CN101282965A - 基于烷氧基吲哚酮的蛋白激酶抑制剂 - Google Patents
基于烷氧基吲哚酮的蛋白激酶抑制剂 Download PDFInfo
- Publication number
- CN101282965A CN101282965A CNA2006800352235A CN200680035223A CN101282965A CN 101282965 A CN101282965 A CN 101282965A CN A2006800352235 A CNA2006800352235 A CN A2006800352235A CN 200680035223 A CN200680035223 A CN 200680035223A CN 101282965 A CN101282965 A CN 101282965A
- Authority
- CN
- China
- Prior art keywords
- compound
- salt
- isomer
- prodrug
- represent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Abstract
基于烷氧基吲哚酮的酸和酰铵衍生物作为蛋白激酶的抑制剂,具有增强的和出乎意料的药物性质,适用于治疗涉及异常蛋白激酶活性的疾病,如癌症。
Description
发明领域:
本发明涉及蛋白激酶抑制剂和它们在治疗涉及异常蛋白激酶活性的疾病方面的用途,如癌症和发炎。尤其特别的,本发明涉及基于烷氧基吲哚酮的衍生物和它们可用作蛋白激酶抑制剂的可药用的盐。
技术背景:
蛋白激酶是催化丝氨酸、酪氨酸和苏氨酸蛋白残基的羟基集团磷酸化作用的酶。细胞生命的很多方面(例如细胞的生长、分化、增殖、细胞周期和存活)依赖蛋白激酶的活性。此外,异常蛋白激酶活性涉及许多疾病,如癌症和发炎。因此,相当大的精力被投入到找出调节蛋白激酶活性的方法上。特别地,作了很多尝试来找出充当蛋白激酶抑制剂的小分子。
一些吡咯-吲哚酮衍生物被证明作为蛋白激酶抑制剂具有出色的活性(Larid等FASEB J.16,681,2002;Smolich等Blood,97,1413,2001;Mendel等Clinical Cancer Res.9,327,2003;Sun等J.Med.Chem.46,1116,2003)。这些化合物的临床效用已被期待,但由于其相对的差水溶性和/或其他的药物性质,部分地受到怀疑。我们需要的是一个改变了的吡咯-吲哚酮衍生物家族,同时具有抑制剂活性和增强的药物性质。
发明概述:
本发明指向基于烷氧基吲哚酮的衍生物以及它们作为蛋白激酶抑制剂的用途。在这里揭示了基于烷氧基吲哚酮的派生物具有增强的和出乎意料的药物性质,从而将这一家族的化合物与已知的具有蛋白激酶抑制活性的吡咯-吲哚酮派生物区分开来。在这里还揭示了基于烷氧基吲哚酮的衍生物有益于治疗涉及异常蛋白激酶活性的疾病,如癌症。
本发明的第一方面指向由式1表示的化合物:

在式1中,R1选自氢、卤素、(C1-C6)烷基、(C3-C8)环烷基、(C1-C6)卤烷基、羟基、(C1-C6)烷氧基、氨基、(C1-C6)烷基氨基、酰胺、磺胺、氰、取代或未取代的(C6-C10)芳香族羟基;R2选自氢、卤素、(C1-C6)烷基、(C3-C8)环烷基、(C1-C6)卤烷基、羟基、(C1-C6)烷氧基、(C2-C8)烷氧基烷基、氨基、(C1-C6)烷基氨基、(C6-C10)芳香族羟基氨基;R3选自氢、(C1-C6)烷基、(C6-C10)芳香族羟基、(C5-C10)杂芳香族羟基和酰胺;R4、R5、R6和R8都独立地选自氢和(C1-C6)烷基;R7是(C1-C6)烷基;R9选自氢基、(C1-C6)O-烷基、(C3-C8)O-环烷基和NR10R11;R10和R11都独立地选自氢、(C1-C6)烷基、(C1-C6)羟基烷基、(C1-C6)二羟基烷基、(C1-C6)烷氧基、(C1-C6)烷基羧酸、(C1-C6)烷基膦酸、(C1-C6)烷基磺酸、(C1-C6)羟基烷基羧酸、(C1-C6)烷基酰胺、(C3-C8)环烷基、(C5-C8)杂环烷基、(C6-C8)芳香族羟基、(C5-C8)杂芳香族羟基、(C3-C8)环烷基羧酸、或者R10和R11以及N组成一个(C5-C8)杂环化合物,该杂环化合物未被取代或被一个或多个羟基、酮基、乙醚和羧酸所取代,且n=1,2,3,m=0,1,2。另外,本发明的该方面还可以指向一种可药用的盐、它的异构体、它的异构体的一种可药用的盐或者式I所述化合物的药物前体。
对于该发明的第一方面,其优选的亚属指向由式2表示的化合物,盐,异构体或者药物前体。

在式2中,R12选自氢、(C1-C6)烷基以及(C3-C8)环烷基,其他基团与式1中定义的一样。在优选实施例中,R1和R2独立地选自氢和氟,R3和R4是甲基,R5、R6、R8、和R12是氢,R7是(C1-C6)烷基,并且n=1或2,m=0或1。优选的种类包括下列化合物:


对于该发明的第一方面,其第二个优选的亚属指向由式3表示的化合物、盐、异构体或者药物前体:

在式3中,各个R基团与式1中的一样。在优选的实施例中,R1和R2独立地选自氢、卤素和氰,R3选自氢、(C1-C6)烷基、(C6-C10)芳香族羟基、(C5-C10)杂芳香族羟基以及酰胺,R4,R5,R6和R8独立地选自氢、(C1-C6))烷基,R7是(C1-C6)烷基,其中n=1或2,m=0或1。R10和R11选自氢、(C1-C6)烷基、(C1-C6)羟基烷基、(C2-C6)二羟基烷基、(C1-C6)烷氧基、(C2-C6)烷基羧酸、(C1-C6)烷基磷酸、(C1-C6)烷基磺酸、(C2-C6)羟基烷基羧酸、(C1-C6)烷基酰基、(C3-C8)环烷基、(C5-C8)杂环烷基、(C6-C8)芳香族羟基、(C5-C8)杂芳香族羟基、(C4-C8)环烷基羧酸、或者R10和R11以及N组成一个(C5-C8)杂环化合物,该杂环化合物未被取代或被一个或多个羟基、酮基、乙醚和羧酸所取代。
在该第二亚属的第一子群中,m=0。第一子群优选的种类如下列结构表示:

在该第二亚属的第二子群中,m=1。第二子群优选的种类如下列结构表示:

本发明第二方面的种类如下列结构表示:

其中R9选自如下列结构表示的自由基:

本发明的另一方面指向一种使用由上述式1-3表示的化合物或盐来调节蛋白激酶催化活性的方法。优先选择的模式是,蛋白激酶选自受体VEGF和PDGF。
附图简要描述:
图1说明了一个用于合成3-烷氧基-4-酰化氨基酰胺派生物与活性酰化剂1-3的方案,其中3-烷氧基-4-酰化氨基酰胺派生物从甲基3-羟基-4-氨基丁酸酯氢氯化物开始。
图2说明了一个用于合成2-烷氧基-3-酰化氨基酰胺派生物与活性酰化剂1-3的方案,其中2-烷氧基-3-酰化氨基酰胺派生物从甲基2-羟基-3-氨基丁酸酯氢氯化物开始。
图3说明了一个用于合成(2S)-2烷氧基-4-酰化氨基酰胺派生物与活性酰化剂1-3的方案,其中(2S)-2烷氧基-4-酰化氨基酰胺派生物从甲基(2S)-2-羟基-4-氨基丁酸酯氢氯化物开始。
详细描述:
例1-8:酸(1-4)和酰胺(1-5)的合成如图1所示。本领域的技术人员能够理解和完成该一般合成过程的变体,因此本发明的化合物能够由这些本领域的技术人员合成。
例1:4-({5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羰基}-氨基)-3-甲氧基-丁酸
向甲基4-氨基-3-羟基丁酸(1.0当量,通过在含有1.2当量HCL的无水甲醇中回流游离氨基酸制备)和DIEA(5当量)的DCM溶液中在25℃分批添加Mmt-CI(1.1当量)。搅拌一整夜后,用减压蒸馏移除DCM。残留物悬浮在乙酸乙酯中,使用盐水(3x)清洗并使用无水Na2SO4干燥。此后,乙酸乙酯被除去,残留物在高度真空中干燥一整夜,通过快速色谱技术就可以得到化合物1-1。在氩气氛下向化合物1-1的无水DMF溶液中加入NaH(1.5当量)。在25℃下搅拌1个小时后,向溶液中加入MeI(5当量),然后将得到的悬浮液在25℃下轻轻摇晃一整夜。在真空下除去DMF,残留物悬浮在乙酸乙酯中,使用盐水(3x)清洗并使用无水Na2SO4干燥。乙酸乙酯通过蒸馏被除去后,将得到的残留物使用1%TFA的DCE/DCM溶液处理30分钟。然后,通过减压蒸馏移除有机溶剂,将得到的残留物使用己烷(3x)捣成粉,从而得到游离氨基酸1-2。该氨基酸可以直接用于下面的步骤,并不需要任何的提纯和表征。在25℃下向1-2(2当量)和DIEA(5当量)的DMF溶液中加入化合物1-3(1当量)。搅拌30分钟后(LC-MS显示1-3完全消耗掉),加入KOH(5当量)的水溶液,然后再搅拌该溶液2个小时(LC-MS显示完全水解)。在减压蒸馏下除去溶剂,加入HCL(1N多)得到沉淀物。收集该沉淀物并使用过滤的方式用水清洗,然后在高度真空下干燥从而得到标题所示化合物(95%基于化合物1-3)。LC-MS:254nm单峰,MH+用C21H22FN3O5计算的值:416,实际获得值:416。1H-NMR(DMSO-de,400MHz),δ13.67(s,1H),12.18(b,1H),10.90(s,1H),7.75(del,J=2.4Hz,J=9.6Hz,1H),7.71(s,1H),7.64(t,J=6.0Hz,1H),6.92(m,1H),6.83(dd,J=4.8Hz,J=8.4Hz,1H),3.73(m,1H),3.43-3.31(m,2H),3.22(s,3H),2.52-2.35(m,2H),2.43(s,3H),2.41(s,3H)。
例2:3-乙氧基-4-({5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羰基}-氨基)-丁酸

制备该标题所示化合物使用的步骤与合成示例1化合物十分类似,只不过是使用碘乙烷而不是碘甲烷来获取3-乙氧基化合物(9.7%基于化合物1-3)。LC-MS:254nm单峰,MH+用C22H24FN3O5计算的值:430,实际获得值:430。
例3-8:合成酰胺(1-5)的一般过程:向含HATU(1.05mmol)和DIEA(5当量)的酸(1-4)的DMF溶液(5ml)中,加入一种胺(2当量)。在25℃下搅拌2小时后,加入HCL水溶液(2ml,1N)。制备高效液相色谱处理,获得纯净的酰胺产品,随后LC-MS和NMR图谱结构表征。
例3:5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羧酸(3-二甲基胺甲醯基-2-乙氧基-丙基)-酰胺
制备高效液相色谱,从30mg起始材料(酸)中,得到13mg标题所示化合物(41%)。LC-MS:254nm单峰,MH+用C24H29FN4O4计算的值:457,实际获得值:457。1H-NMR(DMSO-d6,400MHz),δ13.68(s,1H),10.89(s,1H),7.76(dd,J=2.4Hz,9.2Hz,1H),7.72(s,1H),7.60(t,J=6.0Hz,1H),6.92(m,1H),6.83(dd,J=4.8Hz,8.4Hz11H),3.89(m,1H),3.58-3.45(m,2H),3.40-3.27(m,2H,buried in water signals),2.97(s,3H),2.82(s,3H),2.43(s,3H),2.41(s,3H),1.07(t,J=7.2Hz,3H)。
例4:5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羧酸(3-二甲基胺甲醯基-2-甲氧基-丙基)-酰胺

制备高效液相色谱,从120mg起始材料(酸)中,得到46mg标题所示化合物(36%)。LC-MS:254nm单峰,MH+用C23H27FN4O4计算的值:443,实际获得值:443。1H-NMR(DMSO-d6,400MHz),δ13.68(s,1H),10.89(s,1H),7.76(dd,J=2.4Hz,9.2Hz,1H),7.71(s,1H),7.63(t,J=5.6Hz,1H),6.92(m,1H),6.83(dd,J=4.8Hz,8.8Hz,1H),3.78(m,1H),3.42-3.31(m,2H)1 3.30(s,3H),2.97(s,3H),2.82(s,3H),2.43(s,3H),2.41(s,3H),2.63-2.43(m,2H)。
例5:5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羧酸(2-甲氧基-4-吗啉-4-基-4-酮基-丁基)-酰胺

制备高效液相色谱,从110mg起始材料(酸)中,得到48mg标题所示化合物(37%)。LC-MS:254nm单峰,MH+用C25H29FN4O6计算的值:485,实际获得值:485。1H-NMR(DMSO-d6,400MHz),δ13.68(s,1H),10.89(s,1H),7.76(dd,J=2.4Hz,9.2Hz,1H),7.71(s,1H),7.63(t,J=5.6Hz,1H),6.92(m,1H),6.83(dd,J=4.8Hz,8.4Hz,1H),3.80(m,1H),3.55(m,4H),3.47(m,4H),3.38(m,2H),3.31(s,3H),2.60(m,1H),2.45(m,1H),2.43(s,3H),2.41(s,3H)。
例6:5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羧酸[4-(4-羟基-哌啶-1-基)-2-甲氧基-4-酮基-丁基]-酰胺

制备高效液相色谱,从50mg起始材料(酸)中,得到20mg标题所示化合物(33%)。LC-MS:254nm单峰,MH+用C26H31FN4O5计算的值:499,实际获得值:499。1H-NMR(DMSO-d6,400MHz),δ13.68(s,1H),10.89(s,1H),7.76(dd,J=2.4Hz,9.6Hz,1H),7.72(s,1H),7.63(t,J=5.6Hz,1H),6.93(m,1H),6.83(dd,J=4.4Hz,8.4Hz,1H),3.92(m,1H),3.78(m,1H),3.68(b,1H),3.30(s,3H),3.15(m,1H),3.01(m,1H),2.60(m,1H),2.55(m,2H),2.50(m,1H),2.45(m,2H),2.43(s,3H),2.41(s,3H),1.70(m,2H),1.30(m,2H)。
例7:5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羧酸(2-甲氧基-4-酮基-4-吡咯啶-1-基-丁基)-酰胺

制备高效液相色谱,从110mg起始材料(酸)中,得到40mg标题所示化合物(32%)。LC-MS:254nm单峰,MH+用C25H29FN4O4计算的值:469,实际获得值:469。1H-NMR(DMSO-d6,400MHz),δ13.68(s,1H),10.89(s,1H),7.76(dd,J=2.4Hz,9.6Hz,1H),7.71(s,1H),7.63(t,J=5.6Hz,1H),6.93(m,1H),6.83(dd,J=4.8Hz,8.8Hz,1H),3.82(m,1H),3.50-3.25(m,6H),3.30(s,3H),2.55-2.45(m,2H),2.43(s,3H),2.41(s,3H),1.86(m,2H),1.76(m,2H)。
例8:5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羧酸[2-甲氧基-3-(甲氧基-甲基-胺甲醯基)-丙基]-酰胺

制备高效液相色谱,从80mg起始材料(酸)中,得到15mg标题所示化合物(15%)。LC-MS:254nm单峰,MH+用C23H27FN4O5计算的值:459,实际获得值:459。1H-NMR(DMSO-d6,400MHz),δ13.68(s,1H),10.90(s,1H),7.76(dd,J=2.4Hz,9.2Hz,1H),7.72(s,1H),7.68(t,J=6.0Hz,1H),6.93(m,1H),6.84(dd,J=4.4Hz,8.4Hz,1H),3.79(m,1H),3.66(s,3H),3.50-3.35(m,2H),3.31(s,3H),3.13(s,3H),2.55-2.45(m,2H),2.43(s,3H),2.41(s,3H)。
例9-15:酸(2-3)和酰胺(2-4)的合成如图2所示。该一般过程的变体可以由本领域的技术人员理解和实现。因此本发明的化合物可以由这些技术人员合成。
例9:3-({5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羰基}-氨基)-2-甲氧基-丙酸
在25℃下向甲基3-氨基-2-羟基丙酸甲酯(1.0当量,通过异丝氨酸在含1.2当量HCL的无水甲醇中回流游离氨基酸制备)和DIEA(5当量)的DCM溶液中分批添加Mmt-CI(1.1当量)。搅拌一整夜后,用减压蒸馏移除DCM。残留物悬浮在乙酸乙酯中,使用盐水(3x)清洗并通过无水Na2SO4干燥。除去乙酸乙酯,将残留物在高度真空下干燥一整夜,使用快速色谱技术得到化合物2-1。在氩气氛下向含有化合物2-1的无水DMF溶液中加入NaH(1.5当量)。在25℃下搅拌1个小时后,加入MeI(5当量),然后将得到的悬浮液在25℃下轻轻搅拌一整夜。在真空下除去DMF,残留物悬浮在乙酸乙酯中,使用盐水(3x)清洗并使用无水Na2SO4干燥。通过蒸馏将乙酸乙酯除去后,将残留物在1%TFA的DCE/DCM溶液中处理30分钟。然后,通过减压蒸馏移除有机溶剂,将得到的残留物使用己烷(3x)磨成粉便得到游离氨基酸2-2。该氨基酸可直接用于下面的步骤,不需要提纯和表征。在25℃下向含有2-2(2当量)和DIEA(5当量)的DMF溶液中加入化合物1-3(1当量)。搅拌30分钟后(LC-MS显示1-3已完全消耗掉),加入KOH(5当量)水溶液,将该溶液再搅拌2小时(LC-MS显示完全水解)。在减压蒸馏下除去溶剂,加入HCL(1N多)得到沉淀物。通过过滤得到沉淀物,使用水清洗并在高度真空下干燥便得到标题所示化合物(99%基于化合物1-3)。LC-MS::254nm单峰,MH+用C20H20FN3O5计算的值:402,实际获得值:402。1H-NMR(DMSO-d6,400MHz),δ13.67(s,1H),12.83(b,1H),10.90(s,1H),7.76(dd,J=2.4Hz,J=9.6Hz,1H),7.71(S,1H),7.69(t,J-6.0Hz,1H),6.92(m,1H),6.82(dd,J=4.8Hz,J=8.4Hz,1H),3.90(m,1H),3.55(m,1H),3.41(m,1H),3.32(s,3H),2.42(s,3H),2.40(s,3H)。
例10:2-乙氧基-3-({5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羰基}-氨基)-丙酸
制备该标题所示化合物使用的步骤与合成示例9化合物十分类似,只不过是使用碘乙烷而不是碘甲烷来获取2-乙氧基化合物(38%基于化合物1-3)。LC-MS:254nm单峰,MH+用C21H22FN4O5计算的值:416,实际获得值:416。1H-NMR(DMSO-d6,400MHz),δ13.67(s,1H),12.80(b,1H),10.89(s,1H)1 7.76(dd,J=2.4Hz,J=9.2Hz,1H),7.71(s,1H),7.68(t,J=6.0Hz,1H),6.92(m,1H),6.83(dd,J=4.8Hz,J=8.4Hz,1H),4.00(dd,J=5.2Hz,J=7.6Hz,1H),3.58(m,2H),3.41(m,2H),2.43(s,3H),2.41(s,3H),1.14(t,J=6.8Hz,3H)。
例11-15:合成酰胺(化合物2-4)的一般过程:向含有酸(化合物2-3)、HATU(1.05mmol)和DIEA(5当量)的DMF溶液(5ml)中加入相应的胺(2当量)。将溶液在25℃下搅拌2小时后,加入HCL水溶液(2mL,1N)。制备高效液相色谱处理便得到纯净的酰胺产品,随后LC-MS和NMR图谱结构表征。
例11:5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羧酸(2-二甲基胺甲醯基-2-乙氧基-乙基)-酰胺

制备高效液相色谱,从70mg起始材料(酸)中,得到46mg标题所示化合物(62%)。LC-MS:254nm单峰,MH+用C23H27FN4O4计算的值:443,实际获得值:443.
例12:5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羧酸(2-乙氧基-3-吗啉-4-yl-基-酮基-丙基)-酰胺

制备高效液相色谱,从70mg起始材料(酸)中,得到40mg标题所示化合物(49%)。LC-MS:254nm单峰,MH+用C25H29FN4O5计算的值:485,实际获得值:485。1H-NMR(DMSO-d6,400MHz),δ13.67(s,1H),10.89(s,1H),7.76(dd,J=2.4Hz,J=9.6Hz,1H),7.71(s,1H),7.70(m,1H),6.93(m,1H),6.83(dd,J=4.8Hz,J=8.4Hz,1H),4.40(m,1H),3.73-3.35(m,12H),2.43(s,3H),2.41(S,3H),1.12(t,J=7.2Hz,3H)。
例13:5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羧酸(2-二甲基胺甲醯基-2-甲氧基-乙基)-酰胺

制备高效液相色谱,从115mg起始材料(酸)中,得到93mg标题所示化合物(76%)。LC-MS:254nm单峰,MH+用C22H25FN4O4计算的值:429,实际获得值:429。1H-NMR(DMSO-d6,400MHz),δ13.68(s,1H),10.90(s,1H),7.75(dd,J=2.4Hz,J=9.6Hz,1H),7.72(m,1H),7.71(s,1H),6.93(m,1H),6.83(dd,J=4.8Hz,J=8.8Hz,1H),4.40(dd,J=4.8Hz,J=7.2Hz,1H),3.50(m,1H),3.32(m,1H),3.24(s,3H),3.10(s,3H),2.86(s,3H),2.43(s,3H),2.41(s,3H)。
例14:5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羧酸(2-甲氧基-3-吗啉-4-基-3-酮基-丙基)-酰胺

制备高效液相色谱,从115mg起始材料(酸)中,得到98mg标题所示化合物(73%)。LC-MS:254nm单峰,MH+用C24H27FN4O5计算的值:471,实际获得值:471。1H-NMR(DMSO-d6,400MHz),δ13.67(s,1H),10.89(s,1H),7.75(dd,J=2.4Hz,J=9.6Hz,1H),7.71(s,1H),7.70(m,1H),6.92(m,1H),6.83(dd,J=4.8Hz,J=8.8Hz,1H),4.34(dd,J=4.8Hz,J=7.2Hz,1H),3.85-3.30(m,10H),3.26(s,3H),2.44(s,3H),2.42(s,3H)。
例15:5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羧酸(2-甲氧基-3-酮基-3-吡咯啶-1-基-丙基)-酰胺

制备高效液相色谱,从115mg起始材料(酸)中,得到86mg标题所示化合物(66%)。LC-MS:254nm单峰,MH+用C24H27FN4O4计算的值:455,实际获得值:455。1H-NMR(DMSO-d6,400MHz),δ13.67(s,1H),10.89(s,1H),7.76(dd,J=2.4Hz,J=9.6Hz,1H),7.70(m,1H),,7.71(s,1H),6.93(m,1H),6.83(dd,J=4.4Hz,J=8.4Hz,1H),4.20(dd,J=5.2Hz,J=7.2Hz,1H),3.60-3.47(m,3H),3.43-3.28(m,3H),3.26(s,3H),2.43(s,3H),2.40(s,3H),1.88(m,2H),1.78(m,2H)。
例16-315:更多的酰胺示例如下面表格所示:



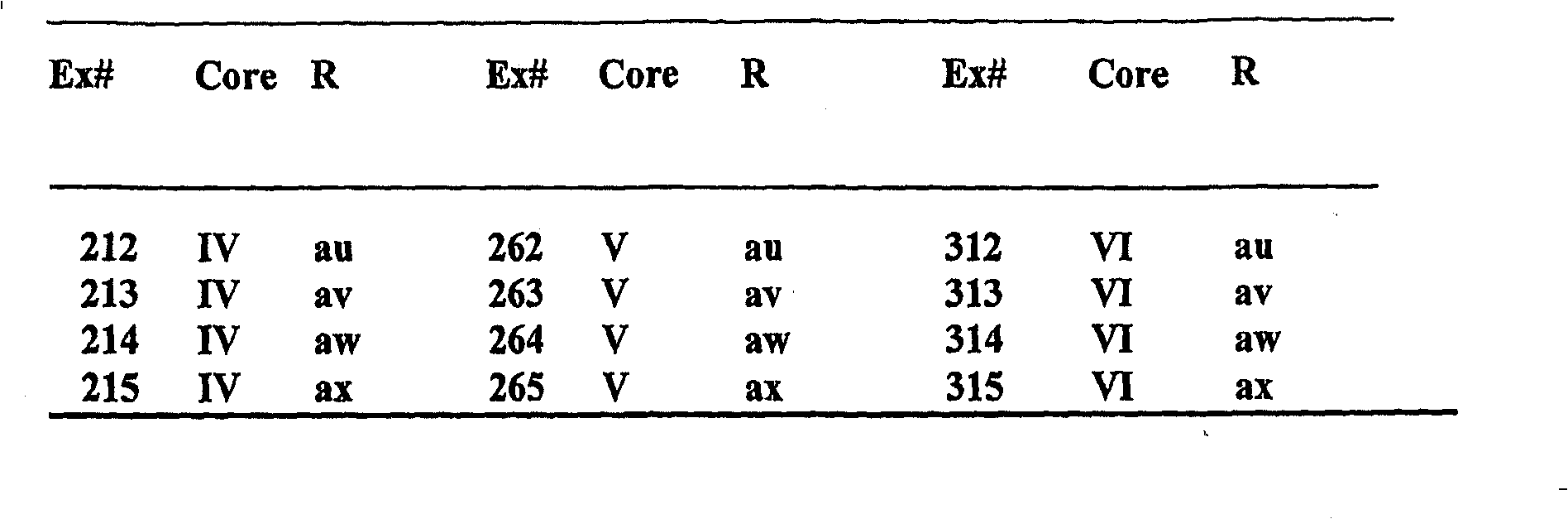
在上述表格中,R9选自如下列结构所示的自由基:

这些酰胺,例16-315,可以按照上述程序和/或已知程序,被本领域的技术人员制造。
例316-320:酸(3-3)和酰胺(3-4)的合成如图3所示。该一般合成过程的变体可以由本领域的技术人员理解和实施,因此本发明的化合物可以由本领域的技术人员合成。
例316:(S)-4-({5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羰基}-氨基)-2-甲氧基-丁酸

在25℃下向甲基4-氨基-2-羟基丁酸(1.0当量,通过在含有1.2当量HCL的无水甲醇中回流游离氨基酸制备)和DIEA(5当量)的DCM溶液中分批加入Mmt-CI(1.1当量)。搅拌一整夜后,用减压蒸馏移除DCM。残留物悬浮在乙酸乙酯中,使用盐水(3x)清洗并使用无水Na2SO4干燥残留物。除去乙酸乙酯,在高度真空下将残留物干燥一整夜,通过快速色谱技术得到化合物3-1。在氩气下向化合物3-1的无水DMF溶液中加入NaH(1.5当量)。在25℃下搅拌1小时后,加入MeI(5当量),然后将得到的悬浮物在25℃下轻轻搅拌一整夜。在真空下除去DMF,残留物悬浮在乙酸乙酯中,使用盐水(3x)清洗并通过无水Na2SO4干燥。通过蒸馏将乙酸乙酯除去后,将得到的残留物使用1%TFA的DCE/DCM溶液处理30分钟。然后,通过减压蒸馏移除有机溶剂,将得到的残留物使用已烷磨成粉便得到游离氨基酸3-2。该氨基酸可直接用于下面的步骤,并不需要提纯和表征。在25℃下向含有3-2(2当量)和DIEA(5当量)的DMF溶液中加入化合物1-3(1当量)。搅拌30分钟后(LC-MS显示1-3完全消耗掉),加入KOH水溶液(5当量),接着搅拌2小时(LC-MS显示完全水解)。在减压蒸馏下除去溶剂,加入HCL(1N多)得到沉淀物。过滤收集该沉淀物并使用水清洗,在高度真空下干燥便得到标题所示化合物(97%基于化合物1-3)。LC-MS:254nm单峰,MH+用C21H22FN3O5计算的值:416,实际获得值:416。1H-NMR(DMSO-d6,400MHz),δ13.68(s,1H),12.80(b,1H),10.90(s,1H),7.76(dd,J-2.4Hz,J=9.6Hz,1H),7.71(s,1H),7.65(t,J=5.6Hz,1H),6.93(m,1H),6.83(dd,J=4.8Hz,J=8.4Hz,1H),3.77(dd,J=4.0Hz,J=8.8Hz,1H),3.40-3.30(m,2H),3.30(s,3H),2.43(s,3H),2.41(s,3H),1.92(m,1H),1.78(m,1H)。
例317:(S)-2-乙氧基-4-({5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羰基}-氨基)-丁酸

使用与合成例316化合物相似的步骤来制备标题所示化合物,只不过使用碘乙烷而不是碘甲烷来获取2-乙氧基化合物(84%基于化合物1-3)。LC-MS:254nm单峰,MH+用C22H24FN3O5计算的值:430,实际获得值:430。1H-NMR(DMSO-d6,400MHz),δ13.68(s,1H),12.70(b,1H),10.89(s,1H),7.76(dd,J=2.4Hz,J=9.6Hz,1H),7.71(s,1H),7.66(t,J=5.6Hz,1H),6.93(m,1H),6.83(dd,J=4.8Hz,J=8.4Hz,1H),3.85(dd,J=4.0Hz,J=8.4Hz,1H),3.58(m,1H),3.40-3.25(m,3H),2.43(s,3H),2.41(s,3H),1.92(m,1H),1.77(m,1H),1.13(t,J=7.2Hz,3H)。
例318-320:合成酰胺(化合物3-4)的通常过程。将相应的胺(2当量)加入含有酸(化合物3-3)、HATU(1.05mmol)和DIEA(5当量)的DMF溶液(5mL)中。将该溶液在25℃下搅拌2小时,加入HCL水溶液(2mL,1N)。制备高效液相色谱处理得到纯净的酰胺产品,随后LC-MS和NMR图谱结构表征。
例318:5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羧酸((S)-3-二甲基胺甲醯基-3-甲氧基-丙基)-酰胺

制备高效液相色谱,从60mg起始材料(酸)中,得到37mg标题所示化合物(58%)。LC-MS:254nm单峰,MH+用C23H27FN4O4计算的值:443,实际获得值:443。1H-NMR(DMSO-d6,400MHz),δ13.68(s,1H),10.89(s,1H),7.76(dd,J=2.4Hz,J=9.6Hz,1H),7.72(s,1H),7.65(t,J=5.6Hz,1H),6.93(m,1H),6.83(dd,J=4.8Hz,J=8.4Hz,1H),4.20(dd,J=4.0Hz,J=8.0Hz,1H),3.30(m,2H),3.27(s,3H),3.04(s,3H),2.88(s,3H)1 2.43(s,3H),2.41(s,3H),1.80(m,2H)。
例319:5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羧酸((S)-3-甲氧基-4-吗啉-4-基-4-酮基-丁基)-酰胺

制备高效液相色谱,从60mg起始材料(酸)中,得到32mg标题所示化合物(46%)。LC-MS:254nm单峰,MH+用C25H29FN4O5计算的值:485,实际获得值:485。1H-NMR(DMSO-d6,400MHz),δ13.68(s,1H),10.89(s,1H),7.76(dd,J=2.4Hz,J=9.6Hz,1H),7.72(s,1H),7.65(t,J=5.6Hz,1H),6.93(m,1H),6.83(dd,J=4.8Hz,J=8.4Hz,1H),4.19(dd,J=4.8Hz,J=8.0Hz,1H),3.57(m,6H),3.47(m,2H),3.28(m,2H),3.23(s,3H),2.44(s,3H),2.41(s,3H),1.79(m,2H)。
例320:5-[5-氟-2-酮基-1,2-二氢-吲哚-(3Z)-亚基甲基]-2,4-二甲基-1H-吡咯-3-羧酸((S)-3-二甲基胺甲醯基-3-乙氧基-丙基)-酰胺

制备高效液相色谱,从120mg起始材料(酸)中,得到67mg标题所示化合物(57%)。LC-MS:254nm单峰,MH+用C24H29FN4O4计算的值:457,实际获得值:457。1H-NMR(DMSOd6,400MHz),δ13.67(s,1H),10.88(s,1H),7.76(dd,J=2.4Hz1 J=9.6Hz,1H),7.71(s,1H),7.56(m,1H),6.91(m,1H),6.83(m,1H),4.25(m,1H),3.45-3.25(m,4H),3.03(s,3H),2.83(s,3H),2.43(s,3H),2.41(s,3H),1.80(m,2H)。
在这里所描述的化合物,是目前首选实例化的代表,是作为例子使用的,并不用于限制发明的范围。一个所属技术领域的专业人员,显而易见的,可以在不离开本发明的范围和精神下,对在这里透露的本发明做出不同的替代和更改。
VEGFR生化测试
该化合物的生化活性,由大不列颠联合王国敦提市的Upstate有限公司,根据以下程序来测试。在最终反应体积25μl中,KDR(h)(5-10mU)和8mM MOPS(3-(N-吗啉)丙磺酸)pH7.0、0.2mM EDTA、0.33mg/ml髓鞘碱蛋白、10mM乙酸镁、以及[γ-33P-ATP](比活度约500cpm/pmol,按要求浓缩)一起培养。加入MgATP混合物,起始反应。在室温下培养40分钟后,加入5μl 3%的磷酸溶液终止反应。然后,10μl反应物在一台P30过滤机上被辨认,并且用75mM磷酸洗三次,每次5分钟,甲醇预干燥一次,闪烁计数。
本发明的化合物使用上述方法进行测试,其IC50在1-5,000nM之间。
PDGFR磷酸化测试
将NIH3T3细胞以96孔板的浓度接种到DMEM+10%FBS中。细胞附着之后将细胞血清饥饿培养一整夜,然后再加入化学测试化合物使终浓度达到0.1%DMSO(二甲基亚砜)。细胞在37℃下被培养1小时后从培养箱中移出,然后使用PDGF-BB在室温下刺激15分钟,不过在这之前可以允许细胞冷却到室温达20分钟。将细胞置于冰上5分钟,除去细胞培养基并使用100μL/孔的溶解液在4℃下溶解细胞1小时。将培养板在4℃下以2000rpm的速度离心转动30分钟,用ELISA(酶联免疫吸附剂测定法)来测定溶解的磷酸化PDGFR。
高结合力的微孔反应板在室温下使用抗小鼠PDGFR-b捕获抗体的PBS溶液培养一整夜后,使用PBS+0.05%Tween20清洗并在室温下使用PBS+1%BSA阻断4小时,然后再次清洗。100μL溶解物/孔在4℃下培养一整夜。清洗培养板并在板孔中以100μL孔的溶解液/浓度在37℃下培养小鼠抗酪氨酸磷酸化HRP抗体2小时。再次清洗培养板并使用TMB作为显色液进行比色检测。
本发明的大部分化合物在该测试中显示IC50小于1μM。
VEGFR磷酸化测试
将NIHT3T细胞过量小鼠VEGFR-2(受体-1)以96孔板的浓度接种到DMEM+10%FBS中。细胞附着4小时后将细胞血清饥饿培养一整夜,然后加入化学测试化合物使终浓度达到0.1%DMSO(二甲基亚砜)。在37℃下培养1小时之后使用VEGF165在37℃下刺激细胞15分钟。将细胞置于冰上5分钟,除去细胞培养基并使用冰冷的PBS清洗一次,然后使用50μL/孔的溶解液在4℃下溶解细胞1小时。将培养板在4℃下以2000rpm的速度离心转动10分钟,用ELISA来测定溶解的磷酸化VEGFR。
高结合力的微孔反应板在室温下使用VEGFR抗体的50μL PBS溶液培养一整夜后,使用PBS+0.05%Tween20清洗并在室温下使用PBS+1%BSA阻断4小时,然后再次清洗。将50μL/孔的溶解物在4℃下培养一整夜。清洗培养板并在板孔中以50μL/孔的浓度在37℃下培养小鼠抗酪氨酸磷酸化HRP抗体2小时。再次清洗培养板并使用TMB作为显色液进行比色检测。
本发明的大部分化合物在该测试中显示IC50小于1μM。
细胞测试:HUVEC(人类脐静脉血管内皮细胞):VEGF诱导下的增殖
该化合物的细胞活性,通过HUVEC细胞在VEGF诱导下的增殖来测试。HUVEC细胞(Cambrex公司,CC-2517)在5%CO2和37℃下,被保持在EGM[培养基](Cambrex公司,CC-3124)中。HUVEC细胞被接种到EGM中,接种浓度5000细胞数/孔(96孔板)。在细胞附着之后(1小时),EGM培养基被替换为EBM[培养基](Cambrex公司,CC-3129)+0.1%FBS(胎牛血清)(ATTC,30-2020),细胞在37℃下培养20小时。培养基被替换为EBM+1%FBS,化合物用DMSO(二甲基亚砜)稀释成一系列浓度,加入到细胞中,终浓度为0-5000nM和1%DMSO。在37℃下1小时预培养之后,用10ng/ml VEGF(Sigma公司,V7259)刺激细胞,37℃下培养45小时。细胞的增殖用BrdU(溴脱氧尿嘧啶核苷)DNA掺入4小时来测量[译注:BrdU将替换DNA中的胸腺嘧啶],BrdU标记用ELISA(酶联免疫吸附剂测定法)(Roche试剂盒,16472229)来测定,用1M H2SO4使反应停止。使用干涉波长690nm,在450nm测量吸光率。
附图详细描述:
图1显示了用于合成从甲基3-羟基-4-氨基丁酸酯氢氯化物开始的3-烷氧基-4-酰化氨基酰胺派生物与活性酰化剂1-3的方案。起始材料氨基酯氢氯化物通过在含有1.2当量HCL的无水甲醇中回流游离氨基酸制备。在与仲羟基生成中性羟基酯1-1时,氨基和它的单甲氧基三苯甲基派生物一样受到保护。羟基使用甲基或者碘乙烷来烷基化从而生成受保护的氨基烷氧基酯。在1%的三氟乙酸中除去Mmt得到氨基氢氯化物或者三氟乙酸化合物1-2。将该化合物使用酰化剂1-3快速酰化并将甲基酯通过氢氧化钾的水/DMF溶液水解后得到1-4。然后将游离酸暴露于HATU、胺以及二异丙基乙胺的DMF溶液中得到烷氧基酸1-5。
图2显示了用于合成从甲基2-羟基-3-氨基丁酸酯氢氯化物开始的2-烷氧基-3-酰化氨基酰胺派生物与活性酰化剂1-3的方案。起始材料氨基酯氢氯化物通过在含有1.2当量HCL的无水甲醇中回流游离氨基酸制备。在与仲羟基生成中性羟基酯1-1时,氨基和它的单甲氧基三苯甲基派生物一样受到保护。羟基使用甲基或者碘乙烷来烷基化从而生成受保护的氨基烷氧基酯。在1%的三氟乙酸中除去Mmt得到氨基氢氯化物或者三氟乙酸化合物2-2。将该化合物使用酰化剂1-3快速酰化并将甲基酯通过氢氧化钾的水/DMF溶液水解后得到2-4。然后将游离酸暴露于HATU、胺以及二异丙基乙胺的DMF溶液中得到烷氧基酸2-5。
图3显示了用于合成从甲基(2S)-2-羟基-4-氨基丁酸酯氢氯化物开始的(2S)-2-烷氧基-4-酰化氨基酰胺派生物与活性酰化剂1-3的方案。起始材料氨基酯氢氯化物通过在含有1.2当量HCL的无水甲醇中回流游离氨基酸制备。在与仲羟基生成中性羟基酯1-1时,氨基和它的单甲氧基三苯甲基派生物一样受到保护。羟基使用甲基或者碘乙烷来烷基化从而生成受保护的氨基烷氧基酯。在1%的三氟乙酸中除去Mmt得到氨基氢氯化物或者三氟乙酸化合物3-2。将该化合物使用酰化剂1-3快速酰化并将甲基酯通过氢氧化钾的水/DMF溶液水解后得到3-4。然后将游离酸暴露于HATU、胺以及二异丙基乙胺的DMF溶液中得到烷氧基酸3-5。
Claims (33)
1. 一种由式1表示的化合物:
其中:
R1选自氢、卤素、(C1-C6)烷基、(C3-C8)环烷基、(C1-C6)卤烷基、羟基、(C1-C6)烷氧基、氨基、(C1-C6)烷基氨基、酰胺、磺胺、氰、取代或未取代的(C6-C10)芳香族羟基;
R2选自氢、卤素、(C1-C6)烷基、(C3-C8)环烷基、(C1-C6)卤烷基、羟基、(C1-C6)烷氧基、(C2-C8)烷氧基烷基、氨基、(C1-C6)烷基氨基、(C6-C10)芳香族羟基氨基;
R3选自氢、(C1-C6)烷基、(C6-C10)芳香族羟基、(C5-C10)杂芳香族羟基和酰胺;
R4、R5、R6和R8都独立地选自氢和(C1-C6)烷基;
R7是(C1-C6)烷基;
R9选自氢基、(C1-C6)O-烷基、(C3-C8)O-环烷基和NR10R11;R10和R11都独立地选自氢、(C1-C6)烷基、(C1-C6)羟基烷基、(C1-C6)二羟基烷基、(C1-C6)烷氧基、(C1-C6)烷基羧酸、(C1-C6)烷基膦酸、(C1-C6)烷基磺酸、(C1-C6)羟基烷基羧酸、(C1-C6)烷基酰胺、(C3-C8)环烷基、(C5-C8)杂环烷基、(C6-C8)芳香族羟基、(C5-C8)杂芳香族羟基、(C3-C8)环烷基羧酸、或者R10和R11以及N组成一个(C5-C8)杂环化合物,该杂环化合物未被取代或被一个或多个羟基、酮基、乙醚和羧酸所取代;
n=1或2或3;并且
m=0或1或2;
或者,一种可药用的盐、它的异构体、它的异构体的一种可药用的盐或者药物前体。
2. 根据权利要求1的化合物、盐、异构体或药物前体,由式2表示:

其中,R12选自氢、(C1-C6)烷基以及(C3-C8)环烷基。
3. 根据权利要求2的化合物、盐、异构体或药物前体,其中:
R1和R2独立地选自氢和氟;
R3和R4是甲基;
R5、R6、R8、和R12是氢;
R7是(C1-C6)烷基;
n=1或2;并且
m=0或1。
4. 根据权利要求3的化合物、盐、异构体或药物前体,选自:

5. 根据权利要求3的化合物、盐、异构体或药物前体,由下面的结构表示:

6. 根据权利要求3的化合物、盐、异构体或药物前体,由下面的结构表示:

7. 根据权利要求3的化合物、盐、异构体或药物前体,由下面的结构表示:

8. 根据权利要求3的化合物、盐、异构体或药物前体,由下面的结构表示:

9. 根据权利要求3的化合物、盐、异构体或药物前体,由下面的结构表示:

10. 根据权利要求3的化合物、盐、异构体或药物前体,由下面的结构表示:

11. 根据权利要求3的化合物、盐、异构体或药物前体,由式3表示:

12. 根据权利要求9的化合物、盐、异构体或药物前体,其中:
R1和R2独立地选自氢、卤素和氰;
R3选自氢、(C1-C6)烷基、(C6-C10)芳香族羟基、(C5-C10)杂芳香族羟基以及酰胺;
R4,R5,R6和R8独立地选自氢、(C1-C6))烷基;
R7是(C1-C6);
n=1或2;
m=0或1;并且
R10和R11选自氢、(C1-C6)烷基、(C1-C6)羟基烷基、(C2-C6)二羟基烷基、(C1-C6)烷氧基、(C2-C6)烷基羧酸、(C1-C6)烷基磷酸、(C1-C6)烷基磺酸、(C2-C6)羟基烷基羧酸、(C1-C6)烷基酰基、(C3-C8)环烷基、(C5-C8)杂环烷基、(C6-C8)芳香族羟基、(C5-C8)杂芳香族羟基、(C4-C8)环烷基羧酸、或者R10和R11以及N组成一个(C5-C8)杂环化合物,该杂环化合物未被取代或被一个或多个羟基、酮基、乙醚和羧酸所取代。
13. 根据权利要求12的化合物、盐、异构体或药物前体,其中m为0。
14. 根据权利要求13的化合物、盐、异构体或药物前体,选自下面的结构:

15. 根据权利要求14的化合物、盐、异构体或药物前体,由下面的结构表示:

16. 根据权利要求14的化合物、盐、异构体或药物前体,由下面的结构表示:
17. 根据权利要求14的化合物、盐、异构体或药物前体,由下面的结构表示:

18. 根据权利要求14的化合物、盐、异构体或药物前体,由下面的结构表示:
19. 根据权利要求14的化合物、盐、异构体或药物前体,由下面的结构表示:

20. 根据权利要求14的化合物、盐、异构体或药物前体,由下面的结构表示:

21. 根据权利要求14的化合物、盐、异构体或药物前体,由下面的结构表示:

22. 根据权利要求14的化合物、盐、异构体或药物前体,由下面的结构表示:
23. 根据权利要求12的化合物、盐、异构体或药物前体,其中m为1。
24. 根据权利要求23的化合物、盐、异构体或药物前体,由下面的结构表示:
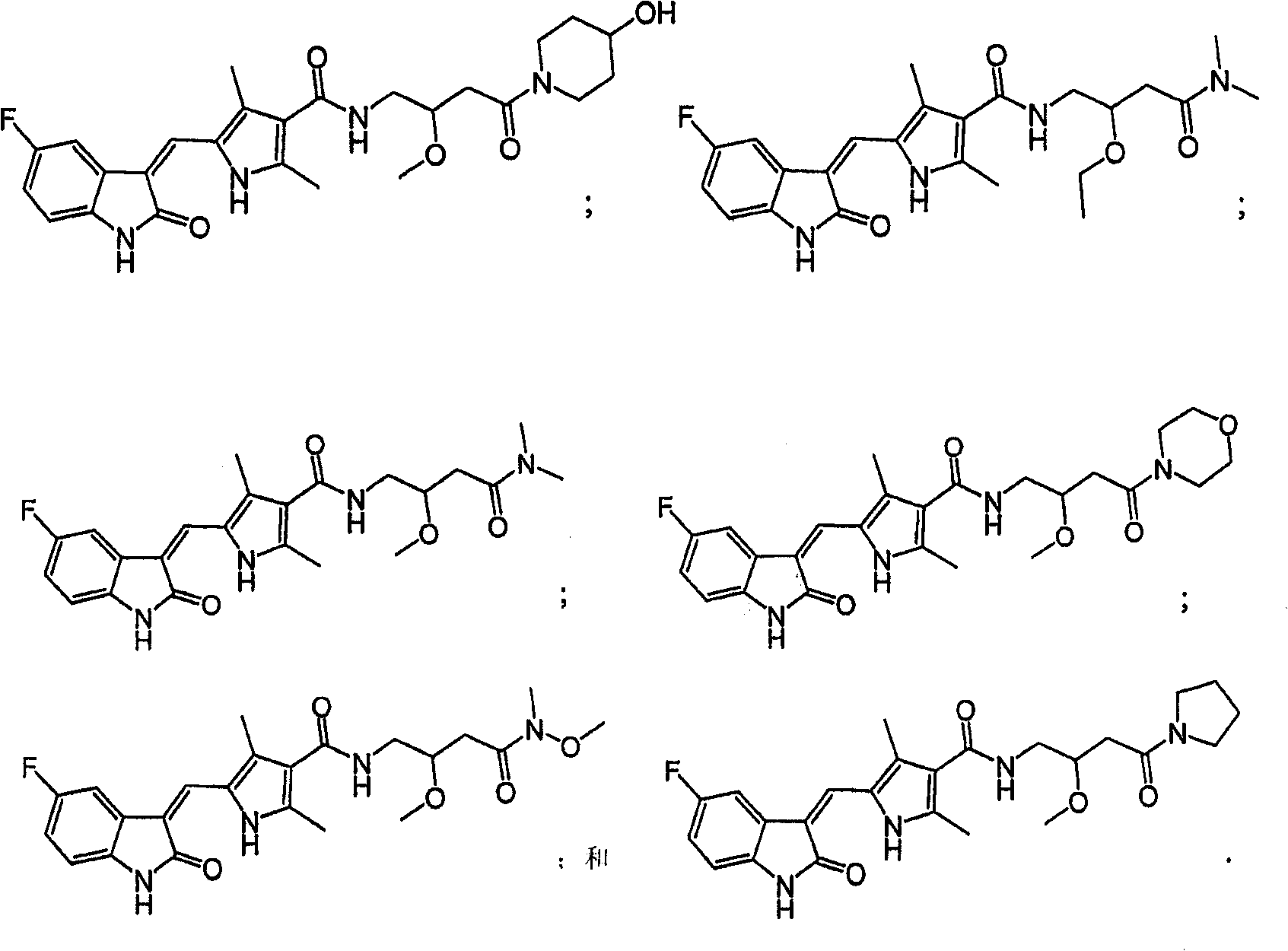
25. 根据权利要求24的化合物、盐、异构体或药物前体,由下面的结构表示:

26. 根据权利要求24的化合物、盐、异构体或药物前体,由下面的结构表示:

27. 根据权利要求24的化合物、盐、异构体或药物前体,由下面的结构表示:

28. 根据权利要求24的化合物、盐、异构体或药物前体,由下面的结构表示:

29. 根据权利要求24的化合物、盐、异构体或药物前体,由下面的结构表示:

30. 根据权利要求24的化合物、盐、异构体或药物前体,由下面的结构表示:
31. 根据权利要求1的化合物、盐、异构体或药物前体,选自下面的结构:

其中R选自如下列结构表示的自由基:

32. 一种使用如权利要求1-31所示的化合物或盐来调节蛋白激酶催化活性的方法。
33. 对于权利要求32的方法,其中所述的蛋白激酶选自VEGF受体和PDGF受体。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US71947405P | 2005-09-22 | 2005-09-22 | |
| US60/719,474 | 2005-09-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN101282965A true CN101282965A (zh) | 2008-10-08 |
Family
ID=37900093
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2006800352235A Pending CN101282965A (zh) | 2005-09-22 | 2006-09-21 | 基于烷氧基吲哚酮的蛋白激酶抑制剂 |
Country Status (10)
| Country | Link |
|---|---|
| US (2) | US20090305382A1 (zh) |
| EP (1) | EP1926725A4 (zh) |
| JP (1) | JP2009508971A (zh) |
| KR (1) | KR20080046701A (zh) |
| CN (1) | CN101282965A (zh) |
| AU (1) | AU2006294600A1 (zh) |
| BR (1) | BRPI0616378A2 (zh) |
| CA (1) | CA2621809A1 (zh) |
| RU (1) | RU2008110083A (zh) |
| WO (1) | WO2007038251A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110016052A (zh) * | 2018-01-08 | 2019-07-16 | 四川海思科制药有限公司 | N-去乙基舒尼替尼的磷酰胺衍生物及其制备方法 |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101282965A (zh) * | 2005-09-22 | 2008-10-08 | 斯克利普斯研究院 | 基于烷氧基吲哚酮的蛋白激酶抑制剂 |
| AU2007296740B2 (en) * | 2006-09-11 | 2012-09-27 | Curis, Inc. | Substituted 2-indolinone as PTK inhibitors containing a zinc binding moiety |
| CN101553482B (zh) | 2006-09-15 | 2013-11-20 | 艾科睿制药公司 | 激酶抑制剂化合物 |
| EP2838538B1 (en) | 2012-04-20 | 2017-03-15 | Annji Pharmaceutical Co., Ltd. | Cyclopropanecarboxylate esters of purine analogues |
| NZ721520A (en) | 2014-03-07 | 2023-03-31 | Hoffmann La Roche | Novel 6-fused heteroaryldihydropyrimidines for the treatment and prophylaxis of hepatitis b virus infection |
| WO2016146598A1 (en) | 2015-03-16 | 2016-09-22 | F. Hoffmann-La Roche Ag | Combined treatment with a tlr7 agonist and an hbv capsid assembly inhibitor |
| KR102497701B1 (ko) | 2016-09-13 | 2023-02-09 | 에프. 호프만-라 로슈 아게 | Tlr7 작용제 및 hbv 캡시드 조립 억제제의 병용 치료 |
| CA3130596A1 (en) | 2019-03-25 | 2020-10-01 | F. Hoffmann-La Roche Ag | Solid forms of a compound of hbv core protein allosteric modifier |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US72934A (en) * | 1867-12-31 | Improvement in marking-gauge fob sswim-mgohines | ||
| AR042586A1 (es) * | 2001-02-15 | 2005-06-29 | Sugen Inc | 3-(4-amidopirrol-2-ilmetiliden)-2-indolinona como inhibidores de la protein quinasa; sus composiciones farmaceuticas; un metodo para la modulacion de la actividad catalitica de la proteinquinasa; un metodo para tratar o prevenir una afeccion relacionada con la proteinquinasa |
| TWI259081B (en) * | 2001-10-26 | 2006-08-01 | Sugen Inc | Treatment of acute myeloid leukemia with indolinone compounds |
| CA2547066A1 (en) * | 2003-11-26 | 2005-06-16 | The Scripps Research Institute | Advanced indolinone based protein kinase inhibitors |
| CN101282965A (zh) * | 2005-09-22 | 2008-10-08 | 斯克利普斯研究院 | 基于烷氧基吲哚酮的蛋白激酶抑制剂 |
-
2006
- 2006-09-21 CN CNA2006800352235A patent/CN101282965A/zh active Pending
- 2006-09-21 EP EP06815164A patent/EP1926725A4/en not_active Withdrawn
- 2006-09-21 RU RU2008110083/04A patent/RU2008110083A/ru not_active Application Discontinuation
- 2006-09-21 AU AU2006294600A patent/AU2006294600A1/en not_active Abandoned
- 2006-09-21 KR KR1020087008492A patent/KR20080046701A/ko not_active Application Discontinuation
- 2006-09-21 CA CA002621809A patent/CA2621809A1/en not_active Abandoned
- 2006-09-21 WO PCT/US2006/036946 patent/WO2007038251A1/en active Application Filing
- 2006-09-21 US US11/991,971 patent/US20090305382A1/en not_active Abandoned
- 2006-09-21 US US11/525,291 patent/US7629339B2/en not_active Expired - Fee Related
- 2006-09-21 BR BRPI0616378-5A patent/BRPI0616378A2/pt not_active IP Right Cessation
- 2006-09-21 JP JP2008532411A patent/JP2009508971A/ja not_active Withdrawn
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110016052A (zh) * | 2018-01-08 | 2019-07-16 | 四川海思科制药有限公司 | N-去乙基舒尼替尼的磷酰胺衍生物及其制备方法 |
| CN110016052B (zh) * | 2018-01-08 | 2021-09-28 | 四川海思科制药有限公司 | N-去乙基舒尼替尼的磷酰胺衍生物及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2621809A1 (en) | 2007-04-05 |
| EP1926725A4 (en) | 2010-10-06 |
| RU2008110083A (ru) | 2009-10-27 |
| WO2007038251A1 (en) | 2007-04-05 |
| US20070072934A1 (en) | 2007-03-29 |
| US20090305382A1 (en) | 2009-12-10 |
| JP2009508971A (ja) | 2009-03-05 |
| KR20080046701A (ko) | 2008-05-27 |
| EP1926725A1 (en) | 2008-06-04 |
| BRPI0616378A2 (pt) | 2011-06-21 |
| AU2006294600A1 (en) | 2007-04-05 |
| US7629339B2 (en) | 2009-12-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101282965A (zh) | 基于烷氧基吲哚酮的蛋白激酶抑制剂 | |
| CN102083828B (zh) | 作为c-kit和pdgfr激酶抑制剂的杂环化合物和组合物 | |
| CN104812747B (zh) | 用于治疗病毒感染的1,2,4‑三嗪衍生物 | |
| JP2024029066A (ja) | ガンの治療方法 | |
| ES2381895T3 (es) | Inhibidores de PI 3-quinasa y métodos para su uso | |
| KR101218926B1 (ko) | 키나제 억제제로서의 5-(4-(할로알콕시)페닐)피리미딘-2-아민 화합물 및 조성물 | |
| CN1863774B (zh) | 用作蛋白激酶抑制剂的化合物和组合物 | |
| JP6611363B2 (ja) | 複素環化合物およびRetinoid−related Orphan Receptor(ROR)ガンマーT阻害剤としてのそれらの使用 | |
| KR20100050554A (ko) | 키나제 억제제로서의 2-헤테로아릴아미노-피리미딘 유도체 | |
| CN101622244A (zh) | 作为蛋白激酶抑制剂的化合物和组合物 | |
| KR20100035635A (ko) | 단백질 키나제 억제제 및 그의 사용 방법 | |
| CN101500574A (zh) | 用作蛋白激酶抑制剂的化合物和组合物 | |
| JPWO2016039408A1 (ja) | 複素環化合物 | |
| CN105837575A (zh) | 3-乙炔基吡唑并嘧啶衍生物及其制备方法和用途 | |
| JP2010529129A (ja) | キナーゼ阻害剤化合物 | |
| Zhao et al. | Structural optimization of diphenylpyrimidine derivatives (DPPYs) as potent Bruton's tyrosine kinase (BTK) inhibitors with improved activity toward B leukemia cell lines | |
| JP2018521029A (ja) | ブルトン型チロシンキナーゼのビアリール阻害剤のアジピン酸塩形態及び組成物 | |
| AU2015274285B2 (en) | Pyrimidine compounds and methods using the same | |
| Quadrelli et al. | N, O-nucleosides from ene reactions of nitrosocarbonyl intermediates with the 3-methyl-2-buten-1-ol | |
| CN101287734B (zh) | 2-氨基-7,8-二氢-6H-吡啶并[4,3-d]嘧啶-5-酮 | |
| JP6659850B2 (ja) | キノリン系化合物の塩、その結晶形、調製方法、組成物及び用途 | |
| CN112110864B (zh) | 一种4-酰胺取代嘧啶类靶向ddr1抑制剂及其制备和抗肿瘤活性的应用 | |
| TW202229278A (zh) | 含氮雜環類衍生物的鹽、晶型及其製備方法和應用 | |
| CN113461661A (zh) | 6-(吡啶-3-基)喹唑啉-4(3h)-酮类衍生物及其制备和应用 | |
| JP2022513844A (ja) | Syk阻害剤の塩及びその結晶形 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Open date: 20081008 |