EP1518710A2 - Support for lithographic printing plate and presensitized plate - Google Patents
Support for lithographic printing plate and presensitized plate Download PDFInfo
- Publication number
- EP1518710A2 EP1518710A2 EP04020031A EP04020031A EP1518710A2 EP 1518710 A2 EP1518710 A2 EP 1518710A2 EP 04020031 A EP04020031 A EP 04020031A EP 04020031 A EP04020031 A EP 04020031A EP 1518710 A2 EP1518710 A2 EP 1518710A2
- Authority
- EP
- European Patent Office
- Prior art keywords
- treatment
- acid
- aqueous solution
- compound
- aluminum
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Images


Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B41—PRINTING; LINING MACHINES; TYPEWRITERS; STAMPS
- B41N—PRINTING PLATES OR FOILS; MATERIALS FOR SURFACES USED IN PRINTING MACHINES FOR PRINTING, INKING, DAMPING, OR THE LIKE; PREPARING SUCH SURFACES FOR USE AND CONSERVING THEM
- B41N3/00—Preparing for use and conserving printing surfaces
- B41N3/03—Chemical or electrical pretreatment
- B41N3/038—Treatment with a chromium compound, a silicon compound, a phophorus compound or a compound of a metal of group IVB; Hydrophilic coatings obtained by hydrolysis of organometallic compounds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B41—PRINTING; LINING MACHINES; TYPEWRITERS; STAMPS
- B41N—PRINTING PLATES OR FOILS; MATERIALS FOR SURFACES USED IN PRINTING MACHINES FOR PRINTING, INKING, DAMPING, OR THE LIKE; PREPARING SUCH SURFACES FOR USE AND CONSERVING THEM
- B41N1/00—Printing plates or foils; Materials therefor
- B41N1/04—Printing plates or foils; Materials therefor metallic
- B41N1/08—Printing plates or foils; Materials therefor metallic for lithographic printing
- B41N1/083—Printing plates or foils; Materials therefor metallic for lithographic printing made of aluminium or aluminium alloys or having such surface layers
Definitions
- the present invention relates to a support for a lithographic printing plate, and to a presensitized plate for lithographic printing.
- the invention relates to a presensitized plate, composed of a support having thereon a thermal type image recording layer, which is made directly into a lithographic printing plate without the use of a film copy by being exposed to laser light scanned over the surface thereof and having text and images to be formed directly on its surface; and relates also to a lithographic printing plate support for use in such a presensitized plate.
- the invention additionally relates to a heat-sensitive presensitized plate which is highly suitable for use in a computer-to-plate system that does not require development, which is capable of image recording by infrared scanning exposure based on digital signals and which, following exposure, can then be mounted on a printing press and printed without passing through a conventional processing step involving the use of a developer; and relates also to a lithographic printing plate support for use in such a presensitized plate.
- thermoplastic polymer particles dispersed in a hydrophilic binder polymer serves as the heat-sensitive, imaging layer.
- the hydrophobic thermoplastic polymer particles fuse, converting the surface of the hydrophilic heat-sensitive layer into oleophilic image areas.
- on-machine development one method for reducing the number of processing steps is referred to as "on-machine development.”
- the presensitized plate is mounted onto the cylinder of a printing press without first processing the plate with a developer. Ink and/or dampening water are then fed to the plate as the cylinder is turned, thereby removing non-image areas of the presensitized plate.
- the plate is directly mounted on the printing press and development processing is completed in the course of a normal printing operation.
- Presensitized plates suitable for such on-machine development must have a heat-sensitive layer that is soluble in the dampening water or the ink solvent. Moreover, they must have daylight handling characteristics suitable for development on a printing press located in a lighted room.
- JP 2938397 B discloses a presensitized plate of this type having a hydrophilic support on which there is provided a heat-sensitive layer composed of a finely divided thermoplastic hydrophobic polymer dispersed in a hydrophilic binder polymer.
- This prior-art describes the exposure of the presensitized plate using an infrared laser and the ensuing coalescence of the thermoplastic hydrophobic polymer particles under the effect of heat to form an image, and how the plate can then be mounted onto the plate cylinder of a printing press and on-machine development carried out by feeding ink and/or dampening water.
- JP 9-127683 A and WO 99/10186 teach the fabrication of a lithographic plate by on-machine development following the coalescence of thermoplastic fine particles under the effect of heat.
- presensitized plates on which an image is formed in this way by the coalescence of fine particles under heating do exhibit a good on-machine developability, they have a poor sensitivity owing to the escape of heat to the metal support. Moreover, when particle coalescence is insufficient, the strength of image areas of the heat-sensitive layer is diminished, which may result in a short press life.
- thermal-type presensitized plates that are not developed "on machine," including what are known as positive-working thermal presensitized plates in which an infrared absorber present in the heat-sensitive layer exhibits a photothermal conversion effect that causes it to generate heat upon exposure to light, rendering exposed areas of the heat-sensitive layer alkali-soluble under the effect of such heat and resulting in the formation of a positive image, and what are known as negative-working thermal presensitized plates in which similar heating causes a radical generator or an acid generator present in the heat-sensitive layer to form radicals or an acid which triggers a radical polymerization reaction and an acid crosslinking reaction, rendering reacted areas of the layer insoluble and forming a negative image.
- Such positive thermal presensitized plates require the use of an infrared absorber capable of photothermal conversion.
- Such absorbers have a relatively large molecular weight, and thus a low solubility. Also, they adhere to and are difficult to remove from microscopic openings that arise from anodization, as a result of which portions of the exposed heat-sensitive layer tend to remain behind (residual coating) after development has been carried out with an alkali developer.
- the support having an anodized layer is sometimes subjected to pore widening treatment to minimize the diffusion into the aluminum support of heat generated within the heat-sensitive layer.
- Pore widening treatment enables the heat-sensitive layer to be rendered fully developer-insoluble near the heat-sensitive layer/support interface, thus enhancing both press life and sensitivity.
- problems associated with such treatment include incomplete removal of the heat-sensitive layer during development and scumming on the press.
- Sealing methods that have been disclosed include treatment with pressurized steam or hot water, silicate treatment, treatment with an aqueous dichromate solution, nitrite treatment, ammonium acetate treatment, electrodeposition sealing, triethanolamine treatment, barium carbonate treatment and treatment with hot water containing a trace amount of phosphate. Yet, although these methods do enhance the resistance to scumming, they worsen the sensitivity and press life of the plate.
- JP 2003-1961 A discloses a support for lithographic printing plates which is obtained by subjecting a grained and anodized metal base to treatment with an aqueous solution containing an inorganic fluorine compound and a silicate compound, and also discloses a presensitized plate in which such a support is used.
- JP 2002-99093 A describes a method of manufacturing lithographic printing plates which is characterized by subjecting a lithographic printing plate composed of an aluminum support having a surface that satisfies the relationship in the formula below and a photosensitive layer or a heat-sensitive layer thereon to development with a silicate-free developer.
- A represents the peak area (counts ⁇ eV/sec) for phosphorus (2P) as measured by electron spectroscopy (X-ray photoelectron spectroscopy) for chemical analysis (ESCA)
- B represents the peak area (counts•eV/sec) for aluminum (2P) as measured by ESCA.
- JP 11-109637 A discloses a positive-working photosensitive lithographic printing plate composed of an aluminum support having a silicon atom coating weight of 0.1 to 8 mg/m 2 and a positive-working photosensitive layer on the support; and also discloses a lithographic printing plate composed of an aluminum support, an intermediate layer containing a polymeric compound having an acid group-bearing component on the support, and a positive-working photosensitive layer on the intermediate layer.
- the objects of the present invention are to provide excellent heat-sensitive presensitized plates for lithography that are endowed with the above properties, and a support for lithographic printing plates which is highly suitable for use in such presensitized plates. More specifically, one object of the invention is to provide a heat-sensitive presensitized plate which, when used as an on-machine development type plate, has a good on-machine developability, a high sensitivity, a long press life and a good resistance to scumming on the press; and which, when used as a conventional positive-working thermal plate or negative-working thermal plate, efficiently utilizes heat in image formation, exhibits a high sensitivity and a long press life, and is resistant to the scumming of non-image areas. Another object of the invention is to provide a support for lithographic printing plates which is suitable for use in such heat-sensitive presensitized plates.
- a lithographic printing plate support composed of a metal base subjected to graining treatment and anodizing treatment, then treated with an aqueous solution containing an inorganic fluorine compound and a phosphate compound and with an aqueous solution containing a silicate compound (which solutions are sometimes referred to collectively herein as "specific aqueous solutions"), and an intermediate layer which lies on the metal base and contains an acid group-bearing polymeric compound, when used to make a lithographic printing plate, has an excellent sensitivity, press life and resistance to scumming.
- the present invention has been completed based on the finding.
- the present invention provides a support for lithographic printing plates that is composed of a metal base subjected to graining treatment and anodizing treatment, then treated with an aqueous solution containing an inorganic fluorine compound and a phosphate compound, which aqueous solution treatment is preceded or followed by treatment with an aqueous solution containing a silicate compound; and an intermediate layer which lies on the treated metal base and contains an acid group-bearing polymeric compound.
- treatment with the aqueous solution containing an inorganic fluorine compound and a phosphate compound may involve separately carrying out treatment with an inorganic fluorine compound-containing aqueous solution and treatment with a phosphate compound-containing aqueous solution.
- the order in which these particular aqueous solution treatments and treatment with the silicate compound-containing aqueous solution are carried out is not subject to any particular limitation. That is, the aqueous solution treatments may be carried out in any suitable order.
- the acid group on the acid group-bearing polymeric compound constituent is an acid group having an acid dissociation constant (pKa) not higher than 7, and more preferably the acid group on the acid group-bearing polymeric compound constituent is selected from the group consisting of -COOH, -SO 3 H, -OSO 3 H, -PO 3 H 2 , -OPO 3 H 2 , -CONHSO 2 and -SO 2 NHSO 2 .
- pKa acid dissociation constant
- the inventor of the present invention has also found that, by having the surface of the metal base used in the support for lithographic printing plates satisfy specific conditions, lithographic printing plates prepared using the support have an excellent sensitivity, press life and resistance to scumming.
- the invention provides a support for lithographic printing plates that is composed of a metal base having a surface which satisfies formula (1) below 0.10 ⁇ (A+B+C)/(A+B+C+D) ⁇ 0.70 and an intermediate layer which lies on the metal base and contains an acid group-bearing polymeric compound.
- A is the peak area (counts•eV/sec) for fluorine (1S) as measured by electron spectroscopy for chemical analysis (ESCA)
- B is the peak area (counts•eV/sec) for silicon (2P) as measured by ESCA
- C is the peak area (counts•eV/sec) for phosphorus (2P) as measured by ESCA
- D is the peak area (counts•eV/sec) for aluminum (2P) as measured by ESCA.
- the method of manufacturing the metal base having a surface which satisfies formula (1) is not subject to any particular limitation.
- the invention provides a presensitized plate composed of a lithographic printing plate support according to the first or second aspect of the invention, and an infrared laser-imageable recording layer which lies on the support.
- the presensitized plates of the invention which are composed of either a metal base treated with an aqueous solution containing an inorganic fluorine compound and a phosphate compound and treated with an aqueous solution containing a silicate compound or a metal base having a specific surface, a specific intermediate layer provided on top of the metal base, and an on-machine developable heat-sensitive layer provided as a recording layer on top of the intermediate layer have a good on-machine developability and a high sensitivity, in addition to which they exhibit a long press life and have a good resistance to scumming on the press.
- Presensitized plates according to the invention which are obtained by providing a specific intermediate layer on either of the above metal bases and providing a positive-working thermal photosensitive layer as the recording layer on the intermediate layer have a high solubility in liquid developers, even when the infrared laser exposure dose is low or the developer has a low sensitivity.
- an excellent press life is achieved, in addition to which the sensitivity is high, the development latitude is broad, residual coating even at low exposure is minimal, and scumming of non-image areas does not readily occur.
- presensitized plates according to the invention which are similarly provided with a negative-working thermal photosensitive layer as the recording layer have a high percent insolubility in the developer in laser-exposed areas, and thus exhibit a high sensitivity, a long press life and excellent scumming resistance.
- the metal base prior to treatment that may be used in the inventive support for lithographic printing plates is not subject to any particular limitation.
- Metal base prior to treatment refers here to the metal base prior to being administered graining treatment, anodizing treatment and other treatment to have the surface satisfy certain specific conditions.
- Exemplary metal bases prior to treatment include iron, stainless steel and aluminum. Of these, aluminum is preferred.
- Aluminum sheet that may be used as the aluminum base is made of a dimensionally stable metal composed primarily of aluminum; that is, aluminum or aluminum alloy. Aside from sheets of pure aluminum, use can also be made of alloy sheets composed primarily of aluminum and small amounts of other elements, or plastic film or paper onto which aluminum or aluminum alloy has been laminated or vapor deposited. Use can also be made of a composite sheet obtained by bonding an aluminum sheet onto a polyethylene terephthalate film as described in JP 48-18327 B.
- Aluminum sheet that may be used in the invention is not subject to any particular limitation, although the use of pure aluminum sheet is preferred. However, because completely pure aluminum is difficult to manufacture for reasons having to do with refining technology, the presence of a small amount of other elements is acceptable. Suitable use can be made of known materials that appear in the 4 th edition of Aluminum Handbook published in 1990 by the Japan Light Metal Association. Examples of such aluminum materials include those having the designations JIS 1050, JIS 1100, JIS 3003, JIS 3103 and JIS 3005.
- suitable aluminum sheets include those containing 0.07 to 0.09 wt% of silicon, 0.20 to 0.40 wt% of iron, 0.000 to 0.030 wt% of copper, up to 0.01 wt% of manganese, up to 0.01 wt% of magnesium, up to 0.01 wt% of chromium, up to 0.01 wt% of zinc, up to 0.04 wt% of titanium and at least 99.5 wt% of aluminum.
- Use can also be made of aluminum sheet made from aluminum alloy, scrap aluminum or secondary aluminum ingots having an aluminum content of 95 to 99.4 wt%, and containing at least five metals from among iron, silicon, copper, magnesium, manganese, zinc, chromium and titanium within the ranges indicated below.
- the aluminum content in this case exceeds 99.4 wt%, the allowable content of impurities decreases, which may diminish the cost-saving effect.
- the impurities content is large, which gives rise to undesirable effects such as crack formation during rolling.
- the aluminum content is more preferably from 95 to 99 wt%, and most preferably from 95 to 97 wt%.
- the iron content is preferably from 0.3 to 1.0 wt%.
- Iron is an element which is present, even within primary aluminum ingots, in a range of 0.1 to 0.2 wt%.
- the amount of iron that enters into a solid solution within aluminum is small; most remains in the form of intermetallic compounds.
- the iron content is most preferably within a range of 0.5 to 1.0 wt%.
- the silicon content is preferably from 0.15 to 1.0 wt%.
- Silicon is an element which is abundant in scrap from JIS 2000, 4000 and 6000 series materials. Silicon is an element present in an amount of about 0.03 to 0.1 wt% even in primary aluminum ingots. It exists in a solid solution within aluminum, or is present as intermetallic compounds. When aluminum sheet is heated during the support manufacturing process, silicon that was present in the aluminum as a solid solution sometimes precipitates out as uncombined silicon. Uncombined silicon and FeSi-based intermetallic compounds are known to have an adverse influence on the resistance to severe ink scumming.
- “severe ink scumming” refers to contamination in the form of spots and rings that appears on the printed medium such as paper as a result of the tendency for ink to adhere to non-image areas of the printing plate surface when printing is carried out with repeated interruptions.
- a silicon content of more than 1.0 wt% such contamination may not be completely eliminated by the subsequently described sulfuric acid treatment (desmutting treatment).
- a silicon content of less than 0.15 wt% the cost-reducing effects are diminished.
- a silicon content within a range of 0.3 to 1.0 wt% is especially preferred.
- the copper content is preferably from 0.1 to 1.0 wt%. Copper is an element which is abundant in scrap from JIS 2000 and 4000 series materials. Copper forms a solid solution in aluminum with relative ease. At a copper content of more than 1.0 wt%, scumming may not be completely eliminated by the subsequently described sulfuric acid treatment. On the other hand, at a copper content of less than 0.1 wt%, the cost-reducing effects are diminished. A copper content within a range of 0.3 to 1.0 wt% is especially preferred.
- the magnesium content is preferably from 0.1 to 1.5 wt%.
- Magnesium is an element which is abundant in scrap from JIS 2000, 3000, 5000 and 7000 series materials. Because it is particularly abundant in can-end material, magnesium is one of the major metal impurities present in aluminum scrap. Magnesium forms a solid solution in aluminum with relative ease, and forms intermetallic compounds with silicon.
- scumming may not be completely eliminated by the subsequently described sulfuric acid treatment.
- the magnesium content is more preferably from 0.5 to 1.5 wt%, and most preferably from 1.0 to 1.5 wt%.
- the manganese content is preferably from 0.1 to 1.5 wt%.
- Manganese is an element which is abundant in scrap from JIS 3000 series materials. Because it is particularly abundant in can-end material, manganese is one of the major metal impurities present in aluminum scrap. Manganese forms a solid solution in aluminum with relative ease, and forms intermetallic compounds with aluminum, iron and silicon. At a manganese content of more than 1.5 wt%, scumming may not be completely eliminated by the subsequently described sulfuric acid treatment. On the other hand, at a manganese content of less than 0.1 wt%, the cost-reducing effects are diminished.
- the manganese content is more preferably from 0.5 to 1.5 wt%, and most preferably from 1.0 to 1.5 wt%.
- the zinc content is preferably from 0.1 to 0.5 wt%.
- Zinc is an element which is abundant particularly in scrap from JIS 7000 series materials. Zinc forms a solid solution in aluminum with relative ease. At a zinc content of more than 0.5 wt%, scumming may not be completely eliminated by the subsequently described sulfuric acid treatment. On the other hand, at a zinc content of less than 0.1 wt%, the cost-reducing effects are diminished. A zinc content within a range of 0.3 to 0.5 wt% is especially preferred.
- the chromium content is preferably from 0.01 to 0.1 wt%.
- Chromium is a metal impurity present in a small quantity in scrap from JIS 5000, 6000 and 7000 series materials.
- contamination may not be completely eliminated by the subsequently described sulfuric acid treatment.
- a chromium content within a range of 0.05 to 0.1 wt% is especially preferred.
- the titanium content is preferably from 0.03 to 0.5 wt%. Titanium is an element which is generally added in a range of 0.01 to 0.04 wt% as a crystal grain refining agent. It is present in a relatively large amount as a metal impurity in scrap from JIS 5000, 6000 and 7000 series materials. At a titanium content of more than 0.5 wt%, scumming may not be completely eliminated by the subsequently described sulfuric acid treatment. On the other hand, at a chromium content of less than 0.03 wt%, the cost-reducing effects are diminished. A titanium content within a range of 0.05 to 0.5 wt% is especially preferred.
- the aluminum sheet used in the invention is manufactured by using a conventional process to cast the above-described raw material, administering suitable rolling treatment and heat treatment to set the thickness to typically 0.1 to 0.7 mm, and applying flatness correcting treatment as required.
- This thickness can be suitably varied according to the size of the printing press, the size of the printing plate, and the desires of the user.
- Processes that may be used to manufacture the above aluminum sheet include direct-chill casting, a process like direct-chill casting but from which soaking treatment and/or annealing treatment has been omitted, and continuous casting.
- the support for lithographic printing plates of the invention is obtained by subjecting a metal base to graining treatment and anodizing treatment, then treating the grained and anodized metal base with specific aqueous solutions, and subsequently forming on the treated metal base an intermediate layer containing a specific compound.
- manufacture of the inventive support for lithographic printing plates may include various other steps as well.
- the support for lithographic printing plates according to the present invention is described below with reference to, for the purpose of illustration, a case in which aluminum sheet is used as the metal base prior to treatment.
- the aluminum sheet preferably passes through a degreasing step to remove rolling oils adhering to the surface of the sheet, a desmutting step to dissolve smut on the surface of the sheet, a graining treatment step to roughen the surface of the sheet, an anodizing treatment step to cover the surface of the sheet with an anodized layer, a pore widening treatment (acid treatment or alkali treatment) step, and steps involving treatment with specific aqueous solutions.
- a degreasing step to remove rolling oils adhering to the surface of the sheet
- a desmutting step to dissolve smut on the surface of the sheet
- a graining treatment step to roughen the surface of the sheet
- an anodizing treatment step to cover the surface of the sheet with an anodized layer
- a pore widening treatment acid treatment or alkali treatment
- Manufacture of the inventive support for lithographic printing plates preferably includes electrochemical graining treatment in which an alternating current is used to electrochemically grain the aluminum sheet in an acidic aqueous solution.
- Manufacture of the inventive lithographic printing plate support may include an aluminum sheet graining treatment step which combines the above-described electrochemical graining treatment with a related operation such as mechanical graining treatment or chemical etching treatment in an acid or alkaline aqueous solution.
- the graining treatment and other steps used to manufacture the inventive support for lithographic printing plates may be carried out as either a continuous or an intermittent process, although the use of a continuous process is industrially advantageous.
- treatment with specific aqueous solutions is also carried out, in addition to which, if necessary, a hydrophilic surface treatment is administered as well, thereby forming the support.
- a specific intermediate layer or "undercoat,” as viewed from the recording layer side) may be provided thereon if necessary.
- the above-described aluminum sheet is administered graining treatment to impart a more desirable surface shape.
- suitable graining methods include mechanical graining like that described in JP 56-28893 A, chemical etching, and electrolytic graining.
- Use can also be made of electrochemical graining and electrolytic graining processes in which the surface is electrochemically grained in an electrolytic solution containing hydrochloric acid or nitric acid; and mechanical graining such as wire brushing in which the aluminum surface is scratched with metal wires, ball graining in which the aluminum surface is grained with abrasive balls and an abrasive compound, and brush graining in which the surface is grained with a nylon brush and an abrasive compound.
- any one or combination of these graining methods may be used.
- mechanical graining with a nylon brush and an abrasive compound may be combined with electrolytic graining using an electrolytic solution of hydrochloric acid or nitric acid, or a plurality of electrolytic graining treatments may be combined.
- electrochemical graining is preferred, although it is also advantageous to carry out a combination of mechanical graining and electrochemical graining. Mechanical graining, followed by electrochemical graining with an electrolytic solution of nitric acid, followed in turn by electrochemical graining with an electrolytic solution of hydrochloric acid, is especially preferred.
- Mechanical graining refers to treatment in which the surface of the aluminum sheet is mechanically grained such as with a brush. It is preferably carried out before electrochemical graining treatment.
- Suitable mechanical graining treatment involves carrying out treatment with a rotating nylon brush roll having a bristle diameter of 0.07 to 0.57 mm and an abrasive compound that is supplied as a slurry to the surface of the aluminum sheet.
- the nylon brush is preferably made of bristles having a low water absorption.
- a preferred example is Nylon Bristle 200T (available from Toray Industries, Inc.), which is made of nylon 6,10, has a softening point of 180°C, a melting point of 212 to 214°C, a specific gravity of 1.08 to 1.09, a water absorption at 20°C and 65% relative humidity of 1.4 to 1.8 and at 20°C and 100% relative humidity of 2.2 to 2.8, a dry tensile strength of 4.5 to 6 g/d, a dry tensile elongation of 20 to 35%, a boiling water shrinkage of 1 to 4%, a dry resistance to stretching of 39 to 45 g/d, and a Young's modulus when dry of 380 to 440 kg/mm 2 .
- Any known abrasive compound may be used, although the use of silica sand, quartz, aluminum hydroxide, or a mixture thereof, mentioned in JP 6-135175 A and JP 50-40047 B is preferred.
- the slurry is preferably one having a specific gravity in a range of 1.05 to 1.3.
- Illustrative examples of methods for supplying the slurry to the surface of the aluminum sheet include blowing the slurry onto the surface, a method involving the use of a wire brush, and a method in which the surface shape of a textured cold rolling roll is transferred to the aluminum sheet.
- the methods described in JP 55-74898 A, JP 61-162351 A and JP 63-104889 A may also be used.
- JP 9-509108 A use can also be made of a method like that described in JP 9-509108 A, wherein the surface of the aluminum sheet is brush grained in an aqueous slurry containing a mixture of particles composed of alumina and quartz in a weight ratio of 95:5 to 5:95.
- the mixture used for this purpose has an average particle size of preferably 1 to 40 ⁇ m, and more preferably 1 to 20 ⁇ m.
- Electrochemical graining differs from the above-described mechanical graining in that it involves graining the surface of the aluminum sheet electrochemically by placing the sheet in an acidic aqueous solution and passing an alternating current through the sheet with the sheet serving as an electrode.
- the ratio Q C /Q A between the amount of electricity when the aluminum sheet serves as the cathode Q C and the amount of electricity when the sheet serves as the anode Q A in the above electrochemical graining treatment is within a range of 0.5 to 2.0, for example, uniform honeycomb pits can be formed on the surface of the aluminum sheet. Non-uniform honeycomb pits tend to form at a Q C /Q A ratio of less than 0.50 or more than 2.0. A Q C /Q A ratio within a range of 0.8 to 1.5 is preferred.
- the alternating current used in electrochemical graining may have a waveform that is, for example, sinusoidal, square, triangular or trapezoidal. Of these, a square or trapezoidal waveform is preferred.
- the alternating current has a frequency which, from the standpoint of the cost of manufacturing the power supply, is preferably from 30 to 200 Hz, and more preferably from 40 to 120 Hz.
- FIG. 1 shows an example of a trapezoidal wave that can be suitably used in the invention.
- the ordinate represents the current value and the abscissa represents time.
- ta is the anode reaction time
- tc is the cathode reaction time
- tp is the time until the current value reaches a peak on the cathode cycle side from zero
- tp' is the time until the current value reaches a peak on the anode cycle side from zero
- Ia is the peak current on the anode cycle side
- Ic is the peak current on the cathode cycle side.
- the respective times tp and tp' until the current reaches a peak from zero are preferably each from 0.1 to 2 msec, and more preferably from 0.3 to 1.5 msec.
- the power circuit impedance has the effect of reducing the power supply voltage required during rise in the current waveform, making it possible to lower the cost of power supply equipment.
- trace ingredients in the acidic aqueous solution have little effect, enabling uniform graining treatment to be carried out.
- the alternating current used in electrochemical graining to have a duty ratio within a range of 0.25 to 0.75, and especially 0.3 to 0.6.
- duty ratio refers to the ratio ta/T, where T is the period of the alternating current and ta is the duration of the anode reaction at the aluminum sheet (anode reaction time).
- smut components composed largely of aluminum hydroxide form on the surface of the aluminum sheet during the cathode reaction, in addition to which oxide film dissolution and breakdown occur, becoming the starting points of pitting reactions during the subsequent anode reaction at the aluminum sheet.
- selection of the alternating current duty cycle has a large effect on providing uniform graining treatment.
- the alternating current has a current density, in the case of a trapezoidal or square waveform, which is preferably such that the current density Iap at the peak on the anode cycle side and the current density Icp at the peak on the cathode cycle side are each from 10 to 200 A/dm 2 . Moreover, the ratio Icp/Iap is preferably within a range of 0.9 to 1.5.
- the total amount of electricity used in the anode reaction on the aluminum sheet when electrochemical graining treatment has been completed is preferably from 50 to 1,000 C/dm 2 .
- the electrochemical graining time is preferably from 1 second to 30 minutes.
- Any acidic aqueous solution used in conventional electrochemical graining treatment involving the use of direct current or alternating current may be employed here in electrochemical graining treatment, although the use of an acidic aqueous solution composed mainly of nitric acid or an acidic aqueous solution composed mainly of hydrochloric acid is preferred.
- "Composed mainly of,” as used here and below, signifies that the main component in an aqueous solution is contained in an amount of at least 30 wt%, and preferably at least 50 wt%, based on all the components within the solution.
- the acidic aqueous solution composed mainly of nitric acid can be one which is employed in conventional electrochemical graining treatment involving the use of direct current or alternating current.
- an aqueous solution with a nitric acid concentration of 5 to 15 g/L in which one or more nitric acid compounds such as aluminum nitrate, sodium nitrate or ammonium nitrate has been added to a concentration of from 0.01 g/L to saturation.
- the acidic aqueous solution composed mainly of nitric acid may contain, dissolved therein, metals which are present in aluminum alloy, such as iron, copper, manganese, nickel, titanium, magnesium and silicon.
- the acidic solution composed mainly of nitric acid used in the invention is one which contains nitric acid, an aluminum salt and a nitrate, and which has been obtained by adding aluminum nitrate and ammonium nitrate to a nitric acid solution having a nitric acid concentration of 5 to 15 g/L so as to set the aluminum ion concentration at 1 to 15 g/L, and preferably 1 to 10 g/L, and the ammonium ion concentration at 10 to 300 ppm.
- the aluminum ions and ammonium ions form spontaneously and thus increase while electrochemical graining is being carried out.
- the liquid temperature at this time is preferably 10 to 95°C, more preferably 20 to 90°C, and most preferably 40 to 80°C.
- electrochemical graining treatment use can be made of a known electrolytic cell apparatus, such as one having a vertical, flat or radial construction.
- a radial electrolytic cell apparatus like that described in JP 5-195300 A is especially preferred.
- FIG. 2 is a schematic view of a radial electrolytic cell apparatus of a type suitable for use in the practice of the invention.
- an aluminum sheet 11 wraps around a radial drum roller 12 situated within a main electrolytic cell 21 and passes through the apparatus while being subjected to electrolytic treatment by means of main electrodes 13a and 13b connected to an AC power supply 20.
- the acidic aqueous solution 14 is supplied from a solution feed inlet 15 through a slit 16, and to a solution channel 17 located between the radial drum roller 12 and the main electrodes 13a and 13b.
- the aluminum sheet 11 treated in an auxiliary anode cell 22 is electrolytically treated in the main electrolytic cell 21.
- an auxiliary anode 18 is situated opposite the aluminum sheet 11 and the acidic aqueous solution 14 is supplied such as to flow between the auxiliary anode 18 and the aluminum sheet 11.
- the current supplied to the auxiliary anode 18 is controlled by thyristors 19a and 19b.
- Main electrodes 13a and 13b may be selected from among carbon, platinum, titanium, niobium, zirconium, stainless steel and electrodes used in fuel cell cathodes, although carbon is especially preferred.
- Examples of carbon that may be used for this purpose include ordinary commercially available impervious graphite for chemical equipment, and resin-impregnated graphite.
- the auxiliary anode 18 may be selected from among known oxygen generating electrodes made of ferrite, iridium oxide, platinum, or platinum that has been clad or plated with a valve metal such as titanium, niobium or zirconium.
- the acidic aqueous solution which passes through the main electrolytic cell 21 and the auxiliary anode cell 22 may be fed in a direction that is either parallel or counter to the direction of advance by the aluminum sheet 11.
- the flow rate of the acidic aqueous solution relative to the aluminum sheet is preferably from 10 to 1,000 cm/s.
- One or more AC power supplies may be connected to a single electrolytic cell apparatus. It is also possible to use two or more electrolytic cell apparatuses, in which case the electrolysis conditions in each apparatus may be the same or different.
- the concentration of the acidic aqueous solution constant by adding nitric acid and water while adjusting the amounts of addition in proportion to the amount of electricity passed through the acidic aqueous solution in which the aluminum sheet within the electrolytic cell apparatus undergoes anodic reaction, and based on the nitric acid and aluminum ion concentrations determined from, for example, (i) the electrical conductivity of the acidic aqueous solution, (ii) the ultrasonic wave propagation velocity of the solution and (iii) the solution temperature, and by successively allowing to overflow and thus discharging from the electrolytic cell apparatus an amount of the acidic aqueous solution equivalent to the volume of nitric acid and water added.
- surface treatment including chemical etching treatment in an acidic aqueous solution or an alkaline aqueous solution, and desmutting treatment shall be described in this order.
- These surface treatments are each carried out either before the above-described electrochemical graining treatment, or after electrochemical graining treatment but before the anodizing treatment described later in the specification. Descriptions of each of the surface treatments are given below, although the invention is not limited to the particular surface treatments as they are described below. Administration of these surface treatments and the other treatments mentioned below is optional.
- Alkali etching treatment is a treatment in which the surface of the aluminum sheet is chemically etched in an alkaline aqueous solution, and is preferably carried out before and after the above-described electrochemical graining treatment. In cases where mechanical graining treatment is carried out before electrochemical graining treatment, it is preferable to carry out alkali etching treatment following mechanical graining treatment. Alkali etching treatment can break down the microstructure in a short time, and is thus more advantageous than the subsequently described acidic etching treatment.
- alkaline aqueous solutions that may be used in alkali etching treatment include aqueous solutions containing one or more of the following: sodium hydroxide, sodium carbonate, sodium aluminate, sodium metasilicate, sodium phosphate, potassium hydroxide and lithium hydroxide.
- An aqueous solution composed mainly of sodium hydroxide is especially preferred.
- the alkaline aqueous solution may contain 0.5 to 10 wt% of aluminum and also alloying ingredients present in the aluminum sheet.
- the alkaline aqueous solution has a concentration of preferably 1 to 50 wt%, and more preferably 1 to 30 wt%.
- alkali etching treatment it is advantageous to carry out alkali etching treatment for 1 to 120 seconds, and preferably 2 to 60 seconds, at an alkaline aqueous solution temperature in a range of 20 to 100°C, and preferably 40 to 80°C.
- the amount of dissolved aluminum is preferably 5 to 20 g/m 2 when alkali etching treatment is carried out after mechanical graining, and preferably 0.01 to 20 g/m 2 when alkali etching treatment is carried out after electrochemical graining.
- a chemical etching solution is initially mixed into the alkaline aqueous solution, it is preferable to prepare the treatment solution using liquid sodium hydroxide and sodium aluminate.
- alkali etching treatment When alkali etching treatment is carried out after electrochemical graining, the smut that forms from electrochemical graining can be removed.
- alkali etching treatments include a method in which the aluminum sheet is brought into contact with 15 to 65 wt% sulfuric acid at a temperature of 50 to 90°C, as described in JP 53-12739 A, and the alkali etching method described in JP 48-28123 B.
- Acidic etching treatment is a treatment in which the aluminum sheet is chemically etched in an acidic aqueous solution. It is preferably carried out after the electrochemical graining treatment described above. In cases where the above-described alkali etching treatment is carried out before and/or after electrochemical graining, it is preferable for acidic etching treatment to be carried out after alkali etching treatment.
- intermetallic compounds which may include silica as a metal and uncombined silicon can be removed from the surface of the aluminum sheet, thus making it possible to eliminate defects in the anodized layer that forms in the subsequent anodizing treatment. As a result, the adherence of ink spots in non-image areas during printing can be prevented.
- the acidic aqueous solution has a concentration of preferably 50 to 500 g/L.
- the acidic aqueous solution may contain aluminum and also the alloying ingredients present in the aluminum sheet.
- the amount of aluminum sheet dissolution at this time is preferably from 0.001 to 0.2 g/m 2 .
- the acid concentration, such as the sulfuric acid concentration and aluminum ion concentration, is preferably selected from a range at which crystallization does not occur at room temperature.
- the aluminum ion concentration is preferably 0.1 to 50 g/L, and more preferably 5 to 15 g/L.
- alkali etching treatment When the above alkali etching treatment is carried out before and/or after electrochemical graining, smut generally forms on the surface of the aluminum sheet as a result of alkali etching treatment. Therefore, following alkali etching treatment, it is desirable to carry out a so-called desmutting treatment in which such smut is dissolved in an acidic solution containing phosphoric acid, nitric acid, sulfuric acid, chromic acid, hydrochloric acid, hydrofluoric acid, fluoroboric acid or a mixture of two or more of these acids. Following alkali etching treatment, it is sufficient to carry out either acidic etching treatment or desmutting.
- the concentration of the acidic solution is preferably 1 to 500 g/L.
- the acidic solution may have dissolved therein 0.001 to 50 g/L of aluminum and also the alloying ingredients present in the aluminum sheet.
- the acidic solution has a liquid temperature of preferably 20 to 95°C, and more preferably 30 to 70°C.
- the treatment time is preferably 1 to 120 seconds, and more preferably 2 to 60 seconds.
- wastewater from the acidic aqueous solution employed in electrochemical graining as the desmutting solution (acidic solution).
- electrochemical graining is carried out, after which (1) acidic etching treatment, (2) alkali etching treatment followed by desmutting, (3) alkali etching treatment followed by acidic etching treatment, and (4) alkali etching treatment following by desmutting or acidic etching treatment are carried out. Then, electrochemical graining or alkali etching treatment followed by desmutting treatment is carried out.
- the aluminum sheet After being subjected to the above-described graining treatment and other types of treatment as needed, the aluminum sheet is administered anodizing treatment.
- Anodizing treatment can be carried out by any suitable method known to be used in the art to which the invention relates. More specifically, an anodizing layer can be formed on the surface of the aluminum sheet by passing a direct current or alternating current through the aluminum sheet in an aqueous or non-aqueous solution of any one or combination of, for example, sulfuric acid, phosphoric acid, chromic acid, oxalic acid, sulfamic acid and benzenesulfonic acid.
- the anodizing treatment conditions vary empirically according to the electrolytic solution used, although it is generally suitable for the solution to have a concentration of 1 to 80 wt% and a temperature of 5 to 70°C, and for the current density to be 0.5 to 60 A/dm 2 , the voltage to be 1 to 200 V, and the electrolysis time to be 1 to 1,000 seconds.
- anodizing process carried out in a sulfuric acid electrolytic solution at a high current density described in GB 1,412,768 B and the anodizing process carried out using phosphoric acid as the electrolytic bath described in US 3,511,661 are preferred. It is also possible to carry out a multi-step anodizing treatment involving, for example, anodizing treatment in sulfuric acid and also anodizing treatment in phosphoric acid.
- the anodized layer has a weight of preferably at least 1.0 g/m 2 , more preferably at least 2.0 g/m 2 , and even more preferably 4.0 g/m 2 .
- the anodized layer has a weight of preferably not more than 100 g/m 2 , more preferably not more than 10.0 g/m 2 , and even more preferably not more than 6.0 g/m 2 .
- micropores Minute depressions called micropores are formed so as to be uniformly distributed over the surface of the anodized layer.
- the density and diameter of the micropores present on the anodized layer can be adjusted by suitable selection of the treatment conditions.
- pore widening treatment which widens the diameter of the micropores.
- This pore widening treatment involves immersion of the aluminum base on which an anodized layer has been formed in an acidic or alkaline aqueous solution to dissolve the anodized layer and widen the diameter of the micropores.
- Pore widening treatment is carried out such that the amount of anodized layer dissolution is in a range of preferably 0.01 to 20 g/m 2 , more preferably 0.1 to 5 g/m 2 , and even more preferably 0.2 to 4 g/m 2 .
- an aqueous solution of an inorganic acid such as sulfuric acid, phosphoric acid, nitric acid or hydrochloric acid, or a mixture thereof, is preferred.
- the acidic aqueous solution has a concentration of preferably 10 to 1,000 g/L, and more preferably 20 to 500 g/L, and has a temperature of preferably 10 to 90°C, and more preferably 30 to 70°C.
- the length of immersion in the acidic aqueous solution is preferably from 1 to 300 seconds, and more preferably from 2 to 100 seconds.
- an aqueous solution of at least one alkali selected from the group consisting of sodium hydroxide, potassium hydroxide and lithium hydroxide it is preferable to use an aqueous solution of at least one alkali selected from the group consisting of sodium hydroxide, potassium hydroxide and lithium hydroxide.
- the alkaline aqueous solution has a pH of preferably 10 to 13, and more preferably 11.5 to 13.0, and has a temperature of preferably 10 to 90°C, and more preferably 30 to 50°C.
- the length of immersion in the alkaline aqueous solution is preferably from 1 to 500 seconds, and more preferably from 2 to 100 seconds.
- Alkali treatment may be followed by treatment with an acidic aqueous solution.
- This treatment is preferably carried out by immersing the anodized metal base in an aqueous solution having a phosphate compound concentration of 0.01 to 20 wt%, an inorganic fluorine compound concentration of 0.01 to 5 wt%, and a pH of 3 to 5. Immersion is carried out at preferably 20 to 100°C, and more preferably 40 to 80°C, and for a period of preferably 1 to 300 seconds, and more preferably 5 to 30 seconds.
- the concentration of the phosphate compound is preferably at least 0.01 wt%, more preferably at least 0.05 wt%, and even more preferably at least 0.1 wt%.
- the concentration is preferably not more than 20 wt%, more preferably not more than 10 wt%, and even more preferably not more than 5 wt%.
- the concentration of the inorganic fluorine compound is preferably at least 0.01 wt%, preferably at least 0.05 wt%, and more preferably at least 0.1 wt%. For a long press life, this concentration is preferably not more than 5 wt%, and more preferably not more than 2 wt%.
- the relative proportions of the respective compounds in the aqueous solution are preferably such that the weight ratio of the inorganic fluorine compound to the phosphate compound is in a range of 1:200 to 200:1.
- Phosphates that may be used in the invention include the phosphoric acid salts of metals such as alkali metals and alkaline earth metals.
- Specific examples include zinc phosphate, aluminum phosphate, ammonium phosphate, diammonium hydrogenphosphate, ammonium dihydrogenphosphate, monoammonium phosphate, monopotassium phosphate, monosodium phosphate, potassium dihydrogenphosphate, dipotassium hydrogenphosphate, calcium phosphate, ammonium sodium hydrogenphosphate, magnesium hydrogenphosphate, magnesium phosphate, iron (II) phosphate, iron (III) phosphate, sodium dihydrogenphosphate, trisodium phosphate, disodium hydrogenphosphate, lead phosphate, diammonium phosphate, calcium dihydrogenphosphate, lithium phosphate, phosphotungstic acid, ammonium phosphotungstate, sodium phosphotungstate, ammonium phosphomolybdate, sodium phosphomolybdate, sodium phosphite, sodium tripolyphosphate and sodium pyrophosphate.
- sodium dihydrogenphosphate, disodium hydrogenphosphate, potassium dihydrogenphosphate and dipotassium hydrogenphosphate are
- Preferred inorganic fluorine compounds that may be used in the aqueous solution containing an inorganic fluorine compound and a phosphate compound include metal fluorides.
- Specific examples include sodium fluoride, potassium fluoride, calcium fluoride, magnesium fluoride, sodium hexafluorozirconate, potassium hexafluorozirconate, sodium hexafluorotitanate, potassium hexafluorotitanate, hexafluorozirconic acid, hexafluorotitanic acid, ammonium hexafluorozirconate, ammonium hexafluorotitanate, hexafluorosilicic acid, nickel fluoride, iron fluoride, fluorophosphoric acid and ammonium fluorophosphate.
- the aqueous solution containing an inorganic fluorine compound and a phosphate compound that is used for treatment may contain one each, or two or more each, of the phosphate and the inorganic fluorine compound.
- treatment with the aqueous solution containing an inorganic fluorine compound and a phosphate compound may be carried out by separate treatments with an aqueous solution containing an inorganic fluorine compound and with an aqueous solution containing a phosphate compound.
- the metal base is dipped in the aqueous solution containing an inorganic fluorine compound and a phosphate compound, following which it is washed such as with water and dried.
- Methods other than dipping include application of the aqueous solution by brush, sponge, spray, wheel coater or some other suitable means.
- Silicate compounds preferred for use in the treatment with a silicate compound-containing aqueous solution that is employed in the invention include silicic acid and silicates. Of these, alkali metal silicates are preferred.
- sodium silicate, potassium silicate and lithium silicate are preferred.
- sodium silicate and potassium silicate are preferred.
- sodium silicate examples include No. 3 sodium silicate, No. 2 sodium silicate, No. 1 sodium silicate, sodium orthosilicate, sodium sesquisilicate and sodium metasilicate.
- potassium silicate examples include No. 1 potassium silicate. Aluminosilicates containing aluminum and borosilicates containing boron may also be used.
- silicic acid examples include orthosilicic acid, metasilicic acid, bisilicic acid, trisilicic acid and tetrasilicic acid.
- the aqueous solution has a silicate compound concentration of preferably at least 0.01 wt%, more preferably at least 0.1 wt%, and even more preferably at least 1 wt%.
- the solution has a concentration of preferably not more than 10 wt%, more preferably not more than 7 wt%, and even more preferably not more than 5 wt%.
- the silicate compound-containing aqueous solution used in the invention may also include a suitable amount of a hydroxide compound such as sodium hydroxide, potassium hydroxide or lithium hydroxide in order to increase the pH.
- a hydroxide compound such as sodium hydroxide, potassium hydroxide or lithium hydroxide in order to increase the pH.
- sodium hydroxide and potassium hydroxide are preferred.
- alkaline earth metal salt or a Group 4 (Group IVA) metal salt may be included.
- alkaline earth metal salts include the following water-soluble salts: nitrates such as calcium nitrate, strontium nitrate, magnesium nitrate and barium nitrate; and also sulfates, hydrochlorides, phosphates, acetates, oxalates, and borates.
- Exemplary Group 4 (Group IVA) metal salts include titanium tetrachloride, titanium trichloride, titanium potassium fluoride, titanium potassium oxalate, titanium sulfate, titanium tetraiodide, zirconium chloride oxide, zirconium dioxide, zirconium oxychloride and zirconium tetrachloride. These alkaline earth metal salts and Group 4 (Group IVA) metal salts may be used singly or in combinations of two or more thereof.
- the silicate compound-containing aqueous solution has a temperature of preferably at least 10°C, and more preferably at least 20°C, but preferably not more than 100°C, and even more preferably not more than 80°C.
- the aqueous solution has a pH of preferably at least 8, and more preferably at least 10, but preferably not more than 13, and more preferably not more than 12.
- the treatment time is preferably at least 1 second, and more preferably at least 3 seconds, but preferably not more than 600 seconds, and more preferably not more than 120 seconds.
- the metal base that has been treated with the above-described silicate compound-containing aqueous solution can, if necessary, be treated with an acidic aqueous solution.
- an acidic aqueous solution include aqueous solutions of sulfuric acid, nitric acid, hydrochloric acid, oxalic acid or phosphoric acid.
- this acidic aqueous solution treatment by dipping the hydrophilized metal base in an aqueous solution containing the acid described above in a concentration of 0.001 to 10 wt%, preferably 0.01 to 1 wt%, at a temperature of 15 to 70°C, preferably 25 to 50°C, and for a period of 0.5 to 120 seconds, preferably 2 to 30 seconds.
- treatment with specific aqueous solutions involves carrying out (1) treatment with an aqueous solution containing an inorganic fluorine compound and a phosphate compound, and (2) treatment with a silicate compound-containing aqueous solution, either before or after treatment (1). That is, either (1) treatment with an aqueous solution containing an inorganic fluorine compound and a phosphate compound is followed by (2) treatment with an aqueous solution containing a silicate compound, or (2) treatment with an aqueous solution containing a silicate compound is followed by (1) treatment with an aqueous solution containing an inorganic fluorine compound and a phosphate compound. These treatments may each be carried out a plurality of times.
- treatment with an aqueous solution containing an inorganic fluorine compound and a phosphate compound involves separately carrying out treatment with an aqueous solution containing an inorganic fluorine compound and treatment with an aqueous solution containing a phosphate compound, these treatments may be combined in any way with the treatment with an aqueous solution containing a silicate compound.
- the metal base obtained by treatment with specific aqueous solutions as described above which metal base is sometimes referred to hereinafter simply as "the metal base of the invention," may be subjected to a hydrophilic surface treatment involving immersion in an aqueous solution containing one or more hydrophilic compounds.
- the hydrophilic compound include polyvinylphosphonic acid, compounds having sulfonic acid groups, and carbohydrate compounds.
- the compound having sulfonic acid groups includes aromatic sulfonic acids, formaldehyde condensation products thereof, derivatives of these, and salts of any of the above.
- aromatic sulfonic acids include phenolsulfonic acid, catecholsulfonic acid, resorcinolsulfonic acid, benzenesulfonic acid, toluenesulfonic acid, ligninsulfonic acid, naphthalenesulfonic acid, acenaphthene-5-sulfonic acid, phenanthrene-2-sulfonic acid, benzaldehyde-2 (or 3)-sulfonic acid, benzaldehyde-2,4 (or 3,5)-disulfonic acid, oxybenzylsulfonic acids, sulfobenzoic acid, sulfanilic acid, naphthionic acid, and taurine.
- benzenesulfonic acid naphthalenesulfonic acid and ligninsulfonic acid are preferred.
- the formaldehyde condensation products of benzenesulfonic acid, naphthalenesulfonic acid and ligninsulfonic acid are also preferred.
- these may be used as their sulfonic acid salts.
- salts include the sodium salts, potassium salts, lithium salts, calcium salts and magnesium salts. Of these, the sodium salts and potassium salts are preferred.
- the aqueous solution containing a sulfonic acid group-bearing compound has a pH of preferably 4 to 6.5.
- Compounds such as sulfuric acid, sodium hydroxide and ammonia may be used to adjust the pH within the above range.
- Exemplary carbohydrate compounds include monosaccharides and sugar alcohols thereof, oligosaccharides, polysaccharides, and glycosides.
- Illustrative examples of monosaccharides and sugar alcohols thereof include trioses and their sugar alcohols, such as glycerol; tetroses and their sugar alcohols, such as threose and erythritol; pentoses and their sugar alcohols, such as arabinose and arabitol; hexoses and their sugar alcohols, such as glucose and sorbitol; heptoses and their sugar alcohols, such as D-glycero-D-galacto-heptose and D-glycero-D-galacto-heptitol; octoses and their sugar alcohols, such as D-erythro-D-galacto-octitol; and nonoses and their sugar alcohols, such as D-erythro-L-gluco-nonulose and its sugar alcohols.
- trioses and their sugar alcohols such as glycerol
- tetroses and their sugar alcohols such as threose and eryth
- oligosaccharides include disaccharides such as saccharose, trehalose and lactose; and trisaccharides such as raffinose.
- polysaccharides include amylose, arabinan, cyclodextrin and cellulose alginate.
- Glycoside refers to compounds in which a sugar constituent and a non-sugar constituent are bonded through, for example, an ether linkage.
- Glycosides can be classified based on the non-sugar constituent. Examples include alkyl glycosides, phenol glycosides, coumarin glycosides, oxycoumarin glycosides, flavonoid glycosides, anthraquinone glycosides, triterpene glycosides, steroid glycosides and mustard oil glycosides.
- Exemplary sugar constituents include the above-mentioned monosaccharides and their sugar alcohols, oligosaccharides, and polysaccharides. Of these, monosaccharides and oligosaccharides are preferred. Monosaccharides and disaccharides are especially preferred.
- glycosides examples include compounds of formula (I) below:
- R represents a linear or branched alkyl, alkenyl or alkynyl group of 1 to 20 carbons.
- alkyl groups having 1 to 20 carbons include methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, tridecyl, tetradecyl, pentadecyl, hexadecyl, heptadecyl, octadecyl, nonadecyl and eicosyl. These may be linear, branched or cyclic alkyl groups.
- alkenyl groups having 1 to 20 carbons include allyl and 2-butenyl. These may be linear, branched or cyclic alkenyl groups.
- alkynyl groups having 1 to 20 carbons include 1-pentynyl. These may be linear, branched or cyclic alkynyl groups.
- compounds of above formula (I) include methyl glucoside, ethyl glucoside, propyl glucoside, isopropyl glucoside, butyl glucoside, isobutyl glucoside, n-hexyl glucoside, octyl glucoside, capryl glucoside, decyl glucoside, 2-ethylhexyl glucoside, 2-pentylnonyl glucoside, 2-hexyldecyl glucoside, lauryl glucoside, myristyl glucoside, stearyl glucoside, cyclohexyl glucoside and 2-butynyl glucoside.
- glucosides one type of glycoside in which the hemiacetalhydroxyl group on glucose is ether bonded to another compound, and can be obtained by a known method involving the reaction of glucose with alcohols.
- Some of these alkyl glucosides are commercially available from Henkel, Germany under the trade name Glucopon, and can be used in the present invention.
- glycosides include saponins, rutin trihydrate, hesperidin methylchalcone, hesperidin, naringin hydrate, phenol- ⁇ -D-glucopyranoside, salicin and 3',5,7-methoxy-7-rutinoside.
- the aqueous solution containing a carbohydrate compound has a pH of preferably 8 to 11.
- the pH can be adjusted within this range using, for example, potassium hydroxide, sulfuric acid, carbonic acid, sodium carbonate, phosphoric acid or sodium phosphate.
- Aqueous solutions of polyvinylphosphonic acid have a concentration of preferably 0.1 to 5 wt%, and more preferably 0.2 to 2.5%.
- the immersion temperature is preferably 10 to 70°C, and more preferably 30 to 60°C.
- the immersion time is preferably 1 to 20 seconds.
- Aqueous solutions of sulfonic acid group-bearing compounds have a concentration of preferably 0.02 to 0.2 wt%.
- the immersion temperature is preferably 60 to 100°C.
- the immersion time is preferably 1 to 300 seconds, and more preferably 10 to 100 seconds.
- Aqueous solutions of carbohydrate compounds have a concentration of preferably 0.5 to 10 wt%.
- the immersion temperature is preferably 40 to 70°C.
- the immersion time is preferably 2 to 300 seconds, and more preferably 5 to 30 seconds.
- the metal base of the invention is immersed in an aqueous solution containing these hydrophilic compounds, following which it is washed such as with water and dried.
- the above hydrophilic surface treatment resolves printing contamination problems such as the poor resistance to contamination after standing (poor ink removability) that arises as a trade-off with improved sensitivity (in the case of a negative-working photosensitive layer, improved press life) due to pore widening treatment following anodizing treatment. That is, because the pore diameter is increased, during printing, and particularly when printing is restarted after a period in which the press was down and the printing plate was left on the press, there is a tendency for the ink to become difficult to remove from the plate (sometimes referred to as "diminished resistance to contamination after standing" or "diminished ink removability"). However, this problem is alleviated by administering hydrophilic surface treatment.
- the metal base of the invention is dipped in these hydrophilic compound-containing aqueous solutions, then is washed such as with water and dried.
- a metal base which has been administered surface treatment and which satisfies formula (1) below is used in the inventive support for lithographic printing plates.
- the surface treatment is not subject to any particular limitation, and may be any treatment or treatments capable of preparing a surface which satisfies formula (1) below.
- Surface treatment that involves administering the above-described specific aqueous solution treatments following the above-described graining treatment and anodizing treatment is preferred. That is, it is preferable for the surface of a metal base prepared by graining treatment and anodizing treatment, followed by administration of the above-described specific aqueous solution treatment, to satisfy formula (1) below.
- A is the peak area (counts ⁇ eV/sec) for fluorine (1S) as measured by electron spectroscopy for chemical analysis (ESCA)
- B is the peak area (counts ⁇ eV/sec) for silicon (2P) as measured by ESCA
- C is the peak area (counts ⁇ eV/sec) for phosphorus (2P) as measured by ESCA
- D is the peak area (counts ⁇ eV/sec) for aluminum (2P) as measured by ESCA.
- (A+B+C)/(A+B+C+D) represents the degree to which the anodized layer is covered with a phosphate compound, an inorganic fluorine compound and a silicate compound. A larger value for (A+B+C)/(A+B+C+D) indicates higher coverage, whereas a smaller value indicates lower coverage.
- the value of (A+B+C)/(A+B+C+D) is preferably at least 0.10, and more preferably at least 0.20.
- Electron spectroscopy for chemical analysis (ESCA) and the procedure involved are described.
- X-ray irradiation is carried out using the Mg-K ⁇ (1253.6 eV) and A1-K ⁇ (1486.6 eV) lines having a small energy range.
- the depth of penetration by such soft x-rays is about several microns from the surface of the specimen.
- the probability is very high that a photoelectron generated from deep areas within the specimen will lose energy from non-elastic scattering with other atoms before it reaches the surface of the specimen.
- Only photoelectrons generated at the surface-most portion of the specimen do not collide with other atoms and can be analyzed after emission with the relationship (I) intact.
- ESCA is capable of measuring the outermost several nanometers (several tens of angstroms) of the specimen surface.
- An intermediate layer containing an acid group-bearing polymeric compound is provided as an intermediate layer (or "undercoat,” as viewed from the recording layer side) on the aluminum support that has been treated as described above. This enables a support for lithographic printing plates to be obtained which, when rendered into a lithographic printing plate, has an excellent sensitivity, press life and resistance to scumming.
- the acid group on the acid group-bearing polymeric compound constituent is preferably an acid group having an acid dissociation constant (pKa) not higher than 7, more preferably -COOH, -SO 3 H, -OSO 3 H, -PO 3 H 2 , -OPO 3 H 2 , -CONHSO 2 or -SO 2 NHSO 2 -, and most preferably -COOH.
- Preferred acid group-bearing constituents include polymerizable compounds of general formula (1) or general formula (2) below.
- A represents a divalent linkage
- B is an aromatic or substituted aromatic group
- D and E are each independently divalent linkages
- G is a trivalent linkage
- X and X' are each independently an acid group having a pKa of 7 or less or an alkali metal salt or ammonium salt
- R 1 represents a hydrogen atom, an alkyl group or a halogen atom.
- the letters a, b, d and e are each independently 0 or 1.
- the letter t is an integer from 1 to 3.
- Preferred acid group-bearing constituents are those wherein A is -COO- or -CONH- and B is a phenylene or substituted phenylene group, with the substituent being a hydroxyl group, a halogen atom or an alkyl group; D and E are each independently an alkylene group or a divalent linkage of the molecular formula C n H 2n O, C n H 2n S or C n H 2n+1 N; G represents a trivalent linkage of the molecular formula C n H 2n-1 , C n H 2n-1 O, C n H 2n-1 S or C n H 2n N, the letter n being an integer from 1 to 12; X and X' are each independently a carboxylic acid, sulfonic acid, phosphonic acid, sulfuric acid monoester or phosphoric acid monoester; R 1 is a hydrogen atom or an alkyl group; and the letters a, b, d and e are
- Especially preferred acid group-bearing constituents are those of the general formula (1) in which B is a phenylene group or a substituted phenylene group, the substituent being a hydroxyl group or an alkyl of 1 to 3 alkyls; D and E are each independently an alkylene group of 1 or 2 carbons or an alkylene group of 1 or 2 carbons bonded through an oxygen atom; R1 is a hydrogen atom or a methyl group; X is a carboxyl group; and the letters a and b are respectively 0 and 1.
- acid group-bearing constituent examples include acrylic acid, methacrylic acid, crotonic acid, isocrotonic acid, itaconic acid, maleic acid and maleic anhydride. Additional examples include compounds having the following formulas.
- the above acid group-bearing constituents may be used singly or as combinations of two or more thereof.
- Preferred onium groups on the onium group-bearing constituent of the polymeric compound that may be used to form the intermediate layer include onium groups composed of atoms of elements belonging to group V or group VI of the Periodic Table. Onium groups containing nitrogen atoms, phosphorus atoms or sulfur atoms are more preferred, and onium groups containing nitrogen atoms are especially preferred.
- This polymeric compound is preferably a vinyl polymer such as one having a backbone structure composed of acrylic acid or methacrylic acid, or polystyrene, or is a urethane resin, a polyester or a polyamide. Of these, vinyl polymers, such as ones having a backbone structure composed of acrylic resin or methacrylic resin, or polystyrene, are especially preferred.
- Especially preferred polymeric compounds are copolymerizable polymers in which the onium group-bearing constituent has general formula (3), (4) or (5) below.
- J is a divalent linkage
- K is an aromatic or substituted aromatic group
- each M independently represents a divalent linkage
- Y 1 is an atom belonging to group V of the Periodic Table
- Y 2 is an atom belonging to group VI of the Periodic Table
- Z- is a counteranion.
- R 2 is a hydrogen atom, an alkyl group or a halogen atom
- R 3 , R 4 , R 5 and R 7 are each independently a hydrogen atom or an alkyl, aromatic or aralkyl group which may have substituents bonded thereto
- R 6 is an alkylidyne or substituted alkylidyne group; wherein R 3 and R 4 or R 6 and R 7 may be mutually bonded to form a ring.
- the letters j, k and m are each independently 0 or 1; and the letter u is an integer from 1 to 3.
- Preferred onium group-bearing constituents include those in which J represents -COO- or -CONH-; K represents a phenylene or substituted phenylene group, wherein the substituent is a hydroxyl group, a halogen atom or an alkyl group; M is an alkylene group, or a divalent linkage of the molecular formula C n H 2n O, C n H 2n S or C n H 2n+1 N, the letter n being an integer from 1 to 12; Y 1 is a nitrogen atom or phosphorus atom; Y 2 is a sulfur atom; and Z- is a halogen ion, PF 6 -, BF 4 - or R 8 SO 3 -.
- R 2 is a hydrogen atom or an alkyl group
- R 3 , R 4 , R 5 and R 7 are each independently hydrogen atoms or C 1-10 alkyl, aromatic or aralkyl groups which may have substituents bonded thereto
- R 6 is a C 1-10 alkylidyne or substituted alkylidyne group; wherein R 3 and R 4 or R 6 and R 7 may be mutually bonded to form a ring.
- the letters j, k and m are each independently 0 or 1, such that j and k are not both 0.
- Especially preferred onium group-bearing constituents include those in which K is a phenylene or substituted phenylene group, the substituent being a hydroxyl group or an alkyl group of 1 to 3 carbons; M is an alkylene group of 1 or 2 carbons or an alkylene group of 1 or 2 carbons that is linked through an oxygen atom; Z - is a chlorine ion or R 8 SO 3 -; R 2 is a hydrogen atom or a methyl group; and the letters j and k are respectively 0 and 1.
- onium group-bearing constituents include the following.
- the polymeric compound used to form the intermediate layer contain at least 1 mol%, and preferably at least 5 mol%, of the above-described onium group-bearing constituent.
- the presence of at least 1 mol% of an onium group-bearing constituent further improves adhesion.
- the onium group-bearing constituent may be of one type or a combination of two or more types.
- the polymeric compound used to form the intermediate layer may also be a mixture of two or more such compounds of differing constituents, compositional ratios or molecular weights.
- polymeric compounds which contain both the above-described acid group and the above-described onium group it is advantageous for the compound to include at least 20 mol%, and preferably at least 40 mol%, of the acid group-bearing constituent, and at least 1 mol%, and preferably at least 5 mol%, of the onium group-bearing constituent.
- the presence of at least 20 mol% of the acid group-bearing constituent further promotes dissolution and removal during alkali development, and also further enhances adhesion due to synergistic effects between the acid groups and the onium groups.
- such polymeric compounds which contain both acid groups and onium groups may of course be a mixture of two or more such compounds of differing constituents, compositional ratios or molecular weights.
- Illustrative examples of polymeric compounds having both the above-described onium groups and acid groups are given below. The compositional ratios in these polymer structures are given in mole percent (mol%).
- polymeric compounds which contain acid groups or both acid groups and onium groups and are used to form the intermediate layer can generally be prepared using a radical chain polymerization process (see Textbook of Polymer Science, 3 rd ed., by F.W. Billmeyer (John Wiley & Sons, 1984).
- These polymeric compounds may have a broad range in molecular weight, although the weight-average molecular weight (Mw) as measured by a light scattering technique is preferably in a range of from 500 to 2,000,000, and more preferably 2,000 to 600,000.
- Mw weight-average molecular weight
- the amount of unreacted monomer present in this polymeric compound may vary over a wide range, although it is preferably not more than 20 wt%, and more preferably not more than 10 wt%.
- a typical example of a polymeric compound containing both acid groups and onium groups is a copolymer of p-vinylbenzoic acid and vinylbenzyltrimethyl ammonium chloride (No. 1 in Table 1 above).
- This can be synthesized as follows. First, 146.9 g (0.99 mol) of p-vinylbenzoic acid (available from Hokko Chemical Industry Co., Ltd.), 44.2 g (0.21 mol) of vinylbenzyltrimethyl ammonium chloride, and 446 g of 2-methoxyethanol were placed in a 1-liter, 3-neck flask, then heated and held at 75°C under a stream of nitrogen and stirring.
- the intermediate layer can be provided by using any of various methods to apply the above-described polymeric compound bearing acid groups or both acid groups and onium groups (referred to hereinafter as simply "the polymeric compound") onto the above-described metal base treated with specific aqueous solutions or treated with specific aqueous solutions, then additionally treated with an acidic aqueous solution.
- One commonly used method for providing the intermediate layer involves applying to the metal base a solution obtained by dissolution of the polymeric compound in an organic solvent such as methanol, ethanol or methyl ethyl ketone, a mixture of such organic solvents, or a mixed solvent composed of any of these organic solvents and water, then drying the applied solution.
- Another method involves dipping the metal base in a solution obtained by dissolution of the polymeric compound in an organic solvent such as methanol, ethanol or methyl ethyl ketone, a mixture of such organic solvents, or a mixed solvent composed of any of these organic solvents and water so as to induce adsorption of the polymeric compound, then washing such as with water and drying.
- an organic solvent such as methanol, ethanol or methyl ethyl ketone, a mixture of such organic solvents, or a mixed solvent composed of any of these organic solvents and water
- the solution concentration is 0.01 to 20 wt%, preferably 0.05 to 5 wt%
- the dipping temperature is 20 to 90°C, and preferably 25 to 50°C
- the dipping time is 0.1 second to 20 minutes, and preferably 2 seconds to 1 minute.
- the above polymeric compound solution can also be used after adjusting the pH within a range of 0 to 12, and preferably 0 to 5, using a basic substance such as ammonia, triethylamine or potassium hydroxide, an inorganic acid such as hydrochloric acid, phosphoric acid, sulfuric acid or nitric acid, any of various organic acidic substances such as organic sulfonic acids (e.g., nitrobenzenesulfonic acid, naphthalenesulfonic acid), organic phosphonic acids (e.g., phenylphosphonic acid) and organic carboxylic acids (e.g., benzoic acid, coumaric acid, malic acid), and organic acid chlorides such as naphthalenesulfonyl chloride and benzenesulfonyl chloride.
- a basic substance such as ammonia, triethylamine or potassium hydroxide
- an inorganic acid such as hydrochloric acid, phosphoric acid, sulfuric acid
- the coating weight after drying of the polymeric compound is generally from 2 to 100 mg/m 2 , and preferably from 5 to 50 mg/m 2 . At a coating weight of less than 2 mg/m 2 or more than 100 mg/m 3 , sufficient effects may not be achieved.
- the support on which an intermediate layer has been formed and which has been obtained as described above may be provided on the back side (the side on which a recording layer is not provided) with a coat (referred to hereinafter as the "back coat") composed of an organic polymeric compound so that scuffing of the recording layer does not occur even when the resulting presensitized plates are stacked on top of one other.
- a coat referred to hereinafter as the "back coat"
- the back coat preferably contains, as the main component, at least one resin which has a glass transition point of at least 20°C and is selected from the group consisting of saturated copolyester resins, phenoxy resins, polyvinyl acetal resins and vinylidene chloride copolymer resins.
- Preferred use can be made of coats composed of metal oxides obtained by the hydrolysis and polycondensation of the organic polymeric compounds mentioned in JP 5-45885 A and the organic or inorganic metal compounds mentioned in JP 6-35174 A.
- alkoxy compounds of silicon such as Si(OCH 3 ) 4 , Si (OC 2 H 5 ) 4 , Si(OC 3 H 7 ) 4 and Si (OC 4 H 9 ) 4 , are preferred because they are inexpensive and readily available, and metal oxide coats obtained from these compounds have an excellent developability.
- the saturated copolyester resin used in the back coat is composed of dicarboxylic acid units and diol units.
- dicarboxylic acid units include aromatic dicarboxylic acids such as phthalic acid, terephthalic acid, isophthalic acid, tetrabromophthalic acid and tetrachlorophthalic acid; and saturated aliphatic dicarboxylic acids such as adipic acid, azelaic acid, succinic acid, oxalic acid, suberic acid, sebacic acid, malonic acid and 1,4-cyclohexanedicarboxylic acid.
- the back coat may additionally include dyes and pigments for coloration, silane coupling agents to improve adhesion to the support, diazo resins composed of diazonium salts, organophosphonic acids, organophosphoric acids, cationic polymers, and the following substances which are commonly used as slip agents: waxes, higher aliphatic acids, higher aliphatic acid amides, silicone compounds made of dimethylsiloxane, modified dimethylsiloxane and polyethylene powder.
- diazo resins composed of diazonium salts, organophosphonic acids, organophosphoric acids, cationic polymers, and the following substances which are commonly used as slip agents: waxes, higher aliphatic acids, higher aliphatic acid amides, silicone compounds made of dimethylsiloxane, modified dimethylsiloxane and polyethylene powder.
- the back coat should have a thickness which is of a degree that will help protect the recording layer from scuffing, even in the absence of a slip sheet.
- a thickness of 0.01 to 8 ⁇ m is preferred. At a thickness of less than 0.01 ⁇ m, it may be difficult to prevent scuffing of the recording layer when a plurality of presensitized plates are stacked and handled together. On the other hand, at a thickness of more than 8 ⁇ m, the chemicals used in the vicinity of the lithographic printing plate during printing cause the back coat to swell and fluctuate in thickness, which may alter the printing pressure and thereby compromise the printability.
- Various methods may be used to provide the back coat on the back side of the support.
- Illustrative examples include preparing the above-mentioned back coat ingredients as a solution in a suitable solvent and applying the solution, or preparing these ingredients as an emulsified dispersion, then applying the dispersion and drying.
- Another method that may be used is to first form a film, then laminate and bond the film to the support using an adhesive or heat.
- Still another method is to form a melt film by a melt extruder and bond the film to the support.
- the especially preferred method for achieving a suitable thickness is to dissolve the back coat-forming ingredients in a suitable solvent, then apply the solution and dry.
- Organic solvents such as those mentioned in JP 62-251739 A may be used singly or in admixture as the media in such methods.
- the presensitized plate it is possible to first provide on the support either the back coat on the back side or the recording layer on the front side. Alternatively, both may be provided at the same time.
- the presensitized plate of the invention can be obtained by providing an infrared laser-imageable recording layer on the inventive support for lithographic printing plates obtained as described above.
- the recording layer is preferably a heat-sensitive layer containing:
- Thermally reactive functional groups common to both (a) and (b) above include ethylenically unsaturated groups which carry out the polymerization reaction (e.g., acryloyl, methacryloyl, vinyl, allyl); isocyanate groups or blocked isocyanate groups which carry out addition reactions, along with active hydrogen atom-bearing functional groups that react therewith (e.g., amino groups, hydroxyl groups, carboxyl groups); epoxy groups that carry out addition reactions, along with amino, carboxyl or hydroxyl groups that react therewith; carboxyl groups that carry out condensation reactions, along with hydroxyl or amino groups that react therewith; and acid anhydrides which carry out ring-opening addition reactions, along with amino or hydroxyl groups that react therewith.
- Thermally reactive functional groups which may be used in the present invention are not limited to the above, and may be any functional group that carries out a reaction, so long as it forms a chemical bond.
- thermally reactive functional groups suitable for the finely divided polymer (a) include acryloyl, methacryloyl, vinyl, allyl, epoxy, amino, hydroxyl, carboxyl, isocyanate and acid anhydride groups, as well as protected forms of these groups.
- the thermally reactive functional groups may be introduced onto the polymer particles when the polymer is polymerized or may be introduced by means of a polymer reaction following polymerization.
- thermally reactive functional groups are introduced onto the polymer during polymerization, it is preferable to carry out emulsion polymerization or suspension polymerization using a monomer having a thermally reactive functional group.
- monomers having a thermally reactive functional group that may be used in the invention include allyl methacrylate, allyl acrylate, vinyl methacrylate, vinyl acrylate, glycidyl methacrylate, glycidyl acrylate, 2-isocyanatoethyl methacrylate and alcohol-blocked isocyanates thereof, 2-isocyanatoethyl acrylate and alcohol-blocked isocyanates thereof, 2-aminoethyl methacrylate, 2-aminoethyl acrylate, 2-hydroxyethyl methacrylate, 2-hydroxyethyl acrylate, acrylic acid, methacrylic acid, maleic anhydride, bifunctional acrylate and bifunctional methacrylate.
- Illustrative, non-limiting, examples of monomers without a thermally reactive functional group that are copolymerizable with these monomers and may be used in the invention include styrene, alkyl acrylates, alkyl methacrylates, acrylonitrile and vinyl acetate.
- Examples of polymer reactions that may be used when the thermally reactive functional groups are introduced after polymerization include the polymer reactions mentioned in WO 96/34316.
- finely divided polymers (a) those in which the fine particles mutually coalesce under heating are preferred, and those which have a hydrophilic surface and disperse in water are especially preferred. It is desirable in this case for a film formed by applying only the finely divided polymer and drying at a temperature below the solidification temperature to have a contact angle (water drop in air) which is smaller than the contact angle (water drop in air) of a film that is similarly formed but dried at a temperature higher than the solidification temperature.
- An illustrative, non-limiting example of a method for making the surface of the polymer fine particles hydrophilic in this way involves the adsorption of a hydrophilic polymer or oligomer such as polyvinyl alcohol or polyethylene glycol, or of a hydrophilic low-molecular-weight compound, onto the surface of the polymer fine particles.
- a hydrophilic polymer or oligomer such as polyvinyl alcohol or polyethylene glycol
- the finely divided polymer (a) prefferably has a solidification temperature of at least 70°C, although a solidification temperature of at least 100°C is especially preferred for good stability over time.
- the finely divided polymer (a) has an average particle size of preferably 0.01 to 20 ⁇ m, more preferably 0.05 to 2.0 ⁇ m, and most preferably 0.1 to 1.0 ⁇ m. Within the above range, good resolution and stability over time can be achieved.
- the amount of finely divided polymer (a) added is preferably at least 50 wt%, and more preferably at least 60 wt%, of the solids in the heat-sensitive layer.
- thermally reactive functional groups on the compound contained within the microcapsules (b) include polymerizable unsaturated groups, hydroxyl groups, carboxyl groups, carboxylate groups, acid anhydride groups, amino groups, epoxy groups, isocyanate groups and blocked isocyanate groups.
- Preferred examples of polymerizable unsaturated group-bearing compounds include compounds with at least one, and preferably at least two, ethylenically unsaturated bond (e.g., acryloyl, methacryloyl, vinyl, allyl). Such compounds are widely used in industrial fields related to the present invention, and may be used herein without any particular limitation. These compounds have a variety of chemical forms, including monomers, prepolymers such as dimers, trimers, oligomers or mixtures thereof, and copolymers of any of the above.
- esters of unsaturated carboxylic acids and aliphatic polyols, and amides of unsaturated carboxylic acids and aliphatic polyamines are preferred.
- the products of addition reactions between an unsaturated carboxylic acid ester or amide having an electrophilic substituent such as an isocyanate or epoxy group and a monofunctional or polyfunctional alcohol, amine or thiol, and the products of substitution reactions between an unsaturated carboxylic acid ester or amide having a removable substituent such as a halogen group or a tosyloxy group and a monofunctional or polyfunctional alcohol, amine or thiol are also preferred.
- polymeric compounds which are esters of unsaturated carboxylic acids and aliphatic polyols are given below.
- Acrylic acid esters include ethylene glycol diacrylate, triethylene glycol diacrylate, 1,3-butanediol diacrylate, tetramethylene glycol diacrylate, propylene glycol diacrylate, neopentyl glycol diacrylate, trimethylolpropane diacrylate, trimethylolpropane triacrylate, trimethylolpropane tris(acryloyloxypropyl) ether, trimethylolethane triacrylate, hexanediol diacrylate, 1,4-cyclohexanediol diacrylate, tetraethylene glycol diacrylate, pentaerythritol diacrylate, pentaerythritol triacrylate, pentaerythritol tetraacrylate, dipentaerythritol diacrylate, dipentaerythritol pentaacrylate, dipentaerythritol hexaacrylate, sorbito
- Methacrylic acid esters include tetramethylene glycol dimethacrylate, triethylene glycol dimethacrylate, neopentyl glycol dimethacrylate, trimethylolpropane trimethacrylate, trimethylolethane trimethacrylate, ethylene glycol dimethacrylate, 1,3-butanediol dimethacrylate, hexanediol dimethacrylate, pentaerythritol dimethacrylate, pentaerythritol trimethacrylate, pentaerythritol tetramethacrylate, dipentaerythritol dimethacrylate, dipentaerythritol hexamethacrylate, sorbitol trimethacrylate, sorbitol tetramethacrylate, bis[p-(3-methacryloyloxy-2-hydroxypropoxy)phenyl] dimethylmethane and bis[p-(
- Itaconic acid esters include ethylene glycol diitaconate, propylene glycol diitaconate, 1,3-butanediol diitaconate, 1,4-butanediol diitaconate, tetramethylene glycol diitaconate, pentaerythritol diitaconate and sorbitol tetraitaconate.
- Crotonic acid esters include ethylene glycol dicrotonate, tetramethylene glycol dicrotonate, pentaerythritol dicrotonate and sorbitol tetradicrotonate.
- Isocrotonic acid esters include ethylene glycol diisocrotonate, pentaerythritol diisocrotonate and sorbitol tetraisocrotonate.
- Maleic acid esters include ethylene glycol dimaleate, triethylene glycol dimaleate, pentaerythritol dimaleate and sorbitol tetramaleate.
- esters examples include the aliphatic alcohol esters mentioned in JP 46-27926 B, JP 51-47334 B and JP 57-196231 A; esters having aromatic skeletons such as those mentioned in JP 59-5240 A, JP 59-5241 A and JP 2-226149 A; and the amino group-bearing esters mentioned in JP 1-165613 A.
- amides of unsaturated carboxylic acids with aliphatic polyamines that may be used as monomers include methylenebisacrylamide, methylenebismethacrylamide, 1,6-hexamethylenebisacrylamide, 1,6-hexamethylenebismethacrylamide, diethylenetriaminetrisacrylamide, xylylenebisacrylamide and xylylenebismethacrylamide.
- Suitable amide-type monomers include those having a cyclohexylene structure mentioned in JP 54-21726 B.
- Urethane-type addition polymerizable compounds prepared using an addition reaction between an isocyanate group and a hydroxyl group are also preferred.
- Specific examples include the urethane compounds having two or more polymerizable unsaturated groups per molecule which are mentioned in JP 48-41708 B and are obtained by adding a hydroxyl group-bearing unsaturated monomer of formula (II) below to a polyisocyanate compound having at least two isocyanate groups per molecule.
- CH 2 C (R 1 ) COOCH 2 CH(R 2 ) OH
- R 1 and R 2 each independently represent H or CH 3 .
- Urethane acrylates such as those mentioned in JP 51-37193 A, JP 2-32293 B and JP 2-16765 B, and the urethane compounds having an ethylene oxide-type skeleton mentioned in JP 58-49860 B, JP 56-17654 B, JP 62-39417 B and JP 62-39418 B are also preferred.
- radical polymerizable compounds having within the molecule an amino structure or a sulfide structure that are mentioned in JP 63-277653 A, JP 63-260909 A and JP 1-105238 A.
- Additional examples include polyfunctional acrylates and methacrylates, including polyester acrylates and epoxy acrylates obtained by reacting an epoxy resin with (meth)acrylic acid, such as those mentioned in JP 48-64183 A, JP 49-43191 B and JP 52-30490 B. Further examples include the specific unsaturated compounds mentioned in JP 46-43946 B, JP 1-40337 B and JP 1-40336 B, and the vinylphosphonic acid compounds mentioned in JP 2-25493 A. In some cases, it will be desirable to use the perfluoroalkyl group-containing compounds mentioned in JP 61-22048 A. Use can also be made of the photocurable monomers and oligomers mentioned in Nippon Setchaku Kyokaishi Vol. 20, No. 7, 300-308 (1984).
- Suitable epoxy compounds include glycerol polyglycidyl ether, polyethylene glycol diglycidyl ether, polypropylene diglycidyl ether, trimethylolpropane polyglycidyl ether, sorbitol polyglycidyl ether, and the polyglycidyl ethers of bisphenols, polyphenols and hydrogenates thereof.
- Suitable isocyanate compounds include tolylene diisocyanate, diphenylmethane diisocyanate, polymethylene polyphenyl polyisocyanate, xylylene diisocyanate, naphthalene diisocyanate, cyclohexanephenylene diisocyanate, isophorone diisocyanate, hexamethylene diisocyanate, cyclohexyl diisocyanate, and compounds obtained by blocking any of the above with alcohol or amine.
- Suitable amine compounds include ethylenediamine, diethylenetriamine, triethylenetetramine, hexamethylenediamine, propylenediamine and polyethyleneimine.
- Suitable hydroxyl group-bearing compounds include compounds having terminal methylol groups, polyols such as pentaerythritol, bisphenols and polyphenols.
- Suitable carboxyl group-bearing compounds include aromatic polycarboxylic acids such as pyromellitic acid, trimellitic acid and phthalic acid; and aliphatic polycarboxylic acids such as adipic acid.
- Suitable acid anhydrides include pyromellitic anhydride and benzophenonetetracarboxylic anhydride.
- Suitable copolymers of ethylenically unsaturated compounds include allyl methacrylate copolymers, specific examples of which include allyl methacrylate/methacrylic acid copolymers, allyl methacrylate/ethyl methacrylate copolymers and allyl methacrylate/butyl methacrylate copolymers.
- Microencapsulation may be carried out by a known method.
- Illustrative, non-limiting examples of techniques for preparing microcapsules that may be used in the invention include the methods involving the use of coacervation described in US 2,800,457 and US 2,800,458; the methods that rely on interfacial polymerization described in GB 990,443 B, US 3,287,154, JP 38-19574 B, JP 42-446 B and JP 42-711 B; the methods involving polymer precipitation disclosed in US 3,418,250 and US 3,660,304; the method that uses an isocyanate polyol wall material described in US 3,796,669; the method that uses an isocyanate wall material described in US 3,914,511; the methods that use a urea-formaldehyde or urea formaldehyde-resorcinol wall-forming material which are described in US 4,001,140, 4,087,376 and 4,089,802; the method which uses wall materials such as
- Microcapsule walls suitable for use in the microcapsules (b) are those which have three-dimensional crosslinkages and are solvent-swellable. Accordingly, it is preferable for the microcapsule wall material to be polyurea, polyurethane, polyester, polycarbonate, polyamide, or a mixture thereof. Polyurea and polyurethane are especially preferred.
- the microcapsule wall may also have introduced therein a compound having thermally reactive functional groups.
- the microcapsules (b) have an average particle size of preferably 0.01 to 20 ⁇ m, more preferably 0.05 to 2.0 ⁇ m, and most preferably 0.10 to 1.0 ⁇ m. Within the above range, it is possible to obtain a good resolution and a good stability over time.
- the microcapsules (b) may or may not mutually coalesce under heating. What is important is that the microcapsules contain a substance which, following application of the recording layer, exudes onto the surface or outside of the microcapsules or penetrates into the microcapsule walls, and induces a chemical reaction under heating. Reaction may take place with a hydrophilic resin that has been added or with a low-molecular-weight compound that has been added. Alternatively, two or more types of microcapsules may each be provided with different functional groups which thermally react with each other, and the different types of microcapsules thereby induced to mutually react.
- microcapsules melt and coalesce with each other under heating.
- the amount of microcapules added to the heat-sensitive layer is preferably 10 to 60 wt%, and more preferably 15 to 40 wt%, based on the solids in the layer. Within this range, a good on-machine developability can be obtained and a good sensitivity and press life can also be achieved.
- a solvent which dissolves the microcapsule contents and causes the wall material to swell may be added to the microcapsule dispersing medium. The presence of such a solvent promotes the diffusion of the encapsulated thermally reactive functional group-bearing compound out of the microcapules.
- solvents include alcohols, ethers, acetals, esters, ketones, polyols, amides, amines and fatty acids.
- Specific examples include but are not limited to methanol, ethanol, t-butanol, n-propanol, tetrahydrofuran, methyl lactate, ethyl lactate, methyl ethyl ketone, propylene glycol monomethyl ether, ethylene glycol diethyl ether, ethylene glycol monomethyl ether, ⁇ -butyrolactone, N,N-dimethylformamide and N,N-dimethylacetamide. It is also possible to use two or more of these solvents together.
- Use can also be made of a solvent which does not dissolve in the microcapsule dispersion itself, but will dissolve therein if the above-described solvent has been admixed.
- a solvent is added in an amount which is selected according to the combination of ingredients, preferably 5 to 95 wt%, more preferably 10 to 90 wt%, and most preferably 15 to 85 wt%, based on the overall amount of the coating fluid.
- thermally reactive functional group-bearing finely divided polymer (a) or thermally reactive functional group-bearing compound-containing microcapsules (b) When a heat-sensitive layer containing the above-described thermally reactive functional group-bearing finely divided polymer (a) or thermally reactive functional group-bearing compound-containing microcapsules (b) is used, a compound which initiates or promotes these reactions may also be added if necessary.
- exemplary reaction-initiating or promoting compounds are compounds which generate radical or cations under heating. Specific examples include the lophine dimer, trihalomethyl compounds, peroxides, azo compounds, onium salts (e.g., diazonium salts, diphenyliodonium salts), acylphosphine and imidosulfonate.
- These compounds are added within a range of preferably 1 to 20 wt%, and more preferably 3 to 10 wt%, of the solids in the heat-sensitive layer. Within the above range, the on-machine developability is not compromised, and good reaction initiating or reaction promoting effects can be obtained.
- a hydrophilic resin may be added to the heat-sensitive layer.
- the addition of a hydrophilic resin provides a good on-machine developability and also enhances the film strength of the heat-sensitive layer itself.
- the hydrophilic resin is preferably one which contains hydrophilic groups such as hydroxyl, carboxyl, hydroxyethyl, hydroxypropyl, amino, aminoethyl, aminopropyl and carboxymethyl.
- hydrophilic resin examples include gum arabic, casein, gelatin, starch derivatives, carboxymethyl cellulose and its sodium salt, cellulose acetate, sodium alginate, vinyl acetate-maleic acid copolymers, styrene-maleic acid copolymers, polyacrylic acids and their salts, polymethacrylic acids and their salts, homopolymers and copolymers of hydroxyethyl methacrylate, homopolymers and copolymers of hydroxyethyl acrylate, homopolymers and copolymers of hydroxypropyl methacrylate, homopolymers and copolymers of hydroxypropyl acrylate, homopolymers and copolymers of hydroxybutyl methacrylate, homopolymers and copolymers of hydroxybutyl acrylate, polyethylene glycols, hydroxypropylene polymers, polyvinyl alcohols, hydrolyzed polyvinyl acetates having a degree of hydrolysis of at least
- the amount of hydrophilic resin added to the heat-sensitive layer is preferably from 5 to 40 wt%, and more preferably from 10 to 30 wt%, based on the solids in the heat sensitive layer. Within the above range, a good on-machine development and a good film strength can be obtained.
- the heat-sensitive layer may include also a photothermal conversion substance which absorbs infrared light and generates heat.
- the photothermal conversion substance may be any light absorbing substance having an absorption band within at least a part of the range from 700 to 1200 nm.
- Various known pigments, dyes and finely divided metals may be used in this way.
- Suitable pigments include black pigments, brown pigments, red pigments, violet pigments, blue pigments, green pigments, fluorescent pigments, metal powder pigments and also polymer-bonded dyes.
- Specific examples include insoluble azo pigments, azo lake pigments, condensed azo pigments, chelate azo pigments, phthalocyanine pigments, anthraquinone pigments, perylene and perinone pigments, thioindigo pigments, quinacridone pigments, dioxazine pigments, isoindolinone pigments, quinophthalone pigments, lake pigments, azine pigments, nitroso pigments, nitro pigments, natural pigments, fluorescent pigments, inorganic pigments and carbon black.
- the pigments may be used without being surface treated or may be used after surface treatment.
- surface treatment methods include surface coating with a hydrophilic resin or an oleophilic resin, surfactant adhesion, and bonding a reactive substance (e.g., a silica sol, alumina sol, silane coupling agent, epoxy compound or isocyanate compound) to the pigment surface.
- a reactive substance e.g., a silica sol, alumina sol, silane coupling agent, epoxy compound or isocyanate compound
- the pigment has a particle size which is in a range of preferably 0.01 to 1 ⁇ m, and more preferably 0.01 to 0.5 ⁇ m.
- Dyes which may be used include commercial dyes, and dyes mentioned in the technical literature, including Senry o Binran [Handbook of Dyes] (The Society of Synthetic Organic Chemistry, Japan, 1970); "Near-infrared absorption dyes," in Kagaku Kogyo (May 1986) pp. 45-51; 90-Nendai Kin o sei Shikiso no Kaihatsu to Shij o D o k o [Functional Dyes: Development and Market Trends in the 1990s], chapter 2, section 2.3 (CMC, 1990); and patents.
- Infrared-absorbing dyes such as azo dyes, metal complex azo dyes, pyrazolone azo dyes, anthraquinone dyes, phthalocyanine dyes, carbonium dyes, quinoneimine dyes, polymethine dyes and cyanine dyes are preferred.
- Suitable dyes include the cyanine dyes mentioned in JP 58-125246 A, JP 59-84356 A and JP 60-78787 A; the methine dyes mentioned in JP 58-173696 A, JP 58-181690 A and JP 58-194595 A; the naphthoquinone dyes mentioned in JP 58-112793 A, JP 58-224793 A, JP 59-48187 A, JP 59-73996 A, JP 60-52940 A and JP 60-63744 A; the squarylium dyes mentioned in JP 58-112792 A; the cyanine dyes mentioned in GB 434,875 B, the dyes mentioned in US 4,756,993; the cyanine dyes mentioned in US 4,973,572; the dyes mentioned in JP 10-268512 A; and the phthalocyanine compounds mentioned in JP 11-235883 A.
- the near-infrared absorbing sensitizers mentioned in US 5,156,938 can also be used advantageously as dyes.
- Other compounds that can be suitably used in this way include the substituted arylbenzo(thio)pyrylium salts mentioned in US 3,881,924; the trimethinethiapyrylium salts mentioned in JP 57-142645 A, the pyrylium compounds mentioned in JP 58-181051 A, JP 58-220143 A, JP 59-41363 A, JP 59-84248 A, JP 59-84249 A, JP 59-146063 A and JP 59-146061 A; the cyanine dyes mentioned in JP 59-216146 A; the pentamethinethiopyrylium salts mentioned in US 4,283,475; the pyrylium compounds mentioned in JP 5-13514 B and JP 5-19702 B; and Epolight III-178, Epolight III-130 and Epolight III-125
- the above-described organic photothermal conversion substance is added to the heat-sensitive layer in a range of preferably up to 30 wt%, more preferably 5 to 25 wt%, and even more preferably 7 to 20 wt%. Within this range, a good sensitivity can be obtained.
- a finely divided metal may also be used as the photothermal conversion substance in the heat-sensitive layer.
- Most finely divided metals are capable of photothermal conversion, and are also self-heating.
- Preferred examples of finely divided metals include finely divided silicon, aluminum, titanium, vanadium, chromium, manganese, iron, cobalt, nickel, copper, zinc, yttrium, zirconium, molybdenum, silver, gold, platinum, palladium, rhodium, indium, tin, tungsten, tellurium, lead, germanium, rhenium and antimony in unalloyed or alloyed form, and finely divided oxides and sulfides thereof.
- These finely divided metals are more preferably metals such as rhenium, antimony, tellurium, gold, silver, copper, germanium, lead and tin which melt at about 1,000°C or less and thus readily coalesce under the influence of heat during light irradiation, and which absorb in the infrared, visible or ultraviolet regions.
- Finely divided metals which have a relatively low melting point and also have a relatively high infrared absorbance, such as silver, gold, copper, antimony, germanium and lead, are especially preferred.
- the most preferred elements include silver, gold and copper.
- These particles have a size of preferably not more than 10 ⁇ m, more preferably from 0.003 to 5 ⁇ m, and even more preferably from 0.01 to 3 ⁇ m. A good sensitivity and good resolution can be attained within this range.
- the amount of addition is preferably at least 10 wt%, more preferably at least 20 wt%, and even more preferably at least 30 wt%, based on the solids in the heat-sensitive layer. A high sensitivity can be obtained within this range.
- the photothermal conversion substance prefferably included within a layer adjacent to the heat-sensitive layer, and specifically an undercoat layer or the subsequently described water-soluble overcoat layer.
- the photothermal conversion substance By including the photothermal conversion substance in at least one layer from among the heat-sensitive layer, undercoat layer and overcoat layer, the infrared absorption efficiency increases, making it possible to enhance sensitivity.
- various compounds other than those mentioned above may also be added to the heat-sensitive layer.
- a polyfunctional monomer may be added to the heat-sensitive layer matrix.
- Polyfunctional monomers suitable for this purpose include those mentioned above as monomers that may be included in the microcapsules.
- One monomer that is especially preferred for this purpose is trimethylolpropane triacrylate.
- Dyes having a large absorption in the visible light region can be used in the heat-sensitive layer as image colorants to enable image areas and non-image areas to be easily distinguished from one another following image formation.
- Specific examples include Oil Yellow #101, Oil Yellow #103, Oil Pink #312, Oil Green BG, Oil Blue BOS, Oil Blue #603, Oil Black BY, Oil Black BS and Oil Black T-505 (all of which are available from Orient Chemical Industries, Ltd.); and also Victoria Pure Blue, Crystal Violet (CI 42555), Methyl Violet (CI 42535), Ethyl Violet, Rhodamine B (CI 145170B), Malachite Green (CI 42000), Methylene Blue (CI 52015), and the dyes mentioned in JP 62-293247 A.
- Preferred use can also be made of pigments such as phthalocyanine pigments, azo pigments, and titanium oxide. The amount of such addition is typically 0.01 to 10 wt %, based on the total solids in the heat-sensitive layer coating fluid.
- thermal polymerization inhibitor examples include hydroquinone, p-methoxyphenol, di-t-butyl-p-cresol, pyrogallol, t-butylcatechol, benzoquinone, 4,4'-thiobis(3-methyl-6-t-butylphenol), 2,2'-methylenebis(4-methyl-6-t-butylphenol) and the aluminum salt of N-nitroso-N-phenylhydroxylamine.
- the thermal polymerization inhibitor is added in an amount of preferably about 0.01 to 5 wt%, based on the weight of the overall composition.
- a higher fatty acid or fatty acid derivative such as behenic acid or behenamide may be added and induced to concentrate primarily at the surface of the heat-sensitive layer as the layer dries after coating.
- the higher fatty acid or fatty acid derivative is added in an amount of preferably about 0.1 to about 10 wt%, based on the total solids in the heat-sensitive layer.
- the heat-sensitive layer may also contain a plasticizer to impart such properties as flexibility to the applied film.
- a plasticizer include polyethylene glycol, tributyl citrate, diethyl phthalate, dibutyl phthalate, dihexyl phthalate, dioctyl phthalate, tricresyl phosphate, tributyl phosphate, trioctyl phosphate and tetrahydrofurfuryl oleate.
- the heat-sensitive layer is formed by dissolving each of the above required components in a solvent to prepare a coating fluid, and applying the fluid onto the support.
- a solvent include ethylene dichloride, cyclohexanone, methyl ethyl ketone, methanol, ethanol, propanol, ethylene glycol monomethyl ether, 1-methoxy-2-propanol, 2-methoxyethyl acetate, 1-methoxy-2-propyl acetate, dimethoxyethane, methyl lactate, ethyl lactate, N,N-dimethylacetamide, N,N-dimethylformamide, tetramethylurea, N-methylpyrrolidone, dimethylsulfoxide, sulfolane, ⁇ -butyrolactone, toluene and water. These solvents may be used singly or as mixtures thereof.
- the coating fluid has a solids concentration of preferably from 1 to 50 wt%
- the coating weight (solids basis) of the heat-sensitive layer obtained on the support after application and drying of the coating fluid will vary depending on the intended application, although a weight of 0.5 to 5.0 g/m 2 is generally preferred. A coating weight below this range will result in a large apparent sensitivity, but diminish the film properties of the heat-sensitive layer whose function is to record an image. Any of various coating methods may be used. Examples of suitable methods include bar coating, spin coating, spray coating, curtain coating, dip coating, air knife coating, blade coating and roll coating.
- a surfactant may be added to the heat-sensitive layer coating fluid to improve the coating properties.
- fluorosurfactants mentioned in JP 62-170950 A may be added for this purpose.
- the amount of addition is preferably 0.01 to 1 wt%, and more preferably 0.05 to 0.5 wt%, based on the total solids in the heat-sensitive layer.
- a water-soluble overcoat layer can be provided on the heat-sensitive layer to protect the surface of the heat-sensitive layer from contamination by oleophilic substances.
- the water-soluble overcoat layer used in the invention can be easily removed during printing, and includes a resin selected from among water-soluble organic polymeric compounds.
- the water-soluble organic polymeric compound is a substance which, when applied as a coat and dried, has film formability.
- Specific examples include polyvinyl acetate having a degree of hydrolysis of at least 65%, polyacrylic acids and alkali metal salts or amine salts thereof, polyacrylic acid copolymers and alkali metal salts or amine salts thereof, polymethacrylic acids and alkali metal salts or amine salts thereof, polymethacrylic acid copolymers and alkali metal salts or amine salts thereof, polyacrylamides and copolymers thereof, polyhydroxyethyl acrylates, polyvinylpyrrolidone and copolymers thereof, polyvinyl methyl ethers, vinyl methyl ether/maleic anhydride copolymers, poly(2-acrylamido-2-methyl-1-propanesulfonic acid) and alkali metal salts or amine salts thereof, poly(2-acrylamido-2-methyl-1-propanesulfonic acid)
- the overcoat layer may also have added to it the above-described water-soluble photothermal conversion substance.
- a nonionic surfactant such as polyoxyethylene nonyl phenyl ether or polyoxyethylene dodecyl ether may be added to the overcoat layer to ensure uniformity of application.
- the overcoat layer has a coating weight when dry of preferably 0.1 to 2.0 g/m 2 .
- a weight within this range can provide good protection of the heat-sensitive layer surface from contamination by oleophilic substances, such as fingerprint contamination, without compromising the on-machine developability of the presensitized plate.
- a recording layer other than the above-described heat-sensitive layer containing (a) a thermally reactive functional group-bearing finely divided polymer or (b) microcapsules containing a thermally reactive functional group-bearing compound.
- Illustrative examples include photosensitive layers which use a negative-working infrared laser recording material, photosensitive layers which use a positive-working infrared laser recording material, and photosensitive layers which use a sulfonate-type infrared laser recording material.
- the heat-sensitive layer may be provided by using a negative-working infrared laser recording material.
- the negative-working infrared laser recording material is preferably a composition made up of (A) a compound which decomposes under the effect of light or heat to generate an acid, (B) a crosslinking agent which induces crosslinking under the effect of an acid, (C) an alkali-soluble resin, (D) an infrared absorber and (E) a compound of the general formula (R 3 -X) n -Ar-(OH) m (wherein R 3 is an alkyl or alkenyl of 6 to 32 carbons; X represents a single bond, oxygen, sulfur, COO or CONH; Ar is an aromatic hydrocarbon group, an aliphatic hydrocarbon group or a heterocyclic group; and the letters m and n are each independently integers from 1 to 3).
- the compound (A) which decomposes under the effect of light or heat to generate an acid is exemplified by compounds which undergo photodecomposition to form a sulfonic acid, such as the iminosulfonates mentioned in Japanese Patent Application No. 3-140109 (JP 4-365048 A), and compounds which form an acid under irradiation at a wavelength of 200 to 500 nm or under heating at a temperature of at least 100°C.
- Preferred acid generators include photocationic polymerization initiators, photoradical polymerization initiators, dye photobleaching agents and photochromogenic substances. These acid generators are preferably added in an amount of 0.01 to 50 wt%, based on the total solids in the recording material.
- Preferred examples of the crosslinking agent (B) which crosslinks under the effect of an acid include (i) aromatic compounds substituted with an alkoxymethyl or hydroxyl group, (ii) compounds having an N-hydroxymethyl, N-alkoxymethyl or N-acyloxymethyl group, and (iii) epoxy compounds.
- alkali-soluble resin (C) examples include novolak resins and polymers having pendant hydroxyaryl groups.
- Illustrative examples of the infrared absorber (D) include commercial dyes (e.g., azo dyes, anthraquinone dyes, phthalocyanine dyes) which effectively absorb infrared light at a wavelength of 760 to 1200 nm; and the black pigments, red pigments, metal powder pigments, phthalocyanine pigments mentioned in the Colour Index. Also, the addition of image colorants such as Oil Yellow and Oil Blue #603 is desirable for improving the visibility of the image. Plasticizers such as polyethylene glycol and phthalic acid esters can be added to improve the flexibility of the photosensitive layer-forming film.
- commercial dyes e.g., azo dyes, anthraquinone dyes, phthalocyanine dyes
- image colorants such as Oil Yellow and Oil Blue #603
- Plasticizers such as polyethylene glycol and phthalic acid esters can be added to improve the flexibility of the photosensitive layer-forming film.
- the presensitized plate of the invention is to be a positive-working presensitized plate intended for exposure with an infrared laser, i.e., a positive thermal presensitized plate, it is advantageous to provide a photosensitive layer composed of a positive-working infrared laser recording material.
- Positive-working infrared laser recording materials suitable for use include those composed of (A) an alkali-soluble polymer, (B) a compound which is compatible with the alkali-soluble polymer and lowers the alkali solubility, and (C) an infrared laser-absorbing compound.
- alkali-soluble polymer (A) examples include (i) polymeric compounds having phenolic hydroxyl groups, such as phenolic resins, cresol resins, novolak resins and pyrogallol resins; (ii) compounds obtained by subjecting sulfonamide group-bearing monomers to homopolymerization or to copolymerization with other polymerizable monomers; and (iii) compounds having active imide groups, such as N-(p-toluenesulfonyl) methacrylamide and N-(p-toluenesulfonyl) acrylamide.
- polymeric compounds having phenolic hydroxyl groups such as phenolic resins, cresol resins, novolak resins and pyrogallol resins
- compounds obtained by subjecting sulfonamide group-bearing monomers to homopolymerization or to copolymerization with other polymerizable monomers and
- compounds having active imide groups such as N-(p-
- Illustrative examples of the compound (B) which is compatible with the alkali-soluble polymer (A) and lowers the alkali solubility include compounds that interact with above component (A), such as sulfone compounds, ammonium salts, sulfonium salts and amide compounds.
- above component (A) is a novolak resin
- a cyanine dye can be suitably used as component (B).
- the compound (C) which absorbs infrared laser light is preferably a material which absorbs in the infrared range of 750 to 1200 nm and is capable of photothermal conversion.
- Compounds having such an ability include squarylium dyes, pyrylium dyes, carbon black, insoluble azo dyes and anthraquinone dyes. These preferably have a particle size in a range of 0.01 to 10 ⁇ m.
- the positive thermal presensitized plate can be obtained by dissolving this positive-working infrared laser recording material in an organic solvent such as methanol or methyl ethyl ketone, adding a dye if necessary, then applying the coating fluid onto the support to a weight when dry of 1 to 3 g/m 2 , and drying.
- an organic solvent such as methanol or methyl ethyl ketone
- a sulfonate-type infrared laser recording material may be used as the recording layer on the inventive presensitized plate.
- Sulfonate-type infrared laser recording materials that may be used include the sulfonate compounds mentioned in JP 270480 B and JP 2704872 B. Use can also be made of photosensitive materials which generate a sulfonic acid under the effect of heat generated by infrared laser irradiation, and which solubilize in water; photosensitive materials in which a styrenesulfonic acid ester is solidified with a sol-gel, following which the surface polarity is changed by irradiation with an infrared laser; and the photosensitive materials which are mentioned in Japanese Patent Application No. 9-89816 (JP 10-282646 A), Japanese Patent Application No. 10-22406 (JP 11-218928 A) and Japanese Patent Application No. 10-027655 (JP 10-282672 A), and in which the hydrophobic surface is rendered hydrophilic by exposure using a laser.
- the accompanying use of the following methods is desirable.
- Illustrative examples include (1) the method described in Japanese Patent Application No. 10-7062 (JP 11-202483 A) which involves use together with an acid or base generator, (2) the method described in Japanese Patent Application No. 9-340358 (JP 11-174685 A) which involves providing a specific intermediate layer, (3) the method described in Japanese Patent Application No. 9-248994 (JP 11-84658 A) which involves the concomitant use of a specific crosslinking agent, and (4) the method described in Japanese Patent Application No. 10-115354 (JP 11-301131 A) which involves use in a solid particle surface-modified form.
- compositions which utilize heat generated by exposure to a laser to effect a hydrophilic/hydrophobic change in a photosensitive layer include the compositions mentioned in US 2,764,085 which include a Werner complex and become hydrophobic under the influence of heat; the compositions described in JP 46-27219 B which include specific sugars and a melamine-formaldehyde resin, and which become hydrophilic under exposure to light; the compositions described in JP 51-63704 A which become hydrophobic under heat-mode exposure; the compositions described in US 4,081,572 which undergo dehydration/hydrophobization under heat, in the manner of phthalyl hydrazide polymers; the compositions described in JP 3-58100 B which have a tetrazolium salt structure and become hydrophilic under the effect of heat; the compositions described in JP 60-132760 A which are composed of a sulfonic acid-modified polymer and become hydrophobic under exposure to light; the compositions described in JP 64-3543
- compositions which may be used to form the recording layer include the compositions described in JP 3-197190 A which include a hydrophobic crystalline polymer and which become hydrophilic under exposure to light; the compositions described in JP 7-186562 A which include both a polymer with insolubilized pendant groups that become hydrophilic under exposure to heat and a photothermal conversion substance; the compositions described in JP 7-1849 A which includes a microcapsule-containing three dimensionally-crosslinked hydrophilic binder and becomes hydrophobic under exposure to light; the compositions described in JP 8-3463 A which undergo valence isomerization or proton transfer isomerization; the compositions described in JP 8-141819 A which give rise to a change in the phase structure (compatibilization) within the layer under the effect of heat, and thus effect a hydrophilic/hydrophobic change; and the compositions described in JP 60-228 B in which the surface shape and the hydrophilicity/hydrophobicity of the surface change under the effect of heat
- compositions which, under so-called heat-mode exposure utilizing heat generated by high-power, high-density laser light, change the adhesive properties between the photosensitive layer and the support include compositions described in JP 44-22957 B which are composed of a heat-fusible substance or a thermally reactive substance.
- the process of manufacturing an aluminum support for lithographic printing plates from an aluminum sheet preferably includes the following steps: (1) playing out a rolled, coil-wound aluminum sheet from a delivery unit composed of a multiple spindle turret; (2) subjecting the aluminum sheet to each of the above-described treatments (mechanical graining, electrochemical graining, alkali etching treatment, acidic etching treatment, desmutting, anodizing treatment, pore widening treatment (acid or alkali treatment), treatment with an aqueous solution containing an inorganic fluorine compound and a phosphate compound, and, before or after such aqueous solution treatment, treatment with a silicate compound-containing aqueous solution), then forming on the treated aluminum sheet an intermediate layer containing an acid group-bearing polymeric compound and subjecting the aluminum sheet to drying treatment; and (3) coil-winding the treated aluminum sheet on a rewinder composed of the above multiple spindle turre
- the above process may additionally include steps in which an undercoat layer and a recording layer are formed and subjected to drying treatment, thus rendering the aluminum sheet into a presensitized plate, in which form it may be rewound into a coil on the above rewinder.
- the manufacture of the aluminum support prefferably includes at least one step in which a device which inspects the surface of the aluminum sheet for defects continuously inspects the sheet and attaches a label at the edge of the sheet whenever a defect is found to mark its position. Also, it is desirable to provide a reserving unit for keeping the traveling speed by the aluminum sheet constant in each of the above steps when travel by the aluminum sheet is halted to change the aluminum coil in the aluminum sheet unwinding and rewinding steps during manufacture of the inventive presensitized plate. For the same reason, it is desirable to include, after the aluminum unwinding step, a step in which successive aluminum sheets are joined by ultrasonic or arc welding.
- the equipment used in the manufacture of the aluminum support preferably includes at least one unit to detect a traveling position on the aluminum sheet and correct the traveling position, at least one drive unit for cutting tension on the aluminum sheet and controlling the traveling speed, and at least one dancer roll unit for controlling tension.
- the aluminum sheets it is preferable for the aluminum sheets to be stacked and held together electrostatically with a slip sheet therebetween, then cut and/or slit to a given length. Moreover, after the sheets have been cut to the predetermined length, or even before such cutting, it is desirable to distinguish non-defective areas from defective areas on the basis of information appearing on the labels affixed to the edge of the aluminum sheet, and to collect only non-defective areas.
- each step including the above-described coil unwinding step, it is important to set the aluminum sheet to the optimal tension for the respective conditions, based on the size of the aluminum sheet (thickness and width), the aluminum material and the traveling speed of the aluminum web.
- a drive unit for cutting tension and controlling the traveling speed and a dancer roll for controlling tension, and provide a plurality of tension controlling units which carry out feedback control using signals from a tension sensing unit.
- the drive unit generally uses a control method which combines a DC motor and a main drive roller.
- the main drive roller is made of ordinary rubber, although rollers made by laminating nonwoven fabric can be used in steps where the aluminum web is in a wet state.
- Each pass roller is generally made of rubber or metal.
- auxiliary drive units can be provided to prevent slippage by the aluminum web in places where slippage tends to occur.
- a motor and a speed reducer may be connected to each pass roller, and rotational control carried out at a constant speed based on signals from the main drive unit.
- the aluminum support used in the invention preferably has a value R 1 -R 2 within 30% of R 1 , and also preferably has an average curvature in the rolling direction of not more than 1.5x10 -3 mm -1 , an average curvature in the width direction of not more than 1.5x10 -3 mm -1 , and an average curvature in the direction perpendicular to the rolling direction of not more than 1.0 ⁇ 10 -3 mm -1 .
- the aluminum support produced by administering the above-described graining treatment is preferably corrected using a correcting roll having a diameter of 20 to 80 mm and a rubber hardness of 50 to 95.
- a correcting roll having a diameter of 20 to 80 mm and a rubber hardness of 50 to 95.
- JP 9-194093 A describes a method and apparatus for measuring curl in a web, a method and apparatus for correcting curl, and an apparatus for cutting the web. These can be used in the present invention as well.
- each step is electrically monitored to determine whether it is operating under the proper conditions, a tracking unit records whether the state in each step agrees with the desired conditions and, before rewinding the aluminum web into a coil, labels are affixed to the edge of the web to make it possible later on to tell from the labels whether a portion of the web meets the desired conditions, thus enabling a decision to be made concerning the acceptability of that portion at the time of cutting and collection.
- the aluminum sheet treatment apparatuses used in the above-described graining treatments preferably determine the liquid composition by measuring one or more parameters from among the liquid temperature, specific gravity, electrical conductivity and ultrasonic wave propagation velocity, then carry out feedback control and/or feed-forward control to keep the liquid concentration constant.
- Aluminum ions and other components present in the aluminum sheet are dissolved in the acidic aqueous solution within the treatment apparatus as surface treatment of the aluminum sheet proceeds.
- the concentration of the acid or alkali added here is preferably from 10 to 98 wt%.
- the following method is preferred for controlling the acid or alkali concentration.
- the electrical conductivity, specific gravity or ultrasonic wave propagation velocity of each component liquid in the concentration range for which use is anticipated is measured at various temperatures and a data table is prepared.
- the concentration is measured by referring to the data table containing temperature data and conductivity, specific gravity or ultrasonic wave propagation velocity data for the solution being measured.
- JP 6-235721 A describes a method for precisely and stably measuring the ultrasonic wave propagation time.
- JP 58-77656 A describes a concentration measurement system that utilizes the above ultrasonic wave propagation velocity.
- JP 4-19559 A describes a method which uses data on a plurality of physical quantities to construct a data table showing the correlations between liquid components, and measures the component concentrations in a multi-component solution by referring to this data table.
- Measurement of the ultrasonic wave propagation velocity is preferably carried out in a pressure range within the pipeline of 1 to 10 kg/cm 2 , and the ultrasonic waves have a frequency of preferably 0.5 to 3 MHz.
- Measurement of the specific gravity, conductivity and ultrasonic wave propagation velocity is temperature sensitive, and so it is preferable to carry out these measurements in a temperature-insulated state and within a line where the temperature fluctuation is controlled to within ⁇ 0.3°C.
- the above measurements are readily affected by slurry, dirt and bubbles, it is preferable to measure the liquid after passing it through a filter, deaerator and the like.
- An image is recorded on the resulting inventive presensitized plate by means of heat.
- Preferred methods for doing so include direct imagewise recording such as with a thermal recording head, scanning-type exposure using an infrared laser, high-intensity flash-type exposure such as with a xenon discharge lamp, or exposure with a solid high-output infrared laser using an infrared lamp.
- the recording layer on the presensitized plate of the invention is an on-machine development type heat-sensitive layer containing (a) a finely divided polymer having thermally reactive functional groups or (b) microcapsules containing a compound having thermally reactive functional groups, following imagewise exposure, the plate can be mounted without further treatment on the printing press and printing carried out by an ordinary procedure using ink and/or dampening water.
- JP 2938398 B after the plate has been mounted on the plate cylinder of the printing press, it can be exposed using a laser mounted on the press, following which ink and/or dampening water can be applied and on-machine development carried out.
- the heat-sensitive layer is removed on the press with ink and/or dampening water, and so there is no need for a separate development operation. Moreover, once development is over, printing can begin without stopping the press; that is, printing can be carried out immediately without interruption once development is complete.
- Platemaking and printing methods for the lithographic printing plate of the invention are characterized by subjecting a presensitized plate provided with an on-machine development type heat-sensitive layer to imagewise exposure with laser light, then mounting the exposed plate on the press and printing, or by mounting the plate on the press then subjecting it to imagewise exposure using laser light and directly carrying out printing in this state.
- a solid laser or semiconductor laser which emits infrared light at a wavelength of 760 to 1200 nm can be used.
- a plate having an on-machine development-type heat-sensitive layer can be used in printing after it has been developed with water or a suitable aqueous solution as the developer.
- the presensitized plate of the invention has a prior-art positive-working or negative-working thermal recording layer, it is subjected to imagewise exposure, then developed with a developer according to a conventional process, and subsequently mounted on the press and furnished for printing.
- Illustrative examples of sources of actinic light that may be used for imagewise exposure include mercury vapor lamps, metal halide lamps, xenon lamps and chemical lamps.
- laser beams that may be used include helium-neon lasers, argon lasers, krypton lasers, helium-cadmium lasers, KrF excimer lasers, semiconductor lasers, YAG lasers and YAG-SHG lasers.
- the liquid developer is preferably an alkali developer, and more preferably an alkaline aqueous solution which is substantially free of organic solvent.
- Liquid developers which are substantially free of alkali metal silicates are also preferred.
- One example of a suitable method of development using a liquid developer that is substantially free of alkali metal silicates is the method described in detail in JP 11-109637 A. Liquid developers containing alkali metal silicates can also be used.
- the aluminum sheets used in the examples of the invention and the comparative examples were produced as follows.
- a melt was prepared using an aluminum alloy composed of 0.073 wt% silicon, 0.270 wt% iron, 0.028 wt% copper, 0.001 wt% manganese, 0.000 wt% magnesium, 0.001 wt% chromium, 0.003 wt% zinc and 0.020 wt% titanium with the balance being aluminum and inadvertent impurities.
- the aluminum alloy melt was subjected to molten metal treatment consisting of degassing and filtration, then was cast into a 500 mm thick ingot by a direct chill casting process.
- the ingot was faced, removing 10 mm of material from the surface.
- the faced ingot was then heated; hot rolling was begun at 400°C without carrying out soaking treatment, and continued to a thickness of 4 mm.
- the ingot was then cold rolled to a thickness of 1.5 mm and intermediate annealed, following which it was again cold rolled, this time to a final thickness of 0.24 mm, after which the flatness was corrected, giving an aluminum sheet.
- Lithographic Printing Plate Support 1 :
- the aluminum sheet obtained as described above was subjected to surface treatment in the sequence (1) to (13) shown below, giving a lithographic printing plate support 1.
- Mechanical graining treatment was carried out using a brush roller having rotating nylon brushes while using a spray line to feed an abrasive slurry consisting of a suspension of silica sand as the abrasive (specific gravity, 1.12; average particle size, 25 ⁇ m) in water to the surface of the aluminum sheet.
- the nylon brush material used was composed of 6,10-nylon, had a bristle length of 50 mm and a bristle diameter of 0.48 mm.
- the nylon brushes were 300 mm diameter stainless steel cylinders in which holes had been formed and bristles densely set therein.
- the brush roller used three nylon brushes and also had two support rollers (200 mm diameter) provided below the brush and spaced 300 mm apart.
- the brush roller controlled the load of the driving motor that rotates the nylon brush relative to the load before the brush is pushed against the aluminum sheet, and pushed the brush against the aluminum sheet such as to give the sheet after graining an average calculated roughness (R a ) of 0.45 ⁇ m.
- the direction of brush rotation was the same as the direction of movement by the aluminum sheet. Rinsing was subsequently carried out.
- the concentration of the abrasive was determined from the temperature and specific gravity by referring to a table prepared beforehand based on the relationship between the abrasive concentration, temperature and specific gravity, and water and abrasive were added under feedback control, thereby holding the concentration of the abrasive constant.
- the abrasive breaks down to a smaller particle size, the surface shape of the grained aluminum sheet changes. Abrasive having a small particle size was thus successively removed from the system with a cyclone.
- the particle size of the abrasive was in a range of 1 to 35 ⁇ m.
- the concentration of the etching solution used for alkali etching treatment was determined from the temperature, specific gravity and electrical conductivity by referring to a table prepared beforehand based on the relationship between the NaOH concentration, aluminum ion concentration, temperature, specific gravity and liquid conductivity, and was held constant under feedback control by adding water and 48 wt% aqueous NaOH. Thereafter, the aluminum sheet was rinsed off with water.
- Aqueous nitric acid having a liquid temperature of 35°C was sprayed onto the aluminum sheet and desmutting was carried out for 10 seconds. Overflow wastewater from the electrolytic cell apparatus employed in the next step was used as the aqueous nitric acid. Next, spray lines for spraying the desmutting liquid were positioned in several places, and the surface of the aluminum sheet was keep from drying until the next operation.
- Electrochemical graining treatment was carried out continuously using alternating current having the trapezoidal waveform shown in FIG. 1 and the two electrolytic cells shown in FIG. 2.
- An aqueous solution containing 1 wt% of nitric acid (and containing also 0.5 wt% of aluminum ions and 0.007 wt% of ammonium ions) was used as the acidic aqueous solution.
- the liquid temperature was 35°C.
- the alternating current had respective times tp and tp' until the current value reached a peak on the cathode cycle side and the anode cycle side, respectively, of 1 msec each.
- a carbon electrode was used as the counterelectrode.
- the alternating current had a current density at the peak, both when the aluminum sheet was the anode and when it was the cathode, of 50 A/dm 2 .
- the ratio Q C /Q A between the amount of electricity when the aluminum sheet served as the cathode Q C and the amount of electricity when the sheet served as the anode Q A was 0.95.
- the duty ratio was 0.50, the frequency was 60 Hz, and the combined amount of electricity when the aluminum sheet served as the anode was 230 C/dm 2 . Following treatment, the aluminum sheet was sprayed to rinse it off.
- Control of the aqueous nitric acid solution concentration was carried out by adding a 67 wt% nitric acid stock solution and water proportional to the amount of current passed and at the same time allowing an amount of the acidic aqueous solution (nitric acid solution) equivalent to the volume of nitric acid and water being added to consecutively overflow from the electrolytic cells and discharge outside of the system.
- the concentration of the nitric acid solution was determined from the temperature, conductivity and ultrasonic wave propagation velocity of the solution by referring to a table prepared beforehand based on the relationship between the nitric acid concentration, aluminum ion concentration, temperature, conductivity of the liquid and ultrasonic wave propagation velocity of the liquid, and the concentration was held constant by carrying out control involving the successive adjustment of the amounts of nitric acid stock solution and water added.
- Alkali etching treatment was carried out by spraying an aqueous solution containing 26 wt% of NaOH and 6.5 wt% of aluminum ions and having a temperature of 45°C onto the aluminum sheet.
- the weight dissolved from the aluminum sheet was 3 g/m 2 .
- the etching solution concentration was determined from the temperature, specific gravity and conductivity by referring to a table prepared beforehand from the relationship between NaOH concentration, aluminum ion concentration, temperature, specific gravity and solution conductivity, and was kept constant under feedback control by the addition of water and 48 wt% aqueous NaOH. Following treatment, the aluminum sheet was rinsed off with water.
- Sulfuric acid (sulfuric acid concentration, 300 g/L; aluminum ion concentration, 15 g/L) was prepared as an acidic etching solution, then sprayed onto the aluminum sheet at 80°C for 7 seconds to carry out acidic etching treatment.
- the acidic etching solution concentration was determined from the temperature, specific gravity and conductivity by referring to a table prepared beforehand based on the relationship between the sulfuric acid concentration, aluminum ion concentration, temperature, specific gravity and solution conductivity, and was kept constant under feedback control by the addition of water and 50 wt% sulfuric acid. Following treatment, the aluminum sheet was rinsed off with water.
- electrochemical graining treatment (II) was carried out in the same way as in (4) Electrochemical Graining Treatment (I) described above.
- Control of the aqueous hydrochloric acid solution concentration was carried out by adding a 30 wt% hydrochloric acid stock solution and water proportional to the amount of current passed and concurrently allowing an amount of the acidic aqueous solution (hydrochloric acid solution) equivalent to the volume of hydrochloric acid and water being added to successively overflow from the electrolytic cells and discharge outside of the system.
- the concentration of the hydrochloric acid solution was determined from the solution temperature, conductivity and ultrasonic wave propagation velocity by referring to a table prepared beforehand based on the relationship between the hydrochloric acid concentration, aluminum ion concentration, temperature, conductivity of the liquid and ultrasonic wave propagation velocity of the liquid, and the concentration was held constant by carrying out control involving the successive adjustment of the amounts of hydrochloric acid stock solution and water added.
- alkali etching treatment (III) was carried out in the same way as in (5) Alkali Etching Treatment (II) described above.
- the weight dissolved from the aluminum sheet was set at 0.2 g/m 2 .
- Acidic etching treatment (II) was carried out in the same way as in (6) Acidic Etching Treatment (I) described above.
- Anodizing treatment of the aluminum sheet was carried out at a current density of 25 A/dm 2 and a temperature of 50°C for 30 seconds using an aqueous solution having a sulfuric acid concentration of 100 g/L (and containing 0.5 wt% of aluminum ions) as the anodizing solution, thereby forming an anodized layer.
- the concentration of the anodizing solution was determined from the temperature, specific gravity and conductivity by referring to a table prepared beforehand based on the relationship between the sulfuric acid concentration, aluminum ion concentration, temperature, specific gravity and conductivity of the solution, and the concentration was held constant by adding water and 50 wt% sulfuric acid under feedback control. Following treatment, the aluminum sheet was sprayed to rinse it off.
- Pore widening treatment was carried out by dipping the anodized aluminum sheet for 1 minute in an aqueous NaOH solution having a pH of 13 and a temperature of 50°C. Following treatment, the aluminum sheet was rinsed with water.
- the aluminum sheet was treated by immersion under the temperature and time conditions shown in Table 2 in respective aqueous solutions prepared using pure water to a sodium fluoride (NaF) concentration of 0.1 wt% and to a sodium dihydrogenphosphate (NaH 2 PO 4 ) concentration of 10 wt%.
- the aluminum sheet was subsequently rinsed with water.
- the aluminum sheet was dipped for 10 seconds and at 20°C in an aqueous solution having a No. 3 sodium silicate concentration of 1.0 wt% for the hydrophilic surface treatment.
- the aluminum sheet was subsequently rinsed with water and dried.
- An intermediate layer coating fluid of the composition indicated below and containing an acid group-bearing polymeric compound was applied, then dried at 80°C for 15 seconds.
- the coating weight after drying of the intermediate layer was 15 mg/m 2 .
- Electron spectroscopy for chemical analysis was carried out on the lithographic printing plate supports 1 to 8 obtained above.
- the ESCA conditions were as follows. Apparatus PHI-5400 MC ESCA spectrometer (manufactured by Ulvac-Phi, Inc.) X-ray source Mg-K ⁇ (400 W) Pulse energy 71.55 eV/178.95 eV Take off angle 45°
- An oil phase component was prepared by dissolving the following in 60 g of ethyl acetate: 40 g of xylene diisocyanate, 10 g of trimethylolpropane diacrylate, 10 g of a copolymer of allyl methacrylate and butyl methacrylate (molar ratio, 7/3), and 0.1 g of surfactant (Pionin A41C, available from Takemoto Oil & Fat Co., Ltd.). In a separate procedure, 120 g of a 4% aqueous solution of polyvinyl alcohol (PVA 205, available from Kuraray Co., Ltd.) was prepared as an aqueous phase component.
- PVA 205 polyvinyl alcohol
- the oil phase component and the aqueous phase component were placed in a homogenizer and emulsified at 10,000 rpm. This was followed by the addition of 40 g of water and stirring, first at room temperature for 30 minutes, then at 40°C for 3 hours, thereby giving a microcapsule liquid.
- the resulting microcapsule liquid had a solids concentration of 20 wt% and an average microcapsule size of 0.2 ⁇ m.
- Heat-sensitive layer (1) was a heat-sensitive layer containing a finely divided polymer, and was formed as follows.
- a coating fluid for heat-sensitive layer (1) of the composition indicated below was applied to a coating weight when dry of 0.5 g/m 2 to the respective lithographic printing plate supports 1 and 5 obtained above, then dried in an oven at 60°C for 150 seconds, giving presensitized plates in Example 1 and Comparative Example 1.
- Heat-sensitive layer (2) was a microcapsule-containing heat-sensitive layer, and was formed as follows.
- a coating fluid for heat-sensitive layer (2) of the composition indicated below was applied to the respective lithographic printing plate supports 2 and 6 obtained above to a coating weight when dry of 0.7 g/m 2 , then dried in an oven at 60°C for 150 seconds, giving presensitized plates in Example 2 and Comparative Example 2.
- Heat-sensitive layer (3) was a two-layer positive-working thermal image recording layer, and was formed as follows.
- a first layer-forming coating liquid of the composition indicated below was prepared and applied to the respective lithographic printing plate supports 3 and 7 obtained above to a coating weight when dry of 0.8 g/m 2 , then dried in an oven at 140°C for 60 seconds, thereby forming a first layer.
- a second layer-forming coating liquid of the composition indicated below was prepared, applied to a coating weight when dry of 0.2 g/m 2 to each of the above lithographic printing plate supports on which the first layer had been formed, then dried in an oven at 140°C for 50 seconds to form a second layer, thereby giving the presensitized plates having a two-layer positive-working thermal image layer in Example 3 and Comparative Example 3.
- Heat-sensitive layer (4) was a negative-working thermal image recording layer, and was formed as follows.
- a coating fluid for heat-sensitive layer (4) of the composition indicated below was applied to the respective lithographic printing plate supports 4 and 8 obtained above to a coating weight when dry (heat-sensitive layer coating weight) of 1.3 g/m 2 , then dried with a hot-air dryer at 122°C for 27 seconds to form a heat-sensitive layer (negative-working thermal image recording layer), giving presensitized plates in Example 4 and Comparative Example 4.
- Infrared absorber IR-1) 0.074 g Polymerization initiator (OS-12) 0.280 g Additive (PM-1) 0.151 g Polymerizable compound (AM-1) 1.00 g Binder polymer (BT-1) 1.00 g Ethyl violet (C-1) 0.04 g Fluorosurfactant (Megafac F-780-F, available from Dainippon Ink & Chemicals; 30 wt% solution in MIBK) 0.015 g Methyl ethyl ketone 10.4 g Methanol 4.83 g 1-Methoxy-2-propanol 10.4 g
- a mixed aqueous solution of polyvinyl alcohol (degree of saponification, 98 mol%; degree of polymerization, 500) and polyvinyl pyrrolidone (Luviskol K-30, available from BASF) was applied with a wiper onto the surface of the above-described heat-sensitive layer, then dried at 125°C for 75 seconds with a hot-air dryer.
- the polyvinyl alcohol/polyvinyl pyrrolidone content ratio was 4/1, and the coating weight (weight of coat when dry) was 2.30 g/m 2 .
- Example 1 The on-machine developable presensitized plates obtained in Example 1 and Comparative Example 1 were exposed using a Trendsetter 3244 VFS (Creo Inc.) equipped with a water-cooled 40 W infrared semiconductor laser, at a resolution of 2,400 dpi.
- the plate surface energy at this time was varied in increments of 5 mJ/cm 2 from 30 to 200 mJ/cm 2 by varying the external drum speed, and the sensitivity was determined from the minimum dose at which image formation was possible.
- the results are shown in Table 2.
- Example 2 and Comparative Example 2 (Microcapsule-Containing Heat-Sensitive Layers)
- Example 2 The on-machine developable presensitized plates obtained in Example 2 and Comparative Example 2 were exposed using a Luxel T-9000 CTP (Fuji Photo Film Co., Ltd.) equipped with a multi-channel laser head, at a resolution of 2,400 dpi.
- the plate surface energy at this time was varied in increments of 5 mJ/cm 2 from 40 to 200 mJ/cm 2 by varying the output per beam and the external drum speed, and the sensitivity was determined from the minimum dose at which image formation was possible.
- the results are shown in Table 2.
- Example 3 and Comparative Example 3 were imagewise exposed at a primary scanning speed of 5 m/s and a surface energy of 140 mJ/cm 2 using a Trendsetter 3244 (Creo Inc.) equipped with a semiconductor laser having an output of 500 mW, a wavelength of 830 nm and a beam diameter of 17 ⁇ m (1/e 2 ).
- a Trendsetter 3244 (Creo Inc.) equipped with a semiconductor laser having an output of 500 mW, a wavelength of 830 nm and a beam diameter of 17 ⁇ m (1/e 2 ).
- samples were prepared by carrying out exposure at surface energies which were varied in increments of 5 mJ/cm 2 from 20 to 150 mJ/cm 2 .
- Development was carried out at a temperature of 30°C for 12 seconds using a PS900NP automated processor (manufactured by Fuji Photo Film Co., Ltd.) filled with the developer DT-2 (1:8) (Fuji Photo Film Co., Ltd.). After development was completed, the developed plate was rinsed with water, then treated with a gum (FG-1, 1:1), thereby giving a completed lithographic printing plate.
- the minimum exposure dose at which image formation could be carried out following development was determined as the sensitivity from samples obtained at varying surface energy levels.
- Example 4 The presensitized plates obtained in Example 4 and Comparative Example 4 were exposed using a Trendsetter 3244 VFS (Creo Inc.) equipped with a water-cooled 40 W infrared semiconductor laser (wavelength, 830 nm) at an output of 9 W, an external drum speed of 150 rpm, a plate surface energy of 100 mJ/cm 2 and a resolution of 175 dpi.
- samples were prepared by carrying out exposure at surface energies which were varied in increments of 5 mJ/cm 2 from 20 to 200 mJ/cm 2 .
- the protective layer was removed by rinsing with tap water, after which development was carried out at 30°C for 12 seconds using an LP-1310HII processor available from Fuji Photo Film Co., Ltd.
- the developer was a 1:4 aqueous dilution of DV-2C (Fuji Photo Film Co., Ltd.), and the finisher was a 1:1 aqueous dilution of GN-2K (Fuji Photo Film Co., Ltd.).
- the presensitized plates obtained in Examples 1 and 2 of the invention and in Comparative Examples 1 and 2 were exposed to light in the same way as described above under "Measurement of Sensitivity.” In each case, the exposed plate was then mounted on the plate cylinder of a SOR-M printing press (Heidelberger Druckmaschinen AG) without first being subjected to development. The plate was supplied first with dampening water containing 1% ALKI and 10% isopropyl alcohol, after which Toyo Vantean Eco red ink and paper were each supplied and a printing test was carried out. In the case of the presensitized plates obtained in Examples 1 and 2, on-machine development was carried out without any difficulty and printing was also possible.
- the press life was evaluated based on the number of clear impressions obtained. A larger number of such impressions indicates a longer press life.
Abstract
Description
- The present invention relates to a support for a lithographic printing plate, and to a presensitized plate for lithographic printing.
- More specifically, the invention relates to a presensitized plate, composed of a support having thereon a thermal type image recording layer, which is made directly into a lithographic printing plate without the use of a film copy by being exposed to laser light scanned over the surface thereof and having text and images to be formed directly on its surface; and relates also to a lithographic printing plate support for use in such a presensitized plate. The invention additionally relates to a heat-sensitive presensitized plate which is highly suitable for use in a computer-to-plate system that does not require development, which is capable of image recording by infrared scanning exposure based on digital signals and which, following exposure, can then be mounted on a printing press and printed without passing through a conventional processing step involving the use of a developer; and relates also to a lithographic printing plate support for use in such a presensitized plate.
- Much research is being done today on presensitized plates for computer-to-plate systems, an area in which remarkable developments have been made in recent years. In particular, to achieve even higher process efficiency and resolve wastewater treatment problems, extensive research has been done on presensitized plates for lithography which, following exposure, can be directly mounted on the printing press and printed.
- One promising technology that has emerged is a heat-sensitive presensitized plate in which a hydrophilic layer composed of hydrophobic thermoplastic polymer particles dispersed in a hydrophilic binder polymer serves as the heat-sensitive, imaging layer. When heat is applied to the heat-sensitive layer in this presensitized plate, the hydrophobic thermoplastic polymer particles fuse, converting the surface of the hydrophilic heat-sensitive layer into oleophilic image areas.
- In such presensitized plates which utilize the thermal fusion of hydrophobic thermoplastic polymer particles, one method for reducing the number of processing steps is referred to as "on-machine development." Following exposure, the presensitized plate is mounted onto the cylinder of a printing press without first processing the plate with a developer. Ink and/or dampening water are then fed to the plate as the cylinder is turned, thereby removing non-image areas of the presensitized plate. In this method, following exposure of the presensitized plate, the plate is directly mounted on the printing press and development processing is completed in the course of a normal printing operation.
- Presensitized plates suitable for such on-machine development must have a heat-sensitive layer that is soluble in the dampening water or the ink solvent. Moreover, they must have daylight handling characteristics suitable for development on a printing press located in a lighted room.
- JP 2938397 B discloses a presensitized plate of this type having a hydrophilic support on which there is provided a heat-sensitive layer composed of a finely divided thermoplastic hydrophobic polymer dispersed in a hydrophilic binder polymer. This prior-art describes the exposure of the presensitized plate using an infrared laser and the ensuing coalescence of the thermoplastic hydrophobic polymer particles under the effect of heat to form an image, and how the plate can then be mounted onto the plate cylinder of a printing press and on-machine development carried out by feeding ink and/or dampening water.
- JP 9-127683 A and WO 99/10186 teach the fabrication of a lithographic plate by on-machine development following the coalescence of thermoplastic fine particles under the effect of heat.
- Although presensitized plates on which an image is formed in this way by the coalescence of fine particles under heating do exhibit a good on-machine developability, they have a poor sensitivity owing to the escape of heat to the metal support. Moreover, when particle coalescence is insufficient, the strength of image areas of the heat-sensitive layer is diminished, which may result in a short press life.
- One solution that has been proposed is to provide a water-insoluble organic polymer between the aluminum support and the heat-sensitive layer (see, for example, JP 2000-23983 A). Unfortunately, although this does increase the sensitivity, plate contamination occurs.
- A number of problems are also associated with conventional thermal-type presensitized plates that are not developed "on machine," including what are known as positive-working thermal presensitized plates in which an infrared absorber present in the heat-sensitive layer exhibits a photothermal conversion effect that causes it to generate heat upon exposure to light, rendering exposed areas of the heat-sensitive layer alkali-soluble under the effect of such heat and resulting in the formation of a positive image, and what are known as negative-working thermal presensitized plates in which similar heating causes a radical generator or an acid generator present in the heat-sensitive layer to form radicals or an acid which triggers a radical polymerization reaction and an acid crosslinking reaction, rendering reacted areas of the layer insoluble and forming a negative image.
- Namely, in such thermal-type image formation, irradiation with laser light causes a photothermal conversion substance within the heat-sensitive layer to generate heat, which in turn triggers an image-forming reaction. However, because an aluminum support that has been subjected to graining treatment and on which an anodized layer has been formed has a much higher heat conductivity that the heat-sensitive layer, the heat generated near the interface between the heat-sensitive layer and the support diffuses into the interior of the support before it can be fully used for image formation. As a result, the following effects occur at the interface between the heat-sensitive layer and the support.
- First, in a positive-working heat-sensitive layer, heat diffusion into the interior of the support results in an insufficient alkali solubilizing reaction, allowing remnants of the heat-sensitive layer to remain in what should be non-image areas (sometimes referred to herein as "residual coating"), and thus a low sensitivity. This is a problem inherent in positive heat-sensitive layers.
- Moreover, such positive thermal presensitized plates require the use of an infrared absorber capable of photothermal conversion. Such absorbers have a relatively large molecular weight, and thus a low solubility. Also, they adhere to and are difficult to remove from microscopic openings that arise from anodization, as a result of which portions of the exposed heat-sensitive layer tend to remain behind (residual coating) after development has been carried out with an alkali developer.
- On the other hand, in a negative-working heat-sensitive layer, heat diffuses into the interior of the support, causing insufficient developer insolubilization of the heat-sensitive layer near the heat-sensitive layer/support interface. As a result, an image may fail to form sufficiently in what should be the image areas, and may be washed off during development. Even if image-wise formation does occur, the image may readily separate from the support during printing.
- To resolve these problems, the support having an anodized layer is sometimes subjected to pore widening treatment to minimize the diffusion into the aluminum support of heat generated within the heat-sensitive layer.
- Pore widening treatment enables the heat-sensitive layer to be rendered fully developer-insoluble near the heat-sensitive layer/support interface, thus enhancing both press life and sensitivity. However, problems associated with such treatment include incomplete removal of the heat-sensitive layer during development and scumming on the press.
- Efforts have hitherto been made to seal micropores in the anodized layer so as to eliminate such problems as residual heat-sensitive film and scumming. Sealing methods that have been disclosed include treatment with pressurized steam or hot water, silicate treatment, treatment with an aqueous dichromate solution, nitrite treatment, ammonium acetate treatment, electrodeposition sealing, triethanolamine treatment, barium carbonate treatment and treatment with hot water containing a trace amount of phosphate. Yet, although these methods do enhance the resistance to scumming, they worsen the sensitivity and press life of the plate.
- To improve the press life, scum resistance and sensitivity, JP 2003-1961 A discloses a support for lithographic printing plates which is obtained by subjecting a grained and anodized metal base to treatment with an aqueous solution containing an inorganic fluorine compound and a silicate compound, and also discloses a presensitized plate in which such a support is used.
- JP 2002-99093 A describes a method of manufacturing lithographic printing plates which is characterized by subjecting a lithographic printing plate composed of an aluminum support having a surface that satisfies the relationship in the formula below and a photosensitive layer or a heat-sensitive layer thereon to development with a silicate-free developer.
- JP 11-109637 A discloses a positive-working photosensitive lithographic printing plate composed of an aluminum support having a silicon atom coating weight of 0.1 to 8 mg/m2 and a positive-working photosensitive layer on the support; and also discloses a lithographic printing plate composed of an aluminum support, an intermediate layer containing a polymeric compound having an acid group-bearing component on the support, and a positive-working photosensitive layer on the intermediate layer.
- Recently, there has existed a desire for presensitized plates in which the press life, scum resistance and sensitivity are all improved to an even higher degree. However, it has not been possible to fully achieve all of these attributes in presensitized plates obtained by the above-described art.
- The objects of the present invention are to provide excellent heat-sensitive presensitized plates for lithography that are endowed with the above properties, and a support for lithographic printing plates which is highly suitable for use in such presensitized plates. More specifically, one object of the invention is to provide a heat-sensitive presensitized plate which, when used as an on-machine development type plate, has a good on-machine developability, a high sensitivity, a long press life and a good resistance to scumming on the press; and which, when used as a conventional positive-working thermal plate or negative-working thermal plate, efficiently utilizes heat in image formation, exhibits a high sensitivity and a long press life, and is resistant to the scumming of non-image areas. Another object of the invention is to provide a support for lithographic printing plates which is suitable for use in such heat-sensitive presensitized plates.
- The inventor of the present invention has conducted intensive studies and found that a lithographic printing plate support composed of a metal base subjected to graining treatment and anodizing treatment, then treated with an aqueous solution containing an inorganic fluorine compound and a phosphate compound and with an aqueous solution containing a silicate compound (which solutions are sometimes referred to collectively herein as "specific aqueous solutions"), and an intermediate layer which lies on the metal base and contains an acid group-bearing polymeric compound, when used to make a lithographic printing plate, has an excellent sensitivity, press life and resistance to scumming. The present invention has been completed based on the finding.
- Accordingly, in a first aspect, the present invention provides a support for lithographic printing plates that is composed of a metal base subjected to graining treatment and anodizing treatment, then treated with an aqueous solution containing an inorganic fluorine compound and a phosphate compound, which aqueous solution treatment is preceded or followed by treatment with an aqueous solution containing a silicate compound; and an intermediate layer which lies on the treated metal base and contains an acid group-bearing polymeric compound.
- Here, treatment with the aqueous solution containing an inorganic fluorine compound and a phosphate compound may involve separately carrying out treatment with an inorganic fluorine compound-containing aqueous solution and treatment with a phosphate compound-containing aqueous solution. In such a case, the order in which these particular aqueous solution treatments and treatment with the silicate compound-containing aqueous solution are carried out is not subject to any particular limitation. That is, the aqueous solution treatments may be carried out in any suitable order. preferably the acid group on the acid group-bearing polymeric compound constituent is an acid group having an acid dissociation constant (pKa) not higher than 7, and more preferably the acid group on the acid group-bearing polymeric compound constituent is selected from the group consisting of -COOH, -SO3H, -OSO3H, -PO3H2, -OPO3H2, -CONHSO2 and -SO2NHSO2.
- As a result of the intensive studies, the inventor of the present invention has also found that, by having the surface of the metal base used in the support for lithographic printing plates satisfy specific conditions, lithographic printing plates prepared using the support have an excellent sensitivity, press life and resistance to scumming.
- Accordingly, in a second aspect, the invention provides a support for lithographic printing plates that is composed of a metal base having a surface which satisfies formula (1) below
- In formula (1), A is the peak area (counts•eV/sec) for fluorine (1S) as measured by electron spectroscopy for chemical analysis (ESCA), B is the peak area (counts•eV/sec) for silicon (2P) as measured by ESCA, C is the peak area (counts•eV/sec) for phosphorus (2P) as measured by ESCA, and D is the peak area (counts•eV/sec) for aluminum (2P) as measured by ESCA.
- The method of manufacturing the metal base having a surface which satisfies formula (1) is not subject to any particular limitation. For example, use may be made of a method which successively administers the treatments described above in connection with the first aspect of the invention.
- In a third aspect, the invention provides a presensitized plate composed of a lithographic printing plate support according to the first or second aspect of the invention, and an infrared laser-imageable recording layer which lies on the support.
- The presensitized plates of the invention which are composed of either a metal base treated with an aqueous solution containing an inorganic fluorine compound and a phosphate compound and treated with an aqueous solution containing a silicate compound or a metal base having a specific surface, a specific intermediate layer provided on top of the metal base, and an on-machine developable heat-sensitive layer provided as a recording layer on top of the intermediate layer have a good on-machine developability and a high sensitivity, in addition to which they exhibit a long press life and have a good resistance to scumming on the press.
- Presensitized plates according to the invention which are obtained by providing a specific intermediate layer on either of the above metal bases and providing a positive-working thermal photosensitive layer as the recording layer on the intermediate layer have a high solubility in liquid developers, even when the infrared laser exposure dose is low or the developer has a low sensitivity. As a result, an excellent press life is achieved, in addition to which the sensitivity is high, the development latitude is broad, residual coating even at low exposure is minimal, and scumming of non-image areas does not readily occur.
- Moreover, presensitized plates according to the invention which are similarly provided with a negative-working thermal photosensitive layer as the recording layer have a high percent insolubility in the developer in laser-exposed areas, and thus exhibit a high sensitivity, a long press life and excellent scumming resistance.
- This application claims priority on Japanese patent application No.2003-331342, the entire contents of which are hereby incorporated by reference. In addition, the entire contents of literatures cited in this specification are incorporated by reference.
-
- FIG. 1 is a waveform diagram showing an example of an alternating current trapezoidal waveform in electrochemical graining treatment which may be advantageously used in the present invention.
- FIG. 2 is a side view showing an example of a radial electrolytic cell apparatus for carrying out electrochemical graining treatment which may be advantageously used in the invention.
-
- The invention is described more fully below in conjunction with the attached diagrams.
- In the first aspect of the invention, the metal base prior to treatment that may be used in the inventive support for lithographic printing plates is not subject to any particular limitation. "Metal base prior to treatment" refers here to the metal base prior to being administered graining treatment, anodizing treatment and other treatment to have the surface satisfy certain specific conditions. Exemplary metal bases prior to treatment include iron, stainless steel and aluminum. Of these, aluminum is preferred.
- Aluminum sheet that may be used as the aluminum base is made of a dimensionally stable metal composed primarily of aluminum; that is, aluminum or aluminum alloy. Aside from sheets of pure aluminum, use can also be made of alloy sheets composed primarily of aluminum and small amounts of other elements, or plastic film or paper onto which aluminum or aluminum alloy has been laminated or vapor deposited. Use can also be made of a composite sheet obtained by bonding an aluminum sheet onto a polyethylene terephthalate film as described in JP 48-18327 B.
- Aluminum sheet that may be used in the invention is not subject to any particular limitation, although the use of pure aluminum sheet is preferred. However, because completely pure aluminum is difficult to manufacture for reasons having to do with refining technology, the presence of a small amount of other elements is acceptable. Suitable use can be made of known materials that appear in the 4th edition of Aluminum Handbook published in 1990 by the Japan Light Metal Association. Examples of such aluminum materials include those having the designations JIS 1050, JIS 1100, JIS 3003, JIS 3103 and JIS 3005.
- Specific examples of suitable aluminum sheets include those containing 0.07 to 0.09 wt% of silicon, 0.20 to 0.40 wt% of iron, 0.000 to 0.030 wt% of copper, up to 0.01 wt% of manganese, up to 0.01 wt% of magnesium, up to 0.01 wt% of chromium, up to 0.01 wt% of zinc, up to 0.04 wt% of titanium and at least 99.5 wt% of aluminum.
- Use can also be made of aluminum sheet made from aluminum alloy, scrap aluminum or secondary aluminum ingots having an aluminum content of 95 to 99.4 wt%, and containing at least five metals from among iron, silicon, copper, magnesium, manganese, zinc, chromium and titanium within the ranges indicated below.
- If the aluminum content in this case exceeds 99.4 wt%, the allowable content of impurities decreases, which may diminish the cost-saving effect. On the other hand, at an aluminum content of less than 95 wt%, the impurities content is large, which gives rise to undesirable effects such as crack formation during rolling. The aluminum content is more preferably from 95 to 99 wt%, and most preferably from 95 to 97 wt%. The various elements included in the aluminum sheet are described below.
- The iron content is preferably from 0.3 to 1.0 wt%. Iron is an element which is present, even within primary aluminum ingots, in a range of 0.1 to 0.2 wt%. The amount of iron that enters into a solid solution within aluminum is small; most remains in the form of intermetallic compounds. At an iron content of more than 1.0 wt%, cracks tend to form easily during rolling, whereas at less than 0.3 wt%, the cost-reducing effects are diminished. The iron content is most preferably within a range of 0.5 to 1.0 wt%.
- The silicon content is preferably from 0.15 to 1.0 wt%. Silicon is an element which is abundant in scrap from JIS 2000, 4000 and 6000 series materials. Silicon is an element present in an amount of about 0.03 to 0.1 wt% even in primary aluminum ingots. It exists in a solid solution within aluminum, or is present as intermetallic compounds. When aluminum sheet is heated during the support manufacturing process, silicon that was present in the aluminum as a solid solution sometimes precipitates out as uncombined silicon. Uncombined silicon and FeSi-based intermetallic compounds are known to have an adverse influence on the resistance to severe ink scumming. Here, "severe ink scumming" refers to contamination in the form of spots and rings that appears on the printed medium such as paper as a result of the tendency for ink to adhere to non-image areas of the printing plate surface when printing is carried out with repeated interruptions. At a silicon content of more than 1.0 wt%, such contamination may not be completely eliminated by the subsequently described sulfuric acid treatment (desmutting treatment). On the other hand, at a silicon content of less than 0.15 wt%, the cost-reducing effects are diminished. A silicon content within a range of 0.3 to 1.0 wt% is especially preferred.
- The copper content is preferably from 0.1 to 1.0 wt%. Copper is an element which is abundant in scrap from JIS 2000 and 4000 series materials. Copper forms a solid solution in aluminum with relative ease. At a copper content of more than 1.0 wt%, scumming may not be completely eliminated by the subsequently described sulfuric acid treatment. On the other hand, at a copper content of less than 0.1 wt%, the cost-reducing effects are diminished. A copper content within a range of 0.3 to 1.0 wt% is especially preferred.
- The magnesium content is preferably from 0.1 to 1.5 wt%. Magnesium is an element which is abundant in scrap from JIS 2000, 3000, 5000 and 7000 series materials. Because it is particularly abundant in can-end material, magnesium is one of the major metal impurities present in aluminum scrap. Magnesium forms a solid solution in aluminum with relative ease, and forms intermetallic compounds with silicon. At a magnesium content of more than 1.5 wt%, scumming may not be completely eliminated by the subsequently described sulfuric acid treatment. On the other hand, at a magnesium content of less than 0.1 wt%, the cost-reducing effects are diminished. The magnesium content is more preferably from 0.5 to 1.5 wt%, and most preferably from 1.0 to 1.5 wt%.
- The manganese content is preferably from 0.1 to 1.5 wt%. Manganese is an element which is abundant in scrap from JIS 3000 series materials. Because it is particularly abundant in can-end material, manganese is one of the major metal impurities present in aluminum scrap. Manganese forms a solid solution in aluminum with relative ease, and forms intermetallic compounds with aluminum, iron and silicon. At a manganese content of more than 1.5 wt%, scumming may not be completely eliminated by the subsequently described sulfuric acid treatment. On the other hand, at a manganese content of less than 0.1 wt%, the cost-reducing effects are diminished. The manganese content is more preferably from 0.5 to 1.5 wt%, and most preferably from 1.0 to 1.5 wt%.
- The zinc content is preferably from 0.1 to 0.5 wt%. Zinc is an element which is abundant particularly in scrap from JIS 7000 series materials. Zinc forms a solid solution in aluminum with relative ease. At a zinc content of more than 0.5 wt%, scumming may not be completely eliminated by the subsequently described sulfuric acid treatment. On the other hand, at a zinc content of less than 0.1 wt%, the cost-reducing effects are diminished. A zinc content within a range of 0.3 to 0.5 wt% is especially preferred.
- The chromium content is preferably from 0.01 to 0.1 wt%. Chromium is a metal impurity present in a small quantity in scrap from JIS 5000, 6000 and 7000 series materials. At a chromium content of more than 0.1 wt%, contamination may not be completely eliminated by the subsequently described sulfuric acid treatment. On the other hand, at a chromium content of less than 0.01 wt%, the cost-reducing effects are diminished. A chromium content within a range of 0.05 to 0.1 wt% is especially preferred.
- The titanium content is preferably from 0.03 to 0.5 wt%. Titanium is an element which is generally added in a range of 0.01 to 0.04 wt% as a crystal grain refining agent. It is present in a relatively large amount as a metal impurity in scrap from JIS 5000, 6000 and 7000 series materials. At a titanium content of more than 0.5 wt%, scumming may not be completely eliminated by the subsequently described sulfuric acid treatment. On the other hand, at a chromium content of less than 0.03 wt%, the cost-reducing effects are diminished. A titanium content within a range of 0.05 to 0.5 wt% is especially preferred.
- The aluminum sheet used in the invention is manufactured by using a conventional process to cast the above-described raw material, administering suitable rolling treatment and heat treatment to set the thickness to typically 0.1 to 0.7 mm, and applying flatness correcting treatment as required. This thickness can be suitably varied according to the size of the printing press, the size of the printing plate, and the desires of the user.
- Processes that may be used to manufacture the above aluminum sheet include direct-chill casting, a process like direct-chill casting but from which soaking treatment and/or annealing treatment has been omitted, and continuous casting.
- The support for lithographic printing plates of the invention is obtained by subjecting a metal base to graining treatment and anodizing treatment, then treating the grained and anodized metal base with specific aqueous solutions, and subsequently forming on the treated metal base an intermediate layer containing a specific compound. In addition to graining treatment, anodizing treatment, treatments with specific aqueous solutions and an intermediate layer forming step, manufacture of the inventive support for lithographic printing plates may include various other steps as well. The support for lithographic printing plates according to the present invention is described below with reference to, for the purpose of illustration, a case in which aluminum sheet is used as the metal base prior to treatment.
- The aluminum sheet preferably passes through a degreasing step to remove rolling oils adhering to the surface of the sheet, a desmutting step to dissolve smut on the surface of the sheet, a graining treatment step to roughen the surface of the sheet, an anodizing treatment step to cover the surface of the sheet with an anodized layer, a pore widening treatment (acid treatment or alkali treatment) step, and steps involving treatment with specific aqueous solutions.
- Manufacture of the inventive support for lithographic printing plates preferably includes electrochemical graining treatment in which an alternating current is used to electrochemically grain the aluminum sheet in an acidic aqueous solution.
- Manufacture of the inventive lithographic printing plate support may include an aluminum sheet graining treatment step which combines the above-described electrochemical graining treatment with a related operation such as mechanical graining treatment or chemical etching treatment in an acid or alkaline aqueous solution. The graining treatment and other steps used to manufacture the inventive support for lithographic printing plates may be carried out as either a continuous or an intermittent process, although the use of a continuous process is industrially advantageous.
- In the invention, treatment with specific aqueous solutions is also carried out, in addition to which, if necessary, a hydrophilic surface treatment is administered as well, thereby forming the support. Moreover, after the support has been formed, a specific intermediate layer (or "undercoat," as viewed from the recording layer side) may be provided thereon if necessary.
- First, graining treatment is described.
- The above-described aluminum sheet is administered graining treatment to impart a more desirable surface shape. Illustrative examples of suitable graining methods include mechanical graining like that described in JP 56-28893 A, chemical etching, and electrolytic graining. Use can also be made of electrochemical graining and electrolytic graining processes in which the surface is electrochemically grained in an electrolytic solution containing hydrochloric acid or nitric acid; and mechanical graining such as wire brushing in which the aluminum surface is scratched with metal wires, ball graining in which the aluminum surface is grained with abrasive balls and an abrasive compound, and brush graining in which the surface is grained with a nylon brush and an abrasive compound. Any one or combination of these graining methods may be used. For example, mechanical graining with a nylon brush and an abrasive compound may be combined with electrolytic graining using an electrolytic solution of hydrochloric acid or nitric acid, or a plurality of electrolytic graining treatments may be combined. Of the above, electrochemical graining is preferred, although it is also advantageous to carry out a combination of mechanical graining and electrochemical graining. Mechanical graining, followed by electrochemical graining with an electrolytic solution of nitric acid, followed in turn by electrochemical graining with an electrolytic solution of hydrochloric acid, is especially preferred.
- Mechanical graining refers to treatment in which the surface of the aluminum sheet is mechanically grained such as with a brush. It is preferably carried out before electrochemical graining treatment.
- Suitable mechanical graining treatment involves carrying out treatment with a rotating nylon brush roll having a bristle diameter of 0.07 to 0.57 mm and an abrasive compound that is supplied as a slurry to the surface of the aluminum sheet.
- The nylon brush is preferably made of bristles having a low water absorption. A preferred example is Nylon Bristle 200T (available from Toray Industries, Inc.), which is made of nylon 6,10, has a softening point of 180°C, a melting point of 212 to 214°C, a specific gravity of 1.08 to 1.09, a water absorption at 20°C and 65% relative humidity of 1.4 to 1.8 and at 20°C and 100% relative humidity of 2.2 to 2.8, a dry tensile strength of 4.5 to 6 g/d, a dry tensile elongation of 20 to 35%, a boiling water shrinkage of 1 to 4%, a dry resistance to stretching of 39 to 45 g/d, and a Young's modulus when dry of 380 to 440 kg/mm2.
- Any known abrasive compound may be used, although the use of silica sand, quartz, aluminum hydroxide, or a mixture thereof, mentioned in JP 6-135175 A and JP 50-40047 B is preferred.
- The slurry is preferably one having a specific gravity in a range of 1.05 to 1.3. Illustrative examples of methods for supplying the slurry to the surface of the aluminum sheet include blowing the slurry onto the surface, a method involving the use of a wire brush, and a method in which the surface shape of a textured cold rolling roll is transferred to the aluminum sheet. The methods described in JP 55-74898 A, JP 61-162351 A and JP 63-104889 A may also be used. Moreover, use can also be made of a method like that described in JP 9-509108 A, wherein the surface of the aluminum sheet is brush grained in an aqueous slurry containing a mixture of particles composed of alumina and quartz in a weight ratio of 95:5 to 5:95. The mixture used for this purpose has an average particle size of preferably 1 to 40 µm, and more preferably 1 to 20 µm.
- Electrochemical graining differs from the above-described mechanical graining in that it involves graining the surface of the aluminum sheet electrochemically by placing the sheet in an acidic aqueous solution and passing an alternating current through the sheet with the sheet serving as an electrode.
- In the practice of the invention, when the ratio QC/QA between the amount of electricity when the aluminum sheet serves as the cathode QC and the amount of electricity when the sheet serves as the anode QA in the above electrochemical graining treatment is within a range of 0.5 to 2.0, for example, uniform honeycomb pits can be formed on the surface of the aluminum sheet. Non-uniform honeycomb pits tend to form at a QC/QA ratio of less than 0.50 or more than 2.0. A QC/QA ratio within a range of 0.8 to 1.5 is preferred.
- The alternating current used in electrochemical graining may have a waveform that is, for example, sinusoidal, square, triangular or trapezoidal. Of these, a square or trapezoidal waveform is preferred. The alternating current has a frequency which, from the standpoint of the cost of manufacturing the power supply, is preferably from 30 to 200 Hz, and more preferably from 40 to 120 Hz.
- FIG. 1 shows an example of a trapezoidal wave that can be suitably used in the invention. In FIG. 1, the ordinate represents the current value and the abscissa represents time. In addition, ta is the anode reaction time, tc is the cathode reaction time, tp is the time until the current value reaches a peak on the cathode cycle side from zero, tp' is the time until the current value reaches a peak on the anode cycle side from zero, Ia is the peak current on the anode cycle side, and Ic is the peak current on the cathode cycle side. When trapezoidal waves are used as the alternating current waveform, the respective times tp and tp' until the current reaches a peak from zero are preferably each from 0.1 to 2 msec, and more preferably from 0.3 to 1.5 msec. When tp and tp' are in the above range, the power circuit impedance has the effect of reducing the power supply voltage required during rise in the current waveform, making it possible to lower the cost of power supply equipment. Moreover, within this range, trace ingredients in the acidic aqueous solution have little effect, enabling uniform graining treatment to be carried out.
- To uniformly grain the surface of the aluminum sheet, it is preferable for the alternating current used in electrochemical graining to have a duty ratio within a range of 0.25 to 0.75, and especially 0.3 to 0.6. As used herein, "duty ratio" refers to the ratio ta/T, where T is the period of the alternating current and ta is the duration of the anode reaction at the aluminum sheet (anode reaction time). In particular, smut components composed largely of aluminum hydroxide form on the surface of the aluminum sheet during the cathode reaction, in addition to which oxide film dissolution and breakdown occur, becoming the starting points of pitting reactions during the subsequent anode reaction at the aluminum sheet. Hence, selection of the alternating current duty cycle has a large effect on providing uniform graining treatment.
- The alternating current has a current density, in the case of a trapezoidal or square waveform, which is preferably such that the current density Iap at the peak on the anode cycle side and the current density Icp at the peak on the cathode cycle side are each from 10 to 200 A/dm2. Moreover, the ratio Icp/Iap is preferably within a range of 0.9 to 1.5.
- The total amount of electricity used in the anode reaction on the aluminum sheet when electrochemical graining treatment has been completed is preferably from 50 to 1,000 C/dm2. The electrochemical graining time is preferably from 1 second to 30 minutes.
- Any acidic aqueous solution used in conventional electrochemical graining treatment involving the use of direct current or alternating current may be employed here in electrochemical graining treatment, although the use of an acidic aqueous solution composed mainly of nitric acid or an acidic aqueous solution composed mainly of hydrochloric acid is preferred. "Composed mainly of," as used here and below, signifies that the main component in an aqueous solution is contained in an amount of at least 30 wt%, and preferably at least 50 wt%, based on all the components within the solution.
- As noted above, the acidic aqueous solution composed mainly of nitric acid can be one which is employed in conventional electrochemical graining treatment involving the use of direct current or alternating current. For example, use can be made of an aqueous solution with a nitric acid concentration of 5 to 15 g/L in which one or more nitric acid compounds such as aluminum nitrate, sodium nitrate or ammonium nitrate has been added to a concentration of from 0.01 g/L to saturation. The acidic aqueous solution composed mainly of nitric acid may contain, dissolved therein, metals which are present in aluminum alloy, such as iron, copper, manganese, nickel, titanium, magnesium and silicon.
- It is advantageous for the acidic solution composed mainly of nitric acid used in the invention to be one which contains nitric acid, an aluminum salt and a nitrate, and which has been obtained by adding aluminum nitrate and ammonium nitrate to a nitric acid solution having a nitric acid concentration of 5 to 15 g/L so as to set the aluminum ion concentration at 1 to 15 g/L, and preferably 1 to 10 g/L, and the ammonium ion concentration at 10 to 300 ppm. The aluminum ions and ammonium ions form spontaneously and thus increase while electrochemical graining is being carried out. The liquid temperature at this time is preferably 10 to 95°C, more preferably 20 to 90°C, and most preferably 40 to 80°C.
- In electrochemical graining treatment, use can be made of a known electrolytic cell apparatus, such as one having a vertical, flat or radial construction. A radial electrolytic cell apparatus like that described in JP 5-195300 A is especially preferred.
- FIG. 2 is a schematic view of a radial electrolytic cell apparatus of a type suitable for use in the practice of the invention. In FIG. 2, an
aluminum sheet 11 wraps around aradial drum roller 12 situated within a mainelectrolytic cell 21 and passes through the apparatus while being subjected to electrolytic treatment by means ofmain electrodes AC power supply 20. The acidicaqueous solution 14 is supplied from asolution feed inlet 15 through aslit 16, and to asolution channel 17 located between theradial drum roller 12 and themain electrodes - The
aluminum sheet 11 treated in anauxiliary anode cell 22 is electrolytically treated in the mainelectrolytic cell 21. In thisauxiliary anode cell 22, anauxiliary anode 18 is situated opposite thealuminum sheet 11 and the acidicaqueous solution 14 is supplied such as to flow between theauxiliary anode 18 and thealuminum sheet 11. The current supplied to theauxiliary anode 18 is controlled bythyristors 19a and 19b. -
Main electrodes - The
auxiliary anode 18 may be selected from among known oxygen generating electrodes made of ferrite, iridium oxide, platinum, or platinum that has been clad or plated with a valve metal such as titanium, niobium or zirconium. - The acidic aqueous solution which passes through the main
electrolytic cell 21 and theauxiliary anode cell 22 may be fed in a direction that is either parallel or counter to the direction of advance by thealuminum sheet 11. The flow rate of the acidic aqueous solution relative to the aluminum sheet is preferably from 10 to 1,000 cm/s. - One or more AC power supplies may be connected to a single electrolytic cell apparatus. It is also possible to use two or more electrolytic cell apparatuses, in which case the electrolysis conditions in each apparatus may be the same or different.
- Following the completion of electrolytic treatment, it is desirable to drain the solution from the treated aluminum sheet with a nip roller and rinse the sheet by spraying it with water to prevent the treatment solution from being carried on to the next step.
- In cases where the above-described electrolytic cell apparatus is used, it is desirable to keep the concentration of the acidic aqueous solution constant by adding nitric acid and water while adjusting the amounts of addition in proportion to the amount of electricity passed through the acidic aqueous solution in which the aluminum sheet within the electrolytic cell apparatus undergoes anodic reaction, and based on the nitric acid and aluminum ion concentrations determined from, for example, (i) the electrical conductivity of the acidic aqueous solution, (ii) the ultrasonic wave propagation velocity of the solution and (iii) the solution temperature, and by successively allowing to overflow and thus discharging from the electrolytic cell apparatus an amount of the acidic aqueous solution equivalent to the volume of nitric acid and water added.
- Next, surface treatment, including chemical etching treatment in an acidic aqueous solution or an alkaline aqueous solution, and desmutting treatment shall be described in this order. These surface treatments are each carried out either before the above-described electrochemical graining treatment, or after electrochemical graining treatment but before the anodizing treatment described later in the specification. Descriptions of each of the surface treatments are given below, although the invention is not limited to the particular surface treatments as they are described below. Administration of these surface treatments and the other treatments mentioned below is optional.
- Alkali etching treatment is a treatment in which the surface of the aluminum sheet is chemically etched in an alkaline aqueous solution, and is preferably carried out before and after the above-described electrochemical graining treatment. In cases where mechanical graining treatment is carried out before electrochemical graining treatment, it is preferable to carry out alkali etching treatment following mechanical graining treatment. Alkali etching treatment can break down the microstructure in a short time, and is thus more advantageous than the subsequently described acidic etching treatment.
- Illustrative examples of alkaline aqueous solutions that may be used in alkali etching treatment include aqueous solutions containing one or more of the following: sodium hydroxide, sodium carbonate, sodium aluminate, sodium metasilicate, sodium phosphate, potassium hydroxide and lithium hydroxide. An aqueous solution composed mainly of sodium hydroxide is especially preferred. The alkaline aqueous solution may contain 0.5 to 10 wt% of aluminum and also alloying ingredients present in the aluminum sheet.
- The alkaline aqueous solution has a concentration of preferably 1 to 50 wt%, and more preferably 1 to 30 wt%.
- It is advantageous to carry out alkali etching treatment for 1 to 120 seconds, and preferably 2 to 60 seconds, at an alkaline aqueous solution temperature in a range of 20 to 100°C, and preferably 40 to 80°C. The amount of dissolved aluminum is preferably 5 to 20 g/m2 when alkali etching treatment is carried out after mechanical graining, and preferably 0.01 to 20 g/m2 when alkali etching treatment is carried out after electrochemical graining. When a chemical etching solution is initially mixed into the alkaline aqueous solution, it is preferable to prepare the treatment solution using liquid sodium hydroxide and sodium aluminate.
- Following the completion of alkali etching treatment, it is desirable to drain the solution from the treated aluminum sheet with a nip roller and rinse the sheet by spraying it with water to prevent the treatment solution from being carried on to the next step.
- When alkali etching treatment is carried out after electrochemical graining, the smut that forms from electrochemical graining can be removed. Preferred examples of such alkali etching treatments include a method in which the aluminum sheet is brought into contact with 15 to 65 wt% sulfuric acid at a temperature of 50 to 90°C, as described in JP 53-12739 A, and the alkali etching method described in JP 48-28123 B.
- Acidic etching treatment is a treatment in which the aluminum sheet is chemically etched in an acidic aqueous solution. It is preferably carried out after the electrochemical graining treatment described above. In cases where the above-described alkali etching treatment is carried out before and/or after electrochemical graining, it is preferable for acidic etching treatment to be carried out after alkali etching treatment.
- When acidic etching treatment is administered following alkali etching treatment of the aluminum sheet, intermetallic compounds which may include silica as a metal and uncombined silicon can be removed from the surface of the aluminum sheet, thus making it possible to eliminate defects in the anodized layer that forms in the subsequent anodizing treatment. As a result, the adherence of ink spots in non-image areas during printing can be prevented.
- Examples of acidic aqueous solutions that may be used in acidic etching treatment include aqueous solutions containing phosphoric acid, nitric acid, sulfuric acid, chromic acid, hydrochloric acid, or a mixture of two or more thereof. Of these, an aqueous solution of sulfuric acid is preferred. The acidic aqueous solution has a concentration of preferably 50 to 500 g/L. The acidic aqueous solution may contain aluminum and also the alloying ingredients present in the aluminum sheet.
- It is advantageous to carry out acidic etching treatment at a liquid temperature of 60 to 90°C, and preferably 70 to 80°C, for a period of 1 to 10 seconds. The amount of aluminum sheet dissolution at this time is preferably from 0.001 to 0.2 g/m2. The acid concentration, such as the sulfuric acid concentration and aluminum ion concentration, is preferably selected from a range at which crystallization does not occur at room temperature. The aluminum ion concentration is preferably 0.1 to 50 g/L, and more preferably 5 to 15 g/L.
- Following the completion of acidic etching treatment, it is desirable to drain the solution from the treated aluminum sheet with a nip roller and rinse the sheet by spraying it with water to prevent the treatment solution from being carried on to the next step.
- When the above alkali etching treatment is carried out before and/or after electrochemical graining, smut generally forms on the surface of the aluminum sheet as a result of alkali etching treatment. Therefore, following alkali etching treatment, it is desirable to carry out a so-called desmutting treatment in which such smut is dissolved in an acidic solution containing phosphoric acid, nitric acid, sulfuric acid, chromic acid, hydrochloric acid, hydrofluoric acid, fluoroboric acid or a mixture of two or more of these acids. Following alkali etching treatment, it is sufficient to carry out either acidic etching treatment or desmutting.
- The concentration of the acidic solution is preferably 1 to 500 g/L. The acidic solution may have dissolved therein 0.001 to 50 g/L of aluminum and also the alloying ingredients present in the aluminum sheet.
- The acidic solution has a liquid temperature of preferably 20 to 95°C, and more preferably 30 to 70°C. The treatment time is preferably 1 to 120 seconds, and more preferably 2 to 60 seconds.
- To reduce the amount of wastewater generated, it is preferable to use wastewater from the acidic aqueous solution employed in electrochemical graining as the desmutting solution (acidic solution).
- Following the completion of desmutting, it is desirable to drain the solution from the treated aluminum sheet with a nip roller and rinse the sheet by spraying it with water to prevent the treatment solution from being carried on to the next step.
- Preferred combinations of these surface treatments in the practice of the invention are indicated below.
- First, mechanical graining treatment and/or alkali etching treatment are carried out, followed by desmutting. Next, electrochemical graining is carried out, after which (1) acidic etching treatment, (2) alkali etching treatment followed by desmutting, (3) alkali etching treatment followed by acidic etching treatment, and (4) alkali etching treatment following by desmutting or acidic etching treatment are carried out. Then, electrochemical graining or alkali etching treatment followed by desmutting treatment is carried out.
- After being subjected to the above-described graining treatment and other types of treatment as needed, the aluminum sheet is administered anodizing treatment.
- Anodizing treatment can be carried out by any suitable method known to be used in the art to which the invention relates. More specifically, an anodizing layer can be formed on the surface of the aluminum sheet by passing a direct current or alternating current through the aluminum sheet in an aqueous or non-aqueous solution of any one or combination of, for example, sulfuric acid, phosphoric acid, chromic acid, oxalic acid, sulfamic acid and benzenesulfonic acid.
- The anodizing treatment conditions vary empirically according to the electrolytic solution used, although it is generally suitable for the solution to have a concentration of 1 to 80 wt% and a temperature of 5 to 70°C, and for the current density to be 0.5 to 60 A/dm2, the voltage to be 1 to 200 V, and the electrolysis time to be 1 to 1,000 seconds.
- Of such anodizing treatments, the anodizing process carried out in a sulfuric acid electrolytic solution at a high current density described in GB 1,412,768 B and the anodizing process carried out using phosphoric acid as the electrolytic bath described in US 3,511,661 are preferred. It is also possible to carry out a multi-step anodizing treatment involving, for example, anodizing treatment in sulfuric acid and also anodizing treatment in phosphoric acid.
- In the practice of the invention, to minimize scuffing and improve the press life of the plate, the anodized layer has a weight of preferably at least 1.0 g/m2, more preferably at least 2.0 g/m2, and even more preferably 4.0 g/m2. Given that a large amount of energy is required to provide a thick layer, the anodized layer has a weight of preferably not more than 100 g/m2, more preferably not more than 10.0 g/m2, and even more preferably not more than 6.0 g/m2.
- Minute depressions called micropores are formed so as to be uniformly distributed over the surface of the anodized layer. The density and diameter of the micropores present on the anodized layer can be adjusted by suitable selection of the treatment conditions.
- To lower the thermal conductivity, following anodizing treatment, it is advantageous to carry out pore widening treatment which widens the diameter of the micropores. This pore widening treatment involves immersion of the aluminum base on which an anodized layer has been formed in an acidic or alkaline aqueous solution to dissolve the anodized layer and widen the diameter of the micropores. Pore widening treatment is carried out such that the amount of anodized layer dissolution is in a range of preferably 0.01 to 20 g/m2, more preferably 0.1 to 5 g/m2, and even more preferably 0.2 to 4 g/m2.
- When pore widening treatment is carried with an acidic aqueous solution, the use of an aqueous solution of an inorganic acid such as sulfuric acid, phosphoric acid, nitric acid or hydrochloric acid, or a mixture thereof, is preferred. The acidic aqueous solution has a concentration of preferably 10 to 1,000 g/L, and more preferably 20 to 500 g/L, and has a temperature of preferably 10 to 90°C, and more preferably 30 to 70°C. The length of immersion in the acidic aqueous solution is preferably from 1 to 300 seconds, and more preferably from 2 to 100 seconds.
- On the other hand, when pore widening treatment is carried out with an alkaline aqueous solution, it is preferable to use an aqueous solution of at least one alkali selected from the group consisting of sodium hydroxide, potassium hydroxide and lithium hydroxide. The alkaline aqueous solution has a pH of preferably 10 to 13, and more preferably 11.5 to 13.0, and has a temperature of preferably 10 to 90°C, and more preferably 30 to 50°C. The length of immersion in the alkaline aqueous solution is preferably from 1 to 500 seconds, and more preferably from 2 to 100 seconds. Alkali treatment may be followed by treatment with an acidic aqueous solution.
- In the practice of the invention, following anodizing treatment as described above, treatments (1) and (2) below are carried out, either in this order or in the reverse order:
- (1) treatment with an aqueous solution containing an inorganic fluorine compound and a phosphate compound;
- (2) treatment with an aqueous solution containing a silicate compound.
-
- Following treatment with the above specific aqueous solutions, by providing on the treated metal base an intermediate layer containing the subsequently described acid group-bearing polymeric compound, there can be obtained a support for lithographic printing plates which, when rendered into a lithographic printing plate, has an excellent sensitivity, press life and scumming resistance.
- This treatment is preferably carried out by immersing the anodized metal base in an aqueous solution having a phosphate compound concentration of 0.01 to 20 wt%, an inorganic fluorine compound concentration of 0.01 to 5 wt%, and a pH of 3 to 5. Immersion is carried out at preferably 20 to 100°C, and more preferably 40 to 80°C, and for a period of preferably 1 to 300 seconds, and more preferably 5 to 30 seconds.
- For good scumming resistance, the concentration of the phosphate compound is preferably at least 0.01 wt%, more preferably at least 0.05 wt%, and even more preferably at least 0.1 wt%. For a long press life, the concentration is preferably not more than 20 wt%, more preferably not more than 10 wt%, and even more preferably not more than 5 wt%.
- For good sealing of the anodized layer, the concentration of the inorganic fluorine compound is preferably at least 0.01 wt%, preferably at least 0.05 wt%, and more preferably at least 0.1 wt%. For a long press life, this concentration is preferably not more than 5 wt%, and more preferably not more than 2 wt%.
- The relative proportions of the respective compounds in the aqueous solution, while not subject to any particular limitation, are preferably such that the weight ratio of the inorganic fluorine compound to the phosphate compound is in a range of 1:200 to 200:1.
- Phosphates that may be used in the invention include the phosphoric acid salts of metals such as alkali metals and alkaline earth metals.
- Specific examples include zinc phosphate, aluminum phosphate, ammonium phosphate, diammonium hydrogenphosphate, ammonium dihydrogenphosphate, monoammonium phosphate, monopotassium phosphate, monosodium phosphate, potassium dihydrogenphosphate, dipotassium hydrogenphosphate, calcium phosphate, ammonium sodium hydrogenphosphate, magnesium hydrogenphosphate, magnesium phosphate, iron (II) phosphate, iron (III) phosphate, sodium dihydrogenphosphate, trisodium phosphate, disodium hydrogenphosphate, lead phosphate, diammonium phosphate, calcium dihydrogenphosphate, lithium phosphate, phosphotungstic acid, ammonium phosphotungstate, sodium phosphotungstate, ammonium phosphomolybdate, sodium phosphomolybdate, sodium phosphite, sodium tripolyphosphate and sodium pyrophosphate. Of these, sodium dihydrogenphosphate, disodium hydrogenphosphate, potassium dihydrogenphosphate and dipotassium hydrogenphosphate are preferred.
- Preferred inorganic fluorine compounds that may be used in the aqueous solution containing an inorganic fluorine compound and a phosphate compound include metal fluorides.
- Specific examples include sodium fluoride, potassium fluoride, calcium fluoride, magnesium fluoride, sodium hexafluorozirconate, potassium hexafluorozirconate, sodium hexafluorotitanate, potassium hexafluorotitanate, hexafluorozirconic acid, hexafluorotitanic acid, ammonium hexafluorozirconate, ammonium hexafluorotitanate, hexafluorosilicic acid, nickel fluoride, iron fluoride, fluorophosphoric acid and ammonium fluorophosphate.
- The aqueous solution containing an inorganic fluorine compound and a phosphate compound that is used for treatment may contain one each, or two or more each, of the phosphate and the inorganic fluorine compound.
- In the practice of the invention, treatment with the aqueous solution containing an inorganic fluorine compound and a phosphate compound may be carried out by separate treatments with an aqueous solution containing an inorganic fluorine compound and with an aqueous solution containing a phosphate compound.
- The metal base is dipped in the aqueous solution containing an inorganic fluorine compound and a phosphate compound, following which it is washed such as with water and dried.
- Methods other than dipping that may be used include application of the aqueous solution by brush, sponge, spray, wheel coater or some other suitable means.
- Silicate compounds preferred for use in the treatment with a silicate compound-containing aqueous solution that is employed in the invention include silicic acid and silicates. Of these, alkali metal silicates are preferred.
- Specific examples include sodium silicate, potassium silicate and lithium silicate. Of these, sodium silicate and potassium silicate are preferred.
- Examples of sodium silicate include No. 3 sodium silicate, No. 2 sodium silicate, No. 1 sodium silicate, sodium orthosilicate, sodium sesquisilicate and sodium metasilicate. Examples of potassium silicate include No. 1 potassium silicate. Aluminosilicates containing aluminum and borosilicates containing boron may also be used.
- Examples of silicic acid include orthosilicic acid, metasilicic acid, bisilicic acid, trisilicic acid and tetrasilicic acid.
- For good scumming resistance, the aqueous solution has a silicate compound concentration of preferably at least 0.01 wt%, more preferably at least 0.1 wt%, and even more preferably at least 1 wt%. For a long press life, the solution has a concentration of preferably not more than 10 wt%, more preferably not more than 7 wt%, and even more preferably not more than 5 wt%.
- The silicate compound-containing aqueous solution used in the invention may also include a suitable amount of a hydroxide compound such as sodium hydroxide, potassium hydroxide or lithium hydroxide in order to increase the pH. Of these, sodium hydroxide and potassium hydroxide are preferred.
- In addition, an alkaline earth metal salt or a Group 4 (Group IVA) metal salt may be included. Examples of alkaline earth metal salts include the following water-soluble salts: nitrates such as calcium nitrate, strontium nitrate, magnesium nitrate and barium nitrate; and also sulfates, hydrochlorides, phosphates, acetates, oxalates, and borates. Exemplary Group 4 (Group IVA) metal salts include titanium tetrachloride, titanium trichloride, titanium potassium fluoride, titanium potassium oxalate, titanium sulfate, titanium tetraiodide, zirconium chloride oxide, zirconium dioxide, zirconium oxychloride and zirconium tetrachloride. These alkaline earth metal salts and Group 4 (Group IVA) metal salts may be used singly or in combinations of two or more thereof.
- The silicate compound-containing aqueous solution has a temperature of preferably at least 10°C, and more preferably at least 20°C, but preferably not more than 100°C, and even more preferably not more than 80°C.
- The aqueous solution has a pH of preferably at least 8, and more preferably at least 10, but preferably not more than 13, and more preferably not more than 12.
- No particular limitation is imposed on the method of treatment with the silicate compound-containing aqueous solution used in the practice of the invention. For example, dipping or spraying may be used. Of these, dipping is preferred. When treatment is carried out by dipping, the treatment time is preferably at least 1 second, and more preferably at least 3 seconds, but preferably not more than 600 seconds, and more preferably not more than 120 seconds.
- In the practice of the invention, the metal base that has been treated with the above-described silicate compound-containing aqueous solution can, if necessary, be treated with an acidic aqueous solution. Illustrative examples of this acidic aqueous solution include aqueous solutions of sulfuric acid, nitric acid, hydrochloric acid, oxalic acid or phosphoric acid. It is advantageous to carry out this acidic aqueous solution treatment by dipping the hydrophilized metal base in an aqueous solution containing the acid described above in a concentration of 0.001 to 10 wt%, preferably 0.01 to 1 wt%, at a temperature of 15 to 70°C, preferably 25 to 50°C, and for a period of 0.5 to 120 seconds, preferably 2 to 30 seconds. By means of this acidic aqueous solution treatment, adhesion with a heat-sensitive layer or other recording layer can be improved, and the press life of the plate can be extended.
- In the invention, treatment with specific aqueous solutions involves carrying out (1) treatment with an aqueous solution containing an inorganic fluorine compound and a phosphate compound, and (2) treatment with a silicate compound-containing aqueous solution, either before or after treatment (1). That is, either (1) treatment with an aqueous solution containing an inorganic fluorine compound and a phosphate compound is followed by (2) treatment with an aqueous solution containing a silicate compound, or (2) treatment with an aqueous solution containing a silicate compound is followed by (1) treatment with an aqueous solution containing an inorganic fluorine compound and a phosphate compound. These treatments may each be carried out a plurality of times.
- In cases where treatment with an aqueous solution containing an inorganic fluorine compound and a phosphate compound involves separately carrying out treatment with an aqueous solution containing an inorganic fluorine compound and treatment with an aqueous solution containing a phosphate compound, these treatments may be combined in any way with the treatment with an aqueous solution containing a silicate compound.
- In the practice of the invention, the metal base obtained by treatment with specific aqueous solutions as described above, which metal base is sometimes referred to hereinafter simply as "the metal base of the invention," may be subjected to a hydrophilic surface treatment involving immersion in an aqueous solution containing one or more hydrophilic compounds. Illustrative examples of the hydrophilic compound include polyvinylphosphonic acid, compounds having sulfonic acid groups, and carbohydrate compounds.
- The compound having sulfonic acid groups includes aromatic sulfonic acids, formaldehyde condensation products thereof, derivatives of these, and salts of any of the above.
- Illustrative examples of aromatic sulfonic acids include phenolsulfonic acid, catecholsulfonic acid, resorcinolsulfonic acid, benzenesulfonic acid, toluenesulfonic acid, ligninsulfonic acid, naphthalenesulfonic acid, acenaphthene-5-sulfonic acid, phenanthrene-2-sulfonic acid, benzaldehyde-2 (or 3)-sulfonic acid, benzaldehyde-2,4 (or 3,5)-disulfonic acid, oxybenzylsulfonic acids, sulfobenzoic acid, sulfanilic acid, naphthionic acid, and taurine. Of these, benzenesulfonic acid, naphthalenesulfonic acid and ligninsulfonic acid are preferred. The formaldehyde condensation products of benzenesulfonic acid, naphthalenesulfonic acid and ligninsulfonic acid are also preferred.
- Moreover, these may be used as their sulfonic acid salts. Examples of such salts include the sodium salts, potassium salts, lithium salts, calcium salts and magnesium salts. Of these, the sodium salts and potassium salts are preferred.
- The aqueous solution containing a sulfonic acid group-bearing compound has a pH of preferably 4 to 6.5. Compounds such as sulfuric acid, sodium hydroxide and ammonia may be used to adjust the pH within the above range.
- Exemplary carbohydrate compounds include monosaccharides and sugar alcohols thereof, oligosaccharides, polysaccharides, and glycosides.
- Illustrative examples of monosaccharides and sugar alcohols thereof include trioses and their sugar alcohols, such as glycerol; tetroses and their sugar alcohols, such as threose and erythritol; pentoses and their sugar alcohols, such as arabinose and arabitol; hexoses and their sugar alcohols, such as glucose and sorbitol; heptoses and their sugar alcohols, such as D-glycero-D-galacto-heptose and D-glycero-D-galacto-heptitol; octoses and their sugar alcohols, such as D-erythro-D-galacto-octitol; and nonoses and their sugar alcohols, such as D-erythro-L-gluco-nonulose and its sugar alcohols.
- Illustrative examples of oligosaccharides include disaccharides such as saccharose, trehalose and lactose; and trisaccharides such as raffinose.
- Illustrative examples of polysaccharides include amylose, arabinan, cyclodextrin and cellulose alginate.
- "Glycoside," as used herein, refers to compounds in which a sugar constituent and a non-sugar constituent are bonded through, for example, an ether linkage.
- Glycosides can be classified based on the non-sugar constituent. Examples include alkyl glycosides, phenol glycosides, coumarin glycosides, oxycoumarin glycosides, flavonoid glycosides, anthraquinone glycosides, triterpene glycosides, steroid glycosides and mustard oil glycosides.
- Exemplary sugar constituents include the above-mentioned monosaccharides and their sugar alcohols, oligosaccharides, and polysaccharides. Of these, monosaccharides and oligosaccharides are preferred. Monosaccharides and disaccharides are especially preferred.
- Examples of preferred glycosides include compounds of formula (I) below:

- In formula (I) above, R represents a linear or branched alkyl, alkenyl or alkynyl group of 1 to 20 carbons.
- Illustrative examples of alkyl groups having 1 to 20 carbons include methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, tridecyl, tetradecyl, pentadecyl, hexadecyl, heptadecyl, octadecyl, nonadecyl and eicosyl. These may be linear, branched or cyclic alkyl groups.
- Illustrative examples of alkenyl groups having 1 to 20 carbons include allyl and 2-butenyl. These may be linear, branched or cyclic alkenyl groups.
- Illustrative examples of alkynyl groups having 1 to 20 carbons include 1-pentynyl. These may be linear, branched or cyclic alkynyl groups.
- Specific examples of compounds of above formula (I) include methyl glucoside, ethyl glucoside, propyl glucoside, isopropyl glucoside, butyl glucoside, isobutyl glucoside, n-hexyl glucoside, octyl glucoside, capryl glucoside, decyl glucoside, 2-ethylhexyl glucoside, 2-pentylnonyl glucoside, 2-hexyldecyl glucoside, lauryl glucoside, myristyl glucoside, stearyl glucoside, cyclohexyl glucoside and 2-butynyl glucoside. These compounds are glucosides, one type of glycoside in which the hemiacetalhydroxyl group on glucose is ether bonded to another compound, and can be obtained by a known method involving the reaction of glucose with alcohols. Some of these alkyl glucosides are commercially available from Henkel, Germany under the trade name Glucopon, and can be used in the present invention.
- Other examples of preferred glycosides include saponins, rutin trihydrate, hesperidin methylchalcone, hesperidin, naringin hydrate, phenol-β-D-glucopyranoside, salicin and 3',5,7-methoxy-7-rutinoside.
- The aqueous solution containing a carbohydrate compound has a pH of preferably 8 to 11. The pH can be adjusted within this range using, for example, potassium hydroxide, sulfuric acid, carbonic acid, sodium carbonate, phosphoric acid or sodium phosphate.
- Aqueous solutions of polyvinylphosphonic acid have a concentration of preferably 0.1 to 5 wt%, and more preferably 0.2 to 2.5%. The immersion temperature is preferably 10 to 70°C, and more preferably 30 to 60°C. The immersion time is preferably 1 to 20 seconds.
- Aqueous solutions of sulfonic acid group-bearing compounds have a concentration of preferably 0.02 to 0.2 wt%. The immersion temperature is preferably 60 to 100°C. The immersion time is preferably 1 to 300 seconds, and more preferably 10 to 100 seconds.
- Aqueous solutions of carbohydrate compounds have a concentration of preferably 0.5 to 10 wt%. The immersion temperature is preferably 40 to 70°C. The immersion time is preferably 2 to 300 seconds, and more preferably 5 to 30 seconds.
- The metal base of the invention is immersed in an aqueous solution containing these hydrophilic compounds, following which it is washed such as with water and dried.
- The above hydrophilic surface treatment resolves printing contamination problems such as the poor resistance to contamination after standing (poor ink removability) that arises as a trade-off with improved sensitivity (in the case of a negative-working photosensitive layer, improved press life) due to pore widening treatment following anodizing treatment. That is, because the pore diameter is increased, during printing, and particularly when printing is restarted after a period in which the press was down and the printing plate was left on the press, there is a tendency for the ink to become difficult to remove from the plate (sometimes referred to as "diminished resistance to contamination after standing" or "diminished ink removability"). However, this problem is alleviated by administering hydrophilic surface treatment.
- The metal base of the invention is dipped in these hydrophilic compound-containing aqueous solutions, then is washed such as with water and dried.
- In the second aspect of the invention, a metal base which has been administered surface treatment and which satisfies formula (1) below is used in the inventive support for lithographic printing plates. The surface treatment is not subject to any particular limitation, and may be any treatment or treatments capable of preparing a surface which satisfies formula (1) below. Surface treatment that involves administering the above-described specific aqueous solution treatments following the above-described graining treatment and anodizing treatment is preferred. That is, it is preferable for the surface of a metal base prepared by graining treatment and anodizing treatment, followed by administration of the above-described specific aqueous solution treatment, to satisfy formula (1) below.
- In above formula (1), "(A+B+C)/(A+B+C+D)" represents the degree to which the anodized layer is covered with a phosphate compound, an inorganic fluorine compound and a silicate compound. A larger value for (A+B+C)/(A+B+C+D) indicates higher coverage, whereas a smaller value indicates lower coverage.
- Here, at a value for (A+B+C)/(A+B+C+D) of 0.10 or more, treatment with the specific aqueous solutions has a larger effect, resulting in a better sensitivity. Accordingly, the value of (A+B+C)/(A+B+C+D) is preferably at least 0.10, and more preferably at least 0.20.
- On the other hand, at a value for (A+B+C)/(A+B+C+D) of 0.70 or less, suitable effects are achieved by treatment with an aqueous solution containing an inorganic fluorine compound and a phosphate compound, resulting in a longer press life. Accordingly, it is preferable for (A+B+C)/(A+B+C+D) to have a value of 0.70 or less.
- Electron spectroscopy for chemical analysis (ESCA) and the procedure involved are described.
- When x-rays having a given energy (hν) are directed at the surface of a specimen in an ultrahigh vacuum, the photoelectric effect causes electrons (photoelectrons) to be emitted within the vacuum from constituent atoms in the specimen. The kinetic energy (Ek) of the emitted photoelectrons is represented by formula (I) below
- X-ray irradiation is carried out using the Mg-Kα (1253.6 eV) and A1-Kα (1486.6 eV) lines having a small energy range. The depth of penetration by such soft x-rays is about several microns from the surface of the specimen. However, the probability is very high that a photoelectron generated from deep areas within the specimen will lose energy from non-elastic scattering with other atoms before it reaches the surface of the specimen. Only photoelectrons generated at the surface-most portion of the specimen do not collide with other atoms and can be analyzed after emission with the relationship (I) intact. For these reasons, ESCA is capable of measuring the outermost several nanometers (several tens of angstroms) of the specimen surface. Intermediate Layer Containing an Acid Group-Bearing Polymeric Compound
- An intermediate layer containing an acid group-bearing polymeric compound is provided as an intermediate layer (or "undercoat," as viewed from the recording layer side) on the aluminum support that has been treated as described above. This enables a support for lithographic printing plates to be obtained which, when rendered into a lithographic printing plate, has an excellent sensitivity, press life and resistance to scumming.
- It is even more preferable to use as the polymeric compound contained in the above intermediate layer a polymeric compound having an acid group or having both an acid group-bearing constituent and an onium group-bearing constituent. The acid group on the acid group-bearing polymeric compound constituent is preferably an acid group having an acid dissociation constant (pKa) not higher than 7, more preferably -COOH, -SO3H, -OSO3H, -PO3H2, -OPO3H2, -CONHSO2 or -SO2NHSO2-, and most preferably -COOH. Preferred acid group-bearing constituents include polymerizable compounds of general formula (1) or general formula (2) below.
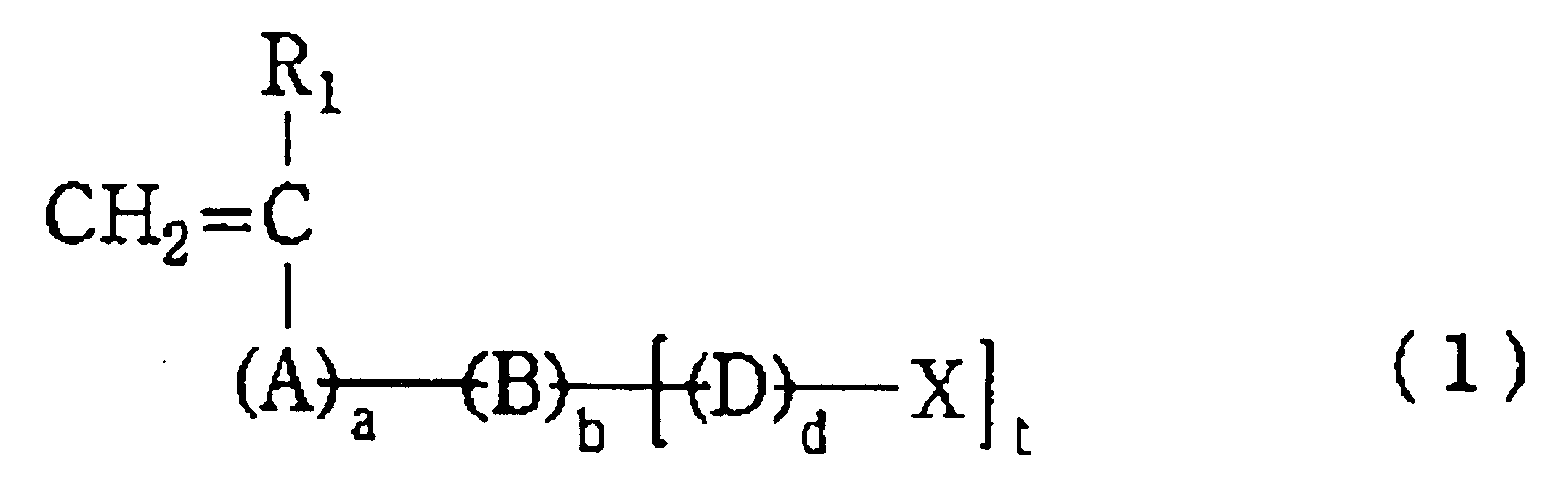

- In the above formulas, A represents a divalent linkage, B is an aromatic or substituted aromatic group, D and E are each independently divalent linkages, G is a trivalent linkage, X and X' are each independently an acid group having a pKa of 7 or less or an alkali metal salt or ammonium salt; and R1 represents a hydrogen atom, an alkyl group or a halogen atom. The letters a, b, d and e are each independently 0 or 1. The letter t is an integer from 1 to 3.
- Preferred acid group-bearing constituents are those wherein A is -COO- or -CONH- and B is a phenylene or substituted phenylene group, with the substituent being a hydroxyl group, a halogen atom or an alkyl group; D and E are each independently an alkylene group or a divalent linkage of the molecular formula CnH2nO, CnH2nS or CnH2n+1N; G represents a trivalent linkage of the molecular formula CnH2n-1, CnH2n-1O, CnH2n-1S or CnH2nN, the letter n being an integer from 1 to 12; X and X' are each independently a carboxylic acid, sulfonic acid, phosphonic acid, sulfuric acid monoester or phosphoric acid monoester; R1 is a hydrogen atom or an alkyl group; and the letters a, b, d and e are each independently 0 or 1; provided that a and b are not both 0.
- Especially preferred acid group-bearing constituents are those of the general formula (1) in which B is a phenylene group or a substituted phenylene group, the substituent being a hydroxyl group or an alkyl of 1 to 3 alkyls; D and E are each independently an alkylene group of 1 or 2 carbons or an alkylene group of 1 or 2 carbons bonded through an oxygen atom; R1 is a hydrogen atom or a methyl group; X is a carboxyl group; and the letters a and b are respectively 0 and 1.
- Specific, non-limiting, examples of the acid group-bearing constituent include acrylic acid, methacrylic acid, crotonic acid, isocrotonic acid, itaconic acid, maleic acid and maleic anhydride. Additional examples include compounds having the following formulas.

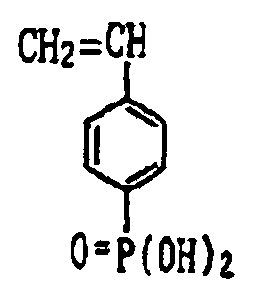







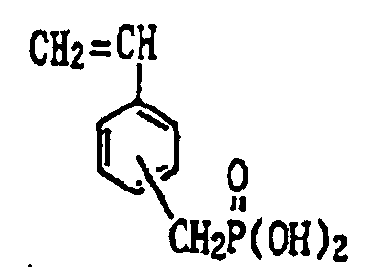















- The above acid group-bearing constituents may be used singly or as combinations of two or more thereof.
- Preferred onium groups on the onium group-bearing constituent of the polymeric compound that may be used to form the intermediate layer include onium groups composed of atoms of elements belonging to group V or group VI of the Periodic Table. Onium groups containing nitrogen atoms, phosphorus atoms or sulfur atoms are more preferred, and onium groups containing nitrogen atoms are especially preferred. This polymeric compound is preferably a vinyl polymer such as one having a backbone structure composed of acrylic acid or methacrylic acid, or polystyrene, or is a urethane resin, a polyester or a polyamide. Of these, vinyl polymers, such as ones having a backbone structure composed of acrylic resin or methacrylic resin, or polystyrene, are especially preferred. Especially preferred polymeric compounds are copolymerizable polymers in which the onium group-bearing constituent has general formula (3), (4) or (5) below.



- In the above formulas, J is a divalent linkage, K is an aromatic or substituted aromatic group, each M independently represents a divalent linkage, Y1 is an atom belonging to group V of the Periodic Table, Y2 is an atom belonging to group VI of the Periodic Table, and Z- is a counteranion. Also, R2 is a hydrogen atom, an alkyl group or a halogen atom; R3, R4, R5 and R7 are each independently a hydrogen atom or an alkyl, aromatic or aralkyl group which may have substituents bonded thereto; and R6 is an alkylidyne or substituted alkylidyne group; wherein R3 and R4 or R6 and R7 may be mutually bonded to form a ring. Moreover, the letters j, k and m are each independently 0 or 1; and the letter u is an integer from 1 to 3.
- Preferred onium group-bearing constituents include those in which J represents -COO- or -CONH-; K represents a phenylene or substituted phenylene group, wherein the substituent is a hydroxyl group, a halogen atom or an alkyl group; M is an alkylene group, or a divalent linkage of the molecular formula CnH2nO, CnH2nS or CnH2n+1N, the letter n being an integer from 1 to 12; Y1 is a nitrogen atom or phosphorus atom; Y2 is a sulfur atom; and Z- is a halogen ion, PF6-, BF4- or R8SO3-. Also, R2 is a hydrogen atom or an alkyl group; R3, R4, R5 and R7 are each independently hydrogen atoms or C1-10 alkyl, aromatic or aralkyl groups which may have substituents bonded thereto; and R6 is a C1-10 alkylidyne or substituted alkylidyne group; wherein R3 and R4 or R6 and R7 may be mutually bonded to form a ring. Moreover, the letters j, k and m are each independently 0 or 1, such that j and k are not both 0.
- Especially preferred onium group-bearing constituents include those in which K is a phenylene or substituted phenylene group, the substituent being a hydroxyl group or an alkyl group of 1 to 3 carbons; M is an alkylene group of 1 or 2 carbons or an alkylene group of 1 or 2 carbons that is linked through an oxygen atom; Z- is a chlorine ion or R8SO3-; R2 is a hydrogen atom or a methyl group; and the letters j and k are respectively 0 and 1.
- Specific, non-limiting, examples of onium group-bearing constituents include the following.








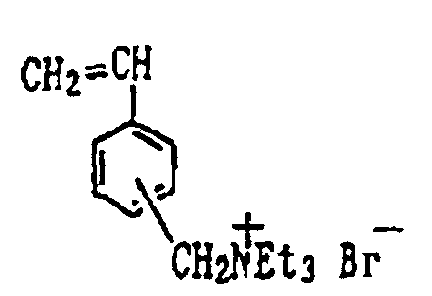








- It is desirable that the polymeric compound used to form the intermediate layer contain at least 1 mol%, and preferably at least 5 mol%, of the above-described onium group-bearing constituent. The presence of at least 1 mol% of an onium group-bearing constituent further improves adhesion. The onium group-bearing constituent may be of one type or a combination of two or more types. Moreover, the polymeric compound used to form the intermediate layer may also be a mixture of two or more such compounds of differing constituents, compositional ratios or molecular weights.
- In polymeric compounds which contain both the above-described acid group and the above-described onium group, it is advantageous for the compound to include at least 20 mol%, and preferably at least 40 mol%, of the acid group-bearing constituent, and at least 1 mol%, and preferably at least 5 mol%, of the onium group-bearing constituent. The presence of at least 20 mol% of the acid group-bearing constituent further promotes dissolution and removal during alkali development, and also further enhances adhesion due to synergistic effects between the acid groups and the onium groups. Moreover, such polymeric compounds which contain both acid groups and onium groups may of course be a mixture of two or more such compounds of differing constituents, compositional ratios or molecular weights. Illustrative examples of polymeric compounds having both the above-described onium groups and acid groups are given below. The compositional ratios in these polymer structures are given in mole percent (mol%).


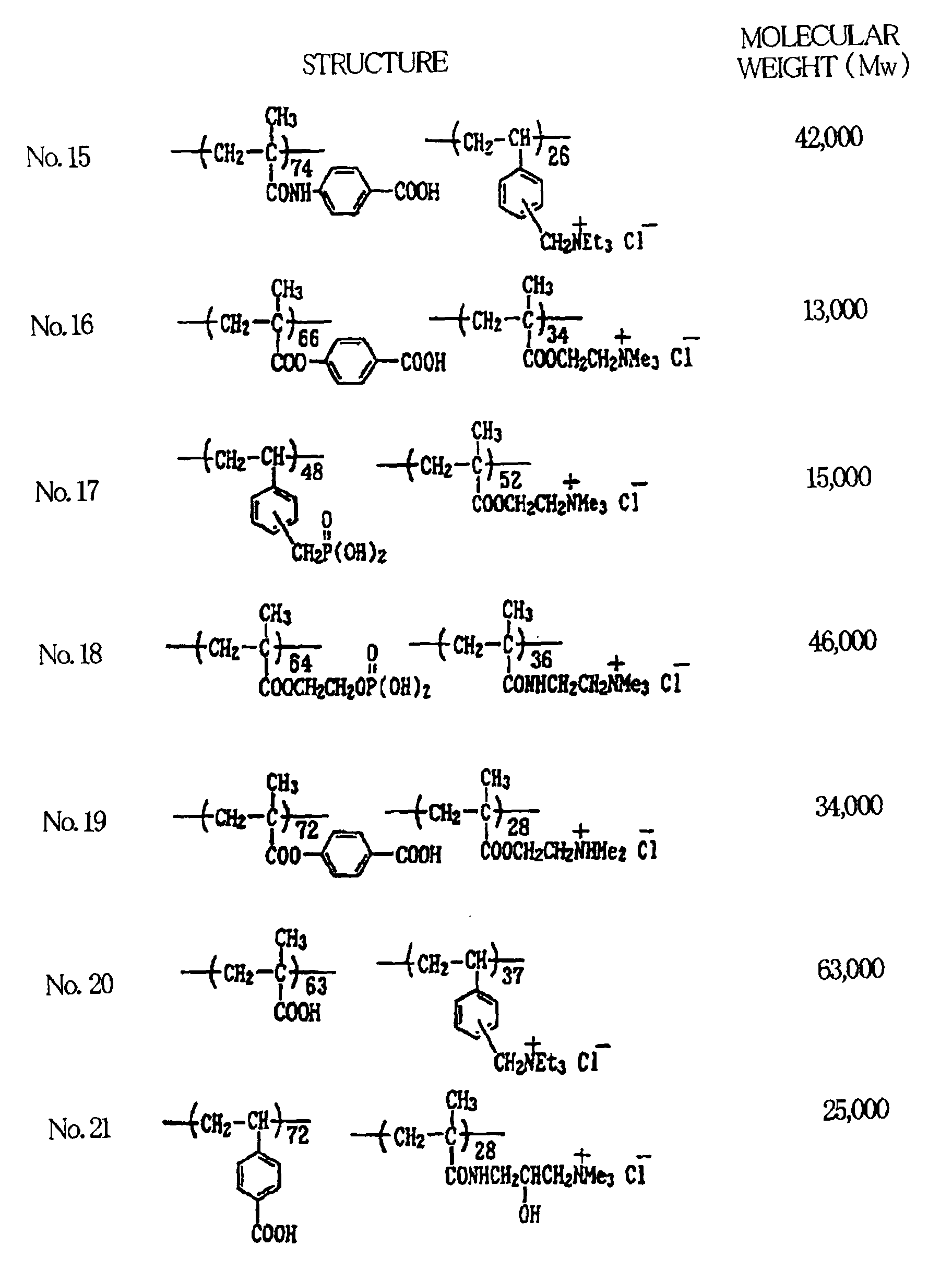

- The above-described polymeric compounds which contain acid groups or both acid groups and onium groups and are used to form the intermediate layer can generally be prepared using a radical chain polymerization process (see Textbook of Polymer Science, 3rd ed., by F.W. Billmeyer (John Wiley & Sons, 1984). These polymeric compounds may have a broad range in molecular weight, although the weight-average molecular weight (Mw) as measured by a light scattering technique is preferably in a range of from 500 to 2,000,000, and more preferably 2,000 to 600,000. The amount of unreacted monomer present in this polymeric compound may vary over a wide range, although it is preferably not more than 20 wt%, and more preferably not more than 10 wt%. A typical example of a polymeric compound containing both acid groups and onium groups is a copolymer of p-vinylbenzoic acid and vinylbenzyltrimethyl ammonium chloride (No. 1 in Table 1 above). This can be synthesized as follows. First, 146.9 g (0.99 mol) of p-vinylbenzoic acid (available from Hokko Chemical Industry Co., Ltd.), 44.2 g (0.21 mol) of vinylbenzyltrimethyl ammonium chloride, and 446 g of 2-methoxyethanol were placed in a 1-liter, 3-neck flask, then heated and held at 75°C under a stream of nitrogen and stirring. Next, 2.76 g (12 mmol) of dimethyl 2,2-azobis(isobutyrate) was added and stirring was continued. After 2 hours, an additional 2.76 g (12 mmol) of dimethyl 2,2-azobis(isobutyrate) was added. Two hours later, yet another 2.76 g (12 mmol) of dimethyl 2,2-azobis(isobutyrate) was added. After 2 hours of stirring, the mixture was allowed to cool to room temperature. The reaction mixture was then poured into 12 L of ethyl acetate under stirring. The solid that precipitated out was removed by filtration and dried, giving a yield of 189.5 g. The molecular weight of the resulting solid was measured by a light scattering technique, yielding a weight-average molecular weight (Mw) of 32,000. The other polymeric compounds were prepared by similar methods.
- The intermediate layer can be provided by using any of various methods to apply the above-described polymeric compound bearing acid groups or both acid groups and onium groups (referred to hereinafter as simply "the polymeric compound") onto the above-described metal base treated with specific aqueous solutions or treated with specific aqueous solutions, then additionally treated with an acidic aqueous solution. One commonly used method for providing the intermediate layer involves applying to the metal base a solution obtained by dissolution of the polymeric compound in an organic solvent such as methanol, ethanol or methyl ethyl ketone, a mixture of such organic solvents, or a mixed solvent composed of any of these organic solvents and water, then drying the applied solution. Another method involves dipping the metal base in a solution obtained by dissolution of the polymeric compound in an organic solvent such as methanol, ethanol or methyl ethyl ketone, a mixture of such organic solvents, or a mixed solvent composed of any of these organic solvents and water so as to induce adsorption of the polymeric compound, then washing such as with water and drying. With the former method, a solution containing the polymeric compound in a concentration of 0.005 to 10 wt% can be applied by any of various methods, such as bar coating, spin coating, spray coating and curtain coating. With the latter method, the solution concentration is 0.01 to 20 wt%, preferably 0.05 to 5 wt%, the dipping temperature is 20 to 90°C, and preferably 25 to 50°C, and the dipping time is 0.1 second to 20 minutes, and preferably 2 seconds to 1 minute.
- The above polymeric compound solution can also be used after adjusting the pH within a range of 0 to 12, and preferably 0 to 5, using a basic substance such as ammonia, triethylamine or potassium hydroxide, an inorganic acid such as hydrochloric acid, phosphoric acid, sulfuric acid or nitric acid, any of various organic acidic substances such as organic sulfonic acids (e.g., nitrobenzenesulfonic acid, naphthalenesulfonic acid), organic phosphonic acids (e.g., phenylphosphonic acid) and organic carboxylic acids (e.g., benzoic acid, coumaric acid, malic acid), and organic acid chlorides such as naphthalenesulfonyl chloride and benzenesulfonyl chloride. Moreover, a yellow dye can be added to improve the tone reproducibility of the photosensitive lithographic printing plate. The coating weight after drying of the polymeric compound is generally from 2 to 100 mg/m2, and preferably from 5 to 50 mg/m2. At a coating weight of less than 2 mg/m2 or more than 100 mg/m3, sufficient effects may not be achieved.
- If necessary, the support on which an intermediate layer has been formed and which has been obtained as described above may be provided on the back side (the side on which a recording layer is not provided) with a coat (referred to hereinafter as the "back coat") composed of an organic polymeric compound so that scuffing of the recording layer does not occur even when the resulting presensitized plates are stacked on top of one other.
- The back coat preferably contains, as the main component, at least one resin which has a glass transition point of at least 20°C and is selected from the group consisting of saturated copolyester resins, phenoxy resins, polyvinyl acetal resins and vinylidene chloride copolymer resins. Preferred use can be made of coats composed of metal oxides obtained by the hydrolysis and polycondensation of the organic polymeric compounds mentioned in JP 5-45885 A and the organic or inorganic metal compounds mentioned in JP 6-35174 A. Of these coats, alkoxy compounds of silicon, such as Si(OCH3) 4, Si (OC2H5) 4, Si(OC3H7)4 and Si (OC4H9) 4, are preferred because they are inexpensive and readily available, and metal oxide coats obtained from these compounds have an excellent developability.
- The saturated copolyester resin used in the back coat is composed of dicarboxylic acid units and diol units. Examples of the dicarboxylic acid units include aromatic dicarboxylic acids such as phthalic acid, terephthalic acid, isophthalic acid, tetrabromophthalic acid and tetrachlorophthalic acid; and saturated aliphatic dicarboxylic acids such as adipic acid, azelaic acid, succinic acid, oxalic acid, suberic acid, sebacic acid, malonic acid and 1,4-cyclohexanedicarboxylic acid.
- The back coat may additionally include dyes and pigments for coloration, silane coupling agents to improve adhesion to the support, diazo resins composed of diazonium salts, organophosphonic acids, organophosphoric acids, cationic polymers, and the following substances which are commonly used as slip agents: waxes, higher aliphatic acids, higher aliphatic acid amides, silicone compounds made of dimethylsiloxane, modified dimethylsiloxane and polyethylene powder.
- The back coat should have a thickness which is of a degree that will help protect the recording layer from scuffing, even in the absence of a slip sheet. A thickness of 0.01 to 8 µm is preferred. At a thickness of less than 0.01 µm, it may be difficult to prevent scuffing of the recording layer when a plurality of presensitized plates are stacked and handled together. On the other hand, at a thickness of more than 8 µm, the chemicals used in the vicinity of the lithographic printing plate during printing cause the back coat to swell and fluctuate in thickness, which may alter the printing pressure and thereby compromise the printability.
- Various methods may be used to provide the back coat on the back side of the support. Illustrative examples include preparing the above-mentioned back coat ingredients as a solution in a suitable solvent and applying the solution, or preparing these ingredients as an emulsified dispersion, then applying the dispersion and drying. Another method that may be used is to first form a film, then laminate and bond the film to the support using an adhesive or heat. Still another method is to form a melt film by a melt extruder and bond the film to the support. The especially preferred method for achieving a suitable thickness is to dissolve the back coat-forming ingredients in a suitable solvent, then apply the solution and dry. Organic solvents such as those mentioned in JP 62-251739 A may be used singly or in admixture as the media in such methods.
- During production of the presensitized plate, it is possible to first provide on the support either the back coat on the back side or the recording layer on the front side. Alternatively, both may be provided at the same time.
- The presensitized plate of the invention can be obtained by providing an infrared laser-imageable recording layer on the inventive support for lithographic printing plates obtained as described above.
- In the presensitized plate of the invention, the recording layer is preferably a heat-sensitive layer containing:
- (a) a finely divided polymer having thermally reactive functional groups, or
- (b) microcapsules containing a compound having thermally reactive functional groups. By using this heat-sensitive layer, there can be obtained an on-machine development type presensitized plate.
-
- Thermally reactive functional groups common to both (a) and (b) above include ethylenically unsaturated groups which carry out the polymerization reaction (e.g., acryloyl, methacryloyl, vinyl, allyl); isocyanate groups or blocked isocyanate groups which carry out addition reactions, along with active hydrogen atom-bearing functional groups that react therewith (e.g., amino groups, hydroxyl groups, carboxyl groups); epoxy groups that carry out addition reactions, along with amino, carboxyl or hydroxyl groups that react therewith; carboxyl groups that carry out condensation reactions, along with hydroxyl or amino groups that react therewith; and acid anhydrides which carry out ring-opening addition reactions, along with amino or hydroxyl groups that react therewith. Thermally reactive functional groups which may be used in the present invention are not limited to the above, and may be any functional group that carries out a reaction, so long as it forms a chemical bond.
- Illustrative examples of thermally reactive functional groups suitable for the finely divided polymer (a) include acryloyl, methacryloyl, vinyl, allyl, epoxy, amino, hydroxyl, carboxyl, isocyanate and acid anhydride groups, as well as protected forms of these groups. The thermally reactive functional groups may be introduced onto the polymer particles when the polymer is polymerized or may be introduced by means of a polymer reaction following polymerization.
- If the thermally reactive functional groups are introduced onto the polymer during polymerization, it is preferable to carry out emulsion polymerization or suspension polymerization using a monomer having a thermally reactive functional group.
- Specific, non-limiting, examples of monomers having a thermally reactive functional group that may be used in the invention include allyl methacrylate, allyl acrylate, vinyl methacrylate, vinyl acrylate, glycidyl methacrylate, glycidyl acrylate, 2-isocyanatoethyl methacrylate and alcohol-blocked isocyanates thereof, 2-isocyanatoethyl acrylate and alcohol-blocked isocyanates thereof, 2-aminoethyl methacrylate, 2-aminoethyl acrylate, 2-hydroxyethyl methacrylate, 2-hydroxyethyl acrylate, acrylic acid, methacrylic acid, maleic anhydride, bifunctional acrylate and bifunctional methacrylate.
- Illustrative, non-limiting, examples of monomers without a thermally reactive functional group that are copolymerizable with these monomers and may be used in the invention include styrene, alkyl acrylates, alkyl methacrylates, acrylonitrile and vinyl acetate.
- Examples of polymer reactions that may be used when the thermally reactive functional groups are introduced after polymerization include the polymer reactions mentioned in WO 96/34316.
- Of the above finely divided polymers (a), those in which the fine particles mutually coalesce under heating are preferred, and those which have a hydrophilic surface and disperse in water are especially preferred. It is desirable in this case for a film formed by applying only the finely divided polymer and drying at a temperature below the solidification temperature to have a contact angle (water drop in air) which is smaller than the contact angle (water drop in air) of a film that is similarly formed but dried at a temperature higher than the solidification temperature.
- An illustrative, non-limiting example of a method for making the surface of the polymer fine particles hydrophilic in this way involves the adsorption of a hydrophilic polymer or oligomer such as polyvinyl alcohol or polyethylene glycol, or of a hydrophilic low-molecular-weight compound, onto the surface of the polymer fine particles.
- It is preferable for the finely divided polymer (a) to have a solidification temperature of at least 70°C, although a solidification temperature of at least 100°C is especially preferred for good stability over time.
- The finely divided polymer (a) has an average particle size of preferably 0.01 to 20 µm, more preferably 0.05 to 2.0 µm, and most preferably 0.1 to 1.0 µm. Within the above range, good resolution and stability over time can be achieved.
- The amount of finely divided polymer (a) added is preferably at least 50 wt%, and more preferably at least 60 wt%, of the solids in the heat-sensitive layer.
- Suitable examples of the thermally reactive functional groups on the compound contained within the microcapsules (b) include polymerizable unsaturated groups, hydroxyl groups, carboxyl groups, carboxylate groups, acid anhydride groups, amino groups, epoxy groups, isocyanate groups and blocked isocyanate groups.
- Preferred examples of polymerizable unsaturated group-bearing compounds include compounds with at least one, and preferably at least two, ethylenically unsaturated bond (e.g., acryloyl, methacryloyl, vinyl, allyl). Such compounds are widely used in industrial fields related to the present invention, and may be used herein without any particular limitation. These compounds have a variety of chemical forms, including monomers, prepolymers such as dimers, trimers, oligomers or mixtures thereof, and copolymers of any of the above.
- Specific examples include unsaturated carboxylic acids (e.g., acrylic acid, methacrylic acid, itaconic acid, crotonic acid, isocrotonic acid, maleic acid), and also esters and unsaturated carboxylic acid amides thereof. Of these, esters of unsaturated carboxylic acids and aliphatic polyols, and amides of unsaturated carboxylic acids and aliphatic polyamines, are preferred.
- Advantageous use can also be made of products of addition reactions between an unsaturated carboxylic acid ester or amide having a nucleophilic substituent such as a hydroxyl, amino or mercapto group and a monofunctional or polyfunctional isocyanate or epoxide, and products of dehydration condensation reactions between such a carboxylic acid ester or amide and a monofunctional or polyfunctional carboxylic acid.
- Moreover, the products of addition reactions between an unsaturated carboxylic acid ester or amide having an electrophilic substituent such as an isocyanate or epoxy group and a monofunctional or polyfunctional alcohol, amine or thiol, and the products of substitution reactions between an unsaturated carboxylic acid ester or amide having a removable substituent such as a halogen group or a tosyloxy group and a monofunctional or polyfunctional alcohol, amine or thiol are also preferred.
- Other preferred examples include compounds like those mentioned above, but in which the unsaturated carboxylic acid is replaced with an unsaturated phosphonic acid or chloromethylstyrene.
- Specific examples of polymeric compounds which are esters of unsaturated carboxylic acids and aliphatic polyols are given below.
- Acrylic acid esters include ethylene glycol diacrylate, triethylene glycol diacrylate, 1,3-butanediol diacrylate, tetramethylene glycol diacrylate, propylene glycol diacrylate, neopentyl glycol diacrylate, trimethylolpropane diacrylate, trimethylolpropane triacrylate, trimethylolpropane tris(acryloyloxypropyl) ether, trimethylolethane triacrylate, hexanediol diacrylate, 1,4-cyclohexanediol diacrylate, tetraethylene glycol diacrylate, pentaerythritol diacrylate, pentaerythritol triacrylate, pentaerythritol tetraacrylate, dipentaerythritol diacrylate, dipentaerythritol pentaacrylate, dipentaerythritol hexaacrylate, sorbitol triacrylate, sorbitol tetraacrylate, sorbitol pentaacrylate, sorbitol hexaacrylate, tris(acryloyloxyethyl) isocyanurate and polyester acrylate oligomers.
- Methacrylic acid esters include tetramethylene glycol dimethacrylate, triethylene glycol dimethacrylate, neopentyl glycol dimethacrylate, trimethylolpropane trimethacrylate, trimethylolethane trimethacrylate, ethylene glycol dimethacrylate, 1,3-butanediol dimethacrylate, hexanediol dimethacrylate, pentaerythritol dimethacrylate, pentaerythritol trimethacrylate, pentaerythritol tetramethacrylate, dipentaerythritol dimethacrylate, dipentaerythritol hexamethacrylate, sorbitol trimethacrylate, sorbitol tetramethacrylate, bis[p-(3-methacryloyloxy-2-hydroxypropoxy)phenyl] dimethylmethane and bis[p-(methacryloyloxyethoxy)phenyl]dimethylmethane.
- Itaconic acid esters include ethylene glycol diitaconate, propylene glycol diitaconate, 1,3-butanediol diitaconate, 1,4-butanediol diitaconate, tetramethylene glycol diitaconate, pentaerythritol diitaconate and sorbitol tetraitaconate.
- Crotonic acid esters include ethylene glycol dicrotonate, tetramethylene glycol dicrotonate, pentaerythritol dicrotonate and sorbitol tetradicrotonate.
- Isocrotonic acid esters include ethylene glycol diisocrotonate, pentaerythritol diisocrotonate and sorbitol tetraisocrotonate.
- Maleic acid esters include ethylene glycol dimaleate, triethylene glycol dimaleate, pentaerythritol dimaleate and sorbitol tetramaleate.
- Preferred examples of other suitable esters include the aliphatic alcohol esters mentioned in JP 46-27926 B, JP 51-47334 B and JP 57-196231 A; esters having aromatic skeletons such as those mentioned in JP 59-5240 A, JP 59-5241 A and JP 2-226149 A; and the amino group-bearing esters mentioned in JP 1-165613 A.
- Specific examples of amides of unsaturated carboxylic acids with aliphatic polyamines that may be used as monomers include methylenebisacrylamide, methylenebismethacrylamide, 1,6-hexamethylenebisacrylamide, 1,6-hexamethylenebismethacrylamide, diethylenetriaminetrisacrylamide, xylylenebisacrylamide and xylylenebismethacrylamide.
- Other suitable amide-type monomers include those having a cyclohexylene structure mentioned in JP 54-21726 B.
- Urethane-type addition polymerizable compounds prepared using an addition reaction between an isocyanate group and a hydroxyl group are also preferred. Specific examples include the urethane compounds having two or more polymerizable unsaturated groups per molecule which are mentioned in JP 48-41708 B and are obtained by adding a hydroxyl group-bearing unsaturated monomer of formula (II) below to a polyisocyanate compound having at least two isocyanate groups per molecule.
- Urethane acrylates such as those mentioned in JP 51-37193 A, JP 2-32293 B and JP 2-16765 B, and the urethane compounds having an ethylene oxide-type skeleton mentioned in JP 58-49860 B, JP 56-17654 B, JP 62-39417 B and JP 62-39418 B are also preferred.
- Other suitable examples include the radical polymerizable compounds having within the molecule an amino structure or a sulfide structure that are mentioned in JP 63-277653 A, JP 63-260909 A and JP 1-105238 A.
- Additional examples include polyfunctional acrylates and methacrylates, including polyester acrylates and epoxy acrylates obtained by reacting an epoxy resin with (meth)acrylic acid, such as those mentioned in JP 48-64183 A, JP 49-43191 B and JP 52-30490 B. Further examples include the specific unsaturated compounds mentioned in JP 46-43946 B, JP 1-40337 B and JP 1-40336 B, and the vinylphosphonic acid compounds mentioned in JP 2-25493 A. In some cases, it will be desirable to use the perfluoroalkyl group-containing compounds mentioned in JP 61-22048 A. Use can also be made of the photocurable monomers and oligomers mentioned in Nippon Setchaku Kyokaishi Vol. 20, No. 7, 300-308 (1984).
- Suitable epoxy compounds include glycerol polyglycidyl ether, polyethylene glycol diglycidyl ether, polypropylene diglycidyl ether, trimethylolpropane polyglycidyl ether, sorbitol polyglycidyl ether, and the polyglycidyl ethers of bisphenols, polyphenols and hydrogenates thereof.
- Suitable isocyanate compounds include tolylene diisocyanate, diphenylmethane diisocyanate, polymethylene polyphenyl polyisocyanate, xylylene diisocyanate, naphthalene diisocyanate, cyclohexanephenylene diisocyanate, isophorone diisocyanate, hexamethylene diisocyanate, cyclohexyl diisocyanate, and compounds obtained by blocking any of the above with alcohol or amine.
- Suitable amine compounds include ethylenediamine, diethylenetriamine, triethylenetetramine, hexamethylenediamine, propylenediamine and polyethyleneimine.
- Suitable hydroxyl group-bearing compounds include compounds having terminal methylol groups, polyols such as pentaerythritol, bisphenols and polyphenols.
- Suitable carboxyl group-bearing compounds include aromatic polycarboxylic acids such as pyromellitic acid, trimellitic acid and phthalic acid; and aliphatic polycarboxylic acids such as adipic acid.
- Suitable acid anhydrides include pyromellitic anhydride and benzophenonetetracarboxylic anhydride.
- Suitable copolymers of ethylenically unsaturated compounds include allyl methacrylate copolymers, specific examples of which include allyl methacrylate/methacrylic acid copolymers, allyl methacrylate/ethyl methacrylate copolymers and allyl methacrylate/butyl methacrylate copolymers.
- Microencapsulation may be carried out by a known method. Illustrative, non-limiting examples of techniques for preparing microcapsules that may be used in the invention include the methods involving the use of coacervation described in US 2,800,457 and US 2,800,458; the methods that rely on interfacial polymerization described in GB 990,443 B, US 3,287,154, JP 38-19574 B, JP 42-446 B and JP 42-711 B; the methods involving polymer precipitation disclosed in US 3,418,250 and US 3,660,304; the method that uses an isocyanate polyol wall material described in US 3,796,669; the method that uses an isocyanate wall material described in US 3,914,511; the methods that use a urea-formaldehyde or urea formaldehyde-resorcinol wall-forming material which are described in US 4,001,140, 4,087,376 and 4,089,802; the method which uses wall materials such as melamine-formaldehyde resins and hydroxycellulose that is described in US 4,025,445; the in situ methods involving monomer polymerization that are taught in JP 36-9163 B and JP 51-9079 B; the spray drying processes described in GB 930,422 B and US 3,111,407; and the electrolytic dispersion cooling processes described in GB 952,807 B and GB 967,074 B.
- Microcapsule walls suitable for use in the microcapsules (b) are those which have three-dimensional crosslinkages and are solvent-swellable. Accordingly, it is preferable for the microcapsule wall material to be polyurea, polyurethane, polyester, polycarbonate, polyamide, or a mixture thereof. Polyurea and polyurethane are especially preferred. The microcapsule wall may also have introduced therein a compound having thermally reactive functional groups.
- The microcapsules (b) have an average particle size of preferably 0.01 to 20 µm, more preferably 0.05 to 2.0 µm, and most preferably 0.10 to 1.0 µm. Within the above range, it is possible to obtain a good resolution and a good stability over time.
- The microcapsules (b) may or may not mutually coalesce under heating. What is important is that the microcapsules contain a substance which, following application of the recording layer, exudes onto the surface or outside of the microcapsules or penetrates into the microcapsule walls, and induces a chemical reaction under heating. Reaction may take place with a hydrophilic resin that has been added or with a low-molecular-weight compound that has been added. Alternatively, two or more types of microcapsules may each be provided with different functional groups which thermally react with each other, and the different types of microcapsules thereby induced to mutually react.
- Accordingly, it is desirable, though not essential, for good image formation that the microcapsules melt and coalesce with each other under heating.
- The amount of microcapules added to the heat-sensitive layer is preferably 10 to 60 wt%, and more preferably 15 to 40 wt%, based on the solids in the layer. Within this range, a good on-machine developability can be obtained and a good sensitivity and press life can also be achieved.
- When microcapsules (b) are included in the heat-sensitive layer, a solvent which dissolves the microcapsule contents and causes the wall material to swell may be added to the microcapsule dispersing medium. The presence of such a solvent promotes the diffusion of the encapsulated thermally reactive functional group-bearing compound out of the microcapules.
- The particular solvent used will depend on the microcapsule dispersing medium, the material making up the microcapsule wall, the wall thickness and the microcapsule contents, but may readily be selected from many commercially available solvents. For example, in the case of water-dispersible microcapsules composed of a crosslinked polyurea or polyurethane wall, preferred solvents include alcohols, ethers, acetals, esters, ketones, polyols, amides, amines and fatty acids.
- Specific examples include but are not limited to methanol, ethanol, t-butanol, n-propanol, tetrahydrofuran, methyl lactate, ethyl lactate, methyl ethyl ketone, propylene glycol monomethyl ether, ethylene glycol diethyl ether, ethylene glycol monomethyl ether, γ-butyrolactone, N,N-dimethylformamide and N,N-dimethylacetamide. It is also possible to use two or more of these solvents together.
- Use can also be made of a solvent which does not dissolve in the microcapsule dispersion itself, but will dissolve therein if the above-described solvent has been admixed. Such a solvent is added in an amount which is selected according to the combination of ingredients, preferably 5 to 95 wt%, more preferably 10 to 90 wt%, and most preferably 15 to 85 wt%, based on the overall amount of the coating fluid.
- When a heat-sensitive layer containing the above-described thermally reactive functional group-bearing finely divided polymer (a) or thermally reactive functional group-bearing compound-containing microcapsules (b) is used, a compound which initiates or promotes these reactions may also be added if necessary. Exemplary reaction-initiating or promoting compounds are compounds which generate radical or cations under heating. Specific examples include the lophine dimer, trihalomethyl compounds, peroxides, azo compounds, onium salts (e.g., diazonium salts, diphenyliodonium salts), acylphosphine and imidosulfonate.
- These compounds are added within a range of preferably 1 to 20 wt%, and more preferably 3 to 10 wt%, of the solids in the heat-sensitive layer. Within the above range, the on-machine developability is not compromised, and good reaction initiating or reaction promoting effects can be obtained.
- A hydrophilic resin may be added to the heat-sensitive layer. The addition of a hydrophilic resin provides a good on-machine developability and also enhances the film strength of the heat-sensitive layer itself.
- The hydrophilic resin is preferably one which contains hydrophilic groups such as hydroxyl, carboxyl, hydroxyethyl, hydroxypropyl, amino, aminoethyl, aminopropyl and carboxymethyl.
- Specific examples of the hydrophilic resin include gum arabic, casein, gelatin, starch derivatives, carboxymethyl cellulose and its sodium salt, cellulose acetate, sodium alginate, vinyl acetate-maleic acid copolymers, styrene-maleic acid copolymers, polyacrylic acids and their salts, polymethacrylic acids and their salts, homopolymers and copolymers of hydroxyethyl methacrylate, homopolymers and copolymers of hydroxyethyl acrylate, homopolymers and copolymers of hydroxypropyl methacrylate, homopolymers and copolymers of hydroxypropyl acrylate, homopolymers and copolymers of hydroxybutyl methacrylate, homopolymers and copolymers of hydroxybutyl acrylate, polyethylene glycols, hydroxypropylene polymers, polyvinyl alcohols, hydrolyzed polyvinyl acetates having a degree of hydrolysis of at least 60 wt%, and preferably at least 80 wt%, polyvinyl formal, polyvinyl butyral, polyvinyl pyrrolidone, the homopolymers and copolymers of acrylamides, the homopolymers and copolymers of methacrylamides, and the homopolymers and copolymers of N-methylolacrylamide.
- The amount of hydrophilic resin added to the heat-sensitive layer is preferably from 5 to 40 wt%, and more preferably from 10 to 30 wt%, based on the solids in the heat sensitive layer. Within the above range, a good on-machine development and a good film strength can be obtained.
- To improve the sensitivity, the heat-sensitive layer may include also a photothermal conversion substance which absorbs infrared light and generates heat. The photothermal conversion substance may be any light absorbing substance having an absorption band within at least a part of the range from 700 to 1200 nm. Various known pigments, dyes and finely divided metals may be used in this way.
- Suitable pigments include black pigments, brown pigments, red pigments, violet pigments, blue pigments, green pigments, fluorescent pigments, metal powder pigments and also polymer-bonded dyes. Specific examples include insoluble azo pigments, azo lake pigments, condensed azo pigments, chelate azo pigments, phthalocyanine pigments, anthraquinone pigments, perylene and perinone pigments, thioindigo pigments, quinacridone pigments, dioxazine pigments, isoindolinone pigments, quinophthalone pigments, lake pigments, azine pigments, nitroso pigments, nitro pigments, natural pigments, fluorescent pigments, inorganic pigments and carbon black.
- The pigments may be used without being surface treated or may be used after surface treatment. Examples of surface treatment methods include surface coating with a hydrophilic resin or an oleophilic resin, surfactant adhesion, and bonding a reactive substance (e.g., a silica sol, alumina sol, silane coupling agent, epoxy compound or isocyanate compound) to the pigment surface. Surface treatment methods that may be used include those described in Kinzoku Sekken no Seishitsu to Oyo [Properties and Applications of Metallic Soaps] (Koshobo), Insatsu Inki Gijutsu [Printing Ink Technology] (CMC Shuppan, 1984), and Saishin Ganryo Oyo Gijutsu [Recent Pigment Applications Technology] (CMC Shuppan, 1986). Of these pigments, a pigment which absorbs infrared light is preferred in that it is suitable for use in lasers that generate infrared light. Carbon black is preferable as this infrared-absorbing pigment.
- The pigment has a particle size which is in a range of preferably 0.01 to 1 µm, and more preferably 0.01 to 0.5 µm.
- Dyes which may be used include commercial dyes, and dyes mentioned in the technical literature, including Senry
o Binran [Handbook of Dyes] (The Society of Synthetic Organic Chemistry, Japan, 1970); "Near-infrared absorption dyes," in Kagaku Kogyo (May 1986) pp. 45-51; 90-Nendai Kino sei Shikiso no Kaihatsu to Shijo Do ko [Functional Dyes: Development and Market Trends in the 1990s], chapter 2, section 2.3 (CMC, 1990); and patents. Infrared-absorbing dyes such as azo dyes, metal complex azo dyes, pyrazolone azo dyes, anthraquinone dyes, phthalocyanine dyes, carbonium dyes, quinoneimine dyes, polymethine dyes and cyanine dyes are preferred. - Other suitable dyes include the cyanine dyes mentioned in JP 58-125246 A, JP 59-84356 A and JP 60-78787 A; the methine dyes mentioned in JP 58-173696 A, JP 58-181690 A and JP 58-194595 A; the naphthoquinone dyes mentioned in JP 58-112793 A, JP 58-224793 A, JP 59-48187 A, JP 59-73996 A, JP 60-52940 A and JP 60-63744 A; the squarylium dyes mentioned in JP 58-112792 A; the cyanine dyes mentioned in GB 434,875 B, the dyes mentioned in US 4,756,993; the cyanine dyes mentioned in US 4,973,572; the dyes mentioned in JP 10-268512 A; and the phthalocyanine compounds mentioned in JP 11-235883 A.
- The near-infrared absorbing sensitizers mentioned in US 5,156,938 can also be used advantageously as dyes. Other compounds that can be suitably used in this way include the substituted arylbenzo(thio)pyrylium salts mentioned in US 3,881,924; the trimethinethiapyrylium salts mentioned in JP 57-142645 A, the pyrylium compounds mentioned in JP 58-181051 A, JP 58-220143 A, JP 59-41363 A, JP 59-84248 A, JP 59-84249 A, JP 59-146063 A and JP 59-146061 A; the cyanine dyes mentioned in JP 59-216146 A; the pentamethinethiopyrylium salts mentioned in US 4,283,475; the pyrylium compounds mentioned in JP 5-13514 B and JP 5-19702 B; and Epolight III-178, Epolight III-130 and Epolight III-125 produced by Epolin, Inc.
- Some specific examples are shown below.








- The above-described organic photothermal conversion substance is added to the heat-sensitive layer in a range of preferably up to 30 wt%, more preferably 5 to 25 wt%, and even more preferably 7 to 20 wt%. Within this range, a good sensitivity can be obtained.
- A finely divided metal may also be used as the photothermal conversion substance in the heat-sensitive layer. Most finely divided metals are capable of photothermal conversion, and are also self-heating. Preferred examples of finely divided metals include finely divided silicon, aluminum, titanium, vanadium, chromium, manganese, iron, cobalt, nickel, copper, zinc, yttrium, zirconium, molybdenum, silver, gold, platinum, palladium, rhodium, indium, tin, tungsten, tellurium, lead, germanium, rhenium and antimony in unalloyed or alloyed form, and finely divided oxides and sulfides thereof.
- These finely divided metals are more preferably metals such as rhenium, antimony, tellurium, gold, silver, copper, germanium, lead and tin which melt at about 1,000°C or less and thus readily coalesce under the influence of heat during light irradiation, and which absorb in the infrared, visible or ultraviolet regions.
- Finely divided metals which have a relatively low melting point and also have a relatively high infrared absorbance, such as silver, gold, copper, antimony, germanium and lead, are especially preferred. The most preferred elements include silver, gold and copper.
- It is also possible to mix fine particles of a low-melting metal such as rhenium, antimony, tellurium, gold, silver, copper, germanium, lead or tin with fine particles of a self-heating metal such as titanium, chromium, iron, cobalt, nickel, tungsten or germanium, and in this way combine two or more different photothermal conversion substances. It is also advantageous to combine and use together microfragments of a type of metal which has a particularly large light absorption in this form, such as silver, platinum or palladium, with microfragments of other metals.
- These particles have a size of preferably not more than 10 µm, more preferably from 0.003 to 5 µm, and even more preferably from 0.01 to 3 µm. A good sensitivity and good resolution can be attained within this range.
- When these finely divided metals are used as the photothermal conversion substance in the practice of the invention, the amount of addition is preferably at least 10 wt%, more preferably at least 20 wt%, and even more preferably at least 30 wt%, based on the solids in the heat-sensitive layer. A high sensitivity can be obtained within this range.
- It is also possible for the photothermal conversion substance to be included within a layer adjacent to the heat-sensitive layer, and specifically an undercoat layer or the subsequently described water-soluble overcoat layer. By including the photothermal conversion substance in at least one layer from among the heat-sensitive layer, undercoat layer and overcoat layer, the infrared absorption efficiency increases, making it possible to enhance sensitivity.
- If necessary, various compounds other than those mentioned above may also be added to the heat-sensitive layer. For example, to further improve the press life, a polyfunctional monomer may be added to the heat-sensitive layer matrix. Polyfunctional monomers suitable for this purpose include those mentioned above as monomers that may be included in the microcapsules. One monomer that is especially preferred for this purpose is trimethylolpropane triacrylate.
- Dyes having a large absorption in the visible light region can be used in the heat-sensitive layer as image colorants to enable image areas and non-image areas to be easily distinguished from one another following image formation. Specific examples include Oil Yellow #101, Oil Yellow #103, Oil Pink #312, Oil Green BG, Oil Blue BOS, Oil Blue #603, Oil Black BY, Oil Black BS and Oil Black T-505 (all of which are available from Orient Chemical Industries, Ltd.); and also Victoria Pure Blue, Crystal Violet (CI 42555), Methyl Violet (CI 42535), Ethyl Violet, Rhodamine B (CI 145170B), Malachite Green (CI 42000), Methylene Blue (CI 52015), and the dyes mentioned in JP 62-293247 A. Preferred use can also be made of pigments such as phthalocyanine pigments, azo pigments, and titanium oxide. The amount of such addition is typically 0.01 to 10 wt %, based on the total solids in the heat-sensitive layer coating fluid.
- To prevent unwanted thermal polymerization of the ethylenically unsaturated compound during preparation or storage of the heat-sensitive layer-forming coating fluid, it is desirable to add a small amount of thermal polymerization inhibitor. Preferred examples of the thermal polymerization inhibitor include hydroquinone, p-methoxyphenol, di-t-butyl-p-cresol, pyrogallol, t-butylcatechol, benzoquinone, 4,4'-thiobis(3-methyl-6-t-butylphenol), 2,2'-methylenebis(4-methyl-6-t-butylphenol) and the aluminum salt of N-nitroso-N-phenylhydroxylamine. The thermal polymerization inhibitor is added in an amount of preferably about 0.01 to 5 wt%, based on the weight of the overall composition.
- If necessary, to prevent the inhibition of polymerization by oxygen, a higher fatty acid or fatty acid derivative such as behenic acid or behenamide may be added and induced to concentrate primarily at the surface of the heat-sensitive layer as the layer dries after coating. The higher fatty acid or fatty acid derivative is added in an amount of preferably about 0.1 to about 10 wt%, based on the total solids in the heat-sensitive layer.
- If necessary, the heat-sensitive layer may also contain a plasticizer to impart such properties as flexibility to the applied film. Illustrative examples of the plasticizer include polyethylene glycol, tributyl citrate, diethyl phthalate, dibutyl phthalate, dihexyl phthalate, dioctyl phthalate, tricresyl phosphate, tributyl phosphate, trioctyl phosphate and tetrahydrofurfuryl oleate.
- The heat-sensitive layer is formed by dissolving each of the above required components in a solvent to prepare a coating fluid, and applying the fluid onto the support. Illustrative, non-limiting examples of the solvent include ethylene dichloride, cyclohexanone, methyl ethyl ketone, methanol, ethanol, propanol, ethylene glycol monomethyl ether, 1-methoxy-2-propanol, 2-methoxyethyl acetate, 1-methoxy-2-propyl acetate, dimethoxyethane, methyl lactate, ethyl lactate, N,N-dimethylacetamide, N,N-dimethylformamide, tetramethylurea, N-methylpyrrolidone, dimethylsulfoxide, sulfolane, γ-butyrolactone, toluene and water. These solvents may be used singly or as mixtures thereof. The coating fluid has a solids concentration of preferably from 1 to 50 wt%.
- The coating weight (solids basis) of the heat-sensitive layer obtained on the support after application and drying of the coating fluid will vary depending on the intended application, although a weight of 0.5 to 5.0 g/m2 is generally preferred. A coating weight below this range will result in a large apparent sensitivity, but diminish the film properties of the heat-sensitive layer whose function is to record an image. Any of various coating methods may be used. Examples of suitable methods include bar coating, spin coating, spray coating, curtain coating, dip coating, air knife coating, blade coating and roll coating.
- A surfactant may be added to the heat-sensitive layer coating fluid to improve the coating properties. For example, fluorosurfactants mentioned in JP 62-170950 A may be added for this purpose. The amount of addition is preferably 0.01 to 1 wt%, and more preferably 0.05 to 0.5 wt%, based on the total solids in the heat-sensitive layer.
- In the presensitized plate of the invention, a water-soluble overcoat layer can be provided on the heat-sensitive layer to protect the surface of the heat-sensitive layer from contamination by oleophilic substances. The water-soluble overcoat layer used in the invention can be easily removed during printing, and includes a resin selected from among water-soluble organic polymeric compounds.
- The water-soluble organic polymeric compound is a substance which, when applied as a coat and dried, has film formability. Specific examples include polyvinyl acetate having a degree of hydrolysis of at least 65%, polyacrylic acids and alkali metal salts or amine salts thereof, polyacrylic acid copolymers and alkali metal salts or amine salts thereof, polymethacrylic acids and alkali metal salts or amine salts thereof, polymethacrylic acid copolymers and alkali metal salts or amine salts thereof, polyacrylamides and copolymers thereof, polyhydroxyethyl acrylates, polyvinylpyrrolidone and copolymers thereof, polyvinyl methyl ethers, vinyl methyl ether/maleic anhydride copolymers, poly(2-acrylamido-2-methyl-1-propanesulfonic acid) and alkali metal salts or amine salts thereof, poly(2-acrylamido-2-methyl-1-propanesulfonic acid) copolymers and alkali metal salts or amine salts thereof, gum arabic, cellulose derivatives (e.g., carboxymethyl cellulose, carboxyethyl cellulose, methyl cellulose) and modified forms thereof, white dextrin, pullulan and enzyme-degraded etherified dextrin. If necessary, two or more of these may be mixed and used together.
- The overcoat layer may also have added to it the above-described water-soluble photothermal conversion substance. Moreover, when the coating fluid used to form the overcoat layer is an aqueous solution, a nonionic surfactant such as polyoxyethylene nonyl phenyl ether or polyoxyethylene dodecyl ether may be added to the overcoat layer to ensure uniformity of application.
- The overcoat layer has a coating weight when dry of preferably 0.1 to 2.0 g/m2. A weight within this range can provide good protection of the heat-sensitive layer surface from contamination by oleophilic substances, such as fingerprint contamination, without compromising the on-machine developability of the presensitized plate.
- In the presensitized plate of the invention, it is also possible to use a recording layer other than the above-described heat-sensitive layer containing (a) a thermally reactive functional group-bearing finely divided polymer or (b) microcapsules containing a thermally reactive functional group-bearing compound. Illustrative examples include photosensitive layers which use a negative-working infrared laser recording material, photosensitive layers which use a positive-working infrared laser recording material, and photosensitive layers which use a sulfonate-type infrared laser recording material.
- If the presensitized plate of the invention is a negative-working presensitized plate designed for exposure to an infrared laser, that is, a negative-working thermal presensitized plate, the heat-sensitive layer may be provided by using a negative-working infrared laser recording material.
- The negative-working infrared laser recording material is preferably a composition made up of (A) a compound which decomposes under the effect of light or heat to generate an acid, (B) a crosslinking agent which induces crosslinking under the effect of an acid, (C) an alkali-soluble resin, (D) an infrared absorber and (E) a compound of the general formula (R3-X)n-Ar-(OH)m (wherein R3 is an alkyl or alkenyl of 6 to 32 carbons; X represents a single bond, oxygen, sulfur, COO or CONH; Ar is an aromatic hydrocarbon group, an aliphatic hydrocarbon group or a heterocyclic group; and the letters m and n are each independently integers from 1 to 3).
- In general, negative-working thermal presensitized plates, after being developed, are easily marred by fingerprints and have a low strength in image areas. However, these drawbacks can be overcome by forming a photosensitive layer of the above composition.
- The compound (A) which decomposes under the effect of light or heat to generate an acid is exemplified by compounds which undergo photodecomposition to form a sulfonic acid, such as the iminosulfonates mentioned in Japanese Patent Application No. 3-140109 (JP 4-365048 A), and compounds which form an acid under irradiation at a wavelength of 200 to 500 nm or under heating at a temperature of at least 100°C.
- Preferred acid generators include photocationic polymerization initiators, photoradical polymerization initiators, dye photobleaching agents and photochromogenic substances. These acid generators are preferably added in an amount of 0.01 to 50 wt%, based on the total solids in the recording material.
- Preferred examples of the crosslinking agent (B) which crosslinks under the effect of an acid include (i) aromatic compounds substituted with an alkoxymethyl or hydroxyl group, (ii) compounds having an N-hydroxymethyl, N-alkoxymethyl or N-acyloxymethyl group, and (iii) epoxy compounds.
- Illustrative examples of the alkali-soluble resin (C) include novolak resins and polymers having pendant hydroxyaryl groups.
- Illustrative examples of the infrared absorber (D) include commercial dyes (e.g., azo dyes, anthraquinone dyes, phthalocyanine dyes) which effectively absorb infrared light at a wavelength of 760 to 1200 nm; and the black pigments, red pigments, metal powder pigments, phthalocyanine pigments mentioned in the Colour Index. Also, the addition of image colorants such as Oil Yellow and Oil Blue #603 is desirable for improving the visibility of the image. Plasticizers such as polyethylene glycol and phthalic acid esters can be added to improve the flexibility of the photosensitive layer-forming film.
- In cases where the presensitized plate of the invention is to be a positive-working presensitized plate intended for exposure with an infrared laser, i.e., a positive thermal presensitized plate, it is advantageous to provide a photosensitive layer composed of a positive-working infrared laser recording material.
- Positive-working infrared laser recording materials suitable for use include those composed of (A) an alkali-soluble polymer, (B) a compound which is compatible with the alkali-soluble polymer and lowers the alkali solubility, and (C) an infrared laser-absorbing compound.
- By using this positive-working infrared laser recording material, the problem of insufficient solubility of non-image areas in the alkali developer can be resolved, and presensitized plates which are scuff resistant, have an excellent resistance to alkali development in image areas and have a good development stability can be obtained.
- Illustrative examples of the alkali-soluble polymer (A) include (i) polymeric compounds having phenolic hydroxyl groups, such as phenolic resins, cresol resins, novolak resins and pyrogallol resins; (ii) compounds obtained by subjecting sulfonamide group-bearing monomers to homopolymerization or to copolymerization with other polymerizable monomers; and (iii) compounds having active imide groups, such as N-(p-toluenesulfonyl) methacrylamide and N-(p-toluenesulfonyl) acrylamide.
- Illustrative examples of the compound (B) which is compatible with the alkali-soluble polymer (A) and lowers the alkali solubility include compounds that interact with above component (A), such as sulfone compounds, ammonium salts, sulfonium salts and amide compounds. For example, when above component (A) is a novolak resin, a cyanine dye can be suitably used as component (B).
- The compound (C) which absorbs infrared laser light is preferably a material which absorbs in the infrared range of 750 to 1200 nm and is capable of photothermal conversion. Compounds having such an ability include squarylium dyes, pyrylium dyes, carbon black, insoluble azo dyes and anthraquinone dyes. These preferably have a particle size in a range of 0.01 to 10 µm.
- The positive thermal presensitized plate can be obtained by dissolving this positive-working infrared laser recording material in an organic solvent such as methanol or methyl ethyl ketone, adding a dye if necessary, then applying the coating fluid onto the support to a weight when dry of 1 to 3 g/m2, and drying.
- A sulfonate-type infrared laser recording material may be used as the recording layer on the inventive presensitized plate.
- Sulfonate-type infrared laser recording materials that may be used include the sulfonate compounds mentioned in JP 270480 B and JP 2704872 B. Use can also be made of photosensitive materials which generate a sulfonic acid under the effect of heat generated by infrared laser irradiation, and which solubilize in water; photosensitive materials in which a styrenesulfonic acid ester is solidified with a sol-gel, following which the surface polarity is changed by irradiation with an infrared laser; and the photosensitive materials which are mentioned in Japanese Patent Application No. 9-89816 (JP 10-282646 A), Japanese Patent Application No. 10-22406 (JP 11-218928 A) and Japanese Patent Application No. 10-027655 (JP 10-282672 A), and in which the hydrophobic surface is rendered hydrophilic by exposure using a laser.
- To further improve the properties of the photosensitive layer composed of a polymeric compound capable of generating a sulfonic acid group under the influence of heat, the accompanying use of the following methods is desirable. Illustrative examples include (1) the method described in Japanese Patent Application No. 10-7062 (JP 11-202483 A) which involves use together with an acid or base generator, (2) the method described in Japanese Patent Application No. 9-340358 (JP 11-174685 A) which involves providing a specific intermediate layer, (3) the method described in Japanese Patent Application No. 9-248994 (JP 11-84658 A) which involves the concomitant use of a specific crosslinking agent, and (4) the method described in Japanese Patent Application No. 10-115354 (JP 11-301131 A) which involves use in a solid particle surface-modified form.
- Other examples of compositions which utilize heat generated by exposure to a laser to effect a hydrophilic/hydrophobic change in a photosensitive layer include the compositions mentioned in US 2,764,085 which include a Werner complex and become hydrophobic under the influence of heat; the compositions described in JP 46-27219 B which include specific sugars and a melamine-formaldehyde resin, and which become hydrophilic under exposure to light; the compositions described in JP 51-63704 A which become hydrophobic under heat-mode exposure; the compositions described in US 4,081,572 which undergo dehydration/hydrophobization under heat, in the manner of phthalyl hydrazide polymers; the compositions described in JP 3-58100 B which have a tetrazolium salt structure and become hydrophilic under the effect of heat; the compositions described in JP 60-132760 A which are composed of a sulfonic acid-modified polymer and become hydrophobic under exposure to light; the compositions described in JP 64-3543 A which are composed of an imide precursor polymer and become hydrophobic under exposure to light; and the compositions described in JP 51-74706 A which are composed of a fluorocarbon polymer and become hydrophilic under exposure to light. The recording layer can be formed using any these compositions.
- Additional examples of compositions which may be used to form the recording layer include the compositions described in JP 3-197190 A which include a hydrophobic crystalline polymer and which become hydrophilic under exposure to light; the compositions described in JP 7-186562 A which include both a polymer with insolubilized pendant groups that become hydrophilic under exposure to heat and a photothermal conversion substance; the compositions described in JP 7-1849 A which includes a microcapsule-containing three dimensionally-crosslinked hydrophilic binder and becomes hydrophobic under exposure to light; the compositions described in JP 8-3463 A which undergo valence isomerization or proton transfer isomerization; the compositions described in JP 8-141819 A which give rise to a change in the phase structure (compatibilization) within the layer under the effect of heat, and thus effect a hydrophilic/hydrophobic change; and the compositions described in JP 60-228 B in which the surface shape and the hydrophilicity/hydrophobicity of the surface change under the effect of heat. The recording layer can be formed using any these compositions.
- Additional examples of preferred recording materials which may be used in the recording layer when working the invention include compositions which, under so-called heat-mode exposure utilizing heat generated by high-power, high-density laser light, change the adhesive properties between the photosensitive layer and the support. These compositions include the compositions described in JP 44-22957 B which are composed of a heat-fusible substance or a thermally reactive substance.
- The process and equipment used to manufacture a support for lithographic printing plates according to a preferred embodiment of the invention are described below.
- The process of manufacturing an aluminum support for lithographic printing plates from an aluminum sheet, which is a preferred embodiment of the inventive support for lithographic printing plates, preferably includes the following steps: (1) playing out a rolled, coil-wound aluminum sheet from a delivery unit composed of a multiple spindle turret; (2) subjecting the aluminum sheet to each of the above-described treatments (mechanical graining, electrochemical graining, alkali etching treatment, acidic etching treatment, desmutting, anodizing treatment, pore widening treatment (acid or alkali treatment), treatment with an aqueous solution containing an inorganic fluorine compound and a phosphate compound, and, before or after such aqueous solution treatment, treatment with a silicate compound-containing aqueous solution), then forming on the treated aluminum sheet an intermediate layer containing an acid group-bearing polymeric compound and subjecting the aluminum sheet to drying treatment; and (3) coil-winding the treated aluminum sheet on a rewinder composed of the above multiple spindle turret or correcting the flatness of the aluminum sheet, then cutting it to a predetermined length and collecting together the cut pieces. If necessary, the above process may additionally include steps in which an undercoat layer and a recording layer are formed and subjected to drying treatment, thus rendering the aluminum sheet into a presensitized plate, in which form it may be rewound into a coil on the above rewinder.
- It is preferable for the manufacture of the aluminum support to include at least one step in which a device which inspects the surface of the aluminum sheet for defects continuously inspects the sheet and attaches a label at the edge of the sheet whenever a defect is found to mark its position. Also, it is desirable to provide a reserving unit for keeping the traveling speed by the aluminum sheet constant in each of the above steps when travel by the aluminum sheet is halted to change the aluminum coil in the aluminum sheet unwinding and rewinding steps during manufacture of the inventive presensitized plate. For the same reason, it is desirable to include, after the aluminum unwinding step, a step in which successive aluminum sheets are joined by ultrasonic or arc welding.
- The equipment used in the manufacture of the aluminum support preferably includes at least one unit to detect a traveling position on the aluminum sheet and correct the traveling position, at least one drive unit for cutting tension on the aluminum sheet and controlling the traveling speed, and at least one dancer roll unit for controlling tension.
- It is also desirable, by means of a tracking unit, to record whether the state of the sheet in each step agrees with the desired conditions and to affix labels to the edge of the aluminum web before the aluminum coil is rewound so as to make it possible to discern later on whether a portion of the web after the label meets the desired conditions.
- In the practice of the invention, it is preferable for the aluminum sheets to be stacked and held together electrostatically with a slip sheet therebetween, then cut and/or slit to a given length. Moreover, after the sheets have been cut to the predetermined length, or even before such cutting, it is desirable to distinguish non-defective areas from defective areas on the basis of information appearing on the labels affixed to the edge of the aluminum sheet, and to collect only non-defective areas.
- In each step, including the above-described coil unwinding step, it is important to set the aluminum sheet to the optimal tension for the respective conditions, based on the size of the aluminum sheet (thickness and width), the aluminum material and the traveling speed of the aluminum web. To this end, it is desirable to utilize a drive unit for cutting tension and controlling the traveling speed and a dancer roll for controlling tension, and provide a plurality of tension controlling units which carry out feedback control using signals from a tension sensing unit. The drive unit generally uses a control method which combines a DC motor and a main drive roller. The main drive roller is made of ordinary rubber, although rollers made by laminating nonwoven fabric can be used in steps where the aluminum web is in a wet state. Each pass roller is generally made of rubber or metal. However, auxiliary drive units can be provided to prevent slippage by the aluminum web in places where slippage tends to occur. For example, a motor and a speed reducer may be connected to each pass roller, and rotational control carried out at a constant speed based on signals from the main drive unit.
- Letting the computed average roughness (Ra) in the rolling direction be R1 and the computed average roughness (Ra) in the width direction be R2 as described in JP 10-114046 A, the aluminum support used in the invention preferably has a value R1-R2 within 30% of R1, and also preferably has an average curvature in the rolling direction of not more than 1.5x10-3 mm-1, an average curvature in the width direction of not more than 1.5x10-3 mm-1, and an average curvature in the direction perpendicular to the rolling direction of not more than 1.0×10-3mm-1.
- The aluminum support produced by administering the above-described graining treatment is preferably corrected using a correcting roll having a diameter of 20 to 80 mm and a rubber hardness of 50 to 95. This makes it possible to supply an aluminum coiled sheet of a flatness which, even in an automated conveying step in a lithographic photosensitive printing press, keeps deviations in exposure from arising on the presensitized plate. JP 9-194093 A describes a method and apparatus for measuring curl in a web, a method and apparatus for correcting curl, and an apparatus for cutting the web. These can be used in the present invention as well.
- In the continuous fabrication of the aluminum support, each step is electrically monitored to determine whether it is operating under the proper conditions, a tracking unit records whether the state in each step agrees with the desired conditions and, before rewinding the aluminum web into a coil, labels are affixed to the edge of the web to make it possible later on to tell from the labels whether a portion of the web meets the desired conditions, thus enabling a decision to be made concerning the acceptability of that portion at the time of cutting and collection.
- The aluminum sheet treatment apparatuses used in the above-described graining treatments preferably determine the liquid composition by measuring one or more parameters from among the liquid temperature, specific gravity, electrical conductivity and ultrasonic wave propagation velocity, then carry out feedback control and/or feed-forward control to keep the liquid concentration constant.
- Aluminum ions and other components present in the aluminum sheet are dissolved in the acidic aqueous solution within the treatment apparatus as surface treatment of the aluminum sheet proceeds. To keep the aluminum ion concentration and the acid or alkali concentration constant, it is preferable to hold the liquid to a fixed composition by intermittently adding water and acid or water and alkali. The concentration of the acid or alkali added here is preferably from 10 to 98 wt%.
- The following method is preferred for controlling the acid or alkali concentration.
- First, the electrical conductivity, specific gravity or ultrasonic wave propagation velocity of each component liquid in the concentration range for which use is anticipated is measured at various temperatures and a data table is prepared. Next, the concentration is measured by referring to the data table containing temperature data and conductivity, specific gravity or ultrasonic wave propagation velocity data for the solution being measured. JP 6-235721 A describes a method for precisely and stably measuring the ultrasonic wave propagation time. JP 58-77656 A describes a concentration measurement system that utilizes the above ultrasonic wave propagation velocity. JP 4-19559 A describes a method which uses data on a plurality of physical quantities to construct a data table showing the correlations between liquid components, and measures the component concentrations in a multi-component solution by referring to this data table.
- When the above method of measuring concentration using the ultrasonic wave propagation velocity is combined with the conductivity and temperature values for the solution being measured and employed in the aluminum support graining operation, process control can be accurately carried out in real time, thus making it possible to manufacture a product of uniform quality and resulting in an improved yield. Similar effects can be obtained by employing in the aluminum support graining treatment operation a method which involves preparing data tables, not only for a combination of temperature, ultrasonic wave propagation velocity and electrical conductivity, but by concentration and temperature for the respective physical quantities, including temperature and specific gravity, temperature and electrical conductivity, and temperature, conductivity and specific gravity; then referring to these data tables to measure the component concentrations in the multi-component solution.
- By measuring the specific gravity and temperature, then referring to the prepared data tables so as to determine the slurry concentrations of the substances to be measured, it is possible to rapidly and accurately measure the slurry concentrations.
- Because measurement of the ultrasonic wave propagation velocity is readily affected by bubbles in the solution, it is preferable to carry out such measurement in a pipeline that is vertically disposed and has a flow velocity directed from below to above. Measurement of the ultrasonic wave propagation velocity is preferably carried out in a pressure range within the pipeline of 1 to 10 kg/cm2, and the ultrasonic waves have a frequency of preferably 0.5 to 3 MHz.
- Measurement of the specific gravity, conductivity and ultrasonic wave propagation velocity is temperature sensitive, and so it is preferable to carry out these measurements in a temperature-insulated state and within a line where the temperature fluctuation is controlled to within ±0.3°C. Moreover, because it is preferable to measure the conductivity and specific gravity, or the conductivity and the ultrasonic wave propagation velocity at the same temperature, it is particularly desirable to carry out the measurements within the same pipeline or within the same pipeline flow. Pressure fluctuations during measurement lead to temperature fluctuations, and should therefore be minimized. It is also desirable to minimize the flow rate distribution within the pipeline used for measurement. Finally, because the above measurements are readily affected by slurry, dirt and bubbles, it is preferable to measure the liquid after passing it through a filter, deaerator and the like.
- An image is recorded on the resulting inventive presensitized plate by means of heat. Preferred methods for doing so include direct imagewise recording such as with a thermal recording head, scanning-type exposure using an infrared laser, high-intensity flash-type exposure such as with a xenon discharge lamp, or exposure with a solid high-output infrared laser using an infrared lamp.
- If the recording layer on the presensitized plate of the invention is an on-machine development type heat-sensitive layer containing (a) a finely divided polymer having thermally reactive functional groups or (b) microcapsules containing a compound having thermally reactive functional groups, following imagewise exposure, the plate can be mounted without further treatment on the printing press and printing carried out by an ordinary procedure using ink and/or dampening water. Moreover, as mentioned in JP 2938398 B, after the plate has been mounted on the plate cylinder of the printing press, it can be exposed using a laser mounted on the press, following which ink and/or dampening water can be applied and on-machine development carried out. In such cases, the heat-sensitive layer is removed on the press with ink and/or dampening water, and so there is no need for a separate development operation. Moreover, once development is over, printing can begin without stopping the press; that is, printing can be carried out immediately without interruption once development is complete.
- Platemaking and printing methods for the lithographic printing plate of the invention are characterized by subjecting a presensitized plate provided with an on-machine development type heat-sensitive layer to imagewise exposure with laser light, then mounting the exposed plate on the press and printing, or by mounting the plate on the press then subjecting it to imagewise exposure using laser light and directly carrying out printing in this state. A solid laser or semiconductor laser which emits infrared light at a wavelength of 760 to 1200 nm can be used.
- Even a plate having an on-machine development-type heat-sensitive layer can be used in printing after it has been developed with water or a suitable aqueous solution as the developer.
- In cases where the presensitized plate of the invention has a prior-art positive-working or negative-working thermal recording layer, it is subjected to imagewise exposure, then developed with a developer according to a conventional process, and subsequently mounted on the press and furnished for printing.
- Illustrative examples of sources of actinic light that may be used for imagewise exposure include mercury vapor lamps, metal halide lamps, xenon lamps and chemical lamps. Examples of laser beams that may be used include helium-neon lasers, argon lasers, krypton lasers, helium-cadmium lasers, KrF excimer lasers, semiconductor lasers, YAG lasers and YAG-SHG lasers.
- Following exposure as described above, it is preferable to carry out development using a liquid developer in order to obtain the lithographic printing plate. The liquid developer is preferably an alkali developer, and more preferably an alkaline aqueous solution which is substantially free of organic solvent. Liquid developers which are substantially free of alkali metal silicates are also preferred. One example of a suitable method of development using a liquid developer that is substantially free of alkali metal silicates is the method described in detail in JP 11-109637 A. Liquid developers containing alkali metal silicates can also be used.
- Examples are given below by way of illustration and not by way of limitation.
- The aluminum sheets used in the examples of the invention and the comparative examples were produced as follows. A melt was prepared using an aluminum alloy composed of 0.073 wt% silicon, 0.270 wt% iron, 0.028 wt% copper, 0.001 wt% manganese, 0.000 wt% magnesium, 0.001 wt% chromium, 0.003 wt% zinc and 0.020 wt% titanium with the balance being aluminum and inadvertent impurities.
- The aluminum alloy melt was subjected to molten metal treatment consisting of degassing and filtration, then was cast into a 500 mm thick ingot by a direct chill casting process. The ingot was faced, removing 10 mm of material from the surface. The faced ingot was then heated; hot rolling was begun at 400°C without carrying out soaking treatment, and continued to a thickness of 4 mm. The ingot was then cold rolled to a thickness of 1.5 mm and intermediate annealed, following which it was again cold rolled, this time to a final thickness of 0.24 mm, after which the flatness was corrected, giving an aluminum sheet. Lithographic Printing Plate Support 1:
- The aluminum sheet obtained as described above, was subjected to surface treatment in the sequence (1) to (13) shown below, giving a lithographic printing plate support 1.
- Following each surface treatment and rinsing with water, the water was drained from the sheet with a nip roller. Rinsing was carried out by spraying with water from a spray line.
- Mechanical graining treatment was carried out using a brush roller having rotating nylon brushes while using a spray line to feed an abrasive slurry consisting of a suspension of silica sand as the abrasive (specific gravity, 1.12; average particle size, 25 µm) in water to the surface of the aluminum sheet.
- The nylon brush material used was composed of 6,10-nylon, had a bristle length of 50 mm and a bristle diameter of 0.48 mm. The nylon brushes were 300 mm diameter stainless steel cylinders in which holes had been formed and bristles densely set therein.
- The brush roller used three nylon brushes and also had two support rollers (200 mm diameter) provided below the brush and spaced 300 mm apart.
- The brush roller controlled the load of the driving motor that rotates the nylon brush relative to the load before the brush is pushed against the aluminum sheet, and pushed the brush against the aluminum sheet such as to give the sheet after graining an average calculated roughness (Ra) of 0.45 µm. The direction of brush rotation was the same as the direction of movement by the aluminum sheet. Rinsing was subsequently carried out.
- The concentration of the abrasive was determined from the temperature and specific gravity by referring to a table prepared beforehand based on the relationship between the abrasive concentration, temperature and specific gravity, and water and abrasive were added under feedback control, thereby holding the concentration of the abrasive constant. When the abrasive breaks down to a smaller particle size, the surface shape of the grained aluminum sheet changes. Abrasive having a small particle size was thus successively removed from the system with a cyclone. The particle size of the abrasive was in a range of 1 to 35 µm.
- An aqueous solution containing 27 wt% of NaOH and 6.5 wt% of aluminum ions and having a temperature of 70°C was sprayed onto the aluminum sheet, thereby carrying out alkali etching treatment. The loss of weight from dissolution on the side of the aluminum sheet to be subsequently electrochemically grained was 8 g/m2, and the loss of weight from dissolution on the back side was 2 g/m2.
- The concentration of the etching solution used for alkali etching treatment was determined from the temperature, specific gravity and electrical conductivity by referring to a table prepared beforehand based on the relationship between the NaOH concentration, aluminum ion concentration, temperature, specific gravity and liquid conductivity, and was held constant under feedback control by adding water and 48 wt% aqueous NaOH. Thereafter, the aluminum sheet was rinsed off with water.
- Aqueous nitric acid having a liquid temperature of 35°C was sprayed onto the aluminum sheet and desmutting was carried out for 10 seconds. Overflow wastewater from the electrolytic cell apparatus employed in the next step was used as the aqueous nitric acid. Next, spray lines for spraying the desmutting liquid were positioned in several places, and the surface of the aluminum sheet was keep from drying until the next operation.
- Electrochemical graining treatment was carried out continuously using alternating current having the trapezoidal waveform shown in FIG. 1 and the two electrolytic cells shown in FIG. 2. An aqueous solution containing 1 wt% of nitric acid (and containing also 0.5 wt% of aluminum ions and 0.007 wt% of ammonium ions) was used as the acidic aqueous solution. The liquid temperature was 35°C. The alternating current had respective times tp and tp' until the current value reached a peak on the cathode cycle side and the anode cycle side, respectively, of 1 msec each. A carbon electrode was used as the counterelectrode. The alternating current had a current density at the peak, both when the aluminum sheet was the anode and when it was the cathode, of 50 A/dm2. The ratio QC/QA between the amount of electricity when the aluminum sheet served as the cathode QC and the amount of electricity when the sheet served as the anode QA was 0.95. The duty ratio was 0.50, the frequency was 60 Hz, and the combined amount of electricity when the aluminum sheet served as the anode was 230 C/dm2. Following treatment, the aluminum sheet was sprayed to rinse it off.
- Control of the aqueous nitric acid solution concentration was carried out by adding a 67 wt% nitric acid stock solution and water proportional to the amount of current passed and at the same time allowing an amount of the acidic aqueous solution (nitric acid solution) equivalent to the volume of nitric acid and water being added to consecutively overflow from the electrolytic cells and discharge outside of the system. At the same time, the concentration of the nitric acid solution was determined from the temperature, conductivity and ultrasonic wave propagation velocity of the solution by referring to a table prepared beforehand based on the relationship between the nitric acid concentration, aluminum ion concentration, temperature, conductivity of the liquid and ultrasonic wave propagation velocity of the liquid, and the concentration was held constant by carrying out control involving the successive adjustment of the amounts of nitric acid stock solution and water added.
- Alkali etching treatment was carried out by spraying an aqueous solution containing 26 wt% of NaOH and 6.5 wt% of aluminum ions and having a temperature of 45°C onto the aluminum sheet. The weight dissolved from the aluminum sheet was 3 g/m2. The etching solution concentration was determined from the temperature, specific gravity and conductivity by referring to a table prepared beforehand from the relationship between NaOH concentration, aluminum ion concentration, temperature, specific gravity and solution conductivity, and was kept constant under feedback control by the addition of water and 48 wt% aqueous NaOH. Following treatment, the aluminum sheet was rinsed off with water.
- Sulfuric acid (sulfuric acid concentration, 300 g/L; aluminum ion concentration, 15 g/L) was prepared as an acidic etching solution, then sprayed onto the aluminum sheet at 80°C for 7 seconds to carry out acidic etching treatment. The acidic etching solution concentration was determined from the temperature, specific gravity and conductivity by referring to a table prepared beforehand based on the relationship between the sulfuric acid concentration, aluminum ion concentration, temperature, specific gravity and solution conductivity, and was kept constant under feedback control by the addition of water and 50 wt% sulfuric acid. Following treatment, the aluminum sheet was rinsed off with water.
- Aside from using as the acidic aqueous solution an aqueous solution of 0.5 wt% hydrochloric acid (containing also 0.5 wt% of ammonium ions) and setting the overall amount of electricity when the aluminum sheet serves as the anode to 50 C/dm2, electrochemical graining treatment (II) was carried out in the same way as in (4) Electrochemical Graining Treatment (I) described above.
- Control of the aqueous hydrochloric acid solution concentration was carried out by adding a 30 wt% hydrochloric acid stock solution and water proportional to the amount of current passed and concurrently allowing an amount of the acidic aqueous solution (hydrochloric acid solution) equivalent to the volume of hydrochloric acid and water being added to successively overflow from the electrolytic cells and discharge outside of the system. At the same time, the concentration of the hydrochloric acid solution was determined from the solution temperature, conductivity and ultrasonic wave propagation velocity by referring to a table prepared beforehand based on the relationship between the hydrochloric acid concentration, aluminum ion concentration, temperature, conductivity of the liquid and ultrasonic wave propagation velocity of the liquid, and the concentration was held constant by carrying out control involving the successive adjustment of the amounts of hydrochloric acid stock solution and water added.
- Using an aqueous solution containing 5 wt% of NaOH and 6.5 wt% of aluminum ions, alkali etching treatment (III) was carried out in the same way as in (5) Alkali Etching Treatment (II) described above. The weight dissolved from the aluminum sheet was set at 0.2 g/m2.
- Acidic etching treatment (II) was carried out in the same way as in (6) Acidic Etching Treatment (I) described above.
- Anodizing treatment of the aluminum sheet was carried out at a current density of 25 A/dm2 and a temperature of 50°C for 30 seconds using an aqueous solution having a sulfuric acid concentration of 100 g/L (and containing 0.5 wt% of aluminum ions) as the anodizing solution, thereby forming an anodized layer. The concentration of the anodizing solution was determined from the temperature, specific gravity and conductivity by referring to a table prepared beforehand based on the relationship between the sulfuric acid concentration, aluminum ion concentration, temperature, specific gravity and conductivity of the solution, and the concentration was held constant by adding water and 50 wt% sulfuric acid under feedback control. Following treatment, the aluminum sheet was sprayed to rinse it off.
- Pore widening treatment was carried out by dipping the anodized aluminum sheet for 1 minute in an aqueous NaOH solution having a pH of 13 and a temperature of 50°C. Following treatment, the aluminum sheet was rinsed with water.
- Following pore widening treatment, the aluminum sheet was treated by immersion under the temperature and time conditions shown in Table 2 in respective aqueous solutions prepared using pure water to a sodium fluoride (NaF) concentration of 0.1 wt% and to a sodium dihydrogenphosphate (NaH2PO4) concentration of 10 wt%. The aluminum sheet was subsequently rinsed with water.
- Following above treatment (i), the aluminum sheet was dipped for 10 seconds and at 20°C in an aqueous solution having a No. 3 sodium silicate concentration of 1.0 wt% for the hydrophilic surface treatment. The aluminum sheet was subsequently rinsed with water and dried.
- An intermediate layer coating fluid of the composition indicated below and containing an acid group-bearing polymeric compound was applied, then dried at 80°C for 15 seconds. The coating weight after drying of the intermediate layer was 15 mg/m2.
Composition of intermediate layer coating fluid containing acid group-bearing polymeric compound: - Polymeric compound No. 2 in above Table 1:
"Typical Examples of Polymeric Compounds" 0.1 g Methanol 100 g Water 1 g - Lithographic Printing Plate Support 2:
Aside from first carrying out above treatment (12)
(ii) following above pore widening treatment (11), then
carrying out above treatment (12) (i) under the conditions
(temperature, time) shown in Table 2 below, a lithographic
printing plate support 2 was obtained by the same method as
that used to obtain lithographic printing plate support 1.
Lithographic Printing Plate Support 3:
- Aside from changing the conditions for above
treatment (12) (i) to the conditions (concentration,
temperature, time) shown in Table 2 below, a lithographic
printing plate support 3 was obtained by the same method as
that used to obtain lithographic printing plate support 1.
Lithographic Printing Plate Support 4:
- Aside from first carrying out above treatment (12) (ii) following above pore widening treatment (11), then carrying out above treatment (12) (i) under the conditions shown in Table 2 below, a lithographic printing plate support 4 was obtained by the same method as that used to obtain lithographic printing plate support 1.
- Lithographic Printing Plate Support 5: Aside from changing the conditions for above treatment (12) (i) to the conditions shown in Table 2 below, not carrying out above treatment (12) (ii), and carrying out above treatment (13) using an intermediate layer coating fluid containing 0.1 g each of the monomers triethanolamine and β-alanine instead of a polymeric compound, a lithographic printing plate support 5 was obtained by the same method as that used to obtain lithographic printing plate support 1.
- Lithographic Printing Plate Support 6: Aside from carrying out above treatment (12) (ii) under the same conditions, but not carrying out above treatment (12) (i), a lithographic printing plate support 6 was obtained by the same method as that used to obtain lithographic printing plate support 1.
- Lithographic Printing Plate Support 7: Aside from changing the conditions for above treatment (12) (i) to the conditions shown in Table 2 below, subsequently carrying out above treatment (12) (ii), and carrying out above treatment (13) using an intermediate layer coating fluid containing 0.1 g each of the monomers triethanolamine and β-alanine instead of a polymeric compound, a lithographic printing plate support 7 was obtained by the same method as that used to obtain lithographic printing plate support 1.
- Lithographic Printing Plate Support 8: Aside from changing the conditions for above treatment (12) (i) to the conditions shown in Table 2 below and subsequently carrying out above treatment (12) (ii), but not carrying out above treatment (13) to form an intermediate layer and thus not providing an intermediate layer, a lithographic printing plate support 8 was obtained by the same method as that used to obtain lithographic printing plate support 1.
-
- Electron spectroscopy for chemical analysis (ESCA) was carried out on the lithographic printing plate supports 1 to 8 obtained above. The ESCA conditions were as follows.
Apparatus PHI-5400 MC ESCA spectrometer (manufactured by Ulvac-Phi, Inc.) X-ray source Mg-Kα (400 W) Pulse energy 71.55 eV/178.95 eV Take off angle 45° - The value (A+B+C)/(A+B+C+D) in above formula (1) was calculated from the resulting peak areas for fluorine (1s), silicon (2p), phosphorus (2p) and aluminum (2p). The peak area values and the calculated results are shown in Table 2.
- Allyl methacrylate (7.5 g), butyl methacrylate (7.5 g) and 200 mL of an aqueous solution of polyoxyethylene nonyl phenol (concentration, 9.84x10-3 mol/L) were added and stirred at 250 rpm while at the same time flushing the system with nitrogen. The resulting liquid was set at 25°C, following which 10 mL of an aqueous solution of ammonium cerium(IV) salt (concentration, 0.984x10-3 mol/L) was added. An aqueous solution of ammonium nitrate (concentration, 58.8 x10-3 mol/L) was added at this time to adjust the pH to 1.3 to 1.4. The system was then stirred for 8 hours, giving a liquid containing finely divided polymer. The resulting liquid had a solids concentration of 9.5%, and the average particle size of the finely divided polymer was 0.2 µm.
- An oil phase component was prepared by dissolving the following in 60 g of ethyl acetate: 40 g of xylene diisocyanate, 10 g of trimethylolpropane diacrylate, 10 g of a copolymer of allyl methacrylate and butyl methacrylate (molar ratio, 7/3), and 0.1 g of surfactant (Pionin A41C, available from Takemoto Oil & Fat Co., Ltd.). In a separate procedure, 120 g of a 4% aqueous solution of polyvinyl alcohol (PVA 205, available from Kuraray Co., Ltd.) was prepared as an aqueous phase component. The oil phase component and the aqueous phase component were placed in a homogenizer and emulsified at 10,000 rpm. This was followed by the addition of 40 g of water and stirring, first at room temperature for 30 minutes, then at 40°C for 3 hours, thereby giving a microcapsule liquid. The resulting microcapsule liquid had a solids concentration of 20 wt% and an average microcapsule size of 0.2 µm.
- Heat-sensitive layer (1) was a heat-sensitive layer containing a finely divided polymer, and was formed as follows.
- A coating fluid for heat-sensitive layer (1) of the composition indicated below was applied to a coating weight when dry of 0.5 g/m2 to the respective lithographic printing plate supports 1 and 5 obtained above, then dried in an oven at 60°C for 150 seconds, giving presensitized plates in Example 1 and Comparative Example 1.
-
Liquid containing finely divided polymer synthesized above 5 g (solids) Polyhydroxyethyl acrylate (weight-average molecular weight, 25,000) 0.5 g Photothermal conversion substance (above structural formula IR-11) 0.3 g Water 100 g - Heat-sensitive layer (2) was a microcapsule-containing heat-sensitive layer, and was formed as follows.
- A coating fluid for heat-sensitive layer (2) of the composition indicated below was applied to the respective lithographic printing plate supports 2 and 6 obtained above to a coating weight when dry of 0.7 g/m2, then dried in an oven at 60°C for 150 seconds, giving presensitized plates in Example 2 and Comparative Example 2.
-
Microcapsule liquid prepared above 5 g (solids) Trimethylolpropane triacrylate 3 g Photothermal conversion substance (above structural formula IR-11) 0.3 g Water 60 g 1-Methoxy-2-propanol 40 g - Heat-sensitive layer (3) was a two-layer positive-working thermal image recording layer, and was formed as follows.
- A first layer-forming coating liquid of the composition indicated below was prepared and applied to the respective lithographic printing plate supports 3 and 7 obtained above to a coating weight when dry of 0.8 g/m2, then dried in an oven at 140°C for 60 seconds, thereby forming a first layer. Next, a second layer-forming coating liquid of the composition indicated below was prepared, applied to a coating weight when dry of 0.2 g/m2 to each of the above lithographic printing plate supports on which the first layer had been formed, then dried in an oven at 140°C for 50 seconds to form a second layer, thereby giving the presensitized plates having a two-layer positive-working thermal image layer in Example 3 and Comparative Example 3.
-


-


- Heat-sensitive layer (4) was a negative-working thermal image recording layer, and was formed as follows.
- A coating fluid for heat-sensitive layer (4) of the composition indicated below was applied to the respective lithographic printing plate supports 4 and 8 obtained above to a coating weight when dry (heat-sensitive layer coating weight) of 1.3 g/m2, then dried with a hot-air dryer at 122°C for 27 seconds to form a heat-sensitive layer (negative-working thermal image recording layer), giving presensitized plates in Example 4 and Comparative Example 4.
-
Infrared absorber (IR-1) 0.074 g Polymerization initiator (OS-12) 0.280 g Additive (PM-1) 0.151 g Polymerizable compound (AM-1) 1.00 g Binder polymer (BT-1) 1.00 g Ethyl violet (C-1) 0.04 g Fluorosurfactant (Megafac F-780-F, available from Dainippon Ink & Chemicals; 30 wt% solution in MIBK) 0.015 g Methyl ethyl ketone 10.4 g Methanol 4.83 g 1-Methoxy-2-propanol 10.4 g - The chemical structures for the infrared absorber (IR-1), additive (PM-1), polymerizable compound (AM-1, wherein m+n = 4 in the formula), binder polymer (BT-1) and ethyl violet (C-1) used in the above heat-sensitive layer-forming coating liquid are shown below.






- A mixed aqueous solution of polyvinyl alcohol (degree of saponification, 98 mol%; degree of polymerization, 500) and polyvinyl pyrrolidone (Luviskol K-30, available from BASF) was applied with a wiper onto the surface of the above-described heat-sensitive layer, then dried at 125°C for 75 seconds with a hot-air dryer. The polyvinyl alcohol/polyvinyl pyrrolidone content ratio was 4/1, and the coating weight (weight of coat when dry) was 2.30 g/m2.
- The on-machine developable presensitized plates obtained in Example 1 and Comparative Example 1 were exposed using a Trendsetter 3244 VFS (Creo Inc.) equipped with a water-cooled 40 W infrared semiconductor laser, at a resolution of 2,400 dpi. The plate surface energy at this time was varied in increments of 5 mJ/cm2 from 30 to 200 mJ/cm2 by varying the external drum speed, and the sensitivity was determined from the minimum dose at which image formation was possible. The results are shown in Table 2.
- The on-machine developable presensitized plates obtained in Example 2 and Comparative Example 2 were exposed using a Luxel T-9000 CTP (Fuji Photo Film Co., Ltd.) equipped with a multi-channel laser head, at a resolution of 2,400 dpi. The plate surface energy at this time was varied in increments of 5 mJ/cm2 from 40 to 200 mJ/cm2 by varying the output per beam and the external drum speed, and the sensitivity was determined from the minimum dose at which image formation was possible. The results are shown in Table 2.
- The presensitized plates obtained in Example 3 and Comparative Example 3 were imagewise exposed at a primary scanning speed of 5 m/s and a surface energy of 140 mJ/cm2 using a Trendsetter 3244 (Creo Inc.) equipped with a semiconductor laser having an output of 500 mW, a wavelength of 830 nm and a beam diameter of 17 µm (1/e2). To evaluate the sensitivity, samples were prepared by carrying out exposure at surface energies which were varied in increments of 5 mJ/cm2 from 20 to 150 mJ/cm2.
- Development was carried out at a temperature of 30°C for 12 seconds using a PS900NP automated processor (manufactured by Fuji Photo Film Co., Ltd.) filled with the developer DT-2 (1:8) (Fuji Photo Film Co., Ltd.). After development was completed, the developed plate was rinsed with water, then treated with a gum (FG-1, 1:1), thereby giving a completed lithographic printing plate. The minimum exposure dose at which image formation could be carried out following development was determined as the sensitivity from samples obtained at varying surface energy levels.
- The presensitized plates obtained in Example 4 and Comparative Example 4 were exposed using a Trendsetter 3244 VFS (Creo Inc.) equipped with a water-cooled 40 W infrared semiconductor laser (wavelength, 830 nm) at an output of 9 W, an external drum speed of 150 rpm, a plate surface energy of 100 mJ/cm2 and a resolution of 175 dpi. To evaluate the sensitivity, samples were prepared by carrying out exposure at surface energies which were varied in increments of 5 mJ/cm2 from 20 to 200 mJ/cm2.
- Following exposure, the protective layer was removed by rinsing with tap water, after which development was carried out at 30°C for 12 seconds using an LP-1310HII processor available from Fuji Photo Film Co., Ltd. The developer was a 1:4 aqueous dilution of DV-2C (Fuji Photo Film Co., Ltd.), and the finisher was a 1:1 aqueous dilution of GN-2K (Fuji Photo Film Co., Ltd.).
- The presensitized plates obtained in Examples 1 and 2 of the invention and in Comparative Examples 1 and 2 were exposed to light in the same way as described above under "Measurement of Sensitivity." In each case, the exposed plate was then mounted on the plate cylinder of a SOR-M printing press (Heidelberger Druckmaschinen AG) without first being subjected to development. The plate was supplied first with dampening water containing 1% ALKI and 10% isopropyl alcohol, after which Toyo Vantean Eco red ink and paper were each supplied and a printing test was carried out. In the case of the presensitized plates obtained in Examples 1 and 2, on-machine development was carried out without any difficulty and printing was also possible.
- The presensitized plates obtained in Examples 3 and 4 of the invention and in Comparative Examples 3 and 4 were subjected to exposure and development in the same way as described above under "Measurement of Sensitivity." The lithographic printing plates obtained in this way were subjected to printing tests under the conditions indicated below.
- The results are shown in Table 2.
- In the printing test, the water level in the press was adjusted and the resistance to scumming was evaluated from the water level at which scumming arises. In Table 2, a water level at which scumming arises of less than 2 was rated as "Very Good", a water level of at least 2 but less than 3 was rated as "Good", a water level of at least 3 but less than 4 was rated as "Fair", and a water level of 4 or more was rated as "Poor."
- The press life was evaluated based on the number of clear impressions obtained. A larger number of such impressions indicates a longer press life.
- It is apparent from Table 2 that the inventive presensitized plates (Examples 1 to 4) in which lithographic printing plate supports according to the invention were used, when rendered into lithographic printing plates, had an excellent sensitivity, were not readily subject to scumming and had a long press life.


Claims (5)
- A support for a lithographic printing plate, comprising:a metal base subjected to graining treatment and anodizing treatment, then treated with an aqueous solution containing an inorganic fluorine compound and a phosphate compound, which aqueous solution treatment is preceded or followed by treatment with an aqueous solution containing a silicate compound; andan intermediate layer which lies on the treated metal base and contains an acid group-bearing polymeric compound.
- The support for a lithographic printing plate according to claim 1, wherein an acid group on the acid group-bearing polymeric compound constituent is an acid group having an acid dissociation constant (pKa) not higher than 7.
- The support for a lithographic printing plate according to claim 1 or 2, wherein an acid group on the acid group-bearing polymeric compound constituent is selected from the group consisting of -COOH, -SO3H, -OSO3H, -PO3H2, -OPO3H2, -CONHSO2 and -SO2NHSO2.
- A support for a lithographic printing plate, comprising:a metal base having a surface which satisfies formula (1) belowA is the peak area (counts•eV/sec) for fluorine (1S) as measured by electron spectroscopy for chemical analysis (ESCA),B is the peak area (counts•eV/sec) for silicon (2P) as measured by ESCA,C is the peak area (counts·eV/sec) for phosphorus (2P) as measured by ESCA,D is the peak area (counts•eV/sec) for aluminum (2P) as measured by ESCA; andan intermediate layer which lies on the metal base and contains an acid group-bearing polymeric compound.
- A presensitized plate comprising:the lithographic printing plate support according to any one of claims 1 to 4, andan infrared laser-imageable recording layer which lies on the support.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003331342 | 2003-09-24 | ||
| JP2003331342A JP2005096184A (en) | 2003-09-24 | 2003-09-24 | Support for lithographic printing plate and original plate for lithographic printing plate |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP1518710A2 true EP1518710A2 (en) | 2005-03-30 |
| EP1518710A3 EP1518710A3 (en) | 2005-06-08 |
Family
ID=34191445
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP04020031A Withdrawn EP1518710A3 (en) | 2003-09-24 | 2004-08-24 | Support for lithographic printing plate and presensitized plate |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP1518710A3 (en) |
| JP (1) | JP2005096184A (en) |
| CN (1) | CN1601382A (en) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009214496A (en) * | 2008-03-12 | 2009-09-24 | Fujifilm Corp | Original lithographic printing plate |
| JP2013026284A (en) * | 2011-07-15 | 2013-02-04 | Toyo Aluminium Kk | Manufacturing method of heat radiation laminated material for mounting substrate |
| CN107499016A (en) * | 2017-09-25 | 2017-12-22 | 浙江康尔达新材料股份有限公司 | A kind of thermosensitive negative planographic printing plate precursor and its method for platemaking |
| CN113567516B (en) * | 2021-06-28 | 2023-05-16 | 滁州职业技术学院 | Sulfamethoxypyrimidine molecularly imprinted electrode and preparation method and application thereof |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0048909A1 (en) * | 1980-09-26 | 1982-04-07 | Hoechst Celanese Corporation | Process for the anodic oxidation of aluminium, and its use as a carrier material for printing plates |
| US4448647A (en) * | 1980-09-26 | 1984-05-15 | American Hoechst Corporation | Electrochemically treated metal plates |
| EP1170149A2 (en) * | 2000-07-07 | 2002-01-09 | Fuji Photo Film Co., Ltd. | Preparation method for lithographic printing plate |
| EP1231074A2 (en) * | 2001-02-07 | 2002-08-14 | Fuji Photo Film Co., Ltd. | Presensitized plate useful for making lithographic printing plate and method for making lithographic printing plate therefrom |
| EP1232878A2 (en) * | 2001-02-20 | 2002-08-21 | Fuji Photo Film Co., Ltd. | Method for producing support for planographic printing plate, support for planographic printing plate, and planographic printing plate precursor |
| EP1251014A2 (en) * | 2001-04-20 | 2002-10-23 | Fuji Photo Film Co., Ltd. | Support for lithographic printing plate and presensitized plate |
| EP1266753A2 (en) * | 2001-06-13 | 2002-12-18 | Fuji Photo Film Co., Ltd. | Presensitized plate for use in making lithographic printing plate |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4174993B2 (en) * | 2002-02-07 | 2008-11-05 | コニカミノルタホールディングス株式会社 | Photosensitive lithographic printing plate material and method for producing photosensitive lithographic printing plate material |
-
2003
- 2003-09-24 JP JP2003331342A patent/JP2005096184A/en active Pending
-
2004
- 2004-08-24 EP EP04020031A patent/EP1518710A3/en not_active Withdrawn
- 2004-09-24 CN CN 200410011875 patent/CN1601382A/en active Pending
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0048909A1 (en) * | 1980-09-26 | 1982-04-07 | Hoechst Celanese Corporation | Process for the anodic oxidation of aluminium, and its use as a carrier material for printing plates |
| US4448647A (en) * | 1980-09-26 | 1984-05-15 | American Hoechst Corporation | Electrochemically treated metal plates |
| EP1170149A2 (en) * | 2000-07-07 | 2002-01-09 | Fuji Photo Film Co., Ltd. | Preparation method for lithographic printing plate |
| EP1231074A2 (en) * | 2001-02-07 | 2002-08-14 | Fuji Photo Film Co., Ltd. | Presensitized plate useful for making lithographic printing plate and method for making lithographic printing plate therefrom |
| EP1232878A2 (en) * | 2001-02-20 | 2002-08-21 | Fuji Photo Film Co., Ltd. | Method for producing support for planographic printing plate, support for planographic printing plate, and planographic printing plate precursor |
| EP1251014A2 (en) * | 2001-04-20 | 2002-10-23 | Fuji Photo Film Co., Ltd. | Support for lithographic printing plate and presensitized plate |
| EP1266753A2 (en) * | 2001-06-13 | 2002-12-18 | Fuji Photo Film Co., Ltd. | Presensitized plate for use in making lithographic printing plate |
Non-Patent Citations (1)
| Title |
|---|
| DATABASE WPI Section Ch, Week 200417 Derwent Publications Ltd., London, GB; Class A89, AN 2004-172077 XP002312070 & JP 2003 233166 A (KONICA CORP) 22 August 2003 (2003-08-22) * |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1518710A3 (en) | 2005-06-08 |
| CN1601382A (en) | 2005-03-30 |
| JP2005096184A (en) | 2005-04-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1247644B1 (en) | Support for lithographic printing plate and original forme for lithographic printing plate | |
| US6637334B2 (en) | Heat-sensitive lithographic printing plate precursor | |
| US7132212B2 (en) | Presensitized plate | |
| US6890700B2 (en) | Lithographic printing plate precursor | |
| JP4015344B2 (en) | Master for lithographic printing plate | |
| US6596455B2 (en) | Lithographic printing plate precursor | |
| JP2001277740A (en) | Original plate for lithographic printing plate | |
| JP2001277742A (en) | Original plate for lithographic printing plate | |
| EP1516724B1 (en) | Presensitized plate and lithographic printing method | |
| EP1281516B1 (en) | Planographic printing plate precursor | |
| JP2004098386A (en) | Method for manufacturing support for lithographic printing plate and support for lithographic printing plate | |
| JP2003103951A (en) | Support for lithographic printing plate and lithographic printing original plate | |
| EP1518710A2 (en) | Support for lithographic printing plate and presensitized plate | |
| JP2003001960A (en) | Original plate for planographic printing plate | |
| JP4764892B2 (en) | Planographic printing plate making method | |
| JP2005271414A (en) | Support for lithographic printing plate and original plate for lithographic printing plate | |
| JP2003034090A (en) | Lithographic printing original plate | |
| JP2005066829A (en) | Support for planographic printing plate and planographic printing original plate | |
| JP2002214764A (en) | Lithographic printing master plate | |
| JP3862636B2 (en) | Lithographic printing plate, lithographic printing plate making method and lithographic printing method | |
| JP2003001961A (en) | Support for planographic printing plate and original plate for planographic printing plate | |
| JP2005262466A (en) | Manufacturing method of aluminum support for lithographic printing form | |
| JP4208380B2 (en) | Master for lithographic printing plate | |
| JP2002067526A (en) | Original plate for thermosensitive lithographic printing | |
| JP4057801B2 (en) | Lithographic printing plate support and planographic printing plate precursor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A2 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LI LU MC NL PL PT RO SE SI SK TR |
|
| AX | Request for extension of the european patent |
Extension state: AL HR LT LV MK |
|
| PUAL | Search report despatched |
Free format text: ORIGINAL CODE: 0009013 |
|
| AK | Designated contracting states |
Kind code of ref document: A3 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LI LU MC NL PL PT RO SE SI SK TR |
|
| AX | Request for extension of the european patent |
Extension state: AL HR LT LV MK |
|
| 17P | Request for examination filed |
Effective date: 20051121 |
|
| AKX | Designation fees paid |
Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LI LU MC NL PL PT RO SE SI SK TR |
|
| RAP1 | Party data changed (applicant data changed or rights of an application transferred) |
Owner name: FUJIFILM CORPORATION |
|
| 17Q | First examination report despatched |
Effective date: 20070411 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE APPLICATION IS DEEMED TO BE WITHDRAWN |
|
| 18D | Application deemed to be withdrawn |
Effective date: 20071023 |