EP0053054A1 - Procédé de récupération de l'uranium (VI) présent dans des solutions d'acide phosphorique - Google Patents
Procédé de récupération de l'uranium (VI) présent dans des solutions d'acide phosphorique Download PDFInfo
- Publication number
- EP0053054A1 EP0053054A1 EP81401686A EP81401686A EP0053054A1 EP 0053054 A1 EP0053054 A1 EP 0053054A1 EP 81401686 A EP81401686 A EP 81401686A EP 81401686 A EP81401686 A EP 81401686A EP 0053054 A1 EP0053054 A1 EP 0053054A1
- Authority
- EP
- European Patent Office
- Prior art keywords
- uranium
- organic solvent
- extraction
- acid
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 title claims abstract description 101
- 229910000147 aluminium phosphate Inorganic materials 0.000 title claims abstract description 50
- 238000000034 method Methods 0.000 title claims abstract description 27
- AAORDHMTTHGXCV-UHFFFAOYSA-N uranium(6+) Chemical compound [U+6] AAORDHMTTHGXCV-UHFFFAOYSA-N 0.000 title claims abstract description 7
- 230000008569 process Effects 0.000 title abstract description 9
- 229960004838 phosphoric acid Drugs 0.000 title 1
- 235000011007 phosphoric acid Nutrition 0.000 title 1
- 239000003960 organic solvent Substances 0.000 claims abstract description 58
- 239000002253 acid Substances 0.000 claims abstract description 34
- 150000002903 organophosphorus compounds Chemical class 0.000 claims abstract description 27
- AUONHKJOIZSQGR-UHFFFAOYSA-N oxophosphane Chemical compound P=O AUONHKJOIZSQGR-UHFFFAOYSA-N 0.000 claims abstract description 18
- 230000007935 neutral effect Effects 0.000 claims abstract description 16
- 125000003118 aryl group Chemical group 0.000 claims abstract description 3
- 229910052770 Uranium Inorganic materials 0.000 claims description 119
- JFALSRSLKYAFGM-UHFFFAOYSA-N uranium(0) Chemical compound [U] JFALSRSLKYAFGM-UHFFFAOYSA-N 0.000 claims description 118
- 238000000605 extraction Methods 0.000 claims description 71
- 239000000243 solution Substances 0.000 claims description 37
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 claims description 30
- -1 alkoxyalkyl radicals Chemical class 0.000 claims description 28
- 229910021529 ammonia Inorganic materials 0.000 claims description 15
- 239000007864 aqueous solution Substances 0.000 claims description 12
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 claims description 11
- 239000001099 ammonium carbonate Substances 0.000 claims description 11
- 235000012501 ammonium carbonate Nutrition 0.000 claims description 11
- 125000004432 carbon atom Chemical group C* 0.000 claims description 11
- 125000000217 alkyl group Chemical group 0.000 claims description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 claims description 7
- 150000003254 radicals Chemical class 0.000 claims description 7
- ZMBHCYHQLYEYDV-UHFFFAOYSA-N trioctylphosphine oxide Chemical group CCCCCCCCP(=O)(CCCCCCCC)CCCCCCCC ZMBHCYHQLYEYDV-UHFFFAOYSA-N 0.000 claims description 6
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 claims description 5
- 150000005840 aryl radicals Chemical class 0.000 claims description 4
- OMLVPEUOCUINNC-UHFFFAOYSA-N 1-(dihexylphosphorylmethoxy)octane Chemical group CCCCCCCCOCP(=O)(CCCCCC)CCCCCC OMLVPEUOCUINNC-UHFFFAOYSA-N 0.000 claims description 3
- MDDUHVRJJAFRAU-YZNNVMRBSA-N tert-butyl-[(1r,3s,5z)-3-[tert-butyl(dimethyl)silyl]oxy-5-(2-diphenylphosphorylethylidene)-4-methylidenecyclohexyl]oxy-dimethylsilane Chemical compound C1[C@@H](O[Si](C)(C)C(C)(C)C)C[C@H](O[Si](C)(C)C(C)(C)C)C(=C)\C1=C/CP(=O)(C=1C=CC=CC=1)C1=CC=CC=C1 MDDUHVRJJAFRAU-YZNNVMRBSA-N 0.000 claims description 3
- WPWHSFAFEBZWBB-UHFFFAOYSA-N 1-butyl radical Chemical compound [CH2]CCC WPWHSFAFEBZWBB-UHFFFAOYSA-N 0.000 claims description 2
- 125000004183 alkoxy alkyl group Chemical group 0.000 abstract 1
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 60
- 229910052742 iron Inorganic materials 0.000 description 29
- 150000001875 compounds Chemical class 0.000 description 24
- 239000002904 solvent Substances 0.000 description 17
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 238000005192 partition Methods 0.000 description 15
- 239000000203 mixture Substances 0.000 description 12
- 239000008346 aqueous phase Substances 0.000 description 10
- 239000012071 phase Substances 0.000 description 10
- 239000000047 product Substances 0.000 description 10
- 230000015572 biosynthetic process Effects 0.000 description 9
- 238000006243 chemical reaction Methods 0.000 description 9
- 150000003333 secondary alcohols Chemical class 0.000 description 9
- 238000009434 installation Methods 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 7
- 230000002378 acidificating effect Effects 0.000 description 7
- 150000002148 esters Chemical class 0.000 description 7
- 239000012074 organic phase Substances 0.000 description 7
- 238000002360 preparation method Methods 0.000 description 7
- DLYUQMMRRRQYAE-UHFFFAOYSA-N tetraphosphorus decaoxide Chemical compound O1P(O2)(=O)OP3(=O)OP1(=O)OP2(=O)O3 DLYUQMMRRRQYAE-UHFFFAOYSA-N 0.000 description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 6
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 6
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 6
- 150000003138 primary alcohols Chemical class 0.000 description 6
- 239000011734 sodium Substances 0.000 description 6
- 229910052708 sodium Inorganic materials 0.000 description 6
- JCMLRUNDSXARRW-UHFFFAOYSA-N trioxouranium Chemical compound O=[U](=O)=O JCMLRUNDSXARRW-UHFFFAOYSA-N 0.000 description 6
- 238000005406 washing Methods 0.000 description 6
- 150000003863 ammonium salts Chemical class 0.000 description 5
- 238000007254 oxidation reaction Methods 0.000 description 5
- 150000003017 phosphorus Chemical class 0.000 description 5
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- 229910019142 PO4 Inorganic materials 0.000 description 4
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 230000003647 oxidation Effects 0.000 description 4
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 4
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 239000004254 Ammonium phosphate Substances 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 229910000148 ammonium phosphate Inorganic materials 0.000 description 3
- 235000019289 ammonium phosphates Nutrition 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- MNNHAPBLZZVQHP-UHFFFAOYSA-N diammonium hydrogen phosphate Chemical compound [NH4+].[NH4+].OP([O-])([O-])=O MNNHAPBLZZVQHP-UHFFFAOYSA-N 0.000 description 3
- SYHPANJAVIEQQL-UHFFFAOYSA-N dicarboxy carbonate Chemical compound OC(=O)OC(=O)OC(O)=O SYHPANJAVIEQQL-UHFFFAOYSA-N 0.000 description 3
- 238000005886 esterification reaction Methods 0.000 description 3
- 239000003350 kerosene Substances 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 238000011084 recovery Methods 0.000 description 3
- 230000002195 synergetic effect Effects 0.000 description 3
- ISIJQEHRDSCQIU-UHFFFAOYSA-N tert-butyl 2,7-diazaspiro[4.5]decane-7-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCCC11CNCC1 ISIJQEHRDSCQIU-UHFFFAOYSA-N 0.000 description 3
- 238000005809 transesterification reaction Methods 0.000 description 3
- 125000005289 uranyl group Chemical group 0.000 description 3
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000005587 bubbling Effects 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 238000010908 decantation Methods 0.000 description 2
- CPUDLRLOFJLISR-UHFFFAOYSA-N diethyl(trihydroxy)-$l^{5}-phosphane Chemical compound CCP(O)(O)(O)CC CPUDLRLOFJLISR-UHFFFAOYSA-N 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- SNRUBQQJIBEYMU-UHFFFAOYSA-N dodecane Chemical compound CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 230000032050 esterification Effects 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 239000003337 fertilizer Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 2
- MPQXHAGKBWFSNV-UHFFFAOYSA-N oxidophosphanium Chemical class [PH3]=O MPQXHAGKBWFSNV-UHFFFAOYSA-N 0.000 description 2
- KJFMBFZCATUALV-UHFFFAOYSA-N phenolphthalein Chemical compound C1=CC(O)=CC=C1C1(C=2C=CC(O)=CC=2)C2=CC=CC=C2C(=O)O1 KJFMBFZCATUALV-UHFFFAOYSA-N 0.000 description 2
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 2
- 229930195734 saturated hydrocarbon Natural products 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 159000000000 sodium salts Chemical class 0.000 description 2
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 2
- YUMNZEWYPUBSQA-UHFFFAOYSA-N 1-(chloromethoxy)octane Chemical compound CCCCCCCCOCCl YUMNZEWYPUBSQA-UHFFFAOYSA-N 0.000 description 1
- DEYGPTFKWXTDDE-UHFFFAOYSA-N 1-(dibutylphosphorylmethoxy)octane Chemical compound CCCCCCCCOCP(=O)(CCCC)CCCC DEYGPTFKWXTDDE-UHFFFAOYSA-N 0.000 description 1
- KMCOLJKZEORYBT-UHFFFAOYSA-N 1-[bis(2-methylpropyl)phosphorylmethoxy]octane Chemical compound CCCCCCCCOCP(=O)(CC(C)C)CC(C)C KMCOLJKZEORYBT-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 101100520660 Drosophila melanogaster Poc1 gene Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 101100520662 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) PBA1 gene Proteins 0.000 description 1
- OCBFFGCSTGGPSQ-UHFFFAOYSA-N [CH2]CC Chemical compound [CH2]CC OCBFFGCSTGGPSQ-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 238000010306 acid treatment Methods 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 238000005266 casting Methods 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- ZAASRHQPRFFWCS-UHFFFAOYSA-P diazanium;oxygen(2-);uranium Chemical compound [NH4+].[NH4+].[O-2].[O-2].[O-2].[O-2].[O-2].[O-2].[O-2].[U].[U] ZAASRHQPRFFWCS-UHFFFAOYSA-P 0.000 description 1
- NZZIMKJIVMHWJC-UHFFFAOYSA-N dibenzoylmethane Chemical compound C=1C=CC=CC=1C(=O)CC(=O)C1=CC=CC=C1 NZZIMKJIVMHWJC-UHFFFAOYSA-N 0.000 description 1
- 125000003963 dichloro group Chemical group Cl* 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 229960004887 ferric hydroxide Drugs 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 239000008240 homogeneous mixture Substances 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- IEECXTSVVFWGSE-UHFFFAOYSA-M iron(3+);oxygen(2-);hydroxide Chemical compound [OH-].[O-2].[Fe+3] IEECXTSVVFWGSE-UHFFFAOYSA-M 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 150000007530 organic bases Chemical group 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 150000003016 phosphoric acids Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 238000004313 potentiometry Methods 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 238000002798 spectrophotometry method Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- RMZAYIKUYWXQPB-UHFFFAOYSA-N trioctylphosphane Chemical compound CCCCCCCCP(CCCCCCCC)CCCCCCCC RMZAYIKUYWXQPB-UHFFFAOYSA-N 0.000 description 1
Images







Classifications
-
- C—CHEMISTRY; METALLURGY
- C22—METALLURGY; FERROUS OR NON-FERROUS ALLOYS; TREATMENT OF ALLOYS OR NON-FERROUS METALS
- C22B—PRODUCTION AND REFINING OF METALS; PRETREATMENT OF RAW MATERIALS
- C22B60/00—Obtaining metals of atomic number 87 or higher, i.e. radioactive metals
- C22B60/02—Obtaining thorium, uranium, or other actinides
- C22B60/0204—Obtaining thorium, uranium, or other actinides obtaining uranium
- C22B60/0217—Obtaining thorium, uranium, or other actinides obtaining uranium by wet processes
- C22B60/0252—Obtaining thorium, uranium, or other actinides obtaining uranium by wet processes treatment or purification of solutions or of liquors or of slurries
- C22B60/026—Obtaining thorium, uranium, or other actinides obtaining uranium by wet processes treatment or purification of solutions or of liquors or of slurries liquid-liquid extraction with or without dissolution in organic solvents
Definitions
- the present invention relates to a process for recovering uranium (VI) present in solutions of phosphoric acid, in particular in solutions of phosphoric acid obtained from phosphate ores.
- the phosphate ores include significant contents of uranium which, during the attack of these ores by a sulfuric solution, pass into the phosphoric acid solution obtained. Also, it is interesting to recover the uranium present in these solutions which constitute an important additional source of uranium.
- the present invention specifically relates to a process for recovering uranium (VI) using an organic solvent which provides better uranium extraction rates than currently known solvents.
- the alkoxyalkyl radical advantageously has from 9 to 23 carbon atoms.
- the acid organophosphorus compound can be constituted by a phosphoric ester of secondary alcohol or by a phosphoric ester of primary alcohol.
- phosphoric esters of primary alcohol these advantageously meet the formula: in which R 6 and R 7 which are identical or different, are alkyl or aryl radicals, and n and n ′ which are identical or different, are numbers equal to 2 or 3.
- n and n ' are preferably equal to 2.
- the alkyl radicals R 6 and R 7 have at least 8 carbon atoms to avoid the formation of a third phase during this re-extraction.
- the acid organophosphorus compound can also consist of a phosphoric ester of secondary alcohol corresponding to formula IV: in which R and R 9 , which are identical or different, represent an alkyl or aryl radical, and p and q which can be identical or different, are equal to 1 or 2.
- p and q are equal to 1, because the extracting power of the system increases when the ether-oxide function of the acid organophosphorus compound is close to the phosphate group.
- the radicals R 8 and R 9 preferably have at least 4 carbon atoms to avoid the formation of a third phase during this re-extraction.
- the acid organophosphorus compounds of formula (III) or of formula (IV) used in the process of the invention can be obtained by transesterification or by esterification of a phosphorus derivative with the corresponding alkoxy alcohol, this reaction being optionally followed by oxidation and / or hydrolysis.
- phosphorus derivatives it is possible to use a dialkyl phosphorous acid, phosphorus oxychloride or also phosphoric anhydride P205.
- the transesterification reaction with the corresponding alkoxy alcohol is followed by an oxidation reaction then by a hydrolysis to give the corresponding acid organophosphorus compound.
- Oxidation can be carried out by the action of sulfuryl chloride SO 2 Cl 2 and hydrolysis by the action of sodium hydroxide and by the action of hydrochloric acid.
- the esterification reaction is carried out with the corresponding alkoxyalcohol in the presence of a base, in part a tertiary organic base, then the product obtained is subjected to hydrolysis, which leads to the production of a mixture of mono and diacids which are then separated.
- the esterification is carried out with the corresponding alkoxyalcohol, away from humidity, a mixture of mono and diacids which may also contain neutral phosphate and impurities such as pyrophosphates and polymers.
- the alkoxy alcohols used as starting materials for the synthesis of acidic organophosphorus compounds can be prepared by reaction of a sodium alcoholate with the dichloro derivative of a secondary alcohol, for example, sodium alcoholate with dichloro-1 , 3-propanol-2, according to the following reaction scheme:
- a sodium alcoholate is reacted with the chlorinated derivative of a primary alcohol.
- the neutral phosphine oxide corresponding to the abovementioned formula I preferably comprises at least one alkoxyalkyl radical, for example, an alkoxymethyl radical having from 4 to 12 carbon atoms.
- alkoxyalkyl radicals for example, an alkoxymethyl radical having from 4 to 12 carbon atoms.
- the other radicals are alkyl radicals, these generally have from 4 to 12 carbon atoms, and they are preferably linear.
- neutral phosphine oxides which may be used, mention may be made of di-isobutyl-octoxymethylphosphine oxide, di-n-butyl-octoxymethylphosphine oxide, di-n- oxide pentyl-octoxymated thylphosphine, and di-n-hexyl-octoxymethylphosphine oxide (POX 11).
- phosphine oxides can be prepared by reacting a halogenated magnesium salt of secondary phosphine oxide with an organic halide of formula RX in which R represents an alkoxyalkyl radical, for example, a chloromethyl n-octyl ether, as described in French patent 2,346,361 filed on 12/13/73.
- trialkylphosphine oxides in which the alkyl radicals have from 4 to 14 carbon atoms, for example tri-n-octylphosphine oxide (TOPO).
- TOPO tri-n-octylphosphine oxide
- the extractant system is generally diluted in an inert organic solvent constituted for example by a saturated hydrocarbon having at least 8 carbon atoms such as dodecane, or by a mixture of hydrocarbons.
- the concentrations of acid organophosphorus compound and of neutral phosphine oxide are such that the molar ratio of the acid organophosphorus compound to the neutral phosphine oxide is between 1 and 9, and preferably from 2 to 4. .
- the method of the invention can be implemented in any conventional extraction device such as batteries of settling mixers, pulsed columns, centrifugal extractors, etc.
- the uranium extracted in the organic solvent can be re-extracted in followed in an aqueous solution of phosphoric acid optionally containing a reducing agent so as to reduce the uranium VI to uranium IV to facilitate its re-extraction.
- the re-extraction of the uranium is carried out in a re-extraction device comprising at least two stages.
- a re-extraction device comprising at least two stages.
- the ammoniated organic solvent leaving the last re-extraction stage is re-acidified by reacting it with an acid to remove the ammonium in the form of the ammonium salt, and the organic solvent thus re-acidified is reused to carry out the extraction of the 'uranium.
- the acid is chosen from the group comprising sulfuric acid, hydrochloric acid and phosphoric acid.
- the ammoniated organic solvent leaving the last re-extraction stage by reacting it with the phosphoric acid recovered at the end of the uranium extraction.
- This preferential mode of uranium re-extraction makes it possible to obtain, at the end of re-extraction, an aqueous uranium solution from which it is easy to recover the uranium directly to the standards defined by the refiners, therefore without additional purification cycle, ie in the form of oxide, either in the form of an alkaline or alkaline-earth uranate, with an overall uranium recovery yield greater than 90%.
- the organic solvent which has been re-acidified by treatment with phosphoric acid can be reused for the extraction of uranium, and the ammonium phosphate obtained during the re-acidification treatment of the organic solvent is a marketable product or recyclable in a fertilizer unit for example.
- the re-extraction of uranium is preferably carried out in three stages.
- the organic solvent containing the uranium is circulated from the first to the third stage and an aqueous solution of ammonium carbonate or a mixture of carbon dioxide and ammonia previously dissolved in the is introduced into the third stage.
- water in the form of carbonate representing 50 to 80% of the stoichiometric quantity necessary to neutralize the acid organophosphorus compound of the organic solvent and to transform the uranium into uranyl ammonium tricarbonate.
- This solution circulates from the third stage to the first stage and ammonia in the form of gas or aqueous solution is added to it before entering the first stage, the amount added being such that the pH of the first stage is maintained at a value between 8 and 8.5.
- the ammonia is added in the form of an aqueous solution having a molar ammonia concentration of 5M to 7.5M.
- the organic solvent charged with uranium and also containing iron is transformed little by little in contact with ammonia into an ammonium salt and the aqueous phase which moves against the current, is enriched in uranium and in iron, the ammonium carbonate forming with uranium the ammonium uranyl tricarbonate which remains in solution and the iron transforming into ferric hydroxide which precipitates and which can be separated by decantation from the aqueous phase.
- the ammoniated organic solvent is preferably re-acidified by treatment with an acid such as sulfuric acid, hydrochloric acid or phosphoric acid, which makes it possible to recover an organic phase which does not contain more ammonium ions and an aqueous phase containing an ammonium salt.
- an acid such as sulfuric acid, hydrochloric acid or phosphoric acid
- a fraction of the phosphoric acid used is used. collected at the end of the uranium extraction stage.
- This example relates to the recovery of uranium present in a 6M phosphoric acid solution containing lg / 1 of uranium (VI) and it illustrates the effect of the temperature and the nature of the extractant system on the uranium extraction rate.
- the various acid organophosphorus compounds in Table 1 are used with trioctylphosphine oxide (TOPO) or with di-n-hexyl-octoxymethylphosphine oxide (POX 11).
- TOPO trioctylphosphine oxide
- POX 11 di-n-hexyl-octoxymethylphosphine oxide
- the two extractants are diluted in hyfrane 120 which is a branched saturated hydrocarbon with an average carbon number equal to 12 and the content of acidic organophosphorus compound in the solvent is 0.5 M and its content of phosphine oxide is 0.125 M .
- the extraction is carried out under the following conditions: one volume of the aqueous phosphoric acid solution with one volume of the organic solvent is brought into contact at 23 ° C or 40 ° C for approximately 15 minutes, the two are mechanically stirred phases present, they are separated by centrifugation, then each phase is sampled and analyzed in order to determine its uranium concentration, the latter being measured by spectrophotometry with dibenzoyl methane, the uranium being previously extracted in an oxide solution trioctylphosphine; the partition coefficient D of the uranium is then determined, which is equal to the ratio of the uranium concentration of the organic phase to the uranium concentration of the aqueous phase.
- the partition coefficient of uranium increases with the number of carbon atoms in the alkoxy chains.
- the organic solvent consists of hyfrane 120 containing a mixture of bis hydrogenphosphate (dibutoxy-1,3-propyl-2), that is to say of compound n ° 8 of table 1 and of oxide of di-n-hexyloctoxymethy .phosphine (POX 11) to recover uranium from a 6N phosphoric acid solution containing 1.1 g per liter of uranium VI.
- a total concentration of 0.5 M in extractants is used and the extraction is carried out under the same conditions as those of Example 1.
- curve (1) of FIG. 1 represents the variations of the partition coefficient D of uranium as a function of the content of acidic organophosphorus compound in the organic solvent.
- curve 2 represents the variations in the partition coefficient D of iron as a function of the content of compound produced extraction under the same conditions from a 6M phosphoric acid solution containing 1.1 g / l of iron III.
- This example illustrates the influence of the concentration of phosphoric acid in the aqueous solution on the extraction of uranium VI, at 23 ° C., using the extractant systems No. I, II and III in Table 2 diluted in of hydrogen 120.
- the extraction is carried out under the same conditions as those of Example 1.
- FIG. 2 represents the variations in the partition coefficient D of uranium VI as a function of the concentration of phosphoric acid in the aqueous phase.
- curves I, II and III respectively illustrate the results obtained with the extractant systems I, II and III in Table 2.
- the partition coefficient D of uranium decreases as a function of the phosphoric acid concentration, but it de grows less significantly with the extractant systems II and III of the invention.
- This example illustrates the influence of temperature on the extraction of uranium VI.
- the extraction is carried out under the same conditions as those of Examples 2 and 3 by varying the temperature from 10 to 60 ° C. and using the extractant systems I to IV of Table 2 diluted in hyfrane 120.
- the results obtained are represented in FIG. 3 where the curves I, II, III and IV relate respectively to the systems I, II, III and IV.
- the uranium is extracted from a 6M phosphoric acid solution containing 1.06 g / 1 of uranium VI and 4.70 g / 1 of iron III, using the system extractants No. III of Table 2 diluted in hyfrane 120 and operating under the same conditions as those of Example 1. but by determining the partition coefficients of uranium VI and iron III after different durations d 'extraction.
- FIG. 4 represents the evolution of the extraction rate (in%) in the organic phase of uranium (curve 1) and iron (curve 2) as a function of the extraction time. (in seconds).
- the uranium is recovered from an industrial phosphoric acid solution titrating 27% P 2 0 5 and 130 m g / 1 uranium, using as extractant systems I, III and IV of Table 2 diluted in the product sold under the brand Escaid 110, which is a dearomatized kerosene, with a concentration of 0.5 M in acidic organophosphorus compound and a concentration of 0.125 M in phosphine oxide.
- FIG. 6 also illustrates the results obtained with regard to the extraction of iron as a function of the number of contacts.
- curve 2 represents the evolution of the iron concentration of the organic solvent as a function of the number of contacts
- curve 1 represents the evolution of the uranium concentration of the organic solvent as a function of the number of contacts, when 'The extractant system III of the invention is used.
- the organic solvent thus obtained which contains 798 mg / 1 of uranium and 775 mg / 1 of iron is brought into contact with a solution of ammonium carbonate at 140 g / 1 and 0.5 M in NH 4 0H for re- milking the uranium in solution and separating the iron as hydroxide.
- the organic phase contains only 0.4 mg / 1 of uranium and 1 mg / 1 of iron.
- the No. III extractant system extracts more than three times better uranium than conventional system of the prior art (system No. I) and more than twice as good as system No. IV of the prior art. Therefore, one can reduce the volume of organic phase and therefore limit the consumption of ammonium carbonate and ammonia during the re-extraction operation.
- the organic solvents used contain the extractant systems I, III, IV or V of Table 2 attached, and a diluent consisting of kerosene known under the trade name ISOPAR L.
- a diluent consisting of kerosene known under the trade name ISOPAR L.
- the phosphine oxide content is 0.125 M and the content of acidic organophosphorus compound is 0.500 M.
- the extraction is carried out by bringing a volume of the aqueous solution into contact at 39 ° C. phosphoric acid with a volume of the organic solvent with stirring for about 5 min; we then separate the two phases then we take them and we analyze them each to obtain their uranium concentration and their iron concentration and then determine the partition coefficient D of uranium and the partition coefficient D of iron.
- the results obtained are given in table 3, attached.
- the organic solvents containing the extractant system of the invention that is to say the organophosphorus acid compound HBIDIBOPP associated with a phosphine oxide such as POX11 or TOPO allow '' obtaining very improved results compared to the extractant systems I and IV of the prior art.
- This example relates to the extraction of uranium contained in industrial phosphoric acid having the same characteristics as that of Example 7, using for the extraction the installation shown in FIG. 7.
- the reference A designates the uranium extraction unit which comprises five extraction stages
- the reference B represents a washing unit for the organic solvent which comprises three stages
- the references C 1 , C 2 and C 3 denote the three stages of uranium re-extraction
- the reference D illustrates the unit for separating uranium
- the reference E denotes the reacidification unit of the organic solvent, which comprises two stages.
- extraction unit A industrial line phosphoric acid is introduced via line la after having flocculated and decanted it. It is specified that this acid was subjected beforehand to an oxidation treatment to bring all of the uranium in the hexavalent form, which also brings the iron to the trivalent state.
- extraction unit A the phosphoric acid is brought into countercurrent contact with an organic solvent introduced via line 3a.
- This organic solvent comprises a system of extractants constituted by an acid organophosphorus compound and by a neutral phosphine oxide diluted in kerosene known under the trade name ISOPAR L, the concentration of acidic organophosphorus compound in the solvent being 0.5 mol.l -1 and the phosphine oxide concentration in the organic solvent being 0.125 mol.l -1 .
- the phosphoric acid solution circulates in the extraction unit at a rate which is maintained at the value of 4 1 / h, and the organic solvent circulates against the current in the extraction unit by being introduced at a flow rate of 1.6 1 / h for extractant system No. I, 1.0 1 / h for extractant system No. IV and 0.54 1 / h for extractant system No. III.
- each extraction stage part of the organic solvent leaving this stage is recycled, which makes it possible to increase the volume of organic phase in contact with phosphoric acid in the extraction unit A, all of the stages of which are maintained at 35 ° C.
- the phosphoric acid containing practically no more uranium is discharged via line 1b and the organic solvent loaded with uranium and iron is discharged via line 3b.
- This solvent then passes into the washing unit comprising three stages where it is washed with water to remove the phosphoric ions entrained by the solvent.
- the water loaded with phosphoric acid which leaves the top floor of the washing unit is recycled in the phosphoric acid manufacturing plant where it is used for washing or rinsing the installations.
- the organic solvent is introduced via line 3c into the first re-extraction stage C 1 , then it circulates in the following stages C 2 and C 3 , stages C 1 , C 2 and C 3 being maintained at 40 ° C.
- stages C 2 and C 3 it is brought into contact against the current with an ammonium carbonate solution at 155 gl -1 introduced in the last stage C 3 by line 4a and in stage C 1 , it is brought into contact against the current with the carbonate solution coming from stage C 2 and with ammonia at 200 g -1 injected by line 5 into the carbonate solution which enters the first stage C 1 .
- the ammonia flow rate is adjusted using a valve controlled by a pH meter so as to maintain the pH of the first stage CI at a value of 8.2.
- the flow rate of the ammonium carbonate solution introduced into the last stage C 3 is regulated by line 4a so that it corresponds to 50 to 80% of the stoichiometric quantity necessary to neutralize, on the one hand , the organo phosphorus acid compound and transform, on the other hand, uranium into uranyl ammonium tri-carbonate.
- the organic solvent loaded with uranium and iron which first comes into contact with ammonia gradually transforms into a hydrated ammonium salt and the aqueous phase which moves against the current is enriched in uranium and iron, the ammonium carbonate reacting with uranium to form uranyl ammonium tricarbonate which remains in solution and the iron being precipitated in the form of hydroxide which is separated by filtration.
- the aqueous phase containing the uranyl ammonium tricarbonate leaves the first stage of re-extraction CI by line 4b and it is then directed to the separation unit D of the uranium.
- the uranium can be separated from the solution, either in the form oxide, either in the form of sodium uranate.
- the solution of ammonium trianyl carbonate uranyl is subjected in an air bubbling reactor, at a temperature between 90 and 100 ° C., for approximately 6 hours, then the precipitate is filtered and washed with water; after drying at 120 ° C and roasting at around 400 ° C, uranium trioxide is thus obtained.
- the ammonium uranyl tricarbonate solution which has been degassed beforehand by bubbling air around 90 ° C.
- the uranium is precipitated by adding sodium hydroxide to the solution, operating at a temperature of 80 ° C for 1 hour. After filtration and washing with water at 50 ° C., the sodium uranate is collected which can be further transformed into ammonium diuranate or uranium trioxide.
- the deuranified organic solvent is removed by line 3d and sent to the reacidification purification unit E comprising two stages in which it is treated with phosphoric acid introduced by line lc, this phosphoric acid constituting a fraction of the phosphoric acid which leaves the extraction unit A via line 1b.
- the reacidification purification unit E comprising two stages in which it is treated with phosphoric acid introduced by line lc, this phosphoric acid constituting a fraction of the phosphoric acid which leaves the extraction unit A via line 1b.
- ammonium phosphate thus recovered can be marketed directly or be used in fertilizer manufacturing units.
Landscapes
- Engineering & Computer Science (AREA)
- General Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Geology (AREA)
- Manufacturing & Machinery (AREA)
- Environmental & Geological Engineering (AREA)
- Materials Engineering (AREA)
- Mechanical Engineering (AREA)
- Metallurgy (AREA)
- Organic Chemistry (AREA)
- Extraction Or Liquid Replacement (AREA)
- Manufacture And Refinement Of Metals (AREA)
Abstract
Description
- La présente invention a pour objet un procédé de récupération de l'uranium (VI) présent dans des solutions d'acide phosphorique, en particulier dans des solutions d'acide phosphorique obtenues à partir de minerais phosphatés.
- On sait que les minerais phosphatés comprennent des teneurs non négligeables en uranium qui, lors de l'attaque de ces minerais par une solution sulfurique, passent dans la solution d'acide phosphorique obtenue. Aussi, il est intéressant de récupérer l'uranium présent dans ces solutions qui constituent une source complémentaire importante d'uranium.
- Les procédés connus actuellement pour récupérer l'uranium présent dans de telles solutions, font généralement appel à l'extraction au moyen de solvants organiques appropriés. Parmi les solvants utilisés jusqu'à présent, de bons résultats ont été obtenus avec des solvants comprenant un mélange synergique d'extractants, par exemple des mélanges d'acide dialkylphosphorique et d'oxyde neutre de phosphine, tels que ceux décrits dans la demande de brevet français n° 2442 796 déposé le 28.11.78 au nom du Commissariat à l'Energie Atomique.
- Bien que ces systèmes d'extractants permettent d'obtenir des résultats satisfaisants, on a développé des recherches pour trouver d'autres systèmes d'extractants capables de conduire à des taux d'extraction en uranium plus importants.
- La présente invention a précisément pour objet un procédé de récupération de l'uranium (VI) au moyen d'un solvant organique qui permet d'obtenir de meilleurs taux d'extraction en uranium que les solvants connus actuellement.
- Le procédé, selon l'invention, de récupération de l'uranium (VI) présent dans une solution d'acide phosphorique par mise en contact de ladite solution avec un solvant organique apte à extraire l'uranium, se caractérise en ce que ledit solvant organique comprend un système d'extractants constitués respectivement par :
- - un oxyde neutre de phosphine de formule :

- - un composé organophosphoré acide répondant à la formule :

- Selon l'invention, le radical alkoxyalkyle a avantageusement de 9 à 23 atomes de carbone.
- Selon l'invention, le composé organophosphoré acide peut être constitué par un ester phosphorique d'alcool secondaire ou par un ester phosphorique d'alcool primaire. Dans le cas des esters phosphoriques d'alcool primaire, ceux-ci répondent avantageusement à la formule :

- En effet, on a trouvé que dans le cas des esters d'alcools primaires, la présence de plusieurs fonctions éther-oxyde n'améliorait pas les résultats obtenus, et que par ailleurs, le pouvoir extractant augmentait lorsque la fonction éther-oxyde était plus proche du groupement phosphate.
- Aussi, n et n' sont de préférence égaux à 2.
- De plus, si l'on veut réaliser la réextraction de l'uranium au moyen d'une solution aqueuse telle qu'une solution de carbonate d'ammonium et d'ammoniaque, il est préférable que les radicaux alkyle R6 et R7 aient au moins 8 atomes de carbone pour éviter la formation d'une troisième phase lors de cette réextraction.
- A titre de composés de ce type susceptibles d'être utilisés, on peut citer le composé dans lequel R6 et R7 représentent le radical éthyl-2-hexyle, et n et n' sont égaux à 2.
- Le composé organophosphoré acide peut aussi être constitué par un ester phosphorique d'alcool secondaire répondant à la formule IV :

- De préférence, p et q sont égaux à 1, car le pouvoir extractant du système augmente lorsque la fonction éther-oxyde du composé organophosphoré acide est proche du groupement phosphate.
- Par ailleurs, pour permettre également la réextraction de l'uranium dans une solution aqueuse de carbonate d'ammonium et d'ammoniaque, les radicaux R8 et R9 ont de préférence au moins 4 atomes de carbone pour éviter la formation d'une troisième phase lors de cette réextraction.
- A titre d'exemple d'ester phosphorique d'alcool secondaire susceptible d'être utilisé, on peut citer le composé de formule (IV) dans laquelle p et q sont égaux à 1 et R 8 et R9,représentent le radical butyle.
- En effet, des essais de réextraction effectués en utilisant comme système d'extractants le composé de formule (IV) dans laquelle R8 et R9 représentent le radical propyle et p et q sont égaux à 1, et de l'oxyde de di-n-hexylocto- xyméthylphosphine (POX 11) ont montré que la solubilité des sels alcalins de ce composé acide est telle qu'elle conduit à la formation d'une troisième phase. En revanche, si l'on augmente le nombre d'atomes de carbone des chaînes alkyle de ce composé acide, on peut éviter ce phénomène.
- Les composés organophosphorés acides de formule (III) ou de formule (IV) utilisés dans le procédé de l'invention peuvent être obtenus par transestérification ou par estérification d'un dérivé du phosphore avec l'alcoxyalcool correspondant, cette réaction étant suivie éventuellement d'une oxydation et/ou d'une hydrolyse.
- Comme dérivés du phosphore, on peut utiliser un acide dialkyl phosphoreux, l'oxychlorure de phosphore ou encore l'anhydride phosphorique P205.
- Lorsque le dérivé du phosphore est un acide dialkyl phosphoreux, la réaction de transestérification avec l'alcoxyalcool correspondant est suivie d'une réaction d'oxydation puis d'une hydrolyse pour donner le composé organophosphoré acide correspondant. L'oxydation peut être réalisée par action du chlorure de sulfuryle SO2Cl2 et l'hydrolyse par action de soude et par action d'acide chlorhydrique.
- Ainsi, dans le cas où l'on utilise l'acide diéthyl phosphoreux pour la préparation des composés de formule IV avec R 8 = R9 et n = p, le schéma réactionnel est le suivant :


- Pour obtenir des composés de formule IV dans lequels R et R9, p et q sont différents, il suffit de remplacer dans le schéma réactionnel précédent, l'alcool secondaire utilisé par l'alcool secondaire de formule suivante : R8-O-(CH2)p-CHOH-(CH2)q-O-R9
- De même, lorsqu'on veut préparer les composés de formule III, il suffit de remplacer dans le schéma réactionnel précédent l'alcool secondaire utilisé par deux molécules d'alcool primaire identiques de formule : R6-O-(CH2)n-1-CH2OH ou par deux molécules d'alcools primaires différents dans le cas où l'on veut obtenir un composé de formule III dans laquelle R 6 et R7, et éventuellement n et n' sont différents.
- Dans ce dernier cas, il est nécessaire de réaliser une purification des produits obtenus pour séparer le composé désiré.
- Lorsque le dérivé de phosphore est l'oxychlorure de phosphore POC13, on réalise la réaction d'estérification avec l'alcoxyalcool correspondant en présence d'une base, en particulier d'une base organique tertiaire, puis on soumet le produit obtenu à une hydrolyse, ce qui conduit à l'obtention d'un mélange de mono et de diacides que l'on sépare ensuite.
- Pour l'obtention de composés de formule IV dans laquelle R8 et R9, p et q sont identiques, le schéma réactionnel est le suivant :

- Dans le cas où l'on veut obtenir les composés de formule IV dans laquelle R 8, R9, p et q sont différents, on utilise comme produit de départ, l'alcool secondaire correspondant de formule :

- Lorsqu'on veut obtenir le composé de formule III dans laquelle R6 et R7 sont différents, on utilise deux molécules des alcools cor- = respondants et on soumet les produits obtenus à une séparation pour isoler le monoacide correspondant du mélange de mono et de diacide obtenus.
- Lorsqu'on part d'un dérivé du phosphore constitué par l'anhydride phosphorique P2O5, on réalise l'estérification avec l'alcoxyalcool correspondant, à l'abri de l'humidité, on obtient ainsi un mélange de mono et de diacides qui peuvent aussi contenir un phosphate neutre et des impuretés telles que des pyrophosphates et des polymères.
- Les alcoxyalcools utilisés comme produits de départ pour la synthèse des composés organo-phosphorés acides peuvent être préparés par réaction d'un alcoolate de sodium sur le dérivé dichloré d'un alcool secondaire, par exemple, d'alcoolate de sodium sur le dichloro-l,3-propanol-2, selon le schéma réactionnel suivant :

- Lorsqu'on veut obtenir des alcoxyalcools primaires, on fait réagir un alcoolate de sodium sur le dérivé chloré d'un alcool primaire.
- Selon l'invention, l'oxyde neutre de phosphine répondant à la formule I précitée comprend, de préférence, au moins un radical alcoxyalkyle, par exemple, un radical alcoxyméthyle ayant de 4 à 12 atomes de carbone. Lorsque les autres radicaux sont des radicaux alkyle, ceux-ci ont généralement de 4 à 12 atomes de carbone, et ils sont de préférence linéaires.
- A titre d'exemples de tels oxydes neutres de phosphine susceptibles d'être utilisés, on peut citer l'oxyde de di-isobutyl-octoxyméthylphosphine, l'oxyde de di-n-butyl-octoxyméthylphosphine, l'oxyde de di-n-pentyl-octoxyméthylphosphine, et l'oxyde de di-n-hexyl-octoxyméthylphosphine (POX 11).
- Ces oxydes de phosphine peuvent être préparés en faisant réagir un sel halogèno magnésien d'oxyde de phosphine secondaire avec un halogénure organique de formule RX dans laquelle R représente un radical alcoxyalkyle, par exemple, un éther chlorométhyl n-octylique, comme cela est décrit dans le brevet français 2.346.361 déposé le 13/12/73.
- On peut également utiliser dans le procédé de l'invention des oxydes de trialkylphos- phine dans lesquels les radicaux alkyle ont de 4 à 14 atomes de carbone, par exemple l'oxyde de tri-n-octylphosphine (TOPO).
- Pour la mise en oeuvre du procédé de l'invention, on dilue généralement le système d'extractants dans un solvant organique inerte constitué par exemple par un hydrocarbure saturé ayant au moins 8 atomes de carbone tel que le dodécane, ou par un mélange d'hydrocarbures.
- Avantageusement, dans le solvant organique les concentrations en composé organophosphoré acide et en oxyde neutre de phosphine sont telles que le rapport molaire du composé organophosphoré acide sur l'oxyde neutre de phosphine, soit compris entre 1 et 9, et de préférence de 2 à 4.
- On précise que le procédé de l'invention peut être mis en oeuvre dans tout appareil classique d'extraction tel que des batteries de mélangeurs décanteurs, des colonnes pulsées, des extracteurs centrifuges, etc...
- Selon l'invention, l'uranium extrait dans le solvant organique peut être réextrait ensuite dans une solution aqueuse d'acide phosphorique contenant éventuellement un agent réducteur de façon à réduire l'uranium VI en uranium IV pour faciliter sa réextraction.
- De préférence selon l'invention, on réalise la réextraction de l'uranium dans un appareil de réextraction comportant au moins deux étages. Dans ce cas, on met en circulation dans lesdits étages, ledit solvant organique contenant l'uranium en l'introduisant dans le premier étage, on met en circulation dans lesdits étages, à contre-courant dudit solvant organique, une solution aqueuse de carbonate d'ammonium en l'introduisant dans le dernier étage en quantité telle qu'elle représente 50 à 80% de la quantité stoechiométrique nécessaire pour neutraliser le composé organophosphoré acide et pour transformer l'uranium présent dans le solvant organique en uranyl tricarbonate d'ammonium, et on ajoute de l'armoniac sous forme de gaz ou de solution aqueuse à la solution de carbonate d'ammonium circulant dans le premier étage pour maintenir le pH dudit premier étage à une valeur comprise entre 8 et 9,5, et de préférence entre 8 et 8,5.
- De préférence, on réacidifie le solvant organique ammonié sortant du dernier étage de réextraction en le faisant réagir avec un acide pour éliminer l'ammonium sous forme de sel d'ammonium, et on réutilise le solvant organique ainsi réacidifié pour réaliser l'extraction de l'uranium.
- Avantageusement, l'acide est choisi dans le groupe comprenant l'acide sulfurique, l'acide chlorhydrique et l'acide phosphorique.
- De préférence, encore, on réacidifie le solvant organique ammonié sortant du dernier étage de réextraction en le faisant réagir avec l'acide phosphorique récupéré à la fin de l'extraction de l'uranium.
- Ce mode préférentiel de réextraction de l'uranium permet d'obtenir en fin de réextraction une solution aqueuse d'uranium à partir de laquelle on peut récupérer facilement l'uranium directement aux normes définies par les raffineurs, donc sans cycle de purification complémentaire, soit sous forme d'oxyde, soit sous la forme d'un uranate alcalin ou alcali- no-terreux, avec un rendement global de récupération en ura nium supérieur à 90%.
- Par ailleurs, il conduit à la formation de produits réutilisables. En effet, le solvant organique qui a été réacidifié par traitement avec de l'acide phosphorique peut être réutilisé pour l'extraction de l'uranium, et le phosphate d'ammonium obtenu lors du traitement de réacidification du solvant organique est un produit commercialisable ou recyclable dans une unité d'engrais par exemple.
- Selon l'invention, on effectue de préférence la réextraction de l'uranium dans trois étages. Dans ce cas, on met en circulation le solvant organique contenant l'uranium du premier au troisième étage et on introduit dans le troisième étage une solution aqueuse de carbonate d'ammonium ou un mélange de gaz carbonique et d'ammoniac préalablement dissous dans l'eau sous forme de carbonate représentant 50 à 80% de la quantité stoechiométrique nécessaire pour neutraliser le composé organophosphoré acide du solvant organique et pour transformer l'uranium en uranyl tricarbonate d'ammonium. Cette solution circule du troisième étage au premier étage et on lui ajoute avant son entrée dans le premier étage de l'ammoniac sous forme de gaz ou de solution aqueuse, la quantité ajoutée étant telle qu'on maintient le pH du premier étage à une valeur comprise entre 8 et 8,5.
- En effet, pour des pH inférieurs à 8, le taux de réextraction d'uranium décroît et pour des pH supérieurs à 8,5, la quantité d'ammoniac introduite, sans permettre d'obtenir une amélioration du taux de réextraction en uranium, conduit à la formation d'émulsions.
- De préférence, on ajoute l'ammoniac sous la forme d'une solution aqueuse ayant une concentration molaire en ammoniac de 5M à 7,5M.
- Dans ces étages, le solvant organique chargé en uranium et contenant également du fer se transforme peu à peu au contact de l'ammoniac en un sel -d'ammonium et la phase aqueuse qui se déplace à contre-courant, s'enrichit en uranium et en fer, le carbonate d'ammonium formant avec l'uranium l'uranyl tricarbonate d'ammonium qui reste en solution et le fer se transformant en hydroxyde ferrique qui précipite et qui peut être séparé par décantation de la phase aqueuse.
- A la sortie du troisième étage, le solvant organique ammonié est de préférence réacidifié par traitement au moyen d'un acide tel que l'acide sulfurique, l'acide chlorhydrique ou l'acide phosphorique, ce qui permet de récupérer une phase organique ne contenant plus d'ions ammonium et une phase aqueuse contenant un sel d'ammonium.
- De préférence, pour ce traitement, on utilise une fraction de l'acide phosphorique récupéré à la fin de l'étape d'extraction de l'uranium.
- D'autres caractéristiques et avantages de l'invention apparaîtront mieux à la lecture des exemples suivants donnés bien entendu à titre illustratif et non limitatif, en référence aux dessins annexés sur lesquels :
- - la figure 1 représente les variations du coefficient de partage D de l'uranium (courbe 1) et du fer (courbe 2) en fonction des teneurs respectives en extractants du solvant organique ;
- - la figure 2 représente les variations du coefficient de partage D de l'uranium en fonction de la concentration en H3PO4 de la phase aqueuse, pour trois systèmes d'extractants ;
- - la figure 3 représente les variations du coefficient de partage D de l'uranium en fonction de la température pour différents systèmes d'extractants ;
- - la figure 4 représente les variations des coefficients de partage de l'uranium (courbe 1) et du fer (courbe 2) en fonction du temps d'extraction ;
- - la figure 5 illustre les variations de la teneur en uranium (mg.l-1) du solvant organique en fonction de la teneur en uranium de la phase aqueuse, pour trois systèmes d'extractants, et
- - la figure 6 illustre les variations de la teneur en uranium (courbe 1) et en fer (courbe 2) du solvant organique en fonction du nombre de contacts ; et
- - la figure 7 représente schématiquement une installation de traitement d'acide phosphorique pour la mise en oeuvre du procédé de l'invention.
- Cet exemple se rapporte à la récupération de l'uranium présent dans une solution d'acide phosphorique 6M contenant lg/1 d'uranium (VI) et il illustre l'effet de la température et de la nature du système d'extractants sur le taux d'extraction en uranium.
- Dans cet exemple, on utilise les différents composés organophosphorés acides du tableau 1 avec de l'oxyde de trioctylphosphine (TOPO) ou avec de l'oxyde de di-n-hexyl-octoxymé- thyl phosphine (POX 11). Les deux extractants sont dilués dans l'hyfrane 120 qui est un hydrocarbure saturé ramifié de nombre de carbones moyen égal à 12 et la teneur en composé organophosphoré acide du solvant est de 0,5 M et sa teneur en oxyde de phosphine est de 0,125 M.
- On réalise l'extraction dans les conditions suivantes : on met en contact à 23°C ou à 40°C pendant environ 15 mn, un volume de la solution aqueuse d'acide phosphorique avec un volume du solvant organique, on agite mécaniquement les deux phases en présence, on les sépare par centrifugation, puis on prélève et on analyse chacune des phases afin de déterminer sa concentration en uranium, celle-ci étant mesurée par spectrophotométrie au dibenzoyl méthane, l'uranium étant préalablement extrait dans une solution d'oxyde de trioctylphosphine ; on détermine ensuite le coefficient de partage D de l'uranium qui est égal au rapport de la concentration en uranium de la phase organique sur la concentration en uranium de la phase aqueuse.
- Les résultats obtenus sont donnés dans le tableau 1, ci-joint.
- Au vu de ces résultats, on constate que l'effet de synergie est le plus important avec les composés 7 et 8 qui sont des esters phosphoriques d'alcools secondaires. Cependant, l'effet de synergie est également très important avec les composés 3 et 5 dans lesquels la fonction éther- oxyde est proche du groupement phosphate.
- On constate par ailleurs que l'introduction de deux fonctions éther-oxyde permet d'améliorer les résultats lorsque ces deux fonctions sont introduites suffisamment près du groupement phosphate comme dans le cas des composés n° 7 et 8, alors que l'effet obtenu avec deux fonctions en ligne (composé n°6) est plutôt néfaste.
- Dans tous les cas, le coefficient de partage de l'uranium augmente avec le nombre d'atomes de carbone des chaînes alcoxy.
- On précise que les composés 7 et 8 du tableau 1 ont été obtenus de la façon suivante. A-Préparation de l'acide bis-(dibutoxy-1,3-pro- pyl-2) phosphorique (composé n°8 dénommé ci-après HBIDIBOPP).
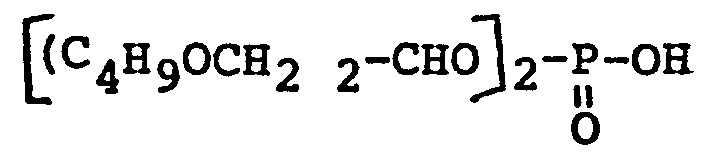
- a) Préparation du dibutoxy-l,3-Propanol-2
- On dissout à chaud tout en agitant 99 g de soude en pastilles à 90% (soit 2,23 moles) dans 1665 g de butanol anhydre (soit 22,5 moles). Lorsque la soude est entièrement solubilisée, on laisse refroidir et, à la température ambiante, on coule 139 g de dichloro-l,3-propanol-2 (soit 1,08 mole). La réaction n'est pas exothermique. La coulée terminée, on chauffe au reflux pendant une heure. On observe la formation d'un précipité de chlorure de sodium. On laisse refroidir, on filtre et on chasse l'excès de butanol. On lave le résidu deux fois à l'eau. On extrait à l'éther, on sèche sur du sulfate de magnésium et on chasse l'éther. On distille le résidu ; point d'ébullition : 110-112°C/15 mm Hg. On récupère 143 g de produit. Rendement : 65%.
- b) Préparation de l'acide bis-(dibuto- xy-l,3-propyl-2) phosphoreux par transestérification :

- On met 143 g de dibutoxy-l,3-propanol-2 (soit 0,7 mole) et 48,36 g d'acide diéthylphos- phoreux (soit 0,35 mole) sous courant d'argon dans un ballon à distiller avec colonne de Vi- greux. On chauffe progressivement jusqu'à environ 150°C dans un bain d'huile et on observe la distillation de l'éthanol. On maintient cette température tant que l'alcool distille puis on l'élève progressivement jusqu'à 180°C. On laisse refroidir et on distille sous vide les produits légers et ceux qui n'ont pas réagi. On récupère 25 g d'éthanol (rendement : 78%) et 150 g d'acide brut (rendement : 94%).
- c) Oxydation et préparation du chlorure d'acide :

- On met dans un réacteur 150 g de l'acide ci-dessus (soit 0,33 mole) et 150 ml de benzène. On refroidit à 0°C et on coule goutte à goutte, tout en agitant, une solution de chlorure de sulfuryle préparée à partir de 44,27 g de SO2Cl2(soit 0,33 mole) et 50 ml de benzène de façon à maintenir la température entre 0 et 5°C. La coulée terminée, on laisse remonter la température progressivement puis on dégaze le mélange au moyen d'azote pendant une heure. On chasse le solvant sous vide et on récupère 160 g d'un résidu visqueux. Rendement : environ 100%.
- d) Préparation de l'acide bis-(dibuto- xy-l,3-propyl-2) phosphorique
-


- On met 160 g du chlorure ci-dessus (soit 0,33 mole) et 150 ml d'eau dans le réacteur (mélange non homogène). On refroidit entre 0 et 5°C et on coule goutte à goutte 165 ml de soude 4N (soit 0,66 mole). La réaction n'est pas exothermique et on observe la formation du sel de sodium qui est insoluble. La coulée terminée, on laisse sous agitation à température ambiante pendant 1 heure, puis on chauffe à 40-50°C pendant une heure. Après refroidissement, on déplace l'acide de son sel de sodium par un acide fort (par exemple HCl). On récupère 140 g de produit. Rendement : 88%.
-
- - Dosage de la pureté par potentiométrie
- - mono-acide : 97,72%
- - di-acide : 2,52%
- - I.R. : ν (P=O) = 1235 cm-1
- - RMN'H : Confirmation de la structure.
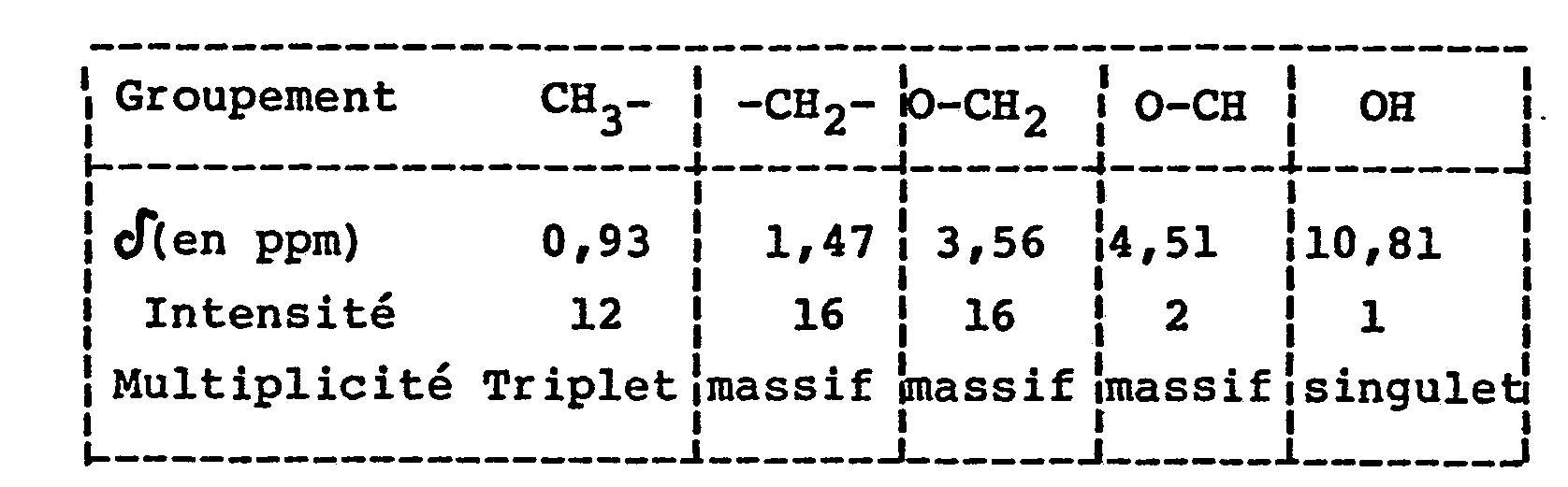
- - Analyse élémentaire

- On utilise le même mode opératoire que dans le cas de la fabrication du composé n° 8 sauf que l'alcool de départ est le propanol au lieu du butanol.
- On récupère ainsi l'acide bis-(dipropoxy-1,3-propyl-2) phosphorique.
-
- - Dosage potentiométrique : 1 seule acidité - pureté 98 %
- - Dosage volumétrique à la soude (phénolphtaléi- ne) - pureté 96,5%
- - RMN du proton : Conforme à la formule.

- Dans cet exemple, on étudie l'influence des teneurs respectives en composé organophosphoré acide et en oxyde neutre de phosphine sur l'extraction de l'uranium. Le solvant organique est constitué par de l'hyfrane 120 contenant un mélange d'hydrogénophosphate de bis(dibutoxy-1,3-propyl-2), c'est-à-dire du composé n°8 du tableau 1 et d'oxyde de di-n-hexyloctoxyméthy .phosphine (POX 11) pour récupérer l'uranium à partir d'une solution d'acide phosphorique 6N contenant 1,1 g par litre d'uranium VI. On utilise dans chaque cas une concentration totale de 0,5 M en extractants et on réalise l'extraction dans les mêmes conditions que celles de l'exemple 1.
- Les résultats obtenus sont donnés sur la courbe (1) de la figure 1, qui représente les variations du coefficient de partage D de l'uranium en fonction de la teneur en composé organophosphoré acide du solvant organique.
- Sur cette figure, la courbe 2 représente les variations du coefficient de partage D du fer en fonction de la teneur en composé réalise l'extraction dans les mêmes conditions à partir d'une solution d'acide phosphorique 6M contenant 1,1 g/1 de fer III.
- On précise que la teneur en fer des deux phases a été déterminée par absorption atomique.
- Au vu de cette figure, on constate que les meilleurs résultats présentant le moins de risque de variation du coefficient d'extraction sont obtenus lorsque le solvant organique contient 0,36 M de composé organophosphoré acide et 0,14 M d'oxyde de phosphine, et que de bons résultats sont obtenus lorsque le rapport molaire du composé organophosphoré acide sur l'oxyde neutre de phosphine est de 1 à 9.
- Cet exemple illustre l'influence de la concentration en acide phosphorique de la solution aqueuse sur l'extraction de l'uranium VI, à 23°C, au moyen des systèmes d'extractants n° I, II et III du tableau 2 dilués dans de l'hyfra- ne 120. Dans cet exemple, on réalise l'extraction dans les mêmes conditions que celles de l'exemple 1.
- Les résultats obtenus sont donnés sur la figure 2 qui représente les variations du coefficient de partage D de l'uranium VI en fonction de la concentration en acide phosphorique de la phase aqueuse. Sur cette figure, les courbes I, II et III illustrent respectivement les résultats obtenus avec les systèmes d'extractants I, II et III du tableau 2.
- Dans tous les cas, le coefficient de partage D de l'uranium décroît en fonction de la concentration en acide phosphorique, mais il décroît de façon moins importante avec les systèmes d'extractants II et III de l'invention.
- Cet exemple illustre l'influence de la température sur l'extraction de l'uranium VI. On réalise l'extraction dans les mêmes conditions que celles des exemples 2 et 3 en faisant varier la température de 10 à 60°C et en utilisant les systèmes d'extractants I à IV du tableau 2 dilués dans de l'hyfrane 120. Les résultats obtenus sont représentés sur la figure 3 où les courbes I, II, III et IV se rapportent respectivement aux systèmes I, II, III et IV.
- Au vu de cette figure, on constate que le coefficient de partage de l'uranium VI décroît lorsque la température augmente. Cependant à 60°C, le coefficient de partage obtenu avec le système n° III selon l'invention, est encore deux fois supérieur à celui obtenu à 40°C avec le système N° I de l'art antérieur.
- Dans cet exemple, on réalise l'extraction de l'uranium à partir d'une solution d'acide phosphorique 6M contenant 1,06 g/1 d'uranium VI et 4,70 g/1 de fer III, en utilisant le système d'extractants n° III du tableau 2 dilué dans l'hyfrane 120 et en opérant dans les mêmes conditions que celles de l'exemple 1. mais en déterminant les coefficients de partage de l'uranium VI et du fer III après différentes durées d'extraction. Les résultats obtenus sont donnés sur la figure 4 qui représente l'évolution du taux d'extraction (en %) dans la phase organique de l'uranium (courbe 1) et du fer (courbe 2) en fonction de la durée d'extraction (en secondes).
- Au vu de cette figure, on constate que l'uranium VI est extrait plus rapidement. La sélectivité à l'équilibre défini par la formule :

- Dans cet exemple, on récupère l'uranium à partir d'une solution d'acide phosphorique industriel titrant 27% en P205 et 130 mg/1 en uranium, en utilisant comme solvant organique les systèmes d'extractants I, III et IV du tableau 2 dilués dans le produit vendu sous la marque Escaid 110, qui est un kérosène désaromatisé, avec une concentration de 0,5 M en composé organophosphoré acide et une concentration de 0,125 M en oxyde de phosphine.
- Pour réaliser l'extraction, on met en contact successivement 8 fractions de l'acide phosphorique avec une fraction de solvant organique, chaque fraction ayant un volume de 50 ml, en opérant dans une ampoule à décanter à double enveloppe, thermostatée à 40°C et agitée manuellement pendant 5 mn. Après décantation des deux phases, on détermine la teneur en uranium du solvant organique et la teneur en uranium de la solution d'acide phosphorique. Les résultats obtenus sont donnés sur la figure 5 dont les courbes I, III et IV illustrent respectivement la teneur en uranium VI (en mg/1) du solvant organique en fonction de la teneur en uranium (mg/1) de l'acide phosphorique, pour les systèmes d'extractants I, III et IV.
- Au vu de cette figure, on constate que le système d'extractants n° III de l'invention est nettement supérieur aux systèmes d'extractants N° I et IV de l'art antérieur.
- Sur la figure 6, on a illustré également les résultats obtenus en ce qui concerne l'extraction du fer en fonction du nombre de contacts. Sur cette figure, la courbe 2 représente l'évolution de la concentration en fer du solvant organique en fonction du nombre de contacts, et la courbe 1 représente l'évolution de la concentration en uranium du solvant organique en fonction du nombre de contacts, lorsqu'on utilise le système d'extractants III de l'invention.
- Le solvant organique ainsi obtenu qui contient 798 mg/1 d'uranium et 775 mg/1 de fer est mis en contact avec une solution de carbonate d'ammonium à 140 g/1 et 0,5 M en NH40H pour réex- traire l'uranium en solution et séparer le fer sous forme d'hydroxyde. A la fin de cette réextraction, la phase organique contient seulement 0,4 mg/1 d'uranium et 1 mg/1 de fer.
- L'exploitation des courbes de la figure 5 selon la méthode de Mac Cabe et Thiele conduit aux conclusions suivantes : avec un taux de récupération en uranium de 99%, on obtient un facteur de concentration de 6,1 pour 5 étages avec le système d'extractants N° III alors que dans le cas des systèmes de l'art antérieur (n° I et IV) on obtient respectivement des facteurs de concentration de 2,3 et de 3,0.
- Ainsi, le système d'extractants n° III extrait plus de trois fois mieux l'uranium que le système classique de l'art antérieur (système n° I) et plus de deux fois mieux que le système n° IV de l'art antérieur. De ce fait, on peut réduire le volume de phase organique et donc limiter les consommations en carbonate d'ammonium et en ammoniaque lors de l'opération de réextraction.
- On utilise différents mélanges d'extractants pour récupérer l'uranium à partir d'acide phosphorique industriel présentant les caractéristiques suivantes :
- - densité : 1,292
- - potentiel redox : 370 mV/E.C.S.
- - viscosité : 4,65 cp
- - teneur en uranium : 135 mg.l-1
- - teneur en PO4 : 484,9 g.l-1 (P2O5=28,04%)
- - teneur en F : 15,8 g.l-1
- - teneur en V : 174 mg.l-1
- - teneur en S04 : 18,26 g.l 1
- - teneur en Fe : 2,12 g.l-1
- - teneur en Al : 1,62 g.l-1
- Les solvants organiques utilisés contiennent les systèmes d'extractants I, III, IV ou V du tableau 2 joint, et un diluant constitué par du kérosène connu sous la dénomination commerciale ISOPAR L. Dans chaque solvant testé, la teneur en oxyde de phosphine est de 0,125 M et la teneur en composé organo phosphoré acide est de 0,500 M.
- On réalise l'extraction en mettant en contact à 39°C un volume de la solution aqueuse . d'acide phosphorique avec un volume du solvant organique sous agitation pendant environ 5 min ; on sépare ensuite les deux phases puis on les prélève et on les analyse chacune pour obtenir leur concentration en uranium et leur concentraction en fer et déterminer ensuite le coefficient de partage D de l'uranium et le coefficient de partage D du fer. Les résultats obtenus sont donnés dans le tableau 3, joint. Au vu de ce tableau, on constate que les solvants organiques contenant le système d'extractants de l'invention, c'est-à-dire le composé organophosphoré acide HBIDIBOPP associé à un oxyde de phosphine tel que le POX11 ou le TOPO permettent d'obtenir des résultats très améliorés par rapport aux systèmes d'extractants I et IV de l'art antérieur.
- Dans une autre série d'essais, on effectue l'extraction en mettant en contact successivement comme dans l'exemple 6, huit fractions d'acide phosphorique industriel avec une fraction de chacun des solvants organiques du tableau 3 pour obtenir les courbes d'équilibre analogues à celles représentées sur la figure 5. Une exploitation de ces courbes pour les différents sol- vants selon la méthode de Mac Cabe et Thiele indique que pour obtenir à la sortie d'une installation d'extraction à 5 étages de l'acide phosphorique ayant une teneur en uranium inférieure ou égale à 3-4 mg.l-1, on charge le solvant d'extraction à :
- - 350 mg.l-l d'uranium et 78 mg.l-1 de fer pour le système d'extractants n°I
- - 450 mg.l-1 d'uranium et 125 mg.l-l de fer pour le système d'extractants n°IV,
- - 1000 mg.l-1 d'uranium et environ 1250 mg.l-1 de fer pour le système d'extractants n°III, et
- - 800 mg.l-1 d'uranium et environ 800 mg.l-1 de fer pour le système d'extractants n°V.
- Ainsi, on remarque que pour la même efficacité de traitement, on obtient avec les systèmes d'extractants de l'invention des solvants beaucoup plus chargés en uranium.
- Cet exemple concerne l'extraction de l'uranium contenu dans de l'acide phosphorique industriel présentant les mêmes caractéristiques que celui de l'exemple 7, en utilisant pour l'extraction l'installation représentée sur la figure 7.
- Sur cette figure 7, la référence A désigne l'unité d'extraction de l'uranium qui comprend cinq étages d'extraction, la référence B représente une unité de lavage du solvant organique qui comprend trois étages, les références C1, C 2 et C3 désignent les trois étages de réextraction de l'uranium, la référence D illustre l'unité de séparation de l'uranium et la référence E désigne l'unité de réacidification du solvant organique, qui comprend deux étages.
- Dans l'unité d'extraction A, on introduit par la ligne la, l'acide phosphorique industriel après avoir floculé et décanté celui-ci. On précise que cet acide a été soumis au préalable à un traitement d'oxydation pour amener la totalité de l'uranium sous la forme hexavalente, ce qui amène également le fer à l'état trivalent. Dans l'unité d'extraction A, l'acide phosphorique est mis en contact à contre-courant avec un solvant organique introduit par la ligne 3a. Ce solvant organique comprend un système d'extractants constitué par un composé organophosphoré acide et par un oxyde neutre de phosphine dilué dans du kérosène connu sous la dénomination commerciale ISOPAR L, la concentration en composé organophosphoré acide du solvant étant de 0,5 mol.l-1 et la concentration en oxyde de phosphine du solvant organique étant de 0,125 mol.l-1.
- La solution d'acide phosphorique circule dans l'unité d'extraction à un débit qui est maintenu à la valeur de 4 1/h, et le solvant organique circule à contre-courant dans l'unité d'extraction en étant introduit à un débit de 1,6 1/h pour le système d'extractants n°I, de 1,0 1/h pour le système d'extractants n°IV et de 0,54 1/h pour le système d'extractants n°III.
- Dans chaque étage d'extraction, on recycle une partie du solvant organique sortant de cet étage, ce qui permet d'augmenter le volume de phase organique en contact avec l'acide phosphorique dans l'unité d'extraction A dont tous les étages sont maintenus à 35°C.
- A la sortie de l'unité d'extraction A, l'acide phosphorique ne contenant pratiquement plus d'uranium est évacué par la ligne lb et le solvant organique chargé en uranium et en fer est évacué par la ligne 3b. Ce solvant passe ensuite dans l'unité de lavage comportant trois étages où il est lavé par de l'eau pour éliminer les ions phosphoriques entraînés par le solvant. L'eau chargée d'acide phosphorique qui quitte le dernier étage de l'unité de lavage est recyclée dans l'usine de fabrication d'acide phosphorique où elle sert au lavage ou au rinçage des installations.
- A la sortie de l'unité de lavage B, le solvant organique est introduit par la ligne 3c dans le premier étage de réextraction C1, puis il circule dans les étages suivants C2 et C3, les étages C 1, C2 et C3 étant maintenus à 40°C.
- Dans les étages C2 et C3, il est mis en contact à contre-courant avec une solution de carbonate d'ammnium à 155 g.l-1 introduite dans le dernier étage C3 par la ligne 4a et dans l'étage C1, il est mis en contact à contre-courant avec la solution de carbonate provenant de l'étage C2 et avec de l'ammoniaque à 200 g.l-1 injectée par la ligne 5 dans la solution de carbonate qui pénètre dans le premier étage C1. On règle le débit d'ammoniaque à l'aide d'une vanne commandée par un pH-mètre de façon à maintenir le pH du premier étage CI à une valeur de 8,2. De même, on règle le débit de la solution de carbonate d'ammonium introduite dans le dernier étage C3 par la ligne 4a de façon telle qu'il corresponde à 50 à 80% de la quantité stoechiométrique nécessaire pour neutraliser, d'une part, le composé organo phosphoré acide et transformer, d'autre part, l'uranium en uranyle tri-carbonate d'ammonium.
- Lors de la réextraction, le solvant organique chargé en uranium et en fer, qui est tout d'abord en contact avec l'ammoniaque se transforme peu à peu en un sel d'ammonium hydraté et la phase aqueuse qui se déplace à contre-courant s'enrichit en uranium et en fer, le carbonate d'ammonium réagissant avec l'uranium pour former de l'uranyle tricarbonate d'ammonium qui reste en solution et le fer étant précipité sous la forme d'hydroxyde que l'on sépare par filtration. La phase aqueuse contenant l'uranyle tricarbonate d'ammonium sort du premier étage de réextraction CI par la ligne 4b et elle est ensuite dirigée vers l'unité de séparation D de l'uranium.
- Dans cette installation, l'uranium peut être séparé de la solution, soit sous forme d'oxyde, soit sous forme d'uranate de sodium. Pour obtenir l'uranium sous forme de trioxyde d'uranium, on soumet la solution d'uranyle tri- carbonate d'ammonium dans un réacteur à barbotage d'air, à température comprise entre 90 et 1000C, pendant environ 6 heures, puis on filtre le précipité et on le lave à l'eau ; après séchage à 120°C et grillage à 400°C environ, on obtient ainsi le trioxyde d'uranium. Pour obtenir l'uranium sous forme d'uranate de sodium, on neutralise au moyen de soude, à environ 80°C la solution d'uranyle tricarbonate d'ammonium qui a été préalablement dégazée par barbotage d'air vers 90°C pour éliminer le gaz carbonique et l'ammoniaque, puis on précipite l'uranium par addition d'hydroxyde de sodium à la solution, en opérant à une température de 80°C pendant 1 heure. Après filtration et lavage à l'eau à 50°C, on recueille l'uranate de sodium qui peut être transformé ultérieurement en diuranate d'ammonium ou en trioxyde d'uranium.
- A la sortie du troisième étage C3 de réextraction, le solvant organique désuranié est évacué par la ligne 3d et envoyé à l'unité purification réacidification E comportant deux étages dans lesquels il est traité au moyen d'acide phosphorique introduit par la ligne lc, cet acide phosphorique constituant une fraction de l'acide phosphorique qui sort de l'unité d'extraction A par la conduite lb. Par mise en contact du solvant organique avec l'acide phosphorique, on décompose le sel d'ammonium de l'agent d'extraction, ce qui conduit à la formation de phosphate d'ammonium évacué par la ligne 6 et à l'obtention de solvant organique acidifié, qui peut être recycle par la ligne 3a pour être réutilisé dans l'unité d'extraction A.
- On précise que le phosphate d'ammonium ainsi récupéré peut être commercialisé directement ou être utilisé dans les unités de fabrication d'engrais.
- On réalise dans cette installation différents essais d'extraction de l'uranium à partir d'acide phosphorique industriel en utilisant des solvants organiques comprenant les systèmes d'extractants I, III ou IV du tableau 3 pour traiter 472 1 d'acide phosphorique, ce qui représente une durée de 118 h. Pendant ces 118 h, on utilise 73 h pour tester le système d'extractants n°III de l'invention, ce qui correspond à 292 1 d'acide phosphorique traité. Dans chaque cas, on prélève des échantillons de phase organique et de phase aqueuse dans chacun des étages d'extraction après avoir atteint l'équilibre et on détermine leur teneur en uranium et éventuellement en fer. Les résultats obtenus sont donnés dans le tableau 4.
- Au vu de ces résultats, on constate qu'il est possible d'atteindre une charge de 1003 mg.l-1 avec le solvant de l'invention en utilisant simplement 5 étages d'extraction. On peut d'ailleurs charger ce solvant à 1220 mg.l-1 en utilisant un sixième étage tout en obtenant à la sortie de l'acide phosphorique contenant moins de 2 mg/1 d'uranium. Dans ce cas, le débit de solvant organique est de 0,43 l/h.
- Dans le cas des solvants de l'art antérieur, une installation d'extraction à 5 étages permet seulement de charger le solvant organique à 300 ou 540 mg.l-1, si l'on veut obtenir à la sortie de l'acide phosphorique contenant moins de 2 mg.l-1 d'uranium.
- Par ailleurs, 'ces résultats montrent que l'on obtient pratiquement la charge théorique en uranium du solvant.


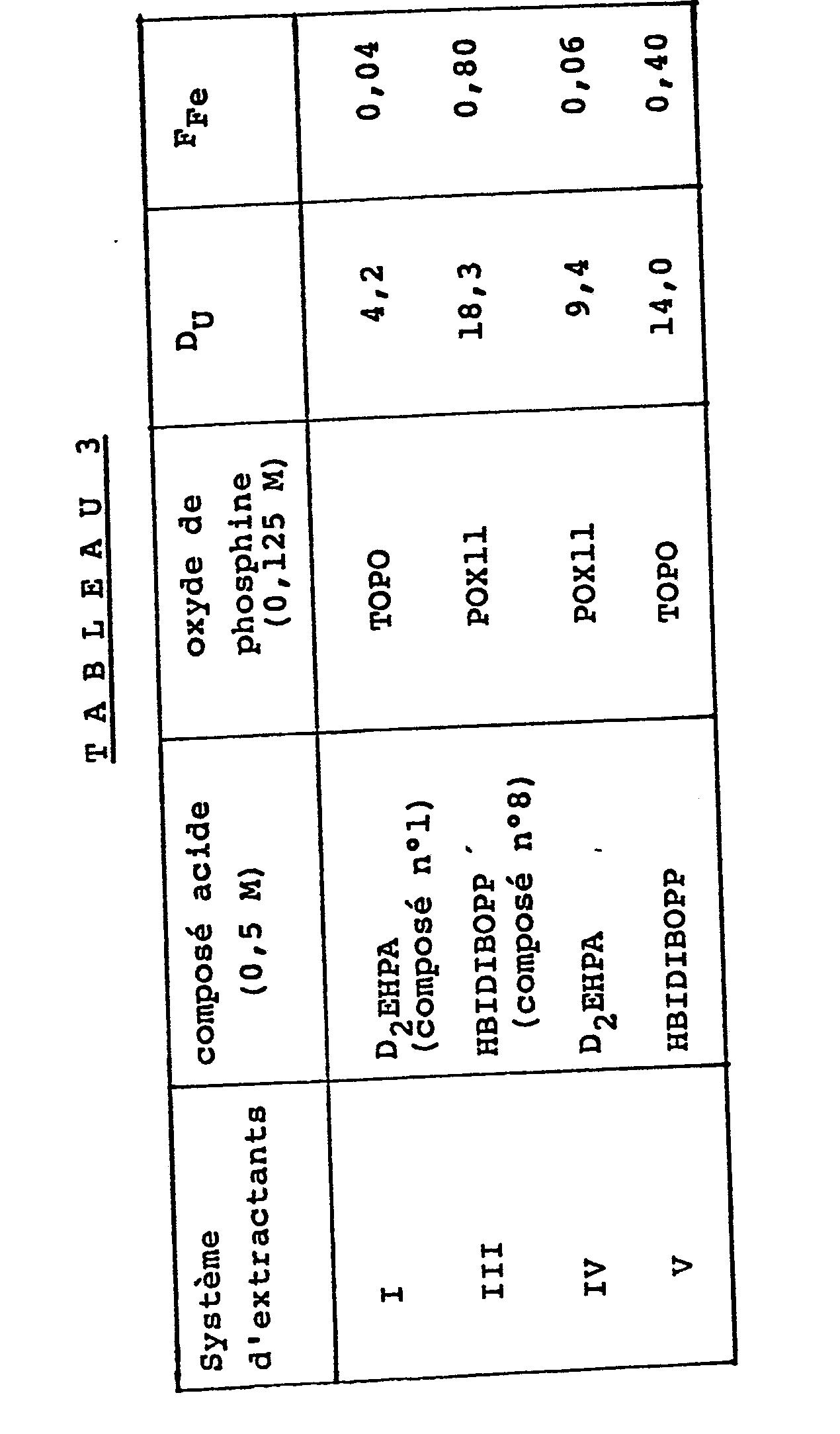


Claims (12)

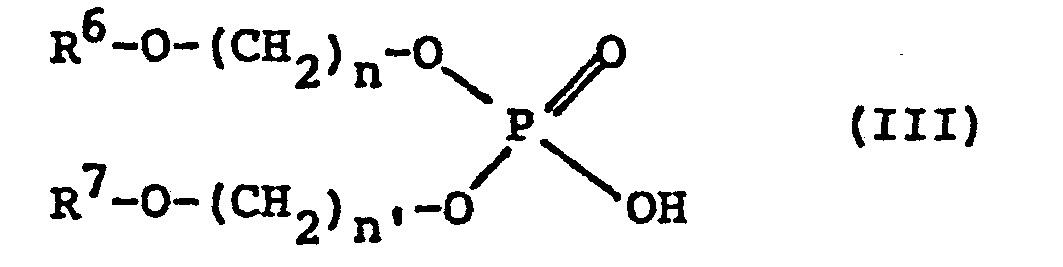

Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR8024253A FR2494258A1 (fr) | 1980-11-14 | 1980-11-14 | Procede de recuperation de l'uranium present dans des solutions d'acide phosphorique |
| FR8024253 | 1980-11-14 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0053054A1 true EP0053054A1 (fr) | 1982-06-02 |
| EP0053054B1 EP0053054B1 (fr) | 1985-01-23 |
Family
ID=9247989
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP81401686A Expired EP0053054B1 (fr) | 1980-11-14 | 1981-10-23 | Procédé de récupération de l'uranium (VI) présent dans des solutions d'acide phosphorique |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US4432946A (fr) |
| EP (1) | EP0053054B1 (fr) |
| JP (1) | JPS57110324A (fr) |
| AU (1) | AU542919B2 (fr) |
| BR (1) | BR8107393A (fr) |
| CA (1) | CA1188106A (fr) |
| DE (1) | DE3168526D1 (fr) |
| EG (1) | EG15457A (fr) |
| ES (1) | ES507131A0 (fr) |
| FR (1) | FR2494258A1 (fr) |
| JO (1) | JO1166B1 (fr) |
| MA (1) | MA19328A1 (fr) |
| OA (1) | OA06944A (fr) |
| YU (1) | YU42740B (fr) |
| ZA (1) | ZA817498B (fr) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0239501A1 (fr) * | 1986-03-28 | 1987-09-30 | Compagnie Generale Des Matieres Nucleaires (Cogema) | Procédé de séparation du fer à partir d'une solution organique contenant de l'uranium |
| WO2017001494A1 (fr) | 2015-06-30 | 2017-01-05 | Areva Mines | Procede de separation du fer d'une phase organique contenant de l'uranium et procede d'extraction de l'uranium d'une solution aqueuse d'acide mineral contenant de l'uranium et du fer |
| WO2019025714A1 (fr) | 2017-07-31 | 2019-02-07 | Orano Mining | Composés bifonctionnels à fonction thiophosphine, utiles comme extractants de l'uranium(vi), leurs procédés de synthèse et leurs utilisations |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5188736A (en) * | 1991-08-27 | 1993-02-23 | Institute Of Nuclear Energy Research | Process for the separation and recovery of extractant from spent solvent |
| US20110226694A1 (en) * | 2010-03-22 | 2011-09-22 | Battelle Energy Alliance, Llc | Methods of reducing radiotoxicity in aqueous acidic solutions and a reaction system for same |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR1193653A (fr) * | 1956-06-29 | 1959-11-04 | Atomic Energy Commission | Agents d'extraction à base d'orthophosphates organiques et leurs procédés d'utilisation |
| FR1303476A (fr) * | 1960-06-03 | 1962-09-14 | Atomic Energy Commission | Procédé d'extraction liquide-liquide pour la récupération de l'uranium |
| FR2364180A1 (fr) * | 1976-09-10 | 1978-04-07 | Westinghouse Electric Corp | Procede de recuperation d'uranium dans le procede de fabrication d'acide phosphorique par voie humide |
| FR2423545A1 (fr) * | 1977-08-25 | 1979-11-16 | Minemet Rech Sa | Procede pour la recuperation de l'uranium contenu dans des solutions phosphatees |
| FR2442796A1 (fr) * | 1978-11-28 | 1980-06-27 | Commissariat Energie Atomique | Procede de recuperation de l'uranium present dans les solutions d'acide phosphorique |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4243637A (en) * | 1977-10-11 | 1981-01-06 | Occidental Petroleum Company | Uranium recovery from pre-treated phosphoric acid |
| IL58726A (en) * | 1978-11-28 | 1982-12-31 | Commissariat Energie Atomique | Recovery of uranium from phosphoric acid solutions |
-
1980
- 1980-11-14 FR FR8024253A patent/FR2494258A1/fr active Granted
-
1981
- 1981-10-23 DE DE8181401686T patent/DE3168526D1/de not_active Expired
- 1981-10-23 EP EP81401686A patent/EP0053054B1/fr not_active Expired
- 1981-10-27 AU AU76847/81A patent/AU542919B2/en not_active Ceased
- 1981-10-29 ZA ZA817498A patent/ZA817498B/xx unknown
- 1981-10-30 US US06/316,941 patent/US4432946A/en not_active Expired - Fee Related
- 1981-11-05 YU YU2627/81A patent/YU42740B/xx unknown
- 1981-11-11 EG EG659/81A patent/EG15457A/xx active
- 1981-11-11 MA MA19532A patent/MA19328A1/fr unknown
- 1981-11-12 CA CA000389920A patent/CA1188106A/fr not_active Expired
- 1981-11-13 ES ES507131A patent/ES507131A0/es active Granted
- 1981-11-13 BR BR8107393A patent/BR8107393A/pt unknown
- 1981-11-13 JP JP56182163A patent/JPS57110324A/ja active Pending
- 1981-11-14 OA OA57540A patent/OA06944A/fr unknown
-
1982
- 1982-02-18 JO JO19821166A patent/JO1166B1/en active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR1193653A (fr) * | 1956-06-29 | 1959-11-04 | Atomic Energy Commission | Agents d'extraction à base d'orthophosphates organiques et leurs procédés d'utilisation |
| FR1303476A (fr) * | 1960-06-03 | 1962-09-14 | Atomic Energy Commission | Procédé d'extraction liquide-liquide pour la récupération de l'uranium |
| FR2364180A1 (fr) * | 1976-09-10 | 1978-04-07 | Westinghouse Electric Corp | Procede de recuperation d'uranium dans le procede de fabrication d'acide phosphorique par voie humide |
| FR2423545A1 (fr) * | 1977-08-25 | 1979-11-16 | Minemet Rech Sa | Procede pour la recuperation de l'uranium contenu dans des solutions phosphatees |
| FR2442796A1 (fr) * | 1978-11-28 | 1980-06-27 | Commissariat Energie Atomique | Procede de recuperation de l'uranium present dans les solutions d'acide phosphorique |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0239501A1 (fr) * | 1986-03-28 | 1987-09-30 | Compagnie Generale Des Matieres Nucleaires (Cogema) | Procédé de séparation du fer à partir d'une solution organique contenant de l'uranium |
| FR2596383A1 (fr) * | 1986-03-28 | 1987-10-02 | Cogema | Procede de separation du fer a partir d'une solution organique contenant de l'uranium |
| US5017344A (en) * | 1986-03-28 | 1991-05-21 | Compagnie Generale Des Matieres Nucleaires (Cogema) | Process for the separation of iron from an organic solution containing uranium |
| WO2017001494A1 (fr) | 2015-06-30 | 2017-01-05 | Areva Mines | Procede de separation du fer d'une phase organique contenant de l'uranium et procede d'extraction de l'uranium d'une solution aqueuse d'acide mineral contenant de l'uranium et du fer |
| WO2019025714A1 (fr) | 2017-07-31 | 2019-02-07 | Orano Mining | Composés bifonctionnels à fonction thiophosphine, utiles comme extractants de l'uranium(vi), leurs procédés de synthèse et leurs utilisations |
Also Published As
| Publication number | Publication date |
|---|---|
| ES8206387A1 (es) | 1982-08-16 |
| AU7684781A (en) | 1982-05-20 |
| YU42740B (en) | 1988-12-31 |
| MA19328A1 (fr) | 1982-07-01 |
| JPS57110324A (en) | 1982-07-09 |
| EG15457A (en) | 1988-10-31 |
| CA1188106A (fr) | 1985-06-04 |
| FR2494258A1 (fr) | 1982-05-21 |
| DE3168526D1 (en) | 1985-03-07 |
| YU262781A (en) | 1983-12-31 |
| OA06944A (fr) | 1983-07-31 |
| EP0053054B1 (fr) | 1985-01-23 |
| JO1166B1 (en) | 1983-11-30 |
| ZA817498B (en) | 1982-10-27 |
| AU542919B2 (en) | 1985-03-21 |
| FR2494258B1 (fr) | 1984-11-02 |
| ES507131A0 (es) | 1982-08-16 |
| BR8107393A (pt) | 1982-08-10 |
| US4432946A (en) | 1984-02-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0210928B1 (fr) | Nouveaux agents d'extraction et nouveaux propanediamides, leur utilisation pour la récuperation des actinides et/ou des lanthanides et leur procédé de préparation | |
| EP0156735B1 (fr) | Procédé de séparation des terres rares par extraction liquide-liquide | |
| US4986742A (en) | Process for the production of high-grade titanium dioxide by sulfate method | |
| FR2990206A1 (fr) | Nouveaux composes bifonctionnels utiles comme ligands de l'uranium(vi), leurs procedes de synthese et leurs utilisations | |
| EP0110789B1 (fr) | Procédé de récupération des actinides et/ou des lanthanides présents à l'état trivalent dans une solution aqueuse acide | |
| US3067010A (en) | Process for preparation of titanium dioxide | |
| CN102753711B (zh) | 用于从天然铀浓缩物中纯化铀的方法 | |
| EP0038764A1 (fr) | Applications à l'échange de cathions de dérivés diphosphoniques, nouveaux dérivés diphosphoniques et leur procédé de fabrication | |
| EP0090692B1 (fr) | Procédé de récuperation et de purification d'un acide sulfurique residuaire contenant des sels de titane | |
| CA2904956A1 (fr) | Utilisation de composes a fonctions amide et phosphonate pour extraire l'uranium(vi) de solutions aqueuses d'acide sulfurique, issues notamment de la lixiviation sulfurique de minerais uraniferes | |
| EP0053054B1 (fr) | Procédé de récupération de l'uranium (VI) présent dans des solutions d'acide phosphorique | |
| FR3062128A1 (fr) | N,n-dialkylamides dissymetriques, utiles notamment pour separer l'uranium(vi) du plutonium(iv), leur synthese et leurs utilisations | |
| Umetani et al. | Solvent extraction of lithium and sodium with 4-benzoyl or 4-perfluoroacyl-5-pyrazolone and TOPO | |
| CA1187706A (fr) | Methode pour separer le germanium d'une solution aqueuse | |
| WO2019002788A1 (fr) | Carbamides pour la séparation de l'uranium(vi) et du plutonium(iv) sans réduction du plutonium(iv) | |
| FR2684670A1 (fr) | Amides a substituant heterocyclique azote, leur procede de preparation et leur utilisation pour extraire selectivement les actinides (iii) et les separer en particulier des lanthanides (iii). | |
| US4256716A (en) | Process for treating loaded extractant from purification of phosphoric acid by extraction | |
| EP0527685B1 (fr) | Procédé pour séparer les actinides des lanthanides par extraction sélective des actinides dans un solvant organique comprenant un propanediamide | |
| EP0129448B1 (fr) | Procédé d'extraction sélective du cuivre utilisant les 4-acyl(3H)-pyrazol-3-ones | |
| EP0266272B1 (fr) | Procédé de récupération de composés organophosphorés acides et/ou d'ions organophosphate présents dans une solution aqueuse et son utilisation pour le traitement d'effluents aqueux | |
| JP3310728B2 (ja) | 希土類金属のための抽出剤及び希土類金属の分離、精製方法 | |
| US4275038A (en) | Process for treating loaded extractant from purification of phosphoric acid by extraction and recovering nutrients | |
| FR2459205A2 (fr) | Procede de recuperation de l'uranium present dans une solution d'acide phosphorique | |
| EP0239501B1 (fr) | Procédé de séparation du fer à partir d'une solution organique contenant de l'uranium | |
| EP0011516A1 (fr) | Procédé pour la récupération de l'uranium contenu dans des composés phosphatés |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Designated state(s): BE DE FR GB IT NL |
|
| 17P | Request for examination filed |
Effective date: 19821109 |
|
| ITF | It: translation for a ep patent filed | ||
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Designated state(s): BE DE FR GB IT NL |
|
| REF | Corresponds to: |
Ref document number: 3168526 Country of ref document: DE Date of ref document: 19850307 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed | ||
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: BE Effective date: 19861031 |
|
| BERE | Be: lapsed |
Owner name: COMMISSARIAT A L'ENERGIE ATOMIQUE ETABLISSEMENT D Effective date: 19861031 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 19871031 Year of fee payment: 7 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Effective date: 19881023 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Effective date: 19890501 |
|
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee | ||
| GBPC | Gb: european patent ceased through non-payment of renewal fee | ||
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 19890925 Year of fee payment: 9 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19910702 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 19991027 Year of fee payment: 19 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20010629 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |