ES2914591T3 - Procedimientos novedosos para la producción de oligonucleótidos - Google Patents
Procedimientos novedosos para la producción de oligonucleótidos Download PDFInfo
- Publication number
- ES2914591T3 ES2914591T3 ES18822043T ES18822043T ES2914591T3 ES 2914591 T3 ES2914591 T3 ES 2914591T3 ES 18822043 T ES18822043 T ES 18822043T ES 18822043 T ES18822043 T ES 18822043T ES 2914591 T3 ES2914591 T3 ES 2914591T3
- Authority
- ES
- Spain
- Prior art keywords
- segment
- oligonucleotide
- product
- ligase
- template
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 108091034117 Oligonucleotide Proteins 0.000 title claims abstract description 364
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 title claims abstract description 138
- 238000000034 method Methods 0.000 title claims abstract description 134
- 238000004519 manufacturing process Methods 0.000 title claims description 20
- 238000006243 chemical reaction Methods 0.000 claims abstract description 199
- 102000003960 Ligases Human genes 0.000 claims abstract description 190
- 108090000364 Ligases Proteins 0.000 claims abstract description 190
- 125000003729 nucleotide group Chemical group 0.000 claims abstract description 143
- 239000002773 nucleotide Substances 0.000 claims abstract description 120
- 230000004048 modification Effects 0.000 claims abstract description 80
- 238000012986 modification Methods 0.000 claims abstract description 80
- 239000012535 impurity Substances 0.000 claims abstract description 52
- 230000015572 biosynthetic process Effects 0.000 claims abstract description 33
- 238000003786 synthesis reaction Methods 0.000 claims abstract description 31
- 235000000346 sugar Nutrition 0.000 claims abstract description 30
- 230000000295 complement effect Effects 0.000 claims abstract description 22
- 230000001225 therapeutic effect Effects 0.000 claims abstract description 16
- 238000005304 joining Methods 0.000 claims abstract description 14
- 230000002255 enzymatic effect Effects 0.000 claims abstract description 12
- 238000000137 annealing Methods 0.000 claims abstract description 10
- 238000004064 recycling Methods 0.000 claims abstract description 5
- 108090000623 proteins and genes Proteins 0.000 claims description 80
- 239000011324 bead Substances 0.000 claims description 63
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 claims description 53
- 102000004169 proteins and genes Human genes 0.000 claims description 50
- 239000000463 material Substances 0.000 claims description 47
- 230000008569 process Effects 0.000 claims description 33
- 102000004357 Transferases Human genes 0.000 claims description 25
- 108090000992 Transferases Proteins 0.000 claims description 25
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 24
- 229910019142 PO4 Inorganic materials 0.000 claims description 21
- 239000010452 phosphate Substances 0.000 claims description 21
- 239000012528 membrane Substances 0.000 claims description 19
- 238000004925 denaturation Methods 0.000 claims description 17
- 230000036425 denaturation Effects 0.000 claims description 17
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 claims description 15
- 239000001913 cellulose Substances 0.000 claims description 12
- 229920002678 cellulose Polymers 0.000 claims description 12
- 108010042407 Endonucleases Proteins 0.000 claims description 11
- 108010090804 Streptavidin Proteins 0.000 claims description 11
- 101710086015 RNA ligase Proteins 0.000 claims description 10
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 claims description 8
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 claims description 8
- PTMHPRAIXMAOOB-UHFFFAOYSA-L phosphoramidate Chemical compound NP([O-])([O-])=O PTMHPRAIXMAOOB-UHFFFAOYSA-L 0.000 claims description 7
- 239000002202 Polyethylene glycol Substances 0.000 claims description 6
- 229920001223 polyethylene glycol Polymers 0.000 claims description 6
- 150000003839 salts Chemical class 0.000 claims description 6
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 claims description 5
- 239000001226 triphosphate Substances 0.000 claims description 5
- YIMATHOGWXZHFX-WCTZXXKLSA-N (2r,3r,4r,5r)-5-(hydroxymethyl)-3-(2-methoxyethoxy)oxolane-2,4-diol Chemical compound COCCO[C@H]1[C@H](O)O[C@H](CO)[C@H]1O YIMATHOGWXZHFX-WCTZXXKLSA-N 0.000 claims description 4
- 102000004533 Endonucleases Human genes 0.000 claims description 4
- 230000008859 change Effects 0.000 claims description 4
- ANCLJVISBRWUTR-UHFFFAOYSA-N diaminophosphinic acid Chemical compound NP(N)(O)=O ANCLJVISBRWUTR-UHFFFAOYSA-N 0.000 claims description 4
- TWGNOYAGHYUFFR-UHFFFAOYSA-N 5-methylpyrimidine Chemical compound CC1=CN=CN=C1 TWGNOYAGHYUFFR-UHFFFAOYSA-N 0.000 claims description 3
- 208000035657 Abasia Diseases 0.000 claims description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 3
- 239000007853 buffer solution Substances 0.000 claims description 3
- 150000001720 carbohydrates Chemical class 0.000 claims description 3
- 235000014633 carbohydrates Nutrition 0.000 claims description 3
- 239000000412 dendrimer Substances 0.000 claims description 3
- 229920000736 dendritic polymer Polymers 0.000 claims description 3
- 239000011521 glass Substances 0.000 claims description 3
- 150000004676 glycans Chemical class 0.000 claims description 3
- 229920001542 oligosaccharide Polymers 0.000 claims description 3
- 150000002482 oligosaccharides Chemical class 0.000 claims description 3
- 229920000620 organic polymer Polymers 0.000 claims description 3
- 229920001282 polysaccharide Polymers 0.000 claims description 3
- 239000005017 polysaccharide Substances 0.000 claims description 3
- 229910052717 sulfur Inorganic materials 0.000 claims description 3
- 239000011593 sulfur Substances 0.000 claims description 3
- 108091027076 Spiegelmer Proteins 0.000 claims description 2
- 125000006239 protecting group Chemical group 0.000 claims description 2
- JRYMOPZHXMVHTA-DAGMQNCNSA-N 2-amino-7-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1h-pyrrolo[2,3-d]pyrimidin-4-one Chemical compound C1=CC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O JRYMOPZHXMVHTA-DAGMQNCNSA-N 0.000 claims 1
- 239000000047 product Substances 0.000 description 183
- 238000004128 high performance liquid chromatography Methods 0.000 description 76
- 102000012410 DNA Ligases Human genes 0.000 description 49
- 238000000926 separation method Methods 0.000 description 49
- 108010061982 DNA Ligases Proteins 0.000 description 48
- 239000000243 solution Substances 0.000 description 40
- 108020004414 DNA Proteins 0.000 description 32
- 150000007523 nucleic acids Chemical class 0.000 description 31
- 239000000872 buffer Substances 0.000 description 28
- 102000004190 Enzymes Human genes 0.000 description 27
- 108090000790 Enzymes Proteins 0.000 description 27
- 238000010438 heat treatment Methods 0.000 description 27
- 108020004707 nucleic acids Proteins 0.000 description 26
- 102000039446 nucleic acids Human genes 0.000 description 26
- 239000006228 supernatant Substances 0.000 description 22
- 241000588921 Enterobacteriaceae Species 0.000 description 21
- 238000002474 experimental method Methods 0.000 description 21
- 239000000725 suspension Substances 0.000 description 21
- 238000010367 cloning Methods 0.000 description 20
- 238000000605 extraction Methods 0.000 description 20
- 239000002953 phosphate buffered saline Substances 0.000 description 19
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 18
- 238000011026 diafiltration Methods 0.000 description 18
- 210000004027 cell Anatomy 0.000 description 17
- 108091030071 RNAI Proteins 0.000 description 15
- 230000009368 gene silencing by RNA Effects 0.000 description 15
- 238000010561 standard procedure Methods 0.000 description 15
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 15
- 241001198387 Escherichia coli BL21(DE3) Species 0.000 description 14
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 14
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 13
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 13
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 13
- 238000001914 filtration Methods 0.000 description 13
- -1 nucleotide triphosphates Chemical class 0.000 description 13
- 239000012466 permeate Substances 0.000 description 13
- 239000011541 reaction mixture Substances 0.000 description 13
- 230000027455 binding Effects 0.000 description 12
- 235000010980 cellulose Nutrition 0.000 description 11
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 11
- 239000007787 solid Substances 0.000 description 11
- BAWFJGJZGIEFAR-NNYOXOHSSA-O NAD(+) Chemical compound NC(=O)C1=CC=C[N+]([C@H]2[C@@H]([C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 BAWFJGJZGIEFAR-NNYOXOHSSA-O 0.000 description 10
- 108091028043 Nucleic acid sequence Proteins 0.000 description 10
- 230000001419 dependent effect Effects 0.000 description 10
- 239000012465 retentate Substances 0.000 description 10
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 9
- 108010008286 DNA nucleotidylexotransferase Proteins 0.000 description 9
- 102100033215 DNA nucleotidylexotransferase Human genes 0.000 description 9
- 238000006209 dephosphorylation reaction Methods 0.000 description 9
- 238000010532 solid phase synthesis reaction Methods 0.000 description 9
- 125000003275 alpha amino acid group Chemical group 0.000 description 8
- 238000005251 capillar electrophoresis Methods 0.000 description 8
- 238000010511 deprotection reaction Methods 0.000 description 8
- 238000002844 melting Methods 0.000 description 8
- 230000008018 melting Effects 0.000 description 8
- 238000000746 purification Methods 0.000 description 8
- 102100031780 Endonuclease Human genes 0.000 description 7
- 239000007983 Tris buffer Substances 0.000 description 7
- 238000005119 centrifugation Methods 0.000 description 7
- 238000003776 cleavage reaction Methods 0.000 description 7
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 7
- 239000000203 mixture Substances 0.000 description 7
- 150000004713 phosphodiesters Chemical class 0.000 description 7
- 230000007017 scission Effects 0.000 description 7
- 239000011780 sodium chloride Substances 0.000 description 7
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 7
- ASJSAQIRZKANQN-CRCLSJGQSA-N 2-deoxy-D-ribose Chemical compound OC[C@@H](O)[C@@H](O)CC=O ASJSAQIRZKANQN-CRCLSJGQSA-N 0.000 description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 6
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 6
- 238000009295 crossflow filtration Methods 0.000 description 6
- 238000007169 ligase reaction Methods 0.000 description 6
- 238000005457 optimization Methods 0.000 description 6
- 230000000717 retained effect Effects 0.000 description 6
- 229930010555 Inosine Natural products 0.000 description 5
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 description 5
- 101710163270 Nuclease Proteins 0.000 description 5
- 229920001213 Polysorbate 20 Polymers 0.000 description 5
- 241001541469 Shigella phage Shf125875 Species 0.000 description 5
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical group OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 5
- 241000700605 Viruses Species 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 238000004587 chromatography analysis Methods 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- 108020001507 fusion proteins Proteins 0.000 description 5
- 102000037865 fusion proteins Human genes 0.000 description 5
- 229960003786 inosine Drugs 0.000 description 5
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 5
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 5
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 5
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- 108091093037 Peptide nucleic acid Proteins 0.000 description 4
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 4
- 239000012634 fragment Substances 0.000 description 4
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 4
- 239000001257 hydrogen Substances 0.000 description 4
- 229910052739 hydrogen Inorganic materials 0.000 description 4
- 238000011534 incubation Methods 0.000 description 4
- 239000013067 intermediate product Substances 0.000 description 4
- 239000013642 negative control Substances 0.000 description 4
- 238000011160 research Methods 0.000 description 4
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 4
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 4
- LRSASMSXMSNRBT-UHFFFAOYSA-N 5-methylcytosine Chemical compound CC1=CNC(=O)N=C1N LRSASMSXMSNRBT-UHFFFAOYSA-N 0.000 description 3
- 241000305071 Enterobacterales Species 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 3
- 241000191967 Staphylococcus aureus Species 0.000 description 3
- 108010076818 TEV protease Proteins 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 238000004185 countercurrent chromatography Methods 0.000 description 3
- 230000030609 dephosphorylation Effects 0.000 description 3
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 3
- 238000010494 dissociation reaction Methods 0.000 description 3
- 230000005593 dissociations Effects 0.000 description 3
- 230000004927 fusion Effects 0.000 description 3
- 239000011544 gradient gel Substances 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- 238000004255 ion exchange chromatography Methods 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 238000004460 liquid liquid chromatography Methods 0.000 description 3
- 239000012160 loading buffer Substances 0.000 description 3
- 229910001629 magnesium chloride Inorganic materials 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 239000007790 solid phase Substances 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 230000002194 synthesizing effect Effects 0.000 description 3
- 235000011178 triphosphate Nutrition 0.000 description 3
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 2
- RYVNIFSIEDRLSJ-UHFFFAOYSA-N 5-(hydroxymethyl)cytosine Chemical compound NC=1NC(=O)N=CC=1CO RYVNIFSIEDRLSJ-UHFFFAOYSA-N 0.000 description 2
- PEHVGBZKEYRQSX-UHFFFAOYSA-N 7-deaza-adenine Chemical compound NC1=NC=NC2=C1C=CN2 PEHVGBZKEYRQSX-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- 108091023037 Aptamer Proteins 0.000 description 2
- 241000195649 Chlorella <Chlorellales> Species 0.000 description 2
- 108010008758 Chlorella virus DNA ligase Proteins 0.000 description 2
- 108010082610 Deoxyribonuclease (Pyrimidine Dimer) Proteins 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 102100037696 Endonuclease V Human genes 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- 229910021380 Manganese Chloride Inorganic materials 0.000 description 2
- GLFNIEUTAYBVOC-UHFFFAOYSA-L Manganese chloride Chemical compound Cl[Mn]Cl GLFNIEUTAYBVOC-UHFFFAOYSA-L 0.000 description 2
- 108700011259 MicroRNAs Proteins 0.000 description 2
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 2
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 2
- 241000607768 Shigella Species 0.000 description 2
- 108020004459 Small interfering RNA Proteins 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical group O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 2
- DRTQHJPVMGBUCF-XVFCMESISA-N Uridine Chemical group O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-XVFCMESISA-N 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 125000000539 amino acid group Chemical group 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 230000001580 bacterial effect Effects 0.000 description 2
- 229920001222 biopolymer Polymers 0.000 description 2
- 239000004202 carbamide Substances 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 238000005112 continuous flow technique Methods 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 2
- NAGJZTKCGNOGPW-UHFFFAOYSA-K dioxido-sulfanylidene-sulfido-$l^{5}-phosphane Chemical compound [O-]P([O-])([S-])=S NAGJZTKCGNOGPW-UHFFFAOYSA-K 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 238000005194 fractionation Methods 0.000 description 2
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- 150000002466 imines Chemical class 0.000 description 2
- 230000003308 immunostimulating effect Effects 0.000 description 2
- 238000011031 large-scale manufacturing process Methods 0.000 description 2
- 239000011565 manganese chloride Substances 0.000 description 2
- 235000002867 manganese chloride Nutrition 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- 125000001360 methionine group Chemical group N[C@@H](CCSC)C(=O)* 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 230000005257 nucleotidylation Effects 0.000 description 2
- 238000002515 oligonucleotide synthesis Methods 0.000 description 2
- PXQPEWDEAKTCGB-UHFFFAOYSA-N orotic acid Chemical compound OC(=O)C1=CC(=O)NC(=O)N1 PXQPEWDEAKTCGB-UHFFFAOYSA-N 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- XUYJLQHKOGNDPB-UHFFFAOYSA-N phosphonoacetic acid Chemical compound OC(=O)CP(O)(O)=O XUYJLQHKOGNDPB-UHFFFAOYSA-N 0.000 description 2
- 150000008300 phosphoramidites Chemical class 0.000 description 2
- 229910000160 potassium phosphate Inorganic materials 0.000 description 2
- 235000011009 potassium phosphates Nutrition 0.000 description 2
- 150000003230 pyrimidines Chemical class 0.000 description 2
- 125000000548 ribosyl group Chemical group C1([C@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 2
- 229920002477 rna polymer Polymers 0.000 description 2
- 238000005070 sampling Methods 0.000 description 2
- 239000011550 stock solution Substances 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 230000003612 virological effect Effects 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- AKVIWWJLBFWFLM-UHFFFAOYSA-N (2-amino-2-oxoethyl)phosphonic acid Chemical compound NC(=O)CP(O)(O)=O AKVIWWJLBFWFLM-UHFFFAOYSA-N 0.000 description 1
- GNYIGHPYQNROKG-MCDZGGTQSA-N (2r,3r,4s,5r)-2-(6-aminopurin-9-yl)-5-(hydroxymethyl)oxolane-3,4-diol;trihydroxy(sulfanylidene)-$l^{5}-phosphane Chemical compound OP(O)(O)=S.C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O GNYIGHPYQNROKG-MCDZGGTQSA-N 0.000 description 1
- NSMOSDAEGJTOIQ-CRCLSJGQSA-N (2r,3s)-2-(hydroxymethyl)oxolan-3-ol Chemical compound OC[C@H]1OCC[C@@H]1O NSMOSDAEGJTOIQ-CRCLSJGQSA-N 0.000 description 1
- MUVQIIBPDFTEKM-IUYQGCFVSA-N (2r,3s)-2-aminobutane-1,3-diol Chemical compound C[C@H](O)[C@H](N)CO MUVQIIBPDFTEKM-IUYQGCFVSA-N 0.000 description 1
- KZVAAIRBJJYZOW-LMVFSUKVSA-N (2r,3s,4s)-2-(hydroxymethyl)oxolane-3,4-diol Chemical compound OC[C@H]1OC[C@H](O)[C@@H]1O KZVAAIRBJJYZOW-LMVFSUKVSA-N 0.000 description 1
- HLHSUNWAPXINQU-GQCTYLIASA-N (E)-3-(3,4-dihydroxyphenyl)-N-prop-2-ynylprop-2-enamide Chemical compound OC=1C=C(C=CC=1O)/C=C/C(=O)NCC#C HLHSUNWAPXINQU-GQCTYLIASA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- MZLSNIREOQCDED-UHFFFAOYSA-N 1,3-difluoro-2-methylbenzene Chemical compound CC1=C(F)C=CC=C1F MZLSNIREOQCDED-UHFFFAOYSA-N 0.000 description 1
- ACIUIZVHXMCTDK-HNPMAXIBSA-N 1-[(2R,4S,5R)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-methylpyrimidine-2,4-dione trihydroxy(sulfanylidene)-lambda5-phosphane Chemical compound OP(O)(O)=S.O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 ACIUIZVHXMCTDK-HNPMAXIBSA-N 0.000 description 1
- VGONTNSXDCQUGY-RRKCRQDMSA-N 2'-deoxyinosine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC2=O)=C2N=C1 VGONTNSXDCQUGY-RRKCRQDMSA-N 0.000 description 1
- MYNLFDZUGRGJES-UHFFFAOYSA-N 2-(cyclopentylamino)-3,7-dihydropurin-6-one Chemical compound N=1C=2N=CNC=2C(=O)NC=1NC1CCCC1 MYNLFDZUGRGJES-UHFFFAOYSA-N 0.000 description 1
- FMYBFLOWKQRBST-UHFFFAOYSA-N 2-[bis(carboxymethyl)amino]acetic acid;nickel Chemical compound [Ni].OC(=O)CN(CC(O)=O)CC(O)=O FMYBFLOWKQRBST-UHFFFAOYSA-N 0.000 description 1
- VWSLLSXLURJCDF-UHFFFAOYSA-N 2-methyl-4,5-dihydro-1h-imidazole Chemical compound CC1=NCCN1 VWSLLSXLURJCDF-UHFFFAOYSA-N 0.000 description 1
- HLPXUVWTMGENBN-UHFFFAOYSA-N 3-methylidenemorpholine Chemical group C=C1COCCN1 HLPXUVWTMGENBN-UHFFFAOYSA-N 0.000 description 1
- WDEISXYUGDKDJQ-OYUVGMAPSA-N 4-amino-1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-methylpyrimidin-2-one trihydroxy(sulfanylidene)-lambda5-phosphane Chemical compound OP(O)(O)=S.Cc1cn([C@@H]2O[C@H](CO)[C@@H](O)[C@H]2O)c(=O)nc1N WDEISXYUGDKDJQ-OYUVGMAPSA-N 0.000 description 1
- YJHUFZQMNTWHBO-UHFFFAOYSA-N 5-(aminomethyl)-1h-pyrimidine-2,4-dione Chemical compound NCC1=CNC(=O)NC1=O YJHUFZQMNTWHBO-UHFFFAOYSA-N 0.000 description 1
- JDBGXEHEIRGOBU-UHFFFAOYSA-N 5-hydroxymethyluracil Chemical compound OCC1=CNC(=O)NC1=O JDBGXEHEIRGOBU-UHFFFAOYSA-N 0.000 description 1
- ZLAQATDNGLKIEV-UHFFFAOYSA-N 5-methyl-2-sulfanylidene-1h-pyrimidin-4-one Chemical compound CC1=CNC(=S)NC1=O ZLAQATDNGLKIEV-UHFFFAOYSA-N 0.000 description 1
- UJBCLAXPPIDQEE-UHFFFAOYSA-N 5-prop-1-ynyl-1h-pyrimidine-2,4-dione Chemical compound CC#CC1=CNC(=O)NC1=O UJBCLAXPPIDQEE-UHFFFAOYSA-N 0.000 description 1
- TVICROIWXBFQEL-UHFFFAOYSA-N 6-(ethylamino)-1h-pyrimidin-2-one Chemical compound CCNC1=CC=NC(=O)N1 TVICROIWXBFQEL-UHFFFAOYSA-N 0.000 description 1
- ZOHFTRWZZPGYIS-UHFFFAOYSA-N 6-amino-5-(aminomethyl)-1h-pyrimidin-2-one Chemical compound NCC1=CNC(=O)N=C1N ZOHFTRWZZPGYIS-UHFFFAOYSA-N 0.000 description 1
- QNNARSZPGNJZIX-UHFFFAOYSA-N 6-amino-5-prop-1-ynyl-1h-pyrimidin-2-one Chemical compound CC#CC1=CNC(=O)N=C1N QNNARSZPGNJZIX-UHFFFAOYSA-N 0.000 description 1
- LHCPRYRLDOSKHK-UHFFFAOYSA-N 7-deaza-8-aza-adenine Chemical compound NC1=NC=NC2=C1C=NN2 LHCPRYRLDOSKHK-UHFFFAOYSA-N 0.000 description 1
- LOSIULRWFAEMFL-UHFFFAOYSA-N 7-deazaguanine Chemical compound O=C1NC(N)=NC2=C1CC=N2 LOSIULRWFAEMFL-UHFFFAOYSA-N 0.000 description 1
- MSSXOMSJDRHRMC-UHFFFAOYSA-N 9H-purine-2,6-diamine Chemical compound NC1=NC(N)=C2NC=NC2=N1 MSSXOMSJDRHRMC-UHFFFAOYSA-N 0.000 description 1
- 231100000582 ATP assay Toxicity 0.000 description 1
- 241000691306 Actia Species 0.000 description 1
- 229930024421 Adenine Natural products 0.000 description 1
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- 108020004634 Archaeal DNA Proteins 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- WSDIDTITDBPVKN-UHFFFAOYSA-N CB1[S+]=P1([O-])O Chemical compound CB1[S+]=P1([O-])O WSDIDTITDBPVKN-UHFFFAOYSA-N 0.000 description 1
- 108091033409 CRISPR Proteins 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- 241000701248 Chlorella virus Species 0.000 description 1
- 108020004635 Complementary DNA Proteins 0.000 description 1
- 125000000824 D-ribofuranosyl group Chemical group [H]OC([H])([H])[C@@]1([H])OC([H])(*)[C@]([H])(O[H])[C@]1([H])O[H] 0.000 description 1
- YTBSYETUWUMLBZ-QWWZWVQMSA-N D-threose Chemical compound OC[C@@H](O)[C@H](O)C=O YTBSYETUWUMLBZ-QWWZWVQMSA-N 0.000 description 1
- GUBGYTABKSRVRQ-WFVLMXAXSA-N DEAE-cellulose Chemical compound OC1C(O)C(O)C(CO)O[C@H]1O[C@@H]1C(CO)OC(O)C(O)C1O GUBGYTABKSRVRQ-WFVLMXAXSA-N 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- 101710148289 DNA ligase 2 Proteins 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 241000701533 Escherichia virus T4 Species 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- 108010058683 Immobilized Proteins Proteins 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 101710203526 Integrase Proteins 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- WXJXBKBJAKPJRN-UHFFFAOYSA-N Methanephosphonothioic acid Chemical compound CP(O)(O)=S WXJXBKBJAKPJRN-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- OVRNDRQMDRJTHS-CBQIKETKSA-N N-Acetyl-D-Galactosamine Chemical compound CC(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@H](O)[C@@H]1O OVRNDRQMDRJTHS-CBQIKETKSA-N 0.000 description 1
- MBLBDJOUHNCFQT-UHFFFAOYSA-N N-acetyl-D-galactosamine Natural products CC(=O)NC(C=O)C(O)C(O)C(O)CO MBLBDJOUHNCFQT-UHFFFAOYSA-N 0.000 description 1
- 208000035823 Non-specific autoimmune cerebellar ataxia without characteristic antibodies Diseases 0.000 description 1
- 108090000119 Nucleotidyltransferases Proteins 0.000 description 1
- 102000003832 Nucleotidyltransferases Human genes 0.000 description 1
- PKUWKAXTAVNIJR-UHFFFAOYSA-N O,O-diethyl hydrogen thiophosphate Chemical compound CCOP(O)(=S)OCC PKUWKAXTAVNIJR-UHFFFAOYSA-N 0.000 description 1
- 108700019535 Phosphoprotein Phosphatases Proteins 0.000 description 1
- 102000045595 Phosphoprotein Phosphatases Human genes 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 108091028664 Ribonucleotide Proteins 0.000 description 1
- 241001287230 Stenotrophomonas phage IME13 Species 0.000 description 1
- 238000010459 TALEN Methods 0.000 description 1
- 108010043645 Transcription Activator-Like Effector Nucleases Proteins 0.000 description 1
- 108020005202 Viral DNA Proteins 0.000 description 1
- 108010017070 Zinc Finger Nucleases Proteins 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- HSULLSUSGAHAOJ-UHFFFAOYSA-N acetic acid;hexan-1-amine Chemical compound CC([O-])=O.CCCCCC[NH3+] HSULLSUSGAHAOJ-UHFFFAOYSA-N 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 238000007259 addition reaction Methods 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- NHQSDCRALZPVAJ-HJQYOEGKSA-N agmatidine Chemical compound NC(=N)NCCCCNC1=NC(=N)C=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NHQSDCRALZPVAJ-HJQYOEGKSA-N 0.000 description 1
- FPIPGXGPPPQFEQ-OVSJKPMPSA-N all-trans-retinol Chemical class OC\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-OVSJKPMPSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- DRTQHJPVMGBUCF-PSQAKQOGSA-N beta-L-uridine Natural products O[C@H]1[C@@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-PSQAKQOGSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 238000010804 cDNA synthesis Methods 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 239000013043 chemical agent Substances 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 230000001143 conditioned effect Effects 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- JADWVLYMWVNVAN-UHFFFAOYSA-N ctk0h5271 Chemical compound NP(N)(O)=S JADWVLYMWVNVAN-UHFFFAOYSA-N 0.000 description 1
- 229940104302 cytosine Drugs 0.000 description 1
- 230000008260 defense mechanism Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 239000005547 deoxyribonucleotide Substances 0.000 description 1
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- BOKOVLFWCAFYHP-UHFFFAOYSA-N dihydroxy-methoxy-sulfanylidene-$l^{5}-phosphane Chemical compound COP(O)(O)=S BOKOVLFWCAFYHP-UHFFFAOYSA-N 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical class [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 238000011067 equilibration Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- ZJXZSIYSNXKHEA-UHFFFAOYSA-N ethyl dihydrogen phosphate Chemical compound CCOP(O)(O)=O ZJXZSIYSNXKHEA-UHFFFAOYSA-N 0.000 description 1
- 125000001475 halogen functional group Chemical group 0.000 description 1
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- 238000002013 hydrophilic interaction chromatography Methods 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000009776 industrial production Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 150000002605 large molecules Chemical class 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 238000003760 magnetic stirring Methods 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 230000013011 mating Effects 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- JWILWRLEBBNTFH-UOWFLXDJSA-N methyl (2r,3r,4r)-2,3,4,5-tetrahydroxypentanoate Chemical compound COC(=O)[C@H](O)[C@H](O)[C@H](O)CO JWILWRLEBBNTFH-UOWFLXDJSA-N 0.000 description 1
- CAAULPUQFIIOTL-UHFFFAOYSA-N methyl dihydrogen phosphate Chemical compound COP(O)(O)=O CAAULPUQFIIOTL-UHFFFAOYSA-N 0.000 description 1
- YACKEPLHDIMKIO-UHFFFAOYSA-N methylphosphonic acid Chemical compound CP(O)(O)=O YACKEPLHDIMKIO-UHFFFAOYSA-N 0.000 description 1
- 108091070501 miRNA Proteins 0.000 description 1
- 239000002679 microRNA Substances 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 238000001728 nano-filtration Methods 0.000 description 1
- 229920002842 oligophosphate Polymers 0.000 description 1
- 229960005010 orotic acid Drugs 0.000 description 1
- 125000003431 oxalo group Chemical group 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 238000004810 partition chromatography Methods 0.000 description 1
- 230000002572 peristaltic effect Effects 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 229920000771 poly (alkylcyanoacrylate) Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000008057 potassium phosphate buffer Substances 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000003531 protein hydrolysate Substances 0.000 description 1
- HBCQSNAFLVXVAY-UHFFFAOYSA-N pyrimidine-2-thiol Chemical compound SC1=NC=CC=N1 HBCQSNAFLVXVAY-UHFFFAOYSA-N 0.000 description 1
- LCDCPQHFCOBUEF-UHFFFAOYSA-N pyrrolidine-1-carboxamide Chemical compound NC(=O)N1CCCC1 LCDCPQHFCOBUEF-UHFFFAOYSA-N 0.000 description 1
- 150000003235 pyrrolidines Chemical class 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 239000002336 ribonucleotide Substances 0.000 description 1
- 125000002652 ribonucleotide group Chemical group 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- IIACRCGMVDHOTQ-UHFFFAOYSA-M sulfamate Chemical compound NS([O-])(=O)=O IIACRCGMVDHOTQ-UHFFFAOYSA-M 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- PCYCVCFVEKMHGA-UHFFFAOYSA-N thiirane 1-oxide Chemical compound O=S1CC1 PCYCVCFVEKMHGA-UHFFFAOYSA-N 0.000 description 1
- ZEMGGZBWXRYJHK-UHFFFAOYSA-N thiouracil Chemical compound O=C1C=CNC(=S)N1 ZEMGGZBWXRYJHK-UHFFFAOYSA-N 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- 229940113082 thymine Drugs 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- 125000002264 triphosphate group Chemical class [H]OP(=O)(O[H])OP(=O)(O[H])OP(=O)(O[H])O* 0.000 description 1
- 238000000108 ultra-filtration Methods 0.000 description 1
- 229940035893 uracil Drugs 0.000 description 1
- DRTQHJPVMGBUCF-UHFFFAOYSA-N uracil arabinoside Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UHFFFAOYSA-N 0.000 description 1
- 229940045145 uridine Drugs 0.000 description 1
- 238000003260 vortexing Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/102—Mutagenizing nucleic acids
- C12N15/1031—Mutagenizing nucleic acids mutagenesis by gene assembly, e.g. assembly by oligonucleotide extension PCR
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/111—General methods applicable to biologically active non-coding nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/93—Ligases (6)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P19/00—Preparation of compounds containing saccharide radicals
- C12P19/26—Preparation of nitrogen-containing carbohydrates
- C12P19/28—N-glycosides
- C12P19/30—Nucleotides
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/11—Antisense
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/31—Chemical structure of the backbone
- C12N2310/315—Phosphorothioates
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/32—Chemical structure of the sugar
- C12N2310/322—2'-R Modification
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/32—Chemical structure of the sugar
- C12N2310/323—Chemical structure of the sugar modified ring structure
- C12N2310/3231—Chemical structure of the sugar modified ring structure having an additional ring, e.g. LNA, ENA
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/34—Spatial arrangement of the modifications
- C12N2310/341—Gapmers, i.e. of the type ===---===
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/34—Spatial arrangement of the modifications
- C12N2310/345—Spatial arrangement of the modifications having at least two different backbone modifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/34—Spatial arrangement of the modifications
- C12N2310/346—Spatial arrangement of the modifications having a combination of backbone and sugar modifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2320/00—Applications; Uses
- C12N2320/30—Special therapeutic applications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2330/00—Production
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Biomedical Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- General Health & Medical Sciences (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Plant Pathology (AREA)
- Crystallography & Structural Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Procedimiento para producir un producto de oligonucleótido terapéutico monocatenario que tiene al menos un residuo de nucleótido modificado, en el que la modificación se selecciona del grupo que consiste en modificación en la posición 2' del resto de azúcar, modificación de la nucleobase y modificación de la estructura principal, y en el que el producto tiene 20-30 nucleótidos de longitud, que comprende: a) proporcionar un conjunto de oligonucleótidos (I) que comprende segmentos de la secuencia del producto, en el que al menos un segmento de la secuencia del producto contiene al menos un residuo de nucleótido modificado, en el que la modificación se selecciona del grupo que consiste en modificación en la posición 2' del resto de azúcar, modificación de la nucleobase y modificación de la estructura principal, y en el que cada segmento se ha producido mediante síntesis enzimática; b) proporcionar un oligonucleótido molde (II) complementario a la secuencia del producto de oligonucleótido monocatenario, teniendo dicho molde una propiedad que permite que se separe del producto y se recicle para su uso en futuras reacciones; c) poner en contacto (I) y (II) en condiciones para permitir el apareamiento de los oligonucleótidos de segmento con el oligonucleótido molde; d) unir los oligonucleótidos de segmento usando una ligasa para formar el producto; e) cambiar las condiciones para desnaturalizar el molde apareado y cualquier hebra de oligonucleótido de impureza, y separar las impurezas; f) cambiar las condiciones para desnaturalizar el molde apareado y las hebras de oligonucleótidos de producto, y separar el producto de oligonucleótido monocatenario; y g) reciclar el oligonucleótido molde para su uso en futuras reacciones.
Description
DESCRIPCIÓN
Procedimientos novedosos para la producción de oligonucleótidos
Campo de la invención
La invención se refiere a procedimientos novedosos que usan enzimas para la producción de oligonucleótidos, en la que dichos procedimientos son adecuados para su uso en la producción de oligonucleótidos modificados químicamente, tal como para su uso en terapia.
Antecedentes de la invención
La síntesis química de oligonucleótidos y oligonucleótidos modificados por medio de química de fosforamidita está bien establecida y ha sido el método de elección para sintetizar estos biopolímeros de secuencia definida durante varias décadas. El procedimiento de síntesis se realiza habitualmente como una síntesis en fase sólida, mediante la cual se añaden secuencialmente nucleótidos individuales requiriendo la adición de cada nucleótido un ciclo de varias etapas químicas para añadir y desproteger el oligonucleótido en crecimiento (“oligo”) en preparación para la etapa posterior. Al final de la adición secuencial de nucleótidos, el oligo se libera del soporte de fase sólida, tiene lugar una desprotección adicional y luego el oligonucleótido en bruto se purifica además mediante cromatografía en columna.
Aunque este método puede considerarse de rutina y puede automatizarse, hay varias deficiencias en esta metodología, especialmente si el objetivo es preparar oligonucleótidos a gran escala tal como sería necesario para productos terapéuticos de oligonucleótidos. Estas deficiencias incluyen, pero no se limitan a:
1) Limitaciones prácticas inherentes al uso de cromatografía que la hacen inadecuada para purificar grandes cantidades de oligonucleótido. El uso de cromatografía a gran escala es caro y difícil de lograr debido a las limitaciones en el tamaño y el rendimiento de la columna.
2) El número de errores se acumula con la longitud del oligonucleótido que está sintetizándose. Por consiguiente, la naturaleza secuencial lineal del procedimiento actual da como resultado una disminución geométrica en el rendimiento. Por ejemplo, si el rendimiento para cada ciclo de adición de nucleótido es del 99 %, en el que el rendimiento de un 20 meros sería del 83 %.
3) Limitaciones de escala con sintetizadores de oligonucleótidos sintéticos y el equipo de purificación y aislamiento posterior: en la actualidad, la cantidad máxima de producto que puede producirse en un solo lote es del orden de 10 kg.
Se necesita, por tanto, tanto reducir (o idealmente eliminar) la cromatografía en columna como realizar la síntesis de un modo que no sea meramente secuencial con el fin de aumentar el rendimiento.
A menudo se usa ADN polimerasa para sintetizar oligonucleótidos para su uso en biología molecular y aplicaciones similares. Sin embargo, la ADN polimerasa no es adecuada para sintetizar oligonucleótidos terapéuticos debido a tanto las longitudes relativamente cortas de los oligonucleótidos como la necesidad de discriminar entre nucleótidos con diferentes modificaciones de desoxirribosa o ribosa. Por ejemplo, los oligonucleótidos terapéuticos están a menudo en el intervalo de 20 a 25 nucleótidos. La ADN polimerasa necesita al menos 7 u 8 nucleótidos, y de manera óptima de 18 a 22 nucleótidos, como cebador en cada dirección, de modo que hay poco que ganar en el intento de sintetizar un oligo terapéutico si los cebadores tienen un tamaño similar al producto deseado. Además, la ADN polimerasa requiere que todos los nucleótidos estén presentes en la reacción y se basa en el apareamiento de bases de Watson-Crick para alinear los nucleótidos entrantes. Por tanto, la ADN polimerasa es incapaz de discriminar entre cualquier orden de modificaciones de desoxirribosa o ribosa, tales como las requeridas por un gapmero, y el resultado sería una mezcla de modificaciones de desoxirribosa o ribosa en una posición dada.
Sumario de la invención
La invención proporciona un procedimiento para producir un producto de oligonucleótido monocatenario que tiene al menos un residuo de nucleótido modificado, que comprende:
a) proporcionar un conjunto de oligonucleótidos (I) que comprende segmentos de la secuencia del producto, en el que al menos un segmento de la secuencia del producto contiene al menos un residuo de nucleótido modificado, y en el que cada segmento se ha producido mediante síntesis enzimática;
b) proporcionar un oligonucleótido molde (II) complementario a la secuencia del producto de oligonucleótido monocatenario, teniendo dicho molde una propiedad que permite que se separe del producto y se recicle para su
uso en futuras reacciones;
c) poner en contacto (I) y (II) en condiciones para permitir el apareamiento de los oligonucleótidos de segmento con el oligonucleótido molde;
d) unir los oligonucleótidos de segmento usando una ligasa para formar el producto;
e) cambiar las condiciones para desnaturalizar el molde apareado y cualquier hebra de oligonucleótido de impureza, y separar las impurezas;
f) cambiar las condiciones para desnaturalizar el molde apareado y las hebras de oligonucleótidos de producto, y separar el producto de oligonucleótido monocatenario; y
g) reciclar el oligonucleótido molde para su uso en futuras reacciones.
Descripción de las figuras
Figura 1. Esquema de la producción enzimática de oligonucleótidos de segmento: a) extensión 3’ para la síntesis de segmentos que comprende una reacción de dos etapas: adición y desprotección. La etapa de adición a modo de ejemplo implica ligamiento dependiente de ATP de nucleótido-3’,5’-bis(tio)fosfato en el 3’-OH de un cebador de ácido nucleico monocatenario y luego desprotección del 3’-fosfato en el oligonucleótido monocatenario mediante una fosfatasa; y b) la extensión 3’ a modo de ejemplo (adición y desprotección) para producir una secuencia de segmento seguido por escisión de la cadena usando una nucleasa específica de sitio (por ejemplo, endonucleasa V: escinde una base después de la inosina, es decir, en el segundo enlace fosfodiéster 3’ con respecto a la inosina) para liberar el segmento.
Figura 2. Esquema del procedimiento de la invención, incluyendo las etapas de ligamiento de los oligonucleótidos de segmento para formar el producto y cambiar las condiciones para eliminar impurezas.
Figura 3. Esquema de múltiples configuraciones de molde.
Figura 4. Esquema básico del procedimiento de la invención que se lleva a cabo en un sistema de flujo.
Figura 5. Esquema detallado del procedimiento de la invención que se lleva a cabo en un sistema de flujo: a) sección de química de ligamiento, b) sección de purificación de ligamiento y c) sección de química de ligamiento alternativa y d) sección de purificación alternativa. N.B. Las secciones a) y b) (alternativamente c) y d)) pueden realizarse en una sola etapa, por ejemplo, recipiente de recogida en a) = salida de la etapa de ligamiento en b). Figura 6. Ejemplos de impurezas que pueden generarse durante el procedimiento de la invención.
Figura 7. Cromatograma que muestra los resultados de una reacción de ligamiento usando ligasa de T4 NEB comercial (SEQ ID NO: 3) y ADN no modificado (ejemplo 1).
Figura 8. Cromatograma que muestra los resultados de una reacción de ligamiento usando ligasa de T4 unida a PERLOZA (SEQ ID NO: 4 ) y ADN no modificado (ejemplo 1).
Figura 9. Cromatograma que muestra los resultados de una reacción de ligamiento usando ligasa de T4 unida a PERLOZA expresada según el ejemplo 2 y fragmentos de oligonucleótidos modificados con 2’-OMe.
Figura 10. Trazos de HPLC para el ejemplo 4: Trazo superior (a) - sin control de ligasa. Trazo inferior (b) - clon A4. El producto y el molde se eluyen conjuntamente en este método de HPLC. Pueden observarse dos fragmentos de ligamiento intermedios (segmentos) en el trazo del clon A4 a los 10,3 y 11,2 minutos.
Figura 11. Esquema del “núcleo de tres moldes” usado en el ejemplo 13, que comprende un material de soporte denominado “núcleo” y tres secuencias de molde.
Figura 12. Esquema de la plataforma de ligamiento semicontinuo usada en el ejemplo 13.
Figura 13. Esquema de la plataforma de filtración sin salida usada en el ejemplo 14.
Figura 14. Esquema de la plataforma de filtración de flujo cruzado usada en el ejemplo 14.
Figura 15. Cromatogramas que muestran los resultados de los experimentos de filtración sin salida usando la membrana NADIR de MWCO de 10 kDa a 60 °C en el ejemplo 14. La figura a) es un cromatograma de la disolución
de material retenido, que permaneció en la celda de filtración y contenía principalmente el núcleo de tres moldes, después de dos volúmenes de diafiltración; y el cromatograma b) es del permeado, disolución enriquecida en el producto, después de dos volúmenes de diafiltración.
Figura 16. Cromatogramas que muestran los resultados de los experimentos de filtración de flujo cruzado usando la membrana Snyder de MWCO de 5 kDa a 50 °C y 3,0 bar de presión, en el ejemplo 14. La figura a) es un cromatograma de la disolución de material retenido, que contenía principalmente el núcleo de tres moldes y producto, después de 20 volúmenes de diafiltración; y la figura b) es un cromatograma del permeado, que contenía principalmente oligonucleótidos de segmento, después de 20 volúmenes de diafiltración.
Figura 17. Cromatogramas que muestran los resultados de los experimentos de filtración de flujo cruzado usando la membrana Snyder de MWCO de 5 kDa a 80 °C y 3,1 bar de presión, en el ejemplo 14. La figura 17 muestra a) un cromatograma de la disolución de material retenido, que contenía el núcleo de tres moldes solo, después de 20 volúmenes de diafiltración; y la figura b) es un cromatograma de la disolución de permeado, que contenía el producto solo, después de 2 volúmenes de diafiltración.
Descripción detallada de la invención
Definiciones
Tal como se usa en el presente documento, el término “oligonucleótido”, u “oligo” para abreviar, significa un polímero de residuos de nucleótidos. Estos pueden ser desoxirribonucleótidos (en los que el oligonucleótido resultante es ADN), ribonucleótidos (en los que el oligonucleótido resultante es ARN), nucleótidos modificados o una mezcla de los mismos. Un oligonucleótido puede estar completamente compuesto por residuos de nucleótidos que se encuentran en la naturaleza o puede contener al menos un nucleótido, o al menos una unión entre nucleótidos, que se ha modificado. Los oligonucleótidos pueden ser monocatenarios o bicatenarios. Un oligonucleótido de la invención puede conjugarse con otra molécula, por ejemplo, N-acetilgalactosamina (GaINAc) o múltiplos de la misma (agrupaciones de GaINAc).
Tal como se usa en el presente documento, el término “oligonucleótido terapéutico” significa un oligonucleótido que tiene una aplicación terapéutica, por ejemplo en la prevención o el tratamiento de una afección o enfermedad en un ser humano o animal. Un oligonucleótido de este tipo contiene normalmente uno o más residuos o uniones de nucleótidos modificados. Los oligonucleótidos terapéuticos actúan por medio de varios mecanismos diferentes, que incluyen, entre otros, antisentido, cambio de corte y empalme o salto de exón, inmunoestimulación e interferencia de ARN (iARN), por ejemplo por medio de microARN (miARN) y ARN de interferencia pequeño (ARNip). Un oligonucleótido terapéutico puede ser un aptámero. Los oligonucleótidos terapéuticos normalmente, pero no siempre, tendrán una secuencia definida.
Tal como se usa en el presente documento, el término “molde” significa un oligonucleótido con una secuencia que es complementaria al 100 % a la secuencia del oligonucleótido diana (o producto).
A menos que se especifique lo contrario, tal como se usa en el presente documento, el término “complementario” significa complementario al 100 %.
Tal como se usa en el presente documento, el término “producto” significa el oligonucleótido deseado, que tiene una secuencia específica, también denominado en el presente documento “oligonucleótido diana”.
Tal como se usa en el presente documento, el término “conjunto” se refiere a un grupo de oligonucleótidos cuya secuencia puede variar, pueden ser más cortos o más largos que la secuencia diana y pueden no tener la misma secuencia que la secuencia diana. El conjunto de oligonucleótidos puede ser el producto de la síntesis de oligonucleótidos, por ejemplo, síntesis enzimática, usada con o sin purificación. El conjunto de oligonucleótidos puede estar compuesto por segmentos de la secuencia diana. Cada segmento, en sí mismo, puede estar presente como un conjunto de ese segmento y puede ser el producto de la síntesis de oligonucleótidos, por ejemplo, síntesis enzimática.
Tal como se usa en el presente documento, el término “apareamiento” significa la hibridación de oligonucleótidos complementarios de una manera específica de secuencia, por ejemplo, el apareamiento de dos oligonucleótidos monocatenarios, por medio de enlaces de hidrógeno del apareamiento de bases de Watson y Crick, para formar un oligonucleótido bicatenario. Las “condiciones para permitir el apareamiento” dependerán de la Tm de los oligonucleótidos complementarios hibridados y será fácilmente evidente para un experto en la técnica. Por ejemplo, la temperatura de apareamiento puede estar por debajo de la Tm de los oligonucleótidos hibridados. Alternativamente, la temperatura de apareamiento puede estar próxima a la Tm de los oligonucleótidos hibridados, por ejemplo, /- 1,2 o 3 °C. La temperatura de apareamiento no es, en general, superior a 10 °C por encima de la Tm de los oligonucleótidos hibridados. Las condiciones específicas para permitir el apareamiento se describen
resumidamente en la sección de ejemplos.
Tal como se usa en el presente documento, el término “desnaturalización” en relación con un oligonucleótido bicatenario se usa para indicar que las cadenas complementarias ya no están apareadas, es decir, el apareamiento de bases de Watson y Crick se ha alterado y las cadenas se han disociado. La desnaturalización se produce como resultado del cambio de las condiciones, por ejemplo, elevando la temperatura, cambiando el pH o cambiando la concentración de sal de la disolución tamponante. Los expertos en la técnica conocen bien las condiciones para la desnaturalización. La desnaturalización de un oligonucleótido bicatenario (un “dúplex”) tal como se describe en el presente documento da como resultado un producto monocatenario o un oligonucleótido de impureza y un oligonucleótido molde monocatenario.
Tal como se usa en el presente documento, el término “impureza” o “impurezas” significa oligonucleótidos que no tienen la secuencia de producto deseada. Estos oligonucleótidos pueden incluir oligonucleótidos que son más cortos que el producto (por ejemplo, 1,2, 3, 4, 5 o más residuos de nucleótido más cortos), o que son más largos que el producto (por ejemplo, 1, 2, 3, 4, 5 o más residuos de nucleótidos más largos). Cuando el proceso de producción incluye una etapa mediante la cual se forman uniones entre segmentos, las impurezas incluyen oligonucleótidos que quedan si no se forman uno o más de los enlaces. Las impurezas también incluyen oligonucleótidos en los que se han incorporado nucleótidos incorrectos, lo que da como resultado un apareamiento erróneo en comparación con el molde. Una impureza puede tener una o más de las características descritas anteriormente. La figura 6 muestra algunas de las impurezas que pueden producirse.
Tal como se usa en el presente documento, el término “segmento” es una porción más pequeña de un oligonucleótido más largo, en particular una porción más pequeña de un producto u oligonucleótido diana. Para un producto dado, cuando todos sus segmentos se aparean con su molde y se unen entre sí, se forma el producto.
Tal como se usa en el presente documento, el término “ligamiento enzimático” significa que la unión entre dos nucleótidos adyacentes se forma enzimáticamente, es decir, mediante una enzima. Esta unión puede ser un enlace fosfodiéster (PO) que se produce de manera natural o una unión modificada que incluye, pero no se limita a, fosforotioato (PS) o fosforamidato (PA).
Tal como se usa en el presente documento, el término “síntesis enzimática” significa la producción de oligonucleótidos, incluyendo los segmentos y el producto final, usando enzimas, por ejemplo, ligasas, transferasas, fosfatasas y nucleasas, en particular endonucleasas. Estas enzimas pueden ser enzimas de tipo natural o enzimas mutantes. Dentro del alcance de la presente invención están enzimas mutantes capaces de actuar sobre sustratos de nucleótidos u oligonucleótidos modificados.
Tal como se usa en el presente documento, el término “ligasa” significa una enzima que cataliza la unión, es decir, la unión covalente, de dos moléculas de oligonucleótidos, por ejemplo, mediante la formación de un enlace fosfodiéster entre el extremo 3’ de un oligonucleótido (o segmento) y el extremo 5’ del mismo u otro oligonucleótido (o segmento). Estas enzimas a menudo se denominan ADN ligasas o ARN ligasas y utilizan cofactores: ATP (ADN ligasas eucariotas, virales y de arqueas) o NAD (ADN ligasas procariotas). A pesar de su presencia en todos los organismos, las ADN ligasas muestran una amplia diversidad de secuencias de aminoácidos, tamaños moleculares y propiedades (Nucleic Acids Research, 2000, vol. 28, n.° 21, 4051-4058). Habitualmente, son miembros de la clase de enzimas EC 6.5 según lo define la Unión Internacional de Bioquímica y Biología Molecular, es decir, ligasas usadas para formar enlaces de éster fosfórico. Dentro del alcance de la invención, se encuentra una ligasa capaz de unir un oligonucleótido no modificado a otro oligonucleótido no modificado, una ligasa capaz de unir un oligonucleótido no modificado a un oligonucleótido modificado (es decir, un oligonucleótido 5’ modificado a un oligonucleótido 3’ no modificado, y/o un oligonucleótido 5’ no modificado a un oligonucleótido 3’ modificado), así como una ligasa capaz de unir un oligonucleótido modificado a otro oligonucleótido modificado.
Tal como se usa en el presente documento, el término “ligasa monocatenaria” o “ligasa mc” significa una enzima, por ejemplo, una ARN ligasa, que es capaz de catalizar el ligamiento dependiente de ATP de (i) ARN monocatenario fosforilado en 5’ al 3’-OH de una hebra de ARN aceptora monocatenaria y (ii) el ligamiento de un solo residuo (incluyendo un residuo modificado), por ejemplo, un nucleótido-3’,5’-bisfosfato, 3’,5’-tiobisfosfato o 3’-fosfato-5’-tiofosfato, al extremo 3’ del ARN o un oligonucleótido modificado (Modified Oligoribonucleotides: 17 (11), 2077-2081, 1978). Un ejemplo de una ligasa mc es la ARN ligasa de T4, que también se ha demostrado que funciona en sustratos de a Dn en ciertas condiciones (Nucleic Acids research 7(2), 453-464, 1979). La función natural de la ARN ligasa de T4 en Escherichia coli infectada con el bacteriófago T4 es reparar los frenos monocatenarios al ARNt bacteriano provocados por los mecanismos de defensa bacterianos contra el ataque viral. Dentro del alcance de la invención se encuentra una ligasa mc capaz de unir un nucleótido no modificado a un oligonucleótido no modificado, una ligasa mc capaz de unir un nucleótido no modificado a un oligonucleótido modificado, una ligasa mc capaz de unir un nucleótido modificado a un oligonucleótido no modificado, así como una ligasa mc capaz de unir un nucleótido modificado a un oligonucleótido modificado. Una ligasa mc según la invención es una ligasa que no requiere un oligonucleótido molde para que se produzca el ligamiento, es decir, la
actividad de ligamiento de la ligasa es independiente del molde.
Tal como se usa en el presente documento, una “ligasa termoestable” es una ligasa que es activa a temperaturas elevadas, es decir, por encima de la temperatura del cuerpo humano, es decir, por encima de 37 °C. Una ligasa termoestable puede ser activa, por ejemplo, a 40 °C - 65 °C; o 40 °C - 90 °C; y así sucesivamente.
Tal como se usa en el presente documento, una “transferasa” significa una enzima que cataliza la unión independiente del molde de un nucleótido a otro nucleótido u oligonucleótido. Una transferasa tal como se describe en el presente documento incluye una nucleotidil transferasa terminal (TdT), también conocida como ADN nucleotidilexotransferasa (DNTT) o transferasa terminal. La TdT es una ADN polimerasa especializada que se expresa en células linfoides pre-B y pre-T inmaduras, donde permite la diversidad de unión del gen de anticuerpo V-D-J. TdT cataliza la adición de nucleótidos al extremo 3’ de una molécula de ADN. Una transferasa tal como se describe en el presente documento incluye una TdT que no se produce de natural o mutante. Dentro del alcance de la invención se encuentra una transferasa capaz de unir un nucleótido no modificado a un oligonucleótido no modificado, una transferasa capaz de unir un nucleótido no modificado a un oligonucleótido modificado, una transferasa capaz de unir un nucleótido modificado a un oligonucleótido no modificado, así como una transferasa capaz de unir un nucleótido modificado a un oligonucleótido modificado.
Tal como se usa en el presente documento, el término “fosfatasa” significa una enzima que cataliza la hidrólisis de un fosfoéster para producir un alcohol. La fosfatasa alcalina cataliza de manera no específica la desfosforilación de los extremos 5’ y 3’ del ADN y el ARN (y oligonucleótidos modificados) y también hidroliza trifosfatos de nucleótidos (NTP y dNTP) y es óptimamente activa en entornos de pH alcalino.
Tal como se usa en el presente documento, el término “endonucleasa específica de secuencia” o “endonucleasa específica de sitio” se usan indistintamente y significan una nucleasa que escinde específicamente un oligonucleótido monocatenario en una posición particular. Por ejemplo, la endonucleasa V escinde el ARN una base después de la inosina, es decir, en el segundo enlace fosfodiéster 3’ con respecto a la inosina. Otros ejemplos de endonucleasas específicas de sitio incluyen la familia de meganucleasas, nucleasas con dedos de zinc, TALEN y Cas9.
Tal como se usa en el presente documento, el término “cebador” significa una secuencia de oligonucleótidos que se usa como punto de partida para sintetizar un oligonucleótido de segmento de la invención. Un cebador comprende al menos 3 nucleótidos y puede unirse a un material de soporte.
Tal como se usa en el presente documento, el término “residuo de nucleótido modificado” u “oligonucleótido modificado” significa un residuo de nucleótido u oligonucleótido que contiene al menos un aspecto de su química que difiere de un residuo de nucleótido u oligonucleótido que se produce de manera natural. Tales modificaciones pueden producirse en cualquier parte del residuo de nucleótido, por ejemplo, azúcar, base o fosfato. A continuación se dan a conocer ejemplos de modificaciones de nucleótidos.
Tal como se usa en el presente documento, el término “ligasa modificada” significa una ligasa que difiere de una ligasa de “tipo natural” que se produce de manera natural en uno o más residuos de aminoácidos. Tales ligasas no se encuentran en la naturaleza. Tales ligasas son útiles en los procesos novedosos de la invención. A continuación se dan a conocer ejemplos de ligasas modificadas. Los términos “ligasa modificada” y “ligasa mutante” se usan indistintamente.
T al como se usa en el presente documento, el término “transferasa modificada” significa una transferasa que difiere de una transferasa de “tipo natural” que se produce de manera natural en uno o más residuos de aminoácidos. Tales transferasas no se encuentran en la naturaleza. Tales transferasas son útiles en los procesos novedosos de la invención. Los términos “transferasa modificada” y “transferasa mutante” se usan indistintamente.
Tal como se usa en el presente documento, el término “gapmero” significa un oligonucleótido que tiene un “segmento de hueco” interno flanqueado por dos “segmentos de ala” externos, en donde el segmento de hueco consiste en una pluralidad de nucleótidos que soportan la escisión de ARNasa H y cada segmento de ala consiste en uno o más nucleótidos que son químicamente distintos a los nucleótidos dentro del segmento de hueco.
Tal como se usa en el presente documento, el término “material de soporte” significa un compuesto o material de alto peso molecular que aumenta el peso molecular de un oligonucleótido, por ejemplo, el molde o el cebador, permitiendo de ese modo que se conserve, por ejemplo, cuando las impurezas y los productos se separan de la mezcla de reacción.
Tal como se usa en el presente documento, el “porcentaje de identidad” entre una secuencia de ácido nucleico de consulta y una secuencia de ácido nucleico sujeto es el valor de “identidades”, expresado como porcentaje, que se calcula mediante el algoritmo BLASTN cuando una secuencia de ácido nucleico sujeto tiene una cobertura de
consulta del 100 % con una secuencia de ácido nucleico de consulta después de realizar una alineación de BLASTN por parejas. Tales alineaciones de BLASTN por parejas entre una secuencia de ácido nucleico de consulta y una secuencia de ácido nucleico sujeto se realizan usando los parámetros por defecto del algoritmo BLASTN disponible en el sitio web del National Center for Biotechnology Institute con el filtro para regiones de baja complejidad desactivado. De manera importante, una secuencia de ácido nucleico de consulta puede describirse mediante una secuencia de ácido nucleico identificada en una o más reivindicaciones del presente documento. La secuencia de consulta puede ser el 100 % idéntica a la secuencia sujeto, o puede incluir hasta un cierto número entero de alteraciones de nucleótidos en comparación con la secuencia sujeto de manera que el % de identidad sea inferior al 100 %. Por ejemplo, la secuencia de consulta es al menos el 80, el 85, el 90, el 95, el 96, el 97, el 98 o el 99 % idéntica a la secuencia sujeto.
Declaración de la invención
En un aspecto de la invención, se proporciona un procedimiento para producir un producto de oligonucleótido monocatenario, que tiene al menos un residuo de nucleótido modificado, comprendiendo dicho procedimiento: a) proporcionar un conjunto de oligonucleótidos (I) que comprende segmentos de la secuencia del producto, en el que al menos un segmento de la secuencia del producto contiene al menos un residuo de nucleótido modificado, y en el que cada segmento se ha producido mediante síntesis enzimática;
b) proporcionar un oligonucleótido molde (II) complementario a la secuencia del producto de oligonucleótido monocatenario, teniendo dicho molde una propiedad que permite que se separe del producto y se recicle para su uso en futuras reacciones;
c) poner en contacto (I) y (II) en condiciones para permitir el apareamiento de los oligonucleótidos de segmento con el oligonucleótido molde;
d) unir los oligonucleótidos de segmento usando una ligasa para formar el producto;
e) cambiar las condiciones para desnaturalizar el molde apareado y cualquier hebra de oligonucleótido de impureza, y separar las impurezas;
f) cambiar las condiciones para desnaturalizar el molde apareado y las hebras de oligonucleótidos de producto, y separar el producto de oligonucleótido monocatenario; y
g) reciclar el oligonucleótido molde para su uso en futuras reacciones.
En una realización de la invención, uno o más o todos los oligonucleótidos de segmento se producen enzimáticamente.
En una realización de la invención, uno o más o todos los oligonucleótidos de segmento se producen usando una ligasa monocatenaria (ligasa mc). En una realización, la ligasa mc es una ARN ligasa. En una realización, la ligasa mc es una ARN ligasa de T4 o ARN ligasa de T4 modificada.
En otra realización, el procedimiento para producir un oligonucleótido de segmento usando una ligasa mc comprende:
(i) añadir un nucleótido, un dinucleótido, un trinucleótido o un tetranucleótido; que tiene un resto o bien fosfato, tiofosfato o bien ditiofosfato en el extremo 3’; y un resto o bien fosfato, tiofosfato o bien ditiofosfato en el extremo 5’; al 3’-OH de un cebador de oligonucleótido monocatenario usando una ligasa mc;
(ii) eliminar el resto 3’-fosfato, 3’-tiofosfato o 3’-ditiofosfato usando una fosfatasa;
(iii) repetir las etapas (i) y (ii) para extender el oligonucleótido para producir la secuencia de segmento; y (iv) liberar el oligonucleótido de segmento del cebador de oligonucleótido usando una endonucleasa específica de secuencia.
En una realización de la invención, el procedimiento para producir un oligonucleótido de segmento, usando una ligasa mc, comprende añadir un 3’,5’-nucleótido bisfosfato, que tiene uno o más de cualquiera de los oxígenos del fosfato sustituidos por azufre, al 3’-OH de un cebador de oligonucleótido monocatenario usando una ligasa mc en la etapa (i).
En una realización de la invención, el procedimiento para producir un oligonucleótido de segmento, usando una
ligasa mc, comprende añadir un 3’,5’-nucleótido-bisfosfato, un 3’,5’-nucleótido-tiofosfato (por ejemplo 3’,5’-bistiofosfato o 3’-fosfato-5’-tiofosfato o 3’-tiofosfato-5’-fosfato) o un 3’,5’-nucleótido-ditiofosfato (por ejemplo 3’,5’-bisditiofosfato o 3’-fosfato-5’-ditiofosfato o 3’-ditiofosfato-5’-fosfato) al 3’-OH de un cebador de oligonucleótido monocatenario usando una ligasa mc en la etapa (i).
En una realización de la invención, se producen uno o más oligonucleótidos de segmento usando una transferasa. En una realización de la invención, el procedimiento para producir un oligonucleótido de segmento usando una transferasa comprende:
(i) añadir un nucleótido-5’-trifosfato, alfa-tiotrifosfato o alfaditiotrifosfato que tiene un grupo protector en su 3’-OH al 3’-OH de un cebador de oligonucleótido monocatenario usando una transferasa;
(ii) desproteger la posición 3’ para regenerar el 3’-OH;
(iii) repetir las etapas (i) y (ii) para extender el oligonucleótido para producir la secuencia de segmento;
(iv) liberar el oligonucleótido de segmento del cebador de oligonucleótido usando una endonucleasa específica de secuencia.
En una realización de la invención, cada segmento se produce mediante síntesis enzimática. En una realización de la invención, cada segmento se produce usando una ligasa mc. En una realización de la invención, cada segmento se produce usando una transferasa.
El uso de una ligasa mc, por ejemplo una ARN ligasa, una transferasa o cualquier otra enzima que sea capaz de añadir un solo nucleótido a un oligonucleótido monocatenario, de una manera independiente del molde, permite la síntesis de un oligonucleótido con una secuencia definida. Un enfoque de este tipo podría usarse para producir el producto de oligonucleótido completo mediante la adición iterativa de bases individuales. Sin embargo, a menos que cada ciclo de síntesis discurra con un rendimiento del 100 %, se incorporarán errores de deleción de secuencia en el producto final. Por ejemplo, si un oligonucleótido se extiende en un nucleótido con un rendimiento del 99 % en un ciclo de síntesis, el 1 % restante estará disponible para reaccionar en ciclos de síntesis posteriores pero el producto formado será un nucleótido más corte que el producto deseado. A medida que el número de ciclos aumenta, entonces la tasa de error se acentúa de modo que, en este ejemplo, un rendimiento del ciclo del 99 % daría como resultado la formación de un 20 % de secuencias acortadas en una sola base para producción de un 20 meros.
Según la presente invención, la acumulación secuencial de errores se evita mediante lo siguiente. En primer lugar, solo se sintetizan secuencias cortas, normalmente de 5 a 8 nucleótidos de longitud, mediante la adición de nucleótidos individuales. Este procedimiento da como resultado secuencias cortas que tienen una mayor pureza que secuencias largas ya que se exponen a menos ciclos de acumulación de errores. En segundo lugar, estas secuencias cortas se ensamblan en un molde de ADN complementario y luego se unen entre sí. El uso de un molde complementario conjuntamente con una ligasa garantiza que solo se ensamblen oligonucleótidos cortos que tienen tanto la longitud correcta como la secuencia correcta. Por consiguiente, los procedimientos de la invención dan como resultado tanto un rendimiento global superior como, de manera importante, una fidelidad de secuencia global superior.
Una realización de la invención proporciona un procedimiento tal como se describió anteriormente en el presente documento, mediante el cual la desnaturalización del dúplex de molde e impureza y/o la desnaturalización del dúplex de molde y producto da como resultado un aumento de temperatura. En una realización, la desnaturalización se produce como resultado del cambio del pH. En una realización adicional, la desnaturalización se produce cambiando la concentración de sal en una disolución tamponante.
Aun otra realización de la invención proporciona un procedimiento tal como se dio a conocer anteriormente en el presente documento, mediante el cual los oligonucleótidos de segmento tienen de 3 a 15 nucleótidos de longitud. En una realización adicional de la invención, los segmentos tienen de 5 a 10 nucleótidos de longitud. En una realización adicional de la invención, los segmentos tienen de 5 a 8 nucleótidos de longitud. En una realización adicional de la invención, los segmentos tienen 4, 5, 6, 7 o 8 nucleótidos de longitud. En una realización particular, hay tres oligonucleótidos de segmento: un segmento 5’ que tiene 7 nucleótidos de longitud, un segmento central que tiene 6 nucleótidos de longitud y un segmento 3’ que tiene 7 nucleótidos de longitud, que cuando se ligan entre sí forman un oligonucleótido que tiene 20 nucleótidos de longitud (un “20 meros”). En una realización particular, hay tres oligonucleótidos de segmento: un segmento 5’ que tiene 6 nucleótidos de longitud, un segmento central que tiene 8 nucleótidos de longitud y un segmento 3’ que tiene 6 nucleótidos de longitud, que cuando se ligan entre sí forman un oligonucleótido que tiene 20 nucleótidos de longitud (un “20 meros”). En una realización particular, hay tres oligonucleótidos de segmento: un segmento 5’ que tiene 5 nucleótidos de longitud, un segmento central que tiene 10 nucleótidos de longitud y un segmento 3’ que tiene 5 nucleótidos de longitud, que cuando se ligan
entre sí forman un oligonucleótido que tiene 20 nucleótidos de longitud (un “20 meros”). En una realización particular, hay tres oligonucleótidos de segmento: un segmento 5’ que tiene 4 nucleótidos de longitud, un segmento central que tiene 12 nucleótidos de longitud y un segmento 3’ que tiene 4 nucleótidos de longitud, que cuando se ligan entre sí forman un oligonucleótido que tiene 20 nucleótidos de longitud (un “20 meros”). En una realización particular, hay cuatro oligonucleótidos de segmento: un segmento 5’ que tiene 5 nucleótidos de longitud, un segmento central 5’ que tiene 5 nucleótidos de longitud, un segmento central 3’ que tiene 5 nucleótidos de longitud y un segmento 3’ que tiene 5 nucleótidos de longitud, que cuando se ligan entre sí forman un oligonucleótido que tiene 20 nucleótidos de longitud (un “20 meros”).
Una realización de la invención proporciona un procedimiento tal como se describió anteriormente en el presente documento, mediante el cual el producto tiene de 10 a 200 nucleótidos de longitud. En una realización adicional de la invención, el producto tiene de 15 a 30 nucleótidos de longitud. En una realización de la invención, el producto tiene 21,22, 23, 24, 25, 26, 27, 28, 29 o 30 nucleótidos de longitud. En una realización de la invención, el producto tiene 20 nucleótidos de longitud, un “20 meros”. En una realización de la invención, el producto tiene 21 nucleótidos de longitud, un “21 meros”. En una realización de la invención, el producto tiene 22 nucleótidos de longitud, un “22 meros”. En una realización de la invención, el producto tiene 23 nucleótidos de longitud, un “23 meros”. En una realización de la invención, el producto tiene 24 nucleótidos de longitud, un “24 meros”. En una realización de la invención, el producto tiene 25 nucleótidos de longitud, un “25 meros”. En una realización de la invención, el producto tiene 26 nucleótidos de longitud, un “26 meros”. En una realización de la invención, el producto tiene 27 nucleótidos de longitud, un “27 meros”. En una realización de la invención, el producto tiene 28 nucleótidos de longitud, un “28 meros”. En una realización de la invención, el producto tiene 29 nucleótidos de longitud, un “29 meros”. En una realización de la invención, el producto tiene 30 nucleótidos de longitud, un “30 meros”.
En una realización de la invención, el procedimiento es un procedimiento para producir un oligonucleótido terapéutico. En una realización de la invención, el procedimiento es un procedimiento para producir un oligonucleótido terapéutico monocatenario. En una realización de la invención, el procedimiento es un procedimiento para producir un oligonucleótido terapéutico bicatenario.
Otra realización de la invención proporciona un procedimiento tal como se dio a conocer anteriormente en el presente documento, en el que la propiedad que permite que el molde se separe del producto es que el molde está unido a un material de soporte. En una realización adicional de la invención, el material de soporte es un material de soporte soluble. En aún una realización adicional de la invención, el material de soporte soluble se selecciona del grupo que consiste en: polietilenglicol, un polímero orgánico soluble, ADN, una proteína, un dendrímero, un polisacárido, un oligosacárido y un hidrato de carbono. En una realización de la invención, el material de soporte es polietilenglicol (PEG). En una realización adicional de la invención, el material de soporte es un material de soporte insoluble. En una realización adicional de la invención, el material de soporte es un material de soporte sólido. En aún una realización adicional, el material de soporte sólido se selecciona del grupo que consiste en: una perla de vidrio, una perla polimérica, un soporte fibroso, una membrana, una perla recubierta con estreptavidina y celulosa. En una realización, el material de soporte sólido es una perla recubierta con estreptavidina. En una realización adicional, el material de soporte sólido forma parte del propio recipiente de reacción, por ejemplo una pared de reacción.
Una realización de la invención proporciona un procedimiento tal como se dio a conocer anteriormente en el presente documento, en el que múltiples copias repetidas del molde se unen de una manera continua por medio de un solo punto de unión al material de soporte. Las múltiples copias repetidas del molde pueden estar separadas por un ligador, por ejemplo, tal como se muestra en la figura 3. Las múltiples copias repetidas del molde pueden ser repeticiones directas, es decir, no están separadas por un ligador.
En otra realización de la invención, el molde se une al material de soporte en múltiples puntos de unión.
Aún otra realización de la invención proporciona un procedimiento tal como se dio a conocer anteriormente en el presente documento, en el que la propiedad que permite que el molde se separe del producto es el peso molecular del molde. Por ejemplo, pueden estar presentes copias repetidas de la secuencia de molde en un solo oligonucleótido, con o sin una secuencia de ligador.
Otra realización de la invención proporciona un procedimiento tal como se dio a conocer anteriormente en el presente documento, en el que el molde, o el molde y el material de soporte, se reciclan para su uso en futuras reacciones, por ejemplo tal como se detalla a continuación. Otra realización de la invención proporciona un procedimiento tal como se dio a conocer anteriormente en el presente documento, en el que la reacción se lleva a cabo usando un procedimiento de flujo continuo o semicontinuo, por ejemplo tal como se muestra en la figura 4 o la figura 5.
En una realización de la invención, el procedimiento es para la fabricación a gran escala de oligonucleótidos, en particular oligonucleótidos terapéuticos. En el contexto de la presente invención, fabricación a gran escala de
oligonucleótidos significa fabricación a una escala mayor de o igual a 1 litro, por ejemplo el procedimiento se lleva a cabo en un reactor de 1 l o mayor. Alternativamente, o además, en el contexto de la presente invención, fabricación a gran escala de oligonucleótidos significa fabricación a escala de gramos del producto, en particular la producción de más de o igual a 10 gramos de producto. En una realización, el producto se produce a una escala de gramos o kilogramos y/o el procedimiento se lleva a cabo en un reactor de al menos 1 l. En una realización de la invención, la cantidad de producto de oligonucleótido producido está en una escala de gramos. En una realización de la invención, la cantidad de producto producido es mayor de o igual a: 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 20, 30, 40, 50, 60, 70, 80, 90 o 100 gramos. En una realización de la invención, la cantidad de producto de oligonucleótido producido es mayor de o igual a: 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950 gramos. En una realización de la invención, la cantidad de producto de oligonucleótido producido es de 500 gramos o mayor. En una realización de la invención, el producto de oligonucleótido producido está en una escala de kilogramos. En una realización de la invención, la cantidad de producto de oligonucleótido producido es de 1 kg o más. En una realización de la invención, la cantidad de producto de oligonucleótido producido es mayor de o igual a: 1,2, 3, 4, 5, 6, 7, 8, 9, 10 kg. En una realización de la invención, la cantidad de producto de oligonucleótido producido es mayor de o igual a: 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95 o 100 kg.
En una realización de la invención, la cantidad de producto producido es de entre 10 gramos y 100 kg. En una realización de la invención, la cantidad de producto producido es de entre 10 gramos y 50 kg. En una realización de la invención, la cantidad de producto producido es de entre 100 gramos y 100 kg. En una realización de la invención, la cantidad de producto producido es de entre 100 gramos y 50 kg. En una realización de la invención, la cantidad de producto producido es de entre 500 gramos y 100 kg. En una realización de la invención, la cantidad de producto producido es de entre 500 gramos y 50 kg. En una realización de la invención, la cantidad de producto producido es de entre 1 kg y 50 kg. En una realización de la invención, la cantidad de producto producido es de entre 10 kg y 50 kg.
En una realización de la invención, la fabricación de oligonucleótidos tienen lugar a una escala mayor de o igual a: 2, 3, 4, 5, 6, 7, 8, 9, 10 litros, por ejemplo en un reactor de 2, 3, 4, 5, 6, 7, 8, 9 o 10 l. En una realización de la invención, la fabricación de oligonucleótidos tienen lugar a una escala mayor de o igual a: 20, 25, 30, 35, 40, 45, 50, 55, 60, 6570, 75, 80, 85, 90, 95, 100 litros, por ejemplo en un reactor de 20, 25, 30, 35, 40, 45, 50, 55, 60, 65 70, 75, 80, 85, 90, 95, 100 l. En una realización de la invención, la fabricación de oligonucleótidos tiene lugar a una escala mayor de o igual a: 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000 litros, por ejemplo en un reactor de 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000 l.
En una realización de la invención, el volumen del reactor es de aproximadamente 10.000 l, aproximadamente 5000 l, aproximadamente 2000 l, aproximadamente 1000 l, aproximadamente 500 l, aproximadamente 125 l, aproximadamente 50 l, aproximadamente 20 l, aproximadamente 10 l o aproximadamente 5 l.
En una realización de la invención, el volumen del reactor es de entre 5 y 10.000 l, entre 10 y 5000 l, entre 20 y 2000 l, o entre 50 y 1000 l.
Un oligonucleótido según la presente invención puede tener al menos una modificación de la estructura principal, y/o al menos una modificación de azúcar y/o al menos una modificación de base en comparación con un oligonucleótido a base de ARN o ADN.
En una realización de la invención, uno o más oligonucleótidos de segmento tienen al menos una modificación de la estructura principal, y/o al menos una modificación de azúcar y/o al menos una modificación de base en comparación con un oligonucleótido a base de ARN o ADN. En una realización, todos los oligonucleótidos de segmento tienen al menos una modificación de la estructura principal. En una realización, al menos uno de los segmentos tiene una estructura principal completamente modificada. En una realización de la invención, todos los segmentos tienen una estructura principal completamente modificada. En una realización, la estructura principal de todos los oligonucleótidos de segmento consiste en uniones fosforotioato. En una realización, dos o más de los segmentos tienen al menos una modificación de azúcar y/o al menos una modificación de base en comparación con un oligonucleótido a base de ARN o ADN. En una realización, los segmentos de “ala” comprenden al menos una modificación de azúcar. En una realización, los segmentos de “ala” consisten totalmente en azúcares modificados. En una realización, los segmentos de “ala” consisten totalmente en azúcares modificados con 2’-MOE.
Una realización de la invención proporciona un procedimiento tal como se dio a conocer anteriormente en el presente documento, en el que el producto contiene al menos 1 residuo de nucleótido modificado. En una realización adicional, la modificación es en la posición 2’ del resto de azúcar.
Los oligonucleótidos usados en el procedimiento de la invención pueden incluir modificaciones de azúcar, es decir,
una versión modificada del resto ribosilo, tal como ARN modificado con 2’-O tal como 2’-O-alquilo o 2’-O-alquilo (sustituido), por ejemplo 2’-O-metilo, 2’-O-(2-cianoetilo), 2’-O-(2-metoxi)etilo (2’-MOE), 2’-O-(2-tiometil)etilo, 2’-O-butirilo, 2’-O-propargilo, 2’-O-alilo, 2’-O-(3-amino)propilo, 2’-O-(3-(dimetilamino)propilo), 2’-O-(2-amino)etilo, 2’-O-(2-(dimetilamino)etilo); 2’-desoxi (ADN); 2’-O-(haloalcoxi)metilo (Arai K. et al. Bioorg. Med. Chem. 2011,21,6285), por ejemplo 2’-O-(2-cloroetoxi)metilo (MCe M), 2’-O-(2,2-dicloroetoxi)metilo (DCEM); 2’-O-alcoxicarbonilo, por ejemplo 2’-O-[2-(metoxicarbonil)etilo] (m Oc E), 2’-O-[2-(N-metilcarbamoil)etilo] (MCE), 2’-O-[2-(N,N-dimetilcarbamoil)etilo] (DCME); 2’-halo, por ejemplo 2’-F, FANA (ácido nucleico de arabinosilo 2’-F); modificaciones de carbazúcar y azazúcar; 3’-O-alquilo por ejemplo 3’-O-metilo, 3’-O-butirilo, 3’-0-propargilo; y sus derivados.
En una realización de la invención, la modificación de azúcar se selecciona del grupo que consiste en 2’-fluoro (2’-F), 2’-O-metilo (2’-OMe), 2’-O-metoxietilo (2’-MOE) y 2’-amino. En aún una realización adicional, la modificación es 2’-MOE.
Otras modificaciones de azúcar incluyen ácido nucleico “con puente” o “bicíclico” (BNA), por ejemplo ácido nucleico bloqueado (LNA), xilo-LNA, a-L-LNA, p-D-LNA, cEt (2’-O,4’-C etilo restringido) LNA, cMOEt (2’-O,4’-C metoxietilo restringido) LNA, ácido nucleico con puente de etileno (ENA), ADN triciclo; ácido nucleico desbloqueado (UNA); ácido nucleico de ciclohexenilo (CeNA), ácido nucleico de altriol (ANA), ácido nucleico de hexitol (HNA), HNA fluorado (F-HNA), piranosil-ARN (p-ARN), 3’-desoxipiranosil-ADN (p-ADN); morfolino (como por ejemplo en PMO, PPMO, PMOPlus, PMO-X); y sus derivados.
Los oligonucleótidos usados en el procedimiento de la invención pueden incluir otras modificaciones, tales como ácido nucleico de base peptídica (PNA), PNA modificado con boro, ácido nucleico de oxi-péptido basado en pirrolidina (POPNA), ácido nucleico basado en glicol o glicerol (GNA), ácido nucleico basado en treosa (TNA), ácido nucleico basado en treoninol acíclico (aTNA), oligonucleótidos con bases y estructuras principales integradas (ONIB), oligonucleótidos de pirrolidin-amida (POM); y sus derivados.
En una realización de la invención, el oligonucleótido modificado comprende un oligómero de fosforodiamidato morfolino (PMO), un ácido nucleico bloqueado (LNA), un ácido nucleico peptídico (PNA), un ácido nucleico con puente (b Na ) tal como (S)-cEt-BNA o un SPIEGELMER.
En una realización adicional, la modificación está en la nucleobase. Las modificaciones de bases incluyen versiones modificadas de las bases de purina y pirimidina naturales (por ejemplo, adenina, uracilo, guanina, citosina y timina), tales como inosina, hipoxantina, ácido orótico, agmatidina, lisidina, 2-tiopirimidina (por ejemplo, 2-tiouracilo, 2-tiotimina), G-clamp y sus derivados, pirimidina sustituida en 5 (por ejemplo 5-metilcitosina, 5-metiluracilo, 5-halouracilo, 5-propiniluracilo, 5-propinilcitosina, 5-aminometiluracilo, 5-hidroximetiluracilo, 5-aminometilcitosina, 5-hidroximetilcitosina, Super T), 2,6-diaminopurina, 7-deazaguanina, 7-deazaadenina, 7-aza-2,6-diaminopurina, 8-aza-7-deazaguanina, 8-aza-7-deazaadenina, 8-aza-7-deaza-2,6-diaminopurina, Super G, Super A y N4-etilcitosina, o derivados de los mismos; N2-ciclopentilguanina (cPent-G), N2-ciclopentil-2-aminopurina (cPent-AP) y N2-propil-2-aminopurina (Pr-AP), o derivados de las mismas; y bases degeneradas o universales, como 2,6-difluorotolueno o bases ausentes como sitios abásicos (por ejemplo 1 -desoxirribosa, 1,2-didesoxirribosa, 1-desoxi-2-O-metilribosa; o derivados de pirrolidina en los que el oxígeno del anillo se ha reemplazado por nitrógeno (azarribosa)). Pueden encontrarse ejemplos de derivados de Super A, Super G y Super T en el documento US6683173. Se demostró que cPent-G, cPent-AP y Pr-AP reducían los efectos inmunoestimuladores cuando se incorporaban en ARNip (Peacock H. et al. J. Am. Chem. Soc. (2011), 133, 9200).
En una realización de la invención, la modificación de nucleobase se selecciona del grupo que consiste en 5-metilpirimidinas, 7-deazaguanosinas y nucleótidos abásicos. En una realización, la modificación es una 5-metilcitosina.
Los oligonucleótidos usados en el procedimiento de esta invención pueden incluir una modificación de la estructura principal, por ejemplo una versión modificada el fosfodiéster presente en el ARN, tal como fosforotioato (PS), fosforoditioato (PS2), fosfonoacetato (PACE), fosfonoacetamida (PACA), tiofosfonoacetato, tiofosfonoacetamida, profármaco de fosforotioato, H-fosfonato, metilfosfonato, metilfosfonotioato, metilfosfato, metilfosforotioato, etilfosfato, etilfosforotioato, boranofosfato, boranofosforotioato, metilboranofosfato, metilboranofosforotioato, metilboranofosfonato, metilboranofosfonotioato, y sus derivados. Otra modificación incluye fosforamidita, fosforamidato, N3’^ P 5 ’ fosforamidato, fosfordiamidato, fosforotiodiamidato, sulfamato, dimetilensulfóxido, sulfonato, triazol, oxalilo, carbamato, metilenimino (MMI) y ácido nucleico de tioacetamido (TANA); y sus derivados.
En una realización adicional, la modificación está en la estructura principal y se selecciona del grupo que consiste en: fosforotioato (PS), fosforamidato (PA) y fosforodiamidato. En una realización de la invención, el oligonucleótido modificado es un oligómero de fosforodiamidato morfolino (PMO). Un PMO tiene una estructura principal de anillos de metilenmorfolina con uniones de fosforodiamidato. En una realización de la invención, el producto tiene una estructura principal de fosforotioato (PS).
En una realización de la invención, el oligonucleótido comprende una combinación de dos o más modificaciones tal como se dio a conocer anteriormente. Un experto en la técnica apreciará que hay muchos derivados sintéticos de oligonucleótidos.
En una realización de la invención, el producto es un gapmero. En una realización de la invención, los segmentos de ala comprenden modificaciones de la estructura principal y de azúcar y el segmento central o ‘hueco’ comprende modificaciones de la estructura principal, pero no modificaciones de azúcar. En una realización de la invención, las alas 5’ y 3’ del gapmero comprenden o consisten en nucleótidos modificados con 2’-MOE. En una realización de la invención, el segmento de hueco del gapmero comprende o consiste en nucleótidos que contienen hidrógeno en la posición 2’ del resto de azúcar, es decir, es similar al ADN. En una realización de la invención, las alas 5’ y 3’ del gapmero consisten en nucleótidos modificados con 2’MOE y el segmento de hueco del gapmero consiste en nucleótidos que contienen hidrógeno en la posición 2’ del resto de azúcar (es decir, desoxinucleótidos). En una realización de la invención, las alas 5’ y 3’ del gapmero consisten en nucleótidos modificados con 2’MOE y el segmento de hueco del gapmero consiste en nucleótidos que contienen hidrógeno en la posición 2’ del resto de azúcar (es decir, desoxinucleótidos) y las uniones entre todos los nucleótidos son uniones de fosforotioato.
Una realización de la invención proporciona un procedimiento tal como se describió anteriormente en el presente documento, en el que el producto resultante es tiene una pureza mayor del 90 %. En una realización adicional, el producto tiene una pureza mayor del 95 %. En una realización adicional, el producto tiene una pureza mayor del 96 %. En una realización adicional, el producto tiene una pureza mayor del 97 %. En una realización adicional, el producto el producto tiene una pureza mayor del 98 %. En una realización adicional, el producto tiene una pureza mayor del 99 % pure. La pureza de un oligonucleótido puede determinarse usando cualquier método adecuado, por ejemplo cromatografía de líquidos de alta resolución (HPLC) o espectrometría de masas (EM), en particular, cromatografía de líquidos-EM (CL-EM), HPLC-EM o electroforesis capilar-espectrometría de masas (CEEM).
En una realización de la invención, el oligonucleótido producido es un oligonucleótido antisentido. En una realización de la invención, el oligonucleótido producido es un aptámero. En una realización de la invención, el oligonucleótido producido es un miARN. En una realización de la invención, el producto es un oligonucleótido terapéutico.
En una realización de la invención, no hay sustancialmente nucleótidos en el recipiente de reacción para ligar los segmentos. En una realización de la invención, no hay nucleótidos en el recipiente de reacción para ligar los segmentos. En otra realización de la invención, el recipiente de reacción para ligar los segmentos no comprende un conjunto de nucleótidos, es decir, la reacción está de sustancialmente libre a completamente libre de nucleótidos.
La invención dada a conocer en el presente documento utiliza las propiedades de oligonucleótidos para proporcionar un procedimiento mejorado para su producción. Al proporcionar un oligonucleótido molde con el 100 % de complementariedad con la secuencia diana, y al controlar las condiciones de reacción de modo que el producto pueda liberarse y separarse en condiciones específicas, puede obtenerse un producto con un alto grado de pureza.
Desnaturalización del dúplex de producto (o impureza):molde y separación del producto (o impureza) del molde
La liberación del producto (o cualquier impureza) del molde requiere que se rompa el apareamiento de bases de Watson-Crick entre la hebra del oligonucleótido molde y el producto (o impureza) (es decir, desnaturalización del dúplex). El producto (o impureza) puede separarse entonces del molde, lo que puede producirse como dos etapas separadas, o como una etapa combinada.
La liberación y separación del producto (o impureza) puede producirse como una etapa, si el procedimiento se lleva a cabo en un reactor de columna. La ejecución en un tampón que altera el pH o la concentración de sal, o que contiene un agente químico que altera el emparejamiento de bases (tal como formamida o urea) provocará la desnaturalización de las cadenas de oligonucleótidos y el producto (o impureza) se eluirá en el tampón.
Cuando el procedimiento se lleva a cabo en otros recipientes de reacción, la liberación y separación del producto (o impureza) puede producirse como un procedimiento de dos etapas. En primer lugar, los pares de bases de Watson-Crick se alteran para desnaturalizar las hebras y luego el producto (o impureza) se separa del molde, por ejemplo se retira del recipiente de reacción. Cuando la liberación y separación del producto se lleva a cabo como un procedimiento de dos etapas, la rotura de los pares de bases Watson-Crick puede lograrse alterando las condiciones del tampón (pH, sal) o introduciendo un agente químico disruptivo (formamida, urea). Alternativamente, elevar la temperatura también provocará la disociación de las dos hebras, es decir, la desnaturalización. El producto (o las impurezas) pueden separarse entonces (y también retirarse del recipiente de reacción, si se desea) por medio de métodos que incluyen separación basada en el peso molecular, separación
basada en la carga, separación basada en la hidrofobicidad, separación basada en secuencias específicas o una combinación de estos métodos.
Cuando el procedimiento se lleva a cabo en un reactor de flujo continuo o semicontinuo, la liberación y separación del producto (o impureza) puede ser en una o dos etapas. Por ejemplo, la liberación y separación del producto (o impureza) en una etapa podría verse afectada por el aumento de la temperatura para provocar la disociación de las dos hebras, y la separación de las hebras liberadas basándose en peso molecular en la misma parte del reactor que se usa para elevar la temperatura. La liberación y separación del producto (o impureza) en dos etapas podría verse afectada por el aumento de la temperatura para provocar la disociación de las dos hebras en una parte del reactor y la separación de las hebras liberadas basándose en el peso molecular en una parte diferente del reactor.
Liberación y separación específicas de las impurezas del molde, pero reteniendo el producto en el molde
Las impurezas surgen cuando se incorpora un nucleótido incorrecto en la hebra de oligonucleótido durante la extensión de la cadena, o cuando la reacción de extensión de la cadena termina antes de tiempo. Las impurezas también surgen cuando la reacción incluye el paso de ligar los oligonucleótidos de segmento y una o más de las etapas de ligamiento fallan. Los tipos de impurezas que pueden surgir se ilustran en la figura 6.
Las propiedades del apareamiento de bases de Watson-Crick pueden aprovecharse para liberar específicamente cualquier impureza unida al molde antes de la liberación del producto. Cada oligonucleótido bicatenario se disociará en condiciones específicas, y esas condiciones son diferentes para secuencias que no tienen un 100 % de complementariedad en comparación con secuencias con un 100 % de complementariedad. La determinación de tales condiciones está dentro del ámbito de competencia de un experto en la técnica.
Un modo común de desnaturalización de oligonucleótidos es elevando la temperatura. La temperatura a la que se disocian la mitad de los pares de bases, es decir, cuando el 50% del dúplex está en estado monocatenario, se denomina temperatura de fusión, Tm. El medio más confiable y preciso para determinar la temperatura de fusión es empíricamente. Sin embargo, esto es engorroso y no es necesario habitualmente. Pueden usarse varias fórmulas para calcular los valores de Tm (Nucleic Acids Research 1987, 15 (13): 5069-5083; PNAS 1986, 83 (11): 3746-3750; Biopolymers 1997, 44 (3): 217-239) y pueden encontrarse numerosas calculadoras de temperatura de fusión en línea, alojadas por proveedores de reactivos y universidades. Se sabe que, para una secuencia de oligonucleótidos dada, una variante con todas las uniones de fosforotioato se fundirá a una temperatura más baja que una variante con todas las uniones de fosfodiéster. El aumento del número de uniones de fosforotioato en un oligonucleótido tiende a disminuir la Tm del oligonucleótido para su objetivo previsto.
Para separar específicamente las impurezas de una mezcla de reacción, en primer lugar se calcula la temperatura de fusión del dúplex de producto:molde. Entonces, el recipiente de reacción se calienta hasta una primera temperatura, por ejemplo, una temperatura por debajo de la temperatura de fusión del dúplex de producto:molde, por ejemplo 1,2, 3, 4, 5, 6, 7, 8, 9 o 10 grados centígrados por debajo de la temperatura de fusión. Esta etapa de calentamiento provoca la desnaturalización de los oligonucleótidos que no son el producto, es decir, no son el 100% complementarios al molde, del molde. Entonces, estos oligonucleótidos desnaturalizados pueden retirarse del recipiente de reacción usando uno de los métodos dados a conocer anteriormente, por ejemplo, separación basada en el peso molecular, separación basada en la carga, separación basada en la hidrofobicidad, separación basada en secuencias específicas o una combinación de estos métodos. Entonces, el recipiente de reacción se elevará hasta una segunda temperatura más alta, por ejemplo, por encima de la temperatura de fusión calculada, por ejemplo 1,2, 3, 4, 5, 6, 7, 8, 9 o 10 grados centígrados por encima de la temperatura de fusión, para provocar la desnaturalización del producto del molde. Entonces, el producto puede separarse (y retirarse del recipiente de reacción) usando uno de los métodos descritos anteriormente, por ejemplo, separación basada en el peso molecular, separación basada en la carga, separación basada en la hidrofobicidad, separación basada en secuencias específicas o una combinación de estos métodos.
Puede usarse un procedimiento similar cuando el agente disruptivo es un agente que provoca un cambio en el pH o la concentración de sal o es un agente disruptivo químico. La concentración del agente disruptivo se aumenta hasta justo por debajo de la concentración a la que se disociaría el producto, para provocar la desnaturalización de los oligonucleótidos que no son el producto del molde. Entonces, estas impurezas pueden retirarse del recipiente de reacción usando uno de los métodos dados a conocer anteriormente. Entonces, se aumenta la concentración del agente disruptivo hasta por encima de la concentración a la que el producto se disocia del molde. Entonces, el producto puede retirarse del recipiente de reacción usando uno de los métodos dados a conocer anteriormente.
El producto obtenido de un procedimiento tal como se dio a conocer anteriormente tiene un alto grado de pureza sin necesidad de etapas de purificación adicionales. Por ejemplo, el producto obtenido tiene una pureza mayor del
Propiedades del molde
El molde requiere una propiedad que le permita quedar retenido en el recipiente de reacción cuando se retira el producto, para evitar que se convierta en una impureza en el producto. En otras palabras, el molde tiene propiedades que permiten separarlo del producto. En una realización de la invención, esta retención se logra acoplando el molde a un material de soporte. Este acoplamiento da como resultado un complejo molde-soporte que tiene un alto peso molecular y, por tanto, puede retenerse en el recipiente de reacción cuando se retiran las impurezas y el producto, por ejemplo, mediante filtración. El molde puede acoplarse a un material de soporte sólido, tal como perlas poliméricas, soportes fibrosos, membranas, perlas recubiertas con estreptavidina y celulosa. El molde también puede acoplarse a un material de soporte soluble tal como polietilenglicol, un polímero orgánico soluble, ADN, una proteína, un dendrímero, un polisacárido, un oligosacárido y un hidrato de carbono.
Cada material de soporte puede tener múltiples puntos donde puede unirse un molde, y cada punto de unión puede tener múltiples moldes unidos, por ejemplo, de la manera mostrada en la figura 3.
El molde puede tener un alto peso molecular por sí mismo, sin estar unido a un material de soporte, por ejemplo, puede ser una molécula con múltiples copias del molde, por ejemplo, separadas por un ligador, de la manera mostrada en la figura 3.
La capacidad de retener el molde en el recipiente de reacción también permite reciclar el molde para futuras reacciones, ya sea recuperándolo o usándolo en un proceso de flujo continuo o semicontinuo.
Métodos de separación del molde del producto (o impurezas)
Las propiedades del molde, tal como se dio a conocer anteriormente, permiten la separación del molde y el producto, o la separación del producto unido al molde y las impurezas. Pueden usarse separación basada en el peso molecular, separación basada en la carga, la separación basada en la hidrofobicidad, separación basada en secuencias específicas o una combinación de estos métodos.
En el caso de que el molde esté unido a un soporte sólido, la separación del molde del producto, o la separación de las impurezas del producto unido al molde, se logra lavando el soporte sólido en condiciones apropiadas, tal como resultaría evidente para un experto en la técnica. En los casos en los que el molde está acoplado a un soporte soluble o está compuesto por secuencias de molde repetidas, la separación del molde del producto o la separación del producto unido al molde de las impurezas puede lograrse por medio de una separación basada en el peso molecular, por ejemplo, usando técnicas tales como ultrafiltración o nanofiltración en las que el material del filtro se elige de modo que la molécula más grande quede retenida por el filtro y la molécula más pequeña pase a través. En los casos en los que una sola etapa de separación de impurezas del complejo molde-producto, o separación del producto del molde, no sea lo suficientemente eficiente, pueden emplearse múltiples etapas de filtración secuenciales para aumentar la eficiencia de separación y así generar un producto que cumpla con la pureza deseada.
Es deseable proporcionar un procedimiento para la separación de tales oligonucleótidos que sea eficiente y aplicable a escala de producción industrial. “Therapeutic oligonucleotides: The state of the art in purification technologies” Sanghvi et al. Current Opinion in Drug Discovery (2004) vol. 7 n.° 8 revisa los procedimientos usados para la purificación de oligonucleótidos.
El documento WO 01/55160 A1 da a conocer la purificación de oligonucleótidos mediante la formación de uniones de imina con contaminantes y luego eliminando las impurezas unidas a imina con cromatografía u otras técnicas. “Size Fractionation of DNA Fragments Ranging from 20 to 30000 Base Pairs by Liquid/Liquid chromatography” Muller et al. Eur. J. Biochem (1982) 128-238 da a conocer el uso de una columna sólida de celulosa microcristalina sobre la que se ha depositado una fase de PEG/dextrano para la separación de secuencias de nucleótidos. “Separation and identification of oligonucleotides by hydrophilic interaction chromatography”. Easter et al. The Analyst (2010); 135(10) da a conocer la separación de oligonucleótidos usando una variante de HPLC que emplea una fase de soporte de sílice sólida. “Fractionation of oligonucleotides of yeast soluble ribonucleic acids by countercurrent Distribution” Doctor et al. Biochemistry (1965) 4(1) 49-54 da a conocer el uso de una columna sólida seca rellena con DEAE-celulosa seca. “Oligonucleotide composition of a yeast lysine transfer ribonucleic acid” Madison et al; Biochemistry, 1974, 13 (3) da a conocer el uso de cromatografía en fase sólida para la separación de secuencias de nucleótidos.
La cromatografía líquido-líquido es un método de separación conocido. “Countercurrent Chromatography The Support-Free Liquid Stationary Phase” Billardello, B.; Berthod, A; Wilson & Wilson's Comprehensive Analytical Chemistry 38; Berthod, A., Ed.; Elsevier Science B.V.: Ámsterdam (2002) págs. 177-200 proporciona una descripción general útil de la cromatografía líquido-líquido. Se conocen diversas técnicas de cromatografía líquidolíquido. Una de tales técnicas es la cromatografía en contracorriente líquido-líquido (denominada en el presente documento “CCC”). Otra técnica conocida es la cromatografía de partición centrífuga (denominada en el presente documento “CPC”).
Los métodos dados a conocer anteriormente y los métodos establecidos en el documento WO 2013/030263 pueden usarse para separar un oligonucleótido de producto, por ejemplo del molde y/o una impureza.
Ligasas para su uso en los procedimientos de la invención, es decir, etapa de ligamiento (d)
En una realización de la invención, la ligasa es una ligasa dependiente de ATP. Las ligasas dependientes de ATP oscilan en tamaño entre 30 y >100 kDa. En una realización de la invención, la ligasa es una ligasa dependiente de NAD. Las enzimas dependientes de NAD son altamente homólogas y son proteínas monoméricas de 70-80 kDa. En una realización de la invención, la ligasa es una ligasa termoestable. Una ligasa termoestable puede derivarse de una bacteria termófila.
En una realización de la invención, la ligasa es una ligasa dependiente de molde.
En una realización de la invención, la ligasa es una ADN ligasa de fago CC31 de enterobacterias de tipo natural (SEQ ID NO: 6) o una ADN ligasa de fago Shf125875 de Shigella de tipo natural (SEQ ID NO: 8).
En una realización de la invención, la ligasa es una ligasa modificada. Por ejemplo, una ligasa modificada incluye una ADN ligasa de T4 modificada, una ligasa de fago CC31 de enterobacterias modificada, una ligasa de fago Shf125875 de Shigella modificada y una ligasa de Chlorella modificada.
En una realización, la ADN ligasa de T4 de tipo natural está modificada en la posición de aminoácido 368 o en la posición de aminoácido 371 de SEQ ID NO: 3.
En una realización, la ligasa mutante comprende o consiste en SEQ ID NO: 3 en la que el aminoácido en la posición 368 es R o K.
En una realización, la ligasa mutante comprende o consiste en SEQ ID NO: 3 en la que el aminoácido en la posición 371 es uno cualquiera de los siguientes aminoácidos: L, K, Q, V, P, R.
En una realización, el/los residuo(s) correspondiente(s) dados a conocer anteriormente en relación con la ADN ligasa de T4 están mutados en una cualquiera de la ligasa de fago CC31 de enterobacterias, la ligasa de fago Shf125875 de Shigella y la ligasa de Chlorella. Se dan a conocer regiones conservadas de ADN ligasas en Chem. Rev. 2006, 106, 687-699 y Nucleic Acids Research, 2000, vol. 28, n.° 21, 4051-4058. En una realización, la ligasa está modifica en una región de ligador.
En una realización de la invención, la ligasa comprende o consiste en SEQ ID NO: 23 o una ligasa con al menos un 90 % de identidad de secuencia con la misma, excluyendo una ligasa de tipo natural, por ejemplo ligasa de fago CC31 de enterobacterias.
En una realización de la invención, la ligasa comprende o consiste en una cualquiera de las siguientes secuencias de aminoácidos: SEQ ID NO: 10-28.
En una realización de la invención, la ligasa se inmoviliza, por ejemplo, sobre una perla.
Oligonucleótidos usados como materiales de partida
Los oligonucleótidos usados como material de partida para los procedimientos de la invención se describen en el presente documento como un “conjunto” y anteriormente se proporcionó una definición de los mismos. El conjunto es un grupo no homogéneo de oligonucleótidos. Los oligonucleótidos que forman el conjunto se habrán producido mediante otros métodos de producción de oligonucleótidos, por ejemplo, síntesis enzimática y, por tanto, es probable que contenga impurezas.
El conjunto está compuesto por segmentos de los oligonucleótidos de producto, que luego se unen entre sí mientras se ensamblan en el molde. Cada segmento será un grupo no homogéneo con impurezas de diferentes longitudes y/o residuos incorporados incorrectamente.
Producción de los oligonucleótidos de segmento usando enzimas
1) Ligasa mc, por ejemplo ARN ligasa
La ligasa mc cataliza la adición impulsada por ATP de, por ejemplo, 3’,5’-nucleótido-bisfosfatos, 3’,5’-nucleótidotiofosfatos (por ejemplo 3’,5’-bistiofosfato o 3’-fosfato-5’-tiofosfato o 3’-tiofosfato-5’-fosfato) o 3’5’-nucleótidoditiofosfatos (por ejemplo 3’,5’-bisditiofosfato o 3’-fosfato-5’-ditiofosfato o 3’-ditiofosfato-5’-fosfato) al 3’-OH de un oligonucleótido corto (cebador) de manera independiente del molde. Un experto apreciaría que también pueden usarse difosfatos (o ditiofosfatos) o trifosfatos (u otros oligofosfatos en donde uno o más átomos de oxígeno se han sustituido por azufre) en la posición 3’ del resto de azúcar, aunque los restos fosfato (o tiofosfato) adicionales no se requieren. Puede usarse un dinucleótido, trinucleótido o tetranucleótido modificado de manera equivalente en lugar de los nucleótidos individuales mencionados anteriormente. El cebador de oligonucleótido tiene habitualmente un mínimo de tres nucleótidos de longitud. El oligonucleótido resultante de esta reacción de adición es un nucleótido más largo que el oligonucleótido de partida (o dos, tres o cuatro nucleótidos más largo que el oligonucleótido de partida si se usa un dinucleótido, trinucleótido o tetranucleótido, respectivamente). La nueva posición 3’ está ahora fosforilada. Con el fin de añadir un nucleótido posterior, el fosfato 3’ del oligonucleótido en crecimiento se elimina para generar un 3’-OH mediante hidrólisis. Esta hidrólisis se realiza normalmente usando una enzima fosfatasa tal como se muestra en la figura 1.
2) T ransferasa
Las enzimas desoxinucleotidil transferasa terminal (TdT) catalizan la adición de nucleótido trifosfatos protegidos en 3’, por ejemplo, protegidos mediante un grupo 3’-O-azidometilo, 3’-aminoxilo o 3’-O-alilo, al 3’-OH de un oligonucleótido corto (cebador) de manera independiente del molde. Este cebador de oligonucleótido tiene habitualmente un mínimo de tres nucleótidos de longitud.
Se exponen métodos adecuados, por ejemplo, en los documentos EP2796552, US8808989, WO16128731 A1 y WO16139477 A1.
El oligonucleótido cebador usado en los métodos de ligasa mc y transferasa descritos anteriormente para producir oligonucleótidos de segmento puede:
(1) quedar retenido como parte del oligonucleótido de segmento si se desea, o
(2) escindirse del oligonucleótido de producto para permitir la separación del producto deseado y permitir la posibilidad de reciclaje para producir oligonucleótidos de segmento adicionales. La escisión del cebador del oligonucleótido de segmento puede realizarse usando una nucleasa específica de secuencia y un diseño apropiado del cebador y el segmento de manera que la escisión sea tanto eficaz como precisa.
EJEMPLOS
Abreviaturas
Ome O-Metilo
MOE O-Metoxietilo (estructura principal de ADN) o metoxietilo (estructura principal de ARN) CBD Dominio de unión a celulosa
HPLC Cromatografía de líquidos de alta resolución
PBS Solución salina tamponada con fosfato
HAA Acetato de hexilamonio
SDSPAGE Electroforesis en gel de poliacrilamida con dodecilsulfato de sodio
CLEM Cromatografía de líquidos-espectrometría de masas
PO Fosfodiéster
DTT Ditiotreitol
Ni-NTA Níquel-ácido nitrilotriacético
/3Phos/ Grupo 3’-fosfato
/5Phos/ Grupo 5’-fosfato
(p) Grupo fosfato
PS o D Fosforotioato
/ideoxil/ 2’-DesoxiInosina
5mC 5-Metilcitosina
BSA Albúmina sérica bovina
LNA Ácido nucleico bloqueado
WT Tipo natural
Ejemplo 1: Ensamblaje de segmentos de oligonucleótidos (ADN) y ligamiento con ADN ligasa de T4 de tipo natural
1.1 Síntesis química de secuencias de partida y de control
Con el fin de demostrar que podrían ensamblarse múltiples oligonucleótidos cortos (“segmentos”) en el orden correcto sobre una hebra molde complementaria y ligarse para dar el producto final deseado (“diana”), los segmentos, las secuencias de molde y diana, tal como se detalla en la tabla 1, se sintetizaron químicamente usando métodos convencionales.
1.2 Análisis de HPLC
Se llevó a cabo el análisis de HPLC usando una columna Agilent ZORBAX Eclipse Plus XDB-C18 (4,6 x 150 mm, 5 pm dp. Agilent P/N 993967-902) ejecutándose a 0,2 ml/min al tiempo que se monitorizó la absorbancia a 258 nm. Se mantuvo la columna a 60 °C. Se inyectaron 20 pl de muestra y se ejecutó un gradiente desde el 20-31 % de tampón B a lo largo de 20 minutos antes de aumentarse hasta el 80 % de tampón B durante 5 minutos.
Tampón A: 75 ml de HAA 1 M, 300 ml de alcohol isopropílico, 200 ml de acetonitrilo, 4425 ml de agua Tampón B: 650 ml de alcohol isopropílico, 350 ml de acetonitrilo
Tabla 1

1.3 Ensamblaje de oligonucleótido y método de ligamiento con ADN ligasa de T4 comercial (SEQ ID NO: 3) El segmento 5’, el segmento central y el segmento 3’ se ensamblaron sobre el molde: cada segmento y el molde se disolvieron en agua a una concentración de 1 mg/ml y luego se mezclaron tal como sigue.
Segmento 5’ 2 pl
Segmento central 2 pl
Segmento 3’ 2 pl
Molde biotinilado 2 pl
H2O 36 pl
Se incubó la disolución de oligonucleótidos combinados a 94 °C durante 5 minutos y se enfrió hasta 37 °C antes
de incubarse a 37 °C durante 5 minutos adicionales para permitir que los segmentos se aparearan con el molde. Entonces se añadieron 2 gl (equivalentes a 2 gg) de ADN ligasa de T4 (NEB) y 4 gl de 1 x tampón de ligamiento de ADN de T4 (NEB) y se incubó la reacción (volumen de reacción total 50 gl) a temperatura ambiente durante una hora. Después de esto, se añadieron 40 gl de perlas magnéticas recubiertas con estreptavidina y se incubó la suspensión a temperatura ambiente durante 10 minutos para permitir que el molde biotinilado se uniera a las perlas de estreptavidina. Se lavaron las perlas de estreptavidina con 2 x 100 gl de PBS para eliminar los segmentos no unidos. Se analizó el lavado mediante HPLC. Entonces se incubó la mezcla de reacción a 94 °C durante 10 minutos para separar los productos de ligamiento unidos (o cualquier segmento unido) del molde antes de enfriarse rápidamente sobre hielo para ‘fundir’ el ADN y detener el reapareamiento de los productos de oligonucleótidos (o segmentos) con el molde. Entonces se llevó a cabo el análisis de la reacción de ligamiento mediante HPLC.
1.4 Ensamblaje de oligonucleótidos y método de ligamiento con suspensión de perlas de ADN ligasa de T4 interna
1.4.1 Generación de la suspensión de perlas
Se produjo ligasa de T4 (SEQ ID NO: 4) fusionada en el extremo N-terminal a un dominio de unión a celulosa (CBD) usando métodos de clonación, expresión y extracción convencionales. Esta secuencia de aminoácidos de ligasa de T4 difiere de la secuencia de ligasa de T4 comercial (SEQ ID NO: 3) en que la metionina N-terminal (M) se ha reemplazado por glicina y serina (GS). Estas sustituciones de aminoácidos se realizaron para ayudar en la generación y expresión de la proteína de fusión de CBD. La proteína de fusión de CBD-ligasa de T4 se expresó en células BL21 A1 (INVITROGEN). Se recogió el sobrenadante y se añadió a 600 gl de perlas PERLOz A 100 (PERLOZA) y se agitaron 26 °C durante 1 hora. Entonces se recogieron las perlas de celulosa PERLOZA y se lavaron con 2 ml de tampón (Tris 50 mM pH 8,0, NaCl 200 mM, Tween 20 al 0,1 %, glicerol al 10 %) seguido por 5 ml de PBS y se resuspendieron finalmente en 200 gl de PBS (PO43- 10 mM, NaCl 137 mM, KCl 2,7 mM pH 7,4). Con el fin de analizar la expresión de proteínas, se mezclaron 15 gl de la suspensión de perlas PERLOZA con 5 gl de tampón de carga SDS y se incubaron a 80 °C durante 10 minutos antes de ejecutarse sobre un gel de gradiente de SDS PAGE (4 - 20 %) según un protocolo convencional.
1.4.2 Ensamblaje de oligonucleótidos y ligamiento usando una suspensión de perlas
Para ligasa de T4 unida a perlas PERLOZA, el método de ensamblaje y ligamiento en 1.3 anterior se modificó tal como sigue. En la mezcla de segmentos inicial, 36 gl de H2O se redujeron a 8 gl de H2O. Tras el apareamiento, se reemplazaron 2 gl de ADN ligasa de T4 comercial por 20 gl de suspensión de perlas PERLOZA. Antes de añadir las perlas magnéticas de estreptavidina, se centrifugaron las perlas PERLOZA y se eliminó el sobrenadante. Se añadieron las perlas magnéticas de estreptavidina al sobrenadante y se incubaron a temperatura ambiente durante 10 minutos para permitir que el molde biotinilado se uniera a las perlas de estreptavidina.
1.5 Resultados y conclusiones
Los oligonucleótidos de producto, molde y los tres de segmento se resolvieron claramente en el cromatograma de control.
El análisis de HPLC de las reacciones de ligasa mostró que quedaban algunos segmentos de oligonucleótidos sin ligar, pero la ADN ligasa de T4 comercial (NEB) fue capaz de catalizar el ligamiento de los tres segmentos para generar el oligonucleótido de producto deseado (figura 7). La ADN ligasa de T4 unida a perlas PERLOZA parecía ser menos eficiente en el ligamiento de los segmentos de oligonucleótidos, apareciendo segmentos de oligonucleótidos tanto en la muestra de lavado de control como en la muestra de reacción (figura 8). Sin embargo, es difícil estar seguro de si se añadió la misma cantidad de enzima sobre las perlas en comparación con la enzima comercial, de modo que no fue posible una comparación directa de la eficiencia de ligamiento.
Ejemplo 2: Ensamblaje de segmentos de oligonucleótidos modificados con 2’-OMe ribosa y ligamiento con ADN ligasa de T4 de tipo natural
2.1 2’OMe en cada posición de nucleótido en cada segmento
Con el fin de determinar si la ADN ligasa de T4 era capaz de ligar segmentos de oligonucleótidos con modificación en la posición 2’ del anillo de ribosa, se sintetizaron segmentos de oligonucleótidos con la misma secuencia que para el ejemplo 1, pero la posición 2’ del anillo de ribosa se sustituyó con un grupo OMe grupo y la timidina se reemplazó por uridina tal como se muestra a continuación.
Tabla 2

El ensamblaje, el ligamiento y el análisis de HPLC se llevaron a cabo usando los métodos del ejemplo 1, con tanto ligasa NEB comercial como fusión de ligasa de T4-CBD unida a perlas PERLOZA. La cantidad de agua usada en la mezcla de reacción para el experimento de ADN ligasa de T4 comercial (NEB) fue de 26 gl, en vez de 36 gl, de modo que el volumen de reacción final fue de 40 gl. La cantidad de agua usada en la mezcla de reacción para el experimento de suspensión de perlas de ADN ligasa de T4 interna fue de 23 gl, y la cantidad de perlas usada fue de 5 gl, de modo que el volumen de reacción final fue también de 40 gl. Se ejecutaron en paralelo experimentos de control usando ADN no modificado en contraposición a 2’-OMe ADN.
Los resultados de los experimentos de control fueron según el ejemplo 1. No se detectó producto usando HPLC para los experimentos de 2’-OMe, lo que indica que la ADN ligasa de T4 es incapaz de ligar segmentos de oligonucleótidos completamente modificados con 2’-OMe independientemente de si se usó una ADN ligasa de T4 comercial o una fusión de ADN ligasa de T4 interna-CBD unida a perlas PERLOZA.
2.2 2’-OMe en cada posición de nucleótido en un solo segmento
Usando una disolución de 1 mg/ml de cada oligonucleótido, se establecieron las reacciones tal como se detalla en la tabla 3.
Tabla 3

El ensamblaje y el ligamiento se llevaron a cabo usando los métodos del ejemplo 1 con ligasa NEB comercial y ADN ligasa de T4 unida a PERLOZA interna.
Las reacciones se incubaron a 94 °C durante 5 minutos, seguido por incubación durante 5 minutos a 37 °C para permitir el apareamiento. Se añadieron 4 gl de 1x tampón de ligamiento de ADN de T4 NEB a cada reacción junto con 5 gl (aproximadamente 2 gg) de ADN ligasa de T4 interna o 2 gl (aproximadamente 2 gg) e ADN ligasa de T4 comercial (aparte del experimento 1 que era un control sin ligasa) y se permitió que la reacción de ligamiento prosiguiera durante 2 horas a temperatura ambiente. Entonces se añadieron perlas magnéticas de estreptavidina a cada reacción y se calentaron las reacciones hasta 94 °C antes de enfriar rápidamente sobre hielo tal como se describe en el ejemplo 1 para separar el molde de los materiales y productos de partida.
Las reacciones procesadas se dividieron en dos: la mitad se analizaron mediante HPLC tal como se describió para el ejemplo 1 (sección 1.2). La otra mitad de la muestra se retuvo para espectrometría de masas para confirmar los resultados de HPLC.
El ligamiento de los segmentos de oligonucleótidos sin modificar (experimento 5) prosiguió tal como se esperaba
para producir el producto de longitud completa. Se observó una pequeña cantidad de ligamiento cuando el segmento 5’ estaba sustituido con 2’-OMe (experimento 3) tal como se muestra en la figura 9, pero no se observó ligamiento cuando el segmento 3’ estaba sustituido con 2’-OMe (experimento 2). No se observó producto significativo cuando los tres segmentos estaban sustituidos con 2’-OMe (experimento 4) según 2.1.
2.3 Conclusión
ADN ligasa de T4 de tipo natural produce un ligamiento deficiente de segmentos de oligonucleótidos sustituidos con 2’-OMe, pero es ligeramente menos sensible a la modificación del segmento de oligonucleótido 5’ que el segmento 3’.
Ejemplo 3: Ensamblaje de segmentos de oligonucleótidos modificados con 2’-OMe ribosa y ligamiento con ligasas de tipo natural y mutantes
3.1 Materiales
Se produjeron ligasa de fago CC31 de enterobacterias de tipo natural (SEQ ID NO: 6), ligasa de fago Shf125875 de Shigella de tipo natural (SEQ ID NO: 8) y 10 ligasas de T4 mutantes de SEQ ID NO: 10-19, cada una fusionada en el extremo N-terminal a un CBD, usando métodos de clonación, expresión y extracción convencionales. Tal como se da a conocer en 1.4.1, con el fin de generar y expresar las proteínas de fusión de CBD, la metionina N-terminal (M) se reemplazó por glicina y serina (GS) en cada caso (por ejemplo SEQ ID NO: 7 para ligasa de fago CC31 de enterobacterias y SEQ ID n O: 9 para ligasa de fago Shf125875 de Shigella).
Se sintetizaron los siguientes oligonucleótidos mediante métodos en fase sólida convencionales.
Tabla 4

3.2 Ensamblaje de oligonucleótidos y método de ligamiento con suspensiones de perlas de ligasa 3.2.1 Generación de la suspensión de perlas
Se unieron ligasas fusionadas a CBD a perlas PERLOZA tal como se describió en 1.4 para generar una suspensión de perlas.
3.2.2 Ensamblaje de oligonucleótidos y ligamiento usando una suspensión de perlas
Se prepararon reacciones de ligamiento con los componentes a continuación hasta un volumen final de 50 pl en una placa de 96 pocillos:
2 pl segmento 5’ (2’-OMe) ~1 mg/ml
2 pl segmento central ~1 mg/ml
2 pl segmento 3’ ~1 mg/ml
2 pl molde ~1 mg/ml
5 pl tampón de ADN ligasa de T4 NEB
22 pl H2O
15 pl suspensión de perlas unidas PERLOZA
Se incubó la reacción durante 15 minutos a temperatura ambiente antes de la adición de la suspensión de perlas PERLOZA para permitir que los segmentos se aparearan con el molde. Se añadió suspensión de perlas PERLOZA
y se incubó la reacción a temperatura ambiente durante 1 hora. Después de una hora de incubación, se transfirió la disolución a una placa de filtro ACOPREP advance 350 (PN 8082) y se colocó la placa de filtro encima de una superplaca ABGENE (Thermo Scientific, n.° AB-2800) y se centrifugó durante 10 minutos a 4.000 rpm para eliminar la suspensión de perlas PERLOZA. Entonces se analizaron las disoluciones mediante HPLC usando el método descrito en el ejemplo 1 (sección 1.2).
Cada ensamblaje y ligamiento de oligonucleótidos se repitió 6 veces para cada ligasa.
3.3 Resultados y conclusiones
La ligasa de fago CC31 de enterobacterias de tipo natural (SEQ ID NO: 6) y la ligasa de fago Shf125875 de Shigella de tipo natural (SEQ ID NO: 8) son capaces de ligar un segmento 5’ sustituido con 2’OMe que contiene cinco nucleobases 2’-OMe y una desoxinucleobase con un segmento que contiene solo ADN no modificado. Además, mientras que la ADN ligasa de T4 de tipo natural (SEQ ID NO: 3 y 4) es deficiente en la realización de esta reacción, tal como se muestra en el ejemplo 2 y se reconfirma en este ejemplo, varias mutaciones en las posiciones 368 y 371 confieren la capacidad de ligar un segmento 5’ sustituido con 2’-OMe que contiene cinco nucleobases 2’ OMe y una desoxinucleobase con un segmento que contiene solo ADN no modificado en la ligasa (SEQ ID NO: 10-19).
Ejemplo 4: Ensamblaje de segmentos de oligonucleótidos modificados con 2’ MOE ribosa y modificados con 5-metilpirimidina y ligamiento con ADN ligasas mutantes
4.1 Materiales
Se sintetizaron segmentos de oligonucleótidos modificados tal como se establece en la tabla 5 a continuación mediante métodos basados en fase sólida convencionales.
Tabla 5

Se fusionaron ADN ligasas mutantes (SEQ ID NO: 20-28) basadas en ligasa de fago CC31 de enterobacterias de tipo natural, ligasa de T4 de tipo natural y ligasa de fago Shf125875 de Shigella de tipo natural cada una en el extremo N-terminal a un dominio de unión a celulosa (CBD), usando métodos de clonación, expresión y extracción convencionales. Las ligasas fusionadas a CBD se unieron a perlas PERLOZA tal como se describió en 1.4 para generar una suspensión de perlas. Con el fin de liberar las ligasas de las perlas PERLOZA, se añadieron 2 gl de proteasa TEV a la suspensión y se incubaron durante la noche at 4 °C. La proteína escindida, que carece ahora del dominio de unión a celulosa, se recogió mediante centrifugación durante 10 min a 4000 rpm.
4.2 Método
Se estableció la reacción tal como sigue:
Segmento central 20 gM final
Segmento 3’ MOE 20 gM final
Segmento 5’ MOE 20 gM final
Molde 20 gM final
Tampón de ADN ligasa de T4 NEB 5 gl
ADN ligasa mutante 15 gl
H2O Para preparar el volumen de reacción final de 50 gl
Se mezclaron todos los componentes y se agitaron con vórtex antes de la adición de la ADN ligasa. Se incubaron las reacciones durante 1 hora a 35 °C. Después de 1 hora, se detuvieron las reacciones calentando a 95 °C durante 5 minutos en un bloque de PCR.
Se analizaron las muestras mediante tanto HPLC como CLEM para confirmar la identidad del producto según el
protocolo de HPLC usado en el ejemplo 1 (sección 1.2). Se incluyeron controles de ADN ligasa de T4 NEB comercial y un control negativo (H2O en lugar de cualquier ligasa).
4.3 Resultados y conclusiones
La figura 10 muestra los trazos de HPLC para la reacción de control y la reacción catalizada por SEQ ID NO: 23 (clon A4 - ligasa de fago CC31 de enterobacterias mutante). El producto y el molde se eluyen conjuntamente usando este método de HPLC de modo que el producto aparece como un aumento en el área de pico del pico del producto molde. En el trazo de la ligasa mutante (b) no solo aumenta el pico del producto molde, sino que aparecen dos nuevos picos a los 10,3 y 11,2 minutos. Estos picos corresponden al ligamiento del segmento central con o bien el segmento 5’ MOE o bien el segmento 3’ MOE. Además, los picos del segmento de entrada son sustancialmente más pequeños que el control en línea con el aumento de los picos intermedios y de producto. El trazo de la ligasa de T4 comercial NEB (a) mostró un ligero aumento en el área de pico para el molde producto, una pequeña cantidad de producto de ligamiento intermedio junto con una disminución concomitante en segmentos de oligonucleótidos de entrada. La ligasa mutante (SEQ ID NO: 23), sin embargo, mostró un área de pico de producto molde sustancialmente mayor y una reducción concomitante en las áreas de pico para segmentos de oligonucleótidos de entrada. Por tanto, la ligasa mutante (SEQ ID NO: 23) es una ligasa mucho más eficaz para los segmentos sustituidos con 2’ MOE que la ADN ligasa de T4 comercial. Se mostraron mejoras similares para las otras ligasas mutantes (SEQ ID NO: 20, 21,24-28).
Ejemplo 5: Efecto de un apareamiento de nucleótidos diferente en el sitio de ligamiento
5.1 Materiales
Se produjo ligasa de fago CC31 de enterobacterias mutante (SEQ ID NO: 23) fusionada en el extremo N-terminal con un dominio de unión a celulosa (CBD) usando métodos de clonación, expresión y extracción convencionales. La proteína de fusión de CBD-ligasa de fago CC31 de enterobacterias mutante extraída se añadió a 25 ml de perlas de celulosa PERLOZA 100 (PERLOZA) y se agitó a 20 °C durante 1 hora. Entonces, se recogieron las perlas PERLOZA y lavaron con 250 ml de tampón (Tris 50 mM pH 8,0, NaCl 200 mM, Tween 20 al 0,1 %, glicerol al 10 %) seguido por 250 ml de PBS y finalmente se resuspendieron en 10 ml de PBS (PO43 10 mM, NaCl 137 mM, KCl 2,7 mM pH 7,4). Con el fin de analizar la expresión de proteínas, se mezclaron 15 ml de la suspensión de perlas PERLOZA con 5 ml de tampón de carga SDS y se incubaron a 80 °C durante 10 minutos antes de procesarlos en un gel de gradiente de SDS PAGE (4 - 20 %) según un protocolo convencional. Para la liberación de la ligasa de las perlas, se añadieron 70 ml de proteasa TEV y se incubaron durante la noche a 4 °C con agitación. La ligasa se recogió lavando las perlas digeridas con 80 ml de PBS. Entonces, se concentró la ligasa hasta 1,2 ml usando un filtro Amicon de Mc O de 30 Kd.
Los siguientes oligonucleótidos molde de ADN biotinilados (tabla 6) y oligonucleótidos de segmento de ADN (tabla 7) se sintetizaron mediante métodos en fase sólida convencionales. Obsérvese que los nucleótidos en negrita son los presentes en el sitio de ligamiento (es decir, los nucleótidos que se unieron en la reacción de ligamiento - tabla 7; y los nucleótidos que son complementarios a los que se unieron por medio de la reacción de ligamiento - tabla 6).
Tabla 6:


Tabla 7

N.B. Obsérvese que, a diferencia de los ejemplos previos, las reacciones de ligamiento en este ejemplo implican la unión de dos segmentos entre sí: un segmento 5’ y un segmento 3’, es decir, no hay segmento central.
5.2 Método
Se establecieron las reacciones tal como sigue:
Tabla 8

Por cada 50 gl de reacción:
Segmento 3’ (disolución madre 1 mM, 20 gM final) 1 gl
Segmento 5’ (disolución madre 1 mM, 20 gM final) 1 gl
Molde (disolución madre 1 mM, 20 gM final) 1 gl
Tampón de ADN ligasa NEB (para ligasa de T4) 5 pl
ADN ligasa de CC31 mutante (disolución madre 0,45 mM, 90 pM final) 10 pl
H2O hasta 50 pl
Se incubó cada mezcla de reacción a 35 °C durante tanto 30 minutos como 1 hora. Cada reacción se terminó calentando a 95 °C durante 5 minutos. Se llevó a cabo el análisis de HPLC.
5.3 Resultados y conclusiones
Todas las reacciones produjeron un pico de producto después de 1 hora de incubación en el análisis de HPLC. Por consiguiente, el método de ligamiento funciona para todas las combinaciones de nucleótidos en las uniones que van a unirse. Es posible una optimización para mejorar el rendimiento del producto, pero no fue necesario ya que los resultados fueron concluyentes y quedó claro que la reacción funcionaba para todas las combinaciones de nucleótidos en las uniones que iban a unirse.
Ejemplo 6: Efecto de diferentes modificaciones en el sitio de ligamiento
6.1 Materiales
Se produjo ligasa de fago CC31 de enterobacterias mutante (SEQ ID NO: 23) tal como se describió en 5.1 y se adquirió ADN ligasa de virus de Chioreiia (SEQ ID NO: 29, comercialmente disponible como ligasa SplintR, NEB). Se sintetizaron los siguientes oligonucleótidos de segmento y oligonucleótido molde biotinilado (tabla 9) mediante métodos en fase sólida convencionales.
Tabla 9

6.2 Método
Se establecieron las reacciones tal como sigue:
Tabla 10


Por cada 50 gl de reacción:
Segmento 3’ (1 mM, disolución madre, 20 gM final) 1 gl
Segmento central (1 mM, disolución madre, 20 gM final) 1 gl
Segmento 5’ (1 mM, disolución madre, 20 gM final) 1 gl
Molde (1 mM, disolución madre, 20 gM final) 1 gl
H2O hasta 50 gl
Para ligasa de fago CC31 de enterobacterias mutante (SEQ ID NO: 23)
Tampón de ADN ligasa (Tris-HCI 50 mM, DTT 1 mM) 5 gl
ADN ligasa de CC31 mutante (0,45 mM) 10 gl MnCl2 (50 mM) 5 gl
ATP (10 mM) 10 gl
Mientras que para la ADN ligasa de virus de Chioreiia (SEQ ID NO: 29, comercialmente disponible como ligasa SplintR, NEB)
Tampón de ADN ligasa NEB (para Chioreiia) 5 gl
ADN ligasa de virus de Chioreiia 2 gl
Se incubó cada mezcla de reacción a 20 °C 1 hora. Se terminó cada reacción calentando a 95 °C durante 10 minutos. Se llevó a cabo análisis de HPLC usando el método del ejemplo 1.
6.3 Resultados y conclusiones
Todas las reacciones produjeron un pico de producto en el análisis de HPLC. Por consiguiente, el método de ligamiento funciona para todas las combinaciones de modificaciones sometidas a prueba en las uniones que van a unirse. Es posible una optimización para mejorar el producto, pero no fue necesario ya que los resultados fueron concluyentes y quedó claro que la reacción funcionaba para todas las combinaciones de modificaciones sometidas a prueba en las uniones que iban a unirse.
Ejemplo 7: Capacidad para usar diferentes números de segmentos para construir oligonucleótidos más grandes
7.1 Materiales
Se produjo ligasa de fago CC31 de enterobacterias mutante (SEQ ID NO: 23) tal como se describió en 5.1. Se sintetizaron los siguientes oligonucleótidos de segmento de ADN y oligonucleótidos de ADN molde biotinilados (tabla 11) mediante métodos en fase sólida convencionales.
Tabla 11

7.2 Método
Se establecieron las reacciones tal como sigue:
Tabla 12

Se ejecutaron las reacciones en solución salina tamponada con fosfato, pH = 7,04 en un volumen total de 100 gl y se establecieron tal como sigue:
Molde (20 gM final)
Cada segmento (20 gM final)
MgCl2 (10 mM final)
ATP (100 gM final)
ADN ligasa de CC31 mutante (25 gM final)
Se incubó cada reacción at 28 °C durante la noche antes de terminarse calentando a 94 °C durante 1 minuto. Se analizaron los productos mediante HPLC-espec. de masas.
7.3 Resultados y conclusiones
La reacción usando 4 segmentos produjo un producto completamente ligado de 27 pares de bases de longitud. La reacción usando 5 segmentos produjo un producto de 33 pares de bases de longitud. En ambos casos, la masa observada del producto estaba de acuerdo con lo esperado para la secuencia deseada. En conclusión, claramente es posible ensamblar múltiples segmentos para generar oligonucleótidos de la longitud y secuencia deseadas tal como se define mediante la secuencia molde complementaria apropiada.
Ejemplo 8: Ensamblaje y ligamiento de 5-10-5 segmentos para formar un gapmero, en el que los segmentos 5’ y 3’ comprenden (i) modificaciones de azúcar de 2’-OMe ribosa, (ii) uniones fosforotioato o (iii) modificaciones de azúcar de 2’-OMe ribosa y uniones fosforotioato; y en los que el segmento central es ADN no modificado
8.1 Materiales
Se produjo ligasa de fago CC31 de enterobacterias mutante (SEQ ID NO: 23) tal como se describió en 5.1, los siguientes oligonucleótidos de segmento y oligonucleótido de ADN molde biotinilado (tabla 13) se sintetizaron mediante métodos en fase sólida convencionales.
Tabla 13

8.2 Método
Se establecieron las reacciones tal como sigue:
Tabla 14

Cada una de las reacciones 1,2 y 3 se estableció en 100 gl de volumen final en solución salina tamponada con fosfato con los siguientes componentes:
Segmento 3’ 20 gM final
Segmento central 20 gM final
Segmento 5’ 20 gM final
Molde 20 gM final
MgCl2 10 mM final
ATP 50 gM final
Enzima 25 gM final
Se incubó cada mezcla de reacción a 20 °C durante la noche. Se terminó cada reacción calentando a 95 °C durante 10 minutos. Se llevó a cabo análisis de HPLC-espec. de masas.
8.3 Resultados y conclusiones
Se produjo oligonucleótido de producto correspondiente al ligamiento satisfactorio de los tres fragmentos en las tres reacciones.
Por consiguiente, la ligasa de fago CC31 de enterobacterias mutante (SEQ ID NO: 23) es capaz de ligar 3 segmentos entre sí formando un ‘gapmero’ donde las ‘alas’ 5’ y 3’ tienen una estructura principal de fosforotioato, mientras que la región central tiene una estructura principal de fosfodiéster, y todos los residuos de azúcar en el gapmero son residuos de desoxirribosa. La ligasa de fago CC31 de enterobacterias (SEQ ID NO: 23) es también capaz de ligar 3 segmentos entre sí formando un ‘gapmero’ donde las ‘alas’ 5’ y 3’ tienen residuos de 2’
metoxirribosa (2’-OMe), mientras que la región central tiene residuos de desoxirribosa, y todas las uniones son uniones fosfodiéster. Finalmente, la ligasa de fago CC31 de enterobacterias (SEQ ID NO: 23) es capaz de ligar 3 segmentos entre sí formando un ‘gapmero’ donde las ‘alas’ 5’ y 3’ tienen las modificaciones combinadas (una estructura principal e fosforotioato y residuos de 2’-metoxirribosa), mientras que la región central tiene residuos de desoxirribosa y uniones fosfodiéster.
Ejemplo 9: Ensamblaje y ligamiento de segmentos que comprenden ácidos nucleicos bloqueados (LNA) 9.1 Materiales
Se produjo ligasa de fago CC31 de enterobacterias mutante (SEQ ID NO: 23) tal como se describió en 5.1. Se produjo una ligasa dependiente de NAD de Staphylococcus aureus mutante (NAD-14) tal como se describe en 13.1.
Se sintetizaron los siguientes oligonucleótidos de segmento y oligonucleótido de ADN molde biotinilado (tabla 15) mediante métodos en fase sólida convencionales.
Tabla 15

9.2 Método
Se establecieron las reacciones tal como sigue:
Volumen de reacción de 100 pl
Molde 20 pM final
Enzima 25 pM final
Todos los segmentos de oligonucleótidos 20 pM final
Se establecieron las reacciones variando la enzima (ligasa de fago CC31 de enterobacterias mutante (SEQ ID NO: 23) o NAD-14), el catión divalente (Mg2+ o Mn2+) y las combinaciones de segmentos de oligonucleótidos tal como se establece en la tabla 16.
Tabla 16

Se incubó cada mezcla de reacción a 28 °C durante la noche. Se terminó cada reacción calentando a 94 °C durante 1 minuto. Se llevó a cabo análisis de HPLC-espec. de masas.
9.3 Resultados y conclusiones
Se produjo el oligonucleótido de producto en reacciones de reacción de control (oligonucleótidos no modificados solo) y donde se incluyó un solo ácido nucleico bloqueado en un segmento en la unión de ligamiento independientemente de si estaba en el lado 3’ o 5’ de la unión. Cuando se incluyeron ácidos nucleicos bloqueados en ambos lados (oligo 1 oligo 2), no se detectó producto. Los datos fueron similares para ambas enzimas e independientemente de si se usó Mg2+ o Mn2+.
Podrían llevarse a cabo mutaciones enzimáticas y/o cribados de selección para identificar una enzima capaz de ligar segmentos con un ácido nucleico bloqueado tanto en el lado 3’ como 5’ de la unión.
Ejemplo 10: Ensamblaje y ligamiento de tres segmentos (7-6-7) para formar un gapmero en el que los segmentos 5’ y 3’ comprenden modificaciones de azúcar de 2’ MOE ribosa y todas las uniones son uniones fosforotioato, usando una variante de ligasa de fago CC31 de enterobacterias en presencia de Mg2+ o Mn2+ 10.1 Formación de enlaces fosforotioato
Con el fin de determinar si una ligasa de fago CC31 de enterobacterias mutante (SEQ ID NO: 23) era capaz de ligar segmentos de oligonucleótidos modificados con una estructura principal de fosforotioato, modificaciones de azúcar de 2’ MOE ribosa y bases de pirimidina 5-metiladas, se realizaron reacciones usando los segmentos de oligonucleótidos mostrados en la tabla 15. Las reacciones se realizaron en presencia de iones Mg2+ y Mn2+.
10.2 Materiales
Se sintetizaron químicamente oligonucleótidos usando métodos convencionales tal como se muestra a continuación:
Tabla 17

N.B. La molécula de 2’MOE PS diana producida mediante ligamiento de los segmentos de la tabla 17, cuando se hibrida con el molde biotinilado mostrado en la tabla 17, es:
5’-mGDmGDmCDmCDmADdADdADdCDdCDdTDdCDdGDdGDdCDdTDmUDmADmCDmCDm ll-3 ’ (SEQ ID NO: 1) Se preparó ligasa de fago CC31 de enterobacterias mutante purificada (SEQ ID NO: 23) tal como se describió en el ejemplo 5.1. Se llevó a cabo el análisis de HPLC.
10.3 Ensamblaje de oligonucleótidos y método de ligamiento con variante de ligasa de fago CC31 de enterobacterias (SEQ ID NO: 23)
Se prepararon las reacciones tal como sigue:
Reacción con MgCl2
10 x tampón de ADN ligasa de T4 (NEB)* 5 pl
Molde Concentración final 20 pM
Segmento 5’ 2’ MOE PS Concentración final 20 pM
Segmento central PS Concentración final 20 pM
Segmento 3’ 2’ MOE PS Concentración final 20 gM
Ligasa (24,3 gM) 10 gl
Agua hasta 50 gl
*1 x tampón contiene Tris-HCI 50 mM, MgCl210 mM, ATP 1 mM, DTT 10 mM, pH 7,5
Reacción con MnCl2
10 x tampón de ligasa* 5 gl
ATP (10 mM) 5 gl
MnCl2 (50 mM) 5 gl
Molde Concentración final 20 gM
Segmento 5’ 2’ MOE PS Concentración final 20 gM
Segmento central PS Concentración final 20 gM
Segmento 3’ 2’ MOE PS Concentración final 20 gM
Ligasa (24,3 gM) 10 gl
Agua hasta 50 gl
*1 x tampón contiene Tris-HCl 50 mM, DTT 10 mM, pH 7,5
Las reacciones finales contenían 20 gM de cada segmento y molde, MgCl25 mM o MnCl25 mM, ATP 1 mM, Tris-HCl 50 mM, DTT 10 mM, pH 7,5 y ligasa 4,9 gM. Se prepararon reacciones adicionales que no contenían enzima y sirvieron como control negativo. Se incubaron las reacciones durante 16 horas a 25 °C y luego se extinguieron calentando hasta 95 °C durante 5 minutos. Se clarificaron las proteínas precipitadas mediante centrifugación y se analizaron las muestras mediante HPLC.
10.4 Resultados y conclusión
Los oligonucleótidos de producto, molde y segmento se resolvieron claramente en el cromatograma de control y no se observó ligamiento. Las reacciones de ligasa realizadas en presencia de MgCl25 mM condujeron a la formación de un producto intermedio formado a partir del ligamiento del segmento 5’ y los segmentos centrales, pero no se detectó producto de longitud completa. Las reacciones de ligasa realizadas en presencia de MnCl2 produjeron tanto el producto de longitud completa como el producto intermedio (producto intermedio de segmento 5’ más segmento central). Ambas reacciones de ligasa mostraron que quedaban segmentos de oligonucleótidos sin ligar. Sin embargo, es posible una optimización del protocolo con el fin de maximizar el rendimiento del producto.
Ejemplo 11: Ensamblaje y ligamiento de tres segmentos (7-6-7) para formar un gapmero en el que los segmentos 5’ y 3’ comprenden modificaciones de azúcar de 2’ MOE ribosa y todas las uniones son uniones fosforotioato, usando ADN ligasa de virus de Chlorella de tipo natural en presencia de Mg2+ nativo
11.1 Materiales
Con el fin de determinar si la ADN ligasa de virus de Chlorella (SEQ ID NO: 29, comercialmente disponible como ligasa SplintR, NEB) era capaz de ligar segmentos de oligonucleótidos modificados con una estructura principal de fosforotioato, se realizaron modificaciones de azúcar de 2’ MOE y reacciones con bases de pirimidina 5-metiladas usando los segmentos de oligonucleótidos mostrados en el ejemplo 10.2, tabla 17. Se realizaron las reacciones a 25 °C, 30 °C y 37 °C para investigar el efecto de la temperatura sobre la actividad enzimática.
11.2 Ensamblaje de oligonucleótidos y método de ligamiento con ADN ligasa de virus de Chlorella comercial (SEQ ID NO: 29)
Cada segmento de oligonucleótido y molde se disolvieron en agua libre de nucleasa tal como se detalla a continuación:
Molde biotinilado 249,6 ng/gl
Segmento 5’ 2’ MOE PS 182,0 ng/gl
Segmento central PS 534,0 ng/gl
Segmento 3’ 2’ MOE PS 531,0 ng/gl
Se prepararon las reacciones tal como sigue:
10 x tampón (NEB)* 6 pl
Molde 3,8 pl
Segmento 5’ 2’ MOE PS 18,1 pl
Segmento central PS 4,8 pl
Segmento 3’ 2’ MOE PS 6,3 pl
Agua 15 pl
Ligasa SplintR (25 U/pl) 6 pl
*1 x tampón contiene Tris-HCl 50 mM, MgCl210 mM, ATP 1 mM, DTT 10 mM, pH 7,5
Las reacciones finales contenían 20 pM de cada segmento y molde, Tris-HCI 50 mM, MgCl210 mM, ATP 1 mM, DTT 10 mM, pH 7,5 y ligasa 2,5 U/pl. Se incubaron las reacciones a 25 °C, 30 °C y 37 °C. Se prepararon reacciones adicionales que no contenían enzima y sirvieron como control negativo. Tras 16 horas de incubación, se extinguieron las reacciones calentando hasta 95 °C durante 10 minutos. Se clarificaron las proteínas precipitadas mediante centrifugación y se analizaron las muestras mediante HPLC.
11.3 Resultados y conclusión
Los oligonucleótidos de producto, molde y segmento se resolvieron claramente en el cromatograma de control y no se observó ligamiento. El análisis de HPLC de las reacciones de ligasa mostró que quedaban segmentos de oligonucleótidos sin ligar, pero la ADN ligasa de virus de Chioreiia fue capaz de ligar satisfactoriamente los segmentos. La actividad de la ligasa aumentó con el aumento de la temperatura. A 25 °C, la ADN ligasa de virus de Chioreiia fue capaz de ligar satisfactoriamente el segmento 5’ y el segmento central pero no se observó producto de longitud completa. A 30 °C y 37 °C, se detectó producto de longitud completa además del producto intermedio formado a partir del segmento 5’ y el segmento central.
Ejemplo 12: Cribado de un panel de 15 ATP y NAD ligasas para determinar la actividad hacia el ligamiento de tres segmentos (7-6-7) para formar un gapmero en el que los segmentos 5’ y 3’ comprenden modificaciones de azúcar de 2’ MOE ribosa y todas las uniones son uniones fosforotioato
12.1 Materiales
Se fusionaron ligasas dependientes de ATP y NAD de tipo natural descritas en las tablas 18 y 19 cada una en el extremo N-terminal con un CBD. Se sintetizaron genes, se clonaron en pET28a y se expresaron en E. coii BL21(DE3) usando métodos de clonación, expresión y extracción convencionales.
Tabla 18

Tabla 19


Se unieron fusiones de CBD-ligasa a perlas PERLOZA tal como se describió en 1.4 con las siguientes modificaciones. Se hicieron crecer proteínas de fusión de CBD-ligasa a partir de una sola colonia de células BL21(DE3) (NEB) y se hicieron crecer en un cultivo de expresión de 50 ml. Se recogieron las células mediante centrifugación, se resuspendieron en 5-10 ml de Tris-HCI (50 mM, pH 7,5) y se lisaron mediante sonicación. Se clarificó el lisado mediante centrifugación y se añadió 1 ml de perlas PERLOZA 100 (PERLOZA) (suspensión al 50 %, preequilibrada con Tris-HCI 50 mM pH 7,5) al sobrenadante que se agitó a 20 °C durante 1 hora. Entonces se recogieron las perlas de celulosa PERLOZA y se lavaron con 30 ml de tampón (Tris 50 mM pH 8,0, NaCl 200 mM, Tween 20 al 0,1 %, glicerol al 10 %) seguido por 10 ml de Tris-HCl (50 mM, pH 7,5) y se resuspendieron finalmente en 1 ml de Tris-HCl (50 mM, pH 7,5). Con el fin de analizar la expresión de proteínas, se mezclaron 20 gl de la suspensión de perlas PERLOZA con 20 gl de tampón de carga SDS y se incubaron a 95 °C durante 5 minutos antes de ejecutarse sobre un gel de gradiente de SDS PAGE (4 - 20 %) según un protocolo convencional.
12.2 Método de ensamblaje y ligamiento de oligonucleótidos modificados con 2’ MOE y fosforotioato
Se usaron segmentos de oligonucleótidos modificados con una estructura principal de fosforotioato, modificaciones de azúcar de 2’ MOE ribosa y bases de pirimidina 5-metiladas que se muestran en el ejemplo 10.2, tabla 17. Cada segmento de oligonucleótido y molde se disolvió en agua libre de nucleasa tal como se detalla a continuación:
Molde biotinilado 1500 ng/gl
Segmento 5’ 2’ MOE PS 1008 ng/gl
Segmento central PS 725 ng/gl
Segmento 3’ 2’ MOE PS 1112 ng/gl
La mezcla de ensayo de ATP se preparó tal como sigue:
Molde 85,6 gl
Segmento 5’ 2’ MOE PS 43,5 gl
Segmento central PS 46,8 gl
Segmento 3’ 2’ MOE PS 40,1 gl
DTT 1 M 8 gl
MgCl21 M 4 gl
ATP (50 mM) 16 gl
Tris 0,5 M 80 gl
Agua 476 gl
La mezcla de ensayo de NAD se preparó tal como sigue:
Molde 85,6 gl
Segmento 5’ 2’ MOE PS 43,5 gl
Segmento central PS 46,8 gl
Segmento 3’ 2’ MOE PS 40,1 gl
DTT 1 M 8 gl
MgCl21 M 4 gl
NAD (50 mM) 1,6 gl
Tris 0,5 M 80 gl
Agua 490,4 gl
Cada proteína inmovilizada (40 pl, suspensión de perlas PERLOZA al 50 %) se pipeteó a un tubo de PCR. Se sedimentaron las perlas mediante centrifugación y se eliminó el sobrenadante mediante pipeteo. Se añadió mezcla de ensayo (40 pl) a cada reacción (las reacciones finales contenían 20 pM de cada segmento y molde, Tris-HCl 50 mM, MgCl210 mM, ATP 1 mM o NAD 100 pM, DTT 10 mM, pH 7,5 y 40 pl de ligasa sobre perlas p Er LOZA). Una reacción que no contenía proteína sirvió como control negativo. Se incubaron las reacciones durante 18 horas a 30 °C y luego se extinguieron calentando hasta 95 °C durante 10 minutos. Se clarificaron las proteínas precipitadas mediante centrifugación y se analizaron las muestras mediante HPLC.
12.3 Resultados y conclusión
Los oligonucleótidos de producto, molde y segmento se resolvieron claramente en el cromatograma de control y no se observó ligamiento. El análisis de HPLC de las reacciones de ligasa mostró que todas las proteínas catalizaron el ligamiento satisfactorio del segmento 5’ y el segmento central para formar un producto intermedio, pero solo algunas ligasas catalizaron el ligamiento de los tres segmentos para producir el producto de longitud completa tal como se describe en la tabla 20. La ligasa dependiente de NAD de Staphylococcus aureus (SaNAD, SEQ ID NO: 61) produjo el producto de longitud más completa. Es posible una optimización para mejorar el rendimiento del producto y está dentro del conjunto de habilidades del experto en la técnica.
Tabla 20
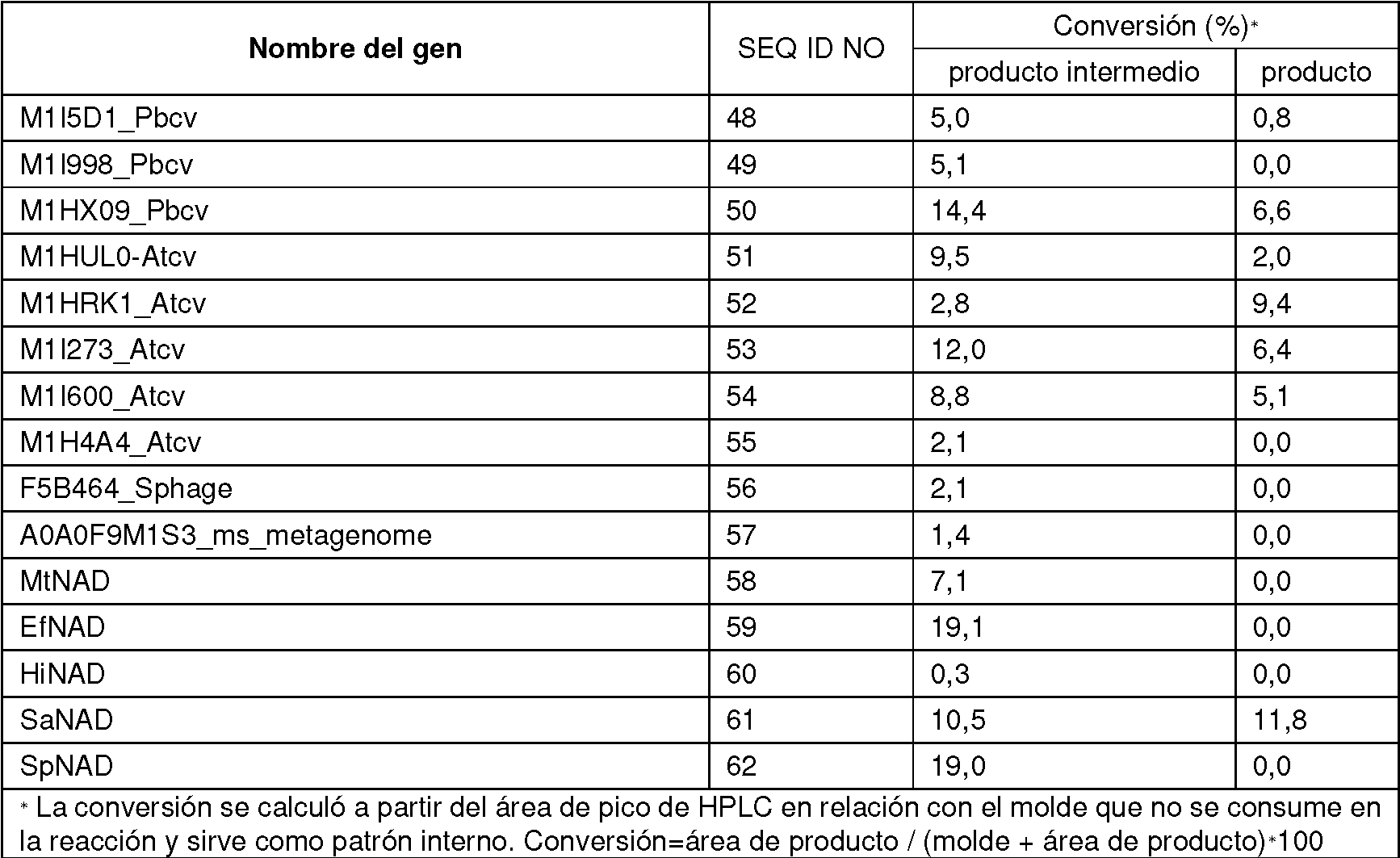
Ejemplo 13: Reacción de ligamiento semicontinuo
13.1 Materiales
Se produjo una ligasa de Staphylococcus aureus mutante (NAD-14) fusionada en el extremo N-terminal a un CBD usando métodos de clonación, expresión y extracción convencionales. Entonces se unió la CBD-ligasa mutante NAD-14 a perlas PERLOZA: se añadieron 50 ml de lisado proteico a 7,5 ml de perlas PERLOZA, se incubaron a temperatura ambiente durante 1 hora y luego se recogieron las perlas en una columna de vidrio (BioRad Econo-Column, 10 cm de longitud, 2,5 cm de diámetro, n.° 7372512). Se lavaron las perlas con 200 ml de tampón Y (Tris8 50 mM, NaCl 500 mM, Tween 20 al 0,1 %, glicerol al 10 %), luego con 200 ml de tampón Z (Tris8 50 mM, NaCl 200 mM, Tween 20 al 0,1 %, glicerol al 10 %) y 200 ml de PBS. La concentración estimada de ligasa NAD-14 mutante sobre las perlas era de 69 pM de ligasa por ml de perlas.
Se sintetizaron los siguientes oligonucleótidos de segmento y oligonucleótido de ADN molde (tabla 21) mediante métodos en fase sólida convencionales.
Tabla 21

Se produjo un “núcleo de tres moldes” (aproximadamente 24 kDa) que comprende un material de soporte denominado “núcleo” y tres secuencias de molde (figura 11). Cada copia del molde de unión covalentemente, en su propio punto de unión individual, al “núcleo”. La molécula de “núcleo de tres moldes” tiene un peso molecular mayor que el oligonucleótido diana (producto) (complementario al 100 % con la secuencia de molde), permitiendo de ese modo que se retenga cuando se separan las impurezas y los productos de la mezcla de reacción. Debe observarse que, en este caso particular, la secuencia de molde era SEQ ID NO: 30 y se unieron tres copias al núcleo. En el siguiente ejemplo, también se produce un núcleo de tres moldes, pero con una secuencia de molde diferente. Por consiguiente, puesto que la secuencia de molde varía entre los ejemplos, también lo hace el núcleo de tres moldes.
Se preparó la siguiente mezcla de reacción (volumen total de 5 ml):
250 pl KH2PO41 M, pH 7,5 (50 mM final)
108 pl Segmento central 0,07011 M (1,5 mM final)
137 pl Segmento 3’ 0,05481 M (1,5 mM final)
168 pl Segmento 5’ 0,04461 M (1,5 mM final)
750 pl Núcleo (molde) 0,00387 M (0,55 mM final)
350 pl NAD+ 50 mM (3,5 mM final)
1000 pl MgCl250 mM (10 mM final)
2237 pl H2O libre de nucleasa
13.2 Métodos
Se estableció un sistema semicontinuo tal como se muestra en la figura 12.
Se empaquetaron 4 ml de perlas PERLOZA y ligasa NAD-14 mutante inmovilizada en una columna Pharmacia XK16 (B). Se usó un baño de agua y una bomba peristáltica (C) para mantener la temperatura de la columna, usando el compartimiento de agua de la columna, a 30 °C. Se equilibraron las perlas procesando 120 ml (30x el volumen de la columna) de tampón que contenía KH2PO450 mM a pH 7,5 durante 120 minutos a 1 ml/min. Se usó una bomba AKTA explorer A1 (A) para crear el flujo a través de la columna Pharmacia XK16.
Tras el equilibrado de la columna, se cargaron sobre la columna la mezcla de reacción de 5 ml (bien mezclada mediante agitación con vórtex), se recogió en el tubo de depósito (D) y se recirculó a través de la columna usando la bomba AKTA explorer A1. Se recirculó la mezcla de reacción a través del sistema a una velocidad de flujo de 1 ml/min en modo de circulación continua durante 16 horas. Se recogieron muestras después de 30 minutos, 60 minutos, 90 minutos, 4 horas, 5 horas, 6 horas, 7 horas, 14 horas y 16 horas para el análisis de HPLC.
13.3 Resultados y conclusiones
Tabla 22
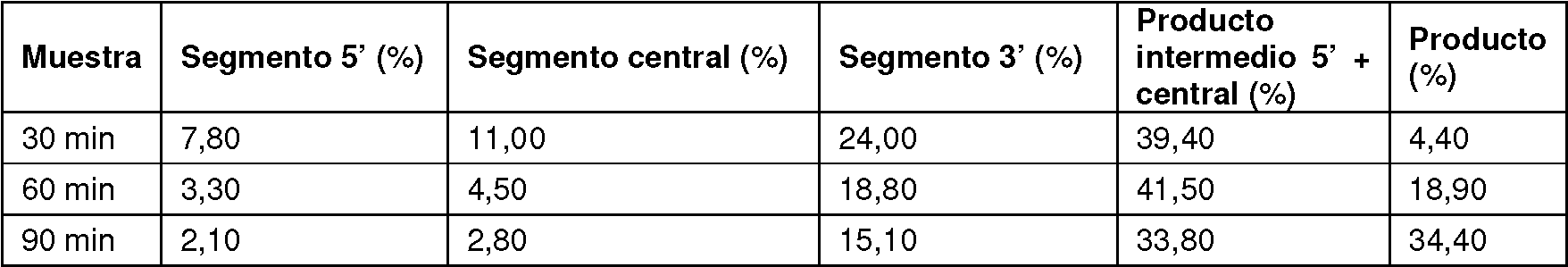

El porcentaje de cada segmento, producto intermedio y producto se expresa como área de pico fraccionaria en relación con el área de pico del núcleo de tres moldes.
En conclusión, la reacción de flujo semicontinuo funcionó y, después de 16 horas, la reacción casi se completó.
Ejemplo 14: Separación de oligonucleótidos de diferentes tamaños mediante filtración: a) separación de un oligonucleótido de 20 meros (SEQ ID NO: 1) y un núcleo que comprende tres oligonucleótidos de 20 meros no complementarios (SEQ ID NO: 30); (b) separación de un oligonucleótido de 20 meros (SEQ ID NO: 1) de oligonucleótidos de segmento de 6 meros y 8 meros (véase la tabla 1) y un núcleo que comprende tres oligonucleótidos de 20 meros complementarios (SEQ ID NO: 2)
14.1 Materiales
Todos los oligonucleótidos usados se sintetizaron mediante métodos en fase sólida convencionales.
Se usó un núcleo de tres moldes, tal como se describió en 13.1 (figura 11).
Se usó una variedad de filtros de puntos de corte de peso molecular variables y de diferentes fabricantes tal como se muestra en las tablas 23 y 24.
14.2 Métodos
14.2.1 Configuración de filtración sin salida y protocolo para el examen de membranas poliméricas: (protocolo 1)
Se estableció una plataforma de filtración sin salida tal como se muestra en la figura 13 que comprende una celda de filtración sin salida MET colocada en un baño de agua que se sitúa sobre un agitador magnético y una placa caliente. La presión dentro de la célula se proporcionó a partir de un flujo de nitrógeno.
La muestra de membrana (14 cm2) que va a someterse a prueba se cortó en primer lugar al tamaño apropiado y se colocó en la celda. La membrana se acondicionó en primer lugar con agua de calidad para HPLC (200 ml) y luego con tampón PBS (200 ml). Entonces se despresurizó la celda, se eliminó la disolución de PBS restante y se reemplazó por una disolución que contenía oligonucleótidos (40 ml de oligonucleótidos en PBS a una concentración de 1 g/l). Se colocó la celda sobre una placa agitadora caliente y se calentó la disolución hasta la temperatura deseada al tiempo que se agitaba usando agitación magnética. Se aplicó presión a la celda (con un objetivo de aproximadamente 3,0 bar; la presión real se registró en cada caso). La agitación de la disolución o bien se detuvo o bien continuó y se recogió disolución de permeado (aproximadamente 20 ml) y se analizó mediante HPLC. Se registró el flujo. Entonces se despresurizó el sistema para permitir el muestreo y el análisis por HPLC de la disolución de material retenido. Entonces se añadió más tampón PBS (20 ml) a la celda de filtración y se repitió el procedimiento anterior 3 veces. Finalmente, se lavó la membrana con tampón PBS.
Se analizaron todas las muestras mediante HPLC sin ninguna dilución.
14.2.2 Configuración de filtración de flujo cruzado y protocolo para el examen de membranas poliméricas: (protocolo 2)
Se estableció una plataforma de filtración de flujo cruzado tal como se muestra en la figura 14. El recipiente de alimentación (1) que consistía en un matraz cónico contenía la disolución de oligonucleótidos que iba a purificarse. Se bombeó la disolución hasta una celda de filtración de flujo cruzado (4) usando una bomba de HPLC (2) al tiempo que se mantuvo la temperatura dentro de la celda usando una placa caliente (3). Se recirculó la disolución dentro de la celda usando una bomba de engranajes (6 ). Un manómetro (5) permitió que se leyera la presión durante el experimento. Se tomaron muestras de la disolución de material retenido a partir de la válvula de muestreo (7) al tiempo que se muestreó la disolución de permeado a partir del recipiente de recogida de permeado (8 ).
La muestra de membrana que va a someterse a prueba se cortó en primer lugar al tamaño apropiado y se colocó
en la celda. Se lavó el sistema con una disolución de PBS (100 ml). Se ajustó la temperatura de la disolución al punto de ajuste deseado. Se alimentó una disolución que contenía productos de oligonucleótidos en PBS (7,5 ml a 1 g/l) al sistema. Entonces se bombeó la disolución de PBS al sistema usando la bomba e HPLC a una velocidad de flujo que coincidía con la velocidad de flujo de la disolución de permeado (normalmente 3 ml/min). Se registró la presión usando el manómetro. Se muestreó la disolución de material retenido para el análisis de HPLC cada 5 volúmenes de diafiltración. Se muestreó la disolución de permeado para el análisis de HPLC cada volumen de diafiltración. Se detuvo el experimento después de 20 volúmenes de diafiltración.
En el caso del experimento que usó la membrana Snyder que tenía un corte de peso molecular de 5 kDa (número de lote 120915R2) y SEQ iD NO: 46 y SEQ ID NO: 30, se modificó la metodología anterior tal como sigue. La muestra de membrana que va a someterse a prueba se cortó en primer lugar al tamaño apropiado y se colocó en la célula. Se lavó el sistema con una disolución de fosfato de potasio (100 ml, 50 mM, pH 7,5). Se ajustó la temperatura de la disolución al punto de ajuste deseado. Se añadió una disolución que contenía productos de oligonucleótidos en fosfato de potasio (aproximadamente 1 g/l) a ácido etilendiaminatetraacético (EDTA) (230 pl de una disolución 500 mM). Entonces se alimentó la disolución al sistema. Luego se bombeó tampón fosfato de potasio al sistema usando la bomba de HPLC a una velocidad de flujo que coincidía con la velocidad de flujo de la disolución de permeado (normalmente 4 ml/min). Se registró la presión usando el manómetro. Se muestreó la disolución de material retenido para el análisis de HPLC cada 5 volúmenes de diafiltración. Se muestreó la disolución de permeado para el análisis de HPLC cada volumen de diafiltración. Se detuvo el experimento después de 15 volúmenes de diafiltración
14.3 Resultados
Tabla 23: Resultados de los experimentos de filtración sin salida siguiendo el protocolo 1 (14.2.1)

En el experimento que usó la membrana NADIR de MWCO de 10 kDa a 60 °C, se demostró una clara separación entre la secuencia de producto (SEQ ID NO: 1) y el núcleo de tres moldes no complementario (que comprende SEQ ID NO: 30). La figura 15 muestra a) un cromatograma de la disolución de material retenido, que permaneció en la celda de filtración y contenía principalmente núcleo de tres moldes, después de dos volúmenes de diafiltración; y b) un cromatograma del permeado, disolución enriquecida en el producto, después de dos volúmenes de diafiltración.

En el experimento que usó la membrana Snyder de MWCO de 5 kDa a 50 °C y 3,0 bar de presión, se demostró una clara separación entre las secuencias de segmentos (véase la tabla 1) y el núcleo de tres moldes complementario (que comprende SEQ ID NO: 2) y el producto (SEQ ID NO: 1). La figura 16 muestra a) un cromatograma de la disolución de material retenido, que contenía principalmente núcleo de tres moldes y producto, después de 20 volúmenes de diafiltración; y b) un cromatograma del permeado, que contenía principalmente oligonucleótidos de segmento, después de 20 volúmenes de diafiltración.
En el experimento que usó la membrana Snyder de MWCO de 5 kDa a 80 °C y 3,1 bar de presión, se demostró una clara separación entre el núcleo de tres moldes complementario (que comprende SEQ ID NO: 2) y el producto (SEQ ID NO: 1). La figura 17 muestra a) un cromatograma de la disolución de material retenido, que contenía núcleo de tres moldes solo, después de 20 volúmenes de diafiltración; y b) un cromatograma de la disolución de permeado, que contenía el producto solo, después de 2 volúmenes de diafiltración.
14.4 Conclusiones
Pueden separarse oligonucleótidos de diferentes longitudes y pesos moleculares usando filtración. Tal como se mostró anteriormente, el tipo de membrana y las condiciones, tales como la temperatura, afectan al nivel de separación. Para un conjunto dado de oligonucleótidos de diferentes longitudes/pesos moleculares, pueden seleccionarse membranas y condiciones adecuadas para permitir la separación requerida. Por ejemplo, se ha demostrado que oligonucleótidos de segmento (meros cortos de 6 y 8 nucleótidos de longitud), tal como se explica resumidamente en la tabla 1 , pueden separarse del oligonucleótido de producto (oligonucleótido de 20 meros que tiene SEQ ID NO: 1) y el núcleo de tres moldes (que comprende 3 x 20 meros de SEQ ID NO: 2 unido a un soporte sólido), y el oligonucleótido de producto y el núcleo de tres moldes, a su vez, pueden separarse entre sí.
Ejemplo 15: Extensión 3’ por ligamiento de una sola base para la síntesis de oligonucleótidos con modificación de base de 2’-OMe y uniones fosfato
Se fusionaron ligasas mc de tipo natural (SEQ ID NO: 63 a 83) cada una en el extremo N-terminal a una etiqueta de histidina que consistía en 6 xHis. Se sintetizaron genes, se clonaron en pET28a y se produjo proteína que codifica el gen en E. coli BL21(DE3) Star usando métodos de clonación, expresión y extracción convencionales. Se purificaron proteínas usando Ni-NTA usando métodos convencionales y se usaron directamente.
Se establecieron las reacciones según la tabla 25 a continuación, con adición de ligasa en último lugar. Se incubaron las reacciones a 25 °C durante 20 horas. Entonces se extinguieron las reacciones calentando hasta 95 °C durante 15 min, se centrifugaron a 4000 x g durante 15 min y se transfirieron 10 pl de sobrenadante a un vial de HPLC. Se analizaron entonces las reacciones mediante HPLC para detectar la presencia de SEQ ID NO: 85.
Tabla 25

Tabla 26: Resultados de las reacciones:


Ejemplo 16: Extensión 3’ mediante ligamiento de una sola base para la síntesis de oligonucleótidos con modificación de base de 2’-OMe y uniones fosforotioato
Se fusionaron ligasas mc de tipo natural (SEQ ID NO: 63 a 83) cada una en el extremo N-terminal a una etiqueta de histidina que consistía en 6 xHis. Se sintetizaron genes, se clonaron en pET28a y se produjo proteína que codifica el gen en E. coli BL21(DE3) Star usando métodos de clonación, expresión y extracción convencionales. Se purificaron proteínas usando Ni-NTA usando métodos convencionales y se usaron directamente.
Se establecieron las reacciones según la tabla 27 a continuación, con adición de ligasa en último lugar. Se incubaron las reacciones a 25 °C durante 20 horas. Entonces se extinguieron las reacciones calentando hasta 95 °C durante 15 min, se centrifugaron a 4000 x g durante 15 min y se transfirieron 10 gl de sobrenadante a un vial de HPLC. Se analizaron entonces las reacciones mediante HPLC para detectar la presencia de SEQ ID NO: 85.
Tabla 27

Tabla 28: Resultados de las reacciones:


Ejemplo 17: Extensión 3’ mediante ligamiento de una sola base para la síntesis de oligonucleótidos con modificación de base de 2’MOE y uniones fosforotioato
Se fusionaron ligasas mc de tipo natural (SEQ ID NO: 63 a 83) cada una en el extremo N-terminal a una etiqueta de histidina que consistía en 6 xHis. Se sintetizaron genes, se clonaron en pET28a y se produjo proteína que codifica el gen en E. coli BL21(DE3) Star usando métodos de clonación, expresión y extracción convencionales. Se purificaron proteínas usando Ni-NTA usando métodos convencionales y se usaron directamente.
Se establecieron las reacciones según la tabla 29 a continuación, con adición de ligasa en último lugar. Se incubaron las reacciones a 25 °C durante 20 horas. Entonces se extinguieron las reacciones calentando hasta 95 °C durante 15 min, se centrifugaron a 4000 x g durante 15 min y se transfirieron 10 gl de sobrenadante a un vial de HPLC. Se analizaron entonces las reacciones mediante HPLC para detectar la presencia de SEQ ID NO: 87.
Tabla 29

Tabla 30: Resultados de las reacciones:


Ejemplo 18: Extensión 3’ mediante ligamiento de una sola base para la síntesis de oligonucleótidos con modificación de base de 2’H y uniones fosfato
Se fusionaron ligasas mc de tipo natural (SEQ ID NO: 63 a 83) cada una en el extremo N-terminal a una etiqueta de histidina que consistía en 6 xHis. Se sintetizaron genes, se clonaron pET28a y se produjo proteína que codifica el gen en E. coli BL21(DE3) Star usando métodos de clonación, expresión y extracción convencionales. Se purificaron proteínas usando Ni-NTA usando métodos convencionales y se usaron directamente.
Se establecieron las reacciones según la tabla 31 a continuación, con adición de ligasa en último lugar. Se incubaron las reacciones a 25 °C durante 20 horas. Entonces se extinguieron las reacciones calentando hasta 95 °C durante 15 min, se centrifugaron a 4000 x g durante 15 min y se transfirieron 10 gl de sobrenadante a un vial de HPLC. Se analizaron entonces las reacciones mediante HPLC para detectar la presencia de SEQ ID NO: 87.
Tabla 31

Tabla 32: Resultados de las reacciones:


Ejemplo 19: Extensión 3’ mediante ligamiento de una sola base para la síntesis de oligonucleótidos con modificación de base de 2’MOE y uniones fosfato
Se fusionaron ligasas mc de tipo natural (SEQ ID NO: 63 a 83) cada una en el extremo N-terminal a 6 xHis. Se sintetizaron genes, se clonaron en pET28a y se produjo proteína que codifica el gen en E. coli BL21(DE3) Star usando métodos de clonación, expresión y extracción convencionales. Se purificaron proteínas usando Ni-NTA usando métodos convencionales y se usaron directamente.
Se establecieron las reacciones según la tabla 33, con adición de ligasa en último lugar. Se incubaron las reacciones a 25 °C durante 20 horas. Entonces se extinguieron las reacciones calentando hasta 95 °C durante 15 min, se centrifugaron a 4000 x g durante 15 min y se transfirieron 10 gl de sobrenadante a un vial de HPLC. Se analizaron entonces las reacciones mediante HPLC para detectar la presencia de SEQ ID NO: 87.
Tabla 33

Tabla 34: Resultados de las reacciones:


Ejemplo 20: Optimización de la extensión 3’ mediante ligamiento de una sola base para la síntesis de oligonucleótidos con modificación de base de 2’OMe y uniones fosfato
Se fusionó ligasa mc de tipo natural (SEQ ID NO: 67) en el extremo N-terminal a 6 xHis. Se sintetizó el gen, se clonó en pET28a y se produjo proteína que codifica el gen en E. co liBL21(DE3) Star usando métodos de clonación, expresión y extracción convencionales. Se purificó la proteína usando Ni-NTA usando métodos convencionales y se usó directamente. Se determinó la concentración de proteína por medio de electroforesis capilar microfluídica. Se establecieron las reacciones según la tabla 35, con adición de ligasa en último lugar. Se incubaron las reacciones a 25 °C durante 18 horas. Entonces se extinguieron las reacciones calentando hasta 95 °C durante 15 min, se centrifugaron a 4000 x g durante 15 min y se transfirieron 10 gl de sobrenadante a un vial de HPLC. Se analizaron entonces las reacciones mediante HPLC para detectar la presencia de SEQ ID NO: 85.
Tabla 35

Tabla 36: Resultados de la reacción:

Ejemplo 21: Optimización de la extensión 3’ mediante ligamiento de una sola base para la síntesis de oligonucleótidos con modificación de base de 2’MOE y uniones fosfato
Se fusionó ligasa mc de tipo natural (SEQ ID NO: 67) en el extremo N-terminal a 6 xHis. Se sintetizó el gen, se clonó en pET28a y se produjo proteína que codifica el gen en E. co liBL21(DE3) Star usando métodos de clonación, expresión y extracción convencionales. Se purificó la proteína usando Ni-NTA usando métodos convencionales y
se usó directamente. Se determinó la concentración de proteína por medio de electroforesis capilar microfluídica.
Se establecieron las reacciones según la tabla 37, con adición de ligasa en último lugar. Se incubaron las reacciones a 25 °C durante 12 horas. Entonces se extinguieron las reacciones calentando hasta 95 °C durante 15 min, se centrifugaron a 4000 x g durante 15 min y se transfirieron 10 pl de sobrenadante a un vial de HPLC. Se analizaron entonces las reacciones mediante HPLC para detectar la presencia de SEQ ID NO: 87.
Tabla 37

Tabla 38: Resultados de la reacción:

Ejemplo 22: Reacciones que comparan WT con mutantes - extensión 3’ mediante ligamiento de una sola base para la síntesis de oligonucleótidos con modificación de base de 2’MOE y uniones fosfato
Se fusionaron ligasas mc de tipo natural o mutantes (SEQ ID NO: 67 y 88 a 92) cada una en el extremo N-terminal a 6 xHis. Se sintetizaron genes, se clonaron en pET28a y se produjo proteína que codifica el gen en E. coli BL21(DE3) Star usando métodos de clonación, expresión y extracción convencionales. Se purificaron proteínas usando Ni-NTA usando métodos convencionales y se usaron directamente. Se determinó la concentración de proteína por medio de electroforesis capilar microfluídica.
Se establecieron las reacciones según la tabla 39, con adición de ligasa en último lugar. Se incubaron las reacciones a 25 °C durante 24 horas. Entonces se extinguieron las reacciones calentando hasta 70 °C durante 30 min, se centrifugaron a 4000 x g durante 15 min y se transfirieron 10 pl de sobrenadante a un vial de HPLC. Se analizaron entonces las reacciones mediante HPLC para detectar la presencia de SEQ ID NO: 87.
Tabla 39

Tabla 40: Resultados de las reacciones:
Conversión (%) en producto* Ligasa de fago IME13 de Stenotrophomonas - ‘RNA8 ' 67 18,34

Ejemplo 23: Reacciones que comparan WT con mutantes - extensión 3’ mediante ligamiento de una sola base para la síntesis de oligonucleótidos con modificación de base de 2’MOE y uniones fosforotioato Se fusionaron ligasas mc de tipo natural o mutantes (SEQ ID NO: 67 y 88 a 92) cada una en el extremo N-terminal a 6 xHis. Se sintetizaron genes, se clonaron en pET28a y se produjo proteína que codifica el gen en E. coli BL21(DE3) Star usando métodos de clonación, expresión y extracción convencionales. Se purificaron proteínas usando Ni-NTA usando métodos convencionales y se usaron directamente. Se determinó la concentración de proteína por medio de electroforesis capilar microfluídica.
Se establecieron las reacciones según la tabla 41, con adición de ligasa en último lugar. Se incubaron las reacciones a 25 °C durante 24 horas. Entonces se extinguieron las reacciones calentando hasta 70 °C durante 30 min, se centrifugaron a 4000 x g durante 15 min y se transfirieron 10 gl de sobrenadante a un vial de HPLC. Se analizaron entonces las reacciones mediante HPLC para detectar la presencia de SEQ ID NO: 87.
Tabla 41
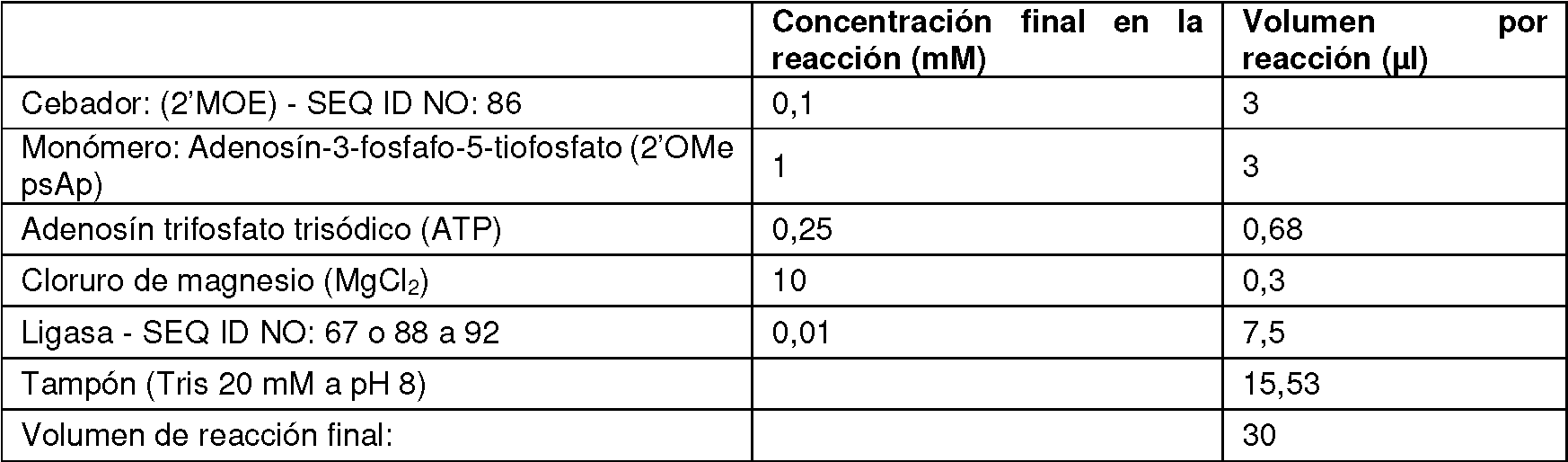
Tabla 42: Resultados de las reacciones:

Ejemplo 24: Extensión 3’ mediante ligamiento de una sola base para la síntesis de oligonucleótidos con modificación de base de 2’MOE y uniones fosforotioato - adenosina
Se fusionó ligasa mc mutante (SEQ ID NO: 88 ) en el extremo N-terminal a 6 xHis. Se sintetizó el gen, se clonó en
pET28a y se produjo proteína que codifica el gen en E. coli BL21(DE3) Star usando métodos de clonación, expresión y extracción convencionales. Se purificaron proteínas usando Ni-NTA usando métodos convencionales y se usaron directamente. Se determinó la concentración de proteína por medio de electroforesis capilar microfluídica.
Se establecieron las reacciones según la tabla 43, con adición de ligasa en último lugar. Se incubaron las reacciones a 25 °C durante 24 horas. Entonces se extinguieron las reacciones calentando hasta 70 °C durante 30 min, se centrifugaron a 4000 x g durante 15 min y se transfirieron 10 gl de sobrenadante a un vial de HPLC. Se analizaron entonces las reacciones mediante HPLC y CLEM para detectar la presencia de SEQ ID NO: 87.
Tabla 43

Tabla 44: Resultados de la reacción:

Ejemplo 25: Extensión 3’ mediante ligamiento de una sola base para la síntesis de oligonucleótidos con modificación de base de 2’MOE y uniones fosforotioato - 5-metilcitidina
Se fusionó ligasa mc mutante (SEQ ID NO: 88 ) en el extremo N-terminal a 6 xHis. Se sintetizó el gen, se clonó en pET28a y se produjo proteína que codifica el gen en E. coli BL21(DE3) Star usando métodos de clonación, expresión y extracción convencionales. Se purificó la proteína usando Ni-NTA usando métodos convencionales y se usó directamente. Se determinó la concentración de proteína por medio de electroforesis capilar microfluídica. Se establecieron las reacciones según la tabla 45, con adición de ligasa en último lugar. Se incubaron las reacciones a 25 °C durante 24 horas. Entonces se extinguieron las reacciones calentando hasta 70 °C durante 30 min, se centrifugaron a 4000 x g durante 15 min y se transfirieron 10 gl de sobrenadante a un vial de HPLC. Se analizaron entonces las reacciones mediante HPLC y CLEM para detectar la presencia de SEQ ID NO: 93.
Tabla 45

Tabla 46: Resultados de la reacción:

Ejemplo 26: Extensión 3’ mediante ligamiento de una sola base para la síntesis de oligonucleótidos con modificación de base de 2’MOE y uniones fosforotioato - timidina
Se fusionó ligasa mc mutante (SEQ ID NO: 88 ) en el extremo N-terminal a 6 xHis. Se sintetizó el gen, se clonó en pET28a y se produjo proteína que codifica el gen en E. coli BL21(DE3) Star usando métodos de clonación, expresión y extracción convencionales. Se purificó la proteína usando Ni-NTA usando métodos convencionales y se usó directamente. Se determinó la concentración de proteína por medio de electroforesis capilar microfluídica.
Se establecieron las reacciones según la tabla 47, con adición de ligasa en último lugar. Se incubaron las reacciones at 25 °C durante 24 horas. Entonces se extinguieron las reacciones calentando hasta 70 °C durante 30 min, se centrifugaron a 4000 x g durante 15 min y se transfirieron 10 pl de sobrenadante a un vial de HPLC. Se analizaron entonces las reacciones mediante HPLC y CLEM para detectar la presencia de SEQ ID NO: 94.
Tabla 47

Tabla 48: Resultados de la reacción:

Ejemplo 27: Extensión 3’ mediante ligamiento de una sola base para la síntesis de oligonucleótidos con modificación de base de 2’MOE y uniones fosforotioato, seguido por defosforilación en 3’ y un segundo ligamiento de una sola base y segunda defosforilación - secuencia de reacción.
Se fusionaron ligasa mc mutante (SEQ ID NO: 88 ) y fosfatasa de tipo natural (SEQ ID NO: 95) en el extremo N-terminal a 6 xHis. Se sintetizaron genes, se clonaron en pET28a y se produjo proteína que codifica el gen en E. coli BL21(DE3) Star usando métodos de clonación, expresión y extracción convencionales. Se purificaron proteínas usando Ni-NTA usando métodos convencionales y se usaron directamente. Se determinó la concentración de proteína por medio de electroforesis capilar microfluídica.
Se estableció la primera reacción de ligamiento según la tabla 49, con adición de ligasa en último lugar. Se incubaron las reacciones a 25 °C durante 24 horas. Entonces se extinguieron las reacciones calentando hasta 70 °C durante 30 min, se centrifugaron a 4000 x g durante 15 min y se transfirieron 10 pl de sobrenadante a un vial de HPLC. Se analizaron entonces las reacciones mediante HPLC y CLEM para detectar la presencia de SEQ ID NO: 87. El resto de las reacciones se purificaron mediante cromatografía de intercambio iónico, se liofilizaron y se usaron directamente en la primera desprotección (es decir desfosforilación).
Tabla 49

Tabla 50: Resultados de la primera reacción de ligamiento:

Se estableció la primera reacción de desprotección/desfosforilación según la tabla 51, con adición de fosfatasa en último lugar. Se incubaron las reacciones a 37 °C durante 24 horas. Entonces se extinguieron las reacciones calentando hasta 70 °C durante 30 min, se centrifugaron a 4000 x g durante 15 min y se transfirieron 10 gl de sobrenadante a un vial de HPLC. Se analizaron entonces las reacciones mediante HPLC y CLEM para detectar la presencia de SEQ ID NO: 96. El resto de las reacciones se purificaron mediante cromatografía de intercambio iónico, se liofilizaron y se usaron directamente en la segunda reacción de ligamiento.
Tabla 51

Tabla 52: Resultados de la primera reacción de desfosforilación:

Se estableció la segunda reacción de ligamiento según la tabla 53, con adición de ligasa en último lugar. Se incubaron las reacciones a 25 °C durante 24 horas. Entonces se extinguieron las reacciones calentando hasta 70 °C durante 30 min, se centrifugaron a 4000 x g durante 15 min y se transfirieron 10 gl de sobrenadante a un vial de HPLC. Se analizaron entonces las reacciones mediante HPLC y CLEM para detectar la presencia de SEQ ID NO: 96. El resto de las reacciones se purificaron mediante cromatografía de intercambio iónico, se liofilizaron y se usaron directamente en la segunda reacción de desprotección/desfosforilación.
Tabla 53


Tabla 54: Resultados de la segunda reacción de ligamiento:

Se estableció la segunda reacción de desprotección/desfosforilación según la tabla 55, con adición de fosfatasa en último lugar. Se incubaron las reacciones a 37 °C durante 24 horas. Entonces se extinguieron las reacciones calentando hasta 70 °C durante 30 min, se centrifugaron a 4000 x g durante 15 min y se transfirieron 10 gl de sobrenadante a un vial de HPLC. Se analizaron entonces las reacciones mediante HPLC y CLEM para detectar la presencia de SEQ ID NO: 98.
Tabla 55

Tabla 56: Resultados de la segunda reacción de desfosforilación:

Ejemplo 28: Escisión específica de la cadena mediante endonucleasa para la liberación de la secuencia de oligonucleótido de segmento o de producto que tiene modificaciones de base de 2’MOE y uniones fosforotioato
Se fusionó endonucleasa de tipo natural (SEQ ID NO: 100) en el extremo N-terminal a CBD. Se sintetizaron genes, se clonaron en pET28a y se produjo proteína que codifica el gen en E. coli BL21(DE3) A1 usando métodos de clonación, expresión y extracción convencionales. Se purificaron las proteínas mediante unión a celulosa en perlas seguido por escisión con proteasa TEV usando métodos convencionales y se usaron directamente.
Se estableció la reacción según la tabla 58, con adición de endonucleasa en último lugar. Se incubó la reacción a 65 °C durante 16 horas. Entonces se extinguió la reacción calentando hasta 95 °C durante 5 min, se centrifugó a 4000 x g durante 15 min y se transfirieron 10 gl de sobrenadante a un vial de HPLC. Se analizó entonces la reacción mediante HPLC y CLEM para detectar la presencia de SEQ ID NO: 101 y ACTIA (modificaciones específicas establecidas en la tabla 57).
Tabla 57

Claims (24)
1. Procedimiento para producir un producto de oligonucleótido terapéutico monocatenario que tiene al menos un residuo de nucleótido modificado, en el que la modificación se selecciona del grupo que consiste en modificación en la posición 2’ del resto de azúcar, modificación de la nucleobase y modificación de la estructura principal, y en el que el producto tiene 20-30 nucleótidos de longitud, que comprende: a) proporcionar un conjunto de oligonucleótidos (I) que comprende segmentos de la secuencia del producto, en el que al menos un segmento de la secuencia del producto contiene al menos un residuo de nucleótido modificado, en el que la modificación se selecciona del grupo que consiste en modificación en la posición 2’ del resto de azúcar, modificación de la nucleobase y modificación de la estructura principal, y en el que cada segmento se ha producido mediante síntesis enzimática;
b) proporcionar un oligonucleótido molde (II) complementario a la secuencia del producto de oligonucleótido monocatenario, teniendo dicho molde una propiedad que permite que se separe del producto y se recicle para su uso en futuras reacciones;
c) poner en contacto (I) y (II) en condiciones para permitir el apareamiento de los oligonucleótidos de segmento con el oligonucleótido molde;
d) unir los oligonucleótidos de segmento usando una ligasa para formar el producto;
e) cambiar las condiciones para desnaturalizar el molde apareado y cualquier hebra de oligonucleótido de impureza, y separar las impurezas;
f) cambiar las condiciones para desnaturalizar el molde apareado y las hebras de oligonucleótidos de producto, y separar el producto de oligonucleótido monocatenario; y
g) reciclar el oligonucleótido molde para su uso en futuras reacciones.
2. Procedimiento según la reivindicación 1, en el que uno o más o todos los oligonucleótidos de segmento se producen usando una ligasa monocatenaria (ligasa mc), opcionalmente una ARN ligasa.
3. Procedimiento según la reivindicación 2, en el que el procedimiento para producir un oligonucleótido de segmento usando una ligasa mc comprende:
(i) añadir un nucleótido, un dinucleótido, un trinucleótido o un tetranucleótido; que tiene un resto o bien fosfato, tiofosfato o bien ditiofosfato en el extremo 3’; y un resto o bien fosfato, tiofosfato o bien ditiofosfato en el extremo 5’; al 3’-OH de un cebador de oligonucleótido monocatenario usando una ligasa mc; (ii) eliminar el 3’-fosfato, 3’-tiofosfato o ditiofosfato usando una fosfatasa;
(iii) repetir las etapas (i) y (ii) para extender el oligonucleótido para producir la secuencia de segmento; y (iv) liberar el oligonucleótido de segmento del cebador de oligonucleótido usando una endonucleasa específica de secuencia.
4. Procedimiento según la reivindicación 3, en el que se añade un 3’,5’-nucleótido-bisfosfato, que tiene uno o más de cualquiera de los oxígenos del fosfato sustituidos por azufre, al 3’-OH de un cebador de oligonucleótido monocatenario usando una ligasa mc en la etapa (i).
5. Procedimiento según la reivindicación 3 o 4, en el que se añade un 3’,5’-bistiofosfato, un 3’-fosfato-5’-tiofosfato, un 3’-tiofosfato-5’-fosfato, un 3’,5’ bisditiofosfato, un 3’-fosfato-5’-ditiofosfato o un 3’-ditiofosfato-5’-fosfato al 3’-OH de un cebador de oligonucleótido monocatenario usando una ligasa mc en la etapa (i).
6. Procedimiento según cualquier reivindicación anterior, en el que uno o más o todos los oligonucleótidos de segmento se producen usando una transferasa.
7. Procedimiento según la reivindicación 6, en el que el procedimiento para producir un oligonucleótido de segmento usando una transferasa comprende:
(i) añadir un nucleótido-5’-trifosfato, alfa-tiotrifosfato o alfaditiotrifosfato, que tiene un grupo protector en su 3’-OH, al 3’-OH de un cebador de oligonucleótido monocatenario, usando una transferasa;
(ii) desproteger la posición 3’ para regenerar el 3’-OH;
(iii) repetir las etapas (i) y (ii) para extender el oligonucleótido para producir la secuencia de segmento; (iv) liberar el oligonucleótido de segmento del cebador de oligonucleótido usando una endonucleasa específica de secuencia.
8 Procedimiento según cualquier reivindicación anterior, en el que el procedimiento es semicontinuo o continuo.
9. Procedimiento según cualquier reivindicación anterior, en el que el producto se produce a una escala de gramos o kilogramos y/o el procedimiento se lleva a cabo en un reactor de al menos 1 l.
10. Procedimiento según cualquier reivindicación anterior, mediante el cual la desnaturalización resulta de un aumento de temperatura, un cambio en el pH o un cambio en la concentración de sal en una disolución tamponante.
11. Procedimiento según la reivindicación 10, en el que el procedimiento incluye dos etapas de aumento de la temperatura: i) para desnaturalizar los dúplex de molde:impureza y ii) para desnaturalizar los dúplex de molde:producto.
12. Procedimiento según cualquier reivindicación anterior, en el que los segmentos tienen de 3 a 15 nucleótidos de longitud.
13. Procedimiento según cualquier reivindicación anterior, en el que la longitud del producto es de 20 a 25 nucleótidos de longitud.
14. Procedimiento según la reivindicación 13, en el que dicho producto tiene 20 nucleótidos de longitud y comprende tres oligonucleótidos de segmento que comprenden:
(i) un segmento 5’ que tiene 7 nucleótidos de longitud, un segmento central que tiene 6 nucleótidos de longitud y un segmento 3’ que tiene 7 nucleótidos de longitud;
(ii) un segmento 5’ que tiene 6 nucleótidos de longitud, un segmento central que tiene 8 nucleótidos de longitud y un segmento 3’ que tiene 6 nucleótidos de longitud;
(iii) un segmento 5’ que tiene 5 nucleótidos de longitud, un segmento central que tiene 10 nucleótidos de longitud y un segmento 3’ que tiene 5 nucleótidos de longitud;
(iv) un segmento 5’ que tiene 4 nucleótidos de longitud, un segmento central que tiene 12 nucleótidos de longitud y un segmento 3’ que tiene 4 nucleótidos de longitud; o
(v) un segmento 5’ que tiene 3 nucleótidos de longitud, un segmento central que tiene 14 nucleótidos de longitud y un segmento 3’ que tiene 3 nucleótidos de longitud.
15. Procedimiento según cualquier reivindicación anterior, en el que el molde está unido a un material de soporte.
16. Procedimiento según la reivindicación 15, en el que el material de soporte es un material de soporte soluble, opcionalmente en el que el material de soporte se selecciona del grupo que consiste en polietilenglicol, un polímero orgánico soluble, ADN, una proteína, un dendrímero, un polisacárido, un oligosacárido y un hidrato de carbono.
17. Procedimiento según la reivindicación 15, en el que el material de soporte es un material de soporte insoluble, opcionalmente en el que el material de soporte insoluble se selecciona del grupo que consiste en: una perla de vidrio, una perla polimérica, un soporte fibroso, una membrana, una perla recubierta con estreptavidina y celulosa, o forma parte del propio recipiente de reacción, por ejemplo una pared de reacción.
18. Procedimiento según una cualquiera de las reivindicaciones 15 a 17, en el que se unen múltiples copias repetidas del molde de manera continua por medio de un solo punto de unión al material de soporte.
19. Procedimiento según una cualquiera de las reivindicaciones 15 a 17, en el que el molde se une al material de soporte en múltiples puntos de unión.
20. Procedimiento según cualquier reivindicación anterior, en el que la modificación es en la posición 2’ del resto de azúcar y se selecciona del grupo que consiste en 2’-F, 2’-OMe, 2’-MOE y 2’-amino, o en el que el oligonucleótido comprende un PMO, un LNA, un PNA, un BNA o un SPIEGELMER.
21. Procedimiento según cualquier reivindicación anterior, en el que la modificación es en la nucleobase y se selecciona del grupo que consiste en una 5-metilpirimidina, una 7-deazaguanosina y un nucleótido abásico.
22. Procedimiento según cualquier reivindicación anterior, en el que la modificación es en la estructura principal y se selecciona del grupo que consiste en fosforotioato, fosforamidato y fosforodiamidato.
23. Procedimiento según cualquier reivindicación anterior, en el que el producto de oligonucleótido monocatenario resultante tiene una pureza de al menos el 90 %, opcionalmente en el que el producto de oligonucleótido monocatenario tiene una pureza de al menos el 95 %, opcionalmente en el que el producto de oligonucleótido monocatenario tiene una pureza de al menos el 98 %.
24. Procedimiento según cualquier reivindicación anterior, en el que el procedimiento produce un gapmero.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1721307.5A GB201721307D0 (en) | 2017-12-19 | 2017-12-19 | Novel processes for the production of oligonucleotides |
| PCT/EP2018/085179 WO2019121500A1 (en) | 2017-12-19 | 2018-12-17 | Novel processes for the production of oligonucleotides |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2914591T3 true ES2914591T3 (es) | 2022-06-14 |
Family
ID=61009182
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18822043T Active ES2914591T3 (es) | 2017-12-19 | 2018-12-17 | Procedimientos novedosos para la producción de oligonucleótidos |
Country Status (10)
| Country | Link |
|---|---|
| EP (2) | EP4015631A1 (es) |
| JP (2) | JP7065970B2 (es) |
| KR (3) | KR102417049B1 (es) |
| CN (1) | CN111479923A (es) |
| BR (1) | BR112020012252A2 (es) |
| CA (1) | CA3085237A1 (es) |
| DK (1) | DK3728585T3 (es) |
| ES (1) | ES2914591T3 (es) |
| GB (1) | GB201721307D0 (es) |
| WO (1) | WO2019121500A1 (es) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020171092A1 (ja) * | 2019-02-18 | 2020-08-27 | 味の素株式会社 | 相補部分を含む修飾オリゴヌクレオチドの製造方法 |
| CN114729390A (zh) * | 2019-11-29 | 2022-07-08 | 深圳华大基因科技有限公司 | 寡核苷酸的酶促合成 |
| US20230295707A1 (en) * | 2022-03-21 | 2023-09-21 | Abclonal Science, Inc. | T4 DNA Ligase Variants with Increased Ligation Efficiency |
| TW202405168A (zh) | 2022-04-08 | 2024-02-01 | 英商葛蘭素史密斯克藍智慧財產發展有限公司 | 製造含有寡核苷酸之聚核苷酸的新穎方法 |
| CN117660374A (zh) * | 2022-09-08 | 2024-03-08 | 凯莱英医药集团(天津)股份有限公司 | Rna连接酶在寡核苷酸制备上的应用 |
| CN115724998A (zh) * | 2022-10-28 | 2023-03-03 | 深圳先进技术研究院 | 一种基于共振能量转移的全遗传编码nmn蛋白探针及其应用 |
| CN117247911A (zh) * | 2023-08-22 | 2023-12-19 | 武汉爱博泰克生物科技有限公司 | 大肠杆菌dna连接酶突变体及其在毕赤酵母中表达纯化方法 |
| CN117106782B (zh) * | 2023-10-16 | 2024-04-26 | 吉林凯莱英医药化学有限公司 | gRNA及其生物合成方法 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5516664A (en) | 1992-12-23 | 1996-05-14 | Hyman; Edward D. | Enzymatic synthesis of repeat regions of oligonucleotides |
| US6683173B2 (en) | 1998-04-03 | 2004-01-27 | Epoch Biosciences, Inc. | Tm leveling methods |
| US6586586B1 (en) | 2000-01-31 | 2003-07-01 | Isis Pharmaceuticals, Inc. | Purification of oligonucleotides |
| WO2001064864A2 (en) | 2000-02-28 | 2001-09-07 | Maxygen, Inc. | Single-stranded nucleic acid template-mediated recombination and nucleic acid fragment isolation |
| US7211654B2 (en) | 2001-03-14 | 2007-05-01 | Regents Of The University Of Michigan | Linkers and co-coupling agents for optimization of oligonucleotide synthesis and purification on solid supports |
| WO2006071568A2 (en) * | 2004-12-29 | 2006-07-06 | Applera Corporation | Methods, compositions, and kits for forming labeled polynucleotides |
| GB201115218D0 (en) | 2011-09-02 | 2011-10-19 | Glaxo Group Ltd | Novel process |
| US8808989B1 (en) | 2013-04-02 | 2014-08-19 | Molecular Assemblies, Inc. | Methods and apparatus for synthesizing nucleic acids |
| GB201502152D0 (en) | 2015-02-10 | 2015-03-25 | Nuclera Nucleics Ltd | Novel use |
| GB201503534D0 (en) | 2015-03-03 | 2015-04-15 | Nuclera Nucleics Ltd | Novel method |
| CN105506125B (zh) * | 2016-01-12 | 2019-01-22 | 上海美吉生物医药科技有限公司 | 一种dna的测序方法及一种二代测序文库 |
| GB201612011D0 (en) * | 2016-07-11 | 2016-08-24 | Glaxosmithkline Ip Dev Ltd | Novel processes for the production of oligonucleotides |
-
2017
- 2017-12-19 GB GBGB1721307.5A patent/GB201721307D0/en not_active Ceased
-
2018
- 2018-12-17 EP EP22154581.7A patent/EP4015631A1/en active Pending
- 2018-12-17 DK DK18822043.8T patent/DK3728585T3/da active
- 2018-12-17 KR KR1020207020495A patent/KR102417049B1/ko active IP Right Grant
- 2018-12-17 CA CA3085237A patent/CA3085237A1/en active Pending
- 2018-12-17 CN CN201880082633.8A patent/CN111479923A/zh active Pending
- 2018-12-17 JP JP2020533192A patent/JP7065970B2/ja active Active
- 2018-12-17 BR BR112020012252-1A patent/BR112020012252A2/pt active Search and Examination
- 2018-12-17 KR KR1020247006894A patent/KR20240033153A/ko active Application Filing
- 2018-12-17 WO PCT/EP2018/085179 patent/WO2019121500A1/en unknown
- 2018-12-17 ES ES18822043T patent/ES2914591T3/es active Active
- 2018-12-17 KR KR1020227022320A patent/KR102643631B1/ko active IP Right Grant
- 2018-12-17 EP EP18822043.8A patent/EP3728585B1/en active Active
-
2022
- 2022-04-26 JP JP2022072032A patent/JP7438259B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CN111479923A (zh) | 2020-07-31 |
| BR112020012252A2 (pt) | 2020-11-24 |
| EP3728585B1 (en) | 2022-03-16 |
| JP2022109986A (ja) | 2022-07-28 |
| JP7438259B2 (ja) | 2024-02-26 |
| WO2019121500A1 (en) | 2019-06-27 |
| KR20220094219A (ko) | 2022-07-05 |
| EP3728585A1 (en) | 2020-10-28 |
| EP4015631A1 (en) | 2022-06-22 |
| JP7065970B2 (ja) | 2022-05-12 |
| JP2021507699A (ja) | 2021-02-25 |
| KR20240033153A (ko) | 2024-03-12 |
| CA3085237A1 (en) | 2019-06-27 |
| DK3728585T3 (da) | 2022-05-23 |
| KR20200099564A (ko) | 2020-08-24 |
| GB201721307D0 (en) | 2018-01-31 |
| KR102417049B1 (ko) | 2022-07-06 |
| KR102643631B1 (ko) | 2024-03-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2914591T3 (es) | Procedimientos novedosos para la producción de oligonucleótidos | |
| ES2831419T3 (es) | Nuevos procedimientos para la producción de oligonucleótidos | |
| US20090298708A1 (en) | 2'-deoxy-2'-fluoro-beta-d-arabinonucleoside 5'-triphosphates and their use in enzymatic nucleic acid synthesis | |
| TW202405168A (zh) | 製造含有寡核苷酸之聚核苷酸的新穎方法 | |
| Anderson | Non-nucleosidic modifications in the selection of thrombin-binding aptamers | |
| Matthew Piperakis | Synthesis and properties of nucleic acid duplexes and quadruplexes containing 3'-S-phosphorothiolate linkages |