CN1820007A - 经芳基取代的苯并[d]异噻唑-3-基胺类似物 - Google Patents
经芳基取代的苯并[d]异噻唑-3-基胺类似物 Download PDFInfo
- Publication number
- CN1820007A CN1820007A CNA2004800197264A CN200480019726A CN1820007A CN 1820007 A CN1820007 A CN 1820007A CN A2004800197264 A CNA2004800197264 A CN A2004800197264A CN 200480019726 A CN200480019726 A CN 200480019726A CN 1820007 A CN1820007 A CN 1820007A
- Authority
- CN
- China
- Prior art keywords
- compound
- alkyl
- pain
- capsaicin receptor
- pharmaceutically acceptable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D275/00—Heterocyclic compounds containing 1,2-thiazole or hydrogenated 1,2-thiazole rings
- C07D275/04—Heterocyclic compounds containing 1,2-thiazole or hydrogenated 1,2-thiazole rings condensed with carbocyclic rings or ring systems
- C07D275/06—Heterocyclic compounds containing 1,2-thiazole or hydrogenated 1,2-thiazole rings condensed with carbocyclic rings or ring systems with hetero atoms directly attached to the ring sulfur atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P39/00—General protective or antinoxious agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pulmonology (AREA)
- Neurology (AREA)
- Rheumatology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Pain & Pain Management (AREA)
- Urology & Nephrology (AREA)
- Immunology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Heart & Thoracic Surgery (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Toxicology (AREA)
- Child & Adolescent Psychology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
本发明涉及一种如右通式的经芳基取代的苯并[d]异噻唑-3-基胺类似物,其中,变量如本发明所述。这种化合物为可用于体内或体外调节特定受体活性的配位体,并且尤其有用于在人类、经驯化的陪伴动物及家畜上治疗与病理受体活化相关的病症。本发明还涉及药学组合物及使用所述化合物治疗疾病的方法,及使用所述配位体进行受体定位研究的方法。
Description
技术领域
本发明大体而言是涉及经芳基取代的苯并[d]异噻唑-3-基胺类似物,所述类似物为辣椒素受体(capsaicin receptor)的调节剂,以及涉及使用所述化合物来治疗与辣椒素受体活化相关的病症。本发明进一步涉及使用所述化合物做为检测和定位辣椒素受体的探针。
背景技术
痛觉或伤害性刺激是由一群特化感觉神经元的周边末稍所介导,称为“伤害性受体(nociceptor)”。各种不同物理和化学刺激会引起哺乳动物所述神经元的活化,导致潜在伤害刺激的认知。然而不适当或过度活化伤害性受体可造成急性或慢性疼痛衰弱。
神经病变性疼痛(亦可简称神经痛)涉及无刺激存在的疼痛讯号传导,一般起因于神经系统的伤害。大部分的例子中,该疼痛的发生被视为由于周边系统初期伤害(例如,直接伤害或全身性疾病)后周边和中枢神经系统的敏化作用(sensitization)。神经痛一般有灼热、刺痛感且其强度持续,有时该神经痛在引发该神经痛的初期伤害或疾病期间更能使人衰弱。
已存在的神经痛治疗方式大部分效果不明显。鸦片类(opiates)如吗啡为强有力的止痛药,但由于有害的副作用使其使用受限,此等副作用会造成例如生理上的成瘾和戒断特性以及呼吸抑制、性情改变和降低肠蠕动并伴随便秘、恶心、呕吐,及内分泌和自律神经系统的改变。另外,神经痛对于已知的鸦片类止痛药疗法经常是无反应的,或仅部分有反应。使用N-甲基-D-天门冬胺酸拮抗剂克他明(ketamine;亦可称为氯胺酮)或α(2)-类肾上腺素促效剂(adrenergic agonist)氯压定(clonidine),可减轻急性或慢性疼痛,并使鸦片消耗量降低,但因其副作用而使所述药剂常有不佳的耐受性。
以辣椒素做局部治疗一直用作慢性或急性疼痛,包括神经痛的治疗方式。辣椒素为来自茄科植物(包括红辣椒)的刺激性物质,可能选择性地作用于被视为介导疼痛的小径传入神经纤维(A-δ和C纤维)。辣椒素反应特性为持续活化在周边组织中的伤害性受体,随后使周边伤害性受体对一或多个刺激脱敏(desensitization)。从动物研究中,辣椒素表现出通过打开钙和钠的阳离子选择性通道而触发C纤维膜去极化作用。
共有相同类香草醇(vanilloid)部分的辣椒素结构类似物也引起相似的反应。一种所述类似物为树胶脂毒素(RTX),为大戟科植物的天然产物。用术语类香草醇受体(VR)来描述辣椒素及其相关刺激化合物的神经细胞膜辨识位置。辣椒素反应会被其它辣椒素类似物(辣椒平(capsazepine;为辣椒素受体阻断剂))竞争性地抑制(而因此拈抗),也可被非选择性阳离子通道阻断剂(钌红;ruthenium red)所抑制。所述拮抗剂使用只以中度的亲和力(一般Ki值不小于140μM)结合至VR。
大鼠和人类的类香草醇受体是从背根神经节细胞克隆出来。经鉴别的第一类型类香草醇受体已知为类香草醇受体1型(VR1),该词”VR1”和”辣椒素受体”在本发明可交换地使用来指称大鼠和/或人类的此类型受体,以及哺乳动物的同源系。VR1在痛觉上所扮演的角色已经通过使用缺乏该受体的小鼠来证实,该小鼠没有表现出类香草醇所引发的疼痛行为,且对热和发炎的反应减弱。VR1为具有开启阈值的非选择性阳离子通道,该阀值对应高温、低pH和辣椒素受体促效剂时降低。例如,该信道通常在大于45℃的温度打开。该辣椒素受体通道通常在发炎肽释放后开启,以增加疼痛反应,所述发炎肽来自于表现该受体的神经元及其邻近神经元。在辣椒素初期活化后,辣椒素受体经由以cAMP依赖型蛋白激酶(cAMP-dependent protein kinase)介导的磷酸化作用而经历快速的脱敏化(desensitization)作用。
VR1促效剂类香草醇化合物一直用作典型的麻醉剂,这是由于其对周边组织的伤害性受体具有脱敏化的能力。然而,促效剂的应用本身可能导致灼热痛,从而限制其治疗上的使用。最近有报导称,VR1拮抗剂,包括非类香草醇化合物,亦有助于疼痛的治疗(请参见PCT国际申请案公开号WO 02/08221,其于2002年1月31日公开)。
因此,与VR1发生反应但不引起VR1促效剂类香草醇化合物的初期痛觉的化合物可用于慢性和急性疼痛(包括神经痛)的治疗。此受体的拮抗剂特别适用于疼痛和下列病症的治疗,例如暴露于催泪瓦斯、发痒及诸如尿失禁和膀胱过动症(overactive bladder)的尿道疾病。本发明达到此需求,并进一步提供相关的优点。
发明内容
本发明提供能改变,优选能抑制辣椒素受体活化的辣椒素受体调节剂。在某些方面,本发明提供如下面通式I所示的化合物或其药学上可接受的形式:

其中:
W、Y和Z独立地为N或CR1;
R1在每种情况下独立地选自氢、卤素、氰基、氨基、任性经取代的C1-C6烷基、任性经取代的卤代C1-C6烷基、任性经取代的C1-C6烷氧基、任性经取代的卤代C1-C6烷氧基或任选经取代的单-与二-C1-C6烷氨基;
Ar1和Ar2独立地选自5-至10-元芳族碳环与杂环,其中每一个任选经取代,优选地经0至3个取代基来取代,所述取代基独立地选自卤素、氰基、硝基或通式为LRa的基团;
L在每种情况下独立地选自单一共价键、O、C(=O)、OC(=O)、C(=O)O、O-C(=O)O、S(O)m、N(Rx)、C(=O)N(Rx)-、N(Rx)C(=O)、N(Rx)S(O)m、S(O)mN(Rx)或N[S(O)mRx]S(O)m;其中m在每种情况下独立地选自0、1或2;而Rx在每种情况下独立地选自氢或任选经取代的C1-C8烷基;及
Ra在每种情况下独立地选自:
(i)氢;及
(ii)C1-C8烷基、C2-C8烯基、C2-C8炔基、卤代C1-C8烷基、C2-C8烷基醚、单-与二-(C1-C8烷基)氨基、及(3-至10-元杂环)C0-C4烷基,其中每一个经0至6个取代基来取代,所述取代基独立地选自(a)羟基、卤素、氨基、氨基羰基、氰基、硝基、酮基或COOH;及(b)C1-C8烷基、C1-C8烯基、C1-C8炔基、C1-C8烷氧基、C1-C8烷硫基、C1-C8烷基醚、C1-C8烷酰基、C1-C8烷酮、C1-C8烷酰基氧基、C1-C8烷氧基羰基、羟基C1-C8烷基、卤代C1-C8烷基、氰基C1-C8烷基、苯基C0-C8烷基、单-与二-(C1-C6烷基)氨基C0-C8烷基、C1-C8烷基磺酰基或(5-至7-元杂环)C0-C8烷基;其中每一个任选地经取代。
在某些方面,本发明所述的VR1调节剂在辣椒素受体结合试验中显现不大于1微摩尔浓度、100纳摩尔浓度、50纳摩尔浓度、10纳摩尔浓度或1纳摩尔浓度的Ki,和/或在测定辣椒素受体促效剂或拮抗剂活性的试验中具有不大于1微摩尔浓度、100纳摩尔浓度、50纳摩尔浓度、10纳摩尔浓度或1纳摩尔浓度的EC50或IC50值。
在某些具体实施例中,本发明所述的VR1调节剂为VRI拮抗剂,且在辣椒素受体活化的体外试验中显示无可检测的促效剂活性。
在某些方面,本发明所述的VR1调节剂用可检测的标记物(例如,放射性标记或荧光标记物)予以标记。
在某些方面,本发明所述的VR1调节剂及其药学上可接受的形式以可检测的标记物(例如,放射性标记或荧光标记物)予以标记。
在其它方面,本发明进一步提供包括至少一种如本发明所述的VR1调节剂(亦即本发明所提供的化合物或其药学上可接受的形式)和生理上可接受的载剂或赋形物的药学组合物。
在其它方面,本发明提供降低细胞辣椒素受体的钙离子传导的方法,所述方法包括使表达辣椒素受体的细胞(例如神经元)与辣椒素受体调节量的至少一种如本发明所述的VR1调节剂接触。所述接触可发生在体内或体外。
本发明进一步提供抑制类香草醇配位体与辣椒素受体结合的方法。在某些此类方面,所述抑制可发生在体外。所述方法包括在足以可检测地抑制类香草醇配位体结合至辣椒素的条件与用量下,使辣椒素受体与至少一种如本发明所述的VR1调节剂接触。在其它此类方面,所述辣椒素受体是在患者体内。所述方法包括在足以体外可检测地抑制类香草醇配位体与表达克隆辣椒素受体的细胞结合的用量下,使表达辣椒素受体的患者细胞与至少一种如本发明所述的VR1调节剂接触,进而抑制类香草醇配位体结合至患者的辣椒素受体。
本发明进一步提供用于治疗患者对辣椒素受体调节敏感的病症的方法,所述包括对患者给药辣椒素受体调节量的至少一种如本发明所述的VR1调节剂。
在其它方面,本发明提供治疗患者疼痛的方法,所述包括对患有疼痛的患者给药辣椒素受体调节量的至少一种如本发明所述的VR1调节剂。
本发明进一步提供用于治疗患者痒、尿失禁、膀胱过动、咳嗽和/或打嗝的方法,所述包括对患有一或多种上述症状的患者给药辣椒素受体调节量的至少一种如本发明所述的VR1调节剂。
本发明进一步提供促使肥胖患者减重的方法,所述方法包括对肥胖患者给药辣椒素受体调节量的至少一种如本发明所述的VR1调节剂。
在其它方面,本发明进一步提供用于测定试样中辣椒素受体存在与否的方法,所述方面包括:(a)在容许VR1调节剂结合至辣椒素受体的条件下,使试样与如本发明所述的VR1调节剂接触;及(b)检测与辣椒素受体结合的VR1调节剂的浓度。
本发明也提供了经包装的药学制剂,所述制剂包括:(a)装于容器中的如本发明所述的药学组合物;及(b)使用该组合物治疗对辣椒素受体调节作用敏感的一种或多种症状(例如疼痛、痒、尿失禁、膀胱过动、咳嗽、打嗝和/或肥胖症)的说明书。
在又另一方面,本发明提供了制备本发明所揭示的化合物(包括中间产物)的方法。
参照下文详细说明后,本发明的上述及其它方面将更为清楚明白。详细说明
如上文所述,本发明提供作为辣椒素受体调节剂的经芳基取代的苯并[d]异噻唑-3-基胺类似物。所述调节剂可用于体外或体内,以调节(优选为抑制)各种背景下的辣椒素受体活性。
术语
本发明中的化合物通常使用标准命名法予以叙述。对于具有不对称中心的化合物,应了解(除非特别指明)所有光学异构体及其混合物均涵盖在内。另外,具有碳-碳双键的化合物可存在Z-与E-型,除非特别指明,否则化合物的所有异构体型均涵盖在本发明范围内。当化合物以多种互变异构体存在时,所述化合物并不受限于任一特定的互变异构体,而是要涵盖所有互变异构体。本发明中某些化合物是使用含有变量(例如,R3、A1、X)的通式进行叙述的。除非特别指明,否则所述通式中每个变量皆独立地界定为任何不同的变量,且在通式中出现超过一次以上的任何变量在每次出现时均各自独立地进行界定。
本发明所使用的术语“经芳基取代的苯并[d]异噻唑-3-基胺类似物”涵盖所有满足通过式I的化合物,以及本发明所提供其它通式的化合物及所述化合物在药学上可接受的形式。所述化合物包括苯并[d]异噻唑核心中环杂原子的数字和/或配置经修改的类似物,以及经各种在下文将做更详细说明的取代基连接至所述核心结构的类似物。在经芳基取代的苯并[d]异噻唑-3-基胺类似物的范围内,具有下述核心结构(具有或不具有进一步环取代)的化合物是做为举例给出的,而非用以限制本发明的范畴:
本发明所述的化合物“药学上可接受的形式”为药学上可接受的盐、水合物、溶剂合物、结晶形式、多态异构物(polymorph)、螯合物、非共价复合物、酯类、晶笼化合物及该化合物的前物(prodrug)。本发明所使用的药学上可接受的盐为酸式或碱式盐,且通常为本技术领域中认为适用于与人类或动物组织接触,而无过度毒性、刺激性、过敏反应或其它问题或并发症的盐。此种盐类包括碱性残基(诸如胺)的无机和有机酸盐,及酸性残基(诸如羧酸)的碱金属或有机盐。特定的药学上的盐包括,但不限于,例如盐酸、磷酸、氢溴酸、苹果酸、乙醇酸、反丁烯二酸、硫酸、胺磺酸、对胺苯磺酸、甲酸、甲苯磺酸、甲烷磺酸、苯磺酸、乙烷二磺酸、2-羟基乙磺酸、硝酸、苯甲酸、2-乙酰氧基苯甲酸、柠檬酸、酒石酸、乳酸、硬脂酸、柳酸、麸胺酸、抗坏血酸、双羟酸、琥珀酸、反丁烯二酸、马来酸、丙酸、羟基马来酸、氢碘酸、苯基乙酸、烷酸如乙酸、HOOC-(CH2)n-COOH(其中n为0至4)等酸的盐。同样地,药学上可接受的阳离子包括但不限于,钠、钾、钙、铝、锂与铵。在此技术领域具有通常知识者可进一步知道本发明提供的化合物的药学上可接受的盐,包括那些由Remington’s PharmaceuticalSciences,17th ed.,Mack Publishing Company,Easton,PA,p.1418(1985)所列举的那些。通常,药学上可接受的酸式或碱式盐可利用任何已知的化学方法,用含碱性或酸性部分的母化合物来合成。简言的,所述盐可通过使游离酸或碱形式的那些化合物与化学计量的适当碱或酸,在水或有机溶剂或二者的混合物中反应来制备;通常,优选使用非水性介质,例如乙醚、乙酸乙酯、乙醇、异丙醇或乙腈。
“前物”是一种化合物,其可能不完全满足本发明提供的化合物的结构需求,然而在给药患者后即在活体内经修饰而产生通式I化合物或本发明所提供的其它通式的化合物。例如,前物可为本发明所提供的化合物的酰基化衍生物。前物包括这样的化合物,所述化合物中羟基、胺或硫氢基与任何基团结合,当给药至哺乳动物对象后,所述基团裂解而分别地形成游离的羟基、氨基或硫氢基。前物的实施例包括,但不限于,本发明所提供化合物中的醇和胺官能基的乙酸盐、甲酸盐及苯甲酸盐衍生物。本发明提供的化合物的前物可通过修饰存在于化合物中的官能基来制备,其中所述修饰为经裂解而成为母化合物。
本发明所用的“烷基”一词是指直链或支链的饱和脂肪族烃。烷基包含具有1至8个碳原子(C1-C8烷基)、1至6个碳原子(C1-C6烷基)及1至4个碳原子(C1-C4烷基)的基团,例如甲基、乙基、丙基、异丙基、正丁基、仲丁基、叔丁基、戊基、2-戊基、异戊基、新戊基、己基、2-己基、3-己基及3-甲基戊基、环丙基、环丙基甲基、环戊基、环戊基甲基、环己基、环庚基及降莰基。“C0-C4烷基”是指单一共价键(C0)或具有1、2、3或4个碳原子的烷基;“C0-C6烷基”是指单一共价键或C1-C6烷基;“C0-C8烷基”是指单一共价键或C1-C8烷基。在某些具体实施例中,优选的烷基为直链或分支状。在本发明某些例子中,烷基的取代基会特别地指明,例如“氰基C1-C4烷基”是指具有至少一个CN取代基的C1-C4烷基。一个代表性的分支状氰基烷基的基团是为-C(CH3)2CN。
同样地,“烯基”是指直链或支链或环状的烯基团,其中至少存在一个不饱和碳-碳双键。烯基包括分别具有2至8个、2至6个或2至4个碳原子的C2-C8烯基、C2-C6烯基及C2-C4烯基,例如乙烯基、烯丙基或异丙烯基。“炔基”是指具有一或多个不饱和碳-碳键且其中至少一者为三键的直链或支链炔基团。炔基包括分别具有2至8个、2至6个或2至4个碳原子的C2-C8炔基、C2-C6炔基及C2-C4炔基。在某些具体实施例中,优选的烯基和炔基为直链或支链。
本发明所用的“烷氧基”意指经由氧桥连接的如上所述的烷基。烷氧基包含分别具有1至6个或1至4个碳原子的C1-C6烷氧基及C1-C4烷氧基。特定的烷氧基为甲氧基、乙氧基、丙氧基、异丙氧基、正丁氧基、仲丁氧基、叔丁氧基、正戊氧基、2-戊氧基、3-戊氧基、异戊氧基、新戊氧基、己氧基、2-己氧基、3-己氧基或3-甲基戊氧基。
同样地,“烷硫基”是指经由硫桥连接的如上所述的烷基、烯基或炔基。优选的烷氧基及烷硫基是经由杂原子桥键连接的烷基。
本发明所用的“酮基(oxo)”一词是指酮(keto)(C=O)基团,所述酮基为非芳族碳原子的取代基,并会使-CH2-转变成-C(=O)-。
“烷酰基”一词是指呈线型或分支型排列的酰基(例如,-(C=O)-烷基),其中经由酮基的碳进行连接。烷酰基包括分别具有2至8个、2至6个或2至4个碳原子的C2-C8烷酰基、C2-C6烷酰基及C2-C4烷酰基。“C1烷酰基”是指-(C=O)-H,其(与C2-C8烷酰基)已由“C1-C8烷酰基”一词所涵盖。乙酰基为C2烷酰基。
“烷酮”为碳原子呈线型或分支型烷基排列的酮基团。“C3-C8烷酮”、“C3-C6烷酮”与“C3-C4烷酮”分别是指具有3至8、6或4个碳原子的烷酮。举例而言,C3烷酮的结构式为-CH2-(C=O)-CH3。
同样地,“烷基醚”是指线型或分支型醚取代基。烷基醚基团包括分别具有2至8、6或4个碳原子的C2-C8烷基醚、C2-C6烷基醚及C2-C4烷基醚等基团。举例而言,C2烷基醚的结构式为-CH2-O-CH3。
“烷氧基羰基”一词是指经由羰基连接的烷氧基(即具有一般结构为-C(=O)-O-烷基)。烷氧基羰基包括分别具有2至8、6或4个碳原子的C2-C8、C2-C6及C2-C4烷氧基羰基。“C1烷氧基羰基”是指-C(=O)-OH,其已由“C1-C8烷氧基羰基”一词所涵盖。
本发明所用的“烷酰基氧基”,是指经由氧桥连接的烷酰基(即具有一般结构为-O-C(=O)-烷基的基团)。烷酰基氧基包括分别具有2至8、6或4个碳原子的C2-C8、C2-C6及C2-C4烷酰基氧基。
“烷基磺酰基”是指通式为-(SO2)-烷基的基团,其中连接点为硫原子。烷基磺酰基包括分别具有1至6或1至4个原子的C1-C6烷基磺酰基与C1-C4烷基磺酰基。甲基磺酰基为一种代表性的烷基磺酰基。
“烷基磺酰氨基”是指通式为-(SO2)-N(R)2的基团,其中连接点为硫原子,且每一个R独立地为氢或烷基。“单-或二-(C1-C6烷基)磺酰氨基”是指该基团中一个R为C1-C6烷基,而另一个R为氢或独立地选自C1-C6烷基。
“烷氨基”是指通式为-NH-烷基或-N(烷基)(烷基)的二级或三级胺,其中每一个烷基相同或不同。此类基团包括,例如,单-及二-(C1-C8烷基)氨基,其中每一个烷基相同或不同且可含1至8个碳原子,以及单-及二-(C1-C6烷基)氨基与单-及二-(C1-C4烷基)氨基。
“烷基胺烷基”是指经由烷基连接的烷氨基(即通式为-烷基-NH-烷基或-烷基-N(烷基)(烷基)的基团),其中各烷基经独立地选定。此类基团包括,例如,单-及二-(C1-C8烷基)氨基C1-C8烷基、单-及二-(C1-C6烷基)氨基C1-C6烷基与单-及二-(C1-C4烷基)氨基C1-C4烷基,其中各烷基相同或不同。“单-或二-(C1-C6烷基)氨基C0-C6烷基”是指经由直接键结或C1-C6烷基连接的单-或二-(C1-C6烷基)氨基。下列为代表性的烷氨基烷基:
“氨基羰基”一词是指酰氨基(即-(C=O)NH2)。“单-或二-(C1-C8烷基)氨基羰基”是其中一或两个氢原子被C1-C8烷基置换的氨基羰基。若两个氢原子均被如此置换时,所述C1-C8烷基可相同或不同。
“卤素”一词是指氟、氯、溴或碘。
“卤烷基”为以1个或多个卤原子取代的支链、直链或环状烷基(例如,“C1-C8卤烷基”具有1至8个碳原子;“C1-C6卤烷基”基团具有1至6个碳原子)。卤烷基的实施例包括但不限于,单-、二-或三-氟甲基;单-、二-或三-氯甲基;单-、二-、三-、四-或五氟乙基;单-、二-、三-、四-或五氯乙基;及1,2,2,2-四氟-1-三氟甲基-乙基。典型的卤烷基为三氟甲基及二氟甲基。“卤烷氧基”是指经由氧桥连接的如上文界定的卤烷基。“C1-C8卤烷氧基”具有1至8个碳原子。
不是介于两个字母或符号间的破折号(-)是用于指示取代基的连接点。例如,-CONH2是经由碳原子连接。
“碳环”或“碳环状基团”包括完全由碳-碳键形成的至少一个环(本发明称为碳环状环),而不包含杂环。除非特别指示,否则碳环内的各碳环状环可为饱和、部分饱和或芳香族。碳环通常具有1至3个稠合环、侧环(pendant ring)或螺环;特定具体实施例中的碳环具有一个环或两个稠合环。典型地,各环含有3至8个环元(即C3-C8);在特定具体实施例中列举出C5-C7环。包括稠合环、侧环或螺环的碳环一般含有9至14个环元。特定代表性碳环为环烷基(即包含饱和和/或部分饱和环的基团,例如环丙基、环丁基、环戊基、环己基、环庚基、环辛基、金刚烷基、十氢-萘基、八氢-茚基,及任何上述基团的部分饱和变体,例如环己烯基)。其它碳环为芳基(即含有至少一个芳族碳环)。所述碳环包括例如,苯基、萘基、芴基、茚满基及1,2,3,4-四氢-萘基。
本发明列举的某些碳环为C6-C10芳基C0-C8烷基(即含有至少一个芳族环的碳环是经由直接键结或C1-C8烷基连接的基团)。此类基团包括,例如,苯基与萘基,以及上述两基团中任一基团经由C1-C8烷基(优选为经由C1-C4烷基)连接的基团。经由直接键结或烷基连接的苯基可记为苯基C0-C8烷基(例如,苄基、1-苯基-乙基、1-苯基-丙基及2-苯基-乙基)。
“杂环”或“杂环基”具有1至3个稠合环、侧环或螺环,其中至少一个为杂环状环(即一或多个环原子为杂原子,其余环原子为碳)。典型地,杂环状环含有1、2、3或4个杂原子;在某些具体实施例中,各杂环状环每环具有1或2个杂原子。各杂环状环通常含有3至8个环元(在某些具体实施例中列举出具有4或5至7个环元的环)及典型地由含有9至14个环元的稠合环、侧出环或螺环构成的杂环。某些杂环包含硫原子作为环元;在某些具体实施例中,硫原子经氧化为SO或SO2。杂环可任选地以各种取代基来取代,如上所述。除非特别指示,否则杂环可为杂环烷基(即各环为饱和或部分饱和)或杂芳基(即该基团内至少一个环为芳族)。杂环基一般可经由任何环或取代基原子来连接,只要能得到稳定的化合物。N-连接的杂环基是经由成份氮原子连接的。
杂环基包括,例如,azepanyl、吖辛因基(azocinyl)、苯并咪唑基、苯并嘧啶基、苯并异噻唑基、苯并异咪唑基、苯并呋喃基、苯并硫代呋喃基、苯并噁唑基、苯并噻唑基、苯并四唑基、苯并二氢吡喃基、苯并吡喃基、噌啉基、十氢喹啉基、二氢呋喃并[2,3-b]四氢呋喃、二氢异喹啉基、二氢四氢呋喃基、1,4-二噁-8-氮杂-螺[4.5]癸基、二噻嗪基、呋喃基、呋呫基、咪唑啉基、咪唑烷基、咪唑基、吲唑基、吲哚烯基(indolenyl)、二氢吲哚基、中氮吲哚基、吲哚基、异苯并呋喃基、异苯并二氢吡喃基、异吲唑基、异二氢吲哚基、异吲哚、异噻唑基、异噁唑基、异喹啉基、吗啉基、萘啶基、八氢异喹啉基、噁二唑基、噁唑啶基、噁唑基、2,3-二氮杂萘基、哌嗪基、哌啶基、哌啶酮基、喋啶基、嘌呤基、吡喃基、吡嗪基、吡唑烷基、吡唑啉基、吡唑基、哒嗪基、吡啶并咪唑基、吡啶并噁唑基、吡啶并噻唑基、吡啶基、嘧啶基、吡咯烷基、吡咯酮基、吡咯啉基、吡咯基、喹唑啉基、喹啉基、喹噁啉基、喹咛环基、四氢异喹啉基、四氢喹啉基、四唑基、噻二嗪基、噻二唑基、噻唑基、噻吩并噻唑基、噻吩并噁唑基、噻吩并咪唑基、噻吩基、苯硫基、硫代吗啉基及其中硫原子经氧化的变体、三嗪基、及以经上述1至4个的取代基所取代的上述任何基团。
“杂环C0-C6烷基”为经由直接键结或C1-C8烷基连接的杂环基团。(3-至10-元杂环)C0-C6烷基为经由单一共价键或具有1至6个碳原子的烷基连接的具有3至10个环元的杂环基。倘若杂环为杂芳基,则该基团记为(5-至10-元杂芳基)C0-C8烷基。(5-至7-元杂环)C0-C8烷基为经由单一共价键或C1-C8烷基连接的具5-至7-元的杂环状环。
某些杂环基为含有1个杂环或2个稠合环或螺环的4-至10-元、5-至10元、3-至7-元、4-至7-元或5-至7-元基团,并任选经取代。4-至10-元杂环烷基包含,例如,哌啶基、哌嗪基、吡咯烷基、azepanyl、1,4-二噁-8-氮杂-螺[4.5]癸-8-基、吗啉基、硫代吗啉基及1,1-二酮基-硫代吗啉-4-基。所述基团可如所示经取代。代表性芳族杂环为吖辛因基、吡啶基、嘧啶基、咪唑基、四唑基及3,4-二氢-1H-异喹啉-2-基。
本发明所用的“取代基”是指与所关注分子的原子共价结合的分子基团。举例而言,“环取代基”可为与环元原子(优选为碳或氮原子)共价结合的部份,例如卤素、烷基、卤烷基或本发明论及的其它基团。“取代”一词是指以如上所述的取代基置换分子结构中的氢原子,且未超出该指定原子的价数,及由该取代产生具有化学稳定性的化合物(即可经分离、鉴定特性、及测试生物活性的化合物)。
“任选取代”的基团为未经取代,或在一或多个可用位置,典型地为1、2、3、4或5个位置,经除了氢以外的一或多个合适基团(可相同或不同)取代。所述任选的取代基包括,例如,羟基、卤素、氰基、硝基、C1-C8烷基、C2-C8烯基、C2-C8炔基、C1-C8烷氧基、C2-C8烷基醚、C3-C8烷酮、C1-C8烷硫基、氨基、单-或二-(C1-C8烷基)氨基、C1-C8卤烷基、C1-C8卤烷氧基、C1-C8烷酰基、C2-C8烷酰基氧基、C1-C8烷氧羰基、-COOH、-CONH2、单-或二-(C1-C8烷基)氨基羰基、-SO2NH2、和/或单-或二-(C1-C8烷基)磺酰氨基,以及碳环与杂环基。任选取代也以“0至X个取代基取代”一词表示,其中X为可能取代基的最大数目。特定的任选取代的基团是以0至2、3或4个独立选定的取代基取代(即未经取代或经达到所述最大数目的取代基取代)。
术语“VR1”与“辣椒素受体”在本发明中可交换使用,它们是指第1型类香草醇受体。除非特别指示,否则这些术语涵盖大鼠及人类的VR1受体(例如,GenBank寄存编号AF327067、AJ277028及NM 018727;特定人类VR1 cDNAs的序列,由美国专利第6,482,611号的SEQ ID NOs:1至3所提供,而其所编码氨基酸序列则示于美国专利第6,482,611号的SEQ ID NOs:4和5),以及在其它物种中发现的同源物。
“VR1调节剂”,在本发明中亦称为“调节剂”,是调节VR1活化作用和/或由VR1介导的讯息传导作用的化合物。具体来说,本发明提供的VR1调节剂为通式I化合物及通式I化合物在药学上可接受的形式。VR1调节剂可为VR1促效剂或拮抗剂。若VR1的Ki小于1微摩尔浓度,优选为小于100纳摩尔浓度、10纳摩尔浓度或1纳摩尔浓度时,则调节剂是以“高亲和力”结合。本发明实施例3提供了用于测定VR1的Ki值的代表性试验。
如果调节剂能可检测地抑制类香草醇配位体结合至VR1和/或由VR1介导的讯息传导作用(使用例如实施例4提供的代表性试验),则此调节剂被视为“拮抗剂”;一般而言,所述拈抗剂在实施例4提供的试验中是以小于1微摩尔浓度,优选为小于100纳摩尔浓度,更优选为小于10纳摩尔浓度或1纳摩尔浓度的IC50值来抑制VR1的活化作用。VR1拮抗剂包括中性拮抗剂及反向促效剂。在特定具体实施例中,本发明提供的辣椒素受体拮抗剂不是类香草醇。
VR1的“反向促效剂”为在不添加类香草醇配位体的情况下,使VR1的活性低于其基础活性量的化合物。VR1的反向促效剂亦可抑制类香草醇配位体在VR1的活性,和/或也可抑制类香草醇配位体结合至VR1。化合物抑制类香草醇配位体结合至VR1的能力可通过结合试验(binding assay)予以测定,例如实施例3所提供的结合试验。VR1的基础活性,以及由于VR1拮抗剂存在下的VR1活性降低,均可由钙离子移动试验(calcium mobilization assay)予以测定,例如实施例4的试验。
VR1的“中性拮抗剂”为抑制类香草醇配位体在VR1的活性,但不显著改变该受体基础活性的化合物(即在类香草醇配位体不存在下所进行的如实施例4所述的钙离子移动试验中,VR1活性的减少不大于10%,更优选为不大于5%,尤其更优选为不大于2%;最优选为没有检测到活性降低)。VR1的中性拮抗剂可抑制类香草醇配位体结合至VR1。
本发明所用的“辣椒素受体促效剂’或“VR1促效剂”是指提升该受体活性至其基础活性量以上(即增进VR1活化作用和/或由VR1介导的讯息传导)的化合物。辣椒素受体促效剂活性可使用实施例4所提供的辣椒素受体促效作用的代表性体外试验予以鉴定。通常,该促效剂在实施例4所提供的试验中具有小于1微摩尔浓度,优选为小于100纳摩尔浓度,更优选为小于10纳摩尔浓度的EC50值。在特定具体实施例中,本发明提供的辣椒素受体促效剂不是类香草醇。
“类香草醇”为辣椒素或任何辣椒素类似物,其含有苯环且该苯环上两个氧原子与相邻环碳原子结合(其中一个碳原子是位于与该苯环结合的第三部分的连接点的对位)。类香草醇若以不大于10μM的Ki(如本发明所述予以测定)与VR1结合,则为“类香草醇配位体”。类香草醇配位体促效剂包括辣椒素、欧瓦尼(olvanil)、N-花生四烯酰基-多巴胺及树胶脂毒素(RTX)。类香草醇配位体拈抗剂包含辣椒平及碘-树胶脂毒素。
“辣椒素受体调节量”是指投药至患者后,在患者体内所达到的VR1调节剂在辣椒素受体的浓度足以改变体外类香草醇配位体与VR1结合(使用实施例3所提供的试验),和/或VR1介导的讯息传导作用(使用实施例4所提供的试验)的量。该辣椒素受体可存在于体液中,例如:血液、血浆、血清、CSF、关节液、淋巴、细胞间质液、眼泪或尿液。
“治疗有效量”是指投药至患者后,可检测到足以提供患者从所治疗的症状中缓解的量。该缓解可使用任何适当准则予以检测,包括一或多种症状(例如疼痛)的缓解。
“患者”是本发明提供的VR1调节剂所治疗的任何个体。患者包括人类,以及其它动物诸如陪伴动物(例如狗与猫)及家畜。患者可能历经对辣椒素受体调节敏感的病症的一种或多种症状(例如,疼痛、暴露于类香草醇配位体、痒、尿失禁、膀胱过动、呼吸性疾病、咳嗽和/或打嗝),或可能无所述症状(即为预防性治疗)。
VR1调节剂
如上所述,本发明提供可用于多种情况下的VR1调节剂,包括治疗疼痛(例如,神经性或周边神经介导的疼痛);暴露于辣椒素下;暴露于酸、热、光、催泪瓦斯空气污染物、辣椒喷雾剂或相关制剂下;呼吸性症状诸如气喘或慢性阻塞性肺疾病;痒;尿失禁或膀胱过动;咳嗽或打嗝;和/或肥胖症。VR1调节剂亦可用于体外试验(例如,受体活性试验)、作为检测与定位VR1的探针以及作为配位体结合与VR1介导的讯息传导试验的标准物。
本发明所提供的VR1调节剂为经芳基取代的苯并[d]异噻唑-3-基胺类似物,所述类似物以纳摩尔浓度(即次微摩尔),优选为以次纳摩尔浓度,更优选为以小于100微微摩尔浓度(picomolar),或甚至小于20微微摩尔浓度检测地调节辣椒素与VR1的结合。所述调节剂优选为非类香草醇。优选的调节剂为VR1拮抗剂,且在实施例4所述及的试验中不具有可检测的促效剂活性。在特定具体实施例中,所述调节剂进一步以高亲和力结合至VR1,且基本上不抑制人类EGF受体酪胺酸激酶(human EGF receptor tyrosine kinase)的活性。
本发明某种程度上是以下述发现为基础:具有上述通式I的小分子(以及其药学上可接受的形式)具有调节VR1活性。通过式I的变数如上述所列举。
式I
如上所述,Ar1与Ar2独立地为任选取代的碳环或杂环。某些取代基选自通式为LRa的那些取代基。如上所述,L在每种情况下独立地选自:单一共价键、O,C(=O)、OC(=O)、C(=O)O、OC(=O)O、S(O)m(即-S-、或
)、N(Rx)(即
 )、C(=O)N(Rx)(即
)、C(=O)N(Rx)(即
 )、N(Rx)C(=O)(即)、N(Rx)S(O)m(即
)、N(Rx)C(=O)(即)、N(Rx)S(O)m(即
 )、S(O)mN(Rx)(即
)、S(O)mN(Rx)(即
 ),以及N[S(O)mRx]S(O)m(即
),以及N[S(O)mRx]S(O)m(即
 )。
)。
在某些具体实施例中,Ar2独立地为任选取代的苯基或任选取代的6-元芳族杂环状环。所述Ar2基团包括,但不限于,苯基、吡啶基或嘧啶基,其中每一个经1或2个取代基取代,所述取代基独立地选自卤素、氰基、C1-C6烷基、卤C1-C6烷基、C1-C6烷氧基、卤C1-C6烷氧基、C1-C6烷基磺酰基或单-与二-(C1-C6烷基)磺酰氨基。代表性Ar2基团包括苯基和吡啶基(任选经卤素、C1-C4烷基或卤C1-C4烷基取代)。在某些具体实施例中,Ar2的其中一个取代基是位于连接点对位的环上位置。换句话说,倘若Ar2为苯基,该取代基是位于4-位置。同样地,倘若Ar2为2-吡啶基,该取代基是位于5-位置。
Ar1,在某些具体实施例中为苯基或吡啶基,经1或2个取代基取代,所述取代基独立地选自卤素(例如,氟或氯)、氰基、COOH、C1-C6烷基(例如,甲基)、卤C1-C6烷基(例如,三氟甲基),C1-C6烷氧基、卤C1-C6烷氧基、C1-C6烷基磺酰基及单-与二-(C1-C6烷基)磺酰氨基。在某些具体实施例中,Ar1是在连接点的邻位做单取代(即若Ar1为苯基则取代于2-位置;若Ar1为2-吡啶基则取代于3-位置。优选的Ar1是2-吡啶基,经1或2个选自卤素、氰基、C1-C6烷基、卤C1-C6烷基、C1-C6烷氧基、卤C1-C6烷氧基、C1-C6烷基磺酰基或单-与二-(C1-C6烷基)磺酰氨基的取代基取代。某些所述Ar1基团是在3-位置取代的2-吡啶基;所述基团的实施例为3-甲基-吡啶-2-基、3-氯-吡啶-2-基、3-氰基-吡啶-2-基和3-三氟甲基-吡啶-2-基。
W、Y、和Z,在某些具体实施例中独立地为N或CH。例如,W、Y、和Z其中的一个可为N,而其它为CH。或者,W、Y、和Z其中的一个可为CH,其它为N。在某些化合物中,W为CH;而Y和Z独立地为N或CH。
在通式I的化合物中,Ar1是苯基或2-吡啶基,经1或2个独立地选自卤素、氰基、C1-C4烷基或卤C1-C4烷基的取代基取代;而Ar2为苯基或吡啶基,经1或2个独立地选自卤素、氰基、C1-C4烷基与卤C1-C4烷基、C1-C6烷基磺酰基或单-与二-(C1-C6烷基)磺酰氨基的取代基取代。
本发明所提供的某些化合物可进一步满足通式II,其中Ar1和Ar2独立地选自经取代的苯基、经取代的吡啶基及经取代的嘧啶基,以及Y及Z如式通I中所述。

本发明所提供的某些化合物可进一步满足通式III,其中A为CH或N,每一个Rb独立地为卤素、氰基、硝基或LRa,而其它变数如式通I中所述。

本发明所提供的代表性化合物包括,但不限于,在本发明实施例1与表1中所述的那些。很明显,此处所特定列举的化合物仅为代表性的,并非要限制本发明范围。此外,如上所述,本发明的所有化合物皆可以游离碱或药学上可接受的形式如酸加成盐形式存在。
当使用体外VR1配位体结合试验和/或功能性试验(例如钙离子移动试验、背根神经节试验或活体内疼痛缓解试验)测定时,本发明所提供的经芳基取代的苯并[d]异噻唑-3-基胺类似物可检测地改变(调节)VR1的活性。本发明中有关“VR1配位体结合试验”,是指例如实施例3所提供的标准体外受体结合试验,以及“钙离子移动试验”(在本发明中亦称为“讯息传导试验”)则可如实施例4所述进行。简而言之,要评估对VR1的结合作用可进行竞争性结合试验,其中,将VR1制剂与会结合至VRI的经标记(例如,125I或3H)的化合物(例如,辣椒素受体促效剂如RTX)以及未标记的测试化合物一起培养。在本发明提供的试验中,所用VR1优选为哺乳动物的VR1,更佳为人类或大鼠的VR1。所述受体可经重组或不经任何修饰(naturally)地得到表达。所述VR1制剂可为,例如,来自重组表达人类VR1的HEK293或CHO细胞的细胞膜制剂。将可检测地调节类香草醇配位体与VR1结合的化合物一起培育,会导致与VR1制剂结合的标记量相对于化合物不存在下结合的标记量降低或增加。这种降低或增加可用于测定如本发明所述VR1的Ki。通常,在所述试验中,优选化合物为能使与VR1制剂结合的标记量降低者。
如上所述,在某些具体实施例中,优选化合物为VR1拮抗剂。所述化合物的IC50值可使用如实施例4提供的标准体外VR1介导的钙离子移动试验予以测定。简而言之,将表达辣椒素受体的细胞与所关注的化合物及细胞内钙离子浓度指示剂(例如,细胞膜可渗透性钙敏感性染料例如Fluo-3或Fura-2(二者均可得自,例如,Molecular Probes,Eugene,OR),其中每一个与Ca++结合时均产生荧光讯号)接触。所述接触优选通过将细胞在缓冲液或培养液中培育一次或多次而进行,所述缓冲液或培养液在溶液中包括化合物和/或指示剂。使接触维持足够长的时间(例如,1至2小时)以容许染料进入细胞中。将细胞洗涤或过滤以去除过量染料,然后再与类香草醇受体促效剂(例如,辣椒素、RTX或欧瓦尼(olvanil))接触,其浓度一般等于EC50浓度,接着测定荧光反应。当接触促效剂的细胞与VR1拮抗剂化合物接触时,与在测试化合物不存在下与促效剂接触的细胞相比,其荧光反应通常减少至少20%,优选为至少50%及更优选为至少80%。本发明提供的VR1拮抗剂的IC50,优选为小于1微摩尔浓度、小于100nM、小于10nM或小于1nM。
在其它具体实施例中,优选化合物为辣椒素受体促效剂。辣椒素受体促效剂的活性通常可如实施例4所述予以测定。当细胞与1微摩尔浓度的VR1促效剂化合物接触时,荧光反应通常增加,而该增加量是细胞与100nM辣椒素接触时观察所得的增加量的至少30%。本发明提供的VR1促效剂的EC50优选为小于1微摩尔浓度、小于100nM或小于10nM。
或者,VR1调节活性也可使用如实施例7提供的经培养的背根神经节试验和/或如实施例8提供的活体内疼痛缓解试验予以评估。本发明提供的化合物优选在本发明提供的一或多个功能性试验中,对于VR1活性具有统计学上显著的特定影响。
在某些具体实施例中,本发明提供的VR1调节剂不会实质上调节配位体与其它细胞表面受体(例如EGF受体酪胺酸激酶或烟碱性乙酰胆碱受体)的结合。换句话说,所述调节剂实质上不会抑制细胞表面受体的活性,例如人类表皮生长因子(EGF)受体酪胺酸激酶或烟碱性乙酰胆碱受体(例如,所述受体的IC50或IC40优选为大于1微摩尔浓度,最优选为大于10微摩尔浓度)。优选地,调节剂在0.5微摩尔浓度、1微摩尔浓度或更优选为10微摩尔浓度下无法可检测地抑制EGF受体活性或烟碱性乙酰胆碱受体活性。测定细胞表面受体活性的试验为市售可得,且包括购自Panvera(Madison,WI)的酪胺酸激酶试验套组。
本发明提供的优选VR1调节剂为非镇静剂。换句话说,在测定疼痛缓解的动物模式(例如本发明实施例8提供的模式)中足以提供止痛的最小剂量的两倍的VR1调节剂剂量,在动物模式镇静试验中(使用Fitzgerald et al.(1988)Toxicology 49(2-3):433-9叙述的方法)只引起短暂(即持续不超过疼痛缓解持续的时间的1/2)或优选为无统计学上显著的镇静作用。优选地,足以提供止痛的最小剂量的五倍剂量不会产生统计学上显著的镇静作用。更优选地,本发明提供的VR1调节剂在小于25mgmg/kg(优选为小于10mgmg/kg)的静脉内剂量或小于140mgmg/kg(优选为小于50mgmg/kg,更佳为小于30mgmg/kg)的口服剂量下不会产生镇静作用。
如果需要,则可对本发明提供的VR1调节剂进行特定药理性质的评估,包括但不限于,口服生物利用度(优选的化合物为经口服生物利用直至使化合物的治疗有效浓度达到小于140mg/kg,优选为小于50mg/kg,更优选为小于30mg/kg,又更优选为小于10mg/kg,更优选为小于1mg/kg,及最优选为小于0.1mg/kg的口服剂量程度)、毒性(优选的VR1调节剂在给药辣椒素受体调节量至对象时没有毒性)、副作用(优选的VR1调节剂在给药治疗有效量的化合物至对象时,会产生相当于安慰剂的副作用)、血清蛋白结合作用及体外与体内半衰期(优选的VR1调节剂具有相当于体内半衰期的体外半衰期,从而容许Q.I.D.给药,优选经T.I.D.给药,更佳为B.I.D.给药,及最优选为一天给药一次)。此外,血脑障壁的差异穿透性(differential penetration)对于通过调节CNS VR1活性来治疗疼痛的VR1调节剂而言可能为所需的,因此使得如上所述的口服总每日剂量提供所述调节作用至治疗有效的程度,然而使用低脑浓度的VR1调节剂在治疗周边神经介导的疼痛可能是优选的(即所述剂量不会提供足以显著地调节VR1活性的脑(例如,CFS)浓度的化合物)。可使用此项技术中已知的常规试验来评估所述性质,及鉴定具有特别用途的优异化合物。例如,用于预测生物利用度的试验包括跨越人类单层肠细胞(包括Caco-2单层细胞)的运送。可由给予(例如,经静脉内)所述化合物的实验室动物中所述化合物的脑浓度来预测在人类中所述化合物的血脑障壁穿透性。血清蛋白结合可由白蛋白结合试验进行预测。化合物半衰期与化合物的剂量频率成反比。化合物的体外半衰期可由本发明实施例5所述的微粒体半衰期试验予以预测。
如上所述,本发明提供的优选VR1调节剂不具有毒性。一般而言,应了解本发明所用的“不具毒性”一词为相对意义,意指由美国食品药物管理局(FDA)认可的可用于给药至哺乳动物(优选为人类)的任何物质,或遵守已建立的准则的任何物质,倾向于被FDA认可的用于给药至哺乳动物(优选为人类)的任何物质。此外,高度优选的不具毒性化合物通常满足一个或多个下述准则:(1)基本上不抑制细胞ATP生产;(2)不显著延长心脏QT间隔;(3)基本上不引起肝脏肿大;且(4)不引起肝脏酵素的大量释放。
本发明所使用“基本上不抑制细胞ATP生产”的VRI调节剂为满足本发明实施例8所述准则的化合物。换句话说,如实施例6所述用100μM的所述化合物处理的细胞表现其ATP浓度为未经处理细胞内所测得的ATP浓度的至少50%。在更高度优选的具体实施例中,所述细胞显现的ATP浓度为未经处理细胞内所测得的ATP浓度的至少80%。
“不显著延长心脏QT间隔”的VR1调节剂为在给药产生活体内治疗有效浓度的最低剂量的两倍剂量后不会造成对天竺鼠、迷你猪或狗在统计学上显著的延长心脏QT间隔(如心电图所测定)的化合物。在某些优选具体实施例中,以非口服或口服形式给药0.01、0.05、0.1、0.5、1、5、10、40或50mg/kg的剂量不会导致统计学上显著的心脏QT间隔延长。所谓“统计学上显著的”意指当使用具统计意义的标准参数试验,例如学生氏T试验(Student’s T test)测定时,偏离控制组在p<0.1标准或更优选在p<0.05标准下的结果。
所谓“基本上不引起肝脏肿大”的VR1调节剂是指,若以产生活体内治疗有效浓度的最低剂量的两倍剂量来每天治疗实验室啮齿类(例如,小鼠或大鼠)5至10天后,造成肝脏对体重比的增加不大于对照组的100%。在更高度优选的具体实施例中,此剂量不引起大于对照组的75%或50%的肝脏肿大。若是使用非啮齿类哺乳动物(例如,狗),此剂量不应造成肝脏对体重比的增加大于对照组的50%,优选为不大于25%,更优选为不大于10%。所述试验中的优选剂量,包括以非口服或口服形式给药0.01、0.05、0.1、0.5、1、5、10、40或50mgmg/kg。
同样地,所谓“不促进肝脏酵素的大量释放”的VR1调节剂是指,若给药产生活体内治疗有效浓度的最低剂量的两倍剂量时不会提升实验室啮齿类动物ALT、LDH或AST的血清浓度至大于模拟处理对照组的100%。在更高度优选的具体实施例中,此剂量不会提升所述血清浓度至大于对照组的75%或50%。或者,所谓“不促进肝脏酵素的大量释放”的VR1调节剂是指,若在体外肝细胞试验中,浓度(在体外与肝细胞接触及培养的培养液或其它所述溶液中)等于该化合物在活体内治疗浓度的最低剂量的两倍时,不会导致任何所述肝脏酵素可检测地释到培养液中,而高于从模拟处理的对照组细胞培养液中观察到的基线量。在更高度优选的具体实施例中,当所述化合物浓度为该化合物在活体内治疗浓度的最低剂量的五倍时,优选为十倍时,仍无任何所述肝脏酵素可检测地释放至培养液中而高于基线量。
于其它具体实施例中,某些优选的VR1调节剂浓度在等于活体内治疗有效浓度的最低剂量时,不会抑制或诱发微粒体细胞色素P450酶活性,例如CYP1A2活性、CYP2A6活性、CYP2C9活性、CYP2C19活性、CYP2D6活性、CYP2E1活性或CYP3A4活性。
某些优选的VR1调节剂浓度在等于活体内治疗有效浓度的最低剂量时不具基因破坏性(clastogenic)(例如,如使用小鼠红血球源祖细胞(erythrocyte precursor cell)微核试验、Ames微核试验、螺旋微核试验等予以测定)。在其它具体实施例中,在所述浓度下,某些优选VR1调节剂不会诱发姊妹染色单体(sister chromatid)交换(例如,于中国仓鼠卵巢细胞中)。
如下文更详细的讨论,为了检测目的,本发明提供的VR1调节剂可为同位素标记或放射性标记。例如,通式I至通III所列举的化合物,可有一个或多个原子被原子质量或质量数与一般在自然界发现的原子质量或质量数不同的相同元素的原子置换。存在于本发明提供的化合物中的同位素实施例包括氢、碳、氮、氧、磷、氟及氯的同位素,例如2H、3H、11C、13C、14C、15N、18O、17O、31P、32P、35S、18F及36Cl。此外,由于代谢稳定性较大,例如活体内半衰期增加或剂量需求减少,所以以重同位素例如氘(亦即,2H)置换可提供特定的治疗优点,因此,在一些情形下可能是优选的。
VR1调节剂的制备
经芳基取代的苯并[d]异噻唑-3-基胺类似物通常可使用标准合成方法予以制备。例如Kwon等人的(1996)Arzneim.Forsch.46:966-971;Boshagen的(1967)Chem.Ber.100:954-960;Boshagen的(1967)Chem.Ber.100:3326-3330(1967);以及Yevich等人的(1986)J.Med.Chem.29:359-369所述那些。一般而言,起始材料来自例如Sigma-Aldrich Corp.(St.Louis,MO)的供货商,通过市场通路即可购得,或使用已建立的实验流程通过市售可得的前驱物予以合成。举例而言,可使用与下文反应图式1至3所示相似的合成途径,以及合成有机化学范畴中已知的合成方法,或在此项技术领域具有通常知识者所知的其变异方法。下文反应图式中的各变量是参照与本发明提供的化合物说明中一致的任何基团。
在下述的反应图示中,“催化剂”一词是指适当的过渡金属催化剂,例如,但不局限于,四(三苯基膦)钯(O)或钯(II)乙酸盐。另外,催化系统可包括配位体,例如,但不局限于,2-(二环己基膦基)联苯及三-叔丁基膦,也可包碱,例如K3PO4、Na2CO3或叔丁醇钠或钾。过渡金属-催化反应可在室温或较高的温度下进行,并使用各种惰性的溶剂包括,但不局限于,甲苯、二甲苯、DMF、N-甲基吡咯烷酮、乙二醇、二甲基醚、二甘醇二甲醚与乙腈。常用的试剂/催化剂对包括芳基硼酸/钯(O)(Suzuki反应;Miyaura与Suzuki的(1995)Chemical Reviews95:2457)以及芳基三烷锡烷/钯(O)(Stille反应;T.N.Mitchell的(1992)Synthesis 9:803-815),芳基锌(arylzinc)/钯(O)及芳基格林纳试剂(Grignard)/镍(II)。
“活化”一词是指合成性的转换,其中酰胺部分的羰基转变成适当的离去基(L)。适于进行此项转换的试剂为有机合成范畴中具有通常知识者所已知的,包括,但不局限于,SOCl2、POCl3及三氟甲磺酸酐。在下述反应图标中标记为″L″的离去基的实施例包括Cl、Br或O(C=O)CF3。
本发明所使用的其它定义如下:
CDCl3 经氚化的氯仿
d 化学位移
DCM 二氯甲烷
DME 乙二醇二甲基醚
DMF 甲基甲酰胺
EtOAc 酸乙酯
EtOH 醇
1H NMR 质子核磁共振
HPLC 高效液相层析
Hz 赫兹
LC/MS 液相层析/质谱
MS 质谱
(M+1) 质量+1
Pd(PPh3)4 四(三苯基膦)钯(O)
反应图示1

反应图示2
反应图示3

在某些具体实施例中,VR1调节剂可包括一或多个不对称碳原子,从而导致化合物以不同的立体异构体形式存在。所述形式为,例如,消旋体或光学活性形式。如上所述,所有立体异构体皆包含于本发明中。但是,希望能获得单一的对映体(即光学活性形式)。制备单一对映体的常规方法包括不对称合成和消旋体的拆分。消旋体的分离可通过例如已知方法予以完成,例如,在拆分剂存在下的结晶作用,或使用例如手性HPLC柱的层析法。
化合物可通过使用含有至少一个放射性同位素原子的前驱物来进行合成,从而被放射性标记。每一个放射性同位素优选为碳(例如,14C)、氢(例如,3H)、硫(例如,35S)、或碘(例如,125I)。以氚标记的化合物也可使用下述方法经催化制备:在氚化乙酸中的铂催化交换、在氚化三氟乙酸中的酸催化交换、或使用化合物作为基质以氚气进行非匀相催化交换。此外,倘若适当,则特定前驱物可使用氚气进行氚-卤素交换、进行不饱和键结的氚气还原、或使用硼氚化钠进行还原。放射性标记化合物的制备亦可方便地向专精于合成放射性标记探针化合物的放射性同位素供货商订购。
药学组合物
本发明也提供含有一或多种VR1调节剂、以及至少一种生理上可接受的载剂或赋形剂的药学组合物。药学组合物可包含,例如一或多种水、缓冲剂(例如,中性缓冲盐液或磷酸盐缓冲盐液)、乙醇、矿物油、植物油、二甲亚砜、碳水化合物(例如,葡萄糖、甘露糖、蔗糖或葡聚糖)、甘露糖醇、蛋白质、佐剂、多肽或例如甘胺酸的氨基酸、抗氧化剂例如EDTA或谷光甘肽的螯合剂和/或防腐剂。此外,本发明提供的药学组合物中也可包含(但并非需要)其它活性成分。
药学组合物可调配成任何适当的给药方式,包括例如局部、口服、经鼻、经直肠或非口服给药。本发明所用的非口服一词包括经皮下、皮内、血管内(例如,静脉内)、肌内、脊髓、颅内、鞘内(intrathecal)和腹腔内注射,以及任何类似注射或灌注的技术。于某些具体实施例中,优选为适用于口服的组合物。所述组合物包括例如,锭剂、片剂、菱形锭剂、水性或油性悬浮液、分散性粉剂或粒剂、乳液、硬或软胶囊、或糖浆或酏剂。又在其它具体实施例中,本发明组合物可调配为冻干物。对于某些症状(例如,于治疗例如灼伤或痒等皮肤症状)供局部给药的配方可能是优选的;治疗尿失禁及膀胱过动时,直接给药至膀胱(膀胱内给药)的配方是优选的。
进行口服用的组合物可进一步包括一或多种成分,例如增甜剂、调味剂、着色剂和/或防腐剂,以提供迎合爱好且美味的制剂。锭剂包括与适用于制造锭剂的生理上可接受的赋形剂掺合的活性成分。所述赋形剂包括,例如,惰性稀释剂(例如,碳酸钙、碳酸钠、乳糖、磷酸钙或磷酸钠)、造粒及崩解剂(例如,玉米淀粉或海藻酸)、黏结剂(例如,淀粉、明胶或阿拉伯胶)及润滑剂(例如,硬脂酸镁、硬脂酸或滑石粉)。锭剂可不用包覆或可利用现有技术包覆以延缓于胃肠道的崩解与吸收,进而提供长期的持续作用。举例而言,可使用时间延缓物质,例如单硬脂酸甘油酯或二硬脂酸甘油酯。
口服用配方也可以硬明胶胶囊形式存在,其中所述活性成分是与惰性固体稀释剂(例如,碳酸钙、磷酸钙或高岭土)混合;或以软明胶胶囊形式存在,其中所述活性成分与水或油介质(例如,花生油、液态石蜡或橄榄油)混合。
水性悬浮液包括与适用于制造水性悬浮液的赋形剂掺合的活性物质。所述赋形剂包括悬浮剂(例如,羧甲基纤维素钠、甲基纤维素、羟丙甲基纤维素、海藻酸钠、聚乙烯吡咯烷酮、黄蓍胶与阿拉伯胶);及分散剂或润湿剂(例如,天然存在的磷脂类例如卵磷脂、烯烃氧化物与脂肪酸的缩合产物例如聚氧乙烯硬脂酸酯、环氧乙烷与长链脂族醇的缩合产物例如十七乙烯氧基鲸蜡醇(heptadecaethyleneoxycatanol)、环氧乙烷与衍生自脂肪酸和己糖醇的部分酯类的缩合产物例如聚氧乙烯山梨糖醇单油酸酯、或环氧乙烷与衍生自脂肪酸和己糖醇的部分酯类的缩合产物例如聚乙烯山梨糖酐单油酸酯)。水性悬浮液也可包括一或多种防腐剂(例如对羟苯甲酸-乙酯或-正丙酯)、一种或多种着色剂、一种或多种调味剂、及一种或多种增甜剂(例如蔗糖或糖精)。
油性悬浮液可通过使活性成分悬浮在植物油(例如,花生油、橄榄油、芝麻油或椰子油)中或矿物油(例如液态石蜡)中进行配制。所述油性悬浮液可包含增稠剂,例如口蜜蜡、硬石蜡或鲸蜡醇。可添加如上所述的甜味剂和/或调味剂以提供美味的口服制剂。所述悬浮液可通过添加抗氧化剂(例如抗坏血酸)予以保存。
适用于通过添加水来制备水性悬浮液的可分散粉剂及粒剂提供了与分散剂或润湿剂、悬浮剂及一或多种防腐剂掺合的活性成分。适当的分散剂或润湿剂及悬浮剂如上所述。也可存在其它赋形剂,例如增甜剂、调味剂与着色剂。
药学组合物亦可调配为水包油型乳液。其油相可为植物油(例如,橄榄油或花生油)、矿物油(例如,液态石蜡)或其混合物。适当的乳化剂包括天然存在的胶类(例如阿拉伯胶或黄蓍胶)、天然存在的磷脂类(例如,大豆卵磷脂、及衍生自脂肪酸与己糖醇的酯类或部分酯类)、酸酐类(例如山梨糖酐单油酸酯)及衍生自脂肪酸与己糖醇的部分酯类与环氧乙烷的缩合产物(例如,聚氧乙烯山梨糖酐单油酸酯)。乳液亦可包括一或多种增甜剂和/或调味剂。
糖浆与酏剂可与增甜剂例如甘油、丙二醇、山梨糖醇或蔗糖一起配制。所述配方亦可包括一或多种缓和剂、防腐剂、调味剂和/或着色剂。
局部给药的配方典型地包括与活性剂结合的局部载剂,含或不含附加的任选成分。合适的局部载剂及附加成分已为此项技术范畴中所熟知,且显而易见地,载剂的选择取决于特定的物理形式及递送方式。局部载剂包括水;有机溶剂例如醇类(例如,乙醇或异丙醇)或甘油;二醇类(例如,丁二醇、异戊二醇或丙二醇);脂族醇类(例如,羊毛脂);水与有机溶剂的混合物,及有机溶剂(例如醇)与甘油的混合物;以脂质为主的物质例如脂肪酸、酰基甘油类(包含油类例如矿物油,及天然或合成来源的脂肪)、磷酸甘油酯类、神经髓鞘脂类及蜡类;以蛋白质为主的物质例如胶原蛋白及明胶;以聚硅氧为主的物质(非挥发性与挥发性二者);及以烃为主的物质例如微囊海绵及聚合物基质。组合物可进一步包括适用于增进所施用配方稳定性或效果的一或多种成分,例如稳定剂、悬浮剂、乳化剂、黏度调整剂、胶凝剂、防腐剂、抗氧化剂、皮肤渗透增强剂、保湿剂及持续释放物质。该成分的实施例如Martindale-- The Extra Pharmacopoeia(Pharmaceutical Press,London1993)及Martin(ed.)Remington’s Pharmaceutical Sciences中所述的那些。配方可包括微胶囊,例如羟甲基纤维素或明胶-微胶囊、微脂粒、白蛋白微球体、微乳液、纳米粒子或纳米胶囊。
局部配方可制备为多种物理形式,包括,例如,固体、糊剂、乳霜、泡沫剂、洗剂、凝胶、粉剂、水性液体或乳液。所述药学可接受形式的物理外观及黏性可由配方中存在的乳化剂及黏度调整剂的存在与否及用量多少予以控制。固体通常坚实且具有不可倾泻性,通常配制成为棒状或条状、或呈微粒状;固体可为不透明的或透明的,且任选地包含溶剂、乳化剂、保湿剂、软化剂、芳香剂、染料/着色剂、防腐剂及增加或加强最终产物效力的其它活性成分。乳霜及洗剂两者通常极为相似,主要不同在于其黏性;洗剂及乳霜两者可为不透明、半透明或透明,并且常含有乳化剂、溶剂、黏度调整剂、以及保湿剂、软化剂、芳香剂、染料/着色剂、防腐剂及增加或加强最终产物效力的其它活性成分。凝胶可制备成具有广范的黏性,从浓稠或高黏性至稀薄或低黏性。这些配方,如洗剂及乳霜,也可含有溶剂、乳化剂、保湿剂、软化剂、芳香剂、染料/着色剂、防腐剂及增加或加强最终产物效力的其它活性成分。液体比乳霜、洗剂、或凝胶稀薄,且通常不含乳化剂。液体局部产品常含有溶剂、乳化剂、保湿剂、软化剂、芳香剂、染料/着色剂、防腐剂及增加或加强最终产物效力的其它活性成分。
局部配方用的合适乳化剂包括,但不限于,离子性乳化剂、鲸蜡醇、非离子乳化剂例如聚氧乙烯油基醚、PEG-40硬脂酸酯、鲸蜡硬脂醇醚(ceteareth)-12、鲸蜡硬脂醇醚-20、鲸蜡硬脂醇醚-30、鲸蜡硬脂醇、PEG-100硬脂酸酯及硬脂酸甘油酯。合适的黏性调整剂包括,但不限于,保护胶体或非离子性胶类例如羟乙基纤维素、黄原胶、硅酸铝镁、硅石、微晶蜡、蜜蜡、石蜡、及棕榈酸鲸蜡酯。凝胶组合物可通过添加胶凝剂例如几丁聚糖、甲基纤维素、乙基纤维素、聚乙烯醇、聚季铵盐类、羟乙基纤维素、羟丙基纤维素、羟丙甲基纤维素、卡波姆(carbomer)或胺化的甘草酸盐而形成。合适的界面活性剂包括,但不限于,非离子性、两性、离子性及阴离子性界面活性剂。举例而言,可于局部配方中使用一或多种二甲基聚硅氧烷共聚物(dimethiconecopolyol)、聚山梨糖醇酯20、聚山梨糖醇酯40、聚山梨糖醇酯60、聚山梨糖醇酯80、月桂酰胺DEA、椰油酰胺DEA与椰油酰胺MEA、油基甜菜碱、椰油酰胺丙基磷脂酰基PG-氯化二甲基铵(PG-dimoniumchloride)、及月桂醇硫酸铵。合适的防腐剂包括,但不限于,抗微生物剂例如对羟苯甲酸甲酯、对羟苯甲酸丙酯、山梨酸、苯甲酸、与甲醛,以及物理性安定剂与抗氧化剂例如维生素E、抗坏血酸钠/抗坏血酸及没食子酸丙酯。合适的保湿剂包括,但不限于,乳酸与其它羟基酸及其盐、甘油、丙二醇与丁二醇。合适的软化剂包括羊毛脂醇、羊毛脂、羊毛脂衍生物、胆固醇、矿脂、新戊酸异硬脂酯及矿物油。合适的芳香剂及着色剂包括,但不限于,FD&C红色40号、FD&C黄色5号。其它可纳入局部配方的合适附加成分包括,但不限于,磨蚀剂、吸收剂、抗结块剂、抗起泡剂、抗静电剂、收敛剂(例如,金缕梅、醇与草本抽出物例如洋甘菊萃取物)、黏结剂/赋形剂、缓冲剂、螯合剂、薄膜形成剂、调理剂、推进剂、遮光剂、pH调整剂及保护剂。
凝胶配方用的合适局部载剂的实例为:羟丙基纤维素(2.1%);70/30异丙醇/水(90.9%);丙二醇(5.1%);及聚山梨糖醇酯80(1.9%)。泡沫剂配方用的合适局部载剂实例为:鲸蜡醇(1.1%);硬脂醇(0.5%);季盐52(1.0%);丙二醇(2.0%);乙醇95PGF3(61.05%);去离子水(30.05%);P75烃推进剂(4.30%)。所有百分比均为重量%。
局部组合物用的典型敷用模式包括使用手指的施敷法;使用物理施敷器(例如布、面纸、纱布、棉棒或刷子)的施敷法;喷雾法(包括水气、气溶胶或泡沫喷雾法);点滴器施敷法;淋洒;浸渍;及冲洗法。也可使用经控制的释放载剂。
药学组合物可制备为无菌注射用水性或油质悬浮液。视所用载体与浓度而定,可使调节剂悬浮或溶解于载剂中。所述组合物可使用如上所述的适当分散剂、润湿剂和/或悬浮剂,根据已知技术进行配制。在可接受的载体与溶剂中,可使用水、1,3-丁二醇、林格氏溶液(Ringer’ssolution)及等张氯化钠溶液。此外,可使用无菌的非挥发性油类作为溶剂或悬浮介质。为了实现此目的,可使用任何厂牌的非挥发性油,包括合成的单-或二甘油酯。另外,脂肪酸例如油酸可用于注射组合物的制备中,而佐剂例如局部麻醉剂、防腐剂和/或缓冲剂可溶于载剂中。
调节剂亦可调配成栓剂(例如,供直肠给药用)。所述组合物可通过将药物与合适的无刺激性赋形剂混合而制备,该赋形剂在常温时为固体,但在直肠温度时为液体,从而在直肠溶解而释出药物。合适的赋形剂包括,例如,可可脂及聚乙二醇类。
药学组合物可配制成持续释放型配方(即给药后缓慢释放调节剂的胶囊配方)。该配方通常可使用已知技术予以制备并通过例如,口服、直肠或皮下植入或在所需目标位置植入来给药。所述配方中所用的载剂具有生物兼容性,亦可具有生物降解性;优选地,所述配方能提供相当固定的调节剂释放量。持续释放型配方中所含调节剂的量取决于,例如,植入位置、释放率与预期持续释放时间及所治疗或预防的症状性质。
除了上述给药模式或与所述模式并用者以外,也可方便地将调节剂添加于食物或饮用水中(例如,供投予非人类动物包括陪伴动物(例如狗与猫)及家畜用)。可配制动物饲料与饮用水以使动物随其膳食一起摄入所述组合物的适当量。亦可方便地以所述组合物的预混物形式将其添加到饲料或饮用水中。
调节剂通常以辣椒素受体调节量,优选以治疗有效量来给药。优选的全身性剂量为每天每公斤体重不高于50mg(例如,每天每公斤体重约0.001mg至约50mg),其中,口服剂量通常比静脉内剂量高出约5至20倍(例如,每天每公斤体重0.01至40mg)。
可与载剂物质组合以产生单一剂量单位的活性成分的量将根据例如所治疗的患者及给药的特定模式而定。剂量单位通常含有约10μg至约500mg之间的活性成分。适当剂量可使用此项技术中已知的常规测试及程序予以建立。
可将药学组合物包装,以用于治疗对VR1调节作用敏感的症状(例如,治疗暴露于类香草醇配位体、疼痛、痒、肥胖症或尿失禁)。经包装的药学组合物可包括:能容纳治疗有效剂量的如本发明所述的至少一种VR1调节剂的容器,及指示所含组合物是用于治疗对VR1调节有敏感症状的患者的说明书(例如,标签)。
使用方法
本发明提供的VR1调节剂可在体外及体内的多种情况下用于改变辣椒素受体的活性和/或活化作用。在某些方面,VR1拮抗剂可用于抑制类香草醇配位体促效剂(例如辣椒素和/或RTX)在体外或体内与辣椒素受体的结合。一般而言,所述方法包括在类香草醇配位体存在于水性溶液中,或存在其它适于该配位体与辣椒素受体结合的条件下使本发明所提供的一或多种VR1调节剂的辣椒素受体调节量与辣椒素受体接触的这一步骤。辣椒素受体可存在于溶液或悬浮液(例如,在分离的细胞膜或细胞制剂)中、或在培养或分离的细胞中。在某些具体实施例中,辣椒素受体是通过存在在患者的神经元细胞所表达的,且所述水性溶液为体液。给药动物的一或多种VR1调节剂的量,优选是使得存在于动物的至少一种体液中的类似物的量在治疗有效浓度,所述治疗有效浓度为1微摩尔浓度或更少;优选为500纳摩尔浓度或更少;又更优选为100纳摩尔浓度或更少、50纳摩尔浓度或更少、20纳摩尔浓度或更少,或10纳摩尔浓度或更少。例如,该化合物可给药的剂量为小于20mg/kg体重,优选为小于5mg/kg体重,于一些情形下为小于1mg/kg体重。
本发明也提供了调节优选为减少,细胞辣椒素受体的讯息传导活性(即钙离子传导)的方法。所述调节可通过在适于使所述调节剂与受体结合的条件下,使本发明提供的一或多种VR1调节剂的辣椒素受体调节量与辣椒素受体接触(在体外或者活体内)而达成。所述受体可存在于溶液或悬浮液中、经培养或分离的细胞制剂中或在患者的细胞中。所述细胞可为例如,在动物体内接触的神经元细胞。或者,所述细胞可为在动物活体内接触的上皮细胞,例如膀胱上皮细胞(泌尿上皮细胞)或气管上皮细胞。讯息传导活性的调节可通过检测对钙离子传导(亦称为钙离子移动或流动)的影响予以评估。讯息传导活性的调节亦可通过检测使用本发明提供的一或多种VR1调节剂治疗的患者症状(例如,疼痛、灼烧感、气管收缩、炎症、咳嗽、打嗝、痒、尿失禁或膀胱过动)的改变予以评估。
本发明提供的VR1调节剂,优选为通过口服或局部给药至患者(例如,人类),并且在调节VR1讯息传导活性的同时,存在于至少一种动物体液中。用于所述方法的优选的VR1调节剂调节体外VR1讯息传导活性的浓度为1纳摩尔浓度或更少,优选为100微微摩尔浓度或更少,更佳为20微微摩尔浓度或更少,于活体内体液中(例如血液中)的浓度是为1微摩尔浓度或更少,500纳摩尔浓度或更少,或100纳摩尔浓度或更少。
本发明进一步提供治疗对VR1调节敏感的症状的方法。在本发明中,“治疗”一词是涵盖疾病改善的治疗和症状的治疗,两者皆可为预防性的(即于症状发作前,为了预防、延缓或减少症状严重性)或治疗性的(即于症状发作后,为了降低症状的严重性和/或持续性)。于下述情况下该症状为“对VR1调节作用敏感”:无论局部存在的类香草醇配位体量的所少,若症状的特征为不适当的辣椒素受体活性,和/或若调节辣椒素受体活性造成其病症或症状的缓解。所述症状包括,例如,在下文详细描述的由于暴露在VR1活化刺激下产生的症状、疼痛、呼吸性疾病(例如气喘及慢性阻塞性肺疾病)、痒、尿失禁、膀胱过动、咳嗽、打嗝、及肥胖症。这些病症可使用此项技术中已建立的准则予以诊断及检测。患者可包括人类、驯养的陪伴动物及家畜,其剂量如上所述。
治疗方法可根据所用的化合物及要进行治疗的特定病症而定。然而,多数疾病的治疗优选采用一天4次或4次以下的给药频率。一般而言,更优选地采用一天2次的剂量疗法,尤其优选为一天给药一次。治疗急性疼痛时,要采用能迅速达到有效浓度的单一剂量。然而,可以理解,对于任何特定患者的特殊剂量标准及治疗方法取决于各项因素,包括所用特定化合物的活性、年龄、体重、一般健康情形、性别、饮食、给药时间、给药途径及排泄率、药物组合及进行治疗的特定疾病严重性。一般而言,优选使用足以提供有效治疗的最低剂量。使用适于所治疗或预防病症的医学或兽医学准则,通常可检测患者治疗的效果。
患有由于暴露在辣椒素受体活化刺激而产生的症状的患者包括经由热、光、催泪瓦斯或酸引起灼伤的个体,及其黏膜暴露(例如,经由摄取、吸入或眼睛接触)在辣椒素(例如,得自辣椒或辣椒喷雾剂)或相关刺激物(例如酸、催泪瓦斯或空气污染物)下的患者。所产生的症状(可使用本发明提供的VR1调节剂,尤其是拮抗剂治疗者)可包括,例如,疼痛、气管收缩及发炎。
使用本发明提供的VR1调节剂治疗的疼痛可为慢性或急性疼痛,包括,但不限于,由周边神经介导的疼痛(尤其是神经痛)。本发明提供的化合物可用于治疗,例如,乳房切除后疼痛症候群、残肢痛、幻觉肢体痛、口腔神经痛、牙痛(牙龈痛)、假牙痛、带状疹后神经痛、糖尿病神经病变、反射性交感神经失养症、三叉神经痛、骨关节炎、风湿性关节炎、纤维肌痛、格巴两氏症候群(Guillain-Barre Syndrome)、感觉异常性股痛、口腔灼热症候群和/或两侧性周边神经病变。其它神经痛症状包括灼热痛(反射性交感神经失养症-RSD,仅次于周边神经伤害)、神经炎(包括,例如,坐骨神经炎、周边神经炎、多神经炎、视神经炎、热病后神经炎、移动性神经炎、分节性神经炎及宫保氏神经炎(Gombault’s neuritis))、神经细胞炎、神经痛(例如,上文所述者、颈臂神经痛、颅部神经痛、膝状神经痛、舌咽神经痛、偏头性神经痛、自发性神经痛、肋间神经痛、乳房神经痛、下颔关节神经痛、摩顿氏神经痛(Morton’s neuralgia)、鼻睫神经痛、枕骨神经痛、红斑性肢痛症、史路德氏神经痛(Sluder’s neuralgia)、蝶腭神经痛、眶上神经痛及翼管神经痛)、与手术相关的疼痛、肌肉与骨骼疼痛、与AIDS相关的神经病变、与MS相关的神经病变、及与脊椎神经受伤相关的疼痛。头痛,包括涉及周边神经活性的头痛,例如窦性、丛发性(即偏头性神经痛)及一些压力性头痛与偏头痛,亦可如本发明所述予以治疗。例如,可在患者一感受到偏头痛前的预兆时,即给药本发明提供的化合物来预防偏头痛。其它疼痛症状可如本发明所述进行治疗,包括“口腔灼热症候群”、产痛、恰可氏疼痛症(Charcot’s pains)、肠气疼痛、经痛、急性与慢性背痛(例如,下背痛)、痔痛、消化不良痛、心绞痛、神经根疼痛、同位性疼痛及异位性疼痛-包括与癌症有关的疼痛(例如,骨癌患者)、与暴露在毒液(例如,由于被蛇咬、被蜘蛛咬、或虫叮)有关的疼痛(及炎症)、及与外伤有关的疼痛(例如,手术后疼痛、由伤口、瘀伤与骨折引起的疼痛、及灼伤痛)。其它的疼痛状症可如本发明所述予以治疗,包括与发炎性肠道疾病相关的疼痛、肠激躁症候群和/或发炎性肠道疾病。
于某些方面,本发明提供的VR1调节剂可用于治疗机械性疼痛。本发明所用的“机械性疼痛”一词是指头痛以外的不是神经性或暴露于热、冷或外在化学刺激下所导致的疼痛。机械性疼痛包括物理外伤(除了热或化学灼烧或有毒化学剂的其它刺激和/或疼痛暴露以外),例如手术后疼痛及由于伤口、瘀伤与骨折导致的疼痛;牙痛;假牙痛;神经根疼痛;骨关节炎;风湿性关节炎;肌纤维痛;感觉异常性股痛;背痛;与癌症相关的疼痛;心绞痛;腕隧道症候群;及由于骨折、生产、痔疮、肠气、消化不良、及月经产生的疼痛。
可治疗的痒症状包括牛皮癣搔痒、由于血液透析引起的痒、过水搔痒症、及与阴道前庭炎、接触性皮肤炎、虫咬及皮肤过敏相关的痒。可如本发明所述予以治疗的尿道症状包括尿失禁(包括满溢性尿失禁、急迫性尿失禁及压力性尿失禁)、以及膀胱过动或不稳定的膀胱症状(包括源自脊椎的迫尿肌过度反射及膀胱过敏症)。在某些该治疗方法中,VR1调节剂是通过导管或类似装置给药的,从而使VR1调节剂直接注射至膀胱中。本发明提供的化合物也可作为止咳剂用(以预防、缓和或压制咳嗽)、用于治疗打嗝、及促进肥胖患者的减重。
在其它方面,本发明提供的VR1调节剂可用于组合疗法以治疗涉及炎性成分的病症。所述病症包括,例如,已知具有炎性成分的自体免疫失调与病理性自体免疫反应,其包括,但不限于,关节炎(尤其是风湿性关节炎)、牛皮癣、克隆氏病症(Crohn’s disease)、红斑狼疮症、肠激躁症候群、组织移植排斥、及移植器官的超急性排斥。其它所述病症包括外伤(例如,对头或脊椎神经的伤害)、心血管与脑血管疾病及特定感染病症。
在所述组合疗法中,将VR1调节剂是与抗炎剂一起给药至患者。VR1调节剂与抗炎剂可存在于相同药学组合物中,或可以以任一顺序分开给药。抗炎剂包括,例如,非类固醇抗炎药物(NSAIDs)、非专一性及环氧化酶-2(COX-2)专一性环氧化酶抑制剂、金化合物、皮质类固醇类、胺甲喋呤、肿瘤坏死因子(TNF)受体拮抗剂、抗-TNFα抗体、抗-C5抗体、及介白素-1(IL-1)受体拮抗剂。NSAIDs的实施例包括,但不限于异丁苯丙酸(例如,ADVILTM、MOTRINTM)、氟联苯丙酸(flurbiprofen)(ANSAIDTM)、甲氧萘丙酸(naproxen)或甲氧萘丙酸钠(naproxen sodium)(例如,NAPROSYN、ANAPROX、ALEVETM)、双氯芬酸(diclofenac)(例如,CATAFLAMTM、VOLTARENTM)、双氯芬酸钠与米索前列醇(misoprostol)的组合(例如,ARTHROTETM)、舒林酸(sulindac)(CLINORILTM)、恶丙秦(oxaprozin)(DAYPROTM)、二氟苯水杨酸(DOLOBIDTM)、匹若西卡(piroxicam)(FELDENETM)、引朵美辛(indomethacin)(INDOCINTM)、伊托多雷(etodolac)(LODINETM)、菲诺洛芬钙(fenoprofen calcium)(NALFONTM)、酮布洛芬(ketoprofen)(例如,ORUDISTM、ORUVAILTM)、萘美丁酮钠(sodium nabumetone)(RELAFENTM)、硫氮磺胺吡啶(sulfasalazine)(AZULFIDINETM)、托美丁钠(tolmetin sodium)(TOLECTINTM)、与羟基氯喹咛(hydroxychloroquine)(PLAQUENILTM)。特定的NSAIDs类别是由抑制环氧化(COX)酶的化合物所构成,例如塞利昔布(celecoxib)(CELEBREXTM)与罗非昔布(rofecoxib)(VIOXXTM)。NSAIDs进一步包括水杨酸盐类例如乙酰基水杨酸或阿司匹灵、水杨酸钠、胆碱与水杨酸镁类(TRILISATETM)、与双水杨酯(salsalate)(DISALCIDTM)、以及皮质类固醇类例如可体松(cortisone)(CORTONETM乙酸盐)、地塞米松(dexamethasone)(例如,DECADRONTM)、甲基泼尼松龙(methylprednisolone)(MEDROLTM)、泼尼松龙(prednisolone)(PRELONETM)、泼尼松龙磷酸钠(prednisolonesodium phosphate)(PEDIAPREDTM)、与泼尼松(例如,PREDNICEN-MTM、DELTASONETM、STERAPREDTM)。
所述组合疗法中,VR1调节剂的适当剂量通常如上述。抗炎剂的给药剂量及方法在,例如,Physician’s Desk Reference中的厂商指示中进行描述。在某些具体实施例中,VR1调节剂与抗炎剂的组合给药导致需要产生治疗效果的抗炎剂剂量减少。因此,优选地,在本发明的组合或组合疗法中抗炎剂的剂量小于由厂商告知的未与VR1拮抗剂组合给药的抗炎剂的最大剂量。更优选地,此剂量小于由厂商告知的未与VR1拮抗剂组合给药的抗炎剂的最大剂量的3/4,又更优选为小于1/2,及非常优选为小于1/4,最优选为小于该最大剂量的10%。显而意见地,达到期望效果所需的组合中VR1拈抗剂成分的剂量同样受该组合抗炎剂成分的剂量与效力影响。
在某些优选具体实施例中,VR1调节剂与抗炎剂的组合给药是通过在相同包装盒中包装一或多种VR1调节剂与一或多种抗炎剂而实现的,其可分别包装在该包装盒的分别容器中,或将含有一或多种VR1调节剂与一或多种抗炎剂的混合物装在相同容器中。优选的混合物是调配成供口服给药形式(例如,呈丸剂、胶囊、锭剂等)。在某些具体实施例中,所述包装含印有指针的标签,说明所述一或多种VR1调节剂及一或多种抗炎剂是一起用于治疗炎性疼痛病症的。高度优选的组合为其中抗炎剂包括下述的至少一种:COX-2专一性环氧化酶抑制剂例如瓦第昔布(valdecoxib)(BEXTRA)、兰拉昔布(lumiracoxib)(PREXIGETM)、依托昔布(etoricoxib)(ARCOXIA)、塞利昔布(CELEBREX)和/或罗非昔布(VIOXX)。
在其它方面,本发明提供的VR1调节剂可用于组合一或多种附加的疼痛缓解药物。特定的疼痛缓解药物也为如上所述的抗炎剂。其它疼痛缓解药物为麻醉止痛剂,其典型地作用于一或多种类鸦片剂受体亚型(例如,μ、K和/或δ),优选为起促效剂或部分促效剂作用。所述制剂包括鸦片剂、鸦片剂衍生物及类鸦片剂,以及其药学上可接受的盐与水合物。在优选具体实施例中,麻醉止痛剂的具体实施例包括阿芬旦尼(alfentanyl)、阿法普鲁汀(alphaprodine)、安尼勒立汀(anileridine)、培集屈密特(bezitramide)、丁基原啡因(buprenorphine)、可待因(codeine)、二乙酰基二氢吗啡、二乙酰基吗啡、二氢可待因、氰苯哌酯(diphenoxylate)、乙基吗啡、芬太尼(fentanyl)、海洛英、氢可酮(hydrocodone)、氢吗啡酮(hydromorphone)、异美沙冬(isomethadone)、左旋甲基吗泛(levomethorphan)、羟甲左吗南(levorphane)、左旋码泛(levorphanol)、麦啶(meperidine)、美他唑新(metazocine)、美沙酮(methadone)、美索芬(methorphan)、美托酮(metopon)、吗啡、鸦片萃取物、鸦片流体萃取物、鸦片粉剂、鸦片粒剂、粗鸦片、鸦片酊、羟二氢可待因酮(oxycodone)、羟二氢吗啡酮(oxymorphone)、复方樟脑酊(paregoric)、潘他唑新(pentazocine)、配西汀(pethidine)、吩那唑新(phenazocine)、匹密诺汀(piminodine)、丙氧吩(propoxyphene)、消旋甲基吗泛(recemethorphan)、消旋吗泛(racemorphan)、蒂巴因(thebaine)及前述制剂药学上可接受的盐与水合物。
麻醉止痛剂的其它实施例包括乙酰托啡因(acetophine)、乙酰基二氢可待因、乙酰美沙多(acetylmethadol)、丙烯普鲁汀(allylprodine)、阿法乙酰美沙多(alphracetylmethadol)、阿法美普鲁汀(alphameprodine)、阿法美沙多(alphamethadol)、苯才西汀(benzethidine)、苄基吗啡、β乙酰美沙多(betacetylmethadol)、贝他美普鲁汀(betameprodine)、贝他美沙多(betamethadol)、贝他普鲁汀(betaprodine)、美妥芬诺(butorphonol)、克罗尼他净(clonitazene)、甲基溴可待因(codeine methylbromide)、N-氧化可待因(codeine-N-oxide)、赛普诺啡(cyprenorphine)、二氢脱氧吗啡(desomorphine)、右旋吗拉密特(dextromoramide)、狄安普鲁密特(diampromide)、二乙胺二噻吩丁烯(diethylthiambutene)、二氢吗啡、狄门诺沙多(dimenoxadol)、狄美菲坦诺(dimepheptanol)、二甲胺二噻吩丁烯(dimethylthiambutene)、吗苯丁酯(dioxaphetyl butyrate)、狄匹潘浓(dipipanone)、托蒂巴醇(drotebanol)、乙醇、甲乙胺二噻吩丁烯(ethylmethylthiambutene)、爱托失立汀(etonitazene)、羟戊甲吗啡(etorphine)、爱托失立汀(etoxeridine)、佛莱西汀(furethidine)、羟二氢吗啡(hydromorphinol)、羟基配西汀(hydroxyprthidine)、酚哌丙酮(ketobemidone)、左旋吗拉密特(levomoramide)、左旋吩纳西吗泛(levophenacylmorphan)、甲基脱氧吗啡(methyldesorphine)、甲基二氢吗啡(methyldihydromorphine)、吗啡里汀(morpheridine)、吗啡甲基溴化物(morphine methylpromide)、甲基磺胺吗啡(morphine methylsulfonate)、N-氧化吗啡(morphine-N-oxide)、密罗啡因(myrophin)、那诺松(naloxone)、那拜芬(nalbuyphine)、那提喝松(naltyhexone)、烟碱酰可待因(nicocodeine)、烟碱酰吗啡(nicomorphine)、去甲基乙酰美沙多(noracymethadol)、左旋原吗泛(norlevorphanol)、原美沙多(normethadone)、原吗啡(normorphine)、原匹潘浓(norpipanone)、戊唑凯因(pentazocaine)、芬那多松(phenadoxone)、吩喃普鲁密特(phenampromide)、吩诺吗泛(phenomorphan)、吩诺配立汀(phenoperidine)、匹立屈密特(piritramide)、福可汀(pholcodine)、普鲁庚唑英(proheptazoine)、普鲁配立汀(properidine)、普鲁匹兰(propiran)、外消旋吗密特(racemoramide)、蒂巴康(thebacon)、屈美配立汀(trimeperidine)及其药学上可接受的盐与水合物。
其它特定的代表性麻醉止痛剂,包括,例如:TALWIN Nx与DEMEROL(二者均得自Sanofi Winthrop Pharmaceuticals;New York,NY);LEVO-DROMORAN;BUPRENEX(Reckitt & ColemanPharmaceuticals,Inc.;Richmond,VA);MSIR(Purdue Pharma L.P.;Norwalk,CT);DILAUDID(Knoll Pharmaceutical Co.;Mount Olive,NJ);SUBLIMAZE;SUFENTA(Janssen Pharmaceutica Inc.;Titusville,NJ);PERCOCET、NUBAIN与NUMORPHAN(所有均得自EndoPharmaceuticals Inc.;Chadds Ford,PA)HYDROSTATIR、MS/S与MS/L(所有均得自Richwood Pharmaceutical Co.Inc;Florence,KY)、ORAMORPHSR与ROXICODONE(二者均得自RoxanneLaboratories;Columbus OH)及STADOL(Bristol-Myers Squibb;NewYork,NY)。另外的麻醉止痛剂包括CB-2受体促效剂,例如AM1241,及与α2δ次单元结合的化合物,例如Neurontin(Gabapentin)(加巴喷丁)与普瑞加巴林(pregabalin)。
在所述组合疗法中,VR1调节剂的适当剂量通常如上所述。其它疼痛缓解药物的给药剂量及方法可见,例如,Physician’s Desk Reference中的厂商说明书。在某些具体实施例中,将VR1调节剂与一或多种附加的疼痛缓解药物组合给药会使产生治疗效果所需的各治疗剂的剂量减少(例如,制剂中一或二者的剂量可能小于厂商告知或上列最大剂量的3/4,小于1/2,小于1/4,或小于该最大剂量的10%)。在某些优选具体实施例中,VR1调节剂与一或多种附加疼痛缓解药物的组合给药,如上所述,是通过在相同包装盒中包装一或多种VR1调节剂与一或多种附加的疼痛缓解药物而实现的。
VR1促效剂的调节剂可进一步用于,例如,群众控制(作为催泪瓦斯的代用品)、私人保护(例如,喷雾调配剂),或通过辣椒素受体去敏化作用作为治疗疼痛、痒、尿失禁或膀胱过动的药学剂用。通常,用于群众控制或私人保护的化合物是根据已知催泪瓦斯或辣椒喷雾剂技术调配及使用的。
在不同方面,本发明为本发明提供的化合物提供多种非药学上的体外及体内用途。例如,所述化合物可进行标记,以用作辣椒素受体(于例如细胞制剂或组织切片、其制剂或其片段的试样中)检测与定位的探针。所述化合物也可在受体活性试验中作为正对照组用、作为测定候选药剂与辣椒素受体结合能力的标准、或作为正子射出断层造影(PET)成像用或单光子射出计算机断层造影(SPECT)用的放射性追踪剂。所述方法可用于鉴定活体对象的辣椒素受体。举例而言,VR1调节剂可使用任何各种已知技术予以标记(例如,如本发明所述以例如氚的放射性核种进行放射性标记),及与试样一起培养适当的培育时间(例如,先进行结合时间进程试验予以决定)。培养后,去除未结合的化合物(例如,通过洗涤),并使用适用于所用标记的任何方法检测结合化合物(例如,进行放射性标记化合物的自动放射线显影或闪烁计数;可使用光谱分析法检测放光基团及荧光基团)。含有标记化合物及较大量(例如,10倍量)未标记化合物的相配试样可进行相同操作,以作为对照组。与对照组相比,较大量的可检测标记残留于测试试样中说明试样中存在辣椒素受体。该检测试验包括培养细胞或组织试样中的受体的自动放射线显影(受体图谱(receptor mapping))可如Kuhar于Current Protocols inPharmacology(1998)John Wiley & Sons,New York中第8.1.1至8.1.9章节所述进行。
本发明提供的调节剂亦可在各种已知的细胞分离方法中使用。例如,调节剂可连接在组织培养盘或其它支持体的内侧表面,作为固定用的亲和配位体,从而于体外分离辣椒素受体(例如,分离受体表达的细胞)。在一优选具体实施例中,使连接在荧光标记(例如荧光黄)的调节剂与细胞接触,然后通过荧光活化细胞分选仪(FACS,亦称流式细胞仪)进行分析(或分离)。
下述实施例是提供用于说明本发明而非用于局限。除非特别指出,否则所有试剂与溶剂均为标准商用级,不需要进一步纯化即可使用。通过运用例行修饰法可将起始物质进行各种变化,并使用其它步骤来产生本发明提供的其它化合物。
实施例
实施例1
代表性化合物的制备
该实施例是说明代表性经芳基取代的苯并[d]异噻唑-3-基胺类似物的制备。
A.[1,1-二酮基-6-(3-三氟甲基-吡啶-2-基)1H-λ6-苯并[D]异噻唑-3-基]-(4-三氟甲基-苯基)-胺
1.2-(4-甲基苯基)-3-(三氟甲基)吡啶

在氮气下的DME(10mL)中,将Pd(PPh3)4(0.09mmol)加到已除气的2-氯-3-(三氟甲基)-吡啶(2.26mmol),4-甲基-苯基硼酸(2.49mmol)及2M Na2CO3(5.65mmol)的混合物中。在80℃搅拌该混合物一夜,浓缩,以EtOAc萃取。以Na2SO4,干燥,在真空下浓缩,并用急骤管柱层析法(4∶1己烷/EtOAc)纯化,得到2-(4-甲基苯基)-3-(三氟甲基)-吡啶。
2.2-甲基-5-(3-三氟甲基-吡啶-2-基)-苯磺酰胺

小心地将2-(4-甲基苯基)-3-(三氟甲基)-吡啶(42mmol)加至冰水冷却的氯磺酸(211mmol)中。在60℃搅拌该混合物一小时。冷却至室温,将该混合物倒至冰水中(200mL),以EtOAc萃取,用Na2SO4,干燥,并在真空下浓缩。用DCM(200mL)溶解该残余物,在NH3气体中起泡30分钟,并于室温下搅拌该混合物1小时。浓缩,在EtOAc与水之间分层,用Na2SO4,干燥,在真空下浓缩。用EtOAc-己烷进行重结晶,获得2-甲基-5-(3-三氟甲基-吡啶-2-基)-苯磺酰胺。
3.1,1-二酮-6-(3-三氟甲基-吡啶-2-基)-1,2-二氢-1λ6-苯并[d]异噻唑-3-酮
将高锰酸钾(14.2mmol)加至2-甲基-5-(3-三氟甲基-吡啶-2-基)-苯磺酰胺(4.75mmol)的1N NaOH(5mL)溶液中。在80℃搅拌该混合物5小时,冷却至室温,过滤,酸化该滤液至pH3,并收集所得沉淀物就得到1,1-二酮基-6-(3-三氟甲烷-吡啶-2-基)-1,2-二氢-1λ6-苯并[d]异噻唑-3-酮。
4.3-氯-6-(3-三氟甲基-吡啶-2-基)-苯并[d]异噻唑1,1-二氧化物

于160℃加热1,1-二酮基-6-(3-三氟甲烷-吡啶-2-基)-1,2-二氢-1λ6-苯并[d]异噻唑-3-酮(1.5mmol)、POCl3(3mL)和PCl5(2.0mmol)的混合物16小时。以冰终止该反应,并以EtOAc萃取,浓缩就获得3-氯-6-(3-三氟甲基-吡啶-2-基)-苯并[异噻唑1,1-二氧化物。
5.[1,1-二酮基-6-(3-三氟甲基-吡啶-2-基)1H-λ6-苯并[d异噻唑-3-基]-(4-三氟甲基-苯基)-胺

B.[1,1-二酮基-6-(3-三氟甲基-苯基)1H-λ6-苯并[D]异噻唑-3-基]-(4-叔丁基-苯基)-胺
1.6-溴-3-氯苯并[d]异噻唑1,1-二氧化物

在160℃加热6-溴-1,1-二酮-1,2-二氢-1λ6-苯并[d]异噻唑-3-酮(Kwon等人的(1996)Arzneim.Forsch.46(10):966-971;1.5mmol)、POCl3(3mL)和PCl5(2.0mmol)的混合物16小时。以冰终止该反应,并以EtOAc萃取,浓缩就获得6-溴-3-氯-苯并[d]异噻唑1,1-二氧化物。
2.(6-溴-1,1-二氧化物-1H-λ6-苯并[d]异噻唑-3-基)-(4-叔丁基-苯基)-胺
在80℃加热6-溴-3-氯-苯并[d]异噻唑1,1-二氧化物(0.33mmol)和4-叔丁基-苯胺(0.33mmol)的吡啶溶液(4mL)16小时。浓缩并用色层分析法(EtOAc∶己烷/1∶1)纯化,得到(6-溴-1,1-二氧化物-1H-λ6-苯并[d]异噻唑-3-基)-(4-叔丁基-苯基)-胺。
3.[1,1-二酮基-6-(3三氟甲基-苯基)1H-λ6-苯并[d]异噻唑-3-基]-(4-叔丁基-苯基)-胺

在氮气下将Pd(PPh3)4(0.012mmol)加至已除气的(6-溴-1,1-二氧化物-1H-λ6-苯并[d]异噻唑-3-基)-(4-叔丁基-苯基)-胺(0.32mmol)、2-三氟甲基-苯基硼酸(0.40mmol)和2M Na2CO3(0.80mmol)的DME(5mL)溶液中。在80℃搅拌该混合物一夜,浓缩并以EtOAc萃取。用Na2SO4,干燥,在真空下浓缩,并用急骤管柱层析法(4∶1己烷/EtOAc)纯化,就得到[1,1-二酮基-6-(3三氟甲基-苯基)1H-λ6-苯并[d]异噻唑-3-基]-(4-叔丁基-苯基)-胺。
C.加成化合物
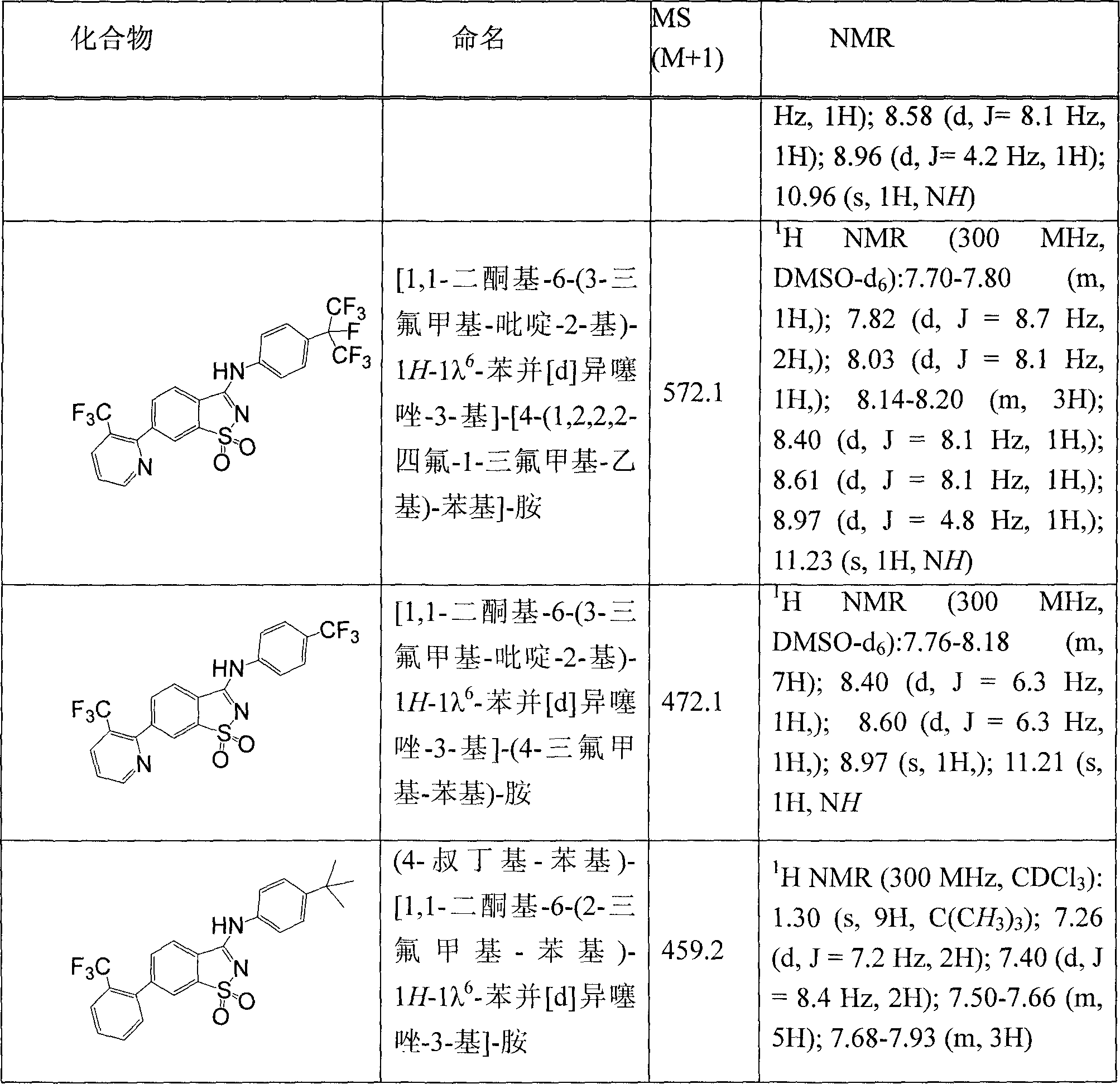
通过常规性的修饰可改变起始物质,并使用另外的步骤来制备本发明所提供的化合物。使用此方法制备了表I列举的化合物。如本发明所述测定的列于表I的化合物的IC501小于毫摩尔。
标示“MS”字段的质谱仪数据为电喷洒值谱(Electrospray MS),是使用Micromass Time-of-Flight LCT,以15V或30V锥电压于正离子模式中所获得的,该Micromass Time-of-Flight LCT装配有Waters 600泵、Waters 996光电二极管数组检测器、Gilson 215自动进样器和Gilson841显微注射仪。MassLynx(Advanced Chemistry Development,Inc;Toronto,Canada)4.0版软件用于数据收集与分析。注射1毫升体积试样于50×4.6mm Chromolith SpeedROD C18管柱,并以6毫升/分钟流速使用2-相线性梯度(2-phase linear gradient)来冲提。在220至340nm紫外光范围内使用全吸收计数(total absorbance count)来测定样品。冲提条件为:一移动相A-95/5/0.05水/甲醇/TFA;移动相B-5/95/0.025水/甲醇/TFA。
梯度:时间
(分钟)
%B
0 10
0.5 100
1.2 100
1.21 10
每次注射间的整个运作时间为2分钟。
Table I-代表性经芳基取代的苯并[d]异噻唑-3-基胺类似物
实施例2
VR1-转染细胞与细胞膜制剂
此实施例说明结合试验(实施例3)用的VR1-转染细胞与含VR1的细胞膜制剂的制备。
将编码人类辣椒素受体全长的cDNA序列(美国专利第6,482,611号的SEQ ID NO:1、2或3)次克隆至用于哺乳动物细胞中重组表现的质体pBK-CMV上(Stratagene,La Jolla,CA)。
使用标准方法将人类胚胎肾脏(HEK293)细胞将表达人类辣椒素受体全长序列的pBK-CMV进行转染。从中选出转染细胞,并放在含G418(400μg/ml)的培养液中两周,就得到一群能稳定转染的细胞。通过限制稀释(limitimg dilution)从该群细胞中分离出独立的无性繁殖系(independent clones),从而获得可稳定无性繁殖的细胞株,并用于下一个试验的使用。
在进行放射性配位体结合试验时,将细胞接种在T175细胞培养烧瓶内不含抗生素的培养液中,生长至约90%融合度(confluency)。再以PBS洗涤烧瓶,并在含5mM EDTA的PBS中收集细胞。通过温和离心使细胞集结成团,并保存在-80℃至分析为止。
用组织均质器将先前的冷冻细胞在冰冷HEPES均质缓冲液中分散(5mM KCl 5、5.8mM NaCl、0.75mM CaCl2、2mM MgCl2、320mM蔗糖与10mM HEPES pH 7.4)。首先将组织均质液在1000xg(4℃)下离心10分钟,以移除核部份及细胞碎片,然后将第一次离心的上清液在35,000xg(4℃)下再离心30分钟,得到部份纯化的膜部份。将膜再悬浮于HEPES均质缓冲液中,再进行分析。取一份膜均质液,利用Bradford方法(BIO-RAD蛋白质分析套组,#500-O001,BIO-RAD,Hercules,CA)测定蛋白质浓度。
实施例3
辣椒素受体结合试验
本实施例说明辣椒素受体结合的代表性试验,可用于测定化合物对辣椒素(VR1)受体的结合亲和性。
与[3H]树胶脂毒素(RTX)的结合试验基本上是依Szallasi与Blumberg(1992)J.Pharmacol.Exp.Ter.262:883-888所述的方法进行。在所述方法中,当结合反应结束后,非专一性的RTX结合会由于添加牛α1酸醣蛋白(每支试管100μg)而下降。
[3H]RTX(37 Ci/mmol)是由国家癌症研究所-费得利克癌症研究与发展中心的化学合成与分析实验室(the Chemical Synthesis and AnalysisLaboratory,National Cancer Institute-Frederick Cancer Research andDevelopment Center,Frederick,MD)合成所得到。[3H]RTX也可为市售购得(例如:Amersham Pharmacia Biotech,Inc.;Piscataway,NJ)。
将实施例2得到的膜均质液如上所述进行离心,并再悬浮在均质缓冲液中以使蛋白质浓度达到333μg/ml。在冰上制备结合试验混合物,该混合物包括[3H]RTX(比活性2200mCi/ml)、2μl非放射活性试验化合物、0.25mg/ml牛血清白蛋白(Cohn V部份)、与5×104至1×105VR1-转染细胞。将上述冰-冷HEPES均质缓冲液(pH 7.4)调整最终体积至500μl(用于竞争结合试验)或1,000μl(用于饱和结合试验)。非专一性结合的定义为在1μM非放射活性RTX(Alexis Corp.;San Diego,CA)的存在下发生的结合性。在进行饱和结合时,使用1比2的稀释,添加的[3H]RTX浓度范围为7至1,000pM。每条饱和结合曲线一般收集11个浓度点。
竞争结合试验是在60pM[3H]RTX及不同浓度的试验化合物存在下进行得。通过将分析混合物移至37℃水浴来使结合反应发生,在培育60分钟后,将试管置于冰上冷却以中止反应。通过在WALLA玻璃纤维滤纸(PERKIN-ELMER,Gaithersburg,MD)上过滤将结合在膜的RTX及任何与α1酸醣蛋白结合的RTX从游离态的分离出来。该玻璃纤维滤纸使用前用1.0%PEI(聚伸乙亚胺)浸泡2小时。将滤纸干燥一夜后,在添加WALLAC BETA SCINT闪烁液后,在WALLAC 1205BETA PLATE计数器上计数。
平衡结合参数的测定是通过代入变构性希尔公式(the allosteric Hillequation),以计算机程序FITP(Biosoft,Ferguson,MO)来辅助计算数据(说明于Szallasi等人的(1993)J.Pharmacol.Exp.Ther.266:678-683)。本发明所提供化合物在此分析法中对辣椒素受体的Ki值小于1μM、100nM、50nM、25nM、10nM或1nM。
实施例4
钙离子移动试验
本实施例说明用于评估试验化合物促效剂与拮抗剂活性的代表性钙离子移动试验。
将用表达质体转染得(如实施例2所述),并进而表达人类辣椒素受体的细胞接种在FALCON黑边、透明底板的96孔分析盘中(#3904,BECTON-DICKINSON,Franklin Lakes,NJ),生长至70至90%融合度。将96孔板中的培养液排空,并在各孔中加入FLUO-3AM钙敏感性染料(Molecular Probes,Eugene,OR)(染料溶液:1mgFLUO-3AM、440μLDMSO与440μl 20%普罗尼克酸(pluronic acid)的DMSO溶液,在克氏-林格氏(Krebs-Ringer)HEPES(KRH)缓冲液(25mM HEPES、5mM KCl、0.96mM NaH2PO4、1mM MgSO4、2mM CaCl2、5mM葡萄糖、1mM羧苯磺胺(probenecid),pH 7.4)中稀释为1∶250,)每孔有50μl稀释溶液)。以铝箔覆盖分析盘,并在37℃在含5%CO2环境下培育1至2小时。培育后,排空分析盘中的染料,并用KRH缓冲液洗涤细胞一次,再悬浮在KRH缓冲液中。
测定辣椒素EC50
为了测量试验化合物促效或拮抗表达辣椒素受体的细胞对辣椒素或其它类香草醇促效剂的钙离子移动反应的能力,先测定促效剂辣椒素的EC50。在各孔如上所述制备的细胞中添加20μl KRH缓冲液与1μlDMSO。通过FLIPR仪器将100μl辣椒素的KRH缓冲液自动加至各孔中。使用FLUOROSKAN ASCENT(Labsystems,Franklin,MA)或FLIPR(荧光成像的分析盘读取系统;Molecular Devices,Sunnyvale,CA)仪器监控辣椒素所诱发的钙离子移动。在促效剂施用后,在30至60秒间获得的数据被用来产生8个点的浓度反应曲线,此时辣椒素终浓度为1nM至3μM。使用KALEIDAGRAPH软件(Synergy Software,Reading,PA)将数据代入公式:
y=a*(1/(1+(b/x)c))
来测定反应的50%刺激浓度(EC-50)。此公式中,y为荧光讯号最大值,x为促效剂或拮抗剂浓度(此例中为辣椒素),a为Emax;b相当于EC50值,而c为希尔是数(Hill coefficient)。
促效剂活性的测定
将试验化合物溶于DMSO中,并用KRH缓冲液稀释,然后立刻加至如上所述制备的细胞中。100nM辣椒素(适当的EC-90浓度)也加至96孔分析盘的细胞中,做为阳性对照组。试验孔中的试验化合物最终浓度为0.1nM至5μM间。
通过测量表达辣椒素受体的细胞的荧光反应来测定该试验化合物作为辣椒素受体促效剂的能力,所述辣椒素受体是通过作为化合物浓度函数的化合物所引发的。该数据填入如上所述的公式中就获得EC50,其一般小于1微摩尔浓度,优选为小于100纳摩尔浓度,更优选为小于10纳摩尔浓度。各试验化合物的效力范围也通过计算所述反应来测定,所述反应由试验化合物浓度(一般为1μM)所引发,与100nM辣椒素所引发的反应相比。此值称为讯号百分比(Percent of Signal,POS),通过下式来计算:
POS=100*试验化合物反应/100nM辣椒素反应
此分析提供了对作为人类辣椒素受体促效剂的试验化合物的功效与效力这两者的定量评估。人类辣椒素受体通常在浓度小于100μM,或优选浓度小于1μM,或更优选为浓度小于10nM下引发可检测的反应。人类辣椒素受体在1μM浓度的效力范围优选为大于30POS,更优选为大于80POS。某些促效剂基本上不具拮抗剂活性,这一点是通过如下所述分析法中,在低于4nM的化合物浓度下没有可检测的促效剂活性所证实得,更优选为低于10μM的浓度,及最优选为低于或等于100μM的浓度。
拮抗剂活性的测定
将试验化合物溶于DMSO中,并稀释在20μl KRH缓冲液,以使分析孔中试验化合物最终浓度在1μM至5μM的间,并加入如上所述制备的细胞。将含有所制备细胞及试验化合物的96孔分析盘在室温下在暗室中培育0.5至6小时,注意不可连续培育超过6小时。在即将测定荧光反应前,通过FLIPR仪器自动添加100μl含辣椒素的KRH缓冲液(其浓度如上述测定所得为EC50浓度的2倍)至96孔分析盘的各孔中,使最终试样体积为200μl,并且最终辣椒素浓度等于EC50。在分析孔中的试验化合物的最终浓度为1μM至5μM间。相对于对应的对照组(即在缺乏试验化合物的情形下,以EC50浓度的2倍的辣椒素进行处理),辣椒素受体的拮抗剂使此反应下降至少约20%,优选为至少约50%,与最优选为至少80%。与观察存在辣椒素且无拮抗剂的反应相比,提供下降50%的拮抗剂所需浓度为所述拮抗剂的IC50,且优选低于1微摩尔浓度、100纳摩尔浓度、10纳摩尔浓度或1纳摩尔浓度。
得某些优选VR1调节剂为基本上无促效剂活性的拮抗剂,这一点是通过如上所述分析法中,在低于4nM的化合物浓度下没有可检测的促效剂活性所证实得,更优选为低于10μM的浓度,及最优选为低于或等于100μM的浓度。
实施例5
活体外微粒体的半衰期
此实施例说明采用代表性肝微粒体半衰期分析来评估化合物半衰期值(t1/2值)。
丛聚的人类肝微粒体是从XenoTech LLC(Kansas City,KS)所获得。该肝微粒体也通过自活体外技术(Baltimore,MD)或组织转形技术(Tissue Transformation Technologies)(Edison,NJ)获得。制备6个试验反应,分别含25μl微粒体、5μl的100μM试验化合物溶液,和399μl 0.1M磷酸盐缓冲液(19mL 0.1M NaH2PO4、81mL 0.1M Na2HPO4,以H3PO4调至pH7.4)。制备第7个反应作为阳性对照组,其中包含25μl微粒体、399μl 0.1M磷酸盐缓冲液与5μl的100μM已知代谢性质的化合物得溶液(例如:DIAZEPAM或CLOZAPINE)。反应在39℃下预培养10分钟。
辅因子混合物(CoFactor Mixture)的制法是将16.2mgNADP与45.4mg葡萄糖-6-磷酸盐(Gluose-6-phosphate)稀释于4mL的100mMMgCl2中。葡萄糖-6-磷酸盐脱氢酶(Gluose-6-phosphate dehydrogenase)溶液是取214.3μl葡萄糖-6-磷酸盐脱氢酶悬浮液(Roche MolecularBiochemicals,Indianapolis,IN)于1285.7μl蒸馏水中稀释制备而成的。将71μl起始反应混合物(3mL辅因子混合物;1.2mL葡萄糖-6-磷酸盐脱氢溶液)添加至6个试验反应中的5个以及阳性对照组中。将71μl的100mM MgCl2添加至第6个试验反应中作为阴性对照组。在各时间点(0、1、3、5、与10分钟),将75μl的各反应混合物滴加至96孔含有75μl冰冷乙腈的深孔分析盘的孔中。将样本涡转混合,在3500rpm下离心10分钟(Sorval T 6000D离心机,H1000B转子)。从每一个反应中取出75μl上清液移至96孔分析盘的孔中,96孔分析盘的每一个孔都含有150μl 0.5μM的具有已知其LCMS图形(内部标准)的化合物溶液。对各样本进行LCMS分析,以AUC测定未代谢试验化合物的含量,画出化合物浓度对时间的关系图,外插就得到试验化合物的t1/2值。
本发明提供的优选化合物在人类肝微粒体活体外具有大于10分钟至小于4小时的t1/2值,优选介于30分钟至1小时之间。
实施例6
MDCK毒性分析
此实施例说明使用Madin Darby犬肾脏(MDCK)细胞的细胞毒性分析评估化合物的毒性。
在透明底板的96孔分析盘(PACKARD,Meriden,CT)的各孔中添加1μl试验化合物,以使分析法中化合物终浓度为10微摩尔浓度、100微摩尔浓度或200微摩尔浓度。向对照组孔中则添加没有试验化合物的溶剂。
取MDCK细胞,ATCC no.CCL-34(美国菌种培养收集处(AmericanType Culture Collection,Manassas,VA)),依ATCC生产资料页的指示,维持在无菌条件下。取融合的MDCK细胞经胰蛋白酶处理,收集后,以温热(37℃)培养液(VITACELL伊格氏最低必需培养液(MinimumEssential Medium Eagle)、ATCC目录#30-2003)稀释至浓度为0.1×106个细胞/毫升。将100μL稀释的细胞加至各孔中,除了5个标准曲线对照组的分析孔中含有不具细胞的100μl温热培养液。再将该分析盘在37℃下,95%O2、5%CO2中,在恒定振荡培育2小时。培育后,在各孔中加入50μL哺乳动物细胞溶胞液,孔上加盖PACKARD TOPSEAL贴纸,将分析盘在约700rpm,在合适振荡器上振荡2分钟。
相与未处理的细胞相比,造成毒性的化合物将会降低ATP产生。ATP-LITE-M冷光ATP检测套组通常是依据制造商的指示使用,以测量已处理及未处理的MDCK细胞中的ATP产量。使PACKARD ATPLITE-M试剂平衡至室温。一旦平衡后,即取冻干的受质溶液在5.5mL受质缓冲液(来自套组)中再组成。冻干的ATP标准溶液于去离子水中再组成,得到10mM母液。关于5个对照组孔,则分别添加10μL连续稀释的PACKARD标准物至各标准曲线对照组孔中,使各连续孔的最终浓度为200nM、100nM、50nM、25nM与12.5nM。将PACKARD受质溶液(50μL)加到所有孔中,然后加盖,将分析盘在合适振荡器上以约700rpm振荡2分钟。将白色PACKARD贴纸黏在各分析盘底部,并用金属箔包裹分析板使试样于黑暗中10分钟。接着在22℃下使用冷光计数器(例如:PACKARD TOPCOUNT微分析板闪烁与冷光计数器或TECAN SPECTRAFLUOR PLUS)量测冷光,并由标准曲线计算ATP含量。比较未处理细胞与经试验化合物处理的细胞中的ATP含量。以10μM优选试验化合物处理的细胞中的ATP含量为未处理细胞的至少80%,优选为至少90%。当试验化合物使用100μM浓度时,以优选试验化合物处理的细胞中检测的ATP含量为未处理细胞的至少50%,优选为至少80%。
实施例7
背根神经节细胞分析法
此实施例说明用于评估化合物的VR1拮抗剂或促效剂活性的代表性背根神经节细胞分析。
DRG是从新生老鼠切下的,使用标准方法(Aguayo与White(1992)Brain Research 570:61-67)进行分离及培养。经过48小时培育后,洗涤细胞一次,并以钙敏感性染料Fluo-4AM(2.5-10μg/ml;TefLabs,Austin,TX)培育30至60分钟,然后再洗涤细胞一次。添加不同浓度化合物至细胞中。将辣椒素加到细胞中会导致细胞外钙离子含量随着VR1而增加,该钙离子含量是通过荧光计监测Fluo-4荧光的变化而检测的。收集60至180秒的数据,以决定荧光讯号的最大值。
在拈抗剂分析中,向细胞中添加各种浓度的化合物,然后以荧光讯号做为化合物浓度的函数画图,从而判别达到抑制50%辣椒素活化反应所需的浓度,或IC50。辣椒素受体拮抗剂优选的IC50为低于1微摩尔浓度、100纳摩尔浓度、10纳摩尔浓度或1纳摩尔浓度。
促效剂分析中,添加各种化合物浓度于细胞中,而不加辣椒素。辣椒素受体促效剂化合物导致细胞外钙离子含量随着VR1而增加,该钙离子含量是以荧光计监测Fluo-4荧光的变化。该EC50,或达到辣椒素活化反应的最大讯号50%所需的浓度,优选为低于1微摩尔浓度、低于100纳摩尔浓度或低于10纳摩尔浓度。
实施例8
用于测定疼痛缓解的动物模式
此实施例说明用于评估化合物所提供的疼痛缓解程度的代表性方法。
A.疼痛缓解试验
下列方法是用于评估疼痛缓解。
机械性异常疼痛
基本上评估机械性异常疼痛(对无害刺激产生的异常反应)是依Chaplan等人的(1994)J.Neurosci.Methods 53:55-63及Tal与Eliav的(1998)Pain 64(3):511至518所述。取一系列不同刚度的凡弗瑞(von Frey)丝线(典型为一系列8至14种丝线)施加在后脚足底表面上,其力量恰足使丝线弯曲。丝线保持此位置不超过3秒或直到大老鼠出现阳性异常疼痛反应为止。阳性异常疼痛反应包括举起处理的后脚,立即舔或摇动脚部。使用狄克森上下分析法(Dixon up-down method)决定各丝线的施加顺序与频率。以此系列中的中等丝线开始试验,随后依向上或向下顺序连续施用,分别依开始时所使用丝线是否出现阴性或阳性反应而定。
若接受此等化合物处理的大老鼠相较于未处理对照组或媒剂处理组大老鼠需要使用较高刚度的凡弗瑞(von Frey)丝线方可引起阳性异常疼痛反应时,表示该化合物可有效逆转或预防类似机械性异常疼痛的症状。或者,或此外,可在给药化合物之前及之后测试动物的慢性疼痛。此等分析法中,相较于处理前诱发反应时所需丝线或未经处理或经媒剂处理且亦具慢性疼痛的动物所需丝线,有效化合物可使处理后诱发反应所需丝线刚度提高。试验化合物是于疼痛发作之前或之后投药。当试验化合物在疼痛发作之后投药时,则在投药后10分钟至3小时进行试验。
机械性痛觉过敏
基本上评估机械性痛觉过敏(对疼痛刺激的反应过度)是依Koch等人的(1996)Analgesia 2(3):157至164所述。取大老鼠置于有温热多孔金属地板的个别笼内。在任一只后脚足底表面上温和针刺后,测定后脚抽回的时间期(亦即动物将其后脚放回地板上之前保持的时间)。
若化合物使后脚抽回的时间期缩短达统计显著性时,则该化合物可降低机械性痛觉过敏。试验化合物可于疼痛发作之前或之后投药。当试验化合物在疼痛发作之后投药时,则在投药后10分钟至3小时进行试验。
热痛觉过敏
基本上评估热痛觉过敏(对有害热刺激的反应过度)是依Hargreaves等人的(1988)Pain.32(1):77至88所述。简言的,在动物任一只后脚的足底表面施加恒定的辐射热源。抽回后脚的时间(亦即动物移动后脚之前的加热时间期),或称为热阈值或潜伏期,即可决定动物后脚对热的敏感性。
若化合物使后脚抽回的时间期增加达统计显著性时(亦即出现反应的热阈值或潜伏期加长),则该化合物可降低热痛觉过敏。试验化合物是于疼痛发作之前或之后投药。当试验化合物在疼痛发作之后投药时,则在投药后10分钟至3小时进行试验。
B.疼痛模式
可采用下述任一种方法诱发疼痛,以测定化合物的止痛效力。一般而言,采用雄性SD大老鼠及下述至少一种模式时,本发明所提供的化合物通过上述至少一种试验方法导致疼痛于统计上显著降低。
急性发炎疼痛模式
急性发炎疼痛基本上是依Field等人的(1997)Br.J.Pharmacol.121(8):1513-1522中的角叉菜胶模式所诱发。取100至200μl的1至2%角叉菜胶溶液注射大老鼠后脚中。注射后3至4小时,依上述方法测定动物对热及机械性刺激的敏感性。在试验前或注射角叉菜胶之前,对动物投与试验化合物(0.01至50mg/kg)。化合物可口服或任何非口服、或局部投药至脚部。在此模式中解除疼痛的化合物可使机械性异常疼痛与/或热痛觉过敏在统计上显著降低。
慢性发炎疼痛模式
慢性发炎疼痛是使用下述其中一种方法所诱发:
1.基本上是依Bertorelli等人的(1999)Br.J.Pharmacol.128(6):1252-1258,及Stein等人的(1998)Pharmacol.Biochem.Behav.31(2):455-51所述,取200μl完全弗洛伊德氏辅剂(Complete Freund′sAdjuvant)(0.1mg热杀死并干燥的结核菌(M.Tuberculosis))注射至大老鼠后脚中:100μl注入足背,100μl注入足底表面。
2.基本上是依Abbadie等人的(1994)J Neurosci.14(10):5865-5871所述,在大老鼠的胫骨-跗骨关节上注射150μlCFA(1.5mg)。
任一方法中,在即将注射CFA之前,先获得各试验动物后脚对机械及热刺激的个别敏感度底线
注射CFA后,依上述测试大老鼠的热痛觉过敏、机械性异常疼痛与机械性痛觉过敏。为了确使其发展出症状,在注射CFA后5、6与7天时才开始进行大老鼠试验。第7天时,以试验化合物、吗啡或媒剂处理动物。以口服剂量为1至5mg/kg的吗啡作为合适的阳性对照组。典型采用的试验化合物剂量为0.01至50mg/kg。化合物可在试验前呈单一剂量投药,或在试验前每天投药1、2或3次,进行数天。药物可口服或任何非口服、或局部投药给动物。
结果如百分比最大值可能效力(Percent Maximum PotentialEfficacy,MPE)表示。0%MPE的定义为媒剂的止痛效力,100%MPE的定义为动物恢复注射CFA前的底线敏感度。在此模式中解除疼痛的化合物所得到的MPE为至少30%。
慢性神经病变性疼痛模式
慢性神经病变性疼痛的诱发基本上是依Bennett与Xie的(1988)Pain 33:87-107所述,采用慢性收缩伤害(CCI)处理大老鼠坐骨神经。麻醉大老鼠(例如:经腹膜内使用剂量50至65mg/kg的戊巴比妥及依需要增加其它剂量)。将各后脚侧面刮干净及消毒。采用无菌技术,切开后脚侧面中股。将股二头肌切成钝端,曝露出坐骨神经。在每只动物的其中一只后脚上,依约1至2毫米的间隔,将四条结扎线松弛地结扎于坐骨神经周围。另一只脚的坐骨神经则没有结扎且不处理。随后盖上肌肉,使用伤口夹或缝合线缝合皮肤。依上述分析大老鼠的机械性异常疼痛、机械性痛觉过敏与热痛觉过敏。
当化合物在此模式中,在即将试验前呈单一剂量投药,或在试验前每天投药1、2或3次,进行数天(0.01至50mg/kg,口服、非口服或局部投药)时,该化合物可在统计上显著降低机械性异常疼痛、机械性痛觉过敏与/或热痛觉过敏。
Claims (53)
1.一种如下面通式的化合物或其药学上可接受的形式:
其中:
W、Y和Z独立地为N或CR1;
R1在每种情况下独立地选自氢、卤素、氰基、氨基、C1-C6烷基、卤代C1-C6烷基、C1-C6烷氧基或卤代C1-C6烷氧基;
Ar1和Ar2独立地选自5-至10-元芳族碳环与杂环,其中每一个经0至3个取代基取代,所述取代基独立地选自卤素、氰基、硝基或通式为LRa的基团;
L在每种情况下独立地选自单一共价键、O、C(=O)、OC(=O),C(=O)O、O-C(=O)O、S(O)m、N(Rx)、C(=O)N(Rx)-、N(Rx)C(=O)、N(Rx)S(O)m、S(O)mN(Rx)或N[S(O)mRx]S(O)m;其中m在每种情况下独立地选自0、1或2;而Rx在每种情况下独立地选自氢或C1-C8烷基;及Ra在每种情况下独立地选自:
(i)氢;及
(ii)C1-C8烷基、C2-C8烯基、C2-C8炔基、卤代C1-C8烷基、C2-C8烷基醚、单-与二-(C1-C8烷基)氨基、及(3-至10-元杂环)C0-C4烷基,其中每一个经0至6个取代基取代,所述取代基独立地选自(a)羟基、卤素、氨基、氨基羰基、氰基、硝基、酮基或COOH;及(b)C1-C8烷基、C1-C8烯基、C1-C8炔基、C1-C8烷氧基、C1-C8烷硫基、C1-C8烷基醚、C1-C8烷酰基、C1-C8烷酮、C1-C8烷酰基氧基、C1-C8烷氧基羰基、羟基C1-C8烷基、卤代C1-C8烷基、氰基C1-C8烷基、苯基C0-C8烷基、单-与二-(C1-C6烷基)氨基C0-C8烷基、C1-C8烷基磺酰基、C1-C8烷基磺酰氨基或(5-至7-元杂环)C0-C8烷基。
2.根据权利要求1所述的化合物或其药学上可接受的形式,其中,Ar2为苯基、吡啶基或嘧啶基,其中每一个经0至2个取代基取代,所述取代基独立地选自卤素、氰基、C1-C6烷基、卤代C1-C6烷基、C1-C6烷氧基、卤代C1-C6烷氧基、C1-C6烷基磺酰基或(C1-C6烷基磺酰氨基)。
3.根据权利要求2所述的化合物或其药学上可接受的形式,其中,Ar2为苯基或吡啶基,并经1或2个取代基取代,所述取代基独立地选自卤素、C1-C4烷基或卤代C1-C4烷基。
4.根据权利要求3所述的化合物或其药学上可接受的形式,其中,Ar2在环的连接点的对位具有一个取代基。
5.根据权利要求1所述的化合物或其药学上可接受的形式,其中,Ar1为苯基或吡啶基,经1或2个取代基取代,所述取代基独立地选自卤素、氰基、COOH、C1-C6烷基、卤代C1-C6烷基、C1-C6烷氧基、卤代C1-C6烷氧基、C1-C6烷基磺酰基或单-或二-(C1-C6烷基)磺酰氨基。
6.根据权利要求5所述的化合物或其药学上可接受的形式,其中,Ar1为2-吡啶基,经1或2个取代基取代,所述取代基独立地选自卤素、C1-C4烷基或卤代C1-C4烷基。
7.根据权利要求5所述的化合物或其药学上可接受的形式,其中,Ar1为3-甲基-吡啶-2-基、3-氯-吡啶-2-基或3-三氟甲基-啶啶-2-基。
8.如权利要求1所述的化合物或其药学上可接受的形式,其中,W为CH;Y与Z独立地为N或CH。
9.根据权利要求1所述的化合物或其药学上可接受的形式,其中,W、Y与Z各为CH。
10.根据权利要求1所述的化合物或其药学上可接受的形式,其中,Ar1为苯基或2-吡啶基,经1或2个取代基取代,所述取代基独立地选自卤素、氰基、C1-C4烷基或卤代C1-C4烷基;Ar2为苯基或吡啶基,经1或2个取代基取代,所述取代基独立地选自卤素、氰基、C1-C4烷基、卤代C1-C4烷基、C1-C6烷基磺酰基或单-或二-(C1-C6烷基)磺酰氨基。
11.根据权利要求1所述的化合物或其药学上可接受的形式,其中,所述化合物具有通式为:
其中A为N或CH,并且每一个Rb独立地为卤素、氰基、硝基或LRa。
12.根据权利要求1所述的化合物或其药学上可接受的形式,其中,所述化合物选自:
(4-叔丁基-苯基)-[1,1-二酮基-6-(3-三氟甲基-吡啶-2-基)-1H-1λ6-苯并[d]异噻唑-3-基]-胺;
[1,1-二酮基-6-(3-三氟甲基-吡啶-2-基)-1H-1λ6-苯并[d]异噻唑-3-基]-(4-异丙基-苯基)-胺;
[1,1-二酮基-6-(3-三氟甲基-吡啶-2-基)-1H-1λ6-苯并[d]异噻唑-3-基]-[4-(1,2,2,2-四氟-1-三氟甲基-乙基)-苯基]-胺;
[1,1-二酮基-6-(3-三氟甲基-吡啶-2-基)-1H-1λ6-苯并[d]异噻唑-3-基]-(4-三氟甲基-苯基)-胺;或
[1,1-二酮基-6-(3-三氟甲基-苯基)-1H-1λ6-苯并[d]异噻唑-3-基]-(4-叔丁基-苯基)-胺。
13.根据权利要求1所述的化合物或其药学上可接受的形式,其中,所述化合物在体外辣椒素受体促效作用试验中没有显示可检测的促效剂活性。
14.根据权利要求1所述的化合物或其药学上可接受的形式,其中,所述化合物在辣椒素受体钙离子移动试验中具有1微摩尔或更小的IC50值。
15.根据权利要求1所述的化合物或其药学上可接受的形式,其中,所述化合物在辣椒素受体钙离子移动试验中具有100纳摩尔或更小的IC50值。
16.一种药学组合物,所述组合物包括如权利要求1所述的至少一种化合物或其药学上可接受的形式,并与生理上可接受的载剂或赋形剂结合。
17.一种降低细胞辣椒素受体钙离子传导的方法,所述方法包括将如权利要求1所述的至少一种化合物或其药学上可接受的形式与表达辣椒素受体的细胞接触,从而降低辣椒素受体的钙离子传导。
18.根据权利要求17所述的方法,其中,所述细胞在动物体内接触。
19.根据权利要求18所述的方法,其中,所述细胞为神经细胞。
20.根据权利要求18所述的方法,其中,所述细胞为尿道上皮细胞。
21.权利要求18所述的方法,其中,在接触期间,所述化合物存在于动物的体液中。
22.根据权利要求18所述的方法,其中,所述化合物以1微摩尔或更小的浓度存在于动物血液中。
23.根据权利要求22所述的方法,其中,所述化合物是以500纳摩尔或更小的浓度存在于动物血液中。
24.根据权利要求23所述的方法,其中,所述化合物是以100纳摩尔或更小的浓度存在于动物血液中。
25.根据权利要求28所述的方法,其中,所述动物为人类。
26.根据权利要求2所述的方法,其中,所述化合物是以口服形式给药。
27.一种体外抑制类香草醇配位体与辣椒素受体结合的方法,所述方法包括在足以可检测地抑制类香草醇配位体与辣椒素受体结合的条件与用量下,将根据权利要求1所述的至少一种化合物或其药学上可接受的形式与辣椒素受体接触。
28.一种在患者体内抑制类香草醇配位体与辣椒素受体结合的方法,所述方法包括在足以可检测地抑制类香草醇配位体与表达克隆的辣椒素受体的细胞在体外结合的用量下,将根据权利要求1所述的至少一种化合物或其药学上可接受的形式与表达辣椒素受体的细胞接触,从而抑制病患体内类香草醇配位体与辣椒素受体的结合。
29.根据权利要求28所述的方法,其中,所述患者为人类。
30.根据权利要求28所述的方法,其中,所述化合物是以1微摩尔或更小的浓度存在于病患血液中。
31.一种治疗患者对辣椒素受体调节敏感的症状的方法,所述方法包括向患者给药辣椒素受体调节量的如权利要求1所述的至少一种化合物或其药学上可接受的形式,从而缓解患者症状。
32.根据权利要求31所述的方法,其中,所述患者患有(i)暴露于辣椒素,(ii)由于暴露在热下所引起的烧伤和刺激,(iii)由于暴露于光下所引起的烧伤和刺激,(iv)由于暴露在催泪瓦斯、空气污染物或辣椒喷雾剂下所引起的烧伤、支气管阻塞或刺激,或(v)由于暴露在酸下所引起的烧伤和刺激。
33.根据权利要求31所述的方法,其中,所述症状为气喘或慢性阻塞性肺疾病。
34.一种治疗病患疼痛的方法,所述方法包括向患者给药辣椒素受体调节量的如权利要求1所述的至少一种化合物或其药学上可接受的形式,从而缓解病患疼痛。
35.根据权利要求34所述的方法,其中,所述化合物是以1微摩尔或更小的浓度存在于病患血液中。
36.根据权利要求35所述的方法,其中,所述化合物是以500纳摩尔或更小的浓度存在于病患血液中。
37.根据权利要求36所述的方法,其中,所述化合物是以100纳摩尔或更小的浓度存在于病患血液中。
38.根据权利要求34所述的方法,其中,所述患者患有神经病变性疼痛。
39.根据权利要求34所述的方法,其中,所述疼痛与选自下列的病症相关:乳房切除后疼痛症候群、残肢痛、幻觉肢体痛、口腔神经痛、牙痛、带状疹后神经痛、糖尿病神经病变、反射性交感神经失养症、三叉神经痛、骨关节炎、风湿性关节炎、纤维肌痛、格巴两氏症候群、感觉异常性股痛、口腔灼热症候群、两侧性末稍神经病变、灼热痛、神经炎、神经细胞炎、神经痛、与AIDS相关的神经病变、与MS相关的神经病变、及与脊椎神经受伤相关的疼痛、与手术相关的疼痛、肌肉与骨骼疼痛、背痛、头痛、偏头痛、心绞痛、产痛、庤痛、消化不良痛、恰可氏疼痛症、肠气疼痛、经痛、癌症、毒液接触、炎性肠疾和/或外伤。
40.根据权利要求34所述的方法,其中,所述患者为人类。
41.一种治疗患者痒症的方法,所述方法包括向患者给药辣椒素受体调节量的如权利要求1所述的至少一种化合物或其药学上可接受的形式,从而缓解患者痒症。
42.一种治疗患者咳嗽或打嗝的方法,所述方法包括向患者给药辣椒素受体调节量的如权利要求1所述的至少一种化合物或其药学上可接受的形式,从而缓解患者咳嗽或打嗝。
43.一种治疗患者尿失禁或膀胱过动的方法,所述方法包括向患者给药辣椒素受体调节量的如权利要求1所述的至少一种化合物或其药学上可接受的形式,从而缓解患者的尿失禁或膀胱过动。
44.一种促进肥胖患者减重的方法,所述方法包括向患者给药辣椒素受体调节量的如权利要求1所述的至少一种化合物或其药学上可接受的形式,从而促进肥胖病患减重。
45.根据权利要求1所述的化合物或其药学上可接受的形式,其中,所述化合物或其药学上可接受的形式是经放射性标记的。
46.一种决定试样中是否有辣椒素受体存在的方法,所述方法包括下列步骤:
(a)在允许化合物与辣椒素受体结合的条件下将如权利要求1所述的化合物或其药学上可接受的形式与试样接触;及
(b)检测与辣椒素受体结合的所述化合物的量,进而测定试样中是否有辣椒素受体存在。
47.根据权利要求46所述的方法,其中,所述化合物为如权利要求45所述的放射性标记化合物,且其中所述检测步骤包括下列步骤:
(i)将未结合的化合物从结合的化合物中分离出来;及
(ii)检测试样中是否有结合的化合物存在。
48.一种经包装的药学制剂,所述制剂包括:
(a)装于容器中的如权利要求16所述的药学组合物;及
(b)使用所述组合物治疗疼痛的说明书。
49.一种经包装的药学制剂,所述制剂包括:
(a)装于容器中的如权利要求16所述的药学组合物;及
(b)使用所述组合物治疗咳嗽或打嗝的说明书。
50.一种经包装的药学制剂,所述制剂包括:
(a)装于容器中的如权利要求16所述的药学组合物;及
(b)使用所述组合物治疗肥胖的说明书。
51.一种经包装的药学制剂,包括:
(a)装于容器中的如权利要求16所述的药学组合物;及
(b)使用所述组合物治疗尿失禁或膀胱过动的说明书。
52.一种根据权利要求1至15中任一权利要求所述的化合物或其药学上可接受形式制造用于治疗对辣椒素受体调节敏感的病症的药品的用途。
53.根据权利要求52所述的用途,其中,所述病症为疼痛、气喘、慢性阻塞性肺疾病、咳嗽、打嗝、肥胖、尿失禁、膀胱过动、暴露于辣椒素下、暴露于热下所引起的烧伤或刺激、暴露于光下所引起的烧伤或刺激、暴露于催泪瓦斯、空气污染或辣椒喷剂下所引起的烧伤、支气管阻塞或刺激,或暴露于酸下所引起的烧伤或刺激。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US48595803P | 2003-07-10 | 2003-07-10 | |
| US60/485,958 | 2003-07-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1820007A true CN1820007A (zh) | 2006-08-16 |
Family
ID=34102670
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2004800197264A Pending CN1820007A (zh) | 2003-07-10 | 2004-07-09 | 经芳基取代的苯并[d]异噻唑-3-基胺类似物 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20080125466A1 (zh) |
| EP (1) | EP1648892B1 (zh) |
| JP (1) | JP2007537978A (zh) |
| CN (1) | CN1820007A (zh) |
| AT (1) | ATE365167T1 (zh) |
| AU (1) | AU2004259676A1 (zh) |
| CA (1) | CA2531515A1 (zh) |
| DE (1) | DE602004007141T2 (zh) |
| ES (1) | ES2285525T3 (zh) |
| WO (1) | WO2005009982A2 (zh) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7615570B2 (en) * | 2004-12-13 | 2009-11-10 | Abbott Laboratories | Antagonists to the vanilloid receptor subtype 1 (VR1) and uses thereof |
| EP2155703A1 (en) * | 2007-05-18 | 2010-02-24 | Inhibox Ltd. | Bicyclosulfonyl acid (bcsa) compounds and their use as therapeutic agents |
| DE102022104759A1 (de) | 2022-02-28 | 2023-08-31 | SCi Kontor GmbH | Co-Kristall-Screening Verfahren, insbesondere zur Herstellung von Co-Kristallen |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5294612A (en) * | 1992-03-30 | 1994-03-15 | Sterling Winthrop Inc. | 6-heterocyclyl pyrazolo [3,4-d]pyrimidin-4-ones and compositions and method of use thereof |
| US6187777B1 (en) * | 1998-02-06 | 2001-02-13 | Amgen Inc. | Compounds and methods which modulate feeding behavior and related diseases |
| AU2001280229B2 (en) * | 2000-08-21 | 2006-12-07 | Pacific Corporation | Novel thiourea derivatives and the pharmaceutical compositions containing the same |
| WO2003062209A2 (en) * | 2002-01-17 | 2003-07-31 | Neurogen Corporation | Substituted quinazolin-4-ylamine analogues as modulators of capsaicin |
| EP1569925A1 (en) * | 2002-12-13 | 2005-09-07 | Neurogen Corporation | 2-substituted quinazolin-4-ylamine analogues as capsaicin receptor modulators |
| US20070197559A1 (en) * | 2004-03-02 | 2007-08-23 | Rajagopal Bakthavatchalam | Aryl substituted purine analogues |
| CA2557852A1 (en) * | 2004-04-08 | 2005-10-27 | Neurogen Corporation | Substituted cinnolin-4-ylamines |
| TW200621251A (en) * | 2004-10-12 | 2006-07-01 | Neurogen Corp | Substituted biaryl quinolin-4-ylamine analogues |
-
2004
- 2004-07-09 AU AU2004259676A patent/AU2004259676A1/en not_active Abandoned
- 2004-07-09 JP JP2006518907A patent/JP2007537978A/ja not_active Withdrawn
- 2004-07-09 EP EP04786051A patent/EP1648892B1/en active Active
- 2004-07-09 CN CNA2004800197264A patent/CN1820007A/zh active Pending
- 2004-07-09 DE DE602004007141T patent/DE602004007141T2/de not_active Expired - Fee Related
- 2004-07-09 CA CA002531515A patent/CA2531515A1/en not_active Abandoned
- 2004-07-09 ES ES04786051T patent/ES2285525T3/es active Active
- 2004-07-09 US US10/564,263 patent/US20080125466A1/en not_active Abandoned
- 2004-07-09 AT AT04786051T patent/ATE365167T1/de not_active IP Right Cessation
- 2004-07-09 WO PCT/US2004/021914 patent/WO2005009982A2/en active IP Right Grant
Also Published As
| Publication number | Publication date |
|---|---|
| ES2285525T3 (es) | 2007-11-16 |
| EP1648892A2 (en) | 2006-04-26 |
| US20080125466A1 (en) | 2008-05-29 |
| WO2005009982A2 (en) | 2005-02-03 |
| WO2005009982A3 (en) | 2005-05-12 |
| CA2531515A1 (en) | 2005-02-03 |
| EP1648892B1 (en) | 2007-06-20 |
| AU2004259676A1 (en) | 2005-02-03 |
| JP2007537978A (ja) | 2007-12-27 |
| DE602004007141T2 (de) | 2007-10-11 |
| DE602004007141D1 (de) | 2007-08-02 |
| ATE365167T1 (de) | 2007-07-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1820001A (zh) | 经取代的杂环二芳基胺类似物 | |
| CN1826328A (zh) | 经取代的吡啶-2-基胺类似物 | |
| CN101014587B (zh) | 经取代的二芳基哌嗪基-吡啶类似物 | |
| CN1890223A (zh) | 辣椒素受体促效剂 | |
| CN101107245A (zh) | 经杂芳基取代的哌嗪-吡啶类似物 | |
| CN101103028A (zh) | 经杂芳基取代的喹啉-4-基氨类似物 | |
| CN1950332A (zh) | 经杂烷基取代的联苯-4-羧酸芳基醯胺类似物 | |
| US20040156869A1 (en) | 2-substituted quinazolin-4-ylamine analogues | |
| CN1964717A (zh) | 经取代∴啉-4-基胺 | |
| CN1823048A (zh) | 作为类香草醇受体配位体的经取代的嘧啶-4-基胺类似物 | |
| CN1349533A (zh) | 杂芳基二氮双环烷烃及其制备方法和应用 | |
| CN1997644A (zh) | 被芳基取代的嘌呤类似物 | |
| CN1656094A (zh) | 作为c-Jun N-末端激酶抑制剂用于治疗神经变性疾病的7-氮杂吲哚类化合物 | |
| US20080175794A1 (en) | Substituted Pyridazinyl- and Pyrimidinyl-Quinolin-4-Ylamine Analogues | |
| CN1950081A (zh) | 芳烷氨基取代的喹唑啉类似物 | |
| JP5421108B2 (ja) | 2−フェノキシピリミジノン類縁体 | |
| CN1820008A (zh) | 经取代的喹啉-4-基胺类似物 | |
| CN1440403A (zh) | 喹唑啉二甲苯磺酸盐化合物 | |
| CN101563349A (zh) | 经卤烷基取代的嘧啶酮衍生物 | |
| CN101080228A (zh) | 哌嗪基-吡啶类似物 | |
| CN101080401A (zh) | 经取代的二芳基类似物 | |
| CN101558069A (zh) | 顺式环己基取代的嘧啶酮衍生物 | |
| CN101068787A (zh) | 8-苯基-5,6,7,8-氢喹啉速激肽受体拮抗剂 | |
| CN1953979A (zh) | 经取代的5,12-二吖-苯幷葱类似物 | |
| CN1820007A (zh) | 经芳基取代的苯并[d]异噻唑-3-基胺类似物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Open date: 20060816 |