WO2014072956A1 - Oxazolidin-2-one-pyrimidine derivatives - Google Patents
Oxazolidin-2-one-pyrimidine derivatives Download PDFInfo
- Publication number
- WO2014072956A1 WO2014072956A1 PCT/IB2013/060052 IB2013060052W WO2014072956A1 WO 2014072956 A1 WO2014072956 A1 WO 2014072956A1 IB 2013060052 W IB2013060052 W IB 2013060052W WO 2014072956 A1 WO2014072956 A1 WO 2014072956A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkoxy
- compound
- amino
- oxazolidin
- trifluoromethyl
- Prior art date
Links
- 0 *C(C(*)O1)NC1=O Chemical compound *C(C(*)O1)NC1=O 0.000 description 2
- PPIBJOQGAJBQDF-IONNQARKSA-N C[C@@H]([C@H](c1ccccc1)O1)NC1=O Chemical compound C[C@@H]([C@H](c1ccccc1)O1)NC1=O PPIBJOQGAJBQDF-IONNQARKSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the present invention relates to oxazolidin-2-one substituted pyrimidine compounds that act as PI3K (phosphatidylinositol-3-kinase) inhibitors, as well as pharmaceutical compositions thereof, methods for their manufacture and uses for the treatment of conditions, diseases and disorders dependent on PI3 kinases.
- PI3K phosphatidylinositol-3-kinase
- the phosphatidylinositol-3-kinases superfamily comprises 4 different PI3K related lipid or protein kinases.
- Class I, II and III are lipid kinases that differ from their substrate specificities whereas class IV PI3K also called PI3-kinase-related protein kinase (PIKK) are protein kinases.
- PIKK PI3-kinase-related protein kinase
- Class I PI3Ks comprise a family of lipid kinases that catalyze the transfer of phosphate to the D-3' position of inositol lipids to produce phosphoinositol-3- phosphate (PIP), phosphoinositol-3,4-diphosphate (PIP 2 ) and phosphoinositol-3,4,5- triphosphate (PIP 3 ) that, in turn, act as second messengers in signaling cascades by docking proteins containing pleckstrin-homology, FYVE, Phox and other phospholipid- binding domains into a variety of signaling complexes often at the plasma membrane
- the tumor suppressor gene PTEN which dephosphorylates phosphoinositides at the 3' position of the inositol ring and in so doing antagonizes PI3K activity, is functionally mutated or deleted in a variety of tumors (Keniry & Parsons, Oncogene 27:5477-5485 (2008)).
- Some studies indicate that the failure to express PTEN can mediate a shift of signaling dependence from the PI3Ka to the ⁇ -isoform (Wee et al, Pore Natl Acad Sci USA 105:13057-13062 (2008)). Therefore, inhibition of both class I PI3K a and ⁇ -isoforms can be particularly advantageous in cancers that are deficient in PTEN phosphatase.
- mTOR is a protein kinase and a member of the class IV PI3K.
- mTORCI protein kinase
- mTORC2 Activation of mTORCI is dependent on active PI3K and Akt kinases. Regulation of mTORC2 activation is more complex.
- mTORC2 is responsible for enhancement of Akt kinase activity via phosphorylation of serine residue 473
- Cutaneous squamous cell carcinoma represents the second most frequent human skin cancer, commonly preceded by actinic keratosis (AK).
- AK actinic keratosis
- the pathogenesis of AK and cutaneous SCC has been associated with chronic UV exposure as one major risk factor (Salasche, J Am Acad Dermatol 42: 4-7 (2000)).
- An enhanced activity of PI3K/Akt/mTOR pathway has been suggested in a previous study (Chen et al., Br J Dermatol; 160 (2):442- 445 (2009)).
- the invention relates to oxazolidin-2-one substituted pyrimidine compounds of the formula (I) and/or pharmaceutically acceptable salts and/or solvates thereof,
- R 1 is methyl, ethyl or hydroxymethyl
- R 2 is phenyl, which is unsubstituted or substituted in meta and/or para positions by 1 or 2 substituents independently selected from D, F or methoxy for the meta position and from D, F, methoxy, CrC 5 -alkoxy, hydroxy-C 2 -C 4 -alkoxy or Ci-C2-alkoxy-C 2 -C 4 -alkoxy for the para position,
- pyridyl which is unsubstituted or substituted in meta and/or para positions by 1 or 2 substituents independently selected from D, F or methoxy for the meta position and from D, F, methoxy, C-rC 5 -alkoxy, hydroxy-C 2 -C 4 -alkoxy or C 1 -C 2 -alkoxy-C 2 -C 4 -alkoxy for the para position,
- a 5 membered monocyclic heteroaryl containing 2 to 3 heteroatoms selected from N, O or S, which is unsubstituted or substituted by 1 to 2 substituents independently selected from D or F;
- R 3 is H or methyl.
- the term "compounds of the present invention” refers to compounds of formula (I) and subformulae thereof, salts of the compound, hydrates or solvates of the compounds and/or salts, as well as all stereoisomers (including diastereoisomers and enantiomers), tautomers and isotopically labeled compounds (including deuterium substitutions).
- Compounds of the present invention further comprise polymorphs of compounds of formula (I) (or subformulae thereof) and salts thereof. Where compounds of formula (I) are mentioned, this is meant to include also the tautomers and N-oxides of the compounds of formula (I).
- the compounds of formula (I) are considered suitable to be used in the treatment of diseases dependent on PI3 kinases.
- the compounds of formula (I) are considered suitable, for example, to be used in the treatment of diseases dependent on class I PI3 kinase or dependent on class I PI3 kinase and mTOR.
- alkoxy refers to a fully saturated branched, including single or multiple branching, or unbranched hydrocarbon moiety attached to the rest of the molecule via an -O- linking group. Unless otherwise provided, alkoxy refers to moieties having 1 to 16 carbon atoms, 1 to 10 carbon atoms, 1 to 7 carbon atoms, or 1 to 4 carbon atoms.
- alkoxy examples include, but are not limited to, methoxy, ethoxy, n- propoxy, / ' so-propoxy, n-butyoxy, sec-butoxy, so-butoxy, iert-butoxy, n-pentoxy, isopentoxy, neopentyl, n-hexyloxy.
- pyridyl refers to 2-pyridyl, 3-pyridyl or 4-pyridyl. Substituents in meta or para position are attached to a carbon atom of the pyridyl. A representative example is 3-pyridyl.
- 5 membered monocyclic heteroaryl containing 2 to 3 heteroatoms selected from N, O or S are attached to a carbon atom of the heteroaryl.
- Examples of 5 membered monocyclic heteroaryl, containing 2 to 3 heteroatoms selected from N, O or S include, but are not limited to thiazolyl, oxazolyl, 1 ,2,4-oxadiazolyl, 1 ,3,4- oxadiazolyl and 1 ,3,4-thiadiazolyl.
- a representative example is thiazolyl.
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, selected from a compound of the formula (la)
- R 1 and R 2 are as defined for a compound of formula (I) above.
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, selected from a compound of the formula (lb)
- R 1 and R 2 are as defined for a compound of formula (I) above.
- the invention provides a compound of the formulae (I), (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 2 is phenyl, which is unsubstituted or substituted in meta and/or para positions by 1 or 2 substituents independently selected from D, F or methoxy for the meta position and from D, F, methoxy, C-pCs-alkoxy, hydroxy-C2-C4-alkoxy or Ci-C2-alkoxy-C2-C4-alkoxy for the para position.
- the invention provides a compound of the formulae (I), (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 2 is pyridyl, which is unsubstituted or substituted in meta and/or para positions by 1 or 2 substituents independently selected from D, F or methoxy for the meta position and from D, F, methoxy, C-pCs-alkoxy, hydroxy-C2-C4-alkoxy or Ci-C2-alkoxy-C2-C4-alkoxy for the para position.
- the invention provides a compound of the formulae (I), (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 2 is a 5 membered monocyclic heteroaryl, containing 2 to 3 heteroatoms selected from N, O or S, which is unsubstituted or substituted by 1 to 2 substituents independently selected from D or F.
- the invention provides a compound of the formulae (I), (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 2 is phenyl, 3-methoxy-phenyl, 4-methoxy-phenyl, 4-(3-hydroxypropoxy)-phenyl, 4- (2-hydroxyethoxy)-phenyl or 4-(2-methoxyethoxy)-phenyl.
- the invention provides a compound of the formulae (I), (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 2 is 3-pyridyl, which is unsubstituted or substituted in meta and/or para positions by 1 or 2 substituents independently selected from D, F or methoxy for the meta position and from D, F, methoxy, CrC 5 -alkoxy, hydroxy-C 2 -C 4 -alkoxy or Ci-C2-alkoxy-C 2 -C 4 - alkoxy for the para position.
- the invention provides a compound of the formulae (I), (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 2 is 3-pyridyl.
- the invention provides a compound of the formulae (I), (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R 2 is thiazolyl, which is unsubstituted or substituted by 1 to 2 substituents
- the invention provides a compound of the formulae (I), (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 2 is 2-thiazolyl which is unsubstituted or substituted by 1 to 2 substituents independently selected from D or F.
- the invention provides a compound of the formulae (I), (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 2 is 2-thiazolyl
- the invention provides a compound of the formulae (I), (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is methyl
- the invention provides a compound of the formulae (I), (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is ethyl
- the invention provides a compound of the formulae (I), (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is hydroxymethyl
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is methyl
- R 2 is phenyl, which is unsubstituted or substituted in meta and/or para positions by 1 or 2 substituents independently selected from D, F or methoxy for the meta position and from D, F, methoxy, CrC 5 -alkoxy, hydroxy-C 2 -C 4 -alkoxy or Ci-C2-alkoxy-C 2 -C 4 -alkoxy for the para position.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R 1 is methyl;
- R 2 is pyridyl, which is unsubstituted or substituted in meta and/or para positions by 1 or 2 substituents independently selected from D, F or methoxy for the meta position and from D, F, methoxy, CrC 5 -alkoxy, hydroxy-C 2 -C 4 -alkoxy or Ci-C 2 -alkoxy-C2-C 4 -alkoxy for the para position.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is methyl
- R 2 is a 5 membered monocyclic heteroaryl, containing 2 to 3 heteroatoms selected from N, O or S, which is unsubstituted or substituted by 1 to 2 substituents independently selected from D or F;.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is methyl
- R 2 is phenyl, 3-methoxy-phenyl, 4-methoxy-phenyl, 4-(3-hydroxypropoxy)-phenyl, 4- (2-hydroxyethoxy)-phenyl or 4-(2-methoxyethoxy)-phenyl.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is methyl
- R 2 is 3-pyridyl, which is unsubstituted or substituted in meta and/or para positions by 1 or 2 substituents independently selected from D, F or methoxy for the meta position and from D, F, methoxy, C-pCs-alkoxy, hydroxy-C2-C4-alkoxy or Ci-C2-alkoxy-C2-C4- alkoxy for the para position.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is methyl
- R 2 is 3-pyridyl.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is methyl;
- R 2 is thiazolyl, which is unsubstituted or substituted by 1 to 2 substituents independently selected from D or F.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is methyl
- R 2 is 2-thiazolyl which is unsubstituted or substituted by 1 to 2 substituents
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is methyl
- R 2 is 2-thiazolyl
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is ethyl
- R 2 is phenyl, which is unsubstituted or substituted in meta and/or para positions by 1 or 2 substituents independently selected from D, F or methoxy for the meta position and from D, F, methoxy, d-C 5 -alkoxy, hydroxy-C 2 -C 4 -alkoxy or C 1 -C 2 -alkoxy-C 2 -C 4 -alkoxy for the para position.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is ethyl
- R 2 is pyridyl, which is unsubstituted or substituted in meta and/or para positions by 1 or 2 substituents independently selected from D, F or methoxy for the meta position and from D, F, methoxy, CrC 5 -alkoxy, hydroxy-C 2 -C 4 -alkoxy or CrC 2 -alkoxy-C 2 -C 4 -alkoxy for the para position.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is ethyl
- R 2 is a 5 membered monocyclic heteroaryl, containing 2 to 3 heteroatoms selected from N, O or S, which is unsubstituted or substituted by 1 to 2 substituents independently selected from D or F.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is ethyl
- R 2 is phenyl, 3-methoxy-phenyl, 4-methoxy-phenyl, 4-(3-hydroxypropoxy)-phenyl, 4-
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is ethyl
- R 2 is 3-pyridyl, which is unsubstituted or substituted in meta and/or para positions by 1 or 2 substituents independently selected from D, F or methoxy for the meta position and from D, F, methoxy, C-i-C 5 -alkoxy, hydroxy-C 2 -C 4 -alkoxy or C -C 2 -alkoxy-C 2 -C 4 - alkoxy for the para position.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is ethyl
- R 2 is 3-pyridyl.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is ethyl
- R 2 is thiazolyl, which is unsubstituted or substituted by 1 to 2 substituents independently selected from D or F.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is ethyl
- R 2 is 2-thiazolyl which is unsubstituted or substituted by 1 to 2 substituents
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is ethyl
- R 2 is 2-thiazolyl
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is hydroxymethyl
- R 2 is phenyl, which is unsubstituted or substituted in meta and/or para positions by 1 or 2 substituents independently selected from D, F or methoxy for the meta position and from D, F, methoxy, C-pCs-alkoxy, hydroxy-C2-C4-alkoxy or Ci-C2-alkoxy-C2-C4-alkoxy for the para position.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is hydroxymethyl
- R 2 is pyridyl, which is unsubstituted or substituted in meta and/or para positions by 1 or 2 substituents independently selected from D, F or methoxy for the meta position and from D, F, methoxy, C-i-C 5 -alkoxy, hydroxy-C 2 -C 4 -alkoxy or C -C 2 -alkoxy-C 2 -C 4 -alkoxy for the para position.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is hydroxymethyl
- R 2 is a 5 membered monocyclic heteroaryl, containing 2 to 3 heteroatoms selected from N, O or S, which is unsubstituted or substituted by 1 to 2 substituents independently selected from D or F.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is hydroxymethyl
- R 2 is phenyl, 3-methoxy-phenyl, 4-methoxy-phenyl, 4-(3-hydroxypropoxy)-phenyl, 4- (2-hydroxyethoxy)-phenyl or 4-(2-methoxyethoxy)-phenyl.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is hydroxymethyl
- R 2 is 3-pyridyl, which is unsubstituted or substituted in meta and/or para positions by
- substituents independently selected from D, F or methoxy for the meta position and from D, F, methoxy, C-pCs-alkoxy, hydroxy-C2-C4-alkoxy or Ci-C2-alkoxy-C2-C4- alkoxy for the para position.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is hydroxymethyl
- R 2 is 3-pyridyl.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is hydroxymethyl
- R 2 is thiazolyl, which is unsubstituted or substituted by 1 to 2 substituents independently selected from D or F.
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is hydroxymethyl
- R 2 is 2-thiazolyl which is unsubstituted or substituted by 1 to 2 substituents
- the invention provides a compound of the formulae (la) or (lb) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein
- R 1 is hydroxymethyl
- R 2 is 2-thiazolyl
- the invention provides a compound and/or a pharmaceutically acceptable salt and/or a solvate therof, selected from
- the invention provides a compound and/or a pharmaceutically acceptable salt and/or a solvate therof, selected from
- an optical isomer or "a stereoisomer” refers to any of the various stereo isomeric configurations which may exist for a given compound of the present invention and includes geometric isomers. It is understood that a substituent may be attached at a chiral center of a carbon atom.
- the term “chiral” refers to molecules which have the property of non-superimposability on their mirror image partner, while the term “achiral” refers to molecules which are superimposable on their mirror image partner. Therefore, the invention includes enantiomers, diastereomers or racemates of the compound. "Enantiomers” are a pair of stereoisomers that are non- superimposable mirror images of each other.
- a 1 :1 mixture of a pair of enantiomers is a "racemic" mixture.
- the term is used to designate a racemic mixture where appropriate.
- "Diastereoisomers” are stereoisomers that have at least two asymmetric atoms, but which are not mirror-images of each other.
- the absolute stereochemistry is specified according to the Cahn- Ingold- Prelog R-S system. When a compound is a pure enantiomer the stereochemistry at each chiral carbon may be specified by either R or S.
- Resolved compounds whose absolute configuration is unknown can be designated (+) or (-) depending on the direction (dextro- or levorotatory) which they rotate plane polarized light at the wavelength of the sodium D line.
- Certain compounds described herein contain one or more asymmetric centers or axes and may thus give rise to enantiomers, diastereomers, and other stereoisomeric forms that may be defined, in terms of absolute stereochemistry, as (R)- or (S)-.
- the compounds can be present in the form of one of the possible isomers or as mixtures thereof, for example as pure optical isomers, or as isomer mixtures, such as racemates and diastereoisomer mixtures, depending on the number of asymmetric carbon atoms.
- the present invention is meant to include all such possible isomers, including racemic mixtures, diasteriomeric mixtures and optically pure forms.
- Optically active (f?)- and (S)- isomers may be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques. If the compound contains a double bond, the substituent may be E or Z configuration. If the compound contains a disubstituted cycloalkyl, the cycloalkyl substituent may have a cis- or trans-configuration. All tautomeric forms are also intended to be included.
- salt refers to an acid addition or base addition salt of a compound of the invention.
- Salts include in particular “pharmaceutical acceptable salts”.
- pharmaceutically acceptable salts refers to salts that retain the biological effectiveness and properties of the compounds of this invention and, which typically are not biologically or otherwise undesirable.
- the compounds of the present invention are capable of forming acid and/or base salts by virtue of the presence of amino and/or carboxyl groups or groups similar thereto.
- Pharmaceutically acceptable acid addition salts can be formed with inorganic acids and organic acids, e.g., acetate, aspartate, benzoate, besylate, bromide/hydrobromide, bicarbonate/carbonate, bi sulfate/sulfate, camphorsulfonate, chloride/hydrochloride, chlortheophyllonate, citrate, ethandisulfonate, fumarate, gluceptate, gluconate, glucuronate, hippurate, hydroiodide/iodide, isethionate, lactate, lactobionate, laurylsulfate, malate, maleate, malonate, mandelate, mesylate, methylsulphate, naphthoate, napsylate, nicotinate, nitrate, octadecanoate, oleate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/di
- Inorganic acids from which salts can be derived include, for example, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like.
- Organic acids from which salts can be derived include, for example, acetic acid, propionic acid, glycolic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, sulfosalicylic acid, and the like.
- Pharmaceutically acceptable base addition salts can be formed with inorganic and organic bases.
- Inorganic bases from which salts can be derived include, for example, ammonium salts and metals from columns I to XII of the periodic table.
- the salts are derived from sodium, potassium, ammonium, calcium, magnesium, iron, silver, zinc, and copper; particularly suitable salts include ammonium, potassium, sodium, calcium and magnesium salts.
- Organic bases from which salts can be derived include, for example, primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and the like.
- Certain organic amines include isopropylamine, benzathine, cholinate, diethanolamine, diethylamine, lysine, meglumine, piperazine and tromethamine.
- the pharmaceutically acceptable salts of the present invention can be synthesized from a basic or acidic moiety, by conventional chemical methods.
- such salts can be prepared by reacting free acid forms of these compounds with a stoichiometric amount of the appropriate base (such as Na, Ca, Mg, or K hydroxide, carbonate, bicarbonate or the like), or by reacting free base forms of these compounds with a stoichiometric amount of the appropriate acid.
- a stoichiometric amount of the appropriate base such as Na, Ca, Mg, or K hydroxide, carbonate, bicarbonate or the like
- Such reactions are typically carried out in water or in an organic solvent, or in a mixture of the two.
- use of non-aqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile is desirable, where practicable. Lists of additional suitable salts can be found, e.g., in "Remington's
- any formula given herein is also intended to represent unlabeled forms as well as isotopically labeled forms of the compounds.
- Isotopically labeled compounds have structures depicted by the formulas given herein except that one or more atoms are replaced by an atom having a selected atomic mass or mass number.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, and chlorine, such as 2 H, 3 H, 11 C, 3 C, 14 C, 15 N, 18 F 31 P, 32 P, 35 S, 36 CI, 125 l respectively.
- the invention includes various isotopically labeled compounds as defined herein, for example those into which radioactive isotopes, such as 3 H and 14 C, or those into which nonradioactive isotopes, such as 2 H and 13 C are present.
- isotopically labelled compounds are useful in metabolic studies (with 14 C), reaction kinetic studies (with, for example 2 H or 3 H), detection or imaging techniques, such as positron emission tomography (PET) or single-photon emission computed tomography (SPECT) including drug or substrate tissue distribution assays, or in radioactive treatment of patients.
- PET positron emission tomography
- SPECT single-photon emission computed tomography
- an 8 F or labeled compound may be particularly desirable for PET or SPECT studies.
- Isotopically-labeled compounds of formula (I) can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically-labeled reagents in place of the non-labeled reagent previously employed.
- isotopic enrichment factor means the ratio between the isotopic abundance and the natural abundance of a specified isotope.
- a substituent in a compound of this invention is denoted deuterium, such compound has an isotopic enrichment factor for each designated deuterium atom of at least 3500 (52.5% deuterium incorporation at each designated deuterium atom), at least 4000 (60% deuterium incorporation), at least 4500 (67.5% deuterium incorporation), at least 5000 (75% deuterium incorporation), at least 5500 (82.5% deuterium incorporation), at least 6000 (90% deuterium incorporation), at least 6333.3 (95% deuterium incorporation), at least 6466.7 (97% deuterium incorporation), at least 6600 (99% deuterium incorporation), or at least 6633.3 (99.5% deuterium incorporation).
- compositions in accordance with the invention include those wherein the solvent of crystallization may be isotopically substituted, e.g. D 2 0, de- acetone, de-DMSO.
- Compounds of the invention i.e. compounds of formula (I) that contain groups capable of acting as donors and/or acceptors for hydrogen bonds may be capable of forming co- crystals with suitable co-crystal formers.
- These co-crystals may be prepared from compounds of formula (I) by known co-crystal forming procedures. Such procedures include grinding, heating, co-subliming, co-melting, or contacting in solution compounds of formula (I) with the co-crystal former under crystallization conditions and isolating co- crystals thereby formed.
- Suitable co-crystal formers include those described in WO 2004/078163. Hence the invention further provides co-crystals comprising a compound of formula (I).
- the term "pharmaceutically acceptable carrier” includes any and all solvents, dispersion media, coatings, surfactants, antioxidants, preservatives (e.g., antibacterial agents, antifungal agents), isotonic agents, absorption delaying agents, salts, preservatives, drug stabilizers, binders, excipients, disintegration agents, lubricants, sweetening agents, flavoring agents, dyes, and the like and combinations thereof, as would be known to those skilled in the art (see, for example, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289- 1329). Except insofar as any conventional carrier is incompatible with the active ingredient, its use in the therapeutic or pharmaceutical compositions is contemplated.
- a therapeutically effective amount of a compound of the present invention refers to an amount of the compound of the present invention that will elicit the biological or medical response of a subject, for example, reduction or inhibition of an enzyme or a protein activity, or ameliorate symptoms, alleviate conditions, slow or delay disease progression, or prevent a disease, etc.
- a therapeutically effective amount refers to the amount of the compound of the present invention that, when administered to a subject, is effective to (1 ) at least partially alleviate, inhibit, prevent and/or ameliorate a condition, or a disorder or a disease (i) mediated by class I PI3 kinase, or (ii) associated with class I PI3 kinase activity, or (iii) characterized by activity (normal or abnormal) of class I PI3 kinase; or (2) reduce or inhibit the activity of class I PI3 kinase.
- a therapeutically effective amount refers to the amount of the compound of the present invention that, when administered to a cell, or a tissue, or a non-cellular biological material, or a medium, is effective to at least partially reducing or inhibiting the activity of class I PI3 kinase.
- a therapeutically effective amount refers to the amount of the compound of the present invention that, when administered to a subject, is effective to (1 ) at least partially alleviate, inhibit, prevent and/or ameliorate a condition, or a disorder or a disease (i) mediated by class I PI3 kinase and mTOR, or (ii) associated with class I PI3 kinase and mTOR activity, or (iii) characterized by activity (normal or abnormal) of class I PI3 kinase and mTOR; or (2) reduce or inhibit the activity of class I PI3 kinase and mTOR.
- a therapeutically effective amount refers to the amount of the compound of the present invention that, when administered to a cell, or a tissue, or a non-cellular biological material, or a medium, is effective to at least partially reducing or inhibiting the activity of class I PI3 kinase and mTOR.
- the term "subject" refers to an animal. Typically the animal is a mammal. A subject also refers to for example, primates (e.g., humans, male or female), cows, sheep, goats, horses, dogs, cats, rabbits, rats, mice, fish, birds and the like. In certain embodiments, the subject is a primate. In yet other embodiments, the subject is a human.
- primates e.g., humans, male or female
- the subject is a primate.
- the subject is a human.
- the term “inhibit”, “inhibition” or “inhibiting” refers to the reduction or suppression of a given condition, symptom, or disorder, or disease, or a significant decrease in the baseline activity of a biological activity or process.
- the term “treat”, “treating” or “treatment” of any disease or disorder refers in one embodiment, to ameliorating the disease or disorder (i.e., slowing or arresting or reducing the development of the disease or at least one of the clinical symptoms thereof).
- “treat”, “treating” or “treatment” refers to alleviating or ameliorating at least one physical parameter including those which may not be discernible by the patient.
- “treat”, “treating” or “treatment” refers in one embodiment, to ameliorating the disease or disorder (i.e., slowing or arresting or reducing the development of the disease or at least one of the clinical symptoms thereof).
- “treat”, “treating” or “treatment” refers to alleviating or ameliorating at least one physical parameter including those which may not be discernible by the patient.
- treatment refers to modulating the disease or disorder, either physically, (e.g., stabilization of a discernible symptom), physiologically, (e.g., stabilization of a physical parameter), or both.
- “treat”, “treating” or “treatment” refers to preventing or delaying the onset or development or progression of the disease or disorder.
- a subject is "in need of” a treatment if such subject would benefit biologically, medically or in quality of life from such treatment.
- any asymmetric atom e.g., carbon or the like
- any asymmetric atom can be present in racemic or enantiomerically enriched, for example the (R)-, (S)- or ⁇ R,S)- configuration.
- each asymmetric atom has at least 50 % enantiomeric excess, at least 60 % enantiomeric excess, at least 70 %
- a compound of the present invention can be in the form of one of the possible isomers, rotamers, atropisomers, tautomers or mixtures thereof, for example, as substantially pure geometric (cis or trans) isomers, diastereomers, optical isomers (antipodes), racemates or mixtures thereof.
- Any resulting mixtures of isomers can be separated on the basis of the physicochemical differences of the constituents, into the pure or substantially pure geometric or optical isomers, diastereomers, racemates, for example, by chromatography and/or fractional crystallization.
- any resulting racemates of final products or intermediates can be resolved into the optical antipodes by known methods, e.g., by separation of the diastereomeric salts thereof, obtained with an optically active acid or base, and liberating the optically active acidic or basic compound.
- a basic moiety may thus be employed to resolve the compounds of the present invention into their optical antipodes, e.g., by fractional crystallization of a salt formed with an optically active acid, e.g., tartaric acid, dibenzoyl tartaric acid, diacetyl tartaric acid, di-0, 0'-p-toluoyl tartaric acid, mandelic acid, malic acid or camphor-10-sulfonic acid.
- Racemic products can also be resolved by chiral chromatography, e.g., high pressure liquid chromatography (HPLC) using a chiral adsorbent.
- HPLC high pressure liquid chromatography
- the compounds of the present invention can also be obtained in the form of their hydrates, or include other solvents used for their crystallization.
- the compounds of the present invention may inherently or by design form solvates with pharmaceutically acceptable solvents (including water); therefore, it is intended that the invention embrace both solvated and unsolvated forms.
- solvate refers to a molecular complex of a compound of the present invention
- solvent molecules are those commonly used in the pharmaceutical art, which are known to be innocuous to the recipient, e.g., water, ethanol, and the like.
- solvent molecules are those commonly used in the pharmaceutical art, which are known to be innocuous to the recipient, e.g., water, ethanol, and the like.
- hydrate refers to the complex where the solvent molecule is water.
- the compounds of the present invention including salts, hydrates and solvates thereof, may inherently or by design form polymorphs.
- Compounds of the present invention may be synthesized by synthetic routes that include processes analogous to those well-known in the chemical arts, particularly in light of the description contained herein.
- the starting materials are generally available from commercial sources or are readily prepared using methods well known to those skilled in the art (e.g., prepared by methods generally described in Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, volumes 1-19, Wiley, New York (1967-1999 ed.), or Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, including supplements (also available via the Beilstein online database)).
- reaction scheme depicted below provides potential routes for synthesizing the compounds of the present invention as well as key intermediates.
- Examples section below For a more detailed description of the individual reaction steps, see the Examples section below.
- Those skilled in the art will appreciate that other synthetic routes may be used to synthesize the inventive compounds.
- specific starting materials and reagents are depicted in the schemes and discussed below, other starting materials and reagents can be easily substituted to provide a variety of derivatives and/or reaction conditions.
- many of the compounds prepared by the methods described below can be further modified in light of this disclosure using conventional chemistry well known to those skilled in the art.
- Suitable hydroxyl protecting groups include trialkylsilyl ethers where one or two of the alkyl groups can be replaced by phenyl.
- the need for such protection is readily determined by one skilled in the art. For a general description of protecting groups and their use, see T. W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
- the invention relates to a process for manufacturing a compound of formula (I) comprising steps a and b. In one embodiment, the invention relates to a process for manufacturing a compound of formula (I) (Method A) comprising step a followed by step b.
- the compound of formula (I) is obtained via the step a) of coupling the compound of formula (A), wherein R 3 is as defined for a compound of formula (I) above
- Step a) is carried out in the presence of a base such as NaH.
- the reaction is carried out in the presence of an organic solvent such as DMF at temperatures from 0 to 80°C for 20 to 30 min.
- the reaction can be carried out under customary Buchwald- Hartwig conditions using a ligand such as Xantphos, X-Phos or 2-di-t-butylphosphino-2'- (N,N-dimethylamino)biphenyl with a palladium catalyst such as Pd 2 (dba) 3 or
- Pd 2 (dba) 3 -CHCI 3 or Pd(OAc) 2 preferably Pd 2 (dba) 3 with Xantphos, in the presence of a base such as preferably Cs 2 C0 3 or ferf-BuONa, in an organic solvent such as an ether, preferably dioxane or THF.
- a base such as preferably Cs 2 C0 3 or ferf-BuONa
- organic solvent such as an ether, preferably dioxane or THF.
- the reaction is preferably stirred at a temperature of approximately 80-120°C.
- the reaction is preferably carried out under an inert gas such as nitrogen or argon. Typical reaction conditions known in the field for Buchwald-Hartwig reactions may be applied to the present process.
- Step b) is carried out in the presence of a catalyst, such as a Pd(0) catalyst, e.g.
- reaction aids such as a base, e.g. an aqueous base, such as aqueous Na 2 C0 3 , optionally in the presence of one or more diluents, particularly polar solvents, e.g. DME.
- a base e.g. an aqueous base, such as aqueous Na 2 C0 3
- diluents particularly polar solvents, e.g. DME.
- the reaction is stirred at a temperature of approximately 80-120°C.
- the reaction may be carried out under an inert gas such as nitrogen or argon. Typical reaction conditions known in the field for Suzuki reactions may be applied to the present process.
- the invention further includes any variant of the present processes, in which an intermediate product obtainable at any stage thereof is used as starting material and the remaining steps are carried out, or in which the starting materials are formed in situ under the reaction conditions, or in which the reaction components are used in the form of their salts or optically pure material.
- Compounds of the invention and intermediates can also be converted into each other according to methods generally known to those skilled in the art.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the present invention, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- the pharmaceutical composition can be formulated for particular routes of administration such as topical administration; enteral administration such as for example oral or rectal administration; and parenteral administration such as for example intravenous, intra- muscular or subcutaneous administration.
- routes of administration such as topical administration; enteral administration such as for example oral or rectal administration; and parenteral administration such as for example intravenous, intra- muscular or subcutaneous administration.
- the pharmaceutical compositions of the present invention can be made up in a solid form (including without limitation capsules, tablets, pills, granules, powders or suppositories), or in a liquid form (including without limitation solutions, suspensions or emulsions).
- the pharmaceutical compositions can be prepared according to methods known in the art.
- compositions can be subjected to conventional pharmaceutical operations such as sterilization and/or can contain conventional inert diluents, lubricating agents, or buffering agents, as well as adjuvants, such as preservatives, stabilizers, wetting agents, emulsifers and buffers, etc.
- suitable compositions for topical application include aqueous solutions, suspensions, ointments, creams, gels, powders, oils or sprayable
- formulations e.g., for delivery by aerosol or the like.
- topical delivery systems will in particular be appropriate for dermal application, e.g., for use in creams, lotions, sprays and the like. They are thus particularly suited for use in topical, including cosmetic, formulations well-known in the art.
- Such may contain solubilizers, stabilizers, tonicity enhancing agents, buffers and preservatives etc. ,
- a topical application may also pertain to an inhalation or to an intranasal application e.g., to the respiratory system. They may be conveniently delivered in the form of a dry powder (either alone, as a mixture, for example a dry blend with lactose, or a mixed component particle, for example with phospholipids) from a dry powder inhaler or an aerosol spray presentation from a pressurised container, pump, spray, atomizer or nebuliser, with or without the use of a suitable propellant.
- a dry powder either alone, as a mixture, for example a dry blend with lactose, or a mixed component particle, for example with phospholipids
- application preferably refers to topical application such as epicutaneous application in a suitable composition comprising a compound of the present invention, or a pharmaceutically acceptable salt thereof, and a
- compositions for epicutaneous application may comprise all pharmaceutical forms normally utilized for this route of administration and are well-known in the art including solutions, gels, lotions, suspensions, creams, powders, oils, ointments, foams, mousses, emulsions, microemulsions, milks, serums, aerosols, sprays, dispersions, microcapsules, vesicles and microparticles thereof. These compositions are formulated according to conventional techniques.
- the term "dermatologically acceptable carrier” is a carrier which is suitable for topical application to the keratinous tissue, has good aesthetic properties, is compatible with the active agents of the present invention and any other components, and will not cause any untoward safety or toxicity concerns.
- the dermatologically acceptable carrier can be in a wide variety of forms.
- emulsion carriers including, but not limited to, oil-in-water, water-in-oil, water-in-oil-in- water, and oil-in-water-in-silicon emulsions, are useful herein.
- a given component will distribute primarily into either the water or oil/silicon phase, depending on the water solubility/dispersibility of the component in the composition.
- the composition suitable for epicutaneous application can contain various known additives such as excipients, binders, lubricants, and disintegrants.
- oily materials such as various fats, oils, waxes, hydrocarbons, fatty acids, higher alcohols, ester oils, metallic soaps, animal or vegetable extracts, hydrophilic or lipophilic gelling agents, hydrophilic or lipophilic active agents, other components such as vitamins, amino acids, surfactants, colorants, dyes, pigments, fragrances, odor absorbers, antiseptics, preservatives, bactericides, humectants, thickeners, solvents, fillers, antioxidants, sequestering agents, sunscreens, and the like and combinations thereof, as would be known to those skilled in the art, as long as these are compatible with the active ingredient.
- oily materials such as various fats, oils, waxes, hydrocarbons, fatty acids, higher alcohols, ester oils, metallic soaps, animal or vegetable extracts, hydrophilic or lipophilic gelling agents, hydrophilic or lipophilic active agents, other components such as vitamins, amino acids, surfactants, colorants, dyes, pigments, fragrances,
- suitable oils includes mineral oils, plant oils such as peanut oil, sesame oil, soybean oil, safflower oil, sunflower oil, animal oils such as lanolin or perhydrosqualene, synthetic oils such as purcellin oil, silicone oils such as cyclomethicome among others.
- mineral oils such as peanut oil, sesame oil, soybean oil, safflower oil, sunflower oil, animal oils such as lanolin or perhydrosqualene, synthetic oils such as purcellin oil, silicone oils such as cyclomethicome among others.
- Fatty alcohols, fatty acids such as stearic acid and waxes such as paraffin wax, carnauba wax or beeswax may also be used as fats.
- composition may also contain emulsifying agents, solvents, hydrophilic gelling agents, lipophilic gelling agents, fatty acid metal salts, hydrophilic acting agents or lipophilic active agents.
- the compounds of formula (I) in free form or in salt form exhibit valuable pharmacological properties, e.g. PI3 kinase modulating properties such as class I PI3 kinase (class I
- PI3K's PI3K's modulating properties or PI3K's modulating properties in conjunction with mTOR modulating properties, e.g. as indicated in in vitro and in vivo tests as provided in the Experimental section, and are therefore indicated for therapy or for use as research chemicals, e.g. as tool compounds.
- the compounds of formula (I) in free form or in salt form are useful in the treatment of class I PI3 kinase dependent diseases, especially diseases depending on class I PI3K a and ⁇ -isoforms, as well as diseases depending on class I PI3 kinases, especially diseases depending on class I PI3K a and ⁇ -isoforms, in conjunction with mTOR.
- Compounds that inhibit the activity of class I PI3K a and ⁇ -isoforms are considered to be of benefit because such compounds are considered to have the ability to avoid adaption mechanisms due to pathway rewiring through the other isoforms, compared to compounds with unique specificity, e.g. specificity for one member of the class I PI3K family.
- equipotent it is meant that the compounds inhibit several isoforms to a comparable extent, e.g. as measured in an enzyme or cellular assay described herein.
- the compounds of formula (I) show sufficient photostability in order to ensure and maximize activity of the compounds of formula (I) when administered epicutaneously to minimize potential irritation and side effects due to degradation products generated. Compounds with superior photostability will simplify technical development and supply due to minimized risks of photodegradation.
- the compounds of formula (I) show good potency in cellular assays using human cell lines derived from cutaneous squamous cell carcinoma. Preferred compounds should demonstrate the ability to penetrate well into skin.
- Compounds of the invention may be useful in the treatment of an indication selected from but not limited to non-melanoma skin cancers such as basal cell carcinoma and squamous cell carcinoma; their pre-malignant stages such as actinic keratosis, solar keratosis and chronically sun damaged skin; and other hyperproliferative skin disorders caused by dysregulation of skin fibroblasts such as skin fibrosis, scleroderma, hypertrophic scars or keloids.
- the compounds of the invention may be particularly useful in the treatment of an indication selected from but not limited to non-melanoma skin cancers.
- the compound of the present invention may be administered separately, by the same or different route of administration, or together in the same pharmaceutical composition as the other agent(s).
- the invention provides a product comprising a compound of formula (I) and at least one other therapeutic agent as a combined preparation for simultaneous, separate or sequential use in therapy.
- the therapy is the treatment of a disease or condition dependent on PI3 kinases.
- Products provided as a combined preparation include a composition comprising the compound of formula (I) and the other therapeutic agent(s) together in the same pharmaceutical composition, or the compound of formula (I) and the other therapeutic agent(s) in separate form, e.g. in the form of a kit.
- the invention provides a pharmaceutical composition comprising a compound of formula (I) and another therapeutic agent(s).
- a pharmaceutical composition comprising a compound of formula (I) and another therapeutic agent(s).
- composition may comprise a pharmaceutically acceptable carrier, as described above.
- the invention provides a kit comprising two or more separate pharmaceutical compositions, at least one of which contains a compound of formula (I).
- the kit comprises means for separately retaining said compositions, such as a container, divided bottle, or divided foil packet.
- An example of such a kit is a blister pack, as typically used for the packaging of tablets, capsules and the like.
- the kit of the invention may be used for administering different dosage forms, for example, oral and parenteral, for administering the separate compositions at different dosage intervals, or for titrating the separate compositions against one another.
- the kit of the invention typically comprises directions for
- the invention provides the use of a compound of formula (I) for treating a disease or condition mediated by PI3 kinases, wherein the medicament is prepared for administration with another therapeutic agent.
- the invention also provides the use of another therapeutic agent for treating a disease or condition mediated by PI3 kinases, wherein the medicament is administered with a compound of formula (I).
- the invention also provides a compound of formula (I) for use in a method of treating a disease or condition mediated by class I PI3 kinase or mediated by class I PI3 kinase and mTOR, wherein the compound of formula (I) is prepared for administration with another therapeutic agent.
- the invention also provides another therapeutic agent for use in a method of treating a disease or condition mediated by class I PI3 kinase or mediated by class I PI3 kinase and mTOR , wherein the other therapeutic agent is prepared for administration with a compound of formula (I).
- the invention also provides a compound of formula (I) for use in a method of treating a disease or condition mediated by class I
- the invention also provides another therapeutic agent for use in a method of treating a disease or condition mediated by class I PI3 kinase or mediated by class I PI3 kinase and mTOR , wherein the other therapeutic agent is administered with a compound of formula (I).
- the invention also provides the use of a compound of formula (I) for treating a disease or condition mediated by class I PI3 kinase or mediated by class I PI3 kinase and mTOR wherein the patient has previously (e.g. within 24 hours) been treated with another therapeutic agent.
- the invention also provides the use of another therapeutic agent for treating a disease or condition mediated by class I PI3 kinase or mediated by class I PI3 kinase and mTOR , wherein the patient has previously (e.g. within 24 hours) been treated with a compound of formula (I).
- the other therapeutic agent is selected from therapeutic agents suitable for the treatment of non-melanoma skin cancers such as basal cell carcinoma and squamous cell carcinoma; their pre-malignant stages such as actinic keratosis, solar keratosis and chronically sun damaged skin.
- these other therapeutic agents can be selected from immunostimulatory compounds for example Toll-like receptor agonists such as imiquimod (Aldara®), or from anti-inflammatory agents such as diclofenac (Solaraze®).
- the pharmaceutical composition or combination of the present invention can be in unit dosage of about 1 to about 1000 mg of active ingredient(s) for a subject of about 50 to about 70 kg, or about 1 to about 500 mg or about 1 to about 250 mg or about 1 to about 150 mg or about 0.5 to about 100 mg, or about 1 to about 50 mg of active ingredients.
- Unit dosage can also be of about 50 to about 1000 mg of active ingredient(s) for a subject of about 50 to about 70 kg, or about 50 to about 500 mg or about 50 to about 250 mg or about 50 to about 150 mg or about 50 to about 100 mg of active ingredients.
- the dosage may depend upon the particular dosage form used for delivering the active ingredient(s).
- the therapeutically effective dosage of a compound, the pharmaceutical composition, or the combinations thereof is dependent on the species of the subject, the body weight, age and individual condition, the disorder or disease or the severity thereof being treated.
- the dosage can also depend on the bioavailability of the active ingredient in the species being treated.
- a physician, pharmacist, clinician or veterinarian of ordinary skill can readily determine the effective amount of each of the active ingredients necessary to prevent, treat or inhibit the progress of the disorder or disease.
- the above-cited dosage properties are demonstrable in vitro and in vivo tests using advantageously mammals, e.g., mice, rats, dogs, monkeys, pigs, mini-pigs or isolated organs, tissues and preparations thereof.
- the compounds of the present invention can be applied in vitro in the form of solutions, e.g., aqueous solutions prepared from e.g. 10 mM DMSO stock solution, and in vivo either enterally, parenterally, advantageously intravenously or topically, e.g., as a suspension, in aqueous solution or other solutions, such as e.g. in propylene glycol based solution.
- the dosage in vitro may range between about 10 "3 molar and about 10 "9 molar concentrations.
- a therapeutically effective amount in vivo may range depending on the route of administration, between about 0.1 to about 500 mg/kg, or between about 1 to about 100 mg/kg.
- starting materials are generally available from commercial sources such as Aldrich Chemicals Co. (Milwaukee, Wis.), Lancaster Synthesis, Inc. (Windham, N.H.), Acros Organics (Fairlawn, N.J.), Maybridge Chemical Company, Ltd. (Cornwall, England), Tyger Scientific (Princeton, N.J.), Chem-lmpex International, Inc. (Wood Dale, IL), and AstraZeneca Pharmaceuticals (London, England).
- boronic ester intermediate used in the preparation of compounds of the present invention is either commercially available or may be prepared as described in the literature, or in an analogous manner, or can be prepared as described hereafter, or in an analogous manner.
- dichloromethane (30 mL) was added dropwise at -30 °C under argon 2- (trimethylsilyl)thiazole (939 ⁇ , 5.97 mmol) over a period of 10 min. Then, the reaction mixture was stirred at -30°C for 20 minutes, allowed to warm to room temperature and stirred at room temperature for 2 hours. The reaction mixture was then concentrated at 30 °C and dissolved in THF (30 mL). TBAF (1 M in THF) (5.97 mL, 5.97 mmol) was added at 0 °C under argon and the reaction mixture was stirred at room temperature for 2 hours. The reaction mixture was concentrated, and the residue was extracted with EtOAc.
- Boc-L-alanal (9.99 g, 57.7 mmol) was dissolved in 200 mL of CH 2 CI 2 .
- 2-(trimethylsilyl)- thiazole (9.90 g, 62.9 mmol) dissolved in 70 mL of CH 2 CI 2 was added to the reaction mixture at -20 °C under argon. After 21 hours at -20 °C, the reaction mixture was concentrated. The residue was then dissolved in 180 mL of THF, when TBAF (1 M in THF, 63.4 mL, 63.4 mmol) was added, and the reaction mixture was stirred for 4 hours at room temperature. The reaction mixture was then concentrated.
- dimethoxyethane was added under argon 5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)-4-(trifluoromethyl)pyrimidin-2-amine (1 .08 g, 3.75 mmol), PdCI 2 (dppf)-CH 2 CI 2 (214 mg, 262 ⁇ ), and 2 M aqueous sodium carbonate (5.11 mL, 10.2 mmol).
- the reaction mixture was stirred at 80 °C for 30 minutes.
- the reaction mixture was then cooled to RT and diluted with 80 mL of EtOAc, the organic solvents were washed with aqueous NH 4 CI and brine, dried over MgS0 4 , filtered, and concentrated.
- Examples 4 to 14 in Table 1 below can be made using procedures analogous to those described in examples 1-3, using the appropriate boronic ester intermediate and oxazolidinone.
- the activity of a compound according to the present invention can be assessed by the following in vitro & in vivo methods
- Test compounds were dissolved in DMSO (10 mM) and transferred into 1.4 mL flat bottom or V-shaped Matrix tubes carrying a unique 2D matrix chip by individual Novartis compound hubs. The numbers of these chips were distinctively linked to Novartis Pharma Numbers. The stock solutions were stored at -20°C if not used immediately. For the test procedure the vials were defrosted and identified by a scanner whereby a working sheet was generated that guided the subsequent working steps.
- Pre-dilution plates 96 polypropylene well plates were used as pre-dilution plates. A total of 4 pre-dilution plates were prepared including 10 test compounds each on the plate positions A1-A10, one standard compound at A1 1 and one DMSO control at A12. The pattern of the dilution steps is summarized in Table 1. Programs have been written to run these pipetting steps on the HamiltonSTAR robots.
- Assay plates Identical "assay plates” were then prepared by pipetting 50nl_ each of compound dilutions of the "master plates” into 384-well “assay plates”. The compounds were mixed with 4.5 ⁇ _ of assays components plus 4.5 ⁇ _ enzyme corresponding to a 1 :181 dilution enabling the final concentration of 10, 3.0, 1.0, 0.3, 0.1 , 0.03, 0.01 and 0.003 ⁇ , respectively. The preparation of the "master plates” was handled by the Matrix PlateMate Plus robot and replication of "assay plates" by the HummingBird robot.
- Catalytically active human ⁇ 3 ⁇ , ⁇ 3 ⁇ , PI3K5, and mTOR were cloned, expressed and purtified as described (Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, de Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, Garcia-Echeverria C (2008), Mol Cancer Ther.
- the luminescence-based ATP detection reagent KinaseGlo was obtained from
- Luminescence is a well established readout to determine ATP concentrations and can thus be used to follow the activity of many kinases regardless of their substrate.
- the Kinase Glo Luminescent Kinase Assay (Promega, Madison/WI, USA) is a homogeneous HTS method of measuring kinase activity by quantifying the amount of ATP remaining in solution following a kinase reaction.
- NBS Non Binding Styrene
- 5 ⁇ _ of a mix of PI/OG with the PI3Ka and Pi3Kb subtypes were added.
- Kinase reactions were started by addition of 5 ⁇ of ATP-mix containing in a final volume 10 ⁇ _ 10 mM TRIS-HCI pH 7.5, 3mM MgCI 2 , 50 mM NaCI, 0.05% CHAPS, 1 mM DTT and 1 ⁇ ATP, and occured at room temperature.
- Reactions were stopped with 10 ⁇ of KinaseGlo and plates were read 10 mins later in a Synergy2 reader using an integration time of 0.1 seconds per well.
- NVP- BGT226 (standard) was added to the assay plates to generate the 100% inhibition of the kinase reaction, and the 0% inhibition was given by the solvent vehicle (90% DMSO in water).
- NVP-BGT226 was used as a reference compound and included in all assay plates in the form of 16 dilution points in duplicate.
- IC 50 values of the percentage inhibition of each compound at 8 concentrations were derived by fitting a sigmoidal dose-response curve to a plot of assay readout over inhibitor concentration as described. All fits were performed with the program XLfit4 (ID Business Solutions, Guildford, UK).
- TR-FRET AdaptaTM Universal Kinase Assay Kit was purchased from Invitrogen
- the kit contains the following reagents: Adapta Eu-anti-ADP Antibody (Europium labeled anti-ADP antibody in HEPES buffered saline, Cat. No. PV5097), Alexa Fluor® 647-labeled ADP tracer (Alexa Fluor® 647-labeled ADP tracer in HEPES buffered saline, Cat. No. PV5098), proprietary TR- FRET dilution buffer pH 7.5 (Cat. No. PV3574).
- Adapta Eu-anti-ADP Antibody Europium labeled anti-ADP antibody in HEPES buffered saline, Cat. No. PV5097
- Alexa Fluor® 647-labeled ADP tracer Alexa Fluor® 647-labeled ADP tracer in HEPES buffered saline, Cat. No. PV5098
- proprietary TR- FRET dilution buffer pH 7.5 Cat. No. PV3574
- PIK3CD substrate Phosphatidylinositol was obtained from Invitrogen (vesicules consisting of 2 mM PI in 50mM HEPES pH7.5; Cat. No. PV5371 ).
- PIK3CG substrate Phosphatidylinositol-4,5-bisphosphate (PIP(4,5)2 was obtained from Invitrogen (PIP2:PS large unilamellar vesicules consisting of 1 mM PIP2: 19mM PS in 50mM HEPES pH7.5, 3mM MgCI2, 1 mM EGTA; Cat. No. PV5100).
- TR-FRET Time-Resolved Fluorescence Resonance Energy Transfer
- TR-FRET assays for protein kinases use a long-lifetime lanthanide Terbium or Europium chelates as the donor species which overcome interference from compound autofluorescence or light scatter from precipitated compounds, by introducing a delay after excitation by a flashlamp excitation source. Results are often expressed as a ratio of the intensities of the acceptor and donor fluorophores. The ratiometric nature of such a value corrects for differences in assay volumes between wells, as well as corrects for quenching effects due to colored compounds.
- the AdaptaTM assay can be divided into two phases : a kinase reaction phase and an ADP detection phase.
- kinase reaction phase all kinase reaction components are added to the well and the reaction is allowed to incubate for a set period of time specific for each kinase.
- a detection solution of Eu-labeled anti-ADP antibody, Alexa Fluor® 647-labeled ADP tracer, and EDTA are added to the assay well.
- ADP formed by the kinase reaction will displace the Alexa Fluor® 647-labeled ADP tracer from the antibody, resulting in a decrease in TR-FRET signal.
- the amount of ADP formed by the kinase reaction is reduced, and the resulting intact antibody-tracer interaction maintains a high TR-FRET signal.
- the donor Europium-anti-ADP antibody
- the acceptor Alexa Fluor® 647-labeled ADP tracer
- the emission from the Alexa Fluor® 647 can be monitored with a filter centered at 665 nm because it is located between the emission peaks of the donor, which is measured at 615/620 nm.
- ADP antibody and the Alexa Fluor® 647-labeled ADP tracer in TR-FRET dilution buffer (proprietary to IVG). Plates are read 15 to 60 mins later in a Synergy2 reader using an integration time of 0.4 seconds and a delay of 0.05 seconds. Control for the 100% inhibition of the kinase reaction was performed by replacing the PI3K by the standard reaction buffer. The control for the 0% inhibition was given by the solvent vehicle of the compounds (90% DMSO in H 2 0). The standard compound NVP-BGT226 was used as a reference compound and included in all assay plates in the form of 16 dilution points in duplicate.
- TR-FRET assays for protein kinases uses a long-lifetime lanthanide Terbium or
- Binding Assays are based on the binding and displacement of an Alexa Fluor® 647- labeled, ATP-competitive kinase inhibitors to the kinase of interest. Invitrogen's "Kinase
- Tracers have been developed to address a wide range of kinase targets and are based on ATP-competitive kinase inhibitors, making them suitable for detection of any compounds that bind to the ATP site or to an allosteric site altering the conformation of the ATP site.
- Inhibitors that bind the ATP site include both Type I kinase inhibitors, which bind solely to the ATP site, and Type II inhibitors (e.g., Gleevec®/lmatinib, Sorafenib,
- Type III inhibitors are compounds that do not compete with ATP are loosely referred to as allosteric inhibitors.
- a study of 15 diverse Type III inhibitors demonstrated that all but one compound was detected in the binding assay with equivalent potency to activity assays. The sole exception was a substrate- competitive compound, and thus not a true allosteric inhibitor.
- LanthaScreen® Eu 3+ Kinase Binding Assays can be read continuously, which facilitates evaluation of compounds with slow binding kinetics. Also, unlike most activity assays, binding assays can be performed using either active or non-activated kinase preparations, which enables characterization of compounds that bind preferentially to non-activated kinases, such as Gleevec®/imatinib and some allosteric inhibitors.
- Alexa Fluor® 647 inhibitor can be monitored with a filter centered at 665 nm because it is located between the emission peaks of the donor, which is measured at 615/620 nm.
- the binding of both, the Tracer-314 and Eu 3+ -anti-GST antibody, to the kinase results in a high degree of FRET from the Eu 3+ -donor fluorophore to the Alexa-Fluor® 647- acceptor fluorophore on the Tracer-314. Binding of an inhibitor to the kinase competes for binding with the tracer, resulting in a loss of FRET.
- the standard reaction buffer for the LanthascreenTM kinase binding assay contained 50mM HEPES pH 7.5, 5mM MgCI2, 1 mM EGTA, 0.01 % Pluronic F-
- Plates are read 60 mins later in a Synergy2 reader using an integration time of 0.2 microseconds and a delay of 0.1 microseconds.
- the signal emitted at 665 nm from the acceptor (Alexa Fluor® 647-labeled Tracer-314) is divided by the signal emitted at 620 nm from the donor (Eu 3+ anti-GST antibody)
- Control for the 0% inhibition was given by the solvent vehicle of the compounds (90% DMSO in H 2 0). Control for the relative 100% inhibition was performed by adding 10 ⁇ in the mix containing GST-mTOR and Europium anti-GST antibody. An additional control for the absolute 0% inhibition is given by Eu 3+ anti-GST antibody without GST-mTOR.
- AlphaScreen (Amplified Luminescent Proximity Homogeneous Assay, ALPHA, Perkin Elmer) is a non-radioactive bead-based proximity assay technology to study
- the brand name SureFire denotes AlphaScreen assays that are adapted to quantify the phosphorylation of endogenous cellular proteins in cell lysates, by using matched antibody pairs, which consist of an anti-phospho-kinase and an anti-kinase antibody.
- the assay allows characterization of kinase signaling in cells as well as measurement of kinase inhibitor effects.
- the AlphaScreen technology provides several advantages over standard assay techniques such as ELISA, as it avoids time-consuming washing procedures and reduces plate handling.
- Rat-1 cell lines stably overexpressing activated PI3K class I isoforms Rat-1 pBABEpuro Myr-HA-hp1 10 delta (Rat-1_PI3Kdelta) and Rat-1 pBABEpuro Myr-HA- hpH Oalpha (Rat-1_PI3Kalpha) and Rat-1 pBABEpuro Myr-HA-hp1 10 beta (Rat- 1_PI3beta) were prepared as described (Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, de Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, Garcia-Echeverria C (2008), Mol Cancer Ther. 7:1851 -63 and Maira SM, Pecchi S, Brueggen J, Huh K, Schnell C, Fritsch
- Dulbecco's modified Eagle's medium (DMEM) high glucose (Gibco Invitrogen, Basel,
- the p-Akt(S473) SureFire assay measures the phosphorylation of endogenous cellular Akt 1/2 at Ser473 in cell lysates.
- Rat-1 cells stably expressing myr-HA-tagged versions of the human PI3Kdelta, PI3Kalpha, or PI3Kbeta p110 catalytic subunit isoforms the assay was developed as a two-plate protocol in a 384-well format.
- the cells were seeded at a density of 4000 (Rat-1_PI3Kdelta), 7500 (Rat-1_PI3Kalpha), or 6200 (Rat-1_PI3Kbeta) cells in 20 ul complete growth medium into cell culture treated 384-well plates and were grown at 37°C / 5 % C0 2 / 90 % humidity for 24 h. Shortly before compound transfer, the complete medium was removed, 30 ul assay buffer (DMEM high glucose, 1x MEM NEAA, 10 mM HEPES, 2 mM L-glutamine, 0.1 % (w/v) BSA) was added and 10 ul of the compound predilutions were transferred to the cells.
- DMEM high glucose, 1x MEM NEAA, 10 mM HEPES, 2 mM L-glutamine, 0.1 % (w/v) BSA was added and 10 ul of the compound predilutions were transferred to the cells.
- IC 50 values of the percentage inhibition of each compound at 8 concentrations were derived by fitting a sigmoidal dose-response curve to a plot of assay readout over inhibitor concentration as described. All fits were performed with the program XLfit4 (ID Business Solutions, Guildford, UK).
- a cell based assay (384-well format) was developed for determination of compound effects on cellular mTOR activity in MEF (mouse embryo fibrobrasts) cells derived from mice lacking TSC1 (Tuberosclerosis Complexl ) a potent suppressor of mTOR activity. Due to lack of TSC1 , mTOR is constitutively activated resulting in permanent phoshorylation of Thr 389 of S6 kinase 1 (S6K1 ) which is one of the downstream targets of mTOR.
- TSC1 -/- MEFs cells were cultured in DMEM high glucose medium supplemented with 10% FBS (Invitrogen), 2mM Glutamine and 1% (w/v) Penicillin/Streptomycin at 37°C, 5% C0 2 .
- the SureFire kit for determination of P70S6kinase phosphorylation was purchased from Perkin Elmer (p70S6K p-T389, #TGR70S50K) and the assay was performed according to the instructions of the supplier and according to the generic method for SureFire assays. Shortly, 5 ⁇ _ cell lysate per well were transferred to 384-well white proxy-plates (for luminescent readout) and mixed with 7 ⁇ _ A and 5 ⁇ _ B (final volume: 12 ⁇ _). After 3 h incubation in the dark at RT luminescence was read with the Envision Reader (Perkin Elmer). Untreated cells were used as control (high control) and cells treated with 3 ⁇ BEZ235 were used as low control. The assay window between the signals obtained for the high and the low controls were defined as 100 % and compound effects were expressed as percent inhibition. IC50 values were calculated from the dose response curves by graphical extrapolation.
- Samples investigated for light stability were treated in a light stability chamber (Atlas CPS+, Serial Number 0704013). Solutions of 1.0mg/ml in ethanol were prepared for each molecule and aliquots of 0.5mL were dispensed into suitable micro tubes. All measurements were done in duplicate and compared to a reference wrapped in aluminium foil during light exposure. Solutions were exposed to 19620kJ/m 2 over 13h.
- the SCL-1 and SCC12B2 are human cutaneous squamous cell carcinoma (SCC) cell lines derived from naturally occurring, poorly differentiated SCCs.
- SCC human cutaneous squamous cell carcinoma
- the SCC12B2 cell line was established as described previously (Rheinwald & Beckett 1981 ) from the facial skin of a male transplant patient who received immunosuppressive therapy for 7 years.
- the human SCC cell line Detroit562 was obtained from American Tissue Culture Collection (ATCC).
- Detroit562 is referred to as Head&Neck squamous cell carcinomas (HNSCC) and was derived from a pharyngeal SCC tumor.
- the Detroit562 cell line bears the activating mutation H1047R of the PIK3CA gene. This mutation is known as one of the "hotspot" mutations of p1 10a -chain rendering this kinase isoform constitutively active and stimulating the Akt/mTOR pathway independent of growth factor receptor activation.
- DMEM Dulbecco modified Eagle's medium
- FCS Gibco; #16000-044
- sodium-pyruvat Gibco; #11360-039
- 10mM HEPES Gibco; #91002-066
- 50U/ml penicillin 50 ⁇ g/ml streptomycin mixture
- Cells of SCC or keratinocyte lines were used for the proliferation assays when they had reached about 80% to 90% confluency. Cells were then harvested after trypsin-mediated dislodgement from culture flasks, washed in culture medium containing 10% FCS, counted in a hemocytometer and diluted complete culture medium with 5% FCS to obtain a cell density of 5x10 4 cells/ml. 100 ⁇ of this cell suspension (e.g. 5000 cells) were transferred into each well of a 96-well white walled tissue culture plate (Corning-Costar article #3917).
- the BrdU reagent was diluted 1 :100 with complete culture medium to obtain a concentration of 100 ⁇ BrdU. From this solution, 20 ⁇ were added to each well and the cell culture was further incubated for 16 to 18 hours at 37°C at 5% C0 2 . The proliferation assay was stopped after 44 to 46 hours by complete removal of the culture medium and addition of 200 ⁇ fixing solution (provided with the ELISA kit) for 30 minutes at room temperature. All further incubations, washings and assay development procedures were performed exactly as described in the ELISA assay manual provided by the
- Anti-proliferative potency of compounds measured in SCC cell lines The data obtained using these SCC and keratinocyte cell lines are shown in the following table. All values indicating the anti-proliferative potency represent the nanomolar IC50
- the compound was applied as 0.5% solutions in propylene glycol mixed with either ethanol or oleylalcohol to pig skin mounted in static Franz-type diffusion cells. At the end of a 48 hours exposure time, drug concentrations were measured in the skin (after removal of stratum corneum) and in the receiver.
- the receiver solution is a mixture of two volume parts of phosphate-buffered saline (PBS) and one volume part of fetal calf serum (FCS).
- PBS phosphate-buffered saline
- FCS fetal calf serum
- Skin concentrations are given as mean ⁇ standard deviation of quadruplicate determinations.
- the compound of example 1 penetrates well into pig skin in vitro, while permeation rate through pig skin is low, indicating a low systemic exposure.
- Pig skin is similar to human skin regarding barrier function and architecture.
- the following table provides dermal concentrations of the compound of example 1 in pig dermis when applied epicutaneously in the identified formulations.
- the data table provides the mean ⁇ standard error of the mean of 8 determinations (e.g. eight skin samples analyzed for each time point).
- Tween®80 polyoxyethylenesorbitan monooleate
- HPMC hydroxypropylmethylcellulose
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description



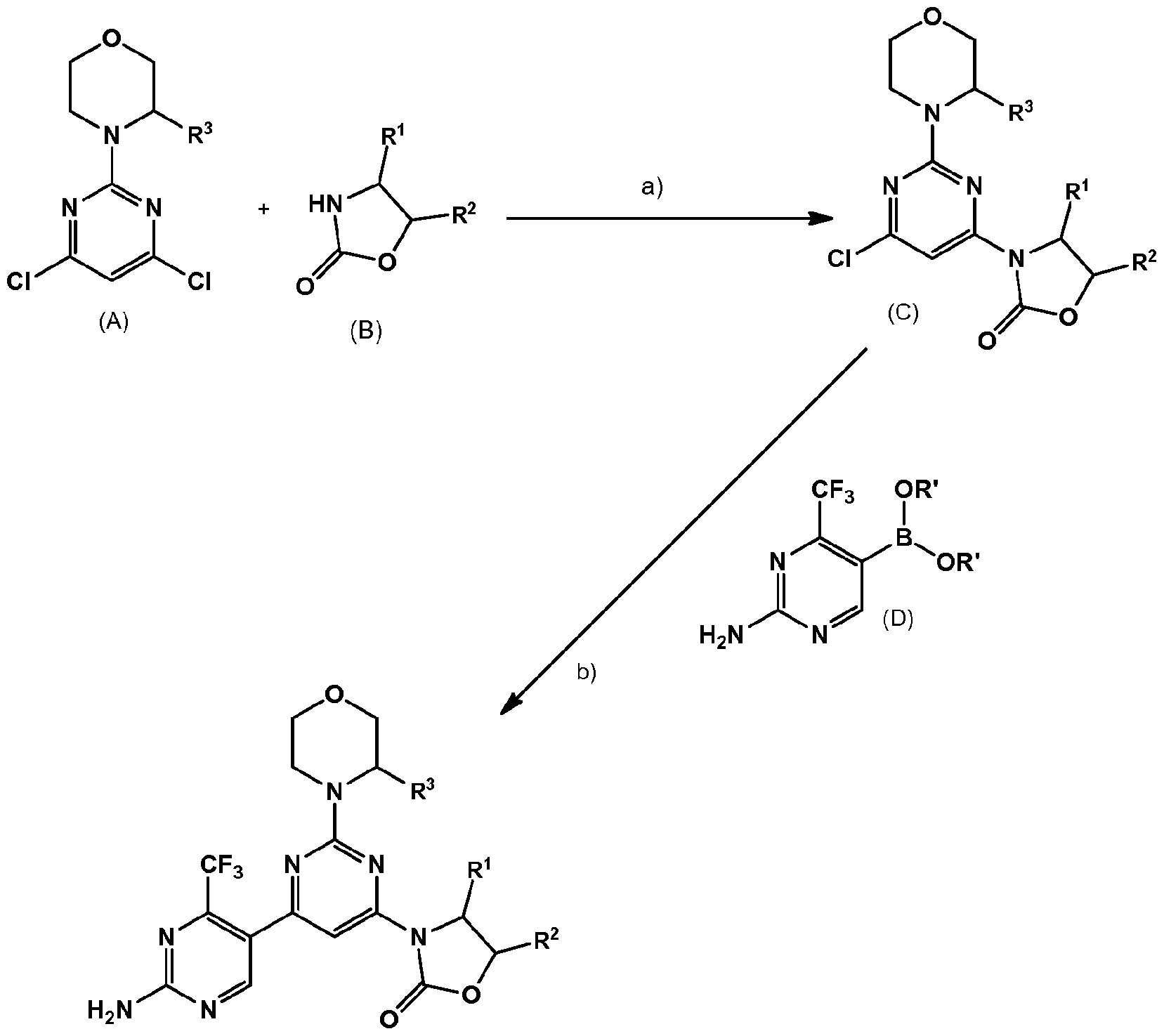







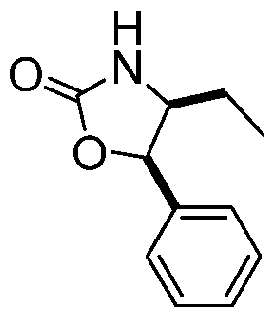














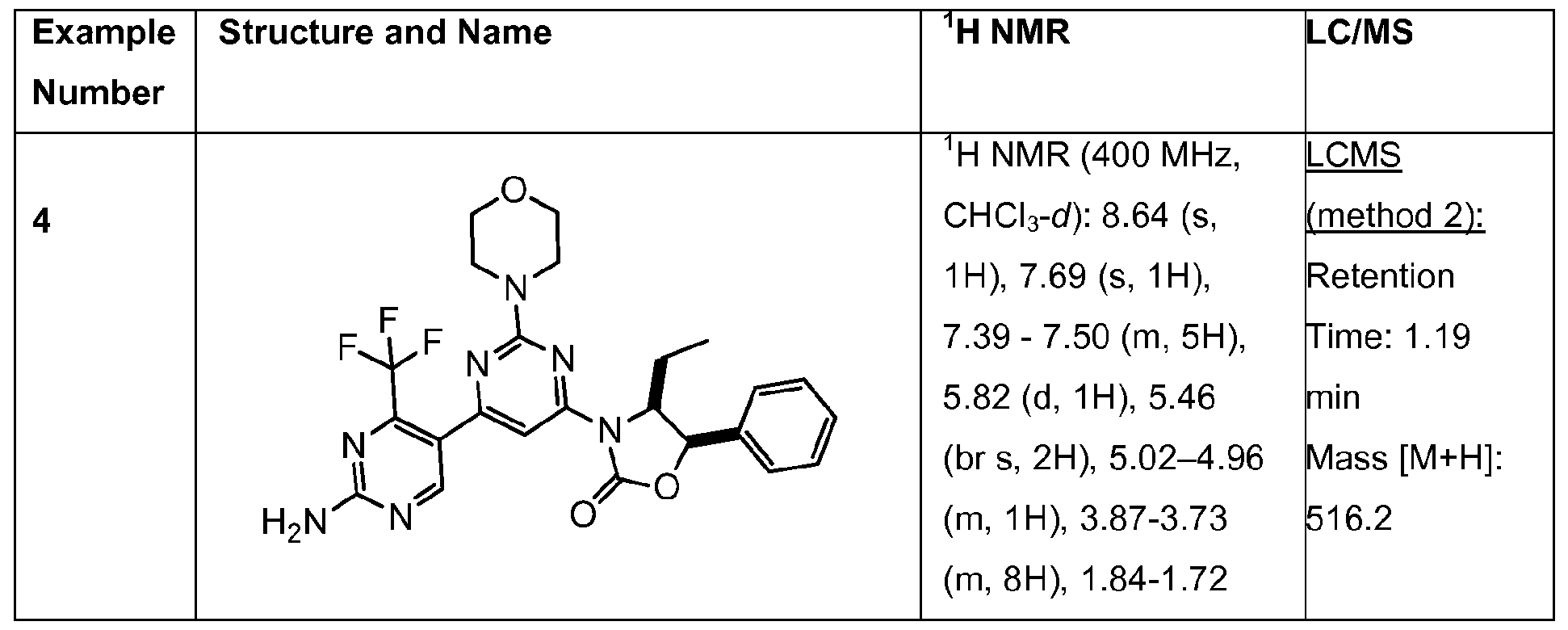








Claims


Priority Applications (31)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MA38076A MA38076A1 (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives |
| IN2912DEN2015 IN2015DN02912A (en) | 2012-11-12 | 2013-11-11 | |
| AU2013343022A AU2013343022B2 (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives |
| DK13795867.4T DK2922848T5 (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives |
| CA2890942A CA2890942C (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives |
| RS20170882A RS56336B1 (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives |
| AP2015008347A AP2015008347A0 (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives |
| ES13795867.4T ES2641820T3 (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives |
| EP17157570.7A EP3199158B1 (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives for use in the treatment of skin fibrosis, scleroderma, hypertrophic scars or keloids |
| CU2015000050A CU24365B1 (en) | 2012-11-12 | 2013-11-11 | DERIVATIVES OF OXAZOLIDIN-2-ONA-PYRIMIDINE |
| JP2015541289A JP6262245B2 (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one pyrimidine derivatives |
| SG11201503447QA SG11201503447QA (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives |
| KR1020157011951A KR102153763B1 (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives |
| EP13795867.4A EP2922848B1 (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives |
| BR112015010136A BR112015010136A2 (en) | 2012-11-12 | 2013-11-11 | oxazolidin-2-one-pyrimidine derivatives |
| LTEP13795867.4T LT2922848T (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives |
| PL13795867T PL2922848T3 (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives |
| EA201590929A EA027987B1 (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives |
| MX2015005914A MX363436B (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives. |
| NZ706591A NZ706591A (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives |
| UAA201502809A UA114001C2 (en) | 2012-11-12 | 2013-11-11 | OXAZOLIDINE-2-ON-PYRIMIDINE DERIVATIVES |
| CN201380052670.1A CN104718204B (en) | 2012-11-12 | 2013-11-11 | The ketone pyrimidine derivatives of oxazolidine 2 |
| SI201330782T SI2922848T1 (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives |
| TNP2015000120A TN2015000120A1 (en) | 2012-11-12 | 2015-03-27 | Oxazolidin-2-one-pyrimidine derivatives |
| ZA2015/02175A ZA201502175B (en) | 2012-11-12 | 2015-03-30 | Oxazolidin-2-one-pyrimidine derivatives |
| IL238239A IL238239A (en) | 2012-11-12 | 2015-04-12 | Oxazolidin-2-one-pyrimidine derivatives |
| PH12015501053A PH12015501053B1 (en) | 2012-11-12 | 2015-05-12 | Oxazolidin-2-one-pyrimidine derivatives |
| CR20150248A CR20150248A (en) | 2012-11-12 | 2015-05-12 | DERIVATIVES OF OXAZOLIDIN-2-ONA-PYRIMIDINE |
| HK15109748.9A HK1209108A1 (en) | 2012-11-12 | 2015-10-06 | Oxazolidin-2-one-pyrimidine derivatives -2-- |
| HRP20171425TT HRP20171425T1 (en) | 2012-11-12 | 2017-09-20 | Oxazolidin-2-one-pyrimidine derivatives |
| CY20171101014T CY1119391T1 (en) | 2012-11-12 | 2017-09-27 | OXAZOLIDIN-2-ONY-PYRIMIDINE PRODUCTS |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261725113P | 2012-11-12 | 2012-11-12 | |
| US61/725,113 | 2012-11-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014072956A1 true WO2014072956A1 (en) | 2014-05-15 |
Family
ID=49667527
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2013/060052 WO2014072956A1 (en) | 2012-11-12 | 2013-11-11 | Oxazolidin-2-one-pyrimidine derivatives |
Country Status (43)
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019087158A2 (en) | 2017-11-06 | 2019-05-09 | Novartis Ag | Formulation comprising oxazolidin-2-one-pyrimidine derivative |
| WO2022053651A2 (en) | 2020-09-10 | 2022-03-17 | Precirix N.V. | Antibody fragment against fap |
| WO2023203135A1 (en) | 2022-04-22 | 2023-10-26 | Precirix N.V. | Improved radiolabelled antibody |
| WO2023213801A1 (en) | 2022-05-02 | 2023-11-09 | Precirix N.V. | Pre-targeting |
| US11918586B2 (en) | 2016-05-18 | 2024-03-05 | Torqur Ag | Treatment of skin lesions |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CU24269B1 (en) | 2011-09-27 | 2017-08-08 | Novartis Ag | 3- PIRIMIDIN- 4-IL- OXAZOLIDIN- 2- INHIBITING WAVES OF THE MUTANT HDI |
| UY34632A (en) | 2012-02-24 | 2013-05-31 | Novartis Ag | OXAZOLIDIN- 2- ONA COMPOUNDS AND USES OF THE SAME |
| US9296733B2 (en) | 2012-11-12 | 2016-03-29 | Novartis Ag | Oxazolidin-2-one-pyrimidine derivative and use thereof for the treatment of conditions, diseases and disorders dependent upon PI3 kinases |
| MX355945B (en) | 2013-03-14 | 2018-05-07 | Novartis Ag | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh. |
| EP3352755A4 (en) | 2015-09-24 | 2019-04-03 | Drexel University | Novel compositions and methods for treating or preventing dermal disorders |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004078163A2 (en) | 2003-02-28 | 2004-09-16 | Transform Pharmaceuticals, Inc. | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2007084786A1 (en) | 2006-01-20 | 2007-07-26 | Novartis Ag | Pyrimidine derivatives used as pi-3 kinase inhibitors |
Family Cites Families (117)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB581334A (en) | 1943-09-29 | 1946-10-09 | Francis Henry Swinden Curd | New pyrimidine compounds |
| JPS4921149B1 (en) | 1970-12-28 | 1974-05-30 | ||
| JPS4921148B1 (en) | 1970-12-28 | 1974-05-30 | ||
| DE2341925A1 (en) | 1973-08-20 | 1975-03-06 | Thomae Gmbh Dr K | 2,4, (opt.5-), 6-substd. pyriminidines as antithrombic agents - e.g. 6-methyl-5-nitro-2-piperazino-4-thiomorpolino-pyrimidine |
| AT340933B (en) | 1973-08-20 | 1978-01-10 | Thomae Gmbh Dr K | PROCESS FOR THE PRODUCTION OF NEW PYRIMIDE DERIVATIVES AND THEIR ACID ADDITIONAL SALTS |
| US4994386A (en) | 1987-07-13 | 1991-02-19 | Pharmacia Diagnostics, Inc. | Production of HBLV virus in the HSB-2 cell line |
| US4929726A (en) | 1988-02-09 | 1990-05-29 | Georgia State University Foundation, Inc. | Novel diazines and their method of preparation |
| EP0330263A1 (en) | 1988-02-25 | 1989-08-30 | Merck & Co. Inc. | Piperazinylalkylpyrimidines as hypoglycemic agents |
| GB9012311D0 (en) | 1990-06-01 | 1990-07-18 | Wellcome Found | Pharmacologically active cns compounds |
| KR950007756B1 (en) | 1990-07-03 | 1995-07-14 | 미쓰이세끼유 가가꾸고오교오 가부시끼가이샤 | Pyrimidine compound and pharmacentically acceptable salt thereof |
| KR100275300B1 (en) | 1995-04-13 | 2000-12-15 | 고바야시 유키오 | Novel 4,6-diarylpyrimidine derivatives and salts thereof |
| JP3734907B2 (en) | 1996-12-19 | 2006-01-11 | 富士写真フイルム株式会社 | Development processing method |
| EP1020462B1 (en) | 1997-07-24 | 2004-02-11 | Zenyaku Kogyo Kabushiki Kaisha | Heterocyclic compounds and antitumor agent containing the same as active ingredient |
| JPH11158073A (en) | 1997-09-26 | 1999-06-15 | Takeda Chem Ind Ltd | Adenosine a3 antagonist |
| US6440965B1 (en) | 1997-10-15 | 2002-08-27 | Krenitsky Pharmaceuticals, Inc. | Substituted pyrimidine derivatives, their preparation and their use in the treatment of neurodegenerative or neurological disorders of the central nervous system |
| US6150362A (en) | 1997-12-12 | 2000-11-21 | Henkin; Jack | Triazine angiogenesis inhibitors |
| EP0930302B1 (en) | 1998-01-16 | 2003-04-02 | F.Hoffmann-La Roche Ag | Benzosulfone derivatives |
| US7045519B2 (en) | 1998-06-19 | 2006-05-16 | Chiron Corporation | Inhibitors of glycogen synthase kinase 3 |
| JP4533534B2 (en) | 1998-06-19 | 2010-09-01 | ノバルティス バクシンズ アンド ダイアグノスティックス,インコーポレーテッド | Inhibitor of glycogen synthase kinase 3 |
| EP1144390A2 (en) | 1999-01-22 | 2001-10-17 | Amgen Inc., | Kinase inhibitors |
| US6495558B1 (en) | 1999-01-22 | 2002-12-17 | Amgen Inc. | Kinase inhibitors |
| JP2003523942A (en) | 1999-06-30 | 2003-08-12 | メルク エンド カムパニー インコーポレーテッド | SRC kinase inhibitor compounds |
| CA2376951A1 (en) | 1999-06-30 | 2001-01-04 | Peter J. Sinclair | Src kinase inhibitor compounds |
| JP2003503351A (en) | 1999-06-30 | 2003-01-28 | メルク エンド カムパニー インコーポレーテッド | SRC kinase inhibitory compounds |
| JP2003505384A (en) | 1999-07-15 | 2003-02-12 | ファーマコピーア,インコーポレーティッド | Bradykinin B1 receptor antagonist |
| JP2001089452A (en) | 1999-09-22 | 2001-04-03 | Sankyo Co Ltd | Pyrimidine derivative |
| HUP0301117A3 (en) | 2000-02-17 | 2004-01-28 | Amgen Inc Thousand Oaks | Imidazole derivatives kinase inhibitors, their use, process for their preparation and pharmaceutical compositions containing them |
| CN100355751C (en) | 2000-03-29 | 2007-12-19 | 西克拉塞尔有限公司 | 2-substd. 4-heteroaryl-pyrimidines and their use in treatment of proliferative disorders |
| ES2360933T3 (en) | 2000-04-27 | 2011-06-10 | Astellas Pharma Inc. | CONDENSED HETEROARILO DERIVATIVES. |
| KR100711042B1 (en) | 2000-04-28 | 2007-04-24 | 다나베 세이야꾸 가부시키가이샤 | Cyclic compounds |
| CA2409762A1 (en) | 2000-06-23 | 2002-01-03 | Donald J.P. Pinto | Heteroaryl-phenyl substituted factor xa inhibitors |
| WO2002020495A2 (en) | 2000-09-06 | 2002-03-14 | Chiron Corporation | Inhibitors of glycogen synthase kinase 3 |
| JP4105948B2 (en) | 2000-09-15 | 2008-06-25 | バーテックス ファーマシューティカルズ インコーポレイテッド | Pyrazole compounds useful as protein kinase inhibitors |
| SE0004053D0 (en) | 2000-11-06 | 2000-11-06 | Astrazeneca Ab | N-type calcium channel antagonists for the treatment of pain |
| KR100909665B1 (en) | 2000-12-21 | 2009-07-29 | 버텍스 파마슈티칼스 인코포레이티드 | Pyrazole Compounds Useful as Protein Kinase Inhibitors and Pharmaceutical Compositions Comprising the Same |
| EP1363890A4 (en) | 2001-02-07 | 2009-06-10 | Ore Pharmaceuticals Inc | Melanocortin-4 receptor binding compounds and methods of use thereof |
| AU2002258400A1 (en) | 2001-02-16 | 2002-08-28 | Tularik Inc. | Methods of using pyrimidine-based antiviral agents |
| CA2448626A1 (en) | 2001-05-25 | 2002-12-05 | Boehringer Ingelheim Pharmaceuticals, Inc. | Carbamate and oxamide compounds as inhibitors of cytokine production |
| WO2002102313A2 (en) | 2001-06-19 | 2002-12-27 | Bristol-Myers Squibb Company | Pyrimidine inhibitors of phosphodiesterase (pde) 7 |
| US6603000B2 (en) | 2001-07-11 | 2003-08-05 | Boehringer Ingelheim Pharmaceuticals, Inc. | Synthesis for heteroarylamine compounds |
| WO2003030909A1 (en) | 2001-09-25 | 2003-04-17 | Bayer Pharmaceuticals Corporation | 2- and 4-aminopyrimidines n-substtituded by a bicyclic ring for use as kinase inhibitors in the treatment of cancer |
| WO2003078427A1 (en) | 2002-03-15 | 2003-09-25 | Vertex Pharmaceuticals, Inc. | Azolylaminoazines as inhibitors of protein kinases |
| AU2003225800A1 (en) | 2002-03-15 | 2003-09-29 | Hayley Binch | Azolylaminoazine as inhibitors of protein kinases |
| WO2003078423A1 (en) | 2002-03-15 | 2003-09-25 | Vertex Pharmaceuticals, Inc. | Compositions useful as inhibitors of protein kinases |
| WO2003077921A1 (en) | 2002-03-15 | 2003-09-25 | Vertex Pharmaceuticals, Inc. | Azinylaminoazoles as inhibitors of protein kinases |
| AU2003245669A1 (en) | 2002-06-21 | 2004-01-06 | Cellular Genomics, Inc. | Certain aromatic monocycles as kinase modulators |
| WO2004029204A2 (en) | 2002-09-27 | 2004-04-08 | Merck & Co., Inc. | Substituted pyrimidines |
| US20040171073A1 (en) | 2002-10-08 | 2004-09-02 | Massachusetts Institute Of Technology | Compounds for modulation of cholesterol transport |
| US7223870B2 (en) | 2002-11-01 | 2007-05-29 | Pfizer Inc. | Methods for preparing N-arylated oxazolidinones via a copper catalyzed cross coupling reaction |
| PL377821A1 (en) | 2002-11-21 | 2006-02-20 | Chiron Corporation | 2,4,6-trisubstituted pyrimidines as phosphotidylinositol (pi) 3-kinase inhibitors and their use in the treatment of cancer |
| CN101468965A (en) | 2003-03-24 | 2009-07-01 | 默克公司 | Biaryl substituted 6-membered heterocyles as sodium channel blockers |
| US20050014753A1 (en) | 2003-04-04 | 2005-01-20 | Irm Llc | Novel compounds and compositions as protein kinase inhibitors |
| CA2520323C (en) | 2003-04-09 | 2013-07-09 | Exelixis, Inc. | Tie-2 modulators and methods of use |
| WO2005009977A1 (en) | 2003-07-15 | 2005-02-03 | Neurogen Corporation | Substituted pyrimidin-4-ylamina analogues as vanilloid receptor ligands |
| JP2007534624A (en) | 2003-07-16 | 2007-11-29 | ニューロジェン・コーポレーション | Biaryl piperazinyl-pyridine analogues |
| AR045944A1 (en) | 2003-09-24 | 2005-11-16 | Novartis Ag | ISOQUINOLINE DERIVATIVES 1.4-DISPOSED |
| DE602005024382D1 (en) | 2004-04-13 | 2010-12-09 | Astellas Pharma Inc | POLYCYCLIC PYRIMIDINES AS CALIUMIONAL CHANNEL MODULATORS |
| GB0415364D0 (en) | 2004-07-09 | 2004-08-11 | Astrazeneca Ab | Pyrimidine derivatives |
| GB0415365D0 (en) | 2004-07-09 | 2004-08-11 | Astrazeneca Ab | Pyrimidine derivatives |
| MY145822A (en) | 2004-08-13 | 2012-04-30 | Neurogen Corp | Substituted biaryl piperazinyl-pyridine analogues |
| MX2007006230A (en) * | 2004-11-30 | 2007-07-25 | Amgen Inc | Quinolines and quinazoline analogs and their use as medicaments for treating cancer. |
| JP2008523156A (en) | 2004-12-13 | 2008-07-03 | ニューロジェン・コーポレーション | Substituted biaryl analogs |
| US20080045525A1 (en) | 2004-12-13 | 2008-02-21 | Rajagopal Bakthavatchalam | Piperazinyl-Pyridine Analogues |
| ATE544748T1 (en) | 2004-12-28 | 2012-02-15 | Kinex Pharmaceuticals Llc | COMPOSITIONS AND METHODS FOR THE TREATMENT OF CELL PROLIFERATION DISEASES |
| EP1838703A2 (en) | 2005-01-19 | 2007-10-03 | Neurogen Corporation | Heteroaryl substituted piperazinyl-pyridine analogues |
| CA2599320A1 (en) | 2005-02-25 | 2006-08-31 | Kudos Pharmaceuticals Limited | Hydrazinomethyl, hydr zonomethyl and 5-membered heterocylic compounds which act as mtor inhibitors and their use as anti cancer agents |
| TW200716594A (en) | 2005-04-18 | 2007-05-01 | Neurogen Corp | Substituted heteroaryl CB1 antagonists |
| GB2431156A (en) | 2005-10-11 | 2007-04-18 | Piramed Ltd | 1-cyclyl-3-substituted- -benzenes and -azines as inhibitors of phosphatidylinositol 3-kinase |
| AU2007204208A1 (en) | 2006-01-11 | 2007-07-19 | Astrazeneca Ab | Morpholino pyrimidine derivatives and their use in therapy |
| EP2012794B1 (en) * | 2006-04-13 | 2014-09-17 | The Trustees of Columbia University in the City of New York | Compositions and intraluminal devices for inhibiting vascular stenosis |
| AR064571A1 (en) | 2006-12-28 | 2009-04-08 | Basf Ag | PYRIMIDINES REPLACED IN POSITION 2 BY NITROGEN HETEROCICLES, MEDICINES AND PHARMACEUTICAL COMPOSITIONS THAT CONTAIN THEM AND USES OF THE SAME IN THE TREATMENT OF CANCER. |
| JP2010518107A (en) | 2007-02-06 | 2010-05-27 | ノバルティス アーゲー | PI3-kinase inhibitors and methods of use thereof |
| US7957951B2 (en) | 2007-03-16 | 2011-06-07 | Robert Bosch Gmbh | Address translation system for use in a simulation environment |
| EA201000092A1 (en) | 2007-07-09 | 2010-06-30 | Астразенека Аб | TRIPLE-SUBSTITUTED PYRIMIDINE DERIVATIVES FOR THE TREATMENT OF PROLIFERATIVE DISEASES |
| WO2009066084A1 (en) | 2007-11-21 | 2009-05-28 | F. Hoffmann-La Roche Ag | 2 -morpholinopyrimidines and their use as pi3 kinase inhibitors |
| EP2591805A1 (en) | 2008-03-05 | 2013-05-15 | Novartis AG | Use of pyrimidine derivatives for the treatment of egfr dependent diseases or diseases that have acquired resistance to agents that target EGFR family members |
| CN101980708B (en) | 2008-03-26 | 2013-03-13 | 诺瓦提斯公司 | Imidazoquinolines and pyrimidine derivatives as potent modulators of VEGF-driven angiogenic processes |
| US20110053907A1 (en) | 2008-03-27 | 2011-03-03 | Auckland Uniservices Limited | Substituted pyrimidines and triazines and their use in cancer therapy |
| EP2280948A1 (en) | 2008-04-09 | 2011-02-09 | Mitsubishi Tanabe Pharma Corporation | Pyrimidine, pyridine and triazine derivatives as maxi-k channel openers. |
| GB0815369D0 (en) | 2008-08-22 | 2008-10-01 | Summit Corp Plc | Compounds for treatment of duchenne muscular dystrophy |
| RU2538683C2 (en) | 2008-10-31 | 2015-01-10 | Новартис Аг | COMBINATION OF PHOSPHATIDYL INOSITOL-3-KINASE (PI3K) INHIBITOR AND mTOR INHIBITOR |
| GB2465405A (en) | 2008-11-10 | 2010-05-19 | Univ Basel | Triazine, pyrimidine and pyridine analogues and their use in therapy |
| WO2010068863A2 (en) | 2008-12-12 | 2010-06-17 | Cystic Fibrosis Foundation Therapeutics, Inc. | Pyrimidine compounds and methods of making and using same |
| ES2453474T3 (en) | 2009-02-06 | 2014-04-07 | Nippon Shinyaku Co., Ltd. | Aminopyrazine derivatives and corresponding medicine |
| JP5747440B2 (en) | 2009-02-06 | 2015-07-15 | 住友化学株式会社 | Hydrazide compound and its use for pest control |
| US20120121515A1 (en) | 2009-03-13 | 2012-05-17 | Lenny Dang | Methods and compositions for cell-proliferation-related disorders |
| WO2010120994A2 (en) | 2009-04-17 | 2010-10-21 | Wyeth Llc | Ureidoaryl-and carbamoylaryl-morpholino- pyrimidine compounds, their use as mtor kinase and pi3 kinase inhibitors, and their synthesis |
| PL2432767T3 (en) | 2009-05-19 | 2013-11-29 | Dow Agrosciences Llc | Compounds and methods for controlling fungi |
| EP2451802A1 (en) | 2009-07-07 | 2012-05-16 | Pathway Therapeutics, Inc. | Pyrimidinyl and 1,3,5-triazinyl benzimidazoles and their use in cancer therapy |
| TWI393716B (en) | 2009-08-04 | 2013-04-21 | Merck Sharp & Dohme | Heterocyclic compounds as factor ixa inhibitors |
| AR077999A1 (en) | 2009-09-02 | 2011-10-05 | Vifor Int Ag | ANTIGONISTS OF PYRIMIDIN AND TRIAZIN-HEPCIDINE |
| NZ598808A (en) | 2009-09-09 | 2014-07-25 | Celgene Avilomics Res Inc | Pi3 kinase inhibitors and uses thereof |
| EP2509600B1 (en) | 2009-12-09 | 2017-08-02 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds for use in the treatment of cancer characterized as having an idh mutation |
| GB201004200D0 (en) | 2010-03-15 | 2010-04-28 | Univ Basel | Spirocyclic compounds and their use as therapeutic agents and diagnostic probes |
| AR081626A1 (en) | 2010-04-23 | 2012-10-10 | Cytokinetics Inc | AMINO-PYRIDAZINIC COMPOUNDS, PHARMACEUTICAL COMPOSITIONS THAT CONTAIN THEM AND USE OF THE SAME TO TREAT CARDIAC AND SKELETIC MUSCULAR DISORDERS |
| US9133123B2 (en) | 2010-04-23 | 2015-09-15 | Cytokinetics, Inc. | Certain amino-pyridines and amino-triazines, compositions thereof, and methods for their use |
| AR081331A1 (en) | 2010-04-23 | 2012-08-08 | Cytokinetics Inc | AMINO- PYRIMIDINES COMPOSITIONS OF THE SAME AND METHODS FOR THE USE OF THE SAME |
| WO2011143160A2 (en) | 2010-05-10 | 2011-11-17 | The Johns Hopkins University | Metabolic inhibitor against tumors having an idh mutation |
| CA2805669C (en) | 2010-07-16 | 2018-08-21 | Agios Pharmaceuticals, Inc. | Therapeutically active compositions and their methods of use |
| MX354482B (en) | 2010-10-01 | 2018-03-07 | Novartis Ag Star | Manufacturing process for pyrimidine derivatives. |
| JP5856177B2 (en) | 2010-10-18 | 2016-02-09 | セレニス セラピューティクス ホールディング エスアー | Compounds, compositions and methods useful for cholesterol mobilization |
| GB201106829D0 (en) | 2011-04-21 | 2011-06-01 | Proximagen Ltd | Heterocyclic compounds |
| RU2013141559A (en) | 2011-02-11 | 2015-03-20 | Дана-Фарбер Кэнсер Инститьют, Инк. | METHOD FOR INHIBITING HAMARTOMA TUMOR CELL CELLS |
| CN102827073A (en) | 2011-06-17 | 2012-12-19 | 安吉奥斯医药品有限公司 | Therapeutically active compositions and application methods thereof |
| EP2750670A1 (en) | 2011-09-01 | 2014-07-09 | Novartis AG | Pi3k inhibitor for use in the treatment of bone cancer or for preventing metastatic dissemination primary cancer cells into the bone |
| CU24269B1 (en) | 2011-09-27 | 2017-08-08 | Novartis Ag | 3- PIRIMIDIN- 4-IL- OXAZOLIDIN- 2- INHIBITING WAVES OF THE MUTANT HDI |
| CA2851082A1 (en) | 2011-10-06 | 2013-04-11 | Merck Sharp & Dohme Corp. | 1,3-substituted azetidine pde10 inhibitors |
| UY34632A (en) | 2012-02-24 | 2013-05-31 | Novartis Ag | OXAZOLIDIN- 2- ONA COMPOUNDS AND USES OF THE SAME |
| SG11201406550QA (en) * | 2012-05-16 | 2014-11-27 | Novartis Ag | Dosage regimen for a pi-3 kinase inhibitor |
| CA2872541A1 (en) | 2012-06-06 | 2013-12-12 | Novartis Ag | Combination of a 17 -alpha -hydroxylase (c17, 20 - lyase) inhibitor and a specific pi-3k inhibitor for treating a tumor disease |
| MX369518B (en) | 2012-08-16 | 2019-11-11 | Novartis Ag | Combination of pi3k inhibitor and c-met inhibitor. |
| PT2912030T (en) | 2012-10-23 | 2016-11-30 | Novartis Ag | Improved process for manufacturing 5-(2,6-di-4-morpholinyl-4-pyrimidinyl)-4-trifluoromethylpyridin-2-amine |
| US9296733B2 (en) | 2012-11-12 | 2016-03-29 | Novartis Ag | Oxazolidin-2-one-pyrimidine derivative and use thereof for the treatment of conditions, diseases and disorders dependent upon PI3 kinases |
| MX355945B (en) | 2013-03-14 | 2018-05-07 | Novartis Ag | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh. |
| WO2014141153A1 (en) | 2013-03-14 | 2014-09-18 | Novartis Ag | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
| CN103483345B (en) | 2013-09-25 | 2016-07-06 | 中山大学 | PI3K inhibitors of kinases, the pharmaceutical composition comprising it and application thereof |
| CN103694218B (en) | 2013-12-05 | 2016-04-27 | 中山大学 | Pyrimidine compound, PI3K inhibitor, the pharmaceutical composition comprising PI3K inhibitor and application |
-
2013
- 2013-11-01 US US14/069,400 patent/US9296733B2/en active Active
- 2013-11-07 UY UY0001035128A patent/UY35128A/en not_active Application Discontinuation
- 2013-11-08 AR ARP130104103A patent/AR093408A1/en unknown
- 2013-11-10 JO JOP/2013/0323A patent/JO3334B1/en active
- 2013-11-11 LT LTEP13795867.4T patent/LT2922848T/en unknown
- 2013-11-11 AU AU2013343022A patent/AU2013343022B2/en not_active Ceased
- 2013-11-11 TW TW102140929A patent/TWI608005B/en not_active IP Right Cessation
- 2013-11-11 DK DK13795867.4T patent/DK2922848T5/en active
- 2013-11-11 ES ES13795867.4T patent/ES2641820T3/en active Active
- 2013-11-11 PE PE2015000615A patent/PE20151006A1/en active IP Right Grant
- 2013-11-11 EP EP17157570.7A patent/EP3199158B1/en active Active
- 2013-11-11 IN IN2912DEN2015 patent/IN2015DN02912A/en unknown
- 2013-11-11 WO PCT/IB2013/060052 patent/WO2014072956A1/en active Application Filing
- 2013-11-11 MY MYPI2015701443A patent/MY174239A/en unknown
- 2013-11-11 KR KR1020157011951A patent/KR102153763B1/en active IP Right Grant
- 2013-11-11 MX MX2015005914A patent/MX363436B/en unknown
- 2013-11-11 CU CU2015000050A patent/CU24365B1/en unknown
- 2013-11-11 CA CA2890942A patent/CA2890942C/en not_active Expired - Fee Related
- 2013-11-11 CN CN201380052670.1A patent/CN104718204B/en not_active Expired - Fee Related
- 2013-11-11 MA MA38076A patent/MA38076A1/en unknown
- 2013-11-11 EA EA201790289A patent/EA029714B1/en not_active IP Right Cessation
- 2013-11-11 BR BR112015010136A patent/BR112015010136A2/en not_active Application Discontinuation
- 2013-11-11 AP AP2015008347A patent/AP2015008347A0/en unknown
- 2013-11-11 JP JP2015541289A patent/JP6262245B2/en not_active Expired - Fee Related
- 2013-11-11 SI SI201330782T patent/SI2922848T1/en unknown
- 2013-11-11 EA EA201590929A patent/EA027987B1/en not_active IP Right Cessation
- 2013-11-11 HU HUE13795867A patent/HUE034492T2/en unknown
- 2013-11-11 EP EP13795867.4A patent/EP2922848B1/en active Active
- 2013-11-11 RS RS20170882A patent/RS56336B1/en unknown
- 2013-11-11 PL PL13795867T patent/PL2922848T3/en unknown
- 2013-11-11 SG SG11201503447QA patent/SG11201503447QA/en unknown
- 2013-11-11 NZ NZ706591A patent/NZ706591A/en not_active IP Right Cessation
- 2013-11-11 UA UAA201502809A patent/UA114001C2/en unknown
- 2013-11-11 PT PT137958674T patent/PT2922848T/en unknown
-
2015
- 2015-03-27 TN TNP2015000120A patent/TN2015000120A1/en unknown
- 2015-03-30 ZA ZA2015/02175A patent/ZA201502175B/en unknown
- 2015-04-12 IL IL238239A patent/IL238239A/en not_active IP Right Cessation
- 2015-05-06 CL CL2015001202A patent/CL2015001202A1/en unknown
- 2015-05-12 CR CR20150248A patent/CR20150248A/en unknown
- 2015-05-12 PH PH12015501053A patent/PH12015501053B1/en unknown
- 2015-05-12 CO CO15107787A patent/CO7380758A2/en unknown
- 2015-06-12 EC ECIEPI201525093A patent/ECSP15025093A/en unknown
- 2015-10-06 HK HK15109748.9A patent/HK1209108A1/en not_active IP Right Cessation
-
2016
- 2016-02-18 US US15/047,574 patent/US10202371B2/en active Active
-
2017
- 2017-09-20 HR HRP20171425TT patent/HRP20171425T1/en unknown
- 2017-09-27 CY CY20171101014T patent/CY1119391T1/en unknown
-
2018
- 2018-12-18 US US16/224,212 patent/US20190382399A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004078163A2 (en) | 2003-02-28 | 2004-09-16 | Transform Pharmaceuticals, Inc. | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2007084786A1 (en) | 2006-01-20 | 2007-07-26 | Novartis Ag | Pyrimidine derivatives used as pi-3 kinase inhibitors |
Non-Patent Citations (22)
| Title |
|---|
| "Beilsteins Handbuch der organischen Chemie", SPRINGER-VERLAG, BERLIN |
| "Remington's Pharmaceutical Sciences", 1985, MACK PUBLISHING COMPANY |
| "Remington's Pharmaceutical Sciences", 1990, MACK PRINTING COMPANY, pages: 1289 - 1329 |
| BAYASCAS; ALESSI, MOL CELL, vol. 18, no. 2, 2005, pages 143 - 145 |
| CHEN ET AL., BR J DERMATOL, vol. 160, no. 2, 2009, pages 442 - 445 |
| DARIDO ET AL., CANCER CELL, vol. 20, no. 5, 2011, pages 635 - 648 |
| HOUBEN-WEYL: "Methods of Organic Synthesis", vol. 21, 1952, THIEME |
| HUANG ET AL., SCIENCE, vol. 318, 2007, pages 1744 - 1748 |
| KENIRY; PARSONS, ONCOGENE, vol. 27, 2008, pages 5477 - 5485 |
| KWIATKOWSKI, D. J.; ZHANG, H.; BANDURA, J. L.; HEIBERGER, K. M.; GLOGAUER, M.; EL-HASHEMITE, N.; ONDA, H., HUM. MOL. GENET., vol. 11, 2002, pages 525 - 534 |
| LIU ET AL., NAT REV DRUG DISCOV, vol. 8, 2009, pages 627 - 644 |
| LOUIS F. FIESER; MARY FIESER: "Reagents for Organic Synthesis", vol. 1-19, 1967, WILEY |
| MAIRA SM; PECCHI S; BRUEGGEN J; HUH K; SCHNELL C; FRITSCH C; NAGEL T; WIESMANN M; BRACHMANN S; DORSCH M, MOL. CANCER THER., 2011 |
| MAIRA SM; STAUFFER F; BRUEGGEN J; FURET P; SCHNELL C; FRITSCH C; BRACHMANN S; CHENE P; DE POVER A; SCHOEMAKER K, MOL CANCER THER., vol. 7, 2008, pages 1851 - 63 |
| MING ET AL., ONCOGENE, vol. 29, no. 4, 2010, pages 492 - 502 |
| SALASCHE, J AM ACAD DERMATOL, vol. 42, 2000, pages 4 - 7 |
| SARBASSOV ET AL., SCIENCE, vol. 307, 2005, pages 1098 - 1101 |
| STAHL; WERMUTH: "Handbook of Pharmaceutical Salts: Properties, Selection, and Use", 2002, WILEY-VCH |
| T. W. GREENE: "Protective Groups in Organic Synthesis", 1991, JOHN WILEY & SONS |
| VANHAESEBROECK ET AL., ANNU. REV. BIOCHEM, vol. 70, 2001, pages 535 |
| WEE ET AL., PORC NATL ACAD SCI USA, vol. 105, 2008, pages 13057 - 13062 |
| ZHAO; VOGT, ONCOGENE, vol. 27, 2008, pages 5486 - 5496 |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11918586B2 (en) | 2016-05-18 | 2024-03-05 | Torqur Ag | Treatment of skin lesions |
| WO2019087158A2 (en) | 2017-11-06 | 2019-05-09 | Novartis Ag | Formulation comprising oxazolidin-2-one-pyrimidine derivative |
| WO2019087158A3 (en) * | 2017-11-06 | 2019-06-20 | Novartis Ag | Formulation comprising oxazolidin-2-one-pyrimidine derivative |
| WO2022053651A2 (en) | 2020-09-10 | 2022-03-17 | Precirix N.V. | Antibody fragment against fap |
| WO2023203135A1 (en) | 2022-04-22 | 2023-10-26 | Precirix N.V. | Improved radiolabelled antibody |
| WO2023213801A1 (en) | 2022-05-02 | 2023-11-09 | Precirix N.V. | Pre-targeting |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10202371B2 (en) | Oxazolidin-2-one-pyrimidine derivatives and the use thereof as phosphatidylinositol-3-kinase inhibitors | |
| US9458177B2 (en) | Oxazolidin-2-one compounds and uses thereof | |
| JP6181173B2 (en) | Selective PI3K delta inhibitor | |
| CA3077383A1 (en) | Substituted pyrimidine piperazine compound and use thereof | |
| CN113773315A (en) | Highly selective deuterated inhibitors of cyclin dependent kinase 2(CDK2) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 12015501053 Country of ref document: PH |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13795867 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 238239 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 2013343022 Country of ref document: AU Date of ref document: 20131111 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20157011951 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 38076 Country of ref document: MA |
|
| ENP | Entry into the national phase |
Ref document number: 2890942 Country of ref document: CA Ref document number: 2015541289 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 000615-2015 Country of ref document: PE Ref document number: MX/A/2015/005914 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15107787 Country of ref document: CO Ref document number: CR2015-000248 Country of ref document: CR |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112015010136 Country of ref document: BR |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013795867 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013795867 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201590929 Country of ref document: EA Ref document number: IDP00201503496 Country of ref document: ID |
|
| ENP | Entry into the national phase |
Ref document number: 112015010136 Country of ref document: BR Kind code of ref document: A2 Effective date: 20150505 |