimidos contra la oxidación. Los comprimidos con cubierta pelicular son comprimidos de capa recubiertos por una capa o película delgada de un material hidrosoluble. Los recubrimientos peliculares incluyen, entre otros, hidroxietilcelulosa, carboximetil celulosa sódica, polietilenglicol 4000 y acetoftalato de celulosa. Los recubrimientos peliculares presentan las mismas características generales que el recubrimiento de azúcar. Los comprimidos multicapa son comprimidos de capa hechos mediante más de un ciclo de compresión, incluyendo comprimidos de varias capas, y comprimidos recubiertos a presión o en seco.
[0101] La forma de dosificación del comprimido se puede preparar a partir del ingrediente activo de forma pulverizada, cristalina o granulada, por sí solo o junto con uno o más portadores o excipientes descritos en la presente memoria, incluyendo aglutinantes, disgregantes, polímeros para liberación controlada, lubrificantes, diluyentes y/o colorantes. Los agentes saborizantes y edulcorantes son especialmente útiles en la formación de comprimidos masticables y pastillas para chupar.
[0102] Las composiciones farmacéuticas proporcionadas en la presente memoria se pueden proporcionar como cápsulas duras o blandas, que se pueden elaborar a partir de gelatina, metilcelulosa, almidón o alginato de calcio. La cápsula de gelatina dura, también conocida como cápsula de llenado en seco (DFC, por sus siglas en inglés), consta de dos secciones, una que se desliza sobre la otra, que por consiguiente rodea el ingrediente activo por completo. La cápsula elástica blanda (SEC, por sus siglas en inglés) es una cubierta blanda globular, como una cubierta de gelatina blanda, que se plastifica mediante la adición de glicerina, sorbitol o un polialcohol similar. Las cubiertas de gelatina blanda pueden contener un conservante para impedir el crecimiento de microorganismos. Los conservantes adecuados son los que se describen en la presente memoria, incluyendo metil- y propilparabenos, y ácido ascórbico. Las formas de dosificación líquidas, semisólidas y sólidas proporcionadas en la presente memoria se pueden encapsular en una cápsula. Las formas de dosificación líquidas y semisólidas adecuadas incluyen soluciones y suspensiones en carbonato de propileno, aceites vegetales o triglicéridos. Las cápsulas que contienen estas soluciones se pueden preparar como se describe en las pat. de EE. UU. N.° 4,328,245; 4,409,239; y 4,410,545. Las cápsulas también se pueden recubrir de formas conocidas por los expertos en la materia para modificar o mantener la disolución del ingrediente activo.
[0103] Las composiciones farmacéuticas proporcionadas en la presente memoria se pueden proporcionar en formas de dosificación líquidas o semisólidas, incluyendo emulsiones, soluciones, suspensiones, elixires y jarabes. Una emulsión es un sistema bifásico en que un líquido se dispersa en forma de pequeños glóbulos en otro líquido, que puede ser aceite en agua o agua en aceite. Las emulsiones pueden incluir un líquido o disolvente no acuosos, un agente emulsionante y un conservante farmacéuticamente aceptables. Las suspensiones pueden incluir un agente de suspensión y conservante farmacéuticamente aceptables. Las soluciones acuosas alcohólicas pueden incluir un acetal farmacéuticamente aceptable, como un di(alquil inferior)acetal de un aldehído de alquilo inferior, p. ej., acetaldehído dietil acetal; y un disolvente miscible en agua que tenga uno o más grupos hidroxi, como propilenglicol y etanol. Los elixires son soluciones claras, edulcoradas e hidroalcohólicas. Los jarabes son soluciones acuosas concentradas de un azúcar, por ejemplo, sacarosa, y que también pueden contener un conservante. Para una forma de dosificación líquida, por ejemplo, una solución en un polietilenglicol se puede diluir con una cantidad suficiente de un portador líquido farmacéuticamente aceptable, p. ej., agua, que se debe medir de forma adecuada para su administración.
[0104] Otras formas de dosificación líquidas y semisólidas incluyen, entre otras, las que contienen el/los ingrediente(s) activo(s) proporcionados en la presente memoria y un mono- o polialquilenglicol dialquilado, incluyendo 1,2-dimetoximetano, diglima, triglima, tetraglima, polietilenglicol-350-dimetiléter, polietilenglicol-550-dimetiléter, polietilenglicol-750-dimetiléter, donde 350, 550, 750 se refieren al peso molecular medio aproximado del polietilenglicol. Estas formulaciones pueden comprender, además, uno o más antioxidantes, como butilhidroxitolunol (BHT), butilhidroxianisol (BHA), galato de propilo, vitamina E, hidroquinona, hidroxicumarinas, etanolamina, lecitina, cefalina, ácido ascórbico, ácido málico, sorbitol, ácido fosfórico, bisulfito, metabisulfito de sodio, tiodipropiónico y sus ésteres, y ditiocarbamatos.
[0105] Si procede, las formulaciones de unidades de dosificación para administración por vía oral se pueden microencapsular. La formulación también se puede preparar para prolongar o mantener la liberación, como, por ejemplo, al recubrir o integrar un material concreto en polímeros, cera o similares.
[0106] Las composiciones farmacéuticas proporcionadas en la presente memoria para administración por vía oral también se pueden proporcionar en forma de liposomas, micelas, microesferas o nanosistemas. Las formas de dosificación micelar se pueden preparar según se describe en la pat. EE. UU. N.° 6,350,458.
[0107] Las composiciones farmacéuticas proporcionadas en la presente memoria también se pueden proporcionar como gránulos o polvos efervescentes o no efervescentes, que se deben reconstituir en forma de dosificación líquida. Los portadores y excipientes farmacéuticamente aceptables usados en los gránulos o polvos no efervescentes pueden incluir diluyentes, edulcorantes y agentes humectantes. Los portadores y excipientes farmacéuticamente aceptables usados en los gránulos o polvos efervescentes pueden incluir ácidos orgánicos y una fuente de dióxido de carbono.
[0108] Los agentes colorantes y saborizantes se pueden usar en todas las formas de dosificación anteriores.
[0109] Los compuestos divulgados en la presente memoria se pueden acoplar a polímeros solubles como portadores de medicamentos dirigidos. Estos polímeros pueden abarcar polivinilpirrolidona, copolímero de pirano, polihidroxipropilmetacrilamidofenol, polihidroxietilaspartamidofenol u óxido de polietileno polilisina, sustituidos por radicales de palmitoilo. Los compuestos además se pueden acoplar a una clase de polímeros biodegradables adecuados para lograr una liberación controlada de un medicamento, por ejemplo, ácido poliláctico, poli-épsilon-caprolactona, ácido polihidroxibutírico, poliortoésteres, poliacetales, polidihidroxipiranos, policianoacrilatos y copolímeros bloque reticulados o anfipáticos de hidrogeles.
[0110] Las composiciones farmacéuticas proporcionadas en la presente memoria se pueden formular como formas de dosificación de liberación inmediata o modificada, incluyendo formas de liberación retardada, sostenida, pulsátil, controlada, dirigida y programada.
[0111] Las composiciones farmacéuticas proporcionadas en la presente memoria se pueden coformular con otros ingredientes activos que no deterioren la acción terapéutica deseada, o con otras sustancias que complementen la acción deseada.
[0112] Las composiciones farmacéuticas proporcionadas en la presente memoria se pueden administrar por vía parenteral mediante inyección, infusión o implantación para administración local o sistémica. La administración por vía parenteral, tal como se usa en la presente memoria, incluye administración por vía intravenosa, intraarterial, intraperitoneal, intratecal, intraventricular, intrauretral, intraesternal, intracraneal, intramuscular, intrasinovial y subcutánea.
[0113] Las composiciones farmacéuticas proporcionadas en la presente memoria se pueden formular en cualquier forma de dosificación que sea adecuada para administración por vía parenteral, incluyendo soluciones, suspensiones, emulsiones, micelas, liposomas, microesferas, nanosistemas y formas sólidas adecuadas para soluciones o suspensiones en líquido antes de inyectarse. Estas formas de dosificación se pueden preparar de acuerdo con métodos convencionales conocidos por los expertos en la materia de las ciencias farmacéuticas (véase Remington: The Science and Practice of Pharmacy, arriba).
[0114] Las composiciones farmacéuticas destinadas a la administración por vía parenteral pueden incluir uno o más portadores y excipientes farmacéuticamente aceptables, incluyendo, entre otros, vehículos acuosos, vehículos miscibles en agua, vehículos no acuosos, agentes antimicrobianos o conservantes contra el crecimiento de microorganismos, estabilizadores, potenciadores de la solubilidad, agentes isotónicos, agentes amortiguadores, antioxidantes, anestésicos locales, agentes de suspensión y dispersión, agentes humectantes o emulsionantes, agentes complejantes, agentes secuestrantes o quelantes, crioprotectores, lioprotectores, agentes espesantes, agentes reguladores del pH y gases inertes.
[0115] Los vehículos acuosos adecuados incluyen, entre otros, agua, suero, suero fisiológico, solución tampón de fosfatos (STF), inyección de cloruro sódico, solución inyectable de Ringer, inyección de dextrosa isotónica, inyección de agua estéril, solución láctica inyectable de Ringer y de dextrosa. Los vehículos no acuosos incluyen, entre otros, aceites fijos de origen vegetal, aceite de ricino, aceite de maíz, aceite de semilla de algodón, aceite de oliva, aceite de cacahuete, aceite de menta, aceite de alazor, aceite de sésamo, aceite de soja, aceites vegetales hidrogenados, aceite de soja hidrogenado, triglicéridos de cadena media de aceite de coco y aceite de palmiste. Los vehículos miscibles en agua incluyen, entre otros, etanol, 1,3-butanodiol, polietilenglicol líquido (p. ej., polietilenglicol 300 y polietilenglicol 400), propilenglicol, glicerina, N-metil-2-pirrolidona, N, N-dimetilacetamida y dimetil sulfóxido.
[0116] Los agentes antimicrobianos o conservantes adecuados incluyen, entre otros, fenoles, cresoles, mercuriales, alcohol bencílico, clorobutanol, metilo y propilo p-hidroxibenzoatos, timerosal, cloruro de benzalconio (p. ej., cloruro de bencetonio), metil- y propilparabenos, y ácido sórbico. Los agentes isotónicos adecuados incluyen, entre otros, cloruro de sodio, glicerina y dextrosa. Los agentes amortiguadores adecuados incluyen, entre otros, fosfato y citrato. Los antioxidantes adecuados son los descritos en la presente memoria, incluyendo bisulfito y metabisulfito de sodio. Los anestésicos locales adecuados incluyen, entre otros, clorhidrato de procaína. Los agentes de suspensión y dispersión son los descritos en la presente memoria, incluyendo carboximetil celulosa sódica, hidroxipropil metilcelulosa y polivinilpirrolidona. Los agentes emulsionantes adecuados incluyen los descritos en la presente memoria, incluyendo monolaurato de sorbitán polioxietilenado, monooleato de sorbitán polioxietilenado 80 y oleato de trietanolamina. Los agentes secuestrantes o quelantes incluyen, entre otros, AEDT. Los agentes reguladores del pH incluyen, entre otros, hidróxido de sodio, ácido clorhídrico, ácido cítrico y ácido láctico. Los agentes complejantes adecuados incluyen, entre otros, ciclodextrinas, incluyendo a-ciclodextrina, S-ciclodextrina, hidroxipropil-S-ciclodextrina, sulfobutiléter-S-ciclodextrina y sulfobutiléter 7-S-ciclodextrina (CAPTISOL®, CyDex, Lenexa, Kans.).
[0117] Las composiciones farmacéuticas proporcionadas en la presente memoria se pueden formular para la administración de dosis únicas o múltiples. Las formulaciones de dosis únicas se empaquetan en una ampolla, un vial o una jeringuilla. Las formulaciones parenterales de dosis múltiples deben contener un agente antimicrobiano en concentraciones bacteriostáticas o fungistáticas. Todas las formulaciones parenterales deben ser estériles, como se conoce y se practica en la materia.
[0118] En un modo de realización, las composiciones farmacéuticas se proporcionan como soluciones estériles listas para usar. En otro modo de realización, las composiciones farmacéuticas se proporcionan como productos secos solubles estériles, incluyendo polvos liofilizados y comprimidos hipodérmicos que se deben reconstituir con un vehículo estéril antes de usarse. En otro modo de realización distinto, las composiciones farmacéuticas se proporcionan como suspensiones estériles listas para usar. En otro modo de realización distinto, las composiciones farmacéuticas se proporcionan como productos secos insolubles estériles que se deben reconstituir con un vehículo antes de usarse. En un modo de realización adicional, las composiciones farmacéuticas se proporcionan como emulsiones estériles listas para usar.
[0119] Las composiciones farmacéuticas se pueden formular como una suspensión, un líquido sólido, semisólido o tixotrópico, para administración como un depósito implantado. En un modo de realización, las composiciones farmacéuticas proporcionadas en la presente memoria se dispersan en una matriz interna sólida, que está rodeada por una membrana polimérica exterior que es insoluble en fluidos corporales, pero que permite que se difunda a través el ingrediente activo en las composiciones farmacéuticas.
[0120] Las matrices internas adecuadas incluyen polimetilmetacrilato, polibutilmetacrilato, cloruro de polivinilo plastificado o no plastificado, nailon plastificado, tereftalato de polietileno plastificado, caucho natural, poliisopreno, poliisobutileno, polibutadieno, polietileno, copolímeros de etilvinilacetato, cauchos de silicona, polidimetilsiloxanos, copolímeros de carbonato de silicona, polímeros hidrofílicos, como hidrogeles de ésteres de ácido acrílico y metacrílico, colágeno, alcohol polivinílico reticulado y acetato de polivinilo hidrolizado parcialmente reticulado.
[0121] Otras membranas poliméricas exteriores adecuadas incluyen polietileno, polipropileno, copolímeros de etileno/propileno, copolímeros de etileno/acrilato de etilo, copolímeros de etileno/acetato de vinilo, cauchos de silicona, polidimetilsiloxanos, caucho de neopreno, polietileno clorado, cloruro de polivinilo, copolímeros de cloruro de vinilo con acetato de vinilo, cloruro de vinilideno, etileno y propileno, ionómero de tereftalato de polietileno, cauchos de epiclorhidrina de caucho butílico, copolímero de etileno/alcohol vinílico, terpolímero de etileno/acetato de vinilo/alcohol vinílico y copolímero de etileno/viniloxietanol.
[0122] La composición farmacéutica divulgada en la presente memoria se prepara en una forma de dosificación adaptada para administrarse a un paciente mediante inhalación como, por ejemplo, un polvo seco, un aerosol, una suspensión o una composición en solución. La presente divulgación proporciona una forma de dosificación adaptada para administrarse a un paciente mediante inhalación como un polvo seco. La presente divulgación proporciona una forma de dosificación adaptada para su administración a un paciente mediante inhalación como un polvo seco. Por lo general, para que las composiciones de polvo seco lleguen al pulmón mediante inhalación comprenden un compuesto divulgado en la presente memoria o una sal farmacéuticamente aceptable de este polvo muy fino junto con uno o más excipientes farmacéuticamente aceptables en forma de polvos muy finos. Los excipientes farmacéuticamente aceptables aptos específicamente para uso en polvos secos son conocidos por los expertos en la materia e incluyen lactosa, almidón, manitol, y mono-, di- y polisacáridos. Se puede preparar polvo muy fino mediante, por ejemplo, micronización y molienda. En general, el compuesto de tamaño reducido (p. ej. micronizado) se puede definir mediante un valor D50 de entre aprox.
1 y aprox. 10 micras (por ejemplo, medido por medio de difracción láser).
[0123] Se pueden formar aerosoles al suspender o disolver un compuesto divulgado en la presente memoria o una sal farmacéuticamente aceptable de este en un propelente licuado. Los propelentes adecuados incluyen halocarbonos, hidrocarburos y otros gases licuados. Los propelentes representativos incluyen: triclorofluorometano (propelente 11), diclorofluorometano (propelente 12), diclorotetrafluoroetano (propelente 114), tetrafluoretano (HFA-134a), 1,1-difluoroetano (HFA-152a), difluorometano (HFA-32), pentafluoroetano (HFA-12), heptafluoropropano (HFA-227a), perfluoropropano, perfluorobutano, perfluoropentano, butano, isobutano y pentano. Por lo general, los aerosoles que comprenden un compuesto de fórmula (I) o una sal farmacéuticamente aceptable de este se administrarán al paciente por medio de un inhalador dosificador (ID). Estos dispositivos son conocidos los expertos en la materia.
[0124] El aerosol puede contener excipientes farmacéuticamente aceptables usados, por lo general, con ID, como tensoactivos, lubrificantes, codisolventes y otros excipientes que mejoran la estabilidad física de la formulación, para mejorar el funcionamiento de la válvula, para mejorar la solubilidad o para mejorar el sabor.
[0125] Las composiciones farmacéuticas adaptadas para la administración por vía transdérmica se pueden presentar como parches discretos destinados a permanecer en contacto estrecho con la epidermis del paciente durante un periodo de tiempo prolongado. Por ejemplo, el ingrediente activo se puede liberar del parche mediante iontoforesis, como se describe de forma general en Pharmaceutical Research, 3(6), 318 (1986).
[0126] Las composiciones farmacéuticas adaptadas para administrarse por vía tópica se pueden formular como pomadas, cremas, suspensiones, lociones, polvos, soluciones, ungüentos, geles, pulverizadores, aerosoles y aceites. Las pomadas, cremas y geles se pueden formular, por ejemplo, con una base acuosa u oleosa con la adición de un agente espesante y/o gelificante y/o disolventes adecuados. Por consiguiente, estas bases pueden, por ejemplo, incluir agua y/o un aceite, como aceite de parafina o un aceite vegetal, como aceite de cacahuete o aceite de ricino, o un disolvente, como polietilenglicol. Los agentes espesantes y gelificantes que se pueden usar de acuerdo con el tipo de base incluyen vaselina, estearato de aluminio, alcohol de cetoestearilo, polietilenglicoles, lanolina, cera de abejas, carboxipolimetileno y derivados de celulosa, y/o monoestearato de glicerilo y/o agentes emulsionantes no iónicos.
[0127] Las lociones se pueden formular con una base acuosa u oleosa y en general también contendrán uno o más agentes emulsionantes, agentes estabilizadores, agentes de dispersión, agentes de suspensión o agentes espesantes.
[0128] Cualquier base en polvo adecuada, como talco, lactosa o almidón, puede servir de soporte para la formación de polvos para aplicación externa. Se pueden formular gotas con una base acuosa o no acuosa que comprendan también uno o más agentes de dispersión, agentes solubilizantes, agentes de suspensión o conservantes.
[0129] Los preparados tópicos se pueden administrar por medio de una o más aplicaciones al día en la zona afectada; el uso de vendajes oclusivos sobre zonas de piel puede ser beneficioso. Se puede lograr la administración continuada o prolongada por medio de un sistema de reservorio adhesivo.
USOS DE LOS COMPUESTOS DE LA INVENCIÓN
[0130] Los compuestos de la invención divulgados en la presente memoria se usan en una cantidad que se puede detectar de forma efectiva para modular la ferroptosis en muestras biológicas o en un trastorno o enfermedad relacionados, en un paciente. Estos usos se pueden incluir en la fabricación de un medicamento para tratar, prevenir, mejorar o mitigar un trastorno o enfermedad en un sujeto, así como otros medicamentos para modular la ferroptosis, y los compuestos de esta invención tienen propiedades farmacocinéticas y farmacodinámicas superiores, menos efectos secundarios tóxicos.
[0131] En concreto, la cantidad del compuesto de la presente invención puede demostrar que modula la ferroptosis de forma detectable y eficaz en muestras biológicas o en un trastorno o una enfermedad relacionados, en un sujeto. Los compuestos de la invención se pueden usar para prevenir, tratar o aliviar enfermedades relacionadas con la ferroptosis en pacientes.
[0132] En un modo de realización, las terapias divulgadas en la presente memoria comprenden la administración de una cantidad segura y eficaz del compuesto de la invención a pacientes que la necesiten. Cada ejemplo divulgado en la presente comprende el método de tratamiento de las enfermedades anteriores que comprende la administración de una cantidad segura y eficaz del compuesto de la invención a pacientes que la necesiten.
[0133] En un modo de realización, el compuesto de la invención se puede administrar por medio de una vía de administración adecuada, incluyendo tanto la administración sistémica como la administración tópica. La administración sistémica incluye la administración por vía oral, la administración por vía parenteral, la administración por vía transdérmica y la administración por vía rectal. La administración por vía parenteral se refiere a vías de administración distintas a la
enteral o transdérmica, y por lo general es mediante inyección o infusión. La administración por vía parenteral incluye inyección o infusión intravenosa, intramuscular y subcutánea. La administración por vía tópica incluye la aplicación en la piel, así como la administración por vía intraocular, intravaginal, inhalada e intranasal. En un modo de realización, el compuesto de la invención se puede administrar por vía oral. En otro modo de realización, el compuesto de la invención se puede administrar mediante inhalación. En un modo de realización adicional, el compuesto de la invención se puede administrar por vía intranasal.
[0134] En un modo de realización, el compuesto de la invención se puede administrar una sola vez o según una pauta posológica donde se administran varias dosis en intervalos de tiempo variables durante un periodo de tiempo dado. Por ejemplo, se pueden administrar las dosis una, dos, tres o cuatro veces al día. En un modo de realización, se administra una dosis una vez al día. En un modo de realización adicional, se administra una dosis dos veces al día. Las dosis se pueden administrar hasta que se logre el efecto terapéutico deseado o se mantenga indefinidamente el efecto terapéutico deseado. Las pautas posológicas adecuadas del compuesto de la invención dependen de las propiedades farmacocinéticas de ese compuesto, como su absorción, distribución y, vidas medias del metabolismo y su eliminación, que puede determinar el experto en la materia. Asimismo, las pautas posológicas, incluyendo la duración en que se administran dichas pautas, del compuesto de la invención dependen del trastorno tratado, la gravedad del trastorno tratado, la edad y la condición física del paciente tratado, la historia clínica del paciente que se debe tratar, el tipo de tratamiento concomitante, el efecto terapéutico deseado y los factores similares dentro de los conocimientos y experiencia del experto en la materia. Estos expertos en la materia también comprenderán que las pautas posológicas adecuadas pueden requerir ajustes debido a la tolerancia de cada paciente a la pauta posológica o a lo largo del tiempo según cambian las necesidades de cada paciente.
[0135] Se pueden administrar los compuestos de la presente invención simultáneamente, o antes o después de uno o más agentes terapéuticos distintos. Los compuestos de la presente invención se pueden administrar por separado, o por la misma o diferente vía de administración, o juntos.
[0136] La combinación de la presente invención puede ser una dosis unitaria de 1-1000 mg aprox. de ingredientes activos para un sujeto de 50-70 kg aprox., preferiblemente 1-500 mg aprox. o 1-250 mg aprox. o 1-150 mg aprox. o 0.5 100 mg o 1-50 mg aprox. de ingredientes activos. Una posología terapéuticamente eficaz de un compuesto o las combinaciones de estos, dependen de la especie del sujeto, del peso corporal, la edad y la condición individual, del trastorno o la enfermedad tratados o de la gravedad de estos. Un médico, clínico o veterinario con habilidades básicas en la materia puede determinar fácilmente la cantidad eficaz de cada uno de los ingredientes activos necesarios para prevenir, tratar o inhibir la evolución del trastorno o enfermedad.
[0137] Las propiedades de dosificación anteriormente citadas se pueden correlacionar con ensayos in vitro e in vivo preferentemente con mamíferos, p. ej., ratones, ratas, perros, primates no humanos, como monos, u órganos, tejidos y preparados aislados de estos. Los compuestos de la presente invención se pueden aplicar in vitro en forma de soluciones, p. ej., preferiblemente soluciones acuosas, e in vivo por vía tópica, inhalatoria, enteral o parenteral, preferentemente intravenosa, p. ej., como una suspensión o en una solución acuosa.
[0138] En un modo de realización, una posología terapéuticamente eficaz del compuesto divulgado en la presente memoria es entre 0.1 mg aprox. y 1,000 mg aprox. al día. Las composiciones farmacéuticas deberían proporcionar una dosis de entre 0.1 mg aprox. y 1,000 mg aprox. del compuesto. En un modo de realización en particular, las formas de dosificaciones farmacéuticas unitarias se preparan para proporcionar entre 1 mg aprox. y 1,000 mg aprox., 10 mg aprox. y 500 mg aprox., 20 mg aprox. y 200 mg aprox., 25 mg aprox. y 100 mg aprox., o 30 mg aprox. y 60 mg aprox. del ingrediente activo o de una combinación de ingredientes esenciales por forma de dosificación unitaria. En un modo de realización en particular, las formas de dosificaciones farmacéuticas unitarias se preparan para proporcionar 1 mg, 5 mg, 10 mg, 20 mg, 25 mg, 50 mg, 100 mg, 250 mg, 500 mg, 1000 mg aprox. del ingrediente activo.
MODO DE REALIZACIÓN PREFERIDO DE LA INVENCIÓN
PROCEDIMIENTOS SINTÉTICOS GENERALES
[0139] Se proporcionan los ejemplos siguientes para que la invención se pueda entender por completo.
[0140] En general, los compuestos divulgados en la presente memoria se pueden preparar mediante los métodos descritos en la presente memoria, donde los sustituyentes son como se definen en la fórmula (I) anteriormente, excepto que se indique más adelante. Se presentan los siguientes esquemas y ejemplos no limitativos para ejemplificar aún más la invención.
[0141] Los profesionales expertos en la materia reconocerán que las reacciones químicas descritas se pueden adaptar fácilmente para preparar varios compuestos distintos divulgados en la presente memoria, y se considerarán dentro del alcance divulgado en la presente memoria los métodos alternativos de preparación de los compuestos divulgados en la presente memoria. Los expertos en la materia reconocerán que los materiales de partida pueden variar y que se pueden emplear pasos adicionales para producir compuestos abarcados por las presentes invenciones, como muestran los siguientes ejemplos. En algunos casos, la protección de ciertas funcionalidades reactivas puede ser necesaria para lograr algunas de las transformaciones anteriores. En general, esta necesidad de grupos protectores, así como las condiciones necesarias para unir y eliminar estos grupos quedarán patentes para los expertos en la materia de síntesis orgánica. Por ejemplo, la síntesis de los compuestos no ejemplificados de acuerdo con la invención se puede llevar a cabo con éxito mediante modificaciones evidentes para los expertos en la materia, al proteger de forma adecuada a los grupos interferentes, al utilizar otros reactivos adecuados conocidos en la materia distintos a los descritos, y/o al hacer modificaciones rutinarias de las condiciones de reacción. Alternativamente, se reconocerá que las condiciones de reacción conocidas o la reacción divulgada en la presente invención son de aplicación para la preparación de otros compuestos divulgados aquí.
[0142] En los ejemplos descritos a continuación, a no ser que se indique lo contrario, todas las temperaturas se expresan en grados Celsius. Los reactivos se adquirieron de proveedores comerciales como Aldrich Chemical Company, Arco Chemical Company y Alfa Chemical Company, y se usaron sin mayor purificación, a no ser que se indique lo contrario.
Preparación de compuestos
[0143] Los compuestos de la presente invención, incluyendo sales, ésteres, hidratos o solvatos de estos, se pueden preparar mediante cualquier técnica de síntesis orgánica conocida y se pueden sintetizar por medio de cualquiera de las numerosas rutas sintéticas posibles.
[0144] Se pueden llevar a cabo las reacciones para preparar compuestos de la presente invención en disolventes adecuados, que un experto en la materia de la síntesis orgánica puede seleccionar fácilmente. Los disolventes adecuados pueden ser esencialmente no reactivos con los materiales de partida (reactivos), productos intermedios o productos a las temperaturas en que se llevan a cabo las reacciones, p. ej., las temperaturas que pueden variar entre el punto de congelación del disolvente y el punto de ebullición del disolvente. Se puede llevar a cabo una reacción dada en un disolvente o una mezcla de más de un disolvente. Dependiendo del paso de reacción concreto, un experto en la materia puede seleccionar los disolventes adecuados para un paso de reacción concreto.
[0145] Se pueden monitorizar las reacciones de acuerdo con cualquier método adecuado conocido en la materia. Por ejemplo, se puede monitorizar la formación del producto mediante medios espectroscópicos, como la espectrometría de resonancia magnética nuclear (p. ej., 1H o 13C), espectroscopia infrarroja, espectrofotometría (p. ej., UV-visible), espectrometría de masas, o mediante métodos cromatográficos como la cromatografía líquida de alta resolución (CLAR), cromatografía líquida-espectroscopia de masas (LC-MS) o cromatografía en capa fina (CCF). Los expertos en la materia pueden purificar compuestos mediante una variedad de métodos, incluyendo la cromatografía líquida de alta resolución (CLAR) ("Preparative LC -MS Purification: Improved Compound Specific Method Optimization" Karl F. Blom, Brian Glass, Richard Sparks, Andrew P. Combs J. Combi. Chem. 2004, 6(6), 874-883) y la cromatografía en columna a presión atmosférica con gel de sílice. CLAR, cromatografía líquida de alta resolución; J, constante de acoplamiento (en RMN); min, minuto(s); h, hora(s); RMN, resonancia magnética nuclear; prep, preparativa.
[0146] Se pueden sintetizar los compuestos de la presente invención por medio de los métodos descritos a continuación, junto con métodos sintéticos conocidos en la materia de la química orgánica sintética, o variaciones de estos, como entienden los expertos en la materia. Los métodos preferidos incluyen, entre otros, los métodos descritos a continuación. Concretamente, se pueden sintetizar los compuestos de la presente invención de fórmula (I) mediante los siguientes pasos resumidos en los esquemas sintéticos generales ejemplares indicados a continuación, y las abreviaciones de los reactivos o de los grupos químicos de los reactivos incluidos en los esquemas sintéticos se resumen en los ejemplos.
Esquema 1: la siguiente secuencia de reacción se usa para sintetizar compuestos de estructura A

[0147] La síntesis hacia la estructura A se puede llevar a cabo de acuerdo con los procedimientos relevantes divulgados en las referencias (1, Journal o f the American Chemical Society, 2005, 127, 12460-12461; 2, Organic Letter, 2011, 13, 2318-2321; Journal o f Organic Chemistry, 2010, 75, 941-944), pero no se limita a estos procedimientos divulgados. Los derivados de sulfonamida 1 condensados con el compuesto 2 en presencia de Cp2ZrHCl para dar el compuesto enantiómero 3 , al que se le retiró más adelante el grupo protector y se oxidó hasta el compuesto ácido 4. Con la ayuda de un auxiliar quiral de Evans, se añadió otro centro quiral al compuesto 4 para proporcionar la estructura A .
Esquema 2: la siguiente secuencia de reacción se usa para sintetizar compuestos de estructura B.

[0148] La síntesis hacia la estructura B se puede llevar a cabo de acuerdo con los procedimientos relevantes divulgados en las referencias (Bulletin o f the Chemical Society o f Japan, 1993, 66, 3113-3115; documento US7528174B2), pero no se limita a estos procedimientos divulgados.
Esquema 3: la siguiente secuencia de reacción se usa para sintetizar compuestos de estructura C .

[0149] La síntesis hacia la estructura C se puede llevar a cabo de acuerdo con los procedimientos relevantes divulgados en las referencias (ACS Central Science, 2016, 2 (9), pp 653-659; Journal o f Medicinal Chemistry, 1986, 959-971; Journal of Medicinal Chemistry, 1984, 27, 684-691), pero no se limita a estos procedimientos divulgados.
Esquema 4: la siguiente secuencia de reacción se usa para sintetizar compuestos de estructura D.
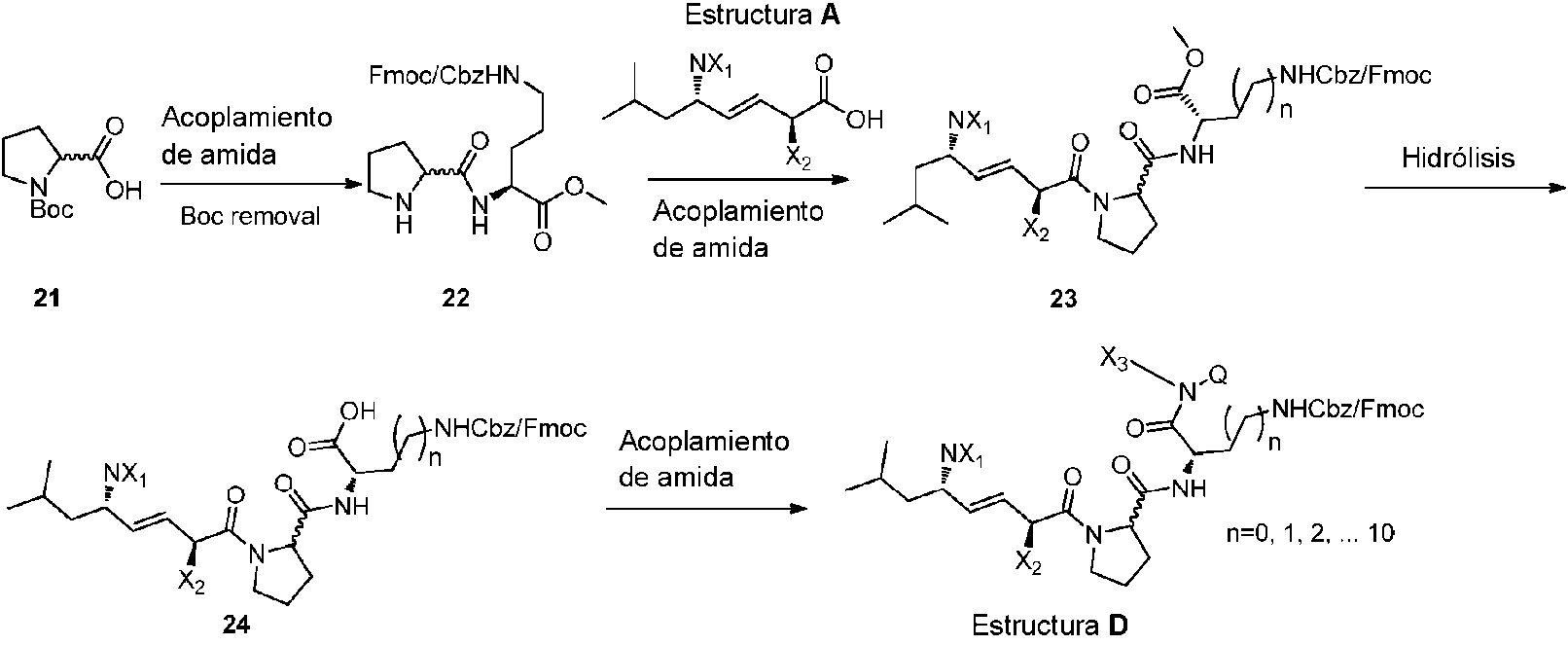
[0150] La síntesis hacia la estructura D se puede llevar a cabo de acuerdo con los procedimientos relevantes divulgados en las referencias (documento US2007161573A1; documento US2009042808A1; Journal o f Medicinal Chemistry, 1984, 27,1351-1354), pero no se limita a estos procedimientos divulgados.
Esquema 5: la siguiente secuencia de reacción se usa para sintetizar compuestos de estructura E.

[0151] La síntesis hacia la estructura E se puede llevar a cabo de acuerdo con los procedimientos relevantes divulgados en las referencias (documento US2007161573A1; documento US2009042808A1; Journal o f the American Chemical Society, 2005, 127, 5742-5743; Journal o f Organic Chemistry, 2004, 69, 7851-7859), pero no se limita a estos procedimientos divulgados.
Esquema 6: cuando PEP (véase la definición) de A es valina, la siguiente secuencia de reacción se usa para sintetizar los compuestos de estructura F.

[0152] La síntesis hacia la estructura F se puede llevar a cabo de acuerdo con los procedimientos relevantes divulgados en las referencias (documento US2007161573A1; Journal o f the American Chemical Society, 2005, 127, 5742-5743; International Journal o f Peptide and Protein Research, 1996; 47, 460-466), pero no se limita a estos procedimientos divulgados.
Esquema 7: cuando PEP de A es valina, la siguiente secuencia de reacción se usa para sintetizar los compuestos de estructura H.

[0153] La síntesis hacia la estructura H se puede llevar a cabo de acuerdo con los procedimientos relevantes divulgados en las referencias (documento US2007161573A1; Journal o f Pharmacology and Experimental Therapeutics, 2007, 320, 1050-1060), pero no se limita a estos procedimientos divulgados.
Preparación y caracterización de compuestos ejemplares
[0154] Los compuestos que abarca la presente divulgación se pueden preparar por medio de diferentes esquemas. A continuación, se describen los procesos de preparación detallados de los compuestos ejemplares por medio de varios esquemas y también se indican los resultados de caracterización.
[0155] A no ser que se indique lo contrario, todos los reactivos se adquirieron de proveedores comerciales sin mayor purificación. Se emplearon métodos normalizados de secado del disolvente cuando fue necesario. Las placas utilizadas para la cromatografía en capa fina (CCF) fueron gel de sílice E. Merck 60F254 (0,24 nm de espesor) prerrevestidas sobre capas de aluminio, y luego visualizadas en luz UV (365 nm y 254 nm) o mediante tinción con un 5 % de ácido dodecamolibdofosforico en etanol y calentamiento posterior. La cromatografía en columna se llevó a cabo con gel de sílice (200-400 malla) de proveedores comerciales. Los espectros de RMN de 1H se registraron en un espectómetro de RMN Agilent 400-MR (400.00 MHz para 1 H) a temperatura ambiente. La señal del disolvente se usó como referencia para la RMN de 1H (CDCl3, 7.26 ppm; CD3o D, 3.31 ppm; DMSO-CÍ6, 2.50 ppm; D2O, 4.79 ppm). Los siguientes acrónimos y abreviaturas de la RMN se usaron para explicar las multiplicidades: s = singlete, d = doblete, t = triplete, c = cuarteto, sa = singlete ancho, dd = doble doblete, td = triple doblete, dt = doble triplete, dc = doble cuarteto, m = multiplete.
Ejemplos
[0156] Cabe destacar que los modos de realización de la presente invención descritos en detalle a continuación son ejemplares para explicar la presente invención solamente, y no se deberían interpretar como limitativos del presente alcance de la invención. Se pueden implementar los ejemplos que carecen de una tecnología o condición específica de acuerdo con la tecnología o condición de la literatura de la materia o de acuerdo con las instrucciones del producto. Los reactivos o instrumentos sin fabricantes están disponibles a través de medios de compra convencionales. Los expertos en la materia reconocerán que los materiales de partida pueden variar y que se pueden emplear pasos adicionales para producir compuestos abarcados por las presentes invenciones, como muestran los siguientes ejemplos.
Ejemplo I de referencia
Bencil N-[(4S)-4-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-4-[(1-oxil-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]butil]carbamato (I)
[0157]

Paso 1: síntesis de (S)-ferc-butil 2-(((S)-5-(((benciloxi)carbonil)amino)-1-metoxi-1-oxopentan-2-il)carbamoil)pirrolidina-1 -carboxilato (61)
[0158] A una solución del compuesto 60 (1.5 g, 6.97 mmol) y (S)-metil 2-amino-5-(((benciloxi)carbonil)amino)pentanoato (2.6 g, 8.37 mmol) en CH2Cl2 (20 mL) a 0 °C se añadieron DIPEA (2.7 g, 20.91 mmol) y Ha Tu (3.4 mL, 9.06 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. La mezcla de la reacción se apagó con solución saturada de NH4Cl (20 mL), se lavó con solución acuosa saturada de salmuera (3 x 10 mL), se secó con MgSO4 y se concentró. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 5 : 1 a 1 : 1) para dar el producto 61 (1.5 g, 56 %) en forma de una espuma blanca.
[0159] RMN de 1H: (400 MHz, CDCl3) 57.35-7.29 (m, 5 H), 5.08 (s, 2 H), 4.56 (s, 1 H), 4.26 (s, 1 H), 3.73 (s, 3 H), 3.45 (s, 2 H), 3.21-3.20 (d, J = 4.0 Hz, 2 H), 2.80 (s, 2 H), 1.93-1.86 (m, 2 H), 1.68-1.63 (m, 2 H), 1.53 (m, 2 H), 1.44 (m, 9 H).
Paso 2: síntesis de (S)-metil 5-(((benciloxi)carbonil)amino)-2-((5)-pirrolidina-2-carboxamido)pentanoato (62)
[0160] A una solución del compuesto 61 (1.4 g, 2.93 mmol) en CH2CI2 (10 mL) a 0 °C se añadió TFA (3.0 mL). La solución resultante se agitó durante 10 min a 0 °C, y 3 h a 25 °C. Se concentró el disolvente al vacío, y se añadieron CH2Cl2 (10 mL) y solución acuosa de Na2CO3 al 5 % hasta alcanzar el pH 8-9. La capa acuosa se extrajo con CH2Ch (3 x 20 mL). La fase orgánica combinada se secó con Na2SO4 y se concentró al vacío para dar el producto 62 (980 mg, 91 %) en forma de un producto oleoso.
Paso 2: síntesis de (S)-metil 2-((S)-1-((2S,5S,E)-2-bencil-5-((ferc-butoxicarboml)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-5-(((benciloxi)carbonil)amino)pentanoato (63)
[0161] A una solución del compuesto 62 (250 mg, 0.66 mmol) y el compuesto 59 (200 mg, 0.55 mmol, el procedimiento fue de acuerdo con las referencias: 1, Journal of the American Chemical Society, 2005, 127, 12460-12461; 2, Organic Letter, 2011, 13, 2318-2321) en CH2Ch (50 mL) a 0 °C se añadieron DIPEA (142 mg, 1.10 mmol) y HATU (272 mg, 0.72 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. La mezcla de la reacción se apagó con solución acuosa saturada de NH4Cl (20 mL), se lavó con solución acuosa saturada de salmuera (3 x 10 mL), se secó con MgSO4 y se concentró para dar el producto bruto 63 (426 mg, 100 %, bruto) en forma de una espuma blanca que se usó en el siguiente paso sin mayor purificación.
[0162] MS (ESI): [M H+] = 721.9.
Paso 3: síntesis de (S)-2-((S)-1-((2S,5S,E)-2-bencil-5-((ferc-butoxicarboml)ammo)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-5-(((benciloxi)carbonil)amino)ácido pentanoico (64)
[0163] Se trató una solución del compuesto 63 (426 mg, 0.55 mmol) en THF/MeOH (4.0/1.0 mL) con LOH.H2O (46 mg, 1.10 mmol). La mezcla de la reacción se agitó a 25 °C durante 2 h, luego el disolvente se eliminó al vacío y la mezcla resultante se trató con EtOAc (20 mL) y HCl acuoso (1 M) hasta alcanzar el pH 3. La fase orgánica se separó de la fase acuosa. La fase acuosa se extrajo con EtOAc (2 x 20 mL), y las capas orgánicas combinadas se lavaron con solución acuosa saturada de salmuera (3 x 20 mL), se secaron con Na2SO4, y se concentraron en vacío para obtener el producto 64 (395 mg, 100 %), que se empleó para el siguiente paso sin purificación.
[0164] MS (ESI): [M H+] = 707.9.
Paso 4: síntesis de bencil N-[(4S)-4-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]amino}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-4-[(1-oxil-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]butil]carbamato (65)
[0165] Se trató una solución del compuesto bruto 64 (395 mg, 0.55 mmol) en CH2Ch seco (5 mL) a 0 °C con HATU (274 mg, 0.72 mmol), DIPEA (142 mg, 1.1 mmol) y 4-AT (113 mg, 0.66 mmol). La mezcla de la reacción se agitó a 25 °C durante 3 h, luego se apagó con solución saturada de NH4Cl (20 mL). La fase acuosa se separó y se extrajo con CH2Cl2 (3 x 20 mL), y las capas orgánicas combinadas se secaron con MgSO4 y se concentraron. El material bruto se purificó mediante pre-CLAR (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1 % en eluyente) para dar el producto I (110 mg, 24 %) en forma de un sólido naranja.
[0166] MS (ESI): [M H+] = 860.7.
Ejemplo II de referencia
íerc-Butil ((4S,7S,£)-7-bencil-8-((S)-2-(((R)-1-((1-oxil-2,2,6,6-tetrametilpiperidin-4-il)amino)-3-metil-1-oxobutan-2-il)carbamoil)pirrolidin-1-il)-2-metil-8-oxooct-5-en-4-il)carbamato (II)
[0167]

Paso 1: síntesis de (5)-ferc-butil 2-(((R)-1-metoxi-3-metiM-oxobutan-2-N)carbamoN)pirroNdma-1-carboxNato (72)
[0168] A una solución del compuesto 71 (4.0 g, 18.58 mmol) y (A)-metil 2-amino-3-metilbutanoato (3.8 g, 22.29 mmol) en CH2Cl2 (40 mL) a 0°C se añadieron DIPEA (2.8 g, 13 mmol) y HATU (5.6 mL, 33 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. La mezcla de la reacción se apagó con solución acuosa saturada de NH4Cl (20 mL), se lavó con solución acuosa saturada de salmuera (3 x 10 mL), se secó con Na2SO4 y se concentró para dar el producto 72 (5.6 g, 92 %) en forma de un sólido de espuma blanca.
[0169] RMN de 1H: (400 MHz, CDCls) 54.53 (s, 1 H), 4.33 (s, 1 H), 3.71 (s, 3 H), 3.48 (s, 2 H), 2.14-2.20 (m, 3 H), 1.89 1.86 (t, J = 4.0 Hz, 2 H), 1.47 (s, 9 H), 0.96-0.94 (d, J = 8.0 Hz, 3 H), 0.88-0.89 (d, J = 4.0 Hz, 3 H).
Paso 2: síntesis de (R)-metil 3-metil-2-((S)-pirrolidina-2-carboxamido)butanoato (73)
[0170] A una solución del compuesto 72 (120 mg, 0.37 mmol) en CH2Cl2 (5 mL) a 0 °C se añadió TFA (1.0 mL). La solución resultante se agitó durante 10 min a 0 °C, y 3 h a 25 °C. Se concentró el disolvente, y se añadieron CH2Cl2 (5 mL) y solución de Na2CO3 al 5 % hasta alcanzar el pH 8-9. Se separaron las capas, y se extrajo la capa acuosa con CH2Ó2 (10 mL). Las capas orgánicas combinadas se secaron con Na2SO4y se concentraron para dar el producto 73 (72 mg, 86 %) en forma de un aceite.
Paso 3: síntesis de (R)-metil 2-((S)-1-((2S,5S,E)-2-bencN-5-((ferc-butoxicarbonN)ammo)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-3-metilbutanoato (74)
[0171] A una solución del compuesto 73 (85 mg, 0.37 mmol) y el compuesto 9 (110 mg, 0.30 mmol) en CH2Cl2 (5 mL) a 0 °C se añadieron DIPEA (80 mg, 0.60 mmol) y HATU (148 mL, 0.39 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. La mezcla de la reacción se apagó con solución saturada de NH4Cl (20 mL), se lavó con solución acuosa saturada de salmuera (3 x 20 mL), se secó con MgSO4 y se concentró para dar el producto bruto 74 (65 mg, 38 %) en forma de una espuma blanca que se usó en el siguiente paso sin mayor purificación.
[0172] MS (ESI): [M H+] = 572.3.
Paso 4: síntesis de (R)-2-((S)-1-((2S, 5S, £)-2-bencN-5-((ferc-butoxicarbonN)ammo)-7-metiloct-3-enoN)pirroNdma-2-carboxamido)-3-ácido metilbutanoico (75)
[0173] Se trató una solución del compuesto 74 (60 mg, 0.11 mmol) en THF/MeOH = 4/1 (2/0.5 mL) con LiOH.H2O (6 mg, 0.14 mmol). La mezcla de la reacción se agitó a 25 °C durante 2 h, luego el disolvente se eliminó al vacío y la mezcla resultante se trató con EtOAc (10 mL) y HCl acuoso (1 M) hasta alcanzar el pH 3. La fase orgánica se separó de la fase acuosa. La fase acuosa se extrajo con EtOAc (2 x 20 mL), y las capas orgánicas combinadas se lavaron con solución acuosa saturada de salmuera (3 x 20 mL), se secaron con Na2SO4, y se concentraron para obtener el producto 75 (51.0 mg, 84 %), que se empleó en bruto para la reacción de acoplamiento.
Paso 5: síntesis de terc-butil ((4S,7S,E)-7-bencN-8-((S)-2-(((R)-1-((1-oxN-2,2,6,6-tetrametilpiperidm-4-N)ammo)-3-metil-1-oxobutan-2-il)carbamoil)pirrolidin-1-il)-2-metil-8-oxooct-5-en-4-il)carbamato (II)
[0174] Se trató una solución del compuesto bruto 75 (50 mg, 0.09 mmol) en CH2Cl2 seco (5 mL) a 0 °C con HATU (45 mg, 0.12 mmol), DIPEA (35.0 mg, 0.27 mmol) y 4-amino-TEMPO (30 mg, 0.18 mmol). La mezcla de la reacción se agitó a 25 °C durante 3 h, luego se apagó con solución acuosa saturada de NH4Cl (20 mL). La fase acuosa se separó y se extrajo con CH2Cl2 (10 mL), y las capas orgánicas combinadas se secaron con MgSO4 y se concentraron. El material en bruto se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 5 : 1 a 1 : 1) para dar el producto II (38 mg, 84 %) en forma de un sólido naranja.
[0175] MS (ESI): [M H+] = 711.5.
Ejemplo III
Metil (2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(terc-butoxi) carboml]ammo}-7-metiloct-3-enoM]pirroMdm-2-M]formamido}-5-({[(1-oxM-2,2,6,6-tetrametilpiperidm-4-il)oxi]carboml}ammo)pentanoato (III)
[0176]

Paso 1: síntesis de metil 2-amino-5-({[(9H-fluoren-9-il)metoxi] carbonil} amino)pentanoato (82)
[0177] Se trató una solución del compuesto 81 (1 g, 2.2 mmol) en metanol (10 mL) a 0 °C con SOCl2 (530 mg, 4.4 mmol), y se agitó la mezcla a 25 °C durante 16 h. Se eliminó el disolvente a presión reducida para dar un producto sólido marrón 82 (800 mg, 98 %).
[0178] MS (ESI): [M H+] = 369.0.
Paso 2: síntesis de terc-butil (2S)-2-{[5-({[(9H-fluoren-9-il)metoxi]carbonil} amino)-1-metoxi-1-oxopentan-2-il]carbamoil}pirrolidina-1 -carboxilato (83)
[0179] A la solución del compuesto 82 (800 mg, 2.2 mmol) en CH2Ch (20 mL) a 0 °C se añadieron Boc-Pro-OH (710 mg, 3.3 mmol) y DIPEA (0.55 mL, 3.3 mmol). Se añadió T3P (50 % en EtOAc, 2.0 mL, 3.3 mmol) lentamente a 0 °C, y se dejó que la mezcla de la reacción se calentara hasta los 25 °C y se agitó durante 16 h. La mezcla de la reacción se lavó con solución de Na2CO3 al 5 %, se secó con MgSO4, y se concentró al vacío para dar un producto sólido de espuma 83 (750 mg, 63 %).
[0180] MS (ESI): [M H+] = 566.3.
Paso 3: síntesis de metil 5-({[(9H-fluoren-9-N)metoxi]carbonN}amino)-2-{[(2S)-pirroNdm-2-il]formamido}pentanoato (84)
[0181] Al compuesto 83 (750 mg, 1.3 mmol) a 0 °C se añadieron HCl en MeOH (4 M, 4 mL, 16 mmol) y CH2CI2 (10 mL). La solución resultante se agitó durante 10 min a 0 °C, y 3 h a 25 °C cuando se usó LC-MS para monitorizar el progreso de la reacción. Se concentró el disolvente en vacío para dar el producto de amina desprotegida 84 (600 mg, 100 %).
[0182] MS (ESI): [M H+] = 466.3.
Paso 4: síntesis de metil 2-[[(2S)-1-[(2R,3E,5S)-2-bencil-5-{[(terc-butoxi) carbonil]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-5-({[(9H-fluoren-9-il)metoxi]carbonil}amino)pentanoato (85)
[0183] A una solución del compuesto 84 (600 mg, 1.3 mmol) y el producto intermedio 59 (0.2 g, 0.6 mmol) suspendido en CH2Cl2 (20 mL) a 0 °C se añadió DIPEA (0.4 mL, 2 mmol). Se añadió T3P (50 % en EtOAc, 0.9 mL, 1 mmol) lentamente a 0 °C, y se dejó que la mezcla de la reacción se calentara hasta los 25 °C y se agitó durante 18 h. La mezcla de la reacción se lavó con solución acuosa de Na2CO3 al 5 % (10 mL) y agua (20 mL), y la capa orgánica se secó con MgSO4 y se concentró al vacío. Los residuos se purificaron mediante cromatografía en columna sobre gel de sílice con disolvente de elución, éter de petróleo/EtOAc = 1 : 3 para obtener un producto oleoso 85 (210 mg, 43 %).
[0184] MS (ESI): [M H+] = 809.6.
Paso 5: síntesis de metil 5-amino-2-{[(2S)-1-[(2R,3E,5S)-2-bencN-5-{[(ferc-butoxi)carbonM]ammol-7-metiloct-3-enoil]pirrolidin-2-il]formamido} pentanoato (86)
[0185] A una solución del compuesto 85 (210 mg, 0.26 mmol) en 10 mL de DMF se añadió Et3N (1 mL, 7.5 mmol), la mezcla se agitó a 25 °C durante 16 h. El producto bruto 86 no se purificó y se usó directamente en el siguiente paso.
[0186] MS (ESI): [M H+] = 587.3.
Paso 6: síntesis de metil (2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(terc-butoxi) carbonil]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-5-({[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi]carboniMamino)pentanoato (87)
[0187] Se trató una solución de 4-hidroxi-TEMPO (180 mg, 1.04 mmol) en 5 mL de DMF con N, N'-carbonato de disuccinimidilo (77) (260 mg, 1.04 mmol) y piridina (200 μL, 0.2 mmol) y la mezcla resultante se calentó a 40 °C durante 16 horas. La solución se enfrío hasta alcanzar la temperatura ambiente y el compuesto 78 se usó directamente en el siguiente paso.
[0188] A una solución del 86 (bruto del paso previo) en 10 mL de DMF se añadió el producto intermedio 78. La solución resultante se agitó a 25 °C durante 16 h, se usó LC-MS para monitorizar el progreso de la reacción. La reacción se apagó con agua (10 mL) y se extrajo con EtOAc (2 * 20 mL), la fase orgánica combinada se lavó con agua (3 * 20 mL), solución acuosa saturada de salmuera (3 * 10 mL) y se secó con Na2SO4, el disolvente se eliminó a presión reducida para dar un residuo bruto y se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar un producto sólido amarillo III (8 mg, 4 %, en dos pasos).
[0189] MS (ESI): [M H+] = 785.6.
Ejemplo IV de referencia
Bencil N-[(4S)-4-[(2E)-2-([(2S)-1-[(2S,3E,5S)-2-bencN-5-{[(ferc-butoxi)carbonN]ammo}-7-metiloct-3-enoN]pirroNdm-2-il]formamido}-3-metilbutanamido]-4-[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil] carbamato (IV)
[0190]

Paso 1: síntesis de (R)-2-((S)-1-(terc-butoxicarboml)pirrolidina-2-carboxamido)-3-ácido metilbutanoico (92)
[0191] Se trató una solución del compuesto 91 (2.0 g, 6.1 mmol) en THF/MeOH = 4 : 1 (10/2.5 mL) con LiOH.HzO (384 mg, 9.14 mmol). La mezcla de la reacción se agitó a 25 °C durante 2 h, luego el disolvente se eliminó al vacío y la mezcla resultante se trató con EtOAc (20 mL) y HCl acuoso (1 M) hasta alcanzar el pH 3. La fase orgánica se separó de la fase acuosa. La fase acuosa se extrajo con EtOAc (2 x 20 mL), y las capas orgánicas combinadas se lavaron con solución acuosa saturada de salmuera (3 x 20 mL), se secaron con Na2SO4, y se concentraron para obtener el producto 92 (1.8 g, 95 %) como una espuma blanca que se empleó para la siguiente reacción de acoplamiento.
Paso 2: síntesis de (S)-terc-butil 2-(((R)-1-(((S)-5-(((benciloxi)carboml)ammo)-1-metoxM-oxopentan-2-il)amino)-3-metil-1-oxobutan-2-il)carbamoil)pirrolidina-1-carboxilato (94)
[0192] A una solución del compuesto 92 (1.5 g, 4.77 mmol) y el compuesto 93 (1.8 g, 5.73 mmol) en CH2Cl2 (20 mL) a 0 °C se añadieron DIPEA (1.8 g, 14.31 mmol) y HATU (2.4 mL, 6.20 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. La mezcla de la reacción se apagó con solución acuosa saturada de NH4Cl (20 mL), se lavó con solución acuosa saturada de salmuera (3 x 10 mL), se secó con MgSO4 y se concentró para dar el producto bruto 94 (1.2 g, 45 %) en forma de una espuma blanca.
[0193] RMN de 1H: (400 MHz, CDCls) 57.35-7.30 (m, 5 H), 5.08 (s, 3 H), 4.55-4.54 (d, J = 4.0 Hz,1 H), 4.30-4.26 (t, J = 8.0 Hz,2 H), 3.72 (s, 3 H), 3.47 (s, 3 H), 3.21-3.18 (t, J = 4.0 Hz,2 H), 2.80 (s, 1 H), 1.89-1.71 (m, 3 H), 1.54 (s, 2 H), 1.54 1.52 (d, J = 8.0 Hz, 2 H), 1.45 (s, 9 H), 1.27-1.24 (m, 2 H), 0.98-0.90 (m, 6 H).
Paso 3: síntesis de (S)-metil 5-(((benciloxi)carboml)ammo)-2-((R)-3-metil-2-((S)-pirrolidma-2-carboxamido)butanamido)pentanoato (95)
[0194] A una solución del compuesto 94 (1.2 g, 2.08 mmol) en CH2Cl2 (9 mL) a 0 °C se añadió TFA (3.0 mL). La solución resultante se agitó durante 10 min a 0 °C, y 3 h a 25 °C. Se concentró el disolvente en vacío, y se añadieron CH2Cl2 (10 mL) y solución acuosa de Na2CO3 al 5 % hasta alcanzar el pH 8-9. La capa acuosa se extrajo con CH2Cl2 (3 x 20 mL). La fase orgánica combinada se lavó con solución acuosa saturada de salmuera (3 x 20 mL), se secó con Na2SO4 y se concentró al vacío para dar el producto 95 (970 mg, 98 %) en forma de una espuma blanca.
[0195] MS (ESI): [M H+] = 477.6.
Paso 4: síntesis de (S)-metil 2-((R)-2-((S)-1-((2S,5S,E)-2-bencil-5-((terc-butoxicarboml)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-3-metilbutanamido)-5-(((benciloxi)carbonil)amino)pentanoato (97)
[0196] A una solución del compuesto 95 (314 mg, 0.66 mmol) y el compuesto 59 (200 mg, 0.55 mmol) en CH2CI2 (5 mL) a 0 °C se añadieron DIPEA (142 mg, 1.1 mmol) y HATU (274 mL, 0.72 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. La mezcla de la reacción se apagó con solución saturada de NH4Cl (20 mL), se lavó con solución acuosa saturada de salmuera (3 * 10 mL), se secó con MgSO4 y se concentró al vacío. Los residuos se purificaron mediante cromatografía en columna sobre gel de sílice con el disolvente de elución éter de petróleo/EtOAc = 1 : 3 para dar el producto 97 (106 mg, 50 %) en forma de una espuma blanca.
[0197] MS (ESI): [M H+] = 820.7.
Paso 5: síntesis de (S)-2-((R)-2-((S)-1-((2S,5S,E)-2-bencN-5-((íerc-butoxicarbonM)ammo)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-3-metilbutanamido)-5-(((benciloxi)carbonil)amino)ácido pentaoico (98)
[0198] Se trató una solución del compuesto 97 (465 mg, 0.55 mmol) en THF/MeOH (4.0/1.0 mL) con LOH.H2O (462 mg, 1.10 mmol en 0.5 mL de agua). La mezcla de la reacción se agitó a 25 °C durante 2 h, luego el disolvente se eliminó al vacío y la mezcla resultante se trató con EtOAc (20 mL) y HCl acuoso (1 M) hasta alcanzar el pH 3. La fase orgánica se separó de la fase acuosa. La fase acuosa se extrajo con EtOAc (2 * 20 mL), y las capas orgánicas combinadas se lavaron con solución acuosa saturada de salmuera (3 * 20 mL), se secaron con Na2SO4, y se concentraron al vacío para obtener el producto 98 (380 mg, 86 %), que se empleó para la reacción de acoplamiento.
[0199] MS (ESI): [M H+] = 806.6.
Paso 6: síntesis de bencil N-[(4S)-4-[(2R)-2-{[(2S)-1-[(2S,3E,5S)-2-bencN-5-{[(íerc-butoxi)carbonN]ammo}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-3-metilbutanamido]-4-[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil]carbamato (99)
[0200] Se trató una solución del compuesto en bruto 98 (380 mg, 0.47 mmol) en CH2Ch seco (5 mL) a 0 °C con HATU (232 mg, 0.61 mmol), DIPEA (121.0 mg, 0.94 mmol) y 4-AT (97 mg, 0.57 mmol). La mezcla de la reacción se agitó a 25 °C durante 3 h, luego se apagó con solución acuosa saturada de NH4Cl (20 mL). La mezcla se extrajo con CH2Ch (2 * 20 mL), y las capas orgánicas combinadas se lavaron con solución acuosa saturada de salmuera (2 * 20 mL), se secaron con Na2SO4, y se concentraron al vacío. El material en bruto se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto 99 (83.0 mg, 18 %) en forma de un sólido naranja.
[0201] MS (ESI): [M H+] = 959.7.
Paso 7: síntesis de bencil N-[(4S)-4-[(2R)-2-{[(2S)-1-[(2S,3E,5S)-2-bencN-5-{[(íerc-butoxi)carbonN]ammo}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-3-metilbutanamido] -4- [(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil]carbamato (IV)
[0202] A una solución del compuesto 99 (30 mg, 0.03 mmol) en MeOH seco (3 mL) se añadió Vc (6 mg, 0.03 mmol). Después de agitar a 25 °C durante 30 min, el disolvente se eliminó al vacío. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto IV (29 mg, 93 %) en forma de un sólido blanco.
[0203] MS (ESI): [M H+] = 960.6.
[0204] RMN de 1H: (400 MHz, CDCla) 88.12-7.96 (m, 5 H), 7.34-7.16 (m, 10 H), 5.43-5.32 (m, 3 H), 5.00 (s, 3 H), 4.30 4.12 (m, 6 H), 2.99-2.92 (m, 4 H), 2.62-2.50 (m, 3 H), 1.97-1.91 (m, 13 H), 1.42-1.23 (m, 21 H), 0.86-0.71 (m, 14 H).
Ejemplo V de referencia
Bencil N-[(4R)-4-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carbonil]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-4-[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil]carbamato (V)
[0205]

Paso 1: síntesis de (R)-metil 2-((S)-1-((2S,5S,E)-2-bencil-5-((ferc-butoxicarboml)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-5-(((benciloxi)carbonil)amino)pentanoato (106)
[0206] A una solución del compuesto 62 (520 mg, 1.38 mmol) y el compuesto 59 (398.0 mg, 1.10 mmol) en CH2CI2 (10 mL) a 0 °C se añadieron DIPEA (356 mg, 2.76 mmol) y HATU (682 mL, 1.79 mmol), y se agitó durante 3 h a 25 °C. La mezcla de la reacción se apagó con solución acuosa saturada de NHaCl (20 mL), se lavó con solución acuosa saturada de salmuera (3 * 10 mL), se secó con Na2SO4 y se concentró al vacío. Los residuos se purificaron mediante cromatografía en columna sobre gel de sílice con el disolvente de elución éter de petróleo/EtOAc = 1 : 3 para dar el producto bruto 106 (830 mg en bruto, 100 %) en forma de una espuma blanca.
[0207] MS (ESI): [M H+] = 721.9.
Paso 2: síntesis de (R)-2-((S)-1-((2S,5S,E)-2-bencil-5-((ferc-butoxicarboml)ammo)-7-metiloct-3-enoil)pirrolidma-2-carboxamido)-5-(((benciloxi)carbonil)amino)ácido pentanoico (107)
[0208] Se trató una solución del compuesto 106 (830 mg, 1.10 mmol) en THF/MeOH = (10/2.5 mL) con LiOH.H2O (56 mg, 1.32 mmol en 0.5 mL de agua). La mezcla de la reacción se agitó a 25 °C durante 2 h, entonces el disolvente se eliminó al vacío y la mezcla resultante se trató con EtOAc (20 mL) y HCl acuoso (1 M) hasta alcanzar el pH 3. La fase orgánica se separó de la fase acuosa. La fase acuosa se extrajo con EtOAc (3 * 20 mL), y las capas orgánicas combinadas se lavaron con solución acuosa saturada de salmuera (3 * 20 mL), se secaron con Na2SO4, y se concentraron al vacío para obtener el producto 107 (730 mg, 94 %), que se empleó para la reacción de acoplamiento.
[0209] MS (ESI): [M H+] = 707.0.
Paso 3: síntesis de bencil N-[(4R)-4-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-4-[(1-oxi¡-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil]carbamato (108)
[0210] Se trató una solución del compuesto bruto 107 (730 mg, 1.03 mmol) en CH2Cl2 seco (5 mL) a 0 °C con HATU (509 mg, 1.34 mmol), DIPEA (266 mg, 2.06 mmol) y 4-a T (212 mg, 1.24 mmol). La mezcla de la reacción se agitó a 25 °C durante 3 h, luego se apagó con solución acuosa saturada de NH4Cl (20 mL). La fase acuosa se separó y se extrajo con CH2Cl2 (2 * 10 mL), y las capas orgánicas combinadas se secaron con Na2SO4 y se concentraron. El material en bruto se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto 108 (160 mg, 18 %) en forma de un sólido naranja.
[0211] MS (ESI): [M H+] = 860.6.
Paso 4: síntesis de bencil N-[(4R)-4-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-4-[(1-hidroxi-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]butil]carbamato (V)
[0212] A una solución del compuesto 108 (152 mg, 0.18 mmol) en MeOH seco (3 mL) se añadió Vc (32 mg, 0.18 mmol). Tras agitar a 25 °C durante 1 h, el disolvente se eliminó al vacío y el residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1 % en eluyente) para dar el producto V (90 mg, 59 %) en forma de un sólido blanco.
[0213] RMN de 1H: (400 MHz, CDCls) 57.28-7.08 (m, 10 H), 5.57-5.52 (m, 1 H), 5.41-5.39 (m, 1 H), 5.09-5.01 (m, 2 H), 4.21 (s, 1 H), 4.16 (s, 2 H), 4.00 (s, 1 H), 3.50 (s, 1 H), 3.35 (s, 1 H), 3.33-3.12 (m, 3 H), 2.67 (s, 1 H), 2.15-1.71 (m, 18 H), 1.55-1.19 (m, 24 H), 0.80-0.79 (d, J = 4.0 Hz, 6 H).
[0214] MS (ESI): [M H+] = 861.9.
Ejemplo VI de referencia
ferc-Butil N-[(4S,5E,7S)-7-bencil-8-[(2R)-2-[(1-hidroxi-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]pirrolidm-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (VI)
[0215]

Paso 1: síntesis de (R)-metil 1-((2S,5SE)-2-bencil-5-((ferc-butoxicarboml)ammo)-7-metiloct-3-enoil)pirrolidma-2-carboxilato (113)
[0216] A una solución del compuesto 111 (165.0 mg, 1.0 mmol) y el compuesto 59 (300 mg, 0.83 mmol) en CH2Cl2 (5 mL) a 0 °C se añadieron DIPEA (268 mg, 2.08 mmol) y HATU (410 mL, 1.08 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. La mezcla de la reacción se apagó con solución acuosa saturada de NH4Cl (20 mL), se lavó con solución acuosa saturada de salmuera (3 x 20 mL), se secó con MgSO4 y se concentró en vacío para dar el producto bruto 113 (410 mg, 100 %) en forma de una espuma blanca que se usó en el siguiente paso sin mayor purificación.
[0217] MS (ESI): [M H+] = 473.6.
Paso 2: síntesis de (R)-1-((2S,5S,E)-2-bencil-5-((ferc-butoxicarboml)ammo)-7-metiloct-3-enoil)pirrolidma-2-ácido carboxílico (114)
[0218] Se trató una solución del compuesto 113 (410 mg, 0.83 mmol) en THF/MeOH (4.0/1.0 mL) con UOH.H2O (42 mg, 1.0 mmol, en 0.5 mL de agua). La mezcla de la reacción se agitó a 25 °C durante 2 h, entonces el disolvente se eliminó al vacío y la mezcla resultante se trató con EtOAc (20 mL) y HCl acuoso (1 M) hasta alcanzar el pH 3. La fase orgánica se separó de la fase acuosa. La fase acuosa se extrajo con EtOAc (2 x 20 mL), y las capas orgánicas combinadas
se lavaron con solución acuosa saturada de salmuera (3 * 20 mL), se secaron con Na2SO4, y se concentraron al vacío para obtener el producto 114 (390 mg, 100 %), que se empleó sin purificación para la reacción de acoplamiento.
[0219] MS (ESI): [M H+] = 459.
Paso 3: síntesis de terc-butil N-[(4S,5E,7S)-7-bencN-8-[(2E)-2-[(1-oxN-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]pirrolidin-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (115)
[0220] Se trató una solución del compuesto en bruto 114 (390 mg, 0.83 mmol) en CH2Ch seco (5 mL) a 0 °C con HATU (410 mg, 1.08 mmol), DIPEA (268 mg, 2.08 mmol) y 4-AT (172 mg, 1.00 mmol). La mezcla de la reacción se agitó a 25 °C durante 3 h, luego se apagó con solución acuosa saturada de NH4Cl (20 mL). La fase acuosa se separó y se extrajo con CH2Cl2 (20 mL), y las capas orgánicas combinadas se secaron con MgSO4 y se concentraron al vacío. El material en bruto se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto 115 (150 mg, 30%) en forma de un sólido naranja.
[0221] MS (ESI): [M H+] = 612.5.
Paso 4: síntesis de terc-butil N-[(4S,5E,7S)-7-bencN-8-[(2E)-2-[(1-hidroxi-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]pirrolidin-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (VI)
[0222] A una solución del compuesto 115 (150 mg, 0.25 mmol) en MeOH seco (3 mL) se añadió Vc (43 mg, 0.25 mmol). Después de agitar a 25 °C durante 30 min, se eliminó el disolvente al vacío. El residuo resultante se disolvió en CH2Ch (10 mL) y se lavó con agua. El residuo se purificó mediante pre-CLAR (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto VI (91 mg, 60 %) en forma de un sólido blanco.
[0223] RMN de 1H: (400 MHz, CDCla) 87.30-7.03 (m, 4 H), 5.63-5.57 (m, 1 H), 5.37-5.32 (m, 1 H), 4.50-4.48 (d, J = 8.0 Hz, 1 H), 4.40 (s, 1 H), 4.15-4.14 (m, 2 H), 3.50-3.43 (m, 3 H), 3.15-2.78 (m, 5 H), 2.07 (s, 1 H), 2.82 (s, 1 H), 1.58-1.25 (m, 30 H), 0.88-0.87 (d, J = 4.0 Hz, 6 H).
[0224] MS (ESI): [M H+] = 613.6.
Ejemplo VII de referencia
Bencil N-[(4S)-4-[(2S)-2-{[(2R)-1-[(2S,3E,5S)-2-bencil-5-{[(terc-butoxi)carboml]ammo}-7-metiloct-3-enoN]pirrolidm-2-il]formamido}-3-metilbutanamido]-4-[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil]carbamato (VII)
[0225]

Paso 1: síntesis de (S)-2-((R)-1-(terc-butoxicarboml)pirrolidma-2-carboxamido)-3-ácido metilbutanoico (122)
[0226] Se trató una solución del compuesto 121 (6.8 g, 18.58 mmol) en THF/MeOH = 4 : 1 (40 mL) con L¡OH.H2O acuoso (940 mg, 22.30 mmol) en agua (2 mL). La mezcla de la reacción se agitó a 25 °C durante 3 h, entonces el disolvente se eliminó al vacío y la mezcla resultante se trató con EtOAc (20 mL) y HCl acuoso (1 M) hasta alcanzar el pH 3. La fase orgánica se separó de la fase acuosa. La fase acuosa se extrajo con EtOAc (2 x 20 mL), y las capas orgánicas combinadas se lavaron con solución acuosa saturada de salmuera (3 x 10 mL), se secaron con Na2SO4, y se concentraron para obtener el producto 122 (4.6 g, 79 %) como una espuma blanca que se empleó para la reacción de acoplamiento.
[0227] MS (ESI): [M H+] = 314.9.
Paso 2: síntesis de (R)-terc-butil 2-(((S)-1-(((S)-5-(((benciloxi)carboml)ammo)-1-metoxi-1-oxopentan-2-il)ammo)-3-metil-1-oxobutan-2-il)carbamoil)pirrolidina-1-carboxilato (124)
[0228] A una solución del compuesto 122 (2.0 g, 6.37 mmol) y el compuesto 123 (2.4 g, 7.64 mmol) en CH2Cl2 (20 mL) a 0 °C se añadieron DIPEA (2.1 g, 15.93 mmol) y HATU (3.1 mL, 8.28 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. La mezcla de la reacción se apagó con solución acuosa saturada de NH4Cl (20 mL), se lavó con solución acuosa saturada de salmuera (3 x 20 mL), se secó con MgSO4 y se concentró al vacío para dar el producto bruto 124 (2.2 g, 61 %) en forma de una espuma blanca que se usó en el siguiente paso sin mayor purificación.
[0229] MS (ESI): [M H+] = 577.7.
Paso 3: síntesis de (S)-metil 5-(((benciloxi)carbonil)amino)-2-((S)-3-metil-2-((R)-pirrolidina-2-carboxamido)butanamido)pentanoato (125)
[0230] A una solución del compuesto 124 (2.2 g, 3.82 mmol) en CH2Cl2 (12 mL) a 0 °C se añadió TFA (3.0 mL). La solución resultante se agitó durante 10 min a 0 °C, y 3 h a 25 °C. Se concentró el disolvente al vacío, y se añadieron CH2Cl2 (10 mL) y solución acuosa de Na2CO3 al 5% hasta alcanzar el pH 8-9. Se separaron las capas, y se extrajo la capa acuosa con CH2Cl2 (20 mL). Las capas orgánicas combinadas se secaron con Na2SO4 y se concentraron para dar el producto 125 (314 mg, 72 %) en forma de una espuma blanca.
[0231] MS (ESI): [M H+] = 477.6.
Paso 4: síntesis de (S)-metil 2-((R)-2-((S)-1-((2S,5S,E)-2-bencil-5-((ferc-butoxicarboml)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-3-metilbutanamido)-5-(((benciloxi)carbonil)amino)pentanoato (126)
[0232] A una solución del compuesto 125 (314 mg, 0.66 mmol) y el compuesto 59 (200 mg, 0.55 mmol) en CH2Cl2 (5 mL) a 0 °C se añadieron DIPEA (142 mg, 1.1 mmol) y HATU (274 mL, 0.72 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. La mezcla de la reacción se apagó con solución acuosa saturada de NH4O (20 mL), se lavó con solución acuosa saturada de salmuera (3 * 10 mL), se secó con MgSO4 y se concentró al vacío. Los residuos se purificaron mediante gel de sílice en cromatografía en columna con disolvente de elución, éter de petróleo/EtOAc = 1 : 3) para dar el producto bruto 126 (460 mg, 100 %) en forma de una espuma blanca.
[0233] MS (ESI): [M H+] = 820.7.
Paso 5: síntesis de (S)-2-((S)-2-((R)-1-((2S,5S,E)-2-bencil-5-((ferc-butoxicarboml)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-3-metilbutanamido)-5-(((benciloxi)carbonil)amino)ácido pentanoico (127)
[0234] Se trató una solución del compuesto 126 (460 mg, 0.55 mmol) en THF/MeOH (4.0/2.0 mL) con LiOHH2O (56 mg, 1.10 mmol, en 0.5 mL de agua). La mezcla de la reacción se agitó a 25 °C durante 2 h, entonces el disolvente se eliminó al vacío y la mezcla resultante se trató con EtOAc (20 mL) y HCl acuoso (1 M) hasta alcanzar el pH 3. La fase orgánica se separó de la fase acuosa. La fase acuosa se extrajo con EtOAc (2 * 20 mL), y las capas orgánicas combinadas se lavaron con solución acuosa saturada de salmuera (3 * 20 mL), se secaron con Na2SO4, y se concentraron para obtener el producto 127 (370 mg, 84 %), que se empleó para la siguiente reacción de acoplamiento.
[0235] MS (ESI): [M H+] = 806.6.
Paso 6: síntesis de bencil N-[(4S)-4-[(2S)-2-{[(2E)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carbonil] amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-3-metilbutanamido]-4-[(1-oxil-2,2,6,6-tetrametipiperidin-4-il)carbamoil]butil]carbamato (128)
[0236] Se trató una solución del compuesto bruto 127 (370 mg, 0.46 mmol) en CH2Ch seco (5 mL) a 0 °C con HATU (227 mg, 0.60 mmol), DIPEA (119 mg, 0.92 mmol) y 4-AT (94 mg, 0.55 mmol). La mezcla de la reacción se agitó a 25 °C durante 3 h, luego se apagó con solución acuosa saturada de NH4Cl (10 mL). La fase acuosa se separó y se extrajo con CH2Cl2 (20 mL), y las capas orgánicas combinadas se secaron con MgSO4 y se concentraron en vacío. El material en bruto se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto 128 (81 mg, 18 %) en forma de un sólido naranja.
[0237] MS (ESI): [M H+] = 959.8.
Paso 7: síntesis de bencil N-[(4S)-4-[(2S)-2-{[(2E)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]amino}-7-metiloct-3-enoil] pirrolidin-2-il]formamido}-3-metilbutanamido] -4- [(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil]carbamato (VII)
[0238] A una solución del compuesto 128 (76 mg, 0.08 mmol) en MeOH seco (3 mL) se añadió Vc (14 mg, 0.08 mmol). Después de agitar a 25 °C durante 1 h, se eliminó el disolvente al vacío. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto VII (32 mg, 42 %) en forma de un sólido blanco.
[0239] RMN de 1H: (400 MHz, CDCla) 87.57-7.03 (m, 8 H), 5.74 (m, 2 H), 5.09-5.07 (d, J = 4.0 Hz, 2 H), 4.40-3.08 (m, 14 H), 2.75-2.04 (m, 16 H), 1.75-1.0 (m, 39 H).
[0240] MS (ESI): [M H+] = 960.7.
Ejemplo VIII de referencia
1-Oxil-2,2,6,6-tetrametilpiperidin-4-il N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-1-{[(1S)-4-{[(benciloxi)carbonil]amino}-1-[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil]carbamoil}-2-metilpropil]carbamoil}pirrolidin-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (VIII)
[0241]

Paso 1: síntesis de bencil ((S)-4-((S)-2-((S)-1-((2S,5S,£)-5-ammo-2-bencil-7-metiloct-3-enoil)pirrolidma-2-carboxamido)-3-metilbutanamido)-5-((1-oxil-2,2,6,6-tetrametilpiperidm-4-il)ammo)-5-oxopentil)carbamato (132) [0242] A una solución del compuesto 131 (200 mg, 0.21 mmol, la síntesis del proceso fue de acuerdo con el Journal of the American Chemical Society, 2005, 127, 12460-12461) en c H2Cl2 (5 mL) a 0 °C se añadió TFA (0.2 mL). La solución resultante se agitó durante 10 min a 0 °C, y 1 h a 25 °C. Se concentró el disolvente, y se añadieron CH2Cl2 (5 mL) y solución acuosa de Na2CO3 al 5 % hasta alcanzar el pH 8-9. Se separaron las capas, y se extrajo la capa acuosa con CH2Cl2 (20 mL). Las orgánicas combinadas se secaron con Na2SO4y se concentraron en vacío para dar el producto 132 (176 mg, 97 %) en forma de un aceite rojo.
Paso 2: síntesis de 1-oxil-2,2,6,6-tetrametilpiperidin-4-il N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-1-{[(1S)-4-{[(benciloxi)carbonil]amino}-1 -[(1-oxil-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]butil]carbamoil}-2-metilpropil]carbamoil}pirrolidin-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (VIII)
[0243] A una solución agitada del compuesto 132 (176 mg, 0.2 mmol) y el compuesto 78 (68.5 mg, 0.25 mmol) en DMF (5.0 mL) se añadió KbP04.3H20 (66.5 mg, 0.25 mmol). La mezcla se agitó luego a 25 °C durante 16 h. La reacción se apagó con agua (10 mL) y se extrajo con EtOAc (3 x 20 mL). La fase orgánica combinada se lavó con solución acuosa saturada de salmuera (3 x 10 mL), se secó con Na2SO4 y se concentró en vacío. Los residuos se purificaron mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 2 : 1 a 1 : 10) para dar el producto VIII (106 mg, 50 %) en forma de un sólido rojo amarronado.
[0244] MS (ESI): [M H+] = 1058.0.
Ejemplo IX de referencia
Bencil N-[(4S)-4-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-4-[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil]carbamato (IX)
[0245]

Paso 1: síntesis de bencil N-[(4S)-4-{[(2S)-1-[(2S,3£,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-4-[(1-hidroxi-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]butil]carbamato (IX)
[0246] A una solución del compuesto I (85 mg, 0.10 mmol) en MeOH seco (3 mL) Se añadió Vc (17 mg, 0.10 mmol). Después de agitar a 25 °C durante 1 h, se eliminó el disolvente al vacío. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1 % en eluyente) para dar el producto IX (47 mg, 55 %) en forma de un sólido blanco.
[0247] RMN de 1H: (400 MHz, CDCls) 57.27-7.07 (m, 10 H), 5.55-5.46 (m, 1 H), 5.35 (s, 1 H), 5.25-5.22 (m, 1 H), 5.01 (s, 2 H), 4.3-4.23 (m, 3 H), 3.63 (s, 1 H), 3.44 (s, 1 H), 3.31 (s, 1 H), 3.30 (s, 1 H), 3.09-3.06 (m, 4 H), 2.47 (s, 1 H), 2.10 (s, 8 H), 1.94 (s, 2 H), 1.84-1.47 (m, 6 H), 1.42-1.10 (m, 23 H), 0.81-0.75 (m, 6 H).
[0248] MS (ESI): [M H+] = 861,7.
Ejemplo X de referencia
Ciclopropilmetil N-[(4S,5£,7S)-7-bencil-8-[(2S)-2-{[(1K)-1-{[(1S)-4-{[(benciloxi)carboml]ammo} -1- [(1-hidroxi-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]butil]carbamoil}-2-metilpropil]carbamoil}pirrolidm-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (X)
[0249]

Paso 1: síntesis de bencil ((S)-4-((ft)-2-((S)-1-((2S,5S,£)-5-ammo-2-bencil-7-metiloct-3-enoil)pirrolidma-2-carboxamido)-3-metilbutanamido)-5-((1-oxil-2,2,6,6-tetrametilpiperidin-4-il)amino)-5-oxopentil)carbamato (132)
[0250] A una solución del compuesto 131 (200 mg, 0.21 mmol) en CH2Cl2 (3 mL) a 0 °C se añadió TFA (1.0 mL). La solución resultante se agitó 10 min a 0 °C, y 3 h a 25 °C. Se concentró el disolvente al vacío, y se añadieron CH2Ch (10 mL) y solución acuosa de Na2CO3 al 5% hasta alcanzar el pH 8-9. Se separaron las capas, y se extrajo la capa acuosa con CH2Cl2(10 mL). La capa orgánica combinada se lavó con solución acuosa saturada de salmuera (2 * 10 mL), se secó con Na2SO4 y se concentró para dar el producto 136 (171 mg, 95 %) en forma de una espuma roja.
Paso 2: síntesis de ciclopropilmetil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1R)-1-{[(1S)-4-{[(benciloxi)carboml]ammo} -1- [(1-oxil-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]butil]carbamoil}-2-metilpropil]carbamoil}pirrolidm-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (138)
[0251] A una solución del compuesto 136 (171 mg, 0.20 mmol) y el compuesto 137 (51 mg, 0.24 mmol, la síntesis del procedimiento fue de acuerdo con el ejemplo del compuesto 78) en DMF (5.0 mL) a 25 °C se añadió K3PO4.3H2O (64.0 mg, 0.24 mmol). La mezcla de la reacción se agitó durante 16 h a 25 °C. La mezcla de la reacción se diluyó con EtOAc (10 mL) y agua (10 mL) La fase orgánica combinada se lavó con solución acuosa saturada de salmuera (3 * 20 mL), se secó con Na2SO4 y se concentró en vacío. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto 138 (76 mg, 40 %) en forma de una espuma roja amarronada.
[0252] MS (ESI): [M H+] = 957.9.
Paso 3: síntesis de ciclopropilmetil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1R)-1-{[(1S)-4-{[(benxiloxi)carboml]amino}-1-[(1-hidroxi-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]butil]carbamoil}-2-metilpropil]carbamoil}pirrolidm-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (X)
[0253] A una solución del compuesto 138 (74 mg, 0.08 mmol) en MeOH seco (3 mL) se añadió Vc (14 mg, 0.08 mmol). Después de agitar a 25 °C durante 1 h, se eliminó el disolvente al vacío. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto X (30 mg, 41 %) en forma de un sólido blanco.
[0254] RMN de 1H: (400 MHz, CDCla) 88.13 (s,1 H), 7.32-7.06 (m, 10 H), 6.76-6.75 (d, J = 4.0 Hz, 1 H), 5.47-5.34 (m, 3 H), 5.04 (m, 2 H), 4.45-3.78 (m, 8 H), 3.38-3.07 (m, 6 H), 2.16-1.81 (m, 19 H), 1.45-0.54 (m, 25 H), 0.61 (s, 2 H), 0.26 (s, 2 H).
[0255] MS (ESI): [M H+] = 959.3.
Ejemplo XI de referencia
Bencil N-[(4S)-4-{[(2S)-1-[(2S)-1-[(2R,3E,5S)-2-bencil-5-{[(terc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidina-2-carbonil] pirrolidm-2-il]formamido}-4-[(1-hidroxi-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]butil]carbamato(XI)
[0256]

Paso 1: síntesis de ferc-butil (2S)-2-[(2S)-2-{[(2S)-5-{[(benciloxi)carboml] amino}-1-metoxi-1-oxopentan-2-il]carbamoil}pirrolidina-1-carbonil] pirrolidina -1-carboxilato (144)
[0257] A la solución del aminocompuesto 62 (1.8 g, 4.4 mmol) en CH2CI2 (20 mL) a 0 °C se añadieron Boc-Pro-OH (1.4 g, 6.6 mmol) y DIPEA (1.2 mL, 6.6 mmol). Se añadió T3P (50 % en EtOAc, 3.7 mL, 6.6 mmol) lentamente a 0 °C, y se dejó que la mezcla de la reacción se calentara hasta los 25 °C y se agitó durante 16 h. La mezcla de la reacción se lavó con solución acuosa de Na2CO3 al 5 % (20 mL), se secó con MgSO4, y se concentró en vacío para dar un producto oleoso 144 (2.2 g, 87 %).
[0258] MS (ESI): [M H+] = 575.3.
Paso 2: síntesis de metil (2S)-5-{[(bencNoxi)carboml]ammo}-2-{[(2S)-1-[(2S)-pirroNdma-2-carboml]pirroNdm-2-il]formamido}pentanoato (145)
[0259] A una solución del compuesto 144 (2.2 g, 3.8 mmol) en CH2Cl2 (10 mL) a 0 °C se añadió HCl en MeOH (4 M, 10 mL, 40 mmol). La solución resultante se agitó durante 10 min a 0 °C, y 2 h a 25 °C cuando se usó LC-MS para monitorizar el progreso de la reacción. Se concentró el disolvente en vacío para dar el producto de amina desprotegida 145 (1.9 g, 90 %).
[0260] MS (ESI): [M H+] = 475.2.
Paso 3: síntesis de metil (2S)-2-{[(2S)-1-[(2S)-1-[(2ft, 3E, 5S)-2-bencN-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidina-2-carbonil]pirrolidin-2-il]formamido}-5-{[(benciloxi)carbonil]amino}pentanoato (146)
[0261] A una solución del compuesto 145 (0.5 g, 1.0 mmol) y el producto intermedio 59 (0.2 g, 0.6 mmol) suspendido en CH2Cl2 (20 mL) a 0 °C se añadió DIPEA (0.4 mL, 2 mmol). Se añadió T3P (50 % en EtOAc, 0.9 mL, 1 mmol) lentamente a 0 °C, y se dejó que la mezcla de la reacción se calentara hasta los 25 °C y se agitó durante 18 h. La mezcla de la reacción se lavó con solución acuosa de Na2CO3 al 5 % (10 mL) y agua (10 mL), y la capa orgánica se secó con MgSO4 y se concentró en vacío. El semisólido residual se purificó mediante cromatografía en columna sobre gel de sílice con el disolvente de elución éter de petróleo/EtOAc = 1 : 3 para obtener el producto 146 (160 mg, 32 %).
[0262] MS (ESI): [M H+] = 818.5.
Paso 4: síntesis de (2S)-2-{[(2S)-1-[(2S)-1-[(2E,3E,5S)-2-bencN-5-{[(íerc-butoxi) carbonM]amino}-7-metiloct-3-enoil]pirrolidina-2-carbonil]pirrolidin-2-il] formamido}-5-{[(benciloxi)carbonil]amino}ácido pentanoico (147) [0263] Se trató una solución del compuesto 146 (160 mg, 0.20 mmol) en THF/MeOH (4.0/1.0 mL) con LiOHHzO (10 mg, 0.22 mmol, en 0.2 mL de agua). La mezcla de la reacción se agitó a 25 °C durante 4 h, luego el disolvente se eliminó al vacío y la mezcla resultante se trató con EtOAc (20 mL) y HCl acuoso (1 M) hasta alcanzar el pH 3-4. La fase orgánica se separó de la fase acuosa. La fase acuosa se extrajo con EtOAc (2 * 20 mL), y las capas orgánicas combinadas se lavaron con solución acuosa saturada de salmuera (2 * 20 mL), se secaron con Na2SO4, y se concentraron en vacío para obtener el producto oleoso 147 (120 mg, 75 %).
[0264] MS (ESI): [M H+] = 804.5.
Paso 5: síntesis de bencil N-[(4S)-4-{[(2S)-1-[(2S)-1-[(2R,3E,5S)-2-bencN-5-{[(ferc-butoxi)carbonM]amino}-7-metiloct-3-enoil]pirrolidina-2-carbonil] pirrolidin-2-il]formamido}-4-[(1-Oxil-2,2,6,6-tetrametilpiperidin-4-il) carbamoil]butil]carbamato (148)
[0265] Se trató una solución del compuesto bruto 147 (120 mg, 0.15 mmol) en CH2Ch seco (5 mL) a 0 °C con HATU (60 mg, 0.15 mmol), DIPEA (25 mg, 0.18 mmol) y 4-AT (30 mg, 0.18 mmol). La mezcla de la reacción se agitó a 25 °C durante 16 h, luego se apagó con solución acuosa saturada de NH4Cl (20 mL). Se separó la capa acuosa y se extrajo con CH2Cl2 (20 mL). Las capas orgánicas combinadas se secaron con MgSO4 y se concentraron en vacío. El material en bruto se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto 148 (80 mg, 56 %) en forma de un sólido naranja.
[0266] MS (ESI): [M H+] = 957.7.
Paso 6: síntesis de bencil N-[(4S)-4-{[(2S)-1-[(2S)-1-[(2R,3E,5S)-2-bencN-5-{[(ferc-butoxi)carbonN]amino}-7-metiloct-3-enoil]pirrolidina-2-carbonil] pirrolidin-2-il]formamido}-4-[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil]carbamato (XI)
[0267] A una solución del compuesto 148 (80 mg, 0.08 mmol) en MeOH seco (3 mL) se añadió Vc (15 mg, 0.08 mmol). Después de agitar a 25 °C durante 60 min, se eliminó el disolvente al vacío. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto XI (15 mg, 19 %) en forma de un sólido blanco.
[0268] RMN de 1H: (400MHz, CDCla) 87.35-7.20 (m, 7 H), 7.20-7.10 (m, 5 H), 5.6-5.30 (m, 2 H), 5.1-4.9 (m, 2 H), 4.6 4.3 (m, 4 H), 3.4-3.2 (m, 6 H), 2.4-1.8 (m, 8 H), 1.8-1.6 (m, 14 H), 1.6-1.2 (m, 22 H), 0.8-0.6 (m, 8 H).
[0269] MS (ESI): [M H+] = 958.8.
Ejemplo XII de referencia
Bencil N-[(4S)-4-[(2S)-2-{[(2S)-1-[(2E,3E, 5S)-2-bencil-5-{[(terc-butoxi)carbonil]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-2-ciclopropilacetamido]-4-[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il) carbamoil] butil]carbamato (XII)
[0270]

Debería ser DIPEA no DIPEA; define EDCI y HOBt en la lista de abreviaturas
Paso 1: síntesis de metil (2S)-5-{[(bencMoxi)carboml]ammo}-2-[(2S)-2-{[(ferc-butoxi)carbonM]ammo}-2-ciclopropilacetamido]pentanoato (152)
[0271] A una solución de Orn(Z)-OMe (151) (1.5 g, 4.7 mmol) y (S)-2-((ferc-butoxicarbonil)amino)-2-ácido cidopropilacético (1.0 g, 4.7 mmol) suspendido en CH2Ch (20 mL) a 0 °C se añadió DIPEA (1.0 mL, 6 mmol). Se añadió T3P (50 % en EtOAc, 4 mL, 6.0 mmol) lentamente a 0 °C, y se dejó que la mezcla de la reacción se calentara hasta los 25 °C y se agitó durante 16 h. La mezcla de la reacción se lavó con solución acuosa de Na2CO3 al 5 % (10 mL) y agua (2 x 10 mL), y la capa orgánica se secó con MgSO4 y se concentró en vacío. Los semisólidos residuales se convierten en una sustancia seca similar al cemento 152 (1.5 g, 68 %).
[0272] MS (ESI): [M H+] = 478.1.
Paso 2: síntesis de metil (25)-2-[(25)-2-amino-2-ciclopropilacetamido]-5-{[(benciloxi)carbonil]amino}pentanoato (153)
[0273] A una solución del compuesto 152 (1.5 g, 3.0 mmol) en CH2Cl2 (10 mL) a 0 °C se añadió HCl en MeOH (4 M, 5 mL, 20 mmol). La solución resultante se agitó durante 10 min a 0 °C, y 2 h a 25 °C cuando se usó LC-MS para monitorizar el progreso de la reacción. Se concentró el disolvente en vacío para dar el producto de amina desprotegida 153 (1.0 g, 83 %).
[0274] MS (ESI): [M H+] = 378.0.
Paso 3: síntesis de terc-butil (2S)-2-{[(S)-{[(2S)-5-{[(benciloxi)carbonil] amino}-1-metoxi-1-oxopentan-2-il]carbamoil}(ciclopropil) metil]carbamoil} pirrolidina-1-carboxilato (154)
[0275] A la solución del compuesto amina 153 (1.0 g, 2.6 mmol) en CH2CI2 (20 mL) a 0 °C se añadieron Boc-Pro-OH (840 mg, 3.9 mmol) y DIPEA (0.64 mL, 3.9 mmol). Se añadió T3P (50 % en EtOAc, 2.2 mL, 3.9 mmol) lentamente, y se dejó que la mezcla de la reacción se calentara hasta los 25 °C y se agitó durante 16 h. La mezcla de la reacción se lavó con solución acuosa de Na2CO3 al 5 % (10 mL), se secó con MgSO4, y se concentró en vacío para dar un producto sólido de espuma 154 (1.1 g, 90 %).
[0276] MS (ESI): [M H+] = 575.3.
Paso 4: síntesis de metil (2S)-5-{[(benciloxi)carbonil]amino}-2-[(2S)-2-ciclopropil-2-{[(2S)-pirrolidin-2-il] formamido} acetamido] pentanoato (155)
[0277] A una solución del compuesto 154 (1.1 g, 1.9 mmol) en CH2Ch (10 mL) a 0 °C se añadió HCl en MeOH (4 M, 5 mL, 20 mmol). La solución resultante se agitó 10 min a 0 °C, y 2 h a 25 °C cuando se usó LC-MS para monitorizar el progreso de la reacción. Se concentró el disolvente en vacío para dar el producto de amina desprotegida 155 (0.8 g, 89 %).
[0278] MS (ESI): [M H+] = 475.3.
Paso 5: síntesis de metil (2S)-2-[(2S)-2-{[(2S)-1-[(2E,3E,5S)-2-bencN-5-{[(terc-butoxi)carbonM]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-2-ciclopropilacetamido] -5- {[(benciloxi)carbonil]amino}pentanoato (156)
[0279] A una solución del compuesto 155 (0.5 g, 1.0 mmol) y el producto intermedio 59 (0.2 g, 0.6 mmol) suspendido en CH2Cl2 (20 mL) a 0 °C se añadió DIPEA (0.4 mL, 2 mmol). Se añadió T3P (50 % en EtOAc, 0.9 mL, 1 mmol) lentamente, y se dejó que la mezcla de la reacción se calentara hasta los 25 °C y se agitó durante 18 h. La mezcla de la reacción se lavó con solución acuosa de Na2CO3 al 5 % (10 mL) y agua (2 * 10 mL), y la capa orgánica se secó con MgSO4 y se concentró en vacío. El residuo se purificó mediante cromatografía en columna sobre gel de sílice con disolvente de eluciónéter de petróleo/EtOAc = 1 : 3, para obtener un producto oleoso 156 (120 mg, 24 %).
[0280] MS (ESI): [M H+] = 818.5.
Paso 6: síntesis de (2S)-2-[(2S)-2-{[(2S)-1-[(2E,3E,5S)-2-bencN-5-{[(terc-butoxi)carbonM] amino}-7-metiloct-3-enoil] pirrolidin-2-il] formamido}-2-ciclopropilacetamido]-5-{[(benciloxi)carbonil] amino} ácido pentanoico (157)
[0281] Se trató una solución del compuesto 156 (120 mg, 0.15 mmol) en THF/MeOH (4.0/1.0 mL) con LOH.H2O (10 mg, 0.22 mmol, en 0.2 mL de agua). La mezcla de la reacción se agitó a 25 °C durante 4 h, luego el disolvente se eliminó al vacío y la mezcla resultante se trató con EtOAc (20 mL) y HCl acuoso (1 M) hasta alcanzar el pH 3-4. La fase orgánica se separó de la fase acuosa. La fase acuosa se extrajo con EtOAc (2 * 20 mL), y las capas orgánicas combinadas se lavaron con solución acuosa saturada de salmuera (3 * 10 mL), se secaron con Na2SO4, y se concentraron en vacío para obtener el producto oleoso 157 (100 mg, 83 %).
[0282] MS (ESI): [M H+] = 804.5.
Paso 7: síntesis de bencil N-[(4S)-4-[(2S)-2-{[(2S)-1-[(2E,3E,5S)-2-bencN-5-{[(terc-butoxi)carbonM] amino}-7-metiloct-3-enoil] pirrolidin-2-il]formamido}-2-ciclopropilacetamido]-4-[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil]carbamato (158)
[0283] Se trató una solución del compuesto bruto 157 (100 mg, 0.12 mmol) en CH2Ch seco (5 mL) a 0 °C con EDCI (35 mg, 0.18 mmol), HOBt (25 mg, 0.18 mmol), DIEA (25 mg, 0.18 mmol) y 4-AT (30 mg, 0.18 mmol). La mezcla de la reacción se agitó a 25 °C durante 16 h, luego se apagó con solución acuosa saturada de NH4Cl (20 mL). La fase acuosa se separó y se extrajo con CH2Ch (20 mL), y las capas orgánicas combinadas se secaron con MgSO4 y se concentraron. El material en bruto se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto 158 (30 mg, 26 %) en forma de un sólido naranja.
[0284] MS (ESI): [M H+] = 957.7.
Paso 8: síntesis de bencil N-[(4S)-4-[(2S)-2-{[(2S)-1-[(2ft,3E,5S)-2-bencil-5-{[(ferc-butoxi)carbonil]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-2-cidopropilacetamido]-4-[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il) carbamoil] butil]carbamato (XII)
[0285] A una solución del compuesto 158 (30 mg, 0.03 mmol) en MeOH seco (3 mL) se añadió Vc (10 mg, 0.05 mmol). Después de agitar a 25 °C durante 60 min, el disolvente se eliminó al vacío. El residuo se purificó mediante pre-CLAR (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1 % en eluyente) para dar el producto XII (4 mg, 13 %) en forma de un sólido blanco.
[0286] RMN de 1H: (400MHz, CDCls) 57.35-7.20 (m, 7 H), 7.20-7.10 (m, 5 H), 5.62-5.30 (m, 2 H), 5.15-4.91 (m, 2 H), 4.6- 4.3 (m, 4 H), 3.40-3.2 (m, 6 H), 2.41-1.82 (m, 8 H), 1.80-1.65 (m, 14 H), 1.60-1.20 (m, 22 H), 0.88-0.61 (m, 8 H).
[0287] MS (ESI): [M H+] = 958.8.
Ejemplo XIII de referencia
Bencil N-[(4ft)-4-{[(2S)-1-[(2S,3E,5S)-5-amino-2-bencil-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-4-[(1-hidroxi-2.2.6.6- tetrametil-piperidin-4-il)carbamoil]butil]carbamato diclorhidrato (XIII)
[0288]

Paso 1: síntesis de bencil N-[(4ft)-4-{[(2S)-1-[(2S,3E,5S)-5-amino-2-bencil-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-4-[(1-hidroxi-2,2,6,6-tetrametil-piperidin-4-il)carbamoil]butil]carbamato diclorhidrato (XÜI)
[0289] A una solución del compuesto V (20 mg, 0.02 mmol) en CH2Cl2 (5 mL) se añadió HCl en MeOH (5 mL, 3M) y se agitó a 25 °C durante 1 h. Se eliminó el disolvente y se purificó mediante CLa R preparativa (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1 % en eluyente) para dar el producto XIII (4.6 mg, 31 %) en forma de un sólido blanco.
[0290] MS (ESI): [M H+] = 761.6.
Ejemplo XIV de referencia
Bencil N-[(4S)-4-[(2S)-2-{[(2S)-1-[(2S,3E, 5S)-2-bencil-5-{[(ferc-butoxi)carbonil]ammo}-7-metiloct-3-enoil]pirrolidin-2-il]formamido} propanamido]-4-[(1 -hidroxi-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil] carbamato (XIV)
[0291]

Define HOBt y EDCI en la lista de abreviaturas
Paso 1: síntesis de metil (2S)-2-[(2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido} propanamido]-5-{[(benciloxi)carbonil]amino}pentanoato (163)
[0292] A una solución del compuesto 161 (465 mg, 0.96 mmol, el procedimiento fue de acuerdo con el Journal o f the American Chemical Society, 2005, 127, 12460-12461) y el compuesto 59 (290 mg, 0.80 mmol) en CH2Cl2 (5 mL) a 0 °C se añadieron HOBt (130.0 mg), e Dc I (185.0 mg, 0.96 mmol) y TEA (242.0 mg, 0.96 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 16 h. La mezcla de la reacción se apagó con HCl acuoso (1M, 5 mL), se lavó con solución acuosa saturada de salmuera (3 x 20 mL), se secó con Na2SO4 y se concentró en vacío. El residuo se purificó por medio de cromatografía en columna (gel de sílice (éter de petróleo/EtOAc = de 3 : 1 a 1 : 1) para dar lugar al producto 163 (210 mg, 28 %) en forma de una espuma blanca.
[0293] MS (ESI): [M H+] = 792.6.
Paso 2: síntesis de (2S)-2-[(2S)-2-{[(2S)-1-[(2S,3E, 5S)-2-bencil-5-{[(ferc-butoxi)carboml]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido} propanamido]-5-{[(benciloxi)carbonil]amino}ácido pentanoico (164)
[0294] Se trató una solución del compuesto 163 (210 mg, 0.26 mmol) en THF/MeOH (4.0/1.0 mL) con UOH.H2O (33 mg, 0.80 mmol, en 0.2 mL de agua). Se agitó la mezcla de la reacción a 25 °C durante 2 h, luego el disolvente se eliminó al vacío. La mezcla resultante se trató con EtOAc (10 mL) y HCl acuoso (1 M) hasta alcanzar el pH 3. La fase orgánica se separó de la fase acuosa. La fase acuosa se extrajo con EtOAc (2 x 20 mL), y las capas orgánicas combinadas se lavaron con solución acuosa saturada de salmuera (2 x 20 mL), se secaron con Na2SO4, y se concentraron para obtener el producto 164 (170 mg, 84 %) en forma de una espuma blanca que se empleó para la siguiente reacción de acoplamiento.
[0295] MS (ESI): [M H+] = 778.4.
Paso 3: síntesis de bencil N-[(4S)-4-[(2S)-2-{[(2S)-1-[(2S,3E,5S)-2-benzyl-5-{[(ferc-butoxy)carbonyl]ammo}-7-metiloct-3-enoil]pirrolidin-2-il]formamido} propanamido]-4-[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil]carbamato (165)
[0296] Se trató una solución del compuesto bruto 164 (170 mg, 0.22 mmol) en CH2Cl2 seco (5 mL) a 0 °C con HOBt (39.0 mg, 0.29 mmol), EDCI (55.0 mg, 0.29 mmol), TEA (44.0 mg, 0.44 mmol), y 4-AT (44.0 mg, 0.26 mmol). La mezcla de la reacción se agitó a 25 °C durante 16 h, luego se lavó la mezcla de la reacción con solución acuosa de NaHCO3 al 5 % (10 mL) y luego HCl acuoso (10 mL, 1M). La fase acuosa se separó y se extrajo con CH2Cl2 (10 mL), y las capas orgánicas combinadas se secaron con Na2SO4 y se concentraron en vacío para dar el producto bruto 165 (230 mg, bruto) en forma de un sólido rojo amarronado.
[0297] MS (ESI): [M H+] = 931.8.
Paso 4: síntesis de bencil N-[(4S)-4-[(2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]amino}-7-m etiloct-3-enoil]pirrolid in-2-il]form am ido} propanamido]-4-[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)carbam oil]butil] carbamato (XIV)
[0298] A una solución del compuesto 165 (230 mg, 0.25 mmol) en MeOH seco (3 mL) se añadió Vc (44 mg, 0.08 mmol). Después de agitar a 25 °C durante 1 h, el disolvente se eliminó al vacío. El residuo se purificó mediante pre-CLAR (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1 % en eluyente) para dar el producto XIV (16 mg, 42 %) en forma de un sólido blanco.
[0299] MS (ESI): [M H+] = 932.8.
Ejemplo XV de referencia
Bencil N-[(4S)-4-[(2S)-2-{[(2S)-1-[(2S,3E,5S)-5-amino-2-bencil-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-3-metilbutanamido]-4-[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil]sal de ácido trifluoroacético de carbamato (XV)
[0300]

Paso 1: síntesis de bencil N-[(4S)-4-[(2S)-2-{[(2S)-1-[(2S,3E,5S)-5-ammo-2-bencil-7-metiloct-3-enoil] p irrolidin-2-il] formamido}-3-metilbutanamido]-4-[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil]sal de ácido trifluoroacético de carbamato (XV)
[0301] A una solución del compuesto 166 (50 mg, 0.05 mmol, el procedimiento fue de acuerdo con el Journal o f the American Chemical Society, 2005, 127, 12460-12461) en CH2Cl2 (5 mL) a 0 °C se añadió TFA (0.2 mL). Se agitó la solución resultante a 0 °C durante 2 h. El disolvente se concentró en vacío y el residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1 % en eluyente) para dar el producto XV (16 mg, 36 %) en forma de un sólido blanco.
[0302] MS (ESI): [M H+] = 859.7.
Ejemplo XVI de referencia
ferc-Butil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-1-[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]-2-(4-hidroxifenil) etil]carbamoil}pirrolidin-1 -il]-2-metil-8-oxooct-5-en-4-il]carbamato (XVI)
[0303]

Paso 1: síntesis de ferc-butil (2S)-2-{[(2S)-3-(4-hidroxifeml)-1-metoxi-1-oxopropan-2-M]carbamoM}pirroMdma-1-carboxilato (173)
[0304] A una solución del compuesto 171 (1.7 g, 8.2 mmol) y el compuesto 172 (1.8 g, 7.8 mmol) en CH2Cl2 (20 mL) a 0 °C se añadieron T3P (2.7 g, 8.6 mmol) y Te a (1.7 g, 17.2 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 16 h. La mezcla de la reacción se lavó con NaHCO3 acuoso al 5 % (10 mL), HCl acuoso (10 mL, 1 M), y solución acuosa saturada de salmuera (3 * 10 mL). La capa orgánica se secó con Na2SO4 y se concentró en vacío. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 5 : 1 a 1 : 1) para dar el producto 173 (1.8 g, 60 %) en forma de una espuma blanca.
[0305] MS (ESI): [M H+] = 393.1.
Paso 2: síntesis de metil (2S)-3-(4-hidroxifenil)-2-t[(2S)-pirrolidin-2-il]formamido}propanoato (174)
[0306] A una solución del compuesto 173 (1.8 g, 4.59 mmol) en CH2Ch (5 mL) se añadió HCl en MeOH (10 mL, 3 M) y se agitó a 25 °C durante 2 h y luego el disolvente se eliminó en vacío para dar el producto 174 (1.5 g, 100 %) como una espuma blanca.
[0307] MS (ESI): [M H+] = 293.1.
Paso 3: síntesis de metil (2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi) carbonil] amino}-7-metiloct-3-enoil] pirrolidin-2-il]formamido}-3-(4-hidroxifenil)propanoato (176)
[0308] A una solución del compuesto 174 (220 mg, 0.67 mmol) y el compuesto 59 (220 mg, 0.61 mmol) en CH2Cl2 (5 mL) a 0 °C se añadieron HOBt (100 mg, 0.73 mmol), EDCI (140 mg, 0.73 mmol) y DlpEA (200 mg, 1.53 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 16 h. La mezcla de la reacción se apagó con solución acuosa saturada de NH4Cl (20 mL), se lavó con HCl acuoso (5 mL, 1 M), solución acuosa saturada de salmuera (2 * 20 mL), se secó con Na2SO4y se concentró en vacío. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 3 : 1 a 1 : 1) para dar lugar al producto 176 (110 mg, 28 %) en forma de una espuma blanca.
[0309] MS (ESI): [M H+] = 636.3.
Paso 4: síntesis de (2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carbonN] amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-3-(4-hidroxifenil) ácido propiónico (177)
[0310] Se trató una solución del compuesto 176 (100 mg, 0.16 mmol) en THF/MeOH (4.0/1.0 mL) con LiOH.H2O (14 mg, 0.32 mmol, en 0.2 mL de agua). Se agitó la mezcla de la reacción a 25 °C durante 2 h, luego el disolvente se eliminó al vacío. La mezcla resultante se trató con EtOAc (10 mL) y HCl acuoso (1 M) hasta alcanzar el pH 2-3. La fase orgánica se separó de la fase acuosa. La fase acuosa se extrajo con EtOAc (2 * 20 mL), y las capas orgánicas combinadas se
lavaron con solución acuosa saturada de salmuera (3 x 10 mL), se secaron con Na2SO4, y se concentraron para obtener el producto 177 (91 mg, 92 %), que se empleó en bruto para la siguiente reacción de acoplamiento.
[0311] MS (ESI): [M H+] = 622.4.
Paso 5: síntesis de ferc-butil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-1-[(1-oxil-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]-2-(4-hidroxifenil)etil]carbamoil} pirrolidin-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (178)
[0312] Se trató una solución del compuesto bruto 177 (90 mg, 0.15 mmol) en CH2Cl2 seco (5 mL) a 0 °C con HOBt (26 mg, 0.18 mmol), EDCI (38 mg, 0.2 mol), DIPEA (39 mg, 0.3 mmol), y 4-AT (31 mg, 0.18 mmol). La mezcla de la reacción se agitó a 25 °C durante 16 h, luego se apagó con solución acuosa saturada de NH4Cl (20 mL). La fase acuosa se separó y se extrajo con CH2Cl2 (20 mL), y las capas orgánicas combinadas se secaron con MgSO4 y se concentraron en vacío para dar el producto bruto 178 (105 mg, 90 %) en forma de un sólido rojo amarronado.
[0313] MS (ESI): [M H+] = 775.6.
Paso 6: síntesis de terc-butil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-1-[(1-hidroxi-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]-2-(4-hidroxifenil) etil] carbamoil}pirrolidin-1 -il]-2-metil-8-oxooct-5-en-4-il]carbamato (XVI)
[0314] A una solución del compuesto 178 (105 mg, 0.14 mmol) en MeOH seco (2 mL) se añadió Vc (27 mg, 0.16 mmol). Después de agitar a 25 °C durante 30 min, el disolvente se eliminó al vacío. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1 % en eluyente) para dar el producto XVI (42 mg, 34 %) en forma de un sólido blanco.
[0315] MS (ESI): [M H+] = 776.6.
Ejemplo XVII de referencia
Bencil N-[(4S)-4-{[(2ft)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-4-[(1 -hidroxi-2,2,6,6-tetrametilpiperidin-4-il)carbamoil] butil] carbamato (XVII)
[0316]

Paso 1: síntesis de ferc-butilo 2-{[(2S)-5-{[(benciloxi)carbonil]amino}-1-metoxi-1-oxopentan-2-il]carbamoil}pirrolidina-1 -carboxilato (183)
[0317] A una solución del compuesto 181 (4.8 g, 15.22 mmol) y el compuesto 182 (3.6 g, 16.74 mmol) en CH2Cl2 (50 mL) a 0 °C se añadieron DIPEA (4.3 g, 33.48 mmol) y T3P (5.8 g, 18.26 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 16 h. La mezcla de la reacción se lavó con NaHCO3 acuoso al 5 % (10 mL), HCl acuoso (5 mL, 1 M) y solución acuosa saturada de salmuera (2 x 20 mL). La capa orgánica se secó con
Na2SO4 y se concentró en vacío. El material bruto se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 5 : 1 a 1 : 1) para dar lugar al producto 183 (5.4 g, 75 %) en forma de una espuma blanca.
Paso 2: síntesis de metil (2S)-5-{[(benc¡lox¡)carboml]ammo}-2-{[(2R)-p¡rrol¡dm-2-¡l]formam¡do}pentanoato (184)
[0318] A una solución del compuesto 183 (5.4 g, 11.31 mmol) en CH2Ch (10 mL) se añadió HCl en MeOH (5 mL, 3 M) y se agitó a 25 °C durante 2 h. El disolvente se eliminó en vacío para dar el producto 184 (3.9 g, 93 %) en forma de un sólido blanco.
[0319] MS (ESI): [M H+] = 378.2.
Paso 3: síntes¡s de met¡l (2S)-2-{[(2R)-1-[(2S,3E,5S)-2-benc¡l-5-{[(íerc-butox¡)carboml] am¡no}-7-met¡loct-3-eno¡l] p¡rrol¡d¡n-2-¡l]formam¡do}-5-{[(benc¡lox¡)carbon¡l]am¡no}pentanoato (186)
[0320] A una solución del compuesto 184 (276 mg, 0.67 mmol) y el compuesto 59 (220 mg, 0.61 mmol) en CH2Ch (5 mL) a 0 °C se añadieron HOBt (99 mg, 0.73 mmol), DIPEA (197 mg, 1.53 mmol) y EDCI (139.0 mg, 0.73 mmol). La mezcla de la reacción se lavó con NaHCO3 acuoso al 5 % (10 mL), HCl acuoso (5 mL, 1 M) y solución acuosa saturada de salmuera (2 * 20 mL). La capa orgánica se secó con Na2SO4 y se concentró en vacío. El material bruto se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 3 : 1 a 1 : 1) para dar el producto 186 (140 mg, 32 %) en forma de un sólido blanco.
[0321] MS (ESI): [M H+] = 721.6.
Paso 4: síntes¡s de (2S)-2-{[(2R)-1-[(2S,3E,5S)-2-benc¡l-5-{[(íerc-butox¡)carboml]ammo}-7-met¡loct-3-eno¡l]p¡rrol¡d¡n-2-¡l]formam¡do}-5-{[(benc¡lox¡) carbon¡l]am¡no}ác¡do pentano¡co (187)
[0322] Se trató una solución del compuesto 186 (140 mg, 0.19 mmol) en THF/MeOH = 4/1 (4/1 mL) con LiOH.HzO (16 mg, 0.38 mmol, en 0.2 mL de agua). Se agitó la mezcla de la reacción a 25 °C durante 2 h, luego el disolvente se eliminó al vacío. La mezcla resultante se trató con EtOAc (10 mL) y HCl acuoso (1 M) hasta alcanzar el pH = 2-3. La fase orgánica se separó de la fase acuosa. La fase acuosa se extrajo con EtOAc (2 * 20 mL), y las capas orgánicas combinadas se lavaron con solución acuosa saturada de salmuera (3 * 10 mL), se secaron con Na2SO4, y se concentraron para obtener el producto 187 (118 mg, 88 %), que se empleó para la siguiente reacción de acoplamiento.
[0323] MS (ESI): [M H+] = 707.5.
Paso 5: síntes¡s de benc¡l N-[(4S)-4-{[(2E)-1-[(2S,3E,5S)-2-benc¡l-5-{[(íerc-butox¡)carboml]ammo}-7-met¡loct-3-eno¡l]p¡rrol¡d¡n-2-¡l]formam¡do}-4-[(1-ox¡l-2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)carbamo¡l]but¡l]carbamato (188)
[0324] A una solución del compuesto 187 (118 mg, 0.17 mmol) y 4-AT (34 mg, 0.20 mmol) en CH2Ch (5 mL) a 0 °C se añadieron HOBt (30 mg, 0.22 mmol), DIPEA (44 mg, 0.34 mmol) y EDCI (42 mg, 0.22 mmol). La mezcla de la reacción se lavó con NaHCO3 acuoso al 5 % (10 mL), HCl acuoso (5 mL, 1 M) y solución acuosa saturada de salmuera (2 * 20 mL). La capa orgánica combinada se secó con Na2SO4 y se concentró para dar el producto bruto 188 (160 mg) en forma de un sólido blanco.
[0325] MS (ESI): [M H+] = 860.7.
Paso 6: síntes¡s de benc¡l N-[(4S)-4-{[(2E)-1-[(2S,3E,5S)-2-benc¡l-5-{[(íerc-butox¡)carboml]ammo}-7-met¡loct-3-eno¡l]p¡rrol¡d¡n-2-¡l]formam¡do}-4-[(1-h¡drox¡-2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)carbamo¡l]but¡l] carbamato (XVII)
[0326] A una solución del compuesto 188 (160.0 mg, 0.18 mmol) en MeOH seco (2 mL) se añadió Vc (32 mg, 0.18 mmol). Después de agitar a 25 °C durante 60 min, el disolvente se eliminó al vacío. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto XVII (48 mg, 31 %) en forma de un sólido blanco.
[0327] MS (ESI): [M H+] = 861.8.
Ejemplo XVIII de referencia
(S)-Metil 2-((S)-1-((2S,5S,E)-2-bencN-5-((ferc-butoxicarbonN)ammo)-7-metiloct-3-enoN)pirroNdma-2-carboxamido)-5-((((1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)oxi)carbonil)amino)pentanoato (XVIII)
[0328]

Paso 1: síntesis de (S)-metil 5-((((9H-fluoren-9-N)metoxi)carbonN)ammo)-2-((S)-1-((2S,5S,E)-2-bencN-5-((fercbutoxicarbonil)amino)-7-metiloct-3-enoil) pirrolidina-2-carboxamido)pentanoato (195)
[0329] A una solución del compuesto 84 (0.5 mg, 1.0 mmol) y (2S,5S,£)-2-bencil-5-((ferc-butoxicarbonil)amino)-7-metiloct-3-ácido enoico (59, 433 mg, 1.2 mmol) en CH2Ch (5 mL) se añadieron T3P (413 mg, 1.3 mmol) y DIPEA (284 mg, 2.2 mmol) a 0 °C. Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 16 h. La mezcla de la reacción se apagó con solución acuosa saturada de NH4Cl (20 mL) y la capa acuosa se extrajo con EtOAc (3 x 10 mL). La capa orgánica combinada se lavó con NaHCO3 acuoso al 5 % (10 mL), solución acuosa saturada de salmuera (3 x 10 mL) y se secó con Na2SO4. Los residuos se concentraron y se purificaron mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 3 : 1 a 1 : 1) para dar el producto 195 (380 mg, 47 %) en forma de un sólido blanco.
[0330] MS (ESI): [M H+] = 809.6.
Paso 2: síntesis de (S)-metil 5-ammo-2-((S)-1-((2S,5S,E)-2-bencN-5-((ferc-butoxicarbonN)ammo)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)pentanoato (196) A una solución del compuesto 195 (380 mg, 0.47 mmol) en DMF (5 mL) a 0 °C se añadió una solución de TBAF (5 % en peso en THF, 6 mL). La reacción se agitó durante 30 min. La mezcla 196 (275 mg, 100 %) se usó para el paso siguiente sin ningún tratamiento.
[0331] MS (ESI): [M H+] = 587.4.
Paso 3: síntesis de (S)-metil 2-((S)-1-((2S,5S,E)-2-bencN-5-((ferc-butoxicarbonN)ammo)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-5-((((1-oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi)carbonil)amino)pentanoato (197)
[0332] A una solución del compuesto 196 (275 mg, 0.47 mmol) y 1, 3-dioxoisoindolin-2-il (1-oxil-2,2,6,6-tetrametilpiperidin-4-il) carbonato (78) (203 mg, 0.56 mmol) en CH2Cl2 (5 mL) a 0 °C se añadió DIPEA (121 mg, 0.94 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 2 h. La mezcla de la reacción se apagó con solución acuosa saturada de NH4Cl (20 mL) y la fase acuosa se extrajo con EtOAc (3 x 10 mL). La capa orgánica combinada se lavó con agua (2 x 10 mL), solución acuosa saturada de salmuera (3 x 20 mL) y se secó
con Na2SO4. El residuo se concentró a presión reducida y se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1 % en eluyente) para dar el producto 197 (30 mg, 8 %) en forma de un sólido rojo pálido.
[0333] MS (ESI): [M H+] = 785.6.
Paso 4: síntesis de (S)-metil 2-((S)-1-((2S,5S,E)-2-bencil-5-((ferc-butoxicarboml)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-5-((((1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)oxi)carbonil)amino)pentanoato (XVIII)
[0334] A una solución del compuesto 197 (25 mg, 0.03 mmol) en MeOH seco (2 mL) se añadió Vc (5.3 mg, 0.03 mmol). Después de agitar a 25 °C durante 30 min, el disolvente se eliminó al vacío. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1 % en eluyente) para dar el producto XVIII (7 mg, 28 %) en forma de un sólido blanco.
[0335] MS (ESI): [M H+] = 787.4.
Ejemplo XX de referencia
ferc-Butil N-(4-{[(2S)-1-[(2ft,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-4-[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil)carbamato (XX)
[0336]
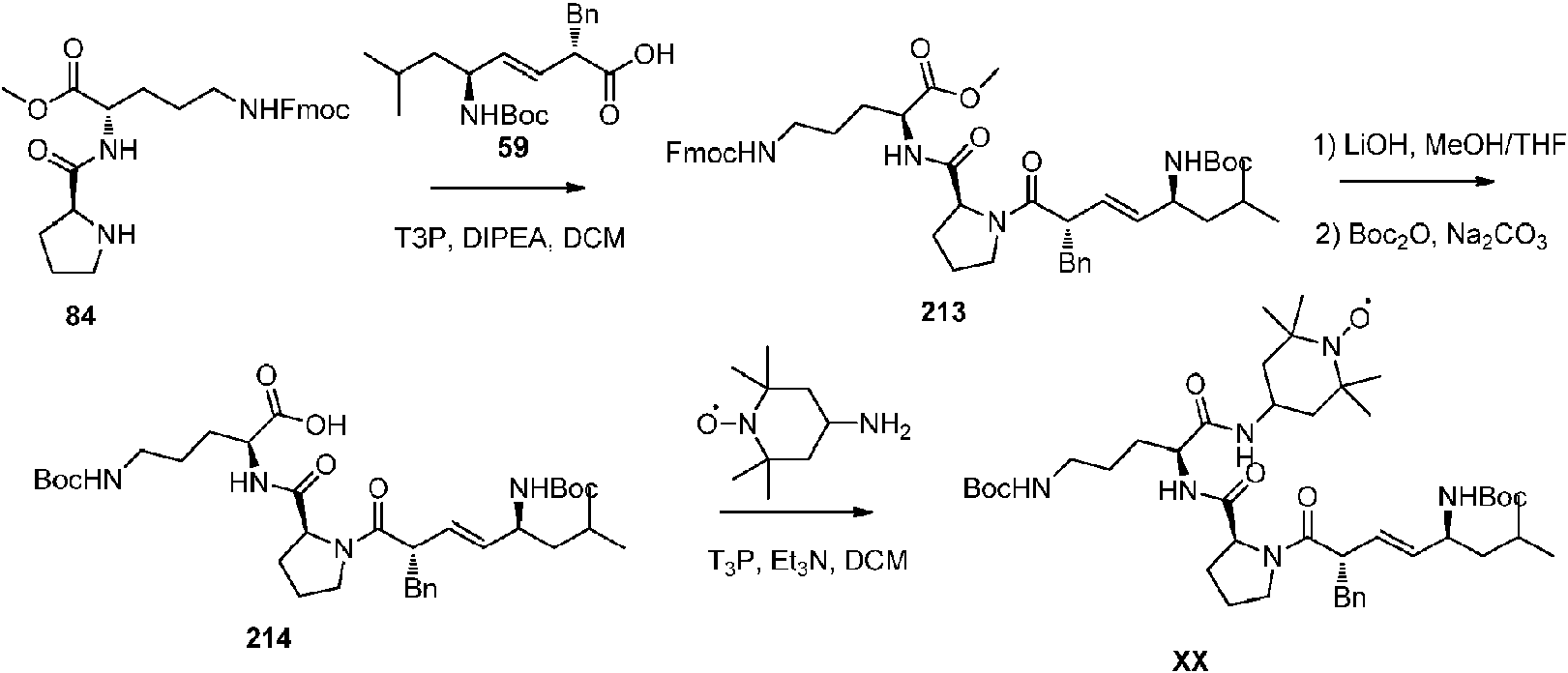
Paso 1: síntesis de metil 2-{[(2S)-1-[(2ft,3E,5S)-2-bencil-5-{[(ferc-butoxi) carbonil]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-5-({[(9H-fluoren-9-il)metoxi]carbonil}amino)pentanoato (213)
[0337] A la solución del compuesto 84 (300 mg, 0.65 mmol) y el producto intermedio 59 (0.2 g, 0.6 mmol) suspendido en CH2Cl2 (20 mL) a 0 °C se añadió DIPeA (0.4 mL, 2 mmol). Se añadió T3P (50 % en EtOAc, 0.9 mL, 1 mmol) lentamente, y se dejó que la mezcla de la reacción se calentara hasta los 25 °C y se agitó durante 18 h. La mezcla de la reacción se lavó con solución acuosa de Na2CO3 al 5 % (10 mL) y agua (2 x 10 mL), y la capa orgánica se secó con MgSO4 y se concentró en vacío. El producto bruto se purificó mediante cromatografía en columna (gel de sílice con disolvente de elución, éter de petróleo/EtOAc = 1 : 3), para obtener un producto oleoso 213 (210 mg, 43 %).
[0338] MS (ESI): [M H+] = 809.6.
Paso 2: síntesis de 2-{[(2S)-1-[(2ft,3E,5S)-2-bencil-5-{[(ferc-butoxi)carbonil] amino}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-5-{[(terc-butoxi) carbonil]amino}ácido pentanoico (214)
[0339] Se trató una solución del compuesto 213 (210 mg, 0.26 mmol) en THF/MeOH (4.0/1.0 mL) con LiOH.HzO (20 mg, 0.40 mmol en 0.2 mL de agua). Se agitó la mezcla de la reacción a 25 °C durante 4 h, luego el disolvente se eliminó al vacío. La mezcla resultante se trató con EtOAc (20 mL) y HCl acuoso (1 M) hasta alcanzar el pH 3-4. La fase orgánica se separó de la fase acuosa. La fase acuosa se extrajo con EtOAc (2 x 20 mL), y las capas orgánicas combinadas se lavaron con solución salina acuosa saturada (3 x 10 mL), se secaron con Na2sO4, y se concentraron para obtener el producto bruto de 100 mg. El producto bruto se añadió a CH2Cl2 (10 mL), luego a Boc2O (60 mg, 0.26 mmol) y DIPEA (20 mg, 0.26 mmol), la mezcla se agitó a 25 °C durante 16 h. La reacción se apagó con HCl (1 M) hasta alcanzar el pH 3-4, y se extrajo con EtOAc (2 x 20 mL). La fase orgánica combinada se lavó con agua (2 x 10 mL), solución salina acuosa saturada (3 x 10 mL), se secó con Na2SO4, y el disolvente se eliminó a presión reducida para dar un producto oleoso 214 (150 mg, 86 %).
[0340] MS (ESI): [M H+] = 673.4.
Paso 3: síntesis de ferc-butil N-(4-{[(2S)-1-[(2ft,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-4-[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil)carbamato (XX)
[0341] A una solución del compuesto de ácido 214 (150 mg, 0.22 mmol) en CH2Cl2 (20 mL) a 0 °C se añadieron 4-AT (75 mg, 0.44 mmol) y DIPEA (60 mg, 0.44 mmol). Se añadió T3P (50 % en EtOAc, 0.2 mL, 0.4 mmol) lentamente, y se dejó que la mezcla de la reacción se calentara hasta los 25 °C y se agitó durante 16 h. La mezcla de la reacción se lavó con solución acuosa de Na2CO3 al 5 % (10 mL), se secó con MgSO4 y se concentró en vacío. El residuo oleoso se purificó mediante CLAR preparativa (Venusil x Bp C18 (Suma, 30 x 250 mm), HCl al 0.1 % en eluyente) para dar el producto XX (6 mg, 3 %)
[0342] MS (ESI): [M H+] = 826.7.
Ejemplo XXI de referencia
ferc-Butil N-[(4S,5E,7S)-7-bencil-8-[(2K)-2-{[(1S)-4-{[(ferc-butoxi)carboml]ammo}-1-[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il) carbamoil] butil] carbamoil} pirrolidin- 1-il] -2-metil-8-oxooct-5-en-4-il] carbamato (XXI) [0343]

Paso 1: síntesis de ferc-butil N-[(4S,5E,7S)-7-bencil-8-[(2ft)-2-{[(1S)-4-{[(ferc-butoxi)carboml]amino}-1-[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il) carbamoil] butil] carbamoil}pirrolidin-1-il]-2-metil-8-oxooct-5-en-4-il] carbamato (216)
[0344] A una solución del compuesto XX (20 mg, 0.02 mmol) en MeOH (10 mL) se añadió Vc (10 mg, 0.10 mmol). Después de agitar a 25 °C durante 60 min, el disolvente se eliminó al vacío. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1 % en eluyente) para dar el producto XXI (15 mg, 75 %) en forma de un sólido blanco.
[0345] MS (ESI): [M H+] = 827.7.
Ejemplo XXII de referencia
(2S)-2-{[(2S)-1-[(2S,3E,5S)-5-amino-2-bencil-7-metiloct-3-enoil] Pirrolidin-2-il]formamido}-5-carbamimidamido-N-(1 -oxil-2,2,6,6-tetrametilpiperidin-4-il)pentanamida (XXII)
[0346]

Paso 1: síntesis de ferc-butil (2S)-2-{[(2S)-1-metoxi-5-[W'-(4-metoxi-2,6-dimetilbencenosulfonil)carbamimidamido]-1-oxopentan-2-il]carbamoil} pirrolidina-1-carboxilato (223)
[0347] A una solución del compuesto 221 (360 mg, 0.9 mmol) y el compuesto 222 (213 g, 0.99 mmol) en CH2Cl2 (3 mL) a 0 °C se añadieron T3P (343 mg, 1.1 mmol) y DIPEA (232 mg, 1.8 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 16 h. La mezcla de la reacción se lavó con NaHCO3 acuoso al 5 %, HCl acuoso (5 mL, 1 M), y solución acuosa saturada de salmuera (3 * 10 mL). La capa orgánica se secó con Na2SO4 y se concentró para dar el producto 223 (390 mg, 72 %) en forma de una espuma blanca.
[0348] MS (ESI): [M H+] = 598.3.
Paso 2: síntesis de metil (2S)-5-[N-(4-metoxi-2,3,6-trimetilbencenosulfoml) carbamimidamido]-2-{[(2S)-pirrolidin-2-il]formamido}pentanoato (224)
[0349] A una solución del compuesto 223 (390 mg, 0.65 mmol) en CH2Cl2 (10 mL) se añadió HCl en MeOH (10 mL, 3 M), se agitó a 25 °C durante 2 h. Luego el disolvente se eliminó para dar el producto 224 (323 mg, 100 %) en forma de una espuma blanca.
[0350] MS (ESI): [M H+] = 498.2.
Paso 3: síntesis de metil (2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml] amino}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-5-[N'-(4-metoxi-2,3,6-trimetilbencenosulfoml)carbamimidamido]pentanoato (226)
[0351] A una solución del compuesto 224 (200 mg, 0.4 mmol) y el compuesto 59 (160 mg, 0.44 mmol) en CH2Cl2 (3 mL) a 0 °C se añadieron HOBt (70 mg, 0.52 mmol), EDCI (100 mg, 0.52 mmol) y DIPeA (103 mg, 0.80 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 16 h. La mezcla de la reacción se apagó con solución acuosa saturada de NH4Cl (20 mL), se lavó con HCl acuoso (10 mL, 1 M), se lavó con solución acuosa saturada de salmuera (2 * 20 mL), se secó con Na2SO4 y se concentró. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 3 : 1 a 1 : 1) para dar el producto 226 (120 mg, 36 %) en forma de una espuma blanca.
[0352] MS (ESI): [M H+] = 841.1.
Paso 4: síntesis de (2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(íerc-butoxi)carboml] amino} - 7-metiloct-3-enoil]pirrolidin-2-il]formamido}-5-[N'-(4-metoxi-2,3,6-trimetil-bencenosulfonil)carbamimidamido]ácido pentanoico (227)
[0353] Se trató una solución del compuesto 226 (120 mg, 0.14 mmol) en THF/MeOH (4.0/2.0 mL) con LOH.H 2O (12 mg, 0.28 mmol en 0.2 mL de agua). La mezcla de la reacción se agitó a 25 °C durante 2 h, entonces el disolvente se eliminó al vacío y la mezcla resultante se trató con EtOAc (20 mL) y HCl acuoso (1 M) hasta alcanzar el pH 3. La fase orgánica se separó de la fase acuosa. La fase acuosa se extrajo con EtOAc (2 * 20 mL), y las capas orgánicas combinadas se lavaron con solución acuosa saturada de salmuera (2 * 20 mL), se secaron con Na2SO4, y se concentraron para obtener el producto 227 (100 mg, 86 %), que se usó para el siguiente paso sin purificación.
[0354] MS (ESI): [M H+] = 827.6.
Paso 5: síntesis de ferc-butil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-1-[(1-oxyiL-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]-4-[N-(4-metoxi-2,3,6-trimetil-bencenosulfoml)carbamimidamido]butil]carbamoil}pirrolidm-1 -il]-2-metil-8-oxooct-5-en-4-il]carbamato (228)
[0355] Se trató una solución del compuesto bruto 227 (100 mg, 0.12 mmol) en CH2Ch seco (3 mL) a 0 °C con HOBt (22 mg, 0.16 mmol), EDCI (31 mg, 0.16 mmol), DIPEA (31.0 mg, 0.24 mmol) y 4-AT (22.0 mg, 0.13 mmol). La mezcla de la reacción se agitó a 25 °C durante 3 h, luego se apagó con solución acuosa saturada de NH4Cl (20 mL). La fase acuosa se separó y se extrajo con CH2Ch (10 mL), y las capas orgánicas combinadas se secaron con MgSO4 y se concentraron para dar el producto bruto 228 (120 mg, bruto) en forma de un sólido naranja.
[0356] MS (ESI): [M H+] = 980.8.
Paso 6 : síntesis de (2S)-2-{[(2S)-1-[(2S,3£,5S)-5-amino-2-bencil-7-metiloct-3-enoil] pirrolidin-2-il]formamido}-5-carbamimidamido-N-(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)pentanamida (XXII)
[0357] Se agitó una solución del compuesto 228 (20 mg, 0.02 mmol) en TFA (1 mL) a 25 °C durante 4 h y luego se eliminó el disolvente. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), TFA al 0.1 % en eluyente) para dar el producto XXII (4.2 mg, 31 %) en forma de una espuma blanca.
[0358] MS (ESI): [M H+] = 699.5.
Ejemplo XXIII de referencia
(2E)-2-{[(2S)-1-[(2S,3E,5S)-5-ammo-2-bencil-7-metiloct-3-enoil] Pirrolidin-2-il]formamido}-N-(1-hidroxi -2,2,6,6-tetrametilpiperidin-4-il)-3-metilbutanamida (XXIII)
[0359]

Paso 1: síntesis de (S)-ferc-butil (2S)-2-{[(2R)-1-metoxi-3-metiM-oxobutan-2-il]carbamoil}pirrolidma-1-carboxilato (233)
[0360] A una solución del compuesto 231 (2.0 g, 9.30 mmol) y el compuesto 232 (1.7.0 g, 10.23 mmol) en CH2CI2 (10 mL) a 0 °C se añadieron T3P (3.5 g) y DIPEA (2.4 g, 18.6 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h y luego el disolvente se eliminó en vacío. La mezcla se disolvió en EtOAc (10mL), se lavó con NaHcO3 acuoso al 5 % (10 mL), solución acuosa saturada de salmuera (2 x 20 mL), se secó con Na2SO4 y se concentró en vacío para dar el producto bruto 233 (2.0 g, 67 %) en forma de una espuma blanca.
[0361] MS (ESI): [M H+] = 329.0.
Paso 2: síntesis de (2R)-2-{[(2S)-1-[(ferc-butoxi)carboml]pirrolidin-2-il] formamido}-3-ácido metilbutanoico (234)
[0362] Se trató una solución del compuesto 233 (2.0 g, 6.1 mmol) en THF/MeOH (12.0/3.0 mL) con LiOH.HzO (33.0 mg, 0.80 mmol en 0.2 mL de agua). La mezcla de la reacción se agitó a 25 °C durante 3 h, luego el disolvente se eliminó al vacío y la mezcla resultante se trató con EtOAc (10 mL) y HCl acuoso (1 M) hasta alcanzar el pH 3-4. La fase orgánica se separó de la fase acuosa. La fase acuosa se extrajo con EtOAc (2 x 20 mL), y las capas orgánicas combinadas se lavaron con solución acuosa saturada de salmuera (2 x 20 mL), se secaron con Na2SO4, y se concentraron en vacío para obtener el producto 234 (1.6 g, 84 %) como una espuma blanca, que se usó para el siguiente paso sin purificación.
[0363] MS (ESI): [M H+] = 315.0.
Paso 3: síntesis de ferc-butil (2S)-2-{[(1R)-1-[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)carbamoil] -2-metilpropil]carbamoil}pirrolidina- 1-carboxilato (235)
[0364] Se trató una solución del compuesto bruto 234 (1.8 g, 5.73 mmol) en CH2Cl2 seco (5 mL) a 0 °C con T3P (2.2 g, 6.88 mmol), DIPEA (1.5 g, 11.46 mmol), y 4-AT (1.1 g, 6.31 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h y luego el disolvente se eliminó en vacío. La mezcla se disolvió en EtOAc (20 mL), se lavó con NaHCO3 acuoso al 5 % (10 mL), y solución acuosa saturada de salmuera (2 x 20 mL), se secó con Na2SO4 y se concentró en vacío para dar el producto bruto 235 (1.4 g, 54 %) en forma de una espuma blanca.
[0365] MS (ESI): [M H+] = 468.3.
Paso 4: síntesis de (2R)-M-(1-hidroxi-2,2,6,6-tetrametilpiperidm-4-il)-3-metil-2-{[(2S)-pirrolidm-2-il]formamido}clorhidrato de butanamida (236)
[0366] Se agitó una solución del compuesto 235 (1.4 g, 3.0 mmol) en HCl en MeOH (10 mL, 3 M) a 25 °C durante 3 h. El disolvente se eliminó en vacío para dar el producto 236 (0.7 g, 64 %) en forma de un aceite rojo.
[0367] MS (ESI): [M H+] = 368.3.
Paso 5: síntesis de (2ft)-2-{[(2S)-1-[(2S,3E,5S)-5-ammo-2-bencil-7-metiloct-3-enoil] pirrolidin-2-il]formamido}-N-(1-hidroxi -2,2,6,6-tetrametilpiperidin-4-il)-3-metilbutanamida (XXIII)
[0368] A una solución del compuesto 236 (223 mg, 0.61 mmol) y el producto intermedio 59 (200 mg, 0.55 mmol) en CH2CI2 (5 mL) a 0 °C se añadieron T3P (210 mg, 0.66 mmol) y DIPEA (142 mg, 1.1 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h y luego el disolvente se eliminó en vacío. La mezcla se disolvió en EtOAc (10 mL), se lavó con NaHCO3 acuoso al 5 % (10 mL), y solución acuosa saturada de salmuera (2 x 10 mL), se secó con Na2SO4 y se concentró en vacío. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1 % en eluyente) para dar el producto XXIII (6.4 mg, rendimiento 4 %) en forma de un sólido blanco.
[0369] MS (ESI): [M H+] = 712.6;
Ejemplo XXIV de referencia
Bencil N-[(4S)-4-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(terc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-4-{[(4-hidroxi-2,6-dimetilfenil)metil]carbamoil}butil]carbamato (XXIV)
[0370]

Paso 1: síntesis de (S)-terc-butil 2-(((S)-1-((4-(benciloxi)-3,5-dimetilbencil)ammo)-5-(((benciloxi)carboml)ammo)-1-oxopentan-2-il)carbamoil)pirrolidina-1-carboxilato (243)
[0371] A una solución del compuesto 241 (1.5 g, 3.25 mmol, este compuesto se sintetizó a partir del compuesto 193) y el compuesto 242 (870 mg, 3.6 mmol) en C ^C h (10 mL) a 0 °C se añadieron T3P (1.3 g, 4.25 mmol) y DlpEA (840 mg, 6.5 mmol). Se dejó que la mezcla de la reacción se calentara a 25°C y se agitó durante 3 h y luego el disolvente se eliminó en vacío. La mezcla se disolvió en EtOAc (10 mL), se lavó con NaHcO3 acuoso al 5 % (10 mL), y solución acuosa saturada de salmuera (2 x 20 mL), se secó con Na2SO4 y se concentró en vacío para dar el producto bruto 243 (1.5 g, 85 %) en forma de una espuma blanca.
[0372] MS (ESI): [M H+] = 687.4.
Paso 2: síntesis de (S)-ferc-butil 2-(((S)-5-amino-1-((4-hidroxi-3,5-dimetilbencil)ammo)-1-oxopentan-2-il)carbamoil)pirrolidina-1-carboxilato (244)
[0373] A una solución del compuesto 243 (1.5 g, 2.19 mmol) en MeOH (10 mL) se añadió Pd/C (0.1 g, 10 %) a 25 °C en una atmósfera de gas de hidrógeno con globo. Se agitó la mezcla a 25 °C durante 16 h. Luego la reacción se filtró y se concentró en vacío para dar el producto 244 (0.75 g, 75 %) en forma de un sólido blanco.
[0374] MS (ESI): [M H+] = 463.2.
Paso 3: síntesis de (S)-ferc-butil 2-(((S)-5-(((benciloxi)carbonil)amino)-1-((4-hidroxi-3,5-dimetilbencil)amino)-1-oxopentan-2-il)carbamoil)pirrolidina-1-carboxilato (245)
[0375] A una solución del compuesto 244 (750 mg, 1.62 mmol) en CH2Cl2 (5 mL) a 0 °C se añadieron TEA (245 mg, 2.43 mmol) y CbzCl (290 mg, 1.7 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. La mezcla de la reacción se apagó con solución acuosa saturada de NH4Cl (10 mL). La fase orgánica combinada se lavó con solución acuosa saturada de salmuera (2 x 10 mL), se secó con Na2SO4y se concentró en vacío. El residuo se purificó por medio de cromatografía en columna (gel de sílice, éter de petróleo/EtoAc = de 3 : 1 a 1 : 1) para dar lugar al producto 245 (580 mg, 60 %) en forma de un sólido blanco.
[0376] MS (ESI): [M H+] = 597.3.
Paso 4: síntesis de bencil ((S)-5-((4-hidroxi-3,5-dimetilbencil)amino)-5-oxo-4-((S)-pirrolidma-2-carboxamido)pentil)carbamato (246)
[0377] Se agitó una solución del compuesto 245 (0.4 g, 0.67 mmol) en HCl en MeOH (5 mL, 3 M) a 25 °C durante 3 h. El disolvente se eliminó en vacío para dar el producto 246 (350 mg, 98 %) en forma de un sólido blanco.
[0378] MS (ESI): [M H+] = 497.3.
Paso 5: síntesis de bencil N-[(4S)-4-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-4-{[(4-hidroxi-2,6-dimetilfeml)metil]carbamoil}butil] carbamato (XXIV)
[0379] A una solución del compuesto 246 (350 mg, 0.66 mmol) y el compuesto 59 (250 mg, 0.69 mmol) en CH2Cl2 (5 mL) a 0 °C se añadieron T3P (251 mg, 0.79 mmol) y DIPEA (170 mg, 1.32 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h y luego el disolvente se eliminó en vacío. La mezcla se disolvió en EtOAc (20 mL), se lavó con NaHCO3 acuoso al 5 % (10 mL), y solución acuosa saturada de salmuera (3 x 10 mL), se secó con Na2SO4 y se concentró. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1 % en eluyente) para dar el producto XXIV (110 mg, rendimiento 20 %) en forma de sólido blanco.
[0380] MS (ESI): [M H+] = 840.7.
Ejemplo XXV de referencia
1-Hidroxi-2,2,6,6-tetrametilpiperidin-4-il N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-1-{[(1S)-4-{[(benciloxi)carboml]amino}-1-[(1-hidroxi-2,2,6,6-tetrametilpiperidm-4-il)carbamoil] butil] carbamoil}-2-metilpropil] carbamoil}pirrolidin-1 -il]-2-metil-8-oxooct-5-en-4-il]carbamato diclorhidrato (XXV)
[0381]

Debería ser VIII no 246
Paso 1: síntesis de 1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il N-[(4S,5E,7S)-7-bencil-8-[(25)-2- {[(15)-1- {[(1S)-4-{[(benciloxi)carbonil] amino} -1- [(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil]carbamoil}-2-metilpropil]carbamoil}pirrolidm-1-il]-2-metil-8-oxooct-5-en-4-il] carbamato diclorhidrato (XXV)
[0382] A una solución del compuesto VIII (30 mg, 0.028 mmol) en MeOH seco (3 mL) se añadió Vc (5 mg, 0.028 mmol). Después de agitar a 25 °C durante 1 h, el disolvente se eliminó al vacío. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1 % en eluyente) para dar el producto XXV (27 mg, 84 %) en forma de un sólido blanco.
[0383] MS (ESI): [M H+] = 1060.0.
Ejemplo XXVI y XXVII de referencia
Bencil N-[(5S)-5-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-([(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-5-[(1-oxil-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]pentil]carbamato (XXVI) y Bencil N-[(5s )-5-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(fercbutoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-5-[(1-hidroxi-2,2,6,6-tetrametilpipe-ridin-4-il)carbamoil]pentil]carbamato (XXVII)
[0384]

Paso 1: síntesis de (S)-metil 2-amino-6-(((benciloxi)carbonil)amino)hexanoato (252)
[0385] A una solución del 251 (2 g, 5.26 mmol) en MeOH (20 mL) se añadió SOCl2 (6.21 g, 52.6 mmol). La mezcla se agitó a 25 °C durante 12 h, y se concentró en vacío para obtener el denominado compuesto 252 (1.74 g, 100 %) en forma de un sólido blanco.
[0386] MS (ESI): [M H+] = 294.9.
Paso 2: síntesis de (S)-íerc-butil 2-(((S)-6-(((benciloxi)carbonil)amino)-1-metoxM-oxohexan-2-il)carbamoil)pirrolidina-1-carboxilato (253)
[0387] A una solución del compuesto 252 (1.74 g, 5.26 mmol), (S)-1-(terc-butoxicarbonil)pirrolidina-2-ácido carboxílico (1.7 g, 7.89 mmol) en CH2Ch (20 mL) se añadieron DIPEA (2.04 g, 15.78 mmol) y T3P (4.35 g, 6.84 mmol, 50 % en EtOAc). Se agitó la mezcla de la reacción a 25 °C durante 10 h, luego la solución orgánica se concentró en vacío. Se añadió EtOAc (100 mL) al residuo y se lavó con H 2O (3 * 20 mL). Se recogió la fase orgánica y se concentró para obtener el denominado compuesto 253 (2.6 g, 100 %) en forma de un sólido blanco.
[0388] MS (ESI): [M H+] = 492.2.
Paso 3: síntesis de (S)-metil 6-(((benciloxi)carbonil)amino)-2-((S)-pirrolidina-2-carboxa-mido)hexanoato (254)
[0389] El compuesto 253 (2.6 g, 5.26 mmol) y HCl en MeOH (40 mL, 3 M) se añadieron a un matraz de un solo cuello de 50 mL, luego se agitó la mezcla a 25 °C durante 1 h. La solución orgánica se concentró y se secó con una bomba de vacío para obtener el denominado compuesto 254 (2.25 g, 100 %) en forma de un sólido blanco.
[0390] MS (ESI): [M H+] = 392.1.
Paso 4: síntesis de (S)-metil 2-((S)-1-((2S,5S,E)-2-bencil-5-((terc-butoxicarboml)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-6-(((benciloxi)carbonil)amino)hexanoato (255)
[0391] A una solución del compuesto 254 (270 mg, 0.63 mmol) se añadió (2S,5S,E)-2-bencil-5-((tercbutoxicarbonil)amino)-7-metiloct-3-ácido enoico (250 mg, 0.69 mmol) y DIPeA (244 mg, 1.89 mmol) en CH2Ch (20 mL). Después de añadir T3P (521 mg, 0.82 mmol, 50 % en EtOAc) a 0 °C, se agitó la mezcla a 25 °C durante 12 h y luego la solución orgánica se concentró en vacío. Se añadió EtOAc (100 mL) al residuo y se lavó con H 2O (3 * 50 mL). La fase orgánica se recogió, se secó con Na2SO4 y se concentró en vacío. Los residuos se purificaron mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 3 : 1 a 1 : 1) para dar el denominado compuesto 255 (463 mg, 100 %) en forma de un sólido blanco.
[0392] MS (ESI): [M H+] = 735.5.
Paso 5: síntesis de (S)-2-((S)-1-((2S,5S,E)-2-bencil-5-((ferc-butoxicarboml)ammo)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-6 -(((benciloxi)carbonil)amino)ácido hexanoico (256)
[0393] A una solución del compuesto 255 (463 mg, 0.63 mmol) en MeOH/THF/H 2O (5 mL/5 mL/5 mL) se añadió L O H H 2O (132 mg, 3.15 mmol). Se agitó la mezcla de la reacción a 25 °C durante 4 h, se concentró en vacío. El residuo se acidificó hasta alcanzar el pH 3 con HCl acuoso (1 M), diluido con EtOAc (50 mL) y la capa orgánica se lavó con H 2O (3 * 25 mL). La fase orgánica se concentró en vacío para obtener el denominado compuesto 256 (454 mg, 100 %) en forma de un sólido blanco.
[0394] MS (ESI): [M H+] = 721.5.
Paso 6 : síntesis de Bencil N-[(5S)-5-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]amino}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-5-[(1-oxil-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]pentil]carbamato (XXVI)
[0395] A una solución del compuesto 256 (454 mg, 0.63 mmol), 4-AT (129 mg, 0.76 mmol), DIPEA (244 mg, 1.89 mmol) en CH2Cl2 (10 mL) se añadió T3P (521 mg, 0.82 mmol, 50 % en EtOAc). Se agitó la mezcla de la reacción a 25 °C durante 12 h, luego la solución orgánica se concentró en vacío. Después de añadir EtOAc (100 mL), la capa orgánica se lavó con H 2O (3 * 50 mL), y se concentró en vacío. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), NH3 H 2O al 0.5% en eluyente) para obtener el denominado producto XXVI (103 mg, 19 %) en forma de un sólido rosa.
[0396] MS (ESI): [M H+] = 874.7.
Paso 7: síntesis de bencil N-[(5S)-5-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-5-[(1-hidroxi-2,2,6,6-tetrametilpipe-ridin-4-il)carbamoil]pentil]carbamato (XXVII)
[0397] A una solución del compuesto XXVI (73 mg, 0.08 mmol) en MeOH (5 mL) se añadió Vc (14 mg, 0.08 mmol). Se agitó la solución resultante a 25 °C durante 30 min, luego la solución orgánica se concentró en vacío y el residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.5% en eluyente) para obtener el producto XXVII (73 mg, 100 %) en forma de un sólido blanco.
[0398] MS (ESI): [M H+] = 875.8.
Ejemplo XXVIII
ferc-Butil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-1-carbamoil-4-({[(1-oxil-2,2,6,6-tetrametilpiperidm-4-il)oxi] carbonil} amino-)butil] carbamoil} pirrolidin- 1-il] -2-metil-8-oxooct-5-en-4-il]carbamato (XXVIII)
[0399]
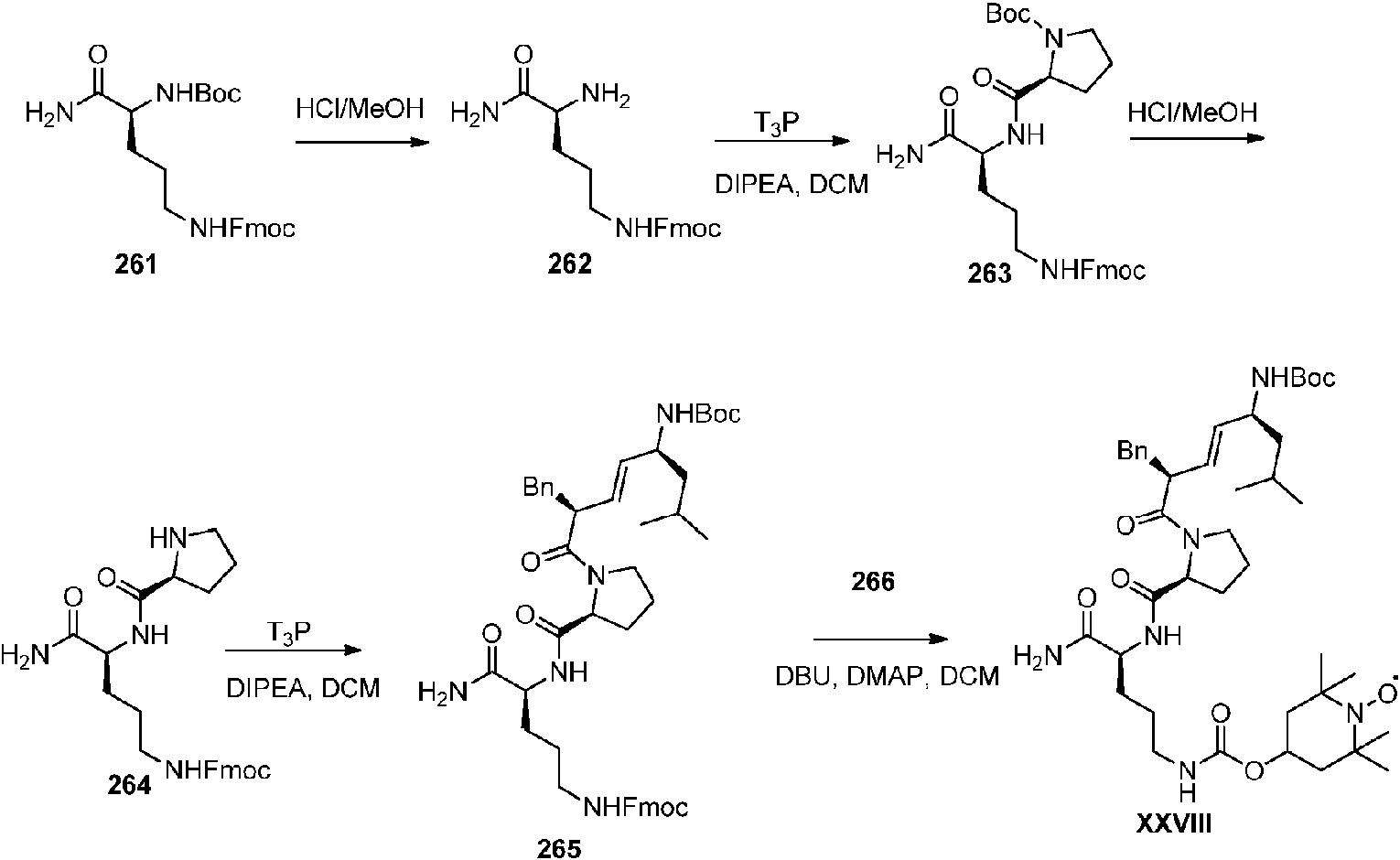
Paso 1: síntesis de (S)-(9H-fluoren-9-il)metil (4,5-diamino-5-oxopentil)carbamato (262)
[0400] Al compuesto 261 (500 mg, 1.1 mmol) se añadió HCl en MeOH (10 mL, 3 M), luego se agitó la mezcla a 25 °C durante 1 h. La solución orgánica se concentró en vacío para obtener el denominado compuesto 262 (428 mg, 100 %) en forma de un sólido blanco.
[0401] MS (ESI): [M H+] = 354.0.
Paso 2: síntesis de (S)-íerc-butil 2-(((S)-5-((((9H-fluoren-9-il)metoxi)carboml)amino)-1-ammo-1-oxopentan-2-il)carbamoil)pirrolidina-1-carboxilato (263)
[0402] A una solución del compuesto 262 (428 mg, 1.1 mmol), (S)-1-(fe/'c-butoxicarbonil)pirrolidina-2-ácido carboxílico (355 mg, 1.5 mmol) en CH2Cl2 (20 mL) se añadieron DIPEA (426 mg, 3.3 mmol) y T3P (910 mg, 1.43 mmol, 50 % en EtOAc) a 0 °C. La mezcla se agitó a 25 °C durante 12 h, luego la solución orgánica se concentró en vacío. Se añadió EtOAc (100 mL) al residuo y la capa orgánica se lavó con H2O (3 x 50 mL). Se recogió la fase orgánica y se secó con Na2SO4, y luego se concentró en vacío para obtener el denominado compuesto 263 (605 mg, 100 %) en forma de un sólido blanco.
[0403] MS (ESI): [M H+] = 551.2.
Paso 3: síntesis de (9H-fluoren-9-il)metil ((S)-5-amino-5-oxo-4-((5)-pirrolidina-2-carboxamido)pentil)carbamato (264)
[0404] Se añadieron el compuesto 263 (135 mg, 0.25 mmol) y HCl en MeOH (5 mL, 3 M) a un matraz de un solo cuello de 50 mL, luego se agitó la mezcla a 25 °C durante 1 h. La solución orgánica se concentró al vacío para obtener el denominado compuesto 264 (110 mg, 100 %) en forma de un sólido blanco.
[0405] MS (ESI): [M H+] = 451.1.
Paso 4: síntesis de (9H-fluoren-9-il)metil N-[(4S)-4-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(íerc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-4-carba-moilbutil]carbamato (265)
[0406] A una solución del compuesto 264 (110 mg, 0.25 mmol), (2S,5S,E)-2-bencil-5-((terc-butoxicarbonil)amino)-7-metiloct-3-ácido enoico (91 mg, 0.25 mmol) en CH2Ch (15 mL) se añadieron DIPEA (97 mg, 0.75 mmol) y T3P (207 mg, 0.33 mmol, 50 % en EtOAc) a 0 °C. La mezcla se agitó a 25 °C durante 12 h, luego la solución orgánica se concentró en vacío. Se añadió EtOAc (100 mL) al residuo y la capa orgánica se lavó con H2O (3 * 20 mL). La fase orgánica se recogió, se secó con Na2SO4 y luego se concentró en vacío. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 3 : 1 a 1 : 1) para obtener el denominado compuesto 265 (153 mg, 77 %) en forma de un sólido blanco.
[0407] MS (ESI): [M H+] = 794.5.
Paso 5: síntesis de íerc-butil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-1-carbamoil-4-({[(1 oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonil}amino-)-butil]carbamoil}pirrolidin-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (XXVIII)
[0408] A una solución del compuesto 265 (150 mg, 0.18 mmol) en CH2Ch (15 mL) se añadió DBU (144 mg, 0.94 mmol). La mezcla se agitó a 25 °C durante 20 min. Se añadieron a la mezcla DMAP (44 mg, 0.36 mmol) y compuesto 266 (130 mg, 0.36 mmol, el procedimiento para el 266 fue de acuerdo con ^cs Central Science, 2016, 2 (9): 653-659). Se agitó la mezcla a 25 °C durante 40 min, se diluyó con CH2Ch (50 mL) y la fase orgánica se lavó con H2O (3 * 20 mL). La fase orgánica se concentró en vacío y se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1% en eluyente) para obtener el denominado producto XXVIII (12 mg, 9 %) en forma de un sólido rosa.
[0409] MS (ESI): [M H+] = 770.6.
Ejemplo XXIX y XXX
Propan-2-il (2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-5-({[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonil}amino)pentanoato (XXIX) y Propan-2-il (2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-5-({[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonil}amino)pentanoato (XXX)
[0410]

Paso 1: síntesis de (S)-isopropil 5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-amino-pentanoato (272)
[0411] A una solución del compuesto 271 (500 mg, 1.1 mmol) en propan-2-ol (10 mL) se añadió SOCl2 (1 mL). La mezcla se agitó a 100 °C durante 24 h, y se concentró al vacío para obtener el denominado compuesto 272 (476 mg, 100 %) en forma de un sólido blanco.
[0412] MS (ESI): [M H+] = 397.1.
Paso 2: síntesis de (S)-terc-butil 2-(((S)-5-((((9H-fluoren-9-N)metoxi)carbonN)ammo)-1-isopropoxi-1-oxopentan-2-il)carbamoil)pirrolidina-1-carboxilato (273)
[0413] A una solución del compuesto 272 (476 mg, 1.1 mmol), (S)-1-(fe/-c-butoxicarbonil)pirrolidina-2-ácido carboxílico (355 mg, 1.65 mmol) en CH2Cl2 (20 mL) se añadieron DIPEA (426 mg, 3.3 mmol) y T3P (910 mg, 1.43 mmol, 50 % en EtOAc). Se agitó la mezcla de la reacción a 25 °C durante 12 h, luego la solución orgánica se concentró en vacío. Se añadió EtOAc (100 mL) al residuo y la capa orgánica se lavó con H2O (3 x 20 mL). Se recogió la fase orgánica y se secó con Na2SO4, y se concentró para obtener el denominado compuesto 273 (653 mg, 100 %) en forma de un sólido blanco.
[0414] MS (ESI): [M H+] = 594.4.
Paso 3: síntesis de (S)-isopropil 5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((S)-pirrolidina-2-carboxamido)pentanoato (274)
[0415] Al 273 (653 mg, 1.1 mmol) se añadió HCl en MeOH (10 mL, 3 M), luego se agitó la mezcla a 25 °C durante 1 h. La solución orgánica se concentró con una bomba de vacío para obtener el denominado compuesto 274 (543 mg, 100 %) en forma de un sólido blanco.
[0416] MS (ESI): [M H+] = 494.2.
Paso 4: síntesis de (S)-isopropil 5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((S)-1-((2S, 5S, E)-2-bencil-5-((tercbutoxicarbonil)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)pentanoato (275)
[0417] A una solución del compuesto 274 (543 mg, 1.1 mmol), (2S, 5S, £)-2-bencil-5-((ferc-butoxicarbonil)amino)-7-metiloct-3-ácido enoico (397 mg, 1.1 mmol) se añadieron DIPEA (426 mg, 3.3 mmol) en c H2CI2 (10 mL) y T3P (910 mg, 1.43 mmol, 50 % en EtOAc) a 0 °C. La mezcla se agitó a 25 °C durante 12 h, luego la solución orgánica se concentró en vacío. Se añadió EtOAc (100 mL) al residuo y la capa orgánica se lavó con H2O (3 x 20 mL). La fase orgánica se secó con Na2SO4, y luego se concentró en vacío y se purificó mediante cromatografía en columna sobre gel de sílice eluido con EtOAc para obtener el denominado compuesto 275 (513 mg, 56 %) en forma de un sólido blanco.
[0418] MS (ESI): [M H+] = 837.6.
Paso 5: síntesis de propan-2-il (2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-5-({[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonil}amino)pentanoato (XXIX)
[0419] A una solución del compuesto 275 (250 mg, 0.3 mmol) en CH2Cl2 (15 mL) se añadió DBU (227 mg, 1.5 mmol). La mezcla se agitó a 25 °C durante 20 min. Se añadieron DMAP (73 mg, 0.6 mmol) y compuesto 266 (217 mg, 0.6 mmol) a la mezcla. La mezcla se agitó a 25 °C durante 40 min, luego la mezcla se diluyó con CH2Cl2 (50 mL) y la capa orgánica se lavó con H2O (3 x 20 mL). La fase orgánica se secó con Na2SO4 y se concentró en vacío, y se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1% en eluyente) para obtener el denominado compuesto XXIX (72 mg, 29 %) en forma de un sólido rosa.
[0420] MS (ESI): [M H+] = 813.6.
Paso 6 : síntesis de propan-2-il (2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-5-({[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonil}amino)pentanoato (XXX)
[0421] A una solución del compuesto XXIX (50 mg, 0.06 mmol) en MeOH (5 mL) se añadió Vc (11 mg, 0.06 mmol). Se agitó la mezcla a 25 °C durante 30 min, se concentró la solución orgánica en vacío y el residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 x 250 mm), NH3 H2O al 0.5% en eluyente) para obtener el denominado compuesto XXIX (13 mg, 27 %) en forma de un sólido blanco.
[0422] MS (ESI): [M H+] = 814.7.
Ejemplo XXXI y XXXII
Propan-2-Il (2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-6-({[(1-oxil-2,2,6,6-tetrametilpiperidm-4-il)oxi]carboml}ammo)hexanoato (XXXI) y Propan-2-il (2S)-2-{[(2 S)-1 -[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-6-[(1-oxil-2,2,5,5-tetrametilpirrolidin-3-il)formamido]-hexanoato (XXXII)
[0423]

Paso 1: síntesis de propan-2-il (2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-6-({[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonil}amino)hexanoato (XXXI)
[0424] A una solución del compuesto 275 (350 mg, 0.41 mmol) en CH2CI2 (15 mL) se añadió DBU (313 mg, 2.1 mmol). La mezcla se agitó a 25 °C durante 20 min. Se añadieron DMAP (100 mg, 0.82 mmol) y compuesto 266 (296 mg, 0.82 mmol) a la mezcla. Después de agitar a 25 °C durante 1 h, la mezcla se diluyó con C ^C h (50 mL) y se lavó con H 2O (3 x 20mL). La fase orgánica se secó con Na2SO4 y se concentró en vacío, y se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1% en eluyente) para obtener el denominado compuesto (80 mg, 22 %) en forma de un sólido rosa.
[0425] MS (ESI): [M H+] = 827.7.
Paso 2: síntesis de propan-2-il (2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(íerc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-6-[(1-oxil-2,2,5,5-tetrametilpirrolidm-3-il)formamido]-hexanoato (XXXII)
[0426] A una solución del compuesto 275 (225 mg, 0.26 mmol) en CH2Ch (15 mL) se añadió DBU (395 mg, 2.6 mmol). La mezcla se agitó a 25 °C durante 20 min. Se añadieron el 287 (45 mg, 0.26 mmol) y T3P (215 mg, 0.34 mmol, 50 % en EtOAc) a la mezcla. Se agitó la mezcla a 25 °C durante 10 h, se diluyó con CH2Ch (50 mL) y se lavó con H 2O (3 x 20 mL). La fase orgánica se secó con Na2SO4 y luego se concentró en vacío, y se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1% en eluyente) para obtener el denominado compuesto x Xx II (10 mg, 5 %) en forma de un sólido blanquecino.
[0427] MS (ESI): [M H+] = 797.7.
Ejemplo XXXIII y XXXIV
Propan-2-il (2R)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-6-({[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonillamino)hexanoato (XXXIII) y (2E)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-6-({[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonil}amino)ácido hexanoico (XXXIV)
[0428]

Paso 1: síntesis de (S)-isopropil 6-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-aminohexanoato (292)
[0429] A una solución del compuesto 291 (500 mg, 1.07 mmol) en propan-2-ol (10 mL) se añadió SOCl2 (0.5 mL). La mezcla se agitó a 100 °C durante 10 h, y se concentró al vacío para obtener el denominado compuesto 292 (439 mg, 100 %) en forma de un sólido blanco.
[0430] MS (ESI): [M H+] = 411.1.
Paso 2: síntesis de (S)-ferc-butil 2-(((ft)-6-((((9H-fluoren-9-il)metoxi)carboml)ammo)-1-isopropoxM-oxohexan-2-il)carbamoil)pirrolidina-1-carboxilato (293)
[0431] A una solución del compuesto 292 (439 mg, 1.07 mmol), (S)-1-(fe/'c-butoxicarbonil)pirrolidina-2-ácido carboxílico (345 mg, 1.61 mmol) en CH2Cl2 (15 mL) se añadieron DIPEA (414 mg, 3.21 mmol) y T3P (885 mg, 1.39 mmol, 50 % en EtOAc) a 0 °C. La mezcla se agitó a 25 °C durante 12 h, luego se concentró. Se añadió EtOAc (100 mL) al residuo y la capa orgánica se lavó con H2O (3 x 20 mL). Se recogió la fase orgánica y se secó con Na2SO4, y luego se concentró en vacío para obtener el denominado compuesto 293 (650 mg, 100 %) en forma de un sólido blanco.
[0432] MS (ESI): [M H+] = 608.3.
Paso 3: síntesis de (ft)-isopropil 6-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((S)-pirrolidina-2-carboxamido)hexanoato (294)
[0433] Al compuesto 293 (650 mg, 1.07 mmol) se añadió HCl en MeOH (5 mL, 3 M). La solución orgánica se concentró al vacío para obtener el denominado compuesto 294 (544 mg, 100 %) en forma de un sólido blanco, después de agitar a 25 °C durante 1 h.
[0434] MS (ESI): [M H+] = 508.2.
Paso 4: síntesis de (R)-isopropil 6-((((9H-fluoren-9-il)metoxi)carboml)ammo)-2-((S)-1-((2S,5S,E)-2-bencil-5-((fercbutoxicarbonil)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)hexanoato (295)
[0435] A una solución del compuesto 294 (544 mg, 1.07 mmol) en CH2CI2 (10 mL) se añadieron (2S, 5S, E)-2-bencil-5-((terc-butoxicarbonil)amino)-7-metiloct-3-ácido enoico (59, 386 mg, 1.07 mmol), DIPEA (414 mg, 3.21 mmol) y T3P (884 mg, 1.4 mmol, 50% en EtOAc) a 0 °C. Se agitó la mezcla a 25 °C durante 12 h, luego se concentró. Se añadió EtOAc (100 mL) al residuo y la capa orgánica se lavó con H 2O (3 * 20 mL). La fase orgánica se recogió y se secó con Na2SO4, y luego se concentró en vacío. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 3 : 1 a 1 : 1) para dar el denominado compuesto 295 (911 mg, 100 %) en forma de un sólido blanco.
[0436] MS (ESI): [M H+] = 851.6.
Paso 5: síntesis de propan-2-il (2R)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(íerc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-6-({[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonil}amino)hexanoato (XXXIII)
[0437] A una solución del compuesto 295 (250 mg, 0.3 mmol) en CH2Ch (15 mL) se añadió DBU (277 mg, 1.5 mmol). La mezcla se agitó a 25 °C durante 20 min. Se añadieron DMAP (73 mg, 0.6 mmol) y compuesto 266 (217 mg, 0.6 mmol). La mezcla se agitó a 25 °C durante 1 h, se diluyó con CH2Ch (50 mL) y se lavó con H 2O (3 * 20 mL). La fase orgánica se secó con Na2SO4 y se concentró en vacío, y se purificó mediante CLAr preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1% en eluyente) para obtener el denominado compuesto XXXIII (100 mg, 40 %) en forma de un sólido rosa.
[0438] MS (ESI): [M H+] = 827.7.
Paso 6 : síntesis (2R)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-6-({[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonil}amino)ácido hexanoico (XXXIV)
[0439] A una solución del compuesto 295 (280 mg, 0.33 mmol) en THF/MeOH/H 2O (10 mL/10 mL/5 mL) se añadió LiOH.HzO (69 mg, 1.65 mmol). Se agitó la mezcla de la reacción a 25 °C durante 10 h, luego la solución orgánica se concentró en vacío. El residuo se diluyó con EtOAc (20 mL) y se acidificó hasta alcanzar el pH 3 con HCl acuoso (1 M), luego se filtró el sólido inorgánico y se concentró el filtrado en vacío para obtener un sólido amarillo claro. A una solución del sólido amarillo en CH2Ch (15 mL) se añadieron DIPEA (426 mg, 3.3 mmol) y compuesto 266 (238 mg, 0.66 mmol). La mezcla se agitó a 25 °C durante 1 h, luego se concentró y se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1% en eluyente) para obtener el denominado producto XXXIV (12 mg, 5 %) en forma de un sólido rosa.
[0440] MS (ESI): [M H+] = 785.6.
Ejemplo XXXV
íerc-Butil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-1-(dietilcarbamoM)-4-({[(1-oxil-2,2,6,6-tetrametilpiperidm-4-il)oxi]carbonil}amino)-butil]carbamoil}pirrolidin-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (XXXV)
[0441]

Paso 1: síntesis de (S)-(9H-fluoren-9-il)metil ferc-butil (5-(dietilamino)-5-oxopentano-1,4-diil)dicarbamato (302) [0442] A una solución del compuesto 301 (500 mg, 1.1 mmol), dietilamina (241 mg, 3.3 mmol) se añadieron DIPEA (426 mg, 3.3 mmol) en CH2Cl2 (15 mL) y T3P (910 mg, 1.43 mmol, 50% en EtOAc). Se agitó la mezcla a 25 °C durante 2 h, luego se concentró en vacío. Se añadió EtOAc (100 mL) al residuo y la capa orgánica se lavó con H2O (3 x 20 mL). Se recogió la fase orgánica y se secó con Na2SO4, y luego se concentró para obtener el denominado compuesto 302 (560 mg, 100 %) en forma de un sólido blanco.
[0443] MS (ESI): [M H+] = 510.2.
Paso 2: síntesis de (S)-(9H-fluoren-9-il)metil (4-amino-5-(dietilamino)-5-oxopentil)carbamato (303)
[0444] Al compuesto 302 (560 mg, 1.1 mmol) se añadieron HCl en MeOH (5 mL, 3 M) y EtOAc (5 mL). Después de agitar a 25 °C durante 1 h, la solución orgánica se concentró al vacío para obtener el denominado compuesto 303 (450 mg, 100 %) en forma de un sólido blanco.
[0445] MS (ESI): [M H+] = 410.1.
Paso 3: síntesis de (S)-ferc-butil 2-(((S)-5-((((9H-fluoren-9-N)metoxi)carbonN)ammo)-1-(dietilammo)-1-oxopentan-2-il)carbamoil)pirrolidina-1-carboxilato (304)
[0446] A una solución del compuesto 303 (450 mg, 1.1 mmol), (S)-1-(ferc-butoxicarbonil)pirrolidina-2-ácido carboxílico (355 mg, 1.65 mmol) en CH2Cl2 (15 mL) se añadieron DIPEA (426 mg, 3.3 mmol) y T3P (910 mg, 1.43 mmol, 50 % en EtOAc). Se agitó la mezcla a 25 °C durante 12 h, luego se concentró en vacío. Se añadió EtOAc (100 mL) al residuo y la capa orgánica se lavó con H2O (3 x 20 mL). Se recogió la fase orgánica y se secó con Na2SO4, y luego se concentró en vacío para obtener el denominado compuesto 304 (667 mg, 100 %) en forma de un sólido blanco.
[0447] MS (ESI): [M H+] = 607.4.
Paso 4: síntesis de (9H-fluoren-9-il)metil ((S)-5-(dietilamino)-5-oxo-4-((S)-pirrolidina-2-carboxamido)pentil)carbamato (305)
[0448] Al compuesto 304 (667 mg, 1.1 mmol) se añadió HCl en MeOH (10 mL, 3 M). La mezcla se agitó a 25 °C durante 30 min, y se concentró al vacío para obtener el denominado compuesto 305 (557 mg, 100 %) en forma de un sólido blanco.
[0449] MS (ESI): [M H+] = 507.2.
Paso 5: síntesis de (9H-fluoren-9-il)metil N-[(4S)-4-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]ammo}-7-metiloct-3-enoil]pirrolidm-2-il]formamido}-4-(dietilcarbamoil)butil]carbamato (306)
[0450] A una solución del compuesto 305 (557 mg, 1.1 mmol) en CH2Cl2 (15 mL) se añadieron ((2S, 5S, EJ-2-bencil-5-((ferc-butoxicarbonil)amino)-7-metiloct-3-ácido enoico (59) (397 mg, 1.1 mmol), DIPEA (426 mg, 3.3 mmol) y T3P (910 mg, 1.43 mmol, 50 % en EtOAc) a 0 °C. La mezcla se agitó a 25 °C durante 12 h, luego se concentró en vacío. Se añadió EtOAc (100 mL) al residuo y la capa orgánica se lavó con H2O (3 * 20 mL). La fase orgánica se recogió y se secó con Na2SO4, y luego se concentró en vacío. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 3 : 1 a 1 : 1) para dar el denominado compuesto 306 (935 mg, 100 %) en forma de un sólido blanco.
[0451] MS (ESI): [M H+] = 850.6.
Paso 6 : síntesis de ferc-butil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-1-(dietilcarbamoil)-4-({[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonil}amino)-butil]carbamoil}pirrolidin-1 -il]-2-metil-8-oxooct-5-en-4-il]carbamato (XXXV)
[0452] A una solución del compuesto 306 (350 mg, 0.41 mmol) en CH2Ch (15 mL) se añadió DBU (313 mg, 2.1 mmol). La mezcla se agitó a 25 °C durante 20 min. Se añadieron DMAP (100 mg, 0.82 mmol) y el 266 (296 mg, 0.82 mmol) a la mezcla. La mezcla se agitó a 25 °C durante 1 h, luego la mezcla se diluyó con CH2Cl2 (50 mL) y la capa orgánica se lavó con H 2O (3 * 20 mL). La fase orgánica se secó con Na2SO4 y se concentró en vacío, y se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1% en eluyente) para obtener el denominado producto XXXV (42 mg, 12 %) en forma de un sólido rosa.
[0453] MS (ESI): [M H+] = 826.9.
Ejemplo XXXVI
ferc-Butil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-1-(terc-butilcarbamoil)-5-({[(1-oxil-2,2,6,6-tetrametilpiperidm-4-il)oxi]carbonil}amino)pentil]-carbamoil}pirrolidin-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (XXXVI)
[0454]

Paso 1: síntesis de (S)-(9H-fluoren-9-il)metil ferc-butil (6-(ferc-butilamino)-6-oxohexano-1,5-diil)dicarbamato (312)
[0455] A una solución del compuesto 311 (500 mg, 1.07 mmol), f-BuNH (234 mg, 3.21 mmol) se añadieron DIPEA (414 mg, 3.21 mmol) en CH2Ch (20 mL) y T3P (885 mg, 1.39 mmol, 50 % en EtOAc) a 0 °C. La mezcla se agitó a 25 °C durante 12 h, luego se concentró en vacío. Se añadió EtOAc (100 mL) al residuo y la capa orgánica se lavó con H 2O (3 * 20 mL). La fase orgánica se secó con Na2SO4 y se concentró en vacío, y se purificó mediante CCF previa (éter de petróleo/EtOAc = 1 : 1) para obtener el denominado compuesto 312 (427 mg, 76 %) en forma de un sólido blanco.
[0456] MS (ESI): [M H+] = 524.2.
Paso 2: síntesis de (S)-(9H-fluoren-9-il)metil (5-ammo-6-(terc-butNammo)-6-oxohexM)carbamato (313)
[0457] Al compuesto 312 (427 mg, 0.81 mmol) se añadió HCl en MeOH (10 mL, 3 M). Después de agitar a 25 °C durante 0.5 h, la solución orgánica se concentró al vacío para obtener el denominado compuesto 313 (343 mg, 100 %) en forma de un sólido blanco.
[0458] MS (ESI): [M H+] = 424.1.
Paso 3: síntesis de (S)-terc-butil 2-(((S)-6-((((9H-fluoren-9-N)metoxi)carbonN)ammo)-1-(terc-butNammo)-1-oxohexan-2-il)carbamoil)pirrolidina-1-carboxilato (314)
[0459] A 0 °C a una solución del compuesto 313 (343 mg, 0.81 mmol) y (S)-1-(terc-butoxicarbonil)pirrolidina-2-ácido carboxílico (261 mg, 1.21 mmol) en C ^C h (15 mL) se añadieron DIPEA (314 mg, 2.43 mmol) y T3P (670 mg, 1.1 mmol, 50 % en EtOAc). La mezcla se agitó a 25 °C durante 12 h, luego se concentró en vacío. Se añadió EtOAc (100 mL) al residuo y la capa orgánica se lavó con H 2O (3 * 20 mL). Se recogió la fase orgánica y se secó con Na2SO4, y luego se concentró para obtener el denominado compuesto 314 (503 mg, 100 %) en forma de un sólido blanco.
[0460] MS (ESI): [M H+] = 621.3.
Paso 4: síntesis de (9H-fluoren-9-il)metil ((S)-6-(terc-butNammo)-6-oxo-5-((S)-pirroNdina-2-carboxamido)hexil)carbamato (315)
[0461] Al compuesto 314 (503 mg, 0.81 mmol) se añadió HCl en MeOH (10 mL, 3 M). Después de agitar a 25 °C durante 30 min, la solución orgánica se concentró al vacío para obtener el denominado compuesto 315 (422 mg, 100 %) en forma de un sólido blanco.
[0462] MS (ESI): [M H+] = 521.2.
Paso 5: síntesis de (9H-fluoren-9-il)metil N-[(5S)-5-{[(2S)-1-[(2S,3E,5S)-2-bencN-5-{[(terc-butoxi)carbonN]ammo}-7-metiloct-3-enoM]pirroMdm-2-M]formamido}-5-(terc-butMcarbamoM)pentM]carbamato (316)
[0463] A una solución del compuesto 315 (422 mg, 0.81 mmol), (2S, 5S, E)-2-bencil-5-((terc-butoxicarbonil)amino)-7-metiloct-3-ácido enoico (293 mg, 0.81 mmol), DIPEA (313 mg, 2.43 mmol) en CH2Ch (15 mL) se añadió T3P (670 mg, 1.05 mmol, 50 % en EtOAc) a 0 °C. Después de agitar a 25 °C durante 12 h, la solución orgánica se concentró en vacío. Se añadió EtOAc (100 mL) al residuo y la capa orgánica se lavó con H 2O (3 * 20 mL). La fase orgánica se concentró en vacío. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 3 : 1 a 1 : 1) para obtener el denominado compuesto 316 (700 mg, 100 %) en forma de un sólido blanco.
[0464] MS (ESI): [M H+] = 864.6.
Paso 6 : síntesis de terc-butil N-[(4S,5E,7S)-7-bencN-8-[(2S)-2-{[(1S)-1-(terc-butMcarbamoM)-5-({[(1-oxN-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonil}amino)pentil]-carbamoil}pirrolidin-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (XXXVI)
[0465] A una solución del compuesto 316 (350 mg, 0.41 mmol) en CH2Ch (15 mL) se añadió DBU (313 mg, 2.1 mmol). La mezcla se agitó a 25 °C durante 20 min. Se añadieron DMAP (100 mg, 0.82 mmol) y compuesto 266 (296 mg, 0.82 mmol) a la mezcla. La mezcla se agitó a 25 °C durante 1 h, se diluyó con CH2Cl2 (50 mL) y se lavó con H 2O (3 * 20 mL). La fase orgánica se concentró en vacío y se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1% en eluyente) para obtener el denominado compuesto XXXVI (84 mg, 24 %) en forma de un sólido rosa.
[0466] MS (ESI): [M H+] = 840.6.
Ejemplo XXXVII
terc-Butil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-5-({[(1-oxil-2,2,6,6-tetrametilpiperidm-4-il)oxi]carboml}ammo)-1-[(propan-2-il)carbamoil]pentil]carbamoil}pirrolidm-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (XXXVII)
[0467]

Paso 1: síntesis de (S)-(9H-fluoren-9-il)metil terc-butil (6-(isopropilamino)-6-oxohexano-1,5-diil)dicarbamato (322)
[0468] A una solución del compuesto 321 (500 mg, 1.07 mmol), propan-2-amina (190 mg, 3.21 mmol), DIPEA (414 mg, 3.21 mmol) en CH2Cl2 (20 mL) se añadió T3P (885 mg, 1.39 mmol, 50 % en EtOAc) a 0 °C. La mezcla se agitó a 25 °C durante 12 h, la solución orgánica se concentró en vacío. Se añadió EtOAc (100 mL) al residuo y la capa orgánica se lavó con H 2O (3 x 20 mL). La fase orgánica se secó con Na2SO4 y luego se concentró en vacío y se purificó mediante CCF preparativa (placa de gel de sílice de 1.5 mm de grosor, éter de petróleo/EtOAc = 1 : 1) para obtener el denominado compuesto 322 (341 mg, 63 %) en forma de un sólido blanco.
[0469] MS (ESI): [M H+] = 510.15
Paso 2: síntesis de (S )-(9H-fluoren-9-il)metil (5-amino-6-(isopropilamino)-6-oxohexil)carbamato (323)
[0470] Al compuesto 322 (341 mg, 0.67 mmol) se añadió HCl en MeOH (10 mL, 3 M). Después de agitar a 25 °C durante 0.5 h, la solución orgánica se concentró en vacío al vacío para obtener el denominado compuesto 323 (274 mg, 100 %) en forma de un sólido blanco.
[0471] MS (ESI): [M H+] = 410.0.
Paso 3: síntesis de (S)-terc-butil 2-(((S)-6-((((9H-fluoren-9-il)metoxi)carboml)ammo)-1-(isopropilammo)-1-oxohexan-2 -il)carbamoil)pirrolidina-1 -carboxilato (324)
[0472] A una solución del compuesto 323 (274 mg, 0.67 mmol), (S)-1-(fe/'c-butoxicarbonil)pirrolidina-2-ácido carboxílico (216 mg, 1.01 mmol) en CH2Cl2 (15 mL) se añadieron DIPEA (260 mg, 2.01 mmol) y T3P (554 mg, 0.87 mmol, 50 % en EtOAc). Se agitó la mezcla a 25 °C durante 12 h, luego la solución orgánica se concentró en vacío. Se añadió EtOAc (100 mL) al residuo y la capa orgánica se lavó con H 2O (3 x 20 mL). Se recogió la fase orgánica y se secó con Na2SO4, y luego se concentró en vacío para obtener el denominado compuesto 324 (407 mg, 100 %) en forma de un sólido blanco.
[0473] MS (ESI): [M H+] = 607.3
Paso 4: síntesis de (9H-fluoren-9-il)metil ((S)-6-(isopropilammo)-6-oxo-5-((S)-pirrolidma-2-carboxamido)hexil)carbamato clorhidrato (325)
[0474] Al compuesto 325 (407 mg, 0.67 mmol) se añadió HCl en MeOH (10 mL, 3 M). Después de agitar a 25 °C durante 30 min, la solución orgánica se concentró en vacío al vacío para obtener el denominado compuesto 325 (340 mg, 100 %) en forma de un sólido blanco.
[0475] MS (ESI): [M H+] = 507.2.
Paso 5: síntesis de (9H-fluoren-9-il)metil N-[(5S)-5-{[(2S)-1-[(2S,3£,5S)-2-bencil-5-{[(ferc-butoxi)carboml]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-5-[(pro-pan-2-il)carbamoil]pentil]carbamato (326)
[0476] A una solución del compuesto 325 (340 mg, 0.67 mmol), (2S, 5S, E)-2-bencil-5-((terc-butoxicarbonil)amino)-7-metiloct-3-ácido enoico (59, 242 mg, 0.67 mmol), DIPEA (259 mg, 2.01 mmol) en CH2Ch (15 mL) se añadió T3P (554 mg, 0.87 mmol, 50 % en EtOAc). Se agitó la mezcla a 25 °C durante 12 h, luego la solución orgánica se concentró en vacío. Se añadió EtOAc (100 mL) al residuo y la capa orgánica se lavó con H 2O (3 * 20 mL). La fase orgánica se recogió y se secó con Na2SO4, y se concentró en vacío. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 3 : 1 a 1 : 1) para dar el denominado compuesto 326 (570 mg, 100 %) en forma de un sólido blanco.
[0477] MS (ESI): [M H+] = 850.5.
[0478] RMN de 1H: (400MHz, CDCla) 8 7.76-7.69 (m, 2 H), 7.61-7.59 (m, 2 H), 7.40-7.36 (m, 2 H), 7.32-7.30 (m, 2 H), 7.25-7.22 (m, 2 H), 7.19-7.12 (m, 3 H), 5.59 (dd, 1 H, J = 15.0, 8.5 Hz), 5.42 (dd,1 H, J = 15.0, 5.0 Hz), 4.42-4.41 (m, 1 H), 4.35-4.34 (m, 2 H), 4.20-4.18 (m, 1 H), 4.12-4.03 (m, 2 H), 3.54-3.47 (m, 1 H), 3.38-3.34 (m, 1 H), 3.18-3.10 (m, 3 H), 2.79 2.74 (m, 1 H), 1.90-1.62 (m, 9 H), 1.45-1.07 (m, 25 H), 0.89-0.85 (m, 6 H).
Paso 6 : síntesis de ferc-butil N-[(4S,5£,7S)-7-bencil-8-[(2S)-2-{[(1S)-5-({[(1-oxil-2,2,6,6-tetrametilpiperidm-4-yl)oxi]carbonil}amino)-1-[(propan-2-il)carbamoil]pentil]carbamoil}pirrolidin-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (XXXVII)
[0479] A una solución del compuesto 326 (350 mg, 0.41 mmol) en CH2Ch (15 mL) se añadió DBU (313 mg, 2.1 mmol). La mezcla se agitó a 25 °C durante 20 min. Se añadieron DMAP (100 mg, 0.82 mmol) y compuesto 266 (296 mg, 0.82 mmol) a la mezcla. La mezcla se agitó a 25 °C durante 1 h, se diluyó con CH2Ch (50 mL) y se lavó con H 2O (3 * 20 mL). La fase orgánica se secó con Na2SO4 y se concentró en vacío, y se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1% en eluyente) para obtener el denominado producto XXXVII (75 mg, 22 %) en forma de un sólido rosa.
[0480] MS (ESI): [M H+] = 826.7.
Ejemplo XXXVIII y XXXIX
(S)-Metil 2-((S)-2-((S)-1-((2S,5S,£)-2-bencil-5-((íerc-butoxicarbonil)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-3-metilbutanamido)-5-((((1-oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi)carbonil)amino)pentanoato (XXXVIII) y (S)-metil 2-((S)-2-((S)-1-((2S,5S,£)-2-bencil-5-((íerc-butoxicarbonii)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-3-metilbutanamido)-5-((((1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)oxi)carbonil)amino)pentanoato (XXXIX)
[0481]

Paso 1: síntesis de (S)-metil 5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((S)-2-((terc-butoxicarbonil)amino)-3-metilbutanamido)pentanoato (332)
[0482] A una solución de (S)-metil 5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-aminopentanoato (331, 1.0 g, 2.5 mmol) y (S)-2-((íerc-butoxicarbonil)amino)-3-ácido metilbutanoico (705 mg, 3.3 mmol) en CH2Cl2 (5 mL) se añadieron T3P (2.2 g, 3.3 mmol) y DIPEA (645 g, 5.0 mmol) a 0 °C. Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante l 6 h. La mezcla de la reacción se extrajo con CH2Cl2 (3 x 10 mL). La mezcla de la reacción se lavó con NaHCO3 acuoso al 5 % (10 mL), HCl acuoso (5 mL, 1 M) y solución acuosa saturada de salmuera (3 x 20 mL). La capa orgánica se secó con Na2SO4y se concentró en vacío. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = 5 : 1 - 1 : 1) para dar el producto 332 (1.0 g, 91 %) en forma de un sólido blanco.
[0483] MS (ESI): [M H+] = 568.4.
Paso 2: síntesis de (S)-metil 5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((S)-2-amino-3-metilbutanamido)pentanoato (333)
[0484] Se agitó una solución del compuesto 332 (1.0 g, 1.8 mmol) en HCl en MeOH (20 mL, 3 M) a 25 °C durante 3 h y luego el disolvente se eliminó en vacío para dar el producto 333 (841 mg, 100 %) en forma de una espuma blanca.
[0485] MS (ESI): [M H+] = 468.2.
Paso 3: síntesis de (S)-ferc-butil 2-(((S)-1-(((S)-5-((((9H-fluoren-9-il)metoxi)carbonil)amino)-1-metoxi-1-oxopentan-2-il)amino)-3-metil-1 -oxobutan-2-il)carbamoil)pirrolidina-1 -carboxilato (334)
[0486] A una solución del compuesto 333 (905 mg, 1.8 mmol) y (S)-1-(fe/'c-butoxicarbonil)pirrolidina-2-ácido carboxílico (464.0 mg, 2.2 mmol) en CH2Ch (10 mL) se añadieron T3P (731 mg, 2.3 mmol) y DIPEA (510 g, 4.0 mmol) a 0 °C. Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 16 h. La mezcla de la reacción se apagó con solución acuosa saturada de NH4Cl (20 mL) y se extrajo con EtOAc (3 x 10 mL). La fase orgánica se lavó con HCl acuoso (10 mL, 1 M), solución acuosa saturada de salmuera (3 x 20 mL) y se secó con Na2SO4. La fase orgánica se concentró a presión reducida para dar el producto 334 (110 mg, 28 %) en forma de una espuma blanca.
[0487] MS (ESI): [M H+] = 665.5.
Paso 4: síntesis de (S)-metil 5-((((9H-fluoren-9-N)metoxi)carbonN)ammo)-2-((S)-3-metil-2-((S)-pirroMdma-2-carboxamido)butanamido)pentanoato (335)
[0488] Se agitó una solución del compuesto 334 (0.96 g, 1.4 mmol) en HCl en MeOH (20 mL, 3 M) a 25 °C durante 3 h y luego el disolvente se eliminó en vacío para dar el producto 335 (840 mg, 100 %) en forma de una espuma blanca.
[0489] MS (ESI): [M H+] = 565.3.
Paso 5: síntesis de (S)-metil 5-((((9H-fluoren-9-il) metoxi) carbonil) amino) -2-((S)-2-((S)-1-((2S,5S,E)-2-bencil-5-((terc-butoxicarbonil)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-3-metilbutanamido)pentanoato (336)
[0490] A una solución del compuesto 335 (0.84 g, 1.4 mmol) y (2S,5S,E)-2-bencil-5-((terc-butoxicarbonil)amino)-7-metiloct-3-ácido enoico (59, 606 mg, 1.7 mmol) en CH2Ch (10 mL) se añadieron T3P (572 g, 1.8 mmol) y DIPeA (400 mg, 3.1 mmol) a 0 °C. Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 16 h. La mezcla de la reacción se apagó con solución acuosa saturada de NH4Cl (20 mL) y se extrajo con EtOAc (3 * 10 mL). La fase orgánica se lavó con NaHCO3 acuoso al 5 % (10 mL), solución acuosa saturada de salmuera (3 * 20 mL), y se secó con Na2SO4. La fase orgánica se concentró y se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 3 : 1 a 1 : 1) para dar el producto 336 (810 mg, 90 %) en forma de un sólido blanco.
[0491] MS (ESI): [M H+] = 908.7.
Paso 6 : síntesis de (S)-metil 5-ammo-2-((S)-2-((S)-1-((2S,5S,E)-2-bencN-5-((íerc-butoxicarbonM)ammo)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-3-metilbutanamido)pentanoato (337)
[0492] Se añadió una solución de TBAF (5 % en peso en THF, 6 mL) a 0 °C a una solución del compuesto 336 (680 mg, 0.75 mmol) en DMF (8 mL). La reacción se agitó durante 30 min. La mezcla del compuesto 337 (514 mg, 100 %) se usó para el siguiente paso sin tratamiento.
[0493] MS (ESI): [M H+] = 686.5.
Paso 7: síntesis de (S)-metil 2-((S)-2-((S)-1-((2S,5S,E)-2-bencN-5-((íerc-butoxicarbonM)ammo)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-3-metilbutanamido)-5-((((1-oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi)carbonil)amino)pentanoato (XXXVIII)
[0494] A una solución del compuesto 337 (514 mg, 0.75 mmol) y 1,3-dioxoisoindolin-2-il (1-oxil-2,2,6,6-tetrametilpiperidin-4-il) carbonato (266) (325 mg, 0.9 mmol) en CH2Ch (8 mL) a 0 °C se añadió DIPEA (194 mg, 1.5 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 2 h. La mezcla de la reacción se apagó con solución acuosa saturada de NH4Cl (20 mL) y la fase acuosa se extrajo con EtOAc (3 * 10 mL). La capa orgánica combinada se secó con Na2SO4 y se concentró en vacío. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto XXXVIII (75 mg, 11 %) en forma de un sólido rojo.
[0495] MS (ESI): [M H+] = 884.7
Paso 8 : (S)-metil 2-((S)-2-((S)-1-((2S,5S,£)-2-bencil-5-((íerc-butoxicarbonil)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-3-metilbutanamido)-5-((((1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)oxi)carbonil)amino)pentanoato (XXXIX)
[0496] A una solución del compuesto XXXVIII (40 mg, 0.05 mmol) en MeOH seco (2 mL) se añadió Vc (8.0 mg, 0.05 mmol). Después de agitar a 25 °C durante 30 min, el disolvente se eliminó en vacío. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto XXXIX (25 mg, 63 %) en forma de un sólido blanco.
[0497] MS (ESI): [M H+] =885.8.
Ejemplo XXXX y XXXXI de referencia
(S)-1-Oxil-2,2,6,6-tetrametilpiperidin-4-il 2-((S)-1-((2S,5S,E)-2-bencil-5-((terc-butoxicarboml)ammo)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-5-(((benciloxi)carbonil)amino)pentanoato (XXXX) y (S)-1 -hidroxi-2,2,6,6-tetrametilpiperidin-4-il 2-((S)-1-((2S,5S,E)-2-bencil-5-((terc-butoxicarboml)ammo)-7-metiloct-3-enoil)pirrolidma-2-carboxamido)-5-(((benciloxi)carbonil)amino)pentanoato (XXXXI)
[0498]

Paso 1: síntesis de (S)-1-oxil-2,2,6,6-tetrametilpiperidin-4-il 2-((S)-1-((2S,5S,E) -2-bencil-5-((tercbutoxicarbonil)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-5-(((benciloxi)carbonil)amino)pentanoato (XXXX)
[0499] A una solución del compuesto 64 (380 mg, 0.54 mmol) y 4-hidroxi-TEMPO (112 mg, 0.65 mmol) en CH2CI2 (5 mL) a 0 °C se añadieron T3P (446 mg, 1.4 mmol), y DIPEA (153 mg, 1.2 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. La mezcla se diluyó con CH2Ch (5 mL) y se lavó con NaHCO3 acuoso al 5 % (10 mL), solución acuosa saturada de salmuera (3 * 20 mL), y se secó con Na2SO4. El disolvente se eliminó a presión reducida y el residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto XXXX (90 mg, 19 %) en forma de un sólido rojo.
[0500] MS (ESI): [M H+] = 861.7.
Paso 2: síntesis de (S)-1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il 2-((S)-1-((2S,5S,E)-2-bencil-5-((tercbutoxicarbonil)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-5-(((benciloxi)carbonil)amino)pentanoato (XXXXI)
[0501] A una solución del compuesto XXXX (60 mg, 0.07 mmol) en MeOH seco (2 mL) se añadió Vc (13 mg, 0.07 mmol). Después de agitar a 25 °C durante 30 min, el disolvente se eliminó al vacío. El residuo resultante se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto XXXXI (36 mg, 60 %) en forma de un sólido blanco.
[0502] MS (ESI): [M H+] = 862.7.
Ejemplo XXXXII y XXXXIII de referencia
1-Oxil-2,2,6,6-tetrametilpiperidin-4-il N-[(4S)-4-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(terc-butoxi)carboml]amino}-7-metiloct-3-enoil] pirrolidm-2-il]formamido}-4-[(1-oxil-2,2,6,6-tetrametilpiperidm-4-il)carbamoil]butil]carbamato (XXXXII) y 1-Hidroxi-2,2,6,6-tetrametilpiperidin-4-il N-[(4S)-4-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(tercbutoxi)carbonil]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-4-[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil]carbamato (XXXXIII)
[0503]

Paso 1: síntesis de (S)-(9H-fluoren-9-il)metil ferc-butil (5-((1-oxN-2,2,6,6-tetrametilpiperidm-4-N)ammo)-5-oxopentano-1,4-diil)dicarbamato (342)
[0504] A una solución del compuesto 341 (1.0 g, 2.2 mmol) y 4-AT (451 mg, 2.64 mmol) en CH2Cl2 (10 mL) se añadieron T3P (909 mg, 2.86 mmol), y DIPEA (369 mg, 2.86 mmol) a 0 °C. Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. La mezcla se diluyó con CH2Cl2 (10 mL), se lavó con NaHCO3 acuoso al 5 % (10 mL), solución acuosa saturada de salmuera (3 x 20 mL), se secó con Na2sO4. El disolvente se eliminó a presión reducida para dar el producto bruto 2 (0.8 g, 62 %) en forma de un sólido rojo.
[0505] MS (ESI): [M H+] = 608.3.
Paso 2: síntesis de (S)-(9H-fluoren-9-il)metil (4-amino-5-((1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)amino)-5-oxopentil)carbamato (343)
[0506] Se agitó una solución del compuesto 342 (0.8 g, 1.3 mmol) en HCl en MeOH (10 mL, 3 M) a 25 °C durante 3 h. El disolvente se eliminó en vacío para dar el producto 343 (654 mg, 100 %) en forma de un sólido blanco.
[0507] MS (ESI): [M H+] = 509.2.
Paso 3: síntesis de (S)-ferc-butil 2-(((S)-5-((((9H-fluoren-9-il)metoxi) carbonil) amino)-1-((1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)amino)-1 -oxopentan-2-il)carbamoil)pirrolidina-1 -carboxilato (344)
[0508] A una solución del compuesto 343 (654 mg, 1.29 mmol) y (S)-1-(fe/'c-butoxicarbonil)pirrolidina-2-ácido carboxílico (333 mg, 1.55 mmol) en CH2Cl2 (5 mL) se añadieron T3P (1.1 g, 1.68 mmol), y DIPEA (366 mg, 2.84 mmol) a 0 °C. Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. La mezcla se diluyó con CH2Q 2 (5 mL), se lavó con NaHCO3 acuoso al 5 % (5 mL), solución acuosa saturada de salmuera (3 x 20 mL), se secó con Na2SO4. El disolvente se eliminó a presión reducida para dar el producto bruto 344 (630 mg, 69 %) en forma de un sólido amarillo.
[0509] MS (ESI): [M H+] = 706.3
Paso 4: síntesis de (9H-fluoren-9-il)metil ((S)-5-((1-hydroxi-2,2,6,6-tetrametilpiperidin-4-il)amino)-5-oxo-4-((S)-pirrolidina-2-carboxamido)pentil)carbamato (345)
[0510] Se agitó una solución del compuesto 344 (630 mg, 0.89 mmol) en HCl en MeOH (10 mL, 3 M) a 25 °C durante 2 h. El disolvente se eliminó en vacío para dar el producto 345 (538 mg, 100 %) en forma de un sólido amarillo.
[0511] MS (ESI): [M H+] = 606.4.
Paso 5: síntesis de (9H-fluoren-9-il)metil N-[(4S)-4-t[(2S)-1-[(2S,3E,5S)-2-bencN-5-{[(íerc-butoxi)carbonN]ammo}-7-metiloct-3-enoil]pirrolidin-2-il] formamido}-4-[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil]carbamato (346)
[0512] A una solución del compuesto 345 (0.3 g, 0.5 mmol) y (2S,5S,E)-2-bencil-5-((terc-butoxicarbonil)amino)-7-metiloct-3-ácido enoico (59) (217 mg, 0.6 mmol) en CH2Ch (5 mL) a 0 °C se añadieron T3P (420 mg, 0.65 mmol, 50 % en EtOAc) y DIPEA (142 mg, 1.1 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. La mezcla de la reacción se diluyó con CH2Ch (10 mL), se lavó con NaHCO3 acuoso, solución acuosa saturada de salmuera (3 * 20 mL), y se secó con Na2SO4. El disolvente se eliminó a presión reducida para dar el producto bruto 346 (220 mg, 46 %) en forma de un sólido amarillo.
[0513] MS (ESI): [M H+] = 949.8.
Paso 6 : síntesis de íerc-butil ((4S,7S,E)-8-((S)-2-(((S)-5-ammo-1-((1-hidroxi-2,2,6,6-tetrametilpiperidm-4-N)ammo)-1-oxopentan-2-il)carbamoil)pirrolidin-1-il)-7-bencil-2-metil-8-oxooct-5-en-4-il)carbamato (347)
[0514] A una solución del compuesto 346 (220 mg, 0.23 mmol) en CH2Ch (5 mL) se añadió DBU (70 mg, 0.46 mmol) a 25 °C. La reacción se agitó a 25 °C durante 30 min. El disolvente se eliminó en vacío para dar el producto 347 (167 mg, 100 %) en forma de un aceite rojo sin purificación para usarse en el siguiente paso.
[0515] MS (ESI): [M H+] = 727.6.
Paso 7: síntesis de 1-oxil-2,2,6,6-tetrametilpiperidin-4-il N-[(4S)-4-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(íercbutoxi)carbonil]amino}-7-metiloct-3-enoil] pirrolidin-2-il]formamido}-4-[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)carbamoil]butil]carbamato (XXXXII)
[0516] A una solución del compuesto 347 (167 mg, 0.23 mmol) y 1, 3-dioxoisoindolin-2-il (1-oxil-2,2,6,6-tetrametilpiperidin-4-il) carbonato (266) (159 mg, 0.46 mmol) en CH2Ch (5 mL) a 0 °C se añadió DIPEA (60 mg, 0.46 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. Después de añadir CH2Ch (10 mL), la fase orgánica se lavó con NaHCO3 acuoso al 5 % (10 mL), solución acuosa saturada de salmuera (3 * 20 mL), y se secó con Na2SO4. El disolvente se eliminó a presión reducida y el residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto XXXXII (80 mg, rendimiento 38 %) en forma de un sólido rojo.
[0517] MS (ESI): [M H+] = 924.8.
Paso 8 : síntesis de 1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il N-[(4S)-4-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(íercbutoxi)carbonil]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-4-[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)carbamoil] butil] carbamato (XXXXIII)
[0518] A una solución del compuesto XXXXII (70 mg, 0.08 mmol) en MeOH seco (2 mL) se añadió Vc (13 mg, 0.08 mmol). Después de agitar a 25 °C durante 30 min, el disolvente se eliminó al vacío. El residuo resultante se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto XXXXIII (28 mg, 38 %) en forma de un sólido blanco.
[0519] MS (ESI): [M H+] = 925.8.
Ejemplo XXXXIV y XXXXV
íerc-Butil N-[(4S,5E,7S)-7-bencN-8-[(2S)-2-{[(1S)-4-({[(1-oxM-2,2,6,6-tetrametilpiperidm-4-N)oxi]carboml}ammo)-1-[(propan-2-il) carbamoil]butil]carbamoil}pirrolidin-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (XXXXIV) y íerc-butil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-4-({[(1-hidroxi-2,2,6,6-tetrametilpiperidm-4-N)oxi]carbonN}ammo)-1-[(propan-2-il) carbamoil]butil]carbamoil}pirrolidin-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (XXXXV)
[0520]

Paso 1: síntesis de (S)-(9H-fluoren-9-il)metil terc-butil (5-(isopropilamino)-5-oxopentano-1,4-diil)dicarbamato (352)
[0521] A una solución del compuesto 351 (700 mg, 1.54 mmol) y propan-2-amina (136 mg, 2.31 mmol) en CH2Cl2 (10 mL) a 0 °C se añadieron T3P (1.3 g, 2.0 mmol) y DIPEA (300 mg, 3.31 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 16 h. La mezcla de la reacción se apagó con solución acuosa saturada de NH4Cl (20 mL) y se extrajo con EtOAc (3 x 10 mL). La capa orgánica combinada se lavó con NaHCO3 acuoso al 5 % (10 mL), HCl acuoso (10 mL, 1 M), solución acuosa saturada de salmuera (3 x 20 mL), y se secó con Na2SO4. El residuo se concentró en vacío y se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 3 : 1 a 1 : 1) para dar el producto 352 (580 mg, 76 %) en forma de una espuma blanca.
[0522] MS (ESI): [M H+] = 496.2.
[0523] RMN de 1H (400 MHz, CDCl3) 57.77-7.76 (d, J = 4.0 Hz, 2 H), 7.59-7.57 (d, J = 8.0 Hz, 2 H), 7.41-7.39 (t, J = 4.0, 8.0 Hz, 2 H), 7.37-7.32 (d, J = 4.0, 4.0 Hz, 2 H), 6.18 (s, 1 H), 5.20-5.19
[0524] (d, J = 4.0 Hz, 1 H), 5.03 (s, 1 H), 4.43-4.0- (m, 5 H), 3.38 (s, 1 H), 3.18-3.15 (m, 1 H), 1.78 (s, 1 H), 1.58 (s, 3 H), 1.44 (s, 9 H), 1.14-1.09 (s, 6 H).
Paso 2: síntesis de (S )-(9H-fluoren-9-il)metil (4-amino-5-(isopropilamino)-5-oxopentil)carbamato (353)
[0525] Se agitó una solución del compuesto 352 (580 mg, 1.17 mmol) en HCl en MeOH (10 mL, 3 M) a 25 °C durante 2 h y luego el disolvente se eliminó en vacío para dar el producto 353 (462 mg, 100 %) en forma de una espuma blanca.
[0526] MS (ESI): [M H+] = 396.1.
Paso 3: síntesis de (S)-terc-butil 2-(((S)-5-((((9H-fluoren-9-il)metoxi)carbonil) amino)-1-(isopropilamino)-1-oxopentan-2-il)carbamoil)pirrolidina-1-carboxilato (354)
[0527] A una solución del compuesto 353 (462 mg, 1.17 mmol) y (S)-1-(fe/'c-butoxicarbonil)pirrolidina-2-ácido carboxílico (302 mg, 1.4 mmol) en CH2Cl2 (10 mL) a 0 °C se añadieron T3P (484 mg, 1.5 mmol) y DlpEA (302 mg, 2.34 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 16 h. La mezcla de la reacción se apagó con solución acuosa saturada de NH4Cl (20 mL) y se extrajo con EtOAc (3 x 10 mL). La capa orgánica combinada se lavó con NaHCO3 acuoso al 5 % (10 mL), HCl acuoso (10 mL, 1 M), solución acuosa saturada de salmuera (3 * 20 mL), y se secó con Na2SO4. El disolvente se eliminó en vacío y el residuo se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 3 : 1 a 1 : 1) para dar el producto 354 (490 mg, 85 %) en forma de una espuma blanca.
[0528] MS (ESI): [M H+] = 593.4.
[0529] RMN de 1H: (400 MHz, CDCh) 87.76-7.74 (d, J = 8.0 Hz, 2 H), 7.58-7.57 (d, J = 4.0 Hz, 2 H), 7.40-7.37 (t, J = 4.0, 8.0 Hz, 2 H), 7.30-7.28 (t, J = 4.0, 4.0 Hz, 2 H), 6.97 (s, 1 H), 6.71 (s, 1 H), 5.25 (s, 1 H), 4.45-4.18 (m, 5 H), 4.0 (s, 1 H), 3.46-3.14 (m, 4 H), 2.11 (s, 3 H), 1.93-1.73 (s, 3 H), 1.62-1.55 (m, 3 H), 1.43 (s, 9 H), 1.12-1.11 (d, J = 4.0 Hz, 6 H).
Paso 4: síntesis de (9H-fluoren-9-il)metil ((S)-5-(isopropilammo)-5-oxo-4-((S)-pirrolidma-2-carboxamido)pentil)carbamato (355)
[0530] Una solución del compuesto 354 (490 mg, 1.0 mmol) en HCl en MeOH (10 mL, 3 M) se agitó a 25 °C durante 2 h y luego el disolvente se eliminó en vacío para dar el producto 355 (392 mg, 100 %) en forma de una espuma blanca.
[0531] MS (ESI): [M H+] = 493.3.
Paso 5: síntesis de (9H-fluoren-9-il)metil N-[(4S)-4-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(ferc-butoxi)carboml]amino}-7-metiloct-3-enoil]pirrolidin-2-il] formamido}-4-[(propan-2-il)carbamoil]butil]carbamato (356)
[0532] A una solución del compuesto 355 (392 mg, 0.80 mmol) y (2S,5S,E)-2-bencil-5-((terc-butoxicarbonil)amino)-7-metiloct-3-ácido enoico (59, 346 mg, 0.96 mmol) en CH2Ch (5 mL) a 0 °C se añadieron T3P (318 mg, 1.0 mmol) y DIPEA (220 g, 1.7 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 16 h. La mezcla de la reacción se apagó con solución acuosa saturada de NH4Cl (20 mL) y se extrajo con EtOAc (3 * 10 mL). La capa orgánica combinada se lavó con HCl acuoso (10 mL, 1 M), solución acuosa saturada de salmuera (3 * 20 mL) y se secó con Na2SO4. Después, el residuo se concentró en vacío y se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 3 : 1 a 1 : 1) para dar el producto 356 (320 mg, 48 %) en forma de una espuma blanca.
[0533] MS (ESI): [M H+] = 836.5.
[0534] RMN de 1H: (400 MHz, CDCh) 87.76-7.75 (d, J = 4.0 Hz, 2 H), 7.59-7.58 (d, J = 4.0 Hz, 2 H), 7.40-7.38 (t, J = 40, 40 Hz, 2 H), 7.31-7.29 (t, J = 40, 40 Hz, 2 H), 7.24-7.23 (d, J = 4.0 Hz, 2 H), 7.19-7.17 (t, J = 4.0, 4.0 Hz, 1 H), 7.14 7.13 (d, J = 4.0 Hz, 2 H), 6.86-6.84 (d, J = 8.0 Hz, 1 H), 6.49-6.48 (d, J = 4.0 Hz, 1 H), 5.61-5.57 (m, 1 H), 5.39-5.34 (m, 1 H), 5.15-5.13 (m, 1 H), 4.43-4.32 (m, 3 H), 4.21-4.19 (t, J = 4.0, 4.0 Hz, 1 H), 4.07-4.03 (m, 1 H), 3.53 (s, 1 H), 3.38-3.32 (m, 1 H), 3.19-3.12 (m, 2 H), 2.78-2.75 (m, 1 H), 2.07-1.18 (m, 4 H), 1.79-1.63 (m, 6 H), 1.54 -1.41 (m, 12 H), 1.28-1.14 (m, 9 H), 0.87-0.82 (m, 6 H).
Paso 6 : síntesis de ferc-butil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-4-({[(1-oxil-2,2,6,6-tetrametilpiperidm-4-il)oxi]carbonil}amino)-1-[(propan-2-il) carbamoil]butil]carbamoil}pirrolidin-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (XXXXIV)
[0535] A una solución del compuesto 356 (320 mg, 0.38 mmol) en CH2Ch (5 mL) se añadió DBU (289 mg, 1.9 mmol) a 25 °C. La mezcla de la reacción se agitó a 25 °C durante 30 min y se usó en el siguiente paso sin purificación. La mezcla se trató con 1,3-dioxoisoindolin-2-il (1-oxil-2,2,6,6-tetrametilpiperidin-4-il) carbonato (266) (262 mg, 0.76 mmol) y DIPEA (98 mg, 0.76 mmol). La mezcla de la reacción se agitó a 25 °C durante 30 min, luego se apagó con solución acuosa saturada de NH4Cl (20 mL). La fase acuosa se separó y se extrajo con CH2Ch (3 * 20 mL), y las capas orgánicas combinadas se secaron con Na2SO4 y se concentraron en vacío. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto XXXXVI (150 mg, 49 %) en forma de un sólido rojo.
[0536] MS (ESI): [M H+] = 812.6.
Paso 7: síntesis de ferc-butil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-4-({[(1-hidroxi-2,2,6,6-tetrametilpiperidm-4-il)oxi]carbonil}amino)-1-[(propan-2-il) carbamoil]butil]carbamoil}pirrolidin-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato (XXXXV)
[0537] A una solución del compuesto XXXXIV (70 mg, 0.09 mmol) en MeOH seco (2 mL) se añadió Vc (16.0 mg, 0.09 mmol). Después de agitar a 25 °C durante 30 min, el disolvente se eliminó al vacío. El residuo resultante se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 x 250 mm), HCl al 0.1 % en eluyente) para dar el producto XXXXV (50 mg, 68 %) en forma de un sólido blanco.
[0538] MS (ESI): [M H+] = 813.6.
Ejemplo XXXXVI
(S)-Ciclohexil 2-((R)-1-((2S,5S,£)-2-bencN-5-((ferc-butoxicarboml)ammo)-7-metiloct-3-enoN)pirroNdma-2-carboxamido)-6-((((1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)oxi)carbonil)amino)hexanoato (XXXXVI)
[0539]

Paso 1: síntesis de (S)-ciclohexil 6-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((tercbutoxicarbonil)amino)hexanoato (362)
[0540] A una solución del compuesto 361 (700 mg, 1.6 mmol) y ciclohexanol (190 mg, 1.9 mmol) en CH2Ch (5 mL) at 0 °C se añadieron DCC, debería estar definido en la lista de abreviaturas, (392 mg, 1.9 mmol), y Dm Ap (232 mg, 1.9 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. La mezcla se apagó con agua (10 mL), y se extrajo con EtOAc (3 x 10 mL), la fase orgánica se lavó con NaHCO3 acuoso, solución acuosa saturada de salmuera (3 x 20 mL), y se secó con Na2SO4. Los residuos se concentraron a presión reducida para dar el producto bruto 362 (570 mg, 65 %) en forma de un sólido blanco.
[0541] MS (ESI): [M H+] = 551.2.
Paso 2: síntesis de (S)-ciclohexil 6-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-aminohexanoato (363)
[0542] Una solución del compuesto 362 (570 mg, 1.04 mmol) en HCl en MeOH (10 mL, 3 M) se agitó a 25 °C durante 2 h y luego el disolvente se eliminó en vacío para dar el producto 363 (468 mg, 100 %) en forma de una espuma blanca.
[0543] MS (ESI): [M H+] = 451.1.
Paso 3: síntesis de (R)-íerc-butil 2-(((S)-6-((((9H-fluoren-9-il)metoxi) carbonil) amino)-1-(ciclohexNoxi)-1-oxohexan-2-il)carbamoil)pirrolidina-1-carboxilato (364)
[0544] A una solución del compuesto 363 (468 mg, 1.04 mmol) y (R)-1-(terc-butoxicarbonil)pirrolidina-2-ácido carboxílico (268 mg, 1.25 mmol) en CH2Ch (10 mL) a 0 °C se añadieron T3P (860 mg, 1.35 mmol, 50 % en EtOAc), y DIPEA (295 mg, 2.3 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. La mezcla de la reacción se diluyó con CH2Ch (10 mL), se lavó con NaHCO3 acuoso al 5 % (5 mL), solución acuosa saturada de salmuera (3 * 20 mL), se secó con Na2SO4 y se concentró en vacío para dar el producto bruto 364 (530 mg, 82 %) en forma de un sólido blanco.
[0545] MS (ESI): [M H+] = 648.3.
Paso 4: síntesis de (S)-ciclohexil 6-((((9H-fluoren-9-il)metoxi)carbonil)amino)-2-((R)-pirrolidina-2-carboxamido)hexanoato (365)
[0546] Se agitó una solución del compuesto 364 (530 mg, 0.82 mmol) en HCl en MeOH (10 mL, 3 M) a 25 °C durante 3 h. El disolvente se eliminó en vacío para dar el producto 365 (449 mg, 100 %) en forma de un aceite incoloro.
[0547] MS (ESI): [M H+] = 548.3.
Paso 5: síntesis de (S)-ciclohexil 6-((((9H-fluoren-9-N)metoxi)carbonM)ammo)-2-((R)-1-((2S,5S,E)-2-bencN-5-((tercbutoxicarbonil)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)hexanoato (366)
[0548] A una solución del compuesto 365 (449 mg, 0.82 mmol) y (2S,5S,E)-2-bencil-5-((terc-butoxicarbonil)amino)-7-metiloct-3-ácido enoico (59, 355 mg, 0.98 mmol) en CH2Ch (10 mL) a 0 °C se añadieron T3P (623 mg, 0.98 mmol, 50 % en EtOAc), y DIPEA (233 mg, 1.8 mmol). Se dejó que la mezcla de la reacción se calentara hasta alcanzar los 25 °C y se agitó durante 3 h. La mezcla se apagó con agua (10 mL) y se extrajo con EtOAc (3 * 10 mL). La fase orgánica combinada se lavó con NaHCO3 acuoso al 5 % (10 mL), solución acuosa saturada de salmuera (3 * 20 mL) y se secó con Na2SO4. Los residuos, después de concentrarse en vacío, se purificaron mediante cromatografía en columna sobre gel de sílice (éter de petróleo/EtOAc = de 3 : 1 a 1 : 1) para dar el producto 366 (380 mg, 52 %) en forma de un sólido blanco.
[0549] MS (ESI): [M H+] = 891.8.
Paso 6: síntesis de (S)-ciclohexil 6-ammo-2-((R)-1-((2S,5S,E)-2-bencN-5-((íerc-butoxicarbonM)ammo)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)hexanoato (367)
[0550] A una solución del compuesto 366 (380 mg, 0.43 mmol) en CH2Ch (5 mL) se añadió DBU (327 mg, 2.15 mmol) a 25 °C, y se agitó la reacción a 25 °C durante 30 min. El disolvente se eliminó para dar el producto 367 (293 mg, 100 %) en forma de un aceite amarillo, que se usó en el siguiente paso sin purificación.
[0551] MS (ESI): [M H+] = 669.9.
Paso 7: síntesis de (S)-ciclohexil 2-((R)-1-((2S,5S,E)-2-bencN-5-((ferc-butoxicarboml)ammo)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-6-((((1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)oxi)carbonil)amino)hexanoato (XXXXVI)
[0552] Se trató una solución del compuesto bruto 367 (293 mg, 0.43 mmol) en CH2Ch seco (5 mL) a 0 °C con 1,3-dioxoisoindolin-2-il (1-oxil-2,2,6,6-tetrametilpiperidin-4-il) carbonato (266) (178 mg, 0.52 mmol) y DIPEA (67 mg, 0.52 mmol). La mezcla de la reacción se agitó a 25 °C durante 30 min, se diluyó con CH2Ch (10 mL) y luego se apagó con solución acuosa saturada de NH4O (20 mL). La fase acuosa se separó y se extrajo con CH2Ch (10 mL), y las capas orgánicas combinadas se secaron con Na2SO4 y se concentraron en vacío. El residuo se purificó mediante CLAR preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), HCl al 0.1 % en eluyente) para dar el producto XXXXVI (35 mg, 10 %) en forma de un sólido rojo.
[0553] MS (ESI): [M H+] = 867.6.
Ejemplo XXXXVII
ferc-Butil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1 S)-1 -{[(1 S)-4-({[(1 -oxil-2,2,6,6-tetrametilpiperidm-4-il)oxi]carbonil}amino)-1 -[(propan-2 -il)carbamoil]butil]carbamoil}-2-metilpropil]carbamoil}pirrolidm-1-il]-2 -metil-8-oxooct-5-en-4-il]carbamato (XXXXVII)
[0554]

Paso 1: síntesis de (S)-(9H-fluoren-9-il)metil ferc-butil (5-(isopropilamino)-5-oxopentano-1,4-diil)dicarbamato (372)
[0555] A una solución del compuesto 371 (1 g, 2.2 mmol), propan-2-amina (389 mg, 6.6 mmol), DIPEA (851 mg, 6.6 mmol) en DCM (30 mL) se añadió T3P (1.82 g, 2.9 mmol) a 0 °C. La mezcla se agitó a 25 °C durante 10 h, y luego se concentró al vacío. Se añadió EA (100 mL) al residuo y se lavó con H2O (3 x 50 mL). Se recogió la fase orgánica después de que se secara con Na2SO4 y se concentró al vacío para obtener el denominado compuesto 372 (1.09 g, 100 %) en forma de un sólido amarillo.
[0556] MS (ESI): [M H+] = 496.1.
Paso 2: síntesis de (S )-(9H-fluoren-9-il)metil (4-amino-5-(isopropilamino)-5-oxopentil)carbamato (373)
[0557] Al compuesto 372 (1.09 g, 2.2 mmol) se añadieron HCl en MeOH (15 mL, 3 M) y EA (15 mL), luego la mezcla se agitó a 25 °C durante 0.5 h. La solución orgánica se concentró al vacío para obtener el denominado compuesto 373 (870 mg, 100 %) en forma de un sólido amarillo.
[0558] MS (ESI): [M H+] = 396.2.
Paso 3: síntesis de (9H-fluoren-9-il) metil N-[(4S)-4-[(2S)-2-{[(terc-butoxi)carboml]ammo}-3-metilbutanamido]-4-[(propan-2-il)carbamoil]butil]carbamato (374)
[0559] A una solución del 373 (870 mg, 2.2 mmol), (S)-2-((ferc-butoxicarbonil)amino)-3-ácido metilbutanoico (477 mg, 2.2 mmol), DIPEA (851 mg, 6.6 mmol) en DCM (30 mL) se añadió T3P (1.82 g, 2.9 mmol) a 0 °C. La mezcla se agitó a 25 °C durante 2 h y se concentró en vacío. Se añadió eA (100 mL) al residuo y la capa orgánica se lavó con H2O (3 x 50 mL). Se recogió la fase orgánica después de que se secara con Na2SO4 y se concentró en vacío para obtener el denominado compuesto 374 (1.31 g, 100 %) en forma de un sólido amarillo.
[0560] MS (ESI): [M H+] = 595.3.
Paso 4: síntesis de (9H-fluoren-9-il)metil N-[(4S)-4-[(2S)-2-ammo-3-met¡lbutanam¡do]-4-[(propan-2-¡l)carbamo¡l] butil] carbamato (375)
[0561] Se añadieron el 374 (1.31 g, 2.2 mmol) y HCl en MeOH (20 mL, 3M) a un matraz de un solo cuello de 50 mL, luego se agitó la mezcla a 25 °C durante 1 h. La solución orgánica se concentró al vacío para obtener el denominado compuesto 5 (1.09 g, 100 %) en forma de un sólido amarillo.
[0562] MS (ESI): [M H+] = 495.3.
Paso 5: síntesis de (S )-terc-butil 2-(((S)-1-(((S)-5-((((9H-fluoren-9-¡l)metox¡)carboml)ammo)-1-(¡soprop¡lammo)-1-oxopentan-2-il)am¡no)-3-met¡l-1-oxobutan-2-¡l)carbamo¡l)p¡rrol¡d¡na-1-carbox¡lato (376)
[0563] A una solución del 375 (1.09 g, 2.2 mmol), (S)-1-(ferc-butoxicarbonil)pirrolidina-2-ácido carboxílico (710 mg, 3.3 mmol), DIPEA (851 mg, 6.6 mmol) en DCM (30 mL) se añadió T3P (1.82 mg, 2.8 mmol) a 0 °C.
[0564] Se agitó la mezcla a 25 °C durante 2 h, luego se concentró al vacío. Se añadió EA (100 mL) al residuo y la capa orgánica se lavó con H 2O (3 * 50 mL). Se recogió la fase orgánica después de que se secara con Na2SO4 y se concentró al vacío para obtener el denominado compuesto 376 (1.52 g, 100 %) en forma de un sólido amarillo.
[0565] MS (ESI): [M H+] = 692.4.
Paso 6 : síntes¡s de (9H-fluoren-9-¡l)met¡l ((S)-5-(¡soprop¡lam¡no)-4-((S)-3-met¡l-2-((S)-p¡rrol¡d¡na-2-carboxam¡do)butanam¡do)-5-oxopent¡l)carbamato (377)
[0566] A la solución de EA (10 mL) del 376 (800 mg, 1.16 mmol) se añadió HCl en MeOH (10 mL, 3 M) y la mezcla se agitó a 25 °C durante 0.5 h. La solución orgánica se concentró al vacío para obtener el denominado compuesto 377 (684 mg, 100 %) en forma de un sólido amarillo.
[0567] MS (ESI): [M H+] = 592.2.
Paso 7: síntes¡s de (9H-fluoren-9-¡l)met¡l N-[(4S)-4-[(2S)-2-{[(2S)-1-[(2S,3E,5S)-2-benc¡l-5-{[(tercbutox¡)carbon¡l]am¡no}-7-met¡loct-3-eno¡l]p¡rrol¡d¡n-2-¡l]formam¡do}-3-met¡lbutanam¡do]-4-[(propan-2-¡l)carbamo¡l]but¡l]carbamato (378)
[0568] A una solución del 377 (592 mg, 1 mmol), (2S, 5S, E)-2-bencil-5-((terc-butoxicarbonil)amino)-7-metiloct-3-ácido enoico (59, 453 mg, 1.25 mmol), DIPEA (387 mg, 3 mmol) en Dc M (30 mL) se añadió T3P (827 mg, 1.3 mmol) a 0 °C. La mezcla se agitó a 25 °C durante 10 h, y luego se concentró en vacío. Se añadió EA (50 mL) al residuo y la capa orgánica se lavó con H 2O (3 * 50 mL). La fase orgánica se secó con Na2SO4 y se concentró en vacío, y se purificó mediante cromatografía en columna sobre gel de sílice (PE/EA = 1 : 1) para obtener el denominado compuesto 378 (580 mg, 62 %) en forma de un sólido amarillo.
[0569] MS (ESI): [M H+] = 935.6.
[0570] RMN de 1H (400 MHz, CDCh) 87.76-7.75 (m, 2 H), 7.59-7.57 (m, 2 H), 7.41-7.37 (m, 2 H), 7.32-7.28 (m, 2 H), 7.24-7.11 (m, 5 H), 5.65-5.30 (m, 2 H), 4.53-3.99 (m, 8 H), 3.74-3.09 (m, 6 H), 2.77-2.75 (m, 1 H), 2.29-1.84 (m, 8 H), 1.52 0.79 (m, 36 H).
Paso 8 : síntes¡s de terc-but¡l N-[(4S,5E,7S)-7-benc¡l-8-[(2S)-2-{[(1S)-1-{[(1S)-4-({[(1-ox¡l-2,2,6,6-tetramet¡lp¡per¡dm-4-¡l)ox¡]carbon¡l}am¡no)-1-[(propan-2-¡l)carbamo¡l]but¡l] carbamo¡l}-2-met¡lprop¡l]carbamo¡l}p¡rrol¡d¡n-1-¡l]-2-met¡l-8-oxooct-5-en-4-¡l] carbamato (XXXXVII)
[0571] A una solución del 378 (200 mg, 0.21 mmol) en DCM (5 mL) se añadió DBU (163 mg, 1.07 mmol). Después de agitar a 25 °C durante 0.5 h, se añadió 1-oxil-2,2,6,6-tetrametilpiperidin-4-il carbonochloridate (56 mg, 0.25 mmol). La mezcla se agitó durante 4 h a 25 °C. Se añadió DCM (50 mL) al residuo y la capa orgánica se lavó con H 2O (3 * 50 mL). La fase orgánica se secó con Na2SO4 y se concentró y se purificó mediante c La R preparativa (Venusil XBP C18 (5 um, 30 * 250 mm), NH3 H 2O al 0.5% en eluyente) para obtener el denominado producto Xx Xx VII (0.4 mg, 0.2 %) en forma de un sólido rosa.
[0572] MS (ESI): [M H+] = 911.6.
Ejemplo XXXXVNI. Cribado primario mediante ensayo de ferroptosis
[0573] Líneas celulares y medios: Se obtuvieron células HT-1080 (fibrosarcoma) de la American Type Culture Collection. Se cultivaron células en medios EMEM, FBS inactivado por calor al 10 % y mezcla de penicilina-estreptomicina (Invitrogen). Se mantuvieron células a 37 °C y CO2 al 5 % en una incubadora de cultivo de tejidos.
[0574] Ensayo de viabilidad celular. Sembrar 10000 células/pocillo de HT-1080 con 195 uL de medio de crecimiento en placas de 96 pocillos de fondo negro y transparente (Cat# Corning3904) y dejar que las células se adhieran durante la noche. Al día siguiente, diluir el compuesto de la prueba mediante dilución doble en 12 puntos con un medio que contenga 400 ^M de erastina. Transferir 5 uL de la solución de los compuestos al medio de cultivo en pocillos por triplicado. 30 horas más tarde, añadir el reactivo de ensayo de viabilidad celular Cell Titer-Glo® Luminescent (Cat#, Promega G8081) para medir la viabilidad de las células HT-1080 de acuerdo con el protocolo de fabricación. En resumen, aspirar 100 uL del medio y añadir 50 uL del reactivo CellTiter-Glo® Luminescent a cada pocillo e incubar durante 30 minutos a temperatura ambiente para estabilizar la señal luminiscente. Sellar las placas y centrifugar durante 1 minuto a 1000 rpm para eliminar las burbujas. Agitar las placas durante 1 minuto en un agitador orbital. Realizar la lectura de la placa para detectar la señal luminiscente mediante el lector EnSpire Multimode Plate Reader (PerkinElmer). El % de actividad restante se usa para evaluar la ferroptosis inducida por erastina en células HT-1080 y se expresa mediante la siguiente fórmula:
% de actividad restante = 100 * [(muestra - fondo
media
)/
(vehículo - fondo
m e d ia
)] Muestra: Lectura del compuesto de ensayo
Vehículo: Lectura de la muestra del vehículo
Fondo: Lectura del tratamiento exclusivo con erastina
[0575] La curva dosis-respuesta se representa gráficamente por medio del análisis de regresión no lineal (ecuación de fórmula 201) en XLFit ( software complementario de Excel) para el % medio de actividad restante de los triplicados, y luego se calculan los valores de EC50. En este ensayo antiferroptosis se incluyen ferrostatina-1 y XJB-5-131 como referencia. Los criterios de aceptación se establecieron en base al resultado histórico, media ± de 2.58* de DE en la escala logarítmica, mientras que Z’ >0.5.
Tabla 1. Resumen de la inhibición de ferroptosis en célula HT-1080 de los compuestos de ejemplo


[0576] El compuesto de fórmula (I) preferiblemente tiene una IC50 menor de 1 j M, preferiblemente menor de 200 nM.
Ejemplo XXXXIX. Ensayo de apoptosis
[0577] Líneas celulares y medios. Se obtuvieron células 3T3-L1 de la American Type Culture Collection. Se cultivaron células en medios completos DMEM, FBS al 10 % y mezcla de penicilina-estreptomicina (Invitrogen). Se mantuvieron células a 37 °C y CO2 al 5 % en una incubadora de cultivo de tejidos.
[0578] Ensayo de apoptosis. El ensayo Caspase-Glo®3/7 de Promega utiliza un sustrato luminogénico que contiene la secuencia DEVD, que ha demostrado ser selectiva de la caspasa-3 y -7. Usamos este ensayo para cribar nuestros compuestos en función de la inhibición de apoptosis. En resumen, se sembraron 40 uL de 3000 células 3T3-L1 por pocillo en placas de 384 pocillos, se dejó que las células se adhirieran durante la noche. Al día siguiente, preparar la actinomicina D como el agente inductor de apoptosis en 100 uM de DMSO madre. Preparar los compuestos de la prueba en 10 mM de DMSO madre. Diluir en serie el compuesto en 10 puntos mediante dilución triple con DMSO. Tratar previamente las células 3T3-L1 con el compuesto de la prueba mediante la transferencia de 40 nL de concentración madre en serie en pocillos por triplicado mediante la transferencia acústica de fluido, incubar durante 2 horas. Después, transferir 40 nL de 100 j M de actinomicina D (la concentración final es de 100 nM) a los pocillos elegidos por medio de la transferencia acústica de fluido. Incubar las placas de ensayo a 37 °C, CO2 al 5 %, incubadora al 95 % de humedad durante 48 horas. Para el ensayo de caspasa, añadir 20 uL/pocillo de reactivo Caspase-Gio® 3/7 (Promega) a los pocillos de la prueba. Incubar durante 30 min a temperatura ambiente para estabilizar la señal luminiscente. Realizar la lectura de la placa para detectar la señal luminiscente mediante el lector EnSpire Multimode Plate Reader (PerkinElmer). Calcular el valor de EC50 por medio de la ecuación de fórmula 201: y = (A ((B - A) / (1 ((x /C)AD)))) del XLFit ( software complementario de Excel), donde
A: Media promedio de las muestras del vehículo
B: Media promedio de las células tratadas con 100 nM AcD
C: E C 50
D: HillSlope
Se usó el factor Z' para describir el rendimiento de la prueba de cribado. El valor del factor Z' fue >0.5 en todas las placas de la prueba, lo que indicó que el ensayo era apto y que el valor de IC50 era fiable para todos los compuestos analizados.
Tabla 2. Resumen de la inhibición de apoptosis en célula 3T3-L1 mediante actinomicina D de los compuestos de ejemplo.

[0579] El compuesto de fórmula (I) preferiblemente tiene una IC50 de menos de 10 j M, preferiblemente menos de 1 |jM.
Ejemplo XXXXXI. Modelo animal de lesión renal aguda (LRA) por isquemia-reperfusión (IR)
[0580] Se anestesiaron ratones con hidrato de cloral al 5 % (400 mg/kg hidrato de cloral al 5 %). Se indujo la isquemia renal bilateral mediante la aplicación de pinzas microvasculares no traumáticas alrededor de los pedículos renales derecho e izquierdo. Se confirmó la isquemia por el blanqueamiento de los riñones. Tras 30 minutos de isquemia, se retiraron las pinzas y se confirmó visualmente la reperfusión. No se sometió a isquemia a los animales intervenidos en cirugías placebo. Durante el intervalo de isquemia, se mantuvo hidratados a los animales con suero fisiológico infundido por vía intraperitoneal y se les mantuvo sobre almohadillas térmicas para que mantuvieran la temperatura corporal.
[0581] Se asignó a los animales al azar a los grupos siguientes: control simulado, IR con suero o IR con artículo de prueba (5.0 mg/kg, 15 mg/kg). El tratamiento se administró por vía subcutánea 2 h antes de la aparición de la isquemia, cuando apareció la reperfusión y 2 horas después de la reperfusión. Se recogieron muestras de suero y orina a las 24 horas y se almacenaron a -20 °C hasta su análisis. Se analizaron los marcadores oxidativos, y la histopatología por microscopía óptica y electrónica de los riñones en momentos diferentes tras la aparición de reperfusión durante 24 horas.
[0582] El efecto de los compuestos de ejemplo no solo disminuye la creatinina y el BUN a un nivel de funcionalidad normal, sino que también ha demostrado la protección frente al daño histológico del riñón en IR renal.
[0583] Por último, cabe señalar que existen otros modos para poner en práctica la invención. Por lo tanto, los modos de realización de la presente invención se deben describir como ejemplos, pero la presente invención no se limita a los contenidos descritos, se pueden hacer modificaciones adicionales dentro del alcance de la presente invención o los equivalentes añadidos en las cláusulas.
[0584] La referencia a lo largo de esta especificación a «un modo de realización», «algunos modos de realización», «otro ejemplo», «un ejemplo», «un ejemplo específico» o «algunos ejemplos» significa que un rasgo, una estructura, un material o una característica concreta descrita en relación con el modo de realización o ejemplo se incluye en al menos un modo de realización o ejemplo de la presente divulgación. Por consiguiente, la aparición de frases como «en algunos modos de realización», «en un modo de realización», «en otro ejemplo», «en un ejemplo», «en un ejemplo específico», o «en algunos ejemplos» en varios apartados a lo largo de esta especificación no se refiere necesariamente al mismo modo de realización o ejemplo de la presente divulgación. Además, se pueden combinar rasgos, estructuras, materiales o características concretos de cualquier manera adecuada en uno o más modos de realización o ejemplos.
[0585] Aunque se han mostrado y descrito modos de realización explicativos, los expertos en la materia entenderán que los modos de realización anteriores no deben interpretarse como limitativos de la presente divulgación, y se pueden realizar cambios, alternativas y modificaciones en los modos de realización sin alejarse del alcance de la presente divulgación.
Claims (10)
1. Compuesto seleccionado de entre Compuesto (III), Compuesto (XXVIII), Compuesto (XXIV), Compuesto (XXX), Compuesto (XXXI), Compuesto (XXXII), Compuesto (XXXIII), Compuesto (XXXIV), Compuesto (XXXV), Compuesto (XXXVI), Compuesto (XXXVII), Compuesto (XXXVIII), Compuesto (XXXIX), Compuesto (XXXXIV), Compuesto (XXXXV), Compuesto (XXXXVI), y Compuesto (XXXXVII), o una sal farmacéuticamente aceptable de cualquiera de los anteriores, para su uso en un método de inhibición de la ferroptosis en un paciente:


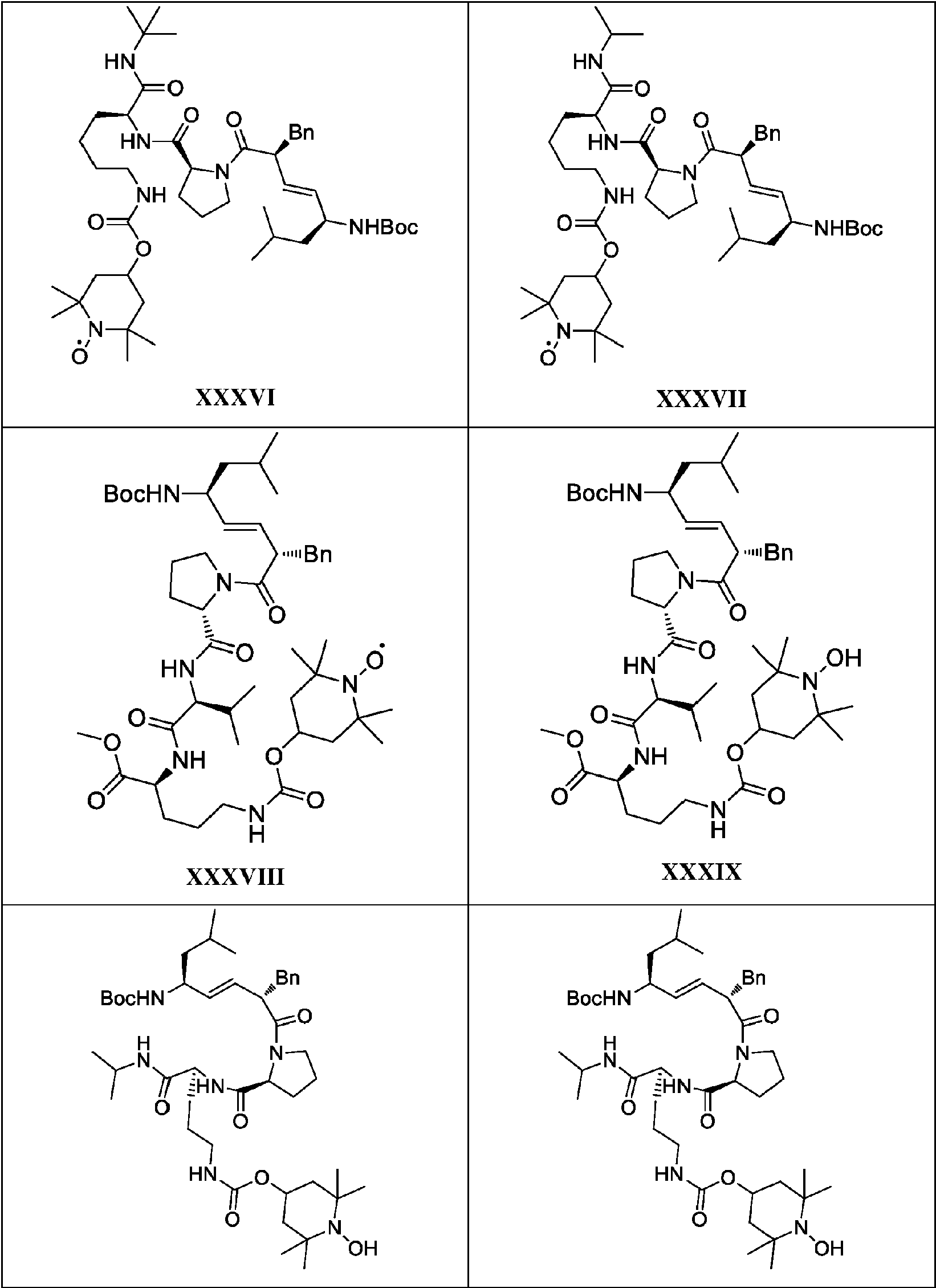

2. Compuesto para su uso de acuerdo con la reivindicación 1, donde la inhibición de la ferroptosis comprende prevenir, tratar o atenuar una enfermedad asociada a la ferroptosis.
3. Compuesto para su uso de acuerdo con cualquier reivindicación anterior, donde el compuesto es (metil (2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(terc-butoxi) carbonil]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-5-({[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonil}amino)pentanoato) (111) o una sal farmacéuticamente aceptable de este.
4. Compuesto para su uso de acuerdo con cualquiera de las reivindicaciones 1 y 2, donde el compuesto es ((2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(terc-butoxi)carbonil]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-5-({[(l-oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonil}amino)pentanoato) (XXIX) o una sal farmacéuticamente aceptable de este.
5. Compuesto para su uso de acuerdo con cualquiera de las reivindicaciones 1 y 2, donde el compuesto es (propan-2-il(2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(terc-butoxi)carbonil]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-5-({[(1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonil}amino)pentanoato") (XXX) o una sal farmacéuticamente aceptable de este.
6. Compuesto para su uso de acuerdo con cualquiera de las reivindicaciones 1 y 2, donde el compuesto es (propan-2-il(2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(terc-butoxi)carbonil]amino}-7-metiloct-3-enoil]pirrolidin-2-il]formamido}-6-({[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonil}amino)hexanoato) (XXXI) o una sal farmacéuticamente aceptable de este.
7. Compuesto para su uso de acuerdo con cualquiera de las reivindicaciones 1 y 2, donde el compuesto es ((S)-metil 2-((S)-2-((S)-1-((2S,5S,E)-2-bencil-5-((terc-butoxicarbonil)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-3-metilbutanamido)-5-((((1-oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi)carbonil)amino)pentanoato (XXXVIII) o una sal farmacéuticamente aceptable de este.
8. Compuesto para su uso de acuerdo con cualquiera de las reivindicaciones 1 y 2, donde el compuesto es ((S)-ciclohexil 2-((E)-1-((2S,5S,E)-2-bencil-5-((terc-butoxicarbonil)amino)-7-metiloct-3-enoil)pirrolidina-2-carboxamido)-6-((((1-hidroxi-2,2,6,6-tetrametilpiperidin-4-il)oxi)carbonil)amino)hexanoato (XXXXVI) o una sal farmacéuticamente aceptable de este.
9. Compuesto para su uso de acuerdo con cualquiera de las reivindicaciones 1 y 2, donde el compuesto se selecciona de entre:
(terc-butil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-1-(dietilcarbamoil)-4-({[(1-oxil-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonil}amino)-butil]carbamoil}pirrolidin-1-il/-2-metil-8-oxooct-5-en-4-il]carbamato) (XXXV);
(terc-butil N-[(4S,5E,7S)-7-benc¡l-8-[(2S)-2-{[(1S)-1-(terc-but¡lcarbamoil)-5-({[(1-ox¡l-2,2,6,6-tetrametilpiperidin-4-il)oxi]carbonil}amino)pentil]-carbamoil}pirrolidin-1-il]-2-metil-8-oxooct-5-en-4-il]carbamato) (XXXVI);
(terc-butil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-4-({[(1-ox¡l-2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)ox¡] carbonil}amino)-1-[(propan-2-il) carbamo¡l]but¡l]carbamo¡l}pirrol¡d¡n-1-¡l]-2-met¡l-8-oxooct-5-en-4-¡l]carbamato) (Xx Xx IV);
(terc-butil N-[(4S,5E,7S)-7-benc¡l-8-[(2S)-2-{[(1S)-4-({[(1-h¡drox¡-2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)ox¡]carbon¡l} am¡no)-1-[(propan-2-¡l) carbamo¡l]but¡l]carbamo¡l}p¡rrol¡d¡n-1-¡l]-2-met¡1-8-oxooct-5-en-4-il]carbamato (XXXXV); y
(terc-butil N-[(4S,5E,7S)-7-benc¡l-8-[(2S)-2-{[(1S)-1-{[(1S)-4-({[(1-ox¡l-2,2,6,6-tetramet¡lp¡perid¡n-4-¡l)ox¡]carbon¡l}am¡no)-1-[(propan-2-¡l)carbamo¡l]but¡l]carbamo¡l}-2-met¡lprop¡l]carbamoil}p¡rrol¡d¡n-1-¡l]-2-metil-8-oxooct-5-en-4-il]carbamato) (XXXXVII);
o una sal farmacéuticamente aceptable de cualquiera de los anteriores.
10. Compuesto para su uso de acuerdo con cualquiera de las reivindicaciones 1 y 2, donde el compuesto se selecciona de entre:
(terc-butil N-[(4S,5E,7S)-7-benc¡l-8-[(2S)-2-{[(1S)-1-carbamo¡l-4-({[(1-ox¡l-2,2,6,6-tetrametilp¡per¡d¡n-4-il)oxi]carbon¡l}am¡no-)but¡l] carbamoil}p¡rrol¡d¡n-1-¡l]-2-metil-8-oxooct-5-en-4-¡l]carbamato) (xXVlII); (propan-2-il (2S)-2-{[(2S)-1-[(2S,3E,5S)-2-bencil-5-{[(terc-butox¡)carbon¡l]am¡no}-7-metiloct-3-eno¡l]p¡rrol¡d¡n-2-il]formam¡do}-6-[(1-ox¡l-2,2,5,5-tetramet¡lp¡rrol¡d¡n-3-¡l)formam¡do]-hexanoato) (XXXII); ((2R)-2-{[(2S)-1-[(2S,3E,5S)-2-benc¡l-5-{[(terc-butox¡)carbon¡l]amino}-7-met¡loct-3-eno¡l]p¡rrol¡d¡n-2-¡l]formam¡do}-6-({[(1-oxil-2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)ox¡]carbon¡l}am¡no)hexanoato) (XXXIII);
((S)-metil 2-((S)-2-((S)-1-((2S,5S,E)-2-bencil-5-((terc-butox¡carbon¡l)am¡no)-7-metiloct-3-eno¡l)p¡rrol¡d¡na-2-carboxam¡do)-3-met¡lbutanam¡do)-5-((((1-h¡drox¡-2,2,6,6-tetrametilp¡per¡d¡n-4-¡l)oxi)carbonil)am¡no)pentanoato) (XXXIX);
((2R)-2-{[(2S)-1-[(2S,3E,5S)-2-benc¡l-5-{[(terc-butox¡)carbon¡l]amino}-7-met¡loct-3-eno¡l]p¡rrol¡d¡n-2-¡l]formam¡do}-6-({[(1-ox¡l-2,2,6,6-tetrametilp¡per¡d¡n-4-¡l)ox¡]carbon¡l}am¡no)ác¡do hexanoico) (XXXIV); y (terc-butil N-[(4S,5E,7S)-7-bencil-8-[(2S)-2-{[(1S)-5-({[(1-ox¡l-2,2,6,6-tetramet¡lpiper¡d¡n-4-¡l)ox¡]carbon¡l}am¡no)-1-[(propan-2-¡l)carbamo¡l]pent¡l]carbamo¡l}p¡rrol¡d¡n-l-¡l]-2-met¡l-8-oxooct-5-en-4-¡l]carbamato (XXXVII); o
una sal farmacéuticamente aceptable de cualquiera de los anteriores.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/CN2017/100431 WO2019041361A1 (en) | 2017-09-04 | 2017-09-04 | PREPARATION AND USE OF A REACTIVE OXYGEN SPECIES TRAPPER |
| EP21212401.0A EP3988561B1 (en) | 2017-09-04 | 2017-09-04 | Preparation and use of reactive oxygen species scavenger |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2947741T3 true ES2947741T3 (es) | 2023-08-17 |
Family
ID=65524901
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17923551T Active ES2909718T3 (es) | 2017-09-04 | 2017-09-04 | Preparación y uso de un barredor de especies reactivas de oxígeno |
| ES21212401T Active ES2947741T3 (es) | 2017-09-04 | 2017-09-04 | Preparación y uso de un barredor de especies reactivas de oxígeno |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17923551T Active ES2909718T3 (es) | 2017-09-04 | 2017-09-04 | Preparación y uso de un barredor de especies reactivas de oxígeno |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US10889572B2 (es) |
| EP (2) | EP3988561B1 (es) |
| JP (1) | JP6976618B2 (es) |
| CN (2) | CN115475229A (es) |
| ES (2) | ES2909718T3 (es) |
| WO (1) | WO2019041361A1 (es) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106866784B (zh) * | 2015-12-11 | 2021-05-18 | 凯瑞康宁生物工程(武汉)有限公司 | 靶向线粒体抗氧化剂及其制备方法和用途 |
| US11602512B1 (en) | 2016-07-22 | 2023-03-14 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12478604B1 (en) | 2016-07-22 | 2025-11-25 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11504347B1 (en) | 2016-07-22 | 2022-11-22 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11602513B1 (en) | 2016-07-22 | 2023-03-14 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12186296B1 (en) | 2016-07-22 | 2025-01-07 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| UY37341A (es) | 2016-07-22 | 2017-11-30 | Flamel Ireland Ltd | Formulaciones de gamma-hidroxibutirato de liberación modificada con farmacocinética mejorada |
| US11986451B1 (en) | 2016-07-22 | 2024-05-21 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| CN113473980A (zh) | 2019-03-01 | 2021-10-01 | 弗拉梅尔爱尔兰有限公司 | 在进食状态下具有改善的药代动力学的γ-羟基丁酸酯组合物 |
| US11779557B1 (en) | 2022-02-07 | 2023-10-10 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11583510B1 (en) | 2022-02-07 | 2023-02-21 | Flamel Ireland Limited | Methods of administering gamma hydroxybutyrate formulations after a high-fat meal |
Family Cites Families (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4328245A (en) | 1981-02-13 | 1982-05-04 | Syntex (U.S.A.) Inc. | Carbonate diester solutions of PGE-type compounds |
| US4410545A (en) | 1981-02-13 | 1983-10-18 | Syntex (U.S.A.) Inc. | Carbonate diester solutions of PGE-type compounds |
| US4409239A (en) | 1982-01-21 | 1983-10-11 | Syntex (U.S.A.) Inc. | Propylene glycol diester solutions of PGE-type compounds |
| WO1997007750A1 (en) | 1995-08-30 | 1997-03-06 | Weg Stuart L | Administration of ketamine to manage pain and to reduce drug dependency |
| AU709088B2 (en) | 1995-10-24 | 1999-08-19 | Merck & Co., Inc. | Thrombin inhibitors |
| US5798377A (en) | 1996-10-21 | 1998-08-25 | Merck & Co., Inc. | Thrombin inhibitors |
| US6350458B1 (en) | 1998-02-10 | 2002-02-26 | Generex Pharmaceuticals Incorporated | Mixed micellar drug deliver system and method of preparation |
| AU2003291037A1 (en) | 2002-11-18 | 2004-06-15 | Yaupon Therapeutics, Inc. | Analgesic uses of norketamine and ketamine/norketamine prodrugs |
| SI2656854T1 (sl) | 2003-02-04 | 2015-09-30 | Cornell Research Foundation, Inc. | Uporabe aromatsko-kationskega peptida |
| US20070161544A1 (en) | 2006-01-06 | 2007-07-12 | Peter Wipf | Selective targeting agents for mitcochondria |
| US7528174B2 (en) * | 2006-01-06 | 2009-05-05 | University Of Pittsburgh-Of The Commonwealth System Of Higher Education | Selective targeting agents for mitochondria |
| US8487079B2 (en) | 2007-08-08 | 2013-07-16 | University of Pittsburgh—of the Commonwealth System of Higher Education | Use of mitochondria-targeted electron scavengers as anti-inflammatory agents |
| TWI535440B (zh) | 2009-10-26 | 2016-06-01 | Lg生命科學有限公司 | 包括吲哚化合物之醫藥組成物 |
| MX2012011460A (es) * | 2010-04-08 | 2012-11-23 | Squibb Bristol Myers Co | Analogos de pirimidinilpiperidiniloxipiridinona como moduladores de gpr119. |
| WO2012068081A1 (en) * | 2010-11-15 | 2012-05-24 | University Of Pittsburgh - Of The Commonwealth System Of Higher Education | Intraesophageal administration of targeted nitroxide agents for protection against ionizing irradiation-induced esophagatis |
| US9217000B2 (en) | 2011-02-18 | 2015-12-22 | Univerisyt of Pittsburgh—of the Commonwealth System of Higher Education | Targeted nitroxide agents |
| EP3904332B1 (en) | 2011-10-14 | 2024-12-04 | The United States of America, as represented by The Secretary, Department of Health and Human Services | The use of (2r, 6r)-hydroxynorketamine, (s)-dehydronorketamine and other stereoisomeric dehydro and hydroxylated metabolites of (r,s)- ketamine in the treatment of depression and neuropathic pain |
| US9580398B2 (en) | 2012-04-02 | 2017-02-28 | The Trustees Of Columbia University In The City Of New York | Compounds, compositions, and methods for modulating ferroptosis and treating excitotoxic disorders |
| WO2014011973A2 (en) * | 2012-07-13 | 2014-01-16 | The Trustees Of Columbia University In The City Of New York | Quinazolinone-based oncogenic-ras-selective lethal compounds and their use |
| EP3791870A1 (en) * | 2013-12-02 | 2021-03-17 | The Trustees of Columbia University in the City of New York | Modulating ferroptosis and treating excitotoxic disorders |
| CN106866784B (zh) * | 2015-12-11 | 2021-05-18 | 凯瑞康宁生物工程(武汉)有限公司 | 靶向线粒体抗氧化剂及其制备方法和用途 |
-
2017
- 2017-09-04 WO PCT/CN2017/100431 patent/WO2019041361A1/en not_active Ceased
- 2017-09-04 JP JP2020512797A patent/JP6976618B2/ja active Active
- 2017-09-04 CN CN202211188161.9A patent/CN115475229A/zh active Pending
- 2017-09-04 ES ES17923551T patent/ES2909718T3/es active Active
- 2017-09-04 ES ES21212401T patent/ES2947741T3/es active Active
- 2017-09-04 CN CN201780096462.XA patent/CN111356699B/zh active Active
- 2017-09-04 EP EP21212401.0A patent/EP3988561B1/en active Active
- 2017-09-04 EP EP17923551.0A patent/EP3679051B1/en active Active
-
2020
- 2020-02-26 US US16/801,286 patent/US10889572B2/en active Active
- 2020-11-12 US US17/096,713 patent/US11312703B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| EP3679051A1 (en) | 2020-07-15 |
| CN111356699A (zh) | 2020-06-30 |
| US20200199108A1 (en) | 2020-06-25 |
| EP3988561B1 (en) | 2023-06-07 |
| US20210061791A1 (en) | 2021-03-04 |
| EP3679051B1 (en) | 2021-12-29 |
| EP3988561A1 (en) | 2022-04-27 |
| EP3679051A4 (en) | 2020-12-30 |
| US10889572B2 (en) | 2021-01-12 |
| JP2020532554A (ja) | 2020-11-12 |
| CN111356699B (zh) | 2022-08-23 |
| US11312703B2 (en) | 2022-04-26 |
| CN115475229A (zh) | 2022-12-16 |
| ES2909718T3 (es) | 2022-05-10 |
| WO2019041361A1 (en) | 2019-03-07 |
| JP6976618B2 (ja) | 2021-12-08 |
| EP3988561C0 (en) | 2023-06-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2947741T3 (es) | Preparación y uso de un barredor de especies reactivas de oxígeno | |
| US11692008B2 (en) | Preparation and uses of reactive oxygen species scavenger derivatives | |
| ES3000422T3 (en) | Nitrile-containing compounds for use as medicaments | |
| KR102483876B1 (ko) | 아미노 피리미딘 ssao 억제제 | |
| WO2021027911A1 (zh) | 新型螺环类K-Ras G12C抑制剂 | |
| JP2020525513A (ja) | 癌および他の疾患を治療するためのatf4阻害剤としてのn−(3−(2−(4−クロロフェノキシ)アセトアミドビシクロ[1.1.1]ペンタン−1−イル)−2−シクロブタン−1−カルボキサミド誘導体および関連化合物 | |
| CN101754762A (zh) | 用于治疗神经炎症性疾病的包含哒嗪化合物的制剂 | |
| CA2986930A1 (en) | Chemical modulators of signaling pathways and therapeutic use | |
| US10501466B2 (en) | WDR5 inhibitors and modulators | |
| CA3155650A1 (en) | Diterpenoid compounds that act on protein kinase c (pkc) | |
| WO2022063297A1 (zh) | 喹唑啉衍生物及其制备方法和用途 | |
| CA3205456A1 (en) | Smac mimetics for treatment of cancer, process for preparation and pharmaceutical composition thereof | |
| JP7152078B2 (ja) | ホウ酸塩ベースの薬物およびその使用 | |
| HK40068053A (en) | Preparation and use of reactive oxygen species scavenger | |
| HK40068053B (en) | Preparation and use of reactive oxygen species scavenger | |
| HK40021802A (en) | Preparation and use of reactive oxygen species scavenger | |
| HK40021802B (en) | Preparation and use of reactive oxygen species scavenger | |
| WO2012169785A9 (en) | Symmetrically structured quinazoline derivatives | |
| BR112020019952A2 (pt) | composto, composição farmacêutica, método para o tratamento ou profilaxia de uma doença ou distúrbio, uso de um composto, produto de combinação, kit-de-partes, e, processo para a preparação de um composto | |
| HK40077904A (en) | Preparation and uses of reactive oxygen species scavenger derivatives | |
| HK40029856B (en) | Preparation and uses of reactive oxygen species scavenger derivatives | |
| HK40029856A (en) | Preparation and uses of reactive oxygen species scavenger derivatives |