CN113905763A - 肿瘤选择性组合治疗 - Google Patents
肿瘤选择性组合治疗 Download PDFInfo
- Publication number
- CN113905763A CN113905763A CN202080037114.7A CN202080037114A CN113905763A CN 113905763 A CN113905763 A CN 113905763A CN 202080037114 A CN202080037114 A CN 202080037114A CN 113905763 A CN113905763 A CN 113905763A
- Authority
- CN
- China
- Prior art keywords
- tumor
- cells
- nqo1
- lap
- dnq
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 206010028980 Neoplasm Diseases 0.000 title claims abstract description 473
- 238000002648 combination therapy Methods 0.000 title abstract description 18
- 238000011282 treatment Methods 0.000 claims abstract description 163
- 239000003814 drug Substances 0.000 claims abstract description 74
- 229940079593 drug Drugs 0.000 claims abstract description 67
- 201000011510 cancer Diseases 0.000 claims abstract description 61
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 claims abstract description 27
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 claims abstract description 27
- 102100022365 NAD(P)H dehydrogenase [quinone] 1 Human genes 0.000 claims abstract description 23
- 238000009169 immunotherapy Methods 0.000 claims abstract description 22
- 231100000518 lethal Toxicity 0.000 claims abstract description 6
- 230000001665 lethal effect Effects 0.000 claims abstract description 6
- 101000973778 Homo sapiens NAD(P)H dehydrogenase [quinone] 1 Proteins 0.000 claims abstract 20
- QZPQTZZNNJUOLS-UHFFFAOYSA-N beta-lapachone Chemical compound C12=CC=CC=C2C(=O)C(=O)C2=C1OC(C)(C)CC2 QZPQTZZNNJUOLS-UHFFFAOYSA-N 0.000 claims description 626
- 238000000034 method Methods 0.000 claims description 78
- -1 DNQ compound Chemical class 0.000 claims description 58
- 150000003839 salts Chemical class 0.000 claims description 43
- 239000003112 inhibitor Substances 0.000 claims description 29
- 239000003795 chemical substances by application Substances 0.000 claims description 27
- 239000002246 antineoplastic agent Substances 0.000 claims description 23
- 238000002560 therapeutic procedure Methods 0.000 claims description 21
- 239000012453 solvate Substances 0.000 claims description 20
- 101001109714 Rhizobium meliloti (strain 1021) NAD(P)H dehydrogenase (quinone) 1 Proteins 0.000 claims description 17
- 102100040678 Programmed cell death protein 1 Human genes 0.000 claims description 15
- 229940127089 cytotoxic agent Drugs 0.000 claims description 15
- 230000033616 DNA repair Effects 0.000 claims description 13
- 238000002512 chemotherapy Methods 0.000 claims description 12
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims description 12
- 230000033590 base-excision repair Effects 0.000 claims description 10
- 230000002401 inhibitory effect Effects 0.000 claims description 10
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 9
- 238000001959 radiotherapy Methods 0.000 claims description 9
- 102000004190 Enzymes Human genes 0.000 claims description 8
- 108090000790 Enzymes Proteins 0.000 claims description 8
- 230000008569 process Effects 0.000 claims description 8
- 238000001356 surgical procedure Methods 0.000 claims description 8
- 206010006187 Breast cancer Diseases 0.000 claims description 7
- 230000007547 defect Effects 0.000 claims description 7
- 230000002147 killing effect Effects 0.000 claims description 7
- 208000026310 Breast neoplasm Diseases 0.000 claims description 6
- 229940045513 CTLA4 antagonist Drugs 0.000 claims description 6
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 6
- 201000002528 pancreatic cancer Diseases 0.000 claims description 6
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 6
- 238000002360 preparation method Methods 0.000 claims description 6
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 5
- 208000029742 colonic neoplasm Diseases 0.000 claims description 5
- 108090000623 proteins and genes Proteins 0.000 claims description 5
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims description 5
- 108010021064 CTLA-4 Antigen Proteins 0.000 claims description 4
- 206010009944 Colon cancer Diseases 0.000 claims description 4
- 206010060862 Prostate cancer Diseases 0.000 claims description 4
- 230000005907 cancer growth Effects 0.000 claims description 4
- 102000004169 proteins and genes Human genes 0.000 claims description 4
- 201000010536 head and neck cancer Diseases 0.000 claims description 3
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 3
- 150000003384 small molecules Chemical class 0.000 claims description 3
- 239000003053 toxin Substances 0.000 claims description 3
- 231100000765 toxin Toxicity 0.000 claims description 3
- 101150042435 Xrcc1 gene Proteins 0.000 claims description 2
- 238000011319 anticancer therapy Methods 0.000 claims 2
- 101150000874 11 gene Proteins 0.000 claims 1
- 102000008203 CTLA-4 Antigen Human genes 0.000 claims 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 16
- 201000010099 disease Diseases 0.000 abstract description 11
- 231100000225 lethality Toxicity 0.000 abstract description 6
- 230000009286 beneficial effect Effects 0.000 abstract 1
- 210000004027 cell Anatomy 0.000 description 241
- 241000699670 Mus sp. Species 0.000 description 159
- 210000001744 T-lymphocyte Anatomy 0.000 description 107
- 125000000217 alkyl group Chemical group 0.000 description 107
- 150000001875 compounds Chemical class 0.000 description 84
- 230000000259 anti-tumor effect Effects 0.000 description 69
- 210000004881 tumor cell Anatomy 0.000 description 56
- 238000011740 C57BL/6 mouse Methods 0.000 description 49
- 108010050904 Interferons Proteins 0.000 description 48
- 102000014150 Interferons Human genes 0.000 description 48
- 229910052739 hydrogen Chemical group 0.000 description 47
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 47
- 239000001257 hydrogen Chemical group 0.000 description 46
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical group [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 45
- 229940079322 interferon Drugs 0.000 description 45
- 230000001404 mediated effect Effects 0.000 description 40
- 229960004400 levonorgestrel Drugs 0.000 description 38
- 239000000427 antigen Substances 0.000 description 36
- 108091007433 antigens Proteins 0.000 description 36
- 102000036639 antigens Human genes 0.000 description 36
- 230000000694 effects Effects 0.000 description 36
- 230000001419 dependent effect Effects 0.000 description 35
- 230000005746 immune checkpoint blockade Effects 0.000 description 35
- 230000004614 tumor growth Effects 0.000 description 33
- 108010074708 B7-H1 Antigen Proteins 0.000 description 32
- 102000008096 B7-H1 Antigen Human genes 0.000 description 32
- 241000282414 Homo sapiens Species 0.000 description 32
- 239000000203 mixture Substances 0.000 description 32
- WWYNJERNGUHSAO-XUDSTZEESA-N (+)-Norgestrel Chemical compound O=C1CC[C@@H]2[C@H]3CC[C@](CC)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=C1 WWYNJERNGUHSAO-XUDSTZEESA-N 0.000 description 30
- 239000003981 vehicle Substances 0.000 description 30
- 230000037449 immunogenic cell death Effects 0.000 description 28
- 239000003642 reactive oxygen metabolite Substances 0.000 description 28
- 238000007920 subcutaneous administration Methods 0.000 description 26
- 125000001424 substituent group Chemical group 0.000 description 26
- 230000004083 survival effect Effects 0.000 description 26
- 102100037907 High mobility group protein B1 Human genes 0.000 description 25
- 238000002474 experimental method Methods 0.000 description 25
- 230000014509 gene expression Effects 0.000 description 25
- 238000001727 in vivo Methods 0.000 description 25
- 210000001519 tissue Anatomy 0.000 description 25
- 101100339431 Arabidopsis thaliana HMGB2 gene Proteins 0.000 description 24
- 108700010013 HMGB1 Proteins 0.000 description 24
- 101150021904 HMGB1 gene Proteins 0.000 description 24
- 125000003118 aryl group Chemical group 0.000 description 23
- 230000011664 signaling Effects 0.000 description 23
- 230000001225 therapeutic effect Effects 0.000 description 23
- 241001529936 Murinae Species 0.000 description 22
- DOBMPNYZJYQDGZ-UHFFFAOYSA-N dicoumarol Chemical compound C1=CC=CC2=C1OC(=O)C(CC=1C(OC3=CC=CC=C3C=1O)=O)=C2O DOBMPNYZJYQDGZ-UHFFFAOYSA-N 0.000 description 22
- 230000001965 increasing effect Effects 0.000 description 22
- 230000003442 weekly effect Effects 0.000 description 22
- 230000030833 cell death Effects 0.000 description 21
- 238000002347 injection Methods 0.000 description 21
- 239000007924 injection Substances 0.000 description 21
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 20
- 238000000338 in vitro Methods 0.000 description 20
- 230000004044 response Effects 0.000 description 20
- 231100000277 DNA damage Toxicity 0.000 description 19
- 230000037396 body weight Effects 0.000 description 19
- 210000001165 lymph node Anatomy 0.000 description 19
- 230000005778 DNA damage Effects 0.000 description 18
- 210000004443 dendritic cell Anatomy 0.000 description 18
- 230000000903 blocking effect Effects 0.000 description 17
- 210000002865 immune cell Anatomy 0.000 description 17
- 230000005867 T cell response Effects 0.000 description 16
- 238000000684 flow cytometry Methods 0.000 description 16
- 230000008447 perception Effects 0.000 description 16
- 239000000243 solution Substances 0.000 description 16
- 108020004414 DNA Proteins 0.000 description 15
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 15
- 230000004913 activation Effects 0.000 description 15
- 229960001912 dicoumarol Drugs 0.000 description 15
- 210000000987 immune system Anatomy 0.000 description 15
- 206010070834 Sensitisation Diseases 0.000 description 14
- 125000006239 protecting group Chemical group 0.000 description 14
- 210000000952 spleen Anatomy 0.000 description 14
- 108010077432 Myeloid Differentiation Factor 88 Proteins 0.000 description 13
- 102000010168 Myeloid Differentiation Factor 88 Human genes 0.000 description 13
- 102100023712 Poly [ADP-ribose] polymerase 1 Human genes 0.000 description 13
- 125000004432 carbon atom Chemical group C* 0.000 description 13
- 238000006243 chemical reaction Methods 0.000 description 13
- 238000003114 enzyme-linked immunosorbent spot assay Methods 0.000 description 13
- 230000015788 innate immune response Effects 0.000 description 13
- 238000004519 manufacturing process Methods 0.000 description 13
- 239000000126 substance Substances 0.000 description 13
- 238000007492 two-way ANOVA Methods 0.000 description 13
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 12
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 12
- 229930182566 Gentamicin Natural products 0.000 description 12
- 108010064218 Poly (ADP-Ribose) Polymerase-1 Proteins 0.000 description 12
- 230000004721 adaptive immunity Effects 0.000 description 12
- 230000014102 antigen processing and presentation of exogenous peptide antigen via MHC class I Effects 0.000 description 12
- 229910052736 halogen Inorganic materials 0.000 description 12
- 150000002367 halogens Chemical class 0.000 description 12
- 230000005764 inhibitory process Effects 0.000 description 12
- 229960004857 mitomycin Drugs 0.000 description 12
- 230000002195 synergetic effect Effects 0.000 description 12
- 238000012360 testing method Methods 0.000 description 12
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 11
- 241000699666 Mus <mouse, genus> Species 0.000 description 11
- 101710089372 Programmed cell death protein 1 Proteins 0.000 description 11
- 239000004480 active ingredient Substances 0.000 description 11
- 125000000623 heterocyclic group Chemical group 0.000 description 11
- 239000007787 solid Substances 0.000 description 11
- 230000008685 targeting Effects 0.000 description 11
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 11
- 102100032912 CD44 antigen Human genes 0.000 description 10
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 description 10
- 101000669447 Homo sapiens Toll-like receptor 4 Proteins 0.000 description 10
- 229930192392 Mitomycin Natural products 0.000 description 10
- 102100039360 Toll-like receptor 4 Human genes 0.000 description 10
- 210000005006 adaptive immune system Anatomy 0.000 description 10
- 210000000612 antigen-presenting cell Anatomy 0.000 description 10
- 239000002585 base Substances 0.000 description 10
- 125000000753 cycloalkyl group Chemical group 0.000 description 10
- 125000000524 functional group Chemical group 0.000 description 10
- 125000001072 heteroaryl group Chemical group 0.000 description 10
- 239000011734 sodium Substances 0.000 description 10
- 239000002904 solvent Substances 0.000 description 10
- 201000009030 Carcinoma Diseases 0.000 description 9
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 9
- 238000011510 Elispot assay Methods 0.000 description 9
- 238000000692 Student's t-test Methods 0.000 description 9
- 239000002253 acid Substances 0.000 description 9
- 125000003545 alkoxy group Chemical group 0.000 description 9
- 238000003556 assay Methods 0.000 description 9
- 125000004404 heteroalkyl group Chemical group 0.000 description 9
- 239000002609 medium Substances 0.000 description 9
- 229910052760 oxygen Inorganic materials 0.000 description 9
- 230000037361 pathway Effects 0.000 description 9
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 9
- 238000009097 single-agent therapy Methods 0.000 description 9
- 238000003786 synthesis reaction Methods 0.000 description 9
- 230000009885 systemic effect Effects 0.000 description 9
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 description 8
- 102000016938 Catalase Human genes 0.000 description 8
- 108010053835 Catalase Proteins 0.000 description 8
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 8
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 8
- 241001465754 Metazoa Species 0.000 description 8
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 8
- 125000003342 alkenyl group Chemical group 0.000 description 8
- 238000004458 analytical method Methods 0.000 description 8
- 230000006378 damage Effects 0.000 description 8
- 230000002950 deficient Effects 0.000 description 8
- 125000005842 heteroatom Chemical group 0.000 description 8
- 230000004048 modification Effects 0.000 description 8
- 238000012986 modification Methods 0.000 description 8
- 210000000440 neutrophil Anatomy 0.000 description 8
- 102000007863 pattern recognition receptors Human genes 0.000 description 8
- 108010089193 pattern recognition receptors Proteins 0.000 description 8
- 230000003389 potentiating effect Effects 0.000 description 8
- 108090000765 processed proteins & peptides Proteins 0.000 description 8
- 230000002829 reductive effect Effects 0.000 description 8
- 238000012353 t test Methods 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 7
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 7
- 102000000849 HMGB Proteins Human genes 0.000 description 7
- 108010001860 HMGB Proteins Proteins 0.000 description 7
- 101710196623 Stimulator of interferon genes protein Proteins 0.000 description 7
- 125000000304 alkynyl group Chemical group 0.000 description 7
- 230000005975 antitumor immune response Effects 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 229910052799 carbon Inorganic materials 0.000 description 7
- 229940088598 enzyme Drugs 0.000 description 7
- 238000009472 formulation Methods 0.000 description 7
- 230000012010 growth Effects 0.000 description 7
- 230000005847 immunogenicity Effects 0.000 description 7
- 239000004615 ingredient Substances 0.000 description 7
- 230000003993 interaction Effects 0.000 description 7
- 230000002601 intratumoral effect Effects 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- 210000002540 macrophage Anatomy 0.000 description 7
- 229950006238 nadide Drugs 0.000 description 7
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 7
- 150000004053 quinones Chemical class 0.000 description 7
- 230000028327 secretion Effects 0.000 description 7
- 150000003573 thiols Chemical group 0.000 description 7
- 238000005406 washing Methods 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- 101000852870 Homo sapiens Interferon alpha/beta receptor 1 Proteins 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 6
- 241000699660 Mus musculus Species 0.000 description 6
- BAWFJGJZGIEFAR-NNYOXOHSSA-N NAD zwitterion Chemical compound NC(=O)C1=CC=C[N+]([C@H]2[C@@H]([C@H](O)[C@@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 BAWFJGJZGIEFAR-NNYOXOHSSA-N 0.000 description 6
- 102000004316 Oxidoreductases Human genes 0.000 description 6
- 108090000854 Oxidoreductases Proteins 0.000 description 6
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 6
- 230000006052 T cell proliferation Effects 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 230000009471 action Effects 0.000 description 6
- 230000005809 anti-tumor immunity Effects 0.000 description 6
- 230000008901 benefit Effects 0.000 description 6
- 230000000973 chemotherapeutic effect Effects 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 238000011284 combination treatment Methods 0.000 description 6
- 125000004122 cyclic group Chemical group 0.000 description 6
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 6
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 6
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 6
- 230000006054 immunological memory Effects 0.000 description 6
- 230000001506 immunosuppresive effect Effects 0.000 description 6
- 230000003902 lesion Effects 0.000 description 6
- 231100000636 lethal dose Toxicity 0.000 description 6
- 210000004698 lymphocyte Anatomy 0.000 description 6
- 230000007246 mechanism Effects 0.000 description 6
- 210000003071 memory t lymphocyte Anatomy 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- 230000002018 overexpression Effects 0.000 description 6
- 239000001301 oxygen Chemical group 0.000 description 6
- 239000002953 phosphate buffered saline Substances 0.000 description 6
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 6
- 238000006722 reduction reaction Methods 0.000 description 6
- 238000007619 statistical method Methods 0.000 description 6
- 229910052717 sulfur Inorganic materials 0.000 description 6
- 230000001988 toxicity Effects 0.000 description 6
- 231100000419 toxicity Toxicity 0.000 description 6
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 6
- 238000011830 transgenic mouse model Methods 0.000 description 6
- 230000003827 upregulation Effects 0.000 description 6
- IOOMXAQUNPWDLL-UHFFFAOYSA-N 2-[6-(diethylamino)-3-(diethyliminiumyl)-3h-xanthen-9-yl]-5-sulfobenzene-1-sulfonate Chemical compound C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=C(S(O)(=O)=O)C=C1S([O-])(=O)=O IOOMXAQUNPWDLL-UHFFFAOYSA-N 0.000 description 5
- 108091033409 CRISPR Proteins 0.000 description 5
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 5
- 102100036714 Interferon alpha/beta receptor 1 Human genes 0.000 description 5
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 5
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 5
- 230000003044 adaptive effect Effects 0.000 description 5
- 230000033289 adaptive immune response Effects 0.000 description 5
- 125000003302 alkenyloxy group Chemical group 0.000 description 5
- 125000003282 alkyl amino group Chemical group 0.000 description 5
- 125000004644 alkyl sulfinyl group Chemical group 0.000 description 5
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 5
- 125000005133 alkynyloxy group Chemical group 0.000 description 5
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 5
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 5
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 5
- 230000001413 cellular effect Effects 0.000 description 5
- 208000035475 disorder Diseases 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 5
- 238000001415 gene therapy Methods 0.000 description 5
- 229960001101 ifosfamide Drugs 0.000 description 5
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 5
- 230000028993 immune response Effects 0.000 description 5
- 230000001771 impaired effect Effects 0.000 description 5
- 230000001939 inductive effect Effects 0.000 description 5
- 230000008595 infiltration Effects 0.000 description 5
- 238000001764 infiltration Methods 0.000 description 5
- 238000002955 isolation Methods 0.000 description 5
- 239000003446 ligand Substances 0.000 description 5
- 230000000670 limiting effect Effects 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 238000010172 mouse model Methods 0.000 description 5
- 230000017074 necrotic cell death Effects 0.000 description 5
- 230000007935 neutral effect Effects 0.000 description 5
- 210000000056 organ Anatomy 0.000 description 5
- 239000008194 pharmaceutical composition Substances 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- 230000035755 proliferation Effects 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 210000004988 splenocyte Anatomy 0.000 description 5
- 230000014567 type I interferon production Effects 0.000 description 5
- 238000001262 western blot Methods 0.000 description 5
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- 108090000672 Annexin A5 Proteins 0.000 description 4
- 102000004121 Annexin A5 Human genes 0.000 description 4
- 238000010354 CRISPR gene editing Methods 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- 102000011727 Caspases Human genes 0.000 description 4
- 108010076667 Caspases Proteins 0.000 description 4
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 4
- 238000002965 ELISA Methods 0.000 description 4
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 4
- 101100220044 Homo sapiens CD34 gene Proteins 0.000 description 4
- 101000889276 Homo sapiens Cytotoxic T-lymphocyte protein 4 Proteins 0.000 description 4
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 4
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 4
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 4
- 230000006044 T cell activation Effects 0.000 description 4
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 4
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 4
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 4
- 230000002378 acidificating effect Effects 0.000 description 4
- 239000013543 active substance Substances 0.000 description 4
- 125000002252 acyl group Chemical group 0.000 description 4
- 208000009956 adenocarcinoma Diseases 0.000 description 4
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 4
- 125000004457 alkyl amino carbonyl group Chemical group 0.000 description 4
- 125000003368 amide group Chemical group 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 235000001014 amino acid Nutrition 0.000 description 4
- 150000001413 amino acids Chemical class 0.000 description 4
- 125000003277 amino group Chemical group 0.000 description 4
- 238000013459 approach Methods 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- 101150092503 batf gene Proteins 0.000 description 4
- 210000000481 breast Anatomy 0.000 description 4
- 230000004663 cell proliferation Effects 0.000 description 4
- 230000003833 cell viability Effects 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 239000012228 culture supernatant Substances 0.000 description 4
- 125000004093 cyano group Chemical group *C#N 0.000 description 4
- 229960004397 cyclophosphamide Drugs 0.000 description 4
- 230000001086 cytosolic effect Effects 0.000 description 4
- 231100000433 cytotoxic Toxicity 0.000 description 4
- 230000001472 cytotoxic effect Effects 0.000 description 4
- 230000003247 decreasing effect Effects 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 230000005782 double-strand break Effects 0.000 description 4
- 230000012361 double-strand break repair Effects 0.000 description 4
- 229960004679 doxorubicin Drugs 0.000 description 4
- 229960003722 doxycycline Drugs 0.000 description 4
- 239000012636 effector Substances 0.000 description 4
- 230000002708 enhancing effect Effects 0.000 description 4
- 229960002258 fulvestrant Drugs 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 4
- 125000005885 heterocycloalkylalkyl group Chemical group 0.000 description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 4
- 230000037417 hyperactivation Effects 0.000 description 4
- 230000001976 improved effect Effects 0.000 description 4
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 4
- 229960003881 letrozole Drugs 0.000 description 4
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 4
- 201000005202 lung cancer Diseases 0.000 description 4
- 208000020816 lung neoplasm Diseases 0.000 description 4
- 230000003211 malignant effect Effects 0.000 description 4
- 201000001441 melanoma Diseases 0.000 description 4
- 229960001924 melphalan Drugs 0.000 description 4
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 4
- 125000002950 monocyclic group Chemical group 0.000 description 4
- 230000003472 neutralizing effect Effects 0.000 description 4
- 230000003647 oxidation Effects 0.000 description 4
- 238000007254 oxidation reaction Methods 0.000 description 4
- 230000007170 pathology Effects 0.000 description 4
- 229960002621 pembrolizumab Drugs 0.000 description 4
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 4
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 4
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 230000008313 sensitization Effects 0.000 description 4
- 230000005783 single-strand break Effects 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- 235000000346 sugar Nutrition 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 4
- 230000001960 triggered effect Effects 0.000 description 4
- 230000005851 tumor immunogenicity Effects 0.000 description 4
- 230000005760 tumorsuppression Effects 0.000 description 4
- 238000012762 unpaired Student’s t-test Methods 0.000 description 4
- 229960003048 vinblastine Drugs 0.000 description 4
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 4
- 229960004528 vincristine Drugs 0.000 description 4
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 4
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 4
- VQVUBYASAICPFU-UHFFFAOYSA-N (6'-acetyloxy-2',7'-dichloro-3-oxospiro[2-benzofuran-1,9'-xanthene]-3'-yl) acetate Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC(Cl)=C(OC(C)=O)C=C1OC1=C2C=C(Cl)C(OC(=O)C)=C1 VQVUBYASAICPFU-UHFFFAOYSA-N 0.000 description 3
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 description 3
- 208000003200 Adenoma Diseases 0.000 description 3
- 102100029822 B- and T-lymphocyte attenuator Human genes 0.000 description 3
- 101150014003 Batf3 gene Proteins 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 3
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 3
- 102000004127 Cytokines Human genes 0.000 description 3
- 108090000695 Cytokines Proteins 0.000 description 3
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 101001137987 Homo sapiens Lymphocyte activation gene 3 protein Proteins 0.000 description 3
- 101000611936 Homo sapiens Programmed cell death protein 1 Proteins 0.000 description 3
- 102000037982 Immune checkpoint proteins Human genes 0.000 description 3
- 108091008036 Immune checkpoint proteins Proteins 0.000 description 3
- 108060003951 Immunoglobulin Proteins 0.000 description 3
- 102100022297 Integrin alpha-X Human genes 0.000 description 3
- 102000002227 Interferon Type I Human genes 0.000 description 3
- 108010014726 Interferon Type I Proteins 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 108020000284 NAD(P)H dehydrogenase (quinone) Proteins 0.000 description 3
- 101710095135 NAD(P)H dehydrogenase [quinone] 1 Proteins 0.000 description 3
- 206010029260 Neuroblastoma Diseases 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 239000012271 PD-L1 inhibitor Substances 0.000 description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- OUUQCZGPVNCOIJ-UHFFFAOYSA-M Superoxide Chemical compound [O-][O] OUUQCZGPVNCOIJ-UHFFFAOYSA-M 0.000 description 3
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 3
- 101150082427 Tlr4 gene Proteins 0.000 description 3
- 229960000473 altretamine Drugs 0.000 description 3
- 230000001093 anti-cancer Effects 0.000 description 3
- 230000006907 apoptotic process Effects 0.000 description 3
- 125000005135 aryl sulfinyl group Chemical group 0.000 description 3
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- 125000002619 bicyclic group Chemical group 0.000 description 3
- 150000001721 carbon Chemical group 0.000 description 3
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 3
- 230000000453 cell autonomous effect Effects 0.000 description 3
- 230000007969 cellular immunity Effects 0.000 description 3
- 229940044683 chemotherapy drug Drugs 0.000 description 3
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 3
- 229960004630 chlorambucil Drugs 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 238000003927 comet assay Methods 0.000 description 3
- 231100000170 comet assay Toxicity 0.000 description 3
- 230000001276 controlling effect Effects 0.000 description 3
- 238000011498 curative surgery Methods 0.000 description 3
- 125000000392 cycloalkenyl group Chemical group 0.000 description 3
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 description 3
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 3
- 230000003013 cytotoxicity Effects 0.000 description 3
- 231100000135 cytotoxicity Toxicity 0.000 description 3
- 230000000779 depleting effect Effects 0.000 description 3
- 238000001514 detection method Methods 0.000 description 3
- 238000001784 detoxification Methods 0.000 description 3
- 230000029087 digestion Effects 0.000 description 3
- 239000002612 dispersion medium Substances 0.000 description 3
- XQTWDDCIUJNLTR-CVHRZJFOSA-N doxycycline monohydrate Chemical compound O.O=C1C2=C(O)C=CC=C2[C@H](C)[C@@H]2C1=C(O)[C@]1(O)C(=O)C(C(N)=O)=C(O)[C@@H](N(C)C)[C@@H]1[C@H]2O XQTWDDCIUJNLTR-CVHRZJFOSA-N 0.000 description 3
- 239000008298 dragée Substances 0.000 description 3
- 239000000975 dye Substances 0.000 description 3
- 239000013604 expression vector Substances 0.000 description 3
- NOOCSNJCXJYGPE-UHFFFAOYSA-N flunixin Chemical compound C1=CC=C(C(F)(F)F)C(C)=C1NC1=NC=CC=C1C(O)=O NOOCSNJCXJYGPE-UHFFFAOYSA-N 0.000 description 3
- 229960000588 flunixin Drugs 0.000 description 3
- 231100000024 genotoxic Toxicity 0.000 description 3
- 230000001738 genotoxic effect Effects 0.000 description 3
- 229940093915 gynecological organic acid Drugs 0.000 description 3
- 125000005843 halogen group Chemical group 0.000 description 3
- 201000002222 hemangioblastoma Diseases 0.000 description 3
- 125000004366 heterocycloalkenyl group Chemical group 0.000 description 3
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 3
- 238000011577 humanized mouse model Methods 0.000 description 3
- 230000002163 immunogen Effects 0.000 description 3
- 102000018358 immunoglobulin Human genes 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 239000000411 inducer Substances 0.000 description 3
- 229940047124 interferons Drugs 0.000 description 3
- 238000011835 investigation Methods 0.000 description 3
- 230000000155 isotopic effect Effects 0.000 description 3
- 125000005647 linker group Chemical group 0.000 description 3
- 150000002632 lipids Chemical class 0.000 description 3
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 3
- 238000011275 oncology therapy Methods 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 235000005985 organic acids Nutrition 0.000 description 3
- 229940121656 pd-l1 inhibitor Drugs 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 3
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 229960004618 prednisone Drugs 0.000 description 3
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 230000002035 prolonged effect Effects 0.000 description 3
- 230000004224 protection Effects 0.000 description 3
- 235000018102 proteins Nutrition 0.000 description 3
- 229950010131 puromycin Drugs 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 description 3
- 230000035484 reaction time Effects 0.000 description 3
- 238000002271 resection Methods 0.000 description 3
- 230000033443 single strand break repair Effects 0.000 description 3
- 230000000638 stimulation Effects 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 3
- 239000011593 sulfur Chemical group 0.000 description 3
- 239000006228 supernatant Substances 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 125000004149 thio group Chemical group *S* 0.000 description 3
- 231100000331 toxic Toxicity 0.000 description 3
- 230000002588 toxic effect Effects 0.000 description 3
- 229960005294 triamcinolone Drugs 0.000 description 3
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 description 3
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 2
- BBMCTIGTTCKYKF-UHFFFAOYSA-N 1-heptanol Chemical compound CCCCCCCO BBMCTIGTTCKYKF-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- IDPUKCWIGUEADI-UHFFFAOYSA-N 5-[bis(2-chloroethyl)amino]uracil Chemical compound ClCCN(CCCl)C1=CNC(=O)NC1=O IDPUKCWIGUEADI-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 description 2
- 101710144268 B- and T-lymphocyte attenuator Proteins 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- 102100038078 CD276 antigen Human genes 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 2
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 2
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- 239000012624 DNA alkylating agent Substances 0.000 description 2
- 108010092160 Dactinomycin Proteins 0.000 description 2
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 2
- BFPYWIDHMRZLRN-SLHNCBLASA-N Ethinyl estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=C1 BFPYWIDHMRZLRN-SLHNCBLASA-N 0.000 description 2
- 201000008808 Fibrosarcoma Diseases 0.000 description 2
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 2
- 108010024636 Glutathione Proteins 0.000 description 2
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 2
- 102100034533 Histone H2AX Human genes 0.000 description 2
- 101000858088 Homo sapiens C-X-C motif chemokine 10 Proteins 0.000 description 2
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 2
- 101001067891 Homo sapiens Histone H2AX Proteins 0.000 description 2
- 101001113483 Homo sapiens Poly [ADP-ribose] polymerase 1 Proteins 0.000 description 2
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 2
- 206010062016 Immunosuppression Diseases 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 102000037984 Inhibitory immune checkpoint proteins Human genes 0.000 description 2
- 108091008026 Inhibitory immune checkpoint proteins Proteins 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 2
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 2
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 2
- 102100020862 Lymphocyte activation gene 3 protein Human genes 0.000 description 2
- 101150053046 MYD88 gene Proteins 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 101001044384 Mus musculus Interferon gamma Proteins 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical compound CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- 240000002853 Nelumbo nucifera Species 0.000 description 2
- 235000006508 Nelumbo nucifera Nutrition 0.000 description 2
- 235000006510 Nelumbo pentapetala Nutrition 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- 239000012270 PD-1 inhibitor Substances 0.000 description 2
- 239000012668 PD-1-inhibitor Substances 0.000 description 2
- 229930012538 Paclitaxel Natural products 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical compound OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 238000002123 RNA extraction Methods 0.000 description 2
- 206010039491 Sarcoma Diseases 0.000 description 2
- 206010041067 Small cell lung cancer Diseases 0.000 description 2
- 108020004459 Small interfering RNA Proteins 0.000 description 2
- 229920002125 Sokalan® Polymers 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- 102000008235 Toll-Like Receptor 9 Human genes 0.000 description 2
- 108010060818 Toll-Like Receptor 9 Proteins 0.000 description 2
- 102000002689 Toll-like receptor Human genes 0.000 description 2
- 108020000411 Toll-like receptor Proteins 0.000 description 2
- 208000027418 Wounds and injury Diseases 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 235000011054 acetic acid Nutrition 0.000 description 2
- 229960000583 acetic acid Drugs 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 229930183665 actinomycin Natural products 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 125000004423 acyloxy group Chemical group 0.000 description 2
- 208000002517 adenoid cystic carcinoma Diseases 0.000 description 2
- 150000001312 aldohexoses Chemical class 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 150000003973 alkyl amines Chemical group 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 150000001408 amides Chemical group 0.000 description 2
- 125000004103 aminoalkyl group Chemical group 0.000 description 2
- 229960003437 aminoglutethimide Drugs 0.000 description 2
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 description 2
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 229960002932 anastrozole Drugs 0.000 description 2
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 2
- MWPLVEDNUUSJAV-UHFFFAOYSA-N anthracene Chemical compound C1=CC=CC2=CC3=CC=CC=C3C=C21 MWPLVEDNUUSJAV-UHFFFAOYSA-N 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000006023 anti-tumor response Effects 0.000 description 2
- 239000003429 antifungal agent Substances 0.000 description 2
- 229940121375 antifungal agent Drugs 0.000 description 2
- 230000030741 antigen processing and presentation Effects 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- GOLCXWYRSKYTSP-UHFFFAOYSA-N arsenic trioxide Inorganic materials O1[As]2O[As]1O2 GOLCXWYRSKYTSP-UHFFFAOYSA-N 0.000 description 2
- 125000001769 aryl amino group Chemical group 0.000 description 2
- 230000002238 attenuated effect Effects 0.000 description 2
- 229940120638 avastin Drugs 0.000 description 2
- 239000011324 bead Substances 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 2
- 229940092714 benzenesulfonic acid Drugs 0.000 description 2
- 125000005605 benzo group Chemical group 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- 229960004365 benzoic acid Drugs 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 230000001488 breeding effect Effects 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 2
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 238000011088 calibration curve Methods 0.000 description 2
- 229940127093 camptothecin Drugs 0.000 description 2
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 2
- 238000002619 cancer immunotherapy Methods 0.000 description 2
- 229960004117 capecitabine Drugs 0.000 description 2
- KBPLFHHGFOOTCA-UHFFFAOYSA-N caprylic alcohol Natural products CCCCCCCCO KBPLFHHGFOOTCA-UHFFFAOYSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 2
- 229960005243 carmustine Drugs 0.000 description 2
- 230000003915 cell function Effects 0.000 description 2
- 230000010261 cell growth Effects 0.000 description 2
- 230000022534 cell killing Effects 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 125000004230 chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 description 2
- 229960004106 citric acid Drugs 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 210000001072 colon Anatomy 0.000 description 2
- 230000001010 compromised effect Effects 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- 229960000684 cytarabine Drugs 0.000 description 2
- 229960003901 dacarbazine Drugs 0.000 description 2
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 2
- MWKFXSUHUHTGQN-UHFFFAOYSA-N decan-1-ol Chemical compound CCCCCCCCCCO MWKFXSUHUHTGQN-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 2
- 229960003668 docetaxel Drugs 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 239000000890 drug combination Substances 0.000 description 2
- 230000004064 dysfunction Effects 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 2
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 2
- 229930013356 epothilone Natural products 0.000 description 2
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical class C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 description 2
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 2
- 229960005420 etoposide Drugs 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 229960000255 exemestane Drugs 0.000 description 2
- 210000004700 fetal blood Anatomy 0.000 description 2
- 229960000961 floxuridine Drugs 0.000 description 2
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 2
- 239000012458 free base Substances 0.000 description 2
- 239000012737 fresh medium Substances 0.000 description 2
- 235000011087 fumaric acid Nutrition 0.000 description 2
- 239000001530 fumaric acid Substances 0.000 description 2
- 229960002598 fumaric acid Drugs 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 2
- 229960005277 gemcitabine Drugs 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 239000004220 glutamic acid Substances 0.000 description 2
- 235000013922 glutamic acid Nutrition 0.000 description 2
- 229960003180 glutathione Drugs 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 2
- 229940022353 herceptin Drugs 0.000 description 2
- ZSIAUFGUXNUGDI-UHFFFAOYSA-N hexan-1-ol Chemical compound CCCCCCO ZSIAUFGUXNUGDI-UHFFFAOYSA-N 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 2
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 2
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 2
- 239000012642 immune effector Substances 0.000 description 2
- 230000036039 immunity Effects 0.000 description 2
- 230000003053 immunization Effects 0.000 description 2
- 238000002649 immunization Methods 0.000 description 2
- 238000010166 immunofluorescence Methods 0.000 description 2
- 229940121354 immunomodulator Drugs 0.000 description 2
- 230000003308 immunostimulating effect Effects 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 102000006639 indoleamine 2,3-dioxygenase Human genes 0.000 description 2
- 108020004201 indoleamine 2,3-dioxygenase Proteins 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 238000011221 initial treatment Methods 0.000 description 2
- 230000000977 initiatory effect Effects 0.000 description 2
- 208000014674 injury Diseases 0.000 description 2
- 238000011081 inoculation Methods 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 229960004768 irinotecan Drugs 0.000 description 2
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 2
- NNPPMTNAJDCUHE-UHFFFAOYSA-N isobutane Chemical compound CC(C)C NNPPMTNAJDCUHE-UHFFFAOYSA-N 0.000 description 2
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 2
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 2
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 2
- 125000005956 isoquinolyl group Chemical group 0.000 description 2
- 125000001786 isothiazolyl group Chemical group 0.000 description 2
- 239000007951 isotonicity adjuster Substances 0.000 description 2
- 125000000468 ketone group Chemical group 0.000 description 2
- 239000004310 lactic acid Substances 0.000 description 2
- 235000014655 lactic acid Nutrition 0.000 description 2
- 229960000448 lactic acid Drugs 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- 238000001325 log-rank test Methods 0.000 description 2
- 229960002247 lomustine Drugs 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 239000011976 maleic acid Substances 0.000 description 2
- 229940098895 maleic acid Drugs 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 230000010534 mechanism of action Effects 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 208000005135 methemoglobinemia Diseases 0.000 description 2
- 229960000485 methotrexate Drugs 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 2
- 210000000822 natural killer cell Anatomy 0.000 description 2
- 229910017604 nitric acid Inorganic materials 0.000 description 2
- ZWRUINPWMLAQRD-UHFFFAOYSA-N nonan-1-ol Chemical compound CCCCCCCCCO ZWRUINPWMLAQRD-UHFFFAOYSA-N 0.000 description 2
- 210000004940 nucleus Anatomy 0.000 description 2
- 229960001756 oxaliplatin Drugs 0.000 description 2
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 2
- 230000001590 oxidative effect Effects 0.000 description 2
- 230000036542 oxidative stress Effects 0.000 description 2
- 229960001592 paclitaxel Drugs 0.000 description 2
- 230000001575 pathological effect Effects 0.000 description 2
- 229940121655 pd-1 inhibitor Drugs 0.000 description 2
- 210000001539 phagocyte Anatomy 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- 239000013612 plasmid Substances 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920005862 polyol Polymers 0.000 description 2
- 150000003077 polyols Chemical class 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 238000007639 printing Methods 0.000 description 2
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 2
- 229960004919 procaine Drugs 0.000 description 2
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 2
- 229960000624 procarbazine Drugs 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 210000002307 prostate Anatomy 0.000 description 2
- 230000001681 protective effect Effects 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 230000000637 radiosensitizating effect Effects 0.000 description 2
- 230000003439 radiotherapeutic effect Effects 0.000 description 2
- 230000006950 reactive oxygen species formation Effects 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 230000008439 repair process Effects 0.000 description 2
- 125000006413 ring segment Chemical group 0.000 description 2
- 229960004641 rituximab Drugs 0.000 description 2
- 201000007416 salivary gland adenoid cystic carcinoma Diseases 0.000 description 2
- 125000005630 sialyl group Chemical group 0.000 description 2
- 238000007086 side reaction Methods 0.000 description 2
- 230000019491 signal transduction Effects 0.000 description 2
- 229910052710 silicon Inorganic materials 0.000 description 2
- 208000000649 small cell carcinoma Diseases 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 235000011121 sodium hydroxide Nutrition 0.000 description 2
- 238000000527 sonication Methods 0.000 description 2
- 230000003393 splenic effect Effects 0.000 description 2
- 206010041823 squamous cell carcinoma Diseases 0.000 description 2
- 210000000130 stem cell Anatomy 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 229960001052 streptozocin Drugs 0.000 description 2
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 2
- TYFQFVWCELRYAO-UHFFFAOYSA-N suberic acid Chemical compound OC(=O)CCCCCCC(O)=O TYFQFVWCELRYAO-UHFFFAOYSA-N 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L sulfate group Chemical group S(=O)(=O)([O-])[O-] QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 2
- 231100000338 sulforhodamine B assay Toxicity 0.000 description 2
- 238000003210 sulforhodamine B staining Methods 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- 229960001603 tamoxifen Drugs 0.000 description 2
- 238000002626 targeted therapy Methods 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 229960001367 tartaric acid Drugs 0.000 description 2
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 2
- 229940063683 taxotere Drugs 0.000 description 2
- 229960004964 temozolomide Drugs 0.000 description 2
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 2
- 229960001278 teniposide Drugs 0.000 description 2
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical group C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 2
- BPEWUONYVDABNZ-DZBHQSCQSA-N testolactone Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(OC(=O)CC4)[C@@H]4[C@@H]3CCC2=C1 BPEWUONYVDABNZ-DZBHQSCQSA-N 0.000 description 2
- 229960005353 testolactone Drugs 0.000 description 2
- 229960003604 testosterone Drugs 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- 229960001196 thiotepa Drugs 0.000 description 2
- 229960003087 tioguanine Drugs 0.000 description 2
- 229960003989 tocilizumab Drugs 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 229960000303 topotecan Drugs 0.000 description 2
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 2
- 108700012359 toxins Proteins 0.000 description 2
- 238000002054 transplantation Methods 0.000 description 2
- ODLHGICHYURWBS-LKONHMLTSA-N trappsol cyclo Chemical compound CC(O)COC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)COCC(O)C)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1COCC(C)O ODLHGICHYURWBS-LKONHMLTSA-N 0.000 description 2
- 229960000575 trastuzumab Drugs 0.000 description 2
- 238000011269 treatment regimen Methods 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 230000005909 tumor killing Effects 0.000 description 2
- 229960001055 uracil mustard Drugs 0.000 description 2
- 230000002485 urinary effect Effects 0.000 description 2
- 238000002255 vaccination Methods 0.000 description 2
- 230000035899 viability Effects 0.000 description 2
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 2
- 229960002066 vinorelbine Drugs 0.000 description 2
- 238000010626 work up procedure Methods 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- JVJGCCBAOOWGEO-RUTPOYCXSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-4-amino-2-[[(2s,3s)-2-[[(2s,3s)-2-[[(2s)-2-azaniumyl-3-hydroxypropanoyl]amino]-3-methylpentanoyl]amino]-3-methylpentanoyl]amino]-4-oxobutanoyl]amino]-3-phenylpropanoyl]amino]-4-carboxylatobutanoyl]amino]-6-azaniumy Chemical compound OC[C@H](N)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(O)=O)CC1=CC=CC=C1 JVJGCCBAOOWGEO-RUTPOYCXSA-N 0.000 description 1
- BDPXTYNYUJHKCI-UFQCMFJCSA-N (8R,9S,10R,13S,14R,17S)-15-(fluoromethyl)-17-hydroxy-10,13-dimethyl-1,2,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-3-one Chemical compound FCC1[C@@H]2[C@]([C@H](C1)O)(C)CC[C@H]1[C@H]2CCC2=CC(=O)CC[C@]12C BDPXTYNYUJHKCI-UFQCMFJCSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- KETQAJRQOHHATG-UHFFFAOYSA-N 1,2-naphthoquinone Chemical compound C1=CC=C2C(=O)C(=O)C=CC2=C1 KETQAJRQOHHATG-UHFFFAOYSA-N 0.000 description 1
- 150000005208 1,4-dihydroxybenzenes Chemical class 0.000 description 1
- 150000004890 1,4-dithianes Chemical class 0.000 description 1
- FUHYSHCCRTWZAY-UHFFFAOYSA-N 1,4-oxathiepane Chemical compound C1COCCSC1 FUHYSHCCRTWZAY-UHFFFAOYSA-N 0.000 description 1
- ZNGWEEUXTBNKFR-UHFFFAOYSA-N 1,4-oxazepane Chemical compound C1CNCCOC1 ZNGWEEUXTBNKFR-UHFFFAOYSA-N 0.000 description 1
- HJTAZXHBEBIQQX-UHFFFAOYSA-N 1,5-bis(chloromethyl)naphthalene Chemical compound C1=CC=C2C(CCl)=CC=CC2=C1CCl HJTAZXHBEBIQQX-UHFFFAOYSA-N 0.000 description 1
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 description 1
- IYTUPMAAMJDPCR-UHFFFAOYSA-N 1-chlorophenanthrene Chemical compound C1=CC2=CC=CC=C2C2=C1C(Cl)=CC=C2 IYTUPMAAMJDPCR-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- GCKMFJBGXUYNAG-UHFFFAOYSA-N 17alpha-methyltestosterone Natural products C1CC2=CC(=O)CCC2(C)C2C1C1CCC(C)(O)C1(C)CC2 GCKMFJBGXUYNAG-UHFFFAOYSA-N 0.000 description 1
- DBPWSSGDRRHUNT-CEGNMAFCSA-N 17α-hydroxyprogesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)C)(O)[C@@]1(C)CC2 DBPWSSGDRRHUNT-CEGNMAFCSA-N 0.000 description 1
- HBEDSQVIWPRPAY-UHFFFAOYSA-N 2,3-dihydrobenzofuran Chemical compound C1=CC=C2OCCC2=C1 HBEDSQVIWPRPAY-UHFFFAOYSA-N 0.000 description 1
- QEBYEVQKHRUYPE-UHFFFAOYSA-N 2-(2-chlorophenyl)-5-[(1-methylpyrazol-3-yl)methyl]-4-[[methyl(pyridin-3-ylmethyl)amino]methyl]-1h-pyrazolo[4,3-c]pyridine-3,6-dione Chemical compound C1=CN(C)N=C1CN1C(=O)C=C2NN(C=3C(=CC=CC=3)Cl)C(=O)C2=C1CN(C)CC1=CC=CN=C1 QEBYEVQKHRUYPE-UHFFFAOYSA-N 0.000 description 1
- RTQWWZBSTRGEAV-PKHIMPSTSA-N 2-[[(2s)-2-[bis(carboxymethyl)amino]-3-[4-(methylcarbamoylamino)phenyl]propyl]-[2-[bis(carboxymethyl)amino]propyl]amino]acetic acid Chemical compound CNC(=O)NC1=CC=C(C[C@@H](CN(CC(C)N(CC(O)=O)CC(O)=O)CC(O)=O)N(CC(O)=O)CC(O)=O)C=C1 RTQWWZBSTRGEAV-PKHIMPSTSA-N 0.000 description 1
- GSXYWVXSJLBESP-UHFFFAOYSA-N 2-bromo-1-phenylpiperazine Chemical compound BrC1CNCCN1C1=CC=CC=C1 GSXYWVXSJLBESP-UHFFFAOYSA-N 0.000 description 1
- HZLCGUXUOFWCCN-UHFFFAOYSA-N 2-hydroxynonadecane-1,2,3-tricarboxylic acid Chemical compound CCCCCCCCCCCCCCCCC(C(O)=O)C(O)(C(O)=O)CC(O)=O HZLCGUXUOFWCCN-UHFFFAOYSA-N 0.000 description 1
- 101710157142 2-methylene-furan-3-one reductase Proteins 0.000 description 1
- BSKHPKMHTQYZBB-UHFFFAOYSA-N 2-methylpyridine Chemical compound CC1=CC=CC=N1 BSKHPKMHTQYZBB-UHFFFAOYSA-N 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- RSEBUVRVKCANEP-UHFFFAOYSA-N 2-pyrroline Chemical compound C1CC=CN1 RSEBUVRVKCANEP-UHFFFAOYSA-N 0.000 description 1
- 150000004881 2H-pyrans Chemical class 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- MCGBIXXDQFWVDW-UHFFFAOYSA-N 4,5-dihydro-1h-pyrazole Chemical class C1CC=NN1 MCGBIXXDQFWVDW-UHFFFAOYSA-N 0.000 description 1
- SHXAUBCOXJAXEI-UHFFFAOYSA-N 4-(1,3-diazepan-1-yl)morpholine Chemical compound O1CCN(CC1)N1CNCCCC1 SHXAUBCOXJAXEI-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 150000000531 4H-pyrans Chemical class 0.000 description 1
- 125000002471 4H-quinolizinyl group Chemical group C=1(C=CCN2C=CC=CC12)* 0.000 description 1
- XYJODUBPWNZLML-UHFFFAOYSA-N 5-ethyl-6-phenyl-6h-phenanthridine-3,8-diamine Chemical compound C12=CC(N)=CC=C2C2=CC=C(N)C=C2N(CC)C1C1=CC=CC=C1 XYJODUBPWNZLML-UHFFFAOYSA-N 0.000 description 1
- ZGMLACURZVGTPK-UHFFFAOYSA-N 5-fluoranyl-1h-pyrimidine-2,4-dione Chemical compound OC1=NC=C(F)C(O)=N1.FC1=CNC(=O)NC1=O ZGMLACURZVGTPK-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- 108700012813 7-aminoactinomycin D Proteins 0.000 description 1
- YXHLJMWYDTXDHS-IRFLANFNSA-N 7-aminoactinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=C(N)C=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 YXHLJMWYDTXDHS-IRFLANFNSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HFDKKNHCYWNNNQ-YOGANYHLSA-N 75976-10-2 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@@H](NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)N)C(C)C)[C@@H](C)O)C1=CC=C(O)C=C1 HFDKKNHCYWNNNQ-YOGANYHLSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- 230000005730 ADP ribosylation Effects 0.000 description 1
- ZKHQWZAMYRWXGA-KQYNXXCUSA-J ATP(4-) Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-J 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N Acrylic acid Chemical compound OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 102000007471 Adenosine A2A receptor Human genes 0.000 description 1
- 108010085277 Adenosine A2A receptor Proteins 0.000 description 1
- 101150051188 Adora2a gene Proteins 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 201000003076 Angiosarcoma Diseases 0.000 description 1
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 206010003571 Astrocytoma Diseases 0.000 description 1
- 241000972773 Aulopiformes Species 0.000 description 1
- OLCWFLWEHWLBTO-HSZRJFAPSA-N BMS-214662 Chemical compound C=1C=CSC=1S(=O)(=O)N([C@@H](C1)CC=2C=CC=CC=2)CC2=CC(C#N)=CC=C2N1CC1=CN=CN1 OLCWFLWEHWLBTO-HSZRJFAPSA-N 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 206010004146 Basal cell carcinoma Diseases 0.000 description 1
- 206010004593 Bile duct cancer Diseases 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 102100025248 C-X-C motif chemokine 10 Human genes 0.000 description 1
- 238000011749 CBA mouse Methods 0.000 description 1
- DCERHCFNWRGHLK-UHFFFAOYSA-N C[Si](C)C Chemical compound C[Si](C)C DCERHCFNWRGHLK-UHFFFAOYSA-N 0.000 description 1
- 101100463133 Caenorhabditis elegans pdl-1 gene Proteins 0.000 description 1
- 102100029968 Calreticulin Human genes 0.000 description 1
- 108090000549 Calreticulin Proteins 0.000 description 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 108010022366 Carcinoembryonic Antigen Proteins 0.000 description 1
- 102100025475 Carcinoembryonic antigen-related cell adhesion molecule 5 Human genes 0.000 description 1
- 201000000274 Carcinosarcoma Diseases 0.000 description 1
- 244000132059 Carica parviflora Species 0.000 description 1
- 235000014653 Carica parviflora Nutrition 0.000 description 1
- 102000003952 Caspase 3 Human genes 0.000 description 1
- 108090000397 Caspase 3 Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical group [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 108010049048 Cholera Toxin Proteins 0.000 description 1
- 102000009016 Cholera Toxin Human genes 0.000 description 1
- 208000005243 Chondrosarcoma Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 102000029816 Collagenase Human genes 0.000 description 1
- 108060005980 Collagenase Proteins 0.000 description 1
- 206010052360 Colorectal adenocarcinoma Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 201000005171 Cystadenoma Diseases 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- WQZGKKKJIJFFOK-CBPJZXOFSA-N D-Gulose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@H](O)[C@H]1O WQZGKKKJIJFFOK-CBPJZXOFSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- WQZGKKKJIJFFOK-WHZQZERISA-N D-aldose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-WHZQZERISA-N 0.000 description 1
- WQZGKKKJIJFFOK-IVMDWMLBSA-N D-allopyranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@H](O)[C@@H]1O WQZGKKKJIJFFOK-IVMDWMLBSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- FEWJPZIEWOKRBE-LWMBPPNESA-L D-tartrate(2-) Chemical compound [O-]C(=O)[C@@H](O)[C@H](O)C([O-])=O FEWJPZIEWOKRBE-LWMBPPNESA-L 0.000 description 1
- 230000005971 DNA damage repair Effects 0.000 description 1
- 230000004543 DNA replication Effects 0.000 description 1
- 108010008532 Deoxyribonuclease I Proteins 0.000 description 1
- 102000007260 Deoxyribonuclease I Human genes 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- 238000008157 ELISA kit Methods 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 102100038595 Estrogen receptor Human genes 0.000 description 1
- 102100029951 Estrogen receptor beta Human genes 0.000 description 1
- 208000006168 Ewing Sarcoma Diseases 0.000 description 1
- 208000007300 Fibrolamellar hepatocellular carcinoma Diseases 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 208000004057 Focal Nodular Hyperplasia Diseases 0.000 description 1
- 229940123457 Free radical scavenger Drugs 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 210000000712 G cell Anatomy 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- 206010018404 Glucagonoma Diseases 0.000 description 1
- 102000005720 Glutathione transferase Human genes 0.000 description 1
- 108010070675 Glutathione transferase Proteins 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 208000002125 Hemangioendothelioma Diseases 0.000 description 1
- 208000001258 Hemangiosarcoma Diseases 0.000 description 1
- 206010019629 Hepatic adenoma Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000971171 Homo sapiens Apoptosis regulator Bcl-2 Proteins 0.000 description 1
- 101000864344 Homo sapiens B- and T-lymphocyte attenuator Proteins 0.000 description 1
- 101000883515 Homo sapiens Chitinase-3-like protein 1 Proteins 0.000 description 1
- 101001010910 Homo sapiens Estrogen receptor beta Proteins 0.000 description 1
- 101001025337 Homo sapiens High mobility group protein B1 Proteins 0.000 description 1
- 101001046686 Homo sapiens Integrin alpha-M Proteins 0.000 description 1
- 101001034314 Homo sapiens Lactadherin Proteins 0.000 description 1
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- 102100022338 Integrin alpha-M Human genes 0.000 description 1
- 108010086140 Interferon alpha-beta Receptor Proteins 0.000 description 1
- 102000006992 Interferon-alpha Human genes 0.000 description 1
- 229930194542 Keto Natural products 0.000 description 1
- PWKSKIMOESPYIA-BYPYZUCNSA-N L-N-acetyl-Cysteine Chemical compound CC(=O)N[C@@H](CS)C(O)=O PWKSKIMOESPYIA-BYPYZUCNSA-N 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VSOAQEOCSA-N L-altropyranose Chemical compound OC[C@@H]1OC(O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-VSOAQEOCSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-L L-tartrate(2-) Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O FEWJPZIEWOKRBE-JCYAYHJZSA-L 0.000 description 1
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 1
- 102000017578 LAG3 Human genes 0.000 description 1
- 102100039648 Lactadherin Human genes 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 108010000851 Laminin Receptors Proteins 0.000 description 1
- 102000002297 Laminin Receptors Human genes 0.000 description 1
- 208000018142 Leiomyosarcoma Diseases 0.000 description 1
- 102100020872 Leucyl-cystinyl aminopeptidase Human genes 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 208000007433 Lymphatic Metastasis Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 235000019759 Maize starch Nutrition 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 208000000172 Medulloblastoma Diseases 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-M Methanesulfonate Chemical compound CS([O-])(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 1
- FQISKWAFAHGMGT-SGJOWKDISA-M Methylprednisolone sodium succinate Chemical compound [Na+].C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCC([O-])=O)CC[C@H]21 FQISKWAFAHGMGT-SGJOWKDISA-M 0.000 description 1
- GCKMFJBGXUYNAG-HLXURNFRSA-N Methyltestosterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@](C)(O)[C@@]1(C)CC2 GCKMFJBGXUYNAG-HLXURNFRSA-N 0.000 description 1
- 206010057269 Mucoepidermoid carcinoma Diseases 0.000 description 1
- 101100026728 Mus musculus Nqo1 gene Proteins 0.000 description 1
- BAWFJGJZGIEFAR-NNYOXOHSSA-O NAD(+) Chemical class NC(=O)C1=CC=C[N+]([C@H]2[C@@H]([C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 BAWFJGJZGIEFAR-NNYOXOHSSA-O 0.000 description 1
- 102000004960 NAD(P)H dehydrogenase (quinone) Human genes 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 206010061309 Neoplasm progression Diseases 0.000 description 1
- 208000034176 Neoplasms, Germ Cell and Embryonal Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 206010029488 Nodular melanoma Diseases 0.000 description 1
- 201000010133 Oligodendroglioma Diseases 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 102000018886 Pancreatic Polypeptide Human genes 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 108010081690 Pertussis Toxin Proteins 0.000 description 1
- 101710114878 Phospholipase A-2-activating protein Proteins 0.000 description 1
- 208000007913 Pituitary Neoplasms Diseases 0.000 description 1
- 208000007452 Plasmacytoma Diseases 0.000 description 1
- 102100022427 Plasmalemma vesicle-associated protein Human genes 0.000 description 1
- 101710193105 Plasmalemma vesicle-associated protein Proteins 0.000 description 1
- 102000015087 Poly (ADP-Ribose) Polymerase-1 Human genes 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 102000007066 Prostate-Specific Antigen Human genes 0.000 description 1
- 108010072866 Prostate-Specific Antigen Proteins 0.000 description 1
- 206010051807 Pseudosarcoma Diseases 0.000 description 1
- LOUPRKONTZGTKE-WZBLMQSHSA-N Quinine Chemical class C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@@H]2[C@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-WZBLMQSHSA-N 0.000 description 1
- 101710189291 Quinone oxidoreductase Proteins 0.000 description 1
- 102100034576 Quinone oxidoreductase Human genes 0.000 description 1
- 101710149118 Quinone oxidoreductase 1 Proteins 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 238000011529 RT qPCR Methods 0.000 description 1
- 231100000991 Reactive Oxygen Species (ROS) Photosafety Assay Toxicity 0.000 description 1
- 206010070308 Refractory cancer Diseases 0.000 description 1
- 208000006265 Renal cell carcinoma Diseases 0.000 description 1
- 201000000582 Retinoblastoma Diseases 0.000 description 1
- 108010039491 Ricin Proteins 0.000 description 1
- 229910007161 Si(CH3)3 Inorganic materials 0.000 description 1
- 102000005157 Somatostatin Human genes 0.000 description 1
- 108010056088 Somatostatin Proteins 0.000 description 1
- ZSJLQEPLLKMAKR-UHFFFAOYSA-N Streptozotocin Natural products O=NN(C)C(=O)NC1C(O)OC(CO)C(O)C1O ZSJLQEPLLKMAKR-UHFFFAOYSA-N 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- 206010042553 Superficial spreading melanoma stage unspecified Diseases 0.000 description 1
- 101000983124 Sus scrofa Pancreatic prohormone precursor Proteins 0.000 description 1
- 230000033540 T cell apoptotic process Effects 0.000 description 1
- 108091008874 T cell receptors Proteins 0.000 description 1
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 1
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 1
- 210000000662 T-lymphocyte subset Anatomy 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 108010040002 Tumor Suppressor Proteins Proteins 0.000 description 1
- 102000001742 Tumor Suppressor Proteins Human genes 0.000 description 1
- 102100039094 Tyrosinase Human genes 0.000 description 1
- 108060008724 Tyrosinase Proteins 0.000 description 1
- 201000005969 Uveal melanoma Diseases 0.000 description 1
- 101710145727 Viral Fc-gamma receptor-like protein UL119 Proteins 0.000 description 1
- 208000008383 Wilms tumor Diseases 0.000 description 1
- 108010000443 X-ray Repair Cross Complementing Protein 1 Proteins 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 239000003070 absorption delaying agent Substances 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 229940022663 acetate Drugs 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 229960004308 acetylcysteine Drugs 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 208000009621 actinic keratosis Diseases 0.000 description 1
- 230000008649 adaptation response Effects 0.000 description 1
- 101150063416 add gene Proteins 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 201000001256 adenosarcoma Diseases 0.000 description 1
- 201000008395 adenosquamous carcinoma Diseases 0.000 description 1
- 201000008424 adenosquamous lung carcinoma Diseases 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 229940040563 agaric acid Drugs 0.000 description 1
- 238000007605 air drying Methods 0.000 description 1
- 150000001299 aldehydes Chemical group 0.000 description 1
- IAJILQKETJEXLJ-RSJOWCBRSA-N aldehydo-D-galacturonic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-RSJOWCBRSA-N 0.000 description 1
- IAJILQKETJEXLJ-QTBDOELSSA-N aldehydo-D-glucuronic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-QTBDOELSSA-N 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 239000012670 alkaline solution Substances 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 125000002877 alkyl aryl group Chemical group 0.000 description 1
- 150000005215 alkyl ethers Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 230000002009 allergenic effect Effects 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 1
- 229960003805 amantadine Drugs 0.000 description 1
- 235000011114 ammonium hydroxide Nutrition 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 229940045799 anthracyclines and related substance Drugs 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000000118 anti-neoplastic effect Effects 0.000 description 1
- 238000011394 anticancer treatment Methods 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 230000009925 apoptotic mechanism Effects 0.000 description 1
- 230000005735 apoptotic response Effects 0.000 description 1
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 1
- 125000003435 aroyl group Chemical group 0.000 description 1
- 229960002594 arsenic trioxide Drugs 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 238000003149 assay kit Methods 0.000 description 1
- 239000005441 aurora Substances 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- 125000003943 azolyl group Chemical group 0.000 description 1
- 229940098166 bactrim Drugs 0.000 description 1
- 238000003287 bathing Methods 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 102000012740 beta Adrenergic Receptors Human genes 0.000 description 1
- 108010079452 beta Adrenergic Receptors Proteins 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000002210 biocatalytic effect Effects 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 238000010170 biological method Methods 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 239000000090 biomarker Substances 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 210000002798 bone marrow cell Anatomy 0.000 description 1
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 201000008274 breast adenocarcinoma Diseases 0.000 description 1
- 238000009395 breeding Methods 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical group BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 235000011116 calcium hydroxide Nutrition 0.000 description 1
- 229940112129 campath Drugs 0.000 description 1
- 208000035269 cancer or benign tumor Diseases 0.000 description 1
- 239000012830 cancer therapeutic Substances 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 229960001631 carbomer Drugs 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 125000004181 carboxyalkyl group Chemical group 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 125000006244 carboxylic acid protecting group Chemical group 0.000 description 1
- 208000002458 carcinoid tumor Diseases 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 229940105657 catalase Drugs 0.000 description 1
- 150000001767 cationic compounds Chemical class 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 239000000460 chlorine Chemical group 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 125000002668 chloroacetyl group Chemical group ClCC(=O)* 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 208000006990 cholangiocarcinoma Diseases 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 208000009060 clear cell adenocarcinoma Diseases 0.000 description 1
- 229960002424 collagenase Drugs 0.000 description 1
- 238000009096 combination chemotherapy Methods 0.000 description 1
- 238000011220 combination immunotherapy Methods 0.000 description 1
- 230000002301 combined effect Effects 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 230000001268 conjugating effect Effects 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- 239000002537 cosmetic Substances 0.000 description 1
- 230000009260 cross reactivity Effects 0.000 description 1
- 238000002681 cryosurgery Methods 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 125000000000 cycloalkoxy group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 1
- 125000006622 cycloheptylmethyl group Chemical group 0.000 description 1
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000004210 cyclohexylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000004851 cyclopentylmethyl group Chemical group C1(CCCC1)C* 0.000 description 1
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 1
- 238000002784 cytotoxicity assay Methods 0.000 description 1
- 231100000263 cytotoxicity test Toxicity 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 238000000432 density-gradient centrifugation Methods 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 125000004663 dialkyl amino group Chemical group 0.000 description 1
- UZVGSSNIUNSOFA-UHFFFAOYSA-N dibenzofuran-1-carboxylic acid Chemical compound O1C2=CC=CC=C2C2=C1C=CC=C2C(=O)O UZVGSSNIUNSOFA-UHFFFAOYSA-N 0.000 description 1
- HIZKPJUTKKJDGA-UHFFFAOYSA-N dicumarol Natural products O=C1OC2=CC=CC=C2C(=O)C1CC1C(=O)C2=CC=CC=C2OC1=O HIZKPJUTKKJDGA-UHFFFAOYSA-N 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- RGLYKWWBQGJZGM-ISLYRVAYSA-N diethylstilbestrol Chemical compound C=1C=C(O)C=CC=1C(/CC)=C(\CC)C1=CC=C(O)C=C1 RGLYKWWBQGJZGM-ISLYRVAYSA-N 0.000 description 1
- 229960000452 diethylstilbestrol Drugs 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 229910001882 dioxygen Inorganic materials 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- NOTIQUSPUUHHEH-UXOVVSIBSA-N dromostanolone propionate Chemical compound C([C@@H]1CC2)C(=O)[C@H](C)C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](OC(=O)CC)[C@@]2(C)CC1 NOTIQUSPUUHHEH-UXOVVSIBSA-N 0.000 description 1
- 229950004683 drostanolone propionate Drugs 0.000 description 1
- 210000003162 effector t lymphocyte Anatomy 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 239000006274 endogenous ligand Substances 0.000 description 1
- 230000002357 endometrial effect Effects 0.000 description 1
- 201000006828 endometrial hyperplasia Diseases 0.000 description 1
- 239000002158 endotoxin Substances 0.000 description 1
- 230000037149 energy metabolism Effects 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 208000037828 epithelial carcinoma Diseases 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 230000008029 eradication Effects 0.000 description 1
- GTTBEUCJPZQMDZ-UHFFFAOYSA-N erlotinib hydrochloride Chemical compound [H+].[Cl-].C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 GTTBEUCJPZQMDZ-UHFFFAOYSA-N 0.000 description 1
- 229960005073 erlotinib hydrochloride Drugs 0.000 description 1
- 108010038795 estrogen receptors Proteins 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- 125000006437 ethyl cyclopropyl group Chemical group 0.000 description 1
- 125000000031 ethylamino group Chemical group [H]C([H])([H])C([H])([H])N([H])[*] 0.000 description 1
- 125000006125 ethylsulfonyl group Chemical group 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 201000004098 fibrolamellar carcinoma Diseases 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 229960000556 fingolimod Drugs 0.000 description 1
- KKGQTZUTZRNORY-UHFFFAOYSA-N fingolimod Chemical compound CCCCCCCCC1=CC=C(CCC(N)(CO)CO)C=C1 KKGQTZUTZRNORY-UHFFFAOYSA-N 0.000 description 1
- 239000000834 fixative Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 229960005304 fludarabine phosphate Drugs 0.000 description 1
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 229940013688 formic acid Drugs 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 239000000446 fuel Substances 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 125000003838 furazanyl group Chemical group 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- 201000000052 gastrinoma Diseases 0.000 description 1
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- 239000003349 gelling agent Substances 0.000 description 1
- 230000030279 gene silencing Effects 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 229950006191 gluconic acid Drugs 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 201000011066 hemangioma Diseases 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 1
- 125000005150 heteroarylsulfinyl group Chemical group 0.000 description 1
- 125000005143 heteroarylsulfonyl group Chemical group 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 150000002402 hexoses Chemical class 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 238000001794 hormone therapy Methods 0.000 description 1
- 102000054350 human CHI3L1 Human genes 0.000 description 1
- 229940042795 hydrazides for tuberculosis treatment Drugs 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 229960002899 hydroxyprogesterone Drugs 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 229960001001 ibritumomab tiuxetan Drugs 0.000 description 1
- 150000002454 idoses Chemical class 0.000 description 1
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 1
- 229960002411 imatinib Drugs 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000005965 immune activity Effects 0.000 description 1
- 238000011575 immunodeficient mouse model Methods 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 229910001411 inorganic cation Inorganic materials 0.000 description 1
- 206010022498 insulinoma Diseases 0.000 description 1
- 230000010468 interferon response Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 208000020082 intraepithelial neoplasia Diseases 0.000 description 1
- 239000011630 iodine Chemical group 0.000 description 1
- 235000014413 iron hydroxide Nutrition 0.000 description 1
- NCNCGGDMXMBVIA-UHFFFAOYSA-L iron(ii) hydroxide Chemical class [OH-].[OH-].[Fe+2] NCNCGGDMXMBVIA-UHFFFAOYSA-L 0.000 description 1
- 230000000622 irritating effect Effects 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 235000013847 iso-butane Nutrition 0.000 description 1
- PHTQWCKDNZKARW-UHFFFAOYSA-N isoamylol Chemical compound CC(C)CCO PHTQWCKDNZKARW-UHFFFAOYSA-N 0.000 description 1
- 125000003384 isochromanyl group Chemical group C1(OCCC2=CC=CC=C12)* 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 150000002574 ketohexoses Chemical class 0.000 description 1
- 150000002576 ketones Chemical group 0.000 description 1
- 238000011813 knockout mouse model Methods 0.000 description 1
- 239000004922 lacquer Substances 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 208000003849 large cell carcinoma Diseases 0.000 description 1
- 238000002430 laser surgery Methods 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 208000011080 lentigo maligna melanoma Diseases 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- DHMTURDWPRKSOA-RUZDIDTESA-N lonafarnib Chemical compound C1CN(C(=O)N)CCC1CC(=O)N1CCC([C@@H]2C3=C(Br)C=C(Cl)C=C3CCC3=CC(Br)=CN=C32)CC1 DHMTURDWPRKSOA-RUZDIDTESA-N 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 201000005296 lung carcinoma Diseases 0.000 description 1
- 201000000966 lung oat cell carcinoma Diseases 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229940099690 malic acid Drugs 0.000 description 1
- 208000030883 malignant astrocytoma Diseases 0.000 description 1
- 208000006178 malignant mesothelioma Diseases 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical class ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- 229960004296 megestrol acetate Drugs 0.000 description 1
- RQZAXGRLVPAYTJ-GQFGMJRRSA-N megestrol acetate Chemical compound C1=C(C)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 RQZAXGRLVPAYTJ-GQFGMJRRSA-N 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 210000004379 membrane Anatomy 0.000 description 1
- 230000015654 memory Effects 0.000 description 1
- 210000001806 memory b lymphocyte Anatomy 0.000 description 1
- 210000002418 meninge Anatomy 0.000 description 1
- 206010027191 meningioma Diseases 0.000 description 1
- 229960001810 meprednisone Drugs 0.000 description 1
- PIDANAQULIKBQS-RNUIGHNZSA-N meprednisone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)CC2=O PIDANAQULIKBQS-RNUIGHNZSA-N 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000004066 metabolic change Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229910052987 metal hydride Inorganic materials 0.000 description 1
- 150000004681 metal hydrides Chemical class 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 208000011645 metastatic carcinoma Diseases 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- GBMDVOWEEQVZKZ-UHFFFAOYSA-N methanol;hydrate Chemical compound O.OC GBMDVOWEEQVZKZ-UHFFFAOYSA-N 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- DGKBWLBTWDDRJX-UHFFFAOYSA-N methoxy-[methoxy(methyl)silyl]oxy-methylsilane Chemical class CO[SiH](C)O[SiH](C)OC DGKBWLBTWDDRJX-UHFFFAOYSA-N 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 125000006431 methyl cyclopropyl group Chemical group 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 229960004584 methylprednisolone Drugs 0.000 description 1
- 125000006216 methylsulfinyl group Chemical group [H]C([H])([H])S(*)=O 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 229960001566 methyltestosterone Drugs 0.000 description 1
- 239000011325 microbead Substances 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- VIJMMQUAJQEELS-UHFFFAOYSA-N n,n-bis(ethenyl)ethenamine Chemical compound C=CN(C=C)C=C VIJMMQUAJQEELS-UHFFFAOYSA-N 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N n-Octanol Natural products CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 230000001338 necrotic effect Effects 0.000 description 1
- 230000019261 negative regulation of glycolysis Effects 0.000 description 1
- 210000005170 neoplastic cell Anatomy 0.000 description 1
- 230000009826 neoplastic cell growth Effects 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 208000007538 neurilemmoma Diseases 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 201000000032 nodular malignant melanoma Diseases 0.000 description 1
- 231100000065 noncytotoxic Toxicity 0.000 description 1
- 125000005187 nonenyl group Chemical group C(=CCCCCCCC)* 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 101150075804 nqo1 gene Proteins 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- OIPZNTLJVJGRCI-UHFFFAOYSA-M octadecanoyloxyaluminum;dihydrate Chemical compound O.O.CCCCCCCCCCCCCCCCCC(=O)O[Al] OIPZNTLJVJGRCI-UHFFFAOYSA-M 0.000 description 1
- 125000004365 octenyl group Chemical group C(=CCCCCCC)* 0.000 description 1
- 230000004768 organ dysfunction Effects 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 239000003791 organic solvent mixture Substances 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- 201000008968 osteosarcoma Diseases 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 229940116315 oxalic acid Drugs 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 230000036284 oxygen consumption Effects 0.000 description 1
- 238000010979 pH adjustment Methods 0.000 description 1
- 238000011499 palliative surgery Methods 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 208000021255 pancreatic insulinoma Diseases 0.000 description 1
- 201000005163 papillary serous adenocarcinoma Diseases 0.000 description 1
- 208000024641 papillary serous cystadenocarcinoma Diseases 0.000 description 1
- 208000003154 papilloma Diseases 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 230000010412 perfusion Effects 0.000 description 1
- 125000005327 perimidinyl group Chemical group N1C(=NC2=CC=CC3=CC=CC1=C23)* 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 125000004934 phenanthridinyl group Chemical group C1(=CC=CC2=NC=C3C=CC=CC3=C12)* 0.000 description 1
- 125000004625 phenanthrolinyl group Chemical group N1=C(C=CC2=CC=C3C=CC=NC3=C12)* 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 208000010916 pituitary tumor Diseases 0.000 description 1
- BLFWHYXWBKKRHI-JYBILGDPSA-N plap Chemical compound N([C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(O)=O)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(=O)[C@@H]1CCCN1C(=O)[C@H](CO)NC(=O)[C@@H](N)CCC(O)=O BLFWHYXWBKKRHI-JYBILGDPSA-N 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 108091033319 polynucleotide Proteins 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 238000010837 poor prognosis Methods 0.000 description 1
- 230000025572 positive regulation of antigen processing and presentation Effects 0.000 description 1
- 235000011118 potassium hydroxide Nutrition 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 230000037452 priming Effects 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 230000005588 protonation Effects 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- ZVJHJDDKYZXRJI-UHFFFAOYSA-N pyrroline Natural products C1CC=NC1 ZVJHJDDKYZXRJI-UHFFFAOYSA-N 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000004151 quinonyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- SBYHFKPVCBCYGV-UHFFFAOYSA-N quinuclidine Chemical compound C1CC2CCN1CC2 SBYHFKPVCBCYGV-UHFFFAOYSA-N 0.000 description 1
- 239000002534 radiation-sensitizing agent Substances 0.000 description 1
- 239000002516 radical scavenger Substances 0.000 description 1
- 238000011127 radiochemotherapy Methods 0.000 description 1
- 229960004622 raloxifene Drugs 0.000 description 1
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000003753 real-time PCR Methods 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 239000012925 reference material Substances 0.000 description 1
- 208000016691 refractory malignant neoplasm Diseases 0.000 description 1
- 210000003289 regulatory T cell Anatomy 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000008261 resistance mechanism Effects 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 1
- 229940100486 rice starch Drugs 0.000 description 1
- 229920002477 rna polymer Polymers 0.000 description 1
- 235000019515 salmon Nutrition 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 206010039667 schwannoma Diseases 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 230000001235 sensitizing effect Effects 0.000 description 1
- 238000011452 sequencing regimen Methods 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 208000004548 serous cystadenocarcinoma Diseases 0.000 description 1
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 210000004872 soft tissue Anatomy 0.000 description 1
- 239000012439 solid excipient Substances 0.000 description 1
- 238000007711 solidification Methods 0.000 description 1
- 230000008023 solidification Effects 0.000 description 1
- NHXLMOGPVYXJNR-ATOGVRKGSA-N somatostatin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N1)[C@@H](C)O)NC(=O)CNC(=O)[C@H](C)N)C(O)=O)=O)[C@H](O)C)C1=CC=CC=C1 NHXLMOGPVYXJNR-ATOGVRKGSA-N 0.000 description 1
- 229960000553 somatostatin Drugs 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 210000004989 spleen cell Anatomy 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 230000010473 stable expression Effects 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 238000011301 standard therapy Methods 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 1
- PXQLVRUNWNTZOS-UHFFFAOYSA-N sulfanyl Chemical compound [SH] PXQLVRUNWNTZOS-UHFFFAOYSA-N 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- JLKIGFTWXXRPMT-UHFFFAOYSA-N sulphamethoxazole Chemical compound O1C(C)=CC(NS(=O)(=O)C=2C=CC(N)=CC=2)=N1 JLKIGFTWXXRPMT-UHFFFAOYSA-N 0.000 description 1
- 229910021653 sulphate ion Inorganic materials 0.000 description 1
- 208000030457 superficial spreading melanoma Diseases 0.000 description 1
- 230000000153 supplemental effect Effects 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 238000011477 surgical intervention Methods 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 230000007761 synergistic anti-cancer Effects 0.000 description 1
- 230000005737 synergistic response Effects 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 101150047061 tag-72 gene Proteins 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- ISIJQEHRDSCQIU-UHFFFAOYSA-N tert-butyl 2,7-diazaspiro[4.5]decane-7-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCCC11CNCC1 ISIJQEHRDSCQIU-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 238000011287 therapeutic dose Methods 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000004627 thianthrenyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3SC12)* 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- RTKIYNMVFMVABJ-UHFFFAOYSA-L thimerosal Chemical compound [Na+].CC[Hg]SC1=CC=CC=C1C([O-])=O RTKIYNMVFMVABJ-UHFFFAOYSA-L 0.000 description 1
- 229940033663 thimerosal Drugs 0.000 description 1
- BRNULMACUQOKMR-UHFFFAOYSA-N thiomorpholine Chemical compound C1CSCCN1 BRNULMACUQOKMR-UHFFFAOYSA-N 0.000 description 1
- PLHJCIYEEKOWNM-HHHXNRCGSA-N tipifarnib Chemical compound CN1C=NC=C1[C@](N)(C=1C=C2C(C=3C=C(Cl)C=CC=3)=CC(=O)N(C)C2=CC=1)C1=CC=C(Cl)C=C1 PLHJCIYEEKOWNM-HHHXNRCGSA-N 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- 229960005267 tositumomab Drugs 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- IUCJMVBFZDHPDX-UHFFFAOYSA-N tretamine Chemical compound C1CN1C1=NC(N2CC2)=NC(N2CC2)=N1 IUCJMVBFZDHPDX-UHFFFAOYSA-N 0.000 description 1
- 229950001353 tretamine Drugs 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 125000006168 tricyclic group Chemical group 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229940086984 trisenox Drugs 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 230000005751 tumor progression Effects 0.000 description 1
- 239000000225 tumor suppressor protein Substances 0.000 description 1
- 210000003171 tumor-infiltrating lymphocyte Anatomy 0.000 description 1
- 238000009281 ultraviolet germicidal irradiation Methods 0.000 description 1
- 208000010576 undifferentiated carcinoma Diseases 0.000 description 1
- 238000011870 unpaired t-test Methods 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 230000001457 vasomotor Effects 0.000 description 1
- 239000013598 vector Substances 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 229940099039 velcade Drugs 0.000 description 1
- 208000008662 verrucous carcinoma Diseases 0.000 description 1
- PXXNTAGJWPJAGM-UHFFFAOYSA-N vertaline Natural products C1C2C=3C=C(OC)C(OC)=CC=3OC(C=C3)=CC=C3CCC(=O)OC1CC1N2CCCC1 PXXNTAGJWPJAGM-UHFFFAOYSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 229940100445 wheat starch Drugs 0.000 description 1
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 1
- 229940053867 xeloda Drugs 0.000 description 1
- 125000004933 β-carbolinyl group Chemical group C1(=NC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
Images








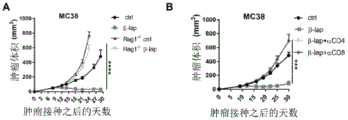
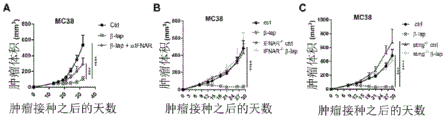


















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/352—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom condensed with carbocyclic rings, e.g. methantheline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/352—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom condensed with carbocyclic rings, e.g. methantheline
- A61K31/353—3,4-Dihydrobenzopyrans, e.g. chroman, catechin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2818—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against CD28 or CD152
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/158—Expression markers
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Immunology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Microbiology (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Endocrinology (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Mycology (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
Abstract
本文中所述治疗可以在对象中使多种不同的癌细胞类型和癌症病症选择性地致死。本文中所述组合治疗可用于疾病的管理、治疗、控制或辅助治疗,其中选择性致死性在免疫治疗中是有益的,特别是在其中疾病伴有升高的NQO1水平的情况下。特别是其中免疫治疗(例如检查点抑制剂)与NQO1生物可激活药物组合的一些实施方案。
Description
优先权要求
本申请要求于2019年3月18日提交的美国临时申请序列号62/819,870的优先权的权益,其全部内容通过引用并入本文。
政府支持
本发明是在国立卫生研究院(National Institutes of Health)授予的合同编号R01 CA102792-18下的政府支持下完成的。政府在本发明中具有一定权利。
背景
I.技术领域
本公开内容的领域总体上涉及药物化学、医学、肿瘤学、化学治疗和免疫治疗。更具体地,其涉及与检查点抑制剂组合的NQO1生物可激活药物(NQO1 bioactivable drug)在治疗癌症中的用途。
II.背景技术
抗程序性细胞死亡蛋白1(Programmed Cell Death Protein 1,PD-1)及其配体程序性死亡配体1(Programmed Death-Ligand 1,PDL1)单克隆抗体在数种不同癌症类型中显示出前所未有的持久应答,并且这些最初的临床成功突出了癌症免疫治疗领域。阻断PD-1与其配体PD-L1之间的相互作用导致T细胞存活和增殖提高并且T细胞耗竭降低,恢复细胞毒性T细胞功能以促进抗肿瘤免疫应答(Topalian et al.,2015;Pauken和Wherry,2015)。遗憾的是,用抗PD1/PD-L1药剂治疗的仅少数患者具有持久应答(Sharma et al.,2017)。虽然其他检查点可促进低应答和复发,但对T细胞的另外的检查点阻断没有提高应答率(Jenkins et al.,2018;Garber,2018)。肿瘤微环境内功能失调的抗原呈递细胞可能会限制T细胞的最佳活化(Lee et al.,2009)。虽然肿瘤抗原可在肿瘤环境中获得,但抗原加工和呈递缺乏适当激活可能会限制肿瘤特异性T细胞被再活化。
有效适应性免疫的产生需要协调的固有免疫应答,包括感知危险或损伤信号以使固有细胞(即树突细胞巨噬细胞和自然杀伤细胞)活化、抗原加工和呈递、I型IFN产生和T细胞的交叉致敏(Woo和Corrales,2015)。一般而言,这些危险或损伤信号被固有免疫细胞表达的胞外和胞内模式识别受体(pattern recognition receptor,PRR)识别,并促进对抗原的摄取,使抗原呈递细胞(antigen-presenting cell,APC)活化,并促进APC与损伤细胞之间的相互作用(Takeuchi和Akira,2010)。然而,正常固有免疫应答的这些关键特性通常在肿瘤微环境中被破坏(Patel和Minn,2018)。例如,癌症普遍有利于缺乏或不能呈递足够新生抗原的肿瘤克隆的存活;肿瘤也更倾向于促进癌症炎症的PRR信号或功能失调的固有免疫细胞而不是致敏适应性应答(Lee et al.,2009;Hernandez et al.,2016;Grivennikovet al.,2010)。
由于肿瘤表现出有利于免疫抑制性微环境的固有感知受损,因此改善检查点阻断的一个重要考虑是增强肿瘤微环境中的固有信号。实现该目标的一种可能方法涉及在肿瘤微环境中诱导免疫原性细胞死亡(immunogenic cell death,ICD)(Patel和Minn,2018)。例如,许多DNA损伤或DNA修复抑制化学治疗(例如放射治疗、蒽环类和奥沙利铂(oxaliplatin))引发ICD(Garg et al.,2017)。ICD的特征在于通过垂死肿瘤细胞释放一系列的免疫刺激性损伤相关分子模式(damage-associated molecular pattem,DAMP),例如高迁移率族框1(high mobility group box 1,HMGB1)蛋白、胞外ATP、胞质钙网蛋白和内源性核酸(Sistigu et al.,2014)。这些DAMP由固有细胞表达的其同源PRR识别。这种DAMP/PRR信号传导改变炎性微环境和/或刺激新生抗原产生,并吸引和活化APC以使T细胞活化,所述T细胞现在可攻击肿瘤(Galluzzi et al.,2015)。因此,免疫刺激性特性使ICD诱导剂成为组合免疫治疗的有吸引力的候选物。然而,仅少数细胞毒性药物被确定为ICD诱导剂,并且一般毒性、免疫抑制性质和缺乏肿瘤选择性限制了它们的使用。因此,高度期望探索“靶向”药剂是否可以更特异性地提高固有感知并随后扩大抗PD1/PD-L1治疗的益处。
NAD(P)H:醌氧化还原酶1(NAD(P)H:quinone oxidoreductase 1,NQO1)是一种胞质双电子氧化还原酶,其在许多人癌症中上调(Li et al.,1995),所述癌症包括结直肠癌、肺癌、黑素瘤、胆管癌和胰腺癌(Oh et al.,2016)。NQO1的高水平表达与晚期临床分期、不良预后和淋巴结转移相关(Li et al.,2015;Ma et al.,2014)。NQO1生物可激活药物(包括β拉帕醌(β-lap,临床形式,ARQ761))具有可被NQO1催化以产生活性氧物质(reactiveoxygen species,ROS)的独特醌结构(Huang et al.,2016)。一般而言,一摩尔β-lap产生约120摩尔的超氧化物,在约2分钟内消耗约60摩尔的NAD(P)H(Pink et al.,2000)。NQO1在肿瘤细胞中过表达,并且与正常组织相比,过氧化氢酶(过氧化氢(H2O2)清除酶)在肿瘤组织中缺失(Doskey et al.,2016)。人癌症中的高NQO1:过氧化氢酶比可为NQO1“生物可激活”药物的使用提供最佳治疗窗,而低表达比保护正常组织。加强的肿瘤特异性ROS产生导致广泛的氧化性DNA病变和肿瘤选择性细胞死亡(Huang et al.,2012)。已经表明NQO1生物可激活β-lap导致未修复的DNA损伤和细胞死亡,并在异种移植模型中与PARP1抑制剂和放射治疗协同(Huang et al.,2016;Li et al.,2016)。β-Lap目前正在患有NQO1+实体瘤的患者中以单一治疗或与其他化学药物组合进行测试(ClinicalTrials.gov标识符NCT02514031和NCT01502800)。然而,β-lap抗肿瘤效力的评价主要在体外和在免疫缺陷型小鼠模型中进行,并且改善治疗通常专注于增强直接肿瘤杀伤,而很少关注适应性免疫。本发明人最近表明,β-lap触发免疫原性细胞死亡(ICD)并诱导损伤相关分子模式(DAMP)释放,其激活宿主TLR4/MyD88/I型干扰素途径和Batf3树突细胞依赖性交叉致敏,以桥接固有免疫应答和针对NQO1阳性肿瘤的适应性免疫应答(Li et al.,2019)。此外,他们发现β-lap触发肿瘤微环境内的固有感知,以在充分建立的肿瘤中克服检查点阻断抗性(Li et al.,2019)。异丁基-脱氧苯醌(isobutyl-deoxynyboquinone,IB-DNQ)是NAD(P)H:醌氧化还原酶(NQO1)(在许多实体瘤中过表达的酶)的新选择性底物。IB-DNQ是靶向NQO1阳性实体癌的有前景且强效的抗癌剂(Lundberg et al.,2017)。
发明概述
因此,根据本公开内容,提供了在患有癌症的患者中杀伤癌细胞或抑制癌细胞生长的方法,其包括施用与检查点抑制剂组合的NQO1生物可激活药物。还提供了与第二药剂组合的NQO1生物可激活药物用于制备用于在具有癌细胞的患者中杀伤癌细胞或抑制癌细胞生长的药物的用途,其中所述第二药剂是检查点抑制剂,其中所述药物包含有效致死或抑制量的NQO1生物可激活药物和检查点抑制剂。还提供了与第二药剂组合的NQO1生物可激活药物在治疗患有癌症的患者中的用途,其中第二药剂是检查点抑制剂。该方法还可包括另外的抗癌治疗,例如化学治疗、放射治疗、免疫治疗、毒素治疗或手术。
所述癌细胞可具有由于错误DNA修复过程引起的碱基切除修复(base excisionrepair,BER)缺陷或脆弱性(vulnerability)。BER缺陷或脆弱性可包含X射线交叉互补1或XRCC1基因/蛋白/酶的水平缺陷。NQO1生物可激活药物可与小分子检查点抑制剂或抗体检查点抑制剂组合使用。NQO1生物可激活药物可与PD-1或CTLA-4的抑制剂组合使用。NQO1生物可激活药物可以是β-拉帕醌或DNQ化合物。该方法可还包括另外的化学治疗剂或放射治疗。癌细胞可具有升高的NQO1水平。癌细胞可以是实体瘤形式。癌细胞可以是非小细胞肺癌细胞、前列腺癌细胞、胰腺癌细胞、乳腺癌细胞、头颈癌细胞或结肠癌细胞。
NQO1生物可激活药物可以是以下或其盐或溶剂合物:


NQO1生物可激活药物可以是DNQ或DNQ-87。
NQO1生物可激活药物可在检查点抑制剂之前、在检查点抑制剂之后或与检查点抑制剂同时施用。NQO1生物可激活药物可施用多于一次。检查点抑制剂可施用多于一次。NQO1生物可激活药物和检查点抑制剂二者均可施用多于一次。
本领域普通技术人员将理解,在本公开内容的实践中可使用除具体例示的那些之外的起始物料、生物材料、试剂、合成方法、纯化方法、分析方法、测定方法和生物方法,而无需借助于过度实验。任何这样的材料和方法的所有本领域已知的功能等效物均旨在包含在本公开内容中。
已采用的术语和表述作为描述而非限制的术语使用,并且使用这样的术语和表述不旨在排除所示出和所描述的特征的任何等同形式或其一部分,而是应认识到,在所要求保护的公开内容的范围内可进行多种修改。
因此,应当理解,尽管已经通过特定实施方案和任选的特征具体公开了本公开内容,但是本领域技术人员可以对本文中公开的概念进行修改和变化,并且这样的修改和变化被认为是在由所附权利要求书限定的本公开内容的范围内。
附图简述
下列附图构成说明书的一部分并被包括在内以进一步说明本发明的某些实施方案或多个方面。在一些情况下,本发明的实施方案可通过参考与本文中呈现的详细描述组合的附图来最好地理解。描述和附图可突出本发明的某个具体实施例或某个方面。然而,本领域技术人员将理解,实施例或方面的一部分可与本发明的其他实施例或方面组合使用。
图1A至J。NQO1生物可激活药物β-lap在体外和在体内以NQO1依赖性方式杀伤鼠肿瘤细胞。(图1A)将在48孔板中培养的NQO1阳性肿瘤细胞系MC38、TC-1和Ag104Ld以及NQO1阴性细胞系Panc02和B16用β-lap(0至8μM)处理3小时,随后洗涤并更换培养基。在4天之后通过磺酰罗丹明B(Sulforhodamine B,SRB)测定来确定细胞生存力。(图1B)将MC38、TC-1和Ag104Ld细胞暴露于4μMβ-lap±双香豆素(DIC,50μM)3小时,并在4天之后评估细胞存活。(图1C)将在96孔板中平板接种的具有基于CRISPR的NQO1敲除的MC38细胞(MC38 NQO1KO#5)暴露于β-lap 3小时,并在48小时之后评估细胞存活。(图1D)将在96孔板中平板接种的稳定携带pCMV-NQO1表达载体的B16细胞(B16 NQO1#1)暴露于β-lap±双香豆素(DIC,50μM)3小时,并在48小时之后评估存活。(图1E)将MC38细胞暴露于致死剂量的β-lap(4μM)指定时间,然后用7-AAD和膜联蛋白V进行染色,随后进行流式细胞术分析。(F至G)将MC38细胞(图1F)或B16和B16NQO1#1细胞(图1G)暴露于β-lap±双香豆素(DIC,50μM)3小时,并随后通过DCFDA细胞ROS测定来确定ROS水平。(图1H)将MC38和B16 NQO1#1细胞暴露于β-lap 3小时。添加过氧化氢酶(1000U/ml),并在48小时之后评估细胞存活。(图1I)将C57BL/6小鼠(n=5只/组)移植MC38细胞并用β-lap(0.03mg、0.1mg或0.3mg,瘤内;或25mg/kg,i.v.)每隔一天处理四次。(图1J)将C57BL/6小鼠移植MC38细胞(NQO1 WT或KO,n=5只/组)并用β-lap(0.3mg,i.t.)每隔一天处理四次。每周两次监测肿瘤生长。数据示出为来自两至三个独立实验的平均值±SEM。**P<0.01,***P<0.001,****P<0.0001,由未配对student t检验(图1F、图1G和图1H)或双因素ANOVA(图1I和图1J)确定。
图2A至F。β-Lap的抗肿瘤作用是CD8+ T细胞依赖性的。(图2A)将MC38细胞分别皮下移植到C57BL/6WT(n=5至6只/组)和Rag1 KO小鼠(n=5只/组)中。将荷瘤小鼠用β-lap(0.3mg,i.t.)每隔一天处理四次。示出了在处理之后无肿瘤小鼠的数目。(图2B)将荷MC38肿瘤的C57BL/6小鼠(n=5只/组)用β-lap(0.3mg,i.t.)每隔一天处理四次。对于CD8+ T细胞耗竭,在处理期间以三天间隔腹膜内注射200μg抗CD8抗体四次。(图2C)在有或没有抗CD8抗体的情况下,将荷TC-1肿瘤的C57BL/6小鼠(n=4至5只/组)用β-lap(0.1mg,i.t.)处理四次。(图2D)在完全排斥之后30天,在距原发性肿瘤的相对位点用3×106个MC38细胞对初始(n=5只/组)和经β-lap治愈的无MC38肿瘤(n=7只/组)C57BL/6小鼠皮下再攻击,并检测肿瘤生长曲线。(图2E)将2×106个A549细胞皮下注射到C57BL/6 Rag1-/-小鼠(对于载剂和β-lap,n=5只/组;对于载剂+OT-1和β-lap+OT-1,n=7只/组)中。在30天之后,将小鼠i.v.过继性转染来自OT-1转基因小鼠的2×106个淋巴结细胞。在第二天,将荷瘤小鼠用β-lap(0.2mg)每隔一天瘤内处理4次。(图2F)将A549细胞皮下注射到NSG-SGM3(n=5只/组)或携带人CD34+造血干细胞的NSG-SGM3中(对于Hu-NSG载剂,n=5只/组;对于Hu-NSGβ-lap,n=6只/组)。将荷瘤小鼠用β-lap(0.2mg,i.t.)每隔一天处理四次。每周两次测量肿瘤生长。数据示出为来自三个独立实验的平均值±SEM。**P<0.01,***P<0.001,****P<0.0001,通过双因素ANOVA确定。
图3A至E。β-lap的抗肿瘤作用需要Batf3依赖性树突细胞介导的T细胞交叉致敏。(图3A)将荷MC38肿瘤的C57BL/6小鼠(n=5只/组)用β-lap(0.3mg,i.t.)每隔一天处理3次,并在第一次处理之后10天,分离来自脾的淋巴细胞并将其用培养基或用60Gy辐照的MC38细胞进行刺激。(图3B)将荷MC38-OVA肿瘤的小鼠(n=4只/组)用β-lap(0.3mg,i.t.)每隔一天处理3次,并在第一次处理之后10天,分离来自脾的淋巴细胞并将其用2.5μg/ml OT-1肽进行刺激。通过ELISPOT测定来确定IFNγ产生细胞。(图3C)将荷MC38肿瘤的C57BL/6小鼠(n=5只/组)用β-lap(0.3mg,i.t.)每隔一天处理四次。在处理期间,以三天间隔瘤内注射100μg抗CSF1R Ab三次。(图3D)将MC38细胞分别皮下移植到C57BL/6 WT(n=5只/组)和Batf3-/-小鼠(对于Batf3-/-载剂,n=5只/组;对于Batf3-/-β-lap,n=6只/组)中。将荷瘤小鼠用β-lap(0.3mg,i.t.)每隔一天处理四次。每周两次监测肿瘤生长。(图3E)将荷MC38-OVA的小鼠(n=3只/组)用β-lap(0.3mg,i.t.)处理1次,并在4天之后,从肿瘤引流淋巴结中纯化CD11c+树突细胞,并将其与从OT-1转基因小鼠的脾中分离的CD8 T细胞共培养。通过流式微珠阵列(Cytometric Bead Array,CBA)小鼠IFNγ测定通过细胞分泌的IFNγ的水平来确定T细胞交叉致敏的活性。数据示出为来自三个独立实验的平均值±SEM。**P<0.01,***P<0.001,****P<0.0001,由未配对student t检验(图3A、3B和3E)或双因素ANOVA(图3C和3D)确定。
图4A至F。β-lap和肿瘤特异性CTL的抗肿瘤作用需要I型IFN和TLR4/MyD88/信号传导。(图4A)将荷MC38肿瘤的C57BL/6小鼠(n=5只/组)用β-lap(0.3mg,i.t.)每隔一天处理四次。在处理期间每四天施用抗IFNAR阻断抗体(150μg,i.t.)3次。(图4B)将荷MC38肿瘤的WT(n=5只/组)和Ifnar1-/-(n=4只/组)C57BL/6小鼠用β-lap(0.3mg,i.t.)每隔一天处理四次。(图4C)将荷MC38肿瘤的WT(n=5只/组)和Myd88-/-(n=3只/组)C57BL/6小鼠用β-lap(0.3mg,i.t.)每隔一天处理四次。(图4D)将荷MC38肿瘤的WT(n=5只/组)和Tlr4-/-(n=4只/组)C57BL/6小鼠用β-lap(0.3mg,i.t.)每隔一天处理四次。(图4E)将荷MC38肿瘤的C57BL/6小鼠(n=5只/组)用β-lap(0.3mg,i.t.)每隔一天处理四次。在处理期间,每三天施用抗HMGB1中和抗体(200μg,i.p)三次。每周两次监测肿瘤生长。(图4F)将荷MC38肿瘤的WT(n=4只/组)或Tlr4-/-(n=4只/组)或MyD88-/-(n=6只/组)C57BL/6小鼠用β-lap(0.3mg,i.t.)每隔一天处理四次。在处理期间,每三天施用抗HMGB1中和抗体(200μg,i.p)三次。在初始处理之后12天,分离来自TdLN的淋巴细胞并将其用60Gy辐照的MC38肿瘤细胞进行刺激。通过ELISPOT测定来确定IFNγ产生细胞。数据示出为来自两至三个独立实验的平均值±SEM。*P<0.05,**P<0.01,***P<0.001,****P<0.0001,通过双因素ANOVA确定。
图5A至F。β-Lap处理诱导的HMGB1释放在体内增强了肿瘤免疫原性并触发了抗肿瘤T细胞免疫。(图5A)将MC38、TC-1和B16(NQO无效(null)和过表达克隆)用β-lap处理3小时,随后洗涤并更换培养基。在24小时之后通过ELISA确定释放到培养上清液中的HMGB1的水平。(图5B)在图5C和图5D中来自β-lap诱导的垂死肿瘤细胞的肿瘤特异性抗原在体内交叉呈递的研究方案。(图5C至D)在有或没有抗-HMGB1抗体的情况下,将活的或β-lap诱导的垂死MC38-OVA细胞皮下接种到WT或Tlr4-/- C57BL/6(n=3至4只/组)小鼠的胁腹中。在5天之后,收集肿瘤引流淋巴结细胞并用OVA蛋白、OT-1肽或经辐照的MC38-OVA细胞再刺激48小时。通过ELISPOT测定来确定IFNγ产生细胞(图5C),并且通过CBA小鼠IFNγ测定对IFNγ分泌水平进行定量(图5D)。(图5E)图5F中免疫原性疫苗测定的研究方案。(图5F)将在体外用β-lap处理的MC38-OVA细胞在有或没有抗HMGB1抗体的情况下s.c.接种到C57BL/6小鼠(n=5至7只/组)的胁腹中。在7天之后,将小鼠用活MC38-OVA细胞通过注射到对侧胁腹中再攻击。示出了再攻击的无肿瘤小鼠的百分比。数据示出为来自至少两至三个独立实验的平均值±SEM。*P<0.05,**P<0.01和***P<0.001,通过双因素ANOVA(图5C和图5D)或对数秩检验(图5F)确定。
图6A至H。β-Lap通过与抗PD-L1治疗组合根除了大的建立的和检查点阻断难治性肿瘤。(图6A)图6B和图6C中基于局部β-lap处理的组合治疗的处理方案。(图6B至C)将MC38肿瘤细胞s.c.接种到C57BL/6小鼠(n=5只/组)的胁腹中。在有或没有基于抗PD-L1的检查点阻断的情况下,将荷小肿瘤(约50mm3,B)或晚期肿瘤(约150至200mm3,图6C)的小鼠用β-lap(0.3mg,i.t.)局部处理四次。每周两次监测肿瘤生长,并示出了在处理之后无肿瘤小鼠的数目。(图6D)图6E和图6F中基于全身β-lap处理的组合治疗的处理方案。(E)将MC38肿瘤细胞s.c.接种到C57BL/6小鼠(n=5至8只/组)的胁腹中。在有或没有基于抗PD-L1的检查点阻断的情况下,将荷瘤小鼠(约50至100mm3)用β-lap(30mg/kg,i.p.)全身处理六次。每周两次监测肿瘤生长,并示出了处理之后无肿瘤小鼠的数目。(图6F)图6E中组合处理下荷MC38肿瘤的小鼠的存活曲线。(图6G、图6H)将荷MC38-OVA肿瘤(约150mm3,n=4只/组)的C57BL/6小鼠用β-lap(0.3mg,i.t.)每隔一天局部处理4次,或用抗PD-L1(100μg,i.p.)处理3次,作为单独或组合。在第一次处理之后12天,通过流式细胞术分析肿瘤浸润性CD45+细胞和淋巴细胞(图6G),并通过IFNγELISPOT测定来确定脾中的OT-1抗原特异性T细胞(图6H)。数据示出为来自两至三个独立实验的平均值±SEM。*P<0.05,**P<0.01,***P<0.001,****P<0.0001,通过双因素ANOVA(图6B、图6C、图6E、图6G和图6H)或对数秩检验(图6F)确定。
图7。用于通过NQO1生物可激活药物提高靶向治疗的抗肿瘤免疫应答激活的拟建模型。
图8A至E。β-lap诱导肿瘤特异性ROS并引起胱天蛋白酶非依赖性程序性坏死。(图8A)如所示对多个鼠肿瘤系中的NQO1表达进行免疫印迹。(图8B)将NQO1+细胞(MC38、TC-1和Ag104Ld)和NQO1-细胞(B16和Pan02)用β-lap±DIC(50μM)处理3小时。去除药物并在5天之后评估存活。(图8C)使用CellRox-Glo在用β-lap以指定剂量处理3小时的多种细胞中评估相对H2O2水平。将值相对于经DMSO处理的对照细胞进行归一化。(图8D)将TC-1细胞暴露于β-lap 12小时,之后通过western印迹测定来分析PARP-1和胱天蛋白酶3水平。(图8E)暴露于β-lap(4μM)12小时的TC-1细胞的照片。
图9A至B。β-lap的抗肿瘤功能依赖于CD8+ T细胞,而不是CD4+ T细胞。(图9A)将C57BL/6WT或rag-/-小鼠(n=5只/组)皮下(s.c.)接种6×105个MC38细胞并在第9天、第12天和第15天用0.3mg的β-lap或载剂进行瘤内(intratumorally,i.t.)处理。(图9B)如上所述将荷MC38肿瘤的C57BL/6小鼠用β-lap处理。从第8天开始,每周两次腹膜内施用CD4耗竭(克隆GK1.5)或CD8耗竭(克隆2.43)抗体(200μg)。每周两次测量肿瘤生长。*P<0.05,**P<0.01,***P<0.001,****P<0.0001,通过双因素ANOVA确定。
图10A至C。β-Lap诱导依赖于STING依赖性DNA感知和I型IFN信号传导来肿瘤消退。(图10A)将C57BL/6WT小鼠(n=5只/组)皮下(s.c.)接种6×105个MC38细胞并在第14天、第16天、第18天和第20天用0.1mg的β-lap或载剂进行瘤内(i.t.)处理。从第13天开始,每周两次i.p.施用抗IFNAR阻断Ab(150μg)。(图10B至C)将C57BL/6WT IFNAR-/-或STINGmut/mut小鼠(n=5只/组)皮下(s.c.)接种MC-38细胞并在第9天、第12天和第15天用0.3mgβ-lap或载剂进行瘤内(i.t.)处理。*P<0.05,**P<0.01,***P<0.001,****P<0.0001,通过双因素ANOVA确定。
图11A至D。β-Lap诱导的嗜中性粒细胞浸润有助于抗肿瘤免疫应答。(图11A至B)在β-lap处理3天之后,从MC38(图11A)或TC-1(图11B)肿瘤组织进行单细胞。然后通过流式细胞术来分析CD11b+Gr1+细胞的频率。(图11C至D)将C57BL/6WT小鼠(n=5只/组)皮下(s.c.)接种6×105个MC38(图11C)或1×105个TC-1(图11D)细胞并在第9天、第12天和第15天用0.3(mg)的β-lap或载剂进行瘤内(i.t.)处理。从第8天开始,每周两次腹膜内施用抗Ly6G阻断Ab(200μg)。每周两次监测肿瘤生长。数据示出为来自两至三个独立实验的平均值±SEM。**P<0.01,***P<0.001,****P<0.0001,通过双因素ANOVA确定。
图12A至B。低剂量的β-Lap与免疫检查点阻断(抗PD-L1)治疗协同。(图12A)将C57BL/6J WT小鼠(n=5只/组)皮下(s.c.)接种6×105个MC38细胞,并用载剂或0.1mgβ-Lap在有或没有抗PD-L1(阿特珠单抗(Atezolizumab))的情况下每三天瘤内(i.t.)处理,4次注射。当肿瘤体积>100mm3时开始处理。在β-Lap处理之前一天,腹膜内(i.p.)注射100μg抗PD-L1(克隆10F.9G2)或同种型对照抗体(克隆LTF-2)。(图12B)将荷相同MC38肿瘤的小鼠用载剂或30mg/μgβ-Lap在有或没有抗PD-L1(阿特珠单抗)的情况下每三天腹膜内(i.p.)处理,6次注射。当肿瘤体积>50mm3时开始处理。每周两次通过卡尺测量肿瘤直径。肿瘤体积表示为平均值±SD。*p<0.05,**p<0.01,***P<0.001。
图13A至B。低剂量β-lap对皮下鼠癌症的辐射敏化。(图13A)将C57BL/6J WT小鼠(n=5只/组)皮下(s.c.)接种6×105个MC38细胞并用载剂、IR(10Gy)或0.1mgβ-lap在有或没有IR的情况下每三天瘤内(i.t.)处理,3次注射。当肿瘤体积>100mm3时开始处理。(图13B)将荷相同MC38肿瘤的小鼠用载剂IR(10Gy)或30mg/kgβ-Lap在有或没有IR的情况下每隔一天腹膜内(i.p.)处理,6次注射。当肿瘤体积>50mm3时开始处理。在β-lap处理之前给予IR。每周两次通过卡尺测量肿瘤直径。肿瘤体积表示为平均值±SD。*p<0.05,**p<0.01,***P<0.001。
图14A至H。IB-DNQ以NQO1依赖性方式杀伤鼠癌细胞并诱导NAD+/ATP耗竭和DNA损伤。(图14A至C)将NQO1+细胞MC38(A)和TC-1(B)和NQO1-细胞B16(C)用不同剂量的IB-DNQ±DIC(50μM)处理2小时。去除药物并在6天之后评估存活。(图14D)如所示对多种鼠肿瘤系中的NQO1表达进行免疫印迹。(图14E至F)在暴露于不同剂量的IB-DNQ 2小时的TC-1细胞中评估相对NAD+(E)或ATP(F)水平。将值相对于经DMSO处理的对照细胞进行归一化。(图14G至H)将TC-1细胞暴露于IB-DNQ(0.25μM)60分钟,使用碱性彗星测定(comet assay)评估细胞的总DNA病变(图14G)。在指定时间监测以a.u.计的彗星尾长度;以及在指定时间使用免疫荧光通过γH2AX病灶/细胞核定量的DSB(图14H)。所绘的是来自图14A至C、图14E和图14F中三个实验的平均值±SD。进行了Student t检验。*p<0.05,**p<0.01,***P<0.001。
图15A至B。IB-DNQ依赖于适应性免疫系统来诱导肿瘤消退。(图15A)将C57BL/6JWT或rag-/-小鼠(n=5只/组)皮下(s.c.)接种6×105个MC38细胞,并在第10天、第12天、第14天和第16天用0.15mg的IB-DNQ或载剂进行瘤内(i.t.)处理。(图15B)将C57BL/6J WT或rag-/-小鼠(n=5只/组)皮下(s.c.)接种1×105个TC-1细胞,并在第10天、第12天、第14天和第16天用0.15mg的IB-DNQ或载剂进行瘤内(i.t.)处理。每周两次通过卡尺测量肿瘤直径。肿瘤体积表示为平均值±SD。*p<0.05,**p<0.01,***P<O.001。
图16A至B。IB-DNQ与免疫检查点阻断(抗PD-L1)治疗协同。将C57BL/6J WT小鼠(n=5只/组)皮下(s.c.)接种6×105个MC38细胞,并在第10天、第13天、第16天和第19天用载剂、抗PD-L1(阿特珠单抗)或0.05mg IB-DNQ在有或没有抗PD-L1的情况下进行瘤内(i.t.)处理。从第9天开始,每三天腹膜内(i.p.)注射100μg的抗PD-L1(克隆10F.9G2)或同种型对照抗体(克隆LTF-2)。每周两次通过卡尺测量肿瘤直径。当肿瘤体积达到1000mm3时处死小鼠。(图16A)代表性的小鼠肿瘤体积(平均值±SD)。(图16B)Kaplan-Meier存活曲线。*p<0.05,**p<0.01,***P<0.001。
图17A至I(与图1A至J相关)。NQO1生物可激活药物β-lap在体外和在体内以NQO1依赖性方式杀伤鼠肿瘤细胞。(图17A)通过western印迹测定来确定不同鼠癌细胞系中的NQO1表达。(图17B)通过western印迹测定来确定具有基于CRISPR的NQO1敲除的MC38细胞的不同克隆中的NQO1表达。(图17C)将MC38细胞(NQO1 WT或KO)用β-lap处理3小时,随后洗涤并更换新鲜培养基。在48小时之后,通过磺酰罗丹明B(Sulforhodamine B,SRB)测定来确定细胞生存力。(图17D)通过western印迹测定来确定稳定携带pCMV-NQO1表达载体的B16细胞的不同克隆中的NQO1表达。(图17E、图17F)将B16细胞(NQO1无效或稳定过表达)用β-lap在有或没有双香豆素(DIC,50μM)的情况下处理3小时,随后洗涤并更换培养基。在48小时之后通过SRB测定来确定细胞生存力。(图17G)将NQO1过表达B16细胞(克隆#3和#4)暴露于β-lap 3小时。添加过氧化氢酶(1000U/ml),并在48小时之后评估细胞生存力。(图17H)将C57BL/6小鼠(n=5只/组)移植TC-1细胞,并用β-lap(0.1或0.3mg)每隔一天瘤内处理4次。每周两次监测肿瘤生长。(图17I)将C57BL/6小鼠(n=4只/组)移植亲本B16细胞(NQO1无效)或NQO1稳定过表达B16细胞(克隆#1和#4),并用β-lap(0.3mg,i.t.)每隔一天处理四次。数据示出为平均值±SEM。**P<0.01,***P<0.001,****P<0.0001,通过未配对student t检验(图17G)或双因素ANOVA(图17H和图17I)确定。
图18A至D(对于图2A至F)。β-Lap介导的抗肿瘤作用依赖于免疫介导的杀伤。(图18A)将B16 NQO1#1细胞分别皮下移植到C57BL/6 WT和Rag1KO小鼠(n=5只/组)中。将荷瘤小鼠用β-lap(0.3mg,i.t.)每隔一天处理四次。(图18B)在β-lap处理之后7天,肿瘤微环境中免疫细胞的变化。将C57BL/6小鼠(n=4只/组)移植MC38细胞并用0.3mgβ-lap瘤内处理两次。在最后一次处理之后7天取出肿瘤组织并消化,并使用流式细胞仪分析免疫细胞(使用未配对student t检验来分析变化的显著性,*P<0.05,**P<0.01)。(图18C)将C57BL/6小鼠移植MC38细胞,并用β-lap(0.3mg,i.t.)每隔一天处理四次。对于T细胞耗竭,在处理期间以三天间隔注射200μg的抗CD4或抗CD8抗体四次。(图18D)将在48孔板中培养的NQO1阳性人肺癌系A549暴露于β-lap(0至6μM)±双香豆素(DIC,50μM)3小时,并在4天之后用SRB测定来评估细胞存活。数据示出为平均值±SEM。
图19A至C(与图4A至F相关)。β-lap的抗肿瘤作用需要TLR4/MyD88途径而非TLR9信号传导。(图19A至B)将荷MC38-OVA的小鼠用β-lap(0.3mg,i.t.)处理两次。在六天之后,收集肿瘤组织用于单细胞消化(图19A)和RNA提取(图19B)二者。(图19A)在24小时培养之后通过ELISA来测量来自悬浮细胞上清液的分泌的IFNβ。(图19B)通过实时PCR测定来确定IFNα1、IFNγ、TNFα和CXCL10的mRNA水平。(图19C)将MC38细胞分别植入到Myd88-/-、Tlr4-/-和Tlr9-/- C57BL/6小鼠中。然后将荷瘤小鼠用β-lap(0.3mg,i.t.)每隔一天处理四次。每周两次监测肿瘤生长。数据示出为平均值±SEM。*P<0.05,**P<0.01,***P<0.001,通过未配对t检验确定。
图20A至F(与图6A至H相关)。局部和全身β-lap处理二者均可与抗PD-L1免疫检查点阻断协同。(图20A)将MC38肿瘤细胞s.c.接种到C57BL/6小鼠(n=5只/组)的胁腹中。将荷晚期肿瘤(约150至200mm3)的小鼠用β-lap(0.3mg,i.t.)在有或没有基于抗PD-L1的检查点阻断(100μg,i.p.)处理三次的情况下局部处理四次。每周两次监测肿瘤生长,并示出了每组中单独小鼠的生长曲线。(图20B)将MC38肿瘤细胞s.c.接种到C57BL/6小鼠(n=5只/组)的胁腹中。将荷晚期肿瘤(约100mm3)的小鼠用低剂量β-lap(0.1mg,i.t.)在有或没有基于PD-L1的检查点阻断的情况下局部处理四次。(图20C、图20D)将荷MC38肿瘤的小鼠(约50至100mm3,n=5至8只/组)用β-lap(30mg/kg,i.p.)在有或没有基于PD-L1的检查点阻断的情况下全身处理六次。每周两次监测肿瘤生长(图20C)和体重(图20D)。示出了每组中单独小鼠的生长曲线。(图20E、图20F)将具有稳定NQO1过表达的B16细胞(混合克隆#1、#3和#4)s.c.接种到C57BL/6小鼠中。将荷瘤小鼠(约100mm3)用β-lap(0.3mg,i.t.)在有或没有基于抗PD-L1的检查点阻断(150μg,i.p.)处理三次的情况下局部处理四次。示出了处理方案(图20E)并监测了肿瘤生长曲线(图20F)。数据示出为来自至少两个独立实验的平均值±SEM。*P<0.05,**P<0.01,***P<0.001,****P<0.0001,通过双因素ANOVA确定。
图21A至F。IB-DNQ通过加强的肿瘤特异性ROS产生和广泛的DNA损伤来选择性地诱导NQO1+肿瘤细胞死亡。(图21A)在IB-DNQ处理4小时之后鼠细胞系的生存力。TC-1、Ag104Ld和MC38细胞表达内源性NQO1,而Pan02和B16细胞对于NQO1呈无效的。(图21B)在IB-DNQ处理4小时之后NQO1 KO(MC38 NQO1-/-)和NQO1过表达(B16 NQO1+)细胞的生存力。(图21C)在暴露于IB-DNQ 1小时之后MC38细胞中的ROS水平。(图21D至E)通过彗星测定(图21D)和γH2AX病灶/细胞核免疫荧光(图21E)来评估DNA双链断裂。(图21F)使用流式细胞术分析通过7AAD和膜联蛋白V染色来确定在暴露于IB-DNQ 4小时之后的MC38细胞死亡。(图21A)、(图21B)数据示出为来自三个独立实验(六个重复/每个实验)的平均值±SD;(图21C至F)数据示出为来自三个独立实验的平均值±SD。使用未配对Student双尾t检验进行统计学分析。*P<0.05;**P<0.01;***P<0.001;NS,不显著。
图22A至F。IB-DNQ介导的抗肿瘤作用涉及免疫系统。将6×105个MC38或8×105个MC38 NQO1 KO细胞分别皮下移植到Rag1-/-、C57BL/6和NSG小鼠中(n=5只/组)。在肿瘤体积达到50mm3之后,将荷瘤小鼠用IB-DNQ(12mg/kg,i.t.)或20%HPβCD(载剂)每隔一天处理四次。评估肿瘤体积和存活。(图22A)WT C57BL/6和NSG小鼠中MC38的肿瘤体积。(图22B)荷MC38肿瘤的WT C57BL/6和NSG小鼠的存活曲线。(图22C至D)分别在WT C57BL/6和Rag1-/-小鼠中MC38和TC-1的肿瘤体积。(图22E至F)C57BL/6小鼠中MC38和MC38 NQO1 KO模型的肿瘤体积和存活分析。数据示出为来自至少两个独立实验的平均值±SD。使用未配对的Student双尾t检验进行统计学分析。*P<0.05;**P<0.01;***P<0.001;NS,不显著。
图23A至D。IB-DNQ介导的抗肿瘤作用影响肿瘤微环境。将6×105个MC38细胞皮下移植到C57BL/6小鼠(n=3只/组)中。在肿瘤体积达到50mm3之后,将荷瘤小鼠用IB-DNQ(12mg/kg,i.t.)或载剂每隔一天处理四次。在最后一次IB-DNQ注射之后24小时之后收集肿瘤。(图23A)来自经载剂和经IB-DNQ处理的肿瘤的肿瘤浸润性免疫细胞的流式细胞术分析。(图23B)来自经载剂和经IB-DNQ处理的肿瘤的免疫群体的定量。(图23C)在存在/不存在IB-DNQ的情况下的T细胞增殖。将经CFSE标记的脾细胞用致死剂量的IB-DNQ处理4小时,随后洗涤并用抗CD3(1μg/ml)和抗CD28(2μg/ml)刺激48小时,然后通过流式细胞术测定分析增殖性CD8 T细胞。(图23D)IB-DNQ对T细胞的作用。将来自初始(n=3只/组)和荷瘤(n=3只/组)小鼠的脾细胞暴露于不同浓度的IB-DNQ 4小时,随后洗涤并更换培养基。在24小时之后,通过流式细胞术分析细胞死亡。使用用相同处理的Ag104细胞作为对照。数据示出为来自两个独立实验的平均值±SD。使用未配对的Student双尾t检验进行统计学分析。*P<0.05;**P<0.01;NS,不显著。
图24A至F。CD8+和CD4+ T细胞对于IB-DNQ介导的抗肿瘤作用是至关重要的。将6×105个MC38细胞皮下移植到C57BL/6小鼠(n=5只/组)中。在肿瘤体积达到50mm3之后,将荷瘤小鼠用IB-DNQ(12mg/kg,i.t.)或载剂每隔一天处理四次。对于CD4+和/或CD8+ T细胞耗竭,在处理期间以三天间隔腹膜内注射200μg的抗CD4和/或CD8+抗体三次。(图24A至B)对用/不用IB-DNQ±抗CD4抗体处理的荷MC38的小鼠的肿瘤体积和存活分析。(图24C至D)对用/不用IB-DNQ±抗CD8抗体处理的荷MC38的小鼠的肿瘤体积和存活分析。(图24E至F)对用/不用IB-DNQ±抗CD4和抗CD8抗体处理的荷MC38的小鼠的肿瘤体积和存活分析。数据示出为来自至少两个独立实验的平均值±SD。使用未配对Student双尾t检验进行统计学分析。*P<0.05;**P<0.01;***P<0.001;NS,不显著。
图25A至E。IB-DNQ诱导肿瘤ICD和树突细胞介导的T细胞交叉致敏。(图25A)在IB-DNQ处理之后释放到细胞培养上清液中的HMGB1的水平。将MC38、MC38 NOQ1-/-、B16和B16NOQ1+细胞用IB-DNQ处理4小时,随后更换培养基并培养24小时,收集培养上清液并通过ELISA确定HMGB1水平。(图25B至C)IFN-α和IFN-β的相对表达。肿瘤样品与图23A的样品相同,根据制造商说明进行总RNA提取,在逆转录成cDNA之后进行qPCR。(图25D至E)IFN-γ指示的T细胞应答。将6×105个MC38或8×105个MC38-OVA细胞移植到C57BL/6小鼠(n=3只/组)中。在肿瘤达到50mm3之后,将小鼠用IB-DNQ或载剂每隔一天处理4次,并且在最后一次施用之后24小时,将从荷瘤小鼠分离的脾细胞用培养基或用60Gy辐照的MC38细胞(图25D)或OT-1(图25E)进行刺激。数据示出为来自至少两个独立实验的平均值±SD。使用未配对的Student双尾t检验进行统计学分析。*P<0.05;**P<0.01;***P<0.001。
图26A至H。IB-DNQ诱导固有免疫记忆而不是经典免疫记忆。在完全排斥之后60天,将初始(n=5只/组)和经IB-DNQ治愈的无MC38肿瘤(n=7只/组)的C57BL/6小鼠用3×106个MC38细胞在距原发性肿瘤的相对位点皮下再攻击。在肿瘤再次根除之后30天收集来自这些无肿瘤小鼠的器官。(图26A至B)MC38模型的肿瘤体积和存活分析。(图26C至D)不同器官中的记忆CD8+ T细胞(图26C)和CD4+ T细胞(图26D)。(图26E)淋巴结(lymph node,LN)中的CD44+ DC。(图26F)将来自无肿瘤(tumor-free,TF)或荷瘤小鼠的LN的DC用IB-DNQ(1μM)诱导的抗原刺激5小时,然后评估DC上的CD44表达。(图26G)将从TF或荷瘤小鼠的脾分离的CD8+ T细胞用CFSE标记并将其与IB-DNQ(1μM)诱导的抗原共培养48小时。通过流式细胞术来确定T细胞增殖。(图26H)在存在抗原、抗CD3(1μg/ml)和抗CD28(2μg/ml)刺激的情况下,将经CFSE标记的脾细胞与来自TF或荷瘤小鼠的LN的细胞共培养48小时,通过流式细胞术确定T细胞增殖。数据示出为来自三个独立实验的平均值±SD。使用未配对的Student双尾t检验进行统计学分析。*P<0.05;**P<0.01;***P<0.001;NS,不显著。
图27A至B。在IB-DNQ处理之后TME中的PD-L1上调。将6×105个MC38细胞皮下移植到C57BL/6小鼠(n=5只/组)中。在肿瘤体积达到50mm3(小)或150mm3(大)之后,将荷瘤小鼠用IB-DNQ(12mg/kg,i.t.)或载剂每隔一天处理四次。(图27A至B)在最后一次IB-DNQ注射之后24小时之后收集小肿瘤(图27A)或晚期肿瘤(图27B)。通过流式细胞术测量CD45+免疫细胞或CD45-肿瘤细胞中的PD-L1蛋白水平。使用未配对的Student双尾t检验进行统计学分析。*P<0.05;NS,不显著。
图28A至F。IB-DNQ与抗PD-L1的组合治疗克服了检查点阻断抗性。(图28A至D)将荷小(50mm3,红线)或晚期(150mm3,蓝线)MC38肿瘤的小鼠用IB-DNQ(12mg/kg,i.v.)或载剂每隔一天处理,4次注射(图28A至B),或用mIgG或抗PD-L1处理,三次注射(图28C至D)。(图28E至F)将荷MC38肿瘤的小鼠(150mm3)用IB-DNQ、抗PD-L1或IB-DNQ+抗PD-L1每隔一天处理,4次注射,载剂是mIgG或HPβCD。测量肿瘤体积(图28A、28C和28E)和总存活(图28B、28D和28F)。每周两次测量肿瘤生长。数据示出为来自三个独立实验的平均值±SD。通过Prism8软件针对总存活来限定Kaplan-Meier存活曲线。*P<0.05,**P<0.01,***P<0.001,通过双因素ANOVA确定。NS,不显著。
发明详述
本发明人先前的研究揭示了,NQO1生物可激活药物(β-lap和IB-DNQ)可导致广泛的DNA损伤和PARP1驱动的肿瘤程序性坏死,同时在免疫缺陷型小鼠中诱导肿瘤抑制。正在寻求提高这些NQO1生物可激活药物的亚致死剂量效力的策略,以提高它们在有力的化学治疗或放射治疗方案中的有效性。在此,本发明人表明,嗜中性粒细胞介导的固有免疫和CD8介导的适应性免疫系统二者均受到刺激,导致NQO1生物可激活药物在免疫活性小鼠中产生更有效的抗肿瘤作用。他们还揭示了NQO1生物可激活药物可触发免疫原性细胞死亡(ICD)并诱导损伤相关分子模式(DAMP)释放和吞噬细胞/APC(抗原呈递细胞)募集,其继而促进细胞毒性T细胞(cytotoxic T cell,CTL)的交叉致敏以用于通过提高抗原/DNA摄取和I型干扰素(IFN)产生来抑制肿瘤生长(图7)。这项研究表明由NQO1生物可激活药物诱导的肿瘤特异性活性氧物质(ROS)和DNA损伤如何可以刺激抗肿瘤免疫,以及这些反应的激活是否可提高肿瘤靶向治疗效力。NQO1生物可激活药物与激活适应性免疫的方法(例如T细胞检查点阻断)的组合将提供持久的效力和患者益处。本公开内容的这些和另一些方面在下面详细阐述。
I.定义
一般而言,本文中使用的术语和短语具有其本领域公认的含义,其可通过参考本领域技术人员已知的标准文本、期刊参考文献和上下文找到。这样的本领域公认的含义可通过参考技术词典获得,例如Hawley’s Condensed Chemical Dictionary第14版,R.J.Lewis,John Wiley&Sons,New York,N.Y.,2001。
BER,碱基切除修复;SSBR,单链断裂修复(single strand break repair);DSBR,双链断裂修复(double strand break repair)。
除非上下文另有明确规定,否则未用数量词修饰的名词包括一个/种或更多个/种。因此,例如,提及“化合物”包括多个这样的化合物,使得化合物X包括多个化合物X。还注意,权利要求书可被制定为排除任何任选要素。因此,本声明旨在用作与权利要求书要素的记载相关的排他性术语例如“单独”、“仅”等的使用或“否定”限制的使用的先行基础。
术语“和/或”意指与该术语相关的任一个项目、任何项目组合或所有的项目。短语“一个/种或更多个/种”是本领域技术人员容易理解的,特别是当在其使用的上下文中阅读时。例如,苯环上的一个或更多个取代基是指一至五个,或一至四个,例如如果苯环是二取代的。
术语“约/大约”可指指定值的±5%、±10%、±20%或±25%的变化。例如在一些实施方案中,“约50%”可带有45%至55%的变化。对于整数范围,术语“约/大约”可包括大于和/或小于在该范围每端所记载整数的一个或两个整数。除非本文中另有说明,否则术语“约/大约”旨在包括接近所记载范围的在单独成分、组合物或实施方案的功能方面是等效的值,例如重量百分比。
如技术人员将理解的,所有数字,包括表述成分量、特性例如分子量、反应条件等的那些均是近似值,并且被理解为在所有情况下被术语“约/大约”任选地修饰。这些值可根据本领域技术人员利用本文中所述的教导寻求获得的期望特性而变化。还应理解,这样的值固有地包含由其各自的测试测量值中存在的标准偏差必然引起的变异性。
虽然本发明可采取多种不同的形式,但出于促进对本发明原理的理解的目的,现在将参考附图中所示出的一些实施方案并且将使用具体语言对其进行描述。然而应当理解,由此不旨在限制本发明的范围。所述实施方案的任何变化方案和另一些修改方案以及如本文中所述的本发明的原理的任何另一些应用均被预期为本发明所涉及的领域的技术人员将通常会想到的。
当本文中公开取代基组时,应理解该组和所有子组的所有单独成员,包括该组成员的任何异构体、对映体和非对映体均分别公开。当在本文中使用马库什(Markush)基团或其他分组时,该组的所有单独成员以及该组的所有组合和可能的子组合均旨在单独地包括在本公开内容中。当在本文中描述化合物使得该化合物的特定异构体、对映体或非对映体未指定(例如以式或以化学名称指定)时,该描述旨在包括单独或以任何组合所述的化合物的每个异构体和对映体。另外,除非另有说明,否则本文中公开的化合物的所有同位素变体均旨在被本公开内容涵盖。例如,应当理解,所公开分子中的任一个或更多个氢可被氘或氚替代。分子的同位素变体通常可用作在分子测定中以及在与分子或其使用相关的化学和生物学研究中的标准。用于制备这样的同位素变体的方法是本领域已知的。化合物的具体名称旨在是示例性的,因为已知本领域普通技术人员可不同地命名同一化合物。
每当说明书中给出范围(例如温度范围、时间范围、碳链范围或组成或浓度范围)时,所有中间范围和子范围以及包含在给定范围内的所有单独值均旨在单独地包括在本公开内容中。应当理解,包括在说明书中的任何子范围或者范围或子范围中的单独值可任选地从本发明的实施方案中排除。
本文中使用的“包含”与“包括”、“含有”或“特征在于”同义,并且是包含性的或开放式的,并且不排除另外的、未记载的要素或方法步骤。本文中使用的“由……组成”排除了权利要求书要素中未指定的任何要素、步骤或成分。本文中使用的“基本上由……组成”不排除对权利要求书的基本特征和新特征没有实质性影响的材料或步骤。在本文中的每个实例中,术语“包含”、“基本上由……组成”和“由……组成”中的任一个均可被其他两个术语中的任一个替换。本文中举例说明性地描述的本发明可以在不存在本文中未具体公开的任何一个或更多个要素、一个或更多个限制的情况下适当地实践。
“化学治疗剂”是指能够降低或阻止癌细胞、癌细胞群、肿瘤或其他恶性组织的生长、增殖或扩散的任何物质。该术语还旨在涵盖任何抗肿瘤或抗癌药剂。
就主题治疗方法而言,化合物的“治疗有效量”是指制剂中化合物的量,该量当根据待治疗的障碍或病症的临床可接受标准或美容目的,例如,以适用于任何医学治疗的合理的益处/风险比,作为所期望剂量方案的一部分施用于(哺乳动物,例如人)时,减轻症状、改善病症或减慢疾病病症的发作。
术语“治疗”包括(i)预防疾病、病理或医学病症免于发生(例如预防);(ii)抑制疾病、病理或医学病症或阻止其发展;(iii)减轻疾病、病理或医学病症;和/或(iv)减弱与疾病、病理或医学病症相关的症状。因此,术语“治疗”可延伸至预防并且可包括预防、降低、停止或逆转所治疗病症或症状的进展或严重程度。因此,术语“治疗”可适当包括医学施用、治疗性和/或预防性施用。术语“治疗”可包括以改善或稳定对象病症的方式逆转、减轻或阻止病症的症状、临床体征和潜在病理状况。
术语“抑制”是指减慢、停止或逆转疾病、感染、病症或细胞群的增长或进展。例如,与在不存在治疗或接触的情况下发生的生长或进展相比,抑制可大于约20%、40%、60%、80%、90%、95%或99%。
术语“接触”是指接触、使接触或使得紧密或密切接近的动作,包括在细胞或分子水平上,例如以引起生理反应、化学反应或物理变化,例如在溶液中、在反应混合物中、在体外或在体内。
术语“暴露”旨在涵盖如本领域广泛理解的定义。在一个实施方案中,该术语意指经受或允许经受动作、影响或条件。例如并仅作为实例,细胞可经受治疗有效量的可药用形式的化学治疗剂的作用、影响或条件。
术语“癌细胞”旨在涵盖如本领域广泛理解的定义。在一个实施方案中,该术语是指可在人或动物中促进癌症的临床病症的异常调节的细胞。在一个实施方案中,该术语可指培养的细胞系或在人或动物身体内或来源于人或动物身体的细胞。如本领域所理解的,癌细胞可以是广泛多种分化的细胞、组织或器官类型。
术语“肿瘤”是指赘生物,通常是包含多个聚集的恶性细胞的块(mass)。
在本文中所述式中,以下基团适当可以是R基团或桥基。
术语“烷基”是指优选地具有1至30个碳原子的单价(monoradical)支链或非支链饱和烃链。短烷基是具有1至12个碳原子的那些,包括甲基、乙基、丙基、丁基、戊基和己基,包括其所有异构体。长烷基是具有12至30个碳原子的那些。该基团可以是端基或桥基。
烷基、杂烷基、芳基、杂芳基和杂环基,及其环状和/或不饱和形式,可以是式I的R基团,并且每个基团可任选地被取代。
术语“经取代的”指示在使用“经取代的”的表述中所指基团上的一个或更多个氢原子被“取代基”替代。由“一个或更多个”所指的数目可从取代基所在部分中明显看出。例如,一个或更多个可指例如1、2、3、4、5或6个;在一些实施方案中,1、2或3个;以及在另一些实施方案中,1或2个。取代基可以是指定基团的选择之一,或者其可以是本领域技术人员已知的合适基团,前提是不超过被取代原子的正常价并且取代产生稳定的化合物。合适的取代基包括例如烷基、烯基、炔基、烷氧基、卤素、卤代烷基、羟基、羟烷基、芳基、芳酰基、(芳基)烷基(例如苄基或苯乙基)、杂芳基、杂环、环烷基、烷酰基、烷氧羰基、氨基、烷基氨基、二烷基氨基、三氟甲基、三氟甲氧基、三氟甲基硫基、二氟甲基、酰氨基、硝基、羧基、羧基烷基、酮基、硫代、烷硫基、烷基亚磺酰基、烷基磺酰基、芳基亚磺酰基、芳基磺酰基、杂芳基亚磺酰基、杂芳基磺酰基、杂环亚磺酰基、杂环磺酰基、磷酸盐/酯、硫酸盐/酯、羟胺、羟基(烷基)胺和氰基。另外,合适的取代基可以是例如-X、-R、-O-、-OR、-SR、-S-、-NR2、-NR3、=NR、-CX3、-CN、-OCN、-SCN、-N=C=O、-NCS、-NO、-NO2、=N2、-N3、-NC(=O)R、-C(=O)R、-C(=O)NRR、-S(=O)2O-、-S(=O)2OH、-S(=O)2R、-OS(=O)2OR、-S(=O)2NR、-S(=O)R、-OP(=O)O2RR、-P(=O)O2RR、-P(=O)(O-)2、-P(=O)(OH)2、-C(=O)R、-C(=O)X、-C(S)R、-C(O)OR、-C(O)O-、-C(S)OR、-C(O)SR、-C(S)SR、-C(O)NRR、-C(S)NRR或-C(NR)NRR,其中每个X独立地是卤素(halogen)(“卤素(halo)”):F、Cl、Br或I;并且每个R独立地是H、烷基、芳基、(芳基)烷基(例如苄基)、杂芳基、(杂芳基)烷基、杂环、杂环(烷基)或保护基。如本领域技术人员将容易理解的,当取代基是酮基(=O)或硫代(=S)等时,则所取代原子上的两个氢原子被替代。在一些实施方案中,以上取代基中的一个或更多个可从所取代基团上的取代基的潜在值的组中排除。
除非另有说明,否则单独地或与另一术语组合的术语“杂烷基”意指稳定的直链或支链或环状烃基或其组合,通常在链中具有2至14个碳,或2至10个碳,包括至少一个碳原子和选自O、N、P、Si和S的至少一个杂原子,并且其中氮和硫原子可任选地被氧化并且氮杂原子可任选地被季铵化。杂原子O、N、P和S和Si可位于杂烷基的任何内部位置或烷基与分子其余部分连接的位置。杂烷基可在链中具有例如1至约20个碳原子。一些实例包括但不限于--CH2--CH2--O--CH3、--CH2--CH2--NH--CH3、--CH2--CH2--N(CH3)--CH3、--CH2--S--CH2--CH3、--CH2--CH2--S(O)--CH3、--CH2--CH2--S(O)2--CH3、--CH=CH--O--CH3、--Si(CH3)3、--CH2-CH2--CH=N--OCH3、--CH=CH--N(CH3)--CH3、O--CH3、--O--CH2--CH3和--CN。杂烷基的另一些实例包括烷基醚、仲和叔烷基胺、酰胺、烷基硫化物等。该基团可以是端基或桥基。本文中使用的当在桥基的上下文中使用时,提及链是指连接桥基的两个末端位置的原子的直链。
本文中使用的术语“醇”可定义为包含在氢原子处被一个羟基取代的C1-12烷基部分的醇。醇包括乙醇、正丙醇、异丙醇、正丁醇、异丁醇、仲丁醇、叔丁醇、正戊醇、异戊醇、正己醇、环己醇、正庚醇、正辛醇、正壬醇、正癸醇等。醇中的碳原子可以是直链、支链或环状的。
“酰基”可定义为烷基-CO-基团,其中烷基如本文中所述。酰基的一些实例包括乙酰基和苯甲酰基。烷基可以是C1-C6烷基。该基团可以是端基或桥基(即二价基团)。
“烷氧基”是指-O-烷基,其中烷基在本文中定义。优选地,烷氧基是C1-C6烷氧基。一些实例包括但不限于甲氧基和乙氧基。该基团可以是端基或桥基。
作为基团或基团的一部分的“烯基”表示包含至少一个碳-碳双键的脂肪族烃基,并且其可以是直链或支链的,优选地在正常链中具有2至14个碳原子,更优选2至12个碳原子,最优选2至6个碳原子。该基团可在正常链中包含多个双键,并且关于每个双键的取向独立地是E或Z。一些示例性烯基包括但不限于乙烯基、丙烯基、丁烯基、戊烯基、己烯基、庚烯基、辛烯基和壬烯基。该基团可以是端基或桥基。
作为基团或基团的一部分的“炔基”可定义为包含碳-碳三键的脂肪族烃基,其链可以为直链或支链的,优选地在正常链中具有2至14个碳原子,更优选2至12个碳原子,更优选2至6个碳原子。一些示例性结构包括但不限于乙炔基和丙炔基。该基团可以是端基或桥基。
“烯氧基”是指--O--烯基基团,其中烯基如本文中所定义。优选烯氧基是C1-C6烯氧基。该基团可以是端基或桥基。
“炔氧基”是指--O-炔基基团,其中炔基如本文中所定义。优选炔氧基是C1-C6炔氧基。该基团可以是端基或桥基。
“烷氧羰基”是指-C(O)--O-烷基基团,其中烷基如本文中所定义。烷基优选地是C1-C6烷基。一些实例包括但不限于甲氧羰基和乙氧羰基。该基团可以是端基或桥基。
“烷基亚磺酰基”可定义为-S(O)-烷基基团,其中烷基如上所定义。烷基优选地是C1-C6烷基。一些示例性烷基亚磺酰基基团包括但不限于甲基亚磺酰基和乙基亚磺酰基。该基团可以是端基或桥基。
“烷基磺酰基”是指-S(O)2-烷基基团,其中烷基如上所定义。烷基优选地是C1-C6烷基。一些实例包括但不限于甲磺酰基和乙磺酰基。该基团可以是端基或桥基。
“氨基”是指-NH2,并且“烷基氨基”是指-NR2,其中至少一个R是烷基,并且第二R是烷基或氢。术语“酰氨基”是指RC(=O)NH-,其中R是烷基或芳基。烷基可以是例如C1-C6烷基。一些实例包括但不限于甲氨基和乙氨基。该基团可以是端基或桥基。
“烷基氨基羰基”是指烷基氨基-羰基基团,其中烷基氨基如上所定义。该基团可以是端基或桥基。
“环烷基”是指3至约30个碳原子的饱和或部分饱和、单环或稠合或螺环多环的碳环,通常每环包含3至约9个碳,例如环丙基、环丁基、环戊基、环己基、环辛基等。其包括单环体系例如环丙基和环己基,双环体系例如十氢化萘,以及多环体系例如金刚烷胺。该基团可以是端基或桥基。
“环烯基”可定义为包含至少一个碳-碳双键并且优选地每个环具有5至10个碳原子的非芳香族单环或多环环体系。一些示例性单环环烯基环包括环戊烯基、环己烯基或环庚烯基。环烯基可被一个或更多个取代基取代。该基团可以是端基或桥基。
烷基和环烷基可以是其他基团的烷基部分上的取代基,例如但不限于烷氧基、烷基胺、烷基酮、芳基烷基、杂芳基烷基、烷基磺酰基和烷基酯取代基等。该基团可以是端基或桥基。
“环烷基烷基”可定义为环烷基-烷基-基团,其中环烷基和烷基部分如前所述。一些示例性单环烷基烷基基团包括环丙基甲基、环戊基甲基、环己基甲基和环庚基甲基。该基团可以是端基或桥基。
“杂环烷基”是指饱和或部分饱和的单环、双环或多环环,其在至少一个环中包含选自氮、硫、氧的至少一个杂原子,优选1至3个杂原子。每个环优选为3至10元,更优选为4至7元。合适的杂环烷基取代基的一些实例包括吡咯烷基、四氢呋喃基、四氢硫代呋喃基、哌啶基、哌嗪基、四氢吡喃基、吗啉代、1,3-二氮杂环庚烷、1,4-二氮杂环庚烷、1,4-氧氮杂环庚烷和1,4-氧硫杂环庚烷。该基团可以是端基或桥基。
“杂环烯基”是指如上所述的但包含至少一个双键的杂环烷基。该基团可以是端基或桥基。
“杂环烷基烷基”是指杂环烷基-烷基基团,其中杂环烷基和烷基部分如前所述。一些示例性杂环烷基烷基基团包括(2-四氢呋喃基)甲基和(2-四氢硫代呋喃基)甲基。该基团可以是端基或桥基。
“卤素”是指卤素取代基,例如氟、氯、溴或碘。
术语“芳基”是指来源于从母体芳环体系的单个碳原子去除一个氢原子的芳香族烃基。该基团可在母体环体系的饱和或不饱和碳原子处。芳基可具有6至18个碳原子。芳基可具有单环(例如苯基)或多个稠合(稠)环,其中至少一个环是芳香族的(例如萘基、二氢菲基、芴基或蒽基)。典型的芳基包括但不限于来源于苯、萘、蒽、联苯基等的基团。芳基可以是未经取代的或任选经取代的,如以上针对烷基所述。
术语“杂芳基”在本文中定义为包含一个、两个或三个芳族环并且在芳族环中包含至少一个氮、氧或硫原子的单环、双环或三环环体系,并且其可以是未经取代或经取代的,例如,被一个或更多个,并且特别是一至三个取代基取代,如以上在“经取代的”定义中所述的。杂芳基的一些实例包括但不限于2H-吡咯基、3H-吲哚基、4H-喹嗪基、吖啶基、苯并[b]噻吩基、苯并噻唑基、β-咔啉基、咔唑基、色烯基(chromenyl)、噌啉基(cinnolinyl)、二苯并[b,d]呋喃基、呋咱基(furazanyl)、呋喃基、咪唑基(imidazolyl)、咪唑基(imidizolyl)、吲唑基、吲嗪基(indolisinyl)、吲哚基、异苯并呋喃基、异吲哚基、异喹啉基、异噻唑基、异 唑基、萘啶基、
唑基、萘啶基、 唑基、咟啶基(perimidinyl)、菲啶基、菲咯啉基(phenanthrolinyl)、吩吡嗪基(phenarsazinyl)、吩嗪基、吩噻嗪基、吩噻
唑基、咟啶基(perimidinyl)、菲啶基、菲咯啉基(phenanthrolinyl)、吩吡嗪基(phenarsazinyl)、吩嗪基、吩噻嗪基、吩噻 基(phenoxathiinyl)、吩
基(phenoxathiinyl)、吩 嗪基、酞嗪基、蝶啶基、嘌呤基、吡喃基、吡嗪基、吡唑基、哒嗪基、吡啶基、嘧啶基(pyrimidinyl)、嘧啶基、吡咯基、喹唑啉基、喹啉基、喹喔啉基、噻二唑基、噻蒽基、噻唑基、噻吩基、三唑基、四唑基和呫吨基。在一个实施方案中,术语“杂芳基”表示包含含有碳和独立地选自非过氧化物氧、硫和N(Z)的1、2、3或4个杂原子的五或六个环原子的单环芳族环,其中Z不存在或者是H、O、烷基、芳基或(C1-C6)烷基芳基。在另一个实施方案中,杂芳基表示由其衍生的约八至十个环原子的邻位稠合双环杂环,特别是苯并衍生物或通过与其稠合丙烯、三亚甲基或四亚甲基双基衍生的杂环。
嗪基、酞嗪基、蝶啶基、嘌呤基、吡喃基、吡嗪基、吡唑基、哒嗪基、吡啶基、嘧啶基(pyrimidinyl)、嘧啶基、吡咯基、喹唑啉基、喹啉基、喹喔啉基、噻二唑基、噻蒽基、噻唑基、噻吩基、三唑基、四唑基和呫吨基。在一个实施方案中,术语“杂芳基”表示包含含有碳和独立地选自非过氧化物氧、硫和N(Z)的1、2、3或4个杂原子的五或六个环原子的单环芳族环,其中Z不存在或者是H、O、烷基、芳基或(C1-C6)烷基芳基。在另一个实施方案中,杂芳基表示由其衍生的约八至十个环原子的邻位稠合双环杂环,特别是苯并衍生物或通过与其稠合丙烯、三亚甲基或四亚甲基双基衍生的杂环。
术语“杂环”是指包含选自氧、氮和硫的至少一个杂原子,并且任选地被如本文中在术语“经取代的”下所定义的一个或更多个基团取代的饱和或部分不饱和的环体系。杂环可以是包含一个或更多个杂原子的单环、双环或三环基团。杂环基团还可包含与环连接的氧代基团(=O)。杂环基团的一些非限制性实例包括1,3-二氢苯并呋喃、1,3-二氧戊环、1,4-二 烷、1,4-二噻烷、2H-吡喃、2-吡唑啉、4H-吡喃、色满基、咪唑烷基、咪唑啉基、吲哚啉基、异色满基、异吲哚啉基、吗啉、哌嗪基、哌啶、哌啶基、吡唑烷、吡唑烷基、吡唑啉基、吡咯烷、吡咯啉、奎宁环和硫代吗啉。
烷、1,4-二噻烷、2H-吡喃、2-吡唑啉、4H-吡喃、色满基、咪唑烷基、咪唑啉基、吲哚啉基、异色满基、异吲哚啉基、吗啉、哌嗪基、哌啶、哌啶基、吡唑烷、吡唑烷基、吡唑啉基、吡咯烷、吡咯啉、奎宁环和硫代吗啉。
本文中使用的缩写“DNQd”是指DNQ的类似物或衍生物。
下面描述了可以是R1、R2、R3和R4的桥基或端基的另外的基团。
术语“碳酸酯”可定义为具有一般性结构R′OC(=O)OR的官能团,其中R′可以是式I的三环核心并且R可如在式I的变量定义中所限定的。
术语“酯”可定义为具有一般性结构RC(=O)OR′的官能团,其中R′可以是式I的三环核心并且R可如在式I的变量定义中所限定的,反之亦然。
“吡啶基”基团可以是2-吡啶基、3-吡啶基或4-吡啶基基团。
术语“巯基”可定义为具有一般性结构-S-H的官能团。
术语“亚磺酰基”可定义为具有一般性结构R-S(=O)-R′的官能团,其中R′可以是式I的三环核心并且R可如在式I的变量定义中所限定的,反之亦然。
术语“磺酰基”可定义为具有一般性结构R-S(=O)2-R′的官能团,其中R′可以是式I的三环核心并且R可如在式I的变量定义中所限定的,反之亦然。
术语“己糖”可定义为具有六个碳原子的具有一般性化学式C6H12O6的单糖,并且可包括在1位具有醛官能团的己醛糖或在2位具有酮官能团的己酮糖。一些示例性己醛糖包括阿洛糖、阿卓糖、葡萄糖、甘露糖、古洛糖、艾杜糖、半乳糖和塔洛糖,呈D或L形式。
方案和实施例中使用的缩写可包括以下:
A549=腺癌人肺泡基底上皮细胞
ATP=三磷酸腺苷
β-lap=β拉帕醌
DHE=二氢乙锭
DNQ=脱氧苯醌
DNQd=脱氧苯醌的任何类似物或衍生物
ELISA=酶联免疫吸附测定
h=小时
H596=[NCI-H596]人肺腺鳞癌细胞系
HT1080=灵长类纤维肉瘤细胞系
LD50=致死概率为50%的致死剂量
LD90=致死概率为90%的致死剂量
LD100=致死概率为100%的致死剂量
MCF-7=人乳腺腺癌细胞系
MDA-MB-231=人乳腺癌细胞系
MIA-PaCa2=胰腺癌细胞系
min=分钟
NADH=烟酰胺腺嘌呤二核苷酸
NQO1=NAD(P)H:醌氧化还原酶1
NSCLC=非小细胞肺癌细胞
OCR=氧消耗速率(oxygen consumption rate)
p53=肿瘤阻抑蛋白
PC-3=人前列腺癌细胞系
ROS=活性氧物质
±SE=标准误差
siRNA=小干扰核糖核酸
shRNA=小发夹核糖核酸
μM=微摩尔
nM=纳摩尔
μmol=微摩尔
II.治疗性醌
DNQ是表现出广泛的治疗窗的强效化学治疗剂,其针对广谱难以治疗的癌症(包括胰腺癌和非小细胞肺癌)的靶向治疗具有巨大的前景。尽管在癌症化学治疗中取得了相当大的进步,但大多数癌症化学治疗缺乏选择性仍然是一个主要的限制因素。存在于大多数实体瘤中,特别是在非小细胞肺癌细胞(non-small-cell lung cancer cell,NSCLC)、前列腺、胰腺和乳腺中的NAD(P)H:醌氧化还原酶-1(NQO1,DT-心肌黄酶,EC 1.6.99.2)水平升高为本文中所述治疗性治疗提供了靶标。NQO1是能够还原大多数醌,形成稳定的氢醌的诱导型II相解毒双电子氧化还原酶。在大多数情况下,谷胱甘肽转移酶然后将氢醌解毒,将其与谷胱甘肽缀合以进行分泌,并有效地避免了更大毒性的半醌。
然而,对于一些稀有化合物,NQO1介导的生物还原可用于抗肿瘤活性。NQO1活性可将特定的醌转化成高度细胞毒性物质,而不是促进解毒。依赖于NQO1的大多数抗肿瘤醌是DNA烷化剂:(a)丝裂霉素C(mitomycin C,MMC);(b)RH1;(c)E09和(d)AZQ。然而,这些DNA烷化剂不仅受解毒途径影响,而且来自升高或诱导性DNA修复途径的抗性限制了其有用性。此外,许多这些药物是在正常组织中普遍表达的单电子氧化还原酶的有效底物。
邻萘醌、β拉帕醌(β-lap,方案1)以NQO1依赖性方式杀伤培养的癌细胞以及体内鼠异种移植物和原位人或小鼠肿瘤模型。与烷基化醌相反,β-lap通过NQO1依赖性活性氧物质(ROS)形成和氧化应激来诱导细胞死亡。β-lap的NQO1代谢产生被两个当量的双氧自发氧化的不稳定的氢醌,产生超氧化物。
方案1.醌化合物的一些实例

因此建立了无效的氧化还原循环,并且升高的超氧化物水平继而导致大量的DNA碱基和单链断裂(single strand break,SSB)病变,其通常容易且快速地修复。然而,在经β-lap处理的NQO1过表达癌细胞中产生的广泛的DNA病变导致聚(ADP-核糖)聚合酶-1(poly(ADP-ribose)polymerase-1,PARP1)(在其他方面必需的碱基和SSB修复酶)超激活。继而,由于ADP核糖基化,PARP1超激活导致NAD+/ATP合并物急剧降低,导致巨大的能耗竭和细胞死亡。因此,β-lap通过独特的程序性坏死机制杀伤NQO1+癌细胞,该机制是:(a)独立于胱天蛋白酶激活或p53状态;(b)独立于bcl-2水平;(c)不受BAX/BAK缺陷的影响;(d)独立于EGFR、Ras或其他组成型信号转导激活;和/或(e)不依赖于增殖,因为NQO1在所有细胞周期阶段中均表达。因此,β-lap是有吸引力的实验性化学治疗剂,并且多种β-lap制剂已经或正在进行I/II期临床试验。
脱氧苯醌(DNQ,方案1)是有前景的抗肿瘤剂。先前的数据表明,DNQ通过氧化应激和ROS形成来杀伤癌细胞。DNQ的细胞毒性部分地被N-乙酰基半胱氨酸(全局自由基清除剂和谷胱甘肽的前体)阻止。现在已经表明,DNQ经历了类似于β-lap的NQO1依赖性无效循环,其中氧被消耗,并且形成了ROS,并且广泛的DNA损伤触发PARP1超激活,以及必需NAD+/ATP核苷酸合并物急剧降低,表明程序性坏死。重要的是,DNQ的效力比β-lap高20至100倍,其中在NQO1+相对于NQO1-NSCLC细胞中,治疗窗显著增强。还在体外乳腺癌、前列腺癌和胰腺癌模型中表明了通过DNQ的有效的NQO1依赖性杀伤。此外,体外NQO1比β-lap更加有效地处理DNQ,表明利用率提高是其效力提高的原因。因此,DNQ作为选择性化学治疗剂为治疗具有升高的NQO1水平的实体瘤提供了显著的前景,然而,由于组合的协同,因此本文中所述的组合治疗可以以多种醌化合物提供有效的治疗。
因为NQO1在大多数实体瘤中过表达,并且多种醌化合物的细胞毒性主要取决于酶NQO1的升高表达,因此奎宁化合物及其衍生物可成为逼近靶向实体瘤的极好手段。本发明提供了可用作新癌症治疗剂的许多新的细胞毒性化合物,如本文中所述。
本公开内容的前述目的和其他目的和特征将通过以下详细描述变得更明显,该详细描述通过参照附图来进行。本申请的另一些实施方案、形式、特征、方面、益处、目的和优点将通过其中提供的详细描述和附图变得明显。
NQO1生物可激活药物(所有β拉帕醌和作为NQO1底物的DNQ衍生物)以NQO1依赖性、肿瘤选择性方式产生极大水平的活性氧物质,从而允许使用DNA修复抑制剂,包括所有PARP1抑制剂、DNA双链断裂修复抑制剂以及碱基切除修复抑制剂,其以肿瘤特异性方式待使用,影响两种药剂的肿瘤选择性效力。因为缺乏肿瘤选择性,DNA修复抑制剂一般均无效。因为这些NQO1生物可激活药物导致肿瘤选择性产生DNA损伤,包括DNA碱基损伤、单链断裂和双链断裂,所以DNA修复抑制剂可用于提供肿瘤选择性抗肿瘤活性。肿瘤选择性活性和响应包括显著抑制糖酵解以及其他肿瘤选择性代谢抑制。
NQO1生物可激活药物可用于以除非知晓DNA病变产生否则不明显的方式,以及以引起代谢变化和细胞死亡响应的方式使DNA修复抑制剂具有肿瘤选择性,所述代谢变化和细胞死亡响应不明显并且取决于所使用的DNA修复抑制剂而变化。例如,与DNQ生物可激活药物一起施用的PARP1抑制剂引起标准的凋亡响应,而无能量损失。相比之下,DNA双链断裂修复、单链断裂修复和碱基切除修复抑制剂均增强PARP1超激活,并随后在能量代谢和程序性坏死中发生损失。
目前唯一使用的DNA修复抑制剂(例如PARP-1抑制剂)是通过对肿瘤特异性合成致死性响应的独特利用(例如,在BRACA1/2突变体肿瘤中使用PARP1抑制剂)。然而,这是DNA修复抑制剂的非常有限的使用——仅大约仅5%的乳腺癌。相比之下,本文中所述的方法可治疗具有升高的NQO1和降低的过氧化氢酶水平的所有癌症,而正常组织具有升高的过氧化氢酶和低水平的NQO1。本文中所述的方法提供了DNA修复抑制剂的新用途,允许其以肿瘤选择性方式使用,同时还增强了NQO1生物可激活药物。两种药剂均可以已无毒剂量使用,以提供协同的、肿瘤选择性的效力响应。
迄今为止,DNA修复抑制剂因缺乏肿瘤选择性响应和效力而失败。本文中所述的方法解决了这些限制,同时极大地增强了NQO1生物可激活药物。该方法还允许使用2至>4倍低的剂量的NQO1生物可激活药物,解决了这些NQO1生物可激活药物的毒性作用(例如,高铁血红蛋白血症)。在治疗方法中,抑制剂可在NQO1生物可激活药物之前和之后添加,至少在之前,并且任选地在之前、同时、之后,或其组合添加。细胞死亡响应取决于所使用的抑制剂。响应是不明显的并且将需要在体内追踪的特异性生物标志物。
本公开内容提供了DNQ化合物、β拉帕醌,及其衍生物,以及NQO1生物可激活药物在治疗癌症的组合治疗中的用途。DNQ化合物的一些实例包括式(I)化合物或其盐或溶剂合物:

其中:
R1、R2、R3和R4各自独立地是-H或-X-R;
每个X独立地是直接键或桥基,其中桥基是-O-、-S-、-NH-、-C(=O)-、-O-C(=O)-、-C(=O)-O-、-O-C(=O)-O-或式-W-A-W-接头,其中:
每个W独立地是-N(R′)C(=O)-、-C(=O)N(R′)-、-OC(=O)-、-C(=O)O-、-O-、-S-、-S(O)-、-S(O)2-、-N(R′)-、-C(=O)-、-(CH2)n-,其中n为1至10,或者直接键,其中每个R′独立地是H、(C1-C6)烷基或氮保护基;并且每个A独立地是(C1-C20)烷基、(C2-C16)烯基、(C2-C16)炔基、(C3-C8)环烷基、(C6-C10)芳基、-(OCH2-CH2)n-,其中n为1至约20、-C(O)NH(CH2)n-,其中n为1至约6、-OP(O)(OH)O-、-OP(O)(OH)O(CH2)n-,其中n为1至约6、或者在两个碳之间或在碳与氧之间间插有环烷基、杂环或芳基的(C1-C20)烷基、(C2-C16)烯基、(C2-C16)炔基或-(OCH2-CH2)n-;
每个R独立地是烷基、烯基、炔基、杂烷基、环烷基、环烯基、杂环烷基、杂环烯基、(环烷基)烷基、(杂环烷基)烷基、(环烷基)杂烷基、(杂环烷基)杂烷基、芳基、杂芳基、(芳基)烷基、(杂芳基)烷基、氢、羟基、羟烷基、烷氧基、(烷氧基)烷基、烯氧基、炔氧基、(环烷基)烷氧基、杂环烷基氧基、氨基、烷基氨基、氨基烷基、酰氨基、芳基氨基、磺酰基氨基、亚磺酰基氨基、-CORx、-COORx、-CONHRx、-NHCORx、-NHCOORx、-NHCONHRx、-N3、-CN、-NC、-NCO、-NO2、-SH、-卤素、烷氧羰基、烷基氨基羰基、磺酸盐/酯、磺酸、烷基磺酰基、烷基亚磺酰基、芳基磺酰基、芳基亚磺酰基、氨基磺酰基、RxS(O)Ry-、RxS(O)2Ry-、RxC(O)N(Rx)Ry-、RxSO2N(Rx)Ry-、RxN(Rx)C(O)Ry-、RxN(Rx)SO2Ry-、RxN(Rx)C(O)N(Rx)Ry-、甲醛、酰基、酰氧基、-OPO3H2、-OPO3Z2,其中Z是无机阳离子、或糖;其中每个Rx独立地是H、OH、烷基或芳基,并且每个Ry独立地是基团W;其中任何烷基或芳基可任选地被一个或更多个羟基、氨基、氰基、硝基或卤素基团取代。
在一些实施方案中,当R1、R2和R3是甲基时,R4不是H或甲基。在另一些实施方案中,当R1、R3和R4是甲基时,R2的基团-X-R不是-CH2-OAc。在某些实施方案中,当R1、R3和R4是甲基时,R2的R基团不是酰氧基。在多个实施方案中,R1至R4不是各自为H。在某些实施方案中,R1至R4不是各自为烷基,例如未经取代的烷基。在一些实施方案中,R1至R4不是各自为甲基。
在一个实施方案中,R1、R2、R3和R4各自为(C1-20)烷基。在一些实施方案中,(C1-20)烷基是(C2-20)烷基、(C3-20)烷基、(C4-20)烷基、(C5-20)烷基,或(C10-20)烷基。烷基可例如被羟基或磷酸基团取代。磷酸基团可以是膦酸或膦酸盐,例如锂盐、钠盐、钾盐或其他已知的膦酸盐。
R1的特定值是H。R2的特定值是H。R3的特定值是H。R4的特定值是H。
R1的特定值是甲基。R2的特定值是甲基。R3的特定值是甲基。R4的特定值是甲基。甲基可如以上针对术语“经取代的”所述被取代。
在式(I)的一些实施方案中:
R1和R2是甲基;R3是氢;并且R4是2-甲基-丙烷;
R1和R2是甲基;R3是氢;并且R4是丁基;
R1和R4是甲基,并且R3是氢;并且R2是乙基;
R1和R2是甲基,并且R3是氢;并且R4是乙基;
R1是甲基;R3是氢;R2是丙基;并且R4是丁基;
R1和R4是甲基;R2是丙基,并且R3是氢;
R1是丙基;R2和R4是甲基,并且R3是氢;
R1和R2是乙基;R3是氢;并且R2是甲基;
R1是丙基;R2是甲基;R3是氢;并且R4是丁基;
R1和R2是丙基;R3是氢;并且R4是丁基;
R1和R2是甲基;R3是氢;并且R4是C12烷基;
R1和R2是甲基;R3是氢;并且R4是叔丁基;
R1和R2是甲基;R3是氢;并且R4是羟丙基;
R1和R2是甲基;R3是氢;并且R4是3,3-二甲基丁基[-CH2CH2C(CH3)2CH3];
R1和R2是甲基;R3是氢;并且R4是3-甲基丁基[-CH2CH2CH(CH3)CH3];
R2和R4是甲基;R3是氢;并且R1是乙基;
R1和R2是甲基;R3是氢;并且R4是丙基;
R1和R2是甲基;R3是氢;并且R4是正戊基;
R1和R2是甲基;R3是氢;并且R4是正己基;
R1和R2是甲基;R3是氢;并且R4是异丙基;
R1和R2是甲基;R3是氢;并且R4是环辛基;
R1和R2是甲基;R3是氢;并且R4是环丙基;
R1和R2是甲基;R3是氢;并且R4是甲基环丙基;
R1和R2是甲基;R3是氢;并且R4为乙基环丙基;
R1是C12烷基;R2和R4是甲基;并且R3是氢;
R1和R4是甲基;R3是氢;并且R2是C12烷基;
R1、R2和R3是甲基;并且R4是-CH2OPO3Na2;
R1是-CH2OPO3Na2;R2和R3是甲基;并且R4是氢;
R1和R3是甲基;R2是-CH2OPO3Na2;并且R4是氢;
R1和R2是甲基;R3是-CH2OPO3Na2;并且R4是氢;
R1和R2是甲基;R3是-CH2CH2OPO3Na2;并且R4是氢;
R1、R2和R3是甲基;并且R4是-CH2OH;
R1是-CH2OH;R2和R3是甲基;并且R4是氢;
R1和R3是甲基;R2是-CH2OH;并且R4是氢;
R1和R2是甲基;R3是-CH2OH;并且R4是氢;或者
R1和R2是甲基;R3是-CH2CH2OH;并且R4是氢。
在式I的某些实施方案中,R1是(C1-4)烷基。在某些情况下,R1是(C1-3)烷基。在某些情况下,R1是(C1-2)烷基。
在式I的某些实施方案中,R2是(C1-4)烷基。在某些情况下,R2是(C1-3)烷基。在某些情况下,R2是(C1-2)烷基。
在式I的某些实施方案中,R3是氢。
在式I的某些实施方案中,R4是任选地经取代的(C1-10)烷基,其中该烷基被羟基、卤素、氨基或硫醇取代。在某些情况下,R4是(C1-10)烷基、(C1-8)烷基、(C1-6)烷基或(C1-4)烷基。在某些情况下,R4是(C2-6)烷基。在某些情况下,R4是经取代的(C1-10)烷基、经取代的(C1-8)烷基、经取代的(C1-6)烷基或经取代的(C1-4)烷基,其中烷基被羟基、卤素、氨基或硫醇取代。在某些情况下,R4是被羟基取代的烷基。在某些情况下,R4是被卤素取代的烷基。在某些情况下,R4是被氨基取代的烷基。在某些情况下,R4是被硫醇取代的烷基。
在式I的某些实施方案中,R1和R2独立地是(C1-4)烷基;R3是氢;并且R4为任选地经取代的(C1-10)烷基,其中烷基被羟基、卤素、氨基和硫醇取代。
在式I的某些实施方案中,R1和R2独立地是(C1-2)烷基;R3是氢;并且R4为任选地经取代的(C1-10)烷基,其中烷基被羟基、卤素、氨基和硫醇取代。
在式I的某些实施方案中,R1和R2独立地是(C1-2)烷基;R3为氢;并且R4为(C1-10)烷基。在式I的某些实施方案中,R1和R2独立地是(C1-2)烷基;R3为氢;并且R4为(C1-8)烷基。在式I的某些实施方案中,R1和R2独立地是(C1-2)烷基;R3为氢;并且R4为(C1-6)烷基。在式I的某些实施方案中,R1和R2独立地是(C1-2)烷基;R3为氢;并且R4为(C1-4)烷基。在式I的某些实施方案中,R1和R2独立地是(C1-2)烷基;R3为氢;并且R4为(C2-6)烷基。在式I的某些实施方案中,R1和R2独立地是(C1-2)烷基;R3为氢;并且R4为经取代的(C1-6)烷基,其中烷基被羟基、卤素、氨基和硫醇取代。在式I的某些实施方案中,R1和R2独立地是(C1-2)烷基;R3为氢;并且R4为经取代的(C1-4)烷基,其中烷基被羟基、卤素、氨基和硫醇取代。
在某些实施方案中,式I化合物是化合物87或其盐或溶剂合物:

在某些实施方案中,式I化合物是化合物9-253或其盐或溶剂合物:

在某些实施方案中,式I化合物是化合物9-251或其盐或溶剂合物:

在某些实施方案中,式I化合物是化合物10-41或其盐或溶剂合物:

在某些实施方案中,式I化合物是化合物109或其盐或溶剂合物:

在某些实施方案中,式I化合物是化合物107或其盐或溶剂合物:

在某些实施方案中,式I化合物是化合物9-281或其盐或溶剂合物:

在某些实施方案中,式I化合物是化合物9-249或其盐或溶剂合物:

在某些实施方案中,式I化合物是化合物9-255或其盐或溶剂合物:

在某些实施方案中,式I化合物是化合物9-257或其盐或溶剂合物:

本公开内容还提供了包含式(I)化合物和可药用稀释剂、赋形剂或载体的药物组合物。载体可以是水,例如在存在羟丙基-β-环糊精(hydroxypropyl-β-cyclodextrin,HPβCD)的情况下。与不含HPβCD的情况下化合物在水中的溶解度相比,该化合物的溶解度可提高约100倍、约200倍、约500倍、约1000倍、约2000倍或约3000倍。国际申请No.PCT/US12/59988(Hergenrother et al.)描述了另外的DNQ化合物和方法。
对于包含一个或更多个取代基的以上式或基团中的任一个,应当理解,这样的基团不包含空间上不切实际和/或合成上不可行的任何取代或取代模式。另外,本公开内容的化合物包括由这些化合物的取代产生的所有立体化学异构体。
本文中所述化合物的选定取代基可以以递归度存在。在该上下文中,“递归取代基(recursive substituent)”意指取代基可引用其自身的另一个实例。由于这样的取代基的递归性质,理论上,在任何给定的权利要求中都可能存在大量取代基。药物化学和有机化学领域的普通技术人员理解,这样的取代基的总数目受到预期化合物的期望特性的合理限制。这样的特性包括,例如且不限于物理特性例如分子量、溶解度或log P,应用特性例如针对预期靶标的活性,以及实用特性例如容易合成。
递归取代基是本公开内容的预期方面。药物化学和有机化学领域的普通技术人员理解这样的取代基的通用性。就递归取代基存在于本公开内容的权利要求书中的程度而言,总数目将如上所述确定。在一些实施方案中,递归取代基仅以这样的程度存在:化合物的分子量为约400至约1600、约450至约1200、约500至约100、约600至约800。在另一些实施方案中,递归取代基仅以这样的程度存在:化合物的分子量小于2000、小于1800、小于1600、小于1500、小于1400、小于1200、小于1000、小于900、小于800、小于750、小于700或小于约600。
患有具有升高的NQO1水平的实体瘤的患者可通过施用有效量的药物活性形式的DNQ和/或DNQd(DNQ化合物)来治疗。以下方案示出了数个更具体的式:

对于DNQd-27和DNQd-28,X可以是式-W-A-W-接头或二价桥基,例如二价烷基、烯基、炔基、杂烷基、无环烷基(acycloalkyl)、环烯基、杂环烷基、杂环烯基、环烷基烷基、杂环烷基烷基、环烷基杂烷基、杂环烷基杂烷基、烷氧基、烷氧基烷基、烯氧基、炔氧基、环烷基氧基、杂环烷基氧基、氨基、烷基氨基、氨基烷基、酰氨基、芳基氨基、磺酰基氨基、亚磺酰基氨基、烷氧羰基、烷基氨基羰基、磺酰基、烷基磺酰基、烷基亚磺酰基、芳基磺酰基、芳基亚磺酰基、氨基磺酰基或酰基,其各自可任选地被取代。
对于DNQd-29,每个X可独立地是如上针对DNQd-27和DNQd-28所述的式-W-A-W-接头或二价桥基;并且每个Y可独立地是:


本文中所述化合物的可药用盐在本公开内容的范围内并且包括保留了期望的药理活性并且不是生物学上不期望的酸或碱加成盐(例如盐没有过度的毒性、变应原性或刺激性,并且是生物可利用的)。当化合物具有碱性基团(例如如氨基)时,可与以下形成可药用盐:无机酸(例如盐酸、氢硼酸、硝酸、硫酸和磷酸)、有机酸(例如褐藻酸、甲酸、乙酸、苯甲酸、葡糖酸、富马酸、草酸、酒石酸、乳酸、马来酸、柠檬酸、琥珀酸、苹果酸、甲磺酸、苯磺酸、萘磺酸和对甲苯磺酸)或酸性氨基酸(例如天冬氨酸和谷氨酸)。当本公开内容的化合物具有酸性基团(例如如羧酸基团)时,其可与以下形成盐:金属,例如碱金属和碱土金属(例如Na+、Li+、K+、Ca2+、Mg2+、Zn2+)、氨或有机胺(例如二环己胺、三甲胺、三乙胺、吡啶、甲基吡啶、乙醇胺、二乙醇胺、三乙醇胺)或碱性氨基酸(例如精氨酸、赖氨酸和鸟氨酸)。这样的盐可在化合物的分离和纯化期间原位制备,或者通过使经纯化的化合物以其游离碱或游离酸形式分别与合适的酸或碱单独反应并分离由此形成的盐来制备。
本文中公开的许多分子包含一个或更多个可电离基团(可从中去除质子(例如-COOH)或添加质子(例如胺)或可被季铵化(例如胺)的基团)。这样的分子及其盐的所有可能的离子形式均旨在单独地包括在本文中公开内容中。关于本文中所述化合物的盐,本领域普通技术人员可从适用于制备本公开内容的盐用于给定应用的多种那些可用反离子中进行选择。在具体应用中,用于制备盐的给定阴离子或阳离子的选择可导致提高或降低该盐的溶解度。
本文中所述化合物的合适盐的一些实例包括其盐酸盐、氢溴酸盐、硫酸盐、甲磺酸盐、硝酸盐、马来酸盐、乙酸盐、柠檬酸盐、富马酸盐、酒石酸盐(例如(+)-酒石酸盐、(-)-酒石酸盐或其混合物包括外消旋混合物)、琥珀酸盐、苯甲酸盐和与氨基酸(例如谷氨酸)一起的盐。这些盐可通过本领域技术人员已知的方法来制备。还包括碱加成盐,例如钠、钾、钙、铵、有机氨或镁盐,或类似的盐。当本公开内容的化合物包含相对碱性的官能团时,酸加成盐可通过使中性形式的这样的化合物与足量的所期望酸(无论是纯的还是在合适的惰性溶剂中)接触来获得。可用酸加成盐的一些实例包括来源于无机酸的那些,所述无机酸例如盐酸、氢溴酸、硝酸、碳酸、一氢碳酸、磷酸、一氢磷酸、二氢磷酸、硫酸、一氢硫酸、氢碘酸或亚磷酸等,以及来源于有机酸的盐,所述有机酸例如乙酸、丙酸、异丁酸、马来酸、丙二酸、苯甲酸、琥珀酸、辛二酸、富马酸、乳酸、扁桃酸、邻苯二甲酸、苯磺酸、对甲苯磺酸、柠檬酸、酒石酸、甲磺酸等。还包括氨基酸(例如精氨酸等)的盐,以及有机酸(例如葡糖醛酸或半乳糖醛酸等)的盐。本公开内容的某些特定化合物可包含允许化合物转化成碱加成盐或酸加成盐的碱性官能团和酸性官能团二者。
本公开内容的某些化合物可以以非溶剂化形式以及溶剂化形式,包括水合形式存在。一般而言,溶剂化形式等同于非溶剂化形式并且涵盖在本公开内容的范围内。本公开内容的某些化合物可以以多种结晶或无定形形式存在。一般而言,所有物理形式对于本公开内容所预期的用途均是等同的,并且旨在在本公开内容的范围内。
术语“溶剂合物”是指这样的固体化合物,其具有与其固体结构缔合的一个或更多个溶剂分子。当化合物从溶剂中结晶时可形成溶剂合物。当一个或更多个溶剂分子在凝固时成为固体结晶基质的整合部分时,就会形成溶剂合物。本文中所述式化合物可以是溶剂合物,例如乙醇溶剂合物。另一类型的溶剂合物是水合物。“水合物”同样是指这样的固体化合物,其具有在分子水平上与其固体或结晶结构密切缔合的一个或更多个水分子。当化合物在水中固化或结晶时可形成水合物,其中一个或更多个水分子成为固体结晶基质的整合部分。本文中所述式化合物可以是水合物。
II.制备DNQ化合物的方法
本发明还涉及制备本发明的化合物和组合物的方法。所述化合物和组合物可通过有机合成的任何适用性技术来制备。许多这样的技术是本领域公知的。然而,许多已知技术在以下中阐述:Compendium of Organic Synthetic Methods(John Wiley&Sons,NewYork),Vol.1,Ian T.Harrison和Shuyen Harrison,1971;Vol.2,Ian T.Harrison和ShuyenHarrison,1974;Vol.3,Louis S.Hegedus和Leroy Wade,1977;Vol.4,Leroy G.Wade,Jr,1980;Vol.5,Leroy G.Wade,Jr,1984;和Vol.6,Michael B.Smith;以及标准的有机参考文本例如March′s Advanced Organic Chemistry:Reactions,Mechanisms,和Structure,5thEd.M.B.Smith和J.March(John Wiley&Sons,New York,2001),Comprehensive OrganicSynthesis;Selectivity,Strategy&Efficiency in Modern Organic Chemistry,在第9卷中,Barry M.Trost,首席编辑(Pergamon Press,New York,1993印刷));Advanced OrganicChemistry,Part B:Reactions and Synthesis,第二版,Cary和Sundberg(1983);Protecting Groups in Organic Synthesis,第二版,Greene,T.W.,和Wutz,P.G.M.,JohnWiley&Sons,New York;以及Comprehensive Organic Transformations,Larock,R.C.,第二版,John Wiley&Sons,New York(1999),其各自通过引用并入在此。
下面提供了用于制备本公开内容的组合物的多个示例性方法。这些方法旨在举例说明这样的制剂的性质,并不旨在限制适用方法的范围。另外的方法和可用技术描述于WO2013/056073(Hergenrother et al.)中。
通常,反应条件例如温度、反应时间、溶剂、后处理程序(work-upprocedure)等将是本领域中对于待进行的特定反应常用的那些。引用的参考材料连同其中引用的材料包含对这样的条件的详细描述。通常来说,温度将为-100℃至200℃,溶剂将取决于所需条件是非质子的或质子的,并且反应时间将为1分钟至10天。后处理通常由以下组成:使任何未反应的试剂淬灭,随后在水/有机层系统之间进行分配(萃取)并分离包含产物的层。氧化和还原反应通常在接近室温的温度(约20℃)下进行,但是对于金属氢化物还原反应温度通常会降至0℃至-100℃。适当时也可使用加热。溶剂对于还原通常是非质子的,并且对于氧化可以是质子的或非质子的。调整反应时间以实现所期望的转化。
缩合反应通常在接近室温的温度下进行,但是对于非平衡、动力学控制的缩合,降低的温度(0℃至-100℃)也很常见。溶剂可以是质子的(常见于平衡反应)或非质子的(常见于动力学控制的反应)。标准合成技术例如共沸去除反应副产物和使用无水反应条件(例如惰性气体环境)在本领域中是常见的并且将在适用时应用。
保护基。术语“保护基”、“阻断基团”或“PG”是指当与羟基或其他杂原子结合时防止在该基团处发生不期望的反应并且可通过常规化学或酶促步骤去除以重建羟基的任何基团。所应用的特定可去除阻断基团并不总是关键的,并且优选的可去除羟基阻断基团包括常规取代基,例如如烯丙基、苄基、乙酰基、氯乙酰基、硫苄基、亚苄基、苯甲酰甲基、甲基甲氧基、甲硅烷基醚(例如三甲基甲硅烷基(TMS)、叔丁基-二苯基甲硅烷基(t-butyl-diphenylsilyl,TBDPS)或叔丁基二甲基甲硅烷基(t-butyldimethylsilyl,TBS))以及可化学引入到羟基官能团上,并随后在与产物性质相容的温和条件下通过化学或酶促方法选择性去除的任何其他基团。式(I)的R基团也可以是保护基,如本文中所述。
合适的羟基保护基是本领域技术人员已知的,并更详细地公开于T.W.Greene,Protecting Groups In Organic Synthesis;Wiley:New York,1981(″Greene″)和其中引用的参考文献,以及Kocienski,Philip J.;Protecting Groups(Georg Thieme VerlagStuttgart,New York,1994)中,其二者均通过引用并入本文。
保护基是可获得的、公知的和使用的,并且任选地用于在合成步骤(即通过本公开内容的方法制备化合物的途径或方法)期间防止与保护基发生副反应。在大多数情况下,决定保护哪些基团、何时保护以及化学保护基“PG”的性质将取决于待保护免于的反应的化学性质(例如酸性、碱性、氧化、还原或其他条件)和合成的预期方向。
如果化合物被多个PG取代,则保护基不必相同,通常也不同。一般而言,PG将用于保护官能团例如羧基、羟基、硫基或氨基,并因此防止副反应或以其他方式促进合成效率。产生游离脱保护基的脱保护顺序取决于合成的预期方向和待遇到的反应条件,并且可按照技术人员确定的任何顺序进行。
本公开内容的化合物的多种官能团可被保护。例如,针对-OH基(无论是羟基、羧酸或其他官能团)的保护基包括“形成醚或酯的基团”。形成醚或酯的基团能够在本文中所述的合成方案中用作化学保护基。然而,如本领域技术人员将理解地,一些羟基和硫基保护基既不是醚形成基团也不是酯形成基团。关于羧酸保护基和针对酸的其他保护基的更多细节,参见以上引用的Greene。这样的基团包括例如但不限于酯、酰胺、酰肼等。
III.检查点抑制剂
检查点抑制剂治疗是目前正在研究的癌症治疗免疫治疗的一种形式。该治疗靶向免疫检查点(即免疫系统的刺激或抑制其作用的关键调节剂),肿瘤可利用所述免疫检查点来保护自身免受免疫系统的攻击。检查点治疗可阻断抑制性检查点,恢复免疫系统功能。靶向免疫检查点的首个抗癌药物是易普利姆玛(ipilimumab),即2011年在美国批准的CTLA-4阻断剂。
目前批准的检查点抑制剂靶向分子CTLA4、PD-1和PD-L1。PD-1是跨膜程序性细胞死亡1蛋白(也称为PDCD1和CD279),其与PD-L1(PD-1配体1,或CD274)相互作用。细胞表面上的PD-L1与免疫细胞表面上的PD1结合,其抑制免疫细胞活性。在PD-L1功能中,对T细胞活性发挥关键的调节作用。显示(癌症介导的)细胞表面上PD-L1的上调可抑制可能以其他方式攻击的T细胞。与PD-1或PD-L1结合并因此阻断相互作用的抗体可允许T细胞攻击肿瘤。
在一些实施方案中,免疫检查点抑制剂治疗可以是靶向以下的分子:腺苷A2A受体(A2A receptor,A2AR)、B7-H3(也称为CD276)、B和T淋巴细胞弱化子(B and T lymphocyteattenuator,BTLA)、细胞毒性T淋巴细胞相关蛋白4(cytotoxic T-lymphocyte-associatedprotein 4,CTLA-4,也称为CD152)、吲哚胺2,3-双加氧酶(indoleamine 2,3-dioxygenase,IDO)、杀伤细胞免疫球蛋白(killer-cell immunoglobulin,KIR)、淋巴细胞活化基因3(lymphocyte activation gene-3,LAG3)、T细胞免疫球蛋白结构域和黏蛋白结构域3(T-cell immunoglobulin domain and mucin domain 3,TIM-3)和T细胞活化的V结构域Ig抑制剂(V-domain Ig suppressor of T cell activation,VISTA)。
免疫检查点抑制剂可以是药物,例如小分子、重组形式的配体或受体、或者特别是抗体,例如人抗体(例如,国际专利公开号No.WO2015016718;二者均通过引用并入本文)。可使用免疫检查点蛋白或其类似物的已知抑制剂,特别地,可使用嵌合的、人源化的或人形式的抗体。如技术人员将知道的,替代和/或等同的名称可用于本公开内容中提及的某些抗体。在本公开内容的上下文中,这样的替代和/或等同的名称是可互换的。例如,已知拉立珠单抗(lambrolizumab)也以替代和等同的名称MK-3475和派姆单抗(pembrolizumab)而为人所知。
IV.药物组合物和治疗方法
A.药物组合物
以下描述了与一些药物和药理学实施方案相关的信息,并由普通技术人员可获得的本领域信息进一步补充。确切的制剂、施用途径和剂量可由个体医师或临床医师根据患者的病症来选择(参见例如Fingl et al.,in The Pharmacological Basis ofTherapeutics,1975,Ch.1)。
应当注意的是,由于毒性或由于器官功能障碍等,主治医师会知道如何并且何时终止、中断或调整施用。相反地,如果临床响应不充分(鉴于或排除毒性方面),则主治医师也会知道将治疗调整到更高水平。管理目的病症的施用剂量的大小可随着待治疗病症的严重程度和施用途径而变化。例如,可例如部分地通过标准预后评价方法来评价病症的严重程度。此外,剂量以及可能剂量频率也可根据情况而变化,例如个体患者的年龄、体重和响应。与上面讨论的那些相当的程序也可用于兽医学。
取决于所治疗的具体病症和选择的靶向方法,可配制这样的药剂并且将其全身或局部地施用。用于配制和施用的技术可见于Alfonso和Gennaro(1995)和本领域其他地方中。
化合物可与可药用的载体、稀释剂或赋形剂组合施用于患者。短语“可药用”是指在合理的医学判断范围内适合于用于与人和动物的组织接触而没有过多毒性、刺激、变应性响应或其他问题或并发症的与合理的益处/风险比相称的那些配体、材料、组合物和/或剂型。
短语“可药用载体″包括任何和所有的溶剂、分散体介质、稀释剂、包衣剂、表面活性剂、抗氧化剂、防腐剂(例如,抗细菌剂、抗真菌剂)、等张剂、吸收延迟剂、盐类、缓冲剂、防腐剂、药物、药物稳定剂、凝胶剂、黏合剂、赋形剂、崩解剂、润滑剂、甜味剂、矫味剂、染料、类似物质及其组合,如本领域普通技术人员所知的(参见,例如,Remington′sPharmaceutical Sciences,18th Ed.Mack Printing Company,1990,pp.1289-1329,其通过引用并入本文)。除非任何常规载体与活性成分不相容,否则考虑其在化学治疗组合物或药物组合物中的用途。
DNQd或DNQ化合物可与不同类型的载体组合,这取决于其是以固体、液体还是以气雾剂形式施用,以及其对于这样的施用途径例如注射是否需要是无菌的。本公开内容可通过静脉内、皮内、动脉内、腹膜内、病灶内、颅内、关节内、前列腺内、胸膜内、气管内、鼻内、玻璃体内、阴道内、直肠内、表面、瘤内、肌内、腹膜内、皮下、结膜下、囊内、经黏膜、心包内、脐内、眼内、经口、经表面、局部、注射、输注、连续输注、直接局部灌注浸浴靶细胞、通过导管、通过灌洗、以脂质组合物(例如,脂质体),或通过本领域普通技术人员已知其他方法或前述的任意组合进行施用(参见,例如,Remington′s Pharmaceutical Sciences,18th Ed.MackPrinting Company,1990,其通过引用并入本文)。
向患者施用的本公开内容组合物的实际剂量可由物理和生理因素确定,所述因素例如体重、病症的严重程度、所治疗疾病的类型、先前或同时治疗干预、患者的特发病和施用途径。在任何情况下,负责施用的实践者将确定组合物中活性成分的浓度和个体对象的合适剂量。
当向对象施用时,有效量将自然取决于所治疗的特定癌症;具体癌症的基因型;癌症的严重程度;个体患者参数,包括年龄、身体状况、大小和体重、同时治疗、治疗频率和施用方式。这些因素对医师来说是公知的,并且可仅仅通过常规实验即可解决。在一些实施方案中,根据合理的医学判断,优选使用最高安全剂量。
在某些实施方案中,药物组合物可包含例如至少约0.1%的DNQd或DNQ化合物。在另一些实施方案中,活性化合物可例如包含单位重量的约2%至约75%,或约25%至约60%,以及其中可推导出的任何范围。在另一些非限制性实例中,剂量还可包含约0.1mg/kg/体重、0.5mg/kg/体重、1mg/kg/体重、约5mg/kg/体重、约10mg/kg/体重、约20mg/kg/体重、约30mg/kg/体重、约40mg/kg/体重、约50mg/kg/体重、约75mg/kg/体重、约100mg/kg/体重、约200mg/kg/体重、约350mg/kg/体重、约500mg/kg/体重、约750mg/kg/体重至约1000mg/kg/体重或更多/施用,以及其中可推导出的任何范围。在从本文中所列数字可推导出的范围的一些非限制性实例中,基于上述数字,可施用约10mg/kg/体重至约100mg/kg/体重等。
在任何情况下,组合物可包含多种抗氧化剂以延迟一种或更多种组分的氧化。另外,通过防腐剂可产生对微生物作用的防止,所述防腐剂例如多种抗细菌剂和抗真菌剂,包括但不限于对羟基苯甲酸酯(例如,对羟基苯甲酸甲酯、对羟基苯甲酸丙酯)、氯丁醇、酚、山梨酸、硫柳汞或其组合。
可将本文中所述的活性物例如DNQd或DNQ化合物配制成以游离碱、中性或盐形式的组合物。可药用盐包括与来源于无机碱(例如如钠、钾、铵、钙或铁的氢氧化物);或这样的有机碱(例如异丙胺、三乙胺、组氨酸或普鲁卡因(procaine))的游离羧基一起形成的盐。
用于经口使用的药物制剂可通过将活性化合物与固体赋形剂组合来获得,如果期望的话,在添加合适的辅料之后,任选地研磨所得混合物并加工颗粒混合物以获得片剂或糖衣丸芯。合适的赋形剂特别是填充剂,例如糖,包括乳糖、蔗糖、甘露糖醇或山梨糖醇;纤维素制剂,例如如玉米淀粉、小麦淀粉、稻米淀粉、马铃薯淀粉、明胶、黄蓍胶、甲基纤维素、羟丙基甲基纤维素、羧甲基纤维素钠和/或聚乙烯吡咯烷酮(polyvinylpyrrolidone,PVP)。如果期望的话,可添加崩解剂,例如交联的聚乙烯吡咯烷酮、琼脂、或者藻酸或其盐(例如藻酸钠)。
糖衣丸芯任选地提供有合适的包衣。出于此目的,可使用浓缩糖溶液,其可任选地包含阿拉伯胶、滑石、聚乙烯吡咯烷酮、卡波姆凝胶(carbopolgel)、聚乙二醇和/或二氧化钛、漆溶液,以及合适的有机溶剂或溶剂混合物。可将染料或色素添加至片剂或糖衣丸剂包衣以用于鉴别或表征活性化合物剂量的不同组合。
在其中组合物为液体形式的一些实施方案中,载体可以是溶剂或分散体介质,包括但不限于水、乙醇、多元醇(例如,甘油、丙二醇、液体聚乙二醇等)、脂质(例如,甘油三酯、植物油、脂质体),及其组合。可维持适当的流动性,例如通过使用包衣例如卵磷脂;通过分散在载体例如如液体多元醇或脂质中维持所需的颗粒尺寸;通过使用表面活性剂,例如如羟丙基纤维素(HPC);或其这样的方法的组合。在许多情况下,将优选包含等张剂,例如如糖、氯化钠、或其组合。
无菌可注射溶液通过根据需要将活性化合物并入在所需量的与以上列举的多种其他成分一起的适当的溶剂中,随后过滤灭菌来制备。通常,通过将多种经灭菌活性成分并入到包含基础分散体介质和/或其他成分的无菌载剂中来制备分散体。在用于制备无菌可注射溶液剂、混悬剂或乳剂的无菌粉末的情况下,优选的制备方法是真空干燥或冷冻干燥技术,这由其经先前无菌过滤的液体介质产生活性成分加任何另外期望成分的粉末。如果必要的话,液体介质应被适当地缓冲,并且在用足够的盐水或葡萄糖注射之前先使液体稀释剂等张。
组合物在制造和储存条件下应当是稳定的,并且防止微生物(例如细菌和真菌)的污染作用。因此,优选组合物具有大于约5,优选约5至约8,更优选约5至约7的pH。将理解,内毒素污染应在安全水平下保持最低,例如小于0.5ng/mg蛋白质。
在一些特定实施方案中,通过在组合物中使用延迟吸收的试剂(例如如单硬脂酸铝、明胶或其组合),可实现可注射组合物的延长吸收。
B.用于体内施用的DNQ化合物的制剂
通过LC-MS测量DNQ在pH 7.4时在磷酸缓冲盐水(PBS)中的水溶解度。将DNQ在PBS中声处理30分钟,然后通过0.45μm注射器式过滤器过滤来去除未溶解的固体,并通过LC-MS(λ=275nm,ESI-TOF负模式)来分析滤液。通过将DNQ声处理1、5、10和30分钟来确定最佳声处理时间。虽然溶液中DNQ的浓度在1、5至10分钟显著提高,但在10至30分钟仅很小的差异。在30分钟的声处理期间,将水浴温热至45℃(将样品冷却至室温,然后过滤)。通过将DNQ溶解在甲醇中至500μM的浓度并在80∶20 水∶甲醇中使该储液(stock)稀释,来从1至100μM生成校准曲线。该校准曲线(通过UV吸光度测量)在此范围内呈线性;1μM大约是检测限。测得DNQ在PBS中的溶解度为115μM。溶液呈非常淡的黄色。
由于DNQ的水溶解度较差,本发明人研究了使用2-羟丙基-β-环糊精(hydroxypropyl-beta-cyclodextrin,HPβCD)(常见赋形剂)来提高DNQ的溶解度。在不存在HPβCD的情况下,在强碱性溶液中DNQ的溶解度显著提高,并且当pH恢复至中性时DNQ会沉淀。然而,在存在足量的HPβCD的情况下,当pH恢复至中性时DNQ不会沉淀。DNQ在HPβCD中的这种相同的中性溶液不能直接制备(即在无pH调整的情况下)。这表明DNQ化合物在碱中去质子化,并且这种去质子化的分子与HPβCD形成紧密的复合体,其足够稳定以防止随着pH值降低而发生质子化。DNQ上可在水性碱中合理去质子化的唯一质子是N-H。尽管尚未测量DNQ的N-H键的酸度,但已对DNQ的衍生物进行了测量,并且发现其pKa为8.0。
用于配制HPβCD中的DNQ化合物的方案如下:将DNQ化合物在HPβCD在pH 7.4 PBS中的20%溶液中制成浆状物,并随后通过添加10M NaOH来提高pH以诱导DNQ化合物溶解。通过小心地添加1M HCl使pH恢复至pH 7.5至8.0。通过这种方法可制备DNQ化合物的3.3mM溶液,该溶液稳定至少24小时。这表明相对于单独的PBS,DNQ的溶解度提高了30倍。本发明人初始选择了20%HPβCD溶液。然而,本发明人发现β-lap被配制为HPβCD的40%溶液用于人临床试验,并且本发明人对DNQ的经验表明DNQ的浓度随HPβCD的浓度而线性提高;因此,40%HPβCD溶液将允许产生DNQ和其他DNQ化合物的6.6mM溶液。
C.治疗
本公开内容还提供了治疗具有有升高的NQO1水平的肿瘤细胞的患者的方法。该方法可包括向具有有升高的NQO1水平的肿瘤细胞的患者施用治疗有效量的式(I)化合物或本文中所述的组合物。本公开内容还提供了治疗具有升高的NQO1水平的肿瘤细胞的方法,其包括将肿瘤细胞暴露于治疗有效量的本文中所述的化合物或组合物,其中肿瘤细胞被治疗、杀伤或抑制生长。肿瘤或肿瘤细胞可以是恶性肿瘤细胞。在一些实施方案中,肿瘤细胞是癌细胞,例如非小细胞肺癌。
因此,本公开内容的方法可用于治疗或预防多种瘤形成病症,包括肢端雀斑样痣黑素瘤、光化性角化病、腺癌、腺样囊性癌(adenoid cystic carcinoma)、腺瘤、腺肉瘤、腺鳞癌、星形细胞肿瘤、前庭大腺癌、基底细胞癌、支气管腺癌、毛细血管类癌、癌、癌肉瘤、海绵状胆管癌、软骨肉瘤(chondosarcoma)、脉络丛乳头状瘤/癌、透明细胞癌、囊腺瘤、内胚窦瘤、子宫内膜增生、子宫内膜间质肉瘤、子宫内膜样腺瘤、室管膜癌、上皮样癌、尤因肉瘤(Ewing′s sarcoma)、纤维板层样癌、局灶性结节性增生、胃泌素瘤、生殖细胞肿瘤、胶质母细胞瘤、胰高血糖素瘤、血管母细胞瘤(hemangiblastomas)、血管内皮瘤、血管瘤、肝腺瘤、肝腺瘤病、肝细胞癌、胰岛素瘤、上皮内瘤形成(intaepithelial neoplasia)、上皮间鳞状细胞瘤形成、浸润性鳞状细胞癌、大细胞癌、平滑肌肉瘤、恶性雀斑样痣黑素瘤、恶性黑素瘤、恶性间皮肿瘤、髓母细胞瘤、髓上皮瘤、黑素瘤、脑膜,间皮,转移癌、黏液表皮样癌、神经母细胞瘤、神经上皮腺癌结节性黑素瘤、燕麦细胞癌、少突胶质肿瘤、骨肉瘤、胰腺多肽,乳头状浆液性腺癌、松果体细胞瘤、垂体瘤、浆细胞瘤、假性肉瘤、肺母细胞瘤、肾细胞癌、视网膜母细胞瘤、横纹肌肉瘤、肉瘤、浆液性癌、小细胞癌、软组织癌、生长抑素分泌肿瘤、鳞状癌、鳞状细胞癌、间皮下,浅表扩散性黑素瘤、未分化癌、葡萄膜黑素瘤、疣状癌、舒血管肠肽瘤、高分化癌和维尔姆斯瘤(Wilm′s tumor)。因此,本文中所述的组合物和方法可用于治疗膀胱癌、脑癌(包括颅内赘生物,例如胶质瘤、脑膜瘤(meninigioma)、神经鞘瘤和腺瘤)、乳腺癌、结肠癌、肺癌(SCLC或NSCLC)卵巢癌、胰腺癌和前列腺癌。
D.组合治疗
本文中所述活性成分(例如式(I)化合物)也可与其他活性成分组合使用。这样的组合基于待治疗的病症、成分的交叉反应性和组合的药物特性来选择。例如,当治疗癌症时,组合物可与其他抗癌化合物(例如紫杉醇(paclitaxel)或雷帕霉素(rapamycin))组合。
还可将本公开内容的化合物与一种或更多种其他活性成分以单一剂型组合以同时或依次施用于患者。组合治疗可以以同时或依次方案来施用。当依次施用时,组合可以以两次或更多次施用来施用。
组合治疗可提供“协同”和“协同性”,即当活性成分一起使用时达到的作用大于由单独使用化合物产生的作用的总和。当活性成分:(1)以组合制剂共配制并同时施用或递送;(2)作为单独制剂交替或并行递送;或(3)通过另一些方案进行时可获得协同作用。当以交替治疗递送时,当化合物依次施用或递送,例如以单独的片剂、丸剂或胶囊剂,或通过在单独注射器中不同注射来依次施用或递送时可获得协同作用。一般而言,在交替治疗期间,有效剂量的每种活性成分依次施用(即连续施用),而在组合治疗中,有效剂量的两种或更多种活性成分一起施用。协同抗癌作用表示比组合的单独化合物的所预测纯累加作用更大的抗癌作用。
美国专利No.6,833,373(McKearn et al.)进一步描述了组合治疗,其包括可与本文中所述的化合物组合的另外的活性剂,以及可用本文中所述化合物治疗的另外的类型的癌症和其他病症。
因此,本公开内容的一个方面是DNQd或DNQ可与另一种药剂或治疗方法,优选另一种癌症治疗组合使用。DNQd或DNQ可在其他药剂治疗之前或之后间隔从数分钟至数周进行。在其中其他药剂和表达构建体单独施加于细胞的一些实施方案中,通常应确保每次递送时间之间的有效时间段没有消逝,使得药剂和表达构建体仍能够对细胞发挥有利的组合作用。例如,在这样的情况下,预期可用两种、三种、四种或更多种方式使细胞、组织或有机体与活性剂基本上同时(即,在少于约一分钟内)接触。在另一些方面中,一种或更多种药剂可在施用活性剂之前和/或之后约1分钟、约5分钟、约10分钟、约20分钟、约30分钟、约45分钟、约60分钟、约2小时、约3小时、约4小时、约6小时、约8小时、约9小时、约12小时、约15小时、约18小时、约21小时、约24小时、约28小时、约31小时、约35小时、约38小时、约42小时、约45小时、至约48小时或更长时间内施用。在某些另一些实施方案中,药剂可在施用活性剂之前和/或之后约1天、约2天、约3天、约4天、约5天、约6天、约8天、约9天、约12天、约15天、约16天、约18天、约20天、至约21天内施用。在一些情况下,可能期望显著延长治疗时间段,然而其中在各施用之间间隔数周(例如约1、约2、约3、约4、约6或约8周或更多)。
将本公开内容的化学治疗组合物施用于患者将遵循用于施用化学治疗剂的一般方案,考虑毒性(如果有的话)。预期将根据需要重复治疗周期。还预期多种标准治疗或辅助癌症治疗以及手术干预可与所述的活性剂组合应用。这些治疗包括但不限于化学治疗、放射治疗、免疫治疗、基因治疗和手术。
i.化学治疗
癌症治疗可还包括多种与化学治疗和基于放射的治疗二者的组合治疗。组合化学治疗包括使用化学治疗剂,例如顺铂、依托泊苷(etoposide)、伊立替康(irinotecan)、camptostar、拓扑替康(topotecan)、紫杉醇、多烯紫杉醇(docetaxel)、埃博霉素(epothilones)、泰索帝(taxotere)、他莫昔芬(tamoxifen)、5-氟尿嘧啶(5-fluorouracil)、甲氨蝶呤、替莫唑胺(temozolomide)、环磷酰胺、SCH 66336、R115777、L778,123、BMS214662、IRESSATM(吉非替尼(gefitinib))、TARCEVATM(埃洛替尼盐酸盐(erlotinib hydrochloride))、针对EGFR的抗体、GLEEVECTM(伊马替尼(imatinib))、内含子、ara-C、阿霉素、环磷酰胺、吉西他滨(gemcitabine)、尿嘧啶氮芥(uracil mustard)、氮芥、异环磷酰胺、美法仑(melphalan)、苯丁酸氮芥、溴丙哌嗪(pipobroman)、三乙撑蜜胺(triethylenemelamine)、三亚乙基硫代磷胺(triethylenethiophosphoramine)、白消安(busulfan)、卡莫司汀(carmustine)、洛莫司丁(lomusitne)、链脲霉素(streptozocin)、达卡巴嗪(dacarbazine)、氟尿苷(floxuridine)、阿糖胞苷(cytarabine)、6-巯基嘌呤(mercaptopurine)、6-硫鸟嘌呤(thioguanine)、磷酸氟达拉滨(fludarabine phosphate)、喷司他汀(pentostatine)、长春花碱(vinblastine)、长春花新碱(vincristine)、长春花碱酰胺(vindesine)、博来霉素(bleomycin)、多柔比星(doxorubicin)、放线菌素D(dactinomycin)、柔红霉素(daunorubicin)、表柔比星(epirubicin)、伊达比星(idarubicin)、光神霉素(mithramycin)、脱氧助间型霉素(deoxycoformycin)、丝裂霉素-C(mitomycin-C)、L-天冬酰胺酶(L-asparaginase)、替尼泊苷(teniposide)、17α-炔雌醇(17α-Ethinylestradiol)、己烯雌酚(Diethylstilbestr0l)、睾酮(testosterone)、泼尼松(prednisone)、氟甲睾酮(fluoxymesterone)、屈他雄酮丙酸酯(dromostanolonepropionate)、睾内酯(testolactone)、乙酸甲地孕酮(megestrolacetate)、甲泼尼龙(methylprednisolone)、甲睾酮(methyltestosterone)、泼尼松龙(prednisolone)、曲安西龙(triamcinolone)、氯烯雌醚(chlorotrianisene)、羟孕酮(hydroxyprogesterone)、氨鲁米特(aminoglutethimide)、雌莫司汀(estramustine)、乙酸甲羟孕酮(medroxyprogesterone acetate)、亮丙瑞林(leuprolide)、氟他胺(flutamide)、托瑞米芬(toremifene)、戈舍瑞林(goserelin)、卡铂、羟基脲(hydroxyurea)、安吖啶(amsacrine)、丙卡巴肼(procarbazine)、米托坦(mitotane)、米托蒽醌(mitoxantrone)、左旋咪唑(levamisole)、诺维本(navelbene)、阿那曲唑(anastrazole)、来曲唑(letrazole)、卡培他滨(capecitabine)、雷洛昔芬(reloxafine)、曲洛昔芬(droloxafine)、六甲蜜胺(hexamethylmelamine)、安维汀(Avastin)、赫塞汀(herceptin)、Bexxar、Velcade、Zevalin、三氧化二砷(Trisenox)、希罗达(Xeloda)、长春瑞滨(Vinorelbine)、卟菲尔钠(Porfimer)、ErbituxTM(西妥昔单抗(cetuximab))、Liposomal、噻替哌(Thiotepa)、六甲蜜胺(Altretamine)、美法仑、曲妥珠单抗(Trastuzumab)、列若唑(Lerozole)、氟维司群(Fulvestrant)、依西美坦(Exemestane)、氟维司群(Fulvestrant)、异环磷酰胺(Ifosfomide)、利妥昔单抗(Rituximab)、C225、坎帕斯(Campath)、卡铂、丙卡巴肼(procarbazine)、二氯甲基二乙胺(mechlorethamine)、环磷酰胺(cyclophosphamide)、喜树碱(camptothecin)、异环磷酰胺(ifosfamide)、美法仑、苯丁酸氮芥(chlorambucil)、白消安、硝基脲(nitrosurea)、放线菌素D、柔红霉素、多柔比星、博来霉素、普卡霉素(plicomycin)、丝裂霉素、依托泊苷(VP 16)、他莫昔芬、雷洛昔芬(raloxifene)、雌激素受体结合剂、紫杉醇、吉西他滨、诺维苯(navelbine)、法尼基-蛋白质转移酶抑制剂、反铂、5-氟尿嘧啶、长春花新碱、长春花碱和甲氨蝶呤,或前述的任何类似物或衍生物变体。
ii.放射治疗
导致DNA损伤并已广泛使用的其他因素包括通常被称为γ射线、X射线和/或向肿瘤细胞定向递送放射性同位素的那些。还考虑了其他形式的DNA损伤因素,例如微波和UV-辐照。很可能所有这些因素均对DNA、DNA前体、DNA的复制和修复以及染色体的组装和维持产生广泛的损害。X射线的剂量范围为从50至200伦琴的日剂量持续延长的时间段(例如3至4周)到2000至6000伦琴的单剂量。放射性同位素的剂量范围变化很大,并且取决于同位素的半衰期、发出的辐射的强度和类型以及赘生性细胞的摄取。当应用于细胞时,在本文中使用的术语″接触″和″暴露″用于描述通过其治疗构建体和化学治疗剂或放射治疗剂被递送至靶细胞或被置于与靶细胞的直接临近处的方法。为了实现细胞杀伤或停滞,将这两种药剂以有效杀伤细胞或阻止细胞分裂的组合量递送至细胞。
iii.免疫治疗
通常来说,免疫治疗依赖于使用免疫效应细胞和分子来靶向和破坏癌细胞。免疫效应物可以是例如对肿瘤细胞表面上的一些标志物具有特异性的抗体。单独的抗体可用作治疗的效应物或者其可募集其他细胞以实际上影响细胞杀伤。抗体还可与药物或毒素(化学治疗剂、放射性核素、蓖麻毒蛋白A链、霍乱毒素、百日咳毒素等)缀合并且仅用作靶向剂。或者,效应物可以是携带直接或间接地与肿瘤细胞靶标相互作用的表面分子的淋巴细胞。多种效应物细胞包括细胞毒性T细胞和NK细胞。
因此,免疫治疗可与基因治疗结合,用作组合治疗的一部分。下面讨论组合治疗的一般方法。通常,肿瘤细胞必须具有适用于靶向(即,不存在于大多数其他细胞上)的一些标志物。存在许多肿瘤标志物,并且这些肿瘤标志物中任一种可适合于在本公开内容的情况下靶向。常见的肿瘤标志物包括癌胚抗原、前列腺特异性抗原、泌尿系统肿瘤相关抗原(urinary tumor associated antigen)、胎儿抗原、酪氨酸酶(p97)、gp68、TAG-72、HMFG、唾液酸路易斯抗原(Sialyl Lewis Antigen)、MucA、MucB、PLAP、雌激素受体、层黏连蛋白受体、erb B和p155。
iv.基因治疗
在又一个实施方案中,二级治疗是二级基因治疗,其中治疗性多核苷酸在第一化学治疗剂之前、之后或同时施用。与编码基因产物的载体结合的化学治疗剂的递送将对靶组织具有组合的抗过度增殖作用。
v.手术
约60%患有癌症的人将经历某种类型的手术,其包括预防性、诊断性或阶段性、治愈性和姑息性手术。治愈性手术是可与其他治疗(例如本公开内容的治疗、化学治疗、放射治疗、激素治疗、基因治疗、免疫治疗和/或替代治疗)结合使用的癌症治疗。治愈性手术包括其中物理去除、切除和/或破坏全部或一部分癌组织的切除术。肿瘤切除术是指物理去除至少一部分肿瘤。除肿瘤切除术之外,手术治疗包括激光手术、冷冻手术,电外科手术和显微控制的手术(莫氏手术(Mohs′surgery))。还考虑了本公开内容可与去除浅表癌、癌前病变(precancer)或附带量的正常组织结合使用。
IV.实施例
以下实施例旨在举例说明以上公开内容并不应被解释为缩小其范围。本领域技术人员将容易地认识到,实施例提出了其中可实践本公开内容的许多其他方式。应当理解,在保持在本公开内容的范围内的同时可进行数种变化和修改。通过以下非限制性实施例可进一步理解本公开内容。
实施例1-材料和方法
小鼠。雌性C57BL/6J和Rag1-/-小鼠购自UT西南小鼠育种中心(UT southwesternmice breeding core)。C57BL/6J背景中的Myd88-/-、Tlr4-/-、Tlr9-/-、Batf3-/-和OT1CD8+ T细胞受体(T cell receptor,TCR)-Tg小鼠和NSG-SMG3小鼠购自Jackson Laboratory。Ifnar1-/-小鼠由来自芝加哥大学(University of Chicago)的Anita Chong博士提供。所有小鼠均维持在无特定病原体的条件下。动物护理和实验根据机构和国立卫生研究院(National Institutes of Health)的方案和指南进行。该研究已通过德克萨斯大学西南医学中心(University of Texas Southwestern Medical Center)机构动物护理和使用委员会(Institutional Animal Care and Use Committee)的批准。
细胞系和试剂。将MC38、TC-1、B16、Panc02、Ag104Ld和A549细胞在补充有10%热灭活胎牛血清、100U/ml青霉素和100U/ml链霉素的DMEM或RPMI 1640培养基中在5%CO2下于37℃下培养。按照描述(Pink et al.,2000)合成β-拉帕醌并将其溶解在DMSO中用于体外研究。过氧化氢酶,双香豆素和FTY720购自Sigma-Aldrich。OT-1肽和OVA蛋白来自ThermoFisher。抗CD4(GK1.5)、抗CSF1R(AFS98)、抗IFNAR1(MAR1-5A3)、抗PD-L1(10F.9G2)、抗CD8(YTS)和抗HMGB1 mAb购自BioXCell。
磺酰罗丹明B(SRB)细胞毒性测定。将4,000个细胞平板接种在96或48孔板中,一式三份。在过夜培养之后,在有或没有NQO1抑制剂双香豆素(50μM)的情况下,将细胞暴露于β拉帕醌进行3小时脉冲。之后,将细胞上清液更换为新鲜培养基,并将板在湿润培养箱中以5%CO2在37℃下孵育另外的2或4天。在处理之后,去除培养上清液,并向每个孔轻轻添加固定试剂。将孔用水洗涤并将板风干过夜。添加100μL SRB Dye溶液并孵育30分钟,随后洗涤并风干。通过在微板阅读器(SpectroSTAR Nano,BMG Labtech)中以560nM处的吸光度来确定细胞生长。%细胞生长=(100×(细胞对照-实验))÷(细胞对照)。
NQO1敲除和过表达。通过CRISPR/Cas9技术敲除MC38细胞中的NQO1基因。将引导序列5’-TTGTGTTCGGCCACAATATC-3’克隆到包含嘌呤霉素(puromysin)选择基因的pSpCas9(BB)-2A-Puro质粒(Addgene,#62988)中。将质粒瞬时转染到MC38细胞中。在转染之后48小时,选择嘌呤霉素抗性细胞并在选择培养基中进行亚克隆。将无NQO1表达的MC38细胞克隆(#1、#2和#5)用于体外和体内研究。对于NQO1过表达,将B16细胞(NQO1无效)用全长小鼠NQO1蛋白表达载体(pCMV3-HA-NQO1)瞬时转染。选择NQO1稳定表达细胞并在含潮霉素的培养基中进行亚克隆。将具有NQO1稳定表达的B16细胞克隆(#1、#3和#4)用于以下研究。通过western印迹测定来确定NQO1表达水平。
肿瘤生长和处理。将约6×105个MC38细胞或1.5×105个TC-1或1.5×105个B16细胞皮下接种到小鼠的右胁腹中。当肿瘤生长至一定尺寸时,将荷瘤小鼠随机分组到处理组中。对于β-lap单一治疗,将荷瘤小鼠用β-lap局部处理(瘤内,0.03mg、0.1mg或0.3mg,每隔一天,4次)或全身处理(静脉内或腹膜内,25或30mg/kg,每隔一天,4或6次)。对于CD4和CD8 T细胞耗竭,以三天间隔腹膜内注射200μg抗体四次。对于巨噬细胞耗竭,在β-lap处理期间以三天间隔瘤内注射100μg抗CSF1R mAb 3次。对于I型IFN阻断实验,以三天间隔瘤内注射150μg抗IFNAR1阻断mAb共3次。以上阻断和耗竭实验在第一次β-lap处理之前一天开始。对于HMGB1阻断实验,从第一次β-lap处理的同一天开始,每三天腹膜内(i.p.)施用200μg抗HMGB1 mAb共3次。对于PD-L1检查点阻断组合治疗,从第一次β-lap处理的同一天开始,每三天向荷瘤小鼠腹膜内施用100μg(对于MC38模型)或150μg抗PD-L1(克隆10F.9G2),共3次。每周至少两次测量肿瘤体积,并计算为0.5×长度×宽度×高度。
免疫重建小鼠模型。对于C57BL/6 Rag1-/-免疫重建模型(Lee et al.,2009;Tanget al.,2016),将2×106个A549细胞s.c.接种到雌性Rag1-/-小鼠中。在肿瘤充分建立(约100mm3)之后,在处理之前一天将来自OTI转基因小鼠的2×106个总LN细胞静脉内注射到荷瘤小鼠中。之后,每隔一天用β-lap将小鼠局部(i.t,0.2mg)处理四次。每周至少两次测量肿瘤体积。
对于NSG-SGM3人源化小鼠模型,在人CD34+细胞转移之前一天,用100cGy(使用X-RAD 320辐照器的X射线辐照)对4周龄NSG-SGM3雌性小鼠进行辐照。将经辐照小鼠用Bactrim(Aurora Pharmaceutical LLC)水处理2周。脐带血获自UT SouthwesternParkland Hospital。通过密度梯度离心( Paque Plus,GE healthcare)从脐带血中纯化人CD34+细胞,随后用抗人CD34微珠(Stem Cell)进行阳性免疫磁珠选择。将105个CD34+细胞静脉内注射到每只接受体小鼠中。在移植之后12周,将具有超过40%人CD45+细胞重建的人源化小鼠和年龄和性别匹配的非人源化小鼠在右胁腹上皮下接种2×106个A549肿瘤细胞。在第19天,将荷瘤小鼠每隔一天用β-lap局部(i.t.,0.2mg)处理四次。每周至少两次测量肿瘤体积。所有实验均按照UTSW人调查委员会协议(UTSW Human InvestigationCommittee protocol)和UTSW机构动物护理和使用委员会(UTSW Institutional AnimalCare and Use Committee)进行。
Paque Plus,GE healthcare)从脐带血中纯化人CD34+细胞,随后用抗人CD34微珠(Stem Cell)进行阳性免疫磁珠选择。将105个CD34+细胞静脉内注射到每只接受体小鼠中。在移植之后12周,将具有超过40%人CD45+细胞重建的人源化小鼠和年龄和性别匹配的非人源化小鼠在右胁腹上皮下接种2×106个A549肿瘤细胞。在第19天,将荷瘤小鼠每隔一天用β-lap局部(i.t.,0.2mg)处理四次。每周至少两次测量肿瘤体积。所有实验均按照UTSW人调查委员会协议(UTSW Human InvestigationCommittee protocol)和UTSW机构动物护理和使用委员会(UTSW Institutional AnimalCare and Use Committee)进行。
HMGB1释放检测。将肿瘤细胞平板接种在6孔板中并培养至70%汇合,并用递增浓度的β-lap处理3小时,随后洗涤并进行培养基更换。在24小时之后使用ELISA试剂盒(Chondrex)测定上清液的胞外HMGB1。
IFNγ酶联免疫吸附斑点测定(Enzyme-Linked Immunosorbent Spot Assay,ELISPOT)。收集来自荷瘤小鼠的肿瘤引流LN和脾并制备单细胞悬液。将经辐照肿瘤细胞或OT-1肽用于再刺激肿瘤特异性T细胞。一般而言,将共2×105至4×105个LN细胞或脾细胞和2×105至4×105个经辐照肿瘤细胞共培养48小时,并使用IFNγELISPOT试剂盒(BDBioscience)根据制造商说明进行ELISPOT测定。通过ImmunoSpot Analyzer(CellularTechnology Limited)计算斑点。
从组织中分离细胞。使用阳性CD11c分离试剂盒或阴性CD8分离试剂盒(Stemcell)根据制造商说明从小鼠的淋巴结或脾中分离CD11c+ DC或CD8+ T细胞。对于肿瘤单细胞悬液,将肿瘤组织切成小块,并将其在37℃摇动培养箱中重悬于消化缓冲液(1.5mg/ml I型胶原酶和100μg/ml DNase I)中45分钟。在消化之后,使细胞通过70μm细胞过滤器。
流式细胞术分析。将肿瘤细胞悬液用抗CD16/32抗体(克隆2.4G2)封闭10分钟,并随后与指定抗体在4℃下在暗中孵育30分钟。使用Fixable viability Dye eFlour 506(eBioscience)来排除死细胞。在cytoFLEX(Backman coulter)流式细胞仪上分析样品。
DCFDA细胞ROS检测测定。通过DCFDA-Cellular ROS Detection Assay试剂盒(Abcam)根据制造商说明来确定细胞ROS的水平。简言之,将细胞平板接种到12孔板中并培养至约70%汇合,并将其用DCFDA在37℃下染色30分钟。之后将细胞用不同浓度的β-lap处理3小时。使用流式细胞术在Ex/Em:485/535nm处确定ROS信号。
统计学分析。所有数据分析均用GraphPad Prism统计学软件进行,并示出为平均值±SEM。*P<0.05、**P<0.01、***P<0.001和****P<0.0001,通过双因素ANOVA或未配对双尾t检验来确定。p<0.05的值被认为是统计学上显著的。
实施例2-结果
β-lap在体外和在体内均以NQO1依赖性方式抑制鼠肿瘤生长。使用多种鼠癌细胞系来检查NQO1在β-lap功能中的作用。表达高水平NQO1的肿瘤细胞系(MC38结直肠腺癌、TC-1肺癌和Ag104Ld纤维肉瘤)(图17A)对3小时β-lap暴露敏感(图1A)。相比之下,NQO1缺陷型细胞系B16(黑素瘤)和Panc02(胰腺癌)(图17A)均对β-lap暴露具有抗性(图1A)。双香豆素(NQO1特异性抑制剂)逆转了NQO1介导的致死性(图1B)。接下来,本发明人确定了NQO1耗竭是否会消除β-lap的细胞毒性。CRISPR介导的NQO1敲除(图17B)赋予MC38细胞对β-lap处理的抗性(图1C;图17C)。类似地,NQO1在B16细胞中的过表达(图17D)导致对β-lap具有敏感性(图1D;图1E),并且双香豆素对NQO1的抑制避免了β-lap的致死性(图17F)。β-lap的致死剂量导致快速的细胞肿胀、膜破裂和膜联蛋白V+/7AAD+细胞死亡而没有胱天蛋白酶激活(图1E并且数据未示出)。NQO1催化β-lap的双电子氧化还原以产生高水平的ROS(即过氧化氢/H2O2),导致大量DNA氧化和细胞死亡(Huang et al.,2016)。事实上,β-lap在NQO1阳性鼠肿瘤系中诱导了高水平的ROS,而在NQO1无效系中则更少(图1F至1G)。双香豆素对NQO1的抑制消除了这种作用(图1F至G)。接下来,本发明人确定使ROS中和是否可抑制β-lap诱导的细胞致死性。过氧化氢酶(H2O2清除酶)显著避免了β-lap介导的致死性(图1H;图17G)。这些结果表明β-lap在体外通过加强的肿瘤特异性ROS产生来诱导NQO1+肿瘤细胞死亡。
本发明人还检查了β-lap在以下三种皮下同基因肿瘤模型中的抗肿瘤效力:MC38、TC-1和B16,其各自具有不同的NQO1水平。在MC38肿瘤模型中,在肿瘤接种之后第7天,将25mg/kg的β-lap全身(静脉内)施用于荷瘤WT C57BL/6小鼠。用β-lap进行处理导致了显著的肿瘤抑制(图1I)。当全身递送时β-Lap可作用于多种细胞。为了从机制上探索β-lap在局部肿瘤环境中是否发挥作用以使肿瘤消退以及如何发挥作用以使肿瘤消退,每隔一天瘤内注射不同剂量的β-lap共4个剂量。局部处理也以剂量依赖性方式显著抑制肿瘤生长。肿瘤生长抑制率(TGI,%)对于全身β-lap处理的小鼠为62.5%,以及对于局部β-lap(1.5mg/kgmg、5mg/kg和15mg/kg mg)处理的小鼠为11.8%、69.4%和89.3%(图1I)。值得注意的是,与全身处理相比用低得多的剂量的β-lap(15mg/kg)进行局部处理诱导了更稳健的肿瘤消退,并且46.7%(7/15)的小鼠实现了完全的肿瘤排斥(图1I),表明主要作用位点可能与局部肿瘤微环境相关。与体外研究一致,β-lap的治疗作用在NQO1敲除MC38小鼠模型中消除。(图1J)。类似地,在TC-1(经HPV E6/E7转化的肿瘤模型)中,β-lap处理也导致显著的肿瘤抑制(图17H)。为了进一步确定β-lap在体内的特异性,本发明人在WT C57BL/6WT小鼠中使用亲本NQO1缺陷型或NQO1过表达B16细胞克隆(#1和#4)建立了皮下异种移植物。正如预期地,NQO1无效肿瘤不响应于β-lap处理(图17I)。与此形成鲜明对比的是,NQO1过表达(克隆#1和#4)荷瘤小鼠显示出在β-lap处理之后显著的肿瘤抑制(图17I)。值得注意的是,过表达NQO1的B16肿瘤的生长比B16亲本细胞快得多,表明NQO1促进体内肿瘤生长(Oh et al.,2016)。这些结果提供了β-lap在体外和在体内选择性地抑制NQO1阳性鼠肿瘤生长的证据。NQO1对于β-lap介导的抗肿瘤作用是必不可少的和足够的。
β-Lap介导的抗肿瘤作用依赖于CD8+ T细胞。关于β-lap如何杀伤肿瘤细胞的大多数研究都集中在癌细胞自主机制上(Huang et al.,2016;Pink et al.,2000;Li et al.,2016)。在此,本发明人探寻β-lap介导的抗肿瘤作用是否涉及免疫系统。他们分别在免疫活性小鼠和免疫缺陷型小鼠中建立了MC38肿瘤,以研究β-lap对适应性免疫的作用。在仅4个剂量的β-lap处理之后,在WT C57BL/6小鼠中MC38肿瘤就被根除,其中50%的小鼠被治愈(图2A)。出乎意料地,β-lap在免疫缺陷型Rag1 KO(Rag1-/-)C57BL/6小鼠中失去了治疗活性(图2A)。在NQO1过表达B16肿瘤模型(图18A)和TC-1模型(数据未示出)中观察到类似的作用,表明β-lap在体内的显著抗肿瘤作用需要适应性免疫系统。事实上,本发明人发现与对照MC38肿瘤相比,经β-lap处理的肿瘤中CD4+和CD8+ T细胞提高(图18B),表明T细胞的一些亚群可有助于适应性免疫。为了测试哪个T细胞亚群是必不可少的,将小鼠用与β-lap处理结合的抗CD4或抗CD8耗竭抗体进行处理。虽然用单独的β-lap或与CD4+ T细胞耗竭组合的β-lap进行处理控制了MC38肿瘤生长,但CD8+ T细胞耗竭消除了β-lap的抗肿瘤作用(图2B;图18C)。该数据表明,β-lap介导的肿瘤消退需要CD8+ T细胞,而不是CD4+ T细胞。在荷TC-1肿瘤的小鼠中获得了类似的结果(图2C)。为了确定β-lap介导的抗肿瘤应答是否导致延长的保护性T细胞免疫,本发明人将在β-lap处理之后经历完全的MC38肿瘤排斥的小鼠用致死剂量的MC38细胞再攻击。所有经β-lap治愈的小鼠都排斥再攻击的肿瘤(图2D),表明在β-lap处理之后产生了记忆T细胞。
为了测试β-lap在控制人肿瘤中的效力,本发明人在免疫重建的C57BL/6 Rag1-/-(图2E)和NSG-SGM3小鼠(图2F)中开发了两种异种移植模型。高表达NQO1的人肺癌细胞系A549在体外对β-lap处理敏感(图18D)。在C57BL/6 Rag1-/-免疫重建模型中(Lee et al.,2009;Tang et al.,2016),s.c.接种A549细胞。在肿瘤充分建立(约100mm3)之后,将来自OTI转基因小鼠的共2×106个淋巴结(LN)细胞转移到荷瘤Rag1-/-小鼠中,以在不降低肿瘤生长的情况下重建少量的T细胞。来自OTI转基因小鼠的LN细胞包含不能响应于人肿瘤抗原但会抑制少量非OTI T细胞的稳态增殖的约98%的OVA特异性T细胞。小部分非OT-1T细胞具有识别人肿瘤抗原的潜力,其有约200至1000个克隆,即人患者中肿瘤反应性T细胞的数目。在没有T细胞转移的情况下,β-lap处理仅部分抑制了A549肿瘤生长。然而,在存在T细胞的情况下,β-lap诱导了更稳健的肿瘤消退,其中50%的肿瘤被完全排斥(图2E),表明β-lap介导的抗肿瘤作用主要依赖于T细胞。为了研究人特异性系统中β-lap介导的肿瘤抑制中的T细胞功能,本发明人还使用NSG-SGM3小鼠开发了下一代人源化异种移植模型,其支持人骨髓细胞增殖并提供了更好地模拟人免疫系统和自然肿瘤微环境的体内条件(Morton etal.,2016)。注射人CD34+造血干细胞(Hu-NSG-SGM3)的NSG-SGM3小鼠显示出稳健的移植效率,通过流式细胞术使用人CD45、CD3、CD4和CD8白细胞标志物测量的。将初始NSG-SGM3和Hu-NSG-SGM3小鼠分别接种A549细胞并用β-lap进行处理。值得注意的是,在存在人免疫系统的情况下,β-lap处理诱导了更好的肿瘤控制,表明重建的免疫系统恢复了β-lap的抗肿瘤作用(图2F)。本发明人还研究了肿瘤微环境中的肿瘤浸润性免疫细胞,并且发现在β-lap处理之后在肿瘤组织中CD45+细胞和CD8+ T细胞显著提高,但CD4+ T细胞则没有。总之,这些数据揭示了T细胞在β-lap诱导的肿瘤控制中的必要性。
β-Lap诱导的抗肿瘤作用依赖于树突细胞介导的T细胞交叉致敏。鉴于CD8+ T细胞对于β-lap的抗肿瘤效力是必不可少的,本发明人假设β-lap处理提高了抗原特异性T细胞应答。为了排除β-lap对CD8 T细胞的直接作用,本发明人评价了NQO1在CD8 T细胞上的表达以及β-lap对CD8 T细胞存活、增殖和功能的作用。结果显示来自脾的初始CD8 T和肿瘤浸润性CD8 T细胞均不表达NQO1。此外,肿瘤致死剂量的β-lap对CD8 T细胞凋亡和对经抗CD3/CD28刺激的细胞增殖和IFNγ产生没有作用。为了测试β-lap是否提高抗原特异性T细胞应答,在初始β-lap处理之后10天收集来自荷MC38肿瘤的小鼠的脾细胞,并进行IFGNγELISPOT测定以测量活化T细胞的效应物功能。如所示地,在存在肿瘤刺激(经辐照肿瘤细胞)的情况下,在β-lap处理组中,IFNγ产生T细胞显著提高(图3A)。本发明人使用OTI肽还产生了MC38-OVA细胞系以更好地追踪T细胞应答。类似地,在IFNγELISPOT测定OTI肽中,在β-lap处理之后在小鼠脾中,OTI特异性T细胞的数目要高得多(图3B)。肿瘤特异性CD8细胞的提高表明β-lap处理可诱导交叉致敏并使T细胞再活化以控制肿瘤生长。通过APC例如树突细胞(dendritic cell,DC)或巨噬细胞的交叉呈递被认为是使肿瘤特异性T细胞活化的主要致敏机制。为了进一步确定哪些APC对于β-lap诱导的抗肿瘤作用是必要的,本发明人首先使用抗CSF1R Ab来耗竭肿瘤组织中的巨噬细胞(Tang et al.,2018)。如所示地,巨噬细胞耗竭不影响荷MC38小鼠对β-lap处理的响应(图3C)。Batf3依赖性DC CD8α+或CD103+ DC专门用于交叉呈递坏死性肿瘤细胞来源表位以直接活化CD8+ T细胞(Sanchez-Paulete etal.,2016;Broz et al.,2014;Salmon et al.,2016)。Baf3-/-小鼠缺乏功能性CD8α+或CD103+ DC,并且细胞交叉呈递活性受损。在WT小鼠中,β-lap处理诱导了稳健的肿瘤消退(图3D)。与此形成鲜明对比的是,在相同处理的Batf3-/-小鼠中未看到治疗作用(图3D)。为了进一步解决β-lap可提高交叉呈递的可能性,本发明人使用抗原特异性系统来追踪肿瘤抗原特异性T细胞的致敏和活化。将荷MC38-OVA肿瘤的WT小鼠用β-lap进行处理,然后从肿瘤引流淋巴结(tumor-drain lymph node,TdLN)中分离DC并将其与来自OTI转基因小鼠的CD8 T细胞共培养。测量IFNγ分泌以评价DC致敏抗原特异性T细胞的能力。事实上,在β-lap处理之后,DC诱导了OTI T细胞产生更多的IFNγ(图3E)。这些结果表明,β-lap诱导的肿瘤消退和肿瘤反应性T细胞应答需要DC介导的交叉致敏。
β-lap和肿瘤特异性CTL的抗肿瘤作用需要I型IFN和TLR4/MyD88信号传导。肿瘤微环境中的APC功能失调,导致T细胞的致敏和活化无效(Lee et al.,2009;Corrales etal.,2017)。I型干扰素(IFN)对T细胞的最佳交叉致敏至关重要(Corrales et al.,2017;Deng et al.,2014)。本发明人还检查了β-lap的治疗作用是否需要I型IFN信号传导。瘤内施用IFNAR1阻断抗体以使肿瘤微环境中的I型IFN信号传导中和。引人注目的是,阻断I型IFN显著降低了β-lap的治疗作用(图4A)。为了确定肿瘤细胞或宿主IFN信号传导是否是必不可少的,将MC38肿瘤接种到WT和IFNAR1缺陷型(Ifnar-/-)C57BL/6小鼠中,随后进行β-lap处理。结果显示,在具有受损的IFNAR1信号传导的小鼠中消除了治疗作用(图4B)。正如预期地,本发明人观察到在经β-lap处理的肿瘤中IFNα/β和IFN应答基因CXCL10如另一些细胞因子例如IFNγ和TNFα上调(图19A至B)。已经表明,MyD88参与I型IFN产生和一些化学治疗剂的抗肿瘤免疫(Sistigu et al.,2014;Zitvogel et al.,2015;Apetoh et al.,2007)。为了确定β-lap处理是否需要MyD88信号传导,将肿瘤细胞s.c.植入到WT和MyD88缺陷型(Myd88-/-)小鼠中。β-lap诱导的肿瘤消退在具有相同治疗方案的Myd88-/-小鼠中消失(图4C),表明宿主MyD88对于β-lap的抗肿瘤作用是必不可少的。由于β-lap可诱导稳健的肿瘤细胞死亡,本发明人假设这继而导致损伤相关分子模式(DAMP)的分泌和肿瘤抗原的暴露,从而加强了抗肿瘤免疫应答。令人感兴趣地,敲除TLR4而非TLR9(这两者均是MyD88信号传导以感知DAMP的主要上游受体)也导致相似的治疗抗性(图4D;图19C)。先前的研究表明,HMGB1(和HMGB1/DNA复合体)(TLR4的内源性配体之一)可用作以MyD88依赖性方式刺激DC交叉致敏的危险信号(Apetoh et al.,2007)。为了确定β-lap诱导的肿瘤消退是否是HMGB1依赖性的,施用抗HMGB1 mAb以中和游离HMGB1以及进行β-lap处理。结果显示,HMGB1信号传导的阻断减弱了β-lap的作用(图4E),表明β-lap可在肿瘤微环境中诱导HMGB1释放以通过TLR4/MyD88途径增强固有应答。为了进一步求解HMGB1的必要作用,本发明人评价了当阻断信号传导结合β-lap处理时的肿瘤特异性T细胞应答。将MC38肿瘤细胞s.c.植入到WT或Tlr4-/-或Myd88-/- C57BL/6小鼠中,并在有或没有抗HMGB1 Ab的情况下将荷瘤小鼠用β-lap进行处理。在处理之后,收集来自肿瘤引流淋巴结(TdLN)的淋巴细胞并进行IFNγELISPOT测定。在WT小鼠中,β-lap处理提高了肿瘤反应性T细胞的数目,并且当与抗HMGB1中和Ab共施用时消除了这种作用(图4F)。类似地,在Tlr4-/-和Myd88-/-小鼠中,与WT小鼠中的肿瘤反应性T细胞相比,对照组中的肿瘤反应性T细胞少得多(图4F)。更重要的是,β-lap处理在这些缺陷型小鼠中不能增强肿瘤特异性T细胞应答(图4F)。这些结果表明,β-lap诱导的固有性和适应性抗肿瘤免疫应答需要TLR4/MyD88/I型IFN信号传导级联。
β-Lap处理在体内诱导肿瘤免疫原性细胞死亡并触发HMGB1依赖性抗肿瘤T细胞免疫。β-lap在体内的HMGB1依赖性肿瘤特异性T细胞应答和HMGB1依赖性抗肿瘤作用表明,β-lap可在肿瘤中潜在性地诱导免疫原性细胞死亡(ICD)。为了检验这一假设,本发明人检查了ICD标志:在体外经β-lap处理的肿瘤细胞中的HMGB1分泌。事实上,他们在NQO1+肿瘤细胞(MC38、TC-1和NQO1过表达B16细胞)中观察到HMGB1的剂量依赖性分泌,但在NQO1-细胞(B16亲本细胞)中没有观察到(图5A)。双香豆素对NQO1的抑制消除了β-lap诱导的HMGB1分泌(数据未示出)。本发明人推测HMGB1暴露可能决定β-lap诱导的肿瘤细胞死亡的免疫原性。在以下两个实验中发现HMGB1对β-lap诱导的免疫原性是至关重要的:(i)将在体外β-lap诱导的垂死MC38-OVA细胞结合或不结合抗HMGB1Ab注射到C57BL/6小鼠的胁腹中(图5B)。确定了TdLN中肿瘤抗原特异性T细胞的数目(图5C)和IFNγ产生(图5D);(ii)将在存在抗HMGB1Ab的情况下接受用β-lap诱导的垂死MC38-OVA细胞疫苗接种的C57BL/6小鼠用MC38-OVA再攻击以评价抗肿瘤保护(图5E至F)。如所示地,与疫苗接种活细胞的小鼠相比,疫苗接种β-lap诱导的垂死细胞的小鼠具有更多的IFNγ产生抗原特异性T细胞(图5C至D)。然而,当疫苗接种垂死细胞和抗HMGB1Ab混合物时,肿瘤特异性T细胞应答减弱(图5C至D)。一致地,当疫苗接种相同的垂死细胞时,与WT小鼠相比,TLR4-/-小鼠也显示出降低的肿瘤反应性T细胞和IFNγ产生(图5C至D)。为了测试β-lap诱导的垂死细胞激活适应性免疫系统的能力,本发明人使用了免疫活性C57BL/6小鼠中的预防性肿瘤疫苗接种模型(图5E)。用β-lap诱导的垂死细胞对小鼠免疫接种防止了再攻击的肿瘤的生长(图5F)。值得注意的是,当小鼠疫苗接种垂死细胞和抗HMGB1中和抗体时,抗肿瘤保护作用降低(图5F)。这些结果表明β-lap以HMGB1依赖性方式诱导ICD并增强抗肿瘤免疫原性。
β-Lap通过与抗PD-L1治疗组合根除了大的已建立的和检查点阻断难治性肿瘤。在临床实践中,患有已充分建立肿瘤的患者可产生复杂的免疫抑制网络,并且通常对于免疫治疗是难治性的(Sharma et al.,2017;Smyth et al.,2016)。类似地,在我们的临床前模型中,仅在β-lap处理之后在荷小肿瘤(约50mm3)的小鼠中实现了完全肿瘤排斥(TGI,93.0%;图6A至B),而大的建立的肿瘤(约150至200mm3)仅部分地由相同的处理方案控制(TGI,59.36%;图6A至B)。β-lap激发固有性和适应性免疫应答作为其作用机制的一部分的发现为β-lap与T细胞检查点阻断的组合(图6A)以根除晚期和检查点阻断难治性肿瘤铺平了道路。为了检验这一点,本发明人接下来在荷建立的大MC38肿瘤的小鼠中将局部β-lap处理(15mg/kg,i.t.)与抗PD-L1处理组合。晚期MC38肿瘤仅显示出对单独的抗PD-L1的中等响应(TGI,63.25%,图6C)。形成鲜明对比的是,组合组中的小鼠显示出稳健的肿瘤控制和消退(TGI,96.86%;图6C;图20A)。值得注意的是,针对组合处理,60%的荷瘤小鼠完全排斥了其肿瘤(图6C;图20A)。令人感兴趣地,局部使用低得多的剂量(5mg/kg,i.t.)的β-lap也观察到了β-lap和免疫治疗的协同作用(图20B)。最近的研究表明,癌症免疫治疗被局部化学治疗处理增强而被全身化学治疗处理消除(Mathios et al.,2016;Ariyan et al.,2018)。为了进一步评价全身β-lap处理是否具有任何免疫抑制作用并损害抗PD-L1免疫治疗,对荷MC38肿瘤的小鼠施用作为单一治疗或与抗PD-L1组合的全身β-lap处理(30mg/kg,i.p.)(图6D)。β-lap或抗PD-L1的单一治疗导致对肿瘤生长的类似中等抑制(图6E;图20C)。组合处理对其抗肿瘤作用具有协同作用(其中25%的肿瘤被完全排斥)(图6E;图20C)并显著提高了荷瘤小鼠的存活(图6F)。B16肿瘤表达PD-L1,但免疫原性较差,并且对PD-L1/PD-1免疫检查点阻断不具有响应性(Chen et al.,2015;Curran et al.,2010)。本发明人使用他们的NQO1过表达B16肿瘤模型来评价β-lap和抗PD-L1组合处理的治疗效力(图20E)。正如预期地,NQO1过表达B16肿瘤未能响应于单独的抗PD-L1 Ab(图20F)。相比之下,β-lap单一治疗在很大程度上抑制了B16-NQO1肿瘤的生长(TGI,68.67%)。引人注目的是,当与PD-L1阻断组合时,β-lap具有显著的协同抗肿瘤作用(TGI,88.76%;图20F)。
为了进一步阐明β-lap与PD-L1阻断之间的协同机制,在MC38-OVA肿瘤模型中在初始处理之后12天,本发明人研究了肿瘤微环境中的肿瘤浸润性免疫细胞并追踪了肿瘤组织和脾中的抗原特异性T细胞。他们发现使用β-lap或抗PD-L1单一治疗,肿瘤组织中CD45+细胞、CD8+ T细胞和CD8+ T细胞:Treg细胞比显著提高(图6G)。重要的是,这些作用在组合组中被显著放大(图6G)。他们进一步在肿瘤组织和脾中追踪了对模型抗原OVA257-264(OT1肽)具有特异性的CD8+ T细胞。如所示地,β-lap或抗PD-L1单一治疗在肿瘤组织(图6G)和脾(图6H)中均未提高OT1抗原特异性T细胞。引人注目的是,组合治疗在肿瘤组织和脾二者中均稳健地扩增了肿瘤抗原特异性T细胞(图6G至H)。这些结果表明,当与PD-L1阻断组合时,β-lap处理增强了肿瘤免疫原性并提高了T细胞浸润和肿瘤特异性T细胞应答。这些数据表明在控制大的建立的和检查点阻断难治性NQO1阳性肿瘤中β-lap与PD-L1阻断之间的强效协同。
p-Lap诱导了肿瘤特异性ROS和DNA损伤,并选择性地促进了NQO1阳性细胞的程序性坏死。NQO1(在多种人癌症(包括NSCLC、胰腺癌、结肠癌、乳腺癌和头颈癌)中特异性且独特升高的酶)可以以肿瘤选择性方式利用以用于治疗。β-lap已显示出显著的抗肿瘤作用,尤其是当与PARP1抑制剂组合时。然而,尚不清楚β-lap介导的肿瘤特异性DNA损伤和细胞死亡是否与免疫系统有一定的相互作用;β-lap是否会引起免疫原性细胞死亡并导致依赖于宿主免疫系统的肿瘤消退。为此,本发明人在体外和在免疫活性小鼠中检查了β-lap的抗肿瘤活性。如所示地,β-lap选择性地杀伤了NQO1+鼠肿瘤细胞系(MC38、TC-1和Ag104Ld),并且这种作用可被双香豆素(DIC,一种相当特异性的NQO1抑制剂)抑制,并且不会在NQO1-细胞(B16,pan02)中发生(图8A至B)。在暴露于β-lap3小时之后在NQO1+细胞中产生了高水平的H2O2,而在NQO1-细胞中未产生(图8C),这表明β-lap具有理想的靶向作用。β-lap通过独特的胱天蛋白酶非依赖性方式触发了NQO1+癌细胞死亡(图8D)并诱导了PARP1介导的程序性坏死(图8E)。
β-lap在小鼠中的抗肿瘤效力需要宿主适应性免疫系统。本发明人分别建立了荷NQO1阳性鼠肺癌细胞TC-1的免疫活性小鼠和免疫缺陷型小鼠。在3个剂量的β-lap处理之后,在荷TC-1肿瘤的WT小鼠中发生了肿瘤消退,但在适应性免疫缺陷型rag-/-小鼠中没有发生(图9A)。该结果表明β-lap在体内的显著抗肿瘤效力需要适应性免疫系统。为了测试哪些T细胞亚群对控制负荷至关重要,将小鼠用与β-lap处理结合的抗CD4或抗CD8耗竭抗体进行处理。虽然用单独的β-lap或与抗CD4抗体组合的β-lap进行处理导致了类似的肿瘤生长抑制,但与单独的β-lap相比,β-lap+抗CD8抗体的组合治疗导致肿瘤生长抑制完全失效(图9B)。这些数据表明β-lap介导的肿瘤消退需要CD8+ T细胞。在荷MC-38细胞的小鼠(鼠结肠癌模型)中获得了类似的结果。
β-Lap依赖于STING依赖性DNA感知和I型IFN信号传导来诱导肿瘤消退。I型IFN已成为潜在的关键危险信号,其在多种抗肿瘤治疗开始之后启动抗肿瘤T细胞应答,桥接固有和适应性免疫。为了测试I型IFN是否参与β-lap诱导的肿瘤消退,本发明人建立了两种模型来阻断I型IFN信号传导:在肿瘤微环境中注射抗IFNAR(干扰素α/β受体)阻断抗体(图10A)以及在小鼠中敲除宿主IFNAR基因(图10)。本发明人发现微环境中I型IFN信号传导的阻断极大地损害了β-lap的抗肿瘤效力(图10A)。与这些结果一致,与WT小鼠中的相比,β-lap诱导的肿瘤消退在IFNAR敲除小鼠中被完全消除(图10B),表明I型IFN可能是β-lap介导的肿瘤消退所必需的细胞因子。最近,已表明TLR/MyD88途径和STING介导的胞质DNA感知级联二者均是I型IFN产生的主要机制。为了研究是否需要MyD88和STING信号传导途径来介导对β-lap处理的响应,将TC-1细胞植入在WT、MyD88或STING缺陷型小鼠中。本发明人发现,在β-lap处理之后,虽然在WT小鼠(图10C)和MyD88缺陷型小鼠中,肿瘤负荷显著降低(数据未示出),但宿主STING的缺乏显著损害了β-lap的抗肿瘤效力(图10C)。这些结果表明,STING依赖性胞质DNA感知和I型IFN产生对于β-lap在体内的治疗作用至关重要。
β-lap在体内的抗肿瘤效力需要肿瘤浸润性嗜中性粒细胞。已知I型IFN桥接固有和适应性免疫,并且对于肿瘤特异性T细胞应答的交叉致敏是至关重要的。本发明人提出β-lap处理可通过提高一些危险信号传导和募集一些吞噬细胞来触发固有感知并导致对肿瘤抗原的交叉呈递。为了验证这一想法,他们在β-lap处理3天之后在肿瘤微环境中详细研究了免疫细胞群。令人感兴趣地,他们发现肿瘤浸润性嗜中性粒细胞(CD11b+Gr1+亚群)显著提高(图11A),而巨噬细胞、DC、CD8+和CD4+ T细胞的百分比受影响较小。为了研究这种提高的嗜中性粒细胞亚群是否参与β-lap诱导的肿瘤消退,使用了特异性地靶向肿瘤浸润性嗜中性粒细胞的抗Ly-6G(克隆1A8)抗体来耗竭该亚群。抗体介导的嗜中性粒细胞耗竭极大地损害了β-lap在体内的治疗作用(图11B),表明新嗜中性粒细胞浸润在抗肿瘤免疫应答中的关键作用。
β-Lap可与免疫检查点阻断(抗PD-L1/PD-1)治疗协同以有效地杀伤NQO1+肿瘤细胞。β-lap可激发固有免疫应答作为其作用机制的一部分的发现对其与适应性基于T细胞的免疫检查点阻断策略(例如PD-L1/PD-1抑制剂)组合以进一步激活适应性免疫具有深远的意义。在肿瘤微环境中I型IFN诱导的PD-L1的上调是对多种治疗产生获得性肿瘤抗性的主要原因之一,这使得为β-lap+PD-L1/PD-1抑制剂之间的组合治疗提供了治疗窗。为了检验这一假设,本发明人然后在C57BL/6J WT小鼠中使用6×105个MC38细胞建立了皮下异种移植物。将小鼠(n=5)用单独的HPβCD载剂、抗PD-L1(阿特珠单抗)或0.1mg(i.t.)或30mg/kg(i.p.)的β-Lap在有或没有抗PD-L1的情况下每三天瘤内(i.t.)(图12A)或腹膜内(i.p.)处理,4次注射。当肿瘤体积>50mm3(i.p.)或>100mm3(i.t.)时开始处理。在β-Lap处理之前一天,腹膜内(i.p.)注射100μg的抗PD-L1(克隆10F.9G2)或同种型对照抗体(克隆LTF-2)。然后监测小鼠的肿瘤体积的变化(图12A至B)。与载剂相比,用低剂量的单独的HPβCD-β-lap或抗PD-L1进行处理导致肿瘤体积略微降低,然而,药物组合治疗导致显著的协同抗肿瘤效力(图12A至B)。
辐照和NQO1生物可激活药物处理在小鼠中协同促进了抗肿瘤免疫。本发明人先前的结果表明β-lap是免疫缺陷型小鼠的辐射增敏剂。在此,本发明人检查了β-lap在免疫活性小鼠中对辐射增敏NQO1+MC38鼠癌细胞的作用。表达高水平NQO1(130+15单位)的MC38NSCLC癌细胞是非常具有响应性的,如在C57BL/6J WT小鼠中在皮下肿瘤(100mm3)中用10Gy+0.1mgβ-lap测试3次瘤内(i.t.)注射的,其中肿瘤生长显著抑制(p<0.01,图13A)。类似地,MC38皮下肿瘤(50mm3)对每隔一天给予10Gy+β-lap(30mg/kg,i.p.),6次注射显示出显著的协同响应(p<0.01,图13B)。小鼠没有显著的高铁血红蛋白血症或体重减轻(数据未示出)。
IB-DNQ以NQO1依赖性方式杀伤鼠癌细胞并诱导NAD+/ATP耗竭和DNA损伤。IB-DNQ是新的更强效的NQO1生物可激活药物。IB-DNQ与β-lap相同,但效力高10至20倍,但与β-lap相比,其引发高铁血红蛋白血症(methemoglobinemia,MH)的能力要弱得多。与β-lap一样,IB-DNQ也经历了NQO1依赖性无效氧化还原循环,其试图使药物解毒,形成其氢醌。尚不清楚类似于β-lap的IB-DNQ介导的肿瘤特异性DNA损伤和细胞死亡是否与免疫系统有一定相互作用。本发明人在体外检查了IB-DNQ的抗肿瘤活性。如所示地,NQO1在鼠肿瘤细胞系(MC38和TC-1)中过表达,而在B16鼠肿瘤细胞系(B16和tubo)中缺乏(图14A)。IB-DNQ选择性地杀伤了NQO1+鼠肿瘤细胞系MC38(图14B)和TC-1(图14C),但不选择性地杀伤NQO1-鼠肿瘤细胞B16(图14D),并且这种作用可被双香豆素(DIC,一种相当特异性的NQO1抑制剂)抑制(图14B至C)并且不会在NQO1-细胞(B16)中发生(图14D)。在TC-1细胞中在IB-DNQ处理2小时之后还注意到必需核苷酸NAD+和ATP耗竭,并且双香豆素可避免这种作用(图14E至F)。在用0.25μMIB-DNQ处理60分钟之后,尾长显著提高,表明DNA总损伤高(图14G),并且DSB标志物γ-H2AX病灶显著提高(图14H),表明双链断裂。
IB-DNQ依赖于适应性免疫系统来诱导肿瘤消退。尚不知晓IB-DNQ是否会刺激抗肿瘤免疫。在此,本发明人分别建立了荷NQO1阳性鼠MC-38结肠癌细胞或TC-1肺癌细胞的免疫活性小鼠(C57BL/6J WT)和免疫缺陷型小鼠(rag-/-)。在4个剂量IB-DNQ(0.15mg)处理之后,在荷MC-38或TC-1肿瘤的WT小鼠中发生了肿瘤消退,但在适应性免疫缺陷型rag-/-小鼠中未发生(图15A至B)。该结果表明,IB-DNQ在体内的显著抗肿瘤效力也需要适应性免疫系统。
来自IB-DNQ和免疫检查点阻断(抗PD-L1)治疗组合的协同。先前的研究揭示了β-lap可与免疫检查点阻断治疗协同。在此,本发明人检查了IB-DNQ是否也可与免疫检查点阻断治疗协同。将荷MC38肿瘤的小鼠(C57BL/6J WT)用单独的HPβCD载剂、抗PD-L1(阿特珠单抗)或0.05mg(i.t.)的IB-DNQ在有或没有抗PD-L1的情况下每三天瘤内(i.t.)处理,4次注射。当肿瘤体积为约100mm3时开始处理。在IB-DNQ处理之前一天,腹膜内(i.p.)注射100μg的抗PD-L1(克隆10F.9G2)或同种型对照抗体(克隆LTF-2)。然后监测小鼠的肿瘤体积和总存活的变化(图16A至B)。类似地,本发明人发现,与载剂相比,用低剂量的单独的HPβCD-IB-DNQ或抗PD-L1进行处理也使得显著降低了肿瘤生长并提高了小鼠寿限,然而,药物组合治疗产生了显著的协同抗肿瘤效力(图16A至B)。
IB-DNQ以NQO1依赖性方式诱导肿瘤特异性ROS形成和广泛的DNA损伤。NAD(P)H:醌氧化还原酶1(NQO1)是在大多数实体癌中升高(>100倍)的双电子氧化还原酶,并且已成为直接肿瘤杀伤的有前景的靶标。NQO1由于其特异性可以以肿瘤选择性方式利用以用于治疗。药物异丁基脱氧苯醌(IB-DNQ)已显示出对NQO1+人实体癌的显著抗肿瘤作用。然而,尚不清楚IB-DNQ介导的肿瘤特异性DNA损伤和细胞死亡是否与免疫系统有一定相互作用。为了确定IB-DNQ是否选择性地靶向NQO1+肿瘤并触发免疫应答,本发明人筛选了多种鼠癌细胞系以研究IB-DNQ在体外的抗肿瘤作用。如图21A至B中所示,IB-DNQ选择性地杀伤了NQO1+鼠肿瘤细胞系(过表达NQO1的TC-1、Ag104Ld、MC38和B16),并且这种作用可通过双香豆素(DIC,一种相当特异性的NQO1抑制剂)或敲除NQO1来抑制(图21B)。此外,在NOQ1+鼠癌细胞中观察到,在暴露于IB-DNQ 1小时之后的高ROS水平(图21C)和在暴露于IB-DNQ 4小时之后的DNA损伤(总断裂或双链断裂)(图21D至E)。进一步的测定表明,在存在IB-DNQ的情况下,观察到膜联蛋白V+指示的凋亡细胞死亡(图21F)。这些结果表明,IB-DNQ在体外通过加强的肿瘤特异性ROS产生和广泛的DNA损伤来选择性地诱导NQO1+肿瘤细胞死亡。
IB-DNQ在小鼠中的抗肿瘤效力需要宿主免疫系统。迄今为止,关注IB-DNQ处理对肿瘤细胞死亡的作用的大多数研究均针对癌细胞自主机制。在此,本发明人假设IB-DNQ介导的抗肿瘤作用涉及免疫应答的激活。为了检查免疫应答在IB-DNQ抗肿瘤作用中的作用,本发明人在免疫活性小鼠和免疫缺陷型小鼠中建立了MC38皮下同基因肿瘤模型。在肿瘤接种之后第7天,将IB-DNQ瘤内施用于荷瘤WT C57BL/6或NOD.Cg-Prkdcscid Il2rgtm1 Wj1/SzJ(NSG)小鼠(图22A至B)。出人意料的是,在荷瘤WT组中在4个剂量的IB-DNQ处理之后肿瘤消退,并且治愈部分为20%。相比之下,与载剂相比,经IB-DNQ处理的NSG小鼠中的肿瘤仅显示出肿瘤尺寸最小的降低。这些结果表明IB-DNQ介导的抗肿瘤作用涉及免疫应答。为了进一步研究IB-DNQ对适应性免疫的作用,将MC38和TC-1肿瘤细胞皮下移植到WT和Rag1 KO(Rag1-/-)C57BL/6小鼠中(图22C至D)。正如所示的和预期地,Rag1 KO阻断了IB-DNQ的抗肿瘤作用,并且肿瘤显著生长,表明IB-DNQ在体内的显著抗肿瘤效力需要适应性免疫系统。此外,与体外研究一致,IB-DNQ治疗作用在NQO1 KO MC38小鼠模型中被消除,即使在开始处理时肿瘤尺寸略微降低(图22E至F),表明NQO1对于IB-DNQ介导的抗肿瘤作用是必不可少的和足够的。
IB-DNQ处理在肿瘤微环境中影响CD45+免疫细胞群。为了测试哪些免疫细胞亚群对于控制肿瘤负荷是必不可少的以及IB-DNQ如何改变肿瘤微环境,本发明人研究了在IB-DNQ处理之后MC38肿瘤的免疫微环境。如所示地,与对照肿瘤相比,在经IB-DNQ处理的肿瘤中观察到CD8+和CD4+ T细胞的显著提高(图23A至B)。此外,IB-DNQ显著提高了MHC II+DC比例但对巨噬细胞浸润没有作用(图23A至B)。这些结果表明CD8+和CD4+ T细胞可能在IB-DNQ的抗肿瘤效力中发挥必不可少的作用。为了排除IB-DNQ对T细胞的直接作用,确定了在暴露于致死剂量的IB-DNQ之后的CD8+ T细胞增殖(图23C)和存活(图23D)。事实上,IB-DNQ对CD8+T细胞凋亡和对经抗CD3/抗CD28刺激的细胞增殖均没有作用。
IB-DNQ介导的抗肿瘤作用依赖于CD8+和CD4+ T细胞。为了研究CD4+和CD8+ T细胞在IB-DNQ作用中的作用,检查了在CD4+和/或CD8+ T细胞耗竭之后IB-DNQ的抗肿瘤作用。如预期地,CD4+和CD8+ T细胞耗竭完全消除了IB-DNQ的抗肿瘤作用(图24E至F),但出人意料的是,单独的CD4+或CD8+ T细胞耗竭部分阻断了IB-DNQ的作用(图24A至D),表明IB-DNQ介导的肿瘤消退需要CD8+和CD4+ T细胞二者。
IB-DNQ诱导肿瘤ICD和树突细胞介导的T细胞交叉致敏。本发明人先前的研究表明,用一些化学放射治疗处理肿瘤诱导了促进肿瘤的抗原性和免疫原性的肿瘤细胞免疫原性细胞死亡(ICD)。由DC将抗原交叉呈递给负责控制肿瘤的抗肿瘤CD8 T细胞,有利于通过ICD垂死的肿瘤细胞的免疫原性。鉴于CD8+ T细胞在IB-DNQ抗肿瘤作用中发挥必不可少的作用,因此,IB-DNQ可在肿瘤中潜在性地诱导ICD,导致将抗原呈递给CD8+ T细胞。为了检验这一假设,在IB-DNQ处理之后检查了ICD标志之一HMGB1。事实上,IB-DNQ靶向的MC38和B16NQO1+细胞分泌了高水平的HMGB1,而不能被IB-DNQ诱导细胞死亡的MC38 NQO1-/-和B16(NQO1缺陷型)细胞分泌较少的HMGB1(图25A)。由于1型干扰素(interferon,IFN)对T细胞的最佳交叉致敏是必不可少的,因此确定了1型IFN其是否参与由IB-DNQ介导的T细胞应答。如所示地,与对照相比,在用IB-DNQ处理的肿瘤中IFN-α和IFN-β的表达显著上调(图25B至C)。同时,还观察到IFN-γ指示的抗原特异性T细胞应答(图25D至E)。在存在肿瘤抗原(经辐照肿瘤细胞)的情况下,在IB-DNQ处理组中,IFN-γ指示的T细胞显著提高(图25D)。类似地,在用OT-1肽进行的IFN-γELISPOT测定中,在IB-DNQ处理组中OT-1特异性T细胞的数目显著提高(图25E)。总之,这些结果表明IB-DNQ诱导ICD进行交叉致敏并使T细胞活化以抑制肿瘤生长。
IB-DNQ诱导固有免疫记忆而不是经典免疫记忆。为了研究IB-DNQ介导的抗肿瘤应答是否导致延长的保护性T细胞免疫,将在IB-DNQ处理之后经历完全MC38肿瘤排斥的小鼠通过MC38细胞(3×106个)再攻击。令人感兴趣地,所有IB-DNQ治愈的小鼠均排斥再攻击的肿瘤(图26A至B),表明在IB-DNQ处理之后可产生记忆T细胞。为了进一步确定记忆T细胞的产生,在治愈的小鼠中检查了CD44+指示的记忆T细胞。在再攻击的肿瘤被排斥之后30天之后,分离来自治愈小鼠的不同器官,并通过流式细胞术分析单细胞。出人意料的是,在脾和LN(图26C至D)和其他器官(数据未示出)中均未观察到CD44+CD8+或CD44+CD4+记忆T细胞。还研究了记忆B细胞,但观察到了与记忆T细胞相似的结果(数据未示出)。令人感兴趣地,在治愈的小鼠中观察到CD44+DC的显著提高(图26E)。数个研究表明,CD44对于DC-T细胞紧密缀合物的形成是重要的,并且DC上的CD44可影响T细胞活化。我们的结果表明,在存在抗原(经辐照或经IB-DNQ处理的肿瘤细胞)的情况下,与对照相比,来自荷瘤LN(TDLN)或无肿瘤LN(tumor-free LN,TF-LN)的CD44+DC受到刺激,而TF-LN组显示出更显著的提高(图26F)。同时,当将从脾分离的CD8+ T细胞与经辐照肿瘤细胞共培养时,T细胞增殖没有显著提高(图26G)。然而,当在存在抗原的情况下,将从初始脾分离的CD8+ T细胞与来自荷瘤或无肿瘤小鼠的LN细胞共培养时,T细胞增殖受到显著刺激(图26H),表明TF-LN中的CD44+DC可在T细胞活化和增殖中发挥关键作用。总之,所有这些结果表明IB-DNQ抗肿瘤作用可诱导固有免疫记忆(记忆样树突细胞应答等),而不是经典免疫记忆。
程序性死亡配体1(PD-L1)表达在具有大肿瘤负荷的小鼠中上调。来自其他研究组的先前研究表明,在肿瘤微环境中I型IFN诱导的PD-L1表达的上调是对多种治疗产生获得性肿瘤抗性的主要原因之一。本发明人的先前研究表明IB-DNQ处理导致肿瘤微环境中I型IFN信号传导的上调(图25B至C)。为了确定IB-DNQ处理在TME中是否提高PD-L1表达,本发明人建立了荷小肿瘤(50mm3)或晚期肿瘤(150mm3)的两种小鼠模型。在最后一次IB-DNQ注射之后24小时之后收集肿瘤用于流式细胞术分析。事实上,他们发现在具有大肿瘤负荷的小鼠中在CD45+免疫细胞中通过IB-DNQ处理(12mg/kg,i.v.)显著上调了PD-L1表达(图27A),但在具有小肿瘤负荷的小鼠中则没有(图27B)。这些发现表明,IB-DNQ处理在TME中导致PD-L1水平上调,这使得为IB-DNQ+PD-L1抑制剂组合治疗晚期NQO1+肿瘤提供了治疗窗。
IB-DNQ克服了检查点阻断抗性。充分建立的肿瘤具有固有抗性机制,并且很难实现完全的肿瘤排斥。为了研究大的建立的肿瘤是否对单独的IB-DNQ或PD-L1阻断处理敏感,本发明人在C57BL/6WT小鼠中产生了小(约50mm3)和大(约150mm3)的肿瘤负荷(12mg/kg IB-DNQ,i.v.或100μg抗PD-L1,i.p.)。监测肿瘤体积和长期存活。发现在单独的IB-DNQ或抗PD-1处理之后,仅在荷小的而非大的建立的肿瘤的小鼠中实现了完全的肿瘤排斥(图28A至D)。基于先前的研究(图27),本发明人提出IB-DNQ与免疫检查点阻断治疗协同以有效地杀伤充分建立的NQO1+肿瘤。为了检验这一假设,他们在荷大的建立的MC38肿瘤的小鼠中将IB-DNQ(12mg/kg,i.v.)处理与抗PD-L1处理(100μg,i.p.)组合。他们发现晚期MC38肿瘤仅对单独的IB-DNQ或抗PD-L1显示出中等响应(图28E至F)。形成鲜明对比的是,组合组中的小鼠显示出稳健的肿瘤控制和消退(图28E至F)。值得注意的是,在通过全身处理的组合处理中,40%的荷瘤小鼠完全排斥了其肿瘤。这些发现表明,在初始响应于IB-DNQ之后,TME中PD-L1提高促进了大肿瘤的肿瘤复发,并且IB-DNQ与免疫检查点阻断的组合治疗根除了晚期和检查点难治性肿瘤。
实施例3-讨论
缺乏适当的固有感知可限制T细胞靶向免疫治疗(Patel et al.,2018;Qiao etal.,2017;Gajewski et al.,2013)。本发明人假设通过一些肿瘤靶向遗传毒性药剂诱导免疫原性固有感知可诱导抗肿瘤免疫并克服免疫检查点阻断抗性。在此,他们使用了数种同基因免疫活性小鼠模型和免疫重建人异种移植模型,所述模型密切概括了人疾病以评价NQO1生物可激活β拉帕醌(β-lapachone,β-lap)的抗肿瘤效力。他们表明β-lap在体内具有令人印象深刻的抗肿瘤作用,这在很大程度上取决于固有和适应性免疫。他们发现,β-lap在被NQO1激活之后导致肿瘤选择性细胞死亡并诱导固有感知以用于适应性抗肿瘤免疫。从机制上讲,肿瘤β-lap通过释放HMGB1触发免疫原性细胞死亡并提高肿瘤免疫原性。这激活了固有免疫应答并以TLR4/MyD88依赖性方式诱导了I型IFN特征,其刺激了抗肿瘤T细胞适应性免疫并抑制了肿瘤生长。重要的是,β-lap克服了免疫治疗抗性。当与抗PDL1 mAb组合时,β-lap进一步增强了CD8 T细胞浸润和抗原特异性T细胞应答,并根除了大的建立的和检查点阻断难治性肿瘤。
在临床前和临床研究中,检查点阻断基本上使免疫系统解除“刹车(brake)”,并且已被证明不足以打破耐受性,除非与某些“燃料(fuel)”共施用以激活局部免疫活性并诱导期望的T细胞应答(Salmon et al.,2016;Kleponis et al.,2015;Kamphorst et al.,2017)。与T细胞检查点免疫治疗组合刺激固有免疫感知可以是一种解决办法。肿瘤的天然固有免疫感知显示通过抗原摄取和呈递、宿主PRR途径、I型IFN产生和T细胞的交叉致敏而发生(Woo et al.,2015)。通常,固有免疫细胞可通过DC活化或产生支持效应T细胞分化的细胞因子来直接或间接地促进肿瘤控制。然而,肿瘤也通过使PRR信号传导沉默或破坏PRR信号以及积累功能失调的固有细胞来避免免疫清除以促进癌症抑制炎症而不是致敏适应性免疫应答(Lee et al.,2009;Hernandez et al.,2016;Givennikov et al.,2010)。因此,肿瘤微环境中存在或缺乏适当的固有感知实际上可以是检查点阻断治疗成功的关键决定因素。
诱导免疫原性固有感知和重塑肿瘤微环境的一种方法是使用遗传毒性药剂,例如已经广泛应用于癌症治疗的化学治疗(Patel et al.,2018;Emens et al.,2015;Pfirschke et al.,2016)。由于癌症治疗而垂死的肿瘤细胞可表达或释放DAMP,以通过特异性固有感知途径来活化免疫细胞,并引发针对肿瘤相关抗原的抗肿瘤免疫应答。事实上,将免疫检查点阻断与化学治疗组合正在临床试验中得到广泛研究(Garg et al.,2017;Langer et al.,2016;Gandhi et al.,2018;Weiss et al.,2017)。然而,使用传统化学治疗药物的主要限制之一是由于它们缺乏选择性和对非靶向组织(尤其是适应性免疫系统)的相关不良毒性。NQO1是在多种肿瘤类型中以高于正常组织5至200倍的水平表达的双电子氧化还原酶,并且是潜在的治疗靶标(Huang et al.,2016;Li et al.,2016)。β-Lap是一种新类别的NQO1靶向药物,其可被NQO1催化以产生活性氧物质(ROS)(Huang et al.,2016;Doskey et al.,2016)。事实上,本发明人发现β-lap在体外和在体内二者选择性地杀伤了高度表达NQO1的肿瘤,并且当本发明人敲除NQO1时这种杀伤作用被消除,表明该药物的理想选择性。尽管长期以来遗传毒性药剂被认为主要通过癌细胞自主机制(即通过直接抑制恶性细胞增殖或触发恶性细胞死亡)来发挥其作用,但越来越多的证据表明,成功应用于临床数十年的多种化学治疗剂还会触发免疫原性细胞死亡(ICD)并引发新的抗癌免疫应答(Sistigu et al.,2014)。遗憾的是,由于缺乏肿瘤选择性和严重的免疫抑制(耗竭快速分裂的免疫细胞群),大多数ICD诱导剂在治疗剂量下具有严重的副作用。重要的是,β-lap本身与其他化学治疗剂的不同之处在于其对NQO1过表达肿瘤的理想靶向作用以及对细胞毒性免疫没有免疫抑制。本发明人证明,即使在肿瘤致死剂量下,肿瘤浸润性CD8β-lap对自然和活化的CD8 T细胞也没有细胞毒性作用。一个新问题是NQO1生物可激活的β-lap介导的肿瘤特异性细胞死亡是否与免疫系统有一定相互作用;这种“靶向”化学治疗药物是否会引起免疫原性细胞死亡并触发固有感知。目前,本发明人发现β-lap诱导的NQO1+肿瘤消退在很大程度上依赖于宿主CD8+ T细胞:该药物在缺乏这些细胞的小鼠中未能控制肿瘤进程(Rag1-/-小鼠以及用抗CD8抗体耗竭的野生型小鼠)。本发明人还证明,β-lap通过HMGB1/TLR4途径诱导免疫原性细胞死亡并激活固有感知,并在肿瘤微环境中上调I型IFN信号传导,使得促进Batf3 DC以交叉致敏T细胞并激活抗肿瘤适应性免疫应答。
降低肿瘤负荷并提高肿瘤免疫原性被认为是改善免疫治疗的两个关键因素(Zappasodi et al,2018)。值得注意的是,β-lap促进了HMGB1依赖性的免疫原性并激活了固有感知以桥接固有和适应性免疫应答,并显著缩小了肿瘤块,并因此是与免疫治疗组合的有前景的搭档。事实上,本发明人表明使β-lap通过与抗PD-L1免疫治疗组合来根除大的建立的和检查点阻断难治性MC38和B16肿瘤,并显著提高了存活率。他们还证明了,与β-lap或抗PDL1单一治疗相比,组合治疗在肿瘤微环境中显著提高了肿瘤浸润性淋巴细胞和肿瘤抗原特异性T细胞以及CD8/Treg比。需要未来的工作来探索化合物的最佳剂量以及这些组合之组合的处理方案和排序。
总体而言,这项研究为β-lap(独特的靶向化学治疗药物)如何通过协调的固有和适应性免疫来诱导抗肿瘤作用以及β-lap治疗如何为免疫治疗建立了理想的微环境提供了新的见解。β-lap目前正在患有NQO1+实体瘤的患者中以单一治疗或与其他化学药物组合进行测试。这项研究指出,β-lap的固有感知能力可使NQO1+患者为成功响应于免疫治疗做好准备。
************
本文中已采用的术语和表述作为描述而非限制的术语使用,并且使用这样的术语和表述不旨在排除所示出和所描述的特征的任何等同形式或其部分,而是应认识到,在所要求保护的公开内容的范围内可进行多种修改。因此,应当理解,尽管已经通过优选实施方案、示例性实施方案和任选的特征具体公开了本公开内容,但是本领域技术人员可以对本文中公开的概念进行修改和变化,并且这样的修改和变化被认为是在如由所附权利要求书限定的本公开内容的范围内。本文中提供的具体实施方案是本公开内容的可用实施方案的一些实例,并且对于本领域技术人员而言将明显的是可使用本说明书中所述的装置、装置组件、方法步骤的大量变化形式来实施本公开内容。本领域技术人员将认识到的是,可用于本方法的方法和装置可包括大量任选的组成以及处理元件和步骤。
V.参考文献
以下参考文献就它们提供补充于本文中所述的那些的示例性程序或其他细节而言特别地通过引用而并入本文:
1.Topalian SL,Drake CG,Pardoll DM.Immune checkpoint blockade:a commondenominator approach to cancer therapy.Cancer cell.2015;27(4):450-61.
2.Pauken KE,Wherry EJ.Overcoming T cell exhaustion in infection andcancer.Trends in immumology.2015;36(4):265-76.
3.Sharma P,Hu-Lieskovan S,Wargo JA,Ribas A.Primary,Adaptive,andAcquired Resistance to Cancer Immunotherapy.Cell.2017:168(4):707-23.
4.Jenkins RW,Barbie DA,Flaherty KT.Mechanisms of resistance to immunecheckpoint inhibitors.British journal of cancer.2018;118(1):9-16.
5.Garber K.A new cancer immunotherapy suffers a setback.Science.2018;360(6389):588.
6.Lee Y,Auh SL,Wang Y,Burnette B,Wang Y,Meng Y,et al.Therapeuticeffects of ablative radiation on local tumor require CD8+ T cells:changingstrategies for cancer treatment.Blood.2009;114(3):589-95.
7.Woo SR,Corrales L,Gajewski TF.Innate immune recognition ofcancer.Annual review of immunology.2015;33:445-74.
8.Takeuchi O,Akira S.Pattern recognition receptors andinflammation.Cell.2010;140(6):805-20.
9.Patel SA,Minn AJ.Combination Cancer Therapy with Immune CheckpointBlockade:Mechanisms and Strategies,Immunity.2018;48(3):417-33.
10.Hernandez C,Huebener P,Schwabe RF.Damage-associated molecularpatterns in cancer:a double-edged sword.Oncogene.2016;35(46):5931-41.
11.Grivennikov SI,Greten FR,Karin M.Immunity,inflammation,andcancer.Cell.2010;140(6):883-99.
12.Garg AD,More S,Rufo N,Mece O,Sassano ML,Agostinis P,et al.Trialwatch;Immunogenic cell death induction by anticancer chemotherapeutics.Oncoimmunology.2017;6(12):e1386829.
13.Sistigu A,Yamazaki T,Vacchelli E,Chaba K,Enot DP,Adam J,etal.Cancer cell-autonomous contributton of type I interferon signaling to theefficacy of chemotherapy.Nature medicine.2014;20(11):1301-9.
14.Galluzzi L,Buque A,Kepp O,Zitvogel L,Kroemer G.ImmunologicalEffects of Conventional Chemotherapy and Targeted Anticancer Agents,Cancercell.2015;28(6):690-714.
15.Li R,Bianchet MA,Talalay P,Amzel LM.The three-dimensionalstructure of NAD(P)H:quinone reductase,a flavoprotein involved in cancerchemoprotection and chemotherapy:mechanism of the two-electronreduction.Proceedings of the National Academy of Sciences of the UnitedStates of America.1995;92(19):8846-50.
16.Oh ET,Kim JW,Kim JM,Kim SJ,Lee JS,Hong SS,et al.NQO1 inhibitsproteasome-mediated degradation of HIF-lalpha.Nature communications.2016;7:13593.
17.Li Z,Zhang Y,Jin T,Men J,Lin Z,Qi P,et al.NQO1 protein expressionpredicts poor prognosis of non-small cell lung cancers.BMC cancer.2015;15:207.
18.Ma Y,Kong J,Yan G,Ren X,Jin D,Jin T,et al.NQO1 overexpression isassociated with poor prognosis in squamous cell carcinoma of the uterinecervix.BMC cancer.2014;14:414.
19.Huang X,Motea EA,Moore ZR,Yao J,Dong Y,Chakrabarti G,etal.Leveraging an NQO1 Bioactivatable Drug for Tumor-Selective Use of Poly(ADP-ribose)Polymerase Inhibitors.Cancer cell.2016;30(6):940-52.
20.Pink JJ,Planchon SM,Tagliarino C,Varnes ME,Siegel D,BoothmanDA.NAD(P)H:Quinone oxidoreductase activity is the principal determinant ofbeta-lapachone cytotoxicity.The Journal of biological chemistry.2000;275(8):5416-24.
21.Doskey CM,Buranasudja V,Wagner BA,Wilkes JG,Du J,Cullen JJ,etal.Tumor cells have decreased ability to metabolize H2O2:Implications forpharmacological ascorbate in cancer therapy.Redox biology.2016;10:274-84.
22.Huang X,Dong Y,Bey EA,Kilgore JA,Bair HS,Li LS,et al.An NQO1substrate with potent antitumor activity that selectively kills by PARP1-induced programmed necrosis.Cancer research.2012;72(12):3038-47.
23.Li LS,Reddy S,Lin ZH,Liu S,Park H,Chun SG,et al,NQO1-MediatedTumor-Selective Lethality and Radiosensitization for Head and NeckCancer.Molecular cancer therapeutics.2016;15(7):1757-67.
24.Tang H,Wang Y,Chlewicki LK,Zhang Y,Guo J,Liang W,etal.Facilitating T Cell Infiltratinn in Tnmor Microenvironment OvercomesResistance to PD-L1 Blockade.Cancer cell.2016;29(3):285-96.
25.Morton JJ,Bird G,Refaeli Y,Jimeno A.Humanized Mouse XenograftModels:Narrowing the Tumor-Microenvironment Gap.Cancer research.2016;76(21):6153-8.
26.Tang H,Liang Y,Anders RA,Taube JM,Qiu X,Mulgaonkar A,et al.PD-L1on host cells is essential for PD-L1 blockade-mediated tumor regression.TheJournal of clinical investigation.2018;128(2):580-8.
27.Sanchez-Paulete AR,Cueto FJ,Martinez-Lopez M,Labiano S,Morales-Kastresana A,Rodriguez-Ruiz ME,et al.Cancer Immunotherapy withImmunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies RequiresBATF3-Dependent Dendritic Cells.Cancer discovery.2016;6(1):71-9.
28.Broz ML,Binnewies M,Boldajipour B,Nelson AE,Pollack JL,,Erle DJ,etal.Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity,Cancer cell.2014;26(6):938.
29.Salmon H,Idoyaga J,Rahman A,Leboeuf M,Remark R,Jordan S,etal.Expansion and Activation of CD103(+)Dendritic Cell Progenitors at theTumor Site Enhances Tumor Responses to Therapeutic PDL1 and BRAFInhibition.Immunity 2016;44(4):924-38.
30.Corrales L,Matson V,Flood B,Spranger S,Gajewski TF.Innate immunesignaling and regulation in cancer immunotherapy.Cell research.2017;27(1):96-108.
31.Deng L,Liang H,Xu M,Yang X,Burnette B,Arina A,et al.STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors.Immunity.2014;41(5):843-52.
32.Zitvogel L,Galluzzi L,Kepp O,Smyth MJ,Kroemer G.Type I interferonsin anticancer immunity.Nature reviews lmmunology.2015;15(7):405-14.
33.Apetoh L,Ghiringhelli F,Tesniere A,Obeid M,Ortiz C,Criollo A,etal.Toll-like receptor 4-dependent contribution of the immune system toanticancer chemotherapy and radiotherapy.Nature medicine.2007;13(9):1050-9.
34.Smyth MJ,Ngiow SF,Ribas A,Teng MW.Combination cancerimmunotherapies tailored to the tumour microenvironment.Nature reviewsClinical oncology.2015;13(3):143-58.
35.Mathios D,Kim JE,Mangraviti A,Phallen J,Park CK,Jackson CM,etal.Anti-PD-1 antitumor immunity is enhanced by local and abrogated bysystemic chemotherapy in GBM.Science translational medicine.2016;8(370):370ra180.
36.Ariyan CE,Brady MS,Siegelbaum RH,Hu J,Bello DM,Rand J,et al.RobustAntitumor Responses Result from Local Chemotherapy and CTLA-4 Blockade.Cancerimmunology research.2018;6(2):189-200.
37.Chen S,Lee LF,Fisher TS,Jessen B,Elliott M,Evering W,etal.Combination of 4-IBB agonist and PD-1 antagonist promotes antitumoreffector/memory CD8 T cells in a poorly immunogenic tumor model.Cancerimmunology research.2015;3(2):149-60.
38.Curran MA,Montalvo W,Yagita H,Allison JP.PD-1 and CTLA-4combination blockade expands infiltrating T cells and reduces regulatory Tand myeloid cells within B16 melanoma tumors.Proceedings of the NationalAcademy of Sciences of the United States of America.2010;107(9):4275-80.
39.Qiao J,Tang H,Fu YX.DNA sensing and immune responses in cancertherapy.Current opinion in immunology.2017;45:16-20.
40.Gajewski TF,Schreiber H,Fu YX.Innate and adaptive immune cells inthe tumor microenvironment.Nature immunology.2013;14(10):1014-22.
41.Kleponis J,Skelton R,Zheng L.Fueling the engine and releasing thebreak:combinational therapy of cancer vaccines and immune checkpointinhibitors.Cancer biology&medicine.2015;12(3):201-8.
42.Kamphorst AO,Wieland A,Nasti T,Yang S,Zhang R,Barber DL,etal.Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent.Science.2017;355(6332):1423-7.
43.Emens LA,Middleton G.The interplay of immunotherapyandchemotherapy:harnessing potential synergies.Cancer immunology research.2015;3(5):436-43.
44.Pfirschke C,Engblom C,Rickelt S,Cortez-Retamozo V,Garris C,PucciF,et al.Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint BlockadeTherapy.Immunity.2016;44(2):343-54.
45.Langer CJ,Gadgeel SM,Borghaei H,Papadimitrakopoulou VA,Patnaik A,Powell SF,et al.Carboplatin and pemetrexed with or without pembrolizumab foradvanced,non-squamous non-small-cell lung cancer:a randomised,phase 2 cohortof the open-label KEYNOTE-021 study.The Lancet Oncology.2016;17(11):1497-508.
46.Gandhi L,Rodriguez-Abreu D,Gadgeel S,Esteban E,Felip E,De AngelisF,et al.Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell LungCancer.The New England journal of medicine.2018;378(22):2078-92.
47.Weiss GJ,Waypa J,Blaydorn L,Coats J,McGahey K,Sangal A,et al.Aphase Ib study of pembrolizumab plus chemotherapy in patients with advancedcancer(PembroPlus).British journal of cancer.2017;117(1):33-40.
48.Zappasodi R,Merghoub T,Wolchok JD.Emerging Concepts for ImmuneCheckpoint Blockade-Based Combination Therapies.Cancer cell.2018;33(4):581-98.
49.Lundberg AP,Francis JM,Pajak M,Parkinson EI,Wycislo KL,Rosol TJ etal.Pharmacokinetics and derivation of an anticancer dosing regimen for thenovel anti-cancer agent isobutyl-deoxynyboquinone(IB-DNQ),a NQO1bioactivatable molecule,in the domestic felid species.Invest New Drugs.2017;35(2):134-44.
50.Li X,Liu Z,Zhang A,Han C,Shen A,Jiang L,et al.NQO1 targetingprodrug triggers innate sensing to overcome checkpoint blockaderesistance.Nat Communications.2019;10(1):3251.
Claims (26)
1.在患有癌症的患者中杀伤癌细胞或抑制癌细胞生长的方法,其包括施用与检查点抑制剂组合的NQO1生物可激活药物。
2.与第二药剂组合的NQO1生物可激活药物用于制备用于在具有癌细胞的患者中杀伤癌细胞或抑制癌细胞生长的药物的用途,其中所述第二药剂是检查点抑制剂,其中所述药物包含有效致死或抑制量的所述NQO1生物可激活药物和所述检查点抑制剂。
3.与第二药剂组合的NQO1生物可激活药物在治疗患有癌症的患者中的用途,其中所述第二药剂是检查点抑制剂。
4.权利要求1所述的方法,其中所述癌细胞具有由于错误DNA修复过程引起的碱基切除修复(BER)缺陷或脆弱性。
5.权利要求4所述的方法,其中所述BER缺陷或脆弱性包含X射线交叉互补1或XRCC1基因/蛋白/酶的水平缺陷。
6.权利要求1所述的方法,其中与小分子检查点抑制剂组合使用所述NQO1生物可激活药物。
7.权利要求1所述的方法,其中与抗体检查点抑制剂组合使用所述NQO1生物可激活药物。
8.权利要求1所述的方法,其中与PD-1或CTLA-4的抑制剂组合使用所述NQO1生物可激活药物。
9.权利要求6所述的方法,其中所述NQO1生物可激活药物是β-拉帕醌或DNQ化合物。
10.权利要求7所述的方法,其中所述NQO1生物可激活药物是β-拉帕醌或DNQ化合物。
11.权利要求8所述的方法,其中所述NQO1生物可激活药物是β-拉帕醌或DNQ化合物。
12.权利要求1至11中任一项所述的方法,其还包括另外的化学治疗剂或放射治疗。
13.权利要求1至12中任一项所述的方法,其中所述癌细胞具有升高的NQO1水平。
14.权利要求1至13中任一项所述的方法,其中所述癌细胞为实体瘤形式。
15.权利要求14所述的方法,其中所述癌细胞是非小细胞肺癌细胞、前列腺癌细胞、胰腺癌细胞、乳腺癌细胞、头颈癌细胞或结肠癌细胞。
16.权利要求1至15所述的方法,其中所述NQO1生物可激活药物是以下或其盐或溶剂合物:

17.权利要求2或3所述的用途,其中所述NQO1生物可激活药物是以下或其盐或溶剂合物:

18.权利要求1至17所述的方法,其中所述NQO1生物可激活药物是DNQ或DNQ-87。
19.权利要求1至18所述的方法,其中所述NQO1生物可激活药物在所述检查点抑制剂之前施用。
20.权利要求1至18所述的方法,其中所述NQO1生物可激活药物在所述检查点抑制剂之后施用。
21.权利要求1至18所述的方法,其中所述NQO1生物可激活药物与所述检查点抑制剂同时施用。
22.权利要求1至21所述的方法,其中所述NQO1生物可激活药物施用多于一次。
23.权利要求1至21所述的方法,其中所述检查点抑制剂施用多于一次。
24.权利要求1至21所述的方法,其中所述NQO1生物可激活药物和所述检查点抑制剂施用多于一次。
25.权利要求1至24所述的方法,其还包括另外的抗癌治疗。
26.权利要求25所述的方法,其中所述另外的抗癌治疗可以是化学治疗、放射治疗、免疫治疗、毒素治疗或手术。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201962819870P | 2019-03-18 | 2019-03-18 | |
| US62/819,870 | 2019-03-18 | ||
| PCT/US2020/023250 WO2020190990A1 (en) | 2019-03-18 | 2020-03-18 | Tumor-selective combination therapy |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN113905763A true CN113905763A (zh) | 2022-01-07 |
Family
ID=72519160
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202080037114.7A Pending CN113905763A (zh) | 2019-03-18 | 2020-03-18 | 肿瘤选择性组合治疗 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20220160703A1 (zh) |
| EP (1) | EP3942061A4 (zh) |
| JP (1) | JP2022525476A (zh) |
| KR (1) | KR20220004025A (zh) |
| CN (1) | CN113905763A (zh) |
| AU (1) | AU2020240035A1 (zh) |
| BR (1) | BR112021018545A2 (zh) |
| CA (1) | CA3130513A1 (zh) |
| MX (1) | MX2021011301A (zh) |
| WO (1) | WO2020190990A1 (zh) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20230128175A (ko) * | 2022-02-25 | 2023-09-04 | 주식회사 온코크로스 | 루복시스타우린을 포함하는 항암용 조성물 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105658809A (zh) * | 2013-04-09 | 2016-06-08 | 伊利诺伊大学董事会 | 肿瘤选择性联合疗法 |
| WO2017025496A1 (en) * | 2015-08-12 | 2017-02-16 | Bayer Pharma Aktiengesellschaft | Pharmaceutical combination for the treatment of cancer |
| WO2017165732A1 (en) * | 2016-03-24 | 2017-09-28 | Tragara Pharmaceuticals, Inc. | Treatment of cancer with tg02 |
| WO2019018757A1 (en) * | 2017-07-21 | 2019-01-24 | Genentech, Inc. | THERAPEUTIC AND DIAGNOSTIC METHODS FOR CANCER |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HUE037382T2 (hu) * | 2010-09-22 | 2018-08-28 | Univ Texas | Eljárás rák kezelésére targeting NQOl alkalmazásával |
-
2020
- 2020-03-18 EP EP20773832.9A patent/EP3942061A4/en active Pending
- 2020-03-18 KR KR1020217033464A patent/KR20220004025A/ko unknown
- 2020-03-18 JP JP2021556462A patent/JP2022525476A/ja active Pending
- 2020-03-18 BR BR112021018545A patent/BR112021018545A2/pt unknown
- 2020-03-18 CA CA3130513A patent/CA3130513A1/en active Pending
- 2020-03-18 US US17/440,787 patent/US20220160703A1/en active Pending
- 2020-03-18 WO PCT/US2020/023250 patent/WO2020190990A1/en unknown
- 2020-03-18 AU AU2020240035A patent/AU2020240035A1/en active Pending
- 2020-03-18 CN CN202080037114.7A patent/CN113905763A/zh active Pending
- 2020-03-18 MX MX2021011301A patent/MX2021011301A/es unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105658809A (zh) * | 2013-04-09 | 2016-06-08 | 伊利诺伊大学董事会 | 肿瘤选择性联合疗法 |
| WO2017025496A1 (en) * | 2015-08-12 | 2017-02-16 | Bayer Pharma Aktiengesellschaft | Pharmaceutical combination for the treatment of cancer |
| WO2017165732A1 (en) * | 2016-03-24 | 2017-09-28 | Tragara Pharmaceuticals, Inc. | Treatment of cancer with tg02 |
| WO2019018757A1 (en) * | 2017-07-21 | 2019-01-24 | Genentech, Inc. | THERAPEUTIC AND DIAGNOSTIC METHODS FOR CANCER |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2020190990A1 (en) | 2020-09-24 |
| CA3130513A1 (en) | 2020-09-24 |
| US20220160703A1 (en) | 2022-05-26 |
| EP3942061A1 (en) | 2022-01-26 |
| MX2021011301A (es) | 2022-01-19 |
| JP2022525476A (ja) | 2022-05-16 |
| BR112021018545A2 (pt) | 2021-12-14 |
| AU2020240035A1 (en) | 2021-10-07 |
| EP3942061A4 (en) | 2022-12-14 |
| KR20220004025A (ko) | 2022-01-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Wang et al. | STAT3 pathway in cancers: Past, present, and future | |
| AU2016276963B2 (en) | Combination therapy of transcription inhibitors and kinase inhibitors | |
| JP2023509881A (ja) | ジアシルグリセロールキナーゼ調節化合物 | |
| JP6719620B2 (ja) | 腫瘍選択的併用療法 | |
| KR20210063475A (ko) | 암치료에 유용한 tie2 키나아제의 억제 방법 | |
| JP2023109974A (ja) | 三置換ベンゾトリアゾール誘導体の使用方法 | |
| CA3144985A1 (en) | Hdac6-activated macrophages, compositions, and uses thereof | |
| CA2987324C (en) | Cerdulatinib for the treatment of b-cell malignancies | |
| WO2014082085A1 (en) | Use of itk inhibitors for the treatment of cancer | |
| US11224608B2 (en) | Compounds and methods for treating cancer | |
| KR20240023628A (ko) | 디아실글리세롤 키나제 조절 화합물 | |
| KR20230059792A (ko) | 암 치료를 위한 조합 | |
| Sai et al. | Basic and translational research on carbon-ion radiobiology | |
| CN113905763A (zh) | 肿瘤选择性组合治疗 | |
| Zhang et al. | Emerging mechanisms and implications of cGAS-STING signaling in cancer immunotherapy strategies | |
| CN113710660B (zh) | Dot1l降解剂及其用途 | |
| CA3191515A1 (en) | Stat-activated macrophages, compositions, and uses thereof | |
| US20240293430A1 (en) | Withaferin a and immune checkpoint blocker combination therapies | |
| US12037327B2 (en) | Compounds and methods targeting GPER for treatment of diseases associated with calcium | |
| KR20230131934A (ko) | 암을 치료하기 위한 조합 치료요법 스케줄 | |
| WO2023069943A1 (en) | Combination decitabine and mps1 inhibitor therapy to prime cancer immunogenicity | |
| JP2022023033A (ja) | ガンの処置に有用なtie2キナーゼの阻害方法 | |
| Zhang et al. | CAFs-Promoted LncRNA DNM3OS Conferred Radioresistance by Regulating DNA Damage Response in a TNFSF4-Dependent Manner in Esophageal Squamous Cell Carcinoma | |
| CN116322674A (zh) | 用于根除癌症干细胞并诱导damp介导的抗肿瘤免疫反应的一组新型copi/arf1-脂解途径抑制剂和化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20220107 |