WO2023190136A1 - 学習システム、決定システム、及び予測システム、並びに学習方法、決定方法、及び予測方法 - Google Patents
学習システム、決定システム、及び予測システム、並びに学習方法、決定方法、及び予測方法 Download PDFInfo
- Publication number
- WO2023190136A1 WO2023190136A1 PCT/JP2023/011772 JP2023011772W WO2023190136A1 WO 2023190136 A1 WO2023190136 A1 WO 2023190136A1 JP 2023011772 W JP2023011772 W JP 2023011772W WO 2023190136 A1 WO2023190136 A1 WO 2023190136A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- biomarker
- sequence
- sequences
- learning
- score
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 84
- 239000000090 biomarker Substances 0.000 claims abstract description 144
- 238000005259 measurement Methods 0.000 claims abstract description 83
- 108090000623 proteins and genes Proteins 0.000 claims abstract description 54
- 238000006243 chemical reaction Methods 0.000 claims abstract description 33
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 claims abstract description 30
- 238000003199 nucleic acid amplification method Methods 0.000 claims abstract description 11
- 239000002773 nucleotide Substances 0.000 claims description 16
- 125000003729 nucleotide group Chemical group 0.000 claims description 16
- 230000003321 amplification Effects 0.000 claims description 10
- 230000011987 methylation Effects 0.000 abstract description 15
- 238000007069 methylation reaction Methods 0.000 abstract description 15
- 230000007067 DNA methylation Effects 0.000 abstract description 9
- 238000011156 evaluation Methods 0.000 abstract description 2
- 239000000126 substance Substances 0.000 abstract description 2
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 16
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 16
- 238000005457 optimization Methods 0.000 description 16
- 238000010586 diagram Methods 0.000 description 11
- 108020004414 DNA Proteins 0.000 description 10
- 102000053602 DNA Human genes 0.000 description 10
- 201000010099 disease Diseases 0.000 description 8
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 8
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 8
- 229940035893 uracil Drugs 0.000 description 8
- 239000008280 blood Substances 0.000 description 7
- 210000004369 blood Anatomy 0.000 description 7
- 238000004364 calculation method Methods 0.000 description 7
- 229940104302 cytosine Drugs 0.000 description 7
- 230000008569 process Effects 0.000 description 7
- 238000012549 training Methods 0.000 description 7
- 206010028980 Neoplasm Diseases 0.000 description 6
- 238000012408 PCR amplification Methods 0.000 description 6
- 201000011510 cancer Diseases 0.000 description 6
- 230000015654 memory Effects 0.000 description 6
- 238000012545 processing Methods 0.000 description 5
- 238000003745 diagnosis Methods 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 3
- 238000013473 artificial intelligence Methods 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- 238000007796 conventional method Methods 0.000 description 3
- 239000003550 marker Substances 0.000 description 3
- 239000000203 mixture Substances 0.000 description 3
- 239000004065 semiconductor Substances 0.000 description 3
- 108091029430 CpG site Proteins 0.000 description 2
- 108091028043 Nucleic acid sequence Proteins 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 239000000091 biomarker candidate Substances 0.000 description 2
- 238000007405 data analysis Methods 0.000 description 2
- 239000003623 enhancer Substances 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 238000012417 linear regression Methods 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 230000000116 mitigating effect Effects 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 229940113082 thymine Drugs 0.000 description 2
- 229930024421 Adenine Natural products 0.000 description 1
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 1
- 108700009124 Transcription Initiation Site Proteins 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 238000013145 classification model Methods 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000013479 data entry Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 210000003494 hepatocyte Anatomy 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000011528 liquid biopsy Methods 0.000 description 1
- 239000004973 liquid crystal related substance Substances 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 230000002906 microbiologic effect Effects 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 238000011524 similarity measure Methods 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12M—APPARATUS FOR ENZYMOLOGY OR MICROBIOLOGY; APPARATUS FOR CULTURING MICROORGANISMS FOR PRODUCING BIOMASS, FOR GROWING CELLS OR FOR OBTAINING FERMENTATION OR METABOLIC PRODUCTS, i.e. BIOREACTORS OR FERMENTERS
- C12M1/00—Apparatus for enzymology or microbiology
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12M—APPARATUS FOR ENZYMOLOGY OR MICROBIOLOGY; APPARATUS FOR CULTURING MICROORGANISMS FOR PRODUCING BIOMASS, FOR GROWING CELLS OR FOR OBTAINING FERMENTATION OR METABOLIC PRODUCTS, i.e. BIOREACTORS OR FERMENTERS
- C12M1/00—Apparatus for enzymology or microbiology
- C12M1/34—Measuring or testing with condition measuring or sensing means, e.g. colony counters
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6869—Methods for sequencing
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
-
- G—PHYSICS
- G06—COMPUTING; CALCULATING OR COUNTING
- G06N—COMPUTING ARRANGEMENTS BASED ON SPECIFIC COMPUTATIONAL MODELS
- G06N20/00—Machine learning
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B30/00—ICT specially adapted for sequence analysis involving nucleotides or amino acids
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B40/00—ICT specially adapted for biostatistics; ICT specially adapted for bioinformatics-related machine learning or data mining, e.g. knowledge discovery or pattern finding
Definitions
- the present invention relates to a technique for measuring the value of a biomarker.
- methylation occurs in DNA (deoxyribonucleic acid). Methylation refers to a modification in which a methyl molecule chemically binds to cytosine. Cytosine (C), together with guanine (G), adenine (A), and thymine (T), constitutes four essential nucleobases that make up DNA. Any sequence of nucleobases is called a “nucleotide sequence,” and nucleotide sequences that encode important information such as proteins are called “genomic sequences" or “genes.”
- methylation is particularly common where cytosine follows guanine on the DNA strand (called "CpG sites"). Methylation status influences gene activation or repression, and the methylation status of CpG sites in certain genes forms important biomarkers for many diseases.
- data obtained from a combination of several biomarker candidate sequences is used to create a quantitative model for disease diagnosis. Therefore, it is important to measure DNA methylation as a biomarker.
- Patent Document 1 describes selecting and evaluating a biomarker set from representative biomarker data. Furthermore, Non-Patent Document 1 describes the measurement and mitigation of PCR bias (PCR: polymerase chain reaction).
- each "signal” is a gene or sequence of interest.
- the number of such sequences is very small, so the derived signal is weak. Therefore, it is possible to increase the number of sequences and amplify the signal by copying the original sequence many times.
- G1_pre the signal intensity of gene 1 before PCR
- G1_post the signal intensity after PCR
- gene 1 has some sequences with CpG as unmethylated, which are converted to another sequence containing uracil. Similarly, sequences where CpGs are methylated are not converted. This is common and seen in a mixture of liver and stomach DNA. In such a mixture, genes important to the liver may be unmethylated in hepatocytes but methylated (and thus repressed) in gastric cells.
- the strength of the pre-PCR signal and the strength of the post-PCR signal are set as G1_U_Pre and G1_U_post (if not methylated), and G1_M_Pre and G1_M_post (if methylated), and the decoded sequence is G1_M_Pre and G1_M_post.
- the present invention is particularly important in cases where simultaneous and highly accurate measurements of DNA methylation from multiple genes are required, such as in liquid biopsies.
- certain cancer cell genes are known to exhibit high methylation compared to the same genes in healthy cells.
- Problem 2 means that in such cases the measurement underestimates the true methylation ratio from the mixture of cancer and normal DNA (negative bias).
- Problems 1 and 3 further exacerbate the degree of underestimation.
- the present invention has been made in view of the above circumstances, and one form thereof provides a learning system and a learning method for learning measurement error characteristics of biomarker sequences. Further, one form of the present invention provides a determination system and determination method that determines a sequence set by reflecting learned error characteristics, and predicts measurement error characteristics of gene sequences using data obtained by the learning system or learning method. A prediction system and a prediction method are provided.
- a learning system is a learning system for learning the relationship between measurement protocol variables and resultant error characteristics of a biomarker array, the learning system comprising a processor, the processor comprising: Enter calibration data designed to ensure that appropriate data are available for the variables, use a probabilistic model to learn the characteristics of the error distribution across each measurement protocol for the variables of interest, and the probabilistic model
- the first parameter is initialized with appropriately selected a priori parameters to model the error of and a third parameter initialized with appropriately selected a priori parameters to model overall PCR bias.
- a learning system is a system that learns a relationship between measurement protocol variables and resulting error characteristics of a biomarker sequence (defined as template-to-product ratio).
- a "variable of significance” is a variable that is known by laboratory experts to affect signal amplification performance;
- the PCR device is calibrated.
- the PCR temperature and the number of PCR cycles as shown in FIG. 2, which will be described later, are examples of "important variables.” If the temperature is too high, the DNA will degrade and the reactions necessary to replicate the target gene sequence will not occur.
- the same parameter may be used as the "appropriately selected a priori parameter”.
- “input of calibration data” for example, in the case of PCR temperature, it is necessary that appropriate display is possible within the temperature range used in normal PCR.
- the second parameter separately acquires counts of methylated sequences and unmethylated sequences of the gene after bisulfite conversion, and uses the acquired counts as These are parameters modeled using a multinomial distribution that allows prior variables to be determined separately for each methylated sequence and non-methylated sequence.
- the second aspect is to specify a specific aspect of the second parameter in order to deal with problem 2 mentioned above, by modeling and correcting the error of bisulfite conversion, and correcting the methylation of the biomarker sequence. It allows for evaluation.
- better a priori variables can be selected from empirical data analysis. Note that the counts obtained in the second aspect can be modeled based on factors such as the GC ratio (guanine to cytosine ratio) of the base sequence.
- the third parameter is an individual count of the counts calculated by a multinomial distribution when a plurality of sequences are simultaneously amplified using a universal primer.
- This parameter is subject to a configuration data constraint such that the sum of the values follows a Gaussian distribution.
- the third aspect is to define a specific aspect of the second parameter in order to deal with the problem 3 mentioned above.
- the sum of each count in multiple distributed counts follows a Gaussian distribution.
- the count values of individual markers are not independent, but the amplification method is such that the total value is approximately constant, so modeling using multinomial distribution as described above is suitable. .
- modeling is performed for the count value of the number of markers x 2.
- a decision system is a decision system comprising a processor, the processor inputting the nucleotide sequence and measurement protocol information of the biomarker sequence of interest for use in the multiplexed panel;
- the learned error characteristics and metadata associated with the error characteristics are input from the learning system according to any one of the third aspects, and the nucleotide sequence and measurement protocol information are input using predetermined criteria.
- the learned error characteristics, and the metadata to output a first score for the set of possible biomarker sequences, and consider the value of the first score for each set to determine the biomarker sequence. Decide on the set.
- a decision system uses the output from the system according to the first aspect to decide whether to use a biomarker sequence in a multiplex panel.
- the first score is a score derived from measurement accuracy, and is a "low error score" which has a higher value as the measurement error is smaller.
- the processor inputs a second score for each biomarker sequence to be determined, and inputs a first score for each biomarker sequence in the biomarker sequence set.
- the best subset of multiplexed panels is selected by optimizing the balance between the first score and the second score.
- the decision system according to the fifth aspect enhances the fourth aspect to enable a more balanced selection of biomarker sequences by considering the ultimate goal of the multiplex panel.
- the second score is, for example, a higher score (relevance score) as the degree of association with the disease to be predicted is greater.
- the "balance between the first score and the second score" is calculated by calculating the third score defined by the arithmetic mean or geometric mean of the first score and the second score, and Optimization can be achieved by maximizing the score.
- a prediction system is a prediction system for predicting measurement error characteristics of a gene sequence, comprising a processor, the processor comprising a nucleotide sequence and a nucleotide sequence of a biomarker sequence of interest to be used in a multiplexed panel.
- the measurement protocol information is input, the learned error characteristics and the metadata associated with the error characteristics are input from the learning system according to any one of the first to third aspects, and the error characteristics between the two gene sequences are input.
- the metric for calculating similarity measures is used to calculate the similarity between the new biomarker sequences and the biomarker sequences previously included in the calibration data, and to apply the calculated similarity to other relevant Used in combination with the input and learned error characteristics to predict error characteristics when measuring biomarker sequences not included in the calibration data.
- the prediction system according to the sixth aspect allows the learning systems according to the first to third aspects to be used for biomarker sequences that were not included in the calibration data.
- other related input means, for example, metadata corresponding to a biomarker sequence.
- the gene type is "promoter or enhancer”
- the CpG type is "island, shore, shelf”
- the CG abundance is "high, low”
- the combination can be represented as a vector "promoter, island, low”.
- the prediction system according to the seventh aspect is the sixth aspect, wherein the processor uses the predicted error characteristics to determine which biomarker sequences are most similar to those available in the calibration data that are not included in the calibration data. information on the obtained biomarker sequence is reflected in the determination of a biomarker sequence set in the determination system according to the fourth or fifth aspect.
- using the determination system according to the fourth or fifth aspect it is possible to use a biomarker sequence that is not included in the calibration data in selecting a biomarker sequence set.
- a learning method is a learning method performed by a learning system that includes a processor and learns a relationship between a measurement protocol variable and an error characteristic that occurs as a result of a biomarker array. enters calibration data designed to ensure that adequate data are available for the variables of interest (calibration data entry step) and uses a probabilistic model to estimate the error distribution across each measurement protocol for the variables of interest. After learning the characteristics (learning step), the probabilistic model uses the interaction of the first parameter initialized with appropriately selected a priori parameters and the amplification of the biomarker sequence to model the error of the bisulfite conversion.
- the second parameter was initialized with an appropriately selected a priori parameter to model the dependence and the second parameter was initialized with an appropriately selected a priori parameter to model overall PCR bias. and a third parameter.
- the eighth aspect defines a learning method corresponding to the first aspect described above.
- the second parameter separately acquires counts of methylated sequences and unmethylated sequences of the gene after bisulfite conversion, and These are parameters modeled using a multinomial distribution that allows prior variables to be determined separately for each methylated sequence and non-methylated sequence.
- the ninth aspect defines a learning method corresponding to the second aspect described above.
- the third parameter is an individual count of the counts calculated by a multinomial distribution when a plurality of sequences are simultaneously amplified using a universal primer.
- This parameter is subject to a configuration data constraint such that the sum of the values follows a Gaussian distribution.
- the tenth aspect defines a learning method corresponding to the third aspect described above.
- a determination method is a determination method performed by a determination system comprising a processor, wherein the processor determines the nucleotide sequence and measurement protocol information of a biomarker sequence of interest to be used in a multiplexed panel.
- input sequence information input step
- input learned error characteristics and metadata associated with the error characteristics obtained as a result of the learning method according to any one of the eighth to tenth aspects learning result input step
- using the input nucleotide sequences using predetermined criteria, measurement protocol information, learned error characteristics, and metadata to create a first set of possible biomarker sequences.
- a score is output (score output step), and a biomarker sequence set is determined by considering the first score value for each set (sequence set determination step).
- the eleventh aspect defines a determination method corresponding to the fourth aspect described above.
- the prediction method according to the fourteenth aspect is the thirteenth aspect, wherein the processor uses the predicted error characteristics to determine which biomarker sequences are available in the calibration data that are most similar to biomarker sequences not included in the calibration data.
- the obtained biomarker sequence is acquired (sequence acquisition step), and the information on the acquired biomarker sequence is reflected in the determination of a biomarker sequence set in the determination method according to the eleventh or twelfth aspect (information reflection step).
- the fourteenth aspect defines a prediction method corresponding to the seventh aspect described above.
- a wet experiment protocol 20 is used to obtain calibration data consisting of important measurement protocol variables such as PCR temperature and number of PCR cycles. It is necessary to create a . Preferably, this calibration data is designed such that appropriate data is available for variables of interest. Finally, the sequence measurement results are stored in a calibration data DB 30 (DB) along with protocol information (hereinafter, the database may be referred to as "DB"). Note that in FIG. 2, a part of the calibration data creation procedure is omitted for clarity.
- the present invention characterizes the measurement error of biomarker sequences by detailing the operation of probabilistic models and estimating the "template-to-product” ratio.
- “Template” refers to the initial amount of a biomarker sequence (amount before PCR amplification)
- product refers to the final amount of the same biomarker sequence after PCR amplification (amount after PCR amplification).
- the processor 110 may include a display control section, a communication control section, an output control section, etc. (not shown).
- the learning system 100 may include a display device (for example, a liquid crystal monitor) and an operation device (for example, a mouse and a keyboard) that are not shown.
- the display device can display calibration data, error distribution data, etc., and the user can perform operations necessary to execute the learning method (learning program) according to the present invention via the operation unit. I can do it.
- FIG. 3 described above shows blood sample data 11, which is any biological data including tissue samples.
- blood sample data 11 is measured using the measurement procedure that includes STEP 1 and STEP 2 described above, plus DNA sequencing. It has several influencing variables (variables of importance). Since it is necessary to obtain data from several values of such variables, the relevant variables are first identified and measurements are taken over a range of these values. For example, if the number of PCR cycles is the only variable of interest, data can be generated for the same blood sample for 5, 10, and 15 PCR cycles. This is so-called calibration data.
- the learning system 100 is tuned through a set of hyperparameters (hyperparameters 40) according to an optimization method (such as a loss function for minimization). Such tuning is done by ascertaining the final performance of the system and selecting hyperparameters that maximize it.
- the learning system 100 described above may be accompanied by a decision system 200 (decision system) and a prediction system 300 (prediction system), as shown in FIG. Adding these decision and prediction systems to the learning system 100 is a recommended option.
- the decision system 200 and the prediction system 300 the best subset of candidate biomarkers can be found by the decision system 200 using, for example, the error characteristics learned by the learning system 100 (including the learning result input step and the score (by carrying out the determination method according to the invention, including an input step, a subset selection step, etc.), thereby informing the selection criteria of the biomarker sequences (including an information reflection step, etc.) prediction system 300), can help effectively utilize the learning system 100.
- the sequence information input unit 212 (processor) of the determination system 200 inputs the nucleotide sequence of the biomarker sequence of interest and measurement protocol information (sequence information input step), and the learning result input unit 214 (processor) inputs the learned error characteristics and metadata associated with the error characteristics from the learning system 100 (learning result input step).
- the score output unit 216 (processor) independently considers the learned measurement error characteristics, and generates a score (measurement error score; first (one example of a score) can be assigned to each biomarker sequence (score output step).
- the sequence set determining unit 218 can sum up the scores (first score) from the order of each biomarker and determine whether to use the combination (biomarker sequence set) (sequence set determining step).
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Organic Chemistry (AREA)
- Biotechnology (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Physics & Mathematics (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- General Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Analytical Chemistry (AREA)
- Biophysics (AREA)
- Immunology (AREA)
- Theoretical Computer Science (AREA)
- Medical Informatics (AREA)
- Software Systems (AREA)
- Medicinal Chemistry (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Evolutionary Biology (AREA)
- Sustainable Development (AREA)
- Hematology (AREA)
- General Physics & Mathematics (AREA)
- Bioinformatics & Computational Biology (AREA)
- Artificial Intelligence (AREA)
- Computer Vision & Pattern Recognition (AREA)
- Data Mining & Analysis (AREA)
- Evolutionary Computation (AREA)
- Urology & Nephrology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Computing Systems (AREA)
- Mathematical Physics (AREA)
- Cell Biology (AREA)
Abstract
本発明の一形態は、学習システム、決定システム、及び予測システム、並びに学習方法、決定方法、及び予測方法を提供する。DNAのメチル化測定においては、バイサルファイト変換の不完全性の問題(問題1)と、いくつかの異なるバイオマーカー配列/遺伝子が一緒に増幅された場合にバイアスが生じる問題(問題2)と、非メチル化シグナルの過剰増幅の程度が、遺伝子配列そのものと、測定に用いられる化学物質とに依存するという問題(問題3)が存在する。本発明の一態様では、3つの問題が存在する中で測定誤差特性を学習し、学習した誤差特性をバイオマーカー選択基準に反映させるシステム及びそのシステムに対応する方法を提供する。問題1~3の組合せが存在する下でのDNAのメチル化に対する測定誤差特性評価の問題に取り組むことは、本発明の主要な新規性を形成する。
Description
本発明は、バイオマーカーの値を測定する技術に関する。
DNA(deoxyribonucleic acid)では、「メチル化」と呼ばれる現象が起きることが知られている。メチル化とは、メチル分子がシトシンに化学的に結合することによる修飾をいう。このシトシン(C:cytosine)は、グアニン(G:guanine)、アデニン(A:adenine)、チミン(T:thymine)と共に、DNAを構成する4つの必須核酸塩基を構成する。核酸塩基の任意の配列は「ヌクレオチド配列」と呼ばれ、タンパク質などの重要な情報をコードするヌクレオチド配列は「ゲノム配列」または「遺伝子」と呼ばれる。
ヒトでは、DNAストランド上でシトシンがグアニンに続く場所(「CpGサイト」と呼ばれる)では、メチル化がとりわけ一般的である。メチル化状態は遺伝子の活性化または抑制化に影響し、ある種の遺伝子のCpGサイトのメチル化状態は、多くの疾患の重要なバイオマーカーを形成する。通常、疾患診断の定量的モデルを作製するために、幾つかのバイオマーカー候補配列の組合せから得られたデータが用いられる。このため、バイオマーカーのDNAメチル化を測定することが重要になる。
DNAの測定過程ではデータに誤りが加わり、どんな推測/予測の信頼性にも影響を与える。バイオマーカーの選択を最適化するための従来の研究は、測定プロセスにおいてほんのわずかな誤りを想定し、利用可能なデータの予測値のみに焦点を当てている。このような方法の例としては、(Artificial Intelligence分類器の性能のような)定量モデルからの出力信号に頼って、分類のための特徴としてバイオマーカー配列を使用するかどうかを決定する特徴選択アルゴリズムが知られている。
このような従来の技術に関し、例えば特許文献1では、代表的なバイオマーカーデーターからバイオマーカーセットを選択して評価することが記載されている。また、非特許文献1には、PCRバイアス(PCR:polymerase chain reaction)の測定及び緩和について記載されている。
"Measuring and Mitigating PCR Bias in Microbiome Data"、 Justin D. Silverman他、[2022年3月22日検索]、インターネット(https://www.biorxiv.org/content/10.1101/604025v1)
次のセクションでは、バイオマーカー配列(シークエンス)の測定誤差特性を学習しようとした先行研究について詳細に議論する。これらの先行研究とそれらに関連する問題について論じ、それぞれの段階の詳細な説明を行う。
[DNAのメチル化測定]
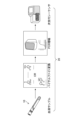
メチル化測定の概要を図1に示す。メチル化の測定では、血液サンプル10がバイサルファイト変換され、PCR装置で遺伝子/シグナルが増幅され、次世代シーケンサー等で測定される。これら一連の測定手順は、湿式実験プロトコル20(wet experiment protocol)を構成する。
メチル化測定の概要を図1に示す。メチル化の測定では、血液サンプル10がバイサルファイト変換され、PCR装置で遺伝子/シグナルが増幅され、次世代シーケンサー等で測定される。これら一連の測定手順は、湿式実験プロトコル20(wet experiment protocol)を構成する。
[STEP1:バイサルファイト変換]
Cm(メチル化シトシン)とCu(非メチル化シトシン)を区別するために、バイサルファイト変換(Bisulfite conversion)の追加ステップが使用される。バイサルファイト変換では、Cuはウラシル(U:uracil)に変換され、CmはCmのままである。変換されたサンプルがシーケンス化されると、CmはC(シトシン)として読み出され、一方、ウラシルはチミンとして読み出される。これにより、シトシンのメチル化状態を区別することが可能になる。
Cm(メチル化シトシン)とCu(非メチル化シトシン)を区別するために、バイサルファイト変換(Bisulfite conversion)の追加ステップが使用される。バイサルファイト変換では、Cuはウラシル(U:uracil)に変換され、CmはCmのままである。変換されたサンプルがシーケンス化されると、CmはC(シトシン)として読み出され、一方、ウラシルはチミンとして読み出される。これにより、シトシンのメチル化状態を区別することが可能になる。
[問題1:バイサルファイト変換における問題]
この手順の理想的な結果は、Cuが100%ウラシルに変換され、Cmは全くウラシルに変換されないこと(変換が0%であり、CmがCmのまま)である。しかし、化学反応の性質上、変換の成功(または不成功)の程度は確率論的であり、定量的な研究は困難である。このような、バイサルファイト変換の不完全性を、以下「問題1」という。
この手順の理想的な結果は、Cuが100%ウラシルに変換され、Cmは全くウラシルに変換されないこと(変換が0%であり、CmがCmのまま)である。しかし、化学反応の性質上、変換の成功(または不成功)の程度は確率論的であり、定量的な研究は困難である。このような、バイサルファイト変換の不完全性を、以下「問題1」という。
[STEP2:PCR増幅]
この段階は、測定のシグナル増幅段階と理解することができる。標準的には(つまり、メチル化のためではなく、バイサルファイト変換をしなければ)、それぞれの「信号」は興味のある遺伝子または配列である。生のデータでは、このような配列の数は非常に少ないので、派生した信号は弱い。そのため、元の配列を何度もコピーすることで、配列数を増やし、シグナルを増幅することが考えられる。例えば、PCR前の遺伝子1のシグナル強度をG1_preと呼び、PCR後のシグナル強度をG1_postと呼ぶことにする。なお、実際には、多くの遺伝子/シグナルを同時に増幅することに焦点を当てる。したがって、遺伝子2に関し、G2_preとG2_postを遺伝子1と同様に定義する。
この段階は、測定のシグナル増幅段階と理解することができる。標準的には(つまり、メチル化のためではなく、バイサルファイト変換をしなければ)、それぞれの「信号」は興味のある遺伝子または配列である。生のデータでは、このような配列の数は非常に少ないので、派生した信号は弱い。そのため、元の配列を何度もコピーすることで、配列数を増やし、シグナルを増幅することが考えられる。例えば、PCR前の遺伝子1のシグナル強度をG1_preと呼び、PCR後のシグナル強度をG1_postと呼ぶことにする。なお、実際には、多くの遺伝子/シグナルを同時に増幅することに焦点を当てる。したがって、遺伝子2に関し、G2_preとG2_postを遺伝子1と同様に定義する。
さて、まず上述のSTEP1を行うと、たった1つの遺伝子でも2つのシグナルが得られる。例えば、遺伝子1は、ウラシルを含む別の配列に変換される、非メチル化としてCpGを有するいくつかの配列を持つ。同様に、CpGがメチル化されている配列は変換されない。これは一般的であり、肝臓と胃のDNAの混合物でみられる。そのような混合物では、肝臓に重要な遺伝子が肝細胞ではメチル化されていないが、胃細胞ではメチル化されている(したがって抑制されている)可能性がある。そこで、遺伝子1に関し、PCR前信号の強さとPCR後信号の強さをG1_U_Pre およびG1_U_postとし(メチル化されていない場合)、G1_M_Pre およびG1_M_postとし(メチル化されている場合)、解読された配列をG1_M_Pre およびG1_M_post とする。
[問題2:単一バイサルファイトプロトコルにおけるPCRバイアス]
同じ遺伝子のシグナルを増幅しても、バイサルファイト変換は2つのシグナルタイプになる。したがって、G1_U_post/G1_U_pre = G1_M_post/G1_M_preは成り立たない。G1_U_pre = G1_M_preの場合であっても、増幅後は、G1_U_post/G1_U_pre > G1_M_post/G1_M_preとなる(すなわち、非メチル化遺伝子が、メチル化遺伝子に対して過剰に増幅される)ことが知られている。しかし、このような非メチル化シグナルの過剰増幅の程度は、遺伝子配列そのものと、測定に用いられる化学物質とに依存する。この問題を、以下「問題2」と呼ぶ。
同じ遺伝子のシグナルを増幅しても、バイサルファイト変換は2つのシグナルタイプになる。したがって、G1_U_post/G1_U_pre = G1_M_post/G1_M_preは成り立たない。G1_U_pre = G1_M_preの場合であっても、増幅後は、G1_U_post/G1_U_pre > G1_M_post/G1_M_preとなる(すなわち、非メチル化遺伝子が、メチル化遺伝子に対して過剰に増幅される)ことが知られている。しかし、このような非メチル化シグナルの過剰増幅の程度は、遺伝子配列そのものと、測定に用いられる化学物質とに依存する。この問題を、以下「問題2」と呼ぶ。
[問題3:PCR増幅における問題]
PCRの理想的な結果は、G1_post/G1_pre = G2_post/G2_preである。しかし実際には、ある種の遺伝子配列は他のものよりも測定しやすく、この等価性は成立しない。このような、いくつかの異なるバイオマーカー配列/遺伝子が一緒に増幅された場合に生じるバイアスを、多重化プロトコルにおける「PCRバイアス」と呼ぶ(以下、「問題3」という)。
PCRの理想的な結果は、G1_post/G1_pre = G2_post/G2_preである。しかし実際には、ある種の遺伝子配列は他のものよりも測定しやすく、この等価性は成立しない。このような、いくつかの異なるバイオマーカー配列/遺伝子が一緒に増幅された場合に生じるバイアスを、多重化プロトコルにおける「PCRバイアス」と呼ぶ(以下、「問題3」という)。
[従来の技術における対応]
上述した問題1~3に対する従来技術での対応を説明する。従来技術では、問題1に関し、定量的な研究にはしばしば極端な正確さが必要とされず、したがって、バイサルファイト変換の成功の程度を考慮していなかった。また、問題3に関し、これまでの微生物学の研究では、PCRの効果を掛け算的に考えていた。すなわち、従来の技術では、もし1回のPCRサイクル後の遺伝子1のシグナル強度がjであれば、2回のサイクル後のシグナル強度はj2であり、x回のサイクル後のシグナル強度は同様にjxであると考えていた。この仮定を用いて、PCRは、多項ロジスティック‐通常線形モデルを用いた対数線形過程としてモデル化された。「バッチ効果」(バッチごとにわずかに異なるバイアス特性を示す標本に対するPCR)などの他の共変量も確率論的な方法で含まれた。モデルは、生成された較正データを「訓練」した後、PCRバイアスの補正に使用される。
上述した問題1~3に対する従来技術での対応を説明する。従来技術では、問題1に関し、定量的な研究にはしばしば極端な正確さが必要とされず、したがって、バイサルファイト変換の成功の程度を考慮していなかった。また、問題3に関し、これまでの微生物学の研究では、PCRの効果を掛け算的に考えていた。すなわち、従来の技術では、もし1回のPCRサイクル後の遺伝子1のシグナル強度がjであれば、2回のサイクル後のシグナル強度はj2であり、x回のサイクル後のシグナル強度は同様にjxであると考えていた。この仮定を用いて、PCRは、多項ロジスティック‐通常線形モデルを用いた対数線形過程としてモデル化された。「バッチ効果」(バッチごとにわずかに異なるバイアス特性を示す標本に対するPCR)などの他の共変量も確率論的な方法で含まれた。モデルは、生成された較正データを「訓練」した後、PCRバイアスの補正に使用される。
また、問題2に関しては、単一プロトコル設定における測定誤差とバイアスの特徴付けはより簡単であるため、一部のPCRデータでは、バイアスの度合いを見出すために線形回帰を行っている。線形回帰推定量を計算した後、この方程式を用いてこのようなバイアスを補正することができる。
DNAメチル化の正確な測定の重要性はすでに述べた。病気診断のような応用分野では、複数のバイオマーカー配列のデータを用い、定量モデルに入力することは珍しいことではない。複数のバイオマーカーのメトリクス値を同時に測定する測定プロセスを設計する際は、問題1、問題2、問題3が組み合わされてしまい、これらの全てが問題となる。このため、誤差の定量化と学習誤差の特性が非常に困難になる。本発明では、3つの問題が存在する中で測定誤差特性を学習し、学習した誤差特性をバイオマーカー選択基準に反映させるシステムを検討する。この問題の組合せが存在する下でのDNAのメチル化に対する測定誤差特性評価の問題に取り組むことは、本発明の主要な新規性を形成する。
本発明は、液体生命学(リキッドバイオプシー)のように、複数の遺伝子からのDNAメチル化の同時的で非常に正確な測定が必要な場合に、特に重要となる。特に、癌のような疾患の正確な同定のために、ある種の癌細胞遺伝子は、健康な細胞における同じ遺伝子と比較して高いメチル化を示すことが知られている。問題2は、そのような場合、測定が、がんと正常なDNAの混合から真のメチル化比を過小評価する(負のバイアス)ことを意味する。問題1と問題3は、過小評価の度合いをさらに悪化させる。
本発明は上記事情に鑑みてなされたもので、その一形態は、バイオマーカー配列の測定誤差特性を学習する学習システム及び学習方法を提供する。また、本発明の一形態は、学習した誤差特性を反映して配列セットを決定する決定システム及び決定方法、並びに学習システムあるいは学習方法により得られたデータを用いて遺伝子配列の測定誤差特性を予測する予測システム及び予測方法を提供する。
本発明の第1の態様に係る学習システムは、測定プロトコル変数と、バイオマーカー配列の結果として生じる誤差特性との関係を学習する学習システムであって、プロセッサを備え、プロセッサは、重要性のある変数について適切なデータが入手できるように設計された較正データを入力し、確率モデルを用いて、重要性のある変数について各測定プロトコルにわたる誤差分布の特性を学習し、確率モデルは、バイサルファイト変換の誤差をモデル化するために、適切に選択された事前パラメータで初期化された第1のパラメータと、バイオマーカー配列の増幅の相互依存性をモデル化するために、適切に選択された事前パラメータで初期化された第2のパラメータと、PCR全体のバイアスをモデル化するために、適切に選択された事前パラメータで初期化された第3のパラメータと、を含む。第1の態様に係る学習システムは、測定プロトコル変数とバイオマーカー配列の結果として生じる誤差特性の間の関係(テンプレート対プロダクト比と定義される)を学習するシステムである。
第1の態様及び以下の各態様において、「重要性のある変数」とは、信号増幅性能に影響を与えることが実験室の専門家によって知られている変数であり、そのような変数について、PCR装置が調整される。例えば、後述する図2に示すようなPCR温度やPCRサイクル数は「重要性のある変数」の一例である。温度が高すぎると、DNAが分解され、標的遺伝子配列を複製するために必要な反応が起こらない。また、第1~第3のパラメータに関し、「適切に選択された事前パラメータ」として同じパラメータを用いてよい。また、「較正データ(キャリブレーションデータ)の入力」に関し、例えばPCR温度の場合、通常のPCRで使用される温度の範囲で適切な表示が可能であることを要する。
第2の態様に係る学習システムは第1の態様において、第2のパラメータは、バイサルファイト変換後の遺伝子のメチル化配列及び非メチル化配列のカウントを別々に取得し、取得されたカウントを、メチル化配列及び非メチル化配列の各配列について、事前変数を別々に決定できる多項分布でモデル化したパラメータである。第2の態様は、上述した問題2に対応するための第2のパラメータの具体的態様を規定するもので、バイサルファイト変換の誤差をモデル化及び修正して、バイオマーカー配列のメチル化を正しく評価できるようにするものである。第2の態様では、経験的なデータ分析から、より優れた事前変数を選択することができる。なお、第2の態様において取得されたカウントは、塩基配列のGC比(グアニンとシトシンの比)のような要因に基づいてモデル化することができる。
第3の態様に係る学習システムは第1または第2の態様において、第3のパラメータは、ユニバーサルプライマーを用いて複数の配列を同時に増幅する場合に、多項分布で計算されたカウントの個々のカウントの合計がガウス分布に従う、という構成データ制約が課されたパラメータである。第3の態様は、上述した問題3に対応するための第2のパラメータの具体的態様を規定するもので、複数のバイオマーカーの数が多く、構成データ制約が計算可能になるようにモデリングパラメータを単純化する場合、複数の分散カウントにおける各カウントの合計はガウス分布に従う。また、複数の配列を同時に増幅する場合、個々のマーカーのカウント値が独立ではなく、合計値がほぼ一定になるような増幅の仕方をするため、上記のような多項分布によるモデリングが適している。さらに、バイサルファイト変換を伴うメチル化計測においては、各マーカーがメチル化、非メチル化の2状態があるため、マーカー数×2のカウント値に対するモデリングになる。
本発明の第4の態様に係る決定システムは、プロセッサを備える決定システムであって、プロセッサは、多重化パネルで使用する、関心バイオマーカー配列のヌクレオチド配列及び測定プロトコル情報を入力し、第1から第3の態様のいずれか1つに係る学習システムから、学習された誤差特性、及び誤差特性に関連付けられたメタデータを入力し、あらかじめ決められた基準を用いて入力したヌクレオチド配列、測定プロトコル情報、学習された誤差特性、及びメタデータを使用して、可能なバイオマーカー配列のセットのための第1のスコアを出力し、各セットについての第1のスコアの値を考慮してバイオマーカー配列セットを決定する。第4の態様に係る決定システムでは、多重パネルでバイオマーカー配列を使用するかどうかを決定するために、第1の態様に係るシステムからの出力を使用する。第1のスコアは測定精度に由来するスコアであり、測定誤差が小さいほど高い値となる「低誤差スコア」である。
第5の態様に係る決定システムは第4の態様において、プロセッサは、決定すべきバイオマーカー配列ごとに、第2のスコアを入力し、バイオマーカー配列セットにおける各バイオマーカー配列についての第1のスコアを考慮して、第1のスコアと第2のスコアとのバランスを最適化することにより多重化パネルのベストなサブセットを選択する。第5の態様に係る決定システムでは、マルチプレックスパネルの最終目標を考慮することにより、バイオマーカー配列の、よりバランスの取れた選択を可能にするために、第4の態様を増強する。第2のスコアは、たとえば予測したい疾患との関連度が大きいほど高いスコア(関連度スコア)である。また、「第1のスコアと第2のスコアのバランス」は、例えば第1のスコアと第2のスコアの相加平均や相乗平均で規定される第3のスコアを算出し、その第3のスコアを最大化することにより、最適化することができる。
本発明の第6の態様に係る予測システムは、遺伝子配列の測定誤差特性を予測する予測システムであって、プロセッサを備え、プロセッサは、多重化パネルで使用する、関心バイオマーカー配列のヌクレオチド配列及び測定プロトコル情報を入力し、第1から第3の態様のいずれか1つに係る学習システムから、学習された誤差特性、及び誤差特性に関連付けられたメタデータを入力し、2つの遺伝子配列間の類似性の尺度を計算するための測定基準を用いて、以前に較正データに含まれていたバイオマーカー配列と新たなバイオマーカー配列との類似度を計算し、計算した類似度を他の関連する入力及び学習された誤差特性と組み合わせて使用して、較正データに含まれていないバイオマーカー配列を測定する際の誤差特性を予測する。第6の態様に係る予測システムは、第1~第3の態様に係る学習システムを、較正データに含まれていなかったバイオマーカー配列に使用することを可能にする。
なお、第6の態様において「他の関連する入力」とは、例えばバイオマーカー配列に対応するメタデータを意味する。例えば、遺伝子タイプが「プロモーターもしくはエンハンサー」であり、CpGタイプが「アイランド、ショア、シェルフ」であり、CGの豊富さが「高、低」であれば、あるバイオマーカー配列G1についてのこれらの情報の組み合わせ(メタデータの一例)は、「プロモーター、アイランド、低」というベクトルとして表すことができる。
第7の態様に係る予測システムは第6の態様において、プロセッサは、予測された誤差特性を使用して、較正データに含まれていないバイオマーカー配列と最も類似する、較正データにおいて利用可能であったバイオマーカー配列を取得し、取得したバイオマーカー配列の情報を、第4または第5の態様に係る決定システムにおけるバイオマーカー配列セットの決定に反映する。第7の態様では、第4または第5の態様に係る決定システムを用いて、バイオマーカー配列セット選択において、較正データに含まれていないバイオマーカー配列を使用できるようにする。
本発明の第8の態様に係る学習方法は、プロセッサを備え、測定プロトコル変数と、バイオマーカー配列の結果として生じる誤差特性との関係を学習する学習システムにより実行される学習方法であって、プロセッサが、重要性のある変数について適切なデータが入手できるように設計された較正データを入力し(較正データ入力ステップ)、確率モデルを用いて、重要性のある変数について各測定プロトコルにわたる誤差分布の特性を学習し(学習ステップ)、確率モデルは、バイサルファイト変換の誤差をモデル化するために、適切に選択された事前パラメータで初期化された第1のパラメータと、バイオマーカー配列の増幅の相互依存性をモデル化するために、適切に選択された事前パラメータで初期化された第2のパラメータと、PCR全体のバイアスをモデル化するために、適切に選択された事前パラメータで初期化された第3のパラメータと、を含む。第8の態様は、上述した第1の態様に対応する学習方法を規定するものである。
第9の態様に係る学習方法は第8の態様において、第2のパラメータは、バイサルファイト変換後の遺伝子のメチル化配列及び非メチル化配列のカウントを別々に取得し、取得されたカウントを、メチル化配列及び非メチル化配列の各配列について、事前変数を別々に決定できる多項分布でモデル化したパラメータである。第9の態様は、上述した第2の態様に対応する学習方法を規定するものである。
第10の態様に係る学習方法は第8または第9の態様において、第3のパラメータは、ユニバーサルプライマーを用いて複数の配列を同時に増幅する場合に、多項分布で計算されたカウントの個々のカウントの合計がガウス分布に従う、という構成データ制約が課されたパラメータである。第10の態様は、上述した第3の態様に対応する学習方法を規定するものである。
本発明の第11の態様に係る決定方法は、プロセッサを備える決定システムにより実行される決定方法であって、プロセッサは、多重化パネルで使用する、関心バイオマーカー配列のヌクレオチド配列及び測定プロトコル情報を入力し(配列情報入力ステップ)、第8から第10の態様のいずれか1つに係る学習方法の結果として得られる、学習された誤差特性、及び誤差特性に関連付けられたメタデータを入力し(学習結果入力ステップ)、あらかじめ決められた基準を用いて入力したヌクレオチド配列、測定プロトコル情報、学習された誤差特性、及びメタデータを使用して、可能なバイオマーカー配列のセットのための第1のスコアを出力し(スコア出力ステップ)、各セットについての第1のスコアの値を考慮してバイオマーカー配列セットを決定する(配列セット決定ステップ)。第11の態様は、上述した第4の態様に対応する決定方法を規定するものである。
第12の態様に係る決定方法は第11の態様において、プロセッサは、決定すべきバイオマーカー配列ごとに、第2のスコアを入力し(スコア入力ステップ)、バイオマーカー配列セットにおける各バイオマーカー配列についての第1のスコアを考慮して、第1のスコアと第2のスコアとのバランスを最適化することにより多重化パネルのベストなサブセットを選択する(サブセット選択ステップ)。第12の態様は、上述した第5の態様に対応する決定方法を規定するものである。
本発明の第13の態様に係る予測方法は、プロセッサを備え、遺伝子配列の測定誤差特性を予測する予測システムにより実行される予測方法であって、プロセッサは、多重化パネルで使用する、関心バイオマーカー配列のヌクレオチド配列及び測定プロトコル情報を入力し(配列情報入力ステップ)、第8から第10の態様のいずれか1つに係る学習方法により得られた、学習された誤差特性、及び誤差特性に関連付けられたメタデータを入力し(学習結果入力ステップ)、2つの遺伝子配列間の類似性の尺度を計算するための測定基準を用いて、以前に較正データに含まれていたバイオマーカー配列と新たなバイオマーカー配列との類似度を計算し(類似度計算ステップ)、計算した類似度を他の関連する入力及び学習された誤差特性と組み合わせて使用して、較正データに含まれていないバイオマーカー配列を測定する際の誤差特性を予測する(誤差特性予測ステップ)。第13の態様は、上述した第6の態様に対応する予測方法を規定するものである。
第14の態様に係る予測方法は第13の態様において、プロセッサは、予測された誤差特性を使用して、較正データに含まれていないバイオマーカー配列と最も類似する、較正データにおいて利用可能であったバイオマーカー配列を取得し(配列取得ステップ)、取得したバイオマーカー配列の情報を、第11または第12の態様に係る決定方法におけるバイオマーカー配列セットの決定に反映する(情報反映ステップ)。第14の態様は、上述した第7の態様に対応する予測方法を規定するものである。
なお、上述した態様の学習方法、決定方法、及び予測方法をプロセッサに実行させるプログラム(学習プログラム、決定プログラム、予測プログラム)、及びそれらプログラムのコンピュータ読み取り可能なコードを記録した非一時的記録媒体も、本発明の範囲に含まれる。
以上説明したように、本発明に係る学習システム、決定システム、及び予測システム、並びに学習方法、決定方法、及び予測方法は、以下の効果を有する。
(1)複数の遺伝子配列を一緒に測定して多重化されたパネルを扱うことができる。
(2)バイサルファイト変換されたサンプルを処理することができる。
(3)配列パラメータとプロトコルパラメータを入力として使用して、測定誤差を予測することができる。
(4)配列を分析/分類の目的に使用するかどうかを決定することができる。
(1)複数の遺伝子配列を一緒に測定して多重化されたパネルを扱うことができる。
(2)バイサルファイト変換されたサンプルを処理することができる。
(3)配列パラメータとプロトコルパラメータを入力として使用して、測定誤差を予測することができる。
(4)配列を分析/分類の目的に使用するかどうかを決定することができる。
以下、本発明の実施形態を説明する。説明においては、必要に応じて添付図面が参照される。なお、添付図面において、説明の便宜上一部の構成要素の記載を省略する場合がある。
[較正データの作成]
本発明においては、図2に示すように、まず、血液サンプル10から、湿式実験プロトコル20(wet experiment protocol)により、PCR温度やPCRサイクル数のような重要な測定プロトコル変数によって構成される較正データを作成することを必要とする。この較正データは、重要性のある変数について適切なデータが入手できるように設計されていることが好ましい。最終的には、配列の測定結果を、プロトコル情報と共に較正データDB30(DB:database)に保存する(以下では、データベースを「DB」と記載する場合がある)。なお、図2では、較正データ作成手順の一部を、明確化のため省略した。
本発明においては、図2に示すように、まず、血液サンプル10から、湿式実験プロトコル20(wet experiment protocol)により、PCR温度やPCRサイクル数のような重要な測定プロトコル変数によって構成される較正データを作成することを必要とする。この較正データは、重要性のある変数について適切なデータが入手できるように設計されていることが好ましい。最終的には、配列の測定結果を、プロトコル情報と共に較正データDB30(DB:database)に保存する(以下では、データベースを「DB」と記載する場合がある)。なお、図2では、較正データ作成手順の一部を、明確化のため省略した。
学習アルゴリズム(学習システム、学習方法)は、このようなプロトコル変数とその測定特性との間の関係を学習するために、この較正データを使用する。次いで、所与の測定プロトコル変数のセット(較正データには含まれていない)に対して、このシステム(予測システム、予測方法)は、所与のバイオマーカー配列の測定誤差特性を予測することができる。この予測を用いて、システム(決定システム、決定方法)はバイオマーカー配列が何らかの定量的研究に使用するのに適しているかどうかを決定することができる。最後に、較正データに存在しないバイオマーカー配列でさえ、本発明のシステム(決定システム、決定方法)は、測定誤差特性が知られている最も類似した配列を見つけ出し、それを用いて新しい配列について類似した決定を行うことができる。
具体的には、本発明は、確率モデルの作用を詳述し、「テンプレート対プロダクト」比を推定することによって、バイオマーカー配列の測定誤差を特徴付ける。「テンプレート」はバイオマーカー配列の最初の量(PCR増幅前の量)を指し、「プロダクト」はPCR増幅後の同じバイオマーカー配列の最終量(PCR増幅後の量)を指す。
[学習システムの構成]
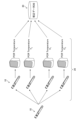
図3は、本発明の一態様に係る学習システム100及びこれに関連するデータ等を示している。多重バイサルファイトPCRプロトコルのためのDNAメチル化測定誤差特性を学習するための、このような学習システムの適用は、本発明の新規性を保証するための最小限の要件である。なお、後述するように、学習システム100には、その結果(学習済み誤差分布DB50)を利用する決定システム200(決定システム)及び予測システム300(予測システム)が付随していてもよい。
図3は、本発明の一態様に係る学習システム100及びこれに関連するデータ等を示している。多重バイサルファイトPCRプロトコルのためのDNAメチル化測定誤差特性を学習するための、このような学習システムの適用は、本発明の新規性を保証するための最小限の要件である。なお、後述するように、学習システム100には、その結果(学習済み誤差分布DB50)を利用する決定システム200(決定システム)及び予測システム300(予測システム)が付随していてもよい。
図4は、学習システム100の構成例を示す図である。図4に示すように、学習システム100は、プロセッサ110(プロセッサ、コンピュータ)、確率モデル120(確率モデル)、記憶部130、ROM140(ROM:Read Only Memory)、RAM150(RAM:Random Access Memory)を備える。プロセッサ110は学習システム100の各部が行う処理の統括制御を行うもので、較正データ入力部112及び学習部114を有する。
プロセッサ110は、図4に示す要素の他に、不図示の表示制御部や通信制御部、出力制御部等を含んでいてよい。
プロセッサ110は、例えば、CPU(Central Processing Unit)、GPU(Graphics Processing Unit)、FPGA(Field Programmable Gate Array)、PLD(Programmable Logic Device)等の各種のプロセッサや電気回路で構成される。これらのプロセッサや電気回路がソフトウェア(プログラム)を実行する際は、実行するソフトウェアのコンピュータ(例えば、プロセッサを構成する各種のプロセッサや電気回路、及び/またはそれらの組み合わせ)で読み取り可能なコードをROM140等の非一時的かつ有体の記録媒体に記憶しておき、コンピュータがそのソフトウェアを参照する。非一時的かつ有体の記録媒体に記憶しておくソフトウェアは、本発明に係る学習方法、予測方法、決定方法を実行するためのプログラム(学習プログラム、予測プログラム、決定プログラム)、及び実行に際して用いられるデータを含む。ROM140ではなく各種の光磁気記録装置、半導体メモリ等の非一時的かつ有体の記録媒体にコードを記録してもよい。ソフトウェアを用いた処理の際には例えばRAM150が一時的記憶領域として用いられ、また例えば不図示のEEPROM(Electronically Erasable and Programmable Read Only Memory)やフラッシュメモリ等の非一時的かつ有体の記録媒体に記憶されたデータを参照することもできる。「非一時的かつ有体の記録媒体」として記憶部130を用いてもよい。
記憶部130はハードディスク、半導体メモリ等の各種記憶デバイス及びその制御部により構成され、上述した較正データや、学習方法の実行条件及び実行結果(学習済み誤差分布のデータ)等を記憶することができる。
学習システム100は、図4に示す要素の他に、不図示の表示装置(例えば、液晶モニタ)や操作装置(例えば、マウスやキーボード)を含んでいてよい。表示装置には、較正データや誤差分布のデータ等を表示することができ、また、ユーザは、操作部を介して、本発明に係る学習方法(学習プログラム)の実行に必要な操作を行うことができる。
上述した図3は、血液サンプルデータ11を示しているが、これは組織サンプルを含むあらゆる生物学的データである。血液サンプルデータ11は、図1のように、上述したSTEP 1,STEP 2,それにDNA配列決定を加えた測定手順で測定するものであり、それ自体が、PCRのサイクル数など、その有効性に影響を与える変数(重要性のある変数)をいくつか持っている。このような変数のいくつかの値からデータを得る必要があるため、関連する変数が最初に識別され、これらの値の範囲で測定が行われる。例えば、もしPCRサイクル数が唯一の重要な変数である場合、5,10および15 PCRサイクルの同じ血液サンプルのデータを生成することができる。これがいわゆる較正データである。
[確率モデル]
学習システム100は、較正データDB30に記憶された較正データ(訓練用データ)を利用して(較正データ入力ステップ)、確率モデルをトレーニングする(学習ステップ)。図5はそのような確率モデルの一例である確率モデル120を、ベイズ階層モデルを通して示している。本発明の重要な新規性は、(i)バイサルファイト変換誤差の事前情報(事前パラメータ;以下同じ)、(ii)バイサルファイト変換の共変量の事前情報、および(iii)バイオマーカー配列の増幅の相互依存性の事前情報を使用することにある。これらの事前情報(i)~(iii)は、本発明の第1~第3のパラメータに対応し、従って上述の問題1~3に対応する。以上の3つの要素を総合すると、本発明は、上述した特許文献1や非特許文献1のような従来のモデルとは異なるものとなっている。
学習システム100は、較正データDB30に記憶された較正データ(訓練用データ)を利用して(較正データ入力ステップ)、確率モデルをトレーニングする(学習ステップ)。図5はそのような確率モデルの一例である確率モデル120を、ベイズ階層モデルを通して示している。本発明の重要な新規性は、(i)バイサルファイト変換誤差の事前情報(事前パラメータ;以下同じ)、(ii)バイサルファイト変換の共変量の事前情報、および(iii)バイオマーカー配列の増幅の相互依存性の事前情報を使用することにある。これらの事前情報(i)~(iii)は、本発明の第1~第3のパラメータに対応し、従って上述の問題1~3に対応する。以上の3つの要素を総合すると、本発明は、上述した特許文献1や非特許文献1のような従来のモデルとは異なるものとなっている。
また、これら3つの要因により、上述した問題1+問題2+問題3が一体となった問題を解決することができる。学習システム100は、最適化方法(最小化のための損失関数など)に従い、一連のハイパーパラメータ(ハイパーパラメータ40)を通して調整される。このような調整は、システムの最終性能を確認し、それを最大化するハイパーパラメータを選択することによって行われる。
なお、上述した第1~第3のパラメータは確率モデル120の一部(したがって、学習システム100の一部)であり、それらパラメータの値は訓練プロセス中に更新される。また、第1~第3のパラメータは学習システム100の一部であるため、図3では表示されていない。
一方、ハイパーパラメータを使用すると、確率モデル120のある側面を制御できる。ただし、ハイパーパラメータの値はユーザが設定するものであり、訓練プロセス中に値が更新されることはない。また、学習システム100と決定システム200(図6を参照)とでは、ハイパーパラメータが異なっている。
より具体的には、二項分布のバイオマーカーについて、バイサルファイト変換の誤差をモデル化することを選択することができる。そこで、バイサルファイト変換誤差の事前確率(事前パラメータの一例)を[0,1]の間の値として選ぶことができる。事前確率が0の場合、そのバイオマーカーの完全な変換(Cuとウラシルとの100%の変換と、Cmの0%の変換)を想定し、事前確率が0より大きい場合はそのバイオマーカーの不完全な変換(Cuの一部のみがウラシルに変換され、Cmの一部もウラシルに変換される)を想定する。理想的には、事前変数は経験的データ分析から設定されるべきである。バイサルファイト共変量には、ナノグラムで測定したサンプル中に加えられた亜硫酸塩の量と初期DNA量が含まれる。このようにして、事前確率で初期化されたバイサルファイト変換誤差が、第1のパラメータである。
同様に、PCR誤差分布は多項分布でモデル化することができ、適切な事前確率を設定することができる。この段階では、PCR後の配列カウント(配列の数)は、選択されたバイオマーカーの数をxとした場合にN1、N2、...、Nxとして表すことができ、その配列カウントは多項分布としてモデル化することができる。ここで、“Ni”は、i番目のバイオマーカーの配列カウントである。PCR共変量には、PCR温度やPCRサイクル数のような、較正データ作成のために選択された要因が含まれることがある。
本発明の新規性は、同じ配列から2つの異なるカウントの可能性を考える能力にある。1つは、バイサルファイト変換後のある配列の塩基化のカウント(メチル化配列のカウント)であり、もう1つは、その配列の非塩基化タイプのカウント(非メチル化配列のカウント)である。これにより、N1_M、N1_U、N2_M、N2_U などの可能性の数が2倍になることが考慮される。ここで“Ni_M”は、i番目のバイオマーカーについてのメチル化配列のカウントを示し、“Ni_U”は、i番目のバイオマーカーについての、非メチル化配列のカウントを示す。Nx_M とNx_U は、互いに自然な制約を課すため(一方の平均の回数が多いことは他方の回数が少ないことを意味する)、このような制約(相互依存性)を用いてモデル化問題を単純化することができる。このようにして、事前確率で初期化されたバイオマーカー配列の増幅の相互依存性が、第2のパラメータである。
最後に、全体分布モデル(PCR全体のバイアスを示すモデル)は、すべてのバイオマーカー、すなわち、N1+N2+....+、Nxの総数の配列を数量化することであり、バイオマーカーカウントを通して相互依存の制約(構成データ制約)を課すためにさえ使用され得る(例えば、N1が高すぎる場合、N3は低すぎる)。N1、N2等の各々(個々のカウント)は多項分布であるため、選択したバイオマーカーの数が多い(例えば、30以上)条件下では、それらの合計(多項分布で計算されたカウントの個々のカウントの合計)は、中心極限定理を満足するために、ガウス分布に従うと考えられる。配列タイプ間のこのような相互依存性(構成データ制約)は、すぐには明らかではないかもしれないが、ユニバーサルプライマーを用いて複数の配列(複数のバイオマーカー配列)を同時に増幅する場合には、このような相互依存性が存在することが知られている。このようにして、事前確率で初期化されたPCR全体のバイアスが、第3のパラメータである。
このようなユニバーサルプライマーの使用は、適切なアダプタ配列が標的バイオマーカー配列の両端に配備された後にのみ可能である。この段階で追加されたユニバーサルプライマーの有限な量は、バイオマーカー配列間の組成依存性を作り出し、純シグナル増幅に影響する。PCR増幅中の多重化パネルに構成データ制約を課し、ユニバーサルプライマーを用いて相対的なバイオマーカー配列の豊富さをモデリングすることは、本発明の第二の新規性を形成する。
[決定システム及び予測システムの位置づけ]
上述した学習システム100には、図6に示すように、決定システム200(決定システム)及び予測システム300(予測システム)が付随していてもよい。これら決定システム及び予測システムを学習システム100に付加することは、選択肢として推奨されるものである。決定システム200及び予測システム300を付加することで、例えば学習システム100により学習された誤差特性を用いて、決定システム200により候補バイオマーカーのベストなサブセットを見つけることができ(学習結果入力ステップやスコア入力ステップ、サブセット選択ステップ等を含む、本発明に係る決定方法の実行による)、これによりバイオマーカー配列の選択基準に情報を与えて(情報反映ステップ等を含む、本発明に係る予測方法の実行による;予測システム300)、学習システム100の効果的な活用を助けることができる。
上述した学習システム100には、図6に示すように、決定システム200(決定システム)及び予測システム300(予測システム)が付随していてもよい。これら決定システム及び予測システムを学習システム100に付加することは、選択肢として推奨されるものである。決定システム200及び予測システム300を付加することで、例えば学習システム100により学習された誤差特性を用いて、決定システム200により候補バイオマーカーのベストなサブセットを見つけることができ(学習結果入力ステップやスコア入力ステップ、サブセット選択ステップ等を含む、本発明に係る決定方法の実行による)、これによりバイオマーカー配列の選択基準に情報を与えて(情報反映ステップ等を含む、本発明に係る予測方法の実行による;予測システム300)、学習システム100の効果的な活用を助けることができる。
上述した学習システム100は、最適化基準を統計的手段によって最大化または最小化することによって学習する確率モデル120を備えており、このようにして学習することは、アルゴリズムを「トレーニング」することの意味を広くカバーしている。一方、決定システム200は、学習システム100がトレーニングを終了した後に機能する。決定システム200自体には、最大化または最小化しようとする定義済みの最適化基準がないため、「トレーニング」されておらず、システムは学習されない。ただし、決定システム200は、システムを「調整可能」にするハイパーパラメータを含んで構成されている。
[決定システム及び予測システムの構成]
図7は、決定システム200の構成を示す図である。同図に示すように、決定システム200は、プロセッサ210(プロセッサ)と、ROM230(非一時的かつ有体の記録媒体)と、RAM240とを備える。プロセッサ210は、配列情報入力部212と、学習結果入力部214と、スコア出力部216と、配列セット決定部218と、を備える。決定システム200は、これら要素の他に、図示せぬ表示制御部や表示装置、記憶装置、操作部等を有していてよい。
図7は、決定システム200の構成を示す図である。同図に示すように、決定システム200は、プロセッサ210(プロセッサ)と、ROM230(非一時的かつ有体の記録媒体)と、RAM240とを備える。プロセッサ210は、配列情報入力部212と、学習結果入力部214と、スコア出力部216と、配列セット決定部218と、を備える。決定システム200は、これら要素の他に、図示せぬ表示制御部や表示装置、記憶装置、操作部等を有していてよい。
図8は、予測システム300の構成を示す図である。同図に示すように、予測システム300は、プロセッサ310(プロセッサ)と、ROM330(非一時的かつ有体の記録媒体)と、RAM340とを備える。プロセッサ310は、配列情報入力部312と、学習結果入力部314と、類似度計算部316と、誤差特性予測部318と、配列情報反映部320と、を備える。予測システム300は、これら要素の他に、図示せぬ表示制御部や表示装置、記憶装置、操作部等を有していてよい。
決定システム200及び予測システム300のこれらの要素は、学習システム100と同様に、例えば、CPU、GPU、FPGA、PLD等の各種のプロセッサや電気回路で構成される。これらのプロセッサや電気回路がソフトウェア(プログラム)を実行する際は、実行するソフトウェアのコンピュータで読み取り可能なコードをROM230やROM330等の非一時的かつ有体の記録媒体に記憶しておき、コンピュータがそのソフトウェアを参照する。非一時的かつ有体の記録媒体に記憶しておくソフトウェアは、本発明に係る予測方法、決定方法を実行するためのプログラム(予測プログラム、決定プログラム)、及び実行に際して用いられるデータを含む。ROM230やROM330ではなく、各種の光磁気記録装置、半導体メモリ等の非一時的かつ有体の記録媒体にコードを記録してもよい。ソフトウェアを用いた処理の際には例えばRAM240,RAM340が一時的記憶領域として用いられ、また例えば不図示のEEPROMやフラッシュメモリ等の非一時的かつ有体の記録媒体に記憶されたデータを参照することもできる。
[スコアに基づくバイオマーカー配列セットの決定]
以下では、「スコアの最適化プロセス(最適化手法)」の2つの大まかな分類である、バイナリーベースのアプローチと、組み合わせベースのアプローチについて説明する。
以下では、「スコアの最適化プロセス(最適化手法)」の2つの大まかな分類である、バイナリーベースのアプローチと、組み合わせベースのアプローチについて説明する。
バイナリーベースの最適化基準では、決定システム200の配列情報入力部212(プロセッサ)が関心バイオマーカー配列のヌクレオチド配列及び測定プロトコル情報を入力し(配列情報入力ステップ)、学習結果入力部214(プロセッサ)が、学習システム100から、学習された誤差特性、及び誤差特性に関連付けられたメタデータを入力する(学習結果入力ステップ)。スコア出力部216(プロセッサ)は、学習した測定誤差特性を独立に考慮し、結果として生じる測定誤差グラフの傾きに基づいて、例えば{+1,0,-1}のスコア(測定誤差スコア;第1のスコアの一例)を各バイオマーカー配列に割当てることができる(スコア出力ステップ)。配列セット決定部218は、各バイオマーカーの順序からスコア(第1のスコア)を合計し、その組み合わせ(バイオマーカー配列セット)を使うかどうかを決定することができる(配列セット決定ステップ)。
これを、よりロバストに実装すると、特徴選択アルゴリズムと同様の方法で、組み合わせベースの最適化基準を設計することができる。特徴選択アルゴリズムは、定量モデルの出力に依存し、定量モデルの性能を最適化するためにそれらの基準を更新する。この従来の特徴選択アルゴリズムの見方は、測定エラー特性から生じるスコア(第1のスコア)を考慮し、与えられたバイオマーカー配列セットからのサブセット選択に最良の情報を与えるために、信号と同じスコア(第1のスコア)を使用するために修正することにより、本発明で用いる組み合わせベースの最適化基準を設計することができる。組み合わせベースの最適化基準の場合も、バイナリーベースの最適化基準の場合と同様に、決定システム200の各要素を用いてバイオマーカー配列セットを決定することができる(配列情報入力ステップ~配列セット決定ステップの実行)。
なお、上述したバイナリーベースの最適化基準では各バイオマーカー配列に独立にスコアを割り当てるのに対して、組み合わせベースの最適化基準の場合は、バイオマーカー配列の組み合わせに対してスコアを与える。このため、測定誤差の小ささを各マーカー配列で独立に扱ってよい場合はバイナリーベースの最適化基準が適しており、相互依存性が特に大きい場合は組み合わせベースの最適化基準が適している。相互依存性とは、例えば、「バイオマーカー配列1はバイオマーカー配列2と同時に測定する場合は測定誤差が小さいが、バイオマーカー配列3と当時に測定する場合は測定誤差が大きい」という場合である。
本発明では、上述した測定誤差スコア(第1のスコア)だけでなく「予測したい疾患との関連度が大きいほど高いスコア」(関連度スコア;第2のスコアの一例)を考慮し、これらスコアのバランスを最適化することによりバイオマーカー配列セットを決定することもできる。このような関連度スコア(第2のスコア)を併せて用いる場合、上述した測定誤差スコア(第1のスコア)と関連度スコア(第2のスコア)とのバランスを最適化する(例えば、測定誤差スコアと関連度スコアとの相加平均や相乗平均を最大化する)ことにより、バイオマーカー配列セットを決定することができる。この場合、関連度スコアも、測定誤差スコアの場合と同様にバイオマーカー配列ごとに独立に割り当てることもできるし、バイオマーカー配列の組み合わせに対して与えることもできる。例えば、マーカー1,2,3がいずれも疾患と関連している場合に、マーカー1,2の相関が小さくマーカー1,3の相関が大きい場合は、マーカー1,2の組み合わせの方が疾患予測に有効であり、関連度スコアが高くなる。
如何なる最適化基準(バイナリーベース、特徴選択アルゴリズム、あるいは組み合わせベースのような)が最良であるかは、応用分野、ユーザ、および時間の制約に依存し、それらの条件に合わせて適宜選択することができる。この出力は、システムが共同で考えるバイオマーカー配列のセットのものであり、多重化PCR配列決定のための与えられた測定誤差のプロトコルには、最小限の誤差がある。実施態様は、本発明の第5,第12の態様を考慮するために、決定システムの実施に基づいて(バランスの取れた配列選択を考慮するか否かにかかわらず)変更することができる。
なお、決定システム200は、学習システム100で得られた誤差分布(図6では、学習済み誤差分布データベース50)に依存しており、それ自体では、元の較正データに含まれないバイオマーカー配列のスコアを計算することができない。このような、較正データに含まれていなかったバイオマーカー配列の測定誤差特性の予測については、図6に示すように、また以下に説明するように、本発明の第6,第7の態様に係る予測システム300(及び、本発明の第13,第14の態様に係る予測方法)が必要である。この予測システム300は、決定システム200について上述したのと同様に本発明に係る学習システム100(学習システム)への追加であり、このような新しいバイオマーカー配列の使用事例、存在、重要性に依存する。
[較正データに含まれていないバイオマーカー配列の測定誤差特性の予測]
関心配列データベース60に含まれる関心バイオマーカー配列が較正データに含まれていないバイオマーカー配列であることが判明した場合(図6の判断「訓練データに含まれている配列か?」でYESの場合)に、その関心バイオマーカー配列の測定誤差特性を予測する手法について説明する。この場合、まず予測システム300に入力を渡す。具体的には、予測システム300の配列情報入力部312(プロセッサ)は、関心バイオマーカー配列のヌクレオチド配列及び測定プロトコル情報を入力し(配列情報入力ステップ)、また学習結果入力部314(プロセッサ)は、学習システム100から、例えば学習済み誤差特性、及び誤差特性に関連付けられたメタデータを入力する(学習結果入力ステップ)。ここで、「メタデータ」は、例えば遺伝子のタイプ(プロモーターかエンハンサーか)、遺伝子の領域(転写開始サイト等)であるが、これらには限定されない。
関心配列データベース60に含まれる関心バイオマーカー配列が較正データに含まれていないバイオマーカー配列であることが判明した場合(図6の判断「訓練データに含まれている配列か?」でYESの場合)に、その関心バイオマーカー配列の測定誤差特性を予測する手法について説明する。この場合、まず予測システム300に入力を渡す。具体的には、予測システム300の配列情報入力部312(プロセッサ)は、関心バイオマーカー配列のヌクレオチド配列及び測定プロトコル情報を入力し(配列情報入力ステップ)、また学習結果入力部314(プロセッサ)は、学習システム100から、例えば学習済み誤差特性、及び誤差特性に関連付けられたメタデータを入力する(学習結果入力ステップ)。ここで、「メタデータ」は、例えば遺伝子のタイプ(プロモーターかエンハンサーか)、遺伝子の領域(転写開始サイト等)であるが、これらには限定されない。
そして、類似度計算部316(プロセッサ)は、レーベンシュタイン距離やGC含量(GC-content;DNA分子中の窒素塩基のうち、グアニンとシトシンの割合)のような、2つの遺伝子配列間の類似度の測定基準(類似性の尺度)を用いて、以前に較正データに含まれていたバイオマーカー配列(較正データにおいて利用可能であったバイオマーカー配列)と新たなバイオマーカー配列(関心バイオマーカー配列)との類似度を計算する(類似度計算ステップ)。類似度計算部316は、学習済み誤差分布データベース50に存在するバイオマーカー配列から、関心バイオマーカー配列と「最も類似する」バイオマーカー配列を見つける(類似度計算ステップ)。予測システム300は、検出された「最も類似する配列」の情報を用いて、その「最も近い配列」に対応する学習済み誤差特性を(学習済み誤差分布データベース50から)取得することができ(誤差特性予測ステップ、配列取得ステップ)、これにより本発明の第6,第13の態様を完全に実装することができる。配列情報反映部320は、この情報を、本発明の第4,第5の態様を実施する決定システム200(及び、本発明の第11,第12の態様に係る決定方法)と併用して、決定システム200におけるバイオマーカー配列セットの決定に反映する(情報反映ステップ)こともできる。
[実施例]
遺伝子配列の候補セットは、まず、GC含量のような測定関連要因の十分な変化を示すと考えられている。例えば、配列GC内容のみを重要と仮定し、「高」の3遺伝子配列および「低」のGC含量の3遺伝子配列を同時測定のために決定することができる。次に、一連の重要な測定プロトコル関連変数を特定し、範囲を考慮する。これに続いて、すべての値とすべての変数の考えられる全範囲について、湿式実験手順を実行する。例えば、{5,10,15}のPCRサイクルを考慮し、メチル化比率を{5%,10%}と考えるならば、同一の生物標本のアリコット(aliquot)から6個の遺伝子の独立測定を3*2=6個行う。続いて、学習システムで用いられる前述の確率モデルを訓練し、決定システムのハイパーパラメータを調整(チューニング)した。さて、がん診断の場合のように、より多くの遺伝子バイオマーカーを探しながら、その遺伝子が測定特性の良否を評価するために決定モデルを用いることができる。
遺伝子配列の候補セットは、まず、GC含量のような測定関連要因の十分な変化を示すと考えられている。例えば、配列GC内容のみを重要と仮定し、「高」の3遺伝子配列および「低」のGC含量の3遺伝子配列を同時測定のために決定することができる。次に、一連の重要な測定プロトコル関連変数を特定し、範囲を考慮する。これに続いて、すべての値とすべての変数の考えられる全範囲について、湿式実験手順を実行する。例えば、{5,10,15}のPCRサイクルを考慮し、メチル化比率を{5%,10%}と考えるならば、同一の生物標本のアリコット(aliquot)から6個の遺伝子の独立測定を3*2=6個行う。続いて、学習システムで用いられる前述の確率モデルを訓練し、決定システムのハイパーパラメータを調整(チューニング)した。さて、がん診断の場合のように、より多くの遺伝子バイオマーカーを探しながら、その遺伝子が測定特性の良否を評価するために決定モデルを用いることができる。
100の遺伝子配列測定で訓練されたArtificial Intelligenceガン分類モデルが70%の感度で行われることを考えると、パフォーマンスが低い理由の一部は、一部の遺伝子で高い測定ノイズである可能性がある。上記のシステムを用いて100の遺伝子を再考し、測定困難なものを取り除くと、測定誤差を回避することにより、Artificial Intelligenceの性能は80%に上昇する可能性があり、より良い頑健性を持つ。
以上説明した実施形態は、以下の効果を有する。
(1)複数の遺伝子配列を一緒に測定して多重化されたパネルを扱うことができる。
(2)バイサルファイト変換されたサンプルを処理することができる。
(3)配列パラメータとプロトコルパラメータを入力として使用して、測定誤差を予測することができる。
(4)配列を分析/分類の目的に使用するかどうかを決定することができる。
(5)適切に学習された誤差特性、適切に選択されたバイオマーカー配列セット(及びそのサブセット)、精度良く予測されたバイオマーカー配列セットの測定誤差により、バイオマーカー配列を用いた分析や診断(例えば、上述したAIによるがんの分類)等を精度良く行うことができる。
(1)複数の遺伝子配列を一緒に測定して多重化されたパネルを扱うことができる。
(2)バイサルファイト変換されたサンプルを処理することができる。
(3)配列パラメータとプロトコルパラメータを入力として使用して、測定誤差を予測することができる。
(4)配列を分析/分類の目的に使用するかどうかを決定することができる。
(5)適切に学習された誤差特性、適切に選択されたバイオマーカー配列セット(及びそのサブセット)、精度良く予測されたバイオマーカー配列セットの測定誤差により、バイオマーカー配列を用いた分析や診断(例えば、上述したAIによるがんの分類)等を精度良く行うことができる。
以上で本発明の実施形態について説明してきたが、本発明は上述した態様に限定されず、種々の変形が可能である。
10 血液サンプル
11 血液サンプルデータ
20 湿式実験プロトコル
30 較正データDB
40 ハイパーパラメータ
50 学習済み誤差分布データベース
60 関心配列データベース
100 学習システム
110 プロセッサ
112 較正データ入力部
114 学習部
120 確率モデル
130 記憶部
200 決定システム
210 プロセッサ
212 配列情報入力部
214 学習結果入力部
216 スコア出力部
218 配列セット決定部
300 予測システム
310 プロセッサ
312 配列情報入力部
314 学習結果入力部
316 類似度計算部
318 誤差特性予測部
320 配列情報反映部
11 血液サンプルデータ
20 湿式実験プロトコル
30 較正データDB
40 ハイパーパラメータ
50 学習済み誤差分布データベース
60 関心配列データベース
100 学習システム
110 プロセッサ
112 較正データ入力部
114 学習部
120 確率モデル
130 記憶部
200 決定システム
210 プロセッサ
212 配列情報入力部
214 学習結果入力部
216 スコア出力部
218 配列セット決定部
300 予測システム
310 プロセッサ
312 配列情報入力部
314 学習結果入力部
316 類似度計算部
318 誤差特性予測部
320 配列情報反映部
Claims (14)
- 測定プロトコル変数と、バイオマーカー配列の結果として生じる誤差特性との関係を学習する学習システムであって、プロセッサを備え、
前記プロセッサは、
重要性のある変数について適切なデータが入手できるように設計された較正データを入力し、
確率モデルを用いて、前記重要性のある変数について各測定プロトコルにわたる誤差分布の特性を学習し、
前記確率モデルは、
バイサルファイト変換の誤差をモデル化するために、適切に選択された事前パラメータで初期化された第1のパラメータと、
バイオマーカー配列の増幅の相互依存性をモデル化するために、適切に選択された事前パラメータで初期化された第2のパラメータと、
PCR全体のバイアスをモデル化するために、適切に選択された事前パラメータで初期化された第3のパラメータと、
を含む学習システム。 - 前記第2のパラメータは、バイサルファイト変換後の遺伝子のメチル化配列及び非メチル化配列のカウントを別々に取得し、取得されたカウントを、前記メチル化配列及び前記非メチル化配列の各配列について、事前変数を別々に決定できる多項分布でモデル化したパラメータである請求項1に記載の学習システム。
- 前記第3のパラメータは、ユニバーサルプライマーを用いて複数の配列を同時に増幅する場合に、多項分布で計算されたカウントの個々のカウントの合計がガウス分布に従う、という構成データ制約が課されたパラメータである、請求項1または2に記載の学習システム。
- プロセッサを備える決定システムであって、
前記プロセッサは、
多重化パネルで使用する、関心バイオマーカー配列のヌクレオチド配列及び測定プロトコル情報を入力し、
請求項1から3のいずれか1項に記載の学習システムから、前記学習された誤差特性、及び前記誤差特性に関連付けられたメタデータを入力し、
あらかじめ決められた基準を用いて前記入力した前記ヌクレオチド配列、前記測定プロトコル情報、前記学習された誤差特性、及び前記メタデータを使用して、可能なバイオマーカー配列のセットのための第1のスコアを出力し、
各セットについての前記第1のスコアの値を考慮してバイオマーカー配列セットを決定する、
決定システム。 - 前記プロセッサは、
決定すべきバイオマーカー配列ごとに、第2のスコアを入力し、
前記バイオマーカー配列セットにおける各バイオマーカー配列についての前記第1のスコアを考慮して、前記第1のスコアと前記第2のスコアとのバランスを最適化することにより前記多重化パネルのベストなサブセットを選択する、
請求項4に記載の決定システム。 - 遺伝子配列の測定誤差特性を予測する予測システムであって、プロセッサを備え、
前記プロセッサは、
多重化パネルで使用する、関心バイオマーカー配列のヌクレオチド配列及び測定プロトコル情報を入力し、
請求項1から3のいずれか1項に記載の学習システムから、前記学習された誤差特性、及び前記誤差特性に関連付けられたメタデータを入力し、
2つの遺伝子配列間の類似性の尺度を計算するための測定基準を用いて、以前に較正データに含まれていたバイオマーカー配列と新たなバイオマーカー配列との類似度を計算し、
前記計算した類似度を他の関連する入力及び前記学習された誤差特性と組み合わせて使用して、前記較正データに含まれていないバイオマーカー配列を測定する際の誤差特性を予測する、
予測システム。 - 前記プロセッサは、
前記予測された誤差特性を使用して、前記較正データに含まれていないバイオマーカー配列と最も類似する、前記較正データにおいて利用可能であったバイオマーカー配列を取得し、
前記取得したバイオマーカー配列の情報を、請求項4または5に記載の決定システムにおけるバイオマーカー配列セットの決定に反映する、
請求項6に記載の予測システム。 - プロセッサを備え、測定プロトコル変数と、バイオマーカー配列の結果として生じる誤差特性との関係を学習する学習システムにより実行される学習方法であって、
前記プロセッサが、
重要性のある変数について適切なデータが入手できるように設計された較正データを入力し、
確率モデルを用いて、前記重要性のある変数について各測定プロトコルにわたる誤差分布の特性を学習し、
前記確率モデルは、
バイサルファイト変換の誤差をモデル化するために、適切に選択された事前パラメータで初期化された第1のパラメータと、
バイオマーカー配列の増幅の相互依存性をモデル化するために、適切に選択された事前パラメータで初期化された第2のパラメータと、
PCR全体のバイアスをモデル化するために、適切に選択された事前パラメータで初期化された第3のパラメータと、
を含む学習方法。 - 前記第2のパラメータは、バイサルファイト変換後の遺伝子のメチル化配列及び非メチル化配列のカウントを別々に取得し、取得されたカウントを、前記メチル化配列及び前記非メチル化配列の各配列について、事前変数を別々に決定できる多項分布でモデル化したパラメータである、請求項8に記載の学習方法。
- 前記第3のパラメータは、ユニバーサルプライマーを用いて複数の配列を同時に増幅する場合に、多項分布で計算されたカウントの個々のカウントの合計がガウス分布に従う、という構成データ制約が課されたパラメータである、請求項8または9に記載の学習方法。
- プロセッサを備える決定システムにより実行される決定方法であって、
前記プロセッサは、
多重化パネルで使用する、関心バイオマーカー配列のヌクレオチド配列及び測定プロトコル情報を入力し、
請求項8から10のいずれか1項に記載の学習方法の結果として得られる、前記学習された誤差特性、及び前記誤差特性に関連付けられたメタデータを入力し、
あらかじめ決められた基準を用いて前記入力した前記ヌクレオチド配列、前記測定プロトコル情報、前記学習された誤差特性、及び前記メタデータを使用して、可能なバイオマーカー配列のセットのための第1のスコアを出力し、
各セットについての前記第1のスコアの値を考慮してバイオマーカー配列セットを決定する、
決定方法。 - 前記プロセッサは、
決定すべきバイオマーカー配列ごとに、第2のスコアを入力し、
前記バイオマーカー配列セットにおける各バイオマーカー配列についての前記第1のスコアを考慮して、前記第1のスコアと前記第2のスコアとのバランスを最適化することにより前記多重化パネルのベストなサブセットを選択する、
請求項11に記載の決定方法。 - プロセッサを備え、遺伝子配列の測定誤差特性を予測する予測システムにより実行される予測方法であって、
前記プロセッサは、
多重化パネルで使用する、関心バイオマーカー配列のヌクレオチド配列及び測定プロトコル情報を入力し、
請求項8から10のいずれか1項に記載の学習方法により得られた、前記学習された誤差特性、及び前記誤差特性に関連付けられたメタデータを入力し、
2つの遺伝子配列間の類似性の尺度を計算するための測定基準を用いて、以前に較正データに含まれていたバイオマーカー配列と新たなバイオマーカー配列との類似度を計算し、
前記計算した類似度を他の関連する入力及び前記学習された誤差特性と組み合わせて使用して、前記較正データに含まれていないバイオマーカー配列を測定する際の誤差特性を予測する、
予測方法。 - 前記プロセッサは、
前記予測された誤差特性を使用して、前記較正データに含まれていないバイオマーカー配列と最も類似する、前記較正データにおいて利用可能であったバイオマーカー配列を取得し、
前記取得したバイオマーカー配列の情報を、請求項11または12に記載の決定方法におけるバイオマーカー配列セットの決定に反映する、
請求項13に記載の予測方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022056626 | 2022-03-30 | ||
| JP2022-056626 | 2022-03-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023190136A1 true WO2023190136A1 (ja) | 2023-10-05 |
Family
ID=88201403
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2023/011772 WO2023190136A1 (ja) | 2022-03-30 | 2023-03-24 | 学習システム、決定システム、及び予測システム、並びに学習方法、決定方法、及び予測方法 |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2023190136A1 (ja) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017523437A (ja) | 2014-06-10 | 2017-08-17 | クレッシェンド バイオサイエンス インコーポレイテッド | 体軸性脊椎関節炎の疾患活動性を測定およびモニタリングするためのバイオマーカーおよび方法 |
| WO2020008192A2 (en) * | 2018-07-03 | 2020-01-09 | Chronomics Limited | Phenotype prediction |
| JP2021521536A (ja) * | 2018-04-13 | 2021-08-26 | フリーノーム・ホールディングス・インコーポレイテッドFreenome Holdings, Inc. | 生体試料の多検体アッセイのための機械学習実装 |
-
2023
- 2023-03-24 WO PCT/JP2023/011772 patent/WO2023190136A1/ja unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017523437A (ja) | 2014-06-10 | 2017-08-17 | クレッシェンド バイオサイエンス インコーポレイテッド | 体軸性脊椎関節炎の疾患活動性を測定およびモニタリングするためのバイオマーカーおよび方法 |
| JP2021521536A (ja) * | 2018-04-13 | 2021-08-26 | フリーノーム・ホールディングス・インコーポレイテッドFreenome Holdings, Inc. | 生体試料の多検体アッセイのための機械学習実装 |
| WO2020008192A2 (en) * | 2018-07-03 | 2020-01-09 | Chronomics Limited | Phenotype prediction |
Non-Patent Citations (1)
| Title |
|---|
| JUSTIN D. SILVERMAN ET AL., MEASURING AND MITIGATING PCR BIAS IN MICROBIOME DATA, 22 March 2022 (2022-03-22), Retrieved from the Internet <URL:https://www.biorxiv.org/content/10.1101/604025v1> |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Janitza et al. | On the overestimation of random forest’s out-of-bag error | |
| US11887696B2 (en) | Systems and methods for classifying, prioritizing and interpreting genetic variants and therapies using a deep neural network | |
| Kayser et al. | Recent advances in Forensic DNA Phenotyping of appearance, ancestry and age | |
| Cao et al. | ROC curves for the statistical analysis of microarray data | |
| JP2021503922A (ja) | ターゲットシーケンシングのためのモデル | |
| JP2005531853A (ja) | Snp遺伝子型クラスタリングのためのシステムおよび方法 | |
| Iqbal et al. | Integrated COVID-19 Predictor: Differential expression analysis to reveal potential biomarkers and prediction of coronavirus using RNA-Seq profile data | |
| Kaplow et al. | Inferring mammalian tissue-specific regulatory conservation by predicting tissue-specific differences in open chromatin | |
| JP7275334B2 (ja) | 個人の生物学的ステータスを予測するためのシステム、方法および遺伝子シグネチャ | |
| CN110191964A (zh) | 确定生物样本中预定来源的游离核酸比例的方法及装置 | |
| WO2023190136A1 (ja) | 学習システム、決定システム、及び予測システム、並びに学習方法、決定方法、及び予測方法 | |
| Shahraki et al. | Robust chromatin state annotation | |
| Pfaffl et al. | Data analysis software | |
| CN101517579A (zh) | 蛋白质查找方法和设备 | |
| Wong et al. | A comparison study for DNA motif modeling on protein binding microarray | |
| ES2937408T3 (es) | Método y producto informático de análisis de ADN fetal por secuenciación masiva | |
| Mboning et al. | BayesAge: A maximum likelihood algorithm to predict epigenetic age | |
| TW202324151A (zh) | 用於分析基因數據之電腦實施的方法及裝置 | |
| Ogunnaike et al. | A probabilistic framework for microarray data analysis: Fundamental probability models and statistical inference | |
| US20040265830A1 (en) | Methods for identifying differentially expressed genes by multivariate analysis of microaaray data | |
| Jung et al. | Identifying Differentially Expressed Genes in Meta‐Analysis via Bayesian Model‐Based Clustering | |
| Lijoi et al. | A Bayesian nonparametric approach for comparing clustering structures in EST libraries | |
| Chong et al. | SeqControl: process control for DNA sequencing | |
| Aliferi et al. | Predicting chronological age from DNA methylation data: a machine learning approach for small datasets and limited predictors | |
| US20200105374A1 (en) | Mixture model for targeted sequencing |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23780146 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2024512336 Country of ref document: JP Kind code of ref document: A |