WO2015164615A1 - Anti-gluten antibodies and uses thereof - Google Patents
Anti-gluten antibodies and uses thereof Download PDFInfo
- Publication number
- WO2015164615A1 WO2015164615A1 PCT/US2015/027315 US2015027315W WO2015164615A1 WO 2015164615 A1 WO2015164615 A1 WO 2015164615A1 US 2015027315 W US2015027315 W US 2015027315W WO 2015164615 A1 WO2015164615 A1 WO 2015164615A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- seq
- antibody
- gliadin
- peptide
- peptides
- Prior art date
Links
Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/564—Immunoassay; Biospecific binding assay; Materials therefor for pre-existing immune complex or autoimmune disease, i.e. systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis, rheumatoid factors or complement components C1-C9
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/16—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from plants
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/569—Immunoassay; Biospecific binding assay; Materials therefor for microorganisms, e.g. protozoa, bacteria, viruses
- G01N33/56961—Plant cells or fungi
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/33—Crossreactivity, e.g. for species or epitope, or lack of said crossreactivity
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/34—Identification of a linear epitope shorter than 20 amino acid residues or of a conformational epitope defined by amino acid residues
Definitions
- the present invention relates to anti-gluten antibodies and methods of using the same. In some embodiments, the present invention relates to the use of anti-gluten antibodies in research, food testing, and diagnostic applications.
- Celiac disease is an autoimmune disorder of the small intestine that occurs in genetically predisposed people of all ages from middle infancy onward. Symptoms include pain and discomfort in the digestive tract, chronic constipation and diarrhoea, failure to thrive (in children), anaemia and fatigue, but these may be absent, and symptoms in other organ systems have been described. Vitamin deficiencies are often noted in people with celiac disease owing to the reduced ability of the small intestine to p-roperly absorb nutrients from food.
- the only effective treatment is a lifelong gluten-free diet.
- Strict adherence to the diet allows the intestines to heal, leading to resolution of all symptoms in most cases and, depending on how soon the diet is begun, can also eliminate the heightened risk of osteoporosis and intestinal cancer and in some cases sterility.
- the diet can be cumbersome; failure to comply with the diet may cause relapse.
- gluten-free is generally used to indicate a supposed harmless level of gluten rather than a complete absence.
- the exact level at which gluten is harmless is uncertain and controversial.
- a recent systematic review tentatively concluded that consumption of less than
- Additonal compositions and methods for accurately and precisely determining the levels of gluten are needed.
- the present invention relates to anti-gluten antibodies and methods of using the same.
- the present invention relates to the use of anti-gluten antibodies in research, food testing, and diagnostic applications.
- the present disclosure provides an isolated monoclonal antibody that binds to gliadin, wherein said antibody recognizes an epitope or motif (e.g., QPXQPFP (SEQ ID NO: 1), wherein X is Q or E; QPQQXFP (SEQ ID NO: 2), wherein X is P, S, T, or Q; QPQ(de)QXFP (SEQ ID NO: 2), wherein X is P, S, T, or Q; PLQPEQPFP (SEQ ID NO: 3); PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4);
- QPXQPFP SEQ ID NO: 1
- QPQQXFP SEQ ID NO: 2
- QPQ(de)QXFP SEQ ID NO: 2
- PLQPEQPFP SEQ ID NO: 3
- PQPEQPFPQPEQPFPQPEQPFPQPEQPFPQP SEQ ID NO: 4
- X1QPQQPX2 (SEQ ID NO: 5), wherein X l is P or S and X 2 is I, L, or F; XiQPQQPX 2 (SEQ ID NO: 5), wherein X l is P or S and X 2 is I, L, or F; XiQPQ(de)QPX 2 (SEQ ID NO: 6), wherein Xi is Q, P, I, or L and X 2 is F, Q, or A;
- LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17); LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18);
- the antibody is an antibody fragment (e.g., Fab, Fab', Fab'-SH, F(ab') 2 , Fv, or scFv variants) or a full length antibody.
- the antibody is fused to a non-antibody molecule (e.g., a label or other molecule).
- the complementarity determining region (CDR) of the antibody is encoded by a nucleic acid described in Table 2 or sequence that are at least 80% (e.g., 85%, 90%, 91 , 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100%) homologous to the sequences shown in Table 2.
- the epitope comprises one or more deamidated amino acids (e.g., represented by E or Q(de)).
- FIG. 1 QPXQPFP (SEQ ID NO: 1), wherein X is Q or E; QPQQXFP (SEQ ID NO: 2), wherein X is P, S, T, or Q; PLQPEQPFP (SEQ ID NO: 3); PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4); XiQPQQPX 2 (SEQ ID NO: 5), wherein X 1 is P or S and X 2 is I, L, or F; XiQPQQPX 2 (SEQ ID NO: 6), wherein X 1 is Q, P, I, or L and X 2 is F, Q, or A; LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17); LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ
- PQPEQPFPQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4); XiQPQQPX 2 (SEQ ID NO: 5), wherein Xj is P or S and X 2 is I, L, or F; XiQPQQPX 2 (SEQ ID NO: 6), wherein X 1 is Q, P, I, or L and X 2 is F, Q, or A; LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17); LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18); QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259); or QPEQPFPEQPEQPEQPFPQPEQPFPW (SEQ ID NO:
- the monoclonal antibody is attached to a solid support (e.g., a bead).
- a solid support e.g., a bead
- at least a portion of the solid support or peptide is labeled.
- the peptide is labeled (e.g., with biotin).
- the label comprises a linker.
- the label is biotin- GSGSGS.
- PLQPEQPFP (SEQ ID NO: 3); PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4);
- X 1 QPQQPX 2 (SEQ ID NO: 5), wherein X l is P or S and X 2 is I, L, or F;
- XiQPQQPX 2 (SEQ ID NO: 6), wherein X 1 is Q, P, I, or L and X 2 is F, Q, or A;
- LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17); LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18);
- the assay is a competitive assay comprising both peptide and antibody and the signal of a signal molecule or label (e.g., fluorescent label) is reduced in the presence of antibodies in the sample that bind to the peptide.
- a signal molecule or label e.g., fluorescent label
- LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17); LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18);
- QPEQPFPEQPEQPEQPFPQPEQPFPW (SEQ ID NO: 260) (e.g., optionally coupled to a solid support and/or a signal molecule or label), with a sample from a subject; and b) measuring the level of binding of the peptide to anti-gliadin antibodies present in the sample.
- the level of binding is compared to the level of binding in a control serum from a subject that does not have celiac disease. In some embodiments, an increased level of binding relative to the level of binding found in the control sample is indicative of celiac disease in the subject. In some embodiments, the the monoclonal antibody and/or the peptide are labeled.
- Additional embodiments provide a method of detecting gluten in a food sample, comprising: a) contacting a food sample with an antibody as described herein; and b) detecting the presence or absence of binding of the antibody to gliadin in the sample.
- the monoclonal antibody is labeled.
- kits comprising the antibody and/or peptides as described herein and a buffer.
- the kit further comprises a solid support.
- the antibody and/or peptide is affixed to the solid support.
- Figure 1 shows survival of intestinal plasma cells in culture, a) Concentration of IgA in supernatants after 0, 1, 2, 3 and 4 weeks culture of single cell suspensions (SCSs) grown with (F+) or without fibroblasts (F-) (n.d. denotes non-detectable), b) Concentration of IgA after 0 and 2 days in supernatants from cultures of fibroblasts and PCs when PCs were added either as isolated IgA PCs (IgA PCs) or as part of single cell suspensions (SCSs). c) Representative flow cytometry plots of SCSs after 4 weeks of co-culture with fibroblasts.
- SCSs single cell suspensions
- Figure 2 shows supernatant reactivity to heat/acid treated chymotrypsin digested gliadin (CT-gliadin) and TG2 by ELISA.
- UCD untreated celiac disease
- b) The ratio of culture supematants with IgA reactivity to TG2 versus CT-gliadin, where the dots represent different subjects with UCD (n 8).
- Horizontal bar indicates mean value
- the background level was defined by signal in supematants of cultures of fibroblasts only.
- Figure 3 shows ELISA reactivity of hmAbs expression cloned from IgA + PCs in culture, a) CT-gliadin. b) BSA control, c) PLQPEQPFP (SEQ ID NO: 3).
- Figure 4 shows staining of intestinal PCs with tetramers of synthetic gluten peptides or TG2 in flow cytometry, a) Representative plots of PCs from SCSs stained with APC- conjugated streptavidin in complex with biotinylated (SEQ ID NO: 3) peptide, b) Frequency of IgA + PCs stained positive with peptide in percentage of total number of IgA + PCs. c)
- Figure 5 shows alphaLISA characterization of gliadin-reactive hmAbs. a) Antibody reactivity to constant concentration of biotinylated PLQPEQPFP (SEQ ID NO: 3) in the presence of competing antigens, b) Reactivity to deamidated gliadin versus native gliadin.
- Figure 6 shows epitopes of gliadin-specific hmAbs. a) Reactivity of hmAbs to PLQPEQPFP (SEQ ID NO: 3) and 33-mer. b) Reactivity of two representative hmAbs to constant concentration of biotinylated PLQPEQPFP (SEQ ID NO: 3) in competition with four different competitive synthetic gliadin peptides.
- Figure 7 shows alphaLISA anti-PLQPEQPFP (SEQ ID NO: 3) immunoglobulin inhibition assay.
- FIG. 8 shows VH/VL usage and somatic hypermutations (SHMs).
- Figure 9 shows frequency of IgA + PCs in SCSs of small intestinal biopsies, a) Representative flow plot of SCSs showing large, viable, CD3 ⁇ GLIADIN ⁇ CD27 + IgA + , defined IgA + PCs. b) Relative frequency of IgA + PC of all cells in SCS in flow cytometry. Each dot represents one subject. Horizontal bar indicates mean value, c) Representative flow plot of SCSs of small intestinal biopsies, d) Relative ratio of IgA + PCs to IgA + memory B cells.
- Figure 10 shows reactivity to antigen as depicted in headline measured by ELISA of 12 hmAbs cloned from single PLQPEQPFP + (SEQ ID NO: 3) (a) or 33-mer + (b) PCs.
- Figure 1 1 shows comparison of PLQPEQPFP (SEQ ID NO: 3) conjugated beads and biotinylated hmAb 1002-1E01 compared to ELISA using streptavidin coated plates, biotinylated PLQPEQPFP (SEQ ID NO: 3)and anti-human IgG as detecting antibody.
- Figure 12 shows estimated concentrations of serum antibodies blocking binding of (a) hmAb 1002-1E01 to PLQPEQPFP (SEQ ID NO: 3) and (b) hmAbs 1002-1E01 and 1002- 1E03 to PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4), by AlphaLISA competitive assay.
- Figure 14 shows ELISA reactivity of four gliadin-specific hmAbs to synthetic gliadin peptides.
- the hmAb tested is depicted in the headline.
- the hmAb was tested in different concentrations as indicated on x-axis.
- Figure 15 shows ELISA reactivity of five gliadin-specific hmAbs to synthetic gliadin peptides.
- the hmAb tested is depicted in the headline.
- the hmAb was tested in different concentrations as indicated on x-axis.
- the different synthetic peptides are presented with different symbols, as described, (a) Initial testing of two of the five hmAbs showed different peptide reactivity pattern than hmAbs tested in Figure 14, with no reactivity to PLQPQQPFP
- Figure 16 shows competitive gliadin AlphaLISA assay for detection of gliadin in flour.
- Figure 17 shows (a) Length of peptides identified by mass spectrometry in fractions of a TG2 -treated digest of gliadin before (grey) and after (black) pull-down by the human monoclonal antibody 1002-1E03. (b) The number of peptides sharing identical sequence motifs, of 3 to 15 residues in length in pre (grey) and post pull-down (black) samples.
- Figure 18 shows alphaLISA anti-c ⁇ 34 Ig assay, (a) Inhibition of 1002-1E03 binding to b-c ⁇ 34 peptide by sera of three test groups as analyzed in AlphaLISA. (b) Reference curve established by serial dilutions of a negative control serum spiked in with known and equimolar concentrations of three gliadin-specific hmAbs. (c) Activity of gliadin-specific serum antibodies expressed as concentration equivalents (mg/L) of reference gliadin-specific hmAbs.
- Figure 19 shows (a) Anti-TG2 IgA and (b) anti-DGP IgG levels of participants from the three test groups.
- Figure 20 shows inhibition of AlphaLISA signals of all three assays with hmAb 1002- 1E03 and the target peptides (a) b-QPEQPFP 3 (SEQ ID NO: 10), (b) b-c ⁇ 26 and (c) b-c ⁇ 34 by sera of the test groups untreated celiac disease patients (celiac disease) and controls (Crohn' disease patients and healthy subjects), (d) Mean of Log AlphaLISA signal for all three peptides as presented in (a-c).
- Figure 21 shows that the target peptide concentration affects the sensitivity, dynamic range and signal/noise-ratios as shown for three different concentrations of b-c ⁇ 34. Mean values for all three concentrations are shown in grey.
- Figure 22 shows that the antibodies pull down long peptides with repeated motifs, a) Kernel density plot of the peptide length identified by mass spectrometry in gliadin fractions pre and post pull-down by the hmAbs 1130-3A02 (b) and 1002-lEOl. c) Mean peptide length pre and post pull-down from different fractions of a gliadin digest with all human monoclonal antibodies, d) Peptides pulled down with hmAb 1 130-3 A02. The most frequent 7mer motif in the peptides is underlined.
- Figure 23 shows common motifs in peptides pulled down by antibody 1130-3B04. a) Percent of peptides sharing identical sequence motifs, of 3 to 15 residues in length, post (triangles/) and pre pull-down (diamonds) from a fraction of digested gliadin by the hmAb 1130-3B04. b) The sequence motifs and the frequency and number of peptides harbouring these motifs
- Figure 24 shows sequence motif of peptides pulled down by antibody 1130-3 BO 1.
- Figure 25 shows the affinity of antibodies to (SEQ ID NO: 10) QPEQPFP-containing peptides depends on residues flanking the motif, a) AlphaLISA affinity of the hmAbs 1002- 1E01, 1002-1E03 and 1130-3B01 to PLQPEQPFP (SEQ ID NO: 3) and the competitive effect of a panel of different synthetic gliadin peptides harbouring the QPEQPFP (SEQ ID NO: 10) sequence motif at different concentrations (M) as indicated on the x-axis.
- B) The XXXQPQQPFPXX (SEQ ID NO: 14) motif (X any amino acid) searched in the Triticum aestivum database using the program "Pattinprot". Sequence logo of resulting 13mer motif generated by WebLOGO 3.0.
- Figure 26 shows that antibodies show better reactivity to gliadin peptides with repeats of epitopes
- c) AlphaLISA competition assay comparing the relative binding of bead-conjugated hmAb 1002-1E03 to the soluble 34mer ⁇ -peptide in the presence of competing soluble whole antibody (grey solid line) or Fab fragment (black stippled line) of the hmAb 1002-1E03.
- Figure 27 shows analysis of factors affecting peptide pull-down by MALDI-TOF.
- a and b 1 130-3 B01 enrichment from the peptide pairs a-gliadin 33mer and PLQPEQPF (SEQ ID NO: 15) peptide (a) or ⁇ -gliadin 33mer and ⁇ -gliadin 26mer (b).
- c) 1 130-3B03 enrichment from the peptide pair ⁇ -gliadin 33mer and ⁇ -gliadin 26mer.
- Figure 28 shows co-localisation of gliadin T-cell and B-cell epitopes in an ⁇ -gliadin protein.
- acceptor human framework for the purposes herein is a framework comprising the amino acid sequence of a light chain variable domain (VL) framework or a heavy chain variable domain (VH) framework derived from a human immunoglobulin framework or a human consensus framework, as defined below.
- An acceptor human framework "derived from” a human immunoglobulin framework or a human consensus framework may comprise the same amino acid sequence thereof, or it may contain amino acid sequence changes. In some embodiments, the number of amino acid changes are 10 or less, 9 or less, 8 or less, 7 or less, 6 or less, 5 or less, 4 or less, 3 or less, or 2 or less.
- the VL acceptor human framework is identical in sequence to the VL human immunoglobulin framework sequence or human consensus framework sequence.
- Binding affinity refers to the strength of the sum total of noncovalent interactions between a single binding site of a molecule (e.g., an antibody) and its binding partner (e.g., an antigen). Unless indicated otherwise, as used herein, "binding affinity” refers to intrinsic binding affinity which reflects a 1 : 1 interaction between members of a binding pair (e.g., antibody and antigen).
- the affinity of a molecule X for its partner Y can generally be represented by the dissociation constant (Kd). Affinity can be measured by common methods known in the art, including those described herein. Specific illustrative and exemplary embodiments for measuring binding affinity are described in the following.
- an “affinity matured” antibody refers to an antibody with one or more alterations in one or more hypervariable regions (HVRs), compared to a parent antibody which does not possess such alterations, such alterations resulting in an improvement in the affinity of the antibody for antigen.
- HVRs hypervariable regions
- antibody is used herein in the broadest sense and encompasses various antibody structures, including but not limited to monoclonal antibodies, polyclonal antibodies, multispecific antibodies (e.g., bispecific antibodies), and antibody fragments so long as they exhibit the desired antigen-binding activity.
- an “antibody fragment” refers to a molecule other than an intact antibody that comprises a portion of an intact antibody and that binds the antigen to which the intact antibody binds.
- antibody fragments include but are not limited to Fv, Fab, Fab', Fab'-SH, F(ab3 ⁇ 4; diabodies; linear antibodies; single-chain antibody molecules (e.g. scFv); and multispecific antibodies formed from antibody fragments.
- An “antibody that binds to the same epitope” as a reference antibody refers to an antibody that blocks binding of the reference antibody to its antigen in a competition assay by 50% or more, and conversely, the reference antibody blocks binding of the antibody to its antigen in a competition assay by 50% or more.
- An exemplary competition assay is provided herein.
- chimeric antibody refers to an antibody in which a portion of the heavy and/or light chain is derived from a particular source or species, while the remainder of the heavy and/or light chain is derived from a different source or species.
- the "class" of an antibody refers to the type of constant domain or constant region possessed by its heavy chain.
- the heavy chain constant domains that correspond to the different classes of immunoglobulins are called ⁇ , ⁇ , ⁇ , ⁇ , and ⁇ , respectively.
- Antibody effector functions refer to those biological activities attributable to the Fc region of an antibody, which vary with the antibody isotype. Examples of antibody effector functions include: Clq binding and complement dependent cytotoxicity (CDC); Fc receptor binding; antibody-dependent cell-mediated cytotoxicity (ADCC); phagocytosis; down regulation of cell surface receptors (e.g. B cell receptor); and B cell activation.
- an "effective amount" of an agent refers to an amount effective, at dosages and for periods of time necessary, to achieve the desired therapeutic or prophylactic result.
- epitope refers to the particular site on an antigen molecule to which an antibody binds.
- Fc region herein is used to define a C-terminal region of an
- immunoglobulin heavy chain that contains at least a portion of the constant region.
- the term includes native sequence Fc regions and variant Fc regions.
- a human IgG heavy chain Fc region extends from Cys226, or from Pro230, to the carboxyl-terminus of the heavy chain.
- the C-terminal lysine (Lys447) of the Fc region may or may not be present.
- numbering of amino acid residues in the Fc region or constant region is according to the EU numbering system, also called the EU index, as described in Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD, 1991.
- FR Framework or "FR” refers to variable domain residues other than hypervariable region (HVR) residues.
- the FR of a variable domain generally consists of four FR domains: FR1, FR2, FR3, and FR4. Accordingly, the HVR and FR sequences generally appear in the following sequence in VH (or VL): FR1-H1(L1)-FR2-H2(L2)-FR3-H3(L3)-FR4.
- full length antibody “intact antibody,” and “whole antibody” are used herein interchangeably to refer to an antibody having a structure substantially similar to a native antibody structure or having heavy chains that contain an Fc region as defined herein.
- host cell "host cell line,” and “host cell culture” are used interchangeably.
- Host cells include “trans formants” and “transformed cells,” which include the primary transformed cell and progeny derived therefrom without regard to the number of passages.
- Progeny may not be completely identical in nucleic acid content to a parent cell, but may contain mutations. Mutant progeny that have the same function or biological activity as screened or selected for in the originally transformed cell are included herein.
- a "human antibody” is one which possesses an amino acid sequence which corresponds to that of an antibody produced by a human or a human cell or derived from a non-human source that utilizes human antibody repertoires or other human antibody- encoding sequences. This definition of a human antibody specifically excludes a humanized antibody comprising non-human antigen-binding residues.
- a "human consensus framework” is a framework which represents the most commonly occurring amino acid residues in a selection of human immunoglobulin VL or VH framework sequences.
- the selection of human immunoglobulin VL or VH sequences is from a subgroup of variable domain sequences.
- the subgroup of sequences is a subgroup as in Kabat et al, Sequences of Proteins of Immunological Interest, Fifth Edition, NIH Publication 91-3242, Bethesda MD (1991), vols. 1-3.
- the subgroup is subgroup kappa I as in Kabat et al, supra.
- the subgroup is subgroup III as in Kabat et al, supra.
- a “humanized” antibody refers to a chimeric antibody comprising amino acid residues from non-human HVRs and amino acid residues from human FRs.
- a humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the HVRs (e.g., CDRs) correspond to those of a non-human antibody, and all or substantially all of the FRs correspond to those of a human antibody.
- a humanized antibody optionally may comprise at least a portion of an antibody constant region derived from a human antibody.
- a "humanized form" of an antibody, e.g., a non-human antibody refers to an antibody that has undergone humanization.
- hypervariable region refers to each of the regions of an antibody variable domain which are hypervariable in sequence and/or form structurally defined loops ("hypervariable loops").
- native four-chain antibodies comprise six HVRs; three in the VH (HI, H2, H3), and three in the VL (LI, L2, L3).
- HVRs generally comprise amino acid residues from the hypervariable loops and/or from the "complementarity determining regions" (CDRs), the latter being of highest sequence variability and/or involved in antigen recognition.
- CDRs complementarity determining regions
- Exemplary hypervariable loops occur at amino acid residues 26-32 (LI), 50-52 (L2), 91-96 (L3), 26-32 (HI), 53-55 (H2), and 96-101 (H3).
- Exemplary CDRs CDR-L1, CDR-L2, CDR-L3, CDR-Hl, CDR-H2, and CDR-H3) occur at amino acid residues 24-34 of LI, 50-56 of L2, 89-97 of L3, 31-35B of HI, 50-65 of H2, and 95-102 of H3.
- CDRs generally comprise the amino acid residues that form the hypervariable loops.
- CDRs also comprise "specificity determining residues,” or "SDRs,” which are residues that contact antigen. SDRs are contained within regions of the CDRs called abbreviated-CD Rs, or a- CDRs.
- Exemplary a-CDRs (a-CDR-Ll, a-CDR-L2, a-CDR-L3, a-CDR-Hl, a-CDR-H2, and a-CDR-H3) occur at amino acid residues 31-34 of LI, 50-55 of L2, 89-96 of L3, 31-35B of HI, 50-58 of H2, and 95-102 of H3.
- HVR residues and other residues in the variable domain are numbered herein according to Kabat et al, supra.
- mammals include, but are not limited to, domesticated animals (e.g., cows, sheep, cats, dogs, and horses), primates (e.g., humans and non-human primates such as monkeys), rabbits, and rodents (e.g., mice and rats).
- domesticated animals e.g., cows, sheep, cats, dogs, and horses
- primates e.g., humans and non-human primates such as monkeys
- rabbits e.g., mice and rats
- rodents e.g., mice and rats.
- the individual or subject is a human.
- an “isolated antibody” is one which has been separated from a component of its natural environment.
- an antibody is purified to greater than 95% or 99% purity as determined by, for example, electrophoretic (e.g., SDS-PAGE, isoelectric focusing (IEF), capillary electrophoresis) or chromatographic (e.g., ion exchange or reverse phase HPLC).
- electrophoretic e.g., SDS-PAGE, isoelectric focusing (IEF), capillary electrophoresis
- chromatographic e.g., ion exchange or reverse phase HPLC.
- An isolated nucleic acid includes a nucleic acid molecule contained in cells that ordinarily contain the nucleic acid molecule, but the nucleic acid molecule is present extrachromosomally or at a chromosomal location that is different from its natural chromosomal location.
- monoclonal antibody refers to an antibody obtained from a population of substantially homogeneous antibodies, i.e., the individual antibodies comprising the population are identical and/or bind the same epitope, except for possible variant antibodies, e.g., containing naturally occurring mutations or arising during production of a monoclonal antibody preparation, such variants generally being present in minor amounts.
- polyclonal antibody preparations typically include different antibodies directed against different determinants (epitopes)
- each monoclonal antibody of a monoclonal antibody preparation is directed against a single determinant on an antigen.
- the modifier "monoclonal” indicates the character of the antibody as being obtained from a substantially homogeneous population of antibodies, and is not to be construed as requiring production of the antibody by any particular method.
- the monoclonal antibodies to be used in accordance with the present invention may be made by a variety of techniques, including but not limited to the hybridoma method, recombinant DNA methods, phage- display methods, and methods utilizing transgenic animals containing all or part of the human immunoglobulin loci, such methods and other exemplary methods for making monoclonal antibodies being described herein.
- Native antibodies refer to naturally occurring immunoglobulin molecules with varying structures.
- native IgG antibodies are heterotetrameric glycoproteins of about 150,000 daltons, composed of two identical light chains and two identical heavy chains that are disulfide-bonded. From N- to C-terminus, each heavy chain has a variable region (VH), also called a variable heavy domain or a heavy chain variable domain, followed by three constant domains (CHI, CH2, and CH3).
- VH variable region
- VL variable region
- the light chain of an antibody may be assigned to one of two types, called kappa ( ⁇ ) and lambda ( ⁇ ), based on the amino acid sequence of its constant domain.
- Percent (%) amino acid sequence identity with respect to a reference polypeptide sequence is defined as the percentage of amino acid residues in a candidate sequence that are identical with the amino acid residues in the reference polypeptide sequence, after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent sequence identity, and not considering any conservative substitutions as part of the sequence identity. Alignment for purposes of determining percent amino acid sequence identity can be achieved in various ways that are within the skill in the art, for instance, using publicly available computer software such as BLAST, BLAST-2, ALIGN or Megalign (DNASTAR) software. Those skilled in the art can determine appropriate parameters for aligning sequences, including any algorithms needed to achieve maximal alignment over the full length of the sequences being compared.
- % amino acid sequence identity values are generated using the sequence comparison computer program ALIGN-2.
- the ALIGN-2 sequence comparison computer program was authored by Genentech, Inc., and the source code has been filed with user documentation in the U.S. Copyright Office,
- the ALIGN-2 program is publicly available from Genentech, Inc., South San Francisco, California, or may be compiled from the source code.
- the ALIGN-2 program should be compiled for use on a UNIX operating system, including digital UNIX V4.0D. All sequence comparison parameters are set by the ALIGN-2 program and do not vary.
- % amino acid sequence identity of a given amino acid sequence A to, with, or against a given amino acid sequence B is calculated as follows:
- pharmaceutical formulation refers to a preparation which is in such form as to permit the biological activity of an active ingredient contained therein to be effective, and which contains no additional components which are unacceptably toxic to a subject to which the formulation would be administered.
- a “pharmaceutically acceptable carrier” refers to an ingredient in a pharmaceutical formulation, other than an active ingredient, which is nontoxic to a subject.
- pharmaceutically acceptable carrier includes, but is not limited to, a buffer, excipient, stabilizer, or preservative.
- treatment refers to clinical intervention in an attempt to alter the natural course of the individual being treated, and can be performed either for prophylaxis or during the course of clinical pathology. Desirable effects of treatment include, but are not limited to, preventing occurrence or recurrence of disease, alleviation of symptoms, diminishment of any direct or indirect pathological consequences of the disease, preventing metastasis, decreasing the rate of disease progression, amelioration or palliation of the disease state, and remission or improved prognosis.
- antibodies of the invention are used to delay development of a disease or to slow the progression of a disease.
- variable region refers to the domain of an antibody heavy or light chain that is involved in binding the antibody to antigen.
- the variable domains of the heavy chain and light chain (VH and VL, respectively) of a native antibody generally have similar structures, with each domain comprising four conserved framework regions (FRs) and three hypervariable regions (HVRs).
- FRs conserved framework regions
- HVRs hypervariable regions
- antibodies that bind a particular antigen may be isolated using a VH or VL domain from an antibody that binds the antigen to screen a library of complementary VL or VH domains, respectively. See, e.g., Portolano et al, J. Immunol. 150:880-887 (1993); Clarkson et al, Nature 352:624-628 (1991).
- vector refers to a nucleic acid molecule capable of propagating another nucleic acid to which it is linked.
- the term includes the vector as a self- replicating nucleic acid structure as well as the vector incorporated into the genome of a host cell into which it has been introduced.
- Certain vectors are capable of directing the expression of nucleic acids to which they are operatively linked. Such vectors are referred to herein as "expression vectors.” DETAILED DESCRIPTION OF CERTAIN EMBODIMENTS OF THE INVENTION
- the present invention relates to anti-gluten antibodies and methods of using the same. In some embodiments, the present invention relates to the use of anti-gluten antibodies in research, food testing, and diagnostic applications.
- CD Celiac disease
- CD4 + T cells recognizing deamidated gluten and by antibodies reactive to gluten or the self-antigen transglutaminase 2 (TG2).
- TG2 self-antigen transglutaminase 2
- the diagnosis of CD has changed the last years, influenced by serological screening of gliadin-specific and TG2-specific antibodies. They have shown potential usage in screening, and in diagnosing CD without endoscopy. Thus, improved serological methods influence the course of examination of patients where CD is suspected.
- hmAbs monoclonal antibodies
- the serological tests based on the characterized hmAbs have the potential to improve the accuracy of serological CD diagnosis, by either replacing or supplementing current tests.
- the hmAbs also find use as reference reagents for established serological tests, and for detection of gluten in food.
- the invention provides isolated antibodies that bind to gliadin.
- the antibodies are monoclonal antibodies.
- the antibodies have variable regions that are specific for gliadin.
- an anti-gliadin antibody is human or humanized.
- an anti-GLIADI antibody comprises a human acceptor framework, e.g. a human immunoglobulin framework or a human consensus framework.
- the human acceptor framework is the human VL kappa IV consensus (VL KIV ) framework and/or the VH framework VHi.
- anit-gliadin antibodies bind to peptide epitopes described herein (e.g., QPXQPFP (SEQ ID NO: 1), wherein X is Q or E; QPQQXFP (SEQ ID NO: 2), wherein X is P, S, T, or Q; PLQPEQPFP (SEQ ID NO: 3);
- PQPEQPFPQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4); XiQPQQPX 2 (SEQ ID NO: 5), wherein X l is P or S and X 2 is I, L, or F; XiQPQQPX 2 (SEQ ID NO: 6), wherein X l is Q, P, I, or L and X 2 is F, Q, or A; LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17); LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18); QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259);
- the invention provides an antibody that binds to the same epitope as an anti-gliadin antibody provided herein.
- the complementarity determining region (CDR) of the antibody is encoded by a nucleic acid described in Table 2 or sequence that are at least 80% (e.g., 85%, 90%, 91, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100%) homologous to the sequences shown in Table 2.
- an anti-gliadin antibody is a monoclonal antibody, including a chimeric, humanized or human antibody.
- an anti-GLIADIN antibody is an antibody fragment, e.g. , a Fv, Fab, Fab', scFv, diabody, or F(ab') 2 fragment.
- the antibody is a substantially full length antibody, e.g., an IgGl antibody or other antibody class or isotype as defined herein.
- a VH or VL sequence described herein contains substitutions (e.g., conservative substitutions), insertions, or deletions relative to the reference sequence, but an anti-gliadin antibody comprising that sequence retains the ability to bind to gliadin.
- substitutions e.g., conservative substitutions
- insertions e.g., insertions, or deletions relative to the reference sequence
- an anti-gliadin antibody comprising that sequence retains the ability to bind to gliadin.
- a total of 1 to 10 amino acids have been substituted, inserted and/or deleted.
- a total of 1 to 5 amino acids have been substituted, inserted and/or deleted.
- an anti- gliadin antibody is a monoclonal antibody, including a human antibody.
- an anti- gliadin antibody is an antibody fragment, e.g., a Fv, Fab, Fab', scFv, diabody, or F(ab') 2 fragment.
- the antibody is a substantially full length antibody, e.g., an IgG2a antibody or other antibody class or isotype as defined herein.
- an antibody provided herein is an antibody fragment.
- Antibody fragments include, but are not limited to, Fab, Fab', Fab'-SH, F(ab')2, Fv, and scFv fragments, and other fragments described below.
- Fab fragment antigen binding protein
- Fab' fragment antigen binding protein
- Fab'-SH fragment antigen binding protein
- F(ab')2 fragment antigen binding protein
- scFv fragments fragment antigen binding protein fragments
- Patent Nos. 5,571,894 and 5,587,458 For discussion of Fab and F(ab3 ⁇ 4 fragments comprising salvage receptor binding epitope residues and having increased in vivo half-life, see U.S.
- Diabodies are antibody fragments with two antigen-binding sites that may be bivalent or bispecific. See, for example, EP 404,097; WO 1993/01161 ; Hudson et al, Nat. Med.
- Single-domain antibodies are antibody fragments comprising all or a portion of the heavy chain variable domain or all or a portion of the light chain variable domain of an antibody.
- a single-domain antibody is a human single-domain antibody (Domantis, Inc., Waltham, MA; see, e.g., U.S. Patent No. 6,248,516 Bl).
- Antibody fragments can be made by various techniques, including but not limited to proteolytic digestion of an intact antibody as well as production by recombinant host cells
- an antibody provided herein is a chimeric antibody.
- Certain chimeric antibodies are described, e.g., in U.S. Patent No. 4,816,567; and Morrison et al, Proc. Natl. Acad. Sci. USA, 81 :6851-6855 (1984)).
- a chimeric antibody comprises a non-human variable region (e.g. , a variable region derived from a mouse, rat, hamster, rabbit, or non-human primate, such as a monkey) and a human constant region.
- a chimeric antibody is a "class switched" antibody in which the class or subclass has been changed from that of the parent antibody. Chimeric antibodies include antigen-binding fragments thereof.
- a chimeric antibody is a humanized antibody.
- a non-human antibody is humanized to reduce immunogenicity to humans, while retaining the specificity and affinity of the parental non-human antibody.
- a humanized antibody comprises one or more variable domains in which VRs, e.g., CDRs, (or portions thereof) are derived from a non-human antibody, and FRs (or portions thereof) are derived from human antibody sequences.
- a humanized antibody optionally will also comprise at least a portion of a human constant region.
- some FR residues in a humanized antibody are substituted with corresponding residues from a non-human antibody (e.g., the antibody from which the VR residues are derived), e.g., to restore or improve antibody specificity or affinity.
- a non-human antibody e.g., the antibody from which the VR residues are derived
- Human framework regions that may be used for humanization include but are not limited to: framework regions selected using the "best- fit" method (see, e.g., Sims et al. J. Immunol. 151 :2296 (1993)); framework regions derived from the consensus sequence of human antibodies of a particular subgroup of light or heavy chain variable regions (see, e.g., Carter et al. Proc. Natl. Acad. Sci. USA, 89:4285 (1992); and Presta et al. J. Immunol, 151 :2623 (1993)); human mature (somatically mutated) framework regions or human germline framework regions (see, e.g., Almagro and Fransson, Front. Biosci.
- an antibody provided herein is a human antibody.
- Human antibodies can be produced using various techniques known in the art. Human antibodies are described generally in van Dijk and van de Winkel, Curr. Opin. Pharmacol. 5: 368-74 (2001) and Lonberg, Curr. Opin. Immunol. 20:450-459 (2008). Human antibodies may be prepared by administering an immunogen to a transgenic animal that has been modified to produce intact human antibodies or intact antibodies with human variable regions in response to antigenic challenge. Such animals typically contain all or a portion of the human immunoglobulin loci, which replace the endogenous
- Human antibodies can also be made by hybridoma-based methods. Human myeloma and mouse-human heteromyeloma cell lines for the production of human monoclonal antibodies have been described. (See, e.g., Kozbor J. Immunol, 133 : 3001 (1984); Brodeur et al, Monoclonal Antibody Production Techniques and Applications, pp. 51-63 (Marcel Dekker, Inc., New York, 1987); and Boerner et al, J. Immunol, 147: 86 (1991).) Human antibodies generated via human B-cell hybridoma technology are also described in Li et al, Proc. Natl. Acad. Sci. USA, 103:3557-3562 (2006).
- Additional methods include those described, for example, in U.S. Patent No. 7,189,826 (describing production of monoclonal human IgM antibodies from hybridoma cell lines) and Ni, Xiandai Mianyixue, 26(4):265-268 (2006) (describing human-human hybridomas).
- Human hybridoma technology Trioma technology
- Vollmers and Brandlein, Histology and Histopathology , 20(3):927-937 (2005) and Vollmers and Brandlein, Methods and Findings in Experimental and Clinical Pharmacology , 27(3): 185-91 (2005).
- Human antibodies may also be generated by isolating Fv clone variable domain sequences selected from human-derived phage display libraries. Such variable domain sequences may then be combined with a desired human constant domain. Techniques for selecting human antibodies from antibody libraries are described below. Library-Derived Antibodies
- Antibodies of the invention may be isolated by screening combinatorial libraries for antibodies with the desired activity or activities. For example, a variety of methods are known in the art for generating phage display libraries and screening such libraries for antibodies possessing the desired binding characteristics. Such methods are reviewed, e.g., in
- repertoires of VH and VL genes are separately cloned by polymerase chain reaction (PCR) and recombined randomly in phage libraries, which can then be screened for antigen-binding phage as described in Winter et al, Ann. Rev. Immunol, 12: 433-455 (1994).
- Phage typically display antibody fragments, either as single- chain Fv (scFv) fragments or as Fab fragments.
- naive repertoire can be cloned (e.g., from human) to provide a single source of antibodies to a wide range of non-self and also self antigens without any immunization as described by Griffiths et al, EMBO J, 12: 725-734 (1993).
- naive libraries can also be made synthetically by cloning unrearranged V-gene segments from stem cells, and using PCR primers containing random sequence to encode the highly variable CDR3 regions and to accomplish rearrangement in vitro, as described by Hoogenboom and Winter, J. Mol. Biol, 227: 381-388 (1992).
- Patent publications describing human antibody phage libraries include, for example: US Patent No. 5,750,373, and US Patent Publication Nos. 2005/0079574, 2005/0119455, 2005/0266000, 2007/01 17126, 2007/0160598,
- Antibodies or antibody fragments isolated from human antibody libraries are considered human antibodies or human antibody fragments herein. Multispecific Antibodies
- an antibody provided herein is a multispecific antibody, e.g. a bispecific antibody.
- Multispecific antibodies are monoclonal antibodies that have binding specificities for at least two different sites. In certain embodiments, one of the binding specificities is for gliadin and the other is for any other antigen.
- bispecific antibodies may bind to two different epitopes of gliadin. Bispecific antibodies may also be used to localize cytotoxic agents to cells which express gliadin. Bispecific antibodies can be prepared as full length antibodies or antibody fragments.
- Multispecific antibodies include, but are not limited to, recombinant co-expression of two immunoglobulin heavy chain-light chain pairs having different specificities (see Milstein and Cuello, Nature 305: 537 (1983)), WO 93/08829, and Traunecker et al, EMBO J. 10: 3655 (1991)), and "knob-in-hole” engineering (see, e.g., U.S. Patent No. 5,731,168).
- Multi-specific antibodies may also be made by engineering electrostatic steering effects for making antibody Fc-heterodimeric molecules
- the antibody or fragment herein also includes a "Dual Acting FAb” or “DAF” comprising an antigen binding site that binds to GLIADIN as well as another, different antigen (see, US 2008/0069820, for example).
- amino acid sequence variants of the antibodies provided herein are contemplated. For example, it may be desirable to improve the binding affinity and/or other biological properties of the antibody.
- Amino acid sequence variants of an antibody may be prepared by introducing appropriate modifications into the nucleotide sequence encoding the antibody, or by peptide synthesis. Such modifications include, for example, deletions from, and/or insertions into and/or substitutions of residues within the amino acid sequences of the antibody. Any combination of deletion, insertion, and substitution can be made to arrive at the final construct, provided that the final construct possesses the desired characteristics, e.g., antigen-binding.
- antibody variants having one or more amino acid substitutions are provided.
- Sites of interest for substitutional mutagenesis include the VRs and FRs.
- Conservative substitutions are shown in the Table below under the heading of "preferred substitutions.” More substantial changes are provided in the Table below under the heading of "exemplary substitutions,” and as further described below in reference to amino acid side chain classes.
- Amino acid substitutions may be introduced into an antibody of interest and the products screened for a desired activity, e.g., retained/improved antigen binding, decreased immunogenicity, or improved ADCC or CDC.
- Amino acids may be grouped according to common side-chain properties:
- Non-conservative substitutions will entail exchanging a member of one of these classes for another class.
- substitutional variant involves substituting one or more hypervariable region residues of a parent antibody (e.g. a humanized or human antibody).
- a parent antibody e.g. a humanized or human antibody
- the resulting variant(s) selected for further study will have modifications (e.g., improvements) in certain biological properties (e.g., increased affinity, reduced immunogenicity) relative to the parent antibody and/or will have substantially retained certain biological properties of the parent antibody.
- An exemplary substitutional variant is an affinity matured antibody, which may be conveniently generated, e.g., using phage display -based affinity maturation techniques such as those described herein. Briefly, one or more residues are mutated and the variant antibodies displayed on phage and screened for a particular biological activity (e.g. binding affinity).
- Alterations may be made e.g., to improve antibody affinity. Such alterations may be made in "hotspots," i.e., residues encoded by codons that undergo mutation at high frequency during the somatic maturation process (see, e.g., Chowdhury, Methods Mol. Biol. 207: 179-196 (2008)), and/or SDRs (a-CDRs), with the resulting variant VH or VL being tested for binding affinity.
- Affinity maturation by constructing and reselecting from secondary libraries has been described, e.g., in Hoogenboom et al.
- affinity maturation diversity is introduced into the variable genes chosen for maturation by any of a variety of methods (e.g. , error-prone PCR, chain shuffling, or oligonucleotide-directed mutagenesis).
- a secondary library is then created. The library is then screened to identify any antibody variants with the desired affinity.
- Another method to introduce diversity involves directed approaches, in which several residues (e.g., 4- 6 residues at a time) are randomized. Residues involved in antigen binding may be specifically identified, e.g., using alanine scanning mutagenesis or modeling.
- substitutions, insertions, or deletions may occur within one or more VRs so long as such alterations do not substantially reduce the ability of the antibody to bind antigen.
- conservative alterations e.g., conservative substitutions as provided herein
- Such alterations may be outside of VR "hotspots" or SDRs.
- each VR either is unaltered, or contains no more than one, two or three amino acid substitutions.
- a useful method for identification of residues or regions of an antibody that may be targeted for mutagenesis is called "alanine scanning mutagenesis" as described by
- a residue or group of target residues e.g., charged residues such as arg, asp, his, lys, and glu
- a neutral or negatively charged amino acid e.g., alanine or polyalanine
- Further substitutions may be introduced at the amino acid locations demonstrating functional sensitivity to the initial substitutions.
- a crystal structure of an antigen-antibody complex is used to identify contact points between the antibody and antigen. Such contact residues and neighboring residues may be targeted or eliminated as candidates for substitution. Variants may be screened to determine whether they contain the desired properties.
- Amino acid sequence insertions include amino- and/or carboxyl-terminal fusions ranging in length from one residue to polypeptides containing a hundred or more residues, as well as intrasequence insertions of single or multiple amino acid residues.
- terminal insertions include an antibody with an N-terminal methionyl residue.
- Other insertional variants of the antibody molecule include the fusion to the N- or C-terminus of the antibody to an enzyme (e.g. for ADEPT) or a polypeptide which increases the serum half-life of the antibody.
- an antibody provided herein is altered to increase or decrease the extent to which the antibody is glycosylated.
- Addition or deletion of glycosylation sites to an antibody may be conveniently accomplished by altering the amino acid sequence such that one or more glycosylation sites is created or removed.
- the carbohydrate attached thereto may be altered.
- Native antibodies produced by mammalian cells typically comprise a branched, biantennary oligosaccharide that is generally attached by an N-linkage to Asn297 of the CH2 domain of the Fc region. See, e.g., Wright et al. TIBTECH 15:26-32 (1997).
- oligosaccharide may include various carbohydrates, e.g., mannose, N-acetyl glucosamine (GlcNAc), galactose, and sialic acid, as well as a fucose attached to a GlcNAc in the "stem" of the biantennary oligosaccharide structure.
- modifications of the oligosaccharide in an antibody of the invention may be made in order to create antibody variants with certain improved properties.
- antibody variants having a carbohydrate structure that lacks fucose attached (directly or indirectly) to an Fc region.
- the amount of fucose in such antibody may be from 1% to 80%, from 1% to 65%, from 5% to 65% or from 20% to 40%.
- the amount of fucose is determined by calculating the average amount of fucose within the sugar chain at Asn297, relative to the sum of all glycostructures attached to Asn 297 (e. g. complex, hybrid and high mannose structures) as measured by MALDI-TOF mass spectrometry, as described in WO 2008/077546, for example.
- Asn297 refers to the asparagine residue located at about position 297 in the Fc region (Eu numbering of Fc region residues); however, Asn297 may also be located about ⁇ 3 amino acids upstream or downstream of position 297, i.e., between positions 294 and 300, due to minor sequence variations in antibodies. Such fucosylation variants may have improved ADCC function. See, e.g., US Patent Publication Nos. US 2003/0157108 (Presta, L.); US 2004/0093621 (Kyowa Hakko Kogyo Co., Ltd).
- knockout cell lines such as alpha- 1,6- fucosyltransferase gene, FUT8, knockout CHO cells (see, e.g., Yamane-Ohnuki et al.
- Antibodies variants are further provided with bisected oligosaccharides, e.g., in which a biantennary oligosaccharide attached to the Fc region of the antibody is bisected by GlcNAc. Such antibody variants may have reduced fucosylation and/or improved ADCC function. Examples of such antibody variants are described, e.g., in WO 2003/01 1878 (Jean- Mairet et al.); US Patent No. 6,602,684 (Umana et al); and US 2005/0123546 (Umana et al.). Antibody variants with at least one galactose residue in the oligosaccharide attached to the Fc region are also provided.
- Such antibody variants may have improved CDC function.
- Such antibody variants are described, e.g., in WO 1997/30087 (Patel et al); WO 1998/58964 (Raju, S.); and WO 1999/22764 (Raju, S.).
- one or more amino acid modifications may be introduced into the Fc region of an antibody provided herein, thereby generating an Fc region variant.
- the Fc region variant may comprise a human Fc region sequence (e.g., a human IgGl, IgG2, IgG3 or IgG4 Fc region) comprising an amino acid modification (e.g. a substitution) at one or more amino acid positions.
- the invention contemplates an antibody variant that possesses some but not all effector functions, which make it a desirable candidate for applications in which the half life of the antibody in vivo is important yet certain effector functions (such as complement and ADCC) are unnecessary or deleterious.
- In vitro and/or in vivo cytotoxicity assays can be conducted to confirm the reduction/depletion of CDC and/or ADCC activities.
- Fc receptor (FcR) binding assays can be conducted to ensure that the antibody lacks FcyR binding (hence likely lacking ADCC activity), but retains FcRn binding ability.
- NK cells express Fc(RIII only, whereas monocytes express Fc(RI, Fc(RII and Fc(RIII.
- FcR expression on hematopoietic cells is summarized in Table 3 on page 464 of Ravetch and Kinet, Annu. Rev. Immunol. 9:457-492 (1991).
- Non- limiting examples of in vitro assays to assess ADCC activity of a molecule of interest is described in U.S. Patent No. 5,500,362 (see, e.g. Hellstrom, I. et al. Proc. Nat'l Acad. Sci.
- non-radioactive assays methods may be employed (see, for example, ACTITM non-radioactive cytotoxicity assay for flow cytometry (CellTechnology, Inc. Mountain View,
- PBMC peripheral blood mononuclear cells
- NK cells Natural Killer (NK) cells.
- ADCC activity of the molecule of interest may be assessed in vivo, e.g., in a animal model such as that disclosed in Clynes et al. Proc. Nat'l Acad. Sci. USA 95:652-656 (1998).
- Clq binding assays may also be carried out to confirm that the antibody is unable to bind Clq and hence lacks CDC activity. See, e.g., Clq and C3c binding ELISA in WO 2006/029879 and WO 2005/100402.
- a CDC assay may be performed (see, for example, Gazzano-Santoro et al., J.
- Antibodies with reduced effector function include those with substitution of one or more of Fc region residues 238, 265, 269, 270, 297, 327 and 329 (U.S. Patent No. 6,737,056).
- Such Fc mutants include Fc mutants with substitutions at two or more of amino acid positions
- an antibody variant comprises an Fc region with one or more amino acid substitutions which improve ADCC, e.g., substitutions at positions 298, 333, and/or 334 of the Fc region (EU numbering of residues).
- alterations are made in the Fc region that result in altered (i.e., either improved or diminished) Clq binding and/or Complement Dependent Cytotoxicity
- FcRn which is responsible for the transfer of maternal IgGs to the fetus
- Those antibodies comprise an Fc region with one or more substitutions therein which improve binding of the Fc region to FcRn.
- Such Fc variants include those with substitutions at one or more of Fc region residues: 238, 256, 265, 272, 286, 303, 305, 307, 31 1, 312, 317, 340, 356, 360, 362, 376, 378, 380, 382, 413, 424 or 434, e.g., substitution of Fc region residue 434 (US Patent No. 7,371,826).
- cysteine engineered antibodies e.g., "thioMAbs”
- one or more residues of an antibody are substituted with cysteine residues.
- the substituted residues occur at accessible sites of the antibody.
- reactive thiol groups are thereby positioned at accessible sites of the antibody and may be used to conjugate the antibody to other moieties, such as drug moieties or linker-drug moieties, or to create an
- an antibody provided herein may be further modified to contain additional nonproteinaceous moieties or non-antibody proteins that are known in the art and readily available.
- the moieties suitable for derivatization of the antibody include but are not limited to water soluble polymers.
- Non-limiting examples of water soluble polymers include, but are not limited to, polyethylene glycol (PEG), copolymers of ethylene glycol/propylene glycol, carboxymethylcellulose, dextran, polyvinyl alcohol, polyvinyl pyrrolidone, poly-1, 3-dioxolane, poly-l,3,6-trioxane, ethylene/maleic anhydride copolymer, polyaminoacids (either homopolymers or random copolymers), and dextran or poly(n- vinyl pyrrolidone)polyethylene glycol, propropylene glycol homopolymers, prolypropylene oxide/ethylene oxide co-polymers, polyoxyethylated polyols (e.g., glycerol), polyvinyl alcohol, and mixtures thereof.
- PEG polyethylene glycol
- copolymers of ethylene glycol/propylene glycol carboxymethylcellulose
- dextran polyvinyl alcohol
- Polyethylene glycol propionaldehyde may have advantages in manufacturing due to its stability in water.
- the polymer may be of any molecular weight, and may be branched or unbranched.
- the number of polymers attached to the antibody may vary, and if more than one polymer are attached, they can be the same or different molecules. In general, the number and/or type of polymers used for derivatization can be determined based on considerations including, but not limited to, the particular properties or functions of the antibody to be improved, whether the antibody derivative will be used in a therapy under defined conditions, etc.
- conjugates of an antibody and nonproteinaceous moiety that may be selectively heated by exposure to radiation are provided.
- the nonproteinaceous moiety is a carbon nanotube (Kam et al, Proc. Natl. Acad. Sci. USA 102: 11600-11605 (2005)).
- the radiation may be of any wavelength, and includes, but is not limited to, wavelengths that do not harm ordinary cells, but which heat the nonproteinaceous moiety to a temperature at which cells proximal to the antibody -nonproteinaceous moiety are killed.
- antibodies or antibody fragments are fused or conjugated to human serum albumin (See e.g., U.S. Pat. No.
- FcRn neonatal Fc receptor
- HSA Human serum albumin
- Albumin has a long serum half-life and because of this property it has been used for drug delivery. Albumin has been conjugated to pharmaceutically beneficial compounds
- Antibodies may be produced using recombinant methods and compositions, e.g., as described in U.S. Patent No. 4,816,567.
- isolated nucleic acid encoding an anti-gliadin antibody described herein is provided.
- Such nucleic acid may encode an amino acid sequence comprising the VL and/or an amino acid sequence comprising the VH of the antibody (e.g., the light and/or heavy chains of the antibody).
- one or more vectors e.g., expression vectors
- a host cell comprising such nucleic acid is provided.
- a host cell comprises (e.g., has been transformed with): (1) a vector comprising a nucleic acid that encodes an amino acid sequence comprising the VL of the antibody and an amino acid sequence comprising the VH of the antibody, or (2) a first vector comprising a nucleic acid that encodes an amino acid sequence comprising the VL of the antibody and a second vector comprising a nucleic acid that encodes an amino acid sequence comprising the VH of the antibody.
- the host cell is eukaryotic, e.g. a Chinese Hamster Ovary (CHO) cell or lymphoid cell (e.g., Y0, NS0, Sp20 cell).
- a method of making an anti- gliadin antibody comprises culturing a host cell comprising a nucleic acid encoding the antibody, as provided above, under conditions suitable for expression of the antibody, and optionally recovering the antibody from the host cell (or host cell culture medium).
- nucleic acid encoding an antibody is isolated and inserted into one or more vectors for further cloning and/or expression in a host cell.
- nucleic acid may be readily isolated and sequenced using conventional procedures (e.g., by using oligonucleotide probes that are capable of binding specifically to genes encoding the heavy and light chains of the antibody).
- Suitable host cells for cloning or expression of antibody-encoding vectors include prokaryotic or eukaryotic cells described herein.
- antibodies may be produced in bacteria, in particular when glycosylation and Fc effector function are not needed.
- U.S. Patent Nos. 5,648,237, 5,789, 199, and 5,840,523. See also Charlton, Methods in Molecular Biology, Vol. 248 (B.K.C. Lo, ed., Humana Press, Totowa, NJ, 2003), pp. 245-254, describing expression of antibody fragments in E. coli.
- the antibody may be isolated from the bacterial cell paste in a soluble fraction and can be further purified.
- eukaryotic microbes such as filamentous fungi or yeast are suitable cloning or expression hosts for antibody-encoding vectors, including fungi and yeast strains whose glycosylation pathways have been "humanized,” resulting in the production of an antibody with a partially or fully human glycosylation pattern. See Gerngross, Nat.
- Suitable host cells for the expression of glycosylated antibody are also derived from multicellular organisms (invertebrates and vertebrates). Examples of invertebrate cells include plant and insect cells. Numerous baculoviral strains have been identified which may be used in conjunction with insect cells, particularly for transfection of Spodoptera frugiperda cells.
- Plant cell cultures can also be utilized as hosts. See, e.g., US Patent Nos. 5,959,177, 6,040,498, 6,420,548, 7, 125,978, and 6,417,429 (describing PLANTIBODIESTM technology for producing antibodies in transgenic plants).
- Vertebrate cells may also be used as hosts.
- mammalian cell lines that are adapted to grow in suspension may be useful.
- useful mammalian host cell lines are monkey kidney CVl line transformed by SV40 (COS-7); human embryonic kidney line (293 or 293 cells as described, e.g., in Graham et al, J. Gen Virol. 36:59 (1977)); baby hamster kidney cells (BHK); mouse Sertoli cells (TM4 cells as described, e.g., in Mather, Biol. Reprod.
- monkey kidney cells (CVl); African green monkey kidney cells (VERO-76); human cervical carcinoma cells (HELA); canine kidney cells (MDCK; buffalo rat liver cells (BRL 3A); human lung cells (W138); human liver cells (Hep G2); mouse mammary tumor (MMT 060562); TRI cells, as described, e.g., in Mather et al, Annals N Y. Acad. Sci. 383 :44-68 (1982); MRC 5 cells; and FS4 cells.
- Other useful mammalian host cell lines include Chinese hamster ovary (CHO) cells, including DHFR " CHO cells (Urlaub et al, Proc. Natl. Acad. Sci.
- Anti- gliadin antibodies provided herein may be identified, screened for, or characterized for their physical/chemical properties and/or biological activities by various assays known in the art. In some embodiments, the experiments described in Example 1 are utilized to screen antibodies for activity.
- an antibody of the invention is tested for its antigen binding activity, e.g., by known methods such as ELISA, BIACore ® , FACS, or Western blot.
- competition assays may be used to identify an antibody that competes with any of the antibodies described herein for binding to gliadin.
- a competing antibody binds to the same epitope (e.g., a linear or a conformational epitope) that is bound by an antibody described herein.
- epitope e.g., a linear or a conformational epitope
- Detailed exemplary methods for mapping an epitope to which an antibody binds are provided in Morris (1996) "Epitope Mapping Protocols," in Methods in Molecular Biology vol. 66 (Humana Press, Totowa, NJ).
- immobilized gliadin is incubated in a solution comprising a first labeled antibody that binds to gliadin (e.g., any of the antibodies described herein) and a second unlabeled antibody that is being tested for its ability to compete with the first antibody for binding to gliadin.
- the second antibody may be present in a hybridoma supernatant.
- immobilized gliadin is incubated in a solution comprising the first labeled antibody but not the second unlabeled antibody. After incubation under conditions permissive for binding of the first antibody to gliadin, excess unbound antibody is removed, and the amount of label associated with immobilized gliadin is measured.
- Embodiments of the present disclosure provide methods and uses diagnosing celiac disease (e.g., using the antibodies described herein). For example, in some embodiments, a sample from a subject suspected of having celiac disease (e.g., exhibiting one or more symptoms of celiac disease) or during routine screening (e.g., newborn screening) is screened for anit-gliadin antibodies using a method described herein.
- a sample from a subject suspected of having celiac disease e.g., exhibiting one or more symptoms of celiac disease
- routine screening e.g., newborn screening
- gliadin-specific autoantibodies are detected using a competitive immunoassay where the binding of a gliadin-specific to a peptide of the corresponding epitope is measured in the presence of serum antibodies in a competitive assay.
- the assay is an ELISA assay.
- An ELISA short for Enzyme-Linked Immunosorbent Assay, is a biochemical technique to detect the presence of an antibody or an antigen in a sample. It utilizes a minimum of two antibodies, one of which is specific to the antigen and the other of which is coupled to an enzyme. The second antibody will cause a chromogenic or fluorogenic substrate to produce a signal. Variations of ELISA include sandwich ELISA, competitive ELISA, and ELISPOT. Because the ELISA can be performed to evaluate either the presence of antigen or the presence of antibody in a sample, it is a useful tool both for determining serum antibody concentrations and also for detecting the presence of antigen.
- the assay is performed on a solid support (e.g., bead based) or in a well based assay. In such assay, both serum IgG and IgA are screened at the same time.
- the peptide is, for example, PLQPEQPFP (SEQ ID NO: 3),
- PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4), or a combination thereof.
- a solid support plate or bead is conjugated with either a gliadin peptide or a gliadin-specific monoclonal antibody.
- Test or control serum, along with either a gliadin peptide or a gliadin-specific monoclonal antibody is contacted with the solid support.
- non-CD control serum is spiked with titrated concentrations of one or more gliadin-specific monoclonal antibodies and used as reference serum (e.g., to obtain absolute concentrations ⁇ g/ml) for test samples).
- Binding is detected using any suitable method (e.g., using a labeled peptide or antibody and appropriate detection reagent).
- Embodiments of the present disclosure further provide research uses (e.g., to study celiac diseae, develop assays for the diagnosis or screening of celiac disease, or develop assays for detection of gluten contamination in food products).
- research uses e.g., to study celiac diseae, develop assays for the diagnosis or screening of celiac disease, or develop assays for detection of gluten contamination in food products.
- the antibodies described herein find use in detection of gluten in food or food products (e.g., gluten contamination from manufacturing or gluten levels naturally found in the food or food product).
- a sample of a food product is contacting with one or more of the monoclonal antibodies described herein. Binding is detected using any suitable method (e.g., using a labeled gliadin-specific antibody or a labeled secondary antibody).
- an article of manufacture e.g., kit or composition
- materials e.g. monoclonal antibodies specific for gliadin
- the article of manufacture comprises a container and a label or package insert on or associated with the container.
- the containers may be formed from a variety of materials such as glass or plastic.
- kits further comprise one or more additional reagents (e.g. buffers, solid supports, controls, etc.) useful in performing immunoassays.
- the non-CD control group consisted of seven HLA-DQ2.5+ subjects, three HLA-DQ8+ subjects and four subjects who were not HLA-typed. All subjects have given written informed consent. The study was approved by Regional Committees for Medical Research Ethics South East Norway (S-97201).
- SCSs Single cell suspensions
- the biopsies After 30 minutes rotation at 37°C, the supernatant was discarded and the biopsies re-suspended in lmg/ml blend collagenase (Sigma, C8051) and 50 ⁇ g/ml DNase (Sigma, DN25) in 2% FCS in Dulbecco's PBS. The biopsies were then incubated under constant rotation at 37°C. After 30 minutes the biopsies were mechanically disrupted with a syringe equipped with a large steel needle. After another 30 minutes constant rotation, a smaller needle was used for the same procedure. After 1-2 hours, the single cell suspension was filtered through 40 ⁇ filter into 50 ml tube and centrifuged at 470 g for 7 minutes.
- Human intestinal stromal cell line Human fibroblast cell lines were derived from small intestinal biopsies as previously described (Roncoroni L, et al. J Transl Med 7, 40 (2009)). Biopsies were transferred to flat-bottomed 6-well plates and gently disrupted with a scalpel for 15-30 seconds. The biopsies were cultured in 1%
- fibroblasts were detached from 25 ml culture flasks and transferred to plates with flat-bottomed wells. After one week, cells from SCSs were seeded on confluent layer of fibroblasts. Fibroblasts and SCSs were incubated in culture medium at 37°C in 5% CO 2 . Different plate formats were suited to different experimental settings; 24-well plates were used for BrdU assays and estimation of total IgA production and 384 well plates were used of single PC cultures.
- 50g was dissolved in 150 ml butanol, vortex mixed and centrifuged at 163 g for 5 minutes. The butanol was decanted and the procedure repeated.
- the wheat flour pellet was dissolved in 350 ml 70% ethanol and incubated at RT overnight under constant stir mixing. The next day, the solution was centrifuged at 650 g for 5 minutes, and the supernatant was mixed with 1.5 M NaCl in ratio 1 :2 and incubated at 4°C for 4 hours to precipitate the gliadin proteins. After centrifugation at 25,000 g for 20 minutes the supernatant was decanted and the gliadin pellet dissolved in 40 ml 8 M urea in 0.01 M ammonium bicarbonate.
- This solution was diluted 1 :4 to give a final urea concentration of 2 M and incubated with 12-24 mg chymotrypsin (CT) overnight at 37°C under constant stir mixing. The next day, chymotrypsin was heat inactivated at 98°C for 5 minutes, and the solution was dialyzed (Spectra/Por® Membrane MWCO 1,000) overnight and dried in speed vacuum concentrator. The digested gliadin was dissolved and incubated in acetic acid pH 1.8 at 95°C for 1 hour to introduce Q to E conversion (deamidation). The final product, heat/acid treated chymotrypsin digested gliadin (CT-gliadin for short), was diluted in distilled H 2 0 and freeze dried before further usage.
- CT chymotrypsin
- ELISA supernatant IgA reactive with CT-gliadin ELISA plates (96 well Nunc 436014) were coated with 75 ⁇ /well of CT-gliadin 40 ⁇ g/ml in carbonate buffer 0.05M pH 9.6 over night at 4°C, washed and subsequently blocked with 0.5% bovine serum albumin (BSA) in PBS and incubated with supernatant from single PC cultures.
- BSA bovine serum albumin
- Anti-human IgA- alkaline phosphatase Sigma, A9669 1 :3000 was used as secondary antibody.
- Anti-human IgG-alkaline phosphatase (Southern Biotech, 2040-04) at concentration 1 :4000 was used when testing gliadin-reactive hmAbs. Plates were developed for approximately 15 min with phosphatase substrate (Sigma-Aldrich) and absorbance was measured at 405 nm.
- Biotin- GSGSGS-PLQPEQPFP SEQ ID NO: 17
- biotin-GSGSGS-PLQPEQPFP SEQ ID NO: 17
- LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18) were produced by GL Biochem (Shanghai). TG2 was expressed in Sf9 insect cells by Phadia and linked to biotin with Sulfo-NHS-LC -biotin (Pierce) as per the manufacturer's protocol.
- GLIADIN SEQ ID NO: 19 1 :40 (BD Biosciences, ⁇ 9), FITC goat anti-human IgA 1 :800 (Southern Biotech, 2050-02) were used for staining of SCS. Propidium iodide for exclusion of dead cells was added just before analysis. Three different flow cytometer instruments were used: Facs Aria, LSRII and Fortessa. The plots (Fig. 1, 3, 9 and 10) are from LSRII. Plasma cells appeared as one homogeneous population of large, CD4 " and GLIADIN " (SEQ ID NO: 19) events, co-expressing CD 138 and CD27, and were previously identified as antibody producing cells (Di Niro R et al.
- variable regions of the heavy and light chain antibody genes of isolated PCs were amplified by RT- PCR and nested PCR, cloned into expression vectors and transfected into a human cell line as IgGl according to previously established protocol (Smith K, et al. Nat Protoc 4, 372-384 (2009)).
- AlphaLISA screening of hmAbs for reactivity to gluten peptides AlphaLISA Acceptor beads (Perkin Elmer, 6772001) were coupled with polyclonal rabbit anti-human IgG (Dako A0423) and stored at a concentration of 2.5 mg/ml according to the
- gliadin-specific hmAbs were produced in human IgGl format.
- Anti-IgG AlphaLISA donor bead solution (1 :400) and gliadin-specific hmAbs 1 ⁇ g/ml in AlphaLISA immunoassay buffer (Perkin Elmer, ALOOOC) were incubated for 1 hour at 4°C in the dark. After incubation, 15 ⁇ were transferred to each well in 384 well plates and mixed with 5 ⁇ analyte. The plates were incubated for 1 hour at RT in the dark.
- AlphaScreen streptavidin donor bead solution (Perkin Elmer, 6760002B) was diluted 1 :200 in AlphaLISA Immunoassay buffer, and 15 ⁇ were added per well before incubation at RT in the dark for 30 minutes.
- the different peptides used in the analyte were the following: biotin-GSGSGS- PLQPEQPFP (SEQ ID NO: 3), PLQPQQPFP (SEQ ID NO: 8), PLQPEQPFP (SEQ ID NO: 3), GIIQPEQPAQL (SEQ ID NO: 20), LQLQPFPQPQLPYPQPQLPYPQPQLPYPQPQPF (SEQ ID NO: 21), FPQPQQPEQSFP (SEQ ID NO: 22), PEQPQQSFPEQERP (SEQ ID NO: 23), LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7),
- LQQPLSQQPEETF (SEQ ID NO: 24) and biotin-GSGSGS- LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18) were purchased from GL Biochem Ltd, Research Genetics, Neosystems or obtained from Burkhard Fleckenstein.
- AlphaLISA Acceptor beads were also coupled to equal amounts of the two gliadin- specific hmAbs UCD1114 1F03 and UCD1143 3B02 and stored at concentration 2.5mg/ml. These beads were used to detect biotin-GSGSGS-PLQPEQPFP (SEQ ID NO: 17) in the presence of serum (Fig 7). First, 5ul biotin-GSGSGS-PLQPEQPFP (SEQ ID NO: 17) was incubated with 10 ⁇ serum at RT for 30 minutes. Then, 30 ⁇ of mixture of AlphaLISA acceptor bead solution (1 :600) and Alphascreen donor bead solution (1 :300) in AlphaLISA immunoassay buffer, was added per well.
- the plate was read after second incubation for 45 minutes at RT in the dark. In pilot experiments several gliadin-specific hmAbs were tested and gave similar results.
- the hmAbs UCD 11 14 1F03 and UCD1143 3B02 were chosen for the final experiments as they were among the commonly used VH/VL pairs VH3-23/VL4-69 and VH3-15/VK4-1, respectively.
- AlphaLISA screening of hmAbs for gliadin specificity and polyreactivity Anti-IgG AlphaLISA donor bead solution (1 :400) and gliadin-specific hmAbs 1 ⁇ g/ml in AlphaLISA immunoassay buffer were incubated in 1.5 ml tube for 1 hour at RT in the dark before 15 ⁇ was transferred to each well in 384 well plate. Then 5 ⁇ analyte was added per well, and the plate subsequently incubated for 30 minutes at RT in the dark.
- the analyte consisted of fixed concentration of 40 nM biotin-GSGSGS-PLQPEQPFP (SEQ ID NO: 17) and titrations of either CT-gliadin, lysate of EBV-transfected B cells or a mixture of LPS (Sigma L-4391), CpG, recombinant TG2 (Phadia) and recombinant Jo-1 antigen (Phadia).
- LPS Sigma L-4391
- CpG CpG
- recombinant TG2 recombinant TG2
- Jo-1 antigen Phadia
- PCs Single cell suspensions (SCSs) from intestinal biopsies were generated, and PCs were cultured either as SCSs, as SCSs in co- culture with human intestinal fibroblasts or as PCs isolated by flow cytometry in co-culture with fibroblasts.
- SCSs Single cell suspensions
- the concentration of IgA in supernatants of cultures with SCSs together with fibroblasts increased with a constant rate over at least 4 weeks (Fig. la), indicating that the majority of PCs survived in these cultures.
- Fig. 9c-d small resting B cells. If one well of SCSs contained just one IgA + PC, there was a high probability that this was the only cell expressing the IgA encoding genes in that well. Where ELISA results indicated gliadin-specific PCs to be present (Fig. 2a), the cells were split into four PCR wells and processed individually to increase the likelihood of having only one PC in each well. Cells were then washed in PBS before snap frozen in DNase containing buffer and subjected to expression cloning of antibodies in a human IgGl format as previously described (Smith K, et al. Nat Protoc 4, 372-384 (2009). Human monoclonal antibodies (hmAbs) were successfully produced from more than half of cultures processed. Of a total of 19 hmAbs produced, nine were reactive to CT-gliadin (Fig. 3a).
- Antigen-specific IgA + PCs were visualized previously by staining with biotinylated antigen bound to fluorescent streptavidin taking advantage of surface IgA and IgM expression of gut PCs (Di Niro R, et al. Nat Med 18, 441-445 (2012), Di Niro R, et al. J Immunol 185, 5377-5383 (2010)).
- the same strategy was used to identify gliadin-specific IgA + PCs by flow cytometry, using fluorescent streptavidin to form tetramer complexes with biotinylated synthetic peptides.
- biotin-(GSGSGS)-PLQPEQPFP SEQ ID NO: 17
- biotin-(PEG)- LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF SEQ ID NO: 7
- Bateman EA et al, Gut 53, 1274-1278 (2004)
- the two peptides represent sequences of gliadin with Q to E transitions in positions targeted by TG2 (Dorum S, et al.
- IgA + PCs stained with synthetic gliadin peptides and TG2 appeared as two separate populations in flow cytometry whereas double positive cells were not detected (Fig. 4c). Also, the populations visualized with TG2 were larger than the populations stained with both the PLQPEQPFP (SEQ ID NO: 3) and the deamidated 33-mer peptides (Fig. 4d). These findings are consistent with the ratio of the frequencies of IgA + PCs reactive with TG2 versus those reactive with CT-gliadin observed in culture (Fig. 2b). The predominance of TG2 + PCs relative to the population stained with gliadin peptides was greater for the IgA + PCs than for the IgM + PCs (Fig. 4d). Based on these experiments it was concluded that there are two distinct populations of gliadin-specific and TG2-specific IgA PCs in the small intestinal lesions of UCD patients.
- IgA + PCs stained with fluorescent streptavidin in complex with either PLQPEQPFP (SEQ ID NO: 3) or deamidated 33-mer were isolated as single cells by flow cytometry for further expression cloning.
- the hmAbs obtained were tested for reactivity to the peptide which they were originally isolated with. Seventeen of 23 hmAbs from PCs isolated with the PLQPEQPFP (SEQ ID NO: 3) peptide, and 13 of 16 hmAbs from IgA + PCs isolated with the deamidated 33-mer peptide were reactive in ELISA to the peptide originally used in sorting. This indicates that the staining specificity of gluten-peptide tetramers was approximately 75- 80%.
- Gliadin-reactive hmAbs specificity and affinity to deamidated and native gliadin
- the eight hmAbs that originated from in vitro PCs cultures reactive with CT-gliadin were tested in ELISA against the two synthetic peptides. Five reacted with PLQPEQPFP (Fig. 3c) and one reacted with deamidated 33-mer. Hence, six out of eight hmAbs were reactive to PLQPEQPFP (SEQ ID NO: 3) and/or deamidated 33-mer. Since cloning of antibody genes from those cultures is unbiased with respect to specific epitope, this result indicated that the epitopes represented by PLQPEQPFP (SEQ ID NO: 3) and the deamidated 33-mer are recognized by IgA of a significant part of gliadin-specific PCs.
- the specificity of all hmAbs reactive to one of the two synthetic peptides in ELISA was further characterized in AlphaLISA.
- the AlphaLISA format detected monovalent binding of hmAbs to soluble biotinylated synthetic peptide.
- Three of the hmAbs (UCD 1002 1D03, UCD 1143 1E01, UCD1130 4A04), originally from IgA + PC sorted with tetramers of PLQPEQPFP (SEQ ID NO: 3) and reactive to PLQPEQPFP (SEQ ID NO: 3) in ELISA, were not reactive in AlphaLISA.
- hmAbs to deamidated versus native gliadin were tested in a competitive AlphaLISA assay. Binding of hmAbs to biotinylated synthetic deamidated gliadin peptide (either PLQPEQPFP (SEQ ID NO: 3) or the deamidated 33-mer) was tested against the homologous peptide in either deamidated or native (non-deamidated) versions. Gliadin-specific hmAbs divided into two groups, either only reactive to deamidated gliadin or reactive with both deamidated and native gliadin (Fig. 5b). None of the hmAbs had higher reactivity to native than to deamidated gliadin. Because some of the hmAbs were not reactive to the native gliadin peptide, this indicates that induction of gliadin-specific B cells in vivo likely happen in the presence of deamidated gliadin.
- Gliadin-specific hmAbs cross-reactivity and repetitive sequences in the gliadin proteome
- a few selected synthetic gliadin peptides could compete with the antibody binding to biotinylated PLQPEQPFP.
- the hmAbs (UCD1143 3B02 and UCD1002 1B06) were tested and both were reactive with PLQPEQPFP (SEQ ID NO: 3) but not PLQPQQPFP (SEQ ID NO: 8) (Table 1).
- the signals were inhibited by peptides containing QPEQ or PEQP. Peptides without these sequences were non-inhibitory (Fig. 6b).
- the cross-reactivity of hmAbs to different gliadin peptides is due to sharing of key sequences.
- the eight hmAbs from in vitro cultured PCs with CT-gliadin were obtained from six subjects (UCD 1050, 1 ; UCD1130, 1; UCD1065, 2; UCD 1186, 2; UCD1163, 1; UCD1030, 1).
- Control IgA + PCs defined as PLQPEQPFP-negative (SEQ ID NO: 3), 33- mer-negative and TG2 -negative (UCD1130, 55; UCD1 143, 54) and TG2-specific PCs (UCD1030, 3; UCD11 14, 4; UCD1010, 2), were isolated by flow cytometry and their antibody genes were cloned and sequenced.
- the variable regions of the hmAbs were analyzed using tools of the IMGT webpage. Two combinations of VH/VL pairing were dominant (Fig. 8a); VH3-15/VK4-1 (seven hmAbs from five different subjects in total) and VH3-23/VL4-69 (15 hmAbs from seven different subjects in total).
- VH3-23/VL4-69 The VH3-23/VL4-69 combination was found in hmAbs from IgA + PCs isolated with both CT-gliadin and synthetic peptides. In addition, VH3-23/VK3-11 was also found in hmAbs generated by both methods. Together, these three combinations made up approximately 75% of the panel of gliadin- specific hmAbs (Fig. 8a, Table 1). Similarity between heavy chain CDR3 sequences of the VH3-15/VK4-1 as well as the VH3-23/VL4-69 hmAbs was investigated. No obvious similarities could be found with variability in D- and J-gene segment usage and CDR3-length among the antibodies of each group (Table 1). The nucleotide sequences of the variable regions of heavy and light chains of all hmAbs are summarized in Table 2.
- VH/VL usage (Table 1) was compared with peptide reactivity pattern characterized in AlphaLISA. All VH3-23/VL4-69 hmAbs showed reactivity to both native and deamidated gliadin, demonstrated by inhibiting effect of both the native PLQPQQPFP and the deamidated PLQPEQPFP (SEQ ID NO: 3). The VH3-15/VK4-1 hmAbs were specific to deamidated gliadin, as the native PLQPQQPFP (SEQ ID NO: 8) showed no inhibitory effect on the binding to PLQPEQPFP (SEQ ID NO: 3).
- VH3-23/VK3-1 1 hmAbs were the only antibodies with reactivity to deamidated 33-mer but not PLQPEQPFP (SEQ ID NO: 3).
- Two hmAbs had reactivity to CT-gliadin but neither to PLQPEQPFP (SEQ ID NO: 3) nor to deamidated 33-mer.
- These two antibodies both used VH3-15/VK3-20. Taken together, this indicated that there is a correlation between VH/VL usage and epitope specificity of the gliadin-specific antibodies.
- VH genes are associated with various levels of SHM (Wang M, et al, J Exp Med 207, 141-153 (2010)).
- the mutation rate in gliadin-specific IgA with VH3-23 was significantly lower than the number of mutations observed in VH3-23 from control population (Fig. 8c). It was concluded that the low mutation rate in gliadin-specific IgA is not a result of selected VH-usage.
- IgA antibodies of TG2-specific PCs found in celiac lesions have limited SHM.
- gluten (gliadin) reactive antibodies expression cloned from IgA + PCs isolated from small intestinal biopsies were studied and it was found that there is also a limited mutation rate in these antibodies, demonstrating an unexpected common feature of the IgA antibody responses to gluten and TG2 in CD.
- T-cell independent B-cell responses have no or little SHM with little restriction of VH usage as often seen in T-cell dependent responses (Maizels N, Bothwell A. Cell 43, 715-720 (1985)).
- T-cell dependent B- cell responses that develop in germinal centers (GCs) typically result in highly mutated antibodies (MacLennan IC. Annu Rev Immunol 12, 1 17-139 (1994)). GC reactions may also happen without involvement of T cells, but then with low level of SHM (Toellner KM, et al. J Exp Med 195, 383-389 (2002)).
- the low mutation rate was contemplated to relate to enzymatic activity of B-cell receptor bound TG2 (Di Niro et al, Nat Med 18, 441-445
- both gluten and gluten-reactive T cells could be such common factors.
- Gluten is a protein antigen, which in contrast to most antigens of the gut has no bacterial or viral origin. Lack of strong concomitant innate signals along with B-cell receptor triggering could result in extrafollicular response, in accordance with a study of the B-cell response to a T-cell dependent antigen, where immunization without adjuvant was insufficient for GC formation (Chappell CP, et al., J Exp Med 209, 1825-1840 (2012)).
- Extrafollicular responses typically give rise to short-lived plasma cells and rapid decline in serum antibodies after the immune response in addition to low SHM (Ho F, et a.l, Eur J Immunol 16, 1297-1301 (1986)).
- TG2-specific PCs were rarely detected in small intestine of CD subjects on a gluten-free diet (Di Niro et al, Nat Med 18, 441-445 (2012)), and levels of serum IgG and IgA antibodies to both gluten and TG2 typically fall below detection level months after the patient commence a gluten-free diet (Sulkanen S, et al.
- T cells influence extrafollicular development of B cells. It has been reported that T cells located at the T-B border in lymph nodes, phenotypically different from GC T cells, are necessary for B-cell priming to extrafollicular antibody responses (Lee et al, J Exp Med 208, 1377-1388 (2011)). Sustained CD40 signaling during B-cell and T-cell interaction has been demonstrated to induce a plasma cell fait rather than GC B-cell development (Sciammas R, et al, Immunity 25, 225-236 (2006).), and administration of a CD40 agonistic antibody was shown to ablate GC reaction and induced a pattern of extrafollicular B-cell differentiation (Erickson LD, et al. J Clin Invest 109, 613-620 (2002)).
- gliadin-specific B cells must have encountered deamidated gliadin.
- gliadin-specific antibodies frequently cross-react with different gliadin peptides. Accordingly, gliadin-specific B cells may take up and display several different T-cell epitopes. This may be beneficial if a B cell receive help from several different T-cell clones, as recently described (Shulman Z, et al. Science 341, 673-677 (2013)).
- Antibodies specific to deamidated variants of gluten peptides had different VH/VL usage than antibodies with reactivity to both the deamidated and native variants.
- VH/VL restriction has been reported in antibodies specific for influenza (Lingwood D, et al. Nature 489, 566-570 (2012)) and HIV (Wu et al, Science 333, 1593-1602 (201 1)) antigens.
- HIV antibodies there has even been reported an allelic preference for antibody usage (Wu et al, Science 333, 1593- 1602 (201 1)).
- Restricted VH/VL usage and limited SHM in antibodies as observed in CD favor influence of VH and/or VL polymorphisms in shaping the antibody response.
- Donor subject patient number
- isolation method sorted by flow cytometry with selecting antigen PLQPEQPFP (SEQ ID NO: 3) or deamidated 33-mer or isolated after in vitro culture with CT-gliadin as selecting antigen
- H/A heat/acid
- H/A-gliadin 40 ⁇ g/ml
- biotinylated synthetic gliadin peptides 500 nM were used as coating antigens in ELISA plates (Nunc, 442404) and streptavidin coated ELISA plates (Nunc, 436014), respectively.
- Alkaline phosphatase conjugated anti-human IgG (Southern Biotech 9040-04) in 1 :4000 dilution was used as the detecting antibody and visualized with phosphatase substrate (Sigma S0942-200TAB) reactivity measured at 405 nm.
- the synthetic gliadin peptides were produced with biotinylated C-terminal spacer (GSGSGS), by GL Biochem.
- PBS pH7.2 was used as buffer for antigens, hmAbs and detecting antibody.
- PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4). Sera, diluted 1 :300 in PBS, was tested in ELISA plates coated with either H/A-gliadin or biotinylated
- PQPEQPFPQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4) in parallel. Serum IgG reactivity was detected as described above.
- AlphaLISA Acceptor beads Perkin Elmer, 6772001
- anti-human IgG Dako, A0423
- the anti-IgG bead solution was diluted 1 :400 together with 0.5 ⁇ g/ml gliadin-specific hmAb in AlphaLISA (AL) buffer (Perking Elmer, AL000C) in 1.5 ml tube, and incubated for 1 hour at 4°C in the dark.
- AlphaLISA Acceptor beads were conjugated either with PLQPEQPFP (SEQ ID NO: 3) or with gliadin-specific hmAbs (1002- 1E01 and 1002-10E3 or 1 130-3B04), according to manufacturers' instructions, and stored at 2.5 mg/ml concentration.
- biotinylated SEQ ID NO: 3
- PQPEQPFPQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4) was dissolved to 25nM in AL buffer with protease inhibitor (Sigma, 1 1714600), and 20 ⁇ was transferred to each well and incubated together with 5 ⁇ serum for 1 hour at 4°C.
- Solution of AL beads conjugated with gliadin-specific hmAbs was diluted 1 :250, 10 ⁇ was added per well, and plate was left for incubation at RT for 30 minutes in the dark.
- Alphascreen streptavidin bead solution was diluted 1 :250, 20 ⁇ was added per well, and the plate was incubated at RT for 45 minutes in the dark before it was analyzed with Envision Multilabel Reader. Positive control, using the same concentrations of anti-FLAG IgG (Sigma, F3165) conjugated AL beads and
- biotinylated FLAG peptide Biotin-GSGSGS-DYKDDDDK (SEQ ID NO: 253)
- Biotin-GSGSGS-DYKDDDDK SEQ ID NO: 253
- PLQPEQPFP SEQ ID NO: 3
- AL bead solution was diluted 1 :300 in AL buffer, and 20 ⁇ transferred each well together with 5 ⁇ serum.
- the plate was incubated at RT for 1 hour at RT in the dark.
- Gliadin-specific hmAb 1002-1E03 that was conjugated with NHS-Biotin (Thermo, 21335) according to manufacturers' instruction, was diluted 50 ng/ml in AL buffer, and 5 ⁇ was transferred to each well.
- Alphascreen streptavidin bead solution was diluted 1 : 150 in AL buffer, and 20 ⁇ was transferred each well. The plate was incubated for 30 minutes at RT in the dark, and then analyzed with Envision 2014 Multilabel reader.
- Reference serum serum of non-CD control subject with known concentrations of gliadin-specific hmAbs was run in all assays to generate a reference curve.
- concentrations of gliadin-specific serum antibodies were estimated using reference serum and Sigmoidal dose-response (variable slope) function in GraphPad Prism 5 (GraphPad Software Inc.). Linear regression analyses and column statistics were calculated using Graphpad Prism 5 (Graphpad Software, Inc.).
- gliadin-specific hmAbs were included in the study; eight hmAbs of IgA + PCs isolated by flow cytometry, where two different synthetic gliadin peptides were used as sorting peptide (Biotin-GSGSGS-PLQPEQPFP (SEQ ID NO: 17), PLQPEQPFP (SEQ ID NO: 3) for short; biotin-GSGSGS-LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18), deamidated 33-mer for short) and three hmAbs of in vitro cultured PCs secreting IgA reactive to H/A-gliadin (Table 3).
- hmAbs were produced with the same IgGl constant region.
- gliadin proteins were digested with the enzymes PTCEC (pepsin, trypsin, chymotrypin, elastase, carboxipeptidase), separated into size fractions by gel filtration, and enzymatically deamidated by transglutaminase 2 (for short: TG2-gliadin), and gliadin-specific hmAbs were used as a matrix to enrich high affinity TG2-gliadin peptides, followed by peptide identification by mass spectrometry (MS).
- PTCEC pepsin, trypsin, chymotrypin, elastase, carboxipeptidase
- MS mass spectrometry
- a rotavirus-specific hmAb (Di Niro, R., et al, J Immunol, 2010. 185(9): p. 5377-83) was used as negative control hmAb.
- gliadin-specific serum antibodies are based on serum IgG or IgA reactivity to synthetic deamidated gliadin peptides (DGP) in ELISA.
- DGP deamidated gliadin peptides
- This method monitored binding of gliadin-specific hmAb to peptide of the corresponding epitope in the presence of serum antibodies in a competitive bead-based AlphaLISA assay. In such assay, both serum IgG and IgA are screened at the same time.
- Two assays were tested, either using beads conjugated with PLQPEQPFP and biotinylated hmAb 1002-1E01, bead conjugated with hmAb 1130-3B04 and biotinylated PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4), or bead conjugated with hmAbs 1002-1E01 and 1002-1E03 and biotinylated PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4).
- Non-CD control serum was spiked with titrated concentrations of two gliadin-specific hmAbs (11 14-1F03 and 1130-3B02) and used as reference serum, such that absolute concentrations ⁇ g/ml) could be estimated for test samples.
- These two hmAbs both showed high reactivity to PLQPEQPFP (SEQ ID NO: 3) and were also reactive to
- PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4).
- the assay with PLQPEQPFP conjugated beads and biotinylated hmAb 1002-1E01 was first compared to ELISA using streptavidin coated plates, biotinylated PLQPEQPFP and anti-human IgG as detecting antibody.
- Testing the reference serum the change in signal from 0 M to 5xl0 "9 M gliadin-specific hmAbs (1 114-1F03 and 1130-3B02), was 50% in the competitive AlphaLISA assay and 20% in ELISA ( Figure 1 1). This shows that the competitive AlphaLISA assay was better at detecting low concentration of gliadin-specific antibodies.
- the inhibition assay results correlated well with QUANTA Gliadin and Celikey TG2.
- the exception was sera screened positive in Celikey TG2 but negative in QUANTA Gliadin, where the inhibition assay correlated better with Celikey TG2.
- the same comparison was done for testing the inhibition assay with beads conjugated with two hmAbs (1002-1E01 and 1002-1E03) and biotin-
- the epitope specificity of hmAbs cloned from the antibody gene of single gliadin-specific IgA + PCs isolated from intestinal CD lesions was investigated.
- the gliadin- specific hmAbs and the characterized epitopes were used in a competitive assay to detect gliadin-specific serum antibodies.
- the assay was tested and compared to anti-TG2 IgA ELISA using sera from 55 patients undergoing endoscopy. None of the patients were previously diagnosed with CD. Using Marsh score as gold standard, the competitive anti- gliadin antibody test had the same specificity but better sensitivity than the anti-TG2 IgA ELISA test.
- CD can now be diagnosed without endoscopy in children with anti-TG2 IgA titers more than ten times above cutoff (Husby, S., et al, J Pediatr Gastroenterol Nutr, 2012. 54(1): p. 136-60). These guidelines have been met with some concerns because of the variation in performance of anti-TG2 IgA kits from different manufacturers (Egner, W., et al, J Pediatr Gastroenterol Nutr, 2012. 55(6): p.
- positive EMA test is included in the ESPHGAN criteria (Husby, S., et al, J Pediatr Gastroenterol Nutr, 2012. 54(1): p. 136-60). EMA testing is highly operator-dependent, because the test is difficult to do and to interpret (Health Quality, O., Ont Health Technol Assess Ser, 2010. 10(21): p. 1-11 1; Rostom, A., J.A. Murray, and M.F. Kagnoff, Gastroenterology, 2006. 131(6): p. 1981-2002).
- Table 3 Overview of the gliadin-specific hmAbs used in the study.
- the first column shows the names of the hmAbs.
- the selecting antigens used in the isolation of the gliadin-specific IgA + PCs are indicated in the second column.
- Column 3-6 is an overview of results that together were used to assess the best binding motifs of each hmAb. These motifs were not always in accordance with the selecting antigens.
- Last two columns describe the VH and VL usage of the hmAbs.
- Table 4 ELISA reactivity of the gliadin-specific hmAbs to synthetic gliadin peptides grouped as positive, low or negative.
- the cutoff is ⁇ 5 for anti-TG2 IgA, and ⁇ 20 for DGP (Quanta Gliadin IgG). **For some patients, HLA genotype was not available.
- Gluten-free diet is the sole treatment in CD patients, but gluten contamination in gluten-free food products is common (Koerner, T.B., et al., Gluten contamination of naturally gluten-free flours and starches used by Canadians with celiac disease. Food Addit Contam Part A Chem Anal Control Expo Risk Assess, 2013. 30(12): p. 2017-21).
- Today, gluten in food is mainly detected using monoclonal mouse antibodies (mAbs) (Haraszi, R., et al., Analytical methods for detection of gluten in food— method developments in support of food labeling legislation. J AOAC Int, 2011. 94(4): p. 1006-25).
- mAbs are used in commercial assays, and they have been generated by immunizing mice with different antigens.
- the 401.21 mAb was developed against gliadin extract of Australian bread wheat cultivar Timgalen (Skerritt, J.H., et al., Journal of Cereal Science, 1984. 2(4): p. 215-224), PN3 mAb against the synthetic 19-mer LGQQQPFPPQQPYPQPQPF (SEQ ID NO: 254) (Ellis, H.J., et al., Gut, 1998. 43(2): p. 190-5.), R5 against a-secalin extract (Sorell, L., et al., FEBS Lett, 1998.
- the epitopes of R5 and HYB 314-01 are QQPFP (SEQ ID NO: 256) (Osman, A.A., et al., Eur J Gastroenterol Hepatol, 2001. 13(10): p. 1189-93) and PELPYPQPQ (SEQ ID NO: 257) (Petersen, N.H., et al., J Immunol Methods, 2011. 365(1-2): p. 174-82), respectively.
- the epitopes of these mouse and human mAbs are different. Since human gliadin-specific IgA + PCs involved in the disease process, hmAbs cloned from these cells recognize immunogenetic gliadin epitopes relevant for CD, in contrast to mAbs generated in mice.
- Gliadin was extracted from wheat flour and treated with heat/acid treated and tested in a competitive assay with synthetic gliadin peptide and gliadin-specific hmAb as previously described.
- AlphaLISA donor beads Perkin Elmer
- PLQPEQPFP PLQPEQPFP
- Gliadin-specific hmAb 1002-lEOl is not dependent on deamidation of gliadin, and will bind to both deamidated and native gliadin.
- the hmAb 1002-1E01 was biotinylated with NHS-biotin (Thermo) according to manufacturers' instructions. Wheat was dissolved 1 mg/ml in buffer and incubated with 0.5 ⁇ g/ml of biotinylated hmAb 1002-1E01 at room temperature (RT) on shaker 1400 rpm for 1 hour.
- PLQPEQPFP SEQ ID NO: 3 bead solution was diluted 1 :400 in AL buffer, 15 ⁇ was transferred to each well in a 384 well plate and incubated together with 5 ⁇ of the analyte of wheat and biotinylated hmAb 1002- 1E01 for 45 minutes at RT in the dark.
- AlphaScreen streptavidin donor bead solution was diluted 1 :200 in AL buffer, 15 ⁇ was transferred to each well, and the plate was incubated for 30 minutes at RT in the dark, and then analyzed.
- the wheat-containing product strongly inhibited the AlphaLISA signal, compared to two gluten-free products, which exhibited robust signals (Figure 16). This demonstrates use the hmAbs for detection of gluten in food.
- hmAbs identified in Example 1, the hmAbs were used in pull-down experiments with peptides of enzymatic digests of wheat gliadins treated with TG2. Results demonstrated that most of the hmAbs preferred binding to long, TG2-deamidated gliadin peptides with several copies of specific motifs.
- a gliadin-specific hmAb and such target peptides identified by pull- down were used in a serologic competition assay. The principle was to measure binding of gliadin-specific hmAb to target peptide, where inhibition of this binding indicated presence of gliadin-specific antibodies in serum.
- the gliadin specific hmAb 1002-1E03 (Example 1) was used to pull down peptides from pepsin, trypsin, chymotrypsin, elastase, and carboxypeptidase digested gliadin treated with TG2. Details of the procedure are described in Example 5.
- the hmAb (40 ⁇ g) was incubated with a gel filtration fraction of TG2 -treated gliadin for 20 min at room temperature (RT), followed by 20 min incubation at RT with Protein G Dynabeads (Novex, Life Technologies). The antibody-enriched peptides were eluted with 0.1% TFA for 10 min at RT.
- a rotavirus specific hmAb (Rota-2B04) was used as negative control.
- the samples were analyzed on a Dionex Ultimate 3000 nano-LC system (Dionex, Sunnyvale, CA, USA) connected to a quadrupole-Orbitrap (QExactive) mass spectrometer (ThermoElectron, Bremen, Germany).
- the peptides were separated on a 250 mm EASY-SPRAY column (CI 8, 75 ⁇ ID, 2 ⁇ particles) using a 60 min linear 5-50% ACN gradient in 0.1% formic acid at a flow rate of 0.3 ⁇ /min.
- the QExactive data was acquired using a data-dependent top 10 method and the LC-MS/MS data were searched in a T. Aestivum database as described in Example 5.
- Biotinylated synthetic gliadin peptides (500 nM) were used as antigens in streptavidin coated ELISA plates (Nunc, 436014). Gliadin-specific hmAbs at 6.67 nM concentration and fourfold dilution were used to generate titration curves. Alkaline phosphatase conjugated anti-human IgG (Southern Biotech 9040-04) in 1 :4000 dilution was used as detecting antibody, and visualized with phosphatase substrate (Sigma S0942-200TAB) reactivity measured at 405 nm.
- the synthetic gliadin peptides were produced with biotinylated C- terminal spacer (GSGSGS) by GL Biochem. PBS pH 7.4 was used as buffer, and the plates were washed three times with 0.05% Tween in PBS between each step. Serologic anti- ⁇ 34 Ig inhibition assay
- the serologic inhibition assay was developed on an amplified luminescent proximity homogeneous assay (Alpha)LISA platform (Perkin Elmer) with customized AlphaLISA acceptor beads (Perkin Elmer) conjugated with the gliadin-specific hmAb (1002-1E03), according to manufacturer's recommendations.
- biotin-QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259) (termed b-c ⁇ 34), biotin-QPEQPFPEQPEQPFPQPEQPFPW (SEQ ID NO: 260) (b- co26), and biotin-PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4) (b-QPEQPFP 3 ), and the following concentrations were used: ⁇ of b-c ⁇ 34 at 2.5 or 1.25 nM per well, b-c ⁇ 26 at 5 or 7.5 nM, and b-QPEQPFP 3 at 4 or 6 nM.
- Target peptide was dissolved in AlphaLISA buffer (0.1% Pluviol in PBS pH 7.4) to the concentrations described above. Subsequently, 10 ⁇ was transferred to each well and incubated with 5 ⁇ serum in 1 hour at RT.
- AlphaLISA 1002-1E03 acceptor bead stock solution was diluted to 3.5 ⁇ g/ml in AlphaLISA buffer, and 15 ⁇ added per well. The plate was incubated in 45 minutes at RT in dark.
- Alphascreen streptavidin donor bead (Perkin Elmer) solution was diluted to 7 ⁇ g/ml in AlphaLISA buffer, 15 ⁇ was added per well, and the plate was incubated in 45 minutes at RT in dark.
- AlphaLISA signal was measured with Envision Multilabel Plate Reader (Perkin Elmer).
- the serum samples were tested in duplicates and the mean value of the two logarithmic AlphaLISA signals were used. Together with a reference curve, generated from healthy control serum spiked with known concentrations of gliadin-specific hmAbs (1002- 1E01, 1002-1E03 and 1 130-3B03), the mean value was used to estimate serum antibody reactivity of equivalent amounts of gliadin-specific hmAbs (mg/L).
- the importmant components of the inhibition assay were the gliadin-specific hmAb and the concomitant target peptide(s).
- the gliadin-specific hmAb 1002-1E03 that showed strong preference for TG2-deamidated gliadin was selected (Example 1).
- the hmAb was incubated with TG2 -treated digest of wheat gliadin, antibody -peptide complexes were pulled down, and the antibody-bound peptides were eluted and analyzed by mass spectrometry. Altogether 381 different peptides were identified in the pull-down analyte (Table 10).
- gliadin proteins are highly similar, and gliadin proteins often contain repeats of motifs. Together with the high hit rate in the pull-down assay, this indicated either promiscuous binding property of the hmAb, or that the hmAb recognized a specific motif expressed in several different gliadin peptides. To address this, the peptide sequences from the pull-down analysis were evaluated by counting the most common motifs within registers of 3-15 residues. This analysis indicated selective enrichment of peptides harboring identical motifs ( Figure 17B, Table 6).
- two representative ⁇ -gliadin derived peptides from the pull-down analysis were synthesized with N-terminal biotin, and selected as target peptides in the serologic inhibition assay (biotin-QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259), b- ⁇ 34 and biotin-QPEQPFPEQPEQPFPQPEQPFPW (SEQ ID NO: 260), b- ⁇ 26).
- Both peptides comprised several copies (three to six) of the 3- to 7-mers identified as most common in the previous analysis.
- the hmAb 1002-1E03 was tested against a panel of synthetic gliadin peptides in ELISA (Table 7; Table 1 1).
- the sequence PEQ was present in all peptides recognized by the hmAb.
- the hmAb was not reactive to all peptides comprising this 3-mer, showing that PEQ was important but not sufficient for binding.
- PQQ was highly abundant in the pull-down analyte, and in most cases (1479 of 1634) flanked by C- terminal proline (PQQP (SEQ ID NO: 262)).
- the hmAb 1002-1E03 was compared to eight different gliadin-specific hmAbs, screened for reactivity to the same synthetic peptides in ELISA (Table 7).
- PQPELPYPQP (SEQ ID NO: 12).
- the nine hmAbs could be divided into four groups based on their peptide reactivity patterns. Seven of the hmAbs (all but 1130-3B03 and 1130-3G05) divided into three groups, but they all recognized peptides harboring QPEQPFP (SEQ ID NO: 10).
- Serum and biotinylated target peptides were first mixed and incubated. Second, AlphaLISA acceptor beads conjugated with hmAb 1002-1E03 were added to the mixture. The binding of the hmAb to the biotinylated peptide was detected with Alphascreen streptavidin donor beads.
- the principle of the assay is that the biotinylated target peptide binds to streptavidin donor beads, which consequently are brought in proximity to the hmAb conjugated acceptor beads given that the antibody binds to the target peptide. Upon light activation, the donor beads release energy that can activate the acceptor beads to emit light signal provided proximity of the beads. Gliadin-reactive serum antibodies can compete with the hmAb for binding to the peptide and hence reduce the signal.
- Three different target peptides (b-QPEQPFP 3 , b-c ⁇ 34, and the shorter variant b-c ⁇ 26) were tested in the assay. Two of them represent peptide fragments that are naturally occurring in the gliadin proteome (b-c ⁇ 34 and b-c ⁇ 26), and the antibody pull-down experiment indicated that these peptides to be of primary targets for the hmAb.
- the third peptide (b-QPEQPFP 3 ) was designed to represent the sequence QPEQPFP three times, a sequence common to many peptides that several gliadin-specific hmAbs reacted with in ELISA (Table 7).
- the peptide b- co34 demonstrated highest diagnostic performance of the three target peptides, evaluated by the area under the curve (AUC) of the receiver operating characteristic (ROC) curve analysis (Table 12).
- AUC area under the curve
- ROC receiver operating characteristic
- the peptide concentrations used in in this study gave dynamic range from approximately 1 mg/L to 20-30 mg/L of the gliadin-specific hmAbs used as reference. Signal/noise-ratio under these conditions spanned approximately two logarithmic units.
- the mean values of the logarithmic AlphaLISA Signal ( Figure 18A) and the reference curve ( Figure 18B) were used to extrapolate the antibody activity in the serum samples to equivalent concentrations (mg/L) of reference gliadin-specific hmAbs ( Figure 18C).
- the serum samples from celiac disease patients and control subjects were also analyzed for anti-TG2 IgA (Varelisa tTG IgA, Phadia) and anti-DGP IgG (QUANTA Lite Gliadin IgG II, INOVA) ( Figure 19A-B).
- the manufacturers operated with two different recommended cut-off values, namely ⁇ 5 or ⁇ 8 U/ml for anti-TG2 IgA, and ⁇ 20 or ⁇ 30 U/ml for anti-DGP IgG. These are termed high ( ⁇ 8 and ⁇ 30 U/ml) and low ( ⁇ 5 and ⁇ 20 U/ml) cut-off values below.
- test results of anti-TG2 IgA and anti-DGP IgG were compared with the results of the serologic anti-c ⁇ 34 Ig inhibition assay (Figure 18C).
- a second cut-off ⁇ 3 mg/L corresponding to the high cut-off values of the two commercial assays was used.
- This example describes a serologic inhibition assay for detection of gliadin-specific antibodies to be used as a diagnostic tool in celiac disease.
- the assay is based on a recombinant human monoclonal antibody, generated from the antibody genes of a single intestinal IgA plasma cell of a celiac disease patient.
- a preferred target for the antibody a deamidated 34-mer ⁇ -gliadin fragment, was identified in TG2-treated enzymatic digest of wheat gluten by antibody pull-down and subsequent mass spectrometry sequencing.
- the combination of this peptide and the monoclonal antibody was employed in an amplified luminescent proximity homogeneous (AlphaLISA) assay whose signal could be specifically inhibited by serum antibodies.
- the inhibition assay was more specific than anti-DGP IgG, more sensitive than anti-TG2 IgA, and detected up to 75% of the patients who scored negative for anti-TG2 IgA in this study.
- this hmAb preferentially recognized by this hmAb.
- the diagnostic performance of the anti-DGP assays is markedly better than former anti-gliadin assays, the anti-DGP assays in general are reported less specific than the anti-TG2 assays (Lewis et al, Aliment Pharmacol Ther 2010, 31 :78-81) and are claimed to have low predictive values in the serologic diagnostics of celiac disease (Vriezinga et al, N Engl J Med 2014, 371 : 1304-15).
- the inhibition assay exhibits better specificity than the anti-DGP IgG assay in the material tested.
- the antigen in the anti-DGP assays typically consists of gluten motifs empirically giving the highest antibody reactivity in patient sera.
- the antigen in the anti-DGP assays typically consists of gluten motifs empirically giving the highest antibody reactivity in patient sera.
- several independent studies have reported strikingly similar results, pointing at peptide sequences highly similar to the 7-mer motif QPQQPFP (SEQ ID NO: 261) enriched by the gliadin-specific hmAb investigated in this study (Osman et al, Clin Exp Immunol 2000, 121 :248-54; Vallejo-Diez et al, PloS One 2013,
- the hmAb clearly demonstrated more complex binding properties, being reactive to several related motifs. This is in line with what was observed for other gliadin-specific hmAbs (Example 1).
- the inhibition assay employed the monoclonal antibody together with a long ⁇ -gliadin peptide with several binding motifs. It is thus particularly suitable for monitoring disease-specific gluten serum antibodies, which may have the same cross-reactive feature.
- hmAbs deriving from celiac disease patients are useful with their natural target peptides in inhibitory serologic assay for diagnostic purpose.
- the assay takes advantage of the specificity introduced by both the hmAb and the antigen.
- Example 1 The human monoclonal antibodies (hmAbs) obtained in Example 1 reacted with deamidated gliadin antigen, and showed no reactivity to other types of antigens.
- the procedures by which these hmAbs were generated did not identify which epitopes in the gluten proteome they are primarily reactive with, as the complex deamidated gliadin antigen used in the first approach represented many different peptides and the synthetic peptides used in the second approach could have lower affinity than similar, but distinct unknown gluten peptide(s) due to cross-reactivity.
- Example 1 The fact that many of the gliadin-specific hmAbs by initial testing were reactive to several synthetic gliadin peptides, and that some of the hmAbs from IgA + plasma cells sorted by flow cytometry showed higher reactivity to other peptides than to the peptide used in cell surface staining (Example 1), indicated that an effort to identify the primary target of the hmAbs would be justified.
- This example describes epitope mapping of gliadin-specific hmAbs by antibody pull-down of fragments from complex proteolytic digests of gliadin followed by sequencing of the isolated peptides by mass spectrometry.
- hmAbs Gliadin-specific human recombinant monoclonal antibodies
- HEK293A cells Gliadin-specific human recombinant monoclonal antibodies
- peptides were purchased from GL Biochem Ltd, except for the y-26mer peptide which was purchased from Peptide 2.0.
- the following peptides were used in the MALDI- TOF experiments: ⁇ -gliadin 26mer; FLQPEQPFPEQPEQPYPEQPFPQ (SEQ ID NO: 15), ⁇ -gliadin 33mer; LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7) and the short ⁇ -gliadin peptide; PLQPEQPFP (SEQ ID NO: 3).
- biotin-GSGSGSPLQPEQPFP SEQ ID NO: 17
- biotin- QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ SEQ ID NO: 259
- FLQPEQPFPEQPEQPYPEQPFPQ (SEQ ID NO: 15), PQPQQPEQPFPQPQ (SEQ ID NO: 561), QQPFPQQPQQPYPQQPEQPFPQP (SEQ ID NO: 562),
- QPQQPFPQPEQPFPWQP (SEQ ID NO: 563), PQQPFPQPEQPFP (SEQ ID NO: 564), FPQPEQPFPWQP(SEQ ID NO: 565), PFPQPEQPFPWQPQQPFPQ (SEQ ID NO: 566), PQQPEQPFP (SEQ ID NO: 9), biotin-GSGSGSPQQPQQPFP (SEQ ID NO: 567), biotin- GSGSGSPQQPEQPFP (SEQ ID NO: 568), biotin-GSGSGSPQQPQQFP (SEQ ID NO:
- biotin-GSGSGSPQQPEQQFP SEQ ID NO: 570
- biotin-GSGSGSPQQPQQSFP SEQ ID NO: 571
- biotin-GSGSGSPQQPEQSFP SEQ ID NO: 572
- biotin- GSGSGSPQQPQQTFP SEQ ID NO: 573
- biotin-GSGSGSPQQPEQTFP SEQ ID NO: 574
- carboxypeptidase was performed as previously described (Dorum et al, J Immunol 2014, 193:4497-506).
- the gliadin digest was further fractionated by size exclusion chromatography using an Akta system with a Superdex peptide 10/300 GL column (GE Healthcare). Fractions of 0.5ml were collected. Two to three adjacent fractions were pooled and treated with transglutaminase 2 (TG2) as described previously (Dorum et al, J Immunol 2014, 193:4497- 506) before subjected to pull-down with hmAbs. The fractions containing the highest molecular weight peptides were not used in the pull-down experiments.
- Rota-2B04 A rotavirus specific hmAb (Rota-2B04) was used as negative control (Di Niro et al, Nat Med 2012, 18:441-5). The few unspecific peptides pulled down with Rota-2B04 were removed from the list of identified hmAb enriched peptides in addition to some few short peptides that derive from actin and other non-gluten proteins.
- the LC -MS/MS data were analysed with the software MaxQuant version 1.5.1.2 using Andromeda to search against a Triticum aestivum database (total of 4722 entries) extracted from the UniprotKB database release September 2013 (European Bioinformatics Institute). In all searches the digestion enzyme specificity was set as none and pyro-glu (N- term Q), deamidation (NQ) and oxidation (M) were selected as variable modifications.
- N- term Q pyro-glu
- NQ deamidation
- M oxidation
- mass error tolerance for MS scans was first searched with an error window of 20 ppm and then with a main search error of 6 ppm. Mass tolerance for MS/MS scans was set to 20 ppm. A false discovery rate of 1% and a PEP score of 0.1 were used.
- Biotinylated synthetic gliadin peptides (500 nM) were used as coating antigens in streptavidin coated ELISA plates (Nunc, 436014).
- the relative affinity of three hmAbs (1002-1E01, 1002-1E03 and 1130-3B01) to a panel of gliadin peptides was investigated using an AlphaLISA platform.
- AlphaLISA acceptor beads were coated with anti-human IgG according to manufacturers' protocol.
- Anti- IgG AlphaLISA beads 6 ⁇ g/ml and hmAb 0.5 ⁇ g/ml were mixed and incubated for 1 hour at RT in dark.
- the binding of whole IgGl versus Fab of gliadin-specific antibody to gliadin peptide was investigated in a similar competitive AlphaLISA assay.
- diluting titrations of IgGl or Fab of hmAb 1002-1E03 were used with either AlphaLISA acceptor beads conjugated with hmAb 1002-1E03 and biotinylated 33mer ⁇ -gliadin (2.5nM), or AlphaLISA acceptor beads conjugated with PLQPEQPFP together with biotinylated hmAb 1002-1E03 (0.1 mg/ml).
- gliadin-specific hmAbs Thirteen gliadin-specific hmAbs were included in this study; nine hmAbs of IgA + plasma cells isolated by flow cytometry, where two different synthetic gliadin peptides were used as sorting peptide (biotin-GSGSGS-PLQPEQPFP (SEQ ID NO: 17), PLQPEQPFP (SEQ ID NO: 3) for short; biotin-GSGSGS-PLQPEQPFP (SEQ ID NO: 17), PLQPEQPFP (SEQ ID NO: 3) for short; biotin-GSGSGS-
- LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18), deamidated 33mer for short) and five hmAbs of in vitro cultured plasma cells secreting IgA reactive to complex deamidated gliadin (Table 13).
- peptide fragments from ⁇ -, ⁇ -, ⁇ -gliadins and low-molecular weight glutenins were pulled down although fragments of a-gliadins were relatively infrequent.
- the gliadin-specific hmAbs pull down long deamidated peptide fragments
- Enriched peptides are not necessarily similar to peptides to which the hmAbs were selected
- the enriched peptides had different peptide sequences than the selecting peptide antigen originally used to isolate the IgA + plasma cell (Table 13). This was particularly observed for the hmAbs of plasma cells sorted with the a-gliadin 33mer peptide LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7) (e.g., mAbs 1130-3B04, 1130-3A02, 1 130-3B01, 1130-3A05, 1 130-3B03, 1 130-3G05).
- the enriched peptide fragments share motifs
- hmAb 1 130-3B04 reacts specifically with peptides that share a motif of no more than 8 amino acids in length. A similar pattern was seen for several of the hmAbs.
- hmAbs 1 114-1G01, 1 130-3B01, 1050-5B05, 1130-3B03, and 1 130-3G05 the frequencies of the most common 7mers were considerably lower. It was investigated whether this was due to reactivity of the hmAbs towards peptides with similar, but not necessarily identical sequences. To evaluate this, the tool "Pattinprot" (PBIL.ibcp.fr) was used to scan all the possible 7mers for motifs of one amino acid substitution (85% similarity) to the
- QPQQPFP (SEQ ID NO: 261) motif among peptides pulled down by these five antibodies.
- hmAbs 1002-lEOl, 1002-1E03 and 1 130-3B01 were tested for reactivity to a panel of deamidated synthetic gliadin peptides harbouring the key sequence QPEQPFP (SEQ ID NO: 261) (Q ⁇ E substitution in position 3) in a competitive AlphaLISA assay (Figure 25). While 1002-1E03 showed similar affinity for all peptides in the panel, the affinity of 1002-lEOl and 1 130-3B01 to the different peptides varied by 1-2 logs ( Figure 25A). This demonstrates that although a "dominant" motif is found for the majority of the gliadin-specific hmAbs, the flanking regions of the sequence motif will affect the binding affinity. In the gluten proteins, the
- QPQQPFP (SEQ ID NO: 261) sequence motif can be found with a variety of different amino acids in the flanking regions ( Figure 25B).
- hmAb 1 130-3B01 was incubated with a synthetic peptide mix containing equimolar amounts of the deamidated a-gliadin 33mer peptide and the peptide PLQPEQPFP (SEQ ID NO: 3), which harbour the deamidated QPQQPFP (SEQ ID NO: 261) 7mer motif.
- the hmAb-peptide complexes were isolated and bound peptides were analysed by MALDI-TOF MS ( Figure 27A). Only the a-gliadin 33mer peptide was enriched by the hmAb.
- Quantitative aspects may also play a role as the target peptide sequences were not equally represented.
- QPQQPFP SEQ ID NO: 261
- the hmAb-enriched peptide fragments harbour several different gliadin T-cell epitopes
- Gliadin-specific hmAbs appear to bind gluten peptides that harbour T-cell epitopes (Example 1).
- the presence of gluten T-cell epitopes in the identified gliadin peptides pre and post hmAb pull-down (Table 14 show post pull-down) were horrged. It was found that more than 80% of all peptides pulled down with the six hmAbs 1002-1E01, 1130-3B04, 1130-3B01, 1002-1E03, 1 130-3A02 and 1 130-2A02, harboured known gluten T-cell epitopes.
- the majority of the T-cell epitope containing peptides contained the DQ2.5-glia- y4c (QQPQQPFPQ (SEQ ID NO: 272)) and/or the DQ2.5-glia-y5 epitope (QQPFPQQPQ (SEQ ID NO: 578)).
- Table 14 Percentage of peptides pulled down with the eleven human monoclonal antibodies that harbour known gluten T-cell epitopes. The number of T-cell epitope per peptide is given in brackets.
- T-cell epitopes are most often overlapping in the gliadin proteins. This is particularly striking in the ⁇ -gliadin protein (Accession number: Q9FUW7) visualised in Figure 24. In this protein, 9 copies of the 7mer motif are present. All copies, except one, are overlapping with one or more T-cell epitopes.
- the DQ2.5-glia-y5 epitope overlaps with four copies of the binding motif, DQ2.5-glia-y4c overlaps with three copies, while DQ2.5-glia-col and DQ2.5- glia-c ⁇ 2 both overlap with one copy.
- This example describes the natural binding targets of eleven gliadin-specific hmAbs made by expression cloning of antibody genes of single intestinal IgA + plasma cells from coeliac disease patients.
- the natural binding targets were identified by isolating and sequencing a large number of fragments pulled down from fractions of gliadin that had been treated with digestive enzymes and TG2.
- the majority of the hmAbs were established from staining plasma cells with labelled synthetic peptides.
- the hmAbs selected for peptides which differed from the selecting peptides.
- the enrichment for long fragments with repeated motifs likely relate to epitope multivalency. This enrichment was observed in experimental settings where the multivalent peptide fragments could engage more than one antibody molecule. This scenario mimics the situation at the surface of a B cell where a multivalent antigen would be able to engage several B-cell receptors on the cell surface. This gives B-cell receptor crosslinking and B-cell activation and thereby causes a strong selection of the B-cell epitopes. This may be a reason why the B-cell epitopes in gliadin are sequence motifs that have multivalent display within long proteolytically resistant fragments.
- the peptide fragments pulled down by the hmAbs typically contained glutamate residues introduced by TG2-mediated deamidation. Further, in general, there was an enrichment of deami dated peptides when comparing pre and post pull-down samples. This was the case even with hmAbs that did not distinguish between synthetic peptides in native and deamidated versions in ELISA.
- the QPQQPFP (SEQ ID NO: 261) motif contains the QXP motif typically targeted by TG2 (Vader et al, J Exp Med 2002, 195:643-9; Fleckenstein et al, J Biol Chem 2002, 277:34109-16), and the hmAbs react with deamidated peptides in the TG2 -treated digests even though the glutamate residue is not necessarily part of the epitope.
- the gliadin-specific hmAbs typically pulled down peptides with multiple gliadin T- cell epitopes, where the hmAb binding motif and the T-cell epitopes overlapped or were in close proximity. This argues for a role for gluten-specific B cells as important antigen presenting cells in coeliac disease. Together with the finding that the hmAbs cross-react with different gliadin peptides, it indicates that the gliadin-specific B-cells may take up and display many different T-cell epitopes and consequently get help from many distinct gliadin- specific T cells, which have been demonstrated to be important for generating B-cell responses (Shulman et al, Science 2013, 341 :673-7).
- the dominant T-cell response in coeliac disease is directed towards a-gliadin and ⁇ - gliadin peptides (Sollid et al, Immunogenetics 2012, 64:455-60; Arentz-Hansen et al, J Exp Med 2000, 191 :603-12; Tye-Din et al, Sci Transl Med 2010, 2:41ra51), while the B-cell response is directed to y/ ⁇ -peptides (Osman et al, Clin Exp Immonol 2000, 121 :248-54; Ballew et al, PNAS 2013, 110: 19330-5).
- the gel filtration fractions of the gliadin digest containing the highest molecular weight peptides were not used in the pull-down experiments to facilitate the identification of motifs recognised by the hmAbs.
- long peptide fragments like the ⁇ -gliadin 33mer may to some extent have been excluded from these analyses.
- the DQ2.5-glia-c ⁇ l epitope is also an immunodominant T-cell epitope (Tye-Din et al, Sci Transl Med 2010, 2:41ra51).
- Table 15 Peptides pulled down with gliadin-specific antibodies. The frequencies of common motifs among pulled down peptides, and the sequences of top 20 peptides abundant peptides (intensity) for each antibody are shown.
Abstract
The present invention relates to anti-gluten antibodies and methods of using the same. In some embodiments, the present invention relates to the use of anti-gluten antibodies in research, food testing, and diagnostic applications.
Description
ANTI-GLUTEN ANTIBODIES AND USES THEREOF
FIELD OF THE INVENTION
The present invention relates to anti-gluten antibodies and methods of using the same. In some embodiments, the present invention relates to the use of anti-gluten antibodies in research, food testing, and diagnostic applications.
BACKGROUND
Celiac disease is an autoimmune disorder of the small intestine that occurs in genetically predisposed people of all ages from middle infancy onward. Symptoms include pain and discomfort in the digestive tract, chronic constipation and diarrhoea, failure to thrive (in children), anaemia and fatigue, but these may be absent, and symptoms in other organ systems have been described. Vitamin deficiencies are often noted in people with celiac disease owing to the reduced ability of the small intestine to p-roperly absorb nutrients from food.
At present, the only effective treatment is a lifelong gluten-free diet. No medication exists that will prevent damage or prevent the body from attacking the gut when gluten is present. Strict adherence to the diet allows the intestines to heal, leading to resolution of all symptoms in most cases and, depending on how soon the diet is begun, can also eliminate the heightened risk of osteoporosis and intestinal cancer and in some cases sterility. The diet can be cumbersome; failure to comply with the diet may cause relapse.
Dietitian input is generally requested to ensure the person is aware which foods contain gluten, which foods are safe, and how to have a balanced diet despite the limitations. Gluten- free products are usually more expensive and harder to find than common gluten- containing foods. Since ready-made products often contain traces of gluten, some celiacs may find it necessary to cook from scratch.
The term gluten-free is generally used to indicate a supposed harmless level of gluten rather than a complete absence. The exact level at which gluten is harmless is uncertain and controversial. A recent systematic review tentatively concluded that consumption of less than
10 mg of gluten per day is unlikely to cause histological abnormalities, although it noted that few reliable studies had been done. Regulation of the label gluten- free varies. In the European
Union, the European Commission issued regulations in 2009 limiting the use of "gluten- free" labels for food products to those with less than 20 mg/kg of gluten, and "very low gluten" labels for those with less than 100 mg/kg. In the United States, the FDA issued regulations in
2013 limiting the use of "gluten-free" labels for food products to those with less than 20 ppm of gluten. The current international Codex Alimentarius standard allows for 20 ppm of gluten in so-called "gluten-free" foods. Several organisations, such as the Gluten-Free Certification Organization (GFCO), the Celiac Sprue Association (CSA), and the National Foundation for Celiac Awareness (NFCA), also certify products and companies as gluten-free.
Additonal compositions and methods for accurately and precisely determining the levels of gluten (e.g., in food products) are needed.
SUMMARY
The present invention relates to anti-gluten antibodies and methods of using the same.
In some embodiments, the present invention relates to the use of anti-gluten antibodies in research, food testing, and diagnostic applications.
For example, in some embodiments, the present disclosure provides an isolated monoclonal antibody that binds to gliadin, wherein said antibody recognizes an epitope or motif (e.g., QPXQPFP (SEQ ID NO: 1), wherein X is Q or E; QPQQXFP (SEQ ID NO: 2), wherein X is P, S, T, or Q; QPQ(de)QXFP (SEQ ID NO: 2), wherein X is P, S, T, or Q; PLQPEQPFP (SEQ ID NO: 3); PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4);
X1QPQQPX2 (SEQ ID NO: 5), wherein Xl is P or S and X2 is I, L, or F; XiQPQQPX2 (SEQ ID NO: 5), wherein Xl is P or S and X2 is I, L, or F; XiQPQ(de)QPX2 (SEQ ID NO: 6), wherein Xi is Q, P, I, or L and X2 is F, Q, or A;
LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17); LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18);
QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259); or
QPEQPFPEQPEQPEQPFPQPEQPFPW (SEQ ID NO: 260) ). In some embodiments, the antibody is an antibody fragment (e.g., Fab, Fab', Fab'-SH, F(ab')2, Fv, or scFv variants) or a full length antibody. In some embodiments, the antibody is fused to a non-antibody molecule (e.g., a label or other molecule). In some embodiments, the complementarity determining region (CDR) of the antibody is encoded by a nucleic acid described in Table 2 or sequence that are at least 80% (e.g., 85%, 90%, 91 , 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100%) homologous to the sequences shown in Table 2. In some embodiments, the epitope comprises one or more deamidated amino acids (e.g., represented by E or Q(de)).
Further embodiments provide a method of detecting a peptide comprising an epitope or motif (e.g., QPXQPFP (SEQ ID NO: 1), wherein X is Q or E; QPQQXFP (SEQ ID NO: 2), wherein X is P, S, T, or Q; PLQPEQPFP (SEQ ID NO: 3);
PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4); XiQPQQPX2 (SEQ ID NO: 5), wherein X1 is P or S and X2 is I, L, or F; XiQPQQPX2 (SEQ ID NO: 6), wherein X1 is Q, P, I, or L and X2 is F, Q, or A; LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17); LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18); QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259); or QPEQPFPEQPEQPEQPFPQPEQPFPW (SEQ ID NO: 260)), comprising: a) a contacting an isolated monoclonal antibody that binds to gliadin, wherein said antibody recognizes an epitope or motif (e.g., QPXQPFP (SEQ ID NO: 1), wherein X is Q or E; QPQQXFP (SEQ ID NO: 2), wherein X is P, S, T, or Q; PLQPEQPFP (SEQ ID NO: 3);
PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4); XiQPQQPX2 (SEQ ID NO: 5), wherein Xj is P or S and X2 is I, L, or F; XiQPQQPX2 (SEQ ID NO: 6), wherein X1 is Q, P, I, or L and X2 is F, Q, or A; LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17); LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18); QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259); or QPEQPFPEQPEQPEQPFPQPEQPFPW (SEQ ID NO: 260)) with a biological sample (e.g., serum, blood, whole blood, urine, saliva, etc.); and b) detecting the binding of the antibody to the epitope. In some embodiments, the monoclonal antibody is attached to a solid support (e.g., a bead). In some embodiments, at least a portion of the solid support or peptide is labeled. In some embodiments, the peptide is labeled (e.g., with biotin). In some
embodiments, the label comprises a linker. In some embodiments, the label is biotin- GSGSGS.
Further embodiments provide a method of diagnosing celiac disease, comprising: a) contacting an antibody as described herein (e.g., conjugated to a solid support or label) or a peptide comprising an epitope or motif selected from, for example, QPXQPFP (SEQ ID NO: 1), wherein X is Q or E; QPQQXFP (SEQ ID NO: 2), wherein X is P, S, T, or Q;
PLQPEQPFP (SEQ ID NO: 3); PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4);
X1QPQQPX2 (SEQ ID NO: 5), wherein Xl is P or S and X2 is I, L, or F; XiQPQQPX2 (SEQ ID NO: 6), wherein X1 is Q, P, I, or L and X2 is F, Q, or A;
LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17); LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18);
QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259); or
QPEQPFPEQPEQPEQPFPQPEQPFPW (SEQ ID NO: 260)) and a sample from a subject; and b) measuring the level of binding of antibodies to the peptide in the sample. In some embodiments, the assay is a competitive assay comprising both peptide and antibody and the
signal of a signal molecule or label (e.g., fluorescent label) is reduced in the presence of antibodies in the sample that bind to the peptide.
Further embodiments provide a method of diagnosing celiac disease, comprising: a) contacting a peptide comprising an epitope or motif, selected from, for example, QPXQPFP (SEQ ID NO: 1), wherein X is Q or E; QPQQXFP (SEQ ID NO: 2), wherein X is P, S, T, or Q; PLQPEQPFP (SEQ ID NO: 3); PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4); X1QPQQPX2 (SEQ ID NO: 5), wherein Xl is P or S and X2 is I, L, or F; XiQPQQPX2 (SEQ ID NO: 6), wherein X1 is Q, P, I, or L and X2 is F, Q, or A;
LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17); LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18);
QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259); or
QPEQPFPEQPEQPEQPFPQPEQPFPW (SEQ ID NO: 260) (e.g., optionally coupled to a solid support and/or a signal molecule or label), with a sample from a subject; and b) measuring the level of binding of the peptide to anti-gliadin antibodies present in the sample.
In some embodiments, the level of binding is compared to the level of binding in a control serum from a subject that does not have celiac disease. In some embodiments, an increased level of binding relative to the level of binding found in the control sample is indicative of celiac disease in the subject. In some embodiments, the the monoclonal antibody and/or the peptide are labeled.
Additional embodiments provide a method of detecting gluten in a food sample, comprising: a) contacting a food sample with an antibody as described herein; and b) detecting the presence or absence of binding of the antibody to gliadin in the sample. In some embodiments, the monoclonal antibody is labeled.
Certain embodiments provide a kit comprising the antibody and/or peptides as described herein and a buffer. In some embodiments, the kit further comprises a solid support. In some embodiments, the antibody and/or peptide is affixed to the solid support.
Additional embodiments are described herein.
BRIEF DESCRIPTION OF THE FIGURES
Figure 1 shows survival of intestinal plasma cells in culture, a) Concentration of IgA in supernatants after 0, 1, 2, 3 and 4 weeks culture of single cell suspensions (SCSs) grown with (F+) or without fibroblasts (F-) (n.d. denotes non-detectable), b) Concentration of IgA after 0 and 2 days in supernatants from cultures of fibroblasts and PCs when PCs were added
either as isolated IgA PCs (IgA PCs) or as part of single cell suspensions (SCSs). c) Representative flow cytometry plots of SCSs after 4 weeks of co-culture with fibroblasts.
Figure 2 shows supernatant reactivity to heat/acid treated chymotrypsin digested gliadin (CT-gliadin) and TG2 by ELISA. b) IgA reactivity to antigen in representative cultures of SCSs from a) subjects with untreated celiac disease (UCD). b) The ratio of culture supematants with IgA reactivity to TG2 versus CT-gliadin, where the dots represent different subjects with UCD (n = 8). Horizontal bar indicates mean value, c) IgA reactivity to antigen in one representative of two tested non-CD controls (Ctr). d) The background level was defined by signal in supematants of cultures of fibroblasts only.
Figure 3 shows ELISA reactivity of hmAbs expression cloned from IgA+ PCs in culture, a) CT-gliadin. b) BSA control, c) PLQPEQPFP (SEQ ID NO: 3).
Figure 4 shows staining of intestinal PCs with tetramers of synthetic gluten peptides or TG2 in flow cytometry, a) Representative plots of PCs from SCSs stained with APC- conjugated streptavidin in complex with biotinylated (SEQ ID NO: 3) peptide, b) Frequency of IgA+ PCs stained positive with peptide in percentage of total number of IgA+ PCs. c)
Representative plots of one subject with UCD of IgM+ PCs and IgA+ PCs from SCSs stained with biotinylated peptides in complex with APC-conjugated strepavidin and biotinylated TG2 in complex with PE-conjugated streptavidin. d) Ratio of PCs stained positive with TG2 compared to synthetic gliadin peptides. IgM+ PCs and IgA+ PCs of the same sample are connected by lines.
Figure 5 shows alphaLISA characterization of gliadin-reactive hmAbs. a) Antibody reactivity to constant concentration of biotinylated PLQPEQPFP (SEQ ID NO: 3) in the presence of competing antigens, b) Reactivity to deamidated gliadin versus native gliadin.
Figure 6 shows epitopes of gliadin-specific hmAbs. a) Reactivity of hmAbs to PLQPEQPFP (SEQ ID NO: 3) and 33-mer. b) Reactivity of two representative hmAbs to constant concentration of biotinylated PLQPEQPFP (SEQ ID NO: 3) in competition with four different competitive synthetic gliadin peptides.
Figure 7 shows alphaLISA anti-PLQPEQPFP (SEQ ID NO: 3) immunoglobulin inhibition assay.
Figure 8 shows VH/VL usage and somatic hypermutations (SHMs). a) VH/VL usage of 38 gliadin-specific hmAbs from PCs either isolated by culture or by flow sorting, b) Frequencies of VH region somatic mutations (y-axis) per sequence in the populations indicated on x-axis. c) Comparison of mutations in VH3-23 genes. Horizontal bars indicate
median values and p-values were obtained by Student's t-test. ns > 0.05. *P < 0.05. **P < 0.01. ***P < 0.001.
Figure 9 shows frequency of IgA+ PCs in SCSs of small intestinal biopsies, a) Representative flow plot of SCSs showing large, viable, CD3~GLIADIN~CD27+IgA+, defined IgA+ PCs. b) Relative frequency of IgA+ PC of all cells in SCS in flow cytometry. Each dot represents one subject. Horizontal bar indicates mean value, c) Representative flow plot of SCSs of small intestinal biopsies, d) Relative ratio of IgA+ PCs to IgA+ memory B cells.
Figure 10 shows reactivity to antigen as depicted in headline measured by ELISA of 12 hmAbs cloned from single PLQPEQPFP+ (SEQ ID NO: 3) (a) or 33-mer+ (b) PCs.
Figure 1 1 shows comparison of PLQPEQPFP (SEQ ID NO: 3) conjugated beads and biotinylated hmAb 1002-1E01 compared to ELISA using streptavidin coated plates, biotinylated PLQPEQPFP (SEQ ID NO: 3)and anti-human IgG as detecting antibody.
Figure 12 shows estimated concentrations of serum antibodies blocking binding of (a) hmAb 1002-1E01 to PLQPEQPFP (SEQ ID NO: 3) and (b) hmAbs 1002-1E01 and 1002- 1E03 to PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4), by AlphaLISA competitive assay.
Figure 13 shows estimated concentrations of serum antibodies blocking binding of hmAbs 1002-1E01 and 1002-1E03 to PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4), showing results from patients (n = 34) with Marsh 2 and 3 scores.
Figure 14 shows ELISA reactivity of four gliadin-specific hmAbs to synthetic gliadin peptides. The hmAb tested is depicted in the headline. The hmAb was tested in different concentrations as indicated on x-axis. The different synthetic peptides are presented with different symbols, as described, (a) Notably, all four hmAbs were similarly reactive to PLQPEQPFP (SEQ ID NO: 3) and PLQPQQPFP (SEQ ID NO: 8) but low or negative to other peptides, (b) Confirmed reactivity to PLQPEQPFP(SEQ ID NO: 3), and all four hmAbs also show reactivity to PQQPEQPFP (SEQ ID NO: 9), showing that the epitope is QPXQPFP (SEQ ID NO: 1) (x = Q or E) for all four hmAbs tested.
Figure 15 shows ELISA reactivity of five gliadin-specific hmAbs to synthetic gliadin peptides. The hmAb tested is depicted in the headline. The hmAb was tested in different concentrations as indicated on x-axis. The different synthetic peptides are presented with different symbols, as described, (a) Initial testing of two of the five hmAbs showed different peptide reactivity pattern than hmAbs tested in Figure 14, with no reactivity to PLQPQQPFP
(SEQ ID NO: 8). (b) Two hmAbs (1002-1E03 and 11 14-1 GO 1) were only reactive to peptides harboring PEQ-motif. (a and b) One hmAb (1 130-3 B01) showed reactivity to peptides either
containing QPEQPFP (SEQ ID NO: 10) or QPELPYP (SEQ ID NO: 11) sequence, (c) whereas two hmAbs (1 130-3B03 and 1130-3G05) were reactive to PQPELPYPQP (SEQ ID NO: 12).
Figure 16 shows competitive gliadin AlphaLISA assay for detection of gliadin in flour.
Figure 17 shows (a) Length of peptides identified by mass spectrometry in fractions of a TG2 -treated digest of gliadin before (grey) and after (black) pull-down by the human monoclonal antibody 1002-1E03. (b) The number of peptides sharing identical sequence motifs, of 3 to 15 residues in length in pre (grey) and post pull-down (black) samples.
Figure 18 shows alphaLISA anti-cω34 Ig assay, (a) Inhibition of 1002-1E03 binding to b-cω34 peptide by sera of three test groups as analyzed in AlphaLISA. (b) Reference curve established by serial dilutions of a negative control serum spiked in with known and equimolar concentrations of three gliadin-specific hmAbs. (c) Activity of gliadin-specific serum antibodies expressed as concentration equivalents (mg/L) of reference gliadin-specific hmAbs.
Figure 19 shows (a) Anti-TG2 IgA and (b) anti-DGP IgG levels of participants from the three test groups.
Figure 20 shows inhibition of AlphaLISA signals of all three assays with hmAb 1002- 1E03 and the target peptides (a) b-QPEQPFP3 (SEQ ID NO: 10), (b) b-cω26 and (c) b-cω34 by sera of the test groups untreated celiac disease patients (celiac disease) and controls (Crohn' disease patients and healthy subjects), (d) Mean of Log AlphaLISA signal for all three peptides as presented in (a-c).
Figure 21 shows that the target peptide concentration affects the sensitivity, dynamic range and signal/noise-ratios as shown for three different concentrations of b-cω34. Mean values for all three concentrations are shown in grey.
Figure 22 shows that the antibodies pull down long peptides with repeated motifs, a) Kernel density plot of the peptide length identified by mass spectrometry in gliadin fractions pre and post pull-down by the hmAbs 1130-3A02 (b) and 1002-lEOl. c) Mean peptide length pre and post pull-down from different fractions of a gliadin digest with all human monoclonal antibodies, d) Peptides pulled down with hmAb 1 130-3 A02. The most frequent 7mer motif in the peptides is underlined.
Figure 23 shows common motifs in peptides pulled down by antibody 1130-3B04. a) Percent of peptides sharing identical sequence motifs, of 3 to 15 residues in length, post (triangles/) and pre pull-down (diamonds) from a fraction of digested gliadin by the hmAb
1130-3B04. b) The sequence motifs and the frequency and number of peptides harbouring these motifs
Figure 24 shows sequence motif of peptides pulled down by antibody 1130-3 BO 1. a) Sequence motif in peptides pulled-down with the hmAb 1 130-3B01, based on 85% similarity with most common 7mer motif QPQQQFP (SEQ ID NO: 13), as generated by WebLOGO 3.4. b) ELISA reactivity of the hmAbs 1 130-3B01 (C) and 1130-3A02 to synthetic gliadin peptides (native sequences in black and deamidated sequences in grey).
Figure 25 shows the affinity of antibodies to (SEQ ID NO: 10) QPEQPFP-containing peptides depends on residues flanking the motif, a) AlphaLISA affinity of the hmAbs 1002- 1E01, 1002-1E03 and 1130-3B01 to PLQPEQPFP (SEQ ID NO: 3) and the competitive effect of a panel of different synthetic gliadin peptides harbouring the QPEQPFP (SEQ ID NO: 10) sequence motif at different concentrations (M) as indicated on the x-axis. B) The XXXQPQQPFPXXX (SEQ ID NO: 14) motif (X=any amino acid) searched in the Triticum aestivum database using the program "Pattinprot". Sequence logo of resulting 13mer motif generated by WebLOGO 3.0.
Figure 26 shows that antibodies show better reactivity to gliadin peptides with repeats of epitopes, a) Pull-down with the hmAb 1130-3B01, 1002-1E03 or 1002-1E01 from samples with equimolar amounts of the PLQPEQPF peptide and the γ-gliadin 26mer peptide, b) Pull-down with hmAb 1002-1E03 from a size fraction of a gliadin digest treated with transglutaminase 2 demonstrating that the hmAb preferentially pull-down long peptides with multiple repeats of epitopes, c) AlphaLISA competition assay comparing the relative binding of bead-conjugated hmAb 1002-1E03 to the soluble 34mer ω-peptide in the presence of competing soluble whole antibody (grey solid line) or Fab fragment (black stippled line) of the hmAb 1002-1E03. d) Inhibition of binding of bead-conjugated hmAb 1002-1E03 to soluble PLQPEQPFP (SEQ ID NO: 3) by FLQPEQPFPEQPEQPYPEQPEQPFPQ (SEQ ID NO: 15) (grey solid line) or PLQPEQPFP (SEQ ID NO: 3) (black stippled line).
Figure 27 shows analysis of factors affecting peptide pull-down by MALDI-TOF. a and b) 1 130-3 B01 enrichment from the peptide pairs a-gliadin 33mer and PLQPEQPF (SEQ ID NO: 15) peptide (a) or α-gliadin 33mer and γ-gliadin 26mer (b). c) 1 130-3B03 enrichment from the peptide pair α-gliadin 33mer and γ-gliadin 26mer.
Figure 28 shows co-localisation of gliadin T-cell and B-cell epitopes in an ω-gliadin protein.
DEFINITIONS
An "acceptor human framework" for the purposes herein is a framework comprising the amino acid sequence of a light chain variable domain (VL) framework or a heavy chain variable domain (VH) framework derived from a human immunoglobulin framework or a human consensus framework, as defined below. An acceptor human framework "derived from" a human immunoglobulin framework or a human consensus framework may comprise the same amino acid sequence thereof, or it may contain amino acid sequence changes. In some embodiments, the number of amino acid changes are 10 or less, 9 or less, 8 or less, 7 or less, 6 or less, 5 or less, 4 or less, 3 or less, or 2 or less. In some embodiments, the VL acceptor human framework is identical in sequence to the VL human immunoglobulin framework sequence or human consensus framework sequence.
"Affinity" refers to the strength of the sum total of noncovalent interactions between a single binding site of a molecule (e.g., an antibody) and its binding partner (e.g., an antigen). Unless indicated otherwise, as used herein, "binding affinity" refers to intrinsic binding affinity which reflects a 1 : 1 interaction between members of a binding pair (e.g., antibody and antigen). The affinity of a molecule X for its partner Y can generally be represented by the dissociation constant (Kd). Affinity can be measured by common methods known in the art, including those described herein. Specific illustrative and exemplary embodiments for measuring binding affinity are described in the following.
An "affinity matured" antibody refers to an antibody with one or more alterations in one or more hypervariable regions (HVRs), compared to a parent antibody which does not possess such alterations, such alterations resulting in an improvement in the affinity of the antibody for antigen.
The term "antibody" is used herein in the broadest sense and encompasses various antibody structures, including but not limited to monoclonal antibodies, polyclonal antibodies, multispecific antibodies (e.g., bispecific antibodies), and antibody fragments so long as they exhibit the desired antigen-binding activity.
An "antibody fragment" refers to a molecule other than an intact antibody that comprises a portion of an intact antibody and that binds the antigen to which the intact antibody binds. Examples of antibody fragments include but are not limited to Fv, Fab, Fab', Fab'-SH, F(ab¾; diabodies; linear antibodies; single-chain antibody molecules (e.g. scFv); and multispecific antibodies formed from antibody fragments.
An "antibody that binds to the same epitope" as a reference antibody refers to an antibody that blocks binding of the reference antibody to its antigen in a competition assay by 50% or more, and conversely, the reference antibody blocks binding of the antibody to its antigen in a competition assay by 50% or more. An exemplary competition assay is provided herein.
The term "chimeric" antibody refers to an antibody in which a portion of the heavy and/or light chain is derived from a particular source or species, while the remainder of the heavy and/or light chain is derived from a different source or species.
The "class" of an antibody refers to the type of constant domain or constant region possessed by its heavy chain. There are five major classes of antibodies: IgA, IgD, IgE, IgG, and IgM, and several of these may be further divided into subclasses (isotypes), e.g., IgGi, IgG2, IgG3, IgG4, IgAi, and IgA2. The heavy chain constant domains that correspond to the different classes of immunoglobulins are called α, δ, ε, γ, and μ, respectively.
"Effector functions" refer to those biological activities attributable to the Fc region of an antibody, which vary with the antibody isotype. Examples of antibody effector functions include: Clq binding and complement dependent cytotoxicity (CDC); Fc receptor binding; antibody-dependent cell-mediated cytotoxicity (ADCC); phagocytosis; down regulation of cell surface receptors (e.g. B cell receptor); and B cell activation.
An "effective amount" of an agent, e.g., a pharmaceutical formulation, refers to an amount effective, at dosages and for periods of time necessary, to achieve the desired therapeutic or prophylactic result.
The term "epitope" refers to the particular site on an antigen molecule to which an antibody binds.
The term "Fc region" herein is used to define a C-terminal region of an
immunoglobulin heavy chain that contains at least a portion of the constant region. The term includes native sequence Fc regions and variant Fc regions. In one embodiment, a human IgG heavy chain Fc region extends from Cys226, or from Pro230, to the carboxyl-terminus of the heavy chain. However, the C-terminal lysine (Lys447) of the Fc region may or may not be present. Unless otherwise specified herein, numbering of amino acid residues in the Fc region or constant region is according to the EU numbering system, also called the EU index, as described in Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD, 1991.
"Framework" or "FR" refers to variable domain residues other than hypervariable region (HVR) residues. The FR of a variable domain generally consists of four FR domains: FR1, FR2, FR3, and FR4. Accordingly, the HVR and FR sequences generally appear in the following sequence in VH (or VL): FR1-H1(L1)-FR2-H2(L2)-FR3-H3(L3)-FR4.
The terms "full length antibody," "intact antibody," and "whole antibody" are used herein interchangeably to refer to an antibody having a structure substantially similar to a native antibody structure or having heavy chains that contain an Fc region as defined herein.
The terms "host cell," "host cell line," and "host cell culture" are used
interchangeably and refer to cells into which exogenous nucleic acid has been introduced, including the progeny of such cells. Host cells include "trans formants" and "transformed cells," which include the primary transformed cell and progeny derived therefrom without regard to the number of passages. Progeny may not be completely identical in nucleic acid content to a parent cell, but may contain mutations. Mutant progeny that have the same function or biological activity as screened or selected for in the originally transformed cell are included herein.
A "human antibody" is one which possesses an amino acid sequence which corresponds to that of an antibody produced by a human or a human cell or derived from a non-human source that utilizes human antibody repertoires or other human antibody- encoding sequences. This definition of a human antibody specifically excludes a humanized antibody comprising non-human antigen-binding residues.
A "human consensus framework" is a framework which represents the most commonly occurring amino acid residues in a selection of human immunoglobulin VL or VH framework sequences. Generally, the selection of human immunoglobulin VL or VH sequences is from a subgroup of variable domain sequences. Generally, the subgroup of sequences is a subgroup as in Kabat et al, Sequences of Proteins of Immunological Interest, Fifth Edition, NIH Publication 91-3242, Bethesda MD (1991), vols. 1-3. In one embodiment, for the VL, the subgroup is subgroup kappa I as in Kabat et al, supra. In one embodiment, for the VH, the subgroup is subgroup III as in Kabat et al, supra.
A "humanized" antibody refers to a chimeric antibody comprising amino acid residues from non-human HVRs and amino acid residues from human FRs. In certain embodiments, a humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the HVRs (e.g., CDRs) correspond to those of a non-human antibody, and all or substantially all of the FRs correspond to those of a human antibody. A humanized antibody optionally may comprise at
least a portion of an antibody constant region derived from a human antibody. A "humanized form" of an antibody, e.g., a non-human antibody, refers to an antibody that has undergone humanization.
The term "hypervariable region" or "HVR," as used herein, refers to each of the regions of an antibody variable domain which are hypervariable in sequence and/or form structurally defined loops ("hypervariable loops"). Generally, native four-chain antibodies comprise six HVRs; three in the VH (HI, H2, H3), and three in the VL (LI, L2, L3). HVRs generally comprise amino acid residues from the hypervariable loops and/or from the "complementarity determining regions" (CDRs), the latter being of highest sequence variability and/or involved in antigen recognition. Exemplary hypervariable loops occur at amino acid residues 26-32 (LI), 50-52 (L2), 91-96 (L3), 26-32 (HI), 53-55 (H2), and 96-101 (H3). (Chothia and Lesk, J. Mol. Biol. 196:901-917 (1987).) Exemplary CDRs (CDR-L1, CDR-L2, CDR-L3, CDR-Hl, CDR-H2, and CDR-H3) occur at amino acid residues 24-34 of LI, 50-56 of L2, 89-97 of L3, 31-35B of HI, 50-65 of H2, and 95-102 of H3. (Kabat et al, Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD (1991).) With the exception of CDR1 in VH, CDRs generally comprise the amino acid residues that form the hypervariable loops. CDRs also comprise "specificity determining residues," or "SDRs," which are residues that contact antigen. SDRs are contained within regions of the CDRs called abbreviated-CD Rs, or a- CDRs. Exemplary a-CDRs (a-CDR-Ll, a-CDR-L2, a-CDR-L3, a-CDR-Hl, a-CDR-H2, and a-CDR-H3) occur at amino acid residues 31-34 of LI, 50-55 of L2, 89-96 of L3, 31-35B of HI, 50-58 of H2, and 95-102 of H3. (See Almagro and Fransson, Front. Biosci. 13: 1619- 1633 (2008).) Unless otherwise indicated, HVR residues and other residues in the variable domain (e.g., FR residues) are numbered herein according to Kabat et al, supra.
An "individual" or "subject" is a mammal. Mammals include, but are not limited to, domesticated animals (e.g., cows, sheep, cats, dogs, and horses), primates (e.g., humans and non-human primates such as monkeys), rabbits, and rodents (e.g., mice and rats). In certain embodiments, the individual or subject is a human.
An "isolated antibody" is one which has been separated from a component of its natural environment. In some embodiments, an antibody is purified to greater than 95% or 99% purity as determined by, for example, electrophoretic (e.g., SDS-PAGE, isoelectric focusing (IEF), capillary electrophoresis) or chromatographic (e.g., ion exchange or reverse phase HPLC). For review of methods for assessment of antibody purity, see, e.g., Flatman et al, J. Chromatogr. B 848:79-87 (2007).
An "isolated nucleic acid" refers to a nucleic acid molecule that has been separated from a component of its natural environment. An isolated nucleic acid includes a nucleic acid molecule contained in cells that ordinarily contain the nucleic acid molecule, but the nucleic acid molecule is present extrachromosomally or at a chromosomal location that is different from its natural chromosomal location.
The term "monoclonal antibody" as used herein refers to an antibody obtained from a population of substantially homogeneous antibodies, i.e., the individual antibodies comprising the population are identical and/or bind the same epitope, except for possible variant antibodies, e.g., containing naturally occurring mutations or arising during production of a monoclonal antibody preparation, such variants generally being present in minor amounts. In contrast to polyclonal antibody preparations, which typically include different antibodies directed against different determinants (epitopes), each monoclonal antibody of a monoclonal antibody preparation is directed against a single determinant on an antigen. Thus, the modifier "monoclonal" indicates the character of the antibody as being obtained from a substantially homogeneous population of antibodies, and is not to be construed as requiring production of the antibody by any particular method. For example, the monoclonal antibodies to be used in accordance with the present invention may be made by a variety of techniques, including but not limited to the hybridoma method, recombinant DNA methods, phage- display methods, and methods utilizing transgenic animals containing all or part of the human immunoglobulin loci, such methods and other exemplary methods for making monoclonal antibodies being described herein.
"Native antibodies" refer to naturally occurring immunoglobulin molecules with varying structures. For example, native IgG antibodies are heterotetrameric glycoproteins of about 150,000 daltons, composed of two identical light chains and two identical heavy chains that are disulfide-bonded. From N- to C-terminus, each heavy chain has a variable region (VH), also called a variable heavy domain or a heavy chain variable domain, followed by three constant domains (CHI, CH2, and CH3). Similarly, from N- to C-terminus, each light chain has a variable region (VL), also called a variable light domain or a light chain variable domain, followed by a constant light (CL) domain. The light chain of an antibody may be assigned to one of two types, called kappa (κ) and lambda (λ), based on the amino acid sequence of its constant domain.
"Percent (%) amino acid sequence identity" with respect to a reference polypeptide sequence is defined as the percentage of amino acid residues in a candidate sequence that are identical with the amino acid residues in the reference polypeptide sequence, after aligning
the sequences and introducing gaps, if necessary, to achieve the maximum percent sequence identity, and not considering any conservative substitutions as part of the sequence identity. Alignment for purposes of determining percent amino acid sequence identity can be achieved in various ways that are within the skill in the art, for instance, using publicly available computer software such as BLAST, BLAST-2, ALIGN or Megalign (DNASTAR) software. Those skilled in the art can determine appropriate parameters for aligning sequences, including any algorithms needed to achieve maximal alignment over the full length of the sequences being compared. For purposes herein, however, % amino acid sequence identity values are generated using the sequence comparison computer program ALIGN-2. The ALIGN-2 sequence comparison computer program was authored by Genentech, Inc., and the source code has been filed with user documentation in the U.S. Copyright Office,
Washington D.C., 20559, where it is registered under U.S. Copyright Registration No.
TXU510087. The ALIGN-2 program is publicly available from Genentech, Inc., South San Francisco, California, or may be compiled from the source code. The ALIGN-2 program should be compiled for use on a UNIX operating system, including digital UNIX V4.0D. All sequence comparison parameters are set by the ALIGN-2 program and do not vary.
In situations where ALIGN-2 is employed for amino acid sequence comparisons, the % amino acid sequence identity of a given amino acid sequence A to, with, or against a given amino acid sequence B (which can alternatively be phrased as a given amino acid sequence A that has or comprises a certain % amino acid sequence identity to, with, or against a given amino acid sequence B) is calculated as follows:
100 times the fraction X/Y where X is the number of amino acid residues scored as identical matches by the sequence alignment program ALIGN-2 in that program's alignment of A and B, and where Y is the total number of amino acid residues in B. It will be appreciated that where the length of amino acid sequence A is not equal to the length of amino acid sequence B, the % amino acid sequence identity of A to B will not equal the % amino acid sequence identity of B to A. Unless specifically stated otherwise, all % amino acid sequence identity values used herein are obtained as described in the immediately preceding paragraph using the ALIGN-2 computer program.
The term "pharmaceutical formulation" refers to a preparation which is in such form as to permit the biological activity of an active ingredient contained therein to be effective,
and which contains no additional components which are unacceptably toxic to a subject to which the formulation would be administered.
A "pharmaceutically acceptable carrier" refers to an ingredient in a pharmaceutical formulation, other than an active ingredient, which is nontoxic to a subject. A
pharmaceutically acceptable carrier includes, but is not limited to, a buffer, excipient, stabilizer, or preservative.
As used herein, "treatment" (and grammatical variations thereof such as "treat" or "treating") refers to clinical intervention in an attempt to alter the natural course of the individual being treated, and can be performed either for prophylaxis or during the course of clinical pathology. Desirable effects of treatment include, but are not limited to, preventing occurrence or recurrence of disease, alleviation of symptoms, diminishment of any direct or indirect pathological consequences of the disease, preventing metastasis, decreasing the rate of disease progression, amelioration or palliation of the disease state, and remission or improved prognosis. In some embodiments, antibodies of the invention are used to delay development of a disease or to slow the progression of a disease.
The term "variable region" or "variable domain" refers to the domain of an antibody heavy or light chain that is involved in binding the antibody to antigen. The variable domains of the heavy chain and light chain (VH and VL, respectively) of a native antibody generally have similar structures, with each domain comprising four conserved framework regions (FRs) and three hypervariable regions (HVRs). (See, e.g., Kindt et al. Kuby Immunology, 6th ed., W.H. Freeman and Co., page 91 (2007).) A single VH or VL domain may be sufficient to confer antigen-binding specificity. Furthermore, antibodies that bind a particular antigen may be isolated using a VH or VL domain from an antibody that binds the antigen to screen a library of complementary VL or VH domains, respectively. See, e.g., Portolano et al, J. Immunol. 150:880-887 (1993); Clarkson et al, Nature 352:624-628 (1991).
The term "vector," as used herein, refers to a nucleic acid molecule capable of propagating another nucleic acid to which it is linked. The term includes the vector as a self- replicating nucleic acid structure as well as the vector incorporated into the genome of a host cell into which it has been introduced. Certain vectors are capable of directing the expression of nucleic acids to which they are operatively linked. Such vectors are referred to herein as "expression vectors."
DETAILED DESCRIPTION OF CERTAIN EMBODIMENTS OF THE INVENTION
The present invention relates to anti-gluten antibodies and methods of using the same. In some embodiments, the present invention relates to the use of anti-gluten antibodies in research, food testing, and diagnostic applications.
Celiac disease (CD), an enteropathy caused by cereal gluten ingestion, is
characterized by CD4+ T cells recognizing deamidated gluten and by antibodies reactive to gluten or the self-antigen transglutaminase 2 (TG2). The diagnosis of CD has changed the last years, influenced by serological screening of gliadin-specific and TG2-specific antibodies. They have shown potential usage in screening, and in diagnosing CD without endoscopy. Thus, improved serological methods influence the course of examination of patients where CD is suspected. Experiments described herin resulted in the development of a new and improved serological assay for detection of gliadin-specific antibodies based on monoclonal antibodies (hmAbs) of gliadin-specific IgA secreting plasma cells isolated from intestinal lesion of patients with untreated CD.
38 monoclonal antibodies were cloned from single PCs of 10 patients either isolated from cultures with reactivity to complex deamidated gluten antigen or by sorting with gluten peptide tetramers. Typically the antibodies bind gluten peptides related to T-cell epitopes and many have higher reactivity to deamidated peptides. Based on the gliadin-specific hmAbs and characterized epitopes, serological bead-based (AlphaLISA) inhibition assays where gliadin-specific serum antibodies competed for binding to a certain gliadin epitope with the representative gliadin-specific hmAb were developed. The serological tests based on the characterized hmAbs have the potential to improve the accuracy of serological CD diagnosis, by either replacing or supplementing current tests. The hmAbs also find use as reference reagents for established serological tests, and for detection of gluten in food.
I. Antibody Compositions
In some embodiments, the invention provides isolated antibodies that bind to gliadin. In some embodiments, the antibodies are monoclonal antibodies. The antibodies have variable regions that are specific for gliadin.
In some embodiments, an anti-gliadin antibody is human or humanized. In one embodiment, an anti-GLIADI antibody comprises a human acceptor framework, e.g. a human immunoglobulin framework or a human consensus framework. In certain
embodiments, the human acceptor framework is the human VL kappa IV consensus (VLKIV) framework and/or the VH framework VHi.
In some embodiments, anit-gliadin antibodies bind to peptide epitopes described herein (e.g., QPXQPFP (SEQ ID NO: 1), wherein X is Q or E; QPQQXFP (SEQ ID NO: 2), wherein X is P, S, T, or Q; PLQPEQPFP (SEQ ID NO: 3);
PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4); XiQPQQPX2 (SEQ ID NO: 5), wherein Xl is P or S and X2 is I, L, or F; XiQPQQPX2 (SEQ ID NO: 6), wherein Xl is Q, P, I, or L and X2 is F, Q, or A; LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17); LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18); QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259);
LQLQPFPQPELPYPQPELPYPQPELPYPQP (SEQ ID NO: 757); or
QPEQPFPEQPEQPEQPFPQPEQPFPW (SEQ ID NO: 260)). In a further aspect, the invention provides an antibody that binds to the same epitope as an anti-gliadin antibody provided herein.
In some embodiments, the complementarity determining region (CDR) of the antibody is encoded by a nucleic acid described in Table 2 or sequence that are at least 80% (e.g., 85%, 90%, 91, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100%) homologous to the sequences shown in Table 2.
In a further aspect of the invention, an anti-gliadin antibody according to any of the above embodiments is a monoclonal antibody, including a chimeric, humanized or human antibody. In one embodiment, an anti-GLIADIN antibody is an antibody fragment, e.g. , a Fv, Fab, Fab', scFv, diabody, or F(ab')2 fragment. In another embodiment, the antibody is a substantially full length antibody, e.g., an IgGl antibody or other antibody class or isotype as defined herein.
In certain embodiments, a VH or VL sequence described herein contains substitutions (e.g., conservative substitutions), insertions, or deletions relative to the reference sequence, but an anti-gliadin antibody comprising that sequence retains the ability to bind to gliadin. In certain embodiments, a total of 1 to 10 amino acids have been substituted, inserted and/or deleted. In certain embodiments, a total of 1 to 5 amino acids have been substituted, inserted and/or deleted.
In a further aspect of the invention, an anti- gliadin antibody according to any of the above embodiments is a monoclonal antibody, including a human antibody. In one embodiment, an anti- gliadin antibody is an antibody fragment, e.g., a Fv, Fab, Fab', scFv, diabody, or F(ab')2 fragment. In another embodiment, the antibody is a substantially full length antibody, e.g., an IgG2a antibody or other antibody class or isotype as defined herein.
Antibody Fragments
In certain embodiments, an antibody provided herein is an antibody fragment.
Antibody fragments include, but are not limited to, Fab, Fab', Fab'-SH, F(ab')2, Fv, and scFv fragments, and other fragments described below. For a review of certain antibody fragments, see Hudson et al. Nat. Med. 9: 129-134 (2003). For a review of scFv fragments, see, e.g.,
Pluckthun, in The Pharmacology of Monoclonal Antibodies, vol. 113, Rosenburg and Moore eds., (Springer-Verlag, New York), pp. 269-315 (1994); see also WO 93/16185; and U.S.
Patent Nos. 5,571,894 and 5,587,458. For discussion of Fab and F(ab¾ fragments comprising salvage receptor binding epitope residues and having increased in vivo half-life, see U.S.
Patent No. 5,869,046.
Diabodies are antibody fragments with two antigen-binding sites that may be bivalent or bispecific. See, for example, EP 404,097; WO 1993/01161 ; Hudson et al, Nat. Med.
9: 129-134 (2003); and Hollinger et al, Proc. Natl. Acad. Sci. USA 90: 6444-6448 (1993). Triabodies and tetrabodies are also described in Hudson et al, Nat. Med. 9: 129-134 (2003).
Single-domain antibodies are antibody fragments comprising all or a portion of the heavy chain variable domain or all or a portion of the light chain variable domain of an antibody. In certain embodiments, a single-domain antibody is a human single-domain antibody (Domantis, Inc., Waltham, MA; see, e.g., U.S. Patent No. 6,248,516 Bl).
Antibody fragments can be made by various techniques, including but not limited to proteolytic digestion of an intact antibody as well as production by recombinant host cells
(e.g. E. coli or phage), as described herein.
Chimeric and Humanized Antibodies
In certain embodiments, an antibody provided herein is a chimeric antibody. Certain chimeric antibodies are described, e.g., in U.S. Patent No. 4,816,567; and Morrison et al, Proc. Natl. Acad. Sci. USA, 81 :6851-6855 (1984)). In one example, a chimeric antibody comprises a non-human variable region (e.g. , a variable region derived from a mouse, rat, hamster, rabbit, or non-human primate, such as a monkey) and a human constant region. In a further example, a chimeric antibody is a "class switched" antibody in which the class or subclass has been changed from that of the parent antibody. Chimeric antibodies include antigen-binding fragments thereof.
In certain embodiments, a chimeric antibody is a humanized antibody. Typically, a non-human antibody is humanized to reduce immunogenicity to humans, while retaining the
specificity and affinity of the parental non-human antibody. Generally, a humanized antibody comprises one or more variable domains in which VRs, e.g., CDRs, (or portions thereof) are derived from a non-human antibody, and FRs (or portions thereof) are derived from human antibody sequences. A humanized antibody optionally will also comprise at least a portion of a human constant region. In some embodiments, some FR residues in a humanized antibody are substituted with corresponding residues from a non-human antibody (e.g., the antibody from which the VR residues are derived), e.g., to restore or improve antibody specificity or affinity.
Humanized antibodies and methods of making them are reviewed, e.g., in Almagro and Fransson, Front. Biosci. 13: 1619-1633 (2008), and are further described, e.g., in
Riechmann et al, Nature 332:323-329 (1988); Queen et al., Proc. Nat Acad. Sci. USA
86: 10029-10033 (1989); US Patent Nos. 5, 821,337, 7,527,791, 6,982,321, and 7,087,409;
Kashmiri et al, Methods 36:25-34 (2005) (describing SDR (a-CDR) grafting); Padlan, Mol.
Immunol. 28:489-498 (1991) (describing "resurfacing"); Dall'Acqua et al, Methods 36:43-60 (2005) (describing "FR shuffling"); and Osbourn et al, Methods 36:61-68 (2005) and Klimka et al, Br. J. Cancer, 83:252-260 (2000) (describing the "guided selection" approach to FR shuffling).
Human framework regions that may be used for humanization include but are not limited to: framework regions selected using the "best- fit" method (see, e.g., Sims et al. J. Immunol. 151 :2296 (1993)); framework regions derived from the consensus sequence of human antibodies of a particular subgroup of light or heavy chain variable regions (see, e.g., Carter et al. Proc. Natl. Acad. Sci. USA, 89:4285 (1992); and Presta et al. J. Immunol, 151 :2623 (1993)); human mature (somatically mutated) framework regions or human germline framework regions (see, e.g., Almagro and Fransson, Front. Biosci. 13 : 1619-1633 (2008)); and framework regions derived from screening FR libraries (see, e.g., Baca et al, J. Biol. Chem. 272: 10678-10684 (1997) and Rosok et al, J. Biol. Chem. 271 :2261 1-22618 (1996)).
Human Antibodies
In certain embodiments, an antibody provided herein is a human antibody. Human antibodies can be produced using various techniques known in the art. Human antibodies are described generally in van Dijk and van de Winkel, Curr. Opin. Pharmacol. 5: 368-74 (2001) and Lonberg, Curr. Opin. Immunol. 20:450-459 (2008).
Human antibodies may be prepared by administering an immunogen to a transgenic animal that has been modified to produce intact human antibodies or intact antibodies with human variable regions in response to antigenic challenge. Such animals typically contain all or a portion of the human immunoglobulin loci, which replace the endogenous
immunoglobulin loci, or which are present extrachromosomally or integrated randomly into the animal's chromosomes. In such transgenic mice, the endogenous immunoglobulin loci have generally been inactivated. For review of methods for obtaining human antibodies from transgenic animals, see Lonberg, Nat. Biotech. 23: 1 117-1125 (2005). See also, e.g., U.S. Patent Nos. 6,075, 181 and 6, 150,584 describing XENOMOUSE™ technology; U.S. Patent No. 5,770,429 describing HUMAB® technology; U.S. Patent No. 7,041,870 describing K-M MOUSE® technology, and U.S. Patent Application Publication No. US 2007/0061900, describing VELOCIMOUSE® technology). Human variable regions from intact antibodies generated by such animals may be further modified, e.g., by combining with a different human constant region.
Human antibodies can also be made by hybridoma-based methods. Human myeloma and mouse-human heteromyeloma cell lines for the production of human monoclonal antibodies have been described. (See, e.g., Kozbor J. Immunol, 133 : 3001 (1984); Brodeur et al, Monoclonal Antibody Production Techniques and Applications, pp. 51-63 (Marcel Dekker, Inc., New York, 1987); and Boerner et al, J. Immunol, 147: 86 (1991).) Human antibodies generated via human B-cell hybridoma technology are also described in Li et al, Proc. Natl. Acad. Sci. USA, 103:3557-3562 (2006). Additional methods include those described, for example, in U.S. Patent No. 7,189,826 (describing production of monoclonal human IgM antibodies from hybridoma cell lines) and Ni, Xiandai Mianyixue, 26(4):265-268 (2006) (describing human-human hybridomas). Human hybridoma technology (Trioma technology) is also described in Vollmers and Brandlein, Histology and Histopathology , 20(3):927-937 (2005) and Vollmers and Brandlein, Methods and Findings in Experimental and Clinical Pharmacology , 27(3): 185-91 (2005).
Human antibodies may also be generated by isolating Fv clone variable domain sequences selected from human-derived phage display libraries. Such variable domain sequences may then be combined with a desired human constant domain. Techniques for selecting human antibodies from antibody libraries are described below.
Library-Derived Antibodies
Antibodies of the invention may be isolated by screening combinatorial libraries for antibodies with the desired activity or activities. For example, a variety of methods are known in the art for generating phage display libraries and screening such libraries for antibodies possessing the desired binding characteristics. Such methods are reviewed, e.g., in
Hoogenboom et al. in Methods in Molecular Biology 178: 1-37 (O'Brien et al, ed., Human Press, Totowa, NJ, 2001) and further described, e.g., in the McCafferty et al, Nature 348:552-554; Clackson et al, Nature 352: 624-628 (1991); Marks et al., J. Mol. Biol. 222: 581-597 (1992); Marks and Bradbury,in Methods in Molecular Biology 248: 161-175 (Lo, ed., Human Press, Totowa, NJ, 2003); Sidhu et al, J. Mol. Biol. 338(2): 299-310 (2004); Lee et al, J. Mol. Biol. 340(5): 1073-1093 (2004); Fellouse, Proc. Natl. Acad. Sci. USA 101(34): 12467-12472 (2004); and Lee et al, J. Immunol. Methods 284(1-2): 119-132(2004).
In certain phage display methods, repertoires of VH and VL genes are separately cloned by polymerase chain reaction (PCR) and recombined randomly in phage libraries, which can then be screened for antigen-binding phage as described in Winter et al, Ann. Rev. Immunol, 12: 433-455 (1994). Phage typically display antibody fragments, either as single- chain Fv (scFv) fragments or as Fab fragments. Libraries from immunized sources provide high-affinity antibodies to the immunogen without the requirement of constructing hybridomas. Alternatively, the naive repertoire can be cloned (e.g., from human) to provide a single source of antibodies to a wide range of non-self and also self antigens without any immunization as described by Griffiths et al, EMBO J, 12: 725-734 (1993). Finally, naive libraries can also be made synthetically by cloning unrearranged V-gene segments from stem cells, and using PCR primers containing random sequence to encode the highly variable CDR3 regions and to accomplish rearrangement in vitro, as described by Hoogenboom and Winter, J. Mol. Biol, 227: 381-388 (1992). Patent publications describing human antibody phage libraries include, for example: US Patent No. 5,750,373, and US Patent Publication Nos. 2005/0079574, 2005/0119455, 2005/0266000, 2007/01 17126, 2007/0160598,
2007/0237764, 2007/0292936, and 2009/0002360.
Antibodies or antibody fragments isolated from human antibody libraries are considered human antibodies or human antibody fragments herein.
Multispecific Antibodies
In certain embodiments, an antibody provided herein is a multispecific antibody, e.g. a bispecific antibody. Multispecific antibodies are monoclonal antibodies that have binding specificities for at least two different sites. In certain embodiments, one of the binding specificities is for gliadin and the other is for any other antigen. In certain embodiments, bispecific antibodies may bind to two different epitopes of gliadin. Bispecific antibodies may also be used to localize cytotoxic agents to cells which express gliadin. Bispecific antibodies can be prepared as full length antibodies or antibody fragments.
Techniques for making multispecific antibodies include, but are not limited to, recombinant co-expression of two immunoglobulin heavy chain-light chain pairs having different specificities (see Milstein and Cuello, Nature 305: 537 (1983)), WO 93/08829, and Traunecker et al, EMBO J. 10: 3655 (1991)), and "knob-in-hole" engineering (see, e.g., U.S. Patent No. 5,731,168). Multi-specific antibodies may also be made by engineering electrostatic steering effects for making antibody Fc-heterodimeric molecules
(WO 2009/089004A1); cross-linking two or more antibodies or fragments (see, e.g., US Patent No. 4,676,980, and Brennan et al, Science, 229: 81 (1985)); using leucine zippers to produce bi-specific antibodies (see, e.g., Kostelny et al, J. Immunol, 148(5): 1547-1553 (1992)); using "diabody" technology for making bispecific antibody fragments (see, e.g., Hollinger et al, Proc. Natl. Acad. Sci. USA, 90:6444-6448 (1993)); and using single-chain Fv (sFv) dimers (see,e.g. Gruber et al, J. Immunol, 152:5368 (1994)); and preparing trispecific antibodies as described, e.g., in Tutt et al. J. Immunol. 147: 60 (1991).
Engineered antibodies with three or more functional antigen binding sites, including "Octopus antibodies," are also included herein (see, e.g. US 2006/0025576A1).
The antibody or fragment herein also includes a "Dual Acting FAb" or "DAF" comprising an antigen binding site that binds to GLIADIN as well as another, different antigen (see, US 2008/0069820, for example).
Antibody Variants
In certain embodiments, amino acid sequence variants of the antibodies provided herein are contemplated. For example, it may be desirable to improve the binding affinity and/or other biological properties of the antibody. Amino acid sequence variants of an antibody may be prepared by introducing appropriate modifications into the nucleotide sequence encoding the antibody, or by peptide synthesis. Such modifications include, for
example, deletions from, and/or insertions into and/or substitutions of residues within the amino acid sequences of the antibody. Any combination of deletion, insertion, and substitution can be made to arrive at the final construct, provided that the final construct possesses the desired characteristics, e.g., antigen-binding.
Substitution, Insertion, and Deletion Variants
In certain embodiments, antibody variants having one or more amino acid substitutions are provided. Sites of interest for substitutional mutagenesis include the VRs and FRs. Conservative substitutions are shown in the Table below under the heading of "preferred substitutions." More substantial changes are provided in the Table below under the heading of "exemplary substitutions," and as further described below in reference to amino acid side chain classes. Amino acid substitutions may be introduced into an antibody of interest and the products screened for a desired activity, e.g., retained/improved antigen binding, decreased immunogenicity, or improved ADCC or CDC.


Amino acids may be grouped according to common side-chain properties:
(1) hydrophobic: Norleucine, Met, Ala, Val, Leu, Ile;
(2) neutral hydrophilic: Cys, Ser, Thr, Asn, Gin;
(3) acidic: Asp, Glu;
(4) basic: His, Lys, Arg;
(5) residues that influence chain orientation: Gly, Pro;
(6) aromatic: Trp, Tyr, Phe.
Non-conservative substitutions will entail exchanging a member of one of these classes for another class.
One type of substitutional variant involves substituting one or more hypervariable region residues of a parent antibody (e.g. a humanized or human antibody). Generally, the resulting variant(s) selected for further study will have modifications (e.g., improvements) in certain biological properties (e.g., increased affinity, reduced immunogenicity) relative to the parent antibody and/or will have substantially retained certain biological properties of the parent antibody. An exemplary substitutional variant is an affinity matured antibody, which may be conveniently generated, e.g., using phage display -based affinity maturation techniques such as those described herein. Briefly, one or more residues are mutated and the variant antibodies displayed on phage and screened for a particular biological activity (e.g. binding affinity).
Alterations (e.g., substitutions) may be made e.g., to improve antibody affinity. Such alterations may be made in "hotspots," i.e., residues encoded by codons that undergo mutation at high frequency during the somatic maturation process (see, e.g., Chowdhury, Methods Mol. Biol. 207: 179-196 (2008)), and/or SDRs (a-CDRs), with the resulting variant
VH or VL being tested for binding affinity. Affinity maturation by constructing and reselecting from secondary libraries has been described, e.g., in Hoogenboom et al. in Methods in Molecular Biology 178: 1-37 (O'Brien et al, ed., Human Press, Totowa, NJ, (2001).) In some embodiments of affinity maturation, diversity is introduced into the variable genes chosen for maturation by any of a variety of methods (e.g. , error-prone PCR, chain shuffling, or oligonucleotide-directed mutagenesis). A secondary library is then created. The library is then screened to identify any antibody variants with the desired affinity. Another method to introduce diversity involves directed approaches, in which several residues (e.g., 4- 6 residues at a time) are randomized. Residues involved in antigen binding may be specifically identified, e.g., using alanine scanning mutagenesis or modeling.
In certain embodiments, substitutions, insertions, or deletions may occur within one or more VRs so long as such alterations do not substantially reduce the ability of the antibody to bind antigen. For example, conservative alterations (e.g., conservative substitutions as provided herein) that do not substantially reduce binding affinity may be made in VRs. Such alterations may be outside of VR "hotspots" or SDRs. In certain embodiments of the variant VH and VL sequences provided above, each VR either is unaltered, or contains no more than one, two or three amino acid substitutions.
A useful method for identification of residues or regions of an antibody that may be targeted for mutagenesis is called "alanine scanning mutagenesis" as described by
Cunningham and Wells (1989) Science, 244: 1081-1085. In this method, a residue or group of target residues (e.g., charged residues such as arg, asp, his, lys, and glu) are identified and replaced by a neutral or negatively charged amino acid (e.g., alanine or polyalanine) to determine whether the interaction of the antibody with antigen is affected. Further substitutions may be introduced at the amino acid locations demonstrating functional sensitivity to the initial substitutions. Alternatively, or additionally, a crystal structure of an antigen-antibody complex is used to identify contact points between the antibody and antigen. Such contact residues and neighboring residues may be targeted or eliminated as candidates for substitution. Variants may be screened to determine whether they contain the desired properties.
Amino acid sequence insertions include amino- and/or carboxyl-terminal fusions ranging in length from one residue to polypeptides containing a hundred or more residues, as well as intrasequence insertions of single or multiple amino acid residues. Examples of terminal insertions include an antibody with an N-terminal methionyl residue. Other insertional variants of the antibody molecule include the fusion to the N- or C-terminus of the
antibody to an enzyme (e.g. for ADEPT) or a polypeptide which increases the serum half-life of the antibody.
Glycosylation variants
In certain embodiments, an antibody provided herein is altered to increase or decrease the extent to which the antibody is glycosylated. Addition or deletion of glycosylation sites to an antibody may be conveniently accomplished by altering the amino acid sequence such that one or more glycosylation sites is created or removed.
Where the antibody comprises an Fc region, the carbohydrate attached thereto may be altered. Native antibodies produced by mammalian cells typically comprise a branched, biantennary oligosaccharide that is generally attached by an N-linkage to Asn297 of the CH2 domain of the Fc region. See, e.g., Wright et al. TIBTECH 15:26-32 (1997). The
oligosaccharide may include various carbohydrates, e.g., mannose, N-acetyl glucosamine (GlcNAc), galactose, and sialic acid, as well as a fucose attached to a GlcNAc in the "stem" of the biantennary oligosaccharide structure. In some embodiments, modifications of the oligosaccharide in an antibody of the invention may be made in order to create antibody variants with certain improved properties.
In one embodiment, antibody variants are provided having a carbohydrate structure that lacks fucose attached (directly or indirectly) to an Fc region. For example, the amount of fucose in such antibody may be from 1% to 80%, from 1% to 65%, from 5% to 65% or from 20% to 40%. The amount of fucose is determined by calculating the average amount of fucose within the sugar chain at Asn297, relative to the sum of all glycostructures attached to Asn 297 (e. g. complex, hybrid and high mannose structures) as measured by MALDI-TOF mass spectrometry, as described in WO 2008/077546, for example. Asn297 refers to the asparagine residue located at about position 297 in the Fc region (Eu numbering of Fc region residues); however, Asn297 may also be located about ± 3 amino acids upstream or downstream of position 297, i.e., between positions 294 and 300, due to minor sequence variations in antibodies. Such fucosylation variants may have improved ADCC function. See, e.g., US Patent Publication Nos. US 2003/0157108 (Presta, L.); US 2004/0093621 (Kyowa Hakko Kogyo Co., Ltd). Examples of publications related to "defucosylated" or "fucose- deficient" antibody variants include: US 2003/0157108; WO 2000/61739; WO 2001/29246; US 2003/01 15614; US 2002/0164328; US 2004/0093621; US 2004/0132140; US
2004/01 10704; US 2004/01 10282; US 2004/0109865; WO 2003/0851 19; WO 2003/084570; WO 2005/035586; WO 2005/035778; WO2005/053742; WO2002/031140; Okazaki et al. J. Mol. Biol. 336: 1239-1249 (2004); Yamane-Ohnuki et al. Biotech. Bioeng. 87: 614 (2004).
Examples of cell lines capable of producing defucosylated antibodies include Led 3 CHO cells deficient in protein fucosylation (Ripka et al. Arch. Biochem. Biophys. 249:533-545 (1986); US Pat Appl No US 2003/0157108 Al, Presta, L; and WO 2004/056312 Al, Adams et al., especially at Example 11), and knockout cell lines, such as alpha- 1,6- fucosyltransferase gene, FUT8, knockout CHO cells (see, e.g., Yamane-Ohnuki et al.
Biotech. Bioeng. 87: 614 (2004); Kanda, Y. et al, Biotechnol. Bioeng., 94(4):680-688 (2006); and WO2003/085107).
Antibodies variants are further provided with bisected oligosaccharides, e.g., in which a biantennary oligosaccharide attached to the Fc region of the antibody is bisected by GlcNAc. Such antibody variants may have reduced fucosylation and/or improved ADCC function. Examples of such antibody variants are described, e.g., in WO 2003/01 1878 (Jean- Mairet et al.); US Patent No. 6,602,684 (Umana et al); and US 2005/0123546 (Umana et al.). Antibody variants with at least one galactose residue in the oligosaccharide attached to the Fc region are also provided. Such antibody variants may have improved CDC function. Such antibody variants are described, e.g., in WO 1997/30087 (Patel et al); WO 1998/58964 (Raju, S.); and WO 1999/22764 (Raju, S.).
Fc region variants
In certain embodiments, one or more amino acid modifications may be introduced into the Fc region of an antibody provided herein, thereby generating an Fc region variant. The Fc region variant may comprise a human Fc region sequence (e.g., a human IgGl, IgG2, IgG3 or IgG4 Fc region) comprising an amino acid modification (e.g. a substitution) at one or more amino acid positions.
In certain embodiments, the invention contemplates an antibody variant that possesses some but not all effector functions, which make it a desirable candidate for applications in which the half life of the antibody in vivo is important yet certain effector functions (such as complement and ADCC) are unnecessary or deleterious. In vitro and/or in vivo cytotoxicity assays can be conducted to confirm the reduction/depletion of CDC and/or ADCC activities. For example, Fc receptor (FcR) binding assays can be conducted to ensure that the antibody lacks FcyR binding (hence likely lacking ADCC activity), but retains FcRn binding ability. The primary cells for mediating ADCC, NK cells, express Fc(RIII only, whereas monocytes express Fc(RI, Fc(RII and Fc(RIII. FcR expression on hematopoietic cells is summarized in Table 3 on page 464 of Ravetch and Kinet, Annu. Rev. Immunol. 9:457-492 (1991). Non- limiting examples of in vitro assays to assess ADCC activity of a molecule of interest is
described in U.S. Patent No. 5,500,362 (see, e.g. Hellstrom, I. et al. Proc. Nat'l Acad. Sci.
USA 83 :7059-7063 (1986)) and Hellstrom, I et al, Proc. Nat'l Acad. Sci. USA 82: 1499-1502
(1985); 5,821,337 (see Bruggemann, M. et al, J. Exp. Med. 166: 1351-1361 (1987)).
Alternatively, non-radioactive assays methods may be employed (see, for example, ACTI™ non-radioactive cytotoxicity assay for flow cytometry (CellTechnology, Inc. Mountain View,
CA; and CytoTox 96® non-radioactive cytotoxicity assay (Promega, Madison, WI). Useful effector cells for such assays include peripheral blood mononuclear cells (PBMC) and
Natural Killer (NK) cells. Alternatively, or additionally, ADCC activity of the molecule of interest may be assessed in vivo, e.g., in a animal model such as that disclosed in Clynes et al. Proc. Nat'l Acad. Sci. USA 95:652-656 (1998). Clq binding assays may also be carried out to confirm that the antibody is unable to bind Clq and hence lacks CDC activity. See, e.g., Clq and C3c binding ELISA in WO 2006/029879 and WO 2005/100402. To assess complement activation, a CDC assay may be performed (see, for example, Gazzano-Santoro et al., J.
Immunol. Methods 202: 163 (1996); Cragg, M.S. et al, Blood 101 : 1045-1052 (2003); and Cragg, M.S. and M.J. Glennie, Blood 103 :2738-2743 (2004)). FcRn binding and in vivo clearance/half life determinations can also be performed using methods known in the art (see, e.g., Petkova, S.B. et al, Int'l. Immunol. 18(12): 1759-1769 (2006)).
Antibodies with reduced effector function include those with substitution of one or more of Fc region residues 238, 265, 269, 270, 297, 327 and 329 (U.S. Patent No. 6,737,056). Such Fc mutants include Fc mutants with substitutions at two or more of amino acid positions
265, 269, 270, 297 and 327, including the so-called "DANA" Fc mutant with substitution of residues 265 and 297 to alanine (US Patent No. 7,332,581).
Certain antibody variants with improved or diminished binding to FcRs are described.
(See, e.g., U.S. Patent No. 6,737,056; WO 2004/056312, and Shields et al, J. Biol. Chem. 9(2): 6591-6604 (2001).)
In certain embodiments, an antibody variant comprises an Fc region with one or more amino acid substitutions which improve ADCC, e.g., substitutions at positions 298, 333, and/or 334 of the Fc region (EU numbering of residues).
In some embodiments, alterations are made in the Fc region that result in altered (i.e., either improved or diminished) Clq binding and/or Complement Dependent Cytotoxicity
(CDC), e.g., as described in US Patent No. 6, 194,551, WO 99/51642, and Idusogie et al. J.
Immunol. 164: 4178-4184 (2000).
Antibodies with increased half lives and improved binding to the neonatal Fc receptor
(FcRn), which is responsible for the transfer of maternal IgGs to the fetus (Guyer et al, J.
Immunol. 1 17:587 (1976) and Kim et al, J. Immunol. 24:249 (1994)), are described in US2005/0014934A1 (Hinton et al). Those antibodies comprise an Fc region with one or more substitutions therein which improve binding of the Fc region to FcRn. Such Fc variants include those with substitutions at one or more of Fc region residues: 238, 256, 265, 272, 286, 303, 305, 307, 31 1, 312, 317, 340, 356, 360, 362, 376, 378, 380, 382, 413, 424 or 434, e.g., substitution of Fc region residue 434 (US Patent No. 7,371,826).
See also Duncan & Winter, Nature 322:738-40 (1988); U.S. Patent No. 5,648,260; U.S. Patent No. 5,624,821 ; and WO 94/29351 concerning other examples of Fc region variants.
Cysteine engineered antibody variants
In certain embodiments, it may be desirable to create cysteine engineered antibodies, e.g., "thioMAbs," in which one or more residues of an antibody are substituted with cysteine residues. In particular embodiments, the substituted residues occur at accessible sites of the antibody. By substituting those residues with cysteine, reactive thiol groups are thereby positioned at accessible sites of the antibody and may be used to conjugate the antibody to other moieties, such as drug moieties or linker-drug moieties, or to create an
immunoconjugate. Antibody Derivatives
In certain embodiments, an antibody provided herein may be further modified to contain additional nonproteinaceous moieties or non-antibody proteins that are known in the art and readily available. The moieties suitable for derivatization of the antibody include but are not limited to water soluble polymers. Non-limiting examples of water soluble polymers include, but are not limited to, polyethylene glycol (PEG), copolymers of ethylene glycol/propylene glycol, carboxymethylcellulose, dextran, polyvinyl alcohol, polyvinyl pyrrolidone, poly-1, 3-dioxolane, poly-l,3,6-trioxane, ethylene/maleic anhydride copolymer, polyaminoacids (either homopolymers or random copolymers), and dextran or poly(n- vinyl pyrrolidone)polyethylene glycol, propropylene glycol homopolymers, prolypropylene oxide/ethylene oxide co-polymers, polyoxyethylated polyols (e.g., glycerol), polyvinyl alcohol, and mixtures thereof. Polyethylene glycol propionaldehyde may have advantages in manufacturing due to its stability in water. The polymer may be of any molecular weight, and may be branched or unbranched. The number of polymers attached to the antibody may vary, and if more than one polymer are attached, they can be the same or different molecules. In
general, the number and/or type of polymers used for derivatization can be determined based on considerations including, but not limited to, the particular properties or functions of the antibody to be improved, whether the antibody derivative will be used in a therapy under defined conditions, etc.
In another embodiment, conjugates of an antibody and nonproteinaceous moiety that may be selectively heated by exposure to radiation are provided. In one embodiment, the nonproteinaceous moiety is a carbon nanotube (Kam et al, Proc. Natl. Acad. Sci. USA 102: 11600-11605 (2005)). The radiation may be of any wavelength, and includes, but is not limited to, wavelengths that do not harm ordinary cells, but which heat the nonproteinaceous moiety to a temperature at which cells proximal to the antibody -nonproteinaceous moiety are killed.
In some embodiments, antibodies or antibody fragments (e.g., antigen binding fragments) are fused or conjugated to human serum albumin (See e.g., U.S. Pat. No.
7,785,599 and 7,550,432). Albumin binds in vivo to the neonatal Fc receptor (FcRn) and this interaction is known to be important for the plasma half-life of albumin (Chaudhury et al 2003; Montoyo et al, 2009). FcRn is a membrane bound protein, and has been found to salvage albumin as well as IgG from intracellular degradation (Roopenian D. C. and Akilesh, S. (2007), Nat.Rev. Immunol 7, 715-725.). Thus, FcRn is a bifunctional molecule that contributes to the maintaining the high level of IgG and albumin in serum of mammals such as humans.
Human serum albumin (HSA) has been well characterised as a polypeptide of 585 amino acids, the sequence of which can be found in Peters, T., Jr. (1996) All about Albumin: Biochemistry, Genetics and Medical, Applications, Academic Press, Inc., Orlando. It has a characteristic binding to its receptor FcRn, where it binds at pH 6.0 but not at pH 7.4. The serum half-life of HSA has been found to be approximately 19 days. A natural variant having lower plasma half-life has been identified (Biochim Biophys Acta. 1991, 1097:49-54) having the substitution D494N. This substitution generated an N-glycosylation site in this variant, which is not present in the wild type HSA.
Albumin has a long serum half-life and because of this property it has been used for drug delivery. Albumin has been conjugated to pharmaceutically beneficial compounds
(WO0069902A), and it was found that conjugate had maintained the long plasma half-life of albumin so the resulting plasma half-life of the conjugate has generally been found to be considerably longer than the plasma half-life of the beneficial therapeutic compound alone.
Further, albumin has been fused to therapeutically beneficial peptides (WO 01/79271 A and WO 03/59934 A) with the typical result that the fusion has the activity of the therapeutically beneficial peptide and a long plasma half-life considerably longer than the plasma half-life of the therapeutically beneficial peptides alone.
Recombinant Methods and Compositions
Antibodies may be produced using recombinant methods and compositions, e.g., as described in U.S. Patent No. 4,816,567. In one embodiment, isolated nucleic acid encoding an anti-gliadin antibody described herein is provided. Such nucleic acid may encode an amino acid sequence comprising the VL and/or an amino acid sequence comprising the VH of the antibody (e.g., the light and/or heavy chains of the antibody). In a further embodiment, one or more vectors (e.g., expression vectors) comprising such nucleic acid are provided. In a further embodiment, a host cell comprising such nucleic acid is provided. In one such embodiment, a host cell comprises (e.g., has been transformed with): (1) a vector comprising a nucleic acid that encodes an amino acid sequence comprising the VL of the antibody and an amino acid sequence comprising the VH of the antibody, or (2) a first vector comprising a nucleic acid that encodes an amino acid sequence comprising the VL of the antibody and a second vector comprising a nucleic acid that encodes an amino acid sequence comprising the VH of the antibody. In one embodiment, the host cell is eukaryotic, e.g. a Chinese Hamster Ovary (CHO) cell or lymphoid cell (e.g., Y0, NS0, Sp20 cell). In one embodiment, a method of making an anti- gliadin antibody is provided, wherein the method comprises culturing a host cell comprising a nucleic acid encoding the antibody, as provided above, under conditions suitable for expression of the antibody, and optionally recovering the antibody from the host cell (or host cell culture medium).
For recombinant production of an anti- gliadin antibody, nucleic acid encoding an antibody, e.g., as described above, is isolated and inserted into one or more vectors for further cloning and/or expression in a host cell. Such nucleic acid may be readily isolated and sequenced using conventional procedures (e.g., by using oligonucleotide probes that are capable of binding specifically to genes encoding the heavy and light chains of the antibody).
Suitable host cells for cloning or expression of antibody-encoding vectors include prokaryotic or eukaryotic cells described herein. For example, antibodies may be produced in bacteria, in particular when glycosylation and Fc effector function are not needed. For expression of antibody fragments and polypeptides in bacteria, see, e.g., U.S. Patent Nos. 5,648,237, 5,789, 199, and 5,840,523. (See also Charlton, Methods in Molecular Biology, Vol.
248 (B.K.C. Lo, ed., Humana Press, Totowa, NJ, 2003), pp. 245-254, describing expression of antibody fragments in E. coli.) After expression, the antibody may be isolated from the bacterial cell paste in a soluble fraction and can be further purified.
In addition to prokaryotes, eukaryotic microbes such as filamentous fungi or yeast are suitable cloning or expression hosts for antibody-encoding vectors, including fungi and yeast strains whose glycosylation pathways have been "humanized," resulting in the production of an antibody with a partially or fully human glycosylation pattern. See Gerngross, Nat.
Biotech. 22: 1409-1414 (2004), and Li et al, Nat. Biotech. 24:210-215 (2006).
Suitable host cells for the expression of glycosylated antibody are also derived from multicellular organisms (invertebrates and vertebrates). Examples of invertebrate cells include plant and insect cells. Numerous baculoviral strains have been identified which may be used in conjunction with insect cells, particularly for transfection of Spodoptera frugiperda cells.
Plant cell cultures can also be utilized as hosts. See, e.g., US Patent Nos. 5,959,177, 6,040,498, 6,420,548, 7, 125,978, and 6,417,429 (describing PLANTIBODIES™ technology for producing antibodies in transgenic plants).
Vertebrate cells may also be used as hosts. For example, mammalian cell lines that are adapted to grow in suspension may be useful. Other examples of useful mammalian host cell lines are monkey kidney CVl line transformed by SV40 (COS-7); human embryonic kidney line (293 or 293 cells as described, e.g., in Graham et al, J. Gen Virol. 36:59 (1977)); baby hamster kidney cells (BHK); mouse Sertoli cells (TM4 cells as described, e.g., in Mather, Biol. Reprod. 23 :243-251 (1980)); monkey kidney cells (CVl); African green monkey kidney cells (VERO-76); human cervical carcinoma cells (HELA); canine kidney cells (MDCK; buffalo rat liver cells (BRL 3A); human lung cells (W138); human liver cells (Hep G2); mouse mammary tumor (MMT 060562); TRI cells, as described, e.g., in Mather et al, Annals N Y. Acad. Sci. 383 :44-68 (1982); MRC 5 cells; and FS4 cells. Other useful mammalian host cell lines include Chinese hamster ovary (CHO) cells, including DHFR" CHO cells (Urlaub et al, Proc. Natl. Acad. Sci. USA 77:4216 (1980)); and myeloma cell lines such as Y0, NS0 and Sp2/0. For a review of certain mammalian host cell lines suitable for antibody production, see, e.g., Yazaki and Wu, Methods in Molecular Biology, Vol. 248 (B.K.C. Lo, ed., Humana Press, Totowa, NJ), pp. 255-268 (2003).
Assays
Anti- gliadin antibodies provided herein may be identified, screened for, or characterized for their physical/chemical properties and/or biological activities by various assays known in the art. In some embodiments, the experiments described in Example 1 are utilized to screen antibodies for activity.
In one aspect, an antibody of the invention is tested for its antigen binding activity, e.g., by known methods such as ELISA, BIACore®, FACS, or Western blot.
In another aspect, competition assays may be used to identify an antibody that competes with any of the antibodies described herein for binding to gliadin. In certain embodiments, such a competing antibody binds to the same epitope (e.g., a linear or a conformational epitope) that is bound by an antibody described herein. Detailed exemplary methods for mapping an epitope to which an antibody binds are provided in Morris (1996) "Epitope Mapping Protocols," in Methods in Molecular Biology vol. 66 (Humana Press, Totowa, NJ).
In an exemplary competition assay, immobilized gliadin is incubated in a solution comprising a first labeled antibody that binds to gliadin (e.g., any of the antibodies described herein) and a second unlabeled antibody that is being tested for its ability to compete with the first antibody for binding to gliadin. The second antibody may be present in a hybridoma supernatant. As a control, immobilized gliadin is incubated in a solution comprising the first labeled antibody but not the second unlabeled antibody. After incubation under conditions permissive for binding of the first antibody to gliadin, excess unbound antibody is removed, and the amount of label associated with immobilized gliadin is measured. If the amount of label associated with immobilized gliadin is substantially reduced in the test sample relative to the control sample, then that indicates that the second antibody is competing with the first antibody for binding to gliadin. See Harlow and Lane (1988) Antibodies: A Laboratory Manual ch.14 (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY).
II. Diagnostic and Research Methods and Compositions
Embodiments of the present disclosure provide methods and uses diagnosing celiac disease (e.g., using the antibodies described herein). For example, in some embodiments, a sample from a subject suspected of having celiac disease (e.g., exhibiting one or more symptoms of celiac disease) or during routine screening (e.g., newborn screening) is screened for anit-gliadin antibodies using a method described herein.
In some embodiments, gliadin-specific autoantibodies are detected using a competitive immunoassay where the binding of a gliadin-specific to a peptide of the corresponding epitope is measured in the presence of serum antibodies in a competitive assay. In some embodiments, the assay is an ELISA assay.
An ELISA, short for Enzyme-Linked Immunosorbent Assay, is a biochemical technique to detect the presence of an antibody or an antigen in a sample. It utilizes a minimum of two antibodies, one of which is specific to the antigen and the other of which is coupled to an enzyme. The second antibody will cause a chromogenic or fluorogenic substrate to produce a signal. Variations of ELISA include sandwich ELISA, competitive ELISA, and ELISPOT. Because the ELISA can be performed to evaluate either the presence of antigen or the presence of antibody in a sample, it is a useful tool both for determining serum antibody concentrations and also for detecting the presence of antigen.
In some embodiments, the assay is performed on a solid support (e.g., bead based) or in a well based assay. In such assay, both serum IgG and IgA are screened at the same time. In some embodiments the peptide is, for example, PLQPEQPFP (SEQ ID NO: 3),
PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4), or a combination thereof.
In some embodiments, a solid support plate or bead is conjugated with either a gliadin peptide or a gliadin-specific monoclonal antibody. Test or control serum, along with either a gliadin peptide or a gliadin-specific monoclonal antibody (e.g., that was not conjugated to the solid support) is contacted with the solid support.
In some embodiments, non-CD control serum is spiked with titrated concentrations of one or more gliadin-specific monoclonal antibodies and used as reference serum (e.g., to obtain absolute concentrations ^g/ml) for test samples).
Binding is detected using any suitable method (e.g., using a labeled peptide or antibody and appropriate detection reagent).
Embodiments of the present disclosure further provide research uses (e.g., to study celiac diseae, develop assays for the diagnosis or screening of celiac disease, or develop assays for detection of gluten contamination in food products).
III. Commerical Applications
In some embodiments, the antibodies described herein find use in detection of gluten in food or food products (e.g., gluten contamination from manufacturing or gluten levels naturally found in the food or food product).
For example, in some embodiments, a sample of a food product is contacting with one or more of the monoclonal antibodies described herein. Binding is detected using any suitable method (e.g., using a labeled gliadin-specific antibody or a labeled secondary antibody). IV. Articles of Manufacture
In another aspect of the invention, an article of manufacture (e.g., kit or composition) containing materials (e.g. monoclonal antibodies specific for gliadin) useful in the diagnostic, research, screening, or commercial applications described herein is provided. The article of manufacture comprises a container and a label or package insert on or associated with the container. The containers may be formed from a variety of materials such as glass or plastic. In some embodiments, kits further comprise one or more additional reagents (e.g. buffers, solid supports, controls, etc.) useful in performing immunoassays.
EXPERIMENTAL
The following example is provided in order to demonstrate and further illustrate certain preferred embodiments and aspects of the present invention and are not to be construed as limiting the scope thereof.
EXAMPLE 1
Methods
Subjects. Small intestinal biopsy specimens were obtained from subjects by esophagogastroduodenoscopy and forceps sampling from the duodenum. UCD patients were referred to endoscopy based on clinical suspicion and positive serologic test, and diagnosed according to the guidelines of the American Gastroenterological Association Institute
(Kagnoff MF. Gastroenterology 131, 1977-1980 (2006)). These subjects were still on a gluten-containing diet at the time when biopsies were obtained, e.g. when the disease was in its active chronic stage. Non-CD controls were appointed to endoscopy, where
histopathology and serology negated CD diagnosis. The non-CD control group consisted of
seven HLA-DQ2.5+ subjects, three HLA-DQ8+ subjects and four subjects who were not HLA-typed. All subjects have given written informed consent. The study was approved by Regional Committees for Medical Research Ethics South East Norway (S-97201).
Single cell suspensions (SCSs). Intestinal biopsies were collected and transported in RPMI 1640 in 50 ml tubes on ice. For preparation of SCSs (Di Niro et al, J Immunol 185, 5377-5383 (2010)), the biopsies were transferred to 15 ml tube and re-suspended in 2 mM EDTA in 2% fetal calf serum (FCS) in PBS. After 30 minutes rotation at 37°C, the supernatant was discarded and the biopsies re-suspended in lmg/ml blend collagenase (Sigma, C8051) and 50 μg/ml DNase (Sigma, DN25) in 2% FCS in Dulbecco's PBS. The biopsies were then incubated under constant rotation at 37°C. After 30 minutes the biopsies were mechanically disrupted with a syringe equipped with a large steel needle. After another 30 minutes constant rotation, a smaller needle was used for the same procedure. After 1-2 hours, the single cell suspension was filtered through 40 μΜ filter into 50 ml tube and centrifuged at 470 g for 7 minutes.
Human intestinal stromal cell line (fibroblasts). Human fibroblast cell lines were derived from small intestinal biopsies as previously described (Roncoroni L, et al. J Transl Med 7, 40 (2009)). Biopsies were transferred to flat-bottomed 6-well plates and gently disrupted with a scalpel for 15-30 seconds. The biopsies were cultured in 1%
penicillin/streptomycin in 10% FCS in RPMI 1640 (culture medium) at 37°C in 5% C02. Medium was changed every second week. After 5-10 weeks, a dense layer of cells attached to the bottom was detectable in the wells. These cells were detached with 0.05% trypsin-EDTA (Gibco, 25300-054) and transferred to 25 ml culture flasks. Culture medium was changed regularly. In these flasks, the cell lines typically survived for 6-9 months. One of these cell lines, Fl 100 from subject UCD1 100, was used in most of the experiments.
In vitro co-culture of SCSs and fibroblasts. First, fibroblasts were detached from 25 ml culture flasks and transferred to plates with flat-bottomed wells. After one week, cells from SCSs were seeded on confluent layer of fibroblasts. Fibroblasts and SCSs were incubated in culture medium at 37°C in 5% CO2. Different plate formats were suited to different experimental settings; 24-well plates were used for BrdU assays and estimation of total IgA production and 384 well plates were used of single PC cultures.
Extraction, enzymatic digestion and deamidation of gliadin proteins. Gliadin, the alcohol soluble component of gluten, was extracted from wheat flour. Wheat flour (M∅llerens
550001) 50g was dissolved in 150 ml butanol, vortex mixed and centrifuged at 163 g for 5 minutes. The butanol was decanted and the procedure repeated. The wheat flour pellet was
dissolved in 350 ml 70% ethanol and incubated at RT overnight under constant stir mixing. The next day, the solution was centrifuged at 650 g for 5 minutes, and the supernatant was mixed with 1.5 M NaCl in ratio 1 :2 and incubated at 4°C for 4 hours to precipitate the gliadin proteins. After centrifugation at 25,000 g for 20 minutes the supernatant was decanted and the gliadin pellet dissolved in 40 ml 8 M urea in 0.01 M ammonium bicarbonate. This solution was diluted 1 :4 to give a final urea concentration of 2 M and incubated with 12-24 mg chymotrypsin (CT) overnight at 37°C under constant stir mixing. The next day, chymotrypsin was heat inactivated at 98°C for 5 minutes, and the solution was dialyzed (Spectra/Por® Membrane MWCO 1,000) overnight and dried in speed vacuum concentrator. The digested gliadin was dissolved and incubated in acetic acid pH 1.8 at 95°C for 1 hour to introduce Q to E conversion (deamidation). The final product, heat/acid treated chymotrypsin digested gliadin (CT-gliadin for short), was diluted in distilled H20 and freeze dried before further usage.
ELISA supernatant IgA reactive with CT-gliadin. ELISA plates (96 well Nunc 436014) were coated with 75 μΐ/well of CT-gliadin 40 μg/ml in carbonate buffer 0.05M pH 9.6 over night at 4°C, washed and subsequently blocked with 0.5% bovine serum albumin (BSA) in PBS and incubated with supernatant from single PC cultures. Anti-human IgA- alkaline phosphatase (Sigma, A9669) 1 :3000 was used as secondary antibody. Anti-human IgG-alkaline phosphatase (Southern Biotech, 2040-04) at concentration 1 :4000 was used when testing gliadin-reactive hmAbs. Plates were developed for approximately 15 min with phosphatase substrate (Sigma-Aldrich) and absorbance was measured at 405 nm.
Isolation of PCs after in vitro culture. Cells from culture wells were transferred to PCR plate and centrifuged at 3750 g for 5 minutes. Supernatant was decanted, and RNAse- inhibiting RT-PCR catch buffer (Smith K, et al. Nat Protoc 4, 372-384 (2009)) was added, 10 μΐ per well. PCR plate was sealed and stored at -70°C until single cell PCR preparation.
Staining antigen-specific plasma cells from SCSs. Two different biotinylated synthetic gliadin peptides were used to stain gliadin-specific PCs from SCSs. Biotin- GSGSGS-PLQPEQPFP (SEQ ID NO: 17) and biotin-GSGSGS-
LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18) were produced by GL Biochem (Shanghai). TG2 was expressed in Sf9 insect cells by Phadia and linked to biotin with Sulfo-NHS-LC -biotin (Pierce) as per the manufacturer's protocol.
The synthetic peptides were incubated on ice with APC-labeled streptavidin
(PhycoLink, PJ27S) and biotinylated TG2 incubated with PE-labeled streptavidin
(Invitrogen, S866) at 4: 1 molar ratio, in the dark for 1 hour. The final tetramer concentration
for staining intestinal PCs was 40nM in staining buffer containing 2% FCS in PBS. PE CD138 1 :40 (eBioscience, DL-101), PE-Cy7 CD27 1 :50 (eBioscience, LG.7F9), Pacific Blue CD 19 1 : 100 (BioLegend, HIB19), PerCP CD3 1 :40 (BD Biosciences, SK7), PerCP
GLIADIN (SEQ ID NO: 19) 1 :40 (BD Biosciences, ΜφΡ9), FITC goat anti-human IgA 1 :800 (Southern Biotech, 2050-02) were used for staining of SCS. Propidium iodide for exclusion of dead cells was added just before analysis. Three different flow cytometer instruments were used: Facs Aria, LSRII and Fortessa. The plots (Fig. 1, 3, 9 and 10) are from LSRII. Plasma cells appeared as one homogeneous population of large, CD4" and GLIADIN" (SEQ ID NO: 19) events, co-expressing CD 138 and CD27, and were previously identified as antibody producing cells (Di Niro R et al. J Immunol 185, 5377-5383 (2010)). CD27 stained with higher intensity and gave a more defined population than CD 138. Thus, PCs were defined as large, viable, CD27+CD3 "GLIADIN" (SEQ ID NO: 19) events.
Cloning and expression of human monoclonal antibodies (hmAbs). The variable regions of the heavy and light chain antibody genes of isolated PCs were amplified by RT- PCR and nested PCR, cloned into expression vectors and transfected into a human cell line as IgGl according to previously established protocol (Smith K, et al. Nat Protoc 4, 372-384 (2009)).
AlphaLISA screening of hmAbs for reactivity to gluten peptides. AlphaLISA Acceptor beads (Perkin Elmer, 6772001) were coupled with polyclonal rabbit anti-human IgG (Dako A0423) and stored at a concentration of 2.5 mg/ml according to the
manufacturer's instructions (as gliadin-specific hmAbs were produced in human IgGl format). Anti-IgG AlphaLISA donor bead solution (1 :400) and gliadin-specific hmAbs 1 μg/ml in AlphaLISA immunoassay buffer (Perkin Elmer, ALOOOC) were incubated for 1 hour at 4°C in the dark. After incubation, 15 μΐ were transferred to each well in 384 well plates and mixed with 5 μΐ analyte. The plates were incubated for 1 hour at RT in the dark. AlphaScreen streptavidin donor bead solution (Perkin Elmer, 6760002B) was diluted 1 :200 in AlphaLISA Immunoassay buffer, and 15 μΐ were added per well before incubation at RT in the dark for 30 minutes. The different peptides used in the analyte were the following: biotin-GSGSGS- PLQPEQPFP (SEQ ID NO: 3), PLQPQQPFP (SEQ ID NO: 8), PLQPEQPFP (SEQ ID NO: 3), GIIQPEQPAQL (SEQ ID NO: 20), LQLQPFPQPQLPYPQPQLPYPQPQLPYPQPQPF (SEQ ID NO: 21), FPQPQQPEQSFP (SEQ ID NO: 22), PEQPQQSFPEQERP (SEQ ID NO: 23), LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7),
LQQPLSQQPEETF (SEQ ID NO: 24) and biotin-GSGSGS-
LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18) were purchased from GL Biochem Ltd, Research Genetics, Neosystems or obtained from Burkhard Fleckenstein.
AlphaLISA Acceptor beads were also coupled to equal amounts of the two gliadin- specific hmAbs UCD1114 1F03 and UCD1143 3B02 and stored at concentration 2.5mg/ml. These beads were used to detect biotin-GSGSGS-PLQPEQPFP (SEQ ID NO: 17) in the presence of serum (Fig 7). First, 5ul biotin-GSGSGS-PLQPEQPFP (SEQ ID NO: 17) was incubated with 10 μΐ serum at RT for 30 minutes. Then, 30 μΐ of mixture of AlphaLISA acceptor bead solution (1 :600) and Alphascreen donor bead solution (1 :300) in AlphaLISA immunoassay buffer, was added per well. The plate was read after second incubation for 45 minutes at RT in the dark. In pilot experiments several gliadin-specific hmAbs were tested and gave similar results. The hmAbs UCD 11 14 1F03 and UCD1143 3B02 were chosen for the final experiments as they were among the commonly used VH/VL pairs VH3-23/VL4-69 and VH3-15/VK4-1, respectively.
AlphaLISA screening of hmAbs for gliadin specificity and polyreactivity. Anti-IgG AlphaLISA donor bead solution (1 :400) and gliadin-specific hmAbs 1 μg/ml in AlphaLISA immunoassay buffer were incubated in 1.5 ml tube for 1 hour at RT in the dark before 15 μΐ was transferred to each well in 384 well plate. Then 5 μΐ analyte was added per well, and the plate subsequently incubated for 30 minutes at RT in the dark. The analyte consisted of fixed concentration of 40 nM biotin-GSGSGS-PLQPEQPFP (SEQ ID NO: 17) and titrations of either CT-gliadin, lysate of EBV-transfected B cells or a mixture of LPS (Sigma L-4391), CpG, recombinant TG2 (Phadia) and recombinant Jo-1 antigen (Phadia). After 45 minutes incubation at RT in the dark, AlphaScreen streptavidin donor bead solution was diluted 1 :200 in AlphaLISA Immunoassay buffer and 15 μΐ added per well. The plate was read after 30 minutes incubation at RT in the dark.
Results
Intestinal PCs secreting IgA reactive with heat/acid treated chymotrypsin digested (CT) gliadin
In vitro culture was an important step for isolation of PCs producing antibodies reactive with heat/acid treated chymotrypsin digested gliadin (hereafter termed CT-gliadin for short). Three conditions of PC cultures were compared. Single cell suspensions (SCSs) from intestinal biopsies were generated, and PCs were cultured either as SCSs, as SCSs in co- culture with human intestinal fibroblasts or as PCs isolated by flow cytometry in co-culture with fibroblasts. The concentration of IgA in supernatants of cultures with SCSs together
with fibroblasts increased with a constant rate over at least 4 weeks (Fig. la), indicating that the majority of PCs survived in these cultures. No increase of IgA in supematants of cultures of SCSs alone (Fig. la) or flow cytometry sorted PCs cultured with fibroblasts (Fig. lb) was observed. In co-cultures of SCSs and fibroblasts, it was observed that intestinal PCs survived for weeks, and viable CD19+CD27+ PCs were detected by flow cytometry after 4 weeks of culture (Fig. lc). PCs did not proliferate in these cultures, as tested in a BrdU incorporation assay. Based on these findings, the system of co-culturing SCSs and fibroblasts was selected for further experiments.
The specificity of antibodies produced at a single PC level were analyzed. Based on the frequency of IgA+ PCs in SCSs that was determined by flow cytometry (mean 3.3%, range 1.0%-5.3%, n = 16) (Fig. 9a-b), 10-20 cells were seeded per well to obtain cultures with less than one IgA+ PC per well. Supematants of single wells were screened for IgA reactivity to CT-gliadin and TG2. Supematants containing IgA reactive to TG2 were three to four times more frequent than IgA reactive to CT-gliadin in cultures from subjects with UCD (Fig. 2a, Fig 2b). IgA reactivity to TG2 and CT-gliadin was not detected in supematants from cultures of non-celiac controls (Fig. 2c) or supematants of cultures containing only fibroblasts (Fig. 2d). Thus, PCs secreting IgA reactive to CT-gliadin are present in small intestinal biopsies of subjects with UCD and are less frequent than PCs secreting TG2- reactive IgA.
Next, the antibody genes of PCs from cultures in which IgA reactivity to CT-gliadin in the supernatant were cloned and expressed. In the SCSs, the large PCs hugely
outnumbered small resting B cells (Fig. 9c-d). If one well of SCSs contained just one IgA+ PC, there was a high probability that this was the only cell expressing the IgA encoding genes in that well. Where ELISA results indicated gliadin-specific PCs to be present (Fig. 2a), the cells were split into four PCR wells and processed individually to increase the likelihood of having only one PC in each well. Cells were then washed in PBS before snap frozen in DNase containing buffer and subjected to expression cloning of antibodies in a human IgGl format as previously described (Smith K, et al. Nat Protoc 4, 372-384 (2009). Human monoclonal antibodies (hmAbs) were successfully produced from more than half of cultures processed. Of a total of 19 hmAbs produced, nine were reactive to CT-gliadin (Fig. 3a).
These were tested for reactivity to BSA and CpG, of which one was found reactive to BSA (Fig. 3b). Thus, eight hmAbs were considered to be gliadin specific and included in further assays.
Intestinal IgA+ PC stained with synthetic gliadin peptides in flow cytometry
Antigen-specific IgA+ PCs were visualized previously by staining with biotinylated antigen bound to fluorescent streptavidin taking advantage of surface IgA and IgM expression of gut PCs (Di Niro R, et al. Nat Med 18, 441-445 (2012), Di Niro R, et al. J Immunol 185, 5377-5383 (2010)). The same strategy was used to identify gliadin-specific IgA+ PCs by flow cytometry, using fluorescent streptavidin to form tetramer complexes with biotinylated synthetic peptides. Two different gliadin peptides with sequences targeted by serum IgA antibodies of CD patients, biotin-(GSGSGS)-PLQPEQPFP (SEQ ID NO: 17) (Osman AA, et al. Clin Exp Immunol 121, 248-254 (2000)) and biotin-(PEG)- LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7) (Bateman EA, et al, Gut 53, 1274-1278 (2004)), were used as antigen. The two peptides represent sequences of gliadin with Q to E transitions in positions targeted by TG2 (Dorum S, et al. PLoS One 5, el4056 (2010)). Both peptides are reported to bind antibodies from UCD patients, primarily in their deamidated versions (Aleanzi et al, Clin Chem 47, 2023-2028 (2001)). PLQPEQPFP (SEQ ID NO: 3) in particular has demonstrated high sensitivity and specificity for CD in serologic assays (Schwertz E, et al. Clin Chem 50, 2370-2375 (2004)). This approach using synthetic peptides, introduced a restricted selection of gliadin-specific PCs compared to screening culture supernatants for IgA reactivity against CT-gliadin.
In flow cytometry the mean percentage of IgA+ PCs stained with the PLQPEQPFP (SEQ ID NO: 3) peptide tetramers was 1.0% (range 0.3-2.6%, n = 10) of total IgA+ PCs in SCSs generated from biopsies taken from UCD patients (Fig. 4a, Fig 4b). In comparison, the mean percentage in non-CD disease controls was 0.2% (range 0-0.3%) (Fig. 4a, Fig 4b). For IgA+ PCs stained with the deamidated 33-mer peptide tetramers, the mean percentage was 0.5% (range 0.4-0.9%, n = 6) for subjects with UCD and 0.1% in controls (range 0-0.3%, n = 4). Background levels were defined by staining IgA+ PCs with fluorescent streptavidin alone (mean 0.3%, range 0.2-0.4%, n = 4) (Fig. 4b).
IgA+ PCs stained with synthetic gliadin peptides and TG2 appeared as two separate populations in flow cytometry whereas double positive cells were not detected (Fig. 4c). Also, the populations visualized with TG2 were larger than the populations stained with both the PLQPEQPFP (SEQ ID NO: 3) and the deamidated 33-mer peptides (Fig. 4d). These findings are consistent with the ratio of the frequencies of IgA+ PCs reactive with TG2 versus those reactive with CT-gliadin observed in culture (Fig. 2b). The predominance of TG2+ PCs relative to the population stained with gliadin peptides was greater for the IgA+ PCs than for the IgM+ PCs (Fig. 4d). Based on these experiments it was concluded that there are two
distinct populations of gliadin-specific and TG2-specific IgA PCs in the small intestinal lesions of UCD patients.
IgA+ PCs stained with fluorescent streptavidin in complex with either PLQPEQPFP (SEQ ID NO: 3) or deamidated 33-mer were isolated as single cells by flow cytometry for further expression cloning. The hmAbs obtained were tested for reactivity to the peptide which they were originally isolated with. Seventeen of 23 hmAbs from PCs isolated with the PLQPEQPFP (SEQ ID NO: 3) peptide, and 13 of 16 hmAbs from IgA+ PCs isolated with the deamidated 33-mer peptide were reactive in ELISA to the peptide originally used in sorting. This indicates that the staining specificity of gluten-peptide tetramers was approximately 75- 80%.
Gliadin-reactive hmAbs: specificity and affinity to deamidated and native gliadin
The eight hmAbs that originated from in vitro PCs cultures reactive with CT-gliadin were tested in ELISA against the two synthetic peptides. Five reacted with PLQPEQPFP (Fig. 3c) and one reacted with deamidated 33-mer. Hence, six out of eight hmAbs were reactive to PLQPEQPFP (SEQ ID NO: 3) and/or deamidated 33-mer. Since cloning of antibody genes from those cultures is unbiased with respect to specific epitope, this result indicated that the epitopes represented by PLQPEQPFP (SEQ ID NO: 3) and the deamidated 33-mer are recognized by IgA of a significant part of gliadin-specific PCs.
The specificity of all hmAbs reactive to one of the two synthetic peptides in ELISA was further characterized in AlphaLISA. In contrast to ELISA, where synthetic peptide was used as a coating antigen, the AlphaLISA format detected monovalent binding of hmAbs to soluble biotinylated synthetic peptide. Three of the hmAbs (UCD 1002 1D03, UCD 1143 1E01, UCD1130 4A04), originally from IgA+ PC sorted with tetramers of PLQPEQPFP (SEQ ID NO: 3) and reactive to PLQPEQPFP (SEQ ID NO: 3) in ELISA, were not reactive in AlphaLISA. One of the hmAbs (UCD1 130 3B04) from IgA+ PC sorted with 33-mer and reactive to deamidated 33-mer in ELISA was not reactive in AlphaLISA. These four antibodies, not reactive in AlphaLISA, gave the lowest signals in the peptide-based ELISA and with signals that were lower than to the CT-gliadin antigen (Fig. 10), indicating that the synthetic peptides may not represent the complete epitope(s) of these four antibodies.
Polyreactivity was assessed by using cell lysate as well as a mixture of Jo-1, TG2, CpG and LPS as competing antigens. In either case, the binding of hmAbs to the synthetic peptides was maintained (Fig. 5a). In contrast, the signal was blocked by CT-gliadin (Fig.
5a). It was concluded that the hmAbs reactive to PLQPEQPFP (SEQ ID NO: 3) or deamidated 33-mer are specific to gliadin and not poly -reactive.
The reactivity of hmAbs to deamidated versus native gliadin was tested in a competitive AlphaLISA assay. Binding of hmAbs to biotinylated synthetic deamidated gliadin peptide (either PLQPEQPFP (SEQ ID NO: 3) or the deamidated 33-mer) was tested against the homologous peptide in either deamidated or native (non-deamidated) versions. Gliadin-specific hmAbs divided into two groups, either only reactive to deamidated gliadin or reactive with both deamidated and native gliadin (Fig. 5b). None of the hmAbs had higher reactivity to native than to deamidated gliadin. Because some of the hmAbs were not reactive to the native gliadin peptide, this indicates that induction of gliadin-specific B cells in vivo likely happen in the presence of deamidated gliadin.
Gliadin-specific hmAbs: cross-reactivity and repetitive sequences in the gliadin proteome
It was observed that some of the hmAbs were reactive with both PLQPEQPFP and the deamidated 33-mer in AlphaLISA (Fig. 6a). Eight of the hmAbs originally from IgA+ PC sorted with tetramers of PLQPEQPFP (SEQ ID NO: 3) were reactive to deamidated 33-mer (Table 1). Of the hmAbs from IgA+ PC sorted with deamidated 33-mer, eleven were reactive to PLQPEQPFP (SEQ ID NO: 3) (Table 1). This indicates that at least some of the gliadin- specific PCs secrete antibodies that cross-react with different gliadin peptides. To test this further, it was investigated whether a few selected synthetic gliadin peptides could compete with the antibody binding to biotinylated PLQPEQPFP. The hmAbs (UCD1143 3B02 and UCD1002 1B06) were tested and both were reactive with PLQPEQPFP (SEQ ID NO: 3) but not PLQPQQPFP (SEQ ID NO: 8) (Table 1). For both hmAbs the signals were inhibited by peptides containing QPEQ or PEQP. Peptides without these sequences were non-inhibitory (Fig. 6b). Thus, the cross-reactivity of hmAbs to different gliadin peptides is due to sharing of key sequences. Reactivity of many hmAbs to PLQPEQPFP (SEQ ID NO: 3) and deamidated 33-mer, both harboring the QPEXP (SEQ ID NO: 25) (X = Q, L) and QPFP (SEQ ID NO: 26) sequences, is consistent with this notion.
Gliadin-specific hmAbs binding to PLQPEQPFP is blocked by patient sera
Assayes were performed to determine if binding of gliadin-specific hmAbs to synthetic gluten peptide was blocked by sera from patients with active CD. Sera were incubated with biotinylated PLQPEQPFP (SEQ ID NO: 3) before incubation with AlpaLISA
beads conjugated with two hmAbs UCD1 1 14 1F03 and UCD1143 3B02. Sera from patients with CD blocked the signal, while sera from control subjects did not (Fig. 7). This indicated that gliadin-specific antibodies from serum and IgA of gliadin-specific PCs in the lamina propria are specific to the same gliadin B-cell epitopes.
Repertoire of gliadin-specific IgA+ PCs
The eight hmAbs from in vitro cultured PCs with CT-gliadin were obtained from six subjects (UCD 1050, 1 ; UCD1130, 1; UCD1065, 2; UCD 1186, 2; UCD1163, 1; UCD1030, 1). The 30 hmAbs derived from single sorted PCs using tetramerized synthetic peptides, originated from five different subjects (UCD1002, 7; UCD1079, 2; UCD 11 14, 2; UCD1 130, 16; UCD1143, 3). Control IgA+ PCs, defined as PLQPEQPFP-negative (SEQ ID NO: 3), 33- mer-negative and TG2 -negative (UCD1130, 55; UCD1 143, 54) and TG2-specific PCs (UCD1030, 3; UCD11 14, 4; UCD1010, 2), were isolated by flow cytometry and their antibody genes were cloned and sequenced. The variable regions of the hmAbs were analyzed using tools of the IMGT webpage. Two combinations of VH/VL pairing were dominant (Fig. 8a); VH3-15/VK4-1 (seven hmAbs from five different subjects in total) and VH3-23/VL4-69 (15 hmAbs from seven different subjects in total). The VH3-23/VL4-69 combination was found in hmAbs from IgA+ PCs isolated with both CT-gliadin and synthetic peptides. In addition, VH3-23/VK3-11 was also found in hmAbs generated by both methods. Together, these three combinations made up approximately 75% of the panel of gliadin- specific hmAbs (Fig. 8a, Table 1). Similarity between heavy chain CDR3 sequences of the VH3-15/VK4-1 as well as the VH3-23/VL4-69 hmAbs was investigated. No obvious similarities could be found with variability in D- and J-gene segment usage and CDR3-length among the antibodies of each group (Table 1). The nucleotide sequences of the variable regions of heavy and light chains of all hmAbs are summarized in Table 2.
VH/VL usage and epitope specificity
VH/VL usage (Table 1) was compared with peptide reactivity pattern characterized in AlphaLISA. All VH3-23/VL4-69 hmAbs showed reactivity to both native and deamidated gliadin, demonstrated by inhibiting effect of both the native PLQPQQPFP and the deamidated PLQPEQPFP (SEQ ID NO: 3). The VH3-15/VK4-1 hmAbs were specific to deamidated gliadin, as the native PLQPQQPFP (SEQ ID NO: 8) showed no inhibitory effect on the binding to PLQPEQPFP (SEQ ID NO: 3). VH3-23/VK3-1 1 hmAbs were the only antibodies with reactivity to deamidated 33-mer but not PLQPEQPFP (SEQ ID NO: 3). Two
hmAbs had reactivity to CT-gliadin but neither to PLQPEQPFP (SEQ ID NO: 3) nor to deamidated 33-mer. These two antibodies both used VH3-15/VK3-20. Taken together, this indicated that there is a correlation between VH/VL usage and epitope specificity of the gliadin-specific antibodies.
Somatic hypermutation within gliadin-specific IgA+ PCs
The mutation rates in the hmAbs from PCs secreting IgA reactive with CT-gliadin (median 8.7, range 3-13, n = 8) as well as IgA+ PCs isolated with synthetic gliadin peptides (median 7.5, range 0-23, n = 30) were lower than in the control PCs (median 15, range 0-37, n = 109) (Fig. 8b). TG2-specific IgA+ PCs were scarcely mutated as previously described (Di Niro et al, Nat Med 18, 441-445 (2012)) (median 4, range 0-15, n = 9).
Different VH genes are associated with various levels of SHM (Wang M, et al, J Exp Med 207, 141-153 (2010)). The mutation rate in gliadin-specific IgA with VH3-23 was significantly lower than the number of mutations observed in VH3-23 from control population (Fig. 8c). It was concluded that the low mutation rate in gliadin-specific IgA is not a result of selected VH-usage.
It was previously reported that IgA antibodies of TG2-specific PCs found in celiac lesions have limited SHM. In this study, gluten (gliadin) reactive antibodies expression cloned from IgA+ PCs isolated from small intestinal biopsies were studied and it was found that there is also a limited mutation rate in these antibodies, demonstrating an unexpected common feature of the IgA antibody responses to gluten and TG2 in CD.
The number of mutations in antibody genes relates to the type of B-cell response elicited (Goodnow CC, et al, Nat Immunol 11, 681-688 (2010)). T-cell independent B-cell responses have no or little SHM with little restriction of VH usage as often seen in T-cell dependent responses (Maizels N, Bothwell A. Cell 43, 715-720 (1985)). T-cell dependent B- cell responses that develop in germinal centers (GCs) typically result in highly mutated antibodies (MacLennan IC. Annu Rev Immunol 12, 1 17-139 (1994)). GC reactions may also happen without involvement of T cells, but then with low level of SHM (Toellner KM, et al. J Exp Med 195, 383-389 (2002)). Low level of SHM is also seen in T-cell dependent extrafollicular responses (MacLennan IC, et al. Immunol Rev 194, 8-18 (2003)) and in GC responses of short duration (Takahashi Y, et al, J Exp Med 187, 885-895 (1998)).
For the TG2-specific IgA antibodies, the low mutation rate was contemplated to relate to enzymatic activity of B-cell receptor bound TG2 (Di Niro et al, Nat Med 18, 441-445
(2012)) or to early termination of auto-reactive B cells in GCs or self-antigen driving an
extrafollicular response, similar to what has been reported in other systems (Goodnow CC, et al, Nat Immunol 11, 681-688 (2010); Chan TD, et al. Immunity 37, 893-904 (2012); Herlands RA, et al, Eur J Immunol 37, 3339-3351 (2007)). None of these mechanisms should apply to IgA antibodies of gluten specific PCs, therefore these PCs should have similar level of SHM compared to control PCs. This is not what was observed. The mutation rate in IgA of gluten- specific PCs was low and similar to TG2-specific PCs. This indicates that the factor(s) causing the limited SHM in IgA of TG2-specific PCs is likely involved in both antibody responses.
The parallel fluctuation of antibodies to gluten and TG2 in response to dietary gluten (Sulkanen S, et al. Gastroenterology 115, 1322-1328 (1998)) indicates that the production of these antibodies is regulated in a coordinated way. In keeping with this notion, it has been contemplated that gluten-specific T cells could provide help to TG2-specific B cells by means of complexes gluten-TG2 acting as hapten-carrier complexes (Maki M. In:
Proceedings of the sixth international symposium coeliac disease (eds Feighery C, O'Farrelly C). 246-252 (Oak Tree Press, 1992); Sollid LM, et al, Gut 41, 851-852 (1997)). This model explains why the TG2 antibodies are produced upon gluten exposure and it why only individuals who express HLA-DQ2 or HLA-DQ8 make these antibodies (Bjorck et al, J Pediatr Gastroenterol Nutr 50, 49-53 (2010 )). Gluten specific T-cells also could provide help to gluten specific B-cells. As gluten specific T cells preferentially recognize deamidated gluten peptides (Sollid et al, Immunogenetics 64, 455-460 (2012)), B cells with surface immunoglobulin that bind and internalize deamidated gluten peptides would be better situated to receive T-cell help. Common sequences of the two gliadin peptides used to isolate IgA+ PCs are found in or adjacent to many T-cell epitopes in gluten proteins. The QPEXP (X = Q, L) sequence is found in the DQ2.5-glia-ala/b, DQ2.5-glia-a2, DQ2.5-glia-y2, DQ2.5- glia-y3, DQ2.5-glia-y4c/d, DQ2.5-glia-rol, DQ2.5-glia-ro2, DQ2.5-hor-l, DQ2.5-hor-2,
DQ2.5-sec-l, DQ2.5-sec-2 and DQ8-glia-yla/b epitopes whereas the QPFP sequence is found in the DQ2.5-glia-y4c, DQ2.5-glia-y5, DQ2.5-glia-ro2, DQ2.5-hor-2, DQ2.5-sec-2 and DQ8- glia-yla epitopes (Sollid et al, Immunogenetics 64, 455-460 (2012))). The low mutation rate in TG2- and gluten-specific PCs compared to other intestinal PCs, indicates that a common factor distinguishing the TG2- and gluten-specific PCs from most other gut PCs could be implicated. Based on the model referred above, both gluten and gluten-reactive T cells could be such common factors.
Gluten is a protein antigen, which in contrast to most antigens of the gut has no bacterial or viral origin. Lack of strong concomitant innate signals along with B-cell receptor triggering could result in extrafollicular response, in accordance with a study of the B-cell response to a T-cell dependent antigen, where immunization without adjuvant was insufficient for GC formation (Chappell CP, et al., J Exp Med 209, 1825-1840 (2012)).
Extrafollicular responses typically give rise to short-lived plasma cells and rapid decline in serum antibodies after the immune response in addition to low SHM (Ho F, et a.l, Eur J Immunol 16, 1297-1301 (1986)). TG2-specific PCs were rarely detected in small intestine of CD subjects on a gluten-free diet (Di Niro et al, Nat Med 18, 441-445 (2012)), and levels of serum IgG and IgA antibodies to both gluten and TG2 typically fall below detection level months after the patient commence a gluten-free diet (Sulkanen S, et al.
Gastroenterology 115, 1322-1328 (1998).). These clinical observations, in addition to the observations of limited SHM in gluten-specific and TG2-specific IgA+ PCs, argue for an extrafollicular origin of gluten and TG2-specific antibodies in CD.
T cells influence extrafollicular development of B cells. It has been reported that T cells located at the T-B border in lymph nodes, phenotypically different from GC T cells, are necessary for B-cell priming to extrafollicular antibody responses (Lee et al, J Exp Med 208, 1377-1388 (2011)). Sustained CD40 signaling during B-cell and T-cell interaction has been demonstrated to induce a plasma cell fait rather than GC B-cell development (Sciammas R, et al, Immunity 25, 225-236 (2006).), and administration of a CD40 agonistic antibody was shown to ablate GC reaction and induced a pattern of extrafollicular B-cell differentiation (Erickson LD, et al. J Clin Invest 109, 613-620 (2002)).
Not only the quality, but also the degree of T-cell help is a relevant factor. Excessive numbers of follicular helper T cells as seen in several mouse models, like the 'san' mutation in the RNA binding protein Roquin (Yu et al, Nature 450, 299-303 (2007)) and PD- 1 deficiency (Kawamoto S, et al. Science 336, 485-489 (2012)), drive survival of GC B cells with low degree of mutation and affinity and cause increased autoreactivity (Pratama A, Vinuesa CG. Immunol Cell Biol OOl 10.1038/icb.2013.69, (2013)). Gluten-reactive T cells of CD patients produce large amounts of interferon-γ (Nilsen et al, Gut 37, 766-776 (1995)) and in mice excess of interferon-γ has been demonstrated to promote accumulation of both follicular helper T cells and GC B cells and to trigger autoimmunity (Lee et al, Immunity 37, 880-892 (2012)). Thus, TG2- and gluten-reactive B cells in such a setting may develop in GC and not extrafollicularly.
Characterization of the specificity of antibodies produced by PCs reactive with gluten gives valuable insights into the pathogenesis in CD. The panel of hmAbs was characterized by AlphaLISA assays. First, it was found that some antibodies are specific to deamidated gliadin peptides and do not recognize non-deamidated counterparts. Gliadin-specific B cells must have encountered deamidated gliadin. Second, it was found that gliadin-specific antibodies frequently cross-react with different gliadin peptides. Accordingly, gliadin-specific B cells may take up and display several different T-cell epitopes. This may be beneficial if a B cell receive help from several different T-cell clones, as recently described (Shulman Z, et al. Science 341, 673-677 (2013)).
In addition to the limited SHM, a restricted VH/VL usage in the panel of gluten- specific antibodies was observed. Limited VH usage is a sign of T-cell dependent responses (Maizels N, Bothwell A. Cell 43, 715-720 (1985)). The same VH/VL pairings appear in different subjects. This phenomenon is similarly seen in TG2-specific antibodies where VH restriction is observed in different patients. The VH usage of TG2-specific antibodies was recently demonstrated to correlate with epitope specificity (Iversen R, et al, J Immunol 190, 5981-5991 (2013)). This was also observed for the gliadin-specific hmAbs. Antibodies specific to deamidated variants of gluten peptides had different VH/VL usage than antibodies with reactivity to both the deamidated and native variants. Such VH/VL restriction has been reported in antibodies specific for influenza (Lingwood D, et al. Nature 489, 566-570 (2012)) and HIV (Wu et al, Science 333, 1593-1602 (201 1)) antigens. For HIV antibodies, there has even been reported an allelic preference for antibody usage (Wu et al, Science 333, 1593- 1602 (201 1)). Restricted VH/VL usage and limited SHM in antibodies as observed in CD favor influence of VH and/or VL polymorphisms in shaping the antibody response. This also supports involvement of VH and VL genes as susceptibility loci in CD, even though these genes have not been indicated by recent genome wide association studies in humans (Trynka G, et al. Nat Genet 43, 1 193-1201 (2011)). In mice, experiments with congenic strains demonstrated that the antibody response to gliadin is chiefly controlled by the MHC and the immunoglobulin heavy chain loci (Kagnoff MF. Nature 296, 158-160 (1982)).
Table 1 : Overview of the monoclonal antibodies from human gliadin-specific
IgA.
Donor subject (patient number) and isolation method (sorted by flow cytometry with selecting antigen PLQPEQPFP (SEQ ID NO: 3) or deamidated 33-mer or isolated after in vitro culture with CT-gliadin as selecting antigen) are indicated as well as are reactivity to antigens, VH and VL usage and number of mutations. H/A: heat/acid
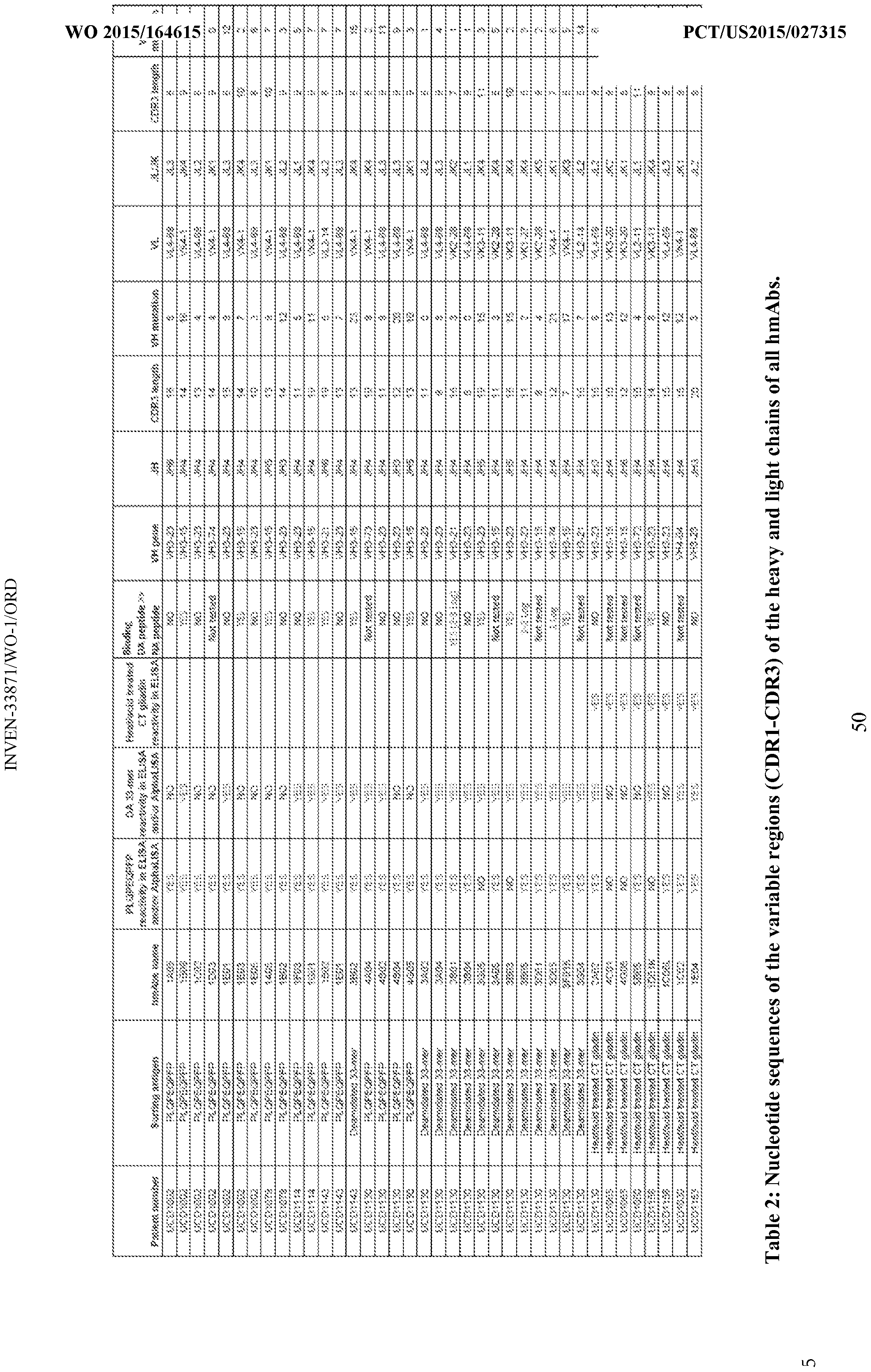

INVEN-33871/WO-l/ORD

INVEN-33871/WO-l/ORD

INVEN-33871/WO-l/ORD

INVEN-33871/WO-l/ORD

INVEN-33871/WO-l/ORD

EXAMPLE 2
Materials and Methods
ELISA reactivity of gliadin-specific hmAbs to H/A-gliadin and synthetic gliadin peptides. H/A-gliadin (40 μg/ml) and biotinylated synthetic gliadin peptides (500 nM) were used as coating antigens in ELISA plates (Nunc, 442404) and streptavidin coated ELISA plates (Nunc, 436014), respectively. Gliadin-specific hmAbs at concentration 2 μg/ml and 1 :4 titrations, were used to generate dilution curves. Alkaline phosphatase conjugated anti-human IgG (Southern Biotech 9040-04) in 1 :4000 dilution was used as the detecting antibody and visualized with phosphatase substrate (Sigma S0942-200TAB) reactivity measured at 405 nm. The synthetic gliadin peptides were produced with biotinylated C-terminal spacer (GSGSGS), by GL Biochem. PBS pH7.2 was used as buffer for antigens, hmAbs and detecting antibody.
ELISA reactivity of serum IgG to H/A-gliadin and
PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4). Sera, diluted 1 :300 in PBS, was tested in ELISA plates coated with either H/A-gliadin or biotinylated
PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4) in parallel. Serum IgG reactivity was detected as described above. The synthetic peptide PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4) was produced with C-terminal biotin but no spacer, by GL Biochem.
Affinity estimates of gliadin-specific hmAbs to PLQPEQPFP (SEQ ID NO: 3) and 33-mer in AlphaLISA. AlphaLISA Acceptor beads (Perkin Elmer, 6772001) were conjugated with anti-human IgG (Dako, A0423) according to the manufacturers' instructions, and stored at concentration 2.5mg/ml. The anti-IgG bead solution was diluted 1 :400 together with 0.5 μg/ml gliadin-specific hmAb in AlphaLISA (AL) buffer (Perking Elmer, AL000C) in 1.5 ml tube, and incubated for 1 hour at 4°C in the dark. Subsequently, 20 μΐ was transferred per well in 384 Optiplates (Perkin Elmer, 6007290) together with 5 μΐ of 40 nM biotinylated synthetic gliadin peptide (PLQPEQPFP (SEQ ID NO: 3) or deamidated 33-mer with biotinylated GSGSGS spacer) and titrations of non-biotinylated PLQPEQPFP (SEQ ID NO: 3) or deamidated 33-mer in AL buffer, and incubated for 1 hour at RT in the dark. Alphascreen Streptavidin Donor beads (Perking Elmer, 6760002B) 5 mg/ml solution was diluted 1 :200 in AL buffer and 20 μΐ was transferred to each well. The plate was left for incubation at RT for 45 minutes in the dark and then analyzed with Envision 2014 Multilabel Reader (Perkin Elmer).
Serological AlphaLISA inhibition assays. AlphaLISA Acceptor beads were conjugated either with PLQPEQPFP (SEQ ID NO: 3) or with gliadin-specific hmAbs (1002- 1E01 and 1002-10E3 or 1 130-3B04), according to manufacturers' instructions, and stored at 2.5 mg/ml concentration. In one assay (Figure 12B and Figure 13), biotinylated
PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4) was dissolved to 25nM in AL buffer with protease inhibitor (Sigma, 1 1714600), and 20 μΐ was transferred to each well and incubated together with 5 μΐ serum for 1 hour at 4°C. Solution of AL beads conjugated with gliadin-specific hmAbs was diluted 1 :250, 10 μΐ was added per well, and plate was left for incubation at RT for 30 minutes in the dark. Alphascreen streptavidin bead solution was diluted 1 :250, 20 μΐ was added per well, and the plate was incubated at RT for 45 minutes in the dark before it was analyzed with Envision Multilabel Reader. Positive control, using the same concentrations of anti-FLAG IgG (Sigma, F3165) conjugated AL beads and
biotinylated FLAG peptide (Biotin-GSGSGS-DYKDDDDK (SEQ ID NO: 253)), was run in parallel for each serum sample. The same was followed when testing 1 130-3B04 conjugated beads.
In the other assay (Figure 12A), PLQPEQPFP (SEQ ID NO: 3) AL bead solution was diluted 1 :300 in AL buffer, and 20 μΐ transferred each well together with 5 μΐ serum. The plate was incubated at RT for 1 hour at RT in the dark. Gliadin-specific hmAb (1002-1E03) that was conjugated with NHS-Biotin (Thermo, 21335) according to manufacturers' instruction, was diluted 50 ng/ml in AL buffer, and 5 μΐ was transferred to each well.
Alphascreen streptavidin bead solution was diluted 1 : 150 in AL buffer, and 20 μΐ was transferred each well. The plate was incubated for 30 minutes at RT in the dark, and then analyzed with Envision 2014 Multilabel reader.
Interpolated concentrations of gliadin-specific serum antibodies and statistical analysis. Reference serum (serum of non-CD control subject with known concentrations of gliadin-specific hmAbs) was run in all assays to generate a reference curve. The
concentrations of gliadin-specific serum antibodies were estimated using reference serum and Sigmoidal dose-response (variable slope) function in GraphPad Prism 5 (GraphPad Software Inc.). Linear regression analyses and column statistics were calculated using Graphpad Prism 5 (Graphpad Software, Inc.).
Results
Previous studies of gliadin B-cell epitopes in CD have focused on serum antibody reactivity to synthetic peptides (Osman, A.A., et al, Clin Exp Immunol, 2000. 121(2): p. 248-
54; Bateman, E.A., et al, Gut, 2004. 53(9): p. 1274-8; Aleanzi, M., et al, Clin Chem, 2001. 47(1 1): p. 2023-8; Ballew, J.T., et al, Proc Natl Acad Sci U S A, 2013. 1 10(48): p. 19330- 19335; Vallejo-Diez, S., et al, PLoS One, 2013. 8(1 1): p. e80982). A method based on gliadin-specific hmAbs binding to wheat gliadins was developed.
Eleven gliadin-specific hmAbs were included in the study; eight hmAbs of IgA+ PCs isolated by flow cytometry, where two different synthetic gliadin peptides were used as sorting peptide (Biotin-GSGSGS-PLQPEQPFP (SEQ ID NO: 17), PLQPEQPFP (SEQ ID NO: 3) for short; biotin-GSGSGS-LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18), deamidated 33-mer for short) and three hmAbs of in vitro cultured PCs secreting IgA reactive to H/A-gliadin (Table 3). All hmAbs were produced with the same IgGl constant region. In brief, gliadin proteins were digested with the enzymes PTCEC (pepsin, trypsin, chymotrypin, elastase, carboxipeptidase), separated into size fractions by gel filtration, and enzymatically deamidated by transglutaminase 2 (for short: TG2-gliadin), and gliadin-specific hmAbs were used as a matrix to enrich high affinity TG2-gliadin peptides, followed by peptide identification by mass spectrometry (MS). To control for potential unspecific binding of gliadin peptides using this approach, a rotavirus-specific hmAb (Di Niro, R., et al, J Immunol, 2010. 185(9): p. 5377-83) was used as negative control hmAb.
Several TG2-gliadin peptides were identified by MS after enrichment with nine of eleven gliadin-specific hmAbs (median = 40, range 6-215) (Table 3). For two of the hmAbs (1065-4C01, 1065-4G05), no TG2-gliadin peptides were identified, and these two hmAbs were excluded from further analyses. No TG2-gliadin peptides were identified after enrichment with the rotavirus-specific hmAbs. Typically, many of the TG2-gliadin peptides identified after enrichment with a gliadin-specific hmAb, contained similar motifs.
Based on these results and the assumed cross-reactivity to similar sequences, a peptide comprising the predicted epitopes of seven hmAbs and similar to the predicted epitopes of two hmAbs (1130-3B03, 1130-3G05) was synthesized (Biotin- PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4)). Even though some hmAbs had higher avidity to other peptides, this was the only tested peptide of which all nine hmAbs were reactive to in ELISA (Table 4).
Gliadin-specific serum antibody inhibition assay in serologic screening
At present, most serological tests for gliadin-specific serum antibodies are based on serum IgG or IgA reactivity to synthetic deamidated gliadin peptides (DGP) in ELISA. This method monitored binding of gliadin-specific hmAb to peptide of the corresponding epitope
in the presence of serum antibodies in a competitive bead-based AlphaLISA assay. In such assay, both serum IgG and IgA are screened at the same time. Two assays were tested, either using beads conjugated with PLQPEQPFP and biotinylated hmAb 1002-1E01, bead conjugated with hmAb 1130-3B04 and biotinylated PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4), or bead conjugated with hmAbs 1002-1E01 and 1002-1E03 and biotinylated PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4).
Non-CD control serum was spiked with titrated concentrations of two gliadin-specific hmAbs (11 14-1F03 and 1130-3B02) and used as reference serum, such that absolute concentrations ^g/ml) could be estimated for test samples. These two hmAbs both showed high reactivity to PLQPEQPFP (SEQ ID NO: 3) and were also reactive to
PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4).
The assay with PLQPEQPFP conjugated beads and biotinylated hmAb 1002-1E01, was first compared to ELISA using streptavidin coated plates, biotinylated PLQPEQPFP and anti-human IgG as detecting antibody. Testing the reference serum, the change in signal from 0 M to 5xl0"9 M gliadin-specific hmAbs (1 114-1F03 and 1130-3B02), was 50% in the competitive AlphaLISA assay and 20% in ELISA (Figure 1 1). This shows that the competitive AlphaLISA assay was better at detecting low concentration of gliadin-specific antibodies.
Next, the PLQPEQPFP (SEQ ID NO: 3) assay was tested with sera (n = 66 anonymous donors with unknown disease history and Marsh score) that were previously screened in ELISA with QUANTA Lite Gliadin IgG II ELISA and Varelisa Celikey TG2 IgA (commercial kits for detection of TG2- and gliadin-antibodies for diagnosing CD). The results were compared to the results of the reference serum, and absolute concentrations of gliadin-specific serum antibodies were calculated for each serum (Figure 12A). Gliadin- specific antibodies were not detected by AlphaLISA competitive assay in any of the sera
(mean 0.2 μg/ml, 95% CI 0.05-0.3 μg/ml, n = 16) that had been negative in both Celikey TG2 (cutoff < 5) and QUANTA Gliadin (cutoff < 20). Of sera screened positive in both ELISAs, all but one were above 0.3 μg/ml (mean 28 μg/ml, range 0-64 μg/ml, n = 44). Some sera were screened positive with Celikey TG2 but negative with QUANTA Gliadin, and most of these were positive in the inhibition assay (mean 15 μg/ml, range 0.3-39 μg/ml, n = 6). In summary, the inhibition assay results correlated well with QUANTA Gliadin and Celikey TG2. The exception was sera screened positive in Celikey TG2 but negative in QUANTA Gliadin, where the inhibition assay correlated better with Celikey TG2.
The same comparison was done for testing the inhibition assay with beads conjugated with two hmAbs (1002-1E01 and 1002-1E03) and biotin-
PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4), by testing sera (n = 80 anonymous donors with unknown disease history and Marsh score) that had been screened with Celikey TG2 and QUANTA Gliadin (Figure 10B). Based on results from sera screened negative in both ELISA assays (95% CI 0.1-0.7 μg/ml, range 0-3 μg/ml, n = 29), cutoff was set at <4 μg/ml. Of sera screened positive in both ELISAs, all but one were above 0 μg/ml (mean 52 μg/ml, range 0-93 μg/ml, n = 47). Most sera screened positive in Celikey TG2 but negative in QUANTA Gliadin were positive in the inhibition assay (mean 12 μg/ml, range 0-20 μg/ml, n = 4).
Next, 1002-1E01 and 1002-1E03 conjugated beads and biotin- PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4) to screen sera from patients (n = 53), where Marsh score and disease history was available. The same cutoff < 4 μg/ml was used here. All patients except one (dyspepsia) were referred to endoscopy based on suspected CD and were on a gluten-containing diet. Serum drawn on the same day as endoscopy was tested for anti-TG2 IgA (Celikey TG2) and for gliadin-specific antibodies using the inhibition assay. The patients were grouped based on screening results (TG2 IgA +/-) and histology evaluation (Marsh score, the gold standard for diagnosing CD) (Table 5).
Of patients with Marsh 0 (mean 1.9 μg/ml, 95% CI of mean 0.4-3.4 μg/ml, median 0 μg/ml, n = 15), one was TG2-IgA positive (CD 1 177) and two were positive in the inhibition assay (CD1177 and CD1220). One patient (CD1 177) was positive in both assays, and the second patient (CD1222) had borderline histology for Marsh 1 (>25 IEL per 100 epithelial cells). Four patients were Marsh 1 and TG2-IgA negative, and one (CD 1225: 6 μg/ml) of these four were positive in the inhibition assay (median 0, n = 4). Gliadin-specific antibodies were detected in all 34 sera from patients with Marsh score 2 or 3 (Figure 11), while TG2 IgA titers were below cutoff in seven of 34 patients. Thus, the gliadin inhibition assay was more sensitive (100%) than the Varelisa TG2 IgA ELISA (80%).
Sera from the same patients (n = 53) were assayed with 1130-3B04 conjugated beads. One Marsh 0 patient were above 4 μg/ml cutoff (CD 1177), and 32 of 34 Marsh 2 and 3 were above cutoff (mean = 14 μg/ml, range 0-38 μg/ml, n = 34) (Table 5). Also this assay was more sensitive (93%) was more sensitive than the Varelisa TG2 IgA ELISA.
This study describes a new serological assay for gliadin-specific serum antibodies in
CD patients. The epitope specificity of hmAbs cloned from the antibody gene of single gliadin-specific IgA+ PCs isolated from intestinal CD lesions was investigated. The gliadin-
specific hmAbs and the characterized epitopes were used in a competitive assay to detect gliadin-specific serum antibodies. The assay was tested and compared to anti-TG2 IgA ELISA using sera from 55 patients undergoing endoscopy. None of the patients were previously diagnosed with CD. Using Marsh score as gold standard, the competitive anti- gliadin antibody test had the same specificity but better sensitivity than the anti-TG2 IgA ELISA test. Only the competitive anti-gliadin antibody test was positive in all patients with Marsh 2 and Marsh 3 (Figure 13). Only six of the patients with Marsh 3 had anti-TG2 IgA titer ten times above cutoff, gliadin-specific serum antibodies were detected in all these six (Table 4).
There has been reported high prevalence of undiagnosed CD (Rubio-Tapia, A., et al,
Am J Gastroenterol, 2012. 107(10): p. 1538-44; quiz 1537, 1545). The results show that many of the CD patients with false negative anti-TG2 IgA would be identified with an initial combined screening.
In Europe, the diagnostic criteria for CD in children have changed the last years. According to ESPHGAN guidelines, CD can now be diagnosed without endoscopy in children with anti-TG2 IgA titers more than ten times above cutoff (Husby, S., et al, J Pediatr Gastroenterol Nutr, 2012. 54(1): p. 136-60). These guidelines have been met with some concerns because of the variation in performance of anti-TG2 IgA kits from different manufacturers (Egner, W., et al, J Pediatr Gastroenterol Nutr, 2012. 55(6): p. 733-5), and because false positive high-titer anti-TG2 IgA results do occur (Swallow, K., et al, Clin Exp Immunol, 2013. 171(1): p. 100-6). Additionally, only half of the CD patients with Marsh 3 have been reported to have high anti-TG2 IgA titers (Hill, P.G. and G.K. Holmes, Aliment Pharmacol Ther, 2008. 27(7): p. 572-7; Zanini, B., et al, Dig Liver Dis, 2012. 44(4): p. 280- 5). To increase the positive predictive value (PPV), positive EMA test is included in the ESPHGAN criteria (Husby, S., et al, J Pediatr Gastroenterol Nutr, 2012. 54(1): p. 136-60). EMA testing is highly operator-dependent, because the test is difficult to do and to interpret (Health Quality, O., Ont Health Technol Assess Ser, 2010. 10(21): p. 1-11 1; Rostom, A., J.A. Murray, and M.F. Kagnoff, Gastroenterology, 2006. 131(6): p. 1981-2002). Thus, a better alternative to high-titer anti-TG2 IgA is to use double positive anti-gliadin and anti-TG2 antibody test results as criteria regardless of anti-TG2 IgA titer, which has been reported to have high PPV (Brusca, I., et al, Clin Chem Lab Med, 2012. 50(1): p. 11 1-7; Anderson, R.P., et al, BMC Med, 2013. 11(1): p. 188) and is less time consuming and operator-dependent.
INVEN-33871/WO- 1/ORD
Table 3: Overview of the gliadin-specific hmAbs used in the study.
The first column shows the names of the hmAbs. The selecting antigens used in the isolation of the gliadin-specific IgA+ PCs are indicated in the second column. Column 3-6 is an overview of results that together were used to assess the best binding motifs of each hmAb. These motifs were not always in accordance with the selecting antigens. Last two columns describe the VH and VL usage of the hmAbs.
INVEN-33871/WO-l/ORD
Table 4: ELISA reactivity of the gliadin-specific hmAbs to synthetic gliadin peptides grouped as positive, low or negative.

INVEN-33871/WO-l/ORD
Table 5: Serum samples (n = 53) tested and compared to Marsh score and anti-TG2 IgA titer (Celikey TG2 IgA).
The cutoff is <5 for anti-TG2 IgA, and <20 for DGP (Quanta Gliadin IgG). **For some patients, HLA genotype was not available.


EXAMPLE 3
Gluten-free diet is the sole treatment in CD patients, but gluten contamination in gluten-free food products is common (Koerner, T.B., et al., Gluten contamination of naturally gluten-free flours and starches used by Canadians with celiac disease. Food Addit Contam Part A Chem Anal Control Expo Risk Assess, 2013. 30(12): p. 2017-21). Today, gluten in food is mainly detected using monoclonal mouse antibodies (mAbs) (Haraszi, R., et al., Analytical methods for detection of gluten in food— method developments in support of food labeling legislation. J AOAC Int, 2011. 94(4): p. 1006-25). Different mAbs are used in commercial assays, and they have been generated by immunizing mice with different antigens. The 401.21 mAb was developed against gliadin extract of Australian bread wheat cultivar Timgalen (Skerritt, J.H., et al., Journal of Cereal Science, 1984. 2(4): p. 215-224), PN3 mAb against the synthetic 19-mer LGQQQPFPPQQPYPQPQPF (SEQ ID NO: 254) (Ellis, H.J., et al., Gut, 1998. 43(2): p. 190-5.), R5 against a-secalin extract (Sorell, L., et al., FEBS Lett, 1998. 439(1-2): p. 46-50), and HYB 314-01 mAb against QPFPQPQLPYPQPQ (SEQ ID NO: 255) (Skovbjerg, H., et al., Biochim Biophys Acta, 2004. 1690(3): p. 220-30). It has been reported that mAb 401.21 primarly reacts with HMW-glutenin subunits and to small degree with a- and γ-gliadin, PN3 reacts with a-gliadins (van Eckert, R., et al., Journal of Cereal Science, 2010. 51(2): p. 198-204), the epitopes of R5 and HYB 314-01 are QQPFP (SEQ ID NO: 256) (Osman, A.A., et al., Eur J Gastroenterol Hepatol, 2001. 13(10): p. 1189-93) and PELPYPQPQ (SEQ ID NO: 257) (Petersen, N.H., et al., J Immunol Methods, 2011. 365(1-2): p. 174-82), respectively. The epitopes of the hmAbs cloned from human IgA+ PCs were QPX1QPFP (SEQ ID NO: 1) (Xi = Q or E), PEQ-dependency, PEX1X2X3P (SEQ ID NO: 258) (Xi= L, Q; X2 = Q, P; X3 = S, F, Y), and PQPELPYPQP (SEQ ID NO: 12) (Table 2, Table 3, Figure 14, Figure 15). Hence, the epitopes of these mouse and human mAbs are different. Since human gliadin-specific IgA+ PCs involved in the disease process, hmAbs cloned from these cells recognize immunogenetic gliadin epitopes relevant for CD, in contrast to mAbs generated in mice.
The use of the hmAbs described herein for application in detection of gluten in food stuffs was thus evaluated. Gliadin was extracted from wheat flour and treated with heat/acid treated and tested in a competitive assay with synthetic gliadin peptide and gliadin-specific hmAb as previously described. In addition, AlphaLISA donor beads (Perkin Elmer) were conjugated with PLQPEQPFP according to manufacturers' instructions and stored at concentration 2.5 mg/ml. Gliadin-specific hmAb 1002-lEOl is not dependent on deamidation
of gliadin, and will bind to both deamidated and native gliadin. The hmAb 1002-1E01 was biotinylated with NHS-biotin (Thermo) according to manufacturers' instructions. Wheat was dissolved 1 mg/ml in buffer and incubated with 0.5 μg/ml of biotinylated hmAb 1002-1E01 at room temperature (RT) on shaker 1400 rpm for 1 hour. PLQPEQPFP (SEQ ID NO: 3) bead solution was diluted 1 :400 in AL buffer, 15 μΐ was transferred to each well in a 384 well plate and incubated together with 5 μΐ of the analyte of wheat and biotinylated hmAb 1002- 1E01 for 45 minutes at RT in the dark. AlphaScreen streptavidin donor bead solution was diluted 1 :200 in AL buffer, 15 μΐ was transferred to each well, and the plate was incubated for 30 minutes at RT in the dark, and then analyzed. As expected, the wheat-containing product strongly inhibited the AlphaLISA signal, compared to two gluten-free products, which exhibited robust signals (Figure 16). This demonstrates use the hmAbs for detection of gluten in food.
EXAMPLE 4
To better identify the preferred epitopes of the human monoclonal antibodies
(hmAbs) identified in Example 1, the hmAbs were used in pull-down experiments with peptides of enzymatic digests of wheat gliadins treated with TG2. Results demonstrated that most of the hmAbs preferred binding to long, TG2-deamidated gliadin peptides with several copies of specific motifs. A gliadin-specific hmAb and such target peptides identified by pull- down were used in a serologic competition assay. The principle was to measure binding of gliadin-specific hmAb to target peptide, where inhibition of this binding indicated presence of gliadin-specific antibodies in serum.
Materials and Methods
Patients and controls
Celiac disease patients (n = 106, mean age 38 years, range 17-72, 69 females and 37 males) with biopsy confirmed diagnosis were enrolled at Oslo University Hospital - Rikshospitalet. The Marsh scores were the following: Marsh 3A: n=17; Marsh 3B n=40; Marsh 3C: n=49. All patients were positive for either HLA-DQ2 and/or HLA-DQ8. Serum was sampled at the day of endoscopy. Exclusion criterion was documented gluten-free diet prior to the endoscopy. None of the enrolled patients had a previous diagnosis of celiac disease. Informed consent for participation was obtained from all patients.
Blood donors (n = 198, mean age 41 years, range 20-60 years, 132 females and 66 males) were recruited from Oslo University Hospital - Ulleval aiming to match the sex and
age distribution of the celiac disease group. Donors with known celiac disease were excluded from the study. Serum and blood samples were de-identified after collection according to the protocol of the ethical approval. For sera giving positive serologic test result(s), the respective blood samples were used for genotyping.
Crohn's disease patients (n = 151, mean age 38 years, range 18-82 years, 66 females and 85 males) under anti-TNF treatment were recruited from Oslo University Hospital - Rikshospitalet.
The protocols were approved by Regional Committees for Medical Research Ethics in South East Norway (ethical approvals 2013/1352, 2014/432 and S-97201).
Peptide pull-down, mass spectrometry and database search
The gliadin specific hmAb 1002-1E03 (Example 1) was used to pull down peptides from pepsin, trypsin, chymotrypsin, elastase, and carboxypeptidase digested gliadin treated with TG2. Details of the procedure are described in Example 5. In brief, the hmAb (40 μg) was incubated with a gel filtration fraction of TG2 -treated gliadin for 20 min at room temperature (RT), followed by 20 min incubation at RT with Protein G Dynabeads (Novex, Life Technologies). The antibody-enriched peptides were eluted with 0.1% TFA for 10 min at RT. A rotavirus specific hmAb (Rota-2B04) was used as negative control. The samples were analyzed on a Dionex Ultimate 3000 nano-LC system (Dionex, Sunnyvale, CA, USA) connected to a quadrupole-Orbitrap (QExactive) mass spectrometer (ThermoElectron, Bremen, Germany). The peptides were separated on a 250 mm EASY-SPRAY column (CI 8, 75 μιη ID, 2 μιη particles) using a 60 min linear 5-50% ACN gradient in 0.1% formic acid at a flow rate of 0.3 μΐ/min. The QExactive data was acquired using a data-dependent top 10 method and the LC-MS/MS data were searched in a T. Aestivum database as described in Example 5.
ELISA peptide reactivity of gliadin-specific hmAbs
Biotinylated synthetic gliadin peptides (500 nM) were used as antigens in streptavidin coated ELISA plates (Nunc, 436014). Gliadin-specific hmAbs at 6.67 nM concentration and fourfold dilution were used to generate titration curves. Alkaline phosphatase conjugated anti-human IgG (Southern Biotech 9040-04) in 1 :4000 dilution was used as detecting antibody, and visualized with phosphatase substrate (Sigma S0942-200TAB) reactivity measured at 405 nm. The synthetic gliadin peptides were produced with biotinylated C- terminal spacer (GSGSGS) by GL Biochem. PBS pH 7.4 was used as buffer, and the plates were washed three times with 0.05% Tween in PBS between each step.
Serologic anti- ω34 Ig inhibition assay
The serologic inhibition assay was developed on an amplified luminescent proximity homogeneous assay (Alpha)LISA platform (Perkin Elmer) with customized AlphaLISA acceptor beads (Perkin Elmer) conjugated with the gliadin-specific hmAb (1002-1E03), according to manufacturer's recommendations. Three different target peptides were tested in separate assays: biotin-QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259) (termed b-cω34), biotin-QPEQPFPEQPEQPEQPFPQPEQPFPW (SEQ ID NO: 260) (b- co26), and biotin-PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4) (b-QPEQPFP3), and the following concentrations were used: ΙΟμΙ of b-cω34 at 2.5 or 1.25 nM per well, b-cω26 at 5 or 7.5 nM, and b-QPEQPFP3 at 4 or 6 nM.
Target peptide was dissolved in AlphaLISA buffer (0.1% Pluviol in PBS pH 7.4) to the concentrations described above. Subsequently, 10 μΐ was transferred to each well and incubated with 5 μΐ serum in 1 hour at RT. AlphaLISA 1002-1E03 acceptor bead stock solution was diluted to 3.5 μg/ml in AlphaLISA buffer, and 15 μΐ added per well. The plate was incubated in 45 minutes at RT in dark. Alphascreen streptavidin donor bead (Perkin Elmer) solution was diluted to 7 μg/ml in AlphaLISA buffer, 15 μΐ was added per well, and the plate was incubated in 45 minutes at RT in dark. AlphaLISA signal was measured with Envision Multilabel Plate Reader (Perkin Elmer).
The serum samples were tested in duplicates and the mean value of the two logarithmic AlphaLISA signals were used. Together with a reference curve, generated from healthy control serum spiked with known concentrations of gliadin-specific hmAbs (1002- 1E01, 1002-1E03 and 1 130-3B03), the mean value was used to estimate serum antibody reactivity of equivalent amounts of gliadin-specific hmAbs (mg/L).
Anti-TG2 IgA and anti-DGP IgG
Celikey Varelisa tTG IgA (Phadia, 181 96) and QUANTA Lite Gliadin IgG II
(INOVA, 704520) were used for serologic testing of anti-TG2 IgA and anti-DGP IgG. The serologic testing was performed by the routine laboratory at the Department of Medical Immunology at Oslo University Hospital - Ulleval.
Statistical analyses
The statistical analyses and graphs of serologic test results were generated with
Graphpad Prism 6 (GraphPad Software Inc.).
Results
Natural binding targets of a patient-derived gliadin-specific antibody
The importmant components of the inhibition assay were the gliadin-specific hmAb and the concomitant target peptide(s). Out of a panel of 38 hmAbs available, the gliadin- specific hmAb 1002-1E03 that showed strong preference for TG2-deamidated gliadin was selected (Example 1). To do an extensive specificity analysis, the hmAb was incubated with TG2 -treated digest of wheat gliadin, antibody -peptide complexes were pulled down, and the antibody-bound peptides were eluted and analyzed by mass spectrometry. Altogether 381 different peptides were identified in the pull-down analyte (Table 10). Most of them were fragments of γ- and ω-gliadins, and the majority contained deamidated residues (85 % of peptides after versus 27 % before pull-down, Fisher's exact test p <0.0001). The exact positions of deamidation could not be ascertained in the grouped analysis, but manual inspection of individual peptides showed preferential deamidation of Q residues in typical QXP motifs. This is in agreement with the reported substrate specificity of TG2 (Vader et al, J Exp Med 2002, 195:643-9). The analysis indicated that the hmAb 1002-1E03 favored long gliadin peptides (28.34±0.4 residues after versus 21.35±0.4 residues before antibody pulldown, Student t test <0.0001) (Figure 17A), similar to what was observed for other of the gliadin-specific hmAbs analyzed by the same method (Example 5).
The primary sequences of different gliadin proteins are highly similar, and gliadin proteins often contain repeats of motifs. Together with the high hit rate in the pull-down assay, this indicated either promiscuous binding property of the hmAb, or that the hmAb recognized a specific motif expressed in several different gliadin peptides. To address this, the peptide sequences from the pull-down analysis were evaluated by counting the most common motifs within registers of 3-15 residues. This analysis indicated selective enrichment of peptides harboring identical motifs (Figure 17B, Table 6). The most common 3 -mers (QQP and PQQ) were counted 1785 and 1634 times in total, and were present in 378 of the 381 peptides (> 99%) pulled down by the hmAb. Similarly, the most common 4-mer was counted 1479 times in 374 peptides, and was given by one amino acid extension of the 3- mers (PQQP). The same was observed for motifs of five, six and seven residues length; more than 95 % (363 of 381 peptides) harbored the same 7-mer motif (QPQQPFP (SEQ ID NO: 261)). In comparison, this 7-mer was found in less than 35% of all peptides identified in the gliadin fraction before pull-down (Table 6). From eight residues the frequency decreased steadily down to 29% (11 1 of 381 peptides) for the most common 15-mer. Altogether, these analyses indicated that long γ- and ω-gliadin peptides with TG2 deamidated glutamate residues and with several copies of certain motif(s) were optimal targets for the hmAb 1002-
1E03. Thus, two representative ω-gliadin derived peptides from the pull-down analysis were synthesized with N-terminal biotin, and selected as target peptides in the serologic inhibition assay (biotin-QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259), b- ω34 and biotin-QPEQPFPEQPEQPEQPFPQPEQPFPW (SEQ ID NO: 260), b-ω26). Both peptides comprised several copies (three to six) of the 3- to 7-mers identified as most common in the previous analysis.
ELISA reactivity of hmAb 1002-1E03 to synthetic gliadin peptides
To evaluate the specificity of the hmAb 1002-1E03 in the light of the identified common motifs, the hmAb 1002-1E03 was tested against a panel of synthetic gliadin peptides in ELISA (Table 7; Table 1 1). The sequence PEQ was present in all peptides recognized by the hmAb. However, the hmAb was not reactive to all peptides comprising this 3-mer, showing that PEQ was important but not sufficient for binding. As mentioned, PQQ was highly abundant in the pull-down analyte, and in most cases (1479 of 1634) flanked by C- terminal proline (PQQP (SEQ ID NO: 262)). In agreement with the substrate specificity of TG2 (Vader et al, J Exp Med 2002, 195:643-9), these motifs would be susceptible to TG2- mediated deamidation (PEQP (SEQ ID NO: 263)). The most common 7-mer in the pull-down was QPQQPFP (SEQ ID NO: 261), and the hmAb showed good reactivity to synthetic peptides comprising the deamidated counterpart QPEQPFP. Both b-cω34 and b-cω26 expressed the QPEQPFP (SEQ ID NO: 10) motif three times. However, one could not exclude that the hmAb recognized other motifs, as PEQ was expressed as many as six and five times in the two peptides, respectively.
The hmAb 1002-1E03 was compared to eight different gliadin-specific hmAbs, screened for reactivity to the same synthetic peptides in ELISA (Table 7). In brief, one hmAb (1 14-1G01) showed almost identical peptide reactivity pattern, four of them (1002-1E01, 1 130-2A02, 1 130-3A02 and 1130-3B04) were reactive to all peptides harboring QPQQPFP (SEQ ID NO: 261) or QPEQPFP (SEQ ID NO: 10), one (1 130-3B01) showed binding to all peptides comprising either QPXiQX2FP (SEQ ID NO: 264) (X1 = Q, E; X2 = Q, S, P), QPEQTFP (SEQ ID NO: 265) or PQPELPYPQP (SEQ ID NO: 12), and the last two hmAbs (1 130-3B03 and 1 130-3G05) only recognized peptides expressing the sequence
PQPELPYPQP (SEQ ID NO: 12). In summary, the nine hmAbs could be divided into four groups based on their peptide reactivity patterns. Seven of the hmAbs (all but 1130-3B03 and 1130-3G05) divided into three groups, but they all recognized peptides harboring QPEQPFP (SEQ ID NO: 10). Based on the ELISA experiments, a peptide comprising three copies of the QPEQPFP (SEQ ID NO: 10) motif (biotin-PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID
NO: 4), b-QPEQPFP3 for short), was synthesized and used as a third target peptide in the serologic inhibition assay.
Serologic inhibition assay
Serum and biotinylated target peptides were first mixed and incubated. Second, AlphaLISA acceptor beads conjugated with hmAb 1002-1E03 were added to the mixture. The binding of the hmAb to the biotinylated peptide was detected with Alphascreen streptavidin donor beads. The principle of the assay is that the biotinylated target peptide binds to streptavidin donor beads, which consequently are brought in proximity to the hmAb conjugated acceptor beads given that the antibody binds to the target peptide. Upon light activation, the donor beads release energy that can activate the acceptor beads to emit light signal provided proximity of the beads. Gliadin-reactive serum antibodies can compete with the hmAb for binding to the peptide and hence reduce the signal.
Sera of untreated celiac disease patients (n = 106) was compared with sera of control subjects (198 healthy blood donors and 151 patients with Crohn's disease). Three different target peptides (b-QPEQPFP3, b-cω34, and the shorter variant b-cω26) were tested in the assay. Two of them represent peptide fragments that are naturally occurring in the gliadin proteome (b-cω34 and b-cω26), and the antibody pull-down experiment indicated that these peptides to be of primary targets for the hmAb. In contrast, the third peptide (b-QPEQPFP3) was designed to represent the sequence QPEQPFP three times, a sequence common to many peptides that several gliadin-specific hmAbs reacted with in ELISA (Table 7). The peptide b- co34 demonstrated highest diagnostic performance of the three target peptides, evaluated by the area under the curve (AUC) of the receiver operating characteristic (ROC) curve analysis (Table 12). The most striking difference, though, was the large variation in negative controls in the assay employing the b-QPEQPFP3 peptide, whereas the two assays using the omega- peptides only gave small variations in the same control groups (Figure 20). There was a modest positive effect of combining the test result of the three target peptides (Table 12), but only the results of the assay with the b-cω34 target peptide (Figure 18A) was used in the further analyses (anti-a>34 Ig, Ig stands for immunoglobulins or antibodies).
Each serum sample was tested in duplicates and thus giving two values of logarithmic AlphaLISA Signal. The mean of these two values was used in the analysis (Figure 18A). In order to establish test characteristics of the assay and to establish a reference curve, a serum from a non-celiac control was spiked with dilutions of known concentrations of gliadin- specific hmAbs (equimolar concentrations of 1002-1E01, 1002-1E03 and 1130-3B03), and tested (Figure 18B). It eas found that the performance of the assay was dependent on the
concentration of target peptide. Low concentrations gave better sensitivity in terms of lower detection limit, but also lower specificity due to reduced signal/noise-ratio (Figure 21). The peptide concentrations used in in this study gave dynamic range from approximately 1 mg/L to 20-30 mg/L of the gliadin-specific hmAbs used as reference. Signal/noise-ratio under these conditions spanned approximately two logarithmic units. The mean values of the logarithmic AlphaLISA Signal (Figure 18A) and the reference curve (Figure 18B) were used to extrapolate the antibody activity in the serum samples to equivalent concentrations (mg/L) of reference gliadin-specific hmAbs (Figure 18C). Optimal cut-off estimated by Youden index (sensitivity - (1 - specificity)) of the extrapolated values (Figure 18C) was < 1.02 mg/L (sensitivity 0.9292, specificity 0.9513), indicating that the detection limit of the test platform was the main limitation of the sensitivity in this assay. This indicates that further
improvements in sensitivity can be obtained by using even lower peptide concentrations. In some configuration, < 1 mg/L as cut-off would be inadequate, as lmg/L of hmAbs in reference serum gave no significant decrease in AlphaLISA Signal compared to background (Figure 18B). In contrast, the reference curve gave a clear difference in signal between lmg/L and 2mg/L (Figure 18B), and hence was cut-off < 2 mg/L selected for the anti-cω34 assay.
Serologic inhibition assay compared to anti-TG2 IgA and anti-DGP IgG
The serum samples from celiac disease patients and control subjects (healthy blood donors and patients with Crohn's disease) were also analyzed for anti-TG2 IgA (Varelisa tTG IgA, Phadia) and anti-DGP IgG (QUANTA Lite Gliadin IgG II, INOVA) (Figure 19A-B). The manufacturers operated with two different recommended cut-off values, namely < 5 or < 8 U/ml for anti-TG2 IgA, and < 20 or < 30 U/ml for anti-DGP IgG. These are termed high (< 8 and < 30 U/ml) and low (< 5 and < 20 U/ml) cut-off values below.
The test results of anti-TG2 IgA and anti-DGP IgG were compared with the results of the serologic anti-cω34 Ig inhibition assay (Figure 18C). In addition to the mentioned cut-off < 2 mg/L, a second cut-off < 3 mg/L, corresponding to the high cut-off values of the two commercial assays was used.
The ROC-estimated AUCs were high (0.9715 - 0.9806) for all three assays (no significant differences, Table 12). In general, all three assays showed very good specificity, whereas the sensitivity was lower for all (Table 8A). There were two noticeable differences between the assays. Anti-TG2 IgA gave more low values in the celiac disease population than the two other assays, and anti-DGP IgG was the only assay giving any high values in the Crohn's disease control group (Figure 18C, Figure 19).
Seropositive controls
The specificity of the assays for the two control groups was evaluated separately. For blood donors, the high and low cut-off values gave identical specificity results. In brief, five of the 198 blood donors were seropositive, and three of them scored positive in two or more assays (Table 9A). One donor had anti-DGP IgG 49 U/ml, but was negative for anti-cω34 Ig (0 mg/L) and anti-TG2 IgA (0.088 U/ml). The fifth donor scored 6.6 mg/L in the anti-cω34 Ig assay, had anti-TG2 IgA value of 2.6 U/ml and was anti-DGP IgG negative (below 5 U/ml). Notably, all five were HLA-DQ2.5 positive (Table 9A). Thus, many of the seropositive blood donors were highly suspicious of having untreated celiac disease, which obviously made the specificity evaluation difficult in the blood donor group.
In contrast, the Crohn's disease control group demonstrated clear differences between the three assays (Table 9B). In total seven patients scored positive using low cut-off values, and none of them were positive in more than one assay (Table 9B). Only three patients had antibody activities above the high cut-off values, all in the anti-DGP IgG assay (McNemar's test p 0.2482). In summary, anti-cω34 Ig (0.9868 - 1.0) and anti-TG2 IgA (0.9934 - 1.0) demonstrated superior specificity as compared to the anti-DGP IgG assay (0.9735 - 0.9801) in the Crohn's disease control group.
Diagnostic odds ratio
The diagnostic odds ratio (DOR), estimating the odds of (true) positive test
(sensitivity / (1 -sensitivity) in subject from disease group, relative to odds of (false) positive test (l-specificity)/specificity in control subject, is a commonly used measurement of the effectiveness of an assay (Glas et al, L Clin Epidemiol 2003, 56: 1 129-35). The DOR results were comparable for all three tests using both high and low cut-off (range 321-453), except for anti-DGP IgG (<30 U/ml) that demonstrated the poorest odds with 167. Although not significant, anti-cω34 Ig (< 2 mg/L) showed highest DOR (Table 8A).
Joint analysis of anti-TG2 IgA and anti-(o34 Ig increases detection rate
Recently, several studies have shown an important role of gliadin-specific antibodies as they allow detection of celiac disease patients who are anti-TG2 IgA negative (Wolf et al,
PloS One 2014, 9:e97853; Mooney et al, Clin Gastroenterol Hepatol 2015; Sugai et al, Clin Chem 2010, 56:661-665; Basso et al, Clin Chim Acta 201 1, 412: 1662-7). Following this argument, it was evaluated whether complemental use of anti-cω34 Ig provided better diagnostic performance than single testing of anti-TG2 IgA. Anti-co34 Ig detected 57% and
75% of the celiac disease patients with negative anti-TG2 IgA titers for high and low cut-off values, respectively (i.e. 86 patients with anti-TG2 IgA > 5 U/ml, against 101 patients with
anti-TG2 IgA > 5 and/or anti-cω34 Ig > 2mg/L). The specificity was modestly decreased, and the DOR was more than doubled (Tables 8A-B). Anti-DGP IgG showed similarly good effect (Table 8B). Thus, the combined use of anti-TG2 IgA and the novel anti-cω34 Ig assay considerably increased the diagnostic performance.
This example describes a serologic inhibition assay for detection of gliadin-specific antibodies to be used as a diagnostic tool in celiac disease. The assay is based on a recombinant human monoclonal antibody, generated from the antibody genes of a single intestinal IgA plasma cell of a celiac disease patient. A preferred target for the antibody, a deamidated 34-mer ω-gliadin fragment, was identified in TG2-treated enzymatic digest of wheat gluten by antibody pull-down and subsequent mass spectrometry sequencing. The combination of this peptide and the monoclonal antibody was employed in an amplified luminescent proximity homogeneous (AlphaLISA) assay whose signal could be specifically inhibited by serum antibodies. The inhibition assay was more specific than anti-DGP IgG, more sensitive than anti-TG2 IgA, and detected up to 75% of the patients who scored negative for anti-TG2 IgA in this study.
Two main characteristics separate the inhibition assay from conventional anti-TG2 and anti-DGP assays. First, it takes advantage of the specificity of the hmAb originating from the plasma cell of a celiac lesion as well as the sequence of the peptide fragment
preferentially recognized by this hmAb. Second, it monitors antibody activity of all antibody isotypes in serum. Although the diagnostic performance of the anti-DGP assays is markedly better than former anti-gliadin assays, the anti-DGP assays in general are reported less specific than the anti-TG2 assays (Lewis et al, Aliment Pharmacol Ther 2010, 31 :78-81) and are claimed to have low predictive values in the serologic diagnostics of celiac disease (Vriezinga et al, N Engl J Med 2014, 371 : 1304-15). The inhibition assay exhibits better specificity than the anti-DGP IgG assay in the material tested. This may relate to the use of the disease-specific hmAb or the different gluten antigens used in the two assays. The antigen in the anti-DGP assays typically consists of gluten motifs empirically giving the highest antibody reactivity in patient sera. Of note, several independent studies have reported strikingly similar results, pointing at peptide sequences highly similar to the 7-mer motif QPQQPFP (SEQ ID NO: 261) enriched by the gliadin-specific hmAb investigated in this study (Osman et al, Clin Exp Immunol 2000, 121 :248-54; Vallejo-Diez et al, PloS One 2013,
8:e80982; Ballew et al, PNAS 2013, 1 10: 19330-5). However, the hmAb clearly demonstrated more complex binding properties, being reactive to several related motifs. This is in line with what was observed for other gliadin-specific hmAbs (Example 1). The inhibition assay
employed the monoclonal antibody together with a long ω-gliadin peptide with several binding motifs. It is thus particularly suitable for monitoring disease-specific gluten serum antibodies, which may have the same cross-reactive feature.
The fact that the assay measures antibody activity of all isotypes in serum is particularly relevant for IgA deficiency. Patients with IgA deficiency is overrepresented among celiac disease patients, and approximately 2% of the celiac disease patients are IgA deficient (Chow et al, J Clin Gastroenterol 2012, 46:850-4). For this reason, screening of anti- gliadin antibodies of IgG isotype is often recommended as the second test to anti-TG2 IgA (Rubio-Tapia et al, Am J Gastroenterol 2013, 108:656-76). IgG and IgA of patients' sera show reactivity to the same motifs of the gluten proteome (Osman et al, Clin Exp Immunol 2000, 121 :248-54; Aleanzi et al, Clin Chem 2001, 47:2023-8; Bateman et al, Gut 2004, 53 : 1274-8), which indicates that the gluten antibodies of the different isotypes have overlapping epitope specificities. In assays where only one isotype is measured, antibodies of other isotypes would compete and potentially reduce the signal. Measuring all isotypes, as done in the inhibition assay of this study, may thus be an advantage.
Taken together this study demonstrates that hmAbs deriving from celiac disease patients are useful with their natural target peptides in inhibitory serologic assay for diagnostic purpose. The assay takes advantage of the specificity introduced by both the hmAb and the antigen.
Table 6. Most frequent motifs of all sequences of 3-15 residues length found in the 381 gliadin peptides pulled down by the hmAb 1002-1E03. Peptides identified by mass spectrometric sequencing here given in native form, although the majority of the peptides harbored one or several deamidated residues.


Table 7. Sequence motifs shared by synthetic peptides recognized by the nine different gliadin-specific hmAbs. * All peptides recognized by the hmAbs 1002-1E03 and 114-1 GO 1 harboured the PEQ sequence, but there were also some peptides with this sequence which were not recognized by the hmAbs.


Table 8. Diagnostic performance of the serologic tests. (A) For each assay estimated separately. (B) Anti-TG2 IgA combined with anti-cω34 Ig or anti-DGP IgG (low cut-off values mean anti-TG2 IgA < 5 U/ml, anti-DGP IgG < 20 U/ml and anti-cω34 Ig < 2 mg/L).
Table 8A:

Table 8B:

Table 9. Seropositive control subjects. (A) Blood donors (n = 5) with positive serology and respective HLA genotypes. (B) Crohn's disease patients (n = 7) with positive serology.
Table 9A:

Table 9B:

Table 10. Peptides pulled down from enzymatically digested and TG2-treated gliadin by hmAb 1002-1E03. Peptides identified by mass spectrometric sequencing here given in native form, although the majority of the 381 peptides harbored one or several deamidated residues.













Table 11. ELISA reactivity of the nine gliadin-specific hmAbs to a panel of synthetic gliadin peptides, showing good reactivity (+), weak reactivity (+/-) or no reactivity (-). The hmAbs were not tested for reactivity to all peptides (empty squares).



EXAMPLE 5
The human monoclonal antibodies (hmAbs) obtained in Example 1 reacted with deamidated gliadin antigen, and showed no reactivity to other types of antigens. Of note, the procedures by which these hmAbs were generated did not identify which epitopes in the gluten proteome they are primarily reactive with, as the complex deamidated gliadin antigen used in the first approach represented many different peptides and the synthetic peptides used in the second approach could have lower affinity than similar, but distinct unknown gluten peptide(s) due to cross-reactivity. The fact that many of the gliadin-specific hmAbs by initial testing were reactive to several synthetic gliadin peptides, and that some of the hmAbs from IgA+ plasma cells sorted by flow cytometry showed higher reactivity to other peptides than to the peptide used in cell surface staining (Example 1), indicated that an effort to identify the primary target of the hmAbs would be justified. This example describes epitope mapping of gliadin-specific hmAbs by antibody pull-down of fragments from complex proteolytic digests of gliadin followed by sequencing of the isolated peptides by mass spectrometry.
Materials and Methods
Gliadin-specific monoclonal antibodies
Gliadin-specific human recombinant monoclonal antibodies (hmAbs) were produced as human IgGl in HEK293A cells as previously reported (Example 1 ; Smith et al, Nat Protoc 2009, 4:372-84). The hmAbs were purified from cell supernatants on protein G sepharose (GE Healthcare).
Peptides
All peptides were purchased from GL Biochem Ltd, except for the y-26mer peptide which was purchased from Peptide 2.0. The following peptides were used in the MALDI- TOF experiments: γ-gliadin 26mer; FLQPEQPFPEQPEQPYPEQPEQPFPQ (SEQ ID NO:
15), α-gliadin 33mer; LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7) and the short γ-gliadin peptide; PLQPEQPFP (SEQ ID NO: 3). In ELISA and AlphaLISA the following peptides were used: biotin-GSGSGSPLQPEQPFP (SEQ ID NO: 17), biotin- QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259),
FLQPEQPFPEQPEQPYPEQPEQPFPQ (SEQ ID NO: 15), PQPQQPEQPFPQPQ (SEQ ID NO: 561), QQPFPQQPQQPYPQQPEQPFPQP (SEQ ID NO: 562),
QPQQPFPQPEQPFPWQP (SEQ ID NO: 563), PQQPFPQPEQPFP (SEQ ID NO: 564), FPQPEQPFPWQP(SEQ ID NO: 565), PFPQPEQPFPWQPQQPFPQ (SEQ ID NO: 566), PQQPEQPFP (SEQ ID NO: 9), biotin-GSGSGSPQQPQQPFP (SEQ ID NO: 567), biotin- GSGSGSPQQPEQPFP (SEQ ID NO: 568), biotin-GSGSGSPQQPQQQFP (SEQ ID NO:
569), biotin-GSGSGSPQQPEQQFP (SEQ ID NO: 570), biotin-GSGSGSPQQPQQSFP (SEQ ID NO: 571), biotin-GSGSGSPQQPEQSFP (SEQ ID NO: 572), biotin- GSGSGSPQQPQQTFP (SEQ ID NO: 573), and biotin-GSGSGSPQQPEQTFP (SEQ ID NO: 574).
Preparation of TG2-treated gliadin
Digestion of gliadin with chymotrypsin, pepsin, trypsin, elastase and
carboxypeptidase was performed as previously described (Dorum et al, J Immunol 2014, 193:4497-506). The gliadin digest was further fractionated by size exclusion chromatography using an Akta system with a Superdex peptide 10/300 GL column (GE Healthcare). Fractions of 0.5ml were collected. Two to three adjacent fractions were pooled and treated with transglutaminase 2 (TG2) as described previously (Dorum et al, J Immunol 2014, 193:4497- 506) before subjected to pull-down with hmAbs. The fractions containing the highest molecular weight peptides were not used in the pull-down experiments.
Antibody pull-down
40 μg hmAb (260 pmol) was incubated with synthetic peptides (1500 pmol) or 2-3 pooled gel filtration fractions of TG2 -treated gliadin for 20 minutes at room temperature (RT). For enrichment, Dynabeads Protein G (Life Technologies) were added to the samples and incubated for 20 minutes at RT (125 μg beads^g IgG). After washing with 5x500 μl PBS/0.1% octylglucoside, the bound peptides were eluted with 0.1% TFA for 10 min at RT. A rotavirus specific hmAb (Rota-2B04) was used as negative control (Di Niro et al, Nat Med 2012, 18:441-5). The few unspecific peptides pulled down with Rota-2B04 were removed
from the list of identified hmAb enriched peptides in addition to some few short peptides that derive from actin and other non-gluten proteins.
Mass spectrometry analysis
Peptides pulled down from complex gliadin fractions were analysed by nano-LC-
MS/MS using a Q Exactive hybrid quadropole-orbitrap mass spectrometer (Thermo
Scientific) coupled to a nano-HPLC (Dionex Ultimate 3000 nano-LC system (Dionex) or EASY-nLC 1000 (Thermo Scientific)). The peptides were separated on a 250 mm EASY- SPRAY column (CI 8, 75 μm ID, 2 μm particles, Thermo Scientific) using a 60 min linear 5- 50% ACN gradient in 0.1% formic acid at a flow rate of 0.3 μΐ/min (60 min or 120 min for the gliadin fractions). The Q Exactive mass spectrometer was operated in the data-dependent acquisition mode using the Xcalibur 2.2 software. Full-scan MS in the Orbitrap were followed by 10 data dependent MS/MS scans. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (Viczaino et al, Nucleic Acids Res 2013, 41 :D1063-9).
Database search
The LC -MS/MS data were analysed with the software MaxQuant version 1.5.1.2 using Andromeda to search against a Triticum aestivum database (total of 4722 entries) extracted from the UniprotKB database release September 2013 (European Bioinformatics Institute). In all searches the digestion enzyme specificity was set as none and pyro-glu (N- term Q), deamidation (NQ) and oxidation (M) were selected as variable modifications. For analysis of QExactive data, the mass error tolerance for MS scans was first searched with an error window of 20 ppm and then with a main search error of 6 ppm. Mass tolerance for MS/MS scans was set to 20 ppm. A false discovery rate of 1% and a PEP score of 0.1 were used.
ELISA reactivity of gliadin-specific hmAbs to synthetic gliadin peptides
Biotinylated synthetic gliadin peptides (500 nM) were used as coating antigens in streptavidin coated ELISA plates (Nunc, 436014). Gliadin-specific hmAb 1130-3B01 or
1130-3 A02 at a concentration of 2 μg/ml and with fourfold dilution were used to generate titration curves. Alkaline phosphatase conjugated anti-human IgG (Southern Biotech 9040-
04) in 1 :4000 dilution was used as the detecting antibody and visualized by phosphatase substrate (Sigma S0942-200TAB) reactivity measured at 405 nm. PBS pH 7.4 was used as
buffer, and the plates were washed three times with PBS with 0.05% Tween between each step.
AlphaLISA assays
The relative affinity of three hmAbs (1002-1E01, 1002-1E03 and 1130-3B01) to a panel of gliadin peptides was investigated using an AlphaLISA platform. AlphaLISA acceptor beads were coated with anti-human IgG according to manufacturers' protocol. Anti- IgG AlphaLISA beads 6 μg/ml and hmAb 0.5 μg/ml were mixed and incubated for 1 hour at RT in dark. Then, 15 μΐ of the solution was transferred to each well in a 384-well AlphaLISA plate, together with 5 μΐ analyte consisting of 40nM biotinylated PLQPEQPFP peptide and diluting concentrations of non-biotinylated competing peptides (5mM, 1 :3 dilution). After incubation for 1 hour at RT in dark, 15 μΐ of 24 μg/ml streptavidin coated Alphascreen donor beads was transferred per well, and the plate incubated for 1 hour at RT in dark. AlphaLISA Signal was measured with Envision Multilabel Plate Reader (Perkin Elmer). PBS pH 7.4 and 0.1% Puviol was used as buffer. The binding of whole IgGl versus Fab of gliadin-specific antibody to gliadin peptide was investigated in a similar competitive AlphaLISA assay. Here diluting titrations of IgGl or Fab of hmAb 1002-1E03 were used with either AlphaLISA acceptor beads conjugated with hmAb 1002-1E03 and biotinylated 33mer ω-gliadin (2.5nM), or AlphaLISA acceptor beads conjugated with PLQPEQPFP together with biotinylated hmAb 1002-1E03 (0.1 mg/ml).
Results
Antibody pull-down and identification of peptides by mass spectrometry
Thirteen gliadin-specific hmAbs were included in this study; nine hmAbs of IgA+ plasma cells isolated by flow cytometry, where two different synthetic gliadin peptides were used as sorting peptide (biotin-GSGSGS-PLQPEQPFP (SEQ ID NO: 17), PLQPEQPFP (SEQ ID NO: 3) for short; biotin-GSGSGS-
LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18), deamidated 33mer for short) and five hmAbs of in vitro cultured plasma cells secreting IgA reactive to complex deamidated gliadin (Table 13).
Table 13. Overview of the gliadin specific hmAbs. Patient number, isolation method (sorting antigen depicts the antigen used to isolate the IgA+ PC of which the hmAb was cloned) and reactivity to antigens is shown as well as the number of hmAb-enriched peptides
identified by MS, the most frequent 7mer motif among these peptides and % deamidated (DA) peptides pre/post hmAb pull-down.


*31 peptides removed from the list as they were identified in the negative control sample
**For this hmAb 28 μg and not 40 μg was used in pull-down experiment *** Tested PLQPEQPFP (SEQ ID NO: 3)/PLQPQQPFP (SEQ ID NO: 8) for all hmAbs except for 3B03 and 3G05 where the DA and native 33mer was tested
a: Example 2
b: Nikiphorou et al, Rheumatology 2014, 53: 1906-7 A method was established to pull down peptides from complex mixtures of enzymatically digested and TG2 -treated gliadin (TG2-gliadin from now). To note, the enzymatic digest of gliadin contained glutenin proteins, hence probing would also be towards this part of the gluten proteome. In brief, the hmAbs were incubated with size-separated fractions of TG2-gliadin, and the formed hmAb-peptide complexes were isolated with magnetic protein G beads. Antibody bound peptides were eluted and analysed using a
QExactive mass spectrometer. The TG2-gliadin fractions were compared pre and post pulldown. To control for unspecific binding, a rotavirus-specific hmAb was included as a negative control.
For eleven of thethirteen hmAbs, several unique peptides (range 19-552) were identified in the pull-down analyses (Table 13). One hmAb (1065-4G05) did not enrich detectable peptides, and only four peptides were identified in the pull-down analyses from the last hmAb (1065-4C01). This can be explained by low affinity of the hmAbs, as observed in ELISA. These hmAbs were hence excluded from further analyses.
In general, peptide fragments from α-, γ-, ω-gliadins and low-molecular weight glutenins were pulled down although fragments of a-gliadins were relatively infrequent.
The gliadin-specific hmAbs pull down long deamidated peptide fragments
The most frequently (based on intensity) pulled down peptides with the various gliadin-specific antibodies are listed in Table 15. A striking observation was that the peptides present in post pull-down samples were generally longer than peptides in the pre pull-down samples. This pattern, as exemplified by the hmAb 1 130-3A02 and hmAb 1002-lEOl (Figure 22A and B), was present for all but two (1 130-3B03 and 1 130-3G05) of the antibodies (Figure 22C). Many of the enriched peptide fragments harboured repeated sequence motifs (Figure 22D). It was further observed that the large majority of the identified hmAb-enriched gliadin peptides had been deamidated by TG2 (Table 13). The exact deamidation sites could not be unambiguously reported by the MS search engine due to the presence of short fragments of proline and glutamine repeats with identical masses in the MS fragment spectra. Manual inspection of MS fragment spectra for some of the peptides confirmed that the deamidation sites typically were in the QXP motif, which is in keeping with the previously reported TG2 substrate specificity (Vader et al, J Exp Med 2002, 195:643-9; Fleckenstein et al, J Biol Chem 2002, 277:34109-16). Because of the ambiguity of the deamidation sites, all peptides are listed with their native sequence, even though the majority were deamidated.
Enriched peptides are not necessarily similar to peptides to which the hmAbs were selected
For some of the hmAbs, the enriched peptides had different peptide sequences than the selecting peptide antigen originally used to isolate the IgA+ plasma cell (Table 13). This was particularly observed for the hmAbs of plasma cells sorted with the a-gliadin 33mer
peptide LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7) (e.g., mAbs 1130-3B04, 1130-3A02, 1 130-3B01, 1130-3A05, 1 130-3B03, 1 130-3G05).
The enriched peptide fragments share motifs
The observation of repeated motifs in the pulled down peptides, prompted a search for sequences of 3-15 residues common to all peptide fragments pulled down by the individual hmAbs. It was found that for the majority of the hmAbs (9 of 11) there was a selective enrichment of peptides harbouring common motifs (Table 13). The results for hmAb 1130- 3B04 are shown as an example in Figure 23. The frequency of the most common 3-15 amino acid motifs in the post pull-down samples was compared to the frequency of the same motifs in the pre pull-down samples. The motifs were typically extensions of a core sequence (Figure 23B). A motif of 8 residues or less could be identified in the majority of the peptides, but beyond this motif length the number of peptides with a shared motif dropped
dramatically. This indicates that the hmAb 1 130-3B04 reacts specifically with peptides that share a motif of no more than 8 amino acids in length. A similar pattern was seen for several of the hmAbs.
The presence of 7mer motifs for all the hmAbs was investigated. The most frequent 7mer motif of the hmAb 1 130-3B04 was the sequence QPQQPFP (SEQ ID NO: 261) (Figure 19 and Table 9). Interestingly, this was the most frequent 7mer motif of five of the other hmAbs (1002-lEOl, 1 130-3A02, 1 130-2A02, 1002-1E03 and 1130-3A05). More than 88% of the peptides pulled down with these hmAbs harboured the shared motif QPQQPFP (SEQ ID NO: 261) (Table 13). The motif was typically present in 2-4 copies in each peptide, as shown for hmAb 1 130-3A02 (Figure 22D). Some hmAbs allow variation in target motif
For hmAbs 1 114-1G01, 1 130-3B01, 1050-5B05, 1130-3B03, and 1 130-3G05 the frequencies of the most common 7mers were considerably lower. It was investigated whether this was due to reactivity of the hmAbs towards peptides with similar, but not necessarily identical sequences. To evaluate this, the tool "Pattinprot" (PBIL.ibcp.fr) was used to scan all the possible 7mers for motifs of one amino acid substitution (85% similarity) to the
QPQQPFP (SEQ ID NO: 261) motif among peptides pulled down by these five antibodies.
The results for the hmAb 1 130-3B01, shown by a sequence logo representation (Figure 24A), indicated sharing of the motif QPQQXFP (SEQ ID NO: 575) (X=P,S,T,Q). This motif was present in 98% of the enriched peptides (Table 15). Reactivity to the motif variants by this
hmAb was also verified by ELISA. The hmAb showed reactivity towards all of the peptides with preferential reactivity to the deamidated versions (Figure 24B). An exception was observed for the peptide variant harbouring a threonine residue, where the hmAb only recognised the deamidated version. In contrast, hmAb 1 130-3A02 only recognised the native and deamidated peptides containing the sequence QPQQPFP (SEQ ID NO: 261) (Figure
24C) which corresponds to the identified peptides pulled down by this hmAb (Table 13). The hmAb 1130-3A02 did not discriminate between native and deamidated versions of the peptides in ELISA and, in fact, less than half of the peptides pulled down with this hmAb were deamidated (Table 13). The most common 7mer motifs for the hmAbs 1050-5B05 and 11 14-1G01 were present in 44% and 73% of the identified peptides, which following the
"Pattinprot" analysis increased to 83% and 100% by allowing for variation at positions 1 and 7 of the 7mer motifs (Table 13). No motif was found at appreciable frequencies among the peptides enriched by 1130-3B03 and 1130-3G05. It is notable that these were the only two hmAbs that did not enrich for long peptide fragments in the pull-down experiment. hmAb affinity for gluten peptides is influenced by sequence flanking the target motif
To further understand the specificity of anti-gliadin antibodies, hmAbs 1002-lEOl, 1002-1E03 and 1 130-3B01 were tested for reactivity to a panel of deamidated synthetic gliadin peptides harbouring the key sequence QPEQPFP (SEQ ID NO: 261) (Q^E substitution in position 3) in a competitive AlphaLISA assay (Figure 25). While 1002-1E03 showed similar affinity for all peptides in the panel, the affinity of 1002-lEOl and 1 130-3B01 to the different peptides varied by 1-2 logs (Figure 25A). This demonstrates that although a "dominant" motif is found for the majority of the gliadin-specific hmAbs, the flanking regions of the sequence motif will affect the binding affinity. In the gluten proteins, the
QPQQPFP (SEQ ID NO: 261) sequence motif can be found with a variety of different amino acids in the flanking regions (Figure 25B).
Pull-down enrichment of peptide fragments is influenced by epitope
multivalency
To test whether the pull-down procedure enriched for long peptides with repeated sequence motifs, a synthetic peptide mix containing equimolar amounts of the deamidated γ- gliadin peptide PLQPEQPFP (SEQ ID NO: 3) (epitope xl underlined) and the longer 26mer γ-gliadin peptide FLOPEOPFPEOPEOPYPEOPEOPFPO (SEQ ID NO: 15) (epitope x2
underlined) was incubated with the hmAbs 1130-3B01, 1002-1E03 and 1002-1E01 before incubation and pull-down with protein G beads, peptide elution and MALDI-TOF MS peptide detection (Figure 26A). For all hmAbs only the long peptide was detectable. Looking at epitope distribution in peptides of the gliadin digests pre and post pull-down, it was noticeable that longer peptides with multiple copies of the epitopes were pulled down (Figure 26B). This indicates that the enrichment for long fragments with repeated motifs can be explained by more efficient binding of peptides harbouring multiple epitopes. When comparing in a competitive AlphaLISA the binding of intact IgGl vs Fab fragment of the hmAb 1002- 1E03 for binding to the ω-gliadin peptide
QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259) (Figure 26C), equal binding was observed, indicating that single peptide molecules, despite harbouring three copies of the epitope, did not engage both antigen binding sites of the IgGl molecule.
However, when hmAb 1002-1E03 was immobilised on beads and binding of the deamidated γ-gliadin peptide PLQPEQPFP (SEQ ID NO: 3) and the longer 26mer γ-gliadin peptide FLQPEQPFPEQPEQPYPEQPEQPFPQ (SEQ ID NO: 15) (epitope underlined) was compared, the longer peptide bound substantially better (Figure 26D). This suggests that the longer peptide with its two repeated epitopes can bind two antibody molecules
simultaneously. Other factors influencing the peptide pull-down
Qualitative and quantitative aspects of the gliadin fractions from which peptides in a competitive fashion were pulled down could influence which peptides were identified. To investigate influence of qualitative aspects, hmAb 1 130-3B01 was incubated with a synthetic peptide mix containing equimolar amounts of the deamidated a-gliadin 33mer peptide and the peptide PLQPEQPFP (SEQ ID NO: 3), which harbour the deamidated QPQQPFP (SEQ ID NO: 261) 7mer motif. The hmAb-peptide complexes were isolated and bound peptides were analysed by MALDI-TOF MS (Figure 27A). Only the a-gliadin 33mer peptide was enriched by the hmAb. In contrast, when incubating the hmAb with an equimolar mix of the deamidated α-gliadin 33mer peptide and a deamidated γ-gliadin 26mer peptide harbouring the QPEQPFP (SEQ ID NO: 10) motif in two copies
(FLQPEQPFPEQPEQPYPEQPEQPFPQ (SEQ ID NO: 15)), only the deamidated γ-gliadin 26mer peptide was pulled down (Figure 27B). This indicates that the hmAb prefer to bind long peptides harbouring multiple copies of the QPEQPFP motif, and if present like in the
competitive environment in the gliadin fractions, these peptides will dominate in the pulldown.
Quantitative aspects may also play a role as the target peptide sequences were not equally represented. A substantial proportion of all peptides (21-39%) in the pre pull-down fractions harboured the QPQQPFP (SEQ ID NO: 261) motif reflecting dominance of fragments from ω-gliadin proteins (about 15%), γ-gliadin proteins (about 45%) and low- molecular weight glutenin proteins (about 25%). In contrast, only 1.5-3% of the identified peptides harboured the typical PQPQLPY (SEQ ID NO: 576) α-gliadin motif, and about 12% of the fragments were derived from α-gliadin proteins.
Of the six hmAbs from IgA+ PC sorted with the deamidated α-gliadin 33mer peptide, a lack of reactivity towards the PLQPEQPFP (SEQ ID NO: 3) peptide in ELISA and AlphaLISA was found for hmAbs 1130-3B03 and 1130-3G05, but not the four remaining hmAbs (Table 9). Notably, no shared motifs were found among the peptides pulled down with these two hmAbs. Of the 19 peptides enriched by hmAb 1 130-3B03, only one single peptide (LQLQPFPQPQLPYPQPHLPYPQPQP (SEQ ID NO: 577), see Table 15) shared a part of its sequence with the α-gliadin 33mer peptide. To investigate whether this could relate to the low abundance of a33mer peptide in the gliadin fractions, a pull-down experiment was performed in a peptide mixture with equimolar amounts of the synthetic γ-gliadin 26mer and the α-gliadin 33mer peptide using hmAb 1 130-3B03. The hmAb-bound peptides were analysed by MS. MALDI-TOF spectra pre and post 1 130-3B03 pull-down, demonstrated a preferential enrichment of the a33mer peptide (Figure 27C). This indicates that the a-gliadin 33mer peptide is a good antigen for 1 130-3B03, although it could not be readily identified in the post pull-down from the gliadin fraction. This may relate to the antibody affinity to the a- gliadin 33mer relative to other gliadin peptides, and the concentration of this peptide in the pre pull-down fraction.
The hmAb-enriched peptide fragments harbour several different gliadin T-cell epitopes
Gliadin-specific hmAbs appear to bind gluten peptides that harbour T-cell epitopes (Example 1). The presence of gluten T-cell epitopes in the identified gliadin peptides pre and post hmAb pull-down (Table 14 show post pull-down) were investiged. It was found that more than 80% of all peptides pulled down with the six hmAbs 1002-1E01, 1130-3B04, 1130-3B01, 1002-1E03, 1 130-3A02 and 1 130-2A02, harboured known gluten T-cell epitopes. The majority of the T-cell epitope containing peptides contained the DQ2.5-glia-
y4c (QQPQQPFPQ (SEQ ID NO: 272)) and/or the DQ2.5-glia-y5 epitope (QQPFPQQPQ (SEQ ID NO: 578)).
A massive enrichment of γ-gliadin T-cell epitope containing peptides was observed. While the DQ2.5-glia-y4c epitope was typically present in 25% of the peptides pre pull- 5 down, it was identified in up to 84% of the peptides post pull-down. The ω-gliadin epitopes were identified in few peptides pre pull-down, but after enrichment they were present in 2- 19% of the peptides. The hmAbs that enriched for the motif QPQQPFP (SEQ ID NO: 261) pulled down peptides that harboured several copies of the T-cell epitopes. This was particularly notable for the hmAb 1130-3B04. The DQ2.5-glia-y4c epitope which was 0 present in 22% of the gliadin peptides in the fraction pre pull-down, was identified in 84% of the pulled down peptides and with an average of 1.7 T-cell epitopes per peptide.
Table 14. Percentage of peptides pulled down with the eleven human monoclonal antibodies that harbour known gluten T-cell epitopes. The number of T-cell epitope per peptide is given in brackets.


*31 peptides removed from the list as they were identified in the negative control sample
5 Gliadin B- and T-cell epitopes appear to be in close proximity or overlap (Osman et al, Clin Exp Immonol 2000, 121 :248-54; Aleanzi et al, Clin Chem 2001, 47:2023-8). Our results confirm this finding. The hmAb binding motif QPQQPFP (SEQ ID NO: 261) and the
T-cell epitopes are most often overlapping in the gliadin proteins. This is particularly striking in the ω-gliadin protein (Accession number: Q9FUW7) visualised in Figure 24. In this
protein, 9 copies of the 7mer motif are present. All copies, except one, are overlapping with one or more T-cell epitopes. The DQ2.5-glia-y5 epitope overlaps with four copies of the binding motif, DQ2.5-glia-y4c overlaps with three copies, while DQ2.5-glia-col and DQ2.5- glia-cω2 both overlap with one copy.
This example describes the natural binding targets of eleven gliadin-specific hmAbs made by expression cloning of antibody genes of single intestinal IgA+ plasma cells from coeliac disease patients. The natural binding targets were identified by isolating and sequencing a large number of fragments pulled down from fractions of gliadin that had been treated with digestive enzymes and TG2. The majority of the hmAbs were established from staining plasma cells with labelled synthetic peptides. In several instances, the hmAbs selected for peptides which differed from the selecting peptides. Several interesting observations emerge from our experiments.
The hmAbs pulled down long peptide fragments of γ-gliadins, ω-gliadins and low molecular weight glutenins that all harboured repeated motifs. For the majority of the hmAbs, this type of motifs could be identified. The motifs all contained a short PQQ sequence, but they differed by a few variations in the flanking residues. While the majority of the hmAbs pulled down peptides that shared the QPQQPFP (SEQ ID NO: 261) motif, some of the hmAbs were more promiscuous and enriched for peptides that harboured up to four different amino acids in certain positions of the 7mer motif. Testing different peptides with the same sequence core (QPEQPFP (SEQ ID NO: 594)), but with various flanking regions in a competitive AlphaLISA assay, revealed that the antibodies' affinity for the different peptides varied greatly. These results indicate that the antibody response to gliadin in coeliac disease is generated in response to a few immunodominant epitopes, typically displayed in repeats, and that flanking amino acids affect the hmAb affinity for variant peptides.
The enrichment for long fragments with repeated motifs likely relate to epitope multivalency. This enrichment was observed in experimental settings where the multivalent peptide fragments could engage more than one antibody molecule. This scenario mimics the situation at the surface of a B cell where a multivalent antigen would be able to engage several B-cell receptors on the cell surface. This gives B-cell receptor crosslinking and B-cell activation and thereby causes a strong selection of the B-cell epitopes. This may be a reason why the B-cell epitopes in gliadin are sequence motifs that have multivalent display within long proteolytically resistant fragments.
The peptide fragments pulled down by the hmAbs typically contained glutamate residues introduced by TG2-mediated deamidation. Further, in general, there was an
enrichment of deami dated peptides when comparing pre and post pull-down samples. This was the case even with hmAbs that did not distinguish between synthetic peptides in native and deamidated versions in ELISA. The reason for this is that the QPQQPFP (SEQ ID NO: 261) motif contains the QXP motif typically targeted by TG2 (Vader et al, J Exp Med 2002, 195:643-9; Fleckenstein et al, J Biol Chem 2002, 277:34109-16), and the hmAbs react with deamidated peptides in the TG2 -treated digests even though the glutamate residue is not necessarily part of the epitope.
The gliadin-specific hmAbs typically pulled down peptides with multiple gliadin T- cell epitopes, where the hmAb binding motif and the T-cell epitopes overlapped or were in close proximity. This argues for a role for gluten-specific B cells as important antigen presenting cells in coeliac disease. Together with the finding that the hmAbs cross-react with different gliadin peptides, it indicates that the gliadin-specific B-cells may take up and display many different T-cell epitopes and consequently get help from many distinct gliadin- specific T cells, which have been demonstrated to be important for generating B-cell responses (Shulman et al, Science 2013, 341 :673-7).
The dominant T-cell response in coeliac disease is directed towards a-gliadin and ω- gliadin peptides (Sollid et al, Immunogenetics 2012, 64:455-60; Arentz-Hansen et al, J Exp Med 2000, 191 :603-12; Tye-Din et al, Sci Transl Med 2010, 2:41ra51), while the B-cell response is directed to y/ ω-peptides (Osman et al, Clin Exp Immonol 2000, 121 :248-54; Ballew et al, PNAS 2013, 110: 19330-5). This study demonstrated a preferential hmAb- enrichment of fragments from y-gliadin, ω-gliadin and LMW glutenin proteins. Few fragments of α-gliadin proteins were identified in the pull down analyses. Epitopes of a- gliadin are important for T cells in coeliac disease (Sollid et al, Immunogenetics 2012, 64:455-60). Testing of synthetic peptides demonstrated that the abundance of the different peptides in the gliadin fractions affected which peptides were pulled down with the hmAbs. Further, the gel filtration fractions of the gliadin digest containing the highest molecular weight peptides were not used in the pull-down experiments to facilitate the identification of motifs recognised by the hmAbs. Thus, long peptide fragments like the α-gliadin 33mer, may to some extent have been excluded from these analyses. Several hmAbs enriched for peptides harbouring the T-cell epitope DQ2.5-glia-cωl, which is similar to the DQ2.5-glia-al epitope and contain the B-cell epitope motif QPQQPFP. The DQ2.5-glia-cωl epitope is also an immunodominant T-cell epitope (Tye-Din et al, Sci Transl Med 2010, 2:41ra51).
Table 15. Peptides pulled down with gliadin-specific antibodies. The frequencies of common motifs among pulled down peptides, and the sequences of top 20 peptides abundant peptides (intensity) for each antibody are shown.

1002-1E01 (Motif: QPQQPFP (SEQ ID NO: 261))


(de): deamidation
(gl): Gln->pyro-Glu 1130-3B04 (Motif: QPQQPFP (SEQ ID NO: 261))


(de): deamidation
(ox): oxidation
(gl): Gln->pyro-Glu
1130-3A02 (Motif: QPQQPFP (SEQ ID NO: 261))

(de): deamidation
1130-2A02 (Motif: QPQQPFP (SEQ ID NO: 261))


(de): deamidation
1002-1E03 (Motif: QPQQPFP (SEQ ID NO: 261))


(de): deamidation
(gl): Gln->pyro-Glu 1130-3B01 (Motif: QPQQXFP, X=P/S/T/Q) (SEQ ID NO: 575)

(de): deamidation
(gl): Gln->pyro-Glu
1130-3A05 (Motif: QPQQPFP (SEQ ID NO: 261))


(de): deamidation
1114-1G01 (Motif: XiQPQQPx2 (Xi=P, S; X2=I,L,F) (SEQ ID NO: 5)

(de): deamidation
(gl): Gln->pyro-Glu
1050-5B05 (Motif: XiQPQQPXz (X^Q P, l/L; X2=F,Q,A)) (SEQ ID NO: 6)


(de): deamidation
1130-3B03 (no common motif)

(de): deamidation
1130-3G03 (no common motif)

(de): deamidation (gl): Gln->pyro-Glu
Claims
1. An isolated monoclonal antibody that binds to gliadin, wherein said antibody recognizes an epitope selected from the group consisting of the peptides QPXQPFP (SEQ ID NO: 1), wherein X is Q or E; QPQQXFP (SEQ ID NO: 2), wherein X is P, S, T, or Q; PLQPEQPFP (SEQ ID NO: 3); PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4); X1QPQQPX2 (SEQ ID NO: 5), wherein Xi is P or S and X2 is I, L, or F; XiQPQQPX2 (SEQ ID NO: 6), wherein X1 is Q, P, I, or L and X2 is F, Q, or A;
LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17); LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18);
QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259); and
QPEQPFPEQPEQPEQPFPQPEQPFPW (SEQ ID NO: 260).
2. The antibody of claim 1, wherein said antibody is an antibody fragment.
3. The antibody of claim 2, wherein said fragment is selected from Fab, Fab', Fab'-SH, F(ab')2, Fv, and scFv variants.
4. The antibody of claim 1 , wherein said antibody is a full length antibody.
5. The antibody of claim 1, wherein said antibody is fused to a non-antibody molecule.
6. The antibody of claim 5, wherein said non-antibody molecule is a label.
7. The antibody of any one of claims 1 to 6, wherein said antibody has a complementarity determining region sequence encoded by a sequence shown in Table 2 or sequences that are at least 80% homologous to the sequences shown in Table 2.
8. The antibody of any one of claims 1 to 7, wherein at least one amino acid of said peptide is deamidated.
9. A method of detecting a peptide comprising an epitope selected from the the group consisting of QPXQPFP (SEQ ID NO: 1), wherein X is Q or E; QPQQXFP (SEQ ID
NO: 2), wherein X is P, S, T, or Q; PLQPEQPFP (SEQ ID NO: 3);
PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4); XiQPQQPX2 (SEQ ID NO: 5), wherein X1 is P or S and X2 is I, L, or F; XiQPQQPX2 (SEQ ID NO: 6), wherein X1 is Q, P, I, or L and X2 is F, Q, or A; LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17); LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18); QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259); and QPEQPFPEQPEQPEQPFPQPEQPFPW (SEQ ID NO: 260) comprising:
a) a contacting an isolated monoclonal antibody that binds to gliadin, wherein said antibody recognizes an epitope selected from the group consisting of QPXQPFP (SEQ ID NO: 1), wherein X is Q or E; QPQQXFP (SEQ ID NO: 2), wherein X is P,
S, T, or Q; PLQPEQPFP (SEQ ID NO: 3); PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4); XiQPQQPX2 (SEQ ID NO: 5), wherein Xi is P or S and X2 is I, L, or F; X1QPQQPX2 (SEQ ID NO: 6), wherein Xi is Q, P, I, or L and X2 is F, Q, or A;
LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17); LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO:
18); QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259); and QPEQPFPEQPEQPEQPFPQPEQPFPW (SEQ ID NO: 260) with a sample; and b) detecting the binding of said antibody to said epitope.
10. The method of claim 9, wherein said monoclonal antibody is attached to a solid support.
1 1. The method of claim 10, wherein said solid support is a bead.
12. The method of claim 10 or 1 1 , wherein at least a portion of said solid support is labeled.
13. The method of any one of claims 9 to 12, wherein said monoclonal antibody and/or said petide are labeled.
14. The method of clailm 13, wherein said label is biotin.
15. The method of claim 13 or 14, wherein said label is attached to said monoclonal antibody or peptide via a linker.
16. The method of claim 15, wherein said linker is GSGSGS.
17. The method of any one of claims 9 to 16, wherein at least one amino acid of said petide is deamidated.
18. A method of diagnosing celiac disease, comprising:
a) contacting an antibody of any one of claims 1 to 8 or a peptide comprising an epitope selected from the group consisting of QPXQPFP (SEQ ID NO: 1), wherein X is Q or E; QPQQXFP (SEQ ID NO: 2), wherein X is P, S, T, or Q; PLQPEQPFP (SEQ ID NO: 3); PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4); XiQPQQPX2 (SEQ ID NO: 5), wherein Xi is P or S and X2 is I, L, or F; XiQPQQPX2 (SEQ ID NO: 6), wherein Xi is Q, P, I, or L and X2 is F, Q, or A; LQLQPFPQPELPYPQPELPYPQPELPYPQP (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17); LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18); QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259); and
QPEQPFPEQPEQPEQPFPQPEQPFPW (SEQ ID NO: 260) with a biological sample from a subject; and
b) measuring the level of binding of antibodies to gliadin in said sample.
19. The method of claim 18, wherein said sample is selected from the group consisting of serum, whole blood, saliva, or urine.
20. The method of claim 18 or 19, wherein said antibody or said peptide is attached to a solid support.
21. The method of any one of claims 18 to 20, wherein both said antibody and said peptide are present and wherein said assay gives a detactable signal when said antibodies to gliadin are not present in said sample and a reduced signal when said antibodies to gliadin are present in said sample.
22. A method of diagnosing celiac disease, comprising:
a) contacting a peptide comprising an epitope selected from the group consisting of QPXQPFP (SEQ ID NO: 1), wherein X is Q or E; QPQQXFP (SEQ ID NO: 2), wherein X is P, S, T, or Q; PLQPEQPFP (SEQ ID NO: 3); PQPEQPFPQPEQPFPQPEQPFPQP (SEQ
ID NO: 4); XiQPQQPX2 (SEQ ID NO: 5), wherein X1 is P or S and X2 is I, L, or F;
X1QPQQPX2 (SEQ ID NO: 6), wherein Xl is Q, P, I, or L and X2 is F, Q, or A;
LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17); LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18);
QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259) and
QPEQPFPEQPEQPEQPFPQPEQPFPW (SEQ ID NO: 260) with a sample from a subject; and
b) measuring the level of binding of said peptide to antibodies to gliadin in said sample.
23. The method of claim 22, wherein said peptide is attached to a solid support.
24. The method of claim 22, wherein said peptide comprise a label.
25. The method of any one of claims 22 to 24, wherein said level of binding is compared to the level of binding in a control serum from a subject that does not have celiac disease.
26. The method of claim 25, wherein an increased level of binding relative to the level of binding found in the control sample is indicative of celiac disease in said subject.
27. The method of any one of claims 22 to 24, wherein said monoclonal antibody and/or said peptide are labeled.
28. A method of detecting gluten in a food sample, comprising:
a) contacting a food sample with an antibody of any one of claims 1 to 8; and
b) detecting the binding of said antibody to gliadin in said sample.
29. The method of claim 28, wherein said monoclonal antibody is labeled.
30. A kit comprising the antibody of any one of claims 1 to 8 or a peptide comprising an epitope selected from the group consisting of QPXQPFP (SEQ ID NO: 1), wherein X is Q or E; QPQQXFP (SEQ ID NO: 2), wherein X is P, S, T, or Q; PLQPEQPFP
(SEQ ID NO: 3); PQPEQPFPQPEQPFPQPEQPFPQP (SEQ ID NO: 4); XiQPQQPX2 (SEQ ID NO: 5), wherein Xi is P or S and X2 is I, L, or F; XiQPQQPX2 (SEQ ID NO: 6), wherein Xi is Q, P, I, or L and X2 is F, Q, or A; LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 7); PLQPEQPFP (SEQ ID NO: 17);
LQLQPFPQPELPYPQPELPYPQPELPYPQPQPF (SEQ ID NO: 18);
QPEQPFPEQPEQPEQPFPQPEQPFPWQPEQPFPQ (SEQ ID NO: 259) and
QPEQPFPEQPEQPEQPFPQPEQPFPW (SEQ ID NO: 260) and a buffer.
31. The kit of claim 30, wherein said kit further comprises a solid support.
32. The kit of claim 30 or 31 , wherein said antibody or said peptide is affixed to said solid support.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461983718P | 2014-04-24 | 2014-04-24 | |
| US61/983,718 | 2014-04-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015164615A1 true WO2015164615A1 (en) | 2015-10-29 |
Family
ID=53053124
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2015/027315 WO2015164615A1 (en) | 2014-04-24 | 2015-04-23 | Anti-gluten antibodies and uses thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2015164615A1 (en) |
Citations (80)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4676980A (en) | 1985-09-23 | 1987-06-30 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Target specific cross-linked heteroantibodies |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| EP0404097A2 (en) | 1989-06-22 | 1990-12-27 | BEHRINGWERKE Aktiengesellschaft | Bispecific and oligospecific, mono- and oligovalent receptors, production and applications thereof |
| WO1993001161A1 (en) | 1991-07-11 | 1993-01-21 | Pfizer Limited | Process for preparing sertraline intermediates |
| WO1993008829A1 (en) | 1991-11-04 | 1993-05-13 | The Regents Of The University Of California | Compositions that mediate killing of hiv-infected cells |
| WO1993016185A2 (en) | 1992-02-06 | 1993-08-19 | Creative Biomolecules, Inc. | Biosynthetic binding protein for cancer marker |
| WO1994029351A2 (en) | 1993-06-16 | 1994-12-22 | Celltech Limited | Antibodies |
| US5500362A (en) | 1987-01-08 | 1996-03-19 | Xoma Corporation | Chimeric antibody with specificity to human B cell surface antigen |
| US5571894A (en) | 1991-02-05 | 1996-11-05 | Ciba-Geigy Corporation | Recombinant antibodies specific for a growth factor receptor |
| US5587458A (en) | 1991-10-07 | 1996-12-24 | Aronex Pharmaceuticals, Inc. | Anti-erbB-2 antibodies, combinations thereof, and therapeutic and diagnostic uses thereof |
| US5624821A (en) | 1987-03-18 | 1997-04-29 | Scotgen Biopharmaceuticals Incorporated | Antibodies with altered effector functions |
| US5648237A (en) | 1991-09-19 | 1997-07-15 | Genentech, Inc. | Expression of functional antibody fragments |
| WO1997030087A1 (en) | 1996-02-16 | 1997-08-21 | Glaxo Group Limited | Preparation of glycosylated antibodies |
| US5731168A (en) | 1995-03-01 | 1998-03-24 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| US5750373A (en) | 1990-12-03 | 1998-05-12 | Genentech, Inc. | Enrichment method for variant proteins having altered binding properties, M13 phagemids, and growth hormone variants |
| US5770429A (en) | 1990-08-29 | 1998-06-23 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5789199A (en) | 1994-11-03 | 1998-08-04 | Genentech, Inc. | Process for bacterial production of polypeptides |
| US5821337A (en) | 1991-06-14 | 1998-10-13 | Genentech, Inc. | Immunoglobulin variants |
| US5840523A (en) | 1995-03-01 | 1998-11-24 | Genetech, Inc. | Methods and compositions for secretion of heterologous polypeptides |
| WO1998058964A1 (en) | 1997-06-24 | 1998-12-30 | Genentech, Inc. | Methods and compositions for galactosylated glycoproteins |
| US5869046A (en) | 1995-04-14 | 1999-02-09 | Genentech, Inc. | Altered polypeptides with increased half-life |
| WO1999022764A1 (en) | 1997-10-31 | 1999-05-14 | Genentech, Inc. | Methods and compositions comprising glycoprotein glycoforms |
| US5959177A (en) | 1989-10-27 | 1999-09-28 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| WO1999051642A1 (en) | 1998-04-02 | 1999-10-14 | Genentech, Inc. | Antibody variants and fragments thereof |
| US6040498A (en) | 1998-08-11 | 2000-03-21 | North Caroline State University | Genetically engineered duckweed |
| US6075181A (en) | 1990-01-12 | 2000-06-13 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| WO2000061739A1 (en) | 1999-04-09 | 2000-10-19 | Kyowa Hakko Kogyo Co., Ltd. | Method for controlling the activity of immunologically functional molecule |
| US6150584A (en) | 1990-01-12 | 2000-11-21 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| WO2000069902A1 (en) | 1999-05-17 | 2000-11-23 | Conjuchem, Inc. | Long lasting fusion peptide inhibitors or viral infection |
| US6194551B1 (en) | 1998-04-02 | 2001-02-27 | Genentech, Inc. | Polypeptide variants |
| WO2001029246A1 (en) | 1999-10-19 | 2001-04-26 | Kyowa Hakko Kogyo Co., Ltd. | Process for producing polypeptide |
| US6248516B1 (en) | 1988-11-11 | 2001-06-19 | Medical Research Council | Single domain ligands, receptors comprising said ligands methods for their production, and use of said ligands and receptors |
| WO2001079271A1 (en) | 2000-04-12 | 2001-10-25 | Principia Pharmaceutical Corporation | Albumin fusion proteins |
| WO2002031140A1 (en) | 2000-10-06 | 2002-04-18 | Kyowa Hakko Kogyo Co., Ltd. | Cells producing antibody compositions |
| US6420548B1 (en) | 1999-10-04 | 2002-07-16 | Medicago Inc. | Method for regulating transcription of foreign genes |
| US20020164328A1 (en) | 2000-10-06 | 2002-11-07 | Toyohide Shinkawa | Process for purifying antibody |
| WO2003011878A2 (en) | 2001-08-03 | 2003-02-13 | Glycart Biotechnology Ag | Antibody glycosylation variants having increased antibody-dependent cellular cytotoxicity |
| US20030115614A1 (en) | 2000-10-06 | 2003-06-19 | Yutaka Kanda | Antibody composition-producing cell |
| WO2003059934A2 (en) | 2001-12-21 | 2003-07-24 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| US6602684B1 (en) | 1998-04-20 | 2003-08-05 | Glycart Biotechnology Ag | Glycosylation engineering of antibodies for improving antibody-dependent cellular cytotoxicity |
| US20030157108A1 (en) | 2001-10-25 | 2003-08-21 | Genentech, Inc. | Glycoprotein compositions |
| WO2003085107A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | Cells with modified genome |
| WO2003085119A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | METHOD OF ENHANCING ACTIVITY OF ANTIBODY COMPOSITION OF BINDING TO FcϜ RECEPTOR IIIa |
| WO2003084570A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | DRUG CONTAINING ANTIBODY COMPOSITION APPROPRIATE FOR PATIENT SUFFERING FROM FcϜRIIIa POLYMORPHISM |
| US20040093621A1 (en) | 2001-12-25 | 2004-05-13 | Kyowa Hakko Kogyo Co., Ltd | Antibody composition which specifically binds to CD20 |
| US6737056B1 (en) | 1999-01-15 | 2004-05-18 | Genentech, Inc. | Polypeptide variants with altered effector function |
| US20040109865A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Antibody composition-containing medicament |
| US20040110282A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Cells in which activity of the protein involved in transportation of GDP-fucose is reduced or lost |
| WO2004056312A2 (en) | 2002-12-16 | 2004-07-08 | Genentech, Inc. | Immunoglobulin variants and uses thereof |
| US20040132140A1 (en) | 2002-04-09 | 2004-07-08 | Kyowa Hakko Kogyo Co., Ltd. | Production process for antibody composition |
| US20050014934A1 (en) | 2002-10-15 | 2005-01-20 | Hinton Paul R. | Alteration of FcRn binding affinities or serum half-lives of antibodies by mutagenesis |
| US20050079574A1 (en) | 2003-01-16 | 2005-04-14 | Genentech, Inc. | Synthetic antibody phage libraries |
| WO2005035778A1 (en) | 2003-10-09 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | PROCESS FOR PRODUCING ANTIBODY COMPOSITION BY USING RNA INHIBITING THE FUNCTION OF α1,6-FUCOSYLTRANSFERASE |
| WO2005035586A1 (en) | 2003-10-08 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | Fused protein composition |
| US20050119455A1 (en) | 2002-06-03 | 2005-06-02 | Genentech, Inc. | Synthetic antibody phage libraries |
| US20050123546A1 (en) | 2003-11-05 | 2005-06-09 | Glycart Biotechnology Ag | Antigen binding molecules with increased Fc receptor binding affinity and effector function |
| WO2005053742A1 (en) | 2003-12-04 | 2005-06-16 | Kyowa Hakko Kogyo Co., Ltd. | Medicine containing antibody composition |
| WO2005100402A1 (en) | 2004-04-13 | 2005-10-27 | F.Hoffmann-La Roche Ag | Anti-p-selectin antibodies |
| US20050266000A1 (en) | 2004-04-09 | 2005-12-01 | Genentech, Inc. | Variable domain library and uses |
| US6982321B2 (en) | 1986-03-27 | 2006-01-03 | Medical Research Council | Altered antibodies |
| US20060025576A1 (en) | 2000-04-11 | 2006-02-02 | Genentech, Inc. | Multivalent antibodies and uses therefor |
| WO2006029879A2 (en) | 2004-09-17 | 2006-03-23 | F.Hoffmann-La Roche Ag | Anti-ox40l antibodies |
| US7041870B2 (en) | 2000-11-30 | 2006-05-09 | Medarex, Inc. | Transgenic transchromosomal rodents for making human antibodies |
| US7087409B2 (en) | 1997-12-05 | 2006-08-08 | The Scripps Research Institute | Humanization of murine antibody |
| US7125978B1 (en) | 1999-10-04 | 2006-10-24 | Medicago Inc. | Promoter for regulating expression of foreign genes |
| US7189826B2 (en) | 1997-11-24 | 2007-03-13 | Institute For Human Genetics And Biochemistry | Monoclonal human natural antibodies |
| US20070061900A1 (en) | 2000-10-31 | 2007-03-15 | Murphy Andrew J | Methods of modifying eukaryotic cells |
| US20070117126A1 (en) | 1999-12-15 | 2007-05-24 | Genentech, Inc. | Shotgun scanning |
| US20070160598A1 (en) | 2005-11-07 | 2007-07-12 | Dennis Mark S | Binding polypeptides with diversified and consensus vh/vl hypervariable sequences |
| US20070237764A1 (en) | 2005-12-02 | 2007-10-11 | Genentech, Inc. | Binding polypeptides with restricted diversity sequences |
| US20070292936A1 (en) | 2006-05-09 | 2007-12-20 | Genentech, Inc. | Binding polypeptides with optimized scaffolds |
| US20080069820A1 (en) | 2006-08-30 | 2008-03-20 | Genentech, Inc. | Multispecific antibodies |
| US7371826B2 (en) | 1999-01-15 | 2008-05-13 | Genentech, Inc. | Polypeptide variants with altered effector function |
| WO2008077546A1 (en) | 2006-12-22 | 2008-07-03 | F. Hoffmann-La Roche Ag | Antibodies against insulin-like growth factor i receptor and uses thereof |
| US20090002360A1 (en) | 2007-05-25 | 2009-01-01 | Innolux Display Corp. | Liquid crystal display device and method for driving same |
| US7527791B2 (en) | 2004-03-31 | 2009-05-05 | Genentech, Inc. | Humanized anti-TGF-beta antibodies |
| US7550432B2 (en) | 1995-12-30 | 2009-06-23 | Novozymes Biopharma Uk Limited | Recombinant fusion proteins to growth hormone and serum albumin |
| WO2009089004A1 (en) | 2008-01-07 | 2009-07-16 | Amgen Inc. | Method for making antibody fc-heterodimeric molecules using electrostatic steering effects |
| WO2012056407A1 (en) * | 2010-10-26 | 2012-05-03 | Technion Research & Development Foundation Ltd. | Antibodies which bind soluble t-cell receptor ligands |
| WO2013185165A1 (en) * | 2012-06-14 | 2013-12-19 | The Walter And Eliza Hall Institute Of Medical Research | Cd-52 antibodies and their use in determining and enhancing an immune response in a subject |
-
2015
- 2015-04-23 WO PCT/US2015/027315 patent/WO2015164615A1/en active Application Filing
Patent Citations (85)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4676980A (en) | 1985-09-23 | 1987-06-30 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Target specific cross-linked heteroantibodies |
| US6982321B2 (en) | 1986-03-27 | 2006-01-03 | Medical Research Council | Altered antibodies |
| US5500362A (en) | 1987-01-08 | 1996-03-19 | Xoma Corporation | Chimeric antibody with specificity to human B cell surface antigen |
| US5624821A (en) | 1987-03-18 | 1997-04-29 | Scotgen Biopharmaceuticals Incorporated | Antibodies with altered effector functions |
| US5648260A (en) | 1987-03-18 | 1997-07-15 | Scotgen Biopharmaceuticals Incorporated | DNA encoding antibodies with altered effector functions |
| US6248516B1 (en) | 1988-11-11 | 2001-06-19 | Medical Research Council | Single domain ligands, receptors comprising said ligands methods for their production, and use of said ligands and receptors |
| EP0404097A2 (en) | 1989-06-22 | 1990-12-27 | BEHRINGWERKE Aktiengesellschaft | Bispecific and oligospecific, mono- and oligovalent receptors, production and applications thereof |
| US5959177A (en) | 1989-10-27 | 1999-09-28 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| US6417429B1 (en) | 1989-10-27 | 2002-07-09 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| US6075181A (en) | 1990-01-12 | 2000-06-13 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US6150584A (en) | 1990-01-12 | 2000-11-21 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US5770429A (en) | 1990-08-29 | 1998-06-23 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5750373A (en) | 1990-12-03 | 1998-05-12 | Genentech, Inc. | Enrichment method for variant proteins having altered binding properties, M13 phagemids, and growth hormone variants |
| US5571894A (en) | 1991-02-05 | 1996-11-05 | Ciba-Geigy Corporation | Recombinant antibodies specific for a growth factor receptor |
| US5821337A (en) | 1991-06-14 | 1998-10-13 | Genentech, Inc. | Immunoglobulin variants |
| WO1993001161A1 (en) | 1991-07-11 | 1993-01-21 | Pfizer Limited | Process for preparing sertraline intermediates |
| US5648237A (en) | 1991-09-19 | 1997-07-15 | Genentech, Inc. | Expression of functional antibody fragments |
| US5587458A (en) | 1991-10-07 | 1996-12-24 | Aronex Pharmaceuticals, Inc. | Anti-erbB-2 antibodies, combinations thereof, and therapeutic and diagnostic uses thereof |
| WO1993008829A1 (en) | 1991-11-04 | 1993-05-13 | The Regents Of The University Of California | Compositions that mediate killing of hiv-infected cells |
| WO1993016185A2 (en) | 1992-02-06 | 1993-08-19 | Creative Biomolecules, Inc. | Biosynthetic binding protein for cancer marker |
| WO1994029351A2 (en) | 1993-06-16 | 1994-12-22 | Celltech Limited | Antibodies |
| US5789199A (en) | 1994-11-03 | 1998-08-04 | Genentech, Inc. | Process for bacterial production of polypeptides |
| US5840523A (en) | 1995-03-01 | 1998-11-24 | Genetech, Inc. | Methods and compositions for secretion of heterologous polypeptides |
| US5731168A (en) | 1995-03-01 | 1998-03-24 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| US5869046A (en) | 1995-04-14 | 1999-02-09 | Genentech, Inc. | Altered polypeptides with increased half-life |
| US7550432B2 (en) | 1995-12-30 | 2009-06-23 | Novozymes Biopharma Uk Limited | Recombinant fusion proteins to growth hormone and serum albumin |
| WO1997030087A1 (en) | 1996-02-16 | 1997-08-21 | Glaxo Group Limited | Preparation of glycosylated antibodies |
| WO1998058964A1 (en) | 1997-06-24 | 1998-12-30 | Genentech, Inc. | Methods and compositions for galactosylated glycoproteins |
| WO1999022764A1 (en) | 1997-10-31 | 1999-05-14 | Genentech, Inc. | Methods and compositions comprising glycoprotein glycoforms |
| US7189826B2 (en) | 1997-11-24 | 2007-03-13 | Institute For Human Genetics And Biochemistry | Monoclonal human natural antibodies |
| US7087409B2 (en) | 1997-12-05 | 2006-08-08 | The Scripps Research Institute | Humanization of murine antibody |
| WO1999051642A1 (en) | 1998-04-02 | 1999-10-14 | Genentech, Inc. | Antibody variants and fragments thereof |
| US6194551B1 (en) | 1998-04-02 | 2001-02-27 | Genentech, Inc. | Polypeptide variants |
| US6602684B1 (en) | 1998-04-20 | 2003-08-05 | Glycart Biotechnology Ag | Glycosylation engineering of antibodies for improving antibody-dependent cellular cytotoxicity |
| US6040498A (en) | 1998-08-11 | 2000-03-21 | North Caroline State University | Genetically engineered duckweed |
| US7332581B2 (en) | 1999-01-15 | 2008-02-19 | Genentech, Inc. | Polypeptide variants with altered effector function |
| US7371826B2 (en) | 1999-01-15 | 2008-05-13 | Genentech, Inc. | Polypeptide variants with altered effector function |
| US6737056B1 (en) | 1999-01-15 | 2004-05-18 | Genentech, Inc. | Polypeptide variants with altered effector function |
| WO2000061739A1 (en) | 1999-04-09 | 2000-10-19 | Kyowa Hakko Kogyo Co., Ltd. | Method for controlling the activity of immunologically functional molecule |
| WO2000069902A1 (en) | 1999-05-17 | 2000-11-23 | Conjuchem, Inc. | Long lasting fusion peptide inhibitors or viral infection |
| US7125978B1 (en) | 1999-10-04 | 2006-10-24 | Medicago Inc. | Promoter for regulating expression of foreign genes |
| US6420548B1 (en) | 1999-10-04 | 2002-07-16 | Medicago Inc. | Method for regulating transcription of foreign genes |
| WO2001029246A1 (en) | 1999-10-19 | 2001-04-26 | Kyowa Hakko Kogyo Co., Ltd. | Process for producing polypeptide |
| US20070117126A1 (en) | 1999-12-15 | 2007-05-24 | Genentech, Inc. | Shotgun scanning |
| US20060025576A1 (en) | 2000-04-11 | 2006-02-02 | Genentech, Inc. | Multivalent antibodies and uses therefor |
| US7785599B2 (en) | 2000-04-12 | 2010-08-31 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| WO2001079271A1 (en) | 2000-04-12 | 2001-10-25 | Principia Pharmaceutical Corporation | Albumin fusion proteins |
| US20020164328A1 (en) | 2000-10-06 | 2002-11-07 | Toyohide Shinkawa | Process for purifying antibody |
| US20030115614A1 (en) | 2000-10-06 | 2003-06-19 | Yutaka Kanda | Antibody composition-producing cell |
| WO2002031140A1 (en) | 2000-10-06 | 2002-04-18 | Kyowa Hakko Kogyo Co., Ltd. | Cells producing antibody compositions |
| US20070061900A1 (en) | 2000-10-31 | 2007-03-15 | Murphy Andrew J | Methods of modifying eukaryotic cells |
| US7041870B2 (en) | 2000-11-30 | 2006-05-09 | Medarex, Inc. | Transgenic transchromosomal rodents for making human antibodies |
| WO2003011878A2 (en) | 2001-08-03 | 2003-02-13 | Glycart Biotechnology Ag | Antibody glycosylation variants having increased antibody-dependent cellular cytotoxicity |
| US20030157108A1 (en) | 2001-10-25 | 2003-08-21 | Genentech, Inc. | Glycoprotein compositions |
| WO2003059934A2 (en) | 2001-12-21 | 2003-07-24 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| US20040093621A1 (en) | 2001-12-25 | 2004-05-13 | Kyowa Hakko Kogyo Co., Ltd | Antibody composition which specifically binds to CD20 |
| US20040109865A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Antibody composition-containing medicament |
| US20040110282A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Cells in which activity of the protein involved in transportation of GDP-fucose is reduced or lost |
| US20040110704A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Cells of which genome is modified |
| WO2003084570A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | DRUG CONTAINING ANTIBODY COMPOSITION APPROPRIATE FOR PATIENT SUFFERING FROM FcϜRIIIa POLYMORPHISM |
| US20040132140A1 (en) | 2002-04-09 | 2004-07-08 | Kyowa Hakko Kogyo Co., Ltd. | Production process for antibody composition |
| WO2003085119A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | METHOD OF ENHANCING ACTIVITY OF ANTIBODY COMPOSITION OF BINDING TO FcϜ RECEPTOR IIIa |
| WO2003085107A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | Cells with modified genome |
| US20050119455A1 (en) | 2002-06-03 | 2005-06-02 | Genentech, Inc. | Synthetic antibody phage libraries |
| US20050014934A1 (en) | 2002-10-15 | 2005-01-20 | Hinton Paul R. | Alteration of FcRn binding affinities or serum half-lives of antibodies by mutagenesis |
| WO2004056312A2 (en) | 2002-12-16 | 2004-07-08 | Genentech, Inc. | Immunoglobulin variants and uses thereof |
| US20050079574A1 (en) | 2003-01-16 | 2005-04-14 | Genentech, Inc. | Synthetic antibody phage libraries |
| WO2005035586A1 (en) | 2003-10-08 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | Fused protein composition |
| WO2005035778A1 (en) | 2003-10-09 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | PROCESS FOR PRODUCING ANTIBODY COMPOSITION BY USING RNA INHIBITING THE FUNCTION OF α1,6-FUCOSYLTRANSFERASE |
| US20050123546A1 (en) | 2003-11-05 | 2005-06-09 | Glycart Biotechnology Ag | Antigen binding molecules with increased Fc receptor binding affinity and effector function |
| WO2005053742A1 (en) | 2003-12-04 | 2005-06-16 | Kyowa Hakko Kogyo Co., Ltd. | Medicine containing antibody composition |
| US7527791B2 (en) | 2004-03-31 | 2009-05-05 | Genentech, Inc. | Humanized anti-TGF-beta antibodies |
| US20050266000A1 (en) | 2004-04-09 | 2005-12-01 | Genentech, Inc. | Variable domain library and uses |
| WO2005100402A1 (en) | 2004-04-13 | 2005-10-27 | F.Hoffmann-La Roche Ag | Anti-p-selectin antibodies |
| WO2006029879A2 (en) | 2004-09-17 | 2006-03-23 | F.Hoffmann-La Roche Ag | Anti-ox40l antibodies |
| US20070160598A1 (en) | 2005-11-07 | 2007-07-12 | Dennis Mark S | Binding polypeptides with diversified and consensus vh/vl hypervariable sequences |
| US20070237764A1 (en) | 2005-12-02 | 2007-10-11 | Genentech, Inc. | Binding polypeptides with restricted diversity sequences |
| US20070292936A1 (en) | 2006-05-09 | 2007-12-20 | Genentech, Inc. | Binding polypeptides with optimized scaffolds |
| US20080069820A1 (en) | 2006-08-30 | 2008-03-20 | Genentech, Inc. | Multispecific antibodies |
| WO2008077546A1 (en) | 2006-12-22 | 2008-07-03 | F. Hoffmann-La Roche Ag | Antibodies against insulin-like growth factor i receptor and uses thereof |
| US20090002360A1 (en) | 2007-05-25 | 2009-01-01 | Innolux Display Corp. | Liquid crystal display device and method for driving same |
| WO2009089004A1 (en) | 2008-01-07 | 2009-07-16 | Amgen Inc. | Method for making antibody fc-heterodimeric molecules using electrostatic steering effects |
| WO2012056407A1 (en) * | 2010-10-26 | 2012-05-03 | Technion Research & Development Foundation Ltd. | Antibodies which bind soluble t-cell receptor ligands |
| WO2013185165A1 (en) * | 2012-06-14 | 2013-12-19 | The Walter And Eliza Hall Institute Of Medical Research | Cd-52 antibodies and their use in determining and enhancing an immune response in a subject |
Non-Patent Citations (181)
| Title |
|---|
| ALEANZI ET AL., CLIN CHEM, vol. 47, 2001, pages 2023 - 2028 |
| ALEANZI ET AL., CLIN CHEM, vol. 47, 2001, pages 2023 - 8 |
| ALEANZI, M. ET AL., CLIN CHEM, vol. 47, no. 11, 2001, pages 2023 - 8 |
| ALMAGRO; FRANSSON, FRONT. BIOSCI., vol. 13, 2008, pages 1619 - 1633 |
| ANDERSON, R.P. ET AL., BMC MED, vol. 11, no. 1, 2013, pages 188 |
| ARENTZ-HANSEN ET AL., J EXP MED, vol. 191, 2000, pages 603 - 12 |
| BACA ET AL., J. BIOL. CHEM., vol. 272, 1997, pages 10678 - 10684 |
| BALLEW ET AL., PNAS, vol. 110, 2013, pages 19330 - 5 |
| BALLEW, J.T. ET AL., PROC NATL ACAD SCI USA, vol. 110, no. 48, 2013, pages 19330 - 19335 |
| BASSO ET AL., CLIN CHIM ACTA, vol. 412, 2011, pages 1662 - 7 |
| BATEMAN EA ET AL., GUT, vol. 53, 2004, pages 1274 - 1278 |
| BATEMAN ET AL., GUT, vol. 53, 2004, pages 1274 - 8 |
| BATEMAN, E.A. ET AL., GUT, vol. 53, no. 9, 2004, pages 1274 - 8 |
| BIOCHIM BIOPHYS ACTA, vol. 1097, 1991, pages 49 - 54 |
| BJORCK ET AL., J PEDIATR GASTROENTEROL NUTR, vol. 50, 2010, pages 49 - 53 |
| BOERNER ET AL., J. IMMUNOL., vol. 147, 1991, pages 86 |
| BRENNAN ET AL., SCIENCE, vol. 229, 1985, pages 81 |
| BRETT G M ET AL: "Epitope mapping studies of broad specificity monoclonal antibodies to cereal prolamins", JOURNAL OF CEREAL SCIENCE, ACADEMIC PRESS LTD, GB, vol. 29, no. 2, 1 March 1999 (1999-03-01), pages 117 - 128, XP002216285, ISSN: 0733-5210, DOI: 10.1006/JCRS.1998.0234 * |
| BRODEUR ET AL.: "Monoclonal Antibody Production Techniques and Applications", 1987, MARCEL DEKKER, INC., pages: 51 - 63 |
| BRUGGEMANN, M. ET AL., J. EXP. MED., vol. 166, 1987, pages 1351 - 1361 |
| BRUSCA, I. ET AL., CLIN CHEM LAB MED, vol. 50, no. 1, 2012, pages 111 - 7 |
| CARTER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 89, 1992, pages 4285 |
| CHAN TD ET AL., IMMUNITY, vol. 37, 2012, pages 893 - 904 |
| CHAPPELL CP ET AL., JEXP MED, vol. 209, 2012, pages 1825 - 1840 |
| CHARLTON: "Methods in Molecular Biology", vol. 248, 2003, HUMANA PRESS, pages: 245 - 254 |
| CHOTHIA; LESK, J. MOL. BIOL., vol. 196, 1987, pages 901 - 917 |
| CHOW ET AL., J CLIN GASTROENTEROL, vol. 46, 2012, pages 850 - 4 |
| CHOWDHURY, METHODS MOL. BIOL., vol. 207, 2008, pages 179 - 196 |
| CLACKSON ET AL., NATURE, vol. 352, 1991, pages 624 - 628 |
| CLARKSON ET AL., NATURE, vol. 352, 1991, pages 624 - 628 |
| CLYNES ET AL., PROC. NAT'L ACAD. SCI. USA, vol. 95, 1998, pages 652 - 656 |
| CRAGG, M.S. ET AL., BLOOD, vol. 101, 2003, pages 1045 - 1052 |
| CRAGG, M.S.; M.J. GLENNIE, BLOOD, vol. 103, 2004, pages 2738 - 2743 |
| CUNNINGHAM; WELLS, SCIENCE, vol. 244, 1989, pages 1081 - 1085 |
| DALL'ACQUA ET AL., METHODS, vol. 36, 2005, pages 43 - 60 |
| DI NIRO ET AL., J IMMUNOL, vol. 185, 2010, pages 5377 - 5383 |
| DI NIRO ET AL., NAT MED, vol. 18, 2012, pages 441 - 445 |
| DI NIRO ET AL., NAT MED, vol. 18, 2012, pages 441 - 5 |
| DI NIRO R ET AL., J IMMUNOL, vol. 185, 2010, pages 5377 - 5383 |
| DI NIRO R ET AL., NAT MED, vol. 18, 2012, pages 441 - 445 |
| DI NIRO, R. ET AL., J IMMUNOL, vol. 185, no. 9, 2010, pages 5377 - 83 |
| DORUM ET AL., J IMMUNOL, vol. 193, 2014, pages 4497 - 506 |
| DORUM S ET AL., PLOS ONE, vol. 5, 2010, pages E14056 |
| DUNCAN; WINTER, NATURE, vol. 322, 1988, pages 738 - 40 |
| EGNER, W. ET AL., J PEDIATR GASTROENTEROL NUTR, vol. 55, no. 6, 2012, pages 733 - 5 |
| ELLIS, H.J. ET AL., GUT, vol. 43, no. 2, 1998, pages 190 - 5 |
| ERICKSON LD ET AL., J CLIN INVEST, vol. 109, 2002, pages 613 - 620 |
| FELLOUSE, PROC. NATL. ACAD. SCI. USA, vol. 101, no. 34, 2004, pages 12467 - 12472 |
| FLATMAN ET AL., J. CHROMATOGR. B, vol. 848, 2007, pages 79 - 87 |
| FLECKENSTEIN ET AL., J BIOL CHEM, vol. 277, 2002, pages 34109 - 16 |
| GAZZANO-SANTORO ET AL., J. IMMUNOL. METHODS, vol. 202, 1996, pages 163 |
| GERNGROSS, NAT. BIOTECH., vol. 22, 2004, pages 1409 - 1414 |
| GLAS ET AL., L CLIN EPIDEMIOL, vol. 56, 2003, pages 1129 - 35 |
| GOODNOW CC ET AL., NAT IMMUNOL, vol. 11, 2010, pages 681 - 688 |
| GRAHAM ET AL., J. GEN VIROL., vol. 36, 1977, pages 59 |
| GRIFFITHS ET AL., EMBO J, vol. 12, 1993, pages 725 - 734 |
| GRUBER ET AL., J. IMMUNOL., vol. 152, 1994, pages 5368 |
| GUYER ET AL., J. IMMUNOL., vol. 117, 1976, pages 587 |
| HARASZI, R. ET AL.: "Analytical methods for detection of gluten in food--method developments in support offood labeling legislation", J AOAC INT, vol. 94, no. 4, 2011, pages 1006 - 25 |
| HARLOW; LANE: "Antibodies: A Laboratory Manual", 1988, COLD SPRING HARBOR LABORATORY, article "ch.14" |
| HEALTH QUALITY, O., ONT HEALTH TECHNOL ASSESS SER, vol. 10, no. 21, 2010, pages 1 - 111 |
| HELLSTROM, I ET AL., PROC. NAT'L ACAD. SCI. USA, vol. 82, 1985, pages 1499 - 1502 |
| HELLSTROM, I. ET AL., PROC. NAT'L ACAD. SCI. USA, vol. 83, 1986, pages 7059 - 7063 |
| HERLANDS RA ET AL., EUR J IMMUNOL, vol. 37, 2007, pages 3339 - 3351 |
| HILL, P.G.; G.K. HOLMES, ALIMENT PHARMACOL THER, vol. 27, no. 7, 2008, pages 572 - 7 |
| HO F, EUR J IMMUNOL, vol. 16, 1986, pages 1297 - 1301 |
| HOLLINGER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 90, 1993, pages 6444 - 6448 |
| HOOGENBOOM ET AL.: "Methods in Molecular Biology", vol. 178, 2001, HUMAN PRESS, pages: 1 - 37 |
| HOOGENBOOM; WINTER, J. MOL. BIOL., vol. 227, 1992, pages 381 - 388 |
| HUDSON ET AL., NAT. MED., vol. 9, 2003, pages 129 - 134 |
| HUSBY, S. ET AL., J PEDIATR GASTROENTEROL NUTR, vol. 54, no. 1, 2012, pages 136 - 60 |
| IDUSOGIE ET AL., J. IMMUNOL., vol. 164, 2000, pages 4178 - 4184 |
| IVERSEN R ET AL., J IMMUNOL, vol. 190, 2013, pages 5981 - 5991 |
| KABAT ET AL.: "Sequences of Proteins of Immunological Interest, 5th Ed.", 1991, NATIONAL INSTITUTES OF HEALTH |
| KABAT ET AL.: "Sequences of Proteins of Immunological Interest, 5th Ed.", vol. 1-3, 1991, NIH PUBLICATION 91-3242 |
| KAGNOFF MF, NATURE, vol. 296, 1982, pages 158 - 160 |
| KAGNOFF MF., GASTROENTEROLOGY, vol. 131, 2006, pages 1977 - 1980 |
| KAM ET AL., PROC. NATL. ACAD. SCI. USA, vol. 102, 2005, pages 11600 - 11605 |
| KANDA, Y. ET AL., BIOTECHNOL. BIOENG., vol. 94, no. 4, 2006, pages 680 - 688 |
| KASHMIRI ET AL., METHODS, vol. 36, 2005, pages 25 - 34 |
| KAWAMOTO S ET AL., SCIENCE, vol. 336, 2012, pages 485 - 489 |
| KIM ET AL., J. IMMUNOL., vol. 24, 1994, pages 249 |
| KINDT ET AL.: "Kuby Immunology 6th ed.,", 2007, W.H. FREEMAN AND CO. |
| KLIMKA ET AL., BR. J. CANCER, vol. 83, 2000, pages 252 - 260 |
| KOERNER, T.B. ET AL.: "Gluten contamination of naturally gluten-free flours and starches used by Canadians with celiac disease", FOOD ADDIT CONTAM PART A CHEM ANAL CONTROL EXPO RISK ASSESS, vol. 30, no. 12, 2013, pages 2017 - 21 |
| KOSTELNY ET AL., J. IMMUNOL., vol. 148, no. 5, 1992, pages 1547 - 1553 |
| KOZBOR, J. IMMUNOL., vol. 133, 1984, pages 3001 |
| LEE ET AL., IMMUNITY, vol. 37, 2012, pages 880 - 892 |
| LEE ET AL., J EXP MED, vol. 208, 2011, pages 1377 - 1388 |
| LEE ET AL., J. IMMUNOL. METHODS, vol. 284, no. 1-2, 2004, pages 119 - 132 |
| LEE ET AL., J. MOL. BIOL., vol. 340, no. 5, 2004, pages 1073 - 1093 |
| LEWIS ET AL., ALIMENT PHARMACOL THER, vol. 31, 2010, pages 78 - 81 |
| LI ET AL., NAT. BIOTECH., vol. 24, 2006, pages 210 - 215 |
| LI ET AL., PROC. NATL. ACAD. SCI. USA, vol. 103, 2006, pages 3557 - 3562 |
| LINGWOOD D ET AL., NATURE, vol. 489, 2012, pages 566 - 570 |
| LONBERG, CURR. OPIN. IMMUNOL., vol. 20, 2008, pages 450 - 459 |
| LONBERG, NAT. BIOTECH., vol. 23, 2005, pages 1117 - 1125 |
| MACLENNAN IC ET AL., IMMUNOL REV, vol. 194, 2003, pages 8 - 18 |
| MACLENNAN IC, ANNU REV IMMUNOL, vol. 12, 1994, pages 117 - 139 |
| MAIZELS N; BOTHWELL A, CELL, vol. 43, 1985, pages 715 - 720 |
| MAKI M: "Proceedings of the sixth international symposium coeliac disease", 1992, OAK TREE PRESS, pages: 246 - 252 |
| MARKS ET AL., J. MOL. BIOL., vol. 222, 1992, pages 581 - 597 |
| MARKS; BRADBURY: "Methods in Molecular Biology", vol. 248, 2003, HUMAN PRESS, pages: 161 - 175 |
| MATHER ET AL., ANNALS N.Y. ACAD. SCI., vol. 383, 1982, pages 44 - 68 |
| MATHER, BIOL. REPROD., vol. 23, 1980, pages 243 - 251 |
| MCCAFFERTY ET AL., NATURE, vol. 348, pages 552 - 554 |
| MILSTEIN; CUELLO, NATURE, vol. 305, 1983, pages 537 |
| MOONEY ET AL., CLIN GASTROENTEROL HEPATOL, 2015 |
| MORRIS: "Methods in Molecular Biology", vol. 66, 1996, HUMANA PRESS, article "Epitope Mapping Protocols" |
| MORRISON ET AL., PROC. NATL. ACAD. SCI. USA, vol. 81, 1984, pages 6851 - 6855 |
| NI, XIANDAI MIANYIXUE, vol. 26, no. 4, 2006, pages 265 - 268 |
| NILSEN ET AL., GUT, vol. 37, 1995, pages 766 - 776 |
| OKAZAKI ET AL., J. MOL. BIOL., vol. 336, 2004, pages 1239 - 1249 |
| OSBOURN ET AL., METHODS, vol. 36, 2005, pages 61 - 68 |
| OSMAN AA ET AL., CLIN EXP IMMUNOL, vol. 121, 2000, pages 248 - 254 |
| OSMAN ET AL., CLIN EXP IMMONOL, vol. 121, 2000, pages 248 - 54 |
| OSMAN ET AL., CLIN EXP IMMUNOL, vol. 121, 2000, pages 248 - 54 |
| OSMAN, A.A. ET AL., CLIN EXP IMMUNOL, vol. 121, no. 2, 2000, pages 248 - 54 |
| OSMAN, A.A. ET AL., EUR J GASTROENTEROL HEPATOL, vol. 13, no. 10, 2001, pages 1189 - 93 |
| PADLAN, MOL. IMMUNOL., vol. 28, 1991, pages 489 - 498 |
| PETERS, T., JR.: "All about Albumin: Biochemistry, Genetics and Medical, Applications", 1996, ACADEMIC PRESS, INC. |
| PETERSEN, N.H. ET AL., J IMMUNOL METHODS, vol. 365, no. 1-2, 2011, pages 174 - 82 |
| PETKOVA, S.B. ET AL., INT'L. IMMUNOL., vol. 18, no. 12, 2006, pages 1759 - 1769 |
| PLUCKTHUN: "The Pharmacology of Monoclonal Antibodies", vol. 113, 1994, SPRINGER-VERLAG, pages: 269 - 315 |
| PORTOLANO ET AL., J. IMMUNOL., vol. 150, 1993, pages 880 - 887 |
| PRATAMA A; VINUESA CG, IMMUNOL CELL BIOL, 2013 |
| PRESTA ET AL., J. IMMUNOL., vol. 151, 1993, pages 2623 |
| QUEEN ET AL., PROC. NAT'L ACAD. SCI. USA, vol. 86, 1989, pages 10029 - 10033 |
| RAVETCH; KINET, ANNU. REV. IMMUNOL., vol. 9, 1991, pages 457 - 492 |
| RIECHMANN ET AL., NATURE, vol. 332, 1988, pages 323 - 329 |
| RIPKA ET AL., ARCH. BIOCHEM. BIOPHYS., vol. 249, 1986, pages 533 - 545 |
| RONCORONI L ET AL., J TRANSL MED, vol. 7, 2009, pages 40 |
| ROOPENIAN D. C.; AKILESH, S., NAT.REV. IMMUNOL, vol. 7, 2007, pages 715 - 725 |
| ROSOK ET AL., J. BIOL. CHEM., vol. 271, 1996, pages 22611 - 22618 |
| ROSTOM, A.; J.A. MURRAY; M.F. KAGNOFF, GASTROENTEROLOGY, vol. 131, no. 6, 2006, pages 1981 - 2002 |
| RUBIO-TAPIA ET AL., AM J GASTROENTEROL, vol. 108, 2013, pages 656 - 76 |
| RUBIO-TAPIA, A. ET AL., AM J GASTROENTEROL, vol. 107, no. 10, 2012, pages 1538 - 44 |
| SCHWERTZ E ET AL., CLIN CHEM, vol. 50, 2004, pages 2370 - 2375 |
| SCIAMMAS R ET AL., IMMUNITY, vol. 25, 2006, pages 225 - 236 |
| SHIELDS ET AL., J. BIOL. CHEM., vol. 9, no. 2, 2001, pages 6591 - 6604 |
| SHULMAN ET AL., SCIENCE, vol. 341, 2013, pages 673 - 7 |
| SHULMAN Z ET AL., SCIENCE, vol. 341, 2013, pages 673 - 677 |
| SIDHU ET AL., J. MOL. BIOL., vol. 338, no. 2, 2004, pages 299 - 310 |
| SIMS ET AL., J. IMMUNOL., vol. 151, 1993, pages 2296 |
| SKERRITT, J.H. ET AL., JOURNAL OF CEREAL SCIENCE, vol. 2, no. 4, 1984, pages 215 - 224 |
| SKOVBJERG, H. ET AL., BIOCHIM BIOPHYS ACTA, vol. 1690, no. 3, 2004, pages 220 - 30 |
| SMITH ET AL., NAT PROTOC, vol. 4, 2009, pages 372 - 84 |
| SMITH K ET AL., NAT PROTOC, vol. 4, 2009, pages 372 - 384 |
| SOLLID ET AL., IMMUNOGENETICS, vol. 64, 2012, pages 455 - 460 |
| SOLLID ET AL., IMMUNOGENETICS, vol. 64, 2012, pages 455 - 60 |
| SOLLID LM ET AL., GUT, vol. 41, 1997, pages 851 - 852 |
| SORELL, L. ET AL., FEBS LETT, vol. 439, no. 1-2, 1998, pages 46 - 50 |
| SUGAI ET AL., CLIN CHEM, vol. 56, 2010, pages 661 - 665 |
| SULKANEN S ET AL., GASTROENTEROLOGY, vol. 115, 1998, pages 1322 - 1328 |
| SWALLOW, K. ET AL., CLIN EXP IMMUNOL, vol. 171, no. 1, 2013, pages 100 - 6 |
| TAKAHASHI Y ET AL., JEXP MED, vol. 187, 1998, pages 885 - 895 |
| TOELLNER KM ET AL., JEXP MED, vol. 195, 2002, pages 383 - 389 |
| TRAUNECKER ET AL., EMBO J., vol. 10, 1991, pages 3655 |
| TRYNKA G ET AL., NAT GENET, vol. 43, 2011, pages 1193 - 1201 |
| TUTT ET AL., J. IMMUNOL., vol. 147, 1991, pages 60 |
| TYE-DIN ET AL., SCI TRANSL MED, vol. 2, 2010, pages 41RA 1 |
| TYE-DIN ET AL., SCI TRANSL MED, vol. 2, 2010, pages 41RA51 |
| URLAUB ET AL., PROC. NATL. ACAD. SCI. USA, vol. 77, 1980, pages 4216 |
| VADER ET AL., J EXP MED, vol. 195, 2002, pages 643 - 9 |
| VALLEJO-DIEZ ET AL., PLOS ONE, vol. 8, 2013, pages E80982 |
| VALLEJO-DIEZ, S. ET AL., PLOS ONE, vol. 8, no. 11, 2013, pages E80982 |
| VAN DIJK; VAN DE WINKEL, CURR. OPIN. PHARMACOL., vol. 5, 2001, pages 368 - 74 |
| VAN ECKERT, R. ET AL., JOURNAL OF CEREAL SCIENCE, vol. 51, no. 2, 2010, pages 198 - 204 |
| VICZAINO ET AL., NUCLEIC ACIDS RES, vol. 41, 2013, pages D1063 - 9 |
| VOLLMERS; BRANDLEIN, HISTOLOGY AND HISTOPATHOLOGY, vol. 20, no. 3, 2005, pages 927 - 937 |
| VOLLMERS; BRANDLEIN, METHODS AND FINDINGS IN EXPERIMENTAL AND CLINICAL PHARMACOLOGY, vol. 27, no. 3, 2005, pages 185 - 91 |
| VRIEZINGA ET AL., N ENGL J MED, vol. 371, 2014, pages 1304 - 15 |
| WANG M ET AL., J EXP MED, vol. 207, 2010, pages 141 - 153 |
| WINTER ET AL., ANN. REV. IMMUNOL, vol. 12, 1994, pages 433 - 455 |
| WOLF ET AL., PLOS ONE, vol. 9, 2014, pages E97853 |
| WRIGHT ET AL., TIBTECH, vol. 15, 1997, pages 26 - 32 |
| WU ET AL., SCIENCE, vol. 333, 2011, pages 1593 - 1602 |
| YAMANE-OHNUKI ET AL., BIOTECH. BIOENG., vol. 87, 2004, pages 614 |
| YAZAKI; WU: "Methods in Molecular Biology", vol. 248, 2003, HUMANA PRESS, pages: 255 - 268 |
| YU ET AL., NATURE, vol. 450, 2007, pages 299 - 303 |
| ZANINI, B. ET AL., DIG LIVER DIS, vol. 44, no. 4, 2012, pages 280 - 5 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20210025890A1 (en) | Methods for detecting and quantifying fgf21 | |
| CN111225925B (en) | anti-HLA-DQ 2.5 antibodies | |
| EP3533459A1 (en) | Anti-pla2-gib antibodies and the uses thereof | |
| CN103097418A (en) | Anti-neuropilin antibodies and methods of us | |
| WO2022173670A1 (en) | Antibodies targeting the spike protein of coronaviruses | |
| Zhou et al. | Focused B cell response to recurring gluten motif with implications for epitope spreading in celiac disease | |
| WO2015164615A1 (en) | Anti-gluten antibodies and uses thereof | |
| JP6626078B2 (en) | How to choose a treatment | |
| US20220099677A1 (en) | Methods for detecting and quantifying membrane-associated proteins |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15720871 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 15720871 Country of ref document: EP Kind code of ref document: A1 |