ES2871140T3 - Derivados bicíclicos de piridina, pirazina y pirimidina como inhibidores de PI3K beta - Google Patents
Derivados bicíclicos de piridina, pirazina y pirimidina como inhibidores de PI3K beta Download PDFInfo
- Publication number
- ES2871140T3 ES2871140T3 ES17732059T ES17732059T ES2871140T3 ES 2871140 T3 ES2871140 T3 ES 2871140T3 ES 17732059 T ES17732059 T ES 17732059T ES 17732059 T ES17732059 T ES 17732059T ES 2871140 T3 ES2871140 T3 ES 2871140T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- substituents
- substituted
- mmol
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 Cc1cccc2c1C(*)(*)C*CC2(*)* Chemical compound Cc1cccc2c1C(*)(*)C*CC2(*)* 0.000 description 8
- QKHJTYGUWNTBPG-UHFFFAOYSA-N CC1OCCN(C)C1 Chemical compound CC1OCCN(C)C1 QKHJTYGUWNTBPG-UHFFFAOYSA-N 0.000 description 2
- SVVZNPXKJXFVNG-UHFFFAOYSA-N CC(C)(C)C(C)(C)[S+](C)OCc1c[n](cc(cc2-c(nc3)c[n]3S(N(C)C)(=O)=O)Br)c2n1 Chemical compound CC(C)(C)C(C)(C)[S+](C)OCc1c[n](cc(cc2-c(nc3)c[n]3S(N(C)C)(=O)=O)Br)c2n1 SVVZNPXKJXFVNG-UHFFFAOYSA-N 0.000 description 1
- JAVICSDLXFEYSD-UHFFFAOYSA-N CC(C)(C)C(C)(C)[S+](C)OCc1c[n](cc(cc2-c(nc3)c[n]3S(N(C)C)(=O)=O)C3=CCOCC3)c2n1 Chemical compound CC(C)(C)C(C)(C)[S+](C)OCc1c[n](cc(cc2-c(nc3)c[n]3S(N(C)C)(=O)=O)C3=CCOCC3)c2n1 JAVICSDLXFEYSD-UHFFFAOYSA-N 0.000 description 1
- NIKPJHALYZMARX-UHFFFAOYSA-N CCC1OCCN(C)C1 Chemical compound CCC1OCCN(C)C1 NIKPJHALYZMARX-UHFFFAOYSA-N 0.000 description 1
- RGNFMQJLAOONTP-UHFFFAOYSA-N CCC1OCCNC1 Chemical compound CCC1OCCNC1 RGNFMQJLAOONTP-UHFFFAOYSA-N 0.000 description 1
- KDIBJRZQZWESCO-UHFFFAOYSA-N CCOC(Cc1c(Cc2cccc(C(F)(F)F)c2C)[n](cc(N2CCOCC2)nc2)c2n1)=O Chemical compound CCOC(Cc1c(Cc2cccc(C(F)(F)F)c2C)[n](cc(N2CCOCC2)nc2)c2n1)=O KDIBJRZQZWESCO-UHFFFAOYSA-N 0.000 description 1
- FAJRZWOLXMTCNP-UHFFFAOYSA-N CCOC(Cc1c[n](cc(N2CCOCC2)nc2)c2n1)=O Chemical compound CCOC(Cc1c[n](cc(N2CCOCC2)nc2)c2n1)=O FAJRZWOLXMTCNP-UHFFFAOYSA-N 0.000 description 1
- NIKPJHALYZMARX-ZETCQYMHSA-N CC[C@@H]1OCCN(C)C1 Chemical compound CC[C@@H]1OCCN(C)C1 NIKPJHALYZMARX-ZETCQYMHSA-N 0.000 description 1
- VGTADGNSGCICTA-UHFFFAOYSA-N CN1CC(CO)OCC1 Chemical compound CN1CC(CO)OCC1 VGTADGNSGCICTA-UHFFFAOYSA-N 0.000 description 1
- SJRJJKPEHAURKC-UHFFFAOYSA-N CN1CCOCC1 Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 1
- VGTADGNSGCICTA-ZCFIWIBFSA-N CN1C[C@H](CO)OCC1 Chemical compound CN1C[C@H](CO)OCC1 VGTADGNSGCICTA-ZCFIWIBFSA-N 0.000 description 1
- CSHMCEYIMFSLSS-UHFFFAOYSA-N COC(C1(CC1)N)=O Chemical compound COC(C1(CC1)N)=O CSHMCEYIMFSLSS-UHFFFAOYSA-N 0.000 description 1
- CWPGLBCSGGKRCS-UHFFFAOYSA-N COC(c1c(Cc2c(C3CCNCC3)c(F)ccc2)[n](cc(cn2)N3CCOCC3)c2n1)=O Chemical compound COC(c1c(Cc2c(C3CCNCC3)c(F)ccc2)[n](cc(cn2)N3CCOCC3)c2n1)=O CWPGLBCSGGKRCS-UHFFFAOYSA-N 0.000 description 1
- QKHJTYGUWNTBPG-LURJTMIESA-N C[C@@H]1OCCN(C)C1 Chemical compound C[C@@H]1OCCN(C)C1 QKHJTYGUWNTBPG-LURJTMIESA-N 0.000 description 1
- WYWHEXLNCXJSKA-UHFFFAOYSA-N C[N]1(CCOCC1)I Chemical compound C[N]1(CCOCC1)I WYWHEXLNCXJSKA-UHFFFAOYSA-N 0.000 description 1
- RADIYRNTDCMSBL-UHFFFAOYSA-N Cc1c(C(F)(F)F)cccc1Cc1c(CC(O)=O)nc2[n]1cc(N1CCOCC1)nc2 Chemical compound Cc1c(C(F)(F)F)cccc1Cc1c(CC(O)=O)nc2[n]1cc(N1CCOCC1)nc2 RADIYRNTDCMSBL-UHFFFAOYSA-N 0.000 description 1
- ZVGDIXBGUJGPDV-UHFFFAOYSA-N Cc1c(C(F)(F)F)cccc1Cc1c(CN2CC3(CNC3)CCC2)nc2ncc(N3CCOCC3)c[n]12 Chemical compound Cc1c(C(F)(F)F)cccc1Cc1c(CN2CC3(CNC3)CCC2)nc2ncc(N3CCOCC3)c[n]12 ZVGDIXBGUJGPDV-UHFFFAOYSA-N 0.000 description 1
- QFKBTJDWMXNCHJ-UHFFFAOYSA-N Cc1c(C(F)(F)F)cccc1Cc1c(CN2CNCCC2)nc(cc2)[n]1cc2N1CCOCC1 Chemical compound Cc1c(C(F)(F)F)cccc1Cc1c(CN2CNCCC2)nc(cc2)[n]1cc2N1CCOCC1 QFKBTJDWMXNCHJ-UHFFFAOYSA-N 0.000 description 1
- PHNXWYHJHAIMAI-UHFFFAOYSA-N Cc1c(C(F)(F)F)cccc1Cc1c(CO)nc(c(-c2ccn[nH]2)c2)[n]1cc2C1=CCOCC1 Chemical compound Cc1c(C(F)(F)F)cccc1Cc1c(CO)nc(c(-c2ccn[nH]2)c2)[n]1cc2C1=CCOCC1 PHNXWYHJHAIMAI-UHFFFAOYSA-N 0.000 description 1
- VNULPLSJQCFGHI-UHFFFAOYSA-N Cc1c(C(F)(F)F)cccc1Cc1c(CO)nc2ncc(N3CCOCC3)c[n]12 Chemical compound Cc1c(C(F)(F)F)cccc1Cc1c(CO)nc2ncc(N3CCOCC3)c[n]12 VNULPLSJQCFGHI-UHFFFAOYSA-N 0.000 description 1
- AWABVLXHPBZYFR-UHFFFAOYSA-N Cc1c[n](cc(cc2CO)[Br]=C)c2n1 Chemical compound Cc1c[n](cc(cc2CO)[Br]=C)c2n1 AWABVLXHPBZYFR-UHFFFAOYSA-N 0.000 description 1
- QJILQVYBRCOUIU-UHFFFAOYSA-N OCC(C1)OCC[N]11[IH]C1 Chemical compound OCC(C1)OCC[N]11[IH]C1 QJILQVYBRCOUIU-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Abstract
Un compuesto de Fórmula (I) un tautómero o una forma estereoisomérica del mismo, en donde X1 representa CH o N; X2 representa CR1 o N; siempre que como máximo uno de X1 y X2 represente N; R1 representa hidrógeno, -C(=O)OH, -C(=O)NH2, -NH2, -CH2OH, **(Ver fórmula)** ; Y representa -CH2- o -NH-; R2 representa **(Ver fórmula)** R3 representa alquilo C1-4; -C(=O)-O-alquilo C1-4; -C(=O)-Het1; -CH(OH)-CH2-Rq; alquilo C1-4 sustituido en el mismo átomo de carbono con un -OH y con un Het1; o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -NH2, -O-(C=O)-alquilo C1-4, -(C=O)-O-alquilo C1-4, -NH-(C=O)-alquilo C1-4, -NH-(SO2)-alquilo C1-4, - N(CH3)-alquil C1-4-SO2-CH3, -NH-alquil C1-4-SO2-CH3, -N(CH3)-alquil C1-4-OH, -N(C=O-alquil C1-4)-alquil C1-4-OH, -(C=O)-NH-alquil C1-4-OH, -O-(C=O)-CH(NH2)-alquilo C1-4, -O-(C=O)-CH(NH2)-alquil C1-4-Ar, , -NH-alquil C1-4-OH, Het1, -O-C(=O)-alquil C1-4-Het1, -C(=O)-Het1, y -NH-C(=O)-Het1; Rq representa Het1, halo, -OH, -NH2, -O-(C=O)-alquilo C1-4, -NH-(C=O)-alquilo C1-4, -NH-(SO2)-alquilo C1-4, -N(CH3)-alquil C1-4-SO2-CH3, -NH-alquil C1-4-SO2-CH3, -N(CH3)-alquil C1-4-OH, -O-(C=O)-CH(NH2)-alquilo C1-4, -O-(C=O)-CH(NH2)-alquil C1-4-Ar, **(Ver fórmula)** o -NH-alquil C1-4-OH; Ar representa fenilo opcionalmente sustituido con un hidroxi; R4a representa hidrógeno, alquilo C1-4, Heta o alquilo C1-4 sustituido con uno o más sustituyentes, cada uno independientemente seleccionado del grupo que consiste en -OH, -NR5R6 y Heta; R4b representa hidrógeno, halo, alquilo C1-4 o alquilo C1-4 sustituido con uno o más sustituyentes halo; o R4a y R4b se toman juntos para formar, junto con el anillo de fenilo al que están unidos, una estructura de Fórmula (a-1), (a-2), (a-3), (a-4) o (a-5): (a-1) (a-2) (a-3) (a-4) (a-5) ; **(Ver fórmula)** X representa -NH-, -O-, -N(alquil C1-3)-, o -N(hidroxialquil C1-3)-; ambos sustituyentes R7 son los mismos y se seleccionan del grupo que consiste en hidrógeno, flúor y metilo; o ambos sustituyentes R7 se toman juntos para formar, junto con el átomo de carbono común al que están unidos, un ciclopropilo, ciclobutilo u oxetanilo; ambos sustituyentes R8 son los mismos y se seleccionan del grupo que consiste en hidrógeno y metilo; o ambos sustituyentes R8 se toman juntos para formar, junto con el átomo de carbono común al que están unidos, un ciclopropilo, ciclobutilo u oxetanilo; R5 representa hidrógeno, alquilo C1-6 o alquilo C1-6 sustituido con un -OH; R6 representa hidrógeno, alquilo C1-6 o alquilo C1-6 sustituido con un -OH; Het1 representa un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo, cada uno independientemente seleccionado de O, S, S(=O)p y N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes, cada uno independientemente seleccionado del grupo que consiste en halo, -NH2, alquilo C1-4, -S(=O)2-alquilo C1-6, -alquil C1-4-S(=O)2-alquilo C1-6, hidroxilo, alquiloxi C1-4, flúor, ciano y alquilo C1-4 sustituido con un hidroxi; o dos sustituyentes en el mismo átomo de carbono de dicho heterociclilo saturado de 4, 5 o 6 miembros se toman juntos para formar junto con el átomo de carbono común al que están unidos el Anillo A; Anillo A representa ciclobutilo, ciclopentilo, ciclohexilo o un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo, cada uno independientemente seleccionado de O, S, S(=O)p y N; dicho ciclobutilo, ciclopentilo, ciclohexilo o heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes alquilo C1-4, con un sustituyente alquilo C1-4 y un sustituyente hidroxi, o con un sustituyente hidroxi; cada Heta representa independientemente un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de O, S, S(=O)p y N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes cada uno seleccionado independientemente del grupo que consiste en alquilo C1-4, -S(=O)2-alquilo C1-6, hidroxi, -alquil C1-4-S(=O)2-alquilo C1-6, y alquilo C1-4 sustituido con un hidroxi; o dos sustituyentes en el mismo átomo de carbono de dicho heterociclilo saturado de 4, 5 o 6 miembros se toman juntos para formar junto con el átomo de carbono común al que están unidos el Anillo B; el Anillo B representa ciclobutilo, ciclopentilo, ciclohexilo, o un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de O, S, S(=O)p y N; dicho ciclobutilo, ciclopentilo, ciclohexilo, o heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes alquilo C1-4, con un alquilo C1-4 y un sustituyente hidroxi, o con un sustituyente hidroxi; p representa 1 o 2; o un N-óxido, una sal de adición farmacéuticamente aceptable o un solvato de los mismos.
Description
DESCRIPCIÓN
Derivados bicíclicos de piridina, pirazina y pirimidina como inhibidores de PI3K beta
Campo de la invención
La presente invención se refiere a derivados de piridina, pirazina y pirimidina bicíclicos útiles como inhibidores de PI3K|3. La invención se refiere además a composiciones farmacéuticas que comprenden dichos compuestos como principio activo, así como también al uso de dichos compuestos como un medicamento.
Antecedentes de la invención
Existen tres clases de fosfoinositida-3-cinasas (PI3K): clase I, clase II y clase III. Las PI3K de clase I son las más asociadas con el cáncer humano [K.D Courtney, R.B. Corcoran y J.A. Engelman (2010), Journal of Clinical Oncology., 28; 1075]. Las fosfoinositida-3-cinasas de clase I (PI3K) se dividen en 2 subclases: PI3K de clase Ia , compuesta de una subunidad catalítica p110 (p110a, p110b o p110d) y una subunidad reguladora p85 (p85a, p55a y p50a, p85b o p55g), y PI3K de clase 1b representada por la subunidad catalítica p110g y las subunidades reguladoras p101 y p84 [B. Vanhaesebroeck y M.D. Waterfield (1999) Experimental Cell Research., 253, 239-254]. Las PI3K de clase Ia se activan en una diversidad de tumores sólidos y no sólidos a través de la mutación o deleción del supresor tumoral PTEN (homólogo de fosfatasa y tensina) o en el caso de p110a activando mutaciones [K.D Courtney, R.B. Corcoran y J.A. Engelman (2010), Journal of Clinical Oncology., 28; 1075]. Las PI3K también se pueden activar por receptores tirosina cinasa (RTK); p110b puede activarse por receptores acoplados a proteína G [K.D Courtney, R.B. Corcoran y J.A. Engelman (2010), Journal of Clinical Oncology., 28; 1075]. Una vez activadas, las fosfoinositida-3-cinasas catalizan la fosforilación de 4,5-difosfato de fosfatidilo, lo que conduce a la generación de fosfatidilo, 3, 4, 5-trifosfato (PIP3) [Zhao L., Vogt P. K.(2008) Oncogene 27, 5486-5496]. PTEN antagoniza la actividad de las PI3K a través de la desfosforilación de PIP3 [Myers M. P., Pass I., Batty I. H., Van der Kaay J., Stolarov J. P., Hemmings B. A., Wigler M. H., Downes C. P., Tonks N. K.(1998) Proc. Natl. Acad. Sci. U.S.A. 95, 13513-13518]. El PIP3 generado por la activación de PI3K o sostenido por la inactivación de PTEN se une a un subconjunto de dominios de unión a lípidos en dianas aguas abajo tales como el dominio de homología a pleckstrina del oncogen Akt, recluyéndolo así a la membrana plasmática [Stokoe D., Stephens L. R., Copeland T., Gaffney P. R., Reese C. B., Painter G. F., Holmes A. B., McCormick F., Hawkins P. T. (1997) Science 277, 567-570]. Una vez en la membrana plasmática, Akt fosforila varias moléculas efectoras que participan en numerosos procesos biológicamente relevantes, tales como el metabolismo, la diferenciación, la proliferación, la longevidad y la apoptosis [D. R. Calnan y A. Brunet (2008) Oncogene 27; 2276)].
Varios estudios sugieren un papel clave para p110b en tumores deficientes en PTEN. Por ejemplo, el knockout genético de p110b, pero no de p110a, es capaz de bloquear la formación de tumores y la activación de Akt provocada por la pérdida de Pten en la próstata anterior en un modelo de ratón [Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, Zhao JJ. Nature 2008; 454:776-9]. Además, otros estudios han demostrado que un subconjunto de líneas celulares tumorales humanas deficientes en PTEN es sensible a la inactivación de p110b en lugar de p110a [Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM, Lengauer C (2008) Proc. Natl. Acad. Sci (USA); 105 13057]. La deficiencia de PTEN ya sea por inactivación genética o expresión reducida ocurre con frecuencia en cánceres humanos tales como GBM, cáncer de endometrio, pulmón, mama y cáncer de próstata, entre otros [K.D Courtney, R.B. Corcoran y J.A. Engelman (2010), Journal of Clinical Oncology., 28; 1075].
Estos estudios sugieren que el tratamiento de cáncer deficiente en PTEN con agentes que inhiben p110b puede ser terapéuticamente beneficioso. Además de su papel en el cáncer, p110b puede ser una diana para la terapia antitrombótica. Se ha informado en modelos de ratón que la inhibición de PI3Kb puede prevenir los contactos de adhesión estable de integrina anbb3 que eliminan la formación de trombos oclusivos sin prolongar el tiempo de sangrado [S. P. Jackson et al. (2005) Nature Medicine, 11,507-514].
Además de ello, la vía fosfatidilinositol-4,5-bisfosfato 3-cinasa (PI3K)/AKT es frecuentemente activada durante el progreso del cáncer de próstata (PCa) a través de la pérdida o mutación del gen homólogo de fosfatasa y tensina (PTEN). Después de la vía del receptor de andrógeno (AR), es el segundo impulsor principal del crecimiento de PCa. La combinación con terapia hormonal mejoró la eficacia de agentes dirigidos a PI3K/AKT en modelos de PCa PTEN-negativos. La regulación ascendente de los genes dirigidos a AR tras la inhibición de PI3K/AKT sugiere una interferencia compensatoria entre las rutas de PI3K-AR que, para un tratamiento de eficacia óptima, podría requerir el codireccionamiento del eje AR [Marques RB, et al., High Efficacy of Combination Therapy Using PI3K/AKT Inhibitors with Androgen Deprivation in Prostate Cancer Preclinical Models. Eur Urol (2014), http://dx.doi.org/10.1016Zj.eururo.2014.08.053]. Por lo tanto, los inhibidores de PI3K|3 pueden combinarse ventajosamente con terapias antiandrógenas que incluyen antagonistas del receptor de andrógenos e inhibidores de la biosíntesis de andrógenos en cánceres de próstata negativos a PTEN.
El documento WO 2012/116237 describe entidades heterocíclicas que modulan la actividad de PI3 cinasa.
El documento WO 2011/123751 describe compuestos heterocíclicos como inhibidores selectivos de la actividad de PI3K. El documento WO 2011/022439 describe entidades heterocíclicas que modulan la actividad de PI3 cinasa.
El documento WO 2008/014219 describe derivados de tiozolidinadiona en calidad de inhibidores de PI3 cinasa.
El documento WO 2013/028263 se refiere a derivados de pirazolopirimidina en calidad de inhibidores de PI3 cinasa. El documento WO 2012/047538 se refiere a derivados de bencimidazol en calidad de inhibidores de PI3 cinasa.
El documento WO 2013/095761 se refiere a derivados de imidazopiridina en calidad de inhibidores de PI3 cinasa.
El documento US 2013/0157977 se refiere a derivados de ácido bencimidazol-borónico en calidad de inhibidores de PI3 cinasa.
El documento WO 2009/021083 describe derivados de quinoxalina como inhibidores de PI3 cinasa.
El documento WO 2007/103756 describe la preparación de tiazolonas para su uso como inhibidores de PI3 cinasa. El documento WO 2011/041399 describe bencimidazolil(morfolinil)purinas y compuestos relacionados como inhibidores de PI3K6 y su preparación y uso para el tratamiento de enfermedades mediadas por PI3K.
El documento WO 2009/088990 describe la preparación de pirazolopirimidinas y otros compuestos heterocíclicos como moduladores de PI3 cinasa terapéuticos. El documento WO2014/09296 describe derivados de imidazo(1,2-a)pirazina útiles como moduladores de TNF alfa.
Por lo tanto, existe una fuerte necesidad de nuevos inhibidores de PI3K|3 cinasa, abriendo con ello nuevas avenidas para el tratamiento o la prevención del cáncer, en particular cánceres deficientes en PTEN, más en particular cáncer de próstata. Por consiguiente, proporcionar tales compuestos es un objeto de la presente invención.
Sumario de la invención
Se ha descubierto que los compuestos de la presente invención son útiles como inhibidores de PI3K|3. Los compuestos de acuerdo con la invención y composiciones de los mismos pueden ser útiles para el tratamiento o la prevención, en particular para el tratamiento, de enfermedades tales como el cáncer, trastornos autoinmunes, enfermedades cardiovasculares, enfermedades inflamatorias, enfermedades neurodegenerativas, alergia, pancreatitis, asma, fallo multiorgánico, enfermedades renales, agregación plaquetaria, motilidad del esperma, rechazo de trasplantes, rechazo de injertos, lesiones pulmonares y similares.
Esta invención concierne a compuestos de Fórmula (I)

sus tautómeros y formas estereoisoméricas, en donde
X1 representa CH o N;
X2 representa CR1 o N;
siempre que como máximo uno de X1 y X2 represente N;
R1 representa hidrógeno, -C(=O)OH, -C(=O)NH2, -NH2, -CH2OH,

o -NH-alquil C1-4-OH;
Ar representa fenilo opcionalmente sustituido con un hidroxi;
R4a representa hidrógeno, alquilo C1-4, Heta o alquilo C1-4 sustituido con uno o más sustituyentes, cada uno independientemente seleccionado del grupo que consiste en -OH, -NR5 R6 y Heta;
R4b representa hidrógeno, halo, alquilo C1-4 o alquilo C1-4 sustituido con uno o más sustituyentes halo;
o R4a y R4b se toman juntos para formar, junto con el anillo de fenilo al que están unidos, una estructura de Fórmula (a-1 ), (a-2 ), (a-3 ), (a-4 ) o (a-5 ):

X representa -NH-, -O-, -N(alquil C1-3)-, o -N(hidroxialquil C1-3)-;
ambos sustituyentes R7 son los mismos y se seleccionan del grupo que consiste en hidrógeno, flúor y metilo; o ambos sustituyentes R7 se toman juntos para formar, junto con el átomo de carbono común al que están unidos, un ciclopropilo, ciclobutilo u oxetanilo;
ambos sustituyentes R8 son los mismos y se seleccionan del grupo que consiste en hidrógeno y metilo; o ambos sustituyentes R8 se toman juntos para formar, junto con el átomo de carbono común al que están unidos, un ciclopropilo, ciclobutilo u oxetanilo;
R5 representa hidrógeno, alquilo C1-6 o alquilo C1-6 sustituido con un -OH;
R6 representa hidrógeno, alquilo C1-6 o alquilo C1-6 sustituido con un -OH;
Het1 representa un heterociclilo saturado de 4 , 5 o 6 miembros que contiene al menos un heteroátomo, cada uno independientemente seleccionado de O, S, S(=O)p y N; dicho heterociclilo saturado de 4 , 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes, cada uno independientemente seleccionado del grupo que consiste en halo, -NH2, alquilo C1-4, -S(=O)2-alquilo C1-6, -alquil C 1-4-S(=O)2-alquilo C1-6, hidroxilo, alquiloxi C1-4, flúor, ciano y alquilo C1-4 sustituido con un hidroxi; o dos sustituyentes en el mismo átomo de carbono de dicho heterociclilo saturad
o de 4 , 5 o 6 miembros se toman juntos para formar junto con el átomo de carbono común al que están unidos el Anillo A;
Anillo A representa ciclobutilo, ciclopentilo, ciclohexilo o un heterociclilo saturado de 4 , 5 o 6 miembros que contiene al menos un heteroátomo, cada uno independientemente seleccionado de O, S, S(=O)p y N; dicho ciclobutilo, ciclopentilo, ciclohexilo o heterociclilo saturado de 4 , 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes alquilo C1-4, con un sustituyente alquilo C1-4 y un sustituyente hidroxi, o con un sustituyente hidroxi;
cada Heta representa independientemente un heterociclilo saturado de 4 , 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de O, S, S(=O)p y N; dicho heterociclilo saturado de 4 , 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes cada uno seleccionado independientemente del grupo que consiste en alquilo C1-4, -S(=O)2-alquilo C1-6, hidroxi, -alquil C 1-4-S(=O)2-alquilo C1-6, y alquilo C1-4 sustituido con un hidroxi; o dos sustituyentes en el mismo átomo de carbono de dicho heterociclilo saturado de 4 , 5 o 6 miembros se toman juntos para formar junto con el átomo de carbono común al que están unidos el Anillo B;
el Anillo B representa ciclobutilo, ciclopentilo, ciclohexilo, o un heterociclilo saturado de 4 , 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de O, S, S(=O)p y N; dicho ciclobutilo, ciclopentilo,
ciclohexilo, o heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes alquilo Ci-4, con un alquilo C1-4 y un sustituyente hidroxi, o con un sustituyente hidroxi;
p representa 1 o 2;
y los N-óxidos, las sales de adición farmacéuticamente aceptables y los solvatos de los mismos.
La presente invención también afecta a métodos para preparar la preparación de compuestos de la presente invención y composiciones farmacéuticas que los comprenden.
Se encontró que los compuestos de la presente invención inhibían PI3K|3 per se o pueden someter el metabolismo a una forma (más) activa in vivo (profármacos) y, por lo tanto, pueden ser útiles en el tratamiento o la prevención, en particular en el tratamiento, de enfermedades tales como el cáncer, trastornos autoinmunes, enfermedades cardiovasculares, enfermedades inflamatorias, enfermedades neurodegenerativas, alergia, pancreatitis, asma, fallo multiorgánico, enfermedades renales, agregación plaquetaria, motilidad del esperma, rechazo de trasplantes, rechazo de injertos, lesiones pulmonares y similares.
A la vista de la farmacología antes mencionada de los compuestos de Fórmula (I) y N-óxidos, sales por adición farmacéuticamente aceptables, y solvatos de los mismos, se deduce que pueden ser adecuados para uso como un medicamento.
En particular, los compuestos de Fórmula (I) y N-óxidos, sales por adición farmacéuticamente aceptables, y solvatos de los mismos, pueden ser adecuados en el tratamiento o la prevención, en particular en el tratamiento del cáncer.
La presente invención concierne también al uso de compuestos de Fórmula (I) y N-óxidos, sales por adición farmacéuticamente aceptables, y solvatos de los mismos, para la fabricación de un medicamento para la inhibición de PI3KP, para el tratamiento o la prevención de cáncer.
La presente invención se describirá ahora adicionalmente. En los siguientes pasajes, se definen más detalladamente diferentes aspectos de la invención. Cada aspecto definido de esta forma puede combinarse con cualquier otro aspecto o aspectos a menos que se indique claramente lo contrario. En particular, cualquier característica indicada como preferida o ventajosa puede combinarse con cualquier otra característica o características indicadas como preferidas o ventajosas.
Descripción detallada
Cuando se describen los compuestos de la invención, los términos utilizados se deben interpretar de acuerdo con las siguientes definiciones, a menos que el contexto indique lo contrario.
Cuando cualquier variable ocurra más de una vez en cualquier componente o en cualquier fórmula (por ejemplo la Fórmula (I)), su definición en cada caso es independiente de su definición en todos los demás casos.
Siempre que se utilice el término "sustituido" en la presente invención, a menos que se indique lo contrario o que sea obvio por el contexto, se pretende que indique que uno o más hidrógenos, en particular de 1 a 3 hidrógenos, preferentemente 1 o 2 hidrógenos, más preferentemente 1 hidrógeno, del átomo o radical indicado en la expresión en la que se utiliza el término "sustituido" son reemplazados por una selección del grupo indicado, siempre que no se exceda la valencia normal y que la sustitución genere un compuesto químicamente estable, es decir, un compuesto que sea lo suficientemente resistente para superar un proceso de aislamiento hasta un grado de pureza útil a partir de una mezcla de reacción y a su formulación en forma de un agente terapéutico.
Cuando dos o más sustituyentes estén presentes en un resto, estos pueden, a menos que se indique lo contrario o que sea obvio por el contexto, reemplazar hidrógenos en el mismo átomo o pueden reemplazar átomos de hidrógeno en átomos diferentes en el resto.
Será evidente para el experto que, a menos que se indique otra cosa o sea evidente por el contexto, un sustituyente en un grupo heterociclilo puede reemplazar cualquier átomo de hidrógeno en un átomo de carbono del anillo o en un heteroátomo del anillo.
El sufijo "Cx-y” (en que x e y son números enteros), tal como se utilizan en el presente documento, se refiere al número de átomos de carbono en un grupo dado. Por lo tanto, un grupo alquilo C1-6 contiene de 1 a 6 átomos de carbono, un grupo alquilo C1-4 contiene de 1 a 4 átomos de carbono, un grupo alquilo C1-3 contiene de 1 a 3 átomos de carbono, un grupo cicloalquilo C3-6 contiene de 3 a 6 átomos de carbono, etc.
El término “halo”, como un grupo o parte de un grupo es genérico para flúor, cloro, bromo, yodo, a menos que se indique de otro modo o resulte claro del contexto.
La expresión "alquilo Ci -6" como un grupo o parte de un grupo, se refiere a un radical hidrocarbilo de Fórmula CnH2n+i en donde n es un número que varía entre 1 y 6. Los grupos alquilo C1-6 comprenden de 1 a 6 átomos de carbono, preferentemente de 1 a 4 átomos de carbono, más preferentemente de 1 a 3 átomos de carbono, aún más preferentemente de 1 a 2 átomos de carbono. Los grupos alquilo pueden ser lineales o ramificados, y pueden estar sustituidos tal como se indica en el presente documento. Cuando se usa en el presente documento un subíndice después de un átomo de carbono, el subíndice se refiere al número de átomos de carbono que el grupo mencionado puede contener. Así, por ejemplo, alquilo C1-6 incluye todos los grupos alquilo lineales o ramificados con entre 1 y 6 átomos de carbono, y por lo tanto incluye, tales como por ejemplo metilo, etilo, n-propilo, /-propilo, 2-metil-etilo, butilo y sus isómeros (por ejemplo n-butilo, /sobutilo y terc-butilo), pentilo y sus isómeros, hexilo y sus isómeros y similares.
La expresión "alquilo C1-4" como un grupo o parte de un grupo, se refiere a un radical hidrocarbilo de Fórmula CnH2n+1 en donde n es un número que varía entre 1 y 4. Los grupos alquilo C1-4 comprenden desde 1 hasta 4 átomos de carbono, preferentemente desde 1 hasta 3 átomos de carbono, más preferentemente desde 1 hasta 2 átomos de carbono. Grupos alquilo C1-4 pueden ser lineales o ramificados y pueden estar sustituidos tal como se indica en el presente documento. Cuando se usa en el presente documento un subíndice después de un átomo de carbono, el subíndice se refiere al número de átomos de carbono que el grupo mencionado puede contener. Alquilo C1-4 incluye todos los grupos alquilo lineales o ramificados con entre 1 y 4 átomos de carbono y, por lo tanto, incluye metilo, etilo, n-propilo, /-propilo, 2-metil-etilo, butilo y sus isómeros (p. ej., n-butilo, /sobutilo y terc.-butilo), y similares.
La expresión "alquilo C1-3" como un grupo o parte de un grupo, se refiere a un radical hidrocarbilo de Fórmula CnH2n+1 en donde n es un número que varía entre 1 y 3. Grupos alquilo C1-3 comprenden de 1 a 3 átomos de carbono, preferiblemente 1 a 2 átomos de carbono. Grupos alquilo C1-3 pueden ser lineales o ramificados y pueden estar sustituidos tal como se indica en el presente documento. Cuando se usa en el presente documento un subíndice después de un átomo de carbono, el subíndice se refiere al número de átomos de carbono que el grupo mencionado puede contener. Alquilo C1-3 incluye todos los grupos alquilo lineales o ramificados con entre 1 y 3 átomos de carbono y, por lo tanto, incluye metilo, etilo, npropilo, /-propilo, 2-metil-etilo, y similares.
En una realización, la expresión "al menos un heteroátomo" está restringida a "1,2 o 3 heteroátomos", en una realización particular a "1 o 2 heteroátomos", en una realización más particular a "1 heteroátomo".
Un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de O, S, S(=O)p y N (como ocurre, por ejemplo, en las definiciones de Het1, Heta, el Anillo A y el Anillo B); en una realización particular es un heterociclilo saturado de 4, 5 o 6 miembros que contiene 1, 2 o 3 heteroátomos seleccionados de O, S, S(=O)p y N; en una realización más particular, un heterociclilo saturado de 4, 5 o 6 miembros que contiene 1 o 2 heteroátomos seleccionados de O, S, S(=O)p y N.
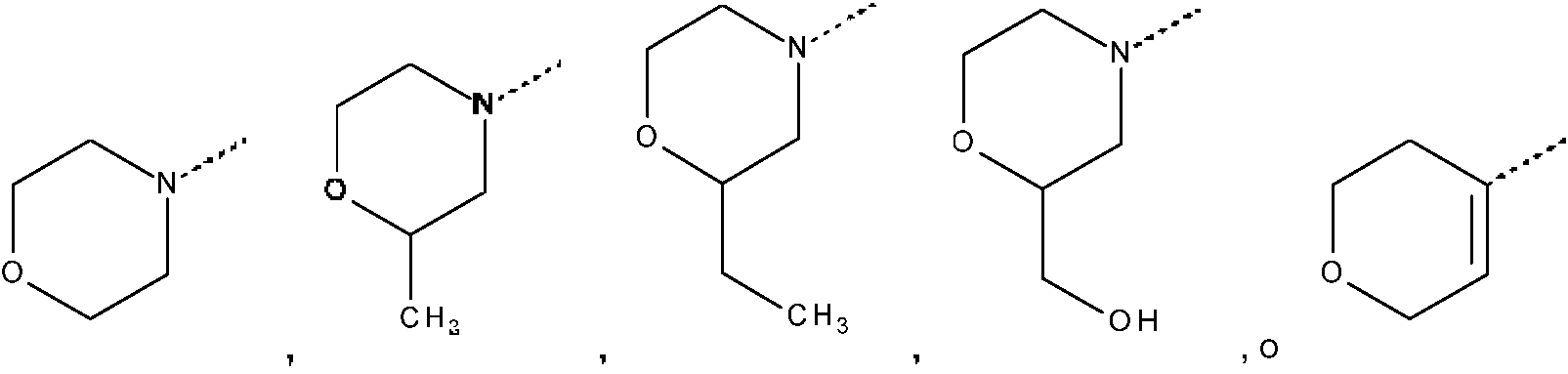
Los ejemplos de un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de O, S, S(=O)p y N, incluyen, pero sin limitación, azetidinilo, morfolinilo, piperidinilo, pirrolidinilo, 1, 1 -dióxido-tietanilo, 1,1-dióxido-tiomorfolinilo, piperazinilo, dioxolanilo, oxazolidinilo, oxetanilo, tetrahidrofuranilo, y similares.
Het1 y Heta pueden estar unidos al resto de la molécula de Fórmula (I) a través de cualquier átomo de carbono del anillo o heteroátomo del anillo, según sea apropiado, si no se especifica de otro modo.
Resultará claro que cuando dos sustituyentes en el mismo átomo de carbono en la definición de Het1 o Heta se toman juntos para forma, junto con el átomo de carbono común al que están unidos el Anillo A o el Anillo B, respectivamente, se forma un resto espiro.
Por ejemplo, cuando Het1 representa 1 -piperidinilo, en donde dos sustituyentes en el átomo de carbono en posición p se toman juntos para formar, junto con el átomo de carbono común al que están unidos, el Anillo A, se forma el siguiente resto espiro:

en particular, si en el ejemplo anterior el anillo A representa 3-azetidinilo, se forma el siguiente resto espiro:

Ejemplos de estos restos espiro incluyen, pero sin limitación,

similares
Siempre que los sustituyentes estén representados por una estructura química, representa el enlace de unión al resto de la molécula de fórmula (I).
Cuando uno de los sistemas de anillo esté sustituido con uno o más sustituyentes, esos sustituyentes pueden reemplazar, a menos que se indique de otro modo o resulte claro del contexto, cualquier átomo de hidrógeno unido a un átomo de carbono o nitrógeno del sistema del anillo.
El término "sujeto", tal como se emplea en la presente, se refiere a un animal, preferentemente un mamífero (p. ej., gato, perro, primate o ser humano), más preferiblemente un ser humano, que es o que ha sido objeto de tratamiento, observación o experimentación.
El término “cantidad terapéuticamente eficaz”, tal como se emplea en la presente, se refiere a la cantidad de compuesto o agente farmacéutico activos que desencadena la respuesta biológica o medicinal en un sistema tisular, animal o ser humano, la cual un investigador, veterinario, médico u otro profesional sanitario desea obtener y que incluye el alivio o anulación de los síntomas de la enfermedad o trastorno que se esté tratando.
Se pretende que el término "composición" abarque un producto que comprende los componentes especificados en las cantidades especificadas, así como también cualquier producto que sea el resultado, directa o indirectamente, de combinaciones de los componentes especificados en las cantidades especificadas.
Se pretende que el término "tratamiento", como se utiliza en el presente documento, se refiera a todos los procesos en los que pueda haber una ralentización, interrupción, detención o parada de la progresión de una enfermedad, pero no indica necesariamente una eliminación total de todos los síntomas.
La expresión "compuestos de la invención", como se utiliza en el presente documento, pretende incluir los compuestos de Fórmula (I) y N-óxidos, sales por adición farmacéuticamente aceptables, y solvatos de los mismos.
Tal como se utiliza en el presente documento, cualquier fórmula química con enlaces mostrados sólo como líneas continuas y no como enlaces en cuña o en cuña con trazo discontinuo, o indicado de otra manera como que tienen una configuración particular (por ejemplo R, S) alrededor de uno o más átomos, contempla cada estereoisómeros posible, o mezcla de dos o más estereoisómeros.
En lo que antecede y en lo que sigue en el presente documento, la expresión “compuesto de Fórmula (I)” pretende incluir los estereoisómeros del mismo y las formas tautoméricas del mismo.
Los términos y expresiones “estereoisómeros”, “formas estereoisoméricas” o “formas estereoquímicamente isoméricas” en lo que antecede y en lo que sigue en el presente documento se utilizan de manera indistinta.
La invención incluye todos los estereoisómeros de los compuestos de la invención, ya sea como un estereoisómero puro o como una mezcla de dos o más estereoisómeros.
Los enantiómeros son estereoisómeros que imágenes especulares no superponibles entre sí. Una mezcla 1 :1 de un par de enantiómeros es un racemato o mezcla racémica.
Los atropisómeros (o atropoisómeros) son estereoisómeros que tienen una configuración espacial particular, que es el resultado de una rotación restringida alrededor de un enlace sencillo, debido a un gran impedimento estérico. Se pretende que todas las formas atropisoméricas de los compuestos de Fórmula (I) estén incluidas dentro del alcance de la presente invención.
Los diastereómeros (o diastereoisómeros) son estereoisómeros que no son enantiómeros, es decir, que no están relacionados como imágenes especulares. Si un compuesto contiene un doble enlace, los sustituyentes pueden estar en la configuración E o la configuración Z. Sustituyentes en radicales cíclicos bivalentes (parcialmente) saturados pueden tener la configuración cis o trans; por ejemplo, si un compuesto contiene un grupo cicloalquilo disustituido, los sustituyentes pueden estar en la configuración cis o trans. Por lo tanto, la invención incluye enantiómeros, atropisómeros, diastereómeros, racematos, isómeros E, isómeros Z, isómeros cis, isómeros trans y sus mezclas, siempre que sea químicamente posible.
Los expertos en la técnica estarán familiarizados con el significado de todos estos términos, es decir, enantiómeros, atropisómeros, diastereómeros, racematos, isómeros E, isómeros Z, isómeros cis, isómeros trans y sus mezclas.
La configuración absoluta se especifica de acuerdo con el sistema de Cahn-Ingold-Prelog. La configuración en un átomo asimétrico se especifica como R o S. Los estereoisómeros resueltos cuya configuración absoluta se desconoce se pueden designar como (+) o (-) dependiendo de la dirección en la que hagan rotar el plano de la luz polarizada. Por ejemplo, los enantiómeros resueltos cuya configuración absoluta se desconozca se pueden designar como (+) o (-) dependiendo de la dirección en la que hagan rotar el plano de la luz polarizada.
Cuando se identifica un estereoisómero específico, esto quiere decir que dicho estereoisómero está sustancialmente exento, es decir, asociado con menos de un 50 % , preferentemente menos de un 20 % , más preferentemente menos de un 10 % , aún más preferentemente menos de un 5 % , en particular menos de un 2 % y aún más preferentemente menos de un 1 % , de los otros estereoisómeros. Por lo tanto, cuando un compuesto de Fórmula (I) se especifica, por ejemplo, como (R), esto significa que el compuesto está sustancialmente libre del isómero (S); cuando un compuesto de Fórmula (I) se especifica, por ejemplo, como E, esto significa que el compuesto está sustancialmente libre del isómero Z; cuando un compuesto de Fórmula (I) se especifica, por ejemplo, como cis, esto significa que el compuesto está sustancialmente libre del isómero trans.
Algunos de los compuestos de Fórmula (I) también pueden existir en su forma tautomérica. Estas formas, en la medida en que puedan existir, pretenden estar incluidas dentro del alcance de la presente invención. Se deduce que un compuesto sencillo puede existir tanto en su forma estereoisomérica como tautomérica.
Por ejemplo, resultará claro para la persona experta que cuando R1 representa

También

se incluye en el alcance de la invención.
Para los usos terapéuticos, las sales de los compuestos de Fórmula (I), N-óxidos y los solvatos de los mismos, son aquellos en donde el contraión es farmacéuticamente aceptable. Sin embargo, sales de ácidos y bases que no son farmacéuticamente aceptables pueden también encontrar uso, por ejemplo, en la preparación o purificación de un compuesto farmacéuticamente aceptable. Todas las sales, ya sean farmacéuticamente aceptables o no, quedan incluidas dentro del ámbito de la presente invención.
Se pretende que las sales de adición farmacéuticamente aceptables tal como se mencionan anteriormente o en lo sucesivo en la presente comprendan las formas salinas de adición de ácidos y bases atóxicas terapéuticamente activas que los compuestos de Fórmula (I), N-óxidos y sus solvatos sean capaces de formar. Sales por adición de ácidos farmacéuticamente aceptables se pueden obtener convenientemente tratando la forma de base con un ácido apropiado de este tipo. Ácidos apropiados comprende, por ejemplo. Ácidos inorgánicos tales como hidrácidos, p. ej., ácido clorhídrico o bromhídrico, sulfúrico, nítrico, fosfórico y ácidos similares; u ácidos orgánicos tales como, por ejemplo, acético, propanoico, hidroxiacético, láctico, pirúvico, oxálico (es decir, etanodioico), malónico, succínico (es decir, ácido butanodioico), maleico, fumárico, málico, tartárico, cítrico, metanosulfónico, etanosulfónico, bencenosulfónico, ptoluenosulfónico, ciclámico, salicílico, p-aminosalicílico, pamoico y ácidos similares. En cambio, dichas formas de sales se pueden convertir en la forma de base libre mediante tratamiento con una base adecuada.
Los compuestos de Fórmula (I), N-óxidos y los solvatos de estos que contengan un protón ácido también se pueden convertir en sus formas salinas de adición de amina o metal atóxicas tratándolos con bases orgánicas e inorgánicas apropiadas. Las formas salinas de bases apropiadas comprenden, por ejemplo, las sales de amonio, las sales de metales alcalinos y alcalinotérreos, por ejemplo, sales de litio, sodio, potasio, magnesio, calcio y similares, sales con bases orgánicas, por ejemplo, aminas alifáticas y aromáticas primarias, secundarias y terciarias tales como metilamina, etilamina, propilamina, isopropilamina, los cuatro isómeros de la butilamina, dimetilamina, dietilamina, dietanolamina, dipropilamina, diisopropilamina, di-n-butilamina, pirrolidina, piperidina, morfolina, trimetilamina, trietilamina, tripropilamina, quinuclidina, piridina, quinolina e isoquinolina; la benzatina, N-metil-D-glucamina, sales de hidrabamina y sales con aminoácidos tales como, por ejemplo, arginina, lisina y similares. Inversamente, la forma de sal se puede convertir, mediante tratamiento con un ácido, en la forma de ácido libre.
El término solvato comprende los hidratos y las formas de adición de disolvente que los compuestos de Fórmula (I) sean capaces de formar, así como N-óxidos y sales de adición farmacéuticamente aceptables de los mismos. Ejemplos de tales formas son, p. ej., hidratos, alcoholatos y similares.
Los compuestos de la invención, tal como se preparan en los procedimientos descritos más adelante, se pueden sintetizar en forma de mezclas de enantiómeros, en particular mezclas racémicas de enantiómeros, que se pueden separar una de otra siguiendo procesos de resolución conocidos en la técnica. Una manera de separar las formas enantioméricas de los compuestos de Fórmula (I), y N-óxidos, sales por adición farmacéuticamente aceptables, y solvatos de los mismos, implica la cromatografía líquida utilizando una fase estacionaria quiral. Dichas formas estereoquímicamente isoméricas puras también pueden derivarse de las formas estereoquímicamente isoméricas puras correspondientes de los materiales de partida apropiados, siempre que la reacción se produzca de manera estereoespecífica. Preferiblemente, si se desea un estereoisómero específico, dicho compuesto se sintetizaría mediante métodos estereoespecíficos de preparación. Estos métodos emplearán ventajosamente materiales de partida enantioméricamente puros.
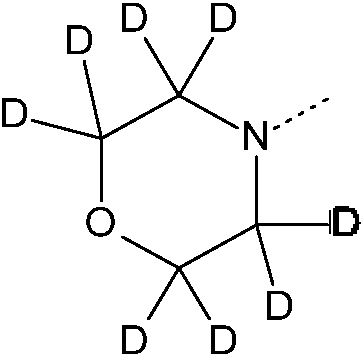
En el marco de esta solicitud, un elemento, en particular cuando se mencione en relación con un compuesto de Fórmula (I), comprenderá todos los isótopos y mezclas isotópicas de este elemento, ya sean de origen natural o producidos de forma sintética, con abundancia natural o en una forma enriquecida isotópicamente. Los compuestos radiomarcados de Fórmula (I) pueden comprender un isótopo radiactivo seleccionado a partir del grupo constituido por 2H, 3H, 11C, 18F, 122I, 123I, 125i, 131i, 75Br, 76Br, 77Br y 82Br. Preferentemente, el isótopo radiactivo se selecciona a partir del grupo constituido por 2H, 3H, 11C y 18F. Más preferiblemente, el isótopo radiactivo es 2H.
En particular, se pretende que los compuestos deuterados estén incluidos dentro del alcance de la presente invención.
En una realización, la presente invención trata sobre compuestos de Fórmula (I) novedosos, sus tautómerosy formas estereoisoméricas, en donde
X1 representa CH o N;
X2 representa CR1 o N;
siempre que como máximo uno de X1 y X2 represente N;
R1 representa hidrógeno, -C(=O)OH, -C(=O)NH2, -NH2, -CH2OH,

Y representa -CH2- o -NH-;
R2 representa
R3 representa alquilo C1-4; -C(=O)-Het1; alquilo C1-4 sustituido en el mismo átomo de carbono con un -OH y con un Het1 ; o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -NH2, -O-(C=O)-alquilo C1-4, -(C=O)-O-alquilo C1-4, -NH-(C=O)-alquilo C1-4, -NH-(SO2)-alquilo C1-4,
-N(CH3)-alquil C1-4-SO2-CH3, -NH-alquil C1-4-SO2-CH3, -N(CH3)-alquil C1-4-OH,
-N(C=O-alquil C1-4)-alquil C1-4-OH, -(C=O)-NH-alquil C1-4-OH,
-O-(C=O)-CH(NH2)-alquilo C1-4,

-NH-alquil C1-4-OH, Het1, -O-C(=O)-alquil C1-4-Het1, -C(=O)-Het1, y -NH-C(=O)-Het1 ;
R4a representa hidrógeno, alquilo C1-4, Heta o alquilo C1-4 sustituido con uno o más sustituyentes, cada uno independientemente seleccionado del grupo que consiste en -OH, -NR5 R6 y Heta;
R4b representa hidrógeno, halo, alquilo C1-4 o alquilo C1-4 sustituido con uno o más sustituyentes halo;
R5 representa hidrógeno, alquilo C1-6 o alquilo C1-6 sustituido con un -OH;
R6 representa hidrógeno, alquilo C1-6 o alquilo C1-6 sustituido con un -OH;
Het1 representa un heterociclilo saturado de 4 , 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de O, S, S(=O)p y N; dicho heterociclilo saturado de 4 , 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes cada uno seleccionado independientemente del grupo que consiste en halo, -NH2, alquilo C1-4, -S(=O)2-alquilo C1-6, -alquil C 1-4-S(=O)2-alquilo C1-6, hidroxilo, alquiloxi C1-4, flúor, ciano y alquilo C1-4 sustituido con un hidroxi; o dos sustituyentes en el mismo átomo de carbono de dicho heterociclilo saturado de 4 , 5 o 6 miembros se toman juntos para formar junto con el átomo de carbono común al que están unidos el Anillo A;
Anillo A representa ciclobutilo, ciclopentilo, ciclohexilo o un heterociclilo saturado de 4 , 5 o 6 miembros que contiene al menos un heteroátomo, cada uno independientemente seleccionado de O, S, S(=O)p y N; dicho ciclobutilo, ciclopentilo, ciclohexilo o heterociclilo saturado de 4 , 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes alquilo C1-4, con un sustituyente alquilo C1-4 y un sustituyente hidroxi, o con un sustituyente hidroxi;
cada Heta representa independientemente un heterociclilo saturado de 4 , 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de O, S, S(=O)p y N; dicho heterociclilo saturado de 4 , 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes cada uno seleccionado independientemente del grupo que consiste en alquilo C1-4, -S(=O)2-alquilo C1-6, hidroxi, -alquil C 1-4-S(=O)2-alquilo C1-6, y alquilo C1-4 sustituido con un hidroxi;
p representa 1 o 2 ;
y los N-óxidos, las sales de adición farmacéuticamente aceptables y los solvatos de los mismos.
En una realización, la presente invención trata sobre compuestos de Fórmula (I) novedosos, sus tautómerosy formas estereoisoméricas, en donde
X 1 representa CH o N;
X2 representa CR1 o N;
siempre que como máximo uno de X 1 y X2 represente N;
R1 representa hidrógeno, -C(=O)OH, -C(=O)NH2, -NH2, -CH2OH,

o
R3 representa alquilo C1-4; o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -O-(C=O)-alquilo C1-4, -(C=O)-O-alquilo C1-4,
-N(CH3)-alquil C1-4-OH, -N(C=O-alquil C 1-4)-alquil C1-4-OH, -NH-alquil C1-4-OH, Het1, y -C(=O)-Het1;
R4a representa alquilo C1-4, o Heta;
R4b representa halo o alquilo C1-4 sustituido con uno o más sustituyentes halo;
Het1 representa un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de S(=O)p y N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes cada uno seleccionado independientemente del grupo que consiste en alquilo C1-4, y alquilo C1-4 sustituido con un hidroxi; o dos sustituyentes en el mismo átomo de carbono de dicho heterociclilo saturado de 4, 5 o 6 miembros se toman juntos para formar junto con el átomo de carbono común al que están unidos el Anillo A;
Anillo A representa ciclobutilo, ciclopentilo, ciclohexilo o un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de S(=O)p y N;
cada Heta representa independientemente un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un átomo de N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes alquilo C1-4
p representa 2;
y los N-óxidos, las sales de adición farmacéuticamente aceptables y los solvatos de los mismos.
En una realización, la presente invención trata sobre compuestos de Fórmula (I) novedosos, sus tautómerosy formas estereoisoméricas, en donde
X1 representa CH o N;
X2 representa CR1 o N;
siempre que como máximo uno de X1 y X2 represente N;
R1 representa hidrógeno, -NH2, -CH2OH,

Y representa -CH2- o -NH-;
R2 representa

o
R3 representa alquilo C1-4; o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en -OH y Het1; R4a representa alquilo C1-4;
R4b representa halo o alquilo C1-4 sustituido con uno o más sustituyentes halo;
Het1 representa

y los N-óxidos, las sales de adición farmacéuticamente aceptables y los solvatos de los mismos.
Otra realización de la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde se aplican una o más de las siguientes limitaciones:
(i) R1 representa hidrógeno, -C(=O)OH, -C(=O)NH2, -NH2, -CH2OH,

(iii) R3 representa alquilo C1-4; o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -O-(C=O)-alquilo C1-4, -(C=O)-O-alquilo C1-4,-N(CH3)-alquil C1-4-OH, -N(C=O-alquil C1 -4)-alquil C1-4-OH, -NH-alquil C1-4-OH, Het1, y -C(=O)-Het1 ;
(iv) R4a representa alquilo C1-4 o Heta;
(v) R4b representa halo o alquilo C1-4 sustituido con uno o más sustituyentes halo;
(vi) Het1 representa un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de S(=O)p y N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes cada uno seleccionado independientemente del grupo que consiste en alquilo C1-4, y alquilo C1-4 sustituido con un hidroxi; o dos sustituyentes en el mismo átomo de carbono de dicho heterociclilo saturado de 4, 5 o 6 miembros se toman juntos para formar junto con el átomo de carbono común al que están unidos el Anillo A;
(vii) Anillo A representa ciclobutilo, ciclopentilo, ciclohexilo o un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno de S(=O)p y N;
(viii) cada Heta representa independientemente un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un átomo de N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes alquilo C1-4
(ix) p representa 2.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R1 representa -NH2;
R2 representa

Y representa -CH2-.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde X1 representa CH, y X2 representa CR1.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde X1 representa CH, y X2 representa N.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en al que X1 representa N, y X2 representa CR1.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde X2 representa CR1 ; en particular en donde X2 representa CH.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde Y representa -CH2-.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde Y representa -NH-.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde Y representa -NH-; y
R3 representa alquilo C1-4; o alquilo C1-4 sustituido con un sustituyente -OH.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R4a representa hidrógeno, alquilo C1-4 o alquilo C1-4 sustituido con uno o más sustituyentes, cada uno independientemente seleccionado del grupo que consiste en -NR5R6 y Heta;
R4b representa hidrógeno, halo, alquilo C1-4 o alquilo C1-4 sustituido con uno o más sustituyentes halo.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R4a representa alquilo C1-4; en particular R4a representa metilo;
R4b representa alquilo C1-4 sustituido con uno o más sustituyentes halo;
en particular R4b representa CF3.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R4a representa alquilo C1-4; en particular, R4a representa metilo.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde R4a representa hidrógeno, alquilo C1-4, Heta o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo consistente en -OH, -NR5 R6 y Heta.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R4b representa alquilo C1-4 sustituido con uno o más sustituyentes halo;
en particular R4b representa CF3.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde R4a y R4b son distintos de hidrógeno.
En una realización, la presente invención se refiere a aquellos compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde R4a y R4b se toman juntos para formar junto con el anillo fenilo al que están unidos una estructura de Fórmula (a-1), (a-2), (a-3), (a-4) o (a-5); en particular una estructura de Fórmula (a-2) o (a-4); más en particular una estructura de Fórmula (a-2).
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R4a representa alquilo C1-4, Heta o alquilo C1-4 sustituido con uno o más sustituyentes, cada uno independientemente seleccionado del grupo que consiste en -OH, -NR5 R6 y Heta;
R4b representa hidrógeno, halo, alquilo C1-4 o alquilo C1-4 sustituido con uno o más sustituyentes halo;
o R4a y R4b se toman juntos para formar, junto con el anillo de fenilo al que están unidos, una estructura de Fórmula (a-2) o (a-4).
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R4a representa alquilo C1-4, Heta o alquilo C1-4 sustituido con uno o más sustituyentes, cada uno independientemente seleccionado del grupo que consiste en -OH, -NR5 R6 y Heta;
R4b representa alquilo C1-4 o alquilo C1-4 sustituido con uno o más sustituyentes halo.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R4a representa alquilo C1-4, Heta o alquilo C1-4 sustituido con uno o más sustituyentes, cada uno independientemente seleccionado del grupo que consiste en -OH, -NR5 R6 y Heta;
R4b representa alquilo C1-4.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R4a representa alquilo C1-4, Heta o alquilo C1-4 sustituido con uno o más sustituyentes, cada uno independientemente seleccionado del grupo que consiste en -OH, -NR5 R6 y Heta;
R4b representa alquilo C1-4 o alquilo C1-4 sustituido con uno o más sustituyentes halo;
o R4a y R4b se toman juntos para formar, junto con el anillo de fenilo al que están unidos, una estructura de Fórmula (a-2) o (a-4).
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde R3 representa alquilo C1-4; -C(=O)-Het1; -CH(OH)-CH2-Rq; alquilo C1-4 sustituido en el mismo átomo de carbono con un -OH y con un Het1 ; o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -NH 2,
-O-(C=O)-alquilo C1-4, -(C=O)-O-alquilo C1-4, -NH-(C=O)-alquilo C1-4, -NH-(SO2)-alquilo C1-4,
-N(CH3)-alquil C1-4-SO2-CH3, -NH-alquil C1-4-SO2-CH3, -N(CH3)-alquil C1-4-OH,
-N(C=O-alquil C1-4)-alquil C1-4-OH, -(C=O)-NH-alquil C1-4-OH,
-O-(C=O)-CH(NH2)-alquilo C1-4, -O-(C=O)-CH(NH2)-alquil C1-4-Ar,

-NH-alquil C1-4-OH, Het1, -O-C(=O)-alquil C1-4-Het1, -C(=O)-Het1, y -NH-C(=O)-Het1.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R3 representa alquilo C1-4; o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -NH 2, -O-(C=O)-alquilo C1-4, -NH-(C=O)-alquilo C1-4,
-NH-(SO2)-alquilo C1-4, -N(CH3)-alquil C1-4-SO2-CH3, -NH-alquil C1-4-SO2-CH3,
-N(CH3)-alquil C1-4-OH, -(C=O)-NH-alquil C1-4-OH, -O-(C=O)-CH(NH2)-alquilo C1-4,
-O-(C=O)-CH(NH2)-alquil C1-4-Ar,

y -NH-alquil C1-4-OH.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R3 representa alquilo C1-4; -CH(OH)-CH2-Rq; o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -NH 2, -NH-(C=O)-alquilo C1-4,
-NH-(SO2)-alquilo C1-4, -N(CH3)-alquil C1-4-SO2-CH3, -NH-alquil C1-4-SO2-CH3,
-N(CH3)-alquil C1-4-OH, -(C=O)-NH-alquil C1-4-OH y -NH-alquil C1-4-OH;
Rq representa -OH o -NH 2.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R3 representa alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -NH2, -O-(C=O)-alquilo C1-4, -NH-(C=O)-alquilo C1-4,
-NH-(SO2)-alquilo C1-4, -N(CH3)-alquil C1-4-SO2-CH3, -NH-alquil C1-4-SO2-CH3,
-N(CH3)-alquil C1-4-OH, -(C=O)-NH-alquil C1-4-OH, -O-(C=O)-CH(NH2)-alquilo C1-4,
-O-(C=O)-CH(NH2)-alquil C1-4-Ar,

y -NH-alquil C1-4-OH.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde R3 representa alquilo C1-4; o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -NH2, -O-(C=O)-alquilo C1-4, -NH-(C=O)-alquilo C1-4,
-N(CH3)-alquil C1-4-SO2-CH3, -NH-alquil C1-4-SO2-CH3, -O-(C=O)-CH(NH2)-alquilo C1-4,
-O-(C=O)-CH(NH2)-alquil C1-4-Ar,

y -NH-alquil C1-4-OH.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde R3 representa -CH(OH)-CH2-Rq; o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -NH 2, -O-(C=O)-alquilo C1-4,
-NH-(C=O)-alquilo C1-4, -NH-(SO2)-alquilo C1-4, -N(CH3)-alquil C1-4-SO2-CH3,
-NH-alquil C1-4-SO2-CH3, -N(CH3)-alquil C1-4-OH, -(C=O)-NH-alquil C1-4-OH,
-O-(C=O)-CH(NH2)-alquilo C1-4, -O-(C=O)-CH(NH2)-alquil C1-4-Ar,

y -NH-alquil C1-4-OH.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R3 representa alquilo C1-4 sustituido con un sustituyente como se define en cualquiera de las otras realizaciones.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R2 representa

En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R2 representa
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R3 representa alquilo C1-4 sustituido con un sustituyente -OH; en particular R3 representa -CH2-OH.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R1 representa -C(=O)NH2, -NH2,

En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R1 representa -C(=O)NH2, -NH2,

farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R1 representa

realización, la presente invención se refiere a aquellos compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde R1 representa -C(=O)OH, -C(=O)NH2 o -NH2.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R1 representa -C(=O)NH2 o -NH2.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R1 representa -NH2.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R1 representa hidrógeno.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R1 representa algo distinto de hidrógeno.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde Rq representa halo, -OH, -NH2, -O-(C=O)-alquilo C1-4, -NH-(C=O)-alquilo C1-4, -NH-(SO2)-alquilo C1-4, -N(CH3)-alquil C1-4-SO2-CH3, -NH-alquil C1-4-SO2-CH3, -N(CH3)-alquil C1-4-OH, -O-(C=O)-CH(NH2)-alquilo C1-4, o -NH-alquil C1-4-OH.
En una realización, la presente invención se refiere a aquellos compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde Rq representa -OH o -NH2; en particular donde Rq representa -NH2.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R3 representa alquilo C1-4; o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -O-(C=O)-alquilo C1-4, -NH-(SO2)-alquilo C1-4,
-N(CH3)-alquil C1-4-SO2-CH3, -NH-alquil C1-4-SO2-CH3, -N(CH3)-alquil C1-4-OH,
-(C=O)-NH-alquil C1-4-OH y -NH-alquil C1-4-OH;
en particular, en donde R3 representa alquilo C1-4; o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH,
-N(CH3)-alquil C1-4-SO2-CH3, -NH-alquil C1-4-SO2-CH3, -N(CH3)-alquil C1-4-OH, y -NH-alquil C1-4-OH; más en particular, en donde R3 representa alquilo C1-4; o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en halo y -OH;
incluso más en particular en donde R3 representa alquilo C1-4; o alquilo C1-4 sustituido con un sustituyente -OH; todavía más en particular en donde R3 representa alquilo C1-4.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
cada Heta representa independientemente

En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
cada Heta representa independientemente un heterociclilo saturado de 4, 5 o 6 miembros que contiene uno o dos heteroátomos seleccionados cada uno de ellos independientemente entre O, S(=O)p y N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes cada uno seleccionado independientemente entre el grupo que consiste en hidroxi y alquilo C1-4 sustituido con un hidroxi;
p representa 1 o 2.
En una realización, la presente invención se refiere a aquellos compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde ambos sustituyentes R7 son hidrógeno; y en donde ambos sustituyentes R8 son hidrógeno.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
ambos sustituyentes R7 son los mismos y se seleccionan del grupo que consiste en hidrógeno, flúor y metilo; y en donde ambos sustituyentes R8 son los mismos y se seleccionan del grupo que consiste en hidrógeno y metilo.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R2 representa

En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R2 representa

En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R2 que representa

se limita a

En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R2 que representa

se limitan respectivamente a

En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R3 representa alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en Het1a, -C(=O)-Het1 y -NH-C(=O)-Het1b; o
alquilo C1-4 sustituido en el mismo átomo de carbono con un -OH y con un Het1b;
Het1 representa un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de O, S, S(=O)p y N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes cada uno seleccionado independientemente del grupo que consiste en halo, -NH2, alquilo C1-4, -S(=O)2-alquilo C1-6, -alquil C1-4-S(=O)2-alquilo C1-6, hidroxi y alquilo C1-4 sustituido con un hidroxi; o dos sustituyentes en el mismo átomo de carbono de dicho heterociclilo saturado de 4, 5 o 6 miembros se toman juntos para formar junto con el átomo de carbono común al que están unidos el Anillo A;
Het1a se define como Het1, con la condición, sin embargo, que Het1a esté siempre unido al resto de R3 a través de un átomo de nitrógeno del anillo;
Het1b se define como Het1, con la condición, sin embargo, que Het1b esté siempre unido al resto de R3 a través de un átomo de carbono del anillo.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R3 representa alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en Het1a, -O-C(=O)-alquil C1-4-Het1a, -C(=O)-Het1 y -NH-C(=O)-Het1b;
-CH(OH)-CH2-Het1a; o alquilo C1-4 sustituido en el mismo átomo de carbono con un -OH y con un Het1b;
Het1 representa un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de O, S, S(=O)p y N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes cada uno seleccionado independientemente del grupo que consiste en halo, -NH2, alquilo C1-4, -S(=O)2-alquilo C1-6, -alquil C1-4-S(=O)2-alquilo C1-6, hidroxi y alquilo C1-4 sustituido con un hidroxi; o dos sustituyentes en el mismo átomo de carbono de dicho heterociclilo saturado de 4, 5 o 6 miembros se toman juntos para formar junto con el átomo de carbono común al que están unidos el Anillo A;
Het1a se define como Het1, con la condición, sin embargo, que Het1a esté siempre unido al resto de R3 a través de un átomo de nitrógeno del anillo;
Het1b se define como Het1, con la condición, sin embargo, que Het1b esté siempre unido al resto de R3 a través de un átomo de carbono del anillo.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R1 representa otra cosa que -C(=O)OH.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R3 representa alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en Het1, -C(=O)-Het1 y -NH-C(=O)-Het1 ; o
alquilo C1-4 sustituido en el mismo átomo de carbono con un -OH y con un Het1.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R3 representa alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en Het1, -C(=O)-Het1, y -NH-C(=O)-Het1 ; -CH(OH)-CH2-Het1; o alquilo C1-4 sustituido en el mismo átomo de carbono con un -OH y con un Het1.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona
en cualquiera de las otras realizaciones, en donde R3 representa alquilo C1-4 sustituido con un sustituyente seleccionado del grupo consistente en Het1, -O-C(=O)-alquil C ^-H et1, -C(=O)-Het1 y -NH-C(=O)-Het1 ; o
-CH(OH)-CH2-Het1.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde R3 representa alquilo C1-4 sustituido con un sustituyente seleccionado del grupo consistente en Het1, -C(=O)-Het1 y -NH-C(=O)-Het1.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R3 representa alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en Het1 y -C(=O)-Het1 ;
en particular, R3 representa alquilo C1-4 sustituido con un Het1.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R3 representa alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en Het1, -C(=O)-Het1 y -NH-C(=O)-Het1.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde R3 representa alquilo C1-4 sustituido con un sustituyente Het1; en particular, R3 representa alquilo C1-4 sustituido con un sustituyente Het1a , en donde Het1a se define como Het1, con la condición, sin embargo, que Het1a esté siempre unido a alquilo C1-4a través de un átomo de nitrógeno del anillo.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde es aplicable la siguiente condición: cuando Y representa -NH-; entonces R3 representa alquilo C1-4 sustituido con un sustituyente Het1 ; en particular, cuando Y representa -NH-, entonces R3 representa alquilo C1-4 sustituido con un sustituyente Het1a, en el que Het1a se define como Het1 siempre que, sin embargo, Het1a esté siempre unido a alquilo C1-4 a través de un átomo de nitrógeno en el anillo.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde puede aplicarse la siguiente condición: cuando Y representa -NH-;
entonces R3 representa alquilo C1-4; o alquilo C1-4 sustituido con un sustituyente -OH.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde Het1 representa un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo, cada uno independientemente seleccionado de S(=O)p y N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes, cada uno independientemente seleccionado del grupo que consiste en -NH2, alquilo C1-4, -S(=O)2-alquilo C1-6, hidroxi y alquilo C1-4 sustituido con un hidroxi; o dos sustituyentes en el mismo átomo de carbono de dicho heterociclilo saturado de 4, 5 o 6 miembros se toman juntos para formar, junto con el átomo de carbono común al que están unidos, el Anillo A.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde Het1 representa un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo, cada uno independientemente seleccionado de S(=O)p y N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes, cada uno independientemente seleccionado del grupo que consiste en -NH2, alquilo C1-4, -S(=O)2-alquilo C1-6, hidroxi y alquilo C1-4 sustituido con un hidroxi.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona
en cualquiera de las otras realizaciones, en donde Anillo A representa un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo, cada uno independientemente seleccionado de S(=O)p y N.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde Anillo A representa ciclobutilo, ciclopentilo, ciclohexilo o un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo, cada uno independientemente seleccionado de S(=O)p y N; dicho ciclobutilo, ciclopentilo, ciclohexilo o heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con un sustituyente hidroxi.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde Het1 representa un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo, cada uno independientemente seleccionado de O, S, S(=O)p y N; y 2 sustituyentes en el mismo átomo de carbono de dicho heterociclilo saturado de 4, 5 o 6 miembros se toman juntos para formar, junto con el átomo de carbono al que están unidos, Anillo A.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde Het1 representa un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de O, S, S(=O)p y N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes cada uno seleccionado independientemente del grupo que consiste en -NH 2, alquilo C1-4, -S(=O)2-alquilo C1-6, -alquil Ci -4-S(=O)2-alquilo C1-6, hidroxi, y alquilo C1-4 sustituido con un hidroxi.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde Het1 representa un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo, cada uno independientemente seleccionado de O, S, S(=O)p y N; p representa 2.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde Het1 representa un heterociclilo saturado de 4, 5 o 6 miembros que contiene un S(=O)p y que contiene también un N; p representa 2.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales de adición farmacéuticamente aceptables, y los solvatos de los mismos, o cualquier subgrupo de los mismos como se menciona en cualquiera de las otras realizaciones, en donde Het1 representa un heterociclilo saturado de 4, 5 o 6 miembros que contiene un S(=O)p y que también contiene un N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes cada uno seleccionado independientemente del grupo que consiste en -NH2, alquilo C1-4, -S(=O)2-alquilo C1-6, -alquil C1-4-S(=O)2-alquilo C1-6, hidroxi y alquilo C1-4 sustituido con un hidroxi;
p representa 2.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
Het1 representa

En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
Het1 representa

opcionalmente sustituido con uno o dos sustituyentes, cada uno seleccionado independientemente del grupo que consiste en —NH2, alquilo 1-4, -S(=O)2-alquilo C1-6, -alquil C i -4-S(=O)2-alquilo C1-6, hidroxi y alquilo C1-4 sustituido con un hidroxi. En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde Het1 representa

En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R3 representa alquilo C1-4; -CH(OH)-CH2-Rq; o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -NH2, -O-(C=O)-alquilo C1-4,
-(C=O)-O-alquilo C1-4, -NH-(C=O)-alquilo C1-4, -NH-(SO2)-alquilo C1-4,
-N(CH 3)-alquil C1-4-SO2-CH3, -NH-alquil C1-4-SO2-CH3, -N(CH3)-alquil C1-4-OH,
-N(C=O-alquil C 1-4)-alquil C1-4-OH, -(C=O)-NH-alquil C1-4-OH,
-O-(C=O)-CH(NH2)-alquilo C1-4, -O-(C=O)-CH(NH2)-alquil C 1-4-Ar,

-NH-alquil C1-4-OH, Het1, y -C(=O)-Het1;
y en donde Het1 representa

En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde Het1 representa

Z1 representa -NH-, -S-, -O- o -S(O)2-; en particular Z1 representa -S(O)2-;
n representa 0 , 1 o 2 ;
m representa 1 , 2 o 3 ; con la condición, sin embargo, de que m no tenga el valor 1 cuando n es 0.
En una realización particular, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde Het1 está unido al resto de la molécula de Fórmula (I) a través de un átomo de nitrógeno.
En una realización particular, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde Het1 está unido al resto de la molécula de Fórmula (I) a través de un átomo de carbono.
En una realización particular, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R3 representa alquilo C1-4; -C(=O)-Het1; alquilo C1-4 sustituido en el mismo átomo de carbono con un -OH y con un Het1 ; o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -NH2, -O-(C=O)-alquilo C1-4, -(C=O)-O-alquilo C1-4, -NH-(C=O)-alquilo C1-4, -NH-(SO2)-alquilo C1-4,
-N(CH3)-alquil C1-4-SO2-CH3, -NH-alquil C1-4-SO2-CH3, -N(CH3)-alquil C1-4-OH,
-N(C=O-alquil C1-4)-alquil C1-4-OH, -(C=O)-NH-alquil C1-4-OH,
-O-(C=O)-CH(NH2)-alquilo C1-4, -O-(C=O)-CH(NH2)-alquil C1-4-Ar,

-NH-alquil C1-4-OH, Het1, y -C(=O)-Het1;
en donde Het1 está unido al resto de la molécula de Fórmula (I) a través de un átomo de nitrógeno.
En una realización, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R3 representa alquilo C1-4; -CH(OH)-CH2-Rq; o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -NH 2, -O-(C=O)-alquilo C1-4,
-(C=O)-O-alquilo C1-4, -NH-(C=O)-alquilo C1-4, -NH-(SO2)-alquilo C1-4,
-N(CH3)-alquil C1-4-SO2-CH3, -NH-alquil C1-4-SO2-CH3, -N(CH3)-alquil C1-4-OH,
-N(C=O-alquil C1-4)-alquil C1-4-OH, -(C=O)-NH-alquil C1-4-OH,
-O-(C=O)-CH(NH2)-alquilo C1-4, -O-(C=O)-CH(NH2)-alquil C1-4-Ar,

-NH-alquil C1-4-OH, Het1, y -C(=O)-Het1;
en donde Het1 está unido al resto de la molécula de Fórmula (I) a través de un átomo de nitrógeno.
En una realización particular, la presente invención se refiere a los compuestos de Fórmula (I) y los N-óxidos, las sales por adición farmacéuticamente aceptables y los solvatos de los mismos, o cualquier subgrupo de los mismos tal como se menciona en cualquiera de las otras realizaciones, en donde
R3 representa alquilo C1-4; -C(=O)-Het1 ; -CH(OH)-CH2-Rq; alquilo C1-4 sustituido en el mismo átomo de carbono con un -OH y con un Het1b; o alquilo C1-4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -NH2,
-O-(C=O)-alquilo C1-4, -(C=O)-O-alquilo C1-4, -NH-(C=O)-alquilo C1-4, -NH-(SO2)-alquilo C1-4, -N(CH3)-alquil C1-4-SO2-CH3, -NH-alquil C1-4-SO2-CH3, -N(CH3)-alquil C1-4-OH,
-N(C=O-alquil C1-4)-alquil C1-4-OH, -(C=O)-NH-alquil C1-4-OH,
-O-(C=O)-CH(NH2)-alquilo C1-4, -O-(C=O)-CH(NH2)-alquil C1-4-Ar,

-NH-alquil C1-4-OH, Het1a, -O-C(=O)-alquil C1-4-Het1a, -C(=O)-Het1, y -NH-C(=O)-Het1b;
Rq representa Het1a, halo, -OH, -NH 2, -O-(C=O)-alquilo C1-4, -NH-(C=O)-alquilo C1-4,
-NH-(SO2)-alquilo C1-4, -N(CH3)-alquil C1-4-SO2-CH3, -NH-alquil C1-4-SO2-CH3,
-N(CH3)-alquil C1-4-OH, -O-(C=O)-CH(NH2)-alquilo C1-4,
-O-(C=O)-CH(NH2)-alquil C1-4-Ar,

o -NH-alquil C1-4-OH;
Het1 representa un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de O, S, S(=O)p y N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes cada uno seleccionado independientemente del grupo que consiste en halo, -NH2, alquilo C1-4, -S(=O)2-alquilo C1-6, -alquil C1-4-S(=O)2-alquilo C1-6, hidroxi y alquilo C1-4 sustituido con un hidroxi; o dos sustituyentes en el mismo átomo de carbono de dicho heterociclilo saturado de 4, 5 o 6 miembros se toman juntos para formar junto con el átomo de carbono común al que están unidos el Anillo A;
Het1a se define como Het1, con la condición, sin embargo, que Het1a esté siempre unido al resto de R3 a través de un átomo de nitrógeno del anillo;
Het1b se define como Het1, con la condición, sin embargo, que Het1b esté siempre unido al resto de R3 a través de un átomo de carbono del anillo.
En una realización, el compuesto de Fórmula (I) se selecciona del grupo que consiste en los compuestos 1, 21, 39 y 46, tautómeros y formas estereoisoméricas de los mismos,
y los N-óxidos, las sales de adición farmacéuticamente aceptables y los solvatos de los mismos.
En una realización el compuesto de Fórmula (I) se selecciona del grupo que consiste en los compuestos 1,21,39 y 46.
Se considera que todas las combinaciones posibles de las realizaciones indicadas anteriormente quedan incluidas en el alcance de esta invención.
Métodos para la preparación de compuestos de fórmula (I)
En esta sección, al igual que en todas las otras secciones, a menos que el contexto lo indique de otro modo, referencias a la Fórmula (I) también incluyen todos los otros sub-grupos y ejemplos de los mismos según se define en el presente documento.
La preparación general de algunos ejemplos típicos de los compuestos de Fórmula (I) se describe aquí en lo que sigue y en los ejemplos específicos, y generalmente se preparan a partir de materiales de partida que están comercialmente disponibles o que se preparan por procedimientos sintéticos convencionales, comúnmente utilizados por los expertos en
la técnica. Se pretende que los siguientes esquemas únicamente representen ejemplos de la invención y no se pretende que limiten la invención de modo alguno.
Alternativamente, compuestos de la presente invención también se pueden preparar mediante protocolos de reacción análogos tal como se describe en los esquemas de reacción que figuran más adelante, combinados con procedimientos sintéticos convencionales, comúnmente utilizados por los expertos en la técnica de la química orgánica. Por ejemplo, el experto en la técnica apreciará que para algunos esquemas generales, la química análoga, tal como se indica para X2 está limitada a N o CH, también puede adaptarse para que X2 sea CR1 en general. Debe entenderse que deberían aplicarse grupos protectores adecuados. Aunque los esquemas a continuación se centran en los compuestos de Fórmula (I), en donde Y representa -CH 2-, los expertos apreciarán que se puede aplicar una química análoga en combinación con procesos sintéticos estándar para sintetizar el compuesto de Fórmula (I), en donde Y representa -NH- (véase también el Esquema 19).
El experto comprenderá que en las reacciones descritas en los Esquemas, puede ser necesario proteger grupos funcionales reactivos, por ejemplo, grupos hidroxi, amino o carboxi, cuando se desea que estén presentes en el producto final, para evitar su participación no deseada en las reacciones. Se pueden utilizar grupos protectores convencionales de acuerdo con la práctica habitual. Esto se ilustra en los ejemplos específicos. Los grupos protectores se podrán eliminar en una etapa posterior conveniente utilizando métodos conocidos en la técnica.
El experto comprenderá que en las reacciones descritas en los Esquemas, puede ser aconsejable o necesario realizar la reacción en una atmósfera inerte, tal como, por ejemplo en una atmósfera de gas de N2.
Será evidente para el experto que puede que sea necesario enfriar la mezcla de reacción antes del tratamiento posterior de la reacción (esto se refiere a la serie de manipulaciones requeridas para aislar y purificar el producto o los productos de una reacción química tal como, por ejemplo, la inactivación, cromatografía en columna, extracción).
El experto comprenderá que calentar la mezcla de reacción con agitación puede mejorar el resultado de la reacción. En algunas reacciones se puede utilizar un calentamiento por microondas en lugar del calentamiento convencional para acortar el tiempo de reacción global.
Las condiciones de reacción en los esquemas generales que se refieren a "condiciones selladas", se refieren a un recipiente de reacción sellado en donde la presión aumenta a medida que el disolvente se vuelve más volátil. Aunque típicamente este no es un requisito absoluto para el éxito en las reacciones de los esquemas a continuación, esto típicamente llevará a tiempos de reacción reducidos.
El experto comprenderá que otra secuencia de las reacciones químicas que se muestran en los siguientes Esquemas también podrá dar como resultado el compuesto de Fórmula (I) deseado.
La persona experta reconocerá que los compuestos intermedios y los compuestos finales mostrados en los esquemas que figuran más adelante se pueden funcionalizar adicionalmente de acuerdo con métodos bien conocidos por la persona experta en la técnica.
Tal como se ha mencionado anteriormente, el prefijo "Cx-y” (en que x e y son números enteros), tal como se utilizan en el presente documento, se refiere al número de átomos de carbono en un grupo dado. La persona experta reconocerá que C0 corresponde a un enlace covalente. Por lo tanto, el término "alquilo C0-3" como un grupo o parte de un grupo, se refiere a un enlace covalente (C0) y un radical hidrocarbilo de Fórmula CnH2n+1 en donde n es un número que varía de 1 a 3.
Algunos compuestos en los esquemas generales podrían ser ejemplos ilustrativos.
En general, los compuestos de Fórmula (I) en donde R1 está restringido a hidrógeno, y en donde las otras variables son como se muestran en la Fórmula (Ia), pueden prepararse de acuerdo con el siguiente Esquema de reacción 1, en donde W1 y W2 representan un grupo saliente tal como Cl, Br o I. Todas las demás variables en el Esquema 1 se definen de acuerdo con el alcance de la presente invención.
Esquema 1

1: en caso de R2H:
- Sin disolvente a una temperatura adecuada tal como 100 °C.
- Como alternativa, en presencia de un ligando adecuado, tal como 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (Ruphos) o 2-Diciclohexilfosfino-2'-(N,N-dimetilamino)bifenilo (Davephos), un catalizador adecuado, tal como, por ejemplo, tris(dibencilidenacetona)dipaladio (Pd2dba3) o acetato de paladio, una base adecuada, tal como, por ejemplo, Cs2CO3 , y un disolvente adecuado, tal como, por ejemplo, 2-metil-2-butanol o dioxano, a una temperatura adecuada tal como, por ejemplo, entre 100 y 120 °C;
en el caso de R2B(OH)2 o R2(4,4,5,5-tetrametil-1,3,2-dioxaborolano), en presencia de un catalizador adecuado, tal como, por ejemplo, aducto de dicloruro de 1,1'-bis(difenilfosfino)ferroceno paladio (II) diclorometano, una base adecuada, tal como, por ejemplo, fosfato de potasio, y un disolvente adecuado, tal como, por ejemplo, una mezcla de dioxano y agua, a una temperatura adecuada que varía entre 80 °C y 105 °C;
2: en presencia de un catalizador adecuado, tal como, por ejemplo, acetato de paladio (Pd(OAc)2), un ligando adecuado, tal como, por ejemplo, tetraquistrifenil fosfina (P(Ph)3), una base adecuada, tal como, por ejemplo, carbonato potásico (K2CO3), en un disolvente adecuado, tal como, por ejemplo, 1,4-dioxano a una temperatura adecuada, tal como, por ejemplo, 100 °C, en condiciones selladas.
En general, los compuestos de Fórmula (I) en donde R1 está restringido a un hidrógeno, y en donde las otras variables son como se muestran en la Fórmula (Ib), (Ica), (Icb) e (Id), pueden prepararse de acuerdo con el siguiente Esquema de reacción 2, en donde W3 representa un grupo saliente, tal como Cl, Br o I. Todas las demás variables en el Esquema 2 se definen de acuerdo con el alcance de la presente invención, o como se han definido anteriormente en el presente documento.
Esquema 2

1: en caso de R2H:
- Sin disolvente a una temperatura adecuada tal como 100 °C.
- Como alternativa, en presencia de un ligando adecuado, tal como 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (Ruphos) o 2-Diciclohexilfosfino-2'-(N,N-dimetilamino)bifenilo (Davephos), un catalizador adecuado, tal como, por ejemplo, tris(dibencilidenacetona)dipaladio (Pd2dba3) o acetato de paladio, una base adecuada, tal como, por ejemplo, Cs2CO3 , y un disolvente adecuado, tal como, por ejemplo, 2-metil-2-butanol o dioxano, a una temperatura adecuada tal como, por ejemplo, entre 100 y 120 °C;
en el caso de R2B(OH)2 o R2(4,4,5,5-tetrametil-1,3,2-dioxaborolano), en presencia de un catalizador adecuado, tal como, por ejemplo, aducto de dicloruro de 1,1'-bis(difenilfosfino)ferroceno paladio (II) diclorometano o paladaciclo RuPhos, una base adecuada, tal como, por ejemplo, fosfato de potasio, y un disolvente adecuado, tal como, por ejemplo, una mezcla de dioxano y agua, a una temperatura adecuada que varía entre 80 °C y 105 °C;
2: en condiciones selladas, en presencia de un tamiz molecular (4 Á), en un disolvente adecuado, tal como, por ejemplo, etilenglicol dimetil éter (DME), a una temperatura adecuada, tal como, por ejemplo, 80 °C;
3: en condiciones selladas, opcionalmente en presencia de una base adecuada, tal como, por ejemplo, hidrogenocarbonato sódico (NaHCO3), en un disolvente adecuado, tal como, por ejemplo, etilenglicol dimetil éter (Dm E) o acetonitrijo (ACN), a una temperatura adecuada que varía entre 60 a 80 °C, opcionalmente en presencia de un tamiz molecular (4 Á);
4: en condiciones selladas, en presencia de un catalizador adecuado, tal como, por ejemplo, acetato de paladio (Pd(OAc)2, un ligando adecuado, tal como, por ejemplo, tetraquistrifenil fosfina (P(Ph)3), una base adecuada, tal como, por ejemplo, carbonato potásico (K2CO3), en un disolvente adecuado, tal como, por ejemplo, 1,4-dioxano a una temperatura adecuada, tal como, por ejemplo, 100 °C;
5 : en presencia de una base adecuada, tal como, por ejemplo, hidróxido de litio, en un disolvente adecuado, tal como, por ejemplo, una mezcla de metanol y agua, a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente;
6 : En presencia de un agente reductor adecuado, tal como, por ejemplo, hidruro de litio y aluminio o borohidruro de litio, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, a una temperatura adecuada que varía entre 0 a la temperatura ambiente.
En general, los compuestos de Fórmula (I) en donde R1 está restringido a COOH, CONH2 y CH2OH, y en donde las otras variables son como se muestran en la Fórmula (Ie), (If) e (Ig), pueden prepararse de acuerdo con el siguiente Esquema de reacción 3 , en donde W4 representa un grupo saliente, tal como Cl o Br. Todas las demás variables en el Esquema 3 se definen de acuerdo con el alcance de la presente invención, o como se han definido anteriormente en el presente documento.
Esquema 3
1: en caso de R2H:
- Sin disolvente a una temperatura adecuada tal como 100 °C.
- Como alternativa, en presencia de un ligando adecuado, tal como, 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (Ruphos) o 2-Diciclohexilfosfino-2'-(N,N-dimetilamino)bifenilo (Davephos), un catalizador adecuado, tal como, por ejemplo, tns(dibencilidenacetona)dipaladio (Pd2dba3) o acetato de paladio, una base adecuada, tal como, por ejemplo, Cs2CO3 , y un disolvente adecuado, tal como, por ejemplo, 2-metil-2-butanol o dioxano, a una temperatura adecuada, tal como, por ejemplo, entre 100 y 120 °C;
en el caso de R2B(OH)2 o R2(4,4,5,5-tetrametil-1,3,2-dioxaborolano), en presencia de un catalizador adecuado, tal como, por ejemplo, aducto de dicloruro de 1,1'-bis(difenilfosfino)ferroceno paladio (II) diclorometano o paladaciclo RuPhos, una base adecuada, tal como, por ejemplo, fosfato de potasio, y un disolvente adecuado, tal como, por ejemplo, una mezcla de dioxano y agua, a una temperatura adecuada que varía entre 80 °C y 105 °C;
2: en un autoclave, en presencia de un catalizador adecuado, tal como, por ejemplo, acetato de paladio, un ligando adecuado, tal como, por ejemplo, 1,3-bis(difenilfosfino)propano, en presencia de una base adecuada, tal como, por ejemplo, acetato de potasio en un disolvente adecuado, tal como, por ejemplo, metanol, a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente;
3: en condiciones selladas, en presencia de un catalizador adecuado, tal como, por ejemplo, acetato de paladio (Pd(OAc)2, un ligando adecuado, tal como, por ejemplo, tetraquistrifenil fosfina (P(Ph)3), una base adecuada, tal como, por ejemplo, carbonato potásico (K2CO3), en un disolvente adecuado, tal como, por ejemplo, 1,4-dioxano a una temperatura adecuada, tal como, por ejemplo, 100 °C;
4: en condiciones selladas, en un disolvente adecuado, tal como, por ejemplo, metanol, a una temperatura adecuada, tal como, por ejemplo, 90 °C;
5: en presencia de una base adecuada, tal como, por ejemplo, hidróxido de litio, en un disolvente adecuado, tal como, por ejemplo, una mezcla de tetrahidrofurano y agua, a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente;
6: en presencia de un agente reductor adecuado, tal como, por ejemplo, hidruro de litio y aluminio, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente.
En general, los compuestos de Fórmula (I), en donde R1 está restringido a NH2 y siendo R1a

y en donde las otras variables son como se muestran en la Fórmula (Ih) e (Ii), pueden prepararse de acuerdo con el siguiente Esquema de reacción 4. PG se define como un grupo protector, tal como, por ejemplo, un resto N,N-dimetilsulfonamidilo o 2-tetrahidropiranilo. Todas las demás variables en el Esquema 4 se definen de acuerdo con el alcance de la presente invención, o como se han definido anteriormente en el presente documento.
Esquema 4

En el esquema 4 , se aplican las siguientes condiciones de reacción:
1: en el caso de (PG)R1aB(OH)2 o (PG)R1a(4,4,5,5-tetrametil-1,3,2-dioxaborolano), en presencia de un catalizador adecuado, tal como, por ejemplo, aducto de dicloruro de 1,1'-bis(difenilfosfino)ferroceno paladio (II) diclorometano, una base adecuada, tal como, por ejemplo, fosfato de potasio, y un disolvente adecuado, tal como, por ejemplo, una mezcla de dioxano y agua, a una temperatura adecuada que varía de 80 a 100 °C;
En el caso de R1a(PG), en primer lugar, en presencia de cloruro de cinc, un agente de desprotonación adecuado, tal como, por ejemplo, butil litio, un disolvente adecuado, tal como, por ejemplo, THF, a una temperatura adecuada, tal como, por ejemplo, -78 °C, seguido de la adición (de/a) esta solución (a) una mezcla del intermedio (L), opcionalmente en solución en THF, y un catalizador adecuado, tal como, por ejemplo, Pd(PPh3)4 , con calentamiento a una temperatura adecuada que varía de 60 a 100 °C;
2: en presencia de un catalizador adecuado, tal como, por ejemplo, acetato de paladio, en presencia de un ligando adecuado, tal como, por ejemplo, 2,2'-bis(difenilfosfino)-1,1 '-binaftilo (BINAP), una base adecuada, tal como, por ejemplo, carbonato de cesio, a una temperatura adecuada, tal como, por ejemplo, 100 °C, en condiciones selladas;
3: en condiciones selladas, en presencia de un catalizador adecuado, tal como, por ejemplo, acetato de paladio (Pd(OAc)2, un ligando adecuado, tal como, por ejemplo, tetraquistrifenil fosfina (P(Ph)3), una base adecuada, tal como, por ejemplo, carbonato potásico (K2CO3), en un disolvente adecuado, tal como, por ejemplo, 1,4-dioxano a una temperatura adecuada, tal como, por ejemplo, 100 °C;
4: en presencia de un ácido adecuado, tal como, por ejemplo, ácido clorhídrico, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, a una temperatura adecuada, tal como, por ejemplo, 60 °C;
5: en presencia de un ácido adecuado, tal como, por ejemplo, ácido clorhídrico, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, a una temperatura adecuada que varía de la temperatura ambiente a 60 °C.
En general, los compuestos de Fórmula (I), en donde las otras variables son como se muestran en la Fórmula (Ii), pueden prepararse de acuerdo con el siguiente Esquema de reacción 5, en donde W5 representa un grupo saliente, tal como Br o I. Todas las demás variables en el Esquema 5 se definen como anteriormente o de acuerdo con el alcance de la presente invención.
Esquema 5

En el esquema 5, se aplican las siguientes condiciones de reacción:
1: en caso de R2H:
- Sin disolvente a una temperatura adecuada, tal como, variando entre 100 °C y 175 °C en condiciones selladas o por irradiación de microondas;
- Como alternativa, en presencia de un ligando adecuado, tal como, 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (Ruphos), un catalizador adecuado, tal como, por ejemplo, cloro[2-(diciclohexilfosfino)-3,6-dimetoxi-2',4',6'-tri-i-propil-1,1'-bifenil][2-(2-aminoetil)fenil]paladio (II) (precatalizador Brettphos primera gen.), una base adecuada, tal como, por ejemplo, íerc-butilato potásico, y un disolvente adecuado, tal como, por ejemplo, dioxano, a una temperatura adecuada, tal como, por ejemplo, entre 120 °C, en condiciones selladas;
en el caso de R2B(OH)2 o R2 (4,4,5,5-tetrametil-1,3,2-dioxaborolano), en presencia de un catalizador adecuado, tal como, por ejemplo, aducto de dicloruro de 1,1'-bis(difenilfosfino)ferroceno paladio (II) diclorometano o paladaciclo RuPhos, una
base adecuada, tal como, por ejemplo, fosfato de potasio, y un disolvente adecuado, tal como, por ejemplo, una mezcla de dioxano y agua, a una temperatura adecuada que varía entre 80 °C y 105 °C;
2 : en presencia de un catalizador adecuado, tal como, por ejemplo, acetato de paladio (Pd(OAc)2, un ligando adecuado, tal como, por ejemplo, tetraquistrifenil fosfina (P(Ph)3), una base adecuada, tal como, por ejemplo, carbonato potásico (K2CO3), en un disolvente adecuado, tal como, por ejemplo, 1 ,4 -dioxano a una temperatura adecuada, tal como, por ejemplo, que varía entre 100 a 140 °C, eventualmente en condiciones de microondas.
En general, los compuestos de Fórmula (I), en donde las otras variables son como se muestran en la Fórmula (Ija), (Ijb) e (Ik), pueden prepararse de acuerdo con el siguiente Esquema de reacción 6. Todas las demás variables en el Esquema 6 se definen según anteriormente o según el alcance de la presente invención.
Esquema 6
1: en un disolvente adecuado, tal como, por ejemplo, dimetilformamida, a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente
2: en caso de R2H:
- En presencia de una base adecuada, sin disolvente a una temperatura adecuada, tal como, temperatura ambiente - Como alternativa, en presencia de un ligando adecuado, tal como, 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (Ruphos) o 2-Diciclohexilfosfino-2'-(N,N-dimetilamino)bifenilo (Davephos), un catalizador adecuado, tal como, por ejemplo, tris(dibencilidenacetona)dipaladio (Pd2dba3) o acetato de paladio o cloro[2-(diciclohexilfosfino)-3,6-dimetoxi-2',4',6'-tri-ipropil-1,1 '-bifenil][2-(2-aminoetil)fenil]paladio (II) (precatalizador Brettphos de primera gen.), una base adecuada, tal como, por ejemplo, Cs2CO3 o terc-butóxido potásico, y un disolvente adecuado, tal como, por ejemplo, 2-metil-2-butanol o dioxano, a una temperatura adecuada, tal como, por ejemplo, entre 100 y 120 °C, opcionalmente en condiciones selladas; en el caso de R2B(OH)2 o R2 (4,4,5,5-tetrametil-1,3,2-dioxaborolano), en presencia de un catalizador adecuado, tal como, por ejemplo, aducto de dicloruro de 1,1'-bis(difenilfosfino)ferroceno paladio (II) diclorometano o paladaciclo RuPhos, una base adecuada, tal como, por ejemplo, fosfato de potasio, y un disolvente adecuado, tal como, por ejemplo, una mezcla de dioxano y agua, a una temperatura adecuada que varía entre 80 °C y 105 °C;
3: en condiciones selladas, en presencia de un catalizador adecuado, tal como, por ejemplo, acetato de paladio (Pd(OAc)2, un ligando adecuado, tal como, por ejemplo, tetraquistrifenil fosfina (P(Ph)3), una base adecuada, tal como, por ejemplo, carbonato potásico (K2CO3), en un disolvente adecuado, tal como, por ejemplo, 1,4-dioxano a una temperatura adecuada, tal como, por ejemplo, 100 °C;
4: En presencia de un agente reductor adecuado, tal como, por ejemplo, hidruro de diisobutilaluminio, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, a una temperatura adecuada, como, por ejemplo, entre 0 °C y temperatura ambiente.
En general, los compuestos de Fórmula (I), en donde R1 está restringido a un hidrógeno, y en donde las otras variables son como se muestran en la Fórmula (Il), pueden prepararse de acuerdo con el siguiente Esquema de reacción 7. Todas las otras variables en el Esquema 7 se definen de acuerdo con el alcance de la presente invención.
Esquema 7

En el esquema 7, se aplican las siguientes condiciones de reacción:
1: en caso de R2H:
- Como alternativa, en presencia de un ligando adecuado, tal como, 2-diciclohexilfosfino-2,,6,-diisopropoxibifenilo (Ruphos), un catalizador adecuado, tal como, por ejemplo, cloro[2-(diciclohexilfosfino)-3,6-dimetoxi-2',4',6'-tri-i-propil-1,1'-bifenil][2-(2-aminoetil)fenil]paladio (II) (precatalizador Brettphos de primera gen.), una base adecuada, tal como, por ejemplo, carbonato de cesio, y un disolvente adecuado, tal como, por ejemplo, 2-metil-2butanol a una temperatura adecuada, tal como, por ejemplo, entre 100 °C, en un reactor schlenk;
en el caso de R2B(OH)2 o R2(4,4,5,5-tetrametil-1,3,2-dioxaborolano), en presencia de un catalizador adecuado, tal como, por ejemplo, aducto de dicloruro de 1,1'-bis(difenilfosfino)ferroceno paladio (II) diclorometano o paladaciclo RuPhos, una base adecuada, tal como, por ejemplo, fosfato de potasio, y un disolvente adecuado, tal como, por ejemplo, una mezcla de dioxano y agua, a una temperatura adecuada que varía entre 80 °C y 105 °C;
2: en presencia de un agente de halogenación, tal como, por ejemplo, W-bromosuccinimida o W-yodosuccinimida, en un disolvente adecuado, tal como, por ejemplo, acetonitrilo a una temperatura adecuada, tal como, por ejemplo, 0 °C;
3: en presencia de un catalizador adecuado, tal como, por ejemplo, ¿>/s(tri-terc-butilfosfina paladio (0), en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano a una temperatura adecuada, tal como, por ejemplo, 60 °C, en un reactor schlenk.
En general, los compuestos de Fórmula (I), en donde R1 está restringido a hidrógeno, y en donde
las otras variables son como se muestran en la Fórmula (Im) e (In), pueden prepararse de acuerdo con el siguiente Esquema de reacción 8. En el esquema 8, Rx y Ry representan alquilo C1-4, y Rz representa alquilo C1-4 o fenilo, por ejemplo, Rx y Ry representan CH3 y Rz representa C(c Hs)3 o fenilo. Todas las otras variables en el Esquema 8 se definen de acuerdo con el alcance de la presente invención.
Esquema 8

En el Esquema 8, se aplican las siguientes condiciones de reacción:
1: en presencia de un reactivo adecuado, tal como, por ejemplo, imidazol, en un disolvente adecuado, tal como, por ejemplo, dimetilformamida, a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente;
2: en presencia de una base, tal como, por ejemplo, hidróxido sódico acuoso, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano o una mezcla de tetrahidrofurano y etanol, a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente;
3 : en presencia de un catalizador adecuado, tal como, por ejemplo, bis(tri-terc-butilfosfinapaladio (0 ), en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano a una temperatura adecuada, tal como, por ejemplo, 60 °C, en un reactor schlenk;
4 : en presencia de un agente de halogenación, tal como, por ejemplo, N-bromosuccinimida o N-yodosuccinimida, en un disolvente adecuado, tal como, por ejemplo, acetonitrilo a una temperatura adecuada, tal como, por ejemplo, 0 °C;
5: en presencia de un agente de desililación adecuado, tal como, por ejemplo, fluoruro de tetrabutilamonio, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente.
En general, los intermedios de Fórmula (XXXVII) y (XXXVIII), en donde R1 está restringido a un hidrógeno, pueden prepararse de acuerdo con el siguiente Esquema de reacción 9. Todas las otras variables en el Esquema 9 se definen de acuerdo con el alcance de la presente invención.
Esquema 9

- Sin disolvente, a una temperatura adecuada, tal como 110 °C;
- Como alternativa, en presencia de una base adecuada, tal como, por ejemplo, trimetilamina o diisopropiletilamina, en un disolvente adecuado, tal como, por ejemplo, dimetilsulfóxido o acetonitrilo, a una temperatura adecuada que varía entre 80 y 120 °C;
- Como alternativa, en presencia de un ligando adecuado, tal como, 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (Ruphos) o 2-Diciclohexilfosfino-2'-(N,N-dimetilamino)bifenilo (Davephos), un catalizador adecuado, tal como, por ejemplo, tris(dibencilidenacetona)dipaladio (Pd2dba3) o acetato de paladio, una base adecuada, tal como, por ejemplo, Cs2CO3, y un disolvente adecuado, tal como, por ejemplo, 2-metil-2-butanol o dioxano, a una temperatura adecuada, tal como, por ejemplo, entre 100 y 120 °C;
en el caso de R2B(OH)2 o R2(4,4,5,5-tetrametil-1,3,2-dioxaborolano), en presencia de un catalizador adecuado, tal como, por ejemplo, cloruro de Bis(trifenilfosfina)-paladio (II) o aducto de dicloruro de 1,1 '-bis(difenilfosfino)ferroceno paladio (II) diclorometano, una base adecuada, tal como, por ejemplo, carbonato de potasio y sodio o fosfato de potasio, y un disolvente adecuado, tal como, por ejemplo, una mezcla de dioxano y agua, a una temperatura adecuada, tal como, por ejemplo, 80 °C;
2: en presencia de un catalizador adecuado, tal como, por ejemplo, paladio sobre carbón, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano o etanol, bajo 1 a 3 bares de hidrógeno,
Como alternativa, en presencia de un metal adecuado, tal como, por ejemplo, cinc, una sal adecuada, tal como, por ejemplo, cloruro de amonio, en un disolvente adecuado, tal como, por ejemplo, metanol, a una temperatura adecuada, tal como, que varía entre 0 a 5 °C;
3 : opcionalmente en presencia de una base adecuada, tal como, por ejemplo, hidrogenocarbonato sódico (NaHCOs), en un disolvente adecuado, tal como, por ejemplo, etilenglicol dimetil éter (DME) o acetonitrilo (ACN) o etanol, a una temperatura adecuada, tal que varía entre 60 a 120 °C, opcionalmente en presencia de un tamiz molecular ( 4 Á), en condiciones selladas o por irradiación de microondas;
4 : en un reactor schlenck, en un disolvente adecuado, tal como, por ejemplo, dimetilformamida, a una temperatura adecuada, tal como, por ejemplo, 1 20 °C;
5 : en presencia de un agente reductor adecuado, tal como, por ejemplo, borohidruro de litio, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, a una temperatura adecuada, tal como, por ejemplo, 5 0 °C; opcionalmente en condiciones selladas;
6 : en presencia de un agente de halogenación, tal como, por ejemplo, N-bromosuccinimida o N-yodosuccinimida, en un disolvente adecuado, tal como, por ejemplo, acetonitrilo a una temperatura adecuada, tal como, por ejemplo, 0 °C.
Como alternativa, los intermedios de Fórmula (XXXIX), en donde R1 está restringido a un hidrógeno, pueden prepararse de acuerdo con el siguiente Esquema de reacción 1 0. Todas las demás variables en el Esquema 10 se definen de acuerdo con el alcance de la presente invención, o como se han definido anteriormente en el presente documento.
Esquema 10

1 : en un reactor schlenck, sin disolvente o en un disolvente adecuado, tal como, por ejemplo, dimetilformamida, a una temperatura adecuada, tal como, por ejemplo, 120 °C;
2: en presencia de una base, tal como, por ejemplo, hidróxido sódico acuoso, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, etanol o una mezcla de tetrahidrofurano y etanol, a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente;
3: en presencia de un reactivo adecuado, tal como, por ejemplo, imidazol, en un disolvente adecuado, tal como, por ejemplo, dimetilformamida o diclorometano, a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente; 4: en caso de R2H:
- Sin disolvente, a una temperatura adecuada, tal como 110 °C;
- Como alternativa, en presencia de una base adecuada, tal como, por ejemplo, trimetilamina o diisopropiletilamina, en un disolvente adecuado, tal como, por ejemplo, dimetilsulfóxido o acetonitrilo, a una temperatura adecuada que varía entre 80 y 120 °C;
- Como alternativa, en presencia de un ligando adecuado, tal como, 2-diciclohexilfosfino-2',6'-diisopropox.ibifenilo (Ruphos) o 2-Diciclohexilfosfino-2'-(N,N-dimetilamino)bifenilo (Davephos), un catalizador adecuado, tal como, por ejemplo, tris(dibencilidenacetona)dipaladio (Pd2dba3) o acetato de paladio, una base adecuada, tal como, por ejemplo, Cs2CO3, y un disolvente adecuado, tal como, por ejemplo, 2-metil-2-butanol o dioxano, a una temperatura adecuada, tal como, por ejemplo, entre 100 y 120 °C;
en el caso de R2B(OH)2 o R2(4,4,5,5-tetrametil-1,3,2-dioxaborolano), en presencia de un catalizador adecuado, tal como, por ejemplo, cloruro de bis(trifenilfosfina)-paladio (II) o aducto de dicloruro de 1,1'-bis(difenilfosfino)ferroceno paladio (II) diclorometano, una base adecuada, tal como, por ejemplo, carbonato de potasio y sodio o fosfato potásico, y un disolvente adecuado, tal como, por ejemplo, una mezcla de dioxano y agua, a una temperatura adecuada, tal como, por ejemplo, 80 °C.
En general, los compuestos de Fórmula (I), en donde R1 está restringido a un hidrógeno, y en donde las otras variables son como se muestran en la Fórmula (Io), pueden prepararse de acuerdo con el siguiente Esquema de reacción 11. Todas las demás variables en el Esquema 11 se definen como anteriormente o de acuerdo con el alcance de la presente invención o como se han definido anteriormente en el presente documento.
Esquema 11

En el esquema 11, se aplican las siguientes condiciones de reacción:
1 :opcionalmente en presencia de una base adecuada, tal como, por ejemplo, hidrogenocarbonato sódico (NaHCO3), en un disolvente adecuado, tal como, por ejemplo, etilenglicol dimetil éter (DME) o acetonitrilo (ACN) o etanol, a una temperatura adecuada tal que varía entre 60 a 120 °C, opcionalmente en presencia de un tamiz molecular (4 Á), en condiciones selladas o por irradiación de microondas;
2: en presencia de un agente de halogenación, tal como, por ejemplo, N-bromosuccinimida o N-yodosuccinimida, en un disolvente adecuado, tal como, por ejemplo, acetonitrilo a una temperatura adecuada, tal como, por ejemplo, 0 °C;
3 : en presencia de un catalizador adecuado, tal como, por ejemplo, bis(tri-terc-butilfosfinapaladio (0 ), en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano a una temperatura adecuada, tal como, por ejemplo, 60 °C, en un reactor schlenk.
En general, los compuestos de Fórmula (I), en donde R1 está restringido a R1a que es

y en donde las otras variables son como se muestran en la Fórmula (Ip), pueden prepararse de acuerdo con el siguiente Esquema de reacción 12. Todas las otras variables en el Esquema 12 se definen de acuerdo con el alcance de la presente invención.
Esquema 12

1 : en caso de R2H:
- Sin disolvente, a una temperatura adecuada, tal como 110 °C;
- Como alternativa, en presencia de una base adecuada, tal como, por ejemplo, trimetilamina o diisopropiletilamina, en un disolvente adecuado, tal como, por ejemplo, dimetilsulfóxido o acetonitrilo, a una temperatura adecuada que varía entre 80 y 120 °C;
- Como alternativa, en presencia de un ligando adecuado, tal como, 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (Ruphos) o 2-Diciclohexilfosfino-2'-(N,N-dimetilamino)bifenilo (Davephos), un catalizador adecuado, tal como, por ejemplo, cloro[2-(diciclohexilfosfino)-3,6-dimetoxi-2',4',6'-tri-i-propil-1,1'-bifenil][2-(2-aminoetil)fenil]paladio (II) (precatalizador Brettphos de primera gen.), una base adecuada, tal como, por ejemplo, Cs2CO3, y un disolvente adecuado, tal como, por ejemplo, 2-metil-2-butanol, a una temperatura adecuada, tal como, por ejemplo, entre 100 y 120 °C, en condiciones selladas;
en el caso de R2B(OH)2 o R2(4,4,5,5-tetrametil-1,3,2-dioxaborolano), en presencia de un catalizador adecuado, tal como, por ejemplo, cloruro de bis(trifenilfosfina)-paladio (II), una base adecuada, tal como, por ejemplo, carbonato de potasio y sodio, y un disolvente adecuado, tal como, por ejemplo, una mezcla de dioxano y agua, a una temperatura adecuada, tal como, por ejemplo, 80 °C;
2: en presencia de un agente de halogenación, tal como, por ejemplo, N-bromosuccinimida o N-yodosuccinimida, en un disolvente adecuado, tal como, por ejemplo, diclorometano a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente;
3: en presencia de un catalizador adecuado, tal como, por ejemplo, bis(tri-terc-butilfosfina)paladio (0), en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano a una temperatura adecuada, tal como, por ejemplo, 60 °C, en un reactor schlenk;
4: en presencia de un ácido adecuado, tal como, por ejemplo, ácido clorhídrico, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, a una temperatura adecuada, tal como, por ejemplo, 60 °C.
En general, los intermedios de Fórmula (L) en donde R1 está restringido a está restringido a R1a que es

pueden prepararse de acuerdo con el siguiente Esquema de reacción 13. Todas las demás variables en el Esquema 13 se definen de acuerdo con el alcance de la presente invención, o como se han definido anteriormente en el presente documento.
Esquema 13

1 : a una temperatura que varía de 6 0 a 8 0 °C, en condiciones selladas;
2 : en el caso de (PG)R1aB(OH)2 o (PG)R1a(4 ,4 ,5 ,5 -tetramet¡l-1 ,3 ,2 -d¡oxaborolano), en presencia de un catalizador adecuado, tal como, por ejemplo, aducto de dicloruro de 1 ,1 '-bis(difenilfosfino)ferroceno paladio (II) diclorometano, una base adecuada, tal como, por ejemplo, fosfato de potasio, y un disolvente adecuado, tal como, por ejemplo, una mezcla de dioxano y agua, a una temperatura adecuada que varía de 8 0 a 100 °C;
En el caso de R1a(PG), en primer lugar, en presencia de cloruro de cinc, un agente de desprotonación adecuado, tal como, por ejemplo, butil litio, un disolvente adecuado, tal como, por ejemplo, THF, a una temperatura adecuada, tal como, por ejemplo, - 78 °C, seguido de la adición (de/a) esta solución (a) una mezcla del intermedio (L), opcionalmente en solución en THF, y un catalizador adecuado, tal como, por ejemplo, Pd(PPh3)4 , con calentamiento a una temperatura adecuada que varía de 6 0 a 100 °C;
3 : en presencia de una base, tal como, por ejemplo, hidróxido sódico acuoso, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, etanol o una mezcla de tetrahidrofurano y etanol, a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente;
4 : en presencia de un reactivo adecuado, tal como, por ejemplo, imidazol, en un disolvente adecuado, tal como, por ejemplo, dimetilformamida o diclorometano, a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente. En general, los compuestos de Fórmula (I), en donde R1 está restringido a R1a que es

y en donde las otras variables son como se muestran en la Fórmula (Iq), pueden prepararse de acuerdo con el siguiente Esquema de reacción 1 4. Todas las otras variables en el Esquema 14 se definen de acuerdo con el alcance de la presente invención.
Esquema 14

1 : a una temperatura que varía de 60 a 8 0 °C, en condiciones selladas;
2 : en el caso de (PG)R1aB(OH)2 o (PG)R1a(4 ,4 ,5 ,5 -tetrametil-1 ,3 ,2 -dioxaborolano), en presencia de un catalizador adecuado, tal como, por ejemplo, aducto de dicloruro de 1 ,1 '-bis(difenilfosfino)ferroceno paladio (II) diclorometano, una base adecuada, tal como, por ejemplo, fosfato de potasio, y un disolvente adecuado, tal como, por ejemplo, una mezcla de dioxano y agua, a una temperatura adecuada que varía de 8 0 a 100 °C;
En el caso de R1a(PG), en primer lugar, en presencia de cloruro de cinc, un agente de desprotonación adecuado, tal como, por ejemplo, butil litio, un disolvente adecuado, tal como, por ejemplo, THF, a una temperatura adecuada, tal como, por ejemplo, - 78 °C, seguido de la adición (de/a) esta solución (a) una mezcla del intermedio (L), opcionalmente en solución
en THF, y un catalizador adecuado, tal como, por ejemplo, Pd(PPh3)4 , con calentamiento a una temperatura adecuada que varía de 60 a 100 °C;
4 : en presencia de un agente de halogenación, tal como, por ejemplo, N-bromosuccinimida o N-yodosuccinimida, en un disolvente adecuado, tal como, por ejemplo, diclorometano a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente;
5: en presencia de un catalizador adecuado, tal como, por ejemplo, bis(tri-terc-butilfosfinapaladio (0), en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano a una temperatura adecuada, tal como, por ejemplo, 60 °C, en un reactor schlenk;
6 : en presencia de un ácido adecuado, tal como, por ejemplo, ácido clorhídrico, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, a una temperatura adecuada, tal como, por ejemplo, 60 °C.
En general, los compuestos de Fórmula (I), en donde R1 está restringido a CH2-OH y en donde las otras variables son como se muestran en la Fórmula (Ir), pueden prepararse de acuerdo con el siguiente Esquema de reacción 15, en donde PG2 es un tetrahidropiranilo o -SiRxRyRz . Todas las otras variables en el Esquema 15 se definen de acuerdo con el alcance de la presente invención.
Esquema 15

En el esquema 15, se aplican las siguientes condiciones de reacción:
1 : en presencia de una base adecuada, tal como, por ejemplo, diisopropiletilamina, en un disolvente adecuado, tal como, por ejemplo, dimetilformamida, a una temperatura que varía de 100 a 130 °C, en condiciones selladas;
2 : en presencia de un ácido adecuado, tal como, por ejemplo, p-tolueno sulfonato de piridinio, en un disolvente adecuado, tal como, por ejemplo, diclorometano, a una temperatura adecuada, tal como 50 °C, o en presencia de un reactivo adecuado, tal como, por ejemplo, imidazol, en un disolvente adecuado, tal como, por ejemplo, dimetilformamida o diclorometano, a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente;
3: en caso de R2H:
- Sin disolvente, a una temperatura adecuada, tal como 110 °C;
- Como alternativa, en presencia de una base adecuada, tal como, por ejemplo, trimetilamina o diisopropiletilamina, en un disolvente adecuado, tal como, por ejemplo, dimetilsulfóxido o acetonitrilo, a una temperatura adecuada que varía entre 80 y 120 °C;
- Como alternativa, en presencia de un ligando adecuado, tal como, 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (Ruphos) o 2-Diciclohexilfosfino-2'-(N,N-dimetilamino)bifenilo (Davephos), un catalizador adecuado, tal como, por ejemplo, cloro[2-(diciclohexilfosfino)-3,6-dimetoxi-2',4',6'-tri-i-propil-1,1 '-bifenil][2-(2-aminoetil)fenil]paladio (II) (precatalizador Brettphos de primera gen.), una base adecuada, tal como, por ejemplo, Cs2CO3, y un disolvente adecuado, tal como, por ejemplo, 2-metil-2-butanol, a una temperatura adecuada, tal como, por ejemplo, entre 100 y 120 °C, en condiciones selladas;
en el caso de R2B(OH)2 o R2(4,4,5,5-tetrametil-1,3,2-dioxaborolano), en presencia de un catalizador adecuado, tal como, por ejemplo, cloruro de bis(trifenilfosfina)-paladio (II), una base adecuada, tal como, por ejemplo, carbonato de potasio y sodio, y un disolvente adecuado, tal como, por ejemplo, una mezcla de dioxano y agua, a una temperatura adecuada, tal como, por ejemplo, 80 °C;
4: en presencia de un agente de halogenación, tal como, por ejemplo, N-bromosuccinimida o N-yodosuccinimida, en un disolvente adecuado, tal como, por ejemplo, acetonitrilo a una temperatura adecuada, tal como, por ejemplo, 0 °C;
5: en presencia de un catalizador adecuado, tal como, por ejemplo, bis(tri-terc-butilfosfinapaladio (0), en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano a una temperatura adecuada, tal como, por ejemplo, 60 °C, en un reactor schlenk;
6: en presencia de un ácido adecuado, tal como, por ejemplo, ácido clorhídrico, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, a una temperatura adecuada que varía de la temperatura ambiente a 60 °C.
En general, los compuestos de Fórmula (I), en donde R1 está restringido a CH2-OH y en donde las otras variables son como se muestran en la Fórmula (Is), pueden prepararse de acuerdo con el siguiente Esquema de reacción 16. Todas las otras variables en el Esquema 16 se definen de acuerdo con el alcance de la presente invención.
Esquema 16

1: en presencia de una base adecuada, tal como, por ejemplo, diisopropiletilamina, en un disolvente adecuado, tal como, por ejemplo, dimetilformamida, a una temperatura que varía de 100 a 130C, en condiciones selladas;
2: en presencia de un ácido adecuado, tal como, por ejemplo, p-tolueno sulfonato de piridinio, en un disolvente adecuado, tal como, por ejemplo, diclorometano, a una temperatura adecuada, tal como 50 °C, o en presencia de un reactivo adecuado, tal como, por ejemplo, imidazol, en un disolvente adecuado, tal como, por ejemplo, dimetilformamida o diclorometano, a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente;
3: en caso de R2H:
- Sin disolvente, a una temperatura adecuada, tal como 110 °C;
- Como alternativa, en presencia de una base adecuada, tal como, por ejemplo, trimetilamina o diisopropiletilamina, en un disolvente adecuado, tal como, por ejemplo, dimetilsulfóxido o acetonitrilo, a una temperatura adecuada que varía entre 80 y 120 °C;
- Como alternativa, en presencia de un ligando adecuado, tal como, 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (Ruphos) o 2-Diciclohexilfosfino-2'-(N,N-dimetilamino)bifenilo (Davephos), un catalizador adecuado, tal como, por ejemplo, cloro[2-(diciclohexirfosfino)-3,6-dimetoxi-2',4',6'-tri-i-propil-1,1 '-bifenil][2-(2-aminoetil)fenil]paladio (II) (precatalizador Brettphos de primera gen.), una base adecuada, tal como, por ejemplo, Cs2CO3, y un disolvente adecuado, tal como, por ejemplo, 2-metil-2-butanol, a una temperatura adecuada, tal como, por ejemplo, entre 100 y 120 °C, en condiciones selladas;
en el caso de R2B(OH)2 o R2(4,4,5,5-tetrametil-1,3,2-dioxaborolano), en presencia de un catalizador adecuado, tal como, por ejemplo, cloruro de bis(trifenilfosfina)-paladio (II), una base adecuada, tal como, por ejemplo, carbonato de potasio y sodio, y un disolvente adecuado, tal como, por ejemplo, una mezcla de dioxano y agua, a una temperatura adecuada, tal como, por ejemplo, 80 °C;
4: en presencia de un agente de halogenación, tal como, por ejemplo, N-bromosuccinimida o N-yodosuccinimida, en un disolvente adecuado, tal como, por ejemplo, acetonitrilo a una temperatura adecuada, tal como, por ejemplo, 0 °C;
5: en presencia de una base, tal como, por ejemplo, hidróxido sódico acuoso, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, etanol o una mezcla de tetrahidrofurano y etanol, a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente;
6: en presencia de un catalizador adecuado, tal como, por ejemplo, bis(tri-terc-butilfosfinapaladio (0), en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano a una temperatura adecuada, tal como, por ejemplo, 60 °C, en un reactor schlenk;
7: en presencia de un ácido adecuado, tal como, por ejemplo, ácido clorhídrico, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, a una temperatura adecuada que varía de la temperatura ambiente a 60 °C.
En general, los compuestos de Fórmula (I), en donde R1 está restringido a NH2 y en donde las otras variables son como se muestran en la Fórmula (It), pueden prepararse de acuerdo con el siguiente Esquema de reacción 17. Todas las otras variables en el Esquema 17 se definen de acuerdo con el alcance de la presente invención.
Esquema 17

En el esquema 17, se aplican las siguientes condiciones de reacción:
1: en presencia de un catalizador adecuado, tal como, por ejemplo, acetato de paladio, en presencia de un ligando adecuado, tal como, por ejemplo, 2,2'-bis(difenilfosfino)-1,1'-binaftilo ( BINAP), una base adecuada, tal como, por ejemplo, carbonato de cesio, a una temperatura adecuada, tal como, por ejemplo, 100 °C, en condiciones selladas;
2: en condiciones selladas, en presencia de un catalizador adecuado, tal como, por ejemplo, acetato de paladio (Pd(OAc)2, un ligando adecuado, tal como, por ejemplo, tetraquistrifenil fosfina (P(Ph)3), una base adecuada, tal como, por ejemplo, carbonato potásico (K2CO3), en un disolvente adecuado, tal como, por ejemplo, 1,4-dioxano a una temperatura adecuada, tal como, por ejemplo, 100 °C;
3: en caso de R2H:
- Sin disolvente, a una temperatura adecuada, tal como 110 °C;
- Como alternativa, en presencia de una base adecuada, tal como, por ejemplo, trimetilamina o diisopropiletilamina, en un disolvente adecuado, tal como, por ejemplo, dimetilsulfóxido o acetonitrilo, a una temperatura adecuada que varía entre 80 y 120 °C;
- Como alternativa, en presencia de un ligando adecuado, tal como, 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (Ruphos) o 2-Diciclohexilfosfino-2'-(N,N-dimetilamino)bifenilo (Davephos), un catalizador adecuado, tal como, por ejemplo, cloro[2-(diciclohexilfosfino)-3,6-dimetoxi-2',4',6'-tri-i-propil-1,1'-bifenil][2-(2-aminoetil)fenil]paladio (II) (paladaciclo Brettphos), una base adecuada, tal como, por ejemplo, Cs2CO3, y un disolvente adecuado, tal como, por ejemplo, 2-metil-2-butanol, a una temperatura adecuada, tal como, por ejemplo, entre 100 y 120 °C, en condiciones selladas;
en el caso de R2B(OH)2 o R2(4,4,5,5-tetrametil-1,3,2-dioxaborolano), en presencia de un catalizador adecuado, tal como, por ejemplo, cloruro de bis(trifenilfosfina)-paladio (II), una base adecuada, tal como, por ejemplo, carbonato de potasio y
sodio, y un disolvente adecuado, tal como, por ejemplo, una mezcla de dioxano y agua, a una temperatura adecuada, tal como, por ejemplo, 80 °C;
4 : en presencia de un ácido adecuado, tal como, por ejemplo, ácido clorhídrico, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, a una temperatura adecuada que varía de la temperatura ambiente a 6 0 °C.
En general, los compuestos de Fórmula (I), en donde R1 está restringido a NH2 y en donde las otras variables son como se muestran en la Fórmula (Iu), pueden prepararse de acuerdo con el siguiente Esquema de reacción 1 8. Todas las otras variables en el Esquema 18 se definen de acuerdo con el alcance de la presente invención.
Esquema 18

En el esquema 1 8 , se aplican las siguientes condiciones de reacción:
1 : en presencia de un catalizador adecuado, tal como, por ejemplo, acetato de paladio, en presencia de un ligando adecuado, tal como, por ejemplo, 2 ,2 '-bis(difenilfosfino)-1 ,1 ’-binaftilo ( BINAP), una base adecuada, tal como, por ejemplo, carbonato de cesio, a una temperatura adecuada, tal como, por ejemplo, 100 °C, en condiciones selladas;
2 : en condiciones selladas, en presencia de un catalizador adecuado, tal como, por ejemplo, acetato de paladio (Pd(OAc)2, un ligando adecuado, tal como, por ejemplo, tetraquistrifenil fosfina (P(Ph)3), una base adecuada, tal como, por ejemplo, carbonato potásico (K2CO3), en un disolvente adecuado, tal como, por ejemplo, 1 ,4 -dioxano a una temperatura adecuada, tal como, por ejemplo, 100 °C;
3 : en caso de R2H:
- Sin disolvente, a una temperatura adecuada, tal como 11 0 °C;
- Como alternativa, en presencia de una base adecuada, tal como, por ejemplo, trimetilamina o diisopropiletilamina, en un disolvente adecuado, tal como, por ejemplo, dimetilsulfóxido o acetonitrilo, a una temperatura adecuada que varía entre 80 y 120 °C;
- Como alternativa, en presencia de un ligando adecuado, tal como, 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (Ruphos) o 2-Diciclohexilfosfino-2'-(N,N-dimetilamino)bifenilo (Davephos), un catalizador adecuado, tal como, por ejemplo, cloro[2-(diciclohexi lfosfino)-3,6-dimetoxi-2',4’,6'-tri-i-propil-1,1'-bifenil][2-(2-aminoetil)fenil]paladio (II) (precatalizador Brettphos de primera gen.), una base adecuada, tal como, por ejemplo, Cs2CÜ3 , y un disolvente adecuado, tal como, por ejemplo, 2-metil-2-butanol, a una temperatura adecuada, tal como, por ejemplo, entre 100 y 120 °C, en condiciones selladas;
en el caso de R2B(OH)2 o R2 (4,4,5,5-tetrametil-1,3,2-dioxaborolano), en presencia de un catalizador adecuado, tal como, por ejemplo, cloruro de bis(trifenilfosfina)-paladio (II), una base adecuada, tal como, por ejemplo, carbonato de potasio y sodio, y un disolvente adecuado, tal como, por ejemplo, una mezcla de dioxano y agua, a una temperatura adecuada, tal como, por ejemplo, 80 °C;
4: en presencia de un ácido adecuado, tal como, por ejemplo, ácido clorhídrico, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, a una temperatura adecuada que varía de la temperatura ambiente a 60 °C.
En general, los compuestos de Fórmula (I), en donde R1 está restringido a un hidrógeno y en donde las otras variables son como se muestran en la Fórmula (Iv), pueden prepararse de acuerdo con el siguiente Esquema de reacción 19. Todas las otras variables en el Esquema 19 se definen de acuerdo con el alcance de la presente invención.
Esquema 19

1: en presencia de un reactivo adecuado, tal como dicloruro de cinc, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, a una temperatura adecuada, tal como 120 °C, con irradiación de microondas;
2: 3: a una temperatura tal como 100 °C o en un microondas a una temperatura de 140 °C, en presencia de un catalizador adecuado, tal como, por ejemplo, tris(dibencilidenacetona)dipaladio(0), un ligando adecuado, tal como, por ejemplo, 2-(diciclohexilfosfino)-3,6-dimetoxi-2',4,,6'-triisopropil-1,1,-bifenilo, una base adecuada, tal como, por ejemplo, carbonato de cesio, y en un disolvente adecuado, tal como, por ejemplo, tolueno;
3: En presencia de un ácido adecuado, tal como, por ejemplo, ácido clorhídrico, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, a una temperatura adecuada que varía de la temperatura ambiente a 60 °C; Como alternativa, en presencia de un agente de desililación adecuado, tal como, por ejemplo, fluoruro de tetrabutilamonio, en un disolvente adecuado, tal como, por ejemplo, tetrahidrofurano, a una temperatura adecuada, tal como, por ejemplo, temperatura ambiente.
En general, los compuestos de Fórmula (I), en donde Z representa

sus tautómeros y formas estereoisoméricas, en donde
X1 representa CH o N;
X2 representa CH o N;
siempre que como máximo uno de X1 y X2 represente N;
en donde las otras variables son como se muestran en la Fórmula (Iwa) e (Iwb), pueden prepararse de acuerdo con el siguiente Esquema de reacción 2 0 , en donde R9 se define como H o CH3 y R10 se define como -alquil C1-4-SO2-CH3 o -alquil C1-4-OH y en donde Het1a se define como Het1 unido a través de un átomo de nitrógeno. Todas las otras variables en el Esquema 20 se definen de acuerdo con el alcance de la presente invención.
Esquema 20

1 : en presencia de reactivos adecuados, tal como, por ejemplo, cloruro de oxalilo y dimetilsulfóxido, una base adecuada, tal como, por ejemplo, trimetilamina, en un disolvente adecuado, tal como, por ejemplo, diclorometano, a una temperatura adecuada que varía entre - 80 °C a temperatura ambiente, o en presencia de un reactivo oxidativo adecuado, tal como, por ejemplo, óxido de manganeso, en un disolvente adecuado, tal como, por ejemplo, diclorometano o tolueno, a una temperatura adecuada que varía de la temperatura ambiente a 80 °C;
2 : en presencia de un agente reductor adecuado, tal como, por ejemplo, triacetoxiborohidruro sódico o borohidruro sódico, en un disolvente adecuado, tal como, por ejemplo, diclorometano o metanol, dicloroetano, opcionalmente en presencia de una base orgánica adecuada, tal como, por ejemplo, acetato sódico o un ácido adecuado, tal como, por ejemplo, ácido acético, a una temperatura adecuada que varía de la temperatura ambiente a 40 °C;
3 : en el caso de un cloruro de acilo o anhídrido de acilo, opcionalmente en presencia de una base adecuada, tal como, por ejemplo, trietilamina, y en un disolvente adecuado, tal como, por ejemplo, diclorometano;
en caso de un ácido carboxílico, en presencia de un reactivo de acoplamiento adecuado, tal como por ejemplo 3 -óxido de 1 -[bis(dimetilamino)metileno] -1 H-1 ,2 ,3 -triazolo[4 ,5 -£>]piridinio, un aditivo adecuado, tal como por ejemplo dimetilaminopiridina, una base adecuada, tal como por ejemplo diisopropiletilamina, y en un disolvente adecuado, tal como por ejemplo DMF.
En general, los compuestos de Fórmula (I), en donde Z representa

sus tautómeros y formas estereoisoméricas, en donde
X1 representa CH o N;
X2 representa CH o N;
siempre que como máximo uno de X1 y X2 represente N;
en donde las otras variables son como se muestran en la Fórmula (Ix), pueden prepararse de acuerdo con el siguiente Esquema de reacción 2 1. Todas las otras variables en el Esquema 21 se definen de acuerdo con el alcance de la presente invención.
Esquema 21
Agente fluorado
Z —alquil C1-4 — O H ---------------------------- 
(Id) o ( lk )o (In) 1
En el esquema 2 1 , se aplican las siguientes condiciones de reacción:
1 : en presencia de un reactivo fluorado adecuado, tal como, por ejemplo, trifluoruro de dietilaminoazufre o tetrafluoroborato de (dietilamino)difluorosulfonio, opcionalmente en presencia de una sal adecuada, tal como, por ejemplo, trifluorhidrato de trietilamina, en un disolvente adecuado, tal como, por ejemplo, diclorometano a una temperatura adecuada, tal como temperatura ambiente;
En general, los compuestos de Fórmula (I), en donde Z representa

sus tautómeros y formas estereoisoméricas, en donde
X 1 representa CH o N;
X2 representa CH o N;
siempre que como máximo uno de X 1 y X2 represente N;
en donde las otras variables son como se muestran en la Fórmula (Iy), pueden prepararse de acuerdo con el siguiente Esquema de reacción 2 2. En el Esquema 2 2 , R11 representa -CH(NH2)-alquilo C1-4,
-CH(NH2)-alquil C 1-4-Ar,

o -alquil C 1-4-Het1, y PG1 representa un grupo protector, tal como, por ejemplo, terc-butoxicarbonilo o benciloxicarbonilo. Todas las otras variables en el Esquema 22 se definen de acuerdo con el alcance de la presente invención.
Esquema 22

En el esquema 2 2 , se aplican las siguientes condiciones de reacción:
1 : en presencia de un reactivo de acoplamiento adecuado, tal como, por ejemplo, 1 -[bis(dimetilamino)metilen]-1 H-1 ,2 ,3 -Triazolo[4 ,5 -¿>]piridinio, 3 -óxido, un aditivo adecuado, tal como, por ejemplo, dimetilaminopiridina, una base adecuada, tal como, por ejemplo, diisopropiletilamina, y en un disolvente adecuado, tal como, por ejemplo, de DMF;
2 : en presencia de un ácido, tal como por ejemplo ácido trifluoroacético o cloruro de hidrógeno en un disolvente adecuado, tal como por ejemplo diclorometano o metanol. Como alternativa, en presencia de paladio sobre carbón, en un disolvente adecuado, tal como metanol en una atmósfera de hidrógeno.
En general, los compuestos de Fórmula (I), en donde Z representa

sus tautómeros y formas estereoisoméricas, en donde
X1 representa CH o N;
X2 representa CH o N;
siempre que como máximo uno de X1 y X2 represente N;
en donde las otras variables son como se muestran en la Fórmula (Iz), pueden prepararse de acuerdo con el siguiente Esquema de reacción 2 3. En el Esquema 2 3 , Het1 está restringido a Het1b que está unido a través del átomo de carbono. Todas las otras variables en el Esquema 23 se definen de acuerdo con el alcance de la presente invención.
Esquema 23

En el esquema 2 3 , se aplican las siguientes condiciones de reacción:
1 : a una temperatura adecuada, tal como, por ejemplo, 0 °C o - 78 °C, en un disolvente adecuado, tal como, por ejemplo, THF;
En general, los compuestos de Fórmula (I), en donde Z representa

sus tautómeros y formas estereoisoméricas, en donde
X1 representa CH o N;
X2 representa CH o N;
siempre que como máximo uno de X 1 y X2 represente N;
en donde las otras variables son como se muestran en la Fórmula (Iza), (Izb) e (Izc), pueden prepararse de acuerdo con el siguiente Esquema de reacción 2 4.
En el Esquema 2 4 , (Id), Ik) e (In) se restringen a (Ida), Ika) e (Ina) en el que R3 está restringido a -CH2OH. Todas las otras variables en el Esquema 24 se definen de acuerdo con el alcance de la presente invención.
Esquema 24

1 : en presencia de reactivos adecuados, tal como, por ejemplo, cloruro de oxalilo y dimetilsulfóxido, una base adecuada, tal como, por ejemplo, trimetilamina, en un disolvente adecuado, tal como, por ejemplo, diclorometano, a una temperatura adecuada que varía entre - 80 °C a temperatura ambiente, o en presencia de un reactivo oxidativo adecuado, tal como, por ejemplo, óxido de manganeso, en un disolvente adecuado, tal como, por ejemplo, diclorometano o tolueno, a una temperatura adecuada que varía de la temperatura ambiente a 80 °C;
2 : en presencia de un reactivo adecuado, tal como, por ejemplo, yoduro de trimetilsulfonio, en presencia de una base adecuada, tal como, por ejemplo, hidróxido potásico, en un disolvente adecuado, tal como, por ejemplo, una mezcla de acetonitrilo y agua, a una temperatura adecuada, tal como, por ejemplo, 60 °C;
3 : en presencia de una base alcalina adecuada, tal como, por ejemplo, hidróxido sódico, en un disolvente adecuado, tal como, por ejemplo, una mezcla de dioxano y agua a una temperatura adecuada, tal como, por ejemplo, 80 °C;
4 : en un disolvente adecuado, tal como, por ejemplo, acetonitrilo o dimetilformamida, a una temperatura adecuada, tal como, por ejemplo, 80 °C, opcionalmente en condiciones selladas;
5 : en un disolvente adecuado, tal como, por ejemplo, acetonitrilo o dimetilformamida, a una temperatura adecuada, tal como, por ejemplo, 80 °C, opcionalmente en condiciones selladas.
En general, los compuestos de Fórmula (I), en donde Z representa

sus tautómeros y formas estereoisoméricas, en donde
X 1 representa CH o N;
X2 representa CH o N;
siempre que como máximo uno de X1 y X2 represente N;
en donde las otras variables son como se muestran en la Fórmula (Izd), (Ize), (Izf) e (Izh), pueden prepararse de acuerdo con el siguiente Esquema de reacción 2 5.
Todas las otras variables en el Esquema 25 se definen de acuerdo con el alcance de la presente invención.
Esquema 25

En el esquema 2 5 , se aplican las siguientes condiciones de reacción:
1 : en presencia de un reactivo adecuado tal como, por ejemplo, di-azodicarboxilato de íerc-butilo, una fosfina adecuada tal como, por ejemplo, trifenilfosfina, y en un disolvente adecuado tal como, por ejemplo, THF;
2 : a una temperatura adecuada tal como, por ejemplo, 80 °C, en un disolvente adecuado tal como, por ejemplo, etanol;
3 : en el caso de un cloruro de acilo, en presencia de una base adecuada, tal como, por ejemplo, diisopropiletilamina, y en un disolvente adecuado, tal como, por ejemplo, diclorometano;
en el caso de un ácido carboxílico, en presencia de un reactivo de acoplamiento adecuado, tal como, por ejemplo, clorhidrato de 1 -(3 -dimetilaminopropil)-3 -etilcarbodiimida, un aditivo adecuado, tal como, por ejemplo, 1 -hidroxibenzotriazol, una base adecuada, tal como, por ejemplo, trietilamina, y en un disolvente adecuado, tal como, por ejemplo, una mezcla de THF y diclorometano;
4 : en presencia de una base adecuada, tal como, por ejemplo, diisopropiletilamina, y en un disolvente adecuado, tal como, por ejemplo, diclorometano;
5 : en presencia de un reactivo de acoplamiento adecuado, tal como, por ejemplo, clorhidrato de 1 -(3 -dimetilaminopropil)-3 -etilcarbodiimida, un aditivo adecuado, tal como, por ejemplo, 1 -hidroxibenzotriazol, una base adecuada, tal como, por ejemplo, trietilamina, y en un disolvente adecuado, tal como, por ejemplo, una mezcla de THF y diclorometano.
En general, los compuestos de Fórmula (I), en donde Z representa

sus tautómeros y formas estereoisoméricas, en donde
X1 representa CH o N;
X2 representa CH o N;
siempre que como máximo uno de X1 y X2 represente N;
en donde las otras variables son como se muestran en la Fórmula (Izi) e (Izj), pueden prepararse de acuerdo con el siguiente Esquema de reacción 2 6. Todas las otras variables en el Esquema 26 se definen de acuerdo con el alcance de la presente invención.
Esquema 26

1: en presencia de una base tal como, por ejemplo, hidróxido de litio acuoso o hidróxido sódico acuoso, en un disolvente adecuado, tal como, por ejemplo, metanol, tetrahidrofurano, etanol;
2: en presencia de un reactivo de acoplamiento adecuado, tal como, por ejemplo, hexafluorofosfato de N,N,N',N'-Tetrametil-O-(1H-benzotriazol-1-il)uronio, hexafluorofosfato de O-(Benzotriazol-1-il)-N,N,N',N'-tetrametiluronio, una base adecuada, tal como, por ejemplo, diisopropiletilamina, en un disolvente adecuado, tal como, por ejemplo, dimetilformamida. En general, los compuestos de Fórmula (I), en donde Z representa
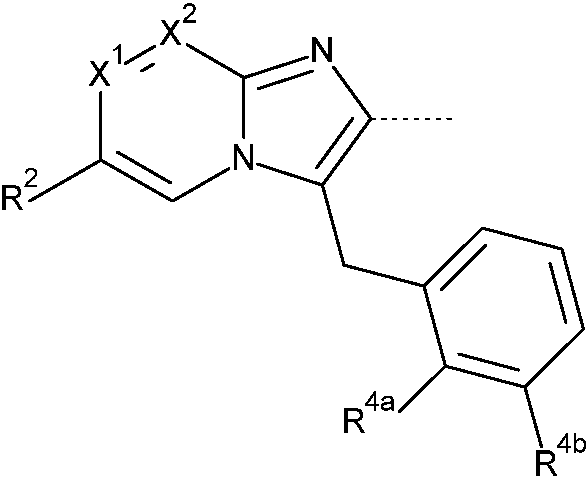
sus tautómeros y formas estereoisoméricas, en donde
X1 representa CH o N;
X2 representa CH o N;
siempre que como máximo uno de X1 y X2 represente N;
en donde las otras variables son como se muestran en la Fórmula (Izk), pueden prepararse de acuerdo con el siguiente Esquema de reacción 27.
En el Esquema 27, (Icb), Ija) e (lo) están restringidos a (Icb1), Ija1) e (loa), en los que R3 está restringido a -C Ü 2alquilo C1-4. Todas las otras variables en el Esquema 27 se definen de acuerdo con el alcance de la presente invención.
Esquema 27
(l 
En el esquema 27, se aplican las siguientes condiciones de reacción:
1: en presencia de una base tal como, por ejemplo, hidróxido de litio acuoso o hidróxido sódico acuoso, en un disolvente adecuado, tal como, por ejemplo, metanol, tetrahidrofurano, etanol;
2: en presencia de un reactivo de acoplamiento adecuado, tal como, por ejemplo, hexafluorofosfato de N,N,N',N'-Tetrametil-O-(1 H-benzotriazol-1-il)uronio, hexafluorofosfato de O-(Benzotriazol-1-il)-N,N,N',N'-tetrametiluronio, una base adecuada, tal como, por ejemplo, diisopropiletilamina, en un disolvente adecuado, tal como, por ejemplo, dimetilformamida. En todas estas preparaciones, los productos de reacción se pueden aislar del medio de reacción y, en caso necesario, purificar posteriormente de acuerdo con metodologías conocidas generalmente en la técnica tales como, por ejemplo, extracción, cristalización, lavado y cromatografía.
Las formas quiralmente puras de los compuestos de Fórmula (I) forman un grupo preferido de compuestos. Por lo tanto, las formas quiralmente puras de los compuestos intermedios y sus formas salinas son particularmente útiles en la preparación de compuestos quiralmente puros de Fórmula (I). También son útiles las mezclas enantioméricas de los productos intermedios en la preparación de compuestos de fórmula (I) con la configuración correspondiente.
Farmacología
Se ha descubierto que los compuestos de la presente invención inhiben la actividad de PI3Kp cinasa, opcionalmente, también tienen actividad inhibidora de PI3K5.
Por lo tanto, se anticipa que los compuestos de acuerdo con la presente invención o composiciones farmacéuticas de los mismos pueden ser útiles para el tratamiento o la prevención, en particular para el tratamiento, de enfermedades tales como el cáncer, trastornos autoinmunes, enfermedades cardiovasculares, enfermedades inflamatorias, enfermedades neurodegenerativas, alergia, pancreatitis, asma, fallo multiorgánico, enfermedades renales, agregación plaquetaria, motilidad del esperma, rechazo de trasplantes, rechazo de injertos, lesiones pulmonares y similares; en particular cáncer. Debido a que los compuestos farmacéuticamente activos de la presente invención son activos como inhibidores de PI3Kp, exhiben utilidad terapéutica en el tratamiento o la prevención, en particular el tratamiento de neoplasias susceptibles, particularmente los neoplasias que exhiben una deficiencia de PTEN.
Tal como se utiliza en el presente documento, la frase “deficiente en PTEN” o “deficiencia en PTEN” debe describir tumores con deficiencias de la función supresora de tumores de PTEN (Homología de Fosfatasa y Tensina). Una deficiencia de este tipo incluye mutación en el gen PTEN, reducción o ausencia de proteínas PTEN cuando se compara con PTEN de tipo salvaje, o mutaciones o ausencia de otros genes que provocan la supresión de la función PTEN.
“Neoplasia susceptible”, tal como se utiliza en el presente documento, se refiere a neoplasias que son susceptibles a tratamiento por parte de un inhibidor de cinasa, y particularmente a neoplasias que son susceptibles al tratamiento por un inhibidor de PI3Kp. Neoplasias que han sido asociados con una actividad inapropiada de la fosfatasa PTEN, y particularmente neoplasias que exhiben mutaciones de PTEN, o una mutación de un activador aguas arriba de la PI3Kp cinasa o la sobre-expresión de un activador aguas arriba de la PI3Kp cinasa y, por lo tanto, son susceptibles a tratamiento con un inhibidor de PI3Kp, son conocidos en la técnica, e incluyen tumores y cánceres tanto primarios como metastásicos. De acuerdo con una realización, se puede utilizar la descripción del tratamiento de una neoplasia susceptible de manera indistinta con la descripción del tratamiento de un cáncer.
De acuerdo con una realización, “neoplasias susceptibles” incluyen pero sin limitación neoplasias deficientes en PTEN enumeradas a continuación: cerebro (gliomas), glioblastomas, leucemias, síndrome de Bannayan-Zonana, enfermedad de Cowden, enfermedad de Lhermitte-Duclos, cáncer de mama, cáncer de mama inflamatorio, cáncer colorrectal tumor de Wilm, sarcoma de Ewing, Rabdomiosarcoma, ependimoma, meduloblastoma, cáncer de colon, cáncer de cabeza y cuello, cáncer de hígado, cáncer de riñón, cáncer de pulmón, melanoma, carcinoma de células escamosas, cáncer ovárico, cáncer pancreático, cáncer de próstata, cáncer sarcoma, osteosarcoma, tumor de células gigantes del hueso, cáncer de tiroides, leucemia linfoblástica de linfocitos T, leucemia mielógena crónica, leucemia linfocítica crónica, leucemia de tricoleucitos,
leucemia linfoblástica aguda, leucemia mielógena aguda, leucemia neutrófila crónica, leucemia de linfocitos T linfoblástica aguda, Plasmacitoma, leucemia Inmunoblástica de células grandes, leucemia de células del Manto, mieloma múltiple, leucemia megacarioblástica, leucemia megacariocítica aguda, leucemia promielocítica, Eritroleucemia, linfoma maligno, linfoma de hodgkin, linfoma no de hodgkin, linfoma linfoblástico de linfocitos T, linfoma de Burkitt, linfoma folicular, neuroblastoma, cáncer de vejiga, cáncer urotelial, cáncer del cuello uterino, cáncer vulvar, cáncer endometrial, cáncer renal, mesotelioma, cáncer esofágico, cáncer de glándulas salivares, cáncer hepatocelular, cáncer gástrico, cáncer nasofaríngeo, cáncer bucal, cáncer de la boca, GIST (tumor del estroma gastrointestinal) y cáncer testicular.
De acuerdo con una realización alternativa, la expresión “neoplasia susceptible” incluye y se limita a cáncer de próstata hormonorrefractario, cáncer de pulmón de células no pequeñas, cáncer endometrial, cáncer gástrico, melanoma, cáncer de cabeza y cuello, cáncer de mama, incluyendo cáncer de mama tripnegativo, y glioma.
En una realización, la expresión “neoplasia susceptible” incluye y se limita a cáncer de próstata, en particular cáncer de próstata hormonorrefractario.
Los compuestos de la presente invención también pueden tener aplicaciones terapéuticas para sensibilizar células tumorales para la radioterapia y quimioterapia.
Así pues, los compuestos de la presente invención pueden usarse como un "radiosensibilizador" y/o "quimiosensibilizador" o pueden proporcionarse combinados con otro "radiosensibilizador" y/o "quimiosensibilizador".
El término "radiosensibilizador", tal como se utiliza en el presente documento, se define como una molécula, preferentemente una molécula con un peso molecular bajo, administrada a animales en cantidades terapéuticamente eficaces para aumentar la sensibilidad de las células a la radiación ionizante y/o para favorecer el tratamiento de enfermedades que sean tratables con radiación ionizante.
El término "quimiosensibilizador", tal como se usa en el presente documento, se define como una molécula, preferentemente una molécula con un peso molecular bajo, administrada a animales en cantidades terapéuticamente eficaces para aumentar la sensibilidad de las células a la quimioterapia y/o favorecer el tratamiento de enfermedades que sean tratables con fármacos quimioterapéuticos.
En la bibliografía se han sugerido varios mecanismos para el modo de acción de radiosensibilizadores, que incluyen: radiosensibilizaciones de células hipóxícas (p. ej., compuestos de 2-nitroimidazol y compuestos de dióxido de benzotriazina) que mimetizan el oxígeno o que, alternativamente, se comportan como agentes biorreductores bajo hipoxia; radiosensibilizaciones de células no hipóxicas (p. ej., pirimidinas halogenadas) pueden ser análogos de bases de ADN y se incorporan preferentemente en el ADN de células cancerosas y, con ello, fomentan la ruptura inducida por radiación de moléculas de ADN y/o previenen los mecanismos normales de reparación del ADN; y diversos otros mecanismos de acción potenciales han sido hipotetizados para radiosensibilizadores en el tratamiento de enfermedades.
Muchos protocolos para el tratamiento del cáncer emplean actualmente radiosensibilizadores en unión con radiación de rayos X. Los ejemplos de radiosensibilizadores activados por rayos X incluyen, sin carácter limitante, los siguientes: metronidazol, misonidazol, desmetilmisonidazol, pimonidazol, etanidazol, nimorazol, mitomicina C, RSU 1 0 6 9 , SR 4 2 3 3 , EO9 ,
RB 6 1 4 5 , nicotinamida, 5 -bromodesoxiuridina (BUdR), 5 -yododesoxiuricina (IUdR), bromodesoxicitidina, fluorodesoxiuridina (FudR), hidroxiurea, cisplatino y sus análogos y derivados terapéuticamente eficaces.
La terapia fotodinámica (PDT, por sus siglas en inglés) de los distintos tipos de cáncer emplea la luz visible como activador de la radiación del agente sensibilizador. Ejemplos de radiosensibilizadores fotodinámicos incluyen los siguientes, pero no se limitan a: derivados de hematoporfirina, fotofrina, derivados de benzoporfirina, etioporfirina de estaño, feoborbida-a, bacterioclorofina-a, naftalocianinas, ftalocicianinas, ftalocianina de zinc y análogos terapéuticamente efectivos y derivados de los mismos.
Los radiosensibilizadores se pueden administrar en unión con una cantidad terapéuticamente efectiva de uno o más de otros compuestos, incluyendo, pero no limitados a: compuestos que fomentan la incorporación de radiosensibilizadores a las células diana; compuestos que controlan el flujo de productos terapéuticos, nutrientes y/u oxígeno a las células diana; agentes quimioterapéuticos que actúan sobre el tumor con o sin radiación adicional; u otros compuestos terapéuticamente efectivos para tratar el cáncer u otras enfermedades.
Los quimiosensibilizadores se pueden administrar junto con una cantidad terapéuticamente eficaz de uno o más compuestos diferentes incluidos, sin carácter limitante: compuestos que favorecen la incorporación de los quimiosensibilizadores a las células diana; compuestos que controlan el flujo de compuestos terapéuticos, nutrientes y/u oxígeno a las células diana; agentes quimioterapéutico que actúan sobre el tumor u otros compuestos terapéuticamente
eficaces para tratar el cáncer u otra enfermedad. Se ha observado que los antagonistas del calcio, por ejemplo, verapamil, son útiles combinados con agentes antineoplásicos para establecer la quimiosensibilidad en las células tumorales resistentes a los agentes quimioterapéuticos aceptados y para favorecer la eficacia de tales compuestos en neoplasias malignas sensibles al fármaco.
La invención se refiere a compuestos de Fórmula (I) y N-óxidos, sales por adición farmacéuticamente aceptables y solvatos de los mismos, para uso como un medicamento.
La invención también se refiere a compuestos de Fórmula (I) y N-óxidos, sales de adición farmacéuticamente aceptables, y solvatos de los mismos, para su uso en la inhibición de la actividad de PI3K|3 cinasa y opcionalmente también para su uso en la inhibición de PI3K5.
Los compuestos de la presente invención pueden ser "agentes anticancerosos", englobando este término también "agentes contra el crecimiento de células tumorales" y "agentes antineoplásicos".
La invención también se refiere a compuestos de fórmula (I) y a N-óxidos, sales de adición farmacéuticamente aceptables y solvatos de los mismos, para su uso en el tratamiento de las enfermedades mencionadas anteriormente.
La invención también se refiere a compuestos de fórmula (I) y a N-óxidos, sales de adición farmacéuticamente aceptables y solvatos de los mismos, para el tratamiento o prevención, en particular para el tratamiento, de dichas enfermedades.
La invención también se refiere a compuestos de Fórmula (I) y N-óxidos, sales por adición farmacéuticamente aceptables, y solvatos de los mismos, para el tratamiento o la prevención, en particular en el tratamiento de enfermedades o afecciones mediadas por PI3K|3.
La invención también se refiere a compuestos de Fórmula (I) y N-óxidos, sales por adición farmacéuticamente aceptables, y solvatos de los mismos, para el tratamiento o la prevención, en particular en el tratamiento de enfermedades o afecciones mediadas por PI3K|3, y opcionalmente PI3K6.
La invención también se refiere al uso de compuestos de fórmula (I) y a N-óxidos, sales de adición farmacéuticamente aceptables y solvatos de los mismos, para la fabricación de un medicamento.
La invención también se refiere al uso de compuestos de fórmula (I) y a N-óxidos, sales de adición farmacéuticamente aceptables y solvatos de los mismos, para la fabricación de un medicamento para la inhibición de PI3K|3.
La invención también se refiere al uso de compuestos de fórmula (I) y a N-óxidos, sales de adición farmacéuticamente aceptables y solvatos de los mismos, para la fabricación de un medicamento para la inhibición de PI3K|3 y opcionalmente también para la inhibición de PI3K6.
La invención también se refiere al uso de compuestos de fórmula (I) y de N-óxidos, sales de adición farmacéuticamente aceptables y solvatos de los mismos para la fabricación de un medicamento para el tratamiento o prevención, en particular para el tratamiento, de una cualquiera de las patologías mencionadas anteriormente en el presente documento.
La invención también se refiere al uso de compuestos de fórmula (I) y de N-óxidos, sales de adición farmacéuticamente aceptables y solvatos de los mismos para la fabricación de un medicamento para el tratamiento de una cualquiera de las patologías mencionadas anteriormente en el presente documento.
Los compuestos de fórmula (I) y los N-óxidos, sales de adición farmacéuticamente aceptables y solvatos de los mismos pueden administrarse a mamíferos, preferentemente a seres humanos, para el tratamiento o prevención de una cualquiera de las enfermedades mencionadas anteriormente en el presente documento.
En vista de la utilidad de los compuestos de fórmula (I) y de los N-óxidos, sales de adición farmacéuticamente aceptables y solvatos de los mismos, se proporciona un método para tratar a animales de sangre caliente, incluyendo seres humanos, que padecen o un método para prevenir que los animales de sangre caliente, incluyendo seres humanos, padezcan una cualquiera de las enfermedades mencionadas anteriormente en el presente documento.
Dichos métodos comprenden la administración, es decir, la administración tópica o sistémica, preferentemente administración oral, de una cantidad eficaz de un compuesto de fórmula (I) o de un N-óxido, una sal de adición farmacéuticamente aceptable o un solvato del mismo, a animales de sangre caliente, incluyendo seres humanos.
Los expertos en el tratamiento de tales enfermedades podrán determinar la cantidad diaria terapéutica eficaz a partir de los resultados de la prueba presentados más adelante en la presente. Una cantidad diaria terapéutica eficaz estaría comprendida entre aproximadamente 0.005 mg/kg y 50 mg/kg, en particular 0.01 mg/kg y 50 mg/kg de peso corporal, más en particular entre 0.01 mg/kg y 25 mg/kg de peso corporal, preferentemente entre aproximadamente 0.01 mg/kg y
aproximadamente 15 mg/kg, más preferentemente entre aproximadamente 0.01 mg/kg y aproximadamente 10 mg/kg, aún más preferentemente entre aproximadamente 0.01 mg/kg y aproximadamente 1 mg/kg, más preferentemente entre aproximadamente 0.05 mg/kg y aproximadamente 1 mg/kg de peso corporal. La cantidad de un compuesto de acuerdo con la presente invención, al que aquí también se alude como el ingrediente activo, que se requiere para conseguir un efecto terapéutico variará naturalmente sobre una base de caso por caso, por ejemplo con el compuesto particular, la vía de administración, la edad y el estado del receptor, y del trastorno o enfermedad particular que esté siendo tratado.
Un método de tratamiento también puede incluir administrar el principio activo en un régimen que comprenda entre una y cuatro tomas al día. En estos métodos de tratamiento, los compuestos de acuerdo con la invención se formulan preferentemente antes de la administración. Tal como se describe a continuación en la presente, las formulaciones farmacéuticas adecuadas se preparan mediante procedimientos conocidos utilizando ingredientes conocidos y de los que se puede disponer fácilmente.
Los compuestos de la presente invención que pueden ser adecuados para tratar o prevenir el cáncer o afecciones relacionadas con el cáncer, se pueden administrar solos o combinados con uno o más agentes terapéuticos adicionales. La terapia combinada incluye la administración de una sola formulación de dosificación farmacéutica que contenga un compuesto de fórmula (I), un N-óxido, una sal de adición farmacéuticamente aceptable o un solvato del mismo y uno o más agentes terapéuticos adicionales, así como la administración del compuesto de fórmula (I), de un N-óxido, una sal de adición farmacéuticamente aceptable o un solvato del mismo y cada agente terapéutico adicional en su propia formulación de dosificación farmacéutica separada. Por ejemplo, puede administrarse al paciente un compuesto de fórmula (I), un N-óxido, una sal de adición farmacéuticamente aceptable o un solvato del mismo y un agente terapéutico al paciente junto con una sola composición de dosificación oral, tal como un comprimido o cápsula o puede administrarse cada agente en formulaciones de dosificación oral separadas.
Aunque es posible administrar el principio activo solo, es preferible presentarlo como una composición farmacéutica.
Por consiguiente, la presente invención proporciona además una composición farmacéutica que comprende un portador farmacéuticamente aceptable y como ingrediente activo, una cantidad terapéuticamente eficaz de un compuesto de fórmula (I), un N-óxido, una sal de adición farmacéuticamente aceptable o un solvato del mismo.
El portador o diluyente debe ser "aceptable" en el sentido de ser compatible con los otros ingredientes de la composición y no nocivos para los receptores de los mismos.
Para facilitar la administración, los compuestos de la presente pueden formularse en varias formas farmacéuticas a efectos de administración. Los compuestos de acuerdo con la invención, en particular, los compuestos de fórmula (I) y los N-óxidos, sales de adición farmacéuticamente aceptables y solvatos del mismo, o cualquier subgrupo o combinación de los mismos, puede formularse en varias formas farmacéuticas para fines de administración. Como composiciones adecuadas, se pueden citar todas las composiciones empleadas normalmente para administrar fármacos por vía sistémica.
Para preparar las composiciones farmacéuticas de esta invención, se combina una cantidad eficaz del compuesto particular como principio activo en mezcla íntima con un portador farmacéuticamente aceptable, pudiendo adoptar dicho portador una gran variedad de formas dependiendo de la forma del preparado que se desee para la administración. Estas composiciones farmacéuticas son deseables en forma de dosificación unitaria, en particular para la administración por vía oral, rectal, percutánea, mediante inyección parenteral o mediante inhalación. Por ejemplo, en la preparación de las composiciones en una forma de dosificación oral se puede emplear cualquiera de los medios farmacéuticos habituales tales como, por ejemplo, agua, glicoles, aceites, alcoholes y similares en el caso de preparaciones líquidas orales tales como suspensiones, jarabes, elixires, emulsiones y soluciones; o vehículos sólidos tales como almidones, azúcares, caolín, diluyentes, lubricantes, aglutinantes, agentes disgregantes y similares en el caso de polvos, pastillas, cápsulas y comprimidos. Dada su facilidad de administración, los comprimidos y las cápsulas representan las formas unitarias de dosificación oral más convenientes, en cuyo caso se emplean obviamente portadores farmacéuticos sólidos. Para las composiciones parenterales, el portador normalmente comprenderá agua esterilizada, al menos en gran parte, aunque puede incluir otros ingredientes, por ejemplo, para incrementar la solubilidad. Se pueden preparar soluciones inyectables, por ejemplo, en las que el portador comprenda solución salina, solución de glucosa o una mezcla de solución salina y de glucosa. Pueden formularse soluciones inyectables que contienen un compuesto de fórmula (I), un N-óxido, una sal de adición farmacéuticamente aceptable o un solvato del mismo en un aceite para una acción prolongada. Aceites apropiados para este fin son, por ejemplo, aceite de cacahuete, aceite de sésamo, aceite de semilla de algodón, aceite de maíz, aceite de soja, ésteres de glicerol sintéticos de ácidos grasos de cadena larga y mezclas de éstos y otros aceites. También se pueden preparar suspensiones inyectables, en cuyo caso se pueden emplear portadores líquidos, agentes de suspensión y similares que sean adecuados. También se incluyen preparaciones en forma sólida que están destinadas a ser convertidas, poco antes del uso, en preparaciones en forma líquida. En las composiciones adecuadas para la administración percutánea, el portador comprende opcionalmente un agente potenciador de la penetración y/o un agente humectante adecuado, combinados opcionalmente con aditivos adecuados de cualquier naturaleza en proporciones
minoritarias, donde los aditivos no provocan ningún efecto perjudicial significativo en la piel. Dichos aditivos pueden facilitar la administración a la piel y/o pueden ser útiles para preparar las composiciones deseadas. Estas composiciones se pueden administrar de diversas maneras, p. ej., como un parche transdérmico, como una unción dorsal puntual, como un ungüento. Sales por adición de ácidos o bases de compuestos de Fórmula (I), debido a su solubilidad incrementada en agua frente a la correspondiente forma de base o ácido, son mas adecuadas en la preparación de composiciones acuosas. Es especialmente ventajoso formular las composiciones farmacéuticas mencionadas anteriormente en forma farmacéutica unitaria por su facilidad de administración y uniformidad de dosificación. La expresión "forma farmacéutica unitaria", tal como se utiliza en la presente, se refiere a unidades físicamente discretas adecuadas como dosis unitarias, donde cada unidad contiene una cantidad predeterminada de principio activo calculada para producir el efecto terapéutico deseado asociado con el portador farmacéutico requerido. Ejemplos de formas de dosificación unitaria de este tipo son comprimidos (incluyendo comprimidos con muesca o revestidos), cápsulas, píldoras, paquetes de polvos, obleas, supositorios, disoluciones o suspensiones inyectables y similares, y múltiples segregados de las mismas.
Para potenciar la solubilidad y/o la estabilidad de los compuestos de fórmula (I) y de los N-óxidos, sales de adición farmacéuticamente aceptables y solvatos de los mismos, en las composiciones farmacéuticas, puede ser ventajoso emplear a , p o Y-ciclodextrinas o sus derivados, en particular, ciclodextrinas sustituidas con hidroxialquilo, por ejemplo, 2-hidroxipropil-p-ciclodextrina o sulfobutil-p-ciclodextrina. Asimismo, los codisolventes, tales como los alcoholes, pueden mejorar la solubilidad y/o la estabilidad de los compuestos de acuerdo con la invención en las composiciones farmacéuticas.
Dependiendo del modo de administración, la composición farmacéutica comprenderá preferentemente entre un 0.05 y un 99 % en peso, más preferentemente entre un 0.1 y un 70 % en peso, aún más preferentemente entre un 0.1 y un 50 % en peso de un compuesto de fórmula (I), un N-óxido, una sal de adición farmacéuticamente aceptable o un solvato del mismo y entre un 1 y un 99.95 % en peso, más preferentemente entre un 30 a un 99.9 % en peso, aún más preferentemente entre un 50 y un 99.9 % en peso de un portador farmacéuticamente aceptable, estando todos los porcentajes basados en el peso total de la composición.
Como otro aspecto de la presente invención, se contempla una combinación de un compuesto de la presente invención con otro agente anticanceroso, especialmente para su uso como un medicamento, más específicamente para su uso en el tratamiento del cáncer o de enfermedades relacionadas.
Para el tratamiento de las afecciones anteriores, los compuestos de la invención se pueden emplear convenientemente combinados con uno o más de otros agentes medicinales, más concretamente, con otros agentes anticancerosos o adyuvantes en la terapia contra el cáncer. Los ejemplos de agentes anticancerosos o adyuvantes (agentes de apoyo en la terapia) incluyen, sin carácter limitante:
- compuestos de coordinación de platino, por ejemplo, cisplatino combinado opcionalmente con amifostina, carboplatino u oxaliplatino;
- compuestos de taxano, por ejemplo paclitaxel, partículas unidas a proteína paclitaxel (Abraxane™) o docetaxel;
- inhibidores de la topoisomerasa I tales como compuestos de camptotecina, por ejemplo, irinotecán, SN-38, topotecán, topotecánHCl;
- inhibidores de la topoisomerasa II tales como epipodofilotoxinas o derivados de podofilotoxinas antitumorales, por ejemplo, etopósido, etopósido fosfato o tenipósido;
- alcaloides de la vinca antitumorales, por ejemplo, vinblastina, vincristina o vinorelbina;
- derivados de nucleósidos antitumorales, por ejemplo, 5-fluorouracilo, leucovorina, gemcitabina, gemcitabina HCl, capecitabina, cladribina, fludarabina, nelarabina;
- agentes alquilantes tales como mostaza nitrogenada o nitrosourea, por ejemplo, ciclofosfamida, clorambucil, carmustina, tiotepa, mefalán (melfalán), lomustina, altretamina, busulfán, dacarbazina, estramustina, ifosfamida opcionalmente en combinación con mesna, pipobromán, procarbazina, estreptozocina, temozolomida, uracilo;
- derivados de antraciclina antitumorales, por ejemplo, daunorubicina, doxorubicina opcionalmente en combinación con dexrazoxano, doxil, idarubicina, mitoxantrona, epirubicina, epirubicinaHCl, valrubicina;
- moléculas que actúan sobre el receptor IG F-1, por ejemplo, picropodofilina;
- derivados de tetracarcina, por ejemplo, terocarcina A;
- glucocorticoides, por ejemplo, prednisona;
- anticuerpos, por ejemplo, trastuzumab (anticuerpo HER2), rituximab (anticuerpo CD20), gemtuzumab, gemtuzumab ozogamicin, cetuximab, pertuzumab, bevacizumab, alemtuzumab, eculizumab, ibritumomab tiuxetan, nofetumomab, panitumumab, tositumomab, CNTO 328;
- antagonistas del receptor de estrógeno o moduladores selectivos del receptor de estrógeno o inhibidores de la síntesis de estrógeno, por ejemplo, tamoxifeno, fulvestrant, toremifeno, droloxifeno, faslodex, raloxifeno o letrozol;
- inhibidores de la aromatasa tales como exemestano, anastrozol, letrazol, testolactona y vorozol;
- agentes de diferenciación tales como retinoides, vitamina D o ácido retinoico y agentes bloqueantes del metabolismo del ácido retinoico (RAMBA, por sus siglas en inglés), por ejemplo, accutane;
- inhibidores de la ADN-metiltransferasa, por ejemplo, azacitidina o decitabina;
- antifolatos, por ejemplo, premetrexed disódico;
- antibióticos, por ejemplo, antinomicina D, bleomicina, mitomicina C, dactinomicina, carminomicina, daunomicina, levamisol, plicamicina, mitramicina;
- antimetabolitos, por ejemplo, clofarabina, aminopterina, arabinósido de citosina o metotrexato, azacitidina, citarabina, floxuridina, pentostatina, tioguanina;
- agentes inductores de la apoptosis y agentes antiangiogénicos tales como inhibidores de Bcl-2, por ejemplo, YC 137, BH 312, ABT 737, gosipol, HA 14-1, TW 37 o ácido decanoico;
- agentes de unión a la tubulina, por ejemplo, combrestatina, colchicinas o nocodazol;
- inhibidores de cinasas (p. ej., inhibidores de EGFR (receptor del factor de crecimiento epitelial), MTKI (inhibidores multicinasa), inhibidores mTOR), por ejemplo, flavoperidol, mesilato de imatinib, erlotinib, gefitinib, dasatinib, lapatinib, lapatinib ditosilato, sorafenib, sunitinib, maleato de sunitinib, temsirolimus;
- inhibidores de la farnesiltransferasa, por ejemplo, tipifarnib;
- inhibidores de la histona-desacetilasa (HDAC, por sus siglas en inglés), por ejemplo, butirato de sodio, suberoilanilida de ácido hidroxámico (SAHA), depsipéptido (FR 901228), NVP-LAQ824, R306465, JNJ-26481585, tricostatina A, vorinostat;
- inhibidores de la ruta de la ubiquitina-proteosoma, por ejemplo, PS-341, MLN .41 o bortezomib;
- yondelis;
- inhibidores de la telomerasa, por ejemplo, telosmetatin;
- inhibidores de la metaloproteinasa de la matriz, por ejemplo, batimastat, marimastat, prinostat o metastat; - interleucinas recombinantes, por ejemplo, aldesleucina, denileucina diftitox, interferón alfa 2a, interferón alfa 2b, peginterferón alfa 2b;
- inhibidores de MAPK;
- retinoides, por ejemplo, alitretinoína, bexaroteno, tretinoína;
- trióxido arsénico;
- Asparaginasa;
- esteroides, por ejemplo, propionato de dromostanolona, acetato de megestrol, nandrolona (decanoato, fenpropionato), dexametasona;
- agonistas o antagonistas de la hormona liberadora de gonadotropina, por ejemplo, abarelix, acetato de goserelina, acetato de histrelina, acetato de leuprolida;
- talidomida, lenalidomida;
- mercaptopurina, mitotano, pamidronato, pegademasa, pegaspargasa, rasburicasa;
- miméticos de BH3, por ejemplo, ABT-737;
- inhibidores de MEK, por ejemplo, PD98059, AZD6244, CI-1040;
- análogos del factor estimulante de colonias, por ejemplo filgrastim, pegfilgrastim, sargramostim; eritropoyetina o análogos de los mismos (p. ej., darbepoyetina alfa); interleuquina 11; oprelvekina; zoledronato, ácido zoledrónico; fentanilo; bisfosfonato; palifermina;
- un inhibidor 17alfa-hidroxilass-17,20-liasa de citocromo esteroide P450 (CYP17), p. ej., abiraterona, acetato de abiraterona;
- inhibidores de la glicólisis, tales como la 2-desoxiglucosa;
- inhibidores de mTOR tales como rapamicinas y rapálogos e inhibidores de la cinasa mTOR;
- inhibidores de PI3K e inhibidores duales de mTOR/PI3K;
- inhibidores de la autofagia, tales como la cloroquina y la hidroxicloroquina;
- anticuerpos que reactivan la respuesta inmunitaria frente a los tumores, por ejemplo, nivolumab (anti-PD-1), lambrolizumab (anti-PD-1), ipilimumab (anti-CTLA4), y MPDL3280A (anti-PD-L1).
Los compuestos de la invención también pueden combinarse ventajosamente con terapias antiandrógenas que incluyen antagonistas del receptor de andrógenos e inhibidores de la biosíntesis de andrógenos en cánceres de próstata negativos a PTEN.
La presente invención se refiere también a un producto que contiene como primer principio activo un compuesto de acuerdo con la invención y como principio activo adicional uno o más agentes anticancerosos, como un preparado combinado para el uso simultáneo, por separado o secuencial en el tratamiento de pacientes que padecen cáncer.
El otro o los otros agentes medicinales y el compuesto de acuerdo con la presente invención se pueden administrar simultáneamente (p. ej., en composiciones unitarias o por separado) o secuencialmente en cualquier orden. En el último caso, los dos o más compuestos se administrarán dentro de un periodo y en una cantidad y modo que sea suficiente para garantizar que se logra un efecto conveniente y sinérgico. Se comprenderá que el método y orden de administración preferidos y las pautas y cantidades posológicas respectivas de cada componente de la combinación dependerán del otro agente medicinal particular y del compuesto de la presente invención que se están administrando, sus vía de administración, el tumor particular que se está tratando y el receptor particular que se está tratando. El experto en la técnica puede determinar fácilmente el método y orden de administración óptimos y las pautas y cantidades posológicas utilizando métodos convencionales y teniendo en cuenta la información expuesta en la presente.
El experto en la técnica puede determinar la relación ponderal del compuesto de acuerdo con la presente invención respecto al otro o a los otros agentes anticancerosos cuando se administran como una combinación. Dicha relación y la dosis exacta y la frecuencia de administración dependen del compuesto particular de acuerdo con la invención y del otro o los otros agentes anticancerosos utilizados, la afección particular que se está tratando, la gravedad de la afección que se está tratando, la edad, el peso, el sexo, la dieta, el momento de administración y el estado físico general del paciente particular, el modo de administración así como también de otra medicación que el individuo pueda estar tomando, como bien sabrán los expertos en la técnica. Además, es obvio que la cantidad diaria eficaz se puede reducir o incrementar dependiendo de la respuesta del sujeto tratado y/o dependiendo de la evaluación del médico que prescriba los compuestos de la presente invención. Una relación ponderal particular entre el presente compuesto de Fórmula (I) y otro agente anticanceroso puede estar comprendido en el intervalo de 1/10 a 10/1, más concretamente de 1/5 a 5/1, aún más concretamente de 1/3 a 3/1.
El compuesto de coordinación de platino se administra convenientemente en una dosis de 1 a 500 mg por metro cuadrado (mg/m2) de área superficial corporal, por ejemplo, de 50 a 400 mg/m2, concretamente para el cisplatino en una dosis de aproximadamente 75 mg/m2 y para el carboplatino de aproximadamente 300 mg/m2 por ciclo de tratamiento.
El compuesto de taxano se administra ventajosamente en una dosis de 50 a 400 mg por metro cuadrado (mg/m2) de área específica corporal, por ejemplo 75 a 250 mg/m2, particularmente para paclitaxel en una dosis de aproximadamente 175 a 250 mg/m2 y para docetaxel en aproximadamente 75 a 150 mg/m2 por ciclo de tratamiento.
El compuesto camptotecina se administra convenientemente en una dosis de 0.1 a
400 mg por metro cuadrado (mg/m2) de área superficial corporal, por ejemplo, de 1 a 300 mg/m2, concretamente para el irinotecán en una dosis de aproximadamente 100 a 350 mg/m2 y para el topotecán de aproximadamente 1 a 2 mg/m2 por ciclo de tratamiento.
El derivado de podofilotoxina antitumoral se administra convenientemente en una dosis de 30 a 300 mg por metro cuadrado (mg/m2) de área superficial corporal, por ejemplo, de 50 a 250 mg/m2, concretamente para el etopósido en una dosis de aproximadamente 35 a 100 mg/m2 y para el tenipósido de aproximadamente 50 a 250 mg/m2 por ciclo de tratamiento. El alcaloide la vinca antitumoral se administra convenientemente en una dosis de 2 a
30 mg por metro cuadrado (mg/m2) de área superficial corporal, concretamente para la vinblastina en una dosis de aproximadamente 3 a 12 mg/m2, para la vincristina en una dosis de aproximadamente 1 a 2 mg/m2 y para la vinorelbina en una dosis de aproximadamente 10 a 30 mg/m2 por ciclo de tratamiento.
El derivado de nucleósidos antitumoral se administra convenientemente en una dosis de 200 a 2500 mg por metro cuadrado (mg/m2) de área superficial corporal, por ejemplo de 700 a 1500 mg/m2, particularmente para 5-FU en una dosis de 200 a 500 mg/m2, para gemcitabina en una dosis de aproximadamente 800 a 1200 mg/m2 y para capecitabina en aproximadamente 1000 a 2500 mg/m2 por ciclo de tratamiento.
Los agentes alquilantes tales como la mostaza nitrogenada o nitrosourea se administran convenientemente en una dosis de 100 a 500 mg por metro cuadrado (mg/m2) de área superficial corporal, por ejemplo, de 120 a 200 mg/m2, concretamente para la ciclofosfamida en una dosis de aproximadamente 100 a 500 mg/m2, para el clorambucilo en una dosis de aproximadamente 0.1 a 0.2 mg/kg, para la carmustina en una dosis de aproximadamente 150 a 200 mg/m2, y para la lomustina en una dosis de aproximadamente 100 a 150 mg/m2 por ciclo de tratamiento.
El derivado de antraciclina antitumoral se administra convenientemente en una dosis de 10 a 75 mg por metro cuadrado (mg/m2) de área superficial corporal, por ejemplo de 15 a 60 mg/m2, particularmente para doxorubicina en una dosis de aproximadamente 40 a 75 mg/m2, para daunorubicina en una dosis de aproximadamente 25 a 45 mg/m2, y para idarubicina en una dosis de aproximadamente 10 a 15 mg/m2, por ciclo de tratamiento.
El agente antiestrogénico se administra convenientemente con una dosificación de aproximadamente 1 a 100 mg diariamente dependiendo del agente particular y de la afección que se está tratando. El tamoxifeno se administra convenientemente por vía oral con una dosificación de 5 a 50 mg, preferentemente de 10 a 20 mg dos veces al día, y se continúa la terapia durante un tiempo suficiente para lograr y mantener un efecto terapéutico. El toremifeno se administra convenientemente por vía oral en una dosis de aproximadamente 60 mg una vez al día, y se continúa la terapia durante un tiempo suficiente para lograr y mantener un efecto terapéutico. El anastrozol se administra convenientemente por vía oral con una dosificación de aproximadamente 1 mg una vez al día. Droloxifeno se administra ventajosamente por vía oral en una dosis de aproximadamente 20-100 mg una vez al día. El raloxifeno se administra ventajosamente por vía oral en una dosis de aproximadamente 60 mg una vez al día. El exemestano se administra ventajosamente por vía oral en una dosis de aproximadamente 25 mg una vez al día.
Los anticuerpos se administran ventajosamente en una dosis de aproximadamente 1 a 5 mg por metro cuadrado (mg/m2) de área de superficie corporal, o tal como se conoce en la técnica, si es diferente. Trastuzumab se administra ventajosamente en una dosis de 1 a 5 mg por metro cuadrado (mg/m2) de área de superficie corporal, particularmente 2 a 4 mg/m2, por ciclo de tratamiento.
Estas dosis se pueden administrar, por ejemplo, una vez, dos veces o más durante el periodo de tratamiento, el cual se puede repetir, por ejemplo, cada 7, 14, 21 o 28 días.
Ejemplos
Los siguientes ejemplos ilustran la presente invención.
Cuando se indica un estereocentro con "RS", esto significa que se obtuvo una mezcla racémica.
En lo sucesivo en el presente documento, el término "DCM" significa diclorometano, "MeOH" significa metanol, "EtOH" significa etanol, "ACN" significa acetonitrilo, "THF" significa tetrahidrofurano, "DMF" significa dimetilformamida, "EtOAc" significa acetato de etilo, "iPrOH" significa isopropanol, "H2O" significa agua, "DME" significa etilenglicol dimetil éter, "DCE" significa dicloroetano, "DIPE" significa diisopropiléter, "K2 CO3 " significa carbonato potásico, "Cs2CO3" significa carbonato de cesio, "K3 PO4" significa fosfato potásico, "NH4OH" significa solución acuosa de amoniaco, "NaHCO3" significa bicarbonato sódico, "NaOH" significa hidróxido sódico, "NaCl" significa cloruro de sodio, "NH4Cl" significa cloruro de amonio, "LiCl" significa cloruro de litio, "NH4 HCO3 " significa bicarbonato de amonio, "HCOONH4" significa formiato de amonio, "KOAc" significa acetato potásico, "DIPEA" significa diisopropiletilamina, "n-BuLi" significa n-butil litio, "iPrNH2 "
significa isopropilamina, "MgSOV' significa sulfato de magnesio, "Na2SO4" significa sulfato sódico, "Na2S2O3" significa tiosulfato sódico, "N2" significa nitrógeno, "HCl" significa ácido clorhídrico, "TFA" significa ácido trifluoroacético, "NaBFU" significa borohidruro sódico, "LiAlH4" significa hidruro de litio y aluminio, "TBAF" significa fluoruro de tetrabutilamonio, "CO2" significa dióxido de carbono, "CO" significa monóxido de carbono, "SFC" significa cromatografía de fluidos supercríticos, "HBTU" significa hexafluorofosfato de W,W,W,W-tetrametil-0-(1H-benzotriazol-1-il)uronio, hexafluorofosfato de O-(Benzotriazol-1-il)-A/,N,A/',A/'-tetrametiluronio, "PPh3" significa trifenilfosfina, "ZnCl2" significa cloruro de cinc, "Pd(PPh3)4" significa tetraquis(trifenilfosfina)paladio (0), "Pd(OAc)2" significa acetato de paladio (II), "PdCl2(dppf).DCM" significa aducto de dicloro[1,1 '-bis(difenilfosfino) ferroceno] paladio (II) diclorometano, "celite®" significa tierra de diatomeas, "RuPhos" significa 2-diciclohexilfosfino-2',6,-diisopropoxi-1,1'-bifenilo, "precatalizador BrettPhos de primera gen." (CAS 1148148-01 -9) significa cloro[2-(diciclohexilfosfino)-3,6-dimetoxi-2,,4,,6,-triisopropil-1,1,-bifenil][2-(2-aminoetil)fenil]-paladio (II), "Binap" significa Rac-bis(difenilfosfino)-1,1 '-binaftilo, "ta" significa temperatura ambiente, "(K)" significa Kofler, "DSC" significa calorimetría diferencial de barrido, "P.F." significa punto de fusión.
A. Preparación de los compuestos intermedios
Ejemplo A1
Preparación del intermedio 1:

Una mezcla de 6-yodo-8-cloro-2-metil-imidazo[1,2-a]pirazina (documento WO 2011/110545) (3 g; 10.22 mmol), 3,6-dihidro-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-2H-pirano (2.41 g; 11.45 mmol), K3 PO4 (6.51 g; 30.67 mmol) en H2 O (18 ml) y 1,4-dioxano (180 ml) se purgó cuidadosamente con N2. Se añadió PdCl2 (dppf).DCM (920 mg; 1.12 mmol) y la mezcla de reacción se purgó una vez más con N2. La mezcla de reacción se calentó a 80 °C durante 24 h. La solución se enfrió, y se vertió en agua enfriada. Se añadió EtOAc y la mezcla se filtró a través de un lecho de celite®. El producto se extrajo con EtOAc, la capa orgánica se secó sobre MgSO4, se filtró y se evaporó a sequedad. El residuo (5 g) se purificó por cromatografía sobre gel de sílice (SiOH Irregular; 20-45 pm; 450 g; fase móvil: 65 % de heptano, 5 % de MeOH (+ NH4OH al 10 %), 35 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 1.6 g (63 %) de compuesto intermedio 1.
Vía alternativa:
Una mezcla de 6-bromo-8-cloro-2-metil-imidazo[1,2-a]pirazina (documento WO2010089292) (1 g; 4.06 mmol), 3,6-dihidro-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-2H-pirano (0.96 g; 4.54 mmol), K3 PO4 (2.6 g; 12.2 mmol) en 1,4-dioxano (30 ml) y H2 O (3 ml) se purgó cuidadosamente con nitrógeno. Se añadió PdCl2 (dppf).DCM (0.37 g; 0.45 mmol) y la mezcla de reacción se purgó una vez más con nitrógeno. La mezcla de reacción se calentó a 80 °C durante 24 h. La solución se enfrió a ta, se vertió en agua enfriada. Se añadió EtOAc y la mezcla se filtró a través de un lecho de celite®. El producto se extrajo con EtOAc, la capa orgánica se secó sobre MgSO4, se filtró y se evaporó a sequedad. El residuo (1.5 g) se purificó por cromatografía sobre gel de sílice (SiOH Irregular; 20-45 pm; 450 g; fase móvil: 65 % de heptano, 5 % de MeOH (+ NH4OH al 10 %), 35 % de EtOAc). Se recogieron las fracciones puras y se evaporó el disolvente para dar 600 mg (59 %) de intermedio 1.
Preparación del intermedio 2:

Una mezcla del intermedio 1 (1 g; 4 mmol), pinacol éster ácido 1-(tetrahidropiran-2-il)-1H-pirazol-5-borónico (1. 7 g; 6 mmol), K3 PO4 (2.55 g; 12 mmol) en 1,4-dioxano (66 ml) y H2 O (6.6 ml) se purgó cuidadosamente con N2. Se añadió
PdCl2 (dppf).DCM (0.36 g; 0.44 mmol) y la mezcla de reacción se purgó una vez más con N2. La mezcla de reacción se calentó a 80 °C durante 24 h. La solución se enfrió a ta, se vertió sobre agua enfriada. Se añadió EtOAc y la mezcla se filtró a través de un lecho de celite®. El producto se extrajo con DCM y la capa orgánica se secó sobre MgSO4, se filtró y se evaporó a sequedad. El residuo (2.2 g) se purificó por cromatografía sobre gel de sílice (15-40 pm; 120 g; eluyente: 99 % de DCM, 1 % de MeOH, 0.1 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo se cristalizó con éter dietílico. El precipitado se filtró y se secó para proporcionar 1.23 g (84 % ) de intermedio 2.
Preparación del intermedio 3:

Una mezcla del intermedio 2 (1.23 g; 3.4 mmol), 1 -(clorometil)-2-metil-3-(trifluorometil-benceno (0.96 g; 4.6 mmol), K2 CO3 (0.7 g; 5 mmol) en 1,4-dioxano (125 ml) se purgó con N2. Después, se añadieron PPh3 (0.35 g; 1.35 mmol) y Pd(OAc)2 (0.15 g; 0.67 mmol) y se calentaron a 100 °C durante una noche en un tubo sellado. La solución se vertió en agua fría. Se añadió EtOAc y la mezcla se filtró a través de un lecho de celite®. El producto se extrajo con EtOAc, la capa orgánica se secó sobre MgSO4, se filtró y se evaporó a sequedad. El residuo (2.55 g) se purificó por cromatografía sobre gel de sílice (15-40 pm; 120 g; eluyente: 60 % de heptano, 5 % de MeOH, 35 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 1.29 g (71 % ) de compuesto intermedio 3.
Ejemplo A2
Preparación del intermedio 4:

El intermedio 1 (1 g; 4 mmol), Pd(OAc)2 (0.19 g; 0.4 mmol), 1,3-bis(difenilfosfino)propano (165 mg; 0.4 mmol), KOAc (0.79 g; 8.01 mmol) en MeOH (70 ml) se calentaron en un autoclave a 120 °C en una atmósfera de CO [gas CO (5 bares)] durante 8 h y a ta durante una noche. La mezcla se filtró a través un lecho de celite® y el filtrado se evaporó a sequedad. El residuo (2.5 g) se purificó por cromatografía sobre gel de sílice (SiOH Irregular; 20-45 pm; 450 g; fase móvil: 0.3 % de NH4OH, 97 % de DCM, 3 % de MeOH). Se recogieron las fracciones puras y se evaporó el disolvente para dar 725 mg (66 %) de intermedio 4.
Preparación del intermedio 5:

El intermedio 5 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del intermedio 3, usando el intermedio 4 y 1 -(clorometil)-2-metil-3-(trifluorometil-benceno como materiales de partida. El producto en bruto se purificó por cromatografía sobre gel de sílice (sílice esencial irregular; 150 g; fase móvil: 98 % de DCM, 2 % de MeOH). Se recogieron las fracciones puras y se evaporó el disolvente para dar 300 mg (25 % ) de intermedio 5.
Ejemplo A3
Preparación del intermedio 6:

Una mezcla del intermedio 1 (0.9 g; 3.6 mmol), A/,A/-dimetil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-Imidazol-1-sulfonamida (1.4 g; 4.7 mmol), K2 CO3 (1 g; 7.2 mmol) en H2O (15.5 ml) y 1,4-dioxano (62 ml) se purgó cuidadosamente con N2. PdCl2 (dppf). Se añadió DCM (0.3 g; 0.36 mmol) y la mezcla de reacción se purgó una vez más con N2. La mezcla de reacción se calentó a 80 °C durante 24 h. La solución se enfrió a ta, se vertió sobre agua enfriada. Se añadió EtOAc y la mezcla se filtró a través de un lecho de celite®. El producto se extrajo con EtOAc, la capa orgánica se secó sobre MgSO4, se filtró y se evaporó a sequedad. El residuo (2.1 g) se purificó por cromatografía sobre gel de sílice (15-40 gm; 80 g; eluyente: 95 % de DCM, 5 % de MeOH, 0.1 % de NH4OH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 1.28 g (91 %) de compuesto intermedio 6.
Preparación del intermedio 7:
El intermedio 7 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del intermedio 3, usando el intermedio 6 y 1 -(clorometil)-2-metil-3-(trifluorometil-benceno como materiales de partida. El producto en bruto se purificó por cromatografía sobre gel de sílice (SiOH irregular; 15-40 gm; 300 g; fase móvil: 0.1 % de NH4OH, 96 % de DCM, 4 % de MeOH). Se recogieron las fracciones puras y se evaporó el disolvente para dar 750 mg (40 %) de intermedio 7. Ejemplo A5
Preparación del intermedio 9:

Una mezcla de 2-amino-5-(morfolino)pirazina (1 g; 2.5 mmol) y cloro-2-propanona (0.9 ml; 11 mmol) en EtOH (50 ml) se calentó a 80 °C durante 4 h en cristalería de vidrio sellada, después durante una noche a ta. La solución se evaporó a sequedad. El residuo (1.2 g) se purificó por cromatografía sobre gel de sílice (15-40 gm; 120 g; eluyente: 98 % de DCM, 2 % de MeOH). Se recogieron las fracciones puras y se evaporó el disolvente para dar 330 mg (27 % ) de intermedio 9. Ejemplo A6
Preparación del intermedio 10:

El experimento se realizó 6 veces en 1 g (5.55 mmol) de 2-amino-5-(morfolino)pirazina.
Una mezcla de2-amino-5-(morfolino)pirazina (1 g; 5.55 mmol), 1-acetoxi-3-cloroacetona (1.15 ml; 9.83 mmol) y tamices moleculares 4 Á (1 g) en DME (30 ml) se calentó a 80 °C durante una noche. La mezcla se enfrió a ta. Se añadió DCM y la mezcla se filtró a través de un lecho de celite®. La capa orgánica se evaporó a sequedad. El residuo (14.15 g) se purificó por cromatografía sobre gel de sílice (SiOH Irregular; 20-45 gm; 450 g; fase móvil: 43 % de heptano, 7 % de MeOH, 50 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 1.1 g (12 %) de compuesto intermedio 10.
Ejemplo A7
Preparación del intermedio 11:

Una mezcla del compuesto 9 (0.36 g; 0.9 mmol) y óxido de manganeso (0.78 g; 9 mmol) en DCM (25 ml) se agitó a ta durante una noche. La mezcla se filtró a través un lecho de celite® y el filtrado se evaporó para dar 360 mg (cuantitativo) del intermedio 11. El producto crudo se utilizó sin purificación en el siguiente paso.
Ejemplo A8
Preparación del intermedio 12:

El experimento se realizó 3 veces en 1 g (5.55 mmol) de 2-amino-5-(morfolino)pirazina.
En un tubo sellado, se añadió 4-cloroacetatoacetato de etilo (1.36 ml; 9.99 mmol) a una mezcla de 2-amino-5-(morfolino)pirazina (1 g; 5.55 mmol) y tamices moleculares 4 Á (1 g) en DME (30 ml). La mezcla de reacción se calentó a 80 °C durante una noche. La mezcla se enfrió a ta, después se filtró a través de un lecho de celite® y el filtrado se evaporó a sequedad. El residuo (6 g) se purificó por cromatografía sobre gel de sílice (15-40 gm; 220 g; fase móvil: 0.1 % de NH4OH, 98 % de DCM, 2 % de MeOH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 1.45 g (30 %) de compuesto intermedio 12.
Preparación del intermedio 13:

Se añadió hidróxido de litio monohidrato (490 mg; 6.49 mmol) a una mezcla del compuesto 17 (0.6 g; 1.3 mmol) en H2O (2 ml) y MeOH (10 ml) a ta durante 24 h. Se eliminó MeOH por evaporación, se añadieron hielo-agua y agua seguido de una solución acuosa 3 N de HCl gota a gota, la solución se agitó a ta durante 3 h. El precipitado se eliminó por filtración, se lavó con agua y después éter dietílico, y se secó al vacío para dar 350 mg (62 %) del intermedio 13.
Ejemplo A9
Preparación del intermedio 14:

En material de vidrio sellado, una mezcla de 2-amino-5-(morfolino)pirazina (2 g; 11.10 mmol), bromopiruvato de etilo (1.39 ml; 11.10 mmol) y NaHCO3 (2.05 g; 24.42 mmol) en Ac N (110 ml) se agitó a 60 °C durante una noche. Después del enfriamiento a ta, la mezcla se filtró a través un lecho de celite® y el filtrado se evaporó a sequedad. El residuo se purificó por cromatografía sobre gel de sílice (SiOH irregular; 120 g; depósito sólido, fase móvil: del 100 % de DCM al 50 % de DCM, 50 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 750 mg (24 % , sólido de color beige) del intermedio 14.
Ejemplo A10
Preparación del intermedio 16:

Se añadió bromopiruvato de etilo (242 g; 1.24 mol) a la mezcla de 2-amino-5-bromopirimidina (180 g; 1.03 mol) en DMF (2 l) y la mezcla de reacción se agitó a ta (25°C) durante 2 días. El disolvente se concentró. Después, el residuo se ajustó a pH 3 con una solución acuosa saturada de NaOH (30 % ) y el precipitado se filtró para dar el producto en bruto 1. El filtrado se extrajo con EtOAc (4 x 500 ml) y la capa orgánica combinada se evaporó. El residuo resultante y el producto en bruto 1 (400 g) se combinaron y se purificaron por cromatografía sobre gel de sílice (gradiente: acetato de éter/éter de petróleo, 0/100 a 30/70) para dar 58.3 g (21 % ) del intermedio 16.
Preparación alternativa del intermedio 16:
Se añadieron 2-amino-5-bromopirimidina (15g; 73.5 mmol) y bromopiruvato de etilo (11.07 ml; 88.2 mmol) a etanol (320 ml). Esta mezcla de reacción se calentó a reflujo durante una noche. Después, se añadió más cantidad de bromopiruvato de etilo (11.07 ml; 88.2 mmol) y la mezcla de reacción se calentó a reflujo una noche más. La mezcla de reacción se enfrió a ta, se diluyó con agua y se basificó hasta pH 9 con carbonato sódico. La fase acuosa se extrajo con DCM. La fase orgánica se secó con MgSO4, se filtró y se concentró. El residuo se purificó por cromatografía sobre gel de sílice (gradiente: DCM al 100 % a DCM al 95 % de MeOH al 5 %). Las fracciones que contenían el producto se mezclaron y se concentraron. El residuo resultante se disolvió en éter dietílico y el precipitado se filtró para proporcionar 7 g (35 % ) del intermedio 16. Preparación del intermedio 18:

Se añadió morfolina (400 ml) a una mezcla del intermedio 16 (20 g; 74.05 mmol) y DIPEA (18.4 ml; 111.08 mmol). La mezcla de reacción se agitó a ta durante 24 h. El producto en bruto se evaporó al vacío (residuo pegajoso de color pardo). El residuo se recogió en DCM y la pasta se eliminó por filtración antes de la purificación. El residuo se purificó por cromatografía sobre gel de sílice (sílice 20-45 pm; 330 g; gradiente: del 100 % de DCM al 90 % de DCM, 10 % de MeOH, 0.1 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo resultante (10 g) se purificó por cromatografía sobre gel de sílice (sílice 20-45 pm; 120 g; gradiente: del 100 % de DCM al 90 % de DCM, 10 % de MeOH, 0.1 % de NH4OH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 3.3 g (16 %) de compuesto intermedio 18.
Ejemplo A11
Preparación del intermedio 21:

de manganeso (2.42 g; 27.81 mmol) en tolueno (23 ml) se calentó a 80 °C durante 12 h. La mezcla se enfrió a ta, se diluyó en DCM y se filtró a través de un lecho de celite® que se lavó con DCM. El filtrado se evaporó a sequedad para dar 0.67 g (76 %) del intermedio 21. El producto se utilizó sin purificación en la siguiente etapa.
Preparación del intermedio 22:

Una mezcla del intermedio 21 (271 mg; 0.67 mmol) y sal oxalato de 2,6-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (453 mg; 1.36 mmol) se disolvieron en DCM (3 ml) y se agitaron a 40 °C durante 1 h. Después, se añadió triacetoxiborohidruro sódico (360 mg; 1.7 mmol) y la mezcla de reacción se agitó a 40 °C durante 1 h 30. La mezcla se vertió en una solución acuosa de NaHCO3. La capa orgánica se lavó con agua y salmuera, se secó sobre MgSO4 , se filtró y se evaporó al vacío. El residuo se purificó por cromatografía sobre gel de sílice (gradiente: del 100 % de DCM al 90 % de DCM, 10 % de MeOH). Las fracciones puras se recogieron y se evaporaron al vacío para dar 176 mg (38 % , sólido) del intermedio 22.
Ejemplo A12
Preparación del intermedio 23:

El Compuesto 51 (176 mg; 0.39 mmol) se disolvió en THF (2 ml) y H2O (1 ml). Después, se añadió NaOH (31 mg; 0.79 mmol). La mezcla de reacción se agitó a ta durante 12 h. El disolvente se eliminó y el residuo (165 mg) se usó sin purificación en la siguiente etapa.
Ejemplo A14
Preparación del intermedio 25:

En un reactor Schlenk, a una solución de 6-bromo-2-metil-imidazo[1,2-a]piridina (1.26 g; 5.95 mmol) en 2-metil-2-butanol seco (25.1 ml) se le añadieron morfolina (1.26 ml; 14.3 mmol) y Cs2CO3 (3.88 g; 11.9 mmol). La mezcla se purgó al vacío y se cargó de nuevo con N2 (x3). Después, se añadieron RuPhos (167 mg; 0.36 mmol) y precatalizador BrettPhos de primera gen. (285 mg; 0.36 mmol). La mezcla se purgó al vacío, se cargó de nuevo con N2 y se calentó a 100 °C durante una noche. Después, se añadieron más morfolina (500 gl; 5.68 mmol), RuPhos (66 mg; 0.14 mmol) y precatalizador BrettPhos de primera gen. (113 mg; 0.14 mmol) y la mezcla se agitó a 100 °C durante una noche. La mezcla se filtró a través un lecho de celite® y la torta se aclaró con DCM (x2). El filtrado se lavó con agua, salmuera, se secó sobre MgSO4, se filtró y se evaporó al vacío. El residuo (1.8 g, goma de color verde oscuro) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 20-45 gm; 450 g; fase móvil: 43 % de heptano, 7 % de MeOH (+10 % de NH4OH), 50 % de DCM). Las fracciones puras se recogieron y el disolvente se evaporó para dar 405 mg (41 % , polvo de color beige) del intermedio 25.
Preparación del intermedio 26:

A una solución del intermedio 25 (405 mg; 1.86 mmol) en ACN (10 ml) a 0 °C se le añadió gota a gota una solución de N-yodosuccinimida (440 mg; 1.96 mmol) en ACN (8.6 ml). La mezcla de reacción se agitó a 0 °C durante 30 min. La mezcla se evaporó al vacío y el residuo se recogió en DCM y una solución acuosa al 10 % de K2 CO3. Las capas se separaron y el producto se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre MgSO4, se filtraron y se evaporaron al vacío. El residuo (1.4 g, polvo de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH irregular 30 gm; 40 g; fase móvil: del 100 % de DCM al 96 % de DCM, 4 % de MeOH, 0.4 % de NH4OH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 633 mg (96 % , polvo de color verde) del intermedio 26. Ejemplo A15
Preparación del intermedio 27:

En un matraz seco, se suspendió polvo de cinc (3.36 g; 51.37 mmol) en THF seco (50 ml) en una atmósfera de N2. La suspensión se calentó a 60 °C y después se añadió 1,2-dibromoetano (171 gl; 1.98 mmol). La mezcla se agitó a 60 °C durante 20 min y se enfrió a ta. Se añadió clorotrimetilsilano (200 gl; 1.58 mmol) y la mezcla de reacción se agitó a ta durante 20 min. A 0 °C, se añadió gota a gota 1-(bromometil)-2-metil-3-(trifluorometil)-benceno (10 g; 39.52 mmol) y la mezcla de reacción se agitó a ta durante 2 h. El producto en bruto se usó (M = 0.565 mol/l) directamente en la siguiente etapa sin purificación adicional.
Ejemplo A16
Preparación del intermedio 28:

En un reactor Schlenk, a una solución de 2-amino-5-(morfolino)piridina (2 g; 11.2 mmol) en DMF (40 ml) se le añadió 1-acetoxi-3-cloroacetona (2.23 ml; 19 mmol). La mezcla de reacción se agitó a 120 °C durante 3 h. La mezcla se evaporó al vacío. El residuo se recogió en DCM y se lavó con una solución saturada de NaHCO3. Las capas se separaron y la capa acuosa se extrajo con DCM (x2). Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre MgSO4, se filtraron y se evaporaron al vacío. El residuo (3.03 g, aceite de color negro) se purificó por cromatografía sobre gel de sílice (SiOH Irregular; 20-45 gm; 450 g; fase móvil: 43 % de heptano, 7 % de MeOH (+10 % de NH4OH), 50 % de DCM). Las fracciones puras se recogieron y el disolvente se evaporó para dar 880 mg (29 % , polvo de color pardo) del intermedio 28. Preparación del intermedio 29:
El compuesto intermedio 29 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis de compuesto intermedio 26, utilizando el compuesto intermedio 28 como material de partida. El producto en bruto (1.4 g, polvo de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH irregular; 15-40 gm; 50 g; gradiente: del 100 % de DCM al 96 % de DCM, 4 % de MeOH, 0.4 % de NH4OH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 1.14 g (79 % , polvo de color verde) del intermedio 29.
Preparación del compuesto intermedio 30 y el compuesto 28:

En un reactor Schlenk, a una solución del intermedio 29 (1.07 g; 2.67 mmol) en THF seco (26.8 ml) se le añadió bis(triterc-butilfosfina)paladio (0) (68 mg; 0.13 mmol). La mezcla se desgasificó cuidadosamente al vacío y se cargó de nuevo con N2 (x3). Después, se añadió el intermedio 27 (8.5 ml; 4.8 mmol) y la mezcla se desgasificó cuidadosamente al vacío y se cargó de nuevo con N2 (x3). La mezcla de reacción se agitó a 60 °C durante 3 h. La mezcla se diluyó en DCM y se filtró sobre sílice. El filtrado se evaporó al vacío y el residuo se recogió en DCM y agua. Las fases se separaron y la fase acuosa se extrajo con DCM. Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo (1.3 g, espuma de color naranja) se purificó por cromatografía sobre gel de sílice (SiOH irregular 15-40 gm; 50 g; gradiente: del 100 % de DCM al 96 % de DCM, 4 % de MeOH (0.4 % de NH4OH)). Las fracciones que contenían el compuesto se recogieron y el disolvente se evaporó. El residuo (750 mg, aceite de color verde) se purificó entonces por cromatografía sobre gel de sílice (SiOH irregular 30 gm; 40 g; gradiente: del 100 % de DCM al 96 % de DCM, 4 % de MeOH, 0.4 % de NH4OH). Las fracciones que contenían el producto se recogieron y el disolvente se evaporó para dar 436 mg (37 % , cristales de color verde) del intermedio 30 y 104 mg (polvo de color beige) de la fracción 1. Esta fracción se trituró en éter dietílico/heptano (2:1). El precipitado se eliminó por filtración y se secó para dar 60 mg (6 % , polvo de color blanco) del compuesto 28.
Ejemplo A17
Preparación del intermedio 31:

El experimento se realizó dos veces en 500 mg (2.79 mmol) de 2-amino-5-(morfolino)piridina.
En un vial para microondas, a una solución de 2-amino-5-(morfolino)piridina (500 mg; 2.79 mmol) en EtOH (12.5 ml) se le añadió bromopiruvato de etilo (0.89 ml; 7.12 mmol). La mezcla de reacción se calentó a 120 °C usando un microondas de un solo modo (Biotage Initiator) con una potencia de salida que variaba de 0 a 400 W durante 30 min de tiempo de mantenimiento fijo. Las dos reacciones se combinaron y se evaporaron al vacío. El residuo se recogió en DCM y una solución saturada de NaHCO3. Las capas se separaron y la capa acuosa se extrajo con DCM (x2). Las capas orgánicas se combinaron, se secaron sobre MgSO4, se filtraron y se evaporaron al vacío. El residuo (2.45 g, aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH irregular 15-40 gm; 80 g; gradiente: del 80 % de DCM, 20 % de EtOAc al 100 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 857 mg (56 % , polvo de color pardo) del intermedio 31.
Preparación del intermedio 32:

En un tubo sellado, a una solución del intermedio 31 (800 mg; 2.91 mmol) en THF seco (29 ml) a 0 °C se le añadió gota a gota borohidruro de litio (4 M en THF) (1.45 ml; 5.81 mmol). Se agitó la mezcla de reacción a 50 °C durante toda la noche. La mezcla se inactivó con una solución acuosa 1 N de HCl y se agitó a ta durante 1 h. La mezcla se basificó con una solución saturada de NaHCO3. La mezcla se concentró, y el concentrado se extrajo con DCM (x8), después con DCM/MeOH (9:1) (x3). Las capas orgánicas se combinaron, se secaron sobre MgSO4, se filtraron y se evaporaron al vacío para dar 613 mg (90 % , polvo de color blanco) del intermedio 32.
Preparación del intermedio 33:
A una solución del intermedio 32 (600 mg; 2.57 mmol) e imidazol (263 mg; 3.86 mmol) en DMF (25.7 ml) se le añadió tercbutildimetilclorosilano (582 mg; 3.86 mmol). La mezcla de reacción se agitó a ta durante 3 h. La mezcla se evaporó al vacío y el residuo se recogió en DCM y agua. Las capas se separaron y el producto se extrajo con DCM (x2). Las capas orgánicas combinadas se lavaron con salmuera (x2), se secaron sobre MgSO4, se filtraron y se evaporaron al vacío. El residuo (677 mg, aceite de color azul) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 |um; 30 g; gradiente: del 100 % de DCM al 20 % de DCM, 80 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 572 mg (64 % , aceite de color azul) del intermedio 33.
Preparación del intermedio 34:

A una solución del intermedio 29 (543 mg; 1.35 mmol) en EtOH (7 ml) y THF (7 ml) se le añadió NaOH (1 M en H2O) (6.77 ml; 6.77 mmol). La solución se agitó a ta durante 3 h, se concentró al vacío y después se neutralizó con una solución acuosa 1 N de HCl. El producto se extrajo con DCM (x2), después DCM/MeOH (95:5) (x2). Las capas orgánicas combinadas se secaron sobre MgSO4, se filtraron y se evaporaron al vacío para dar 461 mg (95 % , sólido de color gris) del intermedio 34. El producto se utilizó sin purificación en la siguiente etapa.
Preparación del intermedio 35:
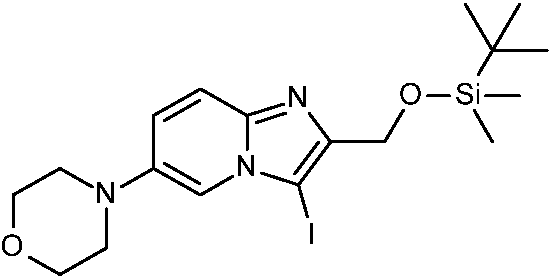
El compuesto intermedio 35 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis de compuesto intermedio 26, utilizando el compuesto intermedio 33 como material de partida. La mezcla de reacción se agitó a ta durante 1 h. El producto en bruto (775 mg, sólido de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH irregular 15-40 |um; 50 g; gradiente: del 100 % de DCM al 70 % de DCM, 30 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 589 mg (76 % , sólido de color rojizo) del intermedio 35.
Vía alternativa:
El compuesto intermedio 35 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis de compuesto intermedio 33, utilizando el compuesto intermedio 34 como material de partida. La mezcla de reacción se agitó a ta durante 18 h. El producto (566 mg, 93 % , sólido de color pardo) se usó sin purificación en la siguiente etapa.
Preparación del intermedio 36:

El intermedio 36 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del intermedio 30, usando el intermedio 35 y el intermedio 27 como materiales de partida. La mezcla de reacción se agitó a 60 °C durante 1 h. El producto en bruto (775 mg, sólido de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH regular 30 gm; 120 g; gradiente: del 100 % de DCM al 96 % de DCM, 4 % de MeOH, 0.4 % de NH4OH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 1.19 g (95 % , sólido de color pardo) del intermedio 36.
Ejemplo A18
Preparación del intermedio 37:

En un reactor sellado, a una solución de 2-amino-5-(morfolino)-piridina (500 mg; 2 .7 9 mmol) en DME (15 ml) se le añadieron 4-cloroacetoacetato de etilo (0.75 ml; 5.58 mmol) y tamices moleculares 4 Á (1 g). La mezcla de reacción se agitó a 80 °C durante 4 h. La mezcla se enfrió a ta, se vertió en hielo-agua y se filtró a través de un lecho de celite®. La torta se aclaró con EtOAc. El filtrado se basificó con una solución acuosa al 10 % de K2 CO3 y el producto se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre MgSO4 y se evaporaron al vacío. El residuo (820 mg, aceite de color negro) se purificó por cromatografía sobre gel de sílice (SiOH irregular 30 gm; 40 g; fase móvil: del 100 % de DCM al 96 % de DCM, 4 % de MeOH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo se recogió con éter dietílico y el disolvente se evaporó para dar 306 mg (38 % , sólido de color pardo) del intermedio 37.
Preparación del intermedio 38:

El compuesto intermedio 38 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis de compuesto intermedio 26, utilizando el compuesto intermedio 37 como material de partida. El producto en bruto (585 mg, aceite de color pardo) se combinó con un lote más pequeño procedente de una reacción preformada en 100 mg del intermedio 37 y el residuo resultante se purificó por cromatografía sobre gel de sílice (SiOH irregular 30 gm; 40 g; gradiente: del 100 % de DCM al 96 % de DCM, 4 % de MeOH, 0.4 % de NH4OH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 528 mg (61 % , polvo de color pardo) del intermedio 38.
Ejemplo A19
Preparación del intermedio 39:

A una solución del compuesto 28 (630 mg; 1.55 mmol) en DCM (36 ml) se le añadió óxido de manganeso (1.35 g; 15.5 mmol). La mezcla se calentó a reflujo durante 18 h. La mezcla se enfrió a ta y se filtró a través de un lecho de celite® que se aclaró con DCM. El filtrado se evaporó al vacío. Después, el residuo se coevaporó con éter dietílico (x2) para dar 566 mg (89 % , polvo de color beige) del intermedio 39.
Preparación del intermedio 40:

En un vial para microondas, a una solución del intermedio 39 (75 mg; 0.19 mmol) en MeOH (1.8 6 ml) se le añadieron ácido 2,6-Diazaespiro[3.5] nonano-2-carboxílico, 1,1-dimetiletil éster, etanodioato (2:1) (101 mg; 0.37 mmol). Se agitó la mezcla de reacción a ta durante la noche. La mezcla se evaporó al vacío. El residuo (180 mg) se recogió en DCE (1.77 ml) y después se añadió acetato de potasio (18 mg; 0.19 mmol). Después de 30 min a ta, se añadió triacetoxiborohidruro sódico (59 mg; 0.28 mmol) y la mezcla se agitó a ta durante 1 h. La mezcla se evaporó al vacío y el residuo se recogió en DCM y agua. Las capas se separaron y la capa orgánica se lavó con salmuera, se secó sobre MgSO4 , se filtró y se evaporó al vacío. El residuo (130 mg, aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH irregular 30 pm; 4 g; fase móvil: 99 % de DCM, 1 % de MeOH, 0.1 % de NH4OH al 95 % de DCM, 5 % de MeOH, 0.5 % de NH4OH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 101 mg (77 % , aceite de color verde) del intermedio 40.
Preparación del intermedio 41:

En un vial para microondas, a una solución del intermedio 39 (80 mg; 0.20 mmol) y sal del ácido 1-N-boc-1,6-diazaespiro[3.3]heptano oxálico (2:1) (53 mg; 0.11 mmol) en DCE (2 ml) se le añadió acetato de potasio (39 mg; 0.40 mmol). La mezcla de reacción se agitó a ta durante 1 h. Se añadió triacetoxiborohidruro sódico (63 mg; 0.30 mmol) y la mezcla se agitó a ta durante 1 h 30. Se añadieron DCM y agua. Las capas se separaron y la capa acuosa se basificó con una solución acuosa al 10 % de NaHCO3. El producto se extrajo con DCM. Las capas orgánicas se combinaron, se secaron sobre MgSO4, se filtraron y se evaporaron al vacío. El residuo (109 mg, aceite de color azul) se purificó por cromatografía
sobre gel de sílice (SiOH irregular 30 gm; 4 g; fase móvil: del 100 % de DCM al 95 % de DCM, 5 % de MeOH, 0.5 % de NH4OH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 85 mg (73 % , sólido de color gris) del intermedio 41.
Ejemplo A20
Preparación del intermedio 42:

En un tubo sellado, una solución de 2-amino-5-bromo-3-yodopiridina (10 g; 33.5 mmol) en 1-acetoxi-3-cloroacetona (35 ml; 298 mmol) se calentó a 60 °C durante 20 h y después a 80 °C durante 3 h. Después del enfriamiento a ta, el producto en bruto se vertió en agua, se neutralizó lentamente con K2 CO3 sólido y se extrajo con EtOAc (x3). La capa orgánica combinada se secaron MgSO4, se filtraron y se evaporaron al vacío. El residuo (32.6 g, aceite de color rosa oscuro) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 gm; 330 g; gradiente: del 100 % de DCM al 90 % de DCM, 10 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 9.64 g (73 % , aceite de color rosa que cristalizó tras reposo) del intermedio 42.
Preparación del intermedio 43:

A una solución del intermedio 42 (9.64 g; 24.4 mmol) en EtOH (120 ml) y THF (120 ml) se le añadió NaOH (1 M en H2O) (122 ml; 122 mmol). La solución se agitó a ta durante 96 h y después se evaporó al vacío. El residuo (sólido de color pardo) se diluyó en agua y se neutralizó con una solución acuosa 1 N de HCl. El sólido se filtró en una frita de vidrio, se lavó con agua y se secó al vacío para dar 5.47 g (64 % , sólido de color pardo pálido) del intermedio 43.
Preparación del intermedio 44:

Se añadió terc-butildimetilclorosilano (3.50 g; 23.2 mmol) a una suspensión del intermedio 43 (5.47 g; 15.5 mmol) e imidazol (1.58 g; 23.2 mmol) en DCM (155 ml) a ta. La mezcla se agitó a ta durante 18 h y después se añadió DMF (50 ml), y la mezcla se agitó durante 5 h (el intermedio 43 no era soluble en DCM). Después, se añadieron imidazol (1.58 g; 23.2 mmol), terc-butildimetilclorosilano (3.50 g; 23.2 mmol) y DMF (50 ml). La mezcla se convirtió en una solución después de unos pocos minutos y se agitó a ta durante 18 h. El producto en bruto se vertió en agua y después se añadieron DCM y una solución saturada de NaHCO3. La capa orgánica se separó y la capa acuosa se extrajo con DCM (x2). Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo (7.72 g, aceite de color pardo) se purificó por cromatografía sobre gel de sílice (Regular SiOH 30 gm; 200 g; gradiente: del 100 % de DCM al 90 % de DCM, 10 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 6.22 g (86 % , sólido de color rosa) del intermedio 44.
Preparación del intermedio 45:

En un tubo Schlenk, una solución del intermedio 42 (1.40 g; 3.54 mmol), pinacol éster del ácido 1-(tetrahidro-2H-piran-2-il)-1H-pirazol-5-borónico (1.18 g; 4.25 mmol) y fosfato potásico (2.26 g; 10.6 mmol) en 1,4-dioxano (25 ml) y H2 O (8 ml) se purgó con N2. Se añadió PdCl2 (dppf).DCM (290 mg; 0.35 mmol). La mezcla de reacción se purgó de nuevo con N2 y se calentó a 80 °C durante 1 h. Después del enfriamiento a ta, el producto en bruto se repartió entre EtOAc y agua. La capa orgánica se separó, se secó sobre MgSO4, se filtró y se evaporó al vacío. El residuo (de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH regular 30 gm; 80 g; gradiente: del 100 % de DCM al 70 % de Dc M, 30 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 1.26 g (85 % , aceite de color naranja pálido) del intermedio 45.
Preparación del intermedio 46:

El compuesto intermedio 46 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis de compuesto intermedio 34, utilizando el compuesto intermedio 45 como material de partida. El producto (461 mg, 95 % , sólido de color gris) se usó directamente sin purificación en la siguiente etapa.
Preparación del intermedio 47:

El intermedio 47 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del intermedio 45, usando el intermedio 44 y pinacol éster del ácido 1 -(tetrahidro-2H-piran-2-il)-1 H-pirazol-5-borónico como materiales de partida. El residuo (787 mg, aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH irregular 15-40 gm; 30 g; gradiente: del 100 % de DCM al 97 % de DCM, 3 % de MeOH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 450 mg (86 % , aceite de color amarillo) del intermedio 47.
Vía alternativa:
El compuesto intermedio 47 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis de compuesto intermedio 44, utilizando el compuesto intermedio 46 como material de partida. Se usó solamente DCM como disolvente. El residuo (aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 gm; 40 g; gradiente: del 100 % de DCM al 90 % de DCM, 10 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 1 g (73 % , aceite de color amarillo) del intermedio 47.
Preparación del intermedio 48:

En un tubo sellado, una mezcla del intermedio 47 (1 g; 2.04 mmol), morfolina (215 pl; 2.44 mmol) y CS2 CO3 (1.33 g; 4.07 mmol) en 2-metil-2-butanol (8.6 ml) se purgó con N2. Se añadieron RuPhos (48 mg; 102 pmol) y precatalizador BrettPhos de primera gen. (81 mg; 102 pmol). La mezcla de reacción se purgó con N2 y se calentó a 100 °C durante 18 h. Después del enfriamiento a ta, el producto en bruto se repartió entre EtOAc y agua. La capa orgánica se separó, se secó sobre MgSO4, se filtró y se evaporó al vacío. El residuo (1.21 g, aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 pm; 50 g; gradiente: del 100 % de DCM al 80 % de DCM, 20 % de acetona). Las fracciones puras se recogieron y el disolvente se evaporó para dar 398 mg (39 % , espuma de color amarillo pálido) del intermedio 48.
Preparación del intermedio 49:

A una solución del intermedio 48 (430 mg; 0.86 mmol) en DCM (8.6 ml) a 0 °C se le añadió N-yodosuccinimida (204 mg; 0.91 mmol). La solución se dejó calentar a ta y se agitó durante 1 h. Se añadieron agua y una solución acuosa al 10 % de Na2S2O3 al producto en bruto. Después, la capa orgánica se separó, se secó sobre MgSO4, se filtró y se evaporó al vacío. El residuo (aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 pm; 30 g; gradiente: del 100 % de DCM al 98 % de DCM, 2 % de iPrOH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 404 mg (75 % , espuma de color amarillo pálido) del intermedio 49.
Preparación del intermedio 50:

El intermedio 50 se preparó de acuerdo con un procedimiento análogo como se describe para la síntesis del intermedio 30, usando el intermedio 49 y el intermedio 27 como material de partida. La mezcla de reacción se agitó a 60 °C durante 1 h. El residuo (aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH irregular 15-40 pm; 30 g; gradiente: del 100 % de DCM al 50 % de DCM, 50 % de EtOAc). Se recogieron las fracciones puras y se evaporó el disolvente para dar 227 mg (53 %) de intermedio 50.
Ejemplo A21
Preparación del intermedio 51:

En un vial para microondas, a una solución de 2-amino-5-(morfolino)piridina (300 mg; 1.67 mmol) en 1,4-dioxano (10 ml) se le añadieron ZnCl2 (1 M en éter dietílico) (0.084 ml; 0.08 mmol), (terc-butildimetilsililoxi)acetaldehído (0.319 ml; 1.67 mmol) y 3-cloro-2-metil fenilisocianuro (0.23 ml; 1.67 mmol). El recipiente se cerró y la mezcla de reacción se calentó a 120 °C usando un microondas de un solo modo (Biotage Initiator e X p 60) con una potencia de salida que variaba de 0 a 400 W durante 15 min [tiempo de mantenimiento fijo]. La reacción se interrumpió con una solución saturada de NaHCO3 y se extrajo con DCM (x3). Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo se purificó por cromatografía sobre gel de sílice (SiOH irregular 15-40 pm; 12 g; fase móvil: del 100 % de DCM al 95 % de DCM, 5 % de MeOH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (300 mg, aceite) se trituró en éter dietílico y se evaporó. El residuo (300 mg, sólido pegajoso) se purificó por cromatografía sobre gel de sílice (SiOH irregular 15-40 pm; 12 g; fase móvil: del 80 % de heptano, 20 % de EtOAc al 60 % de heptano, 40 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 144 mg (aceite que cristalizó tras el reposo) del intermedio 51.
Ejemplo A22
Preparación del intermedio 52:

En un reactor Schlenk, se añadió 1-acetoxi-3-cloroacetona (5.78 ml; 49.1 mmol) a una solución de 5-bromo-2-piridinamina (5 g; 28.9 mmol) en DMF (110 ml). La solución se calentó a 120 °C durante 3 h y después a 80 °C durante 18 h. Después del enfriamiento a ta, el disolvente se eliminó al vacío. El residuo se recogió en DCM y se lavó con una solución acuosa al 10 % de NaHCO3. Las capas se separaron y la capa acuosa se extrajo con DCM (x2). Las capas orgánicas combinadas se lavaron con una solución acuosa saturada de NaCl, se secó sobre MgSO4, se filtró y se evaporó al vacío. El residuo (9.10 g, aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 pm; 330 g; fase móvil: 100 % de DCM al 95 % de DCM, 5 % de MeOH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 4.80 g (62 % , sólido de color rojo) del intermedio 52.
Preparación del intermedio 53:

El compuesto intermedio 53 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis de compuesto intermedio 43, utilizando el compuesto intermedio 52 como material de partida. La mezcla de reacción se agitó a ta durante 1 h, y después se evaporó al vacío. El residuo se neutralizó con una solución acuosa 1 N de HCl y se extrajo con DCM (x2). La mezcla se filtró en una frita de vidrio para dar 348 mg (17 % , sólido de color blanquecino) del intermedio 29. El filtrado se transfirió a un embudo de decantación, la capa orgánica se separó y la capa acuosa se extrajo con DCM (x2). Las capas orgánicas combinadas se secaron sobre MgSO4, se filtraron y se evaporaron al vacío para dar 1.34 g (67 % , sólido de color blanquecino) del intermedio 53. El producto (84 % , rendimiento global) se usó sin purificación en la siguiente etapa.
Preparación del intermedio 54:

El compuesto intermedio 54 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis de compuesto intermedio 44, utilizando el compuesto intermedio 53 como material de partida. Solamente se usó DMF como disolvente. La mezcla de reacción se agitó a ta durante 18 h. El residuo (2.96 g) se purificó por cromatografía sobre gel de sílice (Regular SiOH 30 gm; 200 g; gradiente: del 100 % de DCM al 90 % de DCM, 10 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 2.11 g (84 % ) de compuesto intermedio 54.
Preparación del intermedio 55:

En un tubo sellado, una solución del intermedio 54 (2.11 g; 6.18 mmol), pinacol éster del ácido 3,6-dihidro-2H-piran-4-borónico (1.95 g; 9.27 mmol) y fosfato potásico (3.94 g; 18.5 mmol) en 1,4-dioxano (41 ml) y H2O (12 ml) se purgó con N2. Se añadió PdCl2 (dppf).DCM (506 mg; 0.62 mmol). La mezcla se purgó de nuevo con N2 y se calentó a 80 °C durante 18 h. Después del enfriamiento a ta, se añadieron agua y EtOAc al producto en bruto y la mezcla se filtró a través un lecho de celite®. El filtrado se transfirió en un embudo de decantación. La capa orgánica se separó y la capa acuosa se extrajo con EtOAc (x2). Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo (4.12 g, aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 gm; 150 g; gradiente: del 100 % de DCM al 70 % de DCM, 30 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 2.49 g (95 % , en forma de un sólido de color pardo pálido) del intermedio 55.
Preparación del intermedio 56:
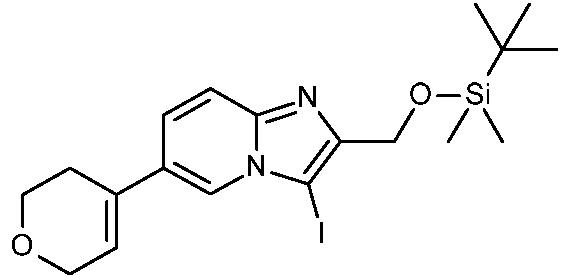
A una solución del intermedio 55 (2.49 g; 5.85 mmol) en ACN (60 ml) a 0 °C se le añadió lentamente N-yodosuccinimida (1.38 g; 6.15 mmol). La mezcla de reacción se dejó calentar a ta y se agitó durante 1 h. La mezcla se evaporó al vacío. Después, el residuo se recogió en DCM y una solución acuosa saturada de NaHCO3. Las capas se separaron y la capa acuosa se extrajo con DCM (x2). Las capas orgánicas combinadas se secaron sobre MgSO4, se filtraron y se evaporaron al vacío para dar 2.92 g (98 % , sólido de color pardo pálido) del intermedio 56 que se usó sin ninguna purificación adicional en la siguiente etapa.
Preparación del intermedio 57:

El intermedio 57 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del intermedio 30, usando el intermedio 56 y el intermedio 27 como material de partida. La mezcla de reacción se agitó a 60 °C durante 2 h. El residuo (aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH irregular 15-40 gm; 220 g; gradiente: del 100 % de DCM al 70 % de d C m, 30 % de EtOAc). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (2.99 g, espuma de color pardo pálido) se purificó por cromatografía sobre gel de sílice (SiOH irregular 40 gm; 120 g; fase móvil: 99 % de DCM, 1 % de MeOH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 1.5 g (51 % , sólido de color pardo pálido) del intermedio 57 y 500 mg (25 % , aceite de color pardo) del intermedio 55.
Ejemplo A23
Preparación del intermedio 59:

El intermedio 59 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del intermedio 39, usando el compuesto 47 y óxido de manganeso como material de partida. El filtrado se evaporó para dar 956 mg (espuma de color pardo) del intermedio 59. El producto se usó directamente sin purificación adicional en la siguiente etapa.
Ejemplo A24
Preparación del intermedio 60:

El intermedio 60 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del intermedio 45, usando el intermedio 44 y pinacol éster del ácido 1-(N,N-dimetilsulfamoil)imidazol-4-borónico como materiales de partida. El residuo (aceite de color amarillo) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 gm; 30 g; depósito sólido: gradiente: del 100 % de DCM al 80 % de DCM, 20 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 414 mg (75 % , sólido de color amarillo) del intermedio 60.
Preparación del intermedio 61:

El intermedio 61 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del intermedio 55, usando el intermedio 60 y pinacol éster del ácido 3,6-dihidro-2H-piran-4-borónico como material de partida. La mezcla de reacción se agitó a 80 °C durante 2 h. El residuo (405 mg, aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 gm; 24 g; gradiente: del 100 % de DCM al 95 % de DCM, 5 % de iPrOH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 213 mg (84 % , aceite de color amarillo) del intermedio 61.
Preparación del intermedio 62:

A una solución del intermedio 61 (152 mg; 0.29 mmol) en DCM (1 ml) a 0 °C se le añadió N-bromosuccinimida (55 mg; 0.31 mmol). La solución se dejó calentar a ta y se agitó durante 3 h. Se añadió una solución saturada de NaHCO3. Después, las capas se separaron y la capa acuosa se extrajo con DCM. Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo (163 mg, aceite de color pardo) se combinó con otro lote procedente de una reacción realizada en 50 mg del intermedio 37 y se purificó por cromatografía sobre gel de sílice (SiOH irregular 15 40 gm; 10 g; gradiente: del 100 % de DCM al 50 % de DCM, 50 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 159 mg (68 % , sólido de color beige) del intermedio 62.
Preparación del intermedio 63:
El intermedio 63 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del intermedio 30, usando el intermedio 62 y el intermedio 27 como materiales de partida. La mezcla de reacción se agitó a ta durante 2 h. El residuo (167 mg, sólido de color blanquecino) se purificó por cromatografía sobre gel de sílice (SiOH irregular 15-40 gm; 10 g; fase móvil: 100 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 68 mg (41 % , sólido de color amarillo) del intermedio 63.
Ejemplo A25
Preparación del intermedio 64:

El intermedio 64 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del intermedio 55, usando el intermedio 47 y pinacol éster del ácido 3,6-dihidro-2H-piran-4-borónico como materiales de partida. La mezcla de reacción se agitó a 80 °C durante 2 h. El residuo (620 mg, residuo de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 gm; 30 g; gradiente: del 100 % de DCM al 90 % de DCM, 10 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 412 mg (68 % , aceite de color amarillo) del intermedio 64.
Preparación del intermedio 65:

A una solución del intermedio 64 (300 mg; 0.61 mmol) en DCM (6 ml) a 0 °C se le añadió N-bromosuccinimida (113 mg; 0.64 mmol). La solución se dejó calentar a ta y se agitó durante 1 h. El producto en bruto se combinó con otro lote procedente de una reacción realizada en 50 mg del intermedio 64 y se lavó con una solución acuosa al 10 % de K2CO3. La capa orgánica se separó, se lavó con agua, se secó sobre MgSO4, se filtró y se evaporó al vacío para dar 338 mg (83 % , sólido de color pardo pálido) del intermedio 65. El producto se usó sin purificación adicional en la siguiente etapa. Preparación del intermedio 66 e intermedio 67:

El intermedio 66 y el intermedio 67 se prepararon de acuerdo con un procedimiento análogo al descrito para la síntesis del intermedio 30, usando el intermedio 65 y el intermedio 27 como materiales de partida. La mezcla de reacción se agitó a 60 °C durante 1 h. El residuo se recogió en DCM/MeOH (50/50) y se filtró a través de un lecho de celite® que se lavó con
DCM/MeOH (50/50). El filtrado se evaporó al vacío para dar 739 mg (residuo de color pardo) de una mezcla de dos intermedios 66 y 67. La mezcla se usó sin purificación adicional en la siguiente etapa.
Ejemplo A26
Preparación del intermedio 68:

Se añadió gota a gota n-BuLi (1.6 M en hexano) (2 ml; 3.21 mmol) a una solución de 1 -(dimetilsulfamoil)imidazol (563 mg; 3.21 mmol) en THF (32 ml) a -78°C. La mezcla de reacción se agitó durante 30 min. Después, se añadió una solución de ZnCl2 (2 M en THF) (3.21 ml; 6.42 mmol). La mezcla de reacción se dejó calentar a ta durante 30 min y se añadió a una mezcla desgasificada previamente del intermedio 44 (1 g; 2.14 mmol) y Pd(PPh3)4 (247 mg; 214 gmol) y la mezcla de reacción se calentó a 90 °C durante 1 h. Después del enfriamiento a ta, se añadieron EtOAc y una mezcla de H2O y una solución acuosa saturada de NaHCO3 (50/50) al producto en bruto. La fase acuosa se separó y se extrajo con EtOAc. Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo (1 .67 g, aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH irregular 15-40 gm; 50 g; gradiente: del 100 % de DCM al 95 % de DCM, 5 % de MeOH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 606 mg (55 % , aceite de color amarillo pálido) del intermedio 68.
Preparación del intermedio 69:

El intermedio 69 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del intermedio 55, usando el intermedio 68 y pinacol éster del ácido 3,6-dihidro-2H-piran-4-borónico como materiales de partida. El residuo (897 mg, aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH irregular 15-40 gm; 50 g; gradiente: del 100 % de DCM al 95 % de d C m, 5 % de MeOH). Se recogieron las fracciones puras y se evaporó el disolvente para dar 390 mg (64 % ) de intermedio 69.
Preparación del intermedio 70:

El compuesto intermedio 70 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis de compuesto intermedio 65, utilizando el compuesto intermedio 69 como material de partida. La capa orgánica se separó, se lavó con agua, se secó sobre MgSO4, se filtró y se evaporó al vacío para dar 444 mg (99 % , espuma de color amarillo pálido) del intermedio 70. El producto se usó sin purificación adicional en la siguiente etapa.
Preparación del intermedio 71:

El intermedio 71 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del intermedio 30, usando el intermedio 70 y el intermedio 27 como materiales de partida. La mezcla de reacción se agitó a 60 °C durante 2 h. Después del enfriamiento a ta, se añadieron de nuevo bis(tri-terc-butilfosfina)paladio (0) (18 mg; 34.4 gmol) y el intermedio 3 (608 gl; 0.344 mmol). La mezcla de reacción se purgó con N2 (x3) y se calentó a 60 °C durante 2 h. El residuo (1.05 g, aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 gm; 40 g; gradiente: del 100 % de DCM al 95 % de dC m , 5 % de MeOH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 386 mg (68 % , sólido de color amarillo) del intermedio 71.
Ejemplo A27
Preparación del intermedio 72:
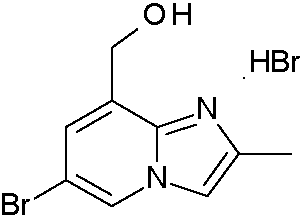
En un tubo sellado, a una solución de bromhidrato de 2-amino-5-bromo-3-piridinametanol (1:1) (4.5 g; 16 mmol) en DMF (50 ml) se le añadieron cloroacetona (3 ml; 17 mmol) y DIPEA (2.2 ml; 27 mmol). La mezcla de reacción se agitó a 130 °C durante 18 h. Después del enfriamiento, la mezcla se evaporó al vacío. El residuo se recogió con DCM lo que condujo a la precipitación. El sólido se eliminó por filtración para dar 2.19 g (43 % , sólido de color beige) del intermedio 72 (sa1HBr).
Preparación del intermedio 73:

En un tubo sellado, a una mezcla del intermedio 72 (600 mg; 1.86 mmol) en DCM (12 ml) y DMF (1.5 ml) se le añadieron p-toluenosulfonato de piridinio (47 mg; 0.19 mmol) y 3,4-dihidro-2H-pirano (2 ml; 22 mmol), y la mezcla de reacción se agitó a 50 °C durante 4 h. Después, la mezcla de reacción se enfrió y se evaporó a presión reducida. El residuo (1.24 g, aceite de color pardo que cristalizó tras el reposo) se recogió con DCM, se lavó dos veces con una solución saturada de NaHCO3, salmuera, se secó sobre MgSO4, se filtró y se evaporó al vacío. El residuo se purificó por cromatografía sobre gel de sílice (SiOH regular 15-40 gm; 40 g; carca seca sobre celite®; gradiente: del 100 % de DCM al 95 % de DCM, 5 % de MeOH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 557 mg (78 % , aceite de color rojo) del intermedio 73 y 241 mg (sólido de color blanco) del intermedio 72.
Preparación del intermedio 74:

El intermedio 74 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del intermedio 55, usando el intermedio 73 y pinacol éster del ácido 3,6-dihidro-2H-piran-4-borónico como materiales de partida. El residuo (1.24 g, aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 30 gm; 80 g; gradiente: del 100 % de DCM al 95 % de DCM, 5 % de acetona). Las fracciones puras se recogieron y el disolvente se evaporó para dar 430 mg (65 % , aceite de color pardo) del intermedio 74.
Preparación del intermedio 75:

El compuesto intermedio 75 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis de compuesto intermedio 56, utilizando el compuesto intermedio 74 como material de partida. La capa orgánica se concentró a 10 ml de una solución de DCM. Se añadió ACN (15 ml) y la solución se evaporó lentamente a 0-5 °C, lo que condujo a la precipitación. El precipitado se eliminó por filtración y se secó para dar 339 mg (67 % , sólido de color pardo pálido) del intermedio 75.
Preparación del intermedio 76:

El intermedio 76 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del intermedio 30, usando el intermedio 75 y 4 equivalentes del intermedio 27 como materiales de partida. La mezcla de reacción se agitó a 60 °C durante 2 h. El residuo (810 mg, aceite de color pardo) se sometió a sonicación en DCM. El sólido se eliminó por filtración y el filtrado se purificó por cromatografía sobre gel de sílice (SiOH regular 30 gm; 80 g; gradiente: del 100 % de DCM al 95 % de DCM, 5 % de MeOH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 196 mg (67 % , sólido de color blanquecino) del intermedio 76.
Ejemplo A28
Preparación del intermedio 77:

Se añadió cloruro de terc-butildimetilsililo (20.83 g; 138.22 mmol) a una solución de 6,8-dicloro-imidazo[1,2-a]piridina-2-metanol (10 g; 46.07 mmol) e imidazol (9.41 g; 138.22 mmol) en DMF (100 ml) a ta. La mezcla de reacción se agitó a ta durante 18 h. La solución se vertió en agua y una solución acuosa al 10 % de NaHCO3 (50/50). Se añadió DCM, la capa orgánica se separó y la capa acuosa se extrajo con DCM (2x). Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo (aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH 20-45 pm; 330 g; gradiente: del 90 % de heptano, 10 % de EtOAc al 70 % de heptano, 30 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 14.6 g (96 %) de compuesto intermedio 77.
Preparación del intermedio 78:

En cristalería sellada, una mezcla del intermedio 77 (14 g; 42.26 mmol), benzofenona imina (6.38 ml; 38.03 mmol), Cs2CO3 (41.3 g; 126.77 mmol), Binap (1.32 g; 2.11 mmol) y Pd(OAc)2 (474 mg; 2.11 mmol) en 1 ,4-dioxano (150 ml) se calentó a 100 °C durante 16 h. Después del enfriamiento a ta, se añadieron agua y EtOAc. La mezcla se extrajo con EtOAc (3X), se secó sobre MgSO4 y se evaporó a sequedad. El residuo (25 g) se purificó por cromatografía sobre gel de sílice (SiOH 20 45 pm; 330 g; gradiente: del 100 % de heptano al 60 % de heptano, 40 % de EtOAc). Las fracciones se recogieron y el disolvente se evaporó. El residuo (17.6 g) se purificó por cromatografía sobre gel de sílice (SiOH 20-45 pm; 220 g; gradiente: del 100 % de heptano al 70 % de heptano, 30 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 1.65 g (8 % ) del intermedio 54 y 12.2 g que se purificó por cromatografía sobre gel de sílice (SiOH 20-45 pm; 220 g; gradiente: del 100 % de heptano al 80 % de heptano, 20 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 8.4 g y 2.4 g (12 % ) del intermedio 78.
Preparación del intermedio 79:

En una atmósfera de nitrógeno en un tubo sellado, una mezcla del intermedio 78 (1.5 g; 3.15 mmol), 1-(clorometil)-2-metil-3-(trifluorometil)-benceno (0.99 g; 4.73 mmol) y K2CO3 (0.65 g; 4.73 mmol) en 1,4-dioxano (11 ml) se desgasificó en una atmósfera de N2. Después, se añadieron PPh3 (0.165 g; 0.63 mmol) y Pd(OAc)2 (71 mg; 0.32 mmol). La mezcla de reacción se calentó a 100°C durante una noche. El residuo (3.3 g) se purificó por cromatografía sobre gel de sílice (SiOH 20-45 pm;
80 g; gradiente: del 100 % de heptano al 70 % de heptano, 30 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 1.2 g (35 % ; 60 % de pureza evaluada por LCMS) del intermedio 79 y 0.4 g (17 % ; 87 % de pureza evaluada por LCMS) del intermedio 79.
Preparación del intermedio 80:

En un tubo sellado, una mezcla del intermedio 79 (600 mg; 0.93 mmol), morfolina (97.7 pl; 1.11 mmol) y Cs2CO3 (603 mg; 1.85 mmol) en 2-metil-2-butanol (4 ml) se desgasificó con N2. Se añadieron Ruphos (21.6 mg; 0.05 mmol) y precatalizador Brettphos de primera gen. (37 mg; 0.05 mmol). La mezcla de reacción se desgasificó con N2 y se calentó a 100 °C durante 18 h. Después del enfriamiento a ta, la mezcla se repartió entre EtOAc y agua. La capa orgánica se separó, se secó sobre MgSO4, se filtró y el disolvente se evaporó al vacío. El residuo (520 mg) se purificó por cromatografía sobre gel de sílice (SiOH 20-45 pm; 24 g; gradiente: del 100 % de heptano al 70 % de heptano, 30 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 68 mg (11 % ) del intermedio 80.
Preparación del intermedio 81:

Una mezcla del intermedio 79 (0.6 g; 0.93 mmol), pinacol éster del ácido 3,6-dihidro-2H-piran-4-borónico (218 mg; 1.04 mmol), fosfato potásico (589 mg; 2.78 mmol) en agua (1.62 ml) y 1,4-dioxano (7.72 ml) se purgó cuidadosamente con N2. Se añadió Pd.Cl2 (dppf).DCM (83 mg; 0.10 mmol) y la mezcla de reacción se purgó una vez más con N2. La mezcla de reacción se calentó a 80 °C durante 18 h. La solución se enfrió, se vertió en agua enfriada y se añadió EtOAc. La mezcla se filtró a través de un lecho de celite®. El producto se extrajo con EtOAc. La fase orgánica se secó sobre MgSO4, se filtró y se evaporó a sequedad. El residuo (1.7 g) se purificó por cromatografía sobre gel de sílice (SiOH 20-45 pm; 24 g; gradiente: del 100 % de heptano al 70 % de heptano, 30 % de EtOAc). Se recogieron las fracciones puras y se evaporó el disolvente para dar 50 mg (8 %) de intermedio 81.
Ejemplo A29
Preparación del intermedio 82:

La reacción se realizó dos veces en la misma cantidad de éster metílico del ácido 2-bromo-3-fluoro-benzoico:
Una mezcla de éster metílico del ácido 2-bromo-3-fluoro-benzoico (24.34 g; 104.45 mmol), 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-5-6-dihidropiridin-1(2H)-carboxilato de terc-butilo (48.44 g; 156.67 mmol) y K3 PO4 (66.5 g; 313.34 mmol) en 1,4-dioxano (250 ml) y agua (75 ml) se desgasificó en una atmósfera de N2. Se añadió PdCl2 (dppf).DCM (4.27 g; 5.22 mmol) y la mezcla de reacción se calentó a 100 °C durante una noche. La mezcla se vertió en agua y se filtró a través de un lecho de celite®. La capa orgánica se extrajo con DCM, se separó, se secó sobre MgSO4, se filtró y se concentró a sequedad. El residuo (55.6 g) se purificó mediante cromatografía sobre gel de sílice (SiOH irregular 15-40 pm; 220 g; fase móvil: DCM al 100 %). Las fracciones puras se recogieron y el disolvente se evaporó a sequedad. El residuo (37.9 g) se cristalizó en pentano. El precipitado se separó por filtración y se secó en vacío para dar 17.6 g (50 %) de compuesto intermedio 82.
Preparación del intermedio 83:

Una mezcla del intermedio 82 (16.5 g; 49.2 mmol) e hidróxido de paladio (1.4 g; 9.84 mmol) en MeOH (170 ml) se hidrógeno en un reactor Parr (2 atmósferas) durante 12 h a ta. Después de la captación de1H2 , el catalizador se filtró a través de un lecho de Celite®, se lavó con DCM y el filtrado se concentró para dar 16.4 g (99 % ) del intermedio 83.
Preparación del intermedio 84:

Se añadió en porciones LiAlH4 (1.85 g; 48.61 mmol) a una mezcla del intermedio 83 (16.4 g; 48.61 mmol) en THF (200 ml) a 5°C en una atmósfera de N2. La mezcla se agitó a 5°C durante 3 h. Se añadió gota a gota EtOAc seguido de H2O a la mezcla a -5°C. La suspensión se filtró a través un lecho de celite®. La capa orgánica se separó, se secó sobre MgSO4, se filtró y el disolvente se evaporó para dar 15.18 g del intermedio 84.
Preparación del intermedio 85:

Se añadió lentamente trietilamina (3.37 ml; 24.24 mmol) seguido de cloruro de metanosulfonilo (1.88 ml; 24.24 mmol) a una solución del intermedio 84 (5 g; 16.16 mmol) en DCM (60 ml) a 0 °C. La mezcla se agitó a ta durante una noche. Se añadió agua y el producto se extrajo con DCM. La capa orgánica se secó sobre MgSO4, se filtró y el disolvente se evaporó. El residuo (5.8 g) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 pm; 40 g; gradiente: del heptano al 80 % , 20 % de EtOAc al 60 % de heptano, 40 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó a sequedad para dar 3.26 g (61 %) del intermedio 85.
Preparación del intermedio 86:

En un vial para microondas, una mezcla del intermedio 19 (1 g; 3.61 mmol), el intermedio 85 (574 mg; 1.75 mmol) y K2 CO3 (0.75 g, 5.43 mmol) en 1,4-dioxano seco (10 ml) se desgasificó y se cargó de nuevo con N2 (3x). Se añadieron Pd(OAc)2 (83 mg, 0.36 mmol) y PPh3 (190 mg, 0.72 mmol). La mezcla se desgasificó, se cargó de nuevo con N2 (3x) y se calentó a 100 °C durante 18 h. Después del enfriamiento a ta, la mezcla se vertió en agua y la capa acuosa resultante se extrajo con DCM. Las capas orgánicas se combinaron, se lavaron con salmuera (2x), se secaron sobre MgSO4, se filtraron y se evaporaron. El residuo (1 g) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 gm; 40 g; gradiente: del 100 % de DCM al 95 % de DCM, 5 % de MeOH). Las fracciones puras se recogieron y el disolvente se evaporó a sequedad para dar 0.672 g (68 % ) del intermedio 86.
B. Preparación de los compuestos finales
Ejemplo B 1:
Preparación del compuesto 1:

La mezcla del intermedio 3 (0.44 g; 0.82 mmol) y HCl (4 M en dioxano) (4 ml) en 1,4-dioxano (44 ml) se calentó a 80 °C durante 1 h 30. La mezcla se enfrió y se añadió éter dietílico. Después, se filtró y se secó un precipitado. El residuo (255 mg) se recogió con DCM y H2 O y se basificó con K2CO3 sólido. La mezcla se agitó a ta durante 30 min. La capa orgánica se extrajo, se secó sobre MgSO4 y se evaporó para dar 40 mg (8 % ) del compuesto 1. P.F.: 240 °C (DSC).
Preparación del compuesto 6:

El intermedio 7 (720 mg; 1.28 mmol) se disolvió en 1,4-dioxano (25 ml) y se añadió HCl (6 M en agua) (8.3 ml). La mezcla de reacción se calentó a 100 °C durante 2 h, se enfrió a ta, se diluyó con EtOAc y se basificó con NH4OH. Se decantó la fase orgánica, se lavó con salmuera, se secó sobre MgSO4 , se filtró y se evaporó hasta sequedad. El residuo (0.5 g) se purificó por cromatografía sobre gel de sílice (SiOH irregular; 15-40 gm; 20 g; fase móvil: 0.5 % de NH4OH, 90 % de d C m, 10 % de MeOH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (115 mg) se cristalizó en éter dietílico. El precipitado se filtró y se secó para dar 90 mg (15 % ) del compuesto 6. P.F.: 197 °C (DSC).
Ejemplo B2:
Preparación del compuesto 2:

El compuesto 2 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del intermedio 3, usando el intermedio 10 y 1-(clorometil)-2-metil-3-(trifluorometil-benceno como materiales de partida. El producto en bruto se purificó por cromatografía sobre gel de sílice (15-40 gm; 120 g; fase móvil: 60 % de heptano, 40 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 400 mg (37 % ) del compuesto 2.
Ejemplo B3:
Preparación del compuesto 3:

Se añadió hidróxido de litio monohidrato (53 mg; 1.3 mmol) a una mezcla del intermedio 5 (113 mg; 0.25 mmol) en H2 O (0.3 ml) y THF (5 ml) a ta. La mezcla de reacción se agitó a ta durante 4 h. Se evaporó THF y se añadió H2 O. La capa acuosa se acidificó con una solución acuosa 3 N de HCl y el producto se extrajo con EtOAc. La fase orgánica se secó con MgSO4 y se evaporó a sequedad. El residuo (152 mg) se purificó por fase inversa (C18 10 gm; 30*150 mm; gradiente: del 80 % de TFA al 0.05 % , 20 % de ACN al 0 % de TFA al 0.05 % , 100 % de ACN). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (41 mg) se liofilizó con 20/80 de ACN/agua para dar 33 mg (30 % , polvo de color blanco) del compuesto 3. P.F.: 80 °C (goma, K).
Ejemplo B4:
Preparación del compuesto 4:

En una atmósfera de N2 a 10 °C, se añadió LiAlH4 (65 mg; 1.7 mmol) a una solución del intermedio 5 (0.1 g; 0.4 mmol) en THF (8 ml). La solución se dejó aclarar lentamente a ta y se agitó durante 20 h. Se añadieron hielo-agua y EtOAc, después la mezcla se filtró a través un lecho de celite®. El producto se extrajo con EtOAc, la capa orgánica se secó sobre MgSO4, se filtró y se evaporó a sequedad. El residuo (110 mg) se purificó por cromatografía sobre gel de sílice (sílice esencial esférica 5 pm 150 x 30.0 mm; gradiente: del 0.2 % de NH4OH, 98 % de DCM, 2 % de MeOH al 1.2 % de NH4OH, 88 % de DCM, 12 % de MeOH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (8 mg) se liofilizó con 20/80 de ACN/agua para dar 7.6 mg (4 % , polvo de color amarillo) del compuesto 4. P.F.: 80 °C (goma, K).
Ejemplo B5:
Preparación del compuesto 5:

En un tubo sellado, el intermedio 5 (110 mg; 0.25 mmol) y amoniaco (7 N en MeOH) (5 ml) se calentaron a 90 °C durante una noche. La mezcla se enfrió a ta y se evaporó a sequedad. El residuo (109 mg) se purificó por cromatografía sobre gel de sílice (SiOH irregular; 15-40 pm; 24 g; fase móvil: 99 % de DCM, 1 % de MeOH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (96 mg) se cristalizó en éter dietílico. El precipitado se filtró y se secó para dar 37 mg (35 % ) del compuesto 5. P.F.: 257 °C (DSC).
Ejemplo B7:
Preparación del compuesto 8:

Una mezcla del intermedio 9 (0.3 g; 1.38 mmol), 1-(bromometil)-2-metil-3-(trifluorometil)-benceno (0.49 g; 1.92 mmol), K2 CO3 (0.29 g; 2.06 mmol) en 1,4-dioxano (50 ml) se purgó con N2. Después, se añadieron PPh3 (0.14 g; 0.55 mmol) y Pd(OAc)2 (62 mg; 0.28 mmol). La mezcla de reacción se agitó a 100 °C durante una noche en un tubo sellado. La solución se enfrió a ta, se vertió en agua enfriada y se añadió EtOAc. La mezcla se filtró a través de un lecho de celite® y el producto se extrajo con EtOAc. La fase orgánica se secó sobre MgSO4, se filtró y se evaporó a sequedad. El residuo (900 mg) se purificó por cromatografía sobre gel de sílice (irregular 15-40 pm; 50 g; fase móvil: 43 % de heptano, 7 % de MeOH (+ NH4OH al 10 %), 50 % de EtOAc). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (175 mg) se purificó por SFC aquiral (NH25 pm; 150*30 mm; fase móvil: 91 % de CO2 , 9 % de MeOH (0.3 % de iPrNH2)). Se recogieron
las fracciones puras y se evaporó el disolvente. El residuo (28 mg) se liofilizó con 20/80 de ACN/agua para dar 26 mg (5 % , polvo de color beige) del compuesto 8. P.F.: 80 °C (goma, K).
Preparación del compuesto 50:

En un tubo sellado, a una solución del intermedio 14 (730 mg; 2.64 mmol) en 1,4-dioxano (26 ml) se añadieron 1-cloro-3-(clorometil)-2-metilbenceno (694 mg; 3.96 mmol) y K2CO3 (1.10 g; 7.93 mmol). La mezcla se desgasificó cuidadosamente al vacío y se cargó de nuevo con N2 (x3). Después, se añadieron Pd(OAc)2 (89 mg; 0.13 mmol) y PPh3 (69 mg; 0.26 mmol) y la mezcla se desgasificó de nuevo cuidadosamente al vacío y se cargó de nuevo con N2 (x3). Se agitó la mezcla de reacción a 100 °C durante toda la noche. La mezcla se combinó con otro lote (de 50 mg del intermedio 14). La mezcla se filtró a través un lecho de celite® y la torta se lavó con DCM. El filtrado se evaporó al vacío y el residuo se recogió en DCM y agua. Las fases se separaron y la fase acuosa se extrajo con DCM. Las capas orgánicas se combinaron, se lavaron son salmuera, se secaron sobre MgSO4, se filtraron y se evaporaron al vacío. El residuo (2.2 g, aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH irregular; 15-40 pm; 80 g; gradiente: del 70 % de DCM, 30 % de EtOAc a 100 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 483 mg (41 % , sólido de color verde) del compuesto 50.
Preparación del compuesto 51:

La reacción se realizó 5 veces en 1.17 g (4.24 mmol) del intermedio 18
En un tubo sellado, una mezcla del intermedio 18 (1.17 g; 4.24 mmol), 1-(clorometil)-2-metil-3-(trifluorometil)-benceno (0.88 g; 4.2 mmol) y K2 CO3 (0.88 g; 6.4 mmol) en 1,4-dioxano seco (10.6 ml) se desgasificó y se cargó de nuevo con N2 (x3). Se añadieron Pd(OAc)2 (97 mg; 0.42 mmol) y PPh3 (220 mg; 0.85 mmol) y la mezcla se desgasificó de nuevo y se cargó de nuevo con N2 (x3). La mezcla de reacción se calentó a 100 °C durante 18 h. Después, la mezcla de reacción se enfrió a ta, después todos los lotes se combinaron y se vertieron en agua (~500 ml). La mezcla acuosa resultante se extrajo con EtOAc (4 x 250 ml). Las capas orgánicas combinadas se lavaron con salmuera (x2), se secaron sobre MgSO4, se filtraron a través de un lecho de celite® que se lavó con DCM y EtOAc. Después, el filtrado se evaporó al vacío. El residuo se trituró con éter dietílico. El sólido resultante se filtró, se aclaró con éter dietílico frío y se secó al vacío para dar 5.05 g (50 % , sólido de color pardo pálido) del compuesto 51.
Preparación del compuesto 52:

La reacción se realizó dos veces en 1.17 g (4.22 mmol) del intermedio 18.
En un vial para microondas, una mezcla del intermedio 18 (1.17 g; 4.22 mmol), 1-(clorometil)-3-fluoro-2-metilbenceno (0.67 g; 4.2 mmol) y K2 CO3 (0.87 g; 6.3 mmol) en 1,4-dioxano seco (10.6 ml) se desgasificó y se cargó de nuevo con N2 (3x). Se añadieron acetato de paladio (II) (97 mg; 0.42 mmol) y PPh3 (221 mg; 0.84 mmol), y después más 1 ,4-dioxano (2.5 ml). La mezcla de reacción se desgasificó de nuevo y se cargó de nuevo con N2 (3x), se calentó a 100 °C durante 18 h y se enfrió a ta. Todos los lotes se combinaron y se vertieron en agua (200 ml). La mezcla acuosa resultante se extrajo con EtOAc (4 x 100 ml). Las capas orgánicas combinadas se lavaron con salmuera (2x), se secaron sobre MgSO4, se filtraron a través de un lecho de celite® que se aclaró con EtOAc y el filtrado se evaporó al vacío. El residuo (sólido de color beige húmedo) se sometió a sonicación y se trituró en éter dietílico. El sólido resultante se filtró, se aclaró con éter dietílico frío y se secó al vacío (30 °C durante 40 h) para dar 2.35 g (70 % , sólido de color blanquecino) del compuesto 52.
Ejemplo B9:
Preparación del compuesto 10:

Una mezcla del intermedio 11 (0.16 g; 0.4 mmol), 4-piperidinmetanol (93 mg; 0.81 mmol) en MeOH (7.5 ml) y THF (4 ml) se agitó a ta durante 1 h 30. Se añadió NaBH4 (8 mg; 0.2 mmol) y la solución se agitó durante 30 min. Se añadieron H2O y DCM. La capa orgánica se extrajo, se secó sobre MgSO4 y se evaporó a sequedad. El residuo (167 mg) se purificó por cromatografía sobre gel de sílice (sílice esencial esférica 5 gm; 150 x 30.0 mm; gradiente: del 95 % de DCM, 5 % de MeOH (+10 % de NH4OH) al 82 % de DCM, 18 % de MeOH (+10 % de NH4OH)). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (81 mg) se cristalizó en éter dietílico. El precipitado se filtró y se secó para dar 51 mg (25 % ) del compuesto 10. P.F.: 198 °C (K).
Preparación del compuesto 11:

El compuesto 11 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del compuesto 10, usando el intermedio 11 y cis-2,6-dimetilpiperazina como materiales de partida. El producto en bruto se purificó por cromatografía sobre gel de sílice (irregular 15-40 gm; 40 g; fase móvil: 94 % de DCM, 6 % de MeOH, 0.6 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (56 mg) se purificó por SFC aquiral (CHIRALPAK IC 5 gm 250 x 20 mm, Fase móvil: 60 % de CO2 , 40 % de MeOH (0.3 % de iPrNH2)). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (42 mg) se purificó de nuevo por SFC aquiral (CYANO 6gm 150x21.2 mm, Fase móvil: 80 % de CO2 , 20 % de MeOH (0.3 % de iPrNH2)). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (22 mg) se liofilizó con 20/80 de ACN/agua para dar 20 mg (9 % , polvo de color blanco) del compuesto 11. P.F.: 80 °C (goma, K).
Preparación del compuesto 12:

El compuesto 12 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del compuesto 10, usando el intermedio 11 y etanolamina como materiales de partida. El producto en bruto se purificó por cromatografía sobre gel de sílice (sílice esencial esférica 5 gm; 150 x 30.0 mm; gradiente: del 95 % de DCM, 5 % de MeOH (+10 % de NH4OH) al 82 % de DCM, 18 % de MeOH (+10 % de NH4OH)). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (121 mg) se purificó por cromatografía sobre gel de sílice (irregular 15-40 gm; 24 g; fase móvil: 90 % de DCM, 10 % de MeOH, 0.1 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (42 mg) se liofilizó con 20/80 de ACN/agua para dar 40 mg (28 % , polvo de color blanco) del compuesto 12. P.F.: 80 °C (goma, K).
Ejemplo B10:
Preparación del compuesto 13:

A una suspensión de LiAlH4 (140 mg; 3.68 mmol) en THF anhidro (5 ml) a 0 °-5 °C en una atmósfera de N2 , se le añadió gota a gota una solución del compuesto 17 (850 mg; 1.84 mmol) en THF anhidro (15 ml) y la mezcla se agitó durante 2 h a 10 °C. Se añadió gota a gota EtOAc seguido cuidadosamente de 2 ml de una solución acuosa 3 N de NaOH y agua (2 ml). Se añadió EtOAc. Después, la mezcla se filtró a través de un lecho de celite®. La fase orgánica se decantó, se secó con MgSO4, se filtró y se evaporó a sequedad. El residuo (0.7 g) se purificó por cromatografía sobre gel de sílice (SiOH irregular; 15-40 gm; 40 g; fase móvil: 0.1 % de NH4OH, 96 % de DCM, 4 % de MeOH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 215 mg (28 % ) del compuesto 13. P.F.: 142 °C (K).
Ejemplo B 11:
Preparación del compuesto 14:

A 10 °C, se añadió HBTU (153 mg; 0.4 mmol) a una mezcla del i
ntermedio 13 (175 mg; 0.4 mmol), cis-2,6-dimetilpiperazina (69 mg; 0.6 mmol), DIPEA (0.21 ml; 1.21 mmol) en DMF seca (5 ml). La mezcla de reacción se agitó durante 48 h. La solución se vertió en H2O y se extrajo con EtOAc (x2). La capa orgánica se lavó con salmuera. La fase orgánica se secó sobre MgSO4, se filtró y se evaporó a sequedad. El producto en bruto se cristalizó con éter dietílico. Después, el precipitado se filtró y se secó. El precipitado (0.34 g) se purificó por cromatografía sobre gel de sílice (sílice esencial irregular 40 g; fase móvil: 0.5 % de NH4OH, 95 % de DCM, 5 % de MeOH).
Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (130 mg) se liofilizó con 20/80 de ACN/agua para dar 106 mg (50 % , polvo de color blanco) del compuesto 14. P.F.: 80 °C (goma, K).
Preparación del compuesto 15:

El compuesto 15 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del compuesto 14, usando el intermedio 13 y 3-(hidroximetil)azetidina como materiales de partida. El producto en bruto se cristalizó en éter dietílico. Después, el precipitado se filtró y se secó. El precipitado (0.21 g) se purificó por cromatografía sobre gel de sílice (sílice esencial irregular 40 g; fase móvil: 0.7 % de NH4 OH, 93 % de DCM, 7 % de MeOH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (60 mg) se liofilizó con 20/80 de ACN/agua para dar 51 mg (25 % , polvo de color blanco) del compuesto 15. P.F.: 80 °C (goma, K).
Ejemplo B12:
Preparación del compuesto 16:

En un tubo sellado, a una solución del compuesto 50 (430 mg; 1.04 mmol) en THF seco (10 ml) a 0 °C se le añadió gota a gota borohidruro de litio (4 M en THF) (518 pl; 2.07 mmol). La mezcla se agitó a ta durante 2 h. La mezcla se diluyó con EtOAc y se inactivó con una solución acuosa al 10 % de NH4Cl. La mezcla se combinó con otro lote procedente de una reacción realizada en 50 mg del compuesto 50. Las capas se separaron y el producto se extrajo con EtOAc (x2). Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo (355 mg, aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH irregular 15-40 pm; 50 g; gradiente: del 100 % de DCM al 92 % de DCM, 8 % de MeOH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (77 mg, sólido de color rojo) se purificó por fase inversa (C18 5 pm; 30*150 mm; gradiente: del 80 % (NH4HCO3 ac. al 0.5 %), 2 0 % de ACN a 100 % de ACN). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (36 mg, aceite incoloro) se liofilizó con MeOH/agua, 20/80, para dar 36 mg (sólido de color blanco). Esta fracción se purificó por cromatografía sobre gel de sílice (sílice esencial esférica 5 pm; 150 x 30.0 mm; gradiente: del 98 % de DCM, 2 % de MeOH, 0.2 % de NH4OH al 87 % de Dc M, 13 % de MeOH, 1.3 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (16 mg, aceite incoloro) se liofilizó con ACN/agua, 23/77 para dar 13 mg (3 % , sólido de color blanco) del compuesto 16. P.F.: 184 °C (DSC).
Ejemplo B13:
Preparación del compuesto 17:

El compuesto 17 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del intermedio 3, usando el intermedio 12 y 1-(clorometil)-2-metil-3-(trifluorometil-benceno como materiales de partida. El producto en bruto se recogió con éter dietílico. El precipitado se separó por filtración y se secó en vacío para dar 530 mg (24 % ) de compuesto 17. P.F.: 135 °C (Mettler Toledo).
Ejemplo B14:
Preparación del compuesto 18:

Se añadió gota a gota hidruro de diisobutilaluminio (1 M en DCM) (54 ml; 54 mmol) a una solución del compuesto 51 (5.04 g; 10.7 mmol) en THF (200 ml) a -5 °C en una atmósfera de N2. Después, la mezcla de color pardo resultante se dejó alcanzar suavemente la ta y se agitó durante 16 h. Se añadió más cantidad de hidruro de diisobutilaluminio (1 M en DCM) (18 ml; 18 mmol) a -5 °C y la mezcla se dejó alcanzar suavemente la ta y se agitó durante 3 h más. La mezcla resultante se vertió suavemente en agua destilada a 0 °C en agitación y las capas acuosas se extrajeron con DCM (4 x 300 ml) y después DCM/MeOH (90/10, 200 ml). Las capas orgánicas combinadas se secaron sobre MgSO4 y la suspensión resultante se filtró a través un lecho de celite® y después se evaporó. El residuo (5.6 g de residuo de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH regular 30 gm; 200 g; carga seca de celite®; gradiente: del 99 % de DCM, 1 % de MeOH al 96 % de DCM, 4 % de MeOH). Las fracciones que contenían producto se combinaron y se evaporó DCM al vacío dando como resultado la precipitación de un sólido en el MeOH restante. Este sólido se filtró (1.41 g, sólido de color blanquecino) y se recristalizó en un mínimo de EtOH caliente (~200 ml) con enfriamiento lento. El sólido se filtró, se aclaró con EtOH frío y se secó a alto vacío a 60 °C durante 4 h para dar 1.16 g (27 % , sólido de color blanco) del compuesto 18. P.F.: 231 °C (DSC).
Preparación del compuesto 25:

Se añadió gota a gota hidruro de diisobutilaluminio (1 M en DCM) (30 ml; 30 mmol) durante 1 h a una solución del compuesto 52 (1.98 g; 4.97 mmol) en THF (93 ml) a -10 °C en agitación en una atmósfera de N2. Después, la mezcla de color pardo resultante se dejó alcanzar suavemente la ta y se agitó durante 18 h. Después, la solución de color pardo se puso a 0 °C, se inactivó mediante la adición gota a gota de EtOAc (50 ml), seguido de una solución acuosa al 15 % de sal de Rochelle (~100 ml). La mezcla se agitó durante 2 h y se extrajo con EtOAc (dos veces). Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo (2.75 g, compuesto pegajoso de color naranja) se combinó con otro lote procedente de una reacción realizada en 350 mg (0.88 mmol) del compuesto 52. La mezcla del residuo se purificó por cromatografía sobre gel de sílice (SiOH regular; 30 gm, 80 g; carga seca (celite®), gradiente: del 100 % de DCM al 90 % de DCM, 10 % de MeOH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (1.1 g, sólido de color blanquecino) se recristalizó en un mínimo de EtOH caliente (~150 ml) con enfriamiento lento a ta (durante ~6 h), después enfriamiento lento a 14 °C durante 2 h para maximizar el rendimiento de cristalización. El sólido resultante se filtró, se lavó con una cantidad mínima de éter dietílico frío y se secó para dar 883 mg (42 % , sólido de color blanco) del compuesto 25. P.F.: 210 °C (DSC).
El filtrado se evaporó al vacío para dar un lote adicional de 228 mg del compuesto 25 (11 % , no totalmente puro, sólido de color beige).
Preparación del compuesto 48:

El compuesto 48 se preparó de acuerdo con un procedimiento análogo como se describe para la síntesis del compuesto 18, usando el intermedio 86 como material de partida (cristalizado en DIPE; 43 mg, 1 %). P.F.: 222 °C (DSC).
Ejemplo B15:
Preparación del compuesto 19:

Una mezcla del intermedio 21 (174 mg; 0.43 mmol) y cis-2,6-dimetilpiperazina en MeOH (3 ml) se agitó a ta durante 2 h. Después, se añadió NaBH4 (24 mg; 0.65 mmol) y la mezcla de reacción se agitó a ta durante una noche. Se añadió más cantidad de cis-2,6-dimetilpiperazina (1.5 equiv.) y la mezcla de reacción se agitó durante 6 h a 30 °C. La mezcla de reacción se calentó a 60 °C durante 1 h. Se añadió NaBH4 y la mezcla se agitó a ta durante 1 h. El disolvente se eliminó, y el producto en bruto se purificó por cromatografía sobre gel de sílice (sílice, gradiente: del 100 % de DCM al 90 % de DCM, 10 % de MeOH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 40 mg (18 % ) del compuesto 19. P.F.: 263 °C (MP50 Mettler Toledo).
Preparación del compuesto 21:

Se añadió triacetoxiborohidruro sódico (0.157 g; 0.74 mmol) a una mezcla del intermedio 21 (0.2 g; 0.50 mmol), 2-tia-6-aza-espiro[3.3]heptano 2,2-dióxido (trifluoroacetato) (0.193 g; 0.74 mmol), acetato sódico (61 mg; 0.74 mmol) en DCE (5 ml). La mezcla de reacción se agitó durante la noche a ta. La solución se vertió en una mezcla de H2 O y NaHCO3, y después se extrajo con DCM. La capa orgánica se secó sobre MgSO4, se filtró y se evaporó a sequedad. El residuo (0.248 g) se purificó por cromatografía sobre gel de sílice (sílice esencial esférica 5 gm 150x30.0 mm; gradiente: del 98 % de DCM, 2 % de MeOH, 0.2 % de NH4OH al 88 % DCM, 12 % de MeO, 1.2 % de NH4OH). Las fracciones puras se recogieron y el disolvente se evaporó a sequedad. El residuo (0.025 g) se cristalizó a partir de DIPE. El precipitado se retiró por filtración y se secó al vacío para dar 0.015 g (6 % ) del compuesto 21. P.F.: 228 °C (kofler).
Preparación del compuesto 22:

El compuesto 22 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del compuesto 21, usando el intermedio 21 y 1,1 -dióxido de tiomorfolina como material de partida. El residuo (286 mg) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 gm; 40 g; gradiente: del 100 % de DCM al 95 % de DCM, 5 % de MeOH, 0.1 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (0.165 g) se cristalizó en DIPE y el 10 % de ACN. El precipitado se separó por filtración y se secó en vacío para dar 0.048 g (15 % ) de compuesto 22. P.F.: 225°C (kofler).
Preparación del compuesto 24 y el compuesto 24a

Una solución del intermedio 21 (250 mg; 0.62 mmol) y clorhidrato de 2,2-dióxido de 2-tia-7-azaespiro[4,4]nonano (130.88 mg; 0.62 mmol) en MeOH (16.6 ml) se agitó a ta. Se añadió gota a gota AcOH (722 gl; 12.61 mmol) seguido de la adición en porciones de borohidruro sódico (39 mg; 0.62 mmol). La mezcla de reacción se agitó a ta durante 16 h. La mezcla de reacción se vertió en una solución acuosa al 10 % de K2CO3 y se extrajo con DCM. La capa orgánica se secó sobre MgSO4, se filtró y el disolvente se evaporó. El residuo se purificó por cromatografía sobre gel de sílice (SiOH 20-45 gm; 24 g; gradiente: 98 % de DCM, 2 % de MeOH, 0.1 % de NH4OH al 95 % de DCM, 5 % de MeOH, 0.1 % de NH4OH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 125 mg del compuesto amorfo 24. Esta fracción se disolvió en ACN (5 ml) a ta, después se añadió gota a gota HCl (4 M en 1,4-dioxano) (500 gl) y la mezcla de reacción se agitó a ta durante 16 h. No tuvo lugar precipitación de sal. Después, el disolvente se evaporó a presión reducida y el sólido resultante se trituró en éter diisopropílico, se filtró y se secó al vacío para dar 75 mg (19 %) del compuesto 24a (1.63 HCl 0.71 H2 O).
Preparación del compuesto 26:
El compuesto 26 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del compuesto 24, usando el intermedio 21 y clorhidrato de 1,1 -dióxido de 1-tia-7-azaespiro[4,4]nonano como material de partida. El residuo se purificó por cromatografía sobre gel de sílice (SiOH 20-45 gm; 24 g; gradiente: 98 % de DCM, 2 % de MeOH, 0.1 % de NH4OH al 95 % de DCM, 5 % de MeOH, 0.1 % de NH4OH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 130 mg del compuesto sólido amorfo 26. Esta fracción se disolvió en ACN (2 ml) y la mezcla se calentó hasta que se disolvió completamente. La mezcla de reacción se enfrió a ta, el precipitado resultante se filtró, se lavó con una pequeña cantidad de éter diisopropílico y se secó para dar 90 mg (26 % , sólido de color blanco) del compuesto 26. P.F.: 200 °C (DSC).
Ejemplo B16:
Preparación del compuesto 20:

Se añadió TFA (1 ml) a una solución del intermedio 22 en DCM (3 ml). La mezcla de reacción se agitó a ta durante 1 h. Los disolventes se eliminaron y el residuo en bruto se lavó dos veces con tolueno. El producto se purificó por cromatografía sobre gel de sílice (sílice; gradiente: del 100 % de DCM al 90 % de DCM, 10 % de MeOH 0.1 % de NH4OH). Las fracciones puras se recogieron y se evaporaron para dar 6 mg (4 % ) del compuesto 20.
Preparación del compuesto 53:

Se añadió gota a gota TFA (1.32 ml; 17.76 mmol) a una solución del intermedio 86 (672 mg; 1.18 mmol) en DCM (10 ml) a 0 °C. La solución se dejó calentar a ta y se agitó a ta durante una noche. La mezcla se vertió en agua, se basificó con una solución acuosa de K2 CO3 al 10 % y el compuesto se extrajo con DCM. La capa orgánica se separó, se secó sobre MgSO4, se filtró y se evaporó. El residuo (0.57 g) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 gm; 40 g; gradiente: del 100 % de DCM al 90 % de DCM, 10 % de MeOH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 0.22 g (40 %) del compuesto 53.
Ejemplo B17:
Preparación del compuesto 23:

La reacción se realizó dos veces en 165 mg (0.39 mmol) del intermedio 23.
Una solución de cis-2,6-dimetilpiperazina (93 mg; 0.79 mmol) en DMF seca se añadió a una solución del intermedio 23 (165 mg; 0.39 mmol), HBTU (447 mg; 1.18 mmol) y DIPEA (0.205 ml; 1.18 mmol) en DMF seca (5 ml). La mezcla de reacción se agitó a ta durante 1 h. Se añadieron gotas de amoniaco (7 N en MeOH) y se vertió EtOAc en la mezcla de reacción. Los dos lotes se combinaron para el tratamiento. La capa orgánica resultante se lavó con agua, y después con salmuera. La capa orgánica se evaporó. El residuo (203 mg) se purificó por cromatografía sobre gel de sílice (SiOH; gradiente: del 100 % de DCM al 90 % de DCM, 10 % de MeOH). Se recogieron las fracciones puras y se evaporaron. El residuo se cristalizó en éter dietílico. El precipitado se eliminó por filtración y se secó para dar 19 mg (5 % ) del compuesto 23. P.F.: 130°C (MP50 Mettler Toledo).
Ejemplo B18:
Preparación del compuesto 27:

En un reactor Schlenk, a una solución del intermedio 26 (630 mg; 1.78 mmol) en THF (17.8 ml) se le añadió bis(tri-tercbutilfosfina)paladio (0) (46 mg; 0.09 mmol). La mezcla se desgasificó cuidadosamente al vacío y se cargó de nuevo con N2 (3 x). Después, se añadió el intermedio 27 (5.67 ml; 3.21 mmol) y la mezcla se desgasificó cuidadosamente al vacío y se cargó de nuevo con N2 (x3). La mezcla de reacción se agitó a 60 °C durante 3 h. La mezcla se diluyó en DCM y se filtró sobre un lecho de gel de sílice. La sílice se aclaró con DCM y el filtrado se evaporó al vacío para dar un residuo que se recogió en DCM y agua. Las fases se separaron y la fase acuosa se extrajo con DCM. Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo (900 mg, aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH irregular 30 pm; 40 g; fase móvil: del 100 % de DCM al 96 % de DCM, 4 % de MeOH, 0.4 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (720 mg, aceite de color verde) se trituró en éter dietílico/heptano. Después, el precipitado se filtró y se secó para dar 605 mg (87 % , polvo de color blanco). Una parte de esta fracción (112 mg) se liofilizó con ACN/agua (20/80) para dar 103 mg. El residuo (103 mg) se purificó por S f C aquiral (CYANO 6 pm 150 x 21.2 mm; fase móvil: 85 % de CO2 , 15 % de MeOH (0.3 % de iPrNH2)). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (36 mg, sólido de color púrpura) se liofilizó con ACN/agua (16/84) para dar 35 mg (5 % , sólido mullido de color blanco) del compuesto 27. P.F.: 162 °C (DSC).
Eje mplo B19:
Preparación del compuesto 28:

A una solución del intermedio 30 (390 mg; 0.87 mmol) en THF (4.3 ml) y EtOH (4.3 ml) se le añadió NaOH (1 M en H2 O) (1.74 ml; 1.74 mmol). Se agitó la mezcla de reacción a ta durante la noche. La mezcla se evaporó al vacío y el residuo se recogió en DCM y agua. La capa acuosa se acidificó con NH4Cl sólido. Las fases se separaron y la fase acuosa se extrajo con DCM. Las capas orgánicas combinadas se secaron sobre MgSO4, se filtraron y el disolvente se evaporó al vacío para dar 343 mg (97 % , sólido de color blanco) del compuesto 28. P.F.: 196 °C (DSC).
Preparación alternativa:
A una solución del intermedio 36 (1.19 g; 2.29 mmol) en THF (23 ml) a 0 °C se le añadió gota a gota fluoruro de tetrabutilamonio (1 M en THF) (2.52 ml; 2.52 mmol). La mezcla de reacción se calentó a ta y se agitó durante 2 h. Después, se añadió más cantidad de fluoruro de tetrabutilamonio (1 M en THF) (4.58 ml; 4.58 mmol) y la mezcla se agitó a ta durante 2 h. La mezcla se vertió sobre una solución saturada de NaHCO3 y la capa acuosa se extrajo con EtOAc (x2). Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo (sólido de color amarillo) se recogió en EtOAc y se lavó con agua (x2). La capa orgánica se secó sobre MgSO4, se filtró y se evaporó al vacío para dar 631 mg (68 % , sólido de color beige) del compuesto 28.
Preparación alternativa: véase A16 (junto con el intermedio 30).
Preparación del compuesto 47:
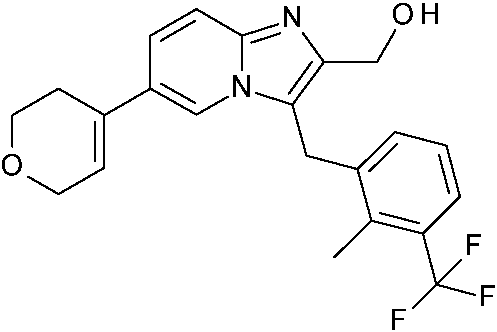
A una solución del intermedio 57 (1.5 g; 2.90 mmol) en THF (29 ml) se le añadió HCl (3 M en H2O) (1.94 ml; 5.81 mmol). La solución se agitó a ta durante 3 h, después se enfrió a 0 °C y se neutralizó lentamente con K2CO3 sólido. La mezcla se extrajo con DCM (x3) y después con EtOAc (x2). Las capas orgánicas combinadas se secaron sobre MgSO4, se filtraron y se evaporaron al vacío para dar 1.05 g (90 % , sólido de color beige) del compuesto 47.
Ejemplo B20:
Preparación del compuesto 30:

En un reactor Schlenk, a una solución del intermedio 38 (475 mg; 1.14 mmol) en THF (11.5 ml) se le añadió bis(tri-tercbutilfosfina)paladio (0) (29 mg; 0.06 mmol). La mezcla se desgasificó cuidadosamente al vacío y se cargó de nuevo con N2 (x3). Después, se añadió el intermedio 27 (3.64 ml; 2.06 mmol) y la mezcla se desgasificó cuidadosamente al vacío y se cargó de nuevo con N2 (x3). Se agitó la mezcla de reacción a ta durante la noche. La mezcla se inactivó con NH4Cl sólido y se filtró a través de un lecho de celite®. El celite® se lavó con EtOAc y el filtrado se evaporó al vacío. El residuo
(600 mg, aceite de color rojo) se combinó con un lote procedente de una reacción realizada 50 mg del intermedio 14 y el residuo resultante se purificó por cromatografía sobre gel de sílice (SiOH irregular 15-40 gm; 50 g; gradiente: del 100 % de DCM al 95 % de DCM, 5 % de MeOH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (376 mg, aceite de color verde) se purificó por cromatografía sobre gel de sílice (sílice esencial esférica; 5 gm 150 x 30.0 mm; gradiente: del 98 % de d C m, 2 % de MeOH (+10 % de NH4OH) al 86 % de Dc M, 14 % de MeOH (+10 % de NH4OH ac.)). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (66 mg) se purificó por fase inversa (X-Bridge-C18 5 gm 30*150 mm; gradiente: del 70 % (NH4 HCO3 ac. al 0.5 %), 30 % de ACN al 100 % de ACN). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (18 mg, aceite incoloro) se liofilizó con ACN/agua 23/77 para dar 17 mg (3 % , sólido mullido de color blanco) del compuesto 30. P.F.: 176 °C (DSC).
Ejemplo B21:
Preparación del compuesto 31:

A una solución del intermedio 40 (101 mg; 0.14 mmol) en DCM (1.44 ml) a 0 °C se le añadió TFA (110 gl; 1.44 mmol). La mezcla se calentó a ta y se agitó a ta durante una noche. Después, se añadió gota a gota más cantidad de TFA (110 gl; 1.44 mmol) y la mezcla se agitó a ta durante 3 h. Se añadió NaOH (1 M en H2 O) (2.88 ml; 2.88 mmol) y la mezcla se agitó a ta durante 1 h. Las capas se separaron y la capa orgánica se lavó con salmuera, se secó sobre MgSO4, se filtró y se evaporó al vacío. El residuo (220 mg, aceite de color verde) se recogió en THF (0.72 ml) y EtOH (0.72 ml) y se añadió NaOH (1 M en H2O) (0.72 ml, 0.72 mmol). La mezcla se agitó a 50 °C durante 2 h. La mezcla se evaporó al vacío y el residuo se recogió en DCM y agua. La capa acuosa se neutralizó con una solución acuosa al 10 % de NH4Cl y el producto se extrajo con DCM (x2). Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo (143 mg, polvo de color amarillo) se purificó por cromatografía sobre gel de sílice (sílice esencial esférica 5 gm; 150 x 30.0 mm; gradiente: del 92 % de DCM, 8 % de MeOH, 0.8 % de NH4OH al 76 % de d C m , 24 % de MeOH, 2.4 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (32 mg, aceite incoloro) se liofilizó con ACN/agua (20/80) para dar 31 mg (42 % , polvo de color blanco) del compuesto 31.
Preparación del compuesto 32:

A una solución del intermedio 41 (85 mg; 0.15 mmol) en DCM (1.45 ml) a 0 °C se le añadió TFA (0.111 ml; 1.45 mmol). La mezcla se calentó a ta y se agitó a ta durante una noche. Se añadió más cantidad de TFA (0.111 ml; 1.45 mmol) y la mezcla se agitó a ta durante el fin de semana. Se añadió NaOH (1 M en H2 O) (3.63 ml; 3.63 mmol) y la mezcla de reacción se agitó a ta durante 2 h. Las capas se separaron y la capa orgánica se lavó con salmuera, se secó sobre MgSO4, se filtró y se evaporó al vacío. El residuo (70 mg, aceite de color verde) se purificó por cromatografía sobre gel de sílice (sílice esencial esférica; 5 gm 150 x 30.0 mm; gradiente: del 92 % de Dc M, 8 % de MeOH, 0.8 % de NH4OH al 76 % de DCM, 24 % de MeOH, 2.4 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (10 mg, aceite incoloro) se liofilizó con ACN/agua 23/77 para dar 8 mg (11 % , sólido mullido de color blanco) del compuesto 32.
Ejemplo B22:
Preparación del compuesto 33:

A una solución del intermedio 39 (305 mg; 0.76 mmol) en MeOH (7.5 ml) se le añadió trifluoroacetato de 2,2-dióxido de 2-tia-6-aza-espiro[3.3]heptano (217 mg; 0.83 mmol). La mezcla se agitó a ta durante 1 h. Después, se añadió triacetoxiborohidruro sódico (481 mg; 2.27 mmol) y la mezcla se agitó a ta durante una noche. La mezcla se recogió en DCM y se añadió una solución saturada de NaHCO3. Las fases se separaron y la fase acuosa se extrajo con DCM. Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo (420 mg, sólido de color verde pálido) se trituró en DCM/éter dietílico (1:9). El precipitado se filtró y se secó al vacío para dar 302 mg (75 % , sólido de color blanco) del compuesto 33. P.F.: 196 °C (DSC).
Preparación del compuesto 34:

En un vial para microondas, a una solución del intermedio 39 (566 mg; 1.40 mmol) en MeOH (14 ml) se le añadió 2-aminoetanol (168 pl; 2.81 mmol). La mezcla se agitó a ta durante 1 h 30. Después, se añadió NaBH4 (27 mg; 0.70 mmol) y la mezcla de reacción se agitó a ta durante una noche. La mezcla se evaporó al vacío. Después, el residuo se recogió en DCM y una solución acuosa 1 N de HCl. Las capas se separaron y la capa acuosa se basificó con una solución saturada de NaHCO3 y se extrajo con DCM (x2). Se secaron las fases orgánicas combinadas sobre MgSO4 , se filtraron y se evaporaron a vacío. El residuo (532 mg, sólido de color beige) se purificó por cromatografía sobre gel de sílice (sílice esencial esférica; 5 pm 150 x 30.0 mm; gradiente: del 96 % de DCM, 4 % de MeOH, 0.4 % de NH4OH al 83 % de DCM, 17 % de MeOH, 1.7 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (436 mg, polvo de color beige) se trituró en éter dietílico/DCM (9:1) y el disolvente se evaporó al vacío. El sólido se secó al vacío (50 °C, 24 h) para dar 400 mg (64 % , polvo de color blanco) del compuesto 34. P.F.: 147 °C (DSC).
Preparación del compuesto 35:

El compuesto 35 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del compuesto 33, usando el intermedio 39 y 3-amino-1 -propanol como material de partida. El residuo (76 mg) se purificó por cromatografía sobre gel de sílice (SiOH irregular 30 pm; 4 g; gradiente: del 100 % de DCM al 96 % de DCM, 4 % de MeOH, 0.4 % de
NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (56 mg, aceite de color verde) se purificó por cromatografía sobre gel de sílice (sílice esencial esférica; 5 pm 150 x 30.0 mm; gradiente: del 97 % de DCM, 3 % de MeOH, 0.3 % de NH4OH al 85 % de DCM, 15 % de MeOH, 1.5 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (28 mg) se purificó por fase inversa (X-Bridge-C18 pm 30*15mm; gradiente: del 80 % (NH4 HCO3 ac. al 0.5 %), 20 % de ACN al 0 % (NH4HCO3 ac. al 0.5 %), 100 % de ACN). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (17 mg, sólido de color blanco) se liofilizó con ACN/agua, 20/80 para dar 16 mg (19 % , polvo mullido de color blanco) del compuesto 35. P.F.: 133 °C (DSC).
Preparación del compuesto 40:

A una solución del intermedio 59 (500 mg; 1.25 mmol) en MeOH (12 ml) se le añadió trifluoroacetato de 2,2-dióxido de 2-tia-6-aza-espiro[3.3]heptano (359 mg; 1.37 mmol) y triacetoxiborohidruro sódico (794 mg; 3.75 mmol). La mezcla de reacción se agitó a ta durante 3 h, y después se evaporó al vacío. El residuo se recogió en DCM y se añadió una solución acuosa saturada de NaHCO3. Las fases se separaron y la fase acuosa se extrajo con DCM. Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo (701 mg; espuma de color pardo pálido) se purificó por cromatografía sobre gel de sílice (SiOH irregular 15-40 pm; 30 g; gradiente: del 100 % de DCM al 95 % de DCM, 5 % de MeOH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (413 mg, sólido de color blanquecino) se purificó por cromatografía sobre gel de sílice (SiOH irregular 15-40 pm; 30 g; gradiente: del 100 % de heptano al 50 % de heptano, 50 % de (iPrOH/NH4OH: 95/5)). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (318 mg, sólido de color blanquecino) se purificó por cromatografía sobre gel de sílice (SiOH irregular 15-40 pm; 24 g; gradiente: del 100 % de DCM al 95 % de Dc M, 5 % de (iPrOH/NH4OH: 95/5)). Las fracciones puras se recogieron y el disolvente se evaporó para dar 287 mg (39 % , sólido de color blanco) del compuesto 40. P.F.: 184 °C (DSC).
Ejemplo B23:
Preparación del compuesto 38:

A una solución del intermedio 50 (227 mg; 0.34 mmol) en THF (3 ml) se le añadió HCl (6 M en H2O) (565 pl; 3.39 mmol). La solución se calentó a 60 °C durante 18 h y después se añadió más cantidad de HCl (6 M en H2O) (395 pl; 2.37 mmol) y la solución se calentó a 60 °C durante 18 h. La solución se neutralizó con una solución acuosa 1 M de NaOH. La capa acuosa se extrajo con DCM (x3) y las capas orgánicas combinadas se secaron sobre MgSO4, se filtraron y se evaporaron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 pm; 10 g; gradiente: del 100 % de DCM al 90 % de DCM, 10 % de MeOH, 0.1 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (48 mg, sólido de color pardo pálido) se purificó por fase inversa (X-Bridge-C185 pm 30*150 mm; gradiente: del 75 % H2O (0.5 % de HCOONH4 pH 4.5), 25 % de ACN al 0 % de H2O (0.5 % de HCOONH4 pH4.5), 100 % de ACN). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (19 mg, sólido de color blanquecino) se purificó por
cromatografía sobre gel de sílice (sílice esencial esférica 5 gm 150x30.0 mm; gradiente: del 50 % de heptano, 3 % de MeOH (+10 % de NH4OH), 47 % de EtOAc al 0 % de heptano, 25 % de MeOH (+10 % de NH4OH), 75 % de EtOAc). Las fracciones puras se recogieron y el disolvente se evaporó para dar 10 mg (6 % , sólido de color blanco) del compuesto 38.
Preparación del compuesto 41:
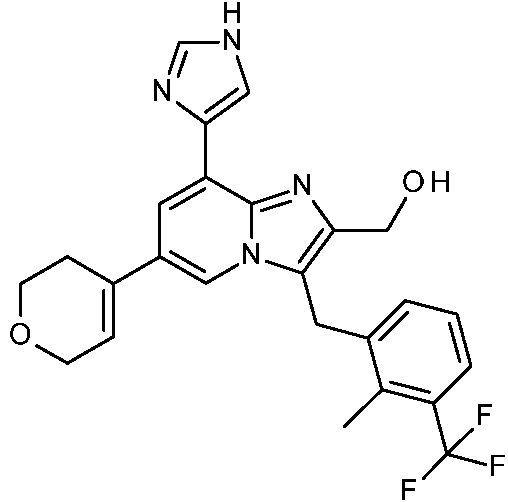
A una mezcla del intermedio 63 (68 mg; 98.6 gmol) en THF (980 gl) se le añadió HCl (6 M en H2O) (82 gl; 0.49 mmol). La mezcla se calentó a 60 °C durante 18 h. Después del enfriamiento a ta, la mezcla de reacción se enfrió a 0 °C, se neutralizó lentamente con K2 CO3 sólido y se transfirió a un embudo de decantación. Se añadieron EtOAc y agua; la capa orgánica se separó y la capa acuosa se extrajo con EtOAc (x2). Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo (43 mg, residuo de color amarillo) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 gm; 4 g; gradiente: del 100 % de DCM al 90 % de DCM, 10 % de MeOH/, 0.1 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (película de color amarillo pálido) se trituró en éter dietílico. El precipitado se filtró y se secó al vacío. El residuo resultante (17 mg, sólido de color pardo pálido) se purificó por fase inversa (X-Bridge-C185 gm; 30*150 mm; gradiente: del 75 % de H2O (NH4HCO3 al 0.5 %), 25 % de ACN al 35 % de H2O (NH4 HCO3 al 0.5 %), 65 % de ACN). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (película incolora) se liofilizó con ACN/agua (20/80) para dar 5 mg (24 % , sólido mullido de color blanco) del compuesto 41.
Preparación del compuesto 42:

A una mezcla del intermedio 66 y el intermedio 67 (739 mg; 1.27 mmol; 33 % de pureza) en THF (3 ml) se le añadió HCl (6 M en H2 O) (0.837 ml; 5.02 mmol). La mezcla se calentó a 50 °C durante 1 h. Se añadió más cantidad de HCl (6 M en H2O) (0.837 ml; 5.02 mmol) y la solución se calentó a 50 °C durante 1 h. Se añadió más cantidad de HCl (6 M en H2O) (1.67 ml; 10.0 mmol) y la solución se calentó a 60 °C durante 96 h. Después, el producto en bruto se enfrió a 0 °C, se neutralizó lentamente con K2 CO3 sólido y se extrajo con DCM (x3). Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo (551 mg, aceite de color pardo) se purificó por cromatografía sobre gel de sílice (SiOH Irregular 15-40 gm; 10 g; gradiente: del 100 % de DCM al 90 % de DCM, 10 % de MeOH, 0.1 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (551 mg, sólido de color blanquecino) se trituró en éter dietílico. El sólido se filtró y se secó al vacío a 50 °C durante 18 h. El residuo (110 mg, sólido de color blanquecino) se solubilizó en una mezcla de acetona y MeOH, se evaporó al vacío y se secó al vacío a 50 °C durante 18 h para dar 65 mg (33 % , sólido de color blanquecino) del compuesto 42. P.F.: 238 °C (DSC).
Preparación del compuesto 43:

El Compuesto 43 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis del compuesto 41, usando el intermedio 71 como material de partida. La mezcla de reacción se agitó a 60 °C durante 1 h. El residuo (241 mg) se purificó por cromatografía sobre gel de sílice (SiOH irregular 15-40 gm; 10 g; gradiente: del 100 % de DCM al 95 % de DCM, 5 % de MeOH, 0.5 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo se trituró con éter dietílico. El precipitado se filtró y se secó al vacío para dar 96 mg (37 % , sólido de color blanquecino) del compuesto 43. P.F.: 247 °C (DSC).
Ejemplo B24:
Preparación del compuesto 39:

A una solución del intermedio 51 (144 mg; 0.30 mmol) en THF (3 ml) se le añadió gota a gota TBAF (1 M en THF) (0.325 ml; 0.33 mmol). La mezcla de reacción se agitó a ta durante 1 h y se vertió sobre una solución saturada de NaHCO3. La capa acuosa se extrajo con EtOAc (x3) y las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre MgSO4 y se filtraron al vacío. El residuo (sólido) se trituró en ACN. El sólido se filtró y se secó para dar 60 mg (54 % sólido de color blanquecino) del compuesto 39. P.F.: 257 °C (DSC).
Ejemplo B25:
Preparación del compuesto 44:

A una solución del intermedio 76 (176 mg; 0.26 mmol) en THF (5 ml) se le añadió HCl (3 M en H2 O) (0.88 ml; 2.6 mmol). La mezcla de reacción se agitó a ta durante 18 h y después se diluyó con EtOAc y la mezcla se basificó lentamente con una solución saturada de NaHCO3 hasta pH = 8. La capa acuosa se extrajo con EtOAc (x3). Las capas orgánicas combinadas se secaron con MgSO4, se filtraron y se evaporaron. El residuo (200 mg, sólido) se purificó por cromatografía sobre gel de sílice (SiOH regular 30 gm; 25 g; carga seca sobre Celite®; gradiente: del 100 % de DCM al 95 % de DCM, 5 % de MeOH). Las fracciones puras se recogieron y el disolvente se evaporó para dar 73 mg (66 % , sólido de color blanco) del compuesto 44. P.F.: 199 °C (DSC).
Ejemplo B26:
Preparación del compuesto 45:

En un matraz de fondo redondo, el compuesto intermedio 80 (68 mg; 0.10 mmol) se diluyó en THF (3.7 ml). Después, se añadió HCl (1 M en H2 O) (0.97 ml; 0.97 mmol) y la mezcla de reacción se agitó durante una noche a ta. La mezcla de reacción se vertió sobre agua enfriada con hielo, se neutralizó con K2CO3 y la capa acuosa se extrajo con EtOAc. Las capas orgánicas se combinaron, se secaron sobre MgSO4, se filtraron y el disolvente se evaporó a sequedad. El residuo (40 mg) se purificó mediante cromatografía sobre gel de sílice (SiOH, 15 pm; 24 g; gradiente: del 98 % de DCM 2 % de MeOH, NH4OH al 0.1 % a 90 % de DCM, 10 % de MeOH, NH4OH al 0.1 %). Las fracciones puras se recogieron y el disolvente se evaporó para dar 18 mg (44 %) del compuesto 45.
Preparación del compuesto 46:

El compuesto 46 se preparó de acuerdo con un procedimiento análogo al descrito para la síntesis de compuesto 45, utilizando el compuesto intermedio 81 como material de partida (5 mg, 28 %). P.F.: 223 °C (K).
C: Conversión
Ejemplo C 1 :
Preparación del compuesto 29:

En un tubo sellado, a una suspensión de tetrafluoroborato de (dietilamino)difluorosulfonio (34 mg; 0.15 mmol) en DCM (0.92 ml) a 0 °C se le añadieron el compuesto 28 (40 mg; 0.10 mmol) y trifluorhidrato de trietilamina (24 pl; 0.15 mmol). La mezcla de reacción se calentó a ta y se agitó durante 2 h. La mezcla se combinó con una reacción realizada en 20 mg del compuesto 28. La mezcla se neutralizó con una solución acuosa al 10 % de K2 CO3. Las capas se separaron y la capa orgánica se secó sobre MgSO4, se filtró y se evaporó al vacío. El residuo (61 mg) se purificó por cromatografía sobre gel de sílice (SiOH irregular 30 pm; 4 g; fase móvil: del 100 % de DCM al 96 % de DCM, 4 % de MeOH, 0.4 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (21 mg, goma de color blanco) se liofilizó con ACN/agua, 20/80, para dar 14 mg (23 % , sólido de color blanco) del compuesto 29. P.F.: 177 °C (DSC).
Preparación del compuesto 36:

A una solución del compuesto 34 (80 mg; 0.18 mmol) en MeOH (1.8 ml) se le añadieron formaldehído (80 gl; 1.07 mmol) y ácido acético (61 gl; 1.07 mmol). La mezcla de reacción se agitó a ta durante 30 min. Después, se añadió triacetoxiborohidruro sódico (227 mg; 1.07 mmol). Se agitó la mezcla de reacción a ta durante la noche. La mezcla se evaporó al vacío, después el residuo se recogió en DCM y se añadió una solución saturada de NaHCO3. Las fases se separaron y la fase acuosa se extrajo con DCM. Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo (91 mg, aceite de color verde) se purificó por cromatografía sobre gel de sílice (sílice esencial irregular 150 g; gradiente: del 95 % de DCM, 5 % de MeOH, 0.5 % de NH4OH al 82 % de DCM, 18 % de MeOH, 1.8 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (51 mg, aceite incoloro) se liofilizó con ACN/agua, 23/77 para dar 41 mg (sólido de color blanco) que se convirtió en un aceite. Esta fracción se solubilizó en EtOAc (5 ml), se transfirió a otro recipiente, se evaporó al vacío y se secó (50 °C) para dar 35 mg (42 % , aceite incoloro) del compuesto 36.
Preparación del compuesto 37:
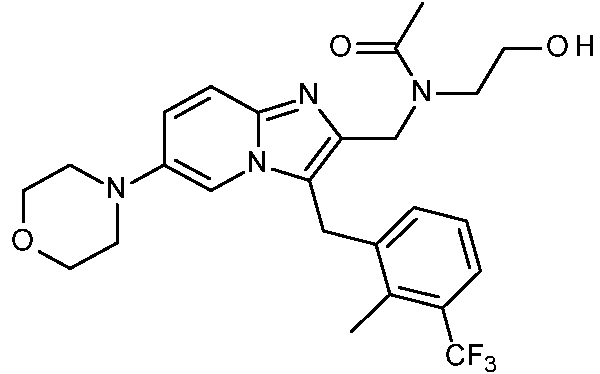
A una solución del compuesto 34(80 mg; 0.18 mmol) en DCM (1.5 ml) a 0 °C se le añadió gota a gota una solución de anhídrido acético (17 gl; 0.18 mmol) en DCM (0.3 ml). La mezcla de reacción se calentó a ta y se agitó durante 1 h 30. Después, se añadió una solución saturada de NaHCO3. Las fases se separaron y la fase acuosa se extrajo con DCM. Se secaron las fases orgánicas combinadas sobre MgSO4, se filtraron y se evaporaron a vacío. El residuo (82 mg, sólido de color azul) se purificó por cromatografía sobre gel de sílice (sílice esencial irregular 150 g; gradiente: del 95 % de DCM, 5 % de MeOH, 0.5 % de NH4OH al 82 % de DCM, 18 % de MeOH, 1.8 % de NH4OH). Se recogieron las fracciones puras y se evaporó el disolvente. El residuo (23 mg, aceite incoloro) se liofilizó con ACN/agua 20/80 para dar 21 mg (24 % , sólido mullido de color blanco) del compuesto 37. P.F.: 172 °C (d Sc ).
Ejemplo C2:
Preparación del compuesto 9:

Se añadió hidróxido de litio monohidrato (35 mg; 0.84 mmol) a una mezcla del compuesto 2 (75 mg; 0.17 mmol) en H2O (0.2 ml) y MeOH (2 ml) a temperatura ambiente y la solución se agitó a ta durante una noche. Se añadieron H2 O y EtOAc.
La fase orgánica se secó con MgSO4 y se evaporó a sequedad. El residuo (117 mg) se recogió con éter dietílico. Después, se filtró un precipitado y se secó para dar 30 mg (44 %) del compuesto 9. P.F.: 195 °C (K).
Ejemplo C3:
Preparación de compuesto 49:

El compuesto 49 se preparó de acuerdo con un procedimiento análogo como se describe para la síntesis del compuesto 18, usando el compuesto 53 como material de partida (cristalizado en DIPE; 19 mg, 9 %). P.F.: 224 °C (DSC).
Parte analítica
LCMS (cromatografía de líquidos/espectrometría de masa)
La medición por cromatografía líquida de alto rendimiento (HPLC) se realizó utilizando una bomba de CL, un haz de diodos (DAD) o un detector de UV y una columna, según se especifique en los métodos respectivos. Cuando fue necesario, se incluyeron detectores adicionales (véase la tabla de los métodos a continuación).
El flujo procedente de la columna se introdujo en un espectrómetro de masas (MS), que estaba configurado con una fuente de iones a presión atmosférica. Es parte del conocimiento de un experto en la materia ajustar los parámetros ajustables (por ejemplo, intervalo de barrido, tiempo de permanencia, etc.) con el fin de obtener iones que permitan la identificación del peso molecular (PM) monoisotópico nominal del compuesto. La adquisición de datos se realizó con el software adecuado.
Los compuestos se describen según sus tiempos de retención (Tr) experimentales e iones. Si no se especifica de otro modo en la tabla de datos, el ion molecular descrito corresponde a [M+H]+ (molécula protonada) y/o [M-H]- (molécula desprotonada). En caso de que el compuesto no se pueda ionizar directamente, se especifica el tipo de aducto (es decir, [M+NH4]+, [M+HCOO]-, etc...). Para moléculas con patrones isotópicos múltiples (Br, Cl, ...), el valor indicado es el obtenido para la masa isotópica más baja. Todos los resultados se obtuvieron con incertidumbres experimentales que están habitualmente asociadas al método usado.
En lo sucesivo en el presente documento, "SQD" significa detector de cuadrupolo único, "TA" temperatura ambiente, "BEH" híbrido en puente de etilsiloxano/sílice, "HSS" sílice de alta resistencia, "DAD" detector por red de diodos.
Tabla: Códigos del método de CLEM (flujo expresado en ml/min; temperatura (T) de la columna en °C; tiempo de ejecución en minutos).


Puntos de fusión
Para varios compuestos, se determinaron los puntos de fusión (PF) con un DSC1 (Mettler-Toledo). Los puntos de fusión se midieron con un gradiente de temperatura de 10 °C/minuto. La temperatura máxima fue de 350 ° C. Los valores son valores máximos."
Para una serie de compuestos, se obtuvieron los puntos de fusión con una placa caliente de Kofler, que consistía en una placa calentada con un gradiente de temperatura lineal, un puntero movible y una escala de temperatura en grados Celsius. Para una diversidad de compuestos, los puntos de fusión se obtuvieron con o un MP50 (Mettler Toledo) con el cual los puntos de fusión se midieron con un gradiente de temperatura de 10 °C/minuto. La temperatura de partida fue 50 °C y la temperatura máxima fue 300 °C.
Los experimentos de RMN se llevaron a cabo utilizando un Bruker Avance 500 III utilizando un bloqueo interno de deuterio y equipado con cabezal de sonda de triple resonancia inversa (1H, 13C,15N TXI). Los desplazamientos químicos (ó) se indican en partes por millón (ppm).
Tabla: N° de Co. significa número de compuesto; tiempo de retención(tr) en min; PF significa punto de fusión (°C); dec significa descomposición; n.d. significa no determinado.












Compuesto 1:1H RMN (500 MHz, DMSO-afe) 5 ppm 13.71 (s a, 1H) 8.04 (s, 1H) 7.71 (s a, 1H) 7.52 - 7.64 (m, 2H) 7.09 -7.31 (m, 2H) 6.86 (d a, J=7.6 Hz, 1H) 4.49 (s, 2H) 4.30 (s a, 2H) 3.82 (t a, J=5.2 Hz, 2H) 2.48 (s a, 3H) 2.44 (s a, 2H) 2.31 (s, 3H)
Compuesto 21:1H RMN (500 MHz, DMSO-afe) 5 ppm 8.59 (d, J=2.8 Hz, 1 H) 7.81 (d, J=2.5 Hz, 1 H) 7.56 (d, J=7.9 Hz, 1 H) 7.23 (t, J=7.7 Hz, 1 H) 6.77 (d, J=7.9 Hz, 1H) 4.41 (s, 2H) 4.20 (s, 4H) 3.63 - 3.80 (m, 4H) 3.54 (s, 2H) 3.28 (s, 4H) 2.95 -3.02 (m, 4H) 2.47 (s, 3H)
Compuesto 46:1H RMN (500 MHz, DMSO-afe) 5 ppm 7.53 (d, J=7.9 Hz, 1H) 7.14 - 7.25 (m, 2H) 6.81 (d, J=7.9 Hz, 1H) 6.42 (s, 1 H) 6.05 (s a, 1H) 5.60 (s, 2H) 4.95 (t, J=5.5 Hz, 1H) 4.52 (d, J=5.4 Hz, 2H) 4.41 (s, 2H) 4.17 (d a, J=2.5 Hz, 2H) 3.74 (t, J=5.4 Hz, 2H) 2.24 (s a, 2H)
Compuesto 39:1H RMN (400 MHz, DMSO-afe) 5 ppm 7.44 (d, J=9.6 Hz, 1H) 7.35 (s, 1H) 7.26 (dd, J=9.6, 2.0 Hz, 1H) 7.14 (d, J=1.5 Hz, 1 H) 6.83 - 6.95 (m, 1H) 6.74 - 6.83 (m, 1H) 5.88 (d, J=8.1 Hz, 1H) 4.81 (t, J=5.6 Hz, 1H) 4.39 (d, J=5.6 Hz, 2H) 3.60 - 3.77 (m, 4H) 2.85 - 2.97 (m, 4H) 2.41 (s, 3H)
Farmacología
Ensayos de Unión a Enzimas (KINOMEscan®)
Las afinidades de unión a enzimas cinasa de compuestos descritos en el presente documento se determinaron utilizando la tecnología KINOMEscan realizada por DiscoveRx Corporation, San Diego, California, Estados Unidos (www.kinomescan.com). La Tabla reseña los valores Kd (nM) obtenidos, siendo Kd la constante de unión al inhibidor:


Ensayos celulares:
La actividad celular de los inhibidores de PI3K|3 se determinó cuantificando la fosforilación de Akt en células PC-3. Los Akt fosforilados en Ser473 y Thr308 se midieron utilizando un ensayo de inmunosorbente ligado a enzimas (ELISA; Meso Scale Discovery (MSD), Gaithersburg, MD) y anticuerpos primarios específicos a partir de MSD.
El día 1, las células PC3 (ATCC # CRL-14351) se sembraron en placas PerkinElmer MW96 a 25.000 células por pocillo, en 75 |jl de medio de cultivo completo (DMEM alta glucosa, AQmedia™, D0819, Sigma-Aldrich) que contenía un 10 % de FCS inactivado por calor e incubado a 37 °C, el 5 % de CO2 durante 24 horas. El día 2, se añadió compuesto o DMSO (0.3 % ) y las células se incubaron adicionalmente durante 60 minutos a 37 °C, 5 % de CO2 en un volumen total de 100 |ul de medio.
El ensayo de fosfoproteína se ejecutó según las instrucciones del proveedor en el Kit de Lisado de Células Completas de Ensayo de Fosfo-Akt (Ser473) (MSD n.° K15100D-3) y el Kit de Lisado de Células Completas de Ensayo de Fosfo-Akt (Thr308) (MSD n.° K151DYD-3) usando el tampón de lisis, bloqueo y lavado proporcionado.
En síntesis, al final del periodo de tratamiento de las células, los medios se separaron mediante aspiración y las células adherentes se lisaron en 50 |ul de tampón de lisis enfriado con hielo. Placas MSD se suministran pre-revestidas con
anticuerpos de captura para Phospho-Akt (Ser473 y Thr308). Después del bloqueo, se añadieron los lisados de placas de cultivo tisular y las placas se lavaron. Después, se añadió una disolución que contenía el anticuerpo de detección (anti-Akt total conjugado con un marcador Sulfo-tag de compuesto-MSD electroquimioluminiscente). Las señales se detectaron utilizando un aparato MSD SECTOR Imager 6000 y son proporcionales a los títulos de fosfo-Akt.
Los datos se procesaron. El porcentaje de inhibición se representó frente a la concentración log de compuestos de ensayo y la curva sigmoidal log de la concentración-efecto de mejor ajuste se calculó mediante análisis de regresión no lineal. A partir de estas curvas de concentración-respuesta se calcularon los valores de CI50. Para el ajuste de las curvas se utilizaron cinco concentraciones.
La Tabla B reseña los valores de CI50 obtenidos (nM):


Ejemplos de composición profética
“Ingrediente activo” (i.a.), tal como se utiliza a lo largo de estos ejemplos, se refiere a un compuesto de Fórmula (I), incluido cualquier tautómero o forma estereoisomérica del mismo, o un N-óxido, una sal por adición farmacéuticamente aceptable o un solvato del mismo; en particular, a uno cualquiera de los compuestos ejemplificados.
Ejemplos típicos de recetas para la formulación de la invención son como sigue:
1. Comprimidos
Principio activo 5-50 mg
Fosfato de dicalcio 20 mg
Lactosa 30 mg
Talco 10 mg
Estearato de magnesio 5 mg
Almidón de papa hasta 200 mg
2. Suspensión
Se prepara una suspensión acuosa para su administración oral de modo que cada mililitro contenga 1-5 mg del principio activo, 50 mg de carboximetilcelulosa de sodio,
1 mg de benzoato de sodio, 500 mg de sorbitol y agua hasta 1 ml.
3. Inyectable
Se prepara una composición parenteral agitando un 1.5 % (peso/volumen) del principio activo en una solución de NaCl al 0.9 % o en un 10 % en volumen de propilenglicol en agua.
4. Pomada
Principio activo 5-1000 mg
Alcohol estearílico 3 g
Lanolina 5 g
Petróleo blanco 15 g
Agua hasta 100 g
En este Ejemplo, el principio activo se puede reemplazar por la misma cantidad de cualquiera de los compuestos de acuerdo con la presente invención, en particular por la misma cantidad de cualquiera de los compuestos empleados como ejemplo.
Claims (15)
1. Un compuesto de Fórmula (I)
un tautómero o una forma estereoisomérica del mismo, en donde
X1 representa CH o N;
X2 representa CR1 o N;
siempre que como máximo uno de X1 y X2 represente N;
R1 representa hidrógeno, -C(=O)OH, -C(=O)NH2 , -NH2 , -CH2OH,

Y representa -CH2 - o -NH-;
R2 representa

R3 representa alquilo C 1 - 4 ; -C(=O)-O-alquilo C 1 - 4 ; -C(=O)-Het1; -CH(OH)-CH2 -Rq; alquilo C 1 -4 sustituido en el mismo átomo de carbono con un -OH y con un Het1; o alquilo C 1 -4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -N H 2 , -O-(C=O)-alquilo C 1 - 4 , -(C=O)-O-alquilo C 1 - 4 , -NH-(C=O)-alquilo C 1 - 4 , -NH-(SO 2 )-alquilo C 1 - 4 , -N(CH3)-alquil C 1 - 4 -SO2 -CH3 , -NH-alquil C 1 - 4 -SO2 -CH3 , -N(CH3)-alquil C 1 - 4 -OH,
-N(C=O-alquil C1-4)-alquil C 1 - 4 -OH, -(C=O)-NH-alquil C 1 - 4 -OH,
-O-(C=O)-CH(NH2)-alquilo C 1 - 4 , -O-(C=O)-CH(NH2)-alquil C1-4-Ar,

-NH-alquil C 1 - 4 -OH, Het1, -O-C(=O)-alquil C1-4-Het1, -C(=O)-Het1, y -NH-C(=O)-Het1;
Rq representa Het1, halo, -OH, -N H 2 , -O-(C=O)-alquilo C 1 - 4 , -NH-(C=O)-alquilo C 1 - 4 ,
-NH-(SO 2)-alquilo C 1 - 4 , -N(CH3)-alquil C 1 - 4 -SO 2 -CH3 , -NH-alquil C 1 - 4 -SO2 -CH3 ,
-N(CH3)-alquil C 1 - 4 -OH, -O-(C=O)-CH(NH2)-alquilo C 1 - 4 ,
-O-(C=O)-CH(NH2)-alquil C1-4-Ar,

o -NH-alquil C 1 - 4 -OH;
Ar representa fenilo opcionalmente sustituido con un hidroxi;
R4a representa hidrógeno, alquilo C 1 - 4 , Heta o alquilo C 1 -4 sustituido con uno o más sustituyentes, cada uno independientemente seleccionado del grupo que consiste en -OH, -NR5R6 y Heta;
R4b representa hidrógeno, halo, alquilo C 1 -4 o alquilo C 1 -4 sustituido con uno o más sustituyentes halo;
o R4a y R4b se toman juntos para formar, junto con el anillo de fenilo al que están unidos, una estructura de Fórmula (a-1), (a-2), (a-3), (a-4) o (a-5):

X representa -NH-, -O-, -N(alquil C 1 - 3)-, o -N(hidroxialquil C 1 - 3)-ambos sustituyentes R7 son los mismos y se seleccionan del grupo que consiste en hidrógeno, flúor y metilo; o ambos sustituyentes R7 se toman juntos para formar, junto con el átomo de carbono común al que están unidos, un ciclopropilo, ciclobutilo u oxetanilo;
ambos sustituyentes R8 son los mismos y se seleccionan del grupo que consiste en hidrógeno y metilo; o ambos sustituyentes R8 se toman juntos para formar, junto con el átomo de carbono común al que están unidos, un ciclopropilo, ciclobutilo u oxetanilo;
R5 representa hidrógeno, alquilo C 1 -6 o alquilo C 1 -6 sustituido con un -OH;
R6 representa hidrógeno, alquilo C 1 -6 o alquilo C 1 -6 sustituido con un -OH;
Het1 representa un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo, cada uno independientemente seleccionado de O, S, S(=O)p y N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes, cada uno independientemente seleccionado del grupo que consiste en halo, -NH2 , alquilo C 1 - 4 ,
-S(=O)2 -alquilo Ci-6, -alquil Ci-4-S(=O)2-alquilo Ci-6, hidroxilo, alquiloxi C 1 - 4 , flúor, ciano y alquilo C 1 -4 sustituido con un hidroxi; o dos sustituyentes en el mismo átomo de carbono de dicho heterociclilo saturado de 4, 5 o 6 miembros se toman juntos para formar junto con el átomo de carbono común al que están unidos el Anillo A;
Anillo A representa ciclobutilo, ciclopentilo, ciclohexilo o un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo, cada uno independientemente seleccionado de O, S, S(=O)p y N; dicho ciclobutilo, ciclopentilo, ciclohexilo o heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes alquilo C 1 - 4 , con un sustituyente alquilo C 1 -4 y un sustituyente hidroxi, o con un sustituyente hidroxi;
cada Heta representa independientemente un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de O, S, S(=O)p y N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes cada uno seleccionado independientemente del grupo que consiste en alquilo C 1 - 4 , -S(=O)2 -alquilo C 1 - 6 , hidroxi, -alquil C1-4-S(=O)2-alquilo C 1 - 6 , y alquilo C 1 -4 sustituido con un hidroxi; o dos sustituyentes en el mismo átomo de carbono de dicho heterociclilo saturado de 4, 5 o 6 miembros se toman juntos para formar junto con el átomo de carbono común al que están unidos el Anillo B;
el Anillo B representa ciclobutilo, ciclopentilo, ciclohexilo, o un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de O, S, S(=O)p y N; dicho ciclobutilo, ciclopentilo, ciclohexilo, o heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes alquilo C 1 - 4 , con un alquilo C 1 -4 y un sustituyente hidroxi, o con un sustituyente hidroxi;
p representa 1 o 2;
o un N-óxido, una sal de adición farmacéuticamente aceptable o un solvato de los mismos.
2. El compuesto de acuerdo con la reivindicación 1, en donde
R3 representa alquilo C 1 - 4 ; -C(=O)-Het1; alquilo C 1 -4 sustituido en el mismo átomo de carbono con un -O H y con un Het1; o alquilo C 1 -4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -N H 2 , -O-(C=O)-alquilo C 1 - 4 , -(C=O)-O-alquilo C 1 - 4 , -NH-(C=O)-alquilo C 1 - 4 , -NH-(SO 2)-alquilo C 1 - 4 ,
-N(CH3)-alquil C 1 - 4 -SO 2 -CH3 , -NH-alquil C 1 - 4 -SO2 -CH3 , -N(CH3)-alquil C 1 - 4 -OH,
-N(C=O-alquil C1-4)-alquil C 1 - 4 -OH, -(C=O)-NH-alquil C 1 - 4 -OH,
-O-(C=O)-CH(NH2)-alquilo C 1 - 4 ,

-NH-alquil C 1 - 4 -OH, Het1, -O-C(=O)-alquil C1-4-Het1, -C(=O)-Het1, y -NH-C(=O)-Het1;
cada Heta representa independientemente un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de O, S, S(=O)p y N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes cada uno seleccionado independientemente del grupo que consiste en alquilo C 1 - 4 , -S(=O)2 -alquilo C 1 - 6 , hidroxi, -alquil C1-4-S(=O)2-alquilo C 1 - 6 , y alquilo C 1 -4 sustituido con un hidroxilo.
3. El compuesto de acuerdo con la reivindicación 1, en donde
R1 representa hidrógeno, -C(=O)OH, -C(=O)NH2 , -NH2 , -CH2OH,

o
R3 representa alquilo C 1 - 4 ; o alquilo C 1 -4 sustituido con un sustituyente seleccionado del grupo que consiste en halo, -OH, -O-(C=O)-alquilo C 1 - 4 , -(C=O)-O-alquilo C 1 - 4 ,
-N(CH3)-alquil C 1 - 4 -OH, -N(C=O-alquil C1-4)-alquil C 1 - 4 -OH, -NH-alquil C 1 - 4 -OH, Het1, y -C(=O)-Het1;
R4a representa alquilo C 1 - 4 , o Heta;
R4b representa halo o alquilo C 1 -4 sustituido con uno o más sustituyentes halo;
Het1 representa un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de S(=O)p y N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes cada uno seleccionado independientemente del grupo que consiste en alquilo C1-4, y alquilo C 1 -4 sustituido con un hidroxi; o dos sustituyentes en el mismo átomo de carbono de dicho heterociclilo saturado de 4, 5 o 6 miembros se toman juntos para formar junto con el átomo de carbono común al que están unidos el Anillo A;
Anillo A representa ciclobutilo, ciclopentilo, ciclohexilo o un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un heteroátomo seleccionado cada uno independientemente de S(=O)p y N;
cada Heta representa independientemente un heterociclilo saturado de 4, 5 o 6 miembros que contiene al menos un átomo de N; dicho heterociclilo saturado de 4, 5 o 6 miembros está opcionalmente sustituido con uno o dos sustituyentes alquilo C1-4
p representa 2.
4. El compuesto de acuerdo con la reivindicación 1, en donde
R1 representa

5. El compuesto de acuerdo con la reivindicación 1, en donde
X2 representa CH.
6. El compuesto de acuerdo con la reivindicación 1, en donde
X1 representa CH, y X2 representa CR1.
7. El compuesto de acuerdo con la reivindicación 1, en donde
X1 representa CH, y X2 representa N.
8. El compuesto de acuerdo con la reivindicación 1, en donde
X1 representa N, y X2 representa CR1.
9. El compuesto de acuerdo con la reivindicación 1, en donde
R2 representa

10. El compuesto de acuerdo con la reivindicación 1, en donde Y representa -CH2 -.
11. Una composición farmacéutica que comprende un portador farmacéuticamente aceptable y, como ingrediente activo, una cantidad terapéuticamente efectiva de un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 10.
12. Un compuesto como se ha definido en una cualquiera de las reivindicaciones 1 a 10 para su uso como medicamento.
13. Un compuesto como se define en una cualquiera de las reivindicaciones 1 a 10 para su uso en el tratamiento o prevención de una enfermedad o afección seleccionada de cáncer, trastornos autoinmunes, enfermedades cardiovasculares, enfermedades inflamatorias, enfermedades neurodegenerativas, alergia, pancreatitis, asma, fallo multiorgánico, enfermedades renales, agregación de plaquetas, motilidad espermática, rechazo a trasplante, rechazo de injerto y lesiones pulmonares.
14. El compuesto para su uso de acuerdo con la reivindicación 13, en donde la enfermedad o afección es cáncer.
15. El compuesto para su uso de acuerdo con la reivindicación 14, en donde la enfermedad o afección es cáncer de próstata.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP16174715 | 2016-06-16 | ||
| PCT/EP2017/064671 WO2017216292A1 (en) | 2016-06-16 | 2017-06-15 | Bicyclic pyridine, pyrazine, and pyrimidine derivatives as pi3k beta inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2871140T3 true ES2871140T3 (es) | 2021-10-28 |
Family
ID=56132835
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17732059T Active ES2871140T3 (es) | 2016-06-16 | 2017-06-15 | Derivados bicíclicos de piridina, pirazina y pirimidina como inhibidores de PI3K beta |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US10894793B2 (es) |
| EP (1) | EP3472160B1 (es) |
| JP (1) | JP7149854B2 (es) |
| KR (1) | KR102472453B1 (es) |
| CN (1) | CN109311875A (es) |
| AU (1) | AU2017286379B2 (es) |
| BR (1) | BR112018075999A2 (es) |
| CA (1) | CA3025594A1 (es) |
| DK (1) | DK3472160T3 (es) |
| EA (1) | EA037361B1 (es) |
| ES (1) | ES2871140T3 (es) |
| HR (1) | HRP20210508T1 (es) |
| HU (1) | HUE054118T2 (es) |
| IL (1) | IL263652A (es) |
| LT (1) | LT3472160T (es) |
| MX (1) | MX2018015707A (es) |
| SI (1) | SI3472160T1 (es) |
| WO (1) | WO2017216292A1 (es) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL272988B (en) | 2017-09-08 | 2022-08-01 | Beigene Ltd | Imidazo-[1,5-a]pyrazine derivatives as pi3k-delta inhibitors |
| CN111039946A (zh) * | 2018-10-15 | 2020-04-21 | 上海轶诺药业有限公司 | 一类咪唑并芳环类化合物的制备和应用 |
Family Cites Families (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007103756A2 (en) | 2006-03-02 | 2007-09-13 | Smithkline Beecham Corporation | Thiazolones for use as pi3 kinase inhibitors |
| JP2009544723A (ja) | 2006-07-24 | 2009-12-17 | スミスクライン・ビーチャム・コーポレイション | Pi3キナーゼ阻害剤としてのチオゾリジンジオン誘導体 |
| US8614215B2 (en) | 2007-02-22 | 2013-12-24 | Merck Serono Sa | Quinoxaline inhibitors of phosphoinositide-3-kinases (PI3Ks) |
| WO2009021083A1 (en) | 2007-08-09 | 2009-02-12 | Smithkline Beecham Corporation | Quinoxaline derivatives as pi3 kinase inhibitors |
| NZ587051A (en) | 2008-01-04 | 2012-12-21 | Intellikine Llc | Isoquinolinone derivatives, compositions and methods of inhibiting phosphatidyl inositol-3 kinase (pi3 kinase) |
| AP2011005779A0 (en) | 2009-02-06 | 2011-08-31 | Ortho Mcneil Janssen Pharm | Novel susbstituted bicyclic heterocyclic compoundsas gamma secretase modulators. |
| US8957064B2 (en) | 2009-02-13 | 2015-02-17 | Bayer Intellectual Property Gmbh | Fused pyrimidines |
| WO2010108074A2 (en) | 2009-03-20 | 2010-09-23 | Amgen Inc. | Inhibitors of pi3 kinase |
| MX2012002066A (es) | 2009-08-17 | 2012-03-29 | Intellikine Inc | Compuestos heterociclicos y usos de los mismos. |
| US8569296B2 (en) | 2009-09-29 | 2013-10-29 | Xcovery Holding Company, Llc | PI3K (delta) selective inhibitors |
| DE102009052575A1 (de) | 2009-11-10 | 2011-05-12 | Siemens Medical Instruments Pte. Ltd. | Verfahren, Hörgerät und Anordnung zur Kalibrierung eines akustischen Anpasssystems |
| AR080754A1 (es) | 2010-03-09 | 2012-05-09 | Janssen Pharmaceutica Nv | Derivados de imidazo (1,2-a) pirazina y su uso como inhibidores de pde10 |
| UY33304A (es) | 2010-04-02 | 2011-10-31 | Amgen Inc | Compuestos heterocíclicos y sus usos |
| EP2624696B1 (en) | 2010-10-06 | 2016-12-21 | Glaxosmithkline LLC | Benzimidazole derivatives as pi3 kinase inhibitors |
| US9127000B2 (en) | 2011-02-23 | 2015-09-08 | Intellikine, LLC. | Heterocyclic compounds and uses thereof |
| WO2013028263A1 (en) | 2011-08-24 | 2013-02-28 | Glaxosmithkline Llc | Pyrazolopyrimidine derivatives as pi3 kinase inhibitors |
| US8906910B2 (en) * | 2011-12-20 | 2014-12-09 | Glaxosmithkline Llc | Imidazopyridine derivatives as PI3 kinase |
| US8778937B2 (en) | 2011-12-20 | 2014-07-15 | Glaxosmithkline Llc | Benzimidazole boronic acid derivatives as PI3 kinase inhibitors |
| GB201212513D0 (en) * | 2012-07-13 | 2012-08-29 | Ucb Pharma Sa | Therapeutic agents |
| CN104619709B (zh) | 2012-07-13 | 2016-11-09 | Ucb生物制药私人有限公司 | 作为tnf活性调节剂的咪唑并吡啶衍生物 |
| TWI574962B (zh) * | 2012-11-14 | 2017-03-21 | 加拓科學公司 | 作爲pi3激酶調節劑的芳雜環化合物及其使用方法和用途 |
| WO2015055071A1 (zh) | 2013-10-16 | 2015-04-23 | 上海璎黎药业有限公司 | 稠合杂环化合物、其制备方法、药物组合物和用途 |
| AU2015366190B2 (en) | 2014-12-19 | 2020-01-23 | Janssen Pharmaceutica Nv | Imidazopyridazine derivatives as P13Kbeta inhibitors |
| CA2967546A1 (en) | 2014-12-19 | 2016-06-23 | Janssen Pharmaceutica Nv | Heterocyclyl linked imidazopyridazine derivatives as pi3k.beta. inhibitors |
| EA037401B1 (ru) | 2015-10-09 | 2021-03-24 | Янссен Фармацевтика Нв | Производные хиноксалина и пиридопиразина в качестве ингибиторов pi3k |
-
2017
- 2017-06-15 MX MX2018015707A patent/MX2018015707A/es active IP Right Grant
- 2017-06-15 DK DK17732059.5T patent/DK3472160T3/da active
- 2017-06-15 SI SI201730708T patent/SI3472160T1/sl unknown
- 2017-06-15 EA EA201990046A patent/EA037361B1/ru not_active IP Right Cessation
- 2017-06-15 WO PCT/EP2017/064671 patent/WO2017216292A1/en unknown
- 2017-06-15 KR KR1020187035198A patent/KR102472453B1/ko active IP Right Grant
- 2017-06-15 BR BR112018075999A patent/BR112018075999A2/pt unknown
- 2017-06-15 US US16/309,999 patent/US10894793B2/en active Active
- 2017-06-15 JP JP2018564783A patent/JP7149854B2/ja active Active
- 2017-06-15 HU HUE17732059A patent/HUE054118T2/hu unknown
- 2017-06-15 LT LTEP17732059.5T patent/LT3472160T/lt unknown
- 2017-06-15 ES ES17732059T patent/ES2871140T3/es active Active
- 2017-06-15 EP EP17732059.5A patent/EP3472160B1/en active Active
- 2017-06-15 CN CN201780037269.9A patent/CN109311875A/zh active Pending
- 2017-06-15 CA CA3025594A patent/CA3025594A1/en active Pending
- 2017-06-15 AU AU2017286379A patent/AU2017286379B2/en active Active
-
2018
- 2018-12-11 IL IL263652A patent/IL263652A/en active IP Right Grant
-
2021
- 2021-03-30 HR HRP20210508TT patent/HRP20210508T1/hr unknown
Also Published As
| Publication number | Publication date |
|---|---|
| SI3472160T1 (sl) | 2021-07-30 |
| KR20190016954A (ko) | 2019-02-19 |
| AU2017286379B2 (en) | 2021-07-01 |
| EP3472160A1 (en) | 2019-04-24 |
| IL263652A (en) | 2019-01-31 |
| DK3472160T3 (da) | 2021-05-10 |
| HRP20210508T1 (hr) | 2021-05-28 |
| LT3472160T (lt) | 2021-04-26 |
| BR112018075999A2 (pt) | 2019-04-02 |
| US20190169199A1 (en) | 2019-06-06 |
| WO2017216292A1 (en) | 2017-12-21 |
| JP7149854B2 (ja) | 2022-10-07 |
| EP3472160B1 (en) | 2021-02-24 |
| CN109311875A (zh) | 2019-02-05 |
| CA3025594A1 (en) | 2017-12-21 |
| HUE054118T2 (hu) | 2021-08-30 |
| US10894793B2 (en) | 2021-01-19 |
| EA201990046A1 (ru) | 2019-05-31 |
| EA037361B1 (ru) | 2021-03-18 |
| MX2018015707A (es) | 2019-03-21 |
| KR102472453B1 (ko) | 2022-11-29 |
| AU2017286379A1 (en) | 2018-12-06 |
| JP2019518032A (ja) | 2019-06-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2818620T3 (es) | Derivados de azabencimidazol como inhibidores de pi3k beta | |
| EP4045151A1 (en) | Bicyclic heterocycles as fgfr inhibitors | |
| BR112018007068B1 (pt) | Derivados de quinoxalina e piridopirazina como inibidores de pi3kbeta, seu uso e composição farmacêutica que os compreende | |
| ES2761051T3 (es) | Derivados de imidazopiridazina como inhibidores de PI3Kbeta | |
| ES2871140T3 (es) | Derivados bicíclicos de piridina, pirazina y pirimidina como inhibidores de PI3K beta | |
| ES2760507T3 (es) | Derivados de imidazopiridazina enlazados a heterociclilo como inhibidores de PI3Kß | |
| ES2925176T3 (es) | Derivados de quinoxalina y piridoprazina como inhibidores de pi3k-beta |