WO2020085441A1 - 改変されたCas9タンパク質及びその用途 - Google Patents
改変されたCas9タンパク質及びその用途 Download PDFInfo
- Publication number
- WO2020085441A1 WO2020085441A1 PCT/JP2019/041751 JP2019041751W WO2020085441A1 WO 2020085441 A1 WO2020085441 A1 WO 2020085441A1 JP 2019041751 W JP2019041751 W JP 2019041751W WO 2020085441 A1 WO2020085441 A1 WO 2020085441A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- protein
- protein according
- amino acid
- sequence
- cells
- Prior art date
Links
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/90—Stable introduction of foreign DNA into chromosome
- C12N15/902—Stable introduction of foreign DNA into chromosome using homologous recombination
- C12N15/907—Stable introduction of foreign DNA into chromosome using homologous recombination in mammalian cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases RNAses, DNAses
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/52—Genes encoding for enzymes or proenzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPRs]
Abstract
Description
従って、本発明者らは、全長タンパク質と実質的に同等のDNA結合親和性を有しながら、より小型化されたdCas9タンパク質の変異体を提供することを目的とする。
欠失及び置換を併せて変異とも称する。
本明細書中、変異を導入する前のdSaCas9タンパク質を野生型dSaCas9(タンパク質)、変異を導入した後のdSaCas9タンパク質をdSaCas9変異体(タンパク質)と称する場合がある。
即ち、本発明は以下の通りである。
[1]配列番号2で表されるアミノ酸配列において、
721~755位の間に連続した欠失領域を有し、
該欠失領域にそれぞれ隣接したアミノ酸が3乃至10個のアミノ酸残基からなるリンカーによって連結しているアミノ酸配列を含む配列からなり、
且つ、ガイドRNAとの結合能を有するタンパク質。
[2]リンカーが、グリシン(G)及びセリン(S)で構成される、5~9アミノ酸長のリンカーである、上記[1]に記載のタンパク質。
[3]リンカーが、以下から選択される、上記[2]に記載のタンパク質:
-SGGGS-
-GGSGGS-
-SGSGSGSG-
-SGSGSGSGS-。
[4]該欠失領域が、721~745位領域
である、上記[1]~[3]のいずれかに記載のタンパク質。
[5]配列番号4で表される上記[4]記載のタンパク質。
[6]該欠失領域が、721~755位領域
である、上記[1]~[3]のいずれかに記載のタンパク質。
[7]配列番号6で表される上記[6]記載のタンパク質。
[8]さらに、45位及び/又は163位のグルタミン酸(E)が他のアミノ酸に置換されている、上記[1]~[7]のいずれかに記載のタンパク質。
[9]他のアミノ酸が塩基性アミノ酸である、上記[8]記載のタンパク質。
[10]塩基性アミノ酸がリジン(K)である、上記[9]記載のタンパク質。
[11]配列番号2の変異及び/又は欠失が施された位置以外の部位において80%以上の同一性を有する、上記[1]~[10]のいずれかに記載のタンパク質。
[12]配列番号2の変異及び/又は欠失が施された位置以外の部位において1~数個のアミノ酸が置換、欠失、挿入及び/又は付加された、上記[1]~[10]のいずれかに記載のタンパク質。
[13]転写制御因子タンパク質又はドメインを連結した、上記[1]~[12]のいずれかに記載のタンパク質。
[14]転写制御因子が転写活性化因子である、上記[13]記載のタンパク質。
[15]転写制御因子が転写サイレンサー又は転写抑制因子である、上記[13]記載のタンパク質。
[16]上記[1]~[15]のいずれかに記載のタンパク質をコードする核酸。
[17]上記[1]~[16]のいずれかに記載のタンパク質と、標的二本鎖ポリヌクレオチド中のPAM(Proto-spacer Adjacent Motif)配列の1塩基上流から20塩基以上24塩基以下上流までの塩基配列に相補的な塩基配列からなるポリヌクレオチドを含むガイドRNAと、を備えるタンパク質-RNA複合体。
[18]標的二本鎖ポリヌクレオチドを部位特異的に改変するための方法であって、
標的二本鎖ポリヌクレオチドと、タンパク質と、ガイドRNAとを混合し、インキュベートする工程と、
前記タンパク質が、PAM配列の上流に位置する結合部位で前記標的二本鎖ポリヌクレオチドを改変する工程と、を備え、
前記タンパク質は、上記[1]~[15]のいずれかに記載のタンパク質であり、
前記ガイドRNAは、前記標的二本鎖ポリヌクレオチド中の前記PAM配列の1塩基上流から20塩基以上24塩基以下上流までの塩基配列に相補的な塩基配列からなるポリヌクレオチドを含むものである方法。
[19]細胞の標的遺伝子の発現を増大させる方法であって、前記細胞内で上記[14]に記載のタンパク質と、前記標的遺伝子に対する1つ又は複数のガイドRNAとを発現させることを含む、方法。
[20]細胞の標的遺伝子の発現を減少させる方法であって、前記細胞内で上記[15]に記載のタンパク質と、前記標的遺伝子に対する1つ又は複数のガイドRNAとを発現させることを含む、方法。
[21]細胞が真核細胞である、上記[19]又は[20]に記載の方法。
[22]細胞が酵母細胞、植物細胞又は動物細胞である、上記[19]又は[20]に記載の方法。
本発明のdSaCas9変異体は、ガイドRNAとの結合能を有しながら、より小型化されたdSaCas9タンパク質である。小型化されたdSaCas9変異体を用いれば、より多くの遺伝子をベクターに搭載することができる。
ガイドRNAは、本発明のdSaCas9変異体と結合して、該タンパク質を標的DNAに導く機能を有する。ガイドRNAは、その5’末端に標的DNAに相補的な配列を有し、該相補的な配列を介して標的DNAに結合することにより、本発明のdSaCas9変異体を標的DNAに導く。dSaCas9変異体はDNAエンドヌクレアーゼを有さない為、標的DNAに結合はするが切断することはない。
ガイドRNAは、標的DNAの配列情報に基づき設計され調製される。具体的には実施例で用いられるような配列が挙げられる。
本明細書中において、「PAM配列」とは、標的二本鎖ポリヌクレオチド中に存在し、Cas9タンパク質により認識可能な配列を意味し、PAM配列の長さや塩基配列は細菌種によって異なる。
721~755位の間に連続した欠失領域を有し、
該欠失領域にそれぞれ隣接したアミノ酸が3乃至10個のアミノ酸残基からなるリンカーによって連結しているアミノ酸配列を含む配列からなり、
且つ、ガイドRNAとの結合能を有するタンパク質(態様1)を提供する。
態様1-1のタンパク質及びそれをコードする遺伝子としては、それぞれ配列番号4で表されるアミノ酸配列からなる蛋白質、及び配列番号3で表される塩基配列からなる遺伝子が挙げられる。当該タンパク質は後述するdSaCas9変異体T10に相当する。
態様1-2のタンパク質及びそれをコードする遺伝子としては、それぞれ配列番号6で表されるアミノ酸配列からなる蛋白質、及び配列番号5で表される塩基配列からなる遺伝子が挙げられる。当該タンパク質は後述するdSaCas9変異体T11に相当する。
45位及び/又は163位における変異は、具体的には、グルタミン酸の塩基性アミノ酸への置換、好ましくはリジン、アルギニン又はヒスチジンへの置換、より好ましくはリジンへの置換である。
「アミノ酸の置換、欠失、挿入及び/又は付加」を人為的に行う場合の手法としては、例えば、所定のアミノ酸配列をコードするDNAに対して慣用の部位特異的変異導入を施し、その後このDNAを常法により発現させる手法が挙げられる。ここで部位特異的変異導入法としては、例えば、アンバー変異を利用する方法(ギャップド・デュプレックス法、Nucleic Acids Res., 12, 9441-9456 (1984))、変異導入用プライマーを用いたPCRによる方法等が挙げられる。また、Q5 Site-Directed Mutagenesis Kit (NEB)を用いて、マニュアルに従い簡便に実施することができる。
前記で改変されるアミノ酸の数については、少なくとも1残基、具体的には1若しくは数個、またはそれ以上である。また前記置換、欠失、挿入または付加のうち、特にアミノ酸の置換が好ましい。当該置換は、疎水性、電荷、pK、立体構造上における特徴等の類似した性質を有するアミノ酸への置換がより好ましい。このような置換としては、例えば、i)グリシン、アラニン;ii)バリン、イソロイシン、ロイシン;iii)アスパラギン酸、グルタミン酸、アスパラギン、グルタミン;iv)セリン、スレオニン;v)リジン、アルギニン;vi)フェニルアラニン、チロシンのグループ内での置換が挙げられる。
-SGGGS-(配列番号7)
-GGSGGS-(配列番号8)
-SGSGSGSG-(配列番号9)
-SGSGSGSGS-(配列番号10)
dSaCas9変異体T10及びT11では該リンカーは-SGGGS-が用いられている。
宿主としては、特に限定されず、動物細胞、植物細胞、昆虫細胞、又は、大腸菌、枯草菌、酵母等の微生物が挙げられる。好ましくは動物細胞である。
一実施形態において、本発明は、上述の<dSaCas9変異体>において示されたタンパク質と、標的二本鎖ポリヌクレオチド中のPAM(Proto-spacer Adjacent Motif)配列の1塩基上流から20塩基以上24塩基以下上流までの塩基配列に相補的な塩基配列からなるポリヌクレオチドを含むガイドRNAと、を備えるタンパク質-RNA複合体を提供する。
一実施形態において、本発明は、上述の<dSaCas9変異体>において示されたタンパク質をコードする遺伝子を含む第1のベクターと、標的二本鎖ポリヌクレオチド中のPAM配列の1塩基上流から20塩基以上24塩基以下上流までの塩基配列に相補的な塩基配列からなるポリヌクレオチドを含むガイドRNAを含む第2のベクターと、を備えるCRISPR-Casベクターシステムを提供する。
別の一実施形態において、本発明は上述の<dSaCas9変異体>において示されたタンパク質をコードする遺伝子と、標的二本鎖ポリヌクレオチド中のPAM配列の1塩基上流から20塩基以上24塩基以下上流までの塩基配列に相補的な塩基配列からなるポリヌクレオチドを含むガイドRNAとを同一のベクター中に有するCRISPR-Casベクターシステムを提供する。
[第1実施形態]
一実施形態において、本発明は、標的二本鎖ポリヌクレオチドを部位特異的に改変するための方法であって、
標的二本鎖ポリヌクレオチドと、タンパク質と、ガイドRNAとを混合し、インキュベートする工程と、前記タンパク質が、PAM配列の上流に位置する結合部位で前記標的二本鎖ポリヌクレオチドを改変する工程と、を備え、
前記標的二本鎖ポリヌクレオチドは、PAM配列を有し、
前記タンパク質は、上述の<dSaCas9変異体>において示されたタンパク質であり、
前記ガイドRNAは、前記標的二本鎖ポリヌクレオチド中の前記PAM配列の1塩基上流から20塩基以上24塩基以下上流までの塩基配列に相補的な塩基配列からなるポリヌクレオチドを含むものである方法を提供する。
まず、前記タンパク質及び前記ガイドRNAを温和な条件で混合し、インキュベートする。温和な条件とは、上述のとおりである。インキュベートする時間は、0.5時間以上1時間以下が好ましい。前記タンパク質及び前記ガイドRNAによる複合体は、安定しており、室温で数時間静置しても安定性を保つことができる。
次に、前記標的二本鎖ポリヌクレオチド上において、前記タンパク質及び前記ガイドRNAは複合体を形成する。前記タンパク質は、PAM配列を認識し、PAM配列の上流に位置する結合部位で、前記標的二本鎖ポリヌクレオチドに結合する。続いて、前記ガイドRNAと前記二本鎖ポリヌクレオチドの相補的結合によって決定される領域において、目的に応じた改変が施された標的二本鎖ポリヌクレオチドを得ることができる。
本発明のdSaCas9変異体は、エンドヌクレアーゼ活性を有さないので該タンパク質はPAM配列の上流に位置する結合部位で前記標的二本鎖ポリヌクレオチドに結合することができるが、そこにとどまって切断することができない。従って、例えば該タンパク質に蛍光タンパク質(例、GFP)等の標識タンパク質を融合させておくと、dSaCas9変異体タンパク質-ガイドRNAを介して標識タンパク質を標的二本鎖ポリヌクレオチドに結合させることができる。dSaCas9変異体に結合させる物質を適宜選択することによって多様な機能を標的二本鎖ポリヌクレオチドに与えることが可能となる。
さらに、dSaCas9変異体タンパク質のN末端あるいはC末端に転写制御因子タンパク質又はドメインを連結することができる。転写制御因子又はそのドメインとしては、転写活性化因子又はそのドメイン(例、VP64、VP160、NF-κB p65)及び転写サイレンサー又はそのドメイン(例、ヘテロクロマチンタンパク質1(HP1))又は転写抑制因子又はそのドメイン(例、クルッペル関連ボックス(KRAB)、ERFリプレッサードメイン(ERD)、mSin3A相互作用ドメイン(SID))が挙げられる。
DNAのメチル化状態を修飾する酵素(例、DNAメチルトランスフェラーゼ(DNMT)、TET)やヒストンサブユニットを修飾する酵素(例、ヒストンアセチルトランスフェラーゼ(HAT)、ヒストンデアセチラーゼ(HDAC)、ヒストンメチルトランスフェラーゼ、ヒストンデメチラーゼ)を連結することもできる。
本実施形態において、インキュベート工程の前に、さらに、上述のCRISPR-Casベクターシステムを用いて、上述の<dSaCas9変異体>において示されたタンパク質と、ガイドRNAとを発現させる発現工程を備えていてもよい。
一実施形態において、本発明は、標的二本鎖ポリヌクレオチドを細胞内において部位特異的に改変するための方法であって、
上述のCRISPR-Casベクターシステムを細胞に導入し、上述の<dSaCas9変異体>において示されたタンパク質と、ガイドRNAとを発現させる発現工程と、
前記タンパク質が、PAM配列の上流に位置する結合部位で前記標的二本鎖ポリヌクレオチドに結合する工程と、
前記ガイドRNAと前記標的二本鎖ポリヌクレオチドの相補的結合によって決定される領域において、改変された前記標的二本鎖ポリヌクレオチドを得る工程と、を備え、
前記ガイドRNAは、前記標的二本鎖ポリヌクレオチド中の前記PAM配列の1塩基上流から20塩基以上24塩基以下上流までの塩基配列に相補的な塩基配列からなるポリヌクレオチドを含むものである方法を提供する。
本実施形態における標的二本鎖ポリヌクレオチドの修飾により、標的二本鎖ポリヌクレオチドが改変された細胞を得ることができる。
(方法)
1.クローニング
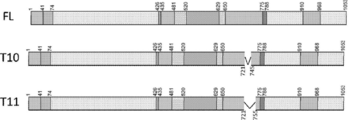
NEB Q5 Site-Directed Mutagenesis Kitを用いて、dSaCas9遺伝子内に、所定の欠失領域を作り、リンカーをコードする遺伝子を導入し、さらに転写制御因子であるKRAB遺伝子を融合させて種々のdSaCas9を作製した(図1)。これらのdSaCas9について、MYD88遺伝子を用いてその発現抑制活性を調べた。全てのdSaCas9の遺伝子構築物をpX601ベクターに組み込んだ(F. Ann Ran et al., Nature 2015; 520(7546); pp.186-191)。各ベクターは、以下のdSaCas9構築物(FL, T10, T11)及びsgRNAを発現する。
[dSaCas9変異体]
FL;野生型dSaCas9(配列番号1、配列番号2)
T10;野生型dSaCas9において欠失領域(721~745位)を有する欠失変異体(配列番号3、配列番号4)
T11;野生型dSaCas9において欠失領域(721~755位)を有する欠失変異体(配列番号5、配列番号6)
T10及びT11における各欠失領域の両端はリンカーペプチドで連結している。
dSaCas9構築物はdSaCas9-KRAB-P2A-sfGFP又はdSaCas9-VR-P2A-sfGFPの融合タンパク質として発現される。
[crRNA配列]
C1;ACGGAGGCUAAGCGUCGCAA(配列番号11)
A2;GGAGCCACAGUUCUUCCACGG(配列番号12)
A3;CUCUACCCUUGAGGUCUCGAG(配列番号13)
上記crRNA配列は以下のtracrRNA配列と融合させsgRNAを形成させベクターから発現させるようにした。
[tracrRNA配列]
GUUUUAGUACUCUGGAAACAGAAUCUACUAAAACAAGGCAAAAUGCCGUGUUUAUCACGUCAACUUGUUGGCGAGAUUUUUUU(配列番号14)
2.細胞トランスフェクション
HEK293FT細胞をトランスフェクションの24時間前に1ウェルあたり75,000細胞の密度で24ウェルプレートに播種し、10%FBS、2mMの新鮮なL-グルタミン、1mMのピルビン酸ナトリウム及び非必須アミノ酸を添加したDMEM培地で培養した。各プラスミド(500ng)を1.5μlのリポフェクトアミン2000(Life technologies)を用い、取扱説明書に従って細胞にトランスフェクトした。遺伝子発現解析の為に、トランスフェクション後48~72時間で細胞を回収しRLTバッファー(Qiagen)に溶解しRNeasy kit (Qiagen)を用いて全RNAを抽出した。
3.遺伝子発現解析
Taqman解析の為に、1.5μgの全RNAから、20μlの容量でTaqManTM High-Capacity RNA-to-cDNA Kit (Applied Biosystems)を用いてcDNAを調製した。調製されたcDNAを20倍に希釈し、Taqman反応あたり6.33μlを用いた。MYD88遺伝子及びHPRT遺伝子に対するTaqmanプライマー及びプローブはApplied Biosystemsより入手した。Roche LightCycler 96又はLightCycler 480中、Taqman gene expression master mix (ThermoFisher)を用いてTaqman反応を行い、LightCycler 96 analysisソフトウェアを用いて解析した。
Taqman probe product IDs:
MYD88: Hs01573837_g1 (FAM)
HPRT: Hs99999909_m1 (FAM, VIC)
Taqman QPCR condition:
Step 1; 95℃ 10 min
Step 2; 95℃ 15 sec
Step 3; 60℃ 30 sec
Repeat Step 2 and 3; 40 times
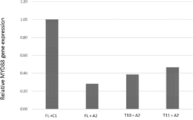
pX601-dSaCas9(FL, T10又はT11)-KRAB-P2A-sfGFP及びsgRNA-A2を発現するプラスミドベクターでHEK293FT細胞をトランスフェクトした。sgRNA-A2の代わりにsgRNA-C1を発現するベクターでトランスフェクトした細胞をネガティブコントロールとして用いた。トランスフェクトした細胞を3日後に回収しRNAを単離した。MYD88遺伝子の発現をTaqmanアッセイで解析し、HPRT遺伝子の発現レベルで標準化した。MYD88遺伝子の相対的な発現レベルの結果を図2に示す。
本発明のdSaCas9変異体におけるMYD88遺伝子発現レベルはコントロールに比べ野生型dSaCas9に匹敵する程度に低かった。この結果より、本発明のdSaCas9変異体は、欠失領域が存在し、全長のdSaCas9に比べて縮小されているにもかかわらず、ガイドRNAとの結合能、ひいてはDNA結合親和性が維持されていることが示された。
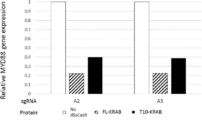
上記結果において、特にDNA結合親和性が高かったT10について、ガイドRNAを変えて、その結合親和性を確認した。pX601-dSaCas9(FL又はT10)-KRAB-P2A-sfGFP及びsgRNA-A2又はsgRNA-A3を発現するプラスミドベクターでHEK293FT細胞をトランスフェクトした。dSaCas9変異体を発現しないベクターでトランスフェクトした細胞をネガティブコントロールとして用いた。トランスフェクトした細胞を3日後に回収しRNAを単離した。MYD88遺伝子の発現をTaqmanアッセイで解析し、HPRT遺伝子の発現レベルで標準化した。MYD88遺伝子の相対的な発現レベルの結果を図3に示す。sgRNA-A3を用いた場合でもsgRNA-A2同様の効果が確認された。
本発明のdSaCas9変異体におけるMYD88遺伝子発現レベルはコントロールに比べ野生型dSaCas9に匹敵する程度に低かった。この結果より、本発明のdSaCas9変異体は、欠失領域が存在し、全長のdSaCas9に比べて縮小されているにもかかわらず、ガイドRNAとの結合能、ひいてはDNA結合親和性が維持されていることが示された。
Claims (22)
- 配列番号2で表されるアミノ酸配列において、
721~755位の間に連続した欠失領域を有し、
該欠失領域にそれぞれ隣接したアミノ酸が3乃至10個のアミノ酸残基からなるリンカーによって連結しているアミノ酸配列を含む配列からなり、
且つ、ガイドRNAとの結合能を有するタンパク質。 - リンカーが、グリシン(G)及びセリン(S)で構成される、5~9アミノ酸長のリンカーである、請求項1に記載のタンパク質。
- リンカーが、以下から選択される、請求項2に記載のタンパク質:
-SGGGS-
-GGSGGS-
-SGSGSGSG-
-SGSGSGSGS-。 - 該欠失領域が、721~745位領域
である、請求項1~3のいずれか1項に記載のタンパク質。 - 配列番号4で表される請求項4記載のタンパク質。
- 該欠失領域が、721~755位領域
である、請求項1~3のいずれか1項に記載のタンパク質。 - 配列番号6で表される請求項6記載のタンパク質。
- さらに、45位及び/又は163位のグルタミン酸(E)が他のアミノ酸に置換されている、請求項1~7のいずれか1項に記載のタンパク質。
- 他のアミノ酸が塩基性アミノ酸である、請求項8記載のタンパク質。
- 塩基性アミノ酸がリジン(K)である、請求項9記載のタンパク質。
- 配列番号2の変異及び/又は欠失が施された位置以外の部位において80%以上の同一性を有する、請求項1~10のいずれか1項に記載のタンパク質。
- 配列番号2の変異及び/又は欠失が施された位置以外の部位において1~数個のアミノ酸が置換、欠失、挿入及び/又は付加された、請求項1~10のいずれか1項に記載のタンパク質。
- 転写制御因子タンパク質又はドメインを連結した、請求項1~12のいずれか1項に記載のタンパク質。
- 転写制御因子が転写活性化因子である、請求項13記載のタンパク質。
- 転写制御因子が転写サイレンサー又は転写抑制因子である、請求項13記載のタンパク質。
- 請求項1~15のいずれか1項に記載のタンパク質をコードする核酸。
- 請求項1~16のいずれか1項に記載のタンパク質と、標的二本鎖ポリヌクレオチド中のPAM(Proto-spacer Adjacent Motif)配列の1塩基上流から20塩基以上24塩基以下上流までの塩基配列に相補的な塩基配列からなるポリヌクレオチドを含むガイドRNAと、を備えるタンパク質-RNA複合体。
- 標的二本鎖ポリヌクレオチドを部位特異的に改変するための方法であって、
標的二本鎖ポリヌクレオチドと、タンパク質と、ガイドRNAとを混合し、インキュベートする工程と、
前記タンパク質が、PAM配列の上流に位置する結合部位で前記標的二本鎖ポリヌクレオチドを改変する工程と、を備え、
前記タンパク質は、請求項1~15のいずれか1項に記載のタンパク質であり、
前記ガイドRNAは、前記標的二本鎖ポリヌクレオチド中の前記PAM配列の1塩基上流から20塩基以上24塩基以下上流までの塩基配列に相補的な塩基配列からなるポリヌクレオチドを含むものである方法。 - 細胞の標的遺伝子の発現を増大させる方法であって、前記細胞内で請求項14に記載のタンパク質と、前記標的遺伝子に対する1つ又は複数のガイドRNAとを発現させることを含む、方法。
- 細胞の標的遺伝子の発現を減少させる方法であって、前記細胞内で請求項15に記載のタンパク質と、前記標的遺伝子に対する1つ又は複数のガイドRNAとを発現させることを含む、方法。
- 細胞が真核細胞である、請求項19又は20に記載の方法。
- 細胞が酵母細胞、植物細胞又は動物細胞である、請求項19又は20に記載の方法。
Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201980069492.0A CN113166738A (zh) | 2018-10-24 | 2019-10-24 | 经改造的Cas9蛋白及其用途 |
| US17/288,379 US20210246473A1 (en) | 2018-10-24 | 2019-10-24 | Modified cas9 protein, and use thereof |
| BR112021007672-7A BR112021007672A2 (pt) | 2018-10-24 | 2019-10-24 | proteína cas9 modificada e uso da mesma |
| AU2019365440A AU2019365440A1 (en) | 2018-10-24 | 2019-10-24 | Modified Cas9 protein, and use thereof |
| CA3116994A CA3116994A1 (en) | 2018-10-24 | 2019-10-24 | Modified cas9 protein, and use thereof |
| KR1020217014297A KR20210082185A (ko) | 2018-10-24 | 2019-10-24 | 개변된 Cas9 단백질 및 그의 용도 |
| JP2020552595A JPWO2020085441A1 (ja) | 2018-10-24 | 2019-10-24 | 改変されたCas9タンパク質及びその用途 |
| EP19876272.6A EP3878956A4 (en) | 2018-10-24 | 2019-10-24 | MODIFIED CAS9 PROTEIN AND RELATED USE |
| SG11202104097TA SG11202104097TA (en) | 2018-10-24 | 2019-10-24 | Modified cas9 protein, and use thereof |
| MX2021004714A MX2021004714A (es) | 2018-10-24 | 2019-10-24 | Proteina cas9 modificada, y uso de la misma. |
| IL282423A IL282423A (en) | 2018-10-24 | 2021-04-19 | CAS9 protein is strange, and its use |
| ZA2021/03259A ZA202103259B (en) | 2018-10-24 | 2021-05-13 | Modified cas9 protein, and use thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862749855P | 2018-10-24 | 2018-10-24 | |
| US62/749,855 | 2018-10-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020085441A1 true WO2020085441A1 (ja) | 2020-04-30 |
Family
ID=70331763
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2019/041751 WO2020085441A1 (ja) | 2018-10-24 | 2019-10-24 | 改変されたCas9タンパク質及びその用途 |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US20210246473A1 (ja) |
| EP (1) | EP3878956A4 (ja) |
| JP (1) | JPWO2020085441A1 (ja) |
| KR (1) | KR20210082185A (ja) |
| CN (1) | CN113166738A (ja) |
| AU (1) | AU2019365440A1 (ja) |
| BR (1) | BR112021007672A2 (ja) |
| CA (1) | CA3116994A1 (ja) |
| IL (1) | IL282423A (ja) |
| MX (1) | MX2021004714A (ja) |
| SG (1) | SG11202104097TA (ja) |
| WO (1) | WO2020085441A1 (ja) |
| ZA (1) | ZA202103259B (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020241903A1 (en) | 2019-05-28 | 2020-12-03 | Astellas Pharma Inc. | Method for treating muscular dystrophy by targeting dmpk gene |
| WO2021230385A1 (en) | 2020-05-15 | 2021-11-18 | Astellas Pharma Inc. | Method for treating muscular dystrophy by targeting utrophin gene |
| WO2022045366A1 (en) * | 2020-08-31 | 2022-03-03 | Modalis Therapeutics Corporation | Method for treating facioscapulohumeral muscular dystrophy (fshd) by targeting dux4 gene |
| WO2022114243A1 (en) | 2020-11-25 | 2022-06-02 | Astellas Pharma Inc. | Method for treating muscular dystrophy by targeting dmpk gene |
| WO2022145495A1 (en) * | 2021-01-04 | 2022-07-07 | Modalis Therapeutics Corporation | Method for treating spinocerebellar ataxias (sca) by targeting atxn7 gene |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023069923A1 (en) * | 2021-10-18 | 2023-04-27 | Duke University | Compositions and methods relating to epigenetic modulation |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016141224A1 (en) | 2015-03-03 | 2016-09-09 | The General Hospital Corporation | Engineered crispr-cas9 nucleases with altered pam specificity |
| WO2016196655A1 (en) * | 2015-06-03 | 2016-12-08 | The Regents Of The University Of California | Cas9 variants and methods of use thereof |
| WO2016205759A1 (en) * | 2015-06-18 | 2016-12-22 | The Broad Institute Inc. | Engineering and optimization of systems, methods, enzymes and guide scaffolds of cas9 orthologs and variants for sequence manipulation |
| WO2017010543A1 (ja) | 2015-07-14 | 2017-01-19 | 国立大学法人東京大学 | 改変されたFnCas9タンパク質及びその使用 |
| JP2017501688A (ja) * | 2013-11-19 | 2017-01-19 | プレジデント アンド フェローズ オブ ハーバード カレッジ | 変異cas9タンパク質 |
| WO2018074979A1 (en) | 2016-10-17 | 2018-04-26 | Nanyang Technological University | Truncated crispr-cas proteins for dna targeting |
| WO2019089910A1 (en) * | 2017-11-01 | 2019-05-09 | Ohio State Innovation Foundation | Highly compact cas9-based transcriptional regulators for in vivo gene regulation |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11674128B2 (en) * | 2016-12-12 | 2023-06-13 | Tsinghua University | Engineering of a minimal SaCas9 CRISPR/Cas system for gene editing and transcriptional regulation optimized by enhanced guide RNA |
| AU2019317066A1 (en) * | 2018-08-07 | 2021-02-18 | Modalis Therapeutics Corporation | Novel transcription activator |
-
2019
- 2019-10-24 US US17/288,379 patent/US20210246473A1/en active Pending
- 2019-10-24 AU AU2019365440A patent/AU2019365440A1/en not_active Abandoned
- 2019-10-24 WO PCT/JP2019/041751 patent/WO2020085441A1/ja unknown
- 2019-10-24 BR BR112021007672-7A patent/BR112021007672A2/pt not_active Application Discontinuation
- 2019-10-24 CN CN201980069492.0A patent/CN113166738A/zh active Pending
- 2019-10-24 SG SG11202104097TA patent/SG11202104097TA/en unknown
- 2019-10-24 MX MX2021004714A patent/MX2021004714A/es unknown
- 2019-10-24 EP EP19876272.6A patent/EP3878956A4/en not_active Withdrawn
- 2019-10-24 CA CA3116994A patent/CA3116994A1/en active Pending
- 2019-10-24 KR KR1020217014297A patent/KR20210082185A/ko unknown
- 2019-10-24 JP JP2020552595A patent/JPWO2020085441A1/ja active Pending
-
2021
- 2021-04-19 IL IL282423A patent/IL282423A/en unknown
- 2021-05-13 ZA ZA2021/03259A patent/ZA202103259B/en unknown
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017501688A (ja) * | 2013-11-19 | 2017-01-19 | プレジデント アンド フェローズ オブ ハーバード カレッジ | 変異cas9タンパク質 |
| WO2016141224A1 (en) | 2015-03-03 | 2016-09-09 | The General Hospital Corporation | Engineered crispr-cas9 nucleases with altered pam specificity |
| WO2016196655A1 (en) * | 2015-06-03 | 2016-12-08 | The Regents Of The University Of California | Cas9 variants and methods of use thereof |
| WO2016205759A1 (en) * | 2015-06-18 | 2016-12-22 | The Broad Institute Inc. | Engineering and optimization of systems, methods, enzymes and guide scaffolds of cas9 orthologs and variants for sequence manipulation |
| WO2017010543A1 (ja) | 2015-07-14 | 2017-01-19 | 国立大学法人東京大学 | 改変されたFnCas9タンパク質及びその使用 |
| WO2018074979A1 (en) | 2016-10-17 | 2018-04-26 | Nanyang Technological University | Truncated crispr-cas proteins for dna targeting |
| WO2019089910A1 (en) * | 2017-11-01 | 2019-05-09 | Ohio State Innovation Foundation | Highly compact cas9-based transcriptional regulators for in vivo gene regulation |
Non-Patent Citations (7)
| Title |
|---|
| F. ANN RAN ET AL., NATURE, vol. 520, no. 7546, 2015, pages 186 - 191 |
| MA, D. ET AL.: "Rational Design of Mini-Cas9 for Transcriptional Activation", ACS SYNTH. BIOL., vol. 7, no. 4, March 2018 (2018-03-01), pages 978 - 985, XP055661686, DOI: 10.1021/acssynbio.7b00404 * |
| MYERSMILLER, CABIOS, vol. 4, 1988, pages 11 - 17 |
| NEEDLEMAN ET AL., J. MOL. BIOL., vol. 48, 1970, pages 444 - 453 |
| NUCLEIC ACIDS RES, vol. 12, 1984, pages 9441 - 9456 |
| RAN, FA. ET AL.: "In vivo genome editing using Staphylococcus aureus Cas9", NATURE, vol. 520, no. 7546, 2015, pages 186 - 191, XP055484527, DOI: 10.1038/nature14299 * |
| See also references of EP3878956A4 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020241903A1 (en) | 2019-05-28 | 2020-12-03 | Astellas Pharma Inc. | Method for treating muscular dystrophy by targeting dmpk gene |
| WO2021230385A1 (en) | 2020-05-15 | 2021-11-18 | Astellas Pharma Inc. | Method for treating muscular dystrophy by targeting utrophin gene |
| WO2022045366A1 (en) * | 2020-08-31 | 2022-03-03 | Modalis Therapeutics Corporation | Method for treating facioscapulohumeral muscular dystrophy (fshd) by targeting dux4 gene |
| WO2022114243A1 (en) | 2020-11-25 | 2022-06-02 | Astellas Pharma Inc. | Method for treating muscular dystrophy by targeting dmpk gene |
| WO2022145495A1 (en) * | 2021-01-04 | 2022-07-07 | Modalis Therapeutics Corporation | Method for treating spinocerebellar ataxias (sca) by targeting atxn7 gene |
Also Published As
| Publication number | Publication date |
|---|---|
| BR112021007672A2 (pt) | 2021-08-10 |
| EP3878956A4 (en) | 2022-07-06 |
| US20210246473A1 (en) | 2021-08-12 |
| ZA202103259B (en) | 2022-08-31 |
| IL282423A (en) | 2021-06-30 |
| AU2019365440A1 (en) | 2021-05-20 |
| JPWO2020085441A1 (ja) | 2021-09-16 |
| MX2021004714A (es) | 2021-06-04 |
| EP3878956A1 (en) | 2021-09-15 |
| SG11202104097TA (en) | 2021-05-28 |
| CA3116994A1 (en) | 2020-04-30 |
| CN113166738A (zh) | 2021-07-23 |
| KR20210082185A (ko) | 2021-07-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11702645B2 (en) | Polynucleotide encoding modified CAS9 protein | |
| WO2020085441A1 (ja) | 改変されたCas9タンパク質及びその用途 | |
| US20230279374A1 (en) | Modified cas9 protein, and use thereof | |
| JP7412001B2 (ja) | 改変されたCas9タンパク質及びその用途 | |
| JPWO2018221685A6 (ja) | 改変されたCas9タンパク質及びその用途 | |
| WO2017010543A1 (ja) | 改変されたFnCas9タンパク質及びその使用 | |
| KR20240052763A (ko) | 스타스민 2 (stmn2)를 표적화하는 rna 가이드를 포함하는 유전자 편집 시스템 및 이의 용도 | |
| WO2019026976A1 (ja) | 改変されたCas9タンパク質及びその用途 | |
| JP2023505234A (ja) | ヌクレアーゼを含む組成物及びその使用 | |
| JP2023539569A (ja) | ヌクレアーゼを含む組成物及びその使用 | |
| TW202246497A (zh) | Cas12i2融合分子及其用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19876272 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2020552595 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 3116994 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112021007672 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 20217014297 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2019365440 Country of ref document: AU Date of ref document: 20191024 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2019876272 Country of ref document: EP Effective date: 20210525 |
|
| ENP | Entry into the national phase |
Ref document number: 112021007672 Country of ref document: BR Kind code of ref document: A2 Effective date: 20210422 |