WO2014080447A1 - データ解析装置、データ解析方法 - Google Patents
データ解析装置、データ解析方法 Download PDFInfo
- Publication number
- WO2014080447A1 WO2014080447A1 PCT/JP2012/080003 JP2012080003W WO2014080447A1 WO 2014080447 A1 WO2014080447 A1 WO 2014080447A1 JP 2012080003 W JP2012080003 W JP 2012080003W WO 2014080447 A1 WO2014080447 A1 WO 2014080447A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- data
- cluster
- clustering
- sample data
- experimental error
- Prior art date
Links
Images














Classifications
-
- G—PHYSICS
- G06—COMPUTING; CALCULATING OR COUNTING
- G06F—ELECTRIC DIGITAL DATA PROCESSING
- G06F16/00—Information retrieval; Database structures therefor; File system structures therefor
- G06F16/20—Information retrieval; Database structures therefor; File system structures therefor of structured data, e.g. relational data
- G06F16/23—Updating
- G06F16/2365—Ensuring data consistency and integrity
-
- G—PHYSICS
- G06—COMPUTING; CALCULATING OR COUNTING
- G06F—ELECTRIC DIGITAL DATA PROCESSING
- G06F16/00—Information retrieval; Database structures therefor; File system structures therefor
- G06F16/20—Information retrieval; Database structures therefor; File system structures therefor of structured data, e.g. relational data
- G06F16/28—Databases characterised by their database models, e.g. relational or object models
- G06F16/284—Relational databases
- G06F16/285—Clustering or classification
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B40/00—ICT specially adapted for biostatistics; ICT specially adapted for bioinformatics-related machine learning or data mining, e.g. knowledge discovery or pattern finding
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B40/00—ICT specially adapted for biostatistics; ICT specially adapted for bioinformatics-related machine learning or data mining, e.g. knowledge discovery or pattern finding
- G16B40/30—Unsupervised data analysis
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B50/00—ICT programming tools or database systems specially adapted for bioinformatics
Definitions
- the present invention relates to a technique for clustering analysis of sample data.
- Non-Patent Document 1 discloses a method that can measure a specific gene expression level with sufficient accuracy by a method using a quantitative PCR apparatus for gene expression analysis in a single cell.
- Non-Patent Document 2 discloses a method for quantifying the expression of almost all genes using a large-scale DNA sequencer (next-generation sequencer) for gene expression analysis in a single cell.
- Non-Patent Document 2 also discloses a data analysis method for specifying a cell type. In the future, genome sequences, intracellular proteins, and various biomolecules in cells are expected to be identified at the single cell level.
- an algorithm for classifying cells using only data and an analysis / diagnosis apparatus using the algorithm do not always have sufficient characteristics to classify cells and use them in medical diagnosis.
- necessary characteristics include, for example, cell grouping (hereinafter referred to as clustering), even if the optimal number of groups (hereinafter referred to as cluster number) is not known in advance. It is conceivable to perform appropriate clustering. In particular, it is difficult for a conventional analysis / diagnosis apparatus to determine whether an exceptional cluster having a small number of data is an independent cluster or a part of another cluster having a large number of data.
- the present invention has been made in view of the above-described problems, and an object of the present invention is to provide a data analysis apparatus that can appropriately perform clustering even when exceptional data resulting from experimental error or the like occurs.
- the data analysis apparatus determines a cluster range parameter for relaxing the cluster boundary in advance according to the range of the experimental error described by the experimental error data.
- the clustering process for exception data that does not belong to any cluster, if any cluster includes an area further away from the exception data by a distance determined by the cluster range parameter, the exception data belongs to that cluster. If it is determined that it is not included in any cluster that is separated by the distance, it is determined that it constitutes an independent cluster.
- the data analysis apparatus can appropriately classify cells using the analysis result of a single cell.
- the number of types of classified cells can be determined with high accuracy.
- FIG. 3 shows the results of calculating logarithmic likelihood representing the likelihood of the number of clusters by sweeping the CR value to 1, 2, and 4 for the simulated sample data described in FIG.
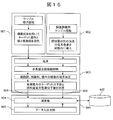
- FIG. 6 is a functional block diagram of a data analysis apparatus 906 according to Embodiment 2.
- FIG. It is a figure which shows the gene expression level of Bglap1, PParg, and Col2a1 of each cluster (indicated by numbers 1 to 5).
- FIG. 13 is a diagram showing expression levels for the three genes described in FIG. 12 with and without clustering, with and without differentiation induction.
- 10 is a functional block diagram of a data analysis apparatus 906 according to Embodiment 3.
- FIG. 10 is a functional block diagram of a data analysis device 906 according to a fourth embodiment.
- Cell data obtained by single cell analysis can be analyzed using, for example, main factor analysis. Data obtained by principal factor analysis is often used to visually determine groups. In order to specifically examine the problems of the conventional clustering method, simulated sample data will be used below. Specifically, assuming that a single cell analysis was performed on four genes and 180 cells, cell data was generated using random numbers on a computer.
- FIG. 1 is a diagram showing a clustering result of simulated sample data.
- FIG. 1A shows the configuration of each cluster in the simulated data.
- ⁇ is a random number and follows a distribution of mean value 0 standard deviation ⁇ .
- the random number ⁇ is assumed to follow an isotropic Gaussian distribution in a four-dimensional space, and the one-sided standard deviation is defined as ⁇ .
- Fig. 1 (b) shows data obtained by conducting a main factor analysis on the simulated data shown in Fig. 1 (a) and visualizing the top two main factors.
- the axis corresponding to the first principal factor was PC1
- the axis corresponding to the second principal factor was PC2.
- the numbers attached to the respective data sets are the cluster numbers described in FIG. As shown in FIG. 1B, the six clusters can be easily visually confirmed.
- the principal factor analysis method is not an algorithm for performing clustering, it does not clearly give a clustering result by means other than visual confirmation.
- the reliability of the clustering result is not given quantitatively.
- the reliability of the clustering result can be used when comparing cell data obtained from a plurality of subjects. For example, when the clustering result of cell data collected from a healthy subject is reliable, the cell data of other subjects can be verified based on the clustering result. However, if the reliability of the clustering result is unknown, it cannot be determined whether it is appropriate to use this as a reference. Therefore, it is very useful to obtain the reliability of clustering results in cell analysis.
- FIG. 2 is a diagram for explaining the hierarchical clustering method.
- the hierarchical clustering method is often used as a method for classifying data in the same manner as the main factor analysis method.
- the result of clustering the same data as in FIG. 1 by the hierarchical clustering method is shown. Since the data of each cell is expressed as a vector in the four-dimensional Euclidean space, the distance between each cell was evaluated using the Euclidean distance.
- the optimal number of clusters cannot be obtained.
- the number of clusters obtained by the hierarchical clustering method does not necessarily match the number of clusters obtained by visual confirmation in the main factor analysis method. Therefore, in the hierarchical clustering method, it is difficult to obtain the optimum number of clusters and the clustering reliability at that time.
- ⁇ To evaluate the reliability of clustering, it is most natural to fit sample data to a probability distribution.
- a method of fitting to a probability distribution a method called a mixed Gaussian model is best known.
- each cell data is assumed to have a Gaussian distribution, and each cell data is considered to belong to any cluster.
- the log likelihood for the Gaussian probability density function is calculated, and the number of clusters, cluster position (average value), and distribution (standard deviation) are determined by the maximum likelihood method.
- FIG. 3 is a diagram showing a result of applying the Akaike information criterion to the same simulated sample data as in FIG.
- various information criterion may be applied to the mixed Gaussian model.
- the Akaike information criterion is well known as a basic information criterion.
- clustering algorithms include support vector machine method and k-means method. However, even if these methods are applied to data whose number of clusters is not known in advance, it is difficult to obtain effective results. Even if the optimum number of clusters is obtained by these methods, it is still difficult to evaluate the reliability of the clustering by numerical values.
- sample data obtained in single cell analysis has a problem that experimental errors cannot be ignored. Since sample data including a large amount of experimental error deviates from the clustering result of sample data with a small error, it is difficult to determine which cluster belongs to, or should be regarded as an independent cluster in the first place. Therefore, it is considered to be important as a cell analysis / diagnosis device to execute clustering with significant resolution on sample data including experimental errors.
- FIG. 4 is a diagram showing a schematic flow of processing for clustering sample data by the data analysis apparatus according to the first embodiment. Hereinafter, each step shown in FIG. 4 will be described. Details of each step will be described again with reference to FIGS.
- the data analysis apparatus acquires sample data in the format described in FIG. 1 (S401).
- the data analysis apparatus acquires data relating to the experimental error when the sample data is acquired (S402). These data may be input to the data analyzer by the operator.
- the data relating to the experimental error is data that numerically indicates how much the sample data varies due to the experimental error. For example, the standard deviation vector value of each gene data can be used as experimental error data.
- the data analysis apparatus relaxes the cluster boundary in the subsequent clustering process in order to include the exception data that does not belong to any cluster due to the experimental error in any cluster. Specifically, when any cluster exists at a position away from the exception data in a predetermined range in the clustering space, the exception data is regarded as belonging to the cluster.
- the predetermined range is called a CR (Clustering Resolution) value in the present invention. Since exceptional data is considered to be caused by experimental error, the CR value is set to a value that is greater than or equal to the experimental error described by the experimental error data. For example, when the experimental error data is represented by the standard deviation ⁇ of the error, the CR value can be a value of about ⁇ to 4 ⁇ .
- Step S404 The data analyzer performs the following step S405 for each CR value while sweeping the CR value.
- the CR value sweep range may be set to ⁇ to 4 ⁇ as described above, for example, when the experimental error data is represented by the standard deviation ⁇ of the error.
- the data analysis apparatus evaluates the optimum number of clusters for each clustering result while temporarily performing clustering by temporarily setting the number k of clusters from 2 to the maximum expected value. Specifically, the likelihood of the current number of clusters is evaluated using the log likelihood of the probability that the sample data belongs to each cluster and the log likelihood of the probability that the sample data does not belong.
- Step S406 When the likelihood of the number of clusters obtained in step S405 takes an extreme value, the data analysis apparatus considers that the number of clusters is optimum, and adopts the number of clusters as the final number of clusters.
- Step S407 The data analysis apparatus outputs the clustering result based on the number of clusters determined in step S406 and the reliability of the clustering result. As the reliability of the clustering result, the likelihood value of the number of clusters obtained in step S405 can be used.
- FIG. 5 is a process flow diagram illustrating the details of step S405. Hereinafter, each step of FIG. 5 will be described.
- the data analysis apparatus provisionally clusters the sample data for a given temporary cluster number k.
- the clustering method in this step may be any method such as a hierarchical clustering method or a k-means method.
- Step S502 The data analysis apparatus replicates k data sets obtained by clustering sample data. Each replicated data set is used to evaluate the likelihood of each clustering result in the following steps.
- the i-th duplicate data set only the i-th cluster is left, all other data are assumed to belong outside the i-th cluster, and clusters other than the i-th cluster are deleted. In other words, it is assumed that all data not belonging to the i-th cluster belongs to a single cluster.
- Step S504 The data analysis device determines whether or not the i-th cluster is configured by exception data. An example of exception data will be described later with reference to FIG. If it is not exception data, steps S505 to S507 are executed, and if it is exception data, steps S508 to S509 are executed.
- Step S504 Judgment Criteria No. 1 of Whether Exception Data or Not
- the i-th cluster holds a sufficient number of sample data, it is determined that the cluster is not due to exception data.
- the data structure determined using the correlation matrix calculated from the sample data belonging to the cluster is recognized as having high reliability.
- a threshold th for determining whether the number of sample data is sufficient is determined in advance. For example, a determination criterion such as considering that the number of sample data in the cluster is 2 or less is regarded as exceptional data can be considered.
- the threshold th can be selected at random within a certain range, for example. Alternatively, an appropriate probability distribution can be assumed and the probability distribution can be selected at random. In this case, the number of clusters located at the center of the probability distribution is most easily selected.
- the parameter of the probability distribution can be arbitrarily determined, or a desired probability distribution can be determined by optimization calculation.
- the data analysis apparatus determines the probability distribution of the sample data using the distribution of the sample data belonging to the i-th cluster.
- the data analysis apparatus evaluates the validity of the judgment whether each sample data belongs to the i-th cluster or not and whether or not the judgment is correct using the probability distribution.
- the specific method is as follows.
- Steps S505 to S507 The data analysis apparatus calculates the standard deviation of the cluster center (average of sample data belonging to the cluster) of the i-th cluster and the sample data in the cluster, and normalizes the sample data (S505).
- the data analysis apparatus calculates an inverse matrix of the correlation matrix of the sample data in the i-th cluster (S506).
- the data analyzer calculates the Mahalanobis distance between each sample data and the i-th cluster center (S507). The reason for calculating the Mahalanobis distance from the cluster center will be described with reference to FIGS.
- Steps S508 to S509 The data analysis apparatus calculates the cluster center of the i-th cluster and normalizes the sample data using the cluster center and the CR value (S508).
- the data analyzer calculates the Euclidean distance between each sample data and the i-th cluster center (S509). The reason for calculating the Euclidean distance from the cluster center will be described with reference to FIGS.
- Step S510 Step 1
- the data analysis apparatus increases the probability that the sample data does not belong to the i-th cluster as the distance from the cluster center increases. Is used to calculate the probability that the sample data does not belong to the i-th cluster.
- the probability distribution function is such that the probability that the sample data belongs to the i-th cluster decreases as the distance from the cluster center increases. The probability that the sample data belongs to the i-th cluster is calculated.
- a probability value is calculated according to a cumulative probability distribution function of an x square distribution with n degrees of freedom
- a probability value is calculated according to a function obtained by subtracting the cumulative probability distribution function from 1.
- An example of this function is illustrated in FIGS. 6 to 7 described later.
- Step S510 Step 2
- the data analysis device calculates the likelihood that each sample data belongs to the i-th cluster or the likelihood that it does not belong by calculating the log likelihood of the probability value calculated according to the above function.
- the likelihood of the current number of clusters k is calculated by calculating the sum of the log likelihoods for all sample data and all clusters and dividing by the number of clusters k. After this step, the same processing is performed from step S503 on the (i + 1) th cluster.
- Step S510 Supplement
- Step S511 Optimization Loop and Cluster Number Sweep Loop
- the data analysis apparatus repeatedly performs clustering using an optimization method such as the Monte Carlo method so that the log likelihood calculated in step S510 is reduced, thereby obtaining an optimal clustering result and an optimal number of clusters. decide.
- an optimization method such as the Monte Carlo method
- the same processing is performed from step S503 while randomly replacing sample data belonging to each cluster.
- the termination condition for the optimization loop may be, for example, when the likelihood of the current number of clusters k calculated in step S510 reaches a predetermined threshold. After completing the optimization loop, the number k of clusters is incremented and the process returns to step S501 to perform the same processing.
- Step S511 CR value sweep loop
- FIG. 6 is a diagram showing a processing image of steps S505 to S507.
- FIG. 1B a clustering space in which axes corresponding to two principal factors are set is illustrated.
- FIG. 6 since a relatively large amount of sample data is collected at the center of the cluster, it is assumed that the data included in the cluster is not exceptional data affected by experimental errors.
- the cluster shape is not necessarily circular in the clustering space
- linear conversion is performed from the data distribution shown on the left of FIG. 6A to the data distribution shown on the right, and each sample data from the cluster center in the cluster shape after conversion is performed.
- Find the distance to. corresponds to obtaining the Mahalanobis distance from the center of each data cluster. In other words, this corresponds to the assumption that the cluster forms a unit space in the Mahalanobis Taguchi method.
- Step S505 In ⁇ S507, for a sample data provisionally determined not to belong to the i-th cluster (e.g. the upper left of the data group in FIG. 6 (a)), with x 2 cumulative probability distribution shown in FIG. 6 (b) Then, the probability that the sample data does not belong to the i-th cluster is calculated. as belonging to the i-th cluster for temporary decision sample data (a data group within a circle in FIG. 6 (a)), using 1-x 2 cumulative probability distribution shown in FIG. 6 (b), the sample data The probability of belonging to the i-th cluster is calculated.
- the probability value increases as it is closer to the cluster center, and for sample data that is provisionally determined not to belong to the i-th cluster, the further away from the cluster center. Probability value increases.
- FIG. 7 is a diagram showing a processing image of steps S508 to S509.
- a clustering space in which axes corresponding to two principal factors are set is illustrated.
- the number of data belonging to the central cluster is small (two in exception 1 and one in exception 2), it is tentatively determined as exception data. This corresponds to the case where the number of data belonging to the i-th cluster is small and the data structure in the cluster cannot be determined sufficiently.
- the CR value is the size of the cluster.
- a predetermined number (exception 1)
- the sample data that does not belong to the exception cluster has a larger probability value.
- the likelihood that the exception data is an independent cluster is highly evaluated.
- the CR value in the present invention has a function of returning sample data deviated from the cluster due to experimental error to the original cluster.
- the CR value is preferably set to a value larger than the experimental error (because the error cannot be corrected if it is smaller than the experimental error), and is set within a limit range obtained from other medical and biological knowledge.
- the data analysis apparatus is tentatively determined not to belong to any cluster using the cluster range parameter (CR value) that relaxes the cluster boundary determined based on the experimental error. It is determined whether the exception data belongs to another cluster. As a result, clustering can be performed with high accuracy even when exceptional data resulting from experimental error occurs.
- CR value cluster range parameter
- the data analysis apparatus evaluates the likelihood of the clustering result while sweeping the CR value with reference to the Mahalanobis distance or the Euclidean distance from the cluster center, and the number of clusters in which the value takes an extreme value Is considered optimal. Thereby, even if the optimum number of clusters is not known in advance, a good clustering result can be obtained.
- the data analysis apparatus outputs the reliability of the clustering result together with the clustering result.
- the diagnostic accuracy by the number of cells belonging to a specific type and the gene expression marker belonging to that type can be improved. That is, conventionally, in a method other than single cell analysis, medical evaluation was performed on a plurality of types of cells.
- a biomarker for a specific population is set. The evaluation is expected to improve the accuracy of determination of the type and condition of a disease using a biomarker.
- the clusters determined by the data analysis apparatus according to the first embodiment correspond to cell types derived from the sample data, respectively.
- a data analysis apparatus that can determine the optimum number of clusters is considered to have the ability to clearly indicate the boundary between one cell type and another cell type. Therefore, the number of cell types can be output from the sample data obtained by cell analysis and diagnosis.
- a population for outputting a statistic such as an average value of a biomarker typified by a specific gene expression level can be determined. Furthermore, since the reliability is given to the determination, it is easy to determine that data having a certain reliability or less is not adopted.
- the data analysis apparatus can be applied to an analysis / diagnosis apparatus in the bio / medical field for analyzing individual cell characteristics one by one.
- the present invention can be applied to an analysis / diagnosis device for analyzing blood cells, an analysis / diagnosis device for cells in urine, or an analysis / diagnosis device for tissue sections. The same applies to the following embodiments.
- a cDNA library in a single cell is prepared on a magnetic bead based on the method described in Non-Patent Document 1, and a single cell A method of quantifying mRNA as small as 0.5 pg in cells with a qPCR apparatus was used.
- FIG. 9 is a functional block diagram of the data analysis apparatus 906 according to the second embodiment.
- the data analysis device 906 includes a calculation unit 904, a data input / output unit 905, a sample data input unit 908, and an experimental error data input unit 909.
- the measurement system for measuring each data is also shown in FIG.
- FIG. 9 shows the configuration of the measurement system in the case where the operation of acquiring experimental error data and the single cell analysis to be clustered are performed in parallel.
- the functional block 901 is a functional unit that acquires sample data to be clustered.
- the functional block 902 is a functional unit that acquires experimental errors.
- a functional block 903 is a functional unit that performs processing common to both. In order to accurately evaluate the experimental error, it is desirable that the data for evaluating the experimental error and the sample data to be clustered have many parts in common. When all or a part of the data related to the experimental error is acquired in advance, the data may be stored in the database 907 and read out by the calculation unit 904 when clustering is performed.
- the functional block 901 collects cells one by one, dispenses each cell into a reaction container, and beads containing poly T probe and a lysis buffer for extracting mRNA into the reaction container into which the cells have been dispensed. Introduce.
- the functional block 902 introduces a large number of cells into the reaction vessel, and then performs mRNA extraction in the same manner as the functional block 901. Thereafter, the mRNA solution is diluted, and an equivalent amount of a single cell is collected. Dispense this solution into the solution.
- the functional block 903 removes the lysis buffer, dispenses a reaction solution containing reverse transcriptase, removes the reaction solution after the reverse transcription reaction, adds mRNA-degrading enzyme, and after washing, introduces qPCR reagent, PCR Quantification is performed by performing fluorescence measurements while applying a temperature cycle for.
- the gene expression level in each sample is calculated based on the cycle number Ct value in which the fluorescence intensity exceeds a certain threshold.
- the Ct value is read as the number of molecules.
- Experimental error includes errors in all processes other than single cell sampling until quantification is performed. Since it is desirable that the quantitative value follows a Gaussian distribution, a value obtained by taking the logarithm of the number of molecules is output to the calculation unit 904 as sample data. The same applies to the experimental error data.
- the sample data input unit 908 acquires sample data via the function blocks 901 and 903.
- the experimental error data input unit 909 acquires experimental error data via the function blocks 902 and 903.
- the calculation unit 904 performs clustering using these data according to the processing flow described in the first embodiment.
- the data input / output unit 905 controls the input / output of these data.
- steps S508 to S509 are performed when the number of elements in the temporary cluster is smaller than 3 (2 or less).
- the expression level (sample data value) of a specific gene is known to fluctuate biologically or medically
- the fluctuation range may be used as the CR value.
- Such a correspondence relationship between a gene and a CR value and a correspondence relationship between a category of sample data and a CR value are stored in the database 907, and the database 907 is selected from the gene and sample data categories input to the data analysis device 906. If there is a corresponding item that matches the one stored in, the calculation unit 906 reads it from the database 907.
- the determination threshold value in step S504 may be stored in advance in the database 907 instead of the experimental error data, and an appropriate value may be used based on the sample data or gene information input to the data analysis device 904.
- the determination threshold value in step S504 is a fixed value corresponding to the number of sample data in the cluster. However, a plurality of determination threshold values are provided and clustering is performed using each threshold value. A threshold having the lowest likelihood (most likely) may be selected. Furthermore, the threshold value may be selected randomly, or may be selected randomly on the assumption that the threshold value follows a certain probability distribution. A plurality of types of probability distribution functions at this time may be stored in the database 907 in advance, and an appropriate probability distribution function may be selected according to the contents of the sample data.
- ⁇ Embodiment 2 Sampling result>
- 92 mesenchymal stem cells (C3H10T1 / 2) of mice were used. That is, such cells were collected in order to know the differentiation induction state of the bone of the mouse from which the cells were obtained.
- other cells for example, immune cells in blood, tissue sections of cancer, etc.
- the gene expression data used as sample data was quantitative values for five types of genes: Bglap1, Col1a1, Pparg, Col2a1, and Eef1g. The kind and number of genes are only one example, and all possible genes may be measured.
- mRNA extracted from a large number of cells of about 5 ⁇ 10 5 or more was sampled for 96 pieces of 0.5 pg which is equivalent to a single cell, and 5 ⁇ 10 5 cells
- the qPCR apparatus was used for quantification.
- the variation in the quantification value is the total amount of experimental errors composed of handling errors when preparing the cDNA library and quantification errors during qPCR quantification. This error varies from gene to gene. This error was evaluated as a five-dimensional vector.
- CR value (vector value) is assumed to be ⁇ .
- the experimental error matches the minimum CR value.
- the expression of a specific gene changes with time even in the same cell state, and there are cases where the fluctuation range of data is known biologically in addition to the variation in data due to experimental errors. If it is not desired to use these variations in clustering, the value of ⁇ may be partially changed based on the sample data. Specifically, for example, when there is a fluctuation of about 10 times with respect to a specific gene, the numerical value of the fluctuation range may be used as the CR value instead of the experimental error only for this gene. However, this is limited to cases where the experimental error is sufficiently smaller than biological or medical fluctuations.
- FIG. 10 shows the result of calculating the log likelihood representing the likelihood of the number of clusters in the second embodiment.
- FIG. 11 is a diagram comparing the result of hierarchical clustering with the result of clustering by the data analysis apparatus 906 according to the second embodiment. Fifteen clusters are represented by square boxes below the tournament diagram corresponding to the hierarchical clustering result. The 15 clusters were found to be composed of 5 significant clusters and 10 exception clusters. Five significant clusters are consistent with the result of hierarchical clustering. It can be seen that the boundary between the significant cluster and the exceptional cluster has been clarified by the second embodiment.
- FIG. 12 is a diagram showing gene expression levels of Bglap1, PPar, and Col2a1 in each cluster (indicated by numbers 1 to 5).
- the vertical axis represents the number of molecules of each gene in a single cell.
- the horizontal axis indicates the cluster number.
- the black bar is the differentiation inducer, and the gray bar is the gene expression level for the corresponding cells without the differentiation inducer.
- the distinction between presence and absence of differentiation inducers is an example of cell information that is known in advance.
- FIG. 13 is a diagram showing the expression levels of the three genes described in FIG. 12 with and without clustering, with and without differentiation induction. As shown in FIG. 13, there is a change corresponding to the prior information for Blap1, but there is no change in the expression level corresponding to the prior information for Pparg and Col2a1, and no information is obtained for these two genes. From the above, it can be seen that more detailed information on gene expression in the living body can be obtained by clustering.
- the data analysis apparatus 906 is an apparatus for estimating the state of a living body by estimating how many cells belonging to what kind (cluster) exist in the living body. is there.
- the second embodiment is effective when the type of cluster and the number of cells belonging to the cluster change when the health state of the living body to be measured changes.
- FIG. 14 is a functional block diagram of the data analysis apparatus 906 according to the third embodiment of the present invention.
- a configuration example using a large-scale DNA sequencer will be described as a gene expression quantification method. Thereby, the number of genes to be measured can be greatly increased.
- the functional block 901 extracts the clustering target samples by dispensing them one by one into a reaction vessel containing poly T probe beads on a plate, crushing cells in the reaction vessel, and trapping mRNA on the bead surface. .
- the function block 902 dispenses known amounts of mRNA for each of a plurality of genes into individual reaction vessels in order to measure experimental errors. By calculating the standard variation deviation from the sequencing data corresponding to the reaction vessel into which a known amount of RNA has been introduced, the experimental error relating to sample processing and sequencing can be quantified.
- Function block 903 performs reverse transcription reaction and mRNA degradation.
- a primer for batch amplification is introduced into the end of the cDNA, and then batch amplification is performed using this primer. Further, fragmentation is performed, and amplification is performed using an amplification primer having a cell recognition tag that is different in sequence for each reaction tank (primer sequence is common to all containers) to construct a sequencing library. Since a tag having a different sequence is inserted for each container, that is, for each cell, at the end of the sequencing library, the sample can be mixed in the following processing. In order to sequence the mixed samples with a large-scale sequencer, sequencing is performed using individual amplification such as emulsion PCR or bridge PCR.
- any device such as a device using fluorescence measurement, a device using FET, or a device using nanopores may be used.
- mapping the sequencing data obtained from the fragmented sample to a known gene sequence it is determined which sequence of which region of which gene can be measured. Thereafter, the data is aggregated for each gene, and the expression level data for each gene is calculated.
- an algorithm generally used by the expert may be used. As a result, expression level data for each cell / gene is obtained.
- the clustering procedure is the same as in Embodiments 1 and 2, but since the number of measured genes is as large as about tens of thousands, cells can be classified in more detail.
- the data input to the data analysis device 906 is the number of mutations obtained by dividing the genome into a plurality of regions and counting each divided region.
- the measurement purpose in this case may be, for example, to elucidate the mechanism of cancer occurrence or metastasis by measuring cells from a tissue section of cancer, or to diagnose for selecting a molecular target drug.
- Supplementary data obtained by analyzing genome data The whole genome or a part thereof is sequence-analyzed for each single cell (assuming that data in mRNA sequence analysis is used), for example, a region is divided every 50 k bases, and mutations are measured.
- the measurement object includes, for example, single base substitution, deletion, insertion, gene copy number abnormality, and the like.
- the input data is the number of each mutation. Experimental error can be assessed by artificially creating a sample without mutation and evaluating it.
- Embodiment 4 of the present invention instead of characterizing a cell by quantifying the expression level (number of molecules) of a gene in the cell, a cell sample obtained by immunostaining or the like is imaged with a fluorescence microscope, and the fluorescence intensity is measured.
- a fluorescence microscope instead of characterizing a cell by quantifying the expression level (number of molecules) of a gene in the cell, a cell sample obtained by immunostaining or the like is imaged with a fluorescence microscope, and the fluorescence intensity is measured.
- An example of a configuration in which cells are classified by quantifying the amount of protein in the cells by the correspondence data of the number of molecules or single molecule fluorescence counting will be described.
- the type of gene corresponds to the type of protein.
- the amount of protein (number of molecules) for each cell is input to the data analyzer 906 as sample data.
- sample preparation such as immunostaining
- an error at the time of fluorescence measurement is evaluated and input to the data analyzer 906 as an experimental error. Thereby, clustering of cells can be executed.
- FIG. 15 is a functional block diagram of the data analysis apparatus 906 according to the fourth embodiment.
- the functional block 901 collects a tissue section as a cell sample, and performs immunostaining after deparaffinization. At this time, a known amount of a fluorescent dye having a fluorescence wavelength different from that of immunostaining is introduced into the cell, and at the same time, nuclear staining and cell membrane staining are performed. By carrying out the measurement of the fluorescence of 7 colors, images corresponding to the target protein (3 types), the experimental error measurement dye, the nucleus, and the cell membrane are obtained. When increasing the type of target protein, it is necessary to increase the measurement color.
- the present invention is not limited to the above-described embodiment, and includes various modifications.
- the above embodiment has been described in detail for easy understanding of the present invention, and is not necessarily limited to the one having all the configurations described.
- a part of the configuration of one embodiment can be replaced with the configuration of another embodiment.
- the configuration of another embodiment can be added to the configuration of a certain embodiment. Further, with respect to a part of the configuration of each embodiment, another configuration can be added, deleted, or replaced.
- the above components, functions, processing units, processing means, etc. may be realized in hardware by designing some or all of them, for example, with an integrated circuit.
- Each of the above-described configurations, functions, and the like may be realized by software by interpreting and executing a program that realizes each function by the processor.
- Information such as programs, tables, and files for realizing each function can be stored in a recording device such as a memory, a hard disk, an SSD (Solid State Drive), or a recording medium such as an IC card, an SD card, or a DVD.
- 906 data analysis device
- 904 calculation unit
- 905 data input / output unit
- 908 sample data input unit
- 909 experimental error data input unit.
Abstract
Description
以下では本発明の理解を促進するため、まず初めに従来のデータ解析手法およびその課題について、具体例を挙げて説明する。その後に、本発明に係るデータ解析装置の具体的な構成について説明する。
以上、従来のデータ解析手法およびその課題について説明した。以下では本発明の実施形態1に係るデータ解析装置について説明する。単一細胞ごとの生体分子に関する解析、特に遺伝子発現解析において得られるデータは、各細胞の生体分子の定量値を要素値とする行列を用いて表現することができる。個々の細胞に対する各遺伝子の発現量のサンプルデータは、遺伝子数をn、細胞数をmとすると、m行n列の行列で表示できる。以下ではこの形式によって記述されたサンプルデータを前提とする。
データ解析装置は、図1で説明した形式のサンプルデータを取得する(S401)。データ解析装置は、サンプルデータを取得したときの実験誤差に関するデータを取得する(S402)。これらのデータは、オペレータがデータ解析装置へ入力してもよい。実験誤差に関するデータとは、サンプルデータが実験誤差に起因してどの程度ばらついているかを数値的に示すデータである。たとえば、各遺伝子データの標準偏差ベクトル値を実験誤差データとすることができる。
データ解析装置は、実験誤差に起因していずれのクラスタにも属さなくなっている例外データをいずれかのクラスタに包含させるため、以後のクラスタリング処理においてクラスタ境界を緩和する。具体的には、クラスタリング空間において例外データから所定範囲離れた位置にいずれかのクラスタが存在する場合は、その例外データは当該クラスタに属するものとみなす。上記所定範囲のことを、本発明においてはCR(Clustering Resolution)値と呼ぶことにする。例外データは実験誤差によって生じると考えられるため、CR値は実験誤差データが記述している実験誤差以上の値として設定する。例えば実験誤差データを誤差の標準偏差σによって表す場合、CR値はσ~4σ程度の値とすることができる。
データ解析装置は、CR値を掃引しながら各CR値について以下のステップS405を実施する。CR値の掃引範囲としては、実験誤差データを誤差の標準偏差σによって表す場合、例えば上述のようにσ~4σなどとすればよい。
データ解析装置は、クラスタ数kを2~予想最大値まで仮設定して実際にクラスタリングを実施しながら、各クラスタリング結果についてクラスタ数の最適度を評価する。具体的には、サンプルデータが各クラスタに属する確率の対数尤度および属さない確率の対数尤度を用いて、現在のクラスタ数の尤もらしさを評価する。
データ解析装置は、ステップS405において求めたクラスタ数の尤もらしさが極値を取るとき、そのクラスタ数が最適であるものとみなし、そのクラスタ数を最終的なクラスタ数として採用する。
データ解析装置は、ステップS406において決定したクラスタ数に基づくクラスタリング結果とともに、そのクラスタリング結果の信頼度を出力する。クラスタリング結果の信頼度としては、ステップS405において求めたクラスタ数の尤もらしさの値を用いることができる。
データ解析装置は、与えられた仮のクラスタ数kについて、サンプルデータを暫定的にクラスタリングする。本ステップにおけるクラスタリング手法は、例えば階層的クラスタリング法やk-means法など任意の手法でよい。
データ解析装置は、サンプルデータをクラスタリングしたデータセットをk個分複製する。複製された各データセットは、それぞれ以下のステップにおいて各クラスタリング結果の尤もらしさを評価するために用いる。データ解析装置は、以下のステップにおいて用いるカウンタiを初期化する(i=1)。
データ解析装置は、i番目(i=1~k)のクラスタについて、i番目の複製データセットを用いて以下のステップを実施する。i番目の複製データセットに関してはi番目のクラスタのみを残し、その他データは全てi番目のクラスタ外に属するものと仮定し、i番目のクラスタ以外のクラスタについては削除する。すなわち、i番目のクラスタに属さないデータについては、全て単一のクラスタに属するものと仮定する。
データ解析装置は、i番目のクラスタが例外データによって構成されているものであるか否かを判定する。例外データの例については後述の図6で説明する。例外データでなければステップS505~S507を実施し、例外データであればステップS508~S509を実施する。
i番目のクラスタがその内部に十分なサンプルデータ数を保持している場合は、そのクラスタは例外データによるものではないと判定する。この場合は、クラスタに属するサンプルデータから計算される相関行列などを用いて決定したデータ構造は、信頼度が高いと認められるからである。サンプルデータ数が十分であるか否かの閾値thは、あらかじめ定めておく。例えばクラスタ内のサンプルデータ数が2個以下であれば例外データとみなす、などの判定基準が考えられる。
閾値thは、例えばある範囲内でランダムに選択することもできる。あるいは、適当な確率分布を仮定して、その確率分布を前提としてランダムに選択することもできる。この場合は、確率分布の中央に位置するクラスタ数が最も選択され易くなる。確率分布のパラメータは任意に定めることもできるし、最適化計算によって望ましい確率分布を決定することもできる。
データ解析装置は、i番目のクラスタ内に属するサンプルデータの分布を用いてそのサンプルデータの確率分布を決定する。データ解析装置は、各サンプルデータがi番目のクラスタに属する、あるいは属さないと判定した判断が正しいか否かについて、上記確率分布を用いてその妥当性を評価する。具体的な手法は以下の通りである。
データ解析装置は、i番目のクラスタのクラスタ中心(クラスタ内に属するサンプルデータの平均)とクラスタ内のサンプルデータの標準偏差を算出し、サンプルデータを規格化する(S505)。データ解析装置は、i番目のクラスタ内のサンプルデータの相関行列の逆行列を計算する(S506)。データ解析装置は、各サンプルデータとi番目のクラスタ中心との間のマハラノビス距離を計算する(S507)。クラスタ中心からのマハラノビス距離を計算する理由については後述の図6~図7で説明する。
データ解析装置は、i番目のクラスタのクラスタ中心を算出し、クラスタ中心とCR値を用いてサンプルデータを規格化する(S508)。データ解析装置は、各サンプルデータとi番目のクラスタ中心との間のユークリッド距離を計算する(S509)。クラスタ中心からのユークリッド距離を計算する理由については後述の図6~図7で説明する。
データ解析装置は、ステップS501においてi番目のクラスタに属さないと判定したサンプルデータについて、クラスタ中心からの距離が離れるほど当該サンプルデータがi番目のクラスタに属さない確率が高くなるような確率分布関数を用いて、当該サンプルデータがi番目のクラスタに属さない確率を算出する。同様にステップS501においてi番目のクラスタに属すると判定したサンプルデータについて、クラスタ中心からの距離が離れるほど当該サンプルデータがi番目のクラスタに属する確率が低くなるような確率分布関数を用いて、当該サンプルデータがi番目のクラスタに属する確率を算出する。例えば、前者については自由度nのx2乗分布の累積確率分布関数にしたがって確率値を計算し、後者については1から上記累積確率分布関数を引いた関数にしたがって確率値を計算する。この関数の例については後述の図6~図7で例示する。
データ解析装置は、上記関数にしたがって計算した確率値の対数尤度を計算することにより、各サンプルデータがi番目のクラスタに属することの尤もらしさ、または属さないことの尤もらしさを計算する。すべてのサンプルデータおよびすべてのクラスタについてこの対数尤度の和を求め、クラスタ数kで除算することにより、現在のクラスタ数kの尤もらしさを計算する。本ステップの次は、i+1番目のクラスタについてステップS503から同様の処理を実施する。
対数尤度の評価に用いているのはクラスタリングが妥当である確率の評価値であるため、最適パラメータが得られた時の対数尤度の値から、クラスタリングの信頼度を確率値で出力することが可能である。
データ解析装置は、例えばモンテカルロ法などのような最適化手法を用いて、ステップS510で計算した対数尤度が小さくなるようにクラスタリングを繰り返し実施することにより、最適なクラスタリング結果と最適なクラスタ数を決定する。最適化手法として例えばモンテカルロ法を用いる場合は、各クラスタに属するサンプルデータをランダムに入れ替えながらステップS503から同様の処理を実施する。最適化ループの終了条件は、例えばステップS510で計算した現在のクラスタ数kの尤もらしさが所定閾値に達した時点などとすればよい。最適化ループを完了した後はクラスタ数kをインクリメントしてステップS501に戻り、同様の処理を実施する。
データ解析装置は、現在のCR値について最適化ループおよびクラスタ数掃引ループを終了した後は、CR値をインクリメントしてステップS501に戻り、同様の処理を実施する。CR値をインクリメントする幅は、想定しているCR値の最小値と最大値の差に応じて適宜定める。
以上のように、本実施形態1に係るデータ解析装置は、実験誤差に基づき定めた、クラスタ境界を緩和するクラスタ範囲パラメータ(CR値)を用いて、いずれのクラスタにも属さないと仮判定された例外データが他のクラスタに属するか否かを判定する。これにより、実験誤差に起因する例外データが生じる場合であっても、精度よくクラスタリングを実施することができる。
本発明の実施形態2では、実施形態1で説明したデータ解析装置の具体的な適用例として、単一細胞中の遺伝子発現解析によるデータによって細胞を分類するデータ解析装置について説明する。
以下では、本実施形態2に係るデータ解析装置906によるサンプリング結果の例を説明する。サンプルとしてマウスの間葉系幹細胞(C3H10T1/2)を92個用いた。すなわち、細胞を取得したマウスの骨の分化誘導状態を知るためにこのような細胞を採取した。もちろん、他の目的のための他の生体(人を含む)から、別の細胞(たとえば、血中の免疫細胞やがんの組織切片など)を採取してもよい。サンプルデータとして用いる遺伝子発現データは、Bglap1、Col1a1、Pparg、Col2a1、Eef1gの5種類の遺伝子についての定量値とした。遺伝子の種類およびその数は1例であり、可能性のある遺伝子すべてを計測対象にしてもかまわない。
図14は、本発明の実施形態3に係るデータ解析装置906の機能ブロック図である。本実施形態3では、遺伝子発現の定量方法として、大規模DNAシーケンサを用いる構成例を説明する。これにより、計測対象とする遺伝子数を大幅に増やすことができる。
本発明の実施形態4では、細胞中の遺伝子の発現量(分子数)を定量することによって細胞を特徴付ける代わりに、免疫染色法などで得られた細胞サンプルを蛍光顕微鏡でイメージングして、蛍光強度と分子数の対応データまたは単分子蛍光カウンティングによって細胞中の蛋白質量を定量することにより細胞を分類する構成例を説明する。
Claims (11)
- 複数のサンプルデータをクラスタリング解析する装置であって、
複数のサンプルデータを受け取るサンプルデータ入力部と、
前記サンプルデータの実験誤差についての情報を記述した実験誤差データを受け取る実験誤差データ入力部と、
前記複数のサンプルデータをクラスタリング空間内においてクラスタリングする演算部と、
前記クラスタリングの結果を出力する出力部と、
を備え、
前記演算部は、
前記実験誤差データが記述している前記実験誤差の範囲に応じて、前記クラスタリングを実施する際におけるクラスタ境界を緩和するためのクラスタ範囲パラメータをあらかじめ取得しておき、
仮設定したクラスタ総数に準じて前記複数のサンプルデータをクラスタリングし、
前記複数のサンプルデータのうちいずれのクラスタにも属さない例外データについては、
前記クラスタリング空間上で前記例外データからさらに前記クラスタ範囲パラメータによって定められる距離だけ離れた領域がいずれかのクラスタに含まれる場合は、前記例外データがそのクラスタに属するものと判定し、前記領域がいずれのクラスタにも含まれない場合は、前記例外データが独立したクラスタを構成するものと判定する
ことを特徴とするデータ解析装置。 - 前記演算部は、
各前記サンプルデータが前記クラスタリングの結果得られた各クラスタに属する確率の尤もらしさを表す第1対数尤度と、各前記サンプルデータが前記クラスタリングの結果得られた各クラスタに属さない確率の尤もらしさを表す第2対数尤度とをそれぞれ算出する処理を、前記第1対数尤度と前記第2対数尤度を用いて算出される前記クラスタリング結果の尤もらしさが所定閾値に達するまで繰り返すことにより、最適なクラスタ総数を求め、
得られた最適なクラスタ総数に準じて、前記複数のサンプルデータの最終的なクラスタリング結果を決定する
ことを特徴とする請求項1記載のデータ解析装置。 - 前記演算部は、
前記仮設定したクラスタ総数に準じて前記複数のサンプルデータをクラスタリングする過程において、前記サンプルデータが仮設定したクラスタに属すると仮定して前記第1対数尤度と前記第2対数尤度を算出し、
前記仮設定したクラスタに属すると仮定した前記サンプルデータについては、前記クラスタリング空間上において、前記仮設定したクラスタの中心からの距離が離れるにしたがって、前記仮設定したクラスタに属する確率を低く評価し、
前記仮設定したクラスタに属さないと仮定した前記サンプルデータについては、前記クラスタリング空間上において、前記仮設定したクラスタの中心からの距離が離れるにしたがって、前記仮設定したクラスタに属さない確率を高く評価する
ことを特徴とする請求項2記載のデータ解析装置。 - 前記演算部は、
クラスタ内に属する前記サンプルデータの個数が所定個数以上であるか否かを基準として、前記サンプルデータが前記例外データであるか否かを判定する
ことを特徴とする請求項1記載のデータ解析装置。 - 前記演算部は、前記所定個数をランダムに選択する
ことを特徴とする請求項4記載のデータ解析装置。 - 前記演算部は、所定の確率分布にしたがって前記所定個数をランダムに選択する
ことを特徴とする請求項4記載のデータ解析装置。 - 前記演算部は、
前記クラスタ範囲パラメータを掃引し、各前記クラスタ範囲パラメータの値を採用した場合における前記クラスタリングによって得られたクラスタ総数を取得し、
前記第1対数尤度と前記第2対数尤度に基づき算出した前記クラスタリング結果の尤もらしさの値が極値となるクラスタ総数を、最適なクラスタ総数として採用する
ことを特徴とする請求項2記載のデータ解析装置。 - 前記演算部は、
前記クラスタリングを実施する過程において得られる情報を用いて、前記クラスタリングの結果の信頼度指標を算出し、
前記出力部は、
前記信頼度指標を前記クラスタリングの結果と併せて出力する
ことを特徴とする請求項1記載のデータ解析装置。 - 前記演算部は、
前記第1対数尤度と前記第2対数尤度に基づき算出した前記クラスタリング結果の尤もらしさの値を、前記クラスタリングの結果の信頼度指標として算出する
ことを特徴とする請求項8記載のデータ解析装置。 - 前記サンプルデータ入力部と前記実験誤差データ入力部は、細胞の解析結果に関するデータをそれぞれ前記サンプルデータおよび前記実験誤差データとして受け取り、
前記演算部は、前記クラスタリングによって前記細胞をグループ化する
ことを特徴とする請求項1記載のデータ解析装置。 - 複数のサンプルデータをクラスタリング解析する方法であって、
複数のサンプルデータを受け取るサンプルデータ入力ステップ、
前記サンプルデータの実験誤差についての情報を記述した実験誤差データを受け取る実験誤差データ入力ステップ、
前記複数のサンプルデータをクラスタリング空間内においてクラスタリングする演算ステップ、
前記クラスタリングの結果を出力する出力ステップ、
を有し、
前記演算ステップにおいては、
前記実験誤差データが記述している前記実験誤差の範囲に応じて、前記クラスタリングを実施する際におけるクラスタ境界を緩和するためのクラスタ範囲パラメータをあらかじめ取得しておき、
仮設定したクラスタ総数に準じて前記複数のサンプルデータをクラスタリングし、
前記複数のサンプルデータのうちいずれのクラスタにも属さない例外データについては、
前記クラスタリング空間上で前記例外データからさらに前記クラスタ範囲パラメータによって定められる距離だけ離れた領域がいずれかのクラスタに含まれる場合は、前記例外データがそのクラスタに属するものと判定し、前記領域がいずれのクラスタにも含まれない場合は、前記例外データが独立したクラスタを構成するものと判定する
ことを特徴とするデータ解析方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/443,118 US20150302042A1 (en) | 2012-11-20 | 2012-11-20 | Data analysis apparatus and data analysis method |
| JP2014548349A JP6029683B2 (ja) | 2012-11-20 | 2012-11-20 | データ解析装置、データ解析プログラム |
| PCT/JP2012/080003 WO2014080447A1 (ja) | 2012-11-20 | 2012-11-20 | データ解析装置、データ解析方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/JP2012/080003 WO2014080447A1 (ja) | 2012-11-20 | 2012-11-20 | データ解析装置、データ解析方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014080447A1 true WO2014080447A1 (ja) | 2014-05-30 |
Family
ID=50775654
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/080003 WO2014080447A1 (ja) | 2012-11-20 | 2012-11-20 | データ解析装置、データ解析方法 |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US20150302042A1 (ja) |
| JP (1) | JP6029683B2 (ja) |
| WO (1) | WO2014080447A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016158576A (ja) * | 2015-03-03 | 2016-09-05 | 富士フイルム株式会社 | 細胞群検出装置および方法並びにプログラム |
| JP2018530815A (ja) * | 2015-08-17 | 2018-10-18 | コーニンクレッカ フィリップス エヌ ヴェKoninklijke Philips N.V. | 生体データにおけるパターン認識のマルチレベルアーキテクチャ |
| WO2022009342A1 (ja) * | 2020-07-08 | 2022-01-13 | 富士通株式会社 | 情報処理プログラム、情報処理方法および情報処理装置 |
| US11327470B2 (en) | 2017-12-21 | 2022-05-10 | Mitsubishi Power, Ltd. | Unit space generating device, plant diagnosing system, unit space generating method, plant diagnosing method, and program |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6274114B2 (ja) * | 2013-01-10 | 2018-02-07 | 富士通株式会社 | 制御方法、制御プログラム、および制御装置 |
| EP3361416A1 (en) | 2017-02-09 | 2018-08-15 | Tata Consultancy Services Limited | Statistical modeling techniques based neural network models for generating intelligence reports |
| US10846311B2 (en) * | 2017-11-01 | 2020-11-24 | Mad Street Den, Inc. | Method and system for efficient clustering of combined numeric and qualitative data records |
| US10635939B2 (en) | 2018-07-06 | 2020-04-28 | Capital One Services, Llc | System, method, and computer-accessible medium for evaluating multi-dimensional synthetic data using integrated variants analysis |
| CN109325059A (zh) * | 2018-12-03 | 2019-02-12 | 枘熠集成电路(上海)有限公司 | 一种数据比较方法及装置 |
| CN110287456A (zh) * | 2019-06-30 | 2019-09-27 | 张家港宏昌钢板有限公司 | 基于数据挖掘的大盘卷轧制表面缺陷分析方法 |
| CN110490229A (zh) * | 2019-07-16 | 2019-11-22 | 昆明理工大学 | 一种基于spark和聚类算法的电能表检定误差诊断方法 |
| CN113418885A (zh) * | 2021-07-22 | 2021-09-21 | 合肥学院 | 一种用于紫外分光光度计实验数据的分析方法 |
| CN113673582B (zh) * | 2021-07-30 | 2023-05-09 | 西南交通大学 | 基于系统聚类分析的铁路动态基准点多层级分群方法 |
| CN116796214B (zh) * | 2023-06-07 | 2024-01-30 | 南京北极光生物科技有限公司 | 一种基于差分特征的数据聚类方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004272350A (ja) * | 2003-03-05 | 2004-09-30 | Nec Corp | クラスタリング装置、クラスタリング方法、クラスタリングプログラム |
| JP2006163894A (ja) * | 2004-12-08 | 2006-06-22 | Hitachi Software Eng Co Ltd | クラスタリングシステム |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2517286C2 (ru) * | 2008-04-25 | 2014-05-27 | Конинклейке Филипс Электроникс Н.В. | Классификация данных выборок |
-
2012
- 2012-11-20 US US14/443,118 patent/US20150302042A1/en not_active Abandoned
- 2012-11-20 WO PCT/JP2012/080003 patent/WO2014080447A1/ja active Application Filing
- 2012-11-20 JP JP2014548349A patent/JP6029683B2/ja not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004272350A (ja) * | 2003-03-05 | 2004-09-30 | Nec Corp | クラスタリング装置、クラスタリング方法、クラスタリングプログラム |
| JP2006163894A (ja) * | 2004-12-08 | 2006-06-22 | Hitachi Software Eng Co Ltd | クラスタリングシステム |
Non-Patent Citations (4)
| Title |
|---|
| DAVE, R.N.: "Characterization and detection of noise in clustering", PATTERN RECOGNITION LETTERS, vol. 12, no. 11, November 1991 (1991-11-01), pages 657 - 664 * |
| ENDO, Y.: "Fuzzy c-Means for Data with Tolerance", PROC. 2005 INTERNATIONAL SYMPOSIUM ON NONLINEAR THEORY AND ITS APPLICATIONS (NOLTA2005), 2005, pages 345 - 348 * |
| HIDETOMO ICHIHASHI: "Fuzziness and Robustness in Fuzzy Clustering : Regularization, Robustification and IRLS", JOURNAL OF JAPAN SOCIETY FOR FUZZY THEORY AND INTELLIGENT INFORMATICS, vol. 17, no. 4, 15 August 2005 (2005-08-15), pages 392 - 397 * |
| YUKIHIRO HAMASUNA: "On Hard Clustering for Data with Tolerance", JOURNAL OF JAPAN SOCIETY FOR FUZZY THEORY AND INTELLIGENT INFORMATICS, vol. 20, no. 3, 15 June 2008 (2008-06-15), pages 388 - 398 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016158576A (ja) * | 2015-03-03 | 2016-09-05 | 富士フイルム株式会社 | 細胞群検出装置および方法並びにプログラム |
| JP2018530815A (ja) * | 2015-08-17 | 2018-10-18 | コーニンクレッカ フィリップス エヌ ヴェKoninklijke Philips N.V. | 生体データにおけるパターン認識のマルチレベルアーキテクチャ |
| JP7041614B2 (ja) | 2015-08-17 | 2022-03-24 | コーニンクレッカ フィリップス エヌ ヴェ | 生体データにおけるパターン認識のマルチレベルアーキテクチャ |
| JP7041614B6 (ja) | 2015-08-17 | 2022-05-31 | コーニンクレッカ フィリップス エヌ ヴェ | 生体データにおけるパターン認識のマルチレベルアーキテクチャ |
| US11327470B2 (en) | 2017-12-21 | 2022-05-10 | Mitsubishi Power, Ltd. | Unit space generating device, plant diagnosing system, unit space generating method, plant diagnosing method, and program |
| WO2022009342A1 (ja) * | 2020-07-08 | 2022-01-13 | 富士通株式会社 | 情報処理プログラム、情報処理方法および情報処理装置 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2014080447A1 (ja) | 2017-01-05 |
| JP6029683B2 (ja) | 2016-11-24 |
| US20150302042A1 (en) | 2015-10-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6029683B2 (ja) | データ解析装置、データ解析プログラム | |
| CN105096225B (zh) | 辅助疾病诊疗的分析系统、装置及方法 | |
| JP6313757B2 (ja) | 統合デュアルアンサンブルおよび一般化シミュレーテッドアニーリング技法を用いてバイオマーカシグネチャを生成するためのシステムおよび方法 | |
| CN112005306A (zh) | 选择、管理和分析高维数据的方法和系统 | |
| US20050159896A1 (en) | Apparatus and method for analyzing data | |
| US9940383B2 (en) | Method, an arrangement and a computer program product for analysing a biological or medical sample | |
| CN109411015A (zh) | 基于循环肿瘤dna的肿瘤突变负荷检测装置及存储介质 | |
| Cao et al. | ROC curves for the statistical analysis of microarray data | |
| US20200227168A1 (en) | Machine learning in functional cancer assays | |
| WO2014024142A2 (en) | Population classification of genetic data set using tree based spatial data structure | |
| CN112634987B (zh) | 一种单样本肿瘤dna拷贝数变异检测的方法和装置 | |
| JP5854346B2 (ja) | トランスクリプトーム解析方法、疾病判定方法、コンピュータプログラム、記憶媒体、及び解析装置 | |
| Raza et al. | A novel anticlustering filtering algorithm for the prediction of genes as a drug target | |
| CN107463797B (zh) | 高通量测序的生物信息分析方法及装置、设备及存储介质 | |
| KR101067352B1 (ko) | 생물학적 네트워크 분석을 이용한 마이크로어레이 실험 자료의 작용기작, 실험/처리 조건 특이적 네트워크 생성 및 실험/처리 조건 관계성 해석을 위한 알고리즘을 포함한 시스템 및 방법과 상기 방법을 수행하기 위한 프로그램을 갖는 기록매체 | |
| KR102111820B1 (ko) | 동적 네트워크 바이오마커의 검출 장치, 검출 방법 및 검출 프로그램 | |
| CN101517579A (zh) | 蛋白质查找方法和设备 | |
| KR20180051333A (ko) | 유전체내 암 특이적 진단 마커 검출 | |
| WO2012149107A2 (en) | Stratifying patient populations through characterization of disease-driving signaling | |
| Wang et al. | Learning dynamics by computational integration of single cell genomic and lineage information | |
| JP6517933B2 (ja) | 検査システム、検査装置、及び検査方法 | |
| CN110751983A (zh) | 一种筛选特征mRNA用于诊断早期肺癌的方法 | |
| CN110475874A (zh) | 脱靶序列在dna分析中的应用 | |
| CN116825367A (zh) | 一种组织硬度预测模型的建立、组织硬度预测方法及装置 | |
| Gulla | An integrated systems biology approach to investigate transcriptomic data of thyroid carcinoma |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12888920 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014548349 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14443118 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 12888920 Country of ref document: EP Kind code of ref document: A1 |