CN116120393A - 一种氧化型谷胱甘肽及其晶型和杂质的制备方法 - Google Patents
一种氧化型谷胱甘肽及其晶型和杂质的制备方法 Download PDFInfo
- Publication number
- CN116120393A CN116120393A CN202211640246.6A CN202211640246A CN116120393A CN 116120393 A CN116120393 A CN 116120393A CN 202211640246 A CN202211640246 A CN 202211640246A CN 116120393 A CN116120393 A CN 116120393A
- Authority
- CN
- China
- Prior art keywords
- oxidized glutathione
- glutathione
- hours
- crystal
- water
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/02—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link
- C07K5/0215—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link containing natural amino acids, forming a peptide bond via their side chain functional group, e.g. epsilon-Lys, gamma-Glu
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/02—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length in solution
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/30—Extraction; Separation; Purification by precipitation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/34—Extraction; Separation; Purification by filtration, ultrafiltration or reverse osmosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/36—Extraction; Separation; Purification by a combination of two or more processes of different types
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/02—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/02—Linear peptides containing at least one abnormal peptide link
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Medicinal Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Analytical Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Water Supply & Treatment (AREA)
- Peptides Or Proteins (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
本发明提供一种制备氧化型谷胱甘肽及其新晶型和杂质的方法,包括以下步骤:利用二甲亚砜(DMSO)作为氧化剂将还原型谷胱甘肽氧化成氧化型谷胱甘肽粗品;在纯化水中重结晶精制得到高纯度的氧化型谷胱甘肽七水合物晶体。
Description
发明领域
本发明属于化学合成领域,具体涉及氧化型谷胱甘肽及其七水合物晶型和杂质的制备方法。
背景技术
氧化型谷胱甘肽与还原型谷胱甘肽的作用类似,但氧化型谷胱甘肽的稳定性优于还原型谷胱甘肽,所以可替代还原型谷胱甘肽,用作保健食品、医药、化妆品等制品的有效成分。
目前已报道的氧化型谷胱甘肽制备方法全是由还原型谷胱甘肽氧化而成,例如:
路线一:过氧化氢氧化法(中国医药工业杂志,2013,44,265)。该方法是将还原型谷胱甘肽溶于水中,调节至合适的pH值后,以过氧化氢作为氧化剂对谷胱甘肽进行氧化,从而制得氧化型谷胱甘肽。该方法的缺点在于:过氧化氢氧化反应的速度虽然相对较快,但反应也较为激烈,所以反应条件比较苛刻,需要严格地控制反应体系的温度、pH、过氧化氢的用量等工艺参数,否则会出现较多的过氧化杂质或者降解杂质,影响产品纯度和收率。另外,氧化剂过氧化氢在我国属于易制爆危险化学品,使用须受到管控。此外,过氧化氢还容易发生自降解,需要密闭低温保存。
路线二:精氨酸催化法(RSC Adv.,2014,4,33399–33407)。该方法将还原型谷胱甘肽溶于水中,以氧气作为氧化剂,精氨酸为催化剂,反应无废料产生,绿色环保,而且氧气和精氨酸比较容易获得,原料获取比较方便。该方法的缺点在于:反应过程中需要使用精氨酸作为催化剂,后处理过程中容易有残留的精氨酸;而且反应需要加热到50℃,产物氧化型谷胱甘肽在高温条件下容易发生部分降解和消旋化,导致收率降低并且纯度下降。
路线三:酶催化法(日本特开平5-146279号公报)。该方法属于生物酶催化法,需要使用特定的生物氧化酶做催化剂,利用空气将还原型谷胱甘肽水溶液氧化,得到氧化型谷胱甘肽。该方法的缺点在于:酶的获取和保存比较困难,不如常用的化学试剂。而且反应结束后还要将酶从反应液中分离出来,需要特定的工艺技术和设备,一般化工原料药企业的现有设备可能不适用。
路线四:溴代丙二酸二乙酯法(Chem.Pharm.Bull.1986,34,486-495)。该方法将还原型谷胱甘肽溶于碱性的水/乙醇溶液,然后将溴代丙二酸二乙酯的乙醇溶液滴加进去,在-16℃反应1个小时。该方法的缺点在于:氧化剂使用溴代丙二酸二乙酯,成本相对较高,而且溴代丙二酸二乙酯生成的副产物较多,操作复杂。反应过程需要-16℃的低温,相对不适宜工业化。
此外,通过上述方法制备的氧化型谷胱甘肽的晶型主要包括非结晶无定型体(CN102858792A)、一水合物晶体(日本特许第4401775号公报)、六水合物晶体(CN102869674A)和八水合物晶体(International Union of Crystallography,pp538,1999)。其中非结晶无定型体水溶性较差限制了其在医药行业的应用;一水合物晶体由于其为针状晶体且易结块,所以晶体分离能力差,而且其中杂质难去除;六水合物晶体在结晶过程中需要调节pH,且析晶时间长达十几个小时,操作较繁琐,得到困难;八水合物晶体的水含量不均匀、稳定性差且需要长达3-4天的长时间来获得晶体。而且,氧化型谷胱甘肽现有的工艺都没有实现大规模的工业化生产,主要还是在于反应的温和性(减少产物降解与消旋)与经济性难以同时实现,需要一个既温和又经济易得而且副产物少的氧化体系和结晶体系,以实现优异的工艺稳定性。
此外,经过液相色谱的检测以及质谱的结构确证,氧化型谷胱甘肽中会含有以下三种杂质:


有关这三种杂质的制备方法报道较少,且所用原料、试剂无法在市场上购得,缺少实用价值。
关于氧化型谷胱甘肽的精制过程没有文献报道,多肽类化合物因为易水解、易消旋、容易生物降解等原因,常用的纯化精制方法为使用离子交换树脂色谱分离或者使用制备液相分离等方法。以上方法的溶剂损耗较高,生产成本较大。
综上,本领域对于具有成本低、反应条件温和、产物纯度高、适用于工业化生产的氧化型谷胱甘肽的合成方法存在需求。
发明内容
相对于现有方法,本申请提供的氧化型谷胱甘肽的合成方法能够解决以上问题。
具体地,本发明涉及以下技术方案:
1.式(I)化合物的七水合物晶体:

2.技术方案1的晶体,其特征在于:在使用CuKα辐射得到的X射线粉末衍射图中,至少包括位于以下°2θ表示的特征峰:8.238±0.2、16.338±0.2和24.551±0.2。
3.技术方案2的晶体,其特征在于:在使用CuKα辐射得到的X射线粉末衍射图中,还至少包括位于以下°2θ表示的特征峰:10.619±0.2、19.539±0.2、26.806±0.2和34.618±0.2。
4.技术方案3的晶体,其特征在于:在使用CuKα辐射得到的X射线粉末衍射图中,还至少包括位于以下°2θ表示的特征峰:9.750±0.2、22.738±0.2和23.200±0.2。
5.技术方案1的晶体,其特征在于:其具有基本上如图5所示的X射线粉末衍射图。
6.技术方案1-5中任一项的晶体,其特征还在于:其具有169±2℃的熔点。
7.技术方案1-6中任一项的晶体,其特征还在于:在热解重量分析中,其在50-100℃失重约14±1%,在100-160℃失重约3±1%。
8.技术方案1-7中任一项的晶体,其特征还在于:其热解重量图如图6所示。
9.一种制备式(I)化合物的方法,

包括将式(II)化合物用DMSO处理,氧化成为式(I)化合物。
10.技术方案9的方法,其中DMSO与式(II)化合物的摩尔比为2:1至25:1,优选2.5:1至5:1,更优选2:1、2.5:1、3:1、3.5:1、4:1、4.5:1或5:1。
11.技术方案9或10的方法,其中额外向反应中加入极性溶剂,促进式(II)化合物溶解;优选地,所述极性溶剂选自水、甲酰胺、三氟乙酸、乙腈、DMF、六甲基磷酰胺、甲醇、乙醇、乙酸、异丙醇、吡啶、四甲基乙二胺、丙酮、三乙胺、正丁醇、二氧六环、四氢呋喃、甲酸甲酯、三丁胺、甲乙酮、乙酸乙酯、氯仿、三辛胺、碳酸二甲酯和乙醚,或其化合物,更优选水。
12.技术方案11的方法,其中所述极性溶剂与式(II)化合物的比例为每100g式(II)化合物添加200至2000mL极性溶剂,优选地250mL至1000mL,更优选250mL、300mL、500mL、750mL或1000mL。
13.技术方案9-12中任一项的方法,其中将反应的pH调节至2至9,优选3至7,更优选5至7,例如5、5.5、6、6.5、7、8或9。
14.技术方案9-13中任一项的方法,其中将反应在-10℃至60℃进行,优选5℃至30℃,例如25℃、40℃或室温。
15.技术方案9-14中任一项的方法,其中所述反应进行5至60h,优选地10至48h,例如10h、15h、20h、25h、30h、35h、40h或45h。
16.技术方案9-15中任一项的方法,其中在反应结束后,将其pH调节至氧化型谷胱甘肽的等电点(pH 2.75至2.90),搅拌析晶至少5h,优选地至少10h。
17.技术方案16的方法,其中所述搅拌析晶在5℃至40℃,优选5℃、10℃、15℃、20℃或室温进行。
18.技术方案9-17中任一项的方法,其中所得式(I)化合物的纯度≧98%,优选地≧98.5%,优选地≧99%,优选地≧99.7%,优选地≧99.8%,优选地≧99.9%;并且其中产物中杂质A、杂质B和杂质C的总含量低于1%,优选低于0.5%,优选低于0.3%,优选低于0.1%,

19.一种精制技术方案9-18中任一项的氧化型谷胱甘肽的方法,包括:
1)将氧化型谷胱甘肽的粗品溶于纯化水中,纯化水用量为氧化型谷胱甘肽粗品质量的3~5倍;
2)溶解后趁热过滤;
3)滤液降温至10~25℃,优选12~20℃析晶。
20.技术方案19的方法,其中在步骤1)中,纯化水的温度为35℃~55℃,优选为40℃~50℃;并且任选地加入氯化亚铁溶液以更好的去除残留的原料,优选地,每公斤氧化型谷胱甘肽加入100mL 5%氯化亚铁溶液。
21.技术方案19或20的方法,其中在步骤3)中,重结晶的析晶时间为3~10小时,优选4~6小时;优选地,采用梯度降温,降温速率为每小时10℃。
22.一种制备技术方案1-8中任一项的氧化型谷胱甘肽七水合物晶体的方法,包括将技术方案9-21中任一项制备的氧化型谷胱甘肽在纯化水中进行重结晶;优选包括以下步骤:
1)将氧化型谷胱甘肽溶于纯化水中,搅拌至溶解;
2)溶解后趁热过滤,然后梯度降温至目标温度;
3)控温析晶。
23.技术方案22的方法,其中在步骤1)中,每公斤氧化型谷胱甘肽使用2L至6L的纯化水,优选3L至6L、更优选3L至5L、最优选4L的纯化水。
24.技术方案22或23的方法,其中在步骤1)中,溶解过程中纯化水的温度为40-60℃,优选40-50℃,更优选50℃。
25.权利要求22-24中任一项的方法,其中在步骤1)中加入氯化亚铁溶液以更好的去除残留的原料,优选地,每公斤氧化型谷胱甘肽加入100mL5%氯化亚铁溶液。
26.技术方案22-25中任一项的方法,其中在步骤2)中,降温梯度为5-25℃/h,优选5-20℃/h,优选5-15℃/h,更优选10℃/h。
27.技术方案22-26中任一项的方法,其中步骤2)中的目标温度为5-25℃,优选10-25℃,更优选15-25℃。
28.技术方案22-27中任一项的方法,其中步骤3)中的温度控制在5-25℃,优选10-25℃,更优选15-25℃。
29.技术方案22-28中任一项的方法,其中步骤3)中的析晶时间为6-12小时,优选6-8小时。
30.化合物,其选自:


31.一种制备技术方案30中的杂质A的方法,

包括将摩尔比1:1的还原型谷胱甘肽和Cys-Gly用DMSO氧化。
32.技术方案31的方法,其中其中DMSO的用量为2~10当量,优选3~5当量。
33.技术方案31或32的方法,包括以下步骤:
1)将摩尔比范围为1:1~1:3的Boc-Cys(Trt)-OH和甘氨酸叔丁酯在1.5~4当量的缩合剂(如HATU、HBTU、PyBOP、DEPBT等,优选HBTU和DEPBT)和2当量的有机叔胺(如N,N-二异丙基乙胺和三乙胺,优选N,N-二异丙基乙胺)催化下进行缩合反应,其中所述反应在DMF或二氯甲烷中进行;
2)用三氟乙酸脱去Trt、Boc和tBu保护基,得到Cys-Gly;
3)将摩尔比1:1的还原型谷胱甘肽和Cys-Gly用3~5当量的DMSO氧化。
34.一种制备技术方案30中的杂质B的方法,

包括将摩尔比1:1的还原型谷胱甘肽和Glu-Cys用DMSO氧化。
35.技术方案34的方法,其中DMSO的用量为2~10当量,优选3~5当量。
36.技术方案34或35的方法,包括以下步骤:
1)将摩尔比范围是1:1~1:2的Boc-Glu-OtBu和H-Cys(Trt)-OtBu在1.5~4当量的缩合剂(如HATU、HBTU、PyBOP、DEPBT等,优选HBTU和DEPBT)和2当量的有机叔胺(如N,N-二异丙基乙胺和三乙胺,优选N,N-二异丙基乙胺)催化下进行缩合反应;
2)用三氟乙酸脱去Boc、Trt和tBu保护基,得到Glu-Cys;
3)将摩尔比1:1的还原型谷胱甘肽和Glu-Cys用3~5当量的DMSO氧化。
37.一种制备技术方案30中的杂质C的方法,包括将摩尔比1:1的还原型谷胱甘肽和半胱氨酸用DMSO氧化。
38.技术方案37的方法,其中DMSO的用量为2~10当量,优选3~5当量。
本发明的有益效果:
该方法得到的氧化型谷胱甘肽粗品中不含有过氧化杂质,水解杂质含量更低,且没有其他固体副产物生成;对其他杂质的结构给予分析和合成后,对后续氧化型谷胱甘肽的质量控制提供了依据和保证。
本发明偶然发现,在氧化型谷胱甘肽的精制过程中,加入少量氯化亚铁水溶液,意想不到的可以更好的提高纯化效果,增加产物纯度;而且采用一定的条件在纯化水中进行重结晶,可以得到氧化型谷胱甘肽七水合物晶体。
附图说明
图1为氧化型谷胱甘肽的ESI-MS图;
图2为氧化型谷胱甘肽杂质A的ESI-MS图;
图3为氧化型谷胱甘肽杂质B的ESI-MS图;
图4为氧化型谷胱甘肽杂质C的ESI-MS图;
图5为氧化型谷胱甘肽七水合物晶体的X射线粉末衍射图;
图6为氧化型谷胱甘肽七水合物晶体的TG和DSC图。
具体实施方式
定义
“极性溶剂”是指含有羟基、羰基、羧基等极性基团的溶剂,即溶剂分子为极性分子的溶剂,由于其分子内正负电荷重心不重合而导致分子产生极性。极性溶剂选自水、甲酰胺、三氟乙酸、乙腈、DMF、六甲基磷酰胺、甲醇、乙醇、乙酸、异丙醇、吡啶、四甲基乙二胺、丙酮、三乙胺、正丁醇、二氧六环、四氢呋喃、甲酸甲酯、三丁胺、甲乙酮、乙酸乙酯、氯仿、三辛胺、碳酸二甲酯和乙醚,或其化合物。
“等电点”是一个分子表面不带电荷时的pH值。分子在等电点时,因为没有相同电荷而互相排斥的影响,所以最不稳定,溶解度最小,极易借静电引力迅速结合成较大的聚集体,因而沉淀析出。
含水量计算方式如下:
含水量=水分子质量*单个结晶分子中水分子数量/(水分子质量*单个结晶分子中水分子数量+氧化型谷胱甘肽分子量)*100%。
例如,一水合物含水量=18.01*1/(18.01*1+612.63)*100%=2.86%,六水合物含水量=18.01*6/(18.01*6+612.63)=14.99%,七水合物含水量=18.01*7/(18.01*7+612.63)=17.07%,八水合物含水量=18.01*8/(18.01*8+612.63)=19.04%。
下面将结合实施例对本发明的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅用于例示本发明,而非用于限制本发明。基于本发明的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
实施例
(一)氧化型谷胱甘肽的合成
实施例1
将1.0kg还原型谷胱甘肽和10.0L水加入三口瓶中,再加入1.0L二甲基亚砜,室温下搅拌反应48小时。有固体析出,过滤,干燥。得到氧化型谷胱甘肽粗品950.2g,收率95.3%,纯度98.4%。ESI-MS[M+H]+=613.3。
实施例2
将1.0kg还原型谷胱甘肽和3.0L水加入至三口瓶中,用NaOH调节pH至6~7,再加入500mL二甲基亚砜,室温下搅拌10小时。调节pH至等电点(2.75至2.90),5℃搅拌析晶10小时。过滤,滤饼用乙醇淋洗,并干燥,得到氧化型谷胱甘肽粗品855.1g,收率86.0%,纯度99.3%。ESI-MS[M+H]+=613.3。
实施例3
将1.0kg还原型谷胱甘肽和2.5L水加入至三口瓶中,用NaOH调节pH至6.0,再加入450mL二甲基亚砜,40℃下搅拌24小时。调节pH至等电点(2.75至2.90),在8℃下析晶10小时。过滤,滤饼用乙醇淋洗,干燥,得到氧化型谷胱甘肽892.7g,收率89.5%,纯度99.0%。ESI-MS[M+H]+=613.3。
实施例4
将1.0kg还原型谷胱甘肽和3.0L水加入至三口瓶中,用NaOH调节pH至5.5,再加入550mL二甲基亚砜,25℃下搅拌48小时。调节pH至等电点(2.75至2.90),在5℃下析晶8小时。过滤,滤饼用乙醇淋洗,干燥,得到氧化型谷胱甘肽912.0g,收率91.5%,纯度99.2%。ESI-MS[M+H]+=613.3。
对比例1:过氧化氢氧化
将10g还原型谷胱甘肽和30mL水加入至三口瓶中,用NaOH调节pH至5.8,在室温水浴条件下加入3.3mL 30%的过氧化氢,反应5小时。然后调节pH至3.0,向体系中加入50mL无水乙醇,10℃下析晶10小时。过滤,滤饼用乙醇淋洗,干燥,得到氧化型谷胱甘肽9.09g,收率91.2%,纯度96.2%,含有过氧化杂质。ESI-MS[M+H]+=613.3。
(二)氧化型谷胱甘肽的精制
对比例2:常规方法的纯化精制
将上述(一)氧化型谷胱甘肽的合成中实施例获得的10g氧化型谷胱甘肽粗品溶于200mL纯化水,然后进样到强酸性阳离子交换树脂,用纯化水进行洗脱。收集洗脱液,然后加入无水乙醇,使固体析出。得到8.43g氧化型谷胱甘肽,单步收率84.3%,纯度99.5%,纯度提升为0.2%。
实施例5
将上述(一)氧化型谷胱甘肽的合成中实施例获得的1.0kg氧化型谷胱甘肽的粗品加入4.0L 50℃的纯化水中,加入100mL 5%氯化亚铁溶液,搅拌至固体溶解。趁热过滤,然后梯度降温至25℃,每小时降温10℃,最后控温在20℃~25℃,析晶8小时。过滤,滤饼用乙醇淋洗,干燥。
其他实验例采用下述表格中的参数,按照上述步骤依次进行。
析出时间的探索
表1:析出时间探索的实验例

表2:析出时间探索的各实验例的结果
| 实施例5 | 实施例6 | 实施例7 | 实施例8 | 实施例9 | |
| 结晶质量(g) | 855.5 | 843.2 | 696.8 | 648.4 | 838.6 |
| 收率(%) | 85.5 | 84.3 | 69.7 | 64.8 | 83.9 |
| 纯度(%) | 99.9 | 99.9 | 99.9 | 99.9 | 99.7 |
| 含水量(%) | 17.2 | 17.3 | 17.2 | 15.8 | 17.2 |
根据上述结果可知,当析晶时间过短时,所得晶体为混晶,含水量不稳定,而当析晶时间过长时,会有杂质析出,导致产品纯度降低,因此,析晶时间为6-12小时为宜,6-8小时最佳。
纯化水倍量的探索
表3:纯化水倍量探索的实验例


表4:纯化水倍量探索的各实验例的结果

根据上述结果可知,以纯化水溶解氧化型谷胱甘肽,水量对收率有较大的影响,当水量过多时,收率过低,当水量过少时,产品不能完全溶解,或溶解再析出后搅拌效果不好,产品纯度降低,因此优选水量为2-6倍,优选3-6倍,更优选3-5倍。
溶解温度的探索
表5:溶解温度探索的实验例
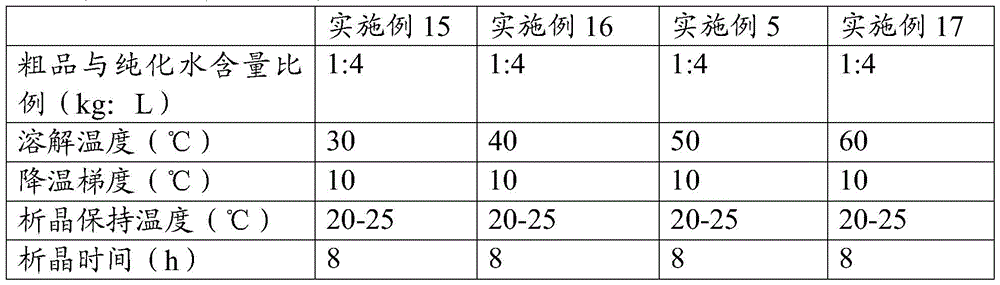
表6:溶解温度探索的各实验例的结果


根据上表可知,当温度高于40℃时均可溶解,但同时发现当温度高于60℃时,会出现消旋现象,而且温度越高、加热时间越长消旋现象越严重,且考虑生产能耗,因此选定温度为40-60℃,优选40-50℃,更优选50℃。
析晶保持温度的探索
表7:析晶保持温度探索的实验例

表8:析晶保持温度探索的各实验例的结果

根据上述实验结果可知,当析晶温度过低的时候,收率较高,但析出杂质也较多,且单杂大于0.1%,而当析晶温度过高,收率会下降很多,综合考虑上述因素,选定析晶温度可为5-25℃,优选10~25℃,更优选15~25℃。
降温梯度的探索
表9:降温梯度探索的实验例


表10:降温梯度探索的各实验例的结果
| 实施例22 | 实施例5 | 实施例23 | 实施例24 | |
| 结晶质量(g) | 883.7 | 855.5 | 848.6 | 857.4 |
| 收率(%) | 88.4 | 85.5 | 84.9 | 85.7 |
| 纯度(%) | 99.9 | 99.9 | 99.8 | 99.7 |
| 含水量(%) | 17.2 | 17.2 | 17.2 | 17.3 |
根据上表可知,当降温速度过快,析晶速度快,会包裹杂质一起快速析出,导致产品纯度降低,当降温速度过慢,会导致析晶时间延长,增加能耗和人工成本,而其收率变化不大,因此综合考虑,降温速率可为5-20℃/h,优选5-15℃/h,更优选10℃/h。
实施例25
将上述(一)氧化型谷胱甘肽的合成中实施例获得的1.0kg氧化型谷胱甘肽的粗品加入4.0L 40℃的纯化水中,搅拌至固体溶解。趁热过滤,然后梯度降温至20℃,每小时降温10℃,最后控温在15℃~20℃,析晶8小时。过滤,滤饼用乙醇淋洗,干燥,得到氧化型谷胱甘肽855.1g,单步收率85.5%,纯度99.7%,纯度提升为0.5%。
实施例26
将上述(一)氧化型谷胱甘肽的合成中实施例获得的1.0kg氧化型谷胱甘肽的粗品加入4.0L 45℃的纯化水中,加入100mL 5%氯化亚铁溶液,搅拌至固体溶解。趁热过滤,然后梯度降温至20℃,每小时降温10℃,最后控温在15℃~20℃,析晶7小时。过滤,滤饼用乙醇淋洗,干燥,得到氧化型谷胱甘肽849.2g,单步收率84.9%,纯度99.9%,纯度提升为0.7%。
实施例27
将上述(一)氧化型谷胱甘肽的合成中实施例获得的1.0kg氧化型谷胱甘肽的粗品加入5.0L 50℃的纯化水中,搅拌至固体溶解。趁热过滤,然后梯度降温至20℃,每小时降温10℃,最后控温在15℃~20℃,析晶8小时。过滤,滤饼用乙醇淋洗,干燥,得到氧化型谷胱甘肽843.3g,单步收率84.3%,纯度99.7%,纯度提升为0.5%。
实施例28
将上述(一)氧化型谷胱甘肽的合成中实施例获得的1.0kg氧化型谷胱甘肽的粗品加入5.0L 50℃的纯化水中,加入100mL 5%氯化亚铁溶液,搅拌至固体溶解。趁热过滤,然后梯度降温至20℃,每小时降温10℃,最后控温在15℃~20℃,析晶8小时。过滤,滤饼用乙醇淋洗,干燥,得到氧化型谷胱甘肽848.6g,单步收率84.9%,纯度99.9%,纯度提升为0.7%。
实施例29
将上述(一)氧化型谷胱甘肽的合成中实施例获得的1.0kg氧化型谷胱甘肽的粗品加入4.0L 50℃的纯化水中,搅拌至固体溶解。趁热过滤,然后梯度降温至20℃,每小时降温10℃,最后控温在20℃~25℃,析晶6小时。过滤,滤饼用乙醇淋洗,干燥,得到氧化型谷胱甘肽851.6g,单步收率85.2%,纯度99.7%,纯度提升为0.5%。
实施例30
杂质A的制备:将4.6g Boc-Cys(Trt)-OH、1.7g盐酸甘氨酸叔丁酯与7.6g HBTU溶于30mL DMF,滴入2mL DIEA。室温下反应24小时,然后加入100mL水,用乙酸乙酯萃取三次,保留有机相,用无水硫酸镁干燥。再蒸干溶剂,然后用30mL二氯甲烷溶解,加入30mL 30%三氟乙酸水溶液脱去Trt、Boc和tBu等保护基,反应结束,分离保留水层,然后向水层加入50mL乙醇析出固体。抽滤,滤饼经过制备液相纯化得到Cys-Gly。再将摩尔比1:1的还原型谷胱甘肽和Cys-Gly溶于20mL水中,加入5当量的DMSO氧化。室温下反应10小时,加40mL乙醇析出固体。得到的固体溶于5mL1%三氟乙酸水溶液后,用制备液相分离,流动相为A相:1%三氟乙酸/水;B相:1%三氟乙酸/乙腈。最后冻干得到2.0g杂质A,总收率42%,纯度99.7%。ESI-MS[M+H]+=484.1。
实施例31
杂质B的制备:将3.0g Boc-Glu-OtBu、4.2g HCl·H-Cys(Trt)-OtBu与7.6g HBTU溶于30mL DMF,滴入3mL DIEA。室温下反应18小时,然后加入100mL水,用乙酸乙酯萃取三次,保留有机相,用无水硫酸镁干燥。再蒸干溶剂。然后用30mL二氯甲烷溶解,加入40mL30%三氟乙酸水溶液脱去Trt、Boc和tBu等保护基,反应结束,分离出水层,然后向水层加入50mL乙醇析出固体。抽滤,滤饼经过制备液相纯化得到Glu-Cys。经过制备液相纯化得到Glu-Cys。再将摩尔比1:1的还原型谷胱甘肽和Glu-Cys溶于20mL水中,加入5当量的DMSO氧化。室温下反应12小时,加入40mL乙醇析出固体。得到的固体溶于5mL纯化水后,用制备液相分离,流动相为A相:1%三氟乙酸/水;B相:1%三氟乙酸/乙腈。最后冻干得到2.3g杂质B。总收率41%,纯度99.7%。ESI-MS[M+H]+=555.2。
实施例32
杂质C的制备:将3.1g还原型谷胱甘肽和1.2g半胱氨酸溶于15mL水中,加入3.9g的DMSO氧化,室温下搅拌反应9小时。反应结束,加入30mL乙醇析出固体。得到的固体溶于5mL纯化水后,用制备液相分离,流动相为A相:1%三氟乙酸/水;B相:1%三氟乙酸/乙腈。最后冻干得到2.1g杂质C。总收率49%,纯度99.8%。ESI-MS[M+H]+=427.1。
(三)晶体表征及分析
经测试,所得晶体的熔点为169℃(显微熔点测定仪X-5,厂家:科瑞仪器),与已报到晶体的熔点均不同,证明该晶体为全新晶型;X射线粉末衍射(规格型号:D8 ADVANCE,生产厂家:德国Bruker公司,该仪器配备了LynxEye检测器,2θ扫描角度从3°到40°,扫描步长为0.02°,扫描速度为0.3秒/步,测定样品时的光管电压和光管电流分别为40KV和40mA)测试所得特征峰(三高峰、八高峰)也与已报到的晶型不一致,而同时其相关峰型证明该晶型为非混晶;经TG、DSC测试(规格型号:STA449F5,生产厂家:德国NETZSCH),通过慢速升温(1.0℃/min)测得样品在50-180℃的失重情况,所得氧化型谷胱甘肽含水量为17.2%,与用卡尔费休水分测定仪(型号:V20S,生产厂家:METTLER TOLEDO)测定了样品的含水量为17.2%(平行三次的结果)含水量基本一致,与上文经计算与氧化型谷胱甘肽七水合物的含水量相对应,因此确定所得晶体为氧化型谷胱甘肽七水合物。
下表11显示如上所述的氧化型谷胱甘肽七水合物的晶体与氧化型谷胱甘肽一水合物和六水合物晶体的饱和溶解度之间的差异。
表11:氧化型谷胱甘肽水合物的饱和溶解度
| <![CDATA[饱和溶解度(g/L)<H<sub>2</sub>O 30℃>]]> | |
| 氧化型谷胱甘肽七水合物 | 198.6 |
| 氧化型谷胱甘肽六水合物 | 173 |
| 氧化型谷胱甘肽一水合物 | 13.5 |
在相同温度下七水合物在水中溶解度明显优于一水合物与六水合物相当,且相较于六水合物的粉状晶体易于产生静电,在分装过程中会有大量损耗,所得七水合物粒状晶体更加适合工业生产。
表12:氧化型谷胱甘肽七水合物晶型的X射线粉末衍射数据


综上,本发明提供一种制备氧化型谷胱甘肽及其新晶型和杂质的方法,简化了氧化型谷胱甘肽的合成方法,其杂质的合成对氧化性谷胱甘肽后续的质量研究提供了保证,同时使用纯化水对合成的氧化型谷胱甘肽进行重结晶,不仅得到了高纯度的氧化型谷胱甘肽,且简化了现有的纯化方式,新发现七水合物的新晶型相较于现有晶型具有更简便的合成方法,更优秀的水溶性,更良好的稳定性,更适合于氧化型谷胱甘肽的工业生产。
Claims (8)
1.式(I)化合物的七水合物晶体:
2.权利要求1的式(I)化合物的七水合物晶体,其特征在于:在使用CuKα辐射得到的X射线粉末衍射图中,至少包括位于以下o2q表示的特征峰:8.238±0.2、16.338±0.2和24.551±0.2。
3.权利要求2的式(I)化合物的七水合物晶体,其特征在于:在使用CuKα辐射得到的X射线粉末衍射图中,还至少包括位于以下o2q表示的特征峰:10.619±0.2、19.539±0.2、26.806±0.2和34.618±0.2。
4.权利要求3的式(I)化合物的七水合物晶体,其特征在于:在使用CuKα辐射得到的X射线粉末衍射图中,还至少包括位于以下o2q表示的特征峰:9.750±0.2、22.738±0.2和23.200±0.2。
5.权利要求1的式(I)化合物的七水合物晶体,其特征在于:其具有基本上如图5所示的X射线粉末衍射图。
6.权利要求1-5中任一项的式(I)化合物的七水合物晶体,其特征还在于:其具有169±2℃的熔点。
7.权利要求1-6中任一项的式(I)化合物的七水合物晶体,其特征还在于:在热解重量分析中,其在50-100℃失重约14±1%,在100-160℃失重约3±1%。
8.权利要求1-7中任一项的式(I)化合物的七水合物晶体,其特征还在于:其热解重量图如图6所示。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202111573909 | 2021-12-21 | ||
| CN2021115739092 | 2021-12-21 | ||
| CN202211572513 | 2022-12-08 | ||
| CN2022115725130 | 2022-12-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN116120393A true CN116120393A (zh) | 2023-05-16 |
Family
ID=86307238
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202211640246.6A Pending CN116120393A (zh) | 2021-12-21 | 2022-12-20 | 一种氧化型谷胱甘肽及其晶型和杂质的制备方法 |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN116120393A (zh) |
| WO (1) | WO2023116664A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114249794A (zh) * | 2021-12-22 | 2022-03-29 | 深圳瑞德林生物技术有限公司 | 一种氧化型谷胱甘肽的合成方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102869674B (zh) * | 2010-04-21 | 2015-07-22 | 协和发酵生化株式会社 | 氧化型谷胱甘肽的晶体及其制造方法 |
-
2022
- 2022-12-20 WO PCT/CN2022/140269 patent/WO2023116664A1/zh unknown
- 2022-12-20 CN CN202211640246.6A patent/CN116120393A/zh active Pending
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114249794A (zh) * | 2021-12-22 | 2022-03-29 | 深圳瑞德林生物技术有限公司 | 一种氧化型谷胱甘肽的合成方法 |
| CN114249794B (zh) * | 2021-12-22 | 2023-06-23 | 深圳瑞德林生物技术有限公司 | 一种氧化型谷胱甘肽的合成方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2023116664A1 (zh) | 2023-06-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2081121C1 (ru) | Способ получения клавулановой кислоты или ее фармацевтически приемлемых солей или эфиров, соль клавулановой кислоты с амином | |
| JP2004534053A (ja) | 結晶性セフジニル酸付加塩及びこれを用いたセフジニルの製造方法 | |
| CN116120393A (zh) | 一种氧化型谷胱甘肽及其晶型和杂质的制备方法 | |
| KR20070095811A (ko) | 퓨린 유도체의 에스테르 제조 방법 | |
| CN105712919B (zh) | 酰胺缩合剂在维格列汀合成方法中的应用 | |
| CN110551144B (zh) | 一种阿莫西林的制备方法 | |
| CN111072660B (zh) | 一种瑞来巴坦的简便制备方法 | |
| JP2013527239A (ja) | イクサベピロンの固体形 | |
| WO2018177048A1 (zh) | 头孢菌素中间体7α-甲氧基头孢噻吩的结晶及其制备方法 | |
| CN112830956B (zh) | 棕榈酰四肽-7的液相合成方法 | |
| CN108033971A (zh) | 一种盐酸头孢卡品酯的合成方法 | |
| KR20120096200A (ko) | 결정형 도세탁셀 및 이의 제조방법 | |
| Noda et al. | A new simple preparation of d-alloisoleucine suitable for large-scale manufacture | |
| US8129536B2 (en) | Method for the purification of lansoprazole | |
| US3821208A (en) | Recovery of cephalosporin c and derivatives thereof | |
| CN105017287B (zh) | 一种头霉素中间体的制备方法 | |
| CN114805220A (zh) | 一种喹唑啉酮类化合物的制备方法 | |
| CN115010723B (zh) | 一种头孢烷酸类亚砜组合物及其制备方法 | |
| CN111808040B (zh) | 多构型2-氧代噁唑烷-4-羧酸类化合物的合成方法 | |
| JP3888402B2 (ja) | 光学活性N−カルボベンゾキシ−tert−ロイシンの製造法 | |
| EP2690097B1 (en) | Method for crystallizing glutamic acid benzyl ester n-carboxylic anhydride | |
| KR100293728B1 (ko) | 결정성세피롬황산염의제조방법 | |
| CN106957311B (zh) | 雷替曲塞的溶剂化物及其制备方法 | |
| WO2006010978A1 (en) | Cefdinir polymorphic forms, and imidazole salt | |
| EP1844041A1 (en) | An improved process for the preparation of mycophenolate mofetil |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| CB02 | Change of applicant information |
Address after: No. 25, New Canal Road, Shenyang Area, China (Liaoning) Pilot Free Trade Zone, Hunnan District, Shenyang, Liaoning 110167 Applicant after: SHENYANG XINGQI PHARMACEUTICAL Co.,Ltd. Address before: 110163 Surabaya street, Dongling District, Shenyang, Liaoning Province, No. 68 Applicant before: SHENYANG XINGQI PHARMACEUTICAL Co.,Ltd. |
|
| CB02 | Change of applicant information |