WO2016189552A2 - Novel recovery and recycling process of unwanted enantiomers of 2-aminopropyl indoline derivatives - Google Patents
Novel recovery and recycling process of unwanted enantiomers of 2-aminopropyl indoline derivatives Download PDFInfo
- Publication number
- WO2016189552A2 WO2016189552A2 PCT/IN2016/050156 IN2016050156W WO2016189552A2 WO 2016189552 A2 WO2016189552 A2 WO 2016189552A2 IN 2016050156 W IN2016050156 W IN 2016050156W WO 2016189552 A2 WO2016189552 A2 WO 2016189552A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- azide
- propyl
- denotes
- process according
- Prior art date
Links
- RQJVVMSIIKSQQB-UHFFFAOYSA-N CC(Cc(cc1CC2)cc(C#N)c1N2C(C)=O)NCCOc(cccc1)c1OCC(F)(F)F Chemical compound CC(Cc(cc1CC2)cc(C#N)c1N2C(C)=O)NCCOc(cccc1)c1OCC(F)(F)F RQJVVMSIIKSQQB-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/08—Indoles; Hydrogenated indoles with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
Definitions
- the present invention relates to a novel recovery and recycling process of unwanted enantiomers of 2-aminopropyl indoline derivatives.
- the enantiomers of 2-aminopropyl indoline derivatives are useful intermediates, particularly, for preparation of Silodosin or salts thereof.
- the present invention also provides novel compounds of Formula-II Formula-Ill, Formula-IV and Formula-V.
- 2-aminopropyl indoline derivatives of Formula -I wherein Rl denotes H, alkyl group, cycloalkyl, hydroxy propyl, phenylmethoxy propyl, and benzoyloxy propyl; R2 denotes CN, halogen and aminocarbonyl (-CONH2) are important raw materials/intermediates for preparing pharmaceutical products, particularly, for preparing Silodosin.
- the compounds of formula I are having one optical center mentioned with asterisk (*), therefore, the compounds exist as R and S enantiomers, mixture of enantiomers and racemic mixture.
- Silodosin is an indoline compound, chemically known as l-(3-Hydroxypropyl)-5- [(2R)-2- ( ⁇ 2- [2-(2,2,2-trifluoroethoxy)phenoxy] ethyl ⁇ amino)propyl] -2 ,3 - dihydro- 1 H- indole-7- carboxamide.
- Silodosin was first disclosed in U.S. Patent No. 5387603 as therapeutic agents for the treatment of dysuria, urinary disturbance associated with benign prostatic hyperplasia.
- the therapeutically active enantiomer of Silodosin is the R enantiomer.
- the desired enantiomers of 2-aminopropyl indoline derivatives of Formula -I are prepared usually by two methods.
- stereo selective synthesis is carried out involving reductive amination using chiral amines and corresponding ketones.
- JP 4634560 patent discloses reaction of (3-benzoyloxy- propyl) -7-cyano-5 -(2-oxopropyl) indoline with L-2-phenylglycinol in presence of hydrogen and platinum oxide to obtain R enantiomer of 5- (2- aminopropyl) -1- (3-benzoyloxy-propyl) -7-cyano-indoline.
- the stereo selective synthesis involves specific chiral amines, which are not available commercially.
- this method suffers due to formation of impurities because of poor selectivity of desired enantiomer leading to yield loss.
- R1 protecting grp
- R2 cyano or carbamoyl using L-(+)tartaric acid to obtain R-enantiomer.
- the resolved R-enantiomer is further converted to Silodosin.
- the object of the invention is to provide an economical and industrially applicable process for recycling the undesired isomers of Formula-I which ameliorates most of the problems associated with the disposal of undesired enantiomers.
- the present invention provides a recovery and recycling process for preparation of an opposite enantiomer or mixture of enantiomers of Formula-I wherein Rl denotes H, alkyl group, cycloalkyl, hydroxy propyl, phenylmethoxy propyl, and benzoyloxy propyl; R2 denotes CN, halogen and aminocarbonyl (-CONH2) which comprises; a) reacting S-enantiomer or enantiomerically enriched S-enantiomer of Formula-I with a chemical agent to obtain compound of Formula- VI , wherein LG is a leaving group;
- the chemical agents selected from sulfonyl agents or pyrylium salts, are reacted with the unwanted enantiomer of Formula-I to form corresponding sulfonylimides and pyridinium compounds respectively as leaving groups.
- present invention provides novel compounds of Formula-II, Formula-Ill, Formula-IV and Formula- V, wherein Rl denotes H, alkyl group, cycloalkyl, hydroxy propyl, phenylmethoxy propyl, and benzoyloxy propyl; R2 denotes CN, halogen and aminocarbonyl; R3 denotes H, alkyl, aryl, substituted aryl and pyridyl; X “ denotes tetrafluoroborates (BF 4 " ), perchlorates (CIO4 “ ) and trifluoromethane sulfonate(CF3SCb " ); and Y denotes methane, p- toluene, p-bromobenzene and p-nitrobenzene.
- Rl denotes H, alkyl group, cycloalkyl, hydroxy propyl, phenylmethoxy propyl, and benzoyloxy propyl
- Mixture of enantiomers means it is mixture of R and S enantiomers in any proportion.
- enantiomeric excess or enatiomeric enriched' refers generally to the concentration of one stereoisomer that exceeds the concentration of another stereoisomer. Typically, the term is used to characterize the optical purity of an optically active compound that exists in the bulk as two or more stereo isomers. For example: enantiomerically enriched R-enantiomer means the enantiomer ratio of R:S varies from 51 :49 to 100:0.
- opposite enantiomer refers, for example, to when S enantiomer is used as reactant, R enantiomer (opposite enantiomer) is obtained as product. Similarly, when R enantiomer is used as reactant and S enantiomer (opposite enantiomer) is obtained as product.
- the Formula-I compounds used for the purpose of this invention may be pure enantiomers or enantiomerically enriched.
- the process comprises reacting unwanted enantiomer or non-racemic mixture of 2-aminopropyl indoline compounds of Formula-I with a chemical agent, selected from sulfonyl agents or pyrylium salts, to form corresponding sulfonylimides and pyridinium compounds respectively as leaving groups.
- a chemical agent selected from sulfonyl agents or pyrylium salts
- the obtained sulfonylimides compounds of Formula-Ill OR pyridinium compounds of Formula- V are further reacted with metal azides to obtain Azide of Formula-II by inversion.
- the azide compounds of Formula - II so obtained are then reduced to obtain enantiomers with opposite configuration. If the starting enantiomer is having S- configuration, then the final amine compounds are obtained having R-configuration. Similarly, if the starting enantiomer is having R-configuration, then the final amine compounds are obtained having S-configuration.
- the invention is depicted in scheme- 1 mentioning unwanted S-enantiomer, for better understanding, as starting material and recovering the R-enantiomer.
- the unwanted enantiomer or non-racemic mixture of 2-aminopropyl indoline compounds of Formula-I are reacted with sulfonyl agents in a suitable solvent medium in presence of base to obtain corresponding sulfonylimides of Formula - III wherein Rl, R2 and Y groups are as defined previously.
- the sulfonyl agents include methanesulfonyl chloride, p-toluenesulfonyl chloride, p-bromobenzene sulfonylchloride and p-nitrobenzene sulfonyl chloride.
- preferred one is p-toluenesulfonyl chloride to obtain corresponding ditosylimide. Formation of mono substituted sulfonyl compounds (sulfonamides) are also observed in minor quantities.
- the solvent medium includes, but not limited to, aliphatic or aromatic hydrocarbons, chlorinated hydrocarbons, esters, ethers, ketones, polar aprotic solvents, nitriles or mixtures thereof.
- Chlorinated hydrocarbons preferably include methylene dichloride, ethylene dichloride, chloroform, carbontetrachloride, and aromatic hydrocarbons preferably selected from toluene, xylene, and aliphatic hydrocarbons include hexane, cyclohexane, heptane etc.
- Esters include ethyl acetate or butyl acetate.
- Ethers include tetrahydrofuran, dioxane, ethyl ether or methyl -t-butyl ether.
- Polar aprotic solvents include dimethylsulfoxide, dimethylacetamide or dimethylformamide. However, preferred solvent is tetrahydrofuran.
- the base may be organic or inorganic base.
- inorganic base is used.
- the inorganic base is selected from the group consisting of alkali metal hydrides, alkali metal hydroxide, alkali metal alkoxides and carbonates.
- more preferred base is sodium hydride or potassium hydride.
- the reaction is conducted in the range of ambient temperature to the boiling temperature of the solvent used.
- the reaction is performed in the temperature range of about 50-90°C. Most preferred temperature range is about 60-75°C.
- the sulfonylimide compounds of Formula- III are further reacted with metal azides, particularly alkaline metal azides or alkaline earth metal azides in suitable solvents to obtain corresponding azides of Formula- II with inversion.
- the reaction optionally, may be conducted in presence of catalyst.
- the alkali metal azides and alkaline earth metal azides include lithium azide, sodium azide, potassium azide, calcium azide, and magnesium azide, strontium azide, and barium azide.
- the preferred azide is sodium azide or potassium azide.
- the solvents include, but not limited to, water, alcohols, esters, aromatic hydrocarbons, nitriles and polar aprotic solvents or mixtures thereof.
- Alcohols include methanol, ethanol, n- propanol, isopropanol, n-butanol, iso butanol and tertiary butanol etc.
- Esters include ethyl acetate, methyl acetate, n-butyl acetate or iso butyl acetate.
- Polar aprotic solvents include dimethyl sulfoxide, dimethyl acetamide, dimethyl formamide or N-methylpyrrolidine.
- Aromatic hydrocarbons include toluene or xylene. However, preferred solvents used for the reaction are dimethyl sulfoxide, dimethyl acetamide, dimethyl formamide or N-methylpyrrolidine.
- Catalyst for forming azide may be selected from sodium bromide, sodium iodide, sodium fluoride, potassium bromide, potassium iodide, potassium fluoride, lithium bromide, lithium iodide and lithium fluoride.
- preferred catalyst is sodium iodide or potassium iodide.
- the azide reaction is conducted in the range of ambient temperature to the boiling temperature of the solvent used.
- the reaction is performed in the temperature range of about 120-135°C.
- the azide is obtained by reacting unwanted enantiomer or non-racemic mixture the 2-aminopropyl indoline with pyrylium salt followed by reaction with metal azide.
- Pyrylium salts The process comprises reacting unwanted enantiomer or non-racemic mixture of 2-aminopropyl indoline compounds of Formula-I wherein Rl and R2 are defined as previously with pyrylium salts in suitable solvents and in presence of organic bases and organic acids to obtain corresponding pyridinium salts of Formula - V.
- the preferred group is phenyl in 2,4,6 position selected from R3 and tetrafluoroborates (BF 4 " ) selected from X "
- the 2-aminopropyl indoline of Formula-I may be reacted as such or they may be reacted with diastereomeric salt.
- the 2-aminopropyl indoline of Formula-I are resolved with optically active organic acids such as L-(+)tartaric acid through diastereomeric salt formation. Therefore, the unwanted enantiomer of 2- aminopropyl indoline with its organic acid salt (diastereomeric salt) may, directly, be reacted with the pyrylium salts.
- the solvents used for formation of pyridinium compounds include, but not limited to, aliphatic or aromatic hydrocarbons, chlorinated hydrocarbons, esters, ethers, ketones, polar aprotic solvents, nitriles or mixtures thereof.
- Chlorinated hydrocarbons preferably include methylene dichloride, ethylene dichloride, chloroform, carbontetrachloride, and aromatic hydrocarbons preferably selected from toluene, xylene, and aliphatic hydrocarbons include hexane, cyclohexane, heptane etc.
- Esters include ethyl acetate or butyl acetate.
- Ethers include tetrahydrofuran, dioxane, ethyl ether or methyl -t-butyl ether.
- Polar aprotic solvents include dimethylsulfoxide, dimethylacetamide or dimethylformamide. However, preferred solvents are methylene dichloride or ethylene dichloride.
- the reaction is conducted in presence of organic bases that are mild in nature.
- the organic bases include triethylamine, diethylamine, methylamine, diisopropyl ethylamine, pyridine, dimethylaminopyridine and tetrabutylammonium hydroxide.
- preferred base is triethylamine.
- Use of organic acids act as catalyst for cyclisation reaction (ring closure) to form pyridinium compound.
- the organic acids are selected from formic acid, acetic acid, propionic acid, lactic acid, fumaric acid, butyric acid, citric acid and benzoic acid. However, preferred organic acid is acetic acid.
- pyridinium compounds usually formation at 10°C to 70°C temperature. However preferred temperature is 25-35°C.
- the reaction completes typically in 10-12 hours. After completion of reaction, added a base dissolved in water to neutralize the reaction mass. Then extracted with suitable solvent and the product, pyridinium compound, is isolated by distilling the solvent.
- the pyridinium compounds are isolated as salts of tetrafluroborates (BF 4 " ), perchlorates (C10 4 " ), bisulfates (HS0 4 " ) and trifluoromethane sulfonate(CF3SCb " ) based on the type of pyrylium salt used for formation of pyridinium compound.
- the starting materials of pyrylium salts may be prepared as per the procedures known in the art - ICS Perkin I, 1982, 117-123.
- the pyridinium compounds are further reacted with metal azides to obtain corresponding azides of Formula-II with inversion.
- the pyridinium compounds of Formula- V are reacted with alkali metal azides or alkaline earth metal azides in suitable solvents to obtain corresponding azides of Formula-II with inversion.
- the alkali metal azides and alkaline earth metal azides include lithium azide, sodium azide, potassium azide, calcium azide, and magnesium azide calcium azide, strontium azide, and barium azide.
- preferred azide is sodium azide or potassium azide.
- a wide range of solvents may be used for forming azide from the pyridinium compounds of Formula- V.
- the solvents include, but not limited to, water, alcohols, esters, aromatic hydrocarbons, nitriles and polar aprotic solvents or mixtures thereof.
- Alcohols include methanol, ethanol, n-propanol, isopropanol, n- butanol, iso butanol and tertiary butanol etc.
- Esters include ethyl acetate, methyl acetate, n-butyl acetate or iso butyl acetate.
- Polar aprotic solvents include dimethyl sulfoxide, dimethyl acetamide, dimethyl formamide or N- methylpyrrolidine.
- Aromatic hydrocarbons include toluene or xylene.
- preferred solvents used for the reaction are dimethyl sulfoxide, dimethyl acetamide, dimethyl formamide or N-methylpyrrolidine.
- the azide reaction from pyridinium compounds of Formula- V is conducted in the range of ambient temperature to the boiling temperature of the solvent used.
- the reaction is performed in the temperature range of about 75-85°C.
- the reaction completes in 3-4 hours.
- the azide compounds of Formula- II are isolated from the reaction mass in the same way as explained above, wherein the azides are prepared via sulfonylimide route.
- the azide compounds of Formula- II are reacted with a reducing agent in suitable solvent to form corresponding inverted amines of Formula-I.
- the reducing agents include, but not limited to, hydrogen in presence of metal catalysts such as Raney nickel, palladium on carbon (Pd/C) and platinum; ammonium formate, tin and tin chloride, hydrazine, iron and iron chloride, zinc, formic acid, hydrogen sulfide, sodium sulfide, and titanium chloride.
- metal catalysts such as Raney nickel, palladium on carbon (Pd/C) and platinum
- Pd/C palladium on carbon
- solvents include, but not limited to, water, alcohols, esters, aromatic hydrocarbons and polar aprotic solvents or mixtures thereof.
- Alcohols include methanol, ethanol, n-propanol, isopropanol, n-butanol, iso butanol and
- Esters include ethyl acetate, methyl acetate, n-butyl acetate or iso butyl acetate.
- Polar aprotic solvents include dimethyl sulfoxide or dimethyl formamide.
- Aromatic hydrocarbons include toluene or xylene. However preferred solvent is methanol or ethanol.
- the reaction may be conducted in atmospheric pressure or under hydrogen pressure in autoclave.
- the hydrogen pressure may be applied from 1 kg/cm 2 and 25 kg/cm 2 .
- the preferred pressure for conducting reduction reaction is 4-5 kg/cm 2 .
- the catalyst if used, is separated by conventional methods such as filtration.
- the filtrate is distilled to isolate the product, inverted enantiomer of Formula-I, which is having opposite configuration compared to the configuration of initial amine subjected for reaction.
- the obtained enantiomerically enriched isomers having opposite configuration are further purified with suitable optically active acids such as L-(+)tartaric acid to obtain pure enantiomer.
- the R-enantiomers of 2- aminopropyl indoline Formula-I, wherein Rl denotes benzoyloxy propyl and R2 denotes CN, prepared as per the present invention mentioned above are converted into Silodosin.
- R-enantiomer of 2-aminopropyl indoline Formula-I wherein Rl denotes benzoyloxy propyl and R2 denotes CN, is reacted with 2- [2-(2,2,2- trifluoroethoxy)phenoxy] ethyl methanesulfonate in presence of potassium carbonate to obtain 3-(5-(R)-2-(2-(2-(2,2,2-trifluoroethoxy)phenoxy] ethyl amino)propyl)-7-cyanoindolin-l-yl)propyl benzoate.
- This compound is hydrolysed using NaOH to obtain 5-((R)-2-(2-(2-(2,2,2- trifluoroethoxy)phenoxy] ethyl amino)propyl))-l-(3-hydroxypropyl)indoline-7-carbonitrile which is further reacted with hydrogen peroxide in dimethylsulfoxide solvent to get Silodosin.
- the intermediate, 2- [2-(2,2,2-trifluoroethoxy)phenoxy] ethyl methanesulfonate is prepared by reacting catechol with potassium carbonate in ethyl acetate solvent at 90°C to form potassium salt of catechol which is further reacted with 2,2,2- trifluoroethyl methane sulfonate in a polar aprotic solvent such as N,N- dimethylformamide in presence of sodium iodide catalyst at 135-145°C.
- a polar aprotic solvent such as N,N- dimethylformamide
- the 2-(2,2,2- trifluoroethoxy)phenol is further reacted with 2-chloro ethanol to obtain 2-[(2,2,2- trifluoroethoxy)phenoxy] ethanol which is reacted with methanesulfonyl chloride to obtain 2- [2-(2,2,2-trifluoroethoxy)phenoxy] ethyl methanesulfonate.
- the starting materials, S-enantiomers of 2-aminopropyl indoline Formula-I are obtained by reducing the corresponding 2-nitropropyl indoline Formula- VII with hydrogen in presence of palladium on carbon or Raney nickel catalyst in solvents such as ethyl acetate and methanol or mixture thereof. This reduction reaction may also be conducted in presence of basic medium such as ammonia or triethylamine to obtain the 2-aminopropyl indoline Formula-I which is further subjected for resolution using L-tartaric acid to obtain corresponding S and R- enantiomers of formula-I.
- present invention provides novel compounds of Formula-II, Formula-Ill, Formula-IV and Formula-V wherein Rl denotes H, alkyl group, cycloalkyl, hydroxy propyl, phenylmethoxy propyl, and benzoyloxy propyl; R2 denotes CN, halogen and aminocarbonyl; R3 denotes H, alkyl, aryl, substituted aryl and pyridyl; X “ denotes tetraflurob orates (BF 4 " ), perchlorates (CIO4 “ ) and trifluoromethane sulfonate(CF3SCb " ); and Y denotes methane, p- toluene, p-bromobenzene and p-nitrobenzene.
- Rl denotes H, alkyl group, cycloalkyl, hydroxy propyl, phenylmethoxy propyl, and benzoyloxy propyl
- Formula-II, Formula-Ill and Formula-V are novel intermediates used for preparation of enantiomer or mixture of enantiomers of Formula-I, whereas the novel compound of Formula-IV is obtained as impurity formed during reaction of enantiomer of Formula-I with p-toluenesulfonyl chloride. The impurity eventually removed by conducting column chromatography.
- Benzoic acid 50 g, 0.41 moles was dissolved in 82.5 ml of dry N, N- dimethylformamide.
- 56.8ml (0.41 moles) of triethylamine was added drop wise to it, followed by the addition of 64.5 g (0.41 moles) of 1 -bromo-3-chloropropane.
- the reaction mixture was stirred overnight at room temperature and then for 6 hours at 50°C.
- the reaction mass was cooled to room temperature and (49 g, 0.41 moles) Indoline, 56.8ml (0.41 moles) of triethylamine and 82.5 ml N, N- dimethylformamide was added.
- the reaction mixture was heated to 100°C and maintained for 14 hours.
- Phosphorus oxy chloride (29.4 ml, 0.315 moles) was added drop wise over a period of about 15 minutes to 73 ml of dry ⁇ , ⁇ -dimethylformamide at 0-5°C and the mixture was allowed to stir for 30 minutes. Then 50 g (0.157 moles) of l-(3- benzoyloxypropyl) indoline hydrochloric acid salt was added lot wise to the mixture and was stirred for 12 hours at room temperature. Poured the reaction mixture into ice water and stirred for 30 minutes. Adjust the pH of reaction mixture to 7 by addition of 20% sodium carbonate solution in water and allowed to stir further for 30 minutes at room temperature.
- reaction mass was extracted with toluene followed by washing with sodium bicarbonate aqueous solution and salt water respectively.
- 48.5 gm (0.646 moles) of nitroethane and ammonium acetate (37.5 gm, 0.486 moles) was added to this toluene layer and heated to 100°C and maintained for 8 hrs.
- Reaction mass was cooled to room temperature and brought the pH 7 by the addition of sodium bicarbonate aqueous solution. Washed the toluene layer with brine and distilled off under reduced pressure.
- Toluene obtained after distillation was analyzed by GC and found to have around 4% nitroethane (by wt; 60-70% recovery) which can be reused/recycled as such further for new reaction.
- Oily mass obtained after complete distillation of toluene was dissolved in 150 ml IPA and stirred at room temperature for 12-14 hrs. Red crystals obtained were filtered and washed with 25 ml IPA. Crystals obtained were dried at 40-45°C for 12 hours to yield 41.5 g of 1 - (3-Benzoyloxypropyl)-5-(2-nitropropenyl) indoline as brown crystals.
- Reaction mass was extracted with ethyl acetate followed by the washing of ethyl acetate layer with brine solution. Distilled off ethyl acetate under reduced pressure in order to obtain 50 gm of 1 - (3- benzoyloxypropyl)-5-(2-nitropropyl) indoline as brown color oil.
- Phosphorus oxychloride (31.6 ml, 0.34 moles) was added dropwise over a period of about 15 minutes to 55 ml of dry ⁇ , ⁇ -dimethylformamide at 0-5°C and the mixture was allowed to stir for 30 minutes. Then to this mixture 50 gm (0.135 moles) of 1 - (3- benzoyloxypropyl)-5-(2-nitropropyl) indoline dissolved in 50 ml of ⁇ , ⁇ -dimethylformamide was added drop wise. Raised the temperature to 50°C and maintained to stir at same temperature for 4-5 hours. Cooled the reaction mass to room temperature and poured into ice water. Adjusted the pH of reaction mixture to 7 by addition of sodium carbonate solution.
- reaction mixture was extracted with ethyl acetate. Distilled off ethyl acetate under reduced pressure and 100 ml methanol was added to obtained semi-solid. Raised the temperature to 60- 65°C and stirred the reaction mass for 20-30 minutes. The reaction mass was gradually cooled to room temperature. The resulting solid was filtered and dried at 45°C to give 34g of 3-(7-formyl-5-(2-nitropropyl)indolin-l-yl)propyl benzoate as light green colored crystals.
- reaction mixture Cooled the reaction mixture to room temperature and 200 ml of water was added. Both organic and aqueous layers were separated. Aqueous layer thus obtained was extracted twice with toluene. All the toluene layers were combined and 350 ml of aqueous acetic acid solution was added. The reaction mixture was stirred at room temperature for 30 minutes. Aqueous acetic acid layer and toluene layers were separated. Dimer impurity formed in the process removed in this toluene layer. 200 ml of fresh toluene was added to obtained aqueous acetic acid layer. pH of this solution was adjusted to 11 by the drop wise addition of NaOH solution.
- Tartrate salt (11 6g, 0.022 moles) of "ACB"-S isomer as obtained at stage-9 was added to the mixture of pyrylium salt (9 g, 0.022 moles) in 50 ml of dichloromethane.
- Triethylamine (7.8 ml, 0.0567 moles) was added to the reaction mixture followed by the addition of dichloromethane (50 ml).
- the reaction mixture was stirred at room temperature for 40-45 minutes.
- acetic acid (3.2 ml, 0.0567 moles) was added and the reaction mixture was allowed to stir at room temperature for 10-12 hours.
- Monitoring of reaction was done on TLC (5% methanol in MDC).
- ACB heartrate salt of "R” isomer
Abstract
The present invention discloses a novel recovery and recycling process of unwanted enantiomers of 2-aminopropyl indoline derivatives, useful intermediates, particularly, for preparation of Silodosin or salts thereof. The present invention also provides novel compounds of Formula-II Formula-III, Formula-IV and Formula-V.
Description
"NOVEL RECOVERY AND RECYCLING PROCESS OF UNWANTED ENANTIOMERS OF 2-AMINOPROPYL INDOLINE DERIVATIVES"
Field of the invention:
The present invention relates to a novel recovery and recycling process of unwanted enantiomers of 2-aminopropyl indoline derivatives. The enantiomers of 2-aminopropyl indoline derivatives are useful intermediates, particularly, for preparation of Silodosin or salts thereof. The present invention also provides novel compounds of Formula-II Formula-Ill, Formula-IV and Formula-V.
Background of Invention:
2-aminopropyl indoline derivatives of Formula -I wherein Rl denotes H, alkyl group, cycloalkyl, hydroxy propyl, phenylmethoxy propyl, and benzoyloxy propyl; R2 denotes CN, halogen and aminocarbonyl (-CONH2) are important raw materials/intermediates for preparing pharmaceutical products, particularly, for preparing Silodosin.

Formula-I
The compounds of formula I are having one optical center mentioned with asterisk (*), therefore, the compounds exist as R and S enantiomers, mixture of enantiomers and racemic mixture.
Silodosin is an indoline compound, chemically known as l-(3-Hydroxypropyl)-5- [(2R)-2- ( { 2- [2-(2,2,2-trifluoroethoxy)phenoxy] ethyl } amino)propyl] -2 ,3 - dihydro- 1 H- indole-7- carboxamide.

Silodosin
Silodosin was first disclosed in U.S. Patent No. 5387603 as therapeutic agents for the treatment of dysuria, urinary disturbance associated with benign prostatic hyperplasia. The therapeutically active enantiomer of Silodosin is the R enantiomer.
The desired enantiomers of 2-aminopropyl indoline derivatives of Formula -I are prepared usually by two methods. In one method, stereo selective synthesis is carried out involving reductive amination using chiral amines and corresponding ketones. For example, JP 4634560 patent discloses reaction of (3-benzoyloxy- propyl) -7-cyano-5 -(2-oxopropyl) indoline with L-2-phenylglycinol in presence of hydrogen and platinum oxide to obtain R enantiomer of 5- (2- aminopropyl) -1- (3-benzoyloxy-propyl) -7-cyano-indoline. However, the stereo selective synthesis involves specific chiral amines, which are not available commercially. Moreover, this method suffers due to formation of impurities because of poor selectivity of desired enantiomer leading to yield loss.
In another common method, first, the racemic compounds of 2-aminopropyl indoline derivatives of Formula -I are prepared followed by resolution with optically active acids to obtain the desired enantiomers. Publications reported this method to prepare the desired isomers are mentioned below.
A Chinese application, CN101759627A, reported that R-enantiomer of 1- acetyls' [ 2- [2- [2- (2, 2, 2- trifluoroethoxy) phenoxy] ethylamino] propyl] indoline -7- cyano is obtained by optical resolution and the R- enantiomer is used for
preparation of Silodosin. The application did not disclose any reuse/recycling of unwanted enantiomer.
Other PCT applications, WO2011124704 and WO2013056842, discloses resolution of the racemic mixture of compound of Formula

R1 = protecting grp, R2 = cyano or carbamoyl using L-(+)tartaric acid to obtain R-enantiomer. The resolved R-enantiomer is further converted to Silodosin. These applications also did not suggest or disclose any reuse/recycling of unwanted enantiomer.
Drugs of the Future 2001, 26(6); 553-560 and Tetrahedron 2013, 69(13); 2834-43 journals report resolution of compound of Formula

with S-(+) mandelic acid to obtain its R-enantiomer. The R-enantiomer is further converted to Silodosin. These journals also did not suggest or disclose any reuse/recycling of unwanted enantiomer.
A cursory review of the prior art confirms that none of the publications disclosed or suggested reuse/recyclization of unwanted enantiomer obtained after resolution. The undesired enantiomer is discarded/incinerated leading to increase in cost of production and causing environmental pollution as well. Therefore, it is essential
to develop an economical and industrially applicable reuse/recycling process of the valuable unwanted enantiomers of 2-aminopropyl indoline derivatives of Formula -I to address the problems of production cost and environmental pollution as well.
Consequently there is a need to recycle and reuse the undesired enantiomer of Formula -I in the preparation of pharmaceutical products, particularly, for preparation of Silodosin.
Therefore, the object of the invention is to provide an economical and industrially applicable process for recycling the undesired isomers of Formula-I which ameliorates most of the problems associated with the disposal of undesired enantiomers.
While screening methods for recycling the unwanted isomer, unexpectedly, the present inventors found an effective recovery and recycling process of the unwanted enantiomers of 2-aminopropyl indoline derivatives of Formula -I, for which protection is sought.
Summary of Invention:
According to one aspect, the present invention provides a recovery and recycling process for preparation of an opposite enantiomer or mixture of enantiomers of Formula-I wherein Rl denotes H, alkyl group, cycloalkyl, hydroxy propyl, phenylmethoxy propyl, and benzoyloxy propyl; R2 denotes CN, halogen and aminocarbonyl (-CONH2) which comprises; a) reacting S-enantiomer or enantiomerically enriched S-enantiomer of Formula-I with a chemical agent to obtain compound of Formula- VI , wherein LG is a leaving group;

S-enanatiomer S-Enanatiomer of
of Formula-I Formula-VI
b) reacting the compound of Formula-VI with metal azide to obtain azide of Formula-II; and

S-Enanatiomer of R-Azide of
Formula-VI Formula-II
c) reducing the azide of Formula -II with a reducing agent to obtain R- enantiomer or mixture of R and S-enantiomers of Formula-I.

R-Azide of R-Enanatiomer of
Formula-II Formula-I
OR a) reacting R-enantiomer or enantiomerically enriched R-enantiomer of Formula-I with a chemical agent to obtain compound of Formula-VI, wherein LG is a leaving group;

R-enanatiomer R-Enanatiomer of
of Formula-I Formula-VI
b) reacting the compound of Formula-VI with metal azide to obtain azide of Formula-II; and

R-Enanatiomer of S-Azide of
Formula-VI Formula-ll
c) reducing the azide of Formula -II with a reducing agent to obtain S-enantiomer or mixture of S and R-enantiomers of Formula-I.

In a preferred embodiment, the chemical agents, selected from sulfonyl agents or pyrylium salts, are reacted with the unwanted enantiomer of Formula-I to form corresponding sulfonylimides and pyridinium compounds respectively as leaving groups.

Pyrylium salts Sulfonylimides Pyridinium compounds
According to another aspect, present invention provides novel compounds of Formula-II, Formula-Ill, Formula-IV and Formula- V, wherein Rl denotes H, alkyl group, cycloalkyl, hydroxy propyl, phenylmethoxy propyl, and benzoyloxy propyl; R2 denotes CN, halogen and aminocarbonyl; R3 denotes H, alkyl, aryl, substituted aryl and pyridyl; X" denotes tetrafluoroborates (BF4 "), perchlorates (CIO4") and trifluoromethane sulfonate(CF3SCb"); and Y denotes methane, p- toluene, p-bromobenzene and p-nitrobenzene.
The novel compounds of Formula-II, Formula-Ill, Formula IV and Formula-V are useful intermediates in the recovery and recycling process of the unwanted enantiomer of Formula-I.

Formula-II Formula-Ill Formula-IV

Formula-V
Detailed description of the invention:
The invention will now be described in detail in connection with certain preferred and optional embodiments, so that various aspects thereof may be more fully understood and appreciated.
Unless specified otherwise, all technical and scientific terms used herein have the same
meaning as commonly understood by one of ordinary skill in the art, to which this invention belongs. To describe the invention, certain terms are defined herein specifically as follows.
Unless stated to the contrary, any of the words, "including", "includes", "comprising", and "comprises" mean "including without limitation" and shall not
be construed to limit any general statement that it follows to the specific or similar items.
The terms "enantiomer", "S" and "R" as used herein, unless specified otherwise, refer to stereoisomer resulting from the spatial arrangement of groups at a chiral centre, and in the present context, the person of ordinary skill will appreciate that the groups attached with the carbon marked with asterisk represents the plane for purposes of determining the configuration.
Mixture of enantiomers means it is mixture of R and S enantiomers in any proportion.
The term 'enantiomeric excess or enatiomeric enriched' as used herein, refers generally to the concentration of one stereoisomer that exceeds the concentration of another stereoisomer. Typically, the term is used to characterize the optical purity of an optically active compound that exists in the bulk as two or more stereo isomers. For example: enantiomerically enriched R-enantiomer means the enantiomer ratio of R:S varies from 51 :49 to 100:0.
The term opposite enantiomer is as used herein refers, for example, to when S enantiomer is used as reactant, R enantiomer (opposite enantiomer) is obtained as product. Similarly, when R enantiomer is used as reactant and S enantiomer (opposite enantiomer) is obtained as product.
The present invention, in one aspect, provides a novel recovery and recycling process for preparation of opposite enantiomer or mixture of enantiomers of 2- aminopropyl indoline Formula-I, wherein Rl denotes H, alkyl group, cycloalkyl, hydroxy propyl, phenylmethoxy propyl, and benzoyloxy propyl; R2 denotes CN, halogen and aminocarbonyl. However, benzoyloxy propyl is preferred from Rl and CN is preferred from R2.

Formula-I
The Formula-I compounds used for the purpose of this invention may be pure enantiomers or enantiomerically enriched.
In a preferred embodiment, the process comprises reacting unwanted enantiomer or non-racemic mixture of 2-aminopropyl indoline compounds of Formula-I with a chemical agent, selected from sulfonyl agents or pyrylium salts, to form corresponding sulfonylimides and pyridinium compounds respectively as leaving groups.
Accordingly, the obtained sulfonylimides compounds of Formula-Ill OR pyridinium compounds of Formula- V are further reacted with metal azides to obtain Azide of Formula-II by inversion. The azide compounds of Formula - II so obtained are then reduced to obtain enantiomers with opposite configuration. If the starting enantiomer is having S- configuration, then the final amine compounds are obtained having R-configuration. Similarly, if the starting enantiomer is having R-configuration, then the final amine compounds are obtained having S-configuration. The invention is depicted in scheme- 1 mentioning unwanted S-enantiomer, for better understanding, as starting material and recovering the R-enantiomer.
Scheme- 1 :

R- Enantiomer of Formula -I
Accordingly, the unwanted enantiomer or non-racemic mixture of 2-aminopropyl indoline compounds of Formula-I are reacted with sulfonyl agents in a suitable solvent medium in presence of base to obtain corresponding sulfonylimides of Formula - III wherein Rl, R2 and Y groups are as defined previously.
The sulfonyl agents include methanesulfonyl chloride, p-toluenesulfonyl chloride, p-bromobenzene sulfonylchloride and p-nitrobenzene sulfonyl chloride. However, preferred one is p-toluenesulfonyl chloride to obtain corresponding ditosylimide. Formation of mono substituted sulfonyl compounds (sulfonamides) are also observed in minor quantities.
The solvent medium includes, but not limited to, aliphatic or aromatic hydrocarbons, chlorinated hydrocarbons, esters, ethers, ketones, polar aprotic solvents, nitriles or mixtures thereof. Chlorinated hydrocarbons preferably include methylene dichloride, ethylene dichloride, chloroform, carbontetrachloride, and aromatic hydrocarbons preferably selected from toluene, xylene, and aliphatic hydrocarbons include hexane, cyclohexane, heptane etc. Esters include ethyl acetate or butyl acetate. Ethers include tetrahydrofuran, dioxane, ethyl ether or
methyl -t-butyl ether. Polar aprotic solvents include dimethylsulfoxide, dimethylacetamide or dimethylformamide. However, preferred solvent is tetrahydrofuran.
The base may be organic or inorganic base. Preferably inorganic base is used. The inorganic base is selected from the group consisting of alkali metal hydrides, alkali metal hydroxide, alkali metal alkoxides and carbonates. However, more preferred base is sodium hydride or potassium hydride.
Typically the reaction is conducted in the range of ambient temperature to the boiling temperature of the solvent used. Preferably, the reaction is performed in the temperature range of about 50-90°C. Most preferred temperature range is about 60-75°C.
After completion of reaction, work up to isolate the sulfonylimide compound of Formula- III is done by extracting with suitable solvent and isolation of the product by distilling the solvent.
In a preferred embodiment, the sulfonylimide compounds of Formula- III are further reacted with metal azides, particularly alkaline metal azides or alkaline earth metal azides in suitable solvents to obtain corresponding azides of Formula- II with inversion. The reaction, optionally, may be conducted in presence of catalyst.
The alkali metal azides and alkaline earth metal azides include lithium azide, sodium azide, potassium azide, calcium azide, and magnesium azide, strontium azide, and barium azide. However, the preferred azide is sodium azide or potassium azide.
A wide range of solvents may be used for forming azide. The solvents include, but not limited to, water, alcohols, esters, aromatic hydrocarbons, nitriles and polar
aprotic solvents or mixtures thereof. Alcohols include methanol, ethanol, n- propanol, isopropanol, n-butanol, iso butanol and tertiary butanol etc. Esters include ethyl acetate, methyl acetate, n-butyl acetate or iso butyl acetate. Polar aprotic solvents include dimethyl sulfoxide, dimethyl acetamide, dimethyl formamide or N-methylpyrrolidine. Aromatic hydrocarbons include toluene or xylene. However, preferred solvents used for the reaction are dimethyl sulfoxide, dimethyl acetamide, dimethyl formamide or N-methylpyrrolidine.
Catalyst for forming azide may be selected from sodium bromide, sodium iodide, sodium fluoride, potassium bromide, potassium iodide, potassium fluoride, lithium bromide, lithium iodide and lithium fluoride. However, preferred catalyst is sodium iodide or potassium iodide.
Usually the azide reaction is conducted in the range of ambient temperature to the boiling temperature of the solvent used. Preferably, the reaction is performed in the temperature range of about 120-135°C.
After completion of reaction, water is added into reaction mass when polar solvents are used for reaction and the product is extracted with suitable solvent followed by distillation of solvent to isolate the azide.
Alternately, in another embodiment, the azide is obtained by reacting unwanted enantiomer or non-racemic mixture the 2-aminopropyl indoline with pyrylium salt followed by reaction with metal azide.

X
Pyrylium salts
The process comprises reacting unwanted enantiomer or non-racemic mixture of 2-aminopropyl indoline compounds of Formula-I wherein Rl and R2 are defined as previously with pyrylium salts in suitable solvents and in presence of organic bases and organic acids to obtain corresponding pyridinium salts of Formula - V. The pyridinium salts, wherein R3 and X" are as mentioned above. However, the preferred group is phenyl in 2,4,6 position selected from R3 and tetrafluoroborates (BF4 ") selected from X"
The 2-aminopropyl indoline of Formula-I may be reacted as such or they may be reacted with diastereomeric salt. Usually the 2-aminopropyl indoline of Formula-I are resolved with optically active organic acids such as L-(+)tartaric acid through diastereomeric salt formation. Therefore, the unwanted enantiomer of 2- aminopropyl indoline with its organic acid salt (diastereomeric salt) may, directly, be reacted with the pyrylium salts.
Typically the solvents used for formation of pyridinium compounds include, but not limited to, aliphatic or aromatic hydrocarbons, chlorinated hydrocarbons, esters, ethers, ketones, polar aprotic solvents, nitriles or mixtures thereof. Chlorinated hydrocarbons preferably include methylene dichloride, ethylene dichloride, chloroform, carbontetrachloride, and aromatic hydrocarbons preferably selected from toluene, xylene, and aliphatic hydrocarbons include hexane, cyclohexane, heptane etc. Esters include ethyl acetate or butyl acetate. Ethers include tetrahydrofuran, dioxane, ethyl ether or methyl -t-butyl ether. Polar aprotic solvents include dimethylsulfoxide, dimethylacetamide or dimethylformamide. However, preferred solvents are methylene dichloride or ethylene dichloride.
The reaction is conducted in presence of organic bases that are mild in nature. The organic bases include triethylamine, diethylamine, methylamine, diisopropyl ethylamine, pyridine, dimethylaminopyridine and tetrabutylammonium hydroxide. However preferred base is triethylamine.
Use of organic acids act as catalyst for cyclisation reaction (ring closure) to form pyridinium compound. The organic acids are selected from formic acid, acetic acid, propionic acid, lactic acid, fumaric acid, butyric acid, citric acid and benzoic acid. However, preferred organic acid is acetic acid.
Usually formation of pyridinium compounds takes place at 10°C to 70°C temperature. However preferred temperature is 25-35°C. The reaction completes typically in 10-12 hours. After completion of reaction, added a base dissolved in water to neutralize the reaction mass. Then extracted with suitable solvent and the product, pyridinium compound, is isolated by distilling the solvent. The pyridinium compounds are isolated as salts of tetrafluroborates (BF4 "), perchlorates (C104 "), bisulfates (HS04 ") and trifluoromethane sulfonate(CF3SCb") based on the type of pyrylium salt used for formation of pyridinium compound.
The starting materials of pyrylium salts may be prepared as per the procedures known in the art - ICS Perkin I, 1982, 117-123.
The pyridinium compounds are further reacted with metal azides to obtain corresponding azides of Formula-II with inversion.
In a preferred embodiment, the pyridinium compounds of Formula- V are reacted with alkali metal azides or alkaline earth metal azides in suitable solvents to obtain corresponding azides of Formula-II with inversion.
The alkali metal azides and alkaline earth metal azides include lithium azide, sodium azide, potassium azide, calcium azide, and magnesium azide calcium azide, strontium azide, and barium azide. However, preferred azide is sodium azide or potassium azide.
A wide range of solvents may be used for forming azide from the pyridinium compounds of Formula- V. The solvents include, but not limited to, water,
alcohols, esters, aromatic hydrocarbons, nitriles and polar aprotic solvents or mixtures thereof. Alcohols include methanol, ethanol, n-propanol, isopropanol, n- butanol, iso butanol and tertiary butanol etc. Esters include ethyl acetate, methyl acetate, n-butyl acetate or iso butyl acetate. Polar aprotic solvents include dimethyl sulfoxide, dimethyl acetamide, dimethyl formamide or N- methylpyrrolidine. Aromatic hydrocarbons include toluene or xylene. However, preferred solvents used for the reaction are dimethyl sulfoxide, dimethyl acetamide, dimethyl formamide or N-methylpyrrolidine.
Typically the azide reaction from pyridinium compounds of Formula- V is conducted in the range of ambient temperature to the boiling temperature of the solvent used. Preferably, the reaction is performed in the temperature range of about 75-85°C. Usually the reaction completes in 3-4 hours.
After completion of reaction, the azide compounds of Formula- II are isolated from the reaction mass in the same way as explained above, wherein the azides are prepared via sulfonylimide route.
In another preferred embodiment, the azide compounds of Formula- II are reacted with a reducing agent in suitable solvent to form corresponding inverted amines of Formula-I.
The reducing agents include, but not limited to, hydrogen in presence of metal catalysts such as Raney nickel, palladium on carbon (Pd/C) and platinum; ammonium formate, tin and tin chloride, hydrazine, iron and iron chloride, zinc, formic acid, hydrogen sulfide, sodium sulfide, and titanium chloride. However, preferred reducing agent is hydrogen in presence of palladium on carbon (Pd/C).
A wide range of solvents may be used for conducting the reduction reaction. The solvents include, but not limited to, water, alcohols, esters, aromatic hydrocarbons and polar aprotic solvents or mixtures thereof.
Alcohols include methanol, ethanol, n-propanol, isopropanol, n-butanol, iso butanol and
tertiary butanol etc. Esters include ethyl acetate, methyl acetate, n-butyl acetate or iso butyl acetate. Polar aprotic solvents include dimethyl sulfoxide or dimethyl formamide.
Aromatic hydrocarbons include toluene or xylene. However preferred solvent is methanol or ethanol.
When the reducing agent is hydrogen, the reaction may be conducted in atmospheric pressure or under hydrogen pressure in autoclave. The hydrogen pressure may be applied from 1 kg/cm2 and 25 kg/cm2. However, the preferred pressure for conducting reduction reaction is 4-5 kg/cm2.
Usually the reduction is conducted between ambient temperature and 100°C. However preferred range of temperature is 30-40°C.
After completion of reaction, the catalyst, if used, is separated by conventional methods such as filtration. The filtrate is distilled to isolate the product, inverted enantiomer of Formula-I, which is having opposite configuration compared to the configuration of initial amine subjected for reaction.
As per the invention, usually enantiomerically enriched opposite isomers are obtained after reduction. Chiral purity of the inverted enantiomer usually found to be >80%ee.
The obtained enantiomerically enriched isomers having opposite configuration are further purified with suitable optically active acids such as L-(+)tartaric acid to obtain pure enantiomer.
In another embodiment of the present invention, the R-enantiomers of 2- aminopropyl indoline Formula-I, wherein Rl denotes benzoyloxy propyl and R2
denotes CN, prepared as per the present invention mentioned above are converted into Silodosin.
R-enantiomer of 2-aminopropyl indoline Formula-I, wherein Rl denotes benzoyloxy propyl and R2 denotes CN, is reacted with 2- [2-(2,2,2- trifluoroethoxy)phenoxy] ethyl methanesulfonate in presence of potassium carbonate to obtain 3-(5-(R)-2-(2-(2-(2,2,2-trifluoroethoxy)phenoxy] ethyl amino)propyl)-7-cyanoindolin-l-yl)propyl benzoate. This compound is hydrolysed using NaOH to obtain 5-((R)-2-(2-(2-(2,2,2- trifluoroethoxy)phenoxy] ethyl amino)propyl))-l-(3-hydroxypropyl)indoline-7-carbonitrile which is further reacted with hydrogen peroxide in dimethylsulfoxide solvent to get Silodosin.
The intermediate, 2- [2-(2,2,2-trifluoroethoxy)phenoxy] ethyl methanesulfonate, is prepared by reacting catechol with potassium carbonate in ethyl acetate solvent at 90°C to form potassium salt of catechol which is further reacted with 2,2,2- trifluoroethyl methane sulfonate in a polar aprotic solvent such as N,N- dimethylformamide in presence of sodium iodide catalyst at 135-145°C. After completion of reaction, the product, 2-(2,2,2-trifluoroethoxy)phenol, is isolated by distillation of solvent followed by extraction methods. The 2-(2,2,2- trifluoroethoxy)phenol is further reacted with 2-chloro ethanol to obtain 2-[(2,2,2- trifluoroethoxy)phenoxy] ethanol which is reacted with methanesulfonyl chloride to obtain 2- [2-(2,2,2-trifluoroethoxy)phenoxy] ethyl methanesulfonate.
The starting materials, S-enantiomers of 2-aminopropyl indoline Formula-I, are obtained by reducing the corresponding 2-nitropropyl indoline Formula- VII with hydrogen in presence of palladium on carbon or Raney nickel catalyst in solvents such as ethyl acetate and methanol or mixture thereof. This reduction reaction may also be conducted in presence of basic medium such as ammonia or triethylamine to obtain the 2-aminopropyl indoline Formula-I which is further subjected for resolution using L-tartaric acid to obtain corresponding S and R- enantiomers of formula-I.
According to a second aspect, present invention provides novel compounds of Formula-II, Formula-Ill, Formula-IV and Formula-V wherein Rl denotes H, alkyl group, cycloalkyl, hydroxy propyl, phenylmethoxy propyl, and benzoyloxy propyl; R2 denotes CN, halogen and aminocarbonyl; R3 denotes H, alkyl, aryl, substituted aryl and pyridyl; X" denotes tetraflurob orates (BF4 "), perchlorates (CIO4") and trifluoromethane sulfonate(CF3SCb"); and Y denotes methane, p- toluene, p-bromobenzene and p-nitrobenzene.
As mentioned above, Formula-II, Formula-Ill and Formula-V are novel intermediates used for preparation of enantiomer or mixture of enantiomers of Formula-I, whereas the novel compound of Formula-IV is obtained as impurity formed during reaction of enantiomer of Formula-I with p-toluenesulfonyl chloride. The impurity eventually removed by conducting column chromatography.
The following examples, which include preferred embodiments, is intended to illustrate the practice of this invention, it being understood that the particulars shown are by way of example and for purpose of illustrative discussion of preferred embodiments of the invention.
Preparation of 1 - (3-benzoyloxypropyl) indoline hydrochloric acid (Stage-1):
Benzoic acid (50 g, 0.41 moles) was dissolved in 82.5 ml of dry N, N- dimethylformamide. 56.8ml (0.41 moles) of triethylamine was added drop wise to it, followed by the addition of 64.5 g (0.41 moles) of 1 -bromo-3-chloropropane. The reaction mixture was stirred overnight at room temperature and then for 6 hours at 50°C. The reaction mass was cooled to room temperature and (49 g, 0.41 moles) Indoline, 56.8ml (0.41 moles) of triethylamine and 82.5 ml N, N- dimethylformamide was added. The reaction mixture was heated to 100°C and maintained for 14 hours. Cooled the reaction mixture to room temperature and 250 ml water was added and extracted with ethyl acetate. Obtained organic layer
was washed successively with sodium bicarbonate aqueous solution and salt water. Ethyl acetate was distilled off under reduced pressure. Thus obtained brown oil residue was dissolved in 300 ml of acetone and cooled to 0-5°C. 40 ml of concentrated hydrochloric acid was added drop wise at 0-5°C. The mixture was allowed to stir at room temperature for overnight. The solid obtained was filtered off and washed with 25 ml of cold acetone. The obtained solid was dried at 45- 50°C for 8-10 hours to achieve 72.5 g of 1 - (3 -benzoyloxypropyl) indoline hydrochloric acid salt as light brown colored crystals.
Preparation of 1 - (3-Benzoyloxypropyl)-5-(2-nitropropenyl) indoline (Stage- 2&3):
Phosphorus oxy chloride (29.4 ml, 0.315 moles) was added drop wise over a period of about 15 minutes to 73 ml of dry Ν,Ν-dimethylformamide at 0-5°C and the mixture was allowed to stir for 30 minutes. Then 50 g (0.157 moles) of l-(3- benzoyloxypropyl) indoline hydrochloric acid salt was added lot wise to the mixture and was stirred for 12 hours at room temperature. Poured the reaction mixture into ice water and stirred for 30 minutes. Adjust the pH of reaction mixture to 7 by addition of 20% sodium carbonate solution in water and allowed to stir further for 30 minutes at room temperature. The reaction mass was extracted with toluene followed by washing with sodium bicarbonate aqueous solution and salt water respectively. 48.5 gm (0.646 moles) of nitroethane and ammonium acetate (37.5 gm, 0.486 moles) was added to this toluene layer and heated to 100°C and maintained for 8 hrs. Reaction mass was cooled to room temperature and brought the pH 7 by the addition of sodium bicarbonate aqueous solution. Washed the toluene layer with brine and distilled off under reduced pressure. Toluene obtained after distillation was analyzed by GC and found to have around 4% nitroethane (by wt; 60-70% recovery) which can be reused/recycled as such further for new reaction. Oily mass obtained after complete distillation of toluene was dissolved in 150 ml IPA and stirred at room temperature for 12-14 hrs. Red crystals obtained were filtered and washed with 25
ml IPA. Crystals obtained were dried at 40-45°C for 12 hours to yield 41.5 g of 1 - (3-Benzoyloxypropyl)-5-(2-nitropropenyl) indoline as brown crystals.
Preparation of 1 - (3- benzoyloxypropyl)-5-(2-nitropropyl) indoline (Stage-4):
Sodium borohydride (7.74 g, 0.204 moles) was added lot wise to the solution of 50 gm (0.136 moles) of 3-(5-((E)-2-nitroprop-l-enyl)indolin-l-yl)propyl benzoate (ENO) in 200 ml of diglyme cooled at 0-5°C. Reaction mixture was stirred at room temperature for 2 hrs. Poured the reaction mass into ice cold water and adjusted the pH 5-6 with the addition of 50% (V/V) acetic acid solution. Reaction mass was stirred at 10-15°C for around 30 minutes. Again the pH was adjusted to 7 with the addition of sodium bicarbonate solution in water. Reaction mass was allowed to stir at room temperature for further 30 minutes. Reaction mass was extracted with ethyl acetate followed by the washing of ethyl acetate layer with brine solution. Distilled off ethyl acetate under reduced pressure in order to obtain 50 gm of 1 - (3- benzoyloxypropyl)-5-(2-nitropropyl) indoline as brown color oil.
Preparation of 3-(7-formyl-5-(2-nitropropyl)indolin-l-yl)propyl benzoate (Stage-5):
Phosphorus oxychloride (31.6 ml, 0.34 moles) was added dropwise over a period of about 15 minutes to 55 ml of dry Ν,Ν-dimethylformamide at 0-5°C and the mixture was allowed to stir for 30 minutes. Then to this mixture 50 gm (0.135 moles) of 1 - (3- benzoyloxypropyl)-5-(2-nitropropyl) indoline dissolved in 50 ml of Ν,Ν-dimethylformamide was added drop wise. Raised the temperature to 50°C and maintained to stir at same temperature for 4-5 hours. Cooled the reaction mass to room temperature and poured into ice water. Adjusted the pH of reaction mixture to 7 by addition of sodium carbonate solution. The reaction mixture was extracted with ethyl acetate. Distilled off ethyl acetate under reduced pressure and 100 ml methanol was added to obtained semi-solid. Raised the temperature to 60- 65°C and stirred the reaction mass for 20-30 minutes. The reaction mass was
gradually cooled to room temperature. The resulting solid was filtered and dried at 45°C to give 34g of 3-(7-formyl-5-(2-nitropropyl)indolin-l-yl)propyl benzoate as light green colored crystals.
Preparation of 1 -(3-Benzoyloxypropyl)-7-cyano-5-(2-nitropropyl) indoline (Stage-6):
1 -(3-Benzoxyloxypropyl)-7-formyl-5-(2-nitropropyl) indoline (50 g, 0.126 moles) was dissolved in 70 ml of dry tetrahydrofuran. 13. lg (0.188 moles) of hydroxylamine hydrochloride and 20.3 ml (0.252 moles) of pyridine was added to it at room temperature and the mixture was allowed to stir at 50°C for 2 hours. Cooled the reaction mass to 15-20°C and acetic anhydride (52.5 ml, 0.558 moles) was added drop wise. Raised the temperature of reaction mass to 70°C and maintained for 4 hours. Reaction mixture was cooled to room temperature. Water (500 ml) was added and the mixture was extracted with toluene, followed by the washing of toluene layers with 1% HC1 solution in water, saturated sodium bicarbonate solution and saturated salt water respectively. Concentrated the toluene layer under reduced pressure to obtained 48 g 1 -(3-Benzoyloxypropyl)-7- cyano-5-(2-nitropropyl) indoline as dark brown oil. This crude residue has been used further for next stage without any purification.
Preparation of 3-(5-(2-aminopropyl)-7-cyanoindolin-l-yl) propyl benzoate (Stage-7):
Mixture of 50 g (0.127 moles) of 1 -(3-Benzoyloxypropyl)-7-cyano-5-(2- nitropropyl) indoline in 150 ml ethyl acetate and 300 ml methanol was hydrogenated over 5% Pd/C (5 g) at 50-55°C and a pressure of 12-15 Kg in the presence of 15 ml of 25% liq. ammonia solution till complete consumption of nitro compound. After the completion of reaction, reaction mixture was passed through hyflo bed and washed with 100 ml methanol. Distilled off the solvent under reduced pressure to obtain 40 g of 3-(5-(2-aminopropyl)-7-cyanoindolin-l-
yl) propyl benzoate (±ACB) as brown colored oil which have been used directly for next stage.
Preparation of 5-[(2R)-2-Aminopropyl]-l-[3-(benzoyloxy) propyl] -2,3- dihydro-lH-indole-7-carbonitrile (2R,3R)-2,3-dihydroxybutanedioate (Stage- 8 & 9):
To the solution of 11.4 g (0.076 moles) L-tartaric acid in 50 ml water, solution of 50 g (0.137 moles) of amine in 150 ml of acetone was added drop wise at 50- 55°C. After complete addition, reaction mixture was heated to 60-65°C and allowed to stir for 30 minutes. Cooled the reaction mixture to room temperature and stirred for 12 hrs. The resulting solid was filtered and washed with solvent mixture (20 ml) of acetone and water (1 : 1) to give 27.5 g of crude 5-[(2R)-2- Aminopropyl]-l-[3-(benzoyloxy) propyl] -2,3-dihydro-lH-indole-7-carbonitrile (2R,3R)-2,3-dihydroxybutanedioate (ACB-tartrate salt). Purification of above salt was carried out by dissolving 27.5 g (0.053 moles) of crude solid in solvent mixture (343 ml) of acetone and water (1 : 1) at 60-65°C. After 30 minutes cooled the reaction mass to room temperature and stirred for 12 hours. The resulting solid was filtered and washed with solvent mixture (14 ml) of acetone and water (1 : 1) to give 20 g of ACB-tartrate salt with chiral purity 90%. Repeated the purification process of the solid obtained to give 12-13 g of ACB-tartrate salt with 95% ee.
The filtrate obtained after filtration of crude L-tartrate salt, was evaporated under reduced pressure to give 37.5 g of oily residue. This residue was dissolved in 225 ml of dichloromethane and 375 ml of water was added to it. Then 113 ml of 10% sodium bicarbonate solution was added to the resulting mixture and allowed to stir at room temperature for 10-15 minutes. Organic layer was separated and distilled out under reduced pressure to give 30 g oily mass. This oily mass was dissolved in 90 ml acetone and added drop wise to the solution of D (-) Tartaric acid (6.8 g) in water (30 ml) at room temperature. Raised the temperature to 65°C and stirred the reaction mass for 10-15 minutes. Gradually cooled the reaction mixture to room
temperature and allowed to stir at the same temperature for 10-12 hours. The solid precipitated was filtered and washed with 30 ml acetone to yield 28 g of crude tartrate salt of "S" isomer. Solvent mixture (518 ml) of acetone and water (1 : 1) was added to the crude tartrate salt and heated to 65-70°C. The reaction mass was stirred at the same temperature for 20-25 minutes. Gradually cooled the reaction mixture to room temperature and stirred for 10-12 hours. The solid obtained was filtered and washed with 14 ml of acetone. Obtained solid was dried at 45-50°C for 8-10 hours to afford 12.5 g of tartrate salt of "S" isomer with ee of 93%. This "S" isomer was directly used for the inversion as described in Method II. But for method I, the obtained salt was dissolved in 125 ml of dichloromethane and to this 100 ml of 10% NaHC03 solution was added at room temperature. After 30 minutes both the organic and aqueous layers were separated. Organic layer was washed with 62 ml water and distilled off under reduced pressure. In this way 8 g of free base of "ACB"-S isomer was obtained which is used further for inversion as described in Method-I.
Preparation of 5-((R)-2-(2-(2-(2,2,2- trifluoroethoxy)phenoxy)ethylamino)propyl)-l-(3-hydroxypropyl)indoline-7- carbonitrile (Stage -10 & 11):
33.6 g of Potassium carbonate was added to the solution of ACB-tartrate salt (50 g), TFS (33.6g) and potassium iodide (1.25 g) in acetonitrile at room temperature. The reaction mixture was heated to 90°C and stirred for 35-40 hours. After completion of reaction, cooled the reaction mixture to 60°C and filtered it through Buckner funnel. Obtained filtrate was distilled off under reduced pressure at 85°C. Oily mass thus obtained was dissolved in toluene (250 ml) followed by the addition of 8 g of NaOH and 25 ml of methanol. The reaction mixture was heated to 50-55°C and stirred at this temperature for 3-4 hours. Cooled the reaction mixture to room temperature and 200 ml of water was added. Both organic and aqueous layers were separated. Aqueous layer thus obtained was extracted twice with toluene. All the toluene layers were combined and 350 ml of aqueous acetic
acid solution was added. The reaction mixture was stirred at room temperature for 30 minutes. Aqueous acetic acid layer and toluene layers were separated. Dimer impurity formed in the process removed in this toluene layer. 200 ml of fresh toluene was added to obtained aqueous acetic acid layer. pH of this solution was adjusted to 11 by the drop wise addition of NaOH solution. Both the organic and aqueous layers were separated and the aqueous layer was extracted twice with toluene. Combined the toluene layers and distilled off under reduced pressure to give 38 g of 5-((R)-2-(2-(2-(2,2,2-trifluoroethoxy)phenoxy)ethylamino)propyl)-l- (3-hydroxypropyl)indoline-7-carbonitrile (HYD) as an oily mass.
Preparation of Si!odosm (Stage-ll):
4.2 g (0.105 moles) of Sodium hydroxide solution was added to the mixture of HYD (50 g, 0.104 moles) and DMSO (250 ml) at room temperature. Cooled the reaction mixture to 15-20°C and 14 g of 30% H202 was added drop wise. The reaction mixture was stirred at room temperature for 6-7 hours. After completion of reaction, the reaction mixture was cooled to 20°C and quenched with sodium sulphite solution. 256 ml of water was added to the reaction mixture and extracted twice with ethyl acetate. Combined the ethyl acetate layers and washed with sodium bicarbonate and brine solution respectively. Distilled out 423 ml of ethyl acetate layer and further charged 256.5 ml of ethyl acetate to obtained residue. This reaction mixture was gradually heated to 70-75°C and stirred at this temperature for 10-15 minutes. The reaction mass was cooled to room temperature and allowed to stir for 3 hours. The resulting solid was filtered and washed twice with ethyl acetate. Dissolved the obtained solid in 192 ml of ethyl acetate at 70-75°C and stirred at same temperature for 10-15 minutes. Cooled the reaction mixture to room temperature and again allowed to stir for 3-4 hours. The resulting solid was filtered and washed with 51 ml of ethyl acetate. The solid was dried at 40°C to yield 35.2 g of pure Silodosin. Polymorph form of this was confirmed as alpha form.
Inversion of unwanted "S" isomer into required "R" isomer (ACB): 5- [(2R)- 2-Aminopropyl]-l-[3-(benzoyloxy) propyl] -2,3-dihydro-lH-indole-7- carbonitrile
Method 1
Preparation of [3-{7-cyano-5-[(2S)-2-{N-[(4-methylbenzene)sulfonyl](4- methylbenzene)sulfonamido}propyl]-2,3-dihydro-lH-indol-l-yl}propyl benzoate]] (Stage-I):

To a mixture of sodium hydride (1.93 g, 0.08 moles) and tetrahydrofuran (24 ml), solution of free base (3.0 g, 0.008 moles) of "ACB-S" isomer as obtained at stage- 9 in THF (24 ml) was added drop wise at 0-5°C. The reaction mixture was stirred at same temperature for 10-15 minutes, /^-toluene sulfonyl chloride (6.30 g, 0.033 moles) dissolved in THF (15 ml) was added drop wise at 0-5°C. Raised the temperature of reaction mixture to 65-70°C and allowed to stir for 6-8 hours. After completion of reaction, cooled the reaction mass to room temperature and methanol (30 ml) was added drop wise. Water (30 ml) was added to the reaction mixture and desired product was extracted with dichloromethane. Solvent was distilled off under reduced pressure and 6.2 g of crude product was obtained which was purified by column chromatography to give 2.0 g of required ditosylated product (NTS) as a light yellow solid.
Preparation of [3-{5-[(2R)-2-azidopropyl]-7-cyano-2,3-dihydro-lH-indol-l- yl} propyl benzoate]] (Stage-

Sodium azide (3.18 g, 0.048 moles) was added to the mixture of NTS (4.0 g, 0.006 moles) and sodium iodide (0.106 g, 0.0007 moles) in N,N- dimethylformamide (40 ml) at room temperature. The reaction mixture was gradually heated to 120°C and stirred for 8-10 hours. The reaction was monitored by TLC (30% ethyl acetate in hexane). The reaction mixture was cooled to room temperature and water (100 ml) was added. The reaction mixture was extracted with dichloromethane. The solvent was evaporated under reduced pressure to give 2.34 g of ACZ as a semisolid.
Preparation of 5-[(2R)-2-Aminopropyl]-l-[3-(benzoyloxy) propyl] dihydro-lH-indole-7-carbonitrile (Stage-Ill):

2 g (0.005 moles) of ACZ was dissolved in 20 ml of methanol and hydrogenated over 10%) Pd/C (0.4 g) at room temperature and a pressure of 7.0 Kg. Reaction was monitored on TLC (100% MDC). After the complete consumption of ACZ, reaction was stopped and the reaction mixture was passed through hyflow. The
solvent was evaporated under reduced pressure to give 1.54 g of "R" isomer of ACB free base with ee of 90%.
Preparation of 5-[(2R)-2-Aminopropyl]-l-[3-(benzoyloxy) propyl] -2,3-dihydro- lH-indole-7-carbonitrile (2R,3R)-2,3-dihydroxybutanedioate (Stage-IV):
Solution of ACB free base (1.5 g, 0.0041 moles) in acetone (4.5 ml) was added drop wise to the mixture of L (+) Tartaric acid (0.34 g, 0.0022 moles) in water (1.5 ml) at 50-55°C. The reaction mixture was heated until a clear solution is obtained. Cooled the reaction mixture to room temperature and stirred for 12 hours. The solid precipitated out was filtered and washed with chilled acetone (0.75 ml). The obtained solid was dried at 40-45°C to yield 0.7 g of ACB with ee of 97%.
Method 2
Preparation of 2,4,6-Triphenylpyrylium tetrafluoroborate (Stage-I)

BF4-
BF3.ethereate (36 ml, 0.291 moles) was added drop wise to the solution of benzaldehyde (10 g, 0.094 moles) and acetophenone (34 g, 0.283 moles) at 5- 10°C and stirred the reaction mass for 10-15 minutes. Gradually raised the temperature to 80°C and continued to stir at the same temperature for 7-8 hours. Reaction was monitored by TLC (20% ethyl acetate in hexane). After complete consumption of benzaldehyde, heating was stopped and the mixture was cooled to 5-10°C. Ethanol (90 ml) was added drop wise to the reaction mixture and was allowed to stir at the same temperature for 2-3 hours. Obtained solid was filtered and washed with 10 ml ethanol. The solid was dried at 70-80°C to afford 15 g of pyrylium salt.
Preparation of [l-[(2S)-l-{l-[3-(benzoyloxy)propyl]-7-cyano-2,3-dihydro-lH- indol-5-yl}propan-2-yl]-2,4,6-triphenylpyridin-l-ium]] tetra fluoroborate (Stage-II):

Tartrate salt (11 6g, 0.022 moles) of "ACB"-S isomer as obtained at stage-9 was added to the mixture of pyrylium salt (9 g, 0.022 moles) in 50 ml of dichloromethane. Triethylamine (7.8 ml, 0.0567 moles) was added to the reaction mixture followed by the addition of dichloromethane (50 ml). The reaction mixture was stirred at room temperature for 40-45 minutes. To this reaction mixture acetic acid (3.2 ml, 0.0567 moles) was added and the reaction mixture was allowed to stir at room temperature for 10-12 hours. Monitoring of reaction was done on TLC (5% methanol in MDC). After the completion of reaction, 20% sodium carbonate (20ml) was added to the reaction mixture to adjust the pH 7. Extract the mixture with dichloromethane and all the organic layers were dried over sodium sulfate. Dichloromethane was distilled off under reduced pressure to give 15 g of crude product which was purified by Column chromatography to obtain 8.6 g of pyridinium salt as desired product.
Preparation of 5-[(2R)-2-Aminopropyl]-l-[3-(benzoyloxy) propyl] -2,3- dihydro-lH-indole-7-carbonitrile (Stage-Ill):

2.6 g (0.04 moles) of sodium azide was added to the mixture of above obtained pyridinium salt (7g, 0.01 moles) in 20 ml of N, N-dimethylformamide at room temperature. The mixture was heated to 80°C and stirred for 3-4 hours. Reaction was monitored on TLC (10% ethyl acetate in hexane). After the complete conversion of pyridinium salt into azide, heating was stopped. The reaction mixture was cooled to room temperature and extracted with ethyl acetate. The ethyl acetate was distilled off under reduced pressure to yield 8 g of crude product. Purification was carried out by column chromatography and 1.5 g of required compound "ACZ" was obtained as sticky mass.
ACB (tartrate salt of "R" isomer) was obtained from this ACZ with the same procedure as described in Method I, Stage-Ill and IV.
Preparation of 2- [2-(2,2,2-trifluoroethoxy)phenoxyl ethyl methanesuloonate
(TFS).
2,2,2- Trifluoroethyl methanesulfonate (Stage-1):
(50 g, 0.499 moles) trifluoroethanol was added to 200 ml of methylene dichloride. Mixture was cooled to 0-5°C and 77 ml (0.55 moles) of triethylamine was added drop wise over a period of 30 minutes followed by the drop wise addition of 42.8 ml (0.31 moles) methanesulfonyl chloride over a period of 90 minutes at same temperature. After completion of reaction (GC analysis), mixture was filtered and
washed with methylene dichloride. Obtained methylene dichloride was washed with water. Methylene Dichloride was distilled off under reduced pressure to give
85.3 g of 2,2,2- Trifluoroethyl methanesulfonate as pale yellow oil.
Monopotassium salt of catechol (Stage-2):
78.4 g (0.57 moles) of potassium carbonate was added to the solution of (50 g, 0.45 moles) catechol in 250 ml ethyl acetate at room temperature. The reaction mixture was heated to 90°C and allowed to stir at same temperature until the TLC (50% Ethyl Acetate in Hexane) showed almost complete consumption of catechol. The reaction mixture was cooled to 50°C and filtered. Obtained solid was washed with hot ethyl acetate and suck dried for around 30 minutes to yield 127.4 g of mixture of monopotassium and dipotassium salt of catechol as light green colored solid.
2-(2,2,2-Trifluoroethoxy)phenol (Stage-3):
80 g (0.45 moles) of 2,2,2- Trifluoroethyl methanesulfonate was added to a solution of 127.4 g of mixture of potassium salt of catechol and 0.8 g (0.005 moles) of sodium iodide in 150 ml of N, N-Dimethylformamide at room temperature. The reaction mixture was stirred at 140°C for 5-6 hrs. After the completion of reaction, DMF was distilled out under reduced pressure and thus the residue obtained was dissolved in 400 ml of water and 240 ml of toluene. Both organic and aqueous layers were separated and obtained aqueous layer was again extracted with toluene. Combined all the toluene layers and washed with water. Toluene layer was then extracted with IN NaOH solution. pH of the NaOH layer was adjusted in the range of 3-4 with the addition of 50% HCl solution in water at 0-5°C. The resulting mixture was allowed to stir for 30 minutes at the same temperature. Solid was precipitated out which was filtered and washed with water. Suck dried the obtained solid to give 31.9 g of 2-(2,2,2-Trifluoroethoxy)phenol as brown colored solid.
2-[2-(2,2,2- Trifluoroethoxy)phenoxy] ethanol (Stage-4):
71.9 g (0.52 moles) of potassium carbonate was added to the mixture of 50 g (0.26 moles) of 2-(2,2,2-Trifluoroethoxy)phenol and 30.9 ml (0.39 moles) of 2- choroethanol in 150 ml of ethanol. The reaction mixture was stirred for 4 hrs at 70°C. Once the reaction is completed, reaction mixture was cooled to room temperature. Reaction mass was filtered and washed with ethanol. Ethanol was distilled out under reduced pressure and the residue obtained was dissolved in 150 ml of methylene dichloride. Organic layer was washed with brine solution and then distilled out under reduced pressure to give 55.3 g of 2-[2-(2,2,2- Trifluoroethoxy)phenoxy]ethanol as brown colored oil.
2-[2-(2,2,2-Trifluoroethoxy)phenoxy]ethyl Methanesulfonate (TFS) (Stage-5):
36.9 ml (0.26 moles) of triethylamine was added to 50 g (0.21 moles) of 2-[2- (2,2,2- Trifluoroethoxy)phenoxy] ethanol dissolved in 150 ml methylene dichloride at room temperature. To this mixture, 20.6 ml (0.26 moles) of methanesulfonyl chloride was added drop wise over a period of 90 minutes at 0- 5°C. Until the reaction is completed, the reaction mixture was allowed to stir at room temperature. Filtered off the reaction mass and washed with methylene dichloride. Obtained Organic layer was washed with water followed by 2% Sodium bicarbonate solution. Methylene dichloride was distilled off under reduced pressure and the oily mass obtained was dissolved in 50 ml of isopropyl alcohol and stirred at room temperature for 30 minutes. Obtained solid was filtered and washed with 25 ml of chilled Isopropyl alcohol. Suck dried the solid to obtained 61.2 g of crude 2-[2-(2,2,2-Trifluoroethoxy)phenoxy]ethyl Methanesulfonate (TFS) as brown colored solid. Purification of crude "TFS" with 50 ml of methanol give 53.2 g of pure "TFS" as white crystals.
Claims
claim;
A recovery and recycling process for preparation of an opposite enantiomer or mixture of non-racemic mixture of enantiomers of Formula- I wherein Rl denotes H, alkyl group, cycloalkyl, hydroxy propyl, phenylmethoxy propyl, and benzoyloxy propyl; R2 denotes CN, halogen and aminocarbonyl (-CONH2) comprising;
reacting S-enantiomer or enantiomerically enriched S-enantiomer of Formula-I with a chemical agent to obtain compound of Formula- VI, wherein LG is a leavin group;

S-enanatiomer S-Enanatiomer of
of Formula-I Formula-VI
b) reacting the compound of Formula-VI with metal azide to obtain azide of
Formula-II and

S-Enanatiomer of R-Azide of
Formula-vl Formula-II
c) reducing the azide of Formula -II with a reducing agent to obtain R- enantiomer or mixture of R and S -enantiomers of Formula-I.

R-Azide of R-Enanatiomer of
Formula-II Formula-I
OR
a) reacting R-enantiomer or enantiomerically enriched R-enantiomer of Formula-I with a chemical agent to obtain compound of Formula- VI, wherein LG is a leavin group;

R-enanatiomer R-Enanatiomer of
of Formula-I Formula-VI
b) reacting the compound of Formula-VI with metal azide to obtain azide of Formula-II and

R-Enanatiomer of S-Azide of
Formula-vl Formula-II
c) reducing the azide of Formula -II with a reducing agent to obtain S-enantiomer or mixture of S and R-enantiomers of Formula-I.

S-Azide of S-Enanatiomer of
Formula-II Formula-I
2) The process according to claim 1, wherein the chemical agent is a sulfonyl agent or pyrylium salt.
3) The process according to claim 1, wherein the of azide of formula -II is prepared by a process comprising;
a) reacting unwanted opposite enantiomer or non-racemic mixture of Formula-I wherein Rl denotes H, alkyl group, cycloalkyl, hydroxy propyl, phenylmethoxy propyl, and benzoyloxy propyl; R2 denotes CN, halogen and aminocarbonyl (- CONH2) with a sulfonyl agent to obtain compound of Formula-Ill

Formula - III
wherein Rl denotes H, alkyl group, cycloalkyl, hydroxyl propyl, phenylmethoxy propyl, and benzoyloxy propyl; R2 denotes CN, halogen and aminocarbonyl (- CONH2); Y denotes methane, p-toluene, p-bromobenzene and p-nitrobenzene; b) reacting the compound of Formula-Ill with metal azides to obtain azide of Formula-II with inversion.
4) The process according to claim 3, wherein the sulfonyl agents are selected from group consisting of methanesulfonyl chloride, p-toluenesulfonyl chloride, p-bromobenzene sulfonylchloride and p-nitrobenzene sulfonyl chloride.
5) The process according to claim 3, wherein the sulfonation reaction of step -a) is performed in a solvent selected from group consisting of aliphatic or aromatic hydrocarbons, chlorinated hydrocarbons, esters, ethers, ketones, polar aprotic solvents, nitriles and mixtures thereof.
6) The process according to claim 5, wherein the ether solvent is
tetrahydrofuran.
7) The process according to claim 3, wherein the sulfonation reaction of step -a) is performed in presence of base selected from sodium hydride or potassium hydride.
8) The process according to claim 1, wherein the of azide of formula -II is prepared by a process comprising;
a) reacting unwanted opposite enantiomer or non-racemic mixture of Formula-I wherein Rl denotes H, alkyl group, cycloalkyl, hydroxy propyl, phenylmethoxy propyl, and benzoyloxy propyl; R2 denotes CN, halogen and aminocarbonyl (- CO H2) with a pyrylium salts to obtain the compounds of pyndinium salts of Formula - V

Formula -V
wherein Rl denotes H, alkyl group, cycloalkyl, hydroxyl propyl, phenylmethoxy propyl, and benzoyloxy propyl; R2 denotes CN, halogen and aminocarbonyl; R3 denotes H, alkyl, aryl, sustituted aryl and pyridyl; X" denotes tetrafluroborates (BF4 "), perchlorates (CIO4") and trifluoromethane sulfonate(CF3SCb"); b) reacting the compound of Formula-V with metal azides to obtain azide of Formula-II with inversion.
9) The process according to claim 1, 3 or 8, wherein, the metal azides are selected from the group consisting of sodium azide, potassium azide, calcium azide, and magnesium azide.
10) The process according to claim 1, 3 or 8, wherein the azide formation reaction is performed in presence of a catalyst selected from the group consisting of sodium bromide, sodium iodide, potassium bromide & potassium iodide.
11) The process according to claim 1, 3 or 8, wherein the azide formation reaction is performed in a solvent selected from the group consisting of water, alcohols, esters, aromatic hydrocarbons, nitriles and polar aprotic solvents and mixtures thereof.
12) The process according to claim 11, wherein the polar aprotic solvent is dimethyl sulfoxide, dimethyl acetamide or dimethyl formamide.
13) The process according to claim 8, wherein the pyrylium

X
wherein R3 denotes H, alkyl, aryl, substituted aryl and pyridyl; X" denotes tetraflurob orates (BF4 "), perchlorates (C104 ") and trifluoromethane sulfonate(CF3S03-).
14) The process according to claim 13, wherein the pyrylium salt is 2,4,6- Triphenylpyry Hum tetrafluorob orate .
15) The process according to claim 8, wherein pyridinium salt-V formation of step a) is conducted in presence of solvent, a base and a catalyst.
16) The process according to claim 15, wherein the solvent is selected from the group consisting of hydrocarbons, chlorinated hydrocarbons, esters, ethers, ketones, polar aprotic solvents, nitriles and mixtures thereof.
17) The process according to claim 16, wherein the chlorinated hydrocarbon solvent is methylene dichloride or ethylene dichloride.
18) The process according to claim 15, wherein the base is selected from the group consisting of triethyl amine, diethylamine, methyl amine, diisopropyl ethylamine, and pyridine.
19) The process according to claim 15, wherein the catalyst is selected from the group consisting of formic acid, acetic acid and propionic acid.
20) The process according to claim 1, wherein the reducing agent is selected from the group consisting of hydrogen in presence of metal catalysts, ammonium formate, tin, tinchloride, hydrazine, iron and iron chloride, zinc, formic acid, hydrogen sulfide, sodium sulfide and titanium chloride.
21) The process according to claim 1, wherein the reducing agent is hydrogen in presence of palladium on carbon catalyst.
22) The process according to claim 1, wherein the reduction is performed in a solvent selected from the group consisting of water, alcohols, esters, aromatic hydrocarbons and polar aprotic solvents or mixtures thereof.
23) The process according to claim 22, wherein the alcohol is methanol or ethanol.
24) A recycling process for the preparation of Silodosin or pharmaceutically acceptable salts thereof comprising;
a) reacting an S-enantiomer or enantiomerically enriched S-enantiomer of Formula-I with a chemical agent to obtain compound of Formula- VI, wherein LG is a leaving group;
b) reacting the compound of Formula- VI with metal azide to obtain azide of Formula-II;
c) reducing the azide of Formula -II with a reducing agent to obtain R- enantiomer of Formula-I or mixture of R and S enantiomers of Formula-I wherein Rl denotes H, alkyl group, cycloalkyl, hydroxy propyl, phenylmethoxy propyl, and benzoyloxy propyl; R2 denotes CN, halogen and aminocarbonyl (-CO H2); and
d) converting the R-enantiomer or mixture of the R and S enantiomers of Formula-I into silodosin by reacting with 2- [2-(2,2,2- trifluoroethoxy)phenoxy] ethyl methanesulfonate.
25) The process according to claim 24, wherein the S-enantiomer of Formula-I is prepared by reducing 2-nitropropyl indoline of Formula-VII with hydrogen in presence of Pd/C or Raney nickel catalyst and optionally in presence of ammonia or tri ethyl amine.
26) The process according to claim 24, wherein the 2- [2-(2,2,2- trifluoroethoxy)phenoxy] ethyl methanesulfonate is prepared by reacting monopotassium salt of catechol with 2,2,2- Trifluoroethyl methanesulfonate.
27) The process according to claim 26, wherein the potassium salt of catechol is prepared by a process comprising reacting catechol with potassium hydroxide followed by isolation of monopotassium salt of catechol.
28) Compounds of Formula-II, Formula-Ill, Formula-IV and Formula- V, wherein Rl denotes H, alkyl group, cycloalkyl, hydroxyl propyl, phenylmethoxy propyl, and benzoyloxy propyl; R2 denotes CN, halogen and aminocarbonyl (-CO H2); Y denotes methane, p-toluene, p- bromobenzene and p-nitrobenzene; R3 denotes H, alkyl, aryl, sustituted aryl and pyridyl; X" denotes tetrafluroborates (BF4 "), perchlorates (C104 ") and trifluoromethane sulfonate(CF3SCb")-

Formula-II Formula-Ill Formula-IV
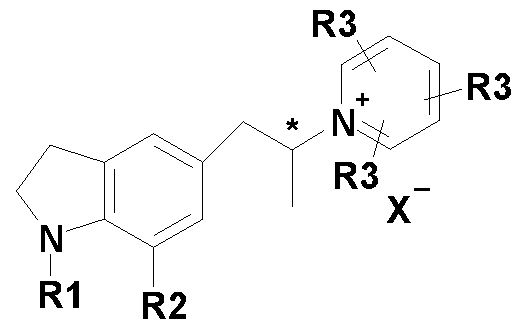
Formula-V
29) A compound according to claim 28 selected from group consisting of a) [3-{5-[2-azidopropyl]-7-cya -2,3-dihydro-lH-indol-l-yl}propyl benzoate]]

b) [3-{7-cyano-5-[2-{N-[(4-methylbenzene)sulfonyl](4- methylbenzene)sulfonamido}propyl]-2,3-dihydro-lH-indol
benzoate]]

c) [3-{7-cyano-5-[-2-[(4-methylbenzene)sulfonamido]propyl]-2,3-dihydro-lH- indol-l-yl} propyl benzoate]]

and
d) [l-[(l-{ l-[3-(benzoyloxy)propyl]-7-cyano-2,3-dihydro-lH-indol-5-yl}propan- 2-yl]-2,4,6-triphenylpyridin-l-ium]] tetra fluoroborate

Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN2047MU2015 | 2015-05-26 | ||
| IN2047/MUM/2015 | 2015-05-26 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2016189552A2 true WO2016189552A2 (en) | 2016-12-01 |
| WO2016189552A3 WO2016189552A3 (en) | 2017-02-09 |
Family
ID=57392212
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2016/050156 WO2016189552A2 (en) | 2015-05-26 | 2016-05-26 | Novel recovery and recycling process of unwanted enantiomers of 2-aminopropyl indoline derivatives |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2016189552A2 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106928118A (en) * | 2017-04-11 | 2017-07-07 | 常州瑞明药业有限公司 | A kind of method for preparing Silodosin intermediate |
| CN108640870A (en) * | 2018-04-12 | 2018-10-12 | 常州大学 | A method of synthesis 6- bromomethyl -3- methoxyl group -2- nitropyridines |
| CN108929259A (en) * | 2018-08-29 | 2018-12-04 | 四川青木制药有限公司 | A method of preparing Silodosin intermediate |
| KR20190016208A (en) * | 2017-08-08 | 2019-02-18 | (주)헥사파마텍 | Novel sulfonamide intermediates and processes for preparing silodosin using the same |
| US10421719B2 (en) | 2015-09-30 | 2019-09-24 | Urquima S.A. | Maleic acid salt of a silodosin intermediate |
| CN114478202A (en) * | 2022-02-16 | 2022-05-13 | 江苏飞宇医药科技股份有限公司 | Method for continuously preparing 2- (2,2, 2-trifluoroethoxy) phenol |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150038727A1 (en) * | 2011-10-21 | 2015-02-05 | Sandoz Ag | Method for preparing silodosin |
-
2016
- 2016-05-26 WO PCT/IN2016/050156 patent/WO2016189552A2/en active Application Filing
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10421719B2 (en) | 2015-09-30 | 2019-09-24 | Urquima S.A. | Maleic acid salt of a silodosin intermediate |
| CN106928118A (en) * | 2017-04-11 | 2017-07-07 | 常州瑞明药业有限公司 | A kind of method for preparing Silodosin intermediate |
| CN106928118B (en) * | 2017-04-11 | 2022-08-23 | 常州瑞明药业有限公司 | Method for preparing silodosin intermediate |
| KR20190016208A (en) * | 2017-08-08 | 2019-02-18 | (주)헥사파마텍 | Novel sulfonamide intermediates and processes for preparing silodosin using the same |
| KR102365411B1 (en) | 2017-08-08 | 2022-02-21 | (주)헥사파마텍 | Novel sulfonamide intermediates and processes for preparing silodosin using the same |
| CN108640870A (en) * | 2018-04-12 | 2018-10-12 | 常州大学 | A method of synthesis 6- bromomethyl -3- methoxyl group -2- nitropyridines |
| CN108929259A (en) * | 2018-08-29 | 2018-12-04 | 四川青木制药有限公司 | A method of preparing Silodosin intermediate |
| CN114478202A (en) * | 2022-02-16 | 2022-05-13 | 江苏飞宇医药科技股份有限公司 | Method for continuously preparing 2- (2,2, 2-trifluoroethoxy) phenol |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2016189552A3 (en) | 2017-02-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2016189552A2 (en) | Novel recovery and recycling process of unwanted enantiomers of 2-aminopropyl indoline derivatives | |
| JP6872500B2 (en) | How to make bribalacetam | |
| JP5632279B2 (en) | Preparation method and polymorph of ivabradine hydrochloride | |
| WO2016119574A1 (en) | Method for preparing sacubitril | |
| TW200808770A (en) | Novel processes for the preparation of DPP IV inhibitors | |
| BG66150B1 (en) | Intermediate compounds during the production of derivatives of the cyclopropylcarboxylic acid | |
| JP2818958B2 (en) | 4- (4-Alkoxyphenyl) -2-butylamine derivative and method for producing the same | |
| US11332460B2 (en) | Process for the preparation of a sulfonamide structured kinase inhibitor | |
| WO2020052545A1 (en) | Method for preparing brivaracetam and intermediates thereof | |
| US10370329B2 (en) | Process for the enantiomeric resolution of apremilast intermediates | |
| WO2015145467A1 (en) | An improved process for preparing vildagliptin | |
| JP2001502329A (en) | Method for producing 2-azadihydroxybicyclo [2.2.1] heptane compound and L-tartrate salt thereof | |
| WO2016042441A1 (en) | A novel process for the preparation of considerably pure silodosin | |
| US20180339964A1 (en) | The process of preparing indoline compounds and a novel indolinesalt | |
| WO2014118606A2 (en) | A novel process for the preparation of silodosin | |
| US10919855B2 (en) | Process to prepare n-[2-[(1s)-1-(3-ethoxy-4-methoxyphenyl)-2-(methylsulphonyl) ethyl]-1, 3-dioxo-2,3-dihydro-1h-isoindol-4yl]acetamide | |
| US10457640B2 (en) | Synthesis of inhibitors of EZH2 | |
| CZ2010997A3 (en) | Process for preparing (2R,3R)-N,N-dimethyl-3-(hydroxyphenyl)-2-methyl pentylamine (tapentadol) | |
| WO2012165607A1 (en) | Method for producing proline compound | |
| US7943784B2 (en) | Process for the preparation of almotriptan | |
| CA2660272A1 (en) | Process for making lactam tachykinin receptor antagonists | |
| WO2013090161A1 (en) | Stereoselective synthesis of tapentadol and its salts | |
| EP0489548B1 (en) | A process for the preparation of 2(R)-methyl-4,4,4-trifluorobutylamine, intermediates, and a process for the preparation of a derivative thereof | |
| JP3902384B2 (en) | Method for purifying optically active α-methyl-bis-3,5- (trifluoromethyl) benzylamines | |
| KR102163068B1 (en) | The manufacturing method of intermediate for synthesis of silodosin and the manufacturing method of silodosin |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16799491 Country of ref document: EP Kind code of ref document: A2 |