-
Die Verwendung der Positronen-Emissions-Tomographie (PET) breitet sich aufgrund der Verfügbarkeit kompakter Cyclotrone für den medizinischen Bereich und automatisierter Chemie für die Produktion von Radiopharmazeutika weltweit schnell aus.
[H. H. Coenen et al., Nucl. Med. Biol. 2010, 10, 727–740.]
-
Unter den Positronen-emittierenden Isotopen besitzt [18F]Fluorid heutzutage eine große Bedeutung als Markierung für Radiotracer, welche als klinische Diagostika und molekulare Sonden in der Wirkstofffindung und -entwicklung eingesetzt werden.
[L. Cai, S. Lu, V. W. Pike, Eur. J. Org. Chem. 2008, 2853–2873.]
-
Hinsichtlich der Konzeption und der Entwicklung von [18F]fluormarkierten PET-Tracern sind Synthesemethoden, die den Zugang zu einer Vielfalt hochselektiver Liganden ermöglichen, von besonderer Bedeutung. In diesem Zusammenhang stellt die Einführung von [18F]Fluorid in aromatische Verbindungen eine besondere Herausforderung dar.
[M. Tredwell, V. Gouverneur, Angew. Chem. Int. Ed. 2012, 51, 11426–11437; R. Littich, P. J. H. Scott; Angew. Chem. Int. Ed. 2012, 51, 1106–1109.]
-
Neuere Entwicklungen beinhalten die Verwendung von Iodoniumsalzen
[T. L. Ross, J. Ermert, C. Hocke, H. H. Coenen, J. Am. Chem. Soc. 2007, 129, 8018–8025.]
wie auch Palladium-katalysierte elektrophile Fluorierungen.
[E. Lee, A. S. Kamlet, D. C. Powers, C. N. Neumann, G. B. Boursalian, T. Furuya, D. C. Choi, J. M. Hooker, T. Ritter, Science 2011, 334, 639–642; Z. Gao, Y. H. Lim, M. Tredwell, L. Li, S. Verhoog, M. Hopkinson, W. Kaluza, T. L. Collier, J. Passchier, M. Huiban, V. Gouverneur, Angew. Chem. Int. Ed. 2012, 51, 6733–6737; I. S. R. Stenhagen, A. K. Kirjavainen, S. J. Forsback, C. G. Jørgensen, E. G. Robins, S. K. Luthra, O. Solin, V. Gouverneur, Chem. Commun. 2013, 49, 1386.]
-
Zur Einführung von 18F-Fluorid mittels nukleophiler aromatischer Substitution sind (wie oben erwähnt) Iodoniumsalze einsetzbar, oder das aromatische System muss für diesen Reaktionstyp durch elektronenziehende Gruppen aktiviert werden.
[J. C. Meleán, J. Ermert, H. H. Coenen, Org. Biomol. Chem. 2011, 9, 765–769; J. C. Meleán, J. Ermert, H. H. Coenen, Tetrahedron 2010, 66, 9996–10001.]
-
Die Reaktionsbedingungen derartiger nukleophiler Substitutionen sind meist jedoch immer noch an erhöhte Temperaturen gebunden und können nicht in sehr kurzen Zeiten bei gleichzeitig hohen Ausbeuten realisiert werden.
-
Vorbereitende, nicht-radioaktive Experimente haben gezeigt, dass Azocarbonsäureester (Alkyloxycarbonylazogruppen) hoch aktivierende Substituenten für nukleophile aromatische Substitutionen sind.
[H. Jasch, S. Höfling, M. R. Heinrich, J. Org. Chem. 2012, 77, 1520–1532; S. B. Höfling, A. L. Bartuschat, M. R. Heinrich, Angew. Chem. Int. Ed. 2010, 49, 9769–9772.]
-
Ein Versuch zur Einführung von [18F]Fluorid in 4-Nitrophenylazocarbonsäure-tert-butylester ergab 4-[18F]Fluorphenylazocarbonsäure-tert-butylester in 74% Ausbeute in 5 Minuten.
[S. Höfling, Entwicklung neuartiger Syntheseverfahren über Arylradikale mit Anwendungen in der Radiopharmazie, Dissertation, TU München 2011, ISBN 978-3-8439-0114-7.]
-
Obwohl diese Reaktion in guter Ausbeute, in einer akzeptablen Reaktionszeit und unter vergleichsweise milden Bedingungen verlief, so wird die Verwendung von 4-Nitrophenylazocarbonsäure-tert-butylester als Markierungsvorläufer signifikant durch die aufwändige Abtrennung des Produkts 4-[18F]Fluorphenylazocarbonsäure-tert-butylester vom Überschuss der Nitroverbindung erschwert.
-
Diese Erfindung basiert auf der Herausforderung, einen hoch aktivierten aromatischen Vorläufer zu konzipieren, der die nukleophile Einführung von [18F]Fluorid in sehr kurzen Reaktionszeiten, unter milden Bedingungen, mit hohen Ausbeuten und einer einfachen Reinigung des markierten Produktes erlauben würde – unter besonderer Beachtung der Abtrennung von seinem unmarkierten Vorläufer.
-
Dieses Ziel wurde wie folgt erreicht.
- 1. Ein Verfahren zur Herstellung von Verbindungen der Formel 1 dadurch gekennzeichnet, dass eine Verbindung der Formel 2 mit [18F]Fluorid zur Reaktion gebracht wird
und
R1 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, tert-Butyl, Trityl, Allyl, Alkyl, Cycloalkyl, Phenyl, Aryl oder Heteroaryl ist;
X Fluorid, Chlorid, Bromid, Iodid, Sulfat, Tetrafluoroborat, Acetat, Trifluoracetat, Hexafluorophosphat, Hexafluoroantimonat, Methansulfonat, Trifluormethansulfonat, Toluol-4-sulfonat oder Monomethylsulfat ist;
R2 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist;
R3 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist;
R5 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist;
R6 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist;
R11 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, n-Butyl oder Phenyl ist;
R12 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, n-Butyl oder Phenyl ist; und
R13 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, n-Butyl oder Phenyl ist.
- 2. Ein Verfahren nach dem vorhergehenden Anspruch, dadurch gekennzeichnet, dass das [18F]Fluoridsalz in Kombination mit einem komplexierenden Liganden eingesetzt wird.
- 3. Ein Verfahren nach den vorhergehenden Ansprüchen, dadurch gekennzeichnet, dass die Reaktion in Gegenwart mindestens eines Lösungsmittels durchgeführt wird.
- 4. Ein Verfahren nach den vorhergehenden Ansprüchen, dadurch gekennzeichnet, dass die Reaktion in Gegenwart mindestens einer Base durchgeführt wird.
- 5. Ein Verfahren nach den vorhergehenden Ansprüchen, dadurch gekennzeichnet, dass die Reaktion unter Schutzgasatmosphäre durchgeführt wird.
- 6. Ein Verfahren nach den vorhergehenden Ansprüchen, dadurch gekennzeichnet, dass die Reaktion in einem Temperaturbereich von 0°C bis 200°C durchgeführt wird.
- 7. Ein Verfahren zur Herstellung von Verbindungen der Formel 3 dadurch gekennezichnet, dass eine Verbindung der Formel 1 mit einer Verbindung der Formel 4 zur Reaktion gebracht wird und
R20 unabhängig voneinander bezeichnet;
und
R1, R2, R3, R5 und R6 wie oben definiert sind.
-
Verbindungen der Formel 1 können desweiteren unter sauren, basischen (über das Azocarboxylatanion) oder thermischen Bedingungen zur Synthese [18F]Fluor-markierter Biarylverbindungen eingesetzt werden.
[H. Jasch, S. Höfling, M. R. Heinrich, J. Org. Chem. 2012, 77, 1520–1532]
-
Unter der Bezeichnung ”Alkyl” versteht man eine geradkettige oder verzweigte Alkylgruppe bestehend aus 1 bis 20 Kohelnstoffatomen, wie beispielsweise Methyl, Ethyl, n-Propyl, 1-Methylethyl (Isopropyl), n-Butyl, 1-Methylpropyl (sec-Butyl), 2-Methylpropyl (Isobutyl), 1,1-Dimethylethyl (tert-Butyl), Pentyl, Hexyl, Heptyl, Octyl, 2-Ethylhexyl, Nonyl, Decyl oder 2-Propylheptyl sowie Konstitutionsisomere davon.
-
Unter der Bezeichnung ”Cycloalkyl” versteht man eine gesättigte aliphatische, cyclische Gruppe bestehen aus 3 bis 10 Kohlenstoffatomen im Ringsystem. Beispiele sind Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl, Cycloheptyl, Cyclooctyl, Cyclononyl und Cyclodecyl. Cycloalkylgruppen können 1, 2 oder 3 weitere Substituenten tragen, die ausgewählt sind unter Alkyl, Alkoxy oder Halogen.
-
Unter der Bezeichnung ”Alkoxy” versteht am eine geradkettige oder verzweigte Alkylgruppe, bestehend aus 1 bis 10 Kohlenstoffatomen, welche über ein Sauerstoffatom gebunden ist, und wobei die Alkylgruppe 1, 2 oder 3 Substituenten tragen darf, die ausgewählte sind aus Halogen, Alkyl, Cycloalkyl oder Alkoxy. Beispiele für ”Alkoxy”-Gruppen sind Methoxy, Ethoxy, n-Propoxy, 1-Methylethoxy (Isopropoxy), n-Butoxy, 1-Methylpropoxy (sec-Butoxy), 2-Methylpropoxy (Isobutoxy) und 1,1-Dimethylethoxy (tert-Butoxy).
-
Unter der Bezeichnung ”Aryl” versteht man carbocyclische aromatische Gruppen, welche 6 bis 14 Kohlenstoffatome umfassen, wobei die Arylgruppe 1, 2, 3, 4 oder 5 weitere Substituenten tragen kann, welche ausgewählt sind aus Halogen, Cyano, Nitro, Alkyl oder Alkoxy. Beispiele sind Phenyl, 4-Chlorphenyl, 4-Methoxyphenyl, Naphthyl, Fluorenyl, Azulenyl, Anthracenyl und Phenanthrenyl.
-
Unter der Bezeichnung ”Heteroaryl” versteht man aromatische Gruppen, welche 1 bis 4 Heteroatome umfassen, welche ausgewählt sind unter O, N, S und SO2. Die Heteroarylgruppe kann 1, 2, 3 oder 4 Substituenten tragen, welche ausgewählt sind unter Halogen, Nitro, Cyano, Alkyl oder Alkoxy. Beispiele sind Pyrrolyl, 5-Methyl-2-pyrrolyl, Furanyl, 3-Methyl-2-furanyl, Thienyl, Pyrazolyl, Imidazolyl, Oxazolyl, Isoxazolyl, Thiazolyl, Isothiazolyl, Triazolyl, Tetrazolyl, Pyridyl, Pyrazinyl, Pyridazinyl, Pyrimidyl oder Triazinyl.
-
Unter der Bezeichnung ”Halogen” versteht man Fluor-, Brom-, Chlor- oder Iod-Substituenten.
-
Die Gruppe R1 wird bevorzugt ausgewählt aus i-Propyl oder t-Butyl.
Die Gruppen R2, R3, R5 and R6 werden bevorzugt ausgewählt aus Wasserstoff, Alkyl oder Halogen.
Die Gruppen R11, R12, R13 werden bevorzugt ausgewählt aus Methyl, Ethyl, n-Propyl or n-Butyl.
Die Gruppe X wird bevorzugt ausgewählt aus Trifluormethansulfonat (Triflat, CF3SO3 –) oder Methansulfonat (Mesylat, CH3SO3 –).
-
Verbindungen der Formel 2 können ausgehend von 4-Fluorophenylazocarbonsäure-tert-butylester in zwei Schritten hergestellt werden. Die entwickelten Vorschriften wurden aus der Literatur abgeleitet.
J. T. Reeves, D. R. Fandrick, Z. Tan, J. J. Song, H. Lee, N. K. Yee, C. H. Senanayake Org. Lett. 2010, 12, 4388–4391.
-
Zur Einführung von [18F]Fluoride in Verbindungen der Formel 2 können Lösungsmittel ausgewählt werden aus Acetonitril, Dimethylformamid, Dimethylsulfoxid, Dimethylacetamid, Tetrahydrofuran, Dioxan, 1,2-Dimethoxyethan, Sulpholan, N-Methylpyrrolidininon, tert-Butanol, oder die Reaktion wird in einer ionischen Flüssigkeit (zum Beispiel 1-Ethyl-3-methylimidazolium-hexafluorophosphat, 1-Butyl-4-methylpyridinium-tetrafluoroborat oder einer Phosphoniumverbindung oder Tetralkylammoniumverbindung) bei nicht extremer Temperatur, beispielsweise, 15°C bis 180°C, bevorzugt bei erhöhten Temperaturen von 80°C bis 150°C, bevorzugt bei 85°C durchgeführt. Geeignete organische Lösungsmittel sind wasserfrei, sie dürfen in Einzelfällen aber auch geringe Mengen an Wasser enthalten.
-
Als Quellen für [18F]Fluorid können Na18F, K18F, Cs18F, Tetraalkylammonium[18F]fluorid oder Tetraalkylphosphonium[18F]fluorid verwendet werden, bevorzugt eine [18F]Fluorid quelle wie Na18F, K18F, Cs18F, Tetraalkylammonium[18F]fluorid (beispielsweise Tetrabutylammonium[18F]fluorid) oder Tetraalkylphosphonium[18F]fluorid. Zur Erhöhung der Reaktivität des [18F]Fluorids kann ein Phasentransferkatalystor wie ein Aminopolyether oder Kronenether, beispielsweise 4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8,8,8]hexacosan (Kryptofix 2.2.2), verwendet werden, um reaktive Fluoridionen zu erhalten.
-
Geeignete Basen (als Zusätze) zur Herstellung von Verbindungen der Formel 1 ausgehend von Verbindungen der Formel 2 unter den oben genannten Bedingungen sind K2CO3, KHCO3, KH2PO4, K2C2O4 und Mischungen davon.
-
Bevorzugte Reaktionstemperaturen für die Herstellung von Verbindungen der Formel 1 ausgehend von Verbindungen der Formel 2 unter den oben genannten Bedingungen liegen im Bereich von 20°C bis 180°C, stärker bevorzugt im Bereich von 80°C bis 150°C, und am stärksten bevorzugt bei 85°C.
-
Zur Herstellung von Verbindungen der Formel 3 ausgehend von Verbindungen der Formel 1 und Verbindungen der Formel 4 können Lösungsmittel ausgewählt werden unter Ethanol, Acetonitril, Ethylacetat, Methanol, Tetrahydrofuran, tert-Butanol sowie Mischungen davon.
-
Zur Herstellung von Verbindungen der Formel 3 ausgehend von Verbindungen der Formel 1 und Verbindungen der Formel sollten Reaktionstemperaturen im Bereich von 0°C bis 100°C, bevorzugt im Bereich von 20°C bis 50°C, gewählt werden.
-
Beispiele:
-
I. Darstellung der Verbindungen mit der Strukturformel 2 ausgehend von 4-Fluorphenylazocarbonsäure-tert-butylester
-
Zu einer gerührten Lösung des 2-(4-Fluorphenyl)azocarbonsäure-tert-butylesters (959 mg, 4.28 mmol) und Na2CO3 (2.95 g, 21.4 mmol) in trockenem DMF (3 mL) wurde Dimethylamin (2 M Lösung in THF, 10.7 mL, 21.4 mmol) unter Argon-Atmosphäre zugegeben und das entstehende Gemisch bei Raumtemperatur gerührt bis 2-(4-Fluorphenyl)azocarbonsäure-tert-butylester nicht mehr mittels Dünnschichtchromatographie detektiert werden konnte. Das Reaktionsgemisch wurde mit gesättigter wässriger NaHCO3 Lösung verdünnt und mit Ethylacetat extrahiert (3 × 70 mL). Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen und über Natriumsulfat getrocknet. Die Aufreinigung durch Säulenchromatographie (Kieselgel, Hexan/EtOAc = 5:1) ergab 2-(4-(Dimethylamino)phenyl)azocarbonsäure-tert-butylester (988 mg, 3.96 mmol, 93%) als orangen Feststoff.
Rf 0.3 (Hexan/EtOAc = 5:1) [UV].
1H-NMR (360 MHz, CDCl3): δ (ppm) = 1.65 (s, 9 H), 3.14 (s, 6 H), 6.71 (d, J = 9.4 Hz, 2 H), 7.94 (d, J = 9.3 Hz, 2 H).
13C-NMR (91 MHz, CDCl3): δ (ppm) = 27.9 (3 × CH3), 40.3 (2 × CH3), 83.2 (Cq), 111.4 (2 × CH), 127.6 (2 × CH), 142.6 (Cq), 154.7 (Cq), 160.9 (Cq).
MS (EI) m/z (%): 249 (10) [M+], 235 (14) [M+-Me], 176 (11), 155 (14), 154 (20), 149 (25), 148 (36), 136 (15), 122 (10), 121 (86), 120 (100), 119 (11), 105 (21), 104 (16), 93 (11), 92 (13), 91 (10), 79 (12), 78 (13), 77 (32), 67 (29), 66 (21), 65 (13), 63 (13), 57 (100), 56 (96), 55 (38), 53 (15), 52 (17), 51 (25), 50 (21), 47 (10), 44 (100), 43 (21), 42 (30), 41 (100).
-
-
Zu einer Lösung von 2-(4-(Dimethylamin)phenyl)azocarbonsäure-tert-butylester (100 mg, 0.40 mmol) in Benzol (2 mL) wurde Trifluormethansulfonsäuremethylester (50 μL, 0.40 mmol) bei 7°C zugegeben. Nach zwei Stunden Rühren bei 7°C wurde das Reaktionsgemisch abfiltriert. Der Filterkuchen wurde zusätzlich mit kaltem Benzol gewaschen. Die Entfernung des Lösungsmittels unter verminderten Druck ergab 2-(4-(N,N,N-Trimethylammonium)phenyl)azocarbonsäure-tert-butylester trifluormethansulfonat (92.0 mg, 0.22 mmol, 56%) als einen orangen Feststoff.
1H-NMR (600 MHz, CD
3CN): δ (ppm) = 1.63 (s, 9 H), 3.59 (s, 9 H), 7.99 (d, J = 9.4 Hz, 2 H), 8.05 (d, J = 9.4 Hz, 2 H).
13C-NMR (151 MHz, CD
3CN): δ (ppm) = 27.9 (3 × CH
3), 58.1 (3 × CH
3), 86.9 (C
q), 122.9 (2 × CH), 125.5 (2 × CH), 130.8 (C
q), 139.9 (C
q), 152.7 (C
q), 161.7 (C
q). II. Repräsentative Versuchsvorschrift zur Einführung von [
18F]Fluorid: Darstellung von [
18F]Fluorphenylazocarbonsäure-tert-butylester ausgehend von 2-(4-(N,N,N-Trimethylammonium)phenyl)azocarbonsäure-tert-butylester trifluormethansulfonat.
-
Die QMA-Kartusche mit [
18F]Fluorid (600–800 MBq) wurde mit einer Lösung aus Kryptofix
® 2.2.2 (15 mg), K
2CO
3 (1.0 M, 15 μL) in Acetonitril (400 μL) eluiert. Das Wasser wurde durch Eindampfen bis zur Trockenheit mithilfe von Acetonitril (3 × 400 μL) und einem Stickstoffstrom bei 85°C entfernt. Der Vorläufer 2-(4-(N,N,N-Trimethylammonium)phenyl)azocarbonsäure-tert-butylester trifluormethansulfonat (2.5 mg, 6.0 μmol) in wasserfreiem Acetonitril (400 μL) wurde zu dem getrockneten K
+/Kryptofix 2.2.2/
18F-Komplex zugegeben und die Lösung wurde für 30 sec. bei 85°C gerührt. Anschließend wurde die gelbe Lösung mit Salzsäure (0.2 M, 15 mL) verdünnt und auf eine Kartusche (tC18, Waters) gegeben. Die Kartusche wurde mit Acetonitril/0.2 M HCl (20:80, 5 mL) und mit Wasser (1 mL) gewaschen und 2-(4-[
18F]Fluorphenyl)azocarbonsäure-tert-butylester (RCY: 85 ± 10% (n = 6), bestimmt durch eine aus dem Reaktionsgemisch entnommene Probe) wurde mit Acetonitril (1 mL) oder Ethylacetat (1 mL) oder Ethanol (1 mL) eluiert, abhängig von dem Lösemittel, das für weitere Reaktionen verwendet wurde. III. Repräsentative Versuchsvorschrift für die Kupplung von [
18F]Fluorphenylazocarbonsäure-tert-butylester mit primären aliphatischen Aminen: Synthese von 2-(4-[
18F]Fluorphenyl)-N-(4-(4-(2-methoxyphenyl)piperazin-1-yl)but-1-yl)azocarboxamid
-
Zu einer Lösung aus 4-(4-(2-Methoxyphenyl)piperazin-1-yl)but-1-ylamin (100 μL) wurde K2CO3 (30 mg) und 2-(4-[18F]Fluorphenyl)azocarbonsäure-tert-butylester in Ethanol (100 μL) oder Ethylacetat (100 μL) zugegeben. Nach 10 min bei 35°C (Ethanol) oder 30 min bei 45°C (Ethylacetat) wurde 2-(4-[18F]Fluorphenyl)-N-(4-(4-(2-methoxyphenyl)piperazin-1-yl)but-1-yl)azocarboxamid durch die Retentionszeit (tR) in einem radio-HPLC System und durch Koinjektion der zugehörigen Referenzverbindung 2-(4-Fluorphenyl)-N-(4-(4-(2-methoxyphenyl)piperazin-1-yl)but-1-yl)azocarboxamid identifiziert (Chromolith RP-18e, 100 × 4.6 mm, 10–90% Acetonitril (0.1% TFA) in Wasser (0.1% TFA) in einem linearen Gradienten über 5 mm, 4 mL/min, tR = 2.15 mm). Die radiochemische Ausbeute von 2-(4-[18F]Fluorphenyl)-N-(4-(4-(2-methoxyphenyl)piperazin-1-yl)but-1-yl)azocarboxamid betrug 100% bestimmt mit analytischer radio-HPLC durch eine aus dem Reaktionsgemisch entnommene Probe. Nach Eindampfen des Lösemittels wurde der Rückstand in Acetonitril/Wasser (0.1% TFA) 50:50 (500 μL) gelöst und 2-(4-[18F]Fluorphenyl)-N-(4-(4-(2-methoxyphenyl)piperazin-1-yl)but-1-yl)azocarboxamid wurde mittels semipräparativer HPLC (Kromasil C8, 125 × 8, 4 mL/min, 25–60% Acetonitrile (0.1% TFA) in Wasser (0.1% TFA) in einem linearem Gradienten über 30 min, tR = 8.5 min) und anschließender Festphasenextraktion (SepPak C18 light, Waters) gereinigt. 2-(4-[18F]Fluorphenyl)-N-(4-(4-(2-methoxyphenyl)piperazin-1-yl)but-1-yl)azocarboxamid (137–180 MBq) wurde von der Kartusche mit Ethanol (1 mL) eluiert, das Lösemittel wurde im Vakuum eingedampft und der Rückstand in einer wässriger Natriumchlorid Lösung für weiter in vitro und in vivo Studien aufgenommen. 2-(4-[18F]Fluorphenyl)-N-(4-(4-(2-methoxyphenyl)-piperazin-1-yl)but-1-yl)azocarboxamid wurde mit einer radiochemischen Gesamtausbeute von 20–24% (zerfallskorrigierte Ausbeute, bezogen auf [18F]Fluorid) in einer Gesamtsynthesezeit von 70–80 min und einer spezifischen Aktivität von 4–10 GBq/μmol synthetisiert (mit Ethylacetat als Lösemittel). Mit Ethanol als Lösemittel wurde 2-(4-[18F]Fluorphenyl)-N-(4-(4-(2-methoxyphenyl)piperazin-1-yl)but-1-yl)azocarboxamid mit einer radiochemischen Gesamtausbeute von 30–35% (zerfallskorrigierte Ausbeute, bezogen auf [18F]Fluorid) in einer Gesamtsynthesezeit von 35–40 min und einer spezifischen Aktivität von 12 GBq/μmol synthetisiert.
-
ZITATE ENTHALTEN IN DER BESCHREIBUNG
-
Diese Liste der vom Anmelder aufgeführten Dokumente wurde automatisiert erzeugt und ist ausschließlich zur besseren Information des Lesers aufgenommen. Die Liste ist nicht Bestandteil der deutschen Patent- bzw. Gebrauchsmusteranmeldung. Das DPMA übernimmt keinerlei Haftung für etwaige Fehler oder Auslassungen.
-
Zitierte Nicht-Patentliteratur
-
- H. H. Coenen et al., Nucl. Med. Biol. 2010, 10, 727–740 [0001]
- L. Cai, S. Lu, V. W. Pike, Eur. J. Org. Chem. 2008, 2853–2873 [0002]
- M. Tredwell, V. Gouverneur, Angew. Chem. Int. Ed. 2012, 51, 11426–11437 [0003]
- R. Littich, P. J. H. Scott; Angew. Chem. Int. Ed. 2012, 51, 1106–1109 [0003]
- T. L. Ross, J. Ermert, C. Hocke, H. H. Coenen, J. Am. Chem. Soc. 2007, 129, 8018–8025 [0004]
- E. Lee, A. S. Kamlet, D. C. Powers, C. N. Neumann, G. B. Boursalian, T. Furuya, D. C. Choi, J. M. Hooker, T. Ritter, Science 2011, 334, 639–642 [0004]
- Z. Gao, Y. H. Lim, M. Tredwell, L. Li, S. Verhoog, M. Hopkinson, W. Kaluza, T. L. Collier, J. Passchier, M. Huiban, V. Gouverneur, Angew. Chem. Int. Ed. 2012, 51, 6733–6737 [0004]
- I. S. R. Stenhagen, A. K. Kirjavainen, S. J. Forsback, C. G. Jørgensen, E. G. Robins, S. K. Luthra, O. Solin, V. Gouverneur, Chem. Commun. 2013, 49, 1386 [0004]
- J. C. Meleán, J. Ermert, H. H. Coenen, Org. Biomol. Chem. 2011, 9, 765–769 [0005]
- J. C. Meleán, J. Ermert, H. H. Coenen, Tetrahedron 2010, 66, 9996–10001 [0005]
- H. Jasch, S. Höfling, M. R. Heinrich, J. Org. Chem. 2012, 77, 1520–1532 [0007]
- S. B. Höfling, A. L. Bartuschat, M. R. Heinrich, Angew. Chem. Int. Ed. 2010, 49, 9769–9772 [0007]
- S. Höfling, Entwicklung neuartiger Syntheseverfahren über Arylradikale mit Anwendungen in der Radiopharmazie, Dissertation, TU München 2011, ISBN 978-3-8439-0114-7 [0008]
- H. Jasch, S. Höfling, M. R. Heinrich, J. Org. Chem. 2012, 77, 1520–1532 [0012]
- J. T. Reeves, D. R. Fandrick, Z. Tan, J. J. Song, H. Lee, N. K. Yee, C. H. Senanayake Org. Lett. 2010, 12, 4388–4391 [0020]
 mit [18F]Fluorid zur Reaktion gebracht wird und R1 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, tert-Butyl, Trityl, Allyl, Alkyl, Cycloalkyl, Phenyl, Aryl oder Heteroaryl ist; X Fluorid, Chlorid, Bromid, Iodid, Sulfat, Tetrafluoroborat, Acetat, Trifluoracetat, Hexafluorophosphat, Hexafluoroantimonat, Methansulfonat, Trifluormethansulfonat, Toluol-4-sulfonat oder Monomethylsulfat ist; R2 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist; R3 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist; R5 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist; R6 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist; R11 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, n-Butyl oder Phenyl ist; R12 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, n-Butyl oder Phenyl ist; und R13 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, n-Butyl oder Phenyl ist.
mit [18F]Fluorid zur Reaktion gebracht wird und R1 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, tert-Butyl, Trityl, Allyl, Alkyl, Cycloalkyl, Phenyl, Aryl oder Heteroaryl ist; X Fluorid, Chlorid, Bromid, Iodid, Sulfat, Tetrafluoroborat, Acetat, Trifluoracetat, Hexafluorophosphat, Hexafluoroantimonat, Methansulfonat, Trifluormethansulfonat, Toluol-4-sulfonat oder Monomethylsulfat ist; R2 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist; R3 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist; R5 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist; R6 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist; R11 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, n-Butyl oder Phenyl ist; R12 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, n-Butyl oder Phenyl ist; und R13 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, n-Butyl oder Phenyl ist.
 mit einer Verbindung der Formel 4 zur Reaktion gebracht wird
mit einer Verbindung der Formel 4 zur Reaktion gebracht wird und R20 unabhängig voneinander
und R20 unabhängig voneinander bezeichnet; und R1, R2, R3, R5 und R6 wie oben definiert sind.
bezeichnet; und R1, R2, R3, R5 und R6 wie oben definiert sind.




 mit [18F]Fluorid zur Reaktion gebracht wird und R1 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, tert-Butyl, Trityl, Allyl, Alkyl, Cycloalkyl, Phenyl, Aryl oder Heteroaryl ist; X Fluorid, Chlorid, Bromid, Iodid, Sulfat, Tetrafluoroborat, Acetat, Trifluoracetat, Hexafluorophosphat, Hexafluoroantimonat, Methansulfonat, Trifluormethansulfonat, Toluol-4-sulfonat oder Monomethylsulfat ist; R2 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist; R3 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist; R5 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist; R6 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist; R11 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, n-Butyl oder Phenyl ist; R12 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, n-Butyl oder Phenyl ist; und R13 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, n-Butyl oder Phenyl ist.
mit [18F]Fluorid zur Reaktion gebracht wird und R1 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, tert-Butyl, Trityl, Allyl, Alkyl, Cycloalkyl, Phenyl, Aryl oder Heteroaryl ist; X Fluorid, Chlorid, Bromid, Iodid, Sulfat, Tetrafluoroborat, Acetat, Trifluoracetat, Hexafluorophosphat, Hexafluoroantimonat, Methansulfonat, Trifluormethansulfonat, Toluol-4-sulfonat oder Monomethylsulfat ist; R2 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist; R3 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist; R5 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist; R6 unabhängig voneinander Wasserstoff, Halogen, Alkyl, Cycloalkyl, Alkoxy, Cyano, Nitro, Aryl oder Heteroaryl ist; R11 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, n-Butyl oder Phenyl ist; R12 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, n-Butyl oder Phenyl ist; und R13 unabhängig voneinander Methyl, Ethyl, n-Propyl, i-Propyl, n-Butyl oder Phenyl ist.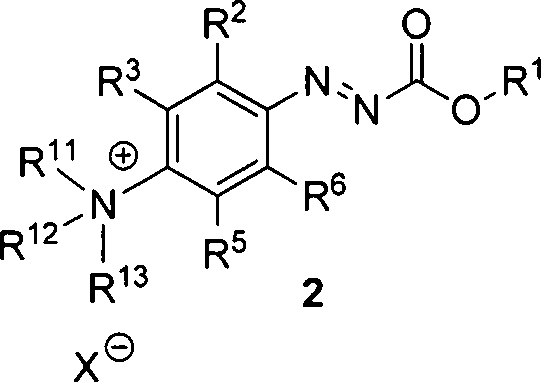
 mit einer Verbindung der Formel 4 zur Reaktion gebracht wird
mit einer Verbindung der Formel 4 zur Reaktion gebracht wird und R20 unabhängig voneinander
und R20 unabhängig voneinander bezeichnet; und R1, R2, R3, R5 und R6 wie oben definiert sind.
bezeichnet; und R1, R2, R3, R5 und R6 wie oben definiert sind.