WO2015125216A1 - 質量分析を用いたタンパク質の検出方法 - Google Patents
質量分析を用いたタンパク質の検出方法 Download PDFInfo
- Publication number
- WO2015125216A1 WO2015125216A1 PCT/JP2014/053780 JP2014053780W WO2015125216A1 WO 2015125216 A1 WO2015125216 A1 WO 2015125216A1 JP 2014053780 W JP2014053780 W JP 2014053780W WO 2015125216 A1 WO2015125216 A1 WO 2015125216A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- protein
- sample
- peptide
- serum
- protease
- Prior art date
Links
Images









Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6803—General methods of protein analysis not limited to specific proteins or families of proteins
- G01N33/6848—Methods of protein analysis involving mass spectrometry
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P21/00—Preparation of peptides or proteins
- C12P21/06—Preparation of peptides or proteins produced by the hydrolysis of a peptide bond, e.g. hydrolysate products
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/34—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving hydrolase
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6803—General methods of protein analysis not limited to specific proteins or families of proteins
- G01N33/6848—Methods of protein analysis involving mass spectrometry
- G01N33/6851—Methods of protein analysis involving laser desorption ionisation mass spectrometry
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y304/00—Hydrolases acting on peptide bonds, i.e. peptidases (3.4)
- C12Y304/21—Serine endopeptidases (3.4.21)
- C12Y304/21014—Microbial serine proteases (3.4.21.14)
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01J—ELECTRIC DISCHARGE TUBES OR DISCHARGE LAMPS
- H01J49/00—Particle spectrometers or separator tubes
Definitions
- the present invention relates to a method for detecting a protein in a sample by mass spectrometry (MS). More specifically, the present invention relates to a method for detecting and identifying a protein by high performance liquid chromatography / mass spectrometry (LC / MS) or matrix-assisted laser desorption / ionization (MALDI) method. .
- MS mass spectrometry
- LC / MS high performance liquid chromatography / mass spectrometry
- MALDI matrix-assisted laser desorption / ionization
- ionization methods such as matrix-assisted laser desorption ionization and electrospray ionization (ESI) are used except for impurities that inhibit ionization from serum samples.
- ESI electrospray ionization
- a system SELDI protein chip (R) system: Patent Document 1
- a plate used for pretreatment can be directly introduced into a mass spectrometer.
- Patent Document 2 a system for directly measuring the target protein after isolating and purifying the target protein from a sample such as serum using an antibody that specifically binds to the target protein.
- a method of analyzing a protein after fragmenting it into short peptide chains by a protein digestion enzyme is one technique established for obtaining protein identification and structural information with high accuracy.
- a protein digestion enzyme protease digestion enzyme
- the mass is determined by mass spectrometry (MS), more preferably the amino acid sequence is determined by tandem MS measurement, and these information are collated with a database.
- MS mass spectrometry
- the method of analogizing the protein present in the original sample is called shotgun proteomics and is widely used in biological research.
- Non-patent Document 1 In shotgun proteomics, a quantitative analysis method for estimating the content of the original protein by quantifying digested peptides has recently been established (Non-patent Document 1). This method has high sensitivity because the target is a peptide that is easily ionized, and accurate quantification is possible by using an internal standard (internal standard) labeled with a stable isotope. Furthermore, by connecting a mass spectrometer to separation of peptides by nanoscale liquid chromatography and measuring continuously as the peptide is eluted, the sensitivity of the measurement system is dramatically improved to a level below femtomole. Yes.
- the concentration range of blood proteins also extends to the 10 9 order.
- the dynamic range that can be captured by the mass spectrometer is about 10 3 to 10 4, it is extremely difficult to detect a trace amount of protein in the blood by the mass spectrometer.
- albumin protein-derived peptides contained in high concentrations such as albumin, saturating the analytical system. Resulting in.
- albumin is known to occupy 50% or more of blood proteins, and when peptides are detected by pretreatment by conventional methods, most of them become albumin-derived peaks and are contained in trace amounts. The protein-derived peak is extremely difficult to detect.
- Non-patent Document 2 the digested peptide is subdivided by multi-step purification, and a small fraction concentrated from the whole is subjected to intensive analysis (Non-patent Document 2), and is subject to quantification.
- concentration at the peptide level such as immunoprecipitation using an antibody that specifically binds to a peptide or ion exchange chromatography (Non-patent Document 3).
- these methods involve very complicated preprocessing, they are out of the throughput and cost constraints required for clinical tests.
- An object of the present invention is a method for pretreatment of a serum or plasma sample for detecting a single or a plurality of target proteins from a serum or plasma sample by mass spectrometry, and a method for detecting the protein in a sample such as albumin. It is intended to provide a method for removing a large amount of protein present in a simple manner and recovering a digested peptide derived from the target protein.
- the present inventors have surprisingly found that serum or plasma can be used in a state where dilution or denaturation of serum or plasma using a buffer is minimized. It was found that by directly adding protease to the digestion, the peptide necessary for quantification of the target protein can be secured while suppressing the production of peptides derived from albumin and the like. Furthermore, by removing undigested protein after protease treatment, the proportion of peptides derived from the target protein can be relatively increased.
- the method described in the present invention is a method of simultaneously performing specific digestion and concentration of a target protein based on the sensitivity to digestion by protease, and in an analysis system for detecting and quantifying the target protein using mass spectrometry, This makes it possible to calibrate a protein having a low blood concentration very easily as compared with the conventional method.
- a sample pretreatment method for detecting a protein in a serum or plasma sample by mass spectrometry comprising a step of digesting a sample by adding protease to the sample under native protein conditions,
- the said pretreatment method including the process of isolate
- a method for detecting a protein in a serum or plasma sample by mass spectrometry comprising a step of digesting a sample by adding a protease to the sample under a native condition of the protein, and converting the obtained peptide into an undigested protein.
- the method as described above comprising a step of separating and a step of subjecting the peptide to mass spectrometry.
- the present invention is surprising for the detection of proteins using mass spectrometry, in contrast to the conventional methods that have sought efficient fragmentation of proteins, making use of conditions that are unlikely to cause digestion by proteases. It is based on the discovery that results can be obtained.
- the method of the present invention eliminates the need for conventional pretreatments such as protein denaturation, reduction, and alkylation, and allows quantitative determination of trace proteins in a sample without the need for a protein or peptide concentration step. It has become possible. Thereby, the time and cost required for pre-processing can be reduced, and a high-throughput quantitative method can be provided.
- the mass spectrum of the serum digestion peptide obtained by the pre-processing method by this invention and the pre-processing by the conventional method is shown. Among the observed peaks, * indicates a peak derived from albumin.
- P14R SEQ ID NO: 12, m / z 1533.9
- the upper result shows the result obtained by the conventional method, and the lower result shows the result obtained by the method of the present invention.
- Peptides derived from ANXA4 are indicated by arrows (A: conventional method, B: method according to the present invention).
- the mass spectrum of the peptide obtained by subjecting Annexin A4 (ANXA4) to trypsin digestion is shown.
- the upper panel shows the results of subjecting ANXA4 alone to trypsin digestion, and the lower panel shows the results of digesting ANXA4 with ⁇ 2 macroglobulin in advance and heating.
- Peptides derived from ANXA4 are indicated by arrows.
- a “serum or plasma sample” is a blood-derived sample intended to be detected for the presence of a protein of interest, and is derived from the serum or plasma itself or from these. It will be understood by those skilled in the art that the sample is included, for example, to which a drug is added for storage of the sample. Subjects include, but are not limited to, mammals, particularly humans.
- the serum or plasma sample is subjected to protein denaturation / reduction treatment and alkylation treatment prior to detection of the target protein, followed by protease treatment.
- the protein / peptide is appropriately concentrated by, for example, immunoprecipitation or ion exchange chromatography.
- an enzyme digestion reaction buffer is added for the purpose of adjusting the reaction solution to an environment having an appropriate pH and ionic strength.
- an ammonium bicarbonate solution, a Tris buffer, or the like is often used for the purpose of pH adjustment.
- the conventional method further requires further processing so that reagents added for pretreatment, particularly high concentrations of urea and surfactant added for denaturation do not affect protease treatment and mass spectrometry.
- denaturation of a protein means that the three-dimensional structure of the protein loses and changes the higher-order structure originally taken in the living body. For example, it is known that, in addition to rapid changes in ionic strength, pH, and temperature, denaturation occurs due to the addition of a high concentration of chaotropic agent or surfactant.
- the present invention relates to a pretreatment method comprising a step of adding a protease to a serum or plasma sample under native protein conditions and digesting, and a step of separating the obtained peptide from undigested protein.
- a method for detecting a protein by mass spectrometry which includes a step of subjecting to mass spectrometry.
- the method of the present invention preferably does not include a protein or peptide enrichment step as conventionally done before and / or after protease treatment.
- Non-denaturing conditions refers to a biological sample such as serum or plasma in addition to the treatment for denaturation usually used in the art to denature proteins, for example, treatment with a denaturing agent such as urea. Means that the state of the composition, temperature, etc. of the constituent components inherently has no change.
- inactivation treatment prior to protease treatment it is preferable to perform inactivation treatment prior to protease treatment as a step of suppressing endogenous protease activity that may slightly affect the detection result and enhancing the detection effect on the target protein.
- the deactivation can be performed, for example, by a heat treatment at 56 ° C. for 30 to 40 minutes. Conditions for deactivation can be easily determined by those skilled in the art.
- protease is directly added to the obtained serum or plasma sample.
- “direct addition” refers to addition to a sample containing protein in a necessary minimum solution composition without subjecting the collected serum or plasma sample to protein denaturation treatment.
- the protease to be added is prepared to a high concentration of 10 mg / mL or higher using ultrapure water or hydrochloric acid having a concentration of 1 mmol / L or less, and this is directly added to about 1/50 volume of the serum or plasma sample volume. By adding, the digestion reaction can be started with almost no change in the original composition of the sample.
- the reaction environment is suitable for many proteases including trypsin without adjusting the pH with a separate buffer. If there is a special need, the pH may be adjusted in the range of pH 7 to 9, more preferably in the range of pH 7.4 to 7.8. Conditions for pH adjustment can be appropriately determined by those skilled in the art, but particularly preferred is buffering with sodium phosphate.
- the “protease” preferably used in the present invention preferably includes trypsin, lysyl endopeptidase, endopeptidase Asp-N, V8 protease, metalloendopeptidase Lys-N, and particularly preferably trypsin.
- Trypsin has high substrate specificity, and since there is Lys or Arg at the C-terminus of the peptide after cleavage, the charge amount and charge localization of the peptide can be homogenized, so that a sample for mass spectrometry can be prepared. Is particularly suitable.
- the amount of protease added to the sample may vary depending on the protease used and the conditions of the sample type, temperature, processing time, etc., but preferably the amount of protease added is 1-50 ⁇ g per 100 ⁇ L of serum or plasma sample. The amount is preferably 20 to 40 ⁇ g. In one embodiment, in the method of the invention, approximately 20 ⁇ g trypsin is added per 100 ⁇ L of serum.
- the reaction temperature in protease treatment is 0 to 40 ° C., preferably 4 ° C. to 37 ° C., and the treatment time is preferably 1 to 24 hours, preferably 3 to 16 hours, more preferably 8 to 12 hours.
- the protease is not particularly limited, but is used as a solution or suspension prepared at a high concentration as described above. After degradation, the protease can be easily removed in the process of separating the protein. Alternatively, a sample can be applied to the immobilized enzyme using a protease bound to a solid support. Methods for producing immobilized enzymes are well known in the art.
- the carrier is not particularly limited.
- agarose, sepharose, polyacrylamide, polyethylene glycol, polystyrene, polyethylene, polypropylene, polyester, polyacrylonitrile, (meth) acrylic acid ester polymer, fluororesin, metal complex resin, Glass, metal, and a magnetic material are mentioned.
- immobilized enzymes can also be removed in the step of separating the protein.
- albumin, globulin and transferrin present in high concentrations in serum or plasma were not digested, and the resulting peptides were subjected to mass spectrometry.
- peaks derived from these proteins were not included in the mass spectrum.
- albumin occupying 50% or more of the protein is not digested, it is possible to detect peptides that were difficult to detect due to the small amount of the conventional method, and detection of the target protein / peptide in the sample is extremely possible. Can be carried out easily and quickly.
- the terms “undigested” and “undigested” used for a protein refer to 90% or more, preferably 95% or more, more preferably 98% or more, more preferably 99% or more of the protein present in the sample. It means that the protein remains as it is without being decomposed by the added protease. In other words, when serum or plasma is used as a sample, degradation of contaminating proteins present in a large amount in the sample, particularly albumin contained in a large amount by protease, is less than 10%, preferably less than 5%, more preferably less than 2%. More preferably, it is less than 1%. Most preferably, in the method of the present invention, albumin does not undergo any protease degradation. Undigested protein is removed by the subsequent separation step of protein and peptide in the method of the present invention.
- An “undigested protein” is an interaction with a protease due to the reason that its tertiary structure and quaternary structure are highly folded in a sample and in a state of being bound to other proteins. Means a protein that is inhibited and does not undergo digestion under the conditions of the present invention.
- the method of the present invention is designed to reverse the original concentration ratio at the level of digested peptide by preferentially proceeding digestion of proteins other than those having such properties.
- A2M ⁇ 2 macroglobulin
- A2M is a protease inhibitor having a function of fixing a wide variety of proteases inside the cage-like A2M molecule, and is contained in serum at a concentration of about 1 to 2 mg / mL. It is known that proteases in the state of being fixed inside A2M remain active, and are continuously digested if they have a small molecular weight accessible to the inside of A2M (Sottrup-Jensen, L , J Biol Chem 1989, 264 (20), 11539-42).
- proteins that are preferentially digested under the conditions of the method of the present invention include those that are digested via A2M molecules in blood. . That is, it is considered that the protease added to a sample such as serum reacts with A2M in the sample and acts on the protein in the sample in a state of being covalently bound inside A2M. Therefore, as the protein to be analyzed, one having a molecular weight of about 10 kDa or less may be more suitable in the present invention.
- the proteins that are digested by the method of the present invention only a part of all peptides that may be generated when the protein is digested may be detected with high sensitivity. This is considered to reflect a situation where a part of the target protein is selectively digested when the protease inside A2M acts.
- the concentration of the protein to be detected in the sample depends on the sensitivity of the measurement system, but a concentration of 1 ⁇ g / mL or more, particularly in the range of 5 to 100 ⁇ g / mL, is advantageously detected by the method of the present invention. . More specifically, the amount of peptide actually detected may be in the range of 10 to 10,000 fmol. When 0.1 ⁇ L of a sample containing 5 ⁇ g / mL protein is used for measurement by MS, the amount of peptide detected is about 15 fmol.
- the undigested protein is separated from the peptide using the difference in properties between the protein and the digested peptide.
- an organic solvent or acid is added to precipitate the protein, and the centrifugal supernatant is collected.
- the protein is fractionated into a high molecular weight protein and a low molecular weight peptide by ultrafiltration or gel filtration. And a method of selectively binding to a carrier using strong hydrophobic interaction.
- pretreatment of a serum or plasma sample according to the method of the present invention and the obtained peptide is measured by a mass spectrometer, so that it is present in a large amount in the sample, for example, interference by proteins such as albumin
- the detection of a trace amount of protein in a sample can be carried out easily and with high throughput without being subjected to.
- Mass spectrometry is performed on peptides ionized using a MALDI method, an ESI method, or the like using an analyzer such as a double focusing type, a quadrupole type, a time-of-flight type (TOF), or a Fourier transform ion sacrotron resonance type. be able to. Moreover, you may analyze by the liquid chromatograph mass spectrometry (LC / MS) combined with the high performance liquid chromatography. Mass spectrometry can be performed by multiple reaction monitoring (MRM) with a triple quadrupole mass spectrometer of an LC / MS system widely used in the art for quantitative detection of peptides. The measurement of MRM can be performed by, for example, LCMS-8080 (Shimadzu Corporation). Measurement of MALDI-TOF MS can be performed by, for example, AXIMA-Resonance (Shimadzu Corporation).
- MRM multiple reaction monitoring
- Measurement of MALDI-TOF MS can be
- Tandem MS (MS / MS) analysis which is used for detailed and precise analysis, selects a precursor ion derived from the target peptide for further mass analysis, and identifies the peptide by the generated product ion. ⁇ Can be quantified. Based on the analysis results obtained, it is possible to confirm that the peptide is the target peptide by referring to the peak information obtained in the same manner using a control sample or information in a known database. A decision can be made.
- the measurement of MS / MS can be performed by, for example, AXIMA-Resonance (Shimadzu Corporation).
- a peptide to be actually analyzed by mass spectrometry in the protein to be detected is selected.
- Information on the amino acid sequence of the protein can be obtained from, for example, http://www.uniprot.org/.
- the type of peptide that can be generated when a protein is decomposed with a protease can be predicted from, for example, http://web.expasy.org/peptide_mass/.
- Data of measurement results by mass spectrometry of peptides generated by protease digestion can be referred to, for example, at http://peptide.nist.gov/.
- amino acid sequence For example, in the selection of peptides that can be subjected to mass spectrometry after digestion with trypsin, several exclusion criteria are known for the amino acid sequence (Nat. Biotechnol. 2009, 27 (2), 190-8). Specifically, for example, it is essential that the same sequence does not exist in a plurality of proteins for the reliability of the detection result, for example, it is not a hydrophilic peptide of less than 8 amino acids or a hydrophobic peptide of more than 20 amino acids.
- the basic amino acid which becomes the carboxy terminus by trypsin digestion is not adjacent (KK, RR, KR, RK); it does not contain a cysteine residue, a methionine residue, an N-terminal glutamine or asparagine.
- a peptide suitable as a marker for detection by mass spectrometry can be predicted on a computer based on such information, and a person skilled in the art can arbitrarily select a peptide to be used for quantification from these.
- annexin A4 (SEQ ID NO: 1), which is known to be highly expressed in some ovarian cancers, is a peptide for detecting in blood, and the following conditions: (Condition 1) The same amino acid sequence does not exist in other proteins; (Condition 2) The sequence does not contain cysteine; (Condition 3) The number of amino acids in the sequence is 8 to 20; (Condition 4) When the N-terminal of the sequence is other than glutamine or asparagine; and those satisfying the above are suitable candidate peptides, the following peptides can be selected.
- AASGFNAMEMQTLR (SEQ ID NO: 2, corresponding to amino acid numbers 10-24 of the amino acid sequence of SEQ ID NO: 1); GLGTDEDAIISVLAYR (SEQ ID NO: 3, corresponding to amino acid numbers 29-44 of the amino acid sequence of SEQ ID NO: 1); ISQTYQQQYGR (SEQ ID NO: 4, corresponding to amino acid numbers 124-134 of the amino acid sequence of SEQ ID NO: 1); SDTSMFMFQR (SEQ ID NO: 5, corresponding to amino acid numbers 142-150 of the amino acid sequence of SEQ ID NO: 1); VLVSLSAGGR (SEQ ID NO: 6, corresponding to amino acid numbers 151-160 of the amino acid sequence of SEQ ID NO: 1); DEGNYLDDALVR (SEQ ID NO: 7, corresponding to amino acid numbers 161-172 of the amino acid sequence of SEQ ID NO: 1); SETSGSFEDALLAIVK (SEQ ID NO: 8, corresponding to amino acid numbers 226 to 241 of the amino acid sequence of
- the peptide predicted on the computer may actually be difficult to be digested with protease depending on conditions such as the position in the three-dimensional structure of the protein.
- a peptide for the detection of annexin A4 particularly preferred in the method of the present invention are peptides having the amino acid sequences shown in SEQ ID NOs: 2, 3 and 7.
- Powdered trypsin (product code 203-09893, Wako Pure Chemical Industries, Ltd.) was dissolved in a 1 mmol / L hydrochloric acid solution to a concentration of 10 mg / mL to obtain a trypsin solution.
- Pretreatment according to the present invention The pretreatment process according to the present invention will be described below.
- 100 ⁇ L of each serum sample prepared as described above was placed in a 1.5 mL Eppendorf tube, 2 ⁇ L of trypsin solution was added, and the mixture was reacted at 37 ° C. for 16 hours (about 20 ⁇ g trypsin for 100 ⁇ L of serum sample).
- the sample after the reaction is transferred to the upper part of an ultrafiltration filter (Amicon 10,000 MWCO filter, Millipore), centrifuged at 10,000 g for 60 minutes, and the filtrate transferred to the lower part of the filter is directly used as a sample for mass spectrometry. It was.
- an ultrafiltration filter Amicon 10,000 MWCO filter, Millipore
- MS / MS measurement For MS / MS measurement, an ion trap was set for the target monoisotopic mass, and a total of 200 profiles were acquired from 200 locations by raster driving under the conditions of Positive Midmass mode and laser intensity of 110, and integrated. . The intensity setting value of CID (Collision Induced Dissociation) was appropriately adjusted according to the target.
- CID collision Induced Dissociation
- FIG. 2 is an enlarged view around the peak of m / z 1379.6. As indicated by the arrows, this peak that was not detected by the pretreatment by the conventional method (A) was detected at 38 ⁇ g / ml or more by the pretreatment (B) according to the present invention. As is clear from this result, no peak derived from ANXA4 was detected by the conventional method, whereas a peak depending on the concentration was observed by the method of the present invention.
- the above peak (m / z 1379.6) was selected as a precursor ion, and a product ion spectrum was obtained by MS / MS measurement.
- the results are shown in FIG. From the result of FIG. 3, it was shown that the peak at m / z 1379.6 was attributed to a peptide derived from ANXA4 (SEQ ID NO: 7). This was confirmed from the fact that the spectrum was the same as that obtained by directly analyzing ANXA4 without adding it to the sample.
- the ANXA4 concentration in serum was detectable at 75 ⁇ g / mL or more, whereas in the method of the present invention (FIG. 3B), detection was possible even at a low concentration of only 5 ⁇ g / mL. Yes, detection sensitivity improved more than 10 times. Furthermore, while many signals that are not derived from ANXA4 are mixed in the conventional method, only the signal derived from ANXA4 is detected in the method according to the present invention, and the S / N ratio is also good.
- Table 1 summarizes the detection of ANXA4-derived peptides by the method according to the present invention and the conventional method.
- the signal intensity derived from ANXA4 after correction was compared with the conventional method involving denaturation and the direct digestion method according to the present invention, it was found that the detection efficiency was remarkably improved in both MS and MS / MS data. That is, the ANXA4-derived m / z 1379 peak could not be quantitatively detected by the conventional method, whereas the peak of the method of the present invention was detected depending on the concentration.
- Example 2 [Selection of peptide for analysis] Although there are peptides of SEQ ID NO: 2-11 listed above as suitable peptide candidates for the method of the present invention, a preliminary experiment using ⁇ 2 macroglobulin (A2M) was conducted in order to select a particularly suitable peptide from these peptides. Carried out.
- A2M ⁇ 2 macroglobulin
- A2M protein (product code M6159, Sigma-Aldrich) and purified recombinant protein ANXA4 obtained by an E. coli expression system were prepared in phosphate buffered saline (PBS) at concentrations of 2 mg / mL and 3 mg / mL, respectively.
- Powdered trypsin (product code 203-09893, Wako Pure Chemical Industries, Ltd.) was dissolved in a 1 mmol / L hydrochloric acid solution to a concentration of 10 mg / mL to obtain a trypsin solution.
- FIG. 4 shows the mass spectrum of the ANXA4 digested peptide in the presence and absence of A2M (final concentration 0.75 mg / mL).
- AASGFNAMEMQTLR SEQ ID NO: 2
- Example 3 [Preparation of sample and protease] The same operation as in Example 1 was performed.
- [MALDI MS measurement] 0.5 ⁇ L of the sample obtained above was desalted with ZipTip, 10 pmol of standard peptide P14R was added to the eluate, and 1/10 of the amount was spotted on a ⁇ Focusing plate (Shimadzu GLC). After air drying, 1 ⁇ L of 3 mg / mL 2,5-dihydroxybenzoic acid was spotted as a matrix, and after crystallization, measurement was performed with MALDI MS (AXIMA-Resonance, Shimadzu Corporation). A mass spectrum was acquired from each spot by acquiring a total of 200 profiles from 200 locations by raster driving under the conditions of Positive MidMass mode and laser intensity of 100.
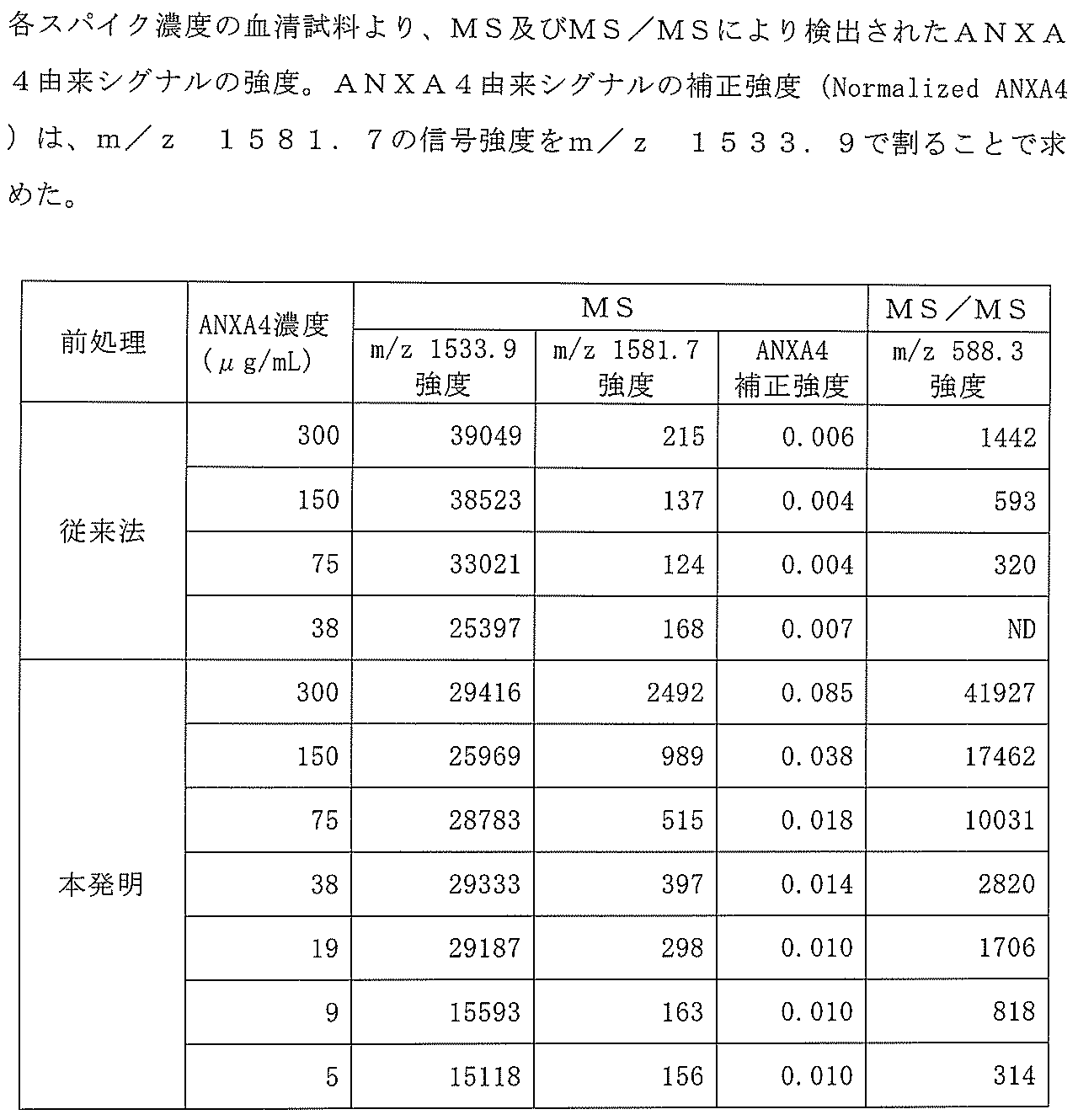
- Peak detection is performed from the mass spectrum with a signal intensity of 0.5 mV as a threshold, and the signal is derived from P14R-derived signal (m / z 1533.9) intensity and ANXA4 (peptide having the amino acid sequence of SEQ ID NO: 2).
- the intensity of each signal (m / z 1581.7) was extracted and tabulated.
- strength of product ion m / z 588.3 (equivalent to y5 ion of sequence number 2) was extracted from the product ion spectrum of m / z 1581.7 acquired by MS / MS mode, and it totaled.
- ND non-detection
- FIG. 5 is an enlarged view around the peak at m / z 1581.7. As indicated by the arrows, this peak that was not detected by the pretreatment by the conventional method (A) was detected at 38 ⁇ g / ml or more by the pretreatment (B) according to the present invention. As is clear from this result, no peak derived from ANXA4 was detected by the conventional method, whereas a peak depending on the concentration was observed by the method of the present invention. In this experimental condition, among the candidate peptides (SEQ ID NO: 2-11) of the ANXA4-derived peptide, only SEQ ID NOs: 2 and 3 were actually detected from the mass spectrum, which is preferable in Example 2. It was confirmed to be consistent with the peptide.
- the above peak (m / z 1581.7) was selected as a precursor ion, and a product ion spectrum was obtained by MS / MS measurement.
- the results are shown in FIG. From the results of FIG. 6, it was shown that the peak at m / z 1581.7 was attributed to the peptide derived from ANXA4 (SEQ ID NO: 2). This was confirmed from the fact that the spectrum was the same as that obtained by directly analyzing ANXA4 without adding it to the sample.
- the ANXA4 concentration in serum was detectable at 75 ⁇ g / mL or more, whereas in the method of the present invention (FIG. 4B), detection was possible even at a low concentration of only 5 ⁇ g / mL. Yes, detection sensitivity improved more than 10 times. Furthermore, while many signals that are not derived from ANXA4 are mixed in the conventional method, only the signal derived from ANXA4 is detected in the method according to the present invention, and the S / N ratio is also good.
- Table 2 summarizes the detection of peptides derived from ANXA4 by the method according to the present invention and the conventional method.
- the pretreatment method of the present invention is effective for obtaining the peptide signal derived from the target protein by eliminating the influence of the protein present in a large amount in the blood such as albumin. It is possible to provide more convenience as a pretreatment for biomarker measurement by analysis.
- the present invention was devised as a method for detecting and identifying proteins in blood by LC / MS or MALDI-TOF MS.
- the present invention provides a blood sample pretreatment method useful for searching for disease biomarkers using these mass spectrometers and for performing clinical tests based on biomarker measurements.
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Chemical & Material Sciences (AREA)
- Physics & Mathematics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Computational Biology (AREA)
- Immunology (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Biomedical Technology (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Microbiology (AREA)
- General Health & Medical Sciences (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Analytical Chemistry (AREA)
- Biophysics (AREA)
- Pathology (AREA)
- Cell Biology (AREA)
- Food Science & Technology (AREA)
- Medicinal Chemistry (AREA)
- General Physics & Mathematics (AREA)
- Genetics & Genomics (AREA)
- General Engineering & Computer Science (AREA)
- Optics & Photonics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Other Investigation Or Analysis Of Materials By Electrical Means (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
Description
(条件1)同じアミノ酸配列が他のタンパク質中に存在しないこと;
(条件2)配列中にシステインを含まないこと;
(条件3)配列のアミノ酸の数が8~20であること;
(条件4)配列のN末端がグルタミンまたはアスパラギン以外であること;を満たすものを好適な候補ペプチドとした場合、以下のペプチドを選択することができる。
GLGTDEDAIISVLAYR(配列番号3、配列番号1のアミノ酸配列のアミノ酸番号29-44に相当);
ISQTYQQQYGR(配列番号4、配列番号1のアミノ酸配列のアミノ酸番号124-134に相当);
SDTSFMFQR(配列番号5、配列番号1のアミノ酸配列のアミノ酸番号142-150に相当);
VLVSLSAGGR(配列番号6、配列番号1のアミノ酸配列のアミノ酸番号151-160に相当);
DEGNYLDDALVR(配列番号7、配列番号1のアミノ酸配列のアミノ酸番号161-172に相当);
SETSGSFEDALLAIVK(配列番号8、配列番号1のアミノ酸配列のアミノ酸番号226-241に相当);
GLGTDDNTLIR(配列番号9、配列番号1のアミノ酸配列のアミノ酸番号260-270に相当);
AEIDMLDIR(配列番号10、配列番号1のアミノ酸配列のアミノ酸番号276-284に相当);
GDTSGDYR(配列番号11、配列番号1のアミノ酸配列のアミノ酸番号301-308に相当)。
[分析対象ペプチドの選定]
本発明の方法により、卵巣癌のバイオマーカーとして期待される、血清中に微量に含まれるアネキシン A4(ANXA4)(配列番号1)の半定量的な検出を行った。
市販のヒト血清(製品コード14-402E、ロンザジャパン)に対して、大腸菌の発現系により得た精製リコンビナントタンパク質ANXA4を3mg/mLの濃度で10%量添加し、それを上記の血清にて段階希釈することで、ANXA4の濃度が300、150、75、38、19、9、及び5μg/mLの血清試料をそれぞれ調製した。血清試料は56℃、40分の加熱処理により非働化し、さらに0.44μmフィルター(ウルトラフリー、ミリポア社)にて清澄化した。粉末状のトリプシン(製品コード203-09893、和光純薬)を1mmol/Lの塩酸溶液にて10mg/mLの濃度に溶解し、トリプシン溶液とした。
本発明による前処理工程を以下に説明する。上述のように調製した血清試料各100μLを1.5mL容量のエッペンドルフチューブに取り、トリプシン溶液2μLを添加し、37℃で16時間反応させた(血清試料100μLに対してトリプシン約20μg)。反応後の試料を限外濾過フィルター(Amicon 10,000MWCOフィルター、ミリポア社)の上部に移し、10,000g、60分の条件で遠心分離し、フィルター下部に移行した濾液をそのまま質量分析用の試料とした。
従来の方法による血清のトリプシン消化ペプチドの調製工程を以下に説明する。1.5mLエッペンドルフチューブに粉末ウレア10mgを量りとり、上述のように調製した血清10μLを添加した(変性)。混合後、50mmol/Lのトリス(2-カルボキシエチル)ホスフィン塩酸塩を2μL加え、37℃,30分間の条件で反応させた(還元)。次に、0.5mol/Lのヨードアセトアミド及び1mol/Lの重炭酸アンモニウムの混合溶液を0.8μL加え、常温で45分間反応させた(アルキル化)。これを160μLの10mmol/Lのトリス塩酸緩衝液(pH8.0)で希釈し、さらに前述のトリプシン溶液を2μL加えて、37℃で16時間反応させた(血清試料10μLに対してトリプシン約20μg)。反応後のペプチド溶液に10%トリフルオロ酢酸を4μL加え、質量分析用の試料とした。
上記で得られた試料0.5μLを、ZipTip(商標、ミリポア社)により脱塩した。この際、試料を1nmol/mLの標準ペプチド(アミノ酸配列PPPPPPPPPPPPPPR(配列番号12)、以下P14R)を含む10μLの0.1%トリフルオロ酢酸にて希釈し、溶出液の1/10をμFocusingプレート(島津GLC)にスポッティングした。風乾後、マトリックスとして3mg/mLの2,5-ジヒドロキシ安息香酸を1μLスポッティングし、結晶化の後、MALDI MS(AXIMA-Resonance、島津製作所)にて測定した。Positive MidMassモード、レーザー強度100の条件でのラスター駆動により200箇所から合計200プロファイルを取得し積算することで、各スポットよりマススペクトルを取得した。
MS/MSの測定は、対象のモノアイソトピックマスに対してイオントラップを設定したうえで、Positive Midmassモード、レーザー強度110の条件でのラスター駆動により200箇所から合計200プロファイルを取得し、積算した。CID(Collision Induced Dissociation、衝突誘起解離)の強度設定値は対象にあわせて適宜調整した。
マススペクトルからP14Rに由来するシグナル(m/z 1533.9)の強度及びANXA4(配列番号7のアミノ酸配列を有するペプチド)に由来するシグナル(m/z 1379.6)の強度をそれぞれ抽出し、集計した。また、MS/MSモードにより取得したm/z 1379.6のプロダクトイオンスペクトルより、プロダクトイオンm/z 458.3の強度を抽出し、集計した。MSモードにおける信号強度はm/z 1533(P14R)に対してノーマライズを行い、イオン化に伴う信号強度の強弱について補正を行った。
本発明による前処理方法及び従来法による前処理により得られた血清消化ペプチドのマススペクトルを図1に示す。観測されるピークのうち、アルブミンに由来するピークを*印により記した。比較すると、従来法(上段)においてはスペクトル中に観測される主要なピークのほぼ全てがアルブミンに由来するのに対して、本発明による方法(下段)ではアルブミン由来のピークの発生が抑えられていることが示された。

[分析対象ペプチドの選定]
本発明の方法に好適なペプチドの候補として上記に挙げた配列番号2-11のペプチドがあるが、この中から特に好適なペプチドを選択するため、α2マクログロブリン(A2M)を用いた予備実験を実施した。
[試料及びプロテアーゼの調製]
実施例1と同様にして行った。
トリプシンによる消化条件を4℃で16時間とする以外は実施例1と同様にして行った。
実施例1と同様にして行った。
上記で得られた試料0.5μLを、ZipTipにより脱塩し、溶出液に10pmolの標準ペプチドP14Rを添加し、その1/10量をμFocusingプレート(島津GLC)にスポッティングした。風乾後、マトリックスとして3mg/mLの2,5-ジヒドロキシ安息香酸を1μLスポッティングし、結晶化の後、MALDI MS(AXIMA-Resonance、島津製作所)にて測定した。Positive MidMassモード、レーザー強度100の条件でのラスター駆動により200箇所から合計200プロファイルを取得し積算することで、各スポットよりマススペクトルを取得した。
実施例1と同様にして行った。
マススペクトルから信号強度0.5mVを閾値にピーク検出を行い、その中からP14Rに由来するシグナル(m/z 1533.9)の強度及びANXA4(配列番号2のアミノ酸配列を有するペプチド)に由来するシグナル(m/z 1581.7)の強度をそれぞれ抽出し、集計した。また、MS/MSモードにより取得したm/z 1581.7のプロダクトイオンスペクトルより、プロダクトイオンm/z 588.3(配列番号2のy5イオンに相当)の強度を抽出し、集計した。この際、閾値0.5mVのピーク検出処理において認識されなかったシグナルを不検出(ND)とした。MSモードにおける信号強度はm/z 1533(P14R)に対してノーマライズを行い、イオン化に伴う信号強度の強弱について補正を行った。
図5は、m/z 1581.7のピーク周辺の拡大図である。矢印で示すように、従来法(A)による前処理では検出されないこのピークは、本発明による前処理(B)により38μg/ml以上で検出された。この結果から明らかなように、従来の方法ではANXA4由来のピークは全く検出されなかったのに対し、本発明の方法では濃度に依存したピークが観察された。なお、本実験条件において、ANXA4由来ペプチドの候補ペプチド(配列番号2-11)のうち、マススペクトル中から実際に検出されたのは配列番号2及び3のみであり、実施例2において好適とされたペプチドと一致していることが確認された。
Claims (9)
- 血清又は血漿試料中のタンパク質を質量分析によって検出するための試料の前処理方法であって、タンパク質の未変性条件下で該試料にプロテアーゼを添加して消化する工程と、得られたペプチドを未消化のタンパク質と分離する工程とを含む、上記前処理方法。
- 血清又は血漿試料中のタンパク質を質量分析によって検出するための方法であって、タンパク質の未変性条件下で該試料にプロテアーゼを添加して消化する工程と、得られたペプチドを未消化のタンパク質と分離する工程と、該ペプチドを質量分析に供する工程とを含む、上記方法。
- プロテアーゼ処理の前及び/又は後にタンパク質又はペプチドの濃縮工程を含まない、請求項1又は2記載の方法。
- プロテアーゼがトリプシンである、請求項1~3のいずれか1項記載の方法。
- プロテアーゼとして固定化酵素を用いる、請求項1~4のいずれか1項記載の方法。
- 試料中にα2マクログロブリンが存在する、請求項1~5のいずれか1項記載の方法。
- LC/MS又はMALDI-TOF法によって分析する、請求項1~6のいずれか1項記載の方法。
- MS/MS分析を行う、請求項1~7のいずれか1項記載の方法。
- アルブミンが消化されない、請求項1~8のいずれか1項記載の方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14882930.2A EP3109630B1 (en) | 2014-02-18 | 2014-02-18 | Protein detection method using mass spectrometry |
| JP2016503809A JP6742235B2 (ja) | 2014-02-18 | 2014-02-18 | 質量分析を用いたタンパク質の検出方法 |
| PCT/JP2014/053780 WO2015125216A1 (ja) | 2014-02-18 | 2014-02-18 | 質量分析を用いたタンパク質の検出方法 |
| US15/119,616 US11378580B2 (en) | 2014-02-18 | 2014-02-18 | Protein detection method using mass spectrometry |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/JP2014/053780 WO2015125216A1 (ja) | 2014-02-18 | 2014-02-18 | 質量分析を用いたタンパク質の検出方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015125216A1 true WO2015125216A1 (ja) | 2015-08-27 |
Family
ID=53877756
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/053780 WO2015125216A1 (ja) | 2014-02-18 | 2014-02-18 | 質量分析を用いたタンパク質の検出方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US11378580B2 (ja) |
| EP (1) | EP3109630B1 (ja) |
| JP (1) | JP6742235B2 (ja) |
| WO (1) | WO2015125216A1 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111965369A (zh) * | 2020-07-28 | 2020-11-20 | 天津国科医工科技发展有限公司 | 一种血浆中血管紧张素ⅰ检测试剂盒及检测方法 |
| CN114609228A (zh) * | 2022-03-08 | 2022-06-10 | 西湖大学 | 一种适用于舌苔样本的高通量蛋白提取方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090148951A1 (en) * | 2007-12-06 | 2009-06-11 | Yanni Zhang | Thyroglobulin quantitation by mass spectrometry |
| JP2010515020A (ja) * | 2006-12-21 | 2010-05-06 | ノバルティス アーゲー | 抗体定量法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5792664A (en) | 1992-05-29 | 1998-08-11 | The Rockefeller University | Methods for producing and analyzing biopolymer ladders |
| US7915008B2 (en) | 2006-04-27 | 2011-03-29 | Syddansk Universitet | Methods for isolation and analysis of sialylated and phosphorylated peptides |
| DE602007011937D1 (de) | 2006-07-03 | 2011-02-24 | Univ Johns Hopkins | Peptidantikörperdepletion und anwendung zur probenvorbereitung für massenspektrometrie |
| EP2088426B1 (en) | 2006-11-29 | 2013-05-15 | Shimadzu Corporation | Mass spectrometry of biological sample using immunoprecipitation |
| US8815315B2 (en) * | 2007-04-10 | 2014-08-26 | Kemin Industries, Inc. | Use of a multi-protease system to improve the protein digestibility of animal feeds containing vegetable meals |
| US8628912B2 (en) | 2007-07-03 | 2014-01-14 | Northeastern University | Biomarkers for diabetes, obesity, and/or hypertension |
-
2014
- 2014-02-18 EP EP14882930.2A patent/EP3109630B1/en active Active
- 2014-02-18 US US15/119,616 patent/US11378580B2/en active Active
- 2014-02-18 WO PCT/JP2014/053780 patent/WO2015125216A1/ja active Application Filing
- 2014-02-18 JP JP2016503809A patent/JP6742235B2/ja active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010515020A (ja) * | 2006-12-21 | 2010-05-06 | ノバルティス アーゲー | 抗体定量法 |
| US20090148951A1 (en) * | 2007-12-06 | 2009-06-11 | Yanni Zhang | Thyroglobulin quantitation by mass spectrometry |
Non-Patent Citations (3)
| Title |
|---|
| See also references of EP3109630A4 * |
| SHI T ET AL.: "Antibody-free, targeted mass- spectrometric approach for quantification of proteins at low picogram per milliliter levels in human plasma/serum", PNAS, PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES OF THE UNITED STATES OF AMERICA, vol. 109, no. 38, 18 September 2012 (2012-09-18), pages 15395 - 15400, XP055221161, ISSN: 0027-8424 * |
| TIRUMALAI R S ET AL.: "Characterization of the low molecular weight human serum proteome", MOLECULAR & CELLULAR PROTEOMICS, vol. 2, no. 10, 13 August 2003 (2003-08-13), pages 1096 - 1103, XP002390543 * |
Also Published As
| Publication number | Publication date |
|---|---|
| US11378580B2 (en) | 2022-07-05 |
| JPWO2015125216A1 (ja) | 2017-03-30 |
| EP3109630A4 (en) | 2017-09-06 |
| EP3109630B1 (en) | 2020-02-12 |
| US20170059579A1 (en) | 2017-03-02 |
| EP3109630A1 (en) | 2016-12-28 |
| JP6742235B2 (ja) | 2020-08-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Virág et al. | Current trends in the analysis of post-translational modifications | |
| Bai et al. | Deep profiling of proteome and phosphoproteome by isobaric labeling, extensive liquid chromatography, and mass spectrometry | |
| US9970943B2 (en) | Mass spectrometric assays for peptides | |
| Guerrera et al. | Application of mass spectrometry in proteomics | |
| Zheng et al. | Analysis of the low molecular weight serum peptidome using ultrafiltration and a hybrid ion trap-Fourier transform mass spectrometer | |
| JP5924455B2 (ja) | ペプチド断片の調製方法およびそれに用いられるペプチド断片調製用キット、ならびに分析方法 | |
| Klemm et al. | Evaluation of the titanium dioxide approach for MS analysis of phosphopeptides | |
| JP2007024631A (ja) | 同位体標識法 | |
| Magni et al. | Biomarkers discovery by peptide and protein profiling in biological fluids based on functionalized magnetic beads purification and mass spectrometry | |
| US11959124B2 (en) | Methods and systems for determining ADAMTS13 enzyme activity | |
| AU2018275881B2 (en) | Methods for absolute quantification of low-abundance polypeptides using mass spectrometry | |
| JP6742235B2 (ja) | 質量分析を用いたタンパク質の検出方法 | |
| US20140051105A1 (en) | Mutant Proteins as Cancer-Specific Biomarkers | |
| Cockrill et al. | Efficient micro-recovery and guanidination of peptides directly from MALDI target spots | |
| US20160238615A1 (en) | Solid phase extraction of global peptides, glycopeptides, and glycans using chemical immobilization in a pipette tip | |
| JP6152908B2 (ja) | ペプチド断片の調製方法および分析方法 | |
| CN109964131B (zh) | 用于测量血浆肾素活性的方法和系统 | |
| WO2024133743A1 (en) | Method for diagnosing and/or classifying the severity of a neurodegenerative disease | |
| Licker et al. | Characterisation of human cerebrospinal fluid (CSF) after tandem mass tag (tmt0) labelling | |
| Chen | Development and Applications of Mass Spectrometric Methods for Proteome Analysis and Protein Sequence Characterization | |
| Cockrill et al. | The Journal of Laboratory Technology for Bioresearch | |
| Chen | An isotope-coding strategy for quantitative proteomics | |
| Patel | A quantitative mass spectrometry-based proteomic analysis of mammalian cell-lines, SH-SY5Y, Homo sapiens neuroblastoma cell line, and PC-12Adh, Rattus norvegicus pheochromocytoma cell line, under hypoxic stress | |
| Kelly | Towards the absolute quantification of protein isoforms through the use of stable-isotope dilution mass spectrometry | |
| Rowell et al. | Serum profiling and biomarker discovery of rat mammary tumors using mass-coded abundance tags (MCAT) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14882930 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2016503809 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15119616 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014882930 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014882930 Country of ref document: EP |