ES2791771B2 - Membrana hidrófila de nanofibras no tejidas para promover la regeneración ósea - Google Patents
Membrana hidrófila de nanofibras no tejidas para promover la regeneración ósea Download PDFInfo
- Publication number
- ES2791771B2 ES2791771B2 ES201930393A ES201930393A ES2791771B2 ES 2791771 B2 ES2791771 B2 ES 2791771B2 ES 201930393 A ES201930393 A ES 201930393A ES 201930393 A ES201930393 A ES 201930393A ES 2791771 B2 ES2791771 B2 ES 2791771B2
- Authority
- ES
- Spain
- Prior art keywords
- membrane
- copolymer
- hydrophilic nonwoven
- bone
- nonwoven nanofiber
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L27/00—Materials for grafts or prostheses or for coating grafts or prostheses
- A61L27/14—Macromolecular materials
- A61L27/16—Macromolecular materials obtained by reactions only involving carbon-to-carbon unsaturated bonds
-
- D—TEXTILES; PAPER
- D04—BRAIDING; LACE-MAKING; KNITTING; TRIMMINGS; NON-WOVEN FABRICS
- D04H—MAKING TEXTILE FABRICS, e.g. FROM FIBRES OR FILAMENTARY MATERIAL; FABRICS MADE BY SUCH PROCESSES OR APPARATUS, e.g. FELTS, NON-WOVEN FABRICS; COTTON-WOOL; WADDING ; NON-WOVEN FABRICS FROM STAPLE FIBRES, FILAMENTS OR YARNS, BONDED WITH AT LEAST ONE WEB-LIKE MATERIAL DURING THEIR CONSOLIDATION
- D04H1/00—Non-woven fabrics formed wholly or mainly of staple fibres or like relatively short fibres
- D04H1/40—Non-woven fabrics formed wholly or mainly of staple fibres or like relatively short fibres from fleeces or layers composed of fibres without existing or potential cohesive properties
- D04H1/42—Non-woven fabrics formed wholly or mainly of staple fibres or like relatively short fibres from fleeces or layers composed of fibres without existing or potential cohesive properties characterised by the use of certain kinds of fibres insofar as this use has no preponderant influence on the consolidation of the fleece
- D04H1/4326—Condensation or reaction polymers
- D04H1/435—Polyesters
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61C—DENTISTRY; APPARATUS OR METHODS FOR ORAL OR DENTAL HYGIENE
- A61C8/00—Means to be fixed to the jaw-bone for consolidating natural teeth or for fixing dental prostheses thereon; Dental implants; Implanting tools
- A61C8/0003—Not used, see subgroups
- A61C8/0004—Consolidating natural teeth
- A61C8/0006—Periodontal tissue or bone regeneration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61C—DENTISTRY; APPARATUS OR METHODS FOR ORAL OR DENTAL HYGIENE
- A61C8/00—Means to be fixed to the jaw-bone for consolidating natural teeth or for fixing dental prostheses thereon; Dental implants; Implanting tools
- A61C8/0012—Means to be fixed to the jaw-bone for consolidating natural teeth or for fixing dental prostheses thereon; Dental implants; Implanting tools characterised by the material or composition, e.g. ceramics, surface layer, metal alloy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/74—Synthetic polymeric materials
- A61K31/765—Polymers containing oxygen
- A61K31/78—Polymers containing oxygen of acrylic acid or derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K6/00—Preparations for dentistry
- A61K6/80—Preparations for artificial teeth, for filling teeth or for capping teeth
- A61K6/884—Preparations for artificial teeth, for filling teeth or for capping teeth comprising natural or synthetic resins
- A61K6/887—Compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L27/00—Materials for grafts or prostheses or for coating grafts or prostheses
- A61L27/14—Macromolecular materials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L27/00—Materials for grafts or prostheses or for coating grafts or prostheses
- A61L27/14—Macromolecular materials
- A61L27/26—Mixtures of macromolecular compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L27/00—Materials for grafts or prostheses or for coating grafts or prostheses
- A61L27/50—Materials characterised by their function or physical properties, e.g. injectable or lubricating compositions, shape-memory materials, surface modified materials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L27/00—Materials for grafts or prostheses or for coating grafts or prostheses
- A61L27/50—Materials characterised by their function or physical properties, e.g. injectable or lubricating compositions, shape-memory materials, surface modified materials
- A61L27/56—Porous materials, e.g. foams or sponges
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L27/00—Materials for grafts or prostheses or for coating grafts or prostheses
- A61L27/50—Materials characterised by their function or physical properties, e.g. injectable or lubricating compositions, shape-memory materials, surface modified materials
- A61L27/58—Materials at least partially resorbable by the body
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L31/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an acyloxy radical of a saturated carboxylic acid, of carbonic acid or of a haloformic acid; Compositions of derivatives of such polymers
- C08L31/02—Homopolymers or copolymers of esters of monocarboxylic acids
-
- D—TEXTILES; PAPER
- D04—BRAIDING; LACE-MAKING; KNITTING; TRIMMINGS; NON-WOVEN FABRICS
- D04H—MAKING TEXTILE FABRICS, e.g. FROM FIBRES OR FILAMENTARY MATERIAL; FABRICS MADE BY SUCH PROCESSES OR APPARATUS, e.g. FELTS, NON-WOVEN FABRICS; COTTON-WOOL; WADDING ; NON-WOVEN FABRICS FROM STAPLE FIBRES, FILAMENTS OR YARNS, BONDED WITH AT LEAST ONE WEB-LIKE MATERIAL DURING THEIR CONSOLIDATION
- D04H1/00—Non-woven fabrics formed wholly or mainly of staple fibres or like relatively short fibres
- D04H1/40—Non-woven fabrics formed wholly or mainly of staple fibres or like relatively short fibres from fleeces or layers composed of fibres without existing or potential cohesive properties
- D04H1/42—Non-woven fabrics formed wholly or mainly of staple fibres or like relatively short fibres from fleeces or layers composed of fibres without existing or potential cohesive properties characterised by the use of certain kinds of fibres insofar as this use has no preponderant influence on the consolidation of the fleece
- D04H1/4382—Stretched reticular film fibres; Composite fibres; Mixed fibres; Ultrafine fibres; Fibres for artificial leather
- D04H1/43838—Ultrafine fibres, e.g. microfibres
-
- D—TEXTILES; PAPER
- D04—BRAIDING; LACE-MAKING; KNITTING; TRIMMINGS; NON-WOVEN FABRICS
- D04H—MAKING TEXTILE FABRICS, e.g. FROM FIBRES OR FILAMENTARY MATERIAL; FABRICS MADE BY SUCH PROCESSES OR APPARATUS, e.g. FELTS, NON-WOVEN FABRICS; COTTON-WOOL; WADDING ; NON-WOVEN FABRICS FROM STAPLE FIBRES, FILAMENTS OR YARNS, BONDED WITH AT LEAST ONE WEB-LIKE MATERIAL DURING THEIR CONSOLIDATION
- D04H1/00—Non-woven fabrics formed wholly or mainly of staple fibres or like relatively short fibres
- D04H1/70—Non-woven fabrics formed wholly or mainly of staple fibres or like relatively short fibres characterised by the method of forming fleeces or layers, e.g. reorientation of fibres
- D04H1/72—Non-woven fabrics formed wholly or mainly of staple fibres or like relatively short fibres characterised by the method of forming fleeces or layers, e.g. reorientation of fibres the fibres being randomly arranged
- D04H1/724—Non-woven fabrics formed wholly or mainly of staple fibres or like relatively short fibres characterised by the method of forming fleeces or layers, e.g. reorientation of fibres the fibres being randomly arranged forming webs during fibre formation, e.g. flash-spinning
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2400/00—Materials characterised by their function or physical properties
- A61L2400/12—Nanosized materials, e.g. nanofibres, nanoparticles, nanowires, nanotubes; Nanostructured surfaces
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2430/00—Materials or treatment for tissue regeneration
- A61L2430/02—Materials or treatment for tissue regeneration for reconstruction of bones; weight-bearing implants
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Oral & Maxillofacial Surgery (AREA)
- Epidemiology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Transplantation (AREA)
- Dermatology (AREA)
- Engineering & Computer Science (AREA)
- Textile Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Dentistry (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Developmental Biology & Embryology (AREA)
- Biomedical Technology (AREA)
- Pharmacology & Pharmacy (AREA)
- Plastic & Reconstructive Surgery (AREA)
- Dispersion Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Ceramic Engineering (AREA)
- Materials For Medical Uses (AREA)
Description
DESCRIPCIÓN
MEMBRANA HIDRÓFILA DE NANOFIBRAS NO TEJIDAS PARA PROMOVER LA
REGENERACIÓN ÓSEA
La invención hace referencia a una membrana hidrófila de nanofibras no tejidas a base de copolímeros de acrilato y metacrilato y a su proceso de preparación. Además, la invención hace referencia a su forma hidrolizada, funcionalizada adicionalmente con un catión divalente seleccionado de Zn+2, Ca+2, Mg+2 y Sr+2, un agente antibacteriano y cualquiera de las combinaciones de los mismos. Más aún, la invención hace referencia a una membrana no reabsorbible para promover la regeneración ósea y una membrana periodontal no reabsorbible que comprende dicha membrana hidrófila de nanofibras no tejidas, su forma hidrolizada o su forma hidrolizada funcionalizada adicionalmente con un catión divalente seleccionado de Zn+2, Ca+2, Mg+2 y Sr+2, un agente antibacteriano y cualquiera de las combinaciones de los mismos.
ESTADO DE LA TÉCNICA
El uso de implantes dentales se ha convertido en una modalidad de tratamiento muy extendida y previsible para la restauración de los dientes que se han perdido y para diversos casos de edentulismo. Es evidente que el uso de una técnica regenerativa con la colocación de un implante dental es un paso importante que ayuda en el proceso de la regeneración ósea. Debido a que el éxito clínico de la terapia de implantes se basa en la osteointegración, definida como el anclaje directo del implante en el tejido óseo sin la interposición de tejido fibroso, se ha realizado una investigación considerable para promover el crecimiento óseo.
El principio básico de la Regeneración Ósea Guiada (ROG) implica la colocación de barreras mecánicas para proteger los coágulos de sangre y para aislar el defecto óseo del tejido conjuntivo circundante, lo que provee de este modo a las células formadoras de hueso de un acceso a un espacio aislado destinado a la regeneración ósea. La ROG tiene, en muchos casos, un resultado clínico imprevisible y sigue siendo un desafío. Una regeneración ósea con éxito requiere: I) cierre primario de la herida para promocionar una curación sin alteraciones e ininterrumpida, II) angiogénesis para proporcionar el suministro necesario de sangre y células mesenquimales indiferenciadas, III) creación de espacio y mantenimiento para facilitar espacio para el
hueso en crecimiento y IV) estabilidad de la herida para inducir la formación de coágulos de sangre y permitir una curación sin incidentes. Se obtiene habitualmente mediante la utilización de membranas barrera que se colocan tras la cirugía. Existen dos tipos de membranas en base a las características de reabsorción.
El uso de matrices de ingeniería tisular reabsorbible para inducir la formación ósea, cuando se necesita soporte adicional, no siempre tiene éxito. Una importante limitación de los materiales reabsorbibles es la incapacidad de ejercer un control espaciotemporal sobre el proceso de curación de la herida. La mayoría de las membranas reabsorbibles empleadas (p. ej., colágeno, poliláctico-co-glicólico, policaprolactona) y sustitutos de injerto óseo (p. ej., hidroxiapatita -HAp- y otros fosfatos cálcicos) muestran una tasa de biodegradación relativamente rápida. Debe tenerse en cuenta que el periodo de curación del hueso alveolar, después de la regeneración periodontal o después de la extracción necesita habitualmente de 6 a 12 meses. Actualmente, los materiales reabsorbibles empleados presentan varias desventajas, ya que sus comportamientos en disolución no son tan duraderos como se requiere. Más aún, algunos productos de degradación de estos materiales reabsorbibles tiene un pH bajo, pueden no ser citocompatibles y podrían también alterar los procesos de remineralización [Ivanovski S, Vaquette C, Gronthos S, Hutmacher DW, Bartold PM (2014) Multiphasic Scaffolds for Periodontal Tissue Engineering. J Dent Res 93(12):1212-1221] [Shimauchi H, Nemoto E, Ishihata H, Shimomura M (2013) Possible functional scaffolds for periodontal regeneration. Japan Dent Sci Rev 49:118-130].
Aunque se está realizando mucha investigación sobre las membranas periodontales reabsorbibles, las membranas sintéticas no reabsorbibles de politetrafluoroetileno (PTFE), aún representan el estándar de oro para los clínicos, debido a la mayor previsibilidad de sus efectos en comparación con las membranas reabsorbibles. Sin embargo, el PTFE posee importantes desventajas: I) baja adhesividad para las células, II) total ausencia de la capacidad de conectarse al tejido óseo y proporcionar osteointegración, sin la formación de una capa intermedia de tejido conjuntivo; se hace necesaria una segunda cirugía para retirar la membrana no integrada, y finalmente III) carencia de propiedades antibacterianas, apareciendo infecciones con frecuencia [Sam G, Pillai BRM (2014) Evolution of Barrier Membranes in Periodontal Regeneration-Are the third Generation Membranes J of Clin Diagn Res 8: 14-17]. Por lo tanto, la membrana ideal para la ROG debería parecerse a la morfología del hueso natural. El hueso natural es un híbrido de tejido inorgánico-orgánico compuesto de
nanocristales de hidroxiapatita y fibras de colágeno (con diámetros que se encuentran en un rango de 50 a 500nm) ensamblados en una malla porosa, con poros interconectados. El hueso está nanoestructurado, de manera que los nanomateriales deberían ser la mejor opción para los sustitutos óseos.
Por las razones expuestas anteriormente, es necesario el desarrollo de nuevas membranas adecuadas para la regeneración ósea.
DESCRIPCIÓN DE LA INVENCIÓN
Un primer aspecto de la presente invención hace referencia a una membrana hidrófila de nanofibras no tejidas (en la presente memoria "la membrana de la invención”) caracterizada por que comprende una mezcla de
o un primer copolímero de (MA)3-co-(HEA)2 con topología estadística en un porcentaje en peso entre 35% y 65%; y
o un segundo copolímero de (MMA)i-co-(HEMA)i en un porcentaje en peso entre 35% y 65%.
El término "membrana hidrófila de nanofibras no tejidas” hace referencia a una membrana formada por fibras largas que tienen un diámetro de entre 150nm y 400nm. Dichas membranas son no tejidas, esto significa que son como un fieltro, que no son tejidas ni tejidas en punto: están realizadas a partir de fibras largas (largo continuo), unidas entre sí mediante tratamiento químico, mecánico, térmico o por disolvente, y tienen carácter hidrófilo.
Idealmente, cada aguja produce una única fibra que es enrrollada en el tambor desde el principio hasta el final del proceso de electrohilado o electrospinning (kilométrico). La realidad es que las fibras se cortan de forma intermitente a lo largo del proceso de electrohilado.
El término "copolímero con topología estadística” hace referencia a copolímeros estadísticos, es decir, un copolímero en el que la distribución de los monómeros en la cadena es aleatoria ya que todos los monómeros presentes en la solución tiene la misma afinidad/probabilidad de reaccionar tanto con monómeros de la misma naturaleza química (con ellos mismos) y con monómeros de una naturaleza química diferente.
En una realización preferida de la membrana de la invención esta comprende una mezcla de
o un primer copolímero de (MA)3-co-(HEA)2 con topología estadística en un 50% en peso; y
o un segundo copolímero de (MMA)1-co-(HEMA)1 en un 50% en peso.
Dicha membrana muestra resistencia a la abrasión, flexibilidad, elasticidad, resistencia al estrés mecánico, y por tanto puede ser manipulada con facilidad; puede cortarse, doblarse y retorcerse.
Uno de los parámetros más importantes en el proceso de electrohilado es el peso molecular del polímero. Generalmente, se prefiere un peso molecular más elevado ya que favorece el entrelazamiento entre cadenas más grandes facilitando la formación de fibras durante el hilado. En contraste, un peso molecular inferior puede dar lugar a gotas que forman perlas o perlas combinadas con fibras cortas, lo que da como resultado materiales heterogéneos con propiedades físicas no deseadas: superficie irregular, baja superficie específica, baja resistencia a la abrasión y al estrés mecánico, pérdida de elasticidad.
En otra realización preferida de la membrana de la invención, el primer copolímero de (MA)3-co-(HEA)2 tiene un peso molecular entre 50000 Da y 3106 Da. Más preferiblemente, el primer copolímero de (MA)3-co-(HEA)2 tiene un peso molecular entre 1106 Da y 3106 Da.
En otra realización preferida de la membrana de la invención, el segundo copolímero de (MMA)i-co-(HEMA)i tiene un peso molecular entre 50000 Da y 1106 Da.
En otra realización preferida de la membrana de la invención, la membrana además comprende nanopartículas de SiO2 (NPs-SiO2), y dichas nanopartículas de SiO2 están • homogéneamente dispersas en la membrana, es decir, atrapadas homogéneamente en el volumen completo de fibras formando una solución sólida (material compuesto) y/o
• físicamente adsorbidas en la superficie de la membrana,
en un porcentaje en peso entre 0,1% y 60% con respecto al peso final de la membrana.
En la presente invención el término "nanopartículas de SÍO2” hace referencia a partículas de tamaños entre 1 nm y >100 nm, preferiblemente tamaños entre 1 nm y 100 nm, más preferiblemente entre 1 nm y 30 nm.
El dióxido de silicio (SiO2) puede mejorar no solamente la bioactividad de los materiales, sino también la adhesión y la proliferación celular en tejidos artificiales, facilitando la diferenciación de células osteogénicas. Se considera que el SiO2 es osteoinductivo y un catalizador para la formación ósea. Por lo tanto, para mejorar la bioactividad de las membranas éstas fueron dopadas con nanopartículas de SiO2.
Las nanopartículas de SiO2 (NPs-SiO2) pueden ser introducidas en las membranas de dos maneras: 1) suspendiéndolas en la solución de electrohilado, y a continuación realizar el proceso de electrohilado. En este caso, las NPs-SiO2 quedan atrapadas homogéneamente en el volumen completo de fibras, formando una solución sólida (material compuesto);
2) por adsorción física en la superficie de las fibras una vez que la membrana se ha fabricado: la membrana se impregna con una suspensión de NPs-SiO2 , y a continuación se evapora el agua.
Se prefiere la opción (1) porque las NPs-SiO2 se retienen en las fibras de forma más eficiente y se minimiza su lixiviación; con la opción (1) las NPs-SiO2 pueden permanecer en la membrana durante más tiempo que con (2).
Un segundo aspecto de la presente invención hace referencia a un proceso de preparación de la membrana de la presente invención (en la presente memoria "el proceso de la invención”) que incluye la preparación de los copolímeros que son sometidos a electrohilado para producir dicha membrana. Los copolímeros de la presente invención pueden prepararse mediante polimerización radical convencional o mediante polimerización radical viva catalizada por metal (MC-LRP) tal como: polimerización radical por transferencia de átomo normal (ATRP normal), polimerización radical por transferencia de átomo inversa (ATRP inversa) y polimerización radical por transferencia de átomo generada por transferencia de electrones (AGET ATRP).
En una realización preferida del proceso de la invención, el proceso está caracterizado por que comprende las siguientes etapas:
a) síntesis del primer copolímero de (MA)3-co-(HEA)2 con topología estadística por polimerización radical viva catalizada por metal, utilizando un sistema catalítico metálico;
b) síntesis del segundo copolímero de (MMA)1-co-(HEMA)1 por polimerización radical por transferencia de átomo inversa utilizando un sistema catalítico metálico;
c) preparación por electrohilado de una membrana de nanofibras que comprende una mezcla, donde dicha mezcla comprende el primer copolímero obtenido en la etapa (a) y el segundo copolímero obtenido en la etapa (b), y d) tratamiento térmico de la membrana de nanofibras obtenida en la etapa (c), en donde el tratamiento térmico se aplica en forma de agua caliente en un rango de temperatura entre 30°C y 80°C, por ejemplo durante al menos 4 horas, y en donde la membrana de nanofibras en la etapa (c) se mantiene tensionada mediante una montura.
La etapa (a) del proceso de la invención hace referencia en la presente memoria a la síntesis del primer copolímero de (MA)3-co-(HEA)2 mediante polimerización radical viva catalizada por metal, utilizando un sistema catalítico metálico y la etapa (b) hace referencia a la síntesis del segundo copolímero de (MMA)i-co-(HEMA)i por polimerización radical por transferencia de átomo inversa, utilizando un sistema catalítico metálico.
El término “polimerización radical viva catalizada por metal” hace referencia a métodos de polimerización basados en establecer un rápido equilibrio dinámico entre una cantidad mínima de radicales en crecimiento y una gran mayoría de especies latentes, en la que un complejo metálico en estado de oxidación bajo actúa como el catalizador.
El término “polimerización radical por transferencia de átomo inversa” hace referencia a los métodos de polimerización basados en establecer un rápido equilibrio dinámico entre una cantidad mínima de radicales en crecimiento y una gran mayoría de especies latentes, en la que un complejo metálico en estado de oxidación bajo actúa como catalizador, donde las especies latentes son halogenuros de alquilo, y la reacción es iniciada por un iniciador convencional de radicales y un complejo de Cu2+
En la presente invención el término "sistema catalítico metálico” hace referencia al catalizador utilizado en la polimerización radical viva catalizada por metal de la etapa (a) y al catalizador utilizado en la polimerización radical por transferencia de átomo inversa de la etapa (b). Dicho sistema catalítico metálico comprende un metal, un ligando y un iniciador y utiliza un disolvente en particular. Preferiblemente, el sistema catalítico metálico de la etapa (a) y la etapa (b) es un complejo aminado de cobre.
Preferiblemente, el metal del sistema catalítico metálico de la etapa (a) y/o la etapa (b) comprende un metal de transición o una mezcla de metales de transición en diferentes estados de oxidación. Más preferiblemente, el metal del sistema catalítico metálico de la etapa (a) y la etapa (b) se selecciona independientemente de la lista que consiste en Cu, Fe, Co, Ni, Ru, Pl, Rh, Re, Cr y Mo.
Preferiblemente, dicho metal del sistema catalítico metálico de la etapa (a) y/o la etapa (b) tiene un porcentaje en peso comprendido entre 0,00001% y 0,1%.
Preferiblemente, el ligando del sistema catalítico metálico de la etapa (a) y/o la etapa (b) es una amina alifática multidentada que puede ser lineal o ramificada. Más preferiblemente, el ligando del sistema catalítico metálico de la etapa (a) y la etapa (b) se selecciona independientemente de la lista que consiste en N,N,N',N",N"-pentametildietilentriamina (PMDETA) tris(2-piridilmetil)amina, tris[2-(dimetilamino)etil]amina, 2,2-bipiridilo, N,N,N',N'-tetrakis(2-piridilmetil)etilendiamina y 1,1,4,7,10,10-hexametiltrietilentetramina.
Preferiblemente, dicho ligando tiene un porcentaje en peso entre 0,0001% y 0,2%.
Ha de mencionarse que el iniciador de la etapa (a) y la etapa (b) son preferiblemente diferentes.
Preferiblemente, el iniciador del sistema catalítico metálico de la etapa (a) se selecciona independientemente de la lista que consiste en dodecil 2-bromoisobutirato, a-bromoisobutirato de etilo, a-bromoisobutirato de etilo, 2-bromoisobutirato de octadecilo, a-bromoisobutirato de metilo, 3-bromopropionato de metilo, 3-bromopropionato de terc-butilo, 2-bromopropionato de etilo.
Preferiblemente, el iniciador de la etapa (b) se selecciona independientemente de la lista que consiste en 1,1’azo-bis(cidohexanocarbonitrilo) (ACHN), 2,2’-didorhidrato de 2,2’-azo-bis (2-metilpropionamidina) (AAPH), 4,4’-azo-bis(ácido 4-cianovalérico) (ACVA), hidroperóxido de terc-butilo, hidroperóxido de cumeno, 2,5-di(tercbutilperóxido)-2,5-dimetil-3-hexino, peróxido de dicumilo y 2,5-bis(terc-butilperóxido)-2,5-dimetilhexano.
El porcentaje en peso del iniciador en la etapa (a) y la etapa (b) se encuentra entre 0.00001% y 0,2%.
Preferiblemente, el disolvente utilizado con el sistema catalítico metálico de la etapa (a) y la etapa (b) se selecciona independientemente de la lista que consiste en acetona, dimetilformamida, poli(etilenglicol), dimetilsulfóxido, 1-4 dioxano, etanol, propanol, hexano, agua, dióxido de carbono, líquido iónico, y una combinación de los mismos.
El porcentaje en peso del disolvente en la etapa (a) y la etapa (b) está por debajo del 90%; preferiblemente el porcentaje en peso del disolvente en la etapa (a) y la etapa (b) está entre 40% y 60%.
En otra realización preferida de la presente invención, el sistema catalítico metálico de la etapa (a) y la etapa (b) no utiliza ningún disolvente. En otras palabras, la etapa (a) y la etapa (b) se realizan sin disolvente ya que los monómeros son líquidos y son miscibles entre sí.
En una realización preferida del proceso de la presente invención, el sistema catalítico metálico de la etapa (a) utiliza Cu°/Cu2+ como metal de transición, tris(2-dimetilaminoetil)amina como ligando, 2-bromopropionato de metilo como iniciador, y dimetilsulfóxido como disolvente.
La etapa (c) del proceso de la invención hace referencia a la preparación por electrohilado de una membrana de nanofibras que comprende una mezcla, donde dicha mezcla comprende el primer copolímero obtenido en la etapa (a) y el segundo copolímero obtenido en la etapa (b). Preferiblemente, la etapa (c) se realiza en presencia de un aditivo capaz de aumentar la conductividad de la solución
mezcla/disolvente. Más preferiblemente, en presencia de ácido clorhídrico (HCl), en donde el porcentaje en peso de HCl en la etapa (c) está entre 0,0001% y 0,2%.
El disolvente de la etapa (c) se selecciona de la lista que consiste en acetona, dimetilformamida, poli(etilenglicol), dimetilsulfóxido, 1-4 dioxano, etanol, propanol, hexano, agua, dióxido de carbono, líquido iónico, y una combinación de los mismos. Más preferiblemente, el disolvente de la etapa (c) es dimetilsulfóxido.
Preferiblemente, el porcentaje en peso del disolvente utilizado en la etapa (c) se encuentra en un rango entre 20% y 98%.
La etapa (d) hace referencia a un tratamiento térmico de la membrana de nanofibras obtenida en la etapa (c), en donde el tratamiento térmico se aplica en forma de agua caliente en un rango de temperatura entre 30°C y 80°C y en donde la membrana de nanofibras obtenida en la etapa (c) se mantiene tensionada mediante una montura. La finalidad de esta etapa (d) es convertir la membrana de nanofibras obtenida en la etapa (c) de hidrófoba a hidrófila; se observa una transformación visual de la membrana cuando se realiza el tratamiento térmico en húmedo de la etapa (d) durante al menos 4 horas. Por favor, ha de señalarse que la membrana se mantiene hidrófila durante días, e incluso años.
Un tercer aspecto de la invención hace referencia a un proceso de preparación de la membrana hidrófila de nanofibras no tejidas que comprende nanopartículas de SiO2 , en donde dichas nanopartículas de SiO2 están homogéneamente dispersas en la membrana, caracterizado por que comprende todas las etapas del proceso de la invención:
a) síntesis del primer copolímero de (MA)3-co-(HEA)2 por polimerización radical viva catalizada por metal, utilizando un sistema catalítico metálico;
b) síntesis del segundo copolímero de (MMA)i-co-(HEMA)i por polimerización radical por transferencia de átomo inversa utilizando un sistema catalítico metálico;
c) preparación por electrohilado de una membrana de nanofibras que comprende una mezcla, donde dicha mezcla comprende el primer copolímero obtenido en la etapa (a) y el segundo copolímero obtenido en la etapa (b), y d) tratamiento térmico de la membrana de nanofibras obtenida en la etapa (c), en donde el tratamiento térmico se aplica en forma de agua caliente en un
rango de temperatura entre 30°C y 80°C, por ejemplo durante al menos 4 horas, y en donde la membrana de nanofibras en la etapa (c) se mantiene tensionada mediante una montura.
y en donde la mezcla de la etapa (c) comprende nanopartículas de SiO2.
Otro aspecto de la invención hace referencia a un proceso de preparación de la membrana hidrófila de nanofibras no tejidas que comprende nanopartículas de SiO2 , en donde dichas nanopartículas de SiO2 son físicamente adsorbidas, caracterizado por que comprende todas las etapas del proceso de la invención:
a) síntesis del primer copolímero de (MA)3-co-(HEA)2 por polimerización radical viva catalizada por metal, utilizando un sistema catalítico metálico;
b) síntesis del segundo copolímero de (MMA)1-co-(HEMA)1 por polimerización radical por transferencia de átomo inversa utilizando un sistema catalítico metálico;
c) preparación por electrohilado de una membrana de nanofibras que comprende una mezcla, donde dicha mezcla comprende el primer copolímero obtenido en la etapa (a) y el segundo copolímero obtenido en la etapa (b) y opcionalmente comprende nanopartículas de SiO2 , y
d) tratamiento térmico de la membrana de nanofibras obtenida en la etapa (c), en donde el tratamiento térmico se aplica en forma de agua caliente en un rango de temperatura entre 30°C y 80°C, por ejemplo durante al menos 4 horas, y en donde la membrana de nanofibras en la etapa (c) se mantiene tensionada mediante una montura.
y una etapa (e) adicional de impregnar la membrana obtenida en la etapa (d) en una suspensión de NPs- SiO2 y evaporar el disolvente.
Otro aspecto de la invención hace referencia a una membrana hidrófila de nanofibras no tejidas hidrolizada (en la presente memoria, la membrana hidrolizada de la presente invención), caracterizada por que comprende la membrana hidrófila de nanofibras no tejidas que comprende grupos carboxilo, en donde la concentración de grupos carboxilo en la membrana se encuentra en un rango entre 20 ^mol/g de la membrana y 3000 ^mol/g de la membrana.
El término "membrana hidrófila de nanofibras no tejidas hidrolizada” hace referencia en la presente memoria a la membrana hidrófila de nanofibras no tejidas mencionada anteriormente que ha sido parcialmente hidrolizada y ahora comprende grupos
carboxilo (COOH) y después secada parcialmente a temperatura ambiente (18-28°C). El número de grupos COOH accesibles en la membrana se encuentra en un rango de entre 20 ^mol/g de la membrana y 3000 ^mol/g de la membrana. Se observó que un tiempo de hidrólisis mayor que 1 hora, producía una alta rigidez en las membranas, volviéndolas frágiles y quebradizas.
En una realización preferida de la membrana hidrolizada de la presente invención, dicha membrana está funcionalizada con un catión divalente seleccionado de Zn+2, Ca+2, Mg+2 y Sr+2, un agente antibacteriano y/o cualquiera de las combinaciones de los mismos. Más preferiblemente, la membrana hidrófila de nanofibras no tejidas está funcionalizada con Zn+2, Ca+2 y doxiciclina.
La funcionalización de la membrana hidrolizada de la presente invención con un catión divalente seleccionado de Zn+2, Ca+2, Mg+2 y Sr+2 comprende una etapa de impregnar el hidrolizado con una solución de un catión divalente seleccionado de Zn+2, Ca+2, Mg+2 y Sr+2, y una etapa de secado a temperatura ambiente (18-28°C).
La funcionalización de la membrana hidrolizada de la presente invención con un agente bacteriano, más preferiblemente doxiciclina, comprende una etapa de impregnar el hidrolizado con una solución de un agente antibacteriano y una etapa de secado a temperatura ambiente (18-28°C).
De acuerdo con los grupos COOH accesibles en la membrana hidrófila de nanofibras no tejidas hidrolizada, la concentración de Ca2+ y Zn2+ cargada en la membrana como (COO-)2 se encuentra en un rango entre 0,0125 ^mol/g de membrana y 1500 ^mol/g de la membrana. Puede cargarse una concentración de calcio y zinc más elevada que 1500 ^mol/g de la membrana en la membrana hidrófila de nanofibras no tejidas hidrolizada, cuando todos los grupos COOH accesibles se coordinen. El exceso de Zn2+ o Ca2 es físicamente adsorbido en la superficie de la membrana en forma de sus respectivas sales (ZnCh y CaCh) durante el secado de la membrana.
La doxiciclina (DOX) se enlazó de forma no covalente en la membrana mediante interacciones ácido-base entre los grupos amino de la DOX y los grupos carboxilo de la membrana, además de por enlaces de hidrógeno entre los grupos hidroxilo de la membrana. Cuando todos los grupos carboxilo e hidroxilo disponibles en la membrana están enlazados a la doxiciclina, el exceso de DOX es físicamente adsorbido en la
superficie de la membrana durante el secado. La concentración de DOX se encuentra en un rango entre 0,01 mg/mg de la membrana y 1 mg/mg de la membrana.
Otro aspecto de la invención hace referencia a una membrana no reabsorbible para promover la regeneración ósea, caracterizado por que comprende la membrana hidrófila de nanofibras no tejidas mencionada anteriormente.
Otro aspecto de la invención hace referencia a una membrana periodontal no reabsorbible caracterizada por que comprende la membrana hidrófila de nanofibras no tejidas mencionada anteriormente.
Las membranas barrera reabsorbibles y no reabsorbibles están disponibles comercialmente, siendo las membranas no reabsorbibles de PFTE el estándar de oro en la regeneración ósea guiada. La principal desventaja de las membranas reabsorbibles es el tiempo de reabsorción imprevisible y las sustancias tóxicas liberadas durante la degradación, lo que afecta a la formación ósea. Entre muchas otras, la principal desventaja de las membranas barrera no reabsorbibles es que no consiguen la osteointegración. Más aún, en el caso de estas últimas membranas, es necesaria una segunda intervención quirúrgica para retirarlas después de la regeneración, lo que puede tener como resultado una herida en el tejido regenerado. Su poca eficacia resulta en un alto grado de recaídas.
En la presente invención la membrana no reabsorbible de la invención es una membrana bioactiva innovadora que permite:
• Una completa osteointegración, evitando la necesidad de una segunda cirugía • Una rápida regeneración ósea mejorando la precipitación de minerales naturales y la activación de las células formadoras de hueso. Sin necesidad de rellenar con precursores de hueso
• Reducir la proliferación de bacterias periodontales cuando comprende un agente antibacteriano
El último aspecto de la invención hace referencia a un recubrimiento para un implante caracterizado por que comprende la membrana hidrófila de nanofibras no tejidas anterior que puede proporcionar una ventaja para su osteointegración. La osteointegración implica contacto directo entre, por ejemplo, un implante de titanio y hueso. La mayoría de los implantes transcutáneos de metal no han tenido éxito,
principalmente debido a las infecciones. Los implantes transcutáneos de aleación de titanio producen partículas de corrosión y fracasan generalmente por mecanismos relacionados con la interacción superficial en el hueso, y provocan una inflamación, junto con un aflojamiento aséptico fibroso o una infección que pueden requerir de la retirada del implante. Además, las bajas concentraciones de oxígeno procedentes de una vascularización deficiente en una interfaz de superficie con un metal extraño, promueve un incremento de electrones relacionados con la célula huésped como en el caso de radicales libres y protones que puede fomentar infección e inflamación que influyen enormemente en el fracaso del implante. Cubrir el implante con la membrana hidrófila de nanofibras no tejidas mencionada anteriormente es una manera eficaz de evitar los riesgos mencionados.
A menos que se defina de otro modo, todos los términos técnicos y científicos utilizados en el presente documento tienen el mismo significado que el que se entiende comúnmente por un experto habitual en la técnica a la que esta invención pertenece. Pueden utilizarse métodos y materiales similares o equivalentes a los descritos en la presente memoria en la práctica de la presente invención. A lo largo de la descripción y las reivindicaciones, el término "comprender” y sus variaciones no pretenden excluir otras características técnicas, aditivos, componentes o etapas. Objetos, ventajas y características adicionales de la presente invención resultarán evidentes para los expertos en la técnica tras el análisis de la descripción, o pueden aprenderse con la práctica de la invención. Los siguientes ejemplos y dibujos se proporcionan a modo de ilustración y no pretenden limitar la presente invención.
BREVE DESCRIPCIÓN DE LOS DIBUJOS
FIG. 1. Modelización teórica de la copolimerización de MA y HEA, Fa vs la conversión de (A) y Fa vs fa (B).
FIG. 2. Perfil cromatográfico de HEA/MA-10/90 (A), HEA/MA-15/85 (B), HEA/MA-25/75 (C), HEA/MA-35/65 (D), HEA/MA-45/55 (E).
FIG. 3. Espectros de H1RMN de HEA/MA-10/90 (A), HEA/MA-15/85 (B), HEA/MA-25/75 (C), HEA/MA-35/75 (D)
FIG. 4. Modelización teórica de la copolimerización de MMA y HEMA, Fa vs la conversión (A) y Fa vs fa (B).
FIG. 5. Perfil cromatográfico de MMA-co-HEMA (A), y espectros de H1RMN de MMA-co-HEMA (B).
FIG. 6. Esquema del sistema de electrohilado: bomba de inyección (1), aguja de inyección (2), tambor colector (3), fuentes de alto voltaje (4), cono de Taylor (5) y eje mecánico con desplazamiento transversal (6).
FIG. 7. Membrana no tejida producida con las mezclas: (A)/(B) 0:100 (A), 100:0 (B), 25:75 (C), 50:50 (D), 75:25 (E).
FIG. 8. Membrana no tejida producida con la mezcla (A)/(B)75:25 (A), y con la mezcla (A)/(B)50:50 (B)
FIG.9. Tiss-OH antes (A) y después del calentamiento (B).
FIG.10. Red de colágeno conjuntivo (A), y membrana no tejida de nanofibras (Tiss-OH) (B).
FIG. 11. Micrografías de FESEM de las membranas después de 7 días de inmersión en SBFS; TissHYD (A), Tiss-Ca2+ (B), Tiss-Zn2+ (C).
FIG. 12. Histomorfometría ósea obtenida después de utilizar Tiss-Zn2+, por tinción con nitrato de plata (tinción von Kossa) para visualizar hueso mineralizado, a las seis semanas del seguimiento: sección de histología que incluye el defecto óseo y la región de interés (ROI) que muestra una gran formación de hueso denso (A). Superficie total (TS) en las ROI; los asteriscos (*) muestran la presencia de tejido de tipo medular y adiposo (B); se observan imágenes de unión ósea (BB). Perímetro del hueso (BPm) en las ROI (C). Grosor del hueso (BTh) con las mediciones trazadas en los ROI (D). Superficie osteoide (OS), en amarillo, en los ROI (E).
FIG. 13. Histomorfometría ósea obtenida por tinción con nitrato de plata (tinción von Kossa) para visualizar hueso mineralizado, a las seis semanas del seguimiento, después de no utilizar ninguna membrana- control (A) y Tiss-Ca2+ (B). Se observó la
formación del hueso trabecular a lo largo del margen del defecto calvárico (flecha), y dentro del defecto. Mbr: membrana, NB: hueso nuevo y OB: hueso antiguo (los punteros muestran islas de hueso dispersas, que corresponden a hueso nuevo).
FIG. 14. Histología ósea obtenida después de utilizar membranas de Tiss-Zn2+ (A) y Tiss-Ca2+ (B) mediante tinción con azul de toluidina para visualizar hueso mineralizado, a las seis semanas de tiempo de curación. Las flechas individuales indican la presencia de osteoblastos; las flechas dobles indican la presencia de osteocitos; las flechas enfrentadas hacen referencia a vasos sanguíneos; los punteros indican tejido conjuntivo fibroso. NB: hueso nuevo, Os: tejido osteoide.
FIG.15. Imagen de microscopio electrónico de barrido de emisión de campo (FE-SEM) de F. nucleatum, S. oralis, A. naeslundii, V. parvula, A. actinomycetemcomitans y P. gingivalis cultivados in vitro como una biopelícula de múltiples especies a las 12 horas de incubación en PTFE (control) (A), discos (B) de hidroxiapatita (HAp) TissHYD (C), Tiss-Ca2+ (D), Tiss-Zn2+ (E) y Tiss-DOX (F).
FIG.16. Número de bacterias [Log CFU/biopelícula media (desviación estandard)] de F. nucleatum, S. oralis, A. naeslundii, V. parvula, A. actinomycetemcomitans y P. gingivalis cultivadas in vitro como una biopelícula de múltiples especies a las 72 horas de incubación, medida por reacción en cadena de la polimerasa cuantitativa en tiempo real (qPCR) (N=3 para cada tiempo de incubación) en discos de Hidroxiapatita (HAp) (A), PTFE (control) (B), T-COOH (C),Tiss-Ca2+ (D), Tiss-Zn2+ (E) y Tiss-DOX (F).
Fig.17. FESEM micrográfías de tejidos después del dopaje con dióxido de silicio e inmersión en SBFS durante 7 días: Tiss-SiO2-COOH (A), Tiss-SiO2-Ca2+ (B), Tiss-SiO2-Zn2+ (C) y Tiss- SiO2-DOX (D).
EJEMPLOS
1. SÍNTESIS DE COPOLÍMEROS
1.1 Preparación de copolímeros de (MA-co-HEA)
En la presente invención, se ha optimizado una variante de la polimerización radical viva catalizada por metal (MC-LRP): la polimerización radical viva mediada por cobre (Cu0-MC-LRP) ha sido optimizada para obtener un copolímero lineal de acrilato
hidrófilo (MA-co-HEA) con topología estadística y con un alto peso molecular (por encima de 1x106 Da).
El sistema Cu0-LRP utilizado fue: 2-bromopropionato de metilo como iniciador, tris(2-dimetilaminoetil)amina como ligando, Cobre/ Cobre(II) como metal de transición: MBP/M6-TREN/Cu0/BrCu2 , y se utilizó dimetil sulfóxido (DMSO) como disolvente. Los monómeros seleccionados fueron: acrilato de metilo (MA), e hidroxietil acrilato (HEA).
En primer lugar, se realizó un análisis teórico de la copolimerización de MA y HEA utilizando el modelo terminal [Mayo, F. R.; Lewis, F. M. J. Am. Chem Soc. 66, (1944), 1594-1601], El modelo terminal asume que la reactividad radical únicamente depende de la unidad terminal de la cadena en crecimiento, de tal manera que la fracción molar del monómero-a en el copolímero (Fa ) depende únicamente de las fracciones molares del monómero (fa and fb, con fa + fb, =1) y las relaciones de la reactividad del monómero:
F rafg f a f b
“ ^a
f a + 2 f a f b rb f ¿
donde ra y rb son las relaciones de reactividad de co-polimerización ra = kpaa/kpab, rb = kpbb/kpba ; kp¡j es el coeficiente de la tasapropagación para la adición del monómero j al radical i, Las relaciones de reactividad de la copolimerización para MA y HEA utilizadas son ra=0,94 y rb=0,90 respectivamente,
La Fig. 1 muestra la modelización teórica de la copolimerización de MA y HEA: Fa vs la conversión (Fa es la fracción molar de MA en el copolímero a lo largo de la polimerización) para diferentes fracciones molares iniciales de MA en la alimentación (f0a), y Fa vs fa (fa es la fracción molar de MA en la alimentación a lo largo de la polimerización).
La modelización teórica de la copolimerización de MA y HEA muestra que Fa es prácticamente igual a la fracción molar inicial de la alimentación (Fig. 1A) para cualquier fracción molar inicial de MA (Fig. 1B), El sistema pasa por una copolimerización casi azeotrópica a cualquier fracción molar inicial de la alimentación, Por lo tanto, la copolimerización teórica de MA y HAE proporciona un copolímero con topología estadística (MA-HEA-MA-HEA-MA-HEA................ ),
La Cu0-LRP es muy sensible a cualquier traza de impurezas: principalmente el inhibidor que contiene ambos monómeros, y los di-acrilatos que se forman en el monómero HEA por condensación de las moléculas de HEA. De este modo, en Cu0-LRP las impurezas a una muy baja concentración proporcionan rendimientos bajos, pesos moleculares bajos y un polímero reticulado, y por tanto es estrictamente necesario purificar de forma adecuada los monómeros.
Protocolo de purificación de HEA:
1. En primer lugar, se purificó HEA en una columna de alúmina básica.
2. Se disolvieron 70 ml de HEA previamente purificado en columna de alúmina básica, en 210 ml de agua destilada, y a continuación se eliminaron trazas de diacrilato de etilenglicol mediante 11 extracciones líquido-líquido con 210 ml de hexano.
3. A continuación se disolvieron 58 g de NaCl en la solución acuosa de HEA, y se extrajo el monómero de HEA mediante 5 extracciones líquido-líquido con 200 ml de éter dietílico.
4. La solución de HEA con éter dietílico se secó con 300g de sulfato de sodio anhidro: la solución se agitó unos pocos minutos y a continuación se filtró para retirar el sulfato de sodio.
5. A continuación, el éter dietílico fue evaporado completamente en un evaporador rotativo, y el HEA purificado se almacenó a -20°C.
Protocolo de purificación de MA:
1. El volumen requerido de MA se hizo pasar a través de una columna de alúmina básica.
Una vez que se purificaron los monómeros, se sintetizaron seis copolímeros de (MA-co-HEA)_con diferente % molar de HEA y MA en la alimentación mediante Cu0-LRP. Los seis valores diferentes de % molar de HEA/MA fueron: a) HEA/MA 10/90 b) HEA/MA 15/85, c) HEA/MA 25/75, d) HEA/MA 34/66, e) HEA/MA 45/55, f) HEA/MA 55/45. La Tabla 1 muestra el % en peso de cada componente en la mezcla de polimerización final para cada % molar HEA/MA.
.
MA.

La masa total de los monómeros (MA HEA) 59,2700 g se añadió en matraces Schlenk, y a continuación se añadió: 59,2700 g de DMSO, 0,0020 g de Cu0, 0,0160 g de tris[2-(dimetilamino)etil]amina (M6-TREN), 0,0012 g de CuBr2 , y 0,0060 g de 2-bromopropionato de metilo (MBP). Los matraces se cerraron con un tapón septum, el oxígeno se retiró burbujeando nitrógeno durante unos pocos minutos, y a continuación se realizaron cuatro ciclos de congelación-vacío-descongelación (después del último ciclo de congelación-vacío-descongelación los matraces se llenaron con nitrógeno). A continuación el matraz sellado se colocó en un baño de aceite termostático a 25°C durante 24h. A continuación los copolímeros se purificaron disolviéndolos en acetona y precipitándolos en agua destilada (dos veces). Después de la purificación los copolímeros se secaron bajo vacío a 80° C hasta un peso constante. Los copolímeros a), b), c), d) tenían un color blanco y una textura de goma, y la conversión final fue entre 90-95% en peso en todos los casos. La copolimerización de e) y f) (Tabla 1) no tuvo lugar de forma apropiada: el rendimiento estuvo por debajo del 40%, y el copolímero no tenía una textura de goma. Por lo tanto el rango de % molar óptimo de HEA en la alimentación estaba entre 10% y 34%.
Los copolímeros de (MA-co-HEA) se caracterizaron por GPC (Viscotek 270max de Malvern) y H1RMN (espectrómetro de Bruker Avance 400 MHz). Las muestras para GPC se prepararon disolviendo 1 mg de cada copolímero en 10 ml de 1-metil-2-pirrolidinona (NMP) y se analizaron por triplicado.
La Fig. 2 muestra un perfil cromatográfico de cada copolímero, y la Tabla 2 muestra el peso molecular (Mw y Mn) y Mw/Mn. La Fig. 3 muestra los espectros de H1RMN de dichos copolímeros.
Tabla 2. Pesos moleculares de cada copolímero sintetizado.

Tabla 3 muestra el % molar real de HEA en cada copolímero: se calculó por la relación de la intensidad entre las señales a (CH3 de MA) y b (CH2-CH 2 de HEA) de los espectros de H1RMN.
Tabla 3. % molar real de HEA en cada copolímero sintetizado.

El análisis de H1RMN muestra que las concentraciones de HEA en los copolímeros son prácticamente las mismas que las concentraciones de alimentación.
La solubilidad de los copolímeros de acrilato sintetizados se sometió a prueba en acetona, dimetilformamida (DMF), dimetilsulfóxido (DMSO), 1-4 dioxano y NMP. Los copolímeros fueron totalmente solubles en todos los disolventes sometidos a ensayo hasta el 6% en peso: por encima de 6%, la viscosidad de las soluciones era extremadamente elevada. La solución al 6% en peso de menor viscosidad fue la solución preparada en DMF, lo que indica que el disolvente DMF es el mejor disolvente para estos copolímeros.
Para tener la máxima concentración de grupos funcionales (grupos OH) en la membrana no tejida de nanofibras, el copolímero de acrilato seleccionado para las formulaciones de la mezcla fue (MA)3-co-(HEA)2 (Tabla 2).
1.2 Preparación de copolímeros de MMA-co-HEMA
La modelización teórica de la copolimerización de MMA y HEMA con topología estadística (MMA-HEMA-MMA-HEMA-MMA-HEMA.................) se muestra en la Fig.
4: Fa vs conversión en donde Fa es la fracción molar de MMA en el copolímero a lo largo de la polimerización, para diferentes fracciones molares iniciales de MMA (foa) en la alimentación, y Fa vs fa en donde fa es la fracción molar de MMA en la alimentación a lo largo de la polimerización.
La técnica Cu0-LRP utilizada para sintetizar los copolímeros de MA-co-HEA no funcionó bien en la copolimerización de los monómeros metacrílicos (MMA y HEMA): se obtuvieron rendimientos y pesos moleculares muy bajos, y también la concentración de HEMA en los copolímeros fue mucho más baja que las concentraciones de la alimentación.
Por lo tanto, para sintetizar un copolímero de metacrilato químicamente miscible con (MA)3-co-(HEA)2 , la copolimerización de MMA y HEMA se llevó a cabo mediante otra variante de MC-LRP como es la polimerización radical por transferencia de átomo inversa (ATRP inversa) [Chem. Rev. 101,(2001),2921-2990.]: No puede obtenerse un peso molecular tan alto como en Cu0-LRP mediante ATRP inversa, pero la ATRP inversa es mucho menos sensible a las impurezas, y por tanto más sencilla de llevar a cabo.
El sistema de ATRP inversa utilizado fue: 2,2'-azo-bis(2-metilpropionitrilo) (AIBN) como iniciador, N,N,N',N'',N''-pentametildietilentriamina (PMDETA) como ligando, cobre(II) como metal de transición, y se utilizó una mezcla de dimetilsulfóxido (DMSO)/xileno como disolvente. Los monómeros seleccionados fueron metacrilato de metilo (MMA), y 2-hidroxietilmetacrilato (HEMA).
Protocolo de purificación de MMA y HEMA:
1. El volumen requerido de MMA y HEMA se hizo pasar a través de una columna de alúmina básica.
En un matraz de dos bocas equipado con un agitador magnético y un reflujo, se añadieron: 95,00ml de DMSO, 0,14g de CuBr2 , 0,24g de N,N,N',N",N"-pentametildietilentriamina (PMDETA), 90,08g de MMA y 40,12g de HEMA previamente purificados, 0,23g de 2,2'-azo-bis(2-metilpropionitrilo) (AIBN) disuelto en 60,02 g de xileno. La mezcla se agitó a 250 rpm, cuando todos los componentes se disolvieron completamente la mezcla de reacción se enfrió a 0° C y se purgó con nitrógeno de alta pureza durante 20 min. A continuación, se realizó la reacción a 80°C en un baño de aceite durante 6 horas. Después de la polimerización, el copolímero fue purificado disolviéndolo en acetona y precipitándolo en agua destilada tres veces. Entonces, el copolímero sólido se lavó con agua destilada 3 veces, y se secó bajo vacío a 80°C hasta un peso constante. El copolímero de metacrilato tenía un color blanco y una textura dura y quebradiza. La conversión fue del 70%.
% en peso de cada componente en la mezcla de polimerización: 33,200% de DMSO, 0,050% de CuBr2 , 0,083% de PMDETA, 14,030% de HEMA, 31,500% de MMA, 0,080% de AIBN y 21,000% xileno.
El copolímero MMA-co-HEMA se caracterizó mediante GPC (Viscotek 270 max de Malvern) y mediante H1RMN (espectrómetro Bruker Avance 400 MHz). Las muestras para GPC se prepararon disolviendo 1 mg de copolímeros en 10 ml 1-Metil-2-pirrolidinona (NMP) y se analizaron por triplicado. La Fig. 5 muestra el perfil cromatográfico y el espectro de H1RMN del copolímero MMA-co-HEMA.
Tabla 4 muestra los pesos moleculares: Mw y Mn y Mw/Mn calculados por GPC.
Table 4. Pesos moleculares de los copolímeros MMA-co-HEMA.
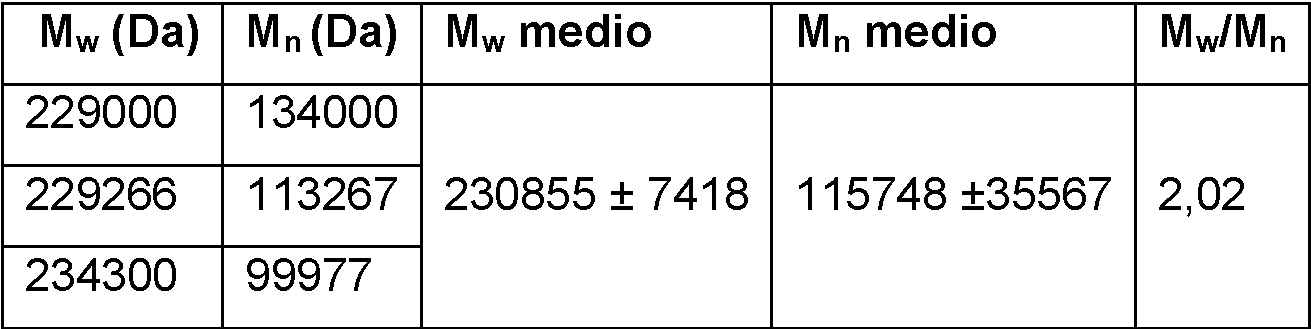
Además, la Tabla 5 muestra el % molar real de HEMA en el copolímero calculado mediante la intensidad de las señales a (CH3 de MMA) y b (CH2-CH 2 de HEMA) de los espectros 1H-NMR.
Tabla.5. % molar real de HEMA en los copolímeros.

Se sometió a ensayo la solubilidad de (MMA)1-co-(HEMA)1 en acetona, dimetilformamida (DMF), dimetilsulfóxido (DMSO), 1-4 dioxano y NMP, y el copolímero era totalmente soluble hasta un porcentaje de 38% en peso; por encima de 38% la viscosidad de las soluciones era extremadamente elevada. Para (MMA)1-co-(HEMA)1 la solución al 38% en peso de menor viscosidad fue también la preparada en el disolvente DMF, lo que indica que la DMF es también el mejor disolvente para este copolímero.
2. PREPARACIÓN DE UNA MEMBRANA NO TEJIDA POR ELECTROHILADO
La solubilidad entre los copolímeros (MMA)1-co-(HEMA)1; (A) y (MA)3-co-(HEA)2 ; (B) se sometió a ensayo en acetona, dimetilformamida (DMF), dimetilsulfóxido (DMSO), 1 4 Dioxano y NMP. Las relaciones (A)/(B) masa/masa sometidas a ensayo fueron: 10/90, 25/75, 50/50, 75/25 y 90/10, y la relación ((A)+(B))/disolvente, masa/masa fue 3/97 en todos los casos. Ambos copolímeros resultaron completamente solubles, a todas las relaciones. Las soluciones de una viscosidad inferior fueron las soluciones de DMF, y por tanto se seleccionó DMF como el disolvente para optimizar el proceso de electrohilado.
Para estudiar y modular las propiedades mecánicas de las membranas no tejidas, las mezclas seleccionadas (A)/(B) masa/masa para ser procesadas por electrohilado fueron: 0/100, 25:75, 50:50, 75:25 y 100/0, y la mezcla /disolvente, masa/masa fue
3/97. Cuando los copolímeros se disolvieron completamente, las soluciones se cargaron en jeringas de 20 cm3 de teflón (Becton & Dickinson) y fueron extruidas a través de un capilar de acero inoxidable, con diámetros externos e internos de 1,5 mm y 1,1 mm, respectivamente. El sistema de inyección se acopló a un sistema mecánico con movimiento axial, se seleccionaron los caudales y las tensiones para permitir la recogida de fibras secas membranas no tejidas, y las fibras se recogieron en un colector de tambor rotativo. La Fig. 6 muestra un esquema del sistema de proceso de electrohilado; y la Tabla 6 muestra los parámetros utilizados en el procesado.
Tabla 6. Parámetros del proceso de electrohilado.

La Fig. 7 muestra un análisis por microscopía SEM de las membranas no tejidas de nanofibras obtenidas con las mezclas (A)/(B) 0:100 (A), 100:0 (B), 25:75 (C), 50:50 (D), 75:25 (E).
El copolímero (B) puro proporciona materiales gomosos elásticos, en los que las fibras están 100% fusionadas entre sí, formando una película (Fig. 7A).
Las membranas no tejidas obtenidas con el copolímero (A) puro, no tenían ningún punto de fusión entre las fibras, éstas estaban completamente sueltas (Fig. 7B), y esto proporcionó materiales con una nula resistencia a la abrasión; unos materiales difícilmente manipulables.
En las membranas no tejidas obtenidas con la mezcla (A)/(B) 25:75 (Fig. 7C), la fusión entre las fibras ya no fue del 100% como en el caso del copolímero (B) puro (Fig. 7A), pero seguía habiendo muchos puntos de fusión entre las fibras (círculos de color negro en la Fig. 7C), y por tanto se obtuvieron membranas no tejidas con una superficie específica muy baja y una elasticidad muy alta.
La mezcla (A)/(B) 75:25 (Fig. 7E) proporcionó membranas no tejidas compactas con una resistencia a la abrasión mejor que la obtenida con el copolímero (A) puro (Fig. 7B), pero las membranas no eran aún flexibles y mostraron una resistencia muy baja a la tensión.
La mezcla (A)/(B) 50:50 masa/masa (denominado Tiss-OH; Fig. 7D) proporcionó membranas no tejidas compactas con excelentes propiedades mecánicas: resistencia a la abrasión muy alta, alta flexibilidad, alta elasticidad, alta resistencia al estrés mecánico, y por tanto fácil de manipular: puede cortarse, doblarse, retorcerse,... etc.
La baja resistencia a la abrasión de la membrana no tejida producida con la mezcla (A)/(B)75:25, en comparación con la membrana no tejida producida con la mezcla (A)/(B)50:50 se muestra en la Fig. 8.
Proceso para el escalado del Electrohilado de Tiss-OH
Para aumentar la producción de Tiss-OH, se estudió la relación mezcla/disolvente (DMF) masa/masa. La relación óptima mezcla/DMF en % masa/masa fue 13,50/86,50: por encima de este valor la viscosidad fue demasiado alta para poder ser procesada por electrohilado.
Para mantener el proceso de electrohilado estable en el tiempo, fue necesario aumentar la conductividad de la solución mezcla/disolvente mediante adición de 0,048g de ácido clorhídrico (HCl).
La solución óptima mezcla/disolvente % masa/masa para el escalado del proceso de electrohilado se preparó de la siguiente forma: se disolvieron 5,000g de (MA)3-co-(HEA)2 (6,246% en peso) y 5,000 g de (MMA)i-co-(HEMA)i (6,246% en peso), en 70,000g de DMF (87,4479% en peso), cuando los copolímeros se disolvieron completamente se añadió 0,048g de ácido clorhídrico (HCl) (0,059% en peso) a la solución.
El sistema de electrohilado fue el mismo que el que se muestra en la Fig. 6, pero para aumentar la producción, el sistema de inyección de una única aguja de la Fig. 6 fue reemplazado por un cabezal de diez agujas.
Los parámetros del proceso de electrohilado optimizados para un cabezal de diez agujas se muestran en la Tabla 7.
Tabla 7. Parámetros del proceso de escalado de electrohilado

El grosor de Tiss-OH fue controlado fácilmente de unas pocas mieras a cientos de mieras, controlando el tiempo de procesado (2h de procesado « 45 ^m de grosor).
2.1. Conversión de Tiss-OH de hidrófobo a hidrófilo
Inicialmente Tiss-OH es hidrófobo; para convertirlo en un material hidrófilo es necesario realizar un tratamiento térmico adicional. El tratamiento térmico se realizó introduciendo Tiss-OH en un baño de agua caliente (40°C) durante 5 horas. Para evitar la contracción de los materiales durante el tratamiento térmico, se mantuvieron tensados utilizando bastidores.
El tratamiento térmico produce una reorientación irreversible de los dominios hidrófobos e hidrófilos presentes en la superficie de las fibras, lo que causa que el material pase de ser completamente hidrófobo a ser altamente hidrófilo: los grupos OH de las fibras se reordenan para interactuar mediante enlaces de hidrógeno con las moléculas de agua, mientras que los grupos hidrófobos se ocultan del agua.
Después del tratamiento térmico, se calculó la capacidad de adsorción de agua (Q) de Tiss-OH, teniendo en cuenta la expresión:
Q = (masa del agua absorbida) / (Masa de la membrana no tejida seca)
Se secaron seis muestras de membranas no tejidas en un horno de vacío a 50°C durante 2h. A continuación, se sumergieron en agua destilada durante 3h a temperatura ambiente; el agua retenida en la superficie de las muestras se retiró utilizando un papel de celulosa. Posteriormente, las muestras se pesaron y la Q calculada fue 2,06 ± 0,15.
Más aún, para calcular aproximadamente la distribución del tamaño de poro de Tiss-OH, se ha realizado un ensayo de filtración a vacío utilizando una serie de suspensiones acuosas de nano y micropartículas hidrófilas monodispersas con tamaños entre 150nm y 5000nm de diámetro. El Tiss-OH permitió el paso de partículas de 800nm a 3000nm de diámetro.
La resistencia térmica del Tiss-OH, se estudió sumergiéndolo en agua a 100°C durante 24h. La estructura interna, las propiedades mecánicas y la masa del Tiss-OH fueron exactamente las mismas antes y después del calentamiento (Fig. 9).
2.2. Hidrólisis de Tiss-OH para obtener Tiss-HYD
La malla de colágeno natural que forma el tejido conjuntivo de los huesos está compuesta de nanofibrillas de aproximadamente 50nm que se agrupan para formar fibras de aproximadamente 500nm con una morfología, propiedades mecánicas y físico-químicas similares a las deTiss-OH.
La Fig. 10 son imágenes de SEM de una red de colágeno conjuntivo (A) y de Tiss-OH (B), que muestran una estructura morfológica muy similar.
Para introducir grupos carboxilo (COOH) en la superficie de las fibras, se realizó una hidrólisis parcial de los grupos éster: R-COOCH3 y RCOOCH2CH2OH de la membrana Tiss-OH de 45 micras de grosor (Fig. 7D ), para obtener TissHYD. La solución de hidrólisis fue carbonato de sodio (333 mM), pH=12,50. Se observó que un tiempo de hidrólisis mayor de 1 hora, producía una rigidez elevada en las membranas, volviéndolas frágiles y quebradizas. Por lo tanto el tiempo seleccionado para la hidrólisis fue 30 min. Por lo tanto, lal hidrólisis se realizó introduciendoTiss-OH en una solución de carbonato de sodio (333 mM), pH=12,50 durante 30 min. A continuación, las membranas (TissHYD) se lavaron 3 veces con agua destilada y se secaron a temperatura ambiente. El número de grupos de COOH calculado mediante el ensayo de adsorción de azul de toluidina O (método del TBO) de acuerdo con Biomateríals.14, (1993), 817-822. El ensayo incluye la incubación de matrices carboxiladas con azul de toluidina O en un tampón alcalino con posterior lavado, seguido de elución y cuantificación de TBO eludido mediante espectrometría UV-Vis. El número de grupos carboxilo accesibles fue de 560±50 ^mol/g de la membrana. Después de la hidrólisis la Q calculada fue 3,06 ± 0,20.
2.2. Funcionalización de TissHYD con Zn+2 (Tiss-Zn2+) y Ca2+ (Tiss-Ca2+) TissHYD se funcionalizó con Zn+2 (Tiss-Zn2+) y Ca2+ (Tiss-Ca2+). La capacidad de los grupos carboxilo para formar complejos con cationes divalentes se utilizó para funcionalizar TissHYD (membranas Tiss) con Zn+2 y Ca2+. TissHYD se impregnó con una solución de Zn2+ y Ca2+, y a continuación el agua se evaporó en vacío a temperatura constante: de este modo se cargó TissHYD con 1,1 ^g/mg, de Ca2+ y Zn2+.
2.3. Funcionalización de TissHYD con doxiciclina (Tiss-DOX)
La Doxiciclina (DOX) se unió de forma no covalente en TissHYD mediante interacciones ácido-base entre los grupos amino de DOX y los grupos carboxilo de TissHYD. El TissHYD se impregnó con una solución de DOX de dos veces su masa, de 40 mg/ml, y a continuación el agua se evaporó a vacío a temperatura constante: de esta forma el TissHYD se cargó con 0,8 mg de DOX/mg Tiss.
3. ENSAYO ESTÁTICO CELULAR DE BIOACTIVIDAD IN VITRO DE TissHYD, Tiss-Zn2+, Tiss-Ca2+
Las membranas deberían potenciar la formación ósea a través de la bioactividad, por lo tanto para dicha aplicación, se realizó el análisis propuesto por Kokubo (ISO 23317:2012. Implants for surgery. In vitro evaluation for apatite-forming ability of implant materials).
Las membranas se impregnaron en 20 ml de solución de fluido corporal simulado (SBFS, del inglés simulated body fluid solution) [pH 7,45] en matraces estériles durante 7 días. Los reactivos por 1000 ml de SBFS fueron: 8,035 g de NaCl, 0,355 g de NaHCO3, 0,225 g de KCl, 0,231 g de K2HPO4 3 H2O, 0,311 g de M gCh^^O , 39 g de 1M HCl, 0,292 g de CaCh, 0,072 g de Na2SO4 , 118 g de Tris, 0 a 5 ml de 1M HCl para el ajuste del pH final.
Después de secar, las superficies se analizaron mediante FESEM a 2,5 Kv, a una distancia de trabajo de 3,5 mm y se realizó un análisis elemental mediante un EDX acoplado al FESEM, a una distancia de trabajo de 15 mm. Los resultados de las imágenes de FESEM de TissHYD, Tiss-Ca2+ y Tiss-Zn2+ después de 7 días de inmersión en SBFS se presentan en la Fig. 11.
Después de la inmersión, las diferencias entre los grupos quedaron evidenciadas:
En TissHYD (Fig. 11A) raramente se observaron algunos depósitosredondeados en las muestras. Se observaron trazas de calcio en los espectros del EDX.
En Tiss-Ca2+ (Fig. 11B), se observó un aumento en el diámetro de las nanofibras, y las nanofibras perdieron su apariencia suave. Unos puntos de depósitos de calcio se distribuyeron de manera uniforme por la superficie de las nanofibras.
En Tiss-Zn2+ (Fig. 11C), el diámetro de las nanofibras se vio sumamente aumentado (de 300 a aproximadamente 500nm), y los depósitos de mineral (100nm) fueron distribuidos debuidos forma aleatoria sobre las superficies de las nanofibras.
Se encontró calcio y fósforo en los espectros del EDX en las superficies de las nanofibras. Se identificaron numerosas aglomeraciones de nanocristales esféricos en la superficie del Tiss-Zn2+. Los SBFS son fluidos con concentraciones de iones prácticamente iguales a las del plasma de la sangre humana y se emplean para evaluar la bioactividad de los biomateriales para la reparación de tejidos duros. El zinc promovió la precipitación biomimética de los depósitos de Ca/P y la formación de nanocristales de Hidroxiapatita (HAp, Ca10(PO4)6 (OH)2) durante su inmersión en SBFS. La formación de complejos de zincen los tejidos facilitó el enlace de grupos fosfato. Estos grupos fosfato, en la superficie, tienen oxígenos infracoordinados, lo que conduce a la creación de superficies reactivas que atraerán iones de calcio de SBFS. Esta deposición biomimética de Ca/P se considera como un método de revestimiento inspirado por el proceso natural de biomineralización. Más aún, debe considerarse que la HAp cristalina es muy lenta de reabsorber, y la mayoría de los sustitutos óseos basados en HAp no se reabsorben o se reabsorben de forma extremadamente lenta. Sin embargo, si se precipita HAp o nano-HAp en las superficies, no se reabsorbe, lo que facilita la regeneración de tejidos duros. La remineralización biomimética de los tejidos sometidos a ensayo facilitarán la regeneración ósea. La HAp facilita la formación de otros materiales similares a esta apatita ósea, tal como es el carbonato-HAp, y es capaz de estimular a las células, lo que conduce a la formación de hueso. Más aún, la HAp promueve la osteoconductividad. Los osteoblastos estimulados con Ca2+ y PO42" extracelular aumentaron la expresión de ARNm de la proteína morfogenética ósea 2. El factor de crecimiento de fibroblastos-2 (FGF-2) y los niveles de expresión de proteínas también aumentan por la concentración de Ca2+ extracelular.
4. FORMACIÓN ÓSEA EN MODELO DE DEFECTO EN CALOTA DE CONEJO
Se sometieron a ensayo tres tipos de membranas, Tiss-Zn2+ (cargada con 1.1 ^g(Zn2+)/mg Tiss), Tiss-Ca2+ (cargada con 1.1 ^g(Ca2+)/mg Tiss) y TissHYD. Se utilizaron defectos desnudos sin ningún tipo de membrana, como control. Se seleccionaron seis conejos para experimentación de raza Nueva Zelanda con idénticas características (edad: 6 mese; peso: 3,5-4 kg) para el estudio, y se alimentaron diariamente con régimen de alimentación Harlan-Teckland Lab Animal Diets (2030). Las intervenciones quirúrgicas se realizaron en centro de cirugía mínimamente invasiva Jesús Usón (CCMI, Cáceres, España). El experimento se desarrolló de acuerdo con las directrices del Instituto Nacional de la Salud (NHI) de EE.UU. y de la directiva europea 86/609/EEC referente al cuidado y uso de animales para la experimentación. El estudio también cumplió con la directiva europea 2010/63/EU sobre la protección de animales utilizados para propósitos científicos y con todas las leyes y regulaciones. Los investigadores obtuvieron la aprobación del Comité de Ética de la Institución. Según se requiere por el marco legislativo, se utilizó el mínimo número de animales por razones éticas. Se han publicado modelos en referencia a los métodos histológicos y de experimentación animal.
Antes de comenzar el procedimiento quirúrgico, se tomaron las constantes vitales y a continuación se procedió a la inmovilización de los conejos. Se administró midazolam (0,25 mg/kg) y propofol (5 mg/kg) por vía intravenosa como anestesia para inducción y también se utilizó una inhalación de gas sevoflurano al 2,8% inspirado. Se proporcionó analgesia con ketorolac (1,5 mg/kg) y tramadol (3 mg/kg). Una vez que los animales se seleccionaron y se prepararon, se realizaron incisiones con un escalpelo del n° 15 entre las ases de sus orejas y entre sus ojos. Se realizó un área quirúrgica triangular después de conectar las dos incisiones con otra en la línea central del cráneo. Se separaron los tejidos epitelial, conjuntivo, y muscular con un periostótomo Prichard del área de operación y se lavó la superficie del cráneo con una solución salina estéril. Se crearon seis defectos óseos no críticos (diámetro: 6 mm; profundidad: 3 mm) en el hueso parietal, a cada lado de la línea central del cráneo, separados 3 mm, utilizando una trefina (Helmut-Zepf Medical Gmbh, Seitingen, Alemania) montada en un micromotor de implantes que opera a 2000 rpm bajo irrigación de suero salino. La trefina tenía un diámetro externo de 6 mm, una longitud de 30 mm, y dientes de 2,35 mm. Se utilizó cirugía piezoeléctrica para retirar la tabla interna y el hueso medular en cada defecto. La profundidad se controló con una sonda periodontal. Una membrana asignada aleatoriamente se utilizó cubrir cada defecto óseo, dejando un defecto
desnudo en cada animal. La secuencia de aleatorización fue generada utilizando un software específico (Research Randomizer, V. 4.0, Urbaniak GC & Plous S, 2013). Las membranas se fijaron con el adhesivo tisular de fibrina Tissucol (Baxter, Hyland S.A. Immuno, Rochester, MI, EE.UU.), que fue situado en los bordes del hueso adyacente a los defectos. La adhesión adecuada y la movilidad limitada de las membranas se confirmaron cuando las aletas se desplazaron nuevamente de vuelta a su posición inicial. Se realizaron suturas en los siguientes planos utilizando material reabsorbible: periósticas (4/0), sub-epidérmicas (4/0) y cutáneas (2/0). Se utilizaron puntos simples tan cerca como fue posible al borde. La herida se limpió cuidadosamente con una solución salina estéril. Se administró analgesia antiinflamatoria (buprenorfina 0,05 mg/kg y carprofeno 1 ml/12,5 kg). Los animales se sacrificaron seis semanas tras la cirugía utilizando una sobredosis intravenosa de una solución de cloruro de potasio. Se obtuvieron muestras del cráneo de cada espécimen, cortándolas en un plano anatómico sagital. Después de que se separara la masa del cerebro y el cráneo se lavara con una solución salina estéril, las muestras de tejido se cortaron y se marcaron individualmente. Se recuperaron los especímenes en bloques craneales y se almacenaron en una solución de formaldehído al 5% (pH 7) y los bloques se recuperaron del defecto óseo regenerado utilizando una sierra de autopsia oscilante (Exakt, Kulzer, Wehrheim, Alemania). Los especímenes diseccionados se sumergieron inmediatamente en una solución de formaldehído al 4% y calcio al 1% y se procesaron para seccionarlas y pulirlas (ground sectioning) siguiendo el método de Donath y Bruener. Para la tinción histológica y un rápido análisis de contraste del tejido (Azul de toluidina de Merck -Merck, Darmstadt, Alemania), se utilizó un colorante metacromático utilizado para evaluar el porcentaje de formación de hueso nuevo. Para visualizar el hueso mineralizado, se utilizó la técnica de tinción de von Kossa (VK) con nitrato de plata (Sigma-Aldrich Chemical Co., Poole, Reino Unido), utilizando el software Image J. Se compilaron los siguientes datos: superficie ósea (BS), superficie osteoide (OS), porcentaje de la superficie osteoide (OS/TS), perímetro óseo (BPm) y grosor del hueso (BTh). Se eligió una solución de azul de toluidina al 1% (TB) con un pH de 3,6 que se ajustó con HCl 1 N. Las muestras fueron expuestas a la tinción durante 10 minutos a temperatura ambiente, se enjuagaron con agua destilada, y se secaron al aire. Se analizaron osteocitos, osteoblastos y vasos sanguíneos en secciones coloreadas con TB.
Se calcularon las medias y las desviaciones estándar (DE) en píxeles, y a continuación se convirtieron a mm o mm2. Se aplicó un ANOVA de una vía y una prueba t de muestras por pares, con un nivel de significación de p < 0,05.
Las membranas implantadas fueron bien toleradas por los tejidos blandos circundantes, sin ninguna evidencia de necrosis, síntomas de alergia, reacciones inmunes, o incompatibilidad. Ningún espécimen mostró señal alguna de inflamación o infección inducida por el uso de los biomateriales.
La técnica de tinción de von Kossa (VK) permitió observar que todos los defectos óseos tratados con membranas mostraron mayor superficie ósea (BS) y grosor del hueso (BTh) que el grupo de control (Tablas 9 y 10). En la Fig. 12 se muestra un defecto óseo con una membrana de Tiss-Zn2+ implantada y teñida con nitrato de plata (tinción von Kossa) para visualizar el hueso mineralizado, a las seis semanas del seguimiento.
La Fig. 12 es una sección histológica que incluye el defecto óseo y la región de interés (ROI), que muestra una gran formación de hueso denso.
La Fig. 12B es la superficie total (TS) en la ROI; los asteriscos (*) muestran la presencia de médula y tejido de tipo adiposo. Se observan imágenes de la unión ósea (BB).
En la Fig. 12C, se observa el perímetro del hueso (BPm) en la ROI, y en la Fig. 12D, se mide el grosor del hueso (BTh) con las mediciones trazadas en la ROI.
En la Fig. 12E, se representa una superficie osteoide (OS). Puede apreciarse cómo las membranas tienen un BTh mayor que el control (Ctr), y por tanto produjeron más superficie osteoide (OS), en comparación con el grupo de control (ver la relación OS/TS en las Tablas 9 y 10).
Tabla 9. Datos histomorfométricos obtenidos dentro del hueso nuevo formado en la región de interés (ROI) (Media±DE)


Abreviaturas:
BS: Superficie ósea, OS: Superficie Osteoide, TS: Superficie Total, BPm: Perímetro del hueso, BTh: grosor del hueso, Ctr: control.
Tiss-Zn2+ logró un perímetro del hueso mayor (BPm) que el producido por TissHYD (Tabla 10).
Tabla 10. Resultados estadísticos de los valores P después del análisis de datos. Las letras en negrita indican la significancia en P < 0,05.

Abreviaturas:
BS: Superficie ósea, OS: Superficie Osteoide, TS: Superficie Total, BPm: Perímetro del hueso, BTh: grosor del hueso, Ctr: control.
Para comparación en la Fig. 13 se muestran secciones de hueso teñidas por la técnica con nitrato de plata de von Kossa a las seis semanas de seguimiento, sin ninguna membrana -control (Fig. 13A)- y membrana Tiss-Ca2+ (Fig. 13B). Se observó formación de hueso trabecular a lo largo del margen del defecto calvárico (cabeza de flecha), y dentro del defecto. Los punteros de la Fig. 13 (Mbr: membrana, NB: hueso nuevo y OB: hueso antiguo) muestran islas de hueso dispersas, en correspondencia con el hueso nuevo. Se observó que el defecto óseo en el grupo de control se rellenó con tejido conjuntivo y unas pocas trabéculas óseas inmaduras (Fig. 13A). También pudieron identificarse áreas de formación de hueso trabecular en los defectos tratados con cualquiera de los tipos de la membrana (Figs. 12B, 13B).
Tabla 11. Células óseas y vasos sanguíneos detectados dentro del hueso nuevo formado en la región de interés (ROI) (Media± Desviación Estándar (DE)).


Abreviaturas: Ctr: control.
Tabla 12. Resultados estadísticos de los valores P después del análisis de datos con pruebas t por parejas. Las letras en negrita indican una significancia en P < 0,05.

Abreviaturas: Ctr: control.
Tanto Tiss-Zn2+ como Tiss-Ca2+ promovieron una cantidad más elevada de osteoblastos que el grupo de control. La cantidad de osteoblastos fue más elevada en sujetos tratados con membranas Tiss-Zn2+ que con membranas no cargadas (Tabla 12). En algunas áreas de todas las muestras se observaron osteoblastos en el proceso de enfrentar el hueso directamente sobre la superficie de la membrana (Figs. 14A y 14B). Tiss-Ca2+ no produjo una cantidad mayor de osteoblastos que el resto de las membranas pero originó una mayor cantidad de vasos sanguíneos que el grupo de control (Tabla 12). Tiss-Ca2+ mostró fibras de colágeno densas y claras que transcurren paralelas al defecto óseo y a la membrana. El grupo de control promovió una cantidad menor de vasos sanguíneos que Tiss-Ca2+. Pudieron detectarse muchos vasos grandes en las muestras tratadas con la membrana Tiss-Zn2+ (Fig. 14A). Se mostraron pequeños vasos sanguíneos en una proximidad cercana al nuevo hueso y al biomaterial Tiss-Ca2+.
Las imágenes obtenidas con TB también permitieron observar que las membranas Tiss-Zn2+ y Tiss-Ca2+ promovieron la formación de la matriz ósea (Fig. 14) sobre la membrana, fuera del defecto quirúrgico. No había células inflamatorias o células gigantes multinuclear presentes en la interfaz con hueso en los animales tratados con Tiss-Zn2+ (Fig. 14A).
En este estudio experimental, se observó regeneración ósea representada en todos los grupos. Al final del estudio el tamaño de los defectos fue menor que su tamaño original. Todos los defectos óseos mostraron superficie ósea mineralizada dentro de la región de interés (Fig. 12A), pero Tiss-Zn2+, Tiss-Ca2+ y TissHYD lograron hueso nuevo formado (BS) significativamente mayor en comparación con los controles (Tablas 9 y 10). El patrón del tejido apareció compuesto por membranas en íntimo contacto con el hueso recién formado y con el tejido osteoide. El aumento significativo de la superficie ósea (BS), es decir, la matriz ósea mineralizada excluyendo el osteoide, y el grosor del hueso (BTh) se asociaron con un aumento generalizado de osteoblastos promovido por todas las membranas, en comparación con el grupo de control, especialmente cuando se utilizaron membranas de Tiss-Zn2+ (Tablas 9 y 10). Se observó hueso nuevo directamente en contacto con las superficies de la membrana Tiss-Zn2+ en las regiones que mostraron una conducción ósea exitosa (Fig. 14A). El hueso recién formado era continuo desde el margen del defecto sin ninguna invasión del tejido blando. El hueso regenerado de forma continua adherido a la membrana Tiss-Zn2+ forma imágenes de unión ósea (Fig. 14B). Se observaron múltiples trabéculas osificadas interconectadas en la región de interés (Fig. 12A).
La aplicación de las membranas Tiss-Zn2+, Tiss-Ca2+ y TissHYD indujeron unos cambios significativos en la remodelación y los índices estructurales del hueso. Este aumento en la remodelación podría tener como resultado el reemplazo de hueso demasiado maduro y antiguo con hueso más joven y más resiliente (Rubin et al., 2018). El osteoide o la matriz ósea que será, pero no aún, mineralizado mostró una mayor superficie que en el grupo de control cuando se utilizaron membranas habituales de huesos jóvenes (La Monaca et al., 2018).
No solamente se observó osteogénesis sino una actividad biológica aumentada después de determinar la cantidad de osteoblastos cuando se utilizaron las membranas Tiss-Zn2+ y Tiss-Ca2+ (Tabla 10). La Tiss-Zn2+ ha mostrado un aumento de la relación proliferación celular/tejido cicatrizado (Augustine et al., 2014). La formación de nuevo hueso indica que las membranas pueden inducir la proliferación de osteoblastos para llenar, o llenar parcialmente poros intracorticales por agregados nucleadores que inducen su posterior fusión para formar fosfato de calcio amorfo y finalmente cristales de apatita, reactivando de este modo la conversión de células mesenquimales a osteoblastos formadores de hueso (Vrahnas et al., 2018). Se ha demostrado que la previa formación de complejos de zinc en los tejidos facilita la unión del grupo fosfato con la precipitación biomimética de depósitos de Ca/P y la formación de nanocristales de hidroxiapatita. Estos grupos fosfato, en la superficie, presentan oxígenos infracoordinados, lo que conduce a superficies reactivas que atraerán los iones de calcio de los medios. Esta deposición biomimética de Ca/P se considera un
método de revestimiento inspirado en el proceso de biomineralización. Además de eso, se cree que las fosfoproteínas suministran fosfato para la mineralización, siendo capaces de modular la nucleación y el crecimiento de cristales, además de unirse a la red de colágeno. Una mayor solubilidad de ZnO cuando está en contacto con sustratos ácidos, como algunas proteínas ácidas no colagenosas, podría también explicar la efectiva liberación de iones de zinc, lo que estimula la fosforilación de proteína, aumenta la deposición de calcio y facilita la precipitación de cristales. Los osteoblastos estimulados con Ca2+, y PO42- extracelular aumentaron la expresión del ARNm de la proteína morfogenética ósea 2 (Shimauchi et al., 2013; Tada et al., 2010).
Más aún, la conectividad de los poros descrita en estas membranas podría influenciar la posibilidad de que un mayor número de osteoblastos puede penetrar en la estructura porosa (Guarnieri et al., 2018). Además, se observó un crecimiento de microvasos cerca de la membrana (Fig. 14A) cuando se utilizaron las membranas Tiss-Zn2+, lo que contribuye a la integración del biomaterial en el tejido (Turri and Dahlin, 2015).
Los actuales descubrimientos proporcionan una evidencia adicional de que estas membranas actúan como un modulador bioactivo de señalización a los defectos subyacentes. Estos resultados proporcionan evidencias de que las membranas son útiles y resultan ventajosas para la regeneración de tejido óseo.
5. EFECTOS ANTIBACTERIANOS DE TissHYD, Tiss-Zn2+, Tiss-Ca2+ Y Tiss-DOX.
Se unieron discos de hidroxiapatita (HAp) con un diámetro de 7 mm y un grosor de 1,8 mm (Clarkson Chromatography Products, Williamsport, PA, EE.UU.) a cuatro tipos de membranas: TissHYD, Tiss-Ca2+(cargada con 1,1 ^g(Ca2+)/mg Tiss), Tiss-Zn2+ (cargada con 1,1 ^g(Zn2+)/mg Tiss), y Tiss-DOX (cargada con 0,8 mg(DOX)/mg Tiss. Se utilizaron discos de HAp desnudos y discos de HAp recubiertos con una membrana de PTFE como control. Se utilizaron todos los especímenes para desarrollar una biopelícula oral de múltiples especies. Se utilizaron las cepas de referencia Streptococcus oralis CECT 907T, Veillonella parvula NCTC 11810, Actinomyces naeslundii ATCC 19039, Fusobacterium nucleatum DMSZ 20482, Aggregatibacter actinomycetemcomitans DSMZ 8324 y Porphyromonas gingivalis ATCC 33277. Brevemente, se hicieron cultivos puros anaeróbicos de cada bacteria en un medio rico en proteínas que contenía caldo de infusión cerebro-corazón. Los discos de HAp de control y los recubiertos con las diferentes membranas se colocaron a continuación en
los pocilios de una placa de cultivo tisular de 24 pocilios. Cada pocilio fue inoculado a continuación con 1,5 ml de una suspensión de una mezcla de bacterias preparada e incubada en condiciones anaeróbica (H2 al 10%, CO2 al 10%, y balance de N2) a 37°C durante 12, 24, 48 y 72 h. Para controlar la esterilidad del sistema, se utilizaron placas que contenían únicamente medio de cultivo.
Se observaron las biopelículas de 12 a 72 horas de evolución mediante Microscopía electrónico de barrido (SEM, por sus siglas en inglés). Para este análisis, los especímenes se fijaron en una solución de paraformaldehído al 4% y glutaraldehído al 2,5% durante 4h a 4°C. Después de eso, los especímenes se desecaron con punto crítico, se recubrieron por deposición catódica con oro y se analizaron.
Para una evaluación cuantitativa del crecimiento de bacterias, se aisló ADN de la biopelícula tras 12, 24, 48 y 72 horas de incubación utilizando un kit comercial (MolYsis Complete5; Molzym GmgH & CoKG, Bremen, Alemania), siguiendo las instrucciones del fabricante (se siguió el protocolo para la extracción de ADN bacteriano a partir de la etapa 6, evitando las etapas preliminares). Se utilizó el método de ensayo de PCR con sonda de hidrólisis 5’ nucleasa para detectar y cuantificar el ADN bacteriano.
Se obtuvieron cebadores y sondas de Life Technologies Invitrogen (Carlsbad, CA, EE.UU.), Applied Biosystems (Carlsbad, CA, EE.UU.) y Roche (Roche Diagnostic GmbH; Mannheim, Alemania) y se establecieron como diana contra el gen 16S ARNr. La amplificación por reacción en cadena de la polimerasa cuantitativa (qPCR) se realizó en un volumen total de la mezcla de reacción de 10 ^L. Se realizaron análisis con un termociclador LightCycler® 480 II (Roche). Las placas utilizadas en el estudio fueron FramStar 480 con estructura transparente y pocillos blancos (4titude; The North Barn; Damphurst Lane, Reino Unido), sellados con QPCR Adhesive Clear Seals (4titude). Cada muestra de ADN se analizó por duplicado. El ciclo de cuantificación (Cq) se determinó utilizando el paquete de software provisto (LC 480 Software 1.5; Roche). La cuantificación de células por qPCR se basó en curvas estándar. La correlación entre los valores del Cq y CFU ml-1 se generó automáticamente mediante el software (LC 480 Software 1.5; Roche).
Se utilizaron ensayos de bondad del ajuste de Shapiro-Wilk y la distribución de datos para evaluar la normalidad. Los datos se expresaron como medias y desviación estándar (DE). Para comparar los efectos de la superficie del material a diferentes
tiempos de exposición en CFU ml-1, se utilizó análisis de variancia y pruebas post-hoc con la corrección T3 de Dunett. Los resultados se consideraron estadísticamente significativos con p<0,05.
Se muestran imágenes de FESEM de formación de diferentes biopelículas en la Fig.
15. Después de 24 h, se observaron cantidades elevadas de bacterias en todos los discos formando una capa gruesa de bacterias, con características iniciales de una biopelícula estructurada. Excepto en el caso de discos Tiss-DOX que mostraron ausencia o pocas bacterias aisladas en las superficies. Pudieron observarse comunidades microbianas intercaladas con canales, lo que sugiere que las bacterias pueden haber alcanzado la fase exponencial de crecimiento. No se observaron diferencias significativas entre los discos de los especímenes en relación con la estructura de la biopelícula, excepto con las biopelículas en los discos Tiss-DOX , que carecían de una estructura organizada.
Los conteos bacterianos (CFU mL-1) para las seis especies a las 72 h de tiempo de incubación en los especímenes sometidos a ensayo se muestran en la Fig. 16. Con el tiempo, la dinámica del crecimiento bacteriano fue similar independientemente del espécimen. Las biopelículas en los discos de HAp recubiertos con Tiss-DOX alcanzaron la menor cantidad de bacterias, en comparación con el resto de los grupos (p<0,01).
6. DOPAJE DE TissHYD CON NANOPARTÍCULAS DE SIO
2
(NPS-SIO
2
)
El dióxido de silicio (SiO2) es capaz de mejorar no sólo la bioactividad de los materiales sino también la adhesión y la proliferación en tejidos artificiales, lo que facilita la diferenciación de células osteogénicas. Se considera que el SiO2 es osteoinductor y un catalizador para la formación ósea. Por lo tanto, para mejorar la bioactividad de las membranas se doparon con nanopartículas de SiO2 (NPs-SiO2) de dos formas diferentes:
1) 1g de NPs-SiO2 se añadió a la solución óptima de para el escalado del electrohilado (ver sección 6), y la solución se preparó de la siguiente forma: 1,000g de NPs-SiO2 (1.219 % en peso) se dispersaron en 70,000 g de DMF (85,316% en peso) mediante 20 min de sonicación, a continuación se disolvieron 5,000g de (MA)5-co-(HEA)5 (6,094 % en peso), y se disolvieron 6,000 g de (MMA)3-co-(HEMA)2 (7,313% en peso) en la suspensión de NPsSÍO2/DMF. Cuando los copolímeros se disolvieron completamente, se añadió 0,048 g de ácido clorhídrico (HCl) (0,058% en peso) a la solución. A continuación, utilizando los parámetros de la Tabla 7, se procesó la solución por electrohilado, y se obtuvo una membrana no tejida de nanofibras dopada con NPs-SiO2 (TissSi-OH): Las propiedades mecánicas de las membranas no se vieron afectadas por la incorporación de NPs-SiO2. A continuación, para introducir grupos carboxilo (COOH) en la superficie de las fibras, se realizó una hidrólisis parcial de los grupos éster: R-COCH3 y RCOCH2CH2OH de TissSi-OH para obtener TissHYDSi. La hidrólisis se realizó introduciendo ambas membranas en una solución de carbonato de sodio (333 mM) pH=12,50 durante 30min. A continuación, las membranas se lavaron 3 veces con agua destilada y se secaron a temperatura ambiente. El número de grupos COOH accesibles calculado por el método de TBO fue 660±50^mol/g.
2) TissHYD se impregnó con una suspensión de 10-20nm de nanopartículas de SiO2 (SiO2-NPs) y a continuación el agua se evaporó bajo vacío a una temperatura constante: de este modo la membrana se cargó con 0,06 mg de SiO2-NPs /mgTiss.
A continuación las membranas dopadas con SiO2-NPs mediante el procedimiento 1) y 2) (TissHYDSi) fueron funcionalizadas con zinc, calcio y DOX a través de los siguientes protocolos:
6.1. Funcionalización de TissHYDSi with Zn+2 (TissSi-Zn2*) y Ca2+ (TissSi-Ca2+).
La capacidad de grupos carboxilo para formar complejos con cationes divalentes fue utilizada para funcionalizar la TissHYDSi con Zn+2 y Ca2+. La TissHYDSi se impregnó con una solución de Zn2+ y Ca2+, y a continuación el agua se evaporó bajo vacío a una temperatura constante: esta forma la TissHYDSi se cargó con 1,1 ^g/mg, de Ca2+ y Zn2+.
6.2. Funcionalización de TissHYDSi con doxiciclina (TissSi-DOX).
La doxiciclina (DOX) se unió a la membrana TissHYDSi de forma no covalente por interacciones ácido-base entre los grupos amino de la DOX y los grupos carboxilo de la TissHYDSi, además de por enlaces de hidrógeno entre los grupos hidroxilo de la membrana y los grupos amino de la DOX. Se impregnó la TissHYDSi con una solución de DOX, y a continuación el agua se evaporó a vacío a una temperatura constante: de este modo la TissHYDSi se cargó con 0,8 mg de DOX/mg Tiss.
7. ENSAYO ESTÁTICO CELULAR DE BIOACTIVIDAD IN VITRO DE TissHYDSi, TissSi-Zn2*’ TissSi-Ca2+ Y TissSi-DOX.
Para investigar el efecto de la inclusión de dióxido de silicio en la bioactividad de las membranas, se realizó el análisis propuesto por Kokubo (ISO 23317:2012. Implants for surgery In vitro evaluation for apatite-forming ability of implant materials), como se ha detallado previamente en la sección 3.1.
Los resultados las imágenes del FESEM de Tiss-SiO2-COOH, Tiss-SiO2-Ca2+, Tiss-SiO2-Zn2+ y Tiss-SiO2-DOX realizadas por el procedimiento 1) después de 7 días de inmersión se presentan en la Fig. 17. Se demostró la bioactividad de los tejidos en todos los casos. La inclusión de dióxido de silicio en los tejidos de muestra ha aumentado drásticamente la aparición de depósitos de mineral en todas las superficies sometidas a ensayo (Fig. 11: sin SiO2 y 17: con SiO2).
Fueron abundantes los depósitos redondeados y distribuidos uniformemente sobre las nanofibras en todos los casos: se obtuvieron resultados similares con los materiales dopados con SiO2 mediante el procedimiento 2).
Las membranas dopadas con SiO2 han demostrado aumentar la bioactividad a través del aumento de la mineralización.
Claims (26)
1. Una membrana hidrófila de nanofibras no tejidas, caracterizada por que comprende una mezcla de
o un primer copolímero de (MA)3-co-(HEA)2 con topología estadística en un porcentaje en peso entre 35% y 65%; y
o un segundo copolímero de (MMA)1-co-(HEMA)1 en un porcentaje en peso entre 35% y 65%.
2. La membrana hidrófila de nanofibras no tejidas según la reivindicación 1, caracterizada por que comprende una mezcla de
o un primer copolímero de (MA)3-co-(HEA)2 con una topología estadística en un 50% en peso; y
o un segundo copolímero de (MMA)1-co-(HEMA)1 en un 50% en peso.
3. La membrana hidrófila de nanofibras no tejidas según cualquiera de las reivindicaciones 1 o 2, caracterizada por que el primer copolímero de (MA)3-co-(HEA)2 tiene un peso molecular entre 50000 Da y 3106 Da.
4. La membrana hidrófila de nanofibras no tejidas según la reivindicación 3, caracterizada por que el primer copolímero de (MA)3-co-(HEA)2 tiene un peso molecular entre 1106 Da y 3106 Da.
5. La membrana hidrófila de nanofibras no tejidas según cualquiera de las reivindicaciones 1 a 4, caracterizada por que el segundo copolímero de (MMA)1-co-(HEMA)1 tiene un peso molecular entre 50000 Da y 1106 Da.
6. La membrana hidrófila de nanofibras no tejidas según cualquiera de las reivindicaciones 1 a 5, caracterizada por que además comprende nanopartículas de SiO2 , en donde dichas nanopartículas de SiO2 están homogéneamente dispersas en la membrana y/o son físicamente adsorbidas en la superficie de la membrana, en un porcentaje en peso de entre 0,1 % y 60 % con respecto al peso final de la membrana.
7. Un proceso de preparación de la membrana hidrófila de nanofibras no tejidas según cualquiera de las reivindicaciones 1 a 5, caracterizada por que comprende las siguientes etapas:
a) síntesis del primer copolímero de (MA)3-co-(HEA)2 por polimerización radical viva catalizada por metal utilizando un sistema catalítico metálico;
b) síntesis del segundo copolímero de (MMA)r co-(HEMA)1 por polimerización radical por transferencia de átomo inversa utilizando un sistema catalítico metálico;
c) preparación por electrohilado de una membrana de nanofibras que comprende una mezcla, donde dicha mezcla comprende el primer copolímero obtenido en la etapa (a) y el segundo copolímero obtenido en la etapa (b), y d) tratamiento térmico de la membrana de nanofibras obtenida en la etapa (c), en donde el tratamiento térmico se aplica en forma de agua caliente en un rango de temperatura entre 30°C y 80°C y en donde la membrana de nanofibras obtenida en la etapa (c) se mantiene tensionada mediante una montura.
8. El proceso según la reivindicación 7, en donde el sistema catalítico de la etapa (a) y la etapa (b) es un complejo aminado de cobre.
9. El proceso según la reivindicación 7, en donde el metal del sistema catalítico metálico de la etapa (a) y/o la etapa (b) comprende un metal de transición o una mezcla de metales de transición en diferentes estados de oxidación.
10. El proceso según la reivindicación 9, en donde el metal del sistema catalítico metálico de la etapa (a) y la etapa (b) se selecciona independientemente de la lista que consiste en Cu, Fe, Co, Ni, Ru, Pl, Rh, Re, Cr y Mo.
11. El proceso según cualquiera de las reivindicaciones 7, 9 o 10, en donde el ligando del sistema catalítico metálico de la etapa (a) y/o la etapa (b) es una amina alifática multidentada.
12. El proceso según cualquiera de las reivindicaciones 7, 9 a 11, en donde el ligando del sistema catalítico metálico de la etapa (a) y la etapa (b) se selecciona independientemente de la lista que consiste en N,N,N',N",N"-pentametildietilentriamina (PMDETA), tris(2-piridilmetil)amina, tris[2-(dimetilamino)etil]amina, 2,2-Bipiridilo, N,N,N',N'-tetrakis(2-piridilmetil)etilendiamina y 1,1,4,7,10,10-hexametiltrietilentetramina.
13. El proceso según cualquiera de las reivindicaciones 7 a 12, en donde el iniciador del sistema catalítico metálico de la etapa (a) se selecciona independientemente de la lista que consiste en dodecil 2-bromoisobutirato, a-bromoisobutirato de etilo, abromoisobutirato de etilo, 2-bromoisobutirato de octadecilo, a-bromoisobutirato de metilo, 3-bromopropionato de metilo, 3-bromopropionato de terc-butilo, 2-bromopropionato de etilo.
14. El proceso según cualquiera de las reivindicaciones 7 a 12, en donde el iniciador del sistema catalítico metálico de la etapa (b) se selecciona independientemente de la lista que consiste en 1,1’azo-bis(ciclohexanocarbonitrilo) (ACHN), 2,2’-diclorhidrato de 2,2’-azo-bis (2-metilpropionamidina) (AAPH), 4,4’-azo-bis(ácido 4-cianovalérico) (ACVA), hidroperóxido de terc-butilo, hidroperóxido de cumeno, 2,5-di(tercbutilperóxido)-2,5-dimetil-3-hexino, peróxido de dicumilo y 2,5-bis(terc-butilperóxido)-2,5-dimetilhexano.
15. El proceso según cualquiera de las reivindicaciones 6 a 14, en donde el disolvente utilizado con el sistema catalítico metálico de la etapa (a) y la etapa (b) se selecciona independientemente de la lista que consiste en acetona, dimetilformamida, poli(etilenglicol), dimetilsulfóxido, 1, 4-dioxane, etanol, propanol, hexano, agua, dióxido de carbono, líquido iónico, y una combinación de los mismos.
16. El proceso según cualquiera de las reivindicaciones 6 a 14, en donde el sistema catalítico metálico de la etapa (a) y la etapa (b) no utiliza disolvente.
17. El proceso según cualquiera de las reivindicaciones 6 a 16, en donde el disolvente utilizado en la etapa (c) es dimetilformamida.
18. El proceso según cualquiera de las reivindicaciones 6 a 17, en donde el porcentaje en peso del disolvente utilizado en la etapa (c) está en un rango entre 20% y 98%.
19. Un proceso para la preparación de la membrana hidrófila de nanofibras no tejidas según la reivindicación 6, en donde dichas nanopartículas de SiO2 están homogéneamente dispersas en la membrana, caracterizado por que comprende todas las etapas del proceso según cualquiera de las reivindicaciones 7 a 18 y en donde la mezcla de la etapa (c) comprende nanopartículas de SiO2.
20. Un proceso de preparación de la membrana hidrófila de nanofibras no tejidas según la reivindicación 6, en donde dichas nanopartículas de SiO2 son físicamente adsorbidas en la superficie de la membrana caracterizado por que comprende todas las etapas del proceso según cualquiera de las reivindicaciones 7 a 19 y una etapa adicional (e) de impregnar la membrana obtenida en la etapa (d) en una suspensión de nanopartículas de SiO2 y evaporar el disolvente.
21. Una membrana hidrófila de nanofibras no tejidas hidrolizada caracterizada por que comprende la membrana hidrófila de nanofibras no tejidas según cualquiera de las reivindicaciones 1 a 6, que comprende grupos carboxilo, en donde la concentración de grupos carboxilo en la membrana está en un rango entre 20 ^mol/g de la membrana y 3000 ^mol/g de la membrana.
22. La membrana hidrófila de nanofibras no tejidas hidrolizada según la reivindicación 21, caracterizada por que está funcionalizada con un catión divalente seleccionado de entre Zn+2, Ca+2, Mg+2 y Sr+2, un agente antibacteriano y/o cualquiera de las combinaciones de los mismos.
23. La membrana hidrófila de nanofibras no tejidas hidrolizada según la reivindicación 21, caracterizado por que la membrana hidrófila de nanofibras no tejidas está funcionalizada con Zn+2, Ca+2 y doxiciclina.
24. Una membrana no reabsorbible para promover la regeneración ósea caracterizada por que comprende la membrana hidrófila de nanofibras no tejidas según cualquiera de las reivindicaciones 1 a 6 o la membrana hidrófila de nanofibras no tejidas hidrolizada según las reivindicaciones 21 a 23.
25. Una membrana periodontal no reabsorbible caracterizada por que comprende la membrana hidrófila de nanofibras no tejidas según cualquiera de las reivindicaciones 1 a 6 o la membrana hidrófila de nanofibras no tejidas hidrolizada según las reivindicaciones 21 a 23.
26. Un recubrimiento para un implante caracterizado en que comprende la membrana hidrófila de nanofibras no tejidas según cualquiera de las reivindicaciones 1 a 6 o la membrana hidrófila de nanofibras no tejidas hidrolizada según las reivindicaciones 21 a 23.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES201930393A ES2791771B2 (es) | 2019-05-03 | 2019-05-03 | Membrana hidrófila de nanofibras no tejidas para promover la regeneración ósea |
| PCT/EP2020/061184 WO2020224960A1 (en) | 2019-05-03 | 2020-04-22 | Hydrophilic nonwoven nanofibers membrane for promoting bone regeneration |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES201930393A ES2791771B2 (es) | 2019-05-03 | 2019-05-03 | Membrana hidrófila de nanofibras no tejidas para promover la regeneración ósea |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| ES2791771A1 ES2791771A1 (es) | 2020-11-05 |
| ES2791771B2 true ES2791771B2 (es) | 2021-05-11 |
Family
ID=70465033
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES201930393A Active ES2791771B2 (es) | 2019-05-03 | 2019-05-03 | Membrana hidrófila de nanofibras no tejidas para promover la regeneración ósea |
Country Status (2)
| Country | Link |
|---|---|
| ES (1) | ES2791771B2 (es) |
| WO (1) | WO2020224960A1 (es) |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2093282C (en) * | 1992-05-04 | 1999-01-12 | Tibor Sipos | Intraoral medicament-releasing device |
| US6153210A (en) * | 1997-08-14 | 2000-11-28 | Periodontix, Inc. | Use of locally delivered metal ions for treatment of periodontal disease |
| EP3086820B1 (en) * | 2013-12-26 | 2020-10-28 | Tepha, Inc. | Medical implants including laminates of poly-4-hydroxybutyrate and copolymers thereof |
| KR101601674B1 (ko) * | 2014-04-23 | 2016-03-09 | 금오공과대학교 산학협력단 | 표면 개질된 골조직 유도 재생용 나노섬유 차폐막 및 그 제조 방법 |
| WO2016049682A1 (en) * | 2014-09-29 | 2016-04-07 | Griffith University | Periodontal tissue grafts |
| CN107930703A (zh) * | 2017-11-27 | 2018-04-20 | 桐乡佳车科技有限公司 | 一种磁场可控阴离子交换膜制备方法 |
-
2019
- 2019-05-03 ES ES201930393A patent/ES2791771B2/es active Active
-
2020
- 2020-04-22 WO PCT/EP2020/061184 patent/WO2020224960A1/en active Application Filing
Also Published As
| Publication number | Publication date |
|---|---|
| WO2020224960A1 (en) | 2020-11-12 |
| ES2791771A1 (es) | 2020-11-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Sowmya et al. | Role of nanostructured biopolymers and bioceramics in enamel, dentin and periodontal tissue regeneration | |
| He et al. | Enhanced healing of rat calvarial defects with MSCs loaded on BMP-2 releasing chitosan/alginate/hydroxyapatite scaffolds | |
| Zhao et al. | Bioinspired mineral hydrogels as nanocomposite scaffolds for the promotion of osteogenic marker expression and the induction of bone regeneration in osteoporosis | |
| Heinemann et al. | Bio-inspired silica–collagen materials: applications and perspectives in the medical field | |
| Chen et al. | Umbilical cord stem cells released from alginate–fibrin microbeads inside macroporous and biofunctionalized calcium phosphate cement for bone regeneration | |
| Place et al. | Strontium-and zinc-alginate hydrogels for bone tissue engineering | |
| Thein-Han et al. | Calcium phosphate cement with biofunctional agents and stem cell seeding for dental and craniofacial bone repair | |
| Nie et al. | Nano-hydroxyapatite mineralized silk fibroin porous scaffold for tooth extraction site preservation | |
| SE1251043A1 (sv) | Hydrogelbelagd scaffold | |
| US20140212468A1 (en) | Preparation method of an implant comprising drug delivery layer and implant composition for living donor transplantation comprising the same | |
| Xiong et al. | The fabrication of a highly efficient hydrogel based on a functionalized double network loaded with magnesium ion and BMP2 for bone defect synergistic treatment | |
| CN107158465B (zh) | 一种骨支架复合材料的制备方法 | |
| Anil et al. | Biomaterials for craniofacial tissue engineering and regenerative dentistry | |
| Skriabin et al. | Titanium membranes with hydroxyapatite/titania bioactive ceramic coatings: Characterization and in vivo biocompatibility testing | |
| Zhu et al. | Periosteum-inspired membranes integrated with bioactive magnesium oxychloride ceramic nanoneedles for guided bone regeneration | |
| Silva-López et al. | The era of biomaterials: smart implants? | |
| Suruagy et al. | Physico-chemical and histomorphometric evaluation of zinc-containing hydroxyapatite in rabbits calvaria | |
| Yang et al. | An engineered lamellar bone mimicking full-scale hierarchical architecture for bone regeneration | |
| ES2791771B2 (es) | Membrana hidrófila de nanofibras no tejidas para promover la regeneración ósea | |
| Peng et al. | Precisely tuning the pore-wall surface composition of bioceramic scaffolds facilitates angiogenesis and orbital bone defect repair | |
| CN110721348B (zh) | 一种天然蚕丝增强羟基磷灰石/壳聚糖复合膜及制备方法 | |
| Song et al. | Structure driven bio-responsive ability of injectable nanocomposite hydrogels for efficient bone regeneration | |
| Razavi et al. | Nanobiomaterials in periodontal tissue engineering | |
| Xu et al. | Cultured epidermal sheet grafting with Hemaseel™ HMN fibrin sealant on nude mice | |
| Jo et al. | Cell recognitive bioadhesive‐based osteogenic barrier coating with localized delivery of bone morphogenetic protein‐2 for accelerated guided bone regeneration |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| BA2A | Patent application published |
Ref document number: 2791771 Country of ref document: ES Kind code of ref document: A1 Effective date: 20201105 |
|
| FG2A | Definitive protection |
Ref document number: 2791771 Country of ref document: ES Kind code of ref document: B2 Effective date: 20210511 |