WO2016202808A2 - NEW SYNTHESIS OF TAPENTADOL-HCl INTERMEDIATES - Google Patents
NEW SYNTHESIS OF TAPENTADOL-HCl INTERMEDIATES Download PDFInfo
- Publication number
- WO2016202808A2 WO2016202808A2 PCT/EP2016/063648 EP2016063648W WO2016202808A2 WO 2016202808 A2 WO2016202808 A2 WO 2016202808A2 EP 2016063648 W EP2016063648 W EP 2016063648W WO 2016202808 A2 WO2016202808 A2 WO 2016202808A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methoxyphenyl
- methyl
- acid
- tapentadol
- azide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- KCDQRMLWPZCJPY-QMMMGPOBSA-N C[C@@H](CC(O)=O)C(c1cc(OC)ccc1)=O Chemical compound C[C@@H](CC(O)=O)C(c1cc(OC)ccc1)=O KCDQRMLWPZCJPY-QMMMGPOBSA-N 0.000 description 2
- KWDQAMIFENFXDB-IINYFYTJSA-N CC[C@@]([C@@H](C)C1)(c2cc(OC)ccc2)OC1=O Chemical compound CC[C@@]([C@@H](C)C1)(c2cc(OC)ccc2)OC1=O KWDQAMIFENFXDB-IINYFYTJSA-N 0.000 description 1
- OBXGWCZNKGUYQM-GXFFZTMASA-N CC[C@H]([C@@H](C)CC(O)=O)c1cccc(OC)c1 Chemical compound CC[C@H]([C@@H](C)CC(O)=O)c1cccc(OC)c1 OBXGWCZNKGUYQM-GXFFZTMASA-N 0.000 description 1
- QLKNTNFWYQKHNF-XJKSGUPXSA-N CC[C@H]([C@@H](C)CNC(OC(C)(C)C)=O)c1cc(OC)ccc1 Chemical compound CC[C@H]([C@@H](C)CNC(OC(C)(C)C)=O)c1cc(OC)ccc1 QLKNTNFWYQKHNF-XJKSGUPXSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/54—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C217/56—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms
- C07C217/62—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms linked by carbon chains having at least three carbon atoms between the amino groups and the six-membered aromatic ring or the condensed ring system containing that ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/02—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton by reactions involving the formation of amino groups from compounds containing hydroxy groups or etherified or esterified hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C215/00—Compounds containing amino and hydroxy groups bound to the same carbon skeleton
- C07C215/46—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C215/48—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by hydroxy groups
- C07C215/54—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by hydroxy groups linked by carbon chains having at least three carbon atoms between the amino groups and the six-membered aromatic ring or the condensed ring system containing that ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/16—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by singly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/09—Preparation of carboxylic acids or their salts, halides or anhydrides from carboxylic acid esters or lactones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/40—Unsaturated compounds
- C07C59/58—Unsaturated compounds containing ether groups, groups, groups, or groups
- C07C59/64—Unsaturated compounds containing ether groups, groups, groups, or groups containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/40—Unsaturated compounds
- C07C59/76—Unsaturated compounds containing keto groups
- C07C59/90—Unsaturated compounds containing keto groups containing singly bound oxygen-containing groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/26—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D307/30—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D307/32—Oxygen atoms
- C07D307/33—Oxygen atoms in position 2, the oxygen atom being in its keto or unsubstituted enol form
Definitions
- the present invention relates to a process for the synthesis of tapentadol intermediates, the process comprising the steps of:
- step b) Stereoselective reacting (3S)-4-(3-methoxyphenyl)-3-methyl-4-oxobutanoic acid obtained in step a) in the presence of an organometallic-reagent to obtain (45,5R)-5-ethyl-5-(3- methoxyphenyl)-4-methyldihydrofuran-2(3H)-one
- the invention is directed to the use of different intermediates in the production of tapentadol and the pharmaceutical use of tapentadol HQ synthesized via the inventive route of synthesis.
- Tapentadol and its salt forms act, among other mode of actions, as opioid agonist and as noradrenalin uptake inhibitor. It has valuable pharmacological and therapeutic properties.
- the drug acts centrally as analgesic.
- Tapentadol is chemically known as 3- [(lR,2R)-3-(dimethylamino)-l-ethyl-2-methylpropyl]phenol (formula I).
- WO 2011 080736 Al describes the preparation of Tapentadol through phenylpent-2-en amide.
- WO 2011 080756 Al consists of the preparation of a cyano-intermediate as key intermediate towards Tapentadol preparation.
- WO 2011 092719 Al describes a method of Tapentadol preparation using the key intermediate (bromopropyl)methoxybenzene .
- WO 2012 001571 Al describes Tapentadol synthesis with a benzyl group as substituent at the amino function.
- WO 2012 089177 Al consists of the description of Tapentadol synthesis of a protected alkene acid as intermediate.
- WO 2012 023147 Al comprises the reaction of (dimethylamino)-2-methylpentan-3-one with anisole-Grignard.
- WO 2012 038974 Al comprises a Tapentadol preparation using l-(3-hydroxyphenyl)propan- 1-one with an appropriate Grignard reagent.
- WO 2012 069004 Al describes the Tapentadol synthesis using Methane sulfonyl esters.
- WO 2012 103799 Al describes the use of Evans auxiliary to generate key chiral intermediates for Tapentadol preparation.
- Such route is inventively achieved by firstly stereoselectively reacting the starting educt, the ketoester (I) in step a) to obtain the stereoselective ketoacid, intermediate (II).
- the starting educt is a C1-C4 alkyl ketoester, wherein the alkyl can for instance be a straight chain methyl, ethyl, propyl, butyl or, if applicable, one of their branched homologues.
- the stereoselective conversion is preferably performed in the presence of a buffered solvent, preferably at least comprising water, and may include one or two process steps addressing the ester hydrolysis and stereoselective conversion.
- Suitable enzymes for performing such reaction and their optimum processing conditions are known to the skilled artisan and can for instance be selected from the group comprising lipases, keto reductases and esterases which may be immobilized on porous organic or inorganic carriers like microporous polypropylene or kaolinite or can be used as soluble enzyme.
- the reaction temperature for the enzymatic conversion may be selected as a function of the used enzyme and may vary from 5 °C up to or below the denaturation temperature of the enzyme, for instance up to 55 °C, preferentially between 20 and 35°C.
- the ester hydrolysis may result as a consequence of the enzymatic reaction conditions or may be achieved separately by acid or base catalyzed hydrolysis before or after the stereoselective conversion.
- the enantiomeric excess (ee) of the desired intermediate (II) obtainable by step a) can be higher than 90%, more specifically higher than 95% or even higher than 98%.
- step b) secondly the stereoselective keto-acid (II) is transformed via the use of an ethyl group transferring organometallic reagent to intermediate (III), a keto-furan-derivative.
- the conversion may be generally performed in a dried aprotic solvent under a protective atmosphere and in general organometallic compounds known as Grignard reagents can be used within this step.
- the cyclisation of the reaction product of the keto-acid (II) and the organometallic compound may be achieved by the addition of a strong acid, resulting in the stereoselective formation of the keto-furan (III).
- Suitable pH- ranges for this conversion may be pH ⁇ 2, preferably ⁇ 1.5 and even more preferred ⁇ 1.0.
- the intermediate (III) can be obtained with diastereomeric ratios of larger than 3:1, preferably larger than 4: 1.
- Step c) thirdly is targeted to the stereoselective conversion of the ketofuran (III) to the (3S,4R)-acid (IV) via metal catalyzed reduction in the presence of an organic base in a hydrogen atmosphere.
- Suitable metal catalysts can be either homogeneous or heterogeneous hydrogenation catalysts, wherein heterogeneous catalysts are preferred. Suitable examples are for instance catalysts comprising Pt, Pd, Rh, Ru, Ni, Co, Fe Cu, Cr or Zn.
- Preferred catalysts comprise platinum, palladium, rhodium, ruthenium or mixtures thereof.
- the metal catalyst may be mounted on a support, preferably a support such as activated carbon, calcium carbonate, barium sulfate, barium carbonate, silicium dioxide, alumina or may for example be present as a colloid in solution.
- the hydrogen can be added just at the beginning of the reaction, added repeatedly, generated in situ or being feed continuously into the reactor, wherein a sequential feeding of the hydrogen is preferred.
- the hydrogen pressure, hydrogenation time and the temperature of conversion is a strong function of the catalyst and the adaption of these reaction conditions is known the skilled artisan.
- Suitable organic bases in step c) may for example be non-nucleophilic bases of moderate strength, i.e. preferably amines and nitrogen heterocycles exhibiting a pKa of the conjugate acid of approximately 10-13.
- Examples for such bases are Diisopropylamine (DIP A), N,N-Diisopropylethylamine (DIPEA), l,8-Diazabicycloundec-7-ene (DBU), 2,6-Di-tert-butylpyridine, or Phosphazene bases, such as t-Bu- P 4 .
- the organic base can be N,N-Diisopropylethylamine.
- the solvent of the reaction in step c) is preferably an alcohol like methanol, ethanol, propanol, isopropanol and butanol, methanol being preferred.
- step d) fourthly the (3S,4R)-acid (IV) is converted to the amine (V).
- This can either be achieved by a modified Curtius- (L), a Hofmann- (ii.) or via a Lossen-reaction, wherein in all three routes an isocyanate reaction intermediate is formed.
- This isocyanate is not stable in aqueous solution and reacts by addition of water to the carbamic acid, followed by decarboxylation to the amine.
- Another possibility in the synthesis route includes the formation of a carbamate by introduction of an alcohol. Therefore, it is also possible to generate a stable intermediate including a protective group. The protective group can later on be removed by the conventional methods known to the skilled in the art.
- an azide in presence of a base and subsequently a pharmaceutically acceptable acid HX are used.
- Suitable azides include, but are not limited to, inorganic or organic azides, for instance alkali salt azide, trimethylsilylazide, tosylazide or trifluoromethylazide.
- Possible pharmaceutically acceptable acids HX can for example be found in the "Handbook of Pharmaceutical Salts: Properties, Selection and Use", Editors P. H. Stahl and C. G. Wermuth, Weinheim/Ziirich:Wiley-VCH/VHCA, 2002.
- hydrochloric acid is used.
- carboxylic acid activating agent a carboxylic acid activating agent
- Common reagents for activation are known to the skilled artisan and can for instance be selected from the group comprising or consisting of thionylchloride, carbodiimide, carbonyldiimidazole, carboxylic acid anhydride, N- Hydroxysuccinimide (NHS) or derivatives thereof.
- Other common coupling/activation reagents can also be used.
- Suitable carbodiimides may for instance be dicyclohexylcarbodiimide (DCC) or 1 -Ethyl - 3-(3-dimethylaminopropyl)carbodiimide (EDC).
- the activated carboxylic acid derivatives are then further reacted with ammonia and a halogen/halogen succinimide according to route ii. or by carboxylic acid chloride or carboxylic acid anhydride according to route iii..
- Suitable halogens are either chloride or bromide and consequently the halogen succinimide may also be a chloride or a bromide succinimide.
- an additional step e) a pharmaceutically acceptable acid HX is added to the compound obtained in step d) in order to obtain the (2R,3R)-3-(3-methoxyphenyl)- 2-methylpentan-l -amine acid salt
- Such reaction step can be advantageous in order to stabilize the amine and for further purification of the intermediate amine V.
- any suitable pharmaceutically acceptable acid HX can be used as mentioned above.
- HX is selected from the group comprising or consisting of HC1, HBr, HI, H 2 S0 4 or mixtures thereof. Such acids result in a significant stabilization of the intermediate and may easily be removed if necessary.
- a further inventive aspect of the process comprises a step a), wherein this step is performed in the presence of a lipase -enzyme.
- this step is performed in the presence of a lipase -enzyme.
- the lipase-enzymes have been found to reliably convert the starting educt to intermediate (II) in high yields and high selectivity.
- the enantiomeric excess (ee) of this reaction step may be larger than 90%, preferably larger than 95% or even 98% and the reaction can for instance be performed using a Lipase PS "Amano" SD in aqueous solution at a pH of 3.5-11.0, preferably at a pH of 7.0-8.0.
- step a) is performed stepwise, wherein in a first step al) R-4-(3-methoxyphenyl)-3-methyl-4-oxobutanoate is stereoselectively reacted in the presence of a keto-reductase enzyme to obtain (35,4R)-4-hydroxy-4-(3- methoxyphenyl)-3-methylbutanoic acid
- keto-ester (I) is subjected to a keto-reductase enzyme solution, preferably also comprising glucose dehydrogenase and a suitable cofactor like NADP. It is preferred to perform the conversion by application of both enzymes and the cofactor at the same time.
- the preferably stereoselectively generated 4-hydroxy ester may afterwards be subjected to an acid or alkaline hydrolysis in order to obtain the 4-hydroxy acid (la). Preferably the alkaline hydrolysis is used for the generation of the free acid.
- the free acid (la) is oxidized via common alcohol oxidizing reagent (e.g. Jones, Collins, Swern, Dess-Martin, Oppenauer or Fetizon procedure) to keto-acid (II).
- common alcohol oxidizing reagent e.g. Jones, Collins, Swern, Dess-Martin, Oppenauer or Fetizon procedure
- free acid (la) is converted in the presence of an oxidation catalyst and preferably a suitable oxidation reagent to obtain the stereoselective preferred keto-acid (II).
- Suitable oxidation catalysts can either be homogeneous or heterogeneous oxidation catalysts or organic peroxides, wherein heterogeneous metallic catalysts are preferred. Suitable examples are for instance catalysts comprising V, Pt, Ag, Ru, Mn, Co, Fe Cu.
- Preferred catalysts comprise platinum, vanadium, ruthenium or mixtures thereof. Especially preferred is ruthenium either as an oxide or halide salt.
- the metal catalyst may be mounted on a support or may be used as a metal salt dissolvable into the reaction medium, the latter alternative being preferred.
- ruthenium has been proven to result in high yields, including products exhibiting a high stereoselectivity and high purity.
- Suitable oxidation reagents are known to the skilled artisan and can for instance be selected from the group comprising hydrogen peroxide, tert.
- oxidation-catalyst in step a2) is selected from the group comprising Ruthenium-halogenides, organic peroxides and mixtures thereof.
- the dissolved ruthenium-salts here especially the halogenides like ruthenium chloride and/or bromide, and the organic peroxides has proven useful for performing the oxidation of the alcohol (la) to the keto-acid (II).
- the ruthenium salts can be used alone or in combination with the organic peroxides.
- Suitable organic peroxides may be hydrogen peroxide, Acetyl acetone peroxide, Acetyl benzoyl peroxide, Ascaridole, tert.
- the oxidation-catalyst in step a2) comprises at least RuCl 3 .
- the ruthenium chloride enables a fast conversion from the alcohol to the ketone resulting in high yields of optical pure product (>90 ee).
- the separation of the catalyst from the reaction mixture is easily achievable and therefore a product of high purity is easily obtainable.
- this process step is performed under acidic conditions.
- the pH may be adjusted to a pH of ⁇ 3.5, preferably to ⁇ 3.0 and even more preferred to ⁇ 2.5.
- An additional embodiment of the invention is related to a process, wherein step d) is performed stepwise, wherein in a first step
- step c) (35,4R)-4-(3-methoxyphenyl)-3-methyl-hexanoic acid obtained in step c) is stereoselectively reacted according to variants i. - iii. at least in the presence of a base and an alcohol Z to obtain Z [(2R,3R)-2-methyl-3-methoxyphenyl pentyl]carbamate
- the alcohol is added in a base catalyzed reaction to the intermediately generated isocyanate.
- These intermediates may for instance be isolated and stored and can easily be further processed afterwards.
- all organic alcohols can be used, preferably C1-C20 alkyl or aryl alcohols.
- the alcohol can either be tert-butanol or benzyl alcohol resulting in the Boc (tert-butoxy carbamate) or CBz (Benzyloxy carbamate) protective groups. These protective groups are preferred, because the deprotection is easily achievable.
- Further protective groups may also be suitable, for instance Trifluoroacetamide, Acetamide (Ac), Formamide, Methyl carbamate, 4-Methoxybenzenesulfonamide, Benzylamine (Bn) protective groups.
- the second step d2) includes the back reaction with an acid induced liberation of the protective group. All pharmaceutical acceptable acids can be used, preferably HC1.
- an extractive workup preferably at least comprising an aqueous phase.
- step c) the metal catalyst is selected from the group consisting of Pt, Pd, Ni, Rh, and Ir and the organic base is selected from the group consisting of primary, secondary or tertiary amines, substituted or non-substituted C1-C50 aliphatic or aromatic N-heterocycles, oxygen containing bases or mixtures thereof.
- the metal catalyst is selected from the group consisting of Pt, Pd, Ni, Rh, and Ir
- the organic base is selected from the group consisting of primary, secondary or tertiary amines, substituted or non-substituted C1-C50 aliphatic or aromatic N-heterocycles, oxygen containing bases or mixtures thereof.
- Such combination of reduction catalyst and organic base has been proven useful in order to achieve high conversion rates at high diastereomeric ratios.
- a Pd catalyst can be used and this catalyst can be mounted on a solid carrier, for example, but not limited to, activated carbon.
- This catalyst enables a reaction at low temperatures and hydrogen pressures, resulting in a high stereoselectivity.
- the base may preferably be selected from the group comprising or consisting of amines, hydroxide salts, carbonate salts or hydrogencarbonate salts.
- the reaction step c) can be performed once or this step can be repeated and the crude product of each cycle can be used as educt for the next cycle. Such cycle can result in the formation of highly pure products exhibiting > 90% ee/de.
- a process can be used, wherein in step b) the organometallic- reagent is a Grignard-reagent selected from the group comprising EtLi, EtMgCl and EtMgBr or mixtures thereof. These group of organometallic -reagents result in a fast and stereoselective cyclisation of the keto-acid (II).
- EtMgBr und EtMgCl can be used.
- step dl) variant i.) the azide is selected from the group comprising Lithium azide, Sodium azide, Potassium azide, Benzyl azide, Tosyl azid, Trifluoromethanesulfonyl azide or diphenylphosphoryl azide or mixtures thereof.
- the azide is selected from the group comprising Lithium azide, Sodium azide, Potassium azide, Benzyl azide, Tosyl azid, Trifluoromethanesulfonyl azide or diphenylphosphoryl azide or mixtures thereof.
- the azide is selected from the group comprising Lithium azide, Sodium azide, Potassium azide, Benzyl azide, Tosyl azid, Trifluoromethanesulfonyl azide or diphenylphosphoryl azide or mixtures thereof.
- Particularly these azides result in a fast conversion with high yields and a low amount of unwanted side-products.
- Another characteristic of the invention is directed to a process, wherein the starting educt (I) R-4-(3- methoxyphenyl)-3-methyl-4-oxobutanoate is obtainable via the synthesis steps i. - iii.: i. Reacting l-(3-methoxyphenyl)propan-l-one and glyoxylic acid in the presence of an acid
- Such pre-steps have been found suitable in order to generate the starting material (I) in an effective and easy way.
- these steps enable an easy work-up of the resulting products, thus clean substances are obtained exhibiting only a low degree of unwanted by-products.
- an intermediate product in the production of tapentadol can comprise (45,5R)-5-ethyl-5- (3 -methoxyphenyl) -4-methyldihydrofur an-2(3H) -one
- an intermediate product in the production of tapentadol can comprise (3S,4R)-4-(3-methoxyphenyl)-3-methylhexanoic acid
- an intermediate product in the production of tapentadol can comprise tert.- butyl [(2R,3R)-2-methyl-3-methoxyphenyl pentyl] carbamate
- Another inventive object reveals the use of a tapentadol HQ synthesized via the inventive intermediates in a pharmaceutical composition.
- Figure 1 depicts the inventive route of synthesis including the pre-steps 1 - 3, which result in the starting educt I of the main synthesis.
- Ketoester (I) 400g
- aq. Keto reductase solution 800g, 2x
- aq. Glucose dehydrogenase solution 400g, lx
- NADP phosphate buffer at pH 7.3
- phosphate buffer at pH 7.3 4.0 L, lOx
- the reaction was monitored with Chiral HPLC analysis (-40% conversion).
- the reaction mixture was extracted with tert.-Butylmethylether iBME (3 x 1.0 L). NaOH solution (10%, 1.05 eq.) was added to above iBME solution to hydrolyze ketoester [(R)-3] and (R,S)-hydroxyl ester.
- the mixture was agitated until all esters were hydrolyzed into the corresponding acids (monitored by HPLC).
- the keto acid was racemized and the aq. layer containing both ketoacid and hydroxyl acid was separated.
- the pH of the aqueous layer was adjusted to pH 2-3 with HC1 solution.
- the (R,S)-(hydroxyl acid) was converted into its corresponding lactone (-1-2 hours, monitoring by HPLC) while the keto acid remained unchanged.
- iBME (3 x 500ml) was charged and the pH of the aqueous layer was adjusted back to pH 8-9 with NaOH solution.
- the organic layer was separated and washed with basic solution to remove the residual keto acid.
- NaOH solution (10%, 0.5-0.6 eq.) was added into the iBME solution and the aqueous solution was separated.
- the pH of the aqueous layer was adjusted to pH 2-3 by addition of HC1.
- the reaction mixture 3 x 500 ml of iBME was added and the pH of the aqueous layer was adjusted to pH 8-9 with NaOH solution.
- the organic layer was separated and washed with basic solution (NaOH solution). Afterwards further NaOH (10% 0.5- 0.6 eq.) was added to the iBME layer and the aqueous layer with intermediate ((R,S)-4) was subjected to the oxidation reaction in next step.
- intermediate (III) 30.4 g
- MeOH 50 mL
- DIPEA 2.6 g
- Pd/C 15.2g, 5 wt% loading (dry basis) on activated carbon, wet support.
- the flask was evacuated and refilled with 1 atm of hydrogen for 4 times.
- the reaction mixture was stirred at room temperature (around 25 °C) for 24 h.
- starting material is less than 1%
- the mixture was filtered, and the Pd/C cake was washed with MeOH (2 x 40 mL).
- the combined filtrate was concentrated to give intermediate (IV) as a yellowish oil (-30.4 g, 100% yield, >99% purity, 93%ee, 94%de).
- Step d) includes for all variants i.-iii. the following product/educt reaction scheme:
- Acetone (3.78 ml, 5.14 mmol) is added at ambient temperature during 30 min and the overall reaction mixture is treated with 20 ml of hexane and cooled to -5°C. The resulting oily solid is isolated. The second crop is obtained by evaporation of the liquids and treating of the remaining residue with hexane.
- the Tapentadol hydrochloride (2.0 g) was charged into a 50 niL flask and then n-butyl alcohol (4 niL) was added. The mixture was heated to reflux for 30min. The mixture was slowly cooled to 5 ⁇ 10°C and stirred for 2h at this temperature. The precipitate was filtered and dried to give 0.8 g of Tapentadol hydrochloride, 40% yield, the main single impurity was decreased from 1.2% to 0.67%.
- the tapentadol hydrochloride (1.0 g) was charged into a 100 mL flask and then MIBC (60 mL) was added. The mixture was heated to reflux for 30min. Then the solvent was concentrated to about 10 mL and the mixture was stirred overnight at rt. The precipitate was filtered and dried to give 0.65 g of Tapentadol hydrochloride, 65% yield, the main single impurity was decreased from 0.97% to 0.71%.
- the tapentadol hydrochloride (1.0 g) was charged into a 50 mL flask and n-butyl alcohol (4 mL) was added. The mixture was heated to reflux for 30min. Then the mixture was stirred at rt overnight. The precipitate was filtered and dried to give 0.8 g of Tapentadol hydrochloride, 80% yield, the main single impurity was decreased from 0.71% to 0.48%.
- the tapentadol hydrochloride (1.0 g) was charged into a 50 mL flask and n-propanol (3 mL) was added. The mixture was heated to reflux for 30min. Then the mixture was stirred at rt overnight. The precipitate was filtered and dried to give 0.7 g of tapentadol hydrochloride, 70% yield, the main single impurity was decreased from 1.5% to 1.1%.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The present invention relates to a process for the synthesis of tapentadol intermediates comprising the following route of synthesis: (I) → (II) → (III) → (IV) → (V).
Description
NEW SYNTHESIS OF TAPENTADOL-HC1 INTERMEDIATES
FIELD OF THE INVENTION
The present invention relates to a process for the synthesis of tapentadol intermediates, the process comprising the steps of:
a) Stereoselective reacting R-4-(3-methoxyphenyl)-3-methyl-4-oxobutanoate, wherein R is a C1-C4 alkyl, in the presence of at least an enzyme to obtain (3S)-4-(3-methoxyphenyl)-3- methyl-4-oxobutanoic acid
b) Stereoselective reacting (3S)-4-(3-methoxyphenyl)-3-methyl-4-oxobutanoic acid obtained in step a) in the presence of an organometallic-reagent to obtain (45,5R)-5-ethyl-5-(3- methoxyphenyl)-4-methyldihydrofuran-2(3H)-one
c) Stereoselective reacting (45,5R)-5-ethyl-5-(3-methoxyphenyl)-4-methyldihydrofuran-2(3H)- one in step b) in the presence of at least a metal catalyst and an organic base to obtain (35,4R)-4-(3-methoxyphenyl)-3-methyl-hexanoic acid
d) Stereoselective reacting (35,4R)-4-(3-methoxyphenyl)-3-methyl-hexanoic acid obtained in step c) by addition of
i. an azide in presence of a base and subsequently addition of a pharmaceutically acceptable acid ΗΧ or
ii. a carboxylic acid activating agent and ammonia followed by addition of a halogen, halogen succinimide or hydroxylamine -O-sulphonic acid; or iii. a carboxylic acid activating agent and hydroxylamine followed by a reaction with a carboxylic acid chloride or carboxylic acid anhydride,
in order to obtain (2R,3R)-3-(3-methoxyphenyl)-2-methylpentan-l-amine or the acid salt thereof. Furthermore, the invention is directed to the use of different intermediates in the production of tapentadol and the pharmaceutical use of tapentadol HQ synthesized via the inventive route of synthesis.
BACKGROUND
Tapentadol and its salt forms act, among other mode of actions, as opioid agonist and as noradrenalin uptake inhibitor. It has valuable pharmacological and therapeutic properties.
For example, the drug acts centrally as analgesic. Tapentadol is chemically known as 3- [(lR,2R)-3-(dimethylamino)-l-ethyl-2-methylpropyl]phenol (formula I). As a hydrochloride- salt it is approved in pharmaceutical compositions.

Multiple processes for the preparation of tapentadol hydrochloride are published, e.g., WO 2008 012047 Al and WO 2008 012046 Al describe a synthesis via Mannich reaction including a diastereomeric salt resolution.
WO 2011 080736 Al describes the preparation of Tapentadol through phenylpent-2-en amide.
WO 2011 080756 Al consists of the preparation of a cyano-intermediate as key intermediate towards Tapentadol preparation.
WO 2011 092719 Al describes a method of Tapentadol preparation using the key intermediate (bromopropyl)methoxybenzene .
WO 2012 001571 Al describes Tapentadol synthesis with a benzyl group as substituent at the amino function.
WO 2012 089177 Al consists of the description of Tapentadol synthesis of a protected alkene acid as intermediate.
WO 2012 023147 Al comprises the reaction of (dimethylamino)-2-methylpentan-3-one with anisole-Grignard.
WO 2012 038974 Al comprises a Tapentadol preparation using l-(3-hydroxyphenyl)propan- 1-one with an appropriate Grignard reagent.
WO 2012 069004 Al describes the Tapentadol synthesis using Methane sulfonyl esters. WO 2012 103799 Al describes the use of Evans auxiliary to generate key chiral intermediates for Tapentadol preparation.
Nevertheless, despite the existing routes for the synthesis of tapentadol or tapentadol intermediates there is still a need for further alternatives, providing easy to handle synthesis steps including high yields and minimizing the formation of unwanted side -products, especially unwanted stereoisomers.
Therefore, it is the task of the present invention to provide such a reliable and high yield route of synthesis, allowing the selective and efficient production of stable tapentadol intermediates, which in turn can be used for the production of tapentadol.
BRIEF DESCRIPTION OF THE INVENTION
It has been found that the above mentioned task is efficiently fulfilled by a process for the synthesis of tapentadol intermediates, the process comprising the steps of:
a) Stereoselective reacting R-4-(3-methoxyphenyl)-3-methyl-4-oxobutanoate, wherein R is a Cl- C4 alkyl, in the presence of at least an enzyme to obtain (35)-4-(3-methoxyphenyl)-3-methyl-4- oxobuta

I I I
b) Stereoselective reacting (35)-4-(3-methoxyphenyl)-3-methyl-4-oxobutanoic acid obtained in step a) in the presence of an organometallic-reagent to obtain (4S,5R)-5-ethyl-5-(3-methoxyphenyl)-4- methyl

c) Stereoselective reacting (45,5R)-5-ethyl-5-(3-methoxyphenyl)-4-methyldihydrofuran-2(3H)- one obtained in step b) in the presence of at least a metal catalyst and an organic base to obtain (3S,4R)-4-(3-methox henyl)-3-methyl-hexanoic acid
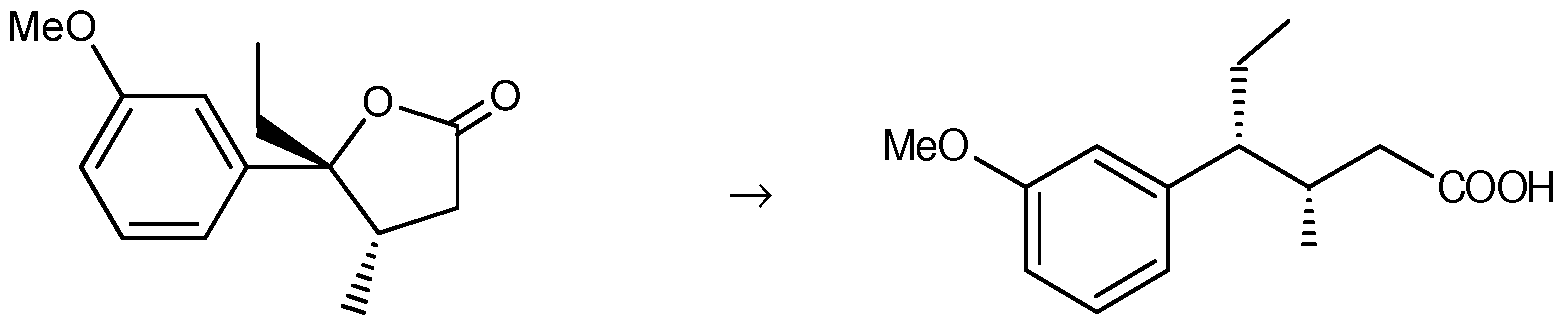
i. an azide in presence of a base and subsequently addition of a pharmaceutically
acceptable acid HX or
ii. a carboxylic acid activating agent and ammonia followed by addition of a halogen, halogen succinimide or hydroxylamine -O-sulphonic acid iii. a carboxylic acid activating agent and hydroxylamine followed by a reaction with a carboxylic acid chloride or carboxylic acid anhydride, in order to obtain (2R,3R)-3-(3-methoxyphenyl)-2-methylpentan-l-amine

IV V
Such route of synthesis has been found to deliver highly selective, stable tapentadol intermediates of defined stereochemistry in high yields, which can easily be processed to tapentadol, either as the free base or in the mineral (inorganic) acid salt form. Furthermore, the process is easy to perform and allows the production of the intermediates with a low level of potential hazardous processing aids.
Such route is inventively achieved by firstly stereoselectively reacting the starting educt, the ketoester (I) in step a) to obtain the stereoselective ketoacid, intermediate (II). The starting educt is a C1-C4 alkyl ketoester, wherein the alkyl can for instance be a straight chain methyl, ethyl, propyl, butyl or, if applicable, one of their branched homologues. The stereoselective conversion is preferably performed in the presence of a buffered solvent, preferably at least comprising water, and may include one or two process steps addressing the ester hydrolysis and stereoselective conversion. Suitable enzymes for performing such reaction and their optimum processing conditions, for instance including pH, are known to the skilled artisan and can for instance be selected from the group comprising lipases, keto reductases and esterases which may be immobilized on porous organic or inorganic carriers like microporous polypropylene or kaolinite or can be used as soluble enzyme. The reaction temperature for the enzymatic conversion may be selected as a function of the used enzyme and may vary from 5 °C up to or below the denaturation temperature of the enzyme, for instance up to 55 °C, preferentially between 20 and 35°C. The ester hydrolysis may result as a consequence of the enzymatic reaction conditions or may be achieved separately by acid or base catalyzed hydrolysis before or after the stereoselective conversion. The enantiomeric excess (ee) of the desired intermediate (II) obtainable by step a) can be higher than 90%, more specifically higher than 95% or even higher than 98%.
In step b) secondly the stereoselective keto-acid (II) is transformed via the use of an ethyl group transferring organometallic reagent to intermediate (III), a keto-furan-derivative. The conversion may be generally performed in a dried aprotic solvent under a protective atmosphere and in general organometallic compounds known as Grignard reagents can be used within this step. The cyclisation of the reaction product of the keto-acid (II) and the organometallic compound may be achieved by the addition of a strong acid, resulting in the stereoselective formation of the keto-furan (III). Suitable pH- ranges for this conversion may be pH < 2, preferably < 1.5 and even more preferred < 1.0. The intermediate (III) can be obtained with diastereomeric ratios of larger than 3:1, preferably larger than 4: 1.
Step c) thirdly is targeted to the stereoselective conversion of the ketofuran (III) to the (3S,4R)-acid (IV) via metal catalyzed reduction in the presence of an organic base in a hydrogen atmosphere. Suitable metal catalysts can be either homogeneous or heterogeneous hydrogenation catalysts, wherein heterogeneous catalysts are preferred. Suitable examples are for instance catalysts comprising Pt, Pd, Rh, Ru, Ni, Co, Fe Cu, Cr or Zn. Preferred catalysts comprise platinum, palladium, rhodium, ruthenium or mixtures thereof. The metal catalyst may be mounted on a support, preferably a support such as activated carbon, calcium carbonate, barium sulfate, barium carbonate, silicium dioxide, alumina or may for example be present as a colloid in solution. The hydrogen can be added just at the beginning of the reaction, added repeatedly, generated in situ or being feed continuously into the reactor, wherein a sequential feeding of the hydrogen is preferred. The hydrogen pressure, hydrogenation time and the temperature of conversion is a strong function of the catalyst and the adaption of these reaction conditions is known the skilled artisan.
Suitable organic bases in step c) may for example be non-nucleophilic bases of moderate strength, i.e. preferably amines and nitrogen heterocycles exhibiting a pKa of the conjugate acid of approximately 10-13. Examples for such bases are Diisopropylamine (DIP A), N,N-Diisopropylethylamine (DIPEA), l,8-Diazabicycloundec-7-ene (DBU), 2,6-Di-tert-butylpyridine, or Phosphazene bases, such as t-Bu- P4. In a preferred embodiment the organic base can be N,N-Diisopropylethylamine. The solvent of the reaction in step c) is preferably an alcohol like methanol, ethanol, propanol, isopropanol and butanol, methanol being preferred.
In step d) fourthly the (3S,4R)-acid (IV) is converted to the amine (V). This can either be achieved by a modified Curtius- (L), a Hofmann- (ii.) or via a Lossen-reaction, wherein in all three routes an isocyanate reaction intermediate is formed. This isocyanate is not stable in aqueous solution and reacts by addition of water to the carbamic acid, followed by decarboxylation to the amine. Another possibility in the synthesis route includes the formation of a carbamate by introduction of an alcohol. Therefore, it is also possible to generate a stable intermediate including a protective group. The protective group can later on be removed by the conventional methods known to the skilled in the art.
Following the first route (L), an azide in presence of a base and subsequently a pharmaceutically acceptable acid HX are used. Suitable azides include, but are not limited to, inorganic or organic azides, for instance alkali salt azide, trimethylsilylazide, tosylazide or trifluoromethylazide. Possible pharmaceutically acceptable acids HX can for example be found in the "Handbook of Pharmaceutical Salts: Properties, Selection and Use", Editors P. H. Stahl and C. G. Wermuth, Weinheim/Ziirich:Wiley-VCH/VHCA, 2002. Preferably hydrochloric acid is used.
In order to generate the isocyanate reaction intermediate it is necessary for the reaction routes ii. and iii. to introduce a carboxylic acid activating agent. Common reagents for activation are known to the skilled artisan and can for instance be selected from the group comprising or consisting of thionylchloride, carbodiimide, carbonyldiimidazole, carboxylic acid anhydride, N- Hydroxysuccinimide (NHS) or derivatives thereof. Other common coupling/activation reagents can also be used. Suitable carbodiimides may for instance be dicyclohexylcarbodiimide (DCC) or 1 -Ethyl - 3-(3-dimethylaminopropyl)carbodiimide (EDC). The activated carboxylic acid derivatives are then further reacted with ammonia and a halogen/halogen succinimide according to route ii. or by carboxylic acid chloride or carboxylic acid anhydride according to route iii.. Suitable halogens are either chloride or bromide and consequently the halogen succinimide may also be a chloride or a bromide succinimide.
In a preferred embodiment of the invention an additional step e) a pharmaceutically acceptable acid HX is added to the compound obtained in step d) in order to obtain the (2R,3R)-3-(3-methoxyphenyl)- 2-methylpentan-l -amine acid salt

V VI
Such reaction step can be advantageous in order to stabilize the amine and for further purification of the intermediate amine V. For this conversion any suitable pharmaceutically acceptable acid HX can be used as mentioned above. In a preferred embodiment of this invention HX is selected from the group comprising or consisting of HC1, HBr, HI, H2S04 or mixtures thereof. Such acids result in a significant stabilization of the intermediate and may easily be removed if necessary.
A further inventive aspect of the process comprises a step a), wherein this step is performed in the presence of a lipase -enzyme. Especially the lipase-enzymes have been found to reliably convert the starting educt to intermediate (II) in high yields and high selectivity. The enantiomeric excess (ee) of this reaction step may be larger than 90%, preferably larger than 95% or even 98% and the reaction can for instance be performed using a Lipase PS "Amano" SD in aqueous solution at a pH of 3.5-11.0, preferably at a pH of 7.0-8.0.
It is further within the scope of the invention to provide a process, wherein step a) is performed stepwise, wherein in a first step al) R-4-(3-methoxyphenyl)-3-methyl-4-oxobutanoate is stereoselectively reacted in the presence of a keto-reductase enzyme to obtain (35,4R)-4-hydroxy-4-(3- methoxyphenyl)-3-methylbutanoic acid

I la
and in a second step
a2) (3S,4R)-4-hydroxy-4-(3-methoxyphenyl)-3-methyibutanoic acid is stereoselectively reacted at least in the presence of an oxidation-catalyst to obtain (3S)-4-(3-methoxyphenyl)-3-methyl-4- oxobutanoic acid

la I I
It is also possible to achieve the stereoselective transformation of the keto-ester (I) to the stereoselectively preferred keto-acid (II) in two steps. Within a first pre-step the keto-ester is subjected to a keto-reductase enzyme solution, preferably also comprising glucose dehydrogenase and a suitable cofactor like NADP. It is preferred to perform the conversion by application of both enzymes and the cofactor at the same time. The preferably stereoselectively generated 4-hydroxy ester may afterwards be subjected to an acid or alkaline hydrolysis in order to obtain the 4-hydroxy acid (la). Preferably the alkaline hydrolysis is used for the generation of the free acid. In the second step the free acid (la) is oxidized via common alcohol oxidizing reagent (e.g. Jones, Collins, Swern, Dess-Martin, Oppenauer or Fetizon procedure) to keto-acid (II). Preferably free acid (la) is converted in the presence of an oxidation catalyst and preferably a suitable oxidation reagent to obtain the stereoselective preferred keto-acid (II). Suitable oxidation catalysts can either be homogeneous or heterogeneous oxidation catalysts or organic peroxides, wherein heterogeneous metallic catalysts are preferred. Suitable examples are for instance catalysts comprising V, Pt, Ag, Ru, Mn, Co, Fe Cu. Preferred catalysts comprise platinum, vanadium, ruthenium or mixtures thereof. Especially preferred is ruthenium either as an oxide or halide salt. The metal catalyst may be mounted on a support or may be used as a metal salt dissolvable into the reaction medium, the latter alternative being preferred. Especially ruthenium has been proven to result in high yields, including products exhibiting a high stereoselectivity and high purity. Suitable oxidation reagents are known to the skilled artisan and can for instance be selected from the group comprising hydrogen peroxide, tert. -butyl hydroperoxide, Di benzoyl peroxide, di- tert.-bvAyl peroxide, l,l-bis(tert.-butylperoxy)-3,3,5-trimethylcyclohexane, di-tert. amyl peroxide, acetyl acetone peroxide, ethyl hydroperoxide, or 2,2,6,6-Tetramethylpiperidinyloxyl (TEMPO). Within this conversion step an optical purity of > 90 ee is achievable.
In another characteristic embodiment a process can be used, wherein the oxidation-catalyst in step a2) is selected from the group comprising Ruthenium-halogenides, organic peroxides and mixtures thereof. The dissolved ruthenium-salts, here especially the halogenides like ruthenium chloride and/or bromide, and the organic peroxides has proven useful for performing the oxidation of the alcohol (la) to the keto-acid (II). The ruthenium salts can be used alone or in combination with the organic peroxides. Suitable organic peroxides may be hydrogen peroxide, Acetyl acetone peroxide, Acetyl benzoyl peroxide, Ascaridole, tert. -Butyl hydroperoxide, Di-(l-naphthoyl)peroxide, Diacetyl peroxide, Ethyl hydroperoxide, Iodoxy compounds, Methyl ethyl ketone peroxide, Methyl isobutyl ketone peroxide or mixtures thereof. Said oxidation catalysts enable a fast and efficient oxidation of intermediate (la) without any impact to the stereochemistry of the intermediate.
Furthermore, a process is inventively feasible, wherein the oxidation-catalyst in step a2) comprises at least RuCl3. The ruthenium chloride enables a fast conversion from the alcohol to the ketone resulting in high yields of optical pure product (>90 ee). The separation of the catalyst from the reaction mixture is easily achievable and therefore a product of high purity is easily obtainable. Preferably this process step is performed under acidic conditions. The pH may be adjusted to a pH of < 3.5, preferably to < 3.0 and even more preferred to < 2.5.
An additional embodiment of the invention is related to a process, wherein step d) is performed stepwise, wherein in a first step
dl) (35,4R)-4-(3-methoxyphenyl)-3-methyl-hexanoic acid obtained in step c) is stereoselectively reacted according to variants i. - iii. at least in the presence of a base and an alcohol Z to obtain Z [(2R,3R)-2-methyl-3-methoxyphenyl pentyl]carbamate

IV IVa and in a second step d2) Z [(2R,3R)-2-methyl-3-methoxyphenyl pentyl] carbamate is stereoselectively reacted in the presence of a pharmaceutically acceptable acid HX, to obtain (2R,3R)-3-(3-methoxyphenyl)-2- methylpentan- 1 -amine

IVa V
Due to the presence of the base and the alcohol it is possible to introduce a protective group in the intermediate IVa. The alcohol is added in a base catalyzed reaction to the intermediately generated isocyanate. These intermediates may for instance be isolated and stored and can easily be further processed afterwards. For the alcohol component generating the group Z all organic alcohols can be used, preferably C1-C20 alkyl or aryl alcohols. Within a preferred embodiment the alcohol can either be tert-butanol or benzyl alcohol resulting in the Boc (tert-butoxy carbamate) or CBz (Benzyloxy carbamate) protective groups. These protective groups are preferred, because the deprotection is easily achievable. Further protective groups may also be suitable, for instance Trifluoroacetamide, Acetamide (Ac), Formamide, Methyl carbamate, 4-Methoxybenzenesulfonamide, Benzylamine (Bn) protective groups. The second step d2) includes the back reaction with an acid induced liberation of the protective group. All pharmaceutical acceptable acids can be used, preferably HC1. In order to purify the resulting product V it is possible to use an extractive workup, preferably at least comprising an aqueous phase.
Another embodiment of the invention is directed to a process, wherein in step c) the metal catalyst is selected from the group consisting of Pt, Pd, Ni, Rh, and Ir and the organic base is selected from the group consisting of primary, secondary or tertiary amines, substituted or non-substituted C1-C50 aliphatic or aromatic N-heterocycles, oxygen containing bases or mixtures thereof. Such combination of reduction catalyst and organic base has been proven useful in order to achieve high conversion rates at high diastereomeric ratios. Preferably a Pd catalyst can be used and this catalyst can be mounted on a solid carrier, for example, but not limited to, activated carbon. This catalyst enables a reaction at low temperatures and hydrogen pressures, resulting in a high stereoselectivity. The base may preferably be selected from the group comprising or consisting of amines, hydroxide salts, carbonate salts or hydrogencarbonate salts. The reaction step c) can be performed once or this step can be repeated and the crude product of each cycle can be used as educt for the next cycle. Such cycle can result in the formation of highly pure products exhibiting > 90% ee/de.
In a further aspect of the invention a process can be used, wherein in step b) the organometallic- reagent is a Grignard-reagent selected from the group comprising EtLi, EtMgCl and EtMgBr or mixtures thereof. These group of organometallic -reagents result in a fast and stereoselective cyclisation of the keto-acid (II). Preferably in step b) EtMgBr und EtMgCl can be used.
Within a preferred embodiment of the invention a process is performed, wherein in step dl) variant i.) the azide is selected from the group comprising Lithium azide, Sodium azide, Potassium azide, Benzyl azide, Tosyl azid, Trifluoromethanesulfonyl azide or diphenylphosphoryl azide or mixtures thereof. Particularly these azides result in a fast conversion with high yields and a low amount of unwanted side-products.
Another characteristic of the invention is directed to a process, wherein the starting educt (I) R-4-(3- methoxyphenyl)-3-methyl-4-oxobutanoate is obtainable via the synthesis steps i. - iii.: i. Reacting l-(3-methoxyphenyl)propan-l-one and glyoxylic acid in the presence of an acid

1 2
Reacting 4-(3-methoxyphenyl)-3-methyl-4-oxo-2-butenoic acid of step a) in the

3 I
Such pre-steps have been found suitable in order to generate the starting material (I) in an effective and easy way. In addition, these steps enable an easy work-up of the resulting products, thus clean substances are obtained exhibiting only a low degree of unwanted by-products.
It is a further object of the invention to provide an intermediate product in the production of tapentadol, comprising methyl (35)-4-(3-methoxyphenyl)-3-methyl-4-oxobutanoate

Furthermore, an intermediate product in the production of tapentadol can comprise (45,5R)-5-ethyl-5- (3 -methoxyphenyl) -4-methyldihydrofur an-2(3H) -one

II I
In another inventive embodiment an intermediate product in the production of tapentadol can comprise (3S,4R)-4-(3-methoxyphenyl)-3-methylhexanoic acid
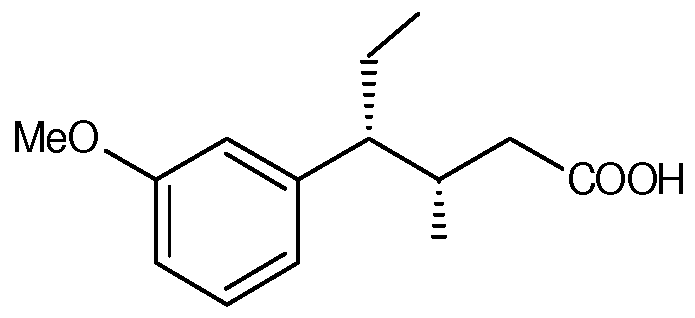
IV
In another inventive aspect an intermediate product in the production of tapentadol can comprise tert.- butyl [(2R,3R)-2-methyl-3-methoxyphenyl pentyl] carbamate

IVa
It is further within the scope of the invention to disclose the use of the inventive tapentadol intermediates in a production process for tapentadol. Within the tapentadol production process one or all of said compounds (II), (III), (IV), (IVa) may be used or generated in order to synthesize tapentadol.
Another inventive object reveals the use of a tapentadol HQ synthesized via the inventive intermediates in a pharmaceutical composition.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 depicts the inventive route of synthesis including the pre-steps 1 - 3, which result in the starting educt I of the main synthesis.
Experimental Examples
I. Pre-Steps
Ste

3'-Methoxypropiophenone (164.8 g, 1.0 mol) and glyoxylic acid (50%, 297 g, 1.5 mol, 1.5 eq.), concentrated H2S04 (50 mL) in dioxane (1 L) was refluxed overnight. After the starting material detected was less than 1 % by HPLC analysis, the mixture was distilled off under vacuum to afford dioxane (1.0-1.2 L, recyclable), and the residue was poured into ice -water (2 L). The solid was collected, washed with H20 and dried under vacuum to give a mixture of intermediate trans-(2) and cis-(2) (trans/cis=3: l), 180.8 g, 81.8% yield with >98% purity.
Ste

2 3
To a solution of mixture of intermediate trans-(2) and cis-(2) (188 g, 0.85 mol) in HOAc (1.7
L) and H20 (680 mL) was added Zinc dust (67 g, 1.03 mol, 1.2 eq). The resulting mixture was refluxed for 1 h, cooled to rt, diluted with brine (800 mL), and extracted with EtOAc (3 x 500 mL). The combined organic layers were washed with brine (1 L), dried over Na2S04 and concentrated under vacuum to give an oil residue, which was diluted with PE (500 mL). After stirred at 0°C for 2 h, the formed solid was collected, washed with PE (500 mL) and dried under vacuum at 40°C overnight to give TP2 as a yellow powder. The crude product was recrystallized from Toluene (0.7x) from 80 °C to 0 °C to give intermediate (3) 140.3 g, >99% pure, 74.6% yield (about 10% product was lost in mother liquor).
Step iii) Preparation of a methylester

3 I
To a solution of intermediate (3) (120.0 g, 0.54 mol) in MeOH (540 niL) at 0°C, H2S04 (64.2 g, 0.54 mol, 1.0 eq.) was added drop wise. The resulting mixture was stirred for 3h at rt and then concentrated under vacuum at 40°C. The residue was dissolved in EtOAc (1 L) and washed with saturated NaHC03 solution (2 x 500 mL) and brine (500 mL) and concentrated under vacuum to give intermediate (I), 140.3 g, > 100% yield, purity: >99%, which was used in next step without any further purification.
II. Step a)
One-step-

II
The mixture of the methylester of intermediate (I) (144 g, 0.6 mol) and Lipase PS (144 g) in phosphate buffer (5.7 L, pH=6.8) and acetone (500 mL) was stirred at rt for 36 h. The resulting mixture was basified to pH 9-10 with saturated Na2C03 solution at 0 °C and washed with MTBE (3 x 1 L). The aqueous layer was acidified to pH 3 with 2 M HCl and extracted with EtOAc (3 x 1 L). The combined organic layers were dried over Na2S04 and concentrated under vacuum to give intermediate (II), 60.0 g, 45% yield, 97.4% pure and 96% ee.
Two-Step-Procedure
Step al)


To an aq. solution of ((R,S)-Ia) from enzymatic reduction (containing ca 100 g of (R,S)-Ia, ca. 0.387 mol) cat. RuCl3 * 3 H20 (0.8 g, 3.8 mmol) was added and tert.-butyl hydroperoxide aq. solution (70%, 1.5eq.) was added at room temperature over 0.5h. The reaction was stirred for additional 0.5 h while monitoring by HPLC analysis for reaction completion (<1.0% starting material was left). After NaHS03 (solid, 1.0 eq.) was added, the reaction mixture was further agitated and was filtered. The filtrate was adjusted to pH 4-5 with 30% H3PO4. The mixture was extracted with Toluene (2x 30 mL) and the combined organic layers were washed with brine and concentrated to give an intermediate (II) as an oil (95.5 g, yield: 96.5%, purity: >99%; optical purity: -93% ee).
III. Step b)

To a solution of intermediate (II) (44.4 g, 0.2 mol, 93% ee) in THF (200 niL) at 10°C under N2, EtMgBr (1 M in THF, 600 mL, 0.6 mol, 3 eq.) was added dropwise over 1 h. The resulting mixture was stirred overnight at 20°C to 25 °C. The reaction was quenched by addition of H20 (50 mL), acidified to pH 1 with 4 M HC1 and stirred for 4h at rt. The resulting mixture was extracted with MTBE (2 x 200 mL). The combined organic layers were washed with 10% K2C03 solution (2 x 200 mL), H20 (200 mL) and brine (200 mL), dried over Na2S04 and concentrated under vacuum to give crude intermediate (III), 49.2 g, >100% yield, 64.8% purity with diastereo ratio 4.5: 1.
IV. Step c)

To a 250 mL flask were added crude intermediate (III) (diasteromer ratio: 4.5: 1 : 49.0 g, ~0.2mol), MeOH (140 mL), DIPEA (4.6g), and Pd/C (9.2 g, 10 wt% loading (dry basis) on activated carbon, wet support). The flask was evacuated and refilled with 1 atm of hydrogen for 4 times. The reaction mixture was stirred at room temperature (around 25 °C) for 12-24h. The reaction mixture was filtered, the Pd/C cake was washed with MeOH (2 x 40 mL). The combined filtrate was concentrated and the residue was dissolved in EtOAc (200 mL). The resulting solution was washed with Na2C03 (10%, 2 x 100 mL). The organic phase was concentrated to give intermediate (III) as yellowish oil (30.4 g, overall yield from last step: yield: 61.5%, diastereomeric ratio: 97:3).
To a 250 mL flask were added intermediate (III) (30.4 g), MeOH (50 mL), DIPEA (2.6 g), and Pd/C (15.2g, 5 wt% loading (dry basis) on activated carbon, wet support). The flask was
evacuated and refilled with 1 atm of hydrogen for 4 times. The reaction mixture was stirred at room temperature (around 25 °C) for 24 h. After the reaction is completed (starting material is less than 1%), the mixture was filtered, and the Pd/C cake was washed with MeOH (2 x 40 mL). The combined filtrate was concentrated to give intermediate (IV) as a yellowish oil (-30.4 g, 100% yield, >99% purity, 93%ee, 94%de).
V. Step d)
Step d) includes for all variants i.-iii. the following product/educt reaction scheme:

IV V
Alternative i.) Azide
Step i.l)

IV IVa
To a tert. -butyl alcoholic solution of intermediate (IV) (630 g, 2.67 mol) under basic conditions DPPA (787 g, 2.86 mol) was added. After completion of the reaction (TLC or HPLC control with related reference standards used to monitor the conversion) the reaction was stopped by water addition. After extractive workup the solution was concentrated to give the crude intermediate (IVa).
Step i.2)

IVa V
The Boc protective group was removed by acidification with HC1 in Methanol to yield the crude intermediate (V) (430 g), 97% pure, 78% yield over two steps.
Alternative ii.) Ammonia and Bromine
Intermediate (IV) (1.260 g, 5.14 mmol) is refluxed for 1 hour in 40 ml thionylchloride. The thionylchloride is distilled off and the residue is 3 times co-evaporated with toluene. 7 ml THF is added to the residue and stirred for 10 min. The resulting mixture is added to a mixture of concentrated methanolic ammonia. (3 g, 51.4 mmol) over a period of 1 hour at ambient temperature. After stirring for 2 hours at ambient temperature, the reaction mixture is evaporated to dryness.
In an icebath sodium hydroxide (3.01 g, 75.42 mmol), water (12 ml) and Bromine (0.8 ml, 15,53 mmol) are added very slowly over period of 30 minutes at below 0 °C under stirring. The reaction was maintained at below 0 °C for another 30 minutes. The residue of the above reaction is added portion wise below 0 °C over a period of 30 minutes. The reaction mixture is warmed to 60 °C and is again cooled below 20 °C. The reaction mass is diluted with dichloromethane (60 ml) and 20 ml water. The organic layer is separated, dried and concentrated under reduced pressure at 40 °C to the complete dryness. The compound (V) is isolated as oily residue.
Alternative iii.) Hydroxylamine
Intermediate (IV) (1.260 g, 5.14 mmol) is refluxed for 1 hour in 40 ml thionylchloride. The thionylchloride is distilled off and the residue is 3 times co-evaporated with toluene. 3 ml THF is added to the residue and stirred for 10 min. The resulting mixture is added to a pre- prepared mixture of aqueous hydroxylamine (50%, 3.4 g, 51.4 mmol) and THF (4 ml) over a period of 2-4 hours at ambient temperature. The reaction mixture is stirred for 2 hours (TLC). Acetone (3.78 ml, 5.14 mmol) is added at ambient temperature during 30 min and the overall reaction mixture is treated with 20 ml of hexane and cooled to -5°C. The resulting oily solid is isolated. The second crop is obtained by evaporation of the liquids and treating of the remaining residue with hexane.
A mixture of this residue, tetrahydrofuran (5 ml) and pyridine (2.086 g, 0.0262 mol), followed by slow addition of acetic anhydride (2.122 g, 0.0208 mol) was stirred for 1 hour while maintaining the temperature at ambient temperature. Ethyl acetate (50 ml) and IN hydrochloric acid (10 ml) is added, followed by phase separation. Aqueous layer is extracted with ethyl acetate (50 ml). The organic layer is washed with water saturated brine (10 ml), aqueous sodium bicarbonate solution (10 ml), followed by drying over sodium sulfate. The mixture is filtered. The filtrate was concentrated under reduced pressure and this residue and
tetrahydrofuran (21ml) was heated to 50°C. To this mixture is given water (1.91 ml) and DBU (1.43 g). This mixture is heated for several hours at reflux until completion of the reaction. After completion of the reaction ethyl acetate (50 ml) and saturated ammonium chloride solution (20 ml) is added. After extraction, phase separation and drying, the title compound 15 is isolated as oily residue.
VI. Step e)

IV V
Crude intermediate (IV) (594 g, 2.9 mol) was dissolved in iPrOH (4.2 L) and cooled to 0 °C before concentrated HCl (36%, 285 mL) is added over 15 min. The mixture is concentrated under vacuum at 40°C to give a clear solution of about 550 mL. Charge MTBE (5.3 L) at 40 °C and agitate for 30 min to obtain a clear solution. Cool the solution to 20°C. Charge seed crystal (1 g) and agitate for 1 h (clear solution). Cool the solution to 15°C. Charge seed crystal (1 g) and agitate for 1 h (small amount solid). Cool the solution to 10°C and agitate for 1 h (large amount solid). Cool the solution to 0 °C and agitate for 2 h to complete the crystallization. Filter the slurry to collect the solid. Wash the cake with MTBE (2 x 2 L). Dry the solid under vacuum at 45°C for 8 h to give a white solid of intermediate (V), 589 g, 99.16% pure (0.84% of diastereomer) and 99.0% ee, 84% yield.
VII. Further Processing - Example for Tapentadol Synthesis using the inventive intermediate
Vila) Liberation of the free base

V IV
Intermediate (V) (17.9 g, 50 mmol) was added to 10% NaOH solution (100 mL) in several batches at 0 °C. The resulted mixture was stirred for 0.5 h and extracted with MTBE (2 x
100 niL). The combined organic layers were washed with brine (100 mL), dried over Na2S04 and concentrated under vacuum to give intermediate (IV), 9.1 g, 88% yield.
Vllb) Methylation

The mixture of intermediate (IV) (29.5 g, 143 mmol), HCOOH (32.8 g, 712 mmol, 5 eq.) and HCHO (37% aq. solution, 25.4 g, 314 mmol, 2.2 eq.) was heated to 85 °C for 3 h. The reaction mixture was cooled to rt, diluted with H20 (120 ml) and acidified to pH 1 with 6 M HQ and washed with MTBE (2 x 100 mL). The aqueous solution was basified to pH 10-11 with 20% NaOH solution and extracted with MTBE (2 x 150 mL). The combined organic layers were washed with brine (100 mL), dried over Na2S04 and concentrated under vacuum to give intermediate (VI), 23.5 g, 70% yield, 95.4% pure.
VIIc) Tapentadol Synthesis

VI VI I
The mixture of intermediate (VI) (23.0 g, 98 mmol) and methionine (43.7 g, 293 mmol, 3 eq.) in MsOH (115 mL, 5 vol) was heated to 85 °C overnight. The reaction mixture was cooled to 10 °C, diluted with H20 (230 mL), and washed with MIBK (100 mL). The aqueous layer was basified to pH 10-11 with 20% NaOH solution and extracted with EtOAc (2 x 200 mL). The combined organic layers were washed with brine (200 mL), dried over Na2S04 and concentrated under vacuum to give crude Tapentadol. HCl-EtOAc (~4 M, 45 mL) was added dropwise to a solution of Tapentadol in acetone (110 mL), the resulting mixture was stirred for 0.5 h and cooled to 5 °C. The formed solid was collected and re-crystallized in IPA (130 mL) to give Tapentadol HC1 salt, 14.2 g, 57% yield, 98.2% pure.
VIIc) Tapentadol Re -Crystallization
The Tapentadol hydrochloride (2.0 g) was charged into a 50 niL flask and then n-butyl alcohol (4 niL) was added. The mixture was heated to reflux for 30min. The mixture was slowly cooled to 5~10°C and stirred for 2h at this temperature. The precipitate was filtered and dried to give 0.8 g of Tapentadol hydrochloride, 40% yield, the main single impurity was decreased from 1.2% to 0.67%.
The tapentadol hydrochloride (1.0 g) was charged into a 100 mL flask and then MIBC (60 mL) was added. The mixture was heated to reflux for 30min. Then the solvent was concentrated to about 10 mL and the mixture was stirred overnight at rt. The precipitate was filtered and dried to give 0.65 g of Tapentadol hydrochloride, 65% yield, the main single impurity was decreased from 0.97% to 0.71%.
The tapentadol hydrochloride (1.0 g) was charged into a 50 mL flask and n-butyl alcohol (4 mL) was added. The mixture was heated to reflux for 30min. Then the mixture was stirred at rt overnight. The precipitate was filtered and dried to give 0.8 g of Tapentadol hydrochloride, 80% yield, the main single impurity was decreased from 0.71% to 0.48%.
The tapentadol hydrochloride (1.0 g) was charged into a 50 mL flask and n-propanol (3 mL) was added. The mixture was heated to reflux for 30min. Then the mixture was stirred at rt overnight. The precipitate was filtered and dried to give 0.7 g of tapentadol hydrochloride, 70% yield, the main single impurity was decreased from 1.5% to 1.1%.
Claims
What is claimed:
1. A process for the synthesis of tapentadol intermediates, the process comprising the steps of: a) Stereoselective reacting R-4-(3-methoxyphenyl)-3-methyl-4-oxobutanoate, wherein R is a Cl- C4 alkyl, in the presence of at least an enzyme to obtain (35)-4-(3-methoxyphenyl)-3-methyl-4- oxobutanoic acid

I b) Stereoselective reacting (3S)-4-(3-methoxyphenyl)-3-methyl-4-oxobutanoic acid obtained in step a) in the presence of an organometallic-reagent to obtain (4S,5R)-5-ethyl-5-(3-methoxyphenyl)-4- methyldihydrofuran-2(3H)-one

I I c) Stereoselective reacting (45,5R)-5-ethyl-5-(3-methoxyphenyl)-4-methyldihydrofuran-2(3H)- one obtained in step b) in the presence of at least a metal catalyst and an organic base to obtain (3S,4R)-4-(3-methoxyphenyl)-3-methyl-hexanoic acid

I I IV d) Stereoselective reacting (35,4R)-4-(3-methoxyphenyl)-3-methyl-hexanoic acid obtained in step c) by addition of
i. an azide in presence of a base and subsequently addition of a pharmaceutically
acceptable acid HX or
ii. a carboxylic acid activating agent and ammonia followed by addition of a halogen, halogen succinimide or hydroxylamine -O-sulphonic acid; or
iii. a carboxylic acid activating agent and hydroxylamine followed by a reaction with a carboxylic acid chloride or carboxylic acid anhydride, in order to obtain (2R,3R)-3-(3-methoxyphenyl)-2-methylpentan-l-amine

IV V
2. Process according to claim 1 , wherein step a) is performed in the presence of a lipase-enzyme.
3. Process according to claim 1, wherein step a) is performed stepwise, wherein in a first step al) R-4-(3-methoxyphenyl)-3-methyl-4-oxobutanoate is stereoselectively reacted in the presence of a keto-reductase enzyme to obtain (3S,4R)-4-hydroxy-4-(3-methoxyphenyl)-3-methyibutanoic acid

I la
and in a second step
a2) (3S,4R)-4-hydroxy-4-(3-methoxyphenyl)-3-methyibutanoic acid is stereoselectively reacted at least in the presence of an oxidation-catalyst to obtain (35)-4-(3-methoxyphenyl)-3-methyl-4- oxobutanoic acid

la
4. Process according to claim 3, wherein the oxidation-catalyst in step a2) is selected from the group comprising Ruthenium-halogenides, organic peroxides and mixtures thereof.
5. Process according to any of the claims 3 or 4, wherein the oxidation-catalyst in step a2) comprises at least RuCl3.
6. Process according to any of the preceding claims, wherein step d) is performed stepwise, wherein in a first step
dl) (3S,4R)-4-(3-methoxyphenyl)-3-methyl-hexanoic acid obtained in step c) is stereoselectively reacted according to variants i. - iii. at least in the presence of a base and an alcohol Z to obtain Z- [(2R,3R)-2-methyl-3-methoxyphenyl pentyl]carbamate

IV IVa and in a second step
d2) Z [(2R,3R)-2-methyl-3-methoxyphenyl pentyl]carbamate is stereoselectively reacted in the presence of a pharmaceutically acceptable acid HX, to obtain (2R,3R)-3-(3-methoxyphenyl)-2- methylpentan- 1 -amine

IVa V
7. Process according to any of the preceding claims, wherein in step c) the metal catalyst is selected from the group consisting of Pt, Pd, Ni, Rh, and Ir and the organic base is selected from the group consisting of primary, secondary or tertiary amines, substituted or non-substituted C1-C50 aliphatic or aromatic N-heterocycles, oxygen containing bases or mixtures thereof.
8. Process according to any of the preceding claims, wherein in step b) the organometallic- reagent is a Grignard-reagent selected from the group comprising EtLi, EtMgCl and EtMgBr or mixtures thereof.
9. Process according to claim 8, wherein in step dl) variant i.) the azide is selected from the group comprising Lithium azide, Sodium azide, Potassium azide, Benzyl azide, Tosyl azid, Trifluoromethanesulfonyl azide or diphenylphosphoryl azide or mixtures thereof.
10. Process according to any of the preceding claims, wherein the starting educt (I) R-4-(3- methoxyphenyl)-3-methyl-4-oxobutanoate is obtainable via the synthesis steps i. - iii.: i.) Reacting l-(3-methoxyphenyl)propan-l-one and glyoxylic acid in the presence of an acid and a solvent to obtain 4-(3-methoxyphenyl)-3-methyl-4-oxo-2-but-enoic acid,

1
ii.) Reacting 4-(3-methoxyphenyl)-3-methyl-4-oxo-2-butenoic acid of step a) in the presence of a reduction catalyst to obtain 4-(3-methoxyphenyl)-3-methyl-4-oxobutanoic acid,

2 3
iii.) Esterification of 4-(3-methoxyphenyl)-3-methyl-4-oxobutanoic acid of step b) to obtain R-4-
(3-methoxyphenyl)-3-methyl-4-oxobutanoate, wherein R is a C1-C4 Alkyl

I
11. Intermediate product in the production of tapentadol, comprising methyl (3S)-4-(3- methoxyphenyl)-3-methyl-4-oxobutanoate

12. Intermediate product in the production of tapentadol, comprising (45,5R)-5-ethyl-5-(3- methoxyphenyl)-4-methyldihydrofuran-2(3H)-one

13. Intermediate product in the production of tapentadol, comprising (35,4R)-4-(3- methoxyphenyl)-3-methylhexanoic acid

IV
14. Intermediate product in the production of tapentadol, comprising tert-butyl [(2R,3R)-2-methyl- 3-methoxyphenyl pentyl] carbamate

IVa
Use of the tapentadol intermediates according to any of the claims 11 - 14 in a production
16. Use of a tapentadol HCl synthesized via the intermediates according to any of the claims 11 - 14 in a pharmaceutical composition.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB1510544.8 | 2015-06-16 | ||
| GB1510544.8A GB2539442A (en) | 2015-06-16 | 2015-06-16 | New synthesis of tapentadol-HCI intermediates |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2016202808A2 true WO2016202808A2 (en) | 2016-12-22 |
| WO2016202808A3 WO2016202808A3 (en) | 2017-01-26 |
Family
ID=53784810
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2016/063648 Ceased WO2016202808A2 (en) | 2015-06-16 | 2016-06-14 | NEW SYNTHESIS OF TAPENTADOL-HCl INTERMEDIATES |
Country Status (2)
| Country | Link |
|---|---|
| GB (1) | GB2539442A (en) |
| WO (1) | WO2016202808A2 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113214195A (en) * | 2021-04-28 | 2021-08-06 | 云南民族大学 | Method for asymmetrically synthesizing dihydrofuran 2- (3H) -ketone compound by nickel catalysis |
| CN118388329A (en) * | 2024-05-16 | 2024-07-26 | 江苏海洋大学 | Preparation method of tapentadol intermediate |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0271287A3 (en) * | 1986-12-11 | 1990-06-13 | Merck Frosst Canada Inc. | Quinoline dioic acids and amides |
| IT1397189B1 (en) * | 2009-12-01 | 2013-01-04 | Archimica Srl | NEW PROCESS FOR THE PREPARATION OF TAPENTADOL AND ITS INTERMEDIATES. |
| CN102617501A (en) * | 2011-01-31 | 2012-08-01 | 中国科学院上海药物研究所 | Substituted valeramide compound, preparation method and application thereof |
| IN2013MU03670A (en) * | 2013-11-21 | 2015-07-31 | Unimark Remedies Ltd | |
| DK3166923T3 (en) * | 2014-07-10 | 2023-04-11 | SpecGx LLC | PROCESS FOR THE PRODUCTION OF SUBSTITUTED PHENYLALKANES |
-
2015
- 2015-06-16 GB GB1510544.8A patent/GB2539442A/en not_active Withdrawn
-
2016
- 2016-06-14 WO PCT/EP2016/063648 patent/WO2016202808A2/en not_active Ceased
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113214195A (en) * | 2021-04-28 | 2021-08-06 | 云南民族大学 | Method for asymmetrically synthesizing dihydrofuran 2- (3H) -ketone compound by nickel catalysis |
| CN113214195B (en) * | 2021-04-28 | 2024-05-10 | 云南民族大学 | Method for asymmetrically synthesizing dihydrofuran 2- (3H) -ketone compound by nickel catalysis |
| CN118388329A (en) * | 2024-05-16 | 2024-07-26 | 江苏海洋大学 | Preparation method of tapentadol intermediate |
Also Published As
| Publication number | Publication date |
|---|---|
| GB201510544D0 (en) | 2015-07-29 |
| GB2539442A (en) | 2016-12-21 |
| WO2016202808A3 (en) | 2017-01-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11767292B2 (en) | Process for making beta 3 agonists and intermediates | |
| EP1720832B1 (en) | Process for preparing n-protected 4-ketoproline derivatives | |
| EP2537825B1 (en) | Method for producing 1-amino-1-alkoxycarbonyl-2-vinylcyclopropane | |
| JP6370712B2 (en) | Method for producing cis-5-hydroxy-2-piperidinecarboxylic acid derivative and method for purifying cis-5-hydroxy-2-piperidinecarboxylic acid | |
| WO2016202808A2 (en) | NEW SYNTHESIS OF TAPENTADOL-HCl INTERMEDIATES | |
| US8431733B2 (en) | Process for the preparation of (3S)-3-amino-N-cyclopropyl-2-hydroxyalkanamide derivatives | |
| Kambourakis et al. | Chemo‐Enzymatic Method for the Synthesis of Statine, Phenylstatine and Analogues | |
| Ojo et al. | Synthesis of the natural product descurainolide and cyclic peptides from lignin-derived aromatics | |
| AU2015308035B2 (en) | Improved process for the preparation of Lacosamide and its novel intermediate | |
| US20090312571A1 (en) | Process for the preparation of (3s)-3-amino-n-cyclopropyl-2-hydroxyalkanamide derivatives | |
| JP7082218B2 (en) | Enzyme method for the production of droxidopa | |
| JPH08311025A (en) | Production of 4-hydroxy-2-pyrrolidone | |
| JP2012116775A (en) | Method for producing ecteinascidin | |
| US20100179335A1 (en) | Process for the preparation of orlistat | |
| US6121487A (en) | Method of producing amino acids and amino-acid derivatives | |
| HK40084722A (en) | Process for the preparation of a chiral piperazine-2-carboxylic acid | |
| JP2023523961A (en) | Method for the preparation of chiral piperazine-2-carboxylic acids | |
| EP0387058A1 (en) | Process for preparing benzylpyruvic acids and esters | |
| WO2004092113A1 (en) | Optically active 2-allylcarboxylic acid derivative and process for producing the same | |
| WO1994027955A1 (en) | Intermediates and process of making dipeptide isosteres | |
| JPWO1998011057A1 (en) | Optically active erythro-3-amino-2-hydroxybutyric acid esters and method for producing said butyric acids | |
| WO2013136265A1 (en) | Synthesis of an intermediate of an antiviral compound | |
| JPH0759553B2 (en) | Method for producing 3-amino-2-azetidinone derivative | |
| JP2002003463A (en) | Method for producing n-protecting n-alkylated amino acid |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16728968 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16728968 Country of ref document: EP Kind code of ref document: A2 |