CN107531602B - 基于泡罩塔反应器的浸煮器及其使用方法 - Google Patents
基于泡罩塔反应器的浸煮器及其使用方法 Download PDFInfo
- Publication number
- CN107531602B CN107531602B CN201680027722.3A CN201680027722A CN107531602B CN 107531602 B CN107531602 B CN 107531602B CN 201680027722 A CN201680027722 A CN 201680027722A CN 107531602 B CN107531602 B CN 107531602B
- Authority
- CN
- China
- Prior art keywords
- slurry
- digestion
- zone
- digester
- terephthalic acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/16—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation
- C07C51/21—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen
- C07C51/255—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen of compounds containing six-membered aromatic rings without ring-splitting
- C07C51/265—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen of compounds containing six-membered aromatic rings without ring-splitting having alkyl side chains which are oxidised to carboxyl groups
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J10/00—Chemical processes in general for reacting liquid with gaseous media other than in the presence of solid particles, or apparatus specially adapted therefor
- B01J10/002—Chemical processes in general for reacting liquid with gaseous media other than in the presence of solid particles, or apparatus specially adapted therefor carried out in foam, aerosol or bubbles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J19/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J19/0053—Details of the reactor
- B01J19/006—Baffles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J19/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J19/24—Stationary reactors without moving elements inside
- B01J19/2415—Tubular reactors
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J19/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J19/24—Stationary reactors without moving elements inside
- B01J19/2415—Tubular reactors
- B01J19/242—Tubular reactors in series
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/16—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation
- C07C51/21—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen
- C07C51/255—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen of compounds containing six-membered aromatic rings without ring-splitting
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
- C07C51/43—Separation; Purification; Stabilisation; Use of additives by change of the physical state, e.g. crystallisation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
- C07C51/487—Separation; Purification; Stabilisation; Use of additives by treatment giving rise to chemical modification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C63/00—Compounds having carboxyl groups bound to a carbon atoms of six-membered aromatic rings
- C07C63/14—Monocyclic dicarboxylic acids
- C07C63/15—Monocyclic dicarboxylic acids all carboxyl groups bound to carbon atoms of the six-membered aromatic ring
- C07C63/26—1,4 - Benzenedicarboxylic acid
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2219/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J2219/00761—Details of the reactor
- B01J2219/00763—Baffles
- B01J2219/00765—Baffles attached to the reactor wall
- B01J2219/00777—Baffles attached to the reactor wall horizontal
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- General Chemical & Material Sciences (AREA)
- Dispersion Chemistry (AREA)
- Crystallography & Structural Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Physical Or Chemical Processes And Apparatus (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
本发明提供一种浸煮从泡罩塔反应器氧化过程中对二甲苯氧化得到的粗对苯二甲酸浆料的方法。该方法在具有由水平挡板限定的一个或多个隔离区域的一个或多个泡罩塔反应器中进行,并控制通过浸煮系统的颗粒流量,以使部分氧化中间体向对苯二甲酸的转化最大化,同时使其它污染物产物的生成最小化。在整个过程中控制温度、氧流量和含量及其它过程和设备变量,以支持对苯二甲酸生成。本发明也提供构造成进行浸煮方法的泡罩塔浸煮系统。
Description
发明领域
本发明涉及氧化浸煮粗对苯二甲酸颗粒以得到可用纯度和粒径分布的纯化对苯二甲酸颗粒的方法,该方法经济上高效,具有减少的能量成本、设备资本成本、维护和可靠性成本,并且有最少或没有有机溶剂和芳族化合物过氧化成碳氧化物(CO和CO2),本发明还涉及进行该方法的设备。
发明背景
对苯二甲酸是用于多种应用的工业重要原料,主要应用是制备聚对苯二甲酸乙二酯(PET)。PET是在全世界大量用于制造诸如瓶、纤维和包装材料产品的熟知塑料。
对苯二甲酸常规通过对二甲苯液相氧化来制备。在典型液相氧化过程中,液相进料流和气相氧化剂流引入初级氧化反应器,并在反应器中形成多相反应介质。引入初级氧化反应器的液相进料流包含对二甲苯,而气相氧化剂流包含分子氧。作为气体引入初级氧化反应器的至少部分分子氧溶入反应介质的液相,为液相反应提供氧可用性。如果多相反应介质的液相的一些部分包含不足浓度分子氧(即,如果某些部分反应介质“贫氧”),则不合需要的副反应就可能产生杂质,并且/或者延迟预期反应速率。如果反应介质的液相包含太少可氧化化合物,则反应速率就可能相对于过氧化反应不合需要地慢。另外,如果反应介质的液相包含过高浓度可氧化化合物,则另外的不合需要副反应可产生杂质。
液相中存在的溶剂一般包含低分子量有机酸(例如乙酸)和水。在其中溶剂循环的制备系统中,溶剂可包含少量杂质,例如,对甲苯甲醛、对苯二醛、4-羧基苯甲醛(4-CBA)、苯甲酸、对甲苯甲酸、对甲苯甲醛(4-甲基苯甲醛)、α-溴-对甲苯甲酸、间苯二甲酸、邻苯二甲酸、偏苯三酸、多环芳族物质和/或悬浮颗粒。
常规用于对二甲苯部分氧化的催化剂系统为包含钴、溴和锰的均质液相催化剂。
泡罩塔反应器用于初级氧化反应提供相对于常规连续搅拌釜反应器的很多优点,利用泡罩塔反应器的氧化方法公开于例如 U.S. 7,355,068、U.S. 7,371,894、U.S. 7,568,361、U.S. 7,829,037、U.S. 7,910,769、U.S. 8,501,986、U.S. 8,685,334和U.S. 8,790,601,这些专利的内容通过引用结合到本文中。泡罩塔反应器提供反应介质搅拌,而不需要昂贵和不可靠的机械设备。泡罩塔反应器一般包括其内包含反应介质的长型直立反应区域。反应区域内反应介质的搅拌主要通过上升通过反应介质液相的自然气泡浮力提供。在泡罩塔反应器中提供的这种自然浮力搅拌相对于机械搅拌反应器减少设施电力、资本和维护成本。另外,基本没有与泡罩塔反应器相关的移动机械部件提供与机械搅拌反应器相比更少倾向于机械故障的氧化系统。
在对二甲苯液相部分氧化中,从初级氧化反应器抽取的产物一般为包含粗对苯二甲酸(CTA)颗粒固相和母液的浆料。CTA包含相对较高水平杂质(例如,4-羧基苯甲醛、对甲苯甲酸、芴酮和其它有色体),使其不适合作为制备PET的原料。因此,CTA一般经过纯化过程,使CTA颗粒转化成可适合用于制备聚对苯二甲酸乙二酯的经纯化对苯二甲酸(PTA)颗粒。在最近工业实施中,进一步纯化CTA经常通过氢化过程或氧化浸煮过程。
使CTA转化成PTA的常规氢化方法可包括以下步骤:(1)用水置换含CTA浆料的母液;(2)加热CTA/水浆料,以使CTA溶于水;(3)催化氢化CTA/水溶液,以使杂质转化成更期望和/或容易分离的化合物;(4)从经氢化溶液通过多个结晶步骤选择性沉淀对苯二甲酸;和(5)从其余液体分离经结晶PTA。虽然有效,但这种类型常规纯化方法可能耗费巨大。对常规CTA纯化方法高成本有贡献的单独因素包括例如促进CTA在水中溶解所需的热能,氢化所需的催化剂,氢化所需的氢流,一些对苯二甲酸氢化导致的产率损失,和多步骤结晶所需的多个容器。
或者,可在一系列另外的氧化反应器中使CTA转化成PTA,一般称为“浸煮器”。一般在这种系统中初始氧化反应母液中的CTA颗粒的初始浆料可包含约10至约50%重量固体CTA颗粒,其余为液体母液。从初级氧化反应器抽取的初始浆料中存在的固体CTA颗粒可包含约400ppmw至约15,000ppmw 4-羧基苯甲醛(4-CBA)。初始氧化反应器系统可包括以下两者:初级氧化反应器,主要用于氧化大部分液相可氧化化合物;和任选的至少一个二级氧化反应器,主要用于在进入浸煮器前精制转化液相可氧化化合物。
一般从初始氧化反应器系统抽取的CTA浆料转移到浸煮器单元系统,其中在略高于至显著高于初级和任选二级氧化反应器中使用的温度进行进一步氧化反应。可任选使CTA颗粒浆料在前进到浸煮器单元之前经过溶剂置换步骤,在此,经置换溶剂具有减小的芳族杂质浓度和/或改变的催化剂和水浓度,使它们重新调节到更适用于浸煮器单元中的氧化催化剂。任选地,在进入浸煮器单元之前,也可利用或不利用溶剂置换调节CTA浆料中固体的质量分数。
由初级氧化反应器中相对快速和不均匀沉淀得到的CTA颗粒一般具有相对较高结晶缺陷度,包括高孔隙率、高表面积和小且不均匀的粒径,且经干燥CTA一般显示比在CSTR氧化中得到的CTA更低的容积密度。另外,相对快速沉淀的CTA颗粒一般包含超平衡浓度的很多氧化杂质,包括偶合的多环芳族种类和不完全氧化中间体。不完全氧化中间体,例如4-CBA和对甲苯甲酸,处理起来特别麻烦,因为它们一般以数百至数千ppmw重量浓度出现。相比之下,在生成缩聚物(例如,PET)中使用PTA需要单羧酸聚合物链终止剂浓度尽可能低,300ppm是PTA中所有单羧酸总和的典型上限。
更优选在送至浸煮之前在二级氧化BCR中处理来自初级氧化的CTA浆料。该二级氧化,也称为后氧化或前氧化浸煮,如U.S. 7,393,973,其主要目的是在进入浸煮的更苛刻氧化条件之前,氧化大部分液相芳族氧化中间体,从初级氧化到TPA。这有用减少在初级氧化后发生的到碳氧化物的过氧化的总量。
为了使经沉淀氧化中间体杂质可用于在系列浸煮器中氧化,使颗粒暴露于比在初级氧化中更高的温度,以至少部分溶解CTA颗粒,并使杂质暴露于包含注入浸煮器的另外分子氧的液相氧化。在使CTA浆料温度适度升高到高于在初级氧化中生成CTA的温度时,小CTA颗粒的高表面积、结晶缺陷和超平衡杂质浓度在动力学和热力学二者上均有利于对苯二甲酸的部分溶解和继续重结晶。
在浸煮器系统中进行的进一步氧化旨在减小CTA颗粒中4-CBA的浓度。浸煮温度可高于初级氧化温度5℃至约90℃,一般可以为约150℃至约280℃。来自氧化浸煮的经纯化产物可结晶并收集在一个或多个结晶和重结晶单元。
在浸煮过程的第二种作用中,对苯二甲酸颗粒可经历奥斯特瓦尔德熟化,这倾向于提供与初级氧化出口流中CTA颗粒比较具有窄化粒径分布的较大颗粒。
在浸煮过程的第三种作用中,重结晶化对苯二甲酸颗粒包含减小浓度抗催化氧化矫正形成对苯二甲酸的很多杂质,杂质如多环芳族羧酸种类,特别是包括很多有色种类,尤其如2,6-DCF和2,7-DCF。这种减小由于更接近固相和液相之间抗氧化杂质的平衡分布而导致,这是由于比在初始氧化中更热的操作温度,也由于在浸煮过程期间延长的重结晶时间。如果任选溶剂置换步骤已使用相对更纯的溶剂,例如,来自用于去除对二甲苯氧化产生的水的溶剂脱水过程的经蒸馏含水乙酸,则进一步促进减小抗氧化杂质的固相浓度。
泡罩塔浸煮器(BCR)提供以上关于泡罩塔氧化反应器所述的机械优势。然而,在利用串联或并联的常规多个BCR时,这导致大的生产设备占位面积(footprint)以及供应和控制多个塔的复杂管道系统。然而,尽管达到浸煮的有利过程目的,但CTA浸煮方法和BCR设备的设计还呈现不同于初始氧化BCR的一些设计困难。
几种这些浸煮方法设计目的在U.S. 7,393,973中提出,其内容通过引用结合到本文中。具体地讲,对本发明值得注意的是,用于在固体入口附近和固体出口附近提供竞争性过程目标的液相和固相的停留时间分布(RTD)的优选范围,减少固体进料从入口到固体出口的短路的措施,和控制过氧化成碳氧化物同时仍实现部分氧化中间体到产物TPA的期望氧化的措施。
如U.S. 7,393,973中披露,由于在公开泡罩塔条件下从初始氧化得到的较小颗粒的较大部分的局部浓度,在到浸煮的固体入口附近,固体溶解速率特别快。因此,与在固体出口附近相反,在固体入口附近,由于来自固相的氧化中间体(例如4-CBA和对甲苯甲酸)溶解的较快速率,局部需要溶解氧从气相进入液相的最大供速。在固体入口附近的一个密切相关的困难是,需要液体混合稀释和同时氧化的恰当组合,使得液相氧化中间体(例如4-CBA和对甲苯甲酸)的静置浓度保持足够小,以防止太多再沉淀进入正形成的较大、更热力学和动力学稳定的PTA颗粒。在组合中,对于这些困难可取的是,在浸煮器固体入口区域更大的局部浆料混合和/或通气度,与固体出口区域相反,以稀释突发的溶解性芳族氧化中间体和提供必需的溶解分子氧局部供速。通过更大摩尔分数气体分子氧和更大通气的不同组合以改善相间传质系数(一般表示为相间薄膜传递作用“kL”和界面面积性质“a”的乘积“kLa”),可提供溶解氧的局部供速。
在U.S. 7,393,973中公开的另一个浸煮过程困难是需要限制从初始氧化送入的固体从其进入浸煮的入口位置到从浸煮的固体出口的短路通道。短路颗粒自身携带大于所需浓度的固相氧化中间体,从而使所有离开固体中的氧化中间体(例如,4-CBA)的加权平均组成不期望地向上偏移。另外,短路颗粒也不期望地加宽粒径分布,并减小离开浸煮过程的固体PTA的各种计算平均粒径,这不利地不同程度影响PTA粉末产物的过滤、洗涤、干燥和大量处理。
涉及在浸煮器中在芳族氧化中间体的期望氧化对比羧酸(例如,乙酸和TPA)过氧化成碳氧化物之间取得平衡的困难,U.S. 7,393,973的公开内容描述,“氧化浸煮的较后级在“缺氧”条件下进行,在此,在气体流出物中存在很低浓度分子氧”。这也与温度差组合,其中较后级的温度大于较前级。另外,根据本发明,“…分子氧在多个高度供到浸煮”,并且“用于提供分子氧到浸煮的分开高度包括在浸煮反应介质上半部的至少一个开口,和在浸煮反应介质下半部的至少一个开口”。
多个浸煮器单元结构描述于U.S. 7,393,973。然而,未描述只基于泡罩塔技术的浸煮器系统。如本文以前提到,与具有高资本成本、高操作成本和高操作和维护需求的机械搅拌反应器比较,泡罩塔反应器(BCR)提供机械和成本效率。
CN 202700501描述一种TPA方法,其中在一个或多个BCR中进行“深氧化” (浸煮)。在泡罩塔氧化反应器中初级氧化后,通过较小直径“排料桶”传导氧化反应浆料,在其中继续初级氧化过程。初级氧化浆料从“排料桶”转移入在BCR底部用含氧气体进料的第一泡罩塔“深氧化反应器”中部。在一个实施方案中,只利用一个深氧化反应器。在第二个实施方案中,从第一深氧化反应器底部抽取的浆料传导入也在底部具有空气入口的第二深氧化反应器中部。将第一深氧化反应器的排气用管送到第二深氧化反应器的底部附近,并且作为含氧气体源用于第二深氧化反应器。在两个实施方案中,在压缩并送到第一深氧化反应器之前,用来自初级氧化反应器的排气稀释环境空气。虽然随同5至8范围的长度-直径比描述与“氧化” BCR (初级氧化器)相比深氧化 BCR 70-80%的具体体积比,但没有公开初级氧化器或深氧化BCR容器相对于液体和固体流速的体积。另外,没有公开与深氧化BCR中CTA浆料流量或容器直径相比在排气循环和环境空气组合中包含的分子氧流量、分子氧摩尔分数或总气体化合物流量的适合比例。提供到深氧化反应器的这些气体流速和分子氧分数对控制能量需求、混合和传质流体动力学、期望的化学反应(包括对甲苯甲酸和4-CBA液相氧化反应成对苯二甲酸)、消耗羧酸生成CO和CO2的碳燃烧反应、没有机械搅拌下生长固体颗粒沉降和深氧化BCR内固体的停留时间分布十分重要。概括地讲,CN 202700501不充分公开放大和设计用于CTA有效浸煮转化成PTA的3-相BCR所必需的特征。
因此,需要用于在泡罩塔单元初始氧化对二甲苯中得到的CTA浆料的浸煮方法和相应结构化反应器,它有效,减少资本设施成本需求、机械维护、过程设施、原料损失,且简单设计成进行浸煮氧化和制备优良品质PTA。
发明概述
因此,本发明的目的是提供一种有效和高效浸煮CTA颗粒的方法,该方法以制备PET所必需的品质水平提供对苯二甲酸。
本发明的目的还提供一种浸煮系统结构,其中使CTA以制备PET所必需的品质水平转化成对苯二甲酸。
这些和其它目的通过本发明提供,其第一实施方案包括纯化粗对苯二甲酸的方法,所述方法包括:
a)得到粗对苯二甲酸颗粒的浆料,该进料浆料包含:在包含含水乙酸的溶剂液体中的对苯二甲酸、4-羧基苯甲醛和对甲苯甲酸,和含至少一种重金属化合物的催化剂系统;
b)将粗对苯二甲酸浆料送到基本没有机械搅拌的泡罩塔系统的第一浸煮区域;
c)在进入第一浸煮区域之前或在第一浸煮区域内时将粗对苯二甲酸浆料加热到约150℃至约280℃温度;
d)将含氧气体提供到第一浸煮区域,在此,在第一浸煮区域顶部附近的上升气体的表观速度为约0.1cm/s至约8cm/s;
e)使粗对苯二甲酸颗粒至少部分溶于乙酸,从而从颗粒释放至少一些4-羧基苯甲醛和对甲苯甲酸,并使溶解的4-羧基苯甲醛和对甲苯甲酸暴露于氧,以氧化成对苯二甲酸,并得到第一级浸煮器浆料;
f)将第一级浸煮器浆料通到第二浸煮区域,第二浸煮区域任选垂直位于第一浸煮区域下,且基本没有机械搅拌;
g)将含氧气体提供到第二浸煮区域下部;
其中到第二浸煮区域的气体的供速小于到第一浸煮区域的供速;和
h)从颗粒溶解并释放另外的4-羧基苯甲醛和对甲苯甲酸,并使溶解的4-羧基苯甲醛和对甲苯甲酸暴露于氧,以另外氧化成对苯二甲酸,并得到第二级浸煮器浆料;
i)任选使第二级浸煮器浆料移动通过结构类似于第二浸煮区域且任选垂直位于第二浸煮区域下的一个或多个另外的浸煮区域;
j)从最后浸煮区域移出所得对苯二甲酸结晶浆料;和
k)分离得到的对苯二甲酸结晶颗粒。
在第二个实施方案中,本发明包括氧化浸煮系统,该系统包括:
在至少一个泡罩塔反应器中布置的一系列至少两个氧化浸煮区域;
位于第一浸煮区域下部的至少一个浆料反应剂入口;
到第一浸煮区域的氧气源入口,和在第一区域后串联的至少一个区域;
各氧气源包括气体分配器单元,气体分配器单元将氧气作为气泡流送入该区域;
在至少一个泡罩塔底部的产物浆料出口。
在一个优选的实施方案中,本发明包括氧化浸煮系统,该系统包括:
在一个泡罩塔反应器中垂直布置的一系列至少两个氧化浸煮区域;
位于第一最上浸煮区域下部的至少一个浆料反应剂入口;
到第一最上浸煮区域的氧气源入口,和垂直位于第一最上区域下的串联的至少一个区域;
位于第一最上区域和垂直处于其下的第二区域之间的至少一个水平挡板;
在第一最上区域下存在多于一个区域时位于各相应垂直相邻区域之间的至少一个水平挡板;
在至少一个泡罩塔底部的产物浆料出口,
其中
各氧气源包括气体分配器单元,气体分配器单元将氧气作为气泡流送入该区域,并且
各水平挡板包括具有多个带有多个开口区域的倒置成形斜面的盘。
在第三个实施方案中,本发明提供泡罩塔浸煮系统,该系统包括:
构造用于对流的第一BCR单元;和
在第一BCR单元后串联的构造用于活塞流的至少一个BCR单元;
其中
第一BCR单元包括:
在塔中心垂直位置的浆料入口;
低于浆料入口的含氧气体入口;
在塔底部的浆料出口;
在塔顶部的装配有氧含量监测器的排气出口;和
任选在气体入口和浆料出口之间的水平挡板;并且
其中
至少一个第二BCR单元包括:
1至5个水平隔离区域,各区域任选装配有氧气入口;
各区域之间的水平挡板;
在最高区域中的浆料入口;和
在BCR单元底部的浆料出口;
其中至少一个区域装配有氧气入口。
在本发明的优选实施方案中,根据以上实施方案的氧化浸煮系统没有机械搅拌。
前面描述旨在提供本发明的一般介绍和总结,而不旨在限于其公开内容,除非另外明确陈述。通过参考以下详述并结合附图,可最好地理解目前优选的实施方案与另外的优点。
附图简述
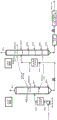
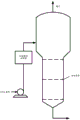
图1为根据本发明的一个实施方案的包括浸煮器氧化单元的对二甲苯氧化系统的示意图。
图2为根据一个任选实施方案的浸煮器氧化单元的示意图,其中两个单独BCR在浆料相上串联流动使用,具有分子氧新鲜进料供到各BCR。
图3为根据本发明的一个实施方案具有比最上区域更小直径的下部区域的浸煮器氧化单元的示意图。
图4A为根据本发明的一个实施方式从水平透视挡板布置的示意图。
图4B为从垂直透视4A的一个挡板的示意图。
图5A为根据本发明的一个实施方式从水平透视另一个挡板的示意图。
图5B为从垂直透视5A的挡板的示意图。
图6为根据本发明的一个实施方案用于浆料进料入口的倒锥形偏转器单元的示意图。
优选实施方案详述
在本发明描述中,词语“一个”(a)、“一种”(an)和“该”意味一个或多个。词语“和/或”在用于两个或更多个项目的列举时意味可利用任何一个所列项目本身,或者可利用两个或更多个所列项目的任何组合。例如,如果描述一种组合物包含组分A、B和/或C,则组合物可只包含A,只包含B,只包含C,包含A和B的组合,包含A和C的组合,包含B和C的组合,或包含A、B和C的组合。词语“包含”为用于从该词语前叙述的对象过渡到该词语后叙述的一个或多个元素的开放式过渡词语,其中在过渡词语后列举的一个或多个元素不必为组成该对象的仅有元素。词语“具有”和词语“包括”具有与以上提供的“包含”相同的开放式含义。
本文所用气体保持率、气泡保持率、气体分数和充气反应介质的气泡分数含义均相同,且简单为气态多相介质中的体积分数。单位为无量纲,例如,为立方米除以立方米,或者可将它们陈述为体积百分数。本文所用表观气体速度为气态多相介质组分的体积流速除以这些气态组分正流动通过的管或容器的横截面面积。表观气体速度的单位为例如立方米/秒/平方米,得到米/秒。分别用浆料体积流速、液体体积流速和固体体积流速代替气体组分体积流速,类似定义表观浆料速度、表观液体速度和表观固体速度。重要的是观察到,气体表观速度为计算值,不必代表任何气泡或气态组分其它等分试样的实际空间速度。类似陈述适用于其它表观速度。重要的是观察到,当与主要局部对流液体湍流比较,液体-固体密度差和重力的影响相对较大时,即,当由于减少液相湍流和/或由于增加固相粒径而存在显著固体沉降时,液体和固体表观速度可相互不同,又可不同于浆料表观速度。本文所用空气指包含任何摩尔分数分子氧的任何气体混合物,除非提供进一步明确修改,尤其如环境空气、压缩环境空气和环境空气+循环排气。
在提供数字范围以量化涉及本发明的某些参数时,应了解,这些范围应解释为对只叙述范围较小值的要求限定和对只叙述范围较大值的要求限定提供文字支持。表达的值也包括在所述限制值内的范围和子范围。
根据本发明,在包括一系列垂直布置区域的塔结构浸煮单元中处理通过初级氧化操作得到的粗对苯二甲酸(CTA)的浆料,如U.S. 7,355,068、U.S. 7,393,973、U.S. 7,829,037、U.S. 7,910,769、U.S. 8,501,986、U.S. 8,685,334和U.S. 8,790,601中所述。CTA浆料包含粗对苯二甲酸颗粒,包括在前述乙酸介质中的对苯二甲酸、4-羧基苯甲醛和对甲苯甲酸。对于浸煮,使从初级氧化系统得到的CTA浆料的温度提高到高于从初始氧化进入的浆料温度至少5℃至约90℃的值,优选约10℃至约60℃,更优选约15℃至约40℃。离开初始氧化的浆料的温度可以为约125℃至约200℃,优选约140℃至约185℃,更优选约150℃至约175℃。因此,在浸煮级浆料的温度为约150℃至约280℃,优选约160℃至约240℃,更优选170℃至210℃。该温度提高可在位于浸煮单元前的加热单元内完成,或者通过适用于提高含水乙酸混合物中颗粒浆料温度的任何适合方法。
为了控制浸煮区域中溶剂的蒸发,在浸煮中提高温度一般包括比在初级和二级氧化中更大的塔顶压力。至少一个浸煮区域的塔顶排气压力相对于初级氧化塔顶排气压力在大约相同至高约4MPa的范围内,优选高约0.5MPa至高3MPa,最优选高约1MPa至高约2MPa。优选至少一个浸煮区域的塔顶排气压力为约0.4MPa至约8MPa,约0.5MPa至约4MPa,或1MPa至2MPa。
发明人已发现,可将浆料从初级氧化器BCR中较高高度较小表压的位置送入二级氧化器BCR中较低高度较大表压的位置,而不用机械泵送装置或任何辅助流动能量,例如喷射器,尽管在二级氧化器BCR中反应介质一般有较大密度。发明人还发现,对于优化增加液相芳族中间体转化率,并具有减少的偶合和有色多环芳族化合物生成同时减少生成碳氧化物的过氧化反应的目的,将CTA浆料从初级氧化器送到二级氧化器BCR较低高度是出乎意料地有利的。
通过分离从初级氧化器较高高度抽取的充气反应介质,并使其脱气生成基本脱气的浆料,优选实现从较小表压到较高表压的转移。更优选根据U.S. 7,371,894的公开内容以最少量时间完成反应介质脱气,由此使包含多环芳族和有色杂质的芳族杂质生成最大限度地减少。内部或外部脱气容器都是优选的。更优选用于反应介质的脱气容器在初级氧化器外部。
从脱气容器下部抽取的经脱气浆料具有比初级或二级氧化器BCR中充气浆料更大的容积密度和位势压头/米高度。因此,当在充气条件操作时,从脱气容器下部的连接管可只通过重力流入初级或二级氧化器BCR的任何较低高度。基本上,初级氧化器BCR中向上上升的气体和溶剂在气体提升类型泵送中使反应介质膨胀,虽然在初级氧化器BCR内保留的浆料连续地通过上升气体按照尤其与粘性和表面张力相互作用的重力向下回落。
然而,作为过程控制实际情况,有用且优选在离开初级氧化器的脱气容器基底高度、二级氧化器中充气反应介质顶部的高度、在二级氧化器BCR内首先提供充气所处的高度和二级氧化器BCR内的充气度之间有某些关系。这是要保证用工业合理尺寸的管和流量控制元件输送足够浆料流。
在初级和二级BCR内不同流量、压力、温度和料面(level)的正常操作条件下,优选二级氧化器BCR中的气体保持率为至少约14%体积,更优选至少约20%体积,更优选至少约26%体积,最优选至少约32%体积。除了考虑支持从初级氧化器BCR上部高度抽取的浆料足够流量和控制外,该充气度可用于支持从气体-液体传质,用于在二级氧化器BCR中提供溶解的分子氧。在BCR中间高度位势、四分之一高度位势和四分之三高度位势测定或计算,优选用至少约0.04米/秒表观气体速度达到该气体保持率,更优选至少约0.08米/秒,更优选至少约0.12米/秒,最优选至少约0.16米/秒。该表观速度用所有上升气体组分的体积流量除以在考虑高度BCR的横截面面积计算,包括真实气体(例如O2、N2、Ar、CO、CO2等)和经蒸发液体蒸气(例如,乙酸蒸气、水蒸气等)二者。
优选在高于气态分子氧供到初级氧化器BCR所处的最低高度至少约12米(更优选至少约18米,更优选至少约24米,最优选约30米)的高度从初级氧化器BCR抽取至少约25%(更优选至少约50%,更优选至少约75%,最优选至少约100%)送到二级氧化器BCR的浆料。
优选使从初级氧化器BCR上部高度抽取的至少约25%质量(更优选至少约50%质量,更优选至少约75%质量,最优选至少约100%质量)浆料在高于气态分子氧供到二级氧化器BCR所处的最低高度小于约12米(更优选小于约8米,更优选小于约4米,最优选小于约2米)的高度供到二级氧化器BCR。
优选二级氧化器BCR的正常操作料面高于气态分子氧供到二级氧化器BCR所处的最低高度小于约45米,更优选小于约40米,更优选小于约35米,最优选小于约30米,且大于约8米,更优选大于约12米,更优选大于约16米,最优选大于约20米。
优选二级氧化器BCR的切线-切线高度小于约50米,更优选小于约45米,更优选小于约40米,最优选小于约35米,且大于约10米,更优选大于约14米,更优选大于约18米,最优选大于约22米。
优选气态分子氧供到二级氧化器BCR所处的最低高度从气态分子氧供到初级氧化器BCR所处的最低高度垂直位移小于约16米,更优选小于约12米,更优选小于约8米,最优选小于约4米。
优选从二级氧化器BCR内正常操作料面和正常气体保持率的以上范围选择组合,使得在浆料流快速停止时,在浆料从初级氧化器进入二级氧化器的最高高度浆料供应管中的表压大于在相同高度二级氧化器内的压力至少约30kPa,优选至少约60kPa,更优选至少约90kPa,最优选至少约120kPa,即,在浆料入口最高高度测定或计算浆料供应管和二级氧化器BCR内之间的压差,在供应管与摩擦流量损失、动压头压损失或控制元件压力损失相关没有任何压力损失。由此,与二级氧化器BCR内的表压比较,在浆料供应管中的过量静态表压(二者均在浆料供到二级氧化器BCR的最高高度测量)可用于动压头发展、在浆料管和进入二级氧化器BCR的摩擦损失和调节浆料流速的控制元件。从初级氧化器进入二级氧化器的浆料流速优选响应至少一个以下过程测定调节,包括初级氧化器BCR中充气反应介质的料面、二级氧化器BCR中充气反应介质的料面、进入初级氧化器的对二甲苯的体积或质量进料速率和进入和/或从二级氧化器的浆料的质量或体积流速。
浆料从初级氧化器BCR送到接近二级氧化器BCR基底的意外效果涉及溶解的分子氧的最大气体-液体传质供速区域与溶解分子氧的最大需求区域与提供浆料进入和离去高度较大轴向分离组合的匹配。例如,当浆料在中间高度进料时,可能使溶解氧峰需求接近气态分子氧新源,或者使溶解芳族中间体的液相浓度向上和向下二者同时出现,以降低溶解分子氧的峰需求速率,因此,BCR底部出口中芳族中间体的浓度也不期望地增加。通过接近基底送入浆料和气态分子氧供应,然后从接近二级氧化器BCR顶部抽取浆料,可同时得到减少偶合多环芳族杂质生成、减少接近二级氧化器BCR顶部过量分子氧的需要、和增加离开二级氧化器的浆料中液相部分氧化中间体转化率的益处。增加液相部分氧化中间体转化率可减少在随后浸煮器操作中碳燃烧,特别在CTA浆料绕过溶剂交换操作直接送到浸煮时。因此,优选供到二级氧化器BCR的气态分子氧的至少约25%(更优选至少约50%,更优选至少约75%,最优选至少约100%)在主圆筒部分直径的1倍内送入,优选在0.5倍内,更优选在0.25倍内,最优选在0.1倍内,且在二级氧化器BCR内最低高度的约0.64米内送入,优选在约0.32米内,更优选在约0.16米内,最优选在约0.08米内。也优选供到二级氧化器BCR的CTA浆料的至少约25%(优选至少约50%,更优选至少约75%内,最优选至少约100%)在主圆筒部分直径的3倍内送入,优选在2倍内,更优选在1倍内,最优选在0.5倍内,且在二级氧化器BCR内最低高度的约6米内送入,优选在约4米内,更优选在约2米内,最优选在约1米内。
由于二级氧化器BCR不是关于反应的完美混合容器,且传质时间常数在对二甲苯部分氧化生成中相关,增加在提高浆料出口的液相氧化中间体的转化率。因此,在浆料的进入和离开点可大大分离时,甚至不改变塔尺寸就提高反应转化率。优选二级BCR的浆料入口和出口垂直分离二级氧化器BCR最大直径的至少8倍,更优选至少约12倍,更优选至少约16倍,最优选至少约20倍。另外,优选二级氧化器BCR的浆料出口位于低于反应介质正常时间平均操作料面的容器主圆筒部分最大直径小于约8倍,更优选小于约6倍,更优选小于约4倍,最优选小于约2倍。对于涉及响应上游和下游流速干扰的气体分离和/或水平波动,可使用高于时间平均操作料面的容器体积部分。在本发明的一个优选实施方案中,二级氧化器BCR中反应介质的正常时间平均操作料面基本位于浆料出口高度,其中容器以浆料溢流模式操作。
然而,通过更宽分离浆料入口和出口位置和/或使二级氧化器BCR体积更大增加轴向分级可达到与在固相颗粒中保持的比较对液相氧化减小返回的状况。
为了避免太多液相对甲苯甲酸转化,可减小二级氧化器BCR的体积,以减少资本成本和过氧化成碳氧化物。在此情况下,通常期望根据容器高度“H”减小容器内径“D”,使得BCR的H:D比增加,从而促进更大分级用于保留的反应体积。当然,体积减小会增加离开二级氧化器BCR的液相氧化中间体的浓度,但通过减小直径减小容器体积提供与通过减小高度减小体积比较更大削弱的偏移。因此,优选二级氧化器BCR中反应介质的高度:直径比为至少6:1,更优选至少12:1,更优选至少16:1,最优选20:1。
根据U.S. 7,568,361的公开内容,通过使用至少约1、2、4、8个“无污染”挡板,可有用增加二级氧化器BCR中液相氧化的轴向分级(应注意到,在说明书的整个其余部分中,如以前句子中多个数值的列举表示从优选到最优选的嵌套范围)。这在其中高H:D比表示过高绝对高度的用于较大生产率大小的生产单元中变得越来越重要。增加无污染挡板可维持二级氧化器BCR中良好的分级性能,即使增加D和减小H:D。
一般优选使这些挡板大部分高于在新鲜分子氧进料中止至少约2、8、16、32小时后实现的固体沉降平面。在CTA固体沉降和密实后,在快速重新开始充气时,可在挡板调节工具上施加很大力。然而,在初级和二级氧化器BCR和浸煮器的总体设计表明该挡板布置可用于减少过氧化成碳氧化物,同时达到用于PTA的目标纯度时,可提供足够机械完整性的无污垢挡板,以经受充气启动力,即使有很好沉降的CTA。
为了缓解或避免在中断期间有沉降固体的以上困难,也优选提供浆料排泄管,浆料排泄管从二级氧化器BCR的底部或很接近底部通到减压出口。更优选该较低排泄管连接到用于将浆料送到浸煮器系统的泵抽吸处。然而,在二级氧化器BCR正常操作期间,该替代管优选完全由关闭阀构成。
利用中断沉降的类似情况存在于将脱气浆料从初级氧化器输送到接近二级氧化器BCR基底的管中。在较小直径浆料管中的操作情况相对比在较大直径BCR中更重要。只要充气在初级氧化器BCR中中断,从脱气容器引出的相对较高细长管就可停止用气升浆料向上供到抽取浆料侧和脱气容器入口的高度。以后不久,在压力与二级氧化器BCR浆料入口位置平衡时,管中的浆料流就将停止。在这发生至少约2、8、16、32小时时,优选通过接近管最低高度送入惰性气体冷却供应管中剩余的浆料。对于CTA固体的这种基本延迟水泥质结合,优选送入足够气体,以蒸发性冷却其余浆料低于正常操作温度至少约10、20、30、40℃。更优选在排泄二级氧化器BCR期间或之后,由其底部出口管通过二级氧化器排泄供应管。任选排泄管从接近浆料供应管最低高度直接提供到用于浆料送到浸煮器系统的泵的抽吸处,但不通过二级氧化器BCR。在二级氧化器BCR正常操作期间,该排泄管也优选完全由关闭阀构成。
尽管这些准备在过程中断和切断后重新开始操作,但优选本发明的很多实施方案以相当顺利和连续的流动方式操作。
优选从二级氧化器BCR抽取的浆料液相中对甲苯甲酸的时间平均浓度小于引入二级氧化器BCR的浆料液相中对甲苯甲酸的时间平均浓度的约50%、10%或5%。优选引入二级氧化器BCR的浆料液相中对甲苯甲酸的时间平均浓度为约50至约10,000、约100至约6,000或500至5,000ppmw。优选从二级氧化器BCR抽取的浆料液相中对甲苯甲酸的时间平均浓度小于约1,000、200或60ppmw。优选从二级氧化器BCR抽取的浆料液相中4-CBA的时间平均浓度小于引入二级氧化器BCR的浆料液相中4-CBA的时间平均浓度的约50%、10%或5%。优选引入二级氧化器BCR的浆料液相中4-CBA的时间平均浓度为约100至约6,000、约200至约4,000或400至3,500ppmw。优选从二级氧化器BCR抽取的浆料液相中4-CBA的时间平均浓度小于约500、100或30ppmw。优选从二级氧化器BCR抽取的浆料固相中4-CBA的时间平均浓度为引入二级氧化器BCR的浆料固相中4-CBA的时间平均浓度的约5%至约95%、约10%至约90%、约20%至约80%或30%至70%。优选引入二级氧化器BCR的浆料固相中4-CBA的时间平均浓度为约100至约15,000、约400至约8,000或1,000至6,000ppmw。优选从二级氧化器BCR抽取的浆料固相中4-CBA的时间平均浓度为约100至约12,000、约300至约8,000或800至1,000ppmw。
初级氧化和二级氧化的组合,如果使用,在本文中称为初始氧化,CTA浆料产物称为初始浆料。
浸煮器进料流液相中存在的催化剂系统优选为能够促进对二甲苯氧化(包括部分氧化)的均匀液相催化剂系统。催化剂系统可包含钴、溴和锰中的一种或更多种。
当催化剂系统中存在钴时,优选液相进料流中存在的钴的量使得反应介质液相中钴的浓度保持在约300至约6,000ppmw,更优选约700至约4,200ppmw,最优选1,200至3,000ppmw。当催化剂系统中存在溴时,优选液相进料流中存在的溴的量使得反应介质液相中溴的浓度保持在约300至约5,000ppmw,更优选约600至约4,000ppmw,最优选900至3,000ppmw。当催化剂系统中存在锰时,优选液相进料流中存在的锰的量使得反应介质液相中锰的浓度保持在约20至约1,000ppmw,更优选约40至约500ppmw,最优选50至200ppmw。
反应介质液相中钴、溴和/或锰的浓度可基于时间平均和体积平均表达。本文所用术语“时间平均”表示经至少100秒连续时间相等进行至少10个测量的平均值。本文所用术语“体积平均”表示在一定体积内均匀3维间隔进行至少10个测量的平均值。引入氧化反应的催化剂系统中钴与镍(Co:Br)的重量比优选为约0.25:1至约4:1,更优选约0.5:1至约3:1,最优选0.75:1至2:1。引入氧化反应的催化剂系统中钴与锰(Co:Mn)的重量比优选为约0.3:1至约40:1,更优选约5:1至约30:1,最优选10:1至25:1。
在送到浸煮之前,可任选在溶剂交换操作或在浆料稠度操作中处理来自初始氧化的CTA的初始浆料。在使用这些任选操作时,过程目标包括调节催化组合物,例如,钴、锰、溴和水的液相组合物,在浸煮前去除额外量液相芳族氧化中间体,去除不容易氧化的液相杂质,例如2,6-DCF和2,7-DCF,以提高经浸煮PTA的纯度和白度,并调节浆料中固体的质量分数,以改变浆料的总体积和/或调节浆料的流体动力学性质。在这些处理后,在本文中将该初始浆料称为CTA的溶剂改良浆料。
在利用这种任选溶剂交换操作时,优选去除在初始浆料液相中发现的溶剂的至少约40、60、80、90%和可溶性化合物。随后,优选用较清洁溶剂提供具有质量分数固体的重组浆料,如本文其它地方所公开。本文所有术语“较清洁溶剂”表示具有小于加较清洁溶剂的浆料液相中总催化剂化合物浓度的总催化剂化合物液相浓度的溶剂。与加较清洁溶剂的浆料液相比较,优选较清洁溶剂包含小于约90、50、10或2%重量总催化剂化合物液相浓度和/或小于约90、50、10或2%重量总芳族化合物液相浓度。
在根据本发明的一个实施方案基本消除初级氧化和氧化浸煮之间的溶剂交换时,可优选在经过氧化浸煮的浆料中保留至少约30、60、80或95%重量从初级氧化抽取的初始浆料中最初存在的初始液体。因此,可优选从经过氧化浸煮的浆料去除小于约70、40、20或5%重量从初级氧化抽取的初始浆料中最初存在的初始液体。优选进入氧化浸煮的浆料中钴、其它催化剂化合物和/或苯甲酸与从初级氧化产生的初始浆料中相同化合物的重量比为至少约0.3、0.6、0.8或0.95。更优选离开氧化浸煮的浆料中钴、其它催化剂化合物和/或苯甲酸与从初级氧化产生的初始浆料中相同化合物的重量比为至少约0.3、0.6、0.8或0.95。在多级进行氧化浸煮时,本段落中的描述可适用于任何或所有氧化浸煮级,最优选包括氧化浸煮的最后级。
因此,送到浸煮器的浆料可以为CTA的初始浆料,或者为CTA的溶剂改良浆料,以后在本文中可称为浸煮器进料浆料。
优选浸煮器进料浆料中固体CTA质量分数大于约0.10、0.20、0.30且小于约0.50、0.45、0.40。
保留在根据本发明提供的浸煮器进料浆料中的芳族中间体氧化所需的分子氧的量相对较小。优选提供到所有浸煮级的分子氧的摩尔数小于送入3、2、1kg压缩环境空气/100kg浸煮器进料浆料中包含的量。比较起来,压缩环境空气到初级氧化器的进料速率通常为约100kg/100kg初始浆料。除了空气进入浸煮的相对较小摩尔和质量流速外,提高浸煮器系统操作温度表明与初始氧化比较显著更大的操作压力,这引起进一步减小供到浸煮器系统的分子氧的体积进料比。在组合下,分子氧到浸煮器系统的小体积进料速率在浸煮器BCR中提供足够气体混合功率以满足过程需要中提出苛刻困难,包括固体悬浮,分子氧从气体-液体的传质,和进入第一浸煮区域的浸煮器进料浆料的相对快速混合。
由于浆料位势压头一般比塔顶绝对压力小得多,浸煮器反应介质内上升气体提供的混合功率接近近似于流动气体体积乘以气体上升通过的浆料的位势压头。本文所用术语“气体混合功率”定义为VdP,等于浸煮器容器内上升气体的体积流速乘以它上升通过的浆料位势压头,且单位适合转化成功率单位。可以理解,浸煮器高度和直径的很多可能组合对浆料特定总停留时间都是可能的,很多不同操作压力都是可能的。因此,对于气体混合功率有很多可能结果,但在所有合理情况下,所得气体混合功率在本发明的浸煮区域内很小。优选在发明所有BCR浸煮器区域中总计的气体混合功率除以这些区域内的浆料质量小于约0.2、0.1、0.05W/kg浆料。关于BCR浸煮器系统不同区域内该限量气体混合功率分布的另外的公开内容在本文其它地方讨论不同区域表观气体速度和尺寸(即,高度和宽度)中揭示。
由于碳燃烧经济和塔顶顶部空间易燃性,一般不希望通过送入比作为实现去除包含对甲苯甲酸和4-CBA的氧化中间体的最低条件所需更大过量的压缩环境空气来增加气体混合功率。由于资本设备成本、压缩能成本和热能成本,通常不希望通过加入或去除尤其包含分子氮、二氧化碳、一氧化碳、分子氢、甲烷、甲基溴和氩的气态惰性稀释剂减小分子氧的摩尔分数。然而,向至少一个浸煮区域送入与环境空气组合物比较富含或贫一定摩尔分数分子氧的压缩空气流在本发明公开内容范围内。
在尤其使用CTA粒径和初始浆料组合物优选范围时,本发明的实施方案排除需要对浸煮器反应介质提供机械轴动力搅拌。然而,发明人也已发现,通过意外少量补充机械轴动力搅拌,可提供气泡流气体动力混合对不太理想情况的适应。例如,关于具有相对较平2:1椭圆形底头的现有浸煮器容器改进本发明可能需要在最低浸煮区域内和缓机械搅拌,以保证浆料固体不在底头内沉淀、停滞和水泥性结块成大的固体块。在使用补充机械搅拌时,优选机械搅拌功率小于关于浸煮器反应介质总体积平均的约0.2、0.1、0.05W/kg浆料,优选气体混合功率和机械混合功率的总组合小于关于浸煮器反应介质总体积平均的约0.30、0.15、0.10W/kg浆料。
在本领域已知的浸煮器浆料加热方法包括对二甲苯以外有机底物的氧化,例如,送入的己烷氧化成碳氧化物和乙酸;其它化学反应,例如乙酸酐水解成乙酸;可冷凝溶剂蒸气提供到浆料,例如水蒸气和乙酸蒸气;和使用热交换器装置浆料和/或气态分子氧源的非接触加热。浆料非接触加热是特别优选的。使用交换器装置非接触加热避免在使用化学加热或溶剂蒸气加热时通常需要的另外的化学复杂性和/或容器体积。
用于浸煮器浆料的交换器加热单元可以为适用于CTA浆料使得单元堵塞和污染避免的任何设计。它也必须由对腐蚀性乙酸混合物稳定的材料构成。热交换器表面可接近位于优选为BCR浸煮器顶部区段的第一浸煮器区域上游,通过初始氧化和浸煮区域之间的浆料管连接。在送到浸煮容器之前,优选用至少一个非接触热交换器装置在来自初始氧化的浆料上提供至少约30、60、90、100%用于浸煮区域的总热负荷。
在一个实施方案中,CTA浆料加热单元可以为垂直管-壳设计,使得浆料在进入浸煮器容器之前以垂直上流方式通过管侧。热交换器表面可任选位于内部,即,在浸煮反应容器自身内。
在另一个优选实施方案中,将用于第一浸煮区域(优选BCR浸煮器顶部或最上区段)的分子氧源的至少约25、50、75或100%送入浸煮器与浸煮器进料浆料混合。优选在首先加热浆料高于在从初始氧化的出口的温度至少约10℃后约8、2、0.5分钟内使该分子氧与浸煮器进料浆料混合。更优选在外部热交换器出口之前,更优选接近外部热交换器的浆料入口,使用于第一浸煮区域(优选BCR浸煮器顶部区段)的分子氧源的至少约25、50、75、100%与浆料混合。加热初始浆料和提供分子氧的这种时间上的密切配合对在初始突发CTA溶解活性期间防止太多液相对甲苯甲酸和4-CBA非反应积累是重要的。例如,CTA浆料从约160℃加热到约210℃使溶解的TPA的平衡质量分数从液相质量的约0.5%增加到液相质量的大于约2%,且最小CTA结晶在此浆料加热后只数秒内溶解。
重要的是,根据本发明从初级氧化操作得到的CTA颗粒的特征为,比尤其用不同反应器混合特性、停留时间、体积氧化率强度、压力和温度分布及溶剂组成操作的很多其它对二甲苯氧化过程产生的CTA颗粒更小和更多孔的颗粒形态结构。在优选的实施方案中,在氧化中得到的固体CTA产物具有20至150微米的平均粒径,优选25至100微米,最优选30至80微米。颗粒的形态结构应使得各颗粒一般由大量小的附聚颗粒形成,因此,CTA颗粒具有约0.6m2/g至4.0m2/g的高BET(Braunauer-Emmett-Teller)表面积,优选0.8至3.0m2/g,最优选0.9至2,0m2/g。粒径、表面积和附聚形态结构的这种组合产生高孔隙率、低密度和低沉降速度的颗粒,这些是有利于本发明浸煮过程的性质。
比较起来,通过一些氧化方法得到的CTA颗粒的特征是约180至220微米的平均粒径,和约0.4至0.8m2/g的BET表面积。这些常规颗粒具有小得多的孔隙率和显著更高的表观密度(在溶剂液体中测定)。
上述本发明的CTA颗粒的物理性质允许通过本发明的泡罩塔浸煮方法有效和高效浸煮,因为具有更大表面积的小颗粒更容易溶解,使夹带的杂质释放到溶液,在此,氧化可使杂质转化成产物。同样重要的是,CTA颗粒的较小粒径和高孔隙率允许在气泡流动环境更抗沉降,因此,可将通过泡罩塔系统的颗粒流控制到足够停留时间,以允许所述纯化。
对串联流中的所有浸煮器区域总计,由于在浸煮器BCR中部分沉降可相互不同的固相和液相的质量平均停留时间分别为约10至约480分钟,优选约20至约360分钟,更优选40至120分钟。
固相的停留时间分布期望用本发明改善。虽然在浆料首先进入浸煮器时需要相当的液相混合,但优选随后用更紧密接近活塞流RTD的RTD使固相移动通过浸煮器系统。在CTA浆料进料中的较小颗粒首先进入浸煮器时,接近浆料入口需要的液相混合涉及4-CBA和对甲苯甲酸初始释放进入液相。在浆料进料位置附近,期望提供足够对流混合,以控制对溶解O2支持在液相中希望芳族氧化的局部液相需求,并控制可在固体颗粒扩大时重新沉淀和埋藏的4-CBA和对甲苯甲酸的局部液相浓度。在浆料进料位置附近满足反应化学混合和气体-液体传质要求后,随后优选对具有较高浓度固体4-CBA和固体对甲苯甲酸的较小颗粒控制和延长在浸煮器中的停留时间。这种RTD通过本发明的不同实施方案提升,包括一定范围表观气体速率、接近容器轴向中线一定范围轴向浆料速度、一定范围粒径分布、H:D比和无污染挡板调节系统。
在以下公开内容中采用符号,其中“t”为时间;时间的停留分布函数为在时间t=0初始供到反应区域然后在时间“t/tavg”前离开反应区域的相的累积质量分数(CMF);其中“tavg”为根据下述计算测定的质量平均停留时间,“t/ tavg”为“约化时间”,指时间除以质量平均停留时间。约化时间为无量纲。“CMF(t/ tavg)”为约化时间的停留分布函数。例如,CMF(0.2)为在时间t=0初始供到反应区域的相然后在约化时间0.2前离开反应区域的累积质量分数。在时间t=0初始送到包围部的质量等分部分的质量平均停留时间(tavg)计算为[(t)*(在时间t离开的等分部分的质量)]/从时间0到等分部分质量的至少约99.9%已离开包围部积分的等分部分总质量。单位tavg简单为任何时间单位。
氧化浸煮级和/或氧化浸煮级系列可在单一流体包围部中或具有流体连接的多个包围部中进行。在本发明的一个实施方案中,优选充分好地混合至少一个氧化浸煮级,更优选其中来自初始氧化的至少约25、50、75、100%浆料首先进入的那个级,最优选BCR浸煮器的顶部或最上区段,使得只对于那个级/区段的CMF(0.5)为至少约0.20、0.25或0.30,并使得对于各固相、液相和组合浆料相,CMF(1.5)也小于约0.95、0.90、0.85。对于CMF的这个值,标准化时间为单一浸煮级中浆料的平均停留时间。
另外,优选在第一氧化浸煮器级后和/或在低于顶部区域的所有BCR浸煮区域中浸煮中的固相接近塞流(活塞流)RTD,使得对于各固相、液相和组合浆料相,CMF(0.5)小于约0.35、0.25、0.20。对于CMF的这个值,标准化时间为不包括第一相对较好混合浸煮级的所有浸煮级中浆料的总平均停留时间。
对于全部所有浸煮级,无论在单一BCR或串联的多个BCR,对于各固相、液相和组合浆料相,都优选全部RTD具有小于约0.35、0.25、0.18的CMF(0.5),和大于约0.80、0.85、0.90且小于约0.98、0.95的CMF(1.5)。对于CMF的这个值,标准化时间为所有浸煮级中浆料的总平均停留时间。
为了在从初始氧化送入浆料的浸煮级达到所需混合,优选接近BCR级中间高度接近轴向中线的浆料的时间平均向上速度为至少约6、8、10cm/s。发明人已发现,这甚至可在具有本文所述表观气体和表观浆料速度范围的起泡流型达到,其条件为BCR级有足够内径。例如,在具有高30cm内径的小型中试规模BCR中利用约0.5cm/s表观气体速度均匀充气达到小于约6cm/s轴向中线浆料向上速度,而在具有2米或更大内径的工业可用BCR中相同表观气体速度达到大于约10cm/s轴向中线浆料向上速度。在BCR壁附近,必然有补充性向下浆料流。具有更大内径的BCR将以更大轴向速度在内部循环,而不用增加表观气体速度。这是多相自然对流复杂能量平衡的意外结果,包括由上升气泡提供的总能;在接近单独气泡的浆料中的滑动、拖动和局部湍流,浆料的诱导总对流,和BCR壁曳力。
为了在更大活塞流分布优选的后面、优选下部浸煮级达到所需混合,优选接近各BCR级中间高度在轴向中线的时间平均向上浆料速度小于约20、15、10cm/s。在BCR壁附近,必然有补充性向下浆料流。通过本公开的适当组合,包括表观气体速率、容器内径、浸煮器级高度和气体进料方法,提供后面、优选下部浸煮级中的浆料的这种减小循环速率。
为了接近浸煮器进料浆料首先进入的浸煮器区域顶部(优选为BCR的最上区域)得到流体动力学混合与适合分子氧溶解配合的所需平衡,发明人已发现,期望表观气体速度大于约0.1、0.2、0.4cm/s且小于约8、4、1cm/s。更优选送入未稀释压缩空气,同时在浸煮器中在引起经蒸发乙酸溶剂包含显著部分这种公开表观速度的压力和温度组合下操作。
为了在优选低于浸煮器进料浆料区域后在至少一个浸煮器区域中得到流体动力学混合与适合分子氧溶解配合的所需平衡,发明人已发现,期望在此以后优选较低区域中表观气体速度大于约0.01、0.02、0.04cm/s且小于约1、4、0.2cm/s。更优选送入未稀释压缩空气,同时在浸煮器中在引起经蒸发乙酸溶剂包含显著部分这种公开表观速度的压力和温度组合下操作。这些是例外地小表观速度,利用初始鼓泡分布需要适当注意,以保证连同所需固体悬浮与所需固体RTD溶解分子氧的充分分布。
优选在浸煮反应介质整个体积平均的表观固体、液体和浆料垂直速度分别大于约0.05、0.1、0.2cm/s且小于约12、8、4cm/s以下。然而,在不同浸煮区域中适当选择表观气体速率和浆料停留时间一般更重要。
本发明的氧化浸煮方法基本减小至少一种芳族反应中间体化合物的量。优选从以后氧化浸煮级抽取的浆料液相中对甲苯甲酸的时间平均浓度小于约50、10或2ppmw。优选从以后氧化浸煮级抽取的浆料液相中4-CBA的时间平均浓度小于约50、10或2ppmw。优选从以后氧化浸煮级抽取的固体PTA产物中对甲苯甲酸的时间平均浓度为约1至约1,000、约1至约500、约5至约125或10至60ppmw。优选从以后氧化浸煮级抽取的固体PTA产物中4-CBA的时间平均浓度为约1至约1,000、约1至约500、约10至约250或20至125ppmw。优选固体PTA产物中4,4'-DCS的时间平均浓度小于约6、4或2ppmw。
本发明的第一实施方案为纯化粗对苯二甲酸的方法,所述方法包括:
a)得到粗对苯二甲酸颗粒的浆料,该进料浆料包含:在包含含水乙酸的溶剂液体中的对苯二甲酸、4-羧基苯甲醛和对甲苯甲酸,和含至少一种重金属化合物的催化剂系统;
b)将粗对苯二甲酸浆料送到基本没有机械搅拌的泡罩塔系统的第一浸煮区域;
c)在进入第一浸煮区域之前或在第一浸煮区域内时将粗对苯二甲酸浆料加热到约150℃至约280℃温度;
d)将含氧气体提供到第一浸煮区域,在此,接近第一浸煮区域顶部上升的气体的表观速度为约0.1cm/s至约8cm/s;
e)使粗对苯二甲酸颗粒至少部分溶于乙酸,从而从颗粒释放至少一些4-羧基苯甲醛和对甲苯甲酸,并使溶解的4-羧基苯甲醛和对甲苯甲酸暴露于氧,以氧化成对苯二甲酸,并得到第一级浸煮器浆料;
f)将第一级浸煮器浆料通到第二浸煮区域,第二浸煮区域任选垂直位于第一浸煮区域下,且基本没有机械搅拌;
g)将含氧气体提供到第二浸煮区域下部;
其中到第二浸煮区域的气体的供速小于到第一浸煮区域的供速;和
h)从颗粒溶解并释放另外的4-羧基苯甲醛和对甲苯甲酸,并使溶解的4-羧基苯甲醛和对甲苯甲酸暴露于氧,以另外氧化成对苯二甲酸,并得到第二级浸煮器浆料;
i)任选使第二级浸煮器浆料移动通过结构类似于第二浸煮区域且任选垂直位于第二浸煮区域下的一个或多个另外的浸煮区域;
j)从最后浸煮区域移出所得对苯二甲酸结晶浆料;和
k)分离得到的对苯二甲酸结晶颗粒。
在第二个实施方案中,本发明包括氧化浸煮系统,该系统包括:
在至少一个泡罩塔反应器中布置的一系列至少两个氧化浸煮区域;
位于第一浸煮区域下部的至少一个浆料反应剂入口;
到第一浸煮区域的氧气源入口,和在第一区域后串联的至少一个区域;
各氧气源包括气体分配器单元,气体分配器单元将氧气作为气泡流送入该区域;
在至少一个泡罩塔底部的产物浆料出口。
在一个优选的实施方案中,本发明包括氧化浸煮系统,该系统包括:
在一个泡罩塔反应器中垂直布置的一系列至少两个氧化浸煮区域;
位于第一最上浸煮区域下部的至少一个浆料反应剂入口;
到第一最上浸煮区域的氧气源入口,和垂直位于第一最上区域下的串联的至少一个区域;
位于第一最上区域和垂直处于其下的第二区域之间的至少一个水平挡板;
在第一最上区域下存在多于一个区域时位于各相应垂直相邻区域之间的至少一个水平挡板;和
在至少一个泡罩塔底部的产物浆料出口;
其中
各氧气源包括气体分配器单元,气体分配器单元将氧气作为气泡流送入该区域,并且
各水平挡板包括具有多个带有多个开口区域的倒置成形斜面的盘。
在本发明的优选实施方案中,氧化浸煮系统可没有机械搅拌。
最上第一浸煮区域和另外的浸煮区域可构造成为一个泡罩塔反应器,其中相应区域与依次在最上区域下的较低另外区域垂直布置。
在此实施方案中,将经加热CTA浆料注入具有在塔反应器中垂直布置的多个区域的塔浸煮反应器的最上区域,且各区域由水平定位挡板布置隔离。在正常操作料面时,优选最上区域包括容器中所有反应介质体积的约10至50%、20至40%、25至35%。优选容器顶部高度提供另外的料面控制缓冲容积(surge volume)和气体分离容积,在正常操作料面时,该容积等于容器中所有反应介质体积的至少约10、15、20%,也优选从正常操作料面到排气出口喷嘴的间隙为高于最大操作料面至少约1米,更优选2米。虽然表观气体速度很低,且气体分离一般不成问题,但可在罐空中使用在本领域已知的任何类型喷雾回流和机械冲击分离装置,以进一步抑制浸煮器排气中浆料的起泡和雾化夹带。
由于在本发明较低另外浸煮区域中利用的很低气体混合功率和总混合功率,优选使用圆锥形底部容器头,底部容器头优选具有在圆锥底部倒置顶点至少约40、60、80°且小于约140、120、100°夹角,且优选具有从倒锥底部顶点垂直向下排料的浆料出口喷嘴。
挡板结构使各区域从直接垂直在下方和垂直在上方的区域分离,且构造各挡板单元,以允许气泡向上通过和颗粒向下通过。最上区域只具有下面挡板,而最下区域只具有上面挡板。各中间区域具有下面挡板和上面挡板二者。挡板单元可包括具有多个带有多个开口区域的倒置成形斜面的盘。
优选挡板的开口面积为挡板总水平面积的约5至约75%,更优选10至35%。
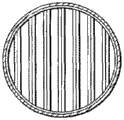
挡板抗污染。具有显著量近水平的面向上平表面区域的挡板可能倾向于在挡板面向上表面积累固体处结垢,并且随着挡板上沉积的固体量增加,沉淀的固体块可能从挡板移走,并向反应器底部落下。这些移走的固体块可堵塞挡板中的孔,导致损害反应器内的固体流模型和停留时间分布。这些固体块也可积累在反应器底部,并且可导致一些问题,包括例如阻止浆料从浸煮器底部排出。在一个实施方案中,挡板未提供面向上的平外表面,并且可由具有圆形横截面的管材构成。这种挡板的实例的示意图在5A和5B从不同透视显示。在图5A中,从水平透视观察挡板,而在图5B中,从垂直透视观察挡板。
除非另外定义,面上向表面为具有高于水平投影的法向向量的表面。在另一个实施方案中,可利用少量基本平表面,只要挡板的总面向上暴露外表面区域的小于约50%包括从水平倾斜小于35°的基本平表面。挡板的面向上暴露外表面更优选具有基本光滑的精整,以进一步阻止结垢。优选挡板的至少基本部分面向上暴露外表面具有小于约125微米RMS表面粗糙度,更优选小于约64微米RMS,最优选小于32微米RMS。电抛光精整和光滑“2B”磨轧精整特别有用。
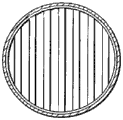
在另一个实施方案中,挡板可包括多个细长单独挡板构件。在此实施方案中,各挡板构件由L-截面构件形成,并提供一般倒置V形面向上暴露外表面。角铁档板构件的数量、间距和取向可基本与关于上述圆柱形挡板构件所述相同。
在本发明的一个优选实施方案中,挡板构造成为由开放间隙分离的一系列水平倒置“V”形截面。倒置“V”各支脚的斜率为约45°,因此,允许向下移动颗粒以缓和流通过挡板,而不堵塞挡板。同时,向上移动气泡可同时通过挡板。这种挡板的示意图在图4A从水平透视显示,在图4B从垂直透视显示。在图4A所示的实施方案中,各平行排列节段的纵轴因挡板而异。在图4A中,交替挡板节段相互以90°定向。可用交替角布置改变固体和气流参数,如本说明书中其它处所述。
图5A和5B显示由一组平行排列隔开管组成的挡板,这些管在BCR塔中连续从一个挡板到下一个挡板相互以一定角度定向。虽然图5A显示垂直的相对取向,但为了改变固体和气流,可安排其它相对角。
在另一个优选实施方案中,挡板可构造成为一系列相邻的圆锥,这些圆锥具有约45°锥角,且各锥终止于孔。
在挡板结构设计中,开口面积为挡板水平面积总面积的约5至约75%,更优选10至35%。这种设计对防止垂直相邻区域之间回混是必要的,同时仍允许颗粒向下流动和气泡向上流动。
除了第一浸煮区域空气入口外,至少一个下部另外的浸煮区域装配有位于区域下部的空气入口。各中间浸煮区域可任选包括空气入口。在本发明的一个实施方案中,所有浸煮器区域装配有空气入口。
优选位于浸煮区域的各空气提供入口包括适合分布器系统,使得空气均匀地分散进入区域,以产生“气泡流”,其中释放的气泡相对较宽地水平隔开并向上移动,且有相对少量气泡结合和破裂。利用适合气体分布器设计,通常可维持气泡流到约4cm/s表观气体速度。具有约4cm/s向上至约10cm/s表观气体速度的BCR通常在泡罩塔流体动力学过渡流型操作。这种过渡流型边界有些随流体性质和系统几何结构变化。从约10cm/s和向上的表观气体速度,泡罩塔流体动力越来越移动进入“搅拌湍流”流型,其中气泡混乱合并和破裂,并形成气泡群。在本发明的一个实施方案中,为了有效提供气泡流,分布器可以星或轮-辐结构构造。关于孔大小、间距、角等的分布器系统的结构通常已知,并且可工业工程化和得到。
各空气分布器系统可装配有“清洗系统”,使得蒸汽和/或高压(HP)含水乙酸液或蒸气与空气混合,并通过分布器,以防止固体污染分布器。在包含催化剂组分的含水乙酸滴落或否则从浆料流到分布器装配内时,连续蒸汽清洗的另外益处是避免过大氧化速率。当然,在局部和整体设备能量和质量平衡中,必须考虑任何清洗流体增加的质量和能量。
将经加热CTA浆料通过位于第一或最上区域的空气分布器上的入口通入最上浸煮器区域。CTA入口和第一或最上区域的空气分布器协同设计,以便在经加热颗粒进入最上区域时,出现CTA颗粒和气泡的最大混合和杂混。如前所述,在较高温度,对苯二甲酸从颗粒表面溶解,且在颗粒中夹带的4-CBA和对甲苯甲酸释放进入溶液,在此与空气氧作用引起进一步氧化。这一过程用本发明的低密度小的多孔高表面积颗粒极大促进,且直接在浆料加热后部分氧化杂质的释放速率特别快。
空气流速在泡罩塔浸煮器中存在的3-相化学系统中提供或引进显著部分混合能量。在CTA浆料注入区域时,第一或最上区域中的总混合能量主要为空气流速和颗粒分散力的函数。区域的高度和直径值是在考虑混合能量/单位体积中必要确定的重要工程设计参数。在使直径更小时,反应介质相同体积所需的高度明显变得更大。另外,由于由气流体积经过的浆料的位势压头增加,VdP气体混合功也变得更大。然而,区域的端至端高度也影响混合循环时间,如同在设定浆料在轴向中线的轴向上升速度中塔直径与表观气体速度的非线性作用一样。由空气源和此区域中颗粒插入能量提供的混合作用必须取得颗粒固相的所需悬浮或移动,以便达到在区域中的足够停留时间。
送入最上或第一浸煮区域的空气,包括用浸煮器进料浆料送入的空气、从下部浸煮器区域上升的空气和直接送到第一或最上浸煮器区域的空气,它们的量必须提供化学计量上超过第一或最上区域中释放的4-CBA和对甲苯甲酸氧化成对苯二甲酸所需的量的氧量,必须支持CTA浆料对流移动,且必须避免提供大过量分子氧。与提供空气的机械压缩能大不相同,在浸煮器所有区域中过量分子氧的主要成本一般为乙酸和CTA浆料中存在的其它可氧化化合物的过量“碳燃烧”。
发明人已认识到,在公开的浸煮器条件下,提供在气泡流下上升的气体提供进入液相的氧溶解速率,可使该速率足够快,以支持所需的溶解氧化中间体氧化,例如4-CBA和对甲苯甲酸。这种提供溶解氧源不需要提供大于规定量气体和总混合功率提升“kLa”传质系数。在泡罩塔中,kLa值紧密与气泡体积分数相关,且气泡体积分数在气泡流区域比在过渡和搅拌湍流区域更强由表观气体速度增加。提供足够溶解分子氧也取决于对该氧的需求速率,包括制备和提供浸煮器进料CTA浆料、用于浸煮的优选加热量和加热和用新鲜空气源化合之间的时间滞后的本文公开内容也是保持适合溶解分子氧浓度而不用借助于更大气体和总混合功率或更大量总空气进料的重要因素。
在浸煮器中芳族氧化中间体希望转化成TPA产物的分子氧的总需量意外地小。一般在浸煮中的化学计量氧需量远小于在初级氧化中对分子氧的需量的1%。当然,在浸煮中不期望碳燃烧反应消耗的分子氧是附加性的,但在浸煮中分子氧的总需流量相对较小,一般小于供到初始氧化的分子氧的0.5%。因此,适合停留时间和适合分子氧流量大小的浸煮器BCR一般在BCR流体动力学的均匀起泡流型内很低操作。
为了得到适合kLa传质速率与适合对流流体动力学的组合,优选在本文其它处公开的范围内选择空气进料速率、容器几何结构、系统温度和压力、表观气体速度和浆料组成,使得接近浸煮器BCR第一最上区域顶部的时间平均、面积平均气体保持分数大于约0.5、1.0、1.5%且小于约6、4、2%。在至少一个随后二级浸煮器BCR区域中,优选时间平均、面积平均气体保持分数小于约2、1、0.5%。
与氧溶解一致,溶剂可通入气泡内的气相,且上升蒸气产生包含空气和有机蒸气的蒸气顶部空间。在公开的过程压力范围,氧分压可能很高。如果不控制,顶部空间蒸气就变得可能爆炸,因为氧含量接近约8%体积,在浓缩出溶剂蒸气从而使氧检测转化成“干基”检测后测定,该检测相当于但不同于在顶部空间和排气中过程条件的实际氧含量。因此,由于过程安全性原因,优选监测顶部空间的氧含量并控制氧含量到干基小于6%体积。
另外,由于与碳燃烧和空气源成本相关的过程经济原因,优选调节浸煮器顶部空间排气中剩余的干基分子氧小于4%体积,更优选小于2%体积,最优选小于约1%体积,均为干基值。这提供控制对甲苯甲酸和4-CBA所需转化成PTA与最小碳燃烧平衡的有效和高效方法。在单一BCR中垂直布置所有浸煮区域时,以过量空气提供到各区域且在顶部排气中约4%体积干基氧开始完成此平衡。然后,减小到最低区域的空气供速,直至PTA中4-CBA的产生浓度开始不期望地升高。向上移动一个区域接一个区域,相对于减小空气提供速率关于产生PTA中最终4-CBA浓度类似滴定到不同区域的空气源。以此方式,可使过量碳燃烧最大限度地减少,同时保持目标PTA纯度。然而,在所有过程变量中一般有小的变化,使得最严格最小化空气供速分布可能不提供优良操作稳定性,因此可将小量增加的过量空气提供到一个或多个区域,以达到以上优选范围对顶部排气氧组成的累积效应。
如上所示,设计在最上浸煮器区域中浆料入口和空气分布器的布置,以使直接在经加热CTA浆料进入区域的入口的颗粒和氧的相互作用达到最大限度。根据本发明的某些实施方案,在任何浸煮器区域没有机械搅动或搅拌。各区域内的混合和流量通过特定区域设计来控制。
优选浸煮器进料浆料在区域下部在稍微高于挡板的位置接近容器轴中线释放进入第一或最上区域。如本文其它处所述,较大直径泡罩塔中的气泡流产生显著自然对流轴向流循环。容器中心芯内的液相/浆相向上流动,这与向下流动的外部环区平衡。在此气泡流中,液体/浆料流的这个芯环转换点一般在容器半径的约0.7倍处。优选设计CTA浆料入口,以使浆料显著水平在中心芯内分散,而不显著投入外部向下流动区域。优选浆料入口可位于高于下部挡板至少约1或2米处,且空气分布器位于浆料入口和下部挡板之间。设计该布置减小更快向下通到挡板、通过挡板和进入下面区域的浸煮器进料浆料颗粒的分数。
在第一浸煮区域中优选的总停留时间可以为10分钟至60分钟,优选15分钟至50分钟,最优选20分钟至40分钟。
优选第一最上区别的高度小于约30、20、10米。这提供合适匹配重结晶和其它化学动力学的第一最上区域端至端混合时间,且轴向芯-环循环速度分布可由很小量本文所述气体混合功率和总混合功率产生。为了提高由本文所述气体小体积流速(V)提供的气体混合功率,优选第一最上区域具有大于约1、2、4米高度,从而提高VdP气体混合功率中dP项。为了提高表观气体速度和可用本文所述气体小体积流速产生的自然对流轴向浆料循环速度分布,优选第一最上区域具有小于约16、12、10米的内径。为了通过避免从容器壁太大壁拖阻力提高自然对流轴向循环速度分布,优选第一最上区域具有大于约0.5、1.0、1.5米的内径。为了平衡这些竞争性流体动力学目的,优选第一最上区域具有大于约0.5、1.0、1.5至1且小于约16、8、4至1的反应介质直径:反应介质高度比。
在一个实施方案中,到第一或最上浸煮区域的浆料提供管入口可包括连接到垂直入口管顶部的偏转器单元。偏转器可以为近似水平的平冲击板、倒置冲击锥或改善浆料从供应管末端向外水平分布的任何形状,更优选在第一或最上浸煮区域下部没有向下朝向挡板的偏转。在图6中显示偏转器单元的一个实施方案的示意图,其中倒锥偏转器位于垂直CTA入口管的顶部。为了关于特定进入流速和与设备设计有关的其它参数优化浆料分布,可改变偏转角。如上所示,通过这种偏转给予系统的能量加到在最上区域的混合能量,并改善区域锥区内颗粒的随机分布。
可任选提供至少一个喷流器装置,以用进入浸煮器进料浆料的动力能接近至少一个浸煮器进料浆料供应管端诱导另外的反应介质循环。
优选离开供应管开口进入浸煮器进料区域的浸煮器进料浆料质量流速的至少约25、50、75、100%具有在相应管开口大于至少约1、3、5m/s且小于约70、50、30m/s的表观速度。
任选浸煮器进料浆料可分成质量流速的两个或更多个近似相等部分,并在分离轴向位置送入。优选这些浆料进料位置分别轴向隔开在浸煮器进料浆料引入的高度泡罩塔浸煮器容器内径的至少约0.5、1.0、1.5倍。浸煮器进料浆料组成和质量流速的某些组合可指示关于控制接近浆料进料开口溶解的对甲苯甲酸、4-CBA和分子O2局部液相组成不充分混合的空气进料速率和容器直径的选择。在这些情况下,在利用主要由起泡流型中气体混合功率提供的反应介质的自然对流整体循环时,浸煮器进料浆料流分离和浸煮器进料浆料引入开口的轴向分离减小混合难度。浸煮器进料浆料流分离可包括位于供应管中的流量检测和变量流量控制元件、供应管设计几何结构(例如,摩擦压降对称与位势压头差调节)和在本领域已知的其它手段。
可使来自最上区域的排气通到系统,以回收溶剂质量和/或热和/或机械轴能量,这在本领域是已知的,或者,该排气可与来自用于回收溶剂质量和/或热和/或机械轴能量的初级和/或二级氧化系统的排气组合。
在低于最上浸煮器区域的区域,空气进料速率和空气进料入口设计结构应使得在浆料的浆料总体表观流继续向下时引起较少芯-环轴向循环。从过程效率的观点,这是有利的,因为期望颗粒以近似活塞流方式通过各个这些较低另外的浸煮区域。然而,深深在气泡流内的甚至少量气体表观速度也仍引起相对于在工业相关大小PTA浸煮器BCR中的停留时间显著的轴向循环。因此,在接收浸煮器进料浆料后(更优选低于),优选用无污染挡板轴向细分浸煮器容积。这些挡板切断接近和跨挡板的轴向循环流的连续性,在区域高度和直径的一些情况下,挡板也可抑制两个挡板之间引起的芯-环循环流的最大速度。在本文其它处提供关于这些挡板机械设计和物理布置另外的详细公开内容。
如前所示,送到浸煮器的空气的主要部分送到第一浸煮区域。因此,注入浸煮器的空气的50至90%质量可注入第一区域。其余部分可以相等部分或者如本文其它处所述调节的不同比在第二和另外的浸煮区域之间分开。
如上所述,使用泡罩塔反应器的优点是减少资本设备成本和与操作和维护机械搅拌器相关的不间断能量需求。与氧化浸煮的现有技术比较,例如U.S. 7,393,973,本发明的实施方案可减少用于CTA浸煮器系统的安装资本成本,包括用钛制成的很大容器、设备和管,按约1,000吨/年TPA目前生产能力大小的设备减少$10,000,000以上。这种节省是现有选择多个容器串联成具有较小总体积的单一容器,而不用机制搅拌和没有用于单独容器的单独加热工具的结果。电机功率节省超过300千瓦。通过避免使用蒸发溶剂,浸煮器浆料加热负荷节省超过10%,而不损害在有利低温纯化CTA的能力。用于循环过滤清洗纯化系统的设计能力和操作成本节省为约10%。
U.S. 7,393,973公开,驱动CSTR浸煮反应器的机械能为0.2至0.8千瓦/立方米(kW/m3)浸煮反应浆料。发明人已计算,在具有串联的两个相等大小容器的现有常规CSTR浸煮器系统中,机械搅拌器能耗和气体混合功率总共为约0.3W/kg。相比之下,处理相同浸煮器进料的本发明的BCR浸煮器的能量功率估计只有达到约0.02W/kg的气体混合功率。因此,本发明方法的能耗成本显著小于CSTR类型方法的成本。
为在最上浸煮区域之下类活塞流颗粒沉降布置的区域的总高度可以为最上区域长度的1.0至5.0倍,优选1.5至4倍,最优选至少2倍。下部另外的浸煮器区域的总高度与下部另外的浸煮器区域的直径之比可以为2/1至12/1,优选3/1至8/1,最优选4/1。
下部另外的浸煮器区域的总数由布置于塔以隔离单独区域的水平挡板数确定。浸煮器中的最低挡板数为其中塔包含一个第一最上更充分混合浸煮区域和一个下部另外的更活塞流浸煮区域的挡板数。优选塔包含多于1个塔板和小于10个塔板,优选2至8个挡板,最优选3至6个挡板,这些提供具有相对于第一最上区域减小表观气体速度的下部另外的浸煮器区域的相应数。
在本发明的一个实施方案中,浸煮器塔的直径从顶部到底部不变。在其它实施方案中,可构造塔,使得直径随区域变化。例如,为了促进控制活塞流以最小气体提升需求通过二级区域,下部区域的直径可小于最上区域的直径,以达到高的高度/直径(H/D)比。另一方面,可构造最上区域,以最少气体提升功率需求有效对流混合。
如前所述,从初级氧化系统得到的浆料的CTA颗粒有不均匀形状和粒径分布。由于在浸煮器中发生的奥斯特瓦尔德熟化,在固体前进通过浸煮器区域时,粒径分布一般可变大和变窄。发明人已发现,为了有效进行本发明的浸煮,必须考虑粒径和形态结构及沉降速率之间的关系。浸煮温度可以是此考虑内的重要参数。发明人已认识到,较高浸煮温度导致增大粒径。例如,在约240至260℃温度浸煮可产生约200微米颗粒,这种粒径的颗粒具有大得多的沉降速率,甚至在通过具有本文所述固体分数的浆料中阻止沉降时。
因此,在本发明的一个优选实施方案中,各区域中浸煮的温度为约180至230℃,优选190至220℃,最优选200至210℃。
任选第一最上浸煮区域可在高于或低于随后浸煮区域至少之一的温度约40、20、10、5℃温差的温度操作。这一温差可用于去除氧化中间体例如4-CBA与碳燃烧反应和/或所得粒径分布平衡。可用任何本文所述加热方法完成随后区域加热。通过加入一定质量冷却剂溶剂液体,通过热交换表面,优选机械刮擦表面,使用冷却流体,和通过蒸发冷却,包括气体进料和减压,可实现随后区域冷却,其条件为涉及表观气体速率、系统压力和操作温度的本发明的其它方面保持在所公开范围。
优选用本文所述一个或多个发明实施方案通过浸煮生成的固体PTA产物基本包括具有至少约30微米平均粒径D(4,3)的颗粒,更优选约35至约200微米,更优选约40至约160微米,最优选45至120微米。优选固体TPA产物基本包括具有约5至约60微米D(v,0.1)测定值的颗粒,更优选约10至约50微米,最优选15至40微米。优选固体TPA产物基本包括具有约25至约160微米中值粒径D(v,0.5)测定值的颗粒,更优选约30至约100微米,最优选35至80微米。优选固体TPA产物基本包括具有约40至约300微米D(v,0.9)测定值的颗粒,更优选约60至约250微米,最优选80至200微米。优选固体TPA产物基本包括具有约0.6至约5.0粒径相对分布测定值的颗粒,更优选约0.9至约4.0,最优选1.2至2.5。优选固体TPA产物基本包括具有约0.25平方米/克(m2/g)平均BET表面积的颗粒,更优选约0.005至约0.2m2/g,最优选0.01至0.18m2/g。
由于本发明中体现的很低气体和总混合功率,优选选择固体粒径分布、浆料中固体的分数和液体介质组成、温度和压力的组合,以提供以下沉降速率范围。优选离开浸煮器区域的PTA颗粒的平均D(4,3)粒径的无阻沉降速率小于约120、100、80米/小时。代替用分离单独适合大小颗粒在实际过程条件物理测定,可利用假定球形颗粒形状,用斯托克斯定律计算这种无阻沉降速率,这在本领域是已知。另外,优选离开浸煮器区域的PTA颗粒的浆料的受阻沉降速率小于约30、20、10米/小时。代替在实际过程条件物理测定,通过平均D(4,3)粒径的以上计算斯托克斯定律终端速度沉降速率乘以浆料浓度校正因子,可计算该受阻沉降速率,通常称为理查森-扎奇(Richardson和Zaki)法,其中ε为在浆料中的液体体积分数:
受阻沉降速率=斯托克斯定律速率*ε ^2 * ε^2.65
在最上区域顶部的排气出口可装配有测定氧含量的氧监测系统。排气出口可将排气转移到冷凝器系统,以回收蒸发的溶剂和存在的其它挥发性有机物质,或者可使排气循环到初级氧化系统。
本发明的泡罩塔浸煮器可具有在最上区域下的1至5个下部二级区域。塔的总高度可以为16至40米,优选20至30米,最优选22至28米。塔的直径可以为1.0米至8.0米,优选2.0至6.0米,最优选约3.0至5.0米。
最上区域的垂直长度可以为4至12米,优选6至10米,最优选约8米。下部浸煮区域的垂直长度可以为2至6米,实际设计基于下部区域中的最大活塞流特性。
各区域可具有相同直径,或者根据特殊设计参数,直径可随区域改变。例如,如图3中所示,最上浸煮区域的直径可大于下部区域的直径。区域数由在塔中布置的挡板数确定。在任何区域机械搅拌装置都是不必要的。挡板结构以前在方法实施方案描述中提供。
在图1中显示本发明的单一BCR实施方案的示意PTA制备系统。根据制备流程,从包含初级BCR氧化单元和二级BCR氧化单元的初级氧化系统得到的CTA浆料流注入包含由水平挡板隔离的5个垂直布置区域的BCR的最上区域。任选布置用于第一区域的空气注入单元由HPA1位置指示,而HPA2位置为用于垂直在第一浸煮区域下的第二和随后浸煮区域的任选空气注入单元。在各情况下,一个或多个HPA1和HPA2入口可存在于所示位置。
或者,在另一个实施方案中,可利用两个或更多个BCR反应器,其中第一容器相当于首先接收浸煮器进料浆料的最上浸煮器区域,而第二BCR反应器从第一BCR接收过程浆料。由于用于BCR的资本成本显著小于CSTR,这种双塔系统成本有效。
根据本发明的这个实施方案,依次区域可分成串联的两个或更多个泡罩塔,第一BCR构造用于如以上关于单一BCR系统第一最上浸煮区域所述的更对流混合,随后一个或多个BCR经构造,以使RTD更紧密接近颗粒浆料的类活塞流通道,如关于下部另外的浸煮区域所述。因此,本发明也包括一种泡罩塔浸煮系统,该系统包括:
构造用于对流的第一BCR单元;和
在第一BCR单元后串联的构造用于活塞流的至少一个BCR单元;
其中
第一BCR单元包括:
在塔下部四分之一圆周垂直位置的浆料入口;
低于浆料入口的含氧气体入口;
在塔底部的浆料出口;
在塔顶部的装配有氧含量监测器的排气出口;和
任选在气体入口和浆料出口之间的水平挡板;并且
其中
至少一个第二BCR单元包括:
1至5个水平隔离区域,各区域任选装配有氧气入口;
各区域之间的水平挡板;
在最高区域中的浆料入口;和
在BCR单元底部的浆料出口;
其中至少一个区域装配有氧气入口。
根据这个实施方案,用于单一BCR实施方案的第一最上区域的浸煮器进料和氧气入口结构和布置的公开内容也适用于多个BCR实施方案的第一BCR的结构和布置。同样,用于垂直在单一BCR系统的第一最上区域下的第二或更多区域的结构和布置的公开内容适用于多个BCR系统的第二和任选随后BCR。
在另一个实施方案中,具有至少两个BCR单元的泡罩塔浸煮系统没有机械搅拌。为了优化对流,构造第一单元具有至少8米的高度和4或更小的H/D比,优选H/D比为3或更小,最优选H/D比为约2。
使第一BCR单元工程化,以具有对流,其中向上气泡流的中心芯与外部向下流动的环区平衡。设计CTA浆料入口,以使浆料快速在中心芯内分散,而不投入外部向下流动区域,在此氧含量会减小。浆料入口可位于高于在BCR底部浆料入口和浆料出口之间空气分布器约1或2米的BCR的下部四分之一圆周处。如上所述,设计这一定位,以使在颗粒向下移到浆料出口之前与氧作用最大的中心芯区的向上颗粒流达到最大限度。
为了有效提供气泡流,分布器可以星或轮-辐结构构造。关于孔大小、间距、角等的分布器系统的结构通常已知,并且可工业工程化和得到。
空气分布器系统可装配有“清洗系统”,使得蒸汽和/或高压(HP)洗酸与空气混合,并通过分布器,以防止固体污染分布器。
在BCR单元顶部的排气出口可装配有测定氧含量的氧监测系统。排气出口可将排气转移到冷凝器系统,以回收蒸发的溶剂和存在的其它挥发性有机物质,或者可使排气循环到初级氧化系统。
如上所述的挡板可位于空气分布器和浆料出口下。
浆料出口通过任选具有输送泵单元的输送管线连接到第二BCR单元的最高区域。如上所述,第二BCR单元关于活塞流设计,并且可具有由前述挡板区分的1至5个水平隔离区域。第二BCR单元的至少一个区域装配有空气入口,且任选各其它区域可独立装配有空气分布器。第二BCR单元的总高度可以为16至40米,优选20至30米,最优选22至28米。塔的直径可以为1.0米至8.0米,优选2.0至6.0米,最优选约3.0至5.0米。
单独活塞流区域的垂直长度可以为2至6米,实际设计基于区域中的最佳活塞流特性。
各区域可具有相同直径,或者根据特殊设计参数,直径可随区域改变。区域数由在塔中布置的挡板数确定。在任何区域机械搅拌装置都是不必要的。然而,在根据本发明改进的系统中,可存在机械搅拌。
在图2中显示本发明的一个实施方案的双BCR浸煮系统的示意图。在此图中,未重复图1中所示的初级氧化系统。如图1中那样,任选布置用于第一区域BCR的空气注入单元由HPA1位置指示,而HPA2位置为用于第二和随后BCR浸煮区域的空气注入单元的任选布置位置。在各情况下,一个或多个HPA1和HPA2入口存在于所示位置。
如前所述,CTA浆料可在进入BCR之前在热交换单元加热,或者可在BCR内加热。
在分析前述说明后,本发明的另外的优点和其它特征对本领域的技术人员变得显而易见,或者可从实施本发明来认识。本发明的优点可如附加权利要求书中特别指出地那样实现和获得。应认识到,本发明可以有其它和不同的实施方案,且其若干细节可在不脱离本发明的要旨下修改。
Claims (30)
1.一种用于纯化对苯二甲酸的氧化浸煮系统,所述氧化浸煮系统包括:
在一个泡罩塔反应器中垂直布置的一系列至少两个氧化浸煮区域;
位于第一最上浸煮区域下部的反应剂入口;
到第一最上浸煮区域的氧气源入口,和垂直位于第一最上区域下的串联的至少一个区域;
位于第一最上区域和垂直处于其下的第二区域之间的至少一个水平挡板;
在第一最上区域下存在多于一个区域时位于各相应垂直相邻区域之间的至少一个水平挡板;
在至少一个泡罩塔底部的产物浆料出口,
其中
所述氧化浸煮系统处于初级氧化反应器的下游反应流,并接收来自初级氧化反应器的粗反应产物,
各氧气源包括气体分配器单元,气体分配器单元将氧气作为具有4cm/s至10cm/s的向上表观气体速度的气泡流送入该区域,并且
各水平挡板包括具有多个带有多个开口区域的倒置成形斜面的盘,以致水平挡板的总的面向上暴露外表面区域的小于50%包括从水平倾斜小于35°的平表面,并且
所述在一个泡罩塔反应器中垂直布置的一系列至少两个氧化浸煮区域没有机械搅拌。
2.权利要求1的氧化浸煮系统,所述氧化浸煮系统进一步包括具有氧含量监测系统的排气出口。
3.权利要求1的氧化浸煮系统,其中所述泡罩塔的总高度为16至40米。
4.权利要求1的氧化浸煮系统,其中所有区域的直径相同,并且为1.0至8.0米。
5.权利要求1的氧化浸煮系统,所述氧化浸煮系统包括在第一最上区域下方垂直布置的3至5个区域。
6.权利要求1的氧化浸煮系统,其中所述第一最上区域的高度:直径比为1/1至4/1。
7.权利要求1的氧化浸煮系统,其中所述水平挡板包括多个横向隔开的挡板构件。
8.权利要求7的氧化浸煮系统,其中所述横向隔开的挡板构件各自包括基本圆柱形的暴露外表面。
9.权利要求1的氧化浸煮系统,其中所述水平挡板包括倒V形面向上的暴露外表面。
10.权利要求1的氧化浸煮系统,其中各水平挡板包括挡板总水平面积的25至75%的开口区域。
11.一种用于纯化对苯二甲酸的泡罩塔浸煮系统,其包括在根据权利要求1所述的氧化浸煮系统中,所述泡罩塔浸煮系统包括:
构造用于对流的第一泡罩塔反应器单元;和
在第一泡罩塔反应器单元后串联的构造用于活塞流的至少一个泡罩塔反应器单元;
其中
所述泡罩塔浸煮系统处于初级氧化反应器的下游反应流,并接收来自初级氧化反应器的粗反应产物,
第一泡罩塔反应器单元包括:
在塔中心垂直位置的浆料入口;
低于浆料入口的含氧气体入口;
在塔底部的浆料出口;
在塔顶部的装配有氧含量监测器的排气出口;和
在气体入口和浆料出口之间的水平挡板;并且
其中
至少一个第二泡罩塔反应器单元包括:
1至5个水平隔离区域;
各区域之间的水平挡板,其中各水平挡板包括具有多个带有多个开口区域的倒置成形斜面的盘,以致水平挡板的总的面向上暴露外表面区域的小于50%包括从水平倾斜小于35°的平表面;
在最高区域中的浆料入口;和
在泡罩塔反应器单元底部的浆料出口;
其中至少一个区域装配有氧气入口,其将氧气作为具有4cm/s至10cm/s的向上表观气体速度的气泡流送入该区域,并且
该系统的各泡罩塔反应器单元没有机械搅拌。
12.一种在根据权利要求1所述的系统中进行的纯化粗对苯二甲酸的方法,所述方法包括:
a)得到粗对苯二甲酸颗粒的浸煮器进料浆料,该进料浆料包含:在由含水乙酸组成的溶剂液体中的对苯二甲酸、4-羧基苯甲醛和对甲苯甲酸,和包含钴和锰中的一种或更多种的均匀液相催化剂系统;
b)将粗对苯二甲酸浆料送到泡罩塔系统的第一浸煮区域;
c)在进入第一浸煮区域之前或在第一浸煮区域内时将粗对苯二甲酸浆料加热到150℃至280℃的温度;
d)将氧气提供到第一浸煮区域,在此,在第一浸煮区域顶部附近的上升气体的表观速度为0.1cm/s至8cm/s;
e)使粗对苯二甲酸颗粒至少部分溶于乙酸,从而从颗粒释放至少一些4-羧基苯甲醛和对甲苯甲酸,并使溶解的4-羧基苯甲醛和对甲苯甲酸暴露于氧,以氧化成对苯二甲酸,并得到第一级浸煮器浆料;
f)将第一级浸煮器浆料通到第二浸煮区域,第二浸煮区域任选垂直位于第一浸煮区域下;
g)将氧气提供到第二浸煮区域下部;
其中到第二浸煮区域的氧气的供速小于到第一浸煮区域的供速;和
h)从颗粒溶解并释放另外的4-羧基苯甲醛和对甲苯甲酸,并使溶解的4-羧基苯甲醛和对甲苯甲酸暴露于氧,以另外氧化成对苯二甲酸,并得到第二级浸煮器浆料;
i)使第二级浸煮器浆料移动通过结构类似于第二浸煮区域且任选垂直位于第二浸煮区域下的一个或多个另外的浸煮区域;
j)从最后浸煮区域移出所得对苯二甲酸结晶浆料;和
k)分离得到的对苯二甲酸结晶颗粒。
13.权利要求12的方法,其中在第二浸煮区域的上升气体的表观速度小于1cm/sec。
14.权利要求12的方法,其中粗对苯二甲酸的平均粒径为20至150微米。
15.权利要求12的方法,其中粗对苯二甲酸的BET表面积为0.6至4.0m2/g。
16.权利要求12的方法,其中在第一浸煮区域中颗粒的停留时间为10至60分钟。
17.权利要求12的方法,其中在第一浸煮区域内粗对苯二甲酸浆料的温度为180至230℃。
18.权利要求12的方法,其中至少一个浸煮区域的温度高于从初级氧化系统得到时粗对苯二甲酸浆料的温度至少10℃。
19.权利要求12的方法,其中在第一和第二浸煮区域中对苯二甲酸颗粒的总停留时间为60至120分钟。
20.权利要求12的方法,其中排气的氧含量为根据干基测定6%体积或更小。
21.权利要求12的方法,其中在浸煮出口对苯二甲酸颗粒的平均粒径为60至100微米。
22.权利要求12的方法,其中关于所有浸煮区域总计的气体混合功率小于0.2W/kg浆料。
23.权利要求22的方法,其中关于所有浸煮区域总计的气体混合功率小于0.05W/kg浆料。
24.权利要求12的方法,其中在泡罩塔内的最大时间平均、面积平均气泡保持率小于6%。
25.权利要求12的方法,其中在泡罩塔的至少一个区域内的最大时间平均、面积平均气泡保持率小于2%。
26.权利要求12的方法,其中关于各固相、液相和组合浆料相的总浸煮停留时间分布具有小于0.35的CMF(0.5)和大于0.80的CMF(1.5)。
27.权利要求12的方法,其中在首先加热浸煮器进料浆料比在从初始氧化的出口的温度高至少10℃后8分钟内,使用于第一浸煮区域的分子氧源的至少25%与粗对苯二甲酸浆料混合。
28.权利要求12的方法,其中将用于第一浸煮区域的分子氧源的至少25%与浸煮器进料浆料混合送入第一浸煮区域。
29.权利要求12的方法,其中用位于泡罩塔外部的至少一个非接触热交换器装置使至少50%的浸煮器进料浆料的温度增加至少10℃。
30.权利要求29的方法,其中在离开外部热交换器前使用于第一浸煮区域的分子氧源的至少25%与浸煮器进料浆料混合。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/657,523 US20160264511A1 (en) | 2015-03-13 | 2015-03-13 | Bubble column reactor based digester and method for its use |
| US14/657523 | 2015-03-13 | ||
| PCT/US2016/021912 WO2016149060A1 (en) | 2015-03-13 | 2016-03-11 | Bubble column reactor based digester andmethod for its use |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN107531602A CN107531602A (zh) | 2018-01-02 |
| CN107531602B true CN107531602B (zh) | 2021-02-12 |
Family
ID=56887404
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201680027722.3A Active CN107531602B (zh) | 2015-03-13 | 2016-03-11 | 基于泡罩塔反应器的浸煮器及其使用方法 |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US20160264511A1 (zh) |
| EP (1) | EP3268341B1 (zh) |
| JP (1) | JP6882182B2 (zh) |
| KR (1) | KR20170128462A (zh) |
| CN (1) | CN107531602B (zh) |
| BR (1) | BR112017019452B1 (zh) |
| CA (1) | CA2979681A1 (zh) |
| EA (1) | EA034735B1 (zh) |
| ES (1) | ES2880688T3 (zh) |
| LT (1) | LT3268341T (zh) |
| MX (1) | MX2017011729A (zh) |
| MY (1) | MY188141A (zh) |
| PL (1) | PL3268341T3 (zh) |
| PT (1) | PT3268341T (zh) |
| SG (2) | SG10201908521RA (zh) |
| WO (1) | WO2016149060A1 (zh) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11203769B1 (en) * | 2017-02-13 | 2021-12-21 | Solugen, Inc. | Hydrogen peroxide and gluconic acid production |
| US10000435B1 (en) * | 2017-02-28 | 2018-06-19 | Grupo Petrotemex, S.A. De C.V. | Energy and environmentally integrated method for production of aromatic dicarboxylic acids by oxidation |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6375921B1 (en) * | 1998-09-23 | 2002-04-23 | Degussa Huls A.G. | Bubble column and the use thereof |
| CN1512978A (zh) * | 2001-06-04 | 2004-07-14 | �Ϻ���ͨ��ѧ | 精制的对苯二甲酸的制备方法 |
| WO2007103029A1 (en) * | 2006-03-01 | 2007-09-13 | Eastman Chemical Company | Polycarboxylic acid production system employing enhanced multistage oxidative digestion |
| US20070293699A1 (en) * | 2004-09-02 | 2007-12-20 | Eastman Chemical Company | Optimized Liquid-Phase Oxidation |
| CN101198578A (zh) * | 2005-06-16 | 2008-06-11 | 伊士曼化工公司 | 优化的液相氧化 |
| CN101410173A (zh) * | 2006-01-04 | 2009-04-15 | 伊士曼化工公司 | 具有内部次级反应器的氧化系统 |
| CN101531588A (zh) * | 2008-03-13 | 2009-09-16 | 周向进 | 一种新的精对苯二甲酸的制造方法 |
| CN102731297A (zh) * | 2012-07-11 | 2012-10-17 | 浙江大学 | 一种逐级升温氧化制备中纯度对苯二甲酸的方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5200557A (en) * | 1991-04-12 | 1993-04-06 | Amoco Corporation | Process for preparation of crude terephthalic acid suitable for reduction to prepare purified terephthalic acid |
| CN202700501U (zh) | 2012-05-15 | 2013-01-30 | 中国昆仑工程公司 | 适于kpta生产的无搅拌氧化和深度氧化反应系统 |
-
2015
- 2015-03-13 US US14/657,523 patent/US20160264511A1/en not_active Abandoned
-
2016
- 2016-03-11 MX MX2017011729A patent/MX2017011729A/es unknown
- 2016-03-11 PL PL16765474T patent/PL3268341T3/pl unknown
- 2016-03-11 ES ES16765474T patent/ES2880688T3/es active Active
- 2016-03-11 JP JP2017548128A patent/JP6882182B2/ja active Active
- 2016-03-11 WO PCT/US2016/021912 patent/WO2016149060A1/en active Application Filing
- 2016-03-11 BR BR112017019452-0A patent/BR112017019452B1/pt active IP Right Grant
- 2016-03-11 SG SG10201908521R patent/SG10201908521RA/en unknown
- 2016-03-11 KR KR1020177029210A patent/KR20170128462A/ko active IP Right Grant
- 2016-03-11 PT PT167654748T patent/PT3268341T/pt unknown
- 2016-03-11 EP EP16765474.8A patent/EP3268341B1/en active Active
- 2016-03-11 CA CA2979681A patent/CA2979681A1/en active Pending
- 2016-03-11 LT LTEP16765474.8T patent/LT3268341T/lt unknown
- 2016-03-11 MY MYPI2017001308A patent/MY188141A/en unknown
- 2016-03-11 EA EA201792031A patent/EA034735B1/ru not_active IP Right Cessation
- 2016-03-11 SG SG11201707399XA patent/SG11201707399XA/en unknown
- 2016-03-11 CN CN201680027722.3A patent/CN107531602B/zh active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6375921B1 (en) * | 1998-09-23 | 2002-04-23 | Degussa Huls A.G. | Bubble column and the use thereof |
| CN1512978A (zh) * | 2001-06-04 | 2004-07-14 | �Ϻ���ͨ��ѧ | 精制的对苯二甲酸的制备方法 |
| US20070293699A1 (en) * | 2004-09-02 | 2007-12-20 | Eastman Chemical Company | Optimized Liquid-Phase Oxidation |
| CN101198578A (zh) * | 2005-06-16 | 2008-06-11 | 伊士曼化工公司 | 优化的液相氧化 |
| CN101410173A (zh) * | 2006-01-04 | 2009-04-15 | 伊士曼化工公司 | 具有内部次级反应器的氧化系统 |
| WO2007103029A1 (en) * | 2006-03-01 | 2007-09-13 | Eastman Chemical Company | Polycarboxylic acid production system employing enhanced multistage oxidative digestion |
| CN101531588A (zh) * | 2008-03-13 | 2009-09-16 | 周向进 | 一种新的精对苯二甲酸的制造方法 |
| CN102731297A (zh) * | 2012-07-11 | 2012-10-17 | 浙江大学 | 一种逐级升温氧化制备中纯度对苯二甲酸的方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| PT3268341T (pt) | 2021-06-30 |
| JP6882182B2 (ja) | 2021-06-02 |
| CA2979681A1 (en) | 2016-09-22 |
| EP3268341A4 (en) | 2018-10-31 |
| PL3268341T3 (pl) | 2021-11-02 |
| MY188141A (en) | 2021-11-23 |
| EA034735B1 (ru) | 2020-03-13 |
| EP3268341B1 (en) | 2021-04-21 |
| CN107531602A (zh) | 2018-01-02 |
| BR112017019452B1 (pt) | 2020-11-17 |
| EP3268341A1 (en) | 2018-01-17 |
| KR20170128462A (ko) | 2017-11-22 |
| MX2017011729A (es) | 2017-11-13 |
| SG10201908521RA (en) | 2019-11-28 |
| JP2018513125A (ja) | 2018-05-24 |
| EA201792031A1 (ru) | 2018-02-28 |
| BR112017019452A2 (pt) | 2018-05-15 |
| ES2880688T3 (es) | 2021-11-25 |
| LT3268341T (lt) | 2021-07-26 |
| SG11201707399XA (en) | 2017-10-30 |
| WO2016149060A1 (en) | 2016-09-22 |
| US20160264511A1 (en) | 2016-09-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101281378B1 (ko) | 최적화된 액상 산화 방법 | |
| KR101281442B1 (ko) | 최적화된 액상 산화 방법 | |
| KR101159409B1 (ko) | 방향족 카르복실산 제조용 산화 반응기 및 이를 이용한 방향족 카르복실산의 제조 방법 | |
| BRPI0708434B1 (pt) | processo para preparar uma composição de ácido tereftálico | |
| CN107522610B (zh) | 用于氧化蒸煮的具有提高的停留时间分布的多元羧酸生产系统 | |
| CN107531602B (zh) | 基于泡罩塔反应器的浸煮器及其使用方法 | |
| RU2581836C2 (ru) | Окислительная система с вторичным реактором для боковой фракции | |
| BRPI0708394A2 (pt) | processo para preparar uma composição de ácido tereftálico | |
| JP2018513125A5 (zh) | ||
| US8968686B2 (en) | Oxidation system with sidedraw secondary reactor | |
| RU2579452C2 (ru) | Окислительная система с вторичным реактором для боковой фракции | |
| KR101281408B1 (ko) | 최적화된 액상 산화 방법 | |
| KR101392653B1 (ko) | 최적화된 액상 산화 방법 | |
| BRPI0708435B1 (pt) | Process for preparing a composition of dicarboxylic acid, and, reactor system |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |