WO2023085232A1 - 改良されたライブラリー調製方法 - Google Patents
改良されたライブラリー調製方法 Download PDFInfo
- Publication number
- WO2023085232A1 WO2023085232A1 PCT/JP2022/041394 JP2022041394W WO2023085232A1 WO 2023085232 A1 WO2023085232 A1 WO 2023085232A1 JP 2022041394 W JP2022041394 W JP 2022041394W WO 2023085232 A1 WO2023085232 A1 WO 2023085232A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acid
- stranded cdna
- rna
- double
- library
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1096—Processes for the isolation, preparation or purification of DNA or RNA cDNA Synthesis; Subtracted cDNA library construction, e.g. RT, RT-PCR
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1068—Template (nucleic acid) mediated chemical library synthesis, e.g. chemical and enzymatical DNA-templated organic molecule synthesis, libraries prepared by non ribosomal polypeptide synthesis [NRPS], DNA/RNA-polymerase mediated polypeptide synthesis
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1093—General methods of preparing gene libraries, not provided for in other subgroups
Definitions
- the present invention relates to a library preparation method.
- next-generation sequencing enables advanced and high-speed processing that enables simultaneous sequencing of multiple individuals, and is a powerful analysis technology that contributes to medical technology such as clinical diagnosis and drug development, and life science fields such as agricultural technology and basic research. is.
- next-generation sequencing it is usually necessary to prepare a library consisting of nucleic acid fragments with adapter sequences added to the ends that can function as binding sites for flow cells or as primer binding sites.
- RNA as a template When preparing a library using RNA as a template, a method of purifying the generated double-stranded cDNA solution and then preparing the library is carried out.
- the RT-RamDA method amplifies cDNA using RNA as a template by incubating a mixture containing template RNA, primers, DNA strand-specific RNA:DNA hybrid strandase, RNase H minus reverse transcriptase, and substrate.
- it is an amplification reverse transcription method that makes a library
- a method in which double-stranded cDNA is generated from single-stranded cDNA and the solution is purified before library preparation is started Patent Document 1, Non-Patent Document 1) Reference 1).
- the double-stranded cDNA solution contains cell-derived contaminants, enzymes, primers, dNTPs, salts, etc. used in the reverse transcription reaction, which can inhibit the reaction in the next step.
- substances in these solutions can significantly inhibit enzymatic reactions used for library preparation.
- purification work is complicated and workability is poor.
- a reduction in yield also occurs due to the presence of the purification step. For these reasons, it is desirable to simplify the purification process.
- a main object of the present invention is to provide a more efficient method for library preparation.
- the inventors of the present invention have found that even with a small amount of template RNA, a sufficient library can be obtained by directly bringing the generated double-stranded cDNA into the library preparation without purifying it. It was found that yields were obtained.
- the present invention was completed by further studies based on the findings.
- a library preparation method comprising the following steps (a), (b), and (c): (a) a step of synthesizing a single-stranded cDNA from 10 pg or more of template RNA; (b) synthesizing double-stranded cDNA from said single-stranded cDNA; and (c) preparing a library using said non-purified double-stranded cDNA.
- step (b) is performed in the presence of 12 mM or less sodium chloride.
- step (b) is performed in the presence of 12 mM or less sodium chloride.
- the nucleic acid polymer is polyinosinic acid, polycytidylic acid, polyguanylic acid, polyadenylic acid, polythymidylic acid, polyuridylic acid, polydeoxyinosinic acid, polydeoxycytidylic acid, polydeoxyguanylic acid, polydeoxyadenylic acid, polydeoxythymidylic acid, poly Item 5.
- the method according to Item 3 or 4, wherein the homopolymer is at least one selected from the group consisting of deoxyuridylic acid and salts thereof.
- nucleic acid polymer is at least one homopolymer selected from the group consisting of polyinosinic acid, polydeoxyinosinic acid, and salts thereof.
- the proteolytic enzyme comprises either proteinase K or subtilisin.
- Item 8 Item 8. The method according to any one of Items 1 to 7, wherein the template RNA is RNA extracted from 1 to 1000 cells.
- Item 9 Item 9. The method according to any one of Items 1 to 8, wherein the non-purified double-stranded cDNA in step (c) is in the form of a solution of 1 ⁇ L or less.
- Item 10 Item 10.
- step (a) is performed by the RT-RamDA method.
- step (b) is performed using Klenow fragment.
- step (c) is performed by either a transposon method or a ligation method.
- step (b) is performed in the presence of a chloride salt of 60mM or less.
- a method for efficiently preparing a library using, for example, non-purified double-stranded cDNA is provided.
- a method for preparing a library with sufficient yield without purifying double-stranded cDNA generated from a small amount of template RNA, such as about 10 pg is provided.
- the method is useful for library preparation for next generation sequencing.
- a library can be efficiently prepared even with a double-stranded cDNA solution of 1 ⁇ L or less, which is much smaller than the amount used in conventional library preparation methods. Therefore, it is possible to reduce the amount of reagents required for library preparation and to reduce costs.
- the amount of the double-stranded cDNA synthesized from the template RNA is usually more than 1 ⁇ L.
- Stranded cDNA solutions are now available for other uses and quality checks. If such other uses and quality checks can be performed, it is possible to decide whether to proceed to the next step, which is beneficial because it saves time, labor, and materials.
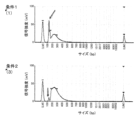
- FIG. 1 is a diagram showing the results of measuring a library with MultiNA (Shimadzu Corporation) in Example 5.
- FIG. 1 is a diagram showing the results of measuring a library with MultiNA (Shimadzu Corporation) in Example 5.
- the library preparation method preferably comprises the following steps (a), (b), and (c): (a) a step of synthesizing a single-stranded cDNA from 10 pg or more of template RNA; (b) synthesizing double-stranded cDNA from said single-stranded cDNA; and (c) preparing a library using said non-purified double-stranded cDNA.
- the template RNA may be purified RNA extracted from tissues or cells (the extracted RNA is further purified (e.g., treated by any purification means known in the art such as ethanol precipitation, column purification, etc.)). may be any RNA such as The type of cells from which template RNA is extracted is not particularly limited, and any type of cells may be used.
- the amount of template RNA is preferably 10 pg or more. Although the upper limit of the amount of template RNA is not particularly limited, it is preferably 10 ng or less, 5 ng or less, 1 ng or less, 500 pg or less, or 100 pg or less.
- a library can be efficiently prepared from the obtained double-stranded cDNA even if the amount of template RNA is small (for example, 10 pg or more and 1 ng or less).
- the amount of template RNA may be, for example, an amount contained in a small number of cells (eg, 1 to 1000, preferably 1 to 100, more preferably 1 to 10, more preferably 1). That is, RNA extracted from the above number of cells can also be used as template RNA. It is said that the standard amount of RNA contained in one cell is about 10 pg. According to the present invention, since a library can be prepared from a small amount of template RNA, it is possible to prepare a library with a sufficient yield even from one cell.
- the template RNA is preferably RNA extracted by lysing cells using a composition for cell lysis.
- the composition for cell lysis contains at least a cell lysing agent.
- Cell lysing agents include, for example, surfactants and chaotropic agents.
- Surfactants include anionic surfactants (e.g. sodium dodecyl sulfate, sodium cholate, sodium deoxycholate), cationic surfactants (e.g. cetyltrimethylammonium bromide), nonionic surfactants (e.g. , octylphenol ethoxylate, polyoxyethylene lauryl ether, polyoxyethylene stearyl ether, polyoxyethylene sorbitan monolaurate), and zwitterionic surfactants (e.g., 3-[(3-cholamidopropyl)dimethylammonio]).
- anionic surfactants e.g. sodium dodecyl sulfate, sodium cholate, sodium deoxycholate
- cationic surfactants e.g. cetyltrimethylammonium bromide
- Chaotropic agents include, for example, urea and lithium salts such as lithium perchlorate.
- Cell lysing agents are usually dissolved in water (preferably nuclease-free water) and used in the form of an aqueous solution. The cell lysing agents may be used singly or in combination of two or more.
- composition for cell lysis may contain additional components.
- Additional components include, for example, proteolytic enzymes (eg, proteinase K and subtilisin), nucleic acid polymers, RNase inhibitors, combinations of two or more thereof.
- Nucleic acid polymers include inosinic acid polymers, cytidylic acid polymers, guanylic acid polymers, adenylic acid polymers, thymidylic acid polymers, uridylic acid polymers, deoxyinosinic acid polymers, deoxycytidylic acid polymers, deoxyguanylic acid polymers, deoxyadenylic acid polymers, At least one selected from the group consisting of deoxythymidylic acid polymers and deoxyuridylic acid polymers is preferred.
- inosinic acid polymers include inosinic acid homopolymers (polyinosinic acid), inosinic acid copolymers (e.g., inosinic acid-derived structural units exceeding 50 mol%, 60 mol% or more, 70 mol% or more, 80 mol% or greater than or equal to 90 mol % but less than 100 mol %), derivatives thereof (e.g.
- the nucleic acid polymer is at least one selected from the group consisting of inosinic acid polymer, cytidylic acid polymer, guanylic acid polymer, deoxyinosinic acid polymer, deoxycytidylic acid polymer, and deoxyguanylic acid polymer. Inosinic acid polymers and/or deoxyinosinic acid polymers are more preferred.
- the nucleic acid polymer is polyinosinic acid, polycytidylic acid, polyguanylic acid, polyadenylic acid, polythymidylic acid, polyuridylic acid, polydeoxyinosinic acid, polydeoxycytidylic acid, polydeoxyguanylic acid, polydeoxyadenylic acid, polydeoxy It is preferably at least one homopolymer selected from the group consisting of thymidylic acid, polydeoxyuridylic acid, and salts thereof, and is preferably selected from the group consisting of polyinosinic acid, polydeoxyinosinic acid, and salts thereof. It is further preferred that at least one homopolymer is
- the nucleic acid polymer can be of any length, but as an example, it can be a nucleic acid polymer with a total length of 30 to 10000 bases.
- the step (a) is preferably performed in the presence of a protease and/or nucleic acid polymer.
- Library yield can be increased by synthesizing single-stranded cDNA in the presence of these components.
- One reason for this is thought to be that these components alleviate the inhibition of library preparation by substances such as cell-derived contaminants and reverse transcriptase used in the reverse transcription reaction.
- the reaction solution does not need to contain the protease and/or the nucleic acid polymer separately.
- the concentration of the protease in the synthesis reaction solution in step (a) is preferably 0.001 mg/mL or more, for example 0.001 to 0.1 mg/mL, preferably 0.001 to 0.01 mg/mL, more preferably 0.001 to 0.01 mg/mL. 0.005 mg/mL.
- the concentration of the protease can also be expressed in activity units, and is preferably 1 U/mL or more, for example 1 U/mL to 100 U/mL, preferably 1 U/mL, in the synthesis reaction solution of step (a). ⁇ 10 U/mL, more preferably 1 U/mL to 5 U/mL.
- proteolytic enzymes The activity of proteolytic enzymes is determined by definitions generally used in the art.
- the definition of activity for proteinase K is 1 unit (1 U) of the enzymatic activity that causes an increase in optical density at 275 nm equivalent to 1 microgram of tyrosine per minute under the conditions described below.
- the activity of other proteases such as subtilisin can be similarly measured.
- [reagent] (A) casein solution (substrate solution): 2.0% (20.0 g of Hammersten milk casein (Merck) is suspended and dissolved in 400 mL of 0.5% NaOH solution. Adjust the pH to 7.2 with 1 N HCl and add 500 mL of 20 mM boron containing 4 mM CaSO4 (pH 7.2).
- TCA solution TCA solution: 20% trichloroacetic acid (TCA)
- C Enzyme Diluent: Concentration 10 mM Borate Buffer in 10 mM Borate Buffer Assay Mix with 2 mM CaSO4 (pH 8.0) Casein 1.0% CaSO4 2mM [procedure] 1. Pipette 1.0 mL Substrate Solution (A) and 0.9 mL Enzyme Diluent (C) into test tubes and equilibrate at 35°C for approximately 5 minutes. 2. Add 0.1 mL of the sample to be measured and mix. 3.
- Weight activity (U/mg) (U/ml) x 1/C
- Vt total volume (4.0 mL)
- Vs sample volume (0.1 mL)
- 0.0074 extinction coefficient of tyrosine ( cm2 /microgram)
- t reaction time (10 minutes)
- df dilution factor
- C Enzyme concentration during dissolution (mg/mL)
- a 0.001 mg/mL solution is about 1 U/mL (1.2 to 1.4 U/mL) when the proteinase K of Promega is measured by the above method.
- the concentration of the nucleic acid polymer in the synthesis reaction solution in step (a) is preferably 1 ng/ ⁇ L or more, for example 1 to 10 ng/ ⁇ L, preferably 1 to 5 ng/ ⁇ L, more preferably 1 to 2 ng/ ⁇ L. be.
- the reaction for synthesizing single-stranded cDNA from template RNA is not particularly limited, and various conventionally known methods can be employed.
- step (a) is performed by reverse transcription, it typically includes a step of incubating a composition for reverse transcription (or a solution for reverse transcription) containing a protein having reverse transcription activity such as a reverse transcriptase.
- a composition for reverse transcription includes, for example, template RNA, protease and/or nucleic acid polymer, primers, deoxyribonucleotides, and reverse transcriptase.
- the composition for reverse transcription may also optionally contain other components such as a DNA polymerase.
- primers include primers specific to template RNA, oligo dT primers, random primers, and combinations of two or more of these.
- INDUSTRIAL APPLICABILITY The present invention is useful when performing reverse transcription reactions using oligo-dT primers and/or random primers, and is particularly beneficial when performing reverse transcription reactions using a combination of oligo-dT primers and random primers.
- the molar ratio of oligo dT primers to random primers is, for example, 1:5 to 1:15, preferably 1:8 to 1:12.
- Random primers include, for example, completely random primers and NSR (Not So Random) primers.
- Completely random primers are a mixture of primers with various base sequences, and each base sequence is a completely random base sequence.
- a fully random primer can contain a sequence that perfectly matches (or is perfectly complementary to) the rRNA sequence.
- completely random primers include completely random pentamers, completely random hexamers, completely random heptamers, completely random octamers, combinations thereof, and the like.
- a completely random hexamer may be a mixture of all possible base sequences (46 types) with four types of nucleotides (A, T, C, G).
- the NSR primer is a complete random primer from which the primer with a sequence completely complementary to the rRNA sequence has been removed.
- the rRNA sequences to be removed include, for example, 18S rRNA sequences, 28S rRNA sequences, 12S rRNA sequences, 16S rRNA sequences, combinations thereof.
- NSR primers include, for example, completely random hexamers excluding hexamers having a sequence completely complementary to the rRNA sequence. NSR primers can also be obtained by excluding those having a sequence completely complementary to the rRNA sequence from a set of primers such as completely random pentamers, completely random heptamers, and completely random octamers.
- the length of the primer is, for example, 5 bases or more, preferably 6 bases or more, and from the viewpoint of synthesis, it is, for example, 30 bases or less, preferably 25 bases or less, more preferably 20 bases or less.
- the concentration of the primer is not particularly limited, but is, for example, 1-10 ⁇ M, preferably 2-6 ⁇ M, more preferably 3-5 ⁇ M.
- Deoxyribonucleoside triphosphates are preferred as deoxyribonucleotides.
- Deoxyribonucleotide triphosphates include, for example, deoxycytidine triphosphate (dCTP), deoxyguanosine triphosphate (dGTP), deoxyadenosine triphosphate (dATP), deoxythymidine triphosphate (dTTP), deoxyuridine triphosphate (dUTP), these and combinations of two or more thereof.
- a mixture of dCTP, dGTP, dATP and dTTP, a mixture of dCTP, dGTP, dATP and dUTP, a mixture of dCTP, dGTP, dATP, dTTP and dUTP, and the like are preferred.
- a reverse transcriptase is any protein (enzyme) that has reverse transcription activity (RNA-dependent DNA polymerase activity), and is not particularly limited, but a polymerase that exhibits reverse transcriptase activity is preferred. Also, the reverse transcriptase preferably has low RNase H activity or no RNase H activity. Examples of reverse transcriptases include, e.g., Avian Myeloblastosis Virus reverse transcriptase (AMV-RT), Moloney Murine Leukemia Virus reverse transcriptase (MMLV-RT), human immune Virus (Human Immunovirus) reverse transcriptase (HIV-RT), EIRV-RT, RAV2-RT, C. hydrogenogormans DNA polymerase, rTth DNA polymerase, SuperScript I, SuperScript )II, variants thereof, and derivatives thereof. Of these, MMLV-RT is preferred.
- AMV-RT Avian Myeloblastosis Virus reverse transcriptase
- MMLV-RT Moloney Murine Le
- composition for reverse transcription may or may not contain the following DNA polymerases: Taq, Tbr, Tfl, Tru, Tth, Tli, Tac, Tne, Tma, Tih, Tfi, Pfu, Pwo , Kod, Bst, Sac, Sso, Poc, Pab, Mth, Pho, ES4, VENTTM, DEEPVENTTM, variants thereof.
- the composition for reverse transcription may contain an RNase inhibitor.
- RNase inhibitors are not particularly limited, and examples thereof include proteins derived from human placenta, rat lung, and pig liver.
- composition for reverse transcription may contain additives.
- Additives include, for example, buffers, salts, and combinations of two or more thereof.
- Buffers include, for example, Tris, Tricine, Bis-Tricine, Hepes, Mops, Tes, Taps, Pipes , Caps, and combinations of two or more thereof.
- the buffer is usually dissolved in water (preferably nuclease-free water) and used in the form of an aqueous solution.
- salts include chlorides (eg, lithium chloride, sodium chloride, potassium chloride, magnesium chloride, manganese chloride), acetates (eg, lithium acetate, sodium acetate, potassium acetate, magnesium acetate, manganese acetate), sulfates. (eg, potassium sulfate, magnesium sulfate, manganese sulfate), and combinations of two or more thereof.
- chlorides eg, lithium chloride, sodium chloride, potassium chloride, magnesium chloride, manganese chloride
- acetates eg, lithium acetate, sodium acetate, potassium acetate, magnesium acetate, manganese acetate
- sulfates eg, potassium sulfate, magnesium sulfate, manganese sulfate
- Conditions for incubation of the composition for reverse transcription are not particularly limited as long as single-stranded cDNA is produced from template RNA, and any conditions known in the art can be adopted.
- the incubation temperature is, for example, 30-65°C, preferably 35-60°C.
- the incubation time is, for example, 5-120 minutes, preferably 10-60 minutes.
- Amplification reverse transcription method (also known as RT-RamDA method) that amplifies cDNA using RNA as a template
- the RT-RamDA method is a nucleic acid amplification method that includes the step of incubating a mixture containing template RNA, primers, DNA strand-specific RNA:DNA hybrid strandase, RNase H minus reverse transcriptase, and a substrate.
- the complementary strand DNA (cDNA) of the template RNA is synthesized by the RNA-dependent DNA polymerase activity of RNase H-minus reverse transcriptase, and the cDNA strand among the hybrid strands of RNA and cDNA is identified as DNA strand-specific.
- Targeted RNA DNA is randomly cleaved by a hybrid chain degrading enzyme, and the cleavage site is the starting point, and the 3' cDNA strand is stripped from the RNA by the strand displacement activity of RNase H minus type reverse transcriptase, resulting in RNase H minus. A new cDNA strand is synthesized in the part stripped off by type reverse transcriptase. Details of the RT-RamDA method are described in US Patent Application Publication No. 2017/0275685, which is incorporated herein by reference in its entirety, and the like.
- step (a) When step (a) is performed by the RT-RamDA method, it includes a step of incubating the composition for reverse transcription containing the DNA strand-specific RNA:DNA hybrid chain degrading enzyme.
- the DNA strand-specific RNA:DNA hybrid strand degrading enzyme is preferably an enzyme that has the activity of cleaving the DNA strand in the hybrid strand of RNA and DNA.
- the enzyme for example, a double-strand-specific DNase and a non-specific DNase can be used. Among these, non-specific DNases are preferred in the present invention.
- double-strand-specific DNase double-strand-specific nuclease; also referred to as DSN
- enzymes derived from prokaryotes or eukaryotes can be used.
- Specific DNases or variants thereof can be used. Specific examples include the following. ⁇ Solenocera melantho DNase ⁇ Penaeus japonicus DNase ⁇ Paralithodes camtschaticus (king crab) DSN ⁇ Pandalus borealis dsDNase ⁇ Chionoecetes opilio (snow crab) DSN ⁇ Other DSN homologues
- the double-strand-specific DNase is preferably an enzyme that has DNase activity even at temperatures below 60°C.
- shrimp-derived double-strand-specific DNA degrading enzymes or modifications thereof are preferred.
- a commercially available product can be used as the double-strand-specific DNA degrading enzyme.
- Commercially available products include, for example, dsDNase (Arctic Zymes), Hl-dsDNase (Arctic Zymes), dsDNase (Thermo scientific), Shrimp Dnase, Recombinant (affymetrix), Atlantis dsDNase (Zymo Research), Thermolabile Nuclease (Roche ) and so on.
- the non-specific DNase has the activity of cleaving the DNA strand of the hybrid strand of RNA and DNA, and substantially has the activity of cleaving the RNA strand and single-stranded RNA of the hybrid strand of RNA and DNA. Enzymes that do not have the ability to cleave single-stranded DNA, but preferably have lower activity to cleave the DNA strand than the activity to cleave the hybrid strand of RNA and DNA.
- the non-specific DNase is preferably an enzyme that has DNase activity even below 60°C. Commercially available products such as DNaseI (manufactured by Thermo Fisher, DNaseI) can be used as such non-specific DNases.
- non-specific DNase enzymes derived from prokaryotes or eukaryotes can be used. Specific DNases or variants thereof can be used.
- the above variant means an enzyme obtained by altering a naturally occurring amino acid sequence. Specifically, an enzyme consisting of an amino acid sequence having a sequence identity of 80% or more (preferably 90% or more, more preferably 95% or more) with a naturally occurring amino acid sequence, and one or a number in the naturally occurring amino acid sequence An enzyme consisting of an amino acid sequence having deletions, substitutions and/or additions of amino acids (eg, 1 to 10, preferably 1 to 5, more preferably 1 to 3) amino acids.
- the composition for reverse transcription may contain a single-stranded DNA binding protein.
- Single-stranded DNA binding proteins are commonly used with DNA strand-specific RNA:DNA hybrid strandases.
- Single-stranded DNA binding proteins include, for example, T4 gene 32 protein, RecA, SSB (Single-Stranded DNA Binding Protein), and combinations of two or more thereof.
- step (a) is performed by the RT-RamDA method
- incubation of the reverse transcription composition may be performed under isothermal conditions or under thermocycling conditions.
- Isothermal conditions When incubation is performed under isothermal conditions, for example, a predetermined temperature between 25°C and less than 50°C, preferably a predetermined temperature between 30 and 45°C, more preferably between 35 and 40°C It can be carried out at a predetermined temperature, eg, 37° C. for a predetermined time (eg, 5 to 180 minutes, preferably 10 to 150 minutes). Incubation at a predetermined temperature between 25°C and 50°C may be performed in two or more steps.
- a predetermined temperature between 25°C and less than 50°C, preferably a predetermined temperature between 30 and 45°C, more preferably between 35 and 40°C
- a predetermined temperature eg, 37° C.
- a predetermined time eg, 5 to 180 minutes, preferably 10 to 150 minutes.
- Incubation at a predetermined temperature between 25°C and 50°C may be performed in two or more steps.
- a predetermined temperature between 25°C and less than 30°C for 5 to 15 minutes then at a predetermined temperature between 30°C and less than 35°C for 5 to 15 minutes, and then at a predetermined temperature between 35°C and less than 50°C.
- Incubation at temperature for a predetermined time eg, 5 to 60 minutes
- incubation at a predetermined temperature between 25°C and less than 50°C incubation at a predetermined temperature between 25°C and less than 50°C
- incubation at a predetermined temperature between, for example, 50°C and less than 100°C may be performed.
- Incubation at a predetermined temperature between 50°C and less than 100°C may be performed in two or more stages. For example, it may be incubated at a predetermined temperature between 50° C. and less than 80° C. for 5 to 15 minutes, followed by incubation at a predetermined temperature between 80 to 100° C. for 5 to 15 minutes.
- thermal cycle conditions When incubation is performed under thermal cycle conditions, for example, a predetermined temperature T1 between 20°C and less than 30°C (eg, 25°C) and a predetermined temperature T2 between 30 and 45°C (eg, 37°C ) in combination with T1 for a predetermined time (eg, 1 to 3 minutes, such as 2 minutes) and T2 for a predetermined time (eg, 1 to 3 minutes, such as 2 minutes) as one cycle, which is preferably may be repeated for 10 to 40 cycles, more preferably 15 to 35 cycles.
- a predetermined temperature T1 between 20°C and less than 30°C (eg, 25°C) and a predetermined temperature T2 between 30 and 45°C (eg, 37°C ) in combination with T1 for a predetermined time (eg, 1 to 3 minutes, such as 2 minutes) and T2 for a predetermined time (eg, 1 to 3 minutes, such as 2 minutes) as one cycle, which is preferably may be repeated for 10 to 40 cycles, more
- a predetermined temperature between 25°C and less than 30°C for a predetermined time (for example, 5 to 15 minutes), and then at a predetermined temperature between 30°C and less than 35°C. for a period of time (eg, 5-15 minutes), followed by incubation at a predetermined temperature between 35° C. and less than 50° C. for a predetermined period of time (eg, 1-5 minutes).
- a predetermined temperature between 50 ° C and less than 80 ° C for a predetermined time (eg, 5 to 15 minutes)
- a predetermined temperature between 80 to 90 ° C for a predetermined time (for example 5-15 minutes).
- Step (b) is preferably carried out in the presence of salt.
- Library yield can be increased by synthesizing double-stranded cDNA in the presence of salt.
- One reason for this is thought to be that the amount of double-stranded cDNA produced by optimizing the binding of the primer to the template was increased.
- salts include chloride salts (e.g., alkali metal chloride salts such as lithium chloride, sodium chloride, and potassium chloride; alkaline earth metal chloride salts such as magnesium chloride; transition metal chloride salts such as manganese chloride). is mentioned. Among them, alkali metal chloride salts and/or alkaline earth metal chloride salts are preferred, and at least one selected from the group consisting of magnesium chloride, potassium chloride and sodium chloride is preferred. Salts may be used singly or in combination of two or more.
- chloride salts e.g., alkali metal chloride salts such as lithium chloride, sodium chloride, and potassium chloride
- alkaline earth metal chloride salts such as magnesium chloride
- transition metal chloride salts such as manganese chloride
- salt When a salt is contained in the reaction product of step (a), salt may not be added separately to the reaction solution of step (b), or salt may be added separately to the reaction solution of step (b). good.
- the salt concentration in the synthesis reaction solution in step (b) is, for example, 60 mM or less, preferably 50 mM or less, more preferably 40 mM or less. Also, the salt concentration in the synthesis reaction solution of step (b) is, for example, 5 to 60 mM, preferably 5 to 50 mM, more preferably 5 to 40 mM. In certain embodiments, the concentration of the salt in the synthesis reaction solution in step (b) is, for example, 20 mM or less (eg, 5 to 20 mM), 15 mM or less (eg, 5 to 15 mM), or 12 mM or less (eg, 5 to 12 mM). ), etc. The concentration of the salt is preferably that of a chloride salt.
- the concentration of sodium chloride is, the better. be.
- the synthesis reaction solution in step (b) may not contain sodium chloride, and when the synthesis reaction solution in step (b) contains sodium chloride, its concentration in the synthesis reaction solution in step (b) is, for example, 5-20 mM, preferably 5-15 mM, more preferably 5-12 mM.
- the concentration of potassium chloride is, for example, 5 mM or higher (eg, 5 to 50 mM), preferably 10 mM or higher (eg, 10 to 40 mM), more preferably 15 mM or higher (eg, : 15-35 mM).
- the concentration of magnesium chloride is, for example, 20 mM or less (eg, 1 to 20 mM), preferably 15 mM or less (eg, 1 to 15 mM), more preferably 10 mM or less (eg, : 1-10 mM).
- the concentration of chloride salt is 60 mM or less (for example, 50 mM or less, preferably 40 mM or less) and the concentration of sodium chloride is 20 mM or less in the synthesis reaction solution of step (b). More preferably, the chloride salt concentration is 60 mM or less (eg, 50 mM or less, preferably 40 mM or less), the sodium chloride concentration is 20 mM or less, and the potassium chloride concentration is 5 mM or more.
- the reaction for synthesizing double-stranded cDNA from single-stranded cDNA is not particularly limited, and various conventionally known methods can be employed.
- the reaction is typically performed using a composition for synthesizing double-stranded cDNA containing an enzyme (double-stranded cDNA synthetase) that catalyzes a nucleotide polymerization reaction in the 5′ ⁇ 3′ direction in the presence of a template DNA and a primer. including the step of incubating the object.
- an enzyme double-stranded cDNA synthetase
- a composition for double-stranded cDNA synthesis includes, for example, single-stranded cDNA, salts, primers, deoxyribonucleotides, and double-stranded cDNA synthetase.
- the composition for double-strand synthesis may also optionally contain other components such as buffers.
- Double-stranded cDNA synthetase refers to an enzyme that catalyzes the polymerization reaction of nucleotides in the 5' ⁇ 3' direction, and is not particularly limited. Examples include the Klenow fragment, T4 DNA polymerase, variants thereof, and derivatives thereof. Of these, the Klenow fragment is preferred.
- primers examples include those exemplified for the reverse transcription composition.
- Conditions for incubation of the composition for double-stranded cDNA synthesis are not particularly limited as long as double-stranded cDNA is produced from single-stranded cDNA, and any conditions known in the art can be adopted. .
- it may be incubated at a predetermined temperature between 10° C. and lower than 35° C. for a predetermined time (eg, 5 to 90 minutes).
- the library preparation method preferably further includes the step of heating the double-stranded cDNA (for example, the reaction solution in step (b)). This step is preferably performed after step (b) and before the next step (c).
- the step may correspond to an inactivation treatment.
- the heating conditions are not particularly limited. minutes or longer).
- the heating temperature is preferably 80°C or higher and 95°C or lower, more preferably 80°C or higher and 90°C or lower, still more preferably 80°C or higher and 85°C or lower.
- the incubation time is preferably 5 minutes or more and 30 minutes or less, more preferably 10 minutes or more and 20 minutes or less, and still more preferably 10 minutes or more and 15 minutes or less.
- step (c) By performing such a heating step, the formation of adapter dimers can be more effectively suppressed in step (c).
- heating at such temperatures unfolds the higher-order structure of the double-stranded cDNA and improves the efficiency of enzymatic reactions (e.g., transposon reactions) in library preparation, thereby promoting the formation of adapter dimers. It is assumed that it can be suppressed more effectively.
- the library is preferably nucleic acid fragments to which sequences necessary for next-generation sequencing have been assigned. Sequences required for next-generation sequencing include flow cell binding sequences and sequences required for sequencing.
- next-generation sequencing typically refers to sequencing technology that can simultaneously perform millions to billions of massive sequencing reactions.
- next-generation sequencing include a method of parallel sequence analysis using amplification techniques such as emulsion PCR and bridge PCR, and highly sensitive detection techniques such as single-molecule observation.
- the device (sequencer) for next-generation sequencing is not particularly limited, but for example, MiSeq, HiSeq, NovaSeq (Illumina); Genetic Analyzer V2.0, Ion Proton (Thermo Fisher Scientific); MinION, PromethION (Nanopore Co.) and the like.
- Next generation sequencing is described, for example, in US Patent Application Publication 2014/178438, which is incorporated herein by reference in its entirety.
- the double-stranded cDNA used for library preparation is in the form of a solution of, for example, 5 ⁇ L or less, preferably 4 ⁇ L or less, more preferably 3 ⁇ L or less, even more preferably 2 ⁇ L or less, and particularly preferably 1 ⁇ L or less.
- the double-stranded cDNA is in the form of a solution of, for example, 0.01 ⁇ L or more, 0.05 ⁇ L or more, or 0.1 ⁇ L or more. In the present invention, a sufficient library yield can be obtained without purification even with a small amount of double-stranded cDNA.
- the library preparation method is not particularly limited.
- transposons are used to fragment and tag template DNA, and nucleic acid amplification of a library composed of tagged DNA fragments is carried out.
- two transposon terminal sequences (equivalent to adapters) and a transposase form a transposome complex, which fragments and tags template DNA in solution, which is necessary for next-generation sequencing such as PCR.
- Sequence analysis can be performed because a unique sequence is added (this is also referred to as the “transposon method”).
- Examples of commercially available kits using transposons include, but are not limited to, Illumina's Nextera XT DNA Library Preparation Kit.
- Another technique is fragmentation of template DNA (e.g., full-length DNA), end-repair of fragmented DNA, and/or adenylation of the ends of fragmented DNA or end-repaired fragmented DNA (this is referred to as "A addition ”), end-repaired and/or end-adenylated DNA fragments added with adapter sequences by ligation with T4 Ligase or the like, and library nucleic acids composed of DNA fragments to which adapters have been added Amplification has been carried out. A sequence necessary for next-generation sequencing is added to the adapter sequence, so sequence analysis can be performed (this is also referred to as a “ligation method”).
- a addition end-repaired and/or end-adenylated DNA fragments added with adapter sequences by ligation with T4 Ligase or the like, and library nucleic acids composed of DNA fragments to which adapters have been added Amplification has been carried out.
- a sequence necessary for next-generation sequencing is added to the adapter sequence, so sequence analysis
- the adapter sequence and length are not particularly limited, and may be, for example, Y-shaped adapters, stem-loop adapters, and the like.
- Examples of commercially available kits using the ligation method include, but are not limited to, GenNext (R) NGS Library Prep Kit from Toyobo Co., Ltd. and KAPA HyperPrep Kit from KAPA.
- the library preparation method is preferably the transposon method.
- purification means separation of contaminants such as the primers contained in a solution or the like from double-stranded cDNA.
- a purification method a method using magnetic beads, a method using a column, a method using phenol or phenol/chloroform, and a method using protein aggregation are known.
- the present invention is characterized by preparing a library using non-purified double-stranded cDNA that has not undergone such purification steps.
- non-purified double-stranded cDNA refers to the double-stranded cDNA synthesized in step (b) (including the double-stranded cDNA solution) itself or heated Examples include those obtained by diluting the main-stranded cDNA with a solvent such as water or a buffer solution, or the products obtained by drying and concentrating the above-mentioned double-stranded cDNA solution, but are limited to these if they have not undergone a purification process. isn't it.
- the library concentration can be increased, eg, 10 nM or higher, 15 nM or higher, 20 nM or higher, 25 nM or higher, 30 nM or higher, 35 nM or higher, or 40 nM or higher.
- the amount of adapter dimers in the library can be reduced.
- the peak intensity of the indicated region is 90% or less, 80% or less, 70% or less, 60% or less, or 50% of the maximum intensity of the region showing the library yield (region present in the range of approximately 150 bp to 5000 bp) You can:
- Example 1 Effect of Sodium Chloride Concentration on Yield
- RNA was diluted, single-stranded cDNA was synthesized by the RT-RamDA method, and double-stranded cDNA was synthesized using Klenow fragment.
- a library was prepared using the Nextera XT DNA Library Preparation Kit (Illumina) using the transposon method, and the library concentration was measured using MultiNA (Shimadzu Corporation). The procedure followed the manufacturer's instructions.
- 10 pg of RNA purified from NIH3T3 cells using RNeasy Mini Kit (Qiagen) was used as the nucleic acid fragment sample used in this example.
- 1 ng/ ⁇ L of RNA was diluted with the RNA diluent shown in Table 1 to prepare a 10 pg/ ⁇ L RNA solution.
- NSR primers (hexamers) were synthesized using Sigma's custom oligos, and were described in Ozsolak et al., Digital transcriptome profiling from attomole-level RNA samples, Genome Research, Vol. 20, 2010, pp. 519-525. They were used by mixing so as to obtain a primer composition.
- Example 2 Influence of addition amount of potassium polyinosinic acid salt and proteinase K on yield
- the following experiment was conducted to examine whether the addition amount of potassium polyinosinic acid salt and proteinase K affects the yield of the library.
- rice field RNA was diluted, single-stranded cDNA was synthesized by the RT-RamDA method, and double-stranded cDNA was synthesized using Klenow fragment.
- a library was prepared using the Nextera XT DNA Library Preparation Kit (Illumina), and the library concentration was measured using MultiNA (Shimadzu Corporation). The procedure followed the manufacturer's instructions.
- RNA purified from NIH3T3 cells using RNeasy Mini Kit As the nucleic acid fragment sample used in this example, 10 pg of RNA purified from NIH3T3 cells using RNeasy Mini Kit (Qiagen) was used. 1 ng/ ⁇ L of RNA was diluted with the RNA diluent shown in Table 6 to prepare a 10 pg/ ⁇ L RNA solution. 1 ⁇ L of this RNA solution was heat-treated at 70° C. for 1 minute and 30 seconds. Then, 2 ⁇ L of the RT-RamDA reaction solution shown in Table 7 was added, and the RT-RamDA reaction was carried out at 3 ⁇ L at 25°C for 10 minutes, 30°C for 10 minutes, 37°C for 30 minutes, 50°C for 5 minutes, and 98°C for 5 minutes.
- Example 3 Yield Improvement by Non-Purification Protocol by Addition of Potassium Polyinosinic Acid Salt
- the following experiment was carried out in order to examine whether the addition of potassium polyinosinic acid salt affects the yield of the library.
- Cells were lysed, single-stranded cDNA was synthesized by RT-RamDA method, and double-stranded cDNA was synthesized using Klenow fragment.
- a library was prepared using the Nextera XT DNA Library Preparation Kit (Illumina), and the library concentration was measured using MultiNA (Shimadzu Corporation). The procedure followed the manufacturer's instructions. Specifically, the following methods were used. Cell samples used in Examples were prepared with the following composition.
- NIH3T3 cells were reacted with a trypsin solution (Nacalai Tesque) at 37°C for 2 minutes to dissociate into single cells. Immediately after dissociation, the reaction was stopped by replacing with PBS(-). Using a FACS Melody cell sorter (BD), PI-negative cells of the dead cell fluorescent marker dye were used as a live cell fraction, and 120 cells were fractionated from this fraction into 3 ⁇ L of the Lysis Buffer shown in Table 11 below. Immediately after fractionation, centrifugation was performed and stored at -80°C.
- BD FACS Melody cell sorter
- Example 4 Evaluation of Amount of Template RNA
- RNA was diluted, single-stranded cDNA was synthesized by RT-RamDA method or conventional reverse transcription method (RT method), and double-stranded cDNA was synthesized using Klenow fragment.
- a library was prepared using the Nextera XT DNA Library Preparation Kit (Illumina), and the library concentration was measured using MultiNA (Shimadzu Corporation). The procedure followed the manufacturer's instructions.
- Mouse ES cell total RNA (Unitech Co., Ltd.) was used as the nucleic acid fragment sample used in this example.
- RNA template amount 10 pg
- 1 ⁇ L of this RNA solution was heat-treated at 70° C. for 1 minute and 30 seconds.
- Conditions 1 and 2 add 2 ⁇ L of the RT-RamDA reaction solution shown in Table 17, and condition 3 adds 2 ⁇ L of the RT reaction solution shown in Table 18, 3 ⁇ L at 25 ° C. 10 minutes, 30 ° C. 10 minutes, 37 RT-RamDA or RT reactions were performed at 30°C, 50°C for 5 minutes, and 98°C for 5 minutes.
- Example 5 Comparison of Inactivation Temperature in Synthesis of Double Strands
- RNA was diluted, single-stranded cDNA was synthesized by the RT-RamDA method, and double-stranded cDNA was synthesized using Klenow fragment. After that, they were inactivated by heating at 70°C or 80°C, then a library was prepared using the Nextera XT DNA Library Preparation Kit (Illumina), and the library concentration was measured using MultiNA (Shimadzu Corporation). The procedure followed the manufacturer's instructions. Mouse ES cell total RNA was used as the nucleic acid fragment sample used in this example.
- RNA template amount 10 pg
- RNA template amount 10 pg
- 1 ⁇ L of this RNA solution was heat-treated at 70° C. for 1 minute and 30 seconds.
- Example 6 Evaluation of Chlorides in Step (b)
- RNA was diluted, single-stranded cDNA was synthesized by the RT-RamDA method, and double-stranded cDNA was synthesized using Klenow fragment.
- a library was prepared using the Nextera XT DNA Library Preparation Kit (Illumina), and the library concentration was measured using MultiNA (Shimadzu Corporation). The procedure followed the manufacturer's instructions. Nucleic acid fragment samples used in this example were total RNA purified from human K562 cells using RNeasy mini kit (QIAGEN).
- RNA template amount 10 pg
- RNA template amount 10 pg
- 1 ⁇ L of this RNA solution was heat-treated at 70° C. for 1 minute and 30 seconds.
- 2 ⁇ L of the RT-RamDA reaction solution shown in Table 2 in Example 1 was added, and 3 ⁇ L was added at 25°C for 10 minutes, 30°C for 10 minutes, 37°C for 30 minutes, 50°C for 5 minutes, and 98°C.
- the RT-RamDA reaction was performed in minutes.
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Organic Chemistry (AREA)
- Zoology (AREA)
- Biomedical Technology (AREA)
- Wood Science & Technology (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Crystallography & Structural Chemistry (AREA)
- Plant Pathology (AREA)
- Molecular Biology (AREA)
- Microbiology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Computational Biology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202280074869.3A CN118318038A (zh) | 2021-11-10 | 2022-11-07 | 改良的文库制备方法 |
| JP2023559618A JPWO2023085232A1 (enExample) | 2021-11-10 | 2022-11-07 | |
| US18/709,418 US20250011759A1 (en) | 2021-10-11 | 2022-11-07 | Improved library preparation method |
| EP22892731.5A EP4431603A4 (en) | 2021-11-10 | 2022-11-07 | IMPROVED BANK PREPARATION METHOD |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021183612 | 2021-11-10 | ||
| JP2021-183612 | 2021-11-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023085232A1 true WO2023085232A1 (ja) | 2023-05-19 |
Family
ID=86336073
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/041394 Ceased WO2023085232A1 (ja) | 2021-10-11 | 2022-11-07 | 改良されたライブラリー調製方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20250011759A1 (enExample) |
| EP (1) | EP4431603A4 (enExample) |
| JP (1) | JPWO2023085232A1 (enExample) |
| CN (1) | CN118318038A (enExample) |
| WO (1) | WO2023085232A1 (enExample) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140178438A1 (en) | 2011-05-24 | 2014-06-26 | Ugur Sahin | Individualized vaccines for cancer |
| US20170275685A1 (en) | 2014-09-30 | 2017-09-28 | Riken | Method for nucleic acid amplification |
| WO2020184551A1 (ja) * | 2019-03-13 | 2020-09-17 | 東洋紡株式会社 | 核酸の生成および増幅 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1038965A1 (en) * | 1999-03-23 | 2000-09-27 | American Cyanamid Company | Method of screening for chemical compounds capable of inducing ERS in plants |
| US10017759B2 (en) * | 2014-06-26 | 2018-07-10 | Illumina, Inc. | Library preparation of tagged nucleic acid |
-
2022
- 2022-11-07 JP JP2023559618A patent/JPWO2023085232A1/ja active Pending
- 2022-11-07 US US18/709,418 patent/US20250011759A1/en active Pending
- 2022-11-07 CN CN202280074869.3A patent/CN118318038A/zh active Pending
- 2022-11-07 WO PCT/JP2022/041394 patent/WO2023085232A1/ja not_active Ceased
- 2022-11-07 EP EP22892731.5A patent/EP4431603A4/en active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140178438A1 (en) | 2011-05-24 | 2014-06-26 | Ugur Sahin | Individualized vaccines for cancer |
| US20170275685A1 (en) | 2014-09-30 | 2017-09-28 | Riken | Method for nucleic acid amplification |
| WO2020184551A1 (ja) * | 2019-03-13 | 2020-09-17 | 東洋紡株式会社 | 核酸の生成および増幅 |
Non-Patent Citations (4)
| Title |
|---|
| HAYASHI T. ET AL.: "Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs", NAT. COMMUN., 2018 |
| HAYASHI TETSUTARO, OZAKI HARUKA, SASAGAWA YOHEI, UMEDA MANA, DANNO HIROKI, NIKAIDO ITOSHI: "Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs", NATURE COMMUNICATIONS, vol. 9, no. 1, 12 February 2018 (2018-02-12), XP093016358, DOI: 10.1038/s41467-018-02866-0 * |
| OZSOLAK ET AL.: "Digital transcriptome profiling from attomole-level RNA samples", GENOME RESEARCH, vol. 20, 2010, pages 519 - 525 |
| See also references of EP4431603A4 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN118318038A (zh) | 2024-07-09 |
| JPWO2023085232A1 (enExample) | 2023-05-19 |
| US20250011759A1 (en) | 2025-01-09 |
| EP4431603A4 (en) | 2025-10-29 |
| EP4431603A1 (en) | 2024-09-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9255291B2 (en) | Oligonucleotide ligation methods for improving data quality and throughput using massively parallel sequencing | |
| JP6583796B2 (ja) | 核酸の増幅方法 | |
| JP7722661B2 (ja) | 核酸の生成および増幅 | |
| EP3071711A1 (en) | Degradable adaptors for background reduction | |
| WO2016135300A1 (en) | Efficiency improving methods for gene library generation | |
| JP7713682B2 (ja) | 長時間の核酸増幅反応の改善 | |
| WO2023085232A1 (ja) | 改良されたライブラリー調製方法 | |
| WO2025024703A1 (en) | Dual-tagmentation single-cell dnaseq | |
| KR20230124636A (ko) | 멀티플렉스 반응에서 표적 서열의 고 감응성 검출을위한 조성물 및 방법 | |
| AU2022407332B2 (en) | A method of capturing crispr endonuclease cleavage products | |
| RU2843259C2 (ru) | Способ захвата продуктов расщепления эндонуклеазы crispr | |
| EP3655572A1 (en) | Rapid library construction for high throughput sequencing | |
| JP2024026695A (ja) | 非特異的な核酸増幅を抑制する方法 | |
| JP2022183762A (ja) | 核内ノンコーディングrnaの抽出方法 | |
| WO2024249591A1 (en) | Methods for double-stranded sequencing by synthesis | |
| JP2025521811A (ja) | 常温での核酸増幅及び検出 | |
| CN120584195A (zh) | 源自核糖体rna的逆转录的抑制 | |
| WO2024206589A2 (en) | Cell fixative agents for single cell sequencing | |
| KR20230080464A (ko) | 전사된 핵산을 생성하는 방법 및 수단 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22892731 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023559618 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18709418 Country of ref document: US Ref document number: 202280074869.3 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022892731 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2022892731 Country of ref document: EP Effective date: 20240610 |