WO2018106738A1 - Brush-arm star polymers, conjugates and particles, and uses thereof - Google Patents
Brush-arm star polymers, conjugates and particles, and uses thereof Download PDFInfo
- Publication number
- WO2018106738A1 WO2018106738A1 PCT/US2017/064784 US2017064784W WO2018106738A1 WO 2018106738 A1 WO2018106738 A1 WO 2018106738A1 US 2017064784 W US2017064784 W US 2017064784W WO 2018106738 A1 WO2018106738 A1 WO 2018106738A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- heteroalkylene
- alkylene
- alkyl
- independently
- arylene
- Prior art date
Links
- 0 C*(C*1)CC2C1C1C=CC2C1 Chemical compound C*(C*1)CC2C1C1C=CC2C1 0.000 description 14
- LCTLQOUCIUNEPY-UHFFFAOYSA-N CC(c1cc(F)c(C(NS)=O)c(F)c1)=O Chemical compound CC(c1cc(F)c(C(NS)=O)c(F)c1)=O LCTLQOUCIUNEPY-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/69—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit
- A61K47/6921—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a particulate, a powder, an adsorbate, a bead or a sphere
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- nanotherapeutics e.g., lipid- or polymeric nanoparticles and viruses
- protein and nucleic acid drugs targeted therapies
- immune therapies e.g., immune checkpoint blockers, vaccines and/or immune cells
- small molecule small molecule
- the present invention discloses, at least in part, macromonomers, conjugates comprising the macromonomers (also referred to alternatively as “BRUSH”, “Brush”, or “BRUSH conjugates”), and brush arm star polymer particles comprising the conjugates (also referred to as “BASPs” or “BASP particles”), each further comprising an agent ⁇ e.g., one or more therapeutic agents, diagnostic agents, or targeting moieties), as well as starting materials (e.g., crosslinkers) and other components (e.g., degradation components) thereof; as well as compositions (e.g., pharmaceutical compositions) comprising the same, and methods of making and using the same.
- Compositions comprising BASP particles are referred to as "BASP- compositions”.
- Compositions comprising BRUSH conjugates are referred to as "BRUSH-compositions”.
- the BASP-compositions or BRUSH-compositions enhance the delivery, distribution, release and/or efficacy of an agent to a desired target site in a subject, e.g., compared to the systemic release of the agent as a free form (e.g., not coupled to a BASP- or BRUSH-composition described herein).
- the agent delivered or released is a therapeutic agent (e.g., an angiotensin receptor blocker (ARB), a vitamin D analog, a bromodomain inhibitor, or an indoleamine 2,3-dioxygenease (IDO) inhibitor, as described herein).
- a therapeutic agent e.g., an angiotensin receptor blocker (ARB), a vitamin D analog, a bromodomain inhibitor, or an indoleamine 2,3-dioxygenease (IDO) inhibitor, as described herein.
- the BASP-compositions or BRUSH- compositions disclosed herein comprise a conjugate of multiple macromonomers, each containing a therapeutic agent linked to the macromonomer, wherein the linker is preferentially labile or preferentially cleavable in a tissue microenvironment.
- the therapeutic agent can be preferentially released in the tissue microenvironment, e.g., compared to the systemic release of the therapeutic agent as a free form, thus allowing for a lower dosage of the agent in the particle.
- the BASP-composition or BRUSH-composition comprises a tissue microenvironment cleavable linker connecting the therapeutic agent to the macromonomer, which preferentially releases the therapeutic agent upon exposure to a set of conditions present in the tissue microenvironment.
- the tissue microenvironment cleavable linker connecting the therapeutic agent to the macromonomer, which preferentially releases the therapeutic agent upon exposure to a set of conditions present in the tissue microenvironment.
- the tissue microenvironment cleavable linker connecting the therapeutic agent to the macromonomer, which preferentially releases the therapeutic agent upon exposure to a set of conditions present in the tissue microenvironment.
- the tissue microenvironment cleavable linker connecting the therapeutic agent to the macromonomer
- microenvironment is a tumor or fibrotic microenvironment, e.g., exhibits one or more of decreased pH, hypoxic condition, or presence of an enzyme, e.g., esterase or protease, e.g., compared to a healthy tissue microenvironment.
- an enzyme e.g., esterase or protease
- BASP- compositions or BRUSH-compositions comprising certain microenvironment labile linkers were surprisingly found to release an agent at a yield between 10 to 100 times greater than other linkers (see e.g., Example 11, Figs. 14A to 14C).
- the BASP-compositions or BRUSH-compositions disclosed herein can result in one or more advantages over systemic release of the free agent, including, but not limited to: (i) increasing the localization, release and/or delivery of the agent to a target tissue, e.g., a cancer or a fibrotic tissue (e.g., a desmoplastic or a fibrotic tumor or tissue chosen from liver, kidney, lung or bone marrow (e.g., myelofibrotic bone marrow)); (ii) exhibiting increased release of the agent in a hypoxic
- a target tissue e.g., a cancer or a fibrotic tissue (e.g., a desmoplastic or a fibrotic tumor or tissue chosen from liver, kidney, lung or bone marrow (e.g., myelofibrotic bone marrow)
- a target tissue e.g., a cancer or a fibrotic tissue (e.g., a desmoplastic or
- microenvironment e.g., in a tumor or a fibrotic tissue (e.g., fibrotic or cirrhotic liver, or a tissue having renal fibrosis, pulmonary fibrosis or myelofibrosis); (iii) reducing a side effect of the agent by having a higher amount of released agent at a target site (e.g., in a hypoxic tumor), relative to other non-target sites (e.g., in intact and/or healthy blood vessels and/or normal or healthy tissues); and/or (iv) increasing the half-life of the agent.
- a target site e.g., in a hypoxic tumor
- non-target sites e.g., in intact and/or healthy blood vessels and/or normal or healthy tissues
- Certain embodiments disclosed herein provide BASP-compositions or BRUSH-compositions for use in methods for treating or preventing a disorder, e.g., a fibrotic disorder, a cancer (e.g., a desmoplastic tumor or metastatic lesion), or an inflammatory disorder.
- a disorder e.g., a fibrotic disorder, a cancer (e.g., a desmoplastic tumor or metastatic lesion), or an inflammatory disorder.
- the disorder is a liver disorder.
- These embodiments comprise administering to a subject a BASP-composition or BRUSH- composition as described herein, as a single agent or as a combination with one or more additional therapeutic agents.
- the present invention features a compound of Formula (I):
- A is C 1 -C 12 alkylene, C 2 -C 12 alkenylene, C 2 -C 12 alkynylene, or C 1 -C 12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R 1 ;
- B is C 1 - C 12 alkylene, C 2 -C 12 alkenylene, C 2 -C 12 alkynylene, or C 1 -C 12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R 1 ;
- ring T is a triazoldiyl moiety (e.g., a 1,2,3-triazoldiyl);
- X is an agent as described herein (e.g., an angiotensin receptor blocker (ARB), vitamin D analog, indoleamine 2,3-dioxygen
- the present invention features a conjugate comprising at least two of a structure according to Formula (III):
- Ring C is a carbocyclyl or heterocyclyl moiety
- Ring T is a triazoldiyl moiety (e.g., a 1,2,3-triazoldiyl)
- A is C 1 -C 12 alkylene, C 2 -C 12 alkenylene, C 2 -C 12 alkynylene, or C 1 -C 12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R 1
- B is C 1 -C 12 alkylene, C 2 -C 12 alkenylene, C 2 -C 12 alkynylene, or C 1 -C 12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R 1
- X is an agent chosen from an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor
- P
- each R 1 and R 2 is independently alkyl, alkenyl, alkynyl, heteroalkyl (e.g., polyethylene glycol), halo, cyano, oxo,–OR A ,–NR B R C ,– NR B C(O)R D , -C(O)NR B R C ,–C(O)R D ,–C(O)OH,–C(O)OR D ,–SR E , or–S(O) m R E ;
- each R A is independently hydrogen, C 1 -C 6 alkyl, C 1 -C 6 heteroalkyl,–C(O)NR B R C ,– C(O)R D ,–C(O)OH, or–C(O)OR D ; each R B and R C is independently hydrogen or C 1 - C 6 alkyl; each R D is independently C 1 -C 6 alkyl, C 1 -C 6 heteroalkyl, or C 1 -C 6 haloalkyl; each R E is independently hydrogen or C 1 -C 6 alkyl; and m is 1 or 2.
- the present invention comprises a BASP particle comprising at least two of a structure of Formula (III) as described herein.
- the present invention features a composition, e.g.,
- composition comprising one or more of a BASP particle or conjugate described herein, and a pharmaceutically acceptable carrier.
- the present invention features a method of treating or preventing a disorder (e.g., a hyperproliferative disorder, a fibrotic disorder, and/or an inflammatory disorder) in a subject, comprising administering the conjugate or BASP particle described herein, e.g., as a single agent or in combination with other agent or therapy, or the composition thereof, to the subject.
- a disorder e.g., a hyperproliferative disorder, a fibrotic disorder, and/or an inflammatory disorder
- the present invention features cross-linkers, which are useful to make the conjugates and BASP particles of the invention, as well as methods of making the conjugates and BASP particles of the invention, the details of which are described below.
- FIG.1 is an exemplary schematic overview of macromonomer, BRUSH and BASP structures and assembly.
- FIG.2 shows the chemical structures of exemplary angiotensin receptor blockers (ARBs), as well as prodrugs and metabolites thereof, that may be
- ARBs angiotensin receptor blockers
- FIG.3 shows the chemical structures of exemplary vitamin D analogs that may be incorporated into BRUSH conjugates and BASP particles.
- FIGs.4A to 4B show the chemical structures of exemplary bromodomain inhibitors that may be incorporated into BRUSH conjugates and BASP particles.
- FIGs.5A to 5B show the characterization of BASP (15) comprising
- FIG.5A depicts a gel permeation
- FIG. 5B depicts a dynamic light scattering plot showing average BASP particle size.
- FIGs.6A to 6B show the characterization of BASP (17) comprising
- FIG.6A depicts a gel permeation chromatogram and FIG.6B depicts a dynamic light scattering plot showing average BASP particle size.
- FIGs.8A to 8F are images of Cy7.5-conjugated Tel-BASP (15) taken in a non-invasive manner in live mice.
- the BASP was dosed at 10 mg/kg i.v in either a 4T1-tumor bearing mouse (FIGs.8A to 8C) or a na ⁇ ve, non-tumor bearing mouse (FIGs.8D-8E) and the biodistribution of the Cy7.5 signal was captured on an IVIS instrument following 30 min, 73 hrs and 10 days post (15) administration.
- FIGs.9A to 9B are images showing the visualization of Tel-BASP (15) in tumor tissue collected 6 days post administration of a single dose (100 mg/kg). Tissue sections (5 um) from (15) treated (+) and non-treated (-) animals were stained by H&E (FIG.9A) and imaged for Cy5 (FIG.9B) confirming significant drug uptake and retention in the tumor tissue.
- FIGs.10A to 10B are graphs showing the quantification of Tel-BASP (15) in tumor tissue collected 6 days post administration of a single dose (100 mg/kg) by two complementary methods.
- FIG.10A shows the pharmacokinetic analysis of polymer- bound telmisartan by LC/MS/MS.
- FIG.10B depicts quantitative image analysis of Cy5.5 signal intensity in tissue sections. Both assays confirm significant, time- dependent drug uptake and retention in the tumor tissue of BASP particles.
- FIGs.11A to 11C are images that show results of a biodistribution study of Tel-BASP (15) in major tissues comparing the signal of the Cy7.5 conjugated BASP particles (FIG.11B, 800 nm) to that of tissue autofluorescence (FIG.11A, 700 nM). Spleen, kidney, lung, heart, skin, liver brain gut and tumor were harvested from the mice and the formalin fixed organs were imaged directly on a Licor Odessey platform. The Heat map shows preferential delivery of Tel-BASP (15) to tumor, spleen and liver (FIG.11C).
- FIGs.12A to 12B depict an in vivo comparison of two different formulations of Tel-BASP that incorporate different PEG lengths (PEG 2000K and PEG 3000K).
- FIG.12A summarizes the results of an LC/MS/MS PK assay, while FIG.12B shows Cy7.5 imaging of both Tel-BASP formulations (formalin-fixed 4T1 tumors). Both formulations show similar high drug retention in tumor (23% of injected dose).
- FIG.13 is a graph showing the results of a long-term pharmacokinetic study of Tel-BASP (15) dosed in 4T1 tumor bearing mice.
- Whole blood, tumor and liver samples were analyzed for free and polymer-bound telmisartan at the indicated timepoints following a single i.v. dose of the BASP (10 mg/kg).
- FIGs.14A to 14C compare the lability of three different telmisartan BASP constructs with resulting in vivo results.
- FIGs.14A to 14B are graphs showing the results of a single-dose pharmacokinetic study in 4T1 tumor bearing mice quantifying the time-dependent concentrations of free and polymer-bound telmisartan in whole blood (FIG.14A) and tumor tissue (FIG.14B) using LC/MS/MS.
- the predicted lability of ester bonds and the rational design of drug linkers (FIG.14C) resulted in increased drug release in whole blood and tumor tissue reaching physiologically relevant levels in the tumor.
- FIGs.16A to 16B are graphs showing the results of a single-dose
- FIGs.17A to 17C show biomarker-based evidence for microenvironmentally active release of paricalcitol from Pari-BASP (17).
- FIG.17A is an image of an in vitro model system of human stellate cells treated with paricalcitol (100 nm), in which the upregulation of VDR protein expression in response to ligand binding is validated by multiplex staining.
- FIG.17B shows the quantification of these data
- FIG. 17C shows the upregulation of VDR protein in vitro, supporting the release of biologically active paricalcitol in the disease tissue.
- FIGs.18A to 18B show the results of dose range finding studies for Pari- BASP (17) in 4T1 tumor bearing Balb/c mice and in na ⁇ ve non-tumor bearing FVB mice.
- FIG.18A shows imaging of Cy7.5-conjugated Pari-BASP particles (17) in formalin fixed tumor tissues collected 72 hrs post i.v. administration of a single dose; the image shows the dose-dependent tumor uptake of BASP nanoparticles.
- Generic paricalcitol (Zemplar) dosed at 10 ⁇ g/kg, is equivalent in terms of the amount of paricalcitol to the dosing of Pari-BASP (17) at 100 ⁇ g/kg with 10% drug loading).
- Pari-BASP (17) doses are shown as 1X (100 ⁇ g/kg), 5X (500 ⁇ g/kg) and 15X (1500 ⁇ g/kg) which is relative to the 1X dose of non-conjugated, generic Paricalcitol (10 ⁇ g/kg).
- FIGs.19A to 19B show the results of an anti-cancer efficacy study in a pancreatic tumor model AK4.4 comparing gemcitabine alone, telmisartan alone, telmisartan plus gemcitabine, Tel-BASP (21) and Tel-BASP (21) plus gemcitabine.
- FIG.19A describes the study design with survival endpoints, while FIG.19B summarizes the dose groups and route of administration.
- FIGs.20A to 20B are graphs showing that Tel-BASP (21) combined with gemcitabine exhibits superior survival benefit compared to generic telmisartan in combination with gemcitabine.
- FIGs.21A to 21D are images showing that Tel-BASP (21) plus gemcitabine enhances the number of AGTR1 positive cells in AK4.4 Tumors as compated to either agent alone.
- FIGs.22A to 22B show the anti-cancer efficacy of Tel-BASP (21) in the pancreatic cancer model AK4.4 in combination with standard of care chemotherapy.
- FIG.22A describes the study design with a single endpoint, while FIG.22B summarizes the dose groups and route of administration.
- FIGs.23A to 23C show efficacy endpoints on tumor size (FIG.23A) and mass (FIGs.23B to 23C) of treatment with gemcitabine alone, telmisartan alone, telmisartan plus gemcitabine, Tel-BASP (21) and Tel-BASP (21) plus gemcitabine.
- FIG.23A is a photo of formalin fixed tumors arranged by dose groups to visualize the effect of drug treatment.
- FIGs.23B to 23C are graphs showing the quantification of average tumor volume and mass at the endpoint of study (Day 19 post dosing).
- FIGs.24A to 24D show the results of an anti-fibrosis efficacy study in a CCl 4 -induced liver fibrosis mouse model, in which Tel-BASP (22) (700 mg/kg; dosed i.v. once weekly) was compared to generic telmisartan (10 mg/kg) dosed daily by oral gavage.
- Liver fibrosis was induced by biweekly i.p. injection of CCl 4 during the 6 week study period with administration of the experimental test agents during the last 2 weeks of the study (Week 4 to Week 6).
- FIGs.25A to 25E show Tel-BASP (22) treatment is more efficacious than generic telmisartan at reducing CCl 4 -induced liver inflammation (FIGs.25A to 25D) and that the enhanced activity of Tel-BASP correlates with an efficient drug delivery (FIG.25E) measured by LC/MS/MS at study endpoint.
- Tissue sections from animals treated as indicated were stained with anti-S100A4 antibody, and the staining was developed with DAB and counterstained with hematoxylin.
- FIGs.26A to 26C show the efficacy of Tel-BASP (22) treatment in a CCl 4 - induced liver fibrosis mouse model (700 mg/kg; dosed i.v.
- telmisartan 10 mg/kg dosed daily by oral gavage.
- FIGs.27A to 27D show that Tel-BASP (22) treatment reverses CCl 4 -induced glycogen depletion. Tissue sections from animals treated as indicated were stained for glycogen using the Periodic-Schiff process.
- FIGs.28A to 28D are images that show Tel-BASP (22) treatment reduces CCl 4 -induced liver fibrosis.
- Tissue sections from animals treated as indicated were stained with anti-BigH3 (TGFBI) antibody, and staining was developed with DAB and counterstained with hematoxylin.
- TGFBI anti-BigH3
- FIGs.29A and 29B show the blood biochemistry results from two
- Score 3 Periportal and interface degeneration/necrosis
- score 4 Pan-lobular degeneration/necrosis. Significant reduction of liver necrosis was observed.
- FIGs.31A and 31B show treatment protects against periportal hepatocyte necrosis.
- FIGs.32A and 32B show the effect of Tel-BASP (22) and Tel-BRUSH (28) on gene expression of indicated genes (listed by their official human genome symbol) in the CCl 4 mouse fibrosis liver study. Tel-BASP and Tel-BRUSH both modulated several genes involved in fibrotic processes with a trend for Tel-BRUSH to be more potent than Tel-BASP.
- FIGs.33A and 33B shows terminal Liver and Blood PK analyses from the CCl 4 liver fibrosis disease model measuring both released (i.e, free) and conjugated Telmisartan and showing the ratio of released to conjugated drug present in each compartment.
- the terminal PK timepoint is 1 week post the 2 nd dosage of Tel- BRUSH (28) and Tel-BASP (22) dosed at 300 mpk i.v. at the beginning of week 1 (1 st dose) and at the beginning of week2 (2 nd dose).
- the PK data for generic telmisart (Telm) is after 14 daily doses and 24 hour post last dose. The data shows that Tel- BRUSH delivers 5 times more active pharmaceutical ingredient (API) to liver & shows 10 times higher fractional release compared to Tel-BASP (Week 6; 300 mg/kg i.v.; total of 2 doses QW)
- FIG.34 shows that plasma levels of conjugated (top curve) and free (bottom curve) telmisartan demonstrated dose proportionality when Tel-BASP (26) was administered to dogs.
- FIG.35 shows the comparative effects of Tel-BASP (21), free telmisartan (“Telm”), and immuno-oncology treatment (a combination of anti-PD1 and anti- CTLA4;“PD-1/CTLA-4”) monotherapy, as well as combinations of immuno- oncology treatment with either Tel-BASP (21) (“Tel-BASP (21) + PD-1/CTLA-4”) or free telmisartan (“Telm + PD-1/CTLA-4”) on tumor size in a mouse breast cancer model.
- Tel-BASP (21) Tel-BASP (21) + PD-1/CTLA-4
- Telm + PD-1/CTLA-4 free telmisartan
- FIGs.36A to 36B show the effects of immuno-oncology treatment (“anti-PD- 1”) alone or in combination with Pari-BASP (27) (“anti-PD-1/Pari-BASP (27)”) on tumor size (FIG.36A) and survival (FIG.36B) in a mouse melanoma model.
- FIGs.37A to 37B show the effects of immuno-oncology treatment (“anti-PD- 1”) alone or in combination with Pari-BASP (27) (“anti-PD-1/Pari-BASP (27)”) on tumor size (FIG.37A) and survival (FIG.37B) in a mouse colon cancer model.
- FIGs.38A to 38C show the experimental protocol (FIG.38A) for and effects of Telmisartan, Tel-BASP (26), and Tel-BRUSH (28) on fibrosis score (FIG.38B) and NAFLD activity (FIG.38C) in the Gubra mouse fatty liver disease/NASH model.
- FIGs.39A and 39B shows terminal Liver and Blood PK Analyses from the Gubra fatty liver disease model measuring both released (i.e, free) and conjugated Telmisartan and showing the ration of released to conjugated drug present in each compartment.
- the terminal PK timepoint is 5 week post the 2 nd dosage of Tel- BRUSH (28) and Tel-BASP (26) dosed at 300 mpk i.v. at the beginning of week 1 (1 st dose) and at the beginning of week 3 (2 nd dose).
- Tel-Brush (28) releases 3-5 times more API and has 10 times higher fractional release in liver compared to Tel-BASP.
- FIGs.40A to 40C show efficient drug delivery of Tel-BASP (26) to mouse liver at 72 hr post dose (300 mpk i.v.) using two different approaches: (FIG. 40A) anti-PEG antibody-based immunohistochemistry (IHC) staining or, (FIG.40B) direct imaging of the Cy7.5 fluorophore tracer revealing regional drug biodistribution to sinusoid areas (white arrows). Moreover, co-staining of IBA-1 macrophage marker with Cy-7.5 (FIG.40C) shows co-location of drug to this cell type (white arrows).
- IHC immunohistochemistry
- conjugates and particles comprising brush-arm star polymers (BASPs) coupled to an agent (e.g., one or more therapeutic agents, diagnostic agents, or targeting moieties), starting materials, intermediates, and degradation components thereof (collectively referred to herein as“BRUSH- compositions” or“BASP-compositions”), as well as pharmaceutical compositions and methods of making and using the same.
- BASPs brush-arm star polymers
- the BRUSH-compositions or BASP-compositions enhance the delivery, distribution, release and/or efficacy of an agent in a subject to a desired target site, e.g., compared to the systemic release of the agent as a free form (e.g., not coupled to a BRUSH-composition or BASP-composition described herein).
- the agent delivered or released is a therapeutic agent (e.g., an angiotensin receptor blocker (ARB), a vitamin D analog, an indoleamine 2,3- dioxigenase 1 (IDO) inhibitor, or a bromodomain inhibitor as described herein).
- a therapeutic agent e.g., an angiotensin receptor blocker (ARB), a vitamin D analog, an indoleamine 2,3- dioxigenase 1 (IDO) inhibitor, or a bromodomain inhibitor as described herein.
- Compounds described herein can comprise one or more asymmetric centers, and thus can exist in various stereoisomeric forms, e.g., enantiomers and/or diastereomers.
- the compounds described herein can be in the form of an individual enantiomer, diastereomer or geometric isomer, or can be in the form of a mixture of stereoisomers, including racemic mixtures and mixtures enriched in one or more stereoisomer.
- Isomers can be isolated from mixtures by methods known to those skilled in the art, including chiral high pressure liquid chromatography (HPLC) and the formation and crystallization of chiral salts; or preferred isomers can be prepared by asymmetric syntheses.
- HPLC high pressure liquid chromatography
- C 1 -C 6 alkyl is intended to encompass, C 1 , C 2 , C 3 , C 4 , C 5 , C 6 , C 1 -C 6 , C 1 -C 5 , C 1 -C 4 , C 1 -C 3 , C 1 -C 2 , C 2 -C 6 , C 2 -C 5 , C 2 -C 4 , C 2 -C 3 , C 3 - C 6 , C 3 -C 5 , C 3 -C 4 , C 4 -C 6 , C 4 -C 5 , and C 5 -C 6 alkyl.
- a range expressed in the form“between XX and YY” or“between XX and YY, inclusive” refers to a range between XX and YY, inclusive, wherein each of XX and YY is independently a number (e.g., integer, fraction, or percentage).
- alkyl refers to a radical of a straight–chain or branched saturated hydrocarbon group.
- an alkyl group has 1 to 10 carbon atoms (“C 1 -C 10 alkyl”), 1 to 9 carbon atoms (“C 1 -C 9 alkyl”), 1 to 8 carbon atoms (“C 1 -C 8 alkyl”), 1 to 7 carbon atoms (“C 1 -C 7 alkyl”), 1 to 6 carbon atoms (“C 1 -C 6 alkyl”), 1 to 5 carbon atoms (“C 1 -C 5 alkyl”), 1 to 4 carbon atoms (“C 1 -C 4 alkyl”), 1 to 3 carbon atoms (“C 1 -C 3 alkyl”), 1 to 2 carbon atoms (“C 1 -C 2 alkyl”), or 1 carbon atom (“C 1 alkyl”).
- C 1 -C 6 alkyl groups include methyl (C 1 ), ethyl (C 2 ), n–propyl (C 3 ), isopropyl (C 3 ), n–butyl (C 4 ), tert–butyl (C 4 ), sec–butyl (C 4 ), iso–butyl (C 4 ), n– pentyl (C 5 ), 3–pentanyl (C 5 ), amyl (C 5 ), neopentyl (C 5 ), 3–methyl–2–butanyl (C 5 ), tertiary amyl (C 5 ), and n–hexyl (C 6 ).
- alkyl groups include n– heptyl (C 7 ), n–octyl (C 8 ) and the like. Unless otherwise specified, each instance of an alkyl group is independently unsubstituted (an“unsubstituted alkyl”) or substituted (a “substituted alkyl”) with one or more substituents.
- alkenyl refers to a radical of a straight–chain or branched hydrocarbon group having from 2 to 10 carbon atoms and one or more carbon-carbon double bonds (e.g., 1, 2, 3, or 4 double bonds).
- an alkenyl group has 2 to 9 carbon atoms (“C 2–9 alkenyl”).
- an alkenyl group has 2 to 8 carbon atoms (“C 2–8 alkenyl”).
- an alkenyl group has 2 to 7 carbon atoms (“C 2–7 alkenyl”).
- an alkenyl group has 2 to 6 carbon atoms (“C 2–6 alkenyl”).
- an alkenyl group has 2 to 5 carbon atoms (“C 2–5 alkenyl”). In some embodiments, an alkenyl group has 2 to 4 carbon atoms (“C 2–4 alkenyl”). In some embodiments, an alkenyl group has 2 to 3 carbon atoms (“C 2–3 alkenyl”). In some embodiments, an alkenyl group has 2 carbon atoms (“C 2 alkenyl”).
- the one or more carbon–carbon double bonds can be internal (such as in 2–butenyl) or terminal (such as in 1–butenyl).
- Examples of C 2–4 alkenyl groups include ethenyl (C 2 ), 1–propenyl (C 3 ), 2–propenyl (C 3 ), 1–butenyl (C 4 ), 2–butenyl (C 4 ), butadienyl (C 4 ), and the like.
- Examples of C 2–6 alkenyl groups include the aforementioned C 2–4 alkenyl groups as well as pentenyl (C 5 ), pentadienyl (C 5 ), hexenyl (C 6 ), and the like. Unless otherwise specified, each instance of an alkenyl group is independently unsubstituted (an“unsubstituted alkenyl”) or substituted (a“substituted alkenyl”) with one or more substituents.
- alkynyl refers to a radical of a straight–chain or branched hydrocarbon group having from 2 to 10 carbon atoms and one or more carbon-carbon triple bonds (e.g., 1, 2, 3, or 4 triple bonds) (“C 2–10 alkynyl”).
- an alkynyl group has 2 to 9 carbon atoms (“C 2–9 alkynyl”), 2 to 8 carbon atoms (“C 2–8 alkynyl”), 2 to 7 carbon atoms (“C 2–7 alkynyl”), 2 to 6 carbon atoms (“C 2–6 alkynyl”), 2 to 5 carbon atoms (“C 2–5 alkynyl”), 2 to 4 carbon atoms (“C 2–4 alkynyl”), 2 to 3 carbon atoms (“C 2–3 alkynyl”), or 2 carbon atoms (“C 2 alkynyl”).
- the one or more carbon–carbon triple bonds can be internal (such as in 2–butynyl) or terminal (such as in 1–butynyl).
- Examples of C 2–4 alkynyl groups include, without limitation, ethynyl (C 2 ), 1–propynyl (C 3 ), 2–propynyl (C 3 ), 1–butynyl (C 4 ), 2–butynyl (C 4 ), and the like.
- Examples of C 2–6 alkenyl groups include the aforementioned C 2–4 alkynyl groups as well as pentynyl (C 5 ), hexynyl (C 6 ), and the like. Unless otherwise specified, each instance of an alkynyl group is independently unsubstituted (an“unsubstituted alkynyl”) or substituted (a“substituted alkynyl”) with one or more substituents.
- heteroalkyl refers to an alkyl group which further includes at least one heteroatom (e.g., 1, 2, 3, or 4 heteroatoms) selected from oxygen, nitrogen, phosphorus, or sulfur within (i.e., inserted between adjacent carbon atoms of) and/or placed at one or more terminal position(s) of the parent chain.
- heteroatom e.g., 1, 2, 3, or 4 heteroatoms
- a heteroalkyl group refers to a saturated group having from 1 to 10 carbon atoms and 1 or more heteroatoms within the parent chain (“C 1– C 10
- heteroalkyl 1 to 9 carbon atoms and 1 or more heteroatoms within the parent chain (“C 1– C 9 heteroalkyl”), 1 to 8 carbon atoms and 1 or more heteroatoms within the parent chain (“C 1– C 8 heteroalkyl”), 1 to 7 carbon atoms and 1 or more heteroatoms within the parent chain (“C 1– C 7 heteroalkyl”), 1 to 6 carbon atoms and 1 or more heteroatoms within the parent chain (“C 1– C 6 heteroalkyl”), 1 to 5 carbon atoms and 1 or more heteroatoms within the parent chain (“C 1– C 5 heteroalkyl”), 1 to 4 carbon atoms and 1or more heteroatoms within the parent chain (“C 1– C 4 heteroalkyl”), 1 to 3 carbon atoms and 1 or more heteroatoms within the parent chain (“C 1– C 3
- heteroalkyl 1 to 2 carbon atoms and 1 heteroatom within the parent chain (“C 1– C 2 heteroalkyl”), or 1 carbon atom and 1 heteroatom (“C 1 heteroalkyl”). Unless otherwise specified, each instance of a heteroalkyl group is independently
- “carbocyclyl” or“carbocyclic” or“cycloalkyl” refers to a radical of a non–aromatic cyclic hydrocarbon group having from 3 to 10 ring carbon atoms (“C 3–10 carbocyclyl”) and zero heteroatoms in the non–aromatic ring system.
- a carbocyclyl group has 3 to 8 ring carbon atoms (“C 3–8 carbocyclyl”), 3 to 7 ring carbon atoms (“C 3–7 carbocyclyl”), 3 to 6 ring carbon atoms (“C 3–6 carbocyclyl”), 4 to 6 ring carbon atoms (“C 4–6 carbocyclyl”), 5 to 6 ring carbon atoms (“C 5–6 carbocyclyl”), or 5 to 10 ring carbon atoms (“C 5–10 carbocyclyl”).
- Exemplary C 3–6 carbocyclyl groups include, without limitation, cyclopropyl (C 3 ), cyclopropenyl (C 3 ), cyclobutyl (C 4 ), cyclobutenyl (C 4 ), cyclopentyl (C 5 ), cyclopentenyl (C 5 ), cyclohexyl (C 6 ), cyclohexenyl (C 6 ), cyclohexadienyl (C 6 ), and the like.
- Exemplary C 3–8 carbocyclyl groups include, without limitation, the aforementioned C 3–6 carbocyclyl groups as well as cycloheptyl (C 7 ), cycloheptenyl (C 7 ), cycloheptadienyl (C 7 ), cycloheptatrienyl (C 7 ), cyclooctyl (C 8 ), cyclooctenyl (C 8 ), bicyclo[2.2.1]heptanyl (C 7 ), bicyclo[2.2.2]octanyl (C 8 ), and the like.
- Exemplary C 3–10 carbocyclyl groups include, without limitation, the aforementioned C 3–8 carbocyclyl groups as well as cyclononyl (C 9 ), cyclononenyl (C 9 ), cyclodecyl (C 10 ), cyclodecenyl (C 10 ), octahydro–1H–indenyl (C 9 ), decahydronaphthalenyl (C 10 ), spiro[4.5]decanyl (C 10 ), and the like.
- the carbocyclyl group is either monocyclic (“monocyclic carbocyclyl”) or polycyclic (e.g., containing a fused, bridged or spiro ring system such as a bicyclic system (“bicyclic carbocyclyl”) or tricyclic system (“tricyclic carbocyclyl”)) and can be saturated or can contain one or more carbon–carbon double or triple bonds.
- Carbocyclyl also includes ring systems wherein the carbocyclyl ring, as defined above, is fused with one or more aryl or heteroaryl groups wherein the point of attachment is on the carbocyclyl ring, and in such instances, the number of carbons continue to designate the number of carbons in the carbocyclic ring system. Unless otherwise specified, each instance of a carbocyclyl group is independently
- heterocyclyl refers to a radical of a 3– to 14– membered non–aromatic ring system having ring carbon atoms and 1 to 4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, phosphorus, and sulfur (“3–14 membered heterocyclyl”).
- heterocyclyl groups that contain one or more nitrogen atoms, the point of attachment can be a carbon or nitrogen atom, as valency permits.
- a heterocyclyl group can either be monocyclic (“monocyclic heterocyclyl”) or polycyclic (e.g., a fused, bridged or spiro ring system such as a bicyclic system (“bicyclic heterocyclyl”) or tricyclic system (“tricyclic heterocyclyl”)), and can be saturated or can contain one or more carbon– carbon double or triple bonds.
- Heterocyclyl polycyclic ring systems can include one or more heteroatoms in one or both rings.“Heterocyclyl” also includes ring systems wherein the heterocyclyl ring, as defined above, is fused with one or more carbocyclyl groups wherein the point of attachment is either on the carbocyclyl or heterocyclyl ring, or ring systems wherein the heterocyclyl ring, as defined above, is fused with one or more aryl or heteroaryl groups, wherein the point of attachment is on the heterocyclyl ring, and in such instances, the number of ring members continue to designate the number of ring members in the heterocyclyl ring system.
- each instance of heterocyclyl is independently unsubstituted (an “unsubstituted heterocyclyl”) or substituted (a“substituted heterocyclyl”) with one or more substituents.
- a heterocyclyl group is a 5–10 membered non– aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, phosphorus, and sulfur (“5–10 membered heterocyclyl”).
- a heterocyclyl group is a 5–8 membered non–aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, phosphorus, and sulfur (“5–8 membered heterocyclyl”).
- a heterocyclyl group is a 5–6 membered non–aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, phosphorus, and sulfur (“5–6 membered heterocyclyl”).
- the 5–6 membered heterocyclyl has 1–3 ring heteroatoms selected from nitrogen, oxygen, phosphorus, and sulfur.
- the 5–6 membered heterocyclyl has 1–2 ring heteroatoms selected from nitrogen, oxygen, phosphorus, and sulfur.
- the 5–6 membered heterocyclyl has 1 ring heteroatom selected from nitrogen, oxygen, phosphorus, and sulfur.
- Exemplary 3–membered heterocyclyl groups containing 1 heteroatom include, without limitation, aziridinyl, oxiranyl, and thiiranyl.
- Exemplary 4–membered heterocyclyl groups containing 1 heteroatom include, without limitation, azetidinyl, oxetanyl and thietanyl.
- Exemplary 5–membered heterocyclyl groups containing 1 heteroatom include, without limitation, tetrahydrofuranyl, dihydrofuranyl,
- Exemplary 5–membered heterocyclyl groups containing 2 heteroatoms include, without limitation, dioxolanyl, oxathiolanyl and dithiolanyl.
- Exemplary 5– membered heterocyclyl groups containing 3 heteroatoms include, without limitation, triazolinyl, oxadiazolinyl, and thiadiazolinyl.
- Exemplary 6–membered heterocyclyl groups containing 1 heteroatom include, without limitation, piperidinyl,
- heterocyclyl groups containing 2 heteroatoms include, without limitation, piperazinyl, morpholinyl, dithianyl, and dioxanyl.
- Exemplary 6–membered heterocyclyl groups containing 3 heteroatoms include, without limitation, triazinyl.
- Exemplary 7– membered heterocyclyl groups containing 1 heteroatom include, without limitation, azepanyl, oxepanyl, and thiepanyl.
- Exemplary 8–membered heterocyclyl groups containing 1 heteroatom include, without limitation, azocanyl, oxecanyl and thiocanyl.
- bicyclic heterocyclyl groups include, without limitation, indolinyl, isoindolinyl, dihydrobenzofuranyl, dihydrobenzothienyl, tetrahydrobenzo- thienyl, tetrahydrobenzofuranyl, tetrahydroindolyl, tetrahydroquinolinyl,
- octahydrochromenyl octahydroisochromenyl, decahydronaphthyridinyl, decahydro– 1,8–naphthyridinyl, octahydropyrrolo[3,2–b]pyrrole, indolinyl, phthalimidyl, naphthalimidyl, chromanyl, chromenyl, 1H–benzo[e][1,4]diazepinyl, 1,4,5,7–tetra- hydropyrano[3,4–b]pyrrolyl, 5,6–dihydro–4H–furo[3,2–b]pyrrolyl, 6,7–dihydro–5H– furo[3,2–b]pyranyl, 5,7–dihydro–4H–thieno[2,3–c]pyranyl, 2,3–dihydro–1H– pyrrolo[2,3–b]pyridinyl, 2,
- aryl refers to a radical of a monocyclic or polycyclic (e.g., bicyclic or tricyclic) 4n+2 aromatic ring system (e.g., having 6, 10, or 14 ⁇ electrons shared in a cyclic array) having 6–14 ring carbon atoms and zero heteroatoms provided in the aromatic ring system (“C 6–14 aryl”).
- aromatic ring system e.g., having 6, 10, or 14 ⁇ electrons shared in a cyclic array
- an aryl group has 6 ring carbon atoms (“C 6 aryl”; e.g., phenyl).
- an aryl group has 10 ring carbon atoms (“C 10 aryl”; e.g., naphthyl such as 1–naphthyl and 2–naphthyl).
- an aryl group has 14 ring carbon atoms (“C 14 aryl”; e.g., anthracyl).“Aryl” also includes ring systems wherein the aryl ring, as defined above, is fused with one or more carbocyclyl or heterocyclyl groups wherein the radical or point of attachment is on the aryl ring, and in such instances, the number of carbon atoms continue to designate the number of carbon atoms in the aryl ring system.
- each instance of an aryl group is independently unsubstituted (an“unsubstituted aryl”) or substituted (a“substituted aryl”) with one or more substituents.
- heteroaryl refers to a radical of a 5–14 membered monocyclic or polycyclic (e.g., bicyclic, tricyclic) 4n+2 aromatic ring system (e.g., having 6, 10, or 14 ⁇ electrons shared in a cyclic array) having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–14 membered heteroaryl”).
- the point of attachment can be a carbon or nitrogen atom, as valency permits.
- Heteroaryl polycyclic ring systems can include one or more heteroatoms in one or both rings.
- Heteroaryl includes ring systems wherein the heteroaryl ring, as defined above, is fused with one or more carbocyclyl or heterocyclyl groups wherein the point of attachment is on the heteroaryl ring, and in such instances, the number of ring members continue to designate the number of ring members in the heteroaryl ring system.“Heteroaryl” also includes ring systems wherein the heteroaryl ring, as defined above, is fused with one or more aryl groups wherein the point of attachment is either on the aryl or heteroaryl ring, and in such instances, the number of ring members designates the number of ring members in the fused polycyclic
- aryl/heteroaryl (aryl/heteroaryl) ring system.
- Polycyclic heteroaryl groups wherein one ring does not contain a heteroatom e.g., indolyl, quinolinyl, carbazolyl, and the like
- the point of attachment can be on either ring, i.e., either the ring bearing a heteroatom (e.g., 2– indolyl) or the ring that does not contain a heteroatom (e.g., 5–indolyl).
- a heteroaryl group be monovalent or may have more than one point of attachment to another moiety (e.g., it may be divalent, trivalent, etc.), although the valency may be specified directly in the name of the group.
- “triazoldiyl” refers to a divalent triazolyl moiety.
- a heteroaryl group is a 5–10 membered aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–10 membered heteroaryl”).
- a heteroaryl group is a 5–8 membered aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–8 membered heteroaryl”).
- a heteroaryl group is a 5–6 membered aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–6 membered heteroaryl”).
- the 5–6 membered heteroaryl has 1–3 ring heteroatoms selected from nitrogen, oxygen, and sulfur.
- the 5–6 membered heteroaryl has 1–2 ring heteroatoms selected from nitrogen, oxygen, and sulfur.
- the 5– 6 membered heteroaryl has 1 ring heteroatom selected from nitrogen, oxygen, and sulfur. Unless otherwise specified, each instance of a heteroaryl group is
- Exemplary 5–membered heteroaryl groups containing 1 heteroatom include, without limitation, pyrrolyl, furanyl, and thiophenyl.
- Exemplary 5–membered heteroaryl groups containing 2 heteroatoms include, without limitation, imidazolyl, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, and isothiazolyl.
- Exemplary 5–membered heteroaryl groups containing 3 heteroatoms include, without limitation, triazolyl, oxadiazolyl, and thiadiazolyl.
- Exemplary 5–membered heteroaryl groups containing 4 heteroatoms include, without limitation, tetrazolyl.
- Exemplary 6–membered heteroaryl groups containing 1 heteroatom include, without limitation, pyridinyl.
- Exemplary 6–membered heteroaryl groups containing 2 heteroatoms include, without limitation, pyridazinyl, pyrimidinyl, and pyrazinyl.
- Exemplary 6–membered heteroaryl groups containing 3 or 4 heteroatoms include, without limitation, triazinyl and tetrazinyl, respectively.
- Exemplary 7–membered heteroaryl groups containing 1 heteroatom include, without limitation, azepinyl, oxepinyl, and thiepinyl.
- Exemplary 5,6–bicyclic heteroaryl groups include, without limitation, indolyl, isoindolyl, indazolyl, benzotriazolyl, benzothiophenyl, isobenzothiophenyl, benzofuranyl, benzoisofuranyl, benzimidazolyl, benzoxazolyl, benzisoxazolyl, benzoxadiazolyl, benzthiazolyl, benzisothiazolyl, benzthiadiazolyl, indolizinyl, and purinyl.
- Exemplary 6,6–bicyclic heteroaryl groups include, without limitation, naphthyridinyl, pteridinyl, quinolinyl, isoquinolinyl, cinnolinyl, quinoxalinyl, phthalazinyl, and quinazolinyl.
- Exemplary tricyclic heteroaryl groups include, without limitation, phenanthridinyl, dibenzofuranyl, carbazolyl, acridinyl, phenothiazinyl, phenoxazinyl and phenazinyl.
- alkyl, alkenyl, alkynyl, carbocyclyl, aryl, and heteroaryl groups are, in certain embodiments, optionally substituted.
- Optionally substituted refers to a group which may be substituted or unsubstituted (e.g., “substituted” or“unsubstituted” alkyl).
- the term“substituted” means that at least one hydrogen present on a group is replaced with a permissible substituent, e.g., a substituent which upon substitution results in a stable compound, e.g., a compound which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, or other reaction.
- a“substituted” group has a substituent at one or more substitutable positions of the group, and when more than one position in any given structure is substituted, the substituent is either the same or different at each position.
- the term“substituted” is contemplated to include substitution with all permissible substituents of organic compounds, any of the substituents described herein that results in the formation of a stable compound.
- the present invention contemplates any and all such combinations in order to arrive at a stable compound.
- heteroatoms such as nitrogen may have hydrogen substituents and/or any suitable substituent as described herein which satisfy the valencies of the heteroatoms and results in the formation of a stable moiety.
- Exemplary carbon atom substituents include, but are not limited to, halogen, ⁇ CN, ⁇ NO 2 , ⁇ N 3 , ⁇ SO 2 H, ⁇ SO 3 H, ⁇ OH, ⁇ OR aa , ⁇ ON(R bb ) 2 , ⁇ N(R bb ) 2 , ⁇ N(R bb ) +
- each instance of R aa is, independently, selected from C 1-10 alkyl, C 1-10 perhaloalkyl, C 2-10 alkenyl, C 2-10 alkynyl, heteroC 1-10 alkyl, heteroC 2-10 alkenyl, heteroC 2-10 alkynyl, C 3-10 carbocyclyl, 3-14 membered heterocyclyl, C 6-14 aryl, and 5- 14 membered heteroaryl, or two R aa groups are joined to form a 3-14 membered heterocyclyl or 5-14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R dd groups;
- each instance of R cc is, independently, selected from hydrogen, C 1-10 alkyl, C 1- 10 perhaloalkyl, C 2-10 alkenyl, C 2-10 alkynyl, heteroC 1-10 alkyl, heteroC 2-10 alkenyl, heteroC 2-10 alkynyl, C 3-10 carbocyclyl, 3-14 membered heterocyclyl, C 6-14 aryl, and 5- 14 membered heteroaryl, or two R cc groups are joined to form a 3-14 membered heterocyclyl or 5-14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R dd groups;
- each instance of R dd is, independently, selected from halogen, ⁇ CN, ⁇ NO 2 , ⁇ N 3 , ⁇ SO 2 H, ⁇ SO 3 H, ⁇ OH, ⁇ OR ee , ⁇ ON(R ff ) 2 , ⁇ N(R ff ) 2 , ⁇ N(R ff ) +
- each instance of R ee is, independently, selected from C 1-6 alkyl, C 1-6 perhaloalkyl, C 2-6 alkenyl, C 2-6 alkynyl, heteroC 1-6 alkyl, heteroC 2-6 alkenyl, heteroC 2-6 alkynyl, C 3-10 carbocyclyl, C 6-10 aryl, 3-10 membered heterocyclyl, and 3-10 membered heteroaryl, wherein each alkyl, alkenyl, alkynyl, heteroalkyl,
- heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R gg groups;
- each instance of R ff is, independently, selected from hydrogen, C 1-6 alkyl, C 1-6 perhaloalkyl, C 2-6 alkenyl, C 2-6 alkynyl, heteroC 1-6 alkyl, heteroC 2-6 alkenyl, heteroC 2- 6 alkynyl, C 3-10 carbocyclyl, 3-10 membered heterocyclyl, C 6-10 aryl and 5-10 membered heteroaryl, or two R ff groups are joined to form a 3-10 membered heterocyclyl or 5-10 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R gg groups; and
- each instance of R gg is, independently, halogen, ⁇ CN, ⁇ NO 2 , ⁇ N 3 , ⁇ SO 2 H, ⁇ SO 3 H, ⁇ OH, ⁇ OC 1-6 alkyl, ⁇ ON(C 1-6 alkyl) 2 , ⁇ N(C 1-6 alkyl) 2 , ⁇ N(C 1-6 alkyl) +
- the carbon atom substituents are independently halogen, substituted or unsubstituted C 1-6 alkyl, ⁇ OR aa , ⁇ SR aa , ⁇ N(R bb ) 2 ,–CN,–SCN, or–NO 2 .
- Nitrogen atoms can be substituted or unsubstituted as valency permits, and include primary, secondary, tertiary, and quaternary nitrogen atoms.
- the substituent present on the nitrogen atom is an nitrogen protecting group (also referred to herein as an“amino protecting group”).
- Nitrogen protecting groups are well known in the art and include those described in detail in Protecting Groups in Organic Synthesis, T. W. Greene and P. G. M. Wuts, 3 rd edition, John Wiley & Sons, 1999, incorporated herein by reference.
- amide groups include, but are not limited to, formamide, acetamide, chloroacetamide, trichloroacetamide, trifluoroacetamide, phenylacetamide, 3-phenylpropanamide, picolinamide, 3-pyridylcarboxamide, N-benzoylphenylalanyl derivative,
- Nitrogen protecting groups such as carbamate groups include, but are not limited to, methyl carbamate, ethyl carbamate, 9-fluorenylmethyl carbamate (Fmoc), 9-(2-sulfo)fluorenylmethyl carbamate, 9-(2,7- dibromo)fluoroenylmethyl carbamate, 2,7-di-t-butyl-[9-(10,10-dioxo-10,10,10,10- tetrahydrothioxanthyl)]methyl carbamate (DBD-Tmoc), 4-methoxyphenacyl carbamate (Phenoc), 2,2,2-trichloroethyl carbamate (Troc), 2-trimethylsilylethyl carbamate (Teoc), 2-phenylethyl carbamate (hZ), 1-(1-adamantyl)-1-methylethyl carbamate
- Nitrogen protecting groups such as sulfonamide groups include, but are not limited to, p-toluenesulfonamide (Ts), benzenesulfonamide, 2,3,6- trimethyl-4-methoxybenzenesulfonamide (Mtr), 2,4,6-trimethoxybenzenesulfonamide (Mtb), 2,6-dimethyl-4-methoxybenzenesulfonamide (Pme), 2,3,5,6-tetramethyl-4- methoxybenzenesulfonamide (Mte), 4-methoxybenzenesulfonamide (Mbs), 2,4,6- trimethylbenzenesulfonamide (Mts), 2,6-dimethoxy-4-methylbenzenesulfonamide (iMds), 2,2,5,7,8-pentamethylchroman-6-sulfonamide (Pmc), methanesulfonamide
- Ts p-toluenesulfonamide
- nitrogen protecting groups include, but are not limited to,
- phenothiazinyl-(10)-acyl derivative N’-p-toluenesulfonylaminoacyl derivative, N’- phenylaminothioacyl derivative, N-benzoylphenylalanyl derivative, N- acetylmethionine derivative, 4,5-diphenyl-3-oxazolin-2-one, N-phthalimide, N- dithiasuccinimide (Dts), N-2,3-diphenylmaleimide, N-2,5-dimethylpyrrole, N-1,1,4,4- tetramethyldisilylazacyclopentane adduct (STABASE), 5-substituted 1,3-dimethyl- 1,3,5-triazacyclohexan-2-one, 5-substituted 1,3-dibenzyl-1,3,5-triazacyclohexan-2- one, 1-substituted 3,5-dinitro-4-pyridone, N-methylamine
- Dpp diphenylphosphinamide
- Mpt dimethylthiophosphinamide
- triphenylmethylsulfenamide triphenylmethylsulfenamide
- 3-nitropyridinesulfenamide Npys
- the substituent present on an oxygen atom is an oxygen protecting group (also referred to herein as an“hydroxyl protecting group”).
- Oxygen protecting groups include, but are not limited to, ⁇ R aa , ⁇ N(R bb ) 2 ,
- Oxygen protecting groups are well known in the art and include those described in detail in Protecting Groups in Organic Synthesis, T. W. Greene and P. G. M. Wuts, 3 rd edition, John Wiley & Sons, 1999, incorporated herein by reference.
- oxygen protecting groups include, but are not limited to, methyl, methoxylmethyl (MOM), methylthiomethyl (MTM), t-butylthiomethyl,
- IPDMS dimethylisopropylsilyl
- DEIPS diethylisopropylsilyl
- TDMS t-butyldimethylsilyl
- TDPS t-butyldiphenylsilyl
- tribenzylsilyl tri-p- xylylsilyl, triphenylsilyl, diphenylmethylsilyl (DPMS), t-butylmethoxyphenylsilyl (TBMPS)
- formate benzoylformate, acetate, chloroacetate, dichloroacetate, trichloroacetate, trifluoroacetate, methoxyacetate, triphenylmethoxyacetate, phenoxyacetate, p-chlorophenoxyacetate, 3-phenylpropionate, 4-oxopentanoate (levulinate), 4,4-(ethylenedithio)pentanoate (levulinoyldi)
- the substituent present on a sulfur atom is a sulfur protecting group (also referred to as a“thiol protecting group”).
- halo or“halogen” refers to fluorine (fluoro,–F), chlorine (chloro, –Cl), bromine (bromo,–Br), or iodine (iodo,–I).
- amine or“amino” refers to the group–NH– or–NH 2 .
- polyethylene glycol or“PEG” refers to an ethylene glycol polymer that contains about 20 to about 2,000,000 linked monomers, typically about 50-1,000 linked monomers, usually about 100-300.
- Polyethylene glycols include ethylene glycol polymer containing various numbers of linked monomers, e.g., PEG20, PEG30, PEG40, PEG60, PEG80, PEG100, PEG115, PEG200, PEG300, PEG400, PEG500, PEG600, PEG1000, PEG1500, PEG2000, PEG3350, PEG4000, PEG4600, PEG5000, PEG6000, PEG8000, PEG11000, PEG12000, PEG2000000 and any mixtures thereof.
- linked monomers e.g., PEG20, PEG30, PEG40, PEG60, PEG80, PEG100, PEG115, PEG200, PEG300, PEG400, PEG500, PEG600, PEG1000, PEG1500, PEG2000, PEG3350, PEG4000, PEG4600, PEG5000, PEG6000, PEG8000, PEG11000, PEG12000, PEG2000000 and any mixtures thereof.
- salt refers to ionic compounds that result from the neutralization reaction of an acid and a base.
- a salt is composed of one or more cations (positively charged ions) and one or more anions (negative ions) so that the salt is electrically neutral (without a net charge).
- Salts of the compounds of this invention include those derived from inorganic and organic acids and bases.
- acid addition salts are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid, and perchloric acid, or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid, or malonic acid or by using other methods known in the art such as ion exchange.
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid, and perchloric acid
- organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid, or malonic acid or by using other methods known in the art such as ion exchange.
- salts include adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2–hydroxy–ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2–naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pectinate, per
- Salts derived from appropriate bases include alkali metal, alkaline earth metal, ammonium and N + (C 1-4 alkyl) 4 salts.
- Representative alkali or alkaline earth metal salts include sodium, lithium, potassium, calcium, magnesium, and the like.
- Further salts include ammonium, quaternary ammonium, and amine cations formed using counterions such as halide, hydroxide, carboxylate, sulfate, phosphate, nitrate, lower alkyl sulfonate, and aryl sulfonate.
- the present invention discloses, at least in part, macromonomers, conjugates comprising the macromonomers (also referred to alternatively as“BRUSH”, or “BRUSH conjugates”), and brush arm star polymer particles comprising the conjugates (also referred to as“BASPs” or“BASP particles”), each further comprising an agent (e.g., one or more therapeutic agents, diagnostic agents, or targeting moieties), Compositions comprising BASP particles are referred to as “BASP-compositions”. Compositions comprising BRUSH conjugates are referred to as“BRUSH-compositions”.
- the macromonomers, BRUSH conjugates, and BASP particles disclosed herein comprise an agent(s), e.g., a first therapeutic agent (e.g., at least one (including, e.g., at least two, at least three) an ARB, a vitamin D analog, IDO inhibitor, or a bromodomain inhibitor).
- a first therapeutic agent e.g., at least one (including, e.g., at least two, at least three) an ARB, a vitamin D analog, IDO inhibitor, or a bromodomain inhibitor.
- the BRUSHs, and BASPs can further comprise a second therapeutic agent, a targeting moiety, a diagnostic moiety, e.g., as described herein.
- the agent(s) can be chemically bound to the BRUSHs, and BASPs.
- the agent(s) can be associated with a BRUSH or BASP.
- a first agent can be coupled to the BRUSH or BASP, and a second agent, targeting moiety, and/or diagnostic moiety can be non-covalently associated with the BRUSH or BASP.
- a second agent, targeting moiety, and/or diagnostic moiety can be non-covalently associated with the BRUSH or BASP.
- agent means a molecule, group of molecules, complex or substance administered to an organism for diagnostic, therapeutic, preventative medical, or veterinary purposes.
- the term“therapeutic agent” includes an agent that is capable of providing a local or systemic biological, physiological, or therapeutic effect in the biological system to which it is applied.
- a therapeutic agent can act to control tumor growth, control infection or inflammation, act as an analgesic, promote anti-cell attachment, and enhance bone growth, among other functions.
- Other suitable therapeutic agents can include anti-viral agents, hormones, antibodies, or therapeutic proteins.
- Other therapeutic agents include prodrugs, which are agents that are not biologically active when administered but, upon administration to a subject are converted to biologically active agents through metabolism or some other mechanism.
- An agent e.g., a therapeutic agent
- polysaccharides polysaccharides; biological macromolecules, e.g., peptides, proteins, and peptide analogs and derivatives; peptidomimetics; antibodies and antigen binding fragments thereof; nucleic acids; nucleic acid analogs and derivatives; an extract made from biological materials such as bacteria, plants, fungi, or animal cells; animal tissues; naturally occurring or synthetic compositions; and any combinations thereof.
- the agent is in the form of a prodrug.
- prodrug refer to a compound that becomes active, e.g., by solvolysis, reduction, oxidation, or under physiological conditions, to provide a pharmaceutically active compound, e.g., in vivo.
- a prodrug can include a derivative of a pharmaceutically active compound, such as, for example, to form an ester by reaction of the acid, or acid anhydride, or mixed anhydrides moieties of the prodrug moiety with the hydroxyl moiety of the pharmaceutical active compound, or to form an amide prepared by the acid, or acid anhydride, or mixed anhydrides moieties of the prodrug moiety with a substituted or unsubstituted amine of the pharmaceutically active compound.
- Simple aliphatic or aromatic esters, amides, and anhydrides derived from acidic groups may comprise prodrugs.
- the conjugate or BASP particle described herein incorporates one therapeutic agent or prodrug thereof. In some embodiments, the conjugate or BASP particle described herein incorporates more than one therapeutic agents or prodrugs.
- the agent e.g., a therapeutic agent
- the agent is a small molecule.
- the term“small molecule” can refer to compounds that are“natural product-like.”
- the term“small molecule” is not limited to“natural product- like” compounds. Rather, a small molecule is typically characterized in that it contains several carbon—carbon bonds, and has a molecular weight of less than 5000 Daltons (5 kDa), preferably less than 3 kDa, still more preferably less than 2 kDa, and most preferably less than 1 kDa. In some cases it is preferred that a small molecule have a molecular weight equal to or less than 700 Daltons.
- Exemplary agents, e.g., a therapeutic agents, in the BASP-compositions include, but are not limited to, those found in Harrison’s Principles of Internal Medicine , 13th Edition, Eds. T.R. Harrison et al. McGraw-Hill N.Y., NY;
- exemplary therapeutic agents in the BASP- compositions or BRUSH-compositions include, but are not limited to, one or more of the agents listed in Paragraph 0148 of U.S. Patent No.9,381,253, incorporated by reference herein.
- exemplary therapeutic agents in the BASP- compositions or BRUSH-compositions include, but are not limited to, one or more of the therapeutic agents listed in WO 2013/169739, including the anti-hypertensive and/or a collagen modifying agents (“AHCM”) disclosed, e.g., in Paragraphs 40-49, 283, 286-295; the microenvironment modulators disclosed, e.g., in Paragraphs 113- 121, of WO 2013/169739, incorporated herein by reference.
- AHCM collagen modifying agents
- the BASP-composition or BRUSH-composition comprising the AHCM and/or the microenvironment modulator causes one or more of: reduces solid stress (e.g., growth-induced solid stress in tumors); decreases tumor fibrosis; reduces interstitial hypertension or interstitial fluid pressure (IFP); increases interstitial tumor transport; increases tumor or vessel perfusion; increases vascular diameters and/or enlarges compressed or collapsed blood vessels; reduces or depletes one or more of: cancer cells, or stromal cells (e.g., tumor associated fibroblasts or immune cells); decreases the level or production of extracellular matrix components, such as fibers (e.g., collagen, procollagen), and/or polysaccharides (e.g., glycosaminoglycans such as hyaluronan or hyaluronic acid); decreases the level or production of collagen or procollagen; decreases the level or production of hyaluronic acid; increases tumor oxygenation; decreases
- cancer stem cells also referred to herein as tumor- initiating cells
- cancer therapy e.g., radiation, photodynamic therapy
- chemotherapeutics and immunotherapies in a tumor or tumor vasculature, in the subject.
- Agents e.g., therapeutic agents, include the herein disclosed categories and specific examples. It is not intended that the category be limited by the specific examples. Those of ordinary skill in the art will recognize also numerous other compounds that fall within the categories and that are useful according to the present disclosure.
- the BASP-compositions or BRUSH-compositions comprise one or more of an ARB, a vitamin D analog, IDO inhibitor, or a
- the BASP-compositions or BRUSH- compositions can further comprise a diagnostic agent, a targeting moiety, or a second therapeutic agent, e.g., an anti-cancer, an anti-inflammatory, or anti-fibrotic therapy, as described herein.
- Angiotensin Receptor Blockers ARBs
- the agent e.g., the therapeutic agent, in the BASP- composition or BRUSH-composition is an angiotensin receptor blocker (ARB), also referred to as angiotensin II receptor blocker, or AT1 blocker or AT1 inhibitor.
- ARB angiotensin receptor blocker
- the ARB comprises losartan (e.g., COZAAR®), candesartan (e.g., ATACAND®), telmisartan (e.g., MICARDIS®), valsartan (e.g., DIOVAN®), olmesartan (e.g., BENICAR®), azilsartan, eprosartan (e.g.,
- ARB is telmisartan or an analogue, prodrug, metabolite, or derivative thereof.
- ARB is losartan or an analogue, prodrug, metabolite, or derivative thereof.
- ARB is candesartan or an analogue, prodrug, metabolite, or derivative thereof.
- ARB is valsartan or an analogue, prodrug, metabolite, or derivative thereof. In some embodiments, ARB is olmesartan or an analogue, prodrug, metabolite, or derivative thereof. In some embodiments, ARB is azilsartan or an analogue, prodrug, metabolite, or derivative thereof. In some embodiments, ARB is eprosartan or an analogue, prodrug, metabolite, or derivative thereof. In some embodiments, ARB is irbesartan or an analogue, prodrug, metabolite, or derivative thereof.
- ARB is saralasin or an analogue, prodrug, metabolite, or derivative thereof. In some embodiments, ARB is EXP 3174 or an analogue, prodrug, metabolite, or derivative thereof. In some embodiments, ARB is L158209 or an analogue, prodrug, metabolite, or derivative thereof.
- the ARB is covalently bound to a macromonomer, conjugate, or particle described herein.
- the ARB comprises the structure of Formula (I-1):
- R 11a and R 11b are independently hydrogen or is taken together to form an oxo group
- R 12 is heteroaryl (e.g., tetrazolyl) or heteroarylalkyl (e.g.,
- R 13 is optionally substituted heteroaryl
- L 4 , L 5 , and L 6 is independently absent, a bond, alkyl, heteroalkyl, carbocyclyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, carbocyclyl, heterocyclyl, aryl, or heteroaryl is optionally substituted.
- the ARB comprises the structure of Formula (I-i), Formula (I-ii), Formula (I-iii), Formula (I-iv), or Formula (I-v):
- m m imn h ARB comprises a structure of Formula (I-i):
- the ARB comprises a structure of Formula (I-ii):
- the ARB com rises a structure of Formula (I-iii):
- the ARB comprises a structure of Formula (I-iv):
- the agent is an ARB, e.g., an ARB shown in FIG.2, and may be covalently coupled to the cleavable linker (e.g., L 3 in Formula (I)) via a hydroxyl group, a sulfonamide, a carboxylic acid, an ester, an amide, an amine, a triazole, or a benzimidazolone.
- the cleavable linker e.g., L 3 in Formula (I)
- the agent is losartan, candesartan, telmisartan, valsartan, olmesartan, azilsartan, eprosartan, irbesartan, saralasin, EXP 3174, L158209, or an analog, a prodrug, a metabolite, or a derivative thereof, and may be covalently coupled to the cleavable linker (e.g., L 3 in Formula (I)) via a hydroxyl group.
- the cleavable linker e.g., L 3 in Formula (I)
- the agent is losartan, candesartan, telmisartan, valsartan, olmesartan, azilsartan, eprosartan, irbesartan, saralasin, EXP 3174, L158209, or an analog, a prodrug, a metabolite, or a derivative thereof, and may be covalently coupled to the cleavable linker (e.g., L 3 in Formula (I)) via a carboxylic acid or ester.
- the cleavable linker e.g., L 3 in Formula (I)
- the agent is losartan, candesartan, telmisartan, valsartan, olmesartan, azilsartan, eprosartan, irbesartan, saralasin, EXP 3174,
- L158209 or an analog, a prodrug, a metabolite, or a derivative thereof, and may be covalently coupled to the cleavable linker (e.g., L 3 in Formula (I)) via an amide.
- Vitamin D Analogs e.g., L 3 in Formula (I)
- the agent e.g., the therapeutic agent, in the BASP- composition or BRUSH-composition is a vitamin D analog.
- Vitamin D analogs may comprise naturally occurring vitamin D analogs and/or non-natural (e.g., synthetically produced) analogs.
- Vitamin D analogs may further comprise a vitamin D receptor (VDR) agonist, a vitamin D receptor ligand, a vitamin D precursor, or derivatives thereof.
- VDR vitamin D receptor
- Exemplary vitamin D analogs include, but are not limited to, paricalcitol, calcipotriol, KH1060 (lexacalcitol), ercalcitriol, EB1089 (seocalcitol), BXL-628 (elocalcitol), MC1288, CB966, BCB 1093, GS 1558, TX527 (19-nor-14,20-bisepi-23- yne-l,25(OH) 2 D 3 ), ED-71 (eldecalcitrol), BXL-01-0029, doxercalciferol, maxacalcitol (OCT), tacalcitol, alfacalcidol, SM-10193, EB1072, EB1129, EB1133, EB1155, EB1270, MC1288, EB1213, CB1093, VD2656, VD2668, VD2708, VD2716, VD2728, VD2736, GS1500,
- the vitamin D analog does not comprise KH1060 (lexacalcitol), seocalcitol (EB 1089), or CB 1093. Additional vitamin D analogs are described by Scolletta et al. (2013) Mediators of Inflammation 2013, Article ID 876319; and Adorini (2005) Cellular Immunology 233: 115-124, which is incorporated herein by reference in its entirety.
- the agent is a vitamin D analog, or a metabolite or derivative thereof, e.g., as described in FIG.3.
- the vitamin D analog comprises paricalcitol, doxercalciferol, falecalcitriol, maxacalcitol, tacalcitol, alfacalcidol, eldecalcitol, seocalcitol, lexicalcitol, CB 1093, CD578, inecalcitol, calcipotriol, TX527, 2MD, WY1112, PRI-2205, ILX23-7553, or a metabolite or derivative thereof, e.g., as shown in FIG.3.
- the vitamin D analog comprises paricalcitol, ergocalciferol, elocalcitol, eldecalcitrol, calcidiol, calcipotriol, cholecalciferol, or a metabolite or derivative thereof.
- the vitamin D analog comprises paricalcitol, ergocalciferol, cholecalciferol, or a metabolite or derivative thereof.
- the vitamin D analog does not comprise seocalcitol (EB 1089), CB 1093, or lexacalcitol (KH 1060). Additional vitamin D analogs and derivatives are described, e.g. in Leyssens, C. et al, Front Physiol (2014), which is incorporated herein by reference in its entirety.
- the vitamin D analog comprises a structure of Formula (I-2):
- R 21 is hydrogen or hydroxyl
- R 22 is hydrogen, alkyl, alkenyl, or hydroxyl
- R 23 is hydrogen, alkyl, or alkenyl
- R 24 is alkyl, alkenyl, alkynyl, or heteroalkyl
- R 25 is hydrogen or absent; is a single bond or absent; and is a single or double bond; wherein each of which the alkyl, alkenyl, alkynyl, and heteroalkyl is optionally substituted.
- the vitamin D analog is attached to L 3 in the macromonomer at a hydroxyl moiety of the vitamin D analog.
- R 21 is hydroxyl, and the vitamin D analog is attached to L 3 at R 21 .
- R 22 is hydroxyl, and the vitamin D analog is attached to L 3 at R 22 .
- the vitamin D analog comprises a structure of Formula (I-vi Formula I-vii or Formula I-viii :
- the vitamin D analog may be attached to the macromonomer, conjugate, or particle through any available atom.
- the vitamin D analog comprises a structure of Formula (I-vi): (Formula I-vi).
- the vitamin D analog comprises a structure of Formula (I-vii):
- the vitamin D analog comprises a structure of Formula (I-viii):
- the vitamin D analog of Formula (I-vi) comprises a structure of Formula (I-ix) or Formula (I-x): .
- Formula (I-ix) Formula (I-x).
- the agent is a vitamin D analog or a metabolite or derivative thereof, e.g., a vitamin D analog or a metabolite or derivative thereof shown in FIG.3, and may be covalently coupled to the cleavable linker (e.g., L 3 in Formula (I)) via a hydroxyl group, a sulfonamide, a carboxylic acid, an ester, an amide, or an amine.
- the cleavable linker e.g., L 3 in Formula (I)
- the agent is paricalcitol, doxercalciferol, falecalcitriol, maxacalcitol, tacalcitol, alfacalcidol, eldecalcitol, seocalcitol, lexicalcitol, CB 1093, CD578, inecalcitol, calcipotriol, TX527, 2MD, WY1112, PRI- 2205, ILX23-7553, or a metabolite or derivative thereof and may be covalently coupled to the cleavable linker (e.g., L 3 in Formula (I)) via a hydroxyl group.
- the cleavable linker e.g., L 3 in Formula (I)
- the agent is paricalcitol, doxercalciferol, falecalcitriol, maxacalcitol, tacalcitol, alfacalcidol, eldecalcitol, seocalcitol, lexicalcitol, CB 1093, CD578, inecalcitol, calcipotriol, TX527, 2MD, WY1112, PRI-2205, ILX23-7553, or a metabolite or derivative thereof and may be covalently coupled to the cleavable linker (e.g., L 3 in Formula (I)) via a carboxylic acid or ester.
- the cleavable linker e.g., L 3 in Formula (I)
- the agent is paricalcitol, doxercalciferol, falecalcitriol, maxacalcitol, tacalcitol, alfacalcidol, eldecalcitol, seocalcitol, lexicalcitol, CB 1093, CD578, inecalcitol, calcipotriol, TX527, 2MD, WY1112, PRI-2205, ILX23-7553, or a metabolite or derivative thereof and may be covalently coupled to the cleavable linker (e.g., L 3 in Formula (I)) macromonomer, conjugate or BASP particle via an amide.
- cleavable linker e.g., L 3 in Formula (I)
- the agent, e.g., the therapeutic agent, in the BASP- composition or BRUSH-composition is an IDO inhibitor (i.e., indoleamine 2,3- dioxygenase (IDO) pathway inhibitor).
- IDO inhibitors include, but are not limited to, GDC-0919, indoximod, 1-methyltryptophan (e.g., 1-methyl- L -tryptophan, 1-methyl-D-tryptophan), NLG8189, INCB024360, NLG919, methylthiohydantoin tryptophan, brassinin, annulin B, exiguamine A, INCB023843, or an analog or derivative thereof.
- IDO inhibitors are described e.g., in Lob, S. et al. Nat Rev Cancer (2009) 9:445-452; Rohrig, U.F. et al. J Med Chem (2015) 58:9421-9437; and U.S. Patent Application No.14/919,184, each of which is incorporated by reference herein in its entirety.
- the IDO inhibitor may be covalently coupled to the cleavable linker (e.g., L 3 in Formula (I)) via a hydroxyl group. In some embodiments, IDO inhibitor may be covalently coupled to the cleavable linker (e.g., L 3 in Formula (I)) via a carboxylic acid or ester. In some embodiments, the IDO inhibitor may be covalently coupled to the cleavable linker (e.g., L 3 in Formula (I)) via an amide. Bromodomain Inhibitors
- the agent e.g., the therapeutic agent, in the BASP- composition or BRUSH-composition is a bromodomain inhibitor (i.e., extra-terminal protein inhibitor (i-BET)).
- exemplary bromodomain inhibitors include, but are not limited to, MS436, PFI-1, I-BET 151, OTX-015, JQ1, CPI-203, bromosporine, RVX- 208, I-BET 762, I-BET 151, OFXBD02, OFXBD03, XD14, MS436, and analogs and derivatives thereof, e.g., as shown in FIGS.6A and 6B.
- bromodomain inhibitors are described e.g., in Haas, M. J. et al SciBX (2014) 7(15); ACS Chem Biol (2015) 10:22-39; Expert Opin Ther Pat (2014) 24:185-199; Clin Cancer Res (2015) 21:1628-1638; Oncotarget (2015) 6:17698-17712; Bioorg Med Chem Lett (2015) 25:1842-1848; Cancer Res (2013) 73:3336-3346; Am J Cardiovasc Drugs (2015) Sep 18; and J Med Chem (2013) 56:9251-9264, each of which is incorporated by reference herein in its entirety.
- the agent is a bromodomain inhibitor (i.e., a bromodomain or an extra-terminal protein inhibitor (i-BET)).
- the bromodomain inhibitor comprises MS436, PFI-1, I-BET 151, OTX-015, JQ1, CPI-203, bromosporine, RVX-208, I-BET 762, I-BET 151, OFXBD02, OFXBD03, XD14, MS436, or an analog or derivative thereof, e.g., as shown in FIGS.3A to 3B. Additional bromodomain inhibitors are described e.g., in Haas, M. J.
- the agent is a bromodomain inhibitor, e.g., a bromodomain inhibitor shown in FIGS.4A to 4B, and may be covalently coupled to the cleavable linker (e.g., L 3 in Formula (I)) via a hydroxyl group, a sulfonamide, a carboxylic acid, an ester, an amide, or an amine.
- the cleavable linker e.g., L 3 in Formula (I)
- the agent is OTX-2015 (5), RVX-208 (7), OXFBD02 (9), OXFBD03 (10), XD14 (18), or dinaciclib (19), e.g., as shown in FIGS.4A to 4B, and may be covalently coupled to the cleavable linker (e.g., L 3 in Formula (I)) via a hydroxyl group.
- the cleavable linker e.g., L 3 in Formula (I)
- the agent is (12), PFI-1 (14), (15), MS436 (16), TG101348 (22), TG101209 (23), or bromosporine, e.g., as shown in FIGS.4A to 4B, and may be covalently coupled to the cleavable linker (e.g., L 3 in Formula (I)) via a sulfonamide.
- the cleavable linker e.g., L 3 in Formula (I)
- the agent is I-BET726 (12), CPI-203 (6), or B12536 (21), e.g., as shown in FIGS.4A to 4B, and may be covalently coupled to the cleavable linker (e.g., L 3 in Formula (I)) via a carboxylic acid, ester, or amide.
- the agent is I-BET151 (11) or B12536 (21), e.g., as shown in FIGS.4A to 4B, and may be covalently coupled to the cleavable linker (e.g., L 3 in Formula (I)) via a benzimidazolone or pyrimidine amine.
- the present invention features, at least in part, BRUSH conjugates and BASP particles coupled to one or more agents.
- the BRUSH conjugates and BASP particles are prepared through the linkage of discrete macromonomer subunits that include the agent, e.g., a therapeutic agent, and optionally a targeting moiety, a diagnostic agent, and/or a second therapeutic agent of interest.
- the brush arm method involves co-polymerization of a macromonomer with a multifunctional crosslinker to generate a unimolecular micelle-like nanostructure (BASP) with a core including the crosslinker and a corona including the
- BASP unimolecular micelle-like nanostructure
- macromonomer refers to a macromolecule with one end-group that enables it to polymerize to form a polymeric structure, e.g., a BRUSH conjugate or BASP particle, e.g., as shown in No. 1 in FIG.1. Macromonomers can contribute a single monomeric unit to a chain of the completed BRUSH conjugate or BASP polymer.
- a macromonomer may range in size from about 3 kDa to about 6 kDa, e.g., as detected by mass spectrometry.
- Macromonomer subunits comprising the agent and optionally the targeting moiety, the diagnostic agent, and/or the second agent of interest may be joined in a number of ways, such as through graft-through ring-opening metathesis polymerization (ROMP) in the presence of a cross-linker (e.g., a bis-norbornene cross-linker described herein).
- a cross-linker e.g., a bis-norbornene cross-linker described herein.
- the invention features a compound (e.g., a macromonomer) of Formula (I):
- Ring C is carbocyclyl or a heterocyclyl moiety
- Ring T is a triazoldiyl moiety (e.g., a 1,2,3-triazoldiyl);
- A is C 1 -C 12 alkylene, C 2 -C 12 alkenylene, C 2 -C 12 alkynylene, or C 1 -C 12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R 1 ;
- B is C 1 -C 12 alkylene, C 2 -C 12 alkenylene, C 2 -C 12 alkynylene, or C 1 -C 12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R 1 ;
- X is an agent as described herein (e.g., an ARB, vitamin D analog,
- IDO indoleamine 2,3-dioxygenease
- P is alkylene or heteroalkylene (e.g., polyethylene glycol);
- each of L 1 and L 2 is independently a bond, C 1 -C 12 alkylene, C 2 -C 12 alkenylene, C 2 -C 12 alkynylene, C 1 -C 12 heteroalkylene, (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 alkylene), (C 0 -C 12 heteroalkylene)-arylene-(C 0 -C 12 alkylene), (C 0 -C 12 alkylene)- arylene-(C 0 -C 12 heteroalkylene), (C 0 -C 12 heteroalkylene)-arylene-(C 0 -C 12 -C 12
- each R A is independently hydrogen, C 1 -C 6 alkyl, C 1 -C 6 heteroalkyl, etc
- each R B and R C is independently hydrogen or C 1 -C 6 alkyl
- n 1 or 2.
- L 1 is a linker connecting B and Ring T, and A is a substituent on L 1 .
- Ring C is a fused carbocyclyl. In some embodiments, Ring C is a fused heterocyclyl.
- Ring C is a structure of Formula (II):
- D is N or CR 4 ;
- Z is C(R 5 ) 2 , O, or S;
- each R 3 is independently C 1 -C 6 alkyl, C 1 - C 6 haloalkyl, oxo, or halo;
- R 4 is hydrogen, C 1 -C 6 alkyl, or halo;
- each R 5 is
- n is 0, 1, or 2
- “1” represents a portion of Ring C bound to B in the macromonomer.
- Ring C is a structure of Formula (II-a):
- Ring C is a structure of Formula (II-b):
- D is N or CR 4 ; each R 3 is independently C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, oxo, or halo; R 4 is hydrogen, C 1 -C 6 alkyl, or halo; n is 0, 1, or 2, and“1” represents a portion of Ring C bound to B in the macromonomer.
- the structure of Formula (II) comprises
- the structure of Formula (II) comprises .
- the structure of Formula (II) comprises .
- the structure of Formula (II) comprises .
- Ring C is a structure of Formula (II-c):
- each R 3’ is independently selected from C 1 -C 6 alkyl, - C(O)-C 1 -C 6 alkyl, -C(O)-O-C 1 -C 6 alkyl, -C(O)-NH-C 1 -C 6 alkyl, C 1 -C 6 heteroalkyl, - C(O)-C 1 -C 6 heteroalkyl, -C(O)-O-C 1 -C 6 heteroalkyl, -C(O)-NH-C 1 -C 6 heteroalkyl, and halo, wherein the any alkyl portion of R 3’ is optionally substituted with halo; each R 5 is independently hydrogen, C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, or halo; s is 0, 1, 2, 3, 4, 5, 6, 7, 8, or 9; and“1” represents a portion of Ring C bound to B in the
- Ring T is 1,2,3 triazoldiyl or 1,2,4-triazoldiyl. In some embodiments, Ring T is 1,2,3 triazoldiyl or 1,2,4-triazoldiyl. In some
- Ring T is , wherein“2” represents a portion of Ring T bound to L 2 in the macromonomer.
- A is C 1 -C 12 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 11 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 10 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 9 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 8 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 7 heteroalkylene optionally substituted with 1-6 independently selected R 1 .
- A is C 1 -C 6 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 5 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 4 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 3 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 2 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 heteroalkylene optionally substituted with 1-6 independently selected R 1 .
- A is C 1 -C 8 heteroalkylene optionally substituted with 1-3 independently selected R 1 .
- R 1 is oxo or heteroalkyl (e.g., polyethylene glycol).
- R 1 is oxo.
- R 1 is heteroalkyl (e.g., polyethylene glycol).
- A is
- R 1 comprises a polyethylene glycol (PEG), a polyethylene oxide (PEO), a polypropylene glycol (PPG), a polyglycerol (PG), a poloxamine (POX), a polybutylene oxide (PBO), polylactic acid (PLA), polyglycolic acid (PGA), poly(lactic-co-glycolic acid) (PLGA), polycaprolactone (PCL), polydioxanone (PDO), a polyanhydride, a polyacrylide, a polyvinyl, or a polyorthoester.
- R 1 comprises a polyethylene glycol (PEG).
- A is C(O)CH 2 CH 2 C(O)NH-PEG. In some embodiments, A is - C(O)CH 2 CH 2 C(O)NH-PEG, optionally wherein the molecular weight of the PEG is between about 200 and about 6000, inclusive, g/mol (Dalton or Da) (e.g., wherein the PEG is PEG200, PEG400, PEG600, PEG800, PEG1000, PEG1500, PEG2000, PEG3000, PEG4000, PEG6000).
- B is C 1 -C 12 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 -C 11 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 -C 10 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 -C 9 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 -C 8 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 -C 7 alkylene optionally substituted with 1-6 independently selected R 1 .
- B is C 1 -C 6 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 -C 5 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 -C 4 alkylene optionally substituted with 1-6 R 1 . In some embodiments, B is C 1 -C 3 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 -C 2 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 alkylene optionally substituted with 1-6 independently selected R 1 .
- B is dodecylene, undecylene, decylene, nonylene, octylene, heptylene, hexylene, pentylene, butylene, propylene, ethylene, or methylene, each of which is optionally substituted with 1-6 independently selected R 1 .
- B is dodecylene, undecylene, decylene, nonylene, octylene, heptylene, hexylene, pentylene, butylene, propylene, ethylene, or methylene.
- B is octylene, heptylene, hexylene, pentylene, butylene, propylene, or ethylene. In some embodiments, B is hexylene, pentylene, butylene, propylene, or ethylene. In some embodiments, B is hexylene. In some embodiments, B is pentylene. In some embodiments, B is butylene. In some embodiments, B is propylene. In some embodiments, B is ethylene. In some embodiments, B is methylene.
- the PEG has a molecular weight of between about 200 and about 6000, g/mol (e.g., PEG200, PEG400, PEG600, PEG800, PEG1000, PEG1500, PEG2000, PEG3000, PEG4000, or PEG6000).
- the PEG is PEG200.
- the PEG is PEG400.
- the PEG is PEG600.
- the PEG is PEG800. In some
- the PEG is PEG1000. In some embodiments, the PEG is PEG2000. In some embodiments, the PEG is PEG3000. In some embodiments, the PEG is
- the PEG is PEG6000.
- each of L 1 and L 2 is independently C 1 -C 12 alkylene, C 1 -C 12 heteroalkylene, (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 alkylene), (C 0 -C 12 heteroalkylene)- arylene-(C 0 -C 12 alkylene), or (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), or (C 0 -C 12 heteroalkylene)-aryl-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 .
- each of L 1 and L 2 is independently C 1 -C 12 alkylene, C 1 -C 12 heteroalkylene, or (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-6 independently selected R 2 .
- L 1 is C 1 -C 12 heteroalkylene and L 2 is C 1 -C 12 alkylene, wherein each alkylene and heteroalkylene is optionally substituted with 1-6 independently selected R 2 .
- each of L 1 and L 2 is independently selected from–CH 2 –,–NCH 2 –, and–CH 2 CH 2 .
- L 1 is–NCH 2 –.
- L 2 is–CH 2 CH 2 – .
- each of L 1 and L 2 is independently–(CH 2 ) 3 –,–(CH 2 ) 4 –,–(CH 2 ) 5 –, or–(CH 2 ) 6 –.
- each of L 1 and L 2 is independently–CH 2 NH–,–(CH 2 ) 2 NH–,–(CH 2 ) 3 NH–,–
- each of L 1 and L 2 is independently–NH(CH 2 ) 2 –,–NH(CH 2 ) 3 –,–NH(CH 2 ) 4 –,–NH(CH 2 ) 5 –, or– NH(CH 2 ) 6 –.
- L 3 is cleavable by an enzyme (e.g., an esterase, a protease), or by hydrolysis at a certain pH (e.g., acidic pH, basic pH).
- an enzyme e.g., an esterase, a protease
- L 3 is C 1 -C 12 heteroalkylene, (C 0 -C 12 heteroalkylene)-arylene-(C 0 -C 12 alkylene), (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), or (C 0 -C 12
- heteroalkylene -aryl-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 .
- L 3 is C 1 -C 12 heteroalkylene, or (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-6 independently selected R 2 .
- R 2 is oxo or halo (e.g., fluoro).
- L 3 is–C(O)–,–OC(O)–,–C(O)O–,– C(O)CH 2 –,–OC(O)CH 2 –,–C(O)OCH 2 –,–CH 2 CH 2 O–, C(O)CH 2 CH 2 O–,
- P is heteroalkylene.
- P comprises polyethylene glycol (PEG), polyethylene oxide (PEO), polypropylene glycol (PPG), polyglycerol (PG), poloxamine (POX), polybutylene oxide (PBO), polylactic acid (PLA), polyglycolic acid (PGA), poly(lactic-co-glycolic acid) (PLGA), polycaprolactone (PCL), polydioxanone (PDO), a polyanhydride, a polyacrylide, a polyvinyl, or a polyorthoester.
- P comprises about 2 to about 200 repeating units (e.g., about 2 to about 150, about 2 to about 100, about 2 to about 50, about 2 to about 25, about 2 to about 15, about 2 to about 10, or about 2 to about 5 repeating units) of polyethylene glycol (PEG), polyethylene oxide (PEO), polypropylene glycol (PPG), polyglycerol (PG), poloxamine (POX), polybutylene oxide (PBO), polylactic acid (PLA), polyglycolic acid (PGA), poly(lactic-co-glycolic acid) (PLGA), polycaprolactone (PCL), polydioxanone (PDO), a polyanhydride, a polyacrylide, a polyvinyl, or a polyorthoester.
- PEG polyethylene glycol
- PEO polyethylene oxide
- PPG polypropylene glycol
- PG polyglycerol
- POX poloxamine
- PBO polybutylene oxide
- PBO polylactic acid
- PLA polygly
- P comprises polyethylene glycol (PEG).
- P comprises between about 2 to about 200 repeating units (e.g., about 2 to about 150, about 2 to about 100, about 2 to about 50, about 2 to about 25, about 2 to about 15, about 2 to about 10, or about 2 to about 5 repeating units) of polyethylene glycol (PEG).
- P comprises between about 2 to about 10 repeating units of polyethylene glycol (PEG).
- the PEG comprises diethylene glycol, triethylene glycol, tetraethylene glycol, or pentaethylene glycol.
- the agent e.g., an agent described herein, e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor
- a cleavable linker e.g., L 3 in Formula (I)
- the agent e.g., an agent described herein, e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor
- a cleavable linker e.g., L 3 in Formula (I)
- a hydroxyl group e.g., a sulfonamide, a carboxylic acid, an ester, an amide, an amine, a triazole, or a benzimidazolone.
- Exemplary macromonomers may be described by a number of properties, including molecular weight (kDa) and hydrodynamic diameter (nm).
- the molecular weight of the macromonomer is between about 1 kDa and about 10 kDa, e.g., between about 2 kDa and about 8 kDa or about 3 kDa and about 6 kDa, e.g., as detected by mass spectrometry.
- the molecular weight of the macromonomer is between about 3 kDa and about 6 kDa.
- the molecular weight of the macromonomer is about 2 kDa, about 3 kDa, about 4 kDa, about 5 kDa, or about 6 kDa.
- the hydrodynamic diameter of the macromonomer is between about 0.5 nm and about 3 nm, e.g., about 1 nm and about 2 nm, e.g., as detected by dynamic light scattering.
- the structure of Formula (I) is a structure of Formula (I- a):
- A is C 1 -C 12 alkylene, C 2 -C 12 alkenylene, C 2 -C 12 alkynylene, or C 1 -C 12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R 1 ;
- B is C 1 -C 12 alkylene, C 2 - C 12 alkenylene, C 2 -C 12 alkynylene, or C 1 -C 12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R 1 ;
- X is an agent (e.g., an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor); each of L 1 and L 2 is independently C 1 -C 12 alkylene, C 1 -C 12 heteroalkylene, (C 0 -C 12 alkylene)-arylene-
- the structure of Formula (I) is a structure of Formula (I- b):
- B is C 1 -C 12 alkylene or C 1 -C 12 heteroalkylene, wherein each alkylene or heteroalkylene is optionally substituted with 1-6 R 1 ;
- X is an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or a bromodomain inhibitor);
- each of L 1 and L 2 is independently C 1 -C 12 alkylene, C 1 -C 12 heteroalkylene, (C 0 -C 12 alkylene)-arylene-(C 0 - C 12 alkylene), (C 0 -C 12 heteroalkylene)-arylene-(C 0 -C 12 alkylene), (C 0 -C 12 alkylene)- arylene-(C 0 -C 12 heteroalkylene), or (C 0 -C 12 heteroalkylene)-aryl-(C 0 -C 12
- heteroalkylene wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 ;
- L 3 is C 1 -C 12 heteroalkylene, (C 0 -C 12 heteroalkylene)-arylene-(C 0 -C 12 alkylene), (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), or (C 0 -C 12 heteroalkylene)-aryl-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 ; each R 1 and R 2 is independently alkyl, heteroalkyl, halo, cyano, oxo,–OR A ,–NR B R C ,–NR B C(O)R D , -C(O)NR B R C ,–C(O)R D ,–C(O)OH,– C(O
- the structure of Formula (I) is a structure of Formula (I- c):
- L 3 is C 1 -C 12 heteroalkylene, (C 0 -C 12 heteroalkylene)- arylene-(C 0 -C 12 alkylene), (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), or (C 0 - C 12 heteroalkylene)-aryl-(C 0 -C 12 heteroalkylene), wherein each alkylene,
- heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 ; each R 2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive.
- the structure of Formula (I) is a structure of Formula (I- d-1) or Formula (I-d-2):
- L 3 is C 1 -C 12 heteroalkylene, (C 0 -C 12 heteroalkylene)-arylene-(C 0 -C 12 alkylene), (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), or (C 0 -C 12
- heteroalkylene -aryl-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 ; each R 2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive.
- the structure of Formula (I) is a structure of Formula (I- e):
- L 3 is C 1 -C 12 heteroalkylene, (C 0 -C 12 heteroalkylene)-arylene-(C 0 -C 12 alkylene), (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), or (C 0 -C 12
- heteroalkylene -aryl-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 ; each R 2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive.
- the structure of any of the formulae described herein e.g., Formulae (I), (I-a). (I-b), (I-c), (I-d-1), (I-d-2), and (I-e)
- comprises a salt e.g., a pharmaceutically acceptable salt.
- BRUSH conjugates and BASP particles comprising one or more agents (e.g., a therapeutic agent, diagnostic agent, or targeting agent). These BRUSH conjugates and BASP particles are prepared through the co-polymerization of discrete macromonomer subunits that include the agent with a multifunctional crosslinker via, e.g., ring-opening metathesis polymerization (ROMP).
- REP ring-opening metathesis polymerization
- the term“conjugate” or“BRUSH conjugate” or“BRUSH” as used herein refers to a molecular structure comprising at least two macromonomers joined together (e.g., directly or indirectly), e.g., as shown in No.2 in FIG.1.
- the macromonomers may be identical, or in some cases, there may be more than one type of macromonomer present within the conjugate.
- the BRUSH conjugate refers to the product of a first round of polymerization (e.g., a bottle-brush polymer) and has an average molecular weight of about 10 kDa to about 100 kDa.
- the macromonomers present within the conjugate are covalently linked.
- the conjugate is prepared from ring opening metathesis polymerization.
- BASP particle refers to a structure comprising at least two conjugates joined together (e.g., directly or indirectly, e.g., by covalent or noncovalent means), e.g., as shown in shown in No.3 in number FIG.1.
- the conjugates may be identical, or in some cases, there may be more than one type of conjugate present within the particle.
- the BASP particle refers to the product of a subsequent round of polymerization (e.g., a second, third, or fourth round of polymerization) and has an average molecular weight of about 100 kDa to about 1,000 kDa.
- the particle is prepared from ring opening metathesis polymerization (ROMP).
- the conjugates present in the particle are noncovalently linked (e.g., are linked through ionic or hydrophobic interactions).
- the conjugate or BASP particle comprises (i) an agent chosen from an angiotension receptor blocker (ARB), a vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor; (ii) a carbocyclyl or heterocyclyl moiety; (iii) a triazole moiety; (iv) a heteroalkyl moiety; and (v) a cleavable linker (e.g., a tissue microenvironment cleavable linker).
- ARB an angiotension receptor blocker
- a vitamin D analog an IDO inhibitor, or a bromodomain inhibitor
- a carbocyclyl or heterocyclyl moiety e.g., a triazole moiety
- a heteroalkyl moiety e.g., a tissue microenvironment cleavable linker
- the agent is an angiotensin receptor blocker (ARB), vitamin D analog, IDO inhibitor, or bromodomain inhibitor, e.g., as described herein.
- ARB angiotensin receptor blocker
- vitamin D analog vitamin D analog
- IDO inhibitor IDO inhibitor
- bromodomain inhibitor e.g., as described herein.
- the carbocyclyl or heterocyclyl moiety comprises a monocyclic or bicyclic carbocyclyl or a monocyclic or bicyclic heterocyclyl. In some embodiments, the carbocyclyl or heterocyclyl moiety comprises a monocyclic carbocyclyl or monocyclic heterocyclyl. In some embodiments, the carbocyclyl or heterocyclyl moiety comprises a bicyclic carbocyclyl or bicyclic heterocyclyl.
- the triazole moiety comprises 1,2,3-triazoldiyl or 1,2,4- triazoldiyl. In some embodiments, the triazole moiety comprises 1,2,3-triazoldiyl. In some embodiments, the triazole moiety comprises 1,2,4-triazoldiyl.
- the heteroalkyl moiety comprises a polyethylene glycol (PEG), a polyethylene oxide (PEO), a polypropylene glycol (PPG), a polyglycerol (PG), a poloxamine (POX), a polybutylene oxide (PBO), polylactic acid (PLA), polyglycolic acid (PGA), poly(lactic-co-glycolic acid) (PLGA), polycaprolactone (PCL), polydioxanone (PDO), a polyanhydride, a polyacrylide, a polyvinyl, or a polyorthoester.
- the heteroalkyl moiety comprises a
- the PEG has a molecular weight of between about 200 and about 6000, g/mol (e.g., PEG200, PEG400, PEG600, PEG800, PEG1000, PEG1500, PEG2000, PEG3000, PEG4000, or
- the PEG is PEG200. In some embodiments, the PEG is PEG400. In some embodiments, the PEG is PEG600. In some embodiments, the PEG is PEG800. In some embodiments, the PEG is PEG1000. In some embodiments,
- the PEG is PEG2000. In some embodiments, the PEG is PEG3000. In some embodiments, the PEG is PEG4000. In some embodiments, the PEG is
- the agent is non-covalently bound to the conjugate (e.g., the agent is associated with the conjugate or BASP particle through ionic bonds or hydrophobic interactions). In some embodiments, the agent is covalently bound to the conjugate or BASP particle through a cleavable linker (e.g., L 3 in Formula (I)). Cleavable Linkers
- the agent e.g., the therapeutic agent, is covalently bound to the BRUSH- composition or BASP-composition through a tissue microenvironment cleavable linker (can also be referred to herein as“sensitive linker”).
- a tissue microenvironment cleavable linker can also be referred to herein as“sensitive linker”.
- the cleavable linker is the L 3 portion of the macromonomer(s) that make up the BRUSH conjugate or BASP particle.
- a cleavable linker is“cleaved” or“degraded” when one or more bonds of the cleavable linker are broken, e.g., resulting in release of an agent (e.g., an angiotension receptor blocker (ARB), a vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor), e.g., from the conjugate or BASP particle.
- an agent e.g., an angiotension receptor blocker (ARB), a vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor
- Linker cleavage or agent release need not be 100%, e.g., a cleavage or release of at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% or higher, e.g., over a period of seconds, minutes, hours (e.g., 6 hours, 12 hours, or 24 hours), days (e.g., 2 days or 7 days), weeks, or months is encompassed by this term.
- the cleavable linker is cleavable by or is sensitive to an enzyme (e.g., an esterase or a protease), pH (e.g., acidic pH, basic pH), light (e.g., ultraviolet light), a nucleophile, reduction, or oxidation.
- the cleavable linker is cleavable by or is sensitive to an enzyme (e.g., an esterase or a protease) or pH (e.g., acidic pH, basic pH).
- the cleavable linker is not cleavable by light (e.g., ultraviolet light).
- the cleavable linker comprises an ester, an acetal, a ketal, a phosphoramidite, a hydrazone, an imine, an oxime, a disulfide, or a silyl moiety, a combination of acetal or ketal with ester group, an oligo-acetal or oligo- ketal group, a combination of the oligo-ketal and silyl ether group, or a combination of the oligo-ketal and vinyl ether group.
- the cleavable linker comprises an ester.
- the cleavable linker comprises an acetal.
- the cleavable linker comprises a phosphoramidite. In some embodiments, the cleavable linker comprises a hydrazine. In some embodiments, the cleavable linker comprises an imine. In some embodiments, the cleavable linker comprises an oxime. In some embodiments, the cleavable linker comprises a silyl moiety. In some embodiments, the cleavable linker comprises a disulfide.
- the cleavable linker is chosen from a combination of acetal or ketal with cis-aconityl, hydrazine, oxime, imidazole or trityl groups. Any of the aforesaid groups or combination of groups can modified to enhance the pH sensitivity of the cleavable linker, e.g., as described herein.
- the cleavable linker is represented by L 3 in a structure of Formula (I) or Formula (III).
- L 3 is C 1 -C 12 heteroalkylene, (C 0 -C 12 heteroalkylene)-arylene-(C 0 -C 12 alkylene), (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), or (C 0 -C 12 heteroalkylene)-aryl-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 .
- L 3 is C 1 -C 12 heteroalkylene, or (C 0 - C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-6 independently selected R 2 .
- R 2 is oxo or halo (e.g., fluoro).
- L 3 is–C(O)–,– OC(O)–,–C(O)O–,–C(O)CH 2 –,–OC(O)CH 2 –,–C(O)OCH 2 –,–CH 2 CH 2 O–,
- the cleavable linker may include an atom or a part of a moiety that is derived in part from the agent (e.g., a therapeutic agent).
- the agent e.g., a therapeutic agent
- the cleavable linker may comprise the carbonyl group derived from the agent.
- the cleavable linker comprises L 3 and an atom or chemical moiety from the agent.
- the cleavable linker comprises L 3 and does not comprise an atom or chemical moiety from the agent.
- the cleavable linker is cleaved or degraded, e.g., preferentially cleaved or degraded, upon exposure to a first set of conditions relative to a second set of conditions.
- the cleavable linker can be“preferentially cleaved” or“preferentially degraded” in a first set of conditions relative to a second set of conditions if at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% or more of a bond or bonds of the cleavable linker are broken, or the agent is released, in the first set of conditions relative to the second set of conditions.
- the cleavable linker is degraded or hydrolyzed at physiological conditions.
- the linker is pH sensitive or cleaved at a certain pH.
- the linker is degraded or hydrolyzed through the action of an enzyme (e.g., a protease or esterase).
- an enzyme e.g., a protease or esterase.
- the cleavable linker is preferentially cleaved in a tissue
- tissue microenvironment cleavable linker e.g., a tumor microenvironment, which is referred to herein as a “tissue microenvironment cleavable linker.”
- the tissue (e.g., tumor) microenvironment cleavable linker is preferentially cleaved or degraded upon exposure to a first desired tissue or tumor microenvironment relative to a second tissue or non-tumor tissue.
- a tissue (e.g., tumor) microenvironment cleavable linker can be preferentially cleaved if at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% or more of a bond or bonds of the linker are broken, or the agent is released, in a desired tissue or tumor microenvironment relative to another tissue or non-tumor tissue.
- the tissue (e.g., tumor) microenvironment cleavable linker is preferentially cleaved or degraded if one or more of the bonds of the linker are broken, or the agent is released, at least 2, 5, 10, 20, 30, 40, 50, 60, 70, 80, or 100 times faster upon exposure to a first desired tissue or tumor
- the tissue (e.g., tumor) microenvironment can have a particular set of conditions, e.g., pH, enzymes, that cause the cleavage or degradation of the linker.
- the cleavable linker is a peptide.
- the linker is a peptide, and the peptide sequence is comprised of naturally occurring amino acids.
- the linker is a peptide, and the peptide sequence comprises at least one synthetically derived amino acids, e.g., at least 2, at least 3, at least 4, at least 5, at least 8, at least 10, at least 15, at least 20, or more synthetically derived amino acids.
- the peptide has a linear structure. In some embodiments, the peptide has a branched structure.
- the peptide has a branched structure with, e.g., at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, or at least 8 branching points. In some embodiments, the peptide has a cyclic structure.
- the cleavable linker is a peptide, and the peptide sequence comprises at least 2 amino acid residues. In some embodiments, the peptide sequence comprises at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or at least 10 amino acid residues. In some embodiments, the peptide sequence is from about 1 to about 10 amino acid residues. In some embodiments, the peptide sequence is from about 1 to about 15, about 20, about 25, about 30, about 40, about 50, about 60, about 70, about 80, about 90, or about 100 amino acid residues. In some embodiments, the peptide sequence is from about 10 to about 100 amino acid residues. In some embodiments, the peptide sequence is from about 25 to about 100 amino acid residues. In some embodiments, the peptide sequence is from about 50 to about 100 amino acid residues.
- the cleavable linker comprises a substrate peptide that is cleaved, e.g., activated, by a matrix metalloprotease (MMP) selected from a sequence disclosed in U.S. Patent Application No.2015/0087810.
- MMP matrix metalloprotease
- the substrate peptide comprises a protease substrate comprising an amino acid sequence selected from the group consisting of SEQ ID NOs: 353-363, 372-375, 376-378, 395-401, 411-419, 426-433, 437-449, 454-456, 459-469, 475-482, 487-495, 318-323, 325-327, 330-335, 341-347, 14-33, and 159, e.g., as described in U.S. Patent Application No.2015/0087810.
- the linker comprises a substrate peptide derived from a sequence disclosed in U.S. Patent No.
- a substrate peptide chosen from an enzyme selected from the group consisting of MMP-1, MMP-2, MMP-3, MMP-8, MMP-9, MMP-14, plasmin, PSA, PSMA, CATHEPSIN D, CATHEPSIN K, CATHEPSIN S, ADAM10, ADAM12, ADAMTS, Ca
- the linker comprises a sequence disclosed in U.S. Patent No.8,513,390. In some embodiments, the linker comprises a sequence disclosed in International Patent Publication No. WO2003/079972. In some embodiments, the linker comprises a sequence disclosed in U.S. Patent No.7,495,099. In some embodiments, the linker comprises a sequence disclosed in U.S. Patent No. 8,580,244. In some embodiments, the linker comprises a sequence disclosed in one of the following articles: van Kempen, et al. Eur Cancer (2006) 42:728-734; Desnoyers, L.R. et al. Sci Transl Med (2013) 5:207ra144; Rice, J.J. et al.
- the cleavable linker comprises a substrate peptide that is cleaved, e.g., activated, by a protease, e.g., a protease present in a tumor or fibrotic microenvironment (e.g., a matrix metalloprotease (MMP), e.g., as described by Desnoyers, L.R. et al. Sci Transl Med (2013) 5:207ra144; Eckhard, U et al Matrix Biol (2015) doi: 10.1016/j.matbio.2015.09.003 (epub ahead of print); and van
- MMP matrix metalloprotease
- the linker includes the amino acid sequence of a substrate for uPA, e.g., comprises the amino acid sequence LSGRSDNH (SEQ ID NO:1), e.g., as described in U.S. Patent No. 8,513,390.
- the linker sequence further includes a Gly-Ser- containing peptide linker, at either end, or both ends to the substrate peptide.
- proteases that may be upregulated in a tumor microenvironment include, but are not limited to, urokinase-type plasminogen activator (uPA), which is upregulated in human carcinomas (S. Ulisse, et al. Curr. Cancer Drug Targets 9, 32– 71 (2009)), membrane-type serine protease 1 (MT-SP1/matriptase) (K. Uhland Cell. Mol. Life Sci.63, 2968–2978 (2006); A. M. LeBeau, et al. Proc. Natl. Acad. Sci.
- uPA urokinase-type plasminogen activator
- MT-SP1/matriptase membrane-type serine protease 1
- K. Uhland Cell. Mol. Life Sci.63, 2968–2978 (2006) A. M. LeBeau, et al. Proc. Natl. Acad. Sci.
- the protease is produced by an inflammatory cell, e.g., a tumor infiltrating leukocyte (e.g., a leukocyte-derived MMP), e.g., as described by van Kempen, et al. Eur Cancer (2006) 42:728-734.
- the MMP is chosen from MMP1, MMP2, MMP3, MMP7, MMP8, MMP9, MMP12, MMP13 or MMP14, e.g., as described by Eckhard, U et al. supra.
- the substrate peptide is derived from a CLiPS library (as described in, e.g., K. T. Boulware, P. S. Daugherty, Proc. Natl. Acad. Sci. U.S.A. 103, 7583–7588 (2006)).
- the substrate peptide specificity is evaluated using combinatorial fluorogenic substrate libraries, e.g., as described by Harris, J.L. Proc Natl Acad Sci USA (2000) 97:7754-7759.
- the substrate peptide is derived from a phage display library (e.g., it is a phase display substrate), e.g., as described by Deperthes, D.
- a phage display substrate is exposed to a plurality of proteases; peptides released through specific cleavage can be amplified in an expression system.
- the substrate peptide is derived from a bacterial display library, e.g., as described by Rice, J.J. et al. Protein Sci (2006) 15:825-836.
- the tissue (e.g., tumor) microenvironment cleavable linker is cleavable by an enzyme.
- the enzyme comprises an esterase or a protease.
- Exemplary proteases include MMP-1, MMP-2, MMP-3, MMP-8, MMP-9, MMP-14, plasmin, PSA, PSMA, CATHEPSIN D, CATHEPSIN K,
- CATHEPSIN S ADAM10, ADAM12, ADAMTS, Caspase-1, Caspase-2, Caspase-3, Caspase-4, Caspase-5, Caspase-6, Caspase-7, Caspase-8, Caspase-9, Caspase-10, Caspase-11, Caspase-12, Caspase-13, Caspase-14, or TACE.
- the tissue microenvironment cleavable linker is cleavable at a particular pH. In some embodiments, the tissue microenvironment cleavable linker is cleavable at a pH between about 5.0 and about 7.4, between 5.0 and 7.0, between 5.0 and 6.5, between 5.0 and 5.5, or between 5.9 and 6.2. In one embodiment, the tissue microenvironment cleavable linker is cleavable at a pH between about 6.0 and about 7.0, between about 6.2 and about 6.9, between about 6.5 and about 6.8, or between about 6.5 and about 6.7.
- the tissue microenvironment cleavable linker is cleavable at a pH between about 5.5 and about 6.5, e.g., between 5.9 and 6.2. In one embodiment, the tissue microenvironment cleavable linker is cleavable at a hypoxic pH, e.g., a pH about 6.7 to 6.9, e.g., compared to a physiological pH of about 7.4.
- the tissue microenvironment cleavable linker is cleavable is cleaved at a pH of no more than 7.4, no more than 7.0, no more than 6.9, no more than 6.8, no more than 6.7, no more than 6.6, no more than 6.5, no more than 6.4, no more than 6.3, no more than 6.2, no more than 6.1, no more than 6.0, no more than 5.5 or lower.
- the tissue microenvironment cleavable linker is preferentially cleaved or degraded upon exposure to a first pH relative to a second pH. In one embodiment, the tissue microenvironment cleavable linker is cleaved or degraded at least 2, 5, 10, 20, 30, 40, 50, 60, 70, 80, or 100 times faster upon exposure to a first pH relative to a second pH. In other embodiments, the tissue
- the tissue microenvironment cleavable linker shows increased pH-sensitivity in a hypoxic microenvironment, e.g., in a tumor, or fibrotic tissue.
- the tissue microenvironment cleavable linker (e.g., L 3 ) exhibits an increased release rate or increased release yield of the agent (e.g., the ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) at a desired site (e.g., a tumor), e.g., relative to the release rate or release yield at another site.
- the tissue microenvironment cleavable linker comprises an electron withdrawing group (e.g., an electron withdrawing group that enhances the cleavage rate or yield, e.g., upon exposure to a first set of conditions relative to a second set of conditions).
- BASP particles comprising a phenylester linker (Tel-PhenylEster) or difluorophenylester linker (Tel- DiFluoroPhenylEster) achieved increased release of an exemplary agent (e.g., telmisartan) over BASP particles comprising an ester linker (Original TelEster).
- a BASP particle comprising a difluorophenylester linker e.g., wherein L 3 is -O-(3,5-difluorophenyl)-C(O)NH
- a BASP particle comprising an ester linker e.g., wherein L 3 is–OCH 2 CH 2 -.
- a BASP particle comprising a phenylester linker e.g., wherein L 3 is -O-phenyl-C(O)NH-
- a BASP particle comprising an ester linker e.g., wherein L 3 is - OCH 2 CH 2 -.
- Exemplary conjugates or BASP particles may be described by a number of properties, including average molecular weight (kDa), average hydrodynamic diameter (nm), and polydispersity.
- average molecular weight may encompass the number average molecular weight (Mn), weight average molecular weight (Mw), higher average molecular weight (Mz or Mz +1), GPC/SEC-determined average molecular weight (Mp), and viscosity average molecular weight (Mv).
- the average molecular weight of the conjugate is between about 2 kDa and about 100 kDa, e.g., between about 15 kDa and about 85 kDa, about 20 kDa and about 60 kDa, or about 30 kDa and about 50 kDa, e.g., as determined by gel permeation chromatography.
- the average molecular weight of the conjugate is less than about 100 kDa (e.g., less than about 95 kDa, about 90 kDa, about 85 kDa, about 80 kDa, about 75 kDa, about 70 kDa, about 65 kDa, about 60 kDa, about 55 kDa, or about 50 kDa), e.g., as determined by gel permeation chromatography. In some embodiments, the average molecular weight of the conjugate is less than about 75 kDa (e.g., less than about 70 kDa, about 65 kDa, about 60 kDa, about 55 kDa, or about 50 kDa).
- the average molecular weight of the conjugate is between about 20 kDa and about 60 kDa. In one embodiment, the average molecular weight of the conjugate is between about 30 kDa and about 50 kDa. In still other embodiments, the average molecular weight of the conjugate is between about 2 kDa and about 6 kDa (e.g., between about 3 kDa and about 5 kDa)
- the average molecular weight of the BASP particle is between about 100 kDa and about 1,000 kDa, e.g., between about 200 kDa and about 700 kDa or about 300 kDa and about 500 kDa, e.g., as determined by gel permeation chromatography. In one embodiment, the average molecular weight of the particle is between about 2000 kDa and about 70 kDa. In one embodiment, the average molecular weight of the particle is between about 300 kDa and about 500 kDa.
- the average molecular weight of the particle is less than about 1,000 kDa (e.g., less than about 950 kDa, about 900 kDa, about 850 kDa, about 800 kDa, about 750 kDa, about 700 kDa, about 650 kDa, about 600 kDa, about 550 kDa, or about 500 kDa), e.g., as determined by gel permeation chromatography.
- the average molecular weight of the particle is less than about 750 kDa (e.g., less than about 700 kDa, about 650 kDa, about 600 kDa, about 550 kDa, or about 500 kDa). In some embodiments, the average molecular weight of the particle is less than about 500 kDa (e.g., less than about 450 kDa, about 400 kDa, about 350 kDa, or 300 kDa).
- average hydrodynamic diameter refers to the average size of a conjugate or BASP particle.
- the average hydrodynamic diameter may or may not encompass the solvation layers of conjugate or BASP particle, and may be determined through a number of methods including dynamic light scattering, electron microscopy (e.g., scanning electron microscopy, transmission electron microscopy), atomic force microscopy, and X-ray diffraction.
- the average hydrodynamic diameter of the conjugate is less than 50 nm (e.g., less than about 45 nm, about 40 nm, about 35 nm, about 25 nm, about 20 nm, about 15 nm, about 10 nm, about 7.5 nm, or less), e.g., as determined by dynamic light scattering. In some embodiments, the average hydrodynamic diameter of the conjugate is between about 1 nm and about 20 nm (e.g., between about 2.5 nm and about 17.5 nm, or about 5 nm and about 15 nm). In some embodiments, the average hydrodynamic diameter of the conjugate is between about 5 nm and about 15 nm.
- the average hydrodynamic diameter of the conjugate is less than about 20 nm (e.g., less than about 15 nm, about 12.5 nm, about 10 nm, about 9 nm, about 8 nm, about 7 nm, about 6 nm, about 5 nm, or less)
- the average hydrodynamic diameter of the BASP particle is less than 100 nm (e.g., less than about 90 nm, about 80 nm, about 75 nm, about 70 nm, about 65 nm, about 60 nm, about 55 nm, about 50 nm, about 45 nm, about 40 nm, about 35 nm, about 25 nm, or less), e.g., as determined by dynamic light scattering.
- the average hydrodynamic diameter of the BASP particle is between about 5 nm and about 100 nm (e.g., between about 7.5 nm and about 75 nm, about 10 nm and about 50 nm, about 12.5 nm and about 40 nm, or about 15 nm and about 30 nm). In some embodiments, the average hydrodynamic diameter of the BASP particle is between about 10 nm and about 50 nm. In some embodiments, the average hydrodynamic diameter of the BASP particle is between about 5 nm and about 50 nm. In some embodiments, the average hydrodynamic diameter of the BASP particle is between about 15 nm and about 30 nm.
- average polydispersity refers to a measure of the distribution of molecular size in a mixture, e.g., as determined by a chromatographic method, such as gel permeation chromatography or size exclusion chromatography, or through dynamic light scattering.
- the average polydispersity of the conjugate or BASP particle is less than about 0.5 (e.g., less than about 0.4, about 0.35, about 0.3, about 0.25, about 0.2, about 0.15, or less).
- the average polydispersity of the conjugate or BASP particle is less than about 0.3.
- the average polydispersity of the conjugate or BASP particle is less than about 0.2.
- the conjugate or BASP particle is monodisperse. In some embodiments, the conjugate or BASP particle is about 50% monodisperse (e.g., about 55%, about 60%, about 65%, about 70%, about 75%, about 80%, about 85%, about 90%, about 95%, about 99%, or about 99.9% monodisperse).
- the conjugate or BASP particle is substantially soluble in water (e.g., hydrophilic). In some embodiments, the conjugate or BASP particle is substantially insoluble in water (e.g., hydrophobic). In some embodiments, the conjugate or BASP particle is substantially insoluble in water and greater than about 10,000 parts water are required to dissolve 1 part polymer. In one embodiment, the conjugate or BASP particle is amphiphilic. In one embodiment, the conjugate or BASP particle comprises a segment that is hydrophobic and a segment that is hydrophilic.
- the conjugate comprises a linear structure. In some embodiments, the conjugate comprises a branched structure. In some embodiments, the conjugate comprises a branched structure, and each repeating unit in the polymer comprises at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, or at least 8 branch points.
- the conjugate or particle comprises a structure according to Formula (III):
- Ring C is a carbocyclyl or heterocyclyl moiety
- Ring T is a triazoldiyl moiety (e.g., 1,2,3-triazoldiyl)
- A is C 1 -C 12 alkylene, C 2 -C 12 alkenylene, C 2 -C 12 alkynylene, C 1 -C 12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or
- heteroalkylene is optionally substituted with 1-6 independently selected R 1 ;
- B is C 1 - C 12 alkylene, C 2 -C 12 alkenylene, C 2 -C 12 alkynylene, C 1 -C 12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R 1 ;
- X is an agent (e.g., an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor);
- P is alkylene or heteroalkylene (e.g., polyethylene glycol); each of L 1 and L 2 is independently a bond, C 1 -C 12 alkylene, C 2 - C 12 alkenylene, C 2 -C 12 alkynylene, C 1 -C 12 heteroalkylene, (C 0 -C 12 alkylene)-arylene- (C 0 -C 12 alkylene), (C 0 -C 12
- each R 1 and R 2 is independently alkyl, alkenyl, alkynyl, heteroalkyl (e.g., polyethylene glycol), halo, cyano, oxo,–OR A ,–NR B R C ,– NR B C(O)R D , -C(O)NR B R C ,–C(O)R D ,–C(O)OH,–C(O)OR D ,–SR E , or–S(O) m R E ; each R A is independently hydrogen, C 1 -C 6 alkyl, C 1 -C 6 heteroalkyl,–C(O)NR B R C ,– C(O)R D ,–C(O)OH, or–C(O)OR D ; each R B and R C is independently hydrogen or C 1 - C 6 alkyl; or each R D is independently C 1 -C 6 alkyl, C 1 -C 6 heteroalkyl, or C 1
- D is N or CR 4 ;
- Z is C(R 5 ) 2 , O, or S;
- each R 3 is independently C 1 -C 6 alkyl, C 1 - C 6 haloalkyl, oxo, or halo;
- R 4 is hydrogen, C 1 -C 6 alkyl, or halo;
- each R 5 is
- n is 0, 1, or 2
- Ring C is a structure of Formula (IV-a):
- structure of Formula (IV) is or. In some embodiments, the structure of Formula (I . In
- the structure of Formula (IV) is .
- the structure of Formula (IV) is .
- Ring C is a structure of Formula (IV-b):
- R 3’’ is selected from hydrogen, C 1 -C 6 alkyl, -C(O)-C 1 -C 6 alkyl, -C(O)-O-C 1 -C 6 alkyl, -C(O)-NH-C 1 -C 6 alkyl, C 1 -C 6 heteroalkyl, -C(O)-C 1 -C 6 heteroalkyl, -C(O)-O-C 1 -C 6 heteroalkyl, -C(O)-NH-C 1 -C 6 heteroalkyl, and halo, wherein the any alkyl portion of R 3’ is optionally substituted with halo; each R 5 is independently hydrogen, C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, or halo;“1” represents a portion of Ring C bound to B; and each“2” represents a portion of Ring C bound to either another macromon
- Ring T is 1,2,3 triazoldiyl or 1,2,4-triazoldiyl. In some embodiments, Ring T is 1,2,3 triazoldiyl. In some embodiments, Ring T is , wherein“2” represents a portion of Ring
- A is C 1 -C 12 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 11 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 10 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 9 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 8 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 7 heteroalkylene optionally substituted with 1-6 independently selected R 1 .
- A is C 1 -C 6 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 5 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 4 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 3 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 -C 2 heteroalkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, A is C 1 heteroalkylene optionally substituted with 1-6 independently selected R 1 .
- A is C 1 -C 8 heteroalkylene optionally substituted with 1-3 independently selected R 1 .
- R 1 is oxo or heteroalkyl (e.g., polyethylene glycol).
- R 1 is oxo.
- R 1 is heteroalkyl (e.g., polyethylene glycol).
- A is
- R 1 comprises a polyethylene glycol (PEG), a polyethylene oxide (PEO), a polypropylene glycol (PPG), a polyglycerol (PG), a poloxamine (POX), a polybutylene oxide (PBO), polylactic acid (PLA), polyglycolic acid (PGA), poly(lactic-co-glycolic acid) (PLGA), polycaprolactone (PCL), polydioxanone (PDO), a polyanhydride, a polyacrylide, a polyvinyl, or a polyorthoester.
- R 1 comprises a polyethylene glycol (PEG).
- A is C(O)CH 2 CH 2 C(O)NH-PEG.
- the PEG has a molecular weight of between about 200 and about 6000, g/mol (e.g., PEG200, PEG400, PEG600, PEG800, PEG1000, PEG1500, PEG2000, PEG3000, PEG4000, or PEG6000).
- the PEG is PEG200.
- the PEG is PEG400.
- the PEG is PEG600.
- the PEG is PEG800. In some
- the PEG is PEG1000. In some embodiments, the PEG is PEG2000. In some embodiments, the PEG is PEG3000. In some embodiments, the PEG is
- the PEG is PEG6000.
- B is C 1 -C 12 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 -C 11 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 -C 10 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 -C 9 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 -C 8 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 -C 7 alkylene optionally substituted with 1-6 independently selected R 1 .
- B is C 1 -C 6 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 -C 5 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 -C 4 alkylene optionally substituted with 1-6 R 1 . In some embodiments, B is C 1 -C 3 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 -C 2 alkylene optionally substituted with 1-6 independently selected R 1 . In some embodiments, B is C 1 alkylene optionally substituted with 1-6 independently selected R 1 .
- B is dodecylene, undecylene, decylene, nonylene, octylene, heptylene, hexylene, pentylene, butylene, propylene, ethylene, or methylene, each of which is optionally substituted with 1-6 independently selected R 1 .
- B is dodecylene, undecylene, decylene, nonylene, octylene, heptylene, hexylene, pentylene, butylene, propylene, ethylene, or methylene.
- B is octylene, heptylene, hexylene, pentylene, butylene, propylene, or ethylene. In some embodiments, B is hexylene, pentylene, butylene, propylene, or ethylene. In some embodiments, B is hexylene. In some embodiments, B is pentylene. In some embodiments, B is butylene. In some embodiments, B is propylene. In some embodiments, B is ethylene. In some embodiments, B is methylene.
- each of L 1 and L 2 is independently C 1 -C 12 alkylene, C 1 - C 12 heteroalkylene, (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 alkylene), (C 0 -C 12
- heteroalkylene -arylene-(C 0 -C 12 alkylene), or (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), or (C 0 -C 12 heteroalkylene)-aryl-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 .
- each of L 1 and L 2 is independently C 1 -C 12 alkylene, C 1 -C 12 heteroalkylene, or (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-6 independently selected R 2 .
- L 1 is C 1 -C 12 heteroalkylene and L 2 is C 1 -C 12 alkylene, wherein each alkylene and heteroalkylene is optionally substituted with 1-6 independently selected R 2 .
- each of L 1 and L 2 is independently selected from–CH 2 –,–NCH 2 –, and–CH 2 CH 2 .
- L 1 is–NCH 2 –.
- L 2 is–CH 2 CH 2 – .
- L 3 is C 1 -C 12 heteroalkylene, (C 0 -C 12 heteroalkylene)- arylene-(C 0 -C 12 alkylene), (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), or (C 0 - C 12 heteroalkylene)-aryl-(C 0 -C 12 heteroalkylene), wherein each alkylene,
- heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 .
- L 3 is C 1 -C 12 heteroalkylene, or (C 0 -C 12 alkylene)-arylene- (C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-6 independently selected R 2 .
- R 2 is oxo or halo (e.g., fluoro).
- L 3 is–C(O)–,–OC(O)–,–C(O)O–, –C(O)CH 2 –,–OC(O)CH 2 –,–C(O)OCH 2 –,–CH 2 CH 2 O–, C(O)CH 2 CH 2 O–,
- P is heteroalkylene.
- P comprises polyethylene glycol (PEG), polyethylene oxide (PEO), polypropylene glycol (PPG), polyglycerol (PG), poloxamine (POX), polybutylene oxide (PBO), polylactic acid (PLA), polyglycolic acid (PGA), poly(lactic-co-glycolic acid)
- P comprises about 2 to about 200 repeating units (e.g., about 2 to about 150, about 2 to about 100, about 2 to about 50, about 2 to about 25, about 2 to about 15, about 2 to about 10, or about 2 to about 5 repeating units) of polyethylene glycol (PEG), polyethylene oxide (PEO), polypropylene glycol (PPG), polyglycerol (PG), poloxamine (POX), polybutylene oxide (PBO), polylactic acid (PLA), polyglycolic acid (PGA), poly(lactic-co-glycolic acid) (PLGA), polycaprolactone (PCL), polydioxanone (PDO), a polyanhydride, a polyacrylide, a polyvinyl, or a polyorthoester.
- PEG polyethylene glycol
- PEO polyethylene oxide
- PPG polypropylene glycol
- PG polyglycerol
- POX poloxamine
- PBO polybutylene oxide
- PBO polylactic acid
- PLA polygly
- P comprises polyethylene glycol (PEG).
- P comprises between about 2 to about 200 repeating units (e.g., about 2 to about 150, about 2 to about 100, about 2 to about 50, about 2 to about 25, about 2 to about 15, about 2 to about 10, or about 2 to about 5 repeating units) of polyethylene glycol (PEG).
- P comprises between about 2 to about 10 repeating units of polyethylene glycol (PEG).
- the PEG comprises diethylene glycol, triethylene glycol, tetraethylene glycol, or pentaethylene glycol.
- the structure of Formula (III) is a structure of Formula (III-a):
- A is C 1 -C 12 alkylene, C 2 -C 12 alkenylene, C 2 -C 12 alkynylene, C 1 -C 12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R 1 ;
- B is alkylene, C 2 - C 12 alkenylene, C 2 -C 12 alkynylene, C 1 -C 12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R 1 ;
- X is an agent (e.g., an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor); each of L 1 and L 2 is independently C 1 -C 12 alkylene, C 1 -C 12 heteroalkylene, (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 alkylene
- each R 1 and R 2 is independently alkyl, alkenyl, alkynyl, heteroalkyl (e.g., polyethylene glycol), halo, cyano, oxo,–OR A ,–NR B R C ,–NR B C(O)R D , -C(O)NR B R C , –C(O)R D ,–C(O)OH,–C(O)OR D ,–SR E , or–S(O) m R E ; each R A is independently hydrogen, C 1 -C 6 alkyl, C 1 -C 6 heteroalkyl,–C(O)NR B R C ,–C(O)R D ,–C(O)OH, or– C(O)OR D ; each R B and R C is independently hydrogen or C 1 -C 6 alkyl; each R D is independently C 1 -C 6 alkyl, C 1 -C 6 alkyl, C 1 -C 6 alkyl, C 1 -C
- the structure of Formula (III) is a structure of Formula (III-b):
- B is C 1 -C 12 alkylene or C 1 -C 12 heteroalkylene, wherein each alkylene or heteroalkylene is optionally substituted with 1-6 R 1 ;
- X is an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or a bromodomain inhibitor);
- each of L 1 and L 2 is independently C 1 -C 12 alkylene, C 1 -C 12 heteroalkylene, (C 0 -C 12 alkylene)-arylene-(C 0 - C 12 alkylene), (C 0 -C 12 heteroalkylene)-arylene-(C 0 -C 12 alkylene), or (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), or (C 0 -C 12 heteroalkylene)-aryl-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12
- the conjugate comprises a structure of Formula (III-c):
- L 3 is C 1 -C 12 heteroalkylene, (C 0 -C 12 heteroalkylene)- arylene-(C 0 -C 12 alkylene), (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), or (C 0 - C 12 heteroalkylene)-aryl-(C 0 -C 12 heteroalkylene), wherein each alkylene,
- heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 ; each R 2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive.
- the structure of Formula (III) is a structure of Formula (III-d-1) or Formula (III-d-2):
- L 3 is C 1 -C 12 heteroalkylene, (C 0 -C 12 heteroalkylene)-arylene-(C 0 -C 12 alkylene), (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), or (C 0 -C 12
- heteroalkylene -aryl-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 ; each R 2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive.
- the structure of Formula (III) is a structure of Formula (III-e):
- L 3 is C 1 -C 12 heteroalkylene, (C 0 -C 12 heteroalkylene)-arylene-(C 0 -C 12 alkylene), or (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 ; each R 2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive.
- the conjugate or BASP particle comprises a structure according to Formula (III-f):
- Ring C’ is a carbocyclyl or heterocyclyl moiety
- Ring T is a triazoldiyl moiety (e.g., 1,2,3-triazoldiyl)
- A is C 1 -C 12 alkylene, C 2 -C 12 alkenylene, C 2 -C 12 alkynylene, C 1 -C 12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R 1
- B is C 1 -C 12 alkylene, C 2 -C 12 alkenylene, C 2 -C 12 alkynylene, C 1 -C 12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R 1
- X is an agent (e.g., an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor);
- each R 1 and R 2 is independently alkyl, alkenyl, alkynyl, heteroalkyl (e.g., polyethylene glycol), halo, cyano, oxo,–OR A ,–NR B R C ,– NR B C(O)R D , -C(O)NR B R C ,–C(O)R D ,–C(O)OH,–C(O)OR D ,–SR E , or–S(O) m R E ; each R A is independently hydrogen, C 1 -C 6 alkyl, C 1 -C 6 heteroalkyl,–C(O)NR B R C ,– C(O)R D ,–C(O)OH, or–C(O)OR D ; each R B and R C is independently hydrogen or C 1 - C 6 alkyl; or each R D is independently C 1 -C 6 alkyl, C 1 -C 6 heteroalkyl, or C 1
- (terminating) macromonomer unit present in the conjugate or BASP particle is selected from hydrogen, phenyl, or C 1 -C 4 alkyl; and q is an integer between 2 and 1,000, inclusive.
- the conjugate or BASP particle comprises the structure according to Formula (III-a) as a repeating unit or as part of a repeating unit.
- the conjugate or BASP particle comprises about 2 to about 200 repeating units (e.g., about 2 to about 150, about 2 to about 100, about 2 to about 50, about 2 to about 25, about 2 to about 15, about 2 to about 10, about 2 to about 5, about 10 to about 200, about 10 to about 100, about 10 to about 50, about 10 to about 25, about 25 to about 200, about 25 to about 100, about 25 to about 50, about 50 to about 200, about 50 to about 100, or about 100 to about 200, repeating units.
- q is an integer between 2 and 500, inclusive (e.g., between 2 and 450, 2 and 400, 2 and 350, 2 and 300, 2 and 250, 2 and 200, 2 and 150, 2 and 100, 2 and 75, and 2 and 50). In some embodiments, q is an integer between 2 and 100, inclusive.
- Ring C’ is a structure of Formula (IV):
- D is N or CR 4 ;
- Z is C(R 5 ) 2 , O, or S;
- each R 3 is independently C 1 -C 6 alkyl, C 1 - C 6 haloalkyl, oxo, or halo;
- R 4 is hydrogen, C 1 -C 6 alkyl, or halo;
- each R 5 is
- Ring C’ is a structure of Formula (IV-b):
- the radicals represents a portion of Ring C’ bound to B
- R 6b an end group
- the structure of Formula (IV) is .
- the structure of Formula (IV) is .
- Ring C’ is a structure of Formula (IV-c):
- R 3’’ is selected from hydrogen, C 1 -C 6 alkyl, -C(O)-C 1 -C 6 alkyl, -C(O)-O-C 1 -C 6 alkyl, -C(O)-NH-C 1 -C 6 alkyl, C 1 -C 6 heteroalkyl, -C(O)-C 1 -C 6 heteroalkyl, -C(O)-O-C 1 -C 6 heteroalkyl, -C(O)-NH-C 1 -C 6 heteroalkyl, and halo, wherein the any alkyl portion of R 3’ is optionally substituted with halo; each R 5 is independently hydrogen, C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, or halo;“1” represents a portion of Ring C’ bound to B; and each“2” represents a portion of ring C’ bound to either
- the agent contains a functional group (e.g., reactive functional group) for conjugation to the conjugate or BASP particle.
- the functional group is chosen from a hydroxyl group, amino group (e.g., a primary or secondary amino group), thiol group, carboxylic acid group, aldehyde group, ketone group, hydrazino group, azido group, vinyl ether group, alkenyl group, isothiocyanate group, or acrylate group.
- the agent can be activated for conjugation to a conjugate or BASP particle or another moiety or polymer (e.g., PEG, PLA, PLGA, PDO, cyclodextrin) through the use of an activating agent, e.g., succinic anhydride, thiophosgene, 4-nitrophenyl chloroformate, ethylenediamine, or cis-aconitic anhydride.
- an activating agent e.g., succinic anhydride, thiophosgene, 4-nitrophenyl chloroformate, ethylenediamine, or cis-aconitic anhydride.
- the structure of Formula (III-f) is a structure of Formula (III-g):
- A is C 1 -C 12 alkylene, C 2 -C 12 alkenylene, C 2 -C 12 alkynylene, C 1 -C 12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R 1 ;
- B is C 1 -C 12 alkylene, C 2 - C 12 alkenylene, C 2 -C 12 alkynylene, C 1 -C 12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R 1 ;
- X is an agent (e.g., an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor); each of L 1 and L 2 is independently C 1 -C 12 alkylene, C 1 -C 12 heteroalkylene, (C 0 -C 12 alkylene)-arylene-(C
- each R 1 and R 2 is independently alkyl, alkenyl, alkynyl, heteroalkyl (e.g., polyethylene glycol), halo, cyano, oxo,–OR A ,–NR B R C ,–NR B C(O)R D , -C(O)NR B R C , –C(O)R D ,–C(O)OH,–C(O)OR D ,–SR E , or–S(O) m R E ; each R A is independently hydrogen, C 1 -C 6 alkyl, C 1 -C 6 heteroalkyl,–C(O)NR B R C ,–C(O)R D ,–C(O)OH, or– C(O)OR D ; each R B and R C is independently hydrogen or C 1 -C 6 alkyl; each R D is independently C 1 -C 6 alkyl, C 1 -C 6 alkyl, C 1 -C 6 alkyl, C 1 -C
- B is C 1 -C 12 alkylene or C 1 -C 12 heteroalkylene, wherein each alkylene or heteroalkylene is optionally substituted with 1-6 R 1 ;
- X is an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or a bromodomain inhibitor);
- each of L 1 and L 2 is independently C 1 -C 12 alkylene, C 1 -C 12 heteroalkylene, (C 0 -C 12 alkylene)-arylene-(C 0 - C 12 alkylene), (C 0 -C 12 heteroalkylene)-arylene-(C 0 -C 12 alkylene), or (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), or (C 0 -C 12 heteroalkylene)-aryl-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12
- the conjugate comprises a structure of Formula (III-h):
- L 3 is C 1 -C 12 heteroalkylene, (C 0 -C 12 heteroalkylene)- arylene-(C 0 -C 12 alkylene), (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), or (C 0 - C 12 heteroalkylene)-aryl-(C 0 -C 12 heteroalkylene), wherein each alkylene,
- heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 ; each R 2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; z is an integer between 1 and 200, inclusive; R 6a is absent or in the first (initiating) macromonomer unit present in the conjugate or BASP particle is selected from hydrogen, phenyl, or C 1 -C 4 alkyl; R 6b is absent or in the last
- (terminating) macromonomer unit present in the conjugate or BASP particle is selected from hydrogen, phenyl, or C 1 -C 4 alkyl; and q is an integer between 2 and 1000, inclusive.
- the structure of Formula (III-a) is a structure of Formula (III-i-1) or Formula (III-i-2):
- L 3 is C 1 -C 12 heteroalkylene, (C 0 -C 12 heteroalkylene)-arylene-(C 0 -C 12 alkylene), (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), or (C 0 -C 12
- heteroalkylene -aryl-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 ; each R 2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; z is an integer between 1 and 200, inclusive; R 6a is absent or in the first (initiating) macromonomer unit present in the conjugate or BASP particle is selected from hydrogen, phenyl, or C 1 -C 4 alkyl; R 6b is absent or in the last (terminating) macromonomer unit present in the conjugate or BASP particle is selected from hydrogen, phenyl, or C 1 -C 4 alkyl; and q is an integer between 2 and 1000, inclusive.
- the structure of Formula (III) is a structure of Formula (III-j):
- heteroalkylene -aryl-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 ; each R 2 is independently alkyl, heteroalkyl, halo, cyano, oxo,–OR A ,–NR B R C ,–
- each R A is independently hydrogen, C 1 -C 6 alkyl, C 1 -C 6 heteroalkyl,–C(O)NR B R C ,– C(O)R D ,–C(O)OH, or–C(O)OR D ;
- each R B and R C is independently hydrogen or C 1 - C 6 alkyl;
- each R D is independently C 1 -C 6 alkyl, C 1 -C 6 heteroalkyl, or C 1 -C 6 haloalkyl;
- each R E is independently hydrogen or C 1 -C 6 alkyl;
- t is an integer between 1 and 10, inclusive;
- z is an integer between 1 and 200, inclusive;
- R 6a is absent or in the first (initiating) macromonomer unit present in the conjugate
- q is an integer between 5 and 100, inclusive.
- R 6a is hydrogen in an initiating macromonomer unit; and R 6b is phenyl in a terminating macromomonomer unit present in the conjugate or BASP particle.
- L 3 is C 1 -C 12
- heteroalkylene (C 0 -C 12 heteroalkylene)-arylene-(C 0 -C 12 alkylene), or (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 ; each R 2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive.
- L 3 is C 1 -C 12
- heteroalkylene or (C 0 -C 12 heteroalkylene)-aryl-(C 0 -C 12 heteroalkylene), or (C 0 -C 12 alkylene)-aryl-(C 0 -C 12 heteroalkylene), wherein each alkylene, heteroalkylene, or aryl is optionally substituted with 1-6 independently selected R 2 .
- “ ” represents a linkage to another portion of the conjugate or particle (e.g., a conjugate or particle described herein) or the terminus of the conjugate or particle (e.g., a conjugate or particle described herein).
- the terminus may be further modified with hydrogen, a protecting group, an agent (e.g., an agent described herein), or other group (e.g., as described herein).
- the structure of Formula (III) or Formula (III-f) is formed from a recursor macromonomer selected from:
- the conjugate or BASP particle comprises a single agent (e.g., one or more of the same agent). In some embodiments, the conjugate or BASP particle comprises multiple agents. In some embodiments, the agent(s) are attached to a conjugate or BASP particle through a covalent bond. In some embodiments, the agent(s) are attached to a conjugate or BASP particle through a non-covalent bond or interaction. In some embodiments, the agents are the same agent. In some embodiments, the agents are different agents. In some embodiments, the conjugate or BASP particle comprises multiple agents (e.g., 2, 3, 4, 5, 6 or more different agents).
- conjugate or BASP particle comprises an agent attached at one end of the conjugate or polymer, e.g., surface exposed end of the conjugate or polymer. In other embodiments, the conjugate or BASP particle comprises an agent in the middle of the conjugate or polymer. In some embodiments, the conjugate or BASP particle comprises an agent attached to at least two conjugates or polymers such that the agent is present between the two conjugates or polymers.
- the conjugate or BASP particle is assembled through the use of a polymerization reaction. In some embodiments, the conjugate or BASP particle is assembled through polymerization of a macromonomer subunit and a cross- linker. In some embodiments, the conjugate or BASP particle is assembled through polymerization of a single type of macromonomer subunit and a cross-linker. In some embodiments, the conjugate or BASP particle is assembled through polymerization of a multiple types of macromonomer subunits and a cross-linker.
- the polymerization reaction comprises ring-opening metathesis polymerization (e.g., ROMP). In some embodiments, multiple rounds of polymerization (e.g., ROMP) are carried out to prepare the conjugate and/or particle.
- the particle can be formed from the conjugates described herein, e.g., by precipitation and/or self-assembly. In some embodiments, the conjugate is not precipitated from solution and/or self-assembled.
- the particle comprises conjugates of the same type, e.g., wherein each conjugate comprises the same agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor).
- the particle comprises conjugates of different types, e.g., wherein each conjugate comprises a different agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor).
- the particle comprises conjugates of different types, e.g., wherein some fraction of the conjugates comprises either an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor and another fraction of the conjugates comprise a second agent.
- the second agent is an anti-cancer agent, an anti-fibrotic therapeutic agent, an anti-inflammatory agent, a liver therapeutic agent, or second therapeutic agent.
- the second agent is a targeting agent.
- the second agent is a diagnostic agent.
- the particle is not selectively delivered or targeted to a target site, e.g., the particle does not include a targeting moiety (e.g., a cell- or liver- targeting agent as described herein).
- a targeting moiety e.g., a cell- or liver- targeting agent as described herein.
- the conjugate or BASP particle e.g., as described herein has a size to include any of the agents described herein, e.g., the ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor.
- the particle is substantially or completely size-excluded from reaching arteriole smooth muscle, which is protected by non-leaky vessels.
- the particle selectively penetrates a leaky vessel, e.g., a leaky vessel of a tumor or liver.
- the agent e.g., ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor
- a second agent e.g., the anti-cancer agent, anti-fibrotic agent, anti-inflammatory agent, or liver therapeutic agent.
- the agent e.g., ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor
- the conjugate or BASP particle without a targeting agent or diagnostic agent.
- the agent e.g., ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor
- the agent is formulated for extended release, e.g., in an extended release formulation for substantially continuous release for hours, days, weeks, months or years, for example, using a cleavable linker (e.g., a tissue
- microenvironment cleavable linker and/or linker of different degradation rates.
- the conjugate or BASP particle may comprise a first agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) and, optionally, a second agent (e.g., an anti-cancer agent, anti-inflammatory agent, anti-fibrotic agent, or liver therapeutic), targeting moiety, or diagnostic agent.
- a first agent e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor
- a second agent e.g., an anti-cancer agent, anti-inflammatory agent, anti-fibrotic agent, or liver therapeutic
- targeting moiety, or diagnostic agent is present in separate entities or in the same entity.
- the first agent e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor
- the second agent e.g., the anti- cancer agent, anti-inflammatory agent, anti-fibrotic agent, or liver therapeutic
- targeting moiety, or diagnostic agent can be provided as a second conjugate or BASP particle (e.g., where the second conjugate particle has a structural property (e.g., size or composition) or a functional property (e.g., release kinetics or a pharmacodynamic property) that differs from the first particle).
- the first agent e.g., the ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor
- the second agent e.g., an anti-cancer agent, anti-inflammatory agent, anti-fibrotic agent, or liver therapeutic
- targeting moiety, or diagnostic agent can be provided in the same entity, e.g., in the same conjugate or BASP particle.
- a conjugate of the present disclosure is a compound shown in Table 2.
- the percentage of the conjugates (e.g., in a particle) that comprise an agent is between about 1 and about 100% (e.g., about 1%, about 2%, about 3%, about 4%, about 5%, about 10%, about 15%, about 20%, about 30%, about 40%, about 50%, about 60%, about 70%, about 80%, about 90%, or about 100%).
- the percentage of the conjugates that comprise an agent is less than about 50%, e.g., less than about 40%, less than about 35%, less than about 30%, less than about 25%, less than about 20%, less than about 15%, or less than about 10%.
- the percentage of the conjugates (e.g., in a particle) that comprise an agent is between about 5% and about 50%, about 5% and about 40%, about 5% and about 30%, about 5% and about 25%, or about 5% and about 20%.
- the percentage of the conjugates (e.g., in a particle) that comprise an agent is between about 5% and 90%. In some embodiments, the percentage of the conjugates (e.g., in a particle) that comprise an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) is between about 5% and about 75%. In some embodiments, the conjugates (e.g., in a particle) that comprise an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) is between about 5% and about 50%.
- the percentage of the conjugates (e.g., in a particle) that comprise an agent is between about 10% and about 25%.
- the total amount of the agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) present in the conjugate or BASP particle is greater than about 5% (e.g., about 6%, about 7%, about 8%, about 9%, about 10%, about 12%, about 15%, about 20%, about 25%, about 30%, or more) of the total size or weight of the conjugate or BASP particle.
- the total amount of the agent present in the conjugate or BASP particle is greater than about 10% (e.g., about 12%, about 15%, about 20%, about 25%, about 30%, or more) of the total size or weight of the conjugate or BASP particle. In some embodiments, the total amount of an ARB present in the conjugate or BASP particle is greater than about 5% (e.g., about 6%, about 7%, about 8%, about 9%, about 10%, about 12%, about 15%, about 20%, about 25%, about 30%, or more) of the total size or weight of the conjugate or BASP particle.
- the total amount of a vitamin D analog present in the conjugate or BASP particle is greater than about 5% (e.g., about 6%, about 7%, about 8%, about 9%, about 10%, about 12%, about 15%, about 20%, about 25%, about 30%, or more) of the total size or weight of the conjugate or BASP particle.
- the total amount of an IDO inhibitor analog present in the conjugate or BASP particle is greater than about 5% (e.g., about 6%, about 7%, about 8%, about 9%, about 10%, about 12%, about 15%, about 20%, about 25%, about 30%, or more) of the total size or weight of the conjugate or BASP particle.
- the total amount of a bromodomain inhibitor analog present in the conjugate or BASP particle is greater than about 5% (e.g., about 6%, about 7%, about 8%, about 9%, about 10%, about 12%, about 15%, about 20%, about 25%, about 30%, or more) of the total size or weight of the conjugate or BASP particle.
- a lower dose or amount of the agent in the conjugates or BASP particles can be administered (e.g., through local sustained delivery) compared to the agent in free form.
- the agent-containing conjugates or BASP particles e.g., conjugates or BASP particles containing an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor
- the agent-containing conjugates or BASP particles are administered at a dose or amount of the agent that is less than the dose or amount of said agent in free form to have a desired effect (e.g., a desired therapeutic effect).
- the agent e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor
- a conjugate or a BASP particle at a dose that is less than the dose or amount of said agent in free form to have a desired effect (e.g., a desired therapeutic effect), e.g., the standard of care dose for the intended use of the free agent.
- the agent are incorporated into the particles at a dose or amount of the agent that is less than the standard of care dose of the agent for a desired therapy (e.g., a dose that is less than about 0.01, about 0.02, about 0.03, about 0.04, about 0.05, about 0.06, about 0.07, about 0.08, about 0.09, about 0.1, about 0.2, about 0.3, about 0.4, about 0.5, about 0.6, about 0.7, about 0.8, about 0.9, or about 0.95 that of the standard of care dose of the agent).
- a dose that is less than about 0.01, about 0.02, about 0.03, about 0.04, about 0.05, about 0.06, about 0.07, about 0.08, about 0.09, about 0.1, about 0.2, about 0.3, about 0.4, about 0.5, about 0.6, about 0.7, about 0.8, about 0.9, or about 0.95 that of the standard of care dose of the agent e.g., a dose that is less than about 0.01, about 0.02, about 0.03,
- the dose is less than the anti-hypertensive or anti-heart failure dose for ARBs such as losartan, candesartan, eprosartan, irbesartan, olmesartan, telmisartan, and valsartan.
- ARBs such as losartan, candesartan, eprosartan, irbesartan, olmesartan, telmisartan, and valsartan.
- the agent e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor
- a conjugate or a BASP particle at a dose equivalent to the dose or amount of said agent in free form to have a desired effect (e.g., a desired therapeutic effect), e.g., the standard of care dose for the intended use of the free agent.
- the conjugate or BASP particle produces a greater therapeutic effect and/or a less adverse effect than the free agent.
- the particle increases the amount of the agent delivered to a tissue or cell in need thereof and reduces the amount of the agent exposed to a non- target tissue or cell, as compared to the free agent.
- the agent e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor
- a conjugate or a BASP particle at a dose higher than the dose or amount of said agent in free form to have a desired effect (e.g., a desired therapeutic effect), e.g., the standard of care dose for the intended use of the free agent.
- the agent is incorporated into a conjugate or a BASP particle at a dose higher than the dose or amount of said agent in free form that would produce an adverse effect by systemic administration (e.g., a reduction in blood pressure).
- the conjugate or BASP particle comprises two or more macromonomers (e.g., as described herein) each bound to an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor).
- an agent e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor.
- the conjugate or BASP particle comprises telmisartan- linked macromonomers.
- the telmisartan-linked telmisartan-linked telmisartan-linked telmisartan-linked telmisartan-linked telmisartan-linked telmisartan-linked telmisartan-linked described by Formulas (III-e) or (III-j). In some embodiments, the telmisartan-linked
- telmisartan-linked macromonomers are those of Formulas (III-e) or (III-j), wherein z is an integer between 25 and 85, inclusive; and L 3 is–CH 2 CH 2 O–.
- the telmisartan-linked macromonomers are those of Formula (III-e), wherein z is 68 and L 3 is–CH 2 CH 2 O–.
- the telmisartan-linked macromonomers are those of Formula (III-e), wherein z is 45 and L 3 is–CH 2 CH 2 O–.
- the telmisartan-linked macromonomers are those of Formulas (III-e) or (III-j),
- telmisartan- l s are those of Formulas (III-e) or (III-j), wherein z is 68 and L 3
- the telmisartan-linked conjugates or BASP particles have a drug-loading level between about 5% and about 25%.
- the conjugate or BASP particle comprises paricalcitol- linked macromonomers, e.g., as described in Formulas (III-d-1), (III-d-2), (III-i-1) or (III-i-2).
- the paricalcitol-linked macromonomers comprises a mixture of regioisomers.
- the paricalcitol-linked macromonomers comprises a mixture of regioisomers.
- macromonomers are those of Formulas (III-d-1), (III-d-2), (III-i-1) or (III-i-2), wherein z is an integer between 25 and 85, inclusive; and L 3 is–C(O)CH 2 CH 2 O–.
- the paricalcitol-linked macromonomers are those of Formulas (III-d-1), (III-d-2), (III-i-1) or (III-i-2), wherein z is 68 and L 3 is–C(O)CH 2 CH 2 O–.
- paricalcitol-linked macromonomers are those of Formulas (III-d-1), (III-d-2), (III-i-1) or (III-i-2), wherein z is 68 and L 3 is–C(O)CH 2 CH 2 O–, and is a mixture of regioisomers.
- paricalcitol-linked conjugate or BASP particle has a drug-loading level between about 5% and about 25%.
- the conjugate or BASP particle comprises an agent (e.g., an agent as described herein, e.g., an agent shown in FIGS.2, 3, 4A to 4B).
- the conjugate or BASP particle additionally comprises one or more of a second agent, a targeting moiety, or a diagnostic moiety.
- the conjugate comprises telmisartan (e.g., as described in Formulas (III-e) or (III-j)) and a second agent (e.g., an anti-cancer agent, anti-inflammatory agent, anti-fibrotic agent, or liver therapeutic).
- the conjugate or BASP particle comprises paricalcitol (e.g., as described in Formulas (III-d-1), (III-d- 2), (III-i-1) or (III-i-2)) and a second agent (e.g., an anti-cancer agent, anti- inflammatory agent, anti-fibrotic agent, or liver therapeutic).
- a second agent e.g., an anti-cancer agent, anti- inflammatory agent, anti-fibrotic agent, or liver therapeutic.
- a conjugate described herein (which is capable of forming a particle) or the BASP particle is a nanoparticle, from about 5 nm to about 100 nm, about 5 nm to about 75 nm, about 5 to about 50 nm, or about 5 to about 30 nm in size.
- the conjugate or BASP particle comprises telmisartan-linked macromonomers, e.g., as described in Formula (III-e) or (III-j), and is present in a particle from about 5 to about 30 nm in size.
- the conjugate or BASP particle comprises paricalcitol-linked monomers, e.g., as described in Formulas (III-d-1), (III-d-2), (III-i-1) or (III-i-2), and is present in a particle from about 5 to about 30 nm in size.
- the conjugate or BASP particle is homogenous and comprises macromonomers of the same type. In another embodiment, the conjugate or BASP particle is heterogeneous and comprises macromonomers of different types. In some embodiments, the conjugate or BASP particle comprises telmisartan-linked macromonomers, e.g., as described in Formula (III-e) or (III-j), and macromonomers linked to a diagnostic moiety (e.g., a Cy3, Cy5, or Cy7.5 dye).
- a diagnostic moiety e.g., a Cy3, Cy5, or Cy7.5 dye
- the conjugate or BASP particle is used for a diagnostic purpose, e.g., to identify the location of a particular structure (e.g., a tumor or cancer cell) within a subject.
- the agent can be a diagnostic agent.
- the diagnostic agent may comprise a fluorescent molecule, a metal chelate, a contrast agent, a radionuclide, or a positron emission tomography (PET) imaging agent, an infrared imaging agent, a near-IR imaging agent, a computer assisted tomography (CAT) imaging agent, a photon emission computerized tomography imaging agent, an X-ray imaging agent, or a magnetic resonance imaging (MRI) agent.
- PET positron emission tomography
- CAT computer assisted tomography
- MRI magnetic resonance imaging
- the diagnostic agent is a fluorescent molecule.
- the fluorescent molecule comprises an acridine dye, a cyanine dye, a rhodamine dye, a BODIPY dye, a fluorescein dye, a dansyl dye, an Alexa dye, an atto dye, a quantum dot, or a fluorescent protein.
- the fluorescent molecule is a cyanine dye (e.g., Cy3, Cy 3.5, Cy5, Cy5.5, Cy7, or Cy7.5).
- the diagnostic agent is an MRI agent (e.g., a contrast agent).
- MRI agents e.g., a contrast agent
- suitable materials for use as MRI agents include gadolinium chelates, as well as iron, magnesium, manganese, copper, and chromium.
- the diagnostic agent is a CAT imaging agent or an X- ray imaging agent.
- materials useful for CAT and X-ray imaging include iodine-based materials.
- the diagnostic agent is a PET imaging agent.
- suitable PET imaging agents include compounds and compositions comprising the positron emitting radioisotopes 18 F, 15 O, 13 N, 11 C, 82 Rb, 64 Cu, and 68 Ga, e.g., fludeoxyglucose ( 18 F-FDG), 68 Ga-DOTA-psuedopeptides (e.g., 68 Ga-DOTA-TOC), 11 C-metomidate, 11 C-acetate, 11 C-methionine, 11 C-choline, 18 F-fluciclovine, 18 F- fluorocholine, 18 F-fluorodeoxysorbitol, 18 F-3’-fluoro-3’-deoxythymidine, 11 C- raclopride, and 18 F-desmethoxyfallypride.
- the diagnostic agent is a near-IR imaging agent.
- near-IR imaging agents examples include Pz 247, DyLight 750, DyLight 800, cyanine dyes (e.g., Cy5, Cy5.5, Cy7), AlexaFluor 680, AlexaFluor 750, IRDye 680, IRDye 800CW, and Kodak X-SIGHT dyes.
- the agent can be a radionuclide, e.g., for use as a therapeutic, diagnostic, or prognostic agents.
- a radionuclide e.g., for use as a therapeutic, diagnostic, or prognostic agents.
- gamma- emitters, positron-emitters, and X-ray emitters are suitable for diagnostic and/or therapy, while beta emitters and alpha-emitters may also be used for therapy.
- Suitable radionuclides for forming use with various embodiments of the present invention include, but are not limited to, 123 I, 125 I, 130 I, 131 I, 133 I, 135 I, 47 Sc, 72 As, 72 Sc, 90 Y, 88 Y, 97 Ru, 100 Pd, 101m Rh, 119 Sb, 128 Ba, 197 Hg, 211 At, 212 Bi, 212 Pb, 109 Pd, 111 In, 67 Ga, 68 Ga, 67 Cu, 75 Br, 77 Br, 99m Tc, 14 C, 13 N, 15 O, 32 P, 33 P, or 18 F.
- the conjugate or BASP particle comprises: i) a diagnostic agent; (ii) a carbocyclyl or heterocyclyl moiety; (iii) a triazole moiety; (iv) a heteroalkyl moiety; and (v) a cleavable linker (e.g., a tissue microenvironment cleavable linker).
- the diagnostic agent is a diagnostic agent described herein (e.g., a fluorescent molecule, a metal chelate, a contrast agent, a radionuclide, or a positron emission tomography (PET) imaging agent, an infrared imaging agent, a near-IR imaging agent, a computer assisted tomography (CAT) imaging agent, a photon emission computerized tomography imaging agent, an X-ray imaging agent, or a magnetic resonance imaging (MRI) agent).
- PET positron emission tomography
- the diagnostic agent is a fluorescent molecule.
- the diagnostic agent is a cyanine dye, e.g., Cy3, Cy 3.5, Cy5, Cy5.5, Cy7, or Cy7.5, e.g., as described herein.
- the diagnostic agent is non-covalently bound to the conjugate or BASP particle (e.g., the diagnostic agent is associated with the conjugate or BASP particle through ionic bonds or hydrophobic interactions). In some embodiments, the diagnostic agent is covalently bound to the conjugate or BASP particle through a cleavable linker (e.g., a cleavable linker described herein). In some embodiments, the diagnostic agent is covalently bound to the conjugate or BASP particle through a non-cleavable linker.
- exemplary diagnostic agents in the BASP- compositions include, but are not limited to, one or more of the diagnostic agents listed in, in Key, J. and Leary, J. Int J Nanomedicine (2014) 9:711-726; Rizzo, L et al Curr Opin Biotechnol (2013) 24:1159-1166; Andresen, T.L.“Liposomes for positron emission tomography (PET) imaging, radiotherapy, and theranostics”
- the BRUSH-composition or BASP-composition can also include a targeting moiety, e.g., a targeting moiety that is specific to a cell type or tissue.
- a targeting moiety e.g., a targeting moiety that is specific to a cell type or tissue.
- PEG polyethylene glycol
- targeting moieties such as ligands, cell surface receptors, glycoproteins, vitamins (e.g., riboflavin), aptamers and monoclonal antibodies, can also be used.
- the targeting moieties can include the entire protein or fragments thereof. Targeting mechanisms generally require that the targeting agents be positioned on the surface of the conjugate or BASP particle in such a manner that the targeting moiety is available for interaction with the target, for example, a cell surface receptor.
- a targeting ligand can be selected from the group consisting of peptides, polypeptides, proteins, enzymes, peptidomimetics, glycoproteins, antibodies
- nucleosides nucleotides, nucleoside and nucleotide analogues
- nucleic acids monosaccharides, disaccharides, trisaccharides, oligosaccharides, polysaccharides, lipopolysaccharides, vitamins, steroids, hormones, cofactors, receptors, receptor ligands, and analogs and derivatives thereof.
- Non-limiting examples of antibodies and other suitable targeting moieties include those that target tumor/cancer-associated antigens, e.g., tumor targeting ligands or antibodies against tumor antigens; antigens that are differentially expressed on inflamed tissue (e.g., EGFR, ICAM-1 VCAM-1); antigens that are differentially expressed during cell maturation or antigens that are expressed on diseased tissues, pathogens or bacteria (e.g., sugar moieties).
- tumor targeting ligands or antibodies against tumor antigens e.g., tumor targeting ligands or antibodies against tumor antigens
- antigens that are differentially expressed on inflamed tissue e.g., EGFR, ICAM-1 VCAM-1
- antigens that are differentially expressed during cell maturation or antigens that are expressed on diseased tissues, pathogens or bacteria e.g., sugar moieties.
- Tumor-antigens include Melan-A/MART-1, Dipeptidyl peptidase IV (DPPIV), adenosine deaminase-binding protein (ADAbp), cyclophilin b, Colorectal associated antigen (CRC)—C017-1A/GA733, Carcinoembryonic Antigen (CEA) and its immunogenic epitopes CAP-1 and CAP-2, etv6, aml1, Prostate Specific Antigen (PSA) and its immunogenic epitopes PSA-1, PSA-2, and PSA-3, prostate-specific membrane antigen (PSMA), T-cell receptor/CD3-zeta chain, MAGE-family of tumor antigens (e.g., MAGE-A1, MAGE-A2, MAGE-A3, MAGE-A4, MAGE-A5, MAGE- A6, MAGE-A7, MAGE-A8, MAGE-A9, MAGE-A10, MAGE-A11, MAGE-A12, MAGE-Xp
- the targeting moieties include peptides that comprise Arg- Gly-Asp motifs (or RGD peptides) that target integrin present on angiogenic tumor vasculature.
- the targeting moiety includes a nucleic acid.
- the targeting moiety can include an aptamer, i.e., a nucleic acid able to specifically bind a specific target molecule, such as a biological moiety.
- aptamers include RNA aptamers and DNA aptamers.
- the size of the aptamer may be at least about 5 kDa, at least about 10 kDa, at least about 15 kDa, or at least about 20 kDa.
- the targeting ligand can be selected from the group consisting of polylysine (PLL), poly Laspartic acid, poly L-glutamic acid, styrene- maleic acid anhydride copolymer, poly(L-lactide-co-glycolide) copolymer, divinyl ether-maleic anhydride copolymer, N-(2-hydroxypropyl)methacrylamide copolymer (HMPA), polyethylene glycol (PEG), polyvinyl alcohol (PVA), polyurethane, poly(2- ethylacryllic acid), N-isopropylacrylamide polymers, polyphosphazine,
- PLL polylysine
- poly Laspartic acid poly L-glutamic acid
- styrene- maleic acid anhydride copolymer poly(L-lactide-co-glycolide) copolymer
- divinyl ether-maleic anhydride copolymer divinyl ether-maleic anhydride copo
- the targeting ligand is mannose-6-phosphate.
- Liver-Targeting Moieties are mannose-6-phosphate.
- any conjugate or BASP particle described herein can be targeted to the liver, e.g., targeted to a liver cell.
- Targeting to the liver or to a specific population of liver cells have the following advantages: reduction of off- target, and potentially toxic, side effects, increase amount of drug delivery to the desired site of action or to the desired cells, increase the specificity of the interaction between the drug delivery system and target cells or tissues, and increase the overall efficacy of the drug.
- a targeting moiety or ligand can be coupled, e.g., covalently or non-covalently, to a component of a BASP particle or a conjugate, e.g., a BASP particle or conjugate as described herein.
- the targeting moiety or ligand specifically can bind to a receptor or surface molecule at the surface membrane of the targeted liver cell, and thus deliver the BASP particle or conjugate to the targeted liver cell.
- liver cells examples include hepatocytes, Kupffer cells, endothelial cells, hepatic stellate cells, bile duct epithelial cells, or hepatocellular carcinoma cells, or any combination thereof.
- a targeting moiety or ligand is present, e.g., covalently or non-covalently attached, to a component of the nanoparticle.
- the targeting moiety or ligand specifically binds to a receptor or surface molecule at the surface membrane of the targeted liver cell.
- targeting moieties antibodies or antigen-binding fragments thereof, lectins, proteins, lipoproteins, hormones, charged molecules, mono-, olio-, and polysaccharides, and low molecular weight ligands such as sugars, folic acids, and peptides.
- the targeting moiety can specifically bind or interact with one or more of the following: asialoglycoprotein receptor (ASGP-R), high density lipoprotein receptor (HDL-R), low density lipoprotein receptor (LDL-R), immunoglobulin A receptor (IgA-R), scavenger receptor (class BI), transferrin receptor, bile acid receptor, insulin receptor, glycyrrhizin receptor (GL receptor), and glycyrrhetinic acid receptor (GA receptor).
- ASGP-R asialoglycoprotein receptor
- HDL-R high density lipoprotein receptor
- LDL-R low density lipoprotein receptor
- IgA-R immunoglobulin A receptor
- scavenger receptor class scavenger receptor
- transferrin receptor transferrin receptor
- bile acid receptor insulin receptor
- GL receptor glycyrrhizin receptor
- GFR glycyrrhetinic acid receptor
- galactosamine lactoferrin, lactobionic acid (LA), asialofetuin ligand (AF), soybean- derived SG ligand (e.g., sterylglucoside), glycyrrhizin (GL), glycyrrhetinic acid (GA), or derivatives thereof.
- LA lactoferrin
- AF asialofetuin ligand
- soybean- derived SG ligand e.g., sterylglucoside
- GL glycyrrhizin
- GA glycyrrhetinic acid
- the targeting moiety can specifically bind or interact with one or more of the following: mannose/N-acetylglucosamine receptor, galactose particle receptor, galactose specific receptor, Fc receptor immune complexes and opsonized material, scavenger receptors (Class AI, BI, BII, MARCO, CD36, and macrosialin), low density lipoprotein receptor matrix compounds
- fibronectin fibronectin
- complement receptor C3b and C1q
- LPS receptor ⁇ 2 LPS receptor ⁇ 2
- targeting moieties for Kupffer cells include D- mannose, cetylmannoside, dexamethasone coupled to mannosylated albumin, and charged molecules with a net negative charge, e.g., albumin with modified lysines such that albumin has a net negative charge, or derivatives thereof.
- the targeting moiety can specifically bind or interact with one or more of the following:
- mannose/N-acetyl glucosamine receptor scavenger receptor (Class A1 and A11)
- Fc Receptor immune complexes e.g., hyaluronan, fibronectin, denatured collagen, PIIINP.
- matrix compounds e.g., hyaluronan, fibronectin, denatured collagen, PIIINP.
- the targeting moiety can be any suitable targeting moiety.
- HSCs hepatic stellate cells
- targeting moieties for HSCs include mannose-6-phosphate (M6P), and cyclic peptide moieties that serve as binding domains of cytokines and growth factors that bind to HSCs, or derivatives thereof.
- M6P mannose-6-phosphate
- cyclic peptide moieties that serve as binding domains of cytokines and growth factors that bind to HSCs, or derivatives thereof.
- the targeting moiety can specifically bind or interact with secretin receptor.
- the targeting moiety can specifically bind or interact with asialoglycoprotein receptor (ASGP-R).
- ASGP-R asialoglycoprotein receptor
- targeting moieties for HCC cells include lactosaminated ligands, galactosamine, galactosylated ligands, e.g., chitosan, N-lactosyl- dioleoylphosphatidylethanolamine (Lac-DOPE), or lactobionic acid, or derivatives thereof.
- the BASP particles and/or conjugates target a hepatocyte.
- the liver targeting moiety can be chosen from an agent that specifically binds to, or interacts with, one or more of the following: asialoglycoprotein receptor (ASGP-R), high density lipoprotein receptor (HDL-R), low density lipoprotein receptor (LDL-R), immunoglobulin A receptor (IgA-R), scavenger receptor (class BI), transferrin receptor, bile acid receptor, insulin receptor, glycyrrhizin receptor (GL receptor), and glycyrrhetinic acid receptor (GA receptor).
- ASGP-R asialoglycoprotein receptor
- HDL-R high density lipoprotein receptor
- LDL-R low density lipoprotein receptor
- IgA-R immunoglobulin A receptor
- scavenger receptor class scavenger receptor
- transferrin receptor transferrin receptor
- bile acid receptor insulin receptor
- GL receptor glycyrrh
- liver targeting moieties for hepatocytes include, but are not limited to, ligands containing galactose, N-acetylgalactosamine, galactosamine, lactoferrin, lactobionic acid (LA), asialofetuin ligand (AF), soybean-derived SG ligand (e.g., sterylglucoside), glycyrrhizin (GL), glycyrrhetinic acid (GA), or derivatives thereof.
- LA lactobionic acid
- AF asialofetuin ligand
- soybean-derived SG ligand e.g., sterylglucoside
- GL glycyrrhizin
- GA glycyrrhetinic acid
- Also provided herein are methods for identifying or selecting a subject that is in need of improved delivery and/or efficacy of a therapy e.g., a cancer therapy, or an anti-fibrotic or anti-inflammatory therapy.
- a therapy e.g., a cancer therapy, or an anti-fibrotic or anti-inflammatory therapy.
- the methods described herein can be used to identify or select a subject that would respond to treatment with a BRUSH-composition or BASP-composition, alone or in combination with other therapies described herein, e.g., such that the delivery of an additional therapy, e.g., a cancer or an anti-fibrotic therapy, is improved.
- Said methods are described in, e.g., on pages 111-113 of WO 2016/140714, incorporated herein by reference.
- the method includes identifying the subject as having a desmoplastic disorder (e.g., a cancer or a fibrotic or inflammatory disorder).
- a desmoplastic disorder e.g., a cancer or a fibrotic or inflammatory disorder.
- Methods for identifying the subject as having a desmoplastic disorder, e.g., a cancer or a fibrotic or inflammatory disorder, are known in the art.
- Such methods include detection of desmoplasia, e.g., fibrosis, such as an increase in the level or production of extracellular matrix components, e.g., collagen, or hyaluronic acid; increased angiotensin II (AngII) type-1 receptor (AT1) signaling; and/or increased expression, production, and/or secretion of pro-inflammatory cytokines, e.g., interleukin-1b (IL- 1b).
- desmoplasia e.g., fibrosis
- extracellular matrix components e.g., collagen, or hyaluronic acid
- AngII angiotensin II
- AT1 angiotensin II type-1 receptor
- pro-inflammatory cytokines e.g., interleukin-1b (IL- 1b).
- the subject is, or is identified as being, overweight or obese, and has a fibrotic or desmoplastic tumor, e.g., a tumor having one or more of: limited tumor perfusion, compressed blood vessels, high interstitial fluid pressure (IFPs), increased hypoxia, or fibrotic tumor interstitium.
- a fibrotic or desmoplastic tumor e.g., a tumor having one or more of: limited tumor perfusion, compressed blood vessels, high interstitial fluid pressure (IFPs), increased hypoxia, or fibrotic tumor interstitium.
- the subject is overweight or obese, and has a tumor having (e.g., elevated levels of) extracellular matrix components, such as fibers (e.g., collagen, procollagen), fibroblasts (e.g., elevated levels of cancer associated fibroblasts (CAFs) or increased activity of CAFs) and/or polysaccharides (e.g., glycosaminoglycans such as hyaluronan or hyaluronic acid).
- extracellular matrix components such as fibers (e.g., collagen, procollagen), fibroblasts (e.g., elevated levels of cancer associated fibroblasts (CAFs) or increased activity of CAFs) and/or polysaccharides (e.g., glycosaminoglycans such as hyaluronan or hyaluronic acid).
- fibers e.g., collagen, procollagen
- fibroblasts e.g., elevated levels of cancer associated fibroblasts (CAFs) or
- the method further includes evaluating, e.g., acquiring a value for, a weight/metabolic-related parameter, for the subject.
- a weight/metabolic-related parameter includes body mass index (BMI).
- the subject is, or is identified as being, overweight or obese.
- Assessment of overweight and obesity can be determined by the classification of body mass index (BMI) as defined by“Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults” from the National Institutes of Health.
- Body mass index is obtained by dividing a subject’s weight, e.g., in kilograms (kg) by the square of the subject’s height, e.g., in meters (m).
- Subjects with a BMI 18.5 to 24.9 are typically classified as normal weight, while subjects with a BMI 25.0 to 29.9 are classified as overweight.
- the subject is overweight, e.g., the subject has a BMI of greater than or equal to 25.0 but less than or equal to 29.9.
- the subject is, or is identified as being, obese, e.g., the subject has a BMI of greater than or equal to 30, e.g., greater than 30, greater than 35, greater than 40, greater than 45, or greater than 50.
- the subject is, or is identified as being, overweight or obese, and has a fibrotic or a hyperproliferative cancerous condition described herein. In an embodiment, the subject is, or is identified as being, overweight or obese and has a fibrotic disorder described herein. In an embodiment, the subject is, or is identified as being, overweight or obese and has an inflammatory disorder described herein.
- the macromonomers, conjugates and BASP particles described herein may be synthesized through the use of specialized reagents and other starting materials as described below.
- Crosslinkers
- the BASP particles and conjugates are prepared through the linkage of discrete macromonomer subunits with a multifunctional crosslinker to generate a nanostructure (BASP particle or BRUSH conjugate).
- the macromonomer subunit and crosslinkers may be joined in a number of ways, such as through graft-through ring-opening metathesis polymerization (ROMP).
- the crosslinker is labile. In some embodiments, the crosslinker is labile at physiological conditions (e.g., biodegradable, e.g., at physiological pH).
- the crosslinker comprises a bis-norbornene derivative. In some embodiments, the crosslinker comprises two norbornene derivatives, separated by an alkylene, heteroalkylene, arylene, or heteroarylene moiety. In some embodiments, the crosslinker comprises a disulfide bond. In some embodiments, the crosslinker comprises an anhydride moiety or ester moiety.
- the crosslinker is a compound of Formula (VI):
- each of Z 1 and Z 2 is independently C 1 -C 6 alkylene or (C 0 -C 6 alkylene)-aryl-(C 0 -C 6 alkylene) (e.g., -CH 2 -, -CH 2 CH 2 CH 2 -, or phenyl).
- each of W 1 and W 2 is independently alkylene or C 1 -C 12 heteroalkylene, wherein each alkylene and heteroalkylene is optionally substituted with 1-6 R 32 .
- R 32 is oxo.
- each of W 1 and W 2 is independently -CH 2 -, -OC(O)CH 2 CH 2 -, -CH 2 OC(O)CH 2 CH 2 -, or–C(O)-.
- G is–O– or–S-S–.
- the com ound is a com ound of Formula (VII-a):
- each of Z 1 and Z 2 is independently CH 2 -, -CH 2 CH 2 CH 2 -, or phenyl; each of W 1 and W 2 is independently -CH 2 -, -OC(O)CH 2 CH 2 -, -CH 2 OC(O)CH 2 CH 2 -, or– C(O)-; and G is–O– or–S-S–.
- Exemplary crosslinkers may be cleavable or susceptible to degradation under certain parameters.
- the crosslinker may be acid labile or sensitive to a reducing agent.
- the crosslinker may be substantially insoluble in water.
- the length or size of the crosslinker (e.g., the length or size of Z 1 , W 1 , G, W 2 , or Z 2 ) is selected to optimize the efficiency of conjugate and/or particle efficiency.
- the length or size of the crosslinker may be empirically chosen depending on the identity of the macromonomer or other brush arm component in order to yield high polymerization efficiency.
- a macromonomer may be assembled in any number of ways.
- the macromonomer is prepared through a process involving at least two steps.
- an agent e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor
- a targeting moiety e.g., a targeting moiety, or a diagnostic agent
- a diagnostic agent is linked to a PEG- azide or a PEG alkyne.
- the agent-linked PEG azide or agent-linked PEG-alkyne is coupled to a norbornene-containing compound, e.g., through copper(I)-mediated azide-alkyne cycloaddition.
- An agent, a targeting moiety, or a diagnostic agent may be linked to a PEG azide or a PEG-alkyne through a reactive group, e.g., a free hydroxyl group or a free carboxylic acid, on the PEG-azide or PEG-alkyne.
- a reactive group e.g., a free hydroxyl group or a free carboxylic acid
- it may be desirable that the agent, targeting moiety, or diagnostic agent have at least one carboxylic acid and/or hydroxyl group.
- the ARB telmisartan with a carboxylic acid group or the vitamin D analog paricalcitol with a hydroxyl group can be conjugated to a PEG-azide or PEG-alkyne via ester bond formation.
- Exemplary coupling agents used for conjugation include EDC, DIC, DCC, HOAt, HOBt, and PyBOP.
- the conjugation reaction further comprises a base (e.g., TEA, pyridine).
- the conjugation reaction further comprises a catalyst (e.g., DMAP).
- the conjugation reaction is carried out in a solvent (e.g., dichloromethane, acetonitrile, tetrahydrofuran, dimethylformamide, or dimethyl sulfoxide).
- the derivatized PEG-alkyne or derivatized PEG-azide i.e., derivatized with an agent, targeting moiety, or diagnostic agent
- macromonomer synthesis may be completed through a second step involving conjugation to a norbornene-containing compound.
- One method for achieving said conjugation may entail copper(I)-mediated azide-alkyne cycloaddition, in which an azide is coupled with an alkyne in the presence of a copper catalyst.
- the copper catalyst is copper(I) acetate.
- the cycloaddition reaction is carried out in a solvent (e.g., dichloromethane).
- the macromonomer is purified by precipitation, trituration,
- the macromonomer comprises a mixture of regioisomers. In some embodiments, the macromonomer is comprised of a single regioisomer.
- Purified macromonomers may be characterized by a variety of methods including NMR, mass spectrometry, and chromatography (e.g., gel-permeation chromatography). Methods of Making Conjugates and BASP Particles
- Conjugates are distinguished from BASP particles in several respects, such as size and hydrodynamic diameter.
- the conjugate refers to the product of a first round of polymerization and has an average molecular weight of about 10 kDa to about 100 kDa and an average hydrodynamic diameter of about 1 nm to about 10 nm.
- the BASP particle refers to the product of a second or subsequent round of polymerization and has an average molecular weight of about 100 kDa to about 1,000 kDa and an average hydrodynamic diameter of about 10 nm to about 100 nm.
- Conjugates and BASP particles described herein may be prepared in any number of methods, including via polymerization reactions.
- the polymerization is ring opening metathesis polymerization (ROMP).
- ROMP is an olefin metathesis chain-growth polymerization reaction that is driven by the relief of ring strain in cyclic olefins (e.g., norbornenes).
- the ROMP may comprise a number of different approaches, including“arm-first” ROMP,“brush-first” ROMP,“graft-to” ROMP,“graft-from” ROMP,“graft-through” ROMP, or combinations thereof.
- ROMP requires use of a metal catalyst.
- Metal catalysts for use in ROMP reactions may comprise ruthenium, molybdenum, or tungsten.
- the metal catalyst is a Grubbs’ catalyst, e.g., first-generation Grubbs’ catalyst, second-generation Grubbs’ catalyst, Hoveyda-Grubbs’ catalyst, or third-generation Grubbs’ catalyst.
- Exemplary catalysts for use in ROMP are described in Grubbs et al., Acc. Chem. Res.1995, 28, 446–452; U.S. Pat. No.5,811,515;
- the ROMP catalyst is a Grubbs catalyst selected from:
- R cyclohexyl (Cy); phenyl (Ph); benzyl (Bn)
- the ROMP catalyst is a Grubbs-Hoveyda catalyst.
- the Grubbs-Hoveyda catalyst is selected from:
- the ROMP catalyst is selected from:
- the ROMP catalyst is selected from:
- ROMP is performed in the absence of a metal catalyst. Polymerization reactions to produce conjugates and BASP particles, e.g., via ROMP, may take place in an oxygen-free and/or water-free environment.
- the ROMP is conducted in one or more aprotic solvents.
- aprotic solvent refers to a non-nucleophilic solvent having a boiling point range above ambient temperature, preferably from about 25 oC to about 190 oC at atmospheric pressure. In some embodiments, the aprotic solvent has a boiling point from about 80 oC to about 160 oC at atmospheric pressure. In certain embodiments, the aprotic solvent has a boiling point from about 80 oC to about 150 oC at
- solvents examples include methylene chloride, acetonitrile, toluene, DMF, diglyme, THF, and DMSO.
- Conjugates and BASP particles generated by ROMP may be terminated or end-capped through the use of a chain-transfer agent.
- a chain-transfer agent may be useful to both to modify the properties of the conjugate or BASP particle termini, and in some embodiments, may also modify the Grubbs’ catalyst. Modifications of the terminus of a conjugate or BASP particle may include increasing or decreasing the hydrophobicity of the BASP particle or conjugate core, increasing or decreasing the degradation rate of the BASP particle or conjugate, and increasing or decreasing the density of the BASP particle or conjugate core.
- Modifications of the Grubbs’ catalyst may include increasing or decreasing catalyst hydrophobicity, increasing or decreasing the transport of the catalyst out of the BASP particle or conjugate core (i.e., thereby altering the ability to remove the coordinated metal), increasing or decreasing the oxidation state of the metal, increasing or decreasing the affinity of the metal for metal scavenging agents, and increasing or decreasing the solubility of the metal in a solvent.
- the chain- transfer agent comprises a reactive olefin.
- Exemplary chain-transfer agents include ethyl vinyl ether (i.e., ethoxyethene), (Z)-oct-4-ene, (Z)-but-2-ene 1,4-diol, (Z)-4,4'- (but-2-ene-1,4-diylbis(oxy))bis(4-oxobutanoic acid), and (Z)-O,O'-(but-2-ene-1,4- diyl) bis(2-(2-(2-methoxyethoxy)ethoxy)ethyl) disuccinate.
- exemplary chain-transfer agents are shown below:
- removal of a contaminant comprises a ruthenium removal procedure. Removal of residual ruthenium from the ROMP reaction may be performed using a compound comprising an amine, phosphine, or thiol.
- a compound used for the removal of a contaminant is N,N-dimethyltryptamine, cysteine, triaminetetraacetate (sodium salt), tris(hydroxymethyl)phosphine, 2-mercaptonicotinic acid, N-acetyl-L-cysteine, imidazole, diethylphenylazothioformamide, lead tetra-acetate, hydrogen peroxide, triphenylphosphine oxide, isocyanide salt, di(ethylene glycol) vinyl ether, acetonitrile, dimethyl sulfoxide, or any reagent described in Vougioukalakis, G. C. Chem. A Eur.
- Conjugates and BASP particles described herein may be prepared through ROMP, e.g., by polymerizing a macromonomer subunit with a cross-linker (e.g., a macromonomer and/or cross-linker described herein).
- a cross-linker e.g., a macromonomer and/or cross-linker described herein.
- the method of making a conjugate or BASP particle comprises:
- X is an agent (e.g., an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor);
- t is an integer between 1 and 10, inclusive;
- z is an integer between 1 and 200, inclusive;
- a metal catalyst e.g., a Grubbs’ catalyst
- each of Z 1 and Z 2 is independently CH 2 -, -CH 2 CH 2 CH 2 -, or phenyl; each of W 1 and W 2 is independently -CH 2 -, -OC(O)CH 2 CH 2 -, -CH 2 OC(O)CH 2 CH 2 -, or– C(O)-; and G is–O– or–S-S– to form a third solution;
- a chain-transfer agent e.g., a chain-transfer agent comprising an olefin
- the amount or concentration of the macromonomer (e.g., a compound of Formula (V-c)) and the amount or concentration of the cross- linker (e.g., a compound of Formula (VII-a)) are present in the reaction mixture in a defined ratio. In some embodiments, the amount or concentration of the macromonomer (e.g., a compound of Formula (V-c)) and the amount or concentration of the cross- linker (e.g., a compound of Formula (VII-a)) are present in the reaction mixture in a defined ratio. In some embodiments, the amount or concentration of the macromonomer (e.g., a compound of Formula (V-c)) and the amount or concentration of the cross- linker (e.g., a compound of Formula (VII-a)) are present in the reaction mixture in a defined ratio. In some embodiments, the amount or concentration of the cross- linker (e.g., a compound of Formula (VII-a)) are present in the reaction mixture in
- m is an integer from 1 to 20
- N is an integer from 1 to 20
- the ratio of the amount or concentration of the macromonomer, cross-linker, and catalyst is m:N:1, wherein m is an integer from 1 to 20 and N is an integer from 1 to 20.
- m is an integer from 3 to 12. In some embodiments, m is 1. In some embodiments, m is 2. In some embodiments, m is 3. In some
- m is 4. In some embodiments, m is 5. In some embodiments, m is 6. In some embodiments, m is 7. In some embodiments, m is 8. In some embodiments, m is 9. In some embodiments, m is 10. In some embodiments, m is 11. In some
- m is 12.
- N is an integer from 1 to 10. In some embodiments, N is 1. In some embodiments, N is 2. In some embodiments, N is 3. In some
- N is 4. In some embodiments, N is 5. In some embodiments, N is 6. In some embodiments, N is 7. In some embodiments, N is 8. In some embodiments, N is 9. In some embodiments, N is 10. Pharmaceutical Compositions, Formulations and Kits
- the BRUSH-compositions and BASP-compositions described herein may comprise a bound therapeutic agent that is preferentially labile or preferentially cleavable in a tissue microenvironment. In some embodiments, this allows for preferential release of the therapeutic agent in the tissue microenvironment, e.g., compared to the systemic release of the therapeutic agent as a free form, thus allowing for a lower dosage of the agent in the conjugate or BASP particle.
- the BRUSH- compositions and BASP-compositions described herein can be incorporated into a variety of formulations for administration. In certain embodiments, the BRUSH- compositions and BASP-compositions further comprise pharmaceutically acceptable carriers or pharmaceutically acceptable diluents. In certain embodiments, the
- BRUSH-compositions and BASP-compositions are in any one of semi-solid, liquid, and gaseous forms; such as capsules, powders, granules, gels, slurries, ointments, solutions, suppositories, injections, inhalants, and aerosols.
- the BRUSH-compositions and BASP-compositions are administered by oral, buccal, rectal, parenteral, intraperitoneal, intradermal, transdermal, or intratracheal administration.
- the BRUSH-compositions and BASP- compositions are administered locally.
- the BRUSH- compositions and BASP-compositions are administered systemically.
- the BRUSH-compositions and BASP-compositions are in a sustained release form.
- BRUSH-compositions and BASP-compositions can be formulated with common excipients, diluents or carriers, and compressed into tablets, or formulated as elixirs or solutions for convenient oral administration, or
- compositions can be administered transdermally, and can be formulated as sustained release dosage forms and the like.
- Compositions can be administered alone, in combination with each other, or they can be used in combination with other known compounds (discussed herein).
- compositions described herein can be manufactured in a manner that is known to those of skill in the art, e.g., by mixing, dissolving, granulating, dragee-making, levigating, emulsifying,
- compositions can be formulated for parenteral administration by injection, e.g., by bolus injection or continuous infusion.
- Formulations for injection can be presented in unit dosage form, e.g., in ampules or in multidose containers, with an added preservative.
- the compositions can take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and can contain formulator agents such as suspending, stabilizing and/or dispersing agents.
- sterile injectable aqueous or oleaginous suspensions can be formulated according to the known art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation can be a sterile injectable solution, suspension, or emulsion in a nontoxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3-butanediol.
- acceptable vehicles and solvents that can be employed are water, Ringer’s solution, U.S.P. and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil can be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid are used in the preparation of injectables.
- the injectable formulations can be sterilized, for example, by filtration through a bacterial-retaining filter, or by incorporating sterilizing agents in the form of sterile solid compositions which can be dissolved or dispersed in sterile water or other sterile injectable medium prior to use.
- compositions described herein include short needle devices such as those described in U.S. Patents 4,886,499; 5,190,521; 5,328,483; 5,527,288; 4,270,537; 5,015,235;
- Intradermal compositions can be administered by devices which limit the effective penetration length of a needle into the skin, such as those described in PCT publication WO 99/34850 and functional equivalents thereof. Jet injection devices which deliver liquid vaccines to the dermis via a liquid jet injector and/or via a needle which pierces the stratum corneum and produces a jet which reaches the dermis are suitable. Jet injection devices are described, for example, in U.S.
- Ballistic powder/particle delivery devices which use compressed gas to accelerate the compound in powder form through the outer layers of the skin to the dermis are suitable.
- conventional syringes can be used in the classical Mantoux method of intradermal administration.
- compositions can also be formulated as a depot preparation.
- Such long acting formulations can be administered by implantation (for example subcutaneously or intramuscularly) or by intramuscular injection.
- the compounds can be formulated with suitable polymeric or hydrophobic materials (for example as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
- Lipid particles e.g., liposomes
- emulsions are known examples of delivery vehicles or carriers for hydrophobic drugs. Long-circulating, e.g., stealth, liposomes can be employed. Such liposomes are generally described in U.S. Pat. No. 5,013,556.
- the compounds of the present invention can also be administered by controlled release means and/or delivery devices such as those described in U.S. Pat. Nos.3,845,770; 3,916,899; 3,536,809; 3,598,123; and 4,008,719.
- compositions can be formulated by combining with pharmaceutically acceptable carriers that are known in the art.
- Such carriers enable the compounds to be formulated as pills, capsules, emulsions, lipophilic and hydrophilic suspensions, liquids, gels, syrups, slurries, suspensions and the like, for oral ingestion by a subject (e.g., patient) to be treated.
- Pharmaceutical preparations for oral use can be obtained by mixing the compositions with an excipient and processing the mixture of granules, after adding suitable auxiliaries, if desired, to obtain tablets or dragee cores.
- Suitable excipients are, in particular, fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol; cellulose preparations such as, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl-cellulose, sodium carboxymethylcellulose, and/or polyvinylpyrrolidone (PVP).
- fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol
- cellulose preparations such as, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl-cellulose, sodium carboxymethylcellulose, and/or polyvinylpyrrolidone (PVP).
- PVP polyvinylpyrrolidone
- compositions for use according to the present invention are conveniently delivered in the form of an aerosol spray presentation from pressurized packs or a nebulizer, with the use of a suitable propellant, e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas, or from propellant- free, dry-powder inhalers.
- a suitable propellant e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas
- propellant- free, dry-powder inhalers e.g., a suitable propellant
- the dosage unit can be determined by providing a valve to deliver a metered amount.
- Capsules and cartridges of, e.g., gelatin for use in an inhaler or insufflator can be formulated containing a powder mix of the compound and a suitable powder base such as lactose or starch.
- the compositions can also be formulated in rectal compositions such as suppositories or retention enemas, e.g., containing conventional suppository bases such as cocoa butter, carbowaxes, polyethylene glycols or other glycerides, all of which melt at body temperature, yet are solidified at room temperature.
- compositions suitable for use in the present invention include compositions wherein the active ingredients are contained in a therapeutically effective amount.
- the amount of composition administered will, of course, be dependent on the subject being treated, on the subject's weight, the severity of the affliction, the manner of administration and the judgment of the prescribing physician. Determination of an effective amount is well within the capability of those skilled in the art, especially in light of the detailed disclosure provided herein.
- the BRUSH-compositions and BASP-compositions can be administered daily, every other day, three times a week, twice a week, weekly, or bi-weekly.
- the dosing schedule can include a“drug holiday,” i.e., the drug can be administered for two weeks on, one week off, or three weeks on, one week off, or four weeks on, one week off, etc., or continuously, without a drug holiday.
- the compounds can be administered orally, intravenously, intraperitoneally, topically, transdermally, intramuscularly, subcutaneously, intranasally, sublingually, or by any other route.
- the BRUSH-composition or BASP-composition (alone or in combination with the therapeutic agents described herein) is formulated for oral, subcutaneous, intravenous or intraperitoneal administration.
- Substantially continuous administration of the BRUSH-composition or BASP- composition can cause a greater reduction in collagen content and/or tumor size than single or pulsatile administration of the agent.
- the BRUSH-composition or BASP-composition (alone or in combination with the therapeutic agents described herein) is administered substantially continuously over a pre-determined period of, or at least 15, 30, 45 minutes; a period of, or at least, 1, 5, 10, 24 hours; a period of, or at least, 2, 5, 10, 14 days; a period of, or at least, 3, 4, 5, 6, 7, 8 weeks; a period of, or at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 months; a period of, or at least, 1, 2, 3, 4, 5 years, or longer.
- the delivery method can be optimized such that a BASP-composition dose as described herein (alone or in combination) is administered and/or maintained in the subject for a pre-determined period (e.g., a period as described herein).
- the BRUSH-composition or BASP-composition can be in a controlled- or extended release formulation, dosage form, or device.
- exemplary formulations and devices for controlled or extended release are known in the art.
- formulations containing polymer matrices, such as hydroxypropylmethyl cellulose, gels, osmotic systems, liposomes and combination thereof can be used to provide the desired release kinetics.
- the BRUSH-composition or BASP-composition (alone or in combination with the therapeutic agents described herein) is administered via an implantable infusion device, e.g., a pump (e.g., a subcutaneous pump), an implant or a depot.
- Implantable infusion devices typically include a housing containing a liquid reservoir which can be filled transcutaneously by a hypodermic needle penetrating a fill port septum. The medication reservoir is generally coupled via an internal flow path to a device outlet port for delivering the liquid through a catheter to a subject body site.
- Typical infusion devices also include a controller and a fluid transfer mechanism, such as a pump or a valve, for moving the liquid from the reservoir through the internal flow path to the device's outlet port.
- kits e.g., pharmaceutical packs.
- the inventive kits may be useful for treating a proliferative disease (e.g., cancer (e.g., leukemia, melanoma, multiple myeloma), benign neoplasm, angiogenesis, inflammatory disease, autoinflammatory disease, or autoimmune disease).
- a proliferative disease e.g., cancer (e.g., leukemia, melanoma, multiple myeloma), benign neoplasm, angiogenesis, inflammatory disease, autoinflammatory disease, or autoimmune disease).
- the kits provided may comprise the BRUSH-compositions or BASP-compositions described herein, or a pharmaceutical composition thereof, and a container (e.g., a vial, ampule, bottle, syringe, and/or dispenser package, or other suitable container).
- kits may optionally further include a second container comprising a pharmaceutical excipient for dilution or suspension of a BRUSH- composition or BASP-composition.
- a second container comprising a pharmaceutical excipient for dilution or suspension of a BRUSH- composition or BASP-composition.
- the BRUSH-composition or BASP-composition provided in the container and the second container are combined to form one unit dosage form.
- Methods described herein comprise administration to a subject of a BRUSH- composition or BASP-composition as a single agent or in combination with one or more other therapeutic agents or modalities (e.g., as a particle or free agent) for treating or preventing a disorder, e.g., a hyperproliferative disorder (e.g., a cancer) or a fibrotic or an inflammatory condition or disorder described herein.
- a disorder e.g., a hyperproliferative disorder (e.g., a cancer) or a fibrotic or an inflammatory condition or disorder described herein.
- the disorder is chosen from one or more of a hyperproliferative disorder, a cancer (e.g., a solid or fibrotic cancer), a fibrotic disorder or condition, an inflammatory disorder or condition, or an autoimmune disorder.
- chemotherapy As used herein,“chemotherapy,”“chemotherapeutic,”“chemotherapeutic agent” and“anti-cancer agent” are synonymous terms.
- the terms“treat,”“treating” and“treatment” refer to both therapeutic treatment and prophylactic or preventative measures, wherein the object is to prevent or slow down (lessen) an undesired physiological change or disorder, such as the development or spread of cancer.
- Beneficial or desired clinical results include, but are not limited to, alleviation of symptoms, diminishment of extent of disease, stabilized (i.e., not worsening) state of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, and remission (whether partial or total), whether detectable or undetectable.“Treatment” can also mean prolonging survival as compared to expected survival if not receiving treatment.
- Those in need of treatment include those already with the condition or disorder as well as those prone to have the condition or disorder or those in which the condition or disorder is to be prevented.
- Treating” a tumor typically refers to one or more of the following:
- therapeutic treatment can refer to inhibiting or reducing tumor growth or progression after administration in accordance with the methods or administration with the BRUSH- compositions or BASP-compositions described herein.
- tumor growth or progression is inhibited or reduced by at least about 10%, at least about 15%, at least about 20%, at least about 30%, at least about 40%, or at least about 50%, after treatment.
- tumor growth or progression is inhibited or reduced by more than 50%, e.g., at least about 60%, or at least about 70%, after treatment.
- tumor growth or progression is inhibited or reduced by at least about 80%, at least about 90% or greater, as compared to a control (e.g. in the absence of the BASP-composition described herein).
- the terms“prevent,”“preventing” and“prevention” contemplate an action that occurs before a subject begins to suffer from the regrowth of the cancer and/or which inhibits or reduces the severity of the cancer.
- a“therapeutically effective amount” of a BRUSH-composition or BASP-composition is an amount sufficient to provide a therapeutic benefit in the treatment of the disorder (e.g., cancer), or to delay or minimize one or more symptoms associated with the disorder (e.g., cancer).
- a therapeutically effective amount of a BRUSH-composition or BASP-composition means an amount of therapeutic agent, alone or in combination with other therapeutic agents, which provides a therapeutic benefit in the treatment or management of the disorder.
- the term“therapeutically effective amount” can encompass an amount that improves overall therapy, reduces or avoids symptoms or causes of the disorder (e.g., cancer), or enhances the therapeutic efficacy of another therapeutic agent.
- a“prophylactically effective amount” of a BRUSH-composition or BASP-composition is an amount sufficient to prevent a disorder (e.g., regrowth of the cancer, or one or more symptoms associated with the cancer, or prevent its recurrence).
- a prophylactically effective amount of a BRUSH-composition or BASP-composition means an amount of the compound, alone or in combination with other therapeutic agents, which provides a prophylactic benefit in the prevention of the disorder.
- the term“prophylactically effective amount” can encompass an amount that improves overall prophylaxis or enhances the prophylactic efficacy of another prophylactic agent.
- A“subject” includes, but is not limited to, humans (i.e., a male or female of any age group, e.g., a pediatric subject (e.g., infant, child, adolescent) or adult subject (e.g., young adult, middle–aged adult, or senior adult)) and/or other non–human animals, for example, mammals (e.g., primates (e.g., cynomolgus monkeys, rhesus monkeys). In certain embodiments, the animal is a mammal. The animal may be a male or female and at any stage of development. A non–human animal may be a transgenic or genetically engineered animal.
- administer refers to implanting, absorbing, ingesting, injecting, inhaling, or otherwise introducing a BRUSH-composition or BASP-composition, or a pharmaceutical composition thereof, or a device incorporating the BRUSH-composition or BASP-composition.
- the disorder, e.g., a cancer, treated is an epithelial, a mesenchymal or a hematologic malignancy.
- the cancer treated is a solid tumor (e.g., carcinoid, carcinoma or sarcoma), a soft tissue tumor (e.g., a heme malignancy), and a metastatic lesion, e.g., a metastatic lesion of any of the cancers disclosed herein.
- the cancer treated is a fibrotic or desmoplastic solid tumor, e.g., a tumor having one or more of: limited tumor perfusion, compressed blood vessels, fibrotic tumor interstitium, or increased interstitial fluid pressure.
- the solid tumor is chosen from one or more of pancreatic (e.g., pancreatic adenocarcinoma or pancreatic ductal adenocarcinoma), breast, colon, colorectal, lung (e.g., small cell lung cancer (SCLC) or non-small cell lung cancer (NSCLC)), skin, ovarian, liver cancer, esophageal cancer, endometrial cancer, gastric cancer, head and neck cancer, kidney, or prostate cancer.
- pancreatic e.g., pancreatic adenocarcinoma or pancreatic ductal adenocarcinoma
- lung e.g., small cell lung cancer (SCLC) or non-small cell lung cancer (NSCLC)
- SCLC small cell lung cancer
- NSCLC non-small cell lung cancer
- the cancer treated is a fibrotic or desmoplastic solid tumor, e.g., a tumor having one or more of: limited tumor perfusion, compressed blood vessels, high interstitial fluid pressure (IFPs), or fibrotic tumor interstitium.
- a fibrotic or desmoplastic solid tumor e.g., a tumor having one or more of: limited tumor perfusion, compressed blood vessels, high interstitial fluid pressure (IFPs), or fibrotic tumor interstitium.
- the solid tumor is chosen from one or more of pancreatic (e.g., pancreatic adenocarcinoma (e.g., pancreatic ductal adenocarcinoma (PDA or PDAC)), breast, gastric, colorectal, lung (e.g., small or non-small cell lung cancer), skin, ovarian, prostate, or liver cancer. Additional examples of cancers treated are described herein below.
- pancreatic e.g., pancreatic adenocarcinoma (e.g., pancreatic ductal adenocarcinoma (PDA or PDAC)
- pancreatic adenocarcinoma e.g., pancreatic ductal adenocarcinoma (PDA or PDAC)
- lung e.g., small or non-small cell lung cancer
- skin ovarian, prostate, or liver cancer. Additional examples of cancers treated are described herein below.
- the cancer treated contains (e.g., has elevated levels of) extracellular matrix components, such as fibers (e.g., collagen, procollagen) and/or polysaccharides (e.g., glycosaminoglycans such as hyaluronan or hyaluronic acid).
- extracellular matrix components such as fibers (e.g., collagen, procollagen) and/or polysaccharides (e.g., glycosaminoglycans such as hyaluronan or hyaluronic acid).
- the levels of the extracellular matrix components in the cancer can vary depending on the particular cancer type, the stage of malignancy, and/or in response to cancer therapy. For example, certain cancer may show elevated levels of extracellular matrix components in response to chemotherapy and/or radiation.
- an agent e.g., an ARB, vitamin D analog, or bromodomain inhibitor
- a second agent as a conjugate, BASP particle or free agent
- the cancer or tumor treated is a solid, fibrotic tumor chosen from one or more of pancreatic (e.g., pancreatic adenocarcinoma or pancreatic ductal adenocarcinoma), breast, colorectal, colon, lung (e.g., small or non-small cell lung cancer), skin, ovarian, prostate, cervix, gastrointestinal (e.g., carcinoid or stromal), stomach, head and neck, kidney, brain cancer or liver cancer (e.g. HCC), or a metastatic lesion thereof. Additional examples of cancers treated are described herein below.
- pancreatic e.g., pancreatic adenocarcinoma or pancreatic ductal adenocarcinoma
- lung e.g., small or non-small cell lung cancer
- skin ovarian
- prostate cervix
- gastrointestinal e.g., carcinoid or stromal
- stomach e.g., head and neck
- kidney
- the disorder is fibrotic or desmoplastic solid tumor, e.g., a tumor having one or more of: limited tumor perfusion, compressed blood vessels, high interstitial fluid pressure (IFPs), or fibrotic tumor interstitium.
- the subject has a tumor having (e.g., elevated levels of) extracellular matrix components, such as fibers (e.g., collagen, procollagen) and/or polysaccharides (e.g., glycosaminoglycans such as hyaluronan or hyaluronic acid).
- the levels of the extracellular matrix components in the tumor can vary depending on the particular cancer type, the stage of malignancy, and/or in response to cancer therapy.
- certain tumors may show elevated levels of extracellular matrix components in response to chemotherapy and/or radiation.
- an agent e.g., an ARB, vitamin D analog, or bromodomain inhibitor
- a second agent as a conjugate, BASP particle, or free agent
- hyperproliferative cancerous disease or disorder all neoplastic cell growth and proliferation, whether malignant or benign, including all transformed cells and tissues and all cancerous cells and tissues.
- Hyperproliferative diseases or disorders include, but are not limited to, precancerous lesions, abnormal cell growths, benign tumors, malignant tumors, and“cancer.”
- the terms“cancer,”“tumor” or“tumor tissue” refer to an abnormal mass of tissue that results from excessive cell division, in certain cases tissue comprising cells which express, over-express, or abnormally express a hyperproliferative cell protein.
- a cancer, tumor or tumor tissue comprises“tumor cells” which are neoplastic cells with abnormal growth properties and no useful bodily function. Cancers, tumors, tumor tissue and tumor cells may be benign or malignant.
- a cancer, tumor or tumor tissue may also comprise“tumor-associated non- tumor cells”, e.g., vascular cells which form blood vessels to supply the tumor or tumor tissue. Non-tumor cells may be induced to replicate and develop by tumor cells, for example, the induction of angiogenesis in a tumor or tumor tissue.
- cancer “malignancy” and“tumor” are synonymous terms.
- cancer examples include, but are not limited to, carcinoma, lymphoma, blastoma, sarcoma, and leukemia or lymphoid malignancies. More particular examples of such cancers are noted below and include: squamous cell cancer (e.g.
- lung cancer including small-cell lung cancer, non- small cell lung cancer, adenocarcinoma of the lung and squamous carcinoma of the lung, cancer of the peritoneum, hepatocellular cancer, gastric or stomach cancer including gastrointestinal cancer, pancreatic cancer, glioblastoma, cervical cancer, ovarian cancer, liver cancer, bladder cancer, hepatoma, breast cancer, colon cancer, rectal cancer, colorectal cancer, endometrial cancer or uterine carcinoma, salivary gland carcinoma, kidney or renal cancer, prostate cancer, vulval cancer, thyroid cancer, hepatic carcinoma, anal carcinoma, penile carcinoma, as well as head and neck cancer.
- lung cancer including small-cell lung cancer, non- small cell lung cancer, adenocarcinoma of the lung and squamous carcinoma of the lung, cancer of the peritoneum, hepatocellular cancer, gastric or stomach cancer including gastrointestinal cancer, pancreatic cancer, glioblastoma, cervical cancer, ovarian cancer, liver cancer
- cancer includes primary malignant cells or tumors (e.g., those whose cells have not migrated to sites in the subject's body other than the site of the original malignancy or tumor) and secondary malignant cells or tumors (e.g., those arising from metastasis, the migration of malignant cells or tumor cells to secondary sites that are different from the site of the original tumor).
- primary malignant cells or tumors e.g., those whose cells have not migrated to sites in the subject's body other than the site of the original malignancy or tumor
- secondary malignant cells or tumors e.g., those arising from metastasis, the migration of malignant cells or tumor cells to secondary sites that are different from the site of the original tumor.
- cancers or malignancies include, but are not limited to: Acute Childhood Lymphoblastic Leukemia, Acute Lymphoblastic Leukemia, Acute Lymphocytic Leukemia, Acute Myeloid Leukemia, Adrenocortical Carcinoma, Adult (Primary) Hepatocellular Cancer, Adult (Primary) Liver Cancer, Adult Acute Lymphocytic Leukemia, Adult Acute Myeloid Leukemia, Adult Hodgkin's Disease, Adult Hodgkin's Lymphoma, Adult Lymphocytic Leukemia, Adult Non-Hodgkin's Lymphoma, Adult Primary Liver Cancer, Adult Soft Tissue Sarcoma, AIDS-Related Lymphoma, AIDS-Related Malignancies, Anal Cancer, Astrocytoma, Bile Duct Cancer, Bladder Cancer, Bone Cancer, Brain Stem Glioma, Brain Tumors, Breast Cancer, Cancer of the Renal Pelvis and Ureter, Central Nervous System (
- Lymphocytic Leukemia Chronic Myelogenous Leukemia, Colon Cancer, Cutaneous T-Cell Lymphoma, Endocrine Pancreas Islet Cell Carcinoma, Endometrial Cancer, Ependymoma, Epithelial Cancer, Esophageal Cancer, Ewing's Sarcoma and Related Tumors, Exocrine Pancreatic Cancer, Extracranial Germ Cell Tumor, Extragonadal Germ Cell Tumor, Extrahepatic Bile Duct Cancer, Eye Cancer, Female Breast Cancer, Gaucher's Disease, Gallbladder Cancer, Gastric Cancer, Gastrointestinal Carcinoid Tumor, Gastrointestinal Tumors, Germ Cell Tumors, Gestational
- Trophoblastic Tumor Hairy Cell Leukemia, Head and Neck Cancer, Hepatocellular Cancer, Hodgkin's Disease, Hodgkin's Lymphoma, Hypergammaglobulinemia, Hypopharyngeal Cancer, Intestinal Cancers, Intraocular Melanoma, Islet Cell Carcinoma, Islet Cell Pancreatic Cancer, Kaposi's Sarcoma, Kidney Cancer,
- Lymphoproliferative Disorders Macroglobulinemia, Male Breast Cancer, Malignant Mesothelioma, Malignant Thymoma, Medulloblastoma, Melanoma, Mesothelioma, Metastatic Occult Primary Squamous Neck Cancer, Metastatic Primary Squamous Neck Cancer, Metastatic Squamous Neck Cancer, Multiple Myeloma, Multiple Myeloma/Plasma Cell Neoplasm, Myelodysplastic Syndrome, Myelogenous
- Pheochromocytoma Pituitary Tumor, Plasma Cell Neoplasm/Multiple Myeloma, Primary Central Nervous System Lymphoma, Primary Liver Cancer, Prostate Cancer, Rectal Cancer, Renal Cell Cancer, Renal Pelvis and Ureter Cancer, Retinoblastoma, Rhabdomyosarcoma, Salivary Gland Cancer, Sarcoidosis Sarcomas, Sezary
- Thymoma Thyroid Cancer, Transitional Cell Cancer of the Renal Pelvis and Ureter, Transitional Renal Pelvis and Ureter Cancer, Trophoblastic Tumors, Ureter and Renal Pelvis Cell Cancer, Urethral Cancer, Uterine Cancer, Uterine Sarcoma, Vaginal Cancer, Visual Pathway and Hypothalamic Glioma, Vulvar Cancer, Waldenstrom's Macroglobulinemia, Wilms' Tumor, and any other hyperproliferative disease, besides neoplasia, located in an organ system listed above.
- the BASP-composition alone or in combination, is used to treat a hyperproliferative disorder, e.g., a hyperproliferative connective tissue disorder (e.g., a hyperproliferative fibrotic disease).
- a hyperproliferative disorder e.g., a hyperproliferative connective tissue disorder (e.g., a hyperproliferative fibrotic disease).
- the hyperproliferative fibrotic disease is multisystemic or organ-specific.
- hyperproliferative fibrotic diseases include, but are not limited to, multisystemic (e.g., systemic sclerosis, multifocal fibrosclerosis, sclerodermatous graft-versus-host disease in bone marrow transplant recipients, nephrogenic systemic fibrosis, scleroderma), and organ-specific disorders (e.g., fibrosis of the eye, lung, liver, heart, kidney, pancreas, skin and other organs).
- the disorder is chosen from liver cirrhosis or tuberculosis.
- the subject treated has a hyperproliferative genetic disorder, e.g., a hyperproliferative genetic disorder chosen from Marfan’s syndrome or Loeys–Dietz syndrome.
- the hyperproliferative disorder e.g., the hetylcholine
- hyperproliferative fibrotic disorder is chosen from one or more of chronic obstructive pulmonary disease, asthma, aortic aneurysm, radiation-induced fibrosis, skeletal- muscle myopathy, diabetic nephropathy, and/or arthritis.
- the disorder is chosen from an inflammatory or an autoimmune disorder chosen from multiple sclerosis, inflammatory bowel disease, scleroderma, lupus, rheumatoid arthritis or osteoarthritis.
- the inflammatory disorder is an inflammatory disorder of: the gastrointestinal tract or a gastrointestinal organ, e.g., colitis, Crohn's disease, inflammatory bowel disease (IBD), Barrett’s esophagus and chronic gastritis; the lung (e.g., asthma, chronic obstructive pulmonary disease (COPD); the skin (e.g., psoriasis), the cardiovascular system (e.g., atherosclerosis, cholesterol metabolic disorders, oxygen free radical injury, ischemia), the nervous system (e.g., Alzheimer's disease, multiple sclerosis), liver (e.g., hepatitis), kidney (e.g., nephritis), and the pancreas (e.g., pancreatitis).
- the gastrointestinal tract or a gastrointestinal organ e.g., colitis, Crohn's disease, inflammatory bowel disease (IBD), Barrett’s esophagus and chronic gastritis
- the lung e.g.,
- the inflammatory disorder is associated with an autoimmune disorder, e.g., arthritis (including rheumatoid arthritis, juvenile rheumatoid arthritis, osteoarthritis, psoriatic arthritis, lupus-associated arthritis, autoimmune thyroiditis or ankylosing spondylitis); scleroderma; lupus; systemic lupus erythematosis; HIV; Sjogren's syndrome; vasculitis; multiple sclerosis; dermatitis (including atopic dermatitis and eczematous dermatitis), myasthenia gravis, inflammatory bowel disease (IBD), Crohn's disease, colitis, diabetes mellitus (type I); acute inflammatory conditions (e.g., endotoxemia, sepsis and septicemia, toxic shock syndrome and infectious disease); transplant rejection and allergy.
- arthritis including rheumatoid arthritis, juvenile rheumatoid arthritis, osteoarthritis, ps
- liver cancers include: hepatocellular carcinoma (HCC), primary liver cell carcinoma, hepatoma, fibrolamellar carcinoma, focal nodular hyperplasia, cholangiosarcoma, intrahepatic bile duct cancer, angiosarcoma or hemangiosarcoma, hepatic adenoma, hepatic hemangiomas, hepatic hamartoma, hepatoblastoma, infantile hemangioendothelioma, mixed tumors of the liver, tumors of mesenchymal tissue, and sarcoma of the liver.
- HCC hepatocellular carcinoma
- primary liver cell carcinoma hepatoma
- fibrolamellar carcinoma focal nodular hyperplasia
- cholangiosarcoma intrahepatic bile duct cancer
- angiosarcoma or hemangiosarcoma intrahepatic bile duct cancer
- the liver disorder is a fibrotic disorder or connective tissue disorder affecting the function or physiology of the liver.
- the fibrotic disorder or connective tissue disorder can be systemic (affecting the whole body), multi-organ, or organ-specific (e.g., liver-specific).
- fibrotic liver disorders include liver fibrosis (hepatic fibrosis), liver cirrhosis, and any disorder associated with accumulation of extracellular matrix proteins, e.g., collagen, in the liver, liver scarring, and/or abnormal hepatic vasculature.
- Liver fibrosis is caused by liver inflammation or damage which triggers the accumulation of extracellular matrix proteins, including collagens, and scar tissue in the liver.
- Liver cirrhosis is the end stage of liver fibrosis, involves regenerative nodules (as a result of repair processes), and is accompanied with the distortion of the hepatic vasculature. Liver fibrotic disorders are most commonly caused by chronic viral infection (e.g., hepatitis B, hepatitis C), alcoholism, and fatty liver disease.
- fatty liver diseases include fatty liver (or FLD), alcoholic liver disease, non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), alcoholic steatohepatitis, simple steatosis, Reye’s syndrome, and any disorder associated with abnormal retention of lipids in liver cells.
- FLD fatty liver
- NAFLD non-alcoholic fatty liver disease
- NASH non-alcoholic steatohepatitis
- alcoholic steatohepatitis simple steatosis
- Reye’s syndrome and any disorder associated with abnormal retention of lipids in liver cells.
- the liver disease is NASH.
- Metabolic disorders can also affect the liver and cause liver damage.
- Examples of metabolic disorders of the liver or affecting the liver include hemachromatosis, diabetes, obesity, hypertension, dyslipidemia, galactosemia, and glycogen storage disease.
- Autoimmune disorders of the liver or affecting the liver can include systemic disorders or disorders that primarily affect an organ other than the liver, but with secondary effects to liver cells or liver function.
- autoimmune disorders include autoimmune hepatitis (AIH), autoimmune liver disease, lupoid hepatitis, systemic lupus erythematosus, primary biliary cirrhosis (PBC), scleroderma, and systemic sclerosis.
- disorders associated with inflammation of the liver include steatohepatitis, primary sclerosing cholangitis (PSC), ulcerative colitis, Crohn’s disease,
- inflammatory bowel disease or any disorder associated with inflammation in the liver.
- the liver disorder is associated with an inherited or congenital disease, e.g., Wilson’s disease, Gilbert’s disease, Byler syndrome,
- Greenland-Eskimo familial cholestasis Zellweger’s syndrome, Alagilles syndrome (ALGS), progressive familial intrahepatic cholestasis (PFIC), or alpha 1-antitrypsin deficiency, cystic fibrosis, Indian childhood cirrhosis, and hereditary
- the liver disorder is associated with pancreatic or biliary tract damage or disorders, e.g., cerebrotendinous, xanthomatosis, gall stones, choledocholithiasis, obstruction of the biliary tree, biliary trauma, biliary atresia, pancreatitis, primary biliary cirrhosis, primary sclerosing cholangitis, cholestasis, cholestasis of pregnancy, or any disorder associated with the obstruction or damage to the biliary system or the pancreas.
- pancreatic or biliary tract damage or disorders e.g., cerebrotendinous, xanthomatosis, gall stones, choledocholithiasis, obstruction of the biliary tree, biliary trauma, biliary atresia, pancreatitis, primary biliary cirrhosis, primary sclerosing cholangitis, cholestasis,
- liver disorders can be induced by infection, for example, by viral infections such as hepatitis A virus, hepatitis B virus, hepatitis C virus, hepatitis D virus (hepatitis delta virus), hepatitis E virus, Epstein-Barr adenovirus, or cytomegalovirus; or parasitic infection, such as schistosomiasis.
- viral infections such as hepatitis A virus, hepatitis B virus, hepatitis C virus, hepatitis D virus (hepatitis delta virus), hepatitis E virus, Epstein-Barr adenovirus, or cytomegalovirus
- parasitic infection such as schistosomiasis.
- liver disorders can be induced by drugs, such as acetaminophen (e.g., paracetamol, TYLENOL®, or PANADOL®), nonsteroidal anti- inflammatory drugs (NSAIDS, e.g., aspirin and phenylbutazone, ibuprofen, piroxicam, diclofenac, sulindac, and indomethacin), glucocorticoids, anti-tuberculosis drugs (e.g., isoniazid), antibiotics, anesthetics, antihypertensives (e.g., statins), oral contraceptives, dietary aids, or herbal supplements (e.g., ackee fruit, bajiaolian, boragecamphor, copaltra, comfrey, cycasin, kava leaves, pyrrolizidine alkaloids, horse chestnut leaves, valerian); or toxins, such as arsenic, carbon tetrachlor
- liver disorders also include disorders or conditions induced by injury to the liver or affecting the liver, including drug toxicity, alcoholism, ischemia, malnutrition, or physical trauma.
- Other liver disorders include hepatic vein thrombosis, Budd-Chiari syndrome, portal hypertension, hepatic encephalopathy, and hepatomegaly (or enlarged liver).
- the invention features a method of treating or preventing a fibrotic condition or disorder in a subject.
- the method includes administering a BRUSH-composition or BASP-composition described herein, alone or in combination with another agent or therapeutic modality, to a subject in need thereof, in an amount sufficient to decrease or inhibit the fibrotic condition in the subject.
- reducing fibrosis, or treatment of a fibrotic condition includes reducing or inhibiting one or more of: formation or deposition of tissue fibrosis; reducing the size, cellularity (e.g., fibroblast or immune cell numbers), composition; or cellular content, of a fibrotic lesion; reducing the collagen or hydroxyproline content, of a fibrotic lesion; reducing expression or activity of a fibrogenic protein; reducing fibrosis associated with an inflammatory response;
- tissue fibrosis includes reducing or inhibiting one or more of: formation or deposition of tissue fibrosis; reducing the size, cellularity (e.g., fibroblast or immune cell numbers), composition; or cellular content, of a fibrotic lesion; reducing the collagen or hydroxyproline content, of a fibrotic lesion; reducing expression or activity of a fibrogenic protein; reducing fibrosis associated with an inflammatory response;
- the fibrotic condition is primary fibrosis. In one embodiment, the fibrotic condition is idiopathic. In other embodiments, the fibrotic condition is associated with (e.g., is secondary to) a disease (e.g., an infectious disease, an inflammatory disease, an autoimmune disease, a malignant or cancerous disease, and/or a connective disease); a toxin; an insult (e.g., an environmental hazard (e.g., asbestos, coal dust, polycyclic aromatic hydrocarbons), cigarette smoking, a wound); a medical treatment (e.g., surgical incision, chemotherapy or radiation), or a combination thereof.
- a disease e.g., an infectious disease, an inflammatory disease, an autoimmune disease, a malignant or cancerous disease, and/or a connective disease
- a toxin e.g., an infectious disease, an inflammatory disease, an autoimmune disease, a malignant or cancerous disease, and/or a connective disease
- an insult e
- the fibrotic condition is a fibrotic condition of the lung, a fibrotic condition of the liver (e.g., as described herein), a fibrotic condition of the heart or vasculature, a fibrotic condition of the kidney, a fibrotic condition of the skin, a fibrotic condition of the gastrointestinal tract, a fibrotic condition of the bone marrow or a hematopoietic tissue, a fibrotic condition of the nervous system, a fibrotic condition of the eye, or a combination thereof.
- a fibrotic condition of the lung a fibrotic condition of the liver (e.g., as described herein), a fibrotic condition of the heart or vasculature, a fibrotic condition of the kidney, a fibrotic condition of the skin, a fibrotic condition of the gastrointestinal tract, a fibrotic condition of the bone marrow or a hematopoietic tissue, a fibrotic condition of the nervous system, a fibrotic
- the fibrotic condition is a fibrotic condition of the lung.
- the fibrotic condition of the lung is chosen from one or more of: pulmonary fibrosis, idiopathic pulmonary fibrosis (IPF), usual interstitial pneumonitis (UIP), interstitial lung disease, cryptogenic fibrosing alveolitis (CFA), bronchiectasis, and scleroderma lung disease.
- the fibrosis of the lung is secondary to a disease, a toxin, an insult, a medical treatment, or a
- the fibrosis of the lung can be associated with (e.g., secondary to) one or more of: a disease process such as asbestosis and silicosis; an occupational hazard; an environmental pollutant; cigarette smoking; an
- autoimmune connective tissue disorders e.g., rheumatoid arthritis, scleroderma and systemic lupus erythematosus (SLE)
- SLE systemic lupus erythematosus
- the fibrotic condition of the lung treated with the methods of the invention is associated with (e.g., secondary to) a cancer treatment, e.g., treatment of a cancer (e.g., squamous cell carcinoma, testicular cancer, Hodgkin’s disease with bleomycin).
- a cancer treatment e.g., treatment of a cancer (e.g., squamous cell carcinoma, testicular cancer, Hodgkin’s disease with bleomycin).
- the fibrotic condition of the lung is associated with an autoimmune connective tissue disorder (e.g., scleroderma or lupus, e.g., SLE).
- Pulmonary fibrosis can occur as a secondary effect in disease processes such as asbestosis and silicosis, and is known to be more prevalent in certain occupations such as coal miner, ship workers and sand blasters where exposure to environmental pollutants is an occupational hazard (Green, FH et al. (2007) Toxicol Pathol.35:136- 47).
- Other factors that contribute to pulmonary fibrosis include cigarette smoking, and autoimmune connective tissue disorders, like rheumatoid arthritis, scleroderma and systemic lupus erythematosus (SLE) (Leslie, KO et al. (2007) Semin Respir Crit Care Med.28:369-78; Swigris, JJ et al.
- sarcoidosis can include pulmonary fibrosis as part of the disease (Paramothayan, S et al. (2008) Respir Med.102:1-9), and infectious diseases of the lung can cause fibrosis as a long term consequence of infection, particularly chronic infections.
- Pulmonary fibrosis can also be a side effect of certain medical treatments, particularly radiation therapy to the chest and certain medicines like bleomycin, methotrexate, amiodarone, busulfan, and nitrofurantoin (Catane, R et al. (1979) Int J Radiat Oncol Biol Phys.5:1513-8; Zisman, DA et al. (2001) Sarcoidosis Vasc Diffuse Lung Dis.18:243-52; Rakita, L et al. (1983) Am Heart J.106:906-16; Twohig, KJ et al. (1990) Clin Chest Med.11:31-54; and Witten CM. (1989) Arch Phys Med Rehabil.
- idiopathic pulmonary fibrosis can occur where no clear causal agent or disease can be identified. Genetic factors can play a significant role in these cases of pulmonary fibrosis (Steele, MP et al. (2007) Respiration 74:601- 8; Brass, DM et al. (2007) Proc Am Thorac Soc.4:92-100 and du Bois RM. (2006) Semin Respir Crit Care Med.27:581-8).
- pulmonary fibrosis includes, but is not limited to, pulmonary fibrosis associated with chronic obstructive pulmonary disease (COPD), acute respiratory distress syndrome, scleroderma, pleural fibrosis, chronic asthma, acute lung syndrome, amyloidosis, bronchopulmonary dysplasia, Caplan's disease, Dressler's syndrome, histiocytosis X, idiopathic pulmonary hemosiderosis, lymphangiomyomatosis, mitral valve stenosis, polymyositis, pulmonary edema, pulmonary hypertension (e.g., idiopathic pulmonary hypertension (IPH)),
- COPD chronic obstructive pulmonary disease
- COPD chronic obstructive pulmonary disease
- scleroderma pulmonary fibrosis
- pleural fibrosis chronic asthma
- acute lung syndrome amyloidosis
- bronchopulmonary dysplasia Caplan's disease
- Dressler's syndrome histi
- pneumoconiosis pneumoconiosis, radiotherapy (e.g., radiation induced fibrosis), rheumatoid disease, Shaver's disease, systemic lupus erythematosus, systemic sclerosis, tropical pulmonary eosinophilia, tuberous sclerosis, Weber-Christian disease, Wegener's granulomatosis, Whipple's disease, or exposure to toxins or irritants (e.g., radiotherapy (e.g., radiation induced fibrosis), rheumatoid disease, Shaver's disease, systemic lupus erythematosus, systemic sclerosis, tropical pulmonary eosinophilia, tuberous sclerosis, Weber-Christian disease, Wegener's granulomatosis, Whipple's disease, or exposure to toxins or irritants (e.g.,
- the pulmonary fibrosis is associated with an inflammatory disorder of the lung, e.g., asthma, and/or COPD.
- the fibrotic condition is a fibrotic condition of the liver.
- the fibrotic condition of the liver is chosen from one or more of: fatty liver disease, steatosis (e.g., nonalcoholic steatohepatitis (NASH), cholestatic liver disease (e.g., primary biliary cirrhosis (PBC)), cirrhosis, alcohol induced liver fibrosis, biliary duct injury, biliary fibrosis, or cholangiopathies.
- fatty liver disease e.g., nonalcoholic steatohepatitis (NASH), cholestatic liver disease (e.g., primary biliary cirrhosis (PBC)), cirrhosis, alcohol induced liver fibrosis, biliary duct injury, biliary fibrosis, or cholangiopathies.
- steatosis e.g., nonalcoholic steatohepatitis (NASH)
- hepatic or liver fibrosis includes, but is not limited to, hepatic fibrosis associated with alcoholism, viral infection, e.g., hepatitis (e.g., hepatitis C, B or D), autoimmune hepatitis, non-alcoholic fatty liver disease (NAFLD), progressive massive fibrosis, exposure to toxins or irritants (e.g., alcohol, pharmaceutical drugs and environmental toxins). Additional examples of liver conditions and disorders are provided in the Sections entitled“Liver Conditions or Disorders,” provided herein.
- the fibrotic condition is a fibrotic condition of the kidney.
- the fibrotic condition of the kidney is chosen from one or more of: renal fibrosis (e.g., chronic kidney fibrosis), nephropathies associated with injury/fibrosis (e.g., chronic nephropathies associated with diabetes (e.g., diabetic nephropathy)), lupus, scleroderma of the kidney, glomerular nephritis, focal segmental glomerular sclerosis, IgA nephropathyrenal fibrosis associated with human chronic kidney disease (CKD), chronic progressive nephropathy (CPN),
- renal fibrosis e.g., chronic kidney fibrosis
- nephropathies associated with injury/fibrosis e.g., chronic nephropathies associated with diabetes (e.g., diabetic nephropathy)
- lupus e.g., chronic nephropathies associated with diabetes (
- tubulointerstitial fibrosis tubulointerstitial fibrosis, ureteral obstruction, chronic uremia, chronic interstitial nephritis, radiation nephropathy, glomerulosclerosis, progressive glomerulonephrosis (PGN), endothelial/thrombotic microangiopathy injury, HIV-associated nephropathy, or fibrosis associated with exposure to a toxin, an irritant, or a chemotherapeutic agent.
- the fibrotic condition of the kidney is scleroderma of the kidney.
- the fibrotic condition of the kidney is transplant nephropathy, diabetic nephropathy, lupus nephritis, focal segmental
- FSGS glomerulosclerosis
- HAVVAN HIV-associated nephropathy
- toxins irritants, or chemotherapeutic agents.
- the fibrotic condition is a fibrotic condition of the bone marrow or a hematopoietic tissue.
- the fibrotic condition of the bone marrow is an intrinsic feature of a chronic myeloproliferative neoplasm of the bone marrow, such as primary myelofibrosis (also referred to herein as agnogenic myeloid metaplasia or chronic idiopathic myelofibrosis).
- the bone marrow fibrosis is associated with (e.g., is secondary to) a malignant condition or a condition caused by a clonal proliferative disease.
- the bone marrow fibrosis is associated with a hematologic disorder (e.g., a hematologic disorder chosen from one or more of polycythemia vera, essential thrombocythemia, myelodysplasia, hairy cell leukemia, lymphoma (e.g., Hodgkin or non-Hodgkin lymphoma), multiple myeloma or chronic myelogenous leukemia (CML)).
- a hematologic disorder e.g., a hematologic disorder chosen from one or more of polycythemia vera, essential thrombocythemia, myelodysplasia, hairy cell leukemia, lymphoma (e.g., Hodgkin or non-Hodgkin lymphoma), multiple myeloma or chronic myelogenous leukemia (CML)).
- a hematologic disorder e.g., a hematologic disorder chosen from one or
- the bone marrow fibrosis is associated with (e.g., secondary to) a non-hematologic disorder (e.g., a non-hematologic disorder chosen from solid tumor metastasis to bone marrow, an autoimmune disorder (e.g., systemic lupus
- the fibrotic condition is idiopathic or drug-induced myelofibrosis.
- the fibrotic condition of the bone marrow or hematopoietic tissue is associated with systemic lupus erythematosus or scleroderma.
- the fibrotic condition is associated with leprosy or tuberculosis.
- the fibrotic condition is a fibrotic condition of the bone marrow.
- the fibrotic condition of the bone marrow is myelofibrosis (e.g., primary myelofibrosis (PMF)), myeloid metaplasia, chronic idiopathic myelofibrosis, or primary myelofibrosis.
- bone marrow fibrosis is associated with a hematologic disorder chosen from one or more of hairy cell leukemia, lymphoma, or multiple myeloma.
- the bone marrow fibrosis is associated with one or more myeloproliferative neoplasms (MPN) chosen from: essential thrombocythemia (ET), polycythemia vera (PV), mastocytosis, chronic eosinophilic leukemia, chronic neutrophilic leukemia, or other MPN.
- MPN myeloproliferative neoplasms
- the fibrotic condition is primary myelofibrosis.
- Primary myelofibrosis (PMF) (also referred to in the literature as idiopathic myeloid metaplasia, and Agnogenic myeloid metaplasia) is a clonal disorder of multipotent hematopoietic progenitor cells (reviewed in Abdel-Wahab, O. et al. (2009) Annu. Rev. Med.60:233-45; Varicchio, L. et al. (2009) Expert Rev. Hematol.2(3):315-334;
- the fibrotic condition is a fibrotic condition of the heart.
- the fibrotic condition of the heart is myocardial fibrosis (e.g., myocardial fibrosis associated with radiation myocarditis, a surgical procedure complication (e.g., myocardial post-operative fibrosis), infectious diseases (e.g., Chagas disease, bacterial, trichinosis or fungal myocarditis)); granulomatous, metabolic storage disorders (e.g., cardiomyopathy, hemochromatosis); developmental disorders (e.g., endocardial fibroelastosis); arteriosclerotic, or exposure to toxins or irritants (e.g., drug induced cardiomyopathy, drug induced cardiotoxicity, alcoholic cardiomyopathy, cobalt poisoning or exposure).
- myocardial fibrosis e.g., myocardial fibrosis associated with radiation myocarditis, a surgical procedure complication (e.g., my
- the myocardial fibrosis is associated with an inflammatory disorder of cardiac tissue (e.g., myocardial sarcoidosis).
- the fibrotic condition is a fibrotic condition associated with a myocardial infarction.
- the fibrotic condition is a fibrotic condition associated with congestive heart failure.
- the fibrotic condition is associated with an autoimmune disease selected from scleroderma or lupus, e.g., systemic lupus erythematosus.
- the fibrotic condition is systemic. In some embodiments, the fibrotic condition is systemic.
- the fibrotic condition is systemic sclerosis (e.g., limited systemic sclerosis, diffuse systemic sclerosis, or systemic sclerosis sine scleroderma), nephrogenic systemic fibrosis, cystic fibrosis, chronic graft vs. host disease, or atherosclerosis.
- systemic sclerosis e.g., limited systemic sclerosis, diffuse systemic sclerosis, or systemic sclerosis sine scleroderma
- nephrogenic systemic fibrosis e.g., cystic fibrosis
- cystic fibrosis e.g., chronic graft vs. host disease
- atherosclerosis e.g., atherosclerosis.
- the fibrotic condition is scleroderma.
- the scleroderma is localized, e.g., morphea or linear scleroderma.
- the condition is a systemic sclerosis, e.g., limited systemic sclerosis, diffuse systemic sclerosis, or systemic sclerosis sine scleroderma.
- the fibrotic condition affects a tissue chosen from one or more of muscle, tendon, cartilage, skin (e.g., skin epidermis or endodermis), cardiac tissue, vascular tissue (e.g., artery, vein), pancreatic tissue, lung tissue, liver tissue, kidney tissue, uterine tissue, ovarian tissue, neural tissue, testicular tissue, peritoneal tissue, colon, small intestine, biliary tract, gut, bone marrow, hematopoietic tissue, or eye (e.g., retinal) tissue.
- skin e.g., skin epidermis or endodermis
- cardiac tissue e.g., vascular tissue (e.g., artery, vein)
- pancreatic tissue e.g., lung tissue, liver tissue, kidney tissue, uterine tissue, ovarian tissue
- neural tissue e.g., testicular tissue, peritoneal tissue, colon, small intestine, biliary tract, gut, bone marrow,
- the fibrotic condition is a fibrotic condition of the eye.
- the fibrotic condition is glaucoma, macular degeneration (e.g., age-related macular degeneration), macular edema (e.g., diabetic macular edema), retinopathy (e.g., diabetic retinopathy), or dry eye disease.
- macular degeneration e.g., age-related macular degeneration
- macular edema e.g., diabetic macular edema
- retinopathy e.g., diabetic retinopathy
- dry eye disease e.g., diabetic retinopathy
- the fibrotic condition is a fibrotic condition of the skin.
- the fibrotic condition of the skin is chosen from one or more of: skin fibrosis (e.g., hypertrophic scarring, keloid), scleroderma, nephrogenic systemic fibrosis (e.g., resulting after exposure to gadolinium (which is frequently used as a contrast substance for MRIs) in subjects with severe kidney failure), and keloid.
- the fibrotic condition is a fibrotic condition of the gastrointestinal tract.
- the fibrotic condition is chosen from one or more of: fibrosis associated with scleroderma; radiation induced gut fibrosis; fibrosis associated with a foregut inflammatory disorder such as Barrett’s esophagus and chronic gastritis, and/or fibrosis associated with a hindgut inflammatory disorder, such as inflammatory bowel disease (IBD), ulcerative colitis and Crohn’s disease.
- the fibrotic condition of the gastrointestinal tract is fibrosis associated with scleroderma.
- the fibrotic condition is a chronic fibrotic condition or disorder. In certain embodiments, the fibrotic condition is associated with an inflammatory condition or disorder.
- the fibrotic and/or inflammatory condition is osteomyelitis, e.g., chronic osteomyelitis.
- the fibrotic condition is an amyloidosis. In certain embodiments, the amyloidosis is associated with chronic osteomyelitis.
- a BRUSH- composition or BASP-composition alone or in combination with a second therapeutic agent or therapeutic modality (e.g., one, two or more therapeutic agents or
- the second agent can be administered in free form, or as part of a conjugate, a BASP particle, a BRUSH-composition or a BASP- composition.
- compositions can be administered concurrently with, prior to, or subsequent to, one or more other additional therapies or therapeutic agents.
- each agent will be administered at a dose and/or on a time schedule determined for that agent.
- the additional therapeutic agent utilized in this combination can be administered together in a single
- compositions or administered separately in different compositions are administered separately in different compositions.
- the particular combination to employ in a regimen will take into account compatibility of the inventive pharmaceutical composition with the additional therapeutically active agent and/or the desired therapeutic effect to be achieved.
- additional therapeutic agents utilized in combination be utilized at levels that do not exceed the levels at which they are utilized individually.
- the levels utilized in combination will be lower than those utilized individually.
- a BRUSH-composition or BASP-composition and a second therapeutic agent or therapeutic modality are administered concurrently (e.g., administration of the two or more agents at the same time or day, or within the same treatment regimen) and/or sequentially (e.g., administration of one agent over a period of time followed by administration of another agent for a second period of time, or within different treatment regimens).
- administration of two or more agents occur in overlapping treatment regimens (e.g., administration of one agent is initiated before the completion of the treatment regimen of another agent, or the administration of one agent is completed before the termination of the treatment regimen of another agent).
- the BRUSH-composition or BASP-composition includes two or more agents (e.g., therapeutic agents).
- the two or more agents include at least a first agent and a second agent.
- a first agent is included in (e.g., as part of) a conjugate or BASP particle, and a second agent is not included in (e.g., not as part of) the conjugate or BASP particle.
- a first agent is included in (e.g., as part of) a first conjugate or first BASP particle, and a second agent is included in (e.g., as part of) a second conjugate or second BASP particle.
- each agent is included in (e.g., as part of) a different conjugate or different BASP particle.
- a first agent and a second agent are included in (e.g., as part of) a conjugate or BASP particle.
- the BRUSH-compositions or BASP-compositions may be able to selectively deliver (e.g., to deliver at different times and/or at different rates) the two or more agents.
- a first cleavable linker which directly or indirectly attaches a first agent to the remaining part of a first conjugate or first particle, is different from a second cleavable linker, which directly or indirectly attaches a second agent to the remaining part of the first conjugate or first particle, or of a second conjugate or second particle.
- the first cleavable linker and the second cleavable linker are cleaved at different rates under the same conditions.
- the second agent or therapeutic modality e.g., the second therapeutic agent
- is a cancer therapy e.g., one or more of anti-cancer agents, photodynamic therapy (PDT), immunotherapy, surgery and/or radiation.
- a cancer therapy e.g., one or more of anti-cancer agents, photodynamic therapy (PDT), immunotherapy, surgery and/or radiation.
- the anti-cancer agent is a small molecule, a kinase inhibitor, an alkylating agent, a vascular disrupting agent, a microtubule targeting agent, a mitotic inhibitor, a topoisomerase inhibitor, an anti-angiogenic agent, or an anti-metabolite.
- the agent e.g., the therapeutic agent
- the anti- cancer agent is an anthracycline (e.g., doxorubicin).
- the anti- cancer agent is a platinum-based agent (e.g., cisplatin or oxaliplatin).
- the anti-cancer agent is a pyrimidine analog (e.g., gemcitabine).
- the anti-cancer agent is chosen from camptothecin, irinotecan, rapamycin, FK506, 5-FU, leucovorin, or a combination thereof.
- the anti-cancer agent is a protein biologic (e.g., an antibody molecule), or a nucleic acid therapy (e.g., an antisense or inhibitory double stranded RNA molecule).
- a protein biologic e.g., an antibody molecule
- a nucleic acid therapy e.g., an antisense or inhibitory double stranded RNA molecule
- the cancer therapy includes one or more of: a cancer therapeutic, including, for example, a nanotherapy (e.g., one or more nanotherapeutic agents, including viral cancer therapeutic agents (e.g., an oncolytic herpes simplex virus (HSV)) a lipid nanoparticle (e.g., a liposomal formulation (e.g., pegylated liposomal doxorubicin (DOXIL ® )), or a polymeric nanoparticle); one or more cancer therapeutic antibodies (e.g., anti-HER2, anti-EGFR, anti-CD20 antibodies); RNAi and antisense RNA agents; one or more chemotherapeutic agents (e.g., low molecular weight chemotherapeutic agents, including a cytotoxic or a cytostatic agent));
- a nanotherapy e.g., one or more nanotherapeutic agents, including viral cancer therapeutic agents (e.g., an oncolytic herpes simplex virus (HSV))
- the chemotherapeutic agent used in combination is a cytotoxic or a cytostatic agent.
- cytotoxic agents include, but are not limited to, antimicrotubule agents, topoisomerase inhibitors (e.g., irinotecan), or taxanes (e.g., docetaxel), antimetabolites, mitotic inhibitors, alkylating agents, intercalating agents, agents capable of interfering with a signal transduction pathway, agents that promote apoptosis and radiation.
- any combination of one or more therapeutic agents or modalities e.g., first, second, third
- exemplary cancer therapeutics include, but are not limited to, nanotherapeutic agents (e.g., one or more lipid nanoparticles (e.g., a liposomal formulation (e.g., pegylated liposomal doxorubicin (DOXIL ® ) or liposomal paclitaxel (e.g., Abraxane®)), or a polymeric nanoparticle); one or more low molecular weight chemotherapeutic s (e.g., gemcitabine, cisplatin, epirubicin, 5-fluorouracil, paclitaxel, oxaliplatin, or leucovorin); one or more antibodies against cancer targets (e.g., growth factor receptor such as HER-2/neu, HER3, VEGF)
- nanotherapeutic agents e.g., one or more lipid nanoparticles (e.g., a
- the methods can be used in combination with immunodulatory agents, e.g., IL- 1, 2, 4, 6, or 12, or interferon alpha or gamma, or immune cell growth factors such as GM-CSF.
- immunodulatory agents e.g., IL- 1, 2, 4, 6, or 12, or interferon alpha or gamma
- immune cell growth factors such as GM-CSF.
- the cancer therapy includes an immune or
- BRUSH-compositions or BASP-compositions can be used to improve the efficacy of said immune therapies.
- immune therapies include, but are not limited to, CTLA-4 blockade (e.g., an anti-CTLA-4 antibody (e.g., ipilimumab)); immune-based therapies (including, e.g., immune or dendritic cell- based vaccines and antagonists of immune inhibitory signals or checkpoints); cancer vaccines, e.g., Sipuleucel-T (APC8015, trade name Provenge); and adoptive T-cell-based therapies.
- Exemplary immune-based therapies include, but are not limited to, e.g., immune or dendritic cell- based vaccines (Seton-Rogers, S. (2012) Nature Reviews Cancer 12:230-231; Palucka, K. et al. (2012) Nature Reviews Cancer 12:265-277); effector memory CD8+ T cells (Bird, L. (2012) Nature Reviews Immunology 12:227);
- TLRs Toll like Receptors
- NLRs Neuroreceptors
- the therapy is a cell-based immunotherapy wherein immune cells are expanded ex vivo and injected into the subject.
- the cancer therapy includes PDT used in combination with the BASP-composition, other cancer therapies, and/or the microenvironment modulator, described herein.
- PDT includes administration of a photosensitizing agent (e.g., a porphyrin, a porphyrin precursor, a corlin, or a phthalocyanine) followed by irradiation at a wavelength corresponding to an absorbance band of the photosensitizing agent.
- a series of events lead to one or more of: cell death (e.g., tumor cell death), damage to the micro vasculature, or induction of a local inflammatory reaction).
- cell death e.g., tumor cell death
- damage to the micro vasculature e.g., or induction of a local inflammatory reaction.
- the cancer therapy includes an inhibitor of a cancer stem cell (also referred to herein as a“cancer initiating cell”), used in combination with the BASP-compositions, or BRUSH-compositions described herein.
- a cancer stem cell also referred to herein as a“cancer initiating cell”
- hypoxia and cancer drugs (including anti- angiogenic drugs) and radiation therapy are believed to increase the number of cancer stem cells.
- BRUSH-composition or BASP-composition alone or in combination with, e.g., an inhibitor of a cancer stem cell, can be used to reduce the production of these stem cells.
- exemplary inhibitors of cancer stem cells include, but are not limited to, hedgehog (e.g., SMO) antagonists; and Wnt pathway antagonists (e.g., antibody, OMP- 18R5).
- the combinations described herein can be further administered in combination with a microenvironment modulator.
- the combined administration of the microenvironment modulator can be used to further enhance the efficacy (e.g., penetration and/or diffusion), of the combination therapies described herein in a tumor or tumor vasculature in a subject.
- Exemplary microenvironment modulators include, but are not limited to, an anti-angiogenic therapy and/or vascular normalization strategy, for example, an inhibitor of vascular endothelial growth factor (VEGF) pathway, an inhibitor of the angiopoietin-Tie-2 pathway (e.g., an Ang-1 or an Ang-2 inhibitor), or sorafenib; an agent that decreases the level or production of hyaluronic acid; an inhibitor of the hedgehog pathway; an agent that improves drug penetration in tumors.
- VEGF vascular endothelial growth factor
- angiopoietin-Tie-2 pathway e.g., an Ang-1 or an Ang-2 inhibitor
- sorafenib e.g., an agent that decreases the level or production of hyaluronic acid
- an inhibitor of the hedgehog pathway e.g., an agent that improves drug penetration in tumors.
- the agent is a disulfide-based cyclic RGD peptide (iRGD) or an analogue thereof; a taxane therapy (e.g., taxane-induced apoptosis); an agent that decreases the level or production of collagen or procollagen; an anti-fibrotic agent and/or a profibrotic pathway inhibitor.
- a taxane therapy e.g., taxane-induced apoptosis
- an agent that decreases the level or production of collagen or procollagen e.g., in Paragraphs 260, 340-346, incorporated herein by reference.
- Agents for anti-angiogenic/vascular normalization strategies as described in Goel et al. (2011) Physiol Rev.91: 1071-1121, and Jain (2014) Cancer Cell 26(5): 605-622, the contents of which are incorporated herein by reference, can also be used as an anti-angiogenic agent for the compositions and methods described herein.
- the anti-angiogenic agent is an inhibitor of vascular endothelial growth factor (VEGF) pathway.
- VEGF vascular endothelial growth factor
- the microenvironment modulator includes an agent that decreases the level or production of hyaluronic acid (HA).
- HA agents used in combination with the combination therapies comprising the BRUSH- compositions and BASP-compositions are described in WO 2013/169739, e.g., in Paragraph 115, incorporated herein by reference.
- the microenvironment modulator includes an inhibitor of the hedgehog pathway.
- hedgehog inhibitors include, but are not limited to, IPI-926, GDC-0449, cyclopamine or an analogue thereof, and GANT58.
- the microenvironment modulator includes an agent that improves drug penetration in tumors.
- the agent is a disulfide- based cyclic RGD peptide (iRGD) or an analogue thereof (e.g., described in Sugahara, KN et al. (2010) Science 328:1031-5; Ye, Y. et al. (2011) Bioorg Med Chem Lett. 21(4):1146-50).
- the microenvironment modulator includes a taxane therapy (e.g. Taxane-induced apoptosis as described in Griffon-Etienne, G. et al. (1999) Cancer Res.59(15):3776-82).
- a taxane therapy e.g. Taxane-induced apoptosis as described in Griffon-Etienne, G. et al. (1999) Cancer Res.59(15):3776-82).
- the microenvironment modulator includes an agent that modulates (e.g., inhibits) a hypoxia inducible factor (HIF), for example, an agent that inhibits hypoxia-inducible factors 1 ⁇ and 2 ⁇ (HIF-1 ⁇ and HIF-2 ⁇ ).
- HIF activity has been shown to be involved in inflammation (e.g., rheumatoid arthritis) and angiogenesis associated with cancer tumor growth.
- HIF inhibitors such as phenethyl isothiocyanate (PEITC) are under investigation for anti-cancer effects (Syed Alwi SS, et al. (2010) Br. J. Nutr.104 (9): 1288–96; Semenza GL (2007). Drug Discov.
- the agent is an antibody against an HIF.
- the agent is an HIF chemical inhibitor, such as phenethyl isothiocyanate (PEITC).
- the microenvironment modulator includes an agent that decreases the level or production of collagen or procollagen. For example, an agent that degrades collagen, e.g., collagenase.
- the combinations described herein can be further administered in combination with a microenvironment modulator chosen from an anti-fibrotic agent or an inhibitor of a profibrotic pathway (a“profibrotic pathway inhibitor”) (e.g., a pathway dependent- or independent of TGF-beta and/or CTGF activation).
- a“profibrotic pathway inhibitor” e.g., a pathway dependent- or independent of TGF-beta and/or CTGF activation.
- the combinations described herein are administered in combination with one or more of: an inhibitor of endothelin-1, PDGF, Wnt/beta- catenin, IGF-1, TNF-alpha, and/or IL-4.
- the combinations described herein are administered in combination with an inhibitor of endothelin-1 and/or PDGF.
- the combinations described herein are administered in combination with an inhibitor of one or more of chemokine receptor type 4 (CXCR4) (e.g., AMD3100, MSX-122); stromal-derived-factor-1(SDF-1) (e.g., tannic acid); hedgehog (e.g., IPI-926, GDC-0449, cyclopamine or an analogue thereof, or GANT58).
- CXCR4 chemokine receptor type 4
- SDF-1 e.g., tannic acid
- hedgehog e.g., IPI-926, GDC-0449, cyclopamine or an analogue thereof, or GANT58.
- an inhibitor of a CXCR4 receptor and/or its ligand, SDF-1 is administered in combination with a therapy (e.g., a cancer or
- Certain embodiments may further include administration of a further AHCM and/or a microenvironment modulator as described herein.
- exemplary SDF-1/CXCR4 inhibitors that can be used include, but are not limited to, 2,2'-bicyclam; 6,6'- bicyclam; AMD3100 (IUPAC name: l,l'-[l,4- phenylene-bis(methylene)]-bis-l,4,8,l 1-tetraazacyclotetradecane), as described in e.g., U.S. Pat.
- compositions and methods described herein can comprise an
- the immunomodulator is an anti- inflammatory agent described herein, e.g., for treating or preventing a disease or disorder, e.g., a cancer or a fibrotic disorder described herein.
- the composition and method can include one, two, three or more anti-inflammatory agents, alone or in combination with one or more therapeutic agents described herein (e.g., an AHCM agent, a microenvironment modulator, an immune-checkpoint inhibitor, or an additional therapy, e.g., a cancer or anti-fibrotic therapy).
- the anti-inflammatory agent is an agent that blocks, inhibits, or reduces inflammation or signaling from an inflammatory signaling pathway.
- the anti-inflammatory agent inhibits or reduces the activity of one or more of any of the following: IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL- 7, IL-8, IL-9, IL-10, IL-12, IL-13, IL-15, IL-18, IL-23, interferons (IFNs), e.g., TNF- ⁇ , TNF- ⁇ , TNF-RI, TNF-RII; CD23, CD30, CD40L, CXCL-1, EGF, G-CSF, GDNF, PDGF-BB, RANTES/CCL5, IKK, NF-kB, TLR2, TLR3, TLR4, TL5, TLR6, TLR7, TLR8, TLR8, TLR9, and/or any cognate receptors thereof.
- IFNs interferons
- the anti-inflammatory agent is an IL-1 or IL-1 receptor antagonist, such as anakinra (KINIRET®), rilonacept, or canakinumab.
- anakinra KINIRET®
- rilonacept rilonacept
- canakinumab canakinumab
- the anti-inflammatory agent is an IL-6 or IL-6 receptor antagonist, e.g., an anti-IL-6 antibody or an anti-IL-6 receptor antibody, such as tocilizumab (ACTEMRA®), olokizumab, clazakizumab, sarilumab, sirukumab, siltuximab, or ALX-0061.
- an anti-IL-6 antibody or an anti-IL-6 receptor antibody such as tocilizumab (ACTEMRA®), olokizumab, clazakizumab, sarilumab, sirukumab, siltuximab, or ALX-0061.
- the anti-inflammatory agent is a TNF-a antagonist, e.g., an anti-TNFa antibody, such as infliximab (REMICADE®), golimumab
- the anti-inflammatory agent is a corticosteroid.
- corticosteroids include, but are not limited to, cortisone (hydrocortisone, hydrocortisone sodium phosphate, hydrocortisone sodium succinate, ALA-CORT®, HYDROCORT ACETATE®, hydrocortone phosphate LANACORT®, SOLU- CORTEF®), decadron (dexamethasone, dexamethasone acetate, dexamethasone sodium phosphate, DEXASONE®, DIODEX®, HEXADROL®, MAXIDEX®), methylprednisolone (6-methylprednisolone, methylprednisolone acetate,
- methylprednisolone sodium succinate DURALONE®, MEDRALONE®,
- MEDROL® M-PREDNISOL®, SOLU-MEDROL®
- prednisolone DELTA- CORTEF®, ORAPRED®, PEDIAPRED®, PRELONE®
- prednisone DELTA- CORTEF®
- the anti-inflammatory agent is a non-steroidal anti- inflammatory drug (NSAID).
- NSAIDs non-steroidal anti-inflammatory drugs
- Exemplary anti-inflammatory agents include, but are not limited to, aspirin, ibuprofen, naproxen, celecoxib, diclofenac, diflunisal, etodolac, fenoprofen, flurbiprofen, ketoprofen, ketorolac, mefenamic acid, meloxicam, nabumetone, oxaprozin, piroxicam, sulindac, and tolmetin.
- the anti-inflammatory agent is an immune selective anti-inflammatory derivative (ImSAID). Immune-Checkpoint Inhibitors
- compositions and methods described herein can comprise an immune- checkpoint inhibitor described herein, e.g., for treating or preventing a disease or disorder, e.g., a cancer, inflammatory or a fibrotic disorder described herein.
- the compositions and methods can include one, two, three or more immune-checkpoint inhibitors, alone or in combination with one or more therapeutic agents described herein (e.g., an AHCM agent, a microenvironment modulator, an anti-inflammatory agent, or an additional therapy, e.g., a cancer or anti-fibrotic therapy).
- Immune checkpoint inhibitors refer to molecules that block, inhibit, or reduce activity of one or more immune checkpoint proteins.
- the inhibitors may be an antibody, an antigen binding fragment thereof, an
- immune- checkpoint molecules include, but are not limited to, PD-1, PD-L 1 , PD-L 2 , CTLA4, B7-H3, B7-H4, HVEM, BTLA, a killer-cell immunoglobulin-like receptor (KIR), LAG3, TIM3, CEACAM-1, CEACAM-3, CEACAM-5, GAL9, VISTA, TIGIT, LAIR1, CD160, 2B4, and A2aR.
- the immune checkpoint inhibitor is a PD-1 inhibitor.
- Antibodies, antibody fragments, and other inhibitors of PD-1 and its ligands are available in the art and may be used combination with metformin as described herein.
- Exemplary anti-PD-1 antibodies include, but are not limited to, nivolumab (also known as MDX-1106 or BMS-936558), pembrolizumab (formerly known as lambrolizumab, also known as Merck 3475 or MK03475), and pidilizumab (also known as CT- 011).
- nivolumab also known as MDX-1106 or BMS-936558
- pembrolizumab formerly known as lambrolizumab, also known as Merck 3475 or MK03475
- pidilizumab also known as CT- 011
- Nivolumab (clone 5C4) and other human monoclonal antibodies that specifically bind to PD-1 are disclosed in US 8,008,449 and
- WO2006/121168 Pidilizumab and other humanized anti-PD-1 monoclonal antibodies are disclosed in WO2009/101611. Pembrolizumab and other humanized anti-PD-1 antibodies are disclosed in US 8,354,509 and WO2009/114335. In some
- the PD-1 inhibitor is an immunoadhesin (e.g., an immunoadhesin comprising an extracellular or PD-1 binding portion of PD-L 1 or PD-L 2 fused to a constant region (e.g., an Fc region of an immunoglobulin sequence), such as AMP- 224.
- an immunoadhesin e.g., an immunoadhesin comprising an extracellular or PD-1 binding portion of PD-L 1 or PD-L 2 fused to a constant region (e.g., an Fc region of an immunoglobulin sequence), such as AMP- 224.
- the immune checkpoint inhibitor is a PD-L 1 inhibitor.
- Antibodies, antibody fragments, and other inhibitors of PD-L 1 are available in the art and may be used combination with metformin as described herein.
- Exemplary anti- PD-L 1 antibodies include, but are not limited to, YW243.55.S70 (as described in PCT Publication No. WO2010/077634), MPDL 3 280A (as described in U.S. Patent No. 7,943,743 and U.S. Publication No.20120039906), MEDI-4736, MSB-0010718C, or MDX-1105 (also referred to as BMS-936559, as described in WO2007/005874).
- the immune checkpoint inhibitor is a TIM3 inhibitor.
- Antibodies, antibody fragments, and other inhibitors of TIM3 and its ligands are available in the art and may be used combination with metformin as described herein.
- antibodies, antibody fragments, small molecules, or peptide inhibitors that target TIM3 binds to the IgV domain of TIM3 to inhibit interaction with its ligands can be administered in combination with a metformin agent as described herein.
- Exemplary TIM3 inhibitors include, but are not limited to the antibodies and peptides disclosed in WO2013/006490 and US20100247521); anti-TIM3 inhibitors such as humanized versions of RMT3-23 (as disclosed in Ngiow et al., 2011, Cancer Res, 71:3540-3551) and clone 8B.2C12 (disclosed in Monney et al., 2002, Nature, 415:536-541). Bi-specific antibodies that inhibit TIM3 and PD-1 are disclosed in US20130156774.
- the immune checkpoint inhibitor is a LAG3 inhibitor.
- Antibodies, antibody fragments, and other inhibitors of LAG3 and its ligands are available in the art and may be used combination with metformin as described herein.
- Exemplaryanti-LAG3 antibodies include, but are not limited to monoclonal antibody BMS-986016 (Bristol-Myers Squib), IMP701 (Immutep), IMP731 (Immutep and GlaxoSmithKline), and antibodies disclosed in WO2010/019570.
- Other LAG3 inhibitors include IMP321 (Immutep), which is a recombinant fusion protein of a soluble portion of LAG3 and Ig that binds to MHC class II molecules and activates antigen presenting cells (APC).
- APC antigen presenting cells
- the immune checkpoint inhibitor is a CEACAM inhibitor, e.g., a CEACAM-1 inhibitor, a CEACAM-3 inhibitor, and/or a CEACAM-5 inhibitor.
- a CEACAM inhibitor e.g., a CEACAM-1 inhibitor, a CEACAM-3 inhibitor, and/or a CEACAM-5 inhibitor.
- Antibodies, antibody fragments, and other inhibitors of CEACAM are available in the art and may be used combination with metformin as described herein.
- Exemplary anti-CEACAM-1 antibodies include, but are not limited to, antibodies described in WO 2010/125571, WO 2013/082366 WO 2014/059251 and WO
- 2014/022332 e.g., a monoclonal antibody 34B1, 26H7, and 5F4; or a recombinant form thereof, as described in, e.g., US 2004/0047858, US 7,132,255 and WO
- the anti-CEACAM antibody binds to CEACAM-5 as described in, e.g., Zheng et al. PLoS One.2010 Sep 2;5(9). pii: e12529
- CEACAM-5 as described in, e.g., WO 2013/054331 and US 2014/0271618.
- a conjugate or BASP particle comprising:
- an agent e.g., a therapeutic agent, (e.g., an agent chosen from an angiotension receptor blocker (ARB), a vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor);
- a therapeutic agent e.g., an agent chosen from an angiotension receptor blocker (ARB), a vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor
- a cleavable linker e.g., a tissue microenvironment cleavable linker.
- carbocyclyl or heterocyclyl moiety comprises a bicyclic carbocyclyl or bicyclic heterocyclyl moiety.
- the triazolyl moiety is 1,2,3-triazoldiyl.
- the heteroalkyl moiety comprises a polyethylene glycol moiety. 5.
- an enzyme e.g., an esterase or a protease
- pH e.g., acidic pH, basic pH
- light e.g., ultraviolet light
- nucleophile reduction, or oxidation.
- the cleavable linker comprises an ester, an acetal, a phosphoramidite, a hydrazone, an imine, an oxime, a disulfide, or a silyl moiety.
- the carbocyclyl or heterocyclyl moiety is linked, e.g., directly or indirectly linked, to the triazole moiety;
- heteroalkyl moiety is linked, e.g., directly or indirectly linked, to the triazolyl moiety
- the agent is linked, e.g., directly or indirectly linked, to the conjugate or BASP particle through the cleavable linker (e.g., the tissue microenvironment cleavable linker).
- the conjugate or BASP particle of any one of embodiments 1-8, wherein the conjugate or BASP particle comprises a structure according to Formula (III) or (III-f) as described herein.
- Ring T is , wherein“2” represents a portion of Ring T bound to L 2 in the conjugate or BASP particle 13.
- A is C 1 -C 12 heteroalkylene, optionally substituted with 1-6 R 1 (e.g., optionally substituted with 1-3 R 1 ). 16.
- the PEG comprises a molecular weight of between about 200 and about 6000, g/mol (e.g., PEG200, PEG400, PEG600, PEG800, PEG1000, PEG1500, PEG2000, PEG3000, PEG4000, PEG6000).
- L 3 is cleaved at a first set of conditions relative to a second set of conditions (e.g., a first pH relative to a second pH). 22.
- an enzyme e.g., an esterase, a protease
- pH e.g., acidic pH, basic pH.
- the enzyme is a protease chosen from MMP-1, MMP-2, MMP-3, MMP-8, MMP-9, MMP-14, plasmin, PSA, PSMA, CATHEPSIN D, CATHEPSIN K, CATHEPSIN S, ADAM10, ADAM12, ADAMTS, Caspase-1, Caspase-2, Caspase-3, Caspase-4, Caspase-5, Caspase-6, Caspase-7, Caspase-8, Caspase-9, Caspase-10, Caspase-11, Caspase-12, Caspase-13, Cas
- each R 2 is oxo or halo (e.g., fluoro).
- each of L 1 and L 2 is independently C 1 -C 12 alkylene, C 1 -C 12 heteroalkylene, (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 alkylene), (C 0 -C 12 heteroalkylene)-arylene-(C 0 -C 12 alkylene), or (C 0 -C 12 alkylene)-arylene-(C 0 -C 12 heteroalkylene), or (C 0 -C 12 heteroalkylene)-aryl-(C 0 -C 12 heteroalkylene), and each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R 2 .
- the agent is an angiotensin receptor blocker (ARB).
- ARB angiotensin receptor blocker
- the ARB comprises losartan, candesartan, telmisartan, valsartan, olmesartan, azilsartan, eprosartan, irbesartan, saralasin, EXP 3174, L158209, or a metabolite or derivative thereof.
- conjugate or BASP particle of any one of embodiments 34-35, wherein the ARB comprises a structure of Formulas (I-i), (I-ii), (I-iii), (I-iv), or (I-v) as described herein.
- the conjugate or BASP particle of any one of embodiments 9-45, wherein the structure of Formula (III) is a structure of Formula (III-c) as described herein. 47.
- the conjugate or BASP particle of embodiment 51 wherein the hydrodynamic diameter of the particle is less than about 90 nm (e.g., less than about 80 nm, about 75 nm, about 70 nm, about 65 nm, about 60 nm, about 55 nm, about 50 nm, about 45 nm, about 40 nm, about 35 nm, about 25 nm, or less).
- 53. The conjugate or BASP particle of any one of embodiments 51-52, wherein the hydrodynamic diameter of the particle is between about 5 nm and 50 nm. 54.
- the conjugate or BASP particle of any one of the preceding embodiments wherein the total amount of the agent present in the conjugate or BASP particle is greater than about 5% (e.g., about 6%, about 7%, about 8%, about 9%, about 10%, about 12%, about 15%, about 20%, about 25%, about 30%, or more).
- the conjugate or BASP particle of claim 60 which comprises a combination of one or more of an ARB, a vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor, and another therapeutic agent.
- the diagnostic agent comprises a fluorescent molecule, a metal chelate, a contrast agent, a radionuclide, or a positron emission tomography (PET) imaging agent, an infrared imaging agent, a near-IR imaging agent, a computer assisted tomography (CAT) imaging agent, a photon emission computerized tomography imaging agent, an X-ray imaging agent, or a magnetic resonance imaging (MRI) agent.
- PET positron emission tomography
- the diagnostic agent comprises a fluorescent molecule, a metal chelate, a contrast agent, a radionuclide, or a positron emission tomography (PET) imaging agent, an infrared imaging agent, a near-IR imaging agent, a computer assisted tomography (CAT) imaging agent, a photon emission computerized tomography imaging agent, an X-ray imaging agent, or a
- the conjugate or BASP particle of embodiment 64 wherein the fluorescent molecule comprises an acridine dye, a cyanine dye, a rhodamine dye, a BODIPY dye, a fluorescein dye, a dansyl dye, an Alexa dye, an atto dye, a quantum dot, or a fluorescent protein.
- the fluorescent molecule comprises an acridine dye, a cyanine dye, a rhodamine dye, a BODIPY dye, a fluorescein dye, a dansyl dye, an Alexa dye, an atto dye, a quantum dot, or a fluorescent protein.
- the cyanine dye comprises Cy3, Cy 3.5, Cy5, Cy5.5, Cy7, or Cy7.5.
- the conjugate or BASP particle of any one of embodiments 1-66, wherein the conjugate or BASP particle further comprises a targeting agent. 68.
- the conjugate or BASP particle of embodiment 67 wherein the targeting agent comprises a ligand, cell surface receptor, a protein (e.g., glycoprotein or antibody), a sugar, a nucleic acid, a cofactor, a vitamin, an aptamer, a small molecule therapeutic, or a metabolite or derivative thereof.
- the targeting agent comprises a liver targeting agent.
- conjugate or BASP particle of embodiment 69 wherein the conjugate or BASP particle is targeting to a hepatocyte, Kupffer cell, an endothelial cell, a hepatic stellate cell, a bile duct cell, or a hepatocarcinoma cell.
- a conjugate or BASP particle comprising:
- a diagnostic agent e.g., a cyanine dye, e.g., Cy3, Cy 3.5, Cy5, Cy5.5, Cy7, or Cy7.5
- a targeting agent e.g., a cyanine dye, e.g., Cy3, Cy 3.5, Cy5, Cy5.5, Cy7, or Cy7.5
- a linker e.g., a cleavable linker, e.g., a tissue microenvironment cleavable linker
- conjugate or BASP particle may be further described by any one of embodiments 1-63.
- 72. A conjugate comprising paricalcitol, wherein the conjugate comprises a structure of Formulas (III-d-1), (III-d-2), (III-i-1), or (III-i-2) as described herein.
- 73. The conjugate of embodiment 72, wherein each R 2 is oxo or halo (e.g., fluoro).
- L 3 is–C(O)–,–C(O)O–,–
- 78. The con u ate of an one of embodiments 75-77 wherein L 3 is–CH 2 CH 2 O–,
- a composition e.g., pharmaceutical composition, comprising one or more of the particles or conjugates of any of the preceding embodiments and a
- a method of treating or preventing a disorder in a subject, comprising administering the particle according to any one of embodiments 50-71, e.g., as a single agent or in combination with other agent or therapy or a composition of embodiment 79 to the subject.
- a disorder e.g., a hyperproliferative disorder, a fibrotic disorder, and/or an inflammatory disorder
- the particle comprising administering the particle according to any one of embodiments 50-71, e.g., as a single agent or in combination with other agent or therapy or a composition of embodiment 79 to the subject.
- the disorder is a cancer, a fibrotic disorder, or a liver disorder.
- the disorder is a fibrotic condition or disorder of the lung, a fibrotic condition of the liver, a fibrotic condition of the heart or vasculature, a fibrotic condition of the kidney, a fibrotic condition of the skin, a fibrotic condition of the gastrointestinal tract, a fibrotic condition of the bone marrow or a hematopoietic tissue, a fibrotic condition of the nervous system, a fibrotic condition of the eye, or a combination thereof.
- any one of embodiments 80-87, wherein the other agent or therapy comprises an anti-cancer, an anti-fibrotic, or anti-inflammatory, agent or therapy.
- the anti-cancer therapy is chosen from one or more of anti-cancer agents, photodynamic therapy, an immunotherapy (e.g., an immune-cell therapy or adoptive immunotherapy), surgery and/or radiation.
- the other agent or therapy e.g., the anti-cancer, anti-fibrotic, or anti-inflammatory
- embodiments 1-79 in combination with the other agent or therapy to the subject under conditions sufficient to treat or prevent the disorder or condition in the subject, or to improve the delivery and/or efficacy of the other agent or therapy provided to the subject.
- the other agent or therapy comprises an anti- cancer, an anti-fibrotic, or an anti-inflammatory, agent or therapy.
- 98. The method of any one of embodiments 8-97, wherein the other agent or therapy is chosen from one or more of:
- a cancer therapeutic chosen from a viral cancer therapeutic agent, a lipid nanoparticle of an anti-cancer therapeutic agent, a polymeric nanoparticle of an anti- cancer therapeutic agent, an antibody against a cancer target, a dsRNA agent, an antisense RNA agent, or a chemotherapeutic agent;
- an immunotherapy e.g., an immune-cell therapy or adoptive
Abstract
Described herein are macromonomers, conjugates comprising the macromonomers (also referred to alternatively as "BRUSH", or "BRUSH conjugates"), and brush arm star polymer particles comprising the conjugates (also referred to as "BASPs" or "BASP particles"), each further comprising an agent (e.g., one or more therapeutic agents, diagnostic agents, or targeting moieties), as well as starting materials (e.g., crosslinkers) and other components (e.g., degradation components) thereof, as well as pharmaceutical compositions and methods of making and using the same.
Description
BRUSH-ARM STAR POLYMERS, CONJUGATES AND PARTICLES, AND
USES THEREOF
RELATED APPLICATIONS
This application claims priority to U.S. Provisional Patent Application No. 62/529,439, filed July 6, 2017; U.S. Provisional Patent Application No. 62/528,940, filed July 5, 2017; and U.S. Provisional Patent Application No. 62/430,257, filed December 5, 2016. The contents of each of the foregoing applications are
incorporated herein by reference in their entirety.
BACKGROUND
Advances in biomedical research have led to the introduction of novel molecular and nanotherapeutic agents in both preclinical and clinical settings (Jones, D. (2007) Nat Rev Drug Discov 6, 174-175; Moghimi, S. M. et al. (2005) Faseb J. 19, 311-330). While these new agents act on unique targets that afford greater specificity to target cells, e.g., tumor cells, or improved pharmacodynamic properties, their efficacy suffers from limitations in delivery options owing to the properties of the target microenvironment (Jain, R. K. {\99%) Nat Med 4, 655-657; Sanhai, W. R. et al. (2008) Nat Nanotechnol 3, 242-244; Chauhan, V et al. (2011) Annu Rev Chem Biomol Eng. 2(l):281-98). Limited approaches are currently available to overcome the delivery barriers for drugs.
Thus, the need exists for developing new agents and formulations that enhance the delivery, distribution, and/or efficacy of therapeutic or diagnostic agents, including nanotherapeutics {e.g., lipid- or polymeric nanoparticles and viruses), protein and nucleic acid drugs, targeted therapies, immune therapies {e.g., immune checkpoint blockers, vaccines and/or immune cells), and small molecule
chemotherapeutic agents.
SUMMARY OF INVENTION
The present invention discloses, at least in part, macromonomers, conjugates comprising the macromonomers (also referred to alternatively as "BRUSH", "Brush", or "BRUSH conjugates"), and brush arm star polymer particles comprising the conjugates (also referred to as "BASPs" or "BASP particles"), each further comprising an agent {e.g., one or more therapeutic agents, diagnostic agents, or
targeting moieties), as well as starting materials (e.g., crosslinkers) and other components (e.g., degradation components) thereof; as well as compositions (e.g., pharmaceutical compositions) comprising the same, and methods of making and using the same. Compositions comprising BASP particles are referred to as "BASP- compositions". Compositions comprising BRUSH conjugates are referred to as "BRUSH-compositions".
In some embodiments, the BASP-compositions or BRUSH-compositions enhance the delivery, distribution, release and/or efficacy of an agent to a desired target site in a subject, e.g., compared to the systemic release of the agent as a free form (e.g., not coupled to a BASP- or BRUSH-composition described herein). In one embodiment, the agent delivered or released is a therapeutic agent (e.g., an angiotensin receptor blocker (ARB), a vitamin D analog, a bromodomain inhibitor, or an indoleamine 2,3-dioxygenease (IDO) inhibitor, as described herein).
Without wishing to be bound by theory, the BASP-compositions or BRUSH- compositions disclosed herein comprise a conjugate of multiple macromonomers, each containing a therapeutic agent linked to the macromonomer, wherein the linker is preferentially labile or preferentially cleavable in a tissue microenvironment. Thus, the therapeutic agent can be preferentially released in the tissue microenvironment, e.g., compared to the systemic release of the therapeutic agent as a free form, thus allowing for a lower dosage of the agent in the particle. In one embodiment, the BASP-composition or BRUSH-composition comprises a tissue microenvironment cleavable linker connecting the therapeutic agent to the macromonomer, which preferentially releases the therapeutic agent upon exposure to a set of conditions present in the tissue microenvironment. In some embodiments, the tissue
microenvironment is a tumor or fibrotic microenvironment, e.g., exhibits one or more of decreased pH, hypoxic condition, or presence of an enzyme, e.g., esterase or protease, e.g., compared to a healthy tissue microenvironment. For example, BASP- compositions or BRUSH-compositions comprising certain microenvironment labile linkers were surprisingly found to release an agent at a yield between 10 to 100 times greater than other linkers (see e.g., Example 11, Figs. 14A to 14C).
Thus, the BASP-compositions or BRUSH-compositions disclosed herein can result in one or more advantages over systemic release of the free agent, including, but not limited to: (i) increasing the localization, release and/or delivery of the agent to a target tissue, e.g., a cancer or a fibrotic tissue (e.g., a desmoplastic or a fibrotic
tumor or tissue chosen from liver, kidney, lung or bone marrow (e.g., myelofibrotic bone marrow)); (ii) exhibiting increased release of the agent in a hypoxic
microenvironment, e.g., in a tumor or a fibrotic tissue (e.g., fibrotic or cirrhotic liver, or a tissue having renal fibrosis, pulmonary fibrosis or myelofibrosis); (iii) reducing a side effect of the agent by having a higher amount of released agent at a target site (e.g., in a hypoxic tumor), relative to other non-target sites (e.g., in intact and/or healthy blood vessels and/or normal or healthy tissues); and/or (iv) increasing the half-life of the agent.
Certain embodiments disclosed herein provide BASP-compositions or BRUSH-compositions for use in methods for treating or preventing a disorder, e.g., a fibrotic disorder, a cancer (e.g., a desmoplastic tumor or metastatic lesion), or an inflammatory disorder. In one embodiment, the disorder is a liver disorder. These embodiments comprise administering to a subject a BASP-composition or BRUSH- composition as described herein, as a single agent or as a combination with one or more additional therapeutic agents.
In one aspect, the present invention features a compound of Formula (I):

Formula (I)
or a salt thereof, wherein A is C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, or C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1; B is C1- C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, or C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1; ring T is a triazoldiyl moiety (e.g., a 1,2,3-triazoldiyl); X is an agent as described herein (e.g., an angiotensin receptor blocker (ARB), vitamin D analog, indoleamine 2,3-dioxygenease (IDO) inhibitor, or bromodomain inhibitor as described herein); P is alkylene or heteroalkylene (e.g., polyethylene glycol); each of L1 and L2 is independently a bond, C1-C12 alkylene, C2- C12 alkenylene, C2-C12 alkynylene, C1-C12 heteroalkylene, (C0-C12 alkylene)-arylene- (C0-C12 alkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 heteroalkylene), (C0-C12 alkylene)-heteroarylene-(C0-C12 alkylene), (C0-C12
heteroalkylene)-heteroarylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)- heteroarylene-(C0-C12 heteroalkylene), (C0-C12 alkylene)-heterocyclylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-heterocyclylene-(C0-C12 alkylene), or (C0-C12 heteroalkylene)-heterocyclylene-(C0-C12 heteroalkylene), wherein each alkylene, alkenylene, alkynylene, heteroalkylene, arylene, heteroarylene, or heterocyclylene is optionally substituted with 1-12 independently selected R2; L3 is a cleavable linker (e.g., a tissue microenvironment cleavable linker); each R1 and R2 is independently alkyl, alkenyl, alkynyl, heteroalkyl (e.g., polyethylene glycol), halo, cyano, oxo,– ORA,–NRBRC,–NRBC(O)RD, -C(O)NRBRC,–C(O)RD,–C(O)OH,–C(O)ORD,–SRE, or–S(O)mRE; each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,– C(O)NRBRC,–C(O)RD,–C(O)OH, or–C(O)ORD; each RB and RC is independently hydrogen or C1-C6 alkyl; each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl; and m is 1 or 2.
In another aspect, the present invention features a conjugate comprising at least two of a structure according to Formula (III):
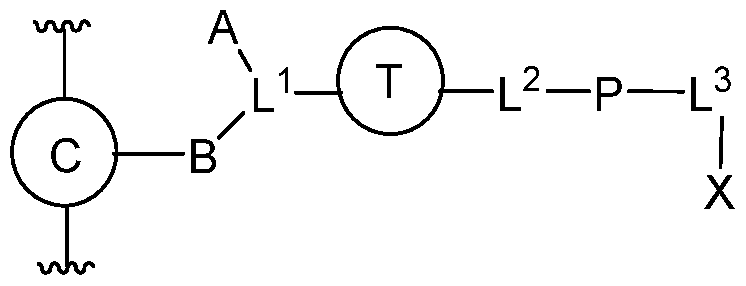
Formula (III)
. wherein Ring C is a carbocyclyl or heterocyclyl moiety; Ring T is a triazoldiyl moiety (e.g., a 1,2,3-triazoldiyl); A is C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, or C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1; B is C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, or C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1; X is an agent chosen from an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor; P is alkylene or heteroalkylene (e.g., polyethylene glycol); each of L1 and L2 is independently a bond, C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, C1-C12 heteroalkylene, (C0- C12 alkylene)-arylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), (C0-C12 heteroalkylene)- arylene-(C0-C12 heteroalkylene), (C0-C12 alkylene)-heteroarylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-heteroarylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-
heteroarylene-(C0-C12 heteroalkylene), (C0-C12 alkylene)-heterocyclylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-heterocyclylene-(C0-C12 alkylene), or (C0-C12 heteroalkylene)-heterocyclylene-(C0-C12 heteroalkylene), wherein each alkylene, alkenylene, alkynylene, heteroalkylene, arylene, heteroarylene, or heterocyclylene is optionally substituted with 1-12 independently selected R2; L3 is a tissue
microenvironment cleavable linker; each R1 and R2 is independently alkyl, alkenyl, alkynyl, heteroalkyl (e.g., polyethylene glycol), halo, cyano, oxo,–ORA,–NRBRC,– NRBC(O)RD, -C(O)NRBRC,–C(O)RD,–C(O)OH,–C(O)ORD,–SRE, or–S(O)mRE;
each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,–C(O)NRBRC,– C(O)RD,–C(O)OH, or–C(O)ORD; each RB and RC is independently hydrogen or C1- C6 alkyl; each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl; and m is 1 or 2.
In another aspect, the present invention comprises a BASP particle comprising at least two of a structure of Formula (III) as described herein.
In another aspect, the present invention features a composition, e.g.,
pharmaceutical composition, comprising one or more of a BASP particle or conjugate described herein, and a pharmaceutically acceptable carrier.
In another aspect, the present invention features a method of treating or preventing a disorder (e.g., a hyperproliferative disorder, a fibrotic disorder, and/or an inflammatory disorder) in a subject, comprising administering the conjugate or BASP particle described herein, e.g., as a single agent or in combination with other agent or therapy, or the composition thereof, to the subject.
In another aspect, the present invention features cross-linkers, which are useful to make the conjugates and BASP particles of the invention, as well as methods of making the conjugates and BASP particles of the invention, the details of which are described below.
Unless otherwise defined, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs. Although methods and materials similar or equivalent to those described herein can be used in the practice or testing of the present invention, suitable methods and materials are described below. All publications, patent applications, patents, and other references mentioned herein are incorporated by reference in their entirety. In addition, the materials, methods, and examples are illustrative only and not intended to be limiting.
Other features and advantages of the invention will be apparent from the detailed description, drawings, and from the claims. BRIEF DESCRIPTION OF THE DRAWINGS FIG.1 is an exemplary schematic overview of macromonomer, BRUSH and BASP structures and assembly.
FIG.2 shows the chemical structures of exemplary angiotensin receptor blockers (ARBs), as well as prodrugs and metabolites thereof, that may be
incorporated into BRUSH conjugates and BASP particles.
FIG.3 shows the chemical structures of exemplary vitamin D analogs that may be incorporated into BRUSH conjugates and BASP particles.
FIGs.4A to 4B show the chemical structures of exemplary bromodomain inhibitors that may be incorporated into BRUSH conjugates and BASP particles.
FIGs.5A to 5B show the characterization of BASP (15) comprising
telmisartan and an imaging agent, Cy7.5. FIG.5A depicts a gel permeation
chromatogram and FIG. 5B depicts a dynamic light scattering plot showing average BASP particle size.
FIGs.6A to 6B show the characterization of BASP (17) comprising
paricalcitol (pari or Pari) and an imaging agent, Cy7.5. FIG.6A depicts a gel permeation chromatogram and FIG.6B depicts a dynamic light scattering plot showing average BASP particle size.
FIGs.7A to 7B show the results of a single-dose pharmacokinetic study measuring free (non-conjugated) and BASP-bound telmisartan in tumor and blood from mice doses with various formulations of telmisartan. All test agents were dosed at equivalent concentrations of telmisartan at 10 mg/kg. The test agents were administered at 0 hr and whole blood and tissue collected at the indicated timepoints (n=3 animals). Generic telmisartan (Tel p.o.) was dosed at 10 mg/kg by oral gavage; BASPs (26) and (15) were both dosed at 100 mg/kg i.v and had similar 10% drug loading.
FIGs.8A to 8F are images of Cy7.5-conjugated Tel-BASP (15) taken in a non-invasive manner in live mice. The BASP was dosed at 10 mg/kg i.v in either a 4T1-tumor bearing mouse (FIGs.8A to 8C) or a naïve, non-tumor bearing mouse (FIGs.8D-8E) and the biodistribution of the Cy7.5 signal was captured on an IVIS instrument following 30 min, 73 hrs and 10 days post (15) administration.
FIGs.9A to 9B are images showing the visualization of Tel-BASP (15) in tumor tissue collected 6 days post administration of a single dose (100 mg/kg). Tissue sections (5 um) from (15) treated (+) and non-treated (-) animals were stained by H&E (FIG.9A) and imaged for Cy5 (FIG.9B) confirming significant drug uptake and retention in the tumor tissue.
FIGs.10A to 10B are graphs showing the quantification of Tel-BASP (15) in tumor tissue collected 6 days post administration of a single dose (100 mg/kg) by two complementary methods. FIG.10A shows the pharmacokinetic analysis of polymer- bound telmisartan by LC/MS/MS. FIG.10B depicts quantitative image analysis of Cy5.5 signal intensity in tissue sections. Both assays confirm significant, time- dependent drug uptake and retention in the tumor tissue of BASP particles.
FIGs.11A to 11C are images that show results of a biodistribution study of Tel-BASP (15) in major tissues comparing the signal of the Cy7.5 conjugated BASP particles (FIG.11B, 800 nm) to that of tissue autofluorescence (FIG.11A, 700 nM). Spleen, kidney, lung, heart, skin, liver brain gut and tumor were harvested from the mice and the formalin fixed organs were imaged directly on a Licor Odessey platform. The Heat map shows preferential delivery of Tel-BASP (15) to tumor, spleen and liver (FIG.11C).
FIGs.12A to 12B depict an in vivo comparison of two different formulations of Tel-BASP that incorporate different PEG lengths (PEG 2000K and PEG 3000K). FIG.12A summarizes the results of an LC/MS/MS PK assay, while FIG.12B shows Cy7.5 imaging of both Tel-BASP formulations (formalin-fixed 4T1 tumors). Both formulations show similar high drug retention in tumor (23% of injected dose).
FIG.13 is a graph showing the results of a long-term pharmacokinetic study of Tel-BASP (15) dosed in 4T1 tumor bearing mice. Whole blood, tumor and liver samples were analyzed for free and polymer-bound telmisartan at the indicated timepoints following a single i.v. dose of the BASP (10 mg/kg).
FIGs.14A to 14C compare the lability of three different telmisartan BASP constructs with resulting in vivo results. FIGs.14A to 14B are graphs showing the results of a single-dose pharmacokinetic study in 4T1 tumor bearing mice quantifying the time-dependent concentrations of free and polymer-bound telmisartan in whole blood (FIG.14A) and tumor tissue (FIG.14B) using LC/MS/MS. The predicted lability of ester bonds and the rational design of drug linkers (FIG.14C) resulted in
increased drug release in whole blood and tumor tissue reaching physiologically relevant levels in the tumor.
FIGs.15A to 15D are graphs showing the pharmacokinetic profile of intermediately labile Tel-BASP (19) and more labile Tel-BASP (20) in liver from a single-dose pharmacokinetic study in 4T1 tumor bearing mice. Graphs shows the time-dependent concentrations of free and polymer-bound telmisartan in liver tissue based on LC/MS/MS measurements (n=3 animals per timepoint).
FIGs.16A to 16B are graphs showing the results of a single-dose
pharmacokinetic study of Pari-BASP (17) (5000 µg/kg i.v.) in 4T-tumor bearing mice. Whole blood and tissues were collected at the indicated timepoints post dosing (n=3 animals per timepoint) and submitted for PK analysis for bound and free paricalcitol. The measurements of polymer-bound paricalcitol is shown as total free drug (FIG.16A) and a percent injection dose/g or /mL (FIG.16B).
FIGs.17A to 17C show biomarker-based evidence for microenvironmentally active release of paricalcitol from Pari-BASP (17). FIG.17A is an image of an in vitro model system of human stellate cells treated with paricalcitol (100 nm), in which the upregulation of VDR protein expression in response to ligand binding is validated by multiplex staining. FIG.17B shows the quantification of these data, while FIG. 17C shows the upregulation of VDR protein in vitro, supporting the release of biologically active paricalcitol in the disease tissue.
FIGs.18A to 18B show the results of dose range finding studies for Pari- BASP (17) in 4T1 tumor bearing Balb/c mice and in naïve non-tumor bearing FVB mice. FIG.18A shows imaging of Cy7.5-conjugated Pari-BASP particles (17) in formalin fixed tumor tissues collected 72 hrs post i.v. administration of a single dose; the image shows the dose-dependent tumor uptake of BASP nanoparticles. Generic paricalcitol (Zemplar), dosed at 10 µg/kg, is equivalent in terms of the amount of paricalcitol to the dosing of Pari-BASP (17) at 100 µg/kg with 10% drug loading). To capture this, the Pari-BASP (17) doses are shown as 1X (100 µg/kg), 5X (500 µg/kg) and 15X (1500 µg/kg) which is relative to the 1X dose of non-conjugated, generic Paricalcitol (10 µg/kg).
FIGs.19A to 19B show the results of an anti-cancer efficacy study in a pancreatic tumor model AK4.4 comparing gemcitabine alone, telmisartan alone, telmisartan plus gemcitabine, Tel-BASP (21) and Tel-BASP (21) plus gemcitabine.
FIG.19A describes the study design with survival endpoints, while FIG.19B summarizes the dose groups and route of administration.
FIGs.20A to 20B are graphs showing that Tel-BASP (21) combined with gemcitabine exhibits superior survival benefit compared to generic telmisartan in combination with gemcitabine.
FIGs.21A to 21D are images showing that Tel-BASP (21) plus gemcitabine enhances the number of AGTR1 positive cells in AK4.4 Tumors as compated to either agent alone.
FIGs.22A to 22B show the anti-cancer efficacy of Tel-BASP (21) in the pancreatic cancer model AK4.4 in combination with standard of care chemotherapy. FIG.22A describes the study design with a single endpoint, while FIG.22B summarizes the dose groups and route of administration.)
FIGs.23A to 23C show efficacy endpoints on tumor size (FIG.23A) and mass (FIGs.23B to 23C) of treatment with gemcitabine alone, telmisartan alone, telmisartan plus gemcitabine, Tel-BASP (21) and Tel-BASP (21) plus gemcitabine. FIG.23A is a photo of formalin fixed tumors arranged by dose groups to visualize the effect of drug treatment. FIGs.23B to 23C are graphs showing the quantification of average tumor volume and mass at the endpoint of study (Day 19 post dosing).
FIGs.24A to 24D show the results of an anti-fibrosis efficacy study in a CCl4-induced liver fibrosis mouse model, in which Tel-BASP (22) (700 mg/kg; dosed i.v. once weekly) was compared to generic telmisartan (10 mg/kg) dosed daily by oral gavage. Liver fibrosis was induced by biweekly i.p. injection of CCl4 during the 6 week study period with administration of the experimental test agents during the last 2 weeks of the study (Week 4 to Week 6). Bodyweight measurements for the duration of the study show that the Tel-BASP (22) formulation is well tolerated (FIG.24A) and that it is more efficacious at reducing elevated liver enzymes ALT (FIG.24B) and AST (FIG.24C) and total bilirubin (FIG.24D) at the 6 week endpoint of the study.
FIGs.25A to 25E show Tel-BASP (22) treatment is more efficacious than generic telmisartan at reducing CCl4-induced liver inflammation (FIGs.25A to 25D) and that the enhanced activity of Tel-BASP correlates with an efficient drug delivery (FIG.25E) measured by LC/MS/MS at study endpoint. Tissue sections from animals treated as indicated were stained with anti-S100A4 antibody, and the staining was developed with DAB and counterstained with hematoxylin.
FIGs.26A to 26C show the efficacy of Tel-BASP (22) treatment in a CCl4- induced liver fibrosis mouse model (700 mg/kg; dosed i.v. once weekly) compared to generic telmisartan (10 mg/kg) dosed daily by oral gavage. The necrosis score was assessed according to the scoring system shown (FIG.26A) and subsequently quantified (FIG.26B, n=9 per group).
FIGs.27A to 27D show that Tel-BASP (22) treatment reverses CCl4-induced glycogen depletion. Tissue sections from animals treated as indicated were stained for glycogen using the Periodic-Schiff process.
FIGs.28A to 28D are images that show Tel-BASP (22) treatment reduces CCl4-induced liver fibrosis. Tissue sections from animals treated as indicated were stained with anti-BigH3 (TGFBI) antibody, and staining was developed with DAB and counterstained with hematoxylin.
FIGs.29A and 29B show the blood biochemistry results from two
independent CCl4 mouse liver fibrosis studies (Study 1 and Study 2) with Study 2 being a head-to-head comparison of Tel-BRUSH (28) vs Tel-BASP (26) vs generic Telmisartan (Tel, tel, or TEL) showing (A) plasma alanine aminotransferase (ALT) and (B) aspartate aminotransferase (AST) levels in response to the indicated drug treatments. The results show superior liver enzyme normalization by Tel-BRUSH (28) and Tel-BASP (26) compared to generic Tel and identifies the Tel-BASP dose of 300mg/kg i.v. q.w. as an minimal efficacious dose (2 doses) in this model.
FIG.30 shows Tel-BASP (26) and Tel-BRUSH (28) treatment protects against periportal hepatocyte necrosis. Histopathology evaluation of paraffin- embedded and H&E-stained liver tissue section from 6 week CCl4 liver efficacy study with drug treatment dusing thet last 2 weeks of CCl4 intoxication. Representative liver tissue sections were score on blinded samples by pathologist according to the following necrosis pathology scoring system (scale of 1 to 4). Score 1 = Scattered foci of coagulative degeneration/necrosis; Score 2 = Coalescing periportal
degeneration/necrosis; Score 3 = Periportal and interface degeneration/necrosis; score 4 = Pan-lobular degeneration/necrosis. Significant reduction of liver necrosis was observed.
FIGs.31A and 31B show treatment protects against periportal hepatocyte necrosis. Molecular histology evaluation and quantification of cleaved caspase-3 on formalin-fixed and paraffin-embedded liver tissue section from 6 week CCl4 liver efficacy study with drug treatment during the last 2 weeks of CCl4 intoxication. (B)
Representative liver tissue sections (20 images per tissue block) were digitally quantified showing (B) significant reduction of hepatocyte liver apoptosis with both Tel-BASP (22) and Tel-BRUSH (28) treatments at 300 mpk dose (total of 2 doses).
FIGs.32A and 32B show the effect of Tel-BASP (22) and Tel-BRUSH (28) on gene expression of indicated genes (listed by their official human genome symbol) in the CCl4 mouse fibrosis liver study. Tel-BASP and Tel-BRUSH both modulated several genes involved in fibrotic processes with a trend for Tel-BRUSH to be more potent than Tel-BASP.
FIGs.33A and 33B shows terminal Liver and Blood PK analyses from the CCl4 liver fibrosis disease model measuring both released (i.e, free) and conjugated Telmisartan and showing the ratio of released to conjugated drug present in each compartment. The terminal PK timepoint is 1 week post the 2nd dosage of Tel- BRUSH (28) and Tel-BASP (22) dosed at 300 mpk i.v. at the beginning of week 1 (1st dose) and at the beginning of week2 (2nd dose). The PK data for generic telmisart (Telm) is after 14 daily doses and 24 hour post last dose. The data shows that Tel- BRUSH delivers 5 times more active pharmaceutical ingredient (API) to liver & shows 10 times higher fractional release compared to Tel-BASP (Week 6; 300 mg/kg i.v.; total of 2 doses QW)
FIG.34 shows that plasma levels of conjugated (top curve) and free (bottom curve) telmisartan demonstrated dose proportionality when Tel-BASP (26) was administered to dogs.
FIG.35 shows the comparative effects of Tel-BASP (21), free telmisartan (“Telm”), and immuno-oncology treatment (a combination of anti-PD1 and anti- CTLA4;“PD-1/CTLA-4”) monotherapy, as well as combinations of immuno- oncology treatment with either Tel-BASP (21) (“Tel-BASP (21) + PD-1/CTLA-4”) or free telmisartan (“Telm + PD-1/CTLA-4”) on tumor size in a mouse breast cancer model.
FIGs.36A to 36B show the effects of immuno-oncology treatment (“anti-PD- 1”) alone or in combination with Pari-BASP (27) (“anti-PD-1/Pari-BASP (27)”) on tumor size (FIG.36A) and survival (FIG.36B) in a mouse melanoma model.
FIGs.37A to 37B show the effects of immuno-oncology treatment (“anti-PD- 1”) alone or in combination with Pari-BASP (27) (“anti-PD-1/Pari-BASP (27)”) on tumor size (FIG.37A) and survival (FIG.37B) in a mouse colon cancer model.
FIGs.38A to 38C show the experimental protocol (FIG.38A) for and effects of Telmisartan, Tel-BASP (26), and Tel-BRUSH (28) on fibrosis score (FIG.38B) and NAFLD activity (FIG.38C) in the Gubra mouse fatty liver disease/NASH model.
FIGs.39A and 39B shows terminal Liver and Blood PK Analyses from the Gubra fatty liver disease model measuring both released (i.e, free) and conjugated Telmisartan and showing the ration of released to conjugated drug present in each compartment. The terminal PK timepoint is 5 week post the 2nd dosage of Tel- BRUSH (28) and Tel-BASP (26) dosed at 300 mpk i.v. at the beginning of week 1 (1st dose) and at the beginning of week 3 (2nd dose). The data shows that Tel-Brush (28) releases 3-5 times more API and has 10 times higher fractional release in liver compared to Tel-BASP.
FIGs.40A to 40C show efficient drug delivery of Tel-BASP (26) to mouse liver at 72 hr post dose (300 mpk i.v.) using two different approaches: (FIG. 40A) anti-PEG antibody-based immunohistochemistry (IHC) staining or, (FIG.40B) direct imaging of the Cy7.5 fluorophore tracer revealing regional drug biodistribution to sinusoid areas (white arrows). Moreover, co-staining of IBA-1 macrophage marker with Cy-7.5 (FIG.40C) shows co-location of drug to this cell type (white arrows).
FIGs.41A to 41B show liver (FIG.41A) and plasma (FIG.41B) PK for Male Golden-Syrian hamsters (6-week old) dosed daily with 10 mg/kg oral telmisartan for three consecutive days or dosed with a single i.v. dose (300 mg/kg) of Tel-BRUSH (29) or Tel-BASP (22). Liver and blood samples were collected for PK analysis (n=3 animals per time-point: 24 hours, 72 hours, and 168 hours for Tel-Brush (29) and Tel- BASP (22), and 2 hours, 4 hours, and 24 hours for oral telmisartan). DETAILED DESCRIPTION OF INVENTION
Disclosed herein, at least in part, are conjugates and particles comprising brush-arm star polymers (BASPs) coupled to an agent (e.g., one or more therapeutic agents, diagnostic agents, or targeting moieties), starting materials, intermediates, and degradation components thereof (collectively referred to herein as“BRUSH- compositions” or“BASP-compositions”), as well as pharmaceutical compositions and methods of making and using the same.
In some embodiments, the BRUSH-compositions or BASP-compositions enhance the delivery, distribution, release and/or efficacy of an agent in a subject to a desired target site, e.g., compared to the systemic release of the agent as a free form
(e.g., not coupled to a BRUSH-composition or BASP-composition described herein). In one embodiment, the agent delivered or released is a therapeutic agent (e.g., an angiotensin receptor blocker (ARB), a vitamin D analog, an indoleamine 2,3- dioxigenase 1 (IDO) inhibitor, or a bromodomain inhibitor as described herein).
Various aspects of the invention are described in further detail in the following subsections. Definitions
For convenience, certain terms employed herein, in the specification, examples and appended claims are collected herein.
Unless otherwise required by context, singular terms shall include pluralities and plural terms shall include the singular.
The following definitions are more general terms used throughout the present application:
The singular terms“a,”“an,” and“the” include plural referents unless context clearly indicates otherwise. Similarly, the word“or” is intended to include“and” unless the context clearly indicates otherwise.
Other than in the operating examples, or where otherwise indicated, all numbers expressing quantities of ingredients or reaction conditions used herein should be understood as modified in all instances by the term“about.”“About” and “approximately” shall generally mean an acceptable degree of error for the quantity measured given the nature or precision of the measurements. Exemplary degrees of error are within 20 percent (%), typically, within 10%, and more typically, within 5%, 4%, 3%, 2% or 1% of a given value or range of values. Selected Chemical Definitions
Definitions of specific functional groups and chemical terms are described in more detail below. The chemical elements are identified in accordance with the Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics, 75th Ed., inside cover, and specific functional groups are generally defined as described therein. Additionally, general principles of organic chemistry, as well as specific functional moieties and reactivity, are described in Organic Chemistry, Thomas Sorrell, University Science Books, Sausalito, 1999; Smith and March March’s Advanced Organic Chemistry, 5th Edition, John Wiley & Sons, Inc., New
York, 2001; Larock, Comprehensive Organic Transformations, VCH Publishers, Inc., New York, 1989; and Carruthers, Some Modern Methods of Organic Synthesis, 3rd Edition, Cambridge University Press, Cambridge, 1987.
Compounds described herein can comprise one or more asymmetric centers, and thus can exist in various stereoisomeric forms, e.g., enantiomers and/or diastereomers. For example, the compounds described herein can be in the form of an individual enantiomer, diastereomer or geometric isomer, or can be in the form of a mixture of stereoisomers, including racemic mixtures and mixtures enriched in one or more stereoisomer. Isomers can be isolated from mixtures by methods known to those skilled in the art, including chiral high pressure liquid chromatography (HPLC) and the formation and crystallization of chiral salts; or preferred isomers can be prepared by asymmetric syntheses. See, for example, Jacques et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Wilen et al., Tetrahedron 33:2725 (1977); Eliel, E.L. Stereochemistry of Carbon Compounds (McGraw–Hill, NY, 1962); and Wilen, S.H. Tables of Resolving Agents and Optical Resolutions p. 268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN 1972). The invention additionally encompasses compounds as individual isomers substantially free of other isomers, and alternatively, as mixtures of various isomers.
When a range of values is listed, it is intended to encompass each value and sub–range within the range. For example,“C1-C6 alkyl” is intended to encompass, C1, C2, C3, C4, C5, C6, C1-C6, C1-C5, C1-C4, C1-C3, C1-C2, C2-C6, C2-C5, C2-C4, C2-C3, C3- C6, C3-C5, C3-C4, C4-C6, C4-C5, and C5-C6 alkyl. Unless otherwise provided, a range expressed in the form“between XX and YY” or“between XX and YY, inclusive” refers to a range between XX and YY, inclusive, wherein each of XX and YY is independently a number (e.g., integer, fraction, or percentage).
The term“alkyl” refers to a radical of a straight–chain or branched saturated hydrocarbon group. In some embodiments, an alkyl group has 1 to 10 carbon atoms (“C1-C10 alkyl”), 1 to 9 carbon atoms (“C1-C9 alkyl”), 1 to 8 carbon atoms (“C1-C8 alkyl”), 1 to 7 carbon atoms (“C1-C7 alkyl”), 1 to 6 carbon atoms (“C1-C6 alkyl”), 1 to 5 carbon atoms (“C1-C5 alkyl”), 1 to 4 carbon atoms (“C1-C4 alkyl”), 1 to 3 carbon atoms (“C1-C3 alkyl”), 1 to 2 carbon atoms (“C1-C2 alkyl”), or 1 carbon atom (“C1 alkyl”). Examples of C1-C6 alkyl groups include methyl (C1), ethyl (C2), n–propyl (C3), isopropyl (C3), n–butyl (C4), tert–butyl (C4), sec–butyl (C4), iso–butyl (C4), n– pentyl (C5), 3–pentanyl (C5), amyl (C5), neopentyl (C5), 3–methyl–2–butanyl (C5),
tertiary amyl (C5), and n–hexyl (C6). Additional examples of alkyl groups include n– heptyl (C7), n–octyl (C8) and the like. Unless otherwise specified, each instance of an alkyl group is independently unsubstituted (an“unsubstituted alkyl”) or substituted (a “substituted alkyl”) with one or more substituents.
The term“alkenyl” refers to a radical of a straight–chain or branched hydrocarbon group having from 2 to 10 carbon atoms and one or more carbon-carbon double bonds (e.g., 1, 2, 3, or 4 double bonds). In some embodiments, an alkenyl group has 2 to 9 carbon atoms (“C2–9 alkenyl”). In some embodiments, an alkenyl group has 2 to 8 carbon atoms (“C2–8 alkenyl”). In some embodiments, an alkenyl group has 2 to 7 carbon atoms (“C2–7 alkenyl”). In some embodiments, an alkenyl group has 2 to 6 carbon atoms (“C2–6 alkenyl”). In some embodiments, an alkenyl group has 2 to 5 carbon atoms (“C2–5 alkenyl”). In some embodiments, an alkenyl group has 2 to 4 carbon atoms (“C2–4 alkenyl”). In some embodiments, an alkenyl group has 2 to 3 carbon atoms (“C2–3 alkenyl”). In some embodiments, an alkenyl group has 2 carbon atoms (“C2 alkenyl”). The one or more carbon–carbon double bonds can be internal (such as in 2–butenyl) or terminal (such as in 1–butenyl).
Examples of C2–4 alkenyl groups include ethenyl (C2), 1–propenyl (C3), 2–propenyl (C3), 1–butenyl (C4), 2–butenyl (C4), butadienyl (C4), and the like. Examples of C2–6 alkenyl groups include the aforementioned C2–4 alkenyl groups as well as pentenyl (C5), pentadienyl (C5), hexenyl (C6), and the like. Unless otherwise specified, each instance of an alkenyl group is independently unsubstituted (an“unsubstituted alkenyl”) or substituted (a“substituted alkenyl”) with one or more substituents.
The term“alkynyl” refers to a radical of a straight–chain or branched hydrocarbon group having from 2 to 10 carbon atoms and one or more carbon-carbon triple bonds (e.g., 1, 2, 3, or 4 triple bonds) (“C2–10 alkynyl”). In some embodiments, an alkynyl group has 2 to 9 carbon atoms (“C2–9 alkynyl”), 2 to 8 carbon atoms (“C2–8 alkynyl”), 2 to 7 carbon atoms (“C2–7 alkynyl”), 2 to 6 carbon atoms (“C2–6 alkynyl”), 2 to 5 carbon atoms (“C2–5 alkynyl”), 2 to 4 carbon atoms (“C2–4 alkynyl”), 2 to 3 carbon atoms (“C2–3 alkynyl”), or 2 carbon atoms (“C2 alkynyl”). The one or more carbon–carbon triple bonds can be internal (such as in 2–butynyl) or terminal (such as in 1–butynyl). Examples of C2–4 alkynyl groups include, without limitation, ethynyl (C2), 1–propynyl (C3), 2–propynyl (C3), 1–butynyl (C4), 2–butynyl (C4), and the like. Examples of C2–6 alkenyl groups include the aforementioned C2–4 alkynyl groups as well as pentynyl (C5), hexynyl (C6), and the like. Unless otherwise specified, each
instance of an alkynyl group is independently unsubstituted (an“unsubstituted alkynyl”) or substituted (a“substituted alkynyl”) with one or more substituents.
The term“heteroalkyl” refers to an alkyl group which further includes at least one heteroatom (e.g., 1, 2, 3, or 4 heteroatoms) selected from oxygen, nitrogen, phosphorus, or sulfur within (i.e., inserted between adjacent carbon atoms of) and/or placed at one or more terminal position(s) of the parent chain. In certain
embodiments, a heteroalkyl group refers to a saturated group having from 1 to 10 carbon atoms and 1 or more heteroatoms within the parent chain (“C1–C10
heteroalkyl”), 1 to 9 carbon atoms and 1 or more heteroatoms within the parent chain (“C1–C9 heteroalkyl”), 1 to 8 carbon atoms and 1 or more heteroatoms within the parent chain (“C1–C8 heteroalkyl”), 1 to 7 carbon atoms and 1 or more heteroatoms within the parent chain (“C1–C7 heteroalkyl”), 1 to 6 carbon atoms and 1 or more heteroatoms within the parent chain (“C1–C6 heteroalkyl”), 1 to 5 carbon atoms and 1 or more heteroatoms within the parent chain (“C1–C5 heteroalkyl”), 1 to 4 carbon atoms and 1or more heteroatoms within the parent chain (“C1–C4 heteroalkyl”), 1 to 3 carbon atoms and 1 or more heteroatoms within the parent chain (“C1–C3
heteroalkyl”), 1 to 2 carbon atoms and 1 heteroatom within the parent chain (“C1–C2 heteroalkyl”), or 1 carbon atom and 1 heteroatom (“C1 heteroalkyl”). Unless otherwise specified, each instance of a heteroalkyl group is independently
unsubstituted (an“unsubstituted heteroalkyl”) or substituted (a“substituted heteroalkyl”) with one or more substituents.
The term“carbocyclyl” or“carbocyclic” or“cycloalkyl” refers to a radical of a non–aromatic cyclic hydrocarbon group having from 3 to 10 ring carbon atoms (“C3–10 carbocyclyl”) and zero heteroatoms in the non–aromatic ring system. In some embodiments, a carbocyclyl group has 3 to 8 ring carbon atoms (“C3–8 carbocyclyl”), 3 to 7 ring carbon atoms (“C3–7 carbocyclyl”), 3 to 6 ring carbon atoms (“C3–6 carbocyclyl”), 4 to 6 ring carbon atoms (“C4–6 carbocyclyl”), 5 to 6 ring carbon atoms (“C5–6 carbocyclyl”), or 5 to 10 ring carbon atoms (“C5–10 carbocyclyl”). Exemplary C3–6 carbocyclyl groups include, without limitation, cyclopropyl (C3), cyclopropenyl (C3), cyclobutyl (C4), cyclobutenyl (C4), cyclopentyl (C5), cyclopentenyl (C5), cyclohexyl (C6), cyclohexenyl (C6), cyclohexadienyl (C6), and the like. Exemplary C3–8 carbocyclyl groups include, without limitation, the aforementioned C3–6 carbocyclyl groups as well as cycloheptyl (C7), cycloheptenyl (C7), cycloheptadienyl (C7), cycloheptatrienyl (C7), cyclooctyl (C8), cyclooctenyl (C8),
bicyclo[2.2.1]heptanyl (C7), bicyclo[2.2.2]octanyl (C8), and the like. Exemplary C3–10 carbocyclyl groups include, without limitation, the aforementioned C3–8 carbocyclyl groups as well as cyclononyl (C9), cyclononenyl (C9), cyclodecyl (C10), cyclodecenyl (C10), octahydro–1H–indenyl (C9), decahydronaphthalenyl (C10), spiro[4.5]decanyl (C10), and the like. As the foregoing examples illustrate, in certain embodiments, the carbocyclyl group is either monocyclic (“monocyclic carbocyclyl”) or polycyclic (e.g., containing a fused, bridged or spiro ring system such as a bicyclic system (“bicyclic carbocyclyl”) or tricyclic system (“tricyclic carbocyclyl”)) and can be saturated or can contain one or more carbon–carbon double or triple bonds.
“Carbocyclyl” also includes ring systems wherein the carbocyclyl ring, as defined above, is fused with one or more aryl or heteroaryl groups wherein the point of attachment is on the carbocyclyl ring, and in such instances, the number of carbons continue to designate the number of carbons in the carbocyclic ring system. Unless otherwise specified, each instance of a carbocyclyl group is independently
unsubstituted (an“unsubstituted carbocyclyl”) or substituted (a“substituted carbocyclyl”) with one or more substituents.
The term“heterocyclyl” or“heterocyclic” refers to a radical of a 3– to 14– membered non–aromatic ring system having ring carbon atoms and 1 to 4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, phosphorus, and sulfur (“3–14 membered heterocyclyl”). In heterocyclyl groups that contain one or more nitrogen atoms, the point of attachment can be a carbon or nitrogen atom, as valency permits. A heterocyclyl group can either be monocyclic (“monocyclic heterocyclyl”) or polycyclic (e.g., a fused, bridged or spiro ring system such as a bicyclic system (“bicyclic heterocyclyl”) or tricyclic system (“tricyclic heterocyclyl”)), and can be saturated or can contain one or more carbon– carbon double or triple bonds. Heterocyclyl polycyclic ring systems can include one or more heteroatoms in one or both rings.“Heterocyclyl” also includes ring systems wherein the heterocyclyl ring, as defined above, is fused with one or more carbocyclyl groups wherein the point of attachment is either on the carbocyclyl or heterocyclyl ring, or ring systems wherein the heterocyclyl ring, as defined above, is fused with one or more aryl or heteroaryl groups, wherein the point of attachment is on the heterocyclyl ring, and in such instances, the number of ring members continue to designate the number of ring members in the heterocyclyl ring system. Unless otherwise specified, each instance of heterocyclyl is independently unsubstituted (an
“unsubstituted heterocyclyl”) or substituted (a“substituted heterocyclyl”) with one or more substituents.
In some embodiments, a heterocyclyl group is a 5–10 membered non– aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, phosphorus, and sulfur (“5–10 membered heterocyclyl”). In some embodiments, a heterocyclyl group is a 5–8 membered non–aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, phosphorus, and sulfur (“5–8 membered heterocyclyl”). In some
embodiments, a heterocyclyl group is a 5–6 membered non–aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, phosphorus, and sulfur (“5–6 membered heterocyclyl”). In some embodiments, the 5–6 membered heterocyclyl has 1–3 ring heteroatoms selected from nitrogen, oxygen, phosphorus, and sulfur. In some embodiments, the 5–6 membered heterocyclyl has 1–2 ring heteroatoms selected from nitrogen, oxygen, phosphorus, and sulfur. In some embodiments, the 5–6 membered heterocyclyl has 1 ring heteroatom selected from nitrogen, oxygen, phosphorus, and sulfur.
Exemplary 3–membered heterocyclyl groups containing 1 heteroatom include, without limitation, aziridinyl, oxiranyl, and thiiranyl. Exemplary 4–membered heterocyclyl groups containing 1 heteroatom include, without limitation, azetidinyl, oxetanyl and thietanyl. Exemplary 5–membered heterocyclyl groups containing 1 heteroatom include, without limitation, tetrahydrofuranyl, dihydrofuranyl,
tetrahydrothiophenyl, dihydrothiophenyl, pyrrolidinyl, dihydropyrrolyl, and pyrrolyl– 2,5–dione. Exemplary 5–membered heterocyclyl groups containing 2 heteroatoms include, without limitation, dioxolanyl, oxathiolanyl and dithiolanyl. Exemplary 5– membered heterocyclyl groups containing 3 heteroatoms include, without limitation, triazolinyl, oxadiazolinyl, and thiadiazolinyl. Exemplary 6–membered heterocyclyl groups containing 1 heteroatom include, without limitation, piperidinyl,
tetrahydropyranyl, dihydropyridinyl, and thianyl. Exemplary 6–membered
heterocyclyl groups containing 2 heteroatoms include, without limitation, piperazinyl, morpholinyl, dithianyl, and dioxanyl. Exemplary 6–membered heterocyclyl groups containing 3 heteroatoms include, without limitation, triazinyl. Exemplary 7– membered heterocyclyl groups containing 1 heteroatom include, without limitation,
azepanyl, oxepanyl, and thiepanyl. Exemplary 8–membered heterocyclyl groups containing 1 heteroatom include, without limitation, azocanyl, oxecanyl and thiocanyl. Exemplary bicyclic heterocyclyl groups include, without limitation, indolinyl, isoindolinyl, dihydrobenzofuranyl, dihydrobenzothienyl, tetrahydrobenzo- thienyl, tetrahydrobenzofuranyl, tetrahydroindolyl, tetrahydroquinolinyl,
tetrahydroisoquinolinyl, decahydroquinolinyl, decahydroisoquinolinyl,
octahydrochromenyl, octahydroisochromenyl, decahydronaphthyridinyl, decahydro– 1,8–naphthyridinyl, octahydropyrrolo[3,2–b]pyrrole, indolinyl, phthalimidyl, naphthalimidyl, chromanyl, chromenyl, 1H–benzo[e][1,4]diazepinyl, 1,4,5,7–tetra- hydropyrano[3,4–b]pyrrolyl, 5,6–dihydro–4H–furo[3,2–b]pyrrolyl, 6,7–dihydro–5H– furo[3,2–b]pyranyl, 5,7–dihydro–4H–thieno[2,3–c]pyranyl, 2,3–dihydro–1H– pyrrolo[2,3–b]pyridinyl, 2,3–dihydrofuro[2,3–b]pyridinyl, 4,5,6,7–tetrahydro–1H– pyrrolo[2,3–b]pyridinyl, 4,5,6,7–tetrahydrofuro[3,2–c]pyridinyl, 4,5,6,7–tetrahydro- thieno[3,2–b]pyridinyl, 1,2,3,4–tetrahydro–1,6–naphthyridinyl, and the like.
The term“aryl” refers to a radical of a monocyclic or polycyclic (e.g., bicyclic or tricyclic) 4n+2 aromatic ring system (e.g., having 6, 10, or 14 π electrons shared in a cyclic array) having 6–14 ring carbon atoms and zero heteroatoms provided in the aromatic ring system (“C6–14 aryl”). In some embodiments, an aryl group has 6 ring carbon atoms (“C6 aryl”; e.g., phenyl). In some embodiments, an aryl group has 10 ring carbon atoms (“C10 aryl”; e.g., naphthyl such as 1–naphthyl and 2–naphthyl). In some embodiments, an aryl group has 14 ring carbon atoms (“C14 aryl”; e.g., anthracyl).“Aryl” also includes ring systems wherein the aryl ring, as defined above, is fused with one or more carbocyclyl or heterocyclyl groups wherein the radical or point of attachment is on the aryl ring, and in such instances, the number of carbon atoms continue to designate the number of carbon atoms in the aryl ring system.
Unless otherwise specified, each instance of an aryl group is independently unsubstituted (an“unsubstituted aryl”) or substituted (a“substituted aryl”) with one or more substituents.
The term“heteroaryl” refers to a radical of a 5–14 membered monocyclic or polycyclic (e.g., bicyclic, tricyclic) 4n+2 aromatic ring system (e.g., having 6, 10, or 14 π electrons shared in a cyclic array) having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–14 membered
heteroaryl”). In heteroaryl groups that contain one or more nitrogen atoms, the point of attachment can be a carbon or nitrogen atom, as valency permits. Heteroaryl polycyclic ring systems can include one or more heteroatoms in one or both rings. “Heteroaryl” includes ring systems wherein the heteroaryl ring, as defined above, is fused with one or more carbocyclyl or heterocyclyl groups wherein the point of attachment is on the heteroaryl ring, and in such instances, the number of ring members continue to designate the number of ring members in the heteroaryl ring system.“Heteroaryl” also includes ring systems wherein the heteroaryl ring, as defined above, is fused with one or more aryl groups wherein the point of attachment is either on the aryl or heteroaryl ring, and in such instances, the number of ring members designates the number of ring members in the fused polycyclic
(aryl/heteroaryl) ring system. Polycyclic heteroaryl groups wherein one ring does not contain a heteroatom (e.g., indolyl, quinolinyl, carbazolyl, and the like) the point of attachment can be on either ring, i.e., either the ring bearing a heteroatom (e.g., 2– indolyl) or the ring that does not contain a heteroatom (e.g., 5–indolyl). A heteroaryl group be monovalent or may have more than one point of attachment to another moiety (e.g., it may be divalent, trivalent, etc.), although the valency may be specified directly in the name of the group. For example,“triazoldiyl” refers to a divalent triazolyl moiety.
In some embodiments, a heteroaryl group is a 5–10 membered aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–10 membered heteroaryl”). In some embodiments, a heteroaryl group is a 5–8 membered aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–8 membered heteroaryl”). In some embodiments, a heteroaryl group is a 5–6 membered aromatic ring system having ring carbon atoms and 1–4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur (“5–6 membered heteroaryl”). In some embodiments, the 5–6 membered heteroaryl has 1–3 ring heteroatoms selected from nitrogen, oxygen, and sulfur. In some embodiments, the 5–6 membered heteroaryl has 1–2 ring heteroatoms selected from nitrogen, oxygen, and sulfur. In some embodiments, the 5– 6 membered heteroaryl has 1 ring heteroatom selected from nitrogen, oxygen, and
sulfur. Unless otherwise specified, each instance of a heteroaryl group is
independently unsubstituted (an“unsubstituted heteroaryl”) or substituted (a “substituted heteroaryl”) with one or more substituents.
Exemplary 5–membered heteroaryl groups containing 1 heteroatom include, without limitation, pyrrolyl, furanyl, and thiophenyl. Exemplary 5–membered heteroaryl groups containing 2 heteroatoms include, without limitation, imidazolyl, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, and isothiazolyl. Exemplary 5–membered heteroaryl groups containing 3 heteroatoms include, without limitation, triazolyl, oxadiazolyl, and thiadiazolyl. Exemplary 5–membered heteroaryl groups containing 4 heteroatoms include, without limitation, tetrazolyl. Exemplary 6–membered heteroaryl groups containing 1 heteroatom include, without limitation, pyridinyl. Exemplary 6–membered heteroaryl groups containing 2 heteroatoms include, without limitation, pyridazinyl, pyrimidinyl, and pyrazinyl. Exemplary 6–membered heteroaryl groups containing 3 or 4 heteroatoms include, without limitation, triazinyl and tetrazinyl, respectively. Exemplary 7–membered heteroaryl groups containing 1 heteroatom include, without limitation, azepinyl, oxepinyl, and thiepinyl. Exemplary 5,6–bicyclic heteroaryl groups include, without limitation, indolyl, isoindolyl, indazolyl, benzotriazolyl, benzothiophenyl, isobenzothiophenyl, benzofuranyl, benzoisofuranyl, benzimidazolyl, benzoxazolyl, benzisoxazolyl, benzoxadiazolyl, benzthiazolyl, benzisothiazolyl, benzthiadiazolyl, indolizinyl, and purinyl. Exemplary 6,6–bicyclic heteroaryl groups include, without limitation, naphthyridinyl, pteridinyl, quinolinyl, isoquinolinyl, cinnolinyl, quinoxalinyl, phthalazinyl, and quinazolinyl. Exemplary tricyclic heteroaryl groups include, without limitation, phenanthridinyl, dibenzofuranyl, carbazolyl, acridinyl, phenothiazinyl, phenoxazinyl and phenazinyl.
As understood from the above, alkyl, alkenyl, alkynyl, carbocyclyl, aryl, and heteroaryl groups are, in certain embodiments, optionally substituted. Optionally substituted refers to a group which may be substituted or unsubstituted (e.g., “substituted” or“unsubstituted” alkyl). In general, the term“substituted” means that at least one hydrogen present on a group is replaced with a permissible substituent, e.g., a substituent which upon substitution results in a stable compound, e.g., a compound which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, or other reaction. Unless otherwise indicated, a“substituted” group has a substituent at one or more substitutable positions of the group, and when more than one position in any given structure is substituted, the
substituent is either the same or different at each position. The term“substituted” is contemplated to include substitution with all permissible substituents of organic compounds, any of the substituents described herein that results in the formation of a stable compound. The present invention contemplates any and all such combinations in order to arrive at a stable compound. For purposes of this invention, heteroatoms such as nitrogen may have hydrogen substituents and/or any suitable substituent as described herein which satisfy the valencies of the heteroatoms and results in the formation of a stable moiety.
Exemplary carbon atom substituents include, but are not limited to, halogen, −CN,−NO2,−N3,−SO2H,−SO3H,−OH,−ORaa,−ON(Rbb)2,−N(Rbb)2,−N(Rbb) +
3 X−, −N(ORcc)Rbb,−SH,−SRaa,−SSRcc,−C(=O)Raa,−CO2H,−CHO,−C(ORcc)2,−CO2Raa, −OC(=O)Raa,−OCO2Raa,−C(=O)N(Rbb)2,−OC(=O)N(Rbb)2,−NRbbC(=O)Raa, −NRbbCO2Raa,−NRbbC(=O)N(Rbb)2,−C(=NRbb)Raa,−C(=NRbb)ORaa,
−OC(=NRbb)Raa,−OC(=NRbb)ORaa,−C(=NRbb)N(Rbb)2,−OC(=NRbb)N(Rbb)2, −NRbbC(=NRbb)N(Rbb)2,−C(=O)NRbbSO2Raa,−NRbbSO2Raa,−SO2N(Rbb)2,−SO2Raa, −SO2ORaa,−OSO2Raa,−S(=O)Raa,−OS(=O)Raa,−Si(Raa)3,−OSi(Raa)3
−C(=S)N(Rbb)2,−C(=O)SRaa,−C(=S)SRaa,−SC(=S)SRaa,−SC(=O)SRaa,
−OC(=O)SRaa,−SC(=O)ORaa,−SC(=O)Raa,−P(=O)(Raa)2,−P(=O)(ORcc)2, −OP(=O)(Raa)2,−OP(=O)(ORcc)2,−P(=O)(N(Rbb)2)2,−OP(=O)(N(Rbb)2)2,
−NRbbP(=O)(Raa)2,−NRbbP(=O)(ORcc)2,−NRbbP(=O)(N(Rbb)2)2,−P(Rcc)2,−P(ORcc)2, −P(Rcc) +
3 X−,−P(ORcc) +
3 X−,−P(Rcc)4,−P(ORcc)4,−OP(Rcc)2,−OP(Rcc) +
3 X−, −OP(ORcc)2,−OP(ORcc) +
3 X−,−OP(Rcc)4,−OP(ORcc)4,−B(Raa)2,−B(ORcc)2, −BRaa(ORcc), C1-10 alkyl, C1-10 perhaloalkyl, C2-10 alkenyl, C2-10 alkynyl, heteroC1-10 alkyl, heteroC2-10 alkenyl, heteroC2-10 alkynyl, C3-10 carbocyclyl, 3-14 membered heterocyclyl, C6-14 aryl, and 5-14 membered heteroaryl, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 Rdd groups; wherein X− is a counterion;
or two geminal hydrogens on a carbon atom are replaced with the group =O, =S, =NN(Rbb)2, =NNRbbC(=O)Raa, =NNRbbC(=O)ORaa, =NNRbbS(=O)2Raa, =NRbb, or =NORcc;
each instance of Raa is, independently, selected from C1-10 alkyl, C1-10 perhaloalkyl, C2-10 alkenyl, C2-10 alkynyl, heteroC1-10 alkyl, heteroC2-10alkenyl, heteroC2-10alkynyl, C3-10 carbocyclyl, 3-14 membered heterocyclyl, C6-14 aryl, and 5-
14 membered heteroaryl, or two Raa groups are joined to form a 3-14 membered heterocyclyl or 5-14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 Rdd groups;
each instance of Rbb is, independently, selected from hydrogen,−OH,−ORaa, −N(Rcc)2,−CN,−C(=O)Raa,−C(=O)N(Rcc)2,−CO2Raa,−SO2Raa,−C(=NRcc)ORaa, −C(=NRcc)N(Rcc)2,−SO2N(Rcc)2,−SO2Rcc,−SO2ORcc,−SORaa,−C(=S)N(Rcc)2, −C(=O)SRcc,−C(=S)SRcc,−P(=O)(Raa)2,−P(=O)(ORcc)2,−P(=O)(N(Rcc)2)2, C1-10 alkyl, C1-10 perhaloalkyl, C2-10 alkenyl, C2-10 alkynyl, heteroC1-10alkyl, heteroC2- 10alkenyl, heteroC2-10alkynyl, C3-10 carbocyclyl, 3-14 membered heterocyclyl, C6-14 aryl, and 5-14 membered heteroaryl, or two Rbb groups are joined to form a 3-14 membered heterocyclyl or 5-14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 Rdd groups; wherein X− is a counterion;
each instance of Rcc is, independently, selected from hydrogen, C1-10 alkyl, C1- 10 perhaloalkyl, C2-10 alkenyl, C2-10 alkynyl, heteroC1-10 alkyl, heteroC2-10 alkenyl, heteroC2-10 alkynyl, C3-10 carbocyclyl, 3-14 membered heterocyclyl, C6-14 aryl, and 5- 14 membered heteroaryl, or two Rcc groups are joined to form a 3-14 membered heterocyclyl or 5-14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 Rdd groups;
each instance of Rdd is, independently, selected from halogen,−CN,−NO2, −N3,−SO2H,−SO3H,−OH,−ORee,−ON(Rff)2,−N(Rff)2,−N(Rff) +
3 X−,−N(ORee)Rff, −SH,−SRee,−SSRee,−C(=O)Ree,−CO2H,−CO2Ree,−OC(=O)Ree,−OCO2Ree, −C(=O)N(Rff)2,−OC(=O)N(Rff)2,−NRffC(=O)Ree,−NRffCO2Ree,−NRffC(=O)N(Rff)2, −C(=NRff)ORee,−OC(=NRff)Ree,−OC(=NRff)ORee,−C(=NRff)N(Rff)2,
−OC(=NRff)N(Rff)2,−NRffC(=NRff)N(Rff)2,−NRffSO2Ree,−SO2N(Rff)2,−SO2Ree, −SO2ORee,−OSO2Ree,−S(=O)Ree,−Si(Ree)3,−OSi(Ree)3,−C(=S)N(Rff)2,
−C(=O)SRee,−C(=S)SRee,−SC(=S)SRee,−P(=O)(ORee)2,−P(=O)(Ree)2,
−OP(=O)(Ree)2,−OP(=O)(ORee)2, C1-6 alkyl, C1-6 perhaloalkyl, C2-6 alkenyl, C2-6 alkynyl, heteroC1-6alkyl, heteroC2-6alkenyl, heteroC2-6alkynyl, C3-10 carbocyclyl, 3-10 membered heterocyclyl, C6-10 aryl, 5-10 membered heteroaryl, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl,
aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 Rgg groups, or two geminal Rdd substituents can be joined to form =O or =S; wherein X− is a counterion;
each instance of Ree is, independently, selected from C1-6 alkyl, C1-6 perhaloalkyl, C2-6 alkenyl, C2-6 alkynyl, heteroC1-6 alkyl, heteroC2-6alkenyl, heteroC2-6 alkynyl, C3-10 carbocyclyl, C6-10 aryl, 3-10 membered heterocyclyl, and 3-10 membered heteroaryl, wherein each alkyl, alkenyl, alkynyl, heteroalkyl,
heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 Rgg groups;
each instance of Rff is, independently, selected from hydrogen, C1-6 alkyl, C1-6 perhaloalkyl, C2-6 alkenyl, C2-6 alkynyl, heteroC1-6alkyl, heteroC2-6alkenyl, heteroC2- 6alkynyl, C3-10 carbocyclyl, 3-10 membered heterocyclyl, C6-10 aryl and 5-10 membered heteroaryl, or two Rff groups are joined to form a 3-10 membered heterocyclyl or 5-10 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 Rgg groups; and
each instance of Rgg is, independently, halogen,−CN,−NO2,−N3,−SO2H, −SO3H,−OH,−OC1-6 alkyl,−ON(C1-6 alkyl)2,−N(C1-6 alkyl)2,−N(C1-6 alkyl) +
3 X−, −NH(C1-6 alkyl) +
2 X−,−NH2(C1-6 alkyl) +X−,−NH +
3 X−,−N(OC1-6 alkyl)(C1-6 alkyl), −N(OH)(C1-6 alkyl),−NH(OH),−SH,−SC1-6 alkyl,−SS(C1-6 alkyl),−C(=O)(C1-6 alkyl),−CO2H,−CO2(C1-6 alkyl),−OC(=O)(C1-6 alkyl),−OCO2(C1-6 alkyl), −C(=O)NH2,−C(=O)N(C1-6 alkyl)2,−OC(=O)NH(C1-6 alkyl),−NHC(=O)( C1-6 alkyl), −N(C1-6 alkyl)C(=O)( C1-6 alkyl),−NHCO2(C1-6 alkyl),−NHC(=O)N(C1-6 alkyl)2, −NHC(=O)NH(C1-6 alkyl),−NHC(=O)NH2,−C(=NH)O(C1-6 alkyl),−OC(=NH)(C1-6 alkyl),−OC(=NH)OC1-6 alkyl,−C(=NH)N(C1-6 alkyl)2,−C(=NH)NH(C1-6 alkyl), −C(=NH)NH2,−OC(=NH)N(C1-6 alkyl)2,−OC(NH)NH(C1-6 alkyl),−OC(NH)NH2, −NHC(NH)N(C1-6 alkyl)2,−NHC(=NH)NH2,−NHSO2(C1-6 alkyl),−SO2N(C1-6 alkyl)2,−SO2NH(C1-6 alkyl),−SO2NH2,−SO2C1-6 alkyl,−SO2OC1-6 alkyl,−OSO2C1-6 alkyl,−SOC1-6 alkyl,−Si(C1-6 alkyl)3,−OSi(C1-6 alkyl)3−C(=S)N(C1-6 alkyl)2, C(=S)NH(C1-6 alkyl), C(=S)NH2,−C(=O)S(C1-6 alkyl),−C(=S)SC1-6 alkyl, −SC(=S)SC1-6 alkyl,−P(=O)(OC1-6 alkyl)2,−P(=O)(C1-6 alkyl)2,−OP(=O)(C1-6 alkyl)2,−OP(=O)(OC1-6 alkyl)2, C1-6 alkyl, C1-6 perhaloalkyl, C2-6 alkenyl, C2-6 alkynyl, heteroC1-6alkyl, heteroC2-6alkenyl, heteroC2-6alkynyl, C3-10 carbocyclyl, C6-10
aryl, 3-10 membered heterocyclyl, 5-10 membered heteroaryl; or two geminal Rgg substituents can be joined to form =O or =S; wherein X− is a counterion.
In certain embodiments, the carbon atom substituents are independently halogen, substituted or unsubstituted C1-6 alkyl,−ORaa,−SRaa,−N(Rbb)2,–CN,–SCN, –NO2,−C(=O)Raa,−CO2Raa,−C(=O)N(Rbb)2,−OC(=O)Raa,−OCO2Raa,
−OC(=O)N(Rbb)2,−NRbbC(=O)Raa,−NRbbCO2Raa, or−NRbbC(=O)N(Rbb)2. In certain embodiments, the carbon atom substituents are independently halogen, substituted or unsubstituted C1-6 alkyl,−ORaa,−SRaa,−N(Rbb)2,–CN,–SCN, or–NO2.
Nitrogen atoms can be substituted or unsubstituted as valency permits, and include primary, secondary, tertiary, and quaternary nitrogen atoms. Exemplary nitrogen atom substituents include, but are not limited to, hydrogen,−OH,−ORaa, −N(Rcc)2,−CN,−C(=O)Raa,−C(=O)N(Rcc)2,−CO2Raa,−SO2Raa,−C(=NRbb)Raa, −C(=NRcc)ORaa,−C(=NRcc)N(Rcc)2,−SO2N(Rcc)2,−SO2Rcc,−SO2ORcc,−SORaa, −C(=S)N(Rcc)2,−C(=O)SRcc,−C(=S)SRcc,−P(=O)(ORcc)2,−P(=O)(Raa)2,
−P(=O)(N(Rcc)2)2, C1-10 alkyl, C1-10 perhaloalkyl, C2-10 alkenyl, C2-10 alkynyl, heteroC1-10alkyl, heteroC2-10alkenyl, heteroC2-10alkynyl, C3-10 carbocyclyl, 3-14 membered heterocyclyl, C6-14 aryl, and 5-14 membered heteroaryl, or two Rcc groups attached to an N atom are joined to form a 3-14 membered heterocyclyl or 5-14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 Rdd groups, and wherein Raa, Rbb, Rcc and Rdd are as defined above.
In certain embodiments, the substituent present on the nitrogen atom is an nitrogen protecting group (also referred to herein as an“amino protecting group”). Nitrogen protecting groups include, but are not limited to,−OH,−ORaa,−N(Rcc)2, −C(=O)Raa,−C(=O)N(Rcc)2,−CO2Raa,−SO2Raa,−C(=NRcc)Raa,−C(=NRcc)ORaa, −C(=NRcc)N(Rcc)2,−SO2N(Rcc)2,−SO2Rcc,−SO2ORcc,−SORaa,−C(=S)N(Rcc)2, −C(=O)SRcc,−C(=S)SRcc, C1-10 alkyl (e.g., aralkyl, heteroaralkyl), C2-10 alkenyl, C2-10 alkynyl, heteroC1-10 alkyl, heteroC2-10 alkenyl, heteroC2-10 alkynyl, C3-10 carbocyclyl, 3-14 membered heterocyclyl, C6-14 aryl, and 5-14 membered heteroaryl groups, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aralkyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 Rdd groups, and wherein Raa, Rbb, Rcc and Rdd are as defined herein. Nitrogen protecting groups are well known in the art and include those
described in detail in Protecting Groups in Organic Synthesis, T. W. Greene and P. G. M. Wuts, 3rd edition, John Wiley & Sons, 1999, incorporated herein by reference.
For example, nitrogen protecting groups such as amide groups (e.g., −C(=O)Raa) include, but are not limited to, formamide, acetamide, chloroacetamide, trichloroacetamide, trifluoroacetamide, phenylacetamide, 3-phenylpropanamide, picolinamide, 3-pyridylcarboxamide, N-benzoylphenylalanyl derivative, benzamide, p-phenylbenzamide, o-nitophenylacetamide, o-nitrophenoxyacetamide,
acetoacetamide, (N’-dithiobenzyloxyacylamino)acetamide, 3-(p- hydroxyphenyl)propanamide, 3-(o-nitrophenyl)propanamide, 2-methyl-2-(o- nitrophenoxy)propanamide, 2-methyl-2-(o-phenylazophenoxy)propanamide, 4- chlorobutanamide, 3-methyl-3-nitrobutanamide, o-nitrocinnamide, N- acetylmethionine derivative, o-nitrobenzamide and o-(benzoyloxymethyl)benzamide.
Nitrogen protecting groups such as carbamate groups (e.g.,−C(=O)ORaa) include, but are not limited to, methyl carbamate, ethyl carbamate, 9-fluorenylmethyl carbamate (Fmoc), 9-(2-sulfo)fluorenylmethyl carbamate, 9-(2,7- dibromo)fluoroenylmethyl carbamate, 2,7-di-t-butyl-[9-(10,10-dioxo-10,10,10,10- tetrahydrothioxanthyl)]methyl carbamate (DBD-Tmoc), 4-methoxyphenacyl carbamate (Phenoc), 2,2,2-trichloroethyl carbamate (Troc), 2-trimethylsilylethyl carbamate (Teoc), 2-phenylethyl carbamate (hZ), 1-(1-adamantyl)-1-methylethyl carbamate (Adpoc), 1,1-dimethyl-2-haloethyl carbamate, 1,1-dimethyl-2,2- dibromoethyl carbamate (DB-t-BOC), 1,1-dimethyl-2,2,2-trichloroethyl carbamate (TCBOC), 1-methyl-1-(4-biphenylyl)ethyl carbamate (Bpoc), 1-(3,5-di-t- butylphenyl)-1-methylethyl carbamate (t-Bumeoc), 2-(2′- and 4′-pyridyl)ethyl carbamate (Pyoc), 2-(N,N-dicyclohexylcarboxamido)ethyl carbamate, t-butyl carbamate (BOC or Boc), 1-adamantyl carbamate (Adoc), vinyl carbamate (Voc), allyl carbamate (Alloc), 1-isopropylallyl carbamate (Ipaoc), cinnamyl carbamate (Coc), 4-nitrocinnamyl carbamate (Noc), 8-quinolyl carbamate, N-hydroxypiperidinyl carbamate, alkyldithio carbamate, benzyl carbamate (Cbz), p-methoxybenzyl carbamate (Moz), p-nitobenzyl carbamate, p-bromobenzyl carbamate, p-chlorobenzyl carbamate, 2,4-dichlorobenzyl carbamate, 4-methylsulfinylbenzyl carbamate (Msz), 9-anthrylmethyl carbamate, diphenylmethyl carbamate, 2-methylthioethyl carbamate, 2-methylsulfonylethyl carbamate, 2-(p-toluenesulfonyl)ethyl carbamate, [2-(1,3- dithianyl)]methyl carbamate (Dmoc), 4-methylthiophenyl carbamate (Mtpc), 2,4-
dimethylthiophenyl carbamate (Bmpc), 2-phosphonioethyl carbamate (Peoc), 2- triphenylphosphonioisopropyl carbamate (Ppoc), 1,1-dimethyl-2-cyanoethyl carbamate, m-chloro-p-acyloxybenzyl carbamate, p-(dihydroxyboryl)benzyl carbamate, 5-benzisoxazolylmethyl carbamate, 2-(trifluoromethyl)-6- chromonylmethyl carbamate (Tcroc), m-nitrophenyl carbamate, 3,5-dimethoxybenzyl carbamate, o-nitrobenzyl carbamate, 3,4-dimethoxy-6-nitrobenzyl carbamate, phenyl(o-nitrophenyl)methyl carbamate, t-amyl carbamate, S-benzyl thiocarbamate, p-cyanobenzyl carbamate, cyclobutyl carbamate, cyclohexyl carbamate, cyclopentyl carbamate, cyclopropylmethyl carbamate, p-decyloxybenzyl carbamate, 2,2- dimethoxyacylvinyl carbamate, o-(N,N-dimethylcarboxamido)benzyl carbamate, 1,1- dimethyl-3-(N,N-dimethylcarboxamido)propyl carbamate, 1,1-dimethylpropynyl carbamate, di(2-pyridyl)methyl carbamate, 2-furanylmethyl carbamate, 2-iodoethyl carbamate, isoborynl carbamate, isobutyl carbamate, isonicotinyl carbamate, p-(p’- methoxyphenylazo)benzyl carbamate, 1-methylcyclobutyl carbamate, 1- methylcyclohexyl carbamate, 1-methyl-1-cyclopropylmethyl carbamate, 1-methyl-1- (3,5-dimethoxyphenyl)ethyl carbamate, 1-methyl-1-(p-phenylazophenyl)ethyl carbamate, 1-methyl-1-phenylethyl carbamate, 1-methyl-1-(4-pyridyl)ethyl carbamate, phenyl carbamate, p-(phenylazo)benzyl carbamate, 2,4,6-tri-t-butylphenyl carbamate, 4-(trimethylammonium)benzyl carbamate, and 2,4,6-trimethylbenzyl carbamate.
Nitrogen protecting groups such as sulfonamide groups (e.g.,−S(=O)2Raa) include, but are not limited to, p-toluenesulfonamide (Ts), benzenesulfonamide, 2,3,6- trimethyl-4-methoxybenzenesulfonamide (Mtr), 2,4,6-trimethoxybenzenesulfonamide (Mtb), 2,6-dimethyl-4-methoxybenzenesulfonamide (Pme), 2,3,5,6-tetramethyl-4- methoxybenzenesulfonamide (Mte), 4-methoxybenzenesulfonamide (Mbs), 2,4,6- trimethylbenzenesulfonamide (Mts), 2,6-dimethoxy-4-methylbenzenesulfonamide (iMds), 2,2,5,7,8-pentamethylchroman-6-sulfonamide (Pmc), methanesulfonamide (Ms), β-trimethylsilylethanesulfonamide (SES), 9-anthracenesulfonamide, 4-(4′,8′- dimethoxynaphthylmethyl)benzenesulfonamide (DNMBS), benzylsulfonamide, trifluoromethylsulfonamide, and phenacylsulfonamide.
Other nitrogen protecting groups include, but are not limited to,
phenothiazinyl-(10)-acyl derivative, N’-p-toluenesulfonylaminoacyl derivative, N’- phenylaminothioacyl derivative, N-benzoylphenylalanyl derivative, N-
acetylmethionine derivative, 4,5-diphenyl-3-oxazolin-2-one, N-phthalimide, N- dithiasuccinimide (Dts), N-2,3-diphenylmaleimide, N-2,5-dimethylpyrrole, N-1,1,4,4- tetramethyldisilylazacyclopentane adduct (STABASE), 5-substituted 1,3-dimethyl- 1,3,5-triazacyclohexan-2-one, 5-substituted 1,3-dibenzyl-1,3,5-triazacyclohexan-2- one, 1-substituted 3,5-dinitro-4-pyridone, N-methylamine, N-allylamine, N-[2- (trimethylsilyl)ethoxy]methylamine (SEM), N-3-acetoxypropylamine, N-(1- isopropyl-4-nitro-2-oxo-3-pyroolin-3-yl)amine, quaternary ammonium salts, N- benzylamine, N-di(4-methoxyphenyl)methylamine, N-5-dibenzosuberylamine, N- triphenylmethylamine (Tr), N-[(4-methoxyphenyl)diphenylmethyl]amine (MMTr), N- 9-phenylfluorenylamine (PhF), N-2,7-dichloro-9-fluorenylmethyleneamine, N- ferrocenylmethylamino (Fcm), N-2-picolylamino N’-oxide, N-1,1- dimethylthiomethyleneamine, N-benzylideneamine, N-p-methoxybenzylideneamine, N-diphenylmethyleneamine, N-[(2-pyridyl)mesityl]methyleneamine, N-(N’,N’- dimethylaminomethylene)amine, N,N’-isopropylidenediamine, N-p- nitrobenzylideneamine, N-salicylideneamine, N-5-chlorosalicylideneamine, N-(5- chloro-2-hydroxyphenyl)phenylmethyleneamine, N-cyclohexylideneamine, N-(5,5- dimethyl-3-oxo-1-cyclohexenyl)amine, N-borane derivative, N-diphenylborinic acid derivative, N-[phenyl(pentaacylchromium- or tungsten)acyl]amine, N-copper chelate, N-zinc chelate, N-nitroamine, N-nitrosoamine, amine N-oxide,
diphenylphosphinamide (Dpp), dimethylthiophosphinamide (Mpt),
diphenylthiophosphinamide (Ppt), dialkyl phosphoramidates, dibenzyl
phosphoramidate, diphenyl phosphoramidate, benzenesulfenamide, o- nitrobenzenesulfenamide (Nps), 2,4-dinitrobenzenesulfenamide,
pentachlorobenzenesulfenamide, 2-nitro-4-methoxybenzenesulfenamide,
triphenylmethylsulfenamide, and 3-nitropyridinesulfenamide (Npys).
In certain embodiments, the substituent present on an oxygen atom is an oxygen protecting group (also referred to herein as an“hydroxyl protecting group”). Oxygen protecting groups include, but are not limited to,−Raa,−N(Rbb)2,
−C(=O)SRaa,−C(=O)Raa,−CO2Raa,−C(=O)N(Rbb)2,−C(=NRbb)Raa,−C(=NRbb)ORaa, −C(=NRbb)N(Rbb)2,−S(=O)Raa,−SO2Raa,−Si(Raa)3,−P(Rcc)2,−P(Rcc) +
3 X−,−P(ORcc)2, −P(ORcc) +
3 X−,−P(=O)(Raa)2,−P(=O)(ORcc)2, and−P(=O)(N(Rbb) 2)2, wherein X−, Raa, Rbb, and Rcc are as defined herein. Oxygen protecting groups are well known in the art and include those described in detail in Protecting Groups in Organic Synthesis, T. W.
Greene and P. G. M. Wuts, 3rd edition, John Wiley & Sons, 1999, incorporated herein by reference.
Exemplary oxygen protecting groups include, but are not limited to, methyl, methoxylmethyl (MOM), methylthiomethyl (MTM), t-butylthiomethyl,
(phenyldimethylsilyl)methoxymethyl (SMOM), benzyloxymethyl (BOM), p- methoxybenzyloxymethyl (PMBM), (4-methoxyphenoxy)methyl (p-AOM), guaiacolmethyl (GUM), t-butoxymethyl, 4-pentenyloxymethyl (POM), siloxymethyl, 2-methoxyethoxymethyl (MEM), 2,2,2-trichloroethoxymethyl, bis(2- chloroethoxy)methyl, 2-(trimethylsilyl)ethoxymethyl (SEMOR), tetrahydropyranyl (THP), 3-bromotetrahydropyranyl, tetrahydrothiopyranyl, 1-methoxycyclohexyl, 4- methoxytetrahydropyranyl (MTHP), 4-methoxytetrahydrothiopyranyl, 4- methoxytetrahydrothiopyranyl S,S-dioxide, 1-[(2-chloro-4-methyl)phenyl]-4- methoxypiperidin-4-yl (CTMP), 1,4-dioxan-2-yl, tetrahydrofuranyl,
tetrahydrothiofuranyl, 2,3,3a,4,5,6,7,7a-octahydro-7,8,8-trimethyl-4,7- methanobenzofuran-2-yl, 1-ethoxyethyl, 1-(2-chloroethoxy)ethyl, 1-methyl-1- methoxyethyl, 1-methyl-1-benzyloxyethyl, 1-methyl-1-benzyloxy-2-fluoroethyl, 2,2,2-trichloroethyl, 2-trimethylsilylethyl, 2-(phenylselenyl)ethyl, t-butyl, allyl, p- chlorophenyl, p-methoxyphenyl, 2,4-dinitrophenyl, benzyl (Bn), p-methoxybenzyl, 3,4-dimethoxybenzyl, o-nitrobenzyl, p-nitrobenzyl, p-halobenzyl, 2,6-dichlorobenzyl, p-cyanobenzyl, p-phenylbenzyl, 2-picolyl, 4-picolyl, 3-methyl-2-picolyl N-oxido, diphenylmethyl, p,p’-dinitrobenzhydryl, 5-dibenzosuberyl, triphenylmethyl, α- naphthyldiphenylmethyl, p-methoxyphenyldiphenylmethyl, di(p- methoxyphenyl)phenylmethyl, tri(p-methoxyphenyl)methyl, 4-(4’- bromophenacyloxyphenyl)diphenylmethyl, 4,4′,4″-tris(4,5- dichlorophthalimidophenyl)methyl, 4,4′,4″-tris(levulinoyloxyphenyl)methyl, 4,4′,4″- tris(benzoyloxyphenyl)methyl, 3-(imidazol-1-yl)bis(4′,4″-dimethoxyphenyl)methyl, 1,1-bis(4-methoxyphenyl)-1′-pyrenylmethyl, 9-anthryl, 9-(9-phenyl)xanthenyl, 9-(9- phenyl-10-oxo)anthryl, 1,3-benzodithiolan-2-yl, benzisothiazolyl S,S-dioxido, trimethylsilyl (TMS), triethylsilyl (TES), triisopropylsilyl (TIPS),
dimethylisopropylsilyl (IPDMS), diethylisopropylsilyl (DEIPS), dimethylthexylsilyl, t-butyldimethylsilyl (TBDMS), t-butyldiphenylsilyl (TBDPS), tribenzylsilyl, tri-p- xylylsilyl, triphenylsilyl, diphenylmethylsilyl (DPMS), t-butylmethoxyphenylsilyl (TBMPS), formate, benzoylformate, acetate, chloroacetate, dichloroacetate, trichloroacetate, trifluoroacetate, methoxyacetate, triphenylmethoxyacetate,
phenoxyacetate, p-chlorophenoxyacetate, 3-phenylpropionate, 4-oxopentanoate (levulinate), 4,4-(ethylenedithio)pentanoate (levulinoyldithioacetal), pivaloate, adamantoate, crotonate, 4-methoxycrotonate, benzoate, p-phenylbenzoate, 2,4,6- trimethylbenzoate (mesitoate), methyl carbonate, 9-fluorenylmethyl carbonate (Fmoc), ethyl carbonate, 2,2,2-trichloroethyl carbonate (Troc), 2-(trimethylsilyl)ethyl carbonate (TMSEC), 2-(phenylsulfonyl) ethyl carbonate (Psec), 2- (triphenylphosphonio) ethyl carbonate (Peoc), isobutyl carbonate, vinyl carbonate, allyl carbonate, t-butyl carbonate (BOC or Boc), p-nitrophenyl carbonate, benzyl carbonate, p-methoxybenzyl carbonate, 3,4-dimethoxybenzyl carbonate, o-nitrobenzyl carbonate, p-nitrobenzyl carbonate, S-benzyl thiocarbonate, 4-ethoxy-1-napththyl carbonate, methyl dithiocarbonate, 2-iodobenzoate, 4-azidobutyrate, 4-nitro-4- methylpentanoate, o-(dibromomethyl)benzoate, 2-formylbenzenesulfonate, 2- (methylthiomethoxy)ethyl, 4-(methylthiomethoxy)butyrate, 2- (methylthiomethoxymethyl)benzoate, 2,6-dichloro-4-methylphenoxyacetate, 2,6- dichloro-4-(1,1,3,3-tetramethylbutyl)phenoxyacetate, 2,4-bis(1,1- dimethylpropyl)phenoxyacetate, chlorodiphenylacetate, isobutyrate, monosuccinoate, (E)-2-methyl-2-butenoate, o-(methoxyacyl)benzoate, α-naphthoate, nitrate, alkyl N,N,N’,N’-tetramethylphosphorodiamidate, alkyl N-phenylcarbamate, borate, dimethylphosphinothioyl, alkyl 2,4-dinitrophenylsulfenate, sulfate, methanesulfonate (mesylate), benzylsulfonate, and tosylate (Ts).
In certain embodiments, the substituent present on a sulfur atom is a sulfur protecting group (also referred to as a“thiol protecting group”). Sulfur protecting groups include, but are not limited to,−Raa,−N(Rbb)2,−C(=O)SRaa,−C(=O)Raa, −CO2Raa,−C(=O)N(Rbb)2,−C(=NRbb)Raa,−C(=NRbb)ORaa,−C(=NRbb)N(Rbb)2, −S(=O)Raa,−SO2Raa,−Si(Raa)3,−P(Rcc)2,−P(Rcc) +
3 X−,−P(ORcc)2,−P(ORcc) +
3 X−, −P(=O)(Raa)2,−P(=O)(ORcc)2, and−P(=O)(N(Rbb) 2)2, wherein Raa, Rbb, and Rcc are as defined herein. Sulfur protecting groups are well known in the art and include those described in detail in Protecting Groups in Organic Synthesis, T. W. Greene and P. G. M. Wuts, 3rd edition, John Wiley & Sons, 1999, incorporated herein by reference.
The term“halo” or“halogen” refers to fluorine (fluoro,–F), chlorine (chloro, –Cl), bromine (bromo,–Br), or iodine (iodo,–I).
The term“hydroxyl” or“hydroxy” refers to the group–OH.
The term“thiol” or“thio” refers to the group–SH.
The term“amine” or“amino” refers to the group–NH– or–NH2.
As used herein, the term“polyethylene glycol” or“PEG” refers to an ethylene glycol polymer that contains about 20 to about 2,000,000 linked monomers, typically about 50-1,000 linked monomers, usually about 100-300. Polyethylene glycols include ethylene glycol polymer containing various numbers of linked monomers, e.g., PEG20, PEG30, PEG40, PEG60, PEG80, PEG100, PEG115, PEG200, PEG300, PEG400, PEG500, PEG600, PEG1000, PEG1500, PEG2000, PEG3350, PEG4000, PEG4600, PEG5000, PEG6000, PEG8000, PEG11000, PEG12000, PEG2000000 and any mixtures thereof.
The term“salt” refers to ionic compounds that result from the neutralization reaction of an acid and a base. A salt is composed of one or more cations (positively charged ions) and one or more anions (negative ions) so that the salt is electrically neutral (without a net charge). Salts of the compounds of this invention include those derived from inorganic and organic acids and bases. Examples of acid addition salts are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid, and perchloric acid, or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid, or malonic acid or by using other methods known in the art such as ion exchange. Other salts include adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2–hydroxy–ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2–naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pectinate, persulfate, 3– phenylpropionate, phosphate, picrate, pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate, p-toluenesulfonate, undecanoate, valerate salts, and the like. Salts derived from appropriate bases include alkali metal, alkaline earth metal, ammonium and N+(C1-4 alkyl)4 salts. Representative alkali or alkaline earth metal salts include sodium, lithium, potassium, calcium, magnesium, and the like. Further salts include ammonium, quaternary ammonium, and amine cations formed using counterions such as halide, hydroxide, carboxylate, sulfate, phosphate, nitrate, lower alkyl sulfonate, and aryl sulfonate.
These and other exemplary substituents are described in more detail in the Detailed Description, Examples, and Claims. The invention is not intended to be limited in any manner by the above exemplary listing of substituents.
Additional terms are defined throughout the specification. Therapeutic Agents
The present invention discloses, at least in part, macromonomers, conjugates comprising the macromonomers (also referred to alternatively as“BRUSH”, or “BRUSH conjugates”), and brush arm star polymer particles comprising the conjugates (also referred to as“BASPs” or“BASP particles”), each further comprising an agent (e.g., one or more therapeutic agents, diagnostic agents, or targeting moieties), Compositions comprising BASP particles are referred to as “BASP-compositions”. Compositions comprising BRUSH conjugates are referred to as“BRUSH-compositions”. The macromonomers, BRUSH conjugates, and BASP particles disclosed herein comprise an agent(s), e.g., a first therapeutic agent (e.g., at least one (including, e.g., at least two, at least three) an ARB, a vitamin D analog, IDO inhibitor, or a bromodomain inhibitor). In some embodiments, the BRUSHs, and BASPs can further comprise a second therapeutic agent, a targeting moiety, a diagnostic moiety, e.g., as described herein. The agent(s) can be chemically bound to the BRUSHs, and BASPs. In other embodiments, the agent(s) can be associated with a BRUSH or BASP. In some embodiments, a first agent can be coupled to the BRUSH or BASP, and a second agent, targeting moiety, and/or diagnostic moiety can be non-covalently associated with the BRUSH or BASP. Any of the agents disclosed herein can be used in the macromonomers, conjugates, particles and other compositions and methods disclosed herein.
As used herein, the term“agent” means a molecule, group of molecules, complex or substance administered to an organism for diagnostic, therapeutic, preventative medical, or veterinary purposes.
As used herein, the term“therapeutic agent” includes an agent that is capable of providing a local or systemic biological, physiological, or therapeutic effect in the biological system to which it is applied. For example, a therapeutic agent can act to control tumor growth, control infection or inflammation, act as an analgesic, promote anti-cell attachment, and enhance bone growth, among other functions. Other suitable therapeutic agents can include anti-viral agents, hormones, antibodies, or therapeutic
proteins. Other therapeutic agents include prodrugs, which are agents that are not biologically active when administered but, upon administration to a subject are converted to biologically active agents through metabolism or some other mechanism.
An agent, e.g., a therapeutic agent, can include a wide variety of different compounds, including chemical compounds and mixtures of chemical compounds, e.g., small organic or inorganic molecules; saccharines; oligosaccharides;
polysaccharides; biological macromolecules, e.g., peptides, proteins, and peptide analogs and derivatives; peptidomimetics; antibodies and antigen binding fragments thereof; nucleic acids; nucleic acid analogs and derivatives; an extract made from biological materials such as bacteria, plants, fungi, or animal cells; animal tissues; naturally occurring or synthetic compositions; and any combinations thereof.
In some embodiments, the agent is in the form of a prodrug. The term “prodrug” refer to a compound that becomes active, e.g., by solvolysis, reduction, oxidation, or under physiological conditions, to provide a pharmaceutically active compound, e.g., in vivo. A prodrug can include a derivative of a pharmaceutically active compound, such as, for example, to form an ester by reaction of the acid, or acid anhydride, or mixed anhydrides moieties of the prodrug moiety with the hydroxyl moiety of the pharmaceutical active compound, or to form an amide prepared by the acid, or acid anhydride, or mixed anhydrides moieties of the prodrug moiety with a substituted or unsubstituted amine of the pharmaceutically active compound. Simple aliphatic or aromatic esters, amides, and anhydrides derived from acidic groups may comprise prodrugs. In some embodiments, the conjugate or BASP particle described herein incorporates one therapeutic agent or prodrug thereof. In some embodiments, the conjugate or BASP particle described herein incorporates more than one therapeutic agents or prodrugs.
In some embodiments, the agent, e.g., a therapeutic agent, is a small molecule. As used herein, the term“small molecule” can refer to compounds that are“natural product-like.” However, the term“small molecule” is not limited to“natural product- like” compounds. Rather, a small molecule is typically characterized in that it contains several carbon—carbon bonds, and has a molecular weight of less than 5000 Daltons (5 kDa), preferably less than 3 kDa, still more preferably less than 2 kDa, and most preferably less than 1 kDa. In some cases it is preferred that a small molecule have a molecular weight equal to or less than 700 Daltons.
Exemplary agents, e.g., a therapeutic agents, in the BASP-compositions include, but are not limited to, those found in Harrison’s Principles of Internal Medicine , 13th Edition, Eds. T.R. Harrison et al. McGraw-Hill N.Y., NY;
Physicians’ Desk Reference, 50th Edition, 1997, Oradell New Jersey, Medical Economics Co.; Pharmacological Basis of Therapeutics, 8th Edition, Goodman and Gilman, 1990; United States Pharmacopeia, The National Formulary, USP XII NF XVII, 1990; current edition of Goodman and Oilman’s The Pharmacological Basis of Therapeutics ; and current edition of The Merck Index , the complete contents of all of which are incorporated herein by reference.
In some embodiments, exemplary therapeutic agents in the BASP- compositions or BRUSH-compositions include, but are not limited to, one or more of the agents listed in Paragraph 0148 of U.S. Patent No.9,381,253, incorporated by reference herein.
In other embodiments, exemplary therapeutic agents in the BASP- compositions or BRUSH-compositions include, but are not limited to, one or more of the therapeutic agents listed in WO 2013/169739, including the anti-hypertensive and/or a collagen modifying agents (“AHCM”) disclosed, e.g., in Paragraphs 40-49, 283, 286-295; the microenvironment modulators disclosed, e.g., in Paragraphs 113- 121, of WO 2013/169739, incorporated herein by reference. In some embodiments, the BASP-composition or BRUSH-composition comprising the AHCM and/or the microenvironment modulator causes one or more of: reduces solid stress (e.g., growth-induced solid stress in tumors); decreases tumor fibrosis; reduces interstitial hypertension or interstitial fluid pressure (IFP); increases interstitial tumor transport; increases tumor or vessel perfusion; increases vascular diameters and/or enlarges compressed or collapsed blood vessels; reduces or depletes one or more of: cancer cells, or stromal cells (e.g., tumor associated fibroblasts or immune cells); decreases the level or production of extracellular matrix components, such as fibers (e.g., collagen, procollagen), and/or polysaccharides (e.g., glycosaminoglycans such as hyaluronan or hyaluronic acid); decreases the level or production of collagen or procollagen; decreases the level or production of hyaluronic acid; increases tumor oxygenation; decreases tumor hypoxia; decreases tumor acidosis; enables immune cell infiltration; decreases immunosuppression; increases antitumor immunity;
decreases the production of cancer stem cells (also referred to herein as tumor- initiating cells); or enhances the efficacy (e.g., penetration or diffusion), of the
therapy, e.g., the cancer therapy (e.g., radiation, photodynamic therapy,
chemotherapeutics and immunotherapies) in a tumor or tumor vasculature, in the subject.
Agents, e.g., therapeutic agents, include the herein disclosed categories and specific examples. It is not intended that the category be limited by the specific examples. Those of ordinary skill in the art will recognize also numerous other compounds that fall within the categories and that are useful according to the present disclosure.
In some embodiments, the BASP-compositions or BRUSH-compositions comprise one or more of an ARB, a vitamin D analog, IDO inhibitor, or a
bromodomain inhibitor, as described herein. The BASP-compositions or BRUSH- compositions can further comprise a diagnostic agent, a targeting moiety, or a second therapeutic agent, e.g., an anti-cancer, an anti-inflammatory, or anti-fibrotic therapy, as described herein. Angiotensin Receptor Blockers (ARBs)
In some embodiments, the agent, e.g., the therapeutic agent, in the BASP- composition or BRUSH-composition is an angiotensin receptor blocker (ARB), also referred to as angiotensin II receptor blocker, or AT1 blocker or AT1 inhibitor.
In some embodiments, the ARB comprises losartan (e.g., COZAAR®), candesartan (e.g., ATACAND®), telmisartan (e.g., MICARDIS®), valsartan (e.g., DIOVAN®), olmesartan (e.g., BENICAR®), azilsartan, eprosartan (e.g.,
TEVETEN®), irbesartan (e.g., AVAPRO®), saralasin, EXP 3174, L158209, or an analogue, prodrug, metabolite, or derivative thereof, e.g., as shown in FIG.2. In some embodiments, ARB is telmisartan or an analogue, prodrug, metabolite, or derivative thereof. In some embodiments, ARB is losartan or an analogue, prodrug, metabolite, or derivative thereof. In some embodiments, ARB is candesartan or an analogue, prodrug, metabolite, or derivative thereof. In some embodiments, ARB is valsartan or an analogue, prodrug, metabolite, or derivative thereof. In some embodiments, ARB is olmesartan or an analogue, prodrug, metabolite, or derivative thereof. In some embodiments, ARB is azilsartan or an analogue, prodrug, metabolite, or derivative thereof. In some embodiments, ARB is eprosartan or an analogue, prodrug, metabolite, or derivative thereof. In some embodiments, ARB is irbesartan or an analogue, prodrug, metabolite, or derivative thereof. In some embodiments, ARB is
saralasin or an analogue, prodrug, metabolite, or derivative thereof. In some embodiments, ARB is EXP 3174 or an analogue, prodrug, metabolite, or derivative thereof. In some embodiments, ARB is L158209 or an analogue, prodrug, metabolite, or derivative thereof.
In some embodiments, the ARB is covalently bound to a macromonomer, conjugate, or particle described herein. In some embodiments, the ARB comprises the structure of Formula (I-1):

wherein each of R11a and R11b is independently hydrogen or is taken together to form an oxo group; R12 is heteroaryl (e.g., tetrazolyl) or heteroarylalkyl (e.g.,
benzoimidazolyl), each of which is optionally substituted by one or more R13; R13 is optionally substituted heteroaryl; and each of L4, L5, and L6 is independently absent, a bond, alkyl, heteroalkyl, carbocyclyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, carbocyclyl, heterocyclyl, aryl, or heteroaryl is optionally substituted.
In some embodiments, the ARB comprises the structure of Formula (I-i), Formula (I-ii), Formula (I-iii), Formula (I-iv), or Formula (I-v):

Formula (I-i) Formula (I-ii)

Formula (I-iii) Formula (I-iv)

Formula (I-v).
In m m imn h ARB comprises a structure of Formula (I-i):

In some embodiments, the ARB comprises a structure of Formula (I-ii):

In some embodiments the ARB com rises a structure of Formula (I-iii):

nts, the ARB comprises a structure of Formula (I-iv):


In some embodiments, the agent is an ARB, e.g., an ARB shown in FIG.2, and may be covalently coupled to the cleavable linker (e.g., L3 in Formula (I)) via a hydroxyl group, a sulfonamide, a carboxylic acid, an ester, an amide, an amine, a triazole, or a benzimidazolone. In some embodiments, the agent is losartan, candesartan, telmisartan, valsartan, olmesartan, azilsartan, eprosartan, irbesartan, saralasin, EXP 3174, L158209, or an analog, a prodrug, a metabolite, or a derivative thereof, and may be covalently coupled to the cleavable linker (e.g., L3 in Formula (I)) via a hydroxyl group. In some embodiments, the agent is losartan, candesartan, telmisartan, valsartan, olmesartan, azilsartan, eprosartan, irbesartan, saralasin, EXP 3174, L158209, or an analog, a prodrug, a metabolite, or a derivative thereof, and may be covalently coupled to the cleavable linker (e.g., L3 in Formula (I)) via a carboxylic acid or ester. In some embodiments, the agent is losartan, candesartan, telmisartan, valsartan, olmesartan, azilsartan, eprosartan, irbesartan, saralasin, EXP 3174,
L158209, or an analog, a prodrug, a metabolite, or a derivative thereof, and may be covalently coupled to the cleavable linker (e.g., L3 in Formula (I)) via an amide. Vitamin D Analogs
In some embodiments, the agent, e.g., the therapeutic agent, in the BASP- composition or BRUSH-composition is a vitamin D analog. Vitamin D analogs may comprise naturally occurring vitamin D analogs and/or non-natural (e.g., synthetically produced) analogs. Vitamin D analogs may further comprise a vitamin D receptor (VDR) agonist, a vitamin D receptor ligand, a vitamin D precursor, or derivatives thereof. Exemplary vitamin D analogs include, but are not limited to, paricalcitol, calcipotriol, KH1060 (lexacalcitol), ercalcitriol, EB1089 (seocalcitol), BXL-628 (elocalcitol), MC1288, CB966, BCB 1093, GS 1558, TX527 (19-nor-14,20-bisepi-23- yne-l,25(OH)2D3), ED-71 (eldecalcitrol), BXL-01-0029, doxercalciferol, maxacalcitol (OCT), tacalcitol, alfacalcidol, SM-10193, EB1072, EB1129, EB1133, EB1155,
EB1270, MC1288, EB1213, CB1093, VD2656, VD2668, VD2708, VD2716, VD2728, VD2736, GS1500, GS1558, KH1060, ZK161422, and combinations or derivatives thereof. Additional exemplary vitamin D analogs are shown in FIG.3. In some embodiments, the vitamin D analog does not comprise KH1060 (lexacalcitol), seocalcitol (EB 1089), or CB 1093. Additional vitamin D analogs are described by Scolletta et al. (2013) Mediators of Inflammation 2013, Article ID 876319; and Adorini (2005) Cellular Immunology 233: 115-124, which is incorporated herein by reference in its entirety.
In some embodiments, the agent is a vitamin D analog, or a metabolite or derivative thereof, e.g., as described in FIG.3. In some embodiments, the vitamin D analog comprises paricalcitol, doxercalciferol, falecalcitriol, maxacalcitol, tacalcitol, alfacalcidol, eldecalcitol, seocalcitol, lexicalcitol, CB 1093, CD578, inecalcitol, calcipotriol, TX527, 2MD, WY1112, PRI-2205, ILX23-7553, or a metabolite or derivative thereof, e.g., as shown in FIG.3. In some embodiments, the vitamin D analog comprises paricalcitol, ergocalciferol, elocalcitol, eldecalcitrol, calcidiol, calcipotriol, cholecalciferol, or a metabolite or derivative thereof. In some
embodiments, the vitamin D analog comprises paricalcitol, ergocalciferol, cholecalciferol, or a metabolite or derivative thereof. In some embodiments, the vitamin D analog does not comprise seocalcitol (EB 1089), CB 1093, or lexacalcitol (KH 1060). Additional vitamin D analogs and derivatives are described, e.g. in Leyssens, C. et al, Front Physiol (2014), which is incorporated herein by reference in its entirety.
In some embodiments, the vitamin D analog comprises a structure of Formula (I-2):

wherein R21 is hydrogen or hydroxyl; R22 is hydrogen, alkyl, alkenyl, or hydroxyl; R23 is hydrogen, alkyl, or alkenyl; R24 is alkyl, alkenyl, alkynyl, or heteroalkyl; R25 is hydrogen or absent; is a single bond or absent; and is a single or double bond;
wherein each of which the alkyl, alkenyl, alkynyl, and heteroalkyl is optionally substituted.
In certain embodiments, the vitamin D analog is attached to L3 in the macromonomer at a hydroxyl moiety of the vitamin D analog. In certain
embodiments, in Formula I-2, above, R21 is hydroxyl, and the vitamin D analog is attached to L3 at R21. In certain embodiments, in Formula I-2, above, R22 is hydroxyl, and the vitamin D analog is attached to L3 at R22.
In some embodiments, the vitamin D analog comprises a structure of Formula (I-vi Formula I-vii or Formula I-viii :
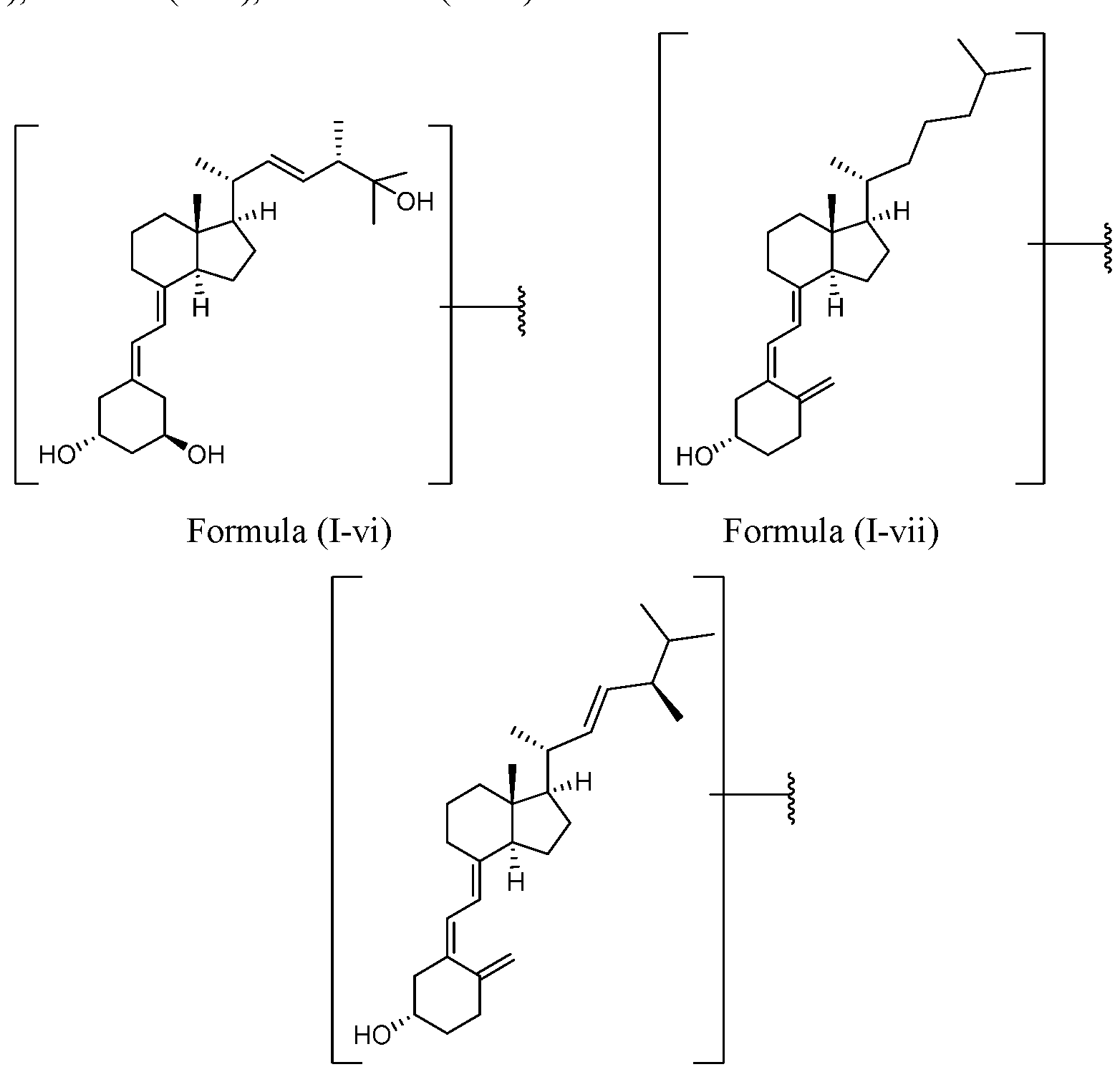
Formula (I-viii),
wherein in each case, as drawn, the vitamin D analog may be attached to the macromonomer, conjugate, or particle through any available atom.
In some embodiments, the vitamin D analog comprises a structure of Formula (I-vi):
 (Formula I-vi).
(Formula I-vi).
In some embodiments, the vitamin D analog comprises a structure of Formula (I-vii):

In some embodiments, the vitamin D analog comprises a structure of Formula (I-viii):

In some embodiments, the vitamin D analog of Formula (I-vi) comprises a structure of Formula (I-ix) or Formula (I-x):
 . Formula (I-ix) Formula (I-x).
. Formula (I-ix) Formula (I-x).
In some embodiments, the agent is a vitamin D analog or a metabolite or derivative thereof, e.g., a vitamin D analog or a metabolite or derivative thereof shown in FIG.3, and may be covalently coupled to the cleavable linker (e.g., L3 in Formula (I)) via a hydroxyl group, a sulfonamide, a carboxylic acid, an ester, an amide, or an amine. In some embodiments, the agent is paricalcitol, doxercalciferol, falecalcitriol, maxacalcitol, tacalcitol, alfacalcidol, eldecalcitol, seocalcitol, lexicalcitol, CB 1093, CD578, inecalcitol, calcipotriol, TX527, 2MD, WY1112, PRI- 2205, ILX23-7553, or a metabolite or derivative thereof and may be covalently coupled to the cleavable linker (e.g., L3 in Formula (I)) via a hydroxyl group. In some embodiments, the agent is paricalcitol, doxercalciferol, falecalcitriol, maxacalcitol, tacalcitol, alfacalcidol, eldecalcitol, seocalcitol, lexicalcitol, CB 1093, CD578, inecalcitol, calcipotriol, TX527, 2MD, WY1112, PRI-2205, ILX23-7553, or a metabolite or derivative thereof and may be covalently coupled to the cleavable linker (e.g., L3 in Formula (I)) via a carboxylic acid or ester. In some embodiments, the agent is paricalcitol, doxercalciferol, falecalcitriol, maxacalcitol, tacalcitol, alfacalcidol, eldecalcitol, seocalcitol, lexicalcitol, CB 1093, CD578, inecalcitol, calcipotriol, TX527, 2MD, WY1112, PRI-2205, ILX23-7553, or a metabolite or derivative thereof and may be covalently coupled to the cleavable linker (e.g., L3 in Formula (I)) macromonomer, conjugate or BASP particle via an amide. IDO Inhibitors
In some embodiments, the agent, e.g., the therapeutic agent, in the BASP- composition or BRUSH-composition is an IDO inhibitor (i.e., indoleamine 2,3- dioxygenase (IDO) pathway inhibitor). Exemplary IDO inhibitors include, but are not limited to, GDC-0919, indoximod, 1-methyltryptophan (e.g., 1-methyl-L-tryptophan, 1-methyl-D-tryptophan), NLG8189, INCB024360, NLG919, methylthiohydantoin
tryptophan, brassinin, annulin B, exiguamine A, INCB023843, or an analog or derivative thereof. Additional IDO inhibitors are described e.g., in Lob, S. et al. Nat Rev Cancer (2009) 9:445-452; Rohrig, U.F. et al. J Med Chem (2015) 58:9421-9437; and U.S. Patent Application No.14/919,184, each of which is incorporated by reference herein in its entirety.
In some embodiments, the IDO inhibitor may be covalently coupled to the cleavable linker (e.g., L3 in Formula (I)) via a hydroxyl group. In some embodiments, IDO inhibitor may be covalently coupled to the cleavable linker (e.g., L3 in Formula (I)) via a carboxylic acid or ester. In some embodiments, the IDO inhibitor may be covalently coupled to the cleavable linker (e.g., L3 in Formula (I)) via an amide. Bromodomain Inhibitors
In some embodiments, the agent, e.g., the therapeutic agent, in the BASP- composition or BRUSH-composition is a bromodomain inhibitor (i.e., extra-terminal protein inhibitor (i-BET)). Exemplary bromodomain inhibitors include, but are not limited to, MS436, PFI-1, I-BET 151, OTX-015, JQ1, CPI-203, bromosporine, RVX- 208, I-BET 762, I-BET 151, OFXBD02, OFXBD03, XD14, MS436, and analogs and derivatives thereof, e.g., as shown in FIGS.6A and 6B. Additional bromodomain inhibitors are described e.g., in Haas, M. J. et al SciBX (2014) 7(15); ACS Chem Biol (2015) 10:22-39; Expert Opin Ther Pat (2014) 24:185-199; Clin Cancer Res (2015) 21:1628-1638; Oncotarget (2015) 6:17698-17712; Bioorg Med Chem Lett (2015) 25:1842-1848; Cancer Res (2013) 73:3336-3346; Am J Cardiovasc Drugs (2015) Sep 18; and J Med Chem (2013) 56:9251-9264, each of which is incorporated by reference herein in its entirety.
In some embodiments, the agent is a bromodomain inhibitor (i.e., a bromodomain or an extra-terminal protein inhibitor (i-BET)). In some embodiments, the bromodomain inhibitor comprises MS436, PFI-1, I-BET 151, OTX-015, JQ1, CPI-203, bromosporine, RVX-208, I-BET 762, I-BET 151, OFXBD02, OFXBD03, XD14, MS436, or an analog or derivative thereof, e.g., as shown in FIGS.3A to 3B. Additional bromodomain inhibitors are described e.g., in Haas, M. J. et al SciBX (2014) 7(15); ACS Chem Biol (2015) 10:22-39; Expert Opin Ther Pat (2014) 24:185- 199; Clin Cancer Res (2015) 21:1628-1638; Oncotarget (2015) 6:17698-17712; Bioorg Med Chem Lett (2015) 25:1842-1848; Cancer Res (2013) 73:3336-3346; Am J
Cardiovasc Drugs (2015) Sep 18; and J Med Chem (2013) 56:9251-9264, each of which is incorporated by reference herein in its entirety.
In some embodiments, the agent is a bromodomain inhibitor, e.g., a bromodomain inhibitor shown in FIGS.4A to 4B, and may be covalently coupled to the cleavable linker (e.g., L3 in Formula (I)) via a hydroxyl group, a sulfonamide, a carboxylic acid, an ester, an amide, or an amine. In some embodiments, the agent is OTX-2015 (5), RVX-208 (7), OXFBD02 (9), OXFBD03 (10), XD14 (18), or dinaciclib (19), e.g., as shown in FIGS.4A to 4B, and may be covalently coupled to the cleavable linker (e.g., L3 in Formula (I)) via a hydroxyl group. In some embodiments, the agent is (12), PFI-1 (14), (15), MS436 (16), TG101348 (22), TG101209 (23), or bromosporine, e.g., as shown in FIGS.4A to 4B, and may be covalently coupled to the cleavable linker (e.g., L3 in Formula (I)) via a sulfonamide. In some embodiments, the agent is I-BET726 (12), CPI-203 (6), or B12536 (21), e.g., as shown in FIGS.4A to 4B, and may be covalently coupled to the cleavable linker (e.g., L3 in Formula (I)) via a carboxylic acid, ester, or amide. In some embodiments, the agent is I-BET151 (11) or B12536 (21), e.g., as shown in FIGS.4A to 4B, and may be covalently coupled to the cleavable linker (e.g., L3 in Formula (I)) via a benzimidazolone or pyrimidine amine. Macromonomers
The present invention features, at least in part, BRUSH conjugates and BASP particles coupled to one or more agents. In some embodiments, the BRUSH conjugates and BASP particles are prepared through the linkage of discrete macromonomer subunits that include the agent, e.g., a therapeutic agent, and optionally a targeting moiety, a diagnostic agent, and/or a second therapeutic agent of interest. The brush arm method involves co-polymerization of a macromonomer with a multifunctional crosslinker to generate a unimolecular micelle-like nanostructure (BASP) with a core including the crosslinker and a corona including the
macromonomer.
As used herein, the term“macromonomer” refers to a macromolecule with one end-group that enables it to polymerize to form a polymeric structure, e.g., a BRUSH conjugate or BASP particle, e.g., as shown in No. 1 in FIG.1. Macromonomers can contribute a single monomeric unit to a chain of the completed BRUSH conjugate or BASP polymer. A macromonomer may range in size from about 3 kDa to about 6
kDa, e.g., as detected by mass spectrometry. Macromonomer subunits comprising the agent and optionally the targeting moiety, the diagnostic agent, and/or the second agent of interest may be joined in a number of ways, such as through graft-through ring-opening metathesis polymerization (ROMP) in the presence of a cross-linker (e.g., a bis-norbornene cross-linker described herein).
In one aspect, the invention features a compound (e.g., a macromonomer) of Formula (I):

Formula (I)
or a salt thereof, wherein Ring C is carbocyclyl or a heterocyclyl moiety;
Ring T is a triazoldiyl moiety (e.g., a 1,2,3-triazoldiyl);
A is C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, or C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1;
B is C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, or C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1;
X is an agent as described herein (e.g., an ARB, vitamin D analog,
indoleamine 2,3-dioxygenease (IDO) inhibitor, or a bromodomain inhibitor as described herein);
P is alkylene or heteroalkylene (e.g., polyethylene glycol);
each of L1 and L2 is independently a bond, C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, C1-C12 heteroalkylene, (C0-C12 alkylene)-arylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)- arylene-(C0-C12 heteroalkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12
heteroalkylene), (C0-C12 alkylene)-heteroarylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-heteroarylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)- heteroarylene-(C0-C12 heteroalkylene), (C0-C12 alkylene)-heterocyclylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-heterocyclylene-(C0-C12 alkylene), or (C0-C12 heteroalkylene)-heterocyclylene-(C0-C12 heteroalkylene), wherein each alkylene, alkenylene, alkynylene, heteroalkylene, arylene, heteroarylene, or heterocyclylene is optionally substituted with 1-12 independently selected R2;
L3 is a cleavable linker (e.g., a tissue microenvironment cleavable linker); each R1 and R2 is independently alkyl, alkenyl, alkynyl, heteroalkyl (e.g., polyethylene glycol), halo, cyano, oxo,–ORA,–NRBRC,–NRBC(O)RD, -C(O)NRBRC, –C(O)RD,–C(O)OH,–C(O)ORD,–SRE, or–S(O)mRE;
each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,–
C(O)NRBRC,–C(O)RD,–C(O)OH, or–C(O)ORD;
each RB and RC is independently hydrogen or C1-C6 alkyl;
each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl; and
m is 1 or 2.
In certain embodiments, L1 is a linker connecting B and Ring T, and A is a substituent on L1. In some embodiments, Ring C is a fused carbocyclyl. In some embodiments, Ring C is a fused heterocyclyl.
In some embodiments, Ring C is a structure of Formula (II):

Formula (II),
wherein D is N or CR4; Z is C(R5)2, O, or S; each R3 is independently C1-C6 alkyl, C1- C6 haloalkyl, oxo, or halo; R4 is hydrogen, C1-C6 alkyl, or halo; each R5 is
independently hydrogen, C1-C6 alkyl, C1-C6 haloalkyl, or halo; n is 0, 1, or 2, and“1” represents a portion of Ring C bound to B in the macromonomer.
In some embodiments, Ring C is a structure of Formula (II-a):

Formula (II-a)
wherein D is N or CR4; each R3 is independently C1-C6 alkyl, C1-C6 haloalkyl, oxo, or halo; R4 is hydrogen, C1-C6 alkyl, or halo; n is 0, 1, or 2, and“1” represents a portion of Ring C bound to B in the macromonomer.
In some embodiments, Ring C is a structure of Formula (II-b):

wherein D is N or CR4; each R3 is independently C1-C6 alkyl, C1-C6 haloalkyl, oxo, or halo; R4 is hydrogen, C1-C6 alkyl, or halo; n is 0, 1, or 2, and“1” represents a portion of Ring C bound to B in the macromonomer.
In some embodiments, the structure of Formula (II) comprises

embodiments, the structure of Formula (II) comprises . In some
embodiments, the structure of Formula (II) comprises . In some
embodiments, the structure of Formula (II) comprises
 .
.
In some embodiment, Ring C is a structure of Formula (II-c):

Formula (II-c)
wherein Z is C(R5)2, O, or S; each R3’ is independently selected from C1-C6 alkyl, - C(O)-C1-C6 alkyl, -C(O)-O-C1-C6 alkyl, -C(O)-NH-C1-C6 alkyl, C1-C6 heteroalkyl, -
C(O)-C1-C6 heteroalkyl, -C(O)-O-C1-C6 heteroalkyl, -C(O)-NH-C1-C6 heteroalkyl, and halo, wherein the any alkyl portion of R3’ is optionally substituted with halo; each R5 is independently hydrogen, C1-C6 alkyl, C1-C6 haloalkyl, or halo; s is 0, 1, 2, 3, 4, 5, 6, 7, 8, or 9; and“1” represents a portion of Ring C bound to B in the
macromonomer.
In some embodiments, Ring T is 1,2,3 triazoldiyl or 1,2,4-triazoldiyl. In some
embodiments, Ring T is
 , wherein“2” represents a portion of Ring T bound to L2 in the macromonomer.
, wherein“2” represents a portion of Ring T bound to L2 in the macromonomer.
In some embodiments, A is C1-C12 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C11 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C10 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C9 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C8 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C7 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C6 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C5 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C4 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C3 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C2 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1 heteroalkylene optionally substituted with 1-6 independently selected R1.
In some embodiments, A is C1-C8 heteroalkylene optionally substituted with 1-3 independently selected R1. In some embodiments, R1 is oxo or heteroalkyl (e.g., polyethylene glycol). In some embodiments, R1 is oxo. In some embodiments, R1 is heteroalkyl (e.g., polyethylene glycol). In some embodiments, A is
C(O)CH2CH2C(O)NH-R1. In some embodiments, R1 comprises a polyethylene glycol (PEG), a polyethylene oxide (PEO), a polypropylene glycol (PPG), a polyglycerol (PG), a poloxamine (POX), a polybutylene oxide (PBO), polylactic acid (PLA), polyglycolic acid (PGA), poly(lactic-co-glycolic acid) (PLGA), polycaprolactone
(PCL), polydioxanone (PDO), a polyanhydride, a polyacrylide, a polyvinyl, or a polyorthoester. In some embodiments, R1 comprises a polyethylene glycol (PEG). In some embodiments, A is C(O)CH2CH2C(O)NH-PEG. In some embodiments, A is - C(O)CH2CH2C(O)NH-PEG, optionally wherein the molecular weight of the PEG is between about 200 and about 6000, inclusive, g/mol (Dalton or Da) (e.g., wherein the PEG is PEG200, PEG400, PEG600, PEG800, PEG1000, PEG1500, PEG2000, PEG3000, PEG4000, PEG6000).
In some embodiments, B is C1-C12 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C11 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C10 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C9 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C8 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C7 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C6 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C5 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C4 alkylene optionally substituted with 1-6 R1. In some embodiments, B is C1-C3 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C2 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1 alkylene optionally substituted with 1-6 independently selected R1.
In some embodiments, B is dodecylene, undecylene, decylene, nonylene, octylene, heptylene, hexylene, pentylene, butylene, propylene, ethylene, or methylene, each of which is optionally substituted with 1-6 independently selected R1. In some embodiments, B is dodecylene, undecylene, decylene, nonylene, octylene, heptylene, hexylene, pentylene, butylene, propylene, ethylene, or methylene. In some
embodiments, B is octylene, heptylene, hexylene, pentylene, butylene, propylene, or ethylene. In some embodiments, B is hexylene, pentylene, butylene, propylene, or ethylene. In some embodiments, B is hexylene. In some embodiments, B is pentylene. In some embodiments, B is butylene. In some embodiments, B is propylene. In some embodiments, B is ethylene. In some embodiments, B is methylene.
In some embodiments, the PEG has a molecular weight of between about 200 and about 6000, g/mol (e.g., PEG200, PEG400, PEG600, PEG800, PEG1000,
PEG1500, PEG2000, PEG3000, PEG4000, or PEG6000). In some embodiments, the PEG is PEG200. In some embodiments, the PEG is PEG400. In some embodiments, the PEG is PEG600. In some embodiments, the PEG is PEG800. In some
embodiments, the PEG is PEG1000. In some embodiments, the PEG is PEG2000. In some embodiments, the PEG is PEG3000. In some embodiments, the PEG is
PEG4000. In some embodiments, the PEG is PEG6000.
In some embodiments, each of L1 and L2 is independently C1-C12 alkylene, C1-C12 heteroalkylene, (C0-C12 alkylene)-arylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)- arylene-(C0-C12 alkylene), or (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2. In some embodiments, each of L1 and L2 is independently C1-C12 alkylene, C1-C12 heteroalkylene, or (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-6 independently selected R2. In some embodiments, L1 is C1-C12 heteroalkylene and L2 is C1-C12 alkylene, wherein each alkylene and heteroalkylene is optionally substituted with 1-6 independently selected R2. In some embodiments, each of L1 and L2 is independently selected from–CH2–,–NCH2–, and–CH2CH2. In some embodiments, L1 is–NCH2–. In some embodiments, L2 is–CH2CH2–. In some embodiments, each of L1 and L2 is independently–(CH2)3–,–(CH2)4–,–(CH2)5–, or–(CH2)6–. In some embodiments, each of L1 and L2 is independently–CH2NH–,–(CH2)2NH–,–(CH2)3NH–,–
(CH2)4NH–,–(CH2)5NH–, or–(CH2)6NH–. In some embodiments, each of L1 and L2 is independently–NH(CH2)2–,–NH(CH2)3–,–NH(CH2)4–,–NH(CH2)5–, or– NH(CH2)6–.
In some embodiments, L3 is cleavable by an enzyme (e.g., an esterase, a protease), or by hydrolysis at a certain pH (e.g., acidic pH, basic pH). In some embodiments, L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12
heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2. In some embodiments, L3 is C1-C12 heteroalkylene, or (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-6 independently selected R2. In some embodiments, R2 is oxo or halo (e.g., fluoro). In some embodiments, L3 is–C(O)–,–OC(O)–,–C(O)O–,–
C(O)CH2–,–OC(O)CH2–,–C(O)OCH2–,–CH2CH2O–, C(O)CH2CH2O–,

In some embodiments, P is heteroalkylene. In some embodiments, P comprises polyethylene glycol (PEG), polyethylene oxide (PEO), polypropylene glycol (PPG), polyglycerol (PG), poloxamine (POX), polybutylene oxide (PBO), polylactic acid (PLA), polyglycolic acid (PGA), poly(lactic-co-glycolic acid) (PLGA), polycaprolactone (PCL), polydioxanone (PDO), a polyanhydride, a polyacrylide, a polyvinyl, or a polyorthoester. In some embodiments, P comprises about 2 to about 200 repeating units (e.g., about 2 to about 150, about 2 to about 100, about 2 to about 50, about 2 to about 25, about 2 to about 15, about 2 to about 10, or about 2 to about 5 repeating units) of polyethylene glycol (PEG), polyethylene oxide (PEO), polypropylene glycol (PPG), polyglycerol (PG), poloxamine (POX), polybutylene oxide (PBO), polylactic acid (PLA), polyglycolic acid (PGA), poly(lactic-co-glycolic acid) (PLGA), polycaprolactone (PCL), polydioxanone (PDO), a polyanhydride, a polyacrylide, a polyvinyl, or a polyorthoester.
In some embodiments, P comprises polyethylene glycol (PEG). In some embodiments, P comprises between about 2 to about 200 repeating units (e.g., about 2 to about 150, about 2 to about 100, about 2 to about 50, about 2 to about 25, about 2 to about 15, about 2 to about 10, or about 2 to about 5 repeating units) of polyethylene glycol (PEG). In some embodiments, P comprises between about 2 to about 10 repeating units of polyethylene glycol (PEG). In some embodiments, the PEG comprises diethylene glycol, triethylene glycol, tetraethylene glycol, or pentaethylene glycol.
In some embodiments, the agent (e.g., an agent described herein, e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) is covalently bound to a cleavable linker (e.g., L3 in Formula (I)). In some embodiments, the agent (e.g., an agent described herein, e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) and may be covalently bound to a cleavable linker (e.g., L3 in
Formula (I)) via a hydroxyl group, a sulfonamide, a carboxylic acid, an ester, an amide, an amine, a triazole, or a benzimidazolone.
Exemplary macromonomers may be described by a number of properties, including molecular weight (kDa) and hydrodynamic diameter (nm). In some embodiments, the molecular weight of the macromonomer is between about 1 kDa and about 10 kDa, e.g., between about 2 kDa and about 8 kDa or about 3 kDa and about 6 kDa, e.g., as detected by mass spectrometry. In some embodiments, the molecular weight of the macromonomer is between about 3 kDa and about 6 kDa. In some embodiments, the molecular weight of the macromonomer is about 2 kDa, about 3 kDa, about 4 kDa, about 5 kDa, or about 6 kDa. In some embodiments, the hydrodynamic diameter of the macromonomer is between about 0.5 nm and about 3 nm, e.g., about 1 nm and about 2 nm, e.g., as detected by dynamic light scattering. Exemplary Macromonomers
In some embodiments, the structure of Formula (I) is a structure of Formula (I- a):

In some embodiments, the structure of Formula (I) is a structure of Formula (I- b):

Formula (I-b)
wherein B is C1-C12 alkylene or C1-C12 heteroalkylene, wherein each alkylene or heteroalkylene is optionally substituted with 1-6 R1; X is an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or a bromodomain inhibitor); each of L1 and L2 is independently C1-C12 alkylene, C1-C12 heteroalkylene, (C0-C12 alkylene)-arylene-(C0- C12 alkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)- arylene-(C0-C12 heteroalkylene), or (C0-C12 heteroalkylene)-aryl-(C0-C12
heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; each R1 and R2 is independently alkyl, heteroalkyl, halo, cyano, oxo,–ORA,–NRBRC,–NRBC(O)RD, -C(O)NRBRC,–C(O)RD,–C(O)OH,– C(O)ORD,–SRE, or–S(O)mRE; each RA is independently hydrogen, C1-C6 alkyl, C1-
C6 heteroalkyl,–C(O)NRBRC,–C(O)RD,–C(O)OH, or–C(O)ORD; each RB and RC is independently hydrogen or C1-C6 alkyl; or each RD is independently C1-C6 alkyl, C1- C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl; t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive.
In some embodiments, the structure of Formula (I) is a structure of Formula (I- c):

Formula (I-c)
wherein X is an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or a bromodomain inhibitor); L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)- arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0- C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene,
heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; each R2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive.
In some embodiments, the structure of Formula (I) is a structure of Formula (I- d-1) or Formula (I-d-2):

Formula (I-d-1)

Formula (I-d-2)
wherein L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12
heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; each R2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive.
In some embodiments, the structure of Formula (I) is a structure of Formula (I- e):

Formula (I-e)
wherein L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12
heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; each R2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive.
In some embodiments, the structure of any of the formulae described herein (e.g., Formulae (I), (I-a). (I-b), (I-c), (I-d-1), (I-d-2), and (I-e)) comprises a salt (e.g., a pharmaceutically acceptable salt).
In some embodiments the structure of Formula I is selected from:


and

Described herein are BRUSH conjugates and BASP particles comprising one or more agents (e.g., a therapeutic agent, diagnostic agent, or targeting agent). These BRUSH conjugates and BASP particles are prepared through the co-polymerization of discrete macromonomer subunits that include the agent with a multifunctional crosslinker via, e.g., ring-opening metathesis polymerization (ROMP).
The term“conjugate” or“BRUSH conjugate” or“BRUSH” as used herein refers to a molecular structure comprising at least two macromonomers joined together (e.g., directly or indirectly), e.g., as shown in No.2 in FIG.1. The macromonomers may be identical, or in some cases, there may be more than one type of macromonomer present within the conjugate. In some embodiments, the BRUSH conjugate refers to the product of a first round of polymerization (e.g., a bottle-brush polymer) and has an average molecular weight of about 10 kDa to about 100 kDa. In some embodiments, the macromonomers present within the conjugate are covalently linked. In some embodiments, the conjugate is prepared from ring opening metathesis polymerization.
The term“BASP particle” or“BASP” as used herein refers to a structure comprising at least two conjugates joined together (e.g., directly or indirectly, e.g., by covalent or noncovalent means), e.g., as shown in shown in No.3 in number FIG.1. The conjugates may be identical, or in some cases, there may be more than one type of conjugate present within the particle. In some embodiments, the BASP particle refers to the product of a subsequent round of polymerization (e.g., a second, third, or fourth round of polymerization) and has an average molecular weight of about 100 kDa to about 1,000 kDa. In some embodiments, the particle is prepared from ring opening metathesis polymerization (ROMP). In some embodiments, the conjugates present in the particle are noncovalently linked (e.g., are linked through ionic or hydrophobic interactions).
In some embodiments, the conjugate or BASP particle comprises (i) an agent chosen from an angiotension receptor blocker (ARB), a vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor; (ii) a carbocyclyl or heterocyclyl moiety; (iii) a triazole moiety; (iv) a heteroalkyl moiety; and (v) a cleavable linker (e.g., a tissue microenvironment cleavable linker).
In some embodiments, the agent is an angiotensin receptor blocker (ARB), vitamin D analog, IDO inhibitor, or bromodomain inhibitor, e.g., as described herein.
In some embodiments, the carbocyclyl or heterocyclyl moiety comprises a monocyclic or bicyclic carbocyclyl or a monocyclic or bicyclic heterocyclyl. In some embodiments, the carbocyclyl or heterocyclyl moiety comprises a monocyclic carbocyclyl or monocyclic heterocyclyl. In some embodiments, the carbocyclyl or heterocyclyl moiety comprises a bicyclic carbocyclyl or bicyclic heterocyclyl.
In some embodiments, the triazole moiety comprises 1,2,3-triazoldiyl or 1,2,4- triazoldiyl. In some embodiments, the triazole moiety comprises 1,2,3-triazoldiyl. In some embodiments, the triazole moiety comprises 1,2,4-triazoldiyl.
In some embodiments, the heteroalkyl moiety comprises a polyethylene glycol (PEG), a polyethylene oxide (PEO), a polypropylene glycol (PPG), a polyglycerol (PG), a poloxamine (POX), a polybutylene oxide (PBO), polylactic acid (PLA), polyglycolic acid (PGA), poly(lactic-co-glycolic acid) (PLGA), polycaprolactone (PCL), polydioxanone (PDO), a polyanhydride, a polyacrylide, a polyvinyl, or a polyorthoester. In some embodiments, the heteroalkyl moiety comprises a
polyethylene glycol (PEG) moiety. In some embodiments, the PEG has a molecular weight of between about 200 and about 6000, g/mol (e.g., PEG200, PEG400, PEG600, PEG800, PEG1000, PEG1500, PEG2000, PEG3000, PEG4000, or
PEG6000). In some embodiments, the PEG is PEG200. In some embodiments, the PEG is PEG400. In some embodiments, the PEG is PEG600. In some embodiments, the PEG is PEG800. In some embodiments, the PEG is PEG1000. In some
embodiments, the PEG is PEG2000. In some embodiments, the PEG is PEG3000. In some embodiments, the PEG is PEG4000. In some embodiments, the PEG is
PEG6000.
In some embodiments, the agent is non-covalently bound to the conjugate (e.g., the agent is associated with the conjugate or BASP particle through ionic bonds or hydrophobic interactions). In some embodiments, the agent is covalently bound to the conjugate or BASP particle through a cleavable linker (e.g., L3 in Formula (I)). Cleavable Linkers
The agent, e.g., the therapeutic agent, is covalently bound to the BRUSH- composition or BASP-composition through a tissue microenvironment cleavable linker (can also be referred to herein as“sensitive linker”). In some embodiments, the cleavable linker is the L3 portion of the macromonomer(s) that make up the BRUSH conjugate or BASP particle. A cleavable linker is“cleaved” or“degraded” when one or more bonds of the cleavable linker are broken, e.g., resulting in release of an agent (e.g., an angiotension receptor blocker (ARB), a vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor), e.g., from the conjugate or BASP particle. Linker cleavage or agent release need not be 100%, e.g., a cleavage or release of at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% or higher, e.g., over a period
of seconds, minutes, hours (e.g., 6 hours, 12 hours, or 24 hours), days (e.g., 2 days or 7 days), weeks, or months is encompassed by this term.
In some embodiments, the cleavable linker is cleavable by or is sensitive to an enzyme (e.g., an esterase or a protease), pH (e.g., acidic pH, basic pH), light (e.g., ultraviolet light), a nucleophile, reduction, or oxidation. In some embodiments, the cleavable linker is cleavable by or is sensitive to an enzyme (e.g., an esterase or a protease) or pH (e.g., acidic pH, basic pH). In some embodiments, the cleavable linker is not cleavable by light (e.g., ultraviolet light).
In some embodiments, the cleavable linker comprises an ester, an acetal, a ketal, a phosphoramidite, a hydrazone, an imine, an oxime, a disulfide, or a silyl moiety, a combination of acetal or ketal with ester group, an oligo-acetal or oligo- ketal group, a combination of the oligo-ketal and silyl ether group, or a combination of the oligo-ketal and vinyl ether group. In some embodiments, the cleavable linker comprises an ester. In some embodiments, the cleavable linker comprises an acetal. In some embodiments, the cleavable linker comprises a phosphoramidite. In some embodiments, the cleavable linker comprises a hydrazine. In some embodiments, the cleavable linker comprises an imine. In some embodiments, the cleavable linker comprises an oxime. In some embodiments, the cleavable linker comprises a silyl moiety. In some embodiments, the cleavable linker comprises a disulfide.
In other embodiments, the cleavable linker is chosen from a combination of acetal or ketal with cis-aconityl, hydrazine, oxime, imidazole or trityl groups. Any of the aforesaid groups or combination of groups can modified to enhance the pH sensitivity of the cleavable linker, e.g., as described herein.
In some embodiments, the cleavable linker is represented by L3 in a structure of Formula (I) or Formula (III). In some embodiments, L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2. In some embodiments, L3 is C1-C12 heteroalkylene, or (C0- C12 alkylene)-arylene-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-6 independently selected R2. In some embodiments, R2 is oxo or halo (e.g., fluoro). In some embodiments, L3 is–C(O)–,– OC(O)–,–C(O)O–,–C(O)CH2–,–OC(O)CH2–,–C(O)OCH2–,–CH2CH2O–,

It should be understood that in some cases, the cleavable linker may include an atom or a part of a moiety that is derived in part from the agent (e.g., a therapeutic agent). For example, if the cleavable linker is an ester, and an agent (e.g., telmisartan) is linked to the macromonomer, conjugate or BASP particle through a carboxylate, the cleavable linker may comprise the carbonyl group derived from the agent. In some embodiments, the cleavable linker comprises L3 and an atom or chemical moiety from the agent. In some embodiments, the cleavable linker comprises L3 and does not comprise an atom or chemical moiety from the agent.
In some embodiments, the cleavable linker is cleaved or degraded, e.g., preferentially cleaved or degraded, upon exposure to a first set of conditions relative to a second set of conditions. For example, the cleavable linker can be“preferentially cleaved” or“preferentially degraded” in a first set of conditions relative to a second set of conditions if at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% or more of a bond or bonds of the cleavable linker are broken, or the agent is released, in the first set of conditions relative to the second set of conditions.
In some embodiments, the cleavable linker is degraded or hydrolyzed at physiological conditions. In some embodiments, the linker is pH sensitive or cleaved at a certain pH. In some embodiments, the linker is degraded or hydrolyzed through the action of an enzyme (e.g., a protease or esterase). For example, in some embodiments, the cleavable linker is preferentially cleaved in a tissue
microenvironment, e.g., a tumor microenvironment, which is referred to herein as a “tissue microenvironment cleavable linker.” In embodiments, the tissue (e.g., tumor) microenvironment cleavable linker is preferentially cleaved or degraded upon exposure to a first desired tissue or tumor microenvironment relative to a second tissue or non-tumor tissue. A tissue (e.g., tumor) microenvironment cleavable linker can be preferentially cleaved if at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% or more of a bond or bonds of the linker are broken, or the agent is
released, in a desired tissue or tumor microenvironment relative to another tissue or non-tumor tissue. In one embodiment, the tissue (e.g., tumor) microenvironment cleavable linker is preferentially cleaved or degraded if one or more of the bonds of the linker are broken, or the agent is released, at least 2, 5, 10, 20, 30, 40, 50, 60, 70, 80, or 100 times faster upon exposure to a first desired tissue or tumor
microenvironment relative to a second tissue or non-tumor tissue. The tissue (e.g., tumor) microenvironment can have a particular set of conditions, e.g., pH, enzymes, that cause the cleavage or degradation of the linker.
In some embodiments, the cleavable linker is a peptide. In some embodiments, the linker is a peptide, and the peptide sequence is comprised of naturally occurring amino acids. In some embodiments, the linker is a peptide, and the peptide sequence comprises at least one synthetically derived amino acids, e.g., at least 2, at least 3, at least 4, at least 5, at least 8, at least 10, at least 15, at least 20, or more synthetically derived amino acids. In some embodiments, the peptide has a linear structure. In some embodiments, the peptide has a branched structure. In some embodiments, the peptide has a branched structure with, e.g., at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, or at least 8 branching points. In some embodiments, the peptide has a cyclic structure.
In some embodiments, the cleavable linker is a peptide, and the peptide sequence comprises at least 2 amino acid residues. In some embodiments, the peptide sequence comprises at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or at least 10 amino acid residues. In some embodiments, the peptide sequence is from about 1 to about 10 amino acid residues. In some embodiments, the peptide sequence is from about 1 to about 15, about 20, about 25, about 30, about 40, about 50, about 60, about 70, about 80, about 90, or about 100 amino acid residues. In some embodiments, the peptide sequence is from about 10 to about 100 amino acid residues. In some embodiments, the peptide sequence is from about 25 to about 100 amino acid residues. In some embodiments, the peptide sequence is from about 50 to about 100 amino acid residues.
In some embodiments, the cleavable linker comprises a substrate peptide that is cleaved, e.g., activated, by a matrix metalloprotease (MMP) selected from a sequence disclosed in U.S. Patent Application No.2015/0087810. In some
embodiments, the substrate peptide comprises a protease substrate comprising an amino acid sequence selected from the group consisting of SEQ ID NOs: 353-363,
372-375, 376-378, 395-401, 411-419, 426-433, 437-449, 454-456, 459-469, 475-482, 487-495, 318-323, 325-327, 330-335, 341-347, 14-33, and 159, e.g., as described in U.S. Patent Application No.2015/0087810. In some embodiments, the linker comprises a substrate peptide derived from a sequence disclosed in U.S. Patent No. 8,541,203, e.g., a substrate peptide chosen from an enzyme selected from the group consisting of MMP-1, MMP-2, MMP-3, MMP-8, MMP-9, MMP-14, plasmin, PSA, PSMA, CATHEPSIN D, CATHEPSIN K, CATHEPSIN S, ADAM10, ADAM12, ADAMTS, Caspase-1, Caspase-2, Caspase-3, Caspase-4, Caspase-5, Caspase-6, Caspase-7, Caspase-8, Caspase-9, Caspase-10, Caspase-11, Caspase-12, Caspase-13, Caspase-14, and TACE. In some embodiments, the linker comprises a sequence disclosed in U.S. Patent No.8,513,390. In some embodiments, the linker comprises a sequence disclosed in International Patent Publication No. WO2003/079972. In some embodiments, the linker comprises a sequence disclosed in U.S. Patent No.7,495,099. In some embodiments, the linker comprises a sequence disclosed in U.S. Patent No. 8,580,244. In some embodiments, the linker comprises a sequence disclosed in one of the following articles: van Kempen, et al. Eur Cancer (2006) 42:728-734; Desnoyers, L.R. et al. Sci Transl Med (2013) 5:207ra144; Rice, J.J. et al. Protein Sci (2006) 15:825-836; Boulware, K.T. and Daugherty, P.S. Proc Natl Acad Sci USA (2006) 103:7583-7588; Deperthes, D. Biol Chem (2002) 383:1107-1112; Harris, J.L. Proc Natl Acad Sci USA (2000) 97:7754-7759; Salmaso S. and Caliceti, P. J Drug Deliv (2013) 2013:1-19; and Eckhard, U et al Matrix Biol (2015) doi:
10.1016/j.matbio.2015.09.003 (epub ahead of print). The contents of any of the publications referenced herein are hereby expressly incorporated by reference.
In some embodiments, the cleavable linker comprises a substrate peptide that is cleaved, e.g., activated, by a protease, e.g., a protease present in a tumor or fibrotic microenvironment (e.g., a matrix metalloprotease (MMP), e.g., as described by Desnoyers, L.R. et al. Sci Transl Med (2013) 5:207ra144; Eckhard, U et al Matrix Biol (2015) doi: 10.1016/j.matbio.2015.09.003 (epub ahead of print); and van
Kempen, et al. Eur Cancer (2006) 42:728-734. In one embodiment, the linker includes the amino acid sequence of a substrate for uPA, e.g., comprises the amino acid sequence LSGRSDNH (SEQ ID NO:1), e.g., as described in U.S. Patent No. 8,513,390. In some embodiment, the linker sequence further includes a Gly-Ser- containing peptide linker, at either end, or both ends to the substrate peptide.
Additional exemplary proteases that may be upregulated in a tumor microenvironment
include, but are not limited to, urokinase-type plasminogen activator (uPA), which is upregulated in human carcinomas (S. Ulisse, et al. Curr. Cancer Drug Targets 9, 32– 71 (2009)), membrane-type serine protease 1 (MT-SP1/matriptase) (K. Uhland Cell. Mol. Life Sci.63, 2968–2978 (2006); A. M. LeBeau, et al. Proc. Natl. Acad. Sci. U.S.A.110, 93–98 (2013)), and legumain, a lysosomal protease found to be released and active in the acidic extracellular tumor microenvironment (C. Liu, et al. Cancer Res.63, 2957–2964 (2003)). In some embodiments, the protease is produced by an inflammatory cell, e.g., a tumor infiltrating leukocyte (e.g., a leukocyte-derived MMP), e.g., as described by van Kempen, et al. Eur Cancer (2006) 42:728-734. In other embodiments, the MMP is chosen from MMP1, MMP2, MMP3, MMP7, MMP8, MMP9, MMP12, MMP13 or MMP14, e.g., as described by Eckhard, U et al. supra.
In some embodiments, the substrate peptide is derived from a CLiPS library (as described in, e.g., K. T. Boulware, P. S. Daugherty, Proc. Natl. Acad. Sci. U.S.A. 103, 7583–7588 (2006)). In other embodiments, the substrate peptide specificity is evaluated using combinatorial fluorogenic substrate libraries, e.g., as described by Harris, J.L. Proc Natl Acad Sci USA (2000) 97:7754-7759. In other embodiments, the substrate peptide is derived from a phage display library (e.g., it is a phase display substrate), e.g., as described by Deperthes, D. Biol Chem (2002) 383:1107-1112. For example, a phage display substrate is exposed to a plurality of proteases; peptides released through specific cleavage can be amplified in an expression system. In other embodiments, the substrate peptide is derived from a bacterial display library, e.g., as described by Rice, J.J. et al. Protein Sci (2006) 15:825-836.
In one embodiment, the tissue (e.g., tumor) microenvironment cleavable linker is cleavable by an enzyme. In some embodiments, the enzyme comprises an esterase or a protease. Exemplary proteases include MMP-1, MMP-2, MMP-3, MMP-8, MMP-9, MMP-14, plasmin, PSA, PSMA, CATHEPSIN D, CATHEPSIN K,
CATHEPSIN S, ADAM10, ADAM12, ADAMTS, Caspase-1, Caspase-2, Caspase-3, Caspase-4, Caspase-5, Caspase-6, Caspase-7, Caspase-8, Caspase-9, Caspase-10, Caspase-11, Caspase-12, Caspase-13, Caspase-14, or TACE.
In other embodiments, the tissue microenvironment cleavable linker is cleavable at a particular pH. In some embodiments, the tissue microenvironment cleavable linker is cleavable at a pH between about 5.0 and about 7.4, between 5.0 and 7.0, between 5.0 and 6.5, between 5.0 and 5.5, or between 5.9 and 6.2. In one
embodiment, the tissue microenvironment cleavable linker is cleavable at a pH between about 6.0 and about 7.0, between about 6.2 and about 6.9, between about 6.5 and about 6.8, or between about 6.5 and about 6.7. In one embodiment, the tissue microenvironment cleavable linker is cleavable at a pH between about 5.5 and about 6.5, e.g., between 5.9 and 6.2. In one embodiment, the tissue microenvironment cleavable linker is cleavable at a hypoxic pH, e.g., a pH about 6.7 to 6.9, e.g., compared to a physiological pH of about 7.4.
In some embodiments, the tissue microenvironment cleavable linker is cleavable is cleaved at a pH of no more than 7.4, no more than 7.0, no more than 6.9, no more than 6.8, no more than 6.7, no more than 6.6, no more than 6.5, no more than 6.4, no more than 6.3, no more than 6.2, no more than 6.1, no more than 6.0, no more than 5.5 or lower.
In one embodiment, the tissue microenvironment cleavable linker is preferentially cleaved or degraded upon exposure to a first pH relative to a second pH. In one embodiment, the tissue microenvironment cleavable linker is cleaved or degraded at least 2, 5, 10, 20, 30, 40, 50, 60, 70, 80, or 100 times faster upon exposure to a first pH relative to a second pH. In other embodiments, the tissue
microenvironment cleavable linker shows a greater release or degradation rate at a first acidic pH (e.g., pH=6.7) relative to a second more basic pH (e.g., pH = 7.4). In one embodiment, ratio of release or degradation rate of the tissue microenvironment cleavable linker at pH=6.7 relative to pH = 7.4 is greater than 1, 1.2, 1.4, 1.6, 1.8, 2, 2.2, 2.4, 2.6, 2.8, 3 or higher. In one embodiment, ratio of release or degradation rate of the tissue microenvironment cleavable linker at pH=6.7 relative to pH = 7.4 is greater than 2.
In one embodiment, the tissue microenvironment cleavable linker shows increased pH-sensitivity in a hypoxic microenvironment, e.g., in a tumor, or fibrotic tissue.
In some embodiments, the tissue microenvironment cleavable linker (e.g., L3) exhibits an increased release rate or increased release yield of the agent (e.g., the ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) at a desired site (e.g., a tumor), e.g., relative to the release rate or release yield at another site. In one embodiment, the tissue microenvironment cleavable linker comprises an electron withdrawing group (e.g., an electron withdrawing group that enhances the cleavage rate or yield, e.g., upon exposure to a first set of conditions relative to a second set of
conditions). Exemplary linkers demonstrating this effect are described, e.g., in Example 11 and FIGS.14A to 14C. For example, BASP particles comprising a phenylester linker (Tel-PhenylEster) or difluorophenylester linker (Tel- DiFluoroPhenylEster) achieved increased release of an exemplary agent (e.g., telmisartan) over BASP particles comprising an ester linker (Original TelEster). In one embodiment, a BASP particle comprising a difluorophenylester linker (e.g., wherein L3 is -O-(3,5-difluorophenyl)-C(O)NH) exhibited greater than a 100-fold increase in release of telmisartan than a BASP particle comprising an ester linker (e.g., wherein L3 is–OCH2CH2-). In another embodiment, a BASP particle comprising a phenylester linker (e.g., wherein L3 is -O-phenyl-C(O)NH-) exhibited roughly a 10-fold increase in release of telmisartan than a BASP comprising an ester linker (e.g., wherein L3 is - OCH2CH2-). BRUSH Conjugate or BASP Particle Properties
Exemplary conjugates or BASP particles may be described by a number of properties, including average molecular weight (kDa), average hydrodynamic diameter (nm), and polydispersity.
The term“average molecular weight” may encompass the number average molecular weight (Mn), weight average molecular weight (Mw), higher average molecular weight (Mz or Mz +1), GPC/SEC-determined average molecular weight (Mp), and viscosity average molecular weight (Mv).
In some embodiments, the average molecular weight of the conjugate is between about 2 kDa and about 100 kDa, e.g., between about 15 kDa and about 85 kDa, about 20 kDa and about 60 kDa, or about 30 kDa and about 50 kDa, e.g., as determined by gel permeation chromatography. In some embodiments, the average molecular weight of the conjugate is less than about 100 kDa (e.g., less than about 95 kDa, about 90 kDa, about 85 kDa, about 80 kDa, about 75 kDa, about 70 kDa, about 65 kDa, about 60 kDa, about 55 kDa, or about 50 kDa), e.g., as determined by gel permeation chromatography. In some embodiments, the average molecular weight of the conjugate is less than about 75 kDa (e.g., less than about 70 kDa, about 65 kDa, about 60 kDa, about 55 kDa, or about 50 kDa). In one embodiment, the average molecular weight of the conjugate is between about 20 kDa and about 60 kDa. In one embodiment, the average molecular weight of the conjugate is between about 30 kDa and about 50 kDa. In still other embodiments, the average molecular weight of the
conjugate is between about 2 kDa and about 6 kDa (e.g., between about 3 kDa and about 5 kDa)
In some embodiments, the average molecular weight of the BASP particle is between about 100 kDa and about 1,000 kDa, e.g., between about 200 kDa and about 700 kDa or about 300 kDa and about 500 kDa, e.g., as determined by gel permeation chromatography. In one embodiment, the average molecular weight of the particle is between about 2000 kDa and about 70 kDa. In one embodiment, the average molecular weight of the particle is between about 300 kDa and about 500 kDa. In some embodiments, the average molecular weight of the particle is less than about 1,000 kDa (e.g., less than about 950 kDa, about 900 kDa, about 850 kDa, about 800 kDa, about 750 kDa, about 700 kDa, about 650 kDa, about 600 kDa, about 550 kDa, or about 500 kDa), e.g., as determined by gel permeation chromatography. In some embodiments, the average molecular weight of the particle is less than about 750 kDa (e.g., less than about 700 kDa, about 650 kDa, about 600 kDa, about 550 kDa, or about 500 kDa). In some embodiments, the average molecular weight of the particle is less than about 500 kDa (e.g., less than about 450 kDa, about 400 kDa, about 350 kDa, or 300 kDa).
The term“average hydrodynamic diameter” as used herein refers to the average size of a conjugate or BASP particle. The average hydrodynamic diameter may or may not encompass the solvation layers of conjugate or BASP particle, and may be determined through a number of methods including dynamic light scattering, electron microscopy (e.g., scanning electron microscopy, transmission electron microscopy), atomic force microscopy, and X-ray diffraction.
In some embodiments, the average hydrodynamic diameter of the conjugate is less than 50 nm (e.g., less than about 45 nm, about 40 nm, about 35 nm, about 25 nm, about 20 nm, about 15 nm, about 10 nm, about 7.5 nm, or less), e.g., as determined by dynamic light scattering. In some embodiments, the average hydrodynamic diameter of the conjugate is between about 1 nm and about 20 nm (e.g., between about 2.5 nm and about 17.5 nm, or about 5 nm and about 15 nm). In some embodiments, the average hydrodynamic diameter of the conjugate is between about 5 nm and about 15 nm. In some embodiments, the average hydrodynamic diameter of the conjugate is less than about 20 nm (e.g., less than about 15 nm, about 12.5 nm, about 10 nm, about 9 nm, about 8 nm, about 7 nm, about 6 nm, about 5 nm, or less)
In some embodiments, the average hydrodynamic diameter of the BASP particle is less than 100 nm (e.g., less than about 90 nm, about 80 nm, about 75 nm, about 70 nm, about 65 nm, about 60 nm, about 55 nm, about 50 nm, about 45 nm, about 40 nm, about 35 nm, about 25 nm, or less), e.g., as determined by dynamic light scattering. In some embodiments, the average hydrodynamic diameter of the BASP particle is between about 5 nm and about 100 nm (e.g., between about 7.5 nm and about 75 nm, about 10 nm and about 50 nm, about 12.5 nm and about 40 nm, or about 15 nm and about 30 nm). In some embodiments, the average hydrodynamic diameter of the BASP particle is between about 10 nm and about 50 nm. In some embodiments, the average hydrodynamic diameter of the BASP particle is between about 5 nm and about 50 nm. In some embodiments, the average hydrodynamic diameter of the BASP particle is between about 15 nm and about 30 nm.
The term“average polydispersity” as used herein refers to a measure of the distribution of molecular size in a mixture, e.g., as determined by a chromatographic method, such as gel permeation chromatography or size exclusion chromatography, or through dynamic light scattering. In some embodiments, the average polydispersity of the conjugate or BASP particle is less than about 0.5 (e.g., less than about 0.4, about 0.35, about 0.3, about 0.25, about 0.2, about 0.15, or less). In some embodiments, the average polydispersity of the conjugate or BASP particle is less than about 0.3. In some embodiments, the average polydispersity of the conjugate or BASP particle is less than about 0.2. In some embodiments, the conjugate or BASP particle is monodisperse. In some embodiments, the conjugate or BASP particle is about 50% monodisperse (e.g., about 55%, about 60%, about 65%, about 70%, about 75%, about 80%, about 85%, about 90%, about 95%, about 99%, or about 99.9% monodisperse).
In some embodiments, the conjugate or BASP particle is substantially soluble in water (e.g., hydrophilic). In some embodiments, the conjugate or BASP particle is substantially insoluble in water (e.g., hydrophobic). In some embodiments, the conjugate or BASP particle is substantially insoluble in water and greater than about 10,000 parts water are required to dissolve 1 part polymer. In one embodiment, the conjugate or BASP particle is amphiphilic. In one embodiment, the conjugate or BASP particle comprises a segment that is hydrophobic and a segment that is hydrophilic.
In some embodiments, the conjugate comprises a linear structure. In some embodiments, the conjugate comprises a branched structure. In some embodiments,
the conjugate comprises a branched structure, and each repeating unit in the polymer comprises at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, or at least 8 branch points.
In some embodiments, the conjugate or particle comprises a structure according to Formula (III):

Formula (III)
wherein Ring C is a carbocyclyl or heterocyclyl moiety; Ring T is a triazoldiyl moiety (e.g., 1,2,3-triazoldiyl); A is C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or
heteroalkylene is optionally substituted with 1-6 independently selected R1; B is C1- C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1; X is an agent (e.g., an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor); P is alkylene or heteroalkylene (e.g., polyethylene glycol); each of L1 and L2 is independently a bond, C1-C12 alkylene, C2- C12 alkenylene, C2-C12 alkynylene, C1-C12 heteroalkylene, (C0-C12 alkylene)-arylene- (C0-C12 alkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 heteroalkylene), (C0-C12 alkylene)-heteroarylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-heteroarylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)- heteroarylene-(C0-C12 heteroalkylene), (C0-C12 alkylene)-heterocyclylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-heterocyclylene-(C0-C12 alkylene), or (C0-C12 heteroalkylene)-heterocyclylene-(C0-C12 heteroalkylene), wherein each alkylene, alkenylene, alkynylene, heteroalkylene, arylene, heteroarylene, or heterocyclylene is optionally substituted with 1-12 independently selected R2; L3 is a tissue
microenvironment cleavable linker; each R1 and R2 is independently alkyl, alkenyl, alkynyl, heteroalkyl (e.g., polyethylene glycol), halo, cyano, oxo,–ORA,–NRBRC,– NRBC(O)RD, -C(O)NRBRC,–C(O)RD,–C(O)OH,–C(O)ORD,–SRE, or–S(O)mRE; each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,–C(O)NRBRC,– C(O)RD,–C(O)OH, or–C(O)ORD; each RB and RC is independently hydrogen or C1-
C6 alkyl; or each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl; and m is 1 or 2. In some embodiments, Ring C is a structure of Formula (IV):

wherein D is N or CR4; Z is C(R5)2, O, or S; each R3 is independently C1-C6 alkyl, C1- C6 haloalkyl, oxo, or halo; R4 is hydrogen, C1-C6 alkyl, or halo; each R5 is
independently hydrogen, C1-C6 alkyl, C1-C6 haloalkyl, or halo; n is 0, 1, or 2,“1” represents a portion of Ring C bound to B; and each“2” represents a portion of Ring C bound to either another macromonomer, an initiating group (R6a), or an end group (R6b), each through a -CH=CH- linker in the conjugate or BASP particle.
In some embodiments, Ring C is a structure of Formula (IV-a):

Formula (IV-a)
wherein D is N or CR4; each R3 is independently C1-C6 alkyl, C1-C6 haloalkyl, oxo, or halo; R4 is hydrogen, C1-C6 alkyl, or halo; n is 0, 1, or 2, and“1” represents a portion of Ring C bound to B; and each“2” represents a portion of Ring C bound to either another macromonomer, an initiating group (R6a), or an end group (R6b), each through a -CH=CH- linker in the conjugate or BASP particle.In some embodiments, the
structure of Formula (IV) is
 or. In some embodiments, the structure of Formula (I . In
or. In some embodiments, the structure of Formula (I . In

some embodiments, the structure of Formula (IV) is . In some

embodiments, the structure of Formula (IV) is .
In some embodiment, Ring C is a structure of Formula (IV-b):

wherein Z is C(R5)2, O, or S; R3’’ is selected from hydrogen, C1-C6 alkyl, -C(O)-C1-C6 alkyl, -C(O)-O-C1-C6 alkyl, -C(O)-NH-C1-C6 alkyl, C1-C6 heteroalkyl, -C(O)-C1-C6 heteroalkyl, -C(O)-O-C1-C6 heteroalkyl, -C(O)-NH-C1-C6 heteroalkyl, and halo, wherein the any alkyl portion of R3’ is optionally substituted with halo; each R5 is independently hydrogen, C1-C6 alkyl, C1-C6 haloalkyl, or halo;“1” represents a portion of Ring C bound to B; and each“2” represents a portion of Ring C bound to either another macromonomer, an initiating group (R6a), or an end group (R6b), each through a -CH=CH- linker in the conjugate or BASP particle.
In some embodiments, Ring T is 1,2,3 triazoldiyl or 1,2,4-triazoldiyl. In some embodiments, Ring T is 1,2,3 triazoldiyl. In some embodiments, Ring T is
, wherein“2” represents a portion of Ring

T bound to L2 in the conjugate.
In some embodiments, A is C1-C12 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C11 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C10 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C9 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C8 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C7 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C6 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C5 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C4 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C3 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1-C2 heteroalkylene optionally substituted with 1-6 independently selected R1. In some embodiments, A is C1 heteroalkylene optionally substituted with 1-6 independently selected R1.
In some embodiments, A is C1-C8 heteroalkylene optionally substituted with 1-3 independently selected R1. In some embodiments, R1 is oxo or heteroalkyl (e.g., polyethylene glycol). In some embodiments, R1 is oxo. In some embodiments, R1 is heteroalkyl (e.g., polyethylene glycol). In some embodiments, A is
C(O)CH2CH2C(O)NH-R1. In some embodiments, R1 comprises a polyethylene glycol (PEG), a polyethylene oxide (PEO), a polypropylene glycol (PPG), a polyglycerol (PG), a poloxamine (POX), a polybutylene oxide (PBO), polylactic acid (PLA), polyglycolic acid (PGA), poly(lactic-co-glycolic acid) (PLGA), polycaprolactone (PCL), polydioxanone (PDO), a polyanhydride, a polyacrylide, a polyvinyl, or a polyorthoester. In some embodiments, R1 comprises a polyethylene glycol (PEG). In some embodiments, A is C(O)CH2CH2C(O)NH-PEG.
In some embodiments, the PEG has a molecular weight of between about 200 and about 6000, g/mol (e.g., PEG200, PEG400, PEG600, PEG800, PEG1000, PEG1500, PEG2000, PEG3000, PEG4000, or PEG6000). In some embodiments, the
PEG is PEG200. In some embodiments, the PEG is PEG400. In some embodiments, the PEG is PEG600. In some embodiments, the PEG is PEG800. In some
embodiments, the PEG is PEG1000. In some embodiments, the PEG is PEG2000. In some embodiments, the PEG is PEG3000. In some embodiments, the PEG is
PEG4000. In some embodiments, the PEG is PEG6000.
In some embodiments, B is C1-C12 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C11 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C10 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C9 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C8 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C7 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C6 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C5 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C4 alkylene optionally substituted with 1-6 R1. In some embodiments, B is C1-C3 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1-C2 alkylene optionally substituted with 1-6 independently selected R1. In some embodiments, B is C1 alkylene optionally substituted with 1-6 independently selected R1.
In some embodiments, B is dodecylene, undecylene, decylene, nonylene, octylene, heptylene, hexylene, pentylene, butylene, propylene, ethylene, or methylene, each of which is optionally substituted with 1-6 independently selected R1. In some embodiments, B is dodecylene, undecylene, decylene, nonylene, octylene, heptylene, hexylene, pentylene, butylene, propylene, ethylene, or methylene. In some
embodiments, B is octylene, heptylene, hexylene, pentylene, butylene, propylene, or ethylene. In some embodiments, B is hexylene, pentylene, butylene, propylene, or ethylene. In some embodiments, B is hexylene. In some embodiments, B is pentylene. In some embodiments, B is butylene. In some embodiments, B is propylene. In some embodiments, B is ethylene. In some embodiments, B is methylene.
In some embodiments, each of L1 and L2 is independently C1-C12 alkylene, C1- C12 heteroalkylene, (C0-C12 alkylene)-arylene-(C0-C12 alkylene), (C0-C12
heteroalkylene)-arylene-(C0-C12 alkylene), or (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein
each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2. In some embodiments, each of L1 and L2 is independently C1-C12 alkylene, C1-C12 heteroalkylene, or (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-6 independently selected R2. In some embodiments, L1 is C1-C12 heteroalkylene and L2 is C1-C12 alkylene, wherein each alkylene and heteroalkylene is optionally substituted with 1-6 independently selected R2. In some embodiments, each of L1 and L2 is independently selected from–CH2–,–NCH2–, and–CH2CH2. In some embodiments, L1 is–NCH2–. In some embodiments, L2 is–CH2CH2–.
In some embodiments, L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)- arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0- C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene,
heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2. In some embodiments, L3 is C1-C12 heteroalkylene, or (C0-C12 alkylene)-arylene- (C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-6 independently selected R2. In some embodiments, R2 is oxo or halo (e.g., fluoro). In some embodiments, L3 is–C(O)–,–OC(O)–,–C(O)O–, –C(O)CH2–,–OC(O)CH2–,–C(O)OCH2–,–CH2CH2O–, C(O)CH2CH2O–,

In some embodiments, P is heteroalkylene. In some embodiments, P comprises polyethylene glycol (PEG), polyethylene oxide (PEO), polypropylene glycol (PPG), polyglycerol (PG), poloxamine (POX), polybutylene oxide (PBO), polylactic acid (PLA), polyglycolic acid (PGA), poly(lactic-co-glycolic acid)
(PLGA), polycaprolactone (PCL), polydioxanone (PDO), a polyanhydride, a polyacrylide, a polyvinyl, or a polyorthoester. In some embodiments, P comprises about 2 to about 200 repeating units (e.g., about 2 to about 150, about 2 to about 100, about 2 to about 50, about 2 to about 25, about 2 to about 15, about 2 to about 10, or about 2 to about 5 repeating units) of polyethylene glycol (PEG), polyethylene oxide
(PEO), polypropylene glycol (PPG), polyglycerol (PG), poloxamine (POX), polybutylene oxide (PBO), polylactic acid (PLA), polyglycolic acid (PGA), poly(lactic-co-glycolic acid) (PLGA), polycaprolactone (PCL), polydioxanone (PDO), a polyanhydride, a polyacrylide, a polyvinyl, or a polyorthoester.
In some embodiments, P comprises polyethylene glycol (PEG). In some embodiments, P comprises between about 2 to about 200 repeating units (e.g., about 2 to about 150, about 2 to about 100, about 2 to about 50, about 2 to about 25, about 2 to about 15, about 2 to about 10, or about 2 to about 5 repeating units) of polyethylene glycol (PEG). In some embodiments, P comprises between about 2 to about 10 repeating units of polyethylene glycol (PEG). In some embodiments, the PEG comprises diethylene glycol, triethylene glycol, tetraethylene glycol, or pentaethylene glycol.
In some embodiments, the structure of Formula (III) is a structure of Formula (III-a):

Formula (III-a)
wherein A is C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1; B is
 alkylene, C2- C12 alkenylene, C2-C12 alkynylene, C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1; X is an agent (e.g., an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor); each of L1 and L2 is independently C1-C12 alkylene, C1-C12 heteroalkylene, (C0-C12 alkylene)-arylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), or (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)- arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0- C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene,
alkylene, C2- C12 alkenylene, C2-C12 alkynylene, C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1; X is an agent (e.g., an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor); each of L1 and L2 is independently C1-C12 alkylene, C1-C12 heteroalkylene, (C0-C12 alkylene)-arylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), or (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)- arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0- C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene,
heteroalkylene, or arylene is optionally substituted with 1-12 independently selected
R2; each R1 and R2 is independently alkyl, alkenyl, alkynyl, heteroalkyl (e.g., polyethylene glycol), halo, cyano, oxo,–ORA,–NRBRC,–NRBC(O)RD, -C(O)NRBRC, –C(O)RD,–C(O)OH,–C(O)ORD,–SRE, or–S(O)mRE; each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,–C(O)NRBRC,–C(O)RD,–C(O)OH, or– C(O)ORD; each RB and RC is independently hydrogen or C1-C6 alkyl; each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl; and t is an integer between 1 and 10, inclusive.
In some embodiments, the structure of Formula (III) is a structure of Formula (III-b):

Formula (III-b)
wherein B is C1-C12 alkylene or C1-C12 heteroalkylene, wherein each alkylene or heteroalkylene is optionally substituted with 1-6 R1; X is an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or a bromodomain inhibitor); each of L1 and L2 is independently C1-C12 alkylene, C1-C12 heteroalkylene, (C0-C12 alkylene)-arylene-(C0- C12 alkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), or (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; each R1 and R2 is independently alkyl, heteroalkyl, halo, cyano, oxo,–ORA,–NRBRC,–NRBC(O)RD, -C(O)NRBRC,–C(O)RD,–C(O)OH,– C(O)ORD,–SRE, or–S(O)mRE; each RA is independently hydrogen, C1-C6 alkyl, C1- C6 heteroalkyl,–C(O)NRBRC,–C(O)RD,–C(O)OH, or–C(O)ORD; each RB and RC is
independently hydrogen or C1-C6 alkyl; or each RD is independently C1-C6 alkyl, C1- C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl; t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive.
In some embodiments, the conjugate comprises a structure of Formula (III-c):

Formula (III-c)
wherein X is an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or a bromodomain inhibitor); L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)- arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0- C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene,
heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; each R2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive.
In some embodiments, the structure of Formula (III) is a structure of Formula (III-d-1) or Formula (III-d-2):

Formula (III-d-1)

Formula (III-d-2)
wherein L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12
heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; each R2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive.
In some embodiments, the structure of Formula (III) is a structure of Formula (III-e):

Formula (III-e)
wherein L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), or (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; each R2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive.
In some embodiments, the conjugate or BASP particle comprises a structure according to Formula (III-f):

wherein Ring C’ is a carbocyclyl or heterocyclyl moiety; Ring T is a triazoldiyl moiety (e.g., 1,2,3-triazoldiyl); A is C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1; B is C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1; X is an agent (e.g., an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor); P is alkylene or heteroalkylene (e.g., polyethylene glycol); each of L1 and L2 is independently a bond, C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, C1-C12 heteroalkylene, (C0-C12 alkylene)-arylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), (C0-C12 heteroalkylene)- arylene-(C0-C12 heteroalkylene), (C0-C12 alkylene)-heteroarylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-heteroarylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)- heteroarylene-(C0-C12 heteroalkylene), (C0-C12 alkylene)-heterocyclylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-heterocyclylene-(C0-C12 alkylene), or (C0-C12 heteroalkylene)-heterocyclylene-(C0-C12 heteroalkylene), wherein each alkylene, alkenylene, alkynylene, heteroalkylene, arylene, heteroarylene, or heterocyclylene is optionally substituted with 1-12 independently selected R2; L3 is a tissue
microenvironment cleavable linker; each R1 and R2 is independently alkyl, alkenyl, alkynyl, heteroalkyl (e.g., polyethylene glycol), halo, cyano, oxo,–ORA,–NRBRC,– NRBC(O)RD, -C(O)NRBRC,–C(O)RD,–C(O)OH,–C(O)ORD,–SRE, or–S(O)mRE; each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,–C(O)NRBRC,–
C(O)RD,–C(O)OH, or–C(O)ORD; each RB and RC is independently hydrogen or C1- C6 alkyl; or each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl; R6a is absent or in the first (initiating) macromonomer unit present in the conjugate or BASP particle is selected from hydrogen, phenyl, or C1-C4 alkyl; R6b is absent or in the last
(terminating) macromonomer unit present in the conjugate or BASP particle is selected from hydrogen, phenyl, or C1-C4 alkyl; and q is an integer between 2 and 1,000, inclusive.
In certain embodiments, the conjugate or BASP particle comprises the structure according to Formula (III-a) as a repeating unit or as part of a repeating unit. In certain embodiments, the conjugate or BASP particle comprises about 2 to about 200 repeating units (e.g., about 2 to about 150, about 2 to about 100, about 2 to about 50, about 2 to about 25, about 2 to about 15, about 2 to about 10, about 2 to about 5, about 10 to about 200, about 10 to about 100, about 10 to about 50, about 10 to about 25, about 25 to about 200, about 25 to about 100, about 25 to about 50, about 50 to about 200, about 50 to about 100, or about 100 to about 200, repeating units. In some embodiments, q is an integer between 2 and 500, inclusive (e.g., between 2 and 450, 2 and 400, 2 and 350, 2 and 300, 2 and 250, 2 and 200, 2 and 150, 2 and 100, 2 and 75, and 2 and 50). In some embodiments, q is an integer between 2 and 100, inclusive.
In some embodiments, Ring C’ is a structure of Formula (IV):

wherein D is N or CR4; Z is C(R5)2, O, or S; each R3 is independently C1-C6 alkyl, C1- C6 haloalkyl, oxo, or halo; R4 is hydrogen, C1-C6 alkyl, or halo; each R5 is
independently hydrogen, C1-C6 alkyl, C1-C6 haloalkyl, or halo; n is 0, 1, or 2,“1” represents a portion of Ring C’ bound to B; and each“2” represents a portion of Ring C’ bound to either another macromonomer, an initiating group (R6a), or an end group (R6b), each through a -CH=CH- linker in the conjugate or BASP particle.
In some embodiments, Ring C’ is a structure of Formula (IV-b):

Formula (IV-b)
wherein D is N or CR4; each R3 is independently C1-C6 alkyl, C1-C6 haloalkyl, oxo, or halo; R4 is hydrogen, C1-C6 alkyl, or halo; n is 0, 1, or 2, and“1” represents a portion of Ring C’ bound to B; and each“2” represents a portion of Ring C’ bound to either another macromonomer, an initiating group (R6a), or an end group (R6b), each through a -CH=CH- linker in the conjugate or BASP particle. In some embodiments, the
structure of Formula (IV) is ,

or. In some embodiments, the structure of Formula (IV) is . In
some embodiments, the structure of Formula (I . In some

embodiments, the structure of Formula (IV) is .
In some embodiment, Ring C’ is a structure of Formula (IV-c):

wherein Z is C(R5)2, O, or S; R3’’ is selected from hydrogen, C1-C6 alkyl, -C(O)-C1-C6 alkyl, -C(O)-O-C1-C6 alkyl, -C(O)-NH-C1-C6 alkyl, C1-C6 heteroalkyl, -C(O)-C1-C6 heteroalkyl, -C(O)-O-C1-C6 heteroalkyl, -C(O)-NH-C1-C6 heteroalkyl, and halo, wherein the any alkyl portion of R3’ is optionally substituted with halo; each R5 is independently hydrogen, C1-C6 alkyl, C1-C6 haloalkyl, or halo;“1” represents a portion of Ring C’ bound to B; and each“2” represents a portion of ring C’ bound to either another macromonomer, an initiating group (R6a), or an end group (R6b), each through a -CH=CH- linker in the conjugate or BASP particle.
In some embodiments, the agent contains a functional group (e.g., reactive functional group) for conjugation to the conjugate or BASP particle. In some embodiments, the functional group is chosen from a hydroxyl group, amino group (e.g., a primary or secondary amino group), thiol group, carboxylic acid group, aldehyde group, ketone group, hydrazino group, azido group, vinyl ether group, alkenyl group, isothiocyanate group, or acrylate group. In other embodiments, the agent can be activated for conjugation to a conjugate or BASP particle or another moiety or polymer (e.g., PEG, PLA, PLGA, PDO, cyclodextrin) through the use of an activating agent, e.g., succinic anhydride, thiophosgene, 4-nitrophenyl chloroformate, ethylenediamine, or cis-aconitic anhydride.
In some embodiments, the structure of Formula (III-f) is a structure of Formula (III-g):

Formula (III-g)
wherein A is C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1; B is C1-C12 alkylene, C2- C12 alkenylene, C2-C12 alkynylene, C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1; X is an agent (e.g., an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor); each of L1 and L2 is independently C1-C12 alkylene, C1-C12 heteroalkylene, (C0-C12 alkylene)-arylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), or (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)- arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0- C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene,
heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; each R1 and R2 is independently alkyl, alkenyl, alkynyl, heteroalkyl (e.g., polyethylene glycol), halo, cyano, oxo,–ORA,–NRBRC,–NRBC(O)RD, -C(O)NRBRC, –C(O)RD,–C(O)OH,–C(O)ORD,–SRE, or–S(O)mRE; each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,–C(O)NRBRC,–C(O)RD,–C(O)OH, or– C(O)ORD; each RB and RC is independently hydrogen or C1-C6 alkyl; each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl; t is an integer between 1 and 10, inclusive; R6a is absent or in the first (initiating) macromonomer unit present in the conjugate or BASP particle is selected from hydrogen, phenyl, or C1-C4 alkyl; R6b is absent or in the last (terminating) macromonomer unit present in the conjugate or BASP particle is selected from hydrogen, phenyl, or C1-C4 alkyl; and q is an integer between 2 and 1000, inclusive.
In some embodiments, the structure of Formula (III-f) is a structure of Formula (III-k):
Formula (III-k)
wherein B is C1-C12 alkylene or C1-C12 heteroalkylene, wherein each alkylene or heteroalkylene is optionally substituted with 1-6 R1; X is an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or a bromodomain inhibitor); each of L1 and L2 is independently C1-C12 alkylene, C1-C12 heteroalkylene, (C0-C12 alkylene)-arylene-(C0- C12 alkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), or (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; each R1 and R2 is independently alkyl, heteroalkyl, halo, cyano, oxo,–ORA,–NRBRC,–NRBC(O)RD, -C(O)NRBRC,–C(O)RD,–C(O)OH,– C(O)ORD,–SRE, or–S(O)mRE; each RA is independently hydrogen, C1-C6 alkyl, C1- C6 heteroalkyl,–C(O)NRBRC,–C(O)RD,–C(O)OH, or–C(O)ORD; each RB and RC is independently hydrogen or C1-C6 alkyl; or each RD is independently C1-C6 alkyl, C1- C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl; t is an integer between 1 and 10, inclusive; z is an integer between 1 and 200, inclusive; R6a is absent or in the first (initiating) macromonomer unit present in the conjugate or BASP particle is selected from hydrogen, phenyl, or C1-C4 alkyl; R6b is absent or in the last (terminating) macromonomer unit present in the conjugate or
BASP particle is selected from hydrogen, phenyl, or C1-C4 alkyl; and q is an integer between 2 and 1000, inclusive.
In some embodiments, the conjugate comprises a structure of Formula (III-h):

Formula (III-h)
wherein X is an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or a bromodomain inhibitor); L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)- arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0- C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene,
heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; each R2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; z is an integer between 1 and 200, inclusive; R6a is absent or in the first (initiating) macromonomer unit present in the conjugate or BASP particle is selected from hydrogen, phenyl, or C1-C4 alkyl; R6b is absent or in the last
(terminating) macromonomer unit present in the conjugate or BASP particle is selected from hydrogen, phenyl, or C1-C4 alkyl; and q is an integer between 2 and 1000, inclusive.
In some embodiments, the structure of Formula (III-a) is a structure of Formula (III-i-1) or Formula (III-i-2):


wherein L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12
heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; each R2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; z is an integer between 1 and 200, inclusive; R6a is absent or in the first (initiating) macromonomer unit present in the conjugate or BASP particle is selected from hydrogen, phenyl, or C1-C4 alkyl; R6b is absent or in the last (terminating) macromonomer unit present in the conjugate or BASP particle is selected from hydrogen, phenyl, or C1-C4 alkyl; and q is an integer between 2 and 1000, inclusive.
In some embodiments, the structure of Formula (III) is a structure of Formula (III-j):

heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; each R2 is independently alkyl, heteroalkyl, halo, cyano, oxo,–ORA,–NRBRC,–
NRBC(O)RD, -C(O)NRBRC,–C(O)RD,–C(O)OH,–C(O)ORD,–SRE, or–S(O)mRE; each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,–C(O)NRBRC,– C(O)RD,–C(O)OH, or–C(O)ORD; each RB and RC is independently hydrogen or C1- C6 alkyl; each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl; t is an integer between 1 and 10, inclusive; z is an integer between 1 and 200, inclusive; R6a is absent or in the first (initiating) macromonomer unit present in the conjugate or BASP particle is selected from hydrogen, phenyl, or C1-C4 alkyl; R6b is absent or in the last (terminating) macromonomer unit present in the conjugate or BASP particle is selected from hydrogen, phenyl, or C1-C4 alkyl; and q is an integer between 2 and 1000, inclusive.
In some embodiments of any of Formulas (III), (III-a), (III-b), (III-c), (III-d-1), (II-d-2), (III-e), (III-f), (III-g), (III-k), (III-h), (III-i-1), (III-i-2), or (III-j), q is an integer between 5 and 100, inclusive.
In some embodiments of any of Formulas (III), (III-a), (III-b), (III-c), (III-d-1), (II-d-2), (III-e), (III-f), (III-g), (III-k), (III-h), (III-i-1), (III-i-2), or (III-j), R6a is hydrogen in an initiating macromonomer unit; and R6b is phenyl in a terminating macromomonomer unit present in the conjugate or BASP particle.
In some embodiments of Formulas (III-e) or (III-j), L3 is C1-C12
heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), or (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; each R2 is independently alkyl, heteroalkyl, halo, or oxo; t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive.
In some embodiments of of Formulas (III-e) or (III-j), L3 is C1-C12
heteroalkylene or (C0-C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), or (C0-C12 alkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or aryl is optionally substituted with 1-6 independently selected R2.
In any and all embodiments,“ ” represents a linkage to another portion of the conjugate or particle (e.g., a conjugate or particle described herein) or the terminus
of the conjugate or particle (e.g., a conjugate or particle described herein). In some embodiments, when“ ” represents a terminus of the conjugate or particle, the terminus may be further modified with hydrogen, a protecting group, an agent (e.g., an agent described herein), or other group (e.g., as described herein).
In some embodiments, the structure of Formula (III) or Formula (III-f) is formed from a recursor macromonomer selected from:


and

In some embodiments, the conjugate or BASP particle comprises a single agent (e.g., one or more of the same agent). In some embodiments, the conjugate or BASP particle comprises multiple agents. In some embodiments, the agent(s) are attached to a conjugate or BASP particle through a covalent bond. In some embodiments, the agent(s) are attached to a conjugate or BASP particle through a non-covalent bond or interaction.
In some embodiments, the agents are the same agent. In some embodiments, the agents are different agents. In some embodiments, the conjugate or BASP particle comprises multiple agents (e.g., 2, 3, 4, 5, 6 or more different agents).
In some embodiments, conjugate or BASP particle comprises an agent attached at one end of the conjugate or polymer, e.g., surface exposed end of the conjugate or polymer. In other embodiments, the conjugate or BASP particle comprises an agent in the middle of the conjugate or polymer. In some embodiments, the conjugate or BASP particle comprises an agent attached to at least two conjugates or polymers such that the agent is present between the two conjugates or polymers.
In some embodiments, the conjugate or BASP particle is assembled through the use of a polymerization reaction. In some embodiments, the conjugate or BASP particle is assembled through polymerization of a macromonomer subunit and a cross- linker. In some embodiments, the conjugate or BASP particle is assembled through polymerization of a single type of macromonomer subunit and a cross-linker. In some embodiments, the conjugate or BASP particle is assembled through polymerization of a multiple types of macromonomer subunits and a cross-linker. In some embodiments, the polymerization reaction comprises ring-opening metathesis polymerization (e.g., ROMP). In some embodiments, multiple rounds of polymerization (e.g., ROMP) are carried out to prepare the conjugate and/or particle.
In some embodiments, the particle can be formed from the conjugates described herein, e.g., by precipitation and/or self-assembly. In some embodiments, the conjugate is not precipitated from solution and/or self-assembled.
In some embodiments, the particle comprises conjugates of the same type, e.g., wherein each conjugate comprises the same agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor). In some embodiments, the particle comprises conjugates of different types, e.g., wherein each conjugate comprises a different agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor). In some embodiments, the particle comprises conjugates of different types, e.g., wherein some fraction of the conjugates comprises either an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor and another fraction of the conjugates comprise a second agent. In some embodiments, the second agent is an anti-cancer agent, an anti-fibrotic therapeutic agent, an anti-inflammatory agent, a liver therapeutic agent, or second therapeutic agent. In some embodiments, the second
agent is a targeting agent. In some embodiments, the second agent is a diagnostic agent.
In one embodiment, the particle is not selectively delivered or targeted to a target site, e.g., the particle does not include a targeting moiety (e.g., a cell- or liver- targeting agent as described herein).
In one embodiment, the conjugate or BASP particle, e.g., as described herein has a size to include any of the agents described herein, e.g., the ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor.
In one embodiment, the particle is substantially or completely size-excluded from reaching arteriole smooth muscle, which is protected by non-leaky vessels. In other embodiments, the particle selectively penetrates a leaky vessel, e.g., a leaky vessel of a tumor or liver.
In an embodiment, the agent (e.g., ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) is present in the conjugate or BASP particle without a second agent (e.g., the anti-cancer agent, anti-fibrotic agent, anti-inflammatory agent, or liver therapeutic agent). In an embodiment, the agent (e.g., ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) is present in the conjugate or BASP particle without a targeting agent or diagnostic agent.
In some embodiments, the agent (e.g., ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) is formulated for extended release, e.g., in an extended release formulation for substantially continuous release for hours, days, weeks, months or years, for example, using a cleavable linker (e.g., a tissue
microenvironment cleavable linker) and/or linker of different degradation rates.
The conjugate or BASP particle may comprise a first agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) and, optionally, a second agent (e.g., an anti-cancer agent, anti-inflammatory agent, anti-fibrotic agent, or liver therapeutic), targeting moiety, or diagnostic agent. In some embodiments, the first agent (e.g., the ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) and the second agent (e.g., an anti-cancer agent, anti-inflammatory agent, anti-fibrotic agent, or liver therapeutic), targeting moiety, or diagnostic agent is present in separate entities or in the same entity. For example, if provided as separate entities, the first agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) can be provided as a first conjugate or BASP particle and the second agent (e.g., the anti- cancer agent, anti-inflammatory agent, anti-fibrotic agent, or liver therapeutic),
targeting moiety, or diagnostic agent can be provided as a second conjugate or BASP particle (e.g., where the second conjugate particle has a structural property (e.g., size or composition) or a functional property (e.g., release kinetics or a pharmacodynamic property) that differs from the first particle). Alternatively, the first agent (e.g., the ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) and the second agent (e.g., an anti-cancer agent, anti-inflammatory agent, anti-fibrotic agent, or liver therapeutic), targeting moiety, or diagnostic agent can be provided in the same entity, e.g., in the same conjugate or BASP particle. In certain embodiments, a conjugate of the present disclosure is a compound shown in Table 2. Conjugate and Particle Drug Loading and Dosage
In some embodiments, the percentage of the conjugates (e.g., in a particle) that comprise an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) is between about 1 and about 100% (e.g., about 1%, about 2%, about 3%, about 4%, about 5%, about 10%, about 15%, about 20%, about 30%, about 40%, about 50%, about 60%, about 70%, about 80%, about 90%, or about 100%). In some embodiments, the percentage of the conjugates that comprise an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) is less than about 50%, e.g., less than about 40%, less than about 35%, less than about 30%, less than about 25%, less than about 20%, less than about 15%, or less than about 10%. In some embodiments, the percentage of the conjugates (e.g., in a particle) that comprise an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) is between about 5% and about 50%, about 5% and about 40%, about 5% and about 30%, about 5% and about 25%, or about 5% and about 20%. In some embodiments, the percentage of the conjugates (e.g., in a particle) that comprise an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) is between about 5% and 90%. In some embodiments, the percentage of the conjugates (e.g., in a particle) that comprise an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) is between about 5% and about 75%. In some embodiments, the conjugates (e.g., in a particle) that comprise an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) is between about 5% and about 50%. In some embodiments, the percentage of the conjugates (e.g., in a particle) that comprise an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) is between about 10% and about 25%.
In some embodiments, the total amount of the agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) present in the conjugate or BASP particle is greater than about 5% (e.g., about 6%, about 7%, about 8%, about 9%, about 10%, about 12%, about 15%, about 20%, about 25%, about 30%, or more) of the total size or weight of the conjugate or BASP particle. In some embodiments, the total amount of the agent present in the conjugate or BASP particle is greater than about 10% (e.g., about 12%, about 15%, about 20%, about 25%, about 30%, or more) of the total size or weight of the conjugate or BASP particle. In some embodiments, the total amount of an ARB present in the conjugate or BASP particle is greater than about 5% (e.g., about 6%, about 7%, about 8%, about 9%, about 10%, about 12%, about 15%, about 20%, about 25%, about 30%, or more) of the total size or weight of the conjugate or BASP particle. In some embodiments, the total amount of a vitamin D analog present in the conjugate or BASP particle is greater than about 5% (e.g., about 6%, about 7%, about 8%, about 9%, about 10%, about 12%, about 15%, about 20%, about 25%, about 30%, or more) of the total size or weight of the conjugate or BASP particle. In some embodiments, the total amount of an IDO inhibitor analog present in the conjugate or BASP particle is greater than about 5% (e.g., about 6%, about 7%, about 8%, about 9%, about 10%, about 12%, about 15%, about 20%, about 25%, about 30%, or more) of the total size or weight of the conjugate or BASP particle. In some embodiments, the total amount of a bromodomain inhibitor analog present in the conjugate or BASP particle is greater than about 5% (e.g., about 6%, about 7%, about 8%, about 9%, about 10%, about 12%, about 15%, about 20%, about 25%, about 30%, or more) of the total size or weight of the conjugate or BASP particle.
Without being bound by theory, due to the localized delivery of the BRUSH compositions or BASP compositions described herein (e.g., the agent-containing particles), a lower dose or amount of the agent in the conjugates or BASP particles can be administered (e.g., through local sustained delivery) compared to the agent in free form. In other embodiments, the agent-containing conjugates or BASP particles (e.g., conjugates or BASP particles containing an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) are administered at a dose or amount of the agent that is less than the dose or amount of said agent in free form to have a desired effect (e.g., a desired therapeutic effect).
In some embodiments, the agent (e.g., an ARB, vitamin D analog, IDO
inhibitor, or bromodomain inhibitor) is incorporated into a conjugate or a BASP particle at a dose that is less than the dose or amount of said agent in free form to have a desired effect (e.g., a desired therapeutic effect), e.g., the standard of care dose for the intended use of the free agent. In one embodiment, the agent are incorporated into the particles at a dose or amount of the agent that is less than the standard of care dose of the agent for a desired therapy (e.g., a dose that is less than about 0.01, about 0.02, about 0.03, about 0.04, about 0.05, about 0.06, about 0.07, about 0.08, about 0.09, about 0.1, about 0.2, about 0.3, about 0.4, about 0.5, about 0.6, about 0.7, about 0.8, about 0.9, or about 0.95 that of the standard of care dose of the agent). In one embodiment, where the agent is an ARB, the dose is less than the anti-hypertensive or anti-heart failure dose for ARBs such as losartan, candesartan, eprosartan, irbesartan, olmesartan, telmisartan, and valsartan.
In some embodiments, the agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) is incorporated into a conjugate or a BASP particle at a dose equivalent to the dose or amount of said agent in free form to have a desired effect (e.g., a desired therapeutic effect), e.g., the standard of care dose for the intended use of the free agent. In these embodiments, the conjugate or BASP particle produces a greater therapeutic effect and/or a less adverse effect than the free agent. In certain embodiments, the particle increases the amount of the agent delivered to a tissue or cell in need thereof and reduces the amount of the agent exposed to a non- target tissue or cell, as compared to the free agent.
In some embodiments, the agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor) is incorporated into a conjugate or a BASP particle at a dose higher than the dose or amount of said agent in free form to have a desired effect (e.g., a desired therapeutic effect), e.g., the standard of care dose for the intended use of the free agent. In some embodiments, the agent is incorporated into a conjugate or a BASP particle at a dose higher than the dose or amount of said agent in free form that would produce an adverse effect by systemic administration (e.g., a reduction in blood pressure). In some embodiments, since the conjugate or BASP particle described herein releases the agent at a target site based on pH
microenvironment, other non-target sites (e.g., blood vessels) with different pH would be less likely to be exposed to the agent.
Exemplary Conjugates and Particles
In one embodiment, the conjugate or BASP particle comprises two or more macromonomers (e.g., as described herein) each bound to an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor).
In some embodiments, the conjugate or BASP particle comprises telmisartan- linked macromonomers. In some embodiments, the telmisartan-linked described by Formulas (III-e) or (III-j). In some embodiments, the telmisartan-linked
macromonomers are those of Formulas (III-e) or (III-j), wherein z is an integer between 25 and 85, inclusive; and L3 is–CH2CH2O–. In some embodiments, the telmisartan-linked macromonomers are those of Formula (III-e), wherein z is 68 and L3 is–CH2CH2O–. In some embodiments, the telmisartan-linked macromonomers are those of Formula (III-e), wherein z is 45 and L3 is–CH2CH2O–. In one embodiment, the telmisartan-linked macromonomers are those of Formulas (III-e) or (III-j),
wherein z is 68 and L3 is
 . In one embodiment, the telmisartan- l s are those of Formulas (III-e) or (III-j), wherein z is 68 and L3
. In one embodiment, the telmisartan- l s are those of Formulas (III-e) or (III-j), wherein z is 68 and L3
i
 In one embodiment, the telmisartan-linked conjugates or BASP particles have a drug-loading level between about 5% and about 25%.
In one embodiment, the telmisartan-linked conjugates or BASP particles have a drug-loading level between about 5% and about 25%.
In some embodiments, the conjugate or BASP particle comprises paricalcitol- linked macromonomers, e.g., as described in Formulas (III-d-1), (III-d-2), (III-i-1) or (III-i-2). In some embodiments, the paricalcitol-linked macromonomers comprises a mixture of regioisomers. In some embodiments, the paricalcitol-linked
macromonomers are those of Formulas (III-d-1), (III-d-2), (III-i-1) or (III-i-2), wherein z is an integer between 25 and 85, inclusive; and L3 is–C(O)CH2CH2O–. In some embodiments, the paricalcitol-linked macromonomers are those of Formulas (III-d-1), (III-d-2), (III-i-1) or (III-i-2), wherein z is 68 and L3 is–C(O)CH2CH2O–. In some embodiments, the paricalcitol-linked macromonomers are those of Formulas (III-d-1), (III-d-2), (III-i-1) or (III-i-2), wherein z is 68 and L3 is–C(O)CH2CH2O–, and is a mixture of regioisomers. In one embodiment, paricalcitol-linked conjugate or BASP particle has a drug-loading level between about 5% and about 25%.
In some embodiments, the conjugate or BASP particle comprises an agent
(e.g., an agent as described herein, e.g., an agent shown in FIGS.2, 3, 4A to 4B). In some embodiments, the conjugate or BASP particle additionally comprises one or more of a second agent, a targeting moiety, or a diagnostic moiety. In some embodiments, the conjugate comprises telmisartan (e.g., as described in Formulas (III-e) or (III-j)) and a second agent (e.g., an anti-cancer agent, anti-inflammatory agent, anti-fibrotic agent, or liver therapeutic). In one embodiments, the conjugate or BASP particle comprises paricalcitol (e.g., as described in Formulas (III-d-1), (III-d- 2), (III-i-1) or (III-i-2)) and a second agent (e.g., an anti-cancer agent, anti- inflammatory agent, anti-fibrotic agent, or liver therapeutic).
In one embodiment, a conjugate described herein (which is capable of forming a particle) or the BASP particle is a nanoparticle, from about 5 nm to about 100 nm, about 5 nm to about 75 nm, about 5 to about 50 nm, or about 5 to about 30 nm in size. In one embodiment, the conjugate or BASP particle comprises telmisartan-linked macromonomers, e.g., as described in Formula (III-e) or (III-j), and is present in a particle from about 5 to about 30 nm in size. In one embodiment, the conjugate or BASP particle comprises paricalcitol-linked monomers, e.g., as described in Formulas (III-d-1), (III-d-2), (III-i-1) or (III-i-2), and is present in a particle from about 5 to about 30 nm in size.
In another embodiment, the conjugate or BASP particle is homogenous and comprises macromonomers of the same type. In another embodiment, the conjugate or BASP particle is heterogeneous and comprises macromonomers of different types. In some embodiments, the conjugate or BASP particle comprises telmisartan-linked macromonomers, e.g., as described in Formula (III-e) or (III-j), and macromonomers linked to a diagnostic moiety (e.g., a Cy3, Cy5, or Cy7.5 dye). In some embodiments, the conjugate or BASP particle comprises paricalcitol-linked macromonomers, e.g., as described in Formulas (III-d-1), (III-d-2), (III-i-1) or (III-i-2), and macromonomers linked to a diagnostic moiety (e.g., a Cy3, Cy5, or Cy7.5 dye). Diagnostic Agents
In some embodiments, the conjugate or BASP particle is used for a diagnostic purpose, e.g., to identify the location of a particular structure (e.g., a tumor or cancer cell) within a subject. In some embodiments, the agent can be a diagnostic agent. For example, the diagnostic agent may comprise a fluorescent molecule, a metal chelate, a contrast agent, a radionuclide, or a positron emission tomography (PET) imaging
agent, an infrared imaging agent, a near-IR imaging agent, a computer assisted tomography (CAT) imaging agent, a photon emission computerized tomography imaging agent, an X-ray imaging agent, or a magnetic resonance imaging (MRI) agent.
In some embodiments, the diagnostic agent is a fluorescent molecule. In some embodiments, the fluorescent molecule comprises an acridine dye, a cyanine dye, a rhodamine dye, a BODIPY dye, a fluorescein dye, a dansyl dye, an Alexa dye, an atto dye, a quantum dot, or a fluorescent protein. In some embodiments, the fluorescent molecule is a cyanine dye (e.g., Cy3, Cy 3.5, Cy5, Cy5.5, Cy7, or Cy7.5).
In some embodiments, the diagnostic agent is an MRI agent (e.g., a contrast agent). Examples of suitable materials for use as MRI agents (e.g., contrast agents) include gadolinium chelates, as well as iron, magnesium, manganese, copper, and chromium.
In some embodiments, the diagnostic agent is a CAT imaging agent or an X- ray imaging agent. Examples of materials useful for CAT and X-ray imaging include iodine-based materials.
In some embodiments, the diagnostic agent is a PET imaging agent. Examples of suitable PET imaging agents include compounds and compositions comprising the positron emitting radioisotopes 18F, 15O, 13N, 11C, 82Rb, 64Cu, and 68Ga, e.g., fludeoxyglucose (18F-FDG), 68Ga-DOTA-psuedopeptides (e.g., 68Ga-DOTA-TOC), 11C-metomidate, 11C-acetate, 11C-methionine, 11C-choline, 18F-fluciclovine, 18F- fluorocholine, 18F-fluorodeoxysorbitol, 18F-3’-fluoro-3’-deoxythymidine, 11C- raclopride, and 18F-desmethoxyfallypride.
In some embodiments, the diagnostic agent is a near-IR imaging agent.
Examples of near-IR imaging agents include Pz 247, DyLight 750, DyLight 800, cyanine dyes (e.g., Cy5, Cy5.5, Cy7), AlexaFluor 680, AlexaFluor 750, IRDye 680, IRDye 800CW, and Kodak X-SIGHT dyes.
In some embodiments, the agent can be a radionuclide, e.g., for use as a therapeutic, diagnostic, or prognostic agents. Among the radionuclides used, gamma- emitters, positron-emitters, and X-ray emitters are suitable for diagnostic and/or therapy, while beta emitters and alpha-emitters may also be used for therapy. Suitable radionuclides for forming use with various embodiments of the present invention include, but are not limited to, 123I, 125I, 130I, 131I, 133I, 135I, 47Sc, 72As, 72Sc, 90Y, 88Y,
97Ru, 100Pd, 101mRh, 119Sb, 128Ba, 197Hg, 211At, 212Bi, 212Pb, 109Pd, 111In, 67Ga, 68Ga, 67Cu, 75Br, 77Br, 99mTc, 14C, 13N, 15O, 32P, 33P, or 18F.
In some embodiments, the conjugate or BASP particle comprises: i) a diagnostic agent; (ii) a carbocyclyl or heterocyclyl moiety; (iii) a triazole moiety; (iv) a heteroalkyl moiety; and (v) a cleavable linker (e.g., a tissue microenvironment cleavable linker).
In some embodiments, the diagnostic agent is a diagnostic agent described herein (e.g., a fluorescent molecule, a metal chelate, a contrast agent, a radionuclide, or a positron emission tomography (PET) imaging agent, an infrared imaging agent, a near-IR imaging agent, a computer assisted tomography (CAT) imaging agent, a photon emission computerized tomography imaging agent, an X-ray imaging agent, or a magnetic resonance imaging (MRI) agent). In some embodiments, the diagnostic agent is a fluorescent molecule. In some embodiments, the diagnostic agent is a cyanine dye, e.g., Cy3, Cy 3.5, Cy5, Cy5.5, Cy7, or Cy7.5, e.g., as described herein.
In some embodiments, the diagnostic agent is non-covalently bound to the conjugate or BASP particle (e.g., the diagnostic agent is associated with the conjugate or BASP particle through ionic bonds or hydrophobic interactions). In some embodiments, the diagnostic agent is covalently bound to the conjugate or BASP particle through a cleavable linker (e.g., a cleavable linker described herein). In some embodiments, the diagnostic agent is covalently bound to the conjugate or BASP particle through a non-cleavable linker.
In some embodiments, exemplary diagnostic agents in the BASP- compositions include, but are not limited to, one or more of the diagnostic agents listed in, in Key, J. and Leary, J. Int J Nanomedicine (2014) 9:711-726; Rizzo, L et al Curr Opin Biotechnol (2013) 24:1159-1166; Andresen, T.L.“Liposomes for positron emission tomography (PET) imaging, radiotherapy, and theranostics”
(www.nanomedicine.dtu.dk/Research/Liposomes); and Paragraph 0149 of U.S. Patent No.9,381,253, each of which is incorporated by reference herein. Targeting Moieties
The BRUSH-composition or BASP-composition can also include a targeting moiety, e.g., a targeting moiety that is specific to a cell type or tissue. Targeting of particles with a surface coating of hydrophilic polymer chains, such as polyethylene glycol (PEG) chains, for targeting has been proposed (Allen, et al., Biochimica et
Biophysica Acta 1237: 99-108 (1995); DeFrees, et al., Journal of the American Chemistry Society 118: 6101-6104 (1996); Blume, et al., Biochimica et Biophysica Acta 1149: 180-184 (1993); Klibanov, et al., Journal of Liposome Research 2: 321- 334 (1992); U.S. Patent No.5,013556; Zalipsky, Bioconjugate Chemistry 4: 296-299 (1993); Zalipsky, FEBS Letters 353: 71-74 (1994); Zalipsky, in Stealth Liposomes Chapter 9 (Lasic and Martin, Eds) CRC Press, Boca Raton Fl (1995). Other targeting moieties, such as ligands, cell surface receptors, glycoproteins, vitamins (e.g., riboflavin), aptamers and monoclonal antibodies, can also be used. The targeting moieties can include the entire protein or fragments thereof. Targeting mechanisms generally require that the targeting agents be positioned on the surface of the conjugate or BASP particle in such a manner that the targeting moiety is available for interaction with the target, for example, a cell surface receptor.
A variety of different targeting ligands and methods are known and available in the art, including those described, e.g., in Sapra, P. and Allen, TM, Prog. Lipid Res. 42(5):439-62 (2003); and Abra, RM et al., J. Liposome Res.12:1-3, (2002). Without limitation, a targeting ligand can be selected from the group consisting of peptides, polypeptides, proteins, enzymes, peptidomimetics, glycoproteins, antibodies
(monoclonal or polyclonal) and portions and fragments thereof (e.g., antigen binding fragments), lectins, nucleosides, nucleotides, nucleoside and nucleotide analogues, nucleic acids, monosaccharides, disaccharides, trisaccharides, oligosaccharides, polysaccharides, lipopolysaccharides, vitamins, steroids, hormones, cofactors, receptors, receptor ligands, and analogs and derivatives thereof.
Non-limiting examples of antibodies and other suitable targeting moieties include those that target tumor/cancer-associated antigens, e.g., tumor targeting ligands or antibodies against tumor antigens; antigens that are differentially expressed on inflamed tissue (e.g., EGFR, ICAM-1 VCAM-1); antigens that are differentially expressed during cell maturation or antigens that are expressed on diseased tissues, pathogens or bacteria (e.g., sugar moieties).
Tumor-antigens include Melan-A/MART-1, Dipeptidyl peptidase IV (DPPIV), adenosine deaminase-binding protein (ADAbp), cyclophilin b, Colorectal associated antigen (CRC)—C017-1A/GA733, Carcinoembryonic Antigen (CEA) and its immunogenic epitopes CAP-1 and CAP-2, etv6, aml1, Prostate Specific Antigen (PSA) and its immunogenic epitopes PSA-1, PSA-2, and PSA-3, prostate-specific membrane antigen (PSMA), T-cell receptor/CD3-zeta chain, MAGE-family of tumor
antigens (e.g., MAGE-A1, MAGE-A2, MAGE-A3, MAGE-A4, MAGE-A5, MAGE- A6, MAGE-A7, MAGE-A8, MAGE-A9, MAGE-A10, MAGE-A11, MAGE-A12, MAGE-Xp2 (MAGE-B2), MAGE-Xp3 (MAGE-B3), MAGE-Xp4 (MAGE-B4), MAGE-C1, MAGE-C2, MAGE-C3, MAGE-C4, MAGE-C5), GAGE-family of tumor antigens (e.g., GAGE-1, GAGE-2, GAGE-3, GAGE-4, GAGE-5, GAGE-6, GAGE-7, GAGE-8, GAGE-9), BAGE, RAGE, LAGE-1, NAG, GnT-V, MUM-1, CDK4, tyrosinase, p53, MUC family, HER2/neu, p21ras, RCAS1, α-fetoprotein, E-cadherin, α-catenin, β-catenin and γ-catenin, p120ctn, gp100Pmel117, PRAME, NY-ESO-1, brain glycogen phosphorylase, SSX-1, SSX-2 (HOM-MEL-40), SSX-1, SSX-4, SSX-5, SCP-1, CT-7, cdc27, adenomatous polyposis coli protein (APC), fodrin, P1A, Connexin 37, Ig-idiotype, p15, gp75, GM2 and GD2 gangliosides, viral products such as human papilloma virus proteins, Smad family of tumor antigens, lmp-1, EBV- encoded nuclear antigen (EBNA)-1, and c-erbB-2.
As other examples, the targeting moieties include peptides that comprise Arg- Gly-Asp motifs (or RGD peptides) that target integrin present on angiogenic tumor vasculature.
In some embodiments, the targeting moiety includes a nucleic acid.
In some embodiments, the targeting moiety can include an aptamer, i.e., a nucleic acid able to specifically bind a specific target molecule, such as a biological moiety. Non-limiting examples of aptamers include RNA aptamers and DNA aptamers. For example, the size of the aptamer may be at least about 5 kDa, at least about 10 kDa, at least about 15 kDa, or at least about 20 kDa.
In some embodiments, the targeting ligand can be selected from the group consisting of polylysine (PLL), poly Laspartic acid, poly L-glutamic acid, styrene- maleic acid anhydride copolymer, poly(L-lactide-co-glycolide) copolymer, divinyl ether-maleic anhydride copolymer, N-(2-hydroxypropyl)methacrylamide copolymer (HMPA), polyethylene glycol (PEG), polyvinyl alcohol (PVA), polyurethane, poly(2- ethylacryllic acid), N-isopropylacrylamide polymers, polyphosphazine,
polyethylenimine, spermine, spermidine, polyamine, pseudopeptide-polyamine, peptidomimetic polyamine, dendrimer polyamine, arginine, amidine, protamine, thyrotropin, melanotropin, lectin, surfactant protein A, mucin, transferrin,
bisphosphonate, polyglutamate, polyaspartate, an aptamer, asialofetuin, hyaluronan, procollagen, insulin, transferrin, albumin, acridines, cross- psoralen, mitomycin C, TPPC4, texaphyrin, Sapphyrin-, polycyclic aromatic hydrocarbons (e.g., phenazine,
dihydrophenazine), bile acids, cholesterol, cholic acid, adamantane acetic acid, 1- pyrene butyric acid, dihydrotestosterone, 1,3-Bis-O(hexadecyl)glycerol,
geranyloxyhexyl group, hexadecylglycerol, borneol, menthol, 1,3-propanediol, heptadecyl group, palmitic acid, myristic acid, O3-(oleoyl)lithocholic acid, O3- (oleoyl)cholenic acid, dimethoxytrityl, or phenoxazine), RGD peptide, radiolabeled markers, haptens, naproxen, aspirin, dinitrophenyl, HRP, AP, lectins, vitamin A, vitamin E, vitamin K, vitamin B, folic acid, B12, riboflavin, biotin, pyridoxal, taxon, vincristine, vinblastine, cytochalasin, nocodazole, jasplakinolide, latrunculin A, phalloidin, swinholide A, indanocine, myoseverin, tumor necrosis factor alpha (TNF alpha), interleukin-1 beta, gamma interferon, GalNAc, galactose, mannose, mannose- 6-phospahte, clusters of sugars such as GalNAc cluster, mannose cluster, galactose cluster, an aptamer, integrin receptor ligands, chemokine receptor ligands, serotonin receptor ligands, PSMA, endothelin, GCPII, somatostatin, cellular adhesion molecules (CAMS), and any combinations thereof.
In one embodiment, the targeting ligand is mannose-6-phosphate. Liver-Targeting Moieties
In some embodiments, any conjugate or BASP particle described herein can be targeted to the liver, e.g., targeted to a liver cell. Targeting to the liver or to a specific population of liver cells have the following advantages: reduction of off- target, and potentially toxic, side effects, increase amount of drug delivery to the desired site of action or to the desired cells, increase the specificity of the interaction between the drug delivery system and target cells or tissues, and increase the overall efficacy of the drug. To target a specific liver cell, a targeting moiety or ligand can be coupled, e.g., covalently or non-covalently, to a component of a BASP particle or a conjugate, e.g., a BASP particle or conjugate as described herein. The targeting moiety or ligand specifically can bind to a receptor or surface molecule at the surface membrane of the targeted liver cell, and thus deliver the BASP particle or conjugate to the targeted liver cell.
Examples of liver cells that can be targeted include hepatocytes, Kupffer cells, endothelial cells, hepatic stellate cells, bile duct epithelial cells, or hepatocellular carcinoma cells, or any combination thereof. To target a specific liver cell, a targeting moiety or ligand is present, e.g., covalently or non-covalently attached, to a
component of the nanoparticle. The targeting moiety or ligand specifically binds to a receptor or surface molecule at the surface membrane of the targeted liver cell.
The following substances can be used as targeting moieties: antibodies or antigen-binding fragments thereof, lectins, proteins, lipoproteins, hormones, charged molecules, mono-, olio-, and polysaccharides, and low molecular weight ligands such as sugars, folic acids, and peptides.
For targeting hepatocytes, the targeting moiety can specifically bind or interact with one or more of the following: asialoglycoprotein receptor (ASGP-R), high density lipoprotein receptor (HDL-R), low density lipoprotein receptor (LDL-R), immunoglobulin A receptor (IgA-R), scavenger receptor (class BI), transferrin receptor, bile acid receptor, insulin receptor, glycyrrhizin receptor (GL receptor), and glycyrrhetinic acid receptor (GA receptor). Examples of targeting moieties for hepatocytes include ligands containing galactose, N-acetylgalactosamine,
galactosamine, lactoferrin, lactobionic acid (LA), asialofetuin ligand (AF), soybean- derived SG ligand (e.g., sterylglucoside), glycyrrhizin (GL), glycyrrhetinic acid (GA), or derivatives thereof.
For targeting Kupffer cells, the targeting moiety can specifically bind or interact with one or more of the following: mannose/N-acetylglucosamine receptor, galactose particle receptor, galactose specific receptor, Fc receptor immune complexes and opsonized material, scavenger receptors (Class AI, BI, BII, MARCO, CD36, and macrosialin), low density lipoprotein receptor matrix compounds
(fibronectin), complement receptor (C3b and C1q), LPS receptor α2, and
macroglobulin receptor. Examples of targeting moieties for Kupffer cells include D- mannose, cetylmannoside, dexamethasone coupled to mannosylated albumin, and charged molecules with a net negative charge, e.g., albumin with modified lysines such that albumin has a net negative charge, or derivatives thereof.
For targeting endothelial cells, e.g., sinusoidal endothelial cells, the targeting moiety can specifically bind or interact with one or more of the following:
mannose/N-acetyl glucosamine receptor, scavenger receptor (Class A1 and A11), Fc Receptor immune complexes, and matrix compounds (e.g., hyaluronan, fibronectin, denatured collagen, PIIINP).
For targeting hepatic stellate cells (HSCs), the targeting moiety can
specifically bind or interact with one or more of the following: mannose-6-phosphate
receptor, insulin growth factor II receptor (IGFII R), α2 macroglobulin receptor, ferritin receptor, uroplasminogen receptor, RBP receptor, and matrix compounds (e.g., integrin, collagen type VI, fibronectin, CD44). Examples of targeting moieties for HSCs include mannose-6-phosphate (M6P), and cyclic peptide moieties that serve as binding domains of cytokines and growth factors that bind to HSCs, or derivatives thereof.
For targeting bile duct epithelial cells, the targeting moiety can specifically bind or interact with secretin receptor.
For targeting hepatocellular carcinoma cells, e.g., liver cancer cells, the targeting moiety can specifically bind or interact with asialoglycoprotein receptor (ASGP-R). Examples of targeting moieties for HCC cells include lactosaminated ligands, galactosamine, galactosylated ligands, e.g., chitosan, N-lactosyl- dioleoylphosphatidylethanolamine (Lac-DOPE), or lactobionic acid, or derivatives thereof.
In some embodiments, the BASP particles and/or conjugates target a hepatocyte. The liver targeting moiety can be chosen from an agent that specifically binds to, or interacts with, one or more of the following: asialoglycoprotein receptor (ASGP-R), high density lipoprotein receptor (HDL-R), low density lipoprotein receptor (LDL-R), immunoglobulin A receptor (IgA-R), scavenger receptor (class BI), transferrin receptor, bile acid receptor, insulin receptor, glycyrrhizin receptor (GL receptor), and glycyrrhetinic acid receptor (GA receptor). Examples of liver targeting moieties for hepatocytes include, but are not limited to, ligands containing galactose, N-acetylgalactosamine, galactosamine, lactoferrin, lactobionic acid (LA), asialofetuin ligand (AF), soybean-derived SG ligand (e.g., sterylglucoside), glycyrrhizin (GL), glycyrrhetinic acid (GA), or derivatives thereof. Patient Selection
Also provided herein are methods for identifying or selecting a subject that is in need of improved delivery and/or efficacy of a therapy (e.g., a cancer therapy, or an anti-fibrotic or anti-inflammatory therapy). In embodiments, the methods described herein can be used to identify or select a subject that would respond to treatment with a BRUSH-composition or BASP-composition, alone or in combination with other therapies described herein, e.g., such that the delivery of an additional therapy, e.g., a
cancer or an anti-fibrotic therapy, is improved. Said methods are described in, e.g., on pages 111-113 of WO 2016/140714, incorporated herein by reference.
In some embodiments, the method includes identifying the subject as having a desmoplastic disorder (e.g., a cancer or a fibrotic or inflammatory disorder). Methods for identifying the subject as having a desmoplastic disorder, e.g., a cancer or a fibrotic or inflammatory disorder, are known in the art. Such methods include detection of desmoplasia, e.g., fibrosis, such as an increase in the level or production of extracellular matrix components, e.g., collagen, or hyaluronic acid; increased angiotensin II (AngII) type-1 receptor (AT1) signaling; and/or increased expression, production, and/or secretion of pro-inflammatory cytokines, e.g., interleukin-1b (IL- 1b).
In one embodiment, the subject is, or is identified as being, overweight or obese, and has a fibrotic or desmoplastic tumor, e.g., a tumor having one or more of: limited tumor perfusion, compressed blood vessels, high interstitial fluid pressure (IFPs), increased hypoxia, or fibrotic tumor interstitium. In certain embodiments, the subject is overweight or obese, and has a tumor having (e.g., elevated levels of) extracellular matrix components, such as fibers (e.g., collagen, procollagen), fibroblasts (e.g., elevated levels of cancer associated fibroblasts (CAFs) or increased activity of CAFs) and/or polysaccharides (e.g., glycosaminoglycans such as hyaluronan or hyaluronic acid).
The method further includes evaluating, e.g., acquiring a value for, a weight/metabolic-related parameter, for the subject. Examples of a weight/metabolic- related parameter includes body mass index (BMI).
In one embodiment, the subject is, or is identified as being, overweight or obese. Assessment of overweight and obesity can be determined by the classification of body mass index (BMI) as defined by“Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults” from the National Institutes of Health. Body mass index is obtained by dividing a subject’s weight, e.g., in kilograms (kg) by the square of the subject’s height, e.g., in meters (m). Subjects with a BMI 18.5 to 24.9 are typically classified as normal weight, while subjects with a BMI 25.0 to 29.9 are classified as overweight. Subjects with a BMI 30.0 or greater are classified as obese, and can be subdivided into three classes: Class I (BMI = 30.0 to 34.9; Class II (BMI = 35.0 to 39.9); and Class III (BMI is greater or equal to 40).
In one embodiment, the subject is overweight, e.g., the subject has a BMI of greater than or equal to 25.0 but less than or equal to 29.9.
In another embodiment, the subject is, or is identified as being, obese, e.g., the subject has a BMI of greater than or equal to 30, e.g., greater than 30, greater than 35, greater than 40, greater than 45, or greater than 50.
In an embodiment, the subject is, or is identified as being, overweight or obese, and has a fibrotic or a hyperproliferative cancerous condition described herein. In an embodiment, the subject is, or is identified as being, overweight or obese and has a fibrotic disorder described herein. In an embodiment, the subject is, or is identified as being, overweight or obese and has an inflammatory disorder described herein. Methods of Making Macromonomers, Conjugates, and Particles
The macromonomers, conjugates and BASP particles described herein may be synthesized through the use of specialized reagents and other starting materials as described below. Crosslinkers
In some embodiments, the BASP particles and conjugates are prepared through the linkage of discrete macromonomer subunits with a multifunctional crosslinker to generate a nanostructure (BASP particle or BRUSH conjugate). The macromonomer subunit and crosslinkers may be joined in a number of ways, such as through graft-through ring-opening metathesis polymerization (ROMP).
In some embodiments, the crosslinker is labile. In some embodiments, the crosslinker is labile at physiological conditions (e.g., biodegradable, e.g., at physiological pH).
In some embodiments, the crosslinker comprises a bis-norbornene derivative. In some embodiments, the crosslinker comprises two norbornene derivatives, separated by an alkylene, heteroalkylene, arylene, or heteroarylene moiety. In some embodiments, the crosslinker comprises a disulfide bond. In some embodiments, the crosslinker comprises an anhydride moiety or ester moiety.
In some embodiments, the crosslinker is a compound of Formula (VI):

Formula (VI)
wherein each of Z1 and Z2 is independently C1-C6 alkylene, C2-C6 alkenylene, C2-C6 alkynylene, C1-C6 heteroalkylene, (C0-C6 alkylene)-aryl-(C0-C6 alkylene), (C0-C6 heteroalkylene)-aryl-(C0-C6 alkylene), (C0-C6 alkylene)-aryl-(C0-C6 heteroalkylene), wherein each alkylene, alkenylene, alkynylene, heteroalkylene, and aryl is optionally substituted with 1-6 R31; each of W1 and W2 is independently C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, or C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, and heteroalkylene is optionally substituted with 1-6 R32; G is –O–,–S–, or–S-S–; each R31 and R32 is independently alkyl, heteroalkyl, halo, cyano, oxo,–ORA,–NRBRC,–NRBC(O)RD, -C(O)NRBRC,–C(O)RD, or–C(O)OH; each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,–C(O)NRBRC,–C(O)RD,– C(O)OH, or–C(O)ORD; each RB and RC is independently hydrogen or C1-C6 alkyl; and each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl.
In some embodiments, each of Z1 and Z2 is independently C1-C6 alkylene or (C0-C6 alkylene)-aryl-(C0-C6 alkylene) (e.g., -CH2-, -CH2CH2CH2-, or phenyl).
In some embodiments, each of W1 and W2 is independently
 alkylene or C1-C12 heteroalkylene, wherein each alkylene and heteroalkylene is optionally substituted with 1-6 R32. In some embodiments, R32 is oxo. In some embodiments, each of W1 and W2 is independently -CH2-, -OC(O)CH2CH2-, -CH2OC(O)CH2CH2-, or–C(O)-.
alkylene or C1-C12 heteroalkylene, wherein each alkylene and heteroalkylene is optionally substituted with 1-6 R32. In some embodiments, R32 is oxo. In some embodiments, each of W1 and W2 is independently -CH2-, -OC(O)CH2CH2-, -CH2OC(O)CH2CH2-, or–C(O)-.
In some embodiments, G is–O– or–S-S–.
In some embodiments the com ound is a com ound of Formula (VII-a):

Formula (VII-a)
wherein each of Z1 and Z2 is independently CH2-, -CH2CH2CH2-, or phenyl; each of W1 and W2 is independently -CH2-, -OC(O)CH2CH2-, -CH2OC(O)CH2CH2-, or– C(O)-; and G is–O– or–S-S–.
In some embodiments the com ound is selected from:

In some embodiments the com ound is selected from:


Exemplary crosslinkers may be cleavable or susceptible to degradation under certain parameters. For example, in some embodiments, the crosslinker may be acid labile or sensitive to a reducing agent. In some embodiments, the crosslinker may be substantially insoluble in water.
In some embodiments, the length or size of the crosslinker (e.g., the length or size of Z1, W1, G, W2, or Z2) is selected to optimize the efficiency of conjugate and/or particle efficiency. The length or size of the crosslinker may be empirically chosen depending on the identity of the macromonomer or other brush arm component in order to yield high polymerization efficiency. Methods of Making Macromonomers
A macromonomer may be assembled in any number of ways. In some embodiments, the macromonomer is prepared through a process involving at least two steps. In a first step, an agent (e.g., an ARB, vitamin D analog, IDO inhibitor, or bromodomain inhibitor), a targeting moiety, or a diagnostic agent is linked to a PEG- azide or a PEG alkyne. In a second step, the agent-linked PEG azide or agent-linked PEG-alkyne is coupled to a norbornene-containing compound, e.g., through copper(I)-mediated azide-alkyne cycloaddition.
An agent, a targeting moiety, or a diagnostic agent may be linked to a PEG azide or a PEG-alkyne through a reactive group, e.g., a free hydroxyl group or a free carboxylic acid, on the PEG-azide or PEG-alkyne. To facilitate this linkage, it may be desirable that the agent, targeting moiety, or diagnostic agent have at least one carboxylic acid and/or hydroxyl group. By way of example only, in one embodiment, the ARB telmisartan with a carboxylic acid group or the vitamin D analog paricalcitol with a hydroxyl group can be conjugated to a PEG-azide or PEG-alkyne via ester bond formation. Exemplary coupling agents used for conjugation include EDC, DIC, DCC, HOAt, HOBt, and PyBOP. In some embodiments, the conjugation reaction
further comprises a base (e.g., TEA, pyridine). In some embodiments, the conjugation reaction further comprises a catalyst (e.g., DMAP). In some embodiments, the conjugation reaction is carried out in a solvent (e.g., dichloromethane, acetonitrile, tetrahydrofuran, dimethylformamide, or dimethyl sulfoxide). In some embodiments, the derivatized PEG-alkyne or derivatized PEG-azide (i.e., derivatized with an agent, targeting moiety, or diagnostic agent) is purified by extraction, precipitation, trituration, chromatography (e.g., silica gel chromatography or HPLC), or a combination thereof.
After preparation of a derivatized PEG-alkyne or derivatized PEG-azide, macromonomer synthesis may be completed through a second step involving conjugation to a norbornene-containing compound. One method for achieving said conjugation may entail copper(I)-mediated azide-alkyne cycloaddition, in which an azide is coupled with an alkyne in the presence of a copper catalyst. In some embodiments, the copper catalyst is copper(I) acetate. In some embodiments, the cycloaddition reaction is carried out in a solvent (e.g., dichloromethane). In some embodiments, the macromonomer is purified by precipitation, trituration,
chromatography (e.g., silica gel chromatography or HPLC), or a combination thereof. In some embodiments, the macromonomer comprises a mixture of regioisomers. In some embodiments, the macromonomer is comprised of a single regioisomer.
Purified macromonomers may be characterized by a variety of methods including NMR, mass spectrometry, and chromatography (e.g., gel-permeation chromatography). Methods of Making Conjugates and BASP Particles
Conjugates are distinguished from BASP particles in several respects, such as size and hydrodynamic diameter. In some embodiments, the conjugate refers to the product of a first round of polymerization and has an average molecular weight of about 10 kDa to about 100 kDa and an average hydrodynamic diameter of about 1 nm to about 10 nm. In some embodiments, the BASP particle refers to the product of a second or subsequent round of polymerization and has an average molecular weight of about 100 kDa to about 1,000 kDa and an average hydrodynamic diameter of about 10 nm to about 100 nm.
Conjugates and BASP particles described herein may be prepared in any number of methods, including via polymerization reactions. In some embodiments,
the polymerization is ring opening metathesis polymerization (ROMP). ROMP is an olefin metathesis chain-growth polymerization reaction that is driven by the relief of ring strain in cyclic olefins (e.g., norbornenes). The ROMP may comprise a number of different approaches, including“arm-first” ROMP,“brush-first” ROMP,“graft-to” ROMP,“graft-from” ROMP,“graft-through” ROMP, or combinations thereof.
In some embodiments, ROMP requires use of a metal catalyst. Metal catalysts for use in ROMP reactions may comprise ruthenium, molybdenum, or tungsten. In some embodiments, the metal catalyst is a Grubbs’ catalyst, e.g., first-generation Grubbs’ catalyst, second-generation Grubbs’ catalyst, Hoveyda-Grubbs’ catalyst, or third-generation Grubbs’ catalyst. Exemplary catalysts for use in ROMP are described in Grubbs et al., Acc. Chem. Res.1995, 28, 446–452; U.S. Pat. No.5,811,515;
Schrock et al., Organometallics (1982) 11645; Gallivan et al., Tetrahedron Letters (2005) 46:2577–2580; Furstner et al., J. Am. Chem. Soc. (1999) 121:9453; and Chem. Eur. J. (2001) 7:5299; the entire contents of each of which are incorporated herein by reference.
In some embodiments, the ROMP catalyst is a Grubbs catalyst selected from:

X = Cl; Br; I
Cy = cyclohexyl
Benzylidenebis– (tricyclohexylphosphine)–dichlororuthenium (X = Cl);
Benzylidenebis– (tricyclohexylphosphine)–dibromoruthenium (X = Br);
Benzylidenebis– (tricyclohexylphosphine)–diiodoruthenium (X = I);

X = Cl; Br; I
R = cyclohexyl (Cy); phenyl (Ph); benzyl (Bn)
1,3–(Bis(mesityl)–2–imidazolidinylidene)dichloro–(phenylmethylene) (tricyclohexyl– phosphine)ruthenium (X = Cl; R = cyclohexyl);
1,3–(Bis(mesityl)–2–imidazolidinylidene)dibromo–(phenylmethylene)
(tricyclohexyl–phosphine)ruthenium (X = Br; R = cyclohexyl);
1,3–(Bis(mesityl)–2–imidazolidinylidene)diiodo–(phenylmethylene) (tricyclohexyl– phosphine)ruthenium (X = I; R = cyclohexyl);
1,3–(Bis(mesityl)–2–imidazolidinylidene)dichloro–(phenylmethylene)
(triphenylphosphine)ruthenium (X = Cl; R = phenyl);
1,3–(Bis(mesityl)–2–imidazolidinylidene)dichloro–(phenylmethylene)
(tribenz l hos hine ruthenium X = Cl R = benz l


In some embodiments, the ROMP catalyst is a Grubbs-Hoveyda catalyst. In some embodiments, the Grubbs-Hoveyda catalyst is selected from:


Furstner
 y
y
In some embodiments, the ROMP catalyst is selected from:
,

In other embodiments, ROMP is performed in the absence of a metal catalyst. Polymerization reactions to produce conjugates and BASP particles, e.g., via ROMP, may take place in an oxygen-free and/or water-free environment. In some embodiments, the ROMP is conducted in one or more aprotic solvents. The term “aprotic solvent” refers to a non-nucleophilic solvent having a boiling point range above ambient temperature, preferably from about 25 ºC to about 190 ºC at
atmospheric pressure. In some embodiments, the aprotic solvent has a boiling point from about 80 ºC to about 160 ºC at atmospheric pressure. In certain embodiments, the aprotic solvent has a boiling point from about 80 ºC to about 150 ºC at
atmospheric pressure. Examples of such solvents are methylene chloride, acetonitrile, toluene, DMF, diglyme, THF, and DMSO.
Conjugates and BASP particles generated by ROMP (e.g., via a Grubbs’ catalyst) may be terminated or end-capped through the use of a chain-transfer agent. A chain-transfer agent may be useful to both to modify the properties of the conjugate or BASP particle termini, and in some embodiments, may also modify the Grubbs’ catalyst. Modifications of the terminus of a conjugate or BASP particle may include increasing or decreasing the hydrophobicity of the BASP particle or conjugate core, increasing or decreasing the degradation rate of the BASP particle or conjugate, and increasing or decreasing the density of the BASP particle or conjugate core.
Modifications of the Grubbs’ catalyst may include increasing or decreasing catalyst hydrophobicity, increasing or decreasing the transport of the catalyst out of the BASP particle or conjugate core (i.e., thereby altering the ability to remove the coordinated metal), increasing or decreasing the oxidation state of the metal, increasing or decreasing the affinity of the metal for metal scavenging agents, and increasing or decreasing the solubility of the metal in a solvent. In some embodiments, the chain- transfer agent comprises a reactive olefin. Exemplary chain-transfer agents include ethyl vinyl ether (i.e., ethoxyethene), (Z)-oct-4-ene, (Z)-but-2-ene 1,4-diol, (Z)-4,4'- (but-2-ene-1,4-diylbis(oxy))bis(4-oxobutanoic acid), and (Z)-O,O'-(but-2-ene-1,4- diyl) bis(2-(2-(2-methoxyethoxy)ethoxy)ethyl) disuccinate. The structures of exemplary chain-transfer agents are shown below:

In certain cases, it may be desirable to perform a step to remove a contaminant (e.g., residual metal catalyst or residual solvent) from the synthesized conjugate or
BASP-particle. In some embodiments, removal of a contaminant comprises a ruthenium removal procedure. Removal of residual ruthenium from the ROMP reaction may be performed using a compound comprising an amine, phosphine, or thiol. In some embodiments, a compound used for the removal of a contaminant (e.g., ruthenium) is N,N-dimethyltryptamine, cysteine, triaminetetraacetate (sodium salt), tris(hydroxymethyl)phosphine, 2-mercaptonicotinic acid, N-acetyl-L-cysteine, imidazole, diethylphenylazothioformamide, lead tetra-acetate, hydrogen peroxide, triphenylphosphine oxide, isocyanide salt, di(ethylene glycol) vinyl ether, acetonitrile, dimethyl sulfoxide, or any reagent described in Vougioukalakis, G. C. Chem. A Eur. J.18, 8868–8880 (2012); Wheeler, P., Phillips, J. H. & Pederson, R. L. Org. Process Res. Dev.20, 1182–1190 (2016); Lambeth, R. H., Pederson, S. J., Baranoski, M. & Rawlett, A. M. J. Polym. Sci. Part A Polym. Chem.48, 5752–5757 (2010), the contents of each of which are incorporated herein by reference in their entirety.
Conjugates and BASP particles described herein may be prepared through ROMP, e.g., by polymerizing a macromonomer subunit with a cross-linker (e.g., a macromonomer and/or cross-linker described herein). In some embodiments, the method of making a conjugate or BASP particle comprises:
(a) preparing a first solution of a compound of Formula (V-c) (e.g., a macromonomer):

Formula (V-c)
wherein X is an agent (e.g., an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor); t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive;
with a metal catalyst (e.g., a Grubbs’ catalyst);
b) mixing the first solution with a second solution comprising a compound of Formula (VII-a) (e.g., a cross-linker):

Formula (VII-a)
wherein each of Z1 and Z2 is independently CH2-, -CH2CH2CH2-, or phenyl; each of W1 and W2 is independently -CH2-, -OC(O)CH2CH2-, -CH2OC(O)CH2CH2-, or– C(O)-; and G is–O– or–S-S– to form a third solution;
(c) allowing the third solution to incubate for a time sufficient to form a conjugate or BASP particle of desired length;
(d) adding a chain-transfer agent (e.g., a chain-transfer agent comprising an olefin); and
(e) purifying the solution,
to thereby make the conjugate or BASP particle.
In some embodiments, the amount or concentration of the macromonomer (e.g., a compound of Formula (V-c)) and the amount or concentration of the cross- linker (e.g., a compound of Formula (VII-a)) are present in the reaction mixture in a defined ratio. In some embodiments, the amount or concentration of the
macromonomer (e.g., a compound of Formula (V-c)) is referred to as“m”, and the amount or concentration of the cross-linker (e.g., a compound of Formula (VII-a)) is referred to as“N”. In some embodiments, m is an integer from 1 to 20, and N is an integer from 1 to 20. In some embodiments, the ratio of the amount or concentration of the macromonomer, cross-linker, and catalyst is m:N:1, wherein m is an integer from 1 to 20 and N is an integer from 1 to 20.
In some embodiments, m is an integer from 3 to 12. In some embodiments, m is 1. In some embodiments, m is 2. In some embodiments, m is 3. In some
embodiments, m is 4. In some embodiments, m is 5. In some embodiments, m is 6. In some embodiments, m is 7. In some embodiments, m is 8. In some embodiments, m is 9. In some embodiments, m is 10. In some embodiments, m is 11. In some
embodiments, m is 12.
In certain embodiments, N is an integer from 1 to 10. In some embodiments, N is 1. In some embodiments, N is 2. In some embodiments, N is 3. In some
embodiments, N is 4. In some embodiments, N is 5. In some embodiments, N is 6. In
some embodiments, N is 7. In some embodiments, N is 8. In some embodiments, N is 9. In some embodiments, N is 10. Pharmaceutical Compositions, Formulations and Kits
The BRUSH-compositions and BASP-compositions described herein may comprise a bound therapeutic agent that is preferentially labile or preferentially cleavable in a tissue microenvironment. In some embodiments, this allows for preferential release of the therapeutic agent in the tissue microenvironment, e.g., compared to the systemic release of the therapeutic agent as a free form, thus allowing for a lower dosage of the agent in the conjugate or BASP particle. The BRUSH- compositions and BASP-compositions described herein can be incorporated into a variety of formulations for administration. In certain embodiments, the BRUSH- compositions and BASP-compositions further comprise pharmaceutically acceptable carriers or pharmaceutically acceptable diluents. In certain embodiments, the
BRUSH-compositions and BASP-compositions are in any one of semi-solid, liquid, and gaseous forms; such as capsules, powders, granules, gels, slurries, ointments, solutions, suppositories, injections, inhalants, and aerosols. In certain embodiments, the BRUSH-compositions and BASP-compositions are administered by oral, buccal, rectal, parenteral, intraperitoneal, intradermal, transdermal, or intratracheal administration. In certain embodiments, the BRUSH-compositions and BASP- compositions are administered locally. In certain embodiments, the BRUSH- compositions and BASP-compositions are administered systemically. In certain embodiments, the BRUSH-compositions and BASP-compositions are in a sustained release form.
In addition, the BRUSH-compositions and BASP-compositions can be formulated with common excipients, diluents or carriers, and compressed into tablets, or formulated as elixirs or solutions for convenient oral administration, or
administered by the intramuscular or intravenous routes. The compositions can be administered transdermally, and can be formulated as sustained release dosage forms and the like. Compositions can be administered alone, in combination with each other, or they can be used in combination with other known compounds (discussed herein).
Suitable formulations for use are found in Remington's Pharmaceutical Sciences (1985). Moreover, for a review of methods for drug delivery, see, Langer (1990) Science 249:1527-1533. The pharmaceutical compositions described herein
can be manufactured in a manner that is known to those of skill in the art, e.g., by mixing, dissolving, granulating, dragee-making, levigating, emulsifying,
encapsulating, entrapping or lyophilizing processes. The following methods and excipients are merely exemplary and are in no way limiting.
The compositions can be formulated for parenteral administration by injection, e.g., by bolus injection or continuous infusion. Formulations for injection can be presented in unit dosage form, e.g., in ampules or in multidose containers, with an added preservative. The compositions can take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and can contain formulator agents such as suspending, stabilizing and/or dispersing agents.
Injectable preparations, for example, sterile injectable aqueous or oleaginous suspensions can be formulated according to the known art using suitable dispersing or wetting agents and suspending agents. The sterile injectable preparation can be a sterile injectable solution, suspension, or emulsion in a nontoxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3-butanediol. Among the acceptable vehicles and solvents that can be employed are water, Ringer’s solution, U.S.P. and isotonic sodium chloride solution. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose any bland fixed oil can be employed including synthetic mono- or diglycerides. In addition, fatty acids such as oleic acid are used in the preparation of injectables.
The injectable formulations can be sterilized, for example, by filtration through a bacterial-retaining filter, or by incorporating sterilizing agents in the form of sterile solid compositions which can be dissolved or dispersed in sterile water or other sterile injectable medium prior to use.
Suitable devices for use in delivering intradermal pharmaceutical
compositions described herein include short needle devices such as those described in U.S. Patents 4,886,499; 5,190,521; 5,328,483; 5,527,288; 4,270,537; 5,015,235;
5,141,496; and 5,417,662. Intradermal compositions can be administered by devices which limit the effective penetration length of a needle into the skin, such as those described in PCT publication WO 99/34850 and functional equivalents thereof. Jet injection devices which deliver liquid vaccines to the dermis via a liquid jet injector and/or via a needle which pierces the stratum corneum and produces a jet which reaches the dermis are suitable. Jet injection devices are described, for example, in U.S. Patents 5,480,381; 5,599,302; 5,334,144; 5,993,412; 5,649,912; 5,569,189;
5,704,911; 5,383,851; 5,893,397; 5,466,220; 5,339,163; 5,312,335; 5,503,627;
5,064,413; 5,520,639; 4,596,556; 4,790,824; 4,941,880; 4,940,460; and PCT publications WO 97/37705 and WO 97/13537. Ballistic powder/particle delivery devices which use compressed gas to accelerate the compound in powder form through the outer layers of the skin to the dermis are suitable. Alternatively or additionally, conventional syringes can be used in the classical Mantoux method of intradermal administration.
In addition, the compositions can also be formulated as a depot preparation. Such long acting formulations can be administered by implantation (for example subcutaneously or intramuscularly) or by intramuscular injection. Thus, for example, the compounds can be formulated with suitable polymeric or hydrophobic materials (for example as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
Lipid particles (e.g., liposomes) and emulsions are known examples of delivery vehicles or carriers for hydrophobic drugs. Long-circulating, e.g., stealth, liposomes can be employed. Such liposomes are generally described in U.S. Pat. No. 5,013,556. The compounds of the present invention can also be administered by controlled release means and/or delivery devices such as those described in U.S. Pat. Nos.3,845,770; 3,916,899; 3,536,809; 3,598,123; and 4,008,719.
For oral administration, the compositions can be formulated by combining with pharmaceutically acceptable carriers that are known in the art. Such carriers enable the compounds to be formulated as pills, capsules, emulsions, lipophilic and hydrophilic suspensions, liquids, gels, syrups, slurries, suspensions and the like, for oral ingestion by a subject (e.g., patient) to be treated. Pharmaceutical preparations for oral use can be obtained by mixing the compositions with an excipient and processing the mixture of granules, after adding suitable auxiliaries, if desired, to obtain tablets or dragee cores. Suitable excipients are, in particular, fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol; cellulose preparations such as, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl-cellulose, sodium carboxymethylcellulose, and/or polyvinylpyrrolidone (PVP).
For administration by inhalation, the compositions for use according to the present invention are conveniently delivered in the form of an aerosol spray presentation from pressurized packs or a nebulizer, with the use of a suitable
propellant, e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas, or from propellant- free, dry-powder inhalers. In the case of a pressurized aerosol the dosage unit can be determined by providing a valve to deliver a metered amount. Capsules and cartridges of, e.g., gelatin for use in an inhaler or insufflator can be formulated containing a powder mix of the compound and a suitable powder base such as lactose or starch. The compositions can also be formulated in rectal compositions such as suppositories or retention enemas, e.g., containing conventional suppository bases such as cocoa butter, carbowaxes, polyethylene glycols or other glycerides, all of which melt at body temperature, yet are solidified at room temperature.
Pharmaceutical compositions suitable for use in the present invention include compositions wherein the active ingredients are contained in a therapeutically effective amount. The amount of composition administered will, of course, be dependent on the subject being treated, on the subject's weight, the severity of the affliction, the manner of administration and the judgment of the prescribing physician. Determination of an effective amount is well within the capability of those skilled in the art, especially in light of the detailed disclosure provided herein.
The BRUSH-compositions and BASP-compositions can be administered daily, every other day, three times a week, twice a week, weekly, or bi-weekly. The dosing schedule can include a“drug holiday,” i.e., the drug can be administered for two weeks on, one week off, or three weeks on, one week off, or four weeks on, one week off, etc., or continuously, without a drug holiday. The compounds can be administered orally, intravenously, intraperitoneally, topically, transdermally, intramuscularly, subcutaneously, intranasally, sublingually, or by any other route.
In certain embodiments, the BRUSH-composition or BASP-composition (alone or in combination with the therapeutic agents described herein) is formulated for oral, subcutaneous, intravenous or intraperitoneal administration.
Substantially continuous administration of the BRUSH-composition or BASP- composition (alone or in combination with the therapeutic agents described herein) can cause a greater reduction in collagen content and/or tumor size than single or pulsatile administration of the agent. Thus, it may be desirable to formulate and/or administered the metformin agent (alone or in combination with the therapies described herein) substantially continuously.
In one embodiment, the BRUSH-composition or BASP-composition (alone or in combination with the therapeutic agents described herein) is administered substantially continuously over a pre-determined period of, or at least 15, 30, 45 minutes; a period of, or at least, 1, 5, 10, 24 hours; a period of, or at least, 2, 5, 10, 14 days; a period of, or at least, 3, 4, 5, 6, 7, 8 weeks; a period of, or at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 months; a period of, or at least, 1, 2, 3, 4, 5 years, or longer. The delivery method can be optimized such that a BASP-composition dose as described herein (alone or in combination) is administered and/or maintained in the subject for a pre-determined period (e.g., a period as described herein).
The BRUSH-composition or BASP-composition (alone or in combination with the therapeutic agents described herein) can be in a controlled- or extended release formulation, dosage form, or device. Exemplary formulations and devices for controlled or extended release are known in the art. For example, formulations containing polymer matrices, such as hydroxypropylmethyl cellulose, gels, osmotic systems, liposomes and combination thereof can be used to provide the desired release kinetics.
In one embodiment, the BRUSH-composition or BASP-composition (alone or in combination with the therapeutic agents described herein) is administered via an implantable infusion device, e.g., a pump (e.g., a subcutaneous pump), an implant or a depot. Implantable infusion devices typically include a housing containing a liquid reservoir which can be filled transcutaneously by a hypodermic needle penetrating a fill port septum. The medication reservoir is generally coupled via an internal flow path to a device outlet port for delivering the liquid through a catheter to a subject body site. Typical infusion devices also include a controller and a fluid transfer mechanism, such as a pump or a valve, for moving the liquid from the reservoir through the internal flow path to the device's outlet port.
Also encompassed by the invention are kits (e.g., pharmaceutical packs). The inventive kits may be useful for treating a proliferative disease (e.g., cancer (e.g., leukemia, melanoma, multiple myeloma), benign neoplasm, angiogenesis, inflammatory disease, autoinflammatory disease, or autoimmune disease). The kits provided may comprise the BRUSH-compositions or BASP-compositions described herein, or a pharmaceutical composition thereof, and a container (e.g., a vial, ampule, bottle, syringe, and/or dispenser package, or other suitable container). In some embodiments, provided kits may optionally further include a second container
comprising a pharmaceutical excipient for dilution or suspension of a BRUSH- composition or BASP-composition. In some embodiments, the BRUSH-composition or BASP-composition provided in the container and the second container are combined to form one unit dosage form. Therapeutic Methods
Methods described herein comprise administration to a subject of a BRUSH- composition or BASP-composition as a single agent or in combination with one or more other therapeutic agents or modalities (e.g., as a particle or free agent) for treating or preventing a disorder, e.g., a hyperproliferative disorder (e.g., a cancer) or a fibrotic or an inflammatory condition or disorder described herein. In certain embodiments, the disorder is chosen from one or more of a hyperproliferative disorder, a cancer (e.g., a solid or fibrotic cancer), a fibrotic disorder or condition, an inflammatory disorder or condition, or an autoimmune disorder.
The terms“condition,”“disease,” and“disorder” are used interchangeably. As used herein,“therapy” and“treatment” are synonymous terms.
As used herein,“chemotherapy,”“chemotherapeutic,”“chemotherapeutic agent” and“anti-cancer agent” are synonymous terms.
As used herein, and unless otherwise specified, the terms“treat,”“treating” and“treatment” refer to both therapeutic treatment and prophylactic or preventative measures, wherein the object is to prevent or slow down (lessen) an undesired physiological change or disorder, such as the development or spread of cancer.
Beneficial or desired clinical results include, but are not limited to, alleviation of symptoms, diminishment of extent of disease, stabilized (i.e., not worsening) state of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, and remission (whether partial or total), whether detectable or undetectable.“Treatment” can also mean prolonging survival as compared to expected survival if not receiving treatment. Those in need of treatment include those already with the condition or disorder as well as those prone to have the condition or disorder or those in which the condition or disorder is to be prevented.
“Treating” a tumor, as used herein, typically refers to one or more of the following:
inhibiting primary or metastatic tumor growth; reducing primary or metastatic tumor mass or volume; reducing size or number of metastatic lesions; inhibiting the
development of new metastatic lesions; reducing one or more of non-invasive tumor volume or metabolism; providing prolonged survival; providing prolonged progression-free survival; or providing prolonged time to progression; and/or enhanced quality of life.
For example, in the case of treating cancer, in some embodiments, therapeutic treatment can refer to inhibiting or reducing tumor growth or progression after administration in accordance with the methods or administration with the BRUSH- compositions or BASP-compositions described herein. For example, tumor growth or progression is inhibited or reduced by at least about 10%, at least about 15%, at least about 20%, at least about 30%, at least about 40%, or at least about 50%, after treatment. In another embodiment, tumor growth or progression is inhibited or reduced by more than 50%, e.g., at least about 60%, or at least about 70%, after treatment. In one embodiment, tumor growth or progression is inhibited or reduced by at least about 80%, at least about 90% or greater, as compared to a control (e.g. in the absence of the BASP-composition described herein).
As used herein, unless otherwise specified, the terms“prevent,”“preventing” and“prevention” contemplate an action that occurs before a subject begins to suffer from the regrowth of the cancer and/or which inhibits or reduces the severity of the cancer.
As used herein, and unless otherwise specified, a“therapeutically effective amount” of a BRUSH-composition or BASP-composition is an amount sufficient to provide a therapeutic benefit in the treatment of the disorder (e.g., cancer), or to delay or minimize one or more symptoms associated with the disorder (e.g., cancer). A therapeutically effective amount of a BRUSH-composition or BASP-composition means an amount of therapeutic agent, alone or in combination with other therapeutic agents, which provides a therapeutic benefit in the treatment or management of the disorder. The term“therapeutically effective amount” can encompass an amount that improves overall therapy, reduces or avoids symptoms or causes of the disorder (e.g., cancer), or enhances the therapeutic efficacy of another therapeutic agent.
As used herein, and unless otherwise specified, a“prophylactically effective amount” of a BRUSH-composition or BASP-composition is an amount sufficient to prevent a disorder (e.g., regrowth of the cancer, or one or more symptoms associated with the cancer, or prevent its recurrence). A prophylactically effective amount of a BRUSH-composition or BASP-composition means an amount of the compound,
alone or in combination with other therapeutic agents, which provides a prophylactic benefit in the prevention of the disorder. The term“prophylactically effective amount” can encompass an amount that improves overall prophylaxis or enhances the prophylactic efficacy of another prophylactic agent.
A“subject” includes, but is not limited to, humans (i.e., a male or female of any age group, e.g., a pediatric subject (e.g., infant, child, adolescent) or adult subject (e.g., young adult, middle–aged adult, or senior adult)) and/or other non–human animals, for example, mammals (e.g., primates (e.g., cynomolgus monkeys, rhesus monkeys). In certain embodiments, the animal is a mammal. The animal may be a male or female and at any stage of development. A non–human animal may be a transgenic or genetically engineered animal.
The terms“administer,”“administering,” or“administration” refer to implanting, absorbing, ingesting, injecting, inhaling, or otherwise introducing a BRUSH-composition or BASP-composition, or a pharmaceutical composition thereof, or a device incorporating the BRUSH-composition or BASP-composition. Hyperproliferative Disorders and Cancer
In some embodiments, the disorder, e.g., a cancer, treated is an epithelial, a mesenchymal or a hematologic malignancy. In an embodiment, the cancer treated is a solid tumor (e.g., carcinoid, carcinoma or sarcoma), a soft tissue tumor (e.g., a heme malignancy), and a metastatic lesion, e.g., a metastatic lesion of any of the cancers disclosed herein. In one embodiment, the cancer treated is a fibrotic or desmoplastic solid tumor, e.g., a tumor having one or more of: limited tumor perfusion, compressed blood vessels, fibrotic tumor interstitium, or increased interstitial fluid pressure. In one embodiment, the solid tumor is chosen from one or more of pancreatic (e.g., pancreatic adenocarcinoma or pancreatic ductal adenocarcinoma), breast, colon, colorectal, lung (e.g., small cell lung cancer (SCLC) or non-small cell lung cancer (NSCLC)), skin, ovarian, liver cancer, esophageal cancer, endometrial cancer, gastric cancer, head and neck cancer, kidney, or prostate cancer.
In one embodiment, the cancer treated is a fibrotic or desmoplastic solid tumor, e.g., a tumor having one or more of: limited tumor perfusion, compressed blood vessels, high interstitial fluid pressure (IFPs), or fibrotic tumor interstitium.
In one embodiment, the solid tumor is chosen from one or more of pancreatic (e.g., pancreatic adenocarcinoma (e.g., pancreatic ductal adenocarcinoma (PDA or
PDAC)), breast, gastric, colorectal, lung (e.g., small or non-small cell lung cancer), skin, ovarian, prostate, or liver cancer. Additional examples of cancers treated are described herein below.
In certain embodiments, the cancer treated contains (e.g., has elevated levels of) extracellular matrix components, such as fibers (e.g., collagen, procollagen) and/or polysaccharides (e.g., glycosaminoglycans such as hyaluronan or hyaluronic acid). The levels of the extracellular matrix components in the cancer can vary depending on the particular cancer type, the stage of malignancy, and/or in response to cancer therapy. For example, certain cancer may show elevated levels of extracellular matrix components in response to chemotherapy and/or radiation. In such cancers, an agent (e.g., an ARB, vitamin D analog, or bromodomain inhibitor) may be administered alone, or in combination with a second agent (as a conjugate, BASP particle or free agent) at any time before, during or after the cancer therapy.
In one embodiment, the cancer or tumor treated is a solid, fibrotic tumor chosen from one or more of pancreatic (e.g., pancreatic adenocarcinoma or pancreatic ductal adenocarcinoma), breast, colorectal, colon, lung (e.g., small or non-small cell lung cancer), skin, ovarian, prostate, cervix, gastrointestinal (e.g., carcinoid or stromal), stomach, head and neck, kidney, brain cancer or liver cancer (e.g. HCC), or a metastatic lesion thereof. Additional examples of cancers treated are described herein below.
In one embodiment, the disorder is fibrotic or desmoplastic solid tumor, e.g., a tumor having one or more of: limited tumor perfusion, compressed blood vessels, high interstitial fluid pressure (IFPs), or fibrotic tumor interstitium. In certain embodiments, the subject has a tumor having (e.g., elevated levels of) extracellular matrix components, such as fibers (e.g., collagen, procollagen) and/or polysaccharides (e.g., glycosaminoglycans such as hyaluronan or hyaluronic acid). The levels of the extracellular matrix components in the tumor can vary depending on the particular cancer type, the stage of malignancy, and/or in response to cancer therapy. For example, certain tumors may show elevated levels of extracellular matrix components in response to chemotherapy and/or radiation. In such cancers an agent (e.g., an ARB, vitamin D analog, or bromodomain inhibitor) may be administered alone, or in combination with a second agent (as a conjugate, BASP particle, or free agent) at any time before, during or after the cancer therapy.
By“hyperproliferative cancerous disease or disorder” is meant all neoplastic cell growth and proliferation, whether malignant or benign, including all transformed cells and tissues and all cancerous cells and tissues. Hyperproliferative diseases or disorders include, but are not limited to, precancerous lesions, abnormal cell growths, benign tumors, malignant tumors, and“cancer.”
As used herein, the terms“cancer,”“tumor” or“tumor tissue” refer to an abnormal mass of tissue that results from excessive cell division, in certain cases tissue comprising cells which express, over-express, or abnormally express a hyperproliferative cell protein. A cancer, tumor or tumor tissue comprises“tumor cells” which are neoplastic cells with abnormal growth properties and no useful bodily function. Cancers, tumors, tumor tissue and tumor cells may be benign or malignant. A cancer, tumor or tumor tissue may also comprise“tumor-associated non- tumor cells”, e.g., vascular cells which form blood vessels to supply the tumor or tumor tissue. Non-tumor cells may be induced to replicate and develop by tumor cells, for example, the induction of angiogenesis in a tumor or tumor tissue. As used herein, “cancer,”“malignancy” and“tumor” are synonymous terms.
Examples of cancer include, but are not limited to, carcinoma, lymphoma, blastoma, sarcoma, and leukemia or lymphoid malignancies. More particular examples of such cancers are noted below and include: squamous cell cancer (e.g. epithelial squamous cell cancer), lung cancer including small-cell lung cancer, non- small cell lung cancer, adenocarcinoma of the lung and squamous carcinoma of the lung, cancer of the peritoneum, hepatocellular cancer, gastric or stomach cancer including gastrointestinal cancer, pancreatic cancer, glioblastoma, cervical cancer, ovarian cancer, liver cancer, bladder cancer, hepatoma, breast cancer, colon cancer, rectal cancer, colorectal cancer, endometrial cancer or uterine carcinoma, salivary gland carcinoma, kidney or renal cancer, prostate cancer, vulval cancer, thyroid cancer, hepatic carcinoma, anal carcinoma, penile carcinoma, as well as head and neck cancer. The term“cancer” includes primary malignant cells or tumors (e.g., those whose cells have not migrated to sites in the subject's body other than the site of the original malignancy or tumor) and secondary malignant cells or tumors (e.g., those arising from metastasis, the migration of malignant cells or tumor cells to secondary sites that are different from the site of the original tumor).
Other examples of cancers or malignancies include, but are not limited to: Acute Childhood Lymphoblastic Leukemia, Acute Lymphoblastic Leukemia, Acute
Lymphocytic Leukemia, Acute Myeloid Leukemia, Adrenocortical Carcinoma, Adult (Primary) Hepatocellular Cancer, Adult (Primary) Liver Cancer, Adult Acute Lymphocytic Leukemia, Adult Acute Myeloid Leukemia, Adult Hodgkin's Disease, Adult Hodgkin's Lymphoma, Adult Lymphocytic Leukemia, Adult Non-Hodgkin's Lymphoma, Adult Primary Liver Cancer, Adult Soft Tissue Sarcoma, AIDS-Related Lymphoma, AIDS-Related Malignancies, Anal Cancer, Astrocytoma, Bile Duct Cancer, Bladder Cancer, Bone Cancer, Brain Stem Glioma, Brain Tumors, Breast Cancer, Cancer of the Renal Pelvis and Ureter, Central Nervous System (Primary) Lymphoma, Central Nervous System Lymphoma, Cerebellar Astrocytoma, Cerebral Astrocytoma, Cervical Cancer, Childhood (Primary) Hepatocellular Cancer,
Childhood (Primary) Liver Cancer, Childhood Acute Lymphoblastic Leukemia, Childhood Acute Myeloid Leukemia, Childhood Brain Stem Glioma, Childhood Cerebellar Astrocytoma, Childhood Cerebral Astrocytoma, Childhood Extracranial Germ Cell Tumors, Childhood Hodgkin's Disease, Childhood Hodgkin's Lymphoma, Childhood Hypothalamic and Visual Pathway Glioma, Childhood Lymphoblastic Leukemia, Childhood Medulloblastoma, Childhood Non-Hodgkin's Lymphoma, Childhood Pineal and Supratentorial Primitive Neuroectodermal Tumors, Childhood Primary Liver Cancer, Childhood Rhabdomyosarcoma, Childhood Soft Tissue Sarcoma, Childhood Visual Pathway and Hypothalamic Glioma, Chronic
Lymphocytic Leukemia, Chronic Myelogenous Leukemia, Colon Cancer, Cutaneous T-Cell Lymphoma, Endocrine Pancreas Islet Cell Carcinoma, Endometrial Cancer, Ependymoma, Epithelial Cancer, Esophageal Cancer, Ewing's Sarcoma and Related Tumors, Exocrine Pancreatic Cancer, Extracranial Germ Cell Tumor, Extragonadal Germ Cell Tumor, Extrahepatic Bile Duct Cancer, Eye Cancer, Female Breast Cancer, Gaucher's Disease, Gallbladder Cancer, Gastric Cancer, Gastrointestinal Carcinoid Tumor, Gastrointestinal Tumors, Germ Cell Tumors, Gestational
Trophoblastic Tumor, Hairy Cell Leukemia, Head and Neck Cancer, Hepatocellular Cancer, Hodgkin's Disease, Hodgkin's Lymphoma, Hypergammaglobulinemia, Hypopharyngeal Cancer, Intestinal Cancers, Intraocular Melanoma, Islet Cell Carcinoma, Islet Cell Pancreatic Cancer, Kaposi's Sarcoma, Kidney Cancer,
Laryngeal Cancer, Lip and Oral Cavity Cancer, Liver Cancer, Lung Cancer,
Lymphoproliferative Disorders, Macroglobulinemia, Male Breast Cancer, Malignant Mesothelioma, Malignant Thymoma, Medulloblastoma, Melanoma, Mesothelioma, Metastatic Occult Primary Squamous Neck Cancer, Metastatic Primary Squamous
Neck Cancer, Metastatic Squamous Neck Cancer, Multiple Myeloma, Multiple Myeloma/Plasma Cell Neoplasm, Myelodysplastic Syndrome, Myelogenous
Leukemia, Myeloid Leukemia, Myeloproliferative Disorders, Nasal Cavity and Paranasal Sinus Cancer, Nasopharyngeal Cancer, Neuroblastoma, Non-Hodgkin's Lymphoma During Pregnancy, Nonmelanoma Skin Cancer, Non-Small Cell Lung Cancer, Occult Primary Metastatic Squamous Neck Cancer, Oropharyngeal Cancer, Osteo-/Malignant Fibrous Sarcoma, Osteosarcoma/Malignant Fibrous Histiocytoma, Osteosarcoma/Malignant Fibrous Histiocytoma of Bone, Ovarian Epithelial Cancer, Ovarian Germ Cell Tumor, Ovarian Low Malignant Potential Tumor, Pancreatic Cancer, Paraproteinemias, Purpura, Parathyroid Cancer, Penile Cancer,
Pheochromocytoma, Pituitary Tumor, Plasma Cell Neoplasm/Multiple Myeloma, Primary Central Nervous System Lymphoma, Primary Liver Cancer, Prostate Cancer, Rectal Cancer, Renal Cell Cancer, Renal Pelvis and Ureter Cancer, Retinoblastoma, Rhabdomyosarcoma, Salivary Gland Cancer, Sarcoidosis Sarcomas, Sezary
Syndrome, Skin Cancer, Small Cell Lung Cancer, Small Intestine Cancer, Soft Tissue Sarcoma, Squamous Neck Cancer, Stomach Cancer, Supratentorial Primitive
Neuroectodermal and Pineal Tumors, T-Cell Lymphoma, Testicular Cancer,
Thymoma, Thyroid Cancer, Transitional Cell Cancer of the Renal Pelvis and Ureter, Transitional Renal Pelvis and Ureter Cancer, Trophoblastic Tumors, Ureter and Renal Pelvis Cell Cancer, Urethral Cancer, Uterine Cancer, Uterine Sarcoma, Vaginal Cancer, Visual Pathway and Hypothalamic Glioma, Vulvar Cancer, Waldenstrom's Macroglobulinemia, Wilms' Tumor, and any other hyperproliferative disease, besides neoplasia, located in an organ system listed above.
In other embodiments, the BASP-composition, alone or in combination, is used to treat a hyperproliferative disorder, e.g., a hyperproliferative connective tissue disorder (e.g., a hyperproliferative fibrotic disease). In one embodiment, the hyperproliferative fibrotic disease is multisystemic or organ-specific. Exemplary hyperproliferative fibrotic diseases include, but are not limited to, multisystemic (e.g., systemic sclerosis, multifocal fibrosclerosis, sclerodermatous graft-versus-host disease in bone marrow transplant recipients, nephrogenic systemic fibrosis, scleroderma), and organ-specific disorders (e.g., fibrosis of the eye, lung, liver, heart, kidney, pancreas, skin and other organs). In other embodiments, the disorder is chosen from liver cirrhosis or tuberculosis.
In other embodiment, the subject treated has a hyperproliferative genetic disorder, e.g., a hyperproliferative genetic disorder chosen from Marfan’s syndrome or Loeys–Dietz syndrome.
In other embodiments, the hyperproliferative disorder (e.g., the
hyperproliferative fibrotic disorder) is chosen from one or more of chronic obstructive pulmonary disease, asthma, aortic aneurysm, radiation-induced fibrosis, skeletal- muscle myopathy, diabetic nephropathy, and/or arthritis.
Additional exemplary hyperproliferative disorders that can be treated by the methods and compositions of the invention are disclosed in Sounni, N.E. et al. (2010) Diseases Models & Mechanisms 3:317-332.
In yet other embodiments, the disorder is chosen from an inflammatory or an autoimmune disorder chosen from multiple sclerosis, inflammatory bowel disease, scleroderma, lupus, rheumatoid arthritis or osteoarthritis.
In certain embodiments, the inflammatory disorder is an inflammatory disorder of: the gastrointestinal tract or a gastrointestinal organ, e.g., colitis, Crohn's disease, inflammatory bowel disease (IBD), Barrett’s esophagus and chronic gastritis; the lung (e.g., asthma, chronic obstructive pulmonary disease (COPD); the skin (e.g., psoriasis), the cardiovascular system (e.g., atherosclerosis, cholesterol metabolic disorders, oxygen free radical injury, ischemia), the nervous system (e.g., Alzheimer's disease, multiple sclerosis), liver (e.g., hepatitis), kidney (e.g., nephritis), and the pancreas (e.g., pancreatitis).
In other embodiments, the inflammatory disorder is associated with an autoimmune disorder, e.g., arthritis (including rheumatoid arthritis, juvenile rheumatoid arthritis, osteoarthritis, psoriatic arthritis, lupus-associated arthritis, autoimmune thyroiditis or ankylosing spondylitis); scleroderma; lupus; systemic lupus erythematosis; HIV; Sjogren's syndrome; vasculitis; multiple sclerosis; dermatitis (including atopic dermatitis and eczematous dermatitis), myasthenia gravis, inflammatory bowel disease (IBD), Crohn's disease, colitis, diabetes mellitus (type I); acute inflammatory conditions (e.g., endotoxemia, sepsis and septicemia, toxic shock syndrome and infectious disease); transplant rejection and allergy. Liver Conditions or Disorders
Examples of liver cancers include: hepatocellular carcinoma (HCC), primary liver cell carcinoma, hepatoma, fibrolamellar carcinoma, focal nodular hyperplasia,
cholangiosarcoma, intrahepatic bile duct cancer, angiosarcoma or hemangiosarcoma, hepatic adenoma, hepatic hemangiomas, hepatic hamartoma, hepatoblastoma, infantile hemangioendothelioma, mixed tumors of the liver, tumors of mesenchymal tissue, and sarcoma of the liver. Examples of cancers that may metastasize to the liver include: breast cancer, colorectal cancer, esophageal cancer, kidney or renal cancer, lung cancer, ovarian cancer, pancreatic cancer, rectal cancer, skin cancer (e.g., melanoma), gastric or stomach cancer (including gastrointestinal cancer), and uterine cancer.
In an embodiment, the liver disorder is a fibrotic disorder or connective tissue disorder affecting the function or physiology of the liver. In one embodiment, the fibrotic disorder or connective tissue disorder can be systemic (affecting the whole body), multi-organ, or organ-specific (e.g., liver-specific). Examples of fibrotic liver disorders include liver fibrosis (hepatic fibrosis), liver cirrhosis, and any disorder associated with accumulation of extracellular matrix proteins, e.g., collagen, in the liver, liver scarring, and/or abnormal hepatic vasculature. Liver fibrosis is caused by liver inflammation or damage which triggers the accumulation of extracellular matrix proteins, including collagens, and scar tissue in the liver. Liver cirrhosis is the end stage of liver fibrosis, involves regenerative nodules (as a result of repair processes), and is accompanied with the distortion of the hepatic vasculature. Liver fibrotic disorders are most commonly caused by chronic viral infection (e.g., hepatitis B, hepatitis C), alcoholism, and fatty liver disease.
Examples of fatty liver diseases include fatty liver (or FLD), alcoholic liver disease, non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), alcoholic steatohepatitis, simple steatosis, Reye’s syndrome, and any disorder associated with abnormal retention of lipids in liver cells.
In one embodiment, the liver disease is NASH.
Metabolic disorders can also affect the liver and cause liver damage. Examples of metabolic disorders of the liver or affecting the liver include hemachromatosis, diabetes, obesity, hypertension, dyslipidemia, galactosemia, and glycogen storage disease.
Autoimmune disorders of the liver or affecting the liver can include systemic disorders or disorders that primarily affect an organ other than the liver, but with secondary effects to liver cells or liver function. Examples of such autoimmune disorders include autoimmune hepatitis (AIH), autoimmune liver disease, lupoid
hepatitis, systemic lupus erythematosus, primary biliary cirrhosis (PBC), scleroderma, and systemic sclerosis.
Disorders associated with inflammation of the liver include steatohepatitis, primary sclerosing cholangitis (PSC), ulcerative colitis, Crohn’s disease,
inflammatory bowel disease, or any disorder associated with inflammation in the liver.
In an embodiment, the liver disorder is associated with an inherited or congenital disease, e.g., Wilson’s disease, Gilbert’s disease, Byler syndrome,
Greenland-Eskimo familial cholestasis, Zellweger’s syndrome, Alagilles syndrome (ALGS), progressive familial intrahepatic cholestasis (PFIC), or alpha 1-antitrypsin deficiency, cystic fibrosis, Indian childhood cirrhosis, and hereditary
hemochromatosis.
In an embodiment, the liver disorder is associated with pancreatic or biliary tract damage or disorders, e.g., cerebrotendinous, xanthomatosis, gall stones, choledocholithiasis, obstruction of the biliary tree, biliary trauma, biliary atresia, pancreatitis, primary biliary cirrhosis, primary sclerosing cholangitis, cholestasis, cholestasis of pregnancy, or any disorder associated with the obstruction or damage to the biliary system or the pancreas.
In an embodiment, liver disorders can be induced by infection, for example, by viral infections such as hepatitis A virus, hepatitis B virus, hepatitis C virus, hepatitis D virus (hepatitis delta virus), hepatitis E virus, Epstein-Barr adenovirus, or cytomegalovirus; or parasitic infection, such as schistosomiasis.
In an embodiment, liver disorders can be induced by drugs, such as acetaminophen (e.g., paracetamol, TYLENOL®, or PANADOL®), nonsteroidal anti- inflammatory drugs (NSAIDS, e.g., aspirin and phenylbutazone, ibuprofen, piroxicam, diclofenac, sulindac, and indomethacin), glucocorticoids, anti-tuberculosis drugs (e.g., isoniazid), antibiotics, anesthetics, antihypertensives (e.g., statins), oral contraceptives, dietary aids, or herbal supplements (e.g., ackee fruit, bajiaolian, boragecamphor, copaltra, comfrey, cycasin, kava leaves, pyrrolizidine alkaloids, horse chestnut leaves, valerian); or toxins, such as arsenic, carbon tetrachloride, vinyl chloride, aflatoxins.
In an embodiment, liver disorders also include disorders or conditions induced by injury to the liver or affecting the liver, including drug toxicity, alcoholism, ischemia, malnutrition, or physical trauma.
Other liver disorders include hepatic vein thrombosis, Budd-Chiari syndrome, portal hypertension, hepatic encephalopathy, and hepatomegaly (or enlarged liver). Fibrotic Conditions or Disorders
In another aspect, the invention features a method of treating or preventing a fibrotic condition or disorder in a subject. The method includes administering a BRUSH-composition or BASP-composition described herein, alone or in combination with another agent or therapeutic modality, to a subject in need thereof, in an amount sufficient to decrease or inhibit the fibrotic condition in the subject.
In certain embodiments, reducing fibrosis, or treatment of a fibrotic condition, includes reducing or inhibiting one or more of: formation or deposition of tissue fibrosis; reducing the size, cellularity (e.g., fibroblast or immune cell numbers), composition; or cellular content, of a fibrotic lesion; reducing the collagen or hydroxyproline content, of a fibrotic lesion; reducing expression or activity of a fibrogenic protein; reducing fibrosis associated with an inflammatory response;
decreasing weight loss associated with fibrosis; or increasing survival.
In certain embodiments, the fibrotic condition is primary fibrosis. In one embodiment, the fibrotic condition is idiopathic. In other embodiments, the fibrotic condition is associated with (e.g., is secondary to) a disease (e.g., an infectious disease, an inflammatory disease, an autoimmune disease, a malignant or cancerous disease, and/or a connective disease); a toxin; an insult (e.g., an environmental hazard (e.g., asbestos, coal dust, polycyclic aromatic hydrocarbons), cigarette smoking, a wound); a medical treatment (e.g., surgical incision, chemotherapy or radiation), or a combination thereof.
In certain embodiments, the fibrotic condition is a fibrotic condition of the lung, a fibrotic condition of the liver (e.g., as described herein), a fibrotic condition of the heart or vasculature, a fibrotic condition of the kidney, a fibrotic condition of the skin, a fibrotic condition of the gastrointestinal tract, a fibrotic condition of the bone marrow or a hematopoietic tissue, a fibrotic condition of the nervous system, a fibrotic condition of the eye, or a combination thereof.
In certain embodiments, the fibrotic condition is a fibrotic condition of the lung. In certain embodiments, the fibrotic condition of the lung is chosen from one or more of: pulmonary fibrosis, idiopathic pulmonary fibrosis (IPF), usual interstitial pneumonitis (UIP), interstitial lung disease, cryptogenic fibrosing alveolitis (CFA),
bronchiectasis, and scleroderma lung disease. In one embodiment, the fibrosis of the lung is secondary to a disease, a toxin, an insult, a medical treatment, or a
combination thereof. For example, the fibrosis of the lung can be associated with (e.g., secondary to) one or more of: a disease process such as asbestosis and silicosis; an occupational hazard; an environmental pollutant; cigarette smoking; an
autoimmune connective tissue disorders (e.g., rheumatoid arthritis, scleroderma and systemic lupus erythematosus (SLE)); a connective tissue disorder such as
sarcoidosis; an infectious disease, e.g., infection, particularly chronic infection; a medical treatment, including but not limited to, radiation therapy, and drug therapy, e.g., chemotherapy (e.g., treatment with as bleomycin, methotrexate, amiodarone, busulfan, and/or nitrofurantoin). In one embodiment, the fibrotic condition of the lung treated with the methods of the invention is associated with (e.g., secondary to) a cancer treatment, e.g., treatment of a cancer (e.g., squamous cell carcinoma, testicular cancer, Hodgkin’s disease with bleomycin). In one embodiment, the fibrotic condition of the lung is associated with an autoimmune connective tissue disorder (e.g., scleroderma or lupus, e.g., SLE).
Pulmonary fibrosis can occur as a secondary effect in disease processes such as asbestosis and silicosis, and is known to be more prevalent in certain occupations such as coal miner, ship workers and sand blasters where exposure to environmental pollutants is an occupational hazard (Green, FH et al. (2007) Toxicol Pathol.35:136- 47). Other factors that contribute to pulmonary fibrosis include cigarette smoking, and autoimmune connective tissue disorders, like rheumatoid arthritis, scleroderma and systemic lupus erythematosus (SLE) (Leslie, KO et al. (2007) Semin Respir Crit Care Med.28:369-78; Swigris, JJ et al. (2008) Chest.133:271-80; and Antoniou, KM et al. (2008) Curr Opin Rheumatol.20:686-91). Other connective tissue disorders such as sarcoidosis can include pulmonary fibrosis as part of the disease (Paramothayan, S et al. (2008) Respir Med.102:1-9), and infectious diseases of the lung can cause fibrosis as a long term consequence of infection, particularly chronic infections.
Pulmonary fibrosis can also be a side effect of certain medical treatments, particularly radiation therapy to the chest and certain medicines like bleomycin, methotrexate, amiodarone, busulfan, and nitrofurantoin (Catane, R et al. (1979) Int J Radiat Oncol Biol Phys.5:1513-8; Zisman, DA et al. (2001) Sarcoidosis Vasc Diffuse Lung Dis.18:243-52; Rakita, L et al. (1983) Am Heart J.106:906-16; Twohig, KJ et al. (1990) Clin Chest Med.11:31-54; and Witten CM. (1989) Arch Phys Med Rehabil.
70:55-7). In other embodiments, idiopathic pulmonary fibrosis can occur where no clear causal agent or disease can be identified. Genetic factors can play a significant role in these cases of pulmonary fibrosis (Steele, MP et al. (2007) Respiration 74:601- 8; Brass, DM et al. (2007) Proc Am Thorac Soc.4:92-100 and du Bois RM. (2006) Semin Respir Crit Care Med.27:581-8).
In other embodiments, pulmonary fibrosis includes, but is not limited to, pulmonary fibrosis associated with chronic obstructive pulmonary disease (COPD), acute respiratory distress syndrome, scleroderma, pleural fibrosis, chronic asthma, acute lung syndrome, amyloidosis, bronchopulmonary dysplasia, Caplan's disease, Dressler's syndrome, histiocytosis X, idiopathic pulmonary hemosiderosis, lymphangiomyomatosis, mitral valve stenosis, polymyositis, pulmonary edema, pulmonary hypertension (e.g., idiopathic pulmonary hypertension (IPH)),
pneumoconiosis, radiotherapy (e.g., radiation induced fibrosis), rheumatoid disease, Shaver's disease, systemic lupus erythematosus, systemic sclerosis, tropical pulmonary eosinophilia, tuberous sclerosis, Weber-Christian disease, Wegener's granulomatosis, Whipple's disease, or exposure to toxins or irritants (e.g.,
pharmaceutical drugs such as amiodarone, bleomycin, busulphan, carmustine, chloramphenicol, hexamethonium, methotrexate, methysergide, mitomycin C, nitrofurantoin, penicillamine, peplomycin, and practolol; inhalation of talc or dust, e.g., coal dust, silica). In certain embodiments, the pulmonary fibrosis is associated with an inflammatory disorder of the lung, e.g., asthma, and/or COPD.
In certain embodiments, the fibrotic condition is a fibrotic condition of the liver. In certain embodiments, the fibrotic condition of the liver is chosen from one or more of: fatty liver disease, steatosis (e.g., nonalcoholic steatohepatitis (NASH), cholestatic liver disease (e.g., primary biliary cirrhosis (PBC)), cirrhosis, alcohol induced liver fibrosis, biliary duct injury, biliary fibrosis, or cholangiopathies. In other embodiments, hepatic or liver fibrosis includes, but is not limited to, hepatic fibrosis associated with alcoholism, viral infection, e.g., hepatitis (e.g., hepatitis C, B or D), autoimmune hepatitis, non-alcoholic fatty liver disease (NAFLD), progressive massive fibrosis, exposure to toxins or irritants (e.g., alcohol, pharmaceutical drugs and environmental toxins). Additional examples of liver conditions and disorders are provided in the Sections entitled“Liver Conditions or Disorders,” provided herein.
In certain embodiments, the fibrotic condition is a fibrotic condition of the kidney. In certain embodiments, the fibrotic condition of the kidney is chosen from
one or more of: renal fibrosis (e.g., chronic kidney fibrosis), nephropathies associated with injury/fibrosis (e.g., chronic nephropathies associated with diabetes (e.g., diabetic nephropathy)), lupus, scleroderma of the kidney, glomerular nephritis, focal segmental glomerular sclerosis, IgA nephropathyrenal fibrosis associated with human chronic kidney disease (CKD), chronic progressive nephropathy (CPN),
tubulointerstitial fibrosis, ureteral obstruction, chronic uremia, chronic interstitial nephritis, radiation nephropathy, glomerulosclerosis, progressive glomerulonephrosis (PGN), endothelial/thrombotic microangiopathy injury, HIV-associated nephropathy, or fibrosis associated with exposure to a toxin, an irritant, or a chemotherapeutic agent. In one embodiment, the fibrotic condition of the kidney is scleroderma of the kidney. In some embodiments, the fibrotic condition of the kidney is transplant nephropathy, diabetic nephropathy, lupus nephritis, focal segmental
glomerulosclerosis (FSGS), endothelial/thrombotic microangiopathy injury, scleroderma of the kidney, HIV-associated nephropathy (HIVVAN), or exposure to toxins, irritants, or chemotherapeutic agents.
In certain embodiments, the fibrotic condition is a fibrotic condition of the bone marrow or a hematopoietic tissue. In certain embodiments, the fibrotic condition of the bone marrow is an intrinsic feature of a chronic myeloproliferative neoplasm of the bone marrow, such as primary myelofibrosis (also referred to herein as agnogenic myeloid metaplasia or chronic idiopathic myelofibrosis). In other embodiments, the bone marrow fibrosis is associated with (e.g., is secondary to) a malignant condition or a condition caused by a clonal proliferative disease. In other embodiments, the bone marrow fibrosis is associated with a hematologic disorder (e.g., a hematologic disorder chosen from one or more of polycythemia vera, essential thrombocythemia, myelodysplasia, hairy cell leukemia, lymphoma (e.g., Hodgkin or non-Hodgkin lymphoma), multiple myeloma or chronic myelogenous leukemia (CML)). In yet other embodiments, the bone marrow fibrosis is associated with (e.g., secondary to) a non-hematologic disorder (e.g., a non-hematologic disorder chosen from solid tumor metastasis to bone marrow, an autoimmune disorder (e.g., systemic lupus
erythematosus, scleroderma, mixed connective tissue disorder, or polymyositis), an infection (e.g., tuberculosis or leprosy), or secondary hyperparathyroidism associated with vitamin D deficiency. In some embodiments, the fibrotic condition is idiopathic or drug-induced myelofibrosis. In some embodiments, the fibrotic condition of the
bone marrow or hematopoietic tissue is associated with systemic lupus erythematosus or scleroderma.
In other embodiments, the fibrotic condition is associated with leprosy or tuberculosis.
In certain embodiments, the fibrotic condition is a fibrotic condition of the bone marrow. In certain embodiments, the fibrotic condition of the bone marrow is myelofibrosis (e.g., primary myelofibrosis (PMF)), myeloid metaplasia, chronic idiopathic myelofibrosis, or primary myelofibrosis. In other embodiments, bone marrow fibrosis is associated with a hematologic disorder chosen from one or more of hairy cell leukemia, lymphoma, or multiple myeloma.
In other embodiments, the bone marrow fibrosis is associated with one or more myeloproliferative neoplasms (MPN) chosen from: essential thrombocythemia (ET), polycythemia vera (PV), mastocytosis, chronic eosinophilic leukemia, chronic neutrophilic leukemia, or other MPN.
In one embodiment, the fibrotic condition is primary myelofibrosis. Primary myelofibrosis (PMF) (also referred to in the literature as idiopathic myeloid metaplasia, and Agnogenic myeloid metaplasia) is a clonal disorder of multipotent hematopoietic progenitor cells (reviewed in Abdel-Wahab, O. et al. (2009) Annu. Rev. Med.60:233-45; Varicchio, L. et al. (2009) Expert Rev. Hematol.2(3):315-334;
Agrawal, M. et al. (2010) Cancer 1-15).
In certain embodiments, the fibrotic condition is a fibrotic condition of the heart. In certain embodiments, the fibrotic condition of the heart is myocardial fibrosis (e.g., myocardial fibrosis associated with radiation myocarditis, a surgical procedure complication (e.g., myocardial post-operative fibrosis), infectious diseases (e.g., Chagas disease, bacterial, trichinosis or fungal myocarditis)); granulomatous, metabolic storage disorders (e.g., cardiomyopathy, hemochromatosis); developmental disorders (e.g., endocardial fibroelastosis); arteriosclerotic, or exposure to toxins or irritants (e.g., drug induced cardiomyopathy, drug induced cardiotoxicity, alcoholic cardiomyopathy, cobalt poisoning or exposure). In certain embodiments, the myocardial fibrosis is associated with an inflammatory disorder of cardiac tissue (e.g., myocardial sarcoidosis). In some embodiments, the fibrotic condition is a fibrotic condition associated with a myocardial infarction. In some embodiments, the fibrotic condition is a fibrotic condition associated with congestive heart failure.
In some embodiments, the fibrotic condition is associated with an autoimmune disease selected from scleroderma or lupus, e.g., systemic lupus erythematosus.
In some embodiments, the fibrotic condition is systemic. In some
embodiments, the fibrotic condition is systemic sclerosis (e.g., limited systemic sclerosis, diffuse systemic sclerosis, or systemic sclerosis sine scleroderma), nephrogenic systemic fibrosis, cystic fibrosis, chronic graft vs. host disease, or atherosclerosis.
In some embodiments, the fibrotic condition is scleroderma. In some embodiments, the scleroderma is localized, e.g., morphea or linear scleroderma. In some embodiments, the condition is a systemic sclerosis, e.g., limited systemic sclerosis, diffuse systemic sclerosis, or systemic sclerosis sine scleroderma.
In other embodiment, the fibrotic condition affects a tissue chosen from one or more of muscle, tendon, cartilage, skin (e.g., skin epidermis or endodermis), cardiac tissue, vascular tissue (e.g., artery, vein), pancreatic tissue, lung tissue, liver tissue, kidney tissue, uterine tissue, ovarian tissue, neural tissue, testicular tissue, peritoneal tissue, colon, small intestine, biliary tract, gut, bone marrow, hematopoietic tissue, or eye (e.g., retinal) tissue.
In some embodiments, the fibrotic condition is a fibrotic condition of the eye. In some embodiments, the fibrotic condition is glaucoma, macular degeneration (e.g., age-related macular degeneration), macular edema (e.g., diabetic macular edema), retinopathy (e.g., diabetic retinopathy), or dry eye disease.
In certain embodiments, the fibrotic condition is a fibrotic condition of the skin. In certain embodiments, the fibrotic condition of the skin is chosen from one or more of: skin fibrosis (e.g., hypertrophic scarring, keloid), scleroderma, nephrogenic systemic fibrosis (e.g., resulting after exposure to gadolinium (which is frequently used as a contrast substance for MRIs) in subjects with severe kidney failure), and keloid.
In certain embodiments, the fibrotic condition is a fibrotic condition of the gastrointestinal tract. In certain embodiments, the fibrotic condition is chosen from one or more of: fibrosis associated with scleroderma; radiation induced gut fibrosis; fibrosis associated with a foregut inflammatory disorder such as Barrett’s esophagus and chronic gastritis, and/or fibrosis associated with a hindgut inflammatory disorder, such as inflammatory bowel disease (IBD), ulcerative colitis and Crohn’s disease. In
some embodiments, the fibrotic condition of the gastrointestinal tract is fibrosis associated with scleroderma.
In one embodiment, the fibrotic condition is a chronic fibrotic condition or disorder. In certain embodiments, the fibrotic condition is associated with an inflammatory condition or disorder.
In some embodiments, the fibrotic and/or inflammatory condition is osteomyelitis, e.g., chronic osteomyelitis.
In some embodiments, the fibrotic condition is an amyloidosis. In certain embodiments, the amyloidosis is associated with chronic osteomyelitis. Combination Therapies
Disclosed herein are methods that comprise administration of a BRUSH- composition or BASP-composition, alone or in combination with a second therapeutic agent or therapeutic modality (e.g., one, two or more therapeutic agents or
modalities), to a subject in need thereof. The second agent can be administered in free form, or as part of a conjugate, a BASP particle, a BRUSH-composition or a BASP- composition.
By“in combination with,” it is not intended to imply that the therapy or the therapeutic agents must be administered at the same time and/or formulated for delivery together, although these methods of delivery are within the scope of the invention. The pharmaceutical compositions can be administered concurrently with, prior to, or subsequent to, one or more other additional therapies or therapeutic agents. In general, each agent will be administered at a dose and/or on a time schedule determined for that agent. In will further be appreciated that the additional therapeutic agent utilized in this combination can be administered together in a single
composition or administered separately in different compositions. The particular combination to employ in a regimen will take into account compatibility of the inventive pharmaceutical composition with the additional therapeutically active agent and/or the desired therapeutic effect to be achieved. In general, it is expected that additional therapeutic agents utilized in combination be utilized at levels that do not exceed the levels at which they are utilized individually. In some embodiments, the levels utilized in combination will be lower than those utilized individually.
In certain embodiments, a BRUSH-composition or BASP-composition and a second therapeutic agent or therapeutic modality are administered concurrently (e.g.,
administration of the two or more agents at the same time or day, or within the same treatment regimen) and/or sequentially (e.g., administration of one agent over a period of time followed by administration of another agent for a second period of time, or within different treatment regimens). In other embodiments, administration of two or more agents occur in overlapping treatment regimens (e.g., administration of one agent is initiated before the completion of the treatment regimen of another agent, or the administration of one agent is completed before the termination of the treatment regimen of another agent).
In certain embodiments, the BRUSH-composition or BASP-composition includes two or more agents (e.g., therapeutic agents). In certain embodiments, the two or more agents include at least a first agent and a second agent. In certain embodiments, a first agent is included in (e.g., as part of) a conjugate or BASP particle, and a second agent is not included in (e.g., not as part of) the conjugate or BASP particle. In certain embodiments, a first agent is included in (e.g., as part of) a first conjugate or first BASP particle, and a second agent is included in (e.g., as part of) a second conjugate or second BASP particle. In certain embodiments, each agent is included in (e.g., as part of) a different conjugate or different BASP particle. In certain embodiments, a first agent and a second agent are included in (e.g., as part of) a conjugate or BASP particle. The BRUSH-compositions or BASP-compositions may be able to selectively deliver (e.g., to deliver at different times and/or at different rates) the two or more agents. In certain embodiments, a first cleavable linker, which directly or indirectly attaches a first agent to the remaining part of a first conjugate or first particle, is different from a second cleavable linker, which directly or indirectly attaches a second agent to the remaining part of the first conjugate or first particle, or of a second conjugate or second particle. In certain embodiments, the first cleavable linker and the second cleavable linker are cleaved at different rates under the same conditions.
In some embodiments, the second agent or therapeutic modality, e.g., the second therapeutic agent, is a cancer therapy (e.g., one or more of anti-cancer agents, photodynamic therapy (PDT), immunotherapy, surgery and/or radiation).
In some embodiments, the anti-cancer agent is a small molecule, a kinase inhibitor, an alkylating agent, a vascular disrupting agent, a microtubule targeting agent, a mitotic inhibitor, a topoisomerase inhibitor, an anti-angiogenic agent, or an anti-metabolite. In one embodiment, the agent, e.g., the therapeutic agent, is a taxane
(e.g., paclitaxel, docetaxel, larotaxel or cabazitaxel). In some embodiments, the anti- cancer agent is an anthracycline (e.g., doxorubicin). In some embodiments, the anti- cancer agent is a platinum-based agent (e.g., cisplatin or oxaliplatin). In some embodiments, the anti-cancer agent is a pyrimidine analog (e.g., gemcitabine). In some embodiments, the anti-cancer agent is chosen from camptothecin, irinotecan, rapamycin, FK506, 5-FU, leucovorin, or a combination thereof. In other
embodiments, the anti-cancer agent is a protein biologic (e.g., an antibody molecule), or a nucleic acid therapy (e.g., an antisense or inhibitory double stranded RNA molecule).
In other embodiments, the cancer therapy includes one or more of: a cancer therapeutic, including, for example, a nanotherapy (e.g., one or more nanotherapeutic agents, including viral cancer therapeutic agents (e.g., an oncolytic herpes simplex virus (HSV)) a lipid nanoparticle (e.g., a liposomal formulation (e.g., pegylated liposomal doxorubicin (DOXIL®)), or a polymeric nanoparticle); one or more cancer therapeutic antibodies (e.g., anti-HER2, anti-EGFR, anti-CD20 antibodies); RNAi and antisense RNA agents; one or more chemotherapeutic agents (e.g., low molecular weight chemotherapeutic agents, including a cytotoxic or a cytostatic agent));
photodynamic therapy; immunotherapy; radiation; or surgery, or any combination thereof. In one embodiment, the chemotherapeutic agent used in combination is a cytotoxic or a cytostatic agent. Exemplary cytotoxic agents include, but are not limited to, antimicrotubule agents, topoisomerase inhibitors (e.g., irinotecan), or taxanes (e.g., docetaxel), antimetabolites, mitotic inhibitors, alkylating agents, intercalating agents, agents capable of interfering with a signal transduction pathway, agents that promote apoptosis and radiation.
Any combination of one or more therapeutic agents or modalities (e.g., first, second, third), e.g., nanotherapeutic agent, antibody agent, low molecular weight chemotherapeutic agent, radiation can be used. Exemplary cancer therapeutics include, but are not limited to, nanotherapeutic agents (e.g., one or more lipid nanoparticles (e.g., a liposomal formulation (e.g., pegylated liposomal doxorubicin (DOXIL®) or liposomal paclitaxel (e.g., Abraxane®)), or a polymeric nanoparticle); one or more low molecular weight chemotherapeutic s (e.g., gemcitabine, cisplatin, epirubicin, 5-fluorouracil, paclitaxel, oxaliplatin, or leucovorin); one or more antibodies against cancer targets (e.g., growth factor receptor such as HER-2/neu, HER3, VEGF)); one or more tyrosine kinase inhibitors, e.g., including low molecular
weight and antibody agents, such as sunitinib, erlotinib, gefitinib, sorafenib, icotinib, lapatinib, neratinib, vandetanib, BIBW 2992 or XL-647, anti-EGFR antibody (e.g., cetuximab, panitumumab, zalutumumab, nimotuzumab necitumumab or
matuzumab)). Additional examples of therapeutic agents or modalities used in combination with the BRUSH-compositions or BASP-compositions are described in WO 2013/169739, e.g., in Paragraphs 352-367, incorporated herein by reference.
In yet other embodiments, the methods can be used in combination with immunodulatory agents, e.g., IL- 1, 2, 4, 6, or 12, or interferon alpha or gamma, or immune cell growth factors such as GM-CSF.
In other embodiments, the cancer therapy includes an immune or
immunotherapy used in combination with the BRUSH-compositions or BASP- compositions, other cancer therapies, and/or the microenvironment modulator, described herein. Without wishing to be bound by theory, factor such as hypoxia and/or limited perfusion are believed to cause immunosuppression and/or limit the efficacy of certain immune therapies. BRUSH-compositions or BASP-compositions, alone or in combination with therapies described herein, can be used to improve the efficacy of said immune therapies. Examples of immune therapies include, but are not limited to, CTLA-4 blockade (e.g., an anti-CTLA-4 antibody (e.g., ipilimumab)); immune-based therapies (including, e.g., immune or dendritic cell- based vaccines and antagonists of immune inhibitory signals or checkpoints); cancer vaccines, e.g., Sipuleucel-T (APC8015, trade name Provenge); and adoptive T-cell-based therapies. Exemplary immune-based therapies include, but are not limited to, e.g., immune or dendritic cell- based vaccines (Seton-Rogers, S. (2012) Nature Reviews Cancer 12:230-231; Palucka, K. et al. (2012) Nature Reviews Cancer 12:265-277); effector memory CD8+ T cells (Bird, L. (2012) Nature Reviews Immunology 12:227);
engineered tumor cells to activate Toll like Receptors (TLRs) and NOD-like
Receptors (NLRs) (Leavy, O. (2012) Nature Reviews Immunology 12:227);
antagonists of immune inhibitory signals or checkpoints (Pardoll, D.M. (2012) Nature Reviews Cancer 12:252-264). In one embodiment, the therapy is a cell-based immunotherapy wherein immune cells are expanded ex vivo and injected into the subject.
In yet other embodiments, the cancer therapy includes PDT used in combination with the BASP-composition, other cancer therapies, and/or the microenvironment modulator, described herein. In certain embodiments, PDT
includes administration of a photosensitizing agent (e.g., a porphyrin, a porphyrin precursor, a corlin, or a phthalocyanine) followed by irradiation at a wavelength corresponding to an absorbance band of the photosensitizing agent. In the presence of oxygen, a series of events lead to one or more of: cell death (e.g., tumor cell death), damage to the micro vasculature, or induction of a local inflammatory reaction). PDT is reviewed in, e.g., Agostinis, P. et al. (2011) CA Cancer J. Clin.61 :250-281.
In other embodiments, the cancer therapy includes an inhibitor of a cancer stem cell (also referred to herein as a“cancer initiating cell”), used in combination with the BASP-compositions, or BRUSH-compositions described herein. Without wishing to be bound by theory, hypoxia and cancer drugs (including anti- angiogenic drugs) and radiation therapy are believed to increase the number of cancer stem cells.
BRUSH-composition or BASP-composition, alone or in combination with, e.g., an inhibitor of a cancer stem cell, can be used to reduce the production of these stem cells. Exemplary inhibitors of cancer stem cells that can be used in combination include, but are not limited to, hedgehog (e.g., SMO) antagonists; and Wnt pathway antagonists (e.g., antibody, OMP- 18R5).
In certain embodiments, the combinations described herein can be further administered in combination with a microenvironment modulator. The combined administration of the microenvironment modulator can be used to further enhance the efficacy (e.g., penetration and/or diffusion), of the combination therapies described herein in a tumor or tumor vasculature in a subject. Exemplary microenvironment modulators include, but are not limited to, an anti-angiogenic therapy and/or vascular normalization strategy, for example, an inhibitor of vascular endothelial growth factor (VEGF) pathway, an inhibitor of the angiopoietin-Tie-2 pathway (e.g., an Ang-1 or an Ang-2 inhibitor), or sorafenib; an agent that decreases the level or production of hyaluronic acid; an inhibitor of the hedgehog pathway; an agent that improves drug penetration in tumors. In one embodiment, the agent is a disulfide-based cyclic RGD peptide (iRGD) or an analogue thereof; a taxane therapy (e.g., taxane-induced apoptosis); an agent that decreases the level or production of collagen or procollagen; an anti-fibrotic agent and/or a profibrotic pathway inhibitor. Additional examples of microenvironment modulators used in combination with the combination therapies comprising the BRUSH-compositions and BASP-compositions are described in WO 2013/169739, e.g., in Paragraphs 260, 340-346, incorporated herein by reference. Agents for anti-angiogenic/vascular normalization strategies as described in Goel et
al. (2011) Physiol Rev.91: 1071-1121, and Jain (2014) Cancer Cell 26(5): 605-622, the contents of which are incorporated herein by reference, can also be used as an anti-angiogenic agent for the compositions and methods described herein.
In other embodiments, the anti-angiogenic agent is an inhibitor of vascular endothelial growth factor (VEGF) pathway. Examples of anti-angiogenic agents used in combination with the combination therapies comprising the BRUSH-compositions and BASP-compositions are described in WO 2013/169739, e.g., in Paragraph 114, incorporated herein by reference.
In another embodiment, the microenvironment modulator includes an agent that decreases the level or production of hyaluronic acid (HA). Examples of HA agents used in combination with the combination therapies comprising the BRUSH- compositions and BASP-compositions are described in WO 2013/169739, e.g., in Paragraph 115, incorporated herein by reference.
In another embodiment, the microenvironment modulator includes an inhibitor of the hedgehog pathway. Exemplary hedgehog inhibitors include, but are not limited to, IPI-926, GDC-0449, cyclopamine or an analogue thereof, and GANT58.
In another embodiment, the microenvironment modulator includes an agent that improves drug penetration in tumors. In one embodiment, the agent is a disulfide- based cyclic RGD peptide (iRGD) or an analogue thereof (e.g., described in Sugahara, KN et al. (2010) Science 328:1031-5; Ye, Y. et al. (2011) Bioorg Med Chem Lett. 21(4):1146-50).
In yet another embodiment, the microenvironment modulator includes a taxane therapy (e.g. Taxane-induced apoptosis as described in Griffon-Etienne, G. et al. (1999) Cancer Res.59(15):3776-82).
In another embodiment, the microenvironment modulator includes an agent that modulates (e.g., inhibits) a hypoxia inducible factor (HIF), for example, an agent that inhibits hypoxia-inducible factors 1α and 2α (HIF-1α and HIF-2α). HIF activity has been shown to be involved in inflammation (e.g., rheumatoid arthritis) and angiogenesis associated with cancer tumor growth. HIF inhibitors, such as phenethyl isothiocyanate (PEITC) are under investigation for anti-cancer effects (Syed Alwi SS, et al. (2010) Br. J. Nutr.104 (9): 1288–96; Semenza GL (2007). Drug Discov. Today 12 (19-20): 853–9; Melillo G (2006). Mol. Cancer Res.4 (9): 601–5. In one embodiment, the agent is an antibody against an HIF. In another embodiment, the agent is an HIF chemical inhibitor, such as phenethyl isothiocyanate (PEITC).
In another embodiment, the microenvironment modulator includes an agent that decreases the level or production of collagen or procollagen. For example, an agent that degrades collagen, e.g., collagenase.
In one embodiment, the combinations described herein can be further administered in combination with a microenvironment modulator chosen from an anti-fibrotic agent or an inhibitor of a profibrotic pathway (a“profibrotic pathway inhibitor”) (e.g., a pathway dependent- or independent of TGF-beta and/or CTGF activation). In one embodiment, the combinations described herein are administered in combination with one or more of: an inhibitor of endothelin-1, PDGF, Wnt/beta- catenin, IGF-1, TNF-alpha, and/or IL-4. In another embodiment, the combinations described herein are administered in combination with an inhibitor of endothelin-1 and/or PDGF. In other embodiments, the combinations described herein are administered in combination with an inhibitor of one or more of chemokine receptor type 4 (CXCR4) (e.g., AMD3100, MSX-122); stromal-derived-factor-1(SDF-1) (e.g., tannic acid); hedgehog (e.g., IPI-926, GDC-0449, cyclopamine or an analogue thereof, or GANT58).
In certain embodiments, an inhibitor of a CXCR4 receptor and/or its ligand, SDF-1, is administered in combination with a therapy (e.g., a cancer or
hyperproliferative therapy as described herein). Certain embodiments may further include administration of a further AHCM and/or a microenvironment modulator as described herein. Exemplary SDF-1/CXCR4 inhibitors that can be used include, but are not limited to, 2,2'-bicyclam; 6,6'- bicyclam; AMD3100 (IUPAC name: l,l'-[l,4- phenylene-bis(methylene)]-bis-l,4,8,l 1-tetraazacyclotetradecane), as described in e.g., U.S. Pat. Nos.5,021,409, 6,001,826 and 5,583,131; Plerixafor (trade name: Mozobil; IUPAC name: 1,1′-[1,4-Phenylenebis-(methylene)]bis [1,4,8,11- tetraazacyclotetradecane); CXCR4 peptide inhibitors or analogs, e.g., T-140 analogs (e.g., 4F-benzoyl-TN14003, TC14012, TE14011, TC14003), CTCE-0214; CTCE- 9908; the peptide antagonist LY2510924; and CP-1221, as well as other inhibitors such as antibodies against SDF-1 or CXCR4, RNA inhibitors (e.g., antisense, siRNAs), among others. Exemplary inhibitors are described in, for example,
Tamamura, H. et al. Org. Biomol. Chem.1:3656-3662, 2003; FEBS Letter 550:1-3 (2003): 79-83; Wong, D. et al. (2008) Clin. Cancer Res.14(24): 7975-7980; US Patent Publications 2010/0055088; 2009/0221683; 2004/0209921, 2005/0059702, 2005/0043367, 2005/0277670, 2010/0178271, and 2003/0220341; US Patent Nos.
5,021,409, 6,001,826, 5,583,131, and Patent Publications WO 03/011277, WO 01/85196; WO 99/50461; WO 01/94420; WO 03/090512, each of which is incorporated by reference. Immunomodulators
The compositions and methods described herein can comprise an
immunomodulator. In one embodiment, the immunomodulator is an anti- inflammatory agent described herein, e.g., for treating or preventing a disease or disorder, e.g., a cancer or a fibrotic disorder described herein. The composition and method can include one, two, three or more anti-inflammatory agents, alone or in combination with one or more therapeutic agents described herein (e.g., an AHCM agent, a microenvironment modulator, an immune-checkpoint inhibitor, or an additional therapy, e.g., a cancer or anti-fibrotic therapy).
In one embodiment, the anti-inflammatory agent is an agent that blocks, inhibits, or reduces inflammation or signaling from an inflammatory signaling pathway. In one embodiment, the anti-inflammatory agent inhibits or reduces the activity of one or more of any of the following: IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL- 7, IL-8, IL-9, IL-10, IL-12, IL-13, IL-15, IL-18, IL-23, interferons (IFNs), e.g., TNF- α, TNF-β, TNF-RI, TNF-RII; CD23, CD30, CD40L, CXCL-1, EGF, G-CSF, GDNF, PDGF-BB, RANTES/CCL5, IKK, NF-kB, TLR2, TLR3, TLR4, TL5, TLR6, TLR7, TLR8, TLR8, TLR9, and/or any cognate receptors thereof.
In one embodiment, the anti-inflammatory agent is an IL-1 or IL-1 receptor antagonist, such as anakinra (KINIRET®), rilonacept, or canakinumab.
In one embodiment, the anti-inflammatory agent is an IL-6 or IL-6 receptor antagonist, e.g., an anti-IL-6 antibody or an anti-IL-6 receptor antibody, such as tocilizumab (ACTEMRA®), olokizumab, clazakizumab, sarilumab, sirukumab, siltuximab, or ALX-0061.
In one embodiment, the anti-inflammatory agent is a TNF-a antagonist, e.g., an anti-TNFa antibody, such as infliximab (REMICADE®), golimumab
(SIMPONI®), adalimumab (HUMIRA®), certolizumab pegol (CIMZIA®) or etanercept.
In one embodiment, the anti-inflammatory agent is a corticosteroid.
Exemplary corticosteroids include, but are not limited to, cortisone (hydrocortisone, hydrocortisone sodium phosphate, hydrocortisone sodium succinate, ALA-CORT®,
HYDROCORT ACETATE®, hydrocortone phosphate LANACORT®, SOLU- CORTEF®), decadron (dexamethasone, dexamethasone acetate, dexamethasone sodium phosphate, DEXASONE®, DIODEX®, HEXADROL®, MAXIDEX®), methylprednisolone (6-methylprednisolone, methylprednisolone acetate,
methylprednisolone sodium succinate, DURALONE®, MEDRALONE®,
MEDROL®, M-PREDNISOL®, SOLU-MEDROL®), prednisolone (DELTA- CORTEF®, ORAPRED®, PEDIAPRED®, PRELONE®), and prednisone
(DELTASONE®, LIQUID PRED®, METICORTEN®, ORASONE®)), and bisphosphonates (e.g., pamidronate (AREDIA®), and zoledronic acid (ZOMETA®).
In another embodiment, the anti-inflammatory agent is a non-steroidal anti- inflammatory drug (NSAID). Exemplary anti-inflammatory agents (e.g., NSAIDs) include, but are not limited to, aspirin, ibuprofen, naproxen, celecoxib, diclofenac, diflunisal, etodolac, fenoprofen, flurbiprofen, ketoprofen, ketorolac, mefenamic acid, meloxicam, nabumetone, oxaprozin, piroxicam, sulindac, and tolmetin. In an embodiment, the anti-inflammatory agent is an immune selective anti-inflammatory derivative (ImSAID). Immune-Checkpoint Inhibitors
The compositions and methods described herein can comprise an immune- checkpoint inhibitor described herein, e.g., for treating or preventing a disease or disorder, e.g., a cancer, inflammatory or a fibrotic disorder described herein. The compositions and methods can include one, two, three or more immune-checkpoint inhibitors, alone or in combination with one or more therapeutic agents described herein (e.g., an AHCM agent, a microenvironment modulator, an anti-inflammatory agent, or an additional therapy, e.g., a cancer or anti-fibrotic therapy).
Immune checkpoint inhibitors, as described herein, refer to molecules that block, inhibit, or reduce activity of one or more immune checkpoint proteins. The inhibitors may be an antibody, an antigen binding fragment thereof, an
immunoadhesin, a fusion protein, or an oligopeptide. Examples of immune- checkpoint molecules include, but are not limited to, PD-1, PD-L1, PD-L2, CTLA4, B7-H3, B7-H4, HVEM, BTLA, a killer-cell immunoglobulin-like receptor (KIR), LAG3, TIM3, CEACAM-1, CEACAM-3, CEACAM-5, GAL9, VISTA, TIGIT, LAIR1, CD160, 2B4, and A2aR.
In some embodiments, the immune checkpoint inhibitor is a PD-1 inhibitor. Antibodies, antibody fragments, and other inhibitors of PD-1 and its ligands (e.g., PD- L1 or PD-L2) are available in the art and may be used combination with metformin as described herein. Exemplary anti-PD-1 antibodies include, but are not limited to, nivolumab (also known as MDX-1106 or BMS-936558), pembrolizumab (formerly known as lambrolizumab, also known as Merck 3475 or MK03475), and pidilizumab (also known as CT- 011). Nivolumab (clone 5C4) and other human monoclonal antibodies that specifically bind to PD-1 are disclosed in US 8,008,449 and
WO2006/121168. Pidilizumab and other humanized anti-PD-1 monoclonal antibodies are disclosed in WO2009/101611. Pembrolizumab and other humanized anti-PD-1 antibodies are disclosed in US 8,354,509 and WO2009/114335. In some
embodiments, the PD-1 inhibitor is an immunoadhesin (e.g., an immunoadhesin comprising an extracellular or PD-1 binding portion of PD-L1 or PD-L2 fused to a constant region (e.g., an Fc region of an immunoglobulin sequence), such as AMP- 224.
In some embodiments, the immune checkpoint inhibitor is a PD-L1 inhibitor. Antibodies, antibody fragments, and other inhibitors of PD-L1 are available in the art and may be used combination with metformin as described herein. Exemplary anti- PD-L1 antibodies include, but are not limited to, YW243.55.S70 (as described in PCT Publication No. WO2010/077634), MPDL3280A (as described in U.S. Patent No. 7,943,743 and U.S. Publication No.20120039906), MEDI-4736, MSB-0010718C, or MDX-1105 (also referred to as BMS-936559, as described in WO2007/005874).
In some embodiments, the immune checkpoint inhibitor is a TIM3 inhibitor. Antibodies, antibody fragments, and other inhibitors of TIM3 and its ligands are available in the art and may be used combination with metformin as described herein. For example, antibodies, antibody fragments, small molecules, or peptide inhibitors that target TIM3 binds to the IgV domain of TIM3 to inhibit interaction with its ligands can be administered in combination with a metformin agent as described herein. Exemplary TIM3 inhibitors include, but are not limited to the antibodies and peptides disclosed in WO2013/006490 and US20100247521); anti-TIM3 inhibitors such as humanized versions of RMT3-23 (as disclosed in Ngiow et al., 2011, Cancer Res, 71:3540-3551) and clone 8B.2C12 (disclosed in Monney et al., 2002, Nature, 415:536-541). Bi-specific antibodies that inhibit TIM3 and PD-1 are disclosed in US20130156774.
In some embodiments, the immune checkpoint inhibitor is a LAG3 inhibitor. Antibodies, antibody fragments, and other inhibitors of LAG3 and its ligands are available in the art and may be used combination with metformin as described herein. Exemplaryanti-LAG3 antibodies include, but are not limited to monoclonal antibody BMS-986016 (Bristol-Myers Squib), IMP701 (Immutep), IMP731 (Immutep and GlaxoSmithKline), and antibodies disclosed in WO2010/019570. Other LAG3 inhibitors include IMP321 (Immutep), which is a recombinant fusion protein of a soluble portion of LAG3 and Ig that binds to MHC class II molecules and activates antigen presenting cells (APC).
In some embodiments, the immune checkpoint inhibitor is a CEACAM inhibitor, e.g., a CEACAM-1 inhibitor, a CEACAM-3 inhibitor, and/or a CEACAM-5 inhibitor. Antibodies, antibody fragments, and other inhibitors of CEACAM are available in the art and may be used combination with metformin as described herein. Exemplary anti-CEACAM-1 antibodies include, but are not limited to, antibodies described in WO 2010/125571, WO 2013/082366 WO 2014/059251 and WO
2014/022332, e.g., a monoclonal antibody 34B1, 26H7, and 5F4; or a recombinant form thereof, as described in, e.g., US 2004/0047858, US 7,132,255 and WO
99/052552. In other embodiments, the anti-CEACAM antibody binds to CEACAM-5 as described in, e.g., Zheng et al. PLoS One.2010 Sep 2;5(9). pii: e12529
(DOI:10:1371/journal.pone.0021146), or cross reacts with CEACAM-1 and
CEACAM-5 as described in, e.g., WO 2013/054331 and US 2014/0271618. SPECIFIC EMBODIMENTS
1. A conjugate or BASP particle comprising:
(i) an agent, e.g., a therapeutic agent, (e.g., an agent chosen from an angiotension receptor blocker (ARB), a vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor);
(ii) a carbocyclyl or heterocyclyl moiety;
(iii) a triazolyl moiety; and
(iv) a heteroalkyl moiety; and
(v) a cleavable linker, e.g., a tissue microenvironment cleavable linker. 2. The conjugate or BASP particle of embodiment 1, wherein the carbocyclyl or heterocyclyl moiety comprises a bicyclic carbocyclyl or bicyclic heterocyclyl moiety.
3. The conjugate or BASP particle of any one of embodiments 1-2, wherein the triazolyl moiety is 1,2,3-triazoldiyl. 4. The conjugate or BASP particle of any one of embodiments 1-3, wherein the heteroalkyl moiety comprises a polyethylene glycol moiety. 5. The conjugate or BASP particle of any one of embodiments 1-4, wherein the cleavable linker is cleavable by or is sensitive to one or more of an enzyme (e.g., an esterase or a protease), pH (e.g., acidic pH, basic pH), light (e.g., ultraviolet light), nucleophile, reduction, or oxidation. 6. The conjugate or BASP particle of any one of embodiments 1-5, wherein the cleavable linker is a tissue microenvironment cleavable linker, e.g., is cleavable by or is sensitive to an enzyme (e.g., an esterase, a protease) or pH (e.g., acidic pH, basic pH). 7. The conjugate or BASP particle of any one of embodiments 1-6, wherein the cleavable linker comprises an ester, an acetal, a phosphoramidite, a hydrazone, an imine, an oxime, a disulfide, or a silyl moiety. 8. The conjugate or BASP particle of any one of embodiments 1-7, wherein:
(i) the carbocyclyl or heterocyclyl moiety is linked, e.g., directly or indirectly linked, to the triazole moiety;
(ii) the heteroalkyl moiety is linked, e.g., directly or indirectly linked, to the triazolyl moiety; and
(iii) the agent is linked, e.g., directly or indirectly linked, to the conjugate or BASP particle through the cleavable linker (e.g., the tissue microenvironment cleavable linker). 9. The conjugate or BASP particle of any one of embodiments 1-8, wherein the conjugate or BASP particle comprises a structure according to Formula (III) or (III-f) as described herein.
10. The conjugate or BASP particle of any one of embodiments 1-9, wherein Ring C is a structure of Formula (IV) as described herein. 11. The conjugate or BASP particle of any one of embodiments 1-10, wherein the

Ring T is
 , wherein“2” represents a portion of Ring T bound to L2 in the conjugate or BASP particle 13. The conjugate or BASP particle of any one of embodiments 9-12, wherein B is C1-C12 alkylene optionally substituted with 1-6 independently selected R1. 14. The conjugate or BASP particle of embodiment 13, wherein B is hexylene (e.g.,–CH2CH2CH2CH2CH2CH2–). 15. The conjugate or BASP particle of any one of embodiments 9-14, wherein A is C1-C12 heteroalkylene, optionally substituted with 1-6 R1 (e.g., optionally substituted with 1-3 R1). 16. The conjugate or BASP particle of embodiment 15, wherein A is 4- aminobutylene, optionally substituted with 1-3 R1. 17. The conjugate or BASP particle of any one of embodiments 15-16, wherein R1 is oxo or heteroalkyl (e.g., polyethylene glycol),
18. The conjugate or BASP particle of any one of embodiments 9-17, wherein A is–C(O)CH2CH2C(O)NH-PEG. 19. The conjugate or BASP particle of embodiments 18, wherein the PEG comprises a molecular weight of between about 200 and about 6000, g/mol (e.g., PEG200, PEG400, PEG600, PEG800, PEG1000, PEG1500, PEG2000, PEG3000, PEG4000, PEG6000). 20. The conjugate or BASP particle of embodiment 19, wherein the PEG is PEG3000. 21. The conjugate or BASP particle of any one of embodiments 9-20, wherein L3 is cleaved at a first set of conditions relative to a second set of conditions (e.g., a first pH relative to a second pH). 22. The conjugate or BASP particle of any one of embodiments 9-21, wherein L3 is cleavable by an enzyme (e.g., an esterase, a protease), or pH (e.g., acidic pH, basic pH). 23. The conjugate or BASP particle of embodiments 22, wherein the enzyme is a protease chosen from MMP-1, MMP-2, MMP-3, MMP-8, MMP-9, MMP-14, plasmin, PSA, PSMA, CATHEPSIN D, CATHEPSIN K, CATHEPSIN S, ADAM10, ADAM12, ADAMTS, Caspase-1, Caspase-2, Caspase-3, Caspase-4, Caspase-5, Caspase-6, Caspase-7, Caspase-8, Caspase-9, Caspase-10, Caspase-11, Caspase-12, Caspase-13, Caspase-14, or TACE. 24. The conjugate or BASP particle of any one of embodiments 9-23, wherein L3 comprises an ester, an acetal, a phosphoramidite, a hydrazone, an imine, an oxime, a disulfide, or a silyl moiety. 25. The conjugate or BASP particle of any one of embodiments 9-24, wherein L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12 heteroalkylene)-aryl-(C0-C12
heteroalkylene), and each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2. 26. The conjugate or BASP particle of embodiment 25, wherein each R2 is oxo or halo (e.g., fluoro). 27. The conjugate or BASP particle of any one of embodiments 8-21, wherein L3 is–C(O)–,–OC(O)–,–C(O)O–,–C(O)CH2–,–OC(O)CH2–,–C(O)OCH2–,–
, wherein“2” represents a portion of Ring T bound to L2 in the conjugate or BASP particle 13. The conjugate or BASP particle of any one of embodiments 9-12, wherein B is C1-C12 alkylene optionally substituted with 1-6 independently selected R1. 14. The conjugate or BASP particle of embodiment 13, wherein B is hexylene (e.g.,–CH2CH2CH2CH2CH2CH2–). 15. The conjugate or BASP particle of any one of embodiments 9-14, wherein A is C1-C12 heteroalkylene, optionally substituted with 1-6 R1 (e.g., optionally substituted with 1-3 R1). 16. The conjugate or BASP particle of embodiment 15, wherein A is 4- aminobutylene, optionally substituted with 1-3 R1. 17. The conjugate or BASP particle of any one of embodiments 15-16, wherein R1 is oxo or heteroalkyl (e.g., polyethylene glycol),
18. The conjugate or BASP particle of any one of embodiments 9-17, wherein A is–C(O)CH2CH2C(O)NH-PEG. 19. The conjugate or BASP particle of embodiments 18, wherein the PEG comprises a molecular weight of between about 200 and about 6000, g/mol (e.g., PEG200, PEG400, PEG600, PEG800, PEG1000, PEG1500, PEG2000, PEG3000, PEG4000, PEG6000). 20. The conjugate or BASP particle of embodiment 19, wherein the PEG is PEG3000. 21. The conjugate or BASP particle of any one of embodiments 9-20, wherein L3 is cleaved at a first set of conditions relative to a second set of conditions (e.g., a first pH relative to a second pH). 22. The conjugate or BASP particle of any one of embodiments 9-21, wherein L3 is cleavable by an enzyme (e.g., an esterase, a protease), or pH (e.g., acidic pH, basic pH). 23. The conjugate or BASP particle of embodiments 22, wherein the enzyme is a protease chosen from MMP-1, MMP-2, MMP-3, MMP-8, MMP-9, MMP-14, plasmin, PSA, PSMA, CATHEPSIN D, CATHEPSIN K, CATHEPSIN S, ADAM10, ADAM12, ADAMTS, Caspase-1, Caspase-2, Caspase-3, Caspase-4, Caspase-5, Caspase-6, Caspase-7, Caspase-8, Caspase-9, Caspase-10, Caspase-11, Caspase-12, Caspase-13, Caspase-14, or TACE. 24. The conjugate or BASP particle of any one of embodiments 9-23, wherein L3 comprises an ester, an acetal, a phosphoramidite, a hydrazone, an imine, an oxime, a disulfide, or a silyl moiety. 25. The conjugate or BASP particle of any one of embodiments 9-24, wherein L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12 heteroalkylene)-aryl-(C0-C12
heteroalkylene), and each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2. 26. The conjugate or BASP particle of embodiment 25, wherein each R2 is oxo or halo (e.g., fluoro). 27. The conjugate or BASP particle of any one of embodiments 8-21, wherein L3 is–C(O)–,–OC(O)–,–C(O)O–,–C(O)CH2–,–OC(O)CH2–,–C(O)OCH2–,–

37. The conjugate or BASP particle of any one of embodiments 1-33, wherein the agent is a vitamin D analog. 38. The conjugate or BASP particle of embodiment 37, wherein the vitamin D analog comprises paricalcitol, ergocalciferol, elocalcitol, eldecalcitrol, calcidiol, calcipotriol, cholecalciferol, or a metabolite or derivative thereof. 39. The conjugate or BASP particle of any one of embodiments 37-38, wherein the vitamin D analog does not comprise seocalcitol (EB 1089), CB 1093, or lexacalcitol (KH 1060). 40. The conjugate or BASP particle of any one of embodiments 37-39, wherein the vitamin D analog comprises a structure of Formulas (I-vi), (I-vii), or (I-viii) as described herein.
41. The conjugate or BASP particle of any one of embodiments 1-33, wherein the agent is a bromodomain inhibitor. 42. The conjugate or BASP particle of embodiment 41, wherein the bromodomain inhibitor is selected from the group depicted in FIGS.4A to 4B. 43. The conjugate or BASP particle of any one of embodiments 1-33, wherein the agent is an IDO inhibitor. 44. The conjugate or BASP particle of any one of embodiments 9-43, wherein the structure of Formula (III) is a structure of Formula (III-a) as described herein.. 45. The conjugate or BASP particle of any one of embodiments 9-44, wherein the structure of Formula (III) is a structure of Formula (III-b) as described herein. 46. The conjugate or BASP particle of any one of embodiments 9-45, wherein the structure of Formula (III) is a structure of Formula (III-c) as described herein. 47. The conjugate or BASP particle of any one of embodiments 1-46, wherein the average molecular weight of the conjugate or BASP particle is less than about 100 kDa (e.g., less than about 90 kDa, about 80 kDa, about 70 kDa, about 60 kDa, or about 50 kDa). 48. The conjugate or BASP particle of any one of embodiments 1-47, wherein the average molecular weight of the conjugate or BASP particle is between about 30 kDa and about 60 kDa. 49. The conjugate or BASP particle of any one of embodiments 1-48, wherein the hydrodynamic diameter of the conjugate or BASP particle is less than about 20 nm (e.g., less than about 15 nm, about 12.5 nm, about 10 nm, about 9 nm, about 8 nm, about 7 nm, about 6 nm, about 5 nm, or less).
51. The conjugate or BASP particle described in any one of embodiments 1-49 and having a hydrodynamic diameter of less than about 100 nm (e.g., less than about 50 nm). 52. The conjugate or BASP particle of embodiment 51, wherein the hydrodynamic diameter of the particle is less than about 90 nm (e.g., less than about 80 nm, about 75 nm, about 70 nm, about 65 nm, about 60 nm, about 55 nm, about 50 nm, about 45 nm, about 40 nm, about 35 nm, about 25 nm, or less). 53. The conjugate or BASP particle of any one of embodiments 51-52, wherein the hydrodynamic diameter of the particle is between about 5 nm and 50 nm. 54. The conjugate or BASP particle of any one of the preceding embodiments, wherein the total amount of the agent present in the conjugate or BASP particle is greater than about 5% (e.g., about 6%, about 7%, about 8%, about 9%, about 10%, about 12%, about 15%, about 20%, about 25%, about 30%, or more). 55. The conjugate or BASP particle described in any one of embodiments 1-54, wherein the total amount of the agent present in the conjugate or BASP particle is greater than about 10% (e.g., about 12%, about 15%, about 20%, about 25%, about 30%, or more). 56. The conjugate or BASP particle of any one of embodiments 51-55, wherein the dispersity of the particle is between about less that 1.5 (e.g., less than about 1.25, about 1.0, about 0.9, about 0.8, about 0.7, about 0.6, about 0.5, about 0.4, about 0.3, or about 0.2). 57. The particle of any one of embodiments 51-56, wherein the particle is monodisperse. 58. The conjugate or BASP particle of any one of the preceding embodiments, wherein the conjugate or BASP particle is substantially hydrophobic.
59. The conjugate or BASP particle of any one of the preceding embodiments, wherein the conjugate or BASP particle comprises a single agent chosen from an ARB, a vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor. 60. The conjugate or BASP particle of any one of the preceding embodiments, wherein the conjugate or BASP particle comprises at least two agents. 61. The conjugate or BASP particle of claim 60, which comprises a combination of two or more of an ARB, a vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor. 62. The conjugate or BASP particle of claim 60, which comprises a combination of one or more of an ARB, a vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor, and another therapeutic agent. 63. The conjugate or BASP particle of any one of the preceding embodiments, wherein the conjugate or BASP particle further comprises a diagnostic agent. 64. The conjugate or BASP particle of embodiment 63, wherein the diagnostic agent comprises a fluorescent molecule, a metal chelate, a contrast agent, a radionuclide, or a positron emission tomography (PET) imaging agent, an infrared imaging agent, a near-IR imaging agent, a computer assisted tomography (CAT) imaging agent, a photon emission computerized tomography imaging agent, an X-ray imaging agent, or a magnetic resonance imaging (MRI) agent. 65. The conjugate or BASP particle of embodiment 64, wherein the fluorescent molecule comprises an acridine dye, a cyanine dye, a rhodamine dye, a BODIPY dye, a fluorescein dye, a dansyl dye, an Alexa dye, an atto dye, a quantum dot, or a fluorescent protein. 66. The conjugate or BASP particle of embodiment 65, wherein the cyanine dye comprises Cy3, Cy 3.5, Cy5, Cy5.5, Cy7, or Cy7.5.
67. The conjugate or BASP particle of any one of embodiments 1-66, wherein the conjugate or BASP particle further comprises a targeting agent. 68. The conjugate or BASP particle of embodiment 67, wherein the targeting agent comprises a ligand, cell surface receptor, a protein (e.g., glycoprotein or antibody), a sugar, a nucleic acid, a cofactor, a vitamin, an aptamer, a small molecule therapeutic, or a metabolite or derivative thereof. 69. The conjugate or BASP particle of any one of embodiments 67-68, wherein the targeting agent comprises a liver targeting agent. 70. The conjugate or BASP particle of embodiment 69, wherein the conjugate or BASP particle is targeting to a hepatocyte, Kupffer cell, an endothelial cell, a hepatic stellate cell, a bile duct cell, or a hepatocarcinoma cell. 71. A conjugate or BASP particle comprising:
(i) a diagnostic agent (e.g., a cyanine dye, e.g., Cy3, Cy 3.5, Cy5, Cy5.5, Cy7, or Cy7.5) or a targeting agent;
(ii) a carbocyclyl or heterocyclyl moiety;
(iii) a triazolyl moiety; and
(iv) a heteroalkyl moiety; and
(v) a linker (e.g., a cleavable linker, e.g., a tissue microenvironment cleavable linker);
wherein the conjugate or BASP particle may be further described by any one of embodiments 1-63. 72. A conjugate comprising paricalcitol, wherein the conjugate comprises a structure of Formulas (III-d-1), (III-d-2), (III-i-1), or (III-i-2) as described herein. 73. The conjugate of embodiment 72, wherein each R2 is oxo or halo (e.g., fluoro).
74. The conjugate of embodiment 73, wherein L3 is–C(O)–,–C(O)O–,–
C(O)CH2–,C(O)CH2CH2O–,
 . 75. A conjugate comprising telmisartan, wherein the conjugate comprises a structure of Formulas (III-e) or (III-j) as described herein. 76. The conjugate of embodiment 75, wherein L3 is C1-C12 heteroalkylene, or (C0- C12 alkylene)-arylene-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or aryl is optionally substituted with 1-12 independently selected R2. 77. The conjugate of any one of embodiments 75-76, wherein each R2 is oxo or halo (e.g., fluoro). 78. The con u ate of an one of embodiments 75-77 wherein L3 is–CH2CH2O–,
. 75. A conjugate comprising telmisartan, wherein the conjugate comprises a structure of Formulas (III-e) or (III-j) as described herein. 76. The conjugate of embodiment 75, wherein L3 is C1-C12 heteroalkylene, or (C0- C12 alkylene)-arylene-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or aryl is optionally substituted with 1-12 independently selected R2. 77. The conjugate of any one of embodiments 75-76, wherein each R2 is oxo or halo (e.g., fluoro). 78. The con u ate of an one of embodiments 75-77 wherein L3 is–CH2CH2O–,
C(O)CH2CH2

pharmaceutically acceptable carrier. 80. A method of treating or preventing a disorder (e.g., a hyperproliferative disorder, a fibrotic disorder, and/or an inflammatory disorder) in a subject, comprising administering the particle according to any one of embodiments 50-71, e.g., as a single agent or in combination with other agent or therapy or a composition of embodiment 79 to the subject. 81. The method of embodiment 80, wherein the disorder is a cancer, a fibrotic disorder, or a liver disorder.
82. The method of any one of embodiments 80-81, wherein the subject has a pre- neoplastic condition or a pre-disposition to cancer. 83. The method of any one of embodiments 80-82, wherein the subject is at risk of having, or has a solid, fibrotic tumor. 84. The method of any one of embodiments 80-83, wherein the subject has a tumor containing an extracellular matrix component chosen from collagen, procollagen and/or hyaluronan (HA). 85. The method any one of embodiments 80-84, wherein the disorder is a cancer chosen from one or more of pancreatic, breast, colorectal, colon, lung, skin, ovarian, prostate, cervix, gastric, gastrointestinal, stomach, head and neck, kidney, brain cancer, liver cancer, or a metastatic lesion thereof. 86. The method any one of embodiments 80-85, wherein the disorder is a fibrotic condition or disorder of the lung, a fibrotic condition of the liver, a fibrotic condition of the heart or vasculature, a fibrotic condition of the kidney, a fibrotic condition of the skin, a fibrotic condition of the gastrointestinal tract, a fibrotic condition of the bone marrow or a hematopoietic tissue, a fibrotic condition of the nervous system, a fibrotic condition of the eye, or a combination thereof. 87. The method of any one of embodiments 80-86, wherein the particle or conjugate is administered prior to and/or in combination with another agent or therapy to the subject. 88. The method of any one of embodiments 80-87, wherein the other agent or therapy comprises an anti-cancer, an anti-fibrotic, or anti-inflammatory, agent or therapy. 89. The method of embodiment 88, wherein the anti-cancer therapy is chosen from one or more of anti-cancer agents, photodynamic therapy, an immunotherapy (e.g., an immune-cell therapy or adoptive immunotherapy), surgery and/or radiation.
90. The method any one of embodiments 80-89, wherein the other agent or therapy (e.g., the anti-cancer, anti-fibrotic, or anti-inflammatory) is administered as a particle or conjugate described in any one of embodiments 1-79. 91. The method of any one of embodiments 80-90, wherein administration of the particle or conjugate is maintained for a preselected portion of the time the subject receives the other agent or therapy. 92. The method of any one of embodiments 80-90, wherein particle or conjugate is maintained for the entire period in which the other agent or therapy is administered. 93. The method of any one of embodiments 80-92, wherein the particle or conjugate is administered continuously, e.g., substantially continuously, over a period of at least 1, 5, 10, or 24 hours; at least 2, 5, 10, or 14 days; at least 2, 3, 4, 5 or 6 weeks; at least 2, 3, 4, 5 or 6 months; or at least 1, 2, 3, 4 or 5 years. 94. The method of any one of embodiments 80-93, wherein the particle or conjugate is administered after cessation of the other agent or therapy. 95. The method of any one of embodiments 80-94, wherein the particle or conjugate is formulated for oral, subcutaneous, intravenous continuous delivery; or is administered as a sustained release formulation. 96. A method of improving the delivery and/or efficacy of other agent or therapy in a subject, the method comprising:
administering the conjugate or BASP particle according to any of
embodiments 1-79 in combination with the other agent or therapy to the subject under conditions sufficient to treat or prevent the disorder or condition in the subject, or to improve the delivery and/or efficacy of the other agent or therapy provided to the subject. 97. The method of embodiment 96, wherein the other agent or therapy comprises an anti- cancer, an anti-fibrotic, or an anti-inflammatory, agent or therapy.
98. The method of any one of embodiments 8-97, wherein the other agent or therapy is chosen from one or more of:
(i) a cancer therapeutic chosen from a viral cancer therapeutic agent, a lipid nanoparticle of an anti-cancer therapeutic agent, a polymeric nanoparticle of an anti- cancer therapeutic agent, an antibody against a cancer target, a dsRNA agent, an antisense RNA agent, or a chemotherapeutic agent;
(ii) an immunotherapy (e.g., an immune-cell therapy or adoptive
immunotherapy);
(iii) radiation,
(iv) surgery,
(v) a photodynamic therapy; or
(vi) any combination of (i)-(v). 105. The method of any one of embodiments 80-98, wherein other agent or therapy is administered to the subject by a systemic administration chosen from oral, parenteral, subcutaneous, intravenous, rectal, intramuscular, intraperitoneal, intranasal, transdermal, or by inhalation or intracavitary installation. 111. A compound of Formula (I) as described herein. 112. The compound of embodiment 111, wherein Ring C is a structure of Formula (II) as described herein. 113. The compound of embodiment 112, wherein the structure of Formula (II) is


C(O)CH2CH2C(O)NH-PEG. 119. The compound of embodiment 118, wherein the PEG comprises a molecular weight of between about 200 and about 6000, g/mol (e.g., PEG200, PEG400,
PEG600, PEG800, PEG1000, PEG1500, PEG2000, PEG3000, PEG4000, PEG6000, e.g., PEG3000). 120. The compound of any one of embodiments 111-119, wherein L3 is cleaved at a first set of conditions relative to a second set of conditions (e.g., a first pH relative to a second pH). 121. The compound of any one of embodiments 111-120, wherein L3 is cleavable by an enzyme (e.g., an esterase, a protease), or pH (e.g., acidic pH, basic pH). 122. The compound of any one of embodiments 111-121, wherein each R2 is oxo or halo (e.g., fluoro). 123. The compound of any one of embodiments 111-122, wherein L3 is–C(O)–,– OC(O)–,–C(O)O–,–C(O)CH2–,–OC(O)CH2–,–C(O)OCH2–,–CH2CH2O–,

CH2CH2–. 128. The compound of any one of embodiments 111-127, wherein P is a polyethylene glycol (PEG) moiety. 129. The compound of embodiment 128, wherein the PEG moiety comprises diethylene glycol, triethylene glycol, tetraethylene glycol, or pentaethylene glycol. 130. The compound of any one of embodiments 111-129, wherein the agent is an angiotensin receptor blocker (ARB) selected from losartan, candesartan, telmisartan,
valsartan, olmesartan, azilsartan, eprosartan, irbesartan, saralasin, EXP 3174, L158209, or a metabolite or derivative thereof. 131. The compound of any one of embodiments 111-129, wherein the agent is a vitamin D analog selected from paricalcitol, ergocalciferol, elocalcitol, eldecalcitrol, calcidiol, calcipotriol, cholecalciferol, or a metabolite or derivative thereof. 132. The compound of any one of embodiments 111-129, wherein the agent is a bromodomain inhibitor selected from the group depicted in FIGS.2 and 3. 133. The compound of any one of embodiments 111-129, wherein the agent is an IDO inhibitor. 134. The compound of any one of embodiments 111-133, wherein the structure of Formula (I) is a structure of Formula (I-a) as described herein. 135. The compound of any one of embodiments 111-134, wherein the structure of Formula (I) is a structure of Formula (I-b) as described herein. 136. A compound comprising paricalcitol of Formula (I-d-i) or Formula (I-d-ii) as described herein.. 137. A compound comprising telmisartan of Formula (I-e) as described herein. 138. A compound of Formula (VII) as described herein. 139. The compound of embodiment 145, wherein each of Z1 and Z2 is
independently C1-C6 alkylene, or (C0-C6 alkylene)-aryl-(C0-C6 alkylene) (e.g., -CH2-, -CH2CH2CH2-, or phenyl). 140. The compound of any one of embodiments 138-139, wherein each of W1 and W2 is independently C1-C12 alkylene or C1-C12 heteroalkylene, wherein each alkylene and heteroalkylene is optionally substituted with 1-6 R32.
141. The compound of any one of embodiments 138-140, wherein R32 is oxo. 142. The compound of any one of embodiments 138-141, wherein each of W1 and W2 is independently -CH2-, -OC(O)CH2CH2-, -CH2OC(O)CH2CH2-, or–C(O)-. 143. The compound of any one of embodiments 138-142, wherein G is–O– or–S- S–. 144. The compound of embodiment 143, wherein the compound is a compound of Formula (VII-a) as described herein. 145. The compound of any one of embodiments 138-144, wherein the compound is


Formula (V-c)
wherein X is an agent (e.g., an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor); t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive; with a metal catalyst (e.g., a Grubbs’ catalyst); b) mixing the first solution with a second solution comprising a compound of Formula (VII-a) (e.g., a cross-linker):

Formula (VII-a)
wherein each of Z1 and Z2 is independently CH2-, -CH2CH2CH2-, or phenyl; each of W1 and W2 is independently -CH2-, -OC(O)CH2CH2-, -CH2OC(O)CH2CH2-, or– C(O)-; and G is–O– or–S-S– to form a third solution;
(c) allowing the third solution to incubate for a time sufficient to form a conjugate or BASP particle of desired length;
(d) adding a chain-transfer agent (e.g., a chain-transfer agent comprising an olefin); and
(e) purifying the solution,
to thereby make a conjugate or BASP particle. 146. The method of embodiment 145, wherein the catalyst is a ruthenium catalyst.
147. The method of any one of embodiments 145-146, wherein the catalyst is of the formula:

. 148. The method of any one embodiments 145-147, further comprising removing the metal catalyst (e.g., a Grubbs’ catalyst) through addition of a compound comprising an amine, phosphine, or thiol (e.g., N,N-dimethyltryptamine, cysteine, triaminetetraacetate (sodium salt), tris(hydroxymethyl)phosphine, 2- mercaptonicotinic acid, N-acetyl-L-cysteine, imidazole,
diethylphenylazothioformamide, lead tetra-acetate, hydrogen peroxide,
triphenylphosphine oxide, isocyanide salt, di(ethylene glycol) vinyl ether, acetonitrile, or dimethyl sulfoxide). 149. The method of any one of embodiments 145-148, wherein the removing the metal catalyst occurs after step (d). EXAMPLES General Protocol for BASP Synthesis
Scheme 1 depicts a general overview of the synthetic protocol for exemplary BRUSHs and BASPs as described herein. Each step is elaborated in more detail below.

MALDI-TOF analysis: Macromonomer samples were submitted to the Koch Institute Biopolymers and Proteomics Core Facility at MIT for matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry using α-cyano-4- hydroxycinnamic acid (CHCA) as the matrix.
Gel Permeation (GPC) Protocol: An analytical GPC system with a PLgel 10um MiniMIX-B Guard 50x4.6mm guard column (Agilent Technologies) and two Shodex KD-806M columns (Shodex) was used, with a Wyatt Technologies Dawn Heleos II for light scattering and an Optilab t-Rex for refractometry. Data collection and analysis was performed on Wyatt’s Astra software (version 6). The mobile phase was 0.025M LiBr in DMF, filtered and stored in a clean, oven dried filtration apparatus. The GPC column compartment was set to 60⁰C and the column equilibrated in the mobile phase at 1mL/min. Samples were dissolved at 1-3mg/mL in 0.025M LiBr DMF and filtered through a PTFE syringe filter (A Chemtek, Cat# 0503-000003, 13mm, 0.20µm).100µL of polymer sample was injected and run for a 30 min method to ensure all material eluted from the column (15mL column volume x2 columns). Dynamic Light Scattering (DLS) Protocol: DLS particle size measurements were obtained using a Wyatt Technologies Mobius DLS instrument with a ~660 nm laser. Samples of particles were prepared in a solution of either nanopure water or PBS at a concentration of approximately 0.1 mg/mL in a 4 mL vial. Samples were left for at least 1 hr to ensure full dissolution of the material, then filtered through a Nylon filter (for example, a ChemTek IC-Nylon Syringe Filter, 0.45 um, 13 mm diameter, catalog
# = 0503-000074) into a clean polystyrene semi-micro cuvette (1 cm path length, 1.5 mL total filling volume). Wyatt Technologies Dynamics software was used to calculate the average hydrodynamic diameter using the DLS correlation function via a regularization fitting method.
Nuclear Magnetic Resonance (NMR) analysis: 1H NMR and 13C-NMR spectra were recorded on Bruker AVANCE-400 MHz NMR spectrometer, VARIAN Inova-500 MHz NMR spectrometer, or a JEOL 500 MHz NMR spectrometer at the Department of Chemistry Instrumentation Facility (DCIF) at MIT. Azide-containing compounds (5-25 mg) or macromonomers (25-40 mg) were dissolved in a deuterated solvent (~0.5 mL), and the solution added to an NMR tube. For 1H NMR, 16-64 scans were performed for azide-containing compounds and 256-1024 scans for macromonomers. Relaxation delay (d1) was set to 2 seconds. Spectra were analyzed on MestReNova NMR software. Chemical shifts are expressed in parts per million (ppm); splitting patterns are designated as s (singlet), d (doublet), t (triplet), m (multiplet), and br (broad); and coupling constants, J, are reported in Hertz (Hz). Example 1: Synthesis of Telmisartan PEG4-Azide (Compound 1)

To a 20-mL scintillation vial charged with a stirbar, telmisartan (180 mg, 0.350 mmol, 1 eq), EDC-HCl (74 mg, 0.386 mmol, 1.1 eq), and DMAP (8.6 mg, 0.070 mmol, 0.2 eq) were dissolved in anhydrous dichloromethane (~0.05-0.1M) under N2, and stirred until solids were fully dissolved in solution. PEG4-azide (100 mg, 0.456 mmol, 1.3 eq) was added, and the reaction was stirred at room temperature overnight. The solution was loaded onto a silica cartridge (50g) and purified by flash chromatography (0% MeOH/DCM to 5% MeOH/DCM) to afford Compound 1 (telmisartan-PEG4- azide, 182 mg, 73%) as a viscous oil.1H NMR (500 MHz, D2Cl2): δ 7.81 (d, J = 7.2 Hz, 1H), 7.70 (d, J = 7.9 Hz, 1H), 7.54-7.24 (m, 10H), 7.13 (d, J = 8.1 Hz, 2H), 5.47
(s, 2H), 4.14 (t, J=4.8 Hz, 2H), 3.79 (s, 3H), 3.59-3.40 (m, 12H), 3.31 (t, J=5.0 Hz, 2H), 2.93 (t, J= 7.8 Hz, 2H), 2.72 (s, 3H), 1.95-1.87 (m, 2H), 1.07 (t, J=7.4 Hz, 3H). Example 2: Synthesis of Telmisartan Phenylester-PEG3-Azide (Compound 2) Synthesis of Phenylester-PEG3-Azide

To a 20-mL scintillation vial charged with a stirbar, PEG3-azide (400 mg, 1.83 mmol, 2 eq), 4-hydroxybenzoic acid (127 mg, 0.920 mmol, 1 eq), and HBTU (348 mg, 0.918 mmol, 1 eq) were dissolved in anhydrous dimethylformamide (10 mL) under N2, followed by addition of DIPEA (0.080 mL, 0.46 mmol, 0.5 eq). The reaction was stirred at room temperature overnight, then added to a separatory funnel with DCM (100 mL). The DCM layer was washed with 100 mL of 0.1M HCl, water, and brine, then dried over magnesium sulfate. After concentration by rotary evaporation, the organic layer was loaded onto a silica cartridge (50g) and purified by flash
chromatography (0% MeOH/DCM to 5% MeOH/DCM) to afford Phenylester-PEG3- Azide (203 mg, 65%) as a viscous oil.1H-NMR (500 MHz, CDCl3): δ 7.70 (m, 2H), 6.85 (d, J = 8.4 Hz, 2H), 6.68 (s, 1 H), 3.69-3.61 (m, 14H), 3.34 (t, J=4.7 Hz, 2H). Synthesis of Telmisartan-Phenylester-PEG3-Azide (Compound 2)

To a 20-mL scintillation vial charged with a stirbar, telmisartan (304 mg, 0.591 mmol, 2 eq), EDC-HCl (113 mg, 0.591 mmol, 2 eq), and DMAP (7.2 mg, 0.059 mmol, 0.2
eq) were dissolved in anhydrous dichloromethane (10 mL) under N2, and stirred until solids were fully dissolved in solution. Phenylester-PEG3-azide (100 mg, 0.296 mmol, 1 eq) was added, and the reaction was stirred at room temperature overnight. The solution was directly loaded onto a silica cartridge (25g) and purified by flash chromatography (0% MeOH/DCM to 5% MeOH/DCM), then dried on high vacuum overnight to afford Compound 2 (telmisartan-phenylester-PEG3-azide, 230 mg, 93%) as a white solid.1H NMR (500 MHz, CD2Cl2): δ 7.95 (d, J = 7.6 Hz, 1H), 7.90 (br, 1H), 7.67 (d, J = 7.6 Hz, 1H), 7.63-7.21 (m, 14H), 6.70 (d, J = 8.6 Hz, 2H), 5.45 (s, 2H), 3.77 (s, 3H), 3.70-3.59 (m, 14H), 3.32 (t, J=4.9 Hz, 2H), 2.89 (t, J= 7.7 Hz, 2H), 2.73 (s, 3H), 1.94-1.87 (m, 2H), 1.06 (t, J=7.5 Hz, 3H). Example 3: Synthesis of Telmisartan Difluorophenylester-PEG3-Azide
(Compound 3)
Synthesis of Difluorophenylester-PEG3-Azide

To a 40-mL scintillation vial charged with a stirbar, amino-PEG-azide (1.0 g, 4.6 mmol, 2 eq), 2,6-difluoro-4-hydroxybenzoic acid (399 mg, 2.3 mmol, 1 eq), and HBTU (742 mg, 2.0 mmol, 0.87 eq) were dissolved in anhydrous dimethylformamide (20 mL) under N2, followed by the addition of DIPEA (0.205 mL, 1.2 mmol, 0.5 eq). The reaction was stirred at room temperature overnight, then added to a separatory funnel with DCM (100 mL). The DCM layer was washed with 100 mL of 0.1M HCl, water, and brine, then dried over magnesium sulfate. After concentration by rotary evaporation, the organic layer was loaded onto a silica cartridge (50g) and purified by flash chromatography (0% MeOH/DCM to 5% MeOH/DCM) to afford
difluorophenylester-PEG3-azide as a viscous oil.1H NMR (500 MHz, CDCl3): δ 6.87 (s, 1H), 6.34 (d, J = 9.8 Hz, 2H), 3.71-3.59 (m, 14H), 3.34 (t, J=4.8 Hz, 2H).

To a 20-mL scintillation vial charged with a stirbar, telmisartan (206 mg, 0.401 mmol, 1.5 equiv), EDC-HCl (76.8 mg, 0.401 mmol,1.5 eq), and DMAP (6.5 mg, 0.053 mmol, 0.2 eq) were dissolved in anhydrous dichloromethane (10 mL) under N2, and stirred until solids were fully dissolved in solution. Difluorophenylester-PEG3-azide (100 mg, 0.267 mmol, 1 eq) was added, and the reaction was stirred at room temperature overnight. The solution was directly loaded onto a silica cartridge (25g) and purified by flash chromatography (0% MeOH/DCM to 5% MeOH/DCM), then dried on high vacuum overnight to afford Compound 3 (telmisartan- difluorophenylester-PEG3-azide, 170 mg, 73%) as a white solid.1H NMR (500 MHz, CD2Cl2): δ8.24 (br, 1H), 7.93 (d, J = 7.7 Hz, 1H), 7.69 (d, J = 7.9 Hz, 1H), 7.63 (t, J=7.5 Hz, 1H), 7.51-7.19 (m, 11H), 6.32 (d, J = 8.4 Hz, 2H), 5.43 (s, 2H), 3.77 (s, 3H), 3.71-3.54 (m, 14H), 3.32 (t, J=5.1 Hz, 2H), 2.81 (t, J= 7.7 Hz, 2H), 2.71 (s, 3H), 1.94-1.86 (m, 2H), 1.06 (t, J=7.3 Hz). Exam le 4: S nthesis of Paricalcitol-PEG3-Azide (Compound 4)

To a dry vial charged with a stirbar and septum cap was added paricalcitol (300 mg, 0.720 mmol, 1.0 equiv), PEG4-azide (218 mg, 0.930 mmol, 1.3 equiv), EDC (2.00 g, 10.8 mmol, 15 equiv), and DMAP (catalytic, ca.20 mg). The vial was purged with
nitrogen, after which anhydrous dichloromethane (5 mL) was added and the reaction mixture was heated to 40⁰C for 17 h. Upon cooling to room temperature, the crude reaction mixture was loaded directly onto silica gel (2 cm x 9 cm) and purified (1– 2% MeOH/DCM then 5– 6% MeOH/DCM) to afford Compound 4 (paricalcitol- PEG3-azide) as a mixture (~ 1:1.3) of regioisomers (130 mg, 43% based on recovered starting material). MS (ESI): calcd for C35H57N3O7 [M– N2– H2O]+: 585.8, found: 586.4. Example 5: Synthesis of Exemplary PEG-alkynes
Exemplary PEG-alkynes were prepared following the procedure outlined in the

Synthesis of (3αR,4S,7R,7αS)-2-(6-Hydroxyhexyl)-3α,4,7,7α-tetrahydro-1H-4,7- methanoisoindole-1,3(2H)-dione (A1)
A stirred mixture of 6-aminohexanol (6.25 g, 53.3 mmol) and norbornene (8.34 g, 50.7 mmol) in toluene (40 mL) was heated to reflux, and water was removed via a Dean-Stark trap. Full consumption of the starting materials After 6 h, 1H-NMR analysis indicated full consumption of norbornene. The mixture was cooled to room temperature and sat. NaHCO3 (20 mL) was added. The organic layer was separated and washed again with sat. NaHCO3 (20 mL), then dried over Na2SO4 filtered concentrated in vacuo. This furnished 13.22 g (98% yield) 3αR,4S,7R,7αS)-2-(6- hydroxyhexyl)-3α,4,7,7α-tetrahydro-1H-4,7-methanoisoindole-1,3(2H)-dione (A1) as a colorless viscous oil (1H-NMR (CDCl3): 6.29 (2H, s), 3.62 (2H, t, J=6.44 Hz), 3.46
(2H, t, J=7.62 Hz), 3.27 (2H, s), 2.67 (2H, s), 1.44-1.66 (m, 5H), 1.24-1.44 (4H, m), 1.22 (1H, d, J=9.96 Hz)). Synthesis of 6-((3αR,4S,7R,7αS)-1,3-Dioxo-1,3,3α,4,7,7α-hexahydro-2H-4,7- methanoisoindol-2-yl)hexyl methanesulfonate (A2)
A solution of alcohol A1 (13.2 g, 50 mmol) and Et3N (10.5 mL, 7.57 g, 75 mmol) in CH2Cl2 (120 mL) at 0 °C was treated with MsCl (4.45 mL, 6.5 g, 57.5 mmol) over approximately 10 min in a dropwise fashion. During addition a precipitate is formed of Et3N·HCl. After 2.5 h, 1H-NMR showed complete conversion. The mixture was poured in water (200 mL) and the organic layer was separated and washed with sat. NaHCO3 (100 mL), then separated, dried over Na2SO4, and concentrated in vacuo to yield 15.8 g (93% yield) of 6-((3αR,4S,7R,7αS)-1,3-dioxo-1,3,3α,4,7,7α-hexahydro- 2H-4,7-methanoisoindol-2-yl)hexyl methanesulfonate (A2) as a viscous oil which solidifies at -18 °C.1H-NMR (CDCl3): 6.29 (2H, s), 4.20 (2H, t, J=6.45 Hz), 3.45 (2H, t, J=7.62 Hz), 3.27 (2H, s), 3.00 (3H, s), 2.67 (2H, s), 1.66-1.82 (2H, m), 1.50- 1.66 (3H, m), 1.28-1.50 (2H, m), 1.24-1.28 (2H, m), 1.21 (1H, d, J = 9.38 Hz)). Synthesis of (3αR,4S,7R,7αS)-2-(6-(Prop-2-yn-1-ylamino)hexyl)-3α,4,7,7α- tetrahydro-1H-4,7-methanoisoindole-1,3(2H)-dione (A3)
A mixture of A2 (7.26 g, 21.2 mmol) and propargylamine (24.7 g, 449 mmol) were stirred at room temperature for 18 h. CH2Cl2 (100 mL) was added to the orange mixture followed by sat. NaHCO3 (100 mL) and the organic layer was separated, dried over Na2SO4, filtered and concentrated in vacuo. This produced 6.6 g of (3αR,4S,7R,7αS)-2-(6-(prop-2-yn-1-ylamino)hexyl)-3α,4,7,7α-tetrahydro-1H-4,7- methanoisoindole-1,3(2H)-dione (A3) as an orange brown oil.1H-NMR (CDCl3): 6.27 (2H, d, J = 1.76 Hz), 3.43 (2H, t, J = 7.62 Hz), 3.41 (2H, d, J = 2.34 Hz), 3.27 (2H, s), 2.67 (2H, t, J = 6.74), 2.66 (2H, s), 2.20 (1H, t, J = 2.34 Hz), 1.40-1.60 (7H, m), 1.15-1.40 (2H, m), 1.22 (1H, d, J = 9.38 Hz)). The material was used as such in the next reaction step. Synthesis of 4-((6-((3αR,4S,7R,7αS)-1,3-dioxo-1,3,3α,4,7,7α-hexahydro-2H-4,7- methanoisoindol-2-yl)(prop-2-yn-1-yl)amino)-4-oxobutanoic acid (A4).
To a stirred solution of A3 (6.6 g, 21.2 mmol) in CH2Cl2 (50 mL) was added succinic anhydride (2.76 g, 27.6 mmol). After 2.5 h, 1H-NMR confirmed full conversion. After
18 h, the mixture washed with 7% KHSO4 (aq., 20 mL), and the organic layer was concentrated to 20 mL and a solution of K2CO3 (2.93 g) in water (50 mL) was added carefully (foaming). At first a clear solution is obtained but upon standing
precipitation occurs. EtOH (10 mL) was added followed by toluene (10 mL) to promote phase separation. The phases were separated and the organic phase was extracted with 10% K2CO3 (aq., 10 mL). The combined aqueous extracts were acidified by KHSO4, then extracted with CH2Cl2/toluene. The organic phase was dried over Na2SO4 and concentrated in vacuo to afford 7.7 g 4-((6-((3αR,4S,7R,7αS)- 1,3-dioxo-1,3,3α,4,7,7α-hexahydro-2H-4,7-methanoisoindol-2-yl)(prop-2-yn-1- yl)amino)-4-oxobutanoic acid (A4) (83%). (1H-NMR (CDCl3): 6.28 (2H, d, J = 1.17 Hz), 4.21/4.04 (ratio 1.2:1) (2H, 2d, both with J = 2.34 Hz), 3.38-3.52 (4H, m), 3.27 (2H, s), 2.60-2.80 (6H, m), 2.32/2.21 (1H, 2t, both with J = 2.34 Hz), 1.42-1.66 (5H, m), 1.24-1.42 (4H, m), 1.21 (1H, d, J = 9.38 Hz)). The remaining two steps were carried out according to the protocols described in Johnson, J et al Macromolecules (2010) 43:10326-10335, which is incorporated herein by reference in its entirety. Synthesis of 2,5-dioxopyrrolidin-1-yl 4-((6-((3aR,4S,7R,7aS)-1,3-dioxo-1,3,3a,4,7,7a- hexahydro-2H-4,7-methanoisoindol-2-yl)hexyl)(prop-2-yn-1-yl)amino)-4- oxobutanoate (A5)
Dichloromethane was added to a flask containing EDCI (1.5 equiv), N- hydroxysuccinimide (1.5 equiv), DMAP (0.1 equiv), and A4 (1 equiv). The resulting solution was stirred under argon at room temperature for 20 h, after which the mixture was transferred to a silica gel column. Elution with 70% EtOAc/hexanes gave the desired product. Synthesis of PEG alkynes (Compound 5 and Compound 6)
O-(2-aminoethyl)polyethylene glycol (0.9 equiv, where z = 45 or 68) and the NHS ester (1 equiv) were dissolved in anhydrous DMF and the resulting solution was stirred at room temperature for 4 h. The reaction mixture was added dropwise to Et2O to precipitate the desired product, which was collected by centrifugation and decanting of the ether before redissolving in CH2Cl2. This last step was repeated five
times, after which the precipitate was dried under vacuum to afford the desired PEG- alkynes as a white powder. Exam le 6: S nthesis of Exem lar Macromonomers

Scheme 2: Overview of macromonomer synthesis
The general protocol outlined in Scheme 2 was applied to the synthesis of exemplary macromonomers described herein, wherein X-L3-PEG-N3 refers to the azide starting materials prepared as exemplified above, e.g., in Examples 1-4 and the alkynes were prepared, e.g., as described in Example 5. Macromonomers comprising a cyanine dye were also prepared using a cyanine dye-azide as the starting material. To a dry 3 dram vial charged with a stirbar and septum cap was added an azide as outlined in Table 1 (0.035 mmol, 1.05 equiv), followed by a PEG-alkyne (0.034 mmol, 1.0 equiv). Two PEG alkynes were used in this step; either Compound 5 (z=68, PEG3000, 110 mg) or Compound 6 (z=45, PEG2000, 77.2mg). Copper(I) acetate (17 mg, 0.14 mmol, 4.0 equiv) and anhydrous dichloromethane (2 mL) were then added under nitrogen, and the mixture was stirred under nitrogen for 1 h, after which the crude reaction mixture was concentrated under vacuum. Purification by reverse-phase preparative HPLC (10– 90% MeCN/0.1% aq AcOH) afforded a white residue that was re-dissolved in dichloromethane and dried over sodium sulfate. The residue was precipitated in cold Et2O to provide the macromonomer (MM) as a white solid.
Table 1: Summary and characterization of macromonomers (MM)


In a nitrogen-filled glovebox, Compound 10 (101.1 mg, 0.0251 mmol, 9.9 equiv) was dissolved in anhydrous 1,4-dioxane (136.2 µL) in a 0.5 dram vial. Compound 14 (2.3 mg) was dissolved in anhydrous 1,4-dioxane (55.3 µL), and 25.4 µL of this stock
solution was added to the solution of Compound 10 to provide 0.1 equiv of
Compound 14 (total equiv of all MM = 10). A solution of Grubbs III (92.1 µL, 1 equiv, 0.02 g/mL in 1,4-dioxane) was added and the resulting mixture was allowed to stir for 15 min. (3aR,4R,7S,7aS)-2-(4-(1-(2-(4-((3aR,4R,7S,7aS)-1,3-dioxo- 1,3,3a,4,7,7a-hexahydro-2H-4,7-methanoisoindol-2- yl)phenoxy)ethoxy)ethoxy)phenyl)-3a,4,7,7a-tetrahydro-1H-4,7-methanoisoindole- 1,3(2H)-dione (Compound 16, acetal cross-linker) was added slowly dropwise (ca.1 drop every 10 sec) in anhydrous 1,4-dioxane (253.6 µL, 10 equiv, 0.1 M). The vial was then sealed and the polymerization was allowed to proceed for ca.1 h before quenching with a drop of ethyl vinyl ether. The crude mixture was diluted with ca.1 mL of ultrapure water and dialyzed against 2 L of ultrapure water for 2-3 hours using a 4kDa MWCO; this step was repeated 3 times. The dialyzed material was filtered through a 0.45 µm syringe filter into a clean and dry 3 dram and lyophilized 60-72 hours to afford an off-white powder, which was characterized by GPC (FIG.5A) and DLS (FIG.5B). The characterization data is summarized in Table 2 below. Example 8: Synthesis of Paracalcitol-PEG3k / Cy7.5-PEG3k BASP (17, m = 10, n = 9):
In a nitrogen-filled glovebox, Compound 7 (41.8 mg, 0.0104 mmol, 9.9 equiv) was dissolved in anhydrous 1,4-dioxane (45 µL) in a 0.5 dram vial. Compound 14 (2.1 mg) was dissolved in anhydrous 1,4-dioxane (100 µL) and 21 µL of this stock solution was added to the solution of Compound 7 to provide 0.1 equiv of Compound 14 (total equiv of all MM = 10). A solution of Grubbs III (38 µL, 1 equiv, 0.02 g/mL in 1,4-dioxane) was added and the resulting mixture was allowed to stir for 15 min. Compound 16 (acetal cross-linker) was added slowly dropwise (ca.1 drop every 10 sec) in anhydrous 1,4-dioxane (94.4 µL, 9 equiv, 0.1 M). The vial was then sealed and the polymerization was allowed to proceed for ca. 1 h before quenching with a drop of ethyl vinyl ether. The crude mixture was diluted with ca.1 mL of ultrapure water and dialyzed against 2 L of ultrapure water for 2-3 hours using a 4kDa MWCO; this step was repeated 3 times. The dialyzed material was filtered through a 0.45 µm syringe filter into a clean and dry 3 dram and lyophilized 60-72 hours to afford an off-white powder, which was characterized by GPC (FIG.6A) and DLS (FIG.6B). The characterization data is summarized in Table 2 below.
Additional BASP particles and BRUSH conjugates were prepared following the procedure outlined in Examples 7, 8 and 23; these particles and characterization thereof is summarized in Table 2. Table 2: Synthesis of exemplary BASP particles and BRUSH conjugates and the characterization thereof. The stoichiometry parameters m and n refer to the molar ratio of total MM to Grubbs 3rd catalyst and crosslinker to Grubbs 3rd catalyst, respectively.

**No DLS size available due to interference of Cy5 with DLS measurement.
“N/A” = Not measured.
† - Compounds 28 and 29 are BRUSH conjugates and therefore n value is not applicable. †† - First value is prior to lyophilization; second value is following lyophilization.
††† - Value obtained from lyophilized compound Example 9: Biodistribution and pharmacokinetic profile of Tel-BASP (15) in mice
The purpose of this study was to evaluate the biodistribution and PK profile of Tel-BASP (15) in mice following administration of a single intravenous dose. The study was conducted in tumor bearing animals to explore tumor drug-uptake, release kinetics and tissue biodistribution of BASP particles. The Tel-BASP formulation was compared directly to generic, non-conjugated telmisartan and to telmisartan entrapped in PEG-PLGA. All formulations were dosed at equivalent concentrations of the active pharmaceutical ingredient (telmisartan; 10 mg/kg). In addition to bioanalytical measurements of both free and polymer-bound drug in biological samples (tumor and whole blood), non-invasive imaging of the fluorophore-conjugated BASP particle (i.e. contains 1% Cy7.5 and 1% Cy5) was used to visualize the whole-body drug biodistribution in intact animals (IVIS imaging; Cy7.5) and in subsequent tissue sections (histology; Cy5 and Cy7.5).
The lyophilized Tel-BASP (15) (30.6 mg @ 9.8% Telmisartan loading), corresponding to 3 mg of Telmisartan, was dissolved in 3 mL of sterile PBS at room temperature by inverting every 5 minutes for 30 min. The resulting solution was sterile filtered through a 0.2 µm filter before dosing.
The mammary fat pads of Balb/C female mice (6-8 weeks old; Taconic) were implanted with syngeneic 4T1 mouse tumor cells (300,000 cells/per animal) obtained from ATCC. Tumors were allowed to develop for 10 days, reaching an average size of 150-450 mm3 (Caliper measurements), before the mice were randomized into four dose groups (Table 3). Following a single administration of the test agents (10 mg/kg) or vehicle alone, whole blood, tumor, and liver tissues were collected at 2, 24, 72 and 144 hrs post dose. In addition, intermediate submandibular bleeds (~100 µL whole blood) were collected at 5 min, 30 min, 4 hrs, and 6 hrs as indicated in the footnotes to Table 3. At the time of sacrifice, each tumor and liver sample was split into two equal parts for PK analysis (flash frozen) and immunohistochemistry (fixed in 10% neutral buffered formalin), respectively. Whole blood was collected directly into EDTA tubes and frozen at -30C.
For PK analysis of whole blood or plasma, each sample was prepared twice, once with hydrolysis and once without. The sample analyzed without hydrolysis
measured released Telmisartan (“Tel”; amount released in vivo). The samples analyzed with hydrolysis measured the total Tel in the samples (sum of both released and conjugated Tel). The released TEL was subtracted from the total Tel to yield the amount of conjugated Tel. For both procedures, 27 µL of blood or plasma was spiked with 3 µL of internal standard (13CD3-Telmisartan) (0.5 µg/mL for released TEL and 100 µg/mL for total TEL).
For non-hydrolyzed sample preparation, 120 µL of ice cold acetonitrile was added to the samples. Vials were vortexed and then spun down at 16,000 rpm at 4 °C for 15 minutes in the centrifuge, and the supernatant was removed and injected (2 µL) into a UPLC/MS. For hydrolyzed sample preparation, 270 µL of 4 M potassium hydroxide in methanol was added to the samples. Vials were vortexed and spun down at 16,000 rpm at 4 °C for 35 minutes in the centrifuge, and the supernatant was removed. After an incubation period of 21 hours, a 10 µL aliquot was removed from each sample and diluted ninety times with methanol. The solution was vortexed, syringe filtered into UPLC vials, and injected (2 µL) into the UPLC/MS.
For PK analysis of tissues, we first combined 50-100 mg of the tissue with 5- fold Dulbecco’s Phosphate Buffered Saline (“DPBS”) and two 3.5 mm UFO stainless steel beads. The sample was then homogenized twice for two minutes at speed 16 in the Next Advance Bullet Blender Gold. The homogenate was left for 10 minutes to settle and then two 90 µL aliquots were transferred to Eppendorf vials– one for released Tel and one for total Tel prior to UPLC/MS analysis.
Five µL of telmisartan (DMSO for non-standards) and 5 µL of internal standard (13CD3-Telmisartan) (3 µg/mL for released Tel and 60 µg/mL for total Tel) was added. For released Tel (non-hydrolyzed) sample preparation, 200 µL of ice cold acetonitrile was added to the sample. Vials were vortexed and then spun down at 16,000 rpm at 4 °C for 15 minutes in the centrifuge, and the supernatant was removed, syringe filtered, and injected (2 µL) into the UPLC/MS.
For total Tel (hydrolyzed) sample preparation, to the 90 µL of tissue homogenate, 200 µL of 4 M KOH in methanol was added. Vials were vortexed and spun down at 16,000 rpm at 4 °C for 35 minutes in the centrifuge, and the supernatant was removed. After an incubation period of 21 hours, a 50 µL aliquot was removed from each sample and diluted ten times with methanol. This solution was vortexed, syringe filtered into UPLC vials, and injected into the UPLC/MS. For both released Tel and Total Tel analysis, samples were diluted 16.7 times. Therefore, any
concentrations detected by the instrument need to be multiplied by the dilution factor of 16.7. Table 3: Summary of study groups used in Example 9. All in vivo doses are e uivalent in terms of amount of telmisartan

blood)
^Intermediate bleed collected at 30 min post dose in EDTA tubes (~100 µL whole blood)
#Intermediate bleed collected at 4 hr post dose in EDTA tubes (~100 µL whole blood) %Intermediate bleed collected at 6 hr post dose in EDTA tubes (~100 µL whole blood) The results of the single-dose PK study are shown in FIGS.7A to 7B. The results indicate a time-dependent, accumulation of Tel-BASP (15) in the tumor tissue with continued drug loading during the 6 days post administration. The accumulated levels of bound telmisartan correspond to 10.6% of the injected dose per gram of tumor (24 hs post dose) to 26% of the injected dose per gram of tumor (144 hrs post dose). Moreover, the Tel-BASP (15) produces lower free telmisartan blood levels than non-conjugated telmisartan for up to 72 hours, while generating much higher tumor telmisartan levels than non-conjugated telmisartan at 72 hours and beyond. In addition, tumor levels of telmisartan still bound to Tel-BASP (15) remain very high even after 144 hours, suggesting that free telmisartan will continue to be released in the tumor for an extended period of time.
In addition to the above studies, optical in vivo imaging in mice was used to visualize the biodistribution of Tel-BASP (15) by tracking the conjugate Cy7.5 contained therein (FIGS.8A to 8F). Optical imaging of mice was performed using
IVIS Spectrum optical imaging systems and a 200 CG filter pair. Although there is not a linear relationship between the apparent signal detected at the surface of an experimental animal and that emanating from an internal organ within the animal, the current results confirmed preferential and effective Cy7.5-conjugated polymer loading in tumor tissue (FIGS.8A to 8C) compared to naïve, non-tumor bearing animals (FIGS.8D-8F).
To further visualize and quantify the BASP particle uptake in tumor tissue, 5 um tissue sections were generated and subjected to H&E stain and imaged for Cy5 content (FIGS.9A to 9B). Again, a robust time dependent tumor accumulation of Tel- BASP (15) was evident from image quantification of the Cy5 signal (FIG.10A) and the kinetics were consistent with the LC/MS/MS based measurements of bound Telmisartan (FIG.10B) in the same tumor samples.
Finally, to evaluate the biodistribution of Tel-BASP (15) among major organs (FIGS.11A to 11C), the spleen, kidney, lung, heart, skin, liver, brain, gut and tumor from animals sacrificed on day 6 were imaged on the Licor Odyssey platform. By comparing the Cy7.5 signal (FIG.11B, 800 nm), originating from the Cy7.5 conjugated BASP particles, to that of tissue auto fluorescence (FIG.11A, 700 nM), this bioanalysis highlighted preferential delivery of Tel-BASP (15) to tumor, spleen and liver as further visualized by the heat map (FIG.11C). Altogether, three independent bioanalytical methods confirm significant Tel-BASP drug uptake and retention in the tumor tissue up to 1 week post dosing. Despite the high drug uptake of Tel-BASP (15), the fractional drug release of free telmisartan is low suggesting the need to look at later timepoints or to engineer more labile drug linkers.
Our results demonstrate favorable BASP-particle biodistribution to disease tumor tissue based on both classical LC/MS/MS PK analysis as well as fluorophore- based imaging of the nanoparticles. We contribute these effects to the enhanced permeability and retention (EPR) effect of the size-optimized BASP particles. Example 10: Biodistribution and pharmacokinetic profile of Tel-BASP (15) in Mice
The purpose of this single-dose PK study was to (i) explore two different PEG formulations of Tel-BASP (15) and Tel-BASP (18) and to (ii) capture PK time points beyond one week of dosing looking at potential delayed release kinetics of telmisartan from BASP particles in vivo. The study was conducted in 4T1 tumor bearing animals
and both formulations were dosed at equivalent concentrations of the active pharmaceutical ingredient (Telmisartan; 10 mg/kg). All samples were subjected to bioanalytical measurements of both free and polymer-bound drug focusing on tumor, liver and whole blood. Imaging of fluorophore-conjugated BASP particle (1% Cy7.5) was used to visualize the drug uptake complementing the classical LC/MS/MS PK analysis.
In this example, two different formulations of telmisartan were prepared and studied in vivo (Table 4). The lyophilized Tel-BASP (15) and Tel-BASP (18) were dissolved in sterile PBS at room temperature by inverting every 5 minutes for 30 min to make a 1 mg/ml dosing solution. The resulting solution was sterile filtered through a 0.2 µm filter before dosing 200 µL per 20 gram mouse.
The mammary fat pads of Balb/C female mice (6-8 weeks old; Taconic) were implanted with syngeneic 4T1 mouse tumor cells (150,000 cells/per animal) obtained from ATCC. Tumors were allowed to develop for 10 days, reaching an average size of 150-450 mm3 (Caliper measurements), before the mice were randomized into four dose groups (Table 4). Following a single administration of the test agents (10 mg/kg) or vehicle alone, whole blood, tumor, and liver tissues were collected at 2, 24, 72 and 144 hrs post dose. In addition, intermediate submandibular bleeds (~100 µL whole blood) were collected at 5 min, 30 min, 4 hrs, and 6 hrs as indicated in the footnotes to Table 4. At the time of sacrifice, each tumor and liver sample was split into two equal parts for PK analysis (flash frozen) and immunohistochemistry (fixed in 10% neutral buffered formalin), respectively. Whole blood was collected directly into EDTA tubes and frozen at -30C. Table 4: Summary of study groups used in Example 10. All in vivo doses are e uivalent in terms of amount of BASP

^Intermediate bleed collected at 30 min post dose in EDTA tubes (~100 µL whole blood)
#Intermediate bleed collected at 24 hr post dose in EDTA tubes (~100 µL whole blood)
%Mice moved to IVIS imaging facility after dosing for non-invasive Cy7.5 imaging (no intermediate bleeds collected)
The results of the single-dose PK study are shown in FIGS.12A to 12B. The results indicate a time-dependent, accumulation of Tel-BASP in the tumor tissue with continued drug loading/presence for 21 days post i.v. administration of a single dose. The accumulated levels of bound telmisartan corresponds to 23% of the injected dose per gram of tumor on week one compared to 17% as described on day 6 of the assay described in Example 8. Again, both Tel-BASP (15) and Tel-BASP (18) produce sustained tumor telmisartan levels that cannot be achieved with a single dose of non- conjugated telmisartan which has a terminal half-life in blood of ~24 hours. In addition, tumor levels of telmisartan still bound to Tel-BASP (15) remain very high even after 3 weeks, suggesting that free telmisartan will continue to be released in the tumor for an extended period of time.
To further visualize the BASP particle uptake in tumor tissue, formalin foxed tissues were imaged on the Licor Odyssey platform (FIG.13). The Cy7.5 signal (800 nm), originating from the Cy7.5 conjugated BASP particles, were evident in all tumor tissues collected post dosing consistent with the long tissue half-life observed by LC/MS/MS (FIG.12A).
Our results demonstrate favorable BASP-particle biodistribution to disease tumor tissue (23% of injected dose) using both forms of PEG. In principal, using lower MW PEG may increase the theoretical drug loading on a per weight basis and potentially afford a different ADME profile as faster clearance of PEGs may be observed for lower MW versions (or other approached to stealth coating of particles). In this study, we did not observe a difference between the 2K and 3K PEGs in terms of in tumor, liver and blood PK profile. Example 11: Comparison of the pharmacokinetic profile of various Tel-BASP polymer constructs in mice
The purpose of this single-dose PK study was to evaluate the in vivo drug release for a series of rationally designed drug polymer linkers focusing on increasing the in vivo drug release kinetics (i.e. the lability of the linker and fractional release). The present PK study was conducted in 4T1 tumor bearing animals and both formulations were dosed at equivalent concentrations of the active pharmaceutical ingredient (Telmisartan; 20 mg/kg). All samples were subjected to bioanalytical measurements of both free and polymer-bound drug focusing on tumor, liver and
whole blood.
In this example, three different BASP comprising telmisartan were compared. The lyophilized Tel-BASP (19) and Tel-BASP (20) were dissolved in sterile PBS at room temperature by inverting every 5 minutes for 30 min to make a 20 mg/ml dosing solution of the polymers. The resulting solution was sterile filtered through a 0.2 µm filter before dosing 200 µL per 20 gram mouse (200 mg/kg polymer @ 10% drug loading).
The mammary fat pads of Balb/C female mice (6-8 weeks old; Taconic) were implanted with syngeneic 4T1 mouse tumor cells (150,000 cells/per animal) obtained from ATCC. Tumors were allowed to develop for 10 days, reaching an average size of 150-450 mm3 (Caliper measurements), before the mice were randomized into three dose groups (Table 5). Following a single administration of the test agents (200 mg/kg) or saline vehicle alone, whole blood, tumor, and liver tissues were collected at 24, 72 and 144 hrs and at week 2 and week 3 post dose. In addition, intermediate submandibular bleeds (~100 µL whole blood) were collected at 5 min, 30 min, 2 hrs, and 6 hrs (n=3 animals per timepoint) following the i.v. dosing. At the time of sacrifice, each tumor and liver sample were split into two equal parts for PK analysis (flash frozen) and immunohistochemistry (fixed in 10% neutral buffered formalin), respectively. Whole blood was collected directly into EDTA tubes and frozen at -30C. Table 5: Summary of study groups used in Example 11. All in vivo doses are equivalent in terms of amount of BASP)

Importantly, this tumor tissue exposure to telmisartan was achieved concomitant with the steady-state blood levels of released free telmisartan being ~100-200-fold below drug-induced blood pressure lowering levels reported in the literature to be ~200-300 ng/ml. With the current formulations, we observe an initial 30 minutes transient peak of free drug released from likely surface-accessible linkers and macromonomers (FIG.14A). However, this fractional release is very low and the drug appears to rapidly distribute into tissue (consistent with the high vol of distribution for telmisartan), hence quickly reducing the effective concentration in the blood to below the blood pressure lowering dose of the angiotensin receptor inhibitor. In preclinical models, telmisartan has shown anti-fibrotic activity in liver disease (i.e. NASH/Cirrhosis). Hence, for the two formulations, we also determined the telmisartan drug concentrations in liver tissue collected during the in vivo single dose PK study (FIG.15A to 15D). Notably, liver levels of released telmisartan are only ~2-fold less for Tel-BASP (19) than for Tel-BASP (20), despite ~5-10-fold lower plasma levels of released telmisartan from Tel-BASP (19) than Tel-BASP (20). In summary, comparing whole blood concentration and tumor concentration of released telmisartan with generic therapeutic levels suggest an opportunity to increase dosage ~ 20-fold of Tel-BASP (19) and ~5 fold of Tel-BASP (20) without eliciting blood pressure lowering effects. Example 12: Pharmacokinetic profile of Pari-BASP (17) in mice and evaluation of biomarker modulation.
Pharmacokinetic and dose range finding studies were carried out to evaluate the in vivo drug release of paricalcitol from the Pari-BASP (17). The PK studies were conducted in 4T1 tumor bearing animals and the Pari-BASP (17) formulation was dosed at API concentrations ranging from 1X to 50X higher than the MTD for generic, non-conjugated paricalcitol. Biological samples (tumor, liver and whole blood) were subjected to bioanalytical LC/MS/MS-based measurements of both free
and polymer-bound drug. In addition tumor tissues were imaged for BASP particle uptake (LiCor imaging of Cy7.5 conjugated particles) and for evidence of drug release (i.e. tissue biomarker modulation) to confirm drug release and target engagement (VDR upregulation) in the tumor disease tissue.
In this example, the Pari-BASP (17) formulation of paricalcitol was as described previously. The lyophilized Pari-BASP (17) was dissolved in sterile PBS at room temperature by inverting every 5 minutes for 30 min to make a 500 mcg/ml dosing solution of the polymer. The resulting solution was sterile filtered through a 0.2 µm filter before dosing 200 µL per 20 gram mouse (5000 mcg/kg). This single dose is a 50X higher concentration of the API compared to the MTD of generic non- conjugated paricalcitol.
The mammary fat pads of Balb/C female mice (6-8 weeks old; Taconic) were implanted with syngeneic 4T1 mouse tumor cells (150,000 cells/per animal) obtained from ATCC. Tumors were allowed to develop for 10 days, reaching an average size of 150-450 mm3 (Caliper measurements), before the mice were randomized into two groups: vehicle control (n=6) and Pari-BASP (17) (n=15). Following a single administration of Pari-BASP (17) (5000 mcg/kg) whole blood, tumor, and liver tissues were collected at 24, 72 and 144 hrs post dose. In addition, intermediate submandibular bleeds (~100 µL whole blood) were collected at 5 min, 30 min, 2 hrs, and 6 hrs (n=3 animals per time point) following the i.v. dosing. At the time of sacrifice, each tumor and liver sample were split into two equal parts for PK analysis (flash frozen) and immunohistochemistry (fixed in 10% neutral buffered formalin), respectively. Whole blood was collected directly into EDTA tubes and frozen at -30C. PK analysis of released and total paricalcitol was performed as described for released and total telmisartan in Example 9.
FIGS.16A to 16B show the results of the single-dose PK study for whole blood, tumor and liver tissue. Upon i.v. dosing of Pari-BASP (17), the polymer-bound fraction of paricalcitol steadily increases in the tumor with the compound distributing away from the blood compartment. Over time the accumulation and retention of Pari- BASP (17) in the target tumor tissue reaches >10% of the injected dose. For all samples, the measured release of free paricalcitol from Pari-BASP (17) was below the level of detection of the LC/MS/MS assay. Likewise, the PK assay was not sensitive enough to measure paricalcitol dosed at 10 µg/kg.
Since paricalcitol is a highly potent compound and hence dosed at very low
concentrations in vivo (mcg/kg vs mg/kg) bioanalytical method developments have been challenging. Therefore, we next wanted to evaluate the microenvironmentally- activated release of paricalcitol from Pari-BASP (17) looking for modulation of drug target engagement biomarkers in the tumor tissue. First, using an in vitro model system of human stellate cells treated with paricalcitol (100 nm), we validated the upregulation of VDR protein expression in response to ligand binding (FIG.17A). This VDR biomarker readout was then applied in a quantitative
immunohistochemistry-based manner to quantify the upregulation of VDR in tumor tissue in response to in vivo drug treatment. Indeed, consistent with drug release from other BASP-delivery systems, we saw robust VDR upregulation in the tumor tissue (FIG.17B) supporting the release of biological active paricalcitol in the disease tissue. Example 13: Dosage and biodistribution studies of Pari-BASP (17) and Pari- BASP (25) in mice.
In Example 12, when Pari-BASP (17) was dosed at 50X, we noted significant >10% bodyweight loss at day 6 (including one death) indicating that the 5000 µg/kg dose was above the MTD for Balb/c mice. Hence, to determine the maximum tolerated dose (MTD) of the Pari-BASP (17) formulation relative to the clinical formulation of paricalcitol (Zemplar 5 mcg/kg), we next conducted dose range finding studies in 4T1 tumor bearing mice (FIG.18A) as well as in naive non-tumor bearing mice (FIG.18B). In 4T1 mice, a single dose of Pari-BASP (17) was tolerated at 1X (100 µg/kg), 5X (500 µg/kg) and 15X (1500 µg/kg) of polymer relative to the 1X of non-conjugated, generic paricalcitol (10 µg/kg). The latter is the well-established MTD for Zemplar in mice. Importantly, imaging of the Cy7.5-conjugated Pari-BASP particles in the tumor tissues confirmed dose-dependent tumor uptake of BASP Nanoparticles (FIG.18A) supporting the higher dose level of Pari-BASP (17).
Given the long half-life of the Pari-BASP (17) formulation in liver and tumor (FIGS.16A to 16B) we also explored repeat weekly i.v. dosing of Pari-BASP (25) in non-disease animals dosed at 500 mcg/kg (5X) and 1000 mcg/kg (10X) relative to the 1X dose of paricalcitol (10 mcg/kg). Again, bodyweight measurements over a three week period (total of 3 weekly i.v. doses of Pari-BASP (25)) confirmed that the Pari- BASP (25) formulation was tolerated and can be dosed above the MTD of generic Paricalcitol (10 µg/kg i.v.; three times weekly).
Altogether, thee MTD studies demonstrate that we have low systemic drug
release of Paricalcitol from our BASP particle formulation, yet achieve high concentration (980 ng/g @ 24hrs and 2940 ng/g @ 144hrs) concentration of polymer- bound paricalcitol in tumor and liver (628 ng/ml @ 24hrs and 918 ng/g @ 144hrs) tissue, respectively. Example 14: Anti-tumor efficacy of Tel-BASP (21) in pancreatic cancer
The tumor microenvironment is in a pro-inflammatory state and becomes stiff through deposition of extracellular matrix components, including collagen and hyaluronan. Moreover, stromal cells, including cancer-associated fibroblasts, stellate cells, and monocyte/macrophage-derived cells, expand and are reprogrammed to an active state. The purpose of this study is to evaluate the tissue normalization and anti- tumor efficacy of the Tel-BASP formulation (21) in a mouse model of human pancreatic (PDAC) cancer.
In this example, the Tel-BASP (21) formulation was prepared for in vivo dosing by dissolving the lyophilized polymer in sterile PBS at room temperature to make a 50 mg/ml dosing solution of the polymer. The resulting solution was sterile filtered through a 0.2 µm filter before dosing 200 µL per 20 gram mouse (500 mg/kg).
The orthotopic murine AK4.4 pancreatic tumor model has been described in Chauhan et al. (Nature Comm (2013) 4:2516). The AK4.4 pancreatic cell line (KRAS mutant p53 null) T were isolated from genetically engineered mice with spontaneous pancreatic tumors (Ptf1-Cre/LSL-KrasG12D/p53Lox/+) and the model is generated by implanting a piece (1 mm3) of viable tissue (from a source tumor collected from donor mice) into the pancreas of a 6–8-week-old male FVB mouse. One week post implantation, survival surgery is performed to measure the tumor size of the implanted fragment and this information is used to randomize all tumor-bearing animals into the treatment groups of n=12 animals per group (FIG.19A). In this study, a total of 72 mice were enrolled to evaluate the effect of combining Tel-BASP (21) with chemotherapy.
Without being bound by theory, Tel-BASP (21) through telmisartan drug release in the tumor microenvironment may lead to pleotropic effects including reprogramming of activated cancer associated fibroblasts, improved vessel perfusion and reduction of solid stress which ultimately may lead to enhanced uptake and activity of chemotherapy and other standard of care agents. Here, the treatment groups (FIG.19B) were designed to compare single agent activity of telmisartan vs Tel-
BASP (21) with and without combination with Gemcitabine. The test agents were dosed for three weeks (or until end of life). The dosing was stopped after 3 weeks of treatment and animals were sacrificed as they became moribund providing survival endpoint data for the Kaplan Meier curve (FIG.20A). As part of the survival study, clinical signs were monitored daily including body weight measurements twice a week (FIG.20B).
The Kaplan Meier curve (FIG.20A) shows that Tel-BASP (21) combined with Gemcitabine exhibits superior survival benefit compared to generic telmisartan combined with the same dose of chemotherapy. The media survival was extended from 12 days to 28 days for the combination compared to 22 days for gemcitabine alone. Moreover, the addition of Tel-BASP (21) to gemcitabine and a profound beneficial effect on body weight (FIG.20B) with the Tel-BASP (21) +GEM group being the only animals on study not to lose weight. Biomarker analysis of the AK4.4 tumor tissue further showed an increase in the presence of AGTR1 positive cells suggesting drug target engagement and biological activity at the receptor level for the Tel-BASP (21) formulation (FIGS.21A to 21D).
To further explore the activity of Tel-BASP (21), we repeated the Gem combination study focusing on a single PD time point as the endpoint for measuring tumor size (measurements and Tumor mass measurement (FIGS.22A to 22B). In this repeat AK4.4 study the average tumor size at start of treatment was 11 mm3 compared to 30 mm3 for the first study (FIGS.23A to 23C). Although tumor growth inhibition (TGI) was close to maximal for all the gemcitabine (Gem or GEM) combination groups, the combination of Gem + Tel-BASP (21) was superior than the combination of Gem with generic telmisartan. Taken together, these anti-tumor efficacy studies demonstrate biological activity of microenvironmentally released telmisartan from Tel-BASP (21). Example 15: CCl4-induced mouse model of liver fibrosis
A. Anti-fibrosis efficacy of Tel-BASP (22)
Carbon tetrachloride (CCl4) induced hepatic fibrosis and cirrhosis in rodents is a widely accepted experimental model for the study of liver fibrosis and cirrhosis. In many aspects, this model mirrors the pattern of human disease progression associated with toxic damages such as viral hepatitis, alcohol abuse, metabolic diseases due to overload of iron or copper, etc. The purpose of this study is to evaluate the efficacy of
Tel-BASP (22) on liver fibrosis induced by CCl4 administration in BALB/c mice. In this example, the Tel-BASP (22) formulation was prepared for in vivo dosing by dissolving the lyophilized polymer in sterile PBS at room temperature to make a 70 mg/ml dosing solution of the polymer. The resulting solution was sterile filtered through a 0.2 µm filter before dosing 200 µL per 20 gram mouse (700 mg/kg). The CCl4 model is a 6 week study with dosing of the experimental test agents during the last two weeks of CCl4 (i.p. bw) administration (FIG.24A). All animals (except healthy control group) was given CCl4 intraperitoneally (4 mL/kg of 25% CCl4 in olive oil) twice per week for a total period of 6 weeks with n=10 animals sacrificed on 4 week to determine baseline fibrosis at start of dosing of test agents. During the drug treatment period, Tel-BASP (22) was administered i.v. once weekly (700 mg/kg; total of two doses) compared to daily oral gavage of generic telmisartan (10 mg/kg p.o. q.d). Hence, over the two week of treatment, equivalent amounts of API are dosed. Body weights were followed during the entire study and all compounds were well tolerated (FIG.24A).
Animal blood samples (non-fasting) was collected at the end of week 4 (start of Tel-BASP (22) dosing) and end of week 6 (end of Tel-BASP (22) dosing) and the samples used for preparing plasma for blood biochemistry analysis. The plasma samples were prepared by refrigerated centrifugation within 30 min of collection (2000 x g for 10 minutes at 4ºC) and the plasma levels of alanine transferase (ALT), aspartate transaminase (AST) and bilirubin at week 4 and week 6 was measured using automatic biochemistry analyzer (HITACHI 7020).
FIGS.24B to 24D show that Tel-BASP (22) is able to significantly reduce the elevated levels of liver enzymes for Alt and AST and elevated levels of bilirubin with an infrequent once a week dosing schedule of the BASP polymer. Moreover, the efficacy observed for Tel-BASP (22) was superior to that observed for generic telmisartan in reducing the enzyme levels, despite being dosed every day for 14 days delivering the same total amount of telmisartan. The ALT and AST blood
biochemical readouts are clinical relevant biomarkers of liver disease and indicates that Tel-BASP (22) is able to normalized liver function in the disease animals.
Histopathology evaluation of liver biopsies is a gold standard/reference point for diagnosis of liver disease and is also used as a regulatory endpoint for assessment of anti-fibrosis activity of experimental test agents. Hence, liver tissue was collected from all treatment groups, processed by standard immunohistochemistry (H&E stain)
and scored in a blinded manner by trained pathologist. The microscopic analysis of H&E-stained tissue sections using the scoring diagram for inflammation shown in FIG.25A reveled significant reduction in liver tissue inflammation by both generic telmisartan (P<0.011) and Tel-BASP (22) (p<0.001). This therapeutic effect is consistent with disease tissue uptake of Tel-BASP (22) and subsequent release of telmisartan in the liver microenvironment using the Tel-BASP (22). Notably, Tel- BASP (22) can reduce the inflammation score beyond the disease level at week 4 (FIG.25B) suggesting a reversal of disease.
Additional pathology examination confirmed potent hepatocyte protective effects of Tel-BASP (22) as evident from significantly reduced levels of necrotic area in the whole mount liver tissue sections as scored by pathology (FIG.26A; scores between 1-4; FIG.26B; quantification) and based on quantification of cleaved caspase 3, a biomarker of apoptosis (FIG.26C; quantified in FIG.26D). Consistent, with the apparent disease normalization, Tel-BASP (22) treatment was also able to reverse CCl4 induced glycogen depletion (FIGS.27A-27D), and the level of fibrosis based on anti-TGFBI IHC staining (FIGS.28A to 28D). In summary, Tel-BASP (22) shows profound biological activity in fibrotic livers consistent with the PK profile described in previous examples. B. Comparative analysis of the anti-fibrosis efficacy of Tel-BASP (22) and Tel-BRUSH (28)
In this example, the Tel-BASP (22) formulation and the Tel-BRUSH (28) formulation were compared head-to-head in the CCl4 mouse model of liver fibrosis described above and the results from the first study (Study 1) were compared with the second repeat study (Study 2). Tel-BASP (22) and Tel-BRUSH (28) were prepared on the day of dosing by dissolving the lyophilized polymers in sterile PBS at room temperature to make a 30 mg/ml dosing solution and dosed at 300 mg/kg i.v. All animals (except the healthy control group) was given CCl4 intraperitoneally (4 mL/kg of 25% CCl4 in olive oil) twice per week for a total period of 6 weeks. During the last two weeks of CCl4 administration, Tel-BASP (22) and Tel-BRUSH (28) were administered i.v. once weekly (300 mg/kg; total of two doses) compared to daily oral gavage of generic telmisartan (10 mg/kg p.o. q.d). Hence, over the two week of drug treatment, a smaller fractional amount (40%) of telmisartan was delivered to teht animal using the polymer formulations relative to the generic Telmisartan dosed
orally once a day.
Animal plasma samples (non-fasting) were collected at the end of week 6 for blood biochemistry analysis (FIG, 29) measuring plasma levels of alanine transferase (ALT) and aspartate transaminase (AST) using automatic biochemistry analyzer (HITACHI 7020). Consistent with the first study (Study 1) the data from the repeat study (Study 2; summarized in FIG.29) shows that Tel-BASP (22) and Tel-BRUSH (28) are both able to significantly reduce the elevated levels of liver enzymes for ALT and AST with an infrequent (once a week) dosing schedule of the Tel-BASP and Tel- BRUSH polymers. Moreover, the efficacy observed for Tel-BASP (22) and Tel- BRUSH (28) was superior to that observed for generic telmisartan in reducing the enzyme levels, despite generic Telmisartan (Tel) being dosed every day for 14 days delivering >200% more of total telmisartan. Notably, the ALT and AST blood biochemical readouts are clinical relevant biomarkers of liver disease and indicates that Tel-BASP (22) and Tel-BRUSH (28) are able to normalize liver function in the disease animals.
Histopathology evaluation of liver biopsies is a gold standard/reference point for diagnosis of liver disease and is also used as a regulatory endpoint for assessment of anti-fibrosis activity of experimental test agents. Hence, liver tissue was collected from all treatment groups, processed by standard H&E stain, and scored in a blinded manner by a trained pathologist (FIG.30). The microscopic analysis of the H&E- stained tissue sections (using a 4-point scale for the necrosis pathology scoring) revealed significant reduction in the necrosis pathology by Tel-BASP (22) (P<0.001) and Tel-BRUSH (28) (P<0.01) compared to generic Tel (FIG.30). Additional pathology examination confirmed the potent hepatocyte protective effects of Tel- BASP (22) and Tel-BRUSH (28) formulations based on quantification of cleaved caspase 3, a biomarker of apoptosis (FIG.31A and 31B), that was significantly reduced in the liver tissue sections in response to drug treatment. Likewise, gene expression analysis of liver tissue samples confirmed inhibition of multiple fibrotic genes, as exemplified in FIG 32.
Consistent, with the apparent disease normalization, terminal liver tissue PK analysis (LC-MS/MS) confirmed significant concentration of telmisartan, both in the form of free, released drug as well as polymer-conjugated drug (FIG 33). Both Tel- BRUSH (28) and Tel-BASP (22) delivered >100 times higher levels of total Tel to the liver (113,656 and 225,523 ng/ml, respectively) compared to daily oral dosing of
generic Tel (mean liver conc of 7,192 ng/mg). Notably, Tel-BRUSH (28) showed a significantly higher (~10 fold) fractional release rate of Tel (1.94%) comparted to Tel BASP (22), where the ratio of free to conjugated Tel was 0.19% (FIG.33BB).
Moreover, the blood PK analysis (FIG 33A) revealed a 5-fold higher concentration of free telmisartan released from Tel-BRUSH (28) compared to Tel BASP (22) with liver concentrations of 2,081 ng/ml for Tel-BRUSH (28) vs 405 ng/ml for Tel-BASP (22) at study endpoint (FIG.7). In summary, Tel-BASP (22) and Tel-BRUSH (28) both shows profound biological activity in fibrotic livers consistent with the liver PK data. Example 16: Synthesis of Telmisartan Phenylester PEG3k/PEG3k BASP (26; m = 10, n = 9)
Under a nitrogen blanket, Compound 10 (100.0 g, 10 eq) was dissolved in anhydrous 1,4-dioxane (150 ml) in a 1000 mL reactor equipped with a mechanical stirrer. The mixture was stirred at 35 °C until a clear solution formed, and
subsequently cooled to 25 °C. A solution of Grubbs III catalyst:

(1.72 g, 1 eq, 50 mL 1,4-dioxane) in addition to a 3 mL solvent rinse were added via syringe with a 16G needle. The resulting mixture was stirred for 20 min. The acetal cross-linker, Compound 16 (12.35 g), was weighed into a 500 ml round-bottom flask and anhydrous 1,4-dioxane (210 ml) was added and stirred at 50-55 °C until fully dissolved. The solution of Compound 16 (218 ml, 9 eq) in addition to a 1 ml 1,4-dioxane rinse was added via syringe with a 12G needle at ca. 1 eq/min stirring at 300 rpm. Post injection, the reaction was allowed to proceed for ca.1 h before adding ethyl vinyl ether (20 mL) to quench the reaction. The solution was further stirred for an additional 15 min. The resultant solution was transferred to a sealed container and frozen at -20 °C.
The above frozen solution was thawed and transferred to a feed tank (5 gallon stainless steel) where it was diluted to ca.5% 1,4-dioxane in DI water. Fifteen diavolumes into water were performed using a Tangential Flow Filtration Column
(Spectrum Labs, 50 kDa MWCO, Cat.# N04-E050-05-N) in order to remove the 1,4- dioxane. The collected retentate was filtered via a pressurized filtration system at 20 psi through a 0.2 µm filter (Supor, Cat.# 01293). The filtered material was aliquoted into 10 mL lyophilization vials each containing 5 mL of retentate. The vials were subsequently frozen and lyophilized on a tray lyophilizer (SP Scientific Virtis Genesis 35EL) to yield off-white cakes. Vials were sealed and stored at 4 °C until use. Example 17: Synthesis of Telmisartan-Phenylester PEG3k/PEG3k-Cy7.5 PEG3k BASP Conjugate (Tel-Brush (28))
To start the process, 148.5 µL of cyanine 7.5 (Cy7.5) macromonomer
(Compound 14; 30 mg (0.1 equiv) in 149.4 µL of 1,4-dioxane; estimated amount of Cy7.5 added to the reaction: 29.8 mg) was added to Tel-phenyl macromonomer (Compound 9; 3.0 g 9.9 equiv in 4414.9 µL of 1,4-dioxane) and stirred for 2-3 minute for complete solubility. Then, the polymerization was initiated by adding 2.60 mL

(1.0 equiv) of the Grubbs III catalyst: (Grubbs III (Lot # LSG-B-152-1) = 55.0 mg in 2.75 mL of 1,4-dioxane; estimated amount of Grubbs III added to the reaction: 52 mg). After 30 min, 20 µL IPC sample was withdrawn for analysis and immediately thereafter the reaction was quenched by adding 0.5 mL (70 equiv) of ethyl vinyl ether and the entire mixture of the resulting Tel-Brush (28) frozen.
To further process the material, the sample was thawed for 1 hour and after total thawing, 10 mL of water was added to the Tel-Brush (28) in dioxane to dilute the sample and decrease its viscosity. Two mL of sample was transferred to each of 9 dialysis cartridges. The vial was then rinsed 2 times with 10 mL of water each time, and the rinses were distributed among the dialysis cartridges as evenly as possible. Each cartridge was then filled to the top with water and placed in 2 L glass beakers full of DI water. These were placed on stir plates with stir bars and allowed to dialyze for 2 hours. Then the dialysis cartridges were momentarily removed from the baths while the water was disposed of in a hazardous waste container, and new water was
added to the baths. The cartridges were then replaced in the baths and the process was repeated again 2 hours later. After 3 baths, the sample from each cartridge was pooled into a sterile plastic bottle yielding 107 mL of retentate.
Sterile filtration required the use of 2 Nylon filters (25 mm diameter, 0.22 µm pore size) with the use of a 6 mL syringe and a 20 mL syringe. The filtered sample was collected in a tared sterile plastic bottle, and 104 mL of filtered retentate was collected.
A total of 20 vials were filled with 5 mL of 3.6 mg/mL Tel-Brush (28), sterile filtered retentate and frozen in liquid nitrogen for at least 20 minutes. These were placed on the bench-top lyophilizer for 3.5 days in glass jars with tissue on the top of each vial. Tel-Brush (28) product cakes of approximately 130 mg each per vial were a white color with hints of light green. Example 18: Pharmacokinetics of Multiple Ascending Doses of Tel-BASP (26) in Dogs and Effect of Systolic Blood Pressure.
Tel-BASP (26) in vehicle (0.9% sodium chloride for injection, USP) was administered once by intravenous (slow push 1–2 minutes) injection to 4 groups (Groups 1, 2, 3, and 4) of Beagle dogs. Dose levels were 36, 100, 500, and 500 mg/kg for Groups 1, 2, 3, and 4, respectively. Group 4 was administered a second dose on Study Day 7 to evaluate effects following repeat dosing. The dose volume was 5 mL/kg for all groups. Groups 1–3 consisted of 1 animal/sex/group and the same animals were used for each dose. Group 4 consisted of 2 animals/sex. A 7-day non- dosing observation period was conducted between doses. Animals were scheduled for necropsy 7 days after their last respective dose. All animals survived to necropsy.
Blood samples were collected from all animals/sex/group/time point prior to dose administration (at time of placement into sling) and at approximately 5 and 30 minutes and 2, 24, 72, and 168 hours after dose administration. The concentration of both BASP-conjugated telmisartan and released, free telmisartan in whole blood was measured and showed dose proportionality for each (FIG.34).
All animals were monitored for food consumption, hematological parameters, serum biochemistry, anatomic pathology (microscopic and macroscopic examination, as well as organ weights), heart rates, arterial blood pressures, pulse pressures, and body temperatures. There were no Tel-BASP (26)-related macroscopic or microscopic findings or effects on body weights, food consumption, clinical pathology parameters,
blood pressure parameters, or organ weights. Example 19: Combination of Immuno-Oncology and Tel-BASP (21) Treatments on Tumor Size in a Mice Model of Human Breast Cancer.
We compared the effects of free telmisartan, Tel-BASP (21), and anti- PD1/anti-CTLA4 alone, as well as combinations of anti-PD1/anti-CTLA4 with either free telmisartan or Tel-BASP (21) in the E0771 orthotopic syngenic mouse model of human breast cancer.
E0771 breast cancer cells were grown in RPMI 1640 with 10% FBS. The orthotopic E0771 breast tumor model is generated by implanting approximately 300,000 cells from the tissue culture cell suspension into the mammary fat pad of a 6– 8-week-old female C57BL/6 mice. This model is described in Chauhan et al., Nature Comm.2013, 4:2516.
Tumor cells were implanted seven days before randomization. Tumors were allowed to grow and randomized on day 0 when the majority of the tumors were between 50 and 100 mm3 in volume. Treatment start was on day 1 for all of the groups. Group 1 mice were given vehicle (i.p.) anti-PD1/anti-CTLA4. Group 2 mice were given anti-PD1/anti-CTLA4 (i.p.) two times per week at a dose of 10 mg/kg and 5 mg/kg respectively, and this was the same dosing regimen given to all groups given the immunotherapy combination. Group 3 mice were given Telmisartan (p.o.) daily at 10 mg/kg. Group 4 mice were given Telmisartan (p.o.) daily at 10 mg/kg in combination with anti-PD1/anti-CTLA4. Group 5 mice were given Tel-BASP (21) (i.v.) weekly at 500 mg/kg. Group 6 mice were given Tel-BASP (21) (i.v.) weekly at 500 mg/kg in combination with PD1/CTLA4.
As shown in FIG.35, telmisartan and Tel-BASP (21) alone had little effect on tumor growth as compared to vehicle. Anti-PD1/anti-CTLA4 treatment showed an inhibition of tumor growth, which was similar to that seen in combination with telmisartan (PD1/CTLA4 + Telmisartan). The anti-PD1/anti-CTLA4 treatment in combination with Tel-BASP (21) showed a trend towards greater inhibition of tumor growth than the anti-PD1/anti-CTLA4 treatment alone or anti-PD1/anti-CTLA4 in combination with telmisartan.
Example 20: Combination of Immuno-Oncology and Pari-BASP (27) Treatment on Tumor Size in a Mice Model of Human Melanoma.
We compared the effects of Pari-BASP (27), and anti-PD1 as single agents, as well as a combination of the two in a Cloudman Melanoma mouse model of human melanoma.
CloudmanS91 melanoma cells were grown to mid-log phase in Kaighn’s modified Ham’s F12 Medium containing 2.5% fetal bovine serum, 15% horse serum, 2 mM glutamine, 100 units/mL sodium penicillin G, 25 µg/mL gentamicin, and 100 µg/mL streptomycin sulfate. The cells were cultured in tissue culture flasks in a humidified incubator at 37 °C, in an atmosphere of 5% CO2 and 95% air. The
CloudmanS91 tumor cells were harvested during exponential growth, and
resuspended at a concentration of 5 x 106 cells/mL in 50% Matrigel™ matrix (BD Biosciences) and 50% phosphate-buffered saline (PBS).
Each mouse (Female DBA/2 mice (DBA/2NCrl, Charles River) six weeks old was inoculated subcutaneously in the right flank with 5 x 105 cells (0.1 mL of cell suspension). Tumors were calipered in two dimensions to monitor growth as their mean volume approached the desired 100– 150 mm3 range. Ten days after tumor implantation, which was designated as Day 1 of the study, animals with individual tumor volumes ranging from 108 to 126 mm3 were sorted into three groups (Groups 1 – 3; n = 10 in each group). The group mean tumor volume was 115 - 117 mm3. Tumor measurements were taken twice a week until the end of the study.
All dosing was adjusted to the body weight of the individual animals. Group 1 mice served as the benchmark group for tumor engraftment and progression, as well as the control and received intraperitoneal (i.p.) doses of Immunotherapy vehicle 1 (phosphate buffered saline;“PBS”) twice a week for two weeks (biwk x 2) and vehicle 2 (isotonic mannitol) intravenously (i.v.), once a week for two weeks (qwk x 2) as vehicle for the test agents. Group 2 received anti-PD-1 i.p. at 5 mg/kg, biwk x 2 and vehicle 2, qwk x 2. Group 3 received anti-PD-1 i.p. at 5 mg/kg, biwk x 2 in combination with Pari-BASP (27) i.v. at 0.35 mg/kg on Day 1 and at 0.116 mg/kg on Day 8. FIG 36A shows median tumor growth. FIG 36B shows Kaplan-Meier survival.
The Group 3 (Pari-BASP (27) + anti-PD1) animals showed significant delay in tumor growth compared to the Group 2 animals (anti-PD1 alone). As shown in Table 6, below, the combination treatment took 38% longer to reach the predefined time to endpoint (“Median TTE”) compared to anti-PD1 alone.
Treatment may cause partial regression (PR) or complete regression (CR) of the tumor in an animal. In a PR response, the tumor volume is 50% or less of its Day 1 volume for three consecutive measurements during the course of the study, and equal to or greater than 13.5 mm3 for one or more of these three measurements. In a CR response, the tumor volume is less than 13.5 mm3 for three consecutive measurements during the course of the study. Animals were scored only once during the study for a PR or CR event and only as CR if both PR and CR criteria were satisfied. All animals were monitored for regression responses. An animal with a CR response at the termination of the study is additionally classified as a tumor-free survivor (TFS). Table 6. Results of Various Treatments in Mouse Melanoma Model

“VEH” = vehicle (negative control);“T-C” = difference between time-to-endpoint (TTE) between the treated (T) vs Control (C) group in days;“%TGD” = Percent Tumor Growth Delay ( [(T-C)/C] x 100);“MTV” = median tumor volume. Likewise, the Pari-BASP (27)/anti-PD1 combination extended the overall survival compared to anti-PD1 alone based on the Kaplan Meier Analysis (FIG.36B). Finally, the median tumor volume (mm3) on Day 43 was 936 (n=3) for the anti-PD1 alone group vs 144 (n=4) for the Pari-BASP (27)/anti-PD1 drug combination group (FIG.36A) showing superior treatment benefit based on tumor volume. Example 21: Combination of Immuno-Oncology and Pari-BASP (27) Treatment on Tumor Size in a Mice Model of Human Colon Cancer.
We compared the effects of Pari-BASP (27), and anti-PD1 as single agents, as well as a combination of the two in a MC30 mouse model of human colon cancer.
MC38 murine colon carcinoma cells were grown to mid-log phase in
Dulbecco’s Modified Eagle’s Medium containing 10% fetal bovine serum, 2 mM glutamine, 100 units/mL, penicillin G, 100 µg/mL streptomycin sulfate and 25 µg/mL gentamicin. The tumor cells were cultured in tissue culture flasks in a humidified incubator at 37 ºC, in an atmosphere of 5% CO2 and 95% air. The MC38 cells were
harvested during log phase growth and resuspended in RPMI media at a concentration of 5 x 106 cells/mL. The MC38 cells (5 x 105 cells; 0.1 mL cell suspension) were implanted subcutaneously in the right flank of each mouse under isoflurane anesthesia. Tumor growth was monitored as the average size approached the target range of 100 - 150 mm3. Sixteen days after tumor implantation, which was designated as Day 1 of the study, animals with individual tumor volumes ranging from 108 to 144 mm3 were sorted into three groups (Groups 1– 3; n = 10 in each Group). The group mean tumor volume was 119 - 121 mm3. Tumor measurements were taken twice a week until the end of the study.
All dosing was adjusted to the body weight of the individual animals. Group 1 mice served as the benchmark group for tumor engraftment and progression, as well as the control and received intraperitoneal (i.p.) doses of Immunotherapy vehicle 1 (phosphate buffered saline;“PBS”) twice a week for two weeks (biwk x 2) and vehicle 2 (isotonic mannitol) intravenously (i.v.), once a week for two weeks (qwk x 2) as vehicle for the test agents. Group 2 received anti-PD-1 i.p. at 5 mg/kg, biwk x 2 and vehicle 2, qwk x 2. Group 3 received anti-PD-1 i.p. at 5 mg/kg, biwk x 2 in combination with Pari-BASP (27) i.v. at 0.35 mg/kg on Day 1 and at 0.116 mg/kg on Day 8. FIG.37A shows median tumor growth. FIG.37B shows Kaplan-Meier survival.
As shown in Table 7 and FIG.32A, Group 3 (Pari-BASP (27) + anti-PD1) animals showed significant delay in tumor growth compared to Group 2 (anti-PD1 treatment alone) with tumors taking almost 2.4 times longer to reach the study endpoints (see T-C and %TGD values). Table 7. Results of Various Treatments in Mouse Colon Cancer Model

”T-C” = difference between time-to-endpoint (TTE) between the treated (T) vs Control (C) group in days;“%TGD” = Percent Tumor Growth Delay ( [(T-C)/C] x 100);“MTV” = median tumor volume. Likewise, the Pari-BASP (27) combination with the anti-PD1 immunotherapy extended the survival based on the Kaplan Meier Analysis compared to anti-PD1 alone (FIG 37B). Finally, there were two complete responses, one partial response
and one tumor free survivor in the Pari-BASP (27) group compared to only one partial response the in anti-PD1 alone group. Example 22: Comparison of Telmisartan, Tel-BASP (26), and Tel-BRUSH (28) in a Mice Model of Fatty Liver Disease
We compared the effects of Telmisartan, Tel-BASP (26), and Tel-BRUSH (28) in a diet-induced mouse model of fatty liver disease and non-alcoholic steatohepatitis (NASH) available from Gubra (www.gubra.dk/). The model exhibit features of steatosis, ballooning degeneration, inflammation, and fibrosis, identified by pre-study liver biopsies and objectively assessed using pathological NAFLD activity score and Fibrosis stage. The model allows for the testing of compounds where treatment efficacy can be assessed by comparing the post-treatment biopsies to the pre-treatment biopsies, which is similar to testing in a clinical setting.
Approximately 45 week old mice that were fed a high fat diet (HFD) for 39 weeks and assessed for fibrosis and NAFLD activity 3 weeks prior to dosing start were separated into multiple groups and given telmisartan (5 mg/kg as a daily oral dose for 8 weeks), a single i.v. bolus of Tel-BASP (26) (300 and 100 mg/kg dose groups; ~10% drug loading), or a single i.v. bolus of Tel-BRUSH (28) (300 mg/kg; ~10% drug loading), at week 1 and week 5. After 8 weeks, mice were reassessed for a number of parameters, and livers were histologically scored for improvement or worsening of fibrosis and NASH. The protocol is shown graphically in FIG.33A.
As shown in FIG.38B, both Telmisartan and Tel-BRUSH (28) demonstrated statistically significant improvement in fibrosis score as compared to vehicle control. In addition, both Telmisartan and Tel-BRUSH (28) demonstrated statistically significant improvement in NAFLD activity as compared to vehicle control. Tel- BASP (26) showed improvement in NAFLD activity in 5/12 mice at the 300 mg/kg dose. Without being bound by theory, we believe that Tel-Brush (28) has improved delivery to the liver as compared to Tel-BASP (26) in this model. To further substantiate this notion, terminal liver tissue PK analysis (LC-MS/MS) confirmed a significantly higher fractional release rate or Tel-BRUSH (28) comparted to Tel BASP (26) resulting in a concomitant 5-fold higher concentration of free telmisartan released from Tel-BRUSH (28) comparted to Tel-BASP (26) at the study endpoint (FIG.39). Notably, the liver PK data is 5 weeks post the last dose of Tel-BRUSH (28) and Tel-BASP (26) compared to 24 hours post the last dose of generic Tel
highlighting the infrequent dosing paradigm for these slow release‘depot-like’ drug polymer conjugates.
Because daily administration of generic Telmisartan (10 mg/kg) resulted in higher drug liver concentrations, yet was less efficacious compared to Tel-BRUSH (28) and Tel-BASP (26), we next studied the liver regional distribution of the Tel- BASP (26) macromolecule drug conjugate using IHC on formalin-fixed, crysectioned mouse livers and the anti-PEG antibody (clone: 5D6-3, Life Diagnostics) (FIG.40). Specifically, we noted a particularly high anti-PEG signal in cells representing parts of the RES (e.g. the reticulum endothelial system including monocyte/macrophages and perisinusoidal endothelial cells; FIG 40A). It is likely that such cell-specific drug uptake will results in the observed superior hepatocyte protective effects compared to animals dosed with generic Tel. Notably, imaging of Cy7.5 flourophore signal (Revolve microscope; Echo Labs), which is present as a tracer in the Tel-BASP formulation, also confirmed the same regional tissue biodistribution of the Tel-BASP (26) polymer (FIG 40B). Co-staing with the macrophage marker IBA-1 (Abcam, cat#ab178846) and Cy7/5 (anti-rabbit IgG-Alexa647; Life Technologies) confirmed partial colocalization of Tel-BASP (26) with this cell specific marker (FIG 40C). Example 23: Synthesis of Telmisartan Phenylester-PEG3-PEG3K BRUSH Conjugate (29, m = 10)


Compound 10 (790 g, 10 eq) and anhydrous 1,4-dioxane (1.2 L) were charged to a 2 L round bottom flask and stirred at 30 °C until a clear solution formed. Under a nitrogen blanket, 1,4-dioxane (0.12 L) was charged to a 5 L jacketed reactor equipped with a mechanical stirrer before addition of the Compound 10 solution in dioxane. The 2 L round bottom flask was rinsed with 0.12 L of 1,4-dioxane, which was also added to the 5 L reactor, and the resulting solution was cooled to 17°C-25°C. To the stirring solution of Compound 10, a freshly prepared solution of Grubbs III catalyst (13.6 g, 1 eq, in 0.67 L of 1,4-dioxane) was added through a funnel in less than 30 s. The resulting mixture was stirred for 25-30 min. before adding ethyl vinyl ether (107 g, 80 eq) to quench the reaction. The solution was further stirred for an additional ca. 30 min at room temperature under nitrogen. DMSO (0.79 L) and SiliaMetS ® Dimercaptotriazine scavenger (316 g) were added to the reaction solution before heating the mixture to 95°C for 72 hours to remove the Grubbs III catalyst. The scavenger was filtered out with a fine-graded fritted funnel and rinsed with three volumes of ca.157 mL of dioxane. The resultant filtrate was transferred to a sealed container, followed by a 50 mL dioxane rinse, then frozen at -20 °C.
The above frozen solution was thawed and transferred to a feed tank where it was diluted to ca.5% 1,4-dioxane in DI water. Fifteen diavolumes into water were performed using a Tangential Flow Filtration Cartridge (Spectrum Labs, 50 kDa MWCO) in order to remove the 1,4-dioxane. The collected retentate was filtered via a pressurized filtration system at 20 psi through a 0.2 µm filter (Supor). The filtered material was aliquoted as 10 mL fills into 20 mL lyophilization vials, each containing
ca.1 g of drug product. The vials were subsequently frozen and lyophilized on a tray lyophilizer (SP Scientific Virtis Genesis 35EL) to yield off-white cakes. Vials were sealed and stored at 4 °C until use.
Characterization data is summarized in Table 2, above, and the recorded plasma and liver PK profile is shown for Tel-BRUSH (29) in FIG 41A and 41B, respectively, confirming a sustained released of Tel in liver compared to the oral gavage of non-conjugated generic Tel with concomitant lower plasma levels of free Tel released for Tel-BRUSH(29) dosed at 300 mg/kg i.v. compared to generic Tel dosed at 10 mg/kg p.o. EQUIVALENTS
It will be apparent to those skilled in the relevant art that various
modifications, additions, substitutions, and the like can be made to the embodiments depicted and described in detail herein without departing from the spirit of the invention and these are therefore considered to be within the scope of the invention as defined in the claims which follow. Further, to the extent not already indicated, it will be understood by those of ordinary skill in the art that any one of the various embodiments herein described and illustrated can be further modified to incorporate features shown in any of the other embodiments disclosed herein.
Claims
CLAIMS We claim: 1. A compound of Formula (I):

Formula (I)
wherein:
A is C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, or C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1;
B is C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, or C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1;
Ring T is a triazolidyl;
X is an agent selected from an ARB, vitamin D analog, indoleamine 2,3- dioxygenease (IDO) inhibitor, and a bromodomain inhibitor;
P is alkylene or heteroalkylene;
each of L1 and L2 is independently a bond,
 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, C1-C12 heteroalkylene, (C0-C12 alkylene)-arylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)- arylene-(C0-C12 heteroalkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12
alkylene, C2-C12 alkenylene, C2-C12 alkynylene, C1-C12 heteroalkylene, (C0-C12 alkylene)-arylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)- arylene-(C0-C12 heteroalkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12
heteroalkylene), (C0-C12 alkylene)-heteroarylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-heteroarylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)- heteroarylene-(C0-C12 heteroalkylene), (C0-C12 alkylene)-heterocyclylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-heterocyclylene-(C0-C12 alkylene), or (C0-C12 heteroalkylene)-heterocyclylene-(C0-C12 heteroalkylene), wherein each alkylene, alkenylene, alkynylene, heteroalkylene, arylene, heteroarylene, or heterocyclylene is optionally substituted with 1-12 independently selected R2;
L3 is a tissue microenvironment cleavable linker;
each R1 and R2 is independently alkyl, alkenyl, alkynyl, heteroalkyl (e.g., polyethylene glycol), halo, cyano, oxo,–ORA,–NRBRC,–NRBC(O)RD, -C(O)NRBRC, –C(O)RD,–C(O)OH,–C(O)ORD,–SRE, or–S(O)mRE;
each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,–
C(O)NRBRC,–C(O)RD,–C(O)OH, or–C(O)ORD;
each RB and RC is independently hydrogen or C1-C6 alkyl;
each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl; and
m is 1 or 2. 2. The compound of claim 1, wherein Ring C is a structure of Formula (II):

Formula (II), or Formula (II-c):
 ,
,
wherein Z is C(R5) 3
2, O, or S; D is N or CR4; each R is independently C1-C6 alkyl, C1- C6 haloalkyl, oxo, or halo; each R3’ is independently selected from C1-C6 alkyl, - C(O)-C1-C6 alkyl, -C(O)-O-C1-C6 alkyl, -C(O)-NH-C1-C6 alkyl, C1-C6 heteroalkyl, - C(O)-C1-C6 heteroalkyl, -C(O)-O-C1-C6 heteroalkyl, -C(O)-NH-C1-C6 heteroalkyl, and halo, wherein the any alkyl portion of R3’ is optionally substituted with halo; R4 is hydrogen, C1-C6 alkyl, or halo; each R5 is independently hydrogen, C1-C6 alkyl, C1-C6 haloalkyl, or halo; n is 0, 1, or 2; s is 0, 1, 2, 3, 4, 5, 6, 7, 8, or 9; and“1” represents a portion of Ring C bound to B in the macromonomer.
3. The compound of claim 2, wherein Ring C is a structure of the formula:

4. The compound of any one of claims 1-2, wherein the structure of Formula (II)

5. The compound of any one of claims 1-4, wherein Ring T is
 , wherein“2” represents a portion of Ring T bound to L2 in the conjugate.
, wherein“2” represents a portion of Ring T bound to L2 in the conjugate.
6. The compound of any one of claims 1-5, wherein B is C1-C12 alkylene optionally substituted with 1-6 independently selected R1.
7. The compound of any one of claims 1-6, wherein A is–
C(O)CH2CH2C(O)NH-PEG comprising a molecular weight of between about 200 and about 6000, g/mol (e.g., PEG200, PEG400, PEG600, PEG800, PEG1000, PEG1500, PEG2000, PEG3000, PEG4000, PEG6000).
8. The compound of any one of claims 1-7, wherein each of L1 and L2 is independently selected from–CH2–,–NCH2–, and–CH2CH2.
9. The compound of any one of claims 1-8, wherein L3 is cleavable by an enzyme (e.g., an esterase, a protease), or pH (e.g., acidic pH, basic pH).
10. The compound of any one of claims 1-9, wherein L3 is–C(O)–,–OC(O)–,– C(O)O–,–C(O)CH2–,–OC(O)CH2–,–C(O)OCH2–,–CH2CH2O–, C(O)CH2CH2O–,

11. The compound of any one of claims 1-10, wherein the ARB comprises a structure of selected from:

13. The compound of any one of claims 1-10, wherein the vitamin D analog com rises a structure selected from:

15. The compound of any one of claims 1-10, wherein the agent is an IDO inhibitor.
16. The compound of any one of claims 1-15, wherein the compound has the structure of Formula I-a :

A is C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, or C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1;
B is C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, or C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1;
X is an agent (e.g., an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor);
each of L1 and L2 is independently C1-C12 alkylene, C1-C12 heteroalkylene, (C0-C12 alkylene)-arylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), or (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2;
L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12
heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2;
each R1 and R2 is independently alkyl, alkenyl, alkynyl, heteroalkyl (e.g., polyethylene glycol), halo, cyano, oxo,–ORA,–NRBRC,–NRBC(O)RD, -C(O)NRBRC, –C(O)RD,–C(O)OH,–C(O)ORD,–SRE, or–S(O)mRE;
each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,–
C(O)NRBRC,–C(O)RD,–C(O)OH, or–C(O)ORD;
each RB and RC is independently hydrogen or C1-C6 alkyl;
each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl; and
t is an integer between 1 and 10, inclusive.
17. The compound of any one of claims 1-16, wherein the compound has the structure of Formula (I-b):

Formula (I-b)
wherein:
B is C1-C12 alkylene or C1-C12 heteroalkylene, wherein each alkylene or heteroalkylene is optionally substituted with 1-6 independently selected R1;
X is an agent (e.g., an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor);
each of L1 and L2 is independently C1-C12 alkylene, C1-C12 heteroalkylene, (C0-C12 alkylene)-arylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), or (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2;
L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12
heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2; each R1 and R2 is independently alkyl, heteroalkyl, halo, cyano, oxo,–ORA,–NRBRC,–
NRBC(O)RD, -C(O)NRBRC,–C(O)RD,–C(O)OH,–C(O)ORD,–SRE, or–S(O)mRE; each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,–
C(O)NRBRC,–C(O)RD,–C(O)OH, or–C(O)ORD;
each RB and RC is independently hydrogen or C1-C6 alkyl;
each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl;
t is an integer between 1 and 10, inclusive; and
z is an integer between 1 and 200, inclusive.
18. A com ound com risin aricalcitol of Formula I-d-i or Formula I-d-ii :

Formula (I-d-i)

Formula (I-d-ii)
wherein:
L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12
heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2;
each R2 is independently alkyl, heteroalkyl, halo, cyano, oxo,–ORA,–NRBRC, –NRBC(O)RD, -C(O)NRBRC,–C(O)RD,–C(O)OH,–C(O)ORD,–SRE, or–S(O)mRE; each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,–
C(O)NRBRC,–C(O)RD,–C(O)OH, or–C(O)ORD;
each RB and RC is independently hydrogen or C1-C6 alkyl;
each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl;
t is an integer between 1 and 10, inclusive; and
z is an integer between 1 and 200, inclusive.
19. A com ound com risin telmisartan of Formula I-e :

Formula (I-e)
wherein:
L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12
heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2;
each R2 is independently alkyl, heteroalkyl, halo, cyano, oxo,–ORA,–NRBRC, –NRBC(O)RD, -C(O)NRBRC,–C(O)RD,–C(O)OH,–C(O)ORD,–SRE, or–S(O)mRE; each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,–
C(O)NRBRC,–C(O)RD,–C(O)OH, or–C(O)ORD;
each RB and RC is independently hydrogen or C1-C6 alkyl;
each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl;
t is an integer between 1 and 10, inclusive; and
z is an integer between 1 and 200, inclusive.
20. A conjugate or BASP particle comprising a structure according to Formula (III-f):

wherein:
Ring C’ is a carbocyclyl or heterocyclyl moiety;
Ring T is a triazoldiyl moiety (e.g., a 1,2,3-triazoldiyl);
A is C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, or C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1;
B is C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, or C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, or heteroalkylene is optionally substituted with 1-6 independently selected R1;
X is an agent chosen from an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor;
P is alkylene or heteroalkylene (e.g., polyethylene glycol);
each of L1 and L2 is independently a bond, C1-C12 alkylene, C2-C12 alkenylene, C2-C12 alkynylene, C1-C12 heteroalkylene, (C0-C12 alkylene)-arylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)- arylene-(C0-C12 heteroalkylene), (C0-C12 heteroalkylene)-arylene-(C0-C12
heteroalkylene), (C0-C12 alkylene)-heteroarylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-heteroarylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)- heteroarylene-(C0-C12 heteroalkylene), (C0-C12 alkylene)-heterocyclylene-(C0-C12 alkylene), (C0-C12 heteroalkylene)-heterocyclylene-(C0-C12 alkylene), or (C0-C12 heteroalkylene)-heterocyclylene-(C0-C12 heteroalkylene), wherein each alkylene, alkenylene, alkynylene, heteroalkylene, arylene, heteroarylene, or heterocyclylene is optionally substituted with 1-12 independently selected R2;
L3 is a tissue microenvironment cleavable linker;
each R1 and R2 is independently alkyl, alkenyl, alkynyl, heteroalkyl (e.g., polyethylene glycol), halo, cyano, oxo,–ORA,–NRBRC,–NRBC(O)RD, -C(O)NRBRC, –C(O)RD,–C(O)OH,–C(O)ORD,–SRE, or–S(O)mRE;
R6a is absent or, in an initiating macromonomer unit present in the conjugate or BASP particle, is selected from hydrogen, phenyl, or C1-C4 alkyl;
R6b is absent or, in a terminating macromonomer unit present in the conjugate or BASP particle, is selected from hydrogen, phenyl, or C1-C4 alkyl;
each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,–
C(O)NRBRC,–C(O)RD,–C(O)OH, or–C(O)ORD;
each RB and RC is independently hydrogen or C1-C6 alkyl;
each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl;
m is 1 or 2; and.
q is an integer between 2 and 200, inclusive.
21. The conjugate or BASP particle of claim 20, wherein:
(i) the average molecular weight of the conjugate is between about 2,000 Da and about 6,000 Da (e.g., between about 3,000 Da and about 5,000 Da); or
(ii) the hydrodynamic diameter of the conjugate is less than about 20 nm (e.g., less than about 15 nm, about 12.5 nm, about 10 nm, about 9 nm, about 8 nm, about 7 nm, about 6 nm, about 5 nm, or less).
22. The conjugate or BASP particle of any one of claims 20-21, wherein the total amount of the agent present in the conjugate or BASP particle is greater than about 10% (e.g., about 12%, about 15%, about 20%, about 25%, about 30%, or more).
23. The conjugate or BASP particle of any one of claims 20-22 comprising a structure of Formula (III-i-1) or Formula (III-i-2):

wherein L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12
heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2;
each R2 is independently alkyl, heteroalkyl, halo, cyano, oxo,–ORA,–NRBRC, –NRBC(O)RD, -C(O)NRBRC,–C(O)RD,–C(O)OH,–C(O)ORD,–SRE, or–S(O)mRE;
R6a is absent or, in an initiating macromonomer unit present in the conjugate or BASP particle, is selected from hydrogen, phenyl, or C1-C4 alkyl;
R6b is absent or, in a terminating macromonomer unit present in the conjugate or BASP particle, is selected from hydrogen, phenyl, or C1-C4 alkyl;
each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,–
C(O)NRBRC,–C(O)RD,–C(O)OH, or–C(O)ORD;
each RB and RC is independently hydrogen or C1-C6 alkyl;
each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl;
each RE is independently hydrogen or C1-C6 alkyl;
t is an integer between 1 and 10, inclusive;
q is an integer between 2 and 200, inclusive; and
z is an integer between 1 and 200, inclusive.
24. The conjugate or BASP particle of any one of claims 20-22 comprising the structure of Formula (III-j):

wherein L3 is C1-C12 heteroalkylene, (C0-C12 heteroalkylene)-arylene-(C0-C12 alkylene), (C0-C12 alkylene)-arylene-(C0-C12 heteroalkylene), or (C0-C12
heteroalkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or arylene is optionally substituted with 1-12 independently selected R2;
each R2 is independently alkyl, heteroalkyl, halo, cyano, oxo,–ORA,–NRBRC, –NRBC(O)RD, -C(O)NRBRC,–C(O)RD,–C(O)OH,–C(O)ORD,–SRE, or–S(O)mRE;
R6a is absent or, in an initiating macromonomer unit present in the conjugate or BASP particle, is selected from hydrogen, phenyl, or C1-C4 alkyl;
R6b is absent or, in a terminating macromonomer unit present in the conjugate or BASP particle, is selected from hydrogen, phenyl, or C1-C4 alkyl;
each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,–
C(O)NRBRC,–C(O)RD,–C(O)OH, or–C(O)ORD;
each RB and RC is independently hydrogen or C1-C6 alkyl;
each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl; each RE is independently hydrogen or C1-C6 alkyl;
t is an integer between 1 and 10, inclusive;
q is an integer between 2 and 200, inclusive; and
z is an integer between 1 and 200, inclusive.
25. The conjugate or BASP particle of any one of claims 20-24, wherein L3 is C1- C12 heteroalkylene or (C0-C12 heteroalkylene)-aryl-(C0-C12 heteroalkylene), or (C0-C12 alkylene)-aryl-(C0-C12 heteroalkylene), wherein each alkylene, heteroalkylene, or aryl is optionally substituted with 1-6 R2.
26. The con u ate or BASP article of claim 20 wherein the con u ate has the

29), wherein“Ph” represents phenyl on a terminating macromonomer in the conjugate or BASP particle.
27. The conjugate or BASP particle of any one of claims 20-26, wherein the conjugate or BASP particle has a hydrodynamic diameter of less than about 100 nm (e.g., between about 5 nm and 50 nm).
28. A pharmaceutical composition comprising a conjugate or BASP particle of any one of claims 20-27, and a pharmaceutically acceptable carrier.
29. A composition for use in treating or preventing a disorder (e.g., a
hyperproliferative disorder, a fibrotic disorder, and/or an inflammatory disorder) in a
subject, comprising administering the conjugate or BASP particle of any one of claims 20-27, or the composition of claim 28 to the subject.
30. The composition of claim 29, wherein:
(i) the disorder is a cancer chosen from one or more of pancreatic, breast, colorectal, colon, lung, skin, ovarian, prostate, cervix, gastric, gastrointestinal, stomach, head and neck, kidney, brain cancer, liver cancer, or a metastatic lesion thereof; or
(ii) the disorder is a fibrotic condition or disorder of the lung, a fibrotic condition of the liver, a fibrotic condition of the heart or vasculature, a fibrotic condition of the kidney, a fibrotic condition of the skin, a fibrotic condition of the gastrointestinal tract, a fibrotic condition of the bone marrow or a hematopoietic tissue, a fibrotic condition of the nervous system, a fibrotic condition of the eye, or a combination thereof.
31. A compound of Formula (VII):

Formula (VII),
or a salt thereof, wherein:
each of Z1 and Z2 is independently C1-C6 alkylene, C2-C6 alkenylene, C2-C6 alkynylene, C1-C6 heteroalkylene, (C0-C6 alkylene)-aryl-(C0-C6 alkylene), (C0-C6 heteroalkylene)-aryl-(C0-C6 alkylene), or (C0-C6 alkylene)-aryl-(C0-C6
heteroalkylene), wherein each alkylene, alkenylene, alkynylene, heteroalkylene, and aryl is optionally substituted with 1-6 independently selected R31;
each of W1 and W2 is independently C1-C12 alkylene, C2-C12 alkenylene, C2- C12 alkynylene, or C1-C12 heteroalkylene, wherein each alkylene, alkenylene, alkynylene, and heteroalkylene is optionally substituted with 1-6 independently selected R32;
G is–O–,–S–, or–S-S–;
each R31 and R32 is independently alkyl, heteroalkyl, halo, cyano, oxo,–ORA, –NRBRC,–NRBC(O)RD, -C(O)NRBRC,–C(O)RD, or–C(O)OH;
each RA is independently hydrogen, C1-C6 alkyl, C1-C6 heteroalkyl,– C(O)NRBRC,–C(O)RD,–C(O)OH, or–C(O)ORD;
each RB and RC is independently hydrogen or C1-C6 alkyl; and
each RD is independently C1-C6 alkyl, C1-C6 heteroalkyl, or C1-C6 haloalkyl.
32. A method of making a conjugate or BASP particle, comprising:
(a) preparing a first solution of a compound of Formula (V-c) (e.g., a macromonomer):

Formula (V-c)
wherein X is an agent (e.g., an ARB, vitamin D analog, an IDO inhibitor, or a bromodomain inhibitor); t is an integer between 1 and 10, inclusive; and z is an integer between 1 and 200, inclusive;
with a metal catalyst (e.g., a Grubbs’ catalyst);
b) mixing the first solution with a second solution comprising a compound of Formula (VII-a e. . a cross-linker :

Formula (VII-a)
wherein each of Z1 and Z2 is independently CH2-, -CH2CH2CH2-, or phenyl; each of W1 and W2 is independently -CH2-, -OC(O)CH2CH2-, -CH2OC(O)CH2CH2-, or– C(O)-; and G is–O– or–S-S– to form a third solution;
(c) allowing the third solution to incubate for a time sufficient to form a conjugate or BASP particle of desired length;
(d) adding a chain-transfer agent (e.g., a chain-transfer agent comprising an olefin); and
(e) purifying the solution,
to thereby make a conjugate or BASP particle.
33. The method of claim 32, further comprising removing the metal catalyst (e.g., a Grubbs’ catalyst) through addition of a compound comprising an amine, phosphine, or thiol (e.g., N,N-dimethyltryptamine, cysteine, triaminetetraacetate (sodium salt), tris(hydroxymethyl)phosphine, 2-mercaptonicotinic acid, N-acetyl-L- cysteine, imidazole, diethylphenylazothioformamide, lead tetra-acetate, hydrogen peroxide, triphenylphosphine oxide, isocyanide salt, di(ethylene glycol) vinyl ether, acetonitrile, or dimethyl sulfoxide).
34. The method of claim 33, wherein the removing the metal catalyst occurs after step (d).
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662430257P | 2016-12-05 | 2016-12-05 | |
| US62/430,257 | 2016-12-05 | ||
| US201762528940P | 2017-07-05 | 2017-07-05 | |
| US62/528,940 | 2017-07-05 | ||
| US201762529439P | 2017-07-06 | 2017-07-06 | |
| US62/529,439 | 2017-07-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018106738A1 true WO2018106738A1 (en) | 2018-06-14 |
Family
ID=60782383
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2017/064784 WO2018106738A1 (en) | 2016-12-05 | 2017-12-05 | Brush-arm star polymers, conjugates and particles, and uses thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2018106738A1 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20180236139A1 (en) * | 2017-02-23 | 2018-08-23 | Medtronic Vascular Inc. | Drug coated medical devices |
| CN109593156A (en) * | 2018-12-14 | 2019-04-09 | 长沙加美乐素化工有限公司 | A kind of technique synthesizing comprehensive water-reducing agent using six carbon monomers |
| WO2021076821A2 (en) | 2019-10-16 | 2021-04-22 | Massachusetts Institute Of Technology | Brush prodrugs and uses thereof |
| WO2022093106A1 (en) * | 2020-10-30 | 2022-05-05 | Agency For Science, Technology And Research | Bioactive synthetic copolymer, bioactive macromolecule and related methods thereof |
| US11376264B2 (en) | 2017-07-24 | 2022-07-05 | Salk Institute For Biological Studies | Use of bromodomain-containing protein 9 antagonists in combination with vitamin D receptor agonists in diabetes treatment |
| US11678976B2 (en) | 2010-04-29 | 2023-06-20 | Adaptilens, Llc | Injectable physiologically adaptive intraocular lenses (IOL's) |
| WO2023121753A1 (en) * | 2021-12-20 | 2023-06-29 | Massachusetts Institute Of Technology | Biomolecule-polymer-pharmaceutical agent conjugates for delivering the pharmaceutical agent |
| US11724985B2 (en) | 2020-05-19 | 2023-08-15 | Cybin Irl Limited | Deuterated tryptamine derivatives and methods of use |
| US11752221B2 (en) | 2017-06-30 | 2023-09-12 | Massachusetts Institute Of Technology | Brush-arm star polymer imaging agents and uses thereof |
| WO2023211384A1 (en) * | 2022-04-28 | 2023-11-02 | Agency For Science, Technology And Research | Hyaluronic acid-based synthetic copolymer, material containing said copolymer and related methods thereof |
| US11897905B2 (en) | 2018-08-17 | 2024-02-13 | Massachusetts Institute Of Technology | Degradable polymers of a cyclic silyl ether and uses thereof |
Citations (77)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3536809A (en) | 1969-02-17 | 1970-10-27 | Alza Corp | Medication method |
| US3598123A (en) | 1969-04-01 | 1971-08-10 | Alza Corp | Bandage for administering drugs |
| US3845770A (en) | 1972-06-05 | 1974-11-05 | Alza Corp | Osmatic dispensing device for releasing beneficial agent |
| US3916899A (en) | 1973-04-25 | 1975-11-04 | Alza Corp | Osmotic dispensing device with maximum and minimum sizes for the passageway |
| US4008719A (en) | 1976-02-02 | 1977-02-22 | Alza Corporation | Osmotic system having laminar arrangement for programming delivery of active agent |
| US4270537A (en) | 1979-11-19 | 1981-06-02 | Romaine Richard A | Automatic hypodermic syringe |
| US4596556A (en) | 1985-03-25 | 1986-06-24 | Bioject, Inc. | Hypodermic injection apparatus |
| US4790824A (en) | 1987-06-19 | 1988-12-13 | Bioject, Inc. | Non-invasive hypodermic injection device |
| US4886499A (en) | 1986-12-18 | 1989-12-12 | Hoffmann-La Roche Inc. | Portable injection appliance |
| US4940460A (en) | 1987-06-19 | 1990-07-10 | Bioject, Inc. | Patient-fillable and non-invasive hypodermic injection device assembly |
| US4941880A (en) | 1987-06-19 | 1990-07-17 | Bioject, Inc. | Pre-filled ampule and non-invasive hypodermic injection device assembly |
| US5013556A (en) | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5015235A (en) | 1987-02-20 | 1991-05-14 | National Carpet Equipment, Inc. | Syringe needle combination |
| US5021409A (en) | 1989-12-21 | 1991-06-04 | Johnson Matthey Plc | Antiviral cyclic polyamines |
| US5064413A (en) | 1989-11-09 | 1991-11-12 | Bioject, Inc. | Needleless hypodermic injection device |
| US5141496A (en) | 1988-11-03 | 1992-08-25 | Tino Dalto | Spring impelled syringe guide with skin penetration depth adjustment |
| US5190521A (en) | 1990-08-22 | 1993-03-02 | Tecnol Medical Products, Inc. | Apparatus and method for raising a skin wheal and anesthetizing skin |
| US5312335A (en) | 1989-11-09 | 1994-05-17 | Bioject Inc. | Needleless hypodermic injection device |
| US5328483A (en) | 1992-02-27 | 1994-07-12 | Jacoby Richard M | Intradermal injection device with medication and needle guard |
| US5334144A (en) | 1992-10-30 | 1994-08-02 | Becton, Dickinson And Company | Single use disposable needleless injector |
| US5339163A (en) | 1988-03-16 | 1994-08-16 | Canon Kabushiki Kaisha | Automatic exposure control device using plural image plane detection areas |
| US5383851A (en) | 1992-07-24 | 1995-01-24 | Bioject Inc. | Needleless hypodermic injection device |
| US5417662A (en) | 1991-09-13 | 1995-05-23 | Pharmacia Ab | Injection needle arrangement |
| US5466220A (en) | 1994-03-08 | 1995-11-14 | Bioject, Inc. | Drug vial mixing and transfer device |
| US5480381A (en) | 1991-08-23 | 1996-01-02 | Weston Medical Limited | Needle-less injector |
| US5527288A (en) | 1990-12-13 | 1996-06-18 | Elan Medical Technologies Limited | Intradermal drug delivery device and method for intradermal delivery of drugs |
| US5569189A (en) | 1992-09-28 | 1996-10-29 | Equidyne Systems, Inc. | hypodermic jet injector |
| US5583131A (en) | 1991-12-16 | 1996-12-10 | Johnson Matthey Public Limited Company | Aromatic-linked polyamine macrocyclic compounds with anti-HIV activity |
| US5599302A (en) | 1995-01-09 | 1997-02-04 | Medi-Ject Corporation | Medical injection system and method, gas spring thereof and launching device using gas spring |
| WO1997013537A1 (en) | 1995-10-10 | 1997-04-17 | Visionary Medical Products Corporation | Gas pressured needle-less injection device |
| US5649912A (en) | 1994-03-07 | 1997-07-22 | Bioject, Inc. | Ampule filling device |
| WO1997037705A1 (en) | 1996-04-11 | 1997-10-16 | Weston Medical Limited | Spring-powered dispensing device for medical purposes |
| US5811515A (en) | 1995-06-12 | 1998-09-22 | California Institute Of Technology | Synthesis of conformationally restricted amino acids, peptides, and peptidomimetics by catalytic ring closing metathesis |
| US5893397A (en) | 1996-01-12 | 1999-04-13 | Bioject Inc. | Medication vial/syringe liquid-transfer apparatus |
| WO1999034850A1 (en) | 1998-01-08 | 1999-07-15 | Fiderm S.R.L. | Device for controlling the penetration depth of a needle, for application to an injection syringe |
| WO1999050461A1 (en) | 1998-03-30 | 1999-10-07 | Northwest Biotherapeutics, Inc. | Therapeutic and diagnostic applications based on the role of the cxcr-4 gene in tumorigenesis |
| WO1999052552A1 (en) | 1998-04-15 | 1999-10-21 | Brigham & Women's Hospital, Inc. | T cell inhibitory receptor compositions and uses thereof |
| US5993412A (en) | 1997-05-19 | 1999-11-30 | Bioject, Inc. | Injection apparatus |
| US6001826A (en) | 1989-12-21 | 1999-12-14 | Anormed, Inc. | Chemical compounds |
| WO2001085196A2 (en) | 2000-05-09 | 2001-11-15 | The University Of British Columbia | Cxcr4 antagonist treatment of hematopoietic cells |
| WO2001094420A1 (en) | 2000-06-05 | 2001-12-13 | The Trustees Of Columbia University In The City Of New York | Identification and use of human bone marrow-derived endothelial progenitor cells to improve myocardial function after ischemic injury |
| WO2003011277A2 (en) | 2001-07-31 | 2003-02-13 | Anormed Inc. | Methods to mobilize progenitor/stem cells |
| WO2003079972A2 (en) | 2002-02-22 | 2003-10-02 | New River Parmaceuticals Inc. | Active agent delivery systems and methods for protecting and administering active agents |
| WO2003090512A2 (en) | 2002-04-23 | 2003-11-06 | The Trustees Of Columbia University In The City Of New York | Regeneration of endogenous myocardial tissue by induction of neovascularization |
| US20030220341A1 (en) | 2001-12-21 | 2003-11-27 | Gary Bridger | Chemokine receptor binding heterocyclic compounds with enhanced efficacy |
| US20040047858A1 (en) | 2002-09-11 | 2004-03-11 | Blumberg Richard S. | Therapeutic anti-BGP(C-CAM1) antibodies and uses thereof |
| US20040209921A1 (en) | 2003-04-11 | 2004-10-21 | Gary Bridger | CXCR4 chemokine receptor binding comounds |
| US20050043367A1 (en) | 2001-07-31 | 2005-02-24 | Gary Bridger | Methods to mobilize progenitor/stem cells |
| US20050059702A1 (en) | 2003-04-22 | 2005-03-17 | Bridger Gary J. | Chemokine receptor binding heterocyclic compounds with enhanced efficacy |
| US20050277670A1 (en) | 2003-12-11 | 2005-12-15 | Anormed Inc. | Chemokine receptor binding compounds |
| WO2006121168A1 (en) | 2005-05-09 | 2006-11-16 | Ono Pharmaceutical Co., Ltd. | Human monoclonal antibodies to programmed death 1(pd-1) and methods for treating cancer using anti-pd-1 antibodies alone or in combination with other immunotherapeutics |
| WO2007005874A2 (en) | 2005-07-01 | 2007-01-11 | Medarex, Inc. | Human monoclonal antibodies to programmed death ligand 1 (pd-l1) |
| US7495099B2 (en) | 2002-10-31 | 2009-02-24 | Nippon Kayaku Kabushiki Kaisha | High-molecular weight derivatives of camptothecins |
| WO2009101611A1 (en) | 2008-02-11 | 2009-08-20 | Curetech Ltd. | Monoclonal antibodies for tumor treatment |
| US20090221683A1 (en) | 2005-10-18 | 2009-09-03 | Caritas St.Elizabeth Medical Center Of Boston, Inc | Combination of CXCR4 Antagonist and Morphogen to Increase Angiogenesis |
| WO2009114335A2 (en) | 2008-03-12 | 2009-09-17 | Merck & Co., Inc. | Pd-1 binding proteins |
| WO2010019570A2 (en) | 2008-08-11 | 2010-02-18 | Medarex, Inc. | Human antibodies that bind lymphocyte activation gene-3 (lag-3), and uses thereof |
| US20100055088A1 (en) | 2005-05-25 | 2010-03-04 | Hadasit Medical Research Services And Development Ltd. | CXCR4 Antagonists for Wound Healing and Re-Epithelialization |
| WO2010077634A1 (en) | 2008-12-09 | 2010-07-08 | Genentech, Inc. | Anti-pd-l1 antibodies and their use to enhance t-cell function |
| US20100178271A1 (en) | 2006-08-07 | 2010-07-15 | Genzyme Corporation | Combination Therapy |
| US20100247521A1 (en) | 2007-10-26 | 2010-09-30 | Jones Richard B | Therapeutic and Diagnostic Methods Using TIM-3 |
| WO2010125571A1 (en) | 2009-04-30 | 2010-11-04 | Tel Hashomer Medical Research Infrastructure And Services Ltd. | Anti ceacam1 antibodies and methods of using same |
| US20120039906A1 (en) | 2009-02-09 | 2012-02-16 | INSER (Institut National de la Recherche Medicale) | PD-1 Antibodies and PD-L1 Antibodies and Uses Thereof |
| WO2013006490A2 (en) | 2011-07-01 | 2013-01-10 | Cellerant Therapeutics, Inc. | Antibodies that specifically bind to tim3 |
| US8354509B2 (en) | 2007-06-18 | 2013-01-15 | Msd Oss B.V. | Antibodies to human programmed death receptor PD-1 |
| WO2013054331A1 (en) | 2011-10-11 | 2013-04-18 | Tel Hashomer Medical Research Infrastructure And Services Ltd. | Antibodies to carcinoembryonic antigen-related cell adhesion molecule (ceacam) |
| WO2013082366A1 (en) | 2011-12-01 | 2013-06-06 | The Brigham And Women's Hospital, Inc. | Anti-ceacam1 recombinant antibodies for cancer therapy |
| US20130156774A1 (en) | 2010-06-18 | 2013-06-20 | The Brigham And Women's Hospital, Inc. | Bi-specific antibodies against tim-3 and pd-1 for immunotherapy in chronic immune conditions |
| US8513390B2 (en) | 2009-01-12 | 2013-08-20 | Cytomx Therapeutics, Inc. | Modified antibody compositions, methods of making and using thereof |
| US8541203B2 (en) | 2007-08-22 | 2013-09-24 | The Regents Of The University Of California | Activatable binding polypeptides and methods of identification and use thereof |
| US8580244B2 (en) | 2002-09-06 | 2013-11-12 | Cerulean Pharma Inc. | Cyclodextrin-based polymers for therapeutics delivery |
| WO2013169739A1 (en) | 2012-05-07 | 2013-11-14 | The General Hospital Corporation | Novel compositions and uses of anti-hypertension agents for cancer therapy |
| WO2014022332A1 (en) | 2012-07-31 | 2014-02-06 | The Brigham And Women's Hospital, Inc. | Modulation of the immune response |
| WO2014059251A1 (en) | 2012-10-12 | 2014-04-17 | The Brigham And Women's Hospital, Inc. | Enhancement of the immune response |
| US20140308234A1 (en) * | 2013-04-09 | 2014-10-16 | Massachusetts Institute Of Technology | Drug delivery polymer and uses thereof |
| US20150087810A1 (en) | 2013-09-25 | 2015-03-26 | Cytomx Therapeutics, Inc. | Matrix Metalloproteinase Substrates And Other Cleavable Moieties And Methods Of Use Thereof |
| WO2016140714A1 (en) | 2015-03-05 | 2016-09-09 | The General Hospital Corporation | Novel compositions and uses of metformin agents |
-
2017
- 2017-12-05 WO PCT/US2017/064784 patent/WO2018106738A1/en active Application Filing
Patent Citations (85)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3536809A (en) | 1969-02-17 | 1970-10-27 | Alza Corp | Medication method |
| US3598123A (en) | 1969-04-01 | 1971-08-10 | Alza Corp | Bandage for administering drugs |
| US3845770A (en) | 1972-06-05 | 1974-11-05 | Alza Corp | Osmatic dispensing device for releasing beneficial agent |
| US3916899A (en) | 1973-04-25 | 1975-11-04 | Alza Corp | Osmotic dispensing device with maximum and minimum sizes for the passageway |
| US4008719A (en) | 1976-02-02 | 1977-02-22 | Alza Corporation | Osmotic system having laminar arrangement for programming delivery of active agent |
| US4270537A (en) | 1979-11-19 | 1981-06-02 | Romaine Richard A | Automatic hypodermic syringe |
| US4596556A (en) | 1985-03-25 | 1986-06-24 | Bioject, Inc. | Hypodermic injection apparatus |
| US4886499A (en) | 1986-12-18 | 1989-12-12 | Hoffmann-La Roche Inc. | Portable injection appliance |
| US5015235A (en) | 1987-02-20 | 1991-05-14 | National Carpet Equipment, Inc. | Syringe needle combination |
| US4790824A (en) | 1987-06-19 | 1988-12-13 | Bioject, Inc. | Non-invasive hypodermic injection device |
| US4940460A (en) | 1987-06-19 | 1990-07-10 | Bioject, Inc. | Patient-fillable and non-invasive hypodermic injection device assembly |
| US4941880A (en) | 1987-06-19 | 1990-07-17 | Bioject, Inc. | Pre-filled ampule and non-invasive hypodermic injection device assembly |
| US5339163A (en) | 1988-03-16 | 1994-08-16 | Canon Kabushiki Kaisha | Automatic exposure control device using plural image plane detection areas |
| US5141496A (en) | 1988-11-03 | 1992-08-25 | Tino Dalto | Spring impelled syringe guide with skin penetration depth adjustment |
| US5013556A (en) | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5064413A (en) | 1989-11-09 | 1991-11-12 | Bioject, Inc. | Needleless hypodermic injection device |
| US5503627A (en) | 1989-11-09 | 1996-04-02 | Bioject, Inc. | Ampule for needleless injection |
| US5312335A (en) | 1989-11-09 | 1994-05-17 | Bioject Inc. | Needleless hypodermic injection device |
| US6001826A (en) | 1989-12-21 | 1999-12-14 | Anormed, Inc. | Chemical compounds |
| US5021409A (en) | 1989-12-21 | 1991-06-04 | Johnson Matthey Plc | Antiviral cyclic polyamines |
| US5190521A (en) | 1990-08-22 | 1993-03-02 | Tecnol Medical Products, Inc. | Apparatus and method for raising a skin wheal and anesthetizing skin |
| US5527288A (en) | 1990-12-13 | 1996-06-18 | Elan Medical Technologies Limited | Intradermal drug delivery device and method for intradermal delivery of drugs |
| US5480381A (en) | 1991-08-23 | 1996-01-02 | Weston Medical Limited | Needle-less injector |
| US5417662A (en) | 1991-09-13 | 1995-05-23 | Pharmacia Ab | Injection needle arrangement |
| US5583131A (en) | 1991-12-16 | 1996-12-10 | Johnson Matthey Public Limited Company | Aromatic-linked polyamine macrocyclic compounds with anti-HIV activity |
| US5328483A (en) | 1992-02-27 | 1994-07-12 | Jacoby Richard M | Intradermal injection device with medication and needle guard |
| US5383851A (en) | 1992-07-24 | 1995-01-24 | Bioject Inc. | Needleless hypodermic injection device |
| US5520639A (en) | 1992-07-24 | 1996-05-28 | Bioject, Inc. | Needleless hypodermic injection methods and device |
| US5704911A (en) | 1992-09-28 | 1998-01-06 | Equidyne Systems, Inc. | Needleless hypodermic jet injector |
| US5569189A (en) | 1992-09-28 | 1996-10-29 | Equidyne Systems, Inc. | hypodermic jet injector |
| US5334144A (en) | 1992-10-30 | 1994-08-02 | Becton, Dickinson And Company | Single use disposable needleless injector |
| US5649912A (en) | 1994-03-07 | 1997-07-22 | Bioject, Inc. | Ampule filling device |
| US5466220A (en) | 1994-03-08 | 1995-11-14 | Bioject, Inc. | Drug vial mixing and transfer device |
| US5599302A (en) | 1995-01-09 | 1997-02-04 | Medi-Ject Corporation | Medical injection system and method, gas spring thereof and launching device using gas spring |
| US5811515A (en) | 1995-06-12 | 1998-09-22 | California Institute Of Technology | Synthesis of conformationally restricted amino acids, peptides, and peptidomimetics by catalytic ring closing metathesis |
| WO1997013537A1 (en) | 1995-10-10 | 1997-04-17 | Visionary Medical Products Corporation | Gas pressured needle-less injection device |
| US5893397A (en) | 1996-01-12 | 1999-04-13 | Bioject Inc. | Medication vial/syringe liquid-transfer apparatus |
| WO1997037705A1 (en) | 1996-04-11 | 1997-10-16 | Weston Medical Limited | Spring-powered dispensing device for medical purposes |
| US5993412A (en) | 1997-05-19 | 1999-11-30 | Bioject, Inc. | Injection apparatus |
| WO1999034850A1 (en) | 1998-01-08 | 1999-07-15 | Fiderm S.R.L. | Device for controlling the penetration depth of a needle, for application to an injection syringe |
| WO1999050461A1 (en) | 1998-03-30 | 1999-10-07 | Northwest Biotherapeutics, Inc. | Therapeutic and diagnostic applications based on the role of the cxcr-4 gene in tumorigenesis |
| WO1999052552A1 (en) | 1998-04-15 | 1999-10-21 | Brigham & Women's Hospital, Inc. | T cell inhibitory receptor compositions and uses thereof |
| US7132255B2 (en) | 1998-04-15 | 2006-11-07 | The Brigham And Women's Hospital, Inc. | Identification of compounds that bind biliary glycoprotein and affect cytotoxic T lymphocyte activity |
| WO2001085196A2 (en) | 2000-05-09 | 2001-11-15 | The University Of British Columbia | Cxcr4 antagonist treatment of hematopoietic cells |
| WO2001094420A1 (en) | 2000-06-05 | 2001-12-13 | The Trustees Of Columbia University In The City Of New York | Identification and use of human bone marrow-derived endothelial progenitor cells to improve myocardial function after ischemic injury |
| US20050043367A1 (en) | 2001-07-31 | 2005-02-24 | Gary Bridger | Methods to mobilize progenitor/stem cells |
| WO2003011277A2 (en) | 2001-07-31 | 2003-02-13 | Anormed Inc. | Methods to mobilize progenitor/stem cells |
| US20030220341A1 (en) | 2001-12-21 | 2003-11-27 | Gary Bridger | Chemokine receptor binding heterocyclic compounds with enhanced efficacy |
| WO2003079972A2 (en) | 2002-02-22 | 2003-10-02 | New River Parmaceuticals Inc. | Active agent delivery systems and methods for protecting and administering active agents |
| WO2003090512A2 (en) | 2002-04-23 | 2003-11-06 | The Trustees Of Columbia University In The City Of New York | Regeneration of endogenous myocardial tissue by induction of neovascularization |
| US8580244B2 (en) | 2002-09-06 | 2013-11-12 | Cerulean Pharma Inc. | Cyclodextrin-based polymers for therapeutics delivery |
| US20040047858A1 (en) | 2002-09-11 | 2004-03-11 | Blumberg Richard S. | Therapeutic anti-BGP(C-CAM1) antibodies and uses thereof |
| US7495099B2 (en) | 2002-10-31 | 2009-02-24 | Nippon Kayaku Kabushiki Kaisha | High-molecular weight derivatives of camptothecins |
| US20040209921A1 (en) | 2003-04-11 | 2004-10-21 | Gary Bridger | CXCR4 chemokine receptor binding comounds |
| US20050059702A1 (en) | 2003-04-22 | 2005-03-17 | Bridger Gary J. | Chemokine receptor binding heterocyclic compounds with enhanced efficacy |
| US20050277670A1 (en) | 2003-12-11 | 2005-12-15 | Anormed Inc. | Chemokine receptor binding compounds |
| WO2006121168A1 (en) | 2005-05-09 | 2006-11-16 | Ono Pharmaceutical Co., Ltd. | Human monoclonal antibodies to programmed death 1(pd-1) and methods for treating cancer using anti-pd-1 antibodies alone or in combination with other immunotherapeutics |
| US8008449B2 (en) | 2005-05-09 | 2011-08-30 | Medarex, Inc. | Human monoclonal antibodies to programmed death 1 (PD-1) and methods for treating cancer using anti-PD-1 antibodies alone or in combination with other immunotherapeutics |
| US20100055088A1 (en) | 2005-05-25 | 2010-03-04 | Hadasit Medical Research Services And Development Ltd. | CXCR4 Antagonists for Wound Healing and Re-Epithelialization |
| WO2007005874A2 (en) | 2005-07-01 | 2007-01-11 | Medarex, Inc. | Human monoclonal antibodies to programmed death ligand 1 (pd-l1) |
| US7943743B2 (en) | 2005-07-01 | 2011-05-17 | Medarex, Inc. | Human monoclonal antibodies to programmed death ligand 1 (PD-L1) |
| US20090221683A1 (en) | 2005-10-18 | 2009-09-03 | Caritas St.Elizabeth Medical Center Of Boston, Inc | Combination of CXCR4 Antagonist and Morphogen to Increase Angiogenesis |
| US20100178271A1 (en) | 2006-08-07 | 2010-07-15 | Genzyme Corporation | Combination Therapy |
| US8354509B2 (en) | 2007-06-18 | 2013-01-15 | Msd Oss B.V. | Antibodies to human programmed death receptor PD-1 |
| US8541203B2 (en) | 2007-08-22 | 2013-09-24 | The Regents Of The University Of California | Activatable binding polypeptides and methods of identification and use thereof |
| US20100247521A1 (en) | 2007-10-26 | 2010-09-30 | Jones Richard B | Therapeutic and Diagnostic Methods Using TIM-3 |
| WO2009101611A1 (en) | 2008-02-11 | 2009-08-20 | Curetech Ltd. | Monoclonal antibodies for tumor treatment |
| WO2009114335A2 (en) | 2008-03-12 | 2009-09-17 | Merck & Co., Inc. | Pd-1 binding proteins |
| WO2010019570A2 (en) | 2008-08-11 | 2010-02-18 | Medarex, Inc. | Human antibodies that bind lymphocyte activation gene-3 (lag-3), and uses thereof |
| WO2010077634A1 (en) | 2008-12-09 | 2010-07-08 | Genentech, Inc. | Anti-pd-l1 antibodies and their use to enhance t-cell function |
| US8513390B2 (en) | 2009-01-12 | 2013-08-20 | Cytomx Therapeutics, Inc. | Modified antibody compositions, methods of making and using thereof |
| US20120039906A1 (en) | 2009-02-09 | 2012-02-16 | INSER (Institut National de la Recherche Medicale) | PD-1 Antibodies and PD-L1 Antibodies and Uses Thereof |
| WO2010125571A1 (en) | 2009-04-30 | 2010-11-04 | Tel Hashomer Medical Research Infrastructure And Services Ltd. | Anti ceacam1 antibodies and methods of using same |
| US20130156774A1 (en) | 2010-06-18 | 2013-06-20 | The Brigham And Women's Hospital, Inc. | Bi-specific antibodies against tim-3 and pd-1 for immunotherapy in chronic immune conditions |
| WO2013006490A2 (en) | 2011-07-01 | 2013-01-10 | Cellerant Therapeutics, Inc. | Antibodies that specifically bind to tim3 |
| US20140271618A1 (en) | 2011-10-11 | 2014-09-18 | Ramot At Tel-Aviv University Ltd. | Antibodies to carcinoembryonic antigen-related cell adhesion molecule (ceacam) |
| WO2013054331A1 (en) | 2011-10-11 | 2013-04-18 | Tel Hashomer Medical Research Infrastructure And Services Ltd. | Antibodies to carcinoembryonic antigen-related cell adhesion molecule (ceacam) |
| WO2013082366A1 (en) | 2011-12-01 | 2013-06-06 | The Brigham And Women's Hospital, Inc. | Anti-ceacam1 recombinant antibodies for cancer therapy |
| WO2013169739A1 (en) | 2012-05-07 | 2013-11-14 | The General Hospital Corporation | Novel compositions and uses of anti-hypertension agents for cancer therapy |
| WO2014022332A1 (en) | 2012-07-31 | 2014-02-06 | The Brigham And Women's Hospital, Inc. | Modulation of the immune response |
| WO2014059251A1 (en) | 2012-10-12 | 2014-04-17 | The Brigham And Women's Hospital, Inc. | Enhancement of the immune response |
| US20140308234A1 (en) * | 2013-04-09 | 2014-10-16 | Massachusetts Institute Of Technology | Drug delivery polymer and uses thereof |
| US9381253B2 (en) | 2013-04-09 | 2016-07-05 | Massachusetts Institute Of Technology | Drug delivery polymer and uses thereof |
| US20150087810A1 (en) | 2013-09-25 | 2015-03-26 | Cytomx Therapeutics, Inc. | Matrix Metalloproteinase Substrates And Other Cleavable Moieties And Methods Of Use Thereof |
| WO2016140714A1 (en) | 2015-03-05 | 2016-09-09 | The General Hospital Corporation | Novel compositions and uses of metformin agents |
Non-Patent Citations (116)
| Title |
|---|
| "Handbook of Chemistry and Physics" |
| "Harrison's Principles of Internal Medicine", MCGRAW-HILL |
| "Physicians' Desk Reference", 1997, MEDICAL ECONOMICS CO. |
| "Remington's Pharmaceutical Sciences", 1985 |
| A. M. LEBEAU ET AL., PROC. NATL. ACAD. SCI. U.S.A., vol. 110, 2013, pages 93 - 98 |
| ABDEL-WAHAB, O. ET AL., ANNU. REV. MED., vol. 60, 2009, pages 233 - 45 |
| ABRA, RM ET AL., J. LIPOSOME RES., vol. 12, 2002, pages 1 - 3 |
| ACS CHEM BIOL, vol. 10, 2015, pages 22 - 39 |
| ADORINI, CELLULAR IMMUNOLOGY, vol. 233, 2005, pages 115 - 124 |
| AGOSTINIS, P. ET AL., CA CANCER J. CLIN., vol. 61, 2011, pages 250 - 281 |
| AGRAWAL, M. ET AL., CANCER, 2010, pages 1 - 15 |
| ALLEN ET AL., BIOCHIMICA ET BIOPHYSICA ACTA, vol. 1237, 1995, pages 99 - 108 |
| AM J CARDIOVASC DRUGS, 18 September 2015 (2015-09-18) |
| ANDRESEN, T.L., LIPOSOMES FOR POSITRON EMISSION TOMOGRAPHY (PET) IMAGING, RADIOTHERAPY, AND THERANOSTICS, Retrieved from the Internet <URL:www.nanomedicine.dtu.dk/Research/Liposomes> |
| ANTONIOU, KM ET AL., CURR OPIN RHEUMATOL., vol. 20, 2008, pages 686 - 91 |
| BARNES JONATHAN C ET AL: "Using an RNAi Signature Assay To Guide the Design of Three-Drug-Conjugated Nanoparticles with Validated Mechanisms, In Vivo Efficacy, and Low Toxicity", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, AMERICAN CHEMICAL SOCIETY, US, vol. 138, no. 38, 28 September 2016 (2016-09-28), pages 12494 - 12501, XP002773230, ISSN: 0002-7863, [retrieved on 20160914], DOI: 10.1021/JACS.6B06321 * |
| BIOORG MED CHEM LETT, vol. 25, 2015, pages 1842 - 1848 |
| BIOORGMED CHEM LETT, vol. 25, 2015, pages 1842 - 1848 |
| BIRD, L., NATURE REVIEWS IMMUNOLOGY, vol. 12, 2012, pages 227 |
| BLUME ET AL., BIOCHIMICA ET BIOPHYSICA ACTA, vol. 1149, 1993, pages 180 - 184 |
| BOIS RM, SEMIN RESPIR CRIT CARE MED, vol. 27, 2006, pages 581 - 8 |
| BOULWARE, K.T.; DAUGHERTY, P.S., PROC NATL ACAD SCI USA, vol. 103, 2006, pages 7583 - 7588 |
| BRASS, DM ET AL., PROC AM THORAC SOC, vol. 4, 2007, pages 92 - 100 |
| C. LIU ET AL., CANCER RES., vol. 63, 2003, pages 2957 - 2964 |
| CANCER RES, vol. 73, 2013, pages 3336 - 3346 |
| CARRUTHERS: "Some Modern Methods of Organic Synthesis", 1987, CAMBRIDGE UNIVERSITY PRESS |
| CATANE, R ET AL., INT J RADIAT ONCOL BIOL PHYS, vol. 5, 1979, pages 1513 - 8 |
| CHANDRAIAH GODUGU: "Abstract 2139: Effect of telmisartan on triple negative breast cancer (TNBC) and lung cancer tumor progression and intratumoral distribution of nanoparticles. | Cancer Research", 1 April 2013 (2013-04-01), XP055452710, Retrieved from the Internet <URL:http://cancerres.aacrjournals.org/content/73/8_Supplement/2139> [retrieved on 20180221] * |
| CHAUHAN ET AL., NATURE COMM, vol. 4, 2013, pages 2516 |
| CHAUHAN ET AL., NATURE COMM., vol. 4, 2013, pages 2516 |
| CHAUHAN, V ET AL., ANNU REV CHEM BIOMOL ENG., vol. 2, no. 1, 2011, pages 281 - 98 |
| CHEM. EUR. J., vol. 7, 2001, pages 5299 |
| CLIN CANCER RES, vol. 21, 2015, pages 1628 - 1638 |
| DEFREES ET AL., JOURNAL OF THE AMERICAN CHEMISTRY SOCIETY, vol. 118, 1996, pages 6101 - 6104 |
| DEPERTHES, D., BIOL CHEM, vol. 383, 2002, pages 1107 - 1112 |
| DESNOYERS, L.R. ET AL., SCI TRANSL MED, vol. 5, 2013, pages 207ra144 |
| ECKHARD, U ET AL., MATRIX BIOL, 2015 |
| ELIEL, E.L.: "Stereochemistry of Carbon Compounds", 1962, MCGRAW-HILL |
| EN-TONG YI ET AL: "Telmisartan attenuates hepatic fibrosis in bile duct-ligated rats", ACTA PHARMACOLOGICA SINICA, vol. 33, no. 12, 29 October 2012 (2012-10-29), GB, pages 1518 - 1524, XP055452708, ISSN: 1671-4083, DOI: 10.1038/aps.2012.115 * |
| EXPERT OPIN THER PAT, vol. 24, 2014, pages 185 - 199 |
| FEBSLETTER, vol. 550, no. 1-3, 2003, pages 79 - 83 |
| FURSTNER ET AL., J. AM. CHEM. SOC., vol. 121, 1999, pages 9453 |
| GALLIVAN ET AL., TETRAHEDRON LETTERS, vol. 46, 2005, pages 2577 - 2580 |
| GOEL ET AL., PHYSIOL REV., vol. 91, 2011, pages 1071 - 1121 |
| GOODMAN; GILMAN: "Pharmacological Basis of Therapeutics", 1990, UNITED STATES PHARMACOPEIA, THE NATIONAL FORMULARY, USP XII NF XVII |
| GREEN, FH ET AL., TOXICOL PATHOL., vol. 35, 2007, pages 136 - 47 |
| GRIFFON-ETIENNE, G. ET AL., CANCER RES., vol. 59, no. 15, 1999, pages 3776 - 82 |
| GRUBBS ET AL., ACC. CHEM. RES., vol. 28, 1995, pages 446 - 452 |
| HAAS, M. J. ET AL., SCIBX, vol. 7, no. 15, 2014 |
| HARRIS, J.L., PROC NATL ACAD SCI USA, vol. 97, 2000, pages 7754 - 7759 |
| J MED CHEM, vol. 56, 2013, pages 9251 - 9264 |
| JACQUES ET AL.: "Enantiomers, Racemates and Resolutions", 1981, WILEY INTERSCIENCE |
| JAIN, CANCER CELL, vol. 26, no. 5, 2014, pages 605 - 622 |
| JAIN, R. K., NAT MED, vol. 4, 1998, pages 655 - 657 |
| JELENA GRAHOVAC: "Abstract B41: The angiotensin receptor blocker telmisartan inhibits the growth of pancreatic ductal adenocarcinoma and improves survival | Cancer Research", 1 May 2016 (2016-05-01), XP055452711, Retrieved from the Internet <URL:http://cancerres.aacrjournals.org/content/76/24_Supplement/B41> [retrieved on 20180221] * |
| JOHNSON, J ET AL., MACROMOLECULES, vol. 43, 2010, pages 10326 - 10335 |
| JONES, D., NAT REV DRUG DISCOV, vol. 6, 2007, pages 174 - 175 |
| K. T. BOULWARE; P. S. DAUGHERTY, PROC. NATL. ACAD. SCI. U.S.A., vol. 103, 2006, pages 7583 - 7588 |
| K. UHLAND, CELL. MOL. LIFE SCI., vol. 63, 2006, pages 2968 - 2978 |
| KEY, J.; LEARY, J., INT J NANOMEDICINE, vol. 9, 2014, pages 711 - 726 |
| KLIBANOV ET AL., JOURNAL OF LIPOSOME RESEARCH, vol. 2, 1992, pages 321 - 334 |
| KUN-CHUN CHIANG ET AL: "Vitamin D for the prevention and treatment of pancreatic cancer", WORLD JOURNAL OF GASTROENTEROLOGY, vol. 15, no. 27, 21 July 2009 (2009-07-21), CN, pages 3349, XP055452714, ISSN: 1007-9327, DOI: 10.3748/wjg.15.3349 * |
| LAMBETH, R. H.; PEDERSON, S. J.; BARANOSKI, M.; RAWLETT, A. M., J. POLYM. SCI. PART A POLYM. CHEM., vol. 48, 2010, pages 5752 - 5757 |
| LANGER, SCIENCE, vol. 249, 1990, pages 1527 - 1533 |
| LAROCK: "Comprehensive Organic Transformations", 1989, VCH PUBLISHERS, INC. |
| LEAVY, O., NATURE REVIEWS IMMUNOLOGY, vol. 12, 2012, pages 227 |
| LESLIE, KO ET AL., SEMIN RESPIR CRIT CARE MED., vol. 28, 2007, pages 369 - 78 |
| LEYSSENS, C. ET AL., FRONT PHYSIOL, 2014 |
| LOB, S. ET AL., NAT REV CANCER, vol. 9, 2009, pages 445 - 452 |
| MELILLO G, MOL. CANCER RES., vol. 4, no. 9, 2006, pages 601 - 5 |
| MOGHIMI, S. M. ET AL., FASEB J., vol. 19, 2005, pages 311 - 330 |
| MONNEY ET AL., NATURE, vol. 415, 2002, pages 536 - 541 |
| MONTSERRAT PÉREZ-SALVIA ET AL: "Bromodomain inhibitors and cancer therapy: From structures to applications", EPIGENETICS, vol. 12, no. 5, 2 December 2016 (2016-12-02), US, pages 323 - 339, XP055452716, ISSN: 1559-2294, DOI: 10.1080/15592294.2016.1265710 * |
| MOON Y W MOON Y W ET AL: "Targeting the indoleamine 2,3-dioxygenase pathway in cancer", JOURNAL FOR IMMUNOTHERAPY OF CANCER, BIOMED CENTRAL LTD, LONDON, UK, vol. 3, no. 1, 15 December 2015 (2015-12-15), pages 1 - 10, XP009503017, ISSN: 2051-1426, DOI: 10.1186/S40425-015-0094-9 * |
| NGIOW ET AL., CANCER RES, vol. 71, 2011, pages 3540 - 3551 |
| NING DING ET AL: "BRD4 is a novel therapeutic target for liver fibrosis", PROCEEDINGS NATIONAL ACADEMY OF SCIENCES PNAS, 7 December 2015 (2015-12-07), US, XP055452715, ISSN: 0027-8424, DOI: 10.1073/pnas.1522163112 * |
| ONCOTARGET, vol. 6, 2015, pages 17698 - 17712 |
| PALUCKA, K. ET AL., NATURE REVIEWS CANCER, vol. 12, 2012, pages 265 - 277 |
| PARAMOTHAYAN, S ET AL., RESPIR MED., vol. 102, 2008, pages 1 - 9 |
| PARDOLL, D.M., NATURE REVIEWS CANCER, vol. 12, 2012, pages 252 - 264 |
| RAKITA, L ET AL., AM HEART J, vol. 106, 1983, pages 906 - 16 |
| RICE, J.J. ET AL., PROTEIN SCI, vol. 15, 2006, pages 825 - 836 |
| RIZZO, L ET AL., CURR OPIN BIOTECHNOL, vol. 24, 2013, pages 1159 - 1166 |
| ROHRIG, U.F. ET AL., JMED CHEM, vol. 58, 2015, pages 9421 - 9437 |
| S. ULISSE ET AL., CURR. CANCER DRUG TARGETS, vol. 9, 2009, pages 32 - 71 |
| SALMASO S.; CALICETI, P. J, DRUG DELIV, vol. 2013, 2013, pages 1 - 19 |
| SANHAI, W. R. ET AL., NAT NANOTECHNOL, vol. 3, 2008, pages 242 - 244 |
| SAPRA, P.; ALLEN, TM, PROG. LIPID RES., vol. 42, no. 5, 2003, pages 439 - 62 |
| SCHROCK ET AL., ORGANOMETALLICS, vol. 1, 1982, pages 1645 |
| SCOLLETTA ET AL., MEDIATORS OF INFLAMMATION, 2013 |
| SEMENZA GL, DRUG DISCOV. TODAY, vol. 12, no. 19-20, 2007, pages 853 - 9 |
| SETON-ROGERS, S., NATURE REVIEWS CANCER, vol. 12, 2012, pages 230 - 231 |
| SMITH; MARCH: "March's Advanced Organic Chemistry", 2001, JOHN WILEY & SONS, INC. |
| SOUNNI, N.E. ET AL., DISEASES MODELS & MECHANISMS, vol. 3, 2010, pages 317 - 332 |
| STEELE, MP ET AL., RESPIRATION, vol. 74, 2007, pages 601 - 8 |
| SUGAHARA, KN ET AL., SCIENCE, vol. 328, 2010, pages 1031 - 5 |
| SWIGRIS, JJ ET AL., CHEST., vol. 133, 2008, pages 271 - 80 |
| SYED ALWI SS ET AL., BR. J. NUTR., vol. 104, no. 9, 2010, pages 1288 - 96 |
| T. W. GREENE; P. G. M. WUTS: "Protecting Groups in Organic Synthesis", 1999, JOHN WILEY & SONS |
| TAMAMURA, H. ET AL., ORG. BIOMOL. CHEM., vol. 1, 2003, pages 3656 - 3662 |
| THOMAS SORRELL: "Organic Chemistry", 1999, UNIVERSITY SCIENCE BOOKS |
| TWOHIG, KJ ET AL., CLIN CHESTMED., vol. 11, 1990, pages 31 - 54 |
| VAN KEMPEN ET AL., EUR CANCER, vol. 42, 2006, pages 728 - 734 |
| VARICCHIO, L. ET AL., EXPERT REV. HEMATOL., vol. 2, no. 3, 2009, pages 315 - 334 |
| VOUGIOUKALAKIS, G. C., CHEM. A EUR. J., vol. 18, 2012, pages 8868 - 8880 |
| WHEELER, P.; PHILLIPS, J. H.; PEDERSON, R. L., ORG. PROCESS RES. DEV., vol. 20, 2016, pages 1182 - 1190 |
| WILEN ET AL., TETRAHEDRON, vol. 33, 1977, pages 2725 |
| WILEN, S.H.: "Tables of Resolving Agents and Optical Resolutions", 1972, UNIV. OF NOTRE DAME PRESS, pages: 268 |
| WITTEN CM, ARCH PHYS MED REHABIL, vol. 70, 1989, pages 55 - 7 |
| WONG, D. ET AL., CLIN. CANCER RES., vol. 14, no. 24, 2008, pages 7975 - 7980 |
| YE, Y. ET AL., BIOORGMED CHEM LETT., vol. 21, no. 4, 2011, pages 1146 - 50 |
| ZALIPSKY, BIOCONJUGATE CHEMISTRY, vol. 4, 1993, pages 296 - 299 |
| ZALIPSKY, FEBSLETTERS, vol. 353, 1994, pages 71 - 74 |
| ZALIPSKY: "Stealth Liposomes", 1995, CRC PRESS |
| ZHENG ET AL., PLOS ONE., vol. 5, no. 9, 2 September 2010 (2010-09-02), pages e12529 |
| ZISMAN, DA ET AL., SARCOIDOSIS VASE DIFFUSE LUNGDIS., vol. 18, 2001, pages 243 - 52 |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11678976B2 (en) | 2010-04-29 | 2023-06-20 | Adaptilens, Llc | Injectable physiologically adaptive intraocular lenses (IOL's) |
| US11701454B2 (en) | 2017-02-23 | 2023-07-18 | Medtronic Vascular, Inc. | Drug-coated medical devices |
| US10772993B2 (en) * | 2017-02-23 | 2020-09-15 | Medtronic Vascular, Inc. | Drug coated medical devices |
| US20180236139A1 (en) * | 2017-02-23 | 2018-08-23 | Medtronic Vascular Inc. | Drug coated medical devices |
| US11752221B2 (en) | 2017-06-30 | 2023-09-12 | Massachusetts Institute Of Technology | Brush-arm star polymer imaging agents and uses thereof |
| US11376264B2 (en) | 2017-07-24 | 2022-07-05 | Salk Institute For Biological Studies | Use of bromodomain-containing protein 9 antagonists in combination with vitamin D receptor agonists in diabetes treatment |
| US11897905B2 (en) | 2018-08-17 | 2024-02-13 | Massachusetts Institute Of Technology | Degradable polymers of a cyclic silyl ether and uses thereof |
| CN109593156A (en) * | 2018-12-14 | 2019-04-09 | 长沙加美乐素化工有限公司 | A kind of technique synthesizing comprehensive water-reducing agent using six carbon monomers |
| WO2021076821A2 (en) | 2019-10-16 | 2021-04-22 | Massachusetts Institute Of Technology | Brush prodrugs and uses thereof |
| US11724985B2 (en) | 2020-05-19 | 2023-08-15 | Cybin Irl Limited | Deuterated tryptamine derivatives and methods of use |
| US11746088B2 (en) | 2020-05-19 | 2023-09-05 | Cybin Irl Limited | Deuterated tryptamine derivatives and methods of use |
| US11958807B2 (en) | 2020-05-19 | 2024-04-16 | Cybin Irl Limited | Deuterated tryptamine derivatives and methods of use |
| US11834410B2 (en) | 2020-05-19 | 2023-12-05 | Cybin Irl Limited | Deuterated tryptamine derivatives and methods of use |
| WO2022093106A1 (en) * | 2020-10-30 | 2022-05-05 | Agency For Science, Technology And Research | Bioactive synthetic copolymer, bioactive macromolecule and related methods thereof |
| WO2023121753A1 (en) * | 2021-12-20 | 2023-06-29 | Massachusetts Institute Of Technology | Biomolecule-polymer-pharmaceutical agent conjugates for delivering the pharmaceutical agent |
| WO2023211384A1 (en) * | 2022-04-28 | 2023-11-02 | Agency For Science, Technology And Research | Hyaluronic acid-based synthetic copolymer, material containing said copolymer and related methods thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2018106738A1 (en) | Brush-arm star polymers, conjugates and particles, and uses thereof | |
| US11338038B2 (en) | Branched multi-functional macromonomers and related polymers and uses thereof | |
| US20200369685A1 (en) | Boronic ester prodrugs and uses thereof | |
| Ai et al. | Biological evaluation of a novel doxorubicin− peptide conjugate for targeted delivery to EGF receptor-overexpressing tumor cells | |
| US11897905B2 (en) | Degradable polymers of a cyclic silyl ether and uses thereof | |
| US20170348431A1 (en) | Drug delivery polymers and uses thereof | |
| Roh et al. | Photodynamic therapy using photosensitizer-encapsulated polymeric nanoparticle to overcome ATP-binding cassette transporter subfamily G2 function in pancreatic cancer | |
| WO2021141662A1 (en) | Proteolysis targeting chimeric molecules (protacs) with functional handles and uses thereof | |
| US20240033364A1 (en) | Castration resistant prostate cancer | |
| US20210386861A1 (en) | Brush prodrugs and uses thereof | |
| US20210113701A1 (en) | Brush prodrugs and uses thereof | |
| Sivák et al. | Polymer-ritonavir derivate nanomedicine with pH-sensitive activation possesses potent anti-tumor activity in vivo via inhibition of proteasome and STAT3 signaling | |
| Carney et al. | Fn14-Directed DART nanoparticles selectively target neoplastic cells in preclinical models of triple-negative breast cancer brain metastasis | |
| Wang et al. | A novel strategy conjugating PD-L1 polypeptide with doxorubicin alleviates chemotherapeutic resistance and enhances immune response in colon cancer | |
| WO2023122599A1 (en) | Glycosylated dendrimers for targeted intracellular delivery | |
| WO2023121753A1 (en) | Biomolecule-polymer-pharmaceutical agent conjugates for delivering the pharmaceutical agent | |
| KR102362265B1 (en) | Drug conjugate having enhanced drug delivery and internalization efficiency | |
| Ahn et al. | Synergistic Approach of Antibody‐Photosensitizer Conjugate Independent of KRAS‐Mutation and Its Downstream Blockade Pathway in Colorectal Cancer | |
| WO2013125730A2 (en) | Cationic graft-copolymer for drug delivery system | |
| Kim et al. | Immune Checkpoint-Blocking Nanocages Cross the Blood–Brain Barrier and Impede Brain Tumor Growth | |
| US20240009321A1 (en) | Immunogenic nanovesicles for cancer immunotherapy | |
| Liu et al. | Enzyme/pH Dual-Responsive Engineered Nanoparticles for Improved Tumor Immuno-Chemotherapy | |
| Zhang | Design of a Private Passageway Fusion Receptor for Sensitive Control of Adoptive Cell Therapies | |
| Goodwin | Treatment of Liver Metastasis Via Nanoparticle Delivery of Engineered Gene Immunotherapies Expressed Locally and Transiently by the Liver Hepatocytes | |
| Teh | Strategic Targeting of Integrin α V β 3 Receptors by Multivalent Expression of RGD Peptidomimetics |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17818395 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 17818395 Country of ref document: EP Kind code of ref document: A1 |