WO2018125548A1 - Methods for the preparation of 6-aminoisoquinoline - Google Patents
Methods for the preparation of 6-aminoisoquinoline Download PDFInfo
- Publication number
- WO2018125548A1 WO2018125548A1 PCT/US2017/065631 US2017065631W WO2018125548A1 WO 2018125548 A1 WO2018125548 A1 WO 2018125548A1 US 2017065631 W US2017065631 W US 2017065631W WO 2018125548 A1 WO2018125548 A1 WO 2018125548A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- aminoisoquinoline
- compound
- nitroisoquinoline
- mol
- reacting
- Prior art date
Links
- NGFCTYXFMDWFRQ-UHFFFAOYSA-N Nc(cc1)cc2c1cncc2 Chemical compound Nc(cc1)cc2c1cncc2 NGFCTYXFMDWFRQ-UHFFFAOYSA-N 0.000 description 1
- DRGUQIQEUWFBDE-UHFFFAOYSA-N Nc1ccc(ccnc2)c2c1 Chemical compound Nc1ccc(ccnc2)c2c1 DRGUQIQEUWFBDE-UHFFFAOYSA-N 0.000 description 1
- SMZLRKAOZOGDFK-UHFFFAOYSA-N [O-][N+](c(cc1CC(N2)=O)ccc1C2=O)=O Chemical compound [O-][N+](c(cc1CC(N2)=O)ccc1C2=O)=O SMZLRKAOZOGDFK-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/02—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with only hydrogen atoms or radicals containing only carbon and hydrogen atoms, directly attached to carbon atoms of the nitrogen-containing ring; Alkylene-bis-isoquinolines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C201/00—Preparation of esters of nitric or nitrous acid or of compounds containing nitro or nitroso groups bound to a carbon skeleton
- C07C201/06—Preparation of nitro compounds
- C07C201/12—Preparation of nitro compounds by reactions not involving the formation of nitro groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/22—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the nitrogen-containing ring
Definitions
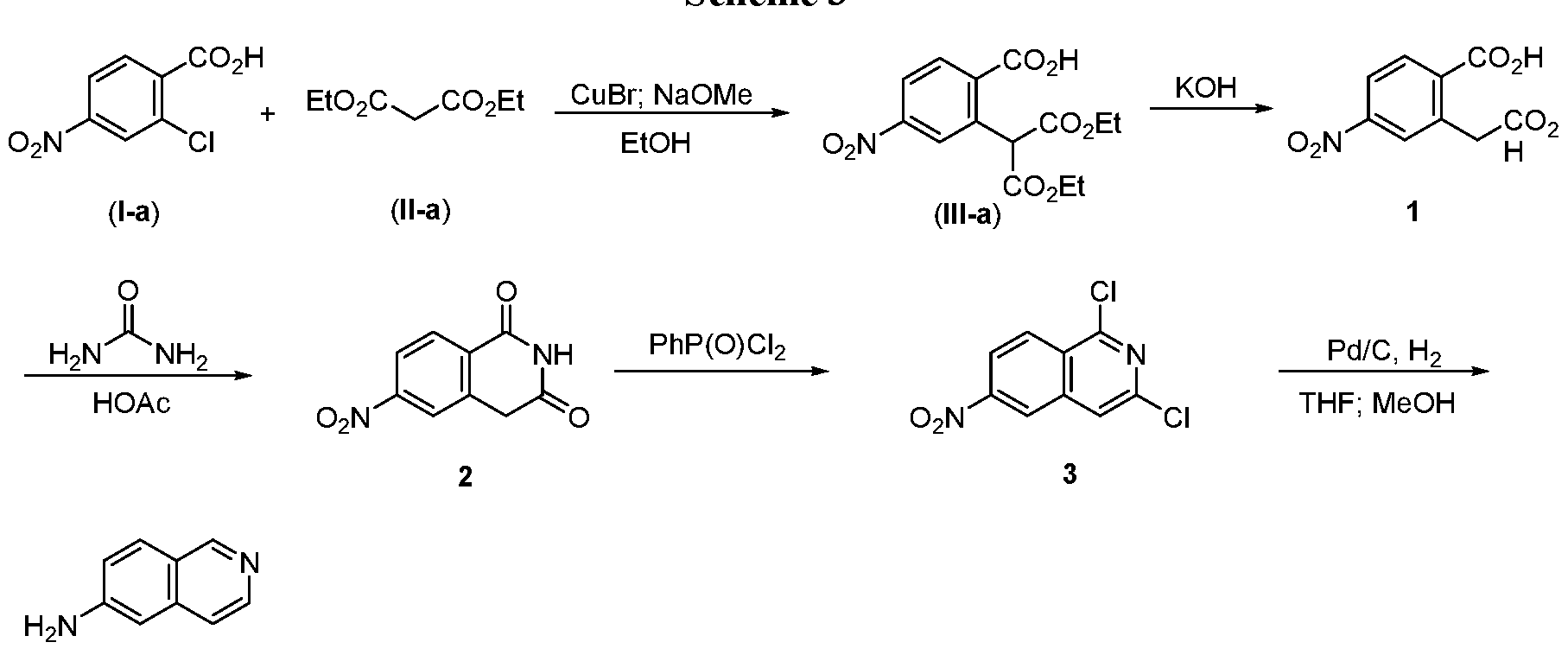
- the present disclosure relates to methods for preparing 6-aminoisoquinoline, an intermediate compound in the synthesis of compounds useful for treating kinase-related diseases and/or disorders.
- the method may comprise converting l,3-dichloro-6-nitroisoquinoline,
- 6- Aminoisoquinoline is a useful intermediate for the synthesis of kinase inhibitors.
- 6- Aminoisoquinoline may be prepared in a manner that efficiently generates large scale quantities and enables the production of kinase inhibitors, which can be used to treat or prevent kinase-related diseases and/or disorders.
- diseases and disorders related to the eye e.g., glaucoma and ocular hypertension, and retinal diseases such as AMD, Diabetic Retinopathy, DME and inflammatory diseases of the retina
- the respiratory system e.g., glaucoma and ocular hypertension
- DME Diabetic Retinopathy
- inflammatory diseases of the retina e.g., AMD, Diabetic Retinopathy, DME and inflammatory diseases of the retina
- the modifier "about” or “at least” used in connection with a quantity is inclusive of the stated value and has the meaning dictated by the context (for example, it includes at least the degree of error associated with the measurement of the particular quantity).
- the modifier “about” or “at least” should also be considered as disclosing the range defined by the absolute values of the two endpoints.
- the expression “from about or least 2 to about or at least 4" also discloses the range “from 2 to 4.”
- the term “about or at least” may refer to plus or minus 10% of the indicated number.
- “about 10%” may indicate a range of 9% to 11%, and “about 1" may mean from 0.9-1.1.
- Other meanings of "about or at least” may be apparent from the context, such as rounding off, so, for example "about 1” may also mean from 0.5 to 1.4.
- alkyl as used herein, means a straight or branched, saturated
- hydrocarbon chain containing from 1 to 10 carbon atoms The term “lower alkyl” or “C ⁇ -C(, alkyl” means a straight or branched chain hydrocarbon containing from 1 to 6 carbon atoms.
- C3-C7 branched alkyl means a branched chain hydrocarbon containing from 3 to 7 carbon atoms.
- C 1 -C4 alkyl means a straight or branched chain hydrocarbon containing from 1 to 4 carbon atoms.
- alkyl include, but are not limited to, methyl, ethyl, ⁇ -propyl, wo-propyl, w-butyl, sec-butyl, z ' so-butyl, fert-butyl, n- pentyl, isopentyl, neopentyl, w-hexyl, 3-methylhexyl, 2,2-dimethylpentyl, 2,3-dimethylpentyl, ft-heptyl, w-octyl, «-nonyl, and w-decyl.
- An alkyl may be substituted or unsubstituted.
- alkylene refers to a divalent group derived from a straight or branched chain hydrocarbon of 1 to 10 carbon atoms, for example, of 2 to 5 carbon atoms.
- Representative examples of alkylene include, but are not limited to, -CH 2 CH 2 -, - CH2CH2CH2-, -CH2CH2CH2CH2-, and -CH2CH2CH2CH2CH2-.
- An alkylene may be substituted or unsubstituted.
- aryl refers to a phenyl group, or a bicyclic fused ring system.
- Bicyclic fused ring systems are exemplified by a phenyl group appended to the parent molecular moiety and fused to a cycloalkyl group, as defined herein, a phenyl group, a heteroaryl group, as defined herein, or a heterocycle, as defined herein.
- Representative examples of aryl include, but are not limited to, indolyl, naphthyl, phenyl, quinolinyl and tetrahydroquinolinyl.
- Aryl groups may be substituted or unsubstituted. Suitable substituents of the aryl groups may be, but are not limited to alkyl, nitro, cyano, halo, haloalkyl and sulfonyl.
- halogen or "halo" as used herein, means CI, Br, I, or F.
- haloalkyl as used herein, means at least one halo group, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- haloalkyl examples include trifluoromethyl and 2,2,2-trifluoroethyl.
- catalyst loading refers to the amount of catalyst used in a reaction, and is typically reported as mol %.
- the catalyst loading is determined as a molar percentage of the limiting reactant of a particular reaction. For example, a reaction with 1 mole of starting material may require 0.1 moles of catalyst, which is equivalent to 10.0 mol % catalyst loading.
- groups and substituents thereof may be selected in accordance with permitted valence of the atoms and the substituents, such that the selections and substitutions result in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc., at normal temperatures and pressures.
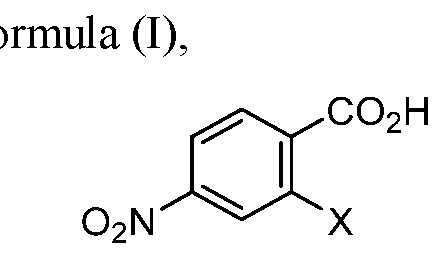
- the methods may include reacting the compound of formula (I), wherein X is halogen or OSC R 3 ; and R a is aryl, alkyl or haloalkyl; with the compound of formula (II), wherein R is alkyl, to form the compound of formula (III), wherein R is alkyl; transforming the compound of formula (III) to 2-(carboxymethyl)-4-nitrobenzoic acid (compound 1) by hydrolysis of the esters and decarboxylation; reacting 2-(carboxymethyl)-4-nitrobenzoic acid (compound 1) with urea to form the bicyclic 6-nitroisoquinoline-l,3(2H, 4H)-dione
- the methods may comprise converting l ,3-dichloro-6-nitroisoquinoline (compound
- converting l ,3-dichloro-6-nitroisoquinoline (compound 3) to 6-aminoisoquinoline (compound 4) comprises hydrogenation of l,3-dichloro-6- nitroisoquinoline (compound 3) in the presence of a metal catalyst.
- the metal catalyst comprises a transition metal.
- the transition metal is selected from the group consisting of palladium, platinum, nickel, rhodium, ruthenium, iridium, cobalt and iron, and combinations thereof.
- the metal catalyst is palladium on carbon.
- the total catalyst loading in the reaction may be about or at least 0.1 mol% to about or at least 5.0 mol%.
- the catalyst loading may be about or at least about or at least 0.1 mol%, about or at least 0.2 mol%, about or at least 0.3 mol%, about or at least 0.4 mol%, about or at least 0.5 mol%, about or at least 0.6 mol%, about or at least 0.7 mol%, about or at least 0.8 mol%, about or at least 0.9 mol%, about or at least 1.0 mol%, about or at least 1.1 mol%, about or at least 1.2 mol%, about or at least 1.3 mol%, about or at least 1.4 mol%, about or at least 1.5 mol%, about or at least 1.6 mol%, about or at least 1.7 mol%, about or at least 1.8 mol%, about or at least 1.9 mol%, about or at least 2.0 mol%, about or at least 2.1 mol%, about or at least 2.2 mol%, about or at least 2.3 mol%, about or at least 2.4 mol%, about or at least 2.5
- l,3-dichloro-6-nitroisoquinoline (compound 3) may be converted to 6- aminoisoquinoline (compound 4) in the presence of a solvent or mixture of solvents.
- a solvent or mixture of solvents Any suitable solvent that is compatible with the components of the reaction mixture may be used.
- a solvent will be selected such that the starting materials will be at least partially soluble (or fully soluble) and will allow the reaction mixture to be heated, if necessary, to a temperature sufficient for the reaction to produce 6-aminoisoquinoline (compound 4).
- the solvents may include, but are not limited to: ethers such as diethyl ether, dibutyl ether, 1,2- dimethoxyethane, diglyme, t-butyl methyl ether, tetrahydrofuran and dioxane; aliphatic or aromatic hydrocarbon solvents such as benzene, xylene, toluene, hexane, and pentane; polar protic solvents such as methanol, ethanol, n-propanol, isopropanol, n-butanol, acetic acid and water; or any combination of two or more solvents.
- ethers such as diethyl ether, dibutyl ether, 1,2- dimethoxyethane, diglyme, t-butyl methyl ether, tetrahydrofuran and dioxane
- aliphatic or aromatic hydrocarbon solvents such as benzene, xylene, toluen
- the solvent is tetrahydrofuran. In certain embodiments, the solvent is a mixture of tetrahydrofuran and methanol.
- reaction mixture may be heated at a temperature greater than ambient or room temperature, wherein ambient or room temperature is about 18°C to about 25°C.
- the reaction mixture may be heated at a temperature of about 25°C to about 60°C, or about 30°C to about
- the reaction mixture may be heated at a temperature of about or at least 25°C, about or at least 30°C, about or at least 35°C, about or at least 40°C, about or at least 45°C, about or at least 50°C, about or at least 55°C, or about or at least 60°C.
- the reaction mixture may be heated at a temperature of less than about 60°C, less than about 55 °C, less than about 50 °C, less than about 45 °C, less than about 40 °C, less than about 35 °C, or less than about 30°C.
- reaction mixture may be pressurized to a pressure greater than atmospheric pressure, wherein ambient or atmospheric pressure is about or at least 0.1 MP.
- the reaction mixture may be pressurized to about 0.1 MP to about 1.0 MP, or about 0.2 MP to about 0.8 MP.
- the reaction mixture may be pressurized to at least 0.1 MP, at least 0.2 MP, at least 0.3 MP, at least 0.4 MP, at least 0.5 MP, at least 0.6 MP, at least 0.7 MP, or at least 0.8 MP.
- the reaction mixture may be pressurized to less than 0.8 MP, less than 0.7 MP, less than 0.6 MP, less than 0.5 MP, less than 0.4 MP, less than 0.3 MP, or less than about 0.2 MP.
- the reaction mixture may be pressurized to about 0.1 MP, about 0.2 MP, about 0.3 MP, about 0.4 MP, about 0.5 MP, about 0.6 MP, about 0.7 MP, or about 0.8 MP.
- reaction mixture other components may also be added to the reaction mixture, such as an acid, a base or a salt.
- potassium carbonate may be added to the reaction mixture.
- Complete hydrogenation of the carbon-chlorine bonds may be achieved by adding a second solvent, such as methanol; adding a salt, such as potassium carbonate; and adding palladium on carbon catalyst, and allowing the reaction to proceed at a pressure of about or at least 0.6 MP and a temperature of about or at least 45°C.
- a second solvent such as methanol
- a salt such as potassium carbonate
- palladium on carbon catalyst and allowing the reaction to proceed at a pressure of about or at least 0.6 MP and a temperature of about or at least 45°C.
- the methods may comprise allowing the conversion of l,3-dichloro-6- nitroisoquinoline (compound 3) to 6-aminoisoquinoline (compound 4) to proceed for a period of time sufficient to form 6-aminoisoquinoline (compound 4).
- the reaction may be allowed to proceed for about 2 hours to about 4 days.
- the reaction may be allowed to proceed for about 2 to about 8 hours for hydrogenation of the nitro group to the amino group.
- the reaction may be allowed to proceed for an additional 2 hours to 4 days for hydrogenation of the carbon-chlorine bonds to be complete.
- the methods may form 6-aminoisoquinoline (compound 4) in a yield of about 20% to about 100%, e.g., about 20% to about 99%.
- the methods may form 6-aminoisoquinoline (compound 4) in about or at least 20%, about or at least 25%, about or at least 30%, about or at least 35%, about or at least 40%, about or at least 45%, about or at least 50%, about or at least 51%, about or at least 52%, about or at least 53%, about or at least 54%, about or at least
- the methods may form 6-aminoisoquinoline (compound 4) with a purity of greater than or equal to about 60%, greater than or equal to about 65%, greater than or equal to about 70%, greater than or equal to about 75%, greater than or equal to about 80%, greater than or equal to about 85%, greater than or equal to about 90%, greater than or equal to about 95%, greater than or equal to about 96%, greater than or equal to about 97%, greater than or equal to about 98%, or greater than or equal to about 99%, or greater than or equal to about 99.5%, or greater than or equal to about 99.9% as determined by HPLC.
- one or more impurities or byproducts may be formed by the methods.
- the following compounds may form while carrying out the disclosed methods (some of them arising from impurities in the starting materials):
- the methods may further comprise reacting 6-nitroisoquinoline-l,3(2H, 4H)-dione (compound 2),
- R 1 -P(0)Cl2 wherein R 1 is aryl, alkyl or chloro; to form l ,3-dichloro-6-nitroisoquinoline (compound 3).
- R 1 is unsubstituted or substituted aryl. In certain embodiments, R 1 is unsubstituted or substituted phenyl. In certain embodiments, R 1 is phenyl.
- the reaction of 6-nitroisoquinoline-l ,3(2H, 4H)-dione (compound 2) with R 1 - P(0)Cl2 may further comprise a solvent or mixture of solvents. Any suitable solvent that is compatible with the components of the reaction mixture may be used. Suitably, a solvent will be selected such that the starting materials will be at least partially soluble (or fully soluble) and will allow the reaction mixture to be heated, if necessary, to a temperature sufficient for the reaction to produce l,3-dichloro-6-nitroisoquinoline (compound 3).
- the reacting of 6- nitroisoquinoline-l,3(2H,4H)-dione (compound 2) with R 1 -P(0)C1 2 may not comprise a solvent. In certain embodiments, R 1 -P(0)Cl 2 is the solvent.
- the reacting of 6-nitroisoquinoline-l,3(2H, 4H)-dione (compound 2) with R 1 -P(0)Cl 2 may further comprise heating.
- the reaction mixture may be heated at a temperature greater than ambient or room temperature, wherein ambient or room temperature is about 18°C to about 25°C.
- the reaction mixture may be heated at a temperature of about 25°C to about 160°C, or about 30°C to about 150°C.
- the reaction mixture may be heated at a temperature of at least 90°C, at least 100°C, at least 110°C, at least 120°C, at least 130°C, or at least 140°C.
- the reaction mixture may be heated at a temperature of less than 140°C, less than 130°C, less than 120°C, less than 110°C, or less than 100°C.
- the reaction mixture may be heated at a temperature of about 90°C, about 100°C, about 110°C, about 120°C, about 130°C, about 140°C, about 150°C, or about 160°C.
- reaction mixture may also be added to the reaction mixture, such as an acid, a base or a salt.
- the methods may comprise allowing the reacting of 6-nitroisoquinoline- l,3(2H, 4H)-dione (compound 2) with R 1 -P(0)Cl 2 to proceed for a period of time sufficient to form l,3-dichloro-6-nitroisoquinoline (compound 3).
- the reaction may be allowed to proceed for about 20 minutes to about 12 hours, or about 1 hour to about 4 hours.
- the methods may form l,3-dichloro-6-nitroisoquinoline (compound 3) in a yield of about or at least 20% to about or at least 100%, e.g., about or at least 20% to about or at least 99%.
- the method may form l,3-dichloro-6-nitroisoquinoline (compound 3) in about or at least 20%, about or at least 25%, about or at least 30%, about or at least 35%, about or at least
- the method may form l ,3-dichloro-6-nitroisoquinoline (compound 3) with a purity of greater than or equal to about 60%, greater than or equal to about 65%, greater than or equal to about 70%, greater than or equal to about 75%, greater than or equal to about 80%, greater than or equal to about 85%, greater than or equal to about 90%, greater than or equal to about 95%, greater than or equal to about 96%, greater than or equal to about 97%, greater than or equal to about 98%, or greater than or equal to about 99%, as determined by HPLC.
- the methods may further comprise reacting 2-(carboxymethyl)-4-nitrobenzoic acid (compound 1),
- the acid is an organic acid selected from the group consisting of formic acid, acetic acid, propionic acid, and combinations thereof. In certain embodiments the acid is acetic acid.
- the reacting of 2-(carboxymethyl)-4-nitrobenzoic acid (compound 1) with urea in the presence of an acid may further comprise a solvent or mixture of solvents. Any suitable solvent that is compatible with the components of the reaction mixture may be used. Suitably, a solvent will be selected such that the starting materials will be at least partially soluble (or fully soluble) and will allow the reaction mixture to be heated, if necessary, to a temperature sufficient for the reaction to produce l,3-dichloro-6-nitroisoquinoline (compound 2).
- the reacting of 2-(carboxymethyl)-4-nitrobenzoic acid (compound 1) with urea in the presence of an acid may not comprise an additional solvent. In certain embodiments, the organic acid (e.g., acetic acid) is added in a sufficient amount to be the solvent.
- the reacting of 2-(carboxymethyl)-4-nitrobenzoic acid (compound 1) with urea in the presence of an acid may further comprise heating.
- the reaction mixture may be heated at a temperature greater than ambient or room temperature, wherein ambient or room temperature is about 18°C to about 25°C.
- the reaction mixture may be heated at a temperature of about 25°C to about 140°C, or about 90°C to about 140°C.
- the reaction mixture may be heated at a temperature of at least 60°C, at least 70°C, at least 80°C, at least 90°C, at least 100°C, or at least 110°C.
- the reaction mixture may be heated at a temperature of less than 110°C, less than 100°C , less than 90°C , less than 80°C, or less than 70°C .
- the reaction mixture may be heated at a temperature of about 70°C, about 80°C, about 90°C, about 100°C, about 110°C, about 120°C, about 130°C, or about 140°C.
- reaction mixture may also be added to the reaction mixture, such as an acid, a base or a salt.
- the methods may comprise allowing the reacting of 2-(carboxymethyl)-4- nitrobenzoic acid (compound 1) with urea in the presence of an acid to proceed for a period of time sufficient to form 6-nitroisoquinoline-l,3(2H, 4H)-dione (compound 2).
- the reaction may be allowed to proceed for about 20 minutes to about 12 hours, or about 1 hour to about 8 hours.
- the methods may form 6-nitroisoquinoline-l,3(2H, 4H)-dione (compound 2) in a yield of about 20% to 100%, e.g., about 20% to about 99%.
- the methods may form 6- nitroisoquinoline-l,3(2H, 4H)-dione (compound 2) in about or at least 20%, about or at least
- the methods may form form form 6-nitroisoquinoline-l,3(2H, 4H)-dione (compound 2) with a purity of greater than or equal to about 60%, greater than or equal to about 65%, greater than or equal to about 70%, greater than or equal to about 75%, greater than or equal to about 80%, greater than or equal to about 85%, greater than or equal to about 90%, greater than or equal to about 95%, greater than or equal to about 96%, greater than or equal to about 97%, greater than or equal to about 98%, or greater than or equal to about 99%, as determined by HPLC.
- the methods may further comprise reacting a compound of formula (III),
- R is alkyl; with a base, to form 2-(carboxymethyl)-4-nitrobenzoic acid (compound 1).
- R is C1-C6 alkyl. In certain embodiments, R is C1-C4 alkyl. In certain embodiments R is ethyl. In certain embodiments, the compound of formula (III) is 2-(l,3-diethoxy-l,3-dioxopropan-2-yl)-4-nitrobenzoic acid.
- the base is an alkali metal hydroxide, an alkaline earth metal hydroxide, or a combination thereof.
- Alkali metal hydroxides include LiOH, NaOH, KOH, RbOH and CsOH.
- Alkaline earth metal hydroxides include Be(OH) 2 , Mg(OH) 2 , Ca(OH) 2 , Sr(OH) 2 , and Ba(OH) 2 .
- the base is KOH.
- the reacting of a compound of formula (III) with a base may further comprise a solvent or mixture of solvents.
- a solvent Any suitable solvent that is compatible with the components of the reaction mixture may be used.
- a solvent will be selected such that the starting materials will be at least partially soluble (or fully soluble) and will allow the reaction mixture to be heated, if necessary, to a temperature sufficient for the reaction to form 2- (carboxymethyl)-4-nitrobenzoic acid (compound 1).
- the solvent is water.
- the reacting of a compound of formula (III) with a base may further comprise heating.
- the reaction mixture may be heated at a temperature greater than ambient or room temperature, wherein ambient or room temperature is about 18°C to about 25°C.
- the reaction mixture may be heated at a temperature of about 25°C to about 100°C.
- the reaction mixture may be heated at a temperature of at least 60°C, at least 70°C, at least 80°C, at least 90°C, or at least 100°C.
- the reaction mixture may be heated at a temperature of less than 100°C, less than 90°C, less than 80°C, less than 70°C, or less than 60°C.
- the reaction mixture may not be heated, but allowed to proceed at ambient or room temperature.
- the methods may comprise allowing the reacting of compound of formula (III) with a base to proceed for a period of time sufficient to form 2-(carboxymethyl)-4-nitrobenzoic acid (compound 1).
- the reaction may be allowed to proceed for about 20 minutes to about 24 hours, about 1 hour to about 18 hours, or about 6 hours to about 16 hours.
- the methods may form 2-(carboxymethyl)-4-nitrobenzoic acid (compound 1) in a yield of about 20% to 100%, or about 20% to about 99%.
- the methods may form 2- (carboxymethyl)-4-nitrobenzoic acid (compound 1) in about or at least 20%, about or at least
- the methods may form 2-(carboxymethyl)-4-nitrobenzoic acid (compound 1) with a purity of greater than or equal to about 60%, greater than or equal to about 65%, greater than or equal to about 70%, greater than or equal to about 75%, greater than or equal to about 80%, greater than or equal to about 85%, greater than or equal to about 90%, greater than or equal to about 95%, greater than or equal to about 96%, greater than or equal to about 97%, greater than or equal to about 98%, or greater than or equal to about 99%, as determined by HPLC.
- the methods may further comprise reacting a compound of formula (I), wherein X is halogen or OS0 2 R a ; and R a is aryl, alkyl or haloalkyl; with a compound of formula (II),
- R is alkyl
- X is halogen. In certain embodiments, X is CI. In certain embodiments, X is S0 2 R a ; and R a is C1-C6 alkyl, aryl, or C1-C6 haloalkyl. In certain embodiments, X is S0 2 R a ; and R a is methyl, 4-methylphenyl, or trifluoromethyl. In certain embodiments, the compound of formula (I) is 2-chloro-4-nitrobenzoic acid.
- R is C1-C6 alkyl. In certain embodiments, R is C1-C4 alkyl. In certain embodiments, R is ethyl. In certain embodiments, the compound of formula (II) is diethyl malonate.
- reacting a compound of formula (I) with a compound of formula (II) comprises adding a base.
- the base is a metal alkoxide.
- An alkoxide is the conjugate base of an alcohol.
- the metal alkoxide may be a lithium alkoxide, a sodium alkoxide, a potassium alkoxide, or a combination thereof.
- the metal alkoxide may comprise a C1-C6 alkyl group, as defined herein.
- the metal alkoxide is sodium methoxide.
- reacting a compound of formula (I) with a compound of formula (II) comprises adding a catalyst.
- the catalyst comprises a transition metal.
- the transition metal is selected from the group consisting of palladium, platinum, nickel, rhodium, ruthenium, iridium, cobalt, iron, copper, gold silver, and combinations thereof.
- the transition metal is copper.
- the catalyst is copper (I) bromide.
- the catalyst loading in the reaction may be about 1 mol% to about 50 mol%, about 1 mol% to about 30 mol%, about 10 mol% to about 30 mol%, or about 15 mol% to about 25 mol%.
- the catalyst loading may be about about or at least 1 mol%, about or at least 2 mol%, about or at least 3 mol%, about or at least 4 mol%, about or at least 5 mol%, about or at least 6 mol%, about or at least 7 mol%, about or at least 8 mol%, about or at least 9 mol%, about or at least 10 mol%, about or at least 11 mol%, about or at least 12 mol%, about or at least 13 mol%, about or at least 14 mol%, about or at least 15 mol%, about or at least 16 mol%, about or at least 17 mol%, about or at least 18 mol%, about or at least 19 mol%, about or at least 20 mol%, about or at least 21 mol%, about or at least 22 mol%, about or at least 23 mol%, about or at least 24 mol%, about or at least 25 mol%, about or at least 26 mol%, about or at least 27 mol%, about or at least 28
- the reacting of a compound of formula (I) with a compound of formula (II) may further comprise adding a solvent or mixture of solvents.
- a solvent Any suitable solvent that is compatible with the components of the reaction mixture may be used.
- a solvent will be selected such that the starting materials will be at least partially soluble (or fully soluble) and will allow the reaction mixture to be heated, if necessary, to a temperature sufficient for the reaction to produce the compound of formula (III).
- the solvents may include, but are not limited to: ethers such as diethyl ether, dibutyl ether, 1,2- dimethoxyethane, diglyme, t-butyl methyl ether, tetrahydrofuran and dioxane; aliphatic or aromatic hydrocarbon solvents such as benzene, xylene, toluene, hexane, and pentane; polar aprotic solvents such as acetonitrile, dimethylformamide, and dimethylsulfoxide; polar protic solvents such as methanol, ethanol, n-propanol, isopropanol, n-butanol, acetic acid and water; or any combination of two or more solvents.
- the solvent is ethanol.
- the reacting of a compound of formula (I) with a compound of formula (II) may further comprise heating.
- the reaction mixture may be heated at a temperature greater than ambient or room temperature, wherein ambient or room temperature is about 18°C to about 25°C.
- the reaction mixture may be heated at a temperature of about 25°C to about 90°C, or about 30°C to about 80°C.
- the reaction mixture may be heated at a temperature of at least 25°C, at least 30°C, at least 35°C, at least 40°C, at least 45°C, at least 50°C, at least 55°C, at least 60°C, at least 65°C, at least 70°C, at least 75°C, at least 78°C, or at least 80°C.
- the reaction mixture may be heated at a temperature less than 90°C, less than 80°C, less than 78°C, less than 75°C, less than 70°C, less than 60°C, less than 55°C, less than 50°C, less than 45°C, less than 40°C, less than 35°C, or less than 30°C.
- the reaction mixture may be heated at a temperature of about 25°C, about 30°C, about 35°C, about 40°C, about 45°C, about 50°C, about 55°C, about 60°C, about 70°C, about 75°C, about 78°C, about 80°C, or about 90°C.
- the methods may comprise allowing the conversion to proceed for a period of time sufficient to form the compound of formula (III). For example, the reaction may be allowed to proceed for about 1 hour to about 24 hours, about 2 hours to about 18 hours, or about 12 hours to about 18 hours.
- the methods may form the compound of formula (III) in a yield of about 20% to 100%, e.g., about 20% to about 99%.
- the methods may provide the compound of formula (III) in about or at least 20%, about or at least 25%, about or at least 30%, about or at least
- the methods may form the compound of formula (III) with a purity of greater than or equal to about 60%, greater than or equal to about 65%, greater than or equal to about 70%, greater than or equal to about 75%, greater than or equal to about 80%, greater than or equal to about 85%, greater than or equal to about 90%, greater than or equal to about 95%, greater than or equal to about 96%, greater than or equal to about 97%, greater than or equal to about 98%, or greater than or equal to about 99%, or greater than or equal to about 99.5% or greater than or equal to about 99.9% as determined by HPLC.
- the methods include reacting 2-chloro-4-nitrobenzoic acid (I-a) with diethylmalonate (Il-a) to form 2-(l,3-diethoxy-l,3-dioxopropan-2-yl)-4-nitrobenzoic acid (IH-a); reacting 2-(l,3-diethoxy-l,3-dioxopropan-2-yl)-4-nitrobenzoic acid (Ill-a) with a base to form 2-(carboxymethyl)-4-nitrobenzoic acid (compound 1); reacting 2-
- the methods include reacting 2-chloro-4-nitrobenzoic acid (I-a) with diethylmalonate (Il-a) in the presence of sodium methoxide and copper (I) bromide to form 2-(l ,3-diethoxy-l,3-dioxopropan-2-yl)-4-nitrobenzoic acid (Ill-a); reacting 2-(l ,3-diethoxy-l ,3-dioxopropan-2-yl)-4-nitrobenzoic acid (Ill-a) with potassium hydroxide to form 2-(carboxymethyl)-4-nitrobenzoic acid (compound 1); reacting 2-(carboxymethyl)-4- nitrobenzoic acid (compound 1) with urea in acetic acid to form 6-nitroisoquinoline- l,3(2H, 4H)-dione (compound 2); reacting 6-nitroisoquinoline-l ,3(2H, 4H)-di
- the compounds and intermediates may be isolated and purified by methods well- known to those skilled in the art of organic synthesis.
- Examples of conventional methods for isolating and purifying compounds can include, but are not limited to, chromatography on solid supports such as silica gel, alumina, or silica derivatized with alkylsilane groups, by recrystallization at high or low temperature with an optional pretreatment with activated carbon, thin-layer chromatography, distillation at various pressures, sublimation under vacuum, and trituration, as described for instance in "Vogel's Textbook of Practical Organic Chemistry", 5th edition (1989), by Furniss, Hannaford, Smith, and Tatchell, pub. Longman Scientific & Technical, Essex CM20 2JE, England.
- 6-aminoisoquinoline may be purified via a multi-step process.
- the multi-step process comprises (1) removal of insoluble impurities via filtration; (2) acid and base extraction; and (3) recrystallization.
- crude 6-aminoisoquinoline may be mixed with activated charcoal for a suitable time period in a solvent or a mixture of solvents to form a first mixture.
- the period of time may be at least about 4 hours.
- Any suitable solvent that is compatible with the components may be used.
- the solvent may be methanol or ethanol.
- the first mixture may be filtered through at least one filter to form a filtrate.
- the resulting filtrate may then be condensed and dissolved in an acidic solution to form a second mixture.
- the acidic solution may be, for example, a citric acid solution such as about or at least 5% citric acid.
- the second mixture is optionally heated to 35 ⁇ 5°C for a period of time of 1 -6 h.
- the resulting solution is washed at least one time with a solvent to form a third mixture. Any suitable solvent that is compatible with the components may be used. In an embodiment, the solvent may be dichloromethane.
- the resulting aqueous layer is mixed with a basic solution to form a precipitate.
- the basic solution may be, for example, concentrated ammonium hydroxide, 10 % aqueous potassium hydroxide, 10 % aqueous potassium carbonate, or combinations thereof.
- the resulting precipitate may be filtered and dried to obtain a solid.
- the solid may be suspended in a solvent to form a suspension.
- a solvent Any suitable solvent that is compatible with the components may be used.
- the solvent may be ethanol.
- the suspension may be heated to a temperature of about 75 °C for a period of time. The period of time may be about 45 minutes. Subsequently, the suspension is gradually cooled over the course of about 16 hours, and filtered to obtain 6-aminoisoquinonline.
- the methods may form 6-aminoisoquinonline with a purity of greater than or equal to 99%, as determined by HPLC. The steps recited in this paragraph may be repeated as necessary, e.g., one time, two times, three times, etc.
- the filters may be nylon or PTFE filters.
- a disclosed compound may have at least one basic nitrogen whereby the compound can be treated with an acid to form a desired salt.
- a compound may be reacted with an acid at or above room temperature to provide the desired salt, which is deposited, and collected by filtration after cooling.
- acids suitable for the reaction include, but are not limited to tartaric acid, lactic acid, succinic acid, as well as mandelic, atrolactic, methanesulfonic, ethanesulfonic, toluenesulfonic, naphthalenesulfonic, benzenesulfonic, carbonic, fumaric, maleic, gluconic, acetic, propionic, salicylic, hydrochloric, hydrobromic, phosphoric, sulfuric, citric, hydroxybutyric, camphorsulfonic, malic, phenylacetic, aspartic, or glutamic acid, combinations thereof and the like.
- reactions can be worked up in the conventional manner, e.g. by eliminating the solvent from the residue and further purified according to methodologies generally known in the art such as, but not limited to, crystallization, distillation, extraction, trituration and chromatography.
- the starting materials and reagents are either commercially available or can be prepared by one skilled in the art from commercially available materials using methods described in the chemical literature. Routine experimentation, including appropriate manipulation of the reaction conditions, reagents and sequence of the synthetic route, protection of any chemical functionality that is not compatible with the reaction conditions, and deprotection at a suitable point in the reaction sequence of the methods are included in the scope of the invention.
- Suitable protecting groups and the methods for protecting and deprotecting different substituents using such suitable protecting groups are well known to those skilled in the art; examples of which can be found in PGM Wuts and TW Greene's book entitled Protective Groups in Organic Synthesis (4 th ed.), John Wiley & Sons, NY (2006), which is incorporated herein by reference in its entirety. It can be appreciated that the synthetic schemes and specific examples as described are illustrative and are not to be read as limiting the scope of the invention as it is defined in the appended claims. All alternatives, modifications, and equivalents of the synthetic methods and specific examples are included within the scope of the claims.
- the present disclosure also includes isotopically-labeled compounds, which are identical to any of those recited in the methods, but for the fact that one or more positions in the molecule areenriched with an isotope of the same atom, having an atomic mass or mass number that is different from the atomic mass or mass number usually found in nature.
- isotopes suitable for inclusion in the compounds of the invention are hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, and chlorine, such as, but not limited to 2 H, H, 1 C, 14 C, 15 N, 18 0, 17 0, 1 P, 2 P, 5 S, 18 F, and 6 C1, respectively.
- isotopes such as deuterium, i.e., 2 H
- the compound may incorporate positron-emitting isotopes for medical imaging and positron-emitting tomography (PET) studies for determining the distribution of receptors.
- PET positron-emitting tomography
- Suitable positron- emitting isotopes that can be incorporated in compounds of formula (I) are n C, 1 N, 15 0, and 18 F.
- Isotopically-labeled compounds of formula (I) can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described herein. Examples using appropriate isotopically-labeled reagent in place of non-isotopically- labeled reagent.
- Basic addition salts may be prepared during the final isolation and purification of the disclosed compounds by reaction of a carboxyl group with a suitable base such as the hydroxide, carbonate, or bicarbonate of a metal cation such as lithium, sodium, potassium, calcium, magnesium, or aluminum, or an organic primary, secondary, or tertiary amine.
- Quaternary amine salts can be prepared, such as those derived from methylamine, dimethylamine, trimethylamine, triethylamine, diethylamine, ethylamine, tributylamine, pyridine, N,N-dimethylaniline, N-methylpiperidine, N-methylmorpholine,
- dicyclohexylamine procaine, dibenzylamine, ⁇ , ⁇ -dibenzylphenethylamine, 1-ephenamine and N,N'-dibenzylethylenediamine, ethylenediamine, ethanolamine, diethanolamine, piperidine, piperazine, and the like.
- temperatures are given in degrees Celsius (°C); synthetic operations were carried out at ambient temperature, "rt,” or “RT,” (typically a range of from about 18-25°C); evaporation of solvents was carried out using a rotary evaporator under reduced pressure (typically, 4.5-30 mm Hg) with a bath temperature of up to 60°C; the course of reactions was typically followed using thin layer chromatography (TLC); all melting points, if given, are uncorrected; all intermediates as well as the final product exhibited satisfactory ⁇ -NMR, HPLC and/or microanalytical data; and the following conventional abbreviations are used: L (liter(s)), mL (milliliters), mmol (millimoles), g (grams), mg (milligrams), min (minutes), and h (hours).
- 6-Nitroisoquinoline-l,3(2H,4H)-dione (2) A suspension of 2-(carboxymethyl)-4- nitrobenzoic acid (1.5 kg, 6.67 mol, 1 eq) in acetic acid (6 L) was heated at 110°C. The suspension became a clear solution when the temperature reached 90°C. Solid urea (2.85 kg, 47.5 mol, 7.1 eq) was then added slowly into the reaction mixture. The reaction was stirred at 110°C for about 4 h. The reaction mixture was combined with another parallel batch (from 1.5 kg of 2-(carboxymethyl)-4-nitrobenzoic acid) and poured into a mixture of ice water (20 kg) and stirred for 0.5 h.
- 6-Aminoisoquinoline (4) l,3-Dichloro-6-nitroisoquinoline (700 g, 2.88 mol, 1 eq) and 10% Pd/C (45 g, 6.5% w/w) were suspended in THF (5.5 L). After purging with a nitrogen/hydrogen cycle three times, the reaction mixture was hydrogenated at 45°C under a pressure of 0.6 MP for about 6 ⁇ 8h until the nitro group was fully reduced to the amino group. Partial formation of the des-chloride intermediate was also indicated by TLC and HPLC analysis. After the pressure of the reactor was released, K 2 CO 3 (1 kg), MeOH (1.5 L), and additional Pd/C (10%, 45 g) were added to the reaction suspension.
- reaction mixture was hydrogenated at 45°C under a pressure of 0.6 MP until completion of the reaction (2-3 days).
- the reaction mixture was filtered through a pad of Celite and washed with MeOH (500 mL x 2). The filtrate was concentrated under reduced pressure.
- the resulting residue (-550 g, wet) was slurried in MeOH (550 mL, 1 vol) for -0.5 h and filtered to produce the title compound as a green solid (380 g).
- the crude 6-aminoisoquinoline was combined with another two parallel batches of crude 6-aminoisoquinoline and purified together.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Other In-Based Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description



















Claims










Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA3048667A CA3048667A1 (en) | 2016-12-30 | 2017-12-11 | Methods for the preparation of 6-aminoisoquinoline |
| AU2017387801A AU2017387801B2 (en) | 2016-12-30 | 2017-12-11 | Methods for the preparation of 6-aminoisoquinoline |
| EP17886003.7A EP3562806A4 (en) | 2016-12-30 | 2017-12-11 | Methods for the preparation of 6-aminoisoquinoline |
| JP2019535803A JP7082980B2 (en) | 2016-12-30 | 2017-12-11 | 6-Method for preparing aminoisoquinoline |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US15/395,068 US9840468B1 (en) | 2016-12-30 | 2016-12-30 | Methods for the preparation of 6-aminoisoquinoline |
| US15/395,068 | 2016-12-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018125548A1 true WO2018125548A1 (en) | 2018-07-05 |
Family
ID=60516487
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2017/065631 WO2018125548A1 (en) | 2016-12-30 | 2017-12-11 | Methods for the preparation of 6-aminoisoquinoline |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9840468B1 (en) |
| EP (1) | EP3562806A4 (en) |
| JP (1) | JP7082980B2 (en) |
| AU (1) | AU2017387801B2 (en) |
| CA (1) | CA3048667A1 (en) |
| WO (1) | WO2018125548A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108929270A (en) * | 2018-08-15 | 2018-12-04 | 上海罕道医药科技有限公司 | A kind of synthesis of the disubstituted nitrogenous heterocyclic aminated compounds of pharmaceutical intermediate |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016123627A1 (en) * | 2015-01-30 | 2016-08-04 | Vanderbilt University | Isoquiniline and napthalene-substituted compounds as mglur4 allosteric potentiators, compounds, and methods of treating neurological dysfunction |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3766472D1 (en) * | 1986-10-28 | 1991-01-10 | Smith Kline French Lab | TETRAHYDROISOCHINOLIN-2-YL DERIVATIVES OF CARBONIC ACIDS AS THROMBOXANE A2 ANTAGONISTS. |
| WO2003045921A1 (en) * | 2001-11-28 | 2003-06-05 | Fujisawa Pharmaceutical Co., Ltd. | Heterocyclic amide compounds as apolipoprotein b inhibitors |
| WO2003068749A1 (en) | 2002-02-15 | 2003-08-21 | Glaxo Group Limited | Vanilloid receptor modulators |
| GB0206876D0 (en) | 2002-03-22 | 2002-05-01 | Merck Sharp & Dohme | Therapeutic agents |
| US8673961B2 (en) | 2008-08-15 | 2014-03-18 | N30 Pharmaceuticals, Inc. | Pyrrole inhibitors of S-nitrosoglutathione reductase as therapeutic agents |
| KR101732907B1 (en) | 2009-06-19 | 2017-05-08 | 가부시키가이샤 디. 웨스턴 세라퓨틱스 겡큐쇼 | Substituted isoquinoline derivative |
| CN105017152B (en) * | 2015-07-17 | 2017-05-10 | 北京六合宁远科技有限公司 | Preparation method of 3-chloro-6-nitroisoquinoline or 3-bromine-6-nitroisoquinoline |
-
2016
- 2016-12-30 US US15/395,068 patent/US9840468B1/en active Active
-
2017
- 2017-12-11 AU AU2017387801A patent/AU2017387801B2/en active Active
- 2017-12-11 WO PCT/US2017/065631 patent/WO2018125548A1/en unknown
- 2017-12-11 CA CA3048667A patent/CA3048667A1/en not_active Abandoned
- 2017-12-11 JP JP2019535803A patent/JP7082980B2/en active Active
- 2017-12-11 EP EP17886003.7A patent/EP3562806A4/en not_active Withdrawn
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016123627A1 (en) * | 2015-01-30 | 2016-08-04 | Vanderbilt University | Isoquiniline and napthalene-substituted compounds as mglur4 allosteric potentiators, compounds, and methods of treating neurological dysfunction |
Non-Patent Citations (2)
| Title |
|---|
| See also references of EP3562806A4 * |
| STURDIVANT JM ET AL.: "Abstrac: MEDI 153 Identification of intermediates in the stepwise reduction of 1, 3- dichloro-6-nitroisoquinoline to 6-aminoisiquinoline", 248TH NATIONAL MEETING OF THE AMERICAN CHEMICAL SOCIETY (ACS), August 2014 (2014-08-01), San Francisco, CA, XP055516779 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108929270A (en) * | 2018-08-15 | 2018-12-04 | 上海罕道医药科技有限公司 | A kind of synthesis of the disubstituted nitrogenous heterocyclic aminated compounds of pharmaceutical intermediate |
Also Published As
| Publication number | Publication date |
|---|---|
| JP7082980B2 (en) | 2022-06-09 |
| US9840468B1 (en) | 2017-12-12 |
| EP3562806A4 (en) | 2020-05-27 |
| JP2020503338A (en) | 2020-01-30 |
| AU2017387801A1 (en) | 2019-06-20 |
| CA3048667A1 (en) | 2018-07-05 |
| EP3562806A1 (en) | 2019-11-06 |
| AU2017387801B2 (en) | 2020-04-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2012209103B2 (en) | Methods and compositions for the synthesis of multimerizing agents | |
| TWI496781B (en) | Process for preparing methyl {4,6-diamino-2-(1-(2-fluorobenzyl)-1h-pyrazolo(3,4-b)pyridin-3-yl)pyrimidin-5-yl}methylcarbamate and its purification for use as pharmaceutically active compound | |
| TWI697494B (en) | Synthesis of copanlisib and its dihydrochloride salt | |
| KR101821090B1 (en) | Process for manufacture of n-acylbiphenyl alanine | |
| KR101420892B1 (en) | Process for the preparation of Imatinib and intermediates thereof | |
| TW201625634A (en) | Synthesis of COPANLISIB and its dihydrochloride salt | |
| CN114805314B (en) | Synthesis method of Entecavir | |
| KR101653025B1 (en) | Method for producing 2-amino-4-(trifluoromethyl)pyridine | |
| AU2017387801B2 (en) | Methods for the preparation of 6-aminoisoquinoline | |
| CN117285537A (en) | Preparation method of Marpatinib | |
| JPH02289563A (en) | Improved process for producing ortho-carboxypyridyl- and ortho-carboxyquinolylimidazolinones | |
| JP4238978B2 (en) | Benzazepine compounds and process for producing the same | |
| EP3906235B1 (en) | Method for preparing sulfonamides drugs | |
| CN111196782B (en) | Dihydronaphthyridine compounds, preparation method and application thereof | |
| JPH04169583A (en) | Phenothiazine derivative and its production | |
| KR101299720B1 (en) | A novel process for preparing 3-amino-5-fluoro-4-dialkoxypetanoic acid ester | |
| CA2484585A1 (en) | A process for the preparation of benazepril hydrochloride | |
| JP4568824B2 (en) | Method for producing diarylsulfonic acid derivative | |
| JP2024150777A (en) | Method for producing prolinamide compound | |
| US8802860B2 (en) | Method for producing substituted pyridin-2-one | |
| WO2022202814A1 (en) | Method for producing pyrimidine compound | |
| JP2022025121A (en) | Method of producing prolinamide compound | |
| CN115785081A (en) | Preparation method of raltitrexed | |
| JP2010502607A (en) | Methods for the synthesis and synthesis of (3-alkyl-5-piperidin-1-yl-3,3a-dihydro-pyrazolo [1,5-a] pyrimidin-7-yl) -amino derivatives and intermediates Intermediate | |
| CN101627041A (en) | The method and the intermediate of synthetic (3-alkyl-5-piperidines-1-base-3,3A-dihydro-pyrazolo [1,5-A] pyrimidin-7-yl)-aminoderivative and intermediate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17886003 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017387801 Country of ref document: AU Date of ref document: 20171211 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 3048667 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2019535803 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2017886003 Country of ref document: EP Effective date: 20190730 |