WO2016199789A1 - Method of manufacturing trifluoromethyl substituted semisquarate, method of manufacturing trifluoromethyl compound starting from trifluoromethyl substituted semisquarate, and trifluoromethyl group-containing compound - Google Patents
Method of manufacturing trifluoromethyl substituted semisquarate, method of manufacturing trifluoromethyl compound starting from trifluoromethyl substituted semisquarate, and trifluoromethyl group-containing compound Download PDFInfo
- Publication number
- WO2016199789A1 WO2016199789A1 PCT/JP2016/066988 JP2016066988W WO2016199789A1 WO 2016199789 A1 WO2016199789 A1 WO 2016199789A1 JP 2016066988 W JP2016066988 W JP 2016066988W WO 2016199789 A1 WO2016199789 A1 WO 2016199789A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- trifluoromethyl
- compound
- butyl
- sec
- Prior art date
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/51—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/68—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C46/00—Preparation of quinones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/753—Unsaturated compounds containing a keto groups being part of a ring containing ether groups, groups, groups, or groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C50/00—Quinones
- C07C50/26—Quinones containing groups having oxygen atoms singly bound to carbon atoms
- C07C50/28—Quinones containing groups having oxygen atoms singly bound to carbon atoms with monocyclic quinoid structure
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C50/00—Quinones
- C07C50/26—Quinones containing groups having oxygen atoms singly bound to carbon atoms
- C07C50/32—Quinones containing groups having oxygen atoms singly bound to carbon atoms the quinoid structure being part of a condensed ring system having two rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C50/00—Quinones
- C07C50/26—Quinones containing groups having oxygen atoms singly bound to carbon atoms
- C07C50/34—Quinones containing groups having oxygen atoms singly bound to carbon atoms the quinoid structure being part of a condensed ring system having three rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/82—Carbazoles; Hydrogenated carbazoles
- C07D209/88—Carbazoles; Hydrogenated carbazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the ring system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/78—Benzo [b] furans; Hydrogenated benzo [b] furans
- C07D307/86—Benzo [b] furans; Hydrogenated benzo [b] furans with an oxygen atom directly attached in position 7
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/91—Dibenzofurans; Hydrogenated dibenzofurans
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/50—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D333/52—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes
- C07D333/54—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/50—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D333/76—Dibenzothiophenes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D455/00—Heterocyclic compounds containing quinolizine ring systems, e.g. emetine alkaloids, protoberberine; Alkylenedioxy derivatives of dibenzo [a, g] quinolizines, e.g. berberine
- C07D455/02—Heterocyclic compounds containing quinolizine ring systems, e.g. emetine alkaloids, protoberberine; Alkylenedioxy derivatives of dibenzo [a, g] quinolizines, e.g. berberine containing not further condensed quinolizine ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
Definitions
- the present invention relates to a novel method for synthesizing a trifluoromethyl-substituted semisquarate, a method for synthesizing various functional trifluoromethyl compounds starting from the compound, and a trifluoromethyl group-containing compound.
- Fluorine-containing compounds are known to have high chemical stability due to the strong carbon-fluorine bond, and are used in fields such as pharmaceuticals, functional materials, and agricultural chemicals. Especially in pharmaceuticals, since it was found that Fludrocortisone has an anti-inflammatory action 10 times higher than Cortisone ⁇ in the 1950s (Non-Patent Documents 1 and 2), many compounds have been synthesized. The development of fluorine-containing drugs such as 5-fluorouracil, which is currently used for chemotherapy, is progressing (Non-patent Documents 3 and 4).
- Non-Patent Documents 5 and 6 Non-Patent Documents 5 and 6
- the polar effect is an effect of having resistance to an electrophilic reaction such as an oxidation reaction due to the influence of the strong electronegativity of fluorine and acquiring a stronger interaction with a protein.
- the mimic effect is an effect in which a fluorine atom is an atom having the next smallest radius after hydrogen, and thus shows a similar behavior without being distinguished in vivo.
- the blocking effect is an effect that stabilizes the suppression of substitution reaction, reduction, and oxidative metabolism in the living body because the spread of the electron orbit of fluorine is close to that of carbon and the carbon-fluorine bond becomes strong.
- the hydrophobicity enhancing effect is an effect of changing the absorption and transport in the living body by acquiring lipophilicity.
- the effect of sustained efficacy, the effect of enhancing drug absorption, and the improvement of selectivity as these combined effects are known as the effects of fluorine. Because of these high effects, the number of fluorine-containing pharmaceuticals has increased to 14% of those on the market.
- Non-patent Documents 3, 4, 7 to 11 aiming at synthesis of target structure using compound having trifluoromethyl group such as trifluoroacetic acid and benzotrifluoride derivative, and trifluoromethyl unit
- This is a direct trifluoromethylation method in which is directly introduced into the molecular skeleton (Non-patent Documents 12 to 21, Patent Document 1).
- the building block method requires many steps to produce the target compound, and further requires separation and purification operations. Since the target compound is synthesized from a simple trifluoromethyl compound through many steps, the more complicated the target compound, the more complicated the synthesis route and the higher the cost. Also, the manufacturing method is poor in generality.
- the direct trifluoromethylation method is a method in which a target skeleton is once constructed and a trifluoromethyl group is introduced at the final stage.
- trifluoromethylation is a special reaction, generally expensive reagents and harsh reaction conditions are required. Therefore, the generality is poor, and the applicable skeleton is limited.
- the present invention provides a method for efficiently synthesizing a trifluoromethyl-substituted semisquarate using a commercially available squalate compound as a starting material in a short process, and a method for producing various functional trifluoromethyl compounds starting from the compound. Let it be an issue.
- the present invention is a method for producing a trifluoromethyl-substituted semisquarate shown below and a novel trifluoromethyl compound.
- a compound represented by the following general formula (1) as a starting material (In the formula, R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group.)
- the production method according to [1], wherein the compound into which the trifluoromethyl group is introduced is a quinone compound.
- R 1 , R 2 , R 3 and R 4 are each independently hydrogen, alkyl Selected from a group, an alkenyl group, an alkoxy group, a chloro group, a fluoro group, and an ester group, any two adjacent substituents of R 1 to R 4 may form a condensed benzene, wherein alkyl
- the hydrocarbon moiety of the group, alkenyl group and alkoxy group may be any of C1 to C8 linear, branched or cyclic.) (Wherein R is selected from isopropyl group, n-propyl group, t-butyl group, isobutyl group and sec-butyl group.
- R 5 and R 6 are each independently hydrogen, alkyl group, alkenyl group, (It is selected from an alkoxy group, a chloro group, and a fluoro group, wherein the hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.)
- the production method of [1], wherein the compound into which the trifluoromethyl group is introduced is a heterocyclic condensed ring compound.
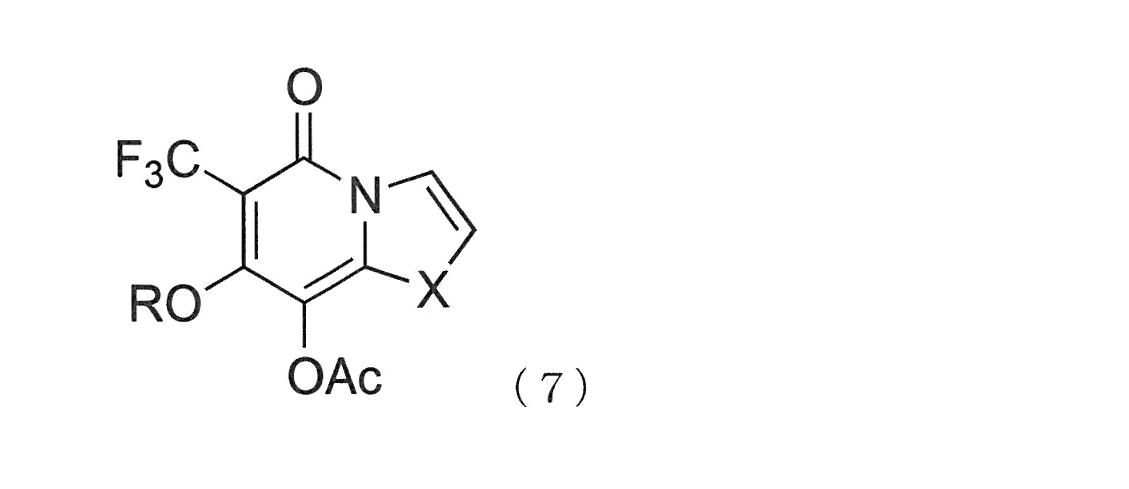
- the heterocyclic condensed ring compound into which the trifluoromethyl group is introduced is a compound represented by the following general formula (6) or (7).
- R is selected from isopropyl group, n-propyl group, t-butyl group, isobutyl group, sec-butyl group.
- R 7 and R 8 are each independently hydrogen, alkyl group, alkenyl group, Selected from an alkoxy group, a chloro group, and a fluoro group, R 7 and R 8 may combine to form a condensed benzene, X is O, S, NP, and P is a carbamate, sulfonamide
- the hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.
- R is selected from an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, and a sec-butyl group.
- X is CH ⁇ CH, S, NCH 3 )
- the production method according to [1], wherein the compound into which the trifluoromethyl group is introduced is a hydroquinone compound.
- the production method of [6], wherein the hydroquinone compound having a trifluoromethyl group introduced therein is a compound represented by any one of the following general formulas (8) to (10). Wherein R is selected from isopropyl, n-propyl, t-butyl, isobutyl and sec-butyl.
- R 9 , R 10 , R 11 and R 12 are each independently hydrogen, Selected from an alkyl group, an alkenyl group, an alkoxy group, a chloro group, a fluoro group, and an ester group, and any two adjacent substituents of R 9 to R 12 may form a condensed benzene, where (The hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.) (Wherein R is selected from isopropyl group, n-propyl group, t-butyl group, isobutyl group, sec-butyl group.
- R 13 and R 14 are each independently hydrogen, alkyl group, alkenyl group, (It is selected from an alkoxy group, a chloro group, and a fluoro group, wherein the hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.) (Wherein R is selected from an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, and a sec-butyl group.
- R 15 and R 16 are each independently hydrogen, an alkyl group, an alkenyl group, Selected from an alkoxy group, a chloro group and a fluoro group, R 15 and R 16 may combine to form a condensed benzene, X is O, S or NP, and P is a carbamate or sulfonamide.
- the hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.
- R 17 , R 18 , R 19 , and R 20 are each independently hydrogen, Selected from an alkyl group, an alkenyl group, an alkoxy group, a chloro group, a fluoro group, and an ester group, and any two adjacent substituents of R 17 to R 20 may form a condensed benzene, where (The hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.) (Wherein R is selected from isopropyl group, n-propyl group, t-butyl group, isobutyl group, sec-butyl group.
- R 21 and R 22 are each independently hydrogen, alkyl group, alkenyl group, Selected from an alkoxy group, a chloro group, and a fluoro group, R 21 and R 22 may combine to form a condensed benzene, X is O, S, NP, and P is a carbamate, sulfonamide.
- the hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.
- R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group, which may be different groups or the same group.
- a compound represented by the following general formula (2) A production method comprising synthesizing by a step of performing an allyl alcohol transfer reaction.
- R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group, which may be different groups or the same group.
- a method for producing a compound represented by the following general formula (2), The squarate represented by the following general formula (3) A production method comprising synthesizing by a trifluoromethylation step.
- R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group, which may be different groups or the same group.
- the trifluoromethylation step comprises a silyl trifluoromethylation reaction using CF 3 Me 3 Si as an organosilicon reagent; A production method comprising a desilylation step.
- R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group.
- a compound of the following general formula (2) (In the formula, R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group, which may be different groups or the same group.)
- Trifluoromethyl compounds represented by the following formulas (12) to (52), (17-1), (10-3a), and (10-3b).
- FIG. 1 schematically shows the trifluoromethyl-substituted semisquarate method of the present invention.
- trifluoromethyl-substituted semisquarate of compound 1 can be synthesized in a short process.
- examples of R include isopropyl, n-propyl, t-butyl, isobutyl, and sec-butyl.
- the squarate of compound 3 and R of compound 2 can have the above functional groups, but the two Rs may be different groups or the same group. However, it is preferable that both are the same groups from the ease of synthesis.
- Squaric acid ester is a useful molecule as a four-membered ring synthesis element because it can be converted into the target skeleton in a short process.
- Various molecules such as quinone and butenolide can be synthesized by ring expansion reaction of hydroxycyclobutenone obtained by adding an organometallic reagent. Therefore, if it is possible to synthesize semisquarates substituted with trifluoromethyl groups, it is possible to efficiently synthesize quinones and butenolides into which trifluoromethyl groups have been introduced by using them as synthesis elements. It becomes.
- R is selected from isopropyl, n-propyl, t-butyl, isobutyl and sec-butyl.
- R 1 , R 2 , R 3 and R 4 are each independently hydrogen, Selected from an alkyl group, an alkenyl group, an alkoxy group, a chloro group, a fluoro group, and an ester group, and any two adjacent substituents of R 1 to R 4 may form a condensed benzene, where (The hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.)
- R is selected from isopropyl group, n-propyl group, t-butyl group, isobutyl group and sec-butyl group.
- R 5 and R 6 are each independently hydrogen, alkyl group, alkenyl group, (It is selected from an alkoxy group, a chloro group, and a fluoro group, wherein the hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.)
- R is selected from isopropyl group, n-propyl group, t-butyl group, isobutyl group, sec-butyl group.
- R 7 and R 8 are each independently hydrogen, alkyl group, alkenyl group, Selected from an alkoxy group, a chloro group, and a fluoro group, R 7 and R 8 may combine to form a condensed benzene, X is O, S, NP, and P is a carbamate, sulfonamide
- the hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.
- R is selected from an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, and a sec-butyl group.
- X is CH ⁇ CH, S, NCH 3 )
- hydroquinones corresponding to the quinones (4), (5) and (6) include (8), (9) and (10).
- R is selected from isopropyl, n-propyl, t-butyl, isobutyl and sec-butyl.
- R 9 , R 10 , R 11 and R 12 are each independently hydrogen, Selected from an alkyl group, an alkenyl group, an alkoxy group, a chloro group, a fluoro group, and an ester group, and any two adjacent substituents of R 9 to R 12 may form a condensed benzene, where (The hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.)
- R is selected from isopropyl group, n-propyl group, t-butyl group, isobutyl group, sec-butyl group.
- R 13 and R 14 are each independently hydrogen, alkyl group, alkenyl group, (It is selected from an alkoxy group, a chloro group, and a fluoro group, wherein the hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.)
- R is selected from an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, and a sec-butyl group.
- R 15 and R 16 are each independently hydrogen, an alkyl group, an alkenyl group, Selected from an alkoxy group, a chloro group and a fluoro group, R 15 and R 16 may combine to form a condensed benzene, X is O, S or NP, and P is a carbamate or sulfonamide.
- the hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.
- examples of butenolides that can be synthesized from trifluoromethyl-substituted semisquarate include compounds represented by the following general formulas (10-1) and (10-2).
- R is selected from isopropyl, n-propyl, t-butyl, isobutyl, and sec-butyl.
- R 17 , R 18 , R 19 , and R 20 are each independently hydrogen, Selected from an alkyl group, an alkenyl group, an alkoxy group, a chloro group, a fluoro group, and an ester group, and any two adjacent substituents of R 17 to R 20 may form a condensed benzene, where (The hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.)
- R 21 and R 22 are each independently hydrogen, alkyl group, alkenyl group, Selected from an alkoxy group, a chloro group, and a fluoro group, R 21 and R 22 may combine to form a condensed benzene, X is O, S, NP, and P is a carbamate, sulfonamide.
- the hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.
- R 23 and R 24 are each independently selected from a C1-C6 linear, branched, cyclic alkyl group or a phenyl group.
- R 25 and R 26 are each independently selected from a C1-C6 linear, branched, cyclic alkyl group or phenyl group.
- R 27 and R 28 are each independently selected from a C1-C6 linear, branched, or cyclic alkyl group or a phenyl group, and R 27 or R 28 may form a ring structure) .
- aminocyclopentenedione represented by the general formula (10-4) can also be synthesized from trifluoromethyl-substituted semisquarate.
- R is selected from isopropyl, n-propyl, t-butyl, isobutyl, sec-butyl, R 29 is a C1-C8 alkyl group, linear, branched, cyclic
- R 30 is selected from a t-butyl group or a 1,1,3,3-tetramethylbutyl group.
- Example 1 ⁇ Method for producing trifluoromethyl-substituted semisquarate (1) ⁇ 1.1 Trifluoromethylation step of squarate ester A method for producing a trifluoromethyl-substituted semisquarate by separately performing two steps of trifluoromethylation and allyl alcohol transfer is shown below.
- a stirring bar was placed in a Schlenk tube and dried by heating under reduced pressure.
- Diisopropyl squarate (3- i Pr) 99.9 mg, 0.50 mmol
- sodium acetate NaOAc
- tetrabutylammonium chloride n Bu 4 NCl
- THF tetrahydrofuran
- the mixture was stirred at 25 ° C. for 15 minutes, and trifluoromethyltrimethylsilane (Me 3 SiCF 3 ) (111 ⁇ L, 0.75 mmol) was added.
- Coupling constants are expressed in Hz.
- the infrared absorption spectrum (IR) was measured using FT / IR-230 manufactured by JASCO Corporation, and all characteristic absorptions were expressed in cm ⁇ 1 .
- MPS100 made by SRS was used for the melting point measurement.
- JMS-T100LP manufactured by JEOL Ltd. was used, and high-resolution mass spectrometry by DART or ESI was performed.
- trifluoromethylation can be carried out in the same manner using other squarate esters as shown below.
- Other trifluoromethylation can be carried out in the same manner using a Schlenk tube or a two-necked eggplant flask. Desilylation can be performed by adding TBAF (2 equivalents) or saturated aqueous potassium fluoride (1 mL).
- Trifluoromethylation was performed using dipropyl squarate as a starting material, and a compound of the following formula (2-1): 4-hydroxy-2,3-dipropyoxy-4- (trifluoromethyl) cyclobut-2-enone (2- n Pr) was synthesized. The yield was 40%. The analysis results of the obtained compound are shown below.
- Trifluoromethylation was performed using disec-butyl squarate as a starting material, and a compound of the following formula (2-3), 2,3-di-sec-butyl-4-hydroxy-4- (trifluoromethyl) cyclobut-2- Oneone (2- s Bu) was synthesized. The yield was 65%. The analysis results of the obtained compound are shown below.
- Tritert-butyl squarate is used as a starting material, and trifluoromethylation is carried out to obtain a compound of the following formula (2-4): 2,3-di-tert-butyl-4-hydroxy-4- (trifluoromethyl) cyclobut-2- en-1-one (2- t Bu) was synthesized. The yield was 90%. The analysis results of the obtained compound are shown below.
- Example 3 ⁇ Synthesis of physiologically active substance analogues (1) ⁇ A method for synthesizing a physiologically active substance analog starting from the compound of the general formula (1) synthesized by the method of Example 1 or 2 is described below.
- a quinone having a trifluoromethyl group introduced therein is synthesized by an addition reaction step, a ring expansion reaction and an oxidation reaction step.
- the production method will be described in detail by taking as an example the production of 2-isopropoxy-3- (trifluoromethyl) naphthalene-1,4-dione (12).
- the following reaction formula is an outline of the production method.
- a Grignard reagent solution is prepared.
- PhMgBr manufactured by Aldrich
- Ring expansion reaction and oxidation reaction are performed as follows. Et 2 O was removed with a rotary evaporator to obtain a p-xylene solution (about 10 mL). The resulting solution was purged with argon and stirred at 140 ° C. for 10 minutes. After confirming the disappearance of 4-hydroxycyclobutenone as an intermediate product, the mixture was cooled to room temperature. Phthalocyanine iron (II) complex [Fe (pc)] (8.4 mg, 0.015 mmol) and acetic acid (0.5 mL) were added and stirred for 1 hour in an oxygen atmosphere. After confirming the disappearance of hydroquinone, cerite filtration and concentration were performed to obtain a crude product.
- Phthalocyanine iron (II) complex [Fe (pc)] (8.4 mg, 0.015 mmol) and acetic acid (0.5 mL) were added and stirred for 1 hour in an oxygen atmosphere. After confirming the disappearance of hydroquinone, cerite filtration and
- Example 4 ⁇ Synthesis of physiologically active substance analogues (2) ⁇ 2-isopropoxy-6-methyl-3- (trifluoromethyl) naphthalene-1,4-dione, whose structural formula is shown in the following formula (13), is obtained by using 4-methylmagnesium bromide (non-non-crystalline) as a Grignard reagent with aryl bromide and magnesium metal. It was manufactured in the same manner as in Example 3 except that Patent Documents 23 and 24) were prepared and used.
- Example 5 ⁇ Synthesis of physiologically active substance analogues (3) ⁇ 2-isopropoxy-6-methoxy-3- (trifluoromethyl) naphthalene-1,4-dione, whose structural formula is shown in the following (14), is obtained by using 4-methoxyphenymagnesium bromide (manufactured by Aldrich) as a Grignard reagent. Was produced in the same manner as in Example 3.
- Example 7 ⁇ Synthesis of physiologically active substance analogues (5) ⁇ 6-chloro-2-isopropyoxy-3- (trifluoromethyl) naphthalene-1,4-dione, whose structural formula is represented by the following formula (16), is obtained by using 4-chlorophenylmagnesium bromide (manufactured by Aldrich) as a Grignard reagent. Was produced in the same manner as in Example 3.
- Example 8 ⁇ Synthesis of physiologically active substance analogues (6) ⁇ 6-fluoro-2-isopropoxy-3- (trifluoromethyl) naphthalene-1,4-dione, whose structural formula is represented by the following formula (17), used 4-fluorophenylmagnesium bromide (manufactured by Aldrich) as a Grignard reagent. Others were produced in the same manner as in Example 3.
- Example 10 ⁇ Synthesis of physiologically active substance analogues (8) ⁇ 3-isopropoxy-2- (trifluoromethyl) phenanthrene-1,4-dione, whose structural formula is represented by the following formula (18), was used in Examples except that 1-naphthylmagnesium bromide (manufactured by Aldrich) was used as a Grignard reagent. 3 was produced in the same manner as in No. 3.
- Example 11 ⁇ Synthesis of bioactive substance analogues (9) ⁇ 2-isopropoxy-3- (trifluoromethyl) phenanthrene-1,4-dione, which has the structural formula shown in the following formula (19), is 2-naphthylmagnesium bromide prepared by using aryl bromide and magnesium metal as a Grignard reagent (non-patent literature) 23 and 24) were used in the same manner as in Example 3.
- Example 12 ⁇ Synthesis of physiologically active substance analogues (10) ⁇ 6-isopropoxy-5- (trifluoromethyl) benzofuran-4,7-dione, which has the structural formula shown in the following formula (20), is 2-furylmagnesium bromide prepared from an organolithium reagent using MgBr 2 as a Grignard reagent. It was manufactured in the same manner as in Example 3 except that Patent Documents 26 and 27) were used.
- Example 13 ⁇ Synthesis of bioactive substance analogues (11) ⁇ 6-isopropoxy-5- (trifluoromethyl) benzo [b] thiophene-4,7-dione, whose structural formula is shown in the following formula (21), was prepared from an organolithium reagent using MgBr 2 as a Grignard reagent. It was manufactured in the same manner as in Example 3 except that thienylmagnesium bromide (Non-patent Documents 26 and 27) was used.
- Example 15 ⁇ Synthesis of bioactive substance analogues (13) ⁇ 3-isopropoxy-2- (trifluoromethyl) dibenzo [b, d] thiophene-1,4-dione, whose structural formula is shown in (23) below, was prepared from an organolithium reagent using MgBr 2 as a Grignard reagent. It was manufactured in the same manner as in Example 3 except that thienylmagnesium bromide (Non-patent Documents 26 and 27) was used.
- Example 18 ⁇ Synthesis of physiologically active substance analogues (16) ⁇ 2-isopropoxy-5,6-dimethyl-3- (trifluoromethyl) cyclohexa-2,5-diene-1,4-dione having the structural formula shown in the following formula (27) is 1-methyl-1- Production was carried out in the same manner as in Example 3 except that propenyl magnesium bromide (manufactured by Aldrich) was used.
- Example 22 ⁇ Synthesis of physiologically active substance analogues (20) ⁇ 7-isopropoxy-1-methyl-5-oxo-6- (trifluoromethyl) -1,5-dihydroimidazo [1,2-a] pyridin-8-yl acetate represented by the following formula (31) is N- This was prepared in the same manner as in Example 20 except that the organolithium reagent prepared from methylimidazole (1-methyl-1H-imidazol-2-yl) lithium (Non-patent Document 30) was used.
- Butenolides can be synthesized by the following method. The method for synthesizing butenolides is described in detail by taking 3-isopropoxy-5-oxo-2-phenyl-4- (trifluoromethyl) -2,5-dihydrofuran-2-yl acetate as a structural formula shown in the following formula (32) as an example. To do.
- PhMgBr 3.0 M ether solution (Aldrich, 415 ⁇ L, 1.25 mmol) was diluted with dehydrated diethyl ether (12.1 mL), adjusted to a concentration of about 0.1 M and used.
- Example 24 ⁇ Synthesis of bioactive substance analogues (22) ⁇ 3-isopropoxy-2- (4-methoxyphenyl) -5-oxo-4- (trifluoromethyl) -2,5-dihydrofuran-2-yl acetate, which has the structural formula shown in the following formula (33), is replaced with PhMgBr. This was prepared in the same manner as in Example 23 except that -MeOC 6 H 4 MgBr was used. The yield was 74%. The analysis results are shown below.
- Bicyclo ring compounds can be synthesized by the following method.
- 2-isopropoxy-7-oxo-1- (trifluoromethyl) bicycle [3.2.0] hept-2-en-3-yl acetate, which is a bicyclo ring compound having the structural formula shown in the following formula (37), is a novel compound.
- the precursors 10-3a and 10-3b were produced as follows.
- Allylmagnesium chloride 2.0 M THF solution (Aldrich, 625 ⁇ L, 1.25 mmol) was diluted in dehydrated diethyl ether (11.9 mL) and used as an approximately 0.1 M solution.
- a precursor compound (73.1 mg, 0.25 mmol) having the structural formula shown in Formula (10-3b) was dissolved in p-xylene (5 mL). This solution was stirred at 120 ° C. for 1 hour.
- the yield of the final product represented by Formula 37 was 88%.
- the analysis results are shown below.
- Example 29 ⁇ Synthesis of bioactive substance analogues (27) ⁇
- the yield of the intermediate compound 10-3a was low. Therefore, the compound 2-isopropoxy-7-oxo-1- (trifluoromethyl) bicycle described in Example 28 was used. 3.2.0] Synthesis was limited to hept-2-en-3-yl acetate. Therefore, by developing the following method using an allyl silicon compound, the yield of the compound represented by 10-3a was successfully improved.
- the heat-dried Schlenk tube was filled with argon, 1- i Pr (52.4 mg, 0.25 mmol) was added, and the mixture was diluted with dehydrated dichloromethane (1.5 mL). This solution was cooled to ⁇ 78 ° C., a 1.0 M solution of tin tetrachloride in dichloromethane (250 ⁇ L, 0.25 mmol) was added, and allylsilane (manufactured by TCI, 60 ⁇ L, 0.375 mmol) was added, and the mixture was added at ⁇ 78 ° C. for 10 minutes. After stirring, the mixture was stirred for 1 hour in an ice bath.
- Example 32 ⁇ Synthesis of bioactive substance analogues (30) ⁇ 2-isopropoxy-7-oxo-5-phenyl-1- (trifluoromethyl) bicyclo [3.2.0] hept-2-en-3-yl acetate is shown in Example 28. It was manufactured by heating for 1 hour in the same manner. The yield was 96%. The analysis results are shown below.
- Example 33 ⁇ Synthesis of bioactive substance analogues (31) ⁇ 4-hydroxy-3-isopropoxy-4- (2-methylenoxyl) -2- (trifluoromethyl) cyclobut-2-enone, which has the structural formula shown in the following formula (41), is trimethyl (2-methylenecylyl) instead of allylsilane. This was manufactured in the same manner as in Example 29 except that (Non-patent Document 31) was used. The yield was 97%. The analysis results are shown below.
- Example 34 ⁇ Synthesis of physiologically active substance analogues (32) ⁇ 2-isopropoxy-1- (2-methylenoxyl) -4-oxo-3- (trifluoromethyl) cyclobut-2-ethyl acetate represented by the following structural formula (42) is the same as compound 10-3b of Example 28. Manufactured. The yield was 98%. The analysis results are shown below.
- Example 35 ⁇ Synthesis of physiologically active substance analogues (33) ⁇ 5-hexyl-2-isopropyoxy-7-oxo-1- (trifluoromethyl) bicycle [3.2.0] hept-2-en-3-yl acetate, which is represented by the following formula (43), is obtained in Example 28. It was manufactured by heating for 1 hour in the same manner. The yield was 95%. The analysis results are shown below.
- Example 36 ⁇ Synthesis of physiologically active substance analogues (34) ⁇ 2-isopropoxy-4-oxo-1- (1-phenylallyl) -3- (trifluoromethyl) cyclobut-2-ethyl acetate is represented by the following formula (44) in the same manner as in Example 29. Instead, titanium tetrachloride was allylated at ⁇ 40 ° C. using cinnamethyltrimethylsilane (Non-patent Document 32) instead of allylsilane, and then the same as compound 10-3b of Example 28 without isolating the resulting alcohol. To be directly acylated. Obtained as a 7: 3 diastereomeric mixture, the yield was 38% in two steps. The analysis results are shown below.
- Example 37 ⁇ Synthesis of physiologically active substance analogues (35) ⁇ 2-isopropoxy-7-oxo-4-phenyl-1- (trifluoromethyl) bicycle [3.2.0] hept-2-en-3-yl acetate represented by the following structural formula (45) is obtained in Example 28. It was manufactured by heating in the same manner for 3 hours. The yield was 75%. The analysis results are shown below.
- Example 38 ⁇ Synthesis of physiologically active substance analogues (36) ⁇ 4-acetoxy-3-isopropoxy-4a, 5,6,7-tetrahydro-2a- (trifluoromethyl) -1H-cyclobuta [c] pentapentalen-2 (2aH) -one, which has the structural formula shown in the following formula (46),
- allylation was carried out using trimethylsilane (Non-patent Document 31) instead of allylsilane.
- the product was acetylated in the same manner as Compound 10-3b of Example 28 without isolating the alcohol, and the resulting ester was prepared by heating for 2 hours in the same manner as in Example 28 without isolation. The yield was 42% in three stages.
- the analysis results are shown below.
- the imidoyllithium reagent was prepared based on the method of Liebeskid et al. (Non-patent Document 33).
- Tert-butyl isocyanide 85 ⁇ L, 0.75 mmol
- diethyl ether 2.02 mL
- n BuLi 15 w / w%, 480 ⁇ L
- stirring was continued at ⁇ 15 ° C. for 30 minutes.
- the resulting solution was cooled to ⁇ 78 ° C. for use in subsequent addition reactions.
- phenylacetylene 120 ⁇ L, 1.1 mmol was dissolved in dehydrated diethyl ether (2.0 mL) and cooled to ⁇ 78 ° C.
- n BuLi 15 w / w%, 640 ⁇ L was added and stirred at room temperature for 2 hours until the solution became cloudy. Thereafter, dehydrated diethyl ether (1.36 mL) was added to give a 0.25 M solution (pale yellow).
- the heat-dried Schlenk tube was filled with argon, 1- i Pr (62.4 mg, 0.30 mmol) was added, and the mixture was diluted with dehydrated dichloromethane (2.0 mL). The solution was cooled to ⁇ 78 ° C., a 1.0 M solution of tin tetrachloride in dichloromethane (300 ⁇ L, 0.30 mmol) was added, and the mixture was stirred for 10 minutes. Trimethyl (1-phenylvinyloxy) silane (184 ⁇ L, 0.90 mmol) separately prepared (Non-patent Document 34) was added to this solution, and the mixture was stirred at ⁇ 78 ° C. for 2 hours.
- Example 42 ⁇ Synthesis of bioactive substance analogues (40) ⁇ Benzyl 2- (1-hydroxy-2-isopropyoxy-4-oxo-3- (trifluoromethyl) cyclobut-2-enyl) acetate having the structural formula shown in the following formula (50) is tin tetrachloride in the same manner as in Example 41. Instead of trimethyl (1-phenylvinyloxy) silane, (1- (benzoyloxy) vinyloxy) trimethylsilane (Non-patent Document 35) was used instead of trimethyl (1-phenylvinyloxy) silane. The yield was 86%. The analysis results are shown below.
- the heat-dried Schlenk tube was filled with argon, and Pb (OAc) 4 (348.6 mg, 0.79 mmol), heat-dried molecular sieves 4A powder (400.2 mg), and dehydrated toluene (4.0 mL) were added.
- Pb (OAc) 4 348.6 mg, 0.79 mmol
- heat-dried molecular sieves 4A powder 400.2 mg
- dehydrated toluene 4.0 mL
- 4-hydroxy-3-isopropoxy-4- (2-oxo-2-phenylethyl) -2- (trifluoromethyl) cyclobut-2-enone 129.0 mg, 0.39 mmol
- the filtrate was extracted with ethyl acetate (10 mL ⁇ 3), and the obtained organic layer was washed with saturated NaHCO 3 (10 mL) and dried over Na 2 SO 4 .
- the crude product obtained by concentrating the organic layer with a rotary evaporator was diluted in THF (8 mL), N-methyl morpholine (48 ⁇ L, 0.43 mmol) was added, and the mixture was stirred at room temperature for 4 hours.
- the analysis results of the obtained compound are shown below.
- Example 44 ⁇ Synthesis of bioactive substance analogues (42) ⁇ Benzyl 2- (3-isopropoxy-5-oxo-4- (trifluoromethyl) furan-2 (5H) -ylidene) acetate having the structural formula shown in the following formula (52) was produced in the same manner as in Example 43. The yield was 84%. The analysis results are shown below.
- Furaquinocins exhibit a wide range of physiological activities such as in vitro cytotoxicity and platelet coagulation inhibitory activity against HeLaS3 and B16 melanoma cells.
- Fraquinocin A, B, and E shown below are synthesized using semisquarate 40 of the reaction formula shown below (Non-patent Document 37). Therefore, the use of trifluoromethyl-substituted semisquarate 1- i Pr makes it possible to synthesize trifluoromethyl analogues of Furaquinocins.
- Non-patent Document 39 The compound represented by the formula (26) synthesized in Example 17 is a trifluoromethyl compound as a precursor when synthesizing Carbazimycin G. Therefore, it is possible to synthesize a trifluoromethyl substituted Carbazimycin G analog using the precursor compound.
- a trifluoromethyl group can be introduced into various compounds by the production method of the present invention.
Abstract
The present invention addresses the problem of efficiently synthesizing a trifluoromethyl substituted semisquarate in a short process, in order to manufacture trifluoromethyl compounds with various functionalities. A method was developed of synthesizing a trifluoromethyl substituted semisquarate with squarate as a starting material, in the two steps of a trifluoromethylation reaction and an allyl alcohol transfer reaction. Furthermore, a method was established of manufacturing trifluoromethyl compounds with various functionalities starting from a trifluoromethyl substituted semisquarate.
Description
トリフルオロメチル置換セミスクアレートを合成する新規の手法、及び該化合物を起点として多様な機能性トリフルオロメチル化合物を合成する方法、及びトリフルオロメチル基含有化合物に関する。
The present invention relates to a novel method for synthesizing a trifluoromethyl-substituted semisquarate, a method for synthesizing various functional trifluoromethyl compounds starting from the compound, and a trifluoromethyl group-containing compound.
含フッ素化合物は、強固な炭素-フッ素結合のため高い化学的安定性を有することが知られており、医薬品をはじめ、機能材料、農薬などの領域において利用されている。特に、医薬品においては、1950年代にFludrocortisoneが、Cortisone と比較して10倍高い抗炎症作用を有することが見出されて以来(非特許文献1、2)、多数の化合物が合成されており、現在でも化学療法に使用されている5-フルオロウラシル等、含フッ素医薬品の開発が進んでいる(非特許文献3、4)。
Fluorine-containing compounds are known to have high chemical stability due to the strong carbon-fluorine bond, and are used in fields such as pharmaceuticals, functional materials, and agricultural chemicals. Especially in pharmaceuticals, since it was found that Fludrocortisone has an anti-inflammatory action 10 times higher than Cortisone に in the 1950s (Non-Patent Documents 1 and 2), many compounds have been synthesized. The development of fluorine-containing drugs such as 5-fluorouracil, which is currently used for chemotherapy, is progressing (Non-patent Documents 3 and 4).
医薬品では、フッ素を導入することによって、極性効果、ミミック効果、ブロック効果、疎水性増強効果と呼ばれる効果がもたらされることが知られている(非特許文献5、6)。極性効果とは、フッ素の強い電気陰性度による影響で酸化反応などの求電子反応に耐性を持つことや、より強いタンパク質との相互作用を獲得する効果である。ミミック効果とは、フッ素原子が水素の次に半径の小さな原子であることから、生体内でこれらが識別されずに類似した挙動を示す効果である。ブロック効果とは、フッ素の電子軌道の広がりが炭素に近いため、炭素-フッ素結合が強固になるため、置換反応や還元、生体内における酸化的代謝を抑制し、安定となる効果である。また、疎水性増強効果とは親油性を獲得することで、生体内での吸収、輸送などに変動を与える効果である。さらに、これらの複合的な効果としての薬効持続効果や薬物吸収増強効果作用、選択性の向上がフッ素の効果として知られている。これら高い効果を備えることから、含フッ素医薬品は、市販されている医薬品の14%にまで増加している。
In pharmaceuticals, it is known that by introducing fluorine, an effect called a polar effect, a mimic effect, a block effect, or a hydrophobic enhancement effect is brought about (Non-Patent Documents 5 and 6). The polar effect is an effect of having resistance to an electrophilic reaction such as an oxidation reaction due to the influence of the strong electronegativity of fluorine and acquiring a stronger interaction with a protein. The mimic effect is an effect in which a fluorine atom is an atom having the next smallest radius after hydrogen, and thus shows a similar behavior without being distinguished in vivo. The blocking effect is an effect that stabilizes the suppression of substitution reaction, reduction, and oxidative metabolism in the living body because the spread of the electron orbit of fluorine is close to that of carbon and the carbon-fluorine bond becomes strong. In addition, the hydrophobicity enhancing effect is an effect of changing the absorption and transport in the living body by acquiring lipophilicity. Furthermore, the effect of sustained efficacy, the effect of enhancing drug absorption, and the improvement of selectivity as these combined effects are known as the effects of fluorine. Because of these high effects, the number of fluorine-containing pharmaceuticals has increased to 14% of those on the market.
含フッ素化合物が、上記のような効果を有することから、医薬品等の開発において、有機フッ素化合物の合成が大きな役割を果たすようになってきている。中でも、芳香族トリフルオロメチル化合物(Ar-CF3)は多くの医薬品に利用されている。
Since fluorine-containing compounds have the effects as described above, synthesis of organic fluorine compounds has played a major role in the development of pharmaceuticals and the like. Among these, aromatic trifluoromethyl compounds (Ar—CF 3 ) are used in many pharmaceuticals.
現在、トリフルオロメチル基導入は、図2に示すように大きく分けて二つの方法により行なわれている。トリフルオロ酢酸やベンゾトリフルオリド誘導体の様な、トリフルオロメチル基を有する化合物を用い、目的構造体の合成を目指すビルディングブロック法(非特許文献3、4、7~11)、及びトリフルオロメチル単位を直接分子骨格に導入する直接トリフルオロメチル化法(非特許文献12~21、特許文献1)である。
At present, the introduction of trifluoromethyl group is performed by two methods as shown in FIG. Building block method (Non-patent Documents 3, 4, 7 to 11) aiming at synthesis of target structure using compound having trifluoromethyl group such as trifluoroacetic acid and benzotrifluoride derivative, and trifluoromethyl unit This is a direct trifluoromethylation method in which is directly introduced into the molecular skeleton (Non-patent Documents 12 to 21, Patent Document 1).
ビルディングブロック法は、目的化合物を製造するのに多くの工程が必要であり、さらに、分離・精製操作を行なう必要がある。単純なトリフルオロメチル化合物から多くの工程を経て、目的化合物を合成することから、目的化合物が複雑であればあるほど、煩雑な合成経路となり、コストも上昇する。また、製造方法も一般性に乏しい。
The building block method requires many steps to produce the target compound, and further requires separation and purification operations. Since the target compound is synthesized from a simple trifluoromethyl compound through many steps, the more complicated the target compound, the more complicated the synthesis route and the higher the cost. Also, the manufacturing method is poor in generality.
そこで、近年、目的とする化合物骨格に直接トリフルオロメチル基を導入する直接トリフルオロメチル化法の開発が進んでいる。直接トリフルオロメチル化法は、目的とする骨格を一旦構築し、最終段階でトリフルオロメチル基を導入する方法である。しかしながら、骨格構築そのものに多段階を必要とすることに加え、トリフルオロメチル化は特殊な反応であるため、一般的に高価な試薬や過酷な反応条件が必要とされる。そのため、一般性に乏しく、適用できる骨格に制限がある。
Therefore, in recent years, development of a direct trifluoromethylation method in which a trifluoromethyl group is directly introduced into a target compound skeleton has been developed. The direct trifluoromethylation method is a method in which a target skeleton is once constructed and a trifluoromethyl group is introduced at the final stage. However, in addition to the fact that the skeleton construction itself requires multiple steps, since trifluoromethylation is a special reaction, generally expensive reagents and harsh reaction conditions are required. Therefore, the generality is poor, and the applicable skeleton is limited.
よって、高価な試薬を必要とせず、汎用性のあるトリフルオロメチル化合物の製造方法の開発が望まれていた。また、例えば、トリフルオロメチル化された複素環化合物など、上記公知の方法によっても合成できないトリフルオロメチル化合物も多数存在する。本発明は市販のスクアレート化合物を出発原料とし、短工程で効率良くトリフルオロメチル置換セミスクアレートを合成する方法、該化合物を起点として多様な機能性トリフルオロメチル化合物の製造方法を提供することを課題とする。
Therefore, it has been desired to develop a method for producing a versatile trifluoromethyl compound without requiring an expensive reagent. There are also many trifluoromethyl compounds that cannot be synthesized by the above-mentioned known methods, such as trifluoromethylated heterocyclic compounds. The present invention provides a method for efficiently synthesizing a trifluoromethyl-substituted semisquarate using a commercially available squalate compound as a starting material in a short process, and a method for producing various functional trifluoromethyl compounds starting from the compound. Let it be an issue.
トリフルオロメチル基が置換したセミスクアレートを合成することが可能になれば、これを合成素子として利用することで、トリフルオロメチル基を導入したキノンやブテノリドを効率よく合成することが可能となる。したがって、トリフルオロメチル基で置換したセミスクアレートを合成することができれば、様々な機能性トリフルオロメチル化合物を提供することが可能となる。本発明は、トリフルオロメチル化合物を効率良く合成するだけではなく、従来法では合成することのできなかった、複素環化合物にトリフルオロメチル基を導入した化合物等、新たな化合物を合成し提供することを課題とする。
If it is possible to synthesize semisquarate substituted with a trifluoromethyl group, it will be possible to efficiently synthesize quinone or butenolide introduced with a trifluoromethyl group by using this as a synthesis element. . Therefore, if semisquarate substituted with a trifluoromethyl group can be synthesized, various functional trifluoromethyl compounds can be provided. The present invention not only synthesizes trifluoromethyl compounds efficiently, but also synthesizes and provides new compounds such as compounds in which a trifluoromethyl group is introduced into a heterocyclic compound that could not be synthesized by conventional methods. This is the issue.
本発明は以下に示すトリフルオロメチル置換セミスクアレートの製造方法、及び新規のトリフルオロメチル化合物である。
[1]下記一般式(1)で表される化合物を出発原料として、

(式中Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基である。)
付加反応工程、及び環拡大工程によってトリフルオロメチル基が導入された化合物を製造する方法。
[2]前記トリフルオロメチル基が導入された化合物が、キノン化合物である[1]記載の製造方法。
[3]トリフルオロメチル基が導入されたキノン化合物が、下記一般式(4)又は(5)で表される化合物である[2]記載の製造方法。

(式中、Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基から選択される。R1、R2、R3、R4はそれぞれ独立して水素、アルキル基、アルケニル基、アルコキシ基、クロロ基、フルオロ基、エステル基から選択され、R1~R4のうち任意の2つの隣接する置換基は縮環ベンゼンを形成してもよい。ここで、アルキル基、アルケニル基、アルコキシ基の炭化水素部位は、C1~C8の直鎖状、分岐状、環状のいずれでも良い。)

(式中、Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基から選択される。R5、R6はそれぞれ独立して、水素、アルキル基、アルケニル基、アルコキシ基、クロロ基、フルオロ基から選択される。ここで、アルキル基、アルケニル基、アルコキシ基の炭化水素部位は、C1~C8の直鎖状、分岐状、環状のいずれでも良い。)
[4]前記トリフルオロメチル基が導入された化合物が、複素環縮環化合物である[1]記載の製造方法。
[5]トリフルオロメチル基が導入された複素環縮環化合物が、下記一般式(6)又は(7)で表される化合物である[4]記載の製造方法。

(式中、Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基から選択される。R7、R8はそれぞれ独立して、水素、アルキル基、アルケニル基、アルコキシ基、クロロ基、フルオロ基から選択され、R7、R8は結合して縮環ベンゼンを形成してもよい。XはO、S、NPであり、Pは、カルバメート、スルホンアミドである。ここで、アルキル基、アルケニル基、アルコキシ基の炭化水素部位は、C1~C8の直鎖状、分岐状、環状のいずれでも良い。)
(式中、Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基から選択される。XはCH=CH、S、NCH3である。)
[6]前記トリフルオロメチル基が導入された化合物が、ヒドロキノン化合物である[1]記載の製造方法。
[7]トリフルオロメチル基が導入されたヒドロキノン化合物が、下記一般式(8)~(10)のいずれかで表される化合物である[6]記載の製造方法。

(式中、Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基から選択される。R9、R10、R11、R12はそれぞれ独立して、水素、アルキル基、アルケニル基、アルコキシ基、クロロ基、フルオロ基、エステル基から選択され、R9~R12のうち任意の2つの隣接する置換基は縮環ベンゼンを形成してもよい。ここで、アルキル基、アルケニル基、アルコキシ基の炭化水素部位は、C1~C8の直鎖状、分岐状、環状のいずれでも良い。)

(式中、Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基から選択される。R13、R14はそれぞれ独立して、水素、アルキル基、アルケニル基、アルコキシ基、クロロ基、フルオロ基から選択される。ここで、アルキル基、アルケニル基、アルコキシ基の炭化水素部位は、C1~C8の直鎖状、分岐状、環状のいずれでも良い。)

(式中、Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基から選択される。R15、R16はそれぞれ独立して、水素、アルキル基、アルケニル基、アルコキシ基、クロロ基、フルオロ基から選択され、R15、R16は結合して縮環ベンゼンを形成してもよい。XはO、S、NPであり、Pは、カルバメート、スルホンアミドである。ここで、アルキル基、アルケニル基、アルコキシ基の炭化水素部位は、C1~C8の直鎖状、分岐状、環状のいずれでも良い。)
[8]前記トリフルオロメチル基が導入された化合物が、ブテノリド化合物である[1]記載の製造方法。
[9]トリフルオロメチル基が導入されたブテノリド化合物が、下記一般式(10-1)、(10-2)で表される化合物である[8]記載の製造方法。

(式中、Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基から選択される。R17、R18、R19、R20はそれぞれ独立して、水素、アルキル基、アルケニル基、アルコキシ基、クロロ基、フルオロ基、エステル基から選択され、R17~R20のうち任意の2つの隣接する置換基は縮環ベンゼンを形成してもよい。ここで、アルキル基、アルケニル基、アルコキシ基の炭化水素部位は、C1~C8の直鎖状、分岐状、環状のいずれでも良い。)

(式中、Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基から選択される。R21、R22はそれぞれ独立して、水素、アルキル基、アルケニル基、アルコキシ基、クロロ基、フルオロ基から選択され、R21、R22は結合して縮環ベンゼンを形成してもよい。XはO、S、NPであり、Pは、カルバメート、スルホンアミドである。ここで、アルキル基、アルケニル基、アルコキシ基の炭化水素部位は、C1~C8の直鎖状、分岐状、環状のいずれでも良い。)
[10]前記トリフルオロメチル基が導入された化合物が、四員環化合物である[1]記載の製造方法。
[11]トリフルオロメチル基が導入された四員環化合物が、下記一般式(10-3c)、(10-3d)で表される化合物である[10]記載の製造方法。

(式中、R23、R24はそれぞれ独立して、C1~C6直鎖状、分岐状、環状のアルキル基またはフェニル基から選択される。)

(式中、R25、R26はそれぞれ独立して、C1~C6直鎖状、分岐状、環状のアルキル基またはフェニル基から選択される。)
[12]前記トリフルオロメチル基が導入された化合物が、ビシクロ環化合物である[1]記載の製造方法。
[13]トリフルオロメチル基が導入されたビシクロ環化合物が、下記一般式(37-1)で表される化合物である[12]記載の製造方法。

(式中、R27、R28はそれぞれ独立して、C1~C6直鎖状、分岐状、環状のアルキル基またはフェニル基から選択され、R27、R28で環構造を形成してもよい。)
[14]前記トリフルオロメチル基が導入された化合物が、アミノシクロペンテンジオン化合物である[1]記載の製造方法。
[15]トリフルオロメチル基が導入されたアミノシクロペンテンジオン化合物が、下記一般式(10-4)で表される化合物である[14]記載の製造方法。

(式中、Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基から選択される。R29はC1~C8のアルキル基で、直鎖状、分岐状、環状のいずれでも良い。R30はt-ブチル基または1,1,3,3-テトラメチルブチル基から選択される。)
[16]下記一般式(1)で表されるトリフルオロメチル置換セミスクアレートの製造方法であって、

下記一般式(3)で表されるスクアレートをトリフルオロメチル化する工程により、
 下記一般式(2)で表される化合物を製造し、
下記一般式(2)で表される化合物を製造し、
 アリルアルコール転移反応を行う工程により合成することを特徴とする製造方法。
アリルアルコール転移反応を行う工程により合成することを特徴とする製造方法。
(式中Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基であり、異なる基であっても、同一の基であってもよい。)
[17]下記一般式(1)で表されるトリフルオロメチル置換セミスクアレートの製造方法であって、

(式中Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基である。)
下記一般式(2)で表される化合物を

アリルアルコール転移反応を行なう工程により合成することを特徴とする製造方法。
(式中Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基であり、異なる基であっても、同一の基であってもよい。)
[18][16]又は[17]記載のトリフルオロメチル置換セミスクアレートの製造方法であって、アリルアルコール転移反応を行なう工程が、Re2O7又はPh3SiReO3を触媒として用いる反応であることを特徴とする製造方法。
[19]下記一般式(2)で表される化合物の製造方法であって、

下記一般式(3)で表されるスクアレートを

トリフルオロメチル化する工程により合成することを特徴とする製造方法。
(式中Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基であり、異なる基であっても、同一の基であってもよい。)
[20][16]又は[19]記載の製造方法であって、前記トリフルオロメチル化する工程が、CF3Me3Siを有機ケイ素試薬として用いてシリルトリフルオロメチル化反応をする工程と、脱シリル化工程からなることを特徴とする製造方法。
[21]下記一般式(1)で表される化合物。

(式中Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基である。)
[22]下記一般式(2)の化合物。

(式中Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基であり、異なる基であっても、同一の基であってもよい。)
[23]下記式(12)~(52)、(17-1)、(10-3a)、(10-3b)で表されるトリフルオロメチル化合物。

The present invention is a method for producing a trifluoromethyl-substituted semisquarate shown below and a novel trifluoromethyl compound.
[1] A compound represented by the following general formula (1) as a starting material,
(In the formula, R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group.)
A method for producing a compound into which a trifluoromethyl group has been introduced by an addition reaction step and a ring expansion step.
[2] The production method according to [1], wherein the compound into which the trifluoromethyl group is introduced is a quinone compound.
[3] The production method according to [2], wherein the quinone compound into which the trifluoromethyl group is introduced is a compound represented by the following general formula (4) or (5).
Wherein R is selected from isopropyl, n-propyl, t-butyl, isobutyl and sec-butyl. R 1 , R 2 , R 3 and R 4 are each independently hydrogen, alkyl Selected from a group, an alkenyl group, an alkoxy group, a chloro group, a fluoro group, and an ester group, any two adjacent substituents of R 1 to R 4 may form a condensed benzene, wherein alkyl The hydrocarbon moiety of the group, alkenyl group and alkoxy group may be any of C1 to C8 linear, branched or cyclic.)
(Wherein R is selected from isopropyl group, n-propyl group, t-butyl group, isobutyl group and sec-butyl group. R 5 and R 6 are each independently hydrogen, alkyl group, alkenyl group, (It is selected from an alkoxy group, a chloro group, and a fluoro group, wherein the hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.)
[4] The production method of [1], wherein the compound into which the trifluoromethyl group is introduced is a heterocyclic condensed ring compound.
[5] The process according to [4], wherein the heterocyclic condensed ring compound into which the trifluoromethyl group is introduced is a compound represented by the following general formula (6) or (7).
(Wherein R is selected from isopropyl group, n-propyl group, t-butyl group, isobutyl group, sec-butyl group. R 7 and R 8 are each independently hydrogen, alkyl group, alkenyl group, Selected from an alkoxy group, a chloro group, and a fluoro group, R 7 and R 8 may combine to form a condensed benzene, X is O, S, NP, and P is a carbamate, sulfonamide Here, the hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.)
(Wherein R is selected from an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, and a sec-butyl group. X is CH═CH, S, NCH 3 )
[6] The production method according to [1], wherein the compound into which the trifluoromethyl group is introduced is a hydroquinone compound.
[7] The production method of [6], wherein the hydroquinone compound having a trifluoromethyl group introduced therein is a compound represented by any one of the following general formulas (8) to (10).
Wherein R is selected from isopropyl, n-propyl, t-butyl, isobutyl and sec-butyl. R 9 , R 10 , R 11 and R 12 are each independently hydrogen, Selected from an alkyl group, an alkenyl group, an alkoxy group, a chloro group, a fluoro group, and an ester group, and any two adjacent substituents of R 9 to R 12 may form a condensed benzene, where (The hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.)
(Wherein R is selected from isopropyl group, n-propyl group, t-butyl group, isobutyl group, sec-butyl group. R 13 and R 14 are each independently hydrogen, alkyl group, alkenyl group, (It is selected from an alkoxy group, a chloro group, and a fluoro group, wherein the hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.)
(Wherein R is selected from an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, and a sec-butyl group. R 15 and R 16 are each independently hydrogen, an alkyl group, an alkenyl group, Selected from an alkoxy group, a chloro group and a fluoro group, R 15 and R 16 may combine to form a condensed benzene, X is O, S or NP, and P is a carbamate or sulfonamide. Here, the hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.)
[8] The production method according to [1], wherein the compound into which the trifluoromethyl group is introduced is a butenolide compound.
[9] The production method according to [8], wherein the butenolide compound into which the trifluoromethyl group is introduced is a compound represented by the following general formulas (10-1) and (10-2).
Wherein R is selected from isopropyl, n-propyl, t-butyl, isobutyl, and sec-butyl. R 17 , R 18 , R 19 , and R 20 are each independently hydrogen, Selected from an alkyl group, an alkenyl group, an alkoxy group, a chloro group, a fluoro group, and an ester group, and any two adjacent substituents of R 17 to R 20 may form a condensed benzene, where (The hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.)
(Wherein R is selected from isopropyl group, n-propyl group, t-butyl group, isobutyl group, sec-butyl group. R 21 and R 22 are each independently hydrogen, alkyl group, alkenyl group, Selected from an alkoxy group, a chloro group, and a fluoro group, R 21 and R 22 may combine to form a condensed benzene, X is O, S, NP, and P is a carbamate, sulfonamide. Here, the hydrocarbon moiety of the alkyl group, alkenyl group, or alkoxy group may be any of C1 to C8 linear, branched, or cyclic.)
[10] The production method of [1], wherein the compound into which the trifluoromethyl group is introduced is a four-membered ring compound.
[11] The production method according to [10], wherein the four-membered ring compound into which the trifluoromethyl group is introduced is a compound represented by the following general formula (10-3c) or (10-3d).
(Wherein R 23 and R 24 are each independently selected from a C1-C6 linear, branched, cyclic alkyl group or a phenyl group.)
(Wherein R 25 and R 26 are each independently selected from a C1-C6 linear, branched, cyclic alkyl group or phenyl group.)
[12] The production method of [1], wherein the compound into which the trifluoromethyl group is introduced is a bicyclo ring compound.
[13] The production method according to [12], wherein the bicyclo ring compound into which the trifluoromethyl group is introduced is a compound represented by the following general formula (37-1).
(Wherein R 27 and R 28 are each independently selected from a C1-C6 linear, branched, or cyclic alkyl group or a phenyl group, and R 27 or R 28 may form a ring structure) .)
[14] The production method of [1], wherein the compound into which the trifluoromethyl group is introduced is an aminocyclopentenedione compound.
[15] The production method of [14], wherein the aminocyclopentenedione compound having a trifluoromethyl group introduced is a compound represented by the following general formula (10-4).
(Wherein R is selected from isopropyl, n-propyl, t-butyl, isobutyl, sec-butyl, R 29 is a C1-C8 alkyl group, linear, branched, cyclic) R 30 is selected from a t-butyl group or a 1,1,3,3-tetramethylbutyl group.)
[16] A method for producing a trifluoromethyl-substituted semisquarate represented by the following general formula (1):
By the step of trifluoromethylating a squarate represented by the following general formula (3):
A compound represented by the following general formula (2) is produced,
A production method comprising synthesizing by a step of performing an allyl alcohol transfer reaction.
(In the formula, R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group, which may be different groups or the same group.)
[17] A method for producing a trifluoromethyl-substituted semisquarate represented by the following general formula (1):
(In the formula, R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group.)
A compound represented by the following general formula (2)
A production method comprising synthesizing by a step of performing an allyl alcohol transfer reaction.
(In the formula, R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group, which may be different groups or the same group.)
[18] The method for producing a trifluoromethyl-substituted semisquarate according to [16] or [17], wherein the allyl alcohol transfer reaction is a reaction using Re 2 O 7 or Ph 3 SiReO 3 as a catalyst. A manufacturing method characterized by being.
[19] A method for producing a compound represented by the following general formula (2),
The squarate represented by the following general formula (3)
A production method comprising synthesizing by a trifluoromethylation step.
(In the formula, R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group, which may be different groups or the same group.)
[20] The production method according to [16] or [19], wherein the trifluoromethylation step comprises a silyl trifluoromethylation reaction using CF 3 Me 3 Si as an organosilicon reagent; A production method comprising a desilylation step.
[21] A compound represented by the following general formula (1).
(In the formula, R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group.)
[22] A compound of the following general formula (2).
(In the formula, R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group, which may be different groups or the same group.)
[23] Trifluoromethyl compounds represented by the following formulas (12) to (52), (17-1), (10-3a), and (10-3b).
[1]下記一般式(1)で表される化合物を出発原料として、
付加反応工程、及び環拡大工程によってトリフルオロメチル基が導入された化合物を製造する方法。
[2]前記トリフルオロメチル基が導入された化合物が、キノン化合物である[1]記載の製造方法。
[3]トリフルオロメチル基が導入されたキノン化合物が、下記一般式(4)又は(5)で表される化合物である[2]記載の製造方法。
[4]前記トリフルオロメチル基が導入された化合物が、複素環縮環化合物である[1]記載の製造方法。
[5]トリフルオロメチル基が導入された複素環縮環化合物が、下記一般式(6)又は(7)で表される化合物である[4]記載の製造方法。
[6]前記トリフルオロメチル基が導入された化合物が、ヒドロキノン化合物である[1]記載の製造方法。
[7]トリフルオロメチル基が導入されたヒドロキノン化合物が、下記一般式(8)~(10)のいずれかで表される化合物である[6]記載の製造方法。
[8]前記トリフルオロメチル基が導入された化合物が、ブテノリド化合物である[1]記載の製造方法。
[9]トリフルオロメチル基が導入されたブテノリド化合物が、下記一般式(10-1)、(10-2)で表される化合物である[8]記載の製造方法。
[10]前記トリフルオロメチル基が導入された化合物が、四員環化合物である[1]記載の製造方法。
[11]トリフルオロメチル基が導入された四員環化合物が、下記一般式(10-3c)、(10-3d)で表される化合物である[10]記載の製造方法。
[12]前記トリフルオロメチル基が導入された化合物が、ビシクロ環化合物である[1]記載の製造方法。
[13]トリフルオロメチル基が導入されたビシクロ環化合物が、下記一般式(37-1)で表される化合物である[12]記載の製造方法。
[14]前記トリフルオロメチル基が導入された化合物が、アミノシクロペンテンジオン化合物である[1]記載の製造方法。
[15]トリフルオロメチル基が導入されたアミノシクロペンテンジオン化合物が、下記一般式(10-4)で表される化合物である[14]記載の製造方法。
[16]下記一般式(1)で表されるトリフルオロメチル置換セミスクアレートの製造方法であって、
(式中Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基であり、異なる基であっても、同一の基であってもよい。)
[17]下記一般式(1)で表されるトリフルオロメチル置換セミスクアレートの製造方法であって、
下記一般式(2)で表される化合物を
(式中Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基であり、異なる基であっても、同一の基であってもよい。)
[18][16]又は[17]記載のトリフルオロメチル置換セミスクアレートの製造方法であって、アリルアルコール転移反応を行なう工程が、Re2O7又はPh3SiReO3を触媒として用いる反応であることを特徴とする製造方法。
[19]下記一般式(2)で表される化合物の製造方法であって、
(式中Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基であり、異なる基であっても、同一の基であってもよい。)
[20][16]又は[19]記載の製造方法であって、前記トリフルオロメチル化する工程が、CF3Me3Siを有機ケイ素試薬として用いてシリルトリフルオロメチル化反応をする工程と、脱シリル化工程からなることを特徴とする製造方法。
[21]下記一般式(1)で表される化合物。
[22]下記一般式(2)の化合物。
[23]下記式(12)~(52)、(17-1)、(10-3a)、(10-3b)で表されるトリフルオロメチル化合物。
[1] A compound represented by the following general formula (1) as a starting material,
A method for producing a compound into which a trifluoromethyl group has been introduced by an addition reaction step and a ring expansion step.
[2] The production method according to [1], wherein the compound into which the trifluoromethyl group is introduced is a quinone compound.
[3] The production method according to [2], wherein the quinone compound into which the trifluoromethyl group is introduced is a compound represented by the following general formula (4) or (5).
[4] The production method of [1], wherein the compound into which the trifluoromethyl group is introduced is a heterocyclic condensed ring compound.
[5] The process according to [4], wherein the heterocyclic condensed ring compound into which the trifluoromethyl group is introduced is a compound represented by the following general formula (6) or (7).
[6] The production method according to [1], wherein the compound into which the trifluoromethyl group is introduced is a hydroquinone compound.
[7] The production method of [6], wherein the hydroquinone compound having a trifluoromethyl group introduced therein is a compound represented by any one of the following general formulas (8) to (10).
[8] The production method according to [1], wherein the compound into which the trifluoromethyl group is introduced is a butenolide compound.
[9] The production method according to [8], wherein the butenolide compound into which the trifluoromethyl group is introduced is a compound represented by the following general formulas (10-1) and (10-2).
[10] The production method of [1], wherein the compound into which the trifluoromethyl group is introduced is a four-membered ring compound.
[11] The production method according to [10], wherein the four-membered ring compound into which the trifluoromethyl group is introduced is a compound represented by the following general formula (10-3c) or (10-3d).
[12] The production method of [1], wherein the compound into which the trifluoromethyl group is introduced is a bicyclo ring compound.
[13] The production method according to [12], wherein the bicyclo ring compound into which the trifluoromethyl group is introduced is a compound represented by the following general formula (37-1).
[14] The production method of [1], wherein the compound into which the trifluoromethyl group is introduced is an aminocyclopentenedione compound.
[15] The production method of [14], wherein the aminocyclopentenedione compound having a trifluoromethyl group introduced is a compound represented by the following general formula (10-4).
[16] A method for producing a trifluoromethyl-substituted semisquarate represented by the following general formula (1):
(In the formula, R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group, which may be different groups or the same group.)
[17] A method for producing a trifluoromethyl-substituted semisquarate represented by the following general formula (1):
A compound represented by the following general formula (2)
(In the formula, R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group, which may be different groups or the same group.)
[18] The method for producing a trifluoromethyl-substituted semisquarate according to [16] or [17], wherein the allyl alcohol transfer reaction is a reaction using Re 2 O 7 or Ph 3 SiReO 3 as a catalyst. A manufacturing method characterized by being.
[19] A method for producing a compound represented by the following general formula (2),
(In the formula, R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group, which may be different groups or the same group.)
[20] The production method according to [16] or [19], wherein the trifluoromethylation step comprises a silyl trifluoromethylation reaction using CF 3 Me 3 Si as an organosilicon reagent; A production method comprising a desilylation step.
[21] A compound represented by the following general formula (1).
[22] A compound of the following general formula (2).
[23] Trifluoromethyl compounds represented by the following formulas (12) to (52), (17-1), (10-3a), and (10-3b).
高価な試薬を必要とせず、汎用性のあるトリフルオロメチル化合物の製造方法を開発したことで、効率よくトリフルオロメチル化合物を合成することができるようになった。さらに、今まで製造することが難しかったトリフルオロメチル化合物を製造することが可能となった。
Developed a versatile method for producing trifluoromethyl compounds without the need for expensive reagents, enabling efficient synthesis of trifluoromethyl compounds. Furthermore, it has become possible to produce trifluoromethyl compounds that have been difficult to produce.
図1に、本発明のトリフルオロメチル置換セミスクアレート法を模式的に示す。化合物3のスクアレートを出発原料として、短工程で化合物1のトリフルオロメチル置換セミスクアレートを合成できる。
FIG. 1 schematically shows the trifluoromethyl-substituted semisquarate method of the present invention. Using squalate of compound 3 as a starting material, trifluoromethyl-substituted semisquarate of compound 1 can be synthesized in a short process.
図1において、Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基が挙げられる。化合物3のスクアレート、及び化合物2のRとしては、上記官能基をとることができるが、2つのRは異なる基であっても同一の基であってもよい。ただし、合成の容易性から両者が同一の基であることが好ましい。
In FIG. 1, examples of R include isopropyl, n-propyl, t-butyl, isobutyl, and sec-butyl. The squarate of compound 3 and R of compound 2 can have the above functional groups, but the two Rs may be different groups or the same group. However, it is preferable that both are the same groups from the ease of synthesis.
スクアリン酸エステルは目的とする骨格へと短工程で変換できるため、四員環合成素子として有用な分子である。有機金属試薬を付加させて得られるヒドロキシシクロブテノンの環拡大反応によりキノンやブテノリドなど多様な分子が合成できる。したがって、トリフルオロメチル基の置換したセミスクアレートを合成することが可能になれば、これを合成素子として利用することで、トリフルオロメチル基を導入したキノンやブテノリドを効率よく合成することが可能となる。
Squaric acid ester is a useful molecule as a four-membered ring synthesis element because it can be converted into the target skeleton in a short process. Various molecules such as quinone and butenolide can be synthesized by ring expansion reaction of hydroxycyclobutenone obtained by adding an organometallic reagent. Therefore, if it is possible to synthesize semisquarates substituted with trifluoromethyl groups, it is possible to efficiently synthesize quinones and butenolides into which trifluoromethyl groups have been introduced by using them as synthesis elements. It becomes.
本発明のトリフルオロメチル置換セミスクアレートより合成可能なナフトキノン類及びベンゾキノン類としては、下記一般式(4)または(5)で表される化合物が挙げられる。
Examples of naphthoquinones and benzoquinones that can be synthesized from the trifluoromethyl-substituted semisquarate of the present invention include compounds represented by the following general formula (4) or (5).


また、トリフルオロメチル置換セミスクアレートより合成可能な複素環誘導体としては、下記一般式(6)、(7)で表される化合物が挙げられる。
In addition, examples of the heterocyclic derivative that can be synthesized from trifluoromethyl-substituted semisquarate include compounds represented by the following general formulas (6) and (7).

また、上記キノン類(4)、(5)、(6)に対応するヒドロキノン類として(8)、(9)、(10)が挙げられる。
Also, hydroquinones corresponding to the quinones (4), (5) and (6) include (8), (9) and (10).



また、トリフルオロメチル置換セミスクアレートより合成可能なブテノリド類としては、下記一般式(10-1)、(10-2)で表される化合物が挙げられる。
In addition, examples of butenolides that can be synthesized from trifluoromethyl-substituted semisquarate include compounds represented by the following general formulas (10-1) and (10-2).


さらに、トリフルオロメチル置換セミスクアレートより、新規化合物である中間体(10-3a、10-3b)を経て、下記式(37)で表されるビシクロ環化合物の合成も可能となった。
Furthermore, it has become possible to synthesize a bicyclo ring compound represented by the following formula (37) from trifluoromethyl-substituted semisquarate via intermediates (10-3a, 10-3b) which are novel compounds.


また、10-3aで表される化合物の収率を向上させる合成方法を開発することができたため、下記一般式10-3cで表される化合物を合成することができるようになった。その結果、下記一般式10-3dで表される化合物の合成、下記一般式37-1で表される化合物の合成も可能になった。
In addition, since a synthesis method for improving the yield of the compound represented by 10-3a could be developed, it became possible to synthesize a compound represented by the following general formula 10-3c. As a result, synthesis of a compound represented by the following general formula 10-3d and synthesis of a compound represented by the following general formula 37-1 became possible.



また、トリフルオロメチル置換セミスクアレートより、一般式(10-4)で表されるアミノシクロペンテンジオンの合成も可能となった。
In addition, aminocyclopentenedione represented by the general formula (10-4) can also be synthesized from trifluoromethyl-substituted semisquarate.

さらに、トリフルオロメチル置換セミスクアレートより、下記式(48)で表される開環化合物である不飽和カルボン酸の合成も可能となった。
Furthermore, it has become possible to synthesize an unsaturated carboxylic acid which is a ring-opening compound represented by the following formula (48) from trifluoromethyl-substituted semisquarate.

[実施例1]
≪トリフルオロメチル置換セミスクアレートの製造方法(1)≫
1.1 スクアリン酸エステルのトリフルオロメチル化工程
トリフルオロメチル化、アリルアルコール転移の2工程を別々に行い、トリフルオロメチル置換セミスクアレートを製造する方法を下記に示す。 [Example 1]
≪Method for producing trifluoromethyl-substituted semisquarate (1) ≫
1.1 Trifluoromethylation step of squarate ester A method for producing a trifluoromethyl-substituted semisquarate by separately performing two steps of trifluoromethylation and allyl alcohol transfer is shown below.
≪トリフルオロメチル置換セミスクアレートの製造方法(1)≫
1.1 スクアリン酸エステルのトリフルオロメチル化工程
トリフルオロメチル化、アリルアルコール転移の2工程を別々に行い、トリフルオロメチル置換セミスクアレートを製造する方法を下記に示す。 [Example 1]
≪Method for producing trifluoromethyl-substituted semisquarate (1) ≫
1.1 Trifluoromethylation step of squarate ester A method for producing a trifluoromethyl-substituted semisquarate by separately performing two steps of trifluoromethylation and allyl alcohol transfer is shown below.
まず、向山らの開発したシリルトリフルオロメチル化法(非特許文献22)を応用して、スクアリン酸エステルをトリフルオロメチル化する工程について説明する。下記反応式で示すスクアリン酸ジイソプロピルに対するトリフルオロメチル化を例に、一般的なトリフルオロメチル化の実験操作を示す。
First, the process of trifluoromethylating a squaric acid ester by applying the silyl trifluoromethylation method developed by Mukaiyama et al. A general experimental procedure for trifluoromethylation is shown by taking trifluoromethylation with respect to diisopropyl squarate shown in the following reaction formula as an example.

シュレンク管に撹拌子を入れ減圧下加熱乾燥し、スクアリン酸ジイソプロピル(3-iPr)(99.9mg、0.50mmol)、酢酸ナトリウム(NaOAc)(4.2mg、0.05mmol)、テトラブチルアンモニウムクロリド(nBu4NCl)(13.8mg、0.05mmol)、テトラヒドロフラン(THF)(1mL)を加えた。25°Cで15分撹拌し、トリフルオロメチルトリメチルシラン(Me3SiCF3)(111μL、0.75mmol)を加えた。20分撹拌後、原料の消失を確認し、テトラブチルアンモニウムフルオリド(TBAF)1M THF溶液(1mL、1mmol)を加え反応を停止させた。水(20mL)を加えEt2O(20mL×3)で抽出し、得られた有機層を飽和食塩水(10mL)で洗浄後、MgSO4で乾燥させ濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=10:1)で精製し白色固体(92.4mg、収率69%)を得た。得られた化合物の解析結果を下記に示す。
A stirring bar was placed in a Schlenk tube and dried by heating under reduced pressure. Diisopropyl squarate (3- i Pr) (99.9 mg, 0.50 mmol), sodium acetate (NaOAc) (4.2 mg, 0.05 mmol), tetrabutylammonium chloride (n Bu 4 NCl) (13.8mg , 0.05mmol), was added tetrahydrofuran (THF) (1mL). The mixture was stirred at 25 ° C. for 15 minutes, and trifluoromethyltrimethylsilane (Me 3 SiCF 3 ) (111 μL, 0.75 mmol) was added. After stirring for 20 minutes, disappearance of the raw materials was confirmed, and tetrabutylammonium fluoride (TBAF) 1M THF solution (1 mL, 1 mmol) was added to stop the reaction. Water (20 mL) was added and the mixture was extracted with Et 2 O (20 mL × 3). The obtained organic layer was washed with saturated brine (10 mL), dried over MgSO 4 and concentrated. The obtained crude product was purified by silica gel column chromatography (hexane: ethyl acetate = 10: 1) to obtain a white solid (92.4 mg, yield 69%). The analysis results of the obtained compound are shown below.
なお、解析は以下の方法で行った。1H・13C・19FNMR測定には日本電子製 ESC-400(400Hz)を使用した。測定溶媒にはCDCl3を用い、NMR測定は25°Cで実施した。1H・13C・19F NMRの化学シフトは全てδ値(ppm)で表記する。1H NMR測定の場合にはCHCl3に由来するシングレット(7.26ppm)を基準ピークとし、13C NMR測定の場合にはCHCl3に由来するトリプレット(77.0ppm)を基準ピークとし、19F NMRの化学シフトは外部標準としてα,α,α-トリフルオロトルエンのシングレットシグナル(-63.7ppm)を基準とした。また、化学シフトの分裂パターンは次のように定義する。s:シングレット、d:ダブレット、t:トリプレット、q:カルテット、quint:クインテット、sext:セクステット、sept:セプテット、m:マルチプレット。カップリング定数はHzで表記した。赤外吸収スペクトル(IR)の測定は日本分光製 FT/IR-230を使用し、特性吸収はすべてcm-1で表記した。融点測定にはSRS製 MPA100を用いた。質量分析には、日本電子製JMS-T100LPを用い、DARTまたはESIによる高分解能質量分析を行った。
The analysis was performed by the following method. For 1 H · 13 C · 19 FNMR measurement, ESC-400 (400 Hz) manufactured by JEOL Ltd. was used. CDCl 3 was used as a measurement solvent, and NMR measurement was performed at 25 ° C. All chemical shifts of 1 H · 13 C · 19 F NMR are expressed in δ values (ppm). In the case of 1 H NMR measurement, a singlet (7.26 ppm) derived from CHCl 3 is used as a reference peak, and in the case of 13 C NMR measurement, a triplet (77.0 ppm) derived from CHCl 3 is used as a reference peak, and 19 F The chemical shift of NMR was based on the singlet signal (-63.7 ppm) of α, α, α-trifluorotoluene as an external standard. The chemical shift splitting pattern is defined as follows. s: singlet, d: doublet, t: triplet, q: quartet, quint: quintet, sext: sextet, sept: septet, m: multiplet. Coupling constants are expressed in Hz. The infrared absorption spectrum (IR) was measured using FT / IR-230 manufactured by JASCO Corporation, and all characteristic absorptions were expressed in cm −1 . MPS100 made by SRS was used for the melting point measurement. For mass spectrometry, JMS-T100LP manufactured by JEOL Ltd. was used, and high-resolution mass spectrometry by DART or ESI was performed.

ここでは、スクアリン酸ジイソプロピルを出発材料とするトリフルオロメチル化の方法を詳細に説明したが、以下に示すように他のスクアリン酸エステルを用いても同様にトリフルオロメチル化を行うことができる。他のトリフルオロメチル化もシュレンク管もしくは二口ナスフラスコを用い同様の操作で行なうことができる。脱シリル化は、TBAF(2当量)またはフッ化カリウム飽和水溶液(1mL)を加え行うことができる。
Here, although the method of trifluoromethylation using diisopropyl squarate as a starting material has been described in detail, trifluoromethylation can be carried out in the same manner using other squarate esters as shown below. Other trifluoromethylation can be carried out in the same manner using a Schlenk tube or a two-necked eggplant flask. Desilylation can be performed by adding TBAF (2 equivalents) or saturated aqueous potassium fluoride (1 mL).
スクアリン酸ジプロピルを出発材料にトリフルオロメチル化を行い、下記式(2-1)の化合物、4-hydroxy-2,3-dipropoxy-4-(trifluoromethyl)cyclobut-2-enone(2-nPr)を合成した。収率は40%であった。得られた化合物の解析結果を下記に示す。
Trifluoromethylation was performed using dipropyl squarate as a starting material, and a compound of the following formula (2-1): 4-hydroxy-2,3-dipropyoxy-4- (trifluoromethyl) cyclobut-2-enone (2- n Pr) Was synthesized. The yield was 40%. The analysis results of the obtained compound are shown below.


スクアリン酸ジイソブチルを出発材料にトリフルオロメチル化を行い、下記式(2-2)の化合物、4-hydroxy-2,3-diisobutoxy-4-(trifluoromethyl)cyclobut-2-enone(2-iBu)を合成した。収率は40%であった。得られた化合物の解析結果を下記に示す。
The squaric acid diisobutyl performed trifluoromethyl into the starting material, the following formula (2-2) compound of, 4-hydroxy-2,3-diisobutoxy -4- (trifluoromethyl) cyclobut-2-enone (2- i Bu) Was synthesized. The yield was 40%. The analysis results of the obtained compound are shown below.


スクアリン酸ジsec-ブチルを出発材料にトリフルオロメチル化を行い、下記式(2-3)の化合物、2,3-di-sec-butoxy-4-hydroxy-4-(trifluoromethyl)cyclobut-2-enone(2-sBu)を合成した。収率は65%であった。得られた化合物の解析結果を下記に示す。
Trifluoromethylation was performed using disec-butyl squarate as a starting material, and a compound of the following formula (2-3), 2,3-di-sec-butyl-4-hydroxy-4- (trifluoromethyl) cyclobut-2- Oneone (2- s Bu) was synthesized. The yield was 65%. The analysis results of the obtained compound are shown below.


スクアリン酸ジtert-ブチルを出発材料にトリフルオロメチル化を行い、下記式(2-4)の化合物、2,3-di-tert-butoxy-4-hydroxy-4-(trifluoromethyl)cyclobut-2-en-1-one(2-tBu)を合成した。収率は90%であった。得られた化合物の解析結果を下記に示す。
Tritert-butyl squarate is used as a starting material, and trifluoromethylation is carried out to obtain a compound of the following formula (2-4): 2,3-di-tert-butyl-4-hydroxy-4- (trifluoromethyl) cyclobut-2- en-1-one (2- t Bu) was synthesized. The yield was 90%. The analysis results of the obtained compound are shown below.


1.2 アリルアルコール転移反応工程
次に、下記反応式で示すアリルアルコール転移反応を行なう。Re2O7触媒を用いた水酸基転移反応による1-iPr(11)の合成を例に、一般的な水酸基転移反応の実験操作を示す。 1.2 Allyl alcohol transfer reaction step Next, an allyl alcohol transfer reaction represented by the following reaction formula is performed. A general experimental procedure for hydroxyl group transfer reaction will be described by taking as an example the synthesis of 1- i Pr (11) by hydroxyl group transfer reaction using a Re 2 O 7 catalyst.
次に、下記反応式で示すアリルアルコール転移反応を行なう。Re2O7触媒を用いた水酸基転移反応による1-iPr(11)の合成を例に、一般的な水酸基転移反応の実験操作を示す。 1.2 Allyl alcohol transfer reaction step Next, an allyl alcohol transfer reaction represented by the following reaction formula is performed. A general experimental procedure for hydroxyl group transfer reaction will be described by taking as an example the synthesis of 1- i Pr (11) by hydroxyl group transfer reaction using a Re 2 O 7 catalyst.

シュレンク管に撹拌子を入れ減圧下加熱乾燥の後、アルゴン置換した。2-iPr(128.5mg、0.48mmol)、Re2O7(7.8mg,0.016mmol)を加え再度アルゴン置換を行った。ジクロロメタン(25mL)を加え、25°Cで10時間撹拌した。反応終了後、アルゴン気流下でアルミナを通してろ過をおこない、ろ液を濃縮し黄色液体(99.2mg、99%)を得た。得られた液体は長時間静置または冷却することで固化した。得られた化合物の解析結果を示す。
A stirring bar was placed in the Schlenk tube, and after heating and drying under reduced pressure, the atmosphere was replaced with argon. 2- i Pr (128.5 mg, 0.48 mmol) and Re 2 O 7 (7.8 mg, 0.016 mmol) were added, and argon substitution was performed again. Dichloromethane (25 mL) was added and stirred at 25 ° C. for 10 hours. After completion of the reaction, the mixture was filtered through alumina under an argon stream, and the filtrate was concentrated to obtain a yellow liquid (99.2 mg, 99%). The obtained liquid was solidified by standing for a long time or cooling. The analysis result of the obtained compound is shown.

ここでは、2-iPrのアリルアルコール転移反応により、トリフルオロメチル置換セミスクアレートを合成する方法を詳細に説明したが、以下に示すように他のヒドロキシシクロブテノンを用いても同様にアリルアルコール転移反応を行うことができる。他のアリルアルコール転移反応もシュレンク管もしくは二口ナスフラスコを用い同様の操作で行なうことができる。
Here, the method for synthesizing a trifluoromethyl-substituted semisquarate by 2- i Pr allylic alcohol transfer reaction has been described in detail. However, as shown below, other hydroxycyclobutenones can be used to form allyl alcohol. Alcohol transfer reactions can be performed. Other allyl alcohol transfer reactions can be performed in the same manner using a Schlenk tube or a two-necked eggplant flask.
2-nPrを出発材料にアリルアルコール転移反応を行い、下記式(11-1)の化合物、3-propoxy-4-(trifluoromethyl)cyclobut-3-ene-1,2-dioneを合成した。精製はAl2O3を充填剤とするショートカラム(展開溶媒はジクロロメタンを用いた。)で行い、収率は62%であった。得られた化合物の解析結果を下記に示す。
The 2-n Pr performed allyl alcohol rearrangement reaction starting materials, compounds of formula (11-1), 3-propoxy- 4- (trifluoromethyl) were synthesized cyclobut-3-ene-1,2- dione. Purification was carried out by a short column using Al 2 O 3 as a filler (developing solvent was dichloromethane), and the yield was 62%. The analysis results of the obtained compound are shown below.


2-iBuを出発材料にアリルアルコール転移反応を行い、下記式(11-2)の化合物、3-isobutoxy-4-(trifluoromethyl)cyclobut-3-ene-1,2-dioneを合成した。精製はAl2O3を充填剤とするショートカラム(展開溶媒はジクロロメタン)で行い、収率は50%であった。得られた化合物の解析結果を下記に示す。
The 2-i Bu performed allyl alcohol rearrangement reaction starting materials, compounds of formula (11-2), 3-isobutoxy- 4- (trifluoromethyl) were synthesized cyclobut-3-ene-1,2- dione. Purification was performed by a short column (developing solvent was dichloromethane) using Al 2 O 3 as a filler, and the yield was 50%. The analysis results of the obtained compound are shown below.


2-sBuを出発材料にアリルアルコール転移反応を行い、下記式(11-3)の化合物、3-sec-butoxy-4-(trifluoromethyl)cyclobut-3-ene-1,2-dioneを合成した。精製はAl2O3を充填剤とするショートカラム(展開溶媒はジクロロメタンを用いた。)で行い、収率は34%であった。得られた化合物の解析結果を下記に示す。
It performed allyl alcohol rearrangement reaction of 2-s Bu starting materials, compounds of formula (11-3) was synthesized 3-sec-butoxy-4- ( trifluoromethyl) cyclobut-3-ene-1,2-dione . Purification was performed by a short column using Al 2 O 3 as a filler (developing solvent was dichloromethane), and the yield was 34%. The analysis results of the obtained compound are shown below.


2-tBuを出発材料にアリルアルコール転移反応を行い、下記式(2-4)の化合物、3-tert-butoxy-4-(trifluoromethyl)cyclobut-3-ene-1,2-dioneを合成した。収率は78%であった。ただし、極めて不安定であり、精製はAl2O3/K2CO3を充填剤とするショートカラム(展開溶媒はジクロロメタンを用いた。)をアルゴン下で行い、濃縮もアルゴン下で行った。得られた化合物の解析結果を下記に示す。
Performed allyl alcohol rearrangement reaction of 2-t Bu in the starting materials, compounds of formula (2-4), was synthesized 3-tert-butoxy-4- ( trifluoromethyl) cyclobut-3-ene-1,2-dione . The yield was 78%. However, it was extremely unstable, and purification was carried out under a short column (developing solvent was dichloromethane) with Al 2 O 3 / K 2 CO 3 as a filler, and concentration was also carried out under argon. The analysis results of the obtained compound are shown below.


[実施例2]
≪トリフルオロメチル置換セミスクアレートの製造方法(2)≫
以下の方法により連続してトリフルオロメチル置換セミスクアレートを製造することも可能である。精製は最後の工程の蒸留操作のみとなり、より簡便に効率よく反応を行うことができる。 [Example 2]
≪Method for producing trifluoromethyl-substituted semisquarate (2) ≫
It is also possible to produce a trifluoromethyl-substituted semisquarate continuously by the following method. Purification is only the distillation operation in the last step, and the reaction can be performed more simply and efficiently.
≪トリフルオロメチル置換セミスクアレートの製造方法(2)≫
以下の方法により連続してトリフルオロメチル置換セミスクアレートを製造することも可能である。精製は最後の工程の蒸留操作のみとなり、より簡便に効率よく反応を行うことができる。 [Example 2]
≪Method for producing trifluoromethyl-substituted semisquarate (2) ≫
It is also possible to produce a trifluoromethyl-substituted semisquarate continuously by the following method. Purification is only the distillation operation in the last step, and the reaction can be performed more simply and efficiently.
市販のジイソプロピルスクアレート(化合物3-iPr)3gより、下記反応式に示す2工程を連続してトリフルオロメチル置換セミスクアレート(化合物1-iPr)を合成した。収率は84%であった。以下、合成方法を詳述する。
A trifluoromethyl-substituted semisquarate (Compound 1- i Pr) was synthesized from 3 g of commercially available diisopropyl squalate (Compound 3- i Pr) by continuously performing the two steps shown in the following reaction formula. The yield was 84%. Hereinafter, the synthesis method will be described in detail.

シュレンク管に撹拌子を入れ減圧下加熱乾燥し、スクアリン酸ジイソプロピル(2.98g、15mmol)、酢酸ナトリウム(61.0mg、0.74mmol)、テトラブチルアンモニウムクロリド(206.2mg、0.74mmol)、THF(15mL)を加えた。25°Cで20分撹拌し、トリフルオロメチルトリメチルシラン(3.1mL、21mmol)を加えた。90分撹拌後、原料の消失を確認し、テトラブチルアンモニウムフルオリド1M THF溶液(21mL、21mmol)を加え、15分攪拌した。水(30mL)を加えEt2O(30mL×3)で抽出し、得られた有機層を飽和食塩水(30mL)で洗浄後、硫酸ナトリウムで乾燥し、溶媒を減圧留去して粗生成物を得た。
A Schlenk tube was charged with a stirring bar and dried under reduced pressure. Diisopropyl squarate (2.98 g, 15 mmol), sodium acetate (61.0 mg, 0.74 mmol), tetrabutylammonium chloride (206.2 mg, 0.74 mmol), THF (15 mL) was added. The mixture was stirred at 25 ° C for 20 minutes, and trifluoromethyltrimethylsilane (3.1 mL, 21 mmol) was added. After stirring for 90 minutes, disappearance of the raw materials was confirmed, tetrabutylammonium fluoride 1M THF solution (21 mL, 21 mmol) was added, and the mixture was stirred for 15 minutes. Water (30 mL) was added and extracted with Et 2 O (30 mL × 3). The obtained organic layer was washed with saturated brine (30 mL) and dried over sodium sulfate, and the solvent was distilled off under reduced pressure to obtain a crude product. Got.
次にアリルアルコール転移反応を行なう。粗生成物を100mL二口フラスコに移し、減圧下で2時間乾燥し、アルゴン置換した。Re2O7(218mg、0.45mmol、3mol%)をグローブバック中で加え、脱水ジクロロメタン(35mL)で希釈し、10時間撹拌した。原料の消失を確認後、濃縮した後にクーゲルロール蒸留器(3hPa、70°C)を用い精製を行い、黄色固体(2.61g、収率84%)を得た。実施例1と同様の化合物を得ることができた。
Next, an allyl alcohol transfer reaction is performed. The crude product was transferred to a 100 mL two-necked flask, dried under reduced pressure for 2 hours, and purged with argon. Re 2 O 7 (218 mg, 0.45 mmol, 3 mol%) was added in the glove bag, diluted with dehydrated dichloromethane (35 mL) and stirred for 10 hours. After confirming the disappearance of the raw materials, after concentration, purification was performed using a Kugelrohr distiller (3 hPa, 70 ° C.) to obtain a yellow solid (2.61 g, yield 84%). A compound similar to Example 1 could be obtained.
[実施例3]
≪生理活性物質類縁体の合成(1)≫
実施例1、又は2の方法によって合成された一般式(1)の化合物を起点として生理活性物質類縁体を合成する方法を次に示す。 [Example 3]
≪Synthesis of physiologically active substance analogues (1) ≫
A method for synthesizing a physiologically active substance analog starting from the compound of the general formula (1) synthesized by the method of Example 1 or 2 is described below.
≪生理活性物質類縁体の合成(1)≫
実施例1、又は2の方法によって合成された一般式(1)の化合物を起点として生理活性物質類縁体を合成する方法を次に示す。 [Example 3]
≪Synthesis of physiologically active substance analogues (1) ≫
A method for synthesizing a physiologically active substance analog starting from the compound of the general formula (1) synthesized by the method of Example 1 or 2 is described below.

付加反応工程、環拡大反応及び酸化反応工程によって、トリフルオロメチル基が導入されているキノンを合成する。2-isopropoxy-3-(trifluoromethyl)naphthalene-1,4-dione(12)の製造を例に、詳細に製造方法を説明する。下記反応式は製造方法の概略である。
A quinone having a trifluoromethyl group introduced therein is synthesized by an addition reaction step, a ring expansion reaction and an oxidation reaction step. The production method will be described in detail by taking as an example the production of 2-isopropoxy-3- (trifluoromethyl) naphthalene-1,4-dione (12). The following reaction formula is an outline of the production method.

まず、グリニヤール試薬溶液を調製する。PhMgBr(Aldrich社製)(1.1M THF溶液910μL又は3.0M Et2O溶液333μL;1mmol)を、減圧下加熱乾燥した容器中に加えドライエーテルで希釈し、30分撹拌し、0.1MのEt2O溶液とした。
First, a Grignard reagent solution is prepared. PhMgBr (manufactured by Aldrich) (910 μL of 1.1 M THF solution or 333 μL of 3.0 M Et 2 O solution; 1 mmol) was added to a container heated and dried under reduced pressure, diluted with dry ether, stirred for 30 minutes, 0.1 M Et 2 O solution.
次に付加反応を行なう。減圧下加熱乾燥した50mL二口ナスフラスコに撹拌子、1-iPr(104.2mg、0.5mmol)、ジエチルエーテル(2mL)を加え、-90°Cに冷却した。温度計で系内の温度を測定し、調製したグリニヤール試薬溶液を-90°C以上の温度にならないよう注意しながら、シリンジポンプを使用し20mL/hの速度で30分かけて滴下した。さらに30分攪拌した後、NMRで原料の消失を確認し、-90°Cのまま飽和塩化アンモニウム水溶液(2mL)を加え反応を停止させ、室温に戻した。Et2O(10mL×3)で抽出後、飽和食塩水(10mL)で洗浄、硫酸ナトリウムで乾燥させ、p-キシレン(10mL)を加えた。
Next, an addition reaction is performed. A stir bar, 1- i Pr (104.2 mg, 0.5 mmol) and diethyl ether (2 mL) were added to a 50 mL two-necked eggplant flask heated and dried under reduced pressure, and cooled to -90 ° C. The temperature in the system was measured with a thermometer, and the prepared Grignard reagent solution was added dropwise at a rate of 20 mL / h over 30 minutes using a syringe pump while taking care not to reach a temperature of −90 ° C. or higher. After further stirring for 30 minutes, disappearance of the raw material was confirmed by NMR, and the reaction was stopped by adding a saturated aqueous ammonium chloride solution (2 mL) while maintaining at −90 ° C., and the temperature was returned to room temperature. The mixture was extracted with Et 2 O (10 mL × 3), washed with saturated brine (10 mL), dried over sodium sulfate, and p-xylene (10 mL) was added.
環拡大反応、及び酸化反応は次のようにして行う。Et2Oをロータリーエバポレーターで除去し、p-キシレン溶液(約10mL)とした。得られた溶液をアルゴン置換し140°Cで10分間攪拌した。中間生成物である4-ヒドロキシシクロブテノンの消失を確認し、室温に冷却した。フタロシアニン鉄(II)錯体 [Fe(pc)](8.4mg、0.015mmol)、酢酸(0.5mL)を加え酸素雰囲気下1時間撹拌した。ヒドロキノンの消失を確認した後、セライト濾過、濃縮し粗生成物を得た。
Ring expansion reaction and oxidation reaction are performed as follows. Et 2 O was removed with a rotary evaporator to obtain a p-xylene solution (about 10 mL). The resulting solution was purged with argon and stirred at 140 ° C. for 10 minutes. After confirming the disappearance of 4-hydroxycyclobutenone as an intermediate product, the mixture was cooled to room temperature. Phthalocyanine iron (II) complex [Fe (pc)] (8.4 mg, 0.015 mmol) and acetic acid (0.5 mL) were added and stirred for 1 hour in an oxygen atmosphere. After confirming the disappearance of hydroquinone, cerite filtration and concentration were performed to obtain a crude product.
シリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=10:1)で精製を行い、黄色液体(103.8mg、収率73%)を得た。得られた化合物、2-isopropoxy-3-(trifluoromethyl)naphthalene-1,4-dioneの解析結果を下記に示す。
Purification was performed by silica gel column chromatography (hexane: ethyl acetate = 10: 1) to obtain a yellow liquid (103.8 mg, yield 73%). The analysis results of the obtained compound, 2-isopropoxy-3- (trifluoromethyl) naphthalene-1,4-dione, are shown below.

[実施例4]
≪生理活性物質類縁体の合成(2)≫
下記式(13)に構造式を示す2-isopropoxy-6-methyl-3-(trifluoromethyl)naphthalene-1,4-dioneは、グリニヤール試薬として、アリールブロマイドと金属マグネシウムにより、4-メチルマグネシウムブロミド(非特許文献23、24)を調製して用いた他は、実施例3と同様にして製造した。 [Example 4]
≪Synthesis of physiologically active substance analogues (2) ≫
2-isopropoxy-6-methyl-3- (trifluoromethyl) naphthalene-1,4-dione, whose structural formula is shown in the following formula (13), is obtained by using 4-methylmagnesium bromide (non-non-crystalline) as a Grignard reagent with aryl bromide and magnesium metal. It was manufactured in the same manner as in Example 3 except that Patent Documents 23 and 24) were prepared and used.
≪生理活性物質類縁体の合成(2)≫
下記式(13)に構造式を示す2-isopropoxy-6-methyl-3-(trifluoromethyl)naphthalene-1,4-dioneは、グリニヤール試薬として、アリールブロマイドと金属マグネシウムにより、4-メチルマグネシウムブロミド(非特許文献23、24)を調製して用いた他は、実施例3と同様にして製造した。 [Example 4]
≪Synthesis of physiologically active substance analogues (2) ≫
2-isopropoxy-6-methyl-3- (trifluoromethyl) naphthalene-1,4-dione, whose structural formula is shown in the following formula (13), is obtained by using 4-methylmagnesium bromide (non-non-crystalline) as a Grignard reagent with aryl bromide and magnesium metal. It was manufactured in the same manner as in Example 3 except that Patent Documents 23 and 24) were prepared and used.


[実施例5]
≪生理活性物質類縁体の合成(3)≫
下記(14)に構造式を示す2-isopropoxy-6-methoxy-3-(trifluoromethyl)naphthalene-1,4-dioneは、グリニヤール試薬として、4-メトキシフェニマグネシウムブロミド(Aldrich社製)を用いた他は、実施例3と同様にして製造した。 [Example 5]
≪Synthesis of physiologically active substance analogues (3) ≫
2-isopropoxy-6-methoxy-3- (trifluoromethyl) naphthalene-1,4-dione, whose structural formula is shown in the following (14), is obtained by using 4-methoxyphenymagnesium bromide (manufactured by Aldrich) as a Grignard reagent. Was produced in the same manner as in Example 3.
≪生理活性物質類縁体の合成(3)≫
下記(14)に構造式を示す2-isopropoxy-6-methoxy-3-(trifluoromethyl)naphthalene-1,4-dioneは、グリニヤール試薬として、4-メトキシフェニマグネシウムブロミド(Aldrich社製)を用いた他は、実施例3と同様にして製造した。 [Example 5]
≪Synthesis of physiologically active substance analogues (3) ≫
2-isopropoxy-6-methoxy-3- (trifluoromethyl) naphthalene-1,4-dione, whose structural formula is shown in the following (14), is obtained by using 4-methoxyphenymagnesium bromide (manufactured by Aldrich) as a Grignard reagent. Was produced in the same manner as in Example 3.


[実施例6]
≪生理活性物質類縁体の合成(4)≫
下記(15)に構造式を示す2-isopropoxy-5-methoxy-3-(trifluoromethyl)naphthalene-1,4-dioneは、グリニヤール試薬として、2-メトキシフェニマグネシウムブロミド(Aldrich社製)を用いた他は、実施例3と同様にして製造した。 [Example 6]
≪Synthesis of bioactive substance analogues (4) ≫
2-isopropoxy-5-methoxy-3- (trifluoromethyl) naphthalene-1,4-dione, whose structural formula is shown in (15) below, is obtained by using 2-methoxyphenymagnesium bromide (manufactured by Aldrich) as a Grignard reagent. Was produced in the same manner as in Example 3.
≪生理活性物質類縁体の合成(4)≫
下記(15)に構造式を示す2-isopropoxy-5-methoxy-3-(trifluoromethyl)naphthalene-1,4-dioneは、グリニヤール試薬として、2-メトキシフェニマグネシウムブロミド(Aldrich社製)を用いた他は、実施例3と同様にして製造した。 [Example 6]
≪Synthesis of bioactive substance analogues (4) ≫
2-isopropoxy-5-methoxy-3- (trifluoromethyl) naphthalene-1,4-dione, whose structural formula is shown in (15) below, is obtained by using 2-methoxyphenymagnesium bromide (manufactured by Aldrich) as a Grignard reagent. Was produced in the same manner as in Example 3.


[実施例7]
≪生理活性物質類縁体の合成(5)≫
下記式(16)に構造式を示す6-chloro-2-isopropoxy-3-(trifluoromethyl)naphthalene-1,4-dioneは、グリニヤール試薬として、4-クロロフェニルマグネシウムブロミド(Aldrich社製)を用いた他は、実施例3と同様にして製造した。 [Example 7]
≪Synthesis of physiologically active substance analogues (5) ≫
6-chloro-2-isopropyoxy-3- (trifluoromethyl) naphthalene-1,4-dione, whose structural formula is represented by the following formula (16), is obtained by using 4-chlorophenylmagnesium bromide (manufactured by Aldrich) as a Grignard reagent. Was produced in the same manner as in Example 3.
≪生理活性物質類縁体の合成(5)≫
下記式(16)に構造式を示す6-chloro-2-isopropoxy-3-(trifluoromethyl)naphthalene-1,4-dioneは、グリニヤール試薬として、4-クロロフェニルマグネシウムブロミド(Aldrich社製)を用いた他は、実施例3と同様にして製造した。 [Example 7]
≪Synthesis of physiologically active substance analogues (5) ≫
6-chloro-2-isopropyoxy-3- (trifluoromethyl) naphthalene-1,4-dione, whose structural formula is represented by the following formula (16), is obtained by using 4-chlorophenylmagnesium bromide (manufactured by Aldrich) as a Grignard reagent. Was produced in the same manner as in Example 3.


[実施例8]
≪生理活性物質類縁体の合成(6)≫
下記式(17)に構造式を示す6-fluoro-2-isopropoxy-3-(trifluoromethyl)naphthalene-1,4-dioneは、グリニヤール試薬として、4-フルオロフェニルマグネシウムブロミド(Aldrich社製)を用いた他は、実施例3と同様にして製造した。 [Example 8]
≪Synthesis of physiologically active substance analogues (6) ≫
6-fluoro-2-isopropoxy-3- (trifluoromethyl) naphthalene-1,4-dione, whose structural formula is represented by the following formula (17), used 4-fluorophenylmagnesium bromide (manufactured by Aldrich) as a Grignard reagent. Others were produced in the same manner as in Example 3.
≪生理活性物質類縁体の合成(6)≫
下記式(17)に構造式を示す6-fluoro-2-isopropoxy-3-(trifluoromethyl)naphthalene-1,4-dioneは、グリニヤール試薬として、4-フルオロフェニルマグネシウムブロミド(Aldrich社製)を用いた他は、実施例3と同様にして製造した。 [Example 8]
≪Synthesis of physiologically active substance analogues (6) ≫
6-fluoro-2-isopropoxy-3- (trifluoromethyl) naphthalene-1,4-dione, whose structural formula is represented by the following formula (17), used 4-fluorophenylmagnesium bromide (manufactured by Aldrich) as a Grignard reagent. Others were produced in the same manner as in Example 3.


[実施例9]
≪生理活性物質類縁体の合成(7)≫
下記式(17-1)に構造式を示すethyl 6-isopropoxy-5,8-dioxo-7-(trifluoromethyl)-5,8-dihydronaphthalene-2-carboxylateは、グリニヤール試薬としてp-エトキシカルボニルフェニルマグネシウムクロリド(非特許文献25)を用いた他は、実施例3と同様にして製造した。 [Example 9]
≪Synthesis of bioactive substance analogues (7) ≫
Ethyl 6-isopropoxy-5,8-dioxo-7- (trifluoromethyl) -5,8-dihydroxynaphthalene-2-carboxylate having the structural formula shown in the following formula (17-1) is p-ethoxycarbonylphenylmagnesium chloride as a Grignard reagent. It was manufactured in the same manner as in Example 3 except that (Non-patent Document 25) was used.
≪生理活性物質類縁体の合成(7)≫
下記式(17-1)に構造式を示すethyl 6-isopropoxy-5,8-dioxo-7-(trifluoromethyl)-5,8-dihydronaphthalene-2-carboxylateは、グリニヤール試薬としてp-エトキシカルボニルフェニルマグネシウムクロリド(非特許文献25)を用いた他は、実施例3と同様にして製造した。 [Example 9]
≪Synthesis of bioactive substance analogues (7) ≫
Ethyl 6-isopropoxy-5,8-dioxo-7- (trifluoromethyl) -5,8-dihydroxynaphthalene-2-carboxylate having the structural formula shown in the following formula (17-1) is p-ethoxycarbonylphenylmagnesium chloride as a Grignard reagent. It was manufactured in the same manner as in Example 3 except that (Non-patent Document 25) was used.


[実施例10]
≪生理活性物質類縁体の合成(8)≫
下記式(18)に構造式を示す3-isopropoxy-2-(trifluoromethyl)phenanthrene-1,4-dioneは、グリニヤール試薬として、1-ナフチルマグネシウムブロミド(Aldrich社製)を用いた他は、実施例3と同様にして製造した。 [Example 10]
≪Synthesis of physiologically active substance analogues (8) ≫
3-isopropoxy-2- (trifluoromethyl) phenanthrene-1,4-dione, whose structural formula is represented by the following formula (18), was used in Examples except that 1-naphthylmagnesium bromide (manufactured by Aldrich) was used as a Grignard reagent. 3 was produced in the same manner as in No. 3.
≪生理活性物質類縁体の合成(8)≫
下記式(18)に構造式を示す3-isopropoxy-2-(trifluoromethyl)phenanthrene-1,4-dioneは、グリニヤール試薬として、1-ナフチルマグネシウムブロミド(Aldrich社製)を用いた他は、実施例3と同様にして製造した。 [Example 10]
≪Synthesis of physiologically active substance analogues (8) ≫
3-isopropoxy-2- (trifluoromethyl) phenanthrene-1,4-dione, whose structural formula is represented by the following formula (18), was used in Examples except that 1-naphthylmagnesium bromide (manufactured by Aldrich) was used as a Grignard reagent. 3 was produced in the same manner as in No. 3.


[実施例11]
≪生理活性物質類縁体の合成(9)≫
下記式(19)に構造式を示す2-isopropoxy-3-(trifluoromethyl)phenanthrene-1,4-dioneは、グリニヤール試薬として、アリールブロマイドと金属マグネシウムにより調製した、2-ナフチルマグネシウムブロミド(非特許文献23、24)を用いた他は、実施例3と同様にして製造した。 [Example 11]
≪Synthesis of bioactive substance analogues (9) ≫
2-isopropoxy-3- (trifluoromethyl) phenanthrene-1,4-dione, which has the structural formula shown in the following formula (19), is 2-naphthylmagnesium bromide prepared by using aryl bromide and magnesium metal as a Grignard reagent (non-patent literature) 23 and 24) were used in the same manner as in Example 3.
≪生理活性物質類縁体の合成(9)≫
下記式(19)に構造式を示す2-isopropoxy-3-(trifluoromethyl)phenanthrene-1,4-dioneは、グリニヤール試薬として、アリールブロマイドと金属マグネシウムにより調製した、2-ナフチルマグネシウムブロミド(非特許文献23、24)を用いた他は、実施例3と同様にして製造した。 [Example 11]
≪Synthesis of bioactive substance analogues (9) ≫
2-isopropoxy-3- (trifluoromethyl) phenanthrene-1,4-dione, which has the structural formula shown in the following formula (19), is 2-naphthylmagnesium bromide prepared by using aryl bromide and magnesium metal as a Grignard reagent (non-patent literature) 23 and 24) were used in the same manner as in Example 3.


[実施例12]
≪生理活性物質類縁体の合成(10)≫
下記式(20)に構造式を示す6-isopropoxy-5-(trifluoromethyl)benzofuran-4,7-dioneは、グリニヤール試薬として、MgBr2を用いて有機リチウム試薬から調製した2-フリルマグネシウムブロミド(非特許文献26、27)を用いた他は、実施例3と同様にして製造した。 [Example 12]
≪Synthesis of physiologically active substance analogues (10) ≫
6-isopropoxy-5- (trifluoromethyl) benzofuran-4,7-dione, which has the structural formula shown in the following formula (20), is 2-furylmagnesium bromide prepared from an organolithium reagent using MgBr 2 as a Grignard reagent. It was manufactured in the same manner as in Example 3 except that Patent Documents 26 and 27) were used.
≪生理活性物質類縁体の合成(10)≫
下記式(20)に構造式を示す6-isopropoxy-5-(trifluoromethyl)benzofuran-4,7-dioneは、グリニヤール試薬として、MgBr2を用いて有機リチウム試薬から調製した2-フリルマグネシウムブロミド(非特許文献26、27)を用いた他は、実施例3と同様にして製造した。 [Example 12]
≪Synthesis of physiologically active substance analogues (10) ≫
6-isopropoxy-5- (trifluoromethyl) benzofuran-4,7-dione, which has the structural formula shown in the following formula (20), is 2-furylmagnesium bromide prepared from an organolithium reagent using MgBr 2 as a Grignard reagent. It was manufactured in the same manner as in Example 3 except that Patent Documents 26 and 27) were used.


[実施例13]
≪生理活性物質類縁体の合成(11)≫
下記式(21)に構造式を示す6-isopropoxy-5-(trifluoromethyl)benzo[b]thiophene-4,7-dioneは、グリニヤール試薬として、MgBr2を用いて有機リチウム試薬から調製した、2-チエニルマグネシウムブロミド(非特許文献26、27)を用いた他は、実施例3と同様にして製造した。 [Example 13]
≪Synthesis of bioactive substance analogues (11) ≫
6-isopropoxy-5- (trifluoromethyl) benzo [b] thiophene-4,7-dione, whose structural formula is shown in the following formula (21), was prepared from an organolithium reagent using MgBr 2 as a Grignard reagent. It was manufactured in the same manner as in Example 3 except that thienylmagnesium bromide (Non-patent Documents 26 and 27) was used.
≪生理活性物質類縁体の合成(11)≫
下記式(21)に構造式を示す6-isopropoxy-5-(trifluoromethyl)benzo[b]thiophene-4,7-dioneは、グリニヤール試薬として、MgBr2を用いて有機リチウム試薬から調製した、2-チエニルマグネシウムブロミド(非特許文献26、27)を用いた他は、実施例3と同様にして製造した。 [Example 13]
≪Synthesis of bioactive substance analogues (11) ≫
6-isopropoxy-5- (trifluoromethyl) benzo [b] thiophene-4,7-dione, whose structural formula is shown in the following formula (21), was prepared from an organolithium reagent using MgBr 2 as a Grignard reagent. It was manufactured in the same manner as in Example 3 except that thienylmagnesium bromide (Non-patent Documents 26 and 27) was used.


[実施例14]
≪生理活性物質類縁体の合成(12)≫
下記式(22)に構造式を示す3-isopropoxy-2-(trifluoromethyl)dibenzo[b,d]furan-1,4-dioneは、グリニヤール試薬として、MgBr2を用いて有機リチウム試薬から調製した、2-ベンゾフリルマグネシウムブロミド(非特許文献26、27)を用いた他は、実施例3と同様にして製造した。 [Example 14]
≪Synthesis of bioactive substance analogues (12) ≫
3-isopropoxy-2- (trifluoromethyl) dibenzo [b, d] furan-1,4-dione, whose structural formula is shown in the following formula (22), was prepared from an organolithium reagent using MgBr 2 as a Grignard reagent. The product was prepared in the same manner as in Example 3 except that 2-benzofurylmagnesium bromide (Non-patent Documents 26 and 27) was used.
≪生理活性物質類縁体の合成(12)≫
下記式(22)に構造式を示す3-isopropoxy-2-(trifluoromethyl)dibenzo[b,d]furan-1,4-dioneは、グリニヤール試薬として、MgBr2を用いて有機リチウム試薬から調製した、2-ベンゾフリルマグネシウムブロミド(非特許文献26、27)を用いた他は、実施例3と同様にして製造した。 [Example 14]
≪Synthesis of bioactive substance analogues (12) ≫
3-isopropoxy-2- (trifluoromethyl) dibenzo [b, d] furan-1,4-dione, whose structural formula is shown in the following formula (22), was prepared from an organolithium reagent using MgBr 2 as a Grignard reagent. The product was prepared in the same manner as in Example 3 except that 2-benzofurylmagnesium bromide (Non-patent Documents 26 and 27) was used.


[実施例15]
≪生理活性物質類縁体の合成(13)≫
下記(23)に構造式を示す3-isopropoxy-2-(trifluoromethyl)dibenzo[b,d]thiophene-1,4-dioneは、グリニヤール試薬として、MgBr2を用いて有機リチウム試薬から調製した、ベンゾチエニルマグネシウムブロミド(非特許文献26、27)を用いた他は、実施例3と同様にして製造した。 [Example 15]
≪Synthesis of bioactive substance analogues (13) ≫
3-isopropoxy-2- (trifluoromethyl) dibenzo [b, d] thiophene-1,4-dione, whose structural formula is shown in (23) below, was prepared from an organolithium reagent using MgBr 2 as a Grignard reagent. It was manufactured in the same manner as in Example 3 except that thienylmagnesium bromide (Non-patent Documents 26 and 27) was used.
≪生理活性物質類縁体の合成(13)≫
下記(23)に構造式を示す3-isopropoxy-2-(trifluoromethyl)dibenzo[b,d]thiophene-1,4-dioneは、グリニヤール試薬として、MgBr2を用いて有機リチウム試薬から調製した、ベンゾチエニルマグネシウムブロミド(非特許文献26、27)を用いた他は、実施例3と同様にして製造した。 [Example 15]
≪Synthesis of bioactive substance analogues (13) ≫
3-isopropoxy-2- (trifluoromethyl) dibenzo [b, d] thiophene-1,4-dione, whose structural formula is shown in (23) below, was prepared from an organolithium reagent using MgBr 2 as a Grignard reagent. It was manufactured in the same manner as in Example 3 except that thienylmagnesium bromide (Non-patent Documents 26 and 27) was used.


[実施例16]
≪生理活性物質類縁体の合成(14)≫
下記式(24)に構造式を示すtert-butyl 2-isopropoxy-1,4-dioxo-3-(trifluoromethyl)-1,4-dihydro-9H-carbazole-9-carboxylate、及び下記(25)に構造式を示すtert-butyl 2-(3-isopropoxy-5-oxo-4-(trifluoromethyl)-2,5-dihydrofuran-2-yl)-1H-indole-1-carboxylateは、グリニヤール試薬として、2-ヨードインドールより調製した、N-Boc-2-インドールマグネシウムブロミド(非特許文献28)を用い、実施例3と同様の方法で製造を行うことにより、同時に得ることができる。 [Example 16]
≪Synthesis of bioactive substance analogues (14) ≫
Tert-butyl 2-isopropoxy-1,4-dioxo-3- (trifluoromethyl) -1,4-dihydro-9H-carbazole-9-carboxylate, which has the structural formula shown in the following formula (24), and a structure shown in the following (25) The tert-butyl 2- (3-isopropoxy-5-oxo-4- (trifluoromethyl) -2,5-dihydrofuran-2-yl) -1H-indole-1-carboxylate having the formula is used as a Grignard reagent as 2-iodo By using N-Boc-2-indole magnesium bromide (Non-patent Document 28) prepared from indole, it can be obtained simultaneously by carrying out the production in the same manner as in Example 3.
≪生理活性物質類縁体の合成(14)≫
下記式(24)に構造式を示すtert-butyl 2-isopropoxy-1,4-dioxo-3-(trifluoromethyl)-1,4-dihydro-9H-carbazole-9-carboxylate、及び下記(25)に構造式を示すtert-butyl 2-(3-isopropoxy-5-oxo-4-(trifluoromethyl)-2,5-dihydrofuran-2-yl)-1H-indole-1-carboxylateは、グリニヤール試薬として、2-ヨードインドールより調製した、N-Boc-2-インドールマグネシウムブロミド(非特許文献28)を用い、実施例3と同様の方法で製造を行うことにより、同時に得ることができる。 [Example 16]
≪Synthesis of bioactive substance analogues (14) ≫
Tert-butyl 2-isopropoxy-1,4-dioxo-3- (trifluoromethyl) -1,4-dihydro-9H-carbazole-9-carboxylate, which has the structural formula shown in the following formula (24), and a structure shown in the following (25) The tert-butyl 2- (3-isopropoxy-5-oxo-4- (trifluoromethyl) -2,5-dihydrofuran-2-yl) -1H-indole-1-carboxylate having the formula is used as a Grignard reagent as 2-iodo By using N-Boc-2-indole magnesium bromide (Non-patent Document 28) prepared from indole, it can be obtained simultaneously by carrying out the production in the same manner as in Example 3.

tert-butyl 2-isopropoxy-1,4-dioxo-3-(trifluoromethyl)-1,4-dihydro-9H-carbazole-9-carboxylate(構造式(24)で表される化合物)の収率は24%であった。得られた化合物の解析結果を下記に示す。
The yield of tert-butyl 2-isopropoxy-1,4-dioxo-3- (trifluoromethyl) -1,4-dihydro-9H-carbazole-9-carboxylate (compound represented by the structural formula (24)) is 24%. Met. The analysis results of the obtained compound are shown below.

tert-butyl 2-(3-isopropoxy-5-oxo-4-(trifluoromethyl)-2,5-dihydrofuran-2-yl)-1H-indole-1-carboxylate(構造式(25)で表される化合物)の収率は23%であった。得られた化合物の解析結果を下記に示す。
tert-butyl 2- (3-isopropoxy-5-oxo-4- (trifluoromethyl) -2,5-dihydrofuran-2-yl) -1H-indole-1-carboxylate (compound represented by structural formula (25)) The yield of was 23%. The analysis results of the obtained compound are shown below.

[実施例17]
≪生理活性物質類縁体の合成(15)≫
下記(26)に構造式を示すtert-butyl 3-isopropoxy-1,4-dioxo-2-(trifluoromethyl)-1,4-dihydro-9H-carbazole-9-carboxylateは、グリニヤール試薬として、3-ヨードインドール誘導体から調製した、N-Boc-3-インドールマグネシウムブロミド(非特許文献29)を用いた他は、実施例3と同様にして製造した。 [Example 17]
≪Synthesis of bioactive substance analogues (15) ≫
The tert-butyl 3-isopropoxy-1,4-dioxo-2- (trifluoromethyl) -1,4-dihydro-9H-carbazole-9-carboxylate having the structural formula shown in the following (26) is a 3-iodo as a Grignard reagent. This was prepared in the same manner as in Example 3 except that N-Boc-3-indole magnesium bromide (Non-patent Document 29) prepared from an indole derivative was used.
≪生理活性物質類縁体の合成(15)≫
下記(26)に構造式を示すtert-butyl 3-isopropoxy-1,4-dioxo-2-(trifluoromethyl)-1,4-dihydro-9H-carbazole-9-carboxylateは、グリニヤール試薬として、3-ヨードインドール誘導体から調製した、N-Boc-3-インドールマグネシウムブロミド(非特許文献29)を用いた他は、実施例3と同様にして製造した。 [Example 17]
≪Synthesis of bioactive substance analogues (15) ≫
The tert-butyl 3-isopropoxy-1,4-dioxo-2- (trifluoromethyl) -1,4-dihydro-9H-carbazole-9-carboxylate having the structural formula shown in the following (26) is a 3-iodo as a Grignard reagent. This was prepared in the same manner as in Example 3 except that N-Boc-3-indole magnesium bromide (Non-patent Document 29) prepared from an indole derivative was used.


[実施例18]
≪生理活性物質類縁体の合成(16)≫
下記式(27)に構造式を示す2-isopropoxy-5,6-dimethyl-3-(trifluoromethyl)cyclohexa-2,5-diene-1,4-dioneは、グリニヤール試薬として、1-メチル-1-プロぺニルマグネシウムブロミド(Aldrich社製)を用いた他は、実施例3と同様にして製造した。 [Example 18]
≪Synthesis of physiologically active substance analogues (16) ≫
2-isopropoxy-5,6-dimethyl-3- (trifluoromethyl) cyclohexa-2,5-diene-1,4-dione having the structural formula shown in the following formula (27) is 1-methyl-1- Production was carried out in the same manner as in Example 3 except that propenyl magnesium bromide (manufactured by Aldrich) was used.
≪生理活性物質類縁体の合成(16)≫
下記式(27)に構造式を示す2-isopropoxy-5,6-dimethyl-3-(trifluoromethyl)cyclohexa-2,5-diene-1,4-dioneは、グリニヤール試薬として、1-メチル-1-プロぺニルマグネシウムブロミド(Aldrich社製)を用いた他は、実施例3と同様にして製造した。 [Example 18]
≪Synthesis of physiologically active substance analogues (16) ≫
2-isopropoxy-5,6-dimethyl-3- (trifluoromethyl) cyclohexa-2,5-diene-1,4-dione having the structural formula shown in the following formula (27) is 1-methyl-1- Production was carried out in the same manner as in Example 3 except that propenyl magnesium bromide (manufactured by Aldrich) was used.


[実施例19]
≪生理活性物質類縁体の合成(17)≫
下記式(28)に構造式を示す3-isopropoxy-2-(trifluoromethyl)dibenzo[b,d]thiophene-1,4-diyl diacetateは、グリニヤール試薬として、MgBr2を用いて有機リチウム試薬から調製したベンゾチエニルマグネシウムブロミド(非特許文献26、27)を用い、付加反応を行なった。付加反応の後に得られたp-キシレン溶液(約10ml)を二口ナスフラスコに移した。アルゴン置換し、140°Cで10分間加熱した。TLC分析で原料消失を確認し、室温に冷却した。 [Example 19]
≪Synthesis of bioactive substance analogues (17) ≫
3-isopropoxy-2- (trifluoromethyl) dibenzo [b, d] thiophene-1,4-diyl diacetate, whose structural formula is shown in the following formula (28), was prepared from an organolithium reagent using MgBr 2 as a Grignard reagent An addition reaction was performed using benzothienylmagnesium bromide (Non-patent Documents 26 and 27). The p-xylene solution (about 10 ml) obtained after the addition reaction was transferred to a two-necked eggplant flask. The atmosphere was replaced with argon and heated at 140 ° C. for 10 minutes. The disappearance of the raw material was confirmed by TLC analysis and cooled to room temperature.
≪生理活性物質類縁体の合成(17)≫
下記式(28)に構造式を示す3-isopropoxy-2-(trifluoromethyl)dibenzo[b,d]thiophene-1,4-diyl diacetateは、グリニヤール試薬として、MgBr2を用いて有機リチウム試薬から調製したベンゾチエニルマグネシウムブロミド(非特許文献26、27)を用い、付加反応を行なった。付加反応の後に得られたp-キシレン溶液(約10ml)を二口ナスフラスコに移した。アルゴン置換し、140°Cで10分間加熱した。TLC分析で原料消失を確認し、室温に冷却した。 [Example 19]
≪Synthesis of bioactive substance analogues (17) ≫
3-isopropoxy-2- (trifluoromethyl) dibenzo [b, d] thiophene-1,4-diyl diacetate, whose structural formula is shown in the following formula (28), was prepared from an organolithium reagent using MgBr 2 as a Grignard reagent An addition reaction was performed using benzothienylmagnesium bromide (Non-patent Documents 26 and 27). The p-xylene solution (about 10 ml) obtained after the addition reaction was transferred to a two-necked eggplant flask. The atmosphere was replaced with argon and heated at 140 ° C. for 10 minutes. The disappearance of the raw material was confirmed by TLC analysis and cooled to room temperature.
無水酢酸(472μL、5mmol)、ピリジン(403μL、5mmol)をシリンジで加え、さらにN,N-ジメチルアミノピリジン(6.3mg、0.05mmol)をアルゴン気流下で加え、アセチル化を行なった。5分間撹拌後、TLC分析によりヒドロキノンの消失を確認した。溶媒を減圧下で除去し、粗生成物を得た。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=10:1)で精製し、薄黄色固体(160.3mg、75%)を得た。
Acetic anhydride (472 μL, 5 mmol) and pyridine (403 μL, 5 mmol) were added with a syringe, and N, N-dimethylaminopyridine (6.3 mg, 0.05 mmol) was further added under an argon stream to perform acetylation. After stirring for 5 minutes, disappearance of hydroquinone was confirmed by TLC analysis. The solvent was removed under reduced pressure to give the crude product. The obtained crude product was purified by silica gel column chromatography (hexane: ethyl acetate = 10: 1) to obtain a pale yellow solid (160.3 mg, 75%).

得られた化合物の解析結果を下記に示す。
The analysis results of the obtained compound are shown below.

[実施例20]
≪生理活性物質類縁体の合成(18)≫
下記式(29)に構造式を示す2-isopropoxy-4-oxo-3-(trifluoromethyl)-4H-quinolizin-1-yl acetateは、2‐ブロモピリジンから調製した有機リチウム試薬、2‐ピリジルリチウム(非特許文献30)を用い、付加反応を行なった後、無水酢酸(2.2当量)を加えて反応を停止した。付加反応の後に得られたp-キシレン溶液(約10ml)を二口ナスフラスコに移した。アルゴン置換し、100°Cで30分間加熱した。TLC分析で原料消失を確認し、室温に冷却した。溶媒を減圧下で除去し、粗生成物を得た。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1)で精製し、褐色液体(88.1mg、54%)を得た。 [Example 20]
≪Synthesis of bioactive substance analogues (18) ≫
2-isopropoxy-4-oxo-3- (trifluoromethyl) -4H-quinolizin-1-yl acetate represented by the following formula (29) is an organolithium reagent prepared from 2-bromopyridine, 2-pyridyllithium ( After performing an addition reaction using Non-Patent Document 30), acetic anhydride (2.2 equivalents) was added to stop the reaction. The p-xylene solution (about 10 ml) obtained after the addition reaction was transferred to a two-necked eggplant flask. The atmosphere was replaced with argon and heated at 100 ° C. for 30 minutes. The disappearance of the raw material was confirmed by TLC analysis and cooled to room temperature. The solvent was removed under reduced pressure to give the crude product. The obtained crude product was purified by silica gel column chromatography (hexane: ethyl acetate = 2: 1) to obtain a brown liquid (88.1 mg, 54%).
≪生理活性物質類縁体の合成(18)≫
下記式(29)に構造式を示す2-isopropoxy-4-oxo-3-(trifluoromethyl)-4H-quinolizin-1-yl acetateは、2‐ブロモピリジンから調製した有機リチウム試薬、2‐ピリジルリチウム(非特許文献30)を用い、付加反応を行なった後、無水酢酸(2.2当量)を加えて反応を停止した。付加反応の後に得られたp-キシレン溶液(約10ml)を二口ナスフラスコに移した。アルゴン置換し、100°Cで30分間加熱した。TLC分析で原料消失を確認し、室温に冷却した。溶媒を減圧下で除去し、粗生成物を得た。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1)で精製し、褐色液体(88.1mg、54%)を得た。 [Example 20]
≪Synthesis of bioactive substance analogues (18) ≫
2-isopropoxy-4-oxo-3- (trifluoromethyl) -4H-quinolizin-1-yl acetate represented by the following formula (29) is an organolithium reagent prepared from 2-bromopyridine, 2-pyridyllithium ( After performing an addition reaction using Non-Patent Document 30), acetic anhydride (2.2 equivalents) was added to stop the reaction. The p-xylene solution (about 10 ml) obtained after the addition reaction was transferred to a two-necked eggplant flask. The atmosphere was replaced with argon and heated at 100 ° C. for 30 minutes. The disappearance of the raw material was confirmed by TLC analysis and cooled to room temperature. The solvent was removed under reduced pressure to give the crude product. The obtained crude product was purified by silica gel column chromatography (hexane: ethyl acetate = 2: 1) to obtain a brown liquid (88.1 mg, 54%).


[実施例21]
≪生理活性物質類縁体の合成(19)≫
下記式(30)に構造式を示す7-isopropoxy-5-oxo-6-(trifluoromethyl)-5H-thiazolo[3,2-a]pyridin-8-yl acetateは、チアゾールから調製した有機リチウム試薬、2‐チアゾリルリチウム(非特許文献30)を用いた他は、実施例20と同様にして製造した。 [Example 21]
≪Synthesis of bioactive substance analogues (19) ≫
7-isopropoxy-5-oxo-6- (trifluoromethyl) -5H-thiazolo [3,2-a] pyridin-8-yl acetate represented by the following formula (30) is an organolithium reagent prepared from thiazole, It was produced in the same manner as in Example 20 except that 2-thiazolyl lithium (Non-patent Document 30) was used.
≪生理活性物質類縁体の合成(19)≫
下記式(30)に構造式を示す7-isopropoxy-5-oxo-6-(trifluoromethyl)-5H-thiazolo[3,2-a]pyridin-8-yl acetateは、チアゾールから調製した有機リチウム試薬、2‐チアゾリルリチウム(非特許文献30)を用いた他は、実施例20と同様にして製造した。 [Example 21]
≪Synthesis of bioactive substance analogues (19) ≫
7-isopropoxy-5-oxo-6- (trifluoromethyl) -5H-thiazolo [3,2-a] pyridin-8-yl acetate represented by the following formula (30) is an organolithium reagent prepared from thiazole, It was produced in the same manner as in Example 20 except that 2-thiazolyl lithium (Non-patent Document 30) was used.


[実施例22]
≪生理活性物質類縁体の合成(20)≫
下記式(31)に構造式を示す7-isopropoxy-1-methyl-5-oxo-6-(trifluoromethyl)-1,5-dihydroimidazo[1,2-a]pyridin-8-yl acetateは、N-メチルイミダゾールから調製した有機リチウム試薬、(1-メチル-1H-イミダゾール-2-イル)リチウム(非特許文献30)を用いた他は、実施例20と同様にして製造した。 [Example 22]
≪Synthesis of physiologically active substance analogues (20) ≫
7-isopropoxy-1-methyl-5-oxo-6- (trifluoromethyl) -1,5-dihydroimidazo [1,2-a] pyridin-8-yl acetate represented by the following formula (31) is N- This was prepared in the same manner as in Example 20 except that the organolithium reagent prepared from methylimidazole (1-methyl-1H-imidazol-2-yl) lithium (Non-patent Document 30) was used.
≪生理活性物質類縁体の合成(20)≫
下記式(31)に構造式を示す7-isopropoxy-1-methyl-5-oxo-6-(trifluoromethyl)-1,5-dihydroimidazo[1,2-a]pyridin-8-yl acetateは、N-メチルイミダゾールから調製した有機リチウム試薬、(1-メチル-1H-イミダゾール-2-イル)リチウム(非特許文献30)を用いた他は、実施例20と同様にして製造した。 [Example 22]
≪Synthesis of physiologically active substance analogues (20) ≫
7-isopropoxy-1-methyl-5-oxo-6- (trifluoromethyl) -1,5-dihydroimidazo [1,2-a] pyridin-8-yl acetate represented by the following formula (31) is N- This was prepared in the same manner as in Example 20 except that the organolithium reagent prepared from methylimidazole (1-methyl-1H-imidazol-2-yl) lithium (Non-patent Document 30) was used.


[実施例23]
≪生理活性物質類縁体の合成(21)≫
以下の方法によりブテノリド類を合成することができる。下記式(32)に構造式を示す3-isopropoxy-5-oxo-2-phenyl-4-(trifluoromethyl)-2,5-dihydrofuran-2-yl acetateを例に、ブテノリド類の合成方法について詳述する。 [Example 23]
≪Synthesis of bioactive substance analogues (21) ≫
Butenolides can be synthesized by the following method. The method for synthesizing butenolides is described in detail by taking 3-isopropoxy-5-oxo-2-phenyl-4- (trifluoromethyl) -2,5-dihydrofuran-2-yl acetate as a structural formula shown in the following formula (32) as an example. To do.
≪生理活性物質類縁体の合成(21)≫
以下の方法によりブテノリド類を合成することができる。下記式(32)に構造式を示す3-isopropoxy-5-oxo-2-phenyl-4-(trifluoromethyl)-2,5-dihydrofuran-2-yl acetateを例に、ブテノリド類の合成方法について詳述する。 [Example 23]
≪Synthesis of bioactive substance analogues (21) ≫
Butenolides can be synthesized by the following method. The method for synthesizing butenolides is described in detail by taking 3-isopropoxy-5-oxo-2-phenyl-4- (trifluoromethyl) -2,5-dihydrofuran-2-yl acetate as a structural formula shown in the following formula (32) as an example. To do.


乾燥させアルゴン雰囲気としたナスフラスコに、トリフルオロメチル置換セミスクアレート(1-iPr)(104.3mg、0.5mmol)、脱水ジエチルエーテル(2mL)を加え-90°Cに冷却した。調製した0.1M PhMgBrエーテル溶液(10mL、1.0mmol)を温度計で-90°C以下に維持し、20mL/hの速度で滴下した。滴下終了後、さらに30分撹拌し、-90°Cの条件下、飽和塩化アンモニウムクロリド水溶液(10mL)を加え反応を停止させた後に、室温へ昇温した。Et2O(10mL×3)で抽出し、得られた有機層を飽和食塩水(10mL)で洗浄後、Na2SO4で乾燥させ濃縮し、粗生成物を得た。
To a dried eggplant flask under an argon atmosphere, trifluoromethyl-substituted semisquarate (1- i Pr) (104.3 mg, 0.5 mmol) and dehydrated diethyl ether (2 mL) were added and cooled to -90 ° C. The prepared 0.1M PhMgBr ether solution (10 mL, 1.0 mmol) was maintained at −90 ° C. or lower with a thermometer and added dropwise at a rate of 20 mL / h. After completion of the dropwise addition, the mixture was further stirred for 30 minutes, and a saturated ammonium chloride aqueous solution (10 mL) was added under a condition of −90 ° C. to stop the reaction, and then the temperature was raised to room temperature. Extraction was performed with Et 2 O (10 mL × 3), and the obtained organic layer was washed with saturated brine (10 mL), dried over Na 2 SO 4 and concentrated to obtain a crude product.
乾燥させアルゴン雰囲気としたシュレンク管に、Pb(OAc)4(445.2mg、1.0mmol)、脱水toluene(2mL)を加え10分撹拌した。この溶液に、上記粗生成物の脱水toluene(1mL)溶液をカニュラーを用いて加えた。室温で一時間撹拌を続け、水(10mL)を加え反応を停止させ、この時生じた褐色沈殿物をジクロロメタンでセライト濾過し、二相の溶液を得た。水層をジクロロメタン(10mL×3)で抽出し、得られた有機層を飽和食塩水(10mL)で洗浄後、Na2SO4で乾燥させた。濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=6:1)で精製し、無色液体(131.9mg、77%)を得た。解析結果を下記に示す。
Pb (OAc) 4 (445.2 mg, 1.0 mmol) and dehydrated toluene (2 mL) were added to a Schlenk tube dried and placed in an argon atmosphere, and stirred for 10 minutes. To this solution, a dehydrated toluene (1 mL) solution of the above crude product was added using a cannula. Stirring was continued at room temperature for 1 hour, the reaction was stopped by adding water (10 mL), and the brown precipitate formed at this time was filtered through celite with dichloromethane to obtain a two-phase solution. The aqueous layer was extracted with dichloromethane (10 mL × 3), and the obtained organic layer was washed with saturated brine (10 mL) and dried over Na 2 SO 4 . The crude product obtained by concentration was purified by silica gel column chromatography (hexane: ethyl acetate = 6: 1) to obtain a colorless liquid (131.9 mg, 77%). The analysis results are shown below.

[実施例24]
≪生理活性物質類縁体の合成(22)≫
下記式(33)に構造式を示す3-isopropoxy-2-(4-methoxyphenyl)-5-oxo-4-(trifluoromethyl)-2,5-dihydrofuran-2-yl acetateは、PhMgBrの代わりに、p-MeOC6H4MgBrを用いた他は実施例23と同様にして製造した。収率は74%であった。解析結果を下記に示す。 [Example 24]
≪Synthesis of bioactive substance analogues (22) ≫
3-isopropoxy-2- (4-methoxyphenyl) -5-oxo-4- (trifluoromethyl) -2,5-dihydrofuran-2-yl acetate, which has the structural formula shown in the following formula (33), is replaced with PhMgBr. This was prepared in the same manner as in Example 23 except that -MeOC 6 H 4 MgBr was used. The yield was 74%. The analysis results are shown below.
≪生理活性物質類縁体の合成(22)≫
下記式(33)に構造式を示す3-isopropoxy-2-(4-methoxyphenyl)-5-oxo-4-(trifluoromethyl)-2,5-dihydrofuran-2-yl acetateは、PhMgBrの代わりに、p-MeOC6H4MgBrを用いた他は実施例23と同様にして製造した。収率は74%であった。解析結果を下記に示す。 [Example 24]
≪Synthesis of bioactive substance analogues (22) ≫
3-isopropoxy-2- (4-methoxyphenyl) -5-oxo-4- (trifluoromethyl) -2,5-dihydrofuran-2-yl acetate, which has the structural formula shown in the following formula (33), is replaced with PhMgBr. This was prepared in the same manner as in Example 23 except that -MeOC 6 H 4 MgBr was used. The yield was 74%. The analysis results are shown below.


[実施例25]
≪生理活性物質類縁体の合成(23)≫
下記式(34)に構造式を示すethyl 4-(2-acetoxy-3-isopropoxy-5-oxo-4-(trifluoromethyl)-2,5-dihydrofuran-2-yl)benzoateは、PhMgBrの代わりに、p-EtO2CC6H4MgBrを用いた他は実施例23と同様にして製造した。収率は65%であった。解析結果を下記に示す。 [Example 25]
≪Synthesis of physiologically active substance analogues (23) ≫
In the following formula (34), ethyl 4- (2-acetoxy-3-isopropoxy-5-oxo-4- (trifluoromethyl) -2,5-dihydrofuran-2-yl) benzoate is substituted for PhMgBr. It was produced in the same manner as in Example 23 except that p-EtO 2 CC 6 H 4 MgBr was used. The yield was 65%. The analysis results are shown below.
≪生理活性物質類縁体の合成(23)≫
下記式(34)に構造式を示すethyl 4-(2-acetoxy-3-isopropoxy-5-oxo-4-(trifluoromethyl)-2,5-dihydrofuran-2-yl)benzoateは、PhMgBrの代わりに、p-EtO2CC6H4MgBrを用いた他は実施例23と同様にして製造した。収率は65%であった。解析結果を下記に示す。 [Example 25]
≪Synthesis of physiologically active substance analogues (23) ≫
In the following formula (34), ethyl 4- (2-acetoxy-3-isopropoxy-5-oxo-4- (trifluoromethyl) -2,5-dihydrofuran-2-yl) benzoate is substituted for PhMgBr. It was produced in the same manner as in Example 23 except that p-EtO 2 CC 6 H 4 MgBr was used. The yield was 65%. The analysis results are shown below.


[実施例26]
≪生理活性物質類縁体の合成(24)≫
下記式(35)に構造式を示す2-(benzofuran-2-yl)-3-isopropoxy-5-oxo-4-(trifluoromethyl)-2,5-dihydrofuran-2-yl acetateは、PhMgBrの代わりに、2-benzofurylMgBrを用いた他は実施例23と同様にして製造した。収率は62%であった。解析結果を下記に示す。 [Example 26]
≪Synthesis of bioactive substance analogues (24) ≫
In the following formula (35), 2- (benzofuran-2-yl) -3-isopropoxy-5-oxo-4- (trifluoromethyl) -2,5-dihydrofuran-2-yl acetate is used instead of PhMgBr. This was prepared in the same manner as in Example 23 except that 2-benzofurylMgBr was used. The yield was 62%. The analysis results are shown below.
≪生理活性物質類縁体の合成(24)≫
下記式(35)に構造式を示す2-(benzofuran-2-yl)-3-isopropoxy-5-oxo-4-(trifluoromethyl)-2,5-dihydrofuran-2-yl acetateは、PhMgBrの代わりに、2-benzofurylMgBrを用いた他は実施例23と同様にして製造した。収率は62%であった。解析結果を下記に示す。 [Example 26]
≪Synthesis of bioactive substance analogues (24) ≫
In the following formula (35), 2- (benzofuran-2-yl) -3-isopropoxy-5-oxo-4- (trifluoromethyl) -2,5-dihydrofuran-2-yl acetate is used instead of PhMgBr. This was prepared in the same manner as in Example 23 except that 2-benzofurylMgBr was used. The yield was 62%. The analysis results are shown below.


[実施例27]
≪生理活性物質類縁体の合成(25)≫
下記式(36)に構造式を示す2-(benzo[b]thiophen-2-yl)-3-isopropoxy-5-oxo-4-(trifluoromethyl)-2,5-dihydrofuran-2-yl acetateは、PhMgBrの代わりに、2-benzothienylMgBrを用いた他は実施例23と同様にして製造した。収率は68%であった。解析結果を下記に示す。 [Example 27]
≪Synthesis of physiologically active substance analogues (25) ≫
2- (benzo [b] thiophen-2-yl) -3-isopropoxy-5-oxo-4- (trifluoromethyl) -2,5-dihydrofuran-2-yl acetate, which is represented by the following formula (36), is: Production was carried out in the same manner as in Example 23 except that 2-benzothienylMgBr was used instead of PhMgBr. The yield was 68%. The analysis results are shown below.
≪生理活性物質類縁体の合成(25)≫
下記式(36)に構造式を示す2-(benzo[b]thiophen-2-yl)-3-isopropoxy-5-oxo-4-(trifluoromethyl)-2,5-dihydrofuran-2-yl acetateは、PhMgBrの代わりに、2-benzothienylMgBrを用いた他は実施例23と同様にして製造した。収率は68%であった。解析結果を下記に示す。 [Example 27]
≪Synthesis of physiologically active substance analogues (25) ≫
2- (benzo [b] thiophen-2-yl) -3-isopropoxy-5-oxo-4- (trifluoromethyl) -2,5-dihydrofuran-2-yl acetate, which is represented by the following formula (36), is: Production was carried out in the same manner as in Example 23 except that 2-benzothienylMgBr was used instead of PhMgBr. The yield was 68%. The analysis results are shown below.


[実施例28]
≪生理活性物質類縁体の合成(26)≫
以下の方法によりビシクロ環化合物を合成することができる。下記式(37)に構造式を示すビシクロ環化合物である2-isopropoxy-7-oxo-1-(trifluoromethyl)bicycle[3.2.0]hept-2-en-3-yl acetateは、新規の前駆体10-3a、10-3bを経て以下のようにして製造した。 [Example 28]
≪Synthesis of physiologically active substance analogues (26) ≫
Bicyclo ring compounds can be synthesized by the following method. 2-isopropoxy-7-oxo-1- (trifluoromethyl) bicycle [3.2.0] hept-2-en-3-yl acetate, which is a bicyclo ring compound having the structural formula shown in the following formula (37), is a novel compound. The precursors 10-3a and 10-3b were produced as follows.
≪生理活性物質類縁体の合成(26)≫
以下の方法によりビシクロ環化合物を合成することができる。下記式(37)に構造式を示すビシクロ環化合物である2-isopropoxy-7-oxo-1-(trifluoromethyl)bicycle[3.2.0]hept-2-en-3-yl acetateは、新規の前駆体10-3a、10-3bを経て以下のようにして製造した。 [Example 28]
≪Synthesis of physiologically active substance analogues (26) ≫
Bicyclo ring compounds can be synthesized by the following method. 2-isopropoxy-7-oxo-1- (trifluoromethyl) bicycle [3.2.0] hept-2-en-3-yl acetate, which is a bicyclo ring compound having the structural formula shown in the following formula (37), is a novel compound. The precursors 10-3a and 10-3b were produced as follows.


加熱乾燥、アルゴン置換した50mLナスフラスコに、1-iPr(104.3mg、0.5mmol)を加え脱水エーテル(2mL)を加えた。この溶液を-90°Cに冷却し、調製した約0.1MアリルマグネシウムクロリドEt2O溶液(12.5mL、1.25mmol)を20mL/hの速度で滴下し、滴下後さらに1時間撹拌した。飽和塩化アンモニウム水溶液(10ml)で反応を停止させ室温へと昇温させた。ジエチルエーテル(10mL×3)で抽出し、得られた有機層を飽和食塩水(10mL)で洗浄後、Na2SO4で乾燥させた。濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=5:1)で精製し、淡黄色固体(10-3a、51.6mg、41%)を得た。
1- i Pr (104.3 mg, 0.5 mmol) was added to a 50 mL eggplant flask that had been heat-dried and purged with argon, and dehydrated ether (2 mL) was added. The solution was cooled to −90 ° C., and the prepared about 0.1 M allylmagnesium chloride Et 2 O solution (12.5 mL, 1.25 mmol) was added dropwise at a rate of 20 mL / h. After the addition, the mixture was further stirred for 1 hour. . The reaction was quenched with saturated aqueous ammonium chloride (10 ml) and allowed to warm to room temperature. Extraction was performed with diethyl ether (10 mL × 3), and the obtained organic layer was washed with saturated brine (10 mL) and then dried over Na 2 SO 4 . The crude product obtained by concentration was purified by silica gel column chromatography (hexane: ethyl acetate = 5: 1) to obtain a pale yellow solid (10-3a, 51.6 mg, 41%).
式(10-3a)に構造式を示す前駆体化合物(196mg、0.78mmol)を30mL二口ナスに加え、脱水ジエチルエーテル(8mL)に希釈した。この溶液に、塩化アセチル(110μL、1.56mmol)およびトリエチルアミン(216μL、1.56mmol)を加え、アルゴン雰囲気下室温で30分撹拌した。H2O(20mL)を加え反応を停止させエーテル(20mL×3)で抽出した。濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=10:1)で精製し、無色液体(10-3b、177.7mg、78%)を得た。
A precursor compound (196 mg, 0.78 mmol) having a structural formula represented by the formula (10-3a) was added to 30 mL double necked eggplant and diluted with dehydrated diethyl ether (8 mL). To this solution were added acetyl chloride (110 μL, 1.56 mmol) and triethylamine (216 μL, 1.56 mmol), and the mixture was stirred at room temperature for 30 minutes under an argon atmosphere. The reaction was quenched with H 2 O (20 mL) and extracted with ether (20 mL × 3). The crude product obtained by concentration was purified by silica gel column chromatography (hexane: ethyl acetate = 10: 1) to obtain a colorless liquid (10-3b, 177.7 mg, 78%).
式(10-3b)に構造式を示す前駆体化合物(73.1mg、0.25mmol)をp-キシレン(5mL)に溶解した。この溶解液を120°Cで1時間撹拌した。室温に冷却し、溶媒を減圧留去して得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン:クロロホルム=1:2)で精製し、式(37)に構造式を示す化合物を無色液体(64.4mg、88%)として得た。式37で表す最終生成物の収率は88%であった。解析結果を下記に示す。
A precursor compound (73.1 mg, 0.25 mmol) having the structural formula shown in Formula (10-3b) was dissolved in p-xylene (5 mL). This solution was stirred at 120 ° C. for 1 hour. The crude product obtained by cooling to room temperature and distilling off the solvent under reduced pressure was purified by silica gel column chromatography (hexane: chloroform = 1: 2), and the compound represented by the structural formula in formula (37) was purified as a colorless liquid ( 64.4 mg, 88%). The yield of the final product represented by Formula 37 was 88%. The analysis results are shown below.

また、中間体化合物10-3a、10-3bの解析結果はそれぞれ下記のとおりであった。
The analysis results of the intermediate compounds 10-3a and 10-3b were as follows.


[実施例29]
≪生理活性物質類縁体の合成(27)≫
アリルグリニヤール試薬を用いて合成を行った場合には、中間体化合物10-3aの収率が低かったために、実施例28に記載の化合物2-isopropoxy-7-oxo-1-(trifluoromethyl)bicycle[3.2.0]hept-2-en-3-yl acetateに合成が限られていた。そこで、アリルケイ素化合物を用いる下記の方法を開発することにより、10-3aで表す化合物の収率を著しく向上することに成功した。 [Example 29]
≪Synthesis of bioactive substance analogues (27) ≫
When the synthesis was carried out using an allyl Grignard reagent, the yield of the intermediate compound 10-3a was low. Therefore, the compound 2-isopropoxy-7-oxo-1- (trifluoromethyl) bicycle described in Example 28 was used. 3.2.0] Synthesis was limited to hept-2-en-3-yl acetate. Therefore, by developing the following method using an allyl silicon compound, the yield of the compound represented by 10-3a was successfully improved.
≪生理活性物質類縁体の合成(27)≫
アリルグリニヤール試薬を用いて合成を行った場合には、中間体化合物10-3aの収率が低かったために、実施例28に記載の化合物2-isopropoxy-7-oxo-1-(trifluoromethyl)bicycle[3.2.0]hept-2-en-3-yl acetateに合成が限られていた。そこで、アリルケイ素化合物を用いる下記の方法を開発することにより、10-3aで表す化合物の収率を著しく向上することに成功した。 [Example 29]
≪Synthesis of bioactive substance analogues (27) ≫
When the synthesis was carried out using an allyl Grignard reagent, the yield of the intermediate compound 10-3a was low. Therefore, the compound 2-isopropoxy-7-oxo-1- (trifluoromethyl) bicycle described in Example 28 was used. 3.2.0] Synthesis was limited to hept-2-en-3-yl acetate. Therefore, by developing the following method using an allyl silicon compound, the yield of the compound represented by 10-3a was successfully improved.

加熱乾燥したシュレンク管をアルゴンで満たし、1-iPr(52.4mg、0.25mmol)を加え脱水ジクロロメタン(1.5mL)で希釈した。この溶液を-78℃に冷却し、四塩化スズの1.0Mジクロロメタン溶液(250μL、0.25mmol)を加え、さらにアリルシラン(TCI製、60μL、0.375mmol)を加え、-78℃で10分撹拌した後、氷浴で1時間撹拌した。精製水(10mL)を加え反応を停止し、室温へと昇温した後に、ジクロロメタン(10mL×3)で抽出し、得られた有機層を飽和食塩水(10mL)で洗浄後、Na2SO4で乾燥した。濃縮して得られた粗生成物に、中性シリカゲル(500mg)およびジクロロメタン(0.5mL)を加え、室温で30分撹拌しシリル基を除去した。シリルエーテル中間体の消失をTLC分析で確認したところで、混合物をロータリーエバポレーターで濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒はヘキサン/酢酸エチル=4:1を用いた。)で精製し、淡黄色固体(10-3a、59.6mg、95%)を得た。
The heat-dried Schlenk tube was filled with argon, 1- i Pr (52.4 mg, 0.25 mmol) was added, and the mixture was diluted with dehydrated dichloromethane (1.5 mL). This solution was cooled to −78 ° C., a 1.0 M solution of tin tetrachloride in dichloromethane (250 μL, 0.25 mmol) was added, and allylsilane (manufactured by TCI, 60 μL, 0.375 mmol) was added, and the mixture was added at −78 ° C. for 10 minutes. After stirring, the mixture was stirred for 1 hour in an ice bath. Purified water (10 mL) was added to stop the reaction, and the temperature was raised to room temperature, followed by extraction with dichloromethane (10 mL × 3). The obtained organic layer was washed with saturated brine (10 mL) and then Na 2 SO 4. And dried. Neutral silica gel (500 mg) and dichloromethane (0.5 mL) were added to the crude product obtained by concentration, and the mixture was stirred at room temperature for 30 minutes to remove the silyl group. When the disappearance of the silyl ether intermediate was confirmed by TLC analysis, the mixture was concentrated by a rotary evaporator, and the obtained crude product was subjected to silica gel column chromatography (developing solvent used hexane / ethyl acetate = 4: 1). To give a pale yellow solid (10-3a, 59.6 mg, 95%).
[実施例30]
≪生理活性物質類縁体の合成(28)≫
上述のように、10-3aで表す化合物の収率が向上したことから以下の化合物を合成することが可能となった。下記式(38)に構造式を示す4-hydroxy-3-isopropoxy-4-(2-phenylallyl)-2-(trifluoromethyl)cyclobut-2-enoneは、アリルシランの代わりに、trimethyl(2-phenylallyl)silane(非特許文献31)を用いた他は実施例29と同様にして製造した。収率は94%であった。解析結果を下記に示す。 [Example 30]
≪Synthesis of bioactive substance analogues (28) ≫
As described above, since the yield of the compound represented by 10-3a was improved, it was possible to synthesize the following compounds. 4-hydroxy-3-isopropyoxy-4- (2-phenylallyl) -2- (trifluoromethyl) cyclobut-2-enone represented by the following formula (38) is trimethyl (2-phenylallyl) silane instead of allylsilane. This was manufactured in the same manner as in Example 29 except that (Non-patent Document 31) was used. The yield was 94%. The analysis results are shown below.
≪生理活性物質類縁体の合成(28)≫
上述のように、10-3aで表す化合物の収率が向上したことから以下の化合物を合成することが可能となった。下記式(38)に構造式を示す4-hydroxy-3-isopropoxy-4-(2-phenylallyl)-2-(trifluoromethyl)cyclobut-2-enoneは、アリルシランの代わりに、trimethyl(2-phenylallyl)silane(非特許文献31)を用いた他は実施例29と同様にして製造した。収率は94%であった。解析結果を下記に示す。 [Example 30]
≪Synthesis of bioactive substance analogues (28) ≫
As described above, since the yield of the compound represented by 10-3a was improved, it was possible to synthesize the following compounds. 4-hydroxy-3-isopropyoxy-4- (2-phenylallyl) -2- (trifluoromethyl) cyclobut-2-enone represented by the following formula (38) is trimethyl (2-phenylallyl) silane instead of allylsilane. This was manufactured in the same manner as in Example 29 except that (Non-patent Document 31) was used. The yield was 94%. The analysis results are shown below.


[実施例31]
≪生理活性物質類縁体の合成(29)≫
下記式(39)に構造式を示す2-isopropoxy-4-oxo-1-(2-phenylallyl)-3-(trifluoromethyl)cyclobut-2-enyl acetateは、実施例28の化合物10-3bと同様にして製造した。収率は93%であった。解析結果を下記に示す。 [Example 31]
≪Synthesis of bioactive substance analogues (29) ≫
2-isopropoxy-4-oxo-1- (2-phenylallyl) -3- (trifluoromethyl) cyclobut-2-ethyl acetate is represented by the following structural formula (39) in the same manner as compound 10-3b of Example 28. Manufactured. The yield was 93%. The analysis results are shown below.
≪生理活性物質類縁体の合成(29)≫
下記式(39)に構造式を示す2-isopropoxy-4-oxo-1-(2-phenylallyl)-3-(trifluoromethyl)cyclobut-2-enyl acetateは、実施例28の化合物10-3bと同様にして製造した。収率は93%であった。解析結果を下記に示す。 [Example 31]
≪Synthesis of bioactive substance analogues (29) ≫
2-isopropoxy-4-oxo-1- (2-phenylallyl) -3- (trifluoromethyl) cyclobut-2-ethyl acetate is represented by the following structural formula (39) in the same manner as compound 10-3b of Example 28. Manufactured. The yield was 93%. The analysis results are shown below.


[実施例32]
≪生理活性物質類縁体の合成(30)≫
下記式(40)に構造式を示す2-isopropoxy-7-oxo-5-phenyl-1-(trifluoromethyl)bicyclo[3.2.0]hept-2-en-3-yl acetateは、実施例28と同様にして1時間加熱することにより製造した。収率は96%であった。解析結果を下記に示す。 [Example 32]
≪Synthesis of bioactive substance analogues (30) ≫
2-isopropoxy-7-oxo-5-phenyl-1- (trifluoromethyl) bicyclo [3.2.0] hept-2-en-3-yl acetate is shown in Example 28. It was manufactured by heating for 1 hour in the same manner. The yield was 96%. The analysis results are shown below.
≪生理活性物質類縁体の合成(30)≫
下記式(40)に構造式を示す2-isopropoxy-7-oxo-5-phenyl-1-(trifluoromethyl)bicyclo[3.2.0]hept-2-en-3-yl acetateは、実施例28と同様にして1時間加熱することにより製造した。収率は96%であった。解析結果を下記に示す。 [Example 32]
≪Synthesis of bioactive substance analogues (30) ≫
2-isopropoxy-7-oxo-5-phenyl-1- (trifluoromethyl) bicyclo [3.2.0] hept-2-en-3-yl acetate is shown in Example 28. It was manufactured by heating for 1 hour in the same manner. The yield was 96%. The analysis results are shown below.


[実施例33]
≪生理活性物質類縁体の合成(31)≫
下記式(41)に構造式を示す4-hydroxy-3-isopropoxy-4-(2-methyleneoctyl)-2-(trifluoromethyl)cyclobut-2-enoneは、アリルシランの代わりに、trimethyl(2-methyleneoctyl)silane(非特許文献31)を用いた他は実施例29と同様にして製造した。収率は97%であった。解析結果を下記に示す。 [Example 33]
≪Synthesis of bioactive substance analogues (31) ≫
4-hydroxy-3-isopropoxy-4- (2-methylenoxyl) -2- (trifluoromethyl) cyclobut-2-enone, which has the structural formula shown in the following formula (41), is trimethyl (2-methylenecylyl) instead of allylsilane. This was manufactured in the same manner as in Example 29 except that (Non-patent Document 31) was used. The yield was 97%. The analysis results are shown below.
≪生理活性物質類縁体の合成(31)≫
下記式(41)に構造式を示す4-hydroxy-3-isopropoxy-4-(2-methyleneoctyl)-2-(trifluoromethyl)cyclobut-2-enoneは、アリルシランの代わりに、trimethyl(2-methyleneoctyl)silane(非特許文献31)を用いた他は実施例29と同様にして製造した。収率は97%であった。解析結果を下記に示す。 [Example 33]
≪Synthesis of bioactive substance analogues (31) ≫
4-hydroxy-3-isopropoxy-4- (2-methylenoxyl) -2- (trifluoromethyl) cyclobut-2-enone, which has the structural formula shown in the following formula (41), is trimethyl (2-methylenecylyl) instead of allylsilane. This was manufactured in the same manner as in Example 29 except that (Non-patent Document 31) was used. The yield was 97%. The analysis results are shown below.


[実施例34]
≪生理活性物質類縁体の合成(32)≫
下記式(42)に構造式を示す2-isopropoxy-1-(2-methyleneoctyl)-4-oxo-3-(trifluoromethyl)cyclobut-2-enyl acetateは、実施例28の化合物10-3bと同様にして製造した。収率は98%であった。解析結果を下記に示す。 [Example 34]
≪Synthesis of physiologically active substance analogues (32) ≫
2-isopropoxy-1- (2-methylenoxyl) -4-oxo-3- (trifluoromethyl) cyclobut-2-ethyl acetate represented by the following structural formula (42) is the same as compound 10-3b of Example 28. Manufactured. The yield was 98%. The analysis results are shown below.
≪生理活性物質類縁体の合成(32)≫
下記式(42)に構造式を示す2-isopropoxy-1-(2-methyleneoctyl)-4-oxo-3-(trifluoromethyl)cyclobut-2-enyl acetateは、実施例28の化合物10-3bと同様にして製造した。収率は98%であった。解析結果を下記に示す。 [Example 34]
≪Synthesis of physiologically active substance analogues (32) ≫
2-isopropoxy-1- (2-methylenoxyl) -4-oxo-3- (trifluoromethyl) cyclobut-2-ethyl acetate represented by the following structural formula (42) is the same as compound 10-3b of Example 28. Manufactured. The yield was 98%. The analysis results are shown below.


[実施例35]
≪生理活性物質類縁体の合成(33)≫
下記式(43)に構造式を示す5-hexyl-2-isopropoxy-7-oxo-1-(trifluoromethyl)bicycle[3.2.0]hept-2-en-3-yl acetateは、実施例28と同様にして1時間加熱することにより製造した。収率は95%であった。解析結果を下記に示す。

[Example 35]
≪Synthesis of physiologically active substance analogues (33) ≫
5-hexyl-2-isopropyoxy-7-oxo-1- (trifluoromethyl) bicycle [3.2.0] hept-2-en-3-yl acetate, which is represented by the following formula (43), is obtained in Example 28. It was manufactured by heating for 1 hour in the same manner. The yield was 95%. The analysis results are shown below.
≪生理活性物質類縁体の合成(33)≫
下記式(43)に構造式を示す5-hexyl-2-isopropoxy-7-oxo-1-(trifluoromethyl)bicycle[3.2.0]hept-2-en-3-yl acetateは、実施例28と同様にして1時間加熱することにより製造した。収率は95%であった。解析結果を下記に示す。
≪Synthesis of physiologically active substance analogues (33) ≫
5-hexyl-2-isopropyoxy-7-oxo-1- (trifluoromethyl) bicycle [3.2.0] hept-2-en-3-yl acetate, which is represented by the following formula (43), is obtained in Example 28. It was manufactured by heating for 1 hour in the same manner. The yield was 95%. The analysis results are shown below.

[実施例36]
≪生理活性物質類縁体の合成(34)≫
下記式(44)に構造式を示す2-isopropoxy-4-oxo-1-(1-phenylallyl)-3-(trifluoromethyl)cyclobut-2-enyl acetateは、実施例29と同様にして四塩化スズの代わりに四塩化チタンを、アリルシランの代わりにcinnamyltrimethylsilane(非特許文献32)を用いて、-40℃でアリル化した後、生成したアルコールを単離することなく実施例28の化合物10-3bと同様にして直接アシル化して製造した。7:3のジアステレオマー混合物として得られ、収率は二段階で38%であった。解析結果を下記に示す。 [Example 36]
≪Synthesis of physiologically active substance analogues (34) ≫
2-isopropoxy-4-oxo-1- (1-phenylallyl) -3- (trifluoromethyl) cyclobut-2-ethyl acetate is represented by the following formula (44) in the same manner as in Example 29. Instead, titanium tetrachloride was allylated at −40 ° C. using cinnamethyltrimethylsilane (Non-patent Document 32) instead of allylsilane, and then the same as compound 10-3b of Example 28 without isolating the resulting alcohol. To be directly acylated. Obtained as a 7: 3 diastereomeric mixture, the yield was 38% in two steps. The analysis results are shown below.
≪生理活性物質類縁体の合成(34)≫
下記式(44)に構造式を示す2-isopropoxy-4-oxo-1-(1-phenylallyl)-3-(trifluoromethyl)cyclobut-2-enyl acetateは、実施例29と同様にして四塩化スズの代わりに四塩化チタンを、アリルシランの代わりにcinnamyltrimethylsilane(非特許文献32)を用いて、-40℃でアリル化した後、生成したアルコールを単離することなく実施例28の化合物10-3bと同様にして直接アシル化して製造した。7:3のジアステレオマー混合物として得られ、収率は二段階で38%であった。解析結果を下記に示す。 [Example 36]
≪Synthesis of physiologically active substance analogues (34) ≫
2-isopropoxy-4-oxo-1- (1-phenylallyl) -3- (trifluoromethyl) cyclobut-2-ethyl acetate is represented by the following formula (44) in the same manner as in Example 29. Instead, titanium tetrachloride was allylated at −40 ° C. using cinnamethyltrimethylsilane (Non-patent Document 32) instead of allylsilane, and then the same as compound 10-3b of Example 28 without isolating the resulting alcohol. To be directly acylated. Obtained as a 7: 3 diastereomeric mixture, the yield was 38% in two steps. The analysis results are shown below.


[実施例37]
≪生理活性物質類縁体の合成(35)≫
下記式(45)に構造式を示す2-isopropoxy-7-oxo-4-phenyl-1-(trifluoromethyl)bicycle[3.2.0]hept-2-en-3-yl acetateは、実施例28と同様にして3時間加熱することにより製造した。収率は75%であった。解析結果を下記に示す。 [Example 37]
≪Synthesis of physiologically active substance analogues (35) ≫
2-isopropoxy-7-oxo-4-phenyl-1- (trifluoromethyl) bicycle [3.2.0] hept-2-en-3-yl acetate represented by the following structural formula (45) is obtained in Example 28. It was manufactured by heating in the same manner for 3 hours. The yield was 75%. The analysis results are shown below.
≪生理活性物質類縁体の合成(35)≫
下記式(45)に構造式を示す2-isopropoxy-7-oxo-4-phenyl-1-(trifluoromethyl)bicycle[3.2.0]hept-2-en-3-yl acetateは、実施例28と同様にして3時間加熱することにより製造した。収率は75%であった。解析結果を下記に示す。 [Example 37]
≪Synthesis of physiologically active substance analogues (35) ≫
2-isopropoxy-7-oxo-4-phenyl-1- (trifluoromethyl) bicycle [3.2.0] hept-2-en-3-yl acetate represented by the following structural formula (45) is obtained in Example 28. It was manufactured by heating in the same manner for 3 hours. The yield was 75%. The analysis results are shown below.


[実施例38]
≪生理活性物質類縁体の合成(36)≫
下記式(46)に構造式を示す4-acetoxy-3-isopropoxy-4a,5,6,7-tetrahydro-2a-(trifluoromethyl)-1H-cyclobuta[c]pentapentalen-2(2aH)-oneは、実施例Iと同様にしてアリルシランの代わりに(cyclopentenylmethyl)trimethylsilane(非特許文献31)を用いてアリル化した。アルコールを単離することなく実施例28の化合物10-3bと同様にしてアセチル化し、得られたエステルを単離することなく実施例28と同様にして2時間加熱して製造した。収率は三段階で42%であった。解析結果を下記に示す。 [Example 38]
≪Synthesis of physiologically active substance analogues (36) ≫
4-acetoxy-3-isopropoxy-4a, 5,6,7-tetrahydro-2a- (trifluoromethyl) -1H-cyclobuta [c] pentapentalen-2 (2aH) -one, which has the structural formula shown in the following formula (46), In the same manner as in Example I, allylation was carried out using trimethylsilane (Non-patent Document 31) instead of allylsilane. The product was acetylated in the same manner as Compound 10-3b of Example 28 without isolating the alcohol, and the resulting ester was prepared by heating for 2 hours in the same manner as in Example 28 without isolation. The yield was 42% in three stages. The analysis results are shown below.
≪生理活性物質類縁体の合成(36)≫
下記式(46)に構造式を示す4-acetoxy-3-isopropoxy-4a,5,6,7-tetrahydro-2a-(trifluoromethyl)-1H-cyclobuta[c]pentapentalen-2(2aH)-oneは、実施例Iと同様にしてアリルシランの代わりに(cyclopentenylmethyl)trimethylsilane(非特許文献31)を用いてアリル化した。アルコールを単離することなく実施例28の化合物10-3bと同様にしてアセチル化し、得られたエステルを単離することなく実施例28と同様にして2時間加熱して製造した。収率は三段階で42%であった。解析結果を下記に示す。 [Example 38]
≪Synthesis of physiologically active substance analogues (36) ≫
4-acetoxy-3-isopropoxy-4a, 5,6,7-tetrahydro-2a- (trifluoromethyl) -1H-cyclobuta [c] pentapentalen-2 (2aH) -one, which has the structural formula shown in the following formula (46), In the same manner as in Example I, allylation was carried out using trimethylsilane (Non-patent Document 31) instead of allylsilane. The product was acetylated in the same manner as Compound 10-3b of Example 28 without isolating the alcohol, and the resulting ester was prepared by heating for 2 hours in the same manner as in Example 28 without isolation. The yield was 42% in three stages. The analysis results are shown below.


[実施例39]
≪生理活性物質類縁体の合成(37)≫
以下の方法によりアミノシクロペンテンジオンを合成することができる。下記式(47)に構造式を示すアミノシクロペンテンジオンである2-butyl-2-(tert-butylamino)-4-isopropoxy-5-(trifluoromethyl)cyclopent-4-ene-1,3-dioneは、以下のようにして製造した。 [Example 39]
≪Synthesis of physiologically active substance analogues (37) ≫
Aminocyclopentenedione can be synthesized by the following method. 2-Butyl-2- (tert-butylamino) -4-isopropoxy-5- (trifluoromethyl) cyclo-4-ene-1,3-dione, which is an aminocyclopentenedione whose structural formula is shown in the following formula (47), is as follows: It manufactured as follows.
≪生理活性物質類縁体の合成(37)≫
以下の方法によりアミノシクロペンテンジオンを合成することができる。下記式(47)に構造式を示すアミノシクロペンテンジオンである2-butyl-2-(tert-butylamino)-4-isopropoxy-5-(trifluoromethyl)cyclopent-4-ene-1,3-dioneは、以下のようにして製造した。 [Example 39]
≪Synthesis of physiologically active substance analogues (37) ≫
Aminocyclopentenedione can be synthesized by the following method. 2-Butyl-2- (tert-butylamino) -4-isopropoxy-5- (trifluoromethyl) cyclo-4-ene-1,3-dione, which is an aminocyclopentenedione whose structural formula is shown in the following formula (47), is as follows: It manufactured as follows.

イミドイルリチウム試薬は、Liebeskidらの方法をもとに調製した(非特許文献33)。tert-ブチルイソシアニド(85μL、0.75mmol)をジエチルエーテル(2.02mL)に溶解し、-15°Cに冷却した。nBuLi(15w/w%、480μL)を加え、-15°Cで30分撹拌を続けた。得られた溶液を続く付加反応に使用するため-78°Cに冷却した。
The imidoyllithium reagent was prepared based on the method of Liebeskid et al. (Non-patent Document 33). Tert-butyl isocyanide (85 μL, 0.75 mmol) was dissolved in diethyl ether (2.02 mL) and cooled to −15 ° C. n BuLi (15 w / w%, 480 μL) was added and stirring was continued at −15 ° C. for 30 minutes. The resulting solution was cooled to −78 ° C. for use in subsequent addition reactions.
1-iPr(104.3mg、0.5mmol)の脱水ジエチルエーテル溶液(10mL)を-78°Cに冷却した。この溶液に、予め-78°Cに冷却した約0.3Mイミドイルリチウム試薬(1.7mL、0.5mmol)をシリンジで加え、30分間撹拌を続けた。飽和塩化アンモニウム水溶液(10mL)で反応を停止させた後、室温へ昇温した。ジエチルエーテル(10mL×3)で抽出し、得られた有機層を飽和食塩水(10mL)で洗浄後、Na2SO4で乾燥させた。濃縮して得られた粗生成物を、シリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=6:1)で精製し、濃赤色固体(87.2mg、50%)を得た。収率は50%であった。解析結果を下記に示す。
A solution of 1- i Pr (104.3 mg, 0.5 mmol) in dehydrated diethyl ether (10 mL) was cooled to -78 ° C. To this solution was added approximately 0.3 M imidoyllithium reagent (1.7 mL, 0.5 mmol) pre-cooled to −78 ° C. with a syringe and stirring was continued for 30 minutes. The reaction was quenched with a saturated aqueous ammonium chloride solution (10 mL), and then the temperature was raised to room temperature. Extraction was performed with diethyl ether (10 mL × 3), and the obtained organic layer was washed with saturated brine (10 mL) and then dried over Na 2 SO 4 . The crude product obtained by concentration was purified by silica gel column chromatography (hexane: ethyl acetate = 6: 1) to obtain a dark red solid (87.2 mg, 50%). The yield was 50%. The analysis results are shown below.

[実施例40]
≪生理活性物質類縁体の合成(38)≫
以下の方法により開環化合物である不飽和カルボン酸を合成することができる。下記式(48)の構造式で示す(Z)-4-acetoxy-3-isopropoxy-6-phenyl-2-(trifluoromethyl)hex-3-en-5-ynoic acidは、以下のようにして製造した。 [Example 40]
≪Synthesis of bioactive substance analogues (38) ≫
The unsaturated carboxylic acid which is a ring-opening compound can be synthesized by the following method. (Z) -4-acetoxy-3-isopropyoxy-6-phenyl-2- (trifluoromethyl) hex-3-en-5-ynoic acid represented by the structural formula of the following formula (48) was produced as follows. .
≪生理活性物質類縁体の合成(38)≫
以下の方法により開環化合物である不飽和カルボン酸を合成することができる。下記式(48)の構造式で示す(Z)-4-acetoxy-3-isopropoxy-6-phenyl-2-(trifluoromethyl)hex-3-en-5-ynoic acidは、以下のようにして製造した。 [Example 40]
≪Synthesis of bioactive substance analogues (38) ≫
The unsaturated carboxylic acid which is a ring-opening compound can be synthesized by the following method. (Z) -4-acetoxy-3-isopropyoxy-6-phenyl-2- (trifluoromethyl) hex-3-en-5-ynoic acid represented by the structural formula of the following formula (48) was produced as follows. .


乾燥させたシュレンク管中で、フェニルアセチレン(120μL、1.1mmol)を脱水ジエチルエーテル(2.0mL)に溶解し、-78°Cに冷却した。nBuLi(15w/w%、640μL)を加え、溶液が白濁するまで室温で二時間撹拌した。その後、脱水ジエチルエーテル(1.36mL)を加え、0.25M溶液(淡黄色)とした。
In a dried Schlenk tube, phenylacetylene (120 μL, 1.1 mmol) was dissolved in dehydrated diethyl ether (2.0 mL) and cooled to −78 ° C. n BuLi (15 w / w%, 640 μL) was added and stirred at room temperature for 2 hours until the solution became cloudy. Thereafter, dehydrated diethyl ether (1.36 mL) was added to give a 0.25 M solution (pale yellow).
50mL二口ナスフラスコに1-iPr(104.2mg、0.5mmol)、脱水ジエチルエーテル(2mL)を加え-90°Cに冷却した。リチウムフェニルアセチリド0.25Mエーテル溶液(2.0mL、0.5mmol)を10mL/hの速度で滴下した。滴下後1時間撹拌し、無水酢酸(105μL、1.11mmol)を加え、さらに30分撹拌を続けた。飽和炭酸水素ナトリウム水溶液(20mL)で反応を停止させ室温へ昇温した。ジエチルエーテル(20mL)で抽出し、得られた水層を10%HCl水溶液で中和した。中和後の水層をジエチルエーテル(20mL×3)で抽出し、合わせた有機層を飽和食塩水 (20mL)で洗浄後、Na2SO4で乾燥させた。濃縮して得られた粗生成物を、硫酸酸性シリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)で精製し、黄色液体(132.1 mg、71%)を得た。解析結果を下記に示す。
1- i Pr (104.2 mg, 0.5 mmol) and dehydrated diethyl ether (2 mL) were added to a 50 mL two-necked eggplant flask and cooled to -90 ° C. Lithium phenylacetylide 0.25 M ether solution (2.0 mL, 0.5 mmol) was added dropwise at a rate of 10 mL / h. After dropping, the mixture was stirred for 1 hour, acetic anhydride (105 μL, 1.11 mmol) was added, and stirring was further continued for 30 minutes. The reaction was stopped with saturated aqueous sodium hydrogen carbonate solution (20 mL) and the temperature was raised to room temperature. Extraction was performed with diethyl ether (20 mL), and the resulting aqueous layer was neutralized with 10% aqueous HCl. The neutralized aqueous layer was extracted with diethyl ether (20 mL × 3), and the combined organic layer was washed with saturated brine (20 mL) and dried over Na 2 SO 4 . The crude product obtained by concentration was purified by sulfuric acid silica gel column chromatography (hexane: ethyl acetate = 1: 1) to obtain a yellow liquid (132.1 mg, 71%). The analysis results are shown below.

[実施例41]
≪生理活性物質類縁体の合成(39)≫
下記式(49)に構造式を記載する4-hydroxy-3-isopropoxy-4-(2-oxo-2-phenylethyl)-2-(trifluoromethyl)cyclobut-2-enoneは、以下のようにして合成した。

[Example 41]
≪Synthesis of physiologically active substance analogues (39) ≫
4-hydroxy-3-isopropyoxy-4- (2-oxo-2-phenylethyl) -2- (trifluoromethyl) cyclobut-2-enone, whose structural formula is shown in the following formula (49), was synthesized as follows. .
≪生理活性物質類縁体の合成(39)≫
下記式(49)に構造式を記載する4-hydroxy-3-isopropoxy-4-(2-oxo-2-phenylethyl)-2-(trifluoromethyl)cyclobut-2-enoneは、以下のようにして合成した。
≪Synthesis of physiologically active substance analogues (39) ≫
4-hydroxy-3-isopropyoxy-4- (2-oxo-2-phenylethyl) -2- (trifluoromethyl) cyclobut-2-enone, whose structural formula is shown in the following formula (49), was synthesized as follows. .

加熱乾燥したシュレンク管をアルゴンで満たし、1-iPr(62.4mg、0.30mmol)を加え脱水ジクロロメタン(2.0mL)で希釈した。この溶液を-78℃に冷却し、四塩化スズの1.0Mジクロロメタン溶液(300μL、0.30mmol)を加え、10分撹拌した。この溶液に別途調製した(非特許文献34)trimethyl(1-phenylvinyloxy)silane(184μL、0.90mmol)を加え、-78℃で2時間攪拌した。精製水(10mL)を加え反応を停止し、室温へと昇温した後に、ジクロロメタン(10mL×3)で抽出し、得られた有機層を飽和食塩水(10mL)で洗浄後、Na2SO4で乾燥した。有機層をロータリーエバポレーターで濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 はヘキサン/酢酸エチル=4:1を用いた。)で精製し、淡黄色液体(84.7mg、86%)を得た。得られた化合物の解析結果を下記に示す。
The heat-dried Schlenk tube was filled with argon, 1- i Pr (62.4 mg, 0.30 mmol) was added, and the mixture was diluted with dehydrated dichloromethane (2.0 mL). The solution was cooled to −78 ° C., a 1.0 M solution of tin tetrachloride in dichloromethane (300 μL, 0.30 mmol) was added, and the mixture was stirred for 10 minutes. Trimethyl (1-phenylvinyloxy) silane (184 μL, 0.90 mmol) separately prepared (Non-patent Document 34) was added to this solution, and the mixture was stirred at −78 ° C. for 2 hours. Purified water (10 mL) was added to stop the reaction, and the temperature was raised to room temperature, followed by extraction with dichloromethane (10 mL × 3). The obtained organic layer was washed with saturated brine (10 mL) and then Na 2 SO 4. And dried. The organic layer was concentrated by a rotary evaporator, and the resulting crude product was purified by silica gel column chromatography (developing solvent was hexane / ethyl acetate = 4: 1), and a pale yellow liquid (84.7 mg, 86 %). The analysis results of the obtained compound are shown below.

[実施例42]
≪生理活性物質類縁体の合成(40)≫
下記式(50)に構造式を示すbenzyl 2-(1-hydroxy-2-isopropoxy-4-oxo-3-(trifluoromethyl)cyclobut-2-enyl)acetateは、実施例41と同様にして四塩化スズの代わりに四塩化チタンを、trimethyl(1-phenylvinyloxy)silaneの代わりに(1-(benzyloxy)vinyloxy)trimethylsilane(非特許文献35)を用いて製造した。収率は86%であった。解析結果を下記に示す。 [Example 42]
≪Synthesis of bioactive substance analogues (40) ≫
Benzyl 2- (1-hydroxy-2-isopropyoxy-4-oxo-3- (trifluoromethyl) cyclobut-2-enyl) acetate having the structural formula shown in the following formula (50) is tin tetrachloride in the same manner as in Example 41. Instead of trimethyl (1-phenylvinyloxy) silane, (1- (benzoyloxy) vinyloxy) trimethylsilane (Non-patent Document 35) was used instead of trimethyl (1-phenylvinyloxy) silane. The yield was 86%. The analysis results are shown below.
≪生理活性物質類縁体の合成(40)≫
下記式(50)に構造式を示すbenzyl 2-(1-hydroxy-2-isopropoxy-4-oxo-3-(trifluoromethyl)cyclobut-2-enyl)acetateは、実施例41と同様にして四塩化スズの代わりに四塩化チタンを、trimethyl(1-phenylvinyloxy)silaneの代わりに(1-(benzyloxy)vinyloxy)trimethylsilane(非特許文献35)を用いて製造した。収率は86%であった。解析結果を下記に示す。 [Example 42]
≪Synthesis of bioactive substance analogues (40) ≫
Benzyl 2- (1-hydroxy-2-isopropyoxy-4-oxo-3- (trifluoromethyl) cyclobut-2-enyl) acetate having the structural formula shown in the following formula (50) is tin tetrachloride in the same manner as in Example 41. Instead of trimethyl (1-phenylvinyloxy) silane, (1- (benzoyloxy) vinyloxy) trimethylsilane (Non-patent Document 35) was used instead of trimethyl (1-phenylvinyloxy) silane. The yield was 86%. The analysis results are shown below.


[実施例43]
≪生理活性物質類縁体の合成(41)≫
下記式(51)に構造式を記載する4-isopropoxy-5-(2-oxo-2-phenylethylidene)-3-(trifluoromethyl)furan-2(5H)-oneは、以下のようにして合成した。

[Example 43]
≪Synthesis of bioactive substance analogues (41) ≫
4-isopropoxy-5- (2-oxo-2-phenylethylidene) -3- (trifluoromethyl) furan-2 (5H) -one, whose structural formula is shown in the following formula (51), was synthesized as follows.
≪生理活性物質類縁体の合成(41)≫
下記式(51)に構造式を記載する4-isopropoxy-5-(2-oxo-2-phenylethylidene)-3-(trifluoromethyl)furan-2(5H)-oneは、以下のようにして合成した。
≪Synthesis of bioactive substance analogues (41) ≫
4-isopropoxy-5- (2-oxo-2-phenylethylidene) -3- (trifluoromethyl) furan-2 (5H) -one, whose structural formula is shown in the following formula (51), was synthesized as follows.

加熱乾燥したシュレンク管をアルゴンで満たし、Pb(OAc)4(348.6mg、0.79mmol)と加熱乾燥したモレキュラーシーブス4A粉末(400.2mg)、および脱水トルエン(4.0mL)を加えた。この懸濁液に、4-hydroxy-3-isopropoxy-4-(2-oxo-2-phenylethyl)-2-(trifluoromethyl)cyclobut-2-enone(129.0mg、0.39mmol)を加え、室温で1時間撹拌した。この溶液に精製水(10mL)を加え反応を停止し、不溶物を濾過した。濾液を酢酸エチル(10mL×3)で抽出し、得られた有機層を飽和NaHCO3(10mL)で洗浄後、Na2SO4で乾燥した。有機層をロータリーエバポレーターで濃縮して得られた粗生成物をTHF(8mL)に希釈し、N-methyl morpholine(48μL、0.43mmol)を加えて室温で4時間攪拌した。反応混合物をエバポレーターで濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒はヘキサン/酢酸エチル=6:1を用いた。)で精製し、黄色固体(97.1mg、76%)を得た。得られた化合物の解析結果を下記に示す。
The heat-dried Schlenk tube was filled with argon, and Pb (OAc) 4 (348.6 mg, 0.79 mmol), heat-dried molecular sieves 4A powder (400.2 mg), and dehydrated toluene (4.0 mL) were added. To this suspension was added 4-hydroxy-3-isopropoxy-4- (2-oxo-2-phenylethyl) -2- (trifluoromethyl) cyclobut-2-enone (129.0 mg, 0.39 mmol) at room temperature. Stir for 1 hour. Purified water (10 mL) was added to this solution to stop the reaction, and insoluble matters were filtered. The filtrate was extracted with ethyl acetate (10 mL × 3), and the obtained organic layer was washed with saturated NaHCO 3 (10 mL) and dried over Na 2 SO 4 . The crude product obtained by concentrating the organic layer with a rotary evaporator was diluted in THF (8 mL), N-methyl morpholine (48 μL, 0.43 mmol) was added, and the mixture was stirred at room temperature for 4 hours. The reaction mixture was concentrated with an evaporator, and the resulting crude product was purified by silica gel column chromatography (developing solvent used was hexane / ethyl acetate = 6: 1) to give a yellow solid (97.1 mg, 76%). Got. The analysis results of the obtained compound are shown below.

[実施例44]
≪生理活性物質類縁体の合成(42)≫
下記式(52)に構造式を示すbenzyl 2-(3-isopropoxy-5-oxo-4-(trifluoromethyl)furan-2(5H)-ylidene)acetateは、実施例43と同様にして製造した。収率は84%であった。解析結果を下記に示す。 [Example 44]
≪Synthesis of bioactive substance analogues (42) ≫
Benzyl 2- (3-isopropoxy-5-oxo-4- (trifluoromethyl) furan-2 (5H) -ylidene) acetate having the structural formula shown in the following formula (52) was produced in the same manner as in Example 43. The yield was 84%. The analysis results are shown below.
≪生理活性物質類縁体の合成(42)≫
下記式(52)に構造式を示すbenzyl 2-(3-isopropoxy-5-oxo-4-(trifluoromethyl)furan-2(5H)-ylidene)acetateは、実施例43と同様にして製造した。収率は84%であった。解析結果を下記に示す。 [Example 44]
≪Synthesis of bioactive substance analogues (42) ≫
Benzyl 2- (3-isopropoxy-5-oxo-4- (trifluoromethyl) furan-2 (5H) -ylidene) acetate having the structural formula shown in the following formula (52) was produced in the same manner as in Example 43. The yield was 84%. The analysis results are shown below.


≪トリフルオロメチル置換セミスクアレートの応用可能性≫
本発明のトリフルオロメチル置換セミスクアレートの合成法を用いれば、生理活性が知られている以下の化合物への応用も期待できる。 ≪Applicability of trifluoromethyl-substituted semisquarate≫
If the method for synthesizing a trifluoromethyl-substituted semisquarate of the present invention is used, application to the following compounds having known physiological activities can be expected.
本発明のトリフルオロメチル置換セミスクアレートの合成法を用いれば、生理活性が知られている以下の化合物への応用も期待できる。 ≪Applicability of trifluoromethyl-substituted semisquarate≫
If the method for synthesizing a trifluoromethyl-substituted semisquarate of the present invention is used, application to the following compounds having known physiological activities can be expected.
≪応用可能性(1)≫
上記式(13)で表す構造の化合物、2-isopropoxy-6-methyl-3-(trifluoromethyl)naphthalene-1,4-dioneは、細胞分裂阻害活性を示す化合物Paravaquinonの類縁体である。Paravaquinoneはアメリカムラサキウニの受精卵の細胞分裂阻害活性を示すことが報告されている。さらに、その類縁体3-ethyl-2-hydroxy-6-methylnaphthalene-1,4-dioneも同様の活性を示すことが報告されている(非特許文献36)。実施例4で合成した前記式(13)で表される化合物のトリフルオロメチル置換ナフトキノンも同様の生理活性を有するものと考えられる。 ≪Applicability (1) ≫
The compound having the structure represented by the above formula (13), 2-isopropoxy-6-methyl-3- (trifluoromethyl) naphthalene-1,4-dione, is an analog of the compound Paravaquinon exhibiting cell division inhibitory activity. Paravaquinone has been reported to exhibit cell division inhibitory activity in fertilized eggs of the sea urchin. Furthermore, it has been reported that its analog 3-ethyl-2-hydroxy-6-methylnaphthalene-1,4-dione exhibits similar activity (Non-patent Document 36). The trifluoromethyl-substituted naphthoquinone of the compound represented by the formula (13) synthesized in Example 4 is considered to have the same physiological activity.
上記式(13)で表す構造の化合物、2-isopropoxy-6-methyl-3-(trifluoromethyl)naphthalene-1,4-dioneは、細胞分裂阻害活性を示す化合物Paravaquinonの類縁体である。Paravaquinoneはアメリカムラサキウニの受精卵の細胞分裂阻害活性を示すことが報告されている。さらに、その類縁体3-ethyl-2-hydroxy-6-methylnaphthalene-1,4-dioneも同様の活性を示すことが報告されている(非特許文献36)。実施例4で合成した前記式(13)で表される化合物のトリフルオロメチル置換ナフトキノンも同様の生理活性を有するものと考えられる。 ≪Applicability (1) ≫
The compound having the structure represented by the above formula (13), 2-isopropoxy-6-methyl-3- (trifluoromethyl) naphthalene-1,4-dione, is an analog of the compound Paravaquinon exhibiting cell division inhibitory activity. Paravaquinone has been reported to exhibit cell division inhibitory activity in fertilized eggs of the sea urchin. Furthermore, it has been reported that its analog 3-ethyl-2-hydroxy-6-methylnaphthalene-1,4-dione exhibits similar activity (Non-patent Document 36). The trifluoromethyl-substituted naphthoquinone of the compound represented by the formula (13) synthesized in Example 4 is considered to have the same physiological activity.

≪応用可能性(2)≫
Furaquinocin類は、HeLaS3およびB16メラノーマ細胞に対するin vitro細胞毒性や血小板凝固作用阻害活性など、幅広い生理活性を示す。それらの中で、下記に示すFraquinocin A、B、およびEが、下記に示す反応式のセミスクアレート40を用いて合成されている(非特許文献37)。したがって、トリフルオロメチル置換セミスクアレート1-iPrを用いれば、Furaquinocin類のトリフルオロメチル類縁体の合成が可能となる。 ≪Applicability (2) ≫
Furaquinocins exhibit a wide range of physiological activities such as in vitro cytotoxicity and platelet coagulation inhibitory activity against HeLaS3 and B16 melanoma cells. Among them, Fraquinocin A, B, and E shown below are synthesized using semisquarate 40 of the reaction formula shown below (Non-patent Document 37). Therefore, the use of trifluoromethyl-substituted semisquarate 1- i Pr makes it possible to synthesize trifluoromethyl analogues of Furaquinocins.
Furaquinocin類は、HeLaS3およびB16メラノーマ細胞に対するin vitro細胞毒性や血小板凝固作用阻害活性など、幅広い生理活性を示す。それらの中で、下記に示すFraquinocin A、B、およびEが、下記に示す反応式のセミスクアレート40を用いて合成されている(非特許文献37)。したがって、トリフルオロメチル置換セミスクアレート1-iPrを用いれば、Furaquinocin類のトリフルオロメチル類縁体の合成が可能となる。 ≪Applicability (2) ≫
Furaquinocins exhibit a wide range of physiological activities such as in vitro cytotoxicity and platelet coagulation inhibitory activity against HeLaS3 and B16 melanoma cells. Among them, Fraquinocin A, B, and E shown below are synthesized using semisquarate 40 of the reaction formula shown below (Non-patent Document 37). Therefore, the use of trifluoromethyl-substituted semisquarate 1- i Pr makes it possible to synthesize trifluoromethyl analogues of Furaquinocins.

≪応用可能性(3)≫
抗マラリア活性物質である(-)-Elisapterosin Bもまた、下記反応式に示すセミスクアレート41からの全合成が達成されている(非特許文献38)。したがって、トリフルオロメチル置換セミスクアレートを用いれば、トリフルオロメチル置換抗マラリア化合物への応用が考えられる。 ≪Applicability (3) ≫
The antimalarial active substance (−)-Elisapterosin B has also been achieved in total synthesis from semisquarate 41 shown in the following reaction formula (Non-patent Document 38). Therefore, if trifluoromethyl-substituted semisquarate is used, application to a trifluoromethyl-substituted antimalarial compound can be considered.
抗マラリア活性物質である(-)-Elisapterosin Bもまた、下記反応式に示すセミスクアレート41からの全合成が達成されている(非特許文献38)。したがって、トリフルオロメチル置換セミスクアレートを用いれば、トリフルオロメチル置換抗マラリア化合物への応用が考えられる。 ≪Applicability (3) ≫
The antimalarial active substance (−)-Elisapterosin B has also been achieved in total synthesis from semisquarate 41 shown in the following reaction formula (Non-patent Document 38). Therefore, if trifluoromethyl-substituted semisquarate is used, application to a trifluoromethyl-substituted antimalarial compound can be considered.

≪応用可能性(4)≫
Carbazomycin類は抗真菌活性や抗菌活性を示し、セミスクアレート1を用いるCarbazimycin Gの合成が最近になって報告された(非特許文献39)。実施例17で合成した式(26)で表される化合物は、Carbazimycin Gを合成する際の前駆物質のトリフルオロメチル化合物である。したがって、該前駆化合物を用いてトリフルオロメチル置換Carbazimycin G類縁体を合成することが可能である。 ≪Possibility of application (4) ≫
Carbazomycins show antifungal activity and antibacterial activity, and recently, synthesis of CarbazimycinG using semisquarate 1 has been reported (Non-patent Document 39). The compound represented by the formula (26) synthesized in Example 17 is a trifluoromethyl compound as a precursor when synthesizing Carbazimycin G. Therefore, it is possible to synthesize a trifluoromethyl substituted Carbazimycin G analog using the precursor compound.
Carbazomycin類は抗真菌活性や抗菌活性を示し、セミスクアレート1を用いるCarbazimycin Gの合成が最近になって報告された(非特許文献39)。実施例17で合成した式(26)で表される化合物は、Carbazimycin Gを合成する際の前駆物質のトリフルオロメチル化合物である。したがって、該前駆化合物を用いてトリフルオロメチル置換Carbazimycin G類縁体を合成することが可能である。 ≪Possibility of application (4) ≫
Carbazomycins show antifungal activity and antibacterial activity, and recently, synthesis of Carbazimycin

以上、本発明の製造方法によって、種々の化合物にトリフルオロメチル基を導入することが可能となる。
As described above, a trifluoromethyl group can be introduced into various compounds by the production method of the present invention.
本発明の合成方法によれば、合成が困難であった多様なトリフルオロメチル化合物が効率よく得られるようになる。
According to the synthesis method of the present invention, various trifluoromethyl compounds that have been difficult to synthesize can be efficiently obtained.
Claims (23)
- 下記一般式(1)で表される化合物を出発原料として、

付加反応工程、
及び環拡大工程によってトリフルオロメチル基が導入された化合物を製造する方法。 Starting from a compound represented by the following general formula (1),
Addition reaction process,
And a method for producing a compound into which a trifluoromethyl group has been introduced by a ring expansion step. - 前記トリフルオロメチル基が導入された化合物が、
キノン化合物である請求項1記載の製造方法。 The compound into which the trifluoromethyl group is introduced is
The production method according to claim 1, which is a quinone compound. - トリフルオロメチル基が導入されたキノン化合物が、
下記一般式(4)又は(5)で表される化合物である請求項2記載の製造方法。

(式中、Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基から選択される。R1、R2、R3、R4はそれぞれ独立して水素、アルキル基、アルケニル基、アルコキシ基、クロロ基、フルオロ基、エステル基から選択され、R1~R4のうち任意の2つの隣接する置換基は縮環ベンゼンを形成してもよい。ここで、アルキル基、アルケニル基、アルコキシ基の炭化水素部位は、C1~C8の直鎖状、分岐状、環状のいずれでも良い。)

The production method according to claim 2, which is a compound represented by the following general formula (4) or (5).
Wherein R is selected from isopropyl, n-propyl, t-butyl, isobutyl and sec-butyl. R 1 , R 2 , R 3 and R 4 are each independently hydrogen, alkyl Selected from a group, an alkenyl group, an alkoxy group, a chloro group, a fluoro group, and an ester group, any two adjacent substituents of R 1 to R 4 may form a condensed benzene, wherein alkyl The hydrocarbon moiety of the group, alkenyl group and alkoxy group may be any of C1 to C8 linear, branched or cyclic.)
- 前記トリフルオロメチル基が導入された化合物が、
複素環縮環化合物である請求項1記載の製造方法。 The compound into which the trifluoromethyl group is introduced is
The production method according to claim 1, which is a heterocyclic condensed ring compound. - トリフルオロメチル基が導入された複素環縮環化合物が、
下記一般式(6)又は(7)で表される化合物である請求項4記載の製造方法。


The production method according to claim 4, which is a compound represented by the following general formula (6) or (7).
- 前記トリフルオロメチル基が導入された化合物が、
ヒドロキノン化合物である請求項1記載の製造方法。 The compound into which the trifluoromethyl group is introduced is
The production method according to claim 1, which is a hydroquinone compound. - トリフルオロメチル基が導入されたヒドロキノン化合物が、
下記一般式(8)~(10)のいずれかで表される化合物である請求項6記載の製造方法。



The production method according to claim 6, which is a compound represented by any one of the following general formulas (8) to (10).
- 前記トリフルオロメチル基が導入された化合物が、
ブテノリド化合物である請求項1記載の製造方法。 The compound into which the trifluoromethyl group is introduced is
The production method according to claim 1, which is a butenolide compound. - トリフルオロメチル基が導入されたブテノリド化合物が、
下記一般式(10-1)、(10-2)で表される化合物である請求項8記載の製造方法。


The production method according to claim 8, which is a compound represented by the following general formula (10-1) or (10-2).
- 前記トリフルオロメチル基が導入された化合物が、
四員環化合物である請求項1記載の製造方法。 The compound into which the trifluoromethyl group is introduced is
The production method according to claim 1, which is a four-membered ring compound. - トリフルオロメチル基が導入された四員環化合物が、
下記一般式(10-3c)、(10-3d)で表される化合物である請求項10記載の製造方法。


The production method according to claim 10, which is a compound represented by the following general formula (10-3c) or (10-3d).
- 前記トリフルオロメチル基が導入された化合物が、
ビシクロ環化合物である請求項1記載の製造方法。 The compound into which the trifluoromethyl group is introduced is
The production method according to claim 1, which is a bicyclo ring compound. - トリフルオロメチル基が導入されたビシクロ環化合物が、
下記一般式(37-1)で表される化合物である請求項12記載の製造方法。

The production method according to claim 12, which is a compound represented by the following general formula (37-1).
- 前記トリフルオロメチル基が導入された化合物が、
アミノシクロペンテンジオン化合物である請求項1記載の製造方法。 The compound into which the trifluoromethyl group is introduced is
The process according to claim 1, which is an aminocyclopentenedione compound. - トリフルオロメチル基が導入されたアミノシクロペンテンジオン化合物が、
下記一般式(10-4)で表される化合物である請求項14記載の製造方法。

The production method according to claim 14, which is a compound represented by the following general formula (10-4).
- 下記一般式(1)で表されるトリフルオロメチル置換セミスクアレートの製造方法であって、



(式中Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基であり、異なる基であっても、同一の基であってもよい。) A method for producing a trifluoromethyl-substituted semisquarate represented by the following general formula (1):
(In the formula, R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group, which may be different groups or the same group.) - 下記一般式(1)で表されるトリフルオロメチル置換セミスクアレートの製造方法であって、

下記一般式(2)で表される化合物を

(式中Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基であり、異なる基であっても、同一の基であってもよい。) A method for producing a trifluoromethyl-substituted semisquarate represented by the following general formula (1):
A compound represented by the following general formula (2)
(In the formula, R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group, which may be different groups or the same group.) - 請求項16又は17記載のトリフルオロメチル置換セミスクアレートの製造方法であって、
アリルアルコール転移反応を行なう工程が、
Re2O7又はPh3SiReO3を触媒として用いる反応であることを特徴とする製造方法。 A method for producing a trifluoromethyl-substituted semisquarate according to claim 16 or 17,
The step of carrying out the allyl alcohol transfer reaction
A production method, which is a reaction using Re 2 O 7 or Ph 3 SiReO 3 as a catalyst. - 下記一般式(2)で表される化合物の製造方法であって、


(式中Rはイソプロピル基、n-プロピル基、t-ブチル基、イソブチル基、sec-ブチル基であり、異なる基であっても、同一の基であってもよい。) A method for producing a compound represented by the following general formula (2),
(In the formula, R is an isopropyl group, an n-propyl group, a t-butyl group, an isobutyl group, or a sec-butyl group, which may be different groups or the same group.) - 請求項16又は19記載の製造方法であって、
前記トリフルオロメチル化する工程が、
CF3Me3Siを有機ケイ素試薬として用いてシリルトリフルオロメチル化反応をする工程と、
脱シリル化工程からなることを特徴とする製造方法。 The manufacturing method according to claim 16 or 19,
The trifluoromethylation step comprises:
Performing a silyl trifluoromethylation reaction using CF 3 Me 3 Si as an organosilicon reagent;
A production method comprising a desilylation step. - 下記一般式(1)で表される化合物。

- 下記一般式(2)の化合物。

- 下記式(12)~(52)、(17-1)、(10-3a)、(10-3b)で表されるトリフルオロメチル化合物。

Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017523658A JPWO2016199789A1 (en) | 2015-06-08 | 2016-06-08 | Method for producing trifluoromethyl-substituted semisquarate, method for producing trifluoromethyl compound starting from trifluoromethyl-substituted semisquarate, and compound containing trifluoromethyl group |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015116151 | 2015-06-08 | ||
| JP2015-116151 | 2015-06-08 | ||
| JP2015150175 | 2015-07-29 | ||
| JP2015-150175 | 2015-07-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016199789A1 true WO2016199789A1 (en) | 2016-12-15 |
Family
ID=57503790
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/066988 WO2016199789A1 (en) | 2015-06-08 | 2016-06-08 | Method of manufacturing trifluoromethyl substituted semisquarate, method of manufacturing trifluoromethyl compound starting from trifluoromethyl substituted semisquarate, and trifluoromethyl group-containing compound |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JPWO2016199789A1 (en) |
| WO (1) | WO2016199789A1 (en) |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH07502007A (en) * | 1991-01-02 | 1995-03-02 | インペリアル ケミカル インダストリーズ ピーエルシー | Benzazapine compounds for treatment |
| JPH09502157A (en) * | 1992-06-10 | 1997-03-04 | ゼネカ リミテッド | 2,5-Dioxo-2,5-dihydro-1H-benzo [b] azepines as NMDA receptor antagonists |
-
2016
- 2016-06-08 JP JP2017523658A patent/JPWO2016199789A1/en active Pending
- 2016-06-08 WO PCT/JP2016/066988 patent/WO2016199789A1/en active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH07502007A (en) * | 1991-01-02 | 1995-03-02 | インペリアル ケミカル インダストリーズ ピーエルシー | Benzazapine compounds for treatment |
| JPH09502157A (en) * | 1992-06-10 | 1997-03-04 | ゼネカ リミテッド | 2,5-Dioxo-2,5-dihydro-1H-benzo [b] azepines as NMDA receptor antagonists |
Non-Patent Citations (4)
| Title |
|---|
| EMMA PACKARD ET AL.: "Organoytterbium Ate Complexes Extend the Value of Cyclobutenediones as Isoprene Equivalents", ANGEWANDTE CHEMIE , INTERNATIONAL EDITION, vol. 52, no. 49, 2013, pages 13076 - 13079, XP055333588 * |
| LANNY S. LIEBESKIND ET AL.: "An Inproved Method for the Synthesis of Substituted Cyclobutenediones", THE JOURNAL OF ORGANIC CHEMISTRY, vol. 53, no. 11, 1988, pages 2482 - 2488, XP055333591 * |
| TERUAKI MUKAIYAMA ET AL.: "Lithium Acetate- catalyzed Trifluoromethylation of Carbonyl Compounds with (Trifluoromethyl)trimethylsilane", CHEMISTRY LETTERS, vol. 34, no. 1, 2005, pages 88 - 89, XP055333596 * |
| YOSHIHIKO YAMAMOTO ET AL.: "Radical-Mediated Ring Enlargement of Cyclobutenones: New Synthetic Potential of Squaric Acid", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 117, no. 38, 1995, pages 9653 - 9661, XP055333590 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2016199789A1 (en) | 2018-03-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Brownbridge | Silyl enol ethers in synthesis-part II | |
| Das et al. | Stereoselective total synthesis of (+)-cryptofolione and (+)-goniothalamin | |
| Vamshikrishna et al. | Total synthesis of (+)-varitriol and (+)-6′-epi-varitriol | |
| Li et al. | Catalytic asymmetric synthesis of the Colorado potato beetle pheromone and its enantiomer | |
| Takeda et al. | Asymmetric total synthesis of enantiopure (−)-methyl jasmonate via catalytic asymmetric intramolecular cyclopropanation of α-diazo-β-keto sulfone | |
| CN108430463B (en) | Improved synthesis of honokiol | |
| Wang et al. | A simple synthesis of fluoroalkyl substituted dihydrofurans by rhodium (II)-catalyzed 1, 3-dipolar reactions | |
| KR20190009326A (en) | Process for preparing eribulin and its intermediates | |
| WO2016199789A1 (en) | Method of manufacturing trifluoromethyl substituted semisquarate, method of manufacturing trifluoromethyl compound starting from trifluoromethyl substituted semisquarate, and trifluoromethyl group-containing compound | |
| Kitazume | Synthesis of optically active β-keto-γ-butyrolactones having a trifluoromethyl group | |
| Liu et al. | A general strategy toward the total synthesis of C17 polyacetylenes virols A and C | |
| Fernandes et al. | Asymmetric dihydroxylation and one-pot epoxidation routes to (+)-and (−)-posticlure: a novel trans-epoxide as a sex pheromone component of Orgyia postica (Walker) | |
| Chen et al. | Total synthesis of γ-trifluoromethylated analogs of goniothalamin and their derivatives | |
| Ghanty et al. | Stereoselective syntheses of siphonarienal and siphonarienone | |
| Sakai et al. | Stereoselective synthesis of 4-fluoro-1, 3-alkadienylboronates and their application in the stereoselective synthesis of fluoropolyenes | |
| JP2010235589A (en) | 4-polyfluoroacylphenylalkyl ketone, and method of producing the same | |
| KR102200558B1 (en) | Preparation of 2-fluoromethyl cyclopentanone derivatives | |
| JPH0892151A (en) | Production of allylquinone | |
| Kvı́cala et al. | Synthesis of fluorinated amphiphiles by the reaction of protected hydroxy carbaldehyde with perfluorinated organomagnesium compounds | |
| Jeong et al. | Preparation of 4-trifluoroethylidene-1, 3-dioxolane derivatives via new stable (trifluoromethyl) ethynylation reagent | |
| Zhu et al. | Photoredox catalyzed difluoro (phenylthio) methylation of 2, 3-allenoic acids with {difluoro (phenylthio) methyl} triphenylphosphonium triflate | |
| CN105198722A (en) | Method for asymmetrically catalyzing and synthesizing (R)-4, 7-dimethyl-1-tetralone | |
| JP4759722B2 (en) | Process for producing aromatic carboxylic acid ester having a substituent | |
| JP4811547B2 (en) | Process for producing optically active cyclohexene compound and optically active cyclohexene derivative used for the production | |
| Ladzik et al. | Regioselective synthesis of 5-ethoxycarbonyl-, 5-acetyl-and 5-trifluoroacetyl-6-trifluoromethylsalicylates by one-pot cyclizations of 1, 3-bis (trimethylsilyloxy)-1, 3-butadienes with 3-alkoxy-2-alken-1-ones |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16807495 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017523658 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16807495 Country of ref document: EP Kind code of ref document: A1 |