ES2829388T3 - Derivados de cis 3,4-dihidroxi-2-(3-metilbutanoil)-5-(-3-metilbutil)-4-(4-metilpentanoil)cyclopent-2-en-1-ona, composiciones sustancialmente puras enantioméricamente y métodos - Google Patents
Derivados de cis 3,4-dihidroxi-2-(3-metilbutanoil)-5-(-3-metilbutil)-4-(4-metilpentanoil)cyclopent-2-en-1-ona, composiciones sustancialmente puras enantioméricamente y métodos Download PDFInfo
- Publication number
- ES2829388T3 ES2829388T3 ES11837235T ES11837235T ES2829388T3 ES 2829388 T3 ES2829388 T3 ES 2829388T3 ES 11837235 T ES11837235 T ES 11837235T ES 11837235 T ES11837235 T ES 11837235T ES 2829388 T3 ES2829388 T3 ES 2829388T3
- Authority
- ES
- Spain
- Prior art keywords
- kdt501
- salt
- humulone
- kdt500
- dihydroxy
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/597—Unsaturated compounds containing a keto groups being part of a ring of a five-membered ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/12—Ketones
- A61K31/122—Ketones having the oxygen directly attached to a ring, e.g. quinones, vitamin K1, anthralin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/703—Unsaturated compounds containing a keto groups being part of a ring containing hydroxy groups
- C07C49/707—Unsaturated compounds containing a keto groups being part of a ring containing hydroxy groups a keto group being part of a three- to five-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Immunology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Epidemiology (AREA)
- Urology & Nephrology (AREA)
- Rheumatology (AREA)
- Endocrinology (AREA)
- Vascular Medicine (AREA)
- Pulmonology (AREA)
- Transplantation (AREA)
- Emergency Medicine (AREA)
- Pain & Pain Management (AREA)
- Heart & Thoracic Surgery (AREA)
- Child & Adolescent Psychology (AREA)
- Gastroenterology & Hepatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Cardiology (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
(+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona que tiene la estructura: **(Ver fórmula)** o una sal o cristal de esta, para su uso en el tratamiento de la diabetes en un sujeto.
Description
DESCRIPCIÓN
Derivados de cis 3,4-dihidroxi-2-(3-metilbutanoil)-5-(-3-metilbutil)-4-(4-metilpentanoil)cydopent-2-en-1-ona, composiciones sustancialmente puras enantioméricamente y métodos
Antecedentes
El miembro más abundante de la familia de los ácidos alfa es (-)-humulona. La configuración absoluta de (-)-humulona se ha informado anteriormente (De Keukeleire 1970), así como también las asignaciones de configuracionales absolutas de cis y trans iso-alfa ácidos que se derivan de (-)-humulona (De Keukeleire 1971).
Los productos llamados 'iso-alfa ácidos reducidos' derivados de ambos los iso alfa ácidos cis y trans también se clasifican. Esta categorización adicional depende de la ubicación y el grado de saturación. La clase de compuestos que resultan de la reducción de solo el carbonilo C6 a un hidroxilo se denomina colectivamente rho iso-alfa ácidos (RIAA). La clase de ácido tetrahidro iso-alfa (THIAA) se refiere colectivamente a aquellos compuestos donde solo ambos restos de isoprenilo están saturados. De manera similar, los hexahidro-iso-alfa ácidos (HIAA) se refieren colectivamente a derivados reducidos de iso-alfa ácidos que contienen el hidroxilo en C6 y la saturación de ambos restos isoprenilos. Además de la existencia de los diastereómeros cis y trans, existen una variedad de congéneres dentro de cada una de las tres clases como resultado de la incorporación de varios ácidos grasos de cadena corta en la ruta biosintética de los correspondientes floroglucinoles (Wang 2008). Los floroglucinoles son precursores comunes de los alfa-ácidos que son precursores de los iso-alfa-ácidos, que a su vez son precursores de los alfa-iso-ácidos reducidos.
Recientemente se ha demostrado que los iso-alfa ácidos reducidos poseen efectos beneficiosos en el tratamiento de la obesidad, la dislipidemia y la inflamación en una variedad de in vitro y modelos murinos. Una mezcla de congéneres y estereoisómeros de rho Los ácidos iso-alfa, denominados RIAA, demostraron actividad antiinflamatoria en el factor de necrosis tumoral alfa (TNF-a), adipocitos 3T3-L1 maduros estimulados y actividad lipogénica en la diferenciación de adipocitos 3T3-L1 (Babish 2010).
Una mezcla de congéneres y estereoisómeros de tetrahidro iso-alfa ácidos, denominados THIAA, inhibió las actividades de la tirosina quinasa del bazo (Syk), la tirosina quinasa de Bruton (Btk), la fosfatidilinositol 3-quinasa (PI 3-quinasa) y la glucógeno sintasa quinasa 3p (GSK3p) así como también la p-fosforilación de catenina in vitro. Adicionalmente, la THIAA inhibió la osteoclastogénesis, como lo indica la transformación disminuida de las células RAW 264.7 en osteoclastos y la actividad TRAP reducida, e inhibió la prostaglandina E activada por IL-1p2, metaloproteinasa 3 de matriz, IL-6, IL-8 y proteína quimiotáctica de monocitos 1 en RASF. Además, en ratones con artritis inducida por colágeno (CIA), esta misma mezcla de congéneres y estereoisómeros (es decir, THIAA) redujo significativamente el índice de artritis y disminuyó la unión de los huesos, la degradación de los cartílagos. Las concentraciones séricas de IL-6 en estos ratones también se inhibieron de manera dependiente de la dosis cuando se trataron con THIAA (Konda 2010).
De acuerdo con estos hallazgos, también se ha informado que RIAA y THIAA, respectivamente, inhibieron la prostaglandina E2 (PGE2) la producción en macrófagos RAW 264,7 estimulados por lipopolisacáridos así como también inhibió la expresión de la proteína ciclooxigenasa-2 (COX-2) inducible. Además de esto, se informa que cada una de estas mezclas, es decir, RIAA y THIAA, respectivamente, se reporta que tienen una translocación nuclear reducida de NF-kB y una abundancia de una manera dependiente de la dosis (Tripp 2009).
La heterogeneidad de RIAA y THIAA impide comprender o conocer la relación entre los diversos congéneres y estereoisómeros presentes tanto en la mezcla RIAA o THIAA con respecto a sus actividades individuales y, por tanto, actividades biológicas relativas. Además, el potencial de sinergia entre los numerosos compuestos presentes en RIAA y THIAA puede explicar gran parte, si no la totalidad, de la actividad biológica observada de estas mezclas.
Con el objetivo de lograr una comprensión y conocer la relación entre los diversos congéneres y estereoisómeros con respecto a su actividad biológica individual y, por tanto, relativa, es imperativo que los compuestos de interés se obtengan como compuestos sustancial y enantioméricamente puros, y que su actividad biológica individual se mide en una forma sustancial y enantioméricamente pura. Este imperativo se deriva necesariamente de la dependencia de la actividad biológica de cualquier sustancia heterogénea que comprende una mezcla de diferentes moléculas (por ejemplo, THIAA, RIAA y HHIAA) sobre porciento de composición, estereoquímica, estructura y otras propiedades de las diferentes moléculas que comprenden la sustancia heterogénea. Por estas razones, existe la necesidad de preparar y purificar sustancialmente derivados de ácidos iso-alfa reducidos sustancialmente puros enantioméricamente para usar en composiciones farmacéuticas y tratamientos.
El uso de extractos de lúpulo, que incluyen tetrahidroisohumulona, para su uso en el tratamiento de la diabetes, se ha descrito en WO2006/053249. WO2010/068731 describe la purificación de humulonas e iso-humulonas a partir de mezclas complejas.
Además de esta necesidad, también existe la necesidad de fabricar un compuesto sustancial y enantioméricamente puro en una forma que sea adecuada para varios procesos que se encuentran habitualmente en el desarrollo farmacéutico. La fabricación de formas cristalinas puras de potenciales candidatos a fármacos es ventajosa para el desarrollo de fármacos.
Una ventaja es la caracterización mejorada de las propiedades químicas y físicas de un candidato a fármaco. No es raro que las formas cristalinas posean una farmacocinética más favorable en comparación con una forma amorfa y, son con frecuencia a menudo, son más fáciles de procesar. La estabilidad de almacenamiento mejorada es una ventaja adicional asociada con frecuencia con las formas cristalinas. Las propiedades físicas inherentes a las formas cristalinas de los posibles candidatos a fármacos son un factor importante a la hora de elegir un ingrediente activo farmacéutico en particular. Un ejemplo es la formulación del fármaco potencial en una composición adecuada para su fabricación, almacenamiento y consumo. Específicamente, la fluidez del sólido cristalino, antes y después de la molienda tiene un gran impacto en la forma en que se maneja el candidato a fármaco durante el procesamiento y la fabricación de la dosis. En los casos donde las partículas de la forma sólida molida no fluyan con facilidad, los científicos de formulaciones se esforzarán por desarrollar una formulación para compensar esta dificultad. Esto a menudo implica el uso de deslizantes tales como dióxido de silicio coloidal, talco y almidón o fosfato cálcico tribásico. Una propiedad adicional del estado sólido de un compuesto farmacéutico potencial es su velocidad de disolución en un fluido acuoso. Las propiedades físicas asociadas con un polimorfo cristalino particular se deben inherentemente a la orientación espacial y a la conformación única de moléculas individuales que comprenden la unidad celular del polimorfo cristalino. Un polimorfo cristalino particular posee propiedades térmicas únicas que generalmente son diferentes sea de una forma amorfa u otro polimorfo.
Las propiedades térmicas de los polimorfos se miden mediante el uso técnicas tales como el punto de fusión capilar, el análisis termogravimétrico (TGA) y la calorimetría diferencial de barrido (DSC). Los resultados de estas mediciones se usan para distinguir e identificar la existencia de formas polimórficas y distinguirlas entre sí. Una forma polimórfica particular generalmente posee distintas propiedades cristalográficas y espectroscópicas que son detectables mediante una variedad de técnicas que incluyen, pero no se limitan a la difracción de rayos X, a la difracción de rayos X en polvo (XRPD), cristalografía monocristalina de rayos X y espectrometría infrarroja.
Resumen
En la presente descripción son cis 3,4-dihidroxi-2-(3-metilbutanoil)-5-(3-metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona ("KDT500") y composiciones sustancialmente y enantioméricamente puras y composiciones farmacéuticas que comprenden estos derivados.
La presente invención proporciona (+)-(4S, 5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3-metilbutil) 4-(4-metilpentanoil) ciclopent-2-en-1-ona ("(+)-KDT500") que tiene la estructura expuesta en la Fórmula I, -1) o una sal o cristal de esta para su uso en el tratamiento de diabetes, obesidad o inflamación en un sujeto:

La invención se expone en las reivindicaciones 1, 2 o 3 y en las reivindicaciones dependientes 4 a 13.
En la presente descripción se describen métodos para inducir lipogénesis, inducir adipogénesis, activar PPARy o activar GPR120 in vitro o in vivo mediante el uso uno o más de los derivados de KDT500 o composiciones de los mismos. Tales composiciones pueden ser composiciones farmacéuticas sustancialmente y enantioméricamente puras que comprenden un derivado de KDT500 y uno o más portador farmacéuticamente aceptables. En la presente descripción se describen métodos para tratar una afección asociada con una lipogénesis disminuida, adipogénesis disminuida o una actividad PPARy o GPR120 disminuida en un sujeto que lo necesite de esta manera.
La presente descripción son métodos para trata el tratamiento de una afección que responde a la modulación de PPARy, el tratamiento de una afección que responde a la modulación de GPR120, el tratamiento de una alteración metabólica, inhibición de la inflamación o el tratamiento de una afección asociada con la inflamación en un sujeto que lo necesita de esta manera mediante la administración al sujeto de una cantidad efectiva de uno o más de los derivados de KDT500 o composiciones de los mismos. Los derivados pueden administrarse mediante una composición farmacéutica sustancialmente pura enantioméricamente como se describe en la presente descripción. La afección que responde a la modulación de PPARy puede ser diabetes tipo II, obesidad, hiperinsulinemia, síndrome metabólico, enfermedad del hígado graso no alcohólico, esteatohepatitis no alcohólica, un trastorno autoinmune o un trastorno proliferativo. La alteración metabólica puede ser diabetes. La administración de los compuestos o composiciones proporcionados en la presente descripción puede resultar en una disminución de los niveles de lípidos y/o glucosa incluidos los niveles de triglicéridos.
Breve descripción de los dibujos
Figura 1: La estructura estereoquímica de (+)-KDT501. Se omiten los iones de potasio y la molécula de agua de la unidad celular.
Figura 2: Un dibujo ORTEP del cristal (+)-KDT501 para su unidad asimétrica.
Figura 3: Difractograma XRPD de (+)-Kd T501.
Figura 4: Termograma TG/DTA de (+)-KDT501.
Figura 5: Efecto de (+)-KDT501 sobre la lipogénesis en adipocitos 3T3-L1. Se usó rosiglitazona como control positivo. Figura 6: Efecto de (+)-KDT501 sobre los niveles de expresión de MMP-9 mediados por TNF-a-(A) y LPS-(B) en células THP-1. Los datos representan la media ± DE de cuatro experimentos.
Figura 7: Efecto de (+)-KDT501 sobre los niveles de expresión de IL-1p mediada por TNF-a-(A) y LPS-(B) en las células THP-1. Los datos representan la media DE de cuatro experimentos.
Figura 8: Efecto de (+)-KDT501 sobre los niveles de expresión de MCP-1 mediada porTNF-a-(A) y LPS-(B) en las células THP-1. Los datos representan la media ± DE de cuatro experimentos.
Figura 9: Efecto de (+)-KDT501 sobre los niveles de expresión de RANTES mediados por TNF-a- (A) y LPS-(B) en las células THP-1. Los datos representan la media ± DE de cuatro experimentos.
Figura 10: Efecto de (+)-KDT501 sobre los niveles de expresión de MIP-1a mediados por TNF-a-(A) y LPS-(B) en las células THP-1. Los datos representan la media DE de cuatro experimentos,
Figura 11: Efecto de (+)-Kd T501 sobre PGE mediada por IL-1p2 niveles en RASF.
Figura 12: Efecto de (+)-KDT501 sobre los niveles de Mm P13 mediados por 1L-1p en RASF.
Figura 13: Efecto de (+)-KDT501 sobre la actividad PPARy. Se usaron rosiglitazona y telmisartán como controles positivos. Figura 14: Efecto de (+)-KDT501 sobre la actividad de PPARa. Se usó GW590735 como control positivo y rosiglitazona como control negativo.
Figura 15: Efecto de (+)-KDT501 sobre la actividad de PPAR8. Se usó GW0742 como control positivo y rosiglitazona como control negativo.
Figura 16: Efecto de (+)-KDT501 y (-)- KDT501 sobre la actividad de PPARy. Se usó rosiglitazona como control positivo. Figura 17: Efecto de (+)-KDT501 sobre la actividad de GPR120. Los datos representan el promedio / desviación estándar de las determinaciones duplicadas. Se usaron DHA, EPA y a-LA como controles.
Figura 18 Efecto de (+)-KDT501 y (-)-KDT501 sobre la actividad de DAPK1 en ensayos libres de células.
Figura 19 Efecto de KDT501 sobre los niveles de glucosa en sangre en ratas ZDF.
Figura 20 Efecto de KDT501 sobre los niveles de triglicéridos en sangre en ratas ZDF.
Figura 21 Efecto de KDT501 sobre los niveles de glucosa en sangre en ratones DIO.
Figura 22 Efecto de KDT501 sobre los niveles de insulina en sangre en ratones DIO.
Figura 23 Efecto de KDT501 sobre los niveles de HbAlC en ratones DIO.
Figura 24 Efecto de KDT501 sobre la masa grasa en ratones DIO.
Descripción detallada
Como se describe en la presente descripción, cis 3,4-dihidroxi-2-(3-metilbutanoil)-5-(3-metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona ("KDT500") se sintetizaron, purificaron y caracterizaron. Por lo tanto, como se describen en la presente descripción son derivados de KDT500 y sales y cristales de los mismos, incluidos los cristales de las sales. También se describen en la presente descripción composiciones que comprenden estos derivados y sales y cristales de los mismos, incluidas sustancialmente composiciones puras enantioméricamente,
El término "sal", como se usa en la presente, puede referirse a cualquier sal farmacéuticamente aceptable, incluidas, por ejemplo, sales de bases inorgánicas tales como sales de potasio, aluminio, calcio, cobre, guanidio, hierro, litio, magnesio, sodio y sales de zinc y sales de bases orgánicas tales como como sales de cinconidina, cinconina y sales de dietanolamina. Pueden encontrarse ejemplos adicionales de sales farmacéuticamente aceptables y preparaciones de acuerdo con la presente invención pueden encontrarse en, por ejemplo, Berge J Pharm Sci 66: 1 (1977).
Un derivado de KDT500 como se describe en la presente descripción tiene la estructura expuesta en la Fórmula I o Fórmula II:


Los compuestos de Fórmula I y Fórmula II representan (+)-(4S, 5R)-3,4-dihidroxi-2-(3-metMbutanoM)-5-(3-metilbutM)-4-(4-metilpentanoil)ciclopent-2-en-1-ona (denominado en la presente descripción como "(+)-KDT500") y (-)-(4R, 5S) -3,4-dihidroxi-2-(3-metilbutanoil)-5-(3-metilbulM)-4-(4-metilpentanoM) ciclopent-2-en-1-ona (denominado en la presente descripción como "(-)-KDT500"), respectivamente. Por lo tanto, un derivado de KDT500 como se describe en la presente descripción es (+)-KDT500 o (-)-KDT500.
Un derivado de KDT500 como se describe en la presente descripción es una sal de KDT500. Las sales de KDT500 incluyen, pero no se limitan a, sales de potasio, sales de magnesio, sales de calcio, sales de zinc, sales de hierro, sales de sodio, sales de cobre, sales de guanidinio, sales de cinconidina y sales de cinconina de KDT500. Por ejemplo, una sal de KDT500 puede ser ((+)-(4S,5R)-3,4-dihidroxi-2-(3-metNbutanoN)-5-(3-metNbutN)-4-(4-metNpentanoN) ciclopent-2-en-1-ona una sal de potasio (denominada en la presente descripción como "(+)-KDT501") tiene la estructura expuesta en la Fórmula III:
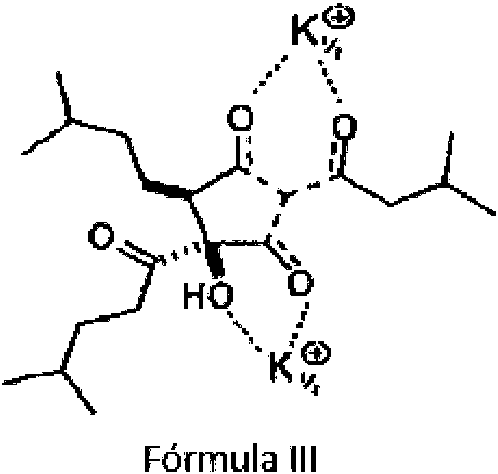
Un derivado de KDT500 como se describe en la presente descripción es un cristal de KDT500 o una sal del mismo, que incluye, por ejemplo, KDT501. En ciertos cristales de (+)-se describen KDT501 que tienen un grupo espacial P 21 21 2 y dimensiones de unidad de célula de a = 23,3110(9) A (a = 90 °), b = 28,9052 (12) A (p =90 °) y c = 13,6845 (5) A (y = 90 °). Esta forma cristalina de (+)-KDT501 se refiere en la presente descripción como "cristal (+)-KDT501".
Cierto cristal (+)-KDT501 tiene las coordenadas atómicas tridimensionales expuestas en la Tabla 2, las longitudes de enlace y los ángulos establecidos en la Tabla 3 y/o los parámetros de desplazamiento anisotrópico establecidos en la Tabla 4, donde el exponente del factor de desplazamiento anisotrópico toma la forma: -2n2[h2a*2U11 ... 2 h k a*b*U12].
En la presente invención en determinadas modalidades, se describen composiciones que comprenden uno o más de los derivados de KDT500 proporcionados en la presente descripción. Ciertas composiciones son sustancialmente puras enantioméricamente. El término "sustancialmente pura enantioméricamente" como se usa en la presente se refiere a una composición en la cual el 90 % o más de un compuesto particular en la composición está en una primera forma enantiomérica, mientras que el 10 % o menos está en una segunda forma enantiomérica. Por ejemplo, en una composición sustancialmente pura enantioméricamente (+)- KDT500, el 90 % o más del KDT500 en la composición es (+)-KDT500 y el 10 % o menos es (-)-KDT500. La "primera forma enantiomérica" de un compuesto puede incluir sales y cristales de esa forma enantiomérica. Por ejemplo, en una composición sustancialmente pura enantioméricamente (+)- KDT500, el 90 % o más del KDT500 en la composición está en forma de (+)-KDT500 o sales o cristales del mismo, mientras que el 10 % o menos está en forma de (-)-KDT500 o sus sales o cristales. Una composición sustancialmente enantioméricamente puede contener 91 %, 92 %, 93 %, 94 %, 95 %, 96 %, 97 %, 98 %, 99 %, 99,5 %, 99,9 %, 99,95 % o 99,99 % o más de una primera forma enantiomérica de un compuesto.
Se describen las composiciones de (+)-KDT500. En algunos de ellos, las composiciones son sustancialmente puras enantioméricamente. En algunos de ellos, un porcentaje del (+)- KDT500 en la composición está en forma de sales o cristales de (+)-KDT500. En algunos de estos, todo el (+)-KDT500 en la composición está en forma de sal o cristal. Por
tanto, en la presente descripción se describen composiciones sustancialmente puras enantioméricamente de sales o cristales de (+)-KDT500. En otras composiciones, ninguno de los (+)-KDT500 de la composición está en forma de sal o cristal.
Se describen ciertas composiciones de (+)-KDT500 en donde las composiciones son sustancialmente puras enantioméricamente. En algunos de ellos, un porcentaje del (-)-KDT500 en la composición está en forma de sales o cristales de (-)-KDT500. En algunos de estos, todo el (-)-KDT500 en la composición está en forma de sal o cristal. Por tanto, en la presente descripción se proporcionan composiciones sustancialmente puras enantioméricamente de sales o cristales de (-)-KDT500. En otras composiciones, ninguno de los (-)- KDT500 de la composición está en forma de sal o cristal.
En la presente descripción se describen composiciones farmacéuticas que comprenden uno o más de los derivados de KDT500 y uno o más vehículos farmacéuticamente aceptables. Ciertas composiciones son sustancialmente puras enantioméricamente. En algunas de estas, las composiciones farmacéuticas sustancialmente pura enantioméricamente comprenden (+)- KDT500, (-)-KDT500, o sales o cristales de los mismos. Un "portador farmacéuticamente aceptable", como se usa en la presente descripción, se refiere a un material, composición, o vehículo farmacéuticamente aceptable que se encarga de llevar o transportar un compuesto de interés de un tejido, órgano, o porción del cuerpo hasta otro tejido, órgano, o porción del cuerpo. Por ejemplo, el portador puede comprender un líquido o sólido de relleno, diluyente, excipiente, solvente, material encapsulante o agente estabilizante, o una combinación de estos. Cada componente del portador debe ser "farmacéuticamente aceptable" en el sentido que debe ser compatible con los demás ingredientes de la composición y debe ser adecuado para entrar en contacto con cualquier tejido, órgano o porción del cuerpo que pueda encontrar, lo que significa que debe no conlleva riesgo de toxicidad, irritación, respuesta alérgica, inmunogenicidad o cualquier otra complicación que supere excesivamente sus beneficios terapéuticos.
Ejemplos de portadores farmacéuticamente aceptables para usar en la presente descripción incluye pero no se limita a (1) azúcares, tales como lactosa, glucosa, sacarosa, o manitol y; (2) almidones, tales como almidón de maíz y almidón de papa; (3) celulosa y sus derivados, tales como carboximetil celulosa de sodio, etil celulosa y acetato de celulosa; (4) tragacanto en polvo; (5) malta; (6) gelatina; (7) talco; (8) excipientes, tales como manteca de cacao y ceras para supositorio; (9) aceites, tales como aceite de maní, aceite de semilla de algodón, aceite de cártamo, aceite de sésamo, aceite de oliva, aceite de maíz y aceite de soja; (10) glicoles, tales como propilenglicol; (11) polioles, tales como glicerina, sorbitol, manitol y polietilenglicol; (12) ésteres, tales como etil oleato y etil laurato; (13) agentes desintegradores tales como el agar o carbonato de calcio (14) agentes tampones, tales como hidróxido de magnesio e hidróxido de aluminio; (15) ácido algínico; (16) agua libre de pirógeno; (17) solución salina isotónica; (18) solución de Ringer; (19) alcohol; tal como etil alcohol y propanol (20) soluciones de tampón fosfato; y (21) parafina; (22) lubricantes tales como talco, estearato de calcio, estearato de magnesio, polietilenglicol sólido, o lauril sulfato de sodio; (23) agentes colorantes; (24) deslizantes tales como dióxido de silicio coloidal, talco y almidón o fosfato de calcio tribásico; y (24) otras sustancias compatibles no tóxicas empleadas en composiciones farmacéuticas tales como la acetona. El portador farmacéuticamente aceptable puede ser un portador acuoso, por ejemplo, solución salina tamponada y similares, o el portador farmacéuticamente aceptable puede ser un disolvente polar, por ejemplo, acetona y alcohol.
Las composiciones farmacéuticas pueden comprender además una o más sustancias auxiliares farmacéuticamente aceptables de acuerdo con como se requiera para aproximarse a las condiciones fisiológicas. Por ejemplo, las composiciones pueden comprender uno o más agentes de ajuste del pH, agentes tamponadores o agentes de ajuste de la toxicidad, que incluyen, por ejemplo, acetato de sodio, cloruro de sodio, cloruro de potasio, cloruro de calcio, lactato de sodio y similares.
Las composiciones farmacéuticas pueden formularse en una forma de dosificación adecuada, que incluye por ejemplo cápsulas, sellos, píldoras, tabletas, comprimidos, (mediante el uso una base saborizada, usualmente sacarosa y acacia o tragacanto), polvos, gránulos, o como una solución o una suspensión en un líquido acuoso o no acuoso, o como una emulsión líquida de aceite en agua o agua en aceite, como un elíxir o jarabe, o como pastillas (mediante el uso de una base inerte, tal como gelatina y glicerina, o sacarosa y acacia) y/o como enjuagues bucales y similares, donde cada uno contiene una cantidad predeterminada de un derivado de KDT500 como un ingrediente activo. Las composiciones pueden formularse como un vehículo de liberación prolongada, tal como, por ejemplo, una cápsula de liberación prolongada. Un "vehículo de liberación prolongada", como se usa en la presente, se refiere a cualquier vehículo de administración que libera el agente activo durante un período de tiempo en lugar de inmediatamente después de la administración. Las composiciones pueden formularse como un vehículo de liberación inmediata.
Una tableta puede fabricarse por compresión o moldeo, opcionalmente con uno o más ingredientes complementarios. Las tabletas comprimidas pueden prepararse mediante el uso aglutinante (por ejemplo, gelatina o hidroxipropilmetil celulosa), lubricante, diluyente inerte, conservante, desintegrante (por ejemplo, almidón glicolato de sodio o carboximetil celulosa de sodio reticulada), agente dispersante o de superficie activa. Las tabletas moldeadas pueden fabricarse por moldeo en una máquina adecuada de una mezcla sustancialmente pura enantioméricamente del derivado de KDT500 en polvo o humedecida adicionalmente con un diluyente líquido inerte. Las tabletas, y otras formas de dosificación sólidas, tales como grageas, cápsulas, píldoras y gránulos, pueden opcionalmente lograrse o prepararse con recubrimientos y capas tales como recubrimientos entéricos y otros recubrimientos bien conocidos en la técnica de la formulación farmacéutica. Pueden formularse además para proporcionar liberación lenta o controlada de un derivado de KDT500 en la misma
mediante el uso de, por ejemplo, hidroxipropilmetil celulosa en proporciones variables para proporcionar el perfil de liberación deseado, otras matrices de polímero, liposomas y/o microesferas. Pueden esterilizarse por, por ejemplo, por filtración a través de un filtro de retención de las bacterias, o mediante la incorporación de agentes esterilizantes en la forma de composiciones sólidas estériles que pueden disolverse en agua estéril, o algún otro medio estéril inyectable inmediatamente antes del uso. Estas composiciones pueden contener, opcionalmente además agentes opacificadores y pueden ser de una composición que libere el(los) derivado(s) de KDT500 solamente, o preferentemente, en una cierta porción del tracto gastrointestinal, opcionalmente, en una manera retardada. Los ejemplos de composiciones de inclusión que pueden usarse incluyen sustancias poliméricas y ceras. El derivado de KDT500 puede estar además en forma microencapsulada, si es adecuado, con uno o más de los excipientes descritos anteriormente.
La concentración de los derivados de KDT500 en las composiciones descritas en la presente descripción puede variar. Las concentraciones pueden seleccionarse en base a los volúmenes de fluido, viscosidades, peso corporal y similares de acuerdo con el modo de administración particular seleccionado y las necesidades del sistema biológico. La concentración de un derivado de KDT500 en una composición descrita en la presente descripción puede ser de aproximadamente 0,0001 % a 100 %, de aproximadamente 0,001 % a aproximadamente 50 %, de aproximadamente 0,01 % a aproximadamente 30 %, de aproximadamente 0,1 % a aproximadamente 20 % o de aproximadamente 1 % a aproximadamente 10 % p/vol.
También se describen en la presente descripción métodos para analizar, sintetizar, purificar y/o cristalizar los derivados de KDT500 proporcionados en la presente descripción, así como también métodos para analizar, sintetizar, purificar y/o cristalizar las composiciones de KDT500 sustancialmente pura enantioméricamente proporcionadas en la presente descripción.
Ciertos métodos de síntesis descritos en la presente descripción generan un solo enantiómero de un derivado de KDT500. Por ejemplo, los métodos de síntesis pueden generar solo (+)-KDT500 o solo (-)-KDT500. Otros métodos de síntesis dan como resultado una mezcla de formas enantioméricas de derivados de KDT500. En estas una o más etapas posteriores de separación y/o purificación pueden realizarse para aislar una única forma enantiomérica o para generar una composición sustancialmente pura enantioméricamente como se describe en la presente descripción.
En algunos de los métodos de síntesis y purificación descritos en la presente descripción, (+)-KDT500 y/o (-)-KDT500 pueden sintetizarse mediante el uso de lupulona como material de partida (véase, por ejemplo, el Ejemplo 11 más abajo). En uno de estos métodos de síntesis/purificación, la lupulona se convierte primero en tetrahidro-desoxihumulona mediante desalquilación reductora, y la tetrahidro-desoxihumulona se convierte en tetrahidro-humulona enantioméricamente pura ((+) y/o (-)) mediante oxidación asimétrica. El enantiómero resultante se purifica y se convierte en el correspondiente cis iso-humulona vía enantio- e isomerización diastereoselectiva.
(+)-KDT500 y/o (-)-KDT500 pueden sintetizarse mediante el uso de desoxihumulona como material de partida (véase, por ejemplo, el Ejemplo 12 más abajo); la desoxihumulona se convierte primero en (+)-humulona y/o (-)-humulona, respectivamente, mediante oxidación asimétrica. La (+)-humulona se convierte en la correspondiente (-)-cis isohumulona enantioméricamente pura mediante enantio e isomerización diastereoselectiva, y luego se convierte en (-)-KDT500 mediante hidrogenación. La (-)-humulona se convierte en el correspondiente (+)- cis isohumulona enantioméricamente pura mediante enantio e isomerización diastereoselectiva, y luego se convierte en (+)-KDT500 mediante hidrogenación.
La (+)-humulona y (-)-humulona enantioméricamente puras pueden purificarse a partir de la (±)- humulona racémica (véase, por ejemplo, el Ejemplo 13 más abajo). La (+)-tetrahidro humulona y (-)-tetrahidro humulona enantioméricamente puras pueden purificarse a partir de la (±)- tetrahidro humulona racémica (véase, por ejemplo, el Ejemplo 14 más abajo).
Como se describe en la presente descripción, (+)-KDT501 se evaluó por sus efectos sobre la lipogénesis y la adipogénesis en un modelo de fibroblasto murino 3T3-L1. Se encontró que el (+)-KDT501 induce tanto la lipogénesis como la adipogénesis de una manera dependiente de la dosis. El fibroblasto murino 3T3-L1 permite la investigación de la replicación de los preadipocitos, la diferenciación de los adipocitos y los efectos de sensibilización a la insulina (Fasshauer 2002; Li 2002; Raz 2005). Se ha informado que un agente que promueve la captación de lípidos en las células grasas mejora la sensibilidad a la insulina. Una hipótesis es que la incorporación de ácidos grasos en el adipocito del plasma provoca un agotamiento relativo de los ácidos grasos en el músculo con una mejora concomitante de la captación de glucosa (Martin 1998). Los efectos desensibilizantes de la insulina de los ácidos grasos libres en el músculo y el hígado se reducirían como consecuencia del tratamiento con tiazolidindiona. Estos resultados in vitro se han confirmado clínicamente (Boden 1997; Stumvoll 2002).
(+)-KDT501 y (-)- KDT501 se evaluaron ambos por su capacidad para unirse competitivamente a PPARy, SCN2A y AGTR2 en presencia de ligandos agonistas para cada uno de estos objetivos. Ambas moléculas exhibieron la capacidad de unirse a las tres dianas de unión en presencia de sus ligandos agonistas, con (+)-KDT501 exhibiendo un mayor grado de afinidad por los tres objetivos. De los objetivos, (+)-KDT501 se unió a PPARy con la mayor afinidad. A continuación, se evaluó el efecto de (+)-KDT501 y (-)- KDT501 sobre la actividad de PPAR. Ambos compuestos aumentaron la actividad de PPARy mientras que tenían poco o ningún efecto sobre la actividad de PPARa o PPAR8. Sin embargo, el efecto de (+)-KDT501 sobre la actividad de PPARy fue al menos tres veces mayor que el de (-)- KDT501 en todas las concentraciones, lo que sugiere que (+)-KDT501 es sustancialmente más eficaz como activador de PPARy. El PPARy es un regulador maestro en la adipogénesis, la homeostasis de la glucosa y la sensibilidad a la insulina, y es el objetivo
molecular de las tiazolidinedionas, que sensibilizan las células a la insulina y tienen efectos antidiabéticos en el hígado, el tejido adiposo y el músculo esquelético (Tontonoz 1994; Tontonoz 1994).
En base a la capacidad de los derivados de KDT500 proporcionados en la presente descripción para inducir lipogénesis, adipogénesis y actividad de PPARy, se proporcionan métodos para inducir lipogénesis, inducir adipogénesis y/o activar PPARy, así como también métodos para tratar una afección que responde a la modulación de PPARy, en un sujeto que lo necesite mediante la administración de una cantidad terapéuticamente eficaz de uno o más de los derivados de KDT500 proporcionados en la presente descripción o sus composiciones farmacéuticas sustancialmente pura enantioméricamente. Los ejemplos de afecciones que responden a la modulación de PPARy incluyen, por ejemplo, afecciones que pueden tratarse mediante una mejora de la homeostasis de la glucosa y la energía, que incluyen, entre otros, diabetes de tipo II, obesidad, hiperinsulinemia, síndrome metabólico, enfermedad del hígado graso no alcohólico, esteatohepatitis no alcohólica, una enfermedad autoinmune y trastorno proliferativo.
Como se discutió anteriormente, se ha demostrado que los agonistas de PPARy previamente identificados suprimen la respuesta inflamatoria. El PPARy no solo activa la transcripción de genes diana para el metabolismo de lípidos, sino que también reduce la expresión de genes de inflamación (Pascual 2005). La supresión de la respuesta inflamatoria por los agonistas de PPARy está estrechamente relacionada con los efectos antidiabéticos y antiateroescleróticos. Para evaluar el papel de KDT501 en la inflamación, se evaluó la capacidad de (+)-KDT501 para inhibir la expresión de varios factores inflamatorios mediados por TNF - a-, LPS- e IL - 1p. Se descubrió que (+)-KDT501 inhibe la expresión mediada por TNF-a y LPS de MMP-9, IL-1p, MCP-1, RANTES y MIP-1a en células THP-1, e inhibe la expresión mediada por IL-1p expresión de PGE2 y MMP-13 en células de fibroblasto sinovial de artritis reumatoide (RASF). Además, se encontró que tanto (+)-KDT501 como (-)- KDT501 inhiben la actividad de DAPK1, mostrando (+)-k Dt 501 la mayor eficacia.
La capacidad de KDT501 para mejorar la sensibilidad a la insulina y la regulación de la glucosa, así como también para reducir las señales proinflamatorias, sugiere un mecanismo que influye en la interacción entre la señalización de la insulina y las vías de inflamación. Un mecanismo potencial para esta actividad es mediante la estimulación de un receptor acoplado a la proteína G. Para evaluar esta posibilidad, se probó el efecto de (+)-KDT501 sobre la actividad del receptor acoplado a proteína G sensibilizante de ácidos grasos w3 GPR120. La activación de GPR120 mejora la sensibilidad a la insulina y reduce la inflamación al inhibir la vía NF-kB (Oh 2010; Talukdar 2011). También se ha demostrado que GPR120 media la secreción de GLP-1 en las células L intestinales (Hirasawa 2005; Hara 2011) y aumenta la lipogénesis en los adipocitos (Goth 2007), lo que contribuyó a los efectos antidiabéticos. Se encontró que (+)-KDT501 agoniza la actividad de GPR120 con un EC50 valor de 30,3 pM, lo que sugiere que (+)-KDT501 puede ejercer sus efectos lipogénicos en parte a través de la activación de GRP120.
En base a la capacidad de los derivados de KDT500 descritos en la presente descripción para introducir la actividad de GPR120, se describen métodos para tratar una afección que responde a la modulación de GPR120 en un sujeto que lo necesita administrando una cantidad terapéuticamente eficaz de uno o más de los derivados de KDT500 proporcionados en la presente descripción o sustancialmente sus composiciones farmacéuticas enantioméricamente puras. Además, en base a la capacidad de los derivados de KDT500 proporcionados en la presente descripción para inducir la actividad de GPR120 y PPARy e inhibir la expresión de varios factores inflamatorios mediados por TNF-a, LPS e IL-1p, se describen métodos para inhibir la inflamación en un sujeto que lo necesita mediante la administración de una cantidad terapéuticamente eficaz de uno o más de los derivados de KDT500 proporcionados en la presente descripción o sus composiciones farmacéuticas sustancialmente pura enantioméricamente. También se describen métodos para tratar diversas afecciones asociadas con la inflamación y/o afecciones asociadas con niveles elevados de uno o más de los factores inflamatorios MMP-9, IL-1p, MCP-1, RANTES, MIP-1a, PGE2y MMP-13 en un sujeto que lo necesite mediante la administración de una cantidad terapéuticamente eficaz de uno o más de los derivados de KDT500 proporcionados en la presente descripción o de sus composiciones farmacéuticas sustancialmente pura enantioméricamente. Dichas afecciones incluyen, por ejemplo, aterosclerosis, inestabilidad de la placa aterosclerótica, enfermedades autoinmunes tales como artritis reumatoide, degradación del cartílago, osteoartritis, afecciones alérgicas, enfermedades de inmunodeficiencia tales como VIH/SIDA, nefropatías, tumores y diabetes.
Como se describe en la presente descripción, se evaluó (+)-KDT501 por su capacidad para alterar los niveles de glucosa y lípidos en un modelo de diabetes en ratas. En una dosis diaria de 200 mg/kg, se encontró que (+)-KDT501 reduce significativamente los niveles de glucosa. Esta disminución fue mucho mayor que la observada en ratas tratadas con metformina, y similar a la observada en ratas tratadas con pioglitazona. De manera similar, se encontró que (+)-KDT501 disminuyó los niveles de triglicéridos en un grado casi igual al observado con pioglitazona.
En un modelo de ratón, la administración dos veces al día de (+)-KDT501 fue eficaz para reducir los niveles circulantes de glucosa e insulina en sangre total, reduciendo el área de glucosa e insulina bajo las curvas de concentración-tiempo (AUC) en la prueba de tolerancia oral a la glucosa (OGTT), y disminución de la masa grasa en ratones obesos inducidos por la dieta (DIO). También se observó una tendencia de reducción en el porcentaje de hemoglobina glucosilada (HbAlc) durante un período de 30 días en los ratones DIO.
En base a la capacidad de los derivados de KDT500 descritos en la presente descripción para reducir los niveles de glucosa, triglicéridos e insulina circulantes en modelos de ratas y ratones, se proporcionan aquí métodos para tratar alteraciones metabólicas mediante la administración de una cantidad terapéuticamente eficaz de uno o más de los
derivados de KDT500 descritos en la presente descripción. o sus composiciones farmacéuticas sustancialmente pura enantioméricamente. Una "alteración metabólica", como se usa en la presente, se refiere a cualquier condición que dé como resultado la pérdida del control metabólico de la homeostasis. Los ejemplos de alteraciones metabólicas incluyen, por ejemplo, diabetes, hiperglucemia, aumento de peso, resistencia a la insulina, dislipidemia e hipercolesterolemia. Se describen ciertos métodos para tratar la diabetes, incluida la diabetes de tipo II, en un sujeto que lo necesite mediante la administración de una cantidad terapéuticamente eficaz de uno o más de los derivados de KDT500 o de las composiciones farmacéuticas sustancialmente pura enantioméricamente descritas en la presente descripción. De manera similar, se describen ciertos métodos para disminuir los niveles de glucosa y/o lípidos en un sujeto que lo necesite mediante la administración de una cantidad terapéuticamente eficaz de uno o más de los derivados de KDT500 o de las composiciones farmacéuticas sustancialmente pura enantioméricamente descritas en la presente descripción. La administración de los derivados de KDT500 o sus composiciones resulta en una disminución de los niveles de glucosa en sangre y/o una disminución de los niveles de triglicéridos, o una disminución de uno o más niveles de lípidos adicionales, incluidos, por ejemplo, los niveles de colesterol total o LDL.
En ciertos métodos descritos en la presente descripción, el sujeto es un mamífero, y en algunas de estas modalidades el sujeto es un ser humano. Un "sujeto que lo necesita" se refiere a un sujeto diagnosticado con una afección que responde a la modulación de PPARy o una alteración metabólica, un sujeto que presenta o ha presentado uno o más síntomas de una afección que responde a la modulación de PPARy o alteraciones metabólicas, o un sujeto que se ha considerado en riesgo de desarrollar una condición que responda a la modulación de PPARy o alteraciones metabólicas en base a en uno o más factores hereditarios o ambientales.
Los términos "tratar", "que trata" o "tratamiento" como se usa en la presente con respecto a una afección se refiere a prevenir la afección, retrasar la aparición o la velocidad de desarrollo de la afección, reducir el riesgo de desarrollar la afección, prevenir o retrasar el desarrollo de los síntomas asociados con la afección, reducir o acabar con los síntomas asociados con la afección, generar una regresión completa o parcial de la afección, o alguna combinación de estos.
Una "cantidad terapéuticamente eficaz" de un derivado de KDT500 o una composición farmacéutica como se usa en la presente es una cantidad de una composición que produce un efecto terapéutico deseado en un sujeto. La cantidad terapéuticamente eficaz precisa es una cantidad del compuesto o composición que producirá los resultados más eficaces en términos de eficacia terapéutica del en un sujeto dado. Esta cantidad variará en dependencia de una variedad de factores que incluyen, pero no se limitan a, las características del compuesto terapéutico (que incluyen, por ejemplo, actividad, farmacocinética, farmacodinámica y biodisponibilidad), la condición fisiológica del sujeto (que incluyen, por ejemplo, edad, sexo, estadio y tipo de enfermedad, condición física general, capacidad de respuesta a una dosis dada, y tipo de medicación), la naturaleza del portador o portadores farmacéuticamente aceptables en la formulación, y la ruta de administración. Un experto en las técnicas clínicas y farmacológicas será capaz de determinar la cantidad terapéuticamente eficaz a través de experimentación habitual, específicamente mediante el monitoreo de la respuesta del sujeto a la administración de un compuesto y el ajuste de la dosis en consecuencia. Para una guía adicional, ver Remington: La ciencia y la práctica de la farmacia 21a edición, Univ. de Ciencias en Filadelfia (USIP), Lippincott Williams & Wilkins, Filadelfia, PA, 2005.
Una cantidad terapéuticamente eficaz de un derivado o composición de KDT500 como se describe en la presente descripción puede seleccionarse de aproximadamente 10-10 g a aproximadamente 100 g, aproximadamente 10-10 g a aproximadamente 10-3 g, aproximadamente 10-9 g a aproximadamente 10-6 g, aproximadamente 10-6 g a aproximadamente 100 g, aproximadamente 0,001 g a aproximadamente 100 g, aproximadamente 0,01 g a aproximadamente 10 g, o aproximadamente 0,1 g a aproximadamente 1 g de ingrediente activo (es decir, del derivado de KDT500) por sujeto y día. Puede calcularse una cantidad terapéuticamente eficaz de un derivado o composición de KDT500 en base al peso corporal del sujeto. Por ejemplo, una cantidad terapéuticamente eficaz puede ser aproximadamente 100 mg/kg o más, aproximadamente 150 mg/kg o más, aproximadamente 200 mg/kg o más, aproximadamente 250 mg/kg o más, o aproximadamente 300 mg/kg o más de ingrediente activo (es decir, del derivado de KDT500) por sujeto por día.
Un compuesto o composición como se describe en la presente descripción puede administrarse una o más veces al día. El compuesto o composición puede administrarse menos de una vez al día. Por ejemplo, el compuesto o la composición puede administrarse una vez a la semana, una vez al mes o una vez cada varios meses. La administración de un compuesto o composición descritos en la presente descripción puede llevarse a cabo durante un período de tratamiento específico determinado de antemano, o puede llevarse a cabo de forma indefinida o hasta que se alcance un punto de referencia terapéutico específico. Por ejemplo, la administración puede llevarse a cabo hasta que los niveles de glucosa y/o lípidos alcancen un umbral predeterminado. La frecuencia de dosificación puede cambiar durante el transcurso del tratamiento. Por ejemplo, un sujeto puede recibir administraciones menos frecuentes durante el curso del tratamiento a medida que se cumplen ciertos parámetros terapéuticos.
Los compuestos y composiciones descritos en la presente descripción pueden administrarse a un sujeto mediante cualquier vía de administración conocida en la técnica, que incluye, entre otros, oral, aerosol, enteral, nasal, oftálmico, parenteral o transdérmico (por ejemplo, crema o ungüento tópico, parche). "Parenteral" se refiere a la vía de administración que se asocia generalmente con la inyección, incluyendo infusión infraorbitaria, intraarterial, intracapsular, intracardiaca, intradérmica, intramuscular, intraperitoneal, intrapulmonar, intraespinal, intraesternal, intratecal, intrauterina, intravenosa,
subaracnoidea, subcapsular, subcutánea, transmucosal, o transtraqueal. Los métodos para preparar composiciones administrares de manera parenteral serán conocidos o evidentes para los expertos en la técnica y se describen en más detalle en algunas publicaciones como Remington: The Science and Practice of Pharmacy 21ra edición, Mack Publishing Company, Easton, Pa. (2005). Una composición puede administrarse además como un bolo, electuario o pasta.
Se describen kits que comprenden uno o más de los derivados de KDT500 o sus composiciones sustancialmente puras enantioméricamente que se proporcionan en la presente descripción. En ciertos kits hay instrucciones de uso, como dosis o instrucciones de administración. Los kits pueden usarse para tratar una afección que responde a la modulación de PPARy o GPR120, tratar una alteración metabólica como la diabetes, disminuir los niveles de glucosa y/o lípidos, inhibir la inflamación o tratar una afección asociada con la inflamación en un sujeto que lo necesite.
Los derivados de KDT500 y las composiciones de derivados de KDT500 sustancialmente puros enantioméricamente proporcionados en la presente descripción pueden usarse como agentes aromatizantes. En algunas de estas modalidades, los compuestos se usan como agentes aromatizantes amargos.
Se proporcionan los siguientes ejemplos
EJEMPLOS
Ejemplo 1: Cristalización y caracterización de derivados (+) -y (-)-KDT500 sustancialmente puros enantioméricamente:
Cristalización de (+)-KDT501 (Esquema I):
100 g de cis tetrahidro-iso alfa ácidos obtenidos predominantemente durante el procesamiento del lúpulo se purificaron mediante el uso de cromatografía en contracorriente de alta velocidad (HSCCC) de acuerdo con un procedimiento informado (Dahlberg 2010). El análisis de área del pico por HPLC y UV mostró que el producto resultante era> 90 % KDT500. El material purificado se convirtió en (+)-KDT501 haciendo reaccionar con 1 equivalente de sal de potasio (por ejemplo, KOH) y se recristalizó varias veces para eliminar la decoloración. Después de lograr suficiente homogeneidad, el (+)-KDT501 se recristalizó adicionalmente para obtener cristales de (+)KDT501 con una dimensión suficiente para experimentos de difracción de rayos X.

Se añadieron agua (450 j L) y (+)-KDT501 (49,5 mg) a un vial de 4 mL, y el vial se selló con una tapa y se calentó mediante el uso un bloque de calentamiento térmico (70 °c ) hasta su disolución. La solución se dejó enfriar a 35 °C durante un período de dos horas. Durante este tiempo, se observó la formación de cristales in situ. La mezcla se dejó reposar a temperatura ambiente (22 °C) durante ocho horas, momento en el que se observó la formación de cristales adicionales. Se identificó un cristal individual adecuado en el análisis por rayos X, se retiró cuidadosamente de la solución, se montó y se sometió al análisis de difracción de rayos X.
Resolución de la estructura cristalina de (+)-KDT501:
Un bloque incoloro de 0,37 x 0,30 x 0,28 mm3 se montó en un capilar de vidrio con aceite. Los datos se recolectaron a -163 °C en un difractómetro de rayos X monocristalino Bruker APEX II, Mo-radiación. La distancia entre el cristal y el detector fue de 40 mm y el tiempo de exposición fue de 10 segundos por grado para todos los conjuntos. El ancho de escaneo fue de 0,5°. La recopilación de datos se completó en un 98,4 % a 25° en 0. Se recogió un total de 68211 reflexiones (fusionadas) que cubren los índices, h = -26 a 28, k = -34 a 34, 1 = -16 a 14. 16681 reflexiones eran independientes de la simetría y la Rint = 0,0386 indicó que los datos eran buenos (calidad promedio 0,07). El índice y el refinamiento de la unidad celular indicaron una red ortorrómbica primitiva. Se encontró que el grupo espacial era P21212 (número 18).
Los datos se integraron y escalaron mediante el uso SAINT, SADABS dentro del paquete de software APEX2 (Bruker 2007 APEX2 (Versión 2.1-4), SAINT (versión 7.34A), SADABS (versión 2007/4), BrukerAXS Inc, Madison, WI).
La Solución por métodos directos (SHELXS; SIR97 (Altomare J Appl Cryst 32: 115 (1999), Alternare J Appl Cryst 26: 343 (1993)) produjo un modelo de fase de átomos pesados completo consistente con la estructura propuesta. La estructura se completó diferentes la síntesis de Fourier con SHELXL97 (Sheldrick, "SHELXL - 97: Programa para el Refinamiento de Estructuras Cristalinas", Universidad de Gottingen, Alemania (1997); Mackay "MaXus: un programa de computadora para la solución y refinamiento de las estructuras cristalinas a partir de datos de difracción", Universidad de Glasgow, Escocia (1997)). Los factores de dispersión son de Waasmairy Kirfel (Waasmaier Acta Crystallogr A 51: 416 (1995)). Los átomos de hidrógeno se colocaron en posiciones geométricamente idealizadas y forzadas a viajar sobre sus átomos progenitores con distancias C—H en el intervalo de 0,95-1,00 Angstrom. Los parámetros térmicos isotrópicos Ueq se fijaron de manera que a que eran 1,2Ueq de su átomo parental Ueq para CH y 1,5 Ueq de su átomo parental Ueq en el caso de los grupos metilo. Todos los átomos que no son de hidrógeno se refinaron anisotrópicamente mediante mínimos cuadrados de matriz completa. La estructura cristalina de (+)-KDT501 se expone en la Figura 1. Los datos cristalográficos se establecen en la Tabla 1, las coordenadas atómicas tridimensionales se exponen en la Tabla 2, los ángulos y las longitudes de enlace se establecen en la Tabla 3 y los parámetros de desplazamiento anisotrópico (donde el exponente del factor de desplazamiento anisotrópico toma la forma: -2n2[h2un*2U11 ... 2 hka * b * U12]) se establecen en la Tabla 4.
La unidad asimétrica de la forma cristalina (+)-KDT501 consta de 4 moléculas de (+)-KDT501 que coordinan cuatro cationes de potasio. Dos cationes de potasio forman un puente con el oxígeno; una molécula de agua desordenada se coordina con las otras dos. Las cetonas de los restos orgánicos se coordinan con los cationes de potasio que conducen a un patrón de empaquetamiento cerrado. La unidad asimétrica de (+)-KDT501 se muestra en la Figura 2. La configuración absoluta se obtuvo a partir de la dispersión anómala (parámetro de estructura absoluta = 0,04(4)).
Difracción de rayos X en polvo (XRPD) y análisis térmico de (+)-KDT501:
Los análisis de XRPD se realizaron mediante el uso un difractómetro Panalytical Xpert Pro equipado con un tubo de rayos X de Cu y un sistema detector Pixcel. Las muestras isotérmicas se analizaron en modo de transmisión y se mantuvieron entre películas de polietileno de baja densidad. El difractograma XRPD mostró que (+)-KDT501 es cristalino (Figura 3).
Analizador de Temperatura Termogravimétrico/Diferencial (TG/DTA):
El análisis térmico se llevó a cabo en un Perkin Elmer Diamond. Los patrones de calibración fueron indio y estaño. Las muestras se colocaron en una bandeja de muestras de aluminio, se insertaron en el horno TG y se pesaron con precisión. Las muestras se calentaron de 30 °C a 300 °C en cable de nitrógeno a una velocidad de 10 °C/minuto. La temperatura del horno se equilibró a 30 °C antes del análisis de las muestras.
El termograma TG/DTA (Figura 4) mostró una pérdida de peso de 1,37 %, lo que indica cierta pérdida de solvente, lo que fue indicativo de un hidrato o solvato. No se presentaron otros eventos térmicos, aparte de la endotermia de fusión con un inicio de 145 °C.
Ejemplo 2: Efecto de (+)-KDT501 sobre la lipogénesis en adipocitos 3T3-L1:
Se evaluó el efecto de (+)-KDT501 sobre la lipogénesis en adipocitos 3T3-L1.
Los preadipocitos murinos 3T3-L1 (ATCC, Rockville, MD) se mantuvieron en DMEM (Invitrogen, Rockville, MD) suplementado con suero bovino fetal al 10 % (FBS; ATCC). Como preadipocitos, las células 3T3-L1 tienen un aspecto fibroblástico. Se replican en cultivo hasta que forman una monocapa confluente, después de lo cual el contacto célulacélula desencadena G0/GRAMO1 detención del crecimiento. La estimulación posterior con 3-isobutil-1-metilxanteno, dexametasona y dosis altas de insulina durante dos días hace que las células experimenten una expansión clonal mitótica posconfluente, salgan del ciclo celular y comiencen a expresar genes específicos de adipocitos. Aproximadamente cinco días después de la inducción de la diferenciación, las células muestran el fenotipo característico de adipocitos lleno de lípidos.
Las células se sembraron a una densidad inicial de aproximadamente 1,5 x 106 células en placas de 24 pocillos y se dejó crecer durante 2 días hasta la confluencia. Las células se trataron con (+)-KDT501 (25, 12,5, 6,25 y 3,25 pM), rosiglitazona (10 pM, control positivo; Cayman Chemicals, Ann Harbour, MI) o DMSO (control negativo) el día 0. Después de este tratamiento inicial, las células se diferenciaron en adipocitos mediante la adición de medio de diferenciación que consiste de 10 % de SFB/DMEM (alto contenido de glucosa), metilisobutilxantina 0,5 mM (Sigm Louis, MO), dexametasona 0,5 pM (Sigma) y 10 pg/mL de insulina (Sigma). Después de dos días, el medio se cambió a un medio de progresión que consiste de 10 pg/ml de insulina en 10 % SFB/d Me M. Después de dos días más, se cambió el medio por un medio de mantenimiento que consiste de FBS al 10 %/DMEM. Las células se volvieron a tratar con (+)-KDT501 o rosiglitazona en DMSO cada dos días durante la fase de maduración (día 6/día 7). Siempre que se añadió medio nuevo, también se añadió material de prueba nuevo.
La evaluación microscópica se realizó el día 0 y cada dos días durante la prueba para evaluar la diferenciación de los preadipocitos a la morfología de los adipocitos. El lípido intracelular se cuantificó con tinción de Aceite Rojo. El medio se desechó cuidadosamente después del día 8/día 9 sin alterar la capa celular. Se añadió formalina al 10 % y se incubó durante 15 minutos, las placas se lavaron con isopropanol al 60 % y se secaron durante 10 minutos a temperatura ambiente. Se añadieron 300 pl de solución de Aceite Rojo O (0,36 % en etanol al 60 %; Millipore, Billerica, MA) y se incubó durante 10 minutos a temperatura ambiente. La placa se lavó con etanol al 70 % seguido de dos lavados con agua. El tinte se extrajo añadiendo 200 pL de isopropanol al 100 % durante 20 minutos. Se transfirieron 150 pL de muestra a una placa de 96 pocillos y se midió la absorbancia a 530 nm mediante el uso un lector de placas (Thermo Electron Corp.). Se usó isopropanol al 100 % como blanco. Los resultados se resumen en la Figura 5. (+)-KDT501 a 6,25, 12,5 y 25 pM aumentó la lipogénesis en los adipocitos 3T3-L1 de manera estadísticamente significativa. El aumento observado fue comparable al observado con rosiglitazona.
Ejemplo 3: Efecto de (+)-KDT501 sobre los factores inflamatorios mediados por TNF- a-y LPS en células THP-1:
Se evaluó el efecto antiinflamatorio de (+)-KDT501 en las células THP-1 monocíticas humanas.
Las células THP-1 (ATCC, Manassas, VA) se mantuvieron en RPMI1640 en presencia de suero al 10 % de acuerdo con las instrucciones del fabricante. Las células se preincubaron con (+)-KDT501 a concentraciones variables (50, 25, 12,5 y 3,125 pM) durante una hora, luego se estimularon con TNF-a (10 ng/mL; Sigma, St. Louis, MO) o E. coli LPS (1 pg/mL; Sigma, St. Louis, MO) durante toda la noche (16-20 horas). Los niveles de MMP-9 en el medio se midieron mediante el uso un kit ELISA (GE Healthcare, Piscataway, NJ), y las citocinas se ensayaron mediante el uso un kit de quimiocinas/citocinas humanas Milliplex MAP (Millipore, Billerica, MA). Los analitos se cuantificaron mediante el uso un Luminex 100™ IS. Los datos se analizaron mediante un método logístico de cinco parámetros.
Los resultados se muestran en las Figuras 6-10. (+)-KDT501 inhibió la expresión inducida por TNF-a y LPS de MMP-9 (Figura 6), IL-1p (Figura 7), MCP-1 (Figura 8), Ra Nt ES (Figura 9) y MIP-1a (Figura 10) en función de la dosis.
Ejemplo 4: Efecto de (+)-KDT501 sobre los factores inflamatorios mediados por IL-1p en RASF:
Se evaluó el efecto antiinflamatorio de (+)-KDT501 en fibroblastos sinoviales de artritis reumatoide (RASF).
Se cultivaron células RASF humanas (Asterand, Detroit, MI) y se mantuvieron en medio DMEM/F12 (1:1) en presencia de 10 % de suero fetal bovino. Las células se subcultivaron en placas de 24 pocillos a una densidad de 1x104 células por pocillo y se dejó alcanzar una confluencia del 70-80 % en dos días. Las células confluentes en medio libre de suero a una concentración final de 0,1 % DMSO se incubaron durante una hora con (+)-KDT501 en concentraciones variables (50, 25, 12,5 y 6,25 pM), luego se estimularon con IL-1p (10 ng/mL) durante 20-24 horas. Los niveles de MMP-13 se midieron en el medio mediante el uso un kit ELISA (GE Healthcare, Piscataway, NJ) y PGE2 se midieron mediante el uso un Immuno Assay Kit (Assay Designs, Ann Arbor, MI).
Los resultados se muestran en las Figuras 11 y 12. (+)-KDT501 inhibió la expresión de PGE inducida por IL-1p2 (Figura 11) y M M P-13 (Figura 12).
Ejemplo 5: Unión competitiva de KDT501 a PPARy, SCN2Ay AGTR2:
Se evaluó la unión competitiva de (+)-KDT501 al receptor gamma activado por proliferador de peroxisomas (PPARy), canal de sodio dependiente de voltaje tipo II (SCN2A) y receptor de angiotensina II tipo II (AGTR2) en presencia de los agonistas rosiglitazona, veratridina y angiotensina-II, respectivamente.
Para PPARy, se incubaron homogenizados de membrana celular (8 pg de proteína) durante 120 minutos a 4 °C con 5 nM [3H] rosiglitazona en presencia o ausencia de (+)-KDT501 en un tampón que contenía Tris-HCl 10 mM (pH 8,2), KCl 50 mM y DTT 1 mM, y se determinó la unión no específica en presencia de 10 pM de rosiglitazona.
Para SCN2A, se incubaron homogenizados de membrana celular de corteza cerebral (200 pg de proteína) durante 60 minutos a 22 °C con 10 nM [3H] batracotoxinina en presencia o ausencia de (+)-KDT501 en un tampón que contiene 50 mM Hepes/Tris (pH 7,4), cloruro de colina 130 mM, Kcl 5,4 mM, MgSO40,8 mM, 1 g/l de glucosa, 0,075 g/l de veneno de escorpión y 0,1 % de BSA, y se determinó la unión inespecífica en presencia de veratridina 300 pM.
Para AGTR2, se incubaron homogenizados de membrana celular (5 pg de proteína) durante 240 min a 37 °C con 0,01 nM [125I] CGP 42112A (agonista del receptor de angiotensina II tipo II) en presencia o ausencia de (+)-KDT501 en un tampón que contiene 50 mM Tris-HCl (pH 7,4), 5 mM MgCl2 , e Dt A 1 mM y BSA al 0,1 %, y se determinó la unión inespecífica en presencia de 1 pM de angiotensina II.
Después de la incubación, las muestras se filtraron rápidamente al vacío a través de filtros de fibra de vidrio (GF/B, Packard) remojados previamente con PEI al 0,3 % y se enjuagaron varias veces con Tris-HCl 50 mM helado mediante el uso un cosechador celular de 96 muestras (Unifilter, Packard). Los filtros se secaron y luego se contó la radioactividad en un contador de centelleo (Topcount, Packard) mediante el uso un cóctel de centelleo (Microscint 0, Packard).
Los resultados se resumen en la Tabla 5. (+)-KDT501 mostró la capacidad de unirse a las tres dianas en presencia de sus ligandos agonistas, la unión de mayor afinidad con ocurrió con PPARy.
Tabla 5:

Los experimentos de unión se repitieron como se describió anteriormente mediante el uso (-)-KDT501 en lugar de (+)-KDT501. Estos resultados se resumen en la Tabla 6. Al igual que (+)-KDT501, (-)-KDT501 mostró la capacidad de unirse a las tres dianas en presencia de sus ligandos agonistas. Sin embargo, el IC50 y Ki de (-)-KDT501 para los tres objetivos fue mayor que el mostrado por (+)-KDT501.
Tabla 6 :

Ejemplo 6 : Efecto de KDT501 sobre la actividad PPAR:
El efecto funcional de (+)-KDT501 sobre la actividad de PPARa, PPAR8 y PPARy se evaluó mediante el uso un ensayo indicador de PPAR (INDIGO Biosciences, PA). Este ensayo usa células de mamíferos no humanos diseñadas para proporcionar un alto nivel de expresión constitutiva de PPARa, PPARS o PPARy que contienen un gen de informe de luciferasa específico para el PPAR apropiado. Después de la activación por la unión de agonistas, PPAR induce la expresión del gen indicador de luciferasa. Por tanto, la actividad de luciferasa proporciona un sustituto para medir la actividad de PPAR en las células tratadas con agonistas.
Las células indicadoras se sembraron en una placa de 96 pocillos a 100 jL por pocillo y se añadieron a cada pocillo 100 |jl de (+)-KDT501 a diversas concentraciones finales (25, 12,5, 6,25, 3,l25, 1 , 5 6 y 0,78 jM ). Para el ensayo de PPARy, se usaron rosiglitazona (1, 0,5, 0,25, 0,125, 0,063 y 0,031 jM ) y telmisartán (10, 5, 2,5, 1,25 y 0,625 jM ) como controles positivos. Para el ensayo PPARa, se usó GW590735 (10, 5, 1, 0,5, 0,25, 0,125, 0,063 y 0,031 jM ) como control positivo y se usó rosiglitazona (1 , 0,5, 0,25, 0,125, 0,063 y 0,031 jM ) como un control negativo. Para el ensayo PPARS, se usó GW0742 (1 , 0,5, 0,25, 0,125, 0,0625, 0,031, 0 , 0 1 6 y 0 , 0 0 8 jM ) como control positivo y se usó rosiglitazona (1 , 0,5, 0,25, 0,125, 0,063 y 0,031 jM ) como un control negativo. También se usó DMSO al 0,1 % como control negativo para cada ensayo. Las placas se incubaron durante 20 horas en una incubadora humidificada a 37 °C y 5 % de CO2. Después de la incubación, se desechó el medio celular y las células se trataron con 100 jL de reactivo de detección de luciferasa durante 15 minutos. Las placas se analizaron mediante el uso un luminómetro (Victor2, Perkin Elmer). La unidad de luz relativa promedio (RLU) y la desviación estándar se analizaron mediante el uso GraphPad Prism.
En el ensayo de PPARy, el control agonista positivo (rosiglitazona) aumentó la actividad de PPARy como se esperaba. Telmisartan, un agonista parcial conocido de PPARy, también aumentó la actividad de PPARy. (+)-KDT501 aumentó la actividad de PPARy de una manera estadísticamente significativa consistente con la actividad como agonista parcial de PPARy (Figura 13). (+)-KDT501 tuvo poco o ningún efecto sobre la actividad de PPARa y PPARS (Figuras 14 y 15).
Los ensayos de actividad PPAR se repitieron como se describió anteriormente mediante el uso tanto (+)-KDT501 (12,5, 6,25, 3,125, 1,56 y 0,78 jM ) como (-)-KDT501 (25, 12,5, 6,25, 3,125, 1,56 y 0,78 jM ). (-)-KDT501 aumentó la actividad de PPARy, pero en un grado significativamente menor que (+)-KDT501 (Figura 16). Los resultados para ambos compuestos se resumen en la Tabla 7.
Tabla 7:

Ejemplo 7: Efecto de (+)-KDT501 sobre la actividad del receptor GPR120:
El efecto de (+)-KDT501 sobre la actividad de GPR120 se evaluó mediante el uso de células CHO - K1/GPR120/Gai5 que expresan de forma estable GPR120. La actividad de GPR120 se midió mediante la respuesta de calcio intracelular.
Las células CHO - K1/GPR120/Gai5 se pasaron regularmente para mantener una viabilidad celular óptima y se cultivaron en F12 de Ham suplementado con 10 %, de suero fetal bovino 200|jg/mL dezeocina e higromicina 100 jg/mL. Las células se sembraron en una placa de fondo transparente de pared negra de 384 pocillos a una densidad de 20.000 células por pocillo en 20 jL de medio de crecimiento 18 horas antes del inicio del experimento y se mantuvieron a 37 °C/5 % de CO2. Las células se cargaron con calcio-4 y se obtuvieron lecturas de fluorescencia de la línea de base de 1 segundo a 20 segundos mediante el uso un lector de placas de imágenes fluorescentes (FLIPR). A los 20 segundos, se añadió (+)-KDT501 (5x concentración final) a la placa de lectura; y se monitoreó la señal de fluorescencia durante 100 segundos (21 segundos a 120 segundos). Se usaron como controles ácido a-linolénico (LA), ácido eicosapentaenoico (EPA) y ácido docosahexaenoico (DHA).
La adquisición y el análisis de datos se realizaron con ScreenWorks (versión 3.1) y se exportaron a Excel. Se calculó el valor medio de fluorescencia para los primeros 20 segundos como lectura de referencia. Los valores de intensidad de la unidad fluorescente relativa (ARFU) se calcularon con las unidades fluorescentes máximas (21s a 120s), restando la lectura de la línea de base. Se usó el asistente de análisis de datos descrito por GenScript para analizar EC50. Las curvas de respuesta a la dosis del antagonista se ajustaron mediante el uso de la ecuación logística de cuatro parámetros GraphPad Prism 4 Y =Inferior (Superior-Inferior)/(1 10A ((LogIC50-X)*HillSlope)), donde X es el logaritmo de concentración y Y es la respuesta.
(+)-KDT501 mostro actividad agonística de GPR120 con un EC50 valor de 30,3 jM (Figura 17). La CE50 los valores para a-LA, DHA y EPA fueron 36,1 jM , 35,3 jM y 116,9 jM , respectivamente.
Ejemplo 8: Efecto de KDT501 sobre la actividad de DAPK1:
Se evaluó el efecto de (+)-KDT501 y (-)-KDT501 sobre la actividad de la proteína quinasa 1 asociada a la muerte (DAPK1).
Se incubó DAPK1 humano con 8 mM MOPS, pH 7,0, EDTA 0,2 mM, sustrato de péptido ZIP 250 jM (KKLNRTLSFAEPG), 10 mM MgOAc y [y-33P-ATP] (actividad específica aproximadamente 500 cpm/pmol, concentración como sea necesaria). Se añadieron (+)-Kd T501 o (-)-KDT501 a concentraciones variables (100, 30, 10, 1, 0,3, 0,1, 0,03, 0,01jM) a la mezcla de reacción. La reacción se inició por medio de la adición de una mezcla de MgATP. Después de la incubación durante 40 minutos a temperatura ambiente, la reacción se detuvo mediante la adición de una solución de ácido fosfórico al 3 %. Se vertieron 10 jL de mezcla de reacción sobre una esterilla filtrante P30, se lavaron tres veces durante cinco minutos en ácido fosfórico 75 mM y una vez en metanol antes, después se secaron y se sometieron a recuento de centelleo.
Los resultados se muestran en la Figura 18. (+)-KDT501 y (-)-KDT501 inhibieron la actividad de DAPK1 de una manera dependiente de la dosis. El IC calculado50 los valores para (+)-KDT501 y (-)-KDT501 fueron 2,65 jM y 40,9 jM , respectivamente, lo que representa aproximadamente una diferencia de 15 veces entre los dos compuestos. Estos datos sugieren que (+)-KDT501 es más efectivo para inhibir la actividad de DAPK1 que (-)-KDT501.
Ejemplo 9: Efecto de (+)-KDT501 sobre los niveles de glucosa y triglicéridos en un modelo de diabetes en ratas:
64 ratas Zucker diabéticas grasas (ZDF) macho de seis semanas de edad con niveles de glucosa de 175 a 300 mg/dL se estudiaron al azar y se dividieron en ocho grupos de dosificación: 1) vehículo solo(metilcelulosa al 0,5 % (p/v), 0,2 % (p/v)de Tween 80; control negativo), 2) (+)-KDT501 25 mg/kg, 3) (+)-KDT501 50 mg/kg, 4) (+)-KDT501 100 mg/kg,5) (+)-KDT501 200 mg/kg, 6) metformina 200 mg/kg (control positivo), 7) metformina 200 mg/kg y (+)-KDT501 100 mg/kg y 8) pioglitazona 30 mg/kg (control positivo). Los fármacos se administraron por vía oral dos veces al día durante 33 días.
Los animales se pesaron aleatoriamente y una vez por semana a partir de entonces, con las dosis de administración calculadas en base a las mediciones de peso corporal más recientes. Las muestras de sangre se recolectaron por sangrado de la cola tres días antes de la aleatorización, en la aleatorización y los días 15 y 29 después del inicio del tratamiento. Estas muestras de sangre se usaron para realizar evaluaciones de glucosa en sangre total mediante el uso un glucómetro, así como también para medir los niveles de triglicéridos en sangre. La composición corporal se evaluó por qNMR los días 2 y 29 después del inicio del tratamiento. Se realizó una prueba de tolerancia a la glucosa oral (OGTT) los días 31 y 32 después del inicio del tratamiento. Las ratas se sometieron a un ayuno nocturno antes de la OGTT, y se realizó la extracción de sangre de la cola para glucosa e insulina antes del bolo de glucosa a los 15, 30, 60, 90 y 120 minutos después del bolo de glucosa. El bolo de glucosa fue de 2 g de dextrosa/kg de peso corporal. El día 33, se realizó una punción cardíaca para el análisis de PK.
Los resultados de glucosa en sangre total se exponen en la Figura 19. Como era de esperar, las ratas del control negativo mostraron un aumento significativo y constante de los niveles de glucosa en sangre total durante el transcurso del estudio. Las ratas del control positivo que recibieron pioglitazona mostraron una disminución significativa en los niveles de glucosa, mientras que las ratas del control positivo que recibieron metformina mostraron un ligero aumento en los niveles de
glucosa. Las ratas que recibieron (+)-KDT501 en dosis de 25, 50 o 100 mg/kg mostraron niveles de glucosa que fueron casi los mismos que los observados en las ratas del control negativo. Sin embargo, las ratas que recibieron (+)-KDT501 a una dosis de 200 mg/kg dos veces al día mostraron una disminución significativa en los niveles de glucosa que fue mucho mayor que la observada en las ratas de control de metformina y casi idéntica a la observada en las ratas de control de pioglitazona.
Los resultados del nivel de triglicéridos se exponen en la Figura 20. Las ratas que recibieron metformina mostraron un aumento en los niveles de triglicéridos en los días 15 y 29 frente a las ratas de control negativo, mientras que las ratas que recibieron pioglitazona mostraron una disminución significativa. Las ratas que recibieron (+)-KDT501 en dosis de 25, 50 o 100 mg/kg mostraron niveles de triglicéridos que fueron similares a los observados en las ratas del control negativo. Sin embargo, las ratas que recibieron (+)-KDT501 una dosis de 200 mg/kg dos veces al día mostraron una disminución significativa en los niveles de triglicéridos que fue casi tan grande que la observada en las ratas del control de pioglitazona.
Ejemplo 10: Efecto de (+)-KDT501 sobre la glucosa, la insulina, la hemoglobina AlC (HbAlC) y la masa grasa en un modelo de ratón con obesidad inducida por una dieta alta en grasas (DIO):
Se seleccionaron al azar ratones machos de 14 semanas de edad y se dividieron en siete grupos de dosificación con 16 animales en cada grupo: 1) vehículo solo(metilcelulosa al 0,5 % (p/v), Tween 80 al 0,2 % (p/v); control negativo), 2) (+)-KDT501 25 mg/kg, 3) (+)-KDT501 50 mg/kg, 4) (+)-KDT501 100 mg/kg, 5) (+)-KDT501 200 mg/kg, 6) 200 mg/kg metformina (control positivo) y 7) 30 mg/kg pioglitazona (control positivo). Los fármacos se administraron por vía oral dos veces al día durante 30 días.
Los animales se pesaron aleatoriamente y una vez por semana a partir de entonces, con las dosis de administración calculadas en base a las mediciones de peso corporal más recientes. Los ratones se mantuvieron con una dieta alta en grasas (40 % Kcal) (TD95217; preparado por Covance Laboratories, Greenfield, IN). La composición corporal se evaluó mediante qNMR los días -5 y 28 después del inicio del tratamiento. Se colecto sangre el día 29 después del inicio del tratamiento y se midieron los niveles de hemoglobina AlC (HbAlC). Se realizó una prueba de tolerancia a la glucosa oral (OGTT) el día 30 después del inicio del tratamiento. Los ratones se sometieron a un ayuno nocturno antes de la OGTT, y se realizó la extracción de sangre de la cola para glucosa e insulina antes del bolo de glucosa y a los 15, 30, 60, 90 y 120 minutos después del bolo de glucosa. El bolo de glucosa fue de 2 g de dextrosa/kg de peso corporal. Los resultados de OGTT se evaluaron de acuerdo con las mediciones del área bajo la curva (AUC) y la prueba de Dunnett.
Los resultados de glucosa en sangre de la OGTT se exponen en la Figura 21. Los ratones del control positivo que recibieron pioglitazona o metformina mostraron una disminución significativa en los niveles de glucosa. De manera similar, los ratones que recibieron (+)-KDT501 en dosis de 100 o 200 mg/kg mostraron una disminución significativa en los niveles de glucosa.
Los resultados de insulina de la OGTT se exponen en la Figura 22. Los ratones del control positivo que recibieron pioglitazona o metformina mostraron una disminución significativa en los niveles de insulina. De manera similar, los ratones que recibieron (+)-KDT501 a una dosis de 200 mg/kg mostraron una disminución significativa en los niveles de insulina.
Los resultados de HbAlC se exponen en la Figura 23. Se observó una diferencia significativa en la HbAlC entre la dosificación por grupos, control negativo vehículo solo y grupos de metformina, control positivo. La administración de (+)-KDT501 dio como resultado una tendencia hacia la inhibición de HbAlC.
Los resultados de masa grasa se exponen en la Figura 24. Los ratones del control positivo que recibieron metformina mostraron una reducción en la masa grasa, mientras que los ratones de control positivo que recibieron pioglitazona no mostraron diferencias. La administración de (+)-KDT501 a 100 o 200 mg/kg dio como resultado una diferencia significativa en la masa grasa frente a los ratones de control negativo.
Ejemplo 11: Síntesis asimétrica de (+)-KDT500 o (-)-KDT500 a partir de lupulona:

Etapal: Síntesis de tetrahidro desoxihumulona (VI) a partir de lupulona (II).
Se prepara una solución al 10 % (p/v) de lupulona en metanol. Se añade una cantidad catalítica de HCl concentrado (ac) y Pd al 10 %/C. La mezcla resultante se agita bajo hidrógeno (1,1 atm) durante 3 horas y la reacción se controla por HPLC. Una vez completada la reacción, el catalizador se elimina por filtración y el filtrado se concentra al vacío para producir tetrahidrodesoxihumulona (VI), que puede usarse para la siguiente reacción sin purificación adicional.
Etapa 2A: Síntesis asimétrica de (-)-tetrahidro humulona ((-)-XI) a partir de tetrahidro desoxihumulona (VI).
Opción 2A(i): Síntesis asimétrica de (-)-tetrahidro humulona ((-)-XI) a partir de tetrahidro desoxihumulona (VI).
Se prepara una solución al 14 % (p/v) de tetrahidro desoxihumulona (VI) en THF anhidro y se enfría a -78 °C mediante el uso un baño de acetona/hielo seco. Se enfría diisopropiletilamina (DIEA) (1,2 eq) y se añade gota a gota a la solución de (VI). Esta solución se transfiere mediante una cánula al complejo [(-)-esparteína]2-Cu2-O2) en THF derivado de 2,2 eq Cu(CH3CN)4PF6 y 2,3 eq de ligando de diamina bajo argón a -78 °C. La mezcla resultante se agita a -78 °C o la temperatura deseada durante 16 horas. La reacción se detiene con 4 volúmenes de ácido sulfúrico acuoso al 5 % (en peso). La mezcla se extrae con acetato de etilo (x3) y los extractos combinados se lavan con ácido sulfúrico al 5 %, agua y salmuera, se secan sobre sulfato de sodio y se concentran al vacío (Dong 2008).
Opción 2A(ii): Síntesis asimétrica de (-)-tetrahidro humulona ((-)-XI) a partir de tetrahidro desoxihumulona (VI).
Se construye y purifica una enzima mutante P450 adecuada capaz de convertir tetrahidro desoxihumulona (VI) en (-)-tetrahidro humulona ((-)- XI) y se purifica como se describe en Patente de Estados Unidos No. 7,704,715 y referencias contenidas en el mismo. Una reacción típica contiene de 1 a 4 pM de enzima de dominio hemo P450 purificada y 1 a 2 mM de tetrahidro desoxihumulona (VI) en 500 pL de tris-HCl 100 mM, pH 8,2. Se combinan los siguientes: 713 pL de agua purificada, 200 pL de tris-HCl 0,100 M pH 8,2, concentración final 100 mM, 2 pL de tetrahidro desoxihumulona (VI) en DMSO (concentración final 1 mM).
La reacción se inicia mediante la adición de 1 a 10 mM de H2O2 y se monitoreo por HPLC para la máxima conversión. La reacción se detiene mediante la adición de 7,5 pL de HCl 6M. La (-)-tetrahidro humulona ((-)-XI) se obtiene mediante un tratamiento extractivo y se purifica como se describió anteriormente.
Opción 2A(iii): Síntesis asimétrica de (-)-tetrahidro humulona ((-)-XI) a partir de tetrahidro desoxihumulona (VI).
Un microbio capaz de convertir tetrahidro desoxihumulona (VI) en (-)-tetrahidro humulona ((-)- XI se produce de acuerdo con la Publicación de Patentes de EE. UU. Número 2010/0144547 y las referencias contenidas en esta. Este microbio se cultiva en presencia de tetrahidro desoxihumulona (VI) de acuerdo con las siguientes condiciones de fermentación: Cultivo de 500 mL con medio LB con 30 mg/L de kanamicina. En una DO600 de 1,0, las células se concentran hasta una DO final600 de 5,0 e inducida con IPTG 1 mM. Se añade tetrahidro desoxihumulona (VI) hasta una concentración final de 1 mM y 4 mM. En diferentes momentos, las muestras de cultivo se colectan, se centrifugan, se filtran y se inyectan en un HPLC (5 pL) y se determina una conversión máxima. Después de la conversión máxima, el caldo se acidifica a un pH de 2,0, se extrae con acetato de etilo, se seca y se redisuelve en hexanos. La (-)-tetrahidro humulona ((-)-XI) se obtiene mediante un tratamiento extractivo y se purifica como se describió anteriormente.
Una variedad de medios de fermentación tales como medios de fermentación LB, F1 o TB bien conocidos en la técnica que pueden usarse o adaptarse para su uso con modalidades de la invención descritas en la presente descripción, incluidos los medios LB, TB y F1. Otros medios adaptados a organismos en crecimiento tales como A. terreus y M. pilosus también son bien conocidos en la técnica (ver, por ejemplo, Miyake 2006; Hajjaj 2001).
Etapa 2B: Síntesis asimétrica de (+)-tetrahidro humulona ((+)-XI) a partir de tetrahidro desoxihumulona (VI).
Opción 2B(i): Síntesis asimétrica de (+)-tetrahidro humulona ((+)-XI) a partir de tetrahidro desoxihumulona (VI).
Se prepara una solución al 14 % (p/v) de tetrahidro desoxihumulona (VI) en THF anhidro y se enfría a -78 °C mediante el uso un baño de acetona/hielo seco. Se enfría diisopropiletilamina (DIEA) (1,2 eq) y se añade gota a gota a la solución de (VI). Esta solución se transfiere mediante una cánula al complejo [(+)-esparteína]2-Cu2-O2) en THF derivado de 2,2 eq de Cu(CH3CN)4PF6 y 2,3 eq de ligando de diamina bajo argón a -78 °C. La mezcla resultante se agita a -78 °C o la temperatura deseada durante 16 horas. La reacción se detiene con 4 volúmenes de ácido sulfúrico acuoso al 5 % (en peso). La mezcla se extrae con acetato de etilo (x3) y los extractos combinados se lavan con ácido sulfúrico al 5 %, agua y salmuera, se secan sobre sulfato de sodio y se concentran al vacío (Dong 2008).
Opción 2B(ii): Síntesis asimétrica de (+)-tetrahidro humulona ((+)-XI) a partir de tetrahidro desoxihumulona (VI).
Una enzima mutante P450 capaz de convertir tetrahidro desoxihumulona (VI) en (+)- tetrahidro humulona ((+)-XI) se construye y purifica como se describe en Patente de Estados Unidos No. 7,704,715 y referencias contenidas en el mismo. Una reacción típica contiene de 1 a 4 pM de enzima de dominio hemo P450 purificada y 1 a 2 mM de tetrahidro desoxihumulona (VI) en 500 pL de Tris-HCl 100 mM, pH 8,2. Se combinan los siguientes: 713 pL de agua purificada, 200 pL de tris-HCl 0,100 M pH 8,2, concentración final 100 mM, 2 pL de tetrahidro desoxihumulona (VI) en DMSO (concentración final 1 mM).
La reacción se inicia mediante la adición de 1 a 10 mM de H2O2 y se monitorea mediante HPLC para la máxima conversión. La reacción se detiene mediante la adición de 7,5 pL de HCl 6M. La (+)-tetrahidro humulona ((+)- XI) se obtiene mediante un tratamiento extractivo y se purifica como se describió anteriormente.
Opción 2B(iii): Síntesis asimétrica de (+)-tetrahidro humulona ((+)-XI) a partir de tetrahidro desoxihumulona (VI).
Un microbio capaz de convertir tetrahidro desoxihumulona (VI) en (+)-tetrahidro humulona ((+)- XI) se produce de acuerdo con la Publicación de Patentes de EE. UU. Número 2010/0144547 y las referencias contenidas en esta. El microbio se cultiva en presencia de tetrahidro desoxihumulona (VI) de acuerdo con las siguientes condiciones de fermentación: Cultivo de 500 mL con medio LB con 30 mg/L de kanamicina. A DO600 de 1,0, las células se concentraron hasta una DO final600 de 5,0 e inducida con 1 mM IPTG. Se añade tetrahidro desoxihumulona (VI) hasta una concentración final de 1 mM y 4 mM. En diferentes momentos, las muestras de cultivo se colectan, se centrifugan, se filtran y se inyectan en un HPLC (5 pL) y se determina una conversión máxima. Después de la conversión máxima, el caldo se acidifica a pH 2,0, se extrae con acetato de etilo, se seca y se vuelve a disolver en hexanos. La (-)-tetrahidro humulona se obtiene mediante un tratamiento extractivo y se purifica como se describió anteriormente.
Una variedad de medios de fermentación tales como medios de fermentación LB, F1 o TB bien conocidos en la técnica pueden usarse o adaptarse con modalidades de la invención descritas en la presente descripción, incluidos los medios LB, TB y F1. Otros medios adaptados a organismos en crecimiento tales como A. terreus y M. pilosus también son bien conocidos en la técnica (ver, por ejemplo, Miyake 2006; Hajjaj 2001).
Etapa 3A: Síntesis asimétrica de (+)-KDT500 a partir de (-)-tetrahidro humulona ((-)-XI).
Se prepara una solución al 50 % (p/v) de (-)- tetrahidrohumulona ((-)- XI) en agua y se calienta a 85 °C con agitación en un recipiente sellado. Se añade MgSO4 (0,6 eq.) a la solución durante 5 minutos con agitación continua. Al recipiente sellado, se le añade gota a gota una solución de KOH (38,25 % (p/v), 1,0 eq) con agitación. La temperatura de reacción se mantiene a 85 °C durante las siguientes 3 horas o hasta que se completa la isomerización de acuerdo con lo juzgado por HPLC. El ácido libre de (+)-KDT - 500 se forma añadiendo 2 volúmenes de isopropanol, 2 volúmenes de agua y 1 equivalente de H2SO4 a la mezcla de reacción. Esta mezcla se mezcla hasta que esté completamente homogénea, momento en el que se usan 2 volúmenes de diclorometano para extraer la mezcla de reacción (3x). El extracto orgánico se concentró al vacío, se disolvió nuevamente en hexanos, se secó sobre Na2SO4, se filtró, se concentró al vacío y se secó durante la noche a 0,070 mbar. El producto final es la forma de ácido libre de (+) DR&T-500. Puede llevarse a cabo una purificación adicional mediante epimerización, cromatografía, formación de complejos y/o cristalización.
Etapa 3B: Síntesis asimétrica de (-)-KDT500 a partir de (+)-tetrahidro humulona ((+)-XI).
Se prepara una solución al 50 % (p/v) de (+)- tetrahidrohumulona ((+)-XI) en agua y se calienta a 85 °C con agitación en un recipiente sellado. Se añade MgSO4 (0,6 eq.) a la solución durante 5 minutos con agitación continua. Al recipiente sellado, se le añade gota a gota una solución de KOH (38,25 % (p/v), 1,0 eq) con agitación. La temperatura de reacción se mantiene a 85 °C durante las siguientes 3 horas o hasta que se completa la isomerización de acuerdo con lo juzgado por HPLC. El ácido libre de (-)-KDT500 se forma añadiendo 2 volúmenes de isopropanol, 2 volúmenes de agua y 1 equivalente de H2SO4 a la mezcla de reacción. Esta mezcla se mezcla hasta que esté completamente homogénea, momento en el que se usan 2 volúmenes de diclorometano para extraer la mezcla de reacción (3x). El extracto orgánico se concentró al vacío, se disolvió nuevamente en hexanos, se secó sobre Na2SO4, se filtró, se concentró al vacío y se
secó durante la noche a 0,070 mbar. El producto final es la forma de ácido libre de (-) KDT-500. Puede llevarse a cabo una purificación adicional mediante epimerización, cromatografía, formación de complejos y/o cristalización.
Ejemplo 12: Síntesis asimétrica de (+)-KDT500 o (-)-KDT500 a partir de desoxihumulona:

Etapa 1A: Síntesis asimétrica de (-)-humulona ((-)-IV) a partir de desoxihumulona (I).
Opción 1A(i): Síntesis asimétrica de (-)-humulona ((-)-IV) a partir de desoxihumulona (I).
Se prepara una solución al 14 % (p/v) de desoxihumulona (I) en THF anhidro y se enfría a -78 °C mediante el uso un baño de acetona/hielo seco. Se enfría diisopropiletilamina (DIEA) (1,2 eq) y se añade gota a gota a la solución de (VI). Esta solución se transfiere mediante una cánula al complejo [(-)-esparteína]2-Cu2-O2) en THF derivado de 2,2 eq Cu(CH3CN)4PF6 y 2,3 eq de ligando de diamina bajo argón a -78 °C. La mezcla resultante se agita a -78 °C o la temperatura deseada durante 16 horas. La reacción se detiene con 4 volúmenes de ácido sulfúrico acuoso al 5 % (en peso). La mezcla se extrae con acetato de etilo (x3) y los extractos combinados se lavan con ácido sulfúrico al 5 %, agua y salmuera, se secan sobre sulfato de sodio y se concentran al vacío (Dong 2008).
Opción 1A(ii): Síntesis asimétrica de (-)-humulona ((-)-IV) a partir de desoxihumulona (I).
Una enzima mutante P450 capaz de convertir desoxihumulona (I) en (-)-humulona ((-)-IV) se construye y purifica como se describe en la Patente de Estados Unidos No. 7,704,715 y las referencias contenidas en la misma. Una reacción típica contiene 1 a 4 pM de enzima de dominio hemo P450 purificada y 1 a 2 mM de humulona (I) en 500 pl de tris-HCl 100 mM, pH 8,2. Se combinan los siguientes: 713 pl de agua purificada, 200 pl de tris-HCl 0,100 M pH 8,2, concentración final 100 mM, 2 pl de desoxihumulona (I) en DMSO (concentración final 1 mM).
La reacción se inicia mediante la adición de 1 a 10 mM de H2O2 y monitorizado mediante HPLC para la máxima conversión. La reacción se detiene mediante la adición de 7,5 pl de HCl 6M. La (-)-humulona ((-)-IV) se obtiene mediante un tratamiento extractivo y se purifica como se describió anteriormente.
Opción 1A(iii): Síntesis asimétrica de (-)-humulona ((-)-IV) a partir de desoxihumulona (I).
Un microbio capaz de convertir humulona (I) en (-)-humulona ((-)-IV) se produce de acuerdo con la Publicación de Patentes de EE. UU. Número 2010/0144547 y las referencias contenidas en esta. Este microbio luego se cultiva en presencia de desoxihumulona (I) de acuerdo con las siguientes condiciones de fermentación: Cultivo de 500 mL con medio LB con 30 mg/L de kanamicina. En una DO600 de 1,0, las células se concentran hasta una DO final600 de 5,0 e inducida con IPTG 1 mM. Se añade desoxihumulona (I) hasta una concentración final de 1 mM y 4 mM. En diferentes momentos, las muestras de cultivo se colectan, se centrifugan, se filtran y se inyectan en un HPLC (5 pl) y se determina una conversión máxima. Después de la conversión máxima, el caldo se acidifica a pH 2,0, se extrae con acetato de etilo, se seca y se vuelve a disolver en hexanos. La (-)-humulona ((-)-IV) se obtiene mediante un tratamiento extractivo y se purifica como se describió anteriormente.
Una variedad de medios de fermentación tales como medios de fermentación LB, F1 o TB bien conocidos en la técnica pueden usarse o adaptarse con modalidades de la invención descritas en la presente descripción, incluidos los medios LB, TB y F1. Otros medios adaptados a organismos en crecimiento tales como A. terreus y M. pilosus también son bien conocidos en la técnica (ver, por ejemplo, Miyake 2006; Hajjaj 2001).
Etapa 1B: Síntesis asimétrica de (+)-humulona ((+)-IV) a partir de desoxihumulona (I).
Opción 1B(i): Síntesis asimétrica de (+)-humulona ((+)-IV) a partir de desoxihumulona (I).
Se prepara una solución al 14 % (p/v) de desoxihumulona (I) en THF anhidro y se enfría a -78 °C mediante el uso un baño de acetona/hielo seco. Se enfría diisopropiletilamina (DIEA) (1,2 eq) y se añade gota a gota a la solución de (VI). Esta solución se transfiere mediante una cánula al complejo [(+)-esparteína]2-Cu2-O2) en THF derivado de 2,2 eq de Cu(CH3CN)4PF6 y 2,3 eq de ligando de diamina bajo argón a -78 °C. La mezcla resultante se agita a -78 °C o la temperatura deseada durante 16 horas. La reacción se detiene con 4 volúmenes de ácido sulfúrico acuoso al 5 % (en peso). La mezcla se extrae con acetato de etilo (x3) y los extractos combinados se lavan con ácido sulfúrico al 5 %, agua y salmuera, se secan sobre sulfato de sodio y se concentran al vacío (Dong 2008).
Opción 1B(ii): Síntesis asimétrica de (+)-humulona ((+)-IV) a partir de desoxihumulona (I).
Una enzima mutante P450 capaz de convertir desoxihumulona (I) en (+)-humulona ((+)-IV) se construye y purifica como se describe en la Patente de Estados Unidos No. 7,704,715 y las referencias contenidas en la mismo. Una reacción típica contiene 1 a 4 pM de enzima de dominio hemo P450 purificada y 1 a 2 mM de humulona (I) en 500 pl de tris-HCl 100 mM, pH 8,2. Se combinan los siguientes: 713 pl de agua purificada, 200 pl de tris-HCl 0,100 M pH 8,2, concentración final 100 mM, 2 pl de desoxihumulona (I) en DMSO (concentración final 1 mM).
La reacción se inicia mediante la adición de 1 a 10 mM de H2O2 y se monitorea mediante HPLC para la máxima conversión. La reacción se detiene mediante la adición de 7,5 pl de HCl 6M. La (+)-humulona ((+)-IV) se obtiene mediante un tratamiento extractivo y se purifica como se describió anteriormente.
Opción 1B(iii): Síntesis asimétrica de (+)-humulona ((+)-IV) a partir de desoxihumulona (I).
Un microbio capaz de convertir desoxihumulona (I) en (+)-humulona ((+)-IV) se produce de acuerdo con la Publicación de Patentes de EE. UU. Número 2010/0144547 y las referencias contenidas en esta. Este microbio se cultiva luego en presencia de (+)-humulona ((+)-IV) de acuerdo con las siguientes condiciones de fermentación: Cultivo de 500 mL con medio LB con 30 mg/L de kanamicina. En una DO600 de 1,0, las células se concentran hasta una DO final600 de 5,0 e inducida con IPTG 1 mM. Se añade desoxihumulona (I) hasta una concentración final de 1 mM y 4 mM. En diferentes momentos, las muestras de cultivo se colectan, se centrifugan, se filtran y se inyectan en un HPLC (5 pl) y se determina una conversión máxima. Después de la conversión máxima, el caldo se acidifica a pH 2,0, se extrae con acetato de etilo, se seca y se vuelve a disolver en hexanos. La (+)-humulona ((+)-IV) se obtiene mediante un tratamiento extractivo y se purifica como se describió anteriormente.
Una variedad de medios de fermentación tales como medios de fermentación LB, F1 o TB bien conocidos en la técnica pueden usarse o adaptarse con modalidades de la invención descritas en la presente descripción, incluidos los medios LB, TB y F1. Otros medios adaptados a organismos en crecimiento tales como A. terreus y M. pilosus también son bien conocidos en la técnica (ver, por ejemplo, Miyake 2006; Hajjaj 2001).
Etapa 2A: Síntesis asimétrica de )-cis isohumulona ((+)-VIII) a partir de (-)-humulona ((-)-IV) (RTP: K008-49)
Se diluyeron 98,9 mg de (-)-humulona ((-)-IV) con 170 pL de agua y se calentó a 85 °C con agitación en un vial de 4 ml tapado. Se añadieron 19,8 mg (0,6 eq) de MgSO4 y se continuó agitando durante 5 minutos. Se añadieron 31 pl (1,03 eq) de una solución de KOH al 38.25 % (p/v) a la solución, y se dejó que la reacción prosiguiera a 85 °C durante las siguientes 3 horas, momento en el cual la reacción se completó de acuerdo con lo juzgado por HPLC. El producto resultante se usó para generar el ácido libre o la sal de Mg de (+)-cis isohumulona ((+)-VIII).
El ácido libre de (+)-cis isohumulona ((+)-VIII) se formó añadiendo 0,5 mL de isopropanol, 0,5 mL de agua y 1 equivalente de H2SO4 a la mezcla de reacción. Esta mezcla se mezcló hasta que fue completamente homogénea, momento en el que se usaron 0,5 ml de diclorometano para extraer la mezcla de reacción 3 veces. El extracto orgánico se concentró al vacío, se volvió a disolver en hexanos, se secó sobre Na2SO4, se filtró y se concentró al vacío y se secó durante la noche a 0,070 mbar, dando como resultado la forma ácida libre de (+)-cis isohumulona ((+)-VIII).
La sal de Mg de (+)-cis isohumulona ((+)-VIII) se formó retirando la mezcla de reacción del calor y añadiendo 0,5 ml de acetato de etilo con agitación hasta que se disolvió por completo. Se añadieron 0,5 ml de agua para separar las capas acuosa y orgánica y para extraer el acetato de etilo. La extracción con acetato de etilo se repitió 2 veces adicionales, y el extracto se concentró al vacío y se secó durante la noche a 0,070 mbar. El producto final fue sal de Mg de (+)-cis isohumulona ((+)-VIII), junto con una pequeña cantidad de impurezas de sal. Puede llevarse a cabo una purificación adicional mediante epimerización, cristalización y/o formación de complejos para eliminar toda la (-)-trans isohumulona.
Etapa 2B: Síntesis asimétrica de (-)-cis isohumulona ((-)-VIII) a partir de (+)-humulona ((+)-IV).
Se diluyeron 1431 mg de (+)-humulona ((+)-IV) con 1 ml de agua y se calentó a 80 °C con agitación en un vial de 4 ml tapado. Se añadieron 273 mg (0,6 eq) de MgSO4 y continuó agitándose durante 5 minutos. Se añadieron 430 pl (1,0 eq) de una solución de KOH al 38,25 % (p/v) a la solución, y se dejó que la reacción prosiguiera a 80 °C durante las siguientes 6 horas, momento en el cual la reacción se completó de acuerdo con lo juzgado por HPLC. El producto resultante puede
usarse para generar el ácido libre o la sal de Mg de (-)-cis isohumulona ((-)-VIII). En este caso, la mezcla de reacción se mantuvo en solución y se llevó directamente a (-) KDT 500 mediante la etapa 3B más abajo.
El ácido libre de (-)-cis isohumulona ((-)-VIII) se formó añadiendo 2 volúmenes de isopropanol, 2 volúmenes de agua y 1 equivalente de H2SO4 a la mezcla de reacción. Esta mezcla se mezcla hasta que esté completamente homogénea, momento en el que se usan 2 volúmenes de diclorometano para extraer la mezcla de reacción (3x). El extracto orgánico se concentró al vacío, se volvió a disolver en hexanos, se secó sobre Na2SO4, y se filtró y se concentró al vacío y se secó durante la noche a 0,070 mbar, dando como resultado la forma ácida libre (-)-cis isohumulona ((-)-VIII).
La sal de Mg de (-)-cis isohumulona ((-)-VIII) se forma retirando la mezcla de reacción del calor y añadiendo 2 volúmenes de acetato de etilo con agitación hasta que se disuelva completamente. Se añaden 2 volúmenes de agua para separar las capas acuosa y orgánica y para extraer el acetato de etilo. Esta extracción con acetato de etilo se repite 2 veces más, y el extracto se concentra al vacío y se seca durante la noche a 0,070 mbar, dando como resultado la sal de Mg de (-)-cis isohumulona ((-)-VIII), junto con una pequeña cantidad de impurezas de sal. Puede llevarse a cabo una purificación adicional mediante epimerización, cristalización, cromatografía y/o formación de complejos para eliminar toda la (+)-trans isohumulona.
Etapa 3A: Síntesis de (+)-KDT500 a partir de (+)-cis isohumulona ((+)-VIII) (RTP: K008-50)
Se disolvieron 96,3 mg de la sal seca de Mg de (+)-cis isohumulona ((+)-VIII) en 1 ml de metanol. Se añadieron 3,1 mg (0,3 eq) de MgO, y la mezcla de reacción se agitó durante cinco minutos. Se añadieron 30 mg de Pd/C al 10 % a la solución y se reanudó la agitación. Se introdujo una atmósfera de hidrógeno burbujeando gas hidrógeno en la solución. Después de 45 minutos, la reacción se consideró completa por HPLC, se detuvo la agitación y se eliminó la atmósfera de hidrógeno abriendo el vial. Se añadió 1 equivalente de ácido sulfúrico a la mezcla de reacción (pH ~1) y la reacción se filtró a través de un filtro de jeringa de 0,2 mm. El recipiente de reacción y los filtros se lavaron 2 veces sucesivamente con acetato de etilo. El filtrado se concentró al vacío, se disolvió nuevamente en hexanos, se secó sobre Na2SO4 , se filtró con una pipeta de algodón, se concentró al vacío y se secó durante la noche a 0,070 mbar. Se produjeron 58,1 mg de (+)-KDT-500. Se llevó a cabo una purificación adicional mediante cromatografía y cristalización.
Etapa 3B: Síntesis de (-)-KDT500 a partir de (-)-cis isohumulona ((-)-VIII).
La mezcla de reacción de la Etapa 2B (~ 1,4 g de (-)-cis isohumulona) se disolvió en 10 ml de metanol y se añadieron 256 mg de MgO y la mezcla de reacción se agitó durante cinco minutos. La solución se filtró y se dividió en 2 soluciones madre iguales en volumen. Se añadieron 34 mg de MgO a la "Reacción 1" seguido de 200 mg de Pd/C al 10 % y se reanudó la agitación. Se introdujo una atmósfera de hidrógeno burbujeando gas hidrógeno en la solución. Después de 1 hora, se consideró que la reacción estaba completa por HPLC, se detuvo la agitación y se eliminó la atmósfera de hidrógeno abriendo el vial. Se añadió 1 equivalente de ácido sulfúrico a la mezcla de reacción (pH ~1) y la reacción se filtró a través de un filtro de jeringa de 0,2 mm. El recipiente de reacción y los filtros se lavaron 2 veces sucesivamente con acetato de etilo. El filtrado se concentró al vacío, se disolvió nuevamente en hexanos, se secó sobre Na2SO4 , se filtró con una pipeta de algodón, se concentró al vacío y se secó durante la noche a 0,070 mbar. Después de la combinación de ambas reacciones se produjeron 933,5 mg de (-)-KDT-500. Se llevó a cabo una purificación adicional mediante cromatografía y cristalización.
Ejemplo 13: Preparación de (+)-humulona y (-)-humulona a partir de (-)-humulona y desoxihumulona, respectivamente:
Etapa 1: Síntesis de (±)-humulona (III) racémica.
Opción 1A: Síntesis de (±)-humulona (III) racémica a partir de (-)-humulona ((-)-IV).
Se disolvió (-)-humulona pura en pineno u otro disolvente adecuado (alcano inerte de alto punto de ebullición, por ejemplo, limoneno, trimetilhexano) para formar una solución del 3 al 10 %. Esta solución se colocó en un tubo de presión, se purgó con argón o nitrógeno para eliminar todo el oxígeno, se selló y se calentó a una temperatura de 125 a 145 °C durante 1 a 20 horas. A continuación, se dejó enfriar la reacción a temperatura ambiente, se eliminó el disolvente a vacío y se purificó (±)-humulona (III) racémica, si era necesario, como se describe en otra parte.
Opción 1B: Síntesis de (±)-humulona (III) racémica a partir de desoxihumulona (I).
La desoxihumulona (I) se disuelve completamente en metanol y se añade 1 eq de acetato de plomo a esta solución seguido de una cantidad catalítica (0,1 eq) de Pd/C al 10 %. La solución se agita y se burbujea con aire. Después de aproximadamente 4 horas, la sal de plomo se separa por filtración y se añade acetato de plomo adicional al filtrado para garantizar la recuperación completa de la (±)-humulona (III) racémica como la sal de plomo. Toda la sal de plomo (amarillo canario) se recoge y se suspende en metanol seguido de la adición de ácido sulfúrico a la solución. El sulfato de plomo forma un precipitado insoluble que se elimina mediante centrifugación. La solución de metanol homogénea se extrae entre hexanos y agua (aproximadamente 2 volúmenes de cada uno). La (±)-humulona (III) racémica se extrae en hexanos y se concentra al vacío para producir la forma de ácido libre de la (±)-humulona (III) racémica.
Etapa 2A: Purificación de (-)-humulona ((-)-IV) a partir de (±)-humulona (III) racémica.
Se formularon soluciones de (±)-humulona (III) racémica en metanol (10 %, p/v) y 1R, 2R-4-ciclohexeno-1,2-diamina en metanol (10 %, p/v), respectivamente. Ambas soluciones se mezclaron en cantidades equimolares (aproximadamente 3:1 en volumen), se evaporó el metanol y el residuo se volvió a disolver en una cantidad mínima de 2-propanool (pueden usarse otros disolventes, como por ejemplo t-butil metil éter, acetonitrilo, acetato de etilo o benceno). Se formaron cristales, que consisten de la sal de amina de (-)-humulona (IV), mientras que la sal de (+)-humulona ((+)-IV) permaneció en solución. Los cristales se filtraron, se secaron y luego se mezclaron con hexanos y ácido sulfúrico ac. para liberar el ácido libre de (-)-humulona ((-)-IV). La capa de hexano se separó, se lavó con salmuera, se secó con sulfato de sodio y se evaporó para producir (-)-humulona ((-)-IV) como se indica por HPLC quiral.
Etapa 2B: Purificación de (+)-humulona ((+)-IV) a partir de (±)-humulona (III) racémica
Se formulan soluciones de ±)-humulona (III) racémica en metanol (10 %, p/v) y 1S,2S-4-ciclohexeno-1,2-diamina en metanol (10 %, p/v), respectivamente. Ambos se mezclan en cantidades equimolares (aproximadamente 3:1 en volumen), el metanol se evapora y el residuo se vuelve a disolver en una cantidad mínima de 2-propanol (pueden usarse otros disolventes, como por ejemplo t-butil metil éter, acetonitrilo, acetato de etilo o benceno). Se formarán cristales, que consisten de la sal de amina de (+)-humulona ((+)-IV), mientras que la sal (-)-humulona ((-)-IV) permanecerá en la solución. Los cristales se filtran, se secan y luego se mezclan con hexanos y ácido sulfúrico ac. para liberar el ácido libre de (-)-humulona ((-)-IV). La capa de hexano se separa, se lava con salmuera, se seca con sulfato de sodio y se evapora para producir (-)-humulona ((-)-IV) como se indica por HPLC quiral.
Ejemplo 14: Preparación de (+)-tetrahidro humulona y (-)-tetrahidro humulona a partir detetrahidro desoxihumulona:

Etapa 1: Síntesis de (±)-tetrahidro humulona racémica (IX) a partir detetrahidro desoxihumulona (VI).
Se prepara una solución al 10 % (p/v) de tetrahidro desoxihumulona (VI) en ácido acético, y se añade una cantidad catalítica de ácido sulfúrico concentrado a la solución. La solución se agita y se añade gota a gota 1 eq de una solución al 30 % (p/p) de peróxido de hidrógeno. Después de 2 horas, la reacción se considera completa mediante el uso de HPLC
y se añaden agua y diclorometano. La (±)-tetrahidro humulona (IX) racémica se extrae en diclorometano y se concentra al vacío para producir la forma de ácido libre de la (±)-tetrahidro humulona (IX) racémica.
Etapa'2A: Purificación de (-)-tetrahidro humulona ((-)-XI) a partir de (±)-tetrahidro humulona (IX) racémica.
Se formulan soluciones de (±)tetrahidro-humulona (IX) racémica en metanol (10 %, p/v) y 1R,2R-4-ciclohexeno-1,2-diamina en metanol (10 %, p/v), respectivamente. Ambas soluciones se mezclan en cantidades equimolares (aproximadamente 3:1 en volumen), el metanol se evapora y el residuo se vuelve a disolver en una cantidad mínima de 2-propanol (pueden usarse otros disolventes, como por ejemplo t-butil metil éter, acetonitrilo, acetato de etilo o benceno). Se formarán cristales, que consisten de la sal de amina de (-)-tetrahidro humulona ((-)-XI), mientras que la sal (+)-tetrahidro humulona ((+)-XI) permanecerá en la solución. Los cristales se filtran, se secan y luego se mezclan con hexanos y ácido sulfúrico ac. para liberar el ácido libre de (-)-tetrahidro humulona ((-)-XI). La capa de hexano se separa, se lava con salmuera, se seca con sulfato de sodio y se evapora a (-)-tetrahidro humulona ((-)-XI) como se indica por HPLC quiral.
Etapa 2B: Purificación de (+)-tetrahidro humulona ((+)-XI) a partir de (±)-tetrahidro humulona racémica
Se formulan soluciones de (±)-tetrahidro humulona (IX) racémica en metanol (10 %, p/v) y 1S, 2S-4-ciclohexeno-1,2-diamina en metanol (10 %, p/v), respectivamente. Ambas soluciones se mezclan en cantidades equimolares (aproximadamente 3:1 en volumen), el metanol se evapora y el residuo se vuelve a disolver en una cantidad mínima de 2-propanol (pueden usarse otros disolventes, como por ejemplo t-butil metil éter, acetonitrilo, acetato de etilo o benceno). Se formarán cristales, que consisten de la sal de amina de (+)-tetrahidro humulona ((+)-XI), mientras que la sal (-)-tetrahidro humulona ((-)-XI) permanecerá en la solución. Los cristales se filtran, se secan y luego se mezclan con hexanos y ácido sulfúrico ac. para liberar el ácido libre de (+)-tetrahidro humulona ((+)-XI). La capa de hexano se separa, se lava con salmuera, se seca con sulfato de sodio y se evapora a (+)-tetrahidro humulona ((+)-XI) como se indica por HPLC quiral.
Ejemplo 15: Preparación de sales adicionales de KDT500:
Preparación de sal de calcio (II) ("KDT505"):
Se añadieron agua (1000 pl) y la sal de potasio de KDT500 (88,2 mg) a un vial de 4 ml, seguido de 26,6 mg de cloruro de calcio (1,10 eq) en 120 pl de agua. La mezcla se agitó durante una hora y el precipitado se filtró, se lavó 4 veces con 1 mL de agua y se secó a alto vacío para obtener 6,9 mg de producto. El producto tenía un punto de fusión de 125,1 °C y un contenido de agua de 1,28 KF.
Preparación de sal de magnesio (II) ("KDT506"):
Se añadieron agua (1200 pl) y la sal de potasio de KDT500 (50,1 mg) a un vial de 4 ml, seguido de 7,8 mg de sulfato de magnesio (0,53 eq) en 200 pl de agua. La mezcla se agitó durante una hora y el precipitado se filtró, se lavó 2 veces con 1 ml de agua y se secó a alto vacío para obtener 47 mg de producto. El producto tenía un punto de fusión de 130,0 °C y un contenido de agua de 1,27 KF.
Preparación de sal de zinc (II) ("KDT507"):
Se añadieron agua (1300 pl) y la sal de potasio de KDT500 (84,7 mg) a un vial de 4 ml, seguido de 31,6 mg de sulfato de zinc heptahidratado (0,53 eq) en 300 pl de agua. La mezcla se agitó durante una hora y el precipitado se filtró, se lavó 2 veces con 1 mL de agua y se secó a alto vacío para obtener 71,6 mg de producto. El producto tenía un punto de fusión de 134,7 °C y un contenido de agua de 2,42 KF.
Preparación de sal de hierro (III) ("KDT508"):
Se añadieron agua (1000 pl) y la sal potásica de KDT500 (61,9 mg) a un vial de 4 ml, seguido de 43,4 mg de cloruro de hierro (III) hexahidratado (0,53 eq) en 300 pl de agua. La mezcla se agitó durante una hora y el precipitado se filtró, se lavó 2 veces con 1 mL de agua y se secó a alto vacío para obtener 71,6 mg de producto. El producto tenía un punto de fusión de 72,9 °C y un contenido de agua de 1,07 KF.
Preparación de sal de sodio (I) ("KDT509"):
Se añadieron agua (1000 pL) y sal libre de KDT500 (109,5 mg) a un vial de 4 ml, seguido de solución de NaOH ag. 1 M (300 pL, 1,00 eq). La mezcla se evaporó y se secó a alto vacío. El producto tenía un punto de fusión de 113,9 °C y un contenido de agua de 1,15 KF.
Preparación de sal de cobre (II) ("KDT510"):
Se añadieron agua (1000 j L) y sal libre de KDT500 (109,5 mg) a un vial de 4 ml, seguido de solución de NaOH ag. 1 M (300 j L, 1,00 eq). La mezcla se evaporó y se secó a alto vacío. El producto tenía un punto de fusión de 104,9 °C y un contenido de agua de 1,18 KF.
Preparación de sal de guanidinio ("KDT511"):
Se añadieron agua (1000 j L) y sal libre de KDT500 (109,5 mg) a un vial de 4 ml, seguido de solución de NaOH ag. 1 M (300 j L, 1,00 eq). La mezcla se evaporó y se secó a alto vacío. El producto tenía un punto de fusión de 140,8 °C y un contenido de agua de 0,94 KF.
Referencias
1. Babish J Med Food 13:535 (2010)
2. Berge J Pharm Sci 66:1 (1977)
3. Boden Diabetes 46:3 (1997)
4. Dahlberg J Sep Sci 33:2828 (2010)
5. De Keukeleire Tetrahedron 26:385 (1970)
6. De Keukeleire Tetrahedron 27:4939 (1971)
7. Dong J Am Chem Soc 130:2738 (2008)
8. Fasshauer Biochem Biophys Res Commun 290:1084 (2002)
9. Gotoh Biochem Biophys Res Commun 354:591 (2007)
10. Hajjaj Appl Environ Microbiol 67:2596 (2001)
11. Hara J Pharmaceut Sci 100:3594 (2011)
12. Hirasawa Nature Med 11:90 (2005)
13. Konda Arthritis Rheum 62:1683 (2010)
14. Li Mol Endocrinol 16:1040 (2002)
15. Martin Atherosclerosis 137:S75 (1998)
16. Miyake Biosci Biotechnol Biochem 70:1154 (2006)
17. Oh Cell 142:687 (2010)
18. Pascual Nature (London) 437:759 (2005)
19. Raz Diabetes Metab Res Rev 21:3 (2005)
20. Stumvoll Ann Med 34:217 (2002)
21. Talukdar Trends Pharmacol Sci 32:543 (2011)
22. TontonozCell 79:1147 (1994)
23. Tontonoz Genes Dev 8:1224 (1994)
24. Tripp Acta Hort (ISHS) 848:221 (2009)
25. Wang Plant Physiol 148:1254 (2008)
Tabla 1: Datos cristalográficos para las estructuras proporcionadas


Tabla 2: Las coordenadas atómicas (x 104), y los parámetros de desplazamiento isotrópico equivalente (Á2x 103) para el cristal (+)-KDT501 (U(eq) se define como 1/3 de la traza del tensor Uij ortogonalizado)



_____ _____ _____ _____
Tabla 3: Longitudes de enlace [A] y ángulos [] para el cristal (+)-KDT501 (transformaciones de simetría usadas para generar átomos equivalentes: #1 -x+3/2,y+1/2,-z+2 #2 -x+3/2,y-1/2,-z+2)




______ ______



Tabla 4: Parámetros de desplazamiento anisotrópico (Á2x103) para el cristal (+)- KDT5.01

___ ___ ___ ___ ____ ____

___ ___ ___ ___ ___ ___ ___ ___ ___ ___
___ ____ ____ ___ ___
Claims (1)
- REIVINDICACIONES(+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)cidopent-2-en-1-ona que tiene la estructura:
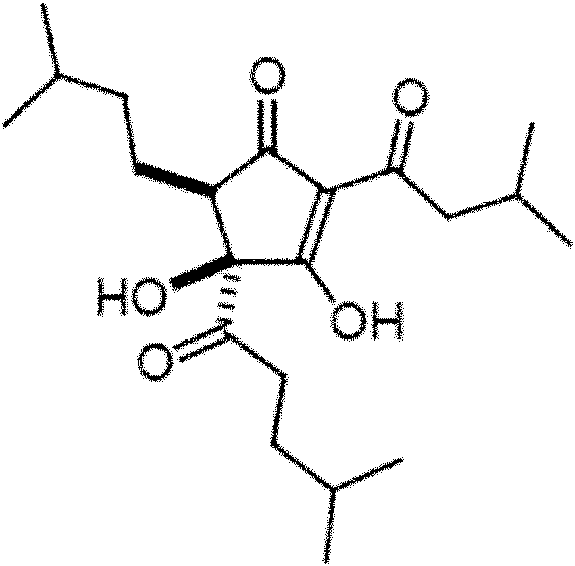 o una sal o cristal de esta, para su uso en el tratamiento de la diabetes en un sujeto.(+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona que tiene la estructura:
o una sal o cristal de esta, para su uso en el tratamiento de la diabetes en un sujeto.(+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona que tiene la estructura: o una sal o cristal de esta, para su uso en el tratamiento de la obesidad en un sujeto.(+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona que tiene la estructura:
o una sal o cristal de esta, para su uso en el tratamiento de la obesidad en un sujeto.(+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona que tiene la estructura: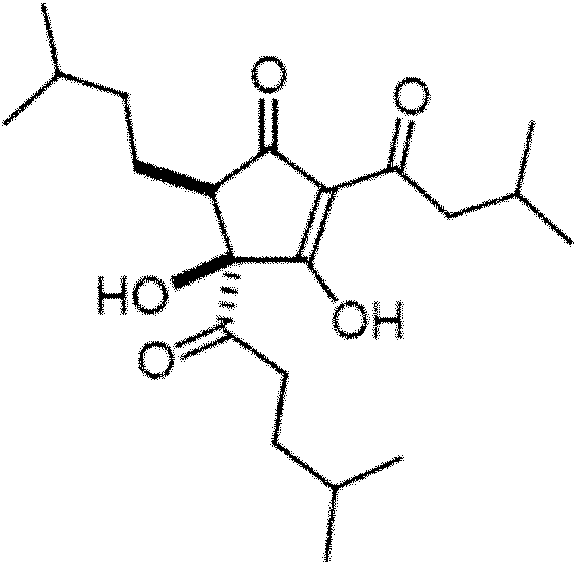 o una sal o cristal de esta, para su uso en el tratamiento de la inflamación en un sujeto.(+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona o una sal o cristal de esta para el uso como se reivindicó en cualquiera de las reivindicaciones 1 a 3 en forma de una composición.5. (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)cidopent-2-en-1-ona o una sal o un cristal de esta para el uso como se reivindicó en la reivindicación 4, que comprende además un portador farmacéuticamente aceptable.6. (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)cidopent-2-en-1-ona o una sal o cristal de esta para para el uso como se reivindicó en la reivindicación 4 o la reivindicación 5, en donde la administración de dicha composición a un sujeto da como resultado una disminución de los niveles de glucosa y/o lípidos.7. (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona o una sal o cristal de esta para para el uso como se reivindicó en cualquier reivindicación anterior, en donde la pureza enantiomérica es del 90 % o superior.8. (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona o una sal o cristal de esta para el uso como se reivindicó en la reivindicación 5, en donde la pureza enantiomérica es del 98 % o superior.9. (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona o una sal o cristal de esta para el uso como se reivindicó en la reivindicación 6, en donde la pureza enantiomérica es del 99 % o superior.10. (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona o una sal o cristal de esta para el uso como se reivindicó en la reivindicación 7, en donde la pureza enantiomérica es del 99,5 % o superior.11. La sal de (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona para el uso como se reivindicó en cualquier reivindicación anterior, en donde la sal se selecciona del grupo que consiste en una sal de potasio, aluminio, calcio, cobre, guanidinio, hierro, litio, magnesio, sodio, zinc, cinconidina, cinconina y dietanolamina.12. (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona o un sal o cristal de esta para el uso como se reivindicó en cualquiera de las reivindicaciones 1 a 8, que es una sal de potasio que tiene la estructura:
o una sal o cristal de esta, para su uso en el tratamiento de la inflamación en un sujeto.(+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona o una sal o cristal de esta para el uso como se reivindicó en cualquiera de las reivindicaciones 1 a 3 en forma de una composición.5. (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)cidopent-2-en-1-ona o una sal o un cristal de esta para el uso como se reivindicó en la reivindicación 4, que comprende además un portador farmacéuticamente aceptable.6. (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)cidopent-2-en-1-ona o una sal o cristal de esta para para el uso como se reivindicó en la reivindicación 4 o la reivindicación 5, en donde la administración de dicha composición a un sujeto da como resultado una disminución de los niveles de glucosa y/o lípidos.7. (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona o una sal o cristal de esta para para el uso como se reivindicó en cualquier reivindicación anterior, en donde la pureza enantiomérica es del 90 % o superior.8. (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona o una sal o cristal de esta para el uso como se reivindicó en la reivindicación 5, en donde la pureza enantiomérica es del 98 % o superior.9. (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona o una sal o cristal de esta para el uso como se reivindicó en la reivindicación 6, en donde la pureza enantiomérica es del 99 % o superior.10. (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona o una sal o cristal de esta para el uso como se reivindicó en la reivindicación 7, en donde la pureza enantiomérica es del 99,5 % o superior.11. La sal de (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona para el uso como se reivindicó en cualquier reivindicación anterior, en donde la sal se selecciona del grupo que consiste en una sal de potasio, aluminio, calcio, cobre, guanidinio, hierro, litio, magnesio, sodio, zinc, cinconidina, cinconina y dietanolamina.12. (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3 -metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona o un sal o cristal de esta para el uso como se reivindicó en cualquiera de las reivindicaciones 1 a 8, que es una sal de potasio que tiene la estructura: 13. (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3-metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona o una sal o cristal de esta para el uso como se reivindicó en la reivindicación 12, en donde el cristal tiene un grupo espacial P 21 21 2 y las dimensiones de la celda unitaria son a = 23,3110(9) Á, a = 90°, b = 28,9052(12) Á , p= 9o°, c = 13,6845(5) Á y y = 90°; opcionalmente en donde el cristal tiene las coordenadas atómicas tridimensionales expuestas en la siguiente tabla:
13. (+)-(4S,5R)-3,4-dihidroxi-2-(3-metilbutanoil)-5-(3-metilbutil)-4-(4-metilpentanoil)ciclopent-2-en-1-ona o una sal o cristal de esta para el uso como se reivindicó en la reivindicación 12, en donde el cristal tiene un grupo espacial P 21 21 2 y las dimensiones de la celda unitaria son a = 23,3110(9) Á, a = 90°, b = 28,9052(12) Á , p= 9o°, c = 13,6845(5) Á y y = 90°; opcionalmente en donde el cristal tiene las coordenadas atómicas tridimensionales expuestas en la siguiente tabla: ____ ____ ____ ____
____ ____ ____ ____ ____ ____
____ ____
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US40857210P | 2010-10-30 | 2010-10-30 | |
| US201161448150P | 2011-03-01 | 2011-03-01 | |
| US201161508434P | 2011-07-15 | 2011-07-15 | |
| PCT/US2011/058477 WO2012058649A1 (en) | 2010-10-30 | 2011-10-28 | Cis 3,4-dihydroxy-2-(3-methylbutanoyl)-5-(-3-methylbutyl)-4-(4-methylpentanoyl)cyclopent-2-en-1-one derivatives, substantially enantiomerically pure compositions and methods |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2829388T3 true ES2829388T3 (es) | 2021-05-31 |
Family
ID=45994458
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES11837235T Active ES2829388T3 (es) | 2010-10-30 | 2011-10-28 | Derivados de cis 3,4-dihidroxi-2-(3-metilbutanoil)-5-(-3-metilbutil)-4-(4-metilpentanoil)cyclopent-2-en-1-ona, composiciones sustancialmente puras enantioméricamente y métodos |
Country Status (9)
| Country | Link |
|---|---|
| US (3) | US8410178B2 (es) |
| EP (1) | EP2632448B1 (es) |
| JP (2) | JP6259287B2 (es) |
| KR (1) | KR20140027910A (es) |
| CN (3) | CN110215442A (es) |
| AU (1) | AU2011320140C1 (es) |
| CA (1) | CA2853974C (es) |
| ES (1) | ES2829388T3 (es) |
| WO (1) | WO2012058649A1 (es) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2725830T3 (es) * | 2012-07-09 | 2019-09-27 | Kindex Pharmaceuticals Inc | Derivados de tetrahidro-isohumulona, métodos para su preparación y uso |
| HK1210825A1 (en) | 2012-07-11 | 2016-05-06 | Elcelyx Therapeutics, Inc. | Compositions comprising statins, biguanides and further agents for reducing cardiometabolic risk |
| AU2015220427B2 (en) | 2014-02-19 | 2017-09-21 | Piramal Enterprises Limited | Compounds for use as GPR120 agonists |
| AU2015293541B2 (en) | 2014-07-25 | 2019-11-21 | Piramal Enterprises Limited | Substituted phenyl alkanoic acid compounds as GPR120 agonists and uses thereof |
| EP3191463B1 (en) | 2014-09-11 | 2019-10-30 | Piramal Enterprises Limited | Fused heterocyclic compounds as gpr120 agonists |
| US10671277B2 (en) * | 2014-12-17 | 2020-06-02 | Datalogic Usa, Inc. | Floating soft trigger for touch displays on an electronic device with a scanning module |
| US10941133B2 (en) | 2015-02-05 | 2021-03-09 | Piramal Enterprises Limited | Compounds containing carbon-carbon linker as GPR120 agonists |
| GB201510758D0 (en) | 2015-06-18 | 2015-08-05 | Ucb Biopharma Sprl | Novel TNFa structure for use in therapy |
| GB201621907D0 (en) | 2016-12-21 | 2017-02-01 | Ucb Biopharma Sprl And Sanofi | Antibody epitope |
| CN111094225A (zh) * | 2017-04-12 | 2020-05-01 | 金戴克斯医药有限责任公司 | 使用异葎草酮及其衍生物治疗多囊卵巢综合征 |
| CN116354520B (zh) * | 2021-05-07 | 2024-10-29 | 茅台学院 | 丛毛红曲菌在处理超高浓度白酒废水中的应用 |
| CN115260032B (zh) * | 2022-10-08 | 2023-01-06 | 中国科学院昆明植物研究所 | 一种利用高速逆流色谱分离纯化对甲氧基肉桂酸乙酯和/或肉桂酸乙酯的方法 |
Family Cites Families (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1266716A (es) * | 1969-09-26 | 1972-03-15 | ||
| US5583262A (en) * | 1994-11-10 | 1996-12-10 | Maye; John P. | Solid salts of hop acids |
| HK1044532A1 (zh) * | 1999-03-01 | 2002-10-25 | 辉瑞产品公司 | 用作甲状腺受体配体的草氨酸及衍生物 |
| US7205151B2 (en) | 2001-06-20 | 2007-04-17 | Metaproteomics, Llc | Complex mixtures exhibiting selective inhibition of cyclooxygenase-2 |
| US7901714B2 (en) | 2001-06-20 | 2011-03-08 | Metaproteomics, Llp | Treatment modalities for autoimmune diseases |
| US20040219240A1 (en) | 2001-06-20 | 2004-11-04 | Babish John G. | Anti-inflammatory pharmaceutical compositions for reducing inflammation and the treatment or prevention of gastric toxicity |
| US8206753B2 (en) | 2001-06-20 | 2012-06-26 | Metaproteomics, Llc | Anti-inflammatory botanical products for the treatment of metabolic syndrome and diabetes |
| US7718198B2 (en) | 2001-06-20 | 2010-05-18 | Metaproteomics, Llc | Treatment modalities for autoimmune diseases |
| US7736677B2 (en) | 2001-06-20 | 2010-06-15 | Metaproteomics, Llc | Xanthohumol and tetrahydro-isoalpha acid based protein kinase modulation cancer treatment |
| US8168234B2 (en) | 2001-06-20 | 2012-05-01 | Metaproteomics, Llc | Compositions that treat or inhibit pathological conditions associated with inflammatory response |
| US7815944B2 (en) | 2001-06-20 | 2010-10-19 | Metaproteomics, Llc | Anti-inflammatory pharmaceutical compositions for reducing inflammation and the treatment of prevention of gastric toxicity |
| US7270835B2 (en) | 2001-06-20 | 2007-09-18 | Metaproteomics, Llc | Compositions that treat or inhibit pathological conditions associated with inflammatory response |
| US7901713B2 (en) | 2001-06-20 | 2011-03-08 | Metaproteomics, Llc | Inhibition of COX-2 and/or 5-LOX activity by fractions isolated or derived from hops |
| US20040115290A1 (en) | 2001-06-20 | 2004-06-17 | Tripp Matthew L. | Modulation of inflammation by hops fractions and derivatives |
| US8142819B2 (en) | 2002-10-21 | 2012-03-27 | Metaproteomics, Llc | Synergistic compositions that treat or inhibit pathological conditions associated with inflammatory response |
| WO2003068205A1 (en) * | 2002-02-14 | 2003-08-21 | Kirin Beer Kabushiki Kaisha | Compositions and foods for improving lipid metabolism |
| AU2003286549B2 (en) | 2002-10-21 | 2006-11-30 | Metaproteomics, Llc | Compositions that treat or inhibit pathological conditions associated with inflammatory response |
| AU2004283065B2 (en) | 2003-05-22 | 2009-11-26 | Metaproteomics, Llc | Anti-inflammatory pharmaceutical compositions for reducing inflammation and the treatment or prevention of gastric toxicity |
| EP1817941A4 (en) * | 2004-11-13 | 2009-11-11 | Metaproteomics Llc | CYCLOOXYGENASE-2 INHIBITION COMPOSITIONS |
| GB0427701D0 (en) * | 2004-12-17 | 2005-01-19 | Astrazeneca Ab | Therapeutic agents |
| US20060287554A1 (en) * | 2005-06-21 | 2006-12-21 | Madsen Kevin K | Process for producing inorganic salts of hop acids |
| WO2007070355A2 (en) | 2005-12-09 | 2007-06-21 | Metaproteomics, Llc | Anti-inflammatory botanical products for the treatment of metabolic syndrome and diabetes |
| JP2009518439A (ja) * | 2005-12-09 | 2009-05-07 | メタプロテオミクス,エルエルシー | ホップ及びアカシア産物によるプロテインキナーゼ調節 |
| JP2010522190A (ja) * | 2007-03-19 | 2010-07-01 | メタプロテオミクス, エルエルシー | 骨及び関節の健康を助成するための方法と組成物 |
| EP2152684A4 (en) * | 2007-05-11 | 2011-05-18 | Metaproteomics Llc | METHOD AND COMPOSITIONS FOR THE DETOXIFICATION OF HEAVY METAL |
| KR20100131969A (ko) * | 2007-12-10 | 2010-12-16 | 메타프로테오믹스, 엘엘씨 | 암, 혈관신생 및 이와 관련된 염증 경로의 치환된 1,3-시클로펜타디온 다중-표적 단백질 키나아제 조절제 |
| WO2010068731A1 (en) * | 2008-12-10 | 2010-06-17 | Metaproteomics, Llc | A method for the purification of substituted cyclopent-2-en-1-one congeners and substituted 1,3-cyclopentadione congeners from a complex mixture using countercurrent separation |
| TW201124132A (en) | 2009-08-14 | 2011-07-16 | Metaproteomics Llc | Tetrahydro-isoalpha acid compositions and methods for weight management |
-
2011
- 2011-10-28 EP EP11837235.8A patent/EP2632448B1/en active Active
- 2011-10-28 AU AU2011320140A patent/AU2011320140C1/en not_active Ceased
- 2011-10-28 CN CN201910333204.XA patent/CN110215442A/zh active Pending
- 2011-10-28 CA CA2853974A patent/CA2853974C/en not_active Expired - Fee Related
- 2011-10-28 CN CN201180063785.1A patent/CN103561730A/zh active Pending
- 2011-10-28 ES ES11837235T patent/ES2829388T3/es active Active
- 2011-10-28 KR KR1020137013796A patent/KR20140027910A/ko not_active Ceased
- 2011-10-28 WO PCT/US2011/058477 patent/WO2012058649A1/en not_active Ceased
- 2011-10-28 CN CN201910332802.5A patent/CN110200955A/zh active Pending
- 2011-10-28 US US13/284,852 patent/US8410178B2/en not_active Expired - Fee Related
- 2011-10-28 JP JP2013536899A patent/JP6259287B2/ja not_active Expired - Fee Related
-
2012
- 2012-03-14 US US13/420,062 patent/US8410179B2/en not_active Expired - Fee Related
-
2013
- 2013-03-18 US US13/846,790 patent/US8829056B2/en not_active Expired - Fee Related
-
2016
- 2016-11-04 JP JP2016216780A patent/JP2017101019A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| JP6259287B2 (ja) | 2018-01-10 |
| US20130018105A1 (en) | 2013-01-17 |
| US20120108671A1 (en) | 2012-05-03 |
| CA2853974C (en) | 2020-11-10 |
| US8410178B2 (en) | 2013-04-02 |
| US8410179B2 (en) | 2013-04-02 |
| WO2012058649A1 (en) | 2012-05-03 |
| US8829056B2 (en) | 2014-09-09 |
| EP2632448A4 (en) | 2014-03-19 |
| JP2013542225A (ja) | 2013-11-21 |
| US20130217781A1 (en) | 2013-08-22 |
| CN110200955A (zh) | 2019-09-06 |
| AU2011320140C1 (en) | 2019-12-12 |
| AU2011320140B2 (en) | 2016-12-01 |
| EP2632448A1 (en) | 2013-09-04 |
| CA2853974A1 (en) | 2012-05-03 |
| CN110215442A (zh) | 2019-09-10 |
| JP2017101019A (ja) | 2017-06-08 |
| EP2632448B1 (en) | 2020-10-21 |
| CN103561730A (zh) | 2014-02-05 |
| KR20140027910A (ko) | 2014-03-07 |
| AU2011320140A1 (en) | 2013-05-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2829388T3 (es) | Derivados de cis 3,4-dihidroxi-2-(3-metilbutanoil)-5-(-3-metilbutil)-4-(4-metilpentanoil)cyclopent-2-en-1-ona, composiciones sustancialmente puras enantioméricamente y métodos | |
| JP2018135343A (ja) | ピラノクロメニルフェノール誘導体、及び代謝症候群または炎症疾患治療用医薬組成物 | |
| JP6916180B2 (ja) | グルコピラノシル誘導体の複合体並びにその調製方法及び使用 | |
| EP3378491A1 (en) | Pharmaceutical composition for treatment or prophylaxis of nash | |
| CN102766097B (zh) | 一种依达拉奉a型晶体及其制备方法 | |
| CN118459439A (zh) | 一种mpc抑制剂及其药物组合物和应用 | |
| CN108947949A (zh) | 抗焦虑氘代化合物及其医药用途 | |
| US20090325904A1 (en) | Maleic acid monosalt of antiviral agent and pharmaceutical composition containing the same | |
| EP2767533B1 (en) | Derivative of butylphthalide and preparation method and use thereof | |
| CA3145694A1 (en) | Pyrimidine-5-carboxamide compound | |
| TW200927090A (en) | Novel sulfamate compounds for medical use | |
| US9828331B2 (en) | Tetrahydro-isohumulone derivatives, methods of making and using | |
| CN106478616B (zh) | 一种gpr40激动剂的结晶形式及其制备方法 | |
| CN115677572B (zh) | 氟代酰胺类衍生物、药物组合物及其应用 | |
| US11312687B2 (en) | 7H-azulene [1,2,3-i,j] isoquinolin-7-one compound, single crystal and use thereof | |
| JP2019529503A (ja) | 置換フェニルプロピオン酸化合物のエナンチオマー及びその製造方法、、組成物並びに応用 | |
| WO2023109761A1 (zh) | 吡唑并嘧啶酮类化合物及其盐的结晶 |