WO2015165930A1 - Enantiomere des n-(2-amino-5-fluor-2-methylpentyl)-8-[(2,6-difluorbenzyl)oxy]-2-methylimidazo[1,2-a]pyridin-3-carboxamids sowie der di- and tri-fluor-derivate zur behandlung von kardiovaskulären erkrankungen - Google Patents
Enantiomere des n-(2-amino-5-fluor-2-methylpentyl)-8-[(2,6-difluorbenzyl)oxy]-2-methylimidazo[1,2-a]pyridin-3-carboxamids sowie der di- and tri-fluor-derivate zur behandlung von kardiovaskulären erkrankungen Download PDFInfo
- Publication number
- WO2015165930A1 WO2015165930A1 PCT/EP2015/059274 EP2015059274W WO2015165930A1 WO 2015165930 A1 WO2015165930 A1 WO 2015165930A1 EP 2015059274 W EP2015059274 W EP 2015059274W WO 2015165930 A1 WO2015165930 A1 WO 2015165930A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- salts
- oxides
- solvates
- compound
- amino
- Prior art date
Links
- 238000011282 treatment Methods 0.000 title claims abstract description 46
- 208000024172 Cardiovascular disease Diseases 0.000 title abstract description 6
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 title description 11
- 238000000034 method Methods 0.000 claims abstract description 68
- 239000003814 drug Substances 0.000 claims abstract description 15
- 238000004519 manufacturing process Methods 0.000 claims abstract description 10
- 150000001875 compounds Chemical class 0.000 claims description 193
- 150000003839 salts Chemical class 0.000 claims description 67
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 61
- -1 2,3,6-trifluorobenzyl Chemical group 0.000 claims description 45
- 239000012453 solvate Substances 0.000 claims description 40
- 239000002904 solvent Substances 0.000 claims description 40
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 claims description 34
- 201000010099 disease Diseases 0.000 claims description 33
- 238000011321 prophylaxis Methods 0.000 claims description 32
- 239000003112 inhibitor Substances 0.000 claims description 19
- 125000006508 2,6-difluorobenzyl group Chemical group [H]C1=C([H])C(F)=C(C(F)=C1[H])C([H])([H])* 0.000 claims description 18
- 206010019280 Heart failures Diseases 0.000 claims description 17
- 239000012442 inert solvent Substances 0.000 claims description 17
- 235000005152 nicotinamide Nutrition 0.000 claims description 17
- 239000011570 nicotinamide Substances 0.000 claims description 17
- 239000002253 acid Substances 0.000 claims description 15
- 230000009897 systematic effect Effects 0.000 claims description 14
- 239000002585 base Substances 0.000 claims description 13
- 239000000460 chlorine Substances 0.000 claims description 13
- 206010020772 Hypertension Diseases 0.000 claims description 12
- 229910052801 chlorine Inorganic materials 0.000 claims description 12
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 11
- 238000002360 preparation method Methods 0.000 claims description 11
- 208000002815 pulmonary hypertension Diseases 0.000 claims description 11
- 241001465754 Metazoa Species 0.000 claims description 10
- 208000001647 Renal Insufficiency Diseases 0.000 claims description 10
- 230000008878 coupling Effects 0.000 claims description 10
- 238000010168 coupling process Methods 0.000 claims description 10
- 238000005859 coupling reaction Methods 0.000 claims description 10
- 201000006370 kidney failure Diseases 0.000 claims description 10
- 206010002383 Angina Pectoris Diseases 0.000 claims description 9
- 229910052739 hydrogen Inorganic materials 0.000 claims description 9
- 239000001257 hydrogen Substances 0.000 claims description 9
- 239000003795 chemical substances by application Substances 0.000 claims description 8
- 208000028867 ischemia Diseases 0.000 claims description 8
- 229910052757 nitrogen Inorganic materials 0.000 claims description 8
- 208000019553 vascular disease Diseases 0.000 claims description 8
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 7
- 150000007513 acids Chemical class 0.000 claims description 7
- 239000003146 anticoagulant agent Substances 0.000 claims description 6
- 230000036772 blood pressure Effects 0.000 claims description 6
- 230000008569 process Effects 0.000 claims description 6
- 206010003210 Arteriosclerosis Diseases 0.000 claims description 5
- 208000011775 arteriosclerosis disease Diseases 0.000 claims description 5
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 5
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 5
- 208000001435 Thromboembolism Diseases 0.000 claims description 4
- 239000004480 active ingredient Substances 0.000 claims description 4
- 150000001412 amines Chemical class 0.000 claims description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims description 4
- 239000003054 catalyst Substances 0.000 claims description 4
- 230000004060 metabolic process Effects 0.000 claims description 4
- 150000001408 amides Chemical class 0.000 claims description 3
- 229960004676 antithrombotic agent Drugs 0.000 claims description 3
- 231100000252 nontoxic Toxicity 0.000 claims description 3
- 230000003000 nontoxic effect Effects 0.000 claims description 3
- 229940082615 organic nitrates used in cardiac disease Drugs 0.000 claims description 3
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 2
- 229910052723 transition metal Inorganic materials 0.000 claims description 2
- 150000003624 transition metals Chemical class 0.000 claims description 2
- 230000002265 prevention Effects 0.000 abstract description 7
- ZMBYQTGAXZOMOO-UHFFFAOYSA-N imidazo[1,2-a]pyridine-3-carboxamide Chemical class C1=CC=CN2C(C(=O)N)=CN=C21 ZMBYQTGAXZOMOO-UHFFFAOYSA-N 0.000 abstract description 2
- 230000006806 disease prevention Effects 0.000 abstract 2
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 141
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 81
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 79
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 78
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 70
- 239000000243 solution Substances 0.000 description 62
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 60
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 50
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 45
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 42
- 238000005481 NMR spectroscopy Methods 0.000 description 41
- 239000003480 eluent Substances 0.000 description 40
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 35
- 239000000203 mixture Substances 0.000 description 35
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 34
- 238000006243 chemical reaction Methods 0.000 description 34
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 32
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 30
- 239000000126 substance Substances 0.000 description 30
- 239000011541 reaction mixture Substances 0.000 description 28
- 208000035475 disorder Diseases 0.000 description 27
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical class Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 26
- 239000012071 phase Substances 0.000 description 24
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 23
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 22
- 238000012360 testing method Methods 0.000 description 21
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 20
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 20
- 239000007787 solid Substances 0.000 description 20
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 19
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 18
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 16
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 16
- 239000012074 organic phase Substances 0.000 description 16
- 238000001514 detection method Methods 0.000 description 15
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 15
- 238000002953 preparative HPLC Methods 0.000 description 15
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 14
- 101000610640 Homo sapiens U4/U6 small nuclear ribonucleoprotein Prp3 Proteins 0.000 description 14
- 101001110823 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 60S ribosomal protein L6-A Proteins 0.000 description 14
- 101000712176 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 60S ribosomal protein L6-B Proteins 0.000 description 14
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 14
- 102100040374 U4/U6 small nuclear ribonucleoprotein Prp3 Human genes 0.000 description 14
- 239000000047 product Substances 0.000 description 14
- 229920006395 saturated elastomer Polymers 0.000 description 14
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 13
- 239000008346 aqueous phase Substances 0.000 description 13
- 230000015572 biosynthetic process Effects 0.000 description 13
- ZOOGRGPOEVQQDX-KHLHZJAASA-N cyclic guanosine monophosphate Chemical compound C([C@H]1O2)O[P@](O)(=O)O[C@@H]1[C@H](O)[C@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-KHLHZJAASA-N 0.000 description 13
- 229910052938 sodium sulfate Inorganic materials 0.000 description 13
- 235000011152 sodium sulphate Nutrition 0.000 description 13
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 12
- 230000000694 effects Effects 0.000 description 12
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 11
- GSNUFIFRDBKVIE-UHFFFAOYSA-N DMF Natural products CC1=CC=C(C)O1 GSNUFIFRDBKVIE-UHFFFAOYSA-N 0.000 description 11
- 239000007821 HATU Substances 0.000 description 11
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 11
- 102000007637 Soluble Guanylyl Cyclase Human genes 0.000 description 11
- 108010007205 Soluble Guanylyl Cyclase Proteins 0.000 description 11
- 235000019253 formic acid Nutrition 0.000 description 11
- 229910052763 palladium Inorganic materials 0.000 description 11
- 230000035699 permeability Effects 0.000 description 11
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 11
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 10
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 10
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 10
- 229910052786 argon Inorganic materials 0.000 description 10
- 210000004027 cell Anatomy 0.000 description 10
- 230000003176 fibrotic effect Effects 0.000 description 10
- 238000000746 purification Methods 0.000 description 10
- 235000017557 sodium bicarbonate Nutrition 0.000 description 10
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 10
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 239000005557 antagonist Substances 0.000 description 9
- 239000000706 filtrate Substances 0.000 description 9
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 9
- 238000010898 silica gel chromatography Methods 0.000 description 9
- AFABGHUZZDYHJO-UHFFFAOYSA-N 2-Methylpentane Chemical compound CCCC(C)C AFABGHUZZDYHJO-UHFFFAOYSA-N 0.000 description 8
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 8
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 8
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 8
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 8
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 8
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 8
- 239000007868 Raney catalyst Substances 0.000 description 8
- 229910000564 Raney nickel Inorganic materials 0.000 description 8
- 241000700159 Rattus Species 0.000 description 8
- 238000002474 experimental method Methods 0.000 description 8
- 208000017169 kidney disease Diseases 0.000 description 8
- 238000010992 reflux Methods 0.000 description 8
- 238000003786 synthesis reaction Methods 0.000 description 8
- 102000004190 Enzymes Human genes 0.000 description 7
- 108090000790 Enzymes Proteins 0.000 description 7
- 101001047090 Homo sapiens Potassium voltage-gated channel subfamily H member 2 Proteins 0.000 description 7
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 7
- 102100022807 Potassium voltage-gated channel subfamily H member 2 Human genes 0.000 description 7
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical class OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 7
- 229910021529 ammonia Inorganic materials 0.000 description 7
- 229940088598 enzyme Drugs 0.000 description 7
- 238000004128 high performance liquid chromatography Methods 0.000 description 7
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 7
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 7
- 239000002002 slurry Substances 0.000 description 7
- 239000011780 sodium chloride Substances 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- 239000003643 water by type Substances 0.000 description 7
- HSINOMROUCMIEA-FGVHQWLLSA-N (2s,4r)-4-[(3r,5s,6r,7r,8s,9s,10s,13r,14s,17r)-6-ethyl-3,7-dihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]-2-methylpentanoic acid Chemical compound C([C@@]12C)C[C@@H](O)C[C@H]1[C@@H](CC)[C@@H](O)[C@@H]1[C@@H]2CC[C@]2(C)[C@@H]([C@H](C)C[C@H](C)C(O)=O)CC[C@H]21 HSINOMROUCMIEA-FGVHQWLLSA-N 0.000 description 6
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 6
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 6
- 108010078321 Guanylate Cyclase Proteins 0.000 description 6
- 102000014469 Guanylate cyclase Human genes 0.000 description 6
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 6
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 6
- 230000001154 acute effect Effects 0.000 description 6
- 235000011114 ammonium hydroxide Nutrition 0.000 description 6
- 239000003613 bile acid Substances 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 238000011534 incubation Methods 0.000 description 6
- 239000002808 molecular sieve Substances 0.000 description 6
- 210000002381 plasma Anatomy 0.000 description 6
- 239000011591 potassium Substances 0.000 description 6
- 238000000926 separation method Methods 0.000 description 6
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 208000011580 syndromic disease Diseases 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- 230000009424 thromboembolic effect Effects 0.000 description 6
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 6
- 206010012289 Dementia Diseases 0.000 description 5
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 5
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical class CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 5
- 208000006011 Stroke Diseases 0.000 description 5
- 208000027418 Wounds and injury Diseases 0.000 description 5
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 230000037396 body weight Effects 0.000 description 5
- 239000012043 crude product Substances 0.000 description 5
- 230000006378 damage Effects 0.000 description 5
- 238000007257 deesterification reaction Methods 0.000 description 5
- 210000002216 heart Anatomy 0.000 description 5
- 150000004677 hydrates Chemical class 0.000 description 5
- 239000000543 intermediate Substances 0.000 description 5
- 230000000155 isotopic effect Effects 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 5
- 229910052700 potassium Inorganic materials 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 125000006239 protecting group Chemical group 0.000 description 5
- 235000015424 sodium Nutrition 0.000 description 5
- 238000000825 ultraviolet detection Methods 0.000 description 5
- JCKMSCHJSIGXNH-UHFFFAOYSA-N 6-chloro-8-[(2,6-difluorophenyl)methoxy]-2-methylimidazo[1,2-a]pyridine-3-carboxylic acid Chemical compound C=1C(Cl)=CN2C(C(O)=O)=C(C)N=C2C=1OCC1=C(F)C=CC=C1F JCKMSCHJSIGXNH-UHFFFAOYSA-N 0.000 description 4
- 201000001320 Atherosclerosis Diseases 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 description 4
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 4
- XKMLYUALXHKNFT-UUOKFMHZSA-N Guanosine-5'-triphosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O XKMLYUALXHKNFT-UUOKFMHZSA-N 0.000 description 4
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical group [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 4
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical class OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 208000004403 Prostatic Hyperplasia Diseases 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 4
- 150000001298 alcohols Chemical class 0.000 description 4
- 239000001569 carbon dioxide Substances 0.000 description 4
- 229910002092 carbon dioxide Inorganic materials 0.000 description 4
- 230000001684 chronic effect Effects 0.000 description 4
- 230000008602 contraction Effects 0.000 description 4
- DDRJAANPRJIHGJ-UHFFFAOYSA-N creatinine Chemical compound CN1CC(=O)NC1=N DDRJAANPRJIHGJ-UHFFFAOYSA-N 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 4
- 150000003278 haem Chemical class 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 235000001968 nicotinic acid Nutrition 0.000 description 4
- 239000011664 nicotinic acid Substances 0.000 description 4
- 230000002093 peripheral effect Effects 0.000 description 4
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 4
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 4
- 235000011181 potassium carbonates Nutrition 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 230000000638 stimulation Effects 0.000 description 4
- 239000006228 supernatant Substances 0.000 description 4
- YYGNTYWPHWGJRM-UHFFFAOYSA-N (6E,10E,14E,18E)-2,6,10,15,19,23-hexamethyltetracosa-2,6,10,14,18,22-hexaene Chemical compound CC(C)=CCCC(C)=CCCC(C)=CCCC=C(C)CCC=C(C)CCC=C(C)C YYGNTYWPHWGJRM-UHFFFAOYSA-N 0.000 description 3
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 3
- IEQAICDLOKRSRL-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-dodecoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO IEQAICDLOKRSRL-UHFFFAOYSA-N 0.000 description 3
- VUJSFQIUJVYUNE-UHFFFAOYSA-N 5-chloro-3-[(2,6-difluorophenyl)methoxy]pyridin-2-amine Chemical compound NC1=NC=C(Cl)C=C1OCC1=C(F)C=CC=C1F VUJSFQIUJVYUNE-UHFFFAOYSA-N 0.000 description 3
- 239000005541 ACE inhibitor Substances 0.000 description 3
- 102000015427 Angiotensins Human genes 0.000 description 3
- 108010064733 Angiotensins Proteins 0.000 description 3
- 102100040214 Apolipoprotein(a) Human genes 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- WCXSKRCUQWZGOR-UHFFFAOYSA-N C(#N)C(C)(CC(F)F)NC(OCC1=CC=CC=C1)=O Chemical compound C(#N)C(C)(CC(F)F)NC(OCC1=CC=CC=C1)=O WCXSKRCUQWZGOR-UHFFFAOYSA-N 0.000 description 3
- DOTHWLHXVOKARQ-UHFFFAOYSA-N C(#N)C(C)(CCCF)NC(OCC1=CC=CC=C1)=O Chemical compound C(#N)C(C)(CCCF)NC(OCC1=CC=CC=C1)=O DOTHWLHXVOKARQ-UHFFFAOYSA-N 0.000 description 3
- 229940127291 Calcium channel antagonist Drugs 0.000 description 3
- 241000282472 Canis lupus familiaris Species 0.000 description 3
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 3
- 0 Cc1c(C(O*)=O)[n](cc(*)cc2OCc(c(F)ccc3)c3F)c2n1 Chemical compound Cc1c(C(O*)=O)[n](cc(*)cc2OCc(c(F)ccc3)c3F)c2n1 0.000 description 3
- 201000003883 Cystic fibrosis Diseases 0.000 description 3
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 3
- 108010033266 Lipoprotein(a) Proteins 0.000 description 3
- 102100031545 Microsomal triglyceride transfer protein large subunit Human genes 0.000 description 3
- 208000009525 Myocarditis Diseases 0.000 description 3
- 208000008589 Obesity Diseases 0.000 description 3
- 206010030113 Oedema Diseases 0.000 description 3
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 3
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 3
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 3
- BHEOSNUKNHRBNM-UHFFFAOYSA-N Tetramethylsqualene Natural products CC(=C)C(C)CCC(=C)C(C)CCC(C)=CCCC=C(C)CCC(C)C(=C)CCC(C)C(C)=C BHEOSNUKNHRBNM-UHFFFAOYSA-N 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 229940083712 aldosterone antagonist Drugs 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 3
- 229940127218 antiplatelet drug Drugs 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 125000004429 atom Chemical group 0.000 description 3
- BZZSIZPHNGARLH-UHFFFAOYSA-N benzyl n-(2-cyano-5,5,5-trifluoropentan-2-yl)carbamate Chemical compound FC(F)(F)CCC(C)(C#N)NC(=O)OCC1=CC=CC=C1 BZZSIZPHNGARLH-UHFFFAOYSA-N 0.000 description 3
- 239000002876 beta blocker Substances 0.000 description 3
- 238000009530 blood pressure measurement Methods 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 150000001735 carboxylic acids Chemical class 0.000 description 3
- 230000000747 cardiac effect Effects 0.000 description 3
- 239000013553 cell monolayer Substances 0.000 description 3
- 239000003354 cholesterol ester transfer protein inhibitor Substances 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000003776 cleavage reaction Methods 0.000 description 3
- CYCGRDQQIOGCKX-UHFFFAOYSA-N dehydroluciferin Chemical compound OC(=O)C1=CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 CYCGRDQQIOGCKX-UHFFFAOYSA-N 0.000 description 3
- 206010012601 diabetes mellitus Diseases 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- PRAKJMSDJKAYCZ-UHFFFAOYSA-N dodecahydrosqualene Natural products CC(C)CCCC(C)CCCC(C)CCCCC(C)CCCC(C)CCCC(C)C PRAKJMSDJKAYCZ-UHFFFAOYSA-N 0.000 description 3
- 150000002170 ethers Chemical class 0.000 description 3
- RDULEYWUGKOCMR-UHFFFAOYSA-N ethyl 2-chloro-3-oxobutanoate Chemical compound CCOC(=O)C(Cl)C(C)=O RDULEYWUGKOCMR-UHFFFAOYSA-N 0.000 description 3
- 239000010408 film Substances 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 238000003304 gavage Methods 0.000 description 3
- 239000008103 glucose Substances 0.000 description 3
- 230000036541 health Effects 0.000 description 3
- 238000007327 hydrogenolysis reaction Methods 0.000 description 3
- 239000007943 implant Substances 0.000 description 3
- 238000002513 implantation Methods 0.000 description 3
- 208000014674 injury Diseases 0.000 description 3
- 229910052742 iron Inorganic materials 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 229910001629 magnesium chloride Inorganic materials 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- 235000020824 obesity Nutrition 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 239000008194 pharmaceutical composition Substances 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- 239000000106 platelet aggregation inhibitor Substances 0.000 description 3
- 230000002685 pulmonary effect Effects 0.000 description 3
- 208000005069 pulmonary fibrosis Diseases 0.000 description 3
- 230000009103 reabsorption Effects 0.000 description 3
- 229940044601 receptor agonist Drugs 0.000 description 3
- 239000000018 receptor agonist Substances 0.000 description 3
- 239000002461 renin inhibitor Substances 0.000 description 3
- 229940086526 renin-inhibitors Drugs 0.000 description 3
- 238000006798 ring closing metathesis reaction Methods 0.000 description 3
- 230000007017 scission Effects 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 238000011699 spontaneously hypertensive rat Methods 0.000 description 3
- 229940031439 squalene Drugs 0.000 description 3
- TUHBEKDERLKLEC-UHFFFAOYSA-N squalene Natural products CC(=CCCC(=CCCC(=CCCC=C(/C)CCC=C(/C)CC=C(C)C)C)C)C TUHBEKDERLKLEC-UHFFFAOYSA-N 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 238000004809 thin layer chromatography Methods 0.000 description 3
- 102000004217 thyroid hormone receptors Human genes 0.000 description 3
- 108090000721 thyroid hormone receptors Proteins 0.000 description 3
- METKIMKYRPQLGS-GFCCVEGCSA-N (R)-atenolol Chemical compound CC(C)NC[C@@H](O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-GFCCVEGCSA-N 0.000 description 2
- PAMIQIKDUOTOBW-UHFFFAOYSA-N 1-methylpiperidine Chemical compound CN1CCCCC1 PAMIQIKDUOTOBW-UHFFFAOYSA-N 0.000 description 2
- OFJRNBWSFXEHSA-UHFFFAOYSA-N 2-(3-amino-1,2-benzoxazol-5-yl)-n-[4-[2-[(dimethylamino)methyl]imidazol-1-yl]-2-fluorophenyl]-5-(trifluoromethyl)pyrazole-3-carboxamide Chemical compound CN(C)CC1=NC=CN1C(C=C1F)=CC=C1NC(=O)C1=CC(C(F)(F)F)=NN1C1=CC=C(ON=C2N)C2=C1 OFJRNBWSFXEHSA-UHFFFAOYSA-N 0.000 description 2
- QOZNHJKSURHSFT-UHFFFAOYSA-N 2-(benzhydrylideneamino)-4,4-difluoro-2-methylbutanenitrile Chemical compound C=1C=CC=CC=1C(=NC(CC(F)F)(C)C#N)C1=CC=CC=C1 QOZNHJKSURHSFT-UHFFFAOYSA-N 0.000 description 2
- QGHJJQCXOHXFCY-UHFFFAOYSA-N 2-(benzhydrylideneamino)-5-fluoro-2-methylpentanenitrile Chemical compound C=1C=CC=CC=1C(=NC(CCCF)(C)C#N)C1=CC=CC=C1 QGHJJQCXOHXFCY-UHFFFAOYSA-N 0.000 description 2
- VRLJFRODHVSTIK-UHFFFAOYSA-N 2-(benzhydrylideneamino)acetonitrile Chemical compound C=1C=CC=CC=1C(=NCC#N)C1=CC=CC=C1 VRLJFRODHVSTIK-UHFFFAOYSA-N 0.000 description 2
- LSXJPJGBWSZHTM-UHFFFAOYSA-N 2-(bromomethyl)-1,3-difluorobenzene Chemical compound FC1=CC=CC(F)=C1CBr LSXJPJGBWSZHTM-UHFFFAOYSA-N 0.000 description 2
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 2
- RPSCIRSLUANCGF-UHFFFAOYSA-N 2-amino-5,5,5-trifluoro-2-methylpentanenitrile Chemical compound N#CC(N)(C)CCC(F)(F)F RPSCIRSLUANCGF-UHFFFAOYSA-N 0.000 description 2
- BMTSZVZQNMNPCT-UHFFFAOYSA-N 2-aminopyridin-3-ol Chemical compound NC1=NC=CC=C1O BMTSZVZQNMNPCT-UHFFFAOYSA-N 0.000 description 2
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 2
- BCQIHJQUPGCSRE-UHFFFAOYSA-N 3-[(2,3,6-trifluorophenyl)methoxy]pyridin-2-amine Chemical compound NC1=NC=CC=C1OCC1=C(F)C=CC(F)=C1F BCQIHJQUPGCSRE-UHFFFAOYSA-N 0.000 description 2
- LBZQPIOLPBOVRI-UHFFFAOYSA-N 3-[(2,6-difluorophenyl)methoxy]pyridin-2-amine Chemical compound NC1=NC=CC=C1OCC1=C(F)C=CC=C1F LBZQPIOLPBOVRI-UHFFFAOYSA-N 0.000 description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical class NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 2
- NCGICGYLBXGBGN-UHFFFAOYSA-N 3-morpholin-4-yl-1-oxa-3-azonia-2-azanidacyclopent-3-en-5-imine;hydrochloride Chemical compound Cl.[N-]1OC(=N)C=[N+]1N1CCOCC1 NCGICGYLBXGBGN-UHFFFAOYSA-N 0.000 description 2
- KSGFMCPOCNISNE-UHFFFAOYSA-N 5,5,5-trifluoropentan-2-one Chemical compound CC(=O)CCC(F)(F)F KSGFMCPOCNISNE-UHFFFAOYSA-N 0.000 description 2
- XRSYKYYBXLVPAJ-UHFFFAOYSA-N 5-chloro-2-nitropyridin-3-ol Chemical compound OC1=CC(Cl)=CN=C1[N+]([O-])=O XRSYKYYBXLVPAJ-UHFFFAOYSA-N 0.000 description 2
- AFWNILVXTVCIJQ-UHFFFAOYSA-N 5-chloro-3-[(2,6-difluorophenyl)methoxy]-2-nitropyridine Chemical compound [O-][N+](=O)C1=NC=C(Cl)C=C1OCC1=C(F)C=CC=C1F AFWNILVXTVCIJQ-UHFFFAOYSA-N 0.000 description 2
- BFYJDRFHLQAKOV-UHFFFAOYSA-N 8-[(2,6-difluorophenyl)methoxy]-2-methylimidazo[1,2-a]pyridine-3-carboxylic acid Chemical compound C=1C=CN2C(C(O)=O)=C(C)N=C2C=1OCC1=C(F)C=CC=C1F BFYJDRFHLQAKOV-UHFFFAOYSA-N 0.000 description 2
- 208000004476 Acute Coronary Syndrome Diseases 0.000 description 2
- 208000009304 Acute Kidney Injury Diseases 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 206010007556 Cardiac failure acute Diseases 0.000 description 2
- 229940127328 Cholesterol Synthesis Inhibitors Drugs 0.000 description 2
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 2
- WACWXJHWXRIXAZ-UHFFFAOYSA-N Cl.CC(N)(CC(F)F)C#N Chemical compound Cl.CC(N)(CC(F)F)C#N WACWXJHWXRIXAZ-UHFFFAOYSA-N 0.000 description 2
- GYJXCRTWVSQDQT-UHFFFAOYSA-N Cl.CC(N)(CCCF)C#N Chemical compound Cl.CC(N)(CCCF)C#N GYJXCRTWVSQDQT-UHFFFAOYSA-N 0.000 description 2
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 2
- 102000003849 Cytochrome P450 Human genes 0.000 description 2
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 2
- 206010012689 Diabetic retinopathy Diseases 0.000 description 2
- 206010014561 Emphysema Diseases 0.000 description 2
- 102000002045 Endothelin Human genes 0.000 description 2
- 108050009340 Endothelin Proteins 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 206010015856 Extrasystoles Diseases 0.000 description 2
- MZXKBYYEROJDAW-UHFFFAOYSA-N FC(F)CC(N=C(c1ccccc1)c1ccccc1)C#N Chemical compound FC(F)CC(N=C(c1ccccc1)c1ccccc1)C#N MZXKBYYEROJDAW-UHFFFAOYSA-N 0.000 description 2
- VASDTJCSAXCJHL-UHFFFAOYSA-N FCCCC(N=C(c1ccccc1)c1ccccc1)C#N Chemical compound FCCCC(N=C(c1ccccc1)c1ccccc1)C#N VASDTJCSAXCJHL-UHFFFAOYSA-N 0.000 description 2
- 206010016654 Fibrosis Diseases 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 208000010412 Glaucoma Diseases 0.000 description 2
- 206010018364 Glomerulonephritis Diseases 0.000 description 2
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- 102000004286 Hydroxymethylglutaryl CoA Reductases Human genes 0.000 description 2
- 108090000895 Hydroxymethylglutaryl CoA Reductases Proteins 0.000 description 2
- 206010020853 Hypertonic bladder Diseases 0.000 description 2
- 102100021711 Ileal sodium/bile acid cotransporter Human genes 0.000 description 2
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 229940127470 Lipase Inhibitors Drugs 0.000 description 2
- 208000026139 Memory disease Diseases 0.000 description 2
- 206010027525 Microalbuminuria Diseases 0.000 description 2
- 208000034486 Multi-organ failure Diseases 0.000 description 2
- 208000010718 Multiple Organ Failure Diseases 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical compound CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 description 2
- GCSPPZHRYVPTMM-UHFFFAOYSA-N NC1=NC=C(Br)C=C1OCC1=C(F)C(F)=CC=C1F Chemical compound NC1=NC=C(Br)C=C1OCC1=C(F)C(F)=CC=C1F GCSPPZHRYVPTMM-UHFFFAOYSA-N 0.000 description 2
- 208000009722 Overactive Urinary Bladder Diseases 0.000 description 2
- 108010015181 PPAR delta Proteins 0.000 description 2
- 108010016731 PPAR gamma Proteins 0.000 description 2
- 102000000536 PPAR gamma Human genes 0.000 description 2
- 102000003728 Peroxisome Proliferator-Activated Receptors Human genes 0.000 description 2
- 108090000029 Peroxisome Proliferator-Activated Receptors Proteins 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- 108010022233 Plasminogen Activator Inhibitor 1 Proteins 0.000 description 2
- 102100039418 Plasminogen activator inhibitor 1 Human genes 0.000 description 2
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 2
- 208000000418 Premature Cardiac Complexes Diseases 0.000 description 2
- 206010064911 Pulmonary arterial hypertension Diseases 0.000 description 2
- 206010037423 Pulmonary oedema Diseases 0.000 description 2
- 241000700157 Rattus norvegicus Species 0.000 description 2
- 208000033626 Renal failure acute Diseases 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- 201000001880 Sexual dysfunction Diseases 0.000 description 2
- 239000012317 TBTU Substances 0.000 description 2
- 208000030886 Traumatic Brain injury Diseases 0.000 description 2
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 2
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 2
- 208000009729 Ventricular Premature Complexes Diseases 0.000 description 2
- 206010052428 Wound Diseases 0.000 description 2
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 2
- 230000009102 absorption Effects 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 206010069351 acute lung injury Diseases 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 208000006682 alpha 1-Antitrypsin Deficiency Diseases 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 150000003863 ammonium salts Chemical class 0.000 description 2
- 238000002399 angioplasty Methods 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 229940127219 anticoagulant drug Drugs 0.000 description 2
- 229940030600 antihypertensive agent Drugs 0.000 description 2
- 239000002220 antihypertensive agent Substances 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 2
- 206010003119 arrhythmia Diseases 0.000 description 2
- 229960002274 atenolol Drugs 0.000 description 2
- 230000006399 behavior Effects 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- 230000001906 cholesterol absorption Effects 0.000 description 2
- 208000020832 chronic kidney disease Diseases 0.000 description 2
- 230000027288 circadian rhythm Effects 0.000 description 2
- 208000019425 cirrhosis of liver Diseases 0.000 description 2
- 208000010877 cognitive disease Diseases 0.000 description 2
- 238000009833 condensation Methods 0.000 description 2
- 230000005494 condensation Effects 0.000 description 2
- 230000001276 controlling effect Effects 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- ZYGHJZDHTFUPRJ-UHFFFAOYSA-N coumarin Chemical compound C1=CC=C2OC(=O)C=CC2=C1 ZYGHJZDHTFUPRJ-UHFFFAOYSA-N 0.000 description 2
- 229940109239 creatinine Drugs 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 230000007850 degeneration Effects 0.000 description 2
- 229910052805 deuterium Inorganic materials 0.000 description 2
- 230000035487 diastolic blood pressure Effects 0.000 description 2
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 239000002934 diuretic Substances 0.000 description 2
- 229940030606 diuretics Drugs 0.000 description 2
- 230000002526 effect on cardiovascular system Effects 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- ZUBDGKVDJUIMQQ-UBFCDGJISA-N endothelin-1 Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(O)=O)NC(=O)[C@H]1NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@@H](CC=2C=CC(O)=CC=2)NC(=O)[C@H](C(C)C)NC(=O)[C@H]2CSSC[C@@H](C(N[C@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N2)=O)NC(=O)[C@@H](CO)NC(=O)[C@H](N)CSSC1)C1=CNC=N1 ZUBDGKVDJUIMQQ-UBFCDGJISA-N 0.000 description 2
- 239000000066 endothelium dependent relaxing factor Substances 0.000 description 2
- 229960001208 eplerenone Drugs 0.000 description 2
- JUKPWJGBANNWMW-VWBFHTRKSA-N eplerenone Chemical compound C([C@@H]1[C@]2(C)C[C@H]3O[C@]33[C@@]4(C)CCC(=O)C=C4C[C@H]([C@@H]13)C(=O)OC)C[C@@]21CCC(=O)O1 JUKPWJGBANNWMW-VWBFHTRKSA-N 0.000 description 2
- 238000010931 ester hydrolysis Methods 0.000 description 2
- FUBBWDWIGBTUPQ-UHFFFAOYSA-N ethyl 2-[4-[3-[(4-fluorophenyl)-hydroxymethyl]-4-hydroxyphenoxy]-3,5-dimethylanilino]-2-oxoacetate Chemical compound CC1=CC(NC(=O)C(=O)OCC)=CC(C)=C1OC1=CC=C(O)C(C(O)C=2C=CC(F)=CC=2)=C1 FUBBWDWIGBTUPQ-UHFFFAOYSA-N 0.000 description 2
- BJZRZBUVFSQJJW-UHFFFAOYSA-N ethyl 8-[(2,6-difluorophenyl)methoxy]-2-methylimidazo[1,2-a]pyridine-3-carboxylate Chemical compound C=1C=CN2C(C(=O)OCC)=C(C)N=C2C=1OCC1=C(F)C=CC=C1F BJZRZBUVFSQJJW-UHFFFAOYSA-N 0.000 description 2
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 230000029142 excretion Effects 0.000 description 2
- 235000013305 food Nutrition 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 238000002290 gas chromatography-mass spectrometry Methods 0.000 description 2
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 2
- 229910052737 gold Inorganic materials 0.000 description 2
- 239000010931 gold Substances 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 208000019622 heart disease Diseases 0.000 description 2
- 210000003494 hepatocyte Anatomy 0.000 description 2
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 2
- 230000002757 inflammatory effect Effects 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 239000006193 liquid solution Substances 0.000 description 2
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 2
- 210000001853 liver microsome Anatomy 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 230000002107 myocardial effect Effects 0.000 description 2
- 208000010125 myocardial infarction Diseases 0.000 description 2
- 201000009925 nephrosclerosis Diseases 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 208000020629 overactive bladder Diseases 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- JLFNLZLINWHATN-UHFFFAOYSA-N pentaethylene glycol Chemical compound OCCOCCOCCOCCOCCO JLFNLZLINWHATN-UHFFFAOYSA-N 0.000 description 2
- 239000003208 petroleum Substances 0.000 description 2
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920000136 polysorbate Polymers 0.000 description 2
- 229950008882 polysorbate Drugs 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 230000000019 pro-fibrinolytic effect Effects 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 2
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 208000005333 pulmonary edema Diseases 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 238000003908 quality control method Methods 0.000 description 2
- 238000011002 quantification Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- QEVHRUUCFGRFIF-MDEJGZGSSA-N reserpine Chemical compound O([C@H]1[C@@H]([C@H]([C@H]2C[C@@H]3C4=C(C5=CC=C(OC)C=C5N4)CCN3C[C@H]2C1)C(=O)OC)OC)C(=O)C1=CC(OC)=C(OC)C(OC)=C1 QEVHRUUCFGRFIF-MDEJGZGSSA-N 0.000 description 2
- 230000000452 restraining effect Effects 0.000 description 2
- 230000033764 rhythmic process Effects 0.000 description 2
- 229960001148 rivaroxaban Drugs 0.000 description 2
- KGFYHTZWPPHNLQ-AWEZNQCLSA-N rivaroxaban Chemical compound S1C(Cl)=CC=C1C(=O)NC[C@@H]1OC(=O)N(C=2C=CC(=CC=2)N2C(COCC2)=O)C1 KGFYHTZWPPHNLQ-AWEZNQCLSA-N 0.000 description 2
- 201000000306 sarcoidosis Diseases 0.000 description 2
- 230000037390 scarring Effects 0.000 description 2
- 231100000872 sexual dysfunction Toxicity 0.000 description 2
- BNRNXUUZRGQAQC-UHFFFAOYSA-N sildenafil Chemical compound CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 BNRNXUUZRGQAQC-UHFFFAOYSA-N 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L sodium carbonate Substances [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 229960002256 spironolactone Drugs 0.000 description 2
- LXMSZDCAJNLERA-ZHYRCANASA-N spironolactone Chemical compound C([C@@H]1[C@]2(C)CC[C@@H]3[C@@]4(C)CCC(=O)C=C4C[C@H]([C@@H]13)SC(=O)C)C[C@@]21CCC(=O)O1 LXMSZDCAJNLERA-ZHYRCANASA-N 0.000 description 2
- 208000010110 spontaneous platelet aggregation Diseases 0.000 description 2
- 230000006641 stabilisation Effects 0.000 description 2
- 238000011105 stabilization Methods 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 238000001356 surgical procedure Methods 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 230000035488 systolic blood pressure Effects 0.000 description 2
- RMMXLENWKUUMAY-UHFFFAOYSA-N telmisartan Chemical compound CCCC1=NC2=C(C)C=C(C=3N(C4=CC=CC=C4N=3)C)C=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C(O)=O RMMXLENWKUUMAY-UHFFFAOYSA-N 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 210000002700 urine Anatomy 0.000 description 2
- 210000002229 urogenital system Anatomy 0.000 description 2
- 235000012431 wafers Nutrition 0.000 description 2
- CEMAWMOMDPGJMB-UHFFFAOYSA-N (+-)-Oxprenolol Chemical compound CC(C)NCC(O)COC1=CC=CC=C1OCC=C CEMAWMOMDPGJMB-UHFFFAOYSA-N 0.000 description 1
- ZXZBJYYDIVJJKZ-UHFFFAOYSA-N (1-amino-5,5,5-trifluoro-2-methylpentan-2-yl) carbamate Chemical compound C(N)(OC(CN)(CCC(F)(F)F)C)=O ZXZBJYYDIVJJKZ-UHFFFAOYSA-N 0.000 description 1
- FFRLMXNUOYHUNP-UHFFFAOYSA-N (1-amino-5-fluoro-2-methylpentan-2-yl) carbamate Chemical compound C(N)(OC(CN)(CCCF)C)=O FFRLMXNUOYHUNP-UHFFFAOYSA-N 0.000 description 1
- FVKQTEQUULVWOH-UHFFFAOYSA-N (2-cyano-4,4-difluorobutan-2-yl) carbamate Chemical compound C(N)(OC(C)(CC(F)F)C#N)=O FVKQTEQUULVWOH-UHFFFAOYSA-N 0.000 description 1
- RLIKGMAAXNSSOD-UHFFFAOYSA-N (2-cyano-5-fluoropentan-2-yl) carbamate Chemical compound C(N)(OC(C)(CCCF)C#N)=O RLIKGMAAXNSSOD-UHFFFAOYSA-N 0.000 description 1
- NXWGWUVGUSFQJC-GFCCVEGCSA-N (2r)-1-[(2-methyl-1h-indol-4-yl)oxy]-3-(propan-2-ylamino)propan-2-ol Chemical compound CC(C)NC[C@@H](O)COC1=CC=CC2=C1C=C(C)N2 NXWGWUVGUSFQJC-GFCCVEGCSA-N 0.000 description 1
- DMYZJLOWGSRVKP-RTBURBONSA-N (2r,4r)-1-n-(4-chlorophenyl)-4-hydroxy-2-n-[4-(3-oxomorpholin-4-yl)phenyl]pyrrolidine-1,2-dicarboxamide Chemical compound N1([C@H](C[C@H](C1)O)C(=O)NC=1C=CC(=CC=1)N1C(COCC1)=O)C(=O)NC1=CC=C(Cl)C=C1 DMYZJLOWGSRVKP-RTBURBONSA-N 0.000 description 1
- AMNXBQPRODZJQR-DITALETJSA-N (2s)-2-cyclopentyl-2-[3-[(2,4-dimethylpyrido[2,3-b]indol-9-yl)methyl]phenyl]-n-[(1r)-2-hydroxy-1-phenylethyl]acetamide Chemical compound C1([C@@H](C=2C=CC=C(C=2)CN2C3=CC=CC=C3C3=C(C)C=C(N=C32)C)C(=O)N[C@@H](CO)C=2C=CC=CC=2)CCCC1 AMNXBQPRODZJQR-DITALETJSA-N 0.000 description 1
- ZXEIEKDGPVTZLD-NDEPHWFRSA-N (2s)-2-dodecylsulfanyl-n-(4-hydroxy-2,3,5-trimethylphenyl)-2-phenylacetamide Chemical compound O=C([C@@H](SCCCCCCCCCCCC)C=1C=CC=CC=1)NC1=CC(C)=C(O)C(C)=C1C ZXEIEKDGPVTZLD-NDEPHWFRSA-N 0.000 description 1
- LJCBAPRMNYSDOP-LVCYMWGESA-N (2s)-3-(7-carbamimidoylnaphthalen-2-yl)-2-[4-[(3s)-1-ethanimidoylpyrrolidin-3-yl]oxyphenyl]propanoic acid;hydron;chloride;pentahydrate Chemical compound O.O.O.O.O.Cl.C1N(C(=N)C)CC[C@@H]1OC1=CC=C([C@H](CC=2C=C3C=C(C=CC3=CC=2)C(N)=N)C(O)=O)C=C1 LJCBAPRMNYSDOP-LVCYMWGESA-N 0.000 description 1
- BIDNLKIUORFRQP-XYGFDPSESA-N (2s,4s)-4-cyclohexyl-1-[2-[[(1s)-2-methyl-1-propanoyloxypropoxy]-(4-phenylbutyl)phosphoryl]acetyl]pyrrolidine-2-carboxylic acid Chemical compound C([P@@](=O)(O[C@H](OC(=O)CC)C(C)C)CC(=O)N1[C@@H](C[C@H](C1)C1CCCCC1)C(O)=O)CCCC1=CC=CC=C1 BIDNLKIUORFRQP-XYGFDPSESA-N 0.000 description 1
- ZGGHKIMDNBDHJB-NRFPMOEYSA-M (3R,5S)-fluvastatin sodium Chemical compound [Na+].C12=CC=CC=C2N(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 ZGGHKIMDNBDHJB-NRFPMOEYSA-M 0.000 description 1
- BHKIPHICFOJGLD-HOFKKMOUSA-N (5s)-4-cyclohexyl-2-cyclopentyl-3-[(s)-fluoro-[4-(trifluoromethyl)phenyl]methyl]-7,7-dimethyl-6,8-dihydro-5h-quinolin-5-ol Chemical compound C1([C@@H](F)C=2C=CC(=CC=2)C(F)(F)F)=C(C2CCCCC2)C([C@@H](O)CC(C2)(C)C)=C2N=C1C1CCCC1 BHKIPHICFOJGLD-HOFKKMOUSA-N 0.000 description 1
- RWIUTHWKQHRQNP-ZDVGBALWSA-N (9e,12e)-n-(1-phenylethyl)octadeca-9,12-dienamide Chemical compound CCCCC\C=C\C\C=C\CCCCCCCC(=O)NC(C)C1=CC=CC=C1 RWIUTHWKQHRQNP-ZDVGBALWSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- TWBNMYSKRDRHAT-RCWTXCDDSA-N (S)-timolol hemihydrate Chemical compound O.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1 TWBNMYSKRDRHAT-RCWTXCDDSA-N 0.000 description 1
- KOHIRBRYDXPAMZ-YHBROIRLSA-N (S,R,R,R)-nebivolol Chemical compound C1CC2=CC(F)=CC=C2O[C@H]1[C@H](O)CNC[C@@H](O)[C@H]1OC2=CC=C(F)C=C2CC1 KOHIRBRYDXPAMZ-YHBROIRLSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- ZUCFGSAENWHFPO-UHFFFAOYSA-N 1-[4-amino-2,6-di(propan-2-yl)phenyl]-3-[1-butyl-4-[3-(3-hydroxypropoxy)phenyl]-2-oxo-1,8-naphthyridin-3-yl]urea;hydrate;hydrochloride Chemical compound O.Cl.CC(C)C=1C=C(N)C=C(C(C)C)C=1NC(=O)NC=1C(=O)N(CCCC)C2=NC=CC=C2C=1C1=CC=CC(OCCCO)=C1 ZUCFGSAENWHFPO-UHFFFAOYSA-N 0.000 description 1
- PMGZJNCIQHGNLT-UHFFFAOYSA-N 1-[bis(2,2-dimethylpropanoyloxymethoxy)phosphoryl]-4-(3-phenoxyphenyl)butane-1-sulfonic acid Chemical compound CC(C)(C)C(=O)OCOP(=O)(OCOC(=O)C(C)(C)C)C(S(O)(=O)=O)CCCC1=CC=CC(OC=2C=CC=CC=2)=C1 PMGZJNCIQHGNLT-UHFFFAOYSA-N 0.000 description 1
- CMCBDXRRFKYBDG-UHFFFAOYSA-N 1-dodecoxydodecane Chemical compound CCCCCCCCCCCCOCCCCCCCCCCCC CMCBDXRRFKYBDG-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- GUSVHVVOABZHAH-OPZWKQDFSA-N 1aw8p77hkj Chemical compound O([C@@H]1[C@@H](CO)O[C@H]([C@@H]([C@H]1O)O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4C[C@H]5[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@@H]([C@]1(OC[C@H](C)CC1)O5)C)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O GUSVHVVOABZHAH-OPZWKQDFSA-N 0.000 description 1
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 1
- JPBWEVKHPSNBCE-UHFFFAOYSA-N 2-(bromomethyl)-1,3,4-trifluorobenzene Chemical compound FC1=CC=C(F)C(CBr)=C1F JPBWEVKHPSNBCE-UHFFFAOYSA-N 0.000 description 1
- DEMLYXMVPJAVFU-UHFFFAOYSA-N 2-(chloromethyl)oxirane;2-methyl-1h-imidazole Chemical compound ClCC1CO1.CC1=NC=CN1 DEMLYXMVPJAVFU-UHFFFAOYSA-N 0.000 description 1
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 1
- HQSRVYUCBOCBLY-XOOFNSLWSA-N 2-[(2r)-butan-2-yl]-4-[4-[4-[4-[[(2s,4r)-2-(4-chlorophenyl)-2-[(4-methyl-1,2,4-triazol-3-yl)sulfanylmethyl]-1,3-dioxolan-4-yl]methoxy]phenyl]piperazin-1-yl]phenyl]-1,2,4-triazol-3-one Chemical compound O=C1N([C@H](C)CC)N=CN1C1=CC=C(N2CCN(CC2)C=2C=CC(OC[C@H]3O[C@@](CSC=4N(C=NN=4)C)(OC3)C=3C=CC(Cl)=CC=3)=CC=2)C=C1 HQSRVYUCBOCBLY-XOOFNSLWSA-N 0.000 description 1
- CMLUGNQVANVZHY-POURPWNDSA-N 2-[1-[2-[(3r,5s)-1-(3-acetyloxy-2,2-dimethylpropyl)-7-chloro-5-(2,3-dimethoxyphenyl)-2-oxo-5h-4,1-benzoxazepin-3-yl]acetyl]piperidin-4-yl]acetic acid Chemical compound COC1=CC=CC([C@@H]2C3=CC(Cl)=CC=C3N(CC(C)(C)COC(C)=O)C(=O)[C@@H](CC(=O)N3CCC(CC(O)=O)CC3)O2)=C1OC CMLUGNQVANVZHY-POURPWNDSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- UOJMJBUYXYEPFX-UHFFFAOYSA-N 2-[4-(4-hydroxy-3-propan-2-ylphenoxy)-3,5-dimethylanilino]-2-oxoacetic acid Chemical compound C1=C(O)C(C(C)C)=CC(OC=2C(=CC(NC(=O)C(O)=O)=CC=2C)C)=C1 UOJMJBUYXYEPFX-UHFFFAOYSA-N 0.000 description 1
- TXIIZHHIOHVWJD-UHFFFAOYSA-N 2-[7-(2,2-dimethylpropanoylamino)-4,6-dimethyl-1-octyl-2,3-dihydroindol-5-yl]acetic acid Chemical compound CC(C)(C)C(=O)NC1=C(C)C(CC(O)=O)=C(C)C2=C1N(CCCCCCCC)CC2 TXIIZHHIOHVWJD-UHFFFAOYSA-N 0.000 description 1
- NOIXNOMHHWGUTG-UHFFFAOYSA-N 2-[[4-[4-pyridin-4-yl-1-(2,2,2-trifluoroethyl)pyrazol-3-yl]phenoxy]methyl]quinoline Chemical class C=1C=C(OCC=2N=C3C=CC=CC3=CC=2)C=CC=1C1=NN(CC(F)(F)F)C=C1C1=CC=NC=C1 NOIXNOMHHWGUTG-UHFFFAOYSA-N 0.000 description 1
- JIVPVXMEBJLZRO-CQSZACIVSA-N 2-chloro-5-[(1r)-1-hydroxy-3-oxo-2h-isoindol-1-yl]benzenesulfonamide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC([C@@]2(O)C3=CC=CC=C3C(=O)N2)=C1 JIVPVXMEBJLZRO-CQSZACIVSA-N 0.000 description 1
- SGUAFYQXFOLMHL-UHFFFAOYSA-N 2-hydroxy-5-{1-hydroxy-2-[(4-phenylbutan-2-yl)amino]ethyl}benzamide Chemical compound C=1C=C(O)C(C(N)=O)=CC=1C(O)CNC(C)CCC1=CC=CC=C1 SGUAFYQXFOLMHL-UHFFFAOYSA-N 0.000 description 1
- YZQLWPMZQVHJED-UHFFFAOYSA-N 2-methylpropanethioic acid S-[2-[[[1-(2-ethylbutyl)cyclohexyl]-oxomethyl]amino]phenyl] ester Chemical compound C=1C=CC=C(SC(=O)C(C)C)C=1NC(=O)C1(CC(CC)CC)CCCCC1 YZQLWPMZQVHJED-UHFFFAOYSA-N 0.000 description 1
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 1
- MPSXGPCFLAGJOM-UHFFFAOYSA-M 2-tert-butyl-5-methyl-1,2-oxazol-2-ium;perchlorate Chemical compound [O-]Cl(=O)(=O)=O.CC1=CC=[N+](C(C)(C)C)O1 MPSXGPCFLAGJOM-UHFFFAOYSA-M 0.000 description 1
- AUYYCJSJGJYCDS-UHFFFAOYSA-N 2/3/6893 Natural products IC1=CC(CC(N)C(O)=O)=CC(I)=C1OC1=CC=C(O)C(I)=C1 AUYYCJSJGJYCDS-UHFFFAOYSA-N 0.000 description 1
- JMTMSDXUXJISAY-UHFFFAOYSA-N 2H-benzotriazol-4-ol Chemical compound OC1=CC=CC2=C1N=NN2 JMTMSDXUXJISAY-UHFFFAOYSA-N 0.000 description 1
- KLDLRDSRCMJKGM-UHFFFAOYSA-N 3-[chloro-(2-oxo-1,3-oxazolidin-3-yl)phosphoryl]-1,3-oxazolidin-2-one Chemical compound C1COC(=O)N1P(=O)(Cl)N1CCOC1=O KLDLRDSRCMJKGM-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- SCJCDNUXDWFVFI-UHFFFAOYSA-N 4,4,4-trifluorobutanal Chemical compound FC(F)(F)CCC=O SCJCDNUXDWFVFI-UHFFFAOYSA-N 0.000 description 1
- WTUCTMYLCMVYEX-UHFFFAOYSA-N 4,4,4-trifluorobutanoic acid Chemical compound OC(=O)CCC(F)(F)F WTUCTMYLCMVYEX-UHFFFAOYSA-N 0.000 description 1
- NOBZETMXGVAWIM-UHFFFAOYSA-N 4-[(2-carbamimidoyl-3,4-dihydro-1h-isoquinolin-7-yl)oxymethyl]-1-pyridin-4-ylpiperidine-4-carboxylic acid;methanesulfonic acid Chemical compound CS(O)(=O)=O.C1=C2CN(C(=N)N)CCC2=CC=C1OCC(CC1)(C(O)=O)CCN1C1=CC=NC=C1 NOBZETMXGVAWIM-UHFFFAOYSA-N 0.000 description 1
- KEWSCDNULKOKTG-UHFFFAOYSA-N 4-cyano-4-ethylsulfanylcarbothioylsulfanylpentanoic acid Chemical compound CCSC(=S)SC(C)(C#N)CCC(O)=O KEWSCDNULKOKTG-UHFFFAOYSA-N 0.000 description 1
- JVVRCYWZTJLJSG-UHFFFAOYSA-N 4-dimethylaminophenol Chemical compound CN(C)C1=CC=C(O)C=C1 JVVRCYWZTJLJSG-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-dimethylaminopyridine Substances CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 1
- OCKGFTQIICXDQW-ZEQRLZLVSA-N 5-[(1r)-1-hydroxy-2-[4-[(2r)-2-hydroxy-2-(4-methyl-1-oxo-3h-2-benzofuran-5-yl)ethyl]piperazin-1-yl]ethyl]-4-methyl-3h-2-benzofuran-1-one Chemical compound C1=C2C(=O)OCC2=C(C)C([C@@H](O)CN2CCN(CC2)C[C@H](O)C2=CC=C3C(=O)OCC3=C2C)=C1 OCKGFTQIICXDQW-ZEQRLZLVSA-N 0.000 description 1
- TUIDQYRZDZRHPQ-UHFFFAOYSA-N 5-chloropyridin-3-ol Chemical compound OC1=CN=CC(Cl)=C1 TUIDQYRZDZRHPQ-UHFFFAOYSA-N 0.000 description 1
- VNHBYKHXBCYPBJ-UHFFFAOYSA-N 5-ethynylimidazo[1,2-a]pyridine Chemical compound C#CC1=CC=CC2=NC=CN12 VNHBYKHXBCYPBJ-UHFFFAOYSA-N 0.000 description 1
- 102100031126 6-phosphogluconolactonase Human genes 0.000 description 1
- 108010029731 6-phosphogluconolactonase Proteins 0.000 description 1
- KKJUPNGICOCCDW-UHFFFAOYSA-N 7-N,N-Dimethylamino-1,2,3,4,5-pentathiocyclooctane Chemical compound CN(C)C1CSSSSSC1 KKJUPNGICOCCDW-UHFFFAOYSA-N 0.000 description 1
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 description 1
- 102000011767 Acute-Phase Proteins Human genes 0.000 description 1
- 108010062271 Acute-Phase Proteins Proteins 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 201000010053 Alcoholic Cardiomyopathy Diseases 0.000 description 1
- UXOWGYHJODZGMF-QORCZRPOSA-N Aliskiren Chemical compound COCCCOC1=CC(C[C@@H](C[C@H](N)[C@@H](O)C[C@@H](C(C)C)C(=O)NCC(C)(C)C(N)=O)C(C)C)=CC=C1OC UXOWGYHJODZGMF-QORCZRPOSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- WSVLPVUVIUVCRA-KPKNDVKVSA-N Alpha-lactose monohydrate Chemical compound O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O WSVLPVUVIUVCRA-KPKNDVKVSA-N 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 1
- 208000000044 Amnesia Diseases 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 206010002199 Anaphylactic shock Diseases 0.000 description 1
- 206010002329 Aneurysm Diseases 0.000 description 1
- 101000988143 Antheraea pernyi Pheromone-binding protein 1 Proteins 0.000 description 1
- 208000019901 Anxiety disease Diseases 0.000 description 1
- 208000003017 Aortic Valve Stenosis Diseases 0.000 description 1
- 206010002915 Aortic valve incompetence Diseases 0.000 description 1
- 206010002921 Aortitis Diseases 0.000 description 1
- QNZCBYKSOIHPEH-UHFFFAOYSA-N Apixaban Chemical compound C1=CC(OC)=CC=C1N1C(C(=O)N(CC2)C=3C=CC(=CC=3)N3C(CCCC3)=O)=C2C(C(N)=O)=N1 QNZCBYKSOIHPEH-UHFFFAOYSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 206010003178 Arterial thrombosis Diseases 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 1
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 1
- 206010003658 Atrial Fibrillation Diseases 0.000 description 1
- 206010003662 Atrial flutter Diseases 0.000 description 1
- 208000006808 Atrioventricular Nodal Reentry Tachycardia Diseases 0.000 description 1
- 229930003347 Atropine Natural products 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 206010061666 Autonomic neuropathy Diseases 0.000 description 1
- PTQXTEKSNBVPQJ-UHFFFAOYSA-N Avasimibe Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1CC(=O)NS(=O)(=O)OC1=C(C(C)C)C=CC=C1C(C)C PTQXTEKSNBVPQJ-UHFFFAOYSA-N 0.000 description 1
- 208000037157 Azotemia Diseases 0.000 description 1
- 239000005552 B01AC04 - Clopidogrel Substances 0.000 description 1
- 239000005528 B01AC05 - Ticlopidine Substances 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- QQSVPKSYAUZMJJ-UHFFFAOYSA-N BrC=1C=C(C=2N(C=1)C(=C(N=2)C)C(=O)O)OCC1=C(C(=CC=C1F)F)F Chemical compound BrC=1C=C(C=2N(C=1)C(=C(N=2)C)C(=O)O)OCC1=C(C(=CC=C1F)F)F QQSVPKSYAUZMJJ-UHFFFAOYSA-N 0.000 description 1
- 206010048962 Brain oedema Diseases 0.000 description 1
- 239000002083 C09CA01 - Losartan Substances 0.000 description 1
- 239000004072 C09CA03 - Valsartan Substances 0.000 description 1
- 239000002053 C09CA06 - Candesartan Substances 0.000 description 1
- 239000005537 C09CA07 - Telmisartan Substances 0.000 description 1
- VBEROFQVLBHEPP-UHFFFAOYSA-N CC(CC(F)F)(CN)NC(OCc1ccccc1)=O Chemical compound CC(CC(F)F)(CN)NC(OCc1ccccc1)=O VBEROFQVLBHEPP-UHFFFAOYSA-N 0.000 description 1
- HRFVDFLTPAPNFA-UHFFFAOYSA-N CC(CCC(F)(F)F)(CN)NC(OCc1ccccc1)=O Chemical compound CC(CCC(F)(F)F)(CN)NC(OCc1ccccc1)=O HRFVDFLTPAPNFA-UHFFFAOYSA-N 0.000 description 1
- RDIOCHPEANSXLR-UHFFFAOYSA-N CC(CCC(F)(F)F)(CNC(c1c(C)nc2[n]1cccc2OCc(c(F)ccc1)c1F)=O)NC(OCc1ccccc1)=O Chemical compound CC(CCC(F)(F)F)(CNC(c1c(C)nc2[n]1cccc2OCc(c(F)ccc1)c1F)=O)NC(OCc1ccccc1)=O RDIOCHPEANSXLR-UHFFFAOYSA-N 0.000 description 1
- WGKMHKFXHTWMSJ-UHFFFAOYSA-N CC(CCCF)(CNC(c1c(C)nc2[n]1cccc2OCc(c(F)ccc1)c1F)=O)N Chemical compound CC(CCCF)(CNC(c1c(C)nc2[n]1cccc2OCc(c(F)ccc1)c1F)=O)N WGKMHKFXHTWMSJ-UHFFFAOYSA-N 0.000 description 1
- KSFOVUSSGSKXFI-GAQDCDSVSA-N CC1=C/2NC(\C=C3/N=C(/C=C4\N\C(=C/C5=N/C(=C\2)/C(C=C)=C5C)C(C=C)=C4C)C(C)=C3CCC(O)=O)=C1CCC(O)=O Chemical compound CC1=C/2NC(\C=C3/N=C(/C=C4\N\C(=C/C5=N/C(=C\2)/C(C=C)=C5C)C(C=C)=C4C)C(C)=C3CCC(O)=O)=C1CCC(O)=O KSFOVUSSGSKXFI-GAQDCDSVSA-N 0.000 description 1
- 201000002829 CREST Syndrome Diseases 0.000 description 1
- 101100296726 Caenorhabditis elegans pde-5 gene Proteins 0.000 description 1
- 206010007558 Cardiac failure chronic Diseases 0.000 description 1
- 206010007559 Cardiac failure congestive Diseases 0.000 description 1
- 206010007572 Cardiac hypertrophy Diseases 0.000 description 1
- 208000006029 Cardiomegaly Diseases 0.000 description 1
- 208000031229 Cardiomyopathies Diseases 0.000 description 1
- 206010007637 Cardiomyopathy alcoholic Diseases 0.000 description 1
- JOATXPAWOHTVSZ-UHFFFAOYSA-N Celiprolol Chemical compound CCN(CC)C(=O)NC1=CC=C(OCC(O)CNC(C)(C)C)C(C(C)=O)=C1 JOATXPAWOHTVSZ-UHFFFAOYSA-N 0.000 description 1
- 206010008120 Cerebral ischaemia Diseases 0.000 description 1
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 1
- JZUFKLXOESDKRF-UHFFFAOYSA-N Chlorothiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O JZUFKLXOESDKRF-UHFFFAOYSA-N 0.000 description 1
- 229940122502 Cholesterol absorption inhibitor Drugs 0.000 description 1
- 102100037637 Cholesteryl ester transfer protein Human genes 0.000 description 1
- 229920001268 Cholestyramine Polymers 0.000 description 1
- 208000026151 Chronic thromboembolic pulmonary hypertension Diseases 0.000 description 1
- 229920002905 Colesevelam Polymers 0.000 description 1
- 229920002911 Colestipol Polymers 0.000 description 1
- 208000031288 Combined hyperlipidaemia Diseases 0.000 description 1
- 208000002330 Congenital Heart Defects Diseases 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 206010056370 Congestive cardiomyopathy Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 208000011990 Corticobasal Degeneration Diseases 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 206010011416 Croup infectious Diseases 0.000 description 1
- 229920001651 Cyanoacrylate Polymers 0.000 description 1
- 208000026292 Cystic Kidney disease Diseases 0.000 description 1
- NBSCHQHZLSJFNQ-GASJEMHNSA-N D-Glucose 6-phosphate Chemical compound OC1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H](O)[C@H]1O NBSCHQHZLSJFNQ-GASJEMHNSA-N 0.000 description 1
- IGXWBGJHJZYPQS-SSDOTTSWSA-N D-Luciferin Chemical compound OC(=O)[C@H]1CSC(C=2SC3=CC=C(O)C=C3N=2)=N1 IGXWBGJHJZYPQS-SSDOTTSWSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- XUIIKFGFIJCVMT-GFCCVEGCSA-N D-thyroxine Chemical compound IC1=CC(C[C@@H](N)C(O)=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-GFCCVEGCSA-N 0.000 description 1
- 206010067889 Dementia with Lewy bodies Diseases 0.000 description 1
- 101000783577 Dendroaspis angusticeps Thrombostatin Proteins 0.000 description 1
- 101000783578 Dendroaspis jamesoni kaimosae Dendroaspin Proteins 0.000 description 1
- 206010012335 Dependence Diseases 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 206010056340 Diabetic ulcer Diseases 0.000 description 1
- 208000003037 Diastolic Heart Failure Diseases 0.000 description 1
- BWLUMTFWVZZZND-UHFFFAOYSA-N Dibenzylamine Chemical compound C=1C=CC=CC=1CNCC1=CC=CC=C1 BWLUMTFWVZZZND-UHFFFAOYSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 201000010046 Dilated cardiomyopathy Diseases 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- 206010052804 Drug tolerance Diseases 0.000 description 1
- 208000032928 Dyslipidaemia Diseases 0.000 description 1
- 208000005189 Embolism Diseases 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 108010061435 Enalapril Proteins 0.000 description 1
- 206010048554 Endothelial dysfunction Diseases 0.000 description 1
- YARKMNAWFIMDKV-UHFFFAOYSA-N Epanolol Chemical compound C=1C=CC=C(C#N)C=1OCC(O)CNCCNC(=O)CC1=CC=C(O)C=C1 YARKMNAWFIMDKV-UHFFFAOYSA-N 0.000 description 1
- 208000010228 Erectile Dysfunction Diseases 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- ZRLNJEZBGAXDCH-UHFFFAOYSA-N FC(C(=O)O)(F)F.C(N)(OC(CNC(=O)C1=C(N=C2N1C=C(C=C2OCC2=C(C=CC=C2F)F)Cl)C)(CCCF)C)=O Chemical compound FC(C(=O)O)(F)F.C(N)(OC(CNC(=O)C1=C(N=C2N1C=C(C=C2OCC2=C(C=CC=C2F)F)Cl)C)(CCCF)C)=O ZRLNJEZBGAXDCH-UHFFFAOYSA-N 0.000 description 1
- RYCHOQIYXUGHTM-UHFFFAOYSA-N FC(C(=O)O)(F)F.C(N)(OC(CNC(=O)C1=C(N=C2N1C=CC=C2OCC2=C(C=CC=C2F)F)C)(CCC(F)(F)F)C)=O Chemical compound FC(C(=O)O)(F)F.C(N)(OC(CNC(=O)C1=C(N=C2N1C=CC=C2OCC2=C(C=CC=C2F)F)C)(CCC(F)(F)F)C)=O RYCHOQIYXUGHTM-UHFFFAOYSA-N 0.000 description 1
- 229940123583 Factor Xa inhibitor Drugs 0.000 description 1
- 206010057671 Female sexual dysfunction Diseases 0.000 description 1
- 108010049003 Fibrinogen Proteins 0.000 description 1
- 102000008946 Fibrinogen Human genes 0.000 description 1
- 108090000331 Firefly luciferases Proteins 0.000 description 1
- BJGNCJDXODQBOB-UHFFFAOYSA-N Fivefly Luciferin Natural products OC(=O)C1CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 BJGNCJDXODQBOB-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VFRROHXSMXFLSN-UHFFFAOYSA-N Glc6P Natural products OP(=O)(O)OCC(O)C(O)C(O)C(O)C=O VFRROHXSMXFLSN-UHFFFAOYSA-N 0.000 description 1
- 208000022461 Glomerular disease Diseases 0.000 description 1
- 206010018366 Glomerulonephritis acute Diseases 0.000 description 1
- 108010018962 Glucosephosphate Dehydrogenase Proteins 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 206010019196 Head injury Diseases 0.000 description 1
- 101000880514 Homo sapiens Cholesteryl ester transfer protein Proteins 0.000 description 1
- 101001057135 Homo sapiens Melanoma-associated antigen H1 Proteins 0.000 description 1
- 208000023105 Huntington disease Diseases 0.000 description 1
- RKUNBYITZUJHSG-UHFFFAOYSA-N Hyosciamin-hydrochlorid Natural products CN1C(C2)CCC1CC2OC(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-UHFFFAOYSA-N 0.000 description 1
- 208000035150 Hypercholesterolemia Diseases 0.000 description 1
- 208000002682 Hyperkalemia Diseases 0.000 description 1
- 208000031226 Hyperlipidaemia Diseases 0.000 description 1
- 206010021036 Hyponatraemia Diseases 0.000 description 1
- 208000001953 Hypotension Diseases 0.000 description 1
- 101710156096 Ileal sodium/bile acid cotransporter Proteins 0.000 description 1
- 206010021518 Impaired gastric emptying Diseases 0.000 description 1
- 206010021639 Incontinence Diseases 0.000 description 1
- 102100025306 Integrin alpha-IIb Human genes 0.000 description 1
- 101710149643 Integrin alpha-IIb Proteins 0.000 description 1
- 206010022562 Intermittent claudication Diseases 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- 108090000862 Ion Channels Proteins 0.000 description 1
- 206010048858 Ischaemic cardiomyopathy Diseases 0.000 description 1
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 1
- UETNIIAIRMUTSM-UHFFFAOYSA-N Jacareubin Natural products CC1(C)OC2=CC3Oc4c(O)c(O)ccc4C(=O)C3C(=C2C=C1)O UETNIIAIRMUTSM-UHFFFAOYSA-N 0.000 description 1
- 208000002260 Keloid Diseases 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 102000007330 LDL Lipoproteins Human genes 0.000 description 1
- 108010007622 LDL Lipoproteins Proteins 0.000 description 1
- 208000020358 Learning disease Diseases 0.000 description 1
- 206010024119 Left ventricular failure Diseases 0.000 description 1
- 201000002832 Lewy body dementia Diseases 0.000 description 1
- 229940086609 Lipase inhibitor Drugs 0.000 description 1
- 208000017170 Lipid metabolism disease Diseases 0.000 description 1
- 108010007859 Lisinopril Proteins 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 208000000185 Localized scleroderma Diseases 0.000 description 1
- DDWFXDSYGUXRAY-UHFFFAOYSA-N Luciferin Natural products CCc1c(C)c(CC2NC(=O)C(=C2C=C)C)[nH]c1Cc3[nH]c4C(=C5/NC(CC(=O)O)C(C)C5CC(=O)O)CC(=O)c4c3C DDWFXDSYGUXRAY-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 102100027256 Melanoma-associated antigen H1 Human genes 0.000 description 1
- 208000001145 Metabolic Syndrome Diseases 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 208000019695 Migraine disease Diseases 0.000 description 1
- 238000006751 Mitsunobu reaction Methods 0.000 description 1
- 206010053236 Mixed incontinence Diseases 0.000 description 1
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 1
- 206010027982 Morphoea Diseases 0.000 description 1
- 208000007101 Muscle Cramp Diseases 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- 108020001621 Natriuretic Peptide Proteins 0.000 description 1
- 102000004571 Natriuretic peptide Human genes 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 206010029164 Nephrotic syndrome Diseases 0.000 description 1
- 208000007256 Nevus Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- SNIOPGDIGTZGOP-UHFFFAOYSA-N Nitroglycerin Chemical compound [O-][N+](=O)OCC(O[N+]([O-])=O)CO[N+]([O-])=O SNIOPGDIGTZGOP-UHFFFAOYSA-N 0.000 description 1
- 239000000006 Nitroglycerin Substances 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 208000036576 Obstructive uropathy Diseases 0.000 description 1
- 208000022873 Ocular disease Diseases 0.000 description 1
- 208000010195 Onychomycosis Diseases 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 206010033645 Pancreatitis Diseases 0.000 description 1
- 206010033799 Paralysis Diseases 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 208000000450 Pelvic Pain Diseases 0.000 description 1
- QGMRQYFBGABWDR-UHFFFAOYSA-M Pentobarbital sodium Chemical compound [Na+].CCCC(C)C1(CC)C(=O)NC(=O)[N-]C1=O QGMRQYFBGABWDR-UHFFFAOYSA-M 0.000 description 1
- 229940122054 Peroxisome proliferator-activated receptor delta agonist Drugs 0.000 description 1
- 229940080774 Peroxisome proliferator-activated receptor gamma agonist Drugs 0.000 description 1
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical class ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 101000622060 Photinus pyralis Luciferin 4-monooxygenase Proteins 0.000 description 1
- 206010063985 Phytosterolaemia Diseases 0.000 description 1
- UJEWTUDSLQGTOA-UHFFFAOYSA-N Piretanide Chemical compound C=1C=CC=CC=1OC=1C(S(=O)(=O)N)=CC(C(O)=O)=CC=1N1CCCC1 UJEWTUDSLQGTOA-UHFFFAOYSA-N 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 206010037596 Pyelonephritis Diseases 0.000 description 1
- 208000003782 Raynaud disease Diseases 0.000 description 1
- 206010038423 Renal cyst Diseases 0.000 description 1
- 206010063897 Renal ischaemia Diseases 0.000 description 1
- 206010063837 Reperfusion injury Diseases 0.000 description 1
- 206010038748 Restrictive cardiomyopathy Diseases 0.000 description 1
- 208000025747 Rheumatic disease Diseases 0.000 description 1
- 206010039163 Right ventricular failure Diseases 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- 108091006614 SLC10A2 Proteins 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- 208000034189 Sclerosis Diseases 0.000 description 1
- 206010040047 Sepsis Diseases 0.000 description 1
- 206010040070 Septic Shock Diseases 0.000 description 1
- 206010040639 Sick sinus syndrome Diseases 0.000 description 1
- 208000002227 Sitosterolemia Diseases 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 208000005392 Spasm Diseases 0.000 description 1
- 208000007718 Stable Angina Diseases 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 206010066218 Stress Urinary Incontinence Diseases 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 208000003734 Supraventricular Tachycardia Diseases 0.000 description 1
- 206010065342 Supraventricular tachyarrhythmia Diseases 0.000 description 1
- 206010051379 Systemic Inflammatory Response Syndrome Diseases 0.000 description 1
- 208000008253 Systolic Heart Failure Diseases 0.000 description 1
- 208000001871 Tachycardia Diseases 0.000 description 1
- 208000001163 Tangier disease Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 101000712605 Theromyzon tessulatum Theromin Proteins 0.000 description 1
- IUJDSEJGGMCXSG-UHFFFAOYSA-N Thiopental Chemical compound CCCC(C)C1(CC)C(=O)NC(=S)NC1=O IUJDSEJGGMCXSG-UHFFFAOYSA-N 0.000 description 1
- 229940122388 Thrombin inhibitor Drugs 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- AUYYCJSJGJYCDS-LBPRGKRZSA-N Thyrolar Chemical compound IC1=CC(C[C@H](N)C(O)=O)=CC(I)=C1OC1=CC=C(O)C(I)=C1 AUYYCJSJGJYCDS-LBPRGKRZSA-N 0.000 description 1
- 208000009205 Tinnitus Diseases 0.000 description 1
- 208000018452 Torsade de pointes Diseases 0.000 description 1
- 208000002363 Torsades de Pointes Diseases 0.000 description 1
- NGBFQHCMQULJNZ-UHFFFAOYSA-N Torsemide Chemical compound CC(C)NC(=O)NS(=O)(=O)C1=CN=CC=C1NC1=CC=CC(C)=C1 NGBFQHCMQULJNZ-UHFFFAOYSA-N 0.000 description 1
- VXFJYXUZANRPDJ-WTNASJBWSA-N Trandopril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@H]2CCCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 VXFJYXUZANRPDJ-WTNASJBWSA-N 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- FNYLWPVRPXGIIP-UHFFFAOYSA-N Triamterene Chemical compound NC1=NC2=NC(N)=NC(N)=C2N=C1C1=CC=CC=C1 FNYLWPVRPXGIIP-UHFFFAOYSA-N 0.000 description 1
- XSTXAVWGXDQKEL-UHFFFAOYSA-N Trichloroethylene Chemical group ClC=C(Cl)Cl XSTXAVWGXDQKEL-UHFFFAOYSA-N 0.000 description 1
- 201000001943 Tricuspid Valve Insufficiency Diseases 0.000 description 1
- 206010044640 Tricuspid valve incompetence Diseases 0.000 description 1
- 206010044642 Tricuspid valve stenosis Diseases 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 208000007814 Unstable Angina Diseases 0.000 description 1
- 208000000921 Urge Urinary Incontinence Diseases 0.000 description 1
- 208000012931 Urologic disease Diseases 0.000 description 1
- SECKRCOLJRRGGV-UHFFFAOYSA-N Vardenafil Chemical compound CCCC1=NC(C)=C(C(N=2)=O)N1NC=2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(CC)CC1 SECKRCOLJRRGGV-UHFFFAOYSA-N 0.000 description 1
- 201000004810 Vascular dementia Diseases 0.000 description 1
- 206010047115 Vasculitis Diseases 0.000 description 1
- 206010047249 Venous thrombosis Diseases 0.000 description 1
- 206010065341 Ventricular tachyarrhythmia Diseases 0.000 description 1
- 201000008485 Wernicke-Korsakoff syndrome Diseases 0.000 description 1
- 201000008803 Wolff-Parkinson-white syndrome Diseases 0.000 description 1
- 206010048215 Xanthomatosis Diseases 0.000 description 1
- VORIUEAZEKLUSJ-UHFFFAOYSA-M [(6-chlorobenzotriazol-1-yl)oxy-(dimethylamino)methylidene]-dimethylazanium;trifluoroborane;fluoride Chemical compound [F-].FB(F)F.C1=C(Cl)C=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 VORIUEAZEKLUSJ-UHFFFAOYSA-M 0.000 description 1
- FGCDPOXCALLWBW-UHFFFAOYSA-N [1-[[8-[(2,6-difluorophenyl)methoxy]-2-methylimidazo[1,2-a]pyridine-3-carbonyl]amino]-5,5,5-trifluoro-2-methylpentan-2-yl] carbamate Chemical compound C(N)(OC(CNC(=O)C1=C(N=C2N1C=CC=C2OCC1=C(C=CC=C1F)F)C)(CCC(F)(F)F)C)=O FGCDPOXCALLWBW-UHFFFAOYSA-N 0.000 description 1
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 1
- 229960000446 abciximab Drugs 0.000 description 1
- 210000000683 abdominal cavity Anatomy 0.000 description 1
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 description 1
- 210000003815 abdominal wall Anatomy 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000036982 action potential Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 231100000851 acute glomerulonephritis Toxicity 0.000 description 1
- 201000011040 acute kidney failure Diseases 0.000 description 1
- 201000005180 acute myocarditis Diseases 0.000 description 1
- 208000012998 acute renal failure Diseases 0.000 description 1
- 125000004442 acylamino group Chemical group 0.000 description 1
- 229950000221 adaprolol Drugs 0.000 description 1
- IPGLIOFIFLXLKR-AXYNENQYSA-N adaprolol Chemical compound C1=CC(OCC(O)CNC(C)C)=CC=C1CC(=O)OCCC1(C2)C[C@@H](C3)C[C@H]2C[C@@H]3C1 IPGLIOFIFLXLKR-AXYNENQYSA-N 0.000 description 1
- 230000001464 adherent effect Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 201000000028 adult respiratory distress syndrome Diseases 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000002170 aldosterone antagonist Substances 0.000 description 1
- 229960004601 aliskiren Drugs 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229960002213 alprenolol Drugs 0.000 description 1
- PAZJSJFMUHDSTF-UHFFFAOYSA-N alprenolol Chemical compound CC(C)NCC(O)COC1=CC=CC=C1CC=C PAZJSJFMUHDSTF-UHFFFAOYSA-N 0.000 description 1
- 229960002414 ambrisentan Drugs 0.000 description 1
- OUJTZYPIHDYQMC-LJQANCHMSA-N ambrisentan Chemical compound O([C@@H](C(OC)(C=1C=CC=CC=1)C=1C=CC=CC=1)C(O)=O)C1=NC(C)=CC(C)=N1 OUJTZYPIHDYQMC-LJQANCHMSA-N 0.000 description 1
- 238000010640 amide synthesis reaction Methods 0.000 description 1
- XSDQTOBWRPYKKA-UHFFFAOYSA-N amiloride Chemical compound NC(=N)NC(=O)C1=NC(Cl)=C(N)N=C1N XSDQTOBWRPYKKA-UHFFFAOYSA-N 0.000 description 1
- 229960002576 amiloride Drugs 0.000 description 1
- 229960000528 amlodipine Drugs 0.000 description 1
- ZPBWCRDSRKPIDG-UHFFFAOYSA-N amlodipine benzenesulfonate Chemical compound OS(=O)(=O)C1=CC=CC=C1.CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1Cl ZPBWCRDSRKPIDG-UHFFFAOYSA-N 0.000 description 1
- 239000001099 ammonium carbonate Substances 0.000 description 1
- 235000012501 ammonium carbonate Nutrition 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- MZZLGJHLQGUVPN-HAWMADMCSA-N anacetrapib Chemical compound COC1=CC(F)=C(C(C)C)C=C1C1=CC=C(C(F)(F)F)C=C1CN1C(=O)O[C@H](C=2C=C(C=C(C=2)C(F)(F)F)C(F)(F)F)[C@@H]1C MZZLGJHLQGUVPN-HAWMADMCSA-N 0.000 description 1
- 229950000285 anacetrapib Drugs 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 230000000202 analgesic effect Effects 0.000 description 1
- 208000003455 anaphylaxis Diseases 0.000 description 1
- 208000007502 anemia Diseases 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000454 anti-cipatory effect Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000003698 antivitamin K Substances 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 210000000709 aorta Anatomy 0.000 description 1
- 210000002376 aorta thoracic Anatomy 0.000 description 1
- 206010002906 aortic stenosis Diseases 0.000 description 1
- 201000002064 aortic valve insufficiency Diseases 0.000 description 1
- 229960003886 apixaban Drugs 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 229940114079 arachidonic acid Drugs 0.000 description 1
- 235000021342 arachidonic acid Nutrition 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 230000006793 arrhythmia Effects 0.000 description 1
- 230000004872 arterial blood pressure Effects 0.000 description 1
- 210000001367 artery Anatomy 0.000 description 1
- 230000001174 ascending effect Effects 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229960005370 atorvastatin Drugs 0.000 description 1
- 230000001746 atrial effect Effects 0.000 description 1
- RKUNBYITZUJHSG-SPUOUPEWSA-N atropine Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)N2C)C(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-SPUOUPEWSA-N 0.000 description 1
- 229960000396 atropine Drugs 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 229950010046 avasimibe Drugs 0.000 description 1
- 229950003799 axitirome Drugs 0.000 description 1
- RQPZNWPYLFFXCP-UHFFFAOYSA-L barium dihydroxide Chemical compound [OH-].[OH-].[Ba+2] RQPZNWPYLFFXCP-UHFFFAOYSA-L 0.000 description 1
- 229910001863 barium hydroxide Inorganic materials 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- RROBIDXNTUAHFW-UHFFFAOYSA-N benzotriazol-1-yloxy-tris(dimethylamino)phosphanium Chemical compound C1=CC=C2N(O[P+](N(C)C)(N(C)C)N(C)C)N=NC2=C1 RROBIDXNTUAHFW-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 229960004324 betaxolol Drugs 0.000 description 1
- CHDPSNLJFOQTRK-UHFFFAOYSA-N betaxolol hydrochloride Chemical compound [Cl-].C1=CC(OCC(O)C[NH2+]C(C)C)=CC=C1CCOCC1CC1 CHDPSNLJFOQTRK-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 229960002781 bisoprolol Drugs 0.000 description 1
- VHYCDWMUTMEGQY-UHFFFAOYSA-N bisoprolol Chemical compound CC(C)NCC(O)COC1=CC=C(COCCOC(C)C)C=C1 VHYCDWMUTMEGQY-UHFFFAOYSA-N 0.000 description 1
- OIRCOABEOLEUMC-GEJPAHFPSA-N bivalirudin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)CNC(=O)CNC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 OIRCOABEOLEUMC-GEJPAHFPSA-N 0.000 description 1
- 229960001500 bivalirudin Drugs 0.000 description 1
- 108010055460 bivalirudin Proteins 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 230000036765 blood level Effects 0.000 description 1
- 238000010241 blood sampling Methods 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 230000004097 bone metabolism Effects 0.000 description 1
- 229960003065 bosentan Drugs 0.000 description 1
- SXTRWVVIEPWAKM-UHFFFAOYSA-N bosentan hydrate Chemical compound O.COC1=CC=CC=C1OC(C(=NC(=N1)C=2N=CC=CN=2)OCCO)=C1NS(=O)(=O)C1=CC=C(C(C)(C)C)C=C1 SXTRWVVIEPWAKM-UHFFFAOYSA-N 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 208000006752 brain edema Diseases 0.000 description 1
- 239000006189 buccal tablet Substances 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 229960004064 bumetanide Drugs 0.000 description 1
- MAEIEVLCKWDQJH-UHFFFAOYSA-N bumetanide Chemical compound CCCCNC1=CC(C(O)=O)=CC(S(N)(=O)=O)=C1OC1=CC=CC=C1 MAEIEVLCKWDQJH-UHFFFAOYSA-N 0.000 description 1
- 229960000330 bupranolol Drugs 0.000 description 1
- HQIRNZOQPUAHHV-UHFFFAOYSA-N bupranolol Chemical compound CC1=CC=C(Cl)C(OCC(O)CNC(C)(C)C)=C1 HQIRNZOQPUAHHV-UHFFFAOYSA-N 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 229960005069 calcium Drugs 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 239000000480 calcium channel blocker Substances 0.000 description 1
- LLSDKQJKOVVTOJ-UHFFFAOYSA-L calcium chloride dihydrate Chemical compound O.O.[Cl-].[Cl-].[Ca+2] LLSDKQJKOVVTOJ-UHFFFAOYSA-L 0.000 description 1
- 229940052299 calcium chloride dihydrate Drugs 0.000 description 1
- ZJKZKKPIKDNHDM-UHFFFAOYSA-L calcium;6-(5-carboxylato-5-methylhexoxy)-2,2-dimethylhexanoate Chemical compound [Ca+2].[O-]C(=O)C(C)(C)CCCCOCCCCC(C)(C)C([O-])=O ZJKZKKPIKDNHDM-UHFFFAOYSA-L 0.000 description 1
- 208000035269 cancer or benign tumor Diseases 0.000 description 1
- SGZAIDDFHDDFJU-UHFFFAOYSA-N candesartan Chemical compound CCOC1=NC2=CC=CC(C(O)=O)=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SGZAIDDFHDDFJU-UHFFFAOYSA-N 0.000 description 1
- 229960000932 candesartan Drugs 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 230000023852 carbohydrate metabolic process Effects 0.000 description 1
- 235000021256 carbohydrate metabolism Nutrition 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 1
- 206010007625 cardiogenic shock Diseases 0.000 description 1
- PUXBGTOOZJQSKH-UHFFFAOYSA-N carprofen Chemical compound C1=C(Cl)C=C2C3=CC=C(C(C(O)=O)C)C=C3NC2=C1 PUXBGTOOZJQSKH-UHFFFAOYSA-N 0.000 description 1
- 229960001222 carteolol Drugs 0.000 description 1
- LWAFSWPYPHEXKX-UHFFFAOYSA-N carteolol Chemical compound N1C(=O)CCC2=C1C=CC=C2OCC(O)CNC(C)(C)C LWAFSWPYPHEXKX-UHFFFAOYSA-N 0.000 description 1
- 229960004195 carvedilol Drugs 0.000 description 1
- NPAKNKYSJIDKMW-UHFFFAOYSA-N carvedilol Chemical compound COC1=CC=CC=C1OCCNCC(O)COC1=CC=CC2=NC3=CC=C[CH]C3=C12 NPAKNKYSJIDKMW-UHFFFAOYSA-N 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 229960002320 celiprolol Drugs 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 206010008118 cerebral infarction Diseases 0.000 description 1
- 230000003788 cerebral perfusion Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- KXZJHVJKXJLBKO-UHFFFAOYSA-N chembl1408157 Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=C(O)C=C1 KXZJHVJKXJLBKO-UHFFFAOYSA-N 0.000 description 1
- NPNSVNGQJGRSNR-UHFFFAOYSA-N chembl73193 Chemical compound N=1C(OC=2C(=CC=C(C=2)C(N)=N)O)=C(F)C(N(CC(O)=O)C)=C(F)C=1OC(C=1)=CC=CC=1C1=NCCN1C NPNSVNGQJGRSNR-UHFFFAOYSA-N 0.000 description 1
- 238000000451 chemical ionisation Methods 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 229960001701 chloroform Drugs 0.000 description 1
- 229960001523 chlortalidone Drugs 0.000 description 1
- 229940125881 cholesteryl ester transfer protein inhibitor Drugs 0.000 description 1
- 208000022831 chronic renal failure syndrome Diseases 0.000 description 1
- 235000019504 cigarettes Nutrition 0.000 description 1
- 230000007882 cirrhosis Effects 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 208000024980 claudication Diseases 0.000 description 1
- GKTWGGQPFAXNFI-HNNXBMFYSA-N clopidogrel Chemical compound C1([C@H](N2CC=3C=CSC=3CC2)C(=O)OC)=CC=CC=C1Cl GKTWGGQPFAXNFI-HNNXBMFYSA-N 0.000 description 1
- 229960003009 clopidogrel Drugs 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- GMRWGQCZJGVHKL-UHFFFAOYSA-N colestipol Chemical compound ClCC1CO1.NCCNCCNCCNCCN GMRWGQCZJGVHKL-UHFFFAOYSA-N 0.000 description 1
- 229960002604 colestipol Drugs 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000004590 computer program Methods 0.000 description 1
- 208000028831 congenital heart disease Diseases 0.000 description 1
- 208000018631 connective tissue disease Diseases 0.000 description 1
- 239000002872 contrast media Substances 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 238000007887 coronary angioplasty Methods 0.000 description 1
- 208000029078 coronary artery disease Diseases 0.000 description 1
- 210000004351 coronary vessel Anatomy 0.000 description 1
- 229960000956 coumarin Drugs 0.000 description 1
- 235000001671 coumarin Nutrition 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 201000010549 croup Diseases 0.000 description 1
- 229960003850 dabigatran Drugs 0.000 description 1
- YBSJFWOBGCMAKL-UHFFFAOYSA-N dabigatran Chemical compound N=1C2=CC(C(=O)N(CCC(O)=O)C=3N=CC=CC=3)=CC=C2N(C)C=1CNC1=CC=C(C(N)=N)C=C1 YBSJFWOBGCMAKL-UHFFFAOYSA-N 0.000 description 1
- 229950004181 dalcetrapib Drugs 0.000 description 1
- IJNIQYINMSGIPS-UHFFFAOYSA-N darexaban Chemical compound C1=CC(OC)=CC=C1C(=O)NC1=CC=CC(O)=C1NC(=O)C1=CC=C(N2CCN(C)CCC2)C=C1 IJNIQYINMSGIPS-UHFFFAOYSA-N 0.000 description 1
- FEJVSJIALLTFRP-LJQANCHMSA-N darusentan Chemical compound COC1=CC(OC)=NC(O[C@H](C(O)=O)C(OC)(C=2C=CC=CC=2)C=2C=CC=CC=2)=N1 FEJVSJIALLTFRP-LJQANCHMSA-N 0.000 description 1
- 229950008833 darusentan Drugs 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 229960002887 deanol Drugs 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 229960005227 delapril Drugs 0.000 description 1
- WOUOLAUOZXOLJQ-MBSDFSHPSA-N delapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N(CC(O)=O)C1CC2=CC=CC=C2C1)CC1=CC=CC=C1 WOUOLAUOZXOLJQ-MBSDFSHPSA-N 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 229960001767 dextrothyroxine Drugs 0.000 description 1
- 201000009101 diabetic angiopathy Diseases 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- ZWWWLCMDTZFSOO-UHFFFAOYSA-N diethoxyphosphorylformonitrile Chemical compound CCOP(=O)(C#N)OCC ZWWWLCMDTZFSOO-UHFFFAOYSA-N 0.000 description 1
- FPUQGCOBYOXAED-UHFFFAOYSA-N diethyl 2-[[2-[3-(dimethylcarbamoyl)-4-[[2-[4-(trifluoromethyl)phenyl]benzoyl]amino]phenyl]acetyl]oxymethyl]-2-phenylpropanedioate Chemical compound C=1C=CC=CC=1C(C(=O)OCC)(C(=O)OCC)COC(=O)CC(C=C1C(=O)N(C)C)=CC=C1NC(=O)C1=CC=CC=C1C1=CC=C(C(F)(F)F)C=C1 FPUQGCOBYOXAED-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- 230000010339 dilation Effects 0.000 description 1
- 229960004166 diltiazem Drugs 0.000 description 1
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 229960002768 dipyridamole Drugs 0.000 description 1
- IZEKFCXSFNUWAM-UHFFFAOYSA-N dipyridamole Chemical compound C=12N=C(N(CCO)CCO)N=C(N3CCCCC3)C2=NC(N(CCO)CCO)=NC=1N1CCCCC1 IZEKFCXSFNUWAM-UHFFFAOYSA-N 0.000 description 1
- XRKMNJXYOFSTBE-UHFFFAOYSA-N disodium;iron(4+);nitroxyl anion;pentacyanide;dihydrate Chemical compound O.O.[Na+].[Na+].[Fe+4].N#[C-].N#[C-].N#[C-].N#[C-].N#[C-].O=[N-] XRKMNJXYOFSTBE-UHFFFAOYSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- GUVUOGQBMYCBQP-UHFFFAOYSA-N dmpu Chemical compound CN1CCCN(C)C1=O GUVUOGQBMYCBQP-UHFFFAOYSA-N 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 238000009513 drug distribution Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 229960000622 edoxaban Drugs 0.000 description 1
- PSMMNJNZVZZNOI-SJILXJHISA-N edoxaban tosylate hydrate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1.N([C@H]1CC[C@@H](C[C@H]1NC(=O)C=1SC=2CN(C)CCC=2N=1)C(=O)N(C)C)C(=O)C(=O)NC1=CC=C(Cl)C=N1 PSMMNJNZVZZNOI-SJILXJHISA-N 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 229950005925 eflucimibe Drugs 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- XFLQIRAKKLNXRQ-UUWRZZSWSA-N elobixibat Chemical compound C12=CC(SC)=C(OCC(=O)N[C@@H](C(=O)NCC(O)=O)C=3C=CC=CC=3)C=C2S(=O)(=O)CC(CCCC)(CCCC)CN1C1=CC=CC=C1 XFLQIRAKKLNXRQ-UUWRZZSWSA-N 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 229960000873 enalapril Drugs 0.000 description 1
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 1
- JJJFUHOGVZWXNQ-UHFFFAOYSA-N enbucrilate Chemical compound CCCCOC(=O)C(=C)C#N JJJFUHOGVZWXNQ-UHFFFAOYSA-N 0.000 description 1
- 206010014665 endocarditis Diseases 0.000 description 1
- 201000010048 endomyocardial fibrosis Diseases 0.000 description 1
- 230000008694 endothelial dysfunction Effects 0.000 description 1
- 230000003511 endothelial effect Effects 0.000 description 1
- 239000002308 endothelin receptor antagonist Substances 0.000 description 1
- 210000003038 endothelium Anatomy 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 229960002711 epanolol Drugs 0.000 description 1
- 229960003745 esmolol Drugs 0.000 description 1
- AQNDDEOPVVGCPG-UHFFFAOYSA-N esmolol Chemical compound COC(=O)CCC1=CC=C(OCC(O)CNC(C)C)C=C1 AQNDDEOPVVGCPG-UHFFFAOYSA-N 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- PUSKHXMZPOMNTQ-UHFFFAOYSA-N ethyl 2,1,3-benzoselenadiazole-5-carboxylate Chemical compound CCOC(=O)C1=CC=C2N=[Se]=NC2=C1 PUSKHXMZPOMNTQ-UHFFFAOYSA-N 0.000 description 1
- YYSVZZWIGYOHLH-UHFFFAOYSA-N ethyl 6-chloro-8-[(2,6-difluorophenyl)methoxy]-2-methylimidazo[1,2-a]pyridine-3-carboxylate Chemical compound C=1C(Cl)=CN2C(C(=O)OCC)=C(C)N=C2C=1OCC1=C(F)C=CC=C1F YYSVZZWIGYOHLH-UHFFFAOYSA-N 0.000 description 1
- SFNALCNOMXIBKG-UHFFFAOYSA-N ethylene glycol monododecyl ether Chemical compound CCCCCCCCCCCCOCCO SFNALCNOMXIBKG-UHFFFAOYSA-N 0.000 description 1
- OLNTVTPDXPETLC-XPWALMASSA-N ezetimibe Chemical compound N1([C@@H]([C@H](C1=O)CC[C@H](O)C=1C=CC(F)=CC=1)C=1C=CC(O)=CC=1)C1=CC=C(F)C=C1 OLNTVTPDXPETLC-XPWALMASSA-N 0.000 description 1
- 229960000815 ezetimibe Drugs 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 210000001105 femoral artery Anatomy 0.000 description 1
- 210000003191 femoral vein Anatomy 0.000 description 1
- 229940012952 fibrinogen Drugs 0.000 description 1
- 229950010034 fidexaban Drugs 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 238000013100 final test Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229960003765 fluvastatin Drugs 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 229960001318 fondaparinux Drugs 0.000 description 1
- KANJSNBRCNMZMV-ABRZTLGGSA-N fondaparinux Chemical compound O[C@@H]1[C@@H](NS(O)(=O)=O)[C@@H](OC)O[C@H](COS(O)(=O)=O)[C@H]1O[C@H]1[C@H](OS(O)(=O)=O)[C@@H](O)[C@H](O[C@@H]2[C@@H]([C@@H](OS(O)(=O)=O)[C@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O[C@@H]4[C@@H]([C@@H](O)[C@H](O)[C@@H](COS(O)(=O)=O)O4)NS(O)(=O)=O)[C@H](O3)C(O)=O)O)[C@@H](COS(O)(=O)=O)O2)NS(O)(=O)=O)[C@H](C(O)=O)O1 KANJSNBRCNMZMV-ABRZTLGGSA-N 0.000 description 1
- 230000002431 foraging effect Effects 0.000 description 1
- 229960002490 fosinopril Drugs 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 210000001652 frontal lobe Anatomy 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 229960003883 furosemide Drugs 0.000 description 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 208000001288 gastroparesis Diseases 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 229940052308 general anesthetics halogenated hydrocarbons Drugs 0.000 description 1
- 230000001434 glomerular Effects 0.000 description 1
- 206010061989 glomerulosclerosis Diseases 0.000 description 1
- 229960003711 glyceryl trinitrate Drugs 0.000 description 1
- 230000002650 habitual effect Effects 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- 210000003709 heart valve Anatomy 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- 229910052734 helium Inorganic materials 0.000 description 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 239000002628 heparin derivative Substances 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- 238000004896 high resolution mass spectrometry Methods 0.000 description 1
- 230000003054 hormonal effect Effects 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 229960002003 hydrochlorothiazide Drugs 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- 201000005991 hyperphosphatemia Diseases 0.000 description 1
- 230000001631 hypertensive effect Effects 0.000 description 1
- 208000006575 hypertriglyceridemia Diseases 0.000 description 1
- 206010020871 hypertrophic cardiomyopathy Diseases 0.000 description 1
- 230000001969 hypertrophic effect Effects 0.000 description 1
- 208000026621 hypolipoproteinemia Diseases 0.000 description 1
- 230000036543 hypotension Effects 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 208000022368 idiopathic cardiomyopathy Diseases 0.000 description 1
- UTCSSFWDNNEEBH-UHFFFAOYSA-N imidazo[1,2-a]pyridine Chemical group C1=CC=CC2=NC=CN21 UTCSSFWDNNEEBH-UHFFFAOYSA-N 0.000 description 1
- 150000005234 imidazo[1,2-a]pyridines Chemical class 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 229950005809 implitapide Drugs 0.000 description 1
- 201000001881 impotence Diseases 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- NDDAHWYSQHTHNT-UHFFFAOYSA-N indapamide Chemical compound CC1CC2=CC=CC=C2N1NC(=O)C1=CC=C(Cl)C(S(N)(=O)=O)=C1 NDDAHWYSQHTHNT-UHFFFAOYSA-N 0.000 description 1
- 229960004569 indapamide Drugs 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 239000001023 inorganic pigment Substances 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- UQSXHKLRYXJYBZ-UHFFFAOYSA-N iron oxide Inorganic materials [Fe]=O UQSXHKLRYXJYBZ-UHFFFAOYSA-N 0.000 description 1
- 235000013980 iron oxide Nutrition 0.000 description 1
- VBMVTYDPPZVILR-UHFFFAOYSA-N iron(2+);oxygen(2-) Chemical class [O-2].[Fe+2] VBMVTYDPPZVILR-UHFFFAOYSA-N 0.000 description 1
- 208000002551 irritable bowel syndrome Diseases 0.000 description 1
- 230000000302 ischemic effect Effects 0.000 description 1
- 229960002725 isoflurane Drugs 0.000 description 1
- DXDRHHKMWQZJHT-FPYGCLRLSA-N isoliquiritigenin Chemical compound C1=CC(O)=CC=C1\C=C\C(=O)C1=CC=C(O)C=C1O DXDRHHKMWQZJHT-FPYGCLRLSA-N 0.000 description 1
- JBQATDIMBVLPRB-UHFFFAOYSA-N isoliquiritigenin Natural products OC1=CC(O)=CC=C1C1OC2=CC(O)=CC=C2C(=O)C1 JBQATDIMBVLPRB-UHFFFAOYSA-N 0.000 description 1
- 235000008718 isoliquiritigenin Nutrition 0.000 description 1
- MOYKHGMNXAOIAT-JGWLITMVSA-N isosorbide dinitrate Chemical compound [O-][N+](=O)O[C@H]1CO[C@@H]2[C@H](O[N+](=O)[O-])CO[C@@H]21 MOYKHGMNXAOIAT-JGWLITMVSA-N 0.000 description 1
- 229960000201 isosorbide dinitrate Drugs 0.000 description 1
- YWXYYJSYQOXTPL-SLPGGIOYSA-N isosorbide mononitrate Chemical compound [O-][N+](=O)O[C@@H]1CO[C@@H]2[C@@H](O)CO[C@@H]21 YWXYYJSYQOXTPL-SLPGGIOYSA-N 0.000 description 1
- 229960003827 isosorbide mononitrate Drugs 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical class C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- 210000004731 jugular vein Anatomy 0.000 description 1
- 210000001117 keloid Anatomy 0.000 description 1
- 229960001632 labetalol Drugs 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 229950005241 landiolol Drugs 0.000 description 1
- WMDSZGFJQKSLLH-RBBKRZOGSA-N landiolol Chemical compound O1C(C)(C)OC[C@H]1COC(=O)CCC(C=C1)=CC=C1OC[C@@H](O)CNCCNC(=O)N1CCOCC1 WMDSZGFJQKSLLH-RBBKRZOGSA-N 0.000 description 1
- 201000003723 learning disability Diseases 0.000 description 1
- 230000007786 learning performance Effects 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- FKDHHVKWGRFRTG-UHFFFAOYSA-N linsidomine Chemical compound [N-]1OC(=N)C=[N+]1N1CCOCC1 FKDHHVKWGRFRTG-UHFFFAOYSA-N 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 229960002394 lisinopril Drugs 0.000 description 1
- CZRQXSDBMCMPNJ-ZUIPZQNBSA-N lisinopril dihydrate Chemical compound O.O.C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 CZRQXSDBMCMPNJ-ZUIPZQNBSA-N 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 230000006742 locomotor activity Effects 0.000 description 1
- 229960003566 lomitapide Drugs 0.000 description 1
- QKVKOFVWUHNEBX-UHFFFAOYSA-N lomitapide mesylate Chemical compound CS(O)(=O)=O.C12=CC=CC=C2C2=CC=CC=C2C1(C(=O)NCC(F)(F)F)CCCCN(CC1)CCC1NC(=O)C1=CC=CC=C1C1=CC=C(C(F)(F)F)C=C1 QKVKOFVWUHNEBX-UHFFFAOYSA-N 0.000 description 1
- 239000002171 loop diuretic Substances 0.000 description 1
- 229960004773 losartan Drugs 0.000 description 1
- KJJZZJSZUJXYEA-UHFFFAOYSA-N losartan Chemical compound CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2[N]N=NN=2)C=C1 KJJZZJSZUJXYEA-UHFFFAOYSA-N 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- DLBFLQKQABVKGT-UHFFFAOYSA-L lucifer yellow dye Chemical compound [Li+].[Li+].[O-]S(=O)(=O)C1=CC(C(N(C(=O)NN)C2=O)=O)=C3C2=CC(S([O-])(=O)=O)=CC3=C1N DLBFLQKQABVKGT-UHFFFAOYSA-L 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- WRUGWIBCXHJTDG-UHFFFAOYSA-L magnesium sulfate heptahydrate Chemical compound O.O.O.O.O.O.O.[Mg+2].[O-]S([O-])(=O)=O WRUGWIBCXHJTDG-UHFFFAOYSA-L 0.000 description 1
- 229940061634 magnesium sulfate heptahydrate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- DKWNMCUOEDMMIN-PKOBYXMFSA-N melagatran Chemical compound C1=CC(C(=N)N)=CC=C1CNC(=O)[C@H]1N(C(=O)[C@H](NCC(O)=O)C2CCCCC2)CC1 DKWNMCUOEDMMIN-PKOBYXMFSA-N 0.000 description 1
- 229960002137 melagatran Drugs 0.000 description 1
- 229950008446 melinamide Drugs 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000006984 memory degeneration Effects 0.000 description 1
- 208000023060 memory loss Diseases 0.000 description 1
- 230000007334 memory performance Effects 0.000 description 1
- 229960003134 mepindolol Drugs 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- YLGXILFCIXHCMC-JHGZEJCSSA-N methyl cellulose Chemical compound COC1C(OC)C(OC)C(COC)O[C@H]1O[C@H]1C(OC)C(OC)C(OC)OC1COC YLGXILFCIXHCMC-JHGZEJCSSA-N 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 229960002237 metoprolol Drugs 0.000 description 1
- IUBSYMUCCVWXPE-UHFFFAOYSA-N metoprolol Chemical compound COCCC1=CC=C(OCC(O)CNC(C)C)C=C1 IUBSYMUCCVWXPE-UHFFFAOYSA-N 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 206010027599 migraine Diseases 0.000 description 1
- 208000027061 mild cognitive impairment Diseases 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 239000002394 mineralocorticoid antagonist Substances 0.000 description 1
- 208000005907 mitral valve insufficiency Diseases 0.000 description 1
- 208000006887 mitral valve stenosis Diseases 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- XLFWDASMENKTKL-UHFFFAOYSA-N molsidomine Chemical compound O1C(N=C([O-])OCC)=C[N+](N2CCOCC2)=N1 XLFWDASMENKTKL-UHFFFAOYSA-N 0.000 description 1
- 229960004027 molsidomine Drugs 0.000 description 1
- VYQNWZOUAUKGHI-UHFFFAOYSA-N monobenzone Chemical compound C1=CC(O)=CC=C1OCC1=CC=CC=C1 VYQNWZOUAUKGHI-UHFFFAOYSA-N 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- 208000029744 multiple organ dysfunction syndrome Diseases 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 206010028537 myelofibrosis Diseases 0.000 description 1
- 208000031225 myocardial ischemia Diseases 0.000 description 1
- 229960004255 nadolol Drugs 0.000 description 1
- VWPOSFSPZNDTMJ-UCWKZMIHSA-N nadolol Chemical compound C1[C@@H](O)[C@@H](O)CC2=C1C=CC=C2OCC(O)CNC(C)(C)C VWPOSFSPZNDTMJ-UCWKZMIHSA-N 0.000 description 1
- YZMHQCWXYHARLS-UHFFFAOYSA-N naphthalene-1,2-disulfonic acid Chemical compound C1=CC=CC2=C(S(O)(=O)=O)C(S(=O)(=O)O)=CC=C21 YZMHQCWXYHARLS-UHFFFAOYSA-N 0.000 description 1
- 239000007923 nasal drop Substances 0.000 description 1
- 229940100662 nasal drops Drugs 0.000 description 1
- 239000000692 natriuretic peptide Substances 0.000 description 1
- 229920005615 natural polymer Polymers 0.000 description 1
- 229960000619 nebivolol Drugs 0.000 description 1
- 229940105631 nembutal Drugs 0.000 description 1
- 201000008383 nephritis Diseases 0.000 description 1
- 230000002580 nephropathic effect Effects 0.000 description 1
- 230000007372 neural signaling Effects 0.000 description 1
- 230000001272 neurogenic effect Effects 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-M nicotinate Chemical compound [O-]C(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-M 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- HYIMSNHJOBLJNT-UHFFFAOYSA-N nifedipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=O HYIMSNHJOBLJNT-UHFFFAOYSA-N 0.000 description 1
- 229960001597 nifedipine Drugs 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- MVPQUSQUURLQKF-MCPDASDXSA-E nonasodium;(2s,3s,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3s,4s,5r,6r)-2-carboxylato-4,5-dimethoxy-6-[(2r,3r,4s,5r,6s)-6-methoxy-4,5-disulfonatooxy-2-(sulfonatooxymethyl)oxan-3-yl]oxyoxan-3-yl]oxy-4,5-disulfonatooxy-2-(sulfonatooxymethyl)oxan-3-yl]oxy-4,5-di Chemical compound [Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[O-]S(=O)(=O)O[C@@H]1[C@@H](OS([O-])(=O)=O)[C@@H](OC)O[C@H](COS([O-])(=O)=O)[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@@H]2[C@@H]([C@@H](OS([O-])(=O)=O)[C@H](O[C@H]3[C@@H]([C@@H](OC)[C@H](O[C@@H]4[C@@H]([C@@H](OC)[C@H](OC)[C@@H](COS([O-])(=O)=O)O4)OC)[C@H](O3)C([O-])=O)OC)[C@@H](COS([O-])(=O)=O)O2)OS([O-])(=O)=O)[C@H](C([O-])=O)O1 MVPQUSQUURLQKF-MCPDASDXSA-E 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 229940126701 oral medication Drugs 0.000 description 1
- 229940100688 oral solution Drugs 0.000 description 1
- 229940100692 oral suspension Drugs 0.000 description 1
- 239000006191 orally-disintegrating tablet Substances 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- AHLBNYSZXLDEJQ-FWEHEUNISA-N orlistat Chemical compound CCCCCCCCCCC[C@H](OC(=O)[C@H](CC(C)C)NC=O)C[C@@H]1OC(=O)[C@H]1CCCCCC AHLBNYSZXLDEJQ-FWEHEUNISA-N 0.000 description 1
- 229960001243 orlistat Drugs 0.000 description 1
- 229950009478 otamixaban Drugs 0.000 description 1
- PFGVNLZDWRZPJW-OPAMFIHVSA-N otamixaban Chemical compound C([C@@H](C(=O)OC)[C@@H](C)NC(=O)C=1C=CC(=CC=1)C=1C=C[N+]([O-])=CC=1)C1=CC=CC(C(N)=N)=C1 PFGVNLZDWRZPJW-OPAMFIHVSA-N 0.000 description 1
- 208000024449 overflow incontinence Diseases 0.000 description 1
- 229960004570 oxprenolol Drugs 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 229950003510 pactimibe Drugs 0.000 description 1
- 230000036407 pain Effects 0.000 description 1
- 208000021090 palsy Diseases 0.000 description 1
- VWMZIGBYZQUQOA-QEEMJVPDSA-N pamaqueside Chemical compound O([C@@H]1[C@@H](CO)O[C@H]([C@@H]([C@H]1O)O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4C[C@H]5[C@@H]([C@]4(CC(=O)[C@@H]3[C@@]2(C)CC1)C)[C@@H]([C@]1(OC[C@H](C)CC1)O5)C)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O VWMZIGBYZQUQOA-QEEMJVPDSA-N 0.000 description 1
- 229950005482 pamaqueside Drugs 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000004963 pathophysiological condition Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 229960002035 penbutolol Drugs 0.000 description 1
- KQXKVJAGOJTNJS-HNNXBMFYSA-N penbutolol Chemical compound CC(C)(C)NC[C@H](O)COC1=CC=CC=C1C1CCCC1 KQXKVJAGOJTNJS-HNNXBMFYSA-N 0.000 description 1
- 230000008447 perception Effects 0.000 description 1
- 208000008494 pericarditis Diseases 0.000 description 1
- 229960002582 perindopril Drugs 0.000 description 1
- IPVQLZZIHOAWMC-QXKUPLGCSA-N perindopril Chemical compound C1CCC[C@H]2C[C@@H](C(O)=O)N(C(=O)[C@H](C)N[C@@H](CCC)C(=O)OCC)[C@H]21 IPVQLZZIHOAWMC-QXKUPLGCSA-N 0.000 description 1
- 208000027232 peripheral nervous system disease Diseases 0.000 description 1
- 208000033808 peripheral neuropathy Diseases 0.000 description 1
- 206010034674 peritonitis Diseases 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- SONNWYBIRXJNDC-VIFPVBQESA-N phenylephrine Chemical compound CNC[C@H](O)C1=CC=CC(O)=C1 SONNWYBIRXJNDC-VIFPVBQESA-N 0.000 description 1
- 229960001802 phenylephrine Drugs 0.000 description 1
- 150000004031 phenylhydrazines Chemical class 0.000 description 1
- PJNZPQUBCPKICU-UHFFFAOYSA-N phosphoric acid;potassium Chemical compound [K].OP(O)(O)=O PJNZPQUBCPKICU-UHFFFAOYSA-N 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 229960002508 pindolol Drugs 0.000 description 1
- JZQKKSLKJUAGIC-UHFFFAOYSA-N pindolol Chemical compound CC(C)NCC(O)COC1=CC=CC2=C1C=CN2 JZQKKSLKJUAGIC-UHFFFAOYSA-N 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 229960001085 piretanide Drugs 0.000 description 1
- 229960002797 pitavastatin Drugs 0.000 description 1
- VGYFMXBACGZSIL-MCBHFWOFSA-N pitavastatin Chemical compound OC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 VGYFMXBACGZSIL-MCBHFWOFSA-N 0.000 description 1
- 230000010118 platelet activation Effects 0.000 description 1
- 229940068918 polyethylene glycol 400 Drugs 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 230000002980 postoperative effect Effects 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 229960000206 potassium canrenoate Drugs 0.000 description 1
- JTZQCHFUGHIPDF-RYVBEKKQSA-M potassium canrenoate Chemical compound [K+].O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@](CC4)(O)CCC([O-])=O)[C@@H]4[C@@H]3C=CC2=C1 JTZQCHFUGHIPDF-RYVBEKKQSA-M 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- KWYUFKZDYYNOTN-UHFFFAOYSA-M potassium hydroxide Inorganic materials [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 1
- 239000008057 potassium phosphate buffer Substances 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 239000003286 potassium sparing diuretic agent Substances 0.000 description 1
- 229940097241 potassium-sparing diuretic Drugs 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 1
- IENZQIKPVFGBNW-UHFFFAOYSA-N prazosin Chemical compound N=1C(N)=C2C=C(OC)C(OC)=CC2=NC=1N(CC1)CCN1C(=O)C1=CC=CO1 IENZQIKPVFGBNW-UHFFFAOYSA-N 0.000 description 1
- 229960001289 prazosin Drugs 0.000 description 1
- 201000011461 pre-eclampsia Diseases 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 208000003476 primary myelofibrosis Diseases 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 229960003712 propranolol Drugs 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 230000006920 protein precipitation Effects 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 229950003776 protoporphyrin Drugs 0.000 description 1
- 208000009138 pulmonary valve stenosis Diseases 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- XKMLYUALXHKNFT-UHFFFAOYSA-N rGTP Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O XKMLYUALXHKNFT-UHFFFAOYSA-N 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 229960003401 ramipril Drugs 0.000 description 1
- HDACQVRGBOVJII-JBDAPHQKSA-N ramipril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@@H]2CCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 HDACQVRGBOVJII-JBDAPHQKSA-N 0.000 description 1
- 229950010535 razaxaban Drugs 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 239000003087 receptor blocking agent Substances 0.000 description 1
- 201000002793 renal fibrosis Diseases 0.000 description 1
- 230000002336 repolarization Effects 0.000 description 1
- 208000015658 resistant hypertension Diseases 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 208000037803 restenosis Diseases 0.000 description 1
- 229940099315 rimadyl Drugs 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 229960000672 rosuvastatin Drugs 0.000 description 1
- BPRHUIZQVSMCRT-VEUZHWNKSA-N rosuvastatin Chemical compound CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC(O)=O BPRHUIZQVSMCRT-VEUZHWNKSA-N 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 201000000980 schizophrenia Diseases 0.000 description 1
- 238000002098 selective ion monitoring Methods 0.000 description 1
- 230000036303 septic shock Effects 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 208000007056 sickle cell anemia Diseases 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 229960003310 sildenafil Drugs 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- 229960002578 sitaxentan Drugs 0.000 description 1
- PHWXUGHIIBDVKD-UHFFFAOYSA-N sitaxentan Chemical compound CC1=NOC(NS(=O)(=O)C2=C(SC=C2)C(=O)CC=2C(=CC=3OCOC=3C=2)C)=C1Cl PHWXUGHIIBDVKD-UHFFFAOYSA-N 0.000 description 1
- 208000019116 sleep disease Diseases 0.000 description 1
- 239000000779 smoke Substances 0.000 description 1
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 229940083618 sodium nitroprusside Drugs 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 229960002370 sotalol Drugs 0.000 description 1
- ZBMZVLHSJCTVON-UHFFFAOYSA-N sotalol Chemical compound CC(C)NCC(O)C1=CC=C(NS(C)(=O)=O)C=C1 ZBMZVLHSJCTVON-UHFFFAOYSA-N 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 230000035882 stress Effects 0.000 description 1
- 208000022170 stress incontinence Diseases 0.000 description 1
- 239000006190 sub-lingual tablet Substances 0.000 description 1
- NCEXYHBECQHGNR-QZQOTICOSA-N sulfasalazine Chemical compound C1=C(O)C(C(=O)O)=CC(\N=N\C=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-QZQOTICOSA-N 0.000 description 1
- 229960001940 sulfasalazine Drugs 0.000 description 1
- NCEXYHBECQHGNR-UHFFFAOYSA-N sulfasalazine Natural products C1=C(O)C(C(=O)O)=CC(N=NC=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-UHFFFAOYSA-N 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 238000011477 surgical intervention Methods 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 206010042772 syncope Diseases 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 230000006794 tachycardia Effects 0.000 description 1
- 229960000835 tadalafil Drugs 0.000 description 1
- IEHKWSGCTWLXFU-IIBYNOLFSA-N tadalafil Chemical compound C1=C2OCOC2=CC([C@@H]2C3=C([C]4C=CC=CC4=N3)C[C@H]3N2C(=O)CN(C3=O)C)=C1 IEHKWSGCTWLXFU-IIBYNOLFSA-N 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229960005187 telmisartan Drugs 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- 230000000542 thalamic effect Effects 0.000 description 1
- 239000003451 thiazide diuretic agent Substances 0.000 description 1
- 229960003279 thiopental Drugs 0.000 description 1
- 239000003868 thrombin inhibitor Substances 0.000 description 1
- 230000002537 thrombolytic effect Effects 0.000 description 1
- PHWBOXQYWZNQIN-UHFFFAOYSA-N ticlopidine Chemical compound ClC1=CC=CC=C1CN1CC(C=CS2)=C2CC1 PHWBOXQYWZNQIN-UHFFFAOYSA-N 0.000 description 1
- 229960005001 ticlopidine Drugs 0.000 description 1
- 229960004605 timolol Drugs 0.000 description 1
- 201000005882 tinea unguium Diseases 0.000 description 1
- 231100000886 tinnitus Toxicity 0.000 description 1
- 229950004437 tiqueside Drugs 0.000 description 1
- COKMIXFXJJXBQG-NRFANRHFSA-N tirofiban Chemical compound C1=CC(C[C@H](NS(=O)(=O)CCCC)C(O)=O)=CC=C1OCCCCC1CCNCC1 COKMIXFXJJXBQG-NRFANRHFSA-N 0.000 description 1
- 229960003425 tirofiban Drugs 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 239000003106 tissue adhesive Substances 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 229960005461 torasemide Drugs 0.000 description 1
- 231100000167 toxic agent Toxicity 0.000 description 1
- 239000003440 toxic substance Substances 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 230000009529 traumatic brain injury Effects 0.000 description 1
- 125000005270 trialkylamine group Chemical group 0.000 description 1
- 229960001288 triamterene Drugs 0.000 description 1
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical compound CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 230000007306 turnover Effects 0.000 description 1
- 238000000870 ultraviolet spectroscopy Methods 0.000 description 1
- 208000009852 uremia Diseases 0.000 description 1
- 206010046494 urge incontinence Diseases 0.000 description 1
- 208000014001 urinary system disease Diseases 0.000 description 1
- 210000001635 urinary tract Anatomy 0.000 description 1
- 201000002327 urinary tract obstruction Diseases 0.000 description 1
- 125000005500 uronium group Chemical group 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 229960004699 valsartan Drugs 0.000 description 1
- SJSNUMAYCRRIOM-QFIPXVFZSA-N valsartan Chemical compound C1=CC(CN(C(=O)CCCC)[C@@H](C(C)C)C(O)=O)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SJSNUMAYCRRIOM-QFIPXVFZSA-N 0.000 description 1
- 229960002381 vardenafil Drugs 0.000 description 1
- 230000001196 vasorelaxation Effects 0.000 description 1
- 230000002883 vasorelaxation effect Effects 0.000 description 1
- 230000002861 ventricular Effects 0.000 description 1
- 208000003663 ventricular fibrillation Diseases 0.000 description 1
- 206010047302 ventricular tachycardia Diseases 0.000 description 1
- 229960001722 verapamil Drugs 0.000 description 1
- 206010047470 viral myocarditis Diseases 0.000 description 1
- 210000001835 viscera Anatomy 0.000 description 1
- 229940019333 vitamin k antagonists Drugs 0.000 description 1
- 238000003260 vortexing Methods 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 238000011706 wistar kyoto rat Methods 0.000 description 1
- 230000029663 wound healing Effects 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
- 230000031143 xenobiotic glucuronidation Effects 0.000 description 1
- ZXIBCJHYVWYIKI-PZJWPPBQSA-N ximelagatran Chemical compound C1([C@@H](NCC(=O)OCC)C(=O)N2[C@@H](CC2)C(=O)NCC=2C=CC(=CC=2)C(\N)=N\O)CCCCC1 ZXIBCJHYVWYIKI-PZJWPPBQSA-N 0.000 description 1
- 229960001522 ximelagatran Drugs 0.000 description 1
- MTZBBNMLMNBNJL-UHFFFAOYSA-N xipamide Chemical compound CC1=CC=CC(C)=C1NC(=O)C1=CC(S(N)(=O)=O)=C(Cl)C=C1O MTZBBNMLMNBNJL-UHFFFAOYSA-N 0.000 description 1
- 229960000537 xipamide Drugs 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/02—Non-specific cardiovascular stimulants, e.g. drugs for syncope, antihypotensives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
Definitions
- the present application relates to novel 6-hydrogen-substituted imidazo [1,2-a] pyridine-3-carboxamides, processes for their preparation, their use alone or in combinations for the treatment and / or prophylaxis of diseases and their use for the production of medicaments for the treatment and / or prophylaxis of diseases, in particular for the treatment and / or prophylaxis of cardiovascular diseases.
- cGMP cyclic guanosine monophosphate
- NO nitric oxide
- GTP guanosine triphosphate
- the soluble guanylate cyclases consist of two subunits and most likely contain one heme per heterodimer, which is part of the regulatory center. This is central to the activation mechanism. NO can bind to the iron atom of the heme and thus significantly increase the activity of the enzyme. On the other hand, heme-free preparations can not be stimulated by NO. Carbon monoxide (CO) is also able to bind to the central iron atom of the heme, with stimulation by CO being markedly lower than by NO.
- CO Carbon monoxide
- guanylate cyclase plays a crucial role in various physiological processes, in particular in the relaxation and proliferation of smooth muscle cells, platelet aggregation and adhesion, neuronal signaling and diseases based on a disturbance of the above operations.
- the NO / cGMP system may be suppressed, leading, for example, to hypertension, platelet activation, increased cell proliferation, endothelial dysfunction, atherosclerosis, angina pectoris, heart failure, myocardial infarction, thrombosis, stroke and sexual dysfunction.
- a NO-independent treatment option for such diseases which is aimed at influencing the cGMP pathway in organisms, is a promising approach on account of the expected high efficiency and low side effects.
- the object of the present invention was to provide new substances which act as stimulators of soluble guanylate cyclase, and as such are suitable for the treatment and / or prophylaxis of diseases.
- the present invention relates to compounds selected from the group consisting of eni-N- (2-amino-5-fluoro-2-methylpentyl) -8 - [(2,6-difluorobenzyl) oxy] -2-methylimidazo [l, 2- a] pyridine-3-carboxamide (enantiomer A)
- Salts used in the context of the present invention are physiologically acceptable salts of the compounds according to the invention. Also included are salts which are themselves unsuitable for pharmaceutical applications but can be used, for example, for the isolation or purification of the compounds of the invention.
- Physiologically acceptable salts of the compounds according to the invention include acid addition salts of mineral acids, carboxylic acids and sulfonic acids, for example salts of hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, methanesulfonic acid, Efhansulfon- acid, toluenesulfonic acid, benzenesulfonic acid, naphthalenedisulfonic acid, formic acid, acetic acid, Trifluoroacetic acid, propionic acid, lactic acid, tartaric acid, malic acid, citric acid, fumaric acid, maleic acid and benzoic acid.
- Physiologically acceptable salts of the compounds according to the invention also include salts of customary bases, such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts) and ammonium salts derived from ammonia or organic amines having from 1 to 16 carbon atoms.
- alkali metal salts for example sodium and potassium salts
- alkaline earth salts for example calcium and magnesium salts
- ammonium salts derived from ammonia or organic amines having from 1 to 16 carbon atoms such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts) and ammonium salts derived from ammonia or organic amines having from 1 to 16 carbon atoms.
- Atoms such as, by way of example and by way of preference, ethylamine, diethylamine, triethylamine, ethyldiisopropylamine, monoethanolamine, diethanolamine, triethanolamine, dicyclohexylamine, dimethylaminoethanol, procaine, dibenzylamine, N-methylmorpholine, arginine, lysine, ethylenediamine and N-methylpiperidine.
- Solvates in the context of the invention are those forms of the compounds according to the invention which form a complex in the solid or liquid state by coordination with solvent molecules. Hydrates are a special form of solvates that coordinate with water. As solvates, hydrates are preferred in the context of the present invention.
- the compounds of the invention may exist in different stereoisomeric forms depending on their structure, i. in the form of configurational isomers or optionally also as conformational isomers (enantiomers and / or diastereomers, including those in atropisomers).
- the present invention therefore includes the enantiomers and diastereomers and their respective mixtures. From such mixtures of enantiomers and / or diastereomers, the stereoisomerically uniform components can be isolated in a known manner; Preferably, chromatographic methods are used for this, in particular HPLC chromatography on achiral or chiral phase.
- the present invention encompasses all tautomeric forms.
- the present invention also includes all suitable isotopic variants of the compounds of the invention.
- An isotopic variant of a compound according to the invention is understood to mean a compound in which at least one atom within the compound according to the invention is exchanged for another atom of the same atomic number but with a different atomic mass than the atomic mass that usually or predominantly occurs in nature.
- isotopes which can be incorporated into a compound of the invention are those of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, chlorine, bromine and iodine, such as 2 H (deuterium), 3 H (tritium), 13 C, 14 C, 15 N, 17 0, 18 0, 32 P, 33 P, 33 S, 34 S, 35 S, 36 S, 18 F, 36 Cl, 82 Br, 123 I, 124 I, 129 I and 131 L
- Certain isotopic variants of a compound according to the invention in particular those in which one or several radioactive isotopes are incorporated, may be useful, for example, to study the mechanism of action or drug distribution in the body; Due to the comparatively easy production and detectability, compounds labeled with 3 H or 14 C isotopes are particularly suitable for this purpose.
- isotopes such as deuterium may result in certain therapeutic benefits as a result of greater metabolic stability of the compound, such as prolonging the body's half-life or reducing the required effective dose;
- isotopic variants of the compounds according to the invention can be prepared by the processes known to the person skilled in the art, for example by the methods described below and the rules given in the exemplary embodiments, by using appropriate isotopic modifications of the respective reagents and / or starting compounds.
- the present invention also comprises prodrugs of the compounds according to the invention.
- prodrugs denotes compounds which themselves may be biologically active or inactive but are converted during their residence time in the body to compounds according to the invention (for example metabolically or hydrolytically).
- treatment includes inhibiting, delaying, arresting, alleviating, attenuating, restraining, reducing, suppressing, restraining or curing a disease, a disease, a disease, an injury or a medical condition , the unfolding, the course or progression of such conditions and / or the symptoms of such conditions.
- therapy is understood to be synonymous with the term “treatment”.
- prevention means the avoidance or reduction of the risk, a disease, a disease, a disease, an injury or a health disorder, a development or a Progression of such conditions and / or to get, experience, suffer or have the symptoms of such conditions.
- the treatment or prevention of a disease, a disease, a disease, an injury or a health disorder can be partial or complete.
- Preferred within the context of the present invention is the compound with the systematic name eni-N- (2-amino-5,5,5-trifluoro-2-methylpentyl) -8 - [(2,6-difluorobenzyl) oxy] -2-methylimidazo [1,2-a] pyridine-3-carboxamide (enantiomer A) and the structural formula
- Preferred within the context of the present invention is the compound with the systematic name eni-N- (2-amino-5,5,5-trifluoro-2-methylpentyl) -8 - [(2,6-difluorobenzyl) oxy] -2-methylimidazo [1,2-a] pyridine-3-carboxamide (enantiomer B) and the structural formula
- the invention further provides a process for the preparation of the compounds according to the invention which comprises [A] a compound of the formula (I)
- R 1 is hydrogen or chlorine, and these are subsequently converted into an inert solvent under amide coupling conditions with an amine selected from the group consisting of
- R 1 is hydrogen or chlorine
- R 2 is (IV-A), (IV-B) or (IV-C)
- Inert solvents for the amide coupling are, for example, ethers, such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons, such as benzene, toluene, xylene, hexane, cyclohexane or petroleum fractions, halogenated hydrocarbons, such as dichloromethane, trichloromethane, carbon tetrachloride, 1,2-dichloroethane, trichlorethylene or chlorobenzene , or other solvents such as acetone, ethyl acetate, acetonitrile, pyridine, dimethyl sulfoxide, A ⁇ -Dimefhylformamid, / V, / V-dimethylacetamide, ⁇ , ⁇ '-dimethylpropyleneurea (DMPU) or / V-methylpyrrolidone
- Suitable condensing agents for amide formation are, for example, carbodiimides such as ⁇ , ⁇ '-diethyl, N, N'-dipropyl, N, N'-diisopropyl, N, N'-dicyclohexylcarbodiimide (DCC) or N- (3-dimethylaminopropyl ) - / V'-ethylcarbodiimide hydrochloride (EDC), phosgene derivatives such as ⁇ , ⁇ '-carbonyldiimidazole (CDI), 1,2-oxazolium compounds such as 2-ethyl-5-phenyl-l, 2-oxazolium-3-sulfate or 2-tert-butyl-5-methylisoxazolium perchlorate, acylamino compounds such as 2-efhoxy-1-ethoxycarbonyl-1,2-dihydroquinoline, or isobutylchloroformate, propanephosphonic anhydr
- TBTU is used in conjunction with N-methylmorpholine, HATU in conjunction with -Diisopropylefhylamin or l-chloro- / V, / V, 2-trimethylprop-l-en-lamin.
- the condensations are generally carried out in a temperature range from -20 ° C to + 100 ° C, preferably at 0 ° C to + 60 ° C.
- the reaction may be carried out at normal, elevated or reduced pressure (e.g., from 0.5 to 5 bar). In general, one works at normal pressure.
- carboxylic acid of the formula (II) can also first be converted into the corresponding carboxylic acid chloride and then reacted directly or in a separate reaction with an amine of the formula (III) to form the compounds according to the invention.
- the formation of carboxylic acid chlorides from carboxylic acids is carried out by the methods known in the art, for example by treatment with thionyl chloride, sulfuryl chloride or oxalyl chloride in the presence of a suitable base, for example in the presence of pyridine, and optionally with the addition of dimethylformamide, optionally in a suitable inert solvent.
- the hydrolysis of the ester group T 1 of the compounds of formula (I) is carried out by conventional methods by treating the esters in inert solvents with acids or bases, wherein in the case of the latter, the initially formed salts are converted into the free carboxylic acids by treatment with acid.
- the tert-butyl ester ester cleavage is preferably carried out with acids.
- the ester cleavage is preferably carried out by hydrogenolysis with palladium on activated carbon or Raney nickel. Suitable inert solvents for this reaction are water or the organic solvents customary for ester cleavage.
- These preferably include alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert-butanol, or ethers such as diethyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, dioxane or glycol dimethyl ether, or other solvents such as acetone, dichloromethane, dimethylformamide or dimethyl sulfoxide , It is likewise possible to use mixtures of the solvents mentioned. In the case of basic ester hydrolysis, preference is given to using mixtures of water with dioxane, tetrahydrofuran, methanol and / or ethanol.
- the usual inorganic bases are suitable. These include preferably alkali or alkaline earth hydroxides such as sodium, lithium, potassium or barium hydroxide, or alkali or alkaline earth metal carbonates such as sodium, potassium or calcium carbonate. Particularly preferred are sodium or lithium hydroxide.
- Suitable acids for the ester cleavage are generally sulfuric acid, hydrochloric acid / hydrochloric acid, hydrobromic / hydrobromic acid, phosphoric acid, acetic acid, trifluoroacetic acid, toluenesulfonic acid, methanesulfonic acid or trifluoromethanesulfonic acid or mixtures thereof, optionally with the addition of water.
- Hydrogen chloride or trifluoroacetic acid are preferred in the case of the tert-butyl esters and hydrochloric acid in the case of the methyl esters.
- the ester cleavage is generally carried out in a temperature range from 0 ° C to + 100 ° C, preferably at + 0 ° C to + 50 ° C.
- the reactions mentioned can be carried out at normal, elevated or reduced pressure (for example from 0.5 to 5 bar). In general, one works at normal pressure.
- the amino-protecting group used is preferably ieri-butoxycarbonyl (Boc) or benzyloxycarbonyl (Z).
- a protective group for a hydroxy or carboxyl function tert-butyl or benzyl is preferably used.
- the cleavage of these protecting groups is carried out by conventional methods, preferably by reaction with a strong acid such as hydrogen chloride, hydrogen bromide or trifluoroacetic acid in an inert solvent such as dioxane, diethyl ether, dichloromethane or acetic acid; optionally, the cleavage can also be carried out without an additional inert solvent.
- a strong acid such as hydrogen chloride, hydrogen bromide or trifluoroacetic acid
- an inert solvent such as dioxane, diethyl ether, dichloromethane or acetic acid
- the cleavage can also be carried out without an additional inert solvent.
- benzyl and benzyloxycarbonyl as a protective group
- these can also be removed by hydrogenolysis in the presence of a palladium catalyst.
- the cleavage of the protective groups mentioned can optionally be carried out simultaneously in a one-pot reaction or in separate reaction steps.
- R 1 is hydrogen or chlorine, and this is then reacted in an inert solvent with a compound of formula (VIII)
- Inert solvents for ring closure to the imidazo [l, 2-a] pyridine backbone are the usual organic solvents. These preferably include alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol, n-pentanol or tert-butanol, or ethers such as diethyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, dioxane or glycol dimethyl ether, or other solvents such as acetone, dichloromethane , 1,2-dichloroethane, acetonitrile, dimethylformamide or dimethyl sulfoxide.
- alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol, n-pentanol or tert-butanol
- ethers such as diethyl ether, tetrahydrofur
- the ring closure is generally carried out in a temperature range from + 50 ° C to + 150 ° C, preferably at + 50 ° C to + 100 ° C, optionally in a microwave.
- the ring closure (VII) + (VIII) -> (I) or (V) + (VIII) -> (IX) is optionally carried out in the presence of water-withdrawing reaction additives, for example in the presence of molecular sieve (4 ⁇ pore size) or by means of water.
- the reaction (VII) + (VIII) -> (I) or (V) + (VIII) -> (IX) is carried out using an excess of the reagent of the formula (VIII), for example with 1 to 20 equivalents of the reagent ( VIII), optionally with the addition of bases (such as sodium bicarbonate) wherein the addition of this reagent can be carried out once or in several portions.
- R 2 is the compounds of the formulas (III-A), (III-B) and (III-C), and in which T 1 has the meanings given above.
- Typical reaction conditions for such Mitsunobu condensations of phenols with alcohols can be found in the literature, eg Hughes, DL Org. Read. 1992, 42, 335; Dembinski, R. Eur. J. Org. Chem. 2004, 2763.
- an activating reagent eg diethylazodicarboxylate (DEAD) or diisopropyl azodicarboxylate (DIAD)
- a phosphine reagent eg triphenylphosphine or tributylphosphine
- an inert solvent eg THF, Dichloromethane, toluene or DMF
- the compounds according to the invention have valuable pharmacological properties and can be used for the prevention and treatment of diseases in humans and animals.
- the compounds according to the invention open up a further treatment alternative and thus represent an enrichment of pharmacy.
- the compounds of the invention cause vasorelaxation and inhibition of platelet aggregation and lead to a reduction in blood pressure and to an increase in coronary blood flow. These effects are mediated by direct stimulation of soluble guanylate cyclase and intracellular cGMP increase.
- the compounds according to the invention enhance the action of substances which increase cGMP levels, such as, for example, endothelium-derived relaxing factor (EDRF), NO donors, protoporphyrin IX, arachidonic acid or phenylhydrazine derivatives.
- EDRF endothelium-derived relaxing factor
- NO donors NO donors
- protoporphyrin IX arachidonic acid or phenylhydrazine derivatives.
- the compounds according to the invention are suitable for the treatment and / or prophylaxis of cardiovascular, pulmonary, thromboembolic and fibrotic disorders.
- the compounds according to the invention can therefore be used in medicaments for the treatment and / or prophylaxis of cardiovascular diseases such as hypertension, resistant hypertension, acute and chronic heart failure, coronary heart disease, stable and unstable angina pectoris, peripheral and cardiac vascular diseases, arrhythmias, rhythm disorders Atrio-ventricular blockades grade ⁇ - ⁇ (AB block I-III), supraventricular tachyarrhythmia, atrial fibrillation, atrial flutter, ventricular fibrillation, ventricular tachyarrhythmia, torsades de pointes tachycardia, extrasystoles of atrial and ventricular atresia Ventricles, AV junctional extrasystoles, sick sinus syndrome, syncope, AV nodal reentrant tachycardia, Wolff-Parkinson-White syndrome, acute
- cardiac failure includes both acute and chronic manifestations of cardiac insufficiency, as well as more specific or related forms of disease such as acute decompensated heart failure, right heart failure, left heart failure, global insufficiency, ischemic cardiomyopathy, dilated cardiomyopathy, hypertrophic cardiomyopathy, idiopathic cardiomyopathy, congenital heart defects.
- Heart failure in heart valve defects mitral valve stenosis, mitral valve insufficiency, aortic valve stenosis, aortic valve insufficiency, tricuspid stenosis, tricuspid insufficiency, pulmonary valve stenosis, pulmonary valvular insufficiency, combined valvular heart failure, myocarditis, chronic myocarditis, acute myocarditis, viral myocarditis, diabetic heart failure, alcoholic cardiomyopathy, cardiac storage disorders, diastolic heart failure as well as systolic heart failure and acute phases de w worsening of heart failure.
- the compounds according to the invention may also be used for the treatment and / or prophylaxis of arteriosclerosis, lipid metabolism disorders, hypolipoproteinemias, dyslipidaemias, hypertriglyceridemias, hyperlipidemias, hypercholesterolemias, abetelipoproteinemia, sitosterolemia, xanthomatosis, Tangier's disease, obesity (obesity) and obesity combined hyperlipidaemias and the metabolic syndrome.
- the compounds according to the invention can be used for the treatment and / or prophylaxis of primary and secondary Raynaud's phenomenon, microcirculatory disorders, claudication, peripheral and autonomic neuropathies, diabetic microangiopathies, diabetic retinopathy, diabetic ulcers on the extremities, gangrenous, CREST syndrome, erythematosis, onychomycosis, rheumatic diseases and to promote wound healing.
- the compounds according to the invention are furthermore suitable for the treatment of urological diseases such as, for example, benign prostate syndrome (BPS), benign prostatic hyperplasia (BPH), benign prostatic hyperplasia (BPE), bladder emptying disorder (BOO), lower urinary tract syndromes (LUTS, including Feiine's urological syndrome ( FUS)), diseases of the urogenital system including neurogenic overactive bladder (OAB) and (IC), incontinence (UI) such as mixed, urge, stress, or overflow incontinence (MUI, UUI, SUI, OUI), Pelvic pain, benign and malignant diseases of the organs of the male and female urogenital system.
- BPS benign prostate syndrome
- BPH benign prostatic hyperplasia
- BPE benign prostatic hyperplasia
- BOO bladder emptying disorder
- LUTS lower urinary tract syndromes
- FUS Feiine's urological syndrome
- diseases of the urogenital system including neurogenic overactive bladder (OAB) and (IC), incon
- kidney diseases in particular of acute and chronic renal insufficiency, as well as of acute and chronic renal failure.
- renal insufficiency includes both acute and chronic manifestations of renal insufficiency, as well as underlying or related renal diseases such as renal hypoperfusion, intradialytic hypotension, obstructive uropathy, glomerulopathies, glomerulonephritis, acute glomerulonephritis, glomerulosclerosis, tubulointerstitial disorders, nephropathic disorders such as primary and congenital kidney disease, nephritis, renal immunological diseases such as renal transplant rejection, immune complex-induced renal disease, toxicant-induced nephropathy, contrast agent-induced nephropathy, diabetic and nondiabetic nephropathy, pyelonephritis, renal cysts, nephrosclerosis, hypertensive
- the present invention also encompasses the use of the compounds of the invention for the treatment and / or prophylaxis of sequelae of renal insufficiency, such as pulmonary edema, cardiac insufficiency, uremia, anemia, electrolyte disorders (eg, hyperkalemia, hyponatremia) and disorders in bone and carbohydrate metabolism.
- sequelae of renal insufficiency such as pulmonary edema, cardiac insufficiency, uremia, anemia, electrolyte disorders (eg, hyperkalemia, hyponatremia) and disorders in bone and carbohydrate metabolism.
- the compounds according to the invention are also suitable for the treatment and / or prophylaxis of asthmatic diseases, pulmonary arterial hypertension (PAH) and other forms of pulmonary hypertension (PH), including left heart disease, HIV, sickle cell disease, thromboembolism (CTEPH), sarcoidosis, COPD or pulmonary fibrosis-associated pulmonary hypertension, chronic obstructive pulmonary disease (COPD), acute respiratory tract syndrome (ARDS), acute lung injury (ALI), alpha-1-antitrypsin deficiency (AATD), pulmonary fibrosis, pulmonary emphysema (eg, cigarette smoke induced emphysema) and cystic fibrosis (CF).
- PAH pulmonary arterial hypertension
- PH pulmonary hypertension
- COPD chronic obstructive pulmonary disease
- ARDS acute respiratory tract syndrome
- ALI acute lung injury
- AATD alpha-1-antitrypsin deficiency
- CF cystic fibros
- the compounds described in the present invention are also agents for controlling diseases in the central nervous system, which are characterized by disorders of the NO / cGMP system.
- they are suitable for improving the perception, concentration performance, learning performance or memory performance after cognitive disorders such as occur in situations / diseases / syndromes such as mild cognitive impairment, age-associated learning and memory disorders, age-associated memory loss, vascular dementia, cranial brain Trauma, post-stroke dementia, post-traumatic traumatic brain injury, generalized concentration disorders, impaired concentration in children with learning and memory problems, Alzheimer's disease, dementia with Lewy bodies, dementia with degeneration of the frontal lobes including Pick's syndrome, Parkinson's disease, progressive nuclear palsy, dementia with corticobasal degeneration, amyolateral sclerosis (ALS), Huntington's disease, demyelinization, multiple sclerosis, thalamic degeneration, Creutzfeld's disease Jacob-Deme nz, HIV dementia, schizophrenia with dementia or Korsakoff's psychosis.
- cognitive disorders such as occur in situations
- the compounds according to the invention are also suitable for the treatment and / or prophylaxis of diseases of the central nervous system such as states of anxiety, tension and depression, central nervous conditional sexual dysfunctions and sleep disorders as well as for the regulation of pathological disorders of food, consumption and addiction.
- the compounds according to the invention are also suitable for regulating cerebral perfusion and are effective agents for combating migraine. They are also suitable for the prophylaxis and control of the consequences of cerebral infarct events (Apoplexia cerebri) such as stroke, cerebral ischaemias and craniocerebral trauma , Likewise, the compounds of the invention can be used to combat pain and tinnitus.
- the compounds of the invention have anti-inflammatory action and can therefore be used as anti-inflammatory agents for the treatment and / or prophylaxis of sepsis (SIRS), multiple organ failure (MODS, MOF), inflammatory diseases of the kidney, chronic inflammatory bowel disease (IBD, Crohn's Disease, UC), pancreatitis , Peritonitis, rheumatoid diseases, inflammatory skin diseases and inflammatory ocular diseases.
- SIRS sepsis
- MODS multiple organ failure
- IBD chronic inflammatory bowel disease
- UC chronic inflammatory bowel disease
- pancreatitis e.g., Peritonitis, rheumatoid diseases, inflammatory skin diseases and inflammatory ocular diseases.
- the compounds of the invention can also be used for the treatment and / or prophylaxis of autoimmune diseases.
- the compounds according to the invention are suitable for the treatment and / or prophylaxis of fibrotic disorders of the internal organs such as, for example, the lung, the heart, the kidney, the bone marrow and in particular the liver, as well as dermatological fibroses and fibrotic disorders of the eye.
- fibrotic disorders includes in particular the following terms: liver fibrosis, cirrhosis, pulmonary fibrosis, endomyocardial fibrosis, nephropathy, glomerulonephritis, interstitial renal fibrosis, fibrotic damage due to diabetes, bone marrow fibrosis and similar fibrotic disorders, scleroderma, morphea, keloids, hypertrophic scarring (also after surgical interventions), nevi, diabetic retinopathy, proliferative vitroretinopathy and connective tissue disorders (eg sarcoidosis).
- the compounds of the invention are useful for controlling postoperative scarring, e.g. as a result of glaucoma surgery.
- the compounds according to the invention can likewise be used cosmetically for aging and keratinizing skin.
- the compounds according to the invention are suitable for the treatment and / or prophylaxis of hepatitis, neoplasm, osteoporosis, glaucoma and gastroparesis.
- Another object of the present invention is the use of compounds according to the invention for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases.
- the present invention further relates to the use of the compounds according to the invention for the treatment and / or prophylaxis of cardiac insufficiency, angina pectoris, hypertension, pulmonary hypertension, ischaemias, vascular disorders, renal insufficiency, thromboembolic disorders, fibrotic disorders and atherosclerosis.
- the present invention furthermore relates to the compounds according to the invention for use in a method for the treatment and / or prophylaxis of cardiac insufficiency, angina pectoris, hypertension, pulmonary hypertension, ischaemias, vascular disorders, renal insufficiency, thromboembolic disorders, fibrotic disorders and atherosclerosis.
- Another object of the present invention is the use of the compounds of the invention for the manufacture of a medicament for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases.
- the present invention further relates to the use of the compounds according to the invention for the production of a medicament for the treatment and / or prophylaxis of cardiac insufficiency, angina pectoris, hypertension, pulmonary hypertension, ischaemias, vascular disorders, renal insufficiency, thromboembolic disorders, fibrotic disorders and arteriosclerosis.
- Another object of the present invention is a method for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases, using an effective amount of at least one of the compounds of the invention.
- the present invention further provides a method for the treatment and / or prophylaxis of cardiac insufficiency, angina pectoris, hypertension, pulmonary hypertension, ischaemias, vascular diseases, renal insufficiency, thromboembolic disorders, fibrotic diseases and atherosclerosis, using an effective amount of at least one of the compounds according to the invention ,
- the compounds of the invention may be used alone or as needed in combination with other agents.
- Another object of the present invention are pharmaceutical compositions containing at least one of the compounds of the invention and one or more other active ingredients, in particular for the treatment and / or prophylaxis of the aforementioned diseases.
- suitable combination active ingredients may be mentioned by way of example and preferably:
- organic nitrates and NO donors such as sodium nitroprusside, nitroglycerin, isosorbide mononitrate, isosorbide dinitrate, molsidomine or SIN-1, and inhaled NO;
- cGMP cyclic guanosine monophosphate
- PDE phosphodiesterases
- Inhibitors such as sildenafil, vardenafil and tadalafil;
- Antithrombotic agents by way of example and preferably from the group of platelet aggregation inhibitors, anticoagulants or profibrinolytic substances;
- Antihypertensive agents by way of example and preferably from the group of calcium antagonists, angiotensin AII antagonists, ACE inhibitors, endothelin antagonists, renin inhibitors, alpha-receptor blockers, beta-receptor blockers, mineralocorticid Receptor antagonists and diuretics; and or Lipid metabolism-altering agents, by way of example and preferably from the group of thyroid receptor agonists, cholesterol synthesis inhibitors such as by way of example and preferably HMG-CoA reductase or squalene synthesis inhibitors, ACAT inhibitors, CETP inhibitors, MTP inhibitors, PPAR inhibitors alpha, PPAR gamma and / or PPAR delta agonists, cholesterol absorption inhibitors, lipase inhibitors, polymeric bile acid adsorbers, bile acid reabsorption inhibitors, and lipoprotein (a) antagonists.
- Lipid metabolism-altering agents by way of example and preferably from the group of thyroid
- Antithrombotic agents are preferably understood as meaning compounds from the group of platelet aggregation inhibitors, anticoagulants or profibrinolytic substances.
- the compounds according to the invention are administered in combination with a platelet aggregation inhibitor, such as, by way of example and by way of preference, aspirin, clopidogrel, ticlopidine or dipyridamole.
- the compounds according to the invention are administered in combination with a thrombin inhibitor, such as, by way of example and by way of preference, ximelagatran, dabigatran, melagatran, bivalirudin or Clexane.
- a thrombin inhibitor such as, by way of example and by way of preference, ximelagatran, dabigatran, melagatran, bivalirudin or Clexane.
- the compounds according to the invention are administered in combination with a GPIIb / IIIa antagonist, such as, by way of example and by way of preference, tirofiban or abciximab.
- a GPIIb / IIIa antagonist such as, by way of example and by way of preference, tirofiban or abciximab.
- the compounds according to the invention are administered in combination with a factor Xa inhibitor, such as by way of example and preferably rivaroxaban (BAY 59-7939), DU-176b, apixaban, otamixaban, fidexaban, razaxaban, fondaparinux, idraparinux , PMD-3112, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 or SSR-128428.
- a factor Xa inhibitor such as by way of example and preferably rivaroxaban (BAY 59-7939), DU-176b, apixaban, otamixaban, fidexaban, razaxaban, fondaparinux, idraparinux , PMD-3112, YM-150, KFA-1982, EMD-503982, MCM
- the compounds according to the invention are administered in combination with heparin or a low molecular weight (LMW) heparin derivative.
- LMW low molecular weight
- the compounds according to the invention are administered in combination with a vitamin K antagonist, such as by way of example and preferably coumarin.
- a vitamin K antagonist such as by way of example and preferably coumarin.
- the antihypertensive agents are preferably compounds from the group of calcium antagonists, angiotensin AII antagonists, ACE inhibitors, endothelin antagonists, Renin inhibitors, alpha-receptor blockers, beta-receptor blockers, mineralocorticoid receptor antagonists and diuretics understood.
- the compounds according to the invention are administered in combination with a calcium antagonist, such as by way of example and preferably nifedipine, amlodipine, verapamil or diltiazem.
- a calcium antagonist such as by way of example and preferably nifedipine, amlodipine, verapamil or diltiazem.
- the compounds according to the invention are administered in combination with an alpha-1-receptor blocker, such as by way of example and preferably prazosin.
- the compounds according to the invention are used in combination with a beta-receptor blocker such as, by way of example and by way of preference, propranolol, atenolol, timolol, pindolol, alprenolol, oxprenolol, penbutolol, bupranolol, metipropanol, nadolol, mepindolol, Caroteneol, sotalol, metoprolol, betaxolol, celiprolol, bisoprolol, carteolol, esmolol, labetalol, carvedilol, adaprolol, landiolol, nebivolol, epanolol or bucinolol.
- a beta-receptor blocker such as, by way of example and by way of preference, propranolol, atenolol,
- the compounds according to the invention are administered in combination with an angiotensin all-antagonist, such as by way of example and preferably losartan, candesartan, valsartan, telmisartan or embursatan.
- an angiotensin all-antagonist such as by way of example and preferably losartan, candesartan, valsartan, telmisartan or embursatan.
- the compounds according to the invention are administered in combination with an ACE inhibitor such as, by way of example and by way of preference, enalapril, captopril, lisinopril, ramipril, delapril, fosinopril, quinopril, perindopril or trandopril.
- an ACE inhibitor such as, by way of example and by way of preference, enalapril, captopril, lisinopril, ramipril, delapril, fosinopril, quinopril, perindopril or trandopril.
- the compounds according to the invention are administered in combination with an endothelin antagonist such as, by way of example and by way of preference, bosentan, darusentan, ambrisentan or sitaxsentan.
- an endothelin antagonist such as, by way of example and by way of preference, bosentan, darusentan, ambrisentan or sitaxsentan.
- the compounds of the invention are administered in combination with a renin inhibitor, such as by way of example and preferably aliskiren, SPP-600 or SPP-800.
- the compounds of the invention are administered in combination with a mineralocorticoid receptor antagonist, such as by way of example and preferably spironolactone or eplerenone.
- a mineralocorticoid receptor antagonist such as by way of example and preferably spironolactone or eplerenone.
- the compounds according to the invention are used in combination with a loop diuretic, such as, for example, furosemide, Torasemide, bumetanide and piretanide, with potassium-sparing diuretics such as amiloride and triamterene, with aldosterone antagonists such as spironolactone, potassium canrenoate and eplerenone, and thiazide diuretics such as hydrochlorothiazide, chlorthalidone, xipamide, and indapamide.
- a loop diuretic such as, for example, furosemide, Torasemide, bumetanide and piretanide
- potassium-sparing diuretics such as amiloride and triamterene
- aldosterone antagonists such as spironolactone
- potassium canrenoate and eplerenone potassium canrenoate and eplerenone
- thiazide diuretics such as hydrochlorothiazide
- lipid metabolizing agents are preferably compounds from the group of CETP inhibitors, thyroid receptor agonists, cholesterol synthesis inhibitors such as HMG-CoA reductase or squalene synthesis inhibitors, the ACAT inhibitors, MTP inhibitors, PPAR alpha- , PPAR gamma and / or PPAR delta agonists, cholesterol absorption inhibitors, polymeric bile acid adsorbers, bile acid reabsorption inhibitors, lipase inhibitors and the lipoprotein (a) - understood antagonists.
- CETP inhibitors such as HMG-CoA reductase or squalene synthesis inhibitors
- ACAT inhibitors such as HMG-CoA reductase or squalene synthesis inhibitors
- MTP inhibitors MTP inhibitors
- PPAR alpha- , PPAR gamma and / or PPAR delta agonists cholesterol absorption inhibitors
- polymeric bile acid adsorbers bile acid
- the compounds according to the invention are administered in combination with a CETP inhibitor, such as by way of example and preferably dalcetrapib, BAY 60-5521, anacetrapib or CETP vaccine (CETi-1).
- a CETP inhibitor such as by way of example and preferably dalcetrapib, BAY 60-5521, anacetrapib or CETP vaccine (CETi-1).
- the compounds according to the invention are administered in combination with a thyroid receptor agonist such as, by way of example and by way of preference, D-thyroxine, 3,5,3'-triiodothyronine (T3), CGS 23425 or axitirome (CGS 26214) ,
- a thyroid receptor agonist such as, by way of example and by way of preference, D-thyroxine, 3,5,3'-triiodothyronine (T3), CGS 23425 or axitirome (CGS 26214) ,
- the compounds according to the invention are administered in combination with an HMG-CoA reductase inhibitor from the class of statins, such as by way of example and preferably lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, rosuvastatin or pitavastatin.
- statins such as by way of example and preferably lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, rosuvastatin or pitavastatin.
- the compounds according to the invention are administered in combination with a squalene synthesis inhibitor, such as by way of example and preferably BMS-188494 or TAK-475.
- a squalene synthesis inhibitor such as by way of example and preferably BMS-188494 or TAK-475.
- the compounds according to the invention are administered in combination with an ACAT inhibitor, such as, for example and preferably, avasimibe, melinamide, pactimibe, eflucimibe or SMP-797.
- an ACAT inhibitor such as, for example and preferably, avasimibe, melinamide, pactimibe, eflucimibe or SMP-797.
- the compounds according to the invention are administered in combination with an MTP inhibitor such as, for example and preferably, implitapide, BMS-201038, R-103757 or JTT-130.
- an MTP inhibitor such as, for example and preferably, implitapide, BMS-201038, R-103757 or JTT-130.
- the compounds of the invention are administered in combination with a PPAR-gamma agonist such as, by way of example and by way of preference, pioglitazone or rosiglitazone.
- a PPAR delta agonist such as by way of example and preferably GW 501516 or BAY 68-5042.
- the compounds according to the invention are administered in combination with a cholesterol absorption inhibitor such as, for example and preferably, ezetimibe, tiqueside or pamaqueside.
- a cholesterol absorption inhibitor such as, for example and preferably, ezetimibe, tiqueside or pamaqueside.
- the compounds according to the invention are administered in combination with a lipase inhibitor, such as, for example and preferably, orlistat.
- a lipase inhibitor such as, for example and preferably, orlistat.
- the compounds of the invention are administered in combination with a polymeric bile acid adsorbent such as, by way of example and by way of preference, cholestyramine, colestipol, colesolvam, cholesta gel or colestimide.
- ASBT IBAT
- the compounds of the invention are administered in combination with a lipoprotein (a) antagonist such as, by way of example and by way of preference, gemcabene calcium (CI-1027) or nicotinic acid.
- a lipoprotein (a) antagonist such as, by way of example and by way of preference, gemcabene calcium (CI-1027) or nicotinic acid.
- Another object of the present invention are pharmaceutical compositions containing at least one compound of the invention, usually together with one or more inert, non-toxic, pharmaceutically suitable excipients, and their use for the purposes mentioned above.
- the compounds according to the invention can act systemically and / or locally.
- they may be applied in a suitable manner, e.g. oral, parenteral, pulmonary, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctival, otic or as an implant or stent.
- the compounds according to the invention can be administered in suitable administration forms.
- the compounds of the invention rapidly and / or modified donating application forms, the compounds of the invention in crystalline and / or amorphized and / or dissolved form such as tablets (uncoated or coated tablets, for example with enteric or delayed-dissolving or insoluble coatings which control the release of the compound of the invention), orally disintegrating tablets or films / wafers, films / lyophilisates, capsules ( hard or soft gelatin capsules, for example), dragees, granules, pellets, powders, emulsions, suspensions, aerosols or solutions.
- Parenteral administration can be accomplished by bypassing a resorption step (e.g., intravenously, intraarterially, intracardially, intraspinal, or intralumbar) or by resorting to absorption (e.g., intramuscularly, subcutaneously, intracutaneously, percutaneously, or intraperitoneally).
- a resorption step e.g., intravenously, intraarterially, intracardially, intraspinal, or intralumbar
- absorption e.g., intramuscularly, subcutaneously, intracutaneously, percutaneously, or intraperitoneally.
- suitable as application forms i.a. Injection and infusion preparations in the form of solutions, suspensions, emulsions, lyophilisates or sterile powders.
- Inhalation medicaments including powder inhalers, nebulizers
- nasal drops solutions or sprays
- lingual, sublingual or buccal tablets films / wafers or capsules
- suppositories ear or ophthalmic preparations
- vaginal capsules aqueous suspensions (lotions, shake mixtures)
- lipophilic suspensions ointments
- creams transdermal therapeutic systems (eg plasters)
- milk pastes, foams, powdered powders, implants or stents.
- the compounds according to the invention can be converted into the stated administration forms. This can be done in a conventional manner by mixing with inert, non-toxic, pharmaceutically suitable excipients.
- excipients for example microcrystalline cellulose, lactose, mannitol
- solvents for example liquid polyethylene glycols
- emulsifiers and dispersants or wetting agents for example sodium dodecyl sulfate, polyoxysorbitanoleate
- binders for example polyvinylpyrrolidone
- synthetic and natural polymers for example albumin
- Stabilizers eg, antioxidants such as ascorbic acid
- dyes eg, inorganic pigments such as iron oxides
- flavor and / or odoriferous include, among others.
- Excipients for example microcrystalline cellulose, lactose, mannitol
- solvents for example liquid polyethylene glycols
- emulsifiers and dispersants or wetting agents for example sodium dodecy
- the dosage is about 0.001 to 2 mg / kg, preferably about 0.001 to 1 mg kg of body weight.
- the multiplicities of proton signals in ⁇ -NMR spectra given in the following paragraphs represent the respective observed signal form and do not take into account higher-order signal phenomena. All data in ⁇ -NMR spectra indicate the chemical shifts ⁇ in ppm.
- the starting compounds, intermediates and embodiments may be present as hydrates. A quantitative determination of the water content was not. The hydrates may have an influence on the ⁇ -NMR spectrum and possibly shift and / or greatly broaden the water signal in ⁇ -NMR.
- the compounds of the invention may be in salt form, for example as trifluoroacetate, formate or ammonium salt, if the Compounds according to the invention contain a sufficiently basic or acidic functionality.
- a salt can be converted into the corresponding free base or acid by various methods known to those skilled in the art.
- a compound in the form of a salt of the corresponding base or acid is listed in the synthesis intermediates and embodiments of the invention described below, the exact stoichiometric composition of such a salt, as according to the respective preparation and / or purification process was received, usually unknown.
- salt-forming components such as “hydrochloride”, “trifluoroacetate”, “sodium salt” or “x HCl”, “x CF 3 COOH”, “x Na +” are not included in such salts stoichiometrically, but are solely descriptive of the salt-forming components contained.
- Example 4A 115 mmol, 1 equivalent
- Example 4A 115 mmol, 1 equivalent
- 550 ml of THF and 700 ml of methanol admixed with 13.8 g of lithium hydroxide (dissolved in 150 ml of water, 577 mmol, 5 equivalents) and stirred at RT overnight. It was treated with 1 N aqueous hydrochloric acid and concentrated in vacuo. The resulting solid was filtered off and washed with water. There was obtained 34 g of the title compound (84% of theory).
- the reaction solution was added first at 0 ° C with water and then with ethyl acetate and washed three times with half-saturated, aqueous sodium chloride solution.
- the combined aqueous phases were reextracted twice with ethyl acetate.
- the combined organic phases were dried over sodium sulfate, filtered and concentrated.
- Enantiomer A Yield: 2.64 g (> 99% ee)
- Enantiomer B Yield: 2.76 g (93% ee)
- Enantiomer A Yield: 5.7 g (> 99% ee)
- Enantiomer B Yield: 5.0 g (> 99% ee)
- the residue was purified by preparative HPLC (RP18 column, eluent: acetonitrile / water gradient with the addition of 0.1% TFA). The product fractions were combined and concentrated. The residue was then taken up in dichloromethane and a little methanol and washed twice with a little saturated aqueous sodium bicarbonate solution. The aqueous phase was extracted twice with dichloromethane. The combined organic phases were dried over sodium sulfate, filtered, concentrated and lyophilized. 53 mg of the target compound (28% of theory) were obtained.
- the residue was purified by preparative HPLC (RP18 column, mobile phase: acetonitrile / water gradient with the addition of 0.1% TFA). The product fractions were combined and concentrated. The residue was then taken up in dichloromethane and a little methanol and washed twice with a little saturated aqueous sodium bicarbonate solution. The aqueous phase was extracted twice with dichloromethane. The combined organic phases were dried over sodium sulfate, filtered, concentrated and lyophilized. 90 mg of the target compound (34% of theory) were obtained.
- the residue was purified by preparative HPLC (RP18 column, eluent: acetonitrile / water gradient with the addition of 0.1% TFA). The product fractions were combined and concentrated. The residue was then taken up in dichloromethane and a little methanol and washed twice with a little saturated aqueous sodium bicarbonate solution. The aqueous phase was extracted twice with dichloromethane. The combined organic phases were dried over sodium sulfate, filtered, concentrated and lyophilized. There were obtained 95 mg of the target compound (95% of theory, purity 93%).
- the residue was purified by preparative HPLC (RP18 column, eluent: acetonitrile / water gradient with the addition of 0.1% TFA). The product fractions were combined and concentrated. The residue was then taken up in dichloromethane and a little methanol and washed twice with a little saturated aqueous sodium bicarbonate solution. The aqueous phase was extracted twice with dichloromethane. The combined organic phases were dried over sodium sulfate, filtered, concentrated and lyophilized. 95 mg of the target compound (98% of theory) were obtained.
- the reaction solution was filtered through a Millipore filter, washed with ethanol and the filtrate evaporated.
- the residue was purified by preparative HPLC (RP18 column, eluent: acetonitrile / water gradient with the addition of 0.1% TFA).
- the product fractions were combined and concentrated.
- the residue was then taken up in dichloromethane and a little methanol and washed twice with a little saturated aqueous sodium bicarbonate solution.
- the aqueous phase was reextracted twice with dichloromethane.
- the combined organic phases were dried over sodium sulfate, filtered, concentrated and lyophilized. There were obtained 138 mg of the target compound (82% of theory, purity 98%).
- Soluble guanylyl cyclase converts GTP to cGMP and pyrophosphate (PPi) upon stimulation.
- PPi is detected by the method described in WO 2008/061626.
- the signal generated in the test increases as the reaction progresses and serves as a measure of the sGC enzyme activity.
- the enzyme can be characterized in a known manner, e.g. in terms of turnover rate, stimulability or Michaelis constant.
- 29 ⁇ M enzyme solution (0-10 nM soluble guanylyl cyclase (prepared according to Hönicka et al., Journal of Molecular Medicine 77 (1999) 14-23), in 50 mM TEA, 2 mM magnesium chloride, 0.1% BSA (fraction V), 0.005% Brij 35, pH 7.5) in the microplate and 1 ⁇ of the stimulator solution (0-10 ⁇ 3-Morpholinosydnonimine, SIN-1, Merck in DMSO) added. It was incubated at RT for 10 min.
- the enzyme reaction was started by the addition of 20 .mu. ⁇ substrate solution (1.25 mM guanosine 5 'triphosphate (Sigma) in 50 mM TEA, 2 mM magnesium chloride, 0.1% BSA (fraction V), 0.005% Brij 35, pH 7.5) and continuously luminometric measured.
- 20 .mu. ⁇ substrate solution (1.25 mM guanosine 5 'triphosphate (Sigma) in 50 mM TEA, 2 mM magnesium chloride, 0.1% BSA (fraction V), 0.005% Brij 35, pH 7.5) and continuously luminometric measured.
- B-2 Effect on recombinant guanylate cyclase reporter cell line
- MEC minimum effective concentration
- aorta Rabbits are stunned and bled by a stroke of the neck.
- the aorta is harvested, detached from adherent tissue, divided into 1.5 mm wide rings and placed individually under bias in 5 ml organ baths with 37 ° C warm, carbogen-gassed Krebs-Henseleit solution of the following composition (in each case mM): Sodium chloride: 119; Potassium chloride: 4.8; Calcium chloride dihydrate: 1; Magnesium sulfate heptahydrate: 1.4; Potassium dihydrogen phosphate: 1.2; Sodium hydrogencarbonate: 25; Glucose: 10.
- the force of contraction is detected with Statham UC2 cells, amplified and digitized via A / D converters (DAS-1802 HC, Keithley Instruments Munich) and registered in parallel on chart recorders.
- DAS-1802 HC A / D converters
- phenylephrine is added cumulatively to the bath in increasing concentration.
- the substance to be examined is added in each subsequent course in increasing dosages and the height of the contraction is compared with the height of the contraction achieved in the last predistortion. This is used to calculate the concentration required to reduce the level of the control value by 50% (IC50 value).
- the standard application volume is 5 ⁇ , the DMSO content in the bath solution corresponds to 0.1%.
- a commercially available telemetry system from DATA SCIENCES INTERNATIONAL DSI, USA is used for the blood pressure measurement on awake rats described below.
- the system consists of 3 main components:
- Implantable transmitters Physiotel® telemetry transmitters
- Physiotel® receivers connected to a data acquisition computer through a multiplexer (DSI Data Exchange Matrix).
- the telemetry system allows a continuous recording of blood pressure heart rate and body movement on awake animals in their habitual habitat.
- the experimental animals are kept individually in macroion cages type 3 after transmitter implantation. You have free access to standard food and water.
- the day - night rhythm in the experimental laboratory is changed by room lighting at 6:00 in the morning and at 19:00 in the evening.
- the TAH PA - C40 telemetry transmitters are surgically implanted into the experimental animals under aseptic conditions at least 14 days before the first trial.
- the animals so instrumented are repeatedly used after healing of the wound and ingrowth of the implant.
- the fasting animals are anaesthetized with pentobabital (Nembutal, Sanofi: 50 mg / kg ip) and shaved and disinfected on the ventral side.
- the system's liquid-filled measuring catheter above the bifurcation is inserted cranially into the descending aorta and secured with tissue adhesive (VetBonD TM, 3M).
- the transmitter housing is fixed intraperitoneally to the abdominal wall musculature and the wound is closed in layers.
- an antibiotic is administered for infection prevention (Tardomyocel COMP Bayer 1ml / kg s.c.)
- a solvent-treated group of animals is used as a control.
- Experimental procedure The existing telemetry measuring device is configured for 24 animals. Each trial is registered under a trial number (VYear month day).
- the instrumented rats living in the plant each have their own receiving antenna (1010 receivers, DSI).
- the implanted transmitters can be activated externally via a built-in magnetic switch. They will be put on the air during the trial run.
- the emitted signals can be recorded online by a data acquisition system (Dataquest TM A.R.T. for Windows, DSI) and processed accordingly. The storage of the data takes place in each case in a folder opened for this purpose which carries the test number.
- SBP Systolic blood pressure
- DBP Diastolic blood pressure
- MAP Mean Arterial Pressure
- HR Heart Rate
- ACT Activity
- the collected individual data are sorted with the analysis software (DATAQUEST TM A.RT. TM ANALYSIS).
- the blank value is assumed here 2 hours before application, so that the selected data record covers the period from 7:00 am on the day of the experiment to 9:00 am on the following day.
- the data is smoothed over a presettable time by means of value determination (15 minutes average) and transferred as a text file to a data medium.
- value determination 15 minutes average
- the presorted and compressed measured values are transferred to Excel templates and displayed in tabular form.
- the filing of the collected data takes place per experiment day in a separate folder that bears the test number. Results and test reports are sorted in folders and sorted by paper.
- the pharmacokinetic parameters of the compounds of the invention are determined in male CD-1 mice, male Wistar rats and female beagle dogs.
- Intravenous administration is in mice and rats using a species-specific plasma / DMSO formulation and in dogs using a water / PEG400 / ethanol formulation.
- Oral administration of the solute by gavage is performed in all species based on a water / PEG400 / ethanol formulation. Rats are placed in the right external jugular vein for ease of blood sampling prior to drug administration. The operation is carried out at least one day before the experiment under isoflurane anesthesia and with the administration of an analgesic (atropine / rimadyl (3/1) 0.1 mL s.c.).
- an analgesic atropine / rimadyl (3/1) 0.1 mL s.c.
- the blood collection (usually more than 10 times) takes place in a time window, which includes terminal times of at least 24 to a maximum of 72 hours after substance administration.
- the blood is transferred to heparinized tubes at collection. So then the blood plasma is recovered by centrifugation and optionally stored at -20 ° C until further processing.
- An internal standard is added to the samples of the compounds according to the invention, calibration samples and qualifiers (this may also be a chemically unrelated substance) and protein precipitation by means of acetonitrile follows in excess.
- the supernatant is measured by LC-MS / MS using C18 reversed-phase columns and variable eluent mixtures.
- the quantification of the substances is based on the peak heights or areas of extracted ion chromatograms of specific selected ion monitoring experiments.
- the pharmacokinetic parameters such as AUC, C ma x (terminal half-life), F (bioavailability), MRT (Mean Residence Time) and CL hn (clearance) by means of a validated pharmacokinetic computer program calculated.
- the blood / plasma distribution of the substance must be determined in order to adjust the pharmacokinetic parameters accordingly.
- a defined amount of substance is incubated in heparinized whole blood of the corresponding species for 20 min in a tumble roll mixer. After centrifugation at 1000 g, the concentration in the plasma is measured (by means of LC-MS / MS, see above) and the quotient formation of the C ⁇ iut / Cpiasma value is determined.
- CYP cytochrome P450
- the compounds of the invention were incubated at a concentration of about 0.1-10 ⁇ .
- stock solutions of the compounds according to the invention with a concentration of 0.01-1 mM in acetonitrile were prepared, and then pipetted with a 1: 100 dilution into the incubation mixture.
- the liver microsomes and recombinant enzymes were incubated in 50 mM potassium phosphate buffer pH 7.4 with and without NADPH-generating system consisting of 1 mM NADP + , 10 mM glucose-6-phosphate and 1 unit glucose-6-phosphate dehydrogenase at 37 ° C.
- Primary hepatocytes were also incubated in suspension in Williams E medium also at 37 ° C.
- the incubation mixtures were stopped with acetonitrile (final concentration about 30%) and the protein was centrifuged off at about 15,000 ⁇ g. The samples thus stopped were either analyzed directly or stored at -20 ° C until analysis.
- the analysis is carried out by high performance liquid chromatography with ultraviolet and mass spectrometric detection (HPLC-UV-MS / MS).
- HPLC-UV-MS / MS ultraviolet and mass spectrometric detection
- the supernatants of the incubation samples are chromatographed with suitable C18-reversed-phase columns and variable eluent mixtures of acetonitrile and 10 mM aqueous ammonium formate solution or 0.05% formic acid.
- the UV chromatograms in combination with mass spectrometry data serve to identify, structure elucidate and quantitatively estimate the metabolites, and quantitative metabolic decrease of the compound of the invention in the incubation approaches.
- the permeability of a test substance was determined using the Caco-2 cell line, an established in vitro model for permeability predictions at the gastrointestinal barrier (Artursson, P. and Karlsson, J. (1991) Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells, Biochem., Biophys. 175 (3), 880-885).
- the Caco-2 cells (ACC No. 169, DSMZ, German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany) were seeded in 24-well plates with use and cultured for 14 to 16 days.
- test substance was dissolved in DMSO and diluted to the final test concentration with transport buffer (Hanks Buffered Salt Solution, Gibco / Invitrogen, with 19.9 mM glucose and 9.8 mM HEPES).
- transport buffer Hanks Buffered Salt Solution, Gibco / Invitrogen, with 19.9 mM glucose and 9.8 mM HEPES.
- P app AB the solution containing the test substance was added to the apical side of the Caco-2 cell monolayer and transport buffer to the basolateral side.
- P app BA the solution containing the test substance was added to the basolateral side of the Caco-2 cell monolayer and transport buffer to the apical side.
- hERG human ether-a-go-go related gene
- the functional hERG assay used here is based on a recombinant HEK293 cell line stably expressing the KCNH2 (HERG) gene (Zhou et al., 1998). These cells are assayed by the whole-cell voltage-clamp technique (Hamill et al., 1981) in an automated system (Patchliner TM, Nanion, Kunststoff, D) which controls membrane voltage and hERG potassium current at room temperature measures.
- the PatchControlHT TM software (Nanion) controls patchliner system, data acquisition and data analysis. The voltage is controlled by 2 EPC-10 quadro amplifiers under the control of the PatchMasterPro TM software (both: HEKA Elektronik, Lambrecht, D).
- NPC-16 medium resistance chips ( ⁇ 2 ⁇ , Nanion) serve as a planar substrate for the voltage-clamp experiments.
- NPC-16 chips are filled with intra- and extracellular solution (see Himmel, 2007) as well as with cell suspension.
- the cell membrane After formation of a giga-ohm seal and production of the whole cell mode (including several automated quality control steps), the cell membrane is clamped to the hold potential -80 mV.
- the following voltage logic protocol changes the command voltage to +20 mV (duration 1000 ms), -120 mV (duration 500 ms), and back to the holding potential -80 mV; this is repeated every 12 seconds.
- an initial stabilization phase about 5-6 minutes
- test substance solution in ascending concentrations (eg, 0.1, 1, and 10 ⁇ / L) (exposure about 5-6 minutes per concentration), followed by several washout steps.
- the amplitude of the inward TaiF current generated by a potential change from +20 mV to -120 mV serves to quantify the hERG potassium current and is plotted as a function of time (IgorPro TM software)
- Periods of time eg, stabilization phase before test substance, first / second / third concentration of test substance serve to produce a concentration-effect curve from which the half-maximal inhibitory concentration IC50 of the test substance is calculated.
- the compounds according to the invention can be converted into pharmaceutical preparations as follows:
- the mixture of compound of the invention, lactose and starch is granulated with a 5% solution (m / m) of the PVP in water.
- the granules are mixed after drying with the magnesium stearate for 5 minutes.
- This mixture is compressed with a conventional tablet press (for the tablet format see above).
- a pressing force of 15 kN is used as a guideline for the compression.
- Composition 1000 mg of the compound of the invention, 1000 mg of ethanol (96%), 400 mg of Rhodigel ® (xanthan gum of the firm FMC, Pennsylvania, USA) and 99 g of water.
- a single dose of 100 mg of the compound of the invention corresponds to 10 ml of oral suspension.
- the compound of the invention is suspended in the mixture of polyethylene glycol and polysorbate with stirring. The stirring is continued until complete dissolution of the compound according to the invention.
- i.v. solution The compound of the invention is dissolved at a concentration below saturation in a physiologically acceptable solvent (e.g., isotonic saline, glucose solution 5% and / or PEG 400 solution 30%). The resulting solution is sterile filtered and filled into sterile and pyrogen-free injection containers.
- a physiologically acceptable solvent e.g., isotonic saline, glucose solution 5% and / or PEG 400 solution 30%.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Epidemiology (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Diabetes (AREA)
- Hospice & Palliative Care (AREA)
- Vascular Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description







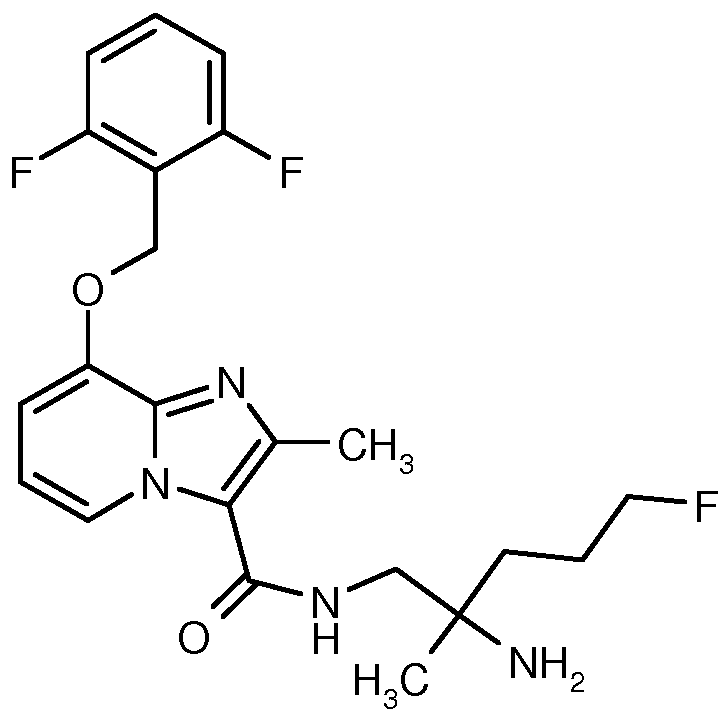




















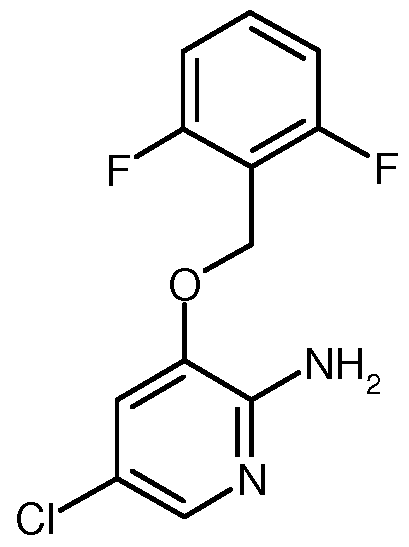







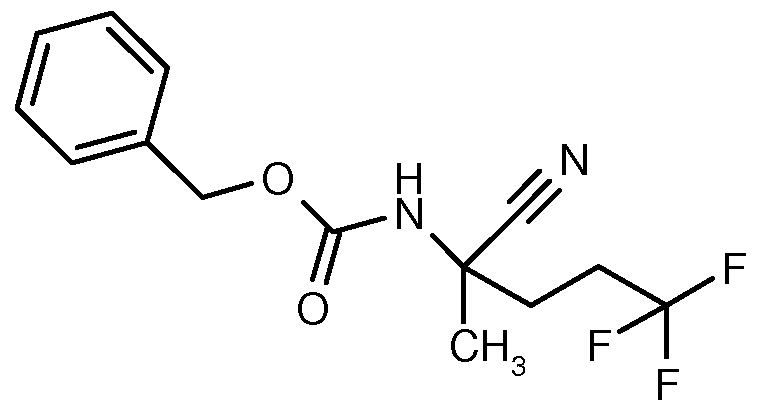


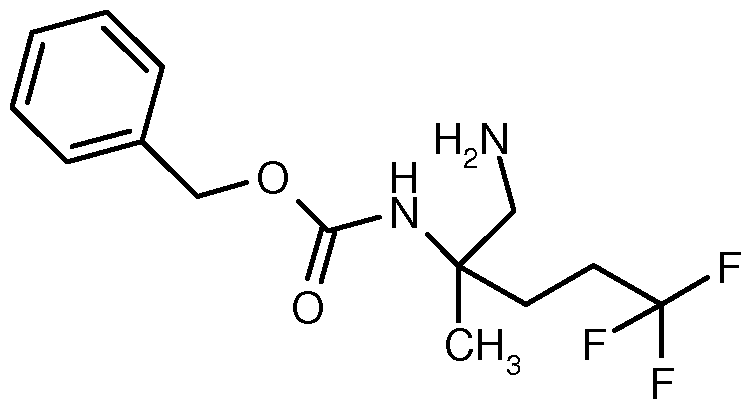




































Claims











Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP15718896.2A EP3137463A1 (de) | 2014-05-02 | 2015-04-29 | Enantiomere des n-(2-amino-5-fluor-2-methylpentyl)-8-[(2,6-difluorbenzyl)oxy]-2-methylimidazo[1,2-a]pyridin-3-carboxamids sowie der di- and tri-fluor-derivate zur behandlung von kardiovaskulären erkrankungen |
| CA2947374A CA2947374A1 (en) | 2014-05-02 | 2015-04-29 | Enantiomers of the n-(2-amino-5-fluoro-2-methylpentyl)-8-[(2,6-difluorobenzyl)oxy]-2-methylimidazo[1,2-a]pyridine-3-carboxamide, as well as of the di- and trifluoro derivatives for the treatment of cardiovascular diseases |
| JP2017508753A JP2017514898A (ja) | 2014-05-02 | 2015-04-29 | 心血管疾患の治療のためのn−(2−アミノ−5−フルオロ−2−メチルペンチル)−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミドの、並びにジ−及びトリフルオロ誘導体のエナンチオマー |
| US15/308,279 US20170057958A1 (en) | 2014-05-02 | 2015-04-29 | Enantiomers of the n-(2-amino-5-fluoro-2-methylpentyl)-8-[(2,6-difluorobenzyl)oxy]-2-methylimidazo[1,2-a]pyridine-3-carboxamide, as well as of the di- and trifluoro derivatives for the treatment of cardiovascular diseases |
| CN201580022270.5A CN106459037A (zh) | 2014-05-02 | 2015-04-29 | 用于治疗心血管疾病的N‑(2‑氨基‑5‑氟‑2‑甲基戊基)‑8‑[(2,6‑二氟苄基)氧基]‑2‑甲基咪唑并[1,2‑a]吡啶‑3‑甲酰胺的对映异构体及其二‑和三氟衍生物的对映异构体 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14166912.7 | 2014-05-02 | ||
| EP14166912 | 2014-05-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015165930A1 true WO2015165930A1 (de) | 2015-11-05 |
Family
ID=50693468
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2015/059274 WO2015165930A1 (de) | 2014-05-02 | 2015-04-29 | Enantiomere des n-(2-amino-5-fluor-2-methylpentyl)-8-[(2,6-difluorbenzyl)oxy]-2-methylimidazo[1,2-a]pyridin-3-carboxamids sowie der di- and tri-fluor-derivate zur behandlung von kardiovaskulären erkrankungen |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20170057958A1 (de) |
| EP (1) | EP3137463A1 (de) |
| JP (1) | JP2017514898A (de) |
| CN (1) | CN106459037A (de) |
| CA (1) | CA2947374A1 (de) |
| WO (1) | WO2015165930A1 (de) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018184976A1 (de) | 2017-04-05 | 2018-10-11 | Bayer Pharma Aktiengesellschaft | Substituierte imidazo[1,2-a]pyridincarboxamide und ihre verwendung |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015165933A2 (de) * | 2014-05-02 | 2015-11-05 | Bayer Pharma Aktiengesellschaft | 6-substituierte imidazo[1,2-a]pyridincarboxamide und ihre verwendung |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0266890A1 (de) | 1986-10-07 | 1988-05-11 | Yamanouchi Pharmaceutical Co. Ltd. | Imidazopyridinderivate, ihre Herstellung und diese enthaltende pharmazeutische Zubereitungen |
| WO1989003833A1 (en) | 1987-10-30 | 1989-05-05 | Aktiebolaget Hässle | IMIDAZO(1,2-a)(PYRIDAZINES OR PYRAZINES) FOR TREATMENT OF DISEASES RELATED TO BONE LOSS |
| JPH01258674A (ja) | 1988-04-08 | 1989-10-16 | Mitsubishi Kasei Corp | イミダゾ〔1,2−a〕ピリジン誘導体 |
| WO1996034866A1 (en) | 1995-05-01 | 1996-11-07 | Fujisawa Pharmaceutical Co., Ltd. | Imidazo 1,2-a pyridine and imidazo 1,2-a pyridezine derivatives and their use as bone resorption inhibitors |
| WO1998016223A1 (de) | 1996-10-14 | 1998-04-23 | Bayer Aktiengesellschaft | Verwendung von 1-benzyl-3-(substituiertes-hetaryl)-kondensierten pyrazol-derivaten zur belandlung von speziellen erkrankungen des herz-kreislaufsystems und des zentralnervensystems |
| EP1277754A1 (de) | 2000-04-27 | 2003-01-22 | Yamanouchi Pharmaceutical Co. Ltd. | Imidazopyridin-derivate |
| WO2006015737A1 (de) | 2004-08-02 | 2006-02-16 | Schwarz Pharma Ag | Carboxamide des indolizins und seiner aza- und diazaderivate |
| WO2008008539A2 (en) | 2006-07-14 | 2008-01-17 | Amgen Inc. | Fused heterocyclic derivatives useful as inhibitors of the hepatocyte growth factor receptor |
| WO2008061626A1 (de) | 2006-11-20 | 2008-05-29 | Bayer Schering Pharma Aktiengesellschaft | Verfahren zum nachweis von pyrophosphat mit biolumineszenz detektion |
| WO2008082490A2 (en) | 2006-12-20 | 2008-07-10 | Schering Corporation | Novel jnk inhibitors |
| WO2008134553A1 (en) | 2007-04-26 | 2008-11-06 | Xenon Pharmaceuticals Inc. | Methods of using bicyclic compounds in treating sodium channel-mediated diseases |
| WO2010030538A2 (en) | 2008-09-11 | 2010-03-18 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| WO2011113606A1 (en) | 2010-03-18 | 2011-09-22 | Institut Pasteur Korea | Anti-infective compounds |
| WO2012165399A1 (ja) | 2011-05-30 | 2012-12-06 | アステラス製薬株式会社 | イミダゾピリジン化合物 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9126998B2 (en) * | 2012-11-05 | 2015-09-08 | Bayer Pharma AG | Amino-substituted imidazo[1,2-a]pyridinecarboxamides and their use |
-
2015
- 2015-04-29 CN CN201580022270.5A patent/CN106459037A/zh active Pending
- 2015-04-29 JP JP2017508753A patent/JP2017514898A/ja active Pending
- 2015-04-29 US US15/308,279 patent/US20170057958A1/en not_active Abandoned
- 2015-04-29 WO PCT/EP2015/059274 patent/WO2015165930A1/de active Application Filing
- 2015-04-29 CA CA2947374A patent/CA2947374A1/en not_active Abandoned
- 2015-04-29 EP EP15718896.2A patent/EP3137463A1/de not_active Withdrawn
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0266890A1 (de) | 1986-10-07 | 1988-05-11 | Yamanouchi Pharmaceutical Co. Ltd. | Imidazopyridinderivate, ihre Herstellung und diese enthaltende pharmazeutische Zubereitungen |
| WO1989003833A1 (en) | 1987-10-30 | 1989-05-05 | Aktiebolaget Hässle | IMIDAZO(1,2-a)(PYRIDAZINES OR PYRAZINES) FOR TREATMENT OF DISEASES RELATED TO BONE LOSS |
| JPH01258674A (ja) | 1988-04-08 | 1989-10-16 | Mitsubishi Kasei Corp | イミダゾ〔1,2−a〕ピリジン誘導体 |
| WO1996034866A1 (en) | 1995-05-01 | 1996-11-07 | Fujisawa Pharmaceutical Co., Ltd. | Imidazo 1,2-a pyridine and imidazo 1,2-a pyridezine derivatives and their use as bone resorption inhibitors |
| WO1998016223A1 (de) | 1996-10-14 | 1998-04-23 | Bayer Aktiengesellschaft | Verwendung von 1-benzyl-3-(substituiertes-hetaryl)-kondensierten pyrazol-derivaten zur belandlung von speziellen erkrankungen des herz-kreislaufsystems und des zentralnervensystems |
| EP1277754A1 (de) | 2000-04-27 | 2003-01-22 | Yamanouchi Pharmaceutical Co. Ltd. | Imidazopyridin-derivate |
| WO2006015737A1 (de) | 2004-08-02 | 2006-02-16 | Schwarz Pharma Ag | Carboxamide des indolizins und seiner aza- und diazaderivate |
| WO2008008539A2 (en) | 2006-07-14 | 2008-01-17 | Amgen Inc. | Fused heterocyclic derivatives useful as inhibitors of the hepatocyte growth factor receptor |
| WO2008061626A1 (de) | 2006-11-20 | 2008-05-29 | Bayer Schering Pharma Aktiengesellschaft | Verfahren zum nachweis von pyrophosphat mit biolumineszenz detektion |
| WO2008082490A2 (en) | 2006-12-20 | 2008-07-10 | Schering Corporation | Novel jnk inhibitors |
| WO2008134553A1 (en) | 2007-04-26 | 2008-11-06 | Xenon Pharmaceuticals Inc. | Methods of using bicyclic compounds in treating sodium channel-mediated diseases |
| WO2010030538A2 (en) | 2008-09-11 | 2010-03-18 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| WO2011113606A1 (en) | 2010-03-18 | 2011-09-22 | Institut Pasteur Korea | Anti-infective compounds |
| WO2012165399A1 (ja) | 2011-05-30 | 2012-12-06 | アステラス製薬株式会社 | イミダゾピリジン化合物 |
| EP2716642A1 (de) * | 2011-05-30 | 2014-04-09 | Astellas Pharma Inc. | Imidazopyridinverbindung |
Non-Patent Citations (27)
| Title |
|---|
| A. A. WUBE ET AL., BIOORGANIC AND MEDICINAL CHEMISTRY, vol. 19, 2011, pages 567 - 579 |
| ARTURSSON, P.; KARLSSON, J.: "Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells", BIOCHEM. BIOPHYS., vol. 175, no. 3, 1991, pages 880 - 885, XP024770797, DOI: doi:10.1016/0006-291X(91)91647-U |
| BITLER; MCELROY, ARCH. BIOCHEM. BIOPHYS., vol. 72, 1957, pages 358 |
| CHEMICAL ABSTRACTS, Columbus, Ohio, US; abstract no. 178986 |
| DEMBINSKI, R., EUR. J. ORG. CHEM., 2004, pages 2763 |
| F. WUNDER ET AL., ANAL. BIOCHEM., vol. 339, 2005, pages 104 - 112 |
| G. M. RUBOTTOM ET AL., JOURNAL OF ORGANIC CHEMISTRY, vol. 48, 1983, pages 1550 - 1552 |
| GOLDBERG ET AL., J. BIOL. CHEM., vol. 252, 1977, pages 1279 |
| HAMILL OP; MARTY A; NEHER E; SAKMANN B; SIGWORTH FJ: "Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches", PFLUEGERS ARCH, vol. 391, 1981, pages 85 - 100, XP000196663, DOI: doi:10.1007/BF00656997 |
| HIMMEL HM: "Suitability of commonly used excipients for electrophysiological in-vitro safety pharmacology assessment of effects on hERG potassium current and on rabbit Purkinje fiber action potential", J PHARMACOL TOXICOL METHODS, vol. 56, 2007, pages 145 - 158, XP022651001, DOI: doi:10.1016/j.vascn.2007.04.004 |
| HÖNICKA ET AL., JOURNAL OF MOLECULAR MEDICINE, vol. 77, 1999, pages 14 - 23 |
| HUGHES, D.L., ORG. REACT., vol. 42, 1992, pages 335 |
| K. HIROI ET AL., SYNLETT, 2001, pages 263 - 265 |
| K. MIKAMI ET AL., CHEMISTRY LETTERS, 1982, pages 1349 - 1352 |
| KLAUS WITTE; KAI HU; JOHANNA SWIATEK; CLAUDIA MÜSSIG; GEORG ERTL; BJÖRN LEMMER: "Experimental heart failure in rats: effects on cardiovascular circadian rhythms and on myocardial ß-adrenergic signaling", CARDIOVASC RES, vol. 47, no. 2, 2000, pages 203 - 405 |
| KOZO OKAMOTO: "Spontaneous hypertension in rats", INT REV EXP PATHOL, vol. 7, 1969, pages 227 - 270 |
| MAARTEN VAN DEN BUUSE: "Circadian Rhythms of Blood Pressure, Heart Rate, and Locomotor Activity in Spontaneously Hypertensive Rats as Measured With Radio-Telemetry", PHYSIOLOGY & BEHAVIOR, vol. 55, no. 4, 1994, pages 783 - 787 |
| MÜLSCH ET AL., BRIT. J. PHARMACOL., vol. 120, 1997, pages 681 |
| PETTIBONE ET AL., EUR. J. PHARMACOL., vol. 116, 1985, pages 307 |
| SCHEEL 0; HIMMEL H; RASCHER-EGGSTEIN G; KNOTT T: "Introduction of a modular automated voltage-clamp platform and its correlation with manual human ether-a-go-go related gene voltage-clamp data", ASSAY DRUG DEV TECHNOL, vol. 9, 2011, pages 600 - 607 |
| STASCH ET AL., BR. J. PHARMACOL., vol. 135, 2002, pages 344 - 355 |
| T. CHEN ET AL., JOURNAL OF ORGANIC CHEMISTRY, vol. 61, 1996, pages 4716 - 4719 |
| T.W. GREENE; P.G.M. WUTS: "Protective Groups in Organic Synthesis", 1999, WILEY |
| WU ET AL., BLOOD, vol. 84, 1994, pages 4226 |
| Y. BAI ET AL., ANGEWANDTE CHEMIE, vol. 51, 2012, pages 4112 - 4116 |
| YU ET AL., BRIT. J. PHARMACOL., vol. 114, 1995, pages 1587 |
| ZHOU ZF; GONG Q; YE B; FAN Z; MAKIELSKI JC; ROBERTSON GA; JANUARY CT: "Properties of hERG channels stably expressed in HEK293 cells studied at physiological temperature", BIOPHYS J, vol. 74, 1998, pages 230 - 241, XP002281087 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018184976A1 (de) | 2017-04-05 | 2018-10-11 | Bayer Pharma Aktiengesellschaft | Substituierte imidazo[1,2-a]pyridincarboxamide und ihre verwendung |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2947374A1 (en) | 2015-11-05 |
| CN106459037A (zh) | 2017-02-22 |
| US20170057958A1 (en) | 2017-03-02 |
| EP3137463A1 (de) | 2017-03-08 |
| JP2017514898A (ja) | 2017-06-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2914596B1 (de) | Hydroxy-substituierte imidazo[1,2-a]pyridincarboxamide und ihre verwendung als stimulatoren der löslichen guanylatcyclase | |
| EP2961755B1 (de) | Trifluormethyl-substituierte annellierte pyrimidine und ihre verwendung | |
| EP2961754B1 (de) | Benzyl-substituierte pyrazolopyridine und ihre verwendung | |
| EP3030562B1 (de) | Substituierte pyrazolo[1,5-a]pyridin-3-carboxamide und ihre verwendung | |
| EP2914595A1 (de) | Carboxy-substituierte imidazo[1,2-a]pyridincarboxamide und ihre verwendung als stimulatoren der löslichen guanylatcyclase | |
| EP3107920B1 (de) | 3-(pyrimidin-2-yl)imidazo[1,2-a]pyridine | |
| EP3119777A1 (de) | Cyano-substituierte imidazo[1,2-a]pyridincarboxamide und ihre verwendung | |
| WO2015018808A1 (de) | Substituierte imidazo[1,2-a]pyrazincarboxamide und ihre verwendung | |
| EP3077394A1 (de) | Aryl- und hetaryl-substituierte imidazo[1,2-a]pyridin-3-carboxamide und ihre verwendung | |
| WO2015140254A1 (de) | Substituierte imidazo[1,2-a]pyridincarboxamide und ihre verwendung | |
| WO2015165931A1 (de) | Imidazo[1,2-a]pyridine als stimulatoren der löslichen guanylatcyclase zur behandlung von kardiovaskulären erkrankungen | |
| EP3227286B1 (de) | Substituierte pyrazolo[1,5-a]pyridine und imidazo[1,2-a]pyrazine und ihre verwendung | |
| EP3137465A1 (de) | 6-chlor-substituierte imidazo[1,2-a]pyridincarboxamide und ihre verwendung als stimulatoren der löslichen guanylatcyclase | |
| WO2015165933A2 (de) | 6-substituierte imidazo[1,2-a]pyridincarboxamide und ihre verwendung | |
| EP3137463A1 (de) | Enantiomere des n-(2-amino-5-fluor-2-methylpentyl)-8-[(2,6-difluorbenzyl)oxy]-2-methylimidazo[1,2-a]pyridin-3-carboxamids sowie der di- and tri-fluor-derivate zur behandlung von kardiovaskulären erkrankungen | |
| EP3227287B1 (de) | Heteroaryl-substituierte imidazo[1,2-a]pyridine und ihre verwendung | |
| EP3180006A1 (de) | Substituierte chinolin-4-carboxamide und ihre verwendung | |
| EP3253758B1 (de) | N-substituierte 8-[(2,6-difluorobenzyl)oxy]-2,6-dimethylimidazo[1,2-a]pyrazin-3-carboxamid-derivate als stimulatoren der löslichen guanylatcyclase (sgc) zur behandlung von kardiovaskulären erkrankungen | |
| WO2018184976A1 (de) | Substituierte imidazo[1,2-a]pyridincarboxamide und ihre verwendung | |
| WO2016124565A1 (de) | Substituierte pyrazolo[1,5-a]pyridin-3-carboxamide und ihre verwendung | |
| WO2017121693A1 (de) | Substituierte thiazol- und thiadiazolamide und ihre verwendung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15718896 Country of ref document: EP Kind code of ref document: A1 |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015718896 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015718896 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2947374 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2017508753 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15308279 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |